Изобретение относится к монолитному катализатору и к его использованию для нефтехимических преобразований, таких как дегидрогенизация, ароматизация, реформинг и сжигание.
Во многих нефтехимических реакциях (например, дегидрогенизации, ароматизации, реформинга и сжигания) используют нанесенные катализаторы из благородных металлов. Применение таких катализаторов очень дорогое вследствие высоких стоимостей благородных металлов. Кроме того, неблагоприятное распределение благородных металлов и длинные пути диффузии в обычных катализаторах приводят к малому использованию благородных металлов.
Патент США 4788371 описывает способ дегидрогенизации с водяным паром способных к дегидрогенизации углеводородов в газовой фазе, объединенной с окислительным подогревом промежуточных соединений, причем используют тот же катализатор и для селективного окисления водорода, и для дегидрогенизации с водяным паром. Здесь водород может быть введен путем одновременной подачи. Используемый катализатор содержит благородный металл группы VIII, щелочной металл и другой металл, выбранный из группы, состоящей из В, Ga, In, Ge, Sn и Pb, на неорганическом оксидном носителе, таком как оксид алюминия. Процесс может быть проведен в одну или несколько стадий в неподвижном или движущемся слое.
Международная заявка WO 94/29021 описывает катализатор, который содержит носитель, состоящий, в основном, из смешанного оксида магния и алюминия Mg(Al)O и также благородного металла группы VIII, предпочтительно, платины, металла группы IVA, предпочтительно, олова, и возможно, щелочного металла, предпочтительно, цезия. Этот катализатор используют в дегидрогенизации углеводородов, которая может быть проведена в присутствии кислорода.
Патент США 5733518 описывают способ селективного окисления водорода кислородом в присутствии углеводородов, таких как н-бутан, над катализатором, содержащим фосфат германия, олова, свинца, мышьяка, сурьмы или висмута, предпочтительно, олова. Сжигание водорода генерирует, по меньшей мере в одной зоне реакции, тепло реакции необходимое для эндотермической дегидрогенизации.
Европейская заявка ЕР-А 0838534 описывает катализатор для гидрогенизации без водяного пара алканов, в частности, изобутана, в присутствии кислорода. Используемый катализатор содержит металл платиновой группы, нанесенный на носитель, содержащий оксид олова/оксид циркония и имеющий содержание олова, по меньшей мере, 10%. Содержание кислорода в потоке, подаваемом для дегидрогенизации, рассчитывают так, чтобы количество тепла, произведенное реакцией сжигания водорода и кислорода, было равно количеству тепла, требуемого для дегидрогенизации.
Международная заявка WO 96/33151 описывает способ дегидрогенизации алканов с 2-5 атомами углерода в отсутствии кислорода над катализатором дегидрогенизации, содержащим Cr, Mo, Ga, Zn или металл из группы VIII, с одновременным окислением получаемого водорода над восстанавливаемым оксидом металла, например, оксидом Bi, In, Sb, Zn, Ti, Pb или Те. Дегидрогенизацию необходимо прерывать через равномерные интервалы, чтобы повторно окислить восстановленный оксид посредством источника кислорода. Патент США 5430209 описывает соответствующий процесс, в котором стадию дегидрогенизации и стадию окисления проводят последовательно и связанные катализаторы отделяют физически друг от друга. Катализаторы, используемые для селективного окисления водорода, представляют собой оксиды Bi, Sb и Те, а также их смешанные оксиды.
Наконец, международная заявка WO 96/33150 описывает способ, в котором алкан с 2-5 атомами углерода дегидрогенируют над катализатором дегидрогенизации в первой стадии, газ, отходящий от стадии дегидрогенизации, смешивают с кислородом и во второй стадии пропускают над катализатором окисления, предпочтительно, Bi2O3, чтобы селективно окислять образующийся водород до воды, и в третьей стадии газ, отходящий от второй стадии, снова пропускают над катализатором дегидрогенизации.
Известно, что ароматические углеводороды могут быть получены каталитической ароматизацией путем дегидрогенизации углеводородов с открытой цепью (смотри, например, Catalysis VI, стр. 535-542, под ред. Р.Н.Emmet, Reinhold Publishing Co., Нью-Йорк, 1958).
Патент США US 3449461 описывает ароматизацию дегидрогенизацией парафинов с 6-20 атомами углерода с открытой цепью в ароматические углеводороды, включая о-ксилол, при помощи серного катализатора, который содержит благородный металл, такой как палладий или платина.
Заявка на патент США 2004/0044261 описывает способ селективного получения п-ксилола путем конверсии изоалкенов или алкенов с 8 атомами углерода над катализатором, который содержит молекулярные сита с нанесенным благородным металлом переходной группы VIII.
Немецкая заявка DE-A 19727021 описывает способ получения ароматических соединений с 8 атомами углерода из бутенов путем дегидрогенизации смесей винильных углеводородов с 8 атомами углерода, получаемых ди-меризацией технических фракций €4 над катализатором, который содержит, по меньшей мере, один элемент платиновой группы на амфотерном керамическом носителе. Главным продуктом реакции является этилбензол, и дополнительно также формируется о-ксилол.
Задача настоящего изобретения состоит в том, чтобы создать способ дегидрогенизации углеводородов, который гарантирует высокие конверсии, объемные производительности и селективности.
Эту задачу достигают с помощью катализатора, содержащего монолит, состоящий из каталитически инертного материала с низкой площадью поверхности по БЭТ, и слой катализатора, который нанесен на этот монолит и содержит, на оксидном материале носителя, по меньшей мере, один благородный металл, выбранный из группы, состоящей из благородных металлов VIII группы периодической таблицы элементов, при необходимости олова и/или рения, и при необходимости других металлов, где толщина слоя катализатора составляет от 5 до 500 микрон.
Изобретение обеспечивает катализаторы неподвижного слоя с существенно пониженным требованием к количеству благородного металла и улучшенными эксплуатационными показателями. В то же время, глубина проникновения катализатора ограничена величиной от 5 до 500 мкм, предпочтительно от 5 до 250 мкм, более предпочтительно от 25 до 250 мкм, и особенно от 50 до 250 мкм. Глубина проникновения катализатора ограничена толщиной слоя катализатора, нанесенного на монолит.
Слой катализатора на монолите содержит, по меньшей мере, керамическую окись в качестве носителя катализатора и, по меньшей мере, один благородный металл, выбранный из элементов переходной группы VIII периодической таблицы элементов, особенно палладий, платину или родий, возможно, рений и/или олово. Носители катализатора представляют собой одну или несколько керамических оксидов элементов из главных подгрупп второй, третьей и четвертой групп, и побочных подгрупп третьей и четвертой групп (подгруппы IVB) элементов и лантанидов, особенно, MgO, СаО, Al2O3, SiO2, ZrO2, TiO2, La2O3 и Се2О3. В особенно предпочтительном варианте выполнения изобретения, носитель катализатора содержит SiO2 и ZrO2, в частности, смешанный оксид SiO2 и ZrO2.
В дополнение к благородным металлам побочной подгруппы VIII группы, возможно использовать другие элементы; в частности, рений и/или олово, которые выступают в качестве добавок к элементам побочной подгруппы VIII группы. Другой составной частью является добавление или легирование либо соединениями элементов главной и побочной подгрупп третьей группы (IIIA или IIIB) или основными соединениями, такими как оксиды щелочных, щелочноземельных или редкоземельных металлов, либо их соединениями, которые могут быть превращены в соответствующие оксиды при температурах выше 400С. Возможно одновременное легирование множеством указанных выше элементов или их соединений. Подходящими примерами являются соединения калия и лантана. Кроме того, катализатор может быть смешан с соединениями серы, теллура, мышьяка, сурьмы или селена, которые во многих случаях вызывают увеличение селективности, возможно, путем частичного "отравления" (замедлители).
Слой катализатора содержит, по меньшей мере, один благородный металл из VIII группы периодической таблицы элементов (Ru, Rh, Pd, Os, Ir, Pt). Предпочтительным благородным металлом является платина. Слой катализатора может, при необходимости, содержать олово и/или рений, предпочтительно олово.
В предпочтительном варианте выполнения изобретения, слой катализатора содержит платину и олово.
Кроме того, слой катализатора может быть легирован другими металлами. В предпочтительном варианте выполнения изобретения, слой катализатора содержит один или несколько металлов побочной подгруппы третьей группы (IIIB) периодической таблицы элементов, включая лантаниды (Sc, Y, La, Се, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu), причем предпочтение отдают церию и лантану, особое предпочтение лантану.
В другом предпочтительном варианте выполнения изобретения, слой катализатора содержит платину, олово и лантан.
Кроме того, слой катализатора может содержать металлы, выбранные из металлов главных подгрупп I и II групп периодической таблицы элементов. В предпочтительном варианте выполнения изобретения, слой катализатора содержит калий и/или цезий. В специфическом варианте выполнения изобретения, слой катализатора содержит платину, олово, лантан и щелочной металл, выбранный из группы, состоящей из калия и цезия.
Слой катализатора, содержащий оксидный материал носителя и, по меньшей мере, один благородный металл VIII группы периодической таблицы элементов, олово и/или рений и, если подходит, другие металлы, нанесен на монолит путем нанесения во влажном состоянии каталитически активного материала. Для этой цели в ином случае также возможно сначала наносить слой носителя катализатора, состоящий из оксидного материала носителя, на монолит путем нанесения во влажном состоянии и импрегнировать этот слой в следующей стадии процесса одним или несколькими различными растворами, содержащими металлы.
Катализаторы согласно изобретению применяют, в частности, для дегидрогенизации алканов в алкены, например, пропана в пропилен или н-бутана в бутены (1- и 2-бутены), для ароматизации при дегидрогенизации и для каталитического сжигания водорода с кислородом.
Монолиты
Подходящие монолитные структуры представляют собой либо металл, либо керамику. Предпочтительно, они состоят из единичных блоков с малыми (0,5-4 мм) параллельными каналами. Предпочтительно, используют монолитные детали либо Corning Incorporated, либо NGK, либо Denso.
Наиболее обычный материал для монолитных структур представляет собой кордиерит (керамический материал, состоящий из оксида магния, ддиоксида кремния и оксида алюминия в отношении 2:5:2). Другие материалы, чьи монолитные структуры коммерчески доступны, представляют собой металлы, муллит (смешанные диоксид кремния и оксид алюминия, отношение 2:3) и карбид кремния. Эти материалы имеют, подобно кордиериту, низкую удельную поверхность по Брунауэру, Эммету и Теллеру (БЭТ) (например, для кордиерита, обычно 0,7 м2/г). Низкая площадь поверхности по БЭТ в контексте этого изобретения представляет собой площадь поверхности по БЭТ<10 м2/г
Предпочтительно, по изобретению используют монолитные детали из кордиерита.
Керамические монолитные элементы доступны с плотностями ячеек 25-1600 cpsi (ячеек на квадратный дюйм, что равно размеру ячеек 5-0,6 мм). При использовании более высокой плотности ячеек, геометрические площади поверхности увеличиваются и, таким образом, катализатор может быть использован более эффективно. Неудобства более высокой плотности ячеек состоят в несколько более трудном процессе производства, более трудном нанесении во влажном состоянии и более высоком падении давления по реактору. Однако падение давления остается очень низким для монолитов с высокой плотностью ячеек (обычно коэффициент ниже 10) по сравнению с реактором с неподвижным слоем, из-за прямых каналов в монолите.
Предпочтительно, плотности ячеек монолитных элементов, используемых по настоящему изобретению, составляют от 100 до 1200 cpsi, наиболее предпочтительно, от 300 до 600 cpsi.
