Область техники, к которой относится изобретение
Настоящее изобретение относится к оксидному катализатору, к способу получения оксидного катализатора и к способу получения ненасыщенной кислоты и способу получения ненасыщенного нитрила с использованием этого оксидного катализатора.
Уровень техники
Способ получения соответствующей ненасыщенной карбоновой кислоты или ненасыщенного нитрила из пропилена или изобутилена с помощью газофазного каталитического окисления или газофазного каталитического аммоксидирования к настоящему времени хорошо известен. В последние годы уделяется внимание способу получения соответствующей ненасыщенной карбоновой кислоты или ненасыщенного нитрила с помощью газофазного каталитического окисления или газофазного каталитического аммоксидирования с использованием пропана или изобутана вместо пропилена или изобутилена.
Предлагаются различные оксидные катализаторы в качестве катализаторов для газофазного каталитического аммоксидирования. Как правило, оксид, полученный посредством смешивания молибдена, ванадия и чего-либо подобного, по потребности, и кальцинирования смеси, непосредственно используется в качестве такого катализатора. В то же время подход к дополнительной последующей обработке катализатора, кальцинированного таким образом, также исследуют для получения ненасыщенной карбоновой кислоты или ненасыщенного нитрила.
Например, патентный документ 1 описывает подход, который включает импрегнирование катализатора раствором Mo-V-Sb/Te, содержащим один или несколько элементов, выбранных из группы, состоящей из вольфрама, молибдена, хрома, циркония, титана, ниобия, тантала, ванадия, бора, висмута, теллура, палладия, кобальта, никеля, железа, фосфора, кремния, редкоземельного элемента, щелочного металла и щелочноземельного металла. Патентный документ 2 описывает подход, который включает смешивание катализатора с добавками, такими как соединение сурьмы, соединение молибдена, соединение теллура и соединение вольфрама, и воздействие на эту смесь реакции или включает смешивание катализатора или предшественника катализатора с добавками, кальцинирование смеси, с последующей реакцией.
Список цитирований
Патентные документы
[Патентный документ 1] Выложенный патент Японии № 10-028862.
[Патентный документ 2] WO 2003-048553.
Сущность изобретения
Техническая проблема
Патентный документ 1 описывает, что катализатор импрегнируют раствором оксида металла или чего-либо подобного. Однако это импрегнирование требует стадий приготовления раствора, содержащего оксид металла или что-либо подобное, импрегнирования катализатора раствором, сушки катализатора, импрегнированного таким образом, и повторного кальцинирования катализатора, высушенного таким образом. Это осложняет стадии получения, по сравнению с теми, которые свободны от таких операций импрегнирования, и является неудобным для крупномасштабного промышленного производства.
Патентный документ 2 описывает, что добавки, такие как соединения металлов, добавляют к катализатору или предшественнику катализатора. Однако этот способ не дает промышленно достаточную селективность или выходы. Автор настоящего изобретения предполагает, что добавление добавок в слишком малом или большом количестве приводит к понижению селективности или выходов продукта, представляющего интерес, даже если этого и не утверждается явным образом в патентном документе 2. Можно потребовать, чтобы количество добавляемых добавок и их формы определялись совместно для получения максимального воздействия добавок.
Таким образом, настоящее изобретение осуществлено с учетом этих обстоятельств. Одна из целей настоящего изобретения заключается в получении оксидного катализатора, который предназначен для использования при реакции газофазного каталитического окисления или газофазного каталитического аммоксидирования пропана или изобутана и может использоваться для получения продукта, представляющего интерес, при высоких выходах. Другая цель настоящего изобретения заключается в создании способа получения этого оксидного катализатора, который является пригодным для эффективного крупномасштабного промышленного производства оксидного катализатора.
Решение проблемы
Настоящее изобретение заключается в следующем:
[1] Оксидный катализатор в форме частиц для использования в газофазной реакции каталитического окисления или в газофазной реакции каталитического аммоксидирования пропана или изобутана,
оксидный катализатор, содержащий соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, соединения W и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных следующей далее формулой (0), где
W концентрируется по поверхности частицы оксидного катализатора и вблизи нее:
CMo:CV:CW:CNb:CX:CZ=1:a:w:c:x:z... (0)
где CMo представляет собой атомное отношение Mo; CV представляет собой атомное отношение V; CW представляет собой атомное отношение W; CNb представляет собой атомное отношение Nb; CX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; CZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла и щелочноземельного металла; и a, w, c, x и z попадают в диапазоны 0,01≤a≤1, 0<w≤2, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно.
[2] Оксидный катализатор в соответствии с [1], где средняя интенсивность для W, присутствующего в области в пределах 5 мкм от поверхности в направлении центра частицы оксидного катализатора, равна или эквивалентна 1,08 величины интенсивности или больше, для W, присутствующего в оксидном катализаторе в целом.
[3] Способ получения оксидного катализатора для использования в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана, способ включает стадии:
(I) получения заготовки исходных материалов, содержащей соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, необязательное соединение W и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных следующей далее формулой (1);
(II) сушки заготовки исходных материалов для получения сухого порошка; и
(III) кальцинирования сухого порошка, где
стадия кальцинирования (III) включает стадию кальцинирования сухого порошка в присутствии соединения, содержащего W, в форме твердого продукта, с получением кальцинированного на предварительной стадии порошка или в основном кальцинированного порошка, или стадию кальцинирования кальцинированного на предварительной стадии порошка, полученного посредством кальцинирования сухого порошка в присутствии соединения, содержащего W, в форме твердого продукта, с получением в основном кальцинированного порошка, и необязательно включает стадию дополнительного кальцинирования в основном кальцинированного порошка в присутствии соединения, содержащего W, в форме твердого продукта,
твердый продукт удовлетворяет условиям, представленным следующей далее формулой (2), и оксидный катализатор содержит каталитический компонент, имеющий композицию, представленную следующей далее общей формулой (3):
AMo:AV:AW:ANb:AX:AZ=1:a:b:c:x:z... (1)
где AMo представляет собой атомное отношение Mo; AV представляет собой атомное отношение V; AW представляет собой атомное отношение W; ANb представляет собой атомное отношение Nb; AX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; AZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла и щелочноземельного металла; и a, b, c, x и z попадают в диапазоны 0,01≤a≤1, 0≤b≤1, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно;
3 м-1<RW/Mo/d<600000 м-1... (2)
где RW/Mo представляет собой атомное отношение W, содержащегося в твердом продукте, к Mo, содержащемуся в сухом порошке; и d представляет собой средний размер частиц твердого продукта; и
Mo1VaWb+b'NbcXxZzOn... (3)
где a, b, c, x и z, каждый, являются такими, как определено выше в формуле (1); X представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Sb и Te; Z представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc; b' попадает в диапазон 0,001≤b'≤0,3; и n представляет собой значение, которое удовлетворяет условию баланса валентностей.
[4] Способ получения оксидного катализатора в соответствии с [3], где твердый продукт удовлетворяет условиям, представленным следующим далее формулам (4) и (5):
0,001<RW/Mo<0,6... (4)
1 мкм<d<300 мкм... (5)
где RW/Mo и d, каждый, являются такими, как определено выше в формуле (2).
[5] Способ получения оксидного катализатора в соответствии с [3] или [4], где в формуле (1), 0<b≤1.
[6] Способ получения оксидного катализатора по любому из [3]-[5], где соединение Mo, соединение V, соединение W, соединение Nb, соединение, представленное X, и соединение, представленное Z, в сухом порошке, в кальцинированном на предварительной стадии порошке или в порошке, в основном кальцинированном, представляют собой, каждое, по меньшей мере, одно соединение, выбранное из группы, состоящей из соли неорганической кислоты, соли органической кислоты, оксида и сложного оксида.
[7] Способ получения оксидного катализатора по любому из [3]-[6], дополнительно включающий стадию сушки распылением раствора или суспензии, содержащей соединение W, с получением твердого продукта.
[8] Способ получения оксидного катализатора по любому из [3]-[7], где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве 10-80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
[9] Способ получения соответствующей ненасыщенной кислоты из пропана или изобутана посредством газофазной реакции каталитического окисления, способ включает использование оксидного катализатора, полученного с помощью способа по любому из [3]-[8].
[10] Способ получения соответствующего ненасыщенного нитрила из пропана или изобутана посредством газофазной реакции каталитического аммоксидирования, способ включает использование оксидного катализатора, полученного с помощью способа получения по любому из [3]-[8].
Преимущественные воздействия изобретения
Настоящее изобретение может предложить оксидный катализатор, который предназначен для использования в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана и может использоваться для получения продукта, представляющего интерес, с высокими выходами. Настоящее изобретение может также предложить способ получения этого оксидного катализатора, который является пригодным для эффективного крупномасштабного промышленного производства оксидного катализатора
Краткое описание чертежей
[Фигура 1] Фиг.1 представляет собой схематическое изображение для иллюстрации средней интенсивности W.
Описание вариантов осуществления
Далее, один из вариантов осуществления настоящего изобретения (упоминаемый далее просто как "настоящий вариант осуществления") будет описываться подробно. Однако, настоящее изобретение, как предполагается, не ограничивается настоящим вариантом осуществления, ниже, и различные изменения или модификации могут быть проделаны без отклонения от духа или основной идеи настоящего изобретения.
(1) Оксидный катализатор
Оксидный катализатор по настоящему варианту осуществления находится в форме частиц и содержит соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, соединения W, и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных формулой (0), показанной ниже. В этом оксидном катализаторе W концентрируется по поверхности частицы и вблизи нее
CMo:CV:CW:CNb:CX:CZ=1:a:w:c:x:z... (0)
В формуле CMo представляет собой атомное отношение Mo; CV представляет собой атомное отношение V; CW представляет собой атомное отношение W; CNb представляет собой атомное отношение Nb; CX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; и CZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла и щелочноземельного металла (далее, они также коллективно упоминаются как "элемент Z");
a, w, c, x и z попадают в диапазоны 0,01≤a≤1, 0<w≤2, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно, с точки зрения получения продукта, представляющего интерес, при высоких выходах. Они предпочтительно составляют 0,01≤a≤1, 0,001≤w≤1, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1, более предпочтительно 0,1≤a≤0,5, 0,005≤w≤1, 0,1≤c≤0,5, 0,01≤x≤0,5 и 0,001≤z≤0,5, еще более предпочтительно 0,1≤a≤0,45, 0,01≤w≤0,5, 0,1≤c≤0,4, 0,01≤x≤0,4 и 0,001≤z≤0,4.
Кроме того, когда оксидный катализатор имеет композицию, представленную общей формулой (3), и его получают с помощью способа получения, описанного далее, является предпочтительным, чтобы w было таким же, как b+b' в формуле (3).
В оксидном катализаторе по настоящему варианту осуществления изобретения, Mo, V, Nb и, по меньшей мере, один элемент, выбранный из группы, состоящей из Sb и Te, образуют сложный оксид, и этот сложный оксид демонстрирует активность в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана. Этот оксид дополнительно образует комплекс с W с получением при этом оксидного катализатора, который может использоваться для получения продукта, представляющего интерес, с более высокими выходами. Является предпочтительным, чтобы каждый элемент имел композицию в диапазоне, описанном выше, с точки зрения облегчения формирования активных кристаллов в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования.
Когда оксидный катализатор содержит элемент Z, этот элемент Z, особенно предпочтительно, представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Mn, B и щелочноземельного металла, с точки зрения, например, подавления горения аммиака, уменьшения образования нежелательных кристаллов (кристаллов, которые ингибируют получение продукта, представляющего интерес) и уменьшения образования активных центров (активных центров разложения), на которых разлагаются продукт, представляющий интерес, промежуточные соединения (например, пропилен) или газообразные исходные материалы (аммиак и пропан). Ce является особенно предпочтительным с точки зрения уменьшения образования активных центров разложения продукта, представляющего интерес.
В оксидном катализаторе по настоящему варианту осуществления, W концентрируется по поверхности частицы и вблизи нее. В настоящем описании, "концентрирование" W означает, что в определенной области W находится при более высоком содержании, чем в других областях. Кроме того, "поверхность частицы" относится к внешней поверхности оксидного катализатора. Когда оксидный катализатор имеет пору на внутренней стороне (внутри) частицы, область, которая располагается внутри этой поры, внутри частицы и может вступать в контакт с газами, жидкостями, и тому подобное, поступающими снаружи, не применяется как "поверхность частицы". Кроме того, "ближнее пространство" около поверхности частицы относится к области, имеющей глубину, соответствующую приблизительно 1/2 от расстояния от поверхности частицы в направлении центра частицы. Кроме того, центр частицы оксидного катализатора определяется как средняя точка самого большого размера частицы, полученного в сечении частицы оксидного катализатора, разрезанной в произвольном направлении.
Предпочтительная область, где концентрируется W, зависит от типа, размера частиц, количества и способа добавления соединения вольфрама (соединение W), добавляемого в способ получения оксидного катализатора, описанный далее. Является в целом предпочтительным, чтобы W концентрировался в области в пределах 5 мкм от поверхности частицы катализатора в направлении центра частицы. В этом контексте, распределение W в настоящем описании должно измеряться для частиц оксидного катализатора, имеющих размер частиц от 40 до 70 мкм. Даже оксидный катализатор, который не имеет среднего размера частиц, находящегося в этом диапазоне, также считается удовлетворяющим состоянию предпочтительного концентрирования, описанному выше, постольку, поскольку он содержит частицы, имеющие размер частиц от 40 до 70 мкм, и в результате измерения распределения W, для частицы, имеющей этот размер частиц, W концентрируется в пределах 5 мкм от поверхности. Авторы настоящего изобретения считают, что частица оксидного катализатора для использования в реакции в псевдоожиженном слое эффективно оказывает каталитическое воздействие, в особенности, когда она имеет размер частиц от 40 до 70 мкм. Таким образом, оксидный катализатор, имеющий, по меньшей мере, этот размер частиц, становится эффективным посредством придания ему предпочтительного распределения W, описанного выше.
Не требуется, чтобы имелась четкая граница между областью, где концентрируется W, и областью, внутренней по отношению к ней (в сторону центра частицы от нее). Например, содержание W может уменьшаться постепенно, при приближении к центру, от области, где концентрируется W (то есть, области в пределах 5 мкм от поверхности частицы в направлении центра частицы; W присутствует при относительно высокой концентрации) к области на внутренней стороне, где W присутствует при относительно низкой концентрации. Альтернативно, содержание W может уменьшаться резко. Когда оксидный катализатор получают с помощью способа получения, который включает добавление соединения W, как описано далее, W концентрируется по поверхности частицы и вблизи нее таким образом, что содержание W имеет тенденцию к понижению при перемещении ближе к центру.
Средний размер частиц оксидного катализатора предпочтительно составляет от 20 до 100 мкм. Для реакции в псевдоожиженном слое, средний размер частиц более предпочтительно составляет от 30 до 90 мкм, особенно предпочтительно, от 40 до 70 мкм, с точки зрения текучести катализатора. Средний размер частиц катализатора измеряют с помощью способа, описанного далее.
Компоненты (элементы) иные, чем W, содержащиеся в оксидном катализаторе, могут распределяться, независимо от распределения W, в области, где концентрация W высокая (например, в области в пределах 5 мкм от поверхности частицы, где концентрируется W), и в области на внутренней стороне (на центральной стороне частицы). Каждый из этих компонентов может распределяться однородно или может концентрироваться вблизи поверхности, как для W. Является предпочтительным, чтобы компоненты иные, чем W, в частности, по меньшей мере, один элемент, выбранный из группы, состоящей из Mo, V, Nb, Sb и Te, присутствовали в области, где концентрируется W, с точки зрения улучшения рабочих характеристик катализатора посредством взаимодействия между W и другими компонентами и/или, например, посредством замещения W другими компонентами во время кальцинирования или реакции катализатора в присутствии соединения вольфрама (соединения W).
Является предпочтительным, чтобы W присутствовал также в центральной области частицы, где W не концентрируется. В настоящем описании, "центральная область" относится к области, внутренней (на центральной стороне частицы от нее) по отношению к области вблизи поверхности частицы. По меньшей мере, часть W, присутствующая в центральной области, как предполагается, должна замещаться на молибденовых (Mo) или ванадиевых (V) активных центрах в активных кристаллах сложного оксида, и как считается, вносить вклад в термостойкость и окислительно-восстановительную стойкость посредством влияния на кристаллическую структуру сложного оксида. По этой причине, частица катализатора, имеющая W в центральной области, имеет тенденцию к тому, чтобы иметь большое время жизни катализатора и быть преимущественной для долговременного промышленного использования.
Для оксидного катализатора по настоящему варианту осуществления, его композиция или структура в области иной, чем поверхность частицы или область вблизи нее, не является как-либо ограниченной постольку, поскольку W концентрируется по поверхности частицы и в области вблизи нее и катализатор удовлетворяет атомным отношениям, представленным формулой (0), как целое. Является предпочтительным, чтобы W был представлен однородно при отношении, полученном посредством вычитания отношения концентрированного W из атомного отношения, представленного формулой (0) (то есть, в полученном отношении, атомное отношение W меньше, чем отношение, представленное формулой (0)), в области иной, чем поверхность частицы или область вблизи нее, с точки зрения улучшения выхода продукта, представляющего интерес.
В настоящем описании, термин "однородный", описанный для распределения и присутствия W, означает, что в результате анализа профиля распределения композиции по сечению частицы оксидного катализатора, интенсивность W в определенном месте в области иной, чем поверхность частицы или область вблизи нее, находится в пределах ±25% по отношению к средней интенсивности во всей области, иной, чем поверхность частицы или область вблизи нее.
Способ получения оксидного катализатора, содержащего W, сконцентрированный по поверхности частицы и в области вблизи нее, будет описан далее.
Концентрируется ли W по поверхности частицы оксидного катализатора и в области вблизи нее или нет, можно определить на основе отношения (Sw0) средней интенсивности W по поверхности частицы и области вблизи нее к отношению для частицы в целом посредством анализа профиля распределения композиции по сечению частицы оксидного катализатора. Это отношение (Sw0) средней интенсивности должно вычисляться в соответствии с формулой (S1), описанной далее. Кроме того, при анализе профиля распределения, описанном далее, W может рассматриваться как концентрируемый по поверхности частицы и в области вблизи нее, когда отношение (Sw) средней интенсивности W, присутствующего в пределах 5 мкм по глубине от поверхности частицы в направлении центра частицы, к отношению для частицы в целом больше, чем 1,05. Более предпочтительно, отношение (Sw) равно или больше, чем 1,08. Это отношение (Sw) средней интенсивности должно вычисляться в соответствии с формулой (S2), описанной далее. В этом случае, средняя интенсивность W, присутствующего в области в пределах 5 мкм от поверхности частицы оксидного катализатора в направлении центра частицы, может превышать более чем в 1,05 раза или равна или эквивалентна 1,08 величины интенсивности или больше для W, присутствующего в оксидном катализаторе в целом. При анализе профиля распределения, используют SEM-EDX, как описано далее. Могут использоваться и другие общие способы анализа композиции, например, EPMA (электронный микрозондовый рентгеновский анализатор). В таком случае, строят калибровочную кривую для получения соответствия численных значений между SEM-EDX и этим способом, и Sw можно определять на основе калибровочной кривой.
В настоящем описании, Sw0 и Sw конкретно измеряют следующим образом с использованием SEM-EDX: сначала, частица, которая должна анализироваться, погружается в соответствующую смолу матрицы (например, в смолу из ненасыщенного полиэстра), которую затем полируют для сошлифовывания ее целиком, пока не будет экспонироваться сечение погруженной в нее частицы катализатора. При полировке можно использовать, например, водную суспензию, содержащую абразив, такой как оксид алюминия. Однако для измерения абразив смывают. После этого, положение образца регулируют таким образом, что экспонируемое сечение частицы катализатора помещается в наблюдаемое поле зрения при измерении SEM-EDX. Затем сечение частицы катализатора облучают пучками электронов. Интенсивность характерного рентгеновского излучения W (то есть, интенсивность W), испускаемую из части, облучаемой электронными пучками, считают, в то время как область, которая должна анализироваться, сканируется с помощью электронных пучков для осуществления при этом анализа профиля распределения. Является предпочтительным, чтобы условия измерения устанавливались при ускоряющем напряжении 0-15 кВ, времени задержки 1,0 мксек, 5000 сканирований, размер пятна 50 и при рабочем расстоянии 10 мм, используя получение изображений с помощью отраженных электронов. Полупроводник Si (Li) используют в качестве детектора. Этот анализ профиля распределения осуществляют в части, имеющей самый большой размер частицы, в сечении частицы катализатора. Из полученных данных анализа профиля распределения, вычисляют Sw0 и Sw в соответствии с формулами (S1) и (S2), показанными ниже. В этом случае, 10 или больше частиц (каждая имеет размер частицы от 40 до 70 мкм), сечение которых экспонируется вплоть до центра частицы или до его окрестностей, исследуют с помощью анализа профиля распределения. Вычисляют Sw0 и Sw каждой частицы, и определяют их среднее значение.
Sw0 = (Средняя интенсивность W на диаметре от каждого края (поверхности) самого большого размера частицы до части, соответствующей 1/4 от размера частицы)/(средняя интенсивность W на самом большом размере частицы в целом)... (S1)
Sw = (Средняя интенсивность W на диаметре в 5 мкм от каждого края (поверхности) самого большого размера частицы)/(средняя интенсивность W на самом большом размере частицы в целом)... (S2)
Когда Sw0 больше чем 1,00, W считается концентрируемым по поверхности частицы оксидного катализатора и в области вблизи нее.
В этом контексте, "средняя интенсивность" относится к среднему значению интенсивности за исключением фона. Является предпочтительным, чтобы данные собирались на интервалах 1 мкм или меньше. Кроме того, Sw оценивают для частиц катализатора, имеющих размер частицы от 40 до 70 мкм, и не оценивают для частиц катализатора, имеющих размер частиц меньше, чем 40 мкм, или больше, чем 70 мкм.
Фиг.1 представляет собой схематическое изображение для иллюстрации средней интенсивности W при анализе профиля распределения средней интенсивности W в частице оксидного катализатора. Фиг.1 показывает пример измерения/вычисления Sw. Как показано на Фиг.1, воздействие настоящего изобретения может осуществляться более эффективно, когда средняя интенсивность W, присутствующего в области в пределах 5 мкм от поверхности частицы, выше чем для W, присутствующего в частице оксидного катализатора в целом, и Sw больше чем 1,05.
Оксидный катализатор, содержащий W, сконцентрированный по поверхности частицы и в области вблизи нее, имеет высокую износостойкость из-за присутствия очень твердого W по поверхности и в области вблизи нее и, особенно предпочтительно, применяется для реакции в псевдоожиженном слое. Кроме того, W имеет более высокую температуру плавления, чем другие компоненты, содержащиеся в катализаторе, и по этой причине демонстрирует воздействие предотвращения осаждения оксида, содержащего Mo, с низкой температурой плавления, по поверхности. Таким образом, такой оксидный катализатор является особенно пригодным для использования, когда температура реакции является высокой. Кроме того, W с высокой температурой плавления, сконцентрированный по поверхности частицы катализатора и в области вблизи нее, как считается, также имеет воздействие предотвращения адгезии катализаторов, приписываемой вытеканию компонентов с низкими температурами плавления, или предотвращения ухудшения текучести катализатора в реакции в псевдоожиженном слое.
Для частицы оксидного катализатора, его поверхность имеет самую высокую частоту контактов между частицами и высокую частоту экспонирования для атмосферы реакции (газ/температура/давление, и тому подобное). Таким образом, W, который концентрируется в области в пределах 5 мкм от поверхности, то есть, в области, относительно близкой к поверхности, может эффективно оказывать свое воздействие, даже если W добавляют к оксидному катализатору в малом количестве. Кроме того, может быть уменьшено отрицательное воздействие на рабочие характеристики, такое как разложение аммиака под действием избыточного добавленного W. В дополнение к этому, при этом можно экономить на количестве добавленного W. По этой причине, такой оксидный катализатор является предпочтительным также и с экономической точки зрения.
Количество W, содержащегося на центральной стороне частицы, где W не концентрируется, при анализе профиля распределения, предпочтительно составляет 0,3≤Iw≤1, более предпочтительно 0,5≤Iw≤0,99,
где Iw = (средняя интенсивность W на диаметре от части, соответствующей 5 мкм от каждого края (поверхности) самого большого размера частиц, и до центра)/(средняя интенсивность W для самого большого размера частиц в целом).
(2) Способ получения оксидного катализатора
Способ получения оксидного катализатора по настоящему варианту осуществления представляет собой способ получения оксидного катализатора для использования в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана, способ включает стадии: (I) получения заготовки исходного материала, содержащей соединение Mo, соединение V, соединение Nb и соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, при конкретных атомных отношениях (далее, упоминается как "стадия приготовления исходных материалов"); (II) сушки заготовки исходных материалов с получением сухого порошка (далее, упоминается как "стадия сушки"); и (III) кальцинирования сухого порошка (далее, упоминается как "стадия кальцинирования").
