Настоящее изобретение относится к усовершенствованному способу получения 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты. Настоящее изобретение также относится к новому соединению, а именно трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбамату, используемому в усовершенствованном способе.
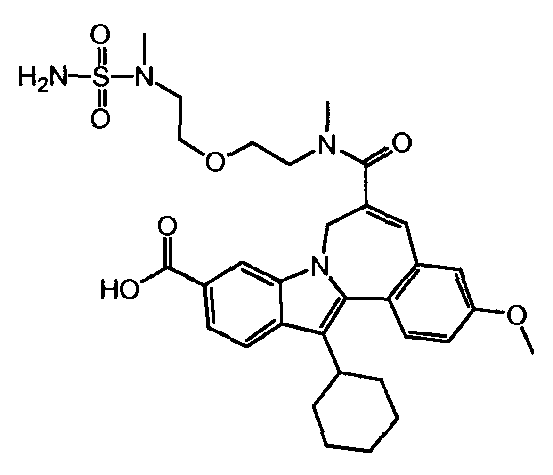
Соединение 'A'
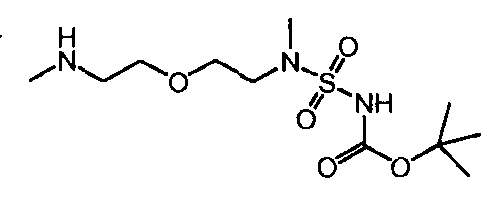
Соединение 'B'
WO 2010/003658 описывает некоторые макроциклические индолы, которые могут быть использованы в качестве ингибиторов вируса гепатита C. Синтез 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты (Соединение Ά) описан в упомянутом документе на страницах 38 и 39 (см. Соединение 1е) как трехстадийный синтез, дающий общий выход 62%.
Стадия 1
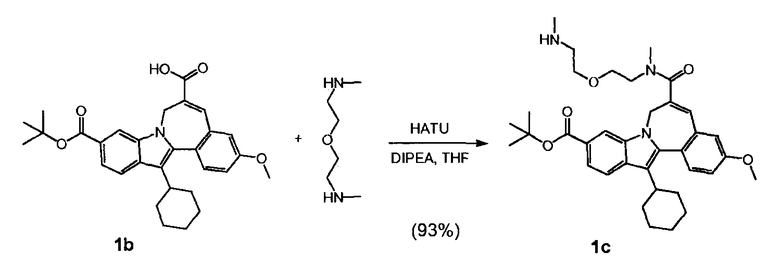
Стадия 2
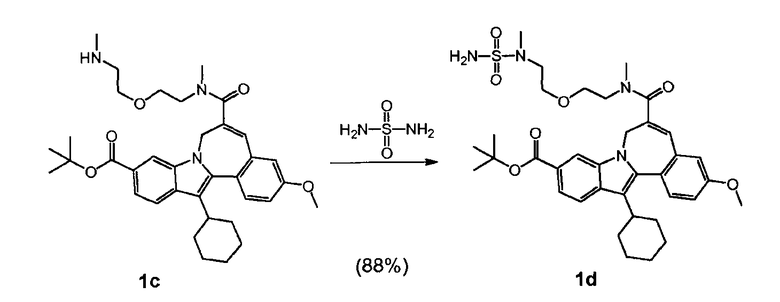
Стадия 3
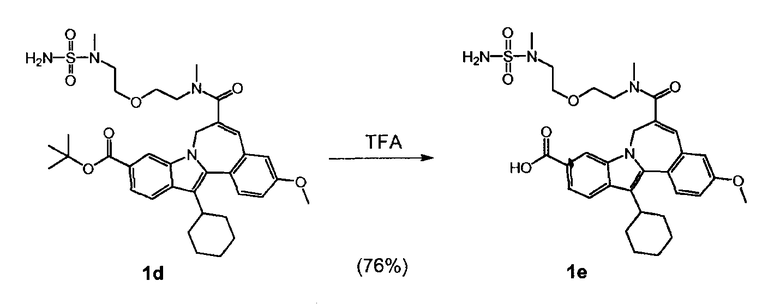
Целью настоящего изобретения является создание усовершенствованного способа синтеза 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты (Соединение 'A'), который легче осуществим и более эффективен, чем способ, известный до сих пор.
