Область техники, к которой относится изобретение
Настоящее изобретение относится к усилителю действия противоопухолевого средства и противоопухолевому лекарственному препарату, содержащему его.
Предпосылки создания изобретения
упоминается как dUTPase (ЕС3.6.1.23)) является ферментом, предотвращающим восстановление ДНК. Указанный фермент специфически Трифосфат дезоксиуридина (далее в данном описании узнает только трифосфат дезоксиуридина, отличая от других трифосфатов природных нуклеиновых кислот, и гидролизует трифосфат дезоксиуридина до монофосфата дезоксиуридина и пирофосфорной кислоты. Известно, что dUTPase является незаменимым ферментом для выживания как прокариотных, так и эукариотных клеток.
Полагают, что в злокачественных опухолях злокачественность ассоциируется с высоким уровнем экспрессии dUTPase (непатентные документы 1 и 2). Также сообщается, что опухоль, в которой экспрессия фермента ускорена, показывает устойчивость к химиотерапии (непатентный документ 3). Более того, наблюдают потенцирование противоопухолевого действия ингибитора тимидилатсинтазы (далее в данном описании упоминается как ингибитор TS), когда уровень экспрессии dUTPase в культивированных раковых клетках снижают с использованием сиРНК (непатентный документ 4). Такие результаты предполагают, что ингибиторы dUTPase человека могут быть применимыми химическими сенсибилизаторами противоопухолевых средств.
Документы известного уровня техники - непатентные документы
Непатентный документ 1: J. Clin. Pathol., 2009, Apr, 62(4): 364-9
Непатентный документ 2: Int. J. Cancer., 1999, Dec 22, 84(6): 614-7
Непатентный документ 3: Cancer Res., 2000, Jul 1, 60(13): 3493-503
Непатентный документ 4: Mol. Pharmacol., 2004, Sep, 66(3): 620-6
Сущность изобретения
Решаемые проблемы
Однако отсутствуют сообщения о том, что низкомолекулярный ингибитор dUTPase человека действительно обнаруживает потенциирующее действие на противоопухолевое действие.
Целью настоящего изобретения является усилитель противоопухолевого действия противоопухолевого средства и содержащее его противоопухолевый лекарственный препарат.
Способы решения проблемы
В результате интенсивных исследований, направленных на достижение вышеуказанной цели, авторы настоящего изобретения обнаружили, что производное урацила с сульфонамидной структурой в положении N-1 урацилового цикла, представленное приведенной ниже формулой (I), или его соль показывают сильное ингибирующее действие в отношении dUTPase человека. Авторы также провели дополнительные исследования и в результате нашли, что вышеуказанное производное урацила или его соль показывают превосходную активность усиления эффективности противоопухолевого средства (в частности, антиметаболита), в связи с этим осуществлено настоящее изобретение.
Настоящее изобретение относится к усилителю противоопухолевого действия противоопухолевого средства, содержащего в качестве активного ингредиента производное урацила, представленное приведенной далее формулой (I)
Формула 1

где Х представляет собой С1-5-алкиленовую группу, и одна из метиленовых групп, составляющих алкиленовую группу, необязательно заменена атомом кислорода;
R1 представляет собой атом водорода или С1-6-алкильную группу; R2 представляет собой атом водорода или атом галогена; и R3 представляет собой С1-6-алкильную группу, С2-6-алкенильную группу, С3-6-циклоалкильную группу, (С3-6-циклоалкил)-С1-6-алкильную группу, галоген-С1-6-алкильную группу или насыщенную гетероциклическую группу,
или его фармацевтически приемлемую соль.
Кроме того, настоящее изобретение относится к противоопухолевому лекарственному препарату включающему комбинацию производного урацила, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли и противоопухолевого средства.
Кроме того, настоящее изобретение относится к производному урацила, представленному приведенной выше формулой (I), или его фармацевтически приемлемой соли для применения при потенцировании противоопухолевого действия.
Кроме того, настоящее изобретение относится к комбинации соединения, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли и противоопухолевого средства для применения при лечении опухолей.
Далее, настоящее изобретение относится к способу усиления противоопухолевого действия, который включает введение эффективного количества соединения, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли.
Далее, настоящее изобретение относится к способу лечения опухолей, который включает введение комбинации соединения, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли и противоопухолевого средства.
Кроме того, настоящее изобретение относится к применению соединения, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли для получения усилителя противоопухолевого действия.
Кроме того, настоящее изобретение относится к применению комбинации соединения, представленного приведенной выше формулой (I), или его фармацевтически приемлемой соли и противоопухолевого средства для получения противоопухолевого средства.
Действие изобретения
Новое производное урацила по настоящему изобретению или его фармацевтически приемлемая соль применимы в качестве усилителя противоопухолевого действия противоопухолевого средства (в частности, антиметаболита) и в содержащем его противоопухолевом лекарственном препарате.
Краткое описание чертежей
Фиг.1 представляет собой диаграмму, показывающую усиливающее действие на противоопухолевое действие TS-1.
Фиг.2 представляет собой диаграмму, показывающую усиливающее действие на противоопухолевое действие TS-1.
Фиг.3 представляет собой диаграмму, показывающую усиливающее действие на противоопухолевое действие 5-FU.
Фиг.4 представляет собой диаграмму, показывающую усиливающее действие на противоопухолевое действие капецитабина, FdUrd, пеметрекседа и UFT.
Фиг.5 представляет собой графики, показывающие на голых мышах, которым трансплантирована линия SC-6 клеток рака желудка человека, изменение массы тела и противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и TS-1.
Фиг.6 представляет собой графики, показывающие на голых мышах, которым трансплантирована линия LS174T клеток рака толстой кишки человека, изменение массы тела и противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и TS-1.
Фиг.7 представляет собой графики, показывающие на голых мышах, которым трансплантирована линия CFPAC-1 клеток рака поджелудочной железы человека, изменение массы тела и противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и TS-1.
Фиг.8 представляет собой диаграмму, показывающую на голых мышах, которым трансплантирована линия МХ-1 клеток рака молочной железы человека, противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и TS-1.
Фиг.9 представляет собой график, показывающий на крысах, которым трансплантирована линия МХ-1 клеток рака молочной железы человека, противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и TS-1.
Фиг.10 представляет собой графики, показывающие на голых мышах, которым трансплантирована линия CFPAC-1 клеток рака поджелудочной железы человека, противоопухолевое действие в случае введения комбинации соединения по настоящему изобретению и капецитабина.
Подробное описание изобретения
В формуле (I) «С1-5-алкиленовая группа», представленная Х, обозначает линейную или разветвленную алкиленовую группу с 1-5 атомами углерода. Конкретные примеры включают метиленовую группу, этиленовую группу, триметиленовую группу, тетраметиленовую группу, пентаметиленовую группу, пропиленовую группу, бутиленовую группу, диметилтриметиленовую группу и этилтриметиленовую группу. Примером С1-5-алкиленовой группы, в которой одна из метиленовых групп, составляющих алкиленовую группу, заменена атомом кислорода, является -О-С1-4-алкиленовая группа.
Х представляет собой предпочтительно этиленовую группу или -О-СН2СН2СН2-.
В формуле (I) «С1-6-алкильная группа», представленная R1, обозначает линейную или разветвленную углеводородную группу с 1-6 атомами углерода. Конкретные примеры включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу и н-гексильную группу. Предпочтительной является С1-3-алкильная группа, и более предпочтительными являются метильная группа и этильная группа.
В формуле (I) примеры «атома галогена», представленного R2, включают атом фтора, атом хлора, атом брома и атом йода. Из них предпочтительным является атом фтора.
В формуле (I) «С1-6-алкильная группа», представленная R3, включает те же группы, какие описаны выше для R1. Предпочтительными являются изобутильная группа и 2-метибутильная группа.
В формуле (I) «С2-6-алкенильная группа», представленная R3, обозначает углеводородную группу с 2-6 атомами углерода, которая содержит углерод-углеродную двойную связь. Ее примеры включают винильную группу, аллильную группу, метилвинильную группу, пропенильную группу, бутенильную группу, пентенильную группу и гексенильную группу. Из них предпочтительной является аллильная группа.
В формуле (I) примеры «С3-6-циклоалкильной группа», представленной R3, включают циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу. Из них предпочтительной является циклопентильная группа.
В формуле (I) «(С3-6-циклоалкил)С1-6-алкильная группа», представленная R3, обозначает алкильную группу с 1-6 атомами углерода, которая имеет описанную выше циклоалкильную группу. Предпочтительной является циклопропилметильная группа.
В формуле (I) «галоген-С1-6-алкильная группа», представленная R3, обозначает алкильную группу с 1-6 атомами углерода, которая имеет описанный выше атом галогена. Предпочтительными являются 2,2-дифторэтильная группа и 2,2,2-трифторэтильная группа.
В формуле (I) «насыщенная гетероциклическая группа», представленная R3, предпочтительно обозначает моноциклическую или бициклическую насыщенную гетероциклическую группу, предпочтительно, с одним или двумя атомами, выбранными из атомов кислорода, азота и серы. Ее примеры включают пирролидинильную группу, пиперидинильную группу, пиперазинильную группу, гексаметилениминогруппу, морфолиногруппу, тиоморфолиногруппу, гомопиперидинильную группу, тетрагидрофурильную группу и тетрагидропирильную группу. Из них предпочтительными являются тетрагидрофурильная группа и тетрагидропирильная группа.
Группа, представленная R3, предпочтительно представляет собой изобутильную группу, 2-метилбутильную группу, аллильную группу, циклопентильную группу, циклопропилметильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, тетрагидрофурильную группу или тетрагидропирильную группу.
Наиболее предпочтительно, когда в формуле (I) Х представляет собой этиленовую группу или -О-С1-4-алкиленовую группу; R1 представляет собой атом водорода или С1-3-алкильную группу; R2 представляет собой атом водорода или атом фтора; и R3 представляет собой С1-6-алкильную группу, С2-6-алкенильную группу, С3-6-циклоалкильную группу, (С3-6-циклоалкил)С1-6-алкильную группу, галоген-С1-6-алкильную группу или насыщенную гетероциклическую группу.
Кроме того, также предпочтительно, когда в формуле (I) Х представляет собой этиленовую группу или -О-С1-4-алкиленовую группу; R1 представляет собой атом водорода или С1-3-алкильную группу; R2 представляет собой атом водорода или атом фтора; и R3 представляет собой С1-6-алкильную группу, С2-6-алкенильную группу, С3-6-циклоалкилалкильную группу, (С3-6-циклоалкил)С1-6-алкильную группу, галоген-С1-6-алкильную группу или тетрагидрофурильную группу или тетрагидропирильную группу.
Более того, особенно предпочтительно, когда в формуле (I) Х представляет собой этиленовую группу или -О-СН2СН2СН2-; R1 представляет собой атом водорода, метильную группу или этильную группу; R2 представляет собой атом водорода или атом фтора; и R3 представляет собой изобутильную группу, 2-метилбутильную группу, аллильную группу, циклопентильную группу, циклопропилметильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, тетрагидрофурильную группу или тетрагидропирильную группу.
Примеры фармацевтически приемлемой соли соединения, представленного формулой (I), включают соли присоединения кислот, таких как хлороводородная кислота, бромоводородная кислота, йодоводородная кислота, серная кислота, азотная кислота или фосфорная кислота, или органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, угольная кислота, пикриновая кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота или глутаминовая кислота; соли с неорганическими основаниями, такими как натрий, калий, магний, кальций или алюминий, или с органическими основаниями, такими как метиламин, этиламин, меглумин или этаноламин, или с основными аминокислотами, такими как лизин, аргинин или орнитин; и аммониевые соли. Кроме того, соединение по настоящему изобретению включает оптический изомер и гидрат(ы).
Производное урацила по настоящему изобретению можно получить согласно реакционным стадиям, описанным далее.
Стадия А
Формула 2

где R3 имеет значения, указанные выше; и Lg представляет собой удаляемую группу, такую как атом галогена, метансульфонилоксигруппа, п-толуолсульфонилоксигруппа или трифторметансульфонилоксигруппа.
[A-1]
(а) На данной стадии коммерчески доступный 3-цианофенол (1) в присутствии основания можно ввести во взаимодействие с алкилгалогенидом, алкилмезилатом, алкилтозилатом, алкилтрифторметансульфонатом, представленными общей формулой (2), и получить соединение, представленной общей формулой (4).
Может быть использован без ограничения любой растворитель для реакционной смеси, который не влияет на реакцию. Примеры растворителя включают диэтиловый эфир, тетрагидрофуран (далее в данном описании упоминаемый как ТГФ), диоксан, ацетон, диметоксиэтан, ацетонитрил, N,N-диметилформамид (далее в данном описании упоминаемый как ДМФА), N,N-диметилацет амид (далее в данном описании упоминаемый как DMA) и диметилсульфоксид (далее в данном описании упоминаемый как ДМСО). Из них предпочтительным является ДМФА.
Примеры основания, используемого в данном случае, включают: неорганические основания, такие как бикарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия, гидрид калия, гидроксид натрия или гидроксид калия; и органические основания, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, лутидин или коллидин. Из них предпочтительным является карбонат калия. Его эквивалентное количество составляет 0,8-10 эквивалентов, и предпочтительно, 1,0-5,0 эквивалентов.
Эквивалентное количество соединения общей формулы (2) составляет 0,8-10 эквивалентов, и предпочтительно, 1,0-5,0 эквивалентов. Температура реакции составляет 20-150°С, и предпочтительно, 50-130°С. Время реакции составляет 0,5-24 часа, и предпочтительно, 1,0-12 часов.
(b) На данной стадии коммерчески доступный 3-цианофенол (1) и спирт, представленный общей формулой (3), можно ввести в реакцию конденсации по Мицунобу и получить соединение, представленное общей формулой (4).
Может быть использован без ограничения любой растворитель для реакционной смеси, который не влияет на реакцию. Примеры растворителя включают дихлорметан, 1,2-дихлорэтан (далее в данном описании упоминаемый как DCE), бензол, ксилол, толуол, этилацетат, пропилацетат, бутилацетат, диэтиловый эфир, ТГФ, диоксан, ацетон, диметоксиэтан, ацетонитрил и ДМФА. Их них предпочтительным является ТГФ.
Любой реагент, который обычно можно использовать в реакции Мицунобу, можно использовать в данном взаимодействии без ограничения. Его примеры включают комбинации ди(низший алкил)азодикарбоксилата (например, диэтилазодикарбоксилата (далее в данном описании упоминаемого как DEAD) или диизопропилазодикарбоксилата (далее в данном описании упоминаемого как DIAD)) или азосоединения (например, азодикарбонила, такого как 1,1-(азодикарбонил)дипепиридин) с триарилфосфином (например, трифенилфосфином) или три(низший алкил)фосфином (например, три-н-бутилфосфином). Предпочтительным является комбинация DEAD с трифенилфосфином.
Эквивалентные количества спирта общей формулы (3), ди(низший алкил)азодикарбоксилата и триарилфосфина составляют, соответственно, 0,8-5,0 эквивалентов, и предпочтительно, 1,0-2,0 эквивалента. Температура реакции составляет от -20°С до 120°С, и предпочтительно, 0-60°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,2-6,0 часов.
[A-2]
На данной стадии цианосоединение, представленное общей формулой (4), можно ввести во взаимодействие с обычным известным восстановителем и получить соединение, представленное общей формулой (5).
Растворитель для реакции различается в зависимости от типа восстановителя. Его примеры включают метанол, этанол, 1-пропанол, 2-пропанол, трет-бутиловый спирт, диметоксиэтан, диметиловый эфир диэтиленгликоля, диизопропиловый эфир, диэтиловый эфир, ТГФ и диоксан. Из них предпочтительным является ТГФ.
Примеры восстановителя, используемого в данном случае, включают гидриды металлов, такие как алюмогидрид лития (далее в данном описании упоминаемый как LAH), диэтоксиалюмогидрид лития, триэтоксиалюмогидрид лития, трет-бутоксиалюмогидрид лития, алюмогидрид магния, гидрид алюминия с хлоридом магния, алюмогидрид натрия, триэтоксиалюмогидрид натрия или бис(2-метоксиэтокси)алюмогидрид натрия; и катализаторы, используемые для гидрирования, такие как палладий-на-угле, гидроксид палладия или платина. Из них предпочтительным является LAH. Эквивалентное количество восстановителя составляет 0,5-5,0 эквивалентов, и предпочтительно, 0,8-2,0 эквивалента. Температура реакции составляет 0°С-100°С, и предпочтительно, 20-60°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,2-6,0 часов.
Стадия В
Формула 3