(I) Стадия приготовления исходных материалов
На стадии приготовления исходных материалов, предпочтительно, соединение, содержащее каждый элемент, составляющий оксидный катализатор, растворяют или диспергируют в растворителе и/или в дисперсионной среде с получением заготовки исходных материалов. Обычно, в качестве растворителя и/или дисперсионной среды можно использовать воду. Далее, будет описываться стадия использования воды в качестве растворителя и/или дисперсионной среды. Количество растворителя и/или дисперсионной среды, содержащееся в заготовке исходных материалов, предпочтительно составляет от 70 до 95% масс., более предпочтительно от 75 до 90% масс., по отношению к общему количеству конечной заготовки исходных материалов, с точки зрения, например, полного растворения и/или диспергирования соединения, содержащего каждый элемент, составляющий оксидный катализатор (далее, также упоминается просто как "элемент составляющий катализатор"), соответствующим образом регулируя окислительно-восстановительное состояние элемента, составляющего катализатор, и регулируя вязкость заготовки исходных материалов непосредственно перед сушкой распылением, чтобы позволить получаемым частицам катализатора иметь предпочтительную форму и/или прочность.
Соединение, содержащее каждый элемент, составляющий оксидный катализатор (также включая соли; далее, упоминается также просто как "соединение составляющего элемента"), может использоваться в качестве компонента, содержащегося в заготовке исходных материалов.
На стадии приготовления исходных материалов, процедуры растворения, процедуры смешивания или процедуры диспергирования для соединений составляющих элементов не являются как-либо ограниченными. Исходные материалы, такие как соединения составляющих элементов, могут растворяться, смешиваться или диспергироваться в одной и той же водной среде. Альтернативно, исходные материалы, такие как соединения составляющих элементов, могут по отдельности растворяться, смешиваться или диспергироваться в различных водных средах, а затем эти водные среды могут смешиваться. Кроме того, нагрев и/или перемешивание могут осуществляться по необходимости. Отношение смешивания каждого соединения составляющего элемента может устанавливаться соответствующим образом с учетом отношения каждого составляющего элемента в конечном получаемом оксидном катализаторе, и количества каждого соединения составляющего элемента (добавляемого на следующих далее стадиях, таких как стадия кальцинирования), содержащегося в нем.
Является предпочтительным, чтобы заготовка исходных материалов содержала Mo, V, Nb и Te и/или Sb. Заготовка исходных материалов может содержать, по меньшей мере, один материал, выбранный из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, в дополнение к этим элементам, и может также содержать W. Примеры соединений составляющих элементов включают, но, не ограничиваясь этим как-либо, соединения, показанные ниже.
Примеры Mo-содержащего соединения (далее, упоминается как "соединение Mo"; это же будет верным для всех других элементов), служащего в качестве исходных материалов для Mo, включают аммоний гептамолибдат [(NH4)6Mo7O24∙4H2O], молибден триоксид [MoO3], фосфорномолибденовую кислоту [H3PMo12O40], кремнемолибденовую кислоту [H4SiMo12O40] и пентахлорид молибдена [MoCl5]. Среди них, аммоний гептамолибдат [(NH4)6Mo7O24∙4H2O] является предпочтительным с точки зрения растворимости, валентности Mo, универсальности, легкой доступности, и тому подобное.
Примеры соединения V, служащего в качестве исходных материалов для V, включают аммоний метаванадат [NH4VO3], ванадий пентоксид [V2O5] и хлорид ванадия [VCl4 или VCl3]. Среди них, аммоний метаванадат [NH4VO3] является предпочтительным с точки зрения растворимости, валентности V, универсальности, легкой доступности, и тому подобное.
Примеры соединения Nb, служащего в качестве исходных материалов для Nb, включают ниобиевую кислоту, соли неорганических кислот и ниобия и соли органических кислот и ниобия. Среди них, ниобиевая кислота является предпочтительной с точки зрения растворимости, валентности Nb, универсальности, легкой доступности, и тому подобное. Ниобиевая кислота представлена химической формулой Nb2O5∙nH2O и также называется гидроксид ниобия или ниобий оксид гидрат. Кроме того, ниобий может растворяться в водном растворе дикарбоновой кислоты и использоваться в качестве соединения Nb. Для этого соединения Nb, молярное отношение дикарбоновая кислота/ниобий предпочтительно составляет 1 к 4. Кроме того, дикарбоновая кислота предпочтительно представляет собой щавелевую кислоту, с точки зрения растворимости и соответствующего образования комплексов с Nb.
Примеры соединения Sb, служащего в качестве исходных материалов для Sb, включают оксид сурьмы [Sb2O3 или Sb2O5], сурмянистую кислоту [HSbO2], сурьмяную кислоту [HSbO3], антимонат аммония [(NH4)SbO3], хлорид сурьмы [Sb2Cl3], соли органических кислот (например, тартрат) и сурьмы и металлическую сурьму. Среди них, дисурьма триоксид [Sb2O3] является предпочтительным, с точки зрения растворимости, валентности Sb, универсальности, легкой доступности, и тому подобное.
Примеры соединения Te, служащего в качестве исходных материалов для Te, включают теллуровую кислоту [H6TeO6] и металлический теллур. Среди них, теллуровая кислота [H6TeO6] является предпочтительной, с точки зрения растворимости, валентности Te, универсальности, легкой доступности, и тому подобное.
Примеры соединения W, служащего в качестве исходных материалов для W, включают аммоний паравольфрамат, аммоний метавольфрамат, вольфрамовую кислоту, триоксид вольфрама, диоксид вольфрама, кремневольфрамовую кислоту, кремнемолибденовольфрамовую кислоту, кремневольфрамованадиевую кислоту, вольфрамат натрия, вольфрамат кальция и вольфрамат кобальта. Среди них, триоксид вольфрама, метавольфрамат аммония и вольфрамат кобальта являются предпочтительными с точки зрения растворимости, валентности W, универсальности, легкой доступности, влияния на элементарные металлы, сосуществующие с ними, и тому подобное.
Соединение, служащее в качестве исходных материалов для Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y или Sc (далее, упоминаются как "соединение, представленное Z"), не является как-либо ограниченным постольку, поскольку оно представляет собой вещество, содержащее каждый из этих элементов. Их примеры включают соединения, содержащие каждый из этих элементов и эти элементарные металлы, солюбилизированные в соответствующих реагентах. Примеры соединений, содержащих каждый из этих элементов, включают соль аммония, нитрат, карбоксилат, карбоксилат аммония, пероксокарбоксилат, пероксокарбоксилат аммония, галогенид аммония, галогенид, ацетилацетонат и алкоксид. Среди них, предпочтительными являются водорастворимые исходные материалы, такие как нитрат и карбоксилат.
Когда оксидный катализатор представляет собой катализатор на носителе из оксида кремния, является предпочтительным, чтобы заготовка исходных материалов содержала исходные материалы для оксида кремния. Золь кремниевой кислоты может использоваться в качестве исходных материалов для оксида кремния. Порошок оксида кремния также может использоваться в виде части исходных материалов для оксида кремния или всего их количества.
Является предпочтительным, чтобы золь кремниевой кислоты предпочтительно содержал 0-270 м.д. масс, более предпочтительно 10-270 м.д. масс ионов азотной кислоты, по отношению к массе SiO2 (оксида кремния) в нем. В настоящем описании, термин "золь кремниевой кислоты" относится к водному раствору кремниевой кислоты в прозрачном состоянии. Продукт, представляющий интерес, получают при более благоприятных выходах посредством регулировки, в конкретном диапазоне концентрации ионов азотной кислоты в золе кремниевой кислоты, служащем в качестве исходных материалов для носителя на основе оксида кремния, и используя такой золь кремниевой кислоты в качестве исходного материала для носителя. В дополнение к этому, получают катализатор на носителе из оксида кремния, дополнительно имеющий еще большее превосходство по физической прочности. Это может быть связано с тем, что агрегированное состояние золя кремниевой кислоты может контролироваться более подходящим образом, хотя причина является неопределенной.
В этом контексте, концентрация ионов азотной кислоты по отношению к массе оксида кремния в золе кремниевой кислоты может определяться с помощью ионной хроматографии. Измерительное устройство и условия измерений показаны ниже. Ионный хроматограф, изготовленный TOSOH CORP. (Торговое наименование "IC-2001"), можно использовать в качестве измерительного устройства. TSKgel superIC-AZ используют в качестве колонки, и TSKguardcolumn superIC-AZ используют в качестве защитной колонки. Кроме того, TSKsuppress A используют в качестве промывочного раствора для защитных клапанов. 1,9 ммоль/л водного раствора NaHCO3 и 3,2 ммоль/л водного раствора Na2CO3 смешивают и используют в качестве элюента. В этом случае, скорость потока устанавливают при 0,8 мл/мин.
Сначала, будет описан способ промышленного получения золя кремниевой кислоты для иллюстрации способа контроля концентрации ионов азотной кислоты в золе кремниевой кислоты. Примеры способа промышленного получения золя кремниевой кислоты включают такие способы, как (1) нейтрализация водорастворимого стекла с последующим диализом, (2) электродиализ, (3) растворение в водном растворе аммиака или амина металлического кремния, (4) пептизация силикагеля, (5) удаление Na из водорастворимого стекла с использованием ионообменной смолы. Среди них, наиболее общий способ получения золя кремниевой кислоты представляет собой способ с использованием удаления Na из водорастворимого стекла с использованием ионообменной смолы. К золю кремниевой кислоты, полученному с помощью этого способа, добавляют стабилизатор, такой как LiOH, NaOH или KOH, для улучшения стабильности при условиях высоких концентраций. Соответственно, область стабильных значений pH для золя кремниевой кислоты, как правило, составляет приблизительно от 8 до 10. Для поддержания стабильного дисперсного состояния золя кремниевой кислоты требуется, чтобы электрические заряды частиц оксида кремния в золе отталкивали друг друга. По этой причине, OH- адсорбируется на поверхности частиц оксида кремния посредством добавления стабилизатора, как описано выше, и предоставления возможности для демонстрации стабилизирующего воздействия на основе его отрицательного заряда, с предотвращением гелеобразования. Однако, известно, что поскольку добавление избытка щелочи (ионов щелочного металла в стабилизаторе) приводит к адсорбции ионов щелочи и к уменьшению отрицательных зарядов, золь кремниевой кислоты становится нестабильным. В последние годы стали коммерчески доступными множество золей кремниевой кислоты, которые имеют эти исходные свойства золя кремниевой кислоты и могут использоваться в различных применениях. Их примеры включают серию SNOWTEX, доступную от NISSAN CHEMICAL INDUSTRIES, LTD., включая: SNOWTEX 30, имеющий концентрацию золя кремниевой кислоты 30%; SNOWTEX C для использования в применениях, которые могли бы дополнительно вызывать гелеобразование; SNOWTEX N, предназначенный для устранения риска, связанного с остатками щелочи, путем использования летучего слабого основания, используемого в качестве стабилизатора; и SNOWTEX O, пригодный для применений, которые требуют использования при кислотных условиях (все они имеют торговые наименования; ссылка: SHOKUBAI KOGAKU KOZA (Lectures on Catalyst Engineering in English) 10, GENSO-BETSU SHOKUBAI BINRAN (Element-By-Element Catalyst Handbook in English), опубликованный 25 февраля 1967).
Частицы оксида кремния в золе кремниевой кислоты, полученном с помощью этого способа получения, классифицируются как кислотный и щелочной тип, с точки зрения поверхности. Однако небольшое количество ионов азотной кислоты присутствуют в золе кремниевой кислоты любого типа. Например, ионы водорода используют в основном в качестве стабилизатора для кислотного типа, в то время как ионы натрия или ионы аммония используют в качестве стабилизатора для щелочного типа. SO4 2-, Cl- или что-либо подобное используют в качестве противоиона для кислотного типа, и OH-, как правило, используют в качестве противоиона для щелочного типа.
Для получения золя кремниевой кислоты, либо кислотного, либо щелочного типа, описанного выше, с помощью массовой доли иона азотной кислоты 0-270 м.д. масс по отношению к массе оксида кремния, является предпочтительным, чтобы количество ионов азотной кислоты устанавливалось как 0-270 м.д. масс по отношению к оксиду кремния посредством добавления азотной кислоты или нитрата (например, нитрата аммония) при нейтрализации водного раствора водорастворимого стекла с помощью серной кислоты или хлористоводородной кислоты, что представляет собой общий способ получения золя кремниевой кислоты. Кроме того, после нейтрализации с помощью серной кислоты или хлористоводородной кислоты, ионы азотной кислоты могут быть заменены в водном растворе водорастворимого стекла анионами с помощью ионного обмена. Альтернативно, количество ионов азотной кислоты может регулироваться посредством добавления ионов азотной кислоты к установившемуся золю кремниевой кислоты с использованием пипетки или чего-либо подобного. Источник азотной кислоты может представлять собой азотную кислоту, а также соль, такую как нитрат аммония.
Исходные материалы для носителя на основе оксида кремния могут представлять собой золь кремниевой кислоты сам по себе. Альтернативно, порошок оксида кремния может быть заменен на часть золя кремниевой кислоты. Использование порошка оксида кремния в качестве исходного материала для носителя на основе оксида кремния, как можно ожидать, даст каталитическую активность и/или такой эффект, как улучшение выхода продукта, представляющего интерес; в противоположность этому, когда катализатор приготавливают с использованием только лишь порошка оксида кремния без использования золя кремниевой кислоты, износостойкость катализатора значительно понижается. В настоящем описании, термин "порошок оксида кремния" относится к мелкодисперсным частицам SiO2 в форме твердого продукта. Если оксид кремния имеет слишком большой основный размер частиц, полученный катализатор имеет тенденцию к охрупчению. Таким образом, предпочтительным является порошок оксида кремния с нанометровыми размерами. Является предпочтительным, чтобы порошок оксида кремния получали с помощью высокотемпературного способа, с точки зрения высокой чистоты оксида кремния, содержащегося в нем, и тому подобное. Конкретные примеры предпочтительного порошка оксида кремния включают Aerosil 200 (торговое наименование), производимый Nippon Aerosil Co., Ltd.
Является предпочтительным, чтобы порошок оксида кремния диспергировался в воде заранее, с точки зрения облегчения добавления к суспензии и перемешивания. Способ диспергирования порошка оксида кремния в воде не является как-либо ограниченным, и порошок оксида кремния может диспергироваться с использованием обычного гомогенизатора, гомомиксера или ультразвукового вибратора, и тому подобное, по отдельности или в сочетании.
Когда золь кремниевой кислоты и порошок оксида кремния в качестве исходных материалов для носителя на основе оксида кремния используют в сочетании, является предпочтительным, чтобы порошок оксида кремния составлял 20-80% масс. от общего количества золя кремниевой кислоты и порошка оксида кремния. Катализатор, имеющий достаточную износостойкость и каталитическую активность, можно проще получить посредством регулировки пропорции порошка оксида кремния в этих пределах, чем посредством регулировки в других пределах. Не требуется, чтобы порошок оксида кремния содержал ион азотной кислоты. Не требуется, чтобы концентрация ионов азотной кислоты, содержащихся в порошке оксида кремния, контролировалась, даже если концентрация ионов азотной кислоты в золе кремниевой кислоты устанавливается как 10-270 м.д. масс по отношению к SiO2 для цели улучшения выхода и/или физической прочности продукта, представляющего интерес.
Стадия приготовления исходных материалов будет описываться, принимая, в качестве примера, получение заготовки исходных материалов, содержащей соединение Mo, соединение V, соединение Nb, соединение(соединения) Te и/или Sb (далее, упоминается как "соединение, представленное X") и соединение, представленное Z, с водой в качестве растворителя и/или дисперсионной среды. Сначала, соединение Mo, соединение V, соединение, представленное X, и соединение, представленное Z, добавляют в воду, и этот раствор нагревают с получением смешанного водного раствора (A). Является предпочтительным, чтобы температура и время нагрева для приготовления смешанного водного раствора (A) регулировались в пределах, которые дают состояние, при котором каждый исходный материал может растворяться и/или диспергироваться полностью. С этой точки зрения, температура нагрева предпочтительно составляет 70-100°C, и время нагрева предпочтительно составляет 30 минут - 5 часов. Подобным же образом, количества оборотов в единицу времени при перемешивании во время нагрева может регулироваться при соответствующем количестве оборотов, при котором каждый исходный материал легко растворяется и/или диспергируется. Когда исходные материалы представляют собой соли металлов, является предпочтительным, чтобы состояние перемешивания поддерживалось в течение приготовления смешанного водного раствора (A), с точки зрения полного растворения солей металлом. В этом случае, атмосфера в контейнере может представлять собой атмосферу воздуха, а также может устанавливаться как атмосфера азота, с точки зрения регулирования числа окисления для полученного оксидного катализатора. Подобным же образом, может добавляться перекись водорода, если это необходимо, в количестве, пригодном для смешанного водного раствора (A), с точки зрения регулировки числа окисления. Например, когда в качестве элемента X используют сурьму, является предпочтительным, чтобы перекись водорода добавлялась к смешанному водному раствору (A) или к раствору, содержащему компоненты для смешанного водного раствора (A), во время его приготовления. H2O2/Sb (молярное отношение) предпочтительно составляет 0,01-5, более предпочтительно 0,5-3, еще более предпочтительно 1-2,5, с точки зрения регулировки числа окисления полученного оксидного катализатора в предпочтительном диапазоне.
Является предпочтительным, чтобы температура и время нагрева после добавления перекиси водорода к смешанному водному раствору (A) регулировались в диапазонах, которые дают возможность для осуществления жидкофазной реакции окисления с помощью перекиси водорода в достаточной степени. С такой точки зрения, температура нагрева предпочтительно составляет 30°C-70°C, а время нагрева предпочтительно составляет 5 минут - 4 часа. Подобным же образом, количество оборотов в единицу времени при перемешивании во время нагрева может регулироваться при соответствующем количестве оборотов, при котором облегчается жидкофазная реакция окисления с помощью перекиси водорода. Является предпочтительным, чтобы состояние перемешивания поддерживалось во время нагрева, с точки зрения предоставления возможности для осуществления жидкофазной реакции окисления с помощью перекиси водорода в достаточной степени.
В дальнейшем, соединение Nb и дикарбоновая кислота по отдельности добавляются в воду, и эти растворы нагревают при перемешивании с получением смешанного раствора (B0). Примеры дикарбоновой кислоты включают щавелевую кислоту [(COOH)2]. Является предпочтительным, чтобы перекись водорода добавлялась затем к смешанному раствору (B0) с получением смешанного водного раствора (C). В этом случае, H2O2/Nb (молярное отношение) предпочтительно составляет 0,5-20, более предпочтительно 1-10, с точки зрения, например, стабилизации состояния растворения посредством образования комплексов с соединением Nb, соответствующим образом регулируя окислительно-восстановительное состояние элемента, составляющего катализатор, и получения соответствующих рабочих характеристик катализатора для получаемого катализатора.
Затем смешанный водный раствор (A) и смешанный водный раствор (C) смешивают при предпочтительном отношении смешивания в соответствии с композицией, представляющей интерес, с получением смешанного водного раствора (D). Полученный смешанный водный раствор (D) состаривают, по потребности, с получением заготовки исходных материалов в форме суспензии.
Состаривание смешанного водного раствора (D) означает, что смешанный водный раствор (D) оставляют стоять или перемешивают в течение заданного времени. Промышленное производство оксидного катализатора может потребовать некоторого времени до завершения сушки распылением всего смешанного водного раствора (D) после сушки распылением части смешанного раствора, поскольку скорость обработки в устройстве для сушки распылением, описанном далее, является стадией, ограничивающей скорость процесса в целом. В это время, состаривание части смешанного раствора, не подвергающейся сушке распылением, можно продолжить. Конкретно, время состаривания включает не только время состаривания до сушки распылением, но также время от начала до завершения сушки распылением.
Время состаривания предпочтительно находится в пределах между 90 минутами и 50 часами включительно, более предпочтительно, между 90 минутами и 6 часами включительно, с точки зрения, например, улучшения рабочих характеристик катализатора из полученного сложного оксида. Если время состаривания короче, чем 90 минут, или больше, чем 50 часов, трудно сформировать смешанный водный раствор (IV), имеющий предпочтительное окислительно-восстановительное состояние (потенциал). В результате, рабочие характеристики катализатора из полученного сложного оксида имеют тенденцию к ухудшению. В этом контексте, промышленное получение оксидного катализатора обычно требует некоторого времени до завершения сушки распылением всего смешанного водного раствора (IV) после сушки распылением части смешанного раствора, поскольку скорость обработки в устройстве для сушки распылением обычно является стадией, ограничивающей скорость процесса в целом. В это время, состаривание части смешанного водного раствора, не подвергающегося сушке распылением, продолжается. Таким образом, время состаривания включает не только время состаривания перед сушкой на стадии (c), описанной далее, но также и время от начала до завершения сушки.
Температура состаривания предпочтительно составляет 25°C или выше с точки зрения предотвращения конденсации компонента Mo или осаждения V. Кроме того, температура состаривания предпочтительно составляет 65°C или ниже с точки зрения предотвращения осуществления слишком большой степени гидролиза комплекса, содержащего Nb и перекись водорода, чтобы при этом сформировать суспензию в предпочтительной форме. Таким образом, температура состаривания предпочтительно находится в пределах между 25°C и 65°C включительно, более предпочтительно, между 30°C и 60°C включительно.
Является предпочтительным, чтобы атмосфера окружающей среды для смешанного водного раствора (D) во время состаривания, например, атмосфера в контейнере для состаривания смешанного водного раствора (D) в ней, имела достаточную концентрацию кислорода. Атмосфера, имеющая достаточную концентрацию кислорода, облегчает существенные изменения в смешанном водном растворе (D). Концентрация кислорода газофазной части в атмосфере окружающей среды, более предпочтительно, составляет 1% объем или выше. Например, является предпочтительным, чтобы смешанный водный раствор (D) состаривался в атмосфере воздуха.
Концентрация кислорода в газовой фазе может быть измерена с помощью общего способа, например, с использованием измерителя концентрации кислорода типа, использующего диоксид циркония. Положение, в котором измеряют концентрацию кислорода в газовой фазе, предпочтительно находится вблизи границы раздела между смешанным водным раствором (D) и газовой фазой, с точки зрения правильного определения концентрации кислорода. Например, является предпочтительным, чтобы концентрация кислорода в газовой фазе измерялась 3 раза в пределах 1 минуты в одной и той же точке, и среднеарифметическое значение результатов 3 измерений должно определяться как концентрация кислорода в газовой фазе.
Примеры разбавляющего газа для уменьшения концентрации кислорода в газовой фазе включают, но, конкретно не ограничиваясь этим, такие газы, как азот, гелий, аргон, диоксид углерода и пары воды. Газообразный азот является промышленно предпочтительным. Кроме того, газ, используемый для увеличения концентрации кислорода в газовой фазе, предпочтительно представляет собой, например, чистый кислород или воздух, имеющий высокую концентрацию кислорода.
Состаривание вероятно вызывает некоторые изменения в окислительно-восстановительном состоянии компонентов, содержащихся в смешанном водном растворе (D). Об этом появлении некоторых изменений также говорят изменения цвета смешанного водного раствора (D), изменения окислительно-восстановительного потенциала, и тому подобное, во время состаривания. В результате, рабочие характеристики получаемого оксидного катализатора также различаются в зависимости от присутствия или отсутствия состаривания в течение времени в пределах между 90 минутами и 50 часами включительно в атмосфере, имеющей концентрацию кислорода от 1 до 25% объем. Конкретно, хотя морфологические изменения компонентов раствора во время состаривания слишком сложно идентифицировать точно, можно предположить, при получении оксидных катализаторов, различающихся по времени состаривания и по оценке их рабочих характеристик, что время состаривания, которое дает оксидный катализатор, имеющий благоприятные рабочие характеристики, является предпочтительным, в течение этого времени суспензия формируется в некоторой предпочтительной форме.
Потенциал смешанного водного раствора (C) (например, 600 мВ/AgCl, когда дикарбоновая кислота в смешанном водном растворе (C) представляет собой щавелевую кислоту) является преобладающим в окислительно-восстановительном потенциале смешанного водного раствора (D). Пероксид дикарбоновой кислоты и Nb, содержащихся в смешанном водном растворе (C), вероятно, вызывает некоторую окислительно-восстановительную реакцию с другими компонентами металлов, уменьшая потенциал со временем. Окислительно-восстановительный потенциал смешанного водного раствора (D) предпочтительно составляет от 450 до 530 мВ/AgCl, более предпочтительно, от 470 до 510 мВ/AgCl.
Концентрация кислорода в атмосфере окружающей среды для смешанного водного раствора (D) во время состаривания предпочтительно составляет 1% объем или выше, с точки зрения предотвращения слишком медленного осуществления окислительно-восстановительной реакции (которая влияет на изменения в окислительно-восстановительном состоянии компонентов, содержащихся в смешанном водном растворе (D)), чтобы тем самым предотвратить переход окислительно-восстановительного состояния в состояние сверхвосстановления на стадии суспензии. С другой стороны, концентрация кислорода в атмосфере окружающей среды для смешанного водного раствора (D) во время состаривания предпочтительно составляет 25% объем или ниже с точки зрения предотвращения сверхокисления суспензии из-за слишком большой степени осуществления окислительно-восстановительной реакции. В любом случае, требуется, чтобы концентрация кислорода поддерживалась в соответствующем диапазоне, поскольку кислород в газовой фазе влияет на окислительно-восстановительное состояние суспензии. Этот диапазон, более предпочтительно, составляет 5-23% объем, еще более предпочтительно 10-20% объем.