Настоящее изобретение достигает этой цели путем предоставления улучшенного способа получения 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты (Соединение 'A'), отличающегося тем, что он включает следующие стадии:
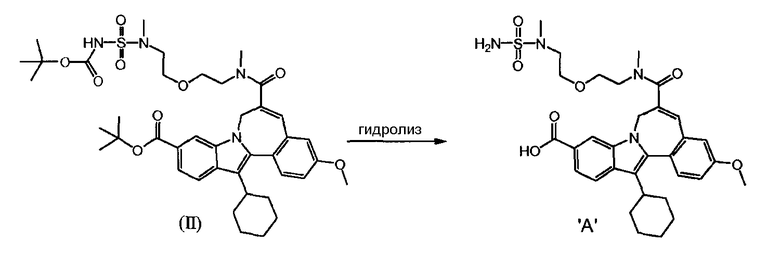
a) 10-(трет-бутоксикарбонил)-13-циклогексил-3-метокси-7H-индоло-[2,1-α]-[2]-бензазепин-6-карбоновая кислота (Соединение I) реагирует с трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбаматом (Соединение 'B') в присутствии связывающего агента в подходящем растворителе


b) и Соединение (II), полученное таким образом, гидролизуют кислотой, так что получается Соединение 'A'.
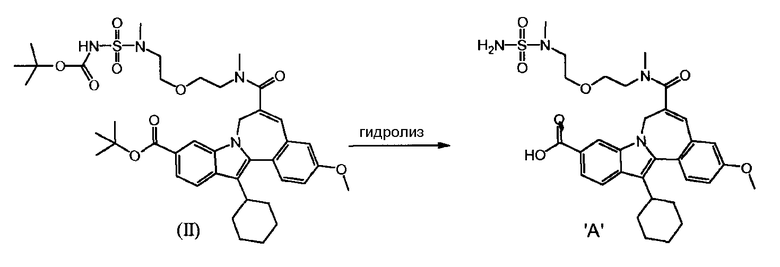
Связывающий агент для стадии а) является, например, карбодиимидазолом (CDI), дициклогексилкарбодиимидом (DCC), гексафторфосфатом O-(7-азабензотриазоло-1-ил)-1,1,3,3-тетраметилурония (HATU), гексафторфосфатом бромотри-(пирролидино)-фосфония (PyBrOP) или комбинацией из гидрата 1-гидроксибензтриазола (HOBT.H20) и гидрохлорида 1-этил-3-(3-диметиламинопропил)-карбодиимида (EDCI).
Подходящим растворителем для стадии a) является, например, дихлорметан, 2-метилтетрагидрофуран, ацетонитрил, ацетон, 2-бутанон, 4-метил-2-пентанон, этилацетат, изопропилацетат или толуол.
Гидролиз на стадии b) может быть осуществлен путем использования трифторуксусной кислоты, метансульфоновой кислоты, хлористого водорода, бромистого водорода, пара-толуолсульфоновой кислоты, серной кислоты или фосфорной кислоты.
Стадии a) и b) могут быть осуществлены как двухстадийный синтез, в котором Соединение (II) выделяется на стадии a) перед проведением стадии b) либо стадии a) и b) проводят как синтез в одном сосуде.
Общий выход, получаемый в стадиях а) и b), находится между 86 и 91%, в зависимости от того, какая из процедур, описанных в примерах 3, 4 и 5 (Эксперимент B), используется для реализации нового способа.
Настоящее изобретение также относится к новому соединению (Соединение 'B') со следующей формулой и к его возможным кислотно-аддитивным солям:
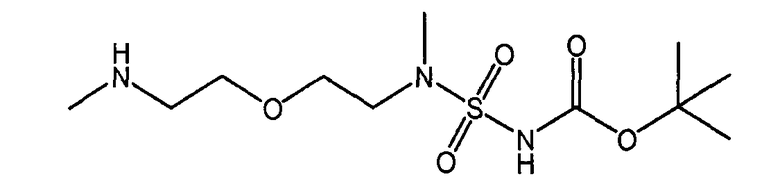
трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбамат (Соединение 'B').