где R1, R2, R3 и Lg имеют значения, указанные выше; Rc представляет собой С1-6-алкильную группу; и Hal представляет собой атом галогена.
[B-1]
На данной стадии карбоксильную группу легко доступного соединения (6) этерифицируют спиртом (7) обычно известным способом, и затем полученное соединение можно ввести во взаимодействие таким же способом, как на стадии [A-1] и получить соединение, представленное общей формулой (8).
[B-2]
На данной стадии соединение, представленное общей формулой (8), можно ввести во взаимодействие с обычно известным восстановителем и получить соединение, представленное общей формулой (9).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают диэтиловый эфир, диизопропиловый эфир, ТГФ и диоксан. Из них предпочтительным является ТГФ.
Примеры восстановителя, используемого в данном случае, включают LAH, диэтоксиалюмогидрид лития, триэтоксиалюмогидрид лития, три-трет-бутоксиалюмогидрид лития, алюмогидрид магния, алюмогидрид с хлоридом магния, алюмогидрид натрия, триэтоксиалюмогидрид натрия, бис(2-метоксиэтокси)алюмогидрид натрия, диизобутилалюмогидрид (далее в данном описании упоминаемый как DIBAL) и борогидрид лития. Из них предпочтительным является борогидрид лития. Эквивалентное количество восстановителя составляет 0,8-10 эквивалентов, и предпочтительно, 1,0-5,0 эквивалента. Температура реакции составляет от 0°С до температуры кипения растворителя, и предпочтительно, температуру кипения растворителя. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,5-12 часов.
[B-3]
На данной стадии соединение, представленное общей формулой (9), можно ввести во взаимодействие с обычно известным окислителем и получить альдегид, представленный общей формулой (10).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают дихлорметан, хлороформ, четыреххлористый углерод, DCE, хлорбензол, толуол и ксилол. Из них предпочтительным является дихлорметан.
Примеры окислителя, используемого в данном случае, включают комплес хромового ангидрида, пиридина и уксусного ангидрида; окислители на основе хрома, такие как хлорхромат пиридиния или дихромат пиридиния; гипервалентные йодные окислители, такие как реагент Десс-Мартина; окислители на основе ДМСО, такие как ДМСО, используемый в комбинации с уксусным ангидридом, оксалилхлоридом, дициклогексилкарбодиимидом (далее в данном описании упоминаемым как DCC) или гидрохлоридом 1-этил-3-(3-диметиламинопропил)карбодиимида (далее в данном описании упоминаемым как EDC-HCl); оксид марганца(IV) и радикалы 2,2,6,6-тетраметипиперидинил-1-оксилы. Из них предпочтительным является оксид марганца(IV). Эквивалентное количество окислителя составляет 0,8-30 эквивалентов, и предпочтительно, 1,0-20 эквивалента. Температура реакции составляет от -20 до 150°С, и предпочтительно, 0-100°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,5-12 часов.
Когда R2 представляет собой атом водорода, коммерчески доступный 3-гидроксибензальдегид можно ввести во взаимодействие как исходный материал таким же путем, как на стадии [A-1], и получить соединение, представленное общей формулой (10). Кроме того, нитрил, представленный общей формулой (4), также можно восстановить обычной известной реакцией восстановления, например, способом восстановления с DIBAL, и получить соединение, представленное общей формулой (10).
[B-4]
На данной стадии соединение, представленное общей формулой (10), или коммерчески доступный альдегид можно ввести во взаимодействие с коммерчески доступным 2-метил-2-пропансульфинамидом в кислой среде и получить соединение, представленное приведенной выше общей формулой (11).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают диэтиловый эфир, диизопропиловый эфир, ТГФ, диоксан, дихлорметан, хлороформ, четыреххлористый углерод, толуол и ксилол. Из них предпочтительным является толуол.
Примеры кислоты, используемой в данном случае, включают хлороводородную кислоту, серную кислоту, п-толуолсульфоновую кислоту и кислоту Льюиса (например, тетраизопропоксид титана или тетраэтоксид титана). Из них предпочтительным является тетраизопропоксид титана. Эквивалентные количества 2-метил-2-пропансульфинамида и тетраизопропоксида титана составляют, соответственно, 0,8-10 эквивалентов, и предпочтительно, 1,0-3,0 эквивалента. Температура реакции составляет 20-150°С, и предпочтительно, 50-120°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,5-6,0 часов.
[B-5]
На данной стадии соединение, представленное общей формулой (11), можно ввести во взаимодействие с реагентом Гриньяра (12), представленным формулой R1MgHal, или литийорганическим реагентом (13), представленным формулой R1Li, и получить диастереоселективно соединение, представленное общей формулой (14).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, циклопентилметиловый эфир, ТГФ, диметоксиэтан, диоксан, дихлорметан, хлороформ, четыреххлористый углерод, толуол и ксилол. Эквивалентное количество реагента Гриньяра или литийорганического реагента составляет 0,8-20 эквивалентов, и предпочтительно, 1,0-10 эквивалентов. Температура реакции составляет от -100°С до 100°С, и предпочтительно, от -78°С до 50°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,5-12 часов.
[B-6]
На данной стадии соединение, представленное общей формулой (14), можно ввести во взаимодействие с кислотой, и получить соединение, представленное общей формулой (15).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают спирты, такие как метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол или 2-бутанол; диоксан и этилацетат. Из них предпочтительным является метанол.
Примеры кислоты, используемой в данном случае, включают хлороводородную кислоту, серную кислоту и фосфорную кислоту. Из них предпочтительной является хлороводородная кислота. Ее эквивалентное количество составляет 0,1-10 эквивалентов, и предпочтительно, 1,0-2,0 эквивалента. Температура реакции составляет от -20°С до 100°С, и предпочтительно, 0-50°С. Время реакции составляет 0,01-24 часа, и предпочтительно, 0,1-1,0 час.
Кроме того, когда R1 представляет собой атом водорода, и R2 представляет собой атом фтора, соединение, представленное общей формулой (9), можно азидировать обычно известным способом и затем обработать обычно известным восстановителем (например, LAH), и получить соединение, представленное общей формулой (15). Кроме того, в случае, где соединение, представленное общей формулой (15), можно получить в виде рацемата, можно соединение, представленное общей формулой (10), превратить в спирт таким же способом, как на стадии [B-5], затем спирт можно азидировать обычно известным способом, и затем полученный азид можно восстановить обычно известным способом, и получить соединение, представленное общей формулой (15).
Стадия С
Формула 4