Во время состаривания, вода может испаряться таким образом, что смешанный водный раствор (D) концентрируется. Однако, хотя состаривание в открытой системе неизбежно вызывает такое испарение воды, является предпочтительным, чтобы состаривание осуществлялось в атмосфере, имеющей концентрацию кислорода от 1 до 25% объем, с точки зрения улучшения рабочих характеристик катализатора.
Для перемешивания во время состаривания, является предпочтительным, чтобы плотность жидкости, количество заготовки исходных материалов, количество оборотов в единицу времени лопастей мешалки, и тому подобное, контролировались с точки зрения предотвращения гелеобразования суспензии и с точки зрения регулировки вязкости получаемой суспензии в соответствующем состоянии. Деформация получаемых частиц или углубления в частицах катализатора на стадии сушки распылением, описанной далее, могут предотвращаться посредством поддержания вязкости суспензии на умеренно высоком уровне, по сравнению со случаем, когда суспензия имеет слишком низкую вязкость. Альтернативно, гелеобразование в заготовке исходных материалов, приводящее к забиванию трубы (которое делает сложным получение сухого порошка) или к ухудшению рабочих характеристик катализатора, можно предотвратить посредством поддержания вязкости суспензии на умеренно низком уровне, по сравнению со случаем, когда суспензия имеет слишком высокую вязкость. Умеренно вязкую суспензию можно получить посредством контроля плотности жидкости, количества заготовки исходных материалов, количества оборотов в единицу времени лопастей мешалки, и тому подобное.
Перемешивание можно осуществлять с использованием, например, обычной лопастной мешалки или импеллера, такого как многолопастной импеллер, якорная крыльчатка, винтовой осевой импеллер, винтовой ленточный импеллер, или лопастной мешалки для растворов с низкой вязкостью, таких как пропеллер, дисковая турбина, лопастная турбина, лопастная турбина с искривленными лопастями, турбина с шевронными шестернями или турбина с наклонными лопастями.
Мощность (далее упоминается как "Pv"), придаваемая с помощью лопастной мешалки в устройстве для перемешивания заготовки исходных материалов на единицу объема в устройстве для получения заготовки исходных материалов, вычисляют в соответствии с формулой, показанной ниже (формула A). Pv предпочтительно составляет от 0,005 до 300 кВт/м3, более предпочтительно, от 0,01 до 280 кВт/м3, еще более предпочтительно, от 0,1 до 250 кВт/м3. Гелеобразование в заготовке исходных материалов, приводящее к забиванию трубы (что делает сложным получение сухого порошка) или ухудшению рабочих характеристик катализатора, может дополнительно предотвращаться посредством перемешивания заготовки исходных материалов при мощности перемешивания Pv 0,005 кВт/м3 или больше. Альтернативно, углубления в частицах катализатора после сушки распылением могут дополнительно предотвращаться с помощью перемешивания заготовки исходных материалов при мощности перемешивания Pv 300 кВт/м3 или меньше. Присутствие углублений отрицательно влияет на прочность катализатора.
Это значение Pv может контролироваться посредством регулировки плотности жидкости, количества заготовки исходных материалов, количества оборотов в единицу времени лопастной мешалки, и тому подобное.
Pv вычисляют в соответствии со следующей формулой (формула A):
Pv=Np×ρ×n3×d5/V... (формула A)
где Np представляет собой число мощности, которое представляет собой безразмерное число, относящееся к мощности, необходимой для перемешивания; ρ представляет собой плотность жидкости (кг/м3); n представляет собой количество оборотов в единицу времени (сек-1) лопастной мешалки; d представляет собой диаметр (м) лопастной мешалки и V представляет собой количество (м3) заготовки исходных материалов.
Np можно вычислить с использованием следующей формулы (формула B1):
Уравнение 1

В этом контексте, символы в формулах (формулы B1-B5), каждый, определяются следующим образом: b представляет собой ширину (м) лопастной мешалки; d представляет собой диаметр (м) лопастной мешалки; D представляет собой диаметр (м) перемешиваемого танка; Z представляет собой глубину (м) жидкости и θ представляет собой угол (°) наклона лопастной мешалки по отношению к горизонтали. Кроме того, вязкость полученной заготовки исходных материалов при комнатной температуре (25°C) предпочтительно составляет 1-100 сП, более предпочтительно 2-90 сП, еще более предпочтительно 2,5-80 сП, с точки зрения, например, дополнительного предотвращения гелеобразования в заготовке исходных материалов, приводящего к забиванию трубы (что делает сложным получение сухого порошка) или к ухудшению рабочих характеристик катализатора, и дополнительного предотвращения появления углублений в частицах катализатора после сушки распылением или деформации частиц катализатора.
Вязкость заготовки исходных материалов можно измерить, например, с помощью метода измерения с использованием коммерчески доступного вискозиметра или способа, который включает определение перепада давления внутри трубы, в которой циркулирует заготовка исходных материалов. Например, когда измеряют вязкость раствора, в котором постепенно происходит гелеобразование, в состоянии без перемешивания, во время измерения с использованием коммерчески доступного вискозиметра, вязкость может постепенно изменяться. Таким образом, является предпочтительным, чтобы вязкость измерялась с помощью способа, который включает определение перепада давления в трубе, в которой циркулирует заготовка исходных материалов, с точки зрения воспроизводимости измеряемых значений.
Когда вязкость заготовки исходных материалов измеряют с помощью способа, который включает определение перепада давления в трубе, в которой циркулирует заготовка исходных материалов, вязкость можно вычислить в соответствии со следующей формулой (формула C1):
Уравнение 2

где ∆P представляет собой перепад давления (ммH2O) в трубе; μ представляет собой вязкость жидкости (сП) при температурах измерения; u представляет собой среднюю скорость (м/сек) циркуляции жидкости; L представляет собой длину (м) трубы и D представляет собой диаметр (м) трубы.
Когда заготовку исходных материалов получают посредством смешивания множества растворов исходных материалов, содержащих каждый компонент, растворенный в них, верхний предел каждой Pv для получения каждого раствора исходных материалов не является как-либо ограниченным. Подобным же образом, нижний предел Pv не является как-либо ограниченным. Однако, является предпочтительным, чтобы значение Pv устанавливалось как равное или большее, чем значение, которое дает состояния, при которых большая часть твердых частиц или все они протекают в устройстве для получения раствора исходных материалов, на некотором расстоянии от нижней части танка в устройстве. Для приготовления растворов исходных материалов, перемешивание может останавливаться после растворения по существу всех твердых частиц в каждом растворе исходных материалов. Кроме того, можно добавлять кислоту и/или щелочь, по необходимости, для регулировки pH суспензии.
Когда оксидный катализатор представляет собой продукт на носителе из оксида кремния, является предпочтительным, чтобы заготовка исходных материалов приготавливалась таким образом, чтобы она содержала золь кремниевой кислоты, с точки зрения, например, полного растворения и/или диспергирования соединения, содержащего каждый элемент, составляющий катализатор, регулируя соответствующим образом окислительно-восстановительное состояние элемента, составляющего катализатор, чтобы позволить полученным частицам катализатора иметь предпочтительную форму и/или прочность, и улучшая рабочие характеристики катализатора из полученного сложного оксида. Золь кремниевой кислоты может добавляться к ним соответствующим образом. Кроме того, часть золя кремниевой кислоты может также заменяться водной дисперсией порошка оксида кремния. Водная дисперсия порошка оксида кремния может также добавляться к ним соответствующим образом.
Стадия приготовления исходных материалов, описанная выше, может осуществляться многократно в соответствии с выходами. Если W содержится в заготовке исходных материалов, полученной на стадии приготовления исходных материалов (I) в способе получения оксидного катализатора, и в сухом порошке, полученном на стадии сушки (II), описанной далее, тогда в формуле (1) b>0. В результате, W, видимо, присутствует также в области на центральной стороне частицы оксидного катализатора, поскольку W, введенный на стадии (I) приготовления исходных материалов, присутствует еще со времени растворенного состояния.
(II) Стадия сушки
Заготовку исходных материалов в форме суспензии, полученной на стадии приготовления исходных материалов, сушат с получением сухого порошка (сухого продукта). Сушку можно осуществлять с помощью способа, известного в данной области, и ее можно также осуществлять, например, с помощью сушки распылением или выпаривания досуха. Когда реакционную систему с псевдоожиженным слоем принимают для газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования, предпочтительно получать сухой порошок или предшественник катализатора в микросферической форме и по этой причине, принимать сушку распылением, с точки зрения, например, получения предпочтительной текучести в реакторе. Атомизацию в способе сушки распылением можно осуществлять с использованием любой системы из системы центрифугирования, системы двухжидкостного сопла и системы сопла высокого давления. Воздух, нагреваемый с использованием водяного пара, электрического нагревателя или чего-либо подобного, можно использовать в качестве источника тепла для сушки. Температура на входе сушилки в устройстве для сушки распылением предпочтительно составляет от 150 до 300°C, с точки зрения, например, предоставления возможности получаемым частицам катализатора, чтобы они имели предпочтительную форму и/или прочность, и улучшения рабочих характеристик катализатора из полученного сложного оксида. Кроме того, выходная температура сушилки предпочтительно составляет 100-160°C.
Скорость распыления, скорость введения раствора в суспензию, количество оборотов в единицу времени атомайзера (в случае системы центрифугирования), и тому подобное, могут регулироваться в соответствии с размером устройства таким образом, чтобы размер частиц получаемого сухого порошка попадал в предпочтительный диапазон. Конкретно, средний размер частиц сухого порошка предпочтительно составляет от 5 мкм до 200 мкм, более предпочтительно, от 10 до 150 мкм.
Средний размер частиц для частиц сухого порошка определяют посредством измерения распределения размеров частиц в соответствии с J IS R 1629-1997 "Determination of particles size distributions for fine ceramic raw powders by laser diffraction method" и определения его среднего значения по отношению к объему. Более конкретно, часть сухого порошка, кальцинируют при 400°C в течение 1 часа на воздухе, и полученные частицы измеряют в качестве субъекта с использованием анализатора распределения размеров частиц на основе метода дифракции/рассеяния лазерного света LS230, производимого BECKMAN COULTER, INC.
Причина для измерения среднего размера частиц после "кальцинирования при 400°C в течение 1 часа на воздухе" части сухого порошка заключается в том, что предотвращается растворение сухого порошка в воде. Это означает, что "кальцинирование при 400°C в течение 1 часа на воздухе" осуществляют в основном для измерения, и оно не связано со стадией кальцинирования, описанной далее. Разумно считать, что размер частиц до и после этого кальцинирования очень мало различается.
Более конкретно, измерение среднего размера частиц осуществляют следующим образом, в соответствии с инструкцией, поставляемой вместе с анализатором распределения размеров частиц на основе метода дифракции/рассеяния лазерного света (торговое наименование "LS230", производится BECKMAN COUITER, INC.): сначала осуществляют измерение фона (количество отсчетов: 60), а затем 0,2 г частиц взвешивают в пробирке с резьбой, имеющей соответствующий размер, в которую затем добавляют 10 см3 воды. Пробирку с резьбой закрывают крышкой (плотно закрывают) и встряхивают в достаточной степени для диспергирования частиц в воде. К ней прикладывают ультразвуковые волны, 300 Вт, с использованием соответствующего устройства, и пробирку с резьбой опять встряхивают в достаточной степени. Затем продолжают приложение ультразвуковых волн, в то время как частицы, диспергируемые в воде, инжектируют в основный корпус устройства с использованием пипетки таким образом, что частицы имеют соответствующую концентрацию (концентрация: 10, PIDS: 60). После подтверждения того, что показатели концентрации стабильны, приложение ультразвуковых волн прекращают. Пробирку с резьбой оставляют стоять в течение 10 секунд, а затем начинают измерение (время измерения: 90 секунд). Величину медианного диаметра в результатах измерений определяют как средний размер частиц.
Также является предпочтительным, чтобы часть сухого порошка, полученного на стадии сушки, собиралась, и его спектры поглощения или отражения измерялись. Спектры поглощения или отражения сухого порошка, полученного на стадии сушки, могут измеряться непрерывно, чтобы предсказать при этом рабочие характеристики полученного конечного оксидного катализатора на основе спектров поглощения или отражения.
Уровень окислительно-восстановительных свойств оксидного катализатора изменяется из-за нагрева на стадии сушки, и рабочие характеристики оксидного катализатора подвергаются изменениям. На стадии сушки, включающей сушку распылением заготовки исходных материалов с получением сухого порошка, часть сухого порошка может прилипать и/или скапливаться на внутренних стенках и/или на дне устройства и при этом оставаться в устройстве в течение продолжительного времени. В таком случае, уровень окислительно-восстановительных свойств изменяется из-за непреднамеренного приложения тепла к сухому порошку. Когда на стадии кальцинирования, описанной далее, кальцинирование осуществляют в атмосфере воздуха, уровень окислительно-восстановительных свойств сухого порошка очень мало влияет на рабочие характеристики полученного катализатора при условии, что окисление осуществляется посредством кальцинирования. С другой стороны, когда на стадии кальцинирования кальцинирование осуществляют в атмосфере инертного газа, уровень окислительно-восстановительных свойств сухого порошка имеет тенденцию к влиянию на рабочие характеристики оксидного катализатора. Конкретно, когда способ получения оптимизируют с учетом уровня окислительно-восстановительных свойств оксидного катализатора, рабочие характеристики катализатора имеют тенденцию к ухудшению, в частности, из-за того, что уровень окислительно-восстановительных свойств сухого порошка находится вне пределов желаемого диапазона. Цвет сухого порошка изменяется при изменении уровня окислительно-восстановительных свойств, хотя подробный механизм неизвестен. Принимая катализатор Mo-V в качестве примера, рабочие характеристики оксидного катализатора имеют повышенную тенденцию к ухудшению, когда сухой порошок становится черным. Это может быть связано, например, с органическими или неорганическими компонентами, содержащимися в сухом порошке, которые термически разлагаются из-за непреднамеренного нагрева, приводя к восстановлению окружающих их элементарных металлов или вызывая окислительно-восстановительную реакцию между элементарными металлами. Таким образом, спектры поглощения или отражения сухого порошка могут измеряться и исследоваться относительно уровня изменения цвета, чтобы тем самым предсказать рабочие характеристики оксидного катализатора.
По этим причинам, является предпочтительным, чтобы устройство для сушки распылением снабжалось, например, устройством для предотвращения накопления сухого порошка, таким как вибратор, который придает вибрацию устройству для сушки распылением, или пневматическим встряхивателем, который придает ему импульс, для цели предотвращения накопления в нем сухого порошка. Кроме того, также является предпочтительным, чтобы внутреннее пространство устройства промывалось водой или чем-либо подобным с умеренной частотой, в то время как сушку распылением временно приостанавливают. Является предпочтительным, чтобы измерение спектра осуществлялось непосредственно после стадии сушки, на которой имеется тенденция к осуществлению непреднамеренного нагрева.
Рабочие условия для пневматического встряхивателя, которым снабжено устройство для сушки, могут регулироваться произвольным образом в зависимости от размера устройства, толщины стенки или степени, до которой отслаивается материал, прилипший к ней. Примеры рабочих условий включают силу удара пневматического встряхивателя, частоту ударов и увеличение или уменьшение количества пневматических встряхивателей, расположенных в нем, и изменение положения, в котором располагается пневматический встряхиватель. Является предпочтительным, чтобы сила удара пневматического встряхивателя была большой, без деформации или разрушения стенки и/или других областей устройства для сушки даже при долговременной работе. С этой точки зрения, частота ударов предпочтительно составляет один или несколько раз за 1 минуту, более предпочтительно, один или несколько раз за 10 секунд. Для количества пневматических встряхивателей, размещенных в нем, и для положения, в котором располагается пневматический встряхиватель, является предпочтительным, чтобы: например, количество пневматических встряхивателей увеличивалось для положения, в котором наблюдают сильную адгезию посредством наблюдения внутреннего пространства после долговременной операции; или пневматический встряхиватель, расположенный в месте, где почти нет адгезии, должен быть перемещен в положение с сильной адгезией.
Способ измерения спектров поглощения или отражения не является как-либо ограниченным. Например, спектры поглощения или отражения определяют на основе коэффициента поглощения сухого порошка, измеренного с использованием спектрофотометра УФ-видимого света. В результате осуществления тщательных исследований авторами настоящего изобретения, продемонстрировано, что сухой порошок, изменяющий цвет на черный, который дает оксидный катализатор, имеющий плохие рабочие характеристики, имеет более высокий коэффициент поглощения при длине волны, равной или большей, чем 500 нм, чем для сухого порошка, который не меняет цвет на черный. Таким образом, коэффициент поглощения на любой длине волны в диапазоне длин волн, равных или больших, чем 500 нм, предпочтительно, в пределах между 500 нм и 800 нм включительно, может быть выбран и использоваться в качестве показателя при измерении.
Является предпочтительным, чтобы спектры поглощения или отражения сухого порошка измерялись непрерывно. В этом контексте, фраза "измерялись непрерывно" означает, что измерение осуществляют один раз в 3 месяца или чаще. Более предпочтительно, спектр измеряют раз в месяц, еще более предпочтительно, раз в неделю, особенно предпочтительно, раз в день или чаще. Чем чаще осуществляют измерения, тем сильнее может быть понижен риск формирования больших количеств сухих порошков, имеющих неприемлемый уровень окислительно-восстановительных свойств. В зависимости от условий получения, спектры поглощения или отражения сухого порошка могут меняться слабо. В таком случае, частые измерения не нужны. Таким образом, частота может устанавливаться соответствующим образом.
Способ получения по настоящему варианту осуществления может необязательно включать следующую стадию (i) определения условий для каждой стадии из стадии получения исходных материалов (I), стадии сушки (II) и стадии кальцинирования (III), описанных далее, в соответствии со спектрами поглощения или отражения, измеренными таким образом.
Стадия (i)
На этой стадии, можно эффективно получать катализатор, имеющий еще большее превосходство по рабочим характеристикам, посредством предсказания рабочих характеристик полученного конечного оксидного катализатора из измеренных спектров поглощения или отражения и контроля рабочих условий на каждой стадии, основываясь на предсказанных рабочих характеристиках оксидного катализатора.
Диаграмма корреляции спектров поглощения или отражения сухих порошков, полученных при различных условиях сушки, в зависимости от рабочих характеристик оксидных катализаторов, полученных из сухих порошков, может быть использована для предсказания рабочих характеристик конечного полученного оксидного катализатора с использованием измеренных спектров поглощения или отражения. Является предпочтительным, чтобы коэффициент поглощения, полученный на конкретной длине волны с использованием спектрофотометра УФ-видимого света, использовался в качестве спектров поглощения или отражения сухих порошков, полученных при различных условиях сушки, с точки зрения получения благоприятной корреляции.
Сухие порошки, полученные при различных условиях сушки, представляют собой сухие порошки, которые могут быть получены посредством изменения, по меньшей мере, одного параметра из количества заготовки исходных материалов (водных исходных материалов), подаваемого в единицу времени, количества воздуха, подаваемого в единицу времени, и температуры подаваемого воздуха на стадии сушки, или изменения рабочих условий или чего-либо подобного, для устройства, предотвращающего накопление сухого порошка или чего-либо подобного, присоединенного к устройству для сушки. Когда атомизацию осуществляют с использованием системы центрифугирования, эти сухие порошки могут также быть получены посредством изменения количества оборотов диска в единицу времени или диаметра диска. Кроме того, сухие порошки могут быть получены посредством нагрева сухих порошков, полученных на стадии сушки, при намеренном изменении температур и/или при намеренном изменении времени, чтобы изменить тем самым цвет сухих порошков.
Спектры поглощения или отражения сухих порошков, полученных при различных условиях сушки, могут измеряться таким же образом, как в способе, описанном выше. Впоследствии, сухие порошки, полученные при различных условиях сушки, каждый, дополнительно кальцинируют при одинаковых условиях, и полученные оксидные катализаторы используют для осуществления газофазной реакции каталитического окисления или каталитического аммоксидирования пропана или изобутана, в течение которого исследуют рабочие характеристики катализаторов. Примеры рабочих характеристик катализатора, которые должны исследоваться, включают выходы, активность, степень преобразования и выходы побочных продуктов. Эти факторы могут объединяться.
Затем создают диаграмму корреляции на основе спектров поглощения или отражения сухих порошков, полученных при различных условиях сушки, и исследуют рабочие характеристики катализатора. Эту диаграмму корреляции можно использовать для предсказания рабочих характеристик конечного полученного оксидного катализатора по измеренным спектрам поглощения или отражения сухого порошка.
В способе получения по настоящему варианту осуществления, оксидный катализатор, имеющий еще большее превосходство по рабочим характеристикам, может быть легко получен посредством изменения рабочих условий на каждой стадии в соответствии с предсказанными значениями рабочих характеристик конечного полученного оксидного катализатора. Промышленно, оксидный катализатор, превосходный по рабочим характеристикам, может быть получен эффективно посредством непрерывного измерения (мониторинга) спектров поглощения или отражения сухого порошка и изменения рабочих условий в соответствии со спектрами поглощения или отражения.
На этой стадии (i), рабочие условия, по меньшей мере, на одной стадии из стадии приготовления исходных материалов (I), стадии сушки (II) и стадии кальцинирования (III), описанных далее, определяют в соответствии с отслеживаемыми спектрами поглощения или отражения. Является предпочтительным, чтобы рабочие условия на стадии сушки (II) определялись в соответствии со спектрами поглощения или отражения, с точки зрения простоты контролирования. Далее, будут описаны примеры стадии определения рабочих условий (стадии (i-1) и (i-2)).
Стадия (i-1)
Стадия (i-1) представляет собой стадию определения условий для стадии приготовления исходных материалов в соответствии с измеренными спектрами поглощения или отражения. На этой стадии, условия получения, которые дают благоприятные рабочие характеристики конечного полученного оксидного катализатора, определяют с использованием диаграммы корреляции спектров поглощения или отражения сухих порошков, полученных при различных условиях сушки, в зависимости от рабочих характеристик оксидных катализаторов, полученных из сухих порошков.
На этой стадии, значения "определение условий получения" не ограничиваются как-либо. Их примеры включают способ, который включает контроль уровня окислительно-восстановительных свойств каталитического компонента в соответствии с процедурами растворения или процедурами смешивания, для растворения и/или диспергирования соединения составляющего элемента в водной среде, и способ, который включает добавление окисляющего агента или восстанавливающего агента.
Стадия (i-2)
Стадия (i-2) представляет собой стадию определения условий сушки для стадии сушки в соответствии с измеренными спектрами поглощения или отражения. На этой стадии, условия сушки, которые дают благоприятные рабочие характеристики конечного полученного оксидного катализатора, определяют с использованием диаграммы корреляции спектров поглощения или отражения для сухих порошков, полученных при различных условиях сушки, в зависимости от рабочих характеристик оксидного катализатора, полученных из сухих порошков.
На этой стадии, значения "определения условий сушки" не ограничиваются как-либо. Например, когда используют устройство для сушки распылением, его примеры включают способ, который включает изменение количества заготовки исходных материалов, подаваемого в единицу времени, количества подаваемого воздуха и температуры воздуха, подаваемого в единицу времени, или рабочих условий, или чего-либо подобного, для устройства для предотвращения накопления сухого порошка, или чего-либо подобного, присоединенного к устройству для сушки. Способ, который включает изменение рабочих условий устройства для предотвращения накопления сухого порошка, является более предпочтительным с точки зрения поддержания рабочих характеристик, физической формы/прочности или чего-либо подобного для полученного катализатора. Когда атомизацию осуществляют с использованием системы центрифугирования, количество оборотов диска в единицу времени или диаметр диска могут изменяться. Кроме того, эти факторы могут объединяться.
Сухой порошок может быть получен таким образом, что содержание частиц, имеющих размер частиц 25 мкм или меньше, составляет 20% масс. или меньше, предпочтительно 15% масс. или меньше, более предпочтительно 10% масс. или меньше, еще более предпочтительно 5% масс. или меньше, особенно предпочтительно, 2% масс. или меньше. Благодаря тому, что 20% масс. или меньше от содержания частиц имеет размер частиц 25 мкм или меньше, у получаемого катализатора имеется тенденция к достижению предотвращения ухудшения его рабочих характеристик и предотвращения уменьшения выхода продукта, представляющего интерес, в устройстве, используемом для реакции в псевдоожиженном слое.
