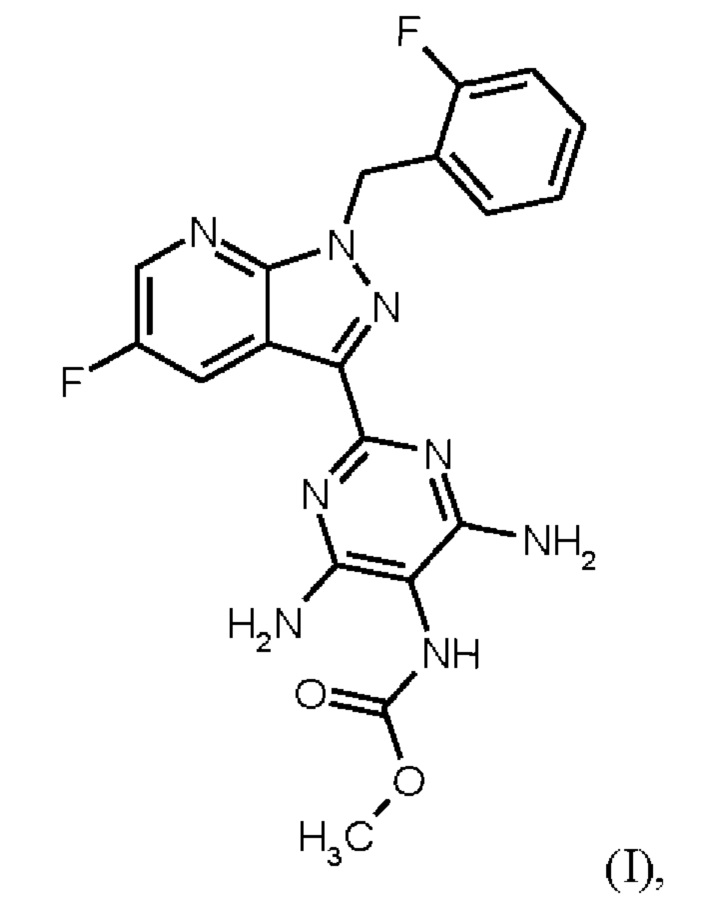
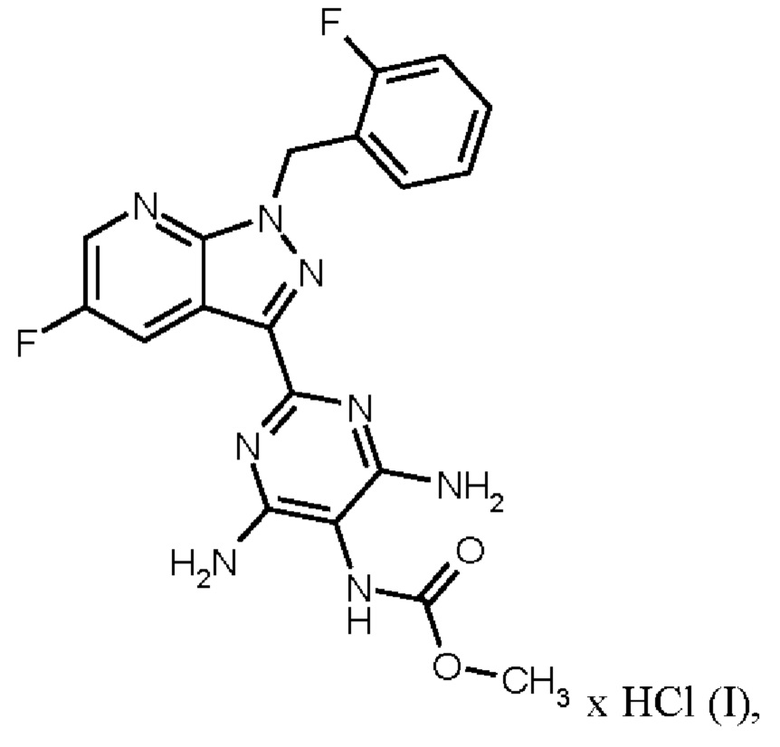
Настоящее изобретение относится к новому и эффективному способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (1) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, в качестве активного соединения с очень высокой чистотой и с улучшенными физическими свойствами для работы с твердыми веществами
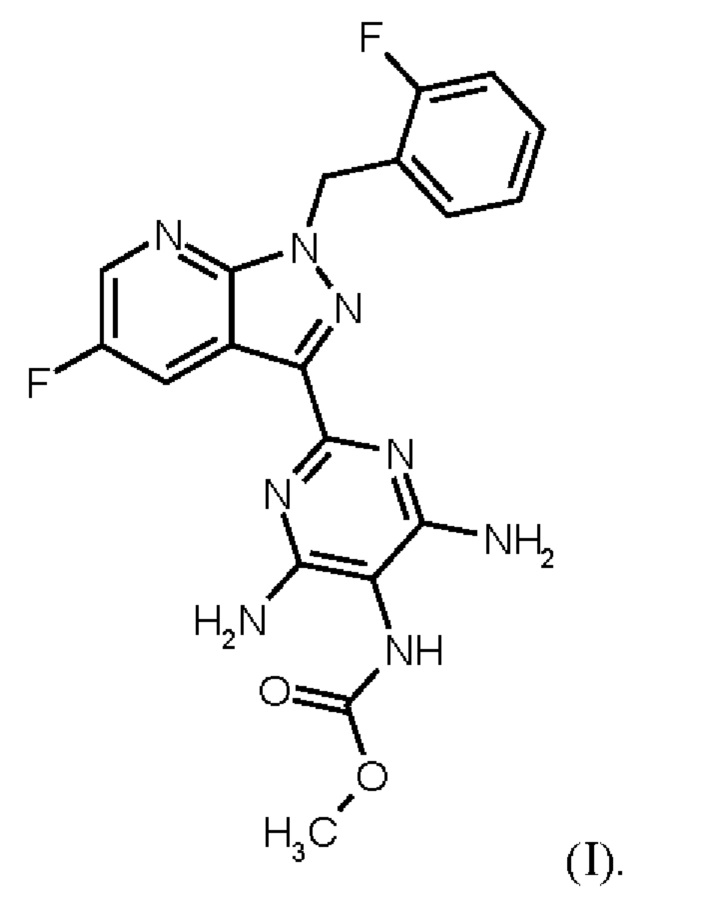
Метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамат является фармацевтически активным соединением, применяемым для лечения и/или профилактики сердечно-сосудистых заболеваний.
Соединение формулы (I) действует как стимулятор растворимой гуанилатциклазы и может быть использовано в качестве средства для профилактики и/или лечения сердечно-сосудистых заболеваний, например, для лечения гипертензии и сердечной недостаточности, включая хроническую сердечную недостаточность, сердечную недостаточность со сниженной фракцией выброса (HFnEF) и сердечную недостаточность с сохраненной фракцией выброса (HFpEF), стабильную и нестабильную стенокардию, нарушения периферических и сердечных сосудов, аритмии, для лечения тромбоэмболических заболеваний и ишемий, таких как инфаркт миокарда, инсульт, транзиторные и ишемические атаки, нарушения периферической перфузии, профилактика рестенозов, например, после терапии тромбозов, чрескожной транслюминальной ангиопластики (РТА), чрескожной транслюминальной коронарной ангиопластики (РТСА), шунтирования, а также для лечения атеросклероза, астматических расстройств и заболеваний мочеполовой системы, например, гипертрофии простаты, эректильной дисфункции, женской сексуальной дисфункции, остеопороза, глаукомы, легочной гипертензии, гастропареза, склеродермии и недержания мочи.
Как описано в WO 2013/076168, соединение формулы (I) может присутствовать в различных кристаллических формах и сольватах. Соединение формулы (I) существует в пяти полиморфных модификациях с температурами плавления 257°С (полиморф I), 253°С (полиморф II), 247°С (полиморф III), 246°С (полиморф IV), 234°С (полиморф V), сольват диметилформамида/воды (содержание DMF 13,6%, содержание воды 0,9%), ди-диметилсульфоксид сольват (стехиометрическое значение: 26,8% DMSO), сольват триуксусной кислоты (ацетат 29,7%), моногидрат (4,1% воды) и дигидрат (7,8% воды). WO 2011/147809 и WO 2013/076168 дополнительно описывают способы получения соединения формулы (I).
В контексте настоящего изобретения «соединение формулы (I) в кристаллической форме модификации I» определяется как метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбаматформулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7.
В контексте настоящего изобретения «соединение формулы (I) в кристаллической форме модификации I» дополнительно характеризуется как модификация соединения формулы (I), которое определяется как кристаллическая форма модификации I в WO 2013/076168; например, со ссылкой на рентгеновскую дифрактограмму, имеющую определенные максимумы пиков угла 2 тета при 5,9, 6,9и 22,7 или при 5,9, 6,9, 16,2, 16,5, 24,1, 22,7 и 24,7; или ИК-спектр, имеющий определенные максимумы полос при 1707, 1633 и 1475 см-1 или при 1707, 1633, 1566, 1475, 1255 и 1223 см-1; или с помощью температуры плавления 257°С.
WO 2020/126983 (опубликована после даты приоритета настоящего изобретения) относится к новому продукту активному соединению метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамату, обладающему улучшенными свойствами, например, в отношении способности к выделению продукта активного соединения, возможности выгрузки продукта активного соединения после выделения и сушки, а также транспортабельности, просеиваемости и микронизируемости продукта активного соединения, а также к способам получения и составления его лекарственной формы. WO 2020/126983 полностью включен в настоящий документ посредством ссылки.
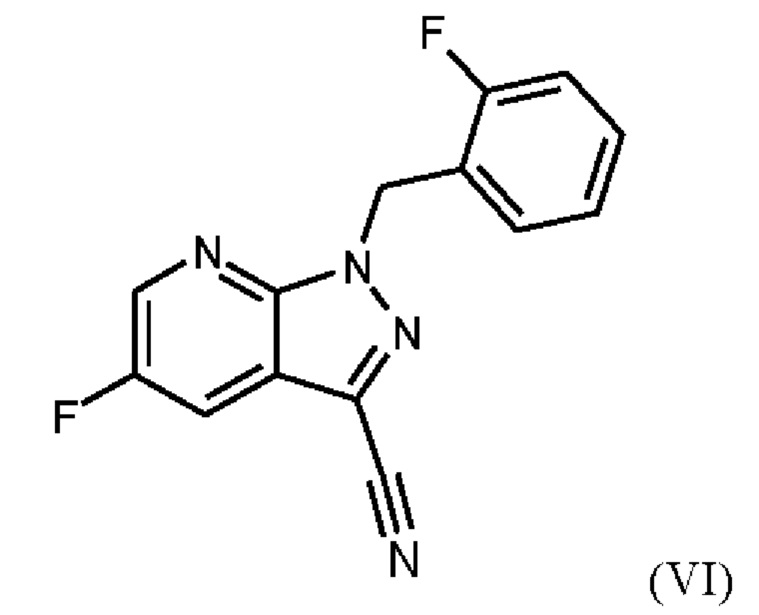
Способ получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, как описано в WO 2013/076168, проиллюстрирован на схеме 1 ниже.
Схема 1
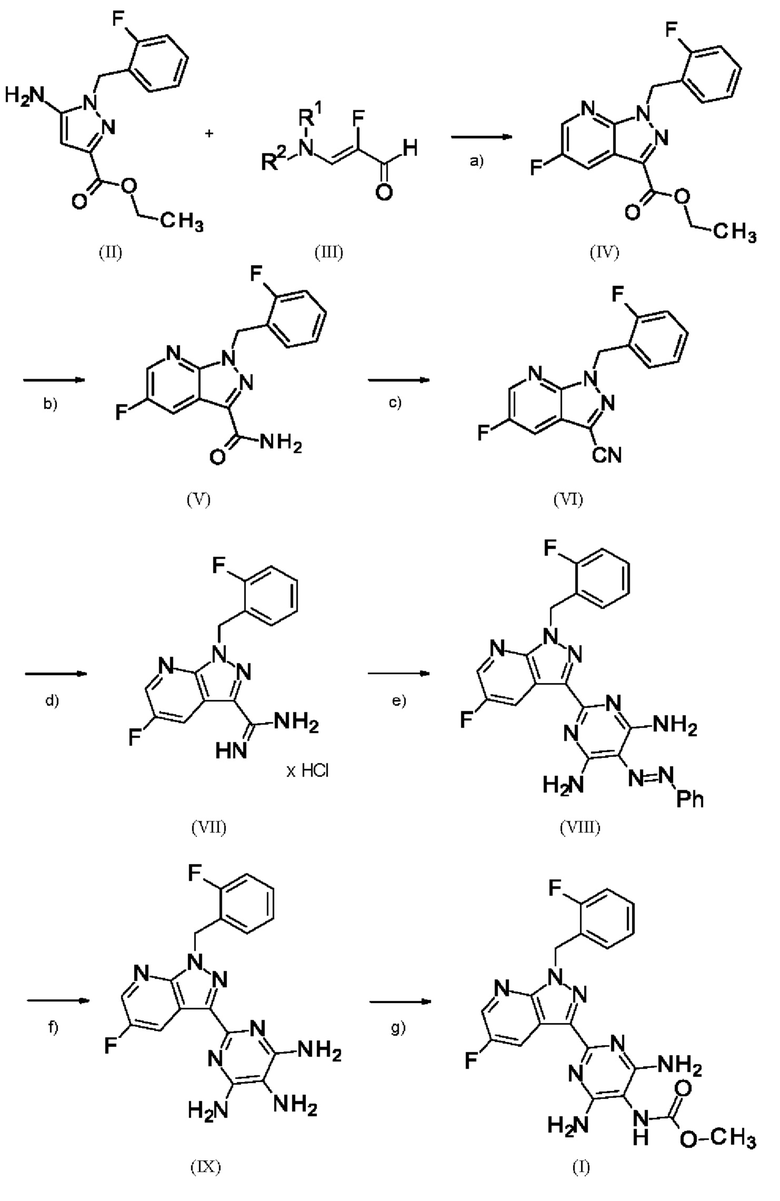
[а): хлорид лития, метансульфокислота, этанол; b) формамид, метоксида натрия/метанол, этанол; с) фосфорилхлорид, ацетонитрил, сульфолан; d) 1. метоксид натрия/метанол, этанол 2. хлорид аммония/этанол; е) DMF, триэтиламин, [(E)-фенилдиазенил]малононитрил; f) Pd/C, водород, DMF; g) изопропанол, метилхлорформиата, триэтиламин].
WO 2013/076168 рассматривается как наиболее близкий документ предшествующего уровня техники. По различным причинам, изложенным ниже, способ, описанный в этом наиболее близком документе предшествующего уровня техники, не подходит для его выполнения в техническом масштабе.
Стадии а) и b) схемы 1 и примеры 6 и 7 WO 2013/076168 проводят в отдельных реакциях с промежуточным выделением соединения формулы (IV). Для осуществления способа в техническом масштабе это имеет тот недостаток, что фракции сложного эфира, которые все еще присутствуют в маточном растворе во время кристаллизации, теряются. Для выделения соединения формулы (IV) должны быть выполнены дополнительные стадии выделения и сушки, что в способе в техническом масштабе имеет тот недостаток, что увеличивается время работы оборудования производственной установки, что приводит к значительному увеличению производственных затрат. Кроме того, при обработке соединения формулы (IV) необходимы сложные стадии промывки изопропанолом для удаления солей метансульфоновой кислоты из промежуточного соединения, что также увеличивает производственные затраты.
При проведении стадии с) схемы 1 и примера 8, как описано в WO 2013/076168, в большем масштабе наблюдается гидролиз продукта (соединение (VI)) до исходного вещества (соединение (V)). Это является еще одним серьезным недостатком выполнения способа в техническом масштабе.
В контексте настоящего изобретения «исходное вещество» используется как синоним «исходного материала» или «эдукта».
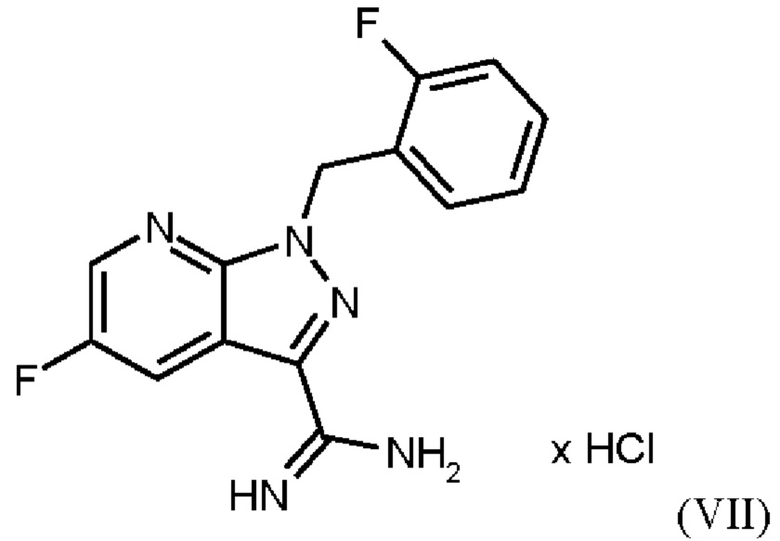
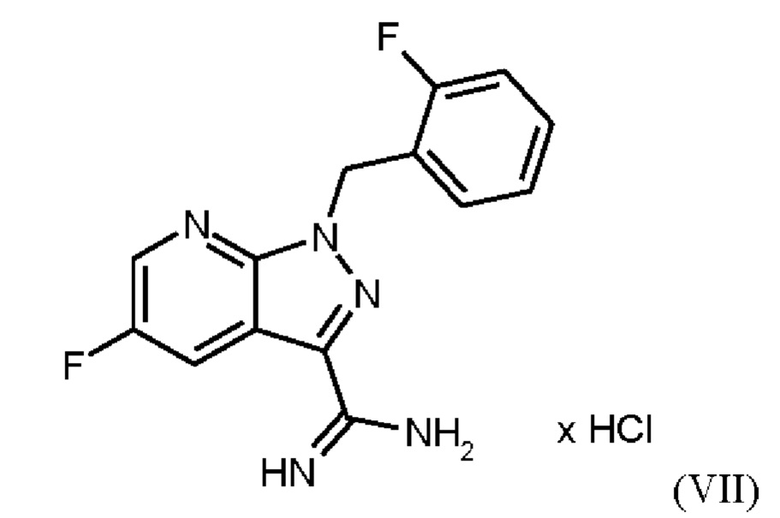
При выполнении превращения (VI)→(VII) согласно стадии d) схемы 1 и примеру 9 WO 2013/076168 на стенках сосуда образуются корки. Это является решающим недостатком при осуществлении способа в техническом масштабе.
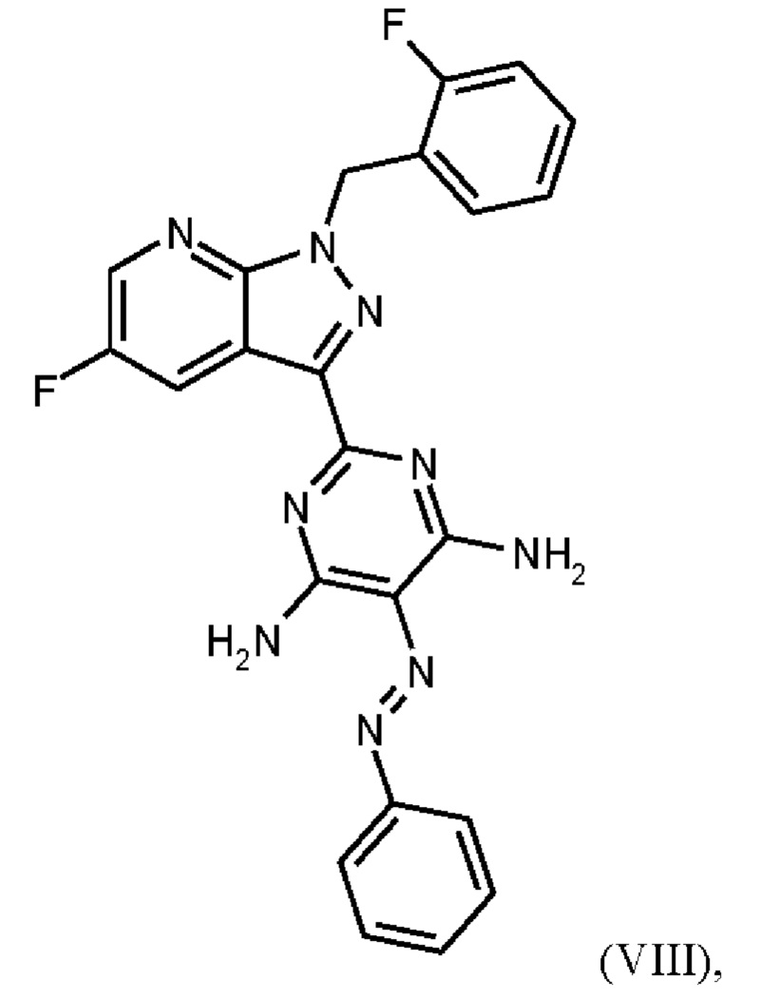
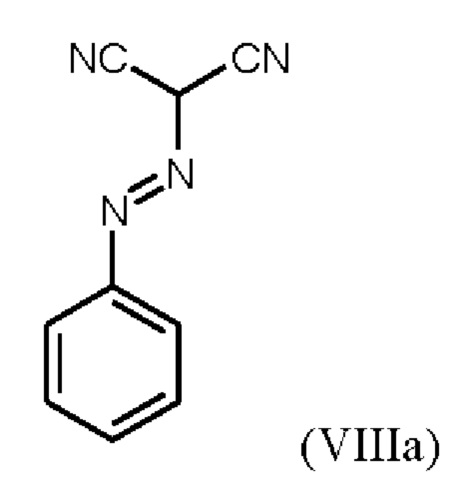
В WO 2013/076168 промежуточное соединение (VIIIa)
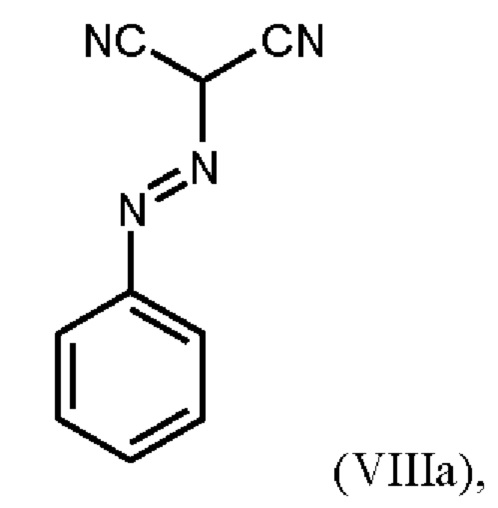
которое применяют в превращении (VII)+(VIIIa)→(VIII) в соответствии со стадией е) схемы 1, синтезируют в соответствии с примером 10 A. [(E)-фенилдиазенил]малононитрил (соединение (VIIIa)) промывают в каждом случае три раза 5,3 л воды на кг анилина и по 4,15 л толуола на кг анилина. Эта процедура промывки невыгодна, так как толуол не смешивается с водой, поэтому замена воды оказывается затруднительной и может привести к неполному удалению солей.