где R1, R2 и R3 имеют значения, указанные выше.
[C-1]
На данной стадии легко доступный 3-хлорпропансульфонилхлорид (16) можно в присутствии основания ввести во взаимодействие с любым амином, представленным общей формулой (5) или (15), и получить соединение, представленное общей формулой (17).
Любой растворитель реакционной смеси, который не влияет на реакцию, можно использовать без ограничений. Примеры растворителя включают ацетон, ТГФ, диэтиловый эфир, диизопропиловый эфир, диоксан, дихлорметан, хлороформ, четыреххлористый углерод, ДМФА, DMA и ацетонитрил. Из них предпочтительным является дихлорметан.
Примеры основания, используемого в данном случае, включают неорганические основания, такие как бикарбонат натрия, карбонат натрия или карбонат калия, и органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, лутидин или коллидин. Из них предпочтительным является триэтиламин. Эквивалентные количества основания и амина составляют, соответственно, 0,5-10 эквивалентов, и предпочтительно, 0,7-5,0 эквивалентов. Температура реакции составляет от -20°С до 100°С, и предпочтительно, 0-50°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,2-6,0 час.
[C-2]
На данной стадии хлорсодержащее соединение, представленное общей формулой (17), можно ацетоксилировать через взаимодействие с ацетоксилирующим агентом обычным способом и затем дезацетилировать обычным способом, и получить соединение, представленное общей формулой (18).
[C-3]
На данной стадии соединение, представленное общей формулой (18), можно метоксиметилировать (индуцировать МОМ) обычным способом, затем обработать кислотой Льюиса и затем в присутствии йода ввести во взаимодействие с 2,4-бис(триметилсилилокси)пиримидином, полученным согласно способу, описанному в документе (Nucleosides & Nucleotides, 4, 565-585 (1985)), и получить соединение, представленное общей формулой (19).
При обработке кислотой Льюиса можно использовать без ограничений любой растворитель реакционной смеси, который не влияет на реакцию. Примеры растворителя включают дихлорметан, хлороформ, четыреххлористый углерод, DCE, толуол и ксилол. Из них предпочтительным является дихлорметан. Примеры кислоты Льюиса включают трихлорид бора (далее в данном описании упоминаемый как BCl3), трифторид бора и трибромид бора. Из них предпочтительным является BCl3. Его эквивалентное количество составляет 0,01-10 эквивалентов, и предпочтительно, 0,2-0,5 эквивалентов. Температура реакции составляет от -20°С до 100°С, и предпочтительно, 0-50°С. Время реакции составляет 0,1-24 часа, и предпочтительно, 0,5-5,0 часов.
При взаимодействии с 2,4-бис(триметилсилилокси)пиримидином можно использовать без ограничений любой растворитель реакционной смеси, который не влияет на реакцию. Примеры растворителя включают дихлорметан, хлороформ, четыреххлористый углерод, DCE, толуол и ксилол. Из них предпочтительным является DCE или толуол. Эквивалентное количество 2,4-бис(триметилсилилокси)пиримидина составляет 0,8-10 эквивалентов, и предпочтительно, 0,9-5,0 эквивалентов. Эквивалентное количество йода составляет 0,001-1,0 эквивалент, и предпочтительно, 0,05-0,5 эквивалентов. Температура реакции составляет от 20-150°С, и предпочтительно, 50-100°С. Время реакции составляет 0,1-120 часов, и предпочтительно, 0,5-100 часов.
Стадия D
Формула 5
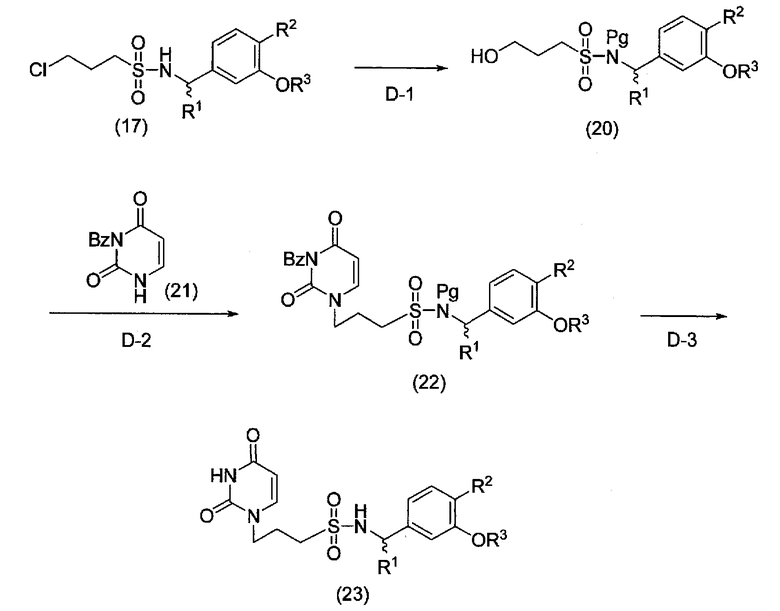
где R1, R2 и R3 имеют значения, указанные выше; Bz представляет собой бензоильную группу; и Pg представляет собой защитную группу для атома азота в сульфонамидной группе.
[D-1]
На данной стадии атом азота в сульфонамидной группе соединения, представленного общей формулой (17), можно обычным способом защитить защитной группой, например, метоксиметильной группой или трет-бутоксикарбонильной группой, и затем полученное соединение можно ввести во взаимодействие со спиртом таким же путем, как на стадии [C-2], и получить соединение, представленное общей формулой (20).
[D-2]
На данной стадии 3-бензоилпиримидин-2,4-(1Н,3Н)-дион (21), полученный согласно способу, описанному в документе (J. Med. Chem., 50, 6032-6038 (2007)), и спирт, представленный общей формулой (20), можно обработать по реакции Мицунобу таким же путем, как на стадии [А-1] (b), и получить соединение, представленное общей формулой (22).
[D-3]
На данной стадии соединение, представленное общей формулой (22), дебензоилируют и удаляют защитную групп Pg обычным способом удаления защитной группы, и получают соединение, представленное общей формулой (23).
Производное урацила, представленное общей формулой (I), обнаруживает сильную активность ингибирования dUTPase человека. Когда производное урацила используют в комбинации с различными противоопухолевыми средствами (далее в данном описании упоминаемых как противоопухолевое средство А), оно показывает активность усиления противоопухолевого действия комбинированного противоопухолевого средства А.
Тип противоопухолевого средства А, эффективность которого усиливается усилителем противоопухолевого действия по настоящему изобретению, особо не ограничивается. Примеры такого противоопухолевого средства А включают алкилирующие агенты, такие как циклофосфамид или нимустин; платинусодержащие средства, такие как цисплатин, карбоплатин или оксалиплатин; антиметаболиты и растительные алкалоиды, такие как паклитаксел, доцетаксел или иринотекан. В качестве противоопухолевого средства А, эффективность которого усиливается усилителем противоопухолевого действия по настоящему изобретению, предпочтителен антиметаболит.
Антиметаболит, используемый в данном случае, обозначает соединение с химической структурой, схожей со структурой природного вещества, используемого для биосинтеза нуклеиновых кислот во время деления клеток и пролиферации раковых клеток, или лекарственное средство, содержащее вышеуказанное соединение в качестве активного фармацевтического ингредиента. Иными словами, антиметаболит обозначает противораковое средство, которое препятствует биосинтезу нуклеиновых кислот или пути биосинтеза нуклеиновых кислот и подавляет пролиферацию раковых клеток. Примеры антиметаболита включают пиримидиновые антиметаболиты, такие как 5-фторурацил (5-FU), тегафур/гимерацил/отерацил калий (TS-1, общее название «смешанное средство тегафур/гимерацил/отерацил калий (известная марка «TS-1»)), тегафур/урацил (UFT; общее название «смешанное средство тегафур/урацил» (известная марка «UFT»)), капецитабин, доксифлуридин, 5-флуоро-2'-дезоксиуридин (FdUrd), гецитабин или цитрабин; пуриновые антиметаболиты, такие как флударабин, кладрибин или неларабин; и антиметаболиты-фолаты, такие как пеметрексед или метотрексат. Из них предпочтительным является ингибитор пути синтеза тимидилата (ТМР). Ингибитор пути синтеза тимидилата обозначает соединение из числа антиметаболитов, которое прямо или косвенно ингибирует фермент, связанный с биосинтезом ТМР, или лекарственное средство, содержащее вышеуказанное соединение в качестве активного фармацевтического ингредиента; при этом ингибитор пути синтеза тимидилата включает, как типичные примеры, ингибитор тимидилатсинтазы и ингибитор дигидрофолатредуктазы. Ингибитор тимидилатсинтазы означает соединение, которое ингибирует тимидилатсинтазу, или лекарственное средство, содержащее вышеуказанное соединение в качестве активного фармацевтического ингредиента. Его примеры включают фторпиримидиновые антиметаболиты, такие как 5-фторурацил (5-FU), тегафур/гимерацил/отерацил калий (TS-1), тегафур/урацил (UFT), капецитабин, доксифлуридин, 5-фтор-2'-дезоксиуридин (FdUrd) или кармофур (ямафул); антиметаболиты-фолаты, такие как пеметрексед, метотрексат или ралтитрексед; и дигидрохлорид нолатрекседа. Кроме того, ингибитор дигидрофолатредуктазы обозначает соединение, которое ингибирует фермент для биосинтеза тетрагидрофолата, существенного для синтеза de novo пуринов, тимидилатов и т.д., или лекарственное средство, содержащее вышеуказанное соединение в качестве активного фармацевтического ингредиента. Его примеры включают антиметаболиты-фолаты, такие как пралатрексат или эдатрексат; пириметамин; бродимоприм и глюкуронат триметатрексата.
Противоопухолевое средство А, действие которого усиливается усилителем противоопухолевого действия по настоящему изобретению, предпочтительнее представляет собой ингибитор тимидилатсинтазы. Среди таких ингибиторов тимидилатсинтазы особенно предпочтительными являются фторурацил (5-FU), тегафур/гимерацил/отерацил калий (TS-1), тегафур/урацил (UFT), капецитабин, 5-фтор-2'-дезоксиуридин (FdUrd) и пеметрексед.
Тип злокачественной опухоли, которую можно лечить комбинацией соединения по настоящему изобретению, как усилителя противоопухолевого действия, и противоопухолевого средства А, эффективность которого усиливается усилителем противоопухолевого действия по настоящему изобретению, особо не ограничивается. Примеры злокачественной опухоли включают рак головы и шеи, эзофагеальный рак, рак желудка, рак толстой кишки, ректальный рак, рак печени, рак желчного пузыря/рак желчных протоков, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, эндометриальный рак, рак почек, рак мочевого пузыря, рак предстательной железы, рак яичка, остеосаркому/саркому мягких тканей, лейкоз, злокачественную лимфому, множественную миелому, рак кожи и опухоль головного мозга.
Противоопухолевый лекарственный препарат с усиленной эффективностью можно получить, объединяя производное урацила формулы (I) или его соль и противоопухолевое средство А. Форма полученного таким образом нового противоопухолевого лекарственного препарата может быть или формой препарата типа единого средства, содержащего производное урацила формулы (I) или его соль и противоопухолевое средство А, или формой отдельных препаратов, которая состоит из препарата, содержащего производное урацила формулы (I) или его соль, и препарата, содержащего противоопухолевое средство А. Кроме того, путь введения композиции, содержащей производное урацила формулы (I), может быть одинаковым с или отличаться от пути для композиции, содержащей противоопухолевое средство А (например, пероральное введение и инъекция).
Противоопухолевое средство А и производное урацила по настоящему изобретению можно получить в виде набора. Отдельные композиции, составляющие набор, могут представлять собой любую известную препаративную форму. Как правило, такие отдельные композиции могут содержаться в обычно используемых контейнерах различного типа, в зависимости от их препаративной формы, с тем, чтобы получить набор для лечения онкозаболеваний у млекопитающих, в том числе людей.
Когда производное урацила по настоящему изобретению или его фармацевтически приемлемая соль содержатся в фармацевтической композиции, они могут быть, при необходимости, смешаны с фармацевтически приемлемым носителем, и могут быть получены в виде форм для введения любых типов, в зависимости от профилактической или лечебной цели. Примеры такой формы включают пероральное средство, инъекцию, супппозиторий, мазь и пэтч. Из них предпочтительным является пероральное средство. Такие формы для введения можно получить способами, обычно используемыми для получения лекарственных средств, которые известны специалистам в данной области техники.
Различные типы органических и неорганических веществ-носителей, которые обычно используют в качестве материалов для фармацевтических препаратов, могут быть использованы в качестве фармацевтически приемлемого носителя. Такой фармацевтически приемлемый носитель может быть введен в смесь в твердых препаратах как разбавитель, связующее вещество, вещество, способствующее рассыпанию, смазывающее вещество и краситель; и в жидких препаратах как растворитель, солюбилизатор, суспендирующее средство, средство для регулирования тоничности, буфер и успокаивающее средство. Кроме того, при необходимости также можно использовать такие фармацевтические добавки, как антисептик, антиоксидант, краситель, подслащивающее вещество и стабилизатор.
Когда получают твердый препарат для перорального введения, к соединению по настоящему изобретению могут быть добавлены, при необходимости, связующее средство, средство, способствующее рассыпанию, смазывающее средство, краситель, корригент/отдушка и т.п., и затем полученная смесь может быть получена в виде таблетки, таблетки с покрытием, гранулы, порошка, капсулы и т.п., согласно обычным способам.
Примеры разбавителя включают лактозу, сахарозу, D-маннит, декстрозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и ангидрид кремниевой кислоты.
Примеры связующего средства включают воду, этанол, 1-пропанол, 2-пропанол, простой сироп, декстрозу в воде, раствор крахмала, набухающего в холодной воде, раствор желатина, D-маннит, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, фосфат кальция и поливинилпирролидон.
Примеры средства, способствующего рассыпанию, включают сухой крахмал, альгинат натрия, порошки агара, бикарбонат натрия, карбонат кальция, лаурилсульфат натрия, стеарат моноглицерида и лактозу.
Примеры смазывающего средства включают очищенный тальк, стеарат натрия, стеарат магния, боракс и полиэтиленгликоль.
Примеры красителя включают оксид титана и оксид железа.
Примеры корригента/отдушки включают сахарозу, апельсиновую кожуру, лимонную кислоту и винную кислоту.
Когда получают жидкий препарат для перорального введения, к соединению по настоящему изобретению могут быть добавлены корригент, буфер, стаблизатор, отдушка и т.п., и затем смесь можно получить в виде жидкого средства для внутреннего употребления, сиропа, эликсира и т.п., согласно обычным способам. В таком случае можно использовать тот же корригент/отдушку, какой описан выше. Примером буфера является цитрат натрия, и примеры стабилизатора включают трагакант, аравийскую камедь и желатин. При необходимости на такие препараты для перорального введения может быть нанесено энтеросолюбильное покрытие или другое покрытие с целью, например, продления действия, согласно способам, известным в области пероральных препаратов. Примеры веществ для такого покрытия включают гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу, полиоксиэтиленгликоль и Твин 80 (зарегистрированный товарный знак).
Когда получают средство для инъекции, к соединению по настоящему изобретению могут быть добавлены регулятор рН, буфер, стабилизатор, средство для регулирования тоничности, местный анестетик и т.п., и смесь может быть переработана в гиподермические, внутримышечные и внутривенные инъекции согласно обычным способам. Примеры регулятора рН и буфера, используемых в таком случае, включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры стабилизатора включают пиросульфит натрия, ЭДТК, тиогликолевую кислоту и тиомолочную кислоту. Примеры местного анестетика включают гидрохлорид прокаина и гидрохлорид лидокаина. Примеры средства для регулирования тоничности включают хлорид натрия, декстрозу, D-маннит и глицерин.
Когда получают суппозиторий, к соединению по настоящему изобретению могут быть добавлены фармацевтически приемлемые носители, известные в данной области техники, такие как полиэтиленгликоль, ланолин, масло какао и триглицерид жирной кислоты, и, при необходимости, поверхностно-активные вещества, такие как Твин 80 (зарегистрированный товарный знак), и затем из полученной смеси можно получить суппозиторий согласно обычному способу.
Когда получают мазь, с соединением по настоящему изобретению могут быть смешаны обычно используемая основа, стабилизатор, смачивающее вещество, консервант и т.п., что необходимо, и полученную смесь можно смешать для получения мази согласно обычному способу. Примеры основы включают жидкий парафин, белый вазелин, белый пчелиный воск, октилдодециловый спирт и парафин. Примеры консерванта включают метил-пара-оксибензоат, этил-пара-оксибензоат и пропил-пара-оксибензоат.
Когда получают пэтч, вышеописанную мазь, крем, гель, пасту или подобную композицию можно нанести на обычную подложку согласно обычному способу. Подходящими в качестве подложки являются тканые материалы или нетканые материалы, состоящие из хлопка, штапельного волокна или химического волокна; и пленка или листовой пенопласт, такой как мягкий винилхлорид, полиэтилен или полиуретан.
Количество производного урацила по настоящему изобретению, добавляемого в каждую вышеописанную стандартную лекарственную форму, не является постоянным и изменяется в зависимости от симптомов у пациента, которому вводят соединение, лекарственной формы и т.д. Как правило, в случае перорального средства количество соединения составляет приблизительно 0,05-1000 мг на стандартную лекарственную форму. В случае инъекции количество соединения составляет приблизительно 0,01-500 мг на стандартную лекарственную форму, и в случае суппозитория количество соединения составляет приблизительно 1-1000 мг на стандартную лекарственную форму.
Кроме того, суточная доза лекарственного средства, имеющего описанную выше лекарственную форму, не является постоянной и может изменяться в зависимости от симптомов, массы тела, возраста, пола пациента и т.д. Как правило, суточная доза лекарственного средства составляет приблизительно 0,05-5000 мг в сутки для взрослого (масса тела 50 кг), и предпочтительно, 0,1-1000 мг в сутки для взрослого (масса тела 50 кг). Такую дозу лекарственного средства предпочтительно вводят один раз в сутки или делят на два или три раза в сутки.
В случае, когда препарат, содержащий производное урацила формулы (I) или его соль, отделен от препарата, содержащего противоопухолевое средство А, два препарата могут вводиться одновременно, или один ингредиент может быть введен в любое время до или после того, как введен другой ингредиент. Предпочтительно два препарата можно вводить одновременно, или один ингредиент можно вводить в пределах 6 часов до или после введения другого ингредиента.
Так как производное урацила по настоящему изобретению способно существенно потенцировать противоопухолевое действие противоопухолевого средства А, количество противоопухолевого средства А на дозу может быть уменьшено по сравнению с обычно используемым количеством в дозе. С другой стороны, количество противоопухолевого средства А на дозу может быть таким же, как обычно используемое количество на дозу.
Соотношение при введении или в комбинации между усилителем противоопухолевого действия по настоящему изобретению или его солью и противоопухолевым средством А особо не ограничивается до тех пор, пока такое соотношение находится в интервале, в котором может быть обеспечено потенцирующее действие на противоопухолевое действие. Соединение по настоящему изобретению или его соль можно использовать в количестве приблизительно 0,01-100 молей, и предпочтительно, приблизительно 0,07-64 молей, относительно 1 моля противоопухолевого средства А. В данном случае пропорцию противоопухолевого средства А при введении или в комбинации можно определить на основании количества активного фармацевтического ингредиента, обладающего противоопухолевым действием. Например, в случае тегафур/гимерацил/отерацила калия (TS-1), тегафур/урацила (UFT) и т.д. соединение по настоящему изобретению или его соль могут быть использованы в количестве приблизительно 0,01-100 молей, и предпочтительно, приблизительно 0,15-64 молей, относительно 1 моля тегафура в сутки. В случае капецитабина соединение по настоящему изобретению или его соль могут быть использованы в количестве приблизительно 0,01-100 молей, и предпочтительно, приблизительно 0,07-8 молей, относительно 1 моля капецитабина.
ПРИМЕРЫ
Далее настоящее изобретение будет описано конкретнее с обращением к ссылочным примерам, примерам и примерам испытания. Однако не предполагается, что настоящее изобретение ограничивается указанными примерами.
Ссылочный пример 1
Синтез (3-(циклопропилметокси)фенил)метанамина
Формула 6

3-Цианофенол (12,4 г) растворяют в N,N-диметилформамиде (далее в данном описании упоминается как ДМФА; 100 мл). К раствор добавляют карбонат калия (30,5 г), йодид калия (1,74 г) и (хлорметил)циклопропан (10,2 мл), и смесь перемешивают при 90°С в течение 4 часов. К реакционной смеси добавляют воду (130 мл), и затем полученную смесь экстрагируют толуолом (130 мл). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в тетрагидрофуране (далее в данном описании упоминается как ТГФ; 60 мл). К раствору постепенно, по каплям, при 0°С добавляют раствор алюмогидрида лития (далее в данном описании упоминается как LAH) в ТГФ (2,4М, 68 мл), и затем реакционную смесь перемешивают при 45°С в течение 4 часов. К реакционной смеси при 0°С постепенно добавляют воду (10 мл), водный раствор гидроксида натрия (1,0М, 10 мл) и воду (5,0 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают смесью 10% метанола/ТГФ (400 мл). Затем объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют воду (50 мл), и затем полученную смесь экстрагируют этилацетатом (50 мл ×3). Органический слой промывают рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении, и получают указанное в заголовке соединение (18,1 г) в виде сырого продукта реакции.
Ссылочный пример 2
Синтез гидрохлорида (R)-1-(3-(циклопентилокси)фенил)этанамина
Формула 7

3-Гидроксибензальдегид (12,2 г) растворяют в ДМФА (120 мл). К раствору добавляют бромциклопентан (32,8 мл), карбонат калия (27,6 г) и йодид калия (1,66 г), и смесь перемешивают при 120°С в течение 3,5 часов. Реакционную смесь охлаждают до комнатной температуры, затем добавляют воду (120 мл), и полученную смесь экстрагируют толуолом (120 мл). Органический слой промывают водой (120 мл), водным раствором гидроксида натрия (1,0 М, 120 мл) и рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в толуоле (250 мл). К раствору добавляют (S)-(-)-2-метил-2-пропансульфинамид (13,3 г) и тетраизопропоксид титана (44,4 мл), и смесь перемешивают при 70°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры, и затем к ней добавляют водный насыщенный раствор бикарбоната натрия (130 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают этилацетатом (200 мл ×4). Объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют рассол (200 мл), и затем полученную смесь экстрагируют этилацетатом (200 мл). Органический слой сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Аликвоту (1,47 г) остатка (29,3 г) растворяют в ТГФ (7,5 мл). К раствору при 0°С по каплям добавляют раствор метилмагнийбромида в диэтиловом эфире (3,0 М, 3,33 мл), и смесь перемешивают при 0°С в течение 4 часов. К реакционной смеси при 0°С в течение 5 минут добавляют водный насыщенный раствор хлорида аммония (6,0 мл), и затем полученную смесь экстрагируют этилацетатом (10 мл). Органический слой промывают рассолом (6,0 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (40% этилацетата/гексан). Полученное соединение (1,09 г) растворяют в метаноле (10 мл). К раствору добавляют раствор хлороводорода в диоксане (4,0 М, 1,1 мл), и смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, затем остаток упаривают совместно с толуолом (5,0 мл ×3), и получают указанное в заголовке соединение (845 мг).
Ссылочный пример 3
Синтез гидрохлорида (R)-1-(3-((R)-тетрагидрофуран-3-илокси)фенил)этанамина
Формула 8