Кислотно-аддитивные соли Соединения 'B' включают соли, которые Соединение 'B' может образовывать с органическими или неорганическими кислотами, такими как минеральные кислоты, сульфоновые кислоты, карбоновые кислоты и фосфорсодержащие кислоты. Примерами солеобразующих минеральных кислот являются плавиковая кислота, соляная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота, хлорная кислота, перхлорная кислота и фосфорная кислота. Солеобразующими сульфоновыми кислотами являются толуолсульфоновая кислота, бензолсульфоновая кислота, метансульфоновая кислота и трифторметансульфоновая кислота. Солеобразующими карбоновыми кислотами являются муравьиная кислота, уксусная кислота, пропионовая кислота, масляная кислота и им подобные. Солеобразующими дикарбоновыми кислотами являются щавелевая кислота, малоновая кислота, янтарная кислота, глутаровая кислота и им подобные. Солеобразующими гидрокси-кислотами являются гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, миндальная кислота и им подобные. Другими солеобразующими карбоновыми кислотами являются трифторуксусная кислота, бензойная кислота, хлоруксусная кислота, фталевая кислота, малеиновая кислота и малоновая кислота. Фосфорсодержащими кислотами являются различные фосфоно-кислоты, фосфоновые кислоты и фосфиновые кислоты.
Это новое Соединение 'B' может быть синтезировано следующим образом:
Стадия 1
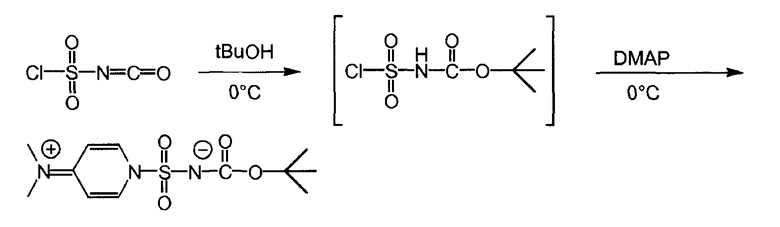
Стадия 2

Экспериментальная часть
Здесь используются следующие сокращения:
A/A: активный выход
CDI: карбонилдиимидазол
CSI: хлоросульфонилизоцианат
DBU: l,8-диаза-бицикло-[5,4,0]-ундецен-7
DIPE: диизопропиловый эфир
DMAP: 4-диметиламинопиридин
DME: 1,2-диметоксиэтан
DMSO-d6: дейтерированный диметилсульфоксид
EDCI: гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида
F/F: физический выход
HOBt: 1-гидроксибензотриазолгидрат
iPrOAc: изопропилацетат
LC: жидкостная хроматография
MeCN: ацетонитрил
MEK: метилэтилкетон (2-бутанон)
MeTHF: 2-метилтетрагидрофуран
MeS03H: метансульфоновая кислота
MIK: метилизопропилкетон
MTBE: метил-трет-бутиловый эфир
NMR: ядерно-магнитный резонанс
tBuOH: трет-бутанол
tBOC: трет-бутоксикарбонил
THF: тетрагидрофуран
Пример 1

Промежуточное соединение (1)
Раствор 8,205 г хлоросульфонилизоцианата (58,0 ммоль, нормальность 1 экв) в 50 мл ацетонитрила (1 л/моль) был охлажден до -2°C на смеси льда с солью в атмосфере азота, с использованием 500-мл четырехгорлой колбы, снабженной термометром, магнитной мешалкой и капельной (для добавления) воронкой. Раствор 4,297 г трет-бутанола (58,0 ммоль, нормальность 1 экв) в 33 мл ацетонитрила (0,5 л/моль) добавлялся по каплям в течение 20 минут, при этом температура оставалась ниже 4°C. Четыре минуты спустя (когда температура упала до 1°C), раствор DMAP (116,0 ммоль, нормальность 2 экв) в 55 мл ацетонитрила (1 л/моль) добавлялся по каплям в течение 24 минут при температуре ниже 5°C.
