УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ДАННОМУ ИЗОБРЕТЕНИЮ
Область техники, к которой относится изобретение
Данное изобретение относится к композитному оксидному катализатору, применяемому для реакции каталитического окисления в паровой фазе или каталитического аммоксидирования в паровой фазе пропана или изобутана, и способу получения композитного оксидного катализатора, и способу получения ненасыщенной кислоты или ненасыщенного нитрила с применением композитного оксидного катализатора.
Описание предшествующего уровня техники
Известен обычный способ подвергания пропилена или бутилена реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе, чтобы получить соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил. В последние годы внимание было направлено на способ подвергания пропана или изобутана вместо пропилена или бутилена каталитическому окислению в паровой фазе или каталитическому аммоксидированию в паровой фазе, чтобы получить соответствующую ненасыщенную карбоновую кислоту или ненасыщенный нитрил.
По этой причине в качестве катализатора каталитического аммоксидирования в паровой фазе пропана или изобутана было предложено множество оксидных катализаторов.
Катализатор аммоксидирования является обычно оксидом металла, полученным посредством смешивания, сушки и обжига молибдена, ванадия, сурьмы, ниобия и т.п., при необходимости. Состав металлов, содержащихся в оксиде металла, исследовали при различных соотношениях компонентов, поскольку соотношение компонентов непосредственным образом влияет на функциональные возможности катализатора. Кроме того, в последнее время было найдено, что физические свойства оксида металла, которые не могут быть представлены лишь соотношением компонентов, также влияют на функциональные возможности катализатора.
Например, Патентный документ 1 описывает оксид металла, содержащий молибден, ванадий, сурьму и ниобий и имеющий степень восстановления от 8 до 12% и удельную поверхность от 5 до 30 м2/г.
Патентный документ 2 описывает образование выступающего вещества, которое сдерживает текучесть, на поверхности катализатора аммоксидирования. В соответствии с Патентным документом 2, за счет удаления этого вещества с поверхности катализатора может поддерживаться выход целевого продукта, и могут быть получены ненасыщенная кислота и ненасыщенный нитрил.
Патентный документ 1: Международная заявка WO 2004-108278
Патентный документ 2: Выложенная заявка на патент Японии № 2007-216212
Проблемы, подлежащие разрешению посредством данного изобретения
Как описано в Патентных документах 1 и 2, если степень восстановления и удельная поверхность оксида металла оптимизируют посредством выбора условий получения катализатора, то, конечно, функциональные возможности катализатора улучшаются. Кроме того, если выступающее вещество (далее в данном документе называемое «выступом»), образованное на поверхности катализатора, удаляют после обжига, то легко поддерживается выход целевого продукта.
В соответствии с исследованиями, проведенными авторами данного изобретения, однако, состав выступа отличается от состава центральной части катализатора. По этой причине, если выступ удаляют, то состав полученного катализатора отличается от состава во время приготовления. А именно, было найдено, что, что даже если состав во время приготовления оптимизируют, и степень восстановления и удельную поверхность определяют на основании состава во время приготовления, часть состава во время приготовления удаляют в виде выступа после приготовления; по этой причине состав после приготовления отклоняется от оптимальной величины.
Принимая во внимание вышеуказанное обстоятельство, целью данного изобретения является предложение комплексного оксидного катализатора, применяемого для реакции каталитического окисления в паровой фазе или каталитического аммоксидирования в паровой фазе пропана или изобутана, в котором металлы, которые образуют комплексный оксидный катализатор, имеют надлежащее соотношение компонентов после стадии удаления выступа, который имеется на поверхности частицы, и способа получения комплексного оксидного катализатора.
Средство для разрешения проблем
В результате интенсивных исследований, проведенных, чтобы разрешить вышеуказанные проблемы, авторы данного изобретения обнаружили, что на состав комплексного оксида стадия удаления выступа особенно сильно влияет через соотношение компонентов ванадия и сурьмы V/Sb и соотношение компонентов ванадия и ниобия V/Nb. Также было найдено, что, если состав во время приготовления оптимизируют на стадии приготовления исходного материала, то получают катализатор, включающий комплексный оксид, имеющий надлежащее соотношение металлических компонентов. Соответственно, может быть достигнута цель данного изобретения.
А именно, данное изобретение состоит в следующем:
[1] Комплексный оксидный катализатор, применяемый для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, где данный катализатор содержит комплексный оксид, представленный приведенной ниже формулой (1):
Mo1VaSbbNbcWdZeOn... (1)
(в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,4, 0,1≤b≤0,4, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3).
[2] Комплексный оксидный катализатор в соответствии с пунктом [1] выше, включающий от 20 до 70 масс.% кремнезема в расчете на SiO2.
[3] Способ получения комплексного оксидного катализатора, содержащего комплексный оксид, представленный приведенной ниже формулой (1):
Mo1VaSbbNbcWdZeOn... (1)
(в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,4, 0,1≤b≤0,4, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3), где данный способ включает следующие стадии от (I) до (V):
(I) приготовление раствора, содержащего исходный материал, содержащий Mo, V, Sb, Nb, W и Z, в котором атомное отношение a V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение c Nb к одному атому Mo составляет 0,01≤c≤0,5, атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение e Z к одному атому Mo составляет 0≤e≤0,2;
(II) сушка раствора, содержащего исходный материал, чтобы получить сухой порошок;
(III) предварительный обжиг сухого порошка, чтобы получить предварительно обожженный продукт;
(IV) основной обжиг предварительно обожженного продукта, чтобы получить обожженный продукт, имеющий выступ на поверхности частиц; и
(V) удаление выступа, имеющегося на поверхности частиц обожженного продукта, посредством воздушного потока,
где степень восстановления предварительно обожженного продукта составляет 8 до 12%, и удельная поверхность обожженного продукта составляет от 7 до 20 м2/г.
[4] Способ получения комплексного оксидного катализатора в соответствии с пунктом [3] выше, в котором содержание частиц, имеющих размер не более чем 25 мкм, в сухом порошке составляет не более чем 20 масс.%, и средний размер частиц сухого порошка составляет от 35 до 75 мкм.
[5] Способ получения комплексного оксидного катализатора в соответствии с пунктом [3] или [4] выше, в котором на стадии (V) выступ удаляют таким образом, что количество выступов, которое имеет обожженный продукт, составляет не более чем 2 масс.%, в расчете на общую массу обожженного продукта.
[6] Способ получения комплексного оксидного катализатора в соответствии с любым из пунктов с [3] по [5] выше, в котором длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с в пересчете на линейную скорость при 15°C и атмосферном давлении.
[7] Способ получения комплексного оксидного катализатора в соответствии с любым из пунктов с [3] по [6] выше, в котором стадия (I) включает следующие стадии с (a) по (d):
(a) приготовление водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) добавление золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) смешивание водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) добавление суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживание раствора.
[8] Способ получения комплексного оксидного катализатора в соответствии с любым из пунктов с [3] по [7] выше, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре ниже, чем температура обжига при основном обжиге.
[9] Способ получения ненасыщенной кислоты с применением комплексного оксидного катализатора в соответствии с пунктом [1] или [2] выше, в котором пропан или изобутан подвергают реакции каталитического окисления в паровой фазе, чтобы получить соответствующую ненасыщенную кислоту.
[10] Способ получения ненасыщенного нитрила с применением комплексного оксидного катализатора в соответствии с пунктом [1] или [2] выше, в котором пропан или изобутан подвергают реакции каталитического аммоксидирования в паровой фазе, чтобы получить соответствующий ненасыщенный нитрил.
Преимущества изобретения
В соответствии с данным изобретением, может быть предложен комплексный оксидный катализатор, имеющий оптимизированное соотношение металлических компонентов для металлов, которые образуют комплексный оксидный катализатор. Комплексный оксидный катализатор в соответствии с данным изобретением проявляет высокие каталитические свойства вследствие оптимизации соотношения металлических компонентов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой чертеж, схематически показывающий один из примеров устройства для удаления выступов в соответствии с представляемым вариантом осуществления;
Фиг. 2 представляет собой чертеж, показывающий поперечное сечение X-X устройства для удаления выступов на Фиг. 1;
Фиг. 3 представляет собой чертеж, показывающий один из примеров ответвлений от трубы в устройстве для удаления выступов в соответствии с представляемым вариантом осуществления;
Фиг. 4 представляет собой чертеж, схематически показывающий один из примеров устройства для удаления выступов в соответствии с представляемым вариантом осуществления;
Фиг. 5 представляет собой чертеж, схематически показывающий один из примеров устройства для удаления выступов в соответствии с представляемым вариантом осуществления;
Фиг. 6 представляет собой чертеж, схематически показывающий один из примеров устройства для удаления выступов в соответствии с представляемым вариантом осуществления; и
Фиг. 7 представляет собой чертеж, схематически показывающий один из примеров устройства для удаления выступов в соответствии с представляемым вариантом осуществления.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
Далее в данном документе будет подробно описан вариант осуществления данного изобретения (далее в данном документе называемый как «представляемый вариант осуществления»). Следует заметить, что данное изобретение не ограничивается приведенным ниже вариантом осуществления, и множество изменений может быть сделано в пределах объема данного изобретения.
На чертежах одни и те же цифровые обозначения будут относиться к одним и тем же элементам, и повторение описания будет опущено. Кроме того, вертикальные и горизонтальные относительные положения основаны на относительных положениях, показанных на чертежах, если не указано иное. Соотношения размеров в устройствах и элементах не ограничиваются соотношениями, показанными на чертежах.
Комплексный оксидный катализатор в соответствии с представляемым вариантом осуществления является комплексным оксидным катализатором, применяемым для реакции каталитического окисления в паровой фазе или реакция каталитического аммоксидирования в паровой фазе пропана или изобутана, причем данный катализатор включает комплексный оксид, представленный приведенной ниже формулой (1):
Mo1VaSbbNbcWdZeOn... (1)
(в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,4, 0,1≤b≤0,4, и 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3).
Комплексный оксидный катализатор в соответствии с представляемым вариантом осуществления проявляет высокие каталитические свойства вследствие оптимизации соотношения металлических компонентов. Способ получения комплексного оксидного катализатора в соответствии с представляемым вариантом осуществления не ограничивается особым образом. Предпочтительно, комплексный оксидный катализатор в соответствии с представляемым вариантом осуществления получают способом, включающим следующие стадии с (I) по (V):
(I) стадию приготовления раствора, содержащего исходный материал, содержащий Mo, V, Sb, Nb, W и Z, в котором атомное отношение a V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение c Nb к одному атому Mo составляет 0,01≤c≤0,5, и атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение e Z к одному атому Mo составляет 0≤e≤0,2;
(II) стадию сушки раствора с составом исходного материала, чтобы получить сухой порошок;
(III) стадию предварительного обжига сухого порошка, чтобы получить предварительно обожженный продукт;
(IV) стадию основного обжига предварительно обожженного продукта, чтобы получить обожженный продукт, имеющий выступ на поверхности частиц; и
(V) стадию удаления выступа, имеющегося на поверхности частиц обожженного продукта, посредством воздушного потока, на которой степень восстановления предварительно обожженного продукта составляет от 8 до 12%, и удельная поверхность обожженного продукта составляет от 7 до 20 м2/г.
(Стадия (I) составления смеси исходных материалов)
Стадия (I) представляет собой стадию приготовления раствора с составом исходного материала, содержащего Mo, V, Sb, Nb, W и Z, в котором атомное отношение a V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение c Nb составляет 0,01≤c≤0,5, и атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение e Z к одному атому Mo составляет 0≤e≤0,2. В данном документе, «составление» и «приготовление» являются синонимами.
На стадии составления смеси исходных материалов элементы, которые образуют комплексный оксидный катализатор, растворяют или диспергируют в растворителе и/или дисперсионной среде в заданном соотношении, чтобы получить раствор с составом исходного материала. В качестве растворителя для раствора с составом исходного материала предпочтительной является водная среда. Обычно может быть использована вода. Раствор с составом исходного материала содержит Mo, V, Sb, Nb, W и Z (Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba). В качестве исходного материала для раствора с составом исходного материала может быть использована соль или соединение, включающие элементы, которые образуют комплексный оксидный катализатор.
На стадии составления смеси исходных материалов раствор с составом исходного материала приготавливают таким образом, что атомное отношение a V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение c Nb к одному атому Mo составляет 0,01≤c≤0,5, атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение e Z к одному атому Mo составляет 0≤e≤0,2. Соотношение компонентов устанавливают при величинах, отличающихся от величин соотношения компонентов в комплексном оксидном катализаторе, получаемом в качестве конечного продукта. Это обусловлено тем, что выступ в катализаторе, описанный далее, имеет состав, отличающийся от состава основной части катализатора, и если выступ удаляют от основной части катализатора, то соотношение компонентов катализатора в целом отклоняется от соотношения компонентов на стадии составления смеси исходных материалов. В данном документе, «выступ» означает объекты, выступающие и/или налипшие на поверхности обожженного продукта, полученного посредством основного обжига, описанного далее, и относится к объектам, выступающим от поверхности обожженного продукта или прилипшим к ней. При этом, многие выступы представляют собой выступающие кристаллы оксидов и других примесей. Особенно в случае обожженного продукта, включающего несколько металлов, оксиды, имеющие состав, отличающийся от состава кристаллов, которые составляют основную часть обожженного продукта, могут быть образованы в такой форме, что данные оксиды выступают из основной части обожженного продукта. В этом случае, выступ часто образуется в виде нескольких выступов (например, высотой от 0,1 мкм до 20 мкм) на поверхности сферического обожженного продукта (например, диаметром от 30 до 150 мкм). Состав выступа и стадия удаления выступа будут описаны подробно далее.
В качестве исходного материала для Mo, могут быть использованы гептамолибдат аммония [(NH4)6Mo7O24·4H2O], триоксид молибдена [MoO3], фосформолибденовая кислота [H3PMo12O40], кремниймолибденовая кислота [H4SiMo12O40], пентахлорид молибдена [MoCl5], и т.п. Особенно предпочтительным является гептамолибдат аммония [(NH4)6Mo7O24·4H2O].
В качестве исходного материала для V, могут быть использованы метаванадат аммония [NH4VO3], пятиокись ванадия [V2O5], хлориды ванадия [VCl4, VCl3] и т.п. Особенно предпочтительным является метаванадат аммония [NH4VO3].
В качестве исходного материала для Sb, могут быть использованы оксиды сурьмы [Sb2O3, Sb2O5], сурьмянистая кислота [HSbO2], сурьмяная кислота [HSbO3], антимонат аммония [(NH4)SbO3], хлорид сурьмы [Sb2Cl3], соли органических кислот, такие как тартрат сурьмы, металлическая сурьма и т.п. Особенно предпочтительным является оксид сурьмы (III) [Sb2O3].
В качестве исходных материалов для Nb могут быть использованы ниобиевая кислота, неорганический ниобат и органический ниобат. Особенно предпочтительной является ниобиевая кислота. Ниобиевая кислота представляет собой соединение формулы Nb2O5·nH2O, и ее также называют гидроксидом ниобия или гидратом оксида ниобия. Кроме того, в качестве исходных материалов для Nb также предпочтительно используют раствор исходного материала для Nb, в котором молярное соотношение дикарбоновая кислота/ниобий составляет от 1 до 4, и в качестве дикарбоновой кислоты предпочтительной является щавелевая кислота.
В качестве исходного материала для W могут быть использованы соли вольфрама, такие как аммониевые соли, соли азотной кислоты, соли карбоновой кислоты, аммониевые соли карбоновой кислоты, соли пероксокарбоновой кислоты, аммониевые соли пероксокарбоновой кислоты, галогенированные аммониевые соли, галогениды, ацетилацетонат, алкоксид, трифенильные соединения, полиоксометалат и аммониевые соли полиоксометалата вольфрама; триоксид вольфрама, диоксид вольфрама, вольфрамовая кислота, водный раствор метавольфрамата аммония, паравольфрамат аммония, вольфрамокремниевая кислота, кремневольфрамомолибденовая кислота, вольфрамокремниевая кислота и т.п. Среди них, предпочтительным является водный раствор метавольфрамата аммония.
Исходные материалы для Z (одного или нескольких элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba) не ограничиваются особым образом, при условии, что исходные материалы содержат эти элементы. Могут быть использованы соединение, содержащее эти элементы, и раствор, в котором металл этих элементов растворен в подходящем реагенте. В качестве соединения, содержащего эти элементы, обычно могут быть использованы аммониевая соль, нитрат, карбоксилат, аммониевая соль карбоновой кислоты, пероксокарбоксилат, аммониевая соль пероксокарбоновой кислоты, галогенированная аммониевая соль, галогенид, ацетилацетат и алкоксид. Предпочтительно, может быть использован водный исходный материал, такой как нитрат и карбоксилат.
При составлении исходного материала процедура растворения исходных материалов для элементов, составляющих катализатор, процедура смешивания исходного материала или процедура диспергирования исходного материала не ограничивается особым образом. Исходные материалы могут быть растворены, смешаны или диспергированы в той же самой водной среде. В качестве альтернативы, исходные материалы могут по отдельности быть растворены, смешаны или диспергированы в водной среде, и водные среды могут быть смешаны. При необходимости, может быть выполнено нагревание и/или перемешивание.
В комплексном оксидном катализаторе, одним из важных моментов является то, что компонент Z равномерно распределен в частицах катализатора. Предпочтительно, катализатор находится в таком состоянии. В данном документе, «равномерность» означает, что распределение компонента Z в частицах катализатора не является неравномерным. Предпочтительно, «равномерность» означает, что не менее чем 80% частиц оксида, содержащих компонент Z (массовая доля), находятся в частицах катализатора в виде тонких частиц, имеющих размер не более чем 1 мкм. В случае, когда комплексный оксидный катализатор содержит кремнезем, подходящим определением «равномерности» является то, что, когда поперечное сечение частицы катализатора подвергается компонентному анализу, величина рассеяния (величина, полученная делением стандартного отклонения на среднюю величину) отношения интенсивности сигнала компонента Z к Si, находится в интервале от 0 до 0,5. В данном документе величина рассеяния обозначается как «Dx».
Обычный метод компонентного анализа может быть использован для анализа состава. Например, могут быть использованы сканирующая электронная микроскопия с энергодисперсионным рентгеновским анализом (SEM-EDX), рентгеновская фотоэлектронная спектроскопия (XPS), масс-спектроскопия вторичных ионов (SIMS), электронно-зондовый микроанализ (EPMA) и т.п. Предпочтительно, может быть использован электронно-зондовый микроанализ (EPMA). Здесь, электронно-зондовым микроанализом (EPMA) называют рентгеноспектральный электронно-зондовый микроанализ (термин «рентгеноспектральный» может быть опущен при ссылке на прибор). Прибор для анализа представляет собой прибор, в котором характеристическое рентгеновское излучение, полученное облучением вещества пучком ускоренных электронов, наблюдается, чтобы выполнить компонентный анализ небольшой области (пятна), облучаемой электронным пучком. Посредством электронно-зондового микроанализа (EPMA) обычно, в отношении поперечного сечения твердой частицы, такой как частицы катализатора и частицы носителя, получают информацию о конкретном элементе, такую как распределение концентрации и изменение в составе.
Величина рассеяния (Dx) отношения интенсивности пика компонента Z к Si при электронно-зондовом микроанализе (EPMA) представляет собой величину, полученную измерением поперечного сечения частицы, подлежащей измерению, и выполнением расчета в соответствии с обычным методом для плоскостного анализа поперечного сечения частицы посредством электронно-зондового микроанализа (EPMA), который выполняется в области катализатора следующим образом. А именно, вначале, распределение интенсивности рентгеновского пика Si (число счетов импульсов ISi) в любой позиции (x, y) в поперечном сечении частицы катализатора измеряют таким образом, что покрывают все поперечное сечение частицы катализатора. Затем аналогичным образом измеряют распределение интенсивности рентгеновского пика (число счетов импульсов IX) компонента Z таким образом, что покрывают все поперечное сечение частицы катализатора. На основании полученных наборов данных (x, y, ISi, IX) для Si и компонента Z определяют отношение интенсивности IR пика компонента Z к Si (IR=IX/ISi) в одной и той же позиции (x, y), и определяют среднее арифметическое (IR)av и стандартное отклонение S для IR. Величину, полученную делением стандартного отклонения S на среднее арифметическое (IR)av, принимают в качестве величины рассеяния (Dx). При этом среднее арифметическое и стандартное отклонение могут быть определены обычным способом. В данном документе «комплексный оксидный катализатор (просто называемый «катализатором» в некоторых случаях)» означает продукт, полученный посредством удаления выступа, образованного на поверхности частиц обожженного продукта после основного обжига. Измерение величины рассеяния выполняется посредством наблюдения поперечного сечения и на него не влияет состояние поверхности катализатора. Соответственно, величина рассеяния означает ту же самую величину, даже если данную величина измеряют после основного обжига и перед стадией удаления выступа.
Предпочтительно, для того, чтобы избежать неопределенности в данных вследствие краевого эффекта поперечного сечения частицы при измерении, область, которая составляет 10% площади поперечного сечения в поперечном сечении частицы катализатора и соответствует внешней периферии частиц, исключают, а область, составляющую 90% от центра в поперечном сечении частица катализатора, используют в качестве эффективной области, и рассчитывают данные для данной эффективной области. Естественно, вначале плоскостной анализ посредством электронно-зондового микроанализа (EPMA) может быть выполнен лишь для внутренней части поперечного сечения частицы катализатора, из которой исключено 10% области, соответствующей внешней периферии частицы, и величина рассеяния Dx может быть определена из этих данных.
Теперь будет описан подходящий способ плоскостного анализа поперечного сечения частиц катализатора.
Вначале, частицы, подлежащие измерению, заделывают в подходящую матричную смолу. Матричную смолу шлифуют и полностью соскабливают до тех пор, пока не будут открыты поперечные сечения заделанных частиц катализатора. Затем выполняют измерение электронно-зондовым микроанализом (EPMA) на частицах катализатора с открытыми поперечными сечениями следующим образом.
(1) Образец позиционируют таким образом, что поперечное сечение частицы катализатора находится в пределах поля наблюдения при измерении электронно-зондовым микроанализом (EPMA).
(2) Поперечное сечение частицы катализатора облучают электронным пучком, подсчитывают интенсивность характеристического рентгеновского излучения Si или компонента Z, поступающего от участка, облученного электронным пучком, и сканируют анализируемую область электронным пучком, чтобы выполнить плоскостной анализ.
В случае, когда комплексный оксидный катализатор в соответствии с представляемым вариантом осуществления является катализатором, включающим кремнезем, и предпочтительно катализатором с носителем из кремнезема, поддерживаемым на кремнеземе, раствор с составом исходного материала предпочтительно приготавливают таким образом, чтобы он содержал исходный материал для кремнезема. В качестве исходного материала для кремнезема может быть использован золь кремниевой кислоты. Порошковый кремнезем может быть использован в качестве части исходного материала для кремнезема или всего исходного материала для кремнезема.
Содержание кремнезема, содержащегося в катализаторе, предпочтительно содержание кремнезем в качестве носителя, составляет предпочтительно не менее чем 20 масс.%, с точки зрения повышения прочности катализатора, и предпочтительно не более чем 70 масс.%, с точки зрения предоставления достаточной активности, в расчете на SiO2 по отношению к общей массе катализатора, включающего комплексный оксид и кремнезем. Содержание более предпочтительно составляет от 40 до 65 масс.% по отношению к общей массе катализатора.
Предпочтительно, золь кремниевой кислоты содержит от 10 до 270 млн-1 по массе и более предпочтительно от 10 до 270 млн-1 по массе ионов азотной кислоты в расчете на массу SiO2 в золе кремниевой кислоты. Хотя причина является неясной, может быть предположен следующий фактор. Фактор не ограничивается этим. А именно, концентрация ионов азотной кислоты в золе кремниевой кислоты, который является исходным материалом кремнезема в качестве носителя, может быть надлежащим образом отрегулирована в конкретном интервале, чтобы должным образом регулировать агрегирование золя кремниевой кислоты. Посредством применения такого золя кремниевой кислоты исходного материала носителя, предоставляется возможность высокого выхода целевого продукта, и получают катализатор, поддерживаемый кремнеземом, обладающий высокой физической прочностью.
При этом концентрация ионов азотной кислоты в расчете на кремнезем в золе кремниевой кислоты может быть определена с помощью ионной хроматографии. Прибор для измерения и условия измерения представлены ниже. В качестве прибора для измерения может быть использован прибор (торговое наименование «IC-2001») производства Tosoh Corporation. В качестве колонки используют TSKgel superIC-AZ (торговое наименование). В качестве защитной колонки используют TSKguardcolumn superIC-AZ (торговое наименование). Кроме того, в качестве жидкости для промывки клапана подавителя используют TSKsupress A (торговое наименование). В качестве элюента смешивают и используют 1,9 ммоль/л водный раствор NaHCO3 и 3,2 ммоль/л водный раствор Na2CO3. Расход при этом составляет 0,8 мл/мин.
Для того, чтобы описать способ регулирования концентрация ионов азотной кислоты в золе кремниевой кислоты, вначале будет описан промышленный способ получения золя кремниевой кислоты. Примеры промышленного способа получения золя кремниевой кислоты включают (1) диализ после нейтрализации жидкого стекла, (2) электродиализ, (3) растворение металлического кремния в водном растворе аммиака или амина, (4) пептизация силикагеля и (5) удаление Na из жидкого стекла с помощью ионообменной смолы. Среди них, наиболее обычным способом получения золя кремниевой кислоты является (5) способ с применением ионообменной смолы (способ с ионообменной смолой). Для того, чтобы улучшить стабильность при высокой концентрации, LiOH, NaOH или KOH в качестве стабилизатора добавляют к золю кремниевой кислоты, полученной способом с ионообменной смолой. По этой причине, обычно, стабильная область pH для золя кремниевой кислоты составляет, соответственно, от 8 до 10. Для того чтобы поддерживать стабильное состояние дисперсии золя кремниевой кислоты, частицы кремнезема в золе необходимо отталкивать одну от другой с помощью электрического заряда. По этой причине, добавляют стабилизатор, как описано выше, ионы OH- адсорбируются на поверхности частиц кремнезема, и посредством отрицательного заряда проявляется стабилизирующий эффект. Тем самым, предотвращается желатинирование. Однако известно, что, если добавляют чрезмерное количество щелочных катионов (ионов щелочных металлов в стабилизаторе), то ионы щелочных металлов адсорбируются и уменьшают отрицательный заряд, что приводит к нестабильному состоянию золя кремниевой кислоты. В последнее время, многие золи кремниевой кислоты, которые имеют свойства, присущие этим золям кремниевой кислоты, и могут быть использованы в различных видах применения, коммерчески доступны. Примеры коммерчески доступного золя кремниевой кислоты включают ряд SNOWTEX производства компании Nissan Chemical Industries, Ltd., особенно, SNOWTEX 30, имеющий концентрацию золя кремниевой кислоты 30%, SNOWTEX C, используемый в видах применения, в которых может происходить желатинирование, SNOWTEX N, используемый для предотвращения сохранения щелочных катионов, с применением летучего слабого основания в качестве стабилизатора, и SNOWTEX O, подходящий для видов применения, в которых обязательным является использование кислотного состояния (ссылка: Shokubai Kougakukouza 10, Gensobetu Shokubai Binran, опубликовано 25 февраля 1967 г. (Сева 42)).
В отношении поверхностей частиц кремнезема золя кремниевой кислоты, полученного способом, указанным выше, золь кремниевой кислоты классифицируют на кислотный тип и щелочной тип. В обоих типах, однако, ионы азотной кислоты практически не могут находиться в золе кремниевой кислоты. Например, в основном ионы водорода используются в качестве стабилизатора в кислотном типе. С другой стороны, ионы натрия или ионы аммония используются в качестве стабилизатора в щелочном типе. В качестве противоаниона в кислотном типе используют SO4 2-, Cl- и т.п. В качестве противоаниона в щелочном типе обычно используют OH-.
В обоих случаях золя кремниевой кислоты, кислотного типа и щелочного типа, чтобы получить золь кремниевой кислоты, в котором массовая доля ионов азотной кислоты составляет от 10 до 270 млн-1 по массе в расчете на массу кремнезема, при применении обычного способа получения золя кремниевой кислоты, такого как нейтрализация водного раствора жидкого стекла серной кислотой или хлористоводородной кислотой, предпочтительно добавляют азотную кислоту или соль азотной кислоты, такую как нитрат аммония, чтобы регулировать количество ионов азотной кислоты по отношению к кремнезему при 10-270 млн-1 по массе. В качестве альтернативы, после нейтрализации серной кислотой или хлористоводородной кислотой анионы в водном растворе жидкого стекла могут быть заменены ионами азотной кислоты посредством ионного обмена. Ионы азотной кислоты могут быть добавлены к имеющемуся золю кремниевой кислоты пипеткой или т.п., чтобы отрегулировать количество ионов азотной кислоты. Источником азотной кислоты может быть азотная кислота и соли, такие как нитрат аммония. Первичные частицы золя кремниевой кислоты являются обычно сферическими. Может быть использован золь кремниевой кислоты с несферическими частицами или золь, имеющий сферические частицы, соединенные одна с другой.