3-Гидроксибензальдегид (1,3 г), трифенилфосфин (3,6 г) и (S)-(+)-тетрагидро-3-фуранол (1,2 мл) растворяют в ТГФ (20 мл). К раствору при 0°С постепенно, по каплям, добавляют раствор диэтилазодикарбоксилата (далее в данном описании упоминается как DEAD) в толуоле (2,2 М, 6,2 мл), и затем смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и затем к остатку добавляют этилацетат (20 мл). Органический слой промывают водным раствором гидроксида натрия (1,0 М, 5,0 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (50% этилацетата/гексан). Полученное соединение растворяют в толуоле (6,5 мл). К раствору добавляют (S)-(-)-2-метил-2-пропансульфинамид (330 мг) и тетраизопропоксид титана (1,1 мл), и смесь перемешивают при 75°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры, и затем к ней добавляют водный насыщенный раствор бикарбоната натрия (10 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают этилацетатом (20 мл ×4). Объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют рассол (30 мл), и затем полученную смесь экстрагируют этилацетатом. Органический слой сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ (7,5 мл). К раствору при 0°С добавляют по каплям раствор метилмагнийбромида в диэтиловом эфире (3,0 М, 1,7 мл), и смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси при 0°С в течение 10 минут добавляют водный насыщенный раствор хлорида аммония (10 мл), и затем полученную смесь экстрагируют этилацетатом (15 мл). Органический слой промывают рассолом (10 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (100% этилацетата). Полученное соединение (1,09 г) растворяют в метаноле (5,0 мл). К раствору добавляют раствор хлороводорода в диоксане (4,0 М, 470 мкл), и смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, затем остаток упаривают совместно с толуолом (4,0 мл ×3), и получают указанное в заголовке соединение (244 мг).
Ссылочный пример 4
Синтез (3-(циклопропилметокси)-4-фторфенил)метанамина
Формула 9
4-Фтор-3-гидроксибензойную кислоту (15,0 г) растворяют в ДМФА (200 мл). К раствору добавляют (хлорметил)циклопропан (18,0 мл), карбонат калия (29,2 г) и йодид калия (1,6 г), и смесь перемешивают при 90°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют воду (120 мл), и затем полученную смесь экстрагируют толуолом (120 мл). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в толуоле (65 мл). К раствору при 0°С добавляют по каплям раствор диизобутилалюмогидрида (далее в данном описании упоминается как DIBAL) в гексане (1,0 М, 130 мл), и реакционную смесь перемешивают при 0°С в течение 2 часов. К реакционной смеси постепенно добавляют воду (10 мл) и водный раствор гидроксида натрия (1,0 М, 10 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают этилацетатом (100 мл ×5). Затем объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют воду (100 мл), и затем полученную смесь экстрагируют этилацетатом (150 мл). Органический слой промывают рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (40% этилацетата/гексан). Полученное соединение растворяют в ТГФ (750 мл). К раствору при комнатной температуре добавляют по каплям дифенилфосфорилазид (12,9 мл) и 1,8-диазабицикло[5.4.0]-7-ундецен (далее в данном описании упоминается как DBU) (9,4 мл), и смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют рассол (100 мл), и водный слой экстрагируют этилацетатом (100 мл ×2). Органический слой промывают рассолом (100 мл), затем сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (20% этилацетата/гексан). Полученное соединение растворяют в ТГФ (80 мл). К раствору при 0°С постепенно, по каплям, добавляют раствор LAH в ТГФ (2,4 М, 40 мл), и смесь перемешивают при 0°С в течение 1 часа. К реакционной смеси при 0°С постепенно, по каплям, добавляют воду (5,0 мл) и водный раствор гидроксида натрия (1,0 М, 5 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают смесью 10% метанола/ТГФ (200 мл). Затем объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют рассол (100 мл), и затем полученную смесь экстрагируют этилацетатом (150 мл). Органический слой сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении, и получают указанное в заголовке соединение (10,5 г) в виде сырого продукта реакции.
Ссылочный пример 5
Синтез гидрохлорида (R)-1-(3-(циклопропилметокси)-4-фторфенил)этанамина
Формула 10

4-Фтор-3-гидроксибензойную кислоту (12,0 г) растворяют в этаноле (200 мл). К раствору добавляют серную кислоту (3,5 мл), и смесь кипятят с обратным холодильником при 105°С в течение 4 часов. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении. К остатку добавляют воду (100 мл) и карбонат натрия (18,0 г), и водный слой экстрагируют этилацетатом (100 мл ×2). Объединенный органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток упаривают совместно с толуолом (15 мл ×2), и затем остаток растворяют в ДМФА (100 мл). К смеси добавляют (хлорметил)циклопропан (6,9 мл), карбонат калия (19,8 г) и йодид калия (1,2 г), и смесь перемешивают при 90°С в течение 3,5 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют воду (200 мл), и затем полученную смесь экстрагируют толуолом (100 мл ×2). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ (75 мл). К смеси при комнатной температуре добавляют по каплям раствор борогидрида лития в ТГФ (2,0 М, 54 мл), и смесь кипятят с обратным холодильником при 80°С в течение 3,5 часов. Реакционную смесь охлаждают до 0°С, затем к ней при той же температуре добавляют по каплям воду (200 мл), и затем полученную смесь экстрагируют этилацетатом (100 мл ×2). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в дихлорметане (250 мл). К смеси при комнатной температуре добавляют диоксид марганца (86 г), и смесь кипятят с обратным холодильником при 45°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры, и выпавшее в осадок вещество удаляют фильтрацией и промывают хлороформом (100 мл ×4). Затем объединенный фильтрат концентрируют. Остаток растворяют в толуоле (150 мл). К раствору добавляют (S)-(-)-2-метил-2-пропансульфинамид (8,5 г) и тетраизопропоксид титана (28,4 мл), и смесь перемешивают при 75°С в течение 6 часов. Реакционную смесь охлаждают до комнатной температуры, и затем к ней добавляют водный насыщенный раствор бикарбоната натрия (150 мл). Полученное выпавшее в осадок вещество удаляют фильтрацией и промывают этилацетатом (200 мл Ч 6). Объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют рассол (150 мл), и затем полученную смесь экстрагируют этилацетатом (200 мл). Органический слой сушат над безводным сульфатом магния и затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ (85 мл). К смеси при 0°С добавляют по каплям раствор метилмагнийбромида в диэтиловом эфире (3,0 М, 42 мл), и смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси при 0°С в течение 10 минут добавляют водный насыщенный раствор хлорида аммония (100 мл), и затем полученную смесь экстрагируют этилацетатом (100 мл ×2). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (50% этилацетата/гексан). Полученное соединение растворяют в метаноле (70 мл). К раствору добавляют раствор хлороводорода в диоксане (4,0 М, 13 мл), и смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, остаток упаривают совместно с толуолом (40 мл ×3), и получают указанное в заголовке соединение (9,09 г).
Ссылочный пример 6
Синтез 1-(3-(циклопропилметокси)фенил)этанамина
Формула 11

3-Гидроксибензальдегид (692 мг) растворяют в ДМФА (25 мл). К раствору добавляют карбонат калия (1,56 г), йодид калия (95 мг) и (хлорметил)циклопропан (578 мкл), и смесь перемешивают при 90°С в течение 4 часов. К реакционной смеси добавляют воду (20 мл), и затем полученную смесь экстрагируют толуолом (20 мл). Органический слой промывают рассолом (20 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в ТГФ (2,5 мл). К смеси при 0°С добавляют по каплям раствор метилмагнийбромида в диэтиловом эфире (1,0 М, 6,5 мл), и смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси при 0°С добавляют водный насыщенный раствор хлорида аммония (10 мл), и затем полученную смесь экстрагируют этилацетатом (20 мл ×2). Органический слой промывают рассолом (20 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (40% этилацетата/гексан). Полученное соединение растворяют в ТГФ (5,0 мл). К раствору при комнатной температуре добавляют по каплям дифенилфосфорилазид (875 мкл) и DBU (592 мкл), и смесь перемешивают в течение 1 часа. К реакционной смеси добавляют рассол (10 мл), и затем полученную смесь экстрагируют этилацетатом (20 мл ×2). Органический слой промывают рассолом (10 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (20% этилацетата/гексан). Полученное соединение растворяют в метаноле (7,5 мл). К раствору добавляют 10% палладий-на-угле (180 мг), и реакционную смесь перемешивают при комнатной температуре в течение 2 часов в атмосфере водорода. Выпавшее в осадок вещество удаляют фильтрацией через слой целита и промывают метанолом (100 мл). Затем объединенный фильтрат концентрируют при пониженном давлении, и получают указанное в заголовке соединение (740 мг) в виде сырого продукта реакции.
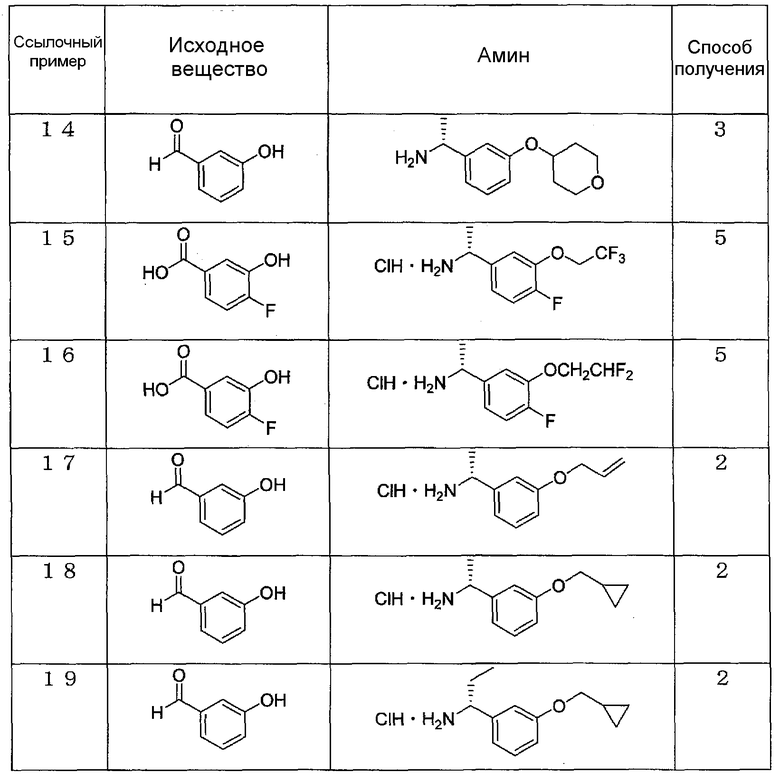
Ссылочные примеры 7-19
Амины, указанные в приведенных далее таблицах, синтезируют согласно любому из ссылочных примеров 1-3 и 5

Ссылочный пример 20
Синтез (N)-(3-(циклопропилметокси)бензил)-3-(метоксиметокси)пропан-1-сульфонамида
Формула 12

(3-(Циклопропилметокси)фенил)метанамин (10,0 г), полученный в ссылочном примере 1, растворяют в дихлорметане (50 мл). К раствору при 0°С добавляют триэтиламин (11,9 г) и 3-хлорпропансульфонилхлорид (10,6 г), и смесь перемешивают при комнатной температуре в течение 12 часов. К реакционной смеси добавляют воду (100 мл), и затем полученную смесь экстрагируют хлороформом (50 мл). Органический слой промывают разбавленной соляной кислотой (1,0 М, 100 мл) и рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток растворяют в ДМФА (100 мл). К смеси добавляют ацетат натрия (10,2 г) и йодид натрия (18,6 г), и смесь перемешивают при 80°С в течение 8 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют воду (100 мл), и затем полученную смесь экстрагируют этилацетатом (80 мл ×2). Органический слой промывают рассолом (100 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (50% этилацетата/гексан). Полученное соединение растворяют в 5-10% растворе хлороводорода в метаноле (100 мл), и раствор кипятят с обратным холодильником при 80°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (66% этилацетата/гексан). Полученное соединение растворяют в дихлорметане (80 мл). К раствору добавляют N,N-диизопропилэтиламин (14,1 мл) и хлорметилметиловый эфир (4,1 мл), и смесь перемешивают при комнатной температуре в течение 1 часа. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония (50 мл), и затем полученную смесь экстрагируют хлороформом (50 мл). Органический слой промывают рассолом (30 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (25% этилацетата/гексан), и получают указанное в заголовке соединение (11,5 г).
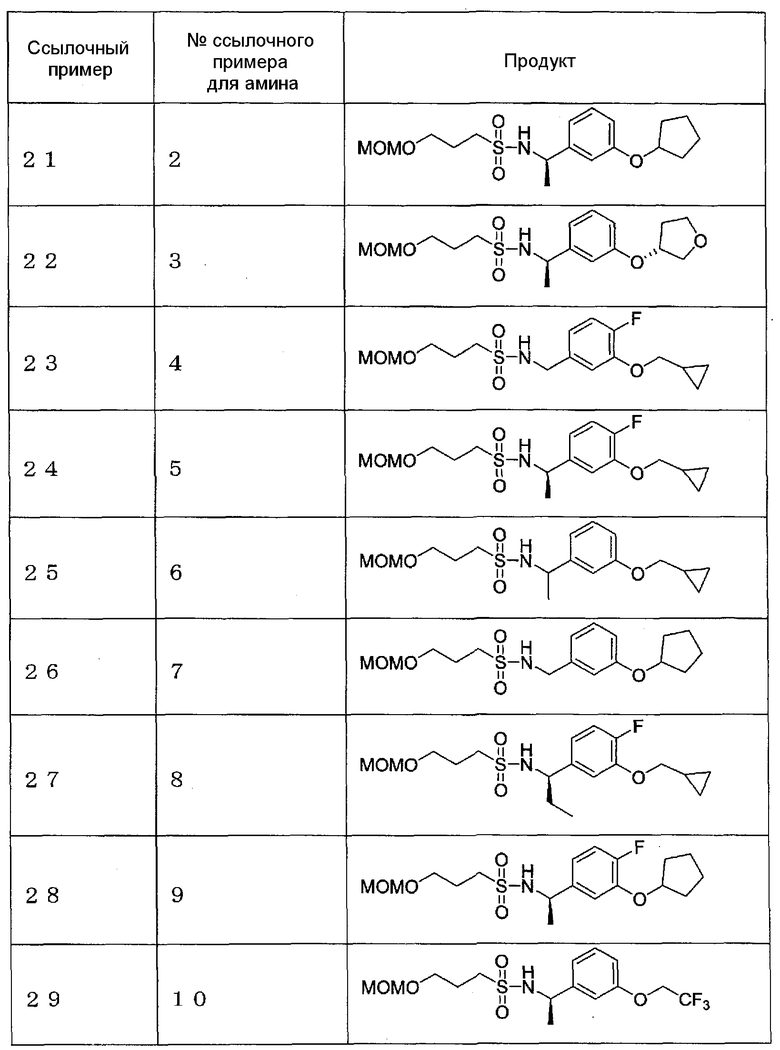
Ссылочные примеры 21-37
Соединения, указанные в приведенных далее таблицах, синтезируют согласно способу ссылочного примера 20