Причина того, почему именно ухудшаются рабочие характеристики катализатора, неясна. Предположительно, это может быть связано с тем, что если содержание частиц, имеющих размер частиц 25 мкм или меньше, превышает 20% масс., имеется тенденция к неоднородному кальцинированию в устройстве для кальцинирования (труба для кальцинирования) из-за уменьшения текучести. В соответствии с более подробным обсуждением, частицы сухого порошка или предшественника катализатора, имеющие малый размер, частиц возвращаются в устройство для кальцинирования, в частности, при непрерывном кальцинировании, и таким образом экспонируются для атмосферы кальцинирования в течение более продолжительного времени, чем это желательно. По этой причине, могут возникнуть проблемы, такие как несоответствующая степень восстановления предшественников катализатора при осуществлении кальцинирования на предварительной стадии, описанной далее, и разложение кристаллов, приписываемое избыточному кальцинированию, при основном кальцинировании. Кроме того, если содержание частиц, имеющих размер частицы 25 мкм или меньше, в сухом порошке превышает 20% масс., получаемые частицы предшественника катализатора прилипают все больше. По этой причине, рабочие характеристики ухудшаются, предположительно, из-за того, что частицы, прилипшие к стенкам устройства для кальцинирования, накапливаются на нем, вызывая риск недостаточного теплопереноса внутри него или загрязнение катализаторами, полученными из частиц, избыточно кальцинированных из-за продолжительной адгезии. В этом контексте, "предшественник катализатора" относится к соединению, сформированному в ходе стадии кальцинирования, описанной далее. Например, соединение, полученное посредством кальцинирования на предварительной стадии, упоминается как предшественник катализатора. По этим причинам, катализатор, имеющий рабочие характеристики (например, выход продукта, представляющего интерес), эквивалентные тем, которые получают при загрузочном кальцинировании, может получаться стабильно при получении катализатора с помощью непрерывного кальцинирования с использованием сухого порошка, полученного так, чтобы он имел содержание частиц, имеющих размер 25 мкм или меньше, 20% масс. или меньше, даже когда композиция катализатора является одинаковой для этих способов.
Кроме того, когда оксидный катализатор содержит Mo, Sb и Te, и тому подобное, во время кальцинирования имеется тенденция к образованию соединения с низкой температурой плавления. Частицы, имеющие размер частицы 25 мкм или меньше, имеют более высокую удельную площадь поверхности, чем частицы, имеющие размер частицы, превышающий 25 мкм, и поэтому, видимо, прилипают в большей степени. Если прилипает слишком много частиц, возникают проблемы, например, нельзя получить достаточную температуру кальцинирования для всего слоя катализатора, и достаточные выходы не могут быть обеспечены. Соответственно, является предпочтительным создавать состояние, в котором частицы, имеющие размер частиц 25 мкм или меньше, составляют малое количество, то есть, регулировать содержание на стадии перед кальцинированием таких частиц до 20% масс. или меньше.
Сухой порошок приготавливают таким образом, что средний размер его частиц предпочтительно составляет от 5 до 200 мкм, более предпочтительно, от 10 до 150 мкм, еще более предпочтительно, от 35 до 70 мкм, еще более предпочтительно, от 40 до 65 мкм, еще более предпочтительно, от 42 до 65 мкм, особенно предпочтительно, от 45 до 63 мкм, еще более предпочтительно, от 45 до 60 мкм. Благодаря тому, что средний размер частиц составляет 5 мкм или больше, имеется тенденция к тому, что получаемый катализатор достигнет предотвращения уменьшения текучести, что приведет к уменьшению выхода продукта реакции, представляющего интерес, в псевдоожиженном слое или предотвращения больших потерь количества катализатора, вызываемых выносом из реактора с псевдоожиженным слоем. Благодаря тому, что средний размер частиц составляет 200 мкм или меньше, имеется тенденция к тому, что получаемый катализатор достигнет предотвращения уменьшения текучести оксидного катализатора и уменьшения эффективности контакта с реакционным газом, приводящего к уменьшению выхода продукта реакции, представляющего интерес, в псевдоожиженном слое.
Степень восстановления предшественника катализатора может регулироваться в предпочтительном диапазоне на стадии кальцинирования, описанной далее, посредством регулировки среднего размера частиц сухого порошка, предпочтительно, до 35-70 мкм и содержания частиц, имеющих размер частиц 25 мкм или меньше, предпочтительно, до 20% масс. или меньше, до осуществления стадии кальцинирования, описанной далее. Этот механизм интерпретируется автором настоящего изобретения следующим образом, хотя и не ограничивается этим.
Сухой порошок обычно содержит, по меньшей мере, одно соединение из исходного аммиака, органической кислоты и неорганической кислоты. Когда сухой порошок кальцинируют с помощью циркуляции инертного газа, элементы, составляющие катализатор, восстанавливаются во время испарения, разложения, или чего-либо подобного, исходного аммиака, органической кислоты и/или неорганической кислоты. Исходный аммиак испаряется с образованием газообразного аммиака, который восстанавливает частицы сухого порошка или предшественника катализатора из газовой фазы. Скорость этого восстановления изменяется в зависимости от времени кальцинирования и температуры кальцинирования, в частности, при осуществлении кальцинирования на предварительной стадии, описанной далее. Продолжительное время кальцинирования или высокая температура кальцинирования облегчает восстановление, приводя к получению высокой степени восстановления. Если сухой порошок обогащен частицами, имеющими относительно малый размер частиц (далее, упоминаются также как "малые частицы"), как правило, когда средний размер частиц меньше, чем 35 мкм, или содержание частиц, имеющих размер частиц 25 мкм или меньше, превышает 20% масс., частицы сухого порошка или предшественника катализатора захватываются в инертном газе или рассеиваются при вращении трубы для кальцинирования, служащей в качестве устройства для кальцинирования. В результате, множество малых частиц возвращается в трубу для кальцинирования, и они могут, таким образом, пребывать в трубе для кальцинирования в течение более продолжительного времени, чем это желательно, делая сложным получение предпочтительного диапазона степеней восстановления. Кроме того, малые частицы также, видимо, восстанавливаются в большей степени, поскольку множество активных центров в поверхностной области вступают в контакт с газообразным аммиаком. В противоположность этому, если средний размер частиц сухого порошка превышает 70 мкм, его частицы являются большими и, таким образом, имеют немного активных центров в поверхностной области, которые вступают в контакт с газообразным аммиаком. Таким образом, такой сухой порошок плохо восстанавливается. В результате, степень восстановления может быть сложным регулировать в предпочтительном диапазоне.
В этом контексте, содержание частиц, имеющих размер частиц 25 мкм или меньше, представляет собой значение, которое определяют посредством кальцинирования части сухого порошка при 400°C в течение 1 часа на воздухе, просеивания 20 г полученных частиц с использованием сита с отверстиями 25 мкм и диаметром 20 см при экспонировании для вибратора (например, Panabrator (торговое наименование), производится National) в течение 3 минут, и измерения массы частиц, проходящих через сито, и массы частиц, остающихся на сите, с последующим вычислением с использованием следующей формулы:
(Содержание (%) 25-мкм или меньших частиц) = (масса частиц, проходящих через сито): {(масса частиц, проходящих через сито) + (масса частиц, остающихся на сите)}×100.
Причина для измерения содержания частиц, имеющих размер частиц 25 мкм или меньше, после "кальцинирования при 400°C в течение 1 часа на воздухе" части сухого порошка заключается в том, что предотвращается растворение сухого порошка в воде. Это означает, что "кальцинирование при 400°C в течение 1 часа на воздухе" осуществляют в основном для измерения, и оно не связано со стадией кальцинирования, описанной далее, и разумно думать, что размер частиц очень мало различается до и после этого кальцинирования. Степень восстановления образца, полученного с помощью этого кальцинирования, может отличаться от других сухих порошков. Обычно, этот образец, количество которого является очень малым, слабо влияет на рабочие характеристики катализатора в целом, даже если образец подвергают воздействию стадии кальцинирования, описанной далее, или не подвергают. Объектом при измерении среднего размера частиц может быть или не быть сухой порошок. Если необходимо, можно измерить средний размер частиц предшественника кальцинированного катализатора.
Примеры способа получения частиц, имеющих содержание частиц, имеющих размер частиц 25 мкм или меньше, равное 20% масс. или меньше, и средний размер частиц от 35 до 70 мкм, включают способ, который включает регулирование условий сушки распылением, например, количества оборотов атомайзера в единицу времени, температуру сушки распылением или количество подаваемого смешанного раствора исходных материалов, и способ, который включает классификацию сухого порошка. Способ классификации не является как-либо ограниченным. Например, может приниматься способ, использующий обычное устройство, такое как центробежный классификатор, воздушный классификатор, гравитационный классификатор, инерционный классификатор, сито и циклон. Среди сухих и влажных типов, классификатор сухого типа можно предпочтительно использовать с точки зрения, например, предотвращения вытекания элементов, составляющих катализатор, в растворитель и устранения отрицательного влияния на рабочие характеристики катализатора. Классификатор регулируют при условиях таких, что степень извлечения сухого порошка при классификации предпочтительно составляет 75% масс. или более, более предпочтительно 80% масс. или более, с точки зрения увеличения выхода катализатора. Альтернативно, является предпочтительным, чтобы для использования выбиралось устройство, которое удовлетворяет этим условиям.
(III) Стадия кальцинирования
Стадия кальцинирования (III) может включать стадию кальцинирования сухого порошка, полученного на стадии сушки, в присутствии соединения, содержащего W, в форме твердого продукта (далее, упоминается как "твердый продукт соединения, содержащего W"), с получением кальцинированного на предварительной стадии порошка или в основном кальцинированного порошка (далее, также упоминается как стадия (b-1)). Альтернативно, стадия кальцинирования (III) может включать стадию кальцинирования сухого порошка и дополнительного кальцинирования полученного кальцинированного на предварительной стадии порошка в присутствии твердого продукта соединения, содержащего W, с получением в основном кальцинированного порошка (далее, также упоминается как стадия (b-2)). Кроме того, стадия кальцинирования (III) может необязательно включать стадию дополнительного кальцинирования в основном кальцинированного порошка в присутствии твердого продукта соединения, содержащего W (далее, также упоминается как стадия (b-3)). Таким путем получают оксидный катализатор. Конкретно, на стадии кальцинирования, кальцинирование можно осуществлять на множестве стадий посредством изменения условий (например, температуры) на каждой стадии, как описано далее. Альтернативно, кальцинирование можно осуществлять на одной стадии при постоянных условиях. Кроме того, временной график, при котором твердый продукт соединения, содержащего W, сосуществует с сухим порошком, может выбираться соответствующим образом. Условия кальцинирования и временной график, при котором твердый продукт соединения, содержащего W, сосуществует с ним, могут устанавливаться, каждое, независимо и могут выбираться, каждое, независимо с точки зрения желаемых рабочих характеристик оксидного катализатора, удобных стадий получения, и тому подобное. Далее, сначала будет описан способ кальцинирования (a) для сухого порошка без учета присутствия или отсутствия твердого продукта соединения, содержащего W. В дальнейшем, стадии (b-1), (b-2) и (b-3) будут описываться отдельно как способ кальцинирования (b), осуществляемый в присутствии твердого продукта соединения, содержащего W.
((a) Способ кальцинирования для сухого порошка)
Например, в качестве устройства для кальцинирования можно использовать вращающуюся обжиговую печь. Форма устройства для кальцинирования не является как-либо ограниченной и предпочтительно представляет собой трубчатую форму (трубу для кальцинирования) принимая во внимание, что можно осуществлять непрерывное кальцинирование. Конкретно, предпочтительной является цилиндрическая форма. Способ нагрева предпочтительно представляет собой способ внешнего нагрева с точки зрения, например, простоты регулирования температуры кальцинирования в виде предпочтительного профиля повышения температуры. Предпочтительно можно использовать электрическую печь. Размер и материал, и тому подобное, трубы для кальцинирования можно выбирать соответствующим образом в соответствии с условиями или выходами кальцинирования. Внутренний диаметр трубы для кальцинирования предпочтительно составляет от 70 до 2000 мм, более предпочтительно, от 100 до 1700 мм, с точки зрения, например, предотвращения неоднородности распределения температуры кальцинирования в слое катализатора и регулирования времени и выходов кальцинирования при соответствующих значениях. Кроме того, длина трубы для кальцинирования предпочтительно составляет от 200 до 10000 мм, более предпочтительно, от 800 до 8000 мм, с точки зрения, например, сведения к минимуму времени пребывания частиц сухого порошка и предшественника катализатора в трубе для кальцинирования, то есть, распределения времен кальцинирования, предотвращения появления деформаций в трубе для кальцинирования и регулировки времени и выходов кальцинирования при соответствующих значениях. Когда по трубе для кальцинирования наносятся удары, толщина ее стенок предпочтительно составляет 2 мм или больше, более предпочтительно 4 мм или больше, с точки зрения поддержания достаточно большой толщины для предотвращения разрушения стенки под действием ударов. Кроме того, толщина стенок предпочтительно составляет 100 мм или меньше, более предпочтительно 50 мм или меньше, с точки зрения достаточного переноса удара во внутреннее пространство трубы для кальцинирования. Кроме того, является предпочтительным, чтобы труба для кальцинирования имела некоторый наклон в направлении протекания порошка и имела меньшую высоту на выходе, чем на входе для порошка, с точки зрения, например, предотвращения неоднородности распределения температур кальцинирования и распределения времен кальцинирования в слое катализатора, и регулирования времен кальцинирования и выходов катализатора при соответствующих значениях. Кроме того, с этой точки зрения, угол наклона θ по отношению к горизонтали предпочтительно составляет 0°<θ<80°, более предпочтительно 1°≤θ≤40°.
Материал устройства для кальцинирования не является как-либо ограниченным постольку, поскольку он предпочтительно имеет термостойкость и является достаточно прочным, чтобы предотвратить разрушение устройства для кальцинирования под действием удара. Например, устройство, изготовленное из SUS (нержавеющей стали), может использоваться предпочтительно.
Отбивная решетка, имеющая в центральной части отверстие, через которое проходит порошок, может располагаться перпендикулярно (или почти перпендикулярно) потоку порошка в трубе для кальцинирования, так что труба для кальцинирования разделяется на две или более областей. Время пребывания порошка в трубе для кальцинирования может обеспечиваться просто посредством расположения отбивной решетки. Количество отбивных решеток может быть равно одной или двум или более. Материал отбивной решетки предпочтительно представляет собой металл с точки зрения улучшения износостойкости, чтобы противостоять атмосфере кальцинирования, и термостойкости. Предпочтительно используют такой же материал, как и для трубы для кальцинирования. Высота отбивной решетки может устанавливаться в соответствии со временем пребывания, которое должно обеспечиваться. Например, когда сухой порошок подается при скорости 250 г/час с использованием вращающейся обжиговой печи, имеющей трубу для кальцинирования, изготовленную из SUS, с внутренним диаметром 150 мм и длиной 1150 мм, высота отбивной решетки предпочтительно составляет 5-50 мм, более предпочтительно 10-40 мм, еще более предпочтительно 13-35 мм. Толщина отбивной решетки не является как-либо ограниченной и предпочтительно регулируется в соответствии с размером трубы для кальцинирования. Например, для вращающейся обжиговой печи, имеющей трубу для кальцинирования, изготовленную из SUS, с внутренним диаметром 150 мм и длиной 1150 мм, толщина отбивной решетки предпочтительно находится в пределах между 0,3 мм и 30 мм включительно, более предпочтительно 0,5 мм и 15 мм включительно.
Для предотвращения разрушения, образования трещин или чего-либо подобного в сухом порошке и для однородного кальцинирования, является предпочтительным, чтобы кальцинирование осуществлялось с помощью трубы для кальцинирования, вращающейся вокруг продольной оси. Скорость вращения трубы для кальцинирования предпочтительно составляет 0,1-30 об/мин, более предпочтительно 0,5-20 об/мин, еще более предпочтительно 1-10 об/мин.
Для кальцинирования сухого порошка, является предпочтительным, чтобы сухой порошок нагревался при температуре, которая начинается при температуре ниже, чем 400°C, и повышается от нее непрерывно или постепенно до температуры в диапазоне от 550 до 800°C, с точки зрения, например, достижения предпочтительного окислительно-восстановительного состояния полученного катализатора и улучшения рабочих характеристик катализатора.
Кальцинирование можно осуществлять в атмосфере воздуха или при циркуляции воздуха. Является предпочтительным, чтобы, по меньшей мере, часть кальцинирования осуществлялась в то время, когда циркулирует инертный газ (например, азот), по существу не содержащий кислорода, с точки зрения, например, простоты регулирования катализатора при предпочтительном окислительно-восстановительном состоянии.
Когда осуществляют загрузочное кальцинирование, подаваемое количество инертного газа предпочтительно составляет 50 Н/литр/час или более, более предпочтительно 50-5000 Н/литр/час, еще более предпочтительно 50-3000 Н/литр/час, на кг сухого порошка, с точки зрения регулирования катализатора при предпочтительном окислительно-восстановительном состоянии. В этом контексте, единица "Н/литр" обозначает литр, измеренный при стандартных условиях температуры и давления, то есть, при 0°C и при давлении 1 атмосфера.
Когда осуществляют непрерывное кальцинирование, количество подаваемого инертного газа предпочтительно составляет 50 Н/литр/час или более, более предпочтительно 50-5000 Н/литр/час, еще более предпочтительно 50-3000 Н/литр/час, на кг сухого порошка, с точки зрения регулирования катализатора при предпочтительном окислительно-восстановительном состоянии. В этом случае, форма контакта между инертным газом и сухим порошком может представлять собой противоточный контакт или может представлять собой спутный контакт. Противоточный контакт является предпочтительным с учетом газообразных компонентов, генерируемых из сухого порошка и воздуха, который может подмешиваться в микроскопическом количестве в сухой порошок. Конкретно, когда способ, который включает добавление перекиси водорода в смешанный водный раствор (A), принимается на стадии приготовления исходных материалов, и молибден и ванадий окисляются до почти самого высокого числа окисления с получением заготовки исходных материалов, является предпочтительным, чтобы сухой порошок, кальцинировался в то время, когда циркулирует инертный газ (например, азот), по существу не содержащий кислорода.
Сухой порошок обычно может содержать исходный аммиак, органическую кислоту, неорганическую кислоту, и тому подобное, в дополнение к воде.
Когда сухой порошок, кальцинируют в то время, когда циркулирует инертный газ, по существу не содержащий кислорода, элементы, составляющие катализатор, содержащиеся в сухом порошке, и предшественники катализатора восстанавливаются во время их испарения, разложения или чего-либо подобного.
Когда элементы, составляющие катализатор, в сухом порошке имеют почти самое большое число окисления, на стадии кальцинирования может осуществляться только лишь восстановление для регулировки степени восстановления оксидного катализатора в желаемом диапазоне. Таким образом, этот подход является удобным для промышленности.
С другой стороны, окисляющий компонент или восстанавливающий компонент может добавляться, как описано далее, в атмосферу кальцинирования таким образом, что степень восстановления регулируется в желаемом диапазоне. Является предпочтительным, чтобы кальцинирование осуществлялось таким образом, чтобы полученный оксидный катализатор имел степень восстановления 8-12% и удельную площадь поверхности от 5 до 30 м2/г. Благодаря тому, что удельная площадь поверхности катализатора составляет от 5 до 30 м2/г, оказывается воздействие получения дополнительной существенной активности, дополнительного предотвращения деградации, а также дополнительного повышения выходов. Кроме того, воздействие, оказываемое посредством добавления соединения молибдена для поддержания выходов во время реакции окисления или реакции аммоксидирования, проявляется более существенно. В дополнение к этому, не наблюдается резкой деградации. Таким образом, количество добавляемого соединения молибдена и частота его добавления могут быть уменьшены. Причина этого неясна. Предположительно, это связано с тем, что если удельная площадь поверхности меньше, чем 5 м2/г, воздействие, оказываемое посредством добавления соединения молибдена, оказывается слабо из-за малой активной поверхности активных частиц, ответственных за реакцию. Кроме того, если удельная площадь поверхности больше, чем 30 м2/г, активные частицы имеют большую активную поверхность. Однако, в этом случае, молибден как предполагается, быстро покидает активную поверхность. Степень восстановления оксидного катализатора или предшественника катализатора вычисляют в соответствии со следующей формулой (6):
Степень восстановления (%) = ((n0-n)/n0)×100... (6)
когда n представляет собой количество атомов кислорода, которое удовлетворяет валентностям составляющих элементов иных, чем кислород, в оксидном катализаторе или предшественнике катализатора; и n0 представляет собой количество атомов кислорода, необходимое для составляющих элементов иных, чем кислород, в оксидном катализаторе или предшественнике катализатора, чтобы они имели их соответствующие самые высокие числа окисления.
Для определения степени восстановления, величина (n0-n) в формуле (6) получается посредством воздействия на образец окислительно-восстановительного титрования с помощью KMnO4. Кроме того, величина (n0-n) может определяться с помощью окислительно-восстановительного титрования, как для предшественника катализатора до завершения кальцинирования, так и для оксидного катализатора после завершения кальцинирования. Однако измерение с помощью окислительно-восстановительного титрования различается по условиям измерения между предшественником катализатора до завершения кальцинирования и катализатором после завершения кальцинирования. Далее, будет показано по одному примеру способа измерения для каждого из предшественников катализатора до завершения кальцинирования и катализатора после завершения кальцинирования.
Для предшественника катализатора до завершения кальцинирования, степень восстановления измеряют, например, следующим образом:
Сначала, приблизительно 200 мг образца точно взвешивают в химическом стакане. Водный раствор KMnO4, имеющий известную концентрацию, добавляют к нему в избыточном количестве. Затем 150 мл чистой воды (70°C) и 2 мл серной кислоты, 1:1 (то есть, водного раствора серной кислоты, полученного посредством смешивания концентрированной серной кислоты и воды при объемном отношении 1/1), добавляют в химический стакан. Затем отверстие химического стакана закрывают наблюдательным стеклом, и раствор в химическом стакане перемешивают в течение 1 часа на горячей водяной бане 70°C±2°C для окисления образца. Поскольку там присутствует избыток KMnO4, цвет раствора, как подтверждается, становится пурпурным из-за присутствия непрореагировавшего KMnO4 в растворе. Затем после завершения окисления, раствор фильтруют через фильтровальную бумагу, и все количество фильтрата собирают. Затем водный раствор оксалата натрия (Na2C2O4), имеющий известную концентрацию, добавляют в избыточном количестве по сравнению с количеством KMnO4, присутствующим в фильтрате. Затем раствор нагревают до температуры 70°C при перемешивании. Раствор, как подтверждается, становится бесцветным и прозрачным, и к нему добавляют 2 мл серной кислоты, 1:1. Кроме того, перемешивание продолжают, поддерживая температуру раствора при 70°C±2°C, с последующим титрованием водным раствором KMnO4, имеющим известную концентрацию. Конечную точку определяют как момент времени, когда цвет раствора становится чуть бледно-розовым из-за титрования KMnO4 и этот цвет присутствует в течение приблизительно 30 секунд. Количество KMnO4, израсходованного при окислении образца, определяют из всего количества KMnO4 и всего количества Na2C2O4. Из этого количества KMnO4, вычисляют величину (n0-n) и степень восстановления определяют на ее основе.
Для оксидного катализатора после завершения кальцинирования, степень восстановления измеряют, например, следующим образом:
Сначала, приблизительно 200 мг катализатора, измельченного с использованием ступки, изготовленной из агата, точно взвешивают в химическом стакане. 150 мл чистой воды (95°C) и 4 мл серной кислоты, 1:1 (то есть, водного раствора серной кислоты, полученного посредством смешивания концентрированной серной кислоты и воды при объемном отношении 1/1), добавляют к ним. Затем раствор перемешивают, поддерживая его температуру при 95°C±2°C, с последующим титрованием водным раствором KMnO4, имеющим известную концентрацию. При этом титровании, KMnO4 постепенно добавляют по каплям малыми порциями таким образом, что цвет раствора временно становится пурпурным из-за добавления по каплям KMnO4 и, однако, этот пурпурный цвет не сохраняется в течение 30 секунд или дольше. Кроме того, количество раствора уменьшается, когда вода испаряется. Таким образом, к нему по потребности добавляют чистую воду (95°C) таким образом, что количество раствора остается постоянным. Конечную точку определяют как момент времени, когда цвет раствора становится чуть бледно-розовым из-за титрования KMnO4, и этот цвет сохраняется в течение приблизительно 30 секунд. Затем определяют количество KMnO4, израсходованного при окислении образца. Из этого количества KMnO4, вычисляют величину (n0-n), и определяют на ее основе степень восстановления.
Кроме того, в дополнение к этим способам измерения, следующее измерение также может осуществляться как для предшественника катализатора до завершения кальцинирования, так и для оксидного катализатора после завершения кальцинирования:
Конкретно, образец полностью окисляется с помощью кислорода посредством нагрева до температуры более высокой, чем температура кальцинирования (при которой кальцинируют предшественник катализатора или катализатор), в атмосфере, содержащей кислород, при условии, что элементы, составляющие образец, не испаряются или не покидают его. Определяют увеличение массы (количество кислорода, связанное с ним). Из этого количества определяют величину (n0-n) и определяют на ее основе степень восстановления.
Для способа кальцинирования для сухого порошка, конкретно, является предпочтительным, чтобы: сухой порошок кальцинировался при условиях кальцинирования, при которых сухой порошок нагревают до температуры, которая начинается при температуре ниже, чем 400°C и повышается от нее непрерывно или постепенно до температуры в пределах 550-700°C; и условия кальцинирования регулировались таким образом, что степень восстановления предшественника катализатора во время кальцинирования составляет 8-12%, когда температура нагрева достигает 400°C.