В качестве исходного материала для кремнезема в качестве носителя может быть использован лишь золь кремниевой кислоты. В качестве альтернативы, часть исходного материала может быть заменена порошковым кремнеземом. Если порошковый кремнезем используют в качестве исходного материала для кремнезема в качестве носителя, то ожидается эффект улучшения активности катализатора и/или выхода целевого продукта. С другой стороны, если используют лишь порошковый кремнезем без применения золя кремниевой кислоты, чтобы приготовить катализатор, износоустойчивость катализатора заметно уменьшается. В данном документе, термин «порошковый кремнезем» обозначает тонкие частицы твердого SiO2. Если размер первичных частиц порошкового кремнезема чрезмерно большой, то полученный катализатор, вероятно, будет хрупким. Соответственно, предпочтительным является наноразмерный порошковый кремнезем. Порошковый кремнезем предпочтительно получают высокотемпературным способом. Конкретные примеры предпочтительного порошкового кремнезема включают Aerosil 200 (торговое наименование) производства компании Nippon Aerosil Co., Ltd.
С точки зрения простоты добавления и смешивания с раствором с составом исходного материала порошковый кремнезем предпочтительно диспергируют предварительно в воде. Метод диспергирования порошкового кремнезема в воде не ограничивается особым образом, и порошковый кремнезем может быть диспергирован с применением обычного гомогенизатора, смесителя-гомогенизатора или ультразвукового вибратора в отдельности или в комбинации одного с другими. Первичная форма порошкового кремнезема при этом может быть сферической или несферической.
В случае, когда золь кремниевой кислоты и порошковый кремнезем используют в качестве исходного материала для кремнезема в качестве носителя, количество порошкового кремнезема составляет предпочтительно от 20 до 70 масс.% от общего количества золя кремниевой кислоты и порошкового кремнезема. Если количество порошкового кремнезема больше чем 70 масс.%, то износоустойчивость катализатора, вероятно, будет уменьшена. Если количество меньше чем 20 масс.%, то активность катализатора и/или выход целевого продукта, вероятно, будут уменьшены. Порошковый кремнезем может не содержать ионы азотной кислоты. Даже если концентрацию ионов азотной кислоты в золе кремниевой кислоты (массовую долю ионов азотной кислоты по отношению к массе кремнезема) регулируют до 10-270 млн-1 по массе в расчете на SiO2 для того, чтобы увеличить выход и/или физическую прочность целевого продукта, не требуется регулирование ионов азотной кислоты, содержащихся в порошковом кремнеземе.
Далее в данном документе стадия составления смеси исходных материалов будет описана в примере, в котором растворителем и/или дисперсионной средой является вода, и приготавливают раствор с составом исходного материала для катализатора, поддерживаемого кремнеземом, включающим соединение Mo, соединение V, соединение Sb, соединение Nb, соединение W и соединение Z. Стадия составления смеси исходных материалов не ограничивается этим.
Вначале приготавливают водный смешанный раствор, содержащий Mo, V, Sb и компонент Z. Более конкретно, соединение Mo, соединение V, соединение Sb и соединение компонента Z добавляют к воде, и нагревают раствор, чтобы приготовить водный смешанный раствор (A). Температуру нагревания и время нагревания во время приготовления водного смешанного раствора (A) предпочтительно регулируют таким образом, что соединения исходных материалов растворяются в достаточной степени. Температура нагревания составляет предпочтительно от 70°C до 100°C, и время нагревания составляет предпочтительно от 30 минут до 5 часов. Число оборотов вращения при перемешивании во время нагревания может быть аналогичным образом отрегулировано до числа оборотов вращения, при котором исходный материал легко растворяется. В случае, когда исходный материал является солью металла, предпочтительно поддерживают состояние перемешивания с целью достаточного растворения соли металла. В это время внутри резервуара может быть воздушная атмосфера. С точки зрения регулирования степени окисления получаемого комплексного оксидного катализатора, может быть использована атмосфера азота. Состояние, когда нагревание водного смешанного раствора (A) завершено, называют водным смешанным раствором (A'). Водный смешанный раствор (A') предпочтительно поддерживают при температуре не менее чем 20°C и не более чем 80°C и более предпочтительно не менее чем 40°C и не более чем 80°C. При температуре водного смешанного раствора (A') меньше чем 20°C, металлический компонент, растворенный в водном смешанном растворе (A'), может выпасть в осадок.
Затем золь кремниевой кислоты добавляют к водному смешанному раствору (A) или водному смешанному раствору (A') после того, как нагревание завершено. Золь кремниевой кислоты функционирует в качестве носителя. Температура, при которой добавляют золь кремниевой кислоты, составляет предпочтительно не более чем 80°C. В случае, когда золь кремниевой кислоты добавляют при температуре более чем 80°C, стабильность золя кремниевой кислоты может быть уменьшена до возможности желатинирования раствора с составом исходного материала. Время, при котором добавляют золь кремниевой кислоты, может быть временем начала выдерживания, описанного выше, временем в течение выдерживания или временем непосредственно перед сушкой раствора с составом исходного материала. Предпочтительно, золь кремниевой кислоты добавляют к водному смешанному раствору (A'). После этого, с целью регулирования степени окисления получаемого комплексного оксида надлежащее количество раствора пероксида водорода предпочтительно добавляют к водному смешанному раствору (A') при необходимости. Раствор пероксида водорода может быть добавлен в любое время; раствор пероксида водорода может быть добавлен к самому водному смешанному раствору (A') или добавлен в то время, когда водный смешанный раствор (A') составляют, или же добавлен перед или после добавления золя кремниевой кислоты. При этом, с точки зрения регулирования степени окисления получаемого комплексного оксидного катализатора в надлежащем интервале, количество добавляемого раствора пероксида водорода составляет предпочтительно от 0,01 до 5, более предпочтительно от 0,5 до 3 и особенно предпочтительно от 1 до 2,5 в отношении H2O2/Sb (молярное отношение).
Температуру нагревания и время нагревания после добавления раствора пероксида водорода к водному смешанному раствору (A') предпочтительно регулируют таким образом, чтобы реакция жидкофазного окисления раствором пероксида водорода могла протекать достаточным образом. Температура нагревания составляет предпочтительно от 30°C до 70°C, и время нагревания составляет предпочтительно от 5 минут до 4 часов. Аналогичным образом, число оборотов вращения при перемешивании во время нагревания может быть отрегулировано при таком числе оборотов вращения, что легко протекает реакция жидкофазного окисления посредством раствора пероксида водорода. С точки зрения достаточного протекания реакции жидкофазного окисления посредством раствора пероксида водорода, перемешивание предпочтительно продолжают во время нагревания. Приготовленный таким образом водный смешанный раствор называют водным смешанным раствором (A").
Вслед за этим, соединение Nb и дикарбоновую кислоту нагревают и перемешивают в воде, чтобы приготовить смешанный раствор (B0). Примеры дикарбоновой кислоты включают щавелевую кислоту [(COOH)2]. Затем раствор пероксида водорода предпочтительно добавляют к смешанному раствору (B0), чтобы приготовить водный смешанный раствор (C). При этом H2O2/Nb (молярное отношение) составляет предпочтительно от 0,5 до 20 и более предпочтительно от 1 до 10, с точки зрения формирования комплекса с соединением Nb и стабилизации комплекса в растворенном состоянии, регулирования надлежащим образом состояния окисления и восстановления элементов, составляющих катализатор, и оптимизации функциональных возможностей получаемого катализатора.
Далее, в зависимости от целевого состава, водный смешанный раствор (A"), водный смешанный раствор (C), соединение W и порошковый кремнезем смешивают соответствующим образом, чтобы получить водный смешанный раствор (D). После этого, полученный водный смешанный раствор (D) выдерживают, чтобы получить раствор с составом исходного материала. Предпочтительно, порошковый кремнезем, используемый здесь, добавляют к раствору, полученному смешиванием водного смешанного раствора (A"), водного смешанного раствора (C) и соединения W, с точки зрения оптимизации функциональных возможностей получаемого катализатора. Порошковый кремнезем может быть добавлен как есть. Более предпочтительно, порошковый кремнезем добавляют в виде раствора, в котором порошковый кремнезем диспергирован, т.е. в виде суспензии, содержащей порошковый кремнезем. Концентрация порошкового кремнезема в суспензии, содержащей порошковый кремнезем, при этом составляет предпочтительно от 1 до 30 масс.% и более предпочтительно от 3 до 20 масс.%. При концентрации порошкового кремнезема меньше чем 1 масс.% вязкость суспензии является чрезмерно низкой. По этой причине, форма получаемых частиц может быть искривлена, и имеет место вероятность образования выемок в частицах катализатора. С другой стороны, при концентрации порошкового кремнезема больше чем 30 масс.%, вязкость раствора с составом исходного материала является чрезмерно высокой, и такой раствор с составом исходного материала может желатинироваться с образованием засорений внутри трубопровода. В результате может быть затруднено получение сухого порошка, и функциональные возможности катализатора могут быть понижены.
Выдерживание водного смешанного раствора (D) означает, что водный смешанный раствор (D) оставляют в неподвижном состоянии или при перемешивании в течение заданного периода времени. Время выдерживания составляет предпочтительно 90 минут или более и 50 часов или менее и более предпочтительно 90 минут или более и 6 часов или менее. При времени выдерживания меньше чем 90 минут или больше чем 50 часов трудно приготовить водный смешанный раствор (D), имеющий подходящее состояние (потенциал) окисления и восстановления, и функциональные возможности получаемого катализатора в виде комплексного оксида, вероятно, будут уменьшены. При этом, в случае, когда комплексный оксидный катализатор производят в промышленном масштабе посредством сушки с применением распылительной сушилки, обычно распылительная сушилка имеет скорость обработки, ограничивающую производительность. При этом, некоторое время занимает распылительная сушка части водного смешанного раствора (D) и полная распылительная сушка смешанного раствора в целом. Во время этой распылительной сушки водный смешанный раствор, не высушенный распылением, непрерывным образом выдерживается. Соответственно, время выдерживания включает не только время выдерживания перед сушкой на стадии (II), описанной далее, но также и время после начала сушки до завершения сушки. Температура выдерживания составляет предпочтительно 25°C или более, с точки зрения предотвращения сгущения компонента Mo и осаждения оксида металла для V и другого металла или нескольких металлов. Температура выдерживания составляет предпочтительно 65°C или менее, с точки зрения предотвращения избыточного протекания гидролиза комплекса, содержащего Nb и пероксид водорода, и образования суспензии в предпочтительной форме. С вышеуказанной точки зрения, температура выдерживания составляет предпочтительно 25°C или более и 65°C или менее и более предпочтительно 30°C или более и 60°C или менее.
Атмосфера в контейнере при выдерживании предпочтительно содержит кислород в достаточной концентрации. Недостаточная концентрация кислорода может почти не вызывать существенного изменения водного смешанного раствора (D). Более конкретно, концентрация кислорода в паровой фазе в резервуаре составляет предпочтительно 1 об.% или более, например, водный смешанный раствор (D) может быть выдержан в воздушной атмосфере. Концентрация кислорода в паровой фазе может быть измерена посредством обычных методов, например, посредством метода измерения с применением измерителя концентрации кислорода на базе диоксида циркония. Место, в котором измеряют концентрация кислорода в паровой фазе, предпочтительно находится вблизи поверхности раздела между водным смешанным раствором (D) и паровой фазой. Например, предпочтительно, концентрацию кислорода в паровой фазе измеряют три раза в одной и той же точке в течение 1 минуты, и среднюю величину результатов трех измерений используют в качестве концентрации кислорода в паровой фазе.
Разбавляющий газ для уменьшения концентрации кислорода в паровой фазе не ограничивается особым образом. Примеры разбавляющего газа включают азот, гелий, аргон, диоксид углерода и пар. В промышленном масштабе предпочтительным является азот. В качестве газа для увеличения концентрации кислорода в паровой фазе, предпочтительным является чистый кислород или воздух с высокой концентрацией кислорода.
Некоторое изменение, как считается, происходит в окислительно-восстановительном состоянии компонента, содержащегося в водном смешанном растворе (D), вследствие выдерживания. Протекание некоторого изменения предполагается на основании возникновения изменения в цвете и изменения в окислительно-восстановительном потенциале или т.п. водного смешанного раствора (D) во время выдерживания. В результате возникает разница в функциональных возможностях между композитными оксидными катализаторами, которые образуются при применении или отсутствии выдерживания в течение 90 минут или более и 50 часов в атмосфере с концентрацией кислорода от 1 до 25 об.%. В частности, чрезвычайно трудно корректным образом идентифицировать изменение в форме компонента в жидкости во время выдерживания. Тем не менее, получают катализаторы, имеющие разное время выдерживания, и оценивают их функциональные возможности, и тем самым можно сделать вывод, что время выдерживания, придающее катализатор высокие функциональные возможности, является предпочтительным, и суспензия, обладающая некоторой предпочтительной формой, образуется в таком случае.
Окислительно-восстановительный потенциал водного смешанного раствора (D) регулируют посредством потенциала (600 мВ/AgCl) водного раствора (C) исходного материала. Например, считается, что, когда водный раствор (C) исходного материала содержит пероксид оксалата Nb, пероксид оксалата Nb и другие металлические компоненты вызывают некоторую окислительно-восстановительную реакцию, обусловливая временное уменьшение потенциала. Окислительно-восстановительный потенциал водного смешанного раствора (D) составляет предпочтительно от 450 до 530 мВ/AgCl и более предпочтительно от 470 до 510 мВ/AgCl.
Концентрация кислорода во время выдерживания составляет предпочтительно 1 об.% или более, с точки зрения предотвращения чрезмерной задержки в протекании окислительно-восстановительной реакции, оказывающей некоторое влияние на окислительно-восстановительное состояние компонентов, содержащихся в водном смешанном растворе (D), и предотвращения какого-либо чрезмерного восстановления в окислительно-восстановительном состоянии раствора с составом исходного материала. С другой стороны, концентрация кислорода во время выдерживания составляет предпочтительно 25 об.% или менее с целью предотвращения некоторого избыточного окисления раствора с составом исходного материала, вызываемого избыточным протеканием окислительно-восстановительной реакции. Так или иначе, необходимо поддерживать концентрацию кислорода в подходящем интервале, поскольку кислород в паровой фазе оказывает влияние на окислительно-восстановительное состояние раствора с составом исходного материала. Интервал составляет более предпочтительно от 5 до 23 об.% и еще более предпочтительно от 10 до 22 об.%.
Во время выдерживания водного смешанного раствора (D), содержащаяся влага может испаряться, создавая сгущение. Если выдерживание выполняют в открытой системе, то содержащаяся влага испаряется естественным образом. Если выдерживание не выполняют в атмосфере с концентрацией кислорода от 1 до 25 об.%, то функциональные возможности катализатора не могут быть улучшены.
При перемешивании во время выдерживания, плотность раствора, количество раствора с составом исходного материала, число оборотов вращения перемешивающей лопасти или т.п. предпочтительно регулируют, с точки зрения предотвращения желатинирования раствора с составом исходного материала и регулирования вязкости раствора с составом исходного материала для его получения в надлежащем состоянии. Если вязкость раствора с составом исходного материала чрезмерно низкая, то форма получаемых частиц может быть искривлена, и имеет место вероятность образования выемок в частицах катализатора на стадии распылительной сушки, описанной далее. С другой стороны, если вязкость чрезмерно высокая, то раствор с составом исходного материала может желатинироваться с образованием засорений внутри трубопровода. В результате может быть затруднено получение сухого порошка, и функциональные возможности катализатора могут быть понижены. Соответственно, предпочтительно, плотность раствора, количество раствора с составом исходного материала, число оборотов вращения перемешивающей лопасти или т.п. регулируют, чтобы получить раствор с составом исходного материала, имеющий надлежащую вязкость.
При перемешивании во время выдерживания в качестве обычных лопастей для перемешивания, крыльчаток для перемешивания и т.п., может быть использована многоступенчатая лопасть, анкерная лопасть, червячная лопасть или лопасть в виде спиральной ленты и т.п. Кроме того, в качестве перемешивающей лопасти для раствора, имеющего низкую вязкость, могут быть использованы пропеллер, дисковая турбина, импеллерная турбина, импеллерная турбина с искривленными лопастями, гребневая турбина, турбина с угловыми лопастями и т.п.
Мощность на единицу объема, прикладываемая к раствору с составом исходного материала перемешивающей лопастью мешалки в устройстве для приготовления раствора с составом исходного материала, (на которую далее в данном документе делается ссылка как на «Pv») составляет предпочтительно от 0,005 до 300 кВт/м3, более предпочтительно от 0,01 до 280 кВт/м3 и еще более предпочтительно от 0,1 до 250 кВт/м3. Если раствор с составом исходного материала перемешивают посредством приложения мощности при перемешивании Pv меньше чем 0,005 кВт/м3, то раствор с составом исходного материала может желатинироваться, создавая засорения внутри трубопровода. В результате может быть затруднено получение сухого порошка, и функциональные возможности катализатора могут быть понижены. Если раствор с составом исходного материала перемешивают посредством приложения мощности при перемешивании Pv больше чем 300 кВт/м3, то имеет место вероятность образования выемок в частицах катализатора после распылительной сушки. Наличие выемок оказывает отрицательное действие на прочность катализатора. Величина Pv представлена приведенным ниже уравнением (A) и может быть установлена посредством регулирования плотности раствора, количества раствора с составом исходного материала, числа оборотов вращения перемешивающей лопасти и т.п.
Pv=Np×ρ×n3×d5/V... (A)
где Np: число мощности (-), которое является безразмерным числом, относящимся к мощности, необходимой для перемешивания (-), ρ: плотность раствора (кг/м3), n: число оборотов вращения перемешивающей лопасти (с-1), d: размер перемешивающей лопасти (м), V: количество раствора с составом исходного материала (м3).
Величина Np может быть вычислена при использовании приведенного ниже уравнения (B1):
[Выражение 1]
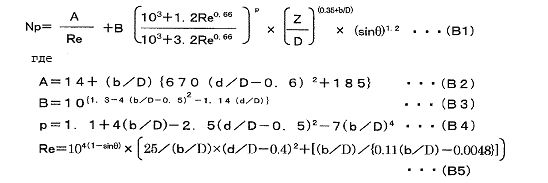
где уравнения с (B1) по (B5) выше являются следующими:
b представляет ширину перемешивающей лопасти (м), d представляет размер перемешивающей лопасти (м), D представляет размер резервуара для перемешивания (м), Z представляет глубину раствора (м), и θ представляет угол наклона (°) перемешивающей лопасти по отношению к горизонтальной поверхности.
Вязкость получаемого раствора с составом исходного материала при комнатной температуре (25°C) составляет предпочтительно от 1 до 100 сП, более предпочтительно от 2 до 90 сП и еще более предпочтительно от 2,5 до 80 сП, с точки зрения сдерживания желатинирования раствора с составом исходного материала, засорения трубопровода и трудностей в получении сухого порошка, дополнительного сдерживания снижения функциональных возможностей катализатора и сдерживания образования выемок в частицах катализатора после распылительной сушки или искривленной формы частиц катализатора.
Вязкость раствора с составом исходного материала может быть измерена, например, методом, в котором измерение выполняют с применением коммерчески доступного вискозиметра, или методом, в котором измеряют потерю давления внутри трубопровода, через который протекает раствор с составом исходного материала. Например, в случае, когда измеряют такую вязкость раствора, при которой желатинирование постепенно прогрессирует без перемешивания, вязкость может постепенно изменяться в ходе измерения вязкости с применением коммерчески доступного вискозиметра. В таком случае, с точки зрения воспроизводимости измеренной величины, вязкость предпочтительно измеряют методом, в котором измеряют потерю давления внутри трубопровода, через который протекает раствор с составом исходного материала.
В случае, когда вязкость раствора с составом исходного материала измеряют методом, в котором измеряют потерю давления внутри трубопровода, через который протекает раствор с составом исходного материала, вязкость раствора может быть вычислена посредством приведенного ниже уравнения (C1):
[Выражение 2]
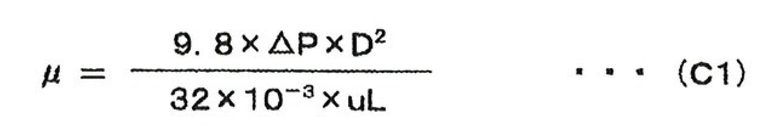
где символы являются следующими:
µ: вязкость раствора (сП), ΔP: потеря давления внутри трубопровода (ммH2O), u: средняя скорость подачи раствора (м/с), L: длина трубопровода (м), D: диаметр трубопровода (м).
В случае, когда несколько растворов исходных материалов, в которых растворены соответствующие компоненты катализатора, смешивают, чтобы получить раствор с составом исходного материала, верхний предел Pv, когда приготавливают каждый из растворов исходных материалов, не ограничивается особым образом. Нижний предел Pv не ограничивается особым образом. Предпочтительно, Pv составляет не менее чем величина Pv, при которой все или большинство твердых частиц удаляются из нижней части резервуара в устройстве для получения раствора исходного материала и втекают в устройство. При приготовлении раствора исходного материала перемешивание может быть остановлено после того, как все твердые частицы в соответствующих растворах исходных материалов по существу растворены.
Для того, чтобы регулировать pH раствора с составом исходного материала, кислота и/или щелочь могут быть добавлены к раствору с составом исходного материала, при необходимости.
В случае, когда комплексный оксидный катализатор является катализатором, поддерживаемым кремнеземом, раствор с составом исходного материала предпочтительно приготавливают таким образом, чтобы он содержал золь кремниевой кислоты, с точки зрения достаточного растворения и/или диспергирования соединения, включающего элемент, составляющий катализатор, надлежащего регулирования состояния окисления и восстановления элемента, составляющего катализатор, предоставления предпочтительной формы и/или прочности получаемым частицам катализатора и улучшения функциональных возможностей получаемого катализатора в виде комплексного оксида. Золь кремниевой кислоты может быть добавлен подходящим образом. Водная дисперсия порошка кремнезема может быть использована в качестве части золя кремниевой кислоты. Водная дисперсия порошка кремнезема может быть также добавлена подходящим образом.
Стадия составления смеси исходных материалов может быть выполнена неоднократно в зависимости от объема производства.
Стадия составления смеси исходных материалов в представляемом варианте осуществления предпочтительно включает следующие стадии (a)-(d):
(a) стадию приготовления водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) стадию добавления золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) стадию смешивания водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) стадию добавления суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживания раствора.
(Стадия (II) сушки)
Стадия (II) представляет собой стадию сушки раствора с составом исходного материала, чтобы получить сухой порошок.
Раствор с составом исходного материала в виде суспензии, подвергнутый обработке на стадии составления смеси исходных материалов, сушат, чтобы получить сухой порошок. Сушка может быть выполнена известным способом. Например, сушка может быть выполнена распылительной сушкой или испарением до сухого состояния. В случае, когда способ с реакцией в псевдоожиженном слое способ используют для реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе, применение распылительной сушки является предпочтительным, поскольку предпочтительно, чтобы был получен сухой порошок в виде микросфер, с точки зрения предпочтительной текучести внутри реактора. Распыление в способе сушки распылением может быть выполнено с помощью центробежного устройства, соплового устройства с двумя текучими средами или соплового устройства высокого давления. Воздух, нагретый паром, и электрический нагреватель или т.п. может быть использован в качестве источника тепла для сушки. Температура источника тепла для сушки на входе распылительной сушилки (далее в данном документе, называемая в некоторых случаях «температурой на входе сушилки») составляет предпочтительно от 150 до 300°C, с точки зрения предоставления предпочтительной формы и/или прочности получаемым частицам катализатора и улучшения функциональных возможностей получаемого катализатора в виде комплексного оксида. Температура выпускаемого воздуха на выходе сушилки (далее в данном документе называемая в некоторых случаях «температурой на выходе сушилки») составляет предпочтительно от 100 до 160°C.
Предпочтительно, скорость распыления, скорость подачи раствора с составом исходного материала и число оборотов вращения распылителя в случае распылителя центробежного типа регулируют таким образом, чтобы получаемый сухой порошок имел подходящий размер. Средний размер частиц сухого порошка составляет предпочтительно от 5 мкм до 200 мкм и более предпочтительно от 10 до 150 мкм.
Средний размер частиц сухого порошка может быть определен следующим образом: в соответствии с JIS R 1629-1997 «Способ измерения распределения размера частиц методом дифракционного рассеяния лазерного излучения для тонкого керамического исходного материала», измеряют распределение размера частиц и усредняют в расчете на объем. Более конкретно, часть сухого порошка обжигают на воздухе при 400°C в течение 1 часа, и полученные частицы измеряют с применением прибора для измерения распределения частиц по размеру с помощью дифракционного рассеяния лазерного излучения LS230 (торговое наименование), производства компании Beckman Coulter, Inc.
Средний размер частиц измеряют после того, как часть сухого порошка «обожжена на воздухе при 400°C в течение 1 часа», поскольку сухой порошок предотвращается от растворения в воде. А именно, «обжиг на воздухе при 400°C в течение 1 часа» предназначен главным образом для измерения и не имеет никакого отношения к стадии обжига, описанной далее. Можно полагать, что размер частиц по существу не изменяется перед и после обжига.
Более конкретно, средний размер частиц сухого порошка измеряют в соответствии с руководством, приложенным к прибору для измерения распределения частиц по размеру с помощью дифракционного рассеяния лазерного излучения (производства компании Beckman Coulter, Inc., торговое наименование «LS230») следующим образом. Вначале, после того как выполнено базовое измерение (скорость прохода 60), отвешивают 0,2 г частиц и размещают в трубку с навинчивающейся крышкой, имеющую надлежащий размер, и добавляют 10 см3 воды. Трубку с навинчивающейся крышкой закупоривают (плотно закрывают) и встряхивают достаточным образом, чтобы диспергировать частицы в воде. Посредством прибора прикладывают ультразвуковую волну мощностью 300 Вт, и встряхивают трубку с навинчивающейся крышкой достаточным образом снова. После этого, наряду с тем, что прикладывают ультразвуковую волну, частицы, диспергированные в воде, впрыскивают в основную часть прибора с помощью пипетки таким образом, чтобы получить надлежащую концентрацию (концентрацию 10, PIDS 60). Когда отображаемая концентрация стабилизируется, приложение ультразвуковой волны прекращают. Трубку с навинчивающейся крышкой оставляют как есть в течение 10 секунд, и начинают измерение (время измерения 90 секунд). Величину (D50) среднего размера в результате измерения определяют как средний размер частиц.
Также, предпочтительно, часть сухого порошка, полученного на стадии (II), извлекают, и измеряют спектр поглощения или отражения. Если спектр поглощения или отражения сухого порошка, полученного на стадии (II), измеряют постоянным образом, то функциональные возможности получаемого комплексного оксидного катализатора могут быть в конечном счете теоретически оценены из спектра поглощения или отражения. При этом степень окисления и восстановления комплексного оксидного катализатора изменяется, например, посредством нагревания на стадии сушки, и это влияет на функциональные возможности получаемого комплексного оксидного катализатора. Соответственно, при использовании этого в качестве примера, будет описана теоретическая оценка функциональных возможностей комплексного оксидного катализатора. Когда водный исходный материал (раствор с составом исходного материала) сушат распылением на стадии (II), чтобы получить сухой порошок, часть сухого порошка прилипает и осаждается на поверхности стенки и/или дне внутри устройства и остается внутри устройства в течение длительного времени. Тем самым, тепло может непреднамеренным образом прикладываться к сухому порошку, что изменяет степень окисления и восстановления. В случае, когда стадию обжига, описанную далее, выполняют в воздушной атмосфере, предполагается, что окисление прогрессирует на стадии обжига. Соответственно, степень окисления и восстановления сухого порошка почти не влияет на функциональные возможности полученного катализатора. С другой стороны, в случае, когда стадию обжига выполняют в атмосфере инертного газа, степень окисления и восстановления сухого порошка, скорее всего, влияет на функциональные возможности комплексного оксидного катализатора. Особенно, в случае, когда способ приготовления оптимизирован с учетом степени окисления и восстановления комплексного оксидного катализатора, естественно, скорее всего, функциональные возможности комплексного оксидного катализатора будут понижены, если степень окисления и восстановления сухого порошка находится вне подходящего интервала. Хотя детальный механизм неясен, цвет сухого порошка изменяется, когда изменяется степень его окисления и восстановления. Если взять в качестве примера катализатор на базе Mo-V, в частности, когда цвет сухого продукта становится темным, функциональные возможности комплексного оксидного катализатора, вероятно, будут ухудшены. В качестве причины этому, полагают, что, например, органический компонент или неорганический компонент, содержащийся в сухом порошке, термически разлагается вследствие непреднамеренного нагревания, восстанавливая металлический элемент в этом месте или вызывая окислительно-восстановительную реакцию между металлическими элементами. Соответственно, выполняют мониторинг спектра поглощения или отражения сухого порошка, чтобы проверить степень изменения цвета, и функциональные возможности комплексного оксидного катализатора могут быть теоретически оценены.