Раствор выдерживался в течение 65 минут для обеспечения лучшей кристаллизации при температуре ниже 3°C. Белая суспензия фильтровалась на воронке Бюхнера с получением нелипкого белого порошка. Осадок высушивался в течение ночи в сушильном шкафу при температуре 40°С под вакуумом, что дало 9,465 г промежуточного соединения (1).
Пример 2
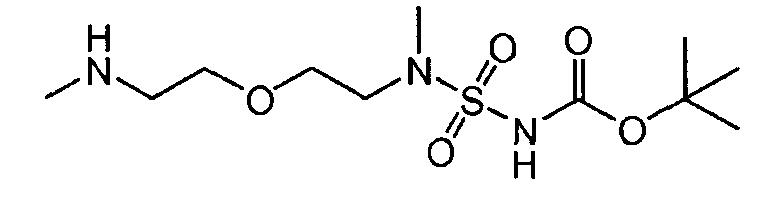
Соединение 'B'
109,1 г (0,825 моль) 1,5-бис-(метиламино)-3-оксапентана и 1,5 л ацетонитрила были введены в реакционный сосуд с инертной атмосферой. Полученный раствор был охлажден до 0°C, и к нему были добавлены 226 г (0,75 моль) промежуточного соединения (1). Полученная таким образом смесь перемешивалась в течение первых 6 часов при 10°C, а затем в течение 3 дней при температуре 0°C. Осадок был отфильтрован и промыт ацетонитрилом. После сушки при 25°C было получено соединение 'B' с выходом 95 г (40,6%, с поправкой на чистоту) в виде белых кристаллов.
Пример 3
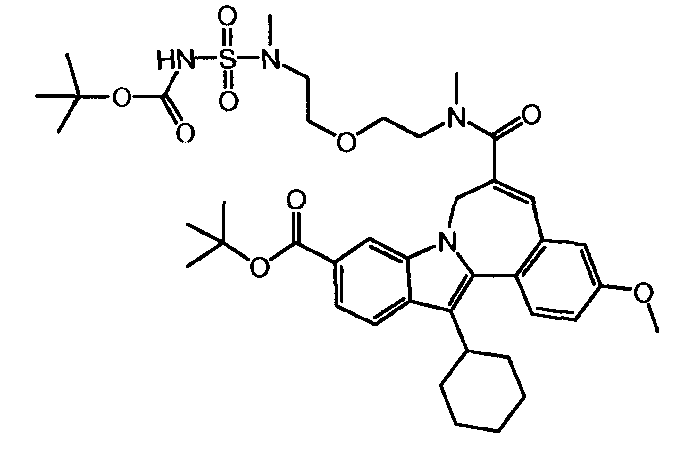
Промежуточное соединение (2)
0,400 г (0,82 ммоль, нормальность 1 экв) 10-(трет-бутоксикарбонил)-13-циклогексил-3-метокси-7H-индоло-[2,1-α]-[2]-бензазепин-6-карбоновой кислоты (названной Соединение 1b на стр. 38 из WO 2010/003658), 0,164 г HOBt (1,07 ммоль, нормальность 1,3 экв) и 0,201 г EDCI (1,07 ммоль, нормальность 1,3 экв) были растворены в 6,5 мл MeTHF (8 л/моль) в закрытой стеклянной колбе. Содержимое колбы перемешивалось в течение 1 часа при комнатной температуре. 0,469 г Соединения 'B' (64,3 масс.%, 0,98 ммоль, нормальность 1,2 экв) было затем добавлено к реакционной смеси, которая после 18 часов реакции была проанализирована. Жидкостно-хроматографический анализ показал, что промежуточное соединение (2) было получено с выходом 93,1%.