Степень восстановления оксидного катализатора, как правило, зависит от количества органического вещества, такого как щавелевая кислота, содержащегося в сухом порошке, количества исходного аммиака, полученного из соли аммония, в качестве исходных материалов, скорости повышения температуры в начале кальцинирования и количества инертного газа (в случае кальцинирования в атмосфере инертного газа) или температуры и времени кальцинирования (в случае кальцинирования в атмосфере воздуха).
Примеры способа установления степени восстановления оксидного катализатора при 8-12% включают способ, который включает повышение температуры, начиная с температуры ниже, чем 400°C, для кальцинирования, чтобы разложить исходную щавелевую кислоту, исходный аммиак, и тому подобное, в сухом порошке и тем самым почти завершить генерирование газа таким образом, что степень восстановления предшественника катализатора во время кальцинирования составляет 8-12%, когда температура нагрева достигает 400°C.
Удельная площадь поверхности катализатора зависит от конечной температуры и времени кальцинирования (нагрева), и количество катализатора, нанесенного на носитель, такой как оксид кремния (в случае катализатора на носителе), и особенно сильно зависит от степени восстановления, когда температура нагрева достигает 400°C, и от конечной температуры кальцинирования. С такой точки зрения, конечная температура кальцинирования предпочтительно составляет 550°C-700°C. При этой температуре, время кальцинирования предпочтительно составляет 0,5 часов - 20 часов. Чем выше конечная температура кальцинирования или чем продолжительнее время кальцинирования, тем меньше получаемая удельная площадь поверхности, как тенденция.
Кроме того, благодаря тому, что степень восстановления попадает в диапазон от 8 до 12%, когда температура нагрева достигает 400°C, имеется тенденция к тому, что получаемый катализатор достигает предотвращения избыточного уменьшения или увеличения его удельной площади поверхности.
Например, для установления удельной площади поверхности катализатора, равной от 5 до 30 м2/г, является предпочтительным, чтобы: степень восстановления попадала в диапазон от 8 до 12%, когда температура нагрева достигает 400°C; и конечная температура кальцинирования устанавливалась при 550°C-700°C.
Кальцинирование можно осуществлять на одной стадии при постоянных условиях. Для эффективного получения оксидного катализатора, имеющего степень восстановления от 8 до 12% и удельную площадь поверхности от 5 до 30 м2/г, является предпочтительным, чтобы стадия кальцинирования включала кальцинирование на предварительной стадии и последующее основное кальцинирование. В этом контексте, температура кальцинирования при осуществлении кальцинирования на предварительной стадии предпочтительно представляет собой температуру более низкую, чем температура кальцинирования при основном кальцинировании. Более конкретно, является предпочтительным, чтобы кальцинирование на предварительной стадии осуществлялось в диапазоне температур от 250 до 400°C, и основное кальцинирование осуществлялось в диапазоне температур от 550 до 700°C. Кальцинированный на предварительной стадии порошок получают с помощью этого кальцинирования на предварительной стадии, в то время как в основном кальцинированный порошок получают с помощью основного кальцинирования.
Основное кальцинирование можно осуществлять непрерывно после кальцинирования на предварительной стадии, то есть, с помощью непосредственного изменения температуры кальцинирования при осуществлении кальцинирования на предварительной стадии до температуры кальцинирования для осуществления основного кальцинирования. Альтернативно, основное кальцинирование можно осуществлять посредством перезапуска после завершения кальцинирования на предварительной стадии, то есть, посредством временного уменьшения температуры от температуры кальцинирования при осуществлении кальцинирования на предварительной стадии, а затем ее увеличения до температуры кальцинирования для осуществления основного кальцинирования. Кроме того, кальцинирование на предварительной стадии и основное кальцинирование могут, каждое, разделяться на множество стадий кальцинирования, различающихся по условиям кальцинирования.
Когда степень восстановления предшественника катализатора измеряют во время кальцинирования, образец может отбираться из устройства для кальцинирования при неизменной температуре. Однако, образец, имеющий высокую температуру, может окисляться при экспонировании для воздуха с изменением степени восстановления. Таким образом, такой образец охлаждают до комнатной температуры, а затем вынимают из устройства для кальцинирования. Полученный образец можно использовать в качестве репрезентативного образца.
Примеры способа контроля степени восстановления в желаемом диапазоне, когда температура нагрева достигает 400°C, конкретно включает способ, который включает регулировку температуры кальцинирования при осуществлении кальцинирования на предварительной стадии, способ, который включает добавление окисляющего компонента, такого как кислород, в атмосферу во время кальцинирования, и способ, который включает добавление восстанавливающего компонента в атмосферу во время кальцинирования. Кроме того, два или более из этих способов могут объединяться.
Способ, который включает изменение температуры кальцинирования при осуществлении кальцинирования на предварительной стадии (далее, упоминается как "температура кальцинирования на предварительной стадии"), представляет собой подход с изменением температуры кальцинирования на предварительной стадии, чтобы тем самым регулировать степень восстановления предшественника катализатора, когда температура нагрева достигает 400°C. Степень восстановления обычно уменьшается посредством понижения температуры кальцинирования на предварительной стадии, и степень восстановления имеет тенденцию к увеличению посредством повышения температуры кальцинирования на предварительной стадии. Таким образом, степень восстановления может контролироваться посредством изменения температуры кальцинирования на предварительной стадии.
Способ, который включает добавление окисляющего компонента, такого как кислород, в атмосферу во время кальцинирования, представляет собой способ, который можно использовать для уменьшения степени восстановления предшественника катализатора, когда температура нагрева достигает 400°C. В этом контексте, кальцинирование в термине "во время кальцинирования" может представлять собой кальцинирование на предварительной стадии или основное кальцинирование, или как то, так и другое.
Окисляющий компонент, добавляемый в атмосферу во время кальцинирования, представляет собой окисляющий компонент, содержащийся в инертном газе, подаваемом в устройство для кальцинирования. Количество добавляемого окисляющего компонента может контролироваться с помощью концентрации в инертном газе, подаваемом в устройство для кальцинирования. Степень восстановления предшественника катализатора, когда температура нагрева достигает 400°C, может контролироваться посредством добавления этого окисляющего компонента. Когда окисляющий компонент представляет собой кислород, воздух (или инертный газ, содержащий воздух) подается в устройство для кальцинирования, в котором кислород в воздухе может затем использоваться в качестве окисляющего компонента.
Способ, который включает добавление восстанавливающего компонента в атмосферу во время кальцинирования, представляет собой способ, который можно использовать для увеличения степени восстановления предшественника катализатора, когда температура нагрева достигает 400°C. В этом контексте, кальцинирование в термине "во время кальцинирования" может представлять собой кальцинирование на предварительной стадии или основное кальцинирование, или как то, так и другое.
Восстанавливающий компонент, добавляемый в атмосферу во время кальцинирования, представляет собой восстанавливающий компонент, содержащийся в инертном газе, подаваемом в устройство для кальцинирования. Количество добавляемого восстанавливающего компонента может контролироваться с помощью концентрации в инертном газе, подаваемом в устройство для кальцинирования. Степень восстановления предшественника катализатора, когда температура нагрева достигает 400°C, может контролироваться посредством добавления этого восстанавливающего компонента. Например, в качестве восстанавливающего компонента может использоваться аммиак.
Когда степень восстановления предшественника катализатора не является желаемой степенью, когда температура нагрева достигает 400°C, общее количество необходимых окисляющих или восстанавливающих компонентов может быть вычислено по разнице между реальной степенью восстановления и желаемой степенью, и оно добавляется в атмосферу во время кальцинирования.
Способ осуществления кальцинирования в атмосфере инертного газа или в предпочтительной окислительной/восстановительной атмосфере не является как-либо ограниченным. Является предпочтительным использовать устройство для кальцинирования, имеющее соответствующую герметизирующую структуру, с помощью которой контакт с внешним воздухом может по существу блокироваться.
Кальцинирование на предварительной стадии осуществляют в диапазоне температур кальцинирования на предварительной стадии, включающем предпочтительно 250°C-400°C, более предпочтительно 300°C-400°C, предпочтительно, при циркуляции инертного газа, с точки зрения, например, простого регулирования полученного катализатора в предпочтительном окислительно-восстановительном состоянии и достижения улучшенных рабочих характеристик катализатора. Является предпочтительным, чтобы температура кальцинирования на предварительной стадии поддерживалась при постоянной температуре в диапазоне температур от 250°C до 400°C. Однако температура может изменяться в диапазоне температур от 250°C до 400°C или может умеренно повышаться или понижаться. Время поддержания температуры нагрева предпочтительно составляет 30 минут или больше, более предпочтительно, от 3 до 12 часов, с точки зрения, например, простой регулировки полученного катализатора в предпочтительном окислительно-восстановительном состоянии и достижения улучшенных рабочих характеристик катализатора. Профиль повышения температуры для достижения температуры кальцинирования на предварительной стадии может представлять собой линейный профиль повышения температуры или может представлять собой выпуклый и вогнутый профиль повышения температуры. Кроме того, во время повышения температуры, температура может понижаться в течение определенного времени или может повышаться и понижаться попеременно. Кроме того, в ходе повышения температуры, эндотермическая реакция может вызываться компонентами, содержащимися в сухом порошке и/или в предшественнике катализатора, временно понижая температуру.
Средняя скорость повышения температуры во время повышения температуры для достижения температуры кальцинирования на предварительной стадии не является как-либо ограниченной. Она может составлять, например, приблизительно от 0,1 до 15°C/мин, а предпочтительно составляет от 0,5 до 5°C/мин, более предпочтительно от 1 до 2°C/мин, с точки зрения, например, простого регулирования полученного катализатора в предпочтительном окислительно-восстановительном состоянии и достижения улучшенных рабочих характеристик катализатора.
Основное кальцинирование осуществляют при температуре кальцинирования, предпочтительно равной от 550 до 800°C, более предпочтительно, от 580 до 750°C, еще более предпочтительно, от 600 до 720°C, особенно предпочтительно, от 620 до 700°C, предпочтительно, при циркуляции инертного газа, с точки зрения, например, простого регулирования полученного катализатора при предпочтительной удельной площади поверхности, для достаточного формирования химически активной кристаллической структуры и для достижения улучшенных рабочих характеристик катализатора. Является предпочтительным, чтобы температура кальцинирования поддерживалась при постоянной температуре в диапазоне температур от 550 до 800°C. Однако температура может изменяться в диапазоне температур от 550 до 800°C или умеренно может увеличиваться или уменьшаться. Кроме того, температура может временно понижаться из-за эндотермической реакции, может понижаться в течение определенного времени во время повышения температуры или может повышаться и понижаться попеременно, как при осуществлении кальцинирования на предварительной стадии. Кроме того, время кальцинирования (время поддержания температуры кальцинирования) при основном кальцинировании предпочтительно равно от 0,5 до 20 часов, более предпочтительно, от 1 до 15 часов, с точки зрения, например, простого регулирования полученного катализатора при предпочтительной удельной площади поверхности, достаточного облегчения образования химически активной кристаллической структуры и достижения улучшенных рабочих характеристик катализатора. Профиль повышения температуры для достижения температуры кальцинирования может представлять собой линейный профиль повышения температуры или может представлять собой выпуклый или вогнутый профиль повышения температуры. Кроме того, средняя скорость повышения температуры во время повышения температуры для достижения температуры кальцинирования не является как-либо ограниченной. Она может составлять, например, от 0,1 до 15°C/мин, а предпочтительно составляет от 0,3 до 10°C/мин, более предпочтительно, от 0,5 до 8°C/мин, с точки зрения, например, простого регулирования полученного катализатора при предпочтительной удельной площади поверхности, достаточного облегчения образования химически активной кристаллической структуры и достижения улучшенных рабочих характеристик катализатора.
Когда труба для кальцинирования разделяется на области с помощью отбивной решетки, сухой порошок, предшественник катализатора или оксидный катализатор (далее, упоминаются как "сухой порошок, и тому подобное") непрерывно проходит, по меньшей мере, через 2 области или через области, предпочтительно изолированные с использованием 2-20, еще более предпочтительно 4-15 отбивных решеток, с точки зрения, например, обеспечения времени пребывания, пригодного для использования для сухого порошка, и тому подобное, в трубе для кальцинирования. Контроль температуры может осуществляться с использованием одного или нескольких контроллеров. Для получения желаемого профиля кальцинирования является предпочтительным, чтобы нагреватель и контроллер располагались для контроля температуры в каждой из этих областей, изолированных с помощью отбивных решеток. Например, 7 отбивных решеток можно разместить для продольного разделения части, присутствующей в нагревательной печи, в трубе для кальцинирования, на 8 одинаковых частей. Когда используют трубу для кальцинирования, разделенную таким образом на 8 областей, является предпочтительным, чтобы установленная температура контролировалась с использованием нагревателя и контроллера, которые располагаются в каждой из этих 8 областей, таким образом, чтобы температура сухого порошка, и тому подобное демонстрировала желаемый профиль температуры кальцинирования. Например, когда используют трубу для кальцинирования, разделенную таким образом на 8 областей, регулировку для получения желаемого профиля кальцинирования можно осуществлять следующим образом: для кальцинирования на предварительной стадии, является предпочтительным, чтобы температура термопары, вставленной в центральную часть, в каждой области для сухого порошка, и тому подобное, находящегося в трубе для кальцинирования, устанавливались для области 1: 100-300°C, области 2: 150-350°C, области 3: 250-400°C, области 4: 250-400°C, области 5: 300-400°C, области 6: 300-400°C, области 7: 310-400°C и области 8: 260-400°C, более предпочтительно, для области 1: 120-280°C, области 2: 180-330°C, области 3: 250-350°C, области 4: 270-380°C, области 5: 300-380°C, области 6: 300-390°C, области 7: 320-390°C и области 8: 260-380°C, где области нумеруются, начиная с 1, в порядке подачи сухого порошка, и тому подобное. Подобным же образом, для осуществления основного кальцинирования, является предпочтительным, чтобы такая температура регулировалась для области 1: 350-600°C, области 2: 400-700°C, области 3: 550-700°C, области 4: 550-700°C, области 5: 550-700°C, области 6: 450-680°C, области 7: 450-650°C и области 8: 350-600°C, более предпочтительно, для области 1: 360-560°C, области 2:450-650°C, области 3: 600-690°C, области 4: 620-690°C, области 5: 580-690°C, области 6: 480-660°C, области 7: 450-630°C и области 8: 370-580°C, где области нумеруются, начиная с 1, в порядке подачи сухого порошка, и тому подобное.
Кроме того, средняя скорость понижения температуры после завершения основного кальцинирования предпочтительно составляет 0,001-1000°C/мин, более предпочтительно 0,005-100°C/мин, еще более предпочтительно 0,01-50°C/мин, особенно предпочтительно, 0,05-20°C/мин, с точки зрения, например, достаточного облегчения образования химически активной кристаллической структуры и улучшения рабочих характеристик катализатора. Кроме того, является также предпочтительным, чтобы температура временно поддерживалась при температуре более низкой, чем температура основного кальцинирования, с точки зрения, например, достаточного облегчения образования химически активной кристаллической структуры и улучшения рабочих характеристик катализатора. С этой точки зрения, температуру предпочтительно поддерживают при температуре, по меньшей мере, на 10°C, более предпочтительно, по меньшей мере, на 50°C, еще более предпочтительно, по меньшей мере, на 100°C ниже, чем температура основного кальцинирования. С этой точки зрения, время поддержания температуры предпочтительно составляет 0,5 часа или больше, более предпочтительно 1 час или больше, еще более предпочтительно 3 часа или больше, особенно предпочтительно, 10 часов или больше.
Когда осуществляют основное кальцинирование посредством перезапуска после завершения кальцинирования на предварительной стадии, можно осуществлять низкотемпературную обработку при основном кальцинировании. Кроме того, низкотемпературная обработка может также осуществляться после основного кальцинирования. В дополнение к этому, дополнительное кальцинирование может осуществляться после низкотемпературной обработки после основного кальцинирования.
Время, необходимое для низкотемпературной обработки, то есть, время, необходимое для понижения температуры порошка, кальцинированного на предварительной стадии, а затем для повышения температуры, чтобы достичь температуры кальцинирования для осуществления основного кальцинирования, может регулироваться соответствующим образом в соответствии с размером, толщиной стенок и материалом устройства для кальцинирования, выходами катализатора, рядом периодов времени во время непрерывного кальцинирования порошка, кальцинированного на предварительной стадии, и/или в основном кальцинированного порошка, скорости адгезии, количеством прилипших частиц, и тому подобное. Например, когда используют трубу для кальцинирования, изготовленную из SUS, с внутренним диаметром 500 мм, длиной 4500 мм и толщиной стенок 20 мм, время, необходимое для низкотемпературной обработки, предпочтительно находится в пределах 30 дней, более предпочтительно, в пределах 15 дней, еще более предпочтительно, в пределах 3 дней, особенно предпочтительно, в пределах 2 дней, в виде ряда периодов времени во время непрерывного кальцинирования порошка, кальцинированного на предварительной стадии, и/или в основном кальцинированного порошка, с точки зрения, например, достаточного отслаивания порошка, кальцинированного на предварительной стадии, и/или в основном кальцинированного порошка, прилипшего к стенкам трубы для кальцинирования, стабильного поддержания температуры оксидного слоя и улучшения рабочих характеристик полученного катализатора. В этом контексте, температура оксидного слоя относится к температуре, измеряемой с использованием термопары, вставленной в порошок, кальцинированный на предварительной стадии, и/или в основном кальцинированный порошок, скапливающийся в устройстве для кальцинирования. Кроме того, например, когда осуществляют основное кальцинирование при температуре кальцинирования 645°C посредством подачи порошка, кальцинированного на предварительной стадии, со скоростью 35 кг/час при вращении при 6 об/мин во вращающейся обжиговой печи, имеющей трубу для кальцинирования, изготовленную из SUS, с внутренним диаметром 500 мм, длиной 4500 мм и толщиной стенок 20 мм, стадия понижения температуры до 400°C перед основным кальцинированием, а затем повышения температуры до 645°C может осуществляться в течение приблизительно 1 дня. Для 1-годичного непрерывного кальцинирования, такая низкотемпературная обработка может осуществляться раз в месяц, чтобы при этом осуществлять стабильное кальцинирование при поддержании температуры оксидного слоя.
В основном кальцинированный порошок, полученный с помощью основного кальцинирования, может использоваться в качестве оксидного катализатора сам по себе или может дополнительно кальцинироваться в присутствии твердого продукта соединения, содержащего W, как описано далее, с получением оксидного катализатора, который может улучшить выход соединения, представляющего интерес. Когда стадию кальцинирования осуществляют в одну стадию, порошок, полученный с помощью этого кальцинирования за одну стадию, используют в качестве в основном кальцинированного порошка.
Кроме того, воздействие растрескивания масс, прилипших к устройству для кальцинирования, имеет тенденцию к увеличению посредством приложения удара к устройству для кальцинирования на стадии кальцинирования. Когда осуществляют низкотемпературную обработку, является предпочтительным, чтобы удары прикладывались к устройству для кальцинирования во время этой низкотемпературной обработки, поскольку растресканные массы имеют тенденцию к легкому выходу из устройства для кальцинирования.
Сила ударов, прикладываемых к устройству для кальцинирования, зависит от глубины сухого порошка и/или предшественника катализатора, подаваемого в устройство для кальцинирования, диаметра, длины, толщины стенок и материала устройства для кальцинирования, материала, типа, формы и положения ударного устройства и частоты ударов, и тому подобное. Таким образом, является предпочтительным, чтобы эта сила устанавливалась соответствующим образом в соответствии с этими факторами.
Вибрационное ускорение в положении, где прикладываются удары (далее, также упоминается как ударная точка), предпочтительно составляет 0,1 м/сек2 или больше, более предпочтительно 1 м/сек2 или больше, еще более предпочтительно 5 м/сек2 или больше, особенно предпочтительно, 10 м/сек2 или больше, с точки зрения достаточного уменьшения адгезии на внутренней стенке устройства для кальцинирования. Кроме того, вибрационное ускорение предпочтительно составляет 3000 м/сек2 или меньше, более предпочтительно 1000 м/сек2 или меньше, еще более предпочтительно 500 м/сек2 или меньше, особенно предпочтительно, 300 м/сек2 или меньше, с точки зрения предотвращения разрушения устройства для кальцинирования и с точки зрения предотвращения возмущения потока порошка, циркулирующего в устройстве для кальцинирования.
В настоящем варианте осуществления, "вибрационное ускорение" ударов, прикладываемых к устройству для кальцинирования, означает среднее значения для величин, измеренных на расстояниях L/4, 3L/8 и L/2 от входа для порошка в устройстве для кальцинирования параллельно с направлением потока порошка, при этом полная длина устройства для кальцинирования представлена как L. Положения измерений будут такими же, как и положения ударных точек в направлении поперечного сечения, ортогонального потоку порошка в устройстве для кальцинирования. Вибрационное ускорение может измеряться с помощью виброметра, прикрепленного к устройству для кальцинирования. В качестве виброметра можно использовать MD220 (торговое наименование), производится ASAHI KASEI TECHNOSYSTEM CO., LTD.
Способ приложения ударов к нему не является как-либо ограниченным, и предпочтительно можно использовать пневматический встряхиватель, молоток, молотковое устройство или что-либо подобное. Материал детали (эта деталь вступает в непосредственный контакт с устройством для кальцинирования) на конце ударной части не является как-либо ограниченным, постольку, поскольку он представляет собой материал, имеющий достаточную термостойкость. Например, можно использовать обычные смолы и металлы, способные противостоять ударам. Среди них, предпочтительными являются металлы. Является предпочтительным, чтобы металлы имели твердость без разрушения или деформирования устройства для кальцинирования. Детали, изготовленные из меди или SUS, могут использоваться предпочтительно. Подобным же образом, положения, в которых прикладываются удары, не являются как-либо ограниченными, и удары могут наноситься в положении, преимущественном с точки зрения работы. Является предпочтительным, чтобы удары прикладывались в положении, которое не закрывается нагревательной печью устройства для кальцинирования, поскольку удар может наноситься непосредственно по устройству для кальцинирования без потерь.
Количество положений, в которых прикладываются удары, может составлять одно или два или более. Для эффективной передачи к ним вибраций, является предпочтительным, чтобы удар прикладывался в направлении, перпендикулярном аксиальному направлению трубы для кальцинирования, когда трубу используют в качестве устройства для кальцинирования. Частота ударов не является как-либо ограниченной, является предпочтительным, чтобы удары прикладывались к устройству для кальцинирования постоянно, поскольку этот подход имеет тенденцию к более благоприятному уменьшению количества порошков, прилипших в устройстве для кальцинирования. В этом контексте, фраза "удары прикладываются постоянно" означает, что удары предпочтительно прикладываются один раз в 1 секунду - 1 час включительно, более предпочтительно, один раз в 1 секунду - 30 минут включительно, еще более предпочтительно, один раз в 1 секунду - 5 минут включительно, особенно предпочтительно, один раз в 1 секунду - 1 минуту включительно. Является предпочтительным, чтобы частота ударов регулировалась соответствующим образом в соответствии с вибрационным ускорением, глубиной сухого порошка и предшественника катализатора, подаваемого в устройство для кальцинирования, диаметром, длиной, толщиной стенок и материалом устройства для кальцинирования и материалом, типом и формой ударного устройства.
Способ кальцинирования, описанный выше, может использоваться независимо от присутствия или отсутствия твердого продукта соединения, содержащего W, описанного далее.
(b) Способ кальцинирования, осуществляемый в присутствии твердого продукта соединения, содержащего W
Кальцинирование в присутствии твердого продукта соединения, содержащего W, дает возможность для твердофазной диффузии W из твердой частицы соединения, содержащего W, в частицу катализатора таким образом, что W может концентрироваться по поверхности частицы катализатора и вблизи нее.
Твердый продукт соединения, содержащего W
Например, соль аммония, нитрат, карбоксилат, карбоксилат аммония, пероксокарбоксилат, пероксокарбоксилат аммония, галогенид аммония, галогенид, ацетилацетонат, алкоксид, трифенильное соединение, полиоксометалат, полиоксометалат аммония, дивольфрам триоксид, диоксид вольфрама, вольфрамовая кислота, паравольфрамат аммония, кремневольфрамовая кислота, кремнемолибденовольфрамовая кислота, кременевольфрамованадиевая кислота и вольфрамат W в форме порошка могут использоваться в качестве твердого продукта соединения, содержащего W, сосуществуя друг с другом.
Размер частиц твердого продукта соединения, содержащего W
При контакте твердого продукта соединения, содержащего W, с сухим порошком, и тому подобное, во время кальцинирования сухого порошка, и тому подобное, часть W включается в поверхностную область и/или внутреннее пространство сухого порошка, и тому подобное. Как описано далее, по большей части, это включаемое количество W вносит вклад в рабочие характеристики катализатора. Чем меньше размер частиц твердого продукта соединения, содержащего W, тем больше становится его площадь поверхности. В результате, W легче включается в сухой порошок, и тому подобное. Средний размер частицы d твердого продукта соединения, содержащего W, предпочтительно составляет d<300 мкм, более предпочтительно d<250 мкм, еще более предпочтительно d<200 мкм, особенно предпочтительно d<150 мкм, с точки зрения простоты включения W и смешиваемости.