Метод измерения спектра поглощения или отражения не ограничивается особым образом. Например, спектр поглощения или отражения определяют из поглощательной способности сухого порошка, измеряемой с применением спектрофотометра для видимой и ультрафиолетовой области. В результате интенсивных исследований, проведенных авторами данного изобретения, было найдено, что темноокрашенный сухой порошок, имеющий низкие функциональные возможности, обладает большей поглощательной способностью при длине волны не менее чем 500 нм, чем сухой порошок, цвет которого не изменен. Соответственно, поглощательная способность при длине волны не менее чем 500 нм и предпочтительно при любой длине волны в интервале не менее чем 500 нм и не более чем 800 нм может быть выбрана и использоваться в качестве показателя для мониторинга.
Предпочтительно, спектр поглощения или отражения сухого порошка измеряют постоянным образом. При этом «измеряют постоянным образом» означает, что измерение выполняют один раз или более каждые три месяца. Измерение выполняют более предпочтительно один раз в месяц, еще более предпочтительно один раз в неделю и особенно предпочтительно один раз или более в день. Когда спектр поглощения или отражения измеряют более часто, риск производства большого количества сухого порошка, имеющего неподходящую степень окисления и восстановления, может быть уменьшен в большей степени. В зависимости от условий производства, спектр поглощения или отражения сухого порошка почти не изменяется, и частое измерение может быть ненужным. Соответственно, частота измерения может быть установлена надлежащим образом.
Кроме того, меры для того, чтобы предотвратить осаждение сухого порошка внутри распылительной сушилки, не ограничиваются. Предпочтительно, присоединяют вибратор, который вызывает вибрацию распылительной сушилки, или пневматический стряхиватель для прикладывания ударного усилия к распылительной сушилке. В качестве альтернативы, предпочтительно, распылительную сушку останавливают при надлежащей частоте, и внутреннее пространство устройства промывают водой или т.п. Спектр поглощения или спектр отражения сухого порошка предпочтительно измеряют сразу же после стадии (II), когда непреднамеренное нагревание может происходить наиболее вероятным образом.
Условия функционирования пневматического стряхивателя, предоставленного в сушилке, могут быть произвольным образом отрегулированы в соответствии с размером сушилки, толщиной стенки, степенью удаления налипших объектов и т.п. Примеры условий функционирования включают силу удара пневматического стряхивателя, частоту ударов, увеличение и уменьшение числа предоставляемых пневматических стряхивателей и изменение местоположения предоставляемого пневматического стряхивателя. Сила удара пневматического стряхивателя предпочтительно является такой силой, что поверхность стенки и/или другой части сушилки не деформируется и не повреждается даже при долговременной эксплуатации. Частота ударов составляет предпочтительно один удар или более в минуту и более предпочтительно один удар или более в 10 секунд. В отношении числа и местоположения предоставляемых пневматических стряхивателей, предпочтительно, число пневматических стряхивателей увеличивают в части, в которой находится большое количество налипших объектов после долговременной эксплуатации, или же пневматический стряхиватель, предоставленный в части, к которой объекты почти не прилипают, перемещают к части, к которой объекты прилипают в значительном количестве.
В соответствии со спектром поглощения или отражения, измеренным таким образом, представленная ниже стадия (A) для определения условий на стадиях (I) и (II) может быть выполнена при необходимости.
[Стадия (A)]
Стадия (A) представляет собой стадию теоретической оценки функциональных возможностей комплексного оксидного катализатора, получаемого в качестве конечного продукта, исходя из измеренного спектра поглощения или отражения сухого порошка, и регулирования рабочих условий на соответствующих стадиях на основании теоретически оцененных функциональных возможностей комплексного оксидного катализатора. Посредством выполнения стадии (A) может быть эффективным образом получен катализатор, обладающий более высокими функциональными возможностями.
Для того чтобы теоретически оценить функциональные возможности комплексного оксидного катализатора, получаемого в качестве конечного продукта, при использовании измеренного спектра поглощения или отражения сухого порошка, может быть использована взаимосвязь между спектром поглощения или отражения сухих порошков, полученных при разных условиях приготовления исходного материала и/или условиях сушки, и функциональными возможностями комплексного оксидного катализатора, полученного из сухого порошка. В качестве спектра поглощения или отражения сухих порошков, полученных при разных условиях приготовления исходного материала и/или условиях сушки, предпочтительно используют поглощательную способность при определенной длине волны, получаемую с применением спектрофотометра для УФ и видимой области.
Сухие порошки, полученные при разных условиях приготовления исходного материала, представляют собой сухие порошки, полученные, например, посредством изменения процедуры растворения или смешивания на стадии (I) или добавления окислителя или восстановителя, когда исходный материал для элемента, составляющего катализатор, растворяют или диспергируют в водной среде, чтобы изменить состояние раствора с составом исходного материала, такое как степень окисления и восстановления компонент катализатора, и дополнительно обработанные на стадии (II). Более конкретно, например, сухие порошки получают изменением состояния раствора с составом исходного материала таким способом, как увеличение количества дикарбоновой кислоты, регулирование концентрации твердого вещества в растворе, увеличение времени обработки или изменение температуры при обработке на стадии (I), и дополнительной обработкой на стадии (II).
Сухие порошки, полученные при разных условиях сушки, представляют собой сухие порошки, полученные изменением рабочих условий и т.п. на стадии (II), например, количества раствора с составом исходного материала, подаваемого в единицу времени, количества источника тепла для сушки, подаваемого в единицу времени, (например, количества подаваемого воздуха) и температуры источника тепла для сушки (например, температуры подаваемого воздуха), и/или узла для предотвращения осаждения сухого порошка, который присоединен к сушилке. В случае метода центробежного распыления могут быть изменены такие условия, как число оборотов вращения диска и размер диска, чтобы получить сухой порошок. В качестве альтернативы, может быть использован сухой порошок, полученный посредством намеренного изменения времени и/или температуры нагревания сухого порошка, полученного на стадии (II), чтобы изменить цвет.
Спектр поглощения или отражения сухих порошков, полученных при разных условиях приготовления исходного материала и/или условиях сушки, может быть измерен таким же методом, что указан выше. Кроме того, функциональные возможности катализатора испытывают, когда реакцию каталитического окисления в паровой фазе или каталитического аммоксидирования в паровой фазе пропана или изобутана выполняют с применением комплексных оксидных катализаторов, полученных обжигом сухих порошков при тех же самых условиях. Примеры функциональных возможностей испытываемого катализатора включают выход, активность, степень конверсии, и выход побочного продукта, и они могут быть использованы в комбинации.
Затем определяют взаимосвязь между спектром поглощения или отражения сухих порошков, полученных при разных условиях приготовления исходного материала и/или условиях сушки, и функциональными возможностями катализатора. С применением такой взаимосвязи, функциональные возможности комплексного оксидного катализатора, получаемого в качестве конечного продукта, могут быть теоретически оценены из измеренного спектра поглощения или отражения сухого порошка.
Посредством стадии (A), рабочие условия на соответствующих стадиях могут быть изменены в соответствии с теоретически оцененными величинами функциональных возможностей комплексного оксидного катализатора, получаемого в качестве конечного продукта, чтобы простым образом получить комплексный оксидный катализатор, обладающий повышенными функциональными возможностями. В промышленных масштабах, спектр поглощения или отражения сухого порошка контролируют постоянным образом, и рабочие условия изменяют в соответствии со спектром поглощения или отражения, как описано выше. Тем самым, может быть эффективным образом получен комплексный оксидный катализатор, обладающий высокими функциональными возможностями.
На стадии (A), в соответствии с контролируемым спектром поглощения или отражения, определяют рабочие условия на стадии (I) составления смеси исходных материалов и стадии (II) сушки, описанных выше. С точки зрения простоты контроля, рабочие условия на стадии (II) сушки предпочтительно определяют в соответствии со спектром поглощения или отражения.
[Стадия (A-1)]
Стадия (A-1) представляет собой стадию установления условий приготовления на стадии (I) в соответствии с измеренным спектром поглощения или отражения. На данной стадии, условия приготовления устанавливают таким образом, чтобы предоставить высокие функциональные возможности комплексного оксидного катализатора, получаемого в качестве конечного продукта, с применением взаимосвязи между спектром поглощения или отражения сухих порошков, полученных на стадии (II) из растворов с составом исходного материала, полученных при разных условиях приготовления на стадии (I), и функциональными возможностями комплексного оксидного катализатора, полученного из сухого порошка.
На данной стадии, способ «установления условий приготовления» не ограничивается особым образом. Примеры способа включают способ регулирования степени окисления и восстановления компонента катализатора посредством процедуры растворения или смешивания, когда исходный материал для элемента, составляющего катализатор, растворяют или диспергируют в водной среде; и способ добавления окислителя или восстановителя. Например, условия приготовления могут быть установлены таким способом, как увеличение или уменьшение количества дикарбоновой кислоты, регулирование концентрации твердого вещества в раствор, увеличение времени обработки или изменение температуры при обработке.
[Стадия (A-2)]
Стадия (A-2) представляет собой стадию установления условий сушки на стадии (II) в соответствии с измеренным спектром поглощения или отражения. На данной стадии, условия сушки устанавливают таким образом, чтобы предоставить высокие функциональные возможности комплексного оксидного катализатора, получаемого в качестве конечного продукта, с применением взаимосвязи между спектром поглощения или отражения сухих порошков, полученных на стадии (II) при разных условиях сушки, и функциональными возможностями комплексных оксидных катализаторов, полученных из сухих порошков.
На данной стадии, способ «установления условий сушки» не ограничивается особым образом. В случае применения распылительной сушилки, примеры способа включают способ изменения рабочих условий, таких как количество водного исходного материала (раствора с составом исходного материала), подаваемого в единицу времени, количество источника тепла для сушки, подаваемого в единицу времени, (например, количество подаваемого воздуха), и температура источника тепла для сушки (например, температура подаваемого воздуха), и/или узла для предотвращения осаждения сухого порошка, который присоединен к сушилке. С точки зрения поддержания функциональных возможностей и физической формы и прочности получаемого катализатора, способ изменения рабочих условий узла для предотвращения осаждения порошка является более предпочтительным. В случае метода центробежного распыления могут быть изменены такие условия, как число оборотов вращения диска и размер диска. В качестве альтернативы, они могут быть использованы в комбинации.
Сухой порошок приготавливают таким образом, что содержание частиц, имеющих размер не более чем 25 мкм составляет предпочтительно не более чем 20 масс.%, более предпочтительно не более чем 15 масс.%, еще более предпочтительно не более чем 10 масс.%, даже еще более предпочтительно не более чем 5 масс.% и особенно предпочтительно не более чем 2 масс.%. Если содержание частиц, имеющих размер не более чем 25 мкм, составляет больше чем 20 масс.%, функциональные возможности получаемого катализатора, вероятно, будут понижены, и выход целевого продукта в реакторе с псевдоожиженным слоем, вероятно, будет уменьшен.
Хотя причина снижения функциональных возможностей катализатора не ясна, полагают, что, если содержание частиц, имеющих размер не более чем 25 мкм, составляет больше чем 20 масс.%, текучесть, вероятно, будет уменьшена, что вызывает неравномерный обжиг в устройстве для обжига. Особенно при непрерывном обжиге частицы сухого порошка, имеющие небольшой размер, возвращаются внутрь устройства для обжига и открыты для воздействия атмосферы обжига в течение времени, более длительного, чем желательное время. По этой причине, не может быть получена надлежащая степень восстановления предварительно обожженного продукта при предварительном обжиге, описанном далее, или же чрезмерный обжиг может быть выполнен при основном обжиге, что приводит к проблемам, таким как разложение кристаллов. Кроме того, частицы предварительно обожженного продукта, вероятнее всего, склонны к прилипанию. По этой причине, налипающие объекты могут осаждаться на поверхности стенки устройства для обжига, уменьшая перенос тепла внутрь устройства, или же может быть примешан катализатор, находившийся в налипшем состоянии в течение длительного времени и чрезмерно обожженный. Таким образом, предполагается такая причина. В данном случае, «предварительно обожженный продукт» относится к соединению, полученному на стадии предварительного обжига, описанной далее. По вышеуказанной причине, обычно, при получении катализатора непрерывным обжигом затруднено стабильное получение катализатора, имеющего такие же функциональные возможности (например, выход целевого продукта), что и катализатор, полученный обжигом в периодическом режиме, даже если состав катализатора является таким же. С другой стороны, в соответствии со способом получения в представляемом варианте осуществления, катализатор, обладающий такими же функциональными возможностями, что и катализатор, полученный периодическим обжигом, может быть получен также и посредством непрерывного обжига. Данный фактор не ограничивается тем, что описано выше.
В случае, когда катализатор содержит Mo, Sb или т.п., вероятно, будет образовано соединение, имеющее низкую температуру плавления. Частицы, имеющие размер не более чем 25 мкм, имеют удельную поверхность больше, чем частицы, имеющие размер больше чем 25 мкм. Соответственно, полагают, что частицы, имеющие размер не более чем 25 мкм, прилипают более вероятным образом. Если прилипает чрезмерное количество частиц, то возникают следующие проблемы: слой катализатора не может быть обожжен при достаточной температуре обжига, объем производства не может быть обеспечен и т.п. Соответственно, долю частиц, имеющих размер не более чем 25 мкм, предпочтительно уменьшают перед обжигом.
Средний размер частиц сухого порошка составляет более предпочтительно от 35 до 75 мкм и еще более предпочтительно от 40 до 65 мкм = мм, и особенно предпочтительно от 45 до 60 мкм. Когда комплексный оксидный катализатор используют в каталитической реакции в псевдоожиженном слое при среднем размере частиц меньше чем 35 мкм, текучесть может быть уменьшена, со снижением выхода целевого продукта, или же катализатор может легко рассеиваться из реактора с псевдоожиженным слоем, что увеличивает потери в количестве катализатора. С другой стороны, при среднем размере частиц больше чем 75 мкм текучесть может быть уменьшена, уменьшая эффективность контакта с реакционным газом, что приводит к снижению выхода целевого продукта.
Если средний размер частиц сухого порошка отрегулирован при 35-75 мкм, и содержание частиц, имеющих размер не более чем 25 мкм, отрегулирован при не более чем 20 масс.%, степень восстановления предварительно обожженного продукта может быть отрегулирована в предпочтительном интервале на стадии обжига. Авторы данного изобретения предполагают следующий механизм.
Сухой порошок обычно содержит радикалы аммония, органические кислоты и неорганические кислоты. В случае, когда сухой порошок обжигают во время протекания инертного газа, элементы, составляющие катализатор, восстанавливаются, когда радикалы аммония, органические кислоты, неорганические кислоты испаряются или разлагаются. Радикалы аммония испаряются и преобразуются в газообразный аммиак, восстанавливая частицы предварительно обожженного продукта из газовой фазы. Степень восстановления предварительно обожженного продукта изменяется в зависимости от времени обжига и температуры обжига, особенно при предварительном обжиге, описанном далее. При более продолжительном времени обжига или при более высокой температуре обжига легко протекает восстановление, и степень восстановления выше. В случае, когда содержится большое количество предшественника, имеющего сравнительно небольшой размер частиц, обычно, если средний размер частиц меньше чем 35 мкм или содержание частиц, имеющих размер не более чем 25 мкм, больше чем 20 масс.%, увеличивается количество частиц меньшего размера, уносимых инертным газом, или находящихся в плавучем состоянии при вращении обжиговой трубы и возвращающихся в обжиговую трубу, когда используется обжиговая труба. Вследствие этого, существуют частицы, которые задерживаются в обжиговой трубе в течение времени, более длительного, чем желательное время, что приводит к затруднениям в предоставлении степени восстановления в предпочтительном интервале. Также полагают, что небольшие частицы имеют большую площадь поверхности, контактирующей с газообразным аммиаком, и такие частицы легко восстанавливаются. Напротив, при среднем размере частиц больше чем 75 мкм частицы являются большими, и имеют небольшую площадь поверхности, контактирующую с газообразным аммиаком. Такие частицы трудно восстанавливать. Вследствие этого, полагают, что затруднено регулирование степени восстановления в предпочтительном интервале.
При этом, содержание частиц, имеющих размер не более чем 25 мкм, может быть определено следующим образом: 20 г сухого порошка просеивают с применением сита, имеющего отверстия 25 мкм и диаметр 20 см при воздействии вибратора (например, Panabrator производства компании Panasonic Corporation) в течение 3 минут; массу частиц, прошедших через сито, и массу частиц, оставшихся на сите измеряют; содержание частиц, имеющих размер не более чем 25 мкм, вычисляют с применением следующего уравнения:
(содержание частиц, имеющих размер не более чем 25 мкм (%)) = (масса частиц, прошедших через сито)/{(масса частиц, прошедших через сито)+(масса частицы, оставшихся на сите)}×100
Содержание частиц, имеющих размер не более чем 25 мкм, измеряют после того, как часть сухого порошка «обожжена на воздухе при 400°C в течение 1 часа», поскольку сухой порошок предотвращается от растворения в воде. А именно, «обжиг на воздухе при 400°C в течение 1 часа» предназначен главным образом для измерения и не имеет никакого отношения к стадии обжига, описанной далее. Можно полагать, что размер частиц по существу не изменяется перед и после обжига. Степень восстановления обожженного образца может отличаться от ее величины для другого сухого порошка. Однако обычно количество образца является чрезвычайно малым количеством. Соответственно, даже если образец подается или же не подается на стадию обжига, описанную далее, это не влияет на функциональные возможности катализатора в целом. Объект, подвергаемый измерению в отношении среднего размера частиц, может не всегда быть сухим порошком. При необходимости, может быть измерен средний размер частиц предварительно обожженного продукта.
Примеры способа приготовления частиц, в которых содержание частиц, имеющих размер не более чем 25 мкм, составляет не более чем 20 масс.%, и средний размер частиц составляет от 35 до 75 мкм, включают способ регулирования условий распылительной сушки, таких как число оборотов вращения распылителя, температура распылительной сушки и количество подаваемого раствора смешанного исходного материала, и способ классификации сухого порошка. Способ классификации сухого порошка не ограничивается особым образом. Например, могут быть использованы обычные способы с применением, например, центробежного классификатора, воздушного классификатора, гравитационного классификатора, инерционного классификатора, сита и циклона. Среди классификаторов сухого и влажного типа, может быть подходящим образом использован классификатор сухого типа, с точки зрения предотвращения вымывания элементов, составляющих катализатор, в растворитель, и отсутствия неблагоприятного воздействия на функциональные возможности катализатора. С точки зрения увеличения количества получаемого катализатора, степень отбора сухого порошка при операции классификации составляет предпочтительно не менее чем 75 масс.% и более предпочтительно не менее чем 80 масс.%. Предпочтительно, выбирают и используют устройство, которое может быть отрегулировано до такого условия или удовлетворяет такому условию.
((III) Стадия предварительного обжига и (IV) стадия основного обжига)
Стадия (III) представляет собой стадию предварительного обжига сухого порошка, чтобы получить предварительно обожженный продукт.
Стадия (IV) представляет собой стадию основного обжига предварительно обожженного продукта, чтобы получить обожженный продукт, имеющий выступ на поверхности частиц.
В данном документе, стадию (III) и стадию (IV) называют в собирательном значении «стадией обжига» в некоторых случаях.
На стадиях (III) и (IV) обжигают сухой порошок, полученный на стадии сушки. Условия, такие как температура обжига, время и атмосфера, могут быть надлежащим образом определены с точки зрения удаления органических компонентов, содержащихся в сухом порошке, или роста кристаллов комплексного оксида, и не ограничиваются особым образом. В способе получения в соответствии с представляемым вариантом осуществления условие, такое как температура, изменяют, и обжиг на нескольких стадиях, таких как предварительный обжиг и основной обжиг, выполняют, как указано далее.
(Способ обжига сухого порошка)
В качестве обжигового устройства для обжига сухого порошка может быть использована, например, вращающаяся печь (вращающаяся обжиговая печь). Форма устройства для обжига, в котором обжигают сухой порошок, не ограничивается особым образом. Трубчатая форма (обжиговая труба) является предпочтительной, и цилиндрическая форма является особенно предпочтительной с точки зрения предоставления возможности непрерывного обжига. В качестве способа нагревания, внешнее нагревание является предпочтительным с точки зрения простоты регулирования температуры обжига в виде предпочтительного профиля повышения температуры. Соответственно, может быть использована электрическая печь. Размер и материал обжиговой трубы могут быть надлежащим образом выбраны в зависимости от условий обжига и объема производства. Внутренний диаметр обжиговой трубы составляет предпочтительно от 70 до 2000 мм и более предпочтительно от 100 до 1200 мм, с точки зрения предоставления равномерного распределения температуры обжига в слое катализатора и регулирования времени обжига и объема производства при надлежащей величине. Длина обжиговой трубы составляет предпочтительно от 200 до 10000 мм и более предпочтительно от 800 до 8000 мм, с точки зрения уменьшения времени задерживания сухого порошка и частиц предшественника катализатора внутри обжиговой трубы, а именно, распределения времени обжига, насколько это возможно, предотвращения деформации обжиговой трубы и регулирования времени обжига и объема производства при надлежащих величинах.
Когда ударное воздействие прикладывается к устройству для обжига (например, обжиговой трубе), толщина устройства для обжига составляет предпочтительно 2 мм или более и более предпочтительно 4 мм или более, с учетом того, что устройство для обжига должно иметь достаточную толщину, чтобы не ломаться при ударном воздействии. Толщина устройства для обжига составляет предпочтительно 100 мм или менее и более предпочтительно 50 мм или менее, с учетом того, что ударное воздействие в достаточной мере передается в устройство для обжига. Материал устройства для обжига не ограничивается особым образом, при условии, что устройство для обжига предпочтительно обладает термостойкостью и такой прочностью, что не ломается при ударном воздействии. Нержавеющая сталь (SUS) может быть использована подходящим образом в качестве материала обжиговой трубы.
В данном документе, «предшественник катализатора» относится к соединению, полученному на промежуточной ступени стадии обжига.
Перегородка, имеющая центральную часть с отверстием, через которое проходит порошок, размещена перпендикулярно (или по существу перпендикулярно) потоку порошка в обжиговой трубе, и тем самым обжиговая труба может быть также разделена на две зоны или более. Время выдерживания порошка в обжиговой трубе легко обеспечивается размещением такой перегородки. Перегородка может быть одна, или их может быть несколько. Материалом перегородки является предпочтительно металл, и перегородка, изготовленная из того же самого материала, что и обжиговая труба, может, соответственно, быть использована, с той точки зрения, что срок службы и термостойкость по отношению к атмосфере для обжига улучшены. Высота перегородки может быть отрегулирована в соответствии со временем выдерживания, которое должно быть обеспечено. Например, когда сухой порошок подают при 250 г/ч при применении барабанной печи с обжиговой трубой, имеющей внутренний диаметр 150 мм и длину 1150 мм и изготовленной из нержавеющей стали (SUS), высота перегородки составляет предпочтительно от 5 до 50 мм, более предпочтительно от 10 до 40 мм и еще более предпочтительно от 13 до 35 мм. Толщина перегородки не ограничивается особым образом и предпочтительно устанавливается в соответствии с размером обжиговой трубы. Например, в случае барабанной печи с обжиговой трубой, имеющей внутренний диаметр 150 мм и длину 1150 мм и изготовленной из нержавеющей стали (SUS), толщина перегородки составляет предпочтительно 0,3 мм или более и 30 мм или менее и более предпочтительно 0,5 мм или более и 15 мм или менее.
Для того, чтобы предотвратить растрескивание частиц сухого порошка и образование в них волосных трещин или т.п. и чтобы равномерно обжечь сухой порошок, обжиговую трубу предпочтительно вращают. Скорость вращения обжиговой трубы составляет предпочтительно от 0,1 до 30 об/мин, более предпочтительно от 0,5 до 20 об/мин и еще более предпочтительно от 1 до 10 об/мин. Во вращающейся печи, имеющей обжиговую трубу, печь предпочтительно имеет наклон оси вращения по отношению к вертикальному направлению. Более предпочтительно, вращающаяся печь наклонена таким образом, что сторона для подачи поднята, а сторона для выпуска порошка после его обжига опущена. Угол возвышения, если смотреть со стороны выпуска порошка, составляет предпочтительно не менее чем 0,3° и не более чем 15° и более предпочтительно не менее чем 0,3° и не более чем 5°.
Для обжига сухого порошка, предпочтительно, температуру нагревания сухого порошка повышают непрерывным или прерывистым образом до температуры в интервале от 550 до 700°C от температуры ниже чем 400°C.
Обжиг может проводиться в воздушной атмосфере или в потоке воздуха. Однако по меньшей мере часть обжига предпочтительно выполняется при протекании инертного газа, который по существу не содержит кислород, такого как азот, с точки зрения регулирования до предпочтительного состояния окисления/восстановления. В случае, когда обжиг выполняют в периодическом режиме, количество подаваемого инертного газа составляет не менее чем 50 норм.литров/ч на 1 кг сухого порошка, с точки зрения регулирования до предпочтительного состояния окисления и восстановления. Подаваемое количество инертного газа составляет предпочтительно от 50 до 5000 норм.литров/ч и более предпочтительно от 50 до 3000 норм.литров/ч. Здесь, «норм.литр» означает литр, измеренный при нормальной температуре и нормальном давлении, а именно, при 0°C и 1 атм.
В случае, когда обжиг выполняют непрерывным образом, количество подаваемого инертного газа составляет не менее чем 50 норм.литров на 1 кг сухого порошка, с точки зрения регулирования до предпочтительного состояния окисления и восстановления. Количество составляет предпочтительно от 50 до 5000 норм.литров и более предпочтительно от 50 до 3000 норм.литров. При этом потоки инертного газа и сухого порошка могут быть противоточными или прямоточными. Однако противоточное контактирование является предпочтительным, принимая во внимание газообразные компоненты, образующиеся из сухого порошка, и следовое количество воздуха, вовлеченного в сухой порошок. Особенно, в случае, когда на вышеуказанной стадии составления смеси исходных материалов используют способ добавления раствора пероксида водорода к водному смешанному раствору (A) и выполняют стадию окисления молибдена, ванадия и сурьмы в растворе до такой высокой степени окисления, насколько это возможно, чтобы получить раствор с составом исходного материала, сухой порошок предпочтительно обжигают при протекании инертного газа, по существу не включающего кислорода, такого как азот.
Помимо влаги, сухой порошок обычно содержит радикалы аммония, органические кислоты, неорганические кислоты и т.п. В случае, когда обжиг выполняют при протекании инертного газа, по существу, не включающего кислород, элементы, составляющие катализатор, восстанавливаются, когда радикалы аммония, органические кислоты неорганические кислоты и т.п. испаряются или разлагаются. В случае, когда элемент, составляющий, катализатор в сухом порошке, имеет почти максимальную степень окисления, для того, чтобы получить степень восстановления катализатора в желательном интервале, лишь восстановление выполняют на стадии обжига, и это несложно выполнить в промышленных масштабах.
С другой стороны, как описано далее, окисляющий компонент или восстанавливающий компонент может быть добавлен в атмосферу для обжига, чтобы получить степень восстановления предварительно обожженного продукта в желательном интервале. В способе получения в соответствии с представляемым вариантом осуществления, степень восстановления получаемого предварительно обожженного продукта составляет от 8 до 12%, предпочтительно от 9 до 11% и более предпочтительно от 9,5 до 11%. Предпочтительно, обжиг выполняют таким образом, что удельная поверхность обожженного продукта составляет от 7 до 20 м2/г. Удельная поверхность более предпочтительно составляет от 10 до 16 м2/г. Если удельная поверхность обожженного продукта составляет от 7 до 20 м2/г, то, вероятно, в результате могут быть достигнуты более высокая активность, подавление ухудшения и более высокий выход. Кроме того, эффект добавления соединения молибдена для того, чтобы поддержать выход в реакции окисления или реакции аммоксидирования, становится более значительным, и не проявляется его быстрое снижение. Соответственно, количество и частота добавления соединения молибдена могут быть уменьшены. Хотя причина этого не ясна, предполагают, что при удельной поверхности обожженного продукта меньше чем 7 м2/г активные компоненты, связанные с реакцией, имеют небольшую активную поверхность, и проявление эффекта добавления соединения молибдена затруднено. Также предполагают, что при удельной поверхности обожженного продукта больше чем 20 м2/г активные компоненты имеют большую активную поверхность, наряду с тем, что молибден быстро освобождается от активной поверхности. Фактор не ограничивается этим.
Степень восстановления предварительно обожженного продукта представлена следующим уравнением (D):
Степень восстановления (%)=(n0-n)/n0)×100... (D)
(где n представляет собой число атомов кислорода, которое удовлетворяет валентности составляющих элементов, иных, чем кислород, в предварительно обожженном продукте, и n0 представляет собой число атомов кислорода, требующихся, когда соответствующие составляющие элементы, иные, чем кислород, в предварительно обожженном продукте имеют наибольшую степень окисления).