Керамические монолитные элементы могут быть получены путем получения смеси талька, глины и компонента, дающего оксид алюминия и ддиоксид кремния, смешиванием этой смеси, чтобы сформировать литьевую композицию, отливкой этой смеси, сушкой сырца и нагреванием его при температуре от 1200 до 1500С, чтобы сформировать керамику, содержащую, главным образом, кордиерит и имеющую низкий коэффициент теплового расширения. Вообще говоря, паста с подходящими реологическими свойствами и составом может быть экструдирована в монолитный носитель. Паста обычно состоит из смеси керамических порошков подходящих размеров, неорганических и/или органических добавок, растворителя (вода), химического пластификатора (кислота), чтобы подобрать рН, и постоянного связующего (коллоидный раствор или золь). Добавки могут быть пластификатором или поверхностно-активным веществом, чтобы подобрать вязкость пасты, либо временным связующим, которое может быть позже выжжено. Иногда добавляют стеклянные или углеродные волокна, чтобы увеличить механическую прочность монолита. Постоянное связующее должно улучшать целостность монолита.
Кордиеритные монолиты могут быть произведены из загрузки, состоящей из талька, каолина, прокаленного каолина и оксида алюминия, которые все вместе дают химическое соединение из SiO2 от 45 до 55, Al2O3 от 32 до 40 и MgO от 12 до 15% масс. Тальк представляет собой материал, состоящий, главным образом, из водного силиката магния, Mg3Si4O10(ОН)2. В зависимости от источника и чистоты талька, он может также быть ассоциирован с другими минералами, такими как тремолит (CaMg3(5SiO3)4), серпентин (3MgO×2SiO2×2H2O), антофиллит (Mg7(OH)2(Si4O11)2), магнезит (MgCO3), слюда и хлорит.
Также может быть использована экструзия для производства монолитов из других материалов, таких как SiC, B4C, Si3N4, BN, AlN, Al2O3, ZrO2, муллит, титанат Al, ZrB2, сталон, перовскит, углерод и TiO2.
В экструзии, в дополнение к качеству фильеры и природе и свойствам материалов, используемых для изготовления литьевых смесей, также имеют значение дополнительные добавки, рН, содержание воды и сила, используемая в экструзии, относительно свойств монолитных продуктов. Добавки, используемые в экструзии, представляют собой, например, целлюлозы, CaCl2, этиленгликоли, диэтиленгликоли, спирты, воск, парафин, кислоты и теплостойкие неорганические волокна. Помимо воды, также могут быть использованы другие растворители, такие как кетоны, спирты и простые эфиры. Добавление добавок может приводить к улучшенным свойствам монолитов, таким как производство микротрещин, которые увеличивает устойчивость к тепловому удару, лучшую пористость и абсорбционную способность, и увеличенную механическую прочность или низкое тепловое расширение.
Процедура нанесения во влажном состоянии
По настоящему изобретению, голую монолитную структуру покрывают слоем носителя катализатора, содержащего одну или несколько керамических окисей, или слоем катализатора, содержащим каталитически активные металлы и при необходимости другие элементы (промоторы), уже нанесенные на керамический оксидный материал носителя, где покрытие производят по процедуре нанесения во влажном состоянии.
Макропористая структура керамических монолитов облегчает закрепление нанесенного во влажном состоянии слоя. Метод, которым производят нанесение во влажном состоянии, может быть разделен на две методики: макропористый носитель может быть (частично) заполнен материалом, нанесенным во влажном состоянии, с высокой поверхностной площадью, или нанесенное во влажном состоянии может быть размещено в качестве слоя в порах на керамическом носителе. Заполнение пор приводит к самому сильному взаимодействию между монолитом и нанесенным во влажном состоянии, когда большая часть слоя покрытия фактически фиксируется внутри пор носителя вместо того, чтобы только присоединяться к внешней поверхности каналов монолита. Этот тип покрытия проводят с использованием раствора (или золя) материала, подлежащего размещению, либо с использованием раствора, содержащего очень малые коллоидные частицы. Недостаток покрытия посредством наполнения пор состоит в том, что количество покрытия, которое может быть нанесено, ограничено, потому что на одной стадии поры будут полностью заполнены, и нанесение во влажном состоянии станет недостижимым.
Предпочтительно, носитель катализатора или слой катализатора наносят на монолитные стенки. Нанесение слоя на монолитные стенки имеет преимущества в том, что возможны более высокие загрузки, и что диффузия в более толстые стенки не влияет на реакцию. Этот тип нанесения проводят путем покрытия суспензией из частиц размера, аналогичного макропорам в монолитных стенках, например, кордиерита (обычно 5 мкм). Принцип, по которому проводят процедуры покрытия из суспензии, следующий. Монолит помещают в жидкость, содержащую суспендированные частицы. Поры в стенке занимают жидкостью, осаждающей частицы на стенках монолита, потому что эти частицы не могут входить в поры, покидая слой нанесенных частиц.
Получают раствор или суспензию для нанесения во влажном состоянии, в которые погружают на короткое время высушенный монолит (окунают). Предпочтительно, окунают в золь или суспензию предварительно высушенную и вакуумированную монолитную деталь. Монолит удаляют из жидкости, и большую часть жидкости стряхивают, причем остаток мягко сдувают сжатым воздухом. Чаще всего, это делают с использованием "воздушного шабера", тонкой щели для продувания сжатого воздуха, потому что этим методом одновременно очищают весь ряд каналов. Затем монолит высушивают в горизонтальном положении при непрерывном вращении вокруг его оси, чтобы предотвращать, чтобы сила тяжести не вызывала неровного распределения при нанесении во влажном состоянии. Наконец, покрытие фиксируют на монолите стадией прокалывания при высокой температуре. Загрузка, получаемая нанесением во влажном состоянии, составляет обычно 5-10% масс. для большинства методов. Если требуется загрузка выше, нужно повторять процедуру нанесения. Это может быть сделано после прокаливания, либо монолит можно окунать снова после сушки.
Чтобы получить слой носителя катализатора, монолит может быть покрыт с помощью подходящего золя влажным нанесением. Этот золь может быть получен гидролитическим путем. Один метод получения золя представляет собой гидролиз подходящего алкоксида. Гидролиз алкоксида металла обычно ускоряется присутствием кислоты или основания. В ходе старения золя, протекает процесс поликонденсации, приводящий к сшивке и формированию соединений, подобных полимеру.
В одном из вариантов выполнения настоящего изобретения, монолитную структуру покрывают влажным нанесением с использованием оксида алюминия в виде золя Al. В дополнение к гидролизу алкоксида, упомянутому выше, золь оксида алюминия может быть получен из других его предшественников, например из псевдо-бемита AlO(ОН)×xH2O или гидролизом AlCl3.
Добавки, например, мочевина или органические амины, такие как гексаме-тилентетрамин, могут быть добавлены к золю, чтобы улучшить качество получаемого оксида алюминия. Кроме того, добавки могут влиять на стабильность золей.
Возможно, катионы, например, La, Mg, Zr, Si, которые ингибируют переход активного оксида алюминия в инертную α-фазу, могут быть включены в золь, чтобы стабилизировать нанесенный во влажном состоянии оксид алюминия от спекания после термической обработки.
В другом варианте выполнения настоящего изобретения, монолитную структуру получают нанесением во влажном состоянии диоксида кремния, используя золь Si. Золь Si может быть получен гидролизом тетраалкоксисиликата (ТАОС), тетраметоксисиликата (ТМОС), тетраэтоксисиликата (ТЭОС) и тетрапропоксисиликата (ТПОС). Вследствие того, что ТАОС обычно не смешиваются с водой, часто добавляют спирты в качестве сорастворителей, чтобы получать гомогенный золь.
Другие оксиды могут быть нанесены во влажном состоянии аналогично. Когда используют в нанесении во влажном состоянии смешанные золи, слой смешанных оксидов может быть сформирован на поверхности монолита.
Диоксид кремния может быть легко нанесен с использованием коммерческих коллоидных растворов диоксида кремния, например, типа Ludox AS. Жидкое стекло может быть добавлено, чтобы увеличить целостность покрытия из диоксида кремния. Коллоидные растворы диоксида кремния также могут быть использованы в качестве постоянного связующего для покрытия цеолитов и других материалов, например, диоксида титана и ддиоксида циркония или полимерного катализатора. Нанесение во влажном состоянии может быть проведено аналогично процедурам, описанным выше для нанесения во влажном состоянии оксида алюминия.
Также возможно покрытие влажным нанесением металлических монолитов с предшествующим окислением или без него. В первом случае, адгезия нанесенного во влажном состоянии слоя лучше.
Вместо золя может быть использована суспензия, чтобы получать слой носителя катализатора или слой катализатора. Это может увеличивать количество оксида, нанесенного каждый раз. Кроме того, для получения монолитных катализаторов при нанесении во влажном состоянии могут быть использованы оптимизированные порошки катализатора. Часто с целью улучшения покрытия необходимо размалывание в шаровой мельнице в течение некоторого периода, чтобы понизить размер твердых частиц до определенного размера.
В предпочтительном варианте выполнения изобретения, размер частиц оксидного материала носителя снижают размалыванием до среднего размера от 1 до 40 мкм, предпочтительно, от 5 до 20 мкм. Данный средний размер частиц определяют как включающий 90% частиц.
Активный компонент, который обычно представляет собой металл побочной подгруппы VIII группы, обычно наносят импрегнированием предшественника в виде подходящей соли металла. Вместо импрегнирования, активный компонент также может быть нанесен другими методами, например, распылением предшественника в виде соли металла на носитель. Подходящие предшественники в виде соли металла представляют собой, например, нитраты, ацетаты и хлориды соответствующих металлов; также возможно использовать гидроксиды или комплексные анионы используемых металлов. Предпочтение отдают использованию платины в виде H2PtCl6 или Pt(NO3)2. Подходящие растворители для прекурсоров в виде солей металлов включают как воду, так и органические растворители. Особенно полезными растворителями являются вода и низшие спирты, такие как метанол и этанол.
Чтобы наносить щелочные и щелочноземельные металлы предпочтительно применяют использование водных растворов соединений, которые могут быть превращены в соответствующие оксиды прокаливанием. Подходящие соединения представляют собой, например, гидроксиды, карбонаты, оксалаты, ацетаты или основные карбонаты щелочных и щелочноземельных металлов. Если носитель катализатора легируют металлами главной и побочной подгруппы III группы, часто используют гидроксиды, карбонаты, нитраты, ацетаты, формиаты или оксалаты, которые могут быть превращены в подходящие оксиды прокаливанием, например, La(OH)3, La3(СО3)2, La(NO3)3, ацетат лантана, формиат лантана или оксалат лантана.
В одном предпочтительном варианте выполнения настоящего изобретения, проводят пост-импрегнирование активных компонентов после нанесения во влажном состоянии и кальцинирования сот монолита следующим образом:
Сырье, то есть материал(ы) носителя и - если необходимо - связующее для стабильного нанесения во влажном состоянии, смешивают в подходящем сосуде, емкости и т.д. и перемешивают или размешивают при суспендировании водой. Полученную суспензию разбавляют до требуемого содержания твердого вещества и доводят до определенного рН с использованием определенных кислот и оснований. Затем суспензию прокачивает через непрерывно действующую мельницу для снижения размера частиц до среднего размера от 1 до 40 мкм, предпочтительно, от 5 до 20 мкм; распределение частиц по размерам может быть проконтролировано автономно лазерной дифракцией. Готовую суспензию используют для покрытия.
Нанесение может быть сделано, например, вручную путем использования ручного сопла для процесса направления воздуха. Правильное полное содержание твердого вещества для покрытия может быть определено в предварительной процедуре, для достижения определенной загрузки влажного нанесения в г/дюйм3 или г/л.