С другой стороны, слишком маленький твердый продукт соединения, содержащего W, имеет тенденцию к низкой смешиваемости с порошком, кальцинированным на предварительной стадии, или с в основном кальцинированным порошком, к тому, что он будет ответственным за забивание трубы или чего-либо подобного из-за адгезии, или к выносу под действием циркулирующих газов. Средний размер частиц d твердого продукта соединения, содержащего W, предпочтительно составляет 1 мкм<d, более предпочтительно 5 мкм<d, еще более предпочтительно 10 мкм<d, особенно предпочтительно, 20 мкм<d, с точки зрения возможности работы стадии кальцинирования.
В настоящем описании, средний размер частиц твердого продукта соединения, содержащего W, определяют посредством измерения распределения размеров частиц в соответствии с JIS R 1629-1997 "Determination of particle size distributions for fine ceramic raw powders by laser diffraction method" и определения его среднего значения по отношению к объему.
После кальцинирования, катализатор также может отделяться от избытка твердых продуктов соединений, содержащих W, с помощью операции классификации или чего-либо подобного. Когда осуществляют эту операцию, является предпочтительным, чтобы катализатор и твердый продукт соединения, содержащего W, отличались по среднему размеру частиц, с точки зрения простоты разделения. В этом случае, является предпочтительным, чтобы средний размер твердого продукта соединения, содержащего W, был равен или меньше, чем 0,8 от среднего размера частиц катализатора или был равен или больше, чем 1,2 от его среднего размера частиц. Средний размер частиц катализатора предпочтительно составляет 5-200 мкм, более предпочтительно 10-150 мкм, с точки зрения, например, получения предпочтительного состояния текучести при реакции в псевдоожиженном слое, легкого достижения соответствующей степени восстановления катализатора и достижения улучшения рабочих характеристик катализатора.
Средний размер частиц катализатора определяют посредством измерения распределения размеров частицы в соответствии с JIS R 1629-1997 "Determination of particle size distributions for fine ceramic raw powders by laser diffraction method" и определения его среднего значения по отношению к объему, таким же путем, как и для среднего размера частиц твердого продукта соединения, содержащего W.
Твердый продукт соединения, содержащего W, может представлять собой коммерчески доступные порошкообразные исходные материалы или коммерчески доступный продукт, у которого размер частиц регулируется посредством механической операции, такой как измельчение. Твердые продукты, полученные посредством сушки распылением водного раствора метавольфрамата аммония с последующим кальцинированием, являются предпочтительными, поскольку они имеют легко регулируемый размер частиц и малое распределение размеров частиц.
Твердый продукт соединения, содержащего W, может получить возможность для присутствия в этом твердом состоянии в устройстве для кальцинирования во время кальцинирования, например, без импрегнирования раствором и/или суспензией, содержащей W. С помощью этого подхода, компонент W может включаться в катализатор удобнее, чем с помощью способа, который требует импрегнирования, сушки и повторного кальцинирования после временного синтеза катализатора.
Количество W, включаемое в катализатор, изменяется в зависимости от количества твердого продукта соединения, содержащего W, сосуществующего с сухим порошком, с порошком, кальцинированным на предварительной стадии, или с кальцинированным в основном порошком. Количество W, включаемое в него, также изменяется в зависимости от типа и формы (например, размера частиц) твердого продукта соединения, содержащего W, и от температуры и времени кальцинирования. По этой причине, трудно обобщить количество W, сосуществующего с ним. Когда атомное отношение W, содержащегося в твердом продукте соединения, содержащего W, к Mo, содержащемуся в сухом порошке, определяют как RW/Mo, твердый продукт предпочтительно удовлетворяет условиям, представленным следующей далее формулой (4), более предпочтительно, удовлетворяет условиям, представленным следующей далее формулой (4a), еще более предпочтительно, удовлетворяет условиям, представленным следующей далее формулой (4b), и особенно предпочтительно, удовлетворяет условиям, представленным следующей далее формулой (4c), с точки зрения, например, улучшения рабочих характеристик катализатора и из эмпирических соображений:
0,001<RW/Mo<0,6... (4)
0,005<RW/Mo<0,4... (4a)
0,01<RW/Mo<0,3... (4b)
0,015<RW/Mo<0,2... (4c)
((b-1) Стадия кальцинирования сухого порошка в присутствии твердого продукта соединения, содержащего W)
На стадии кальцинирования сухого порошка в присутствии твердого продукта соединения, содержащего W, в устройстве для кальцинирования, сухой порошок и твердый продукт соединения, содержащего W, смешивают заранее, до кальцинирования, а затем эту смесь кальцинируют. В качестве смешивающих устройств можно использовать различные смесители, такие как цилиндрический смеситель, V-образный смеситель, шнековый смеситель и проточный смеситель.
Кроме того, кальцинирование с использованием вращающейся обжиговой печи, обжиговой печи с псевдоожиженным слоем или чего-либо подобного, не обязательно требует предварительного перемешивания компонентов. Каждый компонент из сухого порошка и твердого продукта соединения, содержащего W, может вводиться по отдельности в устройство для кальцинирования и кальцинироваться с перемешиванием в устройстве для кальцинирования. Этот способ устраняет необходимость в устройстве для смешивания сухого порошка и твердого продукта соединения, содержащего W, и таким образом, является более предпочтительным, чем способ, который включает их предварительное перемешивание.
Посредством кальцинирования в состоянии контакта между сухим порошком и твердым продуктом соединения, содержащего W, твердый продукт соединения, содержащего W, включается, в состоянии прилипания к поверхности сухого порошка, в порошок, кальцинированный на предварительной стадии, и/или в кальцинированный в основном порошок, в то время как порошок, кальцинируют. На этой стадии (b-1), кальцинирование может осуществляться таким же путем, как способ кальцинирования (a) для сухого порошка, за исключением того, что смесь сухого порошка и твердого продукта соединения, содержащего W, добавляют в устройство для кальцинирования и твердый продукт соединения, содержащего W, смешивают с ними. Порошок, кальцинированный на предварительной стадии, может быть получен с помощью стадии (b-1) и использоваться на стадии (b-2). Альтернативно, в основном кальцинированный порошок может быть получен с помощью стадии (b-1).
((b-2) Стадия кальцинирования порошка, кальцинированного на предварительной стадии, в присутствии твердого продукта соединения, содержащего W)
На этой стадии, твердый продукт соединения, содержащего W, добавляют и подмешивают в порошок, кальцинированный на предварительной стадии, полученный с помощью кальцинирования на предварительной стадии, и эту смесь затем подвергают воздействию основного кальцинирования.
Является предпочтительным, чтобы стадия добавления и подмешивания твердого продукта соединения, содержащего W, в порошок, кальцинированный на предварительной стадии, осуществлялась в атмосфере инертного газа, с точки зрения, например, простого регулирования окислительно-восстановительного состояния катализатора при соответствующем состоянии и достижения улучшения рабочих характеристик катализатора. В качестве смешивающего устройства можно использовать различные смесители, такие как цилиндрический смеситель, V-образный смеситель, шнековый смеситель и проточный смеситель. Кроме того, для кальцинирования с использованием вращающейся обжиговой печи, обжиговой печи с псевдоожиженным слоем или чего-либо подобного, порошок, кальцинированный на предварительной стадии, и твердый продукт соединения, содержащего W, могут также вводиться по отдельности в устройство для кальцинирования, с целью основного кальцинирования, и кальцинироваться при перемешивании в устройстве для кальцинирования. Этот способ устраняет необходимость в устройстве для смешивания сухого порошка и твердого продукта соединения, содержащего W, и таким образом, является более предпочтительным, чем способ, который включает их предварительное смешивание.
Основное кальцинирование может осуществляться таким же путем, как способ кальцинирования (a) для сухого порошка, за исключением того, что твердый продукт соединения, содержащего W, смешивают с ним. В основном кальцинированный порошок может быть получен с помощью стадии (b-2) и использоваться в качестве оксидного катализатора. Альтернативно, в основном кальцинированный порошок может дополнительно использоваться на стадии (b-3).
((b-3) Способ кальцинирования в основном кальцинированного порошка в присутствии твердого продукта соединения, содержащего W)
В этом способе, твердый продукт соединения, содержащего W, добавляют и подмешивают в кальцинированный в основном порошок, полученный с помощью основного кальцинирования, а затем эту смесь кальцинируют опять. В основном кальцинированный порошок может быть получен таким же путем, как в способе кальцинирования (a) для сухого порошка.
В качестве смешивающего устройства можно использовать различные смесители, такие как цилиндрический смеситель, V-образный смеситель, шнековый смеситель и проточный смеситель. Для кальцинирования с использованием вращающейся обжиговой печи, обжиговой печи с псевдоожиженным слоем или чего-либо подобного, в основном кальцинированный порошок и твердый продукт соединения, содержащего W, могут также вводиться по отдельности в устройство для кальцинирования и кальцинироваться при перемешивании в устройстве для кальцинирования. Этот способ устраняет необходимость в устройстве для перемешивания сухого порошка и твердого продукта соединения, содержащего W, и таким образом является более предпочтительным, чем способ, который включает их предварительное перемешивание.
Смесь в основном кальцинированного порошка и твердого продукта соединения, содержащего W, может также кальцинироваться в присутствии кислорода. Однако является предпочтительным, чтобы смесь кальцинировалась в отсутствие кислорода, с точки зрения, например, простого регулирования окислительно-восстановительного состояния катализатора при соответствующем состоянии и достижения улучшения рабочих характеристик катализатора. Температура кальцинирования предпочтительно составляет от 300 до 700°C, более предпочтительно, от 400 до 600°C, и время кальцинирования предпочтительно составляет от 0,5 до 100 часов, более предпочтительно, от 1 до 50 часов, с точки зрения, например, простой и соответствующей ситуации регулировки удельной площади поверхности катализатора и достижения улучшения рабочих характеристик катализатора. Кальцинирование может представлять собой кальцинирование с использованием вращающейся обжиговой печи или чего-либо подобного. Альтернативно, в основном кальцинированный порошок и твердый продукт соединения, содержащего W, могут смешиваться и нагреваться в реакторе. Этот подход также включается в рамки кальцинирования на этой стадии.
Подобным же образом, твердый продукт соединения, содержащего W, добавляют и подмешивают в порошок катализатора, полученный на стадии удаления ингибитора текучести, описанной далее, а затем эту смесь можно также кальцинировать снова. Кроме того, порошок, полученный на стадии (b-3), можно использовать в качестве оксидного катализатора. Твердый продукт соединения, содержащего W, дополнительно добавляют и подмешивают в него таким же путем, как на стадии (b-3), после стадии удаления ингибитора потока, описанной далее, а затем эту смесь можно также кальцинировать снова.
Среди стадий (b-1), (b-2) и (b-3), стадия (b-1), имеющая меньшее количество процессов, является предпочтительной с точки зрения удобного получения катализатора. Кроме того, стадия (b-2) является предпочтительной с точки зрения улучшения рабочих характеристик катализатора. Однако способ получения оксидного катализатора по настоящему варианту осуществления может включать две или более таких стадий.
W вводят в частицу катализатора с помощью твердофазной диффузии из твердого продукта соединения, содержащего W, сосуществующего с ним на стадии кальцинирования (III). В результате, W концентрируется по поверхности частицы катализатора и вблизи нее, в то время как избыток W, вероятно, уменьшается, когда он подходит ближе к центру частицы. Соответственно, градиент, при котором понижается концентрация W, когда он подходит ближе к центру от поверхности частицы оксидного катализатора, больше, когда стадию кальцинирования осуществляют в присутствии твердого продукта соединения, содержащего W, чем когда частица катализатора содержит W, содержащийся только в сухом порошке. Таким образом, сосуществует ли твердый продукт соединения, содержащего W, с ним на стадии кальцинирования, или нет, можно быстро определить на основе этого градиента.
Разделение катализатора и твердого продукта соединения, содержащего W
В результате кальцинирования сухого порошка, порошка, кальцинированного на предварительной стадии, или в основном кальцинированного порошка вместе с твердым продуктом соединения, содержащего W, часть твердого продукта соединения, содержащего W, включается в порошок, кальцинированный на предварительной стадии, в основном кальцинированный порошок или оксидный катализатор. Однако твердый продукт соединения, содержащего W, не включается полностью в порошок, кальцинированный на предварительной стадии, в основном кальцинированный порошок или катализатор. Таким образом, конечный полученный катализатор находится в состоянии, смешанном с избытком твердого продукта соединения, содержащего W. Избыточные твердые продукты соединения, содержащего W, могут быть отделены от катализатора с использованием устройства для классификации. В качестве устройств для классификации можно использовать различные устройства для классификации, такие как вибрационное сито, воздушный классификатор и устройство, использующее в сочетании воздушную классификацию и разделение на фильтре.
Когда оксидный катализатор используют в реакции в псевдоожиженном слое, воздушная классификация может ускоряться посредством протекания оксидного катализатора в реакторе с псевдоожиженным слоем для удаления твердого продукта соединения, содержащего W. Этот способ устраняет необходимость в устройстве для классификации и, таким образом, является более предпочтительным, чем способ разделения с использованием устройства для классификации. Меньшее количество избытка твердых продуктов соединений, содержащих W, является более предпочтительным с точки зрения текучести катализатора, нагрузок катализатора и загрязнений в трубе или в устройстве. Масса избыточных твердых продуктов соединений, содержащих W, предпочтительно составляет 20% масс. или меньше, более предпочтительно, 10% масс. или меньше, по отношению к массе катализатора. В этом случае, твердый продукт соединения, содержащего W, имеющий размер частиц меньше, чем у оксидного катализатора, легко отделяется с помощью воздушной классификации и, таким образом, является более предпочтительным.
Стадия удаления ингибитора текучести
Оксидный катализатор, полученный таким образом, может содержать ингибитор текучести, выступающий из поверхности частицы. Ингибитор текучести формируют в форме, выступающей и/или выдающейся наружу из поверхности оксидного катализатора. Оксидный катализатор, имеющий такой ингибитор текучести, когда его используют в реакции в псевдоожиженном слое, очень слабо демонстрирует достаточную текучесть и, в дополнение к этому, может приводить к понижению выхода продукта, представляющего интерес, по сравнению с выходом для оксидного катализатора, не содержащего ингибитора текучести. Таким образом, является предпочтительным, чтобы содержание ингибитора текучести было сокращено до 2% масс. или меньше по отношению к массе оксидного катализатора посредством его удаления из катализатора.
Способ удаления ингибитора текучести предпочтительно представляет собой способ, который включает удаление посредством контакта между частицами катализатора, и тому подобное, при циркуляции газа. Более конкретные его примеры включают способ, который включает циркуляцию газа, например, в бункере, в котором хранят катализатор, и способ, который включает циркуляцию газа в реакторе с псевдоожиженным слоем, содержащем оксидный катализатор. Использование реактора с псевдоожиженным слоем устраняет необходимость в специальном устройстве для удаления ингибитора текучести. После того, как газ циркулирует в устройстве, таком как реактор с псевдоожиженным слоем, загруженный оксидным катализатором, частицы оксидного катализатора вступают в контакт друг с другом для удаления ингибитора текучести, выступающего из них. Ингибитор текучести, после диссоциации с оксидным катализатором, становится гораздо меньше, чем сферический оксидный катализатор, и таким образом, высвобождается вместе с циркулирующим газом из системы.
Является предпочтительным, чтобы ингибитор текучести удалялся таким образом, чтобы плотность оксидного катализатора составляла 300-1300 кг/м3, в контейнере, таком как бункер или реактор с псевдоожиженным слоем, содержащий оксидный катализатор, во время удаления ингибитора текучести, с точки зрения, например, эффективного удаления ингибиторов текучести и получения предпочтительного состояния контакта между частицами катализатора. Кроме того, площадь поперечного сечения, ортогонального к направлению газофазного потока в контейнере, предпочтительно составляет 0,1-100 м2, более предпочтительно 0,2-85 м2, с точки зрения, например, получения предпочтительного состояния контакта между частицами катализатора и регулирования удаляемого количества ингибитора текучести при предпочтительном значении. Газ, циркулирующий, чтобы дать возможность для протекания катализатора в контейнере, предпочтительно представляет собой инертный газ (например, азот) или воздух с точки зрения, например, устранения отрицательного воздействия на катализатор. Линейная скорость газа в контейнере предпочтительно составляет 0,03 м/сек - 5 м/сек, более предпочтительно 0,05-1 м/сек, с точки зрения, например, эффективного удаления ингибитора текучести и получения предпочтительного состояния контакта между частицами катализатора. Кроме того, время циркуляции газа предпочтительно составляет 1-50 часов.
Композиция составляющих, добавляемых на стадии приготовления исходных материалов, и композиция сухого порошка
Для композиции каждого соединения составляющего элемента, добавляемого на стадии приготовления исходных материалов, используемого в настоящем варианте осуществления, является предпочтительным, чтобы композиция заготовки исходных материалов удовлетворяла условиям, представленным формулой (1), показанной ниже, с точки зрения приготовления катализатора, имеющего благоприятные рабочие характеристики катализатора и время жизни катализатора. С этой точки зрения, является предпочтительным, чтобы композиция сухого порошка также удовлетворяла условиям, представленным следующей далее формулой (1):
AMo:AV:AW:ANb:AX:AZ=1:a:b:c:x:z... (1)
где AMo представляет собой атомное отношение Mo; AV представляет собой атомное отношение V; AW представляет собой атомное отношение W; ANb представляет собой атомное отношение Nb; AX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; AZ представляет собой атомное отношение по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла и щелочноземельного металла; и a, b, c, x и z попадают в диапазоны 0,01≤a≤1, 0≤b≤1, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно.
Диапазоны a, b, c, x и z предпочтительно представляют собой 0,01≤a≤1, 0≤b≤1, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно, более предпочтительно 0,1≤a≤0,5, 0,005≤b≤0,5, 0,1≤c≤0,5, 0,01≤x≤0,5 и 0,001≤z≤0,5 соответственно, еще более предпочтительно 0,1≤a≤0,45, 0,01≤b≤0,4, 0,1≤c≤0,4, 0,01≤x≤0,4 и 0,001≤z≤0,4 соответственно.
Композиция катализатора
Количество W, включаемое в сухой порошок, в порошок, кальцинированный на предварительной стадии, или в кальцинированный в основном порошок, изменяется в соответствии с типом и формой (например, размером частиц) твердого продукта соединения, содержащего W, температурой и временем кальцинирования, и тому подобное. Для контроля количества W, включаемого в сухой порошок, порошок, кальцинированный на предварительной стадии, или в кальцинированный в основном порошок, является предпочтительным, чтобы количество добавляемого твердого продукта соединения, содержащего W, регулировалось таким образом, чтобы композиция W имела конкретное количество оксидного катализатора, с точки зрения высокоселективного получения соединения, представляющего интерес, с помощью каталитической реакции. Конкретно, является предпочтительным, чтобы оксидный катализатор, из которого удаляют избыток твердых продуктов соединений, содержащих W, содержал каталитический компонент, имеющий композицию, представленную нижеследующей общей формулой (3):
Mo1VaWb+b'NbcXxZzOn... (3)
где a, b, c, x и z, каждый, являются такими, как определено выше в формуле (1); X представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Sb и Te; Z представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла и щелочноземельного металла; b' представляет собой измененную часть композиции W после кальцинирования в присутствии твердого продукта соединения, содержащего W, на стадии кальцинирования, по сравнению с композицией W в заготовке исходных материалов; и n представляет собой значение, которое удовлетворяет условию баланса валентностей и зависит от валентности составляющих элементарных металлов.
Диапазон b' предпочтительно составляет 0,001≤b'≤0,3, более предпочтительно 0,005≤b'≤0,2, еще более предпочтительно 0,01≤b'≤0,15, особенно предпочтительно 0,015≤b'≤0,1.
Посредством кальцинирования в присутствии твердого продукта соединения, содержащего W, получают оксидный катализатор, который демонстрирует более высокую селективность соединения, представляющего интерес, чем при кальцинировании без добавления твердого продукта соединения, содержащего W. С другой стороны, это воздействие не оказывается, даже если композиция W увеличивается на стадии приготовления исходных материалов. Точная причина этого неясна. Однако это предположительно связано с тем, что расположение W в конкретной области вблизи поверхности оксидных кристаллов ингибирует последующую деградацию продукта, представляющего интерес, или промежуточных продуктов при каталитической реакции. При обстоятельствах, когда кристаллы сложного оксида, которые вносят вклад в каталитическую реакцию, сформировались или формируются на стадии кальцинирования, W, как предполагается, компенсирует дефекты решетки на поверхности кристалла посредством постепенной твердофазной диффузии. По этой причине, твердофазная диффузия W, видимо, является важной.
Кроме того, оксидный катализатор, который демонстрирует высокую селективность (выход) соединения, представляющего интерес, может быть получен простым путем при высоких уровнях эффективности с помощью стадии кальцинирования в присутствии твердого продукта соединения, содержащего W. Конкретно, добавление соединения, содержащего W, в уже полученный сухой порошок и/или предшественник катализатора, может улучшить рабочие характеристики полученного катализатора без повторения процесса со стадии приготовления исходных материалов.
Количество W, диффундирующего посредством твердофазной диффузии, зависит от множества факторов, таких как: температура кальцинирования; время кальцинирования; размеры частиц сухого порошка, порошка, кальцинированного на предварительной стадии, в основном кальцинированного порошка и твердого продукта соединения, содержащего W; и от добавляемого количества твердого продукта соединения, содержащего W. Если температура кальцинирования, время кальцинирования или размеры частиц сухого порошка, порошка, кальцинированного на предварительной стадии, и в основном кальцинированного порошка изменяются, при этом изменяются и состояние роста кристаллов сложного оксида или удельная площадь поверхности катализатора. В результате, воздействие, оказываемое посредством добавления твердого продукта соединения, содержащего W, уменьшается. По этой причине, для получения более благоприятных рабочих характеристик катализатора, является предпочтительным контролировать размер частиц твердого продукта соединения, содержащего W, и количество добавляемого твердого продукта соединения, содержащего W.
При таких же условиях кальцинирования, чем больше количество добавляемого твердого продукта соединения, содержащего W, тем большее количество W диффундирует посредством твердофазной диффузии в катализатор. Кроме того, чем меньше средний размер частиц твердого продукта соединения, содержащего W, тем большее количество W диффундирует в катализатор посредством твердофазной диффузии. По этой причине, если атомное отношение W, содержащегося в твердом продукте соединения, содержащего W, к Mo, содержащемуся в сухом порошке, представлено как RW/Mo и средний размер частиц твердого продукта соединения, содержащего W, представлен как d, в качестве показателей количества твердого продукта соединения, содержащего W, добавляемого к катализатору, тогда количество W, диффундирующего посредством твердофазной диффузии, имеет положительную корреляцию с RW/Mo/d. Когда кальцинируют сухой порошок, содержащий каждый элементарный металл, при атомном отношении, представленном формулой (1), твердый продукт предпочтительно удовлетворяет условиям, представленным следующей формулой (2), более предпочтительно, удовлетворяет условиям, представленным следующей формулой (2a), еще более предпочтительно, удовлетворяет условиям, представленным следующей формулой (2b), и особенно предпочтительно, удовлетворяет условиям, представленным следующей формулой (2c), из эмпирических соображений:
3 м-1<RW/Mo/d<600000 м-1... (2)
20 м-1<RW/Mo/d<80000 м-1... (2a)
50 м-1<RW/Mo/d<30000 м-1... (2b)
100 м-1<RW/Mo/d<10000 м-1... (2c)
Оксидный катализатор предпочтительно представляет собой катализатор на носителе из оксида кремния. Когда оксидный катализатор в соответствии с настоящим вариантом осуществления представляет собой катализатор на носителе из оксида кремния, он имеет высокую механическую прочность и, таким образом, является пригодным для использования при реакции окисления и реакции аммоксидирования с использованием реактора с псевдоожиженным слоем. Содержание оксида кремния в качестве носителя предпочтительно составляет 10-80% масс., более предпочтительно 20-70% масс., еще более предпочтительно 30-60% масс., в терминах SiO2 по отношению к общей массе оксидного катализатора на носителе из оксида кремния, содержащего каталитический компонент и оксид кремния в качестве носителя, с точки зрения получения катализатора, имеющего высокую прочность, и придания катализатору достаточной активности.
Далее будет описана газофазная реакция каталитического окисления или газофазная каталитическая реакция аммоксидирования пропана или изобутана с использованием оксидного катализатора по настоящему варианту осуществления.
Пропан, изобутан и исходные материалы для аммиака, подаваемые в случае реакции аммоксидирования, не обязательно должны иметь высокую степень чистоты, и могут использоваться газы промышленных сортов. Например, воздух, чистый кислород или воздух, обогащенный чистым кислородом, можно использовать в качестве источника для подачи кислорода. Кроме того, гелий, неон, аргон, CO2, пары воды, азот или что-либо подобное можно также использовать в качестве газообразного разбавителя.
Газофазная реакция каталитического окисления пропана или изобутана может осуществляться, например, при условиях, показанных ниже.
Молярное отношение кислорода, подаваемого в реакцию пропана или изобутана, предпочтительно составляет 0,1-6, более предпочтительно 0,5-4. Температура реакции предпочтительно равна 300-500°C, более предпочтительно 350°C. Давление реакции предпочтительно составляет 5×104-5×105 Па, более предпочтительно 1×105-3×105 Па. Время контакта предпочтительно составляет 0,1-10 (сек∙г/см3), более предпочтительно 0,5-5 (сек∙г/см3).