Более конкретно, сухой порошок обжигают при условиях обжига следующим образом: температуру нагревания сухого порошка повышают от температуры ниже чем 400°C и непрерывным или прерывистым образом до температуры в интервале от 450 до 700°C. При этом условия обжига регулируют таким образом, что степень восстановления предварительно обожженного продукта, обжигаемого, когда температура нагревания достигает 400°C, составляет от 8 до 12%.
На степень восстановления предварительно обожженного продукта обычно влияет количество органических компонентов щавелевой кислоты, содержащихся в сухом порошке, количество радикалов аммония, производных от аммониевых солей в исходном материале, скорость повышения температуры в начале обжига, количество инертного газа в случае обжига в атмосфере инертного газа и температура и время в случае обжига в воздушной атмосфере. Для того чтобы получить степень восстановления предварительно обожженного продукта от 8 до 12%, важно повышать температуру при обжиге от температуры ниже чем 400°C и разлагать радикалы щавелевой кислоты и радикалы аммония в сухом порошке до почти полного образования газа; соответственно, степень восстановления предварительно обожженного продукта, обжигаемого, когда температура нагревания достигает 400°C, получают в интервале от 8 до 12%.
На удельную поверхность обожженного продукта влияет температура и время конечного обжига (нагревания) и количество кремнезема для поддерживания катализатора в случае, когда катализатор поддерживается на кремнеземе, и особенно сильно влияет степень восстановления, когда температура нагревания достигает 400°C, и конечная температура обжига. Обжиг на конечном этапе выполняют при температуре от 450°C до 700°C в течение от 0,5 часа до 20 часов. Когда конечная температура обжига выше, и время больше, то удельная поверхность меньше. Если степень восстановления, когда температура нагревания достигает 400°C, ниже, то удельная поверхность обожженного продукта, вероятнее всего, будет меньше. Если степень восстановления, когда температура нагревания достигает 400°C, выше, то удельная поверхность, вероятнее всего, будет больше. Хотя причина этого не ясна, в случае, когда обжиг выполняют в виде двух стадий, предварительного обжига и основного обжига, и температуру при основном обжиге поддерживают постоянной, то, чем выше наибольшая температура обжига при предварительном обжиге, тем больше удельная поверхность. Чем ниже наибольшая температура обжига при предварительном обжиге, тем меньше удельная поверхность.
В случае, когда обжиг выполняют посредством вращающейся обжиговой печи, количество подаваемого сухого порошка может быть отрегулировано во время обжиг, чтобы регулировать удельную поверхность обожженного продукта. Если подают небольшое количество сухой сухого порошка, то сухой порошок задерживается внутри системы в течение более длительного времени. По этой причине, восстановление сухого порошка прогрессирует под действием восстанавливающего газа, такого как аммиак, который образуется вследствие нагревания сухого порошка в обжиговой трубе. В результате, степень восстановления является более высокой, и удельная поверхность катализатора, получаемого после основного обжига, больше. Напротив, если подают большое количество сухого порошка, то степень восстановления является более низкой, и удельная поверхность катализатора меньше. В качестве альтернативы, удельная поверхность может быть отрегулирована посредством количества азота во время предварительного обжига. Если количество азота увеличивают, то газообразный компонент, который восстанавливает предварительно обожженный продукт во время обжига, быстро выпускается из системы. Соответственно, полагают, что предварительно обожженный продукт трудно восстановить, и это приводит к небольшой удельной поверхности обожженного продукта. Напротив, если количество азота уменьшают, то степень восстановления является более высокой, и удельная поверхность обожженного продукта больше.
Для того, чтобы получить удельную поверхность обожженного продукта от 7 до 20 м2/г, предпочтительно, степень восстановления, когда температура нагревания достигает 400°C, находится в интервале от 8 до 12%, и конечная температура обжига составляет от 450°C до 700°C.
Стадия обжига включает стадию предварительного обжига и стадию основного обжига. Предпочтительно, предварительный обжиг выполняют при температуре в интервале от 250 до 400°C, и основной обжиг выполняют при температуре в интервале от 450 до 700°C. Предварительный обжиг и основной обжиг могут быть выполнены последовательно; или же предварительный обжиг может быть завершен, и основной обжиг может быть выполнен заново. В качестве альтернативы, как предварительный обжиг, так и основной обжиг могут иметь несколько этапов.
В случае, когда степень восстановления предварительно обожженного продукта во время обжига измеряют, образец может быть извлечен из устройства для обжига при соответствующей температуре. Однако образец может контактировать с воздухом при высокой температура и окисляться, и степень восстановления может быть изменена. Предпочтительно, после охлаждения устройство для обжига до комнатной температуры, предварительно обожженный продукт извлекают из устройства для обжига и используют в качестве представительной выборки. Примеры способа регулирования степени восстановления, когда температура нагревания достигает 400°C, в желательном интервале, в частности, включают способ изменения температуры при предварительном обжиге, способ добавления окисляющего компонента, такого как кислород, к атмосфере во время обжига или способ добавления восстанавливающего компонента к атмосфере во время обжига. Кроме того, они могут быть использованы в комбинации.
Способ изменения температуры при предварительном обжиге представляет собой способ изменения степени восстановления, когда температура нагревания достигает 400°C, посредством изменения температуры при предварительном обжиге. Обычно, степень восстановления, вероятнее всего, должна быть уменьшена посредством снижения температуры при предварительном обжиге и увеличена посредством повышения температуры при предварительном обжиге. По этой причине, температура при предварительном обжиге может быть изменена, чтобы регулировать степень восстановления. Степень восстановления может также регулироваться посредством увеличения или уменьшения количества подаваемого азота в случае с применением азота, увеличения или уменьшения количества подаваемого сухого порошка, и увеличения или уменьшения числа оборотов вращения вращающейся обжиговой печи при обжиге с применением вращающейся обжиговой печи. Полагают, что если количество подаваемого азота увеличивают, то в окисленных компонентах, испаренных из сухого порошка посредством нагревания печью, доля окисленных компонентов, выпускаемых из системы без окисления оксидом металла, который присутствует в обжиговой печи, (при восстановлении оксида металла), становится выше, и поэтому обожженный продукт трудно восстановить. Также полагают, что, если количество подаваемого сухого порошка уменьшают, то восстановление прогрессирует во вращающейся обжиговой печи, поскольку катализатор задерживается в течение более длительного времени во вращающейся обжиговой печи. Также полагают, что в случае вращающейся обжиговой печи, если число оборотов ее вращения уменьшают, то скорость перемещения катализатора внутри вращающейся обжиговой печи уменьшается; по этой причине, восстановление прогрессирует, поскольку катализатор контактирует с увеличенным количеством окисленных компонентов в течение более длительного времени.
Способ добавления окисляющего компонента, такого как кислород, к атмосфере во время обжига для того, чтобы регулировать степень восстановления, когда температура нагревания достигает 400°C, в желательном интервале, является способом, который может быть использован, когда степень восстановления уменьшена. Обжиг на этой стадии является предварительным обжигом. На окисляющий компонент, добавляемый в атмосферу во время обжига, ссылаются как на окисляющий компонент в инертном газе, подаваемом в устройство для обжига. Количество добавляемого окисляющего компонента регулируют посредством концентрации инертного газа, подаваемого в устройство для обжига. Посредством добавления окисляющего компонента может регулироваться степень восстановления. В случае, когда окисляющим компонентом является кислород, воздух (или инертный газ, включающий воздух) может подаваться в устройство для обжига, и кислород в воздухе может быть использован в качестве окисляющего компонента.
Способ добавления восстанавливающего компонента к атмосфере во время обжига для того, чтобы регулировать степень восстановления, когда температура нагревания достигает 400°C, в желательном интервале, является способом, который может быть использован, когда степень восстановления увеличена. На восстанавливающий компонент, добавляемый в атмосферу во время обжига, ссылаются как на восстанавливающий компонент в инертном газе, подаваемом в устройство для обжига. Количество добавляемого восстанавливающего компонента регулируют посредством концентрации в инертном газе, подаваемом в устройство для обжига. Посредством добавления восстанавливающего компонента может регулироваться степень восстановления. Обычно примеры восстанавливающего компонента включают водород, синильную кислоту, метан, этан, пропан, монооксид углерода, монооксид азота и аммиак. Среди них, один из них может быть использован в отдельности, или же несколько газом могут быть использованы в комбинации. Среди них, предпочтительно добавляют газ, включающий аммиак в качестве основного компонента.
В случае, когда степень восстановления предварительно обожженного продукта, когда температура нагревания достигает 400°C, не является желательной степенью восстановления, общее количество необходимого окисляющего вещества или восстанавливающего вещества может быть вычислено из разницы между фактической степенью восстановления и желательной степенью восстановления и добавлено к атмосфере во время обжига. Окислительный компонент (например, кислород) или восстановительный компонент (например, аммиак) может быть добавлен в атмосферу для обжига при протекании инертного газа, если это необходимо, для того, чтобы регулировать степень восстановления. При определении степени восстановления величину (n0-n) в уравнении (2) получают окислительно-восстановительным титрованием образца с помощью KMnO4. Как для предварительно обожженного продукта перед завершением обжига, так и для обожженного продукта после завершения обжига величина (n0-n) может быть определены окислительно-восстановительным титрованием. При измерении окислительно-восстановительным титрованием, однако, условия для предварительно обожженного продукта перед завершением основного обжига отличаются от условий для обожженного продукта после завершения основного обжига. Для каждого из предварительно обожженного продукта перед завершением основного обжига и обожженного продукта после завершения основного обжига пример метода измерения представлен ниже.
Для предварительно обожженного продукта перед завершением обжига измерение выполняют следующим образом.
Примерно 200 мг предварительно обожженного продукта отвешивают и помещают в лабораторный стакан. Затем добавляют избыточное количество водного раствора KMnO4, имеющего известную концентрацию. Затем добавляют 150 мл чистой воды при 70°C и 2 мл раствора 1:1 серной кислоты (а именно, водного раствора серной кислоты, полученного смешиванием концентрированной серной кислоты с водой в объемном отношении 1/1) и закрывают лабораторный стакан предметным стеклом. Смешанный раствор перемешивают в горячей водяной бане при 70°C±2°C в течение 1 ч, чтобы окислить образец. При этом имеется избыток KMnO4, и непрореагировавший KMnO4 находится в растворе. В этом отношении, проверяют, чтобы цвет раствора был фиолетовым. После того, как окисление завершено, раствор фильтруют с помощью фильтровальной бумаги, чтобы получить общее количество фильтрата. Избыточное количество водного раствора оксалата натрия, имеющего известную концентрацию, добавляют к KMnO4, который находится в фильтрате, и нагревают и перемешивают таким образом, что температура раствора составляет 70°C. Проверяют, чтобы раствор стал бесцветным и прозрачным, и добавляют 2 мл раствора 1:1 серной кислоты. Перемешивание продолжают при поддержании температуры раствора при 70°C±2°C, и титруют с помощью водного раствора KMnO4, имеющего известную концентрацию. Когда цвет раствора поддерживается бледно-розовым в течение примерно 30 секунд посредством KMnO4, это является конечной точкой.
Из общего количества KMnO4 и общего количества Na2C2O4 определяют количество KMnO4, потребленного для окисления образца. Из данной величины рассчитывают (n0-n) и определяют степень восстановления на основании полученной величины.
Для обожженного продукта после завершения основного обжига измерение выполняют следующим образом.
Примерно 200 мг обожженного продукта, измельченного с помощью агатовой ступки, отвешивают и помещают в лабораторный стакан. Затем добавляют 150 мл чистой воды при 95°C и 4 мл раствора 1:1 серной кислоты (а именно, водного раствора серной кислоты, полученного смешиванием концентрированной серной кислоты с водой в объемном отношении 1/1). Перемешивание продолжают при поддержании температуры раствора при 95°C±2°C, и титруют с помощью водного раствора KMnO4, имеющего известную концентрацию. При этом, хотя цвет раствора временно становится фиолетовым вследствие титрования KMnO4, KMnO4 титруют медленно понемногу, таким образом, чтобы фиолетовый цвет не сохранялся в течение не менее чем 30 секунд. Количество раствора уменьшается вследствие испарения воды. По этой причине, чистую воду при 95°C добавляют таким образом, чтобы количество раствора поддерживалось постоянным. Когда цвет раствора поддерживается бледно-розовым в течение примерно 30 секунд посредством KMnO4, это является конечной точкой.
Соответственно, определяют количество KMnO4, потребленное для окисления образца. Из данной величины рассчитывают (n0-n) и определяют степень восстановления на основании полученной величины.
Помимо данного метода измерения, измерение может быть выполнено для предварительно обожженного продукта перед завершением основного обжига и обожженного продукта после завершения основного обжига следующим образом.
При условии, когда составляющие элементы в образце не улетучиваются и не теряются, образец нагревают до температуры выше, чем температура обжига, при которой предварительно обожженный продукт или обожженный продукт обжигают, и выполняют полное окисление кислородом. Увеличение массы (количество присоединенного кислорода) определяют. Из этого определяют величину (n0-n). На основании этого определяют степень восстановления.
Способ выполнения обжига в инертном газе или предпочтительной окислительно-восстановительной атмосфере не ограничивается особым образом. Предпочтительно, используют устройство для обжига, которое имеет надлежащую герметизируемую структуру и может в достаточной мере предотвращать контакт с наружным воздухом.
Предварительный обжиг выполняют предпочтительно при протекании инертного газа при температуре предварительного обжига в интервале предпочтительно от 250°C до 400°C и более предпочтительно от 300°C до 400°C, с точки зрения простоты регулирования катализатора для его получения в предпочтительном состоянии окисления и восстановления и улучшения функциональных возможностей катализатора. Предпочтительно, температуру при предварительном обжиге поддерживают при постоянной величине в интервале от 250°C до 400°C. Температура может быть изменена в интервале от 250°C до 400°C или умеренно повышаться или понижаться. Время удержания температуры нагревания составляет предпочтительно не менее чем 30 минут и более предпочтительно от 3 до 12 часов, с точки зрения простоты регулирования катализатора для его получения в предпочтительном состоянии окисления и восстановления и улучшения функциональных возможностей катализатора. В отношении профиля температуры, который достигает температуры при предварительном обжиге, температура может быть линейно возрастающей или же может возрастать в виде дугообразного профиля с выпуклостью в верхнем или в нижнем направлении. Кроме того, температура может быть понижена в какой-то момент во время повышения температуры, или же температура может быть неоднократно повышена и понижена. Кроме того, эндотермическая реакция происходит во время повышения температуры вследствие компонента, содержащегося в сухом порошке и/или предшественнике катализатора, и температура может быть временно понижена.
Средняя скорость повышения температуры во время повышения температуры до температуры при предварительном обжиге не ограничивается особым образом. Средняя скорость повышения температуры составляет обычно примерно от 0,1 до 15°C/мин, предпочтительно от 0,5 до 5°C/мин и более предпочтительно от 1 до 2°C/мин, с точки зрения простоты регулирования катализатора для его получения в предпочтительном состоянии окисления и восстановления и улучшения функциональных возможностей катализатора.
Основной обжиг может быть выполнен предпочтительно при протекании инертного газа при температуре предпочтительно от 450 до 700°C и более предпочтительно от 620 до 700°C, с точки зрения простоты регулирования катализатора для его получения в предпочтительном состоянии окисления и восстановления, достаточного формирования кристаллической структуры, активной в отношении реакции, и улучшения функциональных возможностей катализатора. Температуру обжига при основном обжиге (температуру основного обжига) предпочтительно поддерживают при постоянной величине в интервале от 620 до 700°C. Температура может быть изменена или умеренно повышаться или понижаться в интервале от 620 до 700°C. Кроме того, температура может быть понижена в какой-то момент во время повышения температуры, или же температура может быть неоднократно повышена и понижена. Эндотермическая реакция происходит во время повышения температуры вследствие компонента, содержащегося в предварительно обожженном продукте, и вследствие этого температура может быть понижена в профиле.
Выступ образуется на поверхности частиц обожженного продукта, подвергаемого стадии основного обжига. В результате исследования, проведенного авторами данного изобретения, выяснено, что, хотя состав выступа зависит от состава во время приготовления и/или условий обжига, относительно большое количество Mo и Sb содержится в составе выступа, и их отношение близко к Mo:Sb=10:2 или 10:4 (атомное отношение). Если выступ, имеющий такой состав, находится в реакторе при реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, то легко вызывается побочная реакция. Желательно удалять выступ перед реакцией. Соответственно, в способе получения в соответствии с представляемым вариантом осуществления, состав образуемого раствора с составом исходного материала рассчитывают в предположении, что выступ удаляют. Для того, чтобы оптимизировать состав катализатора, получаемого в качестве конечного продукта, важно в достаточной мере образовывать выступ на стадии обжига и в достаточной мере удалять выступ.
В соответствии с исследованием, проведенным авторами данного изобретения, степень выступания образуемого выступа имеет взаимосвязь с удельной поверхностью обожженного продукта. Хотя причина этого не ясна, количество выступов, вероятно, будет больше, когда удельная поверхность меньше. Как описано выше, если компонент выступов, который легко вызывает побочную реакцию, удаляют, то функциональные возможности катализатора улучшаются. В таком случае, чтобы в достаточной степени удалить выступ на стадии удаления выступа, описанной далее, предпочтительно, регулирование выполняют таким образом, что надлежащее количество выступов образуется на стадии обжига. Предпочтительно, регулирование выполняют таким образом, что комплексный оксид, имеющий такой же состав, что и выступ, не остается внутри катализатора, и обожженный продукт получают при небольшой площади поверхности и надлежащей удельной поверхности, при которой не уменьшены функциональные возможности катализатора. Катализатор, отрегулированный при надлежащей удельной поверхности, может поддерживать стабильным образом высокие функциональные возможности не только в течение короткого периода времени, но также и в течение длительного периода времени. Хотя причина этого не ясна, в случае, когда выступ не может быть в достаточной степени удален, полагают, что компоненты выступа, которые остаются внутри катализатора или на его поверхности, могут частично или полностью плавиться в ходе реакции, уменьшая текучесть катализатора или его функциональные возможности вследствие засорения пор на поверхности, поскольку компоненты выступа имеют низкую температуру плавления. В соответствии с наибольшей температурой обжига при основном обжиге и количеством предварительно обожженного продукта, подаваемого для основного обжига, удельная поверхность может быть отрегулирована. Если наибольшая температура обжига при основном обжиге повышается, спекание кремнеземистого компонента, содержащегося в предварительно обожженном продукте, прогрессирует. Соответственно, удельная поверхность уменьшается. Если количество подаваемого предварительно обожженного продукта уменьшается, то предварительно обожженный продукт задерживается во вращающейся обжиговой печи в течение более длительного времени. Соответственно, спекание кремнезема прогрессирует, и удельная поверхность уменьшается. Хотя причина этого не ясна, при чрезмерно малой удельной поверхности обожженного продукта количество выступов возрастает, что уменьшает объем производства. При чрезмерно большой удельной поверхности обожженного продукта количество выступов уменьшается, и функциональные возможности катализатора также уменьшаются. С вышеуказанной точки зрения, в способе получения в соответствии с представляемым вариантом осуществления удельную поверхность обожженного продукта регулируют предпочтительно от 7 до 20 м2/г и более предпочтительно от 10 до 16 м2/г.
Удельную поверхность определяют одноточечным методом БЭТ с применением Gemini 2360 (торговое наименование) производства компании Micrometrics Instrument Corporation.
Удельная поверхность обожженного продукта может быть отрегулирована посредством температуры обжига. Обожженный продукт, имеющий определенную удельную поверхность, может быть получен посредством увеличения или уменьшения температуры предварительного обжига или удельной поверхности. Предпочтительно, температуру обжига при основном обжиге, на которую легко влияет удельная поверхность, регулируют, чтобы получить обожженный продукт, имеющий целевую удельную поверхность.
Время основного обжига составляет предпочтительно от 0,5 до 20 часов и более предпочтительно от 1 до 15 часов. В случае, когда обжиговая труба разделена перегородкой, предварительно обожженный продукт и/или обожженный продукт постоянным образом проходит через по меньшей мере две зоны, предпочтительно от 2 до 20 зон и более предпочтительно от 4 до 15 зон, с точки зрения обеспечения времени задерживания сухого порошка или т.п. в обжиговой трубе. Температура может регулироваться с помощью одного или нескольких контроллеров. Для того чтобы получить желательный температурный профиль обжига, нагреватель и контроллер предпочтительно размещают в каждой из зон, разделенных этими перегородками, чтобы регулировать температуру. Например, в случае, когда семь перегородок установлены таким образом, что длина части обжиговой трубы, размещенной внутри нагревательной печи, разделена равным образом на восемь зон, и используется обжиговая труба, имеющая восемь разделенных зон, заданную температуру каждой из восьми зон предпочтительно регулируют нагревателем и контроллером, установленными в каждой из восьми зон, таким образом, что температуру предварительно обожженного продукта и/или продукта, обожженного при основном обжиге, регулируют при желательном температурном профиле обжига. Например, в случае, когда семь перегородок установлены таким образом, что длина части обжиговой трубы, размещенной внутри нагревательной печи, разделена равным образом на восемь зон, и используется устройство для обжига, имеющее восемь разделенных зон, регулирование может быть выполнена указанным ниже образом для того, чтобы получить желательный температурный профиль обжига. При предварительном обжиге, предпочтительно, температуру термопары, введенной в центральную часть предварительно обожженного продукта, который задерживается внутри каждой из зон в устройстве для обжига, регулируют таким образом, что температура составляет для зоны 1: от 120 до 280°C, зоны 2: от 180 до 330°C, зоны 3: от 250 до 350°C, зоны 4: от 270 до 380°C, зоны 5: от 300 до 380°C, зоны 6: от 300 до 390°C, зоны 7: от 320 до 390°C, и зоны 8: от 260 до 380°C от стороны подачи предварительно обожженного продукта. Аналогичным образом, при основном обжиге регулирование предпочтительно выполняют таким образом, что температура составляет для зоны 1: от 360 до 560°C, зоны 2: от 450 до 650°C, зоны 3: от 600 до 700°C, зоны 4: от 620 до 700°C, зоны 5: от 580 до 700°C, зоны 6: от 480 до 690°C, зоны 7: от 450 до 630°C, и зоны 8: от 370 до 580°C.
Здесь взят пример, в котором состав во время приготовления представляет собой Mo1V0,209Sb0,236Nb0,091W0,027Ce0,005Ox, сухой порошок, имеющий содержание кремнезема в качестве носителя 47,0 масс.% в расчете на общую массу сухого порошка постоянным образом обжигают с применением обжиговой трубы, и температура предварительного обжига составляет 350°C. Для того, чтобы получить обожженный продукт, имеющий удельную поверхность от 7 до 20 м2/г, температура при основном обжиге составляет предпочтительно от 600 до 700°C. Для того, чтобы получить обожженный продукт, имеющий удельную поверхность от 10 до 16 м2/г, температура при основном обжиге составляет предпочтительно от 640 до 700°C. Обожженный продукт, имеющий удельную поверхность от 7 до 20 м2/г, имеет большое количество частиц с выступом на их внешней поверхности, и большее количество выступов может быть удалено на стадии удаления выступа. Соответственно, состав катализатора, получаемого в качестве конечного продукта, легко регулируют до оптимальной величины в соответствии с расчетом.
Например, когда обжиговую трубу из нержавеющей стали (SUS), имеющую внутренний диаметр 500 мм, длину 4500 мм и толщину 20 мм и имеющую внутреннее пространство, разделенное перегородками на восемь частей, вращают при 6 об/мин, наибольшая температура обжига при предварительном обжиге составляет 350°C. Кроме того, в то время, как инертный газ (газообразный азот) протекает внутри системы в количестве 1050 норм.л/мин в целом, сухой порошок подают при расходе 35 кг/ч, чтобы выполнить предварительный обжиг. После этого, наряду с тем, что эту же самую обжиговую трубу из нержавеющей стали (SUS) вращают при 6 об/мин, ударные усилия прикладывают к трубе для основного обжига (обжиговой трубе для выполнения основного обжига) при 6 ударах/мин с применением молота или т.п., и наибольшая температура обжига при основном обжиге составляет 650°C. Кроме того, в то время, как инертный газ (газообразный азот) протекает внутри системы в количестве 667 норм.л/мин в целом, предварительно обожженный продукт подают при расходе 20 кг/ч, чтобы выполнить основной обжиг. В этом случае, степень восстановления предварительно обожженного продукта может составлять от 8 до 12%, и удельная поверхность обожженного продукта может составлять от 7 до 20 м2/г и более предпочтительно от 10 до 16 м2/г. В этом случае, если количество подаваемого сухого порошка или предварительно обожженного продукта изменяют, для того, чтобы получить степень восстановления предварительно обожженного продукта от 8 до 12% и удельную поверхность обожженного продукта от 7 до 20 м2/г и более предпочтительно от 10 до 16 м2/г, то, например, условия предпочтительно изменяют следующим образом.
Для предварительного обжига, будет представлен пример способа оптимизации в основном степени восстановления. В случае, когда количество подаваемого сухого порошка увеличивают от 35 кг/ч до 60 кг/ч, для того, чтобы получить степень восстановления от 8 до 12%, расход инертного газа, вводимого в систему, предпочтительно увеличивают до 1500-1800 норм.л/мин. В случае, когда количество подаваемого инертного газа не увеличивают, время задерживания сухого порошка в обжиговой трубе предпочтительно увеличивают, при сочетании способов, посредством увеличения высоты перегородки, увеличения числа перегородок, увеличения длины обжиговой трубы и увеличения внутреннего диаметра обжиговой трубы. Также, предпочтительно, увеличивают наибольшую температуру обжига при предварительном обжиге. Полагают, что если число оборотов вращения вращающейся обжиговой печи уменьшается, то для сухого порошка поблизости от поверхности раздела газа, протекающего в обжиговой трубе, занимает более длительное время перемещение в нижнюю часть слоя порошка, в которой затруднено контактирование сухого порошка с протекающим газом, и реакция восстановления посредством восстанавливающего газа легко протекает в верхней части слоя порошка. Соответственно, посредством уменьшения числа оборотов вращения вращающейся обжиговой печи степень восстановления предварительно обожженного продукта может также быть увеличена.
Напротив, в случае, когда количество подаваемого сухого порошка уменьшают от 45 кг/ч до 20 кг/ч, для того, чтобы получить степень восстановления от 8 до 12%, расход инертного газа, вводимого в систему, предпочтительно уменьшают до 400-800 норм.л/мин. В случае, когда количество подаваемого инертного газа не уменьшают, время задерживания сухого порошка в обжиговой трубе предпочтительно уменьшают посредством уменьшения высоты перегородки, уменьшения числа перегородок, уменьшения длины обжиговой трубы и уменьшения внутреннего диаметра обжиговой трубы. Также, предпочтительно, уменьшают наибольшую температуру обжига при предварительном обжиге. Полагают, что если число оборотов вращения вращающейся обжиговой печи увеличивается, то для сухого порошка поблизости от поверхности раздела газа, протекающего в обжиговой трубе, занимает более короткое время перемещение в нижнюю часть слоя порошка, в которой затруднено контактирование сухого порошка с протекающим газом, и затруднено протекание реакции восстановления посредством восстанавливающего газа в верхней части слоя порошка. Соответственно, посредством увеличения числа оборотов вращения вращающейся обжиговой печи степень восстановления предварительно обожженного продукта может также быть уменьшена.
Удельная поверхность предварительно обожженного продукта может быть отрегулирована до некоторой степени в соответствии с условиями предварительного обжига, однако не так значительно как в случае удельной поверхности обожженного продукта. Хотя причина этого не ясна, степень восстановления пропорциональна удельной поверхности, и посредством выполнения таких же операций, что указаны выше, легко оптимизируют интервал удельной поверхности. Однако регулирование удельной поверхности обожженного продукта в значительной степени зависит от способа обжига при основном обжиге.
Теперь далее будет представлен пример, в котором обожженный продукт, имеющий надлежащую удельную поверхность, получают из предварительно обожженного продукта, имеющего степень восстановления от 8 до 12%, при основном обжиге.