Детали погружают по длине, предпочтительно, на 80-90%, но не полностью, в суспензию и - после вынимания их оттуда - поворачивают так, чтобы суспензия стекала через ячейки. Конечную загрузку нанесенного во влажном состоянии определяют путем использования пневматического пульверизатора, чтобы распределять суспензию по каналу и выдувать чрезмерные количества суспензии. Стадии покрытия могут быть повторены для достижения полной целевой загрузки нанесения во влажном состоянии. Опытные образцы сушат после каждой стадии нанесения при температуре от 100 до 200°С, предпочтительно при 120-140°С и прокаливают при температуре 400-750°С, предпочтительно 550-650°С перед следующей стадией.
Толщину слоя определяют заданным полным количеством нанесенного во влажном состоянии покрытия, исходящим из плотности нанесенных во влажном состоянии ингредиентов и распределения их частиц по размерам. В зависимости от реологии суспензии полное количество наносимого во влажном состоянии покрытия должно быть нанесено в более чем одну стадию нанесения.
Для импрегнирования активных компонентов поглощение воды определяют с использованием репрезентативного прототипа. Активные компоненты растворяют в нужной концентрации в воде, и деталь погружают в этот раствор на определенное количество времени, в основном, несколько секунд, затем избыточное количество воды выдувают воздушным соплом. Эти стадии импрегнирования повторяют так часто, как требуется по методике.
После каждой стадии импрегнирования прототипы могут быть высушены и прокалены, как описано выше.
В ином варианте выполнения изобретения активные компоненты уже импрегнируют в материал(-ы) носителя и/или добавляют в суспензию перед или после размалывания. Регулирование полного содержания твердого вещества или рН проводят, как описано выше. Стадии нанесения могут быть повторены для достижения полной заданной загрузки наносимого во влажном состоянии покрытия. Прототипы могут быть высушены после каждой стадии покрытия при температуре 100-200°С, предпочтительно при 120-140°С и прокалены при температуре 400-750°С, предпочтительно 500-650°С перед следующей стадией.
Глубина проникновения (dWC) может быть определена из загрузки наносимого во влажном состоянии покрытия (WCL), плотности наносимого во влажном состоянии покрытия (ρWC) и геометрической площади поверхности (GSA) монолита:
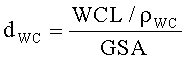
Плотность наносимого во влажном состоянии покрытия (ρWC) может быть определена из плотностей конечного монолитного катализатора (ρкатал.) и субстрата (ρсубстр. ), и загрузки наносимого во влажном состоянии покрытия и удельного веса (SW: общий вес катализатора относительно его объема) монолитного катализатора:
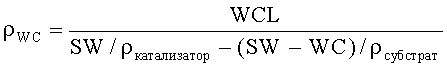
Плотности монолитного катализатора и субстрата могут быть определены посредством измерений плотности с помощью ртути или гелия.
Катализаторы для ароматизации при дегидрогенизации и самой дегидрогенизации, которые наносят в качестве слоя на монолиты
Чтобы получить слой катализатора, пригодный для ароматизации при дегидрогенизации и дегидрогенизации, могут быть использованы так называемые амфотерные керамические окиси, то есть, в частности, оксиды циркония и титана или их смеси; также подходящими являются соответствующие соединения, которые могут быть превращены в эти оксиды прокаливанием. Они могут быть получены известными способами, например золь-гель процессом, осаждением солей, обезвоживанием соответствующих кислот, сухим смешиванием, суспендированием или сушкой распылением.
Подходящие керамические окиси для носителя представляют собой все модификации оксида циркония и оксида титана. Однако было обнаружено, что для получения катализаторов на основе ZrO2 предпочтительно, когда соотношение моноклинной ZrO2 обнаруживаемой дифракцией рентгеновских лучей, более 90%. Моноклинная ZrO2 характеризуется в рентгеновской дифрактограмме двумя сильными сигналами в два-тета диапазоне около 28.2 и 31,5.
Легирование основным соединением может быть произведено в ходе получения, например, соосаждением, или потом, например, импрегнирова-нием керамической окиси соединением щелочного металла или щелочноземельного металла или соединением элемента побочной подгруппы третьей группы или соединением редкоземельного металла.
Содержание щелочного металла или щелочноземельного металла, металла главной или побочной подгруппы III группы, редкоземельного металла или цинка обычно составляет до 20% масс., предпочтительно между 0,1 и 15% масс., более предпочтительно между 0,1 и 10% масс. В качестве источника щелочного и щелочноземельного металла обычно используют соединения, которые могут быть превращены в соответствующие оксиды прокаливанием. Подходящими примерами являются гидроксиды, карбонаты, оксалаты, ацетаты, нитраты или смешанные гидрокарбонаты щелочных и щелочноземельных металлов.
Когда керамический носитель легируют дополнительно металлом главной или побочной подгруппы третьей группы, исходные материалы в этом случае также должны быть соединениями, которые могут быть превращены в соответствующие оксиды прокаливанием. Когда используют, например, лантан, пригодны соединения лантана, которые содержат органические анионы, такие как ацетат лантана, формиат лантана или оксалат лантана.
Компоненты благородного металла могут быть нанесены различными способами. Например, керамический материал носителя катализатора или слой носителя катализатора на монолите может быть импрегнирован или распылен при помощи раствора соответствующего соединения благородного металла, или рения, или олова. Подходящие соли металла для получения таких растворов представляют собой, например, нитраты, галогениды, формиаты, оксалаты, ацетаты благородного металла. Также возможно использовать комплексные анионы, или кислоты этих комплексных анионов, такие как H2PtCl6. Особенно подходящими соединениями для получения катализаторов согласно изобретению, как было обнаружено, являются PdCl2, Pd(OAc)2, Pd(NO3)2 и Pt(NO3)2.
Также могут быть использованы золи благородных металлов с одним или несколькими компонентами, в которых активный компонент присутствует в уже полностью или частично восстановленном состоянии.
Когда используют золи благородных металлов, их получают заранее общепринятым образом, например, восстановлением соли металла или смеси из множества солей металла в присутствии стабилизатора, такого как поливинилпирролидон, и затем его нанесением, либо импрегнированием, либо распылением материала керамического носителя катализатора, либо слоя носителя катализатора. Методика получения раскрыта в немецкой заявке на патент 19500366.7.
Содержание в катализаторе элементов побочной подгруппы VIII группы и, возможно, рения или олова может, например, быть от 0,005 до 5% масс., предпочтительно от 0,01 до 2% масс., более предпочтительно от 0,05 до 1,5% масс. Когда в дополнение используют рений или олово, их отношение к компоненту из благородного металла может быть, например, от 0,1:1 до 20:1, предпочтительно, от 1:1 до 10:1.
Используемые замедляющие добавки (по обычной концепции частичного отравления катализатора) могут, если требуется, представлять собой соединения серы, теллура, мышьяка или селена. Добавление монооксида углерода в ходе работы катализатора также возможно. Было обнаружено, что использование серы особенно предпочтительно, ее удобно применять в форме сульфида аммония (NH4)2S. Мольное отношение компонентов благородного металла к замедляющему соединению может быть от 1:0 до 1:10, предпочтительно, от 1:1 до 1:0,05.
Материал катализатора обычно имеет площадь поверхности по БЭТ до 500 м2/г, обычно от 2 до 300 м2/г, более предпочтительно от 5 до 300 м2/г. Объем пор лежит обычно между 0,1 и 1 мл/г, предпочтительно, от 0,15 до 0,6 мл/г, более предпочтительно от 0,2 до 0,4 мл/г. Средний диаметр пор в мезопорах, определяемый анализом проникновения Hg, лежит обычно между 8 и 60 нм, предпочтительно между 10 и 40 нм. Доля пор, имеющих ширину более 20 нм, варьируется обычно между 0 и 90%; было обнаружено, что предпочтительно использовать носители, имеющие фракцию макропор (то есть пор шириной более 20 нм) более 10%.
Один пример ароматизации при дегидрогенизации, для которой могут быть использованы катализаторы согласно изобретению, представляет собой ароматизацию при дегидрогенизации 3,4- или 2,3-диметилгексена в о-ксилол.
В одном варианте выполнения изобретения материал катализатора, формирующий слой катализатора на монолите, имеет бимодальное распределение радиуса пор и содержит
а) от 10 до 99,9% масс. диоксида циркония и
б) от 0 до 60% масс. оксида алюминия, оксида кремния и/или оксида титана и
в) от 0,1 до 30% масс., по меньшей мере, одного элемента главной подгруппы I или II группы, элемента побочной подгруппы III группы, включая церий и лантан, благородного металла побочной подгруппы VIII группы периодической таблицы элементов, и при необходимости олова,
при условии, что сумма процентов по массе составляет 100. Этот материал катализатора особенно пригоден для дегидрогенизации алканов в алкены и ароматизации при дегидрогенизации, например, 3,4- или 2,3-диметилгексена в о-ксилол.
Этот материал катализатора, формирующий слой катализатора на монолите, предпочтительно содержит
а) от 10 до 99,9% масс., более предпочтительно от 20 до 98% масс., особенно предпочтительно от 30 до 95% масс. диоксида циркония, из которой от 50 до 100% масс., предпочтительно от 60 до 99% масс., особенно предпочтительно от 70 до 98% масс. составляют моноклинная и/или тетрагональная модификации, и
б) от 0,1 до 60% масс., предпочтительно, от 0,1 до 50% масс., особенно предпочтительно, от 1 до 40% масс., в особенности, от 5 до 30% масс. оксида алюминия, диоксида кремния и/или диоксида титана в форме рутила или анатаза, и
в) от 0,1 до 10% масс., предпочтительно от 0 до 8% масс., особенно предпочтительно от 0,1 до 5% масс., по меньшей мере, одного элемента, выбранного из главной подгруппы I или II группы и из побочных подгрупп III и VIII групп периодической таблицы элементов, церия, лантана и/или олова,
где сумма процентов по массе составляет 100.
В одном особенно предпочтительном варианте выполнения, компонент б) состоит из 0,1-30% масс., предпочтительно 0,5-25% масс., особенно предпочтительно 1-20% масс. диоксида кремния.
Материал катализатора предпочтительно состоит из композиции, состав которой указан выше.
Материал катализатора, формирующий слой катализатора на монолите, содержит от 70 до 100%, предпочтительно, от 75 до 98%, особенно предпочтительно, от 80 до 95% пор более 20 нм, предпочтительно между 40 и 5000 нм.
Для получения материала катализатора, формирующего слой катализатора на монолите, могут быть использованы предшественники окидов циркония, титана, лантана, церия, кремния и алюминия (формирующих носитель), которые могут быть преобразованы прокаливанием в оксиды. Они могут быть получены известными способами, например, золь-гель процессом, осаждением солей, дегидратацией соответствующих кислот, сухим смешиванием, суспендированием или сушкой распылением. Например, смешанный оксид ZrO2×xAl2O3×xSiO2 может быть приготовлен изначально получением обогащенного водой оксида циркония общей формулы ZrO2×xH2O при осаждении подходящего предшественника, содержащего цирконий. Подходящие предшественники циркония представляют собой, например, Zr(NO3)4, ZrOCl2 или ZrCL4. Само осаждение проводят добавлением основания, такого как NaOH, Na2CO3 и NH3, оно описано, например, в европейской заявке ЕР-А 849224.
Для получения смешанного оксида ZrO2×xSiO2, предшественник Zr, полученный как указано выше, может быть смешан с Si-содержащим предшественником. Хорошо подходящими предшественниками SiO2 являются, например, содержащие воду золи SiO2, такие как Ludox®. Эти два компонента могут быть смешаны, например, простым механическим смешиванием или сушкой распылением в скруббере с распылителем.
При использовании смешанных оксидов возможно влиять на структуру пор в необходимом направлении. Размеры частиц различных предшественников влияют на структуру пор. Таким образом, например, макропоры в микроструктуре могут быть генерированы использованием Al2O3, имеющим низкую потерю при сжигании и определенное распределение частиц по размерам. Оксид алюминия, который, как было обнаружено, предпочтителен для этой цели, представляет собой Puralox (Al2O3, имеющий потерю при сжигании около 3%).