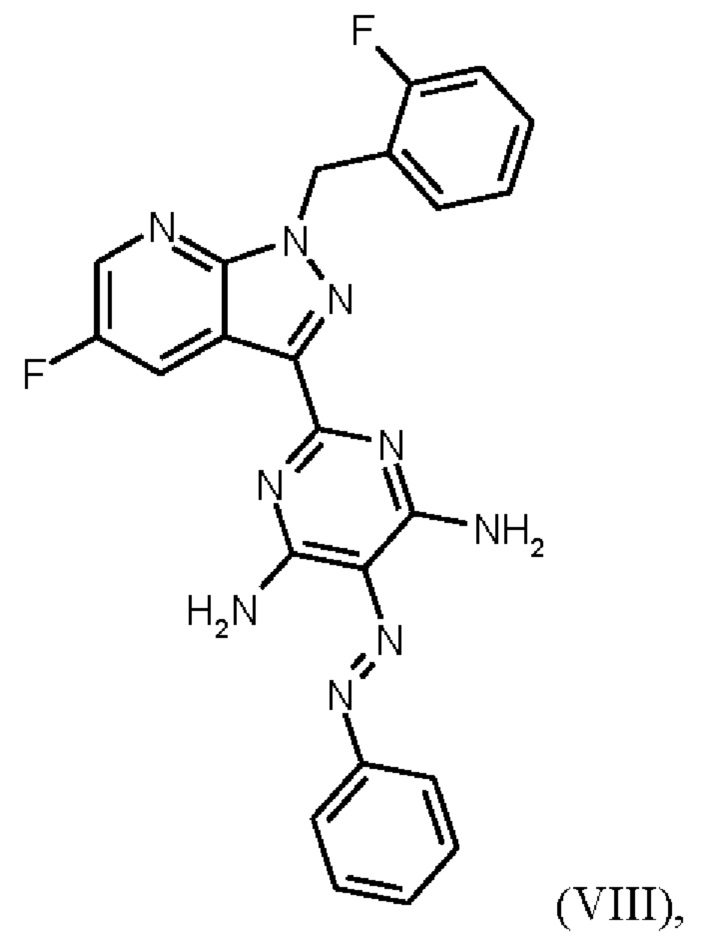
Превращение (VII)+(VIIIa) → (VIII) в соответствии со стадией е) схемы 1 осуществляют в соответствии с примером 11 A WO 2013/076168. В этой реакции один эквивалент соединения (VII), полученного в примере 9 WO 2013/076168, нагревают в DMF. Затем 1,7 экв. соединения (VIIIa) на 1,1 экв. триэтиламина в DMF добавляют в течение 30 мин. Общее количество DMF составляет5,8 кг/кг соединения (VII).
В этих условиях побочный продукт формулы (VIIIb)
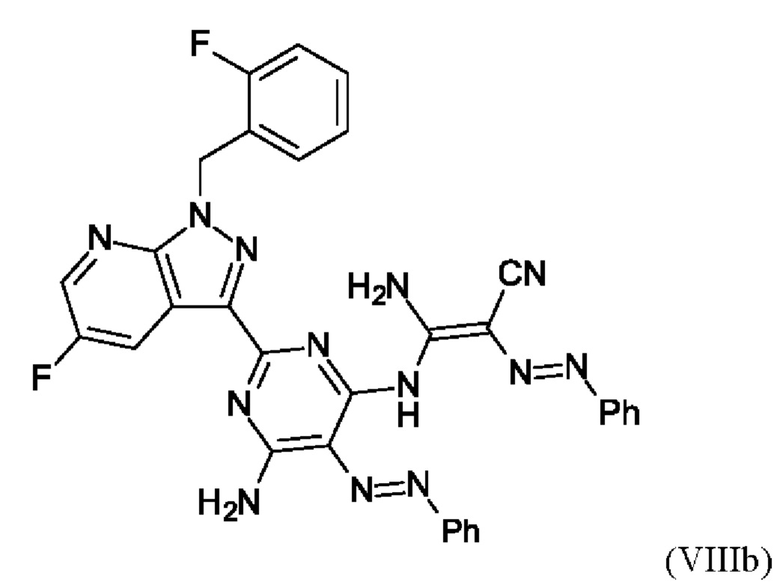
получают реакцией двух молекул соединения (VIIIa) с соединением (VII) в присутствии триэтиламина (пример IIA). Этот побочный продукт необходимо тщательно удалять, что является основным недостатком способа согласно примеру IIA WO 2013/076168.
В способе согласно примеру 12 WO 2013/076168 превращение (VIII) → (IX), стадия f) схемы 1 путем гидрирования проводят в 10 л DMF/кг исходного вещества (соединение (VIII)). Это имеет недостаток, что продукт (соединение (IX)) образует сольват с DMF, который необходимо перевести в свободную от сольватов форму с помощью горячей воды и больших усилий. Оставшийся DMF будет образовывать формильный побочный продукт (соединение (1b), пример 13А) на следующей реакционной стадии g) Схемы 1 путем взаимодействия остаточного DMF с метилхлорформиатом и соединением формулы (IX) из Примера 12 вместо гидрохлорида соединения формулы (I). Эта примесь должна быть удалена с большим усилием. Еще одним недостатком способа согласно примеру 12 WO 2013/076168 является низкая растворимость продукта (соединение (IX)) в DMF. Во время фильтрации для удаления катализатора кристаллизация продукта вызывает препятствия, которые очень неблагоприятны для осуществления способа в техническом масштабе.
Еще одним недостатком стадии f) примера 12 способа согласно WO 2013/076168 является необходимость удаления основной части DMF путем отгонки после гидрирования, что является сложной стадией из-за высокой температуры кипения DMF (162°С). Отсутствие перегонки DMF перед кристаллизацией требует повышенного количества воды и приводит к снижению выхода, что еще более невыгодно.
Способ согласно примеру 13 WO 2013/076168, стадия g) схемы 1, ведущий к выделению гидрохлорида соединения (I), проводят в изопропаноле с использованием триэтиламина в качестве основания. В этом способе исходное вещество (соединение (IX)) суспендируют в изопропаноле и вводят в реакцию с 1,3 экв. (по отношению к исходному веществу) метилхлорформиата, который растворяют в изопропаноле в течение продолжительного времени реакции, составляющего 20 ч, с получением суспензии продукта (гидрохлорида соединения (I)). Как описано выше, оставшийся DMF со стадии f) схемы 1 будет образовывать формильный побочный компонент (соединение (lb), пример 13А) в результате реакции остаточного DMF с метилхлорформиатом и соединением формулы (IX), пример 12, вместо гидрохлорида соединения формулы (I). Длительное время реакции и относительно высокий избыток метилхлорформиата по отношению к исходному веществу являются невыгодными для проведения способа в техническом масштабе. Избыток метилхлорформиата необходимо разрушить добавлением метанола. Продукт способа (гидрохлорид соединения (I)) подвергают непосредственному взаимодействию с триэтиламином с получением соединения (I) без выделения. При фильтровании гидрохлорида триэтиламина кристаллизация продукта приводит к закупорке и возможной блокировке фильтрующего оборудования. Это приводит к потерям продукта и недостаткам в выполнении способа. Выход на этой стадии реакции составляет всего 70% от теоретического.
Как указано в WO 2020/126983 (опубликованной после даты приоритета настоящего изобретения), получение соединения формулы (I) в кристаллической форме модификации I, как описано в WO 2013/076168, приводит к очень тонким волос-подобным структурам, которые при выделении с помощью фильтрации с перепадом давления или в фильтрующих центрифугах образуют очень плотную, похожую на войлок фильтровальную корку, обладающую очень высокой прочностью на разрыв из-за всенаправленного наслоения кристаллов. Можно ожидать, что этот эффект будет более выражен в центробежном поле, чем при фильтрации с перепадом давления, из-за более компактной конфигурации фильтрационной корки. Это приводит к длительному времени выделения, а во время выгрузки из промышленных установок выделения могут возникнуть проблемы, когда фильтровальная корка не ломается и не разрушатся, блокируя, таким образом, путь выгрузки. Можно ожидать, что эти войлочные структуры фильтрационной корки приведут к проблемному поведению сыпучего материала на всех последующих стадиях способа, таких как сушка в вакуумной контактной сушилке, просеивание или микронизация. Из-за частого засорения сита просеивание на промышленных просеивающих машинах может осуществляться только при очень низкой производительности и, следовательно, проблематично. Транспортировка твердых частиц перед последующей микронизацией затруднена из-за высокого электростатического заряда и связанного с этим прилипания к частям установки (например, транспортному каналу).
Задачей настоящего изобретения является обеспечение нового и эффективного способа, который можно осуществить в техническом масштабе для получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (1) в кристаллической форме модификации I, как определено выше, в качестве активного соединения с очень высокой чистотой и очень высоким выходом, с улучшенными физическими свойствами для работы с твердыми веществами, что значительно более экономически выгодно и позволяет избежать недостатков способа, известного из уровня техники, и который может быть осуществлен на обычном опытном и производственном оборудовании (емкость с мешалкой/устройства для выделения).
Еще одной задачей настоящего изобретения является получение продукта активного соединения формулы (I) в кристаллической форме модификации I, как определено выше, который по сравнению с продуктом активного соединения формулы (I) в кристаллической форме модификации I, полученной с помощью способа, описанного в WO 2013/076168, проявляет лучшие свойства, среди прочего, в отношении способности к выделению продукта активного соединения, возможности выгрузки продукта активного соединения после выделения и сушки, а также транспортабельности, просеиваемости и микронизируемости, и поэтому подходит для промышленного производства фармацевтических активных соединений в твердой лекарственной форме.
В контексте настоящего изобретения «продукт активного соединения» определяется как соединение формулы (I) в кристаллической форме модификации I, как определено выше, в твердой форме, которое получают в результате способа, описанного в WO 2013/076168, или способа согласно настоящему изобретению, включающему стадию i) приведенной ниже схемы 2 или примера 15.
В контексте настоящего изобретения и как указано в WO 2020/126983 «улучшенные физические свойства при работе с твердыми веществами», например, в отношении способности к выделению продукта активного соединения формулы (I) в кристаллической форме модификации I, как определено выше, способности к выгрузке продукта активного соединения после выделения и сушки, а также способности к транспортировке, способности к просеиванию и микронизируемость, определяются как улучшение указанных свойств продукта активного соединения формулы (I) в кристаллической форме модификации I, полученного способом согласно настоящему изобретению, по сравнению со свойствами продукта активного соединения формулы (I) в кристаллической форме модификации I, полученного способом согласно WO 2013/076168.
Еще одной задачей настоящего изобретения является получение соединения формулы (I) в определенной модификации, в частности, в кристаллической форме модификации I, определенной выше. Еще одной задачей настоящего изобретения является предотвращение образования гидратов или дигидратов соединения формулы (I) в кристаллической форме модификации I в ходе способа получения согласно настоящему изобретению. Кроме того, по сравнению с твердой лекарственной формой, содержащей соединение формулы (I) в кристаллической форме модификации I, полученной способом, описанным в WO 2013/076168, соединение формулы (I) в кристаллической форме модификации I, полученное способом согласно настоящему изобретению, в твердой лекарственной форме, полученной из него, должно проявлять фармацевтические свойства, которые по меньшей мере являются в равной степени хорошими.
Эта задача достигается в соответствии с настоящим изобретением следующим образом. Схема 2 ниже иллюстрирует отдельные стадии реакции в качестве примера.
Схема 2
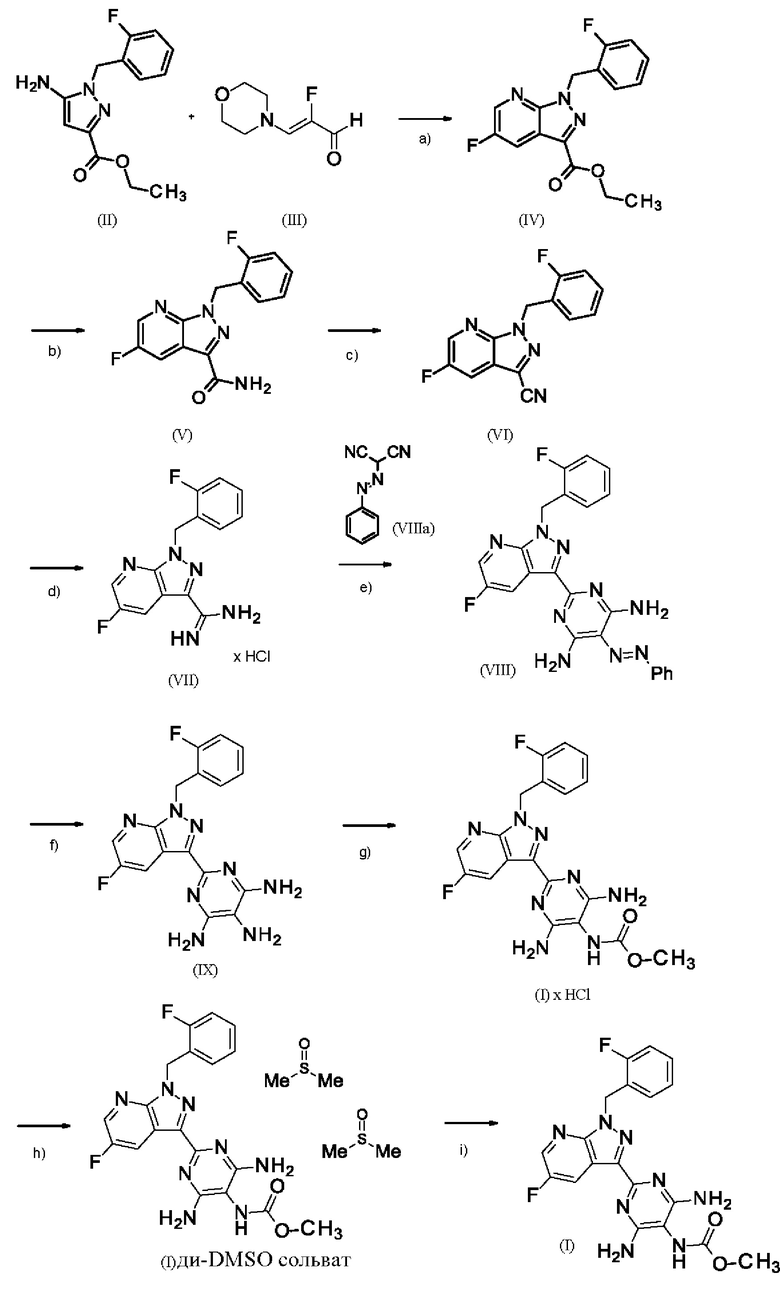
[а): хлорид лития, хлортриметилсилан, этанол; b) формамид, метоксид натрия/метанол, стадии а) + b) проводят в виде реакции в одном сосуде; с) 1. фосфорилхлорид, ацетонитрил, сульфолан, 2. воды; d) 1. суспендирование в метаноле, добавление метоксида натрия/метанола, 2. хлорид аммония/метанол; е) DMF, триэтиламин; f) 1. Pd/C, водород, NMP, 2. вода; g) THF, метилхлорформиат, выделение (I) × НС1; h) 1. DMSO, три-н-бутиламин, 2. Этил ацетат; выделение (I-ди-DMSO сольват); i) растворение в DMSO, постадийное добавление этанола, воды, изопропилацетата].