На стадии основного обжига, когда обжиговую трубу из нержавеющей стали (SUS), имеющую внутренний диаметр 500 мм, длину 4500 мм и толщину 20 мм и имеющую внутреннее пространство, разделенное перегородками на восемь частей, вращают при 6 об/мин, наибольшая температура обжига при основном обжиге составляет 650°C. Кроме того, в то время, как инертный газ (газообразный азот) протекает внутри системы в количестве 667 норм.л/мин в целом, предварительно обожженный продукт подают при расходе 20 кг/ч, чтобы выполнить основной обжиг. В этом случае, если удельную поверхность регулируют в интервале от 7 до 20 м2/г, то, например, операцию предпочтительно выполняют следующим образом. Например, в случае, когда количество предварительно обожженного продукта, подаваемого в трубу для основного обжига, уменьшают до 10 кг/ч, для того, чтобы поддерживать удельную поверхность в надлежащем интервале, расход инертного газа (газообразного азота) предпочтительно регулируют до 250-400 норм.л/мин. Кроме того, удельная поверхность может быть отрегулирована в целевом интервале более простым образом с помощью одного из способов уменьшения высоты перегородки, уменьшения диаметра обжиговой трубы, уменьшения длины обжиговой трубы, увеличения угла наклона предоставляемой обжиговой трубы (однако при более низком расположении выпускной стороны), уменьшения ширины области с наибольшей температурой внутри обжиговой трубы («область с наибольшей температурой» здесь относят к температурному интервалу 300-390°C во время предварительного обжига и температурному интервалу 600-700°C при основном обжиге) в продольном направлении и уменьшения наибольшей температуры при основном обжиге, или произвольной их комбинации. С другой стороны, в случае, когда количество предварительно обожженного продукта, подаваемого в трубу для основного обжига, увеличивают до 40 кг/ч, расход инертного газа (газообразного азота) предпочтительно регулируют до 1000-1600 норм.л/мин. Кроме того, удельная поверхность может быть отрегулирована в целевом интервале с помощью одного из способов увеличения высоты перегородки, увеличения диаметра обжиговой трубы, увеличения длины обжиговой трубы, уменьшения угла наклона предоставляемой обжиговой трубы (однако при более низком расположении выпускной стороны), уменьшения ширины области с наибольшей температурой внутри обжиговой трубы в продольном направлении, увеличения наибольшей температуры обжига при основном обжиге, или произвольной их комбинации. Таким образом, даже если условия изменяют при предварительном обжиге, условия при основном обжиге могут быть отрегулированы таким образом, чтобы поддерживать надлежащую степень восстановления и надлежащую удельную поверхность.
Температура обжига 650°C значительно превышает температуры плавления составляющих оксидов металлов. По этой причине, большое количество оксидов прилипает к поверхности стенки обжиговой трубы. Кроме того, время задерживания предварительно обожженного продукта предпочтительно увеличивают посредством приложения ударных усилий к трубе для основного обжига с применением молота или т.п., увеличения числа ударов или увеличения числа оборотов вращения вращающейся обжиговой печи (вращающейся печи). Увеличиваемые частота ударов или число оборотов вращения могут быть произвольным образом установлены на основании баланса масс между количеством предварительно обожженного продукта, подаваемого в трубу для основного обжига, и количеством обожженного продукта, выпускаемого из трубы для основного обжига. Окислительный компонент (например, кислород) или восстановительный компонент (например, аммиак) может быть добавлен в атмосферу для обжига в потоке инертного газа, если это необходимо.
В отношении профиля повышения температуры до температуры основного обжига, температура может быть линейно возрастающей или же может возрастать в виде дугообразного профиля с выпуклостью в верхнем или в нижнем направлении. Кроме того, температура может быть понижена в какой-то момент во время повышения температуры, или же температура может быть неоднократно повышена и понижена. Эндотермическая реакция происходит во время повышения температуры вследствие компонента, содержащегося в предварительно обожженном продукте, и вследствие этого температура может быть понижена в профиле.
Средняя скорость повышения температуры во время повышения температуры, при котором температура достигает температуры основного обжига, не ограничивается особым образом и предпочтительно составляет от 0,5 до 8°C/мин. Средняя скорость понижения температуры после завершения основного обжига, составляет предпочтительно от 0,05 до 50°C/мин и более предпочтительно от 0,05 до 20°C/мин, с точки зрения достаточного формирования кристаллической структуры, активной в отношении реакции, и улучшения функциональных возможностей катализатора.
Предпочтительно, температуру поддерживают однократно при температуре ниже, чем температура основного обжига, и отжиг выполняют с точки зрения достаточного формирования кристаллической структуры, активной в отношении реакции, и улучшения функциональных возможностей катализатора. Поддерживаемой температурой является температура, которая на 5°C, предпочтительно на 10°C и более предпочтительно на 50°C ниже, чем температура основного обжига. С той же самой точки зрения, что указана выше, время поддержания составляет предпочтительно не менее чем 0,5 часов, более предпочтительно не менее чем 1 час, еще более предпочтительно не менее чем 3 часа и особенно предпочтительно не менее чем 10 часов. Что касается стадии отжига, обожженный продукт может быть отожжен после основного обжига.
Когда основной обжиг выполняют сразу же после завершения предварительного обжига, низкотемпературная обработка может быть выполнена перед основным обжигом. Время, требующееся для низкотемпературной обработки, а именно, время, требующееся для снижения температуры предварительно обожженного продукта и/или обожженного продукта и повышения температуры до температуры обжига, может надлежащим образом регулироваться посредством размера, толщины и материалов устройства для обжига, количества производимого катализатора, последовательности периодов для непрерывного обжига предварительно обожженного продукта и/или обожженного продукта и постоянной скорости и постоянного количества или т.п. Время, необходимое для низкотемпературной обработки составляет предпочтительно 30 дней или менее, более предпочтительно 15 дней или менее, еще более предпочтительно 3 дня или менее и особенно предпочтительно 2 дня или менее во время последовательности непрерывного обжига обожженного продукта, с точки зрения удаления в достаточной степени предварительно обожженного продукта и/или обожженного продукта, прилипшего к поверхности стенки обжиговой трубы, стабильного поддержания температуры оксидного слоя и улучшения функциональных возможностей получаемого катализатора. Температура оксидного слоя относится к температуре, измеренной посредством термопары, введенной в предварительно обожженный продукт и/или продукт, обожженный при основном обжиге, в виде частиц, размещенный внутри устройства для обжига. Кроме того, например, когда предварительно обожженный продукт подают при расходе 35 кг/ч, при том, что вращающуюся обжиговую печь, имеющую обжиговую трубу с внутренним диаметром 500 мм, длиной 4500 мм и толщиной 20 мм, изготовленную из нержавеющей стали (SUS), вращают при 6 об/мин, и температура основного обжига составляет 645°C, стадия снижения температуры до 400°C и повышения температуры до 645°C может быть выполнена в течение примерно 1 дня после предварительного обжига. Когда обжиг выполняют непрерывно в течение 1 года, обжиг может быть выполнен посредством проведения такой низкотемпературной обработки раз в месяц, наряду с тем, что температура слоя оксида поддерживается стабильным образом.
Кроме того, если ударное воздействие прикладывают к устройству для обжига на стадии обжига, эффект растрескивания прилипших комков, вероятно, будет улучшен. В случае, когда выполняют низкотемпературную обработку, ударное воздействие, прикладываемое к устройству для обжига, является предпочтительным, поскольку растрескавшиеся комки, скорее всего, будут легко удаляться из устройства для обжига.
Ударное воздействие, прикладываемое к устройству для обжига, зависит от глубины слоя предварительно обожженного продукта, подаваемого в устройство для обжига, диаметра, длины, толщины и материала устройства для обжига (например, обжиговой трубы), материала, вида, формы и расположения устройства, к которому прикладывают ударное воздействие, и частоты приложения ударного воздействия. Предпочтительно, ударное воздействие, прикладываемое к устройству для обжига, устанавливают надлежащим образом в зависимости от этих параметров.
Вибрационное ускорение в положении, в котором прикладывают ударное воздействие (далее в данном документе, называемое точкой удара) составляет предпочтительно не менее чем 0,1 м/с2 и более предпочтительно не менее чем 10 м/с2, с точки зрения уменьшения в достаточной степени налипания объектов на внутреннюю поверхность стенки устройства для обжига. Кроме того, вибрационное ускорение составляет предпочтительно не более чем 3000 м/с2 и более предпочтительно не более чем 300 м/с2, с точки зрения предотвращения повреждения устройства для обжига и предотвращения нарушения потока порошка, протекающего внутри устройства для обжига.
В представляемом варианте осуществления «вибрационное ускорение» ударного воздействия, прикладываемого к устройству для обжига, означает среднее значение величин, измеренных в позициях, расположенных параллельно направлению потока порошка на расстоянии L/4, 3L/8 и L/2 от впускного отверстия для порошка в устройстве для обжига по отношению к полной длине L устройства для обжига в случае, когда устройством для обжига является обжиговая труба. Позиция для измерения является той же самой позицией, что и точка удара в направлении поперечного сечения устройства для обжига. Вибрационное ускорение может быть измерено виброметром, присоединенным к устройству для обжига. В качестве виброметра может быть использован MD220 (торговое наименование) производства компании ASAHI KASEI TECHNOSYSTEM CO., LTD.
Способ приложение ударных усилий не ограничивается особым образом, и пневматический стряхиватель, молот, ударное устройство и т.п. могут быть использованы соответствующим образом. Материал для части, непосредственно соприкасающейся с устройством для обжига на конце для приложения ударных усилий, не ограничивается особым образом, при условии, что материал обладает достаточной термостойкостью. Например, обычно могут быть использованы ударостойкие смолы и металлы. Среди них, предпочтительными являются металлы. Металлы, обладающие твердостью в той степени, что не повреждают и не деформируют устройство для обжига, являются предпочтительными. Соответственно, могут быть использованы медь и нержавеющая сталь (SUS). Место, к которому прикладывают ударное воздействие, не ограничивается особым образом, и ударное воздействие может быть приложено в любом месте, подходящем для функционирования. Предпочтительным является место, которое не закрыто нагревательной печью в устройстве для обжига, поскольку ударное воздействие может быть приложено непосредственно к устройству для обжига без потерь.
Место, к которому прикладывают ударное воздействие, может быть одним или несколькими местами. В случае, когда обжиговую трубу используют в качестве устройства для обжига, для того, чтобы эффективно проводить колебание, ударное воздействие предпочтительно прикладывают в направлении, перпендикулярном оси вращения обжиговой трубы. Частота приложения ударного воздействия не ограничивается особым образом. Предпочтительно, ударное воздействие постоянно прикладывают к устройству для обжига, поскольку налипшие объекты внутри устройства для обжига будут, скорее всего, удаляться в достаточной степени. Здесь, «ударное воздействие прикладывают постоянно» означает, что ударное воздействие прикладывают предпочтительно один раз не менее чем в 1 секунду и не более чем в 1 час, более предпочтительно один раз не менее чем в 1 секунду и не более чем в 1 минуту. Предпочтительно, частоту приложения ударного воздействия надлежащим образом регулируют в зависимости от вибрационного ускорения, глубины слоя предварительно обожженного продукта, подаваемого в устройство для обжига, диаметра, длины, толщины и материала устройства для обжига (например, обжиговой трубы), и материала, вида и формы устройства, к которому прикладывают ударное воздействие.
(V) Стадия удаления выступа
Стадия (V) в способе получения в соответствии с представляемым вариантом осуществления представляет собой стадию удаления выступа, который имеется на поверхности частиц обожженного продукта, посредством воздушного потока.
Выступ имеется на поверхности частиц обожженного продукта, подвергаемого стадии основного обжига. На стадии (V) выступ удаляют, и количество выступа, который имеет обожженный продукт, составляет предпочтительно не более чем 2 масс.% в расчете на общую массу обожженного продукта. В качестве способа удаления выступа может быть рассмотрено несколько способов. Среди них, предпочтительным является способ, в котором выступ удаляют контактированием обожженных продуктов в потоке газа. Примеры способа включают способ с протеканием газа в бункер или т.п., в котором хранят обожженный продукт, и способ с размещением обожженного продукта в реакторе с псевдоожиженным слоем и протеканием в нем газа. Способ с применением реактора с псевдоожиженным слоем является предпочтительным, поскольку отсутствует необходимость в каком-либо специальном устройстве для удаления выступа, однако он не является устройством, первоначально специально сконструированным для контактирования обожженных продуктов (катализаторов). Вероятно по этой причине, выступ может не быть в достаточной степени удален, в соответствии с условиями, такими как количество подаваемого обожженного продукта, время протекания обожженного продукта и количество газа, если не предприняты какие-либо меры, например, подача небольшого количества обожженного продукта, и требуется время для протекания обожженного продукта. В соответствии с исследованиями, проведенными авторами данного изобретения, воздушный поток при достаточной скорости может контактировать с обожженным продуктом, имеющим выступ, при эффективном удалении выступа. Если надлежащая скорость предоставляется в устройстве, имеющем конструкцию, в которой воздушный поток контактирует с обожженным продуктом, выступ может быть эффективно удален даже в больших масштабах.
Например, устройство может эффективно удалять выступ в больших масштабах, и данное устройство включает основной корпус, в котором размещается обожженный продукт, извлекающий узел для извлечения обожженного продукта, предоставленный в верхней части основного корпуса, и возвратный узел для возвращения обожженного продукта, соединенный с извлекающим узлом. Возвратный узел размещен таким образом, что его нижний конец находится в контакте с воздушным потоком. Часть обожженного продукта, контактирующего с воздушным потоком внутри основного корпуса, извлекают посредством извлекающего узла и возвращают в основной корпус посредством возвратного узла.
Газ протекает через устройство, заполненное обожженным продуктом, такое как реактор с псевдоожиженным слоем. Тем самым, обожженные продукты взаимно контактируют с удалением выступа. Выступ, удаленный от обожженного продукта, много меньше, чем обожженный продукт, и выводится протекающим газом из реактора с псевдоожиженным слоем. Предпочтительно, устройство заполняют обожженным продуктом таким образом, что плотность обожженного продукта при этом составляет от 300 до 1300 кг/м3. Площадь поперечного сечения основной части используемого устройства составляет предпочтительно от 0,1 до 100 м2 и более предпочтительно от 0,2 до 85 м2.
Протекающий газ является предпочтительно инертным газом, таким как азот, и воздухом. Линейная скорость (линейная скорость) газа, протекающего через основную часть устройства, заполненного обожженным продуктом, такого как бункер и реактор с псевдоожиженным слоем, составляет предпочтительно от 0,03 м/с до 5 м/с и более предпочтительно от 0,05 до 1 м/с. Время протекания газа составляет предпочтительно от 1 до 168 часов. Более конкретно, устройство для удаления выступа в соответствии с представляемым вариантом осуществления включает основной корпус, при этом обожженный продукт, размещенный в основном корпусе, контактирует с воздушным потоком, или частицы в текучем состоянии, создаваемом воздушным потоком, взаимно контактируют с удалением выступа на поверхности обожженного продукта от обожженного продукта. Предпочтительно, длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с в расчете на линейную скорость при 15°C и атмосферном давлении.
В случае, когда катализатор, используемый для реакции в псевдоожиженном слое, имеет выступ, или выступ, удаленный из обожженного продукта, и катализатор находятся вместе, текучесть катализатора, вероятно, будет уменьшена. Уменьшение текучести катализатора вызывает то, что катализатор будет локализован внутри реактора. Вследствие этого эффективность удаления тепла снижается. Тепло может накапливаться, вызывая аномальную реакцию, или же реакция разложения целевого продукта может происходить в случае некоторых реакций. Кроме того, в случае, когда часть выступа удаляют посредством контактирования обожженных продуктов внутри устройства для удаления выступа, такого как реактор с псевдоожиженным слоем, и выпускают за пределы системы из внутреннего пространства устройства, полагают, что выступ смешивается на последующей стадии, увеличивая нагрузку на данной стадии. Соответственно, предпочтительно, чтобы обожженный продукт и выступ не находились вместе внутри устройства.
Фиг. 1 представляет собой чертеж, схематически показывающий пример подходящего устройства для удаления выступа от обожженного продукта в больших масштабах. Устройство, показанное на Фиг. 1 включает основной корпус 1, впускную трубу 2 для газа, введенную через боковую стенку основного корпуса 1, и выпускную трубу 3, расположенную на верхней стенке основного корпуса 1 и соединенную с циклоном 4.
Основной корпус 1 имеет примерно цилиндрическую форму, и его нижняя часть имеет форму перевернутого конуса. В основном корпусе 1 размещается обожженный продукт. С точки зрения эффективного удаления выступа, размещаемый обожженный продукт предпочтительно помещают в основной корпус 1 в таком количестве, пока впускное отверстие для газа, расположенное в самой высокой позиции в вертикальном направлении впускной трубы 2 для газа внутри основного корпуса 1, не будет покрыто обожженным продуктом в состоянии, когда обожженный продукт неподвижен. Большое количество обожженного продукта может быть помещено в основной корпус 1. В этом случае, следует учитывать способность к отделению узла для отделения, такого как циклон.
Впускная труба 2 для газа введена горизонтально на высоте примерно половины от высоты основного корпуса 1. Как показано на Фиг. 2, впускная труба 2 для газа разветвлена вблизи центра основного корпуса 1 и, кроме того, вытянута вниз с образованием ответвленной трубы 21. В примере, показанном на Фиг. 1, несколько труб 21, ответвленных от впускной трубы 2 для газа вытянуто вниз в вертикальном направлении. Направление ответвленной трубы 21 не ограничивается этим, и может быть направлением вверх, направлениями вверх и вниз или горизонтальным направлением. Как показано в частично увеличенном виде на Фиг. 1, каждая из ответвленных труб 21 имеет несколько сопел 210. Газ, подаваемый через впускную трубу 2 для газа, выпускается из соответствующих сопел 210. Структура ответвленной трубы 21 не ограничивается той, что имеет сопла 210. Как показано в виде (A) на Фиг. 3, ответвленная труба 21 может иметь множество отверстий 211. Как показано в виде (B) на Фиг. 3, перпендикулярное дополнительное ответвление 22 может быть предоставлено в ответвленной трубе 21, и дополнительное ответвление 22 может иметь множество отверстий 220. Множество нижних сопел 6 для введения газа закреплено в конической нижней части основного корпуса. В примере, показанном на Фиг. 1, сопло 6 для введения газа имеет L-образную форму. После того как сопло 6 для введения газа введено под прямым углом в основной корпус, сопло 6 для введения газа открыто с наклоном вниз. По этой причине, обожженный продукт, находящийся внутри основного корпуса, протекает под действием газа, вводимого из сопла 6, в направлении к нижней части основного корпуса 1. Нижний конец основного корпуса 1 открыт и соединен с впускной трубой 7 для второго газа. Соответственно, обожженный продукт, собираемый в нижнем конце посредством газа, подаваемого из сопла 6 для введения газа, протекает внутрь основного корпуса 1 посредством газа, подаваемого из впускной трубы 7 для второго газа. Форма переднего конца 61 сопла для введения газа не ограничивается L-формой и может быть I-формой. В качестве альтернативы, сопло, выступающее от внутренней поверхности основного корпуса 1, может не быть предоставлено, и поверхность стенки может быть открытой. В случае L-образного сопла, сопло может не быть открыто вниз и может быть надлежащим образом установлено с учетом газа, подаваемого из другой впускной трубы 7 для второго газа, или формы основного корпуса 1. Например, сопло может быть открыто вверх или в горизонтальном направлении.
Один конец выпускной трубы 3 присоединен к центральной части верхней поверхности основного корпуса 1, а другой ее конец соединен с циклоном 4. Циклон 4 отделяет обожженный продукт от выступов, удаляемых из обожженного продукта посредством центробежной силы. Относительно большие частицы обожженного продукта, от которых отделен выступ, проходят из нижнего конца циклона через возвратную трубу 5 для возвращения в основной корпус 1. С другой стороны, поскольку выступы являются легкими, то выступы проходят через выпускной трубопровод 8, открытый на верхней стороне циклона, и удаляются. Предпочтительно, фильтр (не показан) предоставлен ниже по течению в выпускном трубопроводе 8, чтобы улавливать удаляемые выступы.
Устройство, показанное на Фиг. 4, имеет такую же конструкцию, как то, что показано в примере на Фиг. 1, за исключением того, что трубопровод 71 для циркуляции обожженного продукта предоставлен в нижнем конце основного корпуса 1. Другой конец трубопровода 71 для циркуляции открыт на боковой поверхности основного корпуса 1. Соответственно, пневматический узел или т.п. установлен в трубопроводе 71, чтобы перемещать обожженный продукт, втекающий в трубопровод 71 для циркуляции, с помощью газа и возвращать обожженный продукт в основной корпус 1.
Устройство, показанное на Фиг. 5, имеет такую же конструкцию, как то, что показано в примере на Фиг. 1, за исключением того, что второй циклон 42 соединен с выпускной трубой 41 первого циклона 4. Возвратная труба 51, установленная в нижнем конце первого циклона 4 и возвратная труба 52, установленная в нижнем конце второго циклона 42, соединены с боковой стенкой основного корпуса 1, и извлеченный обожженный продукт возвращается в основной корпус 1.
Устройство, показанное на Фиг. 6, включает основной корпус 1, имеющий двухслойную структуру, включающую внешнюю трубу 11 и внутреннюю трубу 12, и газ вводится между внешней трубой 11 и внутренней трубой 12 из впускной трубы 2 для газа. За исключением этого, устройство, показанное на Фиг. 6 имеет примерно такую же конструкцию, как то, что на Фиг. 1, и лишь различия будут описаны ниже. Внутренняя труба 12 имеет множество отверстий 13, и газ, подаваемый между внешней трубой 11 и внутренней трубой 12, выпускается в основной корпус 1 из отверстий 13. Внутренняя труба 12 открыта в выпускную трубу 3 и возвратную трубу 5, в то время как внешняя труба 11 не соединена с выпускной трубой 3 и возвратной трубой 5. Соответственно, обожженный продукт не вводится в пространство между внешней трубой 11 и внутренней трубой 12 и проходит через выпускную трубу 3 для введения в циклон 4 и возвращается из возвратной трубы 5 в основной корпус 1. Впускная труба 7 для второго газа открыта лишь во внутреннюю трубу 12. Тем самым, надлежащее количество газа может быть подано из впускной трубы 7 для газа, для того, чтобы предотвратить задерживание обожженного продукта в нижней части основного корпуса 1.
В примерах, показанных на Фиг. 6, впускная труба 2 для газа, имеющая множество ответвленных труб 21, не предоставлена. В случае, когда устройство включает основной корпус 1, имеющий двухслойную структуру, впускная труба 2 для газа, имеющая ответвленные трубы 21, может также быть предоставлена, как показано на Фиг. 1.
Устройство, показанное на Фиг. 7, включает выпускную трубу 3 и возвратную трубу 5, каждая из которых имеет двухслойную структуру. За исключением этого, устройство, показанное на Фиг. 7 имеет примерно такую же конструкцию, как то, что на Фиг. 1, и лишь различия будут описаны ниже. Выпускная труба 3 включает внешнюю трубу 31 и внутреннюю трубу 32, и газ подается между этими трубами из сопла 33. Возвратная труба 5 включает внешнюю трубу 53 и внутреннюю трубу 54, и газ подается между этими трубами из сопла 55. Устройство, показанное на Фиг. 7 может быть использовано в комбинации с устройством, показанным на Фиг. 6.
Предпочтительно, число отверстий для протекания воздушного потока на единицу обожженного продукта является большим, с точки зрения увеличения эффективности контакта между газом и выступом. Отверстие для протекания воздушного потока может быть предоставлено посредством непосредственного формирования отверстия в стенке основного корпуса для размещения обожженного продукта, чтобы приводить обожженный продукт в контакт с воздушным потоком, или же отверстие для протекания воздушного потока может быть предоставлено в основном корпусе с помощью трубопровода или трубы посредством образования отверстия в трубопроводе или в трубе. Однако, в случае, когда воздушные потоки контактируют друг с другом, обожженные продукты могут взаимно контактировать, и обожженный продукт может иметь трещины или сколы. Вследствие этого, предпочтительно проектировать конструкцию таким образом, чтобы воздушные потоки не пересекались. С точки зрения предотвращения растрескивания обожженного продукта и износа трубопровода и основного корпуса, предпочтительно, воздушный поток не контактирует непосредственным образом со стенками трубы и основного корпуса.
При этом отверстие для протекания воздушного потока означает отверстие, через которое воздушный поток поступает в основной корпус, в котором длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80,0 м/с и не более чем 500 м/с в расчете на линейную скорость при 15°C и атмосферном давлении. Например, отверстие для протекания воздушного потока относится к отверстиям, представленным цифровым обозначением 210 на Фиг. 1, цифровыми обозначениями 211 и 220 на Фиг. 3, цифровым обозначением 13 на Фиг. 6 и цифровыми обозначениями 32 и 54 на Фиг. 7.
Что касается размера отверстия для протекания воздушного потока, его диаметр составляет предпочтительно примерно от 0,04 мм до 20 см и более предпочтительно примерно от 0,04 мм до 5 см. Отверстие для протекания может иметь любой профиль. Кроме того, диаметр отверстия для воздушного потока может не быть одинаковым. Помимо этого, число отверстий для протекания воздушного потока является предпочтительно большим, однако полагают, что взаимно контактирующие обожженные продукты будут растрескиваться в случае, когда отверстия расположены на расстоянии, при котором воздушные потоки взаимно контактируют, как описано выше. Соответственно, желательно, учитывая размер воздушного потока, длину воздушного потока, объем воздушного потока и т.п., рассчитанные посредством уравнений в соответствии с литературным источником (1) Horio et al. и литературным источником (2) YATES et al. ниже, предоставлять интервал между отверстиями для протекания потока, чтобы предотвратить взаимное контактирование обожженных продуктов. При этом, с точки зрения эффективности контакта и текучести катализатора, длина воздушного потока в направлении протекания воздушного потока желательно составляет не менее чем 55 мм, при условии, что воздушный поток не контактирует с устройством, таким как стенка основного корпуса и труба. Длина воздушного потока может быть вычислена при использовании уравнения YATES et al., и размер воздушного потока может быть вычислен при использовании уравнения Horio et al.
Длина воздушного потока представлена следующим уравнением:
hj/dor=21.2·(uor2/(g·dp))0.37·(dor·uor·ρg/µ)0.05×(ρg/ρp)0.68·(dp/dor)0.24·dor,
где hj: длина воздушного потока [м], dor: диаметр отверстия [м], dp: размер частиц катализатора [м], uor: скорость в отверстии [м/с], µ: вязкость газа [кг·м/с], ρg: плотность газа [кг/м3], ρp: плотность частиц катализатора [кг/м3], g: ускорение силы тяжести [м/с2].
С другой стороны, размер воздушного потока представлен следующим уравнением:
(dj/dor)=1.56·(fj·Frj)/(k0.5·tanϕr))0.3·(dor/lor)0.2,
где dj: размер воздушного потока [м], fj: 0.02 (константа), Frj=ρg·uor2/((1-εmf)·ρp·dp·g), k=(1-sinϕr)/(1+sinϕr), ϕr: угол естественного откоса катализатора (здесь, близкий к 30°), lor: шаг [м].
(1) Horio, M., T. Yamada, and I. Muchi: Preprints of the 14th Fall Meeting of Soc. of Chem. Engrs., Japan, p. 760 (1980)
(2) Yates, J.G., P.N. Rowe and D.J. Cheesman: AIChE J., 30, 890 (1984).
Скорость воздушного потока вычисляют из площади отверстия для протекания воздушного потока и расхода газа. Для того чтобы эффективно удалять выступ с поверхности обожженного продукта, средняя скорость воздушных потоков, выпускаемых из соответствующих отверстий для потока, составляет не менее чем 80 м/с и не более чем 500 м/с и предпочтительно не менее чем 200 м/с и не более чем 341 м/с в расчете на линейную скорость при 15°C и атмосферном давлении.
При этом, расход выпуска Y (м3/ч) и скорость воздушного потока u (м/с) могут быть определены посредством уравнений ниже, где внутреннее давление трубопровода сопла представляет собой a (кг/см2 (изб.давл.)), давление сопла представляет собой b (кг/см2 (изб.давл.)), температура газа в это время представляет собой k (°C), давление представляет собой p (кПа) и площадь отверстия для протекания воздушного потока представляет собой S (м2). Определенная линейная скорость может быть усреднена, чтобы определить среднюю скорость воздушного потока.
[Выражение 3]
[Выражение 4]
Время, в течение которого газ контактирует с обожженным продуктом при линейной скорости, указанной выше, составляет предпочтительно не менее чем 10 часов и не более чем 100 часов. При времени контактирования меньше чем 10 часов, выступ, вероятно, останется на поверхности обожженного продукта. При времени контактирования больше чем 100 часов, поверхность обожженного продукта, вероятно, будет ободрана, что уменьшает технологическую эффективность катализатора. Время контактирования между газом и обожженным продуктом составляет предпочтительно не менее чем 15 часов и не более чем 60 часов. Для того, чтобы улучшить циркуляцию обожженного продукта и более эффективно удалять выступ, может быть предоставлен, например, механизм, в котором обожженный продукт перемещается и циркулирует посредством пневматического узла или т.п. для контактирования с воздушным потоком. В качестве альтернативы, вращающийся элемент в виде пропеллера или элемент в виде вращающегося стержня может быть введен в основной корпус устройства, и приведен во вращение, чтобы перемешивать обожженный продукт. Тем самым, эффективность контакта между обожженным продуктом и воздушным потоком может быть увеличена.