Чтобы получить смешанный оксид ZrO2×xSiO2×Al2O3, смесь порошков SiO2×ZrO2, полученная, как описано выше, может быть смешана с предшественником, содержащим Al. Это может быть проведено, например, простым механическим смешиванием в смесителе. Однако смешанный оксид ZrO2×xSiO2×Al2O3 также может быть получен в одну стадию сухим смешиванием индивидуальных предшественников.
Другой возможный путь производства носителя, имеющего специфическое распределение радиуса пор для указанных выше катализаторов, в целевом способе, состоит в добавлении в ходе получения различных полимеров, которые могут быть частично или полностью удалены прокаливанием так, чтобы формировать поры в определенных интервалах радиуса пор. Смешивание полимеров и предшественников оксидов может быть проведено, например, простым механическим смешиванием или сушкой распылением в скруббере с распылителем.
Было обнаружено, что использование ПВП (поливинилпирролидона) особенно предпочтительно для производства носителей, имеющих бимодальное распределение радиуса пор. Если ПВП добавляют в ходе стадии производства к одному или нескольким предшественникам оксидов элементов Zr, Ti, La, Се, Al или Si, после прокаливания формируются макропоры в интервале от 200 до 5000 нм. Другое преимущество использования ПВП состоит в том, что носитель может быть сформирован более легко. Таким образом, экструдаты, имеющие хорошие механические свойства, могут быть произведены из свежеосажденного, содержащего воду ZrO2×xH2O, который предварительно был высушен при 120°С, с добавлением ПВП и муравьиной кислоты, даже без других предшественников оксидов.
Смешанные оксидные носители катализаторов обычно имеют более высокие площади поверхности по БЭТ после прокаливания, чем чистые носители из ZrO2. Площади поверхности по БЭТ смешанных оксидных носителей обычно лежат в интервалах от 40 до 300 м2/г, предпочтительно от 50 до 200 м2/г, особенно предпочтительно, от 60 до 150 м2/г. Объем пор катализаторов, применяемых согласно настоящему изобретению, составляет обычно от 0,1 до 0,8 мл/г, предпочтительно, от 0,2 до 0,6 мл/г.Средний диаметр пор катализаторов согласно настоящему изобретению, который может быть определен Hg-порометрией, составляет от 5 до 30 нм, предпочтительно, от 8 до 25 нм. Кроме того, предпочтительно, чтобы от 10 до 80% объема пространства пор был составлен порами >40 нм.
Прокаливание смешанных оксидных носителей предпочтительно проводят после нанесения активных компонентов и проводят при температуре от 400 до 750°С, предпочтительно от 500 до 700°С, особенно предпочтительно от 550 до 650°С. Температура прокалывания должна обычно быть, по меньшей мере, столь же высокой, как температура реакции дегидрогенизации.
Материал катализатора имеет бимодальное распределение радиуса пор. Поры лежат в интервале, главным образом, до 20 нм и в интервале от 40 до 5000 нм. Относительно объема пор, эти поры составляют, по меньшей мере, 70% пор. Доля пор, которые менее 20 нм, составляет обычно от 20 до 60%, в то время как доля пор в интервале от 40 до 5000 нм составляет обычно аналогично от 20 до 60%.
Легирование смешанных оксидов основным соединением может быть проведено либо при их получении, например, соосаждением, либо затем, например, импрегнированием смешанного оксида соединением щелочного металла, или соединением щелочноземельного металла, или соединением элемента побочной подгруппы III группы, или соединением редкоземельного металла. Особенно подходящими легирующими добавками являются К, Cs и La.
Нанесение компонента, активного в дегидрогенизации, который представляет собой благородный металл побочной подгруппы VIII группы, обычно проводят импрегнированием подходящим предшественником в виде соли металла, которая может быть превращена в соответствующий оксид металла прокаливанием. В качестве альтернативы импрегнированию, компонент активный в дегидрогенизации также может быть нанесен среди других методов, например, распылением предшественника в виде соли металла на носитель. Подходящие предшественники в виде соли металла представляют собой, например, нитраты, ацетаты и хлориды соответствующих металлов, или комплексные анионы используемых металлов. Предпочтение отдают использованию платины в виде H2PtCl6 или Pt(NO3)2. Растворители, которые могут быть использованы для предшественников в виде соли металла, представляют собой воду и органические растворители. Особенно подходящими растворителями являются низшие спирты, такие как метанол и этанол.
Другие подходящие предшественники при использовании благородных металлов в качестве компонента, активного в дегидрогенизации, представляют собой подходящие золи благородных металлов, которые могут быть получены одним из известных методов, например, восстановлением соли металла восстанавливающим агентом в присутствии стабилизатора, такого как ПВП. Методика получения всесторонне описана, например, в немецкой заявке DE-A 19500366.
В качестве предшественников щелочного металла и щелочноземельного металла обычно используют соединения, которые могут быть превращены в соответствующие оксиды прокаливанием, Примерами подходящих предшественников являются гидроксиды, карбонаты, оксалаты, ацетаты или смешанные гидрокарбонаты щелочных и щелочноземельных металлов.
Если смешанный оксидный носитель дополнительно или исключительно легируют металлом главной или побочной подгруппы группы III, исходные материалы в этом случае также должны быть соединениями, которые могут быть превращены в соответствующие оксиды прокаливанием. Если используют лантан, подходящие исходные соединения представляют собой, например, оксикарбонат лантана, La(ОН)3, La2(СО3)3, La(NO3)з или соединения лантана, содержащие органические анионы, например, ацетат лантана, формиат лантана или оксалат лантана.
Катализаторы дегидрогенизации, которые наносят в качестве слоев на монолиты
Другие подходящие материалы катализаторов дегидрогенизации содержат обычно оксид металла, выбранный из группы, состоящей из диоксида циркония, оксида цинка, оксида алюминия, диоксида кремния, диоксида титана, оксида магния, оксида лантана, оксида церия и их смесей, в качестве керамического материала носителя. Предпочтительные носители представляют собой диоксид циркония и/или диоксид кремния; специфическое предпочтение отдают смесям из диоксида циркония и диоксида кремния.
Активная композиция материала катализатора дегидрогенизации обычно содержит один или несколько благородных металлов побочной подгруппы VIII группы, предпочтительно, платину и/или палладий, особенно предпочтительно платину. Кроме того, катализатор дегидрогенизации может далее содержать один или несколько элементов главных подгрупп I и/или II групп, предпочтительно, калий и/или цезий. Катализатор дегидрогенизации также может далее содержать один или несколько элементов побочной подгруппы III группы, включая лантаниды и актиниды, предпочтительно лантан и/или церий. Наконец, катализатор дегидрогенизации может также содержать олово, и предпочтительно олово содержит.
В предпочтительном варианте выполнения изобретения, катализатор дегидрогенизации содержит, по меньшей мере, один элемент побочной подгруппы VIII группы, по меньшей мере, один элемент главных подгрупп I и/или II групп, по меньшей мере, один элемент побочной подгруппы III группы, включая лантаниды и актиниды, и олово.
Для получения материала носителя катализатора дегидрогенизации, наносимого во влажном состоянии на монолит в виде слоя, возможно использовать предшественники оксидов циркония, кремния, алюминия, титана, магния, лантана или церия, которые могут быть преобразованы в оксиды прокаливанием. Они могут быть произведены известными методами, например золь-гель процессом, осаждением солей, дегидратацией соответствующих кислот, сухим смешиванием, суспендированием или сушкой распылением. Для получения смешанного оксида ZrO2×SiO2, содержащий цирконий предшественник, полученный выше, может быть смешан с содержащим кремний предшественником. Хорошо подходящие предшественники SiO2 представляют собой, например, содержащие воду золи SiO2, такие как Ludox™, либо метоксифункциональные метилполисилоксаны, такие как SILRES® MSE 100. Эти два компонента могут быть смешаны, например, простым механическим смешиванием или сушкой распылением в сушилке с распылителем.
Материалы керамических носителей катализатора для катализаторов дегидрогенизации, которые наносят во влажном состоянии в виде слоев на монолиты согласно настоящему изобретению, обычно имеют высокие площади поверхности по БЭТ после прокаливания. Площади поверхности по БЭТ обычно более 40 м2/г предпочтительно более 50 м2/г, особенно предпочтительно более 70 м2/г. Объем пространства пор катализаторов дегидрогенизации, используемых согласно настоящему изобретению, составляет обычно от 0,2 до 0,6 мл/г, предпочтительно от 0,25 до 0,5 мл/г. Средний диаметр пор катализаторов дегидрогенизации, используемых по настоящему изобретению, который может быть определен Hg порометрией, составляет от 3 до 30 нм, предпочтительно от 4 до 25 нм.
Кроме того, материалы катализатора дегидрогенизации согласно настоящему изобретению имеют бимодальное распределение радиуса пор. Поры имеют размеры в интервале до 20 нм и в интервале от 40 до 5000 нм. Эти поры все вместе составляют, по меньшей мере, 70% всего объема пространства пор катализатора дегидрогенизации. Доля пор менее 20 нм лежит обычно в интервале от 20 до 60%, в то время как доля пор в интервале от 40 до 5000 нм составляет обычно аналогично от 20 до 60%.
Активный в дегидрогенизации компонент, который представляет собой благородный металл побочной подгруппы VIII группы, обычно наносят импрегнированием подходящим предшественником в виде соли металла.
Вместо импрегнирования компонент, активный в дегидрогенизации, также может быть нанесен другими методами, например, распылением предшественника в виде соли металла на носитель. Подходящими предшественниками в виде соли металла являются, например, нитраты, ацетаты и хлориды соответствующих металлов; также возможно использовать комплексные анионы данных металлов. Предпочтение отдают использованию платины в виде H2PtCl6 или Pt(NO3)2. Подходящие растворители для предшественников в виде соли металла включают как воду, так и органические растворители. Особенно предпочтительные растворители представляют собой воду и низшие спирты, такие как метанол и этанол.
Для нанесения щелочных и щелочноземельных металлов используют предпочтительно водные растворы соединений, которые могут быть превращены в соответствующие оксиды прокаливанием. Подходящие соединения представляют собой, например, гидроксиды, карбонаты, оксалаты, ацетаты или основные карбонаты щелочных и щелочноземельных металлов. Если носитель катализатора легируют металлами главной или побочной подгруппы III группы, часто используют гидроксиды, карбонаты, нитраты, ацетаты, формиаты или оксалаты, которые могут быть превращены в соответствующие оксиды прокаливанием, например, La(ОН)3, La2(СО3)3, La(NO3)3, ацетат лантана, формиат лантана или оксалат лантана.
При использовании благородных металлов в качестве компонентов, активных в дегидрогенизации, подходящие предшественники также включают соответствующие золи благородных металлов, которые могут быть получены одним из известных методов, например, восстановлением соли металла путем использования восстанавливающего агента в присутствии стабилизатора, такого как ПВП. Методика получения всесторонне описана, например, в немецкой заявке DE-A 19500366.
Количество благородного металла, присутствующего в качестве компонента, активного в дегидрогенизации, в катализаторах дегидрогенизации, используемых согласно настоящему изобретению, составляет от 0 до 5% масс., предпочтительно от 0,05 до 1% масс., особенно предпочтительно, от 0,05 до 0,5% масс.
Другие компоненты активной композиции могут быть нанесены либо в ходе производства носителя, например, соосаждением, либо потом, например, импрегнированием носителя подходящими соединениями-предшественниками. Используемые соединения-предшественники представляют собой обычно соединения, которые могут быть превращены в соответствующие оксиды прокаливанием. Подходящими предшественниками являются, например, гидроксиды, карбонаты, оксалаты, ацетаты, хлориды или смешанные гидрокарбонаты соответствующих металлов.