Пример 4
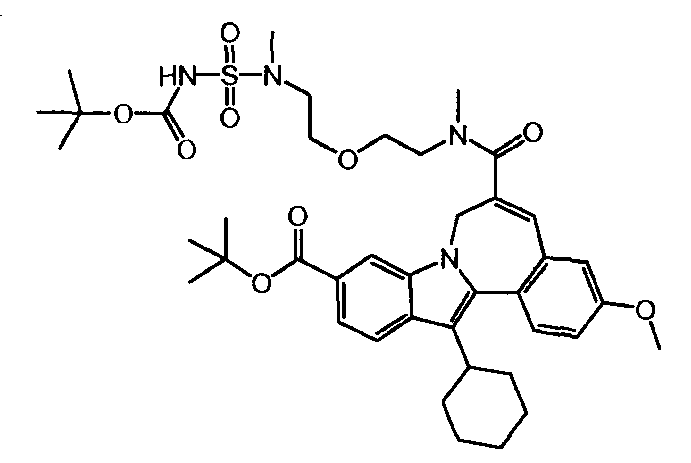
Промежуточное соединение (2)
12,00 г (24,61 ммоль, нормальность 1 экв) 10-(трет-бутоксикарбонил)-13-циклогексил-3-метокси-7H-индоло-[2,1-α]-[2]-бензазепин-6-карбоновой кислоты, 4,92 г HOBt (31,99 ммоль, нормальность 1,3 экв) и 6,03 г EDCI (31,99 ммоль, нормальность 1,3 экв) были введены в 100-мл колбу и растворены в 200 мл MeTHF (8 л/моль). Содержимое колбы перемешивалось в течение 1 часа при комнатной температуре. Затем было добавлено 11,663 г Соединения 'B' (77,4 масс.%, 29,53 ммоль, нормальность 1,2 экв), и после того, как время реакции составило 18 часов, реакционную смесь проанализировали. Жидкостно-хроматографический анализ показал, что промежуточное соединение (2) было получено с выходом 98,3%.
Затем реакционная смесь была экстрагирована и промыта сначала двумя 180-мл порциями H20 (15 л/моль), а затем двумя 180-мл порциями раствора NaHCO3 (15 л/моль). Органический слой был высушен с 2,4 г Na2S04 и отфильтрован, после чего был определен объем полученного фильтрата. 60 мл MeTHF было добавлено, чтобы довести объем до 200 мл (8 л/моль). Жидкостно-хроматографический анализ показал, что промежуточное соединение (2) было получено с выходом 93,7%.
Пример 5
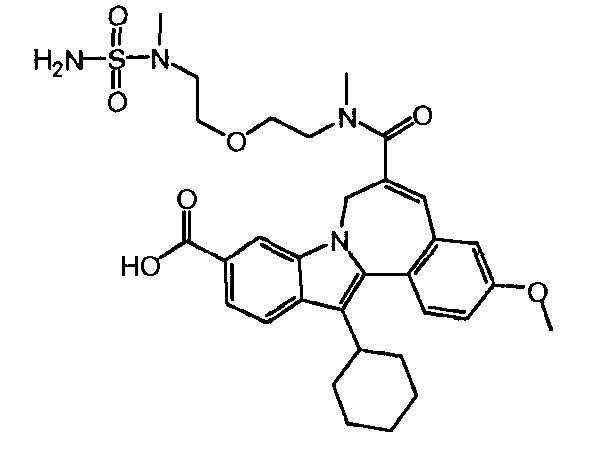
Соединение 'A'
Эксперимент A
К 13,722 г промежуточного соединения (2) (16,4 масс.%, нормальность 1 экв, 2,88 ммоль), растворенного в MeTHF (8 л/моль) в 100-мл колбе без атмосферы азота (этот раствор содержал 2,17 масс.% воды), было добавлено 251 мкл воды, чтобы довести содержание воды до 4 масс.%. После добавления 1,9 мл MeS03H (нормальность 1 экв, 28,8 ммоль) реакционная смесь была помещена в масляную ванну, нагретую до 50°C. После 5 часов реакции был взят и проанализирован образец. После 22 часов реакции реакционная смесь была доведена до комнатной температуры, и снова был взят образец для анализа. Вся реакционная смесь весила 15,022 г.