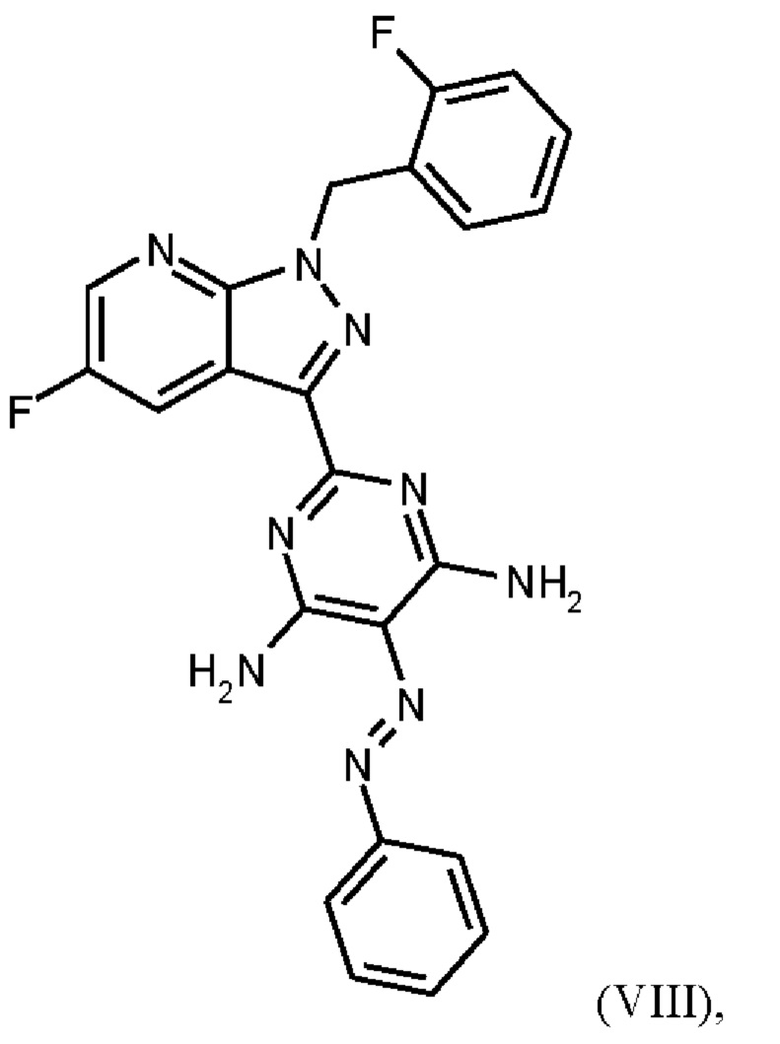
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I)
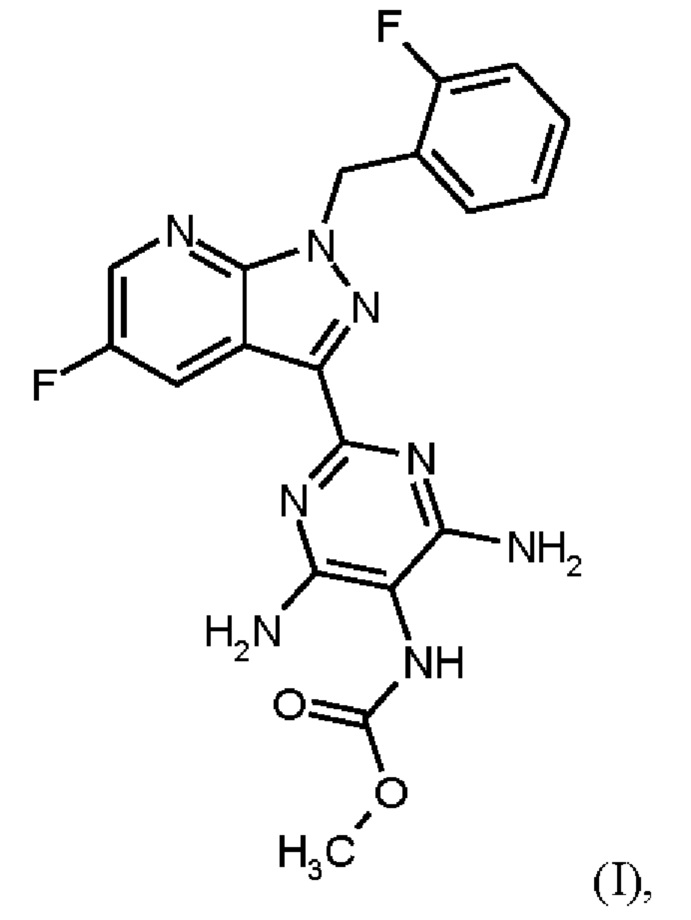
в кристаллической форме модификации I, отличающегося тем, что рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7,
где гидрохлорид соединения формулы (I)
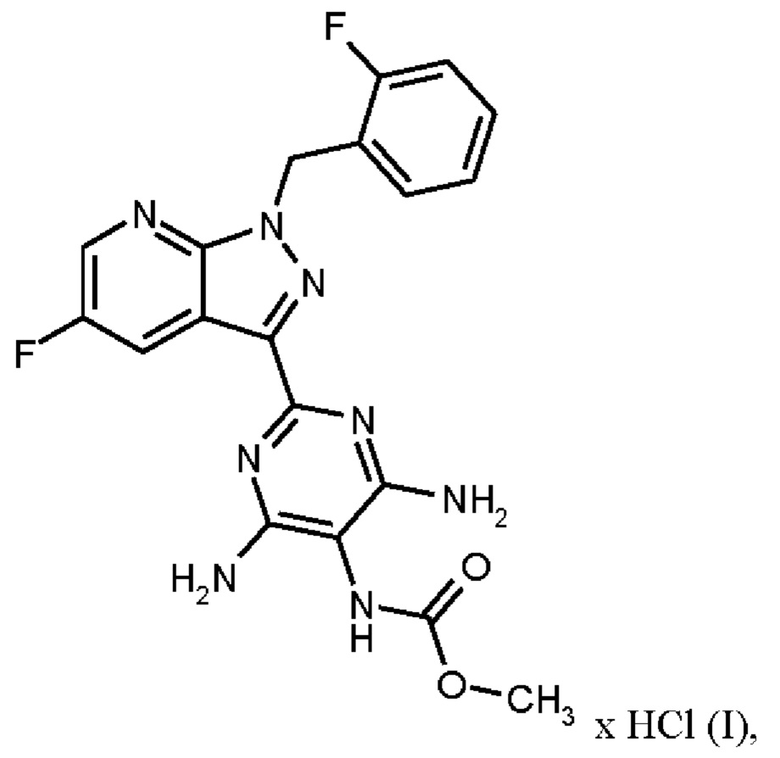
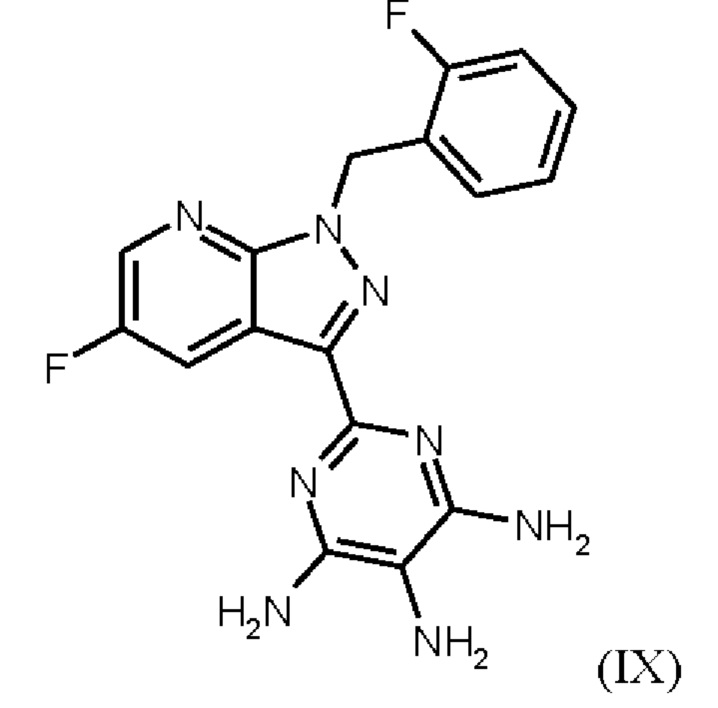
получают путем нагревания соединения формулы (IX)
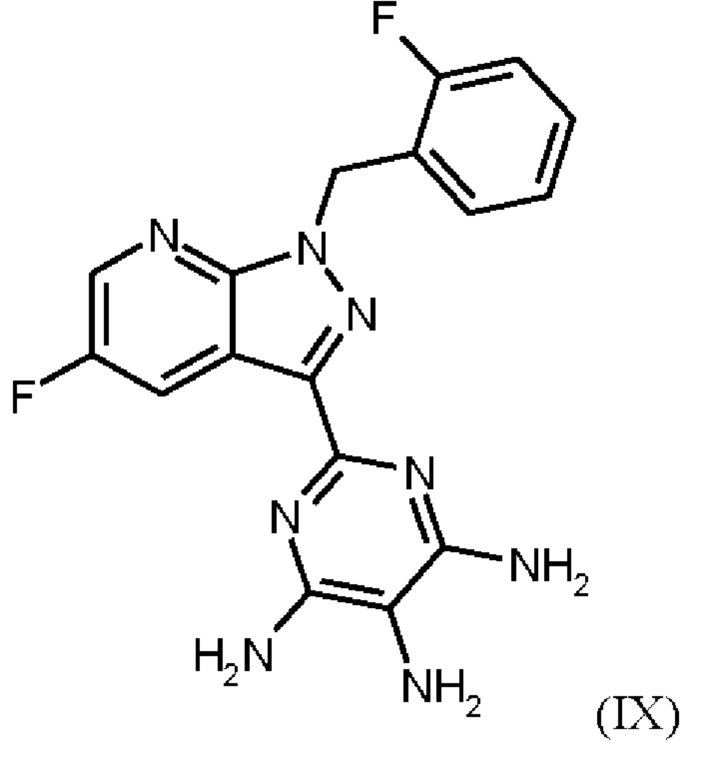
в тетрагидрофуране в качестве растворителя, добавления от 1,0 экв. до 1,2 экв. метилхлорформиата, перемешивания в течение времени реакции от 1 ч до 10 ч и выделения гидрохлорида соединения формулы (I),
затем ди-DMSO сольват соединения формулы (I)
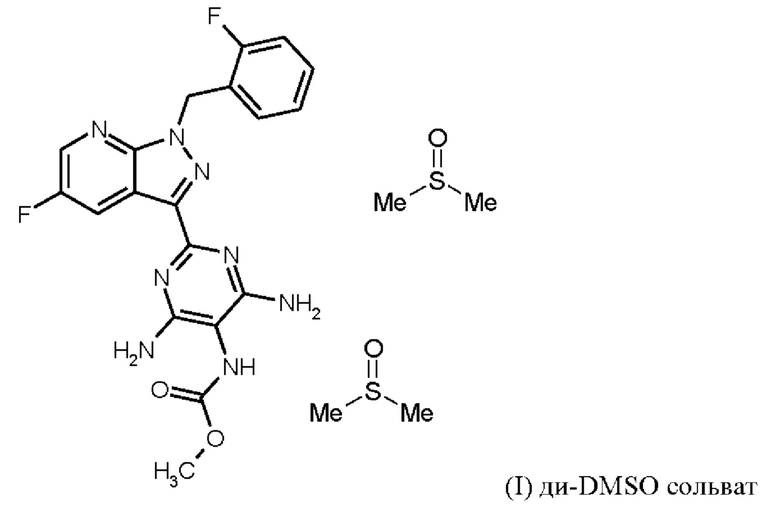
получают путем растворения гидрохлорида соединения формулы (I) в DMSO, добавления три-н-бутиламина и активированного углерода, удаления активированного углерода, кристаллизации ди-DMSO сольвата путем охлаждения и добавления этилацетата, выделения ди-DMSO сольвата в кристаллической форме и промывки смесью DMSO и этил ацетата,
затем соединение формулы (I) в кристаллической форме модификации 1 получают,
где
1.1 ди-DMSO сольват соединения формулы (I) растворяют в DMSO и этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас.,
1.2 растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды;
1.3 образованную суспензию затем охлаждают до температуры от 5°С до 50°С и
1.4 образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
Последовательность реакций соединения (IX) → соединение (I), описанная выше, соответствует стадиям g)-i) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, отличающемуся тем, что рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7,
где гидрохлорид соединения формулы (1) получают путем нагревания соединения формулы (IX) в тетрагидрофуране в качестве растворителя до от 30°С до 66°С, добавления от 1,0 экв. до 1,2 экв. метилхлорформиата в течение от 1 мин до 30 мин, перемешивания при температуре от 30°С до 66°С и в течение времени реакции от 1 ч до 10 ч, выделения гидрохлорида соединения формулы (I) и сушки,
затем ди-DMSO сольват соединения формулы (I) получают путем перемешивания гидрохлорида соединения формулы (I) в течение от 1 ч до 3 ч при от 70°С до 90°С в DMSO, добавления три-н-бутиламина и активированного углерода, перемешивания при от 70°С до 90°С, удаления активированного углерода, промывки с DMSO, охлаждения до от -3°С до +20°С, кристаллизации ди-DMSO сольвата путем добавления этилацетата, выделения ди-DMSO в кристаллической форме, промывки смесью DMSO и этилацетата и сушки,
затем соединение формулы (I) в кристаллической форме модификации I получают,
где
1.1 ди-DMSO сольват соединения формулы (I) суспендируют в DMSO и нагревают до от 70°С до 80°С, этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас., и смесь перемешивают при от 65°С до 85°С в течение от 15 мин до 21 ч,
1.2 растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды при температуре от 15°С до 85°С и в течение от 0,1 мин до 30 мин;
1.3 образованную суспензию затем охлаждают до температуры от 5°С до 50°С в течение от 1 ч до 4 ч, и
1.4 образованные кристаллы на стадии b) затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, отличающегося тем, что рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, включающему реакционные стадии получения соединений формул (I, HCl), (I, ди-DMSO сольват), и (I) в кристаллической форме модификации I согласно настоящему изобретению, как описано в настоящем документе, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,95% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,97% (как измерено посредством % площади по ВЭЖХ) или более.
Согласно одному варианту осуществления настоящего изобретения соединение формулы (IX) нагревают в от 3,00 л до 4,60 л тетрагидрофурана на моль соединения (IX) или в от 3,20 л до 4,30 л тетрагидрофурана на моль соединения (IX). Согласно одному варианту осуществления настоящего изобретения соединение (IX) нагревают до от 30°С до 66°С или от 50°С до 66°С.
Согласно варианту осуществления настоящего изобретения от 0,95 экв. до 1,40 экв. , или от 1,00 экв. до 1,30 экв. , или от 1,0 экв. до 1,2 экв. метилхлорформиата, по отношению к количеству соединения (IX), добавляют. Согласно варианту осуществления настоящего изобретения метилхлорформиат добавляют в течение от 1 мин до 30 мин, или в течение от 10 мин до 20 мин.
Согласно варианту осуществления настоящего изобретения смесь соединения формулы (IX), тетрагидрофурана и метилхлорформиата перемешивают при от 30°С до 66°С, или при от 50°С до 66°С. Согласно варианту осуществления настоящего изобретения эту смесь перемешивают в течение от 1 ч до 10 ч, или от 1 ч до 6 ч, или от 1 ч до 4 ч, или от 2 ч до 3 ч, или 2 ч.
Согласно варианту осуществления настоящего изобретения твердые вещества выделяют и перемешивают с от 1,60 л до 3,00 л тетрагидрофурана на моль соединения (IX), или с от 1,90 л до 2,80 л тетрагидрофурана на моль соединения (IX), изначально дозированного. Согласно варианту осуществления настоящего изобретения твердые вещества выделяют и перемешивают с тетрагидрофураном при от 40°С до 66°С, или при от 45°С до 63°С, в течение от 15 мин до 60 мин. Согласно варианту осуществления настоящего изобретения стадии выделения и перемешивания твердых веществе от 1,60 л до 3,00 л тетрагидрофурана на моль соединения (IX), или с от 1,90 л до 2,80 л тетрагидрофурана на моль соединения (IX), при от 40°С до 66°С, или при от 45°С до 63°С, в течение от 15 мин до 60 мин, повторяют. Согласно варианту осуществления настоящего изобретения твердые вещества собирают при от 40°С до 69°С, или при от 45°С до 63°С, и сушат при от 30°С до 80°С.
Согласно одному варианту осуществления настоящего изобретения промывку кристаллизованного ди-DMSO сольвата проводят смесью DMSO и этилацетата при соотношении от 1:4,5 до 1:5,5 DMSO:этилацетат.
Согласно одному варианту осуществления настоящего изобретения стадию 1.1 способа получения соединения формулы (I) в кристаллической модификации I с последующей фильтрацией.
Согласно одному варианту осуществления настоящего изобретения на стадии 1.2 способа получения соединения формулы (I) в кристаллической модификации I соотношение воды и этанола составляет 2:1-12:1 мас./мас.
Согласно одному варианту осуществления настоящего изобретения стадия 1.4 способа получения соединения формулы (I) в кристаллической модификации I сопровождается выделением, сушкой, просеиванием и измельчением продукта.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, и где соединение формулы (IX) 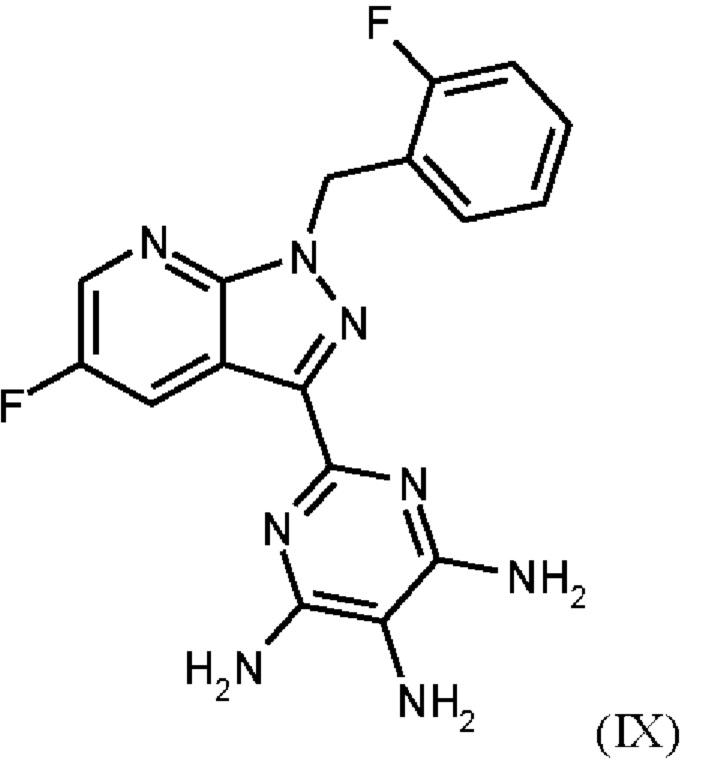
получают путем гидрирования соединения формулы (VIII)
в NMP в качестве растворителя в присутствии водорода, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и выделения с получением соединения формулы (IX).
Реакция соединения (VIII)→соединение (IX), раскрытая выше, соответствует стадии f) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, и где соединение формулы (IX) получают путем гидрирования соединения формулы (VIII) в NMP в качестве растворителя в присутствии водорода при давлении от 50 бар до 90 бар и при температуре от 50°С до 80°С, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и затем выделения и сушки с получением соединения формулы (IX).
Согласно одному варианту осуществления настоящего изобретения гидрирование проводят при концентрации от 4,8 л до 6,8 л NMP/кг исходного вещества (соединение (VIII)). Согласно одному варианту осуществления настоящего изобретения гидрирование проводят при концентрации от 5,1 л до 6,3 л NMP/кг исходного вещества (соединение (VIII)). Согласно другому варианту осуществления настоящего изобретения гидрирование проводят в присутствии водорода при давлении от 50 бар до 90 бар или от 60 бар до 80 бар и при температуре от 50°С до 80°С или от 60°С до 70°С. Согласно другому варианту осуществления настоящего изобретения гидрирование катализируют катализатором палладий на активированном углероде. Согласно другому варианту осуществления настоящего изобретения от 13 г до 48 г 5% Pd/C (50% влажность) или от 15 г до 44 г 5% Pd/C (50% влажность) добавляют на кг исходного вещества (соединение (VIII)).
Согласно другому варианту осуществления настоящего изобретения смесь после гидрирования фильтруют для удаления отработанного катализатора и фильтрующую линию промывают от 0,39 л до 0,58 л NMP на кг исходного вещества (соединение (VIII)), или от 0,44 л до 0,53 л NMP на кг исходного вещества (соединение (VIII)). Согласно другому варианту осуществления настоящего изобретения фильтрат охлаждают до температуры от 10°С до 40°С, затем добавляют от 1,34 л до 2,50 л воды или от 1,50 л до 2,30 л воды в течение 3 ч или более и смесь перемешивают в течение от 0,5 ч до 13 ч, или от 1 ч до 6 ч. Согласно другому варианту осуществления настоящего изобретения твердые вещества выделяют, промывают водой от 2x0,21 л до 0,63 л на кг исходного вещества (соединение (VIII)) или водой от 2×0,24 л до 0,60 л на кг исходного вещества (соединение (VIII)) и затем сушат при температуре от 40°С до 120°С.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, включающему реакционные стадии получения соединений формул (IX), (I, HCl), (I, ди-DMSO сольват) и (I) в кристаллической форме модификации I согласно настоящему изобретению, как описано в настоящем документе, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством %площади по ВЭЖХ) или более, или чистотой 99,95% (как измерено посредством %площади по ВЭЖХ) или более, или чистотой 99,97% (как измерено посредством % площади по ВЭЖХ) или более.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, и где соединение формулы (VIII)
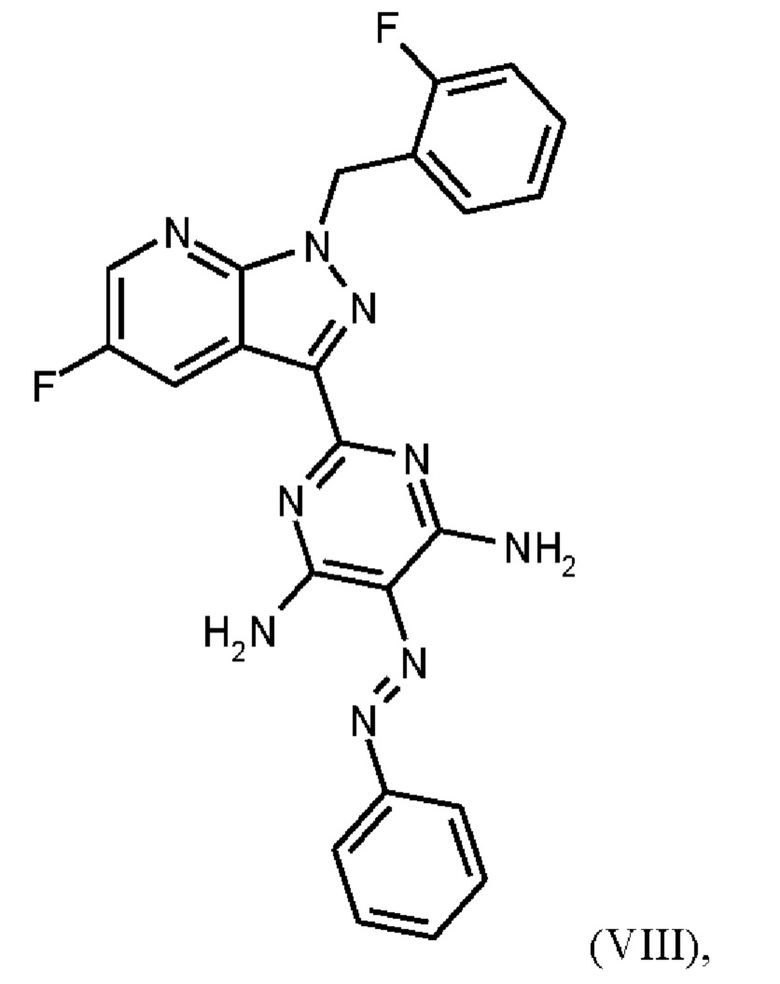
получают путем начального получения соединения формулы (VIIIa)
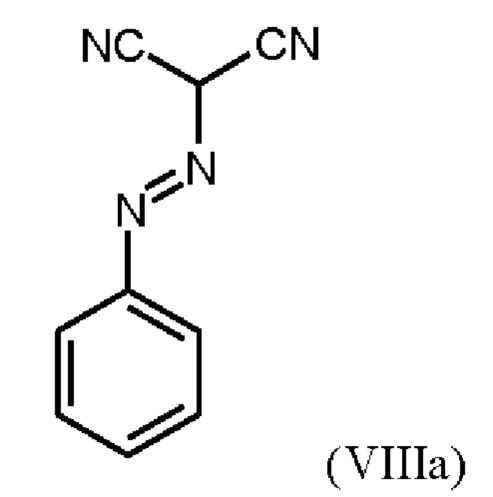
путем добавления концентрированной соляной кислоты в воде к анилину в воде, затем последовательного добавления раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII)
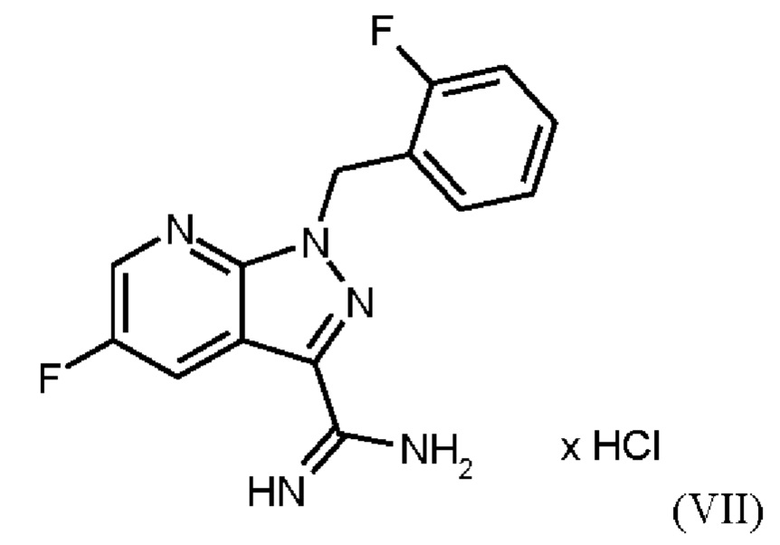
в DMF, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы VII), добавления метанола и выделения соединения формулы (VIII).
Реакция соединения (VII)+(VIIIa) → соединение(VIII) соответствует стадии е) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VIII) получают путем начального получения соединения формулы (VIIIa) путем добавления концентрированной соляной кислоты в воде к анилину в воде при от -3 до 12°С, затем последовательного добавления при той же температуре раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII) в DMF до от 85°С до 115°С, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), в течение от 5 ч до 15 ч, охлаждения до от 77°С до 88°С, добавления метанола и выделения соединения формулы (VIII).