В предпочтительных вариантах выполнения изобретения, активная композиция далее содержит следующие дополнительные компоненты:
- по меньшей мере, один элемент главных подгрупп I или II группы, предпочтительно цезий и/или калий в количестве от 0 до 20% масс., предпочтительно от 0,1 до 15% масс., особенно предпочтительно, от 0,1 до 10% масс.;
- по меньшей мере, один элемент побочной подгруппы III группы, включая лантаниды и актиниды, предпочтительно, лантан и/или церий, в количестве от 0 до 20% масс., предпочтительно от 0,1 до 15% масс., особенно предпочтительно, от 0,2 до 10% масс.;
- олово в количестве от 0 до 10% масс.
Катализатор дегидрогенизации, предпочтительно, не содержащий галогена.
Прокаливание носителей катализатора, импрегнированных рассматриваемым раствором соли металла, обычно проводят при температуре от 400 до 750°С, предпочтительно от 500 до 700°С, особенно предпочтительно от 550 до 650°С за период от 0,5 до 6 часов.
Катализатор сжигания водорода
Предпочтительный катализатор, который катализирует сжигание водорода, содержит благородный металл побочной подгруппы VIII и/или I группы периодической таблицы и/или олово. Особенно предпочтительными являются катализаторы, содержащие платину, возможно, в комбинации с оловом. В качестве материалов носителя для этих катализаторов могут быть использованы оксиды металлов, выбранные из группы, состоящей из диоксида циркония, оксида цинка, оксида алюминия, диоксида кремния, диоксида титана, оксида магния, оксида лантана, оксида церия, цеолитов и их смесей. Предпочтительные носители из оксидов металлов представляют собой диоксид циркония, оксид магния, диоксид кремния, оксид цинка и оксид алюминия или их смеси.
Водородный катализатор может быть использован для селективного окисления водорода, чтобы подавать тепло для дегидрогенизации, как описано выше. В другом варианте выполнения процесса, катализатор сжигания водорода также может быть использован в качестве катализатора очистки, чтобы селективно удалять кислород из содержащих углеводороды потоков.
Каталитическая дегидрогенизация
Дегидрогенизация может быть проведена как окислительная или неокислительная дегидрогенизация. Неокислительная дегидрогенизация может быть проведена автотермически или не автотермически. Дегидрогенизация может быть проведена изотермически или адиабатически.
Неокислительную каталитическую дегидрогенизацию алканов, предпочтительно, проводят автотермически. В этом случае, кислород дополнительно подмешивают к реакционной газовой смеси дегидрогенизации в, по меньшей мере, одной реакционной зоне, и водород и/или углеводород, присутствующий в реакционной газовой смеси, сжигают, по меньшей мере, частично, что производит, по меньшей мере, часть тепла дегидрогенизации, требуемого по меньшей мере в одной реакционной зоне непосредственно в реакционной газовой смеси.
В предпочтительном варианте выполнения катализатор согласно изобретению используют для дегидрогенизации пропана до пропилена или для дегидрогенизации бутана до бутена.
Одна особенность неокислительного метода по сравнению с окислительным методом состоит, по меньшей мере, в промежуточном формировании водорода, которое проявляется в присутствии водорода в газообразном продукте дегидрогенизации. При окислительной дегидрогенизации свободного водорода не находят в газообразном продукте дегидрогенизации.
Подходящей формой реактора является трубчатый реактор с неподвижным слоем или кожухотрубный реактор. В этих реакторах катализатор (катализатор дегидрогенизации и, если подходит, специализированный катализатор окисления) расположен в виде неподвижного слоя в реакционной трубе или в пучке реакционных труб. Обычные внутренние диаметры реакционных труб составляют от около 10 до 15 см. Типичный кожухотрубный реактор дегидрогенизации содержит от около 300 до 1000 реакционных труб. Внутренняя температура в реакционных трубах обычно варьируется в интервале от 300 до 1200°С, предпочтительно, в интервале от 500 до 1000°С. Рабочее давление составляет обычно от 0,5 до 8 бар, часто от 1 до 2 бар, когда используют низкое разбавление паром, или еще от 3 до 8 бар, когда используют высокое разбавление паром (соответствующее процессу активного реформинга с водяным паром (STAR процесс) или процессу Линде) для дегидрогенизации пропана или бутана Phillips Petroleum Со. Обычная часовая объемная скорость подачи газа (GHSV) составляет от 500 до 2000 ч-1 относительно используемого углеводорода. Геометрия катализатора может, например, быть сферической или цилиндрической (полое или твердое тело). Также возможно эксплуатировать множество трубчатых реакторов или реакторов из пучка труб с неподвижным слоем рядом с друг другом, из которых, по меньшей мере, каждый находится поочередно в состоянии регенерации.
Неокислительная каталитическая автотермическая дегидрогенизация также может быть проведена при гетерогенном катализе в псевдоожиженном слое, по процессу Snamprogetti/Yarsintez-FBD. Соответственно, два псевдоожиженных слоя эксплуатируют параллельно, из которых один находится обычно в состоянии регенерации. Рабочее давление составляет обычно от 1 до 2 бар, температура дегидрогенизации обычно от 550 до 600°С. Тепло, требуемое для дегидрогенизации, может быть введено в реакционную систему предварительным нагреванием катализатора дегидрогенизации до температуры реакции. Добавление параллельного потока, содержащего кислород, позволяет обходиться без подогревателя и генерировать требуемое тепло непосредственно в реакторной системе, сжигая водород и/или углеводороды в присутствии кислорода. Если это допустимо, можно добавлять дополнительный поток, содержащий водород.
Неокислительную автотермическую дегидрогенизацию, предпочтительно, проводят в тарельчатом реакторе. Этот реактор содержит один или несколько последовательных слоев катализатора. Число слоев катализатора может быть от 1 до 20, предпочтительно от 1 до 6, более предпочтительно от 1 до 4, и в особенности от 1 до 3. Слои катализатора предпочтительно продувают радиально или вдоль оси реакционным газом. Обычно такой тарельчатый реактор эксплуатируют путем использования неподвижного слоя катализатора. В самом простом случае, неподвижные слои катализатора расположены вдоль оси в шахтном реакторе или в кольцевых зазорах концентрических цилиндрических решеток. Шахтный реактор соответствует одной тарелке. Проведение дегидрогенизации в одном шахтном реакторе соответствуют одному из вариантов выполнения изобретения. В другом, предпочтительном варианте выполнения изобретения дегидрогенизацию проводят в тарельчатом реакторе, имеющем 3 слоя катализатора.
Обычно количество газообразного кислорода, добавляемое к реакционной газовой смеси, выбирают таким образом, чтобы требуемое для дегидрогенизации алкана (например, пропана и/или н-бутана) количество тепла генерировалось за счет сгорания водорода, присутствующего в реакционной газовой смеси, и любых углеводородов, присутствующих в реакционной газовой смеси, и/или углерода, присутствующего в форме кокса. Обычно полное количество подаваемого кислорода относительно общего количества алкана составляет от 0,001 до 0,5 моля/моль, предпочтительно от 0,005 до 0,25 моля/моль, более предпочтительно от 0,01 до 0,25 моля/моль. Кислород может быть использован либо в форме чистого кислорода, либо в форме кислородсодержащего газа, который содержит инертные газы. Для предотвращения потери высших алканов и алкенов при работе (см. ниже), может быть предпочтительно, когда содержание кислорода в кислородсодержащем газе является высоким, и составляет по меньшей мере 50% объемн., предпочтительно по меньшей мере 80% объемн., более предпочтительно по меньшей мере 90% объемн.. Особенно предпочтительным кислородсодержащим газом является кислород технической чистоты с содержанием O2, приблизительно 99% объемн. Кроме того, возможен способ, при котором в качестве кислородсодержащего газа подают воздух.
Водород, сжигаемый для производства тепла, представляет собой водород, формируемый при каталитической дегидрогенизации алкана, а также любой водород, дополнительно добавляемый к реакционной газовой смеси в качестве газообразного водорода. Количество присутствующего водорода должно быть предпочтительно таким, чтобы мольное отношение Н2/O2 в реакционной газовой смеси немедленно после подачи кислорода составляло от 1 до 10 молей/моль, предпочтительно, от 2 до 6 молей/моль. В многоступенчатых реакторах это относится к каждой промежуточной подаче газов, содержащих кислород и водород.
Водород сжигают каталитически. Обычно используемый катализатор дегидрогенизации также катализирует сжигание углеводородов и водорода с кислородом так, чтобы, в принципе, не требовалось никакого специального катализатора окисления, кроме этого. В одном варианте выполнения изобретения, операцию производят в присутствии одного или нескольких катализаторов окисления, которые селективно катализируют сжигание водорода с кислородом в присутствии углеводородов. Сжигание этих углеводородов с кислородом дает СО, CO2 и воду, следовательно, происходит только в малой степени. Катализатор дегидрогенизации и катализатор окисления, предпочтительно, присутствуют в различных зонах реакции.
Когда реакцию проводят в более чем одну стадию, катализатор окисления может присутствовать только в одной, в более чем одной или во всех зонах реакции.
Предпочтение отдают размещению катализатора, который селективно катализирует окисление водорода в точках, где наблюдается более высокое парциальное давления кислорода, чем в других точках в реакторе, в особенности около точки подачи кислородсодержащего газа. Газообразные кислород и/или водород можно подавать в одну или несколько точек в реакторе.
В одном из вариантов выполнения способа согласно изобретению, есть промежуточная подача кислородсодержащего газа и водорода выше каждой тарелки реактора. В другом варианте выполнения способа согласно изобретению, кислородсодержащий газ и водород подают выше каждой тарелки, кроме первой тарелки. В одном из вариантов выполнения изобретения, слой специального катализатора окисления присутствует ниже по ходу потока каждой точки подачи и сопровождается слоем катализатора дегидрогенизации. В другом варианте выполнения изобретения, нет специального катализатора окисления. Температура дегидрогенизации составляет обычно от 400 до 1100°С; давление в последнем слое катализатора тарельчатого реактора составляет обычно от 0,2 до 5 бар абсолютных, предпочтительно от 1 до 3 бар абсолютных. GHSV (часовая объемная скорость подачи газа) составляет обычно от 500 до 2000 ч-1 и при работе с высокой нагрузкой даже до 100000 ч-1, предпочтительно от 4000 до 16000 ч-1.
Каталитическая ароматизация при дегидрогенизации
Ароматизацию при дегидрогенизации обычно проводят при температурах от 300 до 800°С, предпочтительно от 400 до 700°С, более предпочтительно от 450 до 650°С и при давлениях от 100 мбар до 100 бар, предпочтительно от 1 до 30 бар, более предпочтительно, от 1 до 10 бар, при LHSV (часовая объемная скорость жидкости) от 0,01 до 100 ч-1, предпочтительно от 0,1 до 20 ч-1. В дополнение к смеси углеводородов, могут присутствовать разбавители, такие как СО2, N2, инертные газы или пар. Аналогично, возможно добавлять, если требуется, водород, когда объемная доля водорода к углеводородам (газообразным) может быть от 0,1 до 100, предпочтительно, от 0,1 до 20. Добавляют водород, либо может быть использован тот, который сформирован при дегидрогенизации и, если подходит, рециклизованный, чтобы удалять углерод, который накапливается на поверхности катализатора, увеличивая время реакции.
В дополнение к постоянному (непрерывному) добавлению газа, который предотвращает осаждение кокса в ходе реакции, имеется возможность периодической регенерации катализатора пропусканием водорода или воздуха над ним. Сама регенерация происходит при температурах в интервале от 300 до 900°С, предпочтительно от 400 до 800°С со свободным окислительным агентом, предпочтительно воздухом или смесями воздуха и азота, и/или в восстановительной атмосфере, предпочтительно водороде. Регенерация может быть проведена при атмосферном давлении, пониженном давлении, либо выше атмосферного. Подходящие давления составляют, например, от 500 мбар до 100 бар.