Ссылочный пример 38
Синтез (R)-N-(1-(3-(циклопропилметокси)фенил)пропил)-3-гидрокси-N-(метоксиметил)пропан-1-сульфонамида
Формула 13

Гидрохлорид (R)-1-(3-(циклопропилметокси)фенил)пропан-1-амина (5,4 г), полученный в ссылочном примере 19, растворяют в дихлорметане (50 мл). К раствору при 0°С добавляют триэтиламин (8,7 мл) и 3-хлорпропансульфонилхлорид (2,9 мл), и смесь перемешивают при комнатной температуре в течение 2 часов. К реакционной смеси добавляют воду (50 мл), и затем полученную смесь экстрагируют хлороформом (100 мл). Органический слой промывают рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (40% этилацетата/гексан). Полученное соединение растворяют в дихлорметане (50 мл). К раствору добавляют N,N-диизопропилэтиламин (22,9 мл) и хлорметилметиловый эфир (6,6 мл), и смесь перемешивают при 40°С в течение 12 часов. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония (50 мл), и затем полученную смесь экстрагируют хлороформом (50 мл). Органический слой промывают водным насыщенным раствором хлорида аммония (50 мл) и рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (20% этилацетата/гексан). Полученное соединение растворяют в ДМФА (50 мл). К смеси добавляют ацетат натрия (3,6 г) и йодид натрия (6,6 г), и смесь перемешивают при 80°С в течение 8 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют воду (100 мл), и затем полученную смесь экстрагируют этилацетатом (75 мл ×2). Органический слой промывают рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (50% этилацетата/гексан). Полученное соединение растворяют в растворе метиламина в метаноле (40%, 100 мл), и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют при пониженном давлении, затем остаток очищают колоночной хроматографией на силикагеле (66% этилацетата/гексан), и получают указанное в заголовке соединение (4,5 г).
Ссылочный пример 39
Синтез 3-(циклопропилметокси)-N-(1-(3-метоксиметокси))пропил)циклопропил)бензолсульфонамида
Формула 14

Гидрохлорид 1-аминоциклопропанпропанола (258 мг), полученный согласно способу, описанному в документе (J. Heterocyclic Chem., 25, 1769-1772 (1988)), растворяют в воде (850 мкл) и ТГФ (3,4 мл). К раствору добавляют оксид марганца (343 мг), триэтиламин (355 мкл) и 3-бензоилоксибензолсульфонилхлорид (504 мг), полученный согласно способу, описанному в документе (J. Pesticide Chem., 13, 107-115 (1988)), и смесь перемешивают при комнатной температуре в течение 1 часа. Выпавшее в осадок вещество удаляют фильтрацией и промывают этилацетатом (50 мл). Затем объединенный фильтрат концентрируют при пониженном давлении. К остатку добавляют воду (15 мл), и затем полученную смесь экстрагируют этилацетатом (20 мл). Органический слой промывают рассолом (10 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (75% этилацетата/гексан). Аликвоту (520 мг) полученного соединения (530 мг) растворяют в дихлорметане (5,0 мл). К раствору добавляют N,N-диизопропилэтиламин (847 мкл) и хлорметилметиловый эфир (264 мкл), и смесь перемешивают при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляют водный насыщенный раствор хлорида аммония (20 мл), и полученную смесь экстрагируют этилацетатом (20 мл). Органический слой промывают водным насыщенным раствором хлорида аммония (20 мл) и рассолом (20 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (20% этилацетата/гексан). Аликвоту (440 мг) полученного соединения (446 мг) растворяют в растворе метиламина в метаноле (40%, 5,0 мл), и смесь перемешивают при комнатной температуре в течение 20 минут. Реакционную смесь концентрируют при пониженном давлении, и затем остаток растворяют в ДМФА (9,0 мл). К раствору добавляют карбонат калия (290 мг), йодид калия (17 мг) и (хлорметил)циклопропан (107 мкл), и смесь перемешивают при 90°С в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры, к ней добавляют воду (30 мл), и полученную смесь экстрагируют этилацетатом (30 мл). Органический слой промывают водой (20 мл) и рассолом (20 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (20% этилацетата/гексан), и получают указанное в заголовке соединение (312 мг) в виде бледно-желтого масла.
Пример 1
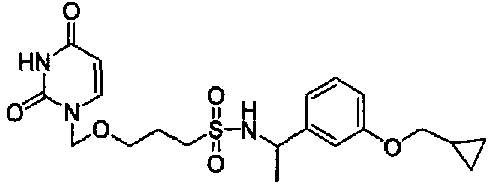
Синтез N-(3-(циклопропилметокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамида
Формула 15

(N)-(3-(Циклопропилметокси)бензил)-3-(метоксиметокси)пропан-1-сульфонамид (6,8 г), полученный в ссылочном примере 20, растворяют в дихлорметане (20 мл). К раствору при 0°С добавляют раствор трихлорида бора (далее в данном описании упоминается как BCl3) в дихлорметане (1,0 М, 6,7 мл), и смесь перемешивают при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрируют при пониженном давлении, и остаток растворяют в 1,2-дихлорэтане (далее в данном описании упоминается как DCE, 25 мл).
2,4-Бис(триметилсилилокси)пиримидин (7,1 г), полученный согласно способу, описанному в документе (Nucleosides & Nucleotides, 4, 565-585 (1985)) растворяют в DCE (150 мл). К раствору добавляют раствор полученного выше остатка в DCE (25 мл) и йод (180 мг), и смесь кипятят с обратным холодильником при 95°С в течение 3,5 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют воду (350 мл) и насыщенный водный раствор тиосульфата натрия (10 мл), и затем полученную смесь экстрагируют смесью 10% метанола/хлороформ (100 мл ×3). Объединенный органический слой промывают рассолом (150 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (100% этилацетата), и получают указанное в заголовке соединение (3,5 г, выход 42%) в виде белого твердого вещества.
1H-ЯМР (CDCl3) δ (м.д.): 0,30-0,39 (2H, м), 0,57-0,68 (2H, м), 1,20-1,31 (1H, м), 1,96-2,09 (2H, м), 3,0 (2H, т, J=7, 2Гц), 3,57-3,64 (2H, м), 3,81 (2H, д, J=6,9 Гц), 4,25 (2H, д, J=6,1 Гц), 4,89 (1H, ушир.с), 5,09 (2H, с), 5,75 (1H, дд, J=7,9, 1,8 Гц), 6,76-6,90 (3H, м), 7,20-7,29 (2H, м), 8,90 (1H, ушир.с).
Пример 2 - пример 18
Перечисленные далее соединения синтезируют согласно способу примера 1 из соединений, полученных в ссылочных примерах 21-37, соответственно. Результаты приводятся в таблицах, следующих далее.
Пример 2
(R)-N-(1-(3-(Циклопентилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 3
3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)-N-((R)-1-(3-((R)-тетрагидрофуран-3-илокси)фенил)этил)пропан-1-сульфонамид
Пример 4
N-(3-(Циклопропилметокси)-4-фторбензил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 5
(R)-N-(1-(3-(Циклопропилметокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 6
N-(1-(3-(Циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 7
N-(3-(Циклопентилокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 8
(R)-N-(1-(3-(Циклопропилметокси)-4-фторфенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 9
(R)-N-(1-(3-(Циклопентилокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 10
(R)-3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)-N-(1-(3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид
Пример 11
(R)-3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)- N-(1-(3-(изобутоксифенил)этил)пропан-1-сульфонамид
Пример 12
3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)-N-((R)-1-(3-((S)-2-метилбутокси)фенил)этил)пропан-1-сульфонамид
Пример 13
(R)-N-(1-(3-(2,2-Дифторэтокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 14
(R)-3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)-N-(1-(3-(тетрагидро-2Н-пиран-4-илокси)фенил)этил)пропан-1-сульфонамид
Пример 15
(R)-3-((2,4-Диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)-N-(1-(4-фтор-3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид
Пример 16
(R)-N-(1-(3-(2,2-Дифторэтокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 17
(R)-N-(1-(3-(Аллилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид
Пример 18
(R)-N-(1-(3-(Циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид

1,53 (3H, д, J=6,8 Гц), 1,56-1,98 (10H, м), 2,67-2,78 (1H, м), 2,80-2,91 (1H, м), 3,42-3,60 (2H, м), 4,51-4,63 (2H, м), 4,74-4,89 (1H, м), 5,05 (2H, c), 5,76 (1H, 1д, J=7,8, 2,2 Гц), 6,77-6,89 (3H, м), 7,20-7,27 (2H, м), 8,31 (1H, ушир.с)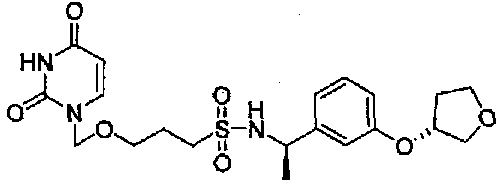
1,52 (3H, д, J=6,8 Гц), 1,85-1,92 (2H, м), 2,10-2,29 (2H, м), 2,68-2,88 (2H, м), 3,43-3,56 (2H, м), 3,89-4,04 (4H, м), 4,53-4,61 (1H, м), 4,92-4,96 (1H, м), 5,05 (2H, c), 5,12 (1H, д, J=7,0 Гц), 5,76 (1H, д, J=8,1 Гц), 6,75-6,92 (3H, м), 7,20-7,29 (2H, м), 9,11 (1H, ушир.с)
0,31-0,40 (2H, м), 0,55-0,69 (2H, м), 1,19-1,36 (1H, м), 1,90-2,10 (2H, м), 2,98 (2H, т, J=7,8 Гц), 3,62 (2H, т, J=5,9 Гц), 3,87 (2H, д, J=6, 8 Гц), 4,21 (2H, д, J=5,9 Гц), 5,09 (2H, с), 5,28-5,39 (1H, м), 5,77 (1H, д, J=7,8 Гц), 6,77-7,09 (3H, м), 7,29 (1H, д, J=8,1 Гц), 9,51 (1H, ушир.с)
0,31-0,38 (2H, м), 0,59-0,69 (2H, м), 1,20-1,38 (1H, м), 1,52 (3H, д, J=6,8 Гц), 1,80-1,98 (2H, м), 2,51-2,88 (2H, м), 3,53 (2H, т, J=5,9 Гц), 3,88 (2H, д, J=7,0 Гц), 4,51-4,62 (1H, м), 5,06 (2H, с), 5,14 (1H, д, J=6,8 Гц), 5,77 (1H, дд, J=8,1 Гц, 1,6 Гц), 6,85-7,11 (3H, м), 7,29 (1H, д, J=7,0 Гц), 9,12 (1H, шир.с)
0,31-0,38 (2H, м), 0,59-0,67 (2H, м), 1,19-1,30 (1H, м), 1,52 (2H, д, J=6,8 Гц), 1,78-2,00 (2H, м), 2,63-2,94 (2H, м), 3,44-3,59 (2H, м), 3,81 (2H, д, J=6,8 Гц), 4,51-4,62 (1H, м), 4,84-4,91 (1H, м), 5,06 (2H, c), 5,14 (1H, д, J=6,8 Гц), 5,77 (1H, дд, J=8, 1, 2,2 Гц), 6,85-7,05 (3H, м), 7,20-7,30 (2H, м), 8,69 (1H, ушир.с)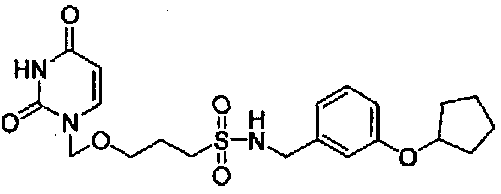
1,68-1,97 (8H, м), 1,98-2,16 (2H, м), 2,92-3,08 (2H, м), 3,60-3,69 (2H, м), 4,25 (2H, д, J=6,1 Гц), 4,74-4,79 (2H, м), 5,01 (2H, c), 5,76 (1H, дд, J=7,9, 2,1 Гц), 6,78-6,90 (3H, м), 7,19-7,29 (2H, м), 8,66 (1H, ушир.с)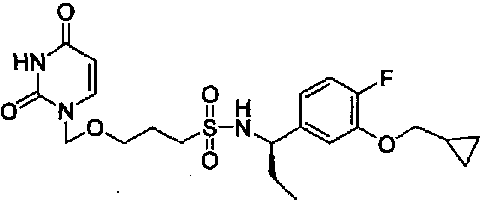
0,32-0,39 (2H, м), 0,63-0,71 (2H, м), 0,89 (3H, т, J=7,3 Гц), 1,20-1,38 (1H, м), 1,71-1,99 (4H, м), 2,53-2,89 (2H, м), 3,41-3,50 (2H, м), 3,88 (2H, д, J=7,1 Гц), 4,21-4,38 (1H, м), 5,04 (2H, c), 5,12 (1H, д, J=7,1 Гц), 5,78 (1H, дд, J=7,9, 2,0 Гц), 6,75-7,09 (3H, м), 7,20 (1H, д, J=7,9 Гц), 8,97 (1H, ушир.с)
1,52 (3H, д, J=7,0 Гц), 1,61-1,70 (2H, м), 1,76-2,00 (8H, м), 2,65-2,90 (2H, м), 3,53 (2H, т, J=5,9 Гц), 4,52-4,61 (1H, м), 4,77-4,85 (1H, м), 5,05 (2H, c), 5,06-5,11 (1H, м), 5,77 (1H, дд, J=8, 1, 2,2 Гц), 6,92-7,04 (3H, м), 7,19 (1H, д, J=8,1 Гц), 9,04 (1H, ушир.с)