Согласно одному варианту осуществления настоящего изобретения соединение формулы (VIIIa)получают путем добавления от 1,9 экв. до 2,2 экв. концентрированной соляной кислоты по отношению к анилину в воде к от 0,9 экв. до 1,1 экв. анилина в воде при температуре от -3°С до +12°С, или при температуре от 0°С до 5°С, затем последовательного добавления при той же температуре раствора от 0,95 экв. до 1,1 экв. по отношению к анилину нитрита натрия в воде в течение от 5 мин до 90 мин, раствора от 1,17 экв. до 1,43 экв. по отношению к анилину ацетата натрия в воде в течение от 5 мин до 90 мин, и раствора от 0,9 до 1,1 экв. по отношению к анилину малононитрила в этаноле в течение от 0,5 ч до 2 ч, выделения твердых веществ, и промывки в каждом случае три раза водой и изопропанолом с получением соединения формулы (VIIIa);
Согласно одному варианту осуществления настоящего изобретения промывку соединения формулы (VIIIa) проводят в каждом случае три раза от 5,2 л до 12,8 л воды на кг анилина и от 3,5 л до 4,8 л изопропанола на кг анилина.
Согласно одному варианту осуществления настоящего изобретения соединение формулы (VII) нагревают DMF до от 85°С до 115°С, соединение формулы (VIIIa), которое растворяют в DMF, и от 1,3 экв. до 1,6 экв. триэтиламина по отношению к соединению формулы (VII) добавляют в течение от 5 ч до 15 ч. Согласно одному варианту осуществления настоящего изобретения смесь далее перемешивают при 100°С в течение от 10,5 ч до 24 ч, охлаждают до от 77°С до 88°С, метанол добавляют по каплям, и полученную смесь охлаждают до от -2°С до+15°С в течение от 4 ч до 10 ч и перемешивают в течение от 0,5 ч до 11 ч, и твердые вещества выделяют.
Согласно одному варианту осуществления настоящего изобретения продукт формулы (VIII) последовательно промывают DMF, метанолом, водой и метанолом.
Согласно одному варианту осуществления настоящего изобретения соединение формулы (VII) суспендируют в общем количестве (включая количество DMF, в котором соединение (VIIIa) растворяют) от 4,7 кг до 6,1 кг DMF на кг соединения формулы (VII).
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, включающему реакционные стадии получения соединений формул (VIII), (IX), (I, НС1), (I, ди-DMSO сольват) и (I) в кристаллической форме модификации I согласно настоящему изобретению, как описано в настоящем документе, где соединение формулы (I) в кристаллической форме модификации I получаю тс чистотой 99,90% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,95% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,97% (как измерено посредством % площади по ВЭЖХ) или более.
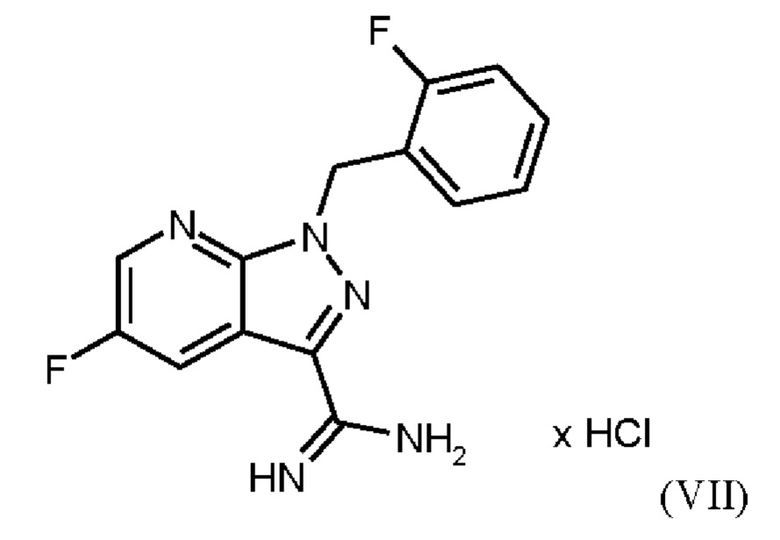
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VII)
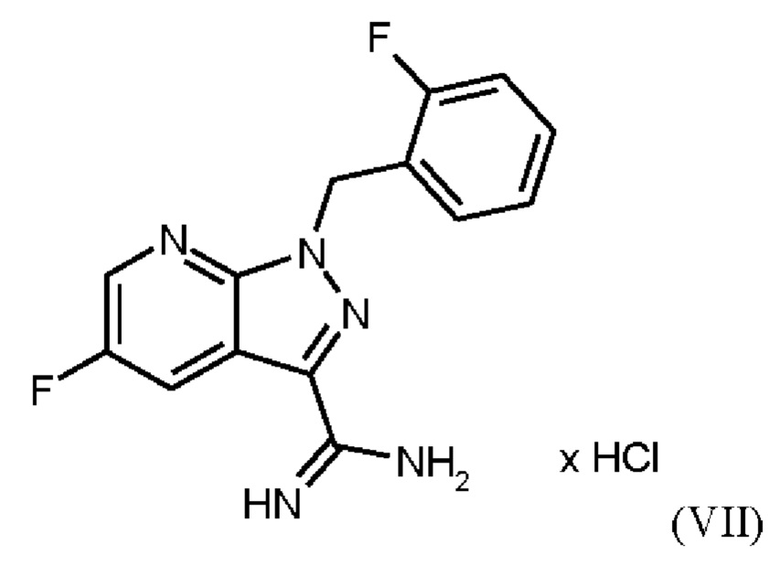
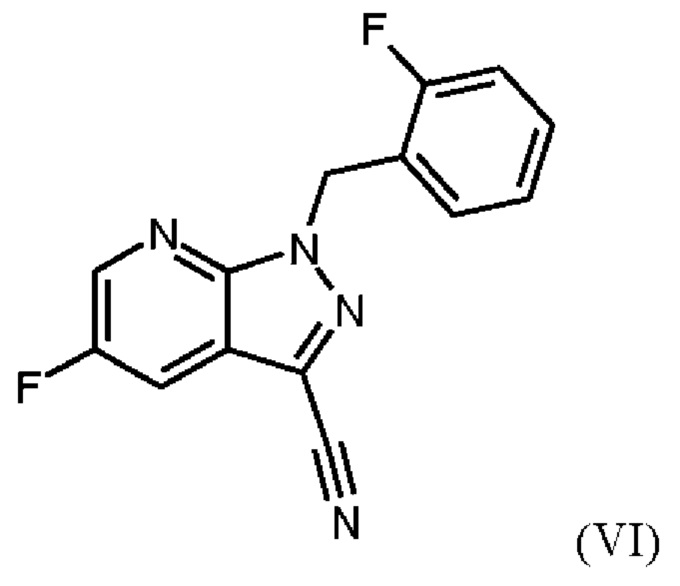
получают путем суспендирования соединения формулы (VI)
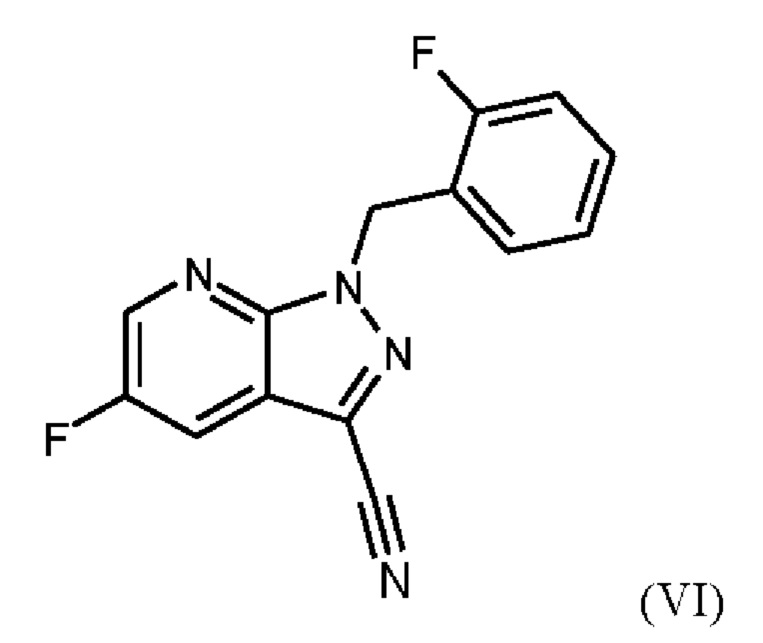
в метаноле, добавления метоксида натрия в метаноле, добавления метанола и хлорида аммония, фильтрации с использованием вспомогательного фильтра, концентрирования и добавления этилацетата, добавления этанола и выделения с получением соединения формулы (VII).
Реакция соединения (VI) → соединение (VII) соответствует стадии d) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VII) получают путем суспендирования соединения формулы (VI) в метаноле, добавления метоксида натрия в метаноле, перемешивания, добавления метанола и хлорида аммония, нагревания с возвратом флегмы, перемешивания, охлаждения, фильтрации с использованием вспомогательного фильтра, концентрирования фильтратов посредством дистилляции, добавления этилацетата, продолжения дистилляции с заполнением отогнанного объема этилацетатом, охлаждения, добавления этанол, выделения, промывки этилацетатом и сушки с получением соединения формулы (VII).
Согласно одному варианту осуществления настоящего изобретения суспензию после добавления метоксида натрия в метаноле, перемешивают в течение 5-10 ч при 15-30°С. Согласно одному варианту осуществления настоящего изобретения суспензию после добавления метанола и хлорида аммония и нагревания c возвратом флегмы перемешивают в течение от 4,5 до 10 ч. Согласно одному варианту осуществления настоящего изобретения суспензию охлаждают до 15-40°С после добавления метанола и хлорида аммония, нагревания с возвратом флегмы и перемешивания.
Согласно одному варианту осуществления настоящего изобретения применяемый вспомогательный фильтр выбирают из диатомовой земли, также называемой диатомитом или кизельгуром. Согласно одному варианту осуществления настоящего изобретения применяемый вспомогательный фильтр представляет собой активированную кальцинированную диатомовую землю. Согласно одному варианту осуществления настоящего изобретения применяемый вспомогательный фильтр представляет собой CLARCEL диатомовую землю. Согласно одному варианту осуществления настоящего изобретения применяемый вспомогательный фильтр представляет собой CLARCEL DICB.
Вспомогательный фильтр CLARCEL® DICB получают путем прокаливания/активации (прокаливание под флюсом) очищенного диатомита. Его цвет белый, а содержание кремнезема (SiO2) составляет около 89%. Этот продукт соответствует спецификациям текущей монографии Кодекса пищевых химикатов США.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, включающему реакционные стадии получения соединений формул (VII), (VIII), (IX), (I, HCl), (I, ди-DMSO сольват) и (I) в кристаллической форме модификации I согласно настоящему изобретению, как описано в настоящем документе, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,95% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,97% (как измерено посредством %площади по ВЭЖХ) или более.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VI)
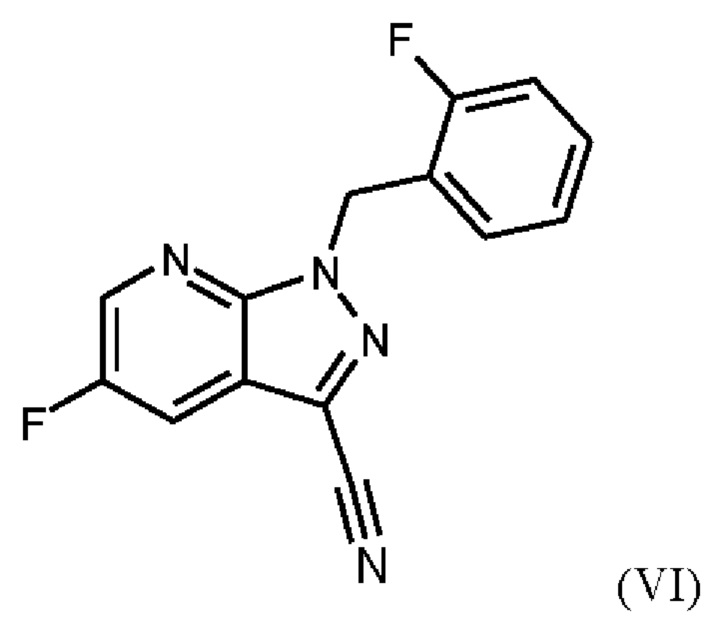
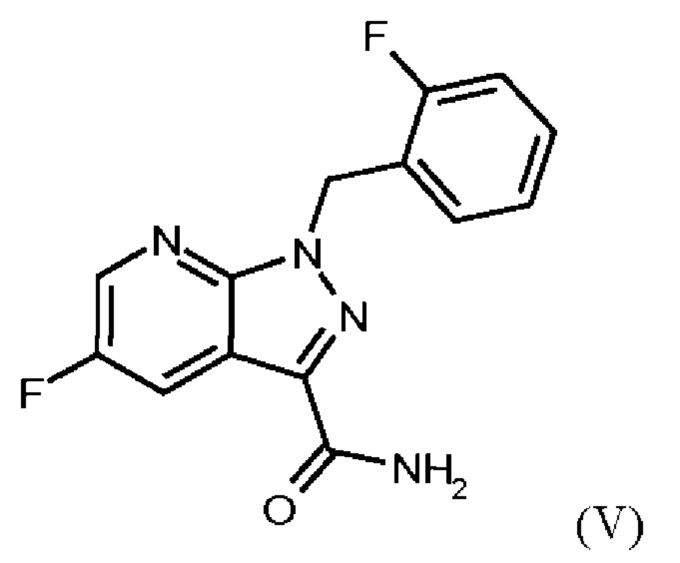
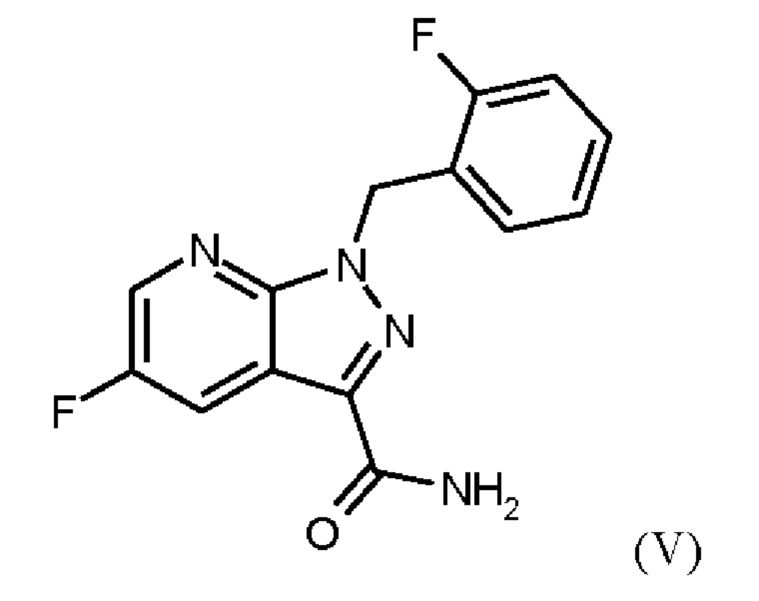
получают путем сначала получения соединения формулы (V)
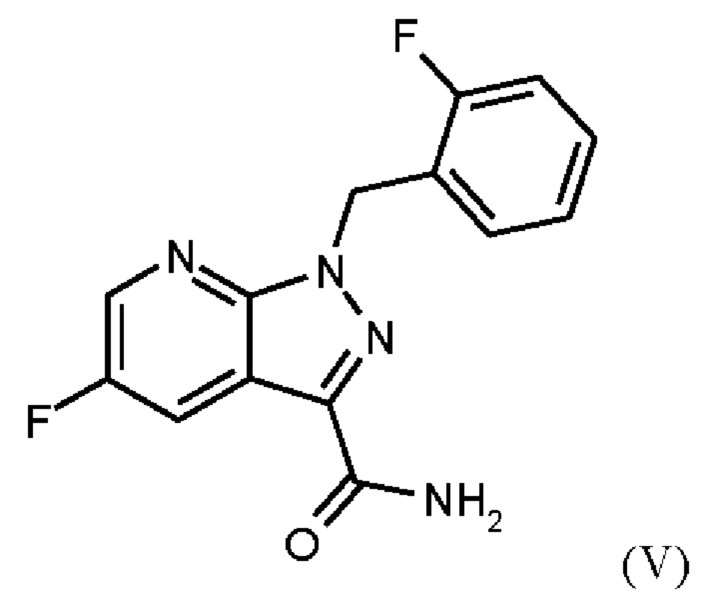
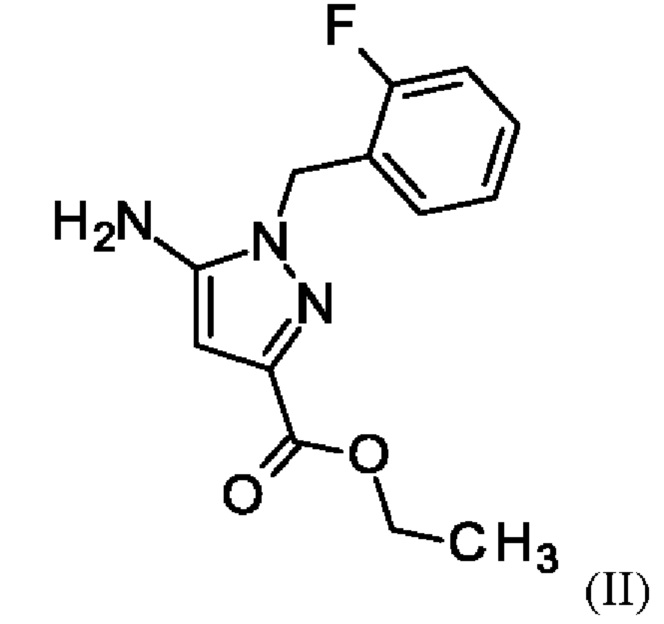
путем начальной загрузки соединения формулы (II)
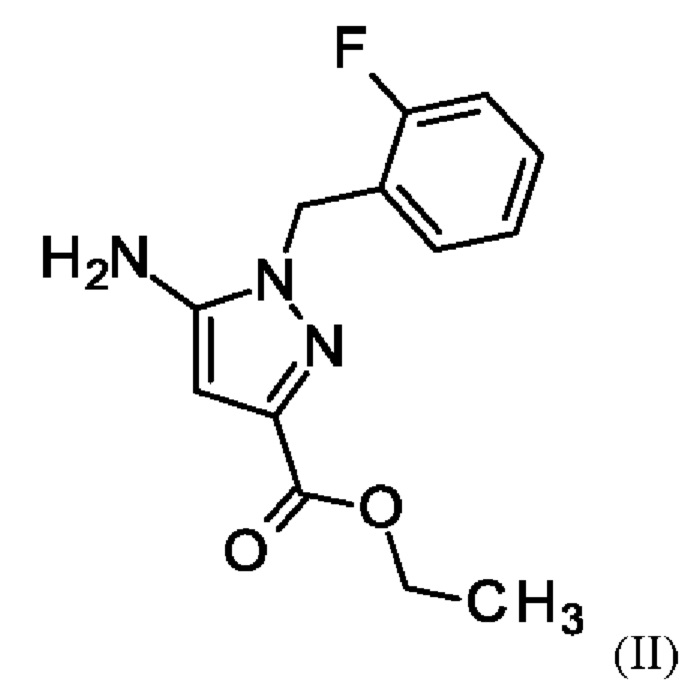
и хлорида лития в этаноле, загрузки соединения формулы (III),
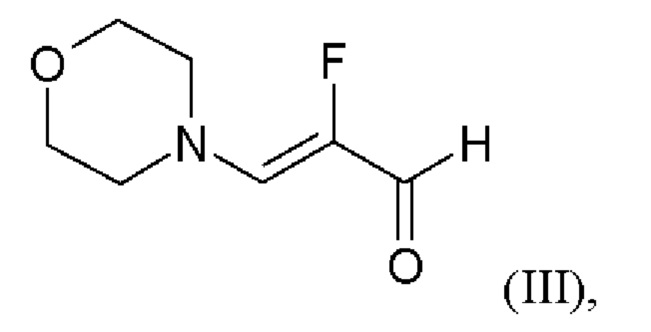
и хлортриметилсилана, и нагревания с получением соединения формулы (IV)
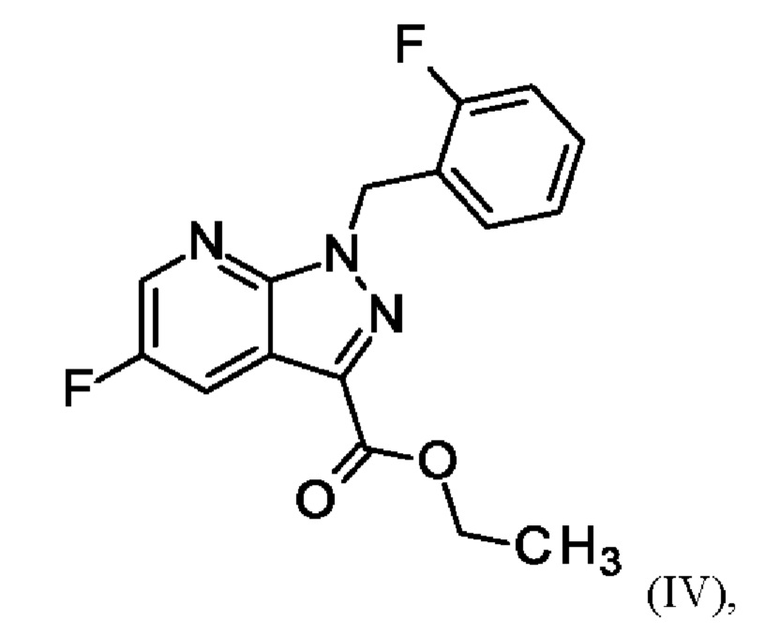
или альтернативно изменяя порядок загрузки любого из исходных веществ, добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V),
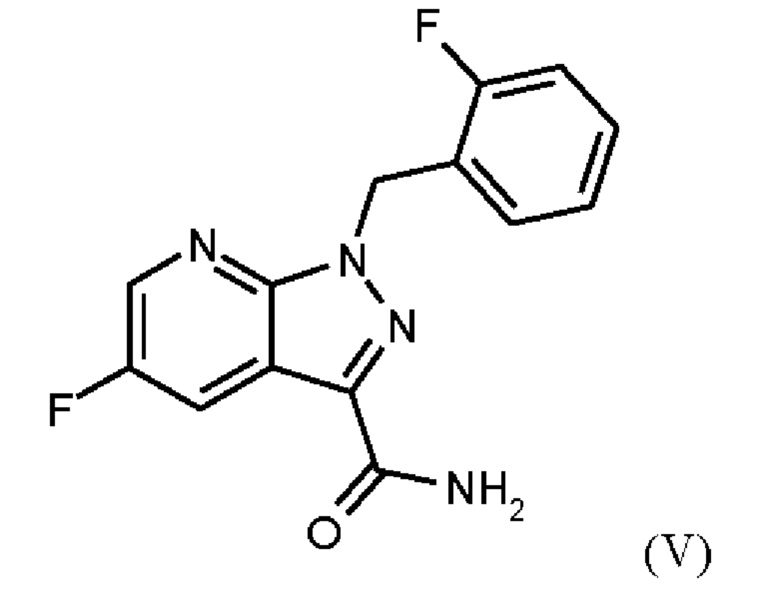
затем дегидратации соединения формулы (V) путем нагревания в сульфолане, ацетонитриле и фосфорилхлориде, добавления ацетонитрила и воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20°С до 50°С, добавления аммиака в воде, и выделения с получением соединения формулы (VI).