Изобретение далее иллюстрируется следующими примерами
Примеры
Примеры 1-12
Получение катализаторов дегидрогенизации/ароматизации согласно изобретению способом А
Способ А описывает пост-импрегнирование активных компонентов после нанесения во влажном состоянии и прокаливания сот монолита.
Получение суспензии для нанесения во влажном состоянии
Для получения суспензии для нанесения во влажном состоянии используют высушенный распылением порошок диоксида циркония BASF SE D9-89. 17000 г этого порошка диспергируют в 15000 мл воды и смешивают с 1200 г SILRES® MSE 100. Полученная суспензия с теоретическим полным содержанием твердого вещества 52% имеет рН от 3,2 до 3,8. Эту полученную суспензию размалывают в непрерывно действующей мельнице в течение около 1 ч до достижения конечного размера частиц 11,5 мкм в среднем с допустимым диапазоном +/-1,5 мкм для 90% частиц. Распределение частиц по размерам многократно контролируют в ходе процесса в режиме офлайн методом лазерной дифракции. Готовая суспензия вновь имеет рН от 3,2 до 3,8. Полученную суспензию используют для покрытия.
Процесс нанесения во влажном состоянии
Эту суспензию затем должным образом разбавляют для нанесения на керамический субстрат при полном содержании твердого вещества около 50% с отклонением +/-1%. Нанесение покрытия проводили посредством процесса с направленной продувкой воздуха. Правильное полное содержание твердого вещества для покрытия должно быть определено в предварительной процедуре, чтобы достичь заданной загрузки наносимого во влажном состоянии покрытия в г/дюйм3 или г/л.
Используют монолитные детали из кордиерита с 400 cpsi (ячеек на квадратный дюйм) от Corning Incorporated. Для создания прототипа эти детали погружают на 80-90% в суспензию; после вынимания их поворачивают, и суспензия течет через ячейки. Загрузки наносимого во влажном состоянии покрытия достигают путем использования пневматического пульверизатора, чтобы распределять суспензию поперек канала и выдувать чрезмерное количество суспензии. Стадии покрытия повторяют, чтобы в итоге достигать определенной конечной загрузки нанесенного во влажном состоянии покрытия 4,5 г/дюйм3. Опытные образцы высушивают после каждой стадии покрытия при температуре от около 120 до 140°С в течение 15 минут с частым реверсированием воздушного потока и прокаливают при 560°С в течение 3 ч перед следующей стадией.
Импрегнирование активными компонентами
Активные компоненты наносят в две стадии впитыванием воды из растворов материалов предшественников. Сначала определяют впитывание воды на репрезентативном образце, и рассчитывают концентрации на основе этого впитывания.
214,9 г (МЭА)2Pt(ОН)6 в виде раствора концентрацией 17,22% в МЭА (МЭА = моноэтаноламин) и 82 г КОН растворяют в 8000 г воды, получая бледно-желтый раствор с рН около 12. Покрытые детали погружают в раствор на время пропитывания 15 секунд. Когда детали вынимают из раствора, их мягко продувают указанным выше воздушным соплом, и высушивают в реверсивном воздушном потоке при температуре от 120 до 140°С в течение еще 15 минут. Прокаливание происходит в печи или устройстве для непрерывного обжига при 560°С в течение около 3 ч.
Для получения второго раствора для импрегнирования CsNO3, SnCl2×Н2О и La(NO3)3 растворяют в 6750 г воды, принимая во внимание то же впитывание воды для влажного импрегнирования. Детали погружают и вынимают из раствора через 15 секунд, свободно продувают умеренным воздушным потоком. Эти детали высушивают в реверсивном воздушном потоке при температуре от 120 до 140°С в течение еще 15 минут. Прокаливание происходит в печи или устройстве для непрерывного обжига при 560°С в течение около 3 ч.
Таблица 1 ниже показывает краткий обзор свойств произведенных катализаторов. Глубину проникновения (dWC) определяли из загрузки наносимого во влажном состоянии покрытия (WCL), плотности наносимого во влажном состоянии покрытия (ρWC) и геометрической площади поверхности (GSA) монолита:
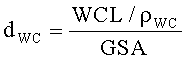
Плотность наносимого во влажном состоянии покрытия (ρWC) определяли из плотностей конечного монолитного катализатора (ркатализатор) и кордиеритного субстрата (рсубстрат), и загрузки наносимого во влажном состоянии покрытия и удельного веса (SW: общий вес катализатора относительно его объема) монолитного катализатора:
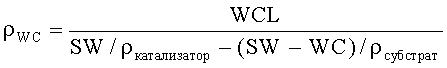
Плотности монолитного катализатора и субстрата определяли посредством измерений плотности с помощью ртути или гелия.
Пример 13
Получение катализаторов дегидрогенизации/ароматизации по изобретению способом Б
Способ Б отличается только слегка от способа А тем, что активные компоненты уже импрегнированы в материал носителя и размолоты для формирования суспензии перед нанесением на монолитный субстрат.
Получение суспензии для нанесения во влажном состоянии и импрегнирование активными компонентами
6220 г диоксида циркония D9-89 импрегнируют 131,4 г раствора соли Pt, используемой в примерах с 1 по 12, и 23 г КОН, растворенного в 1730 г воды. Эту импрегнированный диоксид циркония затем импрегнируют раствором 45 г SnCWW, 35 г CsNOs и 237 г La(NO3)3, растворенных в 3270 г воды, содержащей 13 г HCl (37%). Полученную суспензию размалывают при рН 3,6 в непрерывно действующей мельнице в течение около 1 ч, чтобы достигнуть конечного размера частиц 10,5 мкм в среднем (с допустимым отклонением +/-1,5 мкм для 90% частиц). Распределение частиц по размерам контролируют несколько раз в ходе процесса в режиме офлайн лазерной дифракцией. Готовая суспензия имеет рН от 3,6 до 4. Полученную суспензию используют для покрытия.
Процесс нанесения во влажном состоянии
Нанесение проводят, как описано выше, в 3 отдельные стадии, сопровождаемые сушкой при температуре около 130°С и прокаливанием при 590°С после каждой стадии покрытия.
Таблица 2 ниже показывает краткий обзор свойств произведенного в примере катализатора. Глубину проникновения определяли, как описано в примерах с 1 по 12.
Сравнительный пример 1
Получение экструдатов катализаторов для дегидрогенизации/ароматизации
Катализатор производили в форме экстудата по примеру 4 из немецкой заявки DE 19937107. Загрузка Pt в этом катализаторе была 4,0 г/л.
Дегидрогенизация пропана в лабораторном масштабе
Примеры с 14 по 19 и сравнительные примеры с 2 по 5
Общая процедура
Реакторы состоят из камеры, изготовленной из стали 1.4841, окруженной изолирующим материалом и кожухом от внешнего давления, изготовленным из стали 1.4541. Этот реактор разработан для адиабатического метода. Сам кожух от давления имеет поддерживающий подвод тепла посредством нагревающего контура, чтобы компенсировать потери тепла из камеры. Камера имеет внутренний диаметр 20 мм. Эту камеру заполняют катализатором. Выше адиабатической области, в верхней трети (выше по ходу относительно катализатора), имеется предварительный нагреватель с медной рубашкой.
Пропан, азот, водород и воздух дозировали в реактор в газообразной форме посредством регуляторов массового расхода (от Brooks). Воздух добавляли отдельно через трубку в 8 см выше по ходу слоя катализатора. Воду перемещали из резервуара посредством насоса для ВЭЖХ в испаритель из стальной трубы, и образующийся пар направляли с нагреванием в реактор.
Отходящие газы вводили через регулятор давления и фильтр в водный сепаратор, охлажденный до 10°С. Ниже сепаратора, отходящий газ пропускали через регулятор выходного давления приблизительно при 1,3 бар (абс.) в работающий в онлайн-режиме газовый хроматограф (НР5890, от Agilent) для анализа и определения конверсии и селективностей.
Для проведения дегидрогенизации пропана катализатор устанавливали в середину адиабатической секции реактора. Катализаторы согласно изобретению представляли собой в каждом случае круглые с сотовыми ячейками стержни длиной 101,6 мм и диаметром 15 мм. В случае катализаторов сравнения, устанавливали экструдаты 1,5 мм на 20 мл. Выше и ниже слоя катализатора устанавливали инертный материал (сферы из стеатита 2-3 мм).
Для первой активации катализатор восстанавливали потоком водорода (12 л при н.у./ч) при 450°С и 3 бар (абс.) в течение 45 минут. В ходе дегидрогенизации температуру предварительного нагревателя устанавливали в 450°С, а давление реакции составляло 1,5 бар (абс). Дегидрогенизацию проводили автотермически, то есть с одновременным сжиганием водорода, чтобы обеспечивать тепло, необходимое для дегидрогенизации. Продолжительность цикла дегидрогенизации была 12 ч.
Между циклами дегидрогенизации, катализатор регенерировали выжиганием кокса и последующим восстановлением водородом. Кокс выжигали заранее при температуре подогревателя 450°С и давлении 3 бар (абс.) в обедненном воздухе (смесь азот/воздух, воздушный поток 15 л при н.у./ч) с содержанием кислорода 1% объемн. После этого содержание воздуха постепенно повышали до 100% (поток воздуха 80 л при н.у./ч) и температуру увеличивали до 550°С. Восстановление водородом проводили при 450°С, 3 бар (абс.) и с потоком водорода 12 л при н.у./ч.
Таблица 3 ниже показывает результаты автотермической дегидрогенизации пропана при загрузке (часовая объемная скорость подачи газа, GHSV) 2000 л (н.у.) (пропана)/л (катализатора)/ч. Состав подаваемой газовой смеси был 41,0% объемн. пропана, 41,0% объемн воды, 5,1% объемн. водорода, 10,3% объемн. азота и 2,6% объемн. кислорода. Указанные величины представляют собой средние значения из трех циклов дегидрогенизации со свежим катализатором. Из этого становится ясно, что катализаторы согласно изобретению (из примеров 8, 12 и 13) достигают существенно более высокой производительности по пропилену относительно Pt, чем экструдаты катализаторов по предшествующей технологии. Это верно для катализаторов, произведенных как по способу А (примеры с 1 по 12), так и способу Б (пример 13).
Таблица 4 ниже показывает результаты автотермической дегидрогенизации пропана при различных загрузках (GHSV) от 1000 до 6000 лпропана (н.у.)/лкатализатора/ч и составе подачи 42,6% объемн. пропана, 42,6% объемн. воды, 4,2% объемн. водорода, 8,5% объемн. азота и 2,1% объемн. кислорода. Эти величины представляют собой средние значения из трех циклов катализатора, сформированные более чем за 35 циклов. Из этого становится ясно, что катализатор согласно изобретению (пример 10), даже после длительного времени работы и при различных загрузках, достигает существенно более высокой производительности по пропилену относительно Pt, чем экструдаты катализаторов по предшествующей технологии.
Дегидрогенизация пропана в масштабе пилотной установки
Испытания дегидрогенизации пропана проводили в адиабатическом реакторе из нержавеющей стали (сталь 1.4841, внутренний диаметр 36 мм, длина 4 м), который состоит из 3 слоев катализатора около 90 см длиной каждый в зависимости от типа катализатора. Температуру внутри слоя катализатора контролировали двумя 14-точечными термоэлементами (внутренний диам. 6 мм), вставленными через середину слоя катализатора, одним сверху и одним снизу. Реакционная среда состоит из смеси пропана, водорода, пара и чистого O2. О2 и пар вводили в виде смеси в три отдельные точки дозирования, по одной перед каждым катализатором. Расстояние между точками дозирования и слоями катализатора варьировало, но обычно оно было между 554 и 150 мм. Кроме того, скорости потока О2 слегка изменяли по ходу цикла дегидрогенизации, в то время как скорости подачи других компонентов поддерживали постоянными. Перед вводом в реактор смесь водорода и пропана предварительно подогревали пропусканием через отдельные нагреватели при 520, 525, 530, 580°С.