Жидкостно-хроматографический анализ
После 5 часов: 90,1% Соединения 'A'
После 22 часов: 75,4% Соединения 'A'
Эксперимент B
119,6 г промежуточного соединения (2) (10,27 масс.%, 15,7 ммоль, нормальность 1 экв) было растворено в MeTHF (8 л/моль) в реакционном сосуде с азотной атмосферой. Раствор содержал 2,21 масс.% воды. Чтобы довести содержание воды до 4 масс.%, было добавлено 2,81 г воды. После добавления 10,31 мл MeSO3H (157 ммоль, нормальность 10 экв) реакционная смесь была нагрета до 50°C. После реакции в течение 5 часов реакционная смесь была охлаждена до комнатной температуры, и ее образец был проанализирован. Вся реакционная смесь весила 119,6 г.
Жидкостно-хроматографический анализ
92,7% Соединения 'A'.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2009 |
|
RU2518471C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ | 2010 |
|
RU2551852C2 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2835082C2 |
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПРОИЗВОДНЫЕ 2-ИМИНОПИРРОЛИДИНА | 2002 |
|
RU2270192C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2095361C1 |
| СОЕДИНЕНИЯ ИНДОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КЛЕТОЧНОГО НЕКРОЗА | 2008 |
|
RU2477282C2 |
Настоящее изобретение относится к способу получения 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты и к новому соединению, используемому в способе, а именно к трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбамату. Способ легче осуществим и более эффективен, чем известный способ. 2 н. и 6 з.п. ф-лы, 5 пр.
1. Способ получения 13-циклогексил-3-метокси-6-[метил-(2-{2-[метил-(сульфамоил)-амино]-этокси}-этил)-карбамоил]-7H-индоло-[2,1-α]-[2]-бензазепин-10-карбоновой кислоты (Соединение 'A'), отличающийся тем, что он включает следующие стадии:
a) 10-(трет-бутоксикарбонил)-13-циклогексил-3-метокси-7H-индоло-[2,1-α]-[2]-бензазепин-6-карбоновая кислота (Соединение I) реагирует с трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбаматом (Соединение 'B') в присутствии связывающего агента в подходящем растворителе, и

b) Соединение (II), полученное таким образом, гидролизуют кислотой с получением Соединения 'A'.
2. Способ по п.1, где стадии а) и б) проводят как синтез в одном сосуде.
3. Способ по п.1 или 2, где связывающий агент выбирают из карбодиимидазола (CDI), дициклогексилкарбодиимида (DCC), гексафторфосфата O-(7-азабензотриазоло-1-ил)-1,1,3,3-тетраметилурония (HATU), гексафторфосфата бромотри-(пирролидино)-фосфония (PyBrOP) и комбинации из гидрата 1-гидроксибензтриазола (HOBT.H20) и гидрохлорида 1-этил-3-(3-диметиламинопропил)-карбодиимида (EDCI).
4. Способ по п.1, в котором кислота, используемая в стадии б), является трифторуксусной кислотой, метансульфоновой кислотой, хлористым водородом, бромистым водородом, пара-толуолсульфоновой кислотой, серной кислотой или фосфорной кислотой.
5. Способ по п.1, в котором получение проводят в атмосфере азота.
6. Способ по п.5, в котором растворитель, используемый в стадии а), представляет собой 2-метилтетрагидрофуран.
7. Способ по п.6, в котором органическая кислота, используемая в стадии б), является метансульфоновой кислотой, температура реакции составляет 50°С, а продолжительность реакции составляет не более 5 часов.
8. Трет-бутил(метил-{2-[2-(метиламино)-этокси]-этил}-сульфамоил)-карбамат и его кислотно-аддитивные соли.

| Вентиль | 1929 |
|
SU19008A1 |
| WO 2007140200 A2, 06.12.2007 | |||