В этом контексте, время контакта в настоящем варианте осуществления определяется следующей формулой (8):
Время контакта (сек∙г/см3)=(W/F)×273/(273 + T)... (8),
где W представляет собой количество (г) загружаемого катализатора; F представляет собой скорость потока (Н·см3/сек) газообразной смеси исходных материалов в нормальном состоянии (0°C, 1,013×105 Па) и T представляет собой температуру реакции (°C).
Газофазная каталитическая реакция аммоксидирования пропана или изобутана может осуществляться, например, при условиях, показанных ниже.
Молярное отношение кислорода, подаваемого в реакцию пропана или изобутана, предпочтительно составляет 0,1-6, более предпочтительно 0,5-4. Молярное отношение аммиака, подаваемого в реакцию пропана или изобутана, предпочтительно составляет 0,3-1,5, более предпочтительно 0,7-1,2. Температура реакции предпочтительно составляет 350-500°C, более предпочтительно 380-470°C. Давление реакции предпочтительно составляет 5×104-5×105 Па, более предпочтительно 1×105-3×105 Па. Время контакта предпочтительно составляет 0,1-10 (сек∙г/см3), более предпочтительно 0,5-5 (сек∙г/см3).
Обычный реактор, такой как реактор с неподвижным слоем, реактор с псевдоожиженным слоем или реактор с подвижным слоем, может приниматься в качестве реактора для использования при газофазной реакции каталитического окисления и газофазной реакции каталитического аммоксидирования. Реактор с псевдоожиженным слоем является предпочтительным, поскольку легко удаляется тепло реакции. Кроме того, газофазная каталитическая реакция аммоксидирования может осуществляться прямоточным образом или рециклизационным образом.
Настоящий вариант осуществления может предложить оксидный катализатор, который может использоваться в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана, и способ получения, пригодный для крупномасштабного промышленного производства оксидного катализатора. В соответствии с этим способом получения, может быть легко получен оксидный катализатор, который может использоваться для получения соединения, представляющего интерес, с высокими выходами.
Примеры
Этот вариант осуществления будет описываться более подробно ниже с помощью Примеров и Сравнительных примеров, но этот вариант осуществления не ограничивается этими Примерами.
В Примерах и Сравнительных примерах, преобразование пропана или изобутана и выход акрилонитрила или метакрилонитрила следуют определениям, представленным следующими формулами соответственно:
преобразование пропана или изобутана (%) = (количество молей прореагировавшего пропана или изобутана)/(количество молей подаваемого пропана или изобутана)×100
выход акрилонитрила или метакрилонитрила (%) = (количество молей получаемого акрилонитрила или метакрилонитрила)/(количество молей подаваемого пропана или изобутана)×100
Приготовление жидкой смеси с ниобием
Жидкую смесь с ниобием приготавливают с помощью следующего далее способа. Сначала, 10 кг воды смешивают с 1,530 кг ниобиевой кислоты, содержащей 79,8% масс. Nb2O5 и 5,266 кг дигидрата щавелевой кислоты [H2C2O4∙2H2O]. Исходное молярное отношение щавелевая кислота/ниобий составляет 5,0, и исходная концентрация ниобия составляет 0,50 (моль-Nb/кг-жидкость). Эту жидкость нагревают и перемешивают при 95°C в течение 2 часов для получения жидкой смеси, в которой растворяют ниобий. Этой жидкой смеси позволяют стоять и охлаждаться на льду, а затем твердый продукт отфильтровывают посредством фильтрования с отсосом с получением однородной жидкой смеси с ниобием. Молярное отношение щавелевая кислота/ниобий в этой жидкой смеси с ниобием составляет 2,68, согласно следующему далее анализу.
10 г этой жидкой смеси с ниобием точно взвешивают в тигле, сушат в течение ночи при 95°C, а затем подвергают термической обработке при 600°C в течение 1 часа с получением 0,7895 г Nb2O5. Из этого результата, концентрация ниобия составляет 0,594 (моль-Nb/кг-жидкость). 3 г этой жидкой смеси с ниобием точно взвешивают в 300-мл стеклянном химическом стакане. Добавляют 200 мл горячей воды примерно при 80°C, а затем добавляют 10 мл серной кислоты 1:1. В то время как полученную жидкую смесь поддерживают при температуре жидкости 70°C на горячем столике с мешалкой, жидкую смесь титруют при перемешивании, с использованием 1/4 нормального KMnO4. Точка, в которой светлый бледно-розовый цвет, связанный с KMnO4, поддерживается в течение примерно 30 секунд или больше, принимается в качестве конечной точки. Концентрацию щавелевой кислоты вычисляют из титра KMnO4 с использованием следующей формулы реакции, и она составляет 1,592 (моль-щавелевая кислота/кг):
2KMnO4+3H2SO4+5H2C2O4→K2SO4+2MnSO4+10CO2+8H2O
Полученную жидкую смесь с ниобием используют в качестве жидкой смеси с ниобием (B0) при получении следующего сложного оксида.
Приготовление оксида вольфрама, имеющего средний размер частиц 40 мкм
Водный раствор 50% масс. метавольфрамата аммония приготавливают из коммерческого (торговое наименование «MW-2», производится NIPPON INORGANIC COLOUR & CHEMICAL CO., LTD.) метавольфрамата аммония [(NH4)6H2W12O40], подаваемого в центробежное устройство для сушки распылением, сушат и формируют в микросферической форме. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
100 г этого микросферического метавольфрамата аммония помещают в кристаллизатор и кальцинируют на воздухе при 500°C в течение 2 часов, с использованием неподвижной обжиговой печи, с получением оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, который представляет собой твердый продукт соединения, содержащего W.
Распределение размеров частиц измеряют с помощью устройства для измерения распределения размеров частиц на основе дифракции/рассеяния лазерного света (торговое наименование "LS230", производится Beckman Coulter), и его среднее по объему значение принимается в качестве среднего размера частиц. Дистиллированную воду используют в качестве дисперсионной среды, и вычисление осуществляют при коэффициенте преломления дистиллированной воды, равном 1,33, и коэффициенте преломления образца, равном 1,6.
Пример 1
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
442,7 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 64,1 г метаванадата аммония [NH4VO3] и 91,2 г триоксида дисурьмы [Sb2O3] добавляют к 1,809 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A1).
С другой стороны, 56,5 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 420,6 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C1).
Полученную водную жидкую смесь (A1) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A1), и дополнительно добавляют 105,9 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C1) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа при мощности перемешивания Pv=1,2 кВт/м3 с получением водной жидкой смеси в виде суспензии (D1), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E1)
Затем водную жидкую смесь (D1), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E1), имеющего средний размер частиц 51 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E1)
500 г сухого порошка (E1), полученного, как описано выше, и 13,8 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, смешивают. Трубу для кальцинирования из SUS, имеющую внутренний диаметр 3 дюйма (76 мм), длину 300 мм и толщину стенок 3 мм, заполняют смесью. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. На предварительной стадии кальцинирования температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 42-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 2
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час с получением кальцинированного на предварительной стадии порошка. Кальцинированный на предварительной стадии порошок охлаждают до комнатной температуры. 16,5 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 3
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 588 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем понижая температуру до 350°C при скорости понижения температуры 1°C/мин. Трубу для кальцинирования охлаждают, а затем 35,3 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в трубу для кальцинирования. В потоке газообразного азота при 5,0 нл/мин, температуру повышают от комнатной температуры до 500°C при скорости повышения температуры 4°C/мин, и дополнительно осуществляют кальцинирование при 500°C в течение 2 часов. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 4
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 588 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем понижая температуру до 350°C при скорости понижения температуры 1°C/мин. 50 г катализатора, полученного после охлаждения трубы для кальцинирования, точно взвешивают и заполняют в вертикальную трубу (внутренний диаметр: 41,6 мм, длина: 70 см), в нижней части которой предусматривают перфорированный диск, имеющий три отверстия диаметром 1/64 дюйма (0, 004 см), и в верхней части предусматривают бумажный фильтр, и воздух получает возможность для протекания с нижней стороны при 380 л/час в течение 12 часов. Эту операцию повторяют три раза, и катализатор, извлеченный из трубы, собирают. Линейная скорость основной части в это время составляет 0,05 м/сек. В дополнение к этому, плотность катализатора составляет 1000 кг/м3. Этот катализатор и 35,3 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, удерживаются в трубе для кальцинирования. В потоке газообразного азота при 5,0 нл/мин, температуру повышают от комнатной температуры до 500°C при скорости повышения температуры 4°C/мин, и дополнительно осуществляют кальцинирование при 500°C в течение 2 часов. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 1
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,8% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
424,3 г гептамолибдата аммония ((NH4)6Mo7O24∙4H2O], 61,4 г метаванадата аммония [NH4VO3] и 87,4 г триоксида дисурьмы [Sb2O3] добавляют к 1,732 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A2).
С другой стороны, к 54,1 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 403,1 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C2).
Полученную водную жидкую смесь (A2) охлаждают до 70°C. Затем к водной жидкой смеси (A2) добавляют 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, и дополнительно добавляют 101,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C2), 44,1 г метавольфрамата аммония, содержащего 50% масс. WO3, и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D2), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E2)
Затем водную жидкую смесь (D2), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E2), имеющего средний размер частиц 51 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E2)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E2), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования температуру повышают от 340°C до 670°C при скорости повышения температуры 3°C/мин, сухой порошок (E2) кальцинируют и поддерживают при 670°C в течение 2 часов, затем температуру понижают до 350°C при скорости понижения температуры 1°C/мин, а затем кальцинированный порошок охлаждают с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Получение оксида вольфрама, имеющего средний размер частиц 500 мкм
Коммерческий (торговое наименование "MW-2", производится NIPPON INORGANIC COLOUR & CHEMICAL CO., LTD.) оксид вольфрама в форме частиц просеивают с получением вольфрамата кобальта, имеющего средний размер частиц 500 мкм.
Распределение размеров частиц измеряют с помощью устройства для измерения распределения размеров частиц на основе дифракции/рассеяния лазерного света (торговое наименование "LS230", производится Beckman Coulter), и его среднее по объему значение принимается в качестве среднего размера частиц. Дистиллированную воду используют в качестве дисперсионной среды, и вычисление осуществляют при коэффициенте преломления дистиллированной воды, равном 1,33, и коэффициенте преломления образца, равном 1,6.
Сравнительный пример 2
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,002Nb0,10Sb0,25On/47,8% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час с получением кальцинированного на предварительной стадии порошка. Кальцинированный на предварительной стадии порошок охлаждают до комнатной температуры. 0,13 г оксида вольфрама [WO3], имеющего средний размер частиц 500 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 3°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 250-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Оксид вольфрама, имеющий средний размер частиц 0,5 мкм
Используют коммерческий (торговое наименование "MW-2", производится NIPPON INORGANIC COLOUR & CHEMICAL CO., LTD.) оксид вольфрама, имеющий средний размер частиц 0,5 мкм.
Распределение размеров частиц измеряют с помощью устройства для измерения распределения размеров частиц на основе дифракции/рассеяния лазерного света (торговое наименование "Coulter LS230", производится Beckman), и его среднее по объему значение принимается в качестве среднего размера частиц. Дистиллированную воду используют в качестве дисперсионной среды, и вычисление осуществляют при коэффициенте преломления дистиллированной воды, равном 1,33, и коэффициенте преломления образца, равном 1,6.
Сравнительный пример 3
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,35Nb0,10Sb0,25On/39,6% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 400 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час с получением кальцинированного на предварительной стадии порошка. Порошок, кальцинированный на предварительной стадии, охлаждают до комнатной температуры. 150,4 г оксида вольфрама [WO3], имеющего средний размер частиц 0,5 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 3°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксида, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Приготовление вольфрамата кобальта, имеющего средний размер частиц 15 мкм
Коммерческий (производится Mitsuwa Chemicals Co., Ltd.) кобальт (II) вольфрамат (дигидрат) измельчают в агатовой ступке с получением вольфрамата кобальта, имеющего средний размер частиц 15 мкм.
Распределение размеров частиц измеряют с помощью устройства для измерения распределения размеров частиц на основе дифракции/рассеяния лазерного света (торговое наименование "Coulter LS230", производится Beckman), и его среднее по объему значение принимается в качестве среднего размера частиц. Дистиллированную воду используют в качестве дисперсионной среды, и вычисление осуществляют при коэффициенте преломления дистиллированной воды, равном 1,33, и коэффициенте преломления образца, равном 1,6.
Пример 5
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,03Nb0,10Sb0,25Co0,03On/46,7% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E1) получают как в Примере 1. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E1). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и необязательное кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа с получением кальцинированного на предварительной стадии порошка. Порошок, кальцинированный на предварительной стадии, охлаждают до комнатной температуры. 21,6 г вольфрамата кобальта [CoWO4], имеющего средний размер частиц 15 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 3°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. Оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 6
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,07Nb0,10Sb0,25Ce0,005On/47,1% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
419,5 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 60,8 г метаванадат аммония [NH4VO3], 5,23 г церия нитрата гексагидрата [CeNO3∙6H2O] и 86,4 г триоксида дисурьмы [Sb2O3] добавляют к 1,712 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A3).
С другой стороны, 53,5 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 398,6 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C3).
Полученную водную жидкую смесь (A3) охлаждают до 70°C. Затем 769,0 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A3) и дополнительно добавляют 100,3 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C3), 43,6 г метавольфрамата аммония, содержащего 50% масс. WO3, и дисперсию, в которой 210,7 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,844 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D3), которая представляет собой заготовку исходных материалов
Получение сухого порошка (E3)
Затем водную жидкую смесь (D3), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E3), имеющего средний размер частиц 50 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E3)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E3), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа с получением кальцинированного на предварительной стадии порошка. Кальцинированный на предварительной стадии порошок охлаждают до комнатной температуры. 11,2 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 4
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25Ce0,005On/47,2% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E3) получают как в Примере 6. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E3). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин, с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 7
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,33W0,05Nb0,11Te0,22On, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
39,0 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 8,47 г метаванадата аммония [NH4VO3] и 11,08 г теллуровой кислоты [H6TeO6] добавляют к 196 г воды, смесь нагревают до 60°C при перемешивании для растворения, а затем охлаждают до 30°C с получением водной жидкой смеси (A4).
С другой стороны, 41,3 г воды с перекисью водорода, содержащей 5% масс. H2O2, добавляют к 40,76 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C4).
Водную жидкую смесь (C4) добавляют для получения водной жидкой смеси (A4), и смесь перемешивают в течение 30 минут для получения водной жидкой смеси (D4).
Получение сухого порошка (E4)
Полученную водную жидкую смесь (D4) распыляют на железную пластинку с покрытием из Teflon (зарегистрированное торговое наименование), нагретую до 140°C, с получением сухого порошка (E4), имеющего средний размер частиц 49 мкм.
Кальцинирование сухого порошка (E4)
Кварцевую трубу для кальцинирования, имеющую внутренний диаметр 20 мм, длину 300 мм и толщину стенок 1 мм, заполняют 5 г сухого порошка (E4), полученного, как описано выше, и 0,15 г WO3, регулируемого при среднем размере 15 мкм. В потоке газообразного азота при 1,0 нл/мин, осуществляют основное кальцинирование при условиях 600°C и 2 часа с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубу из SUS с неподвижным слоем, имеющую внутренний диаметр 4 мм, заполняют 0,30 г оксидного катализатора. Температуру реакции устанавливают при 420°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1,2:3:14,8, подают при времени контакта 0,79 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 5
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,33Nb0,11Te0,22On, приготавливают, как описано ниже.
Сухой порошок (E4) получают как в Примере 7. Такую же кварцевую трубу для кальцинирования, как используют в Примере 7, заполняют 5 г сухого порошка (E4). В потоке газообразного азота при 1,0 нл/мин осуществляют основное кальцинирование при условиях 600°C и 2 часа с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 7, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 8
Реакция окисления пропана
Пропан подвергают воздействию газофазной реакции окисления с помощью следующего способа, с использованием оксидного катализатора, полученного в Примере 7. Реакционную трубу из SUS с неподвижным слоем, имеющую внутренний диаметр 4 мм, заполняют 0,35 г оксидного катализатора. Температуру реакции устанавливают при 380°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:кислород:пары воды:гелий = 1:3,1:14,0:10,0, подают при времени контакта 1,2 (сек∙г/см3). Преобразование пропана и выход акриловой кислоты после реакции показаны в Таблице 2.
Сравнительный пример 6
Реакция окисления пропана
Пропан подвергают воздействию газофазной реакции окисления, как в Примере 8, с использованием оксидного катализатора, полученного в Сравнительном примере 5. Преобразование пропана и выход акриловой кислоты после реакции показаны в Таблице 2.
Пример 9
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,07Nb0,10Sb0,25On/47,1% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E2) получают как в Сравнительном примере 1. Смешивают 500 г сухого порошка (E2) и 13,8 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют смесью. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем понижая температуру до 350°C при скорости понижения температуры 1°C/мин, с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 10
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,07Nb0,10Sb0,25Ce0,005On/47,1% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E3), получают как в Примере 6. Смешивают 500 г сухого порошка (E3) и 13,8 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют смесью. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем понижая температуру до 350°C при скорости понижения температуры 1°C/мин, с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 11
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,09Nb0,10Sb0,25Ce0,005On/46,9% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E3) получают как в Примере 6. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 588 г сухого порошка (E3). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем понижая температуру до 350°C при скорости понижения температуры 1°C/мин. Трубу для кальцинирования охлаждают, а затем 15,0 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в трубу для кальцинирования. В потоке газообразного азота при 5,0 нл/мин, температуру повышают от комнатной температуры до 500°C при скорости повышения температуры 4°C/мин, и дополнительно осуществляют кальцинирование при 500°C в течение 2 часов. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 12
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,12Nb0,10Sb0,25Ce0,005On/46,7% масс. SiO2, приготавливают, как описано ниже.
Сухой порошок (E3) получают как в Примере 6. Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E3). В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час с получением кальцинированного на предварительной стадии порошка. Кальцинированный на предварительной стадии порошок охлаждают до комнатной температуры. 20,0 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота со скоростью 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 7
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,042Nb0,10Sb0,25On/47,8% масс. SiO2, приготавливают, как описано ниже.
Оксидный катализатор получают как в Сравнительном примере 2, за исключением того, что сухой порошок (E3) используют вместо сухого порошка (E1).
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 8
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,39Nb0,10Sb0,25On/39,6% масс. SiO2, приготавливают, как описано ниже.
Оксидный катализатор получают как в Сравнительном примере 3, за исключением того, что сухой порошок (E3) используют вместо сухого порошка (E1).
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования, как в Примере 1, с использованием оксидного катализатора, полученного, как описано выше. Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 13
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,30W0,04Nb0,10Sb0,22On/46,8% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
436,8 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 86,2 г метаванадата аммония [NH4VO3] и 79,2 г триоксида дисурьмы [Sb2O3] добавляют к 2,451 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A5).
С другой стороны, 55,7 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 414,9 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C5).
Полученную водную жидкую смесь (A5) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A5), и дополнительно добавляют 91,9 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C5) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D5), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E5)
Затем водную жидкую смесь (D5), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E5), имеющего средний размер частиц 51 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E5)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E5), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час с получением кальцинированного на предварительной стадии порошка. Кальцинированный на предварительной стадии порошок охлаждают до комнатной температуры. 4,4 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 14
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,15W0,04Nb0,10Sb0,30On/46,6% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
440,8 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 43,5 г метаванадата аммония [NH4VO3] и 108,9 г триоксида дисурьмы [Sb2O3] добавляют к 1,212 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A6).
С другой стороны, 56,2 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 418,8 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C6).
Полученную водную жидкую смесь (A6) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A6), и дополнительно добавляют 126,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C6) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D6), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E6)
Затем водную жидкую смесь (D6), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E6), имеющего средний размер частиц 52 мкм. Температура на входе сушилки составляет 210°C, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E6)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E6), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 час. Трубу для кальцинирования охлаждают до комнатной температуры. 13,4 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного кальцинированного в основном порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 15
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,24W0,04Nb0,20Sb0,25On/46,8% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
413,4 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 65,3 г метаванадат аммония [NH4VO3] и 85,1 г триоксида дисурьмы [Sb2O3] добавляют к 1,84 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A7).
С другой стороны, 105,5 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 785,6 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C7).
Полученную водную жидкую смесь (A7) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A7), и дополнительно добавляют 98,9 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C7) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D7), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E7)
Затем водную жидкую смесь (D7), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E7), имеющего средний размер частицы 50 мкм. Температура на входе сушилки составляет 210°C, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E7)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E7), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 12,9 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 16
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/54,9% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
374,5 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 54,2 г метаванадата аммония [NH4VO3] и 77,1 г триоксида дисурьмы [Sb2O3] добавляют к 1,523 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A8).
С другой стороны, 47,8 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 355,7 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C8).
Полученную водную жидкую смесь (A8) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A8), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C8) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D8), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E8)
Затем водную жидкую смесь (D8), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E8), имеющего средний размер частиц 49 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E8)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E8), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 10,4 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 17
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25B0,15On/46,9% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
432,1 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 62,6 г метаванадата аммония [NH4VO3] и 89,0 г триоксида дисурьмы [Sb2O3] добавляют к 1,890 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A9).
С другой стороны, 55,1 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 410,5 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C9).
Полученную водную жидкую смесь (A9) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A9), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C9), дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, и 22,8 г борной кислоты [H3BO3], а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D9), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E9)
Затем водную жидкую смесь (D9), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E9), имеющего средний размер частиц 51 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E9)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E9), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 15,2 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rjgaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 18
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25Mn0,003On/46,8% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
440,2 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 63,7 г метаванадата аммония [NH4VO3], 90,7 г триоксида дисурьмы [Sb2O3] и 6,58 г нитрата церия [Ce(NO3)3∙6H2O] добавляют к 1913 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A10).
С другой стороны, 56,1 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 418,2 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C10).
Полученную водную жидкую смесь (A10) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A10), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C10), дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, и 2,13 г нитрата марганца [Mn(NO3)2∙6H2O], а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D10), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E10)
Затем водную жидкую смесь (D10), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E10), имеющего средний размер частиц 53 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E10)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E10), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 13,2 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 19
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25Ta0,001Ce0,005On/46,9% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
436,5 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 63,2 г метаванадата аммония [NH4VO3], 89,8 г триоксида дисурьмы [Sb2O3] и 5,44 г нитрата церия [Ce(NO3)3∙6H2O] добавляют к 1,896 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A11).
С другой стороны, 55,7 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 414,7 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C11).
Полученную водную жидкую смесь (A11) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A11), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C11), дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, и 6,20 г танталовой кислоты, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D11), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E11)
Затем водную жидкую смесь (D11), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E11), имеющего средний размер частиц 48 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E11)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E11), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 14,9 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, труб currant: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 20
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25Al0,005Ti0,009On/46,8% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
440,7 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 63,8 г метаванадата аммония [NH4VO3] и 90,8 г триоксида дисурьмы [Sb2O3] добавляют к 1,978 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A12).
С другой стороны, 56,2 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 418,7 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C12).
Полученную водную жидкую смесь (A12) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A12), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C12), дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, 0,632 г оксида алюминия [Al2O3] и 1,78 г оксида титана [TiO2], а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D12), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E12)
Затем водную жидкую смесь (D12), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E12), имеющего средний размер частиц 52 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E12)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E12), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 14,7 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 21
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25La0,003On/46,7% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
441,7 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 64,0 г метаванадата аммония [NH4VO3], 91,0 г триоксида дисурьмы [Sb2O3] и 1,86 г нитрата лантана [La(NO3)3∙6H2O] добавляют к 1,933 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A13).
56,3 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 419,6 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C13).
Полученную водную жидкую смесь (A13) охлаждают до 70°C. Затем 762,5 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A13), и дополнительно добавляют 89,5 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C13) и дисперсию, в которой 208,9 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,820 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (E3), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E13)
Затем водную жидкую смесь (E3), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E13), имеющего средний размер частиц 50 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E13)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E13), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 14,0 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита для получения оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 22
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,06Nb0,10Sb0,25Bi0,015On/47,1% масс. SiO2, приготавливают, как описано ниже.
Приготовление заготовок исходных материалов
426,5 г гептамолибдата аммония [(NH4)6Mo7O24∙4H2O], 61,8 г метаванадата аммония [NH4VO3], 87,8 г триоксида дисурьмы [Sb2O3] и 17,6 г нитрата висмута [Bi(NO3)3∙5H2O] добавляют к 1,914 кг воды, и смесь нагревают при 95°C в течение 1 часа при перемешивании с получением водной жидкой смеси (A14).
54,4 г воды с перекисью водорода, содержащей 30% масс. H2O2, добавляют к 405,1 г жидкой смеси с ниобием (B0), и смесь перемешивают и смешивают при комнатной температуре в течение 10 минут с получением водной жидкой смеси (C14).