Когда испытание проводили с катализатором в форме экструдата, использовали 3 мм экструдат катализатора из сравнительного примера 1. Когда испытание проводили с монолитным катализатором, использовали катализатор из примера 11. Всего укладывали вместе 6 монолитов (34 мм ди-ам. на 15 см длины), чтобы сформировать один слой. В центре каждого монолита просверливали 6 миллиметровое отверстие вдоль направления оси, что обеспечивало возможность установки в него термоэлементов. Периметры верхней и нижней части монолита уплотняли стекловолокном для предотвращения возникновения обходного потока. В каждом случае промежуток между двумя слоями катализатора заполняли слоем стеатита.
Испытания активности проводили непрерывно, чередуя циклы дегидрогенизации и регенерации (примерно по 10 часов каждый). После каждого цикла дегидрогенизации реактор продували N2. Процедуру регенерации начинали при давлении 4,5 бар разбавленной смесью воздуха в течение 240 мин., а затем чистым воздухом в течение 8 часов. В конце цикла регенерации проводили 6 циклов повышения давления и его снижения (чередуя между давлением 0,5 и 4 бар каждые 10 мин.), чтобы удалять остаточные следы кокса на поверхности. После продува реактора N2 восстанавливали катализатор, начиная с разбавленного H2 в течение 30 мин., затем чистым H2 в течение 30 мин. при 500°С. Затем реакционную смесь вновь вводили в реактор для следующего цикла дегидрогенизации.
Конверсия и селективность приводимые для каждого цикла являются усредненными величинами по данному циклу. Объемную скорость определяли как скорость подачи пропана, деленную на объем катализатора, где объем катализатора определяют как объем слоя, занятого катализатором, включая объем пустот.
Пример 20
Всего 18 монолитов из примера 11 с общим весом 1448 г (2,30 л) равномерно распределяли в три слоя. Каждый слой состоял из 6 монолитов, и содержание Pt было 2,0 г Pt /л объема реактора. 6360 г/ч пропана (GHSVC3=1400 лC3/лкат./ч) и 75 г/ч H2 подавали в реактор в виде реакционной смеси с O2 и H2O (пар), подаваемой в каждой точке подачи. Пар подавали со скоростью 1000 г/ч в каждую точку подачи, в то время как скорость O2 варьировали по всему циклу в порядке, соответствующем таблице 5 ниже:
Средняя конверсия пропана и селективность по пропилену по циклу составляет 35% и 95%, соответственно, с объемной производительностью 420 кгпропилена./кгPt/ч.
Пример 21
Все условия оставались теми же, как в примере 20, но с 8600 г/ч пропана и подачей О2 по таблице 6 ниже:
Средняя конверсия пропана и селективность по пропилену по циклу были 28% и 95%, соответственно, с объемной производительностью 446 кг (пропилена)/кг(Pt)/ч.
Сравнительный пример 6
Всего 3,27 кг (2,49 л) 3 мм экструдатов равномерно распределяли в три слоя. Каждый слой состоял из 1090 г катализатора (830 мл). Загрузка Pt была 4,0 гPt/л объема реактора. 6360 г/ч пропан (GHSVC3=1300 лC3/лкат./ч) и 75 г/ч Н2 вводили в реактор в качестве реакционной смеси с O2 и H2O, подаваемой в каждую точку подачи. Пар подавали со скоростью 1000 г/ч в каждую точку подачи, в то время как скорость Oz варьировали по всему циклу, согласно по таблице 7 ниже:
Средняя конверсия пропана и селективность по пропилену по циклу составляют 35% и 95%, соответственно, с объемной производительностью 210 кг (пропилена)/кг (Pt)/ч.
Сравнительный пример 7
Все условия оставались теми же, как в сравнительном примере 7, но с 8600 г/ч пропана и подачей O2 по таблице 8 ниже:
Средняя конверсия пропана и селективность по пропилену по циклу составляют 29% и 96%, соответственно, с объемной производительностью 242 кг (пропилена)/кг (Pt)/ч.
Дегидрогенизация н-бутана в лабораторном масштабе Пример 22 и сравнительный пример 8
Дегидрогенизацию н-бутана проводили в электрически нагреваемом трубчатом реакторе (сталь 1.4841, внутренний диаметр 18 мм), н-бутан, азот, водород и воздух дозировали в реактор в газообразной форме посредством регуляторов массового расхода. Воздух добавляли отдельно через 10 см трубку выше по ходу слоя катализатора. Воду перемещали из резервуара посредством насоса для ВЭЖХ в испаритель из стальной трубы, и образующийся пар направляли в реактор.
Отходящие газы вводили через регулятор давления в водный сепаратор, охлажденный до 10°С. Ниже по ходу потока сепаратора, отходящий газ пропускали в работающий в онлайн режиме газовый хроматограф (µGC, от Varian) для анализа и определения конверсии и селективностей.
Чтобы проводить дегидрогенизацию н-бутана, катализатор устанавливали в середину изотермической секции реактора. В случае катализатора по изобретению, устанавливали круглый с сотовыми ячейками стержень длиной 101,6 мм и диаметром 15 мм. В случае сравнительного катализатора, устанавливали круглый с сотовыми ячейками стержень длиной 101,6 мм и диаметром 15 мм. В случае сравнительного катализатора, устанавливали 20 мл 1,5 мм экструдата. Выше и ниже по ходу потока слоя катализатора вводили инертный материал (сферы стеатита 2-3 мм).
Для первой активации катализатор восстанавливали потоком водорода 10 л (н.у.)/ч при 500°С и 2,5 бар (абс.) в течение 30 минут. В ходе дегидрогенизации температуру в реакторной печи устанавливали в 570°С. Давление в реакторе было 1,5 бар (абс). Продолжительность цикла дегидрогенизации составила 12 ч. Дегидрогенизацию проводили при загрузке (часовая объемная скорость потока газа, GHSV) 500 л (н.у.) (н-бутана)/л (катализатора)/ч со следующим составом подаваемой газовой смеси: 41,4% объемн. н-бутана, 16,6% объемн. воды, 18,6% объемн. водорода, 18,7% объемн. азота и 4,7% объемн. кислорода.
Между циклами катализатор регенерировали выжиганием кокса и затем восстановлением водородом при 2,5 бар (абс). Кокс был заранее выжжен обедненном воздухом (азот/воздушная смесь, поток воздуха 5 л (н.у.)/ч) с содержанием кислорода 4% объемн. при температуре в печи 400°С. После этого содержание воздуха постепенно увеличивали до 100% (поток воздуха 33 л (н.у.)/ч) и температуру повышали до 500°С. В ходе восстановления более 30 минут температура была 500°С, и поток водорода был 10 л (н.у.)/ч.
Таблица 9 ниже показывает средние значения из трех циклов катализаторов, сформированные по меньшей мере по 20 циклам. Также в случае дегидрогенизации бутана, становится ясно, что катализатор по изобретению из примера 8, с подобными конверсией и селективностью, достигает существенно более высокой, относительно платины, производительности по пропилену, чем экструдаты катализаторов по предшествующей технологии.
Пример 23
Дегидрогенизация н-бутана в масштабе пилотной установки
Реакторы состоят из стальной камеры, изготовленной из стали 1.4841, которую окружают изолирующим материалом и кожухом защиты от внешнего давления, изготовленным из стали 1.4541. Реактор предназначен для адиабатического метода. Кожух защиты от давления сам имеет поддерживающий подвод тепла посредством нагревательной манжеты, чтобы компенсировать потери тепла из камеры. Камера имеет внутренний диаметр 52,5 мм и длину 2,25 м. Камеру заполняли катализатором. Выше по ходу относительно адиабатического реактора имеется стальной предварительный нагреватель, осуществляющий подачу тепла посредством нагревающей манжеты.
н-Бутан, азот, водород, кислород и воздух дозировали в реактор в газообразной форме через регуляторы массового расхода. Кислород для дегидрогенизации добавляли отдельно через трубки в 25 см выше слоя катализатора. Воду аналогично подавали в стальные трубы испарителя в жидкой форме через регуляторы массового расхода, а водяной пар, который формировался, направляли в реактор в нагретой форме.
Отходящие газы вводили через регулятор давления и фильтр в водный сепаратор, охлажденный до 10°С. Ниже по ходу сепаратора отходящий газ направляли в работающий в онлайн режиме газовый хроматограф (НР5890, от Agilent) для анализа и определения конверсии и селективностей.
Для проведения дегидрогенизации н-бутана катализатор укладывали в три слоя в адиабатической секции реактора, каждые отделенные друг от друга инертным слоем стеатита (кольца, высота × наружный диаметр × внутренний диаметр = 5×3×2 мм). Полный объем установленного катализатора был 1 л; его разделяли на три слоя катализатора одинакового объема. В случае катализатора согласно изобретению устанавливали сотовые стержни диаметром 5 см и заданных длин. В случае сравнительного катализатора использовали 3,0 мм экструдат.
Для первой активации катализатор восстанавливали потоком водорода 200 л (н.у.)/ч при 500°С и 2,5 бар (абс.) в течение 45 минут. В ходе дегидрогенизации температуру подогревателя устанавливали 450°С, и давление реакции было 2,25 бар (абс). Дегидрогенизацию проводили автотермически, то есть с одновременным сжиганием водорода для обеспечения необходимого тепла дегидрогенизации. Продолжительность цикла дегидрогенизации была 12 ч.
Дегидрогенизацию проводили при загрузке (часовая объемная скорость потока газа, GHSV) 650 л (н.у.) (н-бутана)/л (катализатора)/ч (1,7 кг/час н-бутана) и со следующим составом подаваемой газовой смеси: 35% объемн. н-бутана, 41% объемн. воды, 16% объемн. водорода, 2,7% объемн. азота и 5,3% объемн. кислорода. Кислород подавался порциями выше по ходу слоев катализатора: 40% выше первого слоя в направлении потока, 35% выше второго слоя и 25% выше третьего слоя. В ходе дегидрогенизации температуру подогревателя устанавливали 470°С. При продолжительности цикла дегидрогенизации 12 ч, давление реакции было 1,5 бар (абс.).
Между циклами дегидрогенизации, катализатор регенерировали выжиганием кокса, и затем восстанавливали водородом. Кокс заранее выжигали при температуре предварительного нагревателя 450°С и давлении 4,8 бар (абс.) обедненном воздухом с содержанием кислорода 1% объемн. (смесь азот/воздух, поток воздуха 250 л (н.у.)/ч). После этого содержание воздуха постепенно увеличивали до 100% (поток воздуха 1500 л (н.у.)/ч) и температуру повышали до 500°С. Восстановление водородом проводили при 500°С, 2,5 бар (абс.) и потоке водорода 200 л (н.у.)/ч.
Таблица 10 ниже показывает средние значения из трех циклов сформированных катализаторов. При дегидрогенизации бутана в масштабе пилотной установки также становится ясно, что катализатор согласно изобретению (PDR7927), с подобными конверсией и селективностью, достигает существенно более высокой, относительно Pt, производительности по бутену, чем объемные катализаторы по предшествующей технологии.
Примеры 24 и 25
Ароматизацию при дегидрогенизации проводили в нагреваемом электричеством трубчатом реакторе (сталь 1.4841, внутренний диаметр 21 мм). 20 мл экструдата катализатора из сравнительного примера 1 или 12 мл катализатора из примера 1 устанавливали в реактор и активировали водородом при 400°С в течение 1 ч. Сырье, дозируемое в реактор, представляло собой водород, воду и фракцию С8 с, приблизительно, 95% масс. предшественников о-ксилола, преимущественно, 3,4- и 2,3-диметилгексена-2 ("ДМГ" для краткости). Водород дозировали в реактор в газообразной форме через регулятор массового расхода. Воду и фракцию С8 направляли из резервуара посредством насосов для ВЭЖХ в стальной трубчатый испаритель, и газовую смесь, которая формировалась, пропускали в реактор в нагретой форме. Ароматизацию при дегидрогенизации проводили при температуре печи 400°С, давлении 1 бар (абс.), 14 г/ч органических компонентов, отношениях H2O/ДМГ 16/1 моль/моль и Н2/ДМГ 3/1 моль/моль.