Последовательность реакций соединения (II)+соединение (III)→соединение (IV) →соединение (V)→соединение (VI) соответствует стадиям а)-с) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VI) получают путем сначала получения соединения формулы (V) путем начальной загрузки соединения формулы (II) и хлорида лития в этаноле, загрузки соединения формулы (III) и хлортриметилсилана, нагревания с возвратом флегмы и охлаждения, с получением соединения формулы (IV) или альтернативно изменяя порядок загрузки любого из исходных веществ, добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V),
затем дегидратации соединения формулы (V) путем нагревания в сульфолане, ацетонитриле и фосфорилхлориде, промывки ацетонитрилом, перемешивания при повышенной температуре, охлаждения, добавления ацетонитрила, добавления воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20°С до 50°С, добавления аммиака в воде, выделения, промывки водой и сушки с получением соединения формулы (VI).
Согласно одному варианту осуществления настоящего изобретения соединение формулы (V) получают путем начальной загрузки соединения формулы (II) и от 2,25 экв. до 2,75 экв. хлорида лития по отношению к соединению формулы (II) в этаноле, загрузки от 0,85 экв. до 1,2 экв. или от 0,85 экв. до 1,0 экв. по отношению к соединению формулы (II) соединения формулы (III), добавления от 1,6 экв. до 2,3 экв. хлортриметилсилана по отношению к соединению формулы (II), нагревания с возвратом флегмы и охлаждения, с получением соединения формулы (IV), где порядок загрузки любого из исходных веществ может быть изменен, добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V), где метоксид натрия применяют в избытке 0,4 экв. или более по отношению к экв. применяемого хлортриметилсилана.
Согласно одному варианту осуществления настоящего изобретения соединение формулы (V) дегидратируют путем нагревания до от 100°С до 120°С в сульфолане и ацетонитриле, по каплям добавления фосфорилхлорида, промывки ацетонитрилом, перемешивания в течение от 4 ч до 10 ч при повышенной температуре, охлаждения, добавления ацетонитрила, добавления воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20°С до 50°С, добавления аммиака в воде, сбора твердых веществ путем фильтрации, промывки водой и сушки с получением соединения формулы (VI).
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, включающему реакционные стадии получения соединений формул (IV), (V), (VI), (VII), (VIII), (IX), (I, НС1), (I, ди-DMSO сольват) и (I) в кристаллической форме модификации I согласно настоящему изобретению, как описано в настоящем документе, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством %площади по ВЭЖХ) или более, или чистотой 99,95% (как измерено посредством % площади по ВЭЖХ) или более, или чистотой 99,97% (как измерено посредством %площади по ВЭЖХ) или более.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (V),
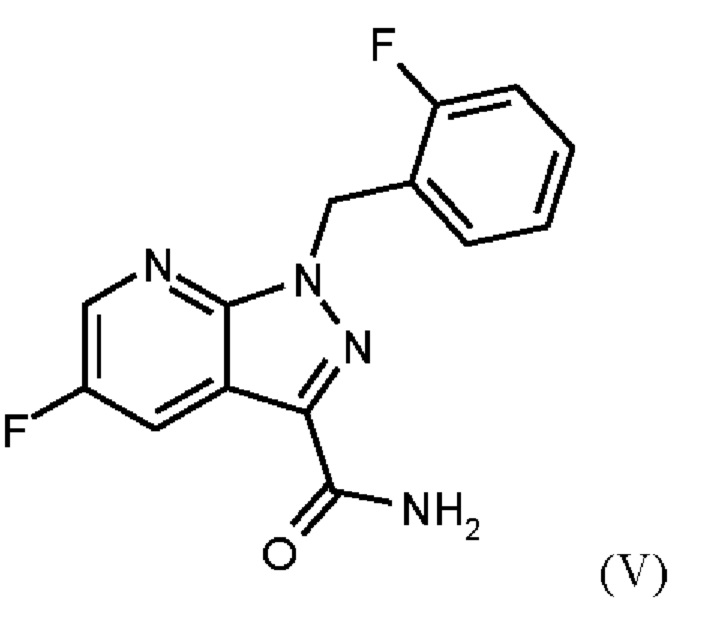
где соединение формулы (II)
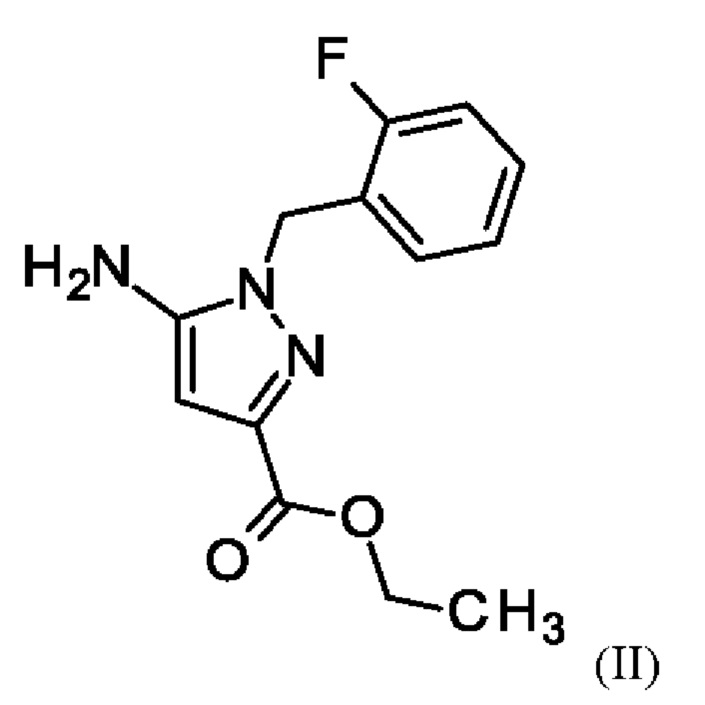
и хлорид лития изначально загружают в этаноле, загружают соединение формулы (III),
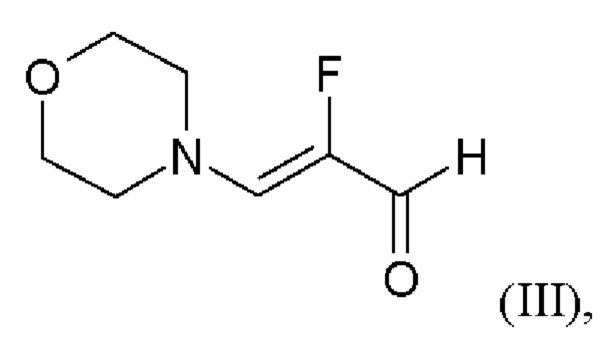
и хлортриметилсилан, и нагревают с получением соединения формулы (IV)
или альтернативно изменяют порядок загрузки любого из исходных веществ,
добавляют формамид и метоксид натрия метаноле, отгоняют низкокипящие соединения, при доливке отогнанного объема формамидом, охлаждают, добавляют воду, выделяют твердые вещества, промывают и сушат с получением соединения формулы (V).
Последовательность реакций соединения (II)+соединение (III) →соединение (IV) →соединение (V) соответствует стадиям а) и b) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (V), где соединение формулы (II) и хлорид лития изначально загружают в этаноле, загружают соединение формулы (III) и хлортриметилсилан, нагревают с возвратом флегмы и охлаждают, с получением соединения формулы (IV), или альтернативно изменяют порядок загрузки любого из исходных веществ, добавляют формамид и метоксид натрия в метаноле, отгоняют низкокипящие соединения, при доливке отогнанного объема формамидом, охлаждают, добавляют воду, выделяют твердые вещества, промывают и сушат с получением соединения формулы (V).
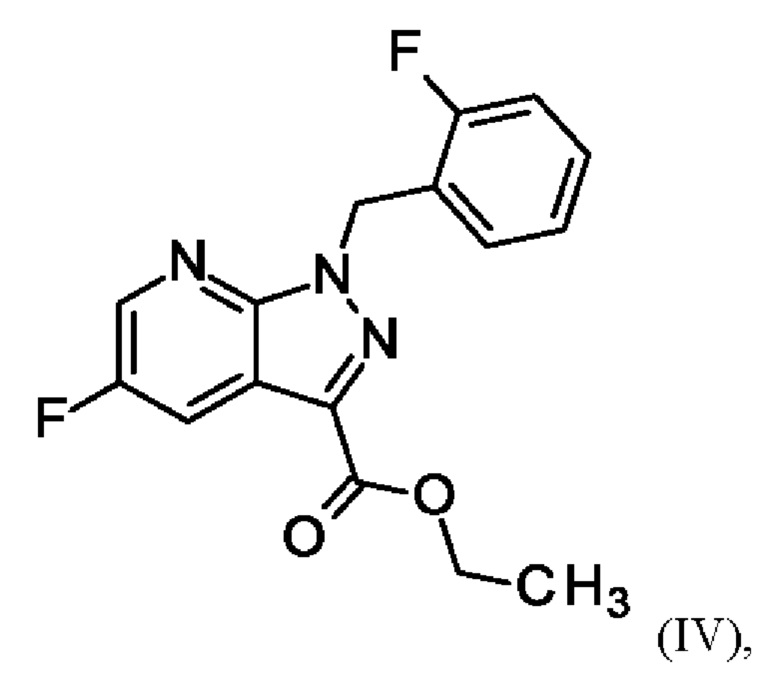
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (VII)
где соединение формулы (VI)
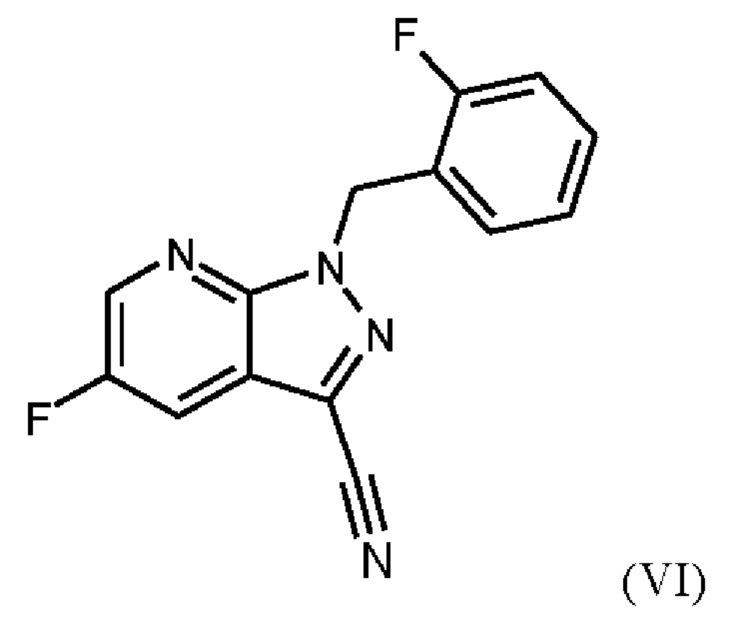
суспендируют в метаноле, добавляют метоксид натрия в метаноле, добавляют метанол и хлорид аммония, фильтруют с использованием вспомогательного фильтра, концентрируют и добавляют этилацетат, добавляют этанол и выделяют с получением соединения формулы (VII).
Реакция соединения (VI) →соединение (VII) соответствует стадии d) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (VII), где соединение формулы (VI) суспендируют в метаноле, добавляют метоксид натрия в метаноле, перемешивают, добавляют метанол и хлорид аммония, нагревают с возвратом флегмы, перемешивают, охлаждают, фильтруют с использованием вспомогательного фильтра, концентрируют фильтраты посредством дистилляции, добавляют этилацетат, продолжают дистилляцию с заполнением отогнанного объема этилацетатом, охлаждают, добавляют этанол, выделяют, промывают этилацетатом и сушат с получением соединения формулы (VII).
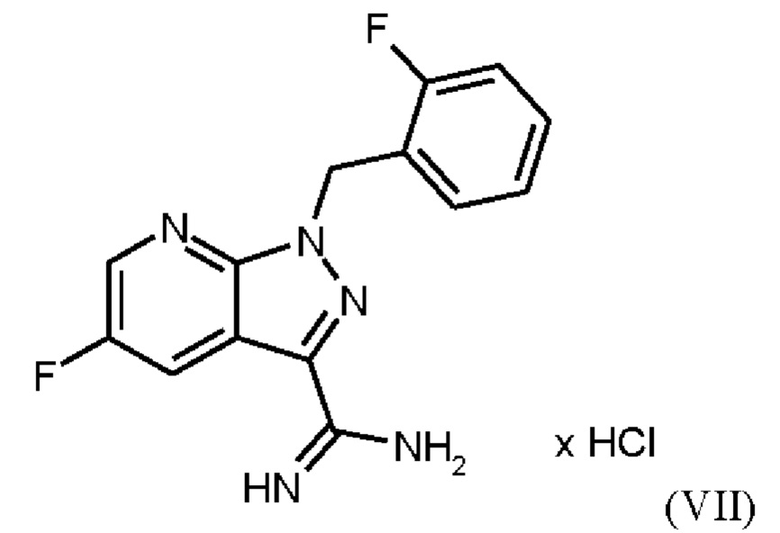
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (VIII),
где соединение формулы (VIIIa)
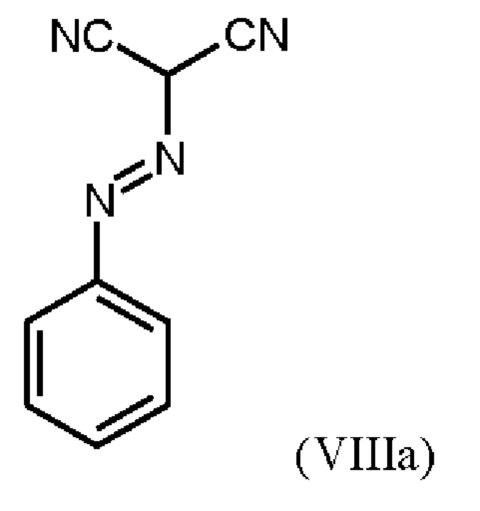
первоначально получают путем добавления концентрированной соляной кислоты в воде к анилину в воде, затем последовательного добавления раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII)
в DMF, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), добавления метанола и выделения соединения формулы (VIII).
Реакция соединения (VII)+соединение (VIIIa) →соединение (VIII) соответствует стадии е) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (VIII), где соединение формулы (VIIIa) первоначально получают путем добавления концентрированной соляной кислоты в воде к анилину в воде при от -3 до +12°С, затем последовательного добавления при той же температуре раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII) в DMF до от 85°С до 115°С, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы (VII) в течение от 5 ч до 15 ч, охлаждения до от 77°С до 88°С, добавления метанола и выделения соединения формулы (VIII).
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (IX)
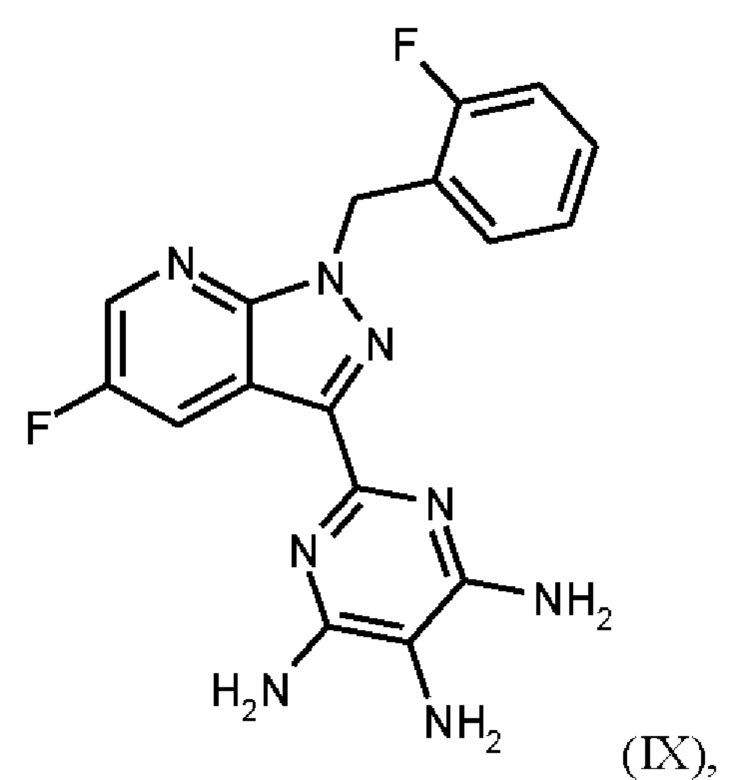
где соединение формулы (VIII)
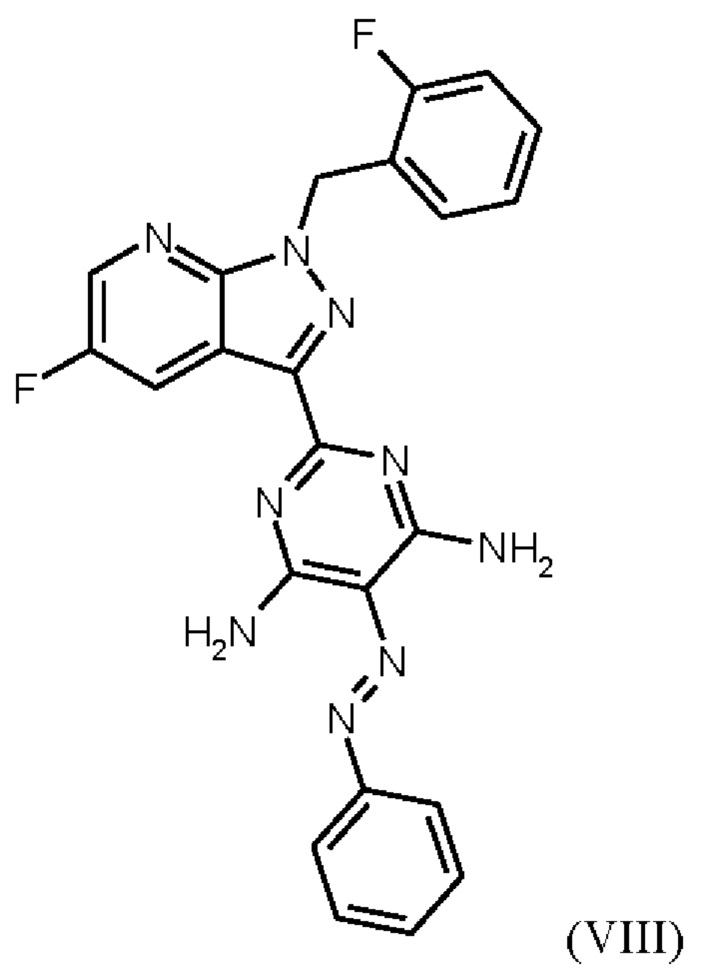
гидрируют в NMP в качестве растворителя в присутствии водорода, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и выделения с получением соединения формулы (IX).
Реакция соединения (VIII) →соединение (IX) соответствует стадии f) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (IX), где соединение формулы (VIII) гидрируют в NMP в качестве растворителя в присутствии водорода при давлении от 50 бар до 90 бар и от 50°С до 80°С, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и затем выделения и сушки с получением соединения формулы (IX).