1,37 (3H, д, J=6,8 Гц), 1,69-1,80 (2H, м), 2,58-2,70 (1H, м), 2,72-2,88 (1H, м), 3,31-3,46 (2H, м), 4,39-4,45 (1H, м), 4,69-4,79 (2H, м), 4,99 (2H, c), 5,60 (1H, дд, J=8,1, 0,8 Гц), 6,91-7,08 (3H, м), 7,26-7,31 (1H, м), 7,63 (1H, дд, J=8,1, 0,8 Гц), 7,73 (1H, д, J=8,6 Гц), 11, 3(1H, ушир.с)
1,01 (6H, д, J=6,8 Гц), 1,52 (3H, д, J=7,0 Гц), 1,82-1,96 (2H, м), 2,00-2,09 (1H, м), 2,65-2,90 (2H, м), 3,48-3,59 (2H, м), 3,71 (2H, д, J=6,5 Гц), 4,50-4,57 (1H, м), 5,04 (2H, c), 5,50 (1H, д, J=7,0 Гц), 5,75 (1H, д, J=7,8 Гц), 6,79-6,90 (3H, м), 7,17-7,29 (2H, м), 8,90 (1H, ушир.с)
0,95 (3H, т, J=7,4 Гц), 1,02 (3H, д, J=6,8 Гц), 1,53 (3H, д, J=6, 8 Гц), 1,54-1,62 (2H, м), 1,80-1,93 (3H, м), 2,67-2,88 (2H, м), 3,47-3,56 (2H, м), 3,71-3,88 (2H, м), 4,53-4,62 (1H, м), 5,05 (2H, c), 5,06 (1H, ушир.с), 5,78 (1H, д, J=7,9 Гц), 6,79-6,92 (3H, м), 7,22-7,31 (2H, м), 9,09 (1H, ушир.с)
1,37 (3H, д, J=6,8 Гц), 1,61-1,84 (2H, м), 2,53-2,67 (1H, м), 2,71-2,90 (1H, м), 3,31-3,40 (2H, м), 4,23-4,46 (3H, м), 4,99 (2H, c), 5,60 (1H, д, J=7,8 Гц), 6,39 (1H, тт, J=54,6, 3,5 Гц), 6,86-7,03 (3H, м), 7,23-7,30 (1H, м), 7,62 (1H, д, J=7,8 Гц), 7,73 (1H, д, J=8,6 Гц), 11,3 (1H, ушир.с)

1,53 (3H, д, J=7,0 Гц), 1,71-2,10 (6H, м), 2,64-2,91 (2H, м), 3,51-3,66 (4H, м), 3,92-4,05 (2H, м), 4,48-4,59 (2H, м), 5,06 (2H, c), 5,16 (1H, д, J=6, 8 Гц), 5,76 (1H, д, J=8, 1 Гц), 6,81-6,92 (3H, м), 7,21-7,27 (2H, м), 9,22 (1H, ушир.с)
1,37 (3H, д, J=6,8 Гц), 1,69-1,80 (2H, м), 2,56-2,90 (2H, м), 3,38-3,43 (2H, м), 4,37-4,48 (1H, м), 4,74-4,89 (2H, м), 5,00 (2H, c), 5,60 (1H, д, J=7,8 Гц), 7,03-7,09 (1H, м), 7,20-7,32 (2H, м), 7,63 (1H, д, J=7,8 Гц), 7,71 (1H, д, J=8,4 Гц), 11,3 (1H, ушир.с)
1,37 (3H, д, J=6,8 Гц), 1,61-1,84 (2H, м), 2,67-2,90 (2H, м), 3,42 (2H, т, J=6,2 Гц), 4,31-4,48 (3H, м), 5,00 (2H, c), 5,60 (1H, д, J=7,8 Гц), 6,42 (1H, тт, J=54, 3,5 Гц), 6,98-7,04 (1H, м), 7,16-7,31 (2H, м), 7,64 (1H, д, J=7,8 Гц), 7,71 (1H, д, J=8,6 Гц), 11,3 (1H, ушир.с)
1,35 (3H, д, J=7,0 Гц), 1,67-1,77 (2H, м), 2,49-2,60 (1H, м), 2,75-2,95 (1H, м), 3,25-3,40 (2H, м), 4,36-4,45 (1H, м), 4,52-4,55 (2H, м), 4,97 (2H, c), 5,24 (1H, д, J=10,5 Гц), 5,38 (1H, д, J=16,7 Гц), 5,59 (1H, д, J=7,8 Гц), 5,95-6,08 (1H, м), 6,78-6,96 (3H, м), 7,17-7,24 (1H, м), 7,61 (1H, д, J=7,8 Гц), 7,72 (1H, д, J=8,6 Гц), 11,3 (1H, ушир.с)

0,31-0,38 (2H, м), 0,59-0,67 (2H, м), 1,19-1,30 (1H, м), 1,52 (3H, д, J=6,8 Гц), 1,78-2,00 (2H, м), 2,51-2,88 (2H, м), 3,44-3,59 (2H, м), 3,88 (2H, д, J=7,0 Гц), 4,51-4,62 (1H, м), 5,06 (2H, c), 5,14 (1H, д, J=7,0 Гц), 5,77 (1H, д, J=7,8 Гц), 6,85-6,99 (3H, м), 7,20-7,30 (2H, м), 9,12 (1H, ушир.с)
Пример 19
Синтез (R)-N-(1-(3-(циклопропилметокси)фенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)пропан-1-сульфонамида
Формула 16

(R)-N-(1-(3-(Циклопропилметокси)фенил)пропил)-3-гидрокси-N-(метоксиметил)пропан-1-сульфонамид (4,5 г), полученный в ссылочном примере 38, растворяют в ТГФ (70 мл). К раствору добавляют трифенилфосфин (4,48 г) и 3-бензоилпиримидин-2,4(1Н,3Н)-дион (3,6 г), полученный согласно способу, описанному в документе (J. Med. Chem., 50, 6032-6038 (2007)), и смесь перемешивают при комнатной температуре в течение 5 минут. К реакционной смеси постепенно, по каплям, добавляют раствор DEAD в толуоле (2,2 М, 7,6 мл), и смесь перемешивают при комнатной температуре в течение 4 часов. Реакционную смесь концентрируют при пониженном давлении, и затем остаток очищают колоночной хроматографией на силикагеле (70% этилацетата/гексан). Полученное соединение растворяют в растворе метиламина в метаноле (40%, 80 мл), и смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь концентрируют при пониженном давлении, и затем остаток очищают колоночной хроматографией на силикагеле (100% этилацетата). Полученное соединение растворяют в диоксане (25 мл). К раствору добавляют раствор хлороводорода в диоксане (4,0 М, 25 мл), и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь нейтрализуют при 0°С путем добавления водного насыщенного раствора бикарбоната натрия (40 мл) и затем экстрагируют этилацетатом (50 мл ×2). Органический слой промывают рассолом (50 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (100% этилацетата), и получают указанное в заголовке соединение (2,0 г, выход 39%).
1H-ЯМР (CDCl3) δ (м.д.): 0,35-0,38 (2H, м), 0,62-0,70 (2H, м), 0, 90 (3H, т, J=7,3 Гц), 1,22-1,32 (1H, м), 1,75-2,01 (4H, м), 2,53-2,64 (2H, м), 3,57-3,79 (2H, м), 3,80 (2H, д, J=6, 8Гц), 4,26-4,32 (1H, м), 4, 80 (1H, ушир.с), 5,65 (1H, д, J=7, 8Гц), 6,82 (2H, д, J=7,0 Гц), 7,10 (1H, д, J=7, 8 Гц), 7,22-7,29 (2H, м), 9,11 (1H, ушир.с).
Соединение для сравнения 1
Синтез 3-(циклопропилметокси)-N-(1-(3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)пропил)циклопропилбензолсульфонамида
Формула 17

3-(Циклопропилметокси)-N-(1-(3-(метоксиметокси)пропил)циклопропил)бензолсульфонамид (308 мг), полученный в ссылочном примере 39, растворяют в дихлорметане (1,0 мл). К раствору при 0°С постепенно добавляют раствор BCl3 в дихлорметане (1,0 М, 300 мкл), и смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют при пониженном давлении, и затем остаток растворяют в DCE (8,0 мл). К смеси добавляют 2,4-бис(триметилсилилокси)пиримидин (319 мг), полученный согласно способу, описанному в документе (Nucleosides & Nucleotides, 4, 565-585 (1985)) и йод (8,0 мг), и смесь кипятят с обратным холодильником при 93°С в течение 1,5 часов. Реакционную смесь охлаждают до комнатной температуры, затем к ней добавляют насыщенный водный раствор бисульфита натрия (5,0 мл), и затем полученную смесь экстрагируют этилацетатом (20 мл). Органический слой промывают рассолом (10 мл), сушат над безводным сульфатом натрия и затем концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле (90% этилацетата/гексан), и получают указанное в заголовке соединение (210 мг, выход 56%) в виде бледно-желтой смолы.
1H-ЯМР (ДМСО-d8) δ (м.д.): 0,30-0,37 (4H, м), 0,50-0,60 (4H, м), 1,18-1,24 (3H, м), 1,46-1,52 (2H, м), 3,21-3,27 (2H, м), 3,85 (2H, д, J=6,9 Гц), 4,97 (2H, с), 5,59 (1H, д, J=7,9 Гц), 7,12-7,16 (1H, м), 7,24-7,32 (2H, м), 7,40-7,46 (1H, м), 7,61 (1H, д, J=7,9 Гц), 8,04 (1H, ушир.с), 11,30 (1H, ушир.с).
Пример испытаний 1 (ингибирующее действие на dUTPase человека)
Ингибирующую активность указанных соединений по настоящему изобретению против dUTPase человека проверяют путем измерения продуцирования [5-3H]дезоксиуридинмонофосфата (далее в данном описании упоминается как [5-3H]dUMP) из [5-3H]дезоксиуридинтрифосфата (далее в данном описании упоминается как [5-3H]dUTP) согласно способу, показанному ниже.
Конкретно, 0,2 мл общего раствора, содержащего 0,02 мл 1 мкМ dUTP (включающего 588 Бк/мл [5-3H]dUTP), 0,05 мл 0,2 М раствора трис-буфера (рН 7,4), 0,05 мл 16 мМ раствора хлорида магния, 0,02 мл 20 мМ раствора 2-меркаптоэтанола, 0,02 мл 1% водного раствора альбумина, полученного из сыворотки плода коровы, 0,02 мл раствора испытываемого соединения в различных концентрациях или чистой воды как контроля и 0,02 мл раствора dUTPase человека, которая экспрессирована в E. coli и затем очищена, инкубируют при 37°С в течение 15 минут. После инкубации раствор греют при 100°С в течение 1 минуты для прекращения реакции и затем центрифугируют при 15000 об/мин течение 2 минут. Аликвоту (150 мкл) супернатанта анализируют с использованием колонки Atlantis dC18 (изготовитель Waters Corp., 4,6×250 мм) и высокоэффективного жидкостного хроматографа (изготовитель Shimadzu Corp., Prominence). Образцы анализируют при скорости потока 0,8 мл/мин в течение 30 минут с градиентом от 4:6 смеси раствора А (10 мМ дигидрофосфата калия (рН 6,7), 10 мМ тетрабутиламмония и 0,25% метанола) и раствора В (50 мМ дигидрофосфата калия (рН 6,7), 5,6 мМ тетрабутиламмония и 30% метанола) до 100% раствора В. Элюат смешивают со сцинтиллятором (изготовитель PerkinElmer Co., Ltd., Ultima-Flo AP) в отношении 1:2, и определяют радиоактивность [5-3H]dUTP (RT, 10,2 мин) анализатором Radiomatic Flow Scintillation (изготовитель PerkinElmer Co., Ltd., 525 TR).
Ингибирующую активность испытываемого соединения определяют согласно формуле, приведенной ниже. Концентрация, при которой испытываемый раствор ингибирует продуцирование [5-3H]dUTP dUTPase человека на 50%, показана в таблице 10 как IC50 (мкМ).