Полученную водную жидкую смесь (A14) охлаждают до 70°C. Затем 826,4 г золя кремниевой кислоты, содержащего 34,0% масс. SiO2, добавляют к водной жидкой смеси (A14), и дополнительно добавляют 102,0 г воды с перекисью водорода, содержащей 30% масс. H2O2. Перемешивание продолжают при 55°C в течение 30 минут. Затем к жидкости последовательно добавляют водную жидкую смесь (C14) и дисперсию, в которой 187,2 г порошка оксида кремния (торговое наименование "AEROSIL 200", производится Nippon Aerosil Co., Ltd.) диспергируют в 2,62 кг воды, а затем смесь перемешивают при 50°C в течение 2,5 часа с получением водной жидкой смеси в виде суспензии (D14), которая представляет собой заготовку исходных материалов.
Получение сухого порошка (E14)
Затем водную жидкую смесь (D14), полученную, как описано выше, подают в центробежное устройство для сушки распылением и сушат с получением микросферического сухого порошка (E14), имеющего средний размер частиц 52 мкм. Температура на входе сушилки составляет 210°С, а температура на выходе составляет 120°C.
Кальцинирование сухого порошка (E14)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E14), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа. Трубу для кальцинирования охлаждают до комнатной температуры. 13,6 г оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, добавляют в эту трубу для кальцинирования. В потоке азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, и осуществляют основное кальцинирование при условиях поддержания температуры при 670°C в течение 2 часов, а затем понижения температуры до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Оксид вольфрама, имеющий средний размер частиц 0,2 мкм
Оксид вольфрама, полученный способом, сходным с тем, с помощью которого получают оксид вольфрама, имеющий средний размер частиц 40 мкм, измельчают в агатовой ступке с получением оксида вольфрама, имеющего средний размер частиц 0,2 мкм.
Распределение размеров частиц измеряют с помощью устройства для измерения распределения размеров частиц на основе дифракции/рассеяния лазерного света (торговое наименование "Coulter LS230", производится Beckman), и его среднее по объему значение принимается в качестве среднего размера частиц. Дистиллированную воду используют в качестве дисперсионной среды, и вычисление осуществляют при коэффициенте преломления дистиллированной воды, равном 1,33, и коэффициенте преломления образца, равном 1,6.
Сравнительный пример 9
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Сухой порошок (E1) получают как в Примере 1. Оксидный катализатор получают с помощью процедуры, сходной с процедурой Примера 3, за исключением того, что используют оксид вольфрама [WO3], имеющий средний размер частиц 0,2 мкм.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 10
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,0002Nb0,10Sb0,25On/47,8% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Сухой порошок (E1) получают как в Примере 1. 500 г сухого порошка (E1) и 0,03 г оксида вольфрама [WO3], полученного с размером частиц 50 мкм, смешивают. Трубу для кальцинирования из SUS, имеющую диаметр 3 дюйма (7,6 см), заполняют смесью. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу вращают, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 час. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Сравнительный пример 11
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W4,0Nb0,10Sb0,25On/14,2% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Сухой порошок (E1) получают как в Примере 1. Смешивают 500 г сухого порошка (E1) и 2309 г оксида вольфрама [WO3], полученного с размером частиц 15 мкм. Трубу для кальцинирования из SUS, имеющую диаметр 3 дюйма (7,6 мм), заполняют смесью. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу вращают, осуществляют кальцинирование на предварительной стадии и основное кальцинирование. При осуществлении кальцинирования на предварительной стадии, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Затем, для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от 340°C до 670°C при скорости повышения температуры 3°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют от полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 23
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Операцию, сходную с операцией Примера 1, повторяют для получения 80 кг сухого порошка (E1). Используя цилиндрическую трубу для кальцинирования из SUS, имеющую внутренний диаметр 150 мм, длину 1150 мм и толщину стенок 7 мм, в которой шесть отбивных решеток, имеющих высоту 30 мм, устанавливают таким образом, чтобы разделить часть длины нагревательной печи на семь равных частей, и в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют кальцинирование на предварительной стадии. При осуществлении кальцинирования на предварительной стадии, сухому порошку дают возможность для протекания через трубу для кальцинирования при скорости 340 г/час, газообразному азоту дают возможность для протекания через трубу для кальцинирования при 10 Н. литр/мин, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и осуществляют кальцинирование при 340°C в течение 1 часа. Полученный порошок, кальцинированный на предварительной стадии, охлаждают до комнатной температуры. Затем, в то время как 6,6 г/час оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, и 200 г/час кальцинированного на предварительной стадии порошка получают возможность для протекания через другую цилиндрическую трубу для кальцинирования из SUS, имеющую внутренний диаметр 150 мм, длину 1150 мм и толщину стенок 7 мм, и в потоке азота при 6 Н. литр/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, осуществляют основное кальцинирование. Для осуществления основного кальцинирования, кальцинирование осуществляют посредством повышения температуры от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, поддерживая температуру при 670°C в течение 2 часов, и затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют и удаляют из полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 24
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Операцию, сходную с операцией Примера 1, повторяют для получения 80 кг сухого порошка (E1). Кальцинирование на предварительной стадии осуществляют с помощью способа, сходного со способом Примера 22, за исключением того, что используют цилиндрическую трубу для кальцинирования из SUS, имеющую внутренний диаметр 80 мм, длину 1300 мм и толщину стенок 2 мм, в которой семь отбивных решеток, имеющих высоту 15 мм устанавливают таким образом, чтобы разделить часть длины нагревательной печи на восемь равных частей, сухой порошок и газообразный азот получают возможность для протекания через трубу для кальцинирования при скорости 86 г/час и при 2,2 Н. литр/мин соответственно. Получают кальцинированный на предварительной стадии порошок. Затем осуществляют основное кальцинирование, как в Примере 22, за исключением того, что используют другую трубу для кальцинирования из SUS, имеющую внутренний диаметр 80 мм, длину 1300 мм и толщину стенок 2 мм, в которой семь отбивных решеток, имеющих высоту 30 мм, устанавливают таким образом, чтобы разделить часть длины нагревательной печи на восемь равных частей, и в то время как 1,65 г/час оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, и 50 г/час кальцинированного на предварительной стадии порошка получают возможность для протекания через трубу для кальцинирования, газообразный азот получает возможность для протекания при скорости 1,7 Н. литр/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют и удаляют из полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Пример 25
Оксидный катализатор, у которого формула композиции представлена как Mo1V0,22W0,04Nb0,10Sb0,25On/46,7% масс. SiO2, приготавливают, как описано ниже.
Кальцинирование сухого порошка (E1)
Операцию, сходную с операцией Примера 1, повторяют для получения 1000 кг сухого порошка (E1). Кальцинирование на предварительной стадии осуществляют способом, сходным со способом Примера 22, за исключением того, что используют цилиндрическую трубу для кальцинирования из SUS, имеющую внутренний диаметр 300 мм, длину 800 мм и толщину стенок 7 мм, в которой четыре отбивных решетки, имеющих высоту 70 мм, устанавливают таким образом, чтобы разделить часть длины нагревательной печи на пять равных частей, сухой порошок и газообразный азот получают возможность для протекания через трубу для кальцинирования при скорости 1,2 кг/час и при 35 Н. литр/мин соответственно. Получают порошок, кальцинированный на предварительной стадии. Затем осуществляют основное кальцинирование, как в Примере 22, за исключением того, что используют другую трубу для кальцинирования из SUS, имеющую внутренний диаметр 300 мм, длину 800 мм и толщину стенок 7 мм, в которой семь отбивных решеток, имеющих высоту 70 мм, устанавливают таким образом, чтобы разделить часть длины нагревательной печи на восемь равных частей, и в то время как 23,1 г/час оксида вольфрама [WO3], имеющего средний размер частиц 40 мкм, и 0,7 кг/час кальцинированного на предварительной стадии порошка получают возможность для протекания через трубу для кальцинирования, газообразный азот получает возможность для протекания при скорости 23 Н. литр/мин. После охлаждения, оксид вольфрама, имеющий малый размер частиц, отделяют и удаляют из полученного в основном кальцинированного порошка с помощью 32-мкм сита с получением оксидного катализатора.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 1.
Реакция аммоксидирования пропана
Пропан подвергают воздействию газофазной реакции аммоксидирования с помощью следующего способа, с использованием оксидного катализатора, полученного, как описано выше. Реакционную трубку из стекла Vycor с псевдоожиженным слоем, имеющую внутренний диаметр 25 мм, заполняют 35 г оксидного катализатора. Температуру реакции устанавливают при 440°C, и давление реакции устанавливают при обычном давлении. Газовую смесь, имеющую молярное отношение пропан:аммиак:кислород:гелий = 1:1:3:18, подают при времени контакта 2,8 (сек∙г/см3). Преобразование пропана и выход акрилонитрила после реакции показаны в Таблице 1.
Анализ профиля распределения поперечных сечений частиц катализатора
Анализ профиля распределения посредством измерения с помощью SEM-EDX осуществляют на полученном оксидном катализаторе с использованием SEM-6060A, производимого JEOL Ltd. Оксидный катализатор погружают в ненасыщенную полиэстровую смолу (смола для холодного погружения № 105, производимая Marumoto Struers K.K.), в которую добавляют отверждающий агент (агент M, производимый той же компанией), и все вместе полируют до тех пор, пока не будут экспонированы поперечные сечения погруженных в смолу частиц катализатора. Полировку осуществляют с помощью полировальной машины (Musashino Denshi MA-150), с использованием наждачной бумаги № 400, наждачной бумаги № 2000 и оберточного листа, по очереди, и наконец, ее полируют с помощью соответствующего количества полирующего агента (полировочная суспензия AP-A на основе оксида алюминия, производимая той же компанией) и воды, помещаемых в полировочную машину. Затем положение образца регулируют таким образом, что поперечные сечения экспонируемых частиц катализатора находятся в наблюдаемом поле зрения при измерении с помощью SEM-EDX. В качестве условий измерения, используют получение изображения с помощью обратного рассеяния электронов и устанавливают напряжение ускорения: 0-15 кВ, время задержки: 1,0 мсек, количество сканирований: 5000, размер пятна: 50 и рабочее расстояние: 10 мм. Полупроводник Si (Li) используют в качестве детектора.
Для вычисления Sw, 10 или более частиц, экспонируемых до сечения в центре частиц или вблизи него и имеющих размер частицы от 40 до 70 мкм, подвергают измерению с помощью анализа профиля распределения, и среднее значение Sw для частиц берут для вычислений.
Пример 26
Для катализатора, полученного в Примере 10, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Пример 27
Для катализатора, полученного в Примере 11, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Пример 28
Для катализатора, полученного в Примере 12, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Пример 29
Для катализатора, полученного в Примере 1, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Пример 30
Для катализатора, полученного в Примере 3, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Пример 31
Для катализатора, полученного в Примере 4, осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Ссылочный пример 1
Способ получения оксидного катализатора
Сухой порошок (E3) получают как в Примере 6.
Кальцинирование сухого порошка (E3)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E3), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа, а затем осуществляют основное кальцинирование посредством повышения температуры от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин. После охлаждения, 100 г полученного оксидного катализатора импрегнируют жидкостью, в которой 3,4 г водного раствора метавольфрамата аммония (содержащего вольфрам (W)), соответствующие 50% масс. в терминах WO3), разбавляют водой до 20 мл, сушат на воздухе при комнатной температуре в течение примерно 1 дня, а затем сушат в сушилке с горячим воздухом при 150°C в течение 1 часа. Для полученного катализатора осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 3.
Ссылочный пример 2
Способ получения оксидного катализатора
Сухой порошок (E1) получают как в Примере 1.
Кальцинирование сухого порошка (E1)
Такую же трубу для кальцинирования из SUS, как используют в Примере 1, заполняют 500 г сухого порошка (E1), полученного, как описано выше. В потоке газообразного азота при 5,0 нл/мин, в то время как трубу для кальцинирования вращают вокруг ее продольной оси, температуру повышают от комнатной температуры до 340°C при скорости повышения температуры 0,75°C/мин, и кальцинирование на предварительной стадии осуществляют при условиях 340°C и 1 часа, а затем осуществляют основное кальцинирование посредством повышения температуры от комнатной температуры до 670°C при скорости повышения температуры 5°C/мин, поддерживая температуру при 670°C в течение 2 часов, а затем, понижая температуру до 350°C при скорости понижения температуры 1°C/мин.
После охлаждения, 100 г полученного оксидного катализатора импрегнируют жидкостью, в которой 4,5 г водного раствора метавольфрамата аммония (содержащего вольфрам (W), соответствующий 50% масс. в терминах WO3), разбавляют водой до 20 мл, сушат на воздухе при комнатной температуре в течение примерно 1 дня, а затем сушат в сушилке с горячим воздухом при 150°C в течение 1 часа. Для полученного катализатора осуществляют анализ профиля распределения, и Sw вычисляют, как описано выше. Результат показан в Таблице 3.
Композицию катализатора измеряют посредством анализа с помощью устройства на основе рентгеновской флуоресценции (торговое наименование "RINT1000", производится Rigaku, Cr трубка, напряжение трубки: 50 кВ, ток трубки: 50 мА).
RW/Mo/d показано в Таблице 3.


[мкм]

Эта заявка основывается на заявке на патент Японии № 2010-248669, поданной 5 ноября 2010 г., содержание которой включается в настоящий документ в качестве ссылки.
Промышленное применение
Настоящее изобретение может предложить оксидный катализатор, который можно использовать для газофазной каталитической реакции окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана, и способ получения, пригодный для промышленного получения оксидного катализатора в большом количестве. Следовательно, настоящее изобретение имеет промышленное применение в его областях.
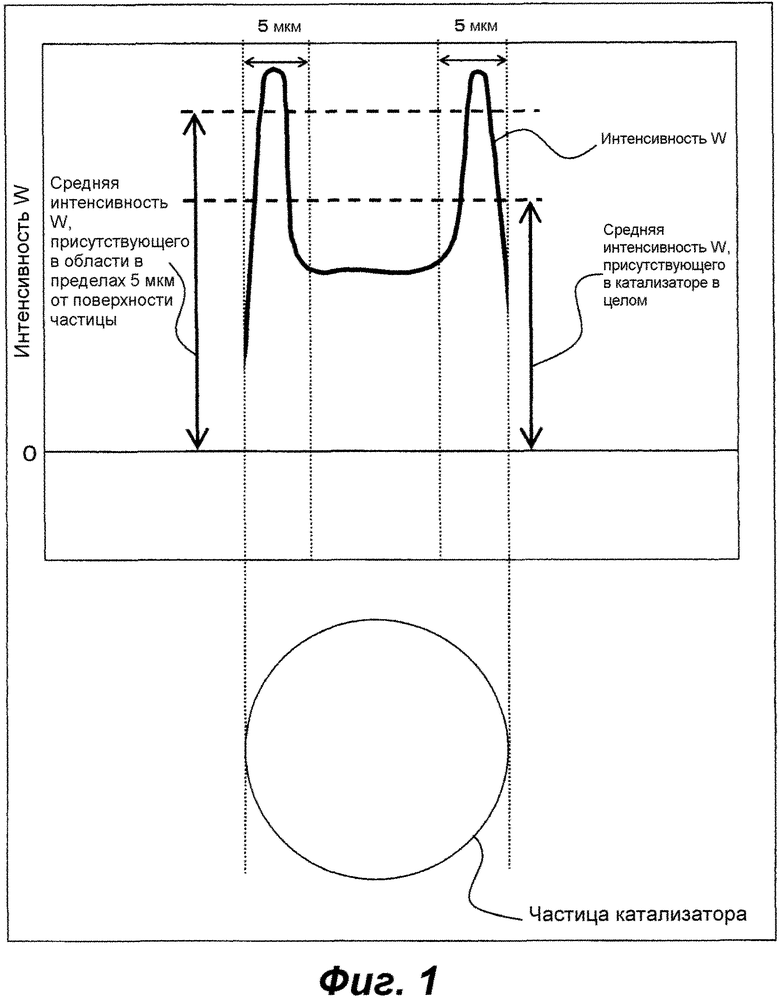
Настоящее изобретение относится к оксидному катализатору в форме частиц для использования в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана. Заявленный катализатор содержит соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, соединения W и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных следующей далее формулой (0), где средняя интенсивность для W, присутствующего в области в пределах 5 мкм от поверхности в направлении центра частицы оксидного катализатора, равна или больше чем в 1,08 раз величины интенсивности для W, присутствующего в оксидном катализаторе в целом, и средняя интенсивность для W, присутствующего в области от части, соответствующей 5 мкм, от поверхности в направлении центра частицы оксидного катализатора, равна или больше чем в 0,3, и менее чем в 1 раз величины интенсивности для W, присутствующего в оксидном катализаторе в целом:
CMo:CV:CW:CNb:CX:CZ=1:a:w:c:x:z ..., (0)
где CMo представляет собой атомное отношение Mo; CV представляет собой атомное отношение V; CW представляет собой атомное отношение W; CNb представляет собой атомное отношение Nb; CX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; CZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc; и a, w, c, x и z попадают в диапазоны 0,01≤a≤1, 0<w≤2, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно. Кроме того, изобретение относится к способу получения оксидного катализатора, а также к способам получения ненасыщенной кислоты и ненасыщенного нитрила с использованием оксидного катализатора. Применение заявленного оксидного катализатора позволяет получить соответствующую ненасыщенную кислоту и соответствующий ненасыщенный нитрил с высоким выходом. 4 н. и 16 з.п. ф-лы, 1 ил., 3 табл., 31 пр.
1. Оксидный катализатор в форме частиц для использования в газофазной реакции каталитического окисления или газофазной реакции каталитического аммоксидирования пропана или изобутана,
содержащий соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, соединения W и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных следующей далее формулой (0), где
средняя интенсивность для W, присутствующего в области в пределах 5 мкм от поверхности в направлении центра частицы оксидного катализатора, равна или больше чем в 1,08 раз величины интенсивности для W, присутствующего в оксидном катализаторе в целом, и средняя интенсивность для W, присутствующего в области от части, соответствующей 5 мкм, от поверхности в направлении центра частицы оксидного катализатора, равна или больше чем в 0,3, и менее чем в 1 раз величины интенсивности для W, присутствующего в оксидном катализаторе в целом:
CMo:CV:CW:CNb:CX:CZ=1:a:w:c:x:z ..., (0)
где CMo представляет собой атомное отношение Mo; CV представляет собой атомное отношение V; CW представляет собой атомное отношение W; CNb представляет собой атомное отношение Nb; CX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; CZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc; и a, w, c, x и z попадают в диапазоны 0,01≤a≤1, 0<w≤2, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1, соответственно.
2. Оксидный катализатор по п.1, где в формуле (0):
CMo:CV:CW:CNb:CX:CZ=1:a:w:c:x:z ..., (0)
где CX представляет собой атомное отношение Sb.
3. Способ получения оксидного катализатора по п.1 или 2 для использования в газофазной реакции каталитического окисления или газофазной реакции аммоксидирования пропана или изобутана, включающий стадии:
(I) получения заготовки исходных материалов, содержащей соединение Mo, соединение V, соединение Nb, соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te, необязательное соединение W и необязательное соединение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc, при атомных отношениях, представленных следующей далее формулой (1);
(II) сушку заготовки исходных материалов для получения сухого порошка; и
(III) кальцинирования сухого порошка, где
стадия кальцинирования (III) включает стадию кальцинирования сухого порошка в присутствии соединения, содержащего W, в форме твердого продукта с получением кальцинированного на предварительной стадии порошка или в основном кальцинированного порошка, или стадию кальцинирования кальцинированного на предварительной стадии порошка, полученного посредством кальцинирования сухого порошка в присутствии соединения, содержащего W, в форме твердого продукта с получением в основном кальцинированного порошка, и необязательно включает стадию дополнительного кальцинирования в основном кальцинированного порошка в присутствии соединения, содержащего W в форме твердого продукта,
твердый продукт удовлетворяет условиям, представленным нижеследующей формулой (2), и оксидный катализатор содержит каталитический компонент, имеющий композицию, представленную нижеследующей общей формулой (3):
AMo:AV:AW:ANb:AX:AZ = 1:a:b:c:x:z ..., (1)
где AMo представляет собой атомное отношение Mo; AV представляет собой атомное отношение V; AW представляет собой атомное отношение W; ANb представляет собой атомное отношение Nb; AX представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Sb и Te; AZ представляет собой атомное отношение, по меньшей мере, одного элемента, выбранного из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc; и a, b, c, x и z попадают в диапазоны 0,01≤a≤1, 0≤b≤1, 0,01≤c≤1, 0,01≤x≤1 и 0≤z≤1 соответственно;
3 м-1<RW/Mo/d<600000 м-1 ..., (2)
где RW/Mo представляет собой атомное отношение W, содержащегося в твердом продукте к Mo, содержащемуся в сухом порошке; и d представляет собой средний размер частиц твердого продукта; и
Mo1VaWb+b'NbcXxZzOn ..., (3)
где a, b, c, x и z, каждый, являются такими, как определено выше в формуле (1); X представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Sb и Te; Z представляет собой, по меньшей мере, один элемент, выбранный из группы, состоящей из Mn, B, Ti, Al, Ta, щелочного металла, щелочноземельного металла, La, Ce, Pr, Yb, Co, Y и Sc; b' попадает в диапазон 0,001≤b'≤0,3 и n представляет собой значение, которое удовлетворяет условию баланса валентностей.
4. Способ получения оксидного катализатора по п.3, где твердый продукт удовлетворяет условиям, представленным следующими далее формулами (4) и (5):
0,001<RW/Mo<0,6 ... (4)
1 мкм<d<300 мкм ..., (5)
где RW/Mo и d, каждый, являются такими, как определено выше в формуле (2).
5. Способ получения оксидного катализатора по п.3, где в формуле (1) 0<b≤1.
6. Способ получения оксидного катализатора по п.3, где соединение Mo, соединение V, соединение W, соединение Nb, соединение, представленное X, и соединение, представленное Z, в сухом порошке, кальцинированном на предварительной стадии порошке или в порошке, в основном кальцинированном, представляют собой, каждое, по меньшей мере, одно соединение, выбранное из группы, состоящей из соли неорганической кислоты, соли органической кислоты, оксида и сложного оксида.
7. Способ получения оксидного катализатора по п.3, дополнительно включающий стадию сушки распылением раствора или суспензии, содержащей соединение W, с получением твердого продукта.
8. Способ получения оксидного катализатора по п.3, где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве 10-80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
9. Способ получения оксидного катализатора по п.4, где в формуле (1) 0<b≤1.
10. Способ получения оксидного катализатора по п.4, где соединение Mo, соединение V, соединение W, соединение Nb, соединение, представленное X, и соединение, представленное Z в сухом порошке, порошке, кальцинированном на предварительной стадии, или в кальцинированном в основном порошке, каждое, представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из соли неорганической кислоты, соли органической кислоты, оксида и сложного оксида.
11. Способ получения оксидного катализатора по п.5, где соединение Mo, соединение V, соединение W, соединение Nb, соединение, представленное X, и соединение, представленное Z в сухом порошке, порошке, кальцинированном на предварительной стадии, или в кальцинированном в основном порошке, каждое, представляет собой, по меньшей мере, одно соединение, выбранное из группы, состоящей из соли неорганической кислоты, соли органической кислоты, оксида и сложного оксида.
12. Способ получения оксидного катализатора по п.4, дополнительно включающий стадию сушки распылением раствора или суспензии, содержащих соединение W, с получением твердого продукта.
13. Способ получения оксидного катализатора по п.5, дополнительно включающий стадию сушки распылением раствора или суспензии, содержащих соединение W, с получением твердого продукта.
14. Способ получения оксидного катализатора по п.6, дополнительно включающий стадию сушки распылением раствора или суспензии, содержащих соединение W, с получением твердого продукта.
15. Способ получения оксидного катализатора по п.4, где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве от 10 до 80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
16. Способ получения оксидного катализатора по п.5, где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве от 10 до 80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
17. Способ получения оксидного катализатора по п.6, где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве от 10 до 80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
18. Способ получения оксидного катализатора по п.7, где оксидный катализатор содержит каталитический компонент на носителе из оксида кремния в количестве от 10 до 80% масс. в терминах SiO2 по отношению к общему количеству каталитического компонента и оксида кремния.
19. Способ получения соответствующей ненасыщенной кислоты из пропана или изобутана посредством газофазной реакции каталитического окисления, включающий использование оксидного катализатора по п.1 или 2, полученного с помощью способа по любому из пп.3-8 и 9-18.
20. Способ получения соответствующего ненасыщенного нитрила из пропана или изобутана посредством газофазной реакции каталитического аммоксидирования, включающий использование оксидного катализатора по п.1 или 2, полученного с помощью способа получения по любому из пп.3-8 и 9-18.
| WO 2009151254A2, 17.12.2009 | |||
| US 20030236163A1, 25.12.2003 | |||
| СПОСОБ ПОЛУЧЕНИЯ АКРОЛЕИНА И/ИЛИ АКРИЛОВОЙ КИСЛОТЫ | 2001 |
|
RU2285690C2 |
| US 7087551B2, 08.08.2006 | |||
| WO 2009048553A2, 16.04.2009 | |||
| JP 2005305428A, 04.11.2005 | |||
| JP 2008221032A, 25.09.2008 | |||
| JP 2009066463A, 02.04.2009 | |||
| JP 2007320847A, 13.12.2007 | |||
| US 20080214863A1, 04.09.2008 | |||