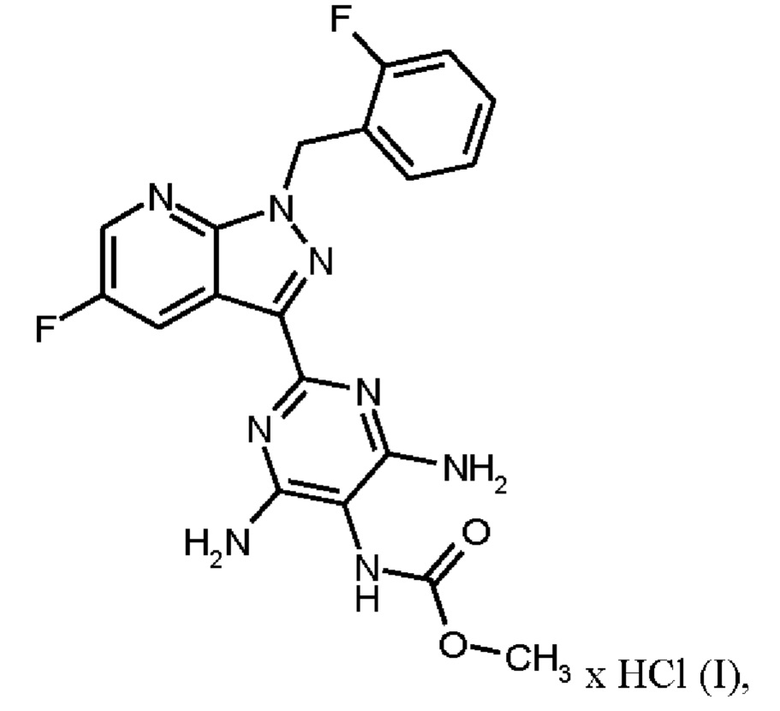
Согласно одному варианту осуществления настоящее изобретение относится к способу получения гидрохлорида соединения формулы (I)
где соединение формулы (IX)
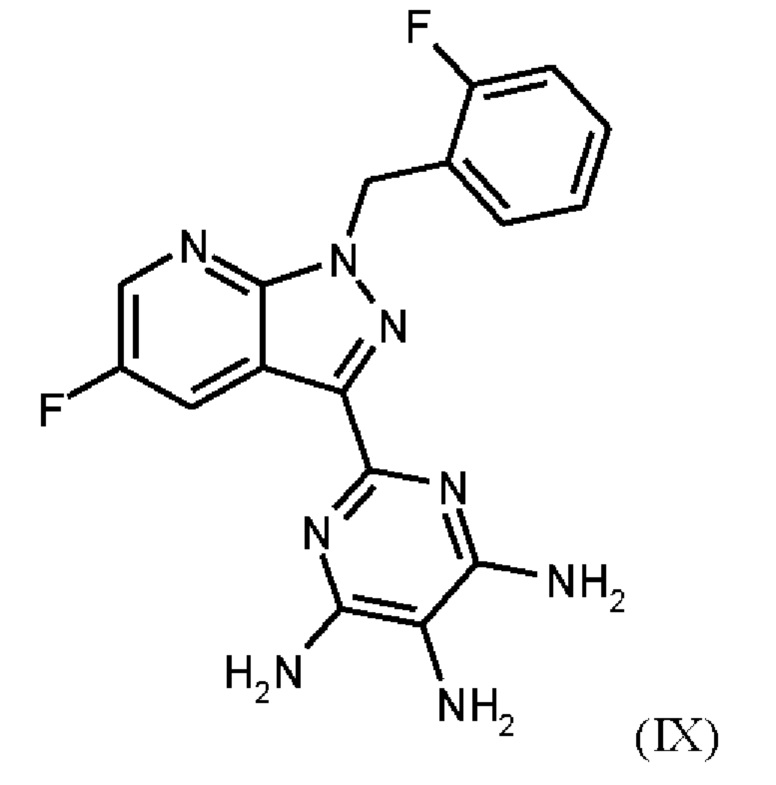
нагревают в тетрагидрофуране в качестве растворителя, добавляют от 1,0 экв. до 1,2 экв. метилхлорформиата, перемешивают в течение времени реакции от 1 ч до 10 ч и выделяют гидрохлорид соединения формулы (I).
Реакция соединения (IX)→соединение (I) х HCl соответствует стадии g) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения гидрохлорида соединения формулы (I), где соединение формулы (IX) нагревают в тетрагидрофуране в качестве растворителя до от 30°С до 66°С, добавляют от 1,0 экв. до 1,2 экв. метилхлорформиата в течение от 1 мин до 30 мин, перемешивают при температуре от 30°С до 66°С и в течение времени реакции от 1 ч до 10 ч, выделяют гидрохлорид соединения формулы (I) и сушат.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (I, ди-DMSO сольват),
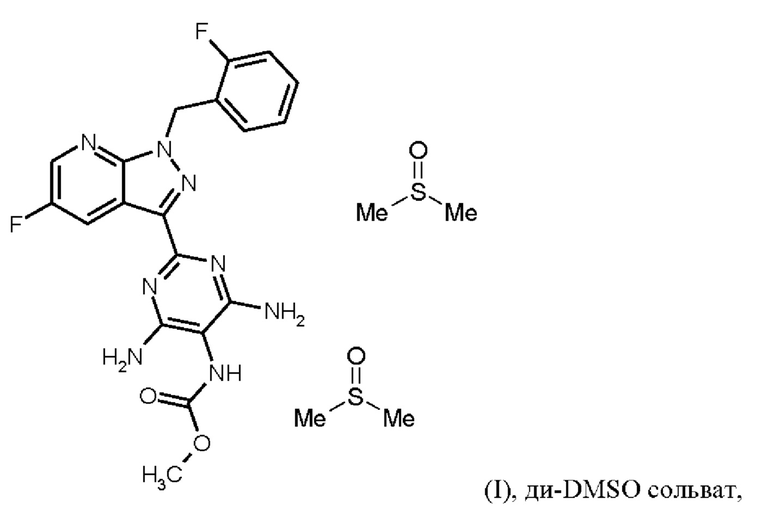
где гидрохлорид соединения формулы (I) растворяют в DMSO, добавляют три-н-бутиламин и активированный углерод, удаляют активированный углерод, кристаллизуют ди-DMSO сольват путем охлаждения и добавления этилацетата, выделяют ди-DMSO в кристаллической форме и промывают смесью DMSO и этилацетата.
Последовательность реакций соединения (I) × HCl →соединение (I) ди-DMSO сольват соответствует стадии h) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения соединения формулы (I, ди-DMSO сольват), где гидрохлорид соединения формулы (I) перемешивают в течение от 1 ч до 3 ч при от 70°С до 90°С в DMSO, добавляют три-н-бутиламин и активированный углерод, перемешивают при от 70°С до 90°С, удаляют активированный углерод, промывают с DMSO, охлаждают до от -3 до +20°С, кристаллизуют ди-DMSO сольват добавлением этилацетата, выделяют ди-DMSO в кристаллической форме, промывают смесью DMSO и этилацетата и сушат.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I),
в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, и
где соединение формулы (VI)
получают путем сначала получения соединения формулы (V)
путем начальной загрузки соединения формулы (II)
и хлорида лития в этаноле, загрузки соединения формулы (III),
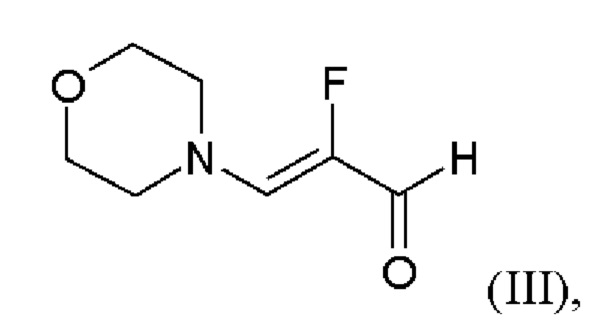
и хлортриметилсилана, и нагревания с получением соединения формулы (IV)
или альтернативно изменяя порядок загрузки любого из исходных веществ, добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V),
затем дегидратации соединения формулы (V) путем нагревания в сульфолане, ацетонитриле и фосфорилхлориде, добавления ацетонитрила и воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20°С до 50°С, добавления аммиака в воде и выделения с получением соединения формулы (VI);
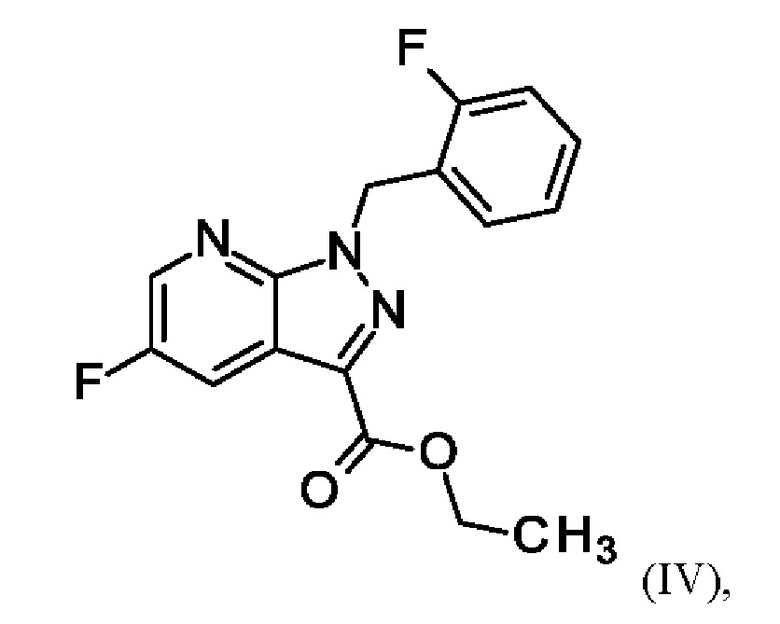
затем соединение формулы (VII)
получают путем суспендирования соединения формулы (VI)
в метаноле, добавления метоксида натрия в метаноле, добавления метанола и хлорида аммония, фильтрации с использованием вспомогательного фильтра, концентрирования и добавления этилацетата, добавления этанола и выделения с получением соединения формулы (VII);
затем соединение формулы (VIII)
получают путем начального получения соединения формулы (VIIIa)
путем добавления концентрированной соляной кислоты в воде к анилину в воде, затем последовательного добавления раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII)
в DMF, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), добавления метанола и выделения соединения формулы (VIII);
затем соединение формулы (IX)
получают путем гидрирования соединения формулы (VIII)
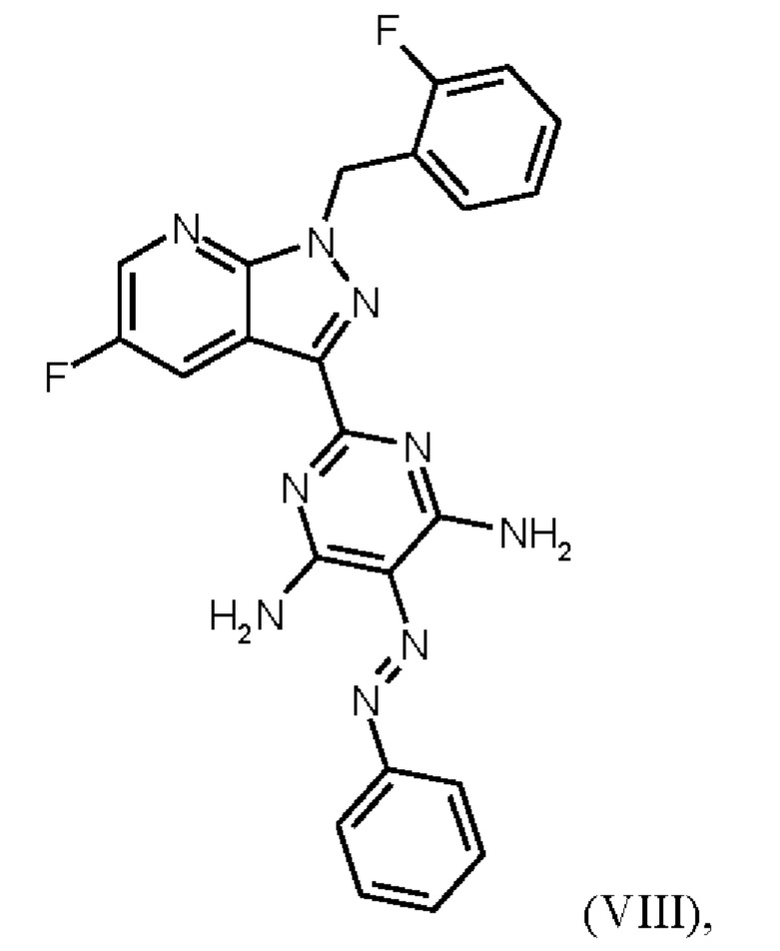
в NMP в качестве растворителя в присутствии водорода, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и выделения с получением соединения формулы (IX);
затем гидрохлорид соединения формулы (I)
получают путем нагревания соединения формулы (IX)
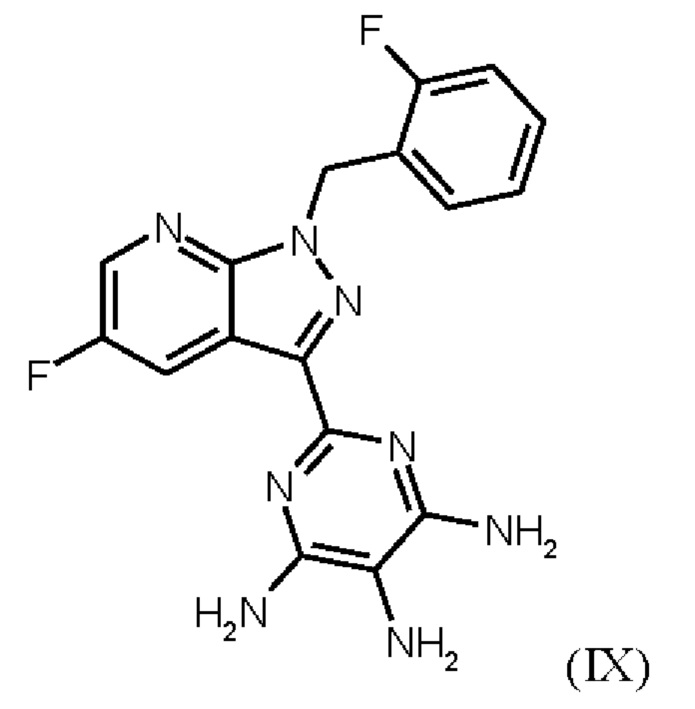
в тетрагидрофуране в качестве растворителя, добавления от 1,0 экв. до 1,2 экв. метилхлорформиата, перемешивания в течение времени реакции от 1 ч до 10 ч и выделения гидрохлорида соединения формулы (I),
затем ди-DMSO сольват соединения формулы (I)
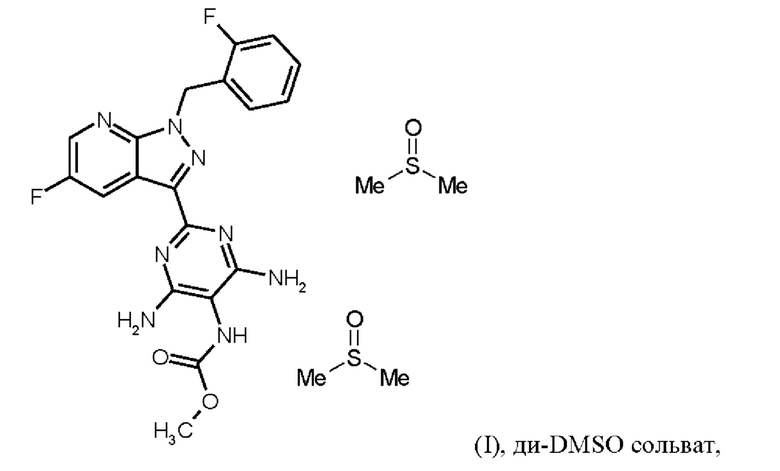
получают путем растворения гидрохлорида соединения формулы (I) в DMSO, добавления три-н-бутиламина и активированного углерода, удаления активированного углерода, кристаллизации ди-DMSO сольвата путем охлаждения и добавления этилацетата, выделения ди-DMSO сольвата в кристаллической форме, и промывки смесью DMSO и этилацетата,
наконец, соединение формулы (I) в кристаллической форме модификации I получают, где
1.1 ди-DMSO сольват соединения формулы (I) растворяют в DMSO и этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас,
1.2 растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды;
1.3 образованную суспензию затем охлаждают до температуры от 5°С до 50°С и
1.4 образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
Эта последовательность реакций соответствует стадиям а) - i) схемы 2.
Согласно одному варианту осуществления настоящее изобретение относится к способу получения кристаллической формы модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7, где соединение формулы (VI) получают путем сначала получения соединения формулы (V) путем начальной загрузки соединения формулы (II) и хлорида лития в этаноле, загрузки соединения формулы (III) и хлортриметилсилана, нагревания с возвратом флегмы и охлаждения, с получением соединения формулы (IV),
или альтернативно изменяя порядок загрузки любого из исходных веществ,
добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V),
затем дегидратации соединения формулы (V) путем нагревания в сульфолане и ацетонитриле, добавления фосфорилхлорида, промывки ацетонитрилом, перемешивания при повышенной температуре, охлаждения, добавления ацетонитрила, добавления воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20°С до 50°С, добавления аммиака в воде, сбора твердых веществ путем фильтрации, промывки водой и сушки с получением соединения формулы (VI);
затем соединение формулы (VII) получают путем суспендирования соединения формулы (VI) в метаноле, добавления метоксида натрия в метаноле, перемешивания, добавления метанола и хлорида аммония, нагревания с возвратом флегмы, перемешивания, охлаждения, фильтрации с использованием вспомогательного фильтра, концентрирования фильтратов посредством дистилляции, добавления этилацетата, продолжения дистилляции с заполнением отогнанного объема этилацетатом, охлаждения, добавления этанола, выделения, промывки этилацетатом и сушки с получением соединения формулы (VII);
затем соединение формулы (VIII) получают путем начального получения соединения формулы (VIIIa) путем добавления концентрированной соляной кислоты в воде к анилину в воде при от -3 до 12°С, затем последовательного добавления при той же температуре раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII) в DMF до от 85°С до 115°С, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 экв. до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), в течение от 5 ч до 15 ч, охлаждения до от 77°С до 88°С, добавления метанола и выделения соединения формулы (VIII);
затем соединение формулы (IX) получают путем гидрирования соединения формулы (VIII) в NMP в качестве растворителя в присутствии водорода при давлении от 50 бар до 90 бар и при температуре от 50°С до 80°С, при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и затем выделения и сушки с получением соединения формулы (IX);
затем гидрохлорид соединения формулы (I) получают путем нагревания соединения формулы (IX) в тетрагидрофуране в качестве растворителя до от 30°С до 66°С, добавления от 1,0 экв. до 1,2 экв. метилхлорформиата в течение от 1 мин до30 мин, перемешивания при температуре от 30°С до 66°С и в течение времени реакции от 1 ч до 10 ч, выделения гидрохлорида соединения формулы (I) и сушки,
затем ди-DMSO сольват соединения формулы (I) получают путем перемешивания гидрохлорида соединения формулы (I) в течение от 1 ч до 3 ч при от 70°С до 90°С в DMSO, добавления три-н-бутиламина и активированного углерода, перемешивания при от 70°С до 90°С, удаления активированного углерода, охлаждения до от -3 до+20°С, кристаллизации ди-DMSO сольвата путем добавления этилацетата, выделения ди-DMSO в кристаллической форме, промывки смесью DMSO и этилацетата и сушки,
наконец, соединение формулы (I) в кристаллической форме модификации I получают, где
1.1 ди-DMSO сольват соединения формулы (I) суспендируют в DMSO и нагревают до от 70°С до 80°С, этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас., и смесь перемешивают при от 65°С до 85°С в течение от 15 мин до 21 ч,
1.2 растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды при температуре от 15°С до 85°С и в течение от 0,1 мин до 30 мин;
1.3 образованную суспензию затем охлаждают до температуры от 5°С до 50°С в течение от 1 ч до 4 ч и
1.4 образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
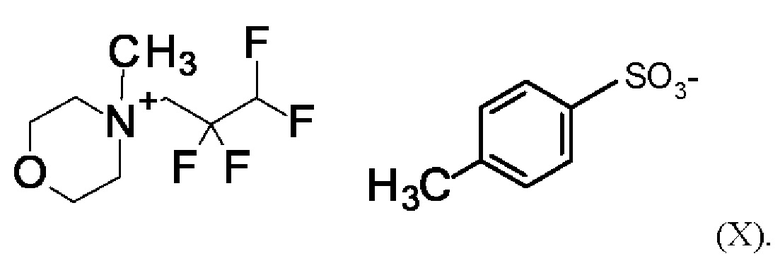
Согласно одному варианту осуществления настоящее изобретение относится к соединению формулы (X)
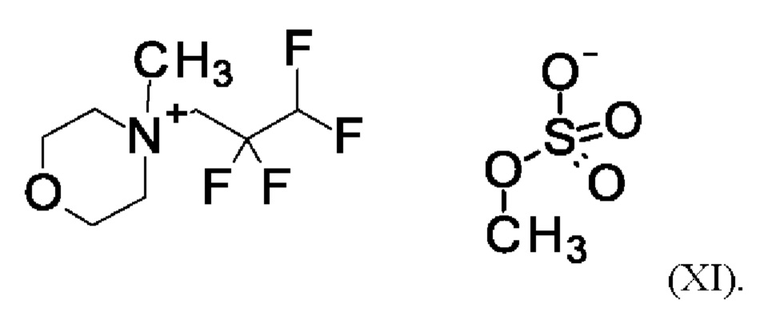
Согласно одному варианту осуществления настоящее изобретение относится к соединению формулы (XI)
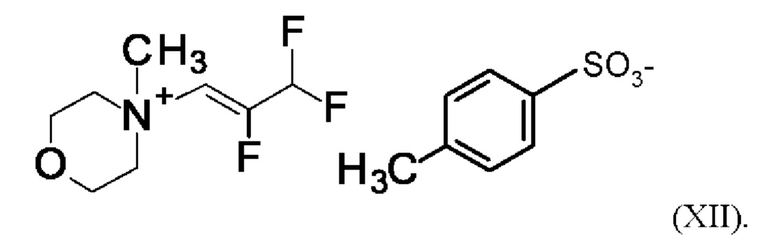
Согласно одному варианту осуществления настоящее изобретение относится к соединению формулы (XII)
4-(2,2,3,3-тетрафторпропил)морфолин формулы (XIII)
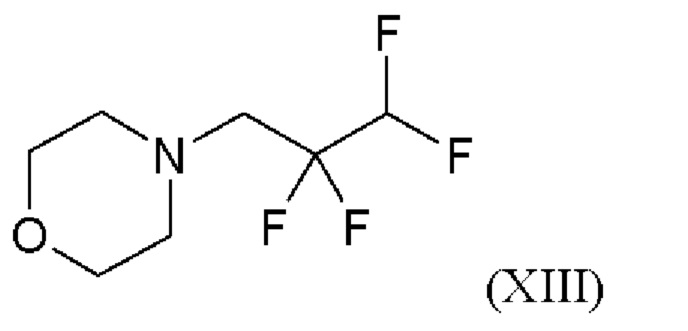
получают в соответствии с Примером 3, как описано в WO2020/152010 (опубликовано после даты приоритета настоящего изобретения). Возможны и другие варианты дозирования.