Активность ингибирования dUTPase человека показана в следующей таблице.
Пример испытаний 2 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия TS-1)
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки 5-6-недельных самцов мышей BALB/cA Jc1-nu. После трансплантации измеряют большую ось (мм) и малую ось (мм) опухоли, и путем вычисления оценивают объем опухоли (TV). Затем с использованием программы разделения на группы MiSTAT мышей делят на отдельные группы так, чтобы средний TV в каждой группе был бы одним и тем же. Дату, когда мышей делят на группы (n=5), обозначают как день 0.
Получают испытываемый раствор тегафур/гимерацил/отерацил калия (TS-1, изготовитель Taiho Pharmaceutical Co., Ltd.) для группы введения одного TS-1, содержащий 0,5% гидроксипропилметилцеллюлозы, 2,5% диметилацетамида, 2,5% Твина 80 и 10% кремофора в конечных концентрациях, обеспечивающий 8,3 мг/кг/день. Дозу TS-1 представляют количеством тегафура (FT).
Испытываемый раствор для группы комбинированного введения (TS-1 + соединение по настоящему изобретению) получают таким же способом, как вышеуказанный испытываемый раствор для группы введения одного TS-1, конкретно, 200 мг/кг/день испытываемого лекарственного средства + TS-1 (8,3 мг/кг/день) в случае соединений по настоящему изобретению 6, 4, 7 и 8, 100 мг/кг/день испытываемого лекарственного средства + TS-1 (8,3 мг/кг/день) в случае соединений по настоящему изобретению 19, 9, 2 и 1, и 200 мг/кг/день испытываемого лекарственного средства + TS-1 (8,3 мг/кг/день) в случае соединения для сравнения 1.
Каждый испытываемый раствор вводят мыши перорально в объеме 10 мл/кг каждый день в течение 14 дней от дня 1.
Измеряют TV в день 15, и вычисляют относительный объем опухоли (RTV). Затем вычисляют Т/С (%) по формуле, приведенной ниже, и оценивают противоопухолевое действие. Результаты показаны на фиг.1. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
TV (мм2) = (большая ось × малая ось2)/2
Т/С (%) = (средняя величина RTV группы введения испытываемого раствора)/(средняя величина RTV контрольной группы) ×100
Пример испытаний 3 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия TS-1
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu и используют так же, как в примере испытаний 2.
Получают испытываемый раствор для группы введения одного TS-1, в котором доза TS-1 (10 мг/кг/день) представлена количеством FT. Испытываемый раствор для группы комбинированного введения (TS-1 + соединение по настоящему изобретению) получают так, чтобы составлялось 300 мг/кг/день соединений по настоящему изобретению + TS-1 (10 мг/кг/день), и усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.2. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
Пример испытаний 4 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия 5-FU)
Линию клеток рака яичников человека OVCAR-3 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu и используют так же, как в примере испытаний 2.
Испытываемый раствор для группы введения одного 5-FU получают, растворяя 5-FU в 7% мейлоне (рН 9,0), и доводят дозу 5-FU до 15 мг/кг/день. Испытываемый раствор, содержащий соединение по настоящему изобретению, получают в виде суспензии в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу соединения по настоящему изобретению доводят до 300 мг/кг/день.
В случае группы введения одного 5-FU испытываемый раствор вводят подкожно непрерывно в течение 14 дней от дня 1 с использованием осмотического мининасоса alzet, модель 2002 (скорость потока 0,5 мкл/час). В случае группы комбинированного введения (5-FU + соединение по настоящему изобретению) 5-FU вводят подкожно непрерывно в течение 14 дней от дня 1 с использованием осмотического мининасоса alzet, модель 2002 (скорость потока 0,5 мкл/час), и каждый день каждой мыши вводят перорально испытываемый раствор, содержащий соединение по настоящему изобретению, в объеме 10 мл/кг. Усиливающую активность оценивают таким же способом, как в примере испытаний 2. Результаты показаны на фигуре 3. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного 5-FU.
Пример испытаний 5 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия капецитабина)
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu и используют так же, как в примере испытаний 2.
Испытываемый раствор для группы введения одного капецитабина получают, суспендируя капецитабин в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу капецитабина доводят до 270 мг/кг/день. Испытываемый раствор, содержащий капецитабин и соединение по настоящему изобретению, получают, суспендируя два соединения в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу двух соединений доводят до 300 мг/кг/день соединения по настоящему изобретению и 270 мг/кг/день капецитабина. Усиливающую активность оценивают таким же способом, как в примере испытаний 2. Результаты показаны на фиг.4. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного капецитабина.
Пример испытаний 6 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия FdUrd)
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu и используют так же, как в примере испытаний 2.
Испытываемый раствор для группы введения одного FdUrd получают, растворяя FdUrd в физиологическом растворе, и дозу FdUrd доводят до 250 мг/кг/день. Испытываемый раствор, содержащий соединение по настоящему изобретению, получают, суспендируя соединение по настоящему изобретению в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу соединения по настоящему изобретению доводят до 300 мг/кг/день.
В случае группы введения одного FdUrd испытываемый раствор вводят внутривенно в течение 3 дней от дня 1. В случае группы комбинированного введения (FdUrd + соединение по настоящему изобретению) FdUrd вводят внутривенно в течение 3 дней от дня 1, и испытываемый раствор, содержащий соединение по настоящему изобретению, вводят перорально каждой мыши в дозе 10 мл/кг каждый день в течение 3 дней от дня 1. В день 15 усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.4. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного FdUrd.
Пример испытаний 7 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия пеметрекседа)
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu и используют так же, как в примере испытаний 2.
Испытываемый раствор для группы введения одного пеметрекседа получают, растворяя пеметрексед в физиологическом растворе, и дозу пеметрекседа доводят до 25 мг/кг/день. Испытываемый раствор, содержащий соединение по настоящему изобретению, получают, суспендируя соединение по настоящему изобретению в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу соединения по настоящему изобретению доводят до 300 мг/кг/день.
Что касается группы введения одного пеметрекседа, то испытываемый раствор вводят внутривенно в день 1 и день 8. Что касается группы комбинированного введения (пеметрексед + соединение по настоящему изобретению), то пеметрексед вводят внутривенно в день 1 и день 8, и испытываемый раствор, содержащий соединение по настоящему изобретению, вводят перорально каждой мыши в объеме 10 мл/кг каждый день в течение 14 дней от дня 1. Усиливающую активность оценивают таким же способом, как в примере испытаний 2. Результаты показаны на фиг.4. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного пеметрекседа.
Пример испытаний 8 (усиливающая активность соединений по настоящему изобретению в отношении противоопухолевого действия UFT)
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца крыс F344N Jc1-rnu и используют так же, как в примере испытаний 2.
Испытываемый раствор для группы введения одного тегафур/урацила (UFT, изготовитель Taiho Pharmaceutical Co., Ltd.) получают, суспендируя UFT в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу UFT доводят до 30 мг/кг/день, которая представлена количеством FT. Смешанный испытываемый раствор, содержащий UFT и соединение по настоящему изобретению, получают, суспендируя два соединения в 0,5% растворе гидроксипропилметилцеллюлозы, и дозу двух соединений доводят до 300 мг/кг/день соединения по настоящему изобретению и 30 мг/кг/день UFT.
Что касается группы введения одного UFT, то испытываемый раствор вводят перорально каждый день в течение 21 дня от дня 1. Что касается группы комбинированного введения (UFT + соединение по настоящему изобретению), также испытываемый раствор, вводят перорально каждый день в течение 21 дня от дня 1. В день 22 усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.4. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного UFT.
Пример испытаний 9 (токсичность и эффективность соединения по настоящему изобретению)
С целью оценки безопасности комбинированного применения противоопухолевого средства с соединением по настоящему изобретению оценивают токсичность и противоопухолевое действие, когда в комбинации с противоопухолевым средством используют высокую дозу соединения по настоящему изобретению.
Линию клеток рака желудка человека SC-6, линию клеток рака толстой кишки человека LS174T и линию клеток рака поджелудочной железы человека CFPAC-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu. После трансплантации измеряют большую ось (мм) и малую ось (мм) каждой опухоли, и путем вычисления оценивают объем опухоли (TV). Затем с использованием программы разделения на группы MiSTAT мышей делят на отдельные группы так, чтобы средний TV в каждой группе был бы одним и тем же. Дату, когда мышей делят на группы (n=5), обозначают как день 0.
Испытываемый раствор для группы введения одного TS-1 получают с использованием 0,5% раствора гидроксипропилметилцеллюлозы, и дозу TS-1 доводят до 10 мг/кг/день как количество FT.
Испытываемый раствор для группы комбинированного введения (TS-1 + соединение по настоящему изобретению) получают таким же способом, как для испытываемого раствора для группы введения одного TS-1, так, чтобы он состоял из соединения по настоящему изобретению (600 мг/кг/день) + TS-1 (10 мг/кг/день).
Испытываемый раствор вводят перорально каждой мыши в объеме 10 мл/кг каждый день в течение 14 дней от дня 1.
При испытании на токсичность измеряют изменение массы тела со временем. Среднюю степень изменения массы тела [изменение массы тела, ВМС (%)] в день 15 относительно дня 0 вычисляют согласно следующей формуле:
ВМС (%)=[(BW в день 15)-(BW в день 0)]/(BW в день 0) Ч 100.
В случае противоопухолевого действия измеряют TV, вычисляют относительный объем опухоли (RTV) ко дню 0, и оценивают противоопухолевое действие. Результаты показаны на фиг.5-7. На фигурах символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
TV (мм2) = (большая ось × малая ось2)/2
Т/С (%) = (средняя величина RTV группы введения испытываемого раствора)/(средняя величина RTV контрольной группы) ×100
Как видно на фиг.4, производное урацила формулы (I) или его соль имеют существенную усиливающую активность в отношении противоопухолевого действия противоопухолевых средств, в частности, антиметаболитов. С другой стороны, как видно из таблицы 10, хотя соединение для сравнения 1 также имеет сильную активность ингибирования dUTPase, усиливающей активности не наблюдают. Более того, как видно на фиг.5-7, потеря массы тела в случае комбинированного применения производного урацила формулы (I) или его соли с противоопухолевым средством не отличается от случая введения одного противоопухолевого средства, и таким образом, обнаружено, что производное урацила формулы (I) или его соль могут усиливать противоопухолевое действие противоопухолевого средства без проявления токсичности.
Пример испытаний 10 (исследование 1, рассматривающее молярное отношение объединенных соединений, необходимое для усиления противоопухолевого действия)
Отношение в комбинации между соединением по настоящему изобретению и TS-1, необходимое для достижения усиливающего действия на противоопухолевое действие, когда два соединения используют в комбинации, оценивают на мышах.
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мышей BALB/cA Jc1-nu. После трансплантации измеряют большую ось (мм) и малую ось (мм) опухоли, и вычисляют объем опухоли (TV). Затем с использованием программы разделения на группы MiSTAT мышей делят на отдельные группы так, чтобы средний TV в каждой группе был бы одним и тем же. Дату, когда мышей делят на группы (n=7), обозначают как день 0.
Дозу TS-1 в испытываемом растворе для группы введения одного TS-1 доводят до 8,3 мг/кг/день, которая представлена количеством FT. Дозу соединения 2 в испытываемом растворе для группы введения одного соединения 2 устанавливают в 1200 мг/кг/день. Указанные испытываемые растворы получают с использованием 0,5% раствора гидроксипропилметилцеллюлозы.
Испытываемый раствор для группы комбинированного введения (TS-1 + соединение по настоящему изобретению) получают таким же способом, как для вышеуказанного испытываемого раствора для группы введения одного TS-1, так, чтобы он состоял из соединения по настоящему изобретению (1200, 600, 300 или 150 мг/кг/день) + TS-1 (8,3 мг/кг/день).
Каждый испытываемый раствор вводят перорально каждой мыши в объеме 10 мл/кг каждый день в течение 14 дней от дня 1, и усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.8. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
Пример испытаний 11 (исследование 2, рассматривающее молярное отношение объединенных соединений, необходимое для усиления противоопухолевого действия)
Отношение в комбинации между соединением по настоящему изобретению и TS-1, необходимое для достижения усиливающего действия на противоопухолевое действие, когда два соединения используют в комбинации, оценивают на крысах.
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца крысы F344N Jc1-rnu. После трансплантации измеряют большую ось (мм) и малую ось (мм) опухоли, и вычисляют объем опухоли (TV). Затем с использованием программы разделения на группы MiSTAT крыс делят на отдельные группы так, чтобы средний TV в каждой группе был бы одним и тем же. Дату, когда крыс делят на группы (n=5 или 6), обозначают как день 0.
Испытываемый раствор для группы введения одного TS-1 получают с использованием 0,5% раствора гидроксипропилметилцеллюлозы, и дозу TS-1 в нем доводят до 18 мг/кг/день в виде количества FT.
Испытываемый раствор для группы комбинированного введения (TS-1 + соединение по настоящему изобретению) получают таким же способом, как для вышеуказанного испытываемого раствора для группы введения одного TS-1, так, чтобы он состоял из соединения по настоящему изобретению (100, 50, 25, 12,5 или 6,25 мг/кг/день) + TS-1 (18 мг/кг/день).
Каждый испытываемый раствор вводят перорально каждой крысе в объеме 10 мл/кг каждый день в течение 28 дней от дня 1, и измеряют TV в день 29. Усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.9.
На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
Пример испытаний 12 (исследование 3, рассматривающее молярное отношение объединенных соединений, необходимое для усиления противоопухолевого действия)
Отношение в комбинации между соединением по настоящему изобретению и капецитабином, необходимое для достижения усиливающего действия на противоопухолевое действие, когда два соединения используют в комбинации, оценивают на мышах.
Линию клеток рака молочной железы человека МХ-1 трансплантируют в правую сторону грудной клетки каждого 5-6-недельного самца мыши BALB/cA Jc1-nu. После трансплантации измеряют большую ось (мм) и малую ось (мм) опухоли, и вычисляют объем опухоли (TV). Затем с использованием программы разделения на группы MiSTAT мышей делят на отдельные группы так, чтобы средний TV в каждой группе был бы одним и тем же. Дату, когда мышей делят на группы (n=5), обозначают как день 0.
Испытываемый раствор для группы введения одного капецитабина получают с использованием 0,5% раствора гидроксипропилметилцеллюлозы, и устанавливают дозу капецитабина в нем 160, 359 или 809 мг/кг/день.
Испытываемый раствор для группы комбинированного введения (капецитабин + соединение по настоящему изобретению) получают таким же способом, как для вышеуказанного испытываемого раствора для группы введения одного капецитабина, так, чтобы он состоял из соединения по настоящему изобретению (75, 300, 600, 1200 или 1600 мг/кг/день) + капецитабин (160, 359 или 809 мг/кг/день), как показано на фигуре для комбинации.
Каждый испытываемый раствор вводят перорально каждой мыши в объеме 10 мл/кг каждый день в течение 14 дней от дня 1, и усиливающую активность оценивают таким же способом, как в примере испытания 2. Результаты показаны на фиг.10. На фигуре символ * показывает, что наблюдают статистически значимое различие относительно группы введения одного TS-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КРИСТАЛЛЫ УРАЦИЛЬНОГО СОЕДИНЕНИЯ | 2016 |
|
RU2686722C1 |
| НОВОЕ УРАЦИЛОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ОТНОСИТЕЛЬНО ДЕЗОКСИУРИДИНТРИФОСФАТАЗЫ ЧЕЛОВЕКА | 2009 |
|
RU2495873C2 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| ПРИМЕНЕНИЕ N′-{ 1-[6-МЕТИЛ-3-(ТИЕТАН-3-ИЛ)-2,4-ДИОКСО-3,4-ДИГИДРОПИРИМИДИН-1(2Н)-ИЛ]ПРОПАН-2-ИЛИДЕН} ИЗОНИКОТИНОГИДРАЗИДА В КАЧЕСТВЕ СРЕДСТВА, СТИМУЛИРУЮЩЕГО РЕГЕНЕРАЦИЮ ТКАНЕЙ | 2023 |
|
RU2813148C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| ТРИАРИЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2840345C2 |
| ПРОИЗВОДНЫЕ УРАЦИЛА, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ SARS-COV-2 | 2021 |
|
RU2769828C1 |
| СУЛЬФОНАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2607081C2 |
| Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения | 2017 |
|
RU2644156C1 |
| ПРОИЗВОДНОЕ СУЛЬФОНАМИДА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2014 |
|
RU2667520C9 |
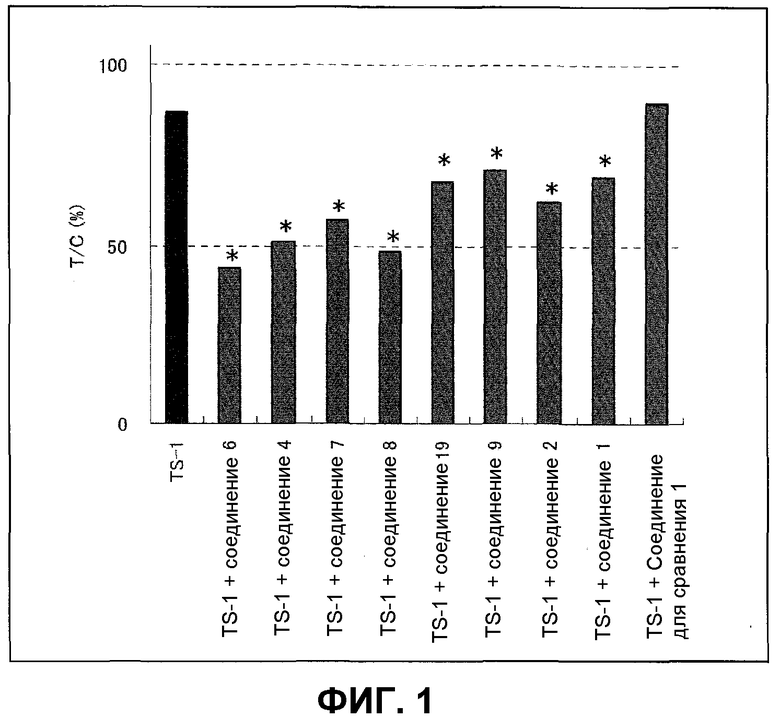