На стадии удаления выступа в соответствии с представляемым вариантом осуществления является предпочтительной комбинация способов, в которой газ (воздушный поток) протекает с высокой скоростью, и воздушный поток контактирует с обожженным продуктом, чтобы привести обожженный продукт в текучее состояние; и в то же самое время, выступ на поверхности обожженного продукта удаляется сдвиговым усилием воздушного потока и одновременно удаляется взаимным контактированием движущихся частиц обожженного продукта. При этом может быть использован любой вид газа. Предпочтительным является сухой воздух или инертный газ, такой как азот.
При этом авторы данного изобретения считают, что величина, полученная посредством умножения объема воздушного потока (V) на число отверстий для протекания воздушного потока (K), отражает общий объем воздушного потока, который может придать скорость обожженному продукту, и полагают, что величина, полученная умножением этой величины на квадрат скорости воздушного потока (u), эквивалентна общей энергии, используемой для удаления выступа. Затем, наряду с тем, что соответствующие переменные изменяли независимым образом, количество остающихся выступов в обожженном продукте, при количестве обожженного продукта M, измеряли с течением времени. Взаимосвязь между величиной, полученной делением u2×V×K на количество обожженного продукта (M), и временем, необходимым для удаления достаточного количества выступов с поверхности обожженного продукта, анализировали. В результате, u2×V×K/M является величиной, примерно обратно пропорциональной времени, которое необходимо для обработки. Соответственно, это позволяет предположить, что u2×V×K/M подходит в качестве показателя энергии (далее в данном документе на u2×V×K/M также делается ссылка как на «величину, преобразуемую в энергию» в некоторых случаях).
В случае, когда катализатор производят в промышленном масштабе, предпочтительно, время, требуемое для каждой из стадий, находится в пределах постоянного интервала, принимая во внимание легкость обработки. Что касается стадии удаления выступа, то, если время обработки устанавливают таким образом, что стадия выполняется, например, в течение одного дня, технологический процесс становится удобнее. Как описано выше, величина, преобразуемая в энергию, примерно обратно пропорциональна времени, требуемого для обработки при удалении. Соответственно, если величина, преобразуемая в энергию, возрастает до некоторой степени, то время, требуемое для обработки при удалении, которое примерно обратно пропорционально величине, преобразуемой в энергию, может находиться в пределах предпочтительного времени или менее. С точки зрения регулирования времени, необходимого для стадии удаления выступа, до одного дня или менее, авторы данного изобретения исследовали предпочтительную величину, преобразуемую в энергию, экспериментально. Было найдено, что u2, V, K, и M предпочтительно устанавливают таким образом, чтобы величина, преобразуемая в энергию, u2×V×K/M (м5/с2/кг), представленная при использовании скорости воздушного потока u (м/с), объема (объема воздушного потока) V (м3), образованного воздушным потоком, который протекает через отверстие для протекания воздушного потока, числа отверстий K для протекания воздушного потока и массы M (кг) обожженного продукта, размещенного внутри системы, удовлетворяла следующему выражению:
14<u2×V×K/M
С другой стороны, с точки зрения предотвращения растрескивания обожженного продукта при контактировании обожженных продуктов и/или обожженного продукта с поверхностью стенки системы, было найдено, что соответствующие численные величины предпочтительно устанавливают таким образом, чтобы величина, преобразуемая в энергию, удовлетворяла следующему выражению:
u2×V×K/M<100
А именно, было найдено, что если уравнение (X):
14<u2×V×K/M<100 (X)
выполняется, то время обработки на стадии удаления может находиться в пределах постоянного интервала, и разламывание обожженного продукта может быть предотвращено; поэтому выступ может быть эффективно удален с поверхности обожженного продукта. Подходящее значение для величины, преобразуемой в энергию, зависит от характеристик устройства для удаления и изменяется в зависимости от формы и размера устройства, направления сопла и контактирования со стенкой. В случае устройства с параметрами для применения в промышленных масштабах, 20<u2×V×K/M<90 является более предпочтительной, и 30<u2×V×K/M<80 является еще более предпочтительной.
Выступ, удаляемый от обожженного продукта устройством для удаления выступа, много меньше, чем сферический катализатор и выпускается вместе с протекающим газом. Соответственно, выступ может быть захвачен фильтром или т.п. В то же время, однако, тонкие (однако больше, чем выступ) частицы катализатора могут быть захвачены фильтром. Соответственно, предпочтительно используют узел для отделения, такой как циклон, чтобы увеличить эффективность отделения. Может быть предоставлено несколько узлов для отделения, таких как циклон, и разные узлы для отделения могут быть использованы в комбинации. Принимая во внимание случай, в котором смесь катализатора, имеющего частицы малого размера, и выступов или т.п. возвращается из циклона в основной корпус, например, трехходовой клапан или т.п. может быть предоставлен в нижней части циклона, и может быть предоставлен механизм для раздельного извлечения смеси для выведения из системы. Отделенный компонент катализатора снова перемещают внутрь основного корпуса. При этом, предпочтительно, компонент катализатора возвращают в место, в котором катализатор снова контактирует с воздушным потоком. Например, в случае, когда весь поток газа в заключение направляется вверх, полагают, что катализатор перемещается вверх вместе с потоком газа. По этой причине, предпочтительно, отверстие для возврата отделенного катализатора предоставляют ниже отверстия для воздушного потока. В случае, когда выступ имеет большой угол естественного откоса, или выступ обладает вязкостью, выступ может прилипать к поверхности стенки внутри основного корпуса и прилипать к трубопроводу, засоряя трубопровод. По этой причине, предпочтительно, стряхиватель, продувочный воздух или т.п. надлежащим образом вводят в систему. Кроме того, для того, чтобы удалить выступы, прилипшие к трубопроводу, может быть предоставлен механизм для промывки с применением жидкости, такой как вода и спирт.
В случае, когда выступ удаляют в граммовом масштабе, может быть использовано следующее устройство. А именно, может быть использована вертикальная труба, включающая пластину с одним или несколькими отверстиями в нижней части и бумажный фильтр в верхней части. Обожженный продукт помещают в вертикальную трубу и пропускают воздух со стороны нижней части вертикальной трубы. Тем самым, воздушные потоки протекают через соответствующие отверстия, что способствует контактированию обожженных продуктов и удалению выступа.
В способе получения в соответствии с представляемым вариантом осуществления, после того, как выступ удален, получаемый комплексный оксидный катализатор содержит комплексный оксид, имеющий состав, представленный формулой (1):
Mo1VaSbbNbcWdZeOn... (1)
(в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr, Yb, Y, Sc, Sr и Ba; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,4, 0,1≤b≤0,4, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, атомное отношение a/c составляет 1,4<a/c<2,3).
В способе получения в соответствии с представляемым вариантом осуществления, в зависимости от состава раствора с составом исходного материала во время приготовления, обычно, посредством удаления выступа, содержание Mo, V и Sb уменьшается, и атомное отношение a/b удовлетворяет условию 0,85≤a/b<1,0, и атомное отношение a/c удовлетворяет условию 1,4<a/c<2,3. Если атомное отношение a/b удовлетворяет условию 0,85≤a/b<1,0, выход существенно не уменьшается посредством сгорания пропана, образуемого избыточным количеством V, и другая кристаллическая фаза, образуемая избыточным количеством Sb, не увеличивается существенным образом. Предпочтительно, 0,88≤a/b<1,0, и более предпочтительно, 0,90≤a/b<1,0. Кроме того, полагают, что, если атомное отношение a/c удовлетворяет условию 1.4<a/c<2.3, промотируется увеличение кристаллической реакционной области для надлежащего аммоксидирования. Если количество Nb невелико, то кристаллическая область для реакции аммоксидирования уменьшается. Если количество Nb чрезмерно велико, то может промотироваться рост кристаллической фазы, иной, чем реакционная область. Предпочтительно, 1,5<a/c<2,3 и более предпочтительно 1,6<a/c<2,3.
Размер кристаллитов катализатора составляет предпочтительно от 20 до 250 нм. При размере кристаллитов меньше чем 20 нм, вследствие того, что кристаллические частицы малы, активные центры в катализаторе также уменьшаются. Кроме того, поскольку ожидается, что даже те, что могут являться по существу активными компонентами, приобретают другую кристаллическую форму или становятся аморфными, то, вероятнее всего, будет вызываться побочная реакция. При размере кристаллитов катализатора более чем 250 нм, одна из плоскостей активных компонентов катализатора представляет собой активный компонент катализатора, а друга их плоскость промотирует побочную реакцию. По этой причине, чрезмерно большой катализатор будет, вероятнее всего, промотировать побочную реакцию. С такой точки зрения, размер кристаллитов катализатора составляет более предпочтительно от 40 до 150 нм.
Обычно размер кристалла может быть измерен с применением рентгеновской дифракции. В данном документе, размер кристаллитов катализатора (L) представляет собой величину, определенную с применением RINT2500 VHF (торговое наименование) производства компании Rigaku Corporation в соответствии с уравнением Шерера, представленным ниже.
L=Kλ/(βCosθ)
Здесь, K представляет собой константу 0,9. λ представляет собой длину волны рентгеновского излучения и равна 1,5418 Å. β представляет собой полуширину при данном угле (единица измерения: радианы) и величину, полученную вычитанием полуширины B для совершенного и хорошо выращенного кристалла (0,1 используется в данном приборе) из фактической полуширины b. θ и β представляют собой входные величины в радианах. Рост кристаллических частиц в значительной степени зависит от температуры обжига и состава катализатора. Если состав является одинаковым, то размер кристаллитов активных компонентов катализатора может быть увеличен посредством повышения температуры обжига. Кроме того, посредством медленного уменьшения скорости снижения температуры, когда температура понижается от температуры основного обжига, размер кристаллитов становится больше. С другой стороны, если температура снижается быстро, то размер кристаллитов меньше. Кроме того, посредством условий окисления/восстановления во время обжига и состава во время приготовления исходного материала, например, увеличения содержания такого элемента, как Nb, размер кристаллитов становится меньше. Соответственно, посредством регулирования различных условий может быть отрегулирован размер кристаллитов.
Комплексный оксидный катализатор в соответствии с представляемым вариантом осуществления содержит комплексный оксид, имеющий состав, представленный формулой (1), на этапе перед тем, как комплексный оксидный катализатор подают к реакции каталитического окисления в паровой фазе или реакции каталитического аммоксидирования в паровой фазе пропана или изобутана. После того как каталитическая реакция началась, соотношение металлических компонентов изменяется посредством сублимации металлического элемента, такого как Mo, или катализатор добавляется заново. Однако для того, чтобы продемонстрировать результат предпочтительной реакции на начальной стадии и при длительной реакции, важно иметь состав, представленный формулой (1) на этапе перед тем, как реакция началась. А именно, поскольку оптимизированный состав комплексного оксида перед началом реакции является составом, представленным формулой (1), комплексный оксид может иметь данный состав во время начала реакции, даже если состав позднее изменяется до другого состава.
Реакция каталитического окисления в паровой фазе и реакция каталитического аммоксидирования в паровой фазе
Реакция каталитического окисления в паровой фазе в соответствии с представляемым вариантом осуществления представляет собой способ получения ненасыщенной кислоты, соответствующей пропану или изобутану, посредством реакции каталитического окисления в паровой фазе пропана или изобутана, в которой используют комплексный оксидный катализатор.
Кроме того, реакция каталитического аммоксидирования в паровой фазе в соответствии с представляемым вариантом осуществления представляет собой способ получения ненасыщенного нитрила, соответствующего пропану или изобутану, посредством реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, в которой используют комплексный оксидный катализатор.
Пропан, изобутан и аммиак, являющиеся подаваемыми исходными материалами, не всегда должны быть высокочистыми материалами, и может быть использован газ технического сорта. В качестве источника подаваемого кислорода может быть использован воздух, чистый кислород или воздух, обогащенный чистым кислородом. Кроме того, в качестве разбавляющего газа могут подаваться гелий, неон, аргон, газообразный диоксид углерода, пар и азот и т.п.
Реакция каталитического окисления в паровой фазе пропана или изобутана может быть выполнена при следующих условиях.
Молярное отношение кислорода, подаваемого в реакцию, к пропану или изобутану составляет предпочтительно от 0,1 до 6 и более предпочтительно от 0,5 до 4. Температура реакции составляет предпочтительно от 300 до 500°C и более предпочтительно от 350 до 500°C. Давление реакции составляет предпочтительно от 5×104 до 5×105 Па и более предпочтительно от 1×105 до 3×105 Па. Время контактирования составляет предпочтительно от 0,1 до 10 (с·г/см3), и более предпочтительно от 0,5 до 5 (с·г/см3).
В представляемом варианте осуществления время контактирования определяют посредством уравнения, приведенного ниже:
Время контактирования (с·г/см3)=(W/F)×273/(273+T)
При этом W, F и T определяются следующим образом:
W = количество загруженного катализатора (г)
F = расход исходного смешанного газообразного материала (норм.куб.см/с) в стандартном состоянии (0°C, 1,013×105 Па)
T = температура реакции (°C)
Реакция каталитического аммоксидирования в паровой фазе пропана или изобутана может быть выполнена при следующих условиях.
Молярное отношение кислорода, подаваемого в реакцию, к пропану или изобутану составляет предпочтительно от 0,1 до 6 и более предпочтительно от 0,5 до 4. Молярное отношение аммиака, подаваемого в реакцию, к пропану или изобутану составляет предпочтительно от 0,3 до 1,5 и более предпочтительно от 0,7 до 1,2. Температура реакции составляет предпочтительно от 350 до 500°C и более предпочтительно от 380 до 470°C. Давление реакции составляет предпочтительно от 5×104 до 5×105 Па и более предпочтительно от 1×105 до 3×105 Па. Время контактирования составляет предпочтительно от 0,1 до 10 (с·г/см3), и более предпочтительно от 0,5 до 5 (с·г/см3).
В качестве способа выполнения реакции для реакции окисления в паровой фазе и реакции аммоксидирования в паровой фазе может быть использован обычный способ, такой как с неподвижным слоем, псевдоожиженным слоем и подвижным слоем. Предпочтительным является реактор с псевдоожиженным слоем, в котором легко удаляется теплота реакции. Реакция каталитического аммоксидирования в паровой фазе может представлять собой либо однопоточную систему, либо систему с рециклингом.
Примеры
Далее в данном документе представляемый вариант осуществления будет дополнительно описан подробно со ссылками на примеры и сравнительные примеры. Однако представляемый вариант осуществления не ограничивается данными примерами.
В примерах и сравнительных примерах степень конверсии пропана или изобутана и выход акрилонитрила или метакрилонитрила, соответственно, определяются указанным ниже образом.
Степень конверсии пропана (%) = (Число молей прореагировавшего пропана)/(Число молей поданного пропана)×100
Выход акрилонитрила (AN)(%) = (Число молей произведенного акрилонитрила)/(Число молей поданного пропана)×100
Метод измерения коэффициента восстановления предварительно обожженного продукта
Примерно 200 мг предварительно обожженного продукта точно отвешивали в лабораторном стакане. Водный раствор KMnO4 с известной концентрацией добавляли к нему в избыточном количестве. Затем дополнительно добавляли 150 мл чистой воды при 70°C и 2 мл раствора 1:1 серной кислоты (а именно, водного раствора серной кислоты, полученного смешиванием концентрированной серной кислоты с водой в объемном отношении 1/1; далее в данном документе, то же самое). После этого лабораторный стакан закрывали предметным стеклом, и раствор перемешивали в горячей водяной бане при 70°C±2°C в течение 1 часа, чтобы окислить образец. При этом поскольку KMnO4 присутствовал в избыточном количестве, и непрореагировавший KMnO4 присутствовал в растворе, было подтверждено, что цвет раствора был пурпурным. После завершения окисления раствор фильтровали с помощью фильтровальной бумаги и извлекали все количество фильтрата. Водный раствор оксалата натрия (Na2C2O4), концентрация которого была известна, добавляли в избыточном количестве по отношению к KMnO4, присутствующему в фильтрате, и нагревали и перемешивали таким образом, что температура раствора становилась равной 70°C. Убеждались в том, что, что раствор становился бесцветным и прозрачным, и затем добавляли 2 мл раствора 1:1 серной кислоты. Раствор перемешивали постоянным образом при температуре раствора, поддерживаемой при 70°C±2°C, и титровали с помощью водного раствора KMnO4, концентрация которого была известна. При этом момент времени, при котором считалось, что цвет раствора становился бледно-розовым посредством KMnO4 и сохранялся в течение примерно 30 секунд, принимали в качестве конечного момента.
Количество KMnO4, необходимое для окисления образца, определяли из общего количества KMnO4 и общего количества Na2C2O4. (n0-n) рассчитывали из данной величины, и коэффициент восстановления определяли на основании (n0-n).
Метод измерения удельной поверхности обожженного продукта
Удельную поверхность обожженного продукта определяли одноточечным методом по БЭТ с применением Gemini 2360 (торговое наименование) производства компании Micromeritics Instrument Corp.
Приготовление смешанного раствора ниобия
Смешанный раствор ниобия приготавливали описанным ниже способом.
К 10 кг воды добавляли 1,530 кг ниобиевой кислоты, содержащей 79,8 масс.% ниобия в расчете на Nb2O5, и 5,266 кг дигидрата щавелевой кислоты [H2C2O4·2H2O]. Молярное соотношение щавелевая кислота/ниобий в качестве исходных материалов составляло 5,0, и концентрация исходного ниобия составляла 0,50 (молей Nb/кг раствора). Результирующий раствор нагревали в течение двух часов при 95°C при перемешивании, посредством чего получали смешанный раствор, в котором был растворен ниобий. Этот смешанный раствор оставляли для выдерживания, охлаждали льдом и подвергали вакуумной фильтрации для удаления твердотельного вещества, посредством чего получали равномерный смешанный раствор ниобия. Молярное соотношение щавелевая кислота/ниобий в этом смешанном растворе ниобия составляло 2,68 в соответствии с анализом, описанным ниже.
10 г этого смешанного раствора ниобия точно отвешивали и помещали в тигель, сушили в течение ночи при 95°C и подвергали термообработке в течение одного часа при 600°C, посредством чего получали 0,7895 г Nb2O5. Из этого следовало, что концентрация ниобия составляла 0,594 (молей Nb/кг раствора). 3 г этого смешанного раствора ниобия точно отвешивали и помещали в стеклянный лабораторный стакан емкостью 300 мл, добавляли 200 мл горячей воды, имеющей температуру примерно 80°C, и затем добавляли 10 мл серной кислоты (1:1). Результирующий смешанный раствор титровали при использовании 1/4 н раствора KMnO4 при перемешивании и поддержании при температуре 70°C на обогреваемой мешалке. Момент времени, при котором бледно-розовый цвет, обусловленный KMnO4, сохранялся в течение примерно 30 секунд или более, определяли как конечный момент. Концентрацию щавелевой кислоты определяли на основании результирующего титра в соответствии с приведенной ниже формулой и, в качестве результата, она составляла 1,592 (молей щавелевой кислоту/кг).
2KMnO4+3H2SO4+5H2C2O4→K2SO4+2MnSO4+10CO2+8H2O
Полученный смешанный раствор ниобия (B0) использовали в качестве раствора ниобиевого исходного материала при получении композитного оксидного катализатора в Примерах с 1 по 14, описанных ниже.
Пример 1
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
К 1,557 кг воды добавляли 432,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 59,9 г метаванадата аммония [NH4VO3], 84,3 г оксида сурьмы (III) [Sb2O3] и дополнительно 4,8 г нитрата церия [Ce(NO3)3·6H2O] и нагревали в течение 1 часа при 95°C при перемешивании, посредством чего приготавливали водный раствор исходного материала (A1).
К 378,4 г смешанного раствора ниобия (B0) добавляли 66,3 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали в течение 10 минут при комнатной температуре при перемешивании, посредством чего приготавливали водный раствор исходного материала (B1).
После охлаждения полученного водного раствора исходного материала (A1) до 70°C к нему добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего результирующую смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку (источником тепла для сушки является воздух; далее в данном документе, аналогично) и сушили, получая таким образом микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,2 масс.%, и средний размер частиц составлял 54 мкм. Содержание частиц и средний размер частиц измеряли посредством LS230 (торговое наименование) производства компании BECKMAN COULTER, Inc. (далее в данном документе, аналогично).
Обжиг сухого порошка (E1)
Полученный сухой порошок (E1) подавали в количестве 80 г/ч в цилиндрическую обжиговую трубу для непрерывного обжига, изготовленную из нержавеющей стали (SUS) и имеющую диаметр (внутренний диаметр; далее в данном документе, аналогично) 3 дюйма (7,62 см) и длину 89 см, во вращающейся печи. Газообразный азот при 1,5 норм.л/мин пропускали как в направлении, противоположном направлению подачи сухого порошка (т.е., в противотоке; далее в данном документе, аналогично), так и в том же самом направлении (т.е., в прямотоке; далее в данном документе, аналогично), и общий расход составлял 3,0 норм.л/мин. Пневматический стряхиватель предоставляли на любом конце обжиговой трубы, изготовленной из нержавеющей стали (SUS), и частоту ударов пневматического стряхивателя устанавливали таким образом, что делать 10 ударов в минуту. Давление воздуха во впускном отверстии пневматического стряхивателя устанавливали таким образом, что вибрационное ускорение на поверхности обжиговой трубы, изготовленной из нержавеющей стали (SUS), посредством ударов становилось равным 50 м/с2. Вибрационное ускорение измеряли с применением виброметра (MD-220 (торговое наименование) производства компании Asahi Kasei Technosystem Co., Ltd.; далее в данном документе, аналогично). Предварительный обжиг выполняли посредством регулирования температуры печи таким образом, что температуру повышали до 370°C, что являлось максимальной температурой обжига, на протяжении четырех часов и поддерживали при 370°C в течение одного часа, в то время как обжиговую трубу вращали при скорости 4 об/мин. Небольшое количество предварительно обожженного продукта извлекали на выходе обжиговой трубы в качестве образца; данный образец нагревали до 400°C в атмосфере азота и после этого измеряли коэффициент восстановления, который составлял 10,2%. Извлеченный предварительно обожженный продукт подавали в количестве 60 г/ч в цилиндрическую обжиговую трубу для непрерывного обжига, изготовленную из нержавеющей стали (SUS) и имеющую диаметр 3 дюйма (7,62 см) и длину 89 см, во вращающейся печи. Газообразный азот при 1,1 норм.л/мин пропускали как в направлении, противоположном направлению подачи сухого порошка, так и в том же самом направлении, и общий расход составлял 2,2 норм.л/мин. Пневматический стряхиватель предоставляли на любом конце обжиговой трубы, изготовленной из нержавеющей стали (SUS), и частоту ударов пневматического стряхивателя устанавливали таким образом, что делать 10 ударов в минуту. Давление воздуха во впускном отверстии пневматического стряхивателя устанавливали таким образом, что вибрационное ускорение на поверхности обжиговой трубы, изготовленной из нержавеющей стали (SUS), посредством ударов становилось равным 50 м/с2. Вибрационное ускорение измеряли с применением виброметра. Основной обжиг выполняли посредством регулирования температуры печи таким образом, что температуру повышали до 680°C на протяжении двух часов, поддерживали при 680°C в течение двух часов и снижали до 300°C на протяжении восьми часов. Удельную поверхность обожженного продукта (F1), полученного на выходе обжиговой трубы измеряли, и она составляла 14,0 м2/г. Удельную поверхность обожженного продукта определяли одноточечным методом по БЭТ с применением Gemini 2360 (торговое наименование) производства компании Micromeritics Instrument Corp. (далее в данном документе, аналогично).
Удаление выступов
50 г обожженного продукта (F1) загружали в вертикальную трубу (внутренний диаметр: 41,6 мм, длина: 70 см), в которой перфорированный диск, имеющий три отверстия диаметром 1/64 дюйма (0,4 мм), был размещен в нижней части трубы, и бумажный фильтр был размещен в ее верхней части. Затем воздух пропускали при его циркулировании при комнатной температуре от нижней стороны вверх в вертикальной трубе через отверстия, способствуя тем самым взаимному контактированию обожженного продукта. Длина воздушного потока в направлении протекания воздуха при этом составляла 56 мм, и средняя линейная скорость воздушного потока составляла 332 м/с. Выступы отсутствовали в композитном оксидном катализаторе (G1), полученном после 24 часов.
Отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли с помощью рентгенофлуоресцентного анализа (прибор: RINT 1000 (торговое наименование), производства компании Rigaku Corp.; трубка Cr, напряжение на трубке: 50 кВ, ток трубки: 50 мА; далее в данном документе, аналогично). Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Рентгеновскую дифрактометрию композитного оксидного катализатора (G1) выполняли с применением Рентгеновского дифрактометра (RINT 2500 VHF (торговое наименование), производства компании Rigaku Corp.; трубка Cu, напряжение на трубке: 40 кВ, ток трубки: 200 мА; далее в данном документе, аналогично). При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 52 нм.
Реакция аммоксидирования пропана
Пропан подвергали реакции аммоксидирования в паровой фазе представленным ниже способом, при использовании полученного, как указано выше, оксидного катализатора. 35 г композитного оксидного катализатора помещали в реакционную трубу с псевдоожиженным слоем из викорового стекла, имеющую внутренний диаметр 25 мм; и смешанный газ, имеющий молярное соотношение пропан:аммиак:кислород:гелий = 1:1:3:18, подавали при времени контактирования 2,8 (с·г/см3), при температуре реакции 440°C и при давлении реакции, являющемся нормальным давлением. Степень конверсии пропана после реакции составляла 89,2%, и выход акрилонитрила составлял 55,5%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 55,4%.
Пример 2
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
К 2,202 кг воды добавляли 611,5 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 84,7 г метаванадата аммония [NH4VO3], 119,3 г оксида сурьмы (III) [Sb2O3] и дополнительно 6,8 г нитрата церия [Ce(NO3)3·6H2O] и нагревали в течение 1 часа при 95°C при перемешивании, посредством чего приготавливали водный раствор исходного материала (A1).
К 535,5 г смешанного раствора ниобия (B0) добавляли 93,8 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали при перемешивании в течение 10 минут при комнатной температуре, посредством чего приготавливали водный раствор исходного материала (B1).
После охлаждения полученного водного раствора исходного материала (A1) до 70°C к нему добавляли 429,7 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего результирующую смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 42,8 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 112,5 г порошка кремнезема было диспергировано в 1519 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала. Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,3%, и удельная поверхность обожженного продукта после основного обжига составляла 12,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,03Ce0,005On/30,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,2%, и выход акрилонитрила составлял 54,5%.
Пример 3
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
К 1,027 кг воды добавляли 285,4 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 39,5 г метаванадата аммония [NH4VO3], 55,7 г оксида сурьмы (III) [Sb2O3] и дополнительно 3,2 г нитрата церия [Ce(NO3)3·6H2O] и нагревали в течение одного часа при 95°C при перемешивании, посредством чего приготавливали водный раствор исходного материала (A1).
К 249,9 г смешанного раствора ниобия (B0) добавляли 43,8 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали при перемешивании в течение 10 минут при комнатной температуре, посредством чего приготавливали водный раствор исходного материала (B1).
После охлаждения полученного водного раствора исходного материала (A1) до 70°C к нему добавляли 1117,2 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего результирующую смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 20,4 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 292,5 г порошка кремнезема было диспергировано в 3948 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 56 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,5%, и удельная поверхность обожженного продукта после основного обжига составляла 17,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,03Ce0,005On/68,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,8%, и выход акрилонитрила составлял 55,1%.
Пример 4
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
К 1,806 кг воды добавляли 432,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 69,4 г метаванадата аммония [NH4VO3], 90,1 г оксида сурьмы (III) [Sb2O3] и дополнительно 4,8 г нитрата церия [Ce(NO3)3·6H2O] и нагревали в течение одного часа при 95°C при перемешивании, посредством чего приготавливали водный раствор исходного материала (A1).
К 504,5 г смешанного раствора ниобия (B0) добавляли 88,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и смешивали при перемешивании в течение 10 минут при комнатной температуре, посредством чего приготавливали водный раствор исходного материала (B1).
После охлаждения полученного водного раствора исходного материала (A1) до 70°C к нему добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2, и затем добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, после чего результирующую смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,5 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,0%, и удельная поверхность обожженного продукта после основного обжига составляла 12,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,240Sb0,250Nb0,120W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,5%, и выход акрилонитрила составлял 55,2%.
Пример 5
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,5 масс.%, и средний размер частиц составлял 58 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения температуры предварительного обжига до 360°C и изменения общего расхода азота при предварительном обжиге до 7,5 норм.л/мин. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,0%, и удельная поверхность обожженного продукта после основного обжига составляла 12,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,8%, и выход акрилонитрила составлял 55,1%.
Пример 6
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,7 масс.%, и средний размер частиц составлял 54 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения общего расхода азота при предварительном обжиге до 2,3 норм.л/мин (каждый из противоточного и прямоточного потоков составлял 1,15 норм.л/мин). Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,3%, и удельная поверхность обожженного продукта после основного обжига составляла 14,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,0%, и выход акрилонитрила составлял 55,1%.