Способ согласно настоящему изобретению, включающий отдельные стадии реакции, последовательности реакций и весь способ в целом, обеспечивает значительные преимущества по сравнению с предшествующим уровнем техники, в частности, по сравнению с WO 2013/076168, и, таким образом, может быть осуществлен в техническом масштабе. Это изложено далее.
В отличие от WO 2013/076168 стадии а) и b) схемы 2 согласно настоящему изобретению проводят в виде реакции в одном сосуде (по сравнению с Примером 8). Для превращения (II)+(III) →(IV) (стадия а) схемы 2, вместо метансульфоновой кислоты используют хлортриметилсилан.
Использование хлортриметилсилана вместо метансульфоновой кислоты приводит к значительным преимуществам реакции согласно настоящему изобретению по сравнению с реакцией предшествующего уровня техники. Хлортриметилсилан реагирует с этанолом с образованием соответствующего триметилсилилэтилового простого эфира и соляной кислоты. Соляная кислота катализирует реакцию этил 5-амино-1-(2-фторбензил)-1Н-пиразол-3-карбоксилата (II) и 2-фтор-3-(морфолин-4-ил)акрилового альдегида (III) в качестве метансульфокислоты в реакции согласно Примеру 6 WO 2013/076168. Триметилсилилэтиловый простой эфир реагирует с водой, образующейся во время конденсации соединения (II) и соединения (III), с получением триметилсиланола. Удаление воды путем образования триметилсиланола положительно влияет на реакцию, поскольку вода гидролизует легко омыляемый сложный эфир (II) с образованием карбоновой кислоты. Опасность омыления соединения (II) также является причиной того, что в реакции нельзя использовать водную соляную кислоту. Таким образом, хлортриметилсилан одновременно действует как кислота и как ловушка для воды на стадии а).
Еще одним преимуществом нового протокола реакции является возможность проведения реакции в одном сосуде. Преимущество этого заключается в том, что до следующей стадии также обрабатывают те фракции сложного эфира (II), которые присутствуют в маточном растворе во время кристаллизации. Реакция в одном сосуде имеет еще одно преимущество, заключающееся в том, что исключаются стадии выделения и сушки. В способе в техническом масштабе это имеет то преимущество, что сокращается время использования производственного оборудования, что приводит к значительному снижению производственных затрат.
Еще одним преимуществом является значительно упрощенная обработка по сравнению со способом уровня техники. Промежуточное перемешивание с изопропанолом и промывание водой для удаления солей метансульфокислоты не проводят. Далее продукт получают без промежуточного выделения, так как хлориды удаляются легче, чем соли метансульфокислоты. Это, в свою очередь, значительно снижает производственные затраты.
Выход этой реакции в одном сосуде (82,9 мас. %) выше, чем общий выход соответствующих двух стадий 79,0% (примеры 6 и 7) согласно WO 2013/076168. Продукт этого способа (соединение (V)) согласно настоящему изобретению получают с высоким выходом (82,9 мас. %) и высокой чистотой (площадь % ВЭЖХ: 99,7%), что является еще одним неожиданным преимуществом по сравнению с предшествующим уровнем техники.
Принимая во внимание предшествующий уровень техники, удивительно, что упомянутые выше различия стадий а) и b) схемы 2 настоящего изобретения по сравнению с ближайшим уровнем техники, т.е. использование хлортриметилсилана вместо метансульфоновой кислоты и проведение стадий а) и b) в виде реакции в одном сосуде, приводят к таким выраженным преимуществам по сравнению с предшествующим уровнем техники.
Для превращения (V)→(VI), стадия с) схемы 2, используют те же исходные вещества, что и в примере 8 WO 2013/076168. Решающее отличие заключается в контроле реакции. В реакции согласно настоящему изобретению воду добавляли при соответствующей агитации и при хорошей скорости и при охлаждении для того, чтобы поддерживать внутреннюю температуру от 20°С до 50°С.
Путем добавления ацетонитрила и воды при соответствующем перемешивании и с хорошей скоростью при охлаждении для поддержания внутренней температуры от 20°С до 50°С гидролиза продукта (соединение (VI)) до исходного вещества (соединение (V)) удивительно удается избежать. Таким образом, другой контроль реакции обеспечивает большое преимущество по сравнению со способом, известным из уровня техники.
Еще одним преимуществом этой реакции согласно настоящему изобретению является то, что продукт (соединение VI) получают с высоким выходом (95,9 мас. %) и высокой чистотой (ВЭЖХ, % площади: 99,4%).
Для превращения (VI)→(VII) стадия d) схемы 1 согласно примеру 9 WO 2013/076168 вводимое вещество суспендируют в этаноле. Напротив, исходное вещество стадии d) схемы 2 суспендируют в метаноле согласно настоящему изобретению.
Удивительно, но корки, образующиеся на стенках сосуда при превращении (VI)->(VII) в соответствии с примером 9 WO 2013/076168, полностью исключаются путем суспендирования исходного вещества (соединение (VI)) в метаноле вместо этанола. Это имеет решающее преимущество при выполнении способа в техническом масштабе.
В качестве дополнительного неожиданного эффекта продукт (соединение (VII)) полностью растворяется при 20°С, и в нерастворенной форме присутствуют только присутствующие в избытке соли и примеси. Это приводит к дополнительному преимуществу, заключающемуся в том, что эти примеси можно легко отделить фильтрованием при добавлении вспомогательных веществ для фильтрации.
С 18 кг вспомогательного вещества для фильтрации (кизельгур Clarcel DICB) по отношению к 190 кг исходного вещества (соединение (VI)), перепадом давления 2 бар и фильтрующей поверхностью 6,5 м и последующим очистным фильтром (также резервный фильтр), время фильтрации составляет менее 30 мин, чего нельзя было ожидать от предшествующего уровня техники. Эта фильтрация считается очень быстрой с технической точки зрения и обеспечивает экономическую выгоду за счет короткого времени работы производственного оборудования.
После замены растворителя на этилацетат продукт получают с высоким выходом (88,6% от теоретического выхода) и высокой чистотой (ВЭЖХ, площадь %: 99,9%).
Принимая во внимание предшествующий уровень техники, удивительно, что суспендирование исходного вещества (соединение (VI)) в метаноле на стадии d) схемы 2 настоящего изобретения вместо использования этанола, как описано в ближайшем предшествующем уровне техники, приводит к таким выраженным преимуществам над предшествующим уровнем техники.
При получении промежуточного соединения (VIIIa) для превращения (VII)+(VIIIa)→(VIII) (стадия е) схемы 2 соединение (VIIIa) промывают иным образом, чем в примере 10А WO 2013/076168. В соответствии с примером 10A WO 2013/076168 промежуточное соединение (VIIIa) промывают три раза по 5,3 л воды на кг анилина и 4,15 л толуола на кг анилина. Согласно настоящему изобретению соединение (VIIIa) промывают трижды от 5,2 л до 12,8 л воды на кг анилина и от 3,5 л до 4,8 л изопропанола (вместо толуола) на кг анилина.
Изменение процедуры промывки соединения (VIIIa) согласно настоящему изобретению (изопропанол вместо толуола и различные соотношения воды) неожиданно имеет главное преимущество, заключающееся в полном удалении всех солей.
В качестве дополнительного преимущества этой процедуры промывки соединения (VIIIa) согласно настоящему изобретению вода эффективно удаляется при промывке изопропанолом, который, в отличие от толуола, используемого в реакции, описанной в примере 10 A WO 2013/076168, смешивается с водой. Путем промывки изопропанолом, таким образом, удаляются дополнительные примеси, и получают соединение (VIIIa) очень высокой чистоты (% площади ВЭЖХ: 100%). Таким образом, процедура промывки согласно настоящему изобретению имеет неожиданное и значительное преимущество по сравнению со способом, известным из уровня техники.
Отличие от примера 11 A WO 2013/076168 (превращение (VII)+(VIIIa)→(VIII)) заключается в доле исходного вещества (соединение (VII), триэтиламин и общее количество DMF: в примере 11 A WO 2013/076168 один эквивалент соединения (VII) нагревают в DMF. Затем добавляют 1,7 экв. соединения (VIIIa) на 1,1 экв. триэтиламина в DMF в течение 30 мин. Общее количество DMF составляет 5,8 кг/кг соединения формулы (VII). Согласно настоящему изобретению один эквивалент соединения (VII) нагревают в DMF.Затем добавляют 1,25 экв. соединения (VIIIa) на 1,45 экв. триэтиламина в DMF в течение 10 ч. Общее количество DMF (включая количество DMF, в котором растворено соединение (VIIIa)) составляет от 4,7 кг до 6,1 кг DMF на кг соединения формулы (VII) или 5,2 кг DMF на кг соединения формулы (VII).
Различное соотношение исходного вещества (соединение (VII), триэтиламин и общее количество DMF) в превращении (VII)+(VIIIa)→(VIII) согласно настоящему изобретению по сравнению с предшествующим уровнем техники неожиданно привело к получению продукта высокой чистоты.
Триэтиламин используют для высвобождения соединения (VII) из гидрохлорида. Обычно немного больше 1 экв. триэтиламина по отношению к соединению (VII) является достаточным. Однако неожиданно использование 1,45 экв. триэтиламина дает продукты более высокой чистоты. Еще одним преимуществом использования более высокого избытка триэтиламина является подавление образования побочного компонента, образующегося в результате реакции двух молекул соединения (VIIIa). Применение условий реакции с менее чем 1,30 экв. триэтиламина дает соединение Примера 11 (VIII) со значительно более высоким содержанием соединения формулы (VIIIb).
Еще одним отличием Примера 11 A WO 2013/076168 от настоящего изобретения является промывка соединения (VIII). В примере 11А WO 2013/076168 промывку проводят водой/DMF, 2х водой/метанолом и метанолом. Согласно настоящему изобретению последовательно проводят промывку DMF, метанолом, водой и метанолом. Эта оптимизация стадии промывки соединения (VIII) неожиданно привела к дополнительной очистке продукта соединения (VIII). Соединение формулы (VIII) получают с высоким выходом (78,1 мас. %) и высокой чистотой (ВЭЖХ, площадь %: 99,0%).
В способе согласно примеру 12 WO 2013/076168 превращение (VIII) →(IX), стадия f) схемы 1, проводят в DMF. В способе согласно настоящему изобретению вместо DMF используют NMP.
Использование DMF в способе согласно примеру 12 WO 2013/076168 (превращение (VIII)→(IX), стадия f) схемы 1 имеет несколько основных недостатков. Продукт (IX) образует сольват с DMF, который необходимо перевести в свободную от сольватов форму с помощью горячей воды и больших усилий. Оставшийся DMF будет образовывать формильный побочный продукт с метилхлорформиатом на следующей стадии (превращение (IX) →гидрохлорид соединения (I)), который необходимо удалить с большими усилиями. Еще одним недостатком способа согласно примеру 12 WO 2013/076168 является низкая растворимость продукта (соединение (IX)) в DMF. Во время фильтрации для удаления катализатора кристаллизация продукта вызывает препятствия, которые очень неблагоприятны для осуществления процесса в техническом масштабе.
В способе по настоящему изобретению вместо DMF используют NMP. Продукт (соединение (IX) имеет значительно более высокую растворимость в NMP, что имеет то преимущество, что гидрирование можно проводить при гораздо более высокой концентрации (от 4,6 л до 6,8 л NMP/кг исходного вещества (соединение (VIII)) по сравнению с 10 л DMF/кг исходного вещества в соответствии с примером 12 WO 2013/076168). В качестве дополнительного преимущества NMP можно легко удалить путем фильтрации маточного раствора во время кристаллизации. Это упрощает способ, например, в отношении сокращения времени работы установки и, следовательно, снижения производственных затрат. Из предшествующего уровня техники нельзя было ожидать, что использование NMP вместо DMF в этой реакции приводит к такому выраженному преимуществу.
Еще одним недостатком стадии f) примера 12 способа согласно WO 2013/076168 является необходимость удаления основной части DMF после гидрирования путем отгонки, что является сложной стадией из-за высокой температуры кипения DMF (162°С). Эта стадия может быть исключена измененным способом согласно настоящему изобретению. Отсутствие отгонки DMF перед кристаллизацией требует повышенного количества воды и приводит к снижению выхода, что еще более невыгодно.
Посредством этого способа согласно настоящему изобретению (соединение (IX)) получают с высоким выходом (95,5% от теоретического выхода) и высокой чистотой (ВЭЖХ, площадь %): 98,6%).
На основании предшествующего уровня техники удивительно, что использование NMP на стадии f) схемы 2 настоящего изобретения в отличие от использования DMF в наиболее близком известном уровне техники приводит к таким выраженным преимуществам по сравнению с предшествующим уровнем техники.
Первую технологическую стадию способа согласно примеру 13 A WO 2013/076168, превращение (IX)->(I) гидрохлорид, проводят в изопропаноле. Исходное вещество (соединение (IX)) суспендируют в изопропаноле и подвергают взаимодействию в течение 20 ч с метилхлорформиатом, растворенным в изопропаноле, с получением суспензии гидрохлорида соединения (I). Избыток метилхлорформиата разрушают добавлением метанола. Гидрохлорид соединения (I) не выделяют.
В способе согласно настоящему изобретению реакцию с получением гидрохлорида соединения (I) (стадия g) схемы 2) проводят в THF вместо изопропанола и выделяют гидрохлорид соединения (I).
В способе согласно настоящему изобретению неожиданно было обнаружено, что при проведении реакции в THF вместо изопропанола реакционная суспензия полностью превращается в раствор, из которого во время реакции кристаллизуется продукт. Благодаря этому время реакции сокращается с 20 часов в изопропаноле до 2 часов в THF, что является важным преимуществом, например, с точки зрения затрат и времени работы оборудования производственной установки. Продукт реакции, соединение (I) гидрохлорид, может быть легко выделен фильтрованием.
Для 120 кг исходного вещества (соединение (IX)), перепад давления 2 бар и поверхность фильтра 2,5 м, время фильтрации составляет менее 30 мин, что было неожиданным. В тех же условиях 2 × 740 л тетрагидрофурана для промывки осадка на фильтре также отделяют менее чем за 30 мин. Эта фильтрация считается очень быстрой с технической точки зрения и обеспечивает экономическую выгоду посредством более короткого времени работы на производственной установке.
На основании предшествующего уровня техники удивительно, что использование THF на стадии g) схемы 2 настоящего изобретения в отличие от использования изопропанола в наиболее близком известном уровне техники приводит к таким выраженным преимуществам в технических характеристиках способа по сравнению с предшествующим уровнем техники.
Еще одним важным преимуществом способа согласно настоящему изобретению является то, что избыток только от 1,0 экв. до 1,2 эквивалентов метилхлорформиата используют в реакции согласно настоящему изобретению, в отличие от 1,3 эквивалентов метилхлорформиата, которые используют в способе согласно примеру 13 A WO 2013/076168.
Кроме того, поскольку метилхлорформиат удаляется при фильтровании маточного раствора в способе согласно настоящему изобретению, нет необходимости разрушать избыток метилхлорформиата добавлением метанола, что является дополнительным преимуществом по сравнению со способом, известным в данной области техники.
Выделение соединения (I) в виде гидрохлорида уже дает продукт с высоким выходом (96,2% от теоретического выхода) и высокой чистотой (ВЭЖХ, площадь%: 99,14%).
Согласно примеру 13 A WO 2013/076168 гидрохлорид соединения (I), образующийся на первой стадии способа, не выделяют. Неочищенный продукт соединения (I) получают обработкой гидрохлорида соединения (I) триэтиламином. Неочищенный продукт соединения (I) затем перемешивают в DMSO, добавляют этилацетат и активированный уголь и кипятят с обратным холодильником. Затем отфильтровывают активированный уголь и остаток на фильтре промывают этилацетатом. Фильтрат, полученный после фильтрования активированного угля, содержащий соединение формулы (I), растворенное в DMSO и этилацетате, дозируют в предварительно нагретый этилацетат с получением соединения (I) в кристаллизованной форме. Таким образом, согласно примеру 13 A WO 2013/076168 ди-DMSO сольват соединения (I) не выделяют.
После выделения неочищенного продукта в соответствии с примером 13 A WO 2013/076168 его три раза промывают этанолом для удаления гидрохлорида триэтиламина, что является трудоемким.
В способе согласно настоящему изобретению гидрохлорид соединения формулы (I) обрабатывают три-н-бутиламином вместо триэтиламина (стадия h) схемы 2).
Согласно настоящему изобретению и в отличие от стадии g) WO 2013/076168 во время высвобождения гидрохлорида соединения (I) с получением соединения (I) образуется неочищенный продукт, гидрохлорид три-н-бутиламина, который полностью растворим в маточном растворе и отделяется при выделении DMSO-сольвата соединения (I). Преимущество состоит в том, что исключается стадия промывки гидрохлорида амина, что приводит к гораздо менее трудоемкой стадии. Такого эффекта нельзя было ожидать на основании предшествующего уровня техники.
Еще одно отличие способа согласно настоящему изобретению от примера 13 А из WO 2013/076168 состоит в том, что после отфильтровывания активированного угля остаток на фильтре промывают DMSO вместо этилацетата, который используется в способе согласно Примеру 13 А из WO 2013/076168.
В качестве дополнительного отличия в способе согласно настоящему изобретению ди-DMSO сольват соединения (I) кристаллизуют добавлением этилацетата и выделяют фильтрованием. Смесь DMSO и этилацетата используют для промывки продукта согласно настоящему изобретению.
Кроме того, фильтрация активированного угля в соответствии с настоящим изобретением улучшена по сравнению с примером 13 A WO 2013/076168 путем проведения разделения активированного угля в чистом DMSO вместо смеси DMSO и этилацетата.
Еще одним преимуществом реакции согласно настоящему изобретению является то, что при последующем добавлении к фильтрату этилацетата сольват ди-DMSO кристаллизуется и может быть выделен фильтрованием.
Согласно настоящему изобретению после фильтрации ди-DMSO сольвата остаток промывают смесью DMSO и этилацетата. Преимущество этого заключается в том, что можно избежать элюирования DMSO из сольвата, что вызвано увеличенным временем контакта при проведении реакции в большем масштабе. Таким образом, промывка смесью DMSO и этилацетата всегда приводит к теоретическому содержанию DMSO в сольвате. При кристаллизации согласно настоящему изобретению очень эффективно удаляются примеси. Это имеет большое значение для следующей стадии, которая ведет к фармацевтическому продукту. Неожиданно продукт реакции стабилен и может быть высушен и впоследствии сохранен в описанных условиях. Ди-DMSO-сольват соединения (I) получают с высоким выходом (77,7% от теоретического выхода) и высокой чистотой (ВЭЖХ, площадь %: 99,92%).
Как указано выше для стадии h) схемы 1 ди-DMSO-сольват соединения (I) не выделяют, а непосредственно кристаллизуют путем добавления этилацетата с получением соединения формулы (I) в кристаллической форме модификации I.
Согласно настоящему изобретению превращение ди-DMSO-сольвата соединения (I) в соединение формулы (I) в кристаллической модификации I осуществляют в основном так, как описано в WO 2020/126983(опубликовано после даты приоритета настоящего изобретения):
Выделенный ди-DMSO сольват соединения формулы (I) суспендируют в DMSO и нагревают, этанол добавляют и смесь перемешивают, где растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды; образованную суспензию затем охлаждают и образованные кристаллы на стадии b) затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата.
Способ стадии i) в соответствии с настоящим изобретением дает продукт активного соединения формулы (I) в кристаллической модификации I с улучшенными свойствами, например, в отношении способности к выделению продукта активного соединения, способности к выгрузке продукта активного соединения после выделения и сушки, а также транспортабельности, просеиваемости и микронизируемости.
В контексте настоящего изобретения и как указано в WO 2020/126983, улучшенная способность к выделению продукта активного соединения, способность к выгрузке продукта активного соединения после выделения и сушки, а также транспортабельность, просеиваемость и микронизируемость продукта активного соединения формулу (I) следует понимать как означающие, например, следующее:
Улучшенная способность к выделению может быть измерена в промышленном масштабе, например, посредством более высокой удельной производительностью в центрифуге с выворачиваемым фильтром (пример 16).
Улучшение способности к выгрузке из устройства для выделения можно измерить, например, по максимальной толщине фильтрационной корки, при которой путь выгрузки, например, из центрифуги с выворачивающим фильтром, не блокируется.
Улучшение сушки можно измерить, например, беспроблемной сушкой в вакуумной контактной сушилке, например, и предотвращением засорения шахты опускания при выгрузке из сушилки.
Улучшение просеиваемости можно измерить, например, посредством улучшенной подачи в промышленную просеивающую машину в результате улучшенной сыпучести продукта активного соединения и посредством меньшего количества забивания сита, например, посредством пропускной способности продукта активного соединения в единицу времени (пример 17).
Улучшение микронизации можно измерить, например, посредством более легкой подачи продукта активного соединения в струйную мельницу.
В контексте настоящего изобретения «промышленный масштаб» определяется как размер партии >10 кг активного соединения.
В контексте настоящего изобретения выделение продукта активного соединения осуществляют с использованием, например, центрифуги с фильтром, например центрифуги с выворачиваемым фильтром.
В контексте настоящего изобретения сушку продукта активного соединения проводят с использованием, например, вакуумной контактной сушилки, например сферической сушилки.
В контексте настоящего изобретения просеивание продукта активного соединения проводят, например, с использованием устройства для просеивания Frewitt Coniwitt ТС200 (диаметр отверстия сита 3 мм) или устройства для просеивания Frewitt Oscillowitt MG-800 (диаметр отверстий сита от 2,5 мм до 4,0 мм).
В контексте настоящего изобретения микронизацию осуществляют, например, путем измельчения в струйной мельнице.