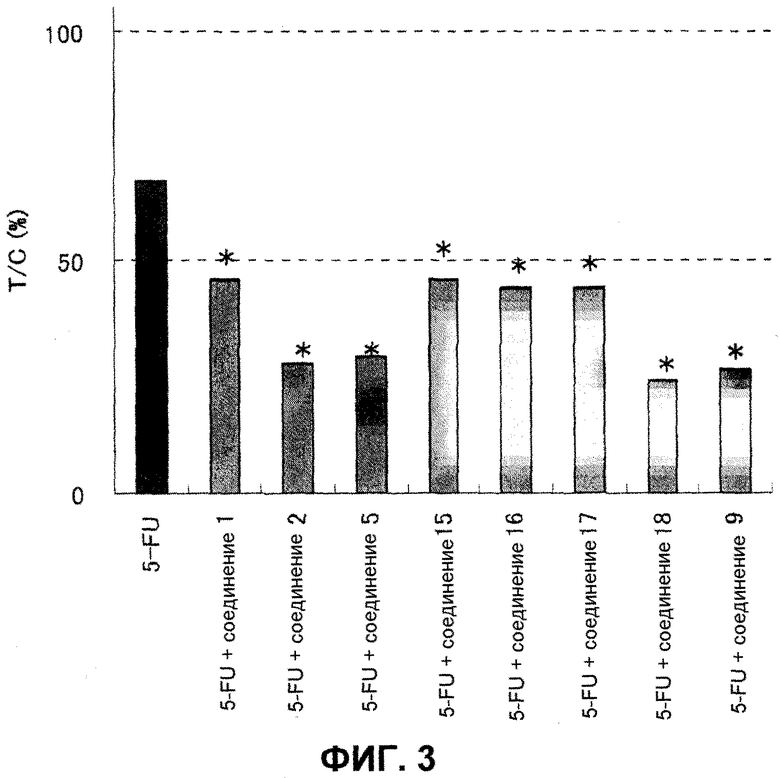




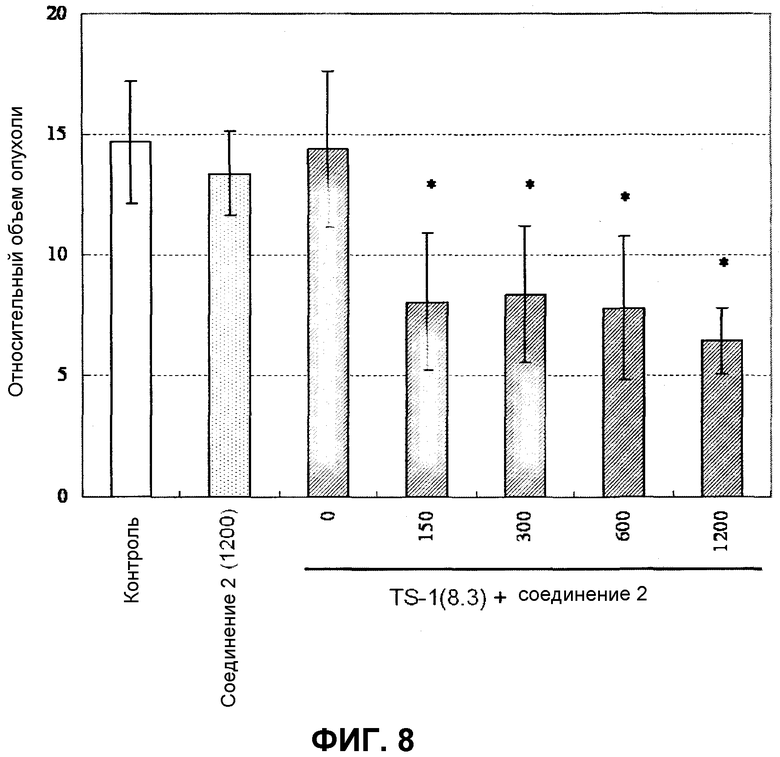
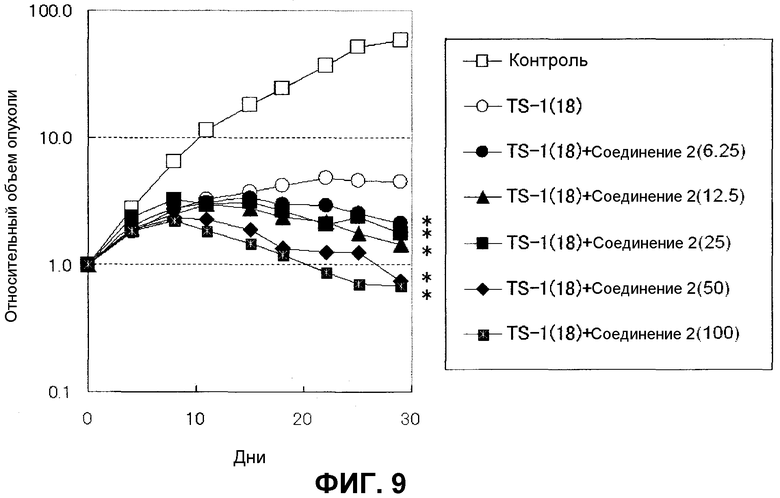

Изобретение относится к новому усилителю противоопухолевого действия, представляющего собой производное урацила общей формулы (I) или его фармацевтически приемлемой соли. В общей формуле (I). 
X представляет собой C1-5-алкиленовую группу, и где одна из метиленовых групп, составляющих алкиленовую группу, необязательно заменена атомом кислорода; R1 представляет собой атом водорода или C1-6-алкильную группу; R2 представляет собой атом водорода или атом галогена; и R3 представляет собой C1-6-алкильную группу, C2-6-алкенильную группу, C3-6-циклоалкильную группу, (C3-6-циклоалкил)-C1-6-алкильную группу, галоген-C1-6-алкильную группу или 5-6-членную насыщенную гетероциклическую группу с атомом кислорода в качестве гетероатома, производное урацила, представленное следующей формулой (I). Изобретение также относится к способу потенцирования противоопухолевого действия или способу лечения опухолей, включающему введение эффективного количества комбинации указанного производного урацила или его фармацевтически приемлемой соли и антиметаболита в эффективном количестве. При этом антиметаболит представляет средство выбранное из 5-фторурацила (5-FU), тегафур/гимерацил/отерацил калия (TS-1), тегафур/урацила (UFT), капецитабина, 5-фтор-2′-дезоксиуридина (FdUrd) и пеметрекседа. 6 н. и 12 з.п. ф-лы, 10 ил., 10 табл., 67 пр.
1. Усилитель противоопухолевого действия, представляющий собой производное урацила общей формулы (I)
где X представляет собой C1-5-алкиленовую группу, и одна из метиленовых групп, составляющих алкиленовую группу,
необязательно заменена атомом кислорода;
R1 представляет собой атом водорода или C1-6-алкильную группу; R2 представляет собой атом водорода или атом галогена; и R3 представляет собой C1-6-алкильную группу, C2-6-алкенильную группу, C3-6-циклоалкильную группу, (C3-6-циклоалкил)-C1-6-алкильную группу, галоген-C1-6-алкильную группу или 5-6-членную насыщенную гетероциклическую группу с атомом кислорода в качестве гетероатома,
или его фармацевтически приемлемую соль.
2. Усилитель противоопухолевого действия по п. 1, представляющий собой производное урацила или его фармацевтически приемлемую соль, при этом в общей формуле (I) X представляет собой этиленовую группу или -O-C1-4-алкиленовую группу;
R1 представляет собой атом водорода или C1-3-алкильную группу; и R2 представляет собой атом водорода или атом фтора.
3. Усилитель противоопухолевого действия по п. 1 или 2, представляющий собой производное урацила или его фармацевтически приемлемую соль, при этом в общей формуле (I) X представляет
собой этиленовую группу или -O-CH2CH2-CH2-;
R1 представляет собой атом водорода, метильную группу или этильную группу; R2 представляет собой атом водорода или атом фтора;
и R3 представляет собой изобутильную группу, 2-метилбутильную группу, аллильную группу, циклопентильную группу, циклопропилметильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, тетрагидрофурильную группу или тетрагидропирильную группу.
4. Усилитель противоопухолевого действия по п. 1, представляющий собой производное урацила или его фармацевтически приемлемую соль, при этом соединение формулы (I) выбирают из группы, в которую входят
- N-(3-(циклопропилметокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-N-(l-(3-(циклопентилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-N-((R)-1-(3-((R)-тетрагидрофуран-3-илокси)фенил)этил)пропан-1-сульфонамид;
- N-(3-(циклопропилметокси)-4-фторбензил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Η)-ил)метокси)пропан-1-сульфонамид;
- Ν-(3-(циклопентилокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопентилокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)ил)метокси)-Ν-(1-(3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)ил)метокси)-Ν-(1-(3-(изобутоксифенил)этил)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-((R)-1-(3-((S)-2-метилбутокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)ил)метокси)-Ν-(1-(3-(тетрагидро-2H-пиран-4-илокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)ил)метокси)-Ν-(1-(4-фтор-3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(аллилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- (R)-N-(l-(3-(циклопропилметокси)фенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)пропан-1-сульфонамид.
5. Противоопухолевый лекарственный препарат, содержащий производное урацила по любому из пп. 1-4 или его фармацевтически приемлемую соль в комбинации с антиметаболитом в эффективном количестве.
6 Противоопухолевый лекарственный препарат по п. 5, где антиметаболит представляет собой любое средство из 5-фторурацила (5-FU), тегафур/гимерацил/отерацил калия (TS-1), тегафур/урацила (UFT), капецитабина, 5-фтор-2′-дезоксиуридина (FdUrd) и пеметрекседа.
7. Производное урацила, представленное общей формулой (I)

где X представляет собой C1-5-алкиленовую группу, и одна из метиленовых групп, составляющих алкиленовую группу, необязательно заменена атомом кислорода;
R1 представляет собой атом водорода или C1-6-алкильную группу; R2 представляет собой атом водорода или атом галогена; и R3 представляет собой C1-6-алкильную группу, C1-6-алкенильную группу, C3-6-циклоалкильную группу, (C3-6-циклоалкил)-C1-6-алкильную группу, галоген-C1-6-алкильную группу или 5-6-членную насыщенную гетероциклическую группу с атомом кислорода в качестве гетероатома,
или его фармацевтически приемлемая соль
для применения при потенцировании противоопухолевого действия.
8. Производное урацила по п. 7 или его фармацевтически приемлемая соль, при этом в общей формуле (I) X представляет собой этиленовую группу или -O-C1-4-алкиленовую группу;
R1 представляет собой атом водорода или C1-3-алкильную группу; и R2 представляет собой атом водорода или атом фтора.
9. Производное урацила по п. 7 или 8 или его фармацевтически приемлемая соль, при этом в общей формуле (I) X представляет собой этиленовую группу или -O-CH2CH2CH2-;
R1 представляет собой атом водорода, метильную группу или этильную группу; R2 представляет собой атом водорода или атом фтора;
и R3 представляет собой изобутильную группу, 2-метилбутильную группу, аллильную группу, циклопентильную группу, циклопропилметильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, тетрагидрофурильную группу или тетрагидропирильную группу.
10. Производное урацила по п. 7 или его фармацевтически приемлемая соль, при этом соединение формулы (I) выбирают из группы, в которую входят
- N-(3-(циклопропилметокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопентилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-N-((R)-1-(3-((R)-тетрагидрофуран-3-илокси)фенил)этил)пропан-1-сульфонамид;
- N-(3-(циклопропилметокси)-4-фторбензил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- N-(3-(циклопентилокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопентилокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(изобутоксифенил)этил)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-((R)-1-(3-((S)-2-метилбутокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(тетрагидро-2H-пиран-4-илокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(4-фтор-3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(аллилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
-(R)-Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
-(R)-Ν-(1-(3-(циклопропилметокси)фенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)пропан-1-сульфонамид.
11. Комбинация, предназначенная для лечения опухолей, содержащая производное урацила по любому из пп. 7-10 или его фармацевтически приемлемую соль и антиметаболит в эффективном количестве.
12. Комбинация по п. 11, где антиметаболит представляет собой любое средство из 5-фторурацила (5-FU), тегафур/гимерацил/отерацил калия (TS-1), тегафур/урацила (UFT), капецитабина, 5-фтор-2′-дезоксиуридина (FdUrd) и пеметрекседа.
13. Способ потенцирования противоопухолевого действия, включающий введение эффективного количества производного урацила, представленного общей формулой (I)

где X представляет собой C1-5-алкиленовую группу, и одна из метиленовых групп, составляющих алкиленовую группу, необязательно заменена атомом кислорода;
R1 представляет собой атом водорода или C1-6-алкильную группу; R2 представляет собой атом водорода или атом галогена; и R3 представляет собой C1-6-алкильную группу, C2-6-алкенильную группу, C3-6-циклоалкильную группу, (C3-6-циклоалкил)-C1-6-алкильную группу, галоген-C1-6-алкильную группу или 5-6-членную насыщенную гетероциклическую группу с атомом кислорода в качестве гетероатома, или его фармацевтически приемлемой соли.
14. Способ по п. 13, при этом в общей формуле (I) X представляет собой этиленовую группу или -O-C1-4-алкиленовую группу;
R1 представляет собой атом водорода или C1-3-алкильную группу; и R2 представляет собой атом водорода или атом фтора.
15. Способ по п. 13 или 14, при этом в общей формуле (I) X представляет собой этиленовую группу или -O-CH2CH2CH2-;
R1 представляет собой атом водорода, метильную группу или этильную группу; R2 представляет собой атом водорода или атом фтора; и R3 представляет собой изобутильную группу, 2-метилбутильную группу, аллильную группу, циклопентильную группу, циклопропилметильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, тетрагидрофурильную группу или тетрагидропирильную группу.
16. Способ по п. 13, при этом соединение формулы (I) выбирают из группы, в которую входят
- N-(3-(циклопропилметокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопентилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-N-((R)-1-(3-((R)-тетрагидрофуран-3-илокси)фенил)этил)пропан-1-сульфонамид;
- N-(3-(циклопропилметокси)-4-фторбензил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- N-(3-(циклопентилокси)бензил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)-4-фторфенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Η)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопентилокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(изобутоксифенил)этил)пропан-1-сульфонамид;
- 3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-((R)-1-(3-((S)-2-метилбутокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(3-(тетрагидро-2H-пиран-4-илокси)фенил)этил)пропан-1-сульфонамид;
- (R)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)-Ν-(1-(4-фтор-3-(2,2,2-трифторэтокси)фенил)этил)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(2,2-дифторэтокси)-4-фторфенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(аллилокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)фенил)этил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)метокси)пропан-1-сульфонамид;
- (R)-Ν-(1-(3-(циклопропилметокси)фенил)пропил)-3-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)пропан-1-сульфонамид.
17. Способ лечения опухолей, включающий введение комбинации производного урацила по любому из пп. 13-16 или его фармацевтически приемлемой соли и антиметаболита в эффективном количестве.
18. Способ по п. 17, при этом антиметаболит представляет собой любое средство из 5-фторурацила (5-FU), тегафур/гимерацил/отерацил калия (TS-1), тегафур/урацила (UFT), капецитабина, 5-фтор-2′-дезоксиуридина (FdUrd) и пеметрекседа.
| НОВОЕ УРАЦИЛОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ОТНОСИТЕЛЬНО ДЕЗОКСИУРИДИНТРИФОСФАТАЗЫ ЧЕЛОВЕКА | 2009 |
|
RU2495873C2 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| WO 2008077453,03.07.2008 | |||
| US 2008300216 A1,04.12.2008 | |||
| Способ окисления боковых цепей ароматических углеводородов и их производных в кислоты и альдегиды | 1921 |
|
SU58A1 |
| ZHAOHUA HUANG et al., A novel kind of antimour drugs using sulfonamide as parent compound EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, 01.01.2001, Vol:36, Nr:11-12, Page(s):863-872 | |||
| N-(6-МЕТИЛ-2,4-ДИОКСО-1,2,3,4-ТЕТРАГИДРОПИРИМИДИНИЛ-5-СУЛЬФОНИЛ) ПИРАЗИН-2-КАРБОКСАМИД, ОБЛАДАЮЩИЙ ПРОТИВОЛЕПРОЗНОЙ, ПРОТИВОТУБЕРКУЛЕЗНОЙ И ИММУНОТРОПНОЙ АКТИВНОСТЬЮ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1996 |
|
RU2102390C1 |