Таблица 11 ниже показывает средние результаты ароматизации за более чем 4 часа времени проведения. Аналогично ароматизации в о-ксилол, становится ясно, что катализатор согласно изобретению (PDR6936), с подобными конверсиями, достигает существенно более высокой, относительно Pt, производительности по о-ксилолу, чем экструдаты катализаторов по предшествующей технологии.
Пример 25
Получение катализатора для селективного окисления водорода кислородом в присутствии углеводородов
Процедура пропитки по влагоемкости для Pt/Sn на носитель из альфа-Al2O3
2-4 мм сферы альфа-оксида алюминия от Axens (оксид алюминия Spheralite 512) использовали в качестве материала носителя. Процедура импрегнирования была следующая:
i чтобы определить начальную влажность материала оксида алюминия, подлежащего импрегнированию,
a) 10 г материала носителя оксида алюминия помещали в пластмассовую ампулу.
b) Добавляли к материалу деионизованную воду (ДИ) на 10% больше полного веса носителя
c) Смешивали воду и носитель. Когда достигали точки начальной влажности, материал носителя комковали.
d) Для образца оксида алюминия 10 г добавляли 4 г воды. Весовой процент воды, подлежащий добавлению к носителю, определили в 40% масс.
ii Определяли количество раствора нитрата Pt, необходимого, для получения желательного уровня Pt. Для 50 г материала носителя из оксида алюминия, 0,52 г раствора нитрата Pt с концентрацией нитрата Pt 13,46% разбавляли до 20 мл раствора и добавляли.
iii Эту жидкость выливали в сухой порошок. Эти два компонента смешивали до получения гомогенной смеси.
iv Смесь сушили при 75°С в течение 16 часов.
v Желательное количество SnCl2×2H2O необходимо было рассчитать и растворить в ДИ воде. Для 50 г материала носителя из оксида алюминия, 0,046 г твердого хлорида олова растворяли в 20 мл воды и добавляли.
vi Смесь сушили при 75°С в течение 16 часов.
vii Прокаливание проводили при 540°С в течение 2 часов.
Процедура нанесения во влажном состоянии Pt/Sn-фритты на монолитный субстрат
Процедура нанесения во влажном состоянии была следующая:
i Размалывание или уменьшение размера частиц фритты.
a) Получали суспензию с содержанием твердых веществ 35%, используя Pt/Sn на носителе из оксида алюминия и ДИ воду.
b) Эту суспензию помещали в керамическую в шаровую мини-мельницу.
c) Эту шаровую мельницу помещали на барабан фирмы US Stoneware, и регулятор скорости устанавливали на скорость около 107 об/мин.
г) Размалывание проводили в течение около 9 часов, пока 90% частиц не станет менее 10 мкм.
ii Суспензию выгружали в емкость для хранения.
ii Вырезали стержни из куска материала Corning, блока необработанной подложки 400 cpsi/6,5 мил. Эти стержни имеют 16 мм в диаметре и 100 мм длины. Полный объем был 324 мл.
iv Суспензию смешивали до однородного состояния. Затем сосуд разгружали в цилиндр, который был немного большего диаметра, чем стержень. Стержень погружали в суспензию и вынимали через 5 секунд. Избыточную суспензию стряхивали со стержня, и каналы продували до чистоты.
v Смесь сушили при температуре от 75 до 90°С в течение 16 часов.
vi Стадии 4 и 5 повторяли, пока высушенное нанесенное во влажном состоянии покрытие не достигало значений между 0,16 и 0,17 г/мл.
vii Прокаливание проводили при 540°С в течение 2 часов.
viii Массу стержня определяли при температуре стержня выше 150°С. При взвешивании стержня в теплом состоянии может быть определен верный вес загрузки нанесенного во влажном состоянии покрытия. Когда стержень остывает, он впитывает воду, что приводит к неверной загрузке нанесенного во влажном состоянии покрытия.
Характеристики полученного катализатора даны в таблице 12 ниже. Глубину проникновения определяли, как описано в примерах с 1 по 12.
Сравнительный пример 9
Получение насыпного катализатора для селективного сжигания Ог с водородом
Катализатор получали по международной заявке WO 2005097715, пример А.
Пример 26 и сравнительный пример 10
Удаление кислорода каталитическим сжиганием с водородом в лабораторном масштабе
Селективное каталитическое сжигание О2 проводили в электрически нагреваемом трубчатом реакторе (сталь 1.4841, внутренний диаметр 18 мм). н-бутан, бутадиен-1,3, азот, водород и воздух дозировали в реактор в газообразной форме через регуляторы массового расхода (от Bronckhorst). Воду перемещали из резервуара посредством насоса для ВЭЖХ в стальной трубчатый испаритель, и образующийся пар направляли в реактор.
Отходящие газы вводили через регулятор давления в водный сепаратор, охлажденный до 10°С. Ниже сепаратора отходящий газ пропускали в работающий в онлайн режиме газовый хроматограф (µGC, от Varian) для анализа и определения конверсии и селективностей.
Для осуществления удаления O2 катализатор устанавливали в середину изотермической секции реактора. В случае катализатора согласно изобретению, устанавливали круглый с сотовыми ячейками стержень длиной 101,6 мм и диаметром 15 мм. В случае сравнительного катализатора, устанавливали 20 мл 1,5 мм экструдата. Выше и ниже слоя катализатора имелся слой инертного материала (сферы стеатита 2-3 мм).
В ходе сжигания O2, температуру реакторной печи устанавливали 300°С.Давление реакции было 1,5 бар (абс.). Сжигание O2 проводили при потоке подачи фракции C4 20 л (н.у.)C4/ч и со следующим составом подаваемой газовой смеси: 11,6% объемн. н-бутана, 9,8% бутадиена-1,3, 23,5% объемн. воды, 6,2% объемн. водорода, 47,0% объемн. азота и 1,9% объемн. кислорода.
Таблица 13 ниже показывает результаты каталитического удаления кислорода через 2 дня проведения процесса. В селективном сжигании O2 с водородом становится ясно, что катализатор согласно изобретению, при существенно более высокой относительно Pt загрузке кислорода (WHSV, часовая весовая скорость подачи газа), достигает конверсии O2, сходной с таковой для насыпных катализаторов по предшествующей технологии.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР ОКИСЛЕНИЯ ВОДОРОДА, ЕГО ПРИМЕНЕНИЕ И СПОСОБ РЕКОМБИНАЦИИ ВОДОРОДА | 2013 |
|
RU2603007C2 |
| КАТАЛИЗАТОР И СПОСОБ РЕФОРМИНГА | 2012 |
|
RU2547466C1 |
| КАТАЛИЗАТОР, СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2005 |
|
RU2292237C1 |
| КАТАЛИЗАТОР РЕФОРМИНГА | 2011 |
|
RU2558150C2 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ЧАСТИЧНОГО ОКИСЛЕНИЯ УГЛЕВОДОРОДОВ | 1994 |
|
RU2123471C1 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ МЕТАНА, ПРОЦЕСС ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2016 |
|
RU2715732C2 |
| ПОДЛОЖКА НОСИТЕЛЯ КАТАЛИЗАТОРА, ПОКРЫТАЯ КОМПОЗИЦИЕЙ ГРУНТОВКИ, СОДЕРЖАЩЕЙ ВОЛОКНИСТЫЙ МАТЕРИАЛ | 2005 |
|
RU2400301C2 |
| КАТАЛИЗАТОРЫ | 2012 |
|
RU2591702C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРСОДЕРЖАЩЕГО КАТАЛИЗАТОРА, ПОЛУЧЕННЫЙ КАТАЛИЗАТОР И ЕГО ИСПОЛЬЗОВАНИЕ | 2014 |
|
RU2669372C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2004 |
|
RU2327519C2 |
Изобретение относится к применению катализатора, содержащего монолит и слой катализатора, для дегидрогенизации алканов до алкенов или ароматизации при дегидрогенизации. При этом монолит состоит из каталитически инертного материала с площадью поверхности по БЭТ <10 м2/г, а слой катализатора, который нанесен на монолит, содержит на материале оксидного носителя платину и олово и/или рений и при необходимости другие металлы, толщина слоя катализатора составляет от 5 до 500 микрон. Кроме того, часовая объемная скорость подачи газа составляет от 500 до 2000 ч-1. Изобретение также относится к способам дегидрогенизации алканов до алкенов и ароматизации при дегидрогенизации с применением определенного выше катализатора. Предлагаемое применение катализатора обеспечивает высокие конверсии, объемные производительности и селективности. 3 н. и 11 з.п. ф-лы, 13 табл., 25 пр.
1. Применение катализатора, содержащего монолит, состоящий из каталитически инертного материала с площадью поверхности по БЭТ <10 м2/г, и слой катализатора, который нанесен на монолит и содержит на материале оксидного носителя платину и олово и/или рений и при необходимости другие металлы, где толщина слоя катализатора составляет от 5 до 500 микрон и где часовая объемная скорость подачи газа составляет от 500 до 16000 ч-1, для дегидрогенизации алканов до алкенов или ароматизации при дегидрогенизации.
2. Применение по п.1, где слой катализатора содержит металл из побочной подгруппы третьей группы периодической таблицы элементов, включая лантаниды, и/или щелочной металл или щелочноземельный металл.
3. Применение по п.1 или 2, где слой катализатора содержит по меньшей мере один из элементов, выбранных из группы, состоящей из олова и лантана.
4. Применение по п.1 или 2, где материал оксидного носителя выбран из оксидов магния, кальция, алюминия, кремния, титана и циркония.
5. Применение по п.1 или 2, которое содержит монолит из кордиерита.
6. Применение по п.1 или 2 для дегидрогенизации пропана до пропилена или н-бутана до бутенов.
7. Способ дегидрогенизации алканов до алкенов, включающий взаимодействие в зоне реакции реакционной газовой смеси, содержащей углеводород, с катализатором, который определен в любом из пп.1-5, где дегидрогенизацию проводят в виде окислительной или неокислительной дегидрогенизации.
8. Способ по п.7, где дегидрогенизацию проводят в виде неокислительной дегидрогенизации.
9. Способ по п.8, где неокислительную дегидрогенизацию проводят автотермически.
10. Способ по п.9, где автотермическую дегидрогенизацию выполняют в тарельчатом реакторе.
11. Способ по п.9 или 10, где этот катализатор также катализирует сжигание углеводородов и водорода, присутствующих в этой реакционной смеси.
12. Способ ароматизации при дегидрогенизации, включающий взаимодействие в зоне реакции газовой реакционной смеси, содержащей углеводород, с катализатором, который определен в любом из пп.1-5, где ароматизацию дегидрогенизацией проводят при температуре от 300 до 800°С.
13. Способ по п.12, где к реакционной газовой смеси, содержащей углеводород, добавляют водород.
14. Способ по п.13, где объемное отношение водорода к углеводороду составляет от 0,1 до 100.
| Способ получения серы из сернистых газов | 1985 |
|
SU1305123A1 |
| US 6365543 B1, 02.04.2002 | |||
| Самоходный фрезерный станок | 1978 |
|
SU878443A2 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 4755359 A, 05.07.1988 | |||
| US 5525570 A, 11.06.1996 | |||
| КАТАЛИЗАТОР ВЫСОКОТЕМПЕРАТУРНОГО СЖИГАНИЯ УГЛЕВОДОРОДНОГО ТОПЛИВА (ВАРИАНТЫ) | 2001 |
|
RU2185238C1 |