Пример 7
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,2 масс.%, и средний размер частиц составлял 53 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения общего расхода азота при основном обжиге до 3,2 норм.л/мин (каждый из противоточного и прямоточного потоков составлял 1,6 норм.л/мин). Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,3%, и удельная поверхность обожженного продукта после основного обжига составляла 13,6 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,6%, и выход акрилонитрила составлял 55,1%.
Пример 8
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения общего расхода азота при основном обжиге до 1,0 норм.л/мин (каждый из противоточного и прямоточного потоков составлял 0,5 норм.л/мин). Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,1%, и удельная поверхность обожженного продукта после основного обжига составляла 12,4 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,1%, и выход акрилонитрила составлял 55,1%.
Пример 9
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,1 масс.%, и средний размер частиц составлял 49 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения частоты вращения обжиговой трубы при предварительном обжиге до 1 об/мин. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,2%, и удельная поверхность обожженного продукта после основного обжига составляла 15,6 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,2%, и выход акрилонитрила составлял 54,0%.
Пример 10
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,6 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге 330°C, частоты вращения обжиговой трубы до 12 об/мин и общего расхода азота при предварительном обжиге до 6,0 норм.л/мин (каждый из противоточного и прямоточного потоков составлял 3,0 норм.л/мин). Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,8%, и удельная поверхность обожженного продукта после основного обжига составляла 12,1 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 90,0%, и выход акрилонитрила составлял 54,2%.
Пример 11
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,5 масс.%, и средний размер частиц составлял 58 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения подаваемого количества сухого порошка (E1) для предварительного обжига до 72 г/ч. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,5%, и удельная поверхность обожженного продукта после основного обжига составляла 14,3 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,9%, и выход акрилонитрила составлял 54,9%.
Пример 12
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 60 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения подаваемого количества сухого порошка (E1) для предварительного обжига до 89 г/ч. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 12,2 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,8%, и выход акрилонитрила составлял 54,6%.
Пример 13
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 62 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения подаваемого количества предварительно обожженного продукта для основного обжига до 36 г/ч. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,3%, и удельная поверхность обожженного продукта после основного обжига составляла 12,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,4%, и выход акрилонитрила составлял 55,2%.
Пример 14
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 58 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения подаваемого количества предварительно обожженного продукта для основного обжига до 84 г/ч. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,4%, и удельная поверхность обожженного продукта после основного обжига составляла 14,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,4%, и выход акрилонитрила составлял 54,3%.
Пример 15
Приготовление раствора ниобиевого исходного материала
Смешанный раствор ниобиевого исходного материала приготавливали описанным ниже способом. 72,2 кг ниобиевой кислоты, содержащей 77,9 масс.% Nb2O5, и 267 кг дигидрата щавелевой кислоты [H2C2O4·2H2O] смешивали в 500 кг воды. Молярное соотношение щавелевая кислота/ниобий в качестве исходных материалов составляло 5,0, и концентрация исходного ниобия составляла 0,552 (молей Nb/кг раствора).
Результирующий раствор нагревали в течение одного часа при 95°C при перемешивании, посредством чего получали водный раствор, в котором было растворено ниобиевое соединение. Этот водный раствор оставляли для выдерживания, охлаждали льдом и подвергали вакуумной фильтрации для удаления твердотельного вещества, посредством чего получали равномерный водный раствор ниобиевого соединения. Ту же самую операцию повторяли несколько раз, и полученные водные растворы ниобиевого соединения объединяли, получая таким образом раствор ниобиевого исходного материала. Молярное соотношение щавелевая кислота/ниобий в растворе ниобиевого исходного материала составляло 2,40 в соответствии с анализом, описанным ниже.
10 г этого раствора ниобиевого исходного материала точно отвешивали и помещали в тигель, сушили в течение ночи при 95°C и подвергали термообработке в течение одного часа при 600°C, посредством чего получали 0,835 г Nb2O5. Из этого следовало, что концентрация ниобия составляла 0,590 (молей Nb/кг раствора).
3 г этого раствора ниобиевого исходного материала точно отвешивали и помещали в стеклянный лабораторный стакан емкостью 300 мл, добавляли 200 мл горячей воды, имеющей температуру примерно 80°C, и затем добавляли 10 мл серной кислоты (1:1). Результирующий раствор титровали при использовании 1/4 н раствора KMnO4 при перемешивании и поддержании при температуре 70°C на обогреваемой мешалке. Момент времени, при котором бледно-розовый цвет, обусловленный KMnO4, сохранялся в течение примерно 30 секунд или более, определяли как конечный момент. Концентрацию щавелевой кислоты определяли на основании результирующего титра в соответствии с приведенной ниже формулой и, в качестве результата, она составляла 1,50 (молей щавелевой кислоту/кг).
2KMnO4+3H2SO4+5H2C2O4→K2SO4+2MnSO4+10CO2+8H2O
Данную стадию повторяли, и полученные растворы ниобиевого исходного материала использовали в качестве растворов ниобиевого исходного материала при последующем получении композитных оксидных катализаторов.
Приготовление сухого порошка
30,24 кг гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 4,19 кг метаванадата аммония [NH4VO3], 5,52 кг оксида сурьмы (III) [Sb2O3] и, кроме того, раствор, в котором 371 г нитрата церия [Ce(NO3)3·6H2O] растворено в 26 кг воды, добавляли к 100 кг воды и нагревали при перемешивании в течение одного часа при 95°C, получая таким образом водный смешанный раствор (A-1).
3,42 кг раствора пероксида водорода, содержащий 30 масс.% H2O2, добавляли к 29,9 кг раствора ниобиевого исходного материала. Смесь перемешивали и смешивали при температуре раствора, поддерживаемой при примерно 20°C, получая таким образом водный раствор (B-1).
Полученный водный смешанный раствор (A-1) охлаждали до 70°C, и после этого добавляли 56,55 кг золя кремниевой кислоты, содержащий 32,0 масс.% SiO2. Затем добавляли 6,44 кг раствора пероксида водорода, содержащего 30 масс.% H2O2, и перемешивали и смешивали в течение одного часа при 50°C; и после этого 2,38 кг водного раствора метавольфрамата аммония растворяли и затем добавляли водный раствор (B-1). Кроме того, добавляли раствор, в котором 14,81 кг тонкодисперсного кремнезема диспергировали в 214,7 кг воды, и выдерживали в течение 2,5 часа при 50°C при перемешивании, получая таким образом водный смешанный раствор (C1) в виде суспензии в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Для того чтобы выполнить стадию «обжига сухого порошка (E1)», описанную далее, в системе непрерывного действия, данную стадию повторяли 38 раз, посредством чего приготавливали в целом примерно 2600 кг сухого порошка (D1).
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,8 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Полученный сухой порошок (E1) подавали при расходе 20 кг/ч в цилиндрическую обжиговую трубу для непрерывного обжига во вращающейся печи, изготовленную из нержавеющей стали (SUS), имеющую диаметр 500 мм, длину 3500 мм и толщину 20 мм и снабженную семью перегородками, имеющими высоту 150 мм, так что длина секции для нагревания была разделена на восемь равных секций. Газообразный азот пропускали при 600 норм.л/мин в обжиговой трубе, и температуру нагревательной печи регулировали таким образом, чтобы иметь температурный профиль, при котором температуру повышали до 370°C на протяжении примерно четырех часов и поддерживали при 370°C в течение трех часов, в то время как обжиговую трубу вращали при 4 об/мин, чтобы подвергнуть сухой порошок предварительному обжигу, получая таким образом предварительно обожженный продукт. Для полученного предварительно обожженного продукта выполняли обжиг при тех же самых условиях основного обжига, что указаны в Примере 1. Коэффициент восстановления предварительно обожженного продукта составлял 10,2%, и удельная поверхность обожженного продукта после основного обжига составляла 13,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксидного катализатора (G1) выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 48 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,1%, и выход акрилонитрила составлял 55,0%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 55,3%.
Пример 16
Раствор ниобиевого исходного материала приготавливали таким же способом, что и способ приготовления, использованный в Примере 15.
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же способом, что и в Примере 15.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 53 мкм.
Обжиг сухого порошка (E1)
Полученный сухой порошок (E1) подавали при расходе 20 кг/ч в цилиндрическую обжиговую трубу для непрерывного обжига во вращающейся печи, изготовленную из нержавеющей стали (SUS), имеющую диаметр 500 мм, длину 3500 мм и толщину 20 мм и снабженную семью перегородками, имеющими высоту 150 мм, так что длина секции для нагревания была разделена на восемь равных секций. Газообразный азот пропускали при 600 норм.л/мин в обжиговой трубе, и температуру нагревательной печи регулировали таким образом, чтобы иметь температурный профиль, при котором температуру повышали до 370°C на протяжении примерно четырех часов и поддерживали при 370°C в течение трех часов, в то время как обжиговую трубу вращали при 4 об/мин, чтобы подвергнуть сухой порошок предварительному обжигу, получая таким образом предварительно обожженный продукт. Затем предварительно обожженный продукт подавали при расходе 15 кг/ч в другую цилиндрическую обжиговую трубу для непрерывного обжига во вращающейся печи, изготовленную из нержавеющей стали (SUS), имеющую диаметр 500 мм, длину 3500 мм и толщину 20 мм и снабженную семью перегородками, имеющими высоту 150 мм, так что длина секции для нагревания была разделена на восемь равных секций, при том, что трубу вращали при 4 об/мин. При этом, наряду с тем, что часть обжиговой трубы на впускной стороне для предварительно обожженного продукта (часть, которая не закрыта нагревательной печью) подвергали ударному воздействию один раз каждые пять секунд с высоты 250 мм над обжиговой трубой в направлении, перпендикулярном оси вращения, посредством молота массой 14 кг, конец ударной части которого был изготовлен из нержавеющей стали (SUS), и протекании газообразного азота при 500 норм.л/мин, температуру нагревательной печи регулировали таким образом, чтобы иметь температурный профиль с повышением температуры до 675°C при 2°C/мин, поддержанием при 675°C в течение двух часов для обжига и снижением при 1°C/мин, чтобы выполнить основной обжиг предварительно обожженного продукта, получая таким образом обожженный продукт. Коэффициент восстановления предварительно обожженного продукта, полученного в этом процессе, составлял 10,1%, и удельная поверхность обожженного продукта после основного обжига составляла 15,2 м2/г.
Удаление выступов
1800 кг обожженного продукта помещали в устройство, что показано на Фиг. 1, которое регулировали, чтобы предоставлять величину, преобразуемую в энергию, в расчете на массу катализатора (м5/с2/кг) при 15°C и 1 атм, составляющую 50, и проводили процесс в течение 24 часов. Длина воздушного потока в направлении протекания воздуха при этом составляла 390 мм, и средняя линейная скорость воздушного потока составляла 341 м/с; и число K отверстий, являющихся отверстиями для протекания воздуха, составляло 350. Отношения a/b и a/c в составе композитного оксидного катализатора (G1) после удаления выступов измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,1%, и выход акрилонитрила составлял 55,2%.
Пример 17
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количества добавляемого водного раствора метавольфрамата аммония до 93,0 г (чистота: 50%).
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,9 масс.%, и средний размер частиц составлял 56 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,2%, и удельная поверхность обожженного продукта после основного обжига составляла 14,2 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,090Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,1%, и выход акрилонитрила составлял 55,2%.
Пример 18
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением того, что не добавляли водный раствор метавольфрамата аммония.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,9 масс.%, и средний размер частиц составлял 57 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 11,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,7%, и выход акрилонитрила составлял 54,4%.
Пример 19
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением того, что не добавляли нитрат церия [Ce(NO3)3·6H2O].
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 15,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,5%, и выход акрилонитрила составлял 54,3%.
Пример 20
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количества добавляемого нитрата церия [Ce(NO3)3·6H2O] до 8,7 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,7%, и удельная поверхность обожженного продукта после основного обжига составляла 12,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,009On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,1%, и выход акрилонитрила составлял 54,6%.
Пример 21
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением того, что добавляли 4,8 г нитрата лантана [La(NO3)3·6H2O] вместо нитрата церия [Ce(NO3)3·6H2O].
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,7 масс.%, и средний размер частиц составлял 51 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 14,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030La0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,8%, и выход акрилонитрила составлял 54,0%.
Пример 22
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением того, что добавляли 4,8 г нитрата празеодима [Pr(NO3)3·6H2O] вместо нитрата церия [Ce(NO3)3·6H2O].
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,7 масс.%, и средний размер частиц составлял 56 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 14,2 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Pr0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,7%, и выход акрилонитрила составлял 54,1%.
Пример 23
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением того, что добавляли 4,6 г нитрата иттербия [Yb(NO3)3·3H2O] вместо нитрата церия [Ce(NO3)3·6H2O].
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,6 масс.%, и средний размер частиц составлял 58 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 14,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Yb0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,8%, и выход акрилонитрила составлял 54,1%.
Пример 24
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества оксида сурьмы (III) [Sb2O3] до 93,5 г; добавляемого количества смешанного раствора ниобия (B0) до 452,6 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 79,3 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,7 масс.%, и средний размер частиц составлял 53 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,8%, и удельная поверхность обожженного продукта после основного обжига составляла 14,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,243Nb0,122W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,1%, и выход акрилонитрила составлял 54,8%.
Пример 25
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количество воды до 1655 г; добавляемого количества гептамолибдата аммония [(NH4)6Mo7O24·4H2O] до 459,2 г; добавляемого количества метаванадата аммония [NH4VO3] до 63,7 г; добавляемого количества оксида сурьмы (III) [Sb2O3] до 99,3 г; добавляемого количества смешанного раствора ниобия (B0) 363,6 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 63,7 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,8 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 350°C и максимальной температуры обжига при основном обжиге до 685°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 11,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,220Sb0,258Nb0,098W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксида выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ = 8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 55 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,3%, и выход акрилонитрила составлял 54,6%.
Пример 26
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же способом, что и в Примере 1, за исключением изменения добавляемого количества оксида сурьмы (III) [Sb2O3] до 80,8 г; добавляемого количества смешанного раствора ниобия (B0) до 337,6 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 59,2 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 49 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 350°C и максимальной температуры обжига при основном обжиге до 690°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,9%, и удельная поверхность обожженного продукта после основного обжига составляла 11,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,210Nb0,091W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксидного катализатора выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 65 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,0%, и выход акрилонитрила составлял 54,5%.
Пример 27
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества оксида сурьмы (III) [Sb2O3] до 81,2 г; добавляемого количества смешанного раствора ниобия (B0) до 445,2 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 78,0 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 54 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при таких же условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,7%, и удельная поверхность обожженного продукта после основного обжига составляла 15,6 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,211Nb0,120W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,0%, и выход акрилонитрила составлял 55,5%.
Пример 28
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества оксида сурьмы (III) [Sb2O3] до 81,2 г; добавляемого количества смешанного раствора ниобия (B0) до 519,4 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 91,0 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,6 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 385°C и максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,0%, и удельная поверхность обожженного продукта после основного обжига составляла 16,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,211Nb0,140W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксидного катализатора выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 46 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,4%, и выход акрилонитрила составлял 54,6%.
Пример 29
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества оксида сурьмы (III) [Sb2O3] до 93,5 г; добавляемого количества смешанного раствора ниобия (B0) до 519,4 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 91,0 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,5 масс.%, и средний размер частиц составлял 53 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 385°C и максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,3%, и удельная поверхность обожженного продукта после основного обжига составляла 15,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,243Nb0,140W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксидного катализатора выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 45 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,2%, и выход акрилонитрила составлял 54,4%.
Пример 30
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количество воды до 1505 г; добавляемого количества гептамолибдата аммония [(NH4)6Mo7O24·4H2O] до 417,5 г; добавляемого количества метаванадата аммония [NH4VO3] до 57,9 г; добавляемого количества оксида сурьмы (III) [Sb2O3] до 87,4 г; добавляемого количества смешанного раствора ниобия (B0) 341,3 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 59,8 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,8 масс.%, и средний размер частиц составлял 50 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,1%, и удельная поверхность обожженного продукта после основного обжига составляла 14,3 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,200Sb0,227Nb0,092W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,3%, и выход акрилонитрила составлял 55,1%.
Пример 31
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количество воды до 1505 г; добавляемого количества гептамолибдата аммония [(NH4)6Mo7O24·4H2O] до 417,5 г; добавляемого количества метаванадата аммония [NH4VO3] до 57,9 г; добавляемого количества оксида сурьмы (III) [Sb2O3] до 87,4 г; добавляемого количества смешанного раствора ниобия (B0) 426,6 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 74,8 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,8 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,0%, и удельная поверхность обожженного продукта после основного обжига составляла 13,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,200Sb0,227Nb0,115W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,3%, и выход акрилонитрила составлял 55,2%.
Пример 32
Приготовление сухого порошка
Приготовление сухого порошка (D1) выполняли таким же способом, что и в Примере 1.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 55 мкм.
Обжиг сухого порошка (E1)
Условия обжига изменяли следующим образом: подаваемое количество сухого порошка (E1) при предварительном обжиге изменяли до 70 г/ч; общий расход азота при предварительном обжиге изменяли до 0,8 норм.л/мин (каждый из противоточного и прямоточного потоков составлял 0,4 норм.л/мин); и максимальную температуру обжига при предварительном обжиге изменяли до 400°C. Подаваемое количество предварительно обожженного продукта для основного обжига изменяли до 51 г/ч; и максимальную температуру обжига при основном обжиге изменяли до 695°C. Обжиг выполняли при таких же условиях обжига, что и в Примере 1, за исключением этих изменений. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,2%, и удельная поверхность обожженного продукта после основного обжига составляла 8,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,102W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,4%, и выход акрилонитрила составлял 53,8%.
Пример 33
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества смешанного раствора ниобия (B0) до 500,8 г, и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 87,8 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 49 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при основном обжиге до 670°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,2%, и удельная поверхность обожженного продукта после основного обжига составляла 14,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,135W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,5%, и выход акрилонитрила составлял 54,4%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 54,6%.
Пример 34
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения количество воды до 1520 г; добавляемого количества гептамолибдата аммония [(NH4)6Mo7O24·4H2O] до 421,7 г; добавляемого количества метаванадата аммония [NH4VO3] до 58,5 г; добавляемого количества смешанного раствора ниобия (B0) до 437,8 г; и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 76,7 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,7 масс.%, и средний размер частиц составлял 50 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях, что и в Примере 1, за исключением изменения максимальной температуры обжига при основном обжиге до 670°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,8%, и удельная поверхность обожженного продукта после основного обжига составляла 14,3 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,202Sb0,219Nb0,118W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,6%, и выход акрилонитрила составлял 52,8%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 54,8%.
Пример 35
Приготовление сухого порошка
Сухой порошок (D1) приготавливали таким же образом, что и в Примере 1, за исключением изменения добавляемого количества смешанного раствора ниобия (B0) до 341,3 г, и добавляемого количества раствора пероксида водорода, который добавляли вместе со смешанным раствором ниобия (B0), содержащего 30 масс.% H2O2, до 59,8 г.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,2 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при основном обжиге до 685°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,5%, и удельная поверхность обожженного продукта после основного обжига составляла 13,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,092W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 89,6%, и выход акрилонитрила составлял 55,4%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 54,5%.
Сравнительный пример 1
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
438,4 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 60,8 г метаванадата аммония [NH4VO3], 107,8 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,580 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
65,0 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 371,0 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1).
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,9 масс.%, и средний размер частиц составлял 50 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 390°C и максимальной температуры обжига при основном обжиге до 695°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,2%, и удельная поверхность обожженного продукта после основного обжига составляла 9,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,210Sb0,280Nb0,100W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 86,0%, и выход акрилонитрила составлял 51,5%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 49,0%.
Сравнительный пример 2
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
480,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 66,6 г метаванадата аммония [NH4VO3], 84,7 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,730 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
58,5 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 333,9 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,9 масс.%, и средний размер частиц составлял 52 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 345°C и максимальной температуры обжига при основном обжиге до 650°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,8%, и удельная поверхность обожженного продукта после основного обжига составляла 11,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,230Sb0,220Nb0,090W0,030Ce0,005On/51,0 масс.%-SiO2.
Для композитного оксидного катализатора выполняли рентгеновскую дифрактометрию. При этом внимание было обращено на пик с 2θ=8,9°, и диаметр кристаллических зерен измеряли по уравнению Шеррера, и он составлял 35 нм.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 84,0%, и выход акрилонитрила составлял 52,3%.
Сравнительный пример 3
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
480,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 66,6 г метаванадата аммония [NH4VO3], 100,1 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,730 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
58,5 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 333,9 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 56 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 390°C и максимальной температуры обжига при основном обжиге до 695°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,6%, и удельная поверхность обожженного продукта после основного обжига составляла 9,5 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,230Sb0,260Nb0,090W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 81,2%, и выход акрилонитрила составлял 52,0%.
Сравнительный пример 4
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
417,5 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 57,9 г метаванадата аммония [NH4VO3], 100,1 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,505 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
91,0 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 519,4 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом самым водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 54 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 340°C и максимальной температуры обжига при основном обжиге до 640°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,8%, и удельная поверхность обожженного продукта после основного обжига составляла 15,2 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,200Sb0,260Nb0,140W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 82,0%, и выход акрилонитрила составлял 51,5%.
Сравнительный пример 5
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
432,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 59,9 г метаванадата аммония [NH4VO3], 84,3 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,557 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
100,8 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 575,0 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 53 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры при предварительном обжиге до 400°C и максимальной температуры при основном обжиге до 700°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,5%, и удельная поверхность обожженного продукта после основного обжига составляла 15,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,219Nb0,155W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 81,7%, и выход акрилонитрила составлял 48,5%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 46,5%.
Сравнительный пример 6
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
432,1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 59,9 г метаванадата аммония [NH4VO3], 75,1 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,557 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
84,5 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 482,3 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,3 масс.%, и средний размер частиц составлял 56 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 370°C и максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 11,2%, и удельная поверхность обожженного продукта после основного обжига составляла 14,2 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,207Sb0,195Nb0,130W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 82,3%, и выход акрилонитрила составлял 51,6%.
Сравнительный пример 7
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
417,5 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 57,9 г метаванадата аммония [NH4VO3], 94,3 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,505 кг воды, и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
53,3 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 304,2 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 57 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 370°C и максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 9,0%, и удельная поверхность обожженного продукта после основного обжига составляла 11,0 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,200Sb0,245Nb0,082W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана)
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 84,2%, и выход акрилонитрила составлял 49,5%. Реакцию выполняли постоянным образом в течение 30 дней с применением катализатора, и выход акрилонитрила составлял 44,0%.
Сравнительный пример 8
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
459,2 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 63,7 г метаванадата аммония [NH4VO3], 80,8 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,655 кг воды, и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
71,5 г раствора пероксида водорода, содержащего 30 масс.% H2O2, добавляли к 408,1 г смешанного раствора ниобия (B0), приготовленного таким же образом, что и в Примере 1, и перемешивали и смешивали в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс.% SiO2; кроме того, добавляли 98,4 г раствора пероксида водорода, содержащего 30 масс.% H2O2, и постоянным образом перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (C1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (E1) составляло 0,4 масс.%, и средний размер частиц составлял 58 мкм.
Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1, за исключением изменения максимальной температуры обжига при предварительном обжиге до 370°C и максимальной температуры обжига при основном обжиге до 680°C. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,5%, и удельная поверхность обожженного продукта после основного обжига составляла 13,8 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения a/b и a/c в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Полученные результаты представлены в Таблице 1. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,220Sb0,210Nb0,110W0,030Ce0,005On/51,0 масс.%-SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 84,3%, и выход акрилонитрила составлял 49,5%.
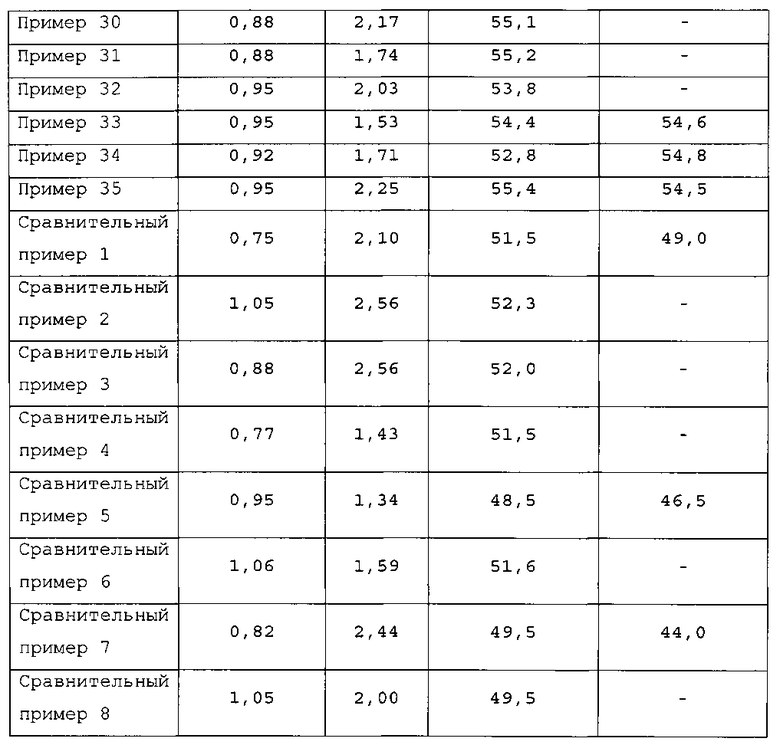
Сравнительный пример 9 Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
432, 1 г гептамолибдата аммония [(NH4)6Mo7O24·4H2O], 72,3 г метаванадата аммония [NH4VO3], 98,2 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Ce(NO3)3·6H2O] добавляли к 1,806 кг воды и нагревали при перемешивании в течение одного часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
78,0 г раствора пероксида водорода, содержащего 30 масс. % H2O2, добавляли к 445,2 г смешанного раствора ниобия (В0), и смешивали посредством перемешивания в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (В1).
Полученный водный раствор исходного материала (A1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс. % SiO2; и далее добавляли 114,5 г раствора пероксида водорода, содержащего 30 масс. % H2O2, и затем полученную смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (B1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (С1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (С1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (Е1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (Е1) составляло 0,3 масс. %, и средний размер частиц составлял 54 мкм. Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,5%, и удельная поверхность обожженного продукта после основного обжига составляла 13,9 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения а/b и а/с в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,250Sb0,255Nb0,120W0,030Ce0,005On/51, 3 масс. % - SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 87,5%, и выход акрилонитрила составлял 51,8%.
Сравнительный пример 10
Приготовление сухого порошка
Сухой порошок (D1) получали следующим образом.
432, 1 г гептамолибдата аммония [(NH4) 6Mo7O24·4H2O], 66,6 г метаванадата аммония [NH4VO3], 103,9 г оксида сурьмы (III) [Sb2O3] и, кроме того, 4,8 г нитрата церия [Се (NO3)3·6H2O] добавляли к 1,806 кг воды и нагревали при перемешивании в течение одного
часа при 95°C, приготавливая таким образом водный раствор исходного материала (A1).
78,0 г раствора пероксида водорода, содержащего 30 масс. % H2O2, добавляли к 445,2 г смешанного раствора ниобия (В0), и смешивали посредством перемешивания в течение 10 минут при комнатной температуре, приготавливая таким образом водный раствор исходного материала (B1).
Полученный водный раствор исходного материала (А1) охлаждали до 70°C и затем добавляли 807,8 г золя кремниевой кислоты, содержащего 34,0 масс. % SiO2; и далее добавляли 121,0 г раствора пероксида водорода, содержащего 30 масс. % H2O2, и затем полученную смесь непрерывно перемешивали в течение 30 минут при 55°C. Затем последовательно добавляли водный раствор исходного материала (В1), 31,0 г водного раствора метавольфрамата аммония (чистота: 50%) и жидкостную дисперсию, в которой 211,5 г порошка кремнезема было диспергировано в 2,855 кг воды, и после этого выдерживали в течение 2,5 часов при 50°C при перемешивании, получая таким образом водный смешанный раствор в виде суспензии (C1) в качестве раствора с составом исходного материала.
Полученный водный смешанный раствор (С1) подавали в центробежную распылительную сушилку и сушили в ней, посредством чего получали микросферический сухой порошок (D1). Температура на входе сушилки составляла 210°C, и температура на ее выходе составляла 120°C.
Операция классификации
Полученный сухой порошок (D1) классифицировали с применением сита, имеющего отверстия 25 мкм, чтобы получить сухой порошок (E1), являющийся классифицированным продуктом. Содержание частиц размером 25 мкм или менее в полученном сухом порошке (Е1) составляло 0,6 масс. %, и средний размер частиц составлял 52 мкм. Обжиг сухого порошка (E1)
Обжиг выполняли при тех же самых условиях обжига, что и в Примере 1. Коэффициент восстановления предварительно обожженного продукта при этом составлял 10,3%, и удельная поверхность обожженного продукта после основного обжига составляла 13,6 м2/г.
Удаление выступов
Выступы удаляли при таких же условиях, что и в Примере 1, и отношения а/b и а/с в составе композитного оксидного катализатора (G1) измеряли посредством рентгенофлуоресцентного анализа. Состав полученного при этом композитного оксидного катализатора (G1) представлял собой Mo1V0,230Sb0,270Nb0,120W0,030Ce0,005On/50,9 масс. % - SiO2.
Реакция аммоксидирования пропана
Реакцию выполняли при таких же условиях, что и в Примере 1; и степень конверсии пропана после реакции составляла 88,5%, и выход акрилонитрила составлял 52,2%.
Результаты испытаний согласно сравнительным примерам 9 и 10, в сопоставлении с Примером 4 по изобретению представлены в Таблице 2 ниже.


Как можно видеть из полученных данных, выход акрилонитрила для образца примера 4 по настоящему изобретению заметно выше, чем выход при использовании катализатора, в котором атомная доля а ванадия V превосходит 0,24 или катализатора, в котором атомная доля b сурьмы Sb составляет свыше 0,25.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Данное изобретение применимо в производственных условиях в качестве композитного оксидного катализатора, применяемого в реакции каталитического окисления в паровой фазе или каталитического аммоксидирования в паровой фазе пропана или изобутана.
ОПИСАНИЕ ОБОЗНАЧЕНИЙ 1: ОСНОВНОЙ КОРПУС
2: ВПУСКНАЯ ТРУБА ДЛЯ ГАЗА, 21: ОТВЕТВЛЕННАЯ ТРУБА, 210: СОПЛО, 211: ОТВЕРСТИЕ, 22: ДОПОЛНИТЕЛЬНОЕ ОТВЕТВЛЕНИЕ, 220: ОТВЕРСТИЕ
3: ВЫПУСКНАЯ ТРУБА, 31: ВНЕШНЯЯ ТРУБА, 32: ВНУТРЕННЯЯ ТРУБА, 33: СОПЛО
4: ЦИКЛОН, 41: ВЫПУСКНАЯ ТРУБА, 42: ЦИКЛОН
5, 51, 52: ВОЗВРАТНАЯ ТРУБА, 53: ВНЕШНЯЯ ТРУБА, 54: ВНУТРЕННЯЯ ТРУБА, 55: СОПЛО
6: СОПЛО, 61: ПЕРЕДНИЙ КОНЕЦ СОПЛА
7: ВПУСКНАЯ ТРУБА ДЛЯ ГАЗА, 71: ТРУБОПРОВОД ДЛЯ ЦИРКУЛЯЦИИ
8: ВЫПУСКНОЙ ТРУБОПРОВОД
11: ВНЕШНЯЯ ТРУБА
12: ВНУТРЕННЯЯ ТРУБА
13: ОТВЕРСТИЕ
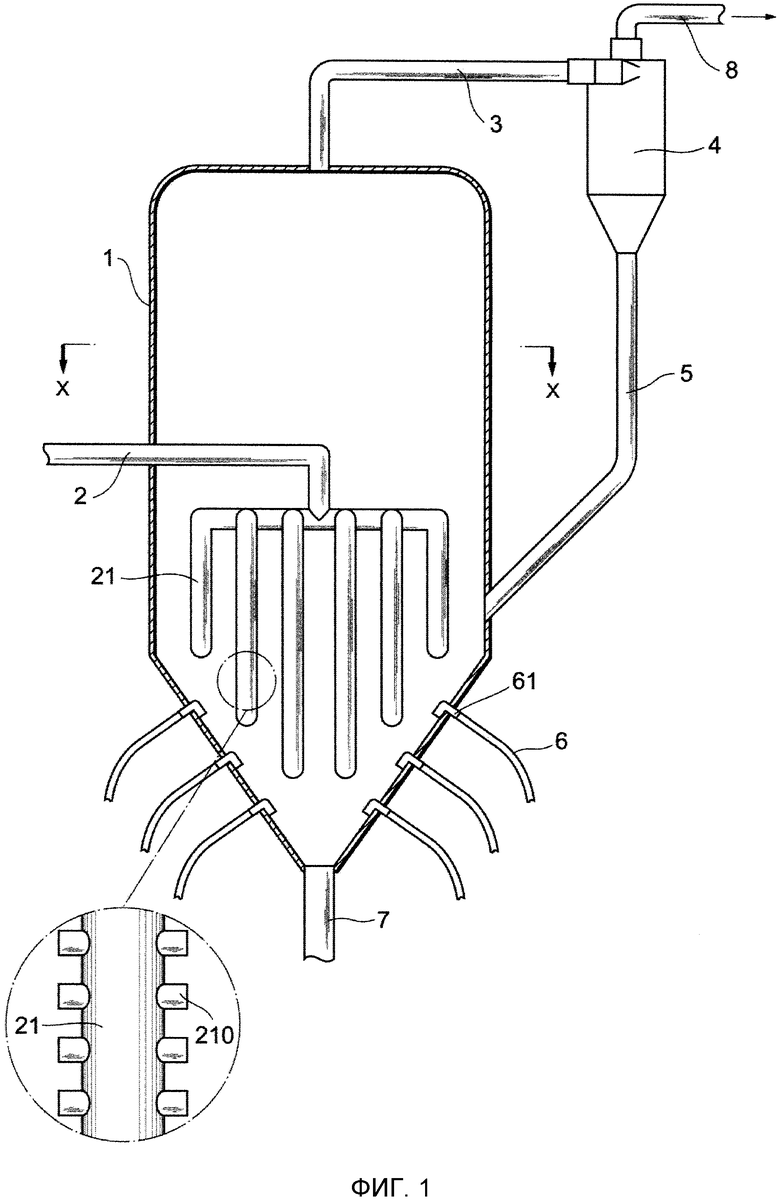
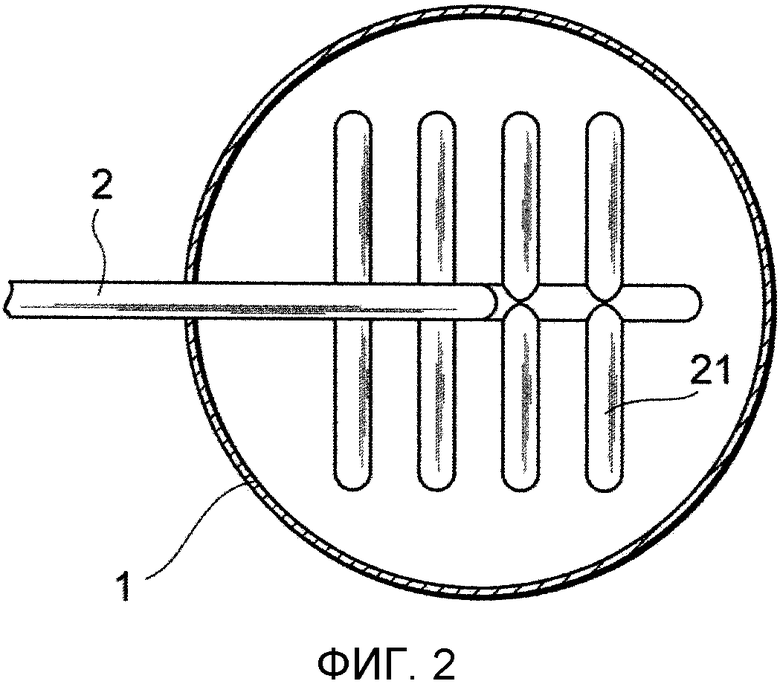
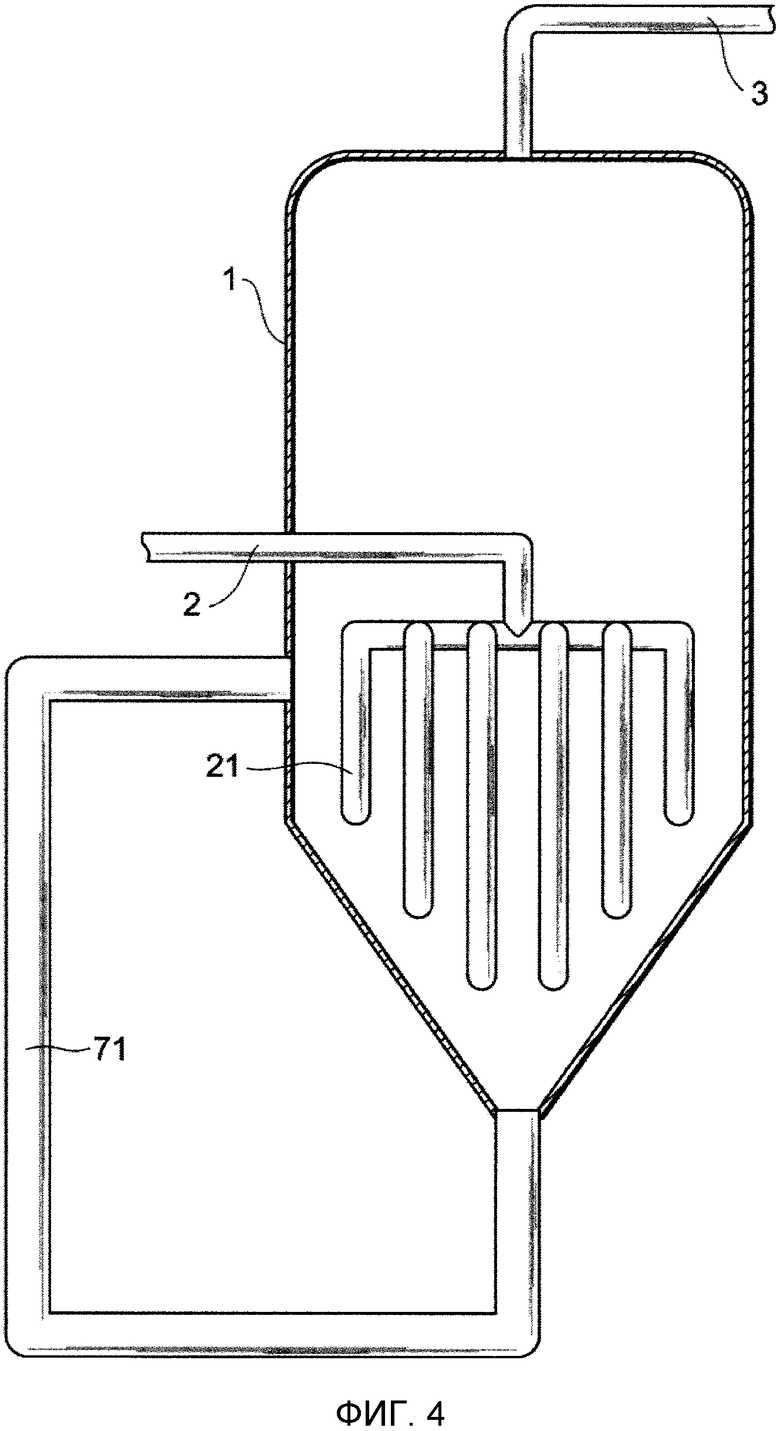
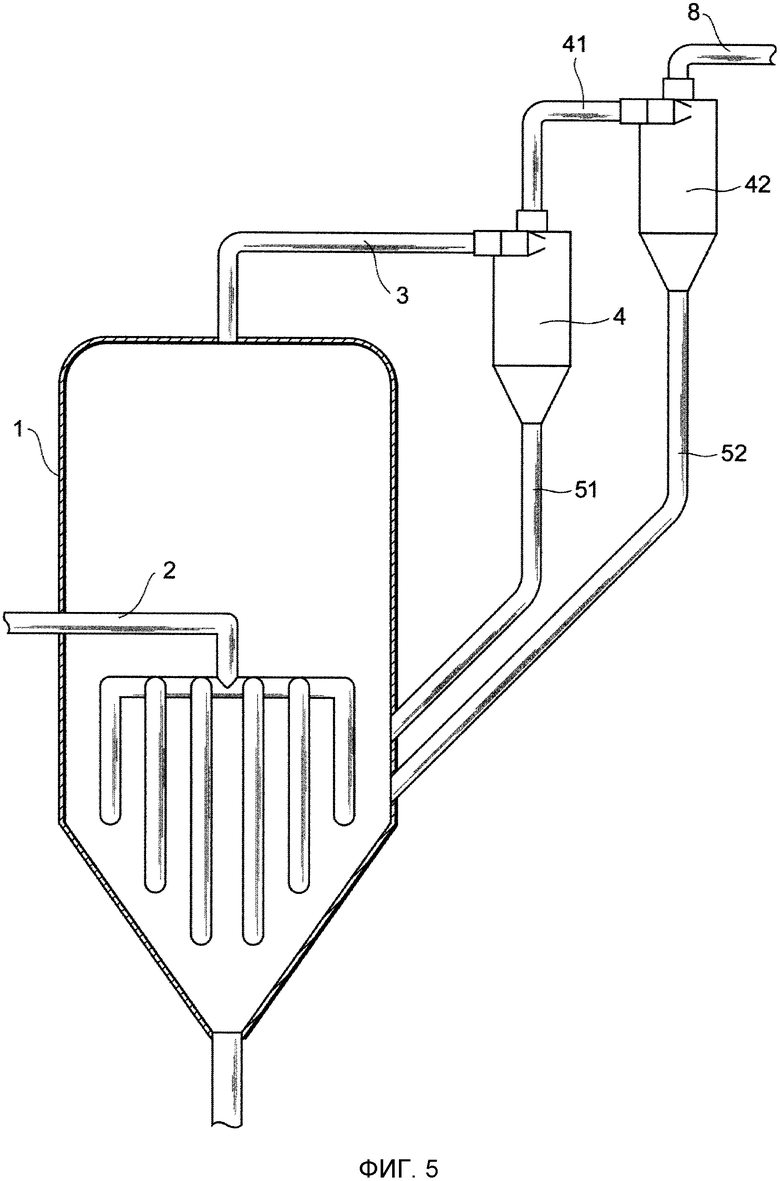
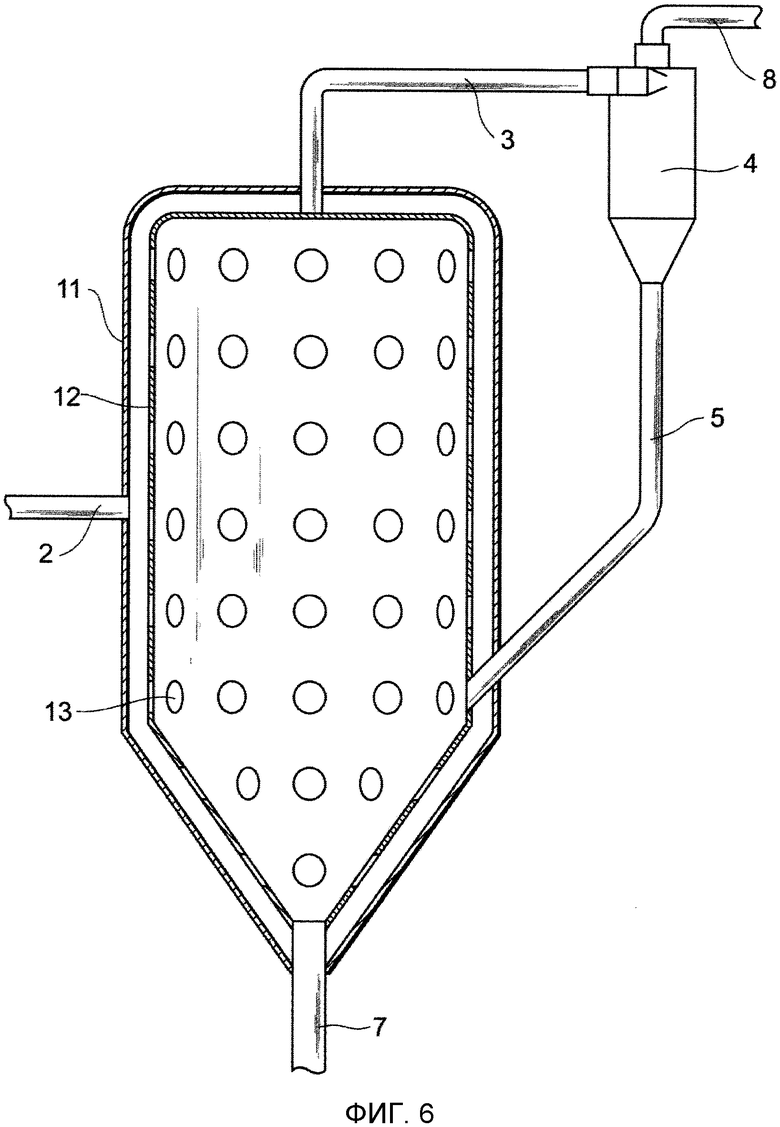
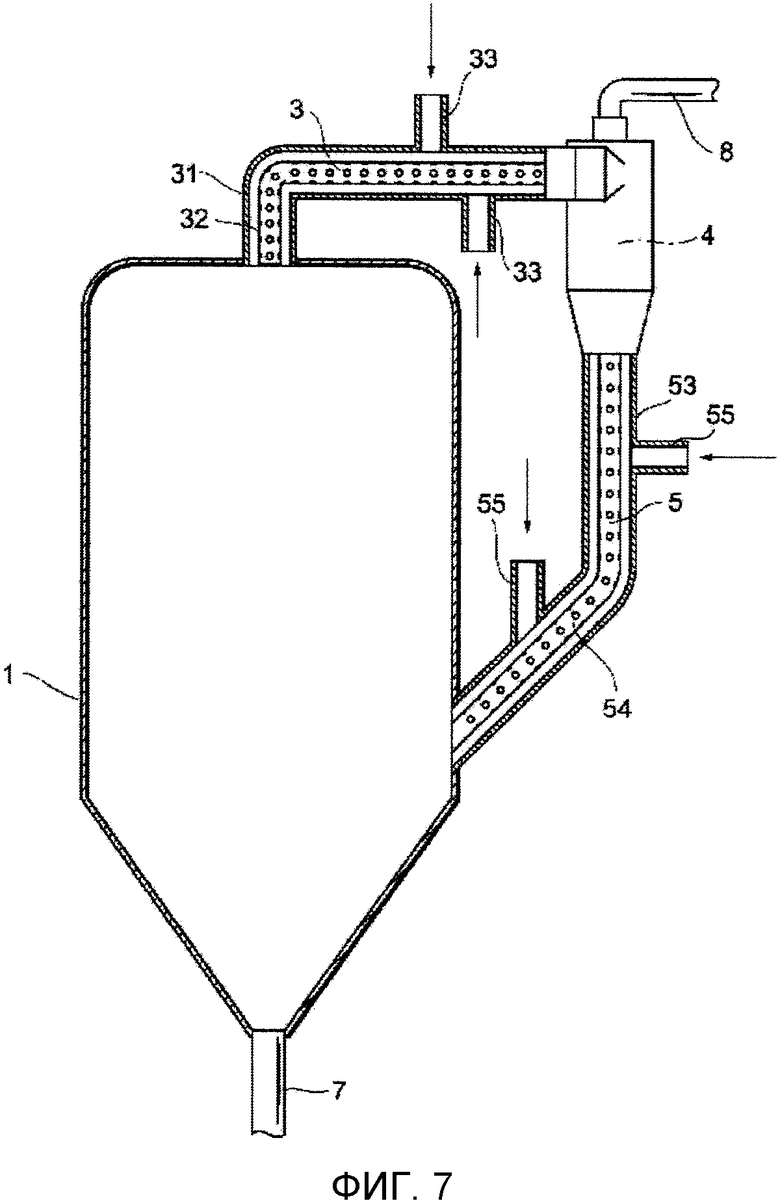
Изобретение относится к комплексному оксидному катализатору, применяемому для реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, где данный катализатор содержит комплексный оксид, представленный приведенной ниже формулой 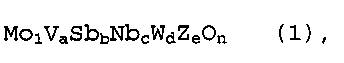 в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr и Yb; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,24, 0,1≤b≤0,25, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3. Изобретение также относится к способу получения комплексного оксидного катализатора, где данный способ включает следующие стадии от (I) до (V): (I) приготовление раствора, содержащего исходный материал, содержащий Mo, V, Sb, Nb, W и Z, в котором атомное отношение а V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение с Nb к одному атому Mo составляет 0,01≤c≤0,5, атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение е Z к одному атому Mo составляет 0≤e≤0,2; (II) сушка раствора, содержащего исходный материал, с получением сухого порошка; (III) предварительный обжиг сухого порошка с получением предварительно обожженного продукта; (IV) основной обжиг предварительно обожженного продукта с получением обожженного продукта, имеющего выступы на поверхности частиц; и (V) удаление выступов, имеющихся на поверхности частиц обожженного продукта, посредством воздушного потока, где степень восстановления предварительно обожженного продукта составляет от 8 до 12%, а удельная поверхность обожженного продукта составляет от 7 до 20 м2/г. Изобретение также относится к способу получения ненасыщенного нитрила с применением вышеуказанного комплексного оксидного катализатора. В изобретении улучшены функциональные возможности катализатора. 4 н. и 16 з.п. ф-лы, 7 ил., 2 табл., 35 пр.
в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr и Yb; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,24, 0,1≤b≤0,25, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3. Изобретение также относится к способу получения комплексного оксидного катализатора, где данный способ включает следующие стадии от (I) до (V): (I) приготовление раствора, содержащего исходный материал, содержащий Mo, V, Sb, Nb, W и Z, в котором атомное отношение а V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение с Nb к одному атому Mo составляет 0,01≤c≤0,5, атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение е Z к одному атому Mo составляет 0≤e≤0,2; (II) сушка раствора, содержащего исходный материал, с получением сухого порошка; (III) предварительный обжиг сухого порошка с получением предварительно обожженного продукта; (IV) основной обжиг предварительно обожженного продукта с получением обожженного продукта, имеющего выступы на поверхности частиц; и (V) удаление выступов, имеющихся на поверхности частиц обожженного продукта, посредством воздушного потока, где степень восстановления предварительно обожженного продукта составляет от 8 до 12%, а удельная поверхность обожженного продукта составляет от 7 до 20 м2/г. Изобретение также относится к способу получения ненасыщенного нитрила с применением вышеуказанного комплексного оксидного катализатора. В изобретении улучшены функциональные возможности катализатора. 4 н. и 16 з.п. ф-лы, 7 ил., 2 табл., 35 пр.
1. Комплексный оксидный катализатор, применяемый для реакции каталитического аммоксидирования в паровой фазе пропана или изобутана, где данный катализатор содержит комплексный оксид, представленный приведенной ниже формулой (1):
в которой компонент Z представляет собой один или несколько элементов, выбранных из La, Ce, Pr и Yb; каждый индекс из a, b, c, d, e и n представляет собой атомную долю элемента в расчете на один атом Mo; 0,1≤a≤0,24, 0,1≤b≤0,25, 0,01≤c≤0,3, 0≤d≤0,2, и 0≤e≤0,1; атомное отношение a/b составляет 0,85≤a/b<1,0, и атомное отношение a/c составляет 1,4<a/c<2,3.
2. Комплексный оксидный катализатор по п. 1, содержащий от 20 до 70 мас. % кремнезема в расчете на SiO2.
3. Способ получения комплексного оксидного катализатора по п. 1, где данный способ включает следующие стадии от (I) до (V):
(I) приготовление раствора, содержащего исходный материал, содержащий Mo, V, Sb, Nb, W и Z, в котором атомное отношение а V к одному атому Mo составляет 0,1≤a≤0,5, атомное отношение b Sb к одному атому Mo составляет 0,1≤b≤0,5, атомное отношение с Nb к одному атому Mo составляет 0,01≤c≤0,5, атомное отношение d W к одному атому Mo составляет 0≤d≤0,4, и атомное отношение е Z к одному атому Mo составляет 0≤e≤0,2;
(II) сушка раствора, содержащего исходный материал, с получением сухого порошка;
(III) предварительный обжиг сухого порошка с получением предварительно обожженного продукта;
(IV) основной обжиг предварительно обожженного продукта с получением обожженного продукта, имеющего выступы на поверхности частиц; и
(V) удаление выступов, имеющихся на поверхности частиц обожженного продукта, посредством воздушного потока,
где степень восстановления предварительно обожженного продукта составляет от 8 до 12%, а удельная поверхность обожженного продукта составляет от 7 до 20 м2/г.
4. Способ получения комплексного оксидного катализатора по п. 3, в котором содержание частиц, имеющих размер не более чем 25 мкм, в сухом порошке составляет не более чем 20 мас. %, и средний размер частиц сухого порошка составляет от 35 до 75 мкм.
5. Способ получения комплексного оксидного катализатора по п. 3, в котором на стадии (V) выступы удаляют таким образом, что количество выступов, которое имеет обожженный продукт, составляет не более чем 2 мас. % в расчете на общую массу обожженного продукта.
6. Способ получения комплексного оксидного катализатора по п. 3, в котором длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с в пересчете на линейную скорость при 15°C и атмосферном давлении.
7. Способ получения комплексного оксидного катализатора по п. 3, в котором стадия (I) включает стадии с (a) по (d):
(a) приготовление водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) добавление золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) смешивание водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) добавление суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживание раствора.
8. Способ получения комплексного оксидного катализатора по п. 3, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает следующие стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре, ниже чем температура обжига при основном обжиге.
9. Способ получения комплексного оксидного катализатора по п. 4, в котором на стадии (V) выступ удаляют таким образом, что количество выступов, которое имеет обожженный продукт, составляет не более чем 2 мас. %, в расчете на общую массу обожженного продукта.
10. Способ получения комплексного оксидного катализатора по п. 4, в котором длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с в пересчете на линейную скорость при 15°C и атмосферном давлении.
11. Способ получения комплексного оксидного катализатора по п. 5, в котором длина воздушного потока в направлении протекания воздушного потока составляет не менее чем 55 мм, и средняя скорость воздушного потока составляет не менее чем 80 м/с и не более чем 500 м/с в пересчете на линейную скорость при 15°C и атмосферном давлении.
12. Способ получения комплексного оксидного катализатора по п. 4, в котором стадия (I) включает следующие стадии с (a) по (d):
(a) приготовление водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) добавление золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) смешивание водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) добавление суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживание раствора.
13. Способ получения комплексного оксидного катализатора по п. 5, в котором стадия (I) включает следующие стадии с (a) по (d):
(a) приготовление водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) добавление золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) смешивание водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) добавление суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживание раствора.
14. Способ получения комплексного оксидного катализатора по п. 6, в котором стадия (I) включает следующие стадии с (a) по (d):
(a) приготовление водного смешанного раствора, содержащего Mo, V, Sb и компонент Z;
(b) добавление золя кремниевой кислоты и раствора пероксида водорода к водному смешанному раствору, полученному на стадии (a);
(c) смешивание водного раствора, содержащего Nb, дикарбоновую кислоту и раствор пероксида водорода, и соединения W с раствором, полученным на стадии (b); и
(d) добавление суспензии, содержащей порошковый кремнезем, к раствору, полученному на стадии (c), и выдерживание раствора.
15. Способ получения комплексного оксидного катализатора по п. 4, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает следующие стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре, ниже чем температура обжига при основном обжиге.
16. Способ получения комплексного оксидного катализатора по п. 5, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает следующие стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре, ниже чем температура обжига при основном обжиге.
17. Способ получения комплексного оксидного катализатора по п. 6, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает следующие стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре, ниже чем температура обжига при основном обжиге.
18. Способ получения комплексного оксидного катализатора по п. 7, в котором стадия предварительного обжига (III) и/или стадия основного обжига (IV) включает следующие стадии (i) и (ii):
(i) приложение ударных усилий к устройству для обжига, в котором обжигают предварительно обожженный продукт и/или обожженный продукт; и
(ii) отжиг предварительно обожженного продукта и/или обожженного продукта при температуре, ниже чем температура обжига при основном обжиге.
19. Способ получения ненасыщенного нитрила с применением комплексного оксидного катализатора по п. 1, в котором пропан или изобутан подвергают реакции каталитического аммоксидирования в паровой фазе с получением соответствующего ненасыщенного нитрила, проводимой в следующих условиях: молярное отношение подаваемого в реакцию кислорода к пропану или изобутану предпочтительно от 0,1 до 6; молярное отношение подаваемого в реакцию аммиака к пропану или изобутану от 0,3 до 1,5; температура реакции от 350 до 500°C; давление реакции от 5×104 до 5×105 Па; и время контактирования от 0,1 до 10 (с·г/см3).
20. Способ получения ненасыщенного нитрила с применением комплексного оксидного катализатора по п. 2, в котором пропан или изобутан подвергают реакции каталитического аммоксидирования в паровой фазе с получением соответствующего ненасыщенного нитрила, проводимой в следующих условиях: молярное отношение подаваемого в реакцию кислорода к пропану или изобутану предпочтительно от 0,1 до 6; молярное отношение подаваемого в реакцию аммиака к пропану или изобутану от 0,3 до 1,5; температура реакции от 350 до 500°C; давление реакции от 5×104 до 5×105 Па; и время контактирования от 0,1 до 10 (с·г/см3).
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| МАССЫ ОКСИДОВ МЕТАЛЛОВ | 2003 |
|
RU2352390C9 |