Способность к выделению материала, полученного способом согласно изобретению, улучшилась по сравнению с материалом, полученным способом согласно WO 2013/076168. Это проявляется, например, в более высокой удельной производительности центрифуги с выворачиваемым фильтром. В промышленном масштабе выделение материала из способа согласно WO 2013/076168 достигло средней производительности по площади 1,6 кг/м2 ч. Средняя удельная производительность материала по способу согласно настоящему изобретению составляла 3,0 кг/м3ч и, таким образом, почти вдвое превышала указанную выше (пример 16).
Улучшенная способность к выгрузке из устройства для выделения: способ в соответствии со способом согласно настоящему изобретению предотвращает образование волос-подобной фильтрационной корки, имеющей высокую прочность на разрыв. Как после выделения в напорном фильтре, так и после выделения в фильтре центрифугирования осадок на фильтре мягкий и пластичный. Это предотвращает закупорку пути выброса. Например, в промышленном масштабе блокировку выпускного тракта центрифуги с выворачиваемым фильтром после выделения материала из способа согласно WO 2013/076168 можно было избежать только посредством уменьшения толщины осадка на фильтре от 8 мм до 9 мм. В отличие от этого при выделении материала в способе согласно настоящему изобретению средняя высота осадка на фильтре 25 мм была получена без наблюдения какой-либо блокировки пути выпуска.
Улучшенная сушка: благодаря мягкой консистенции и хорошей деформируемости осадка на фильтре, получаемого в результате способа согласно настоящему изобретению, сушка в вакуумной контактной сушилке (например, сферической сушилке) не вызывает проблем. Высушенный материал образует легкотекучий сыпучий материал, который также не приводит к засорению вала при выходе из сушилки.
Улучшенная способность к просеиванию: материал, полученный способом согласно настоящему изобретению, легко подается в устройство для просеивания благодаря его хорошей сыпучести. Просеивание приводит к заметно меньшему количеству засорений сита, чем для материала, полученного в способе согласно пути 1. Например, в промышленном масштабе 65 кг материала, полученного в результате способа согласно настоящему изобретению, можно просеять за <5 мин в устройстве для просеивания Frewitt Coniwitt ТС200 (диаметр отверстия сита 3 мм). Это соответствует >13 кг/мин. Для сравнения просеивание материала согласно пути 1 через устройство для просеивания Frewitt Oscillowitt MG-800 (диаметр отверстий сита от 2,5 мм до 4,0 мм) достигало пропускной способности только <10 кг/ч. Это соответствуем 0,17 кг/мин (Пример 17). Наблюдается очень большая разница почти в 100 раз в пропускной способности сита продукта активного соединения согласно пути 1 по сравнению с продуктом активного соединения согласно настоящему изобретению. Эта очень большая разница в пропускной способности сита обусловлена главным образом характеристиками материала продукта активного соединения и не может быть объяснена различными типами устройств.
Значительно улучшены характеристики обработки и транспортировки твердых веществ.
Улучшенная микронизация: материал, полученный способом согласно настоящему изобретению, легко подается в струйную мельницу благодаря его хорошей сыпучести.
Дополнительные преимущества по сравнению со способом согласно WO 2013/076168, касающиеся, например, грануляции, описаны в WO 2020/126983.
На основании предшествующего уровня техники нельзя было ожидать, что способ согласно настоящему изобретению приводит к продукту активного соединения, который по сравнению с продуктом способа предшествующего уровня техники демонстрирует такие заметно улучшенные свойства при промышленном производстве фармацевтически активного соединения формула (I) в твердой лекарственной форме. На основании известного уровня техники также нельзя было ожидать, что способ согласно настоящему изобретению приводит к определенной модификации активного соединения формулы (I), предпочтительно в кристаллической форме модификации I. Также неожиданным было отсутствие гидратов или дигидраты активного соединения формулы (I), образующихся в ходе получения продукта активного соединения согласно настоящему изобретению. При определенных условиях активное соединение образует гидраты при контакте с водой. Это неожиданно предотвращается в способе согласно настоящему изобретению. Кроме того, способ согласно настоящему изобретению приводит к определенной модификации продукта активного соединения формулы (I), а именно соединения (I) в кристаллической форме модификации I. Кроме того, способ согласно настоящему изобретению не приводит к образованию гидратов или дигидратов продукта активного соединения формулы (I).
Примеры
Аббревиатуры:
Ас - Ацетил
вод. - Водный
конц. - Концентрированный
DMF - Диметилформамид
DMSO - Диметилсульфоксид
экв. - эквивалент (эквиваленты)
ESI - ионизация электрораспылением (в MS)
Et - Этил
нас. - Насыщенный
Ч - час (часы)
HCl - соляная кислота
HPLC - высокоэффективная жидкостная хроматография
Me - Метил
Мин - минута (с)
MS - Масс-спектрометрия
NMP - М-метил-2-пирролидон
ЯМР - спектроскопия ядерного магнитного резонанса
от теор. выхода - От теоретического выхода
Pd/C - палладий на активированном углероде
Rf - коэффициент удерживания (в тонкослойной хроматографии на силикагеле)
Rt - время удерживания (в ВЭЖХ)
THF - Тетрагидрофуран
мас./мас - Соотношение массы к массе
Условия/способы ВЭЖХ
Способ А
Zorbax Bonus RP; 150 мм ×3,00 мм; 3,5 мкм
температура колонки: 35°С; объем впрыска: 5,0 мкл; скорость потока: 0,6 мл/мин
подвижная фаза А: 1,0 мл трифторуксусной кислоты в воде (1 л);
подвижная фаза В: 1,0 мл трифторуксусной кислоты в метаноле (1 л);
растворитель образца: ацетонитрил / диметилсульфоксид / воды (4:4:2)
градиент: 0,0':65% А; 2,0':65% А; 23,0':10% А; 25,0':10% А; 25,1':65% А
УФ-обнаружение: 236 нм.
Способ В
Zorbax Bonus RP; 100 мм×4,6 мм; 1,8 мкм
температура колонки: 60°С; объем впрыска: 3,0 мкл; скорость потока: 0,6 мл/мин
подвижная фаза А: 1,0 мл трифторуксусной кислоты в воде (1 л);
подвижная фаза В: 1,0 мл трифторуксусной кислоты в ацетонитриле(1 л);
растворитель образца: диметилформамид
градиент: 0,0':78% А; 17,0':60% А; 34,0':10% А; 40,0':10% А; 40,1':78% А; 50,1':78% А
УФ-обнаружение: 260 нм.
Способ С
Zorbax Bonus RP; 100 мм ×4,6 мм; 1,8 мкм
температура колонки: 40°С; объем впрыска: 4,0 мкл; скорость потока: 0,5 мл/мин
подвижная фаза А: 1,0 мл трифторуксусной кислоты в воде (1 л);
подвижная фаза В: 1,0 мл трифторуксусной кислоты в ацетонитриле (1 л);
растворитель образца: диметилсульфоксид / ацетонитрил (1:1)
градиент: 0':85% А; 1,0':85% А; 24,0:15% А; 36,0:5% А; 36,1:85% А; 46,1:85% А
УФ-обнаружение: 260 нм.
Способ D
Poroshell 120 Bonus-RP; 250 × 4,00 мм; 2,7 мкм,
Температура колонки: 30°С; объем впрыска: 5,0 мкл; скорость потока: 0,5 мл/мин
подвижная фаза А: 1,0 мл трифторуксусной кислоты в воде (1 л);
подвижная фаза В: 0,7 мл трифторуксусной кислоты в ацетонитриле (1 л);
растворитель образца: диметилсульфоксид
градиент: 0,0':95% А; 13,0':74% А; 24,0':25% А; 30,0':10% А; 33,0:10% А; 33,1:95% А; 40,1:95% А
УФ-обнаружение: 310 нм.
Способ Е
XBridge Shield RP 18, 150 мм × 3,00 мм; 3,5 мкм
температура колонки: 10°С; объем впрыска: 5,0 мкл; скорость потока: 0,5 мл/мин
подвижная фаза А: 1,15 г (NH4) H2PO4+0,69 мл H3PO4 (85%) /лводы; подвижная фаза В: ацетонитрил;
растворитель образца: буфер как для элюента А / Ацетонитрил (1:1) градиент: 0,0':75% А; 8,0:60% А; 15,0:55% А; 22,0:20% А; 30,0':20% А УФ-обнаружение: 210 нм.
Способ F
XBridge Shield RP 18, 150 мм × 3,00 мм; 3,5 мкм
температура колонки: 10°С; объем впрыска: 5,0 мкл; скорость потока: 0,5 мл/мин
подвижная фаза а: 1,13 г {sstu) H2PO4 + 0,69 мл H3 PO4 (85%) /лводы; подвижная фаза В: ацетонитрил;
растворитель образца: буфер как для элюента А / Ацетонитрил (1:1)
градиент: 0,0':60% А; 8,0:50% А; 15,0:50% А; 22,0:20% А; 30,0':20% А
УФ-обнаружение: 210 нм.
Пример 1
4-(2,2,3,3-Тетрафторпропил)морфолин
Получают согласно Примеру 3, как описано в WO 2020/152010 (опубликована после даты приоритета настоящего изобретения).
Перемешанную смесь 2,2,3,3-тетрафторпропилтозилата формулы (II) (330,0 г, 1,10 моль) и морфолин (208,0 г, 2,39 моль) нагревали медленно в автоклаве до 130°С и перемешивали при этой температуре в течение 18 ч. Автоклав охлаждали до 80°С, открывали и реакционную смесь разбавляли с 110 мл воды и далее охлаждали до комнатной температуры. Нижний слой продукта отделяли и водный слой промывали метил трет-бутиловым простым эфиром (2×83 мл). Органические слои объединяли и растворитель выпаривали при нормальном давлении.
Соединение формулы (I) (4-(2,2,3,3-тетрафторпропил)морфолин) получали в виде бесцветной жидкости при отгонке в вакууме 185 мм рт. ст. при 115°С.
Точка кипения 115°С/185 мбар, выход 188,0 г (85% от теоретического выхода).
1Н ЯМР (400 МГц, CDCl3): δ=5,83-6,22 (m, 1H), 3,61-3,78 (m, 4Н), 2,89 (tt, J=14,0, 1,7 Гц, 2Н), 2,53-2,70 (m, 4Н).
Пример 2
4-Метил-4-(2,2,3,3-тетрафторпропил)морфолин-4-ия метансульфонат
Способ А:
20,0 г (181,3 ммоль) метил метансульфоната нагревали до 135°С и при этой температуре по каплям добавляли 35,1 г (172,7 ммоль) соединения из примера 1. Смесь перемешивали при 135°С в течение 3 часов и затем добавляли 40 мл воды. После охлаждения до 50°С водный раствор указанного в заголовке соединения использовали на следующей стадии (см. Пример 5).
1Н ЯМР (400 МГц, D2O): δ=2,81 (s, 3Н) 3,55 (s, 3Н) 3,68-3,93 (m, 4Н) 4,01-4,24 (m, 4Н) 4,33-4,51 (m, 2Н) 6,13-6,48 (m, 1Н) ppm.
Способ В:
Метилметансульфонат (143,7 г, 1,31 моль) нагревали до 135°С и при этой температуре 250,0 г (1,24 моль) соединения согласно Примеру 1 добавляли по каплям. Смесь перемешивали при 100°С в течение 22 ч, затем охлаждали до 85°С и изопропанол (375 мл) добавляли. После охлаждения до от 0 до 5°С смесь перемешивали в течение еще 30 мин. Продукт собирали фильтрованием с отсасыванием, промывали изопропанолом (3 × 125 мл) и сушили при 45°С в слабом потоке азота в вакуумном сушильном шкафу. Выход: 336,8 г (87% от теоретического выхода).
1Н ЯМР (400 МГц, D2O): δ=6,13-6,48 (m, 1H), 4,33-4,51 (m, 2Н), 4,01-4,24 (m, 4Н), 3,68-3,93 (m, 4Н), 3,55 (s, 3Н), 2,81 (s, 3Н).
Пример 3
4-Метил-4-(2,2,3,3-тетрафторпропил)морфолин-4-ия тозилат
Смесь метил-4-толуолсульфоната (17,0 г, 91,3 ммоль) и соединения Примера 1 (1,8,4 г, 91,3 ммоль) нагревали до 130°С и перемешивали при этой температуре в течение 5 часов. Затем смесь охлаждали до 80°С и добавляли изопропанол (20 мл). К раствору добавляли 140 мл диэтилового простого эфира и перемешивали в течение 10 часов. Осажденный продукт собирали фильтрацией с отсасыванием, промывали 50 мл диэтилового простого эфира и сушили при 55°С в слабом потоке азота в вакуумном сушильном шкафу. Выход: 33,5 г (86% от теоретического выхода).
1Н ЯМР (500 МГц, D2O): δ=7,69 (d, 2Н), 7,36 (d, 2Н), 6,17-6,40 (m, 1H), 4,39 (m, 2H), 4,05^,16 (m, 4H), 3,80-3,85 (m, 2H), 3,71-3,74 (m, 2H), 3,52 (s, 3Н), 2,39 (s, 3Н).
Пример 4
4-Метил-4-(2,2,3,3-тетрафторпропил)морфолин-4-ия метилсульфат
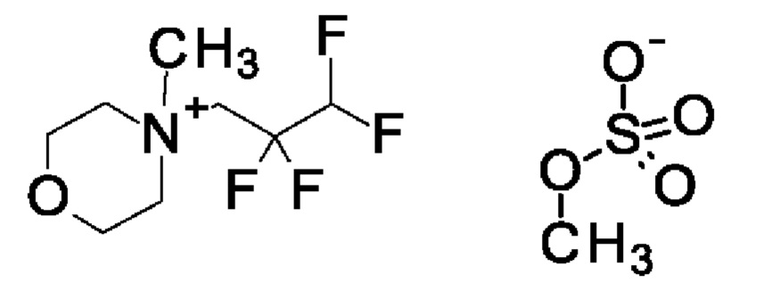
Смесь диметилсульфата (0,66 г, 5,2 ммоль) и соединения Примера 1 (1,0 г, 4,97 ммоль) нагревали до 130°С и перемешивали при 100°С в течение 2 часов. Затем смесь охлаждали до 20°С и выделяли в виде масла. Выход: 1,6 г (98% от теоретического выхода).
1H ЯМР (500 МГц, D2O): δ=6,16-6,40 (m, 1H), 4,37^1,43 (m, 2Н), 3,72-4,18 (m, 8Н), 3,74 (s, 3Н), 3,53 (s, 3Н).
Пример 5
4-Метил-4-[2,3,3-трифторпроп-1-ен-1-ил]морфолин-4-ия метансульфонат
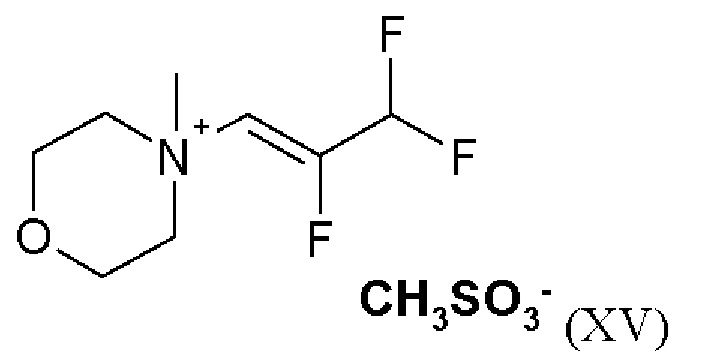
16,9 г (189,9 ммоль) 45%-ного раствора гидроксида натрия дозировали в водный раствор соединения Примера 2, способ А (макс. 172,7 ммоль) при температуре от 50°С до 55°С и смесь перемешивали при 50°С. С в течение 1 часа. Реакционную смесь охлаждали до 20°С, выпавшие в осадок соли отсасывали и промывали 5 мл воды. Водный раствор продукта (102,1 г; макс. 172,7 ммоль) использовали на следующей стадии (см. пример 7).
Для аналитических целей образец концентрировали и сушили.
1Н ЯМР (400 МГц, D2O): δ=2,81 (s, 3Н) 3,59 (s, 3Н) 3,76-3,85 (m, 2Н) 3,97-4,09 (m, 4Н) 4,12-4,20 (m, 2Н) 6,39-6,69 (m, 1H) 6,74-6,83 (m, 1H) ppm.
Пример 6
4-Метил-4-[2,3,3-трифторпроп-1-ен-1-ил]морфолин-4-ия тозилат
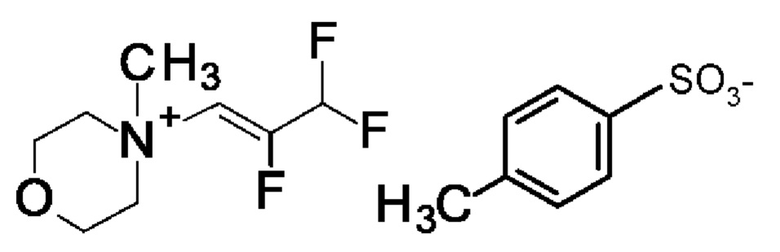
0,55 г (6,2 ммоль) 45%-ного раствора гидроксида натрия дозировали в водный раствор 2,0 г (5,2 ммоль) соединения Примера 3 при 50°С и смесь перемешивали при 50°С в течение 1 часа. Реакционную смесь охлаждали до 20°С, выпавшие в осадок соли отсасывали и промывали 1 мл воды. Добавляли 10 мл дихлорметана и концентрировали в вакууме. К остатку снова добавляли 10 мл дихлорметана и концентрировали в вакууме с получением 1,55 г неочищенного продукта.
Пример 7
2-Фтор-3-(морфолин-4-ил)акрилальдегид
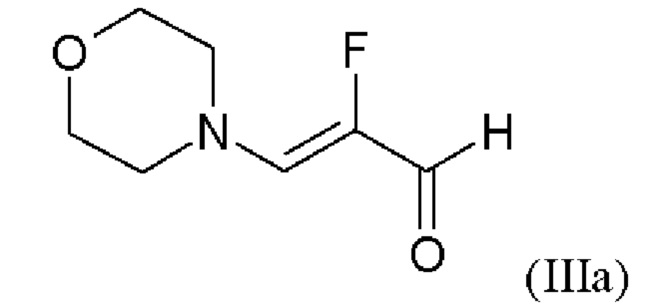
Смесь 43,8 г (503 ммоль) морфолина и 76,3 г (755 ммоль) триэтиламина нагревали до 75°С и добавляли по каплям в течение 25 мин водный раствор соединения Примера 5 (макс. 251,5 ммоль). Затем смесь перемешивали при 75°С в течение 2 ч, охлаждали до 23°С и добавляли 290 мл дихлорметана и 100 мл триэтиламина. Смесь фильтровали, фазы разделяли, водную фазу промывали смесью 290 мл дихлорметана и 100 мл триэтиламина, объединенные органические фазы промывали 250 мл насыщ. водного раствора карбоната калия и концентрировали на роторном испарителе при 40°С, добавляли 50 мл толуола и смесь дополнительно концентрировали. Получили 35,3 г (83,4% от теоретического выхода) указанного в заголовке соединения.
1Н ЯМР (500 МГц, CDCl3): δ=3,51-3,60 (m, 4Н) 3,72-3,83 (m, 4Н) 6,16 (d, J=27,l Гц, 1H) 8,59 (d, J=18,9 Гц, 1H) ppm.
Пример 8
5- Фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-карбоксамид
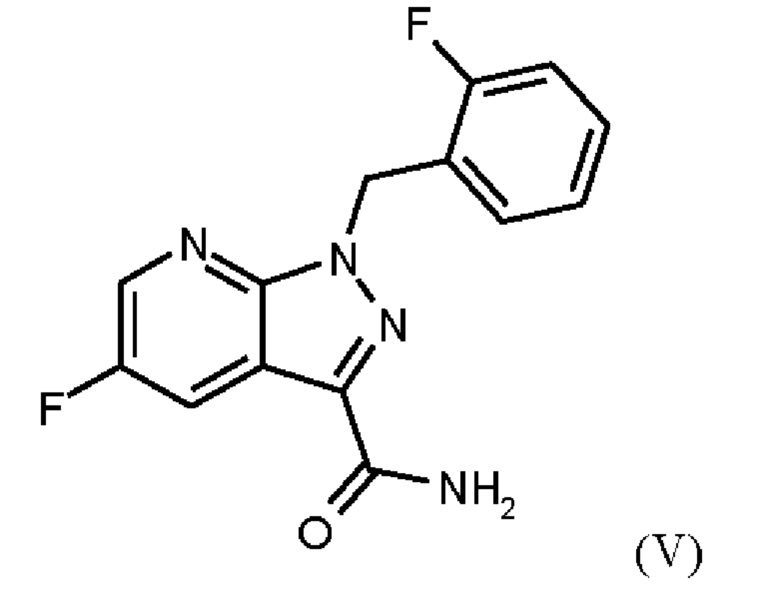
Хлорид лития (40,3 г, 0,95 моль) и этил 5-амино-1-(2-фторбензил)-1H-пиразол-3-карбоксилат (II), получение описано для Примера 20А в WO 00/06569) (100,0 г, 0,38 моль) сначала загружали в этанол (денатурированный толуолом, 361 мл) и соединение Примера 7, 0,95 экв. по отношению к соединению (II) добавляли. В течение 10 мин добавляли хлортриметилсилан (74,3 г, 0,68 моль) и смесь нагревали до температуры кипения с обратным холодильником, перемешивали в течение 2 ч и охлаждали до 65°С. При этой температуре добавляли формамид (303 мл) и добавляли 30% метоксид натрия в метаноле (191,5 г, 1,1 моль) в течение 2 часов. Внутреннюю температуру повышали не более чем до 110°С и низкокипящие продукты отгоняли до достижения внутренней температуры от 105°С до 107°С. Во время отгонки непрерывно добавляли формамид (439 мл), чтобы поддерживать постоянный уровень наполнения. Перемешивали еще 0,5 ч и охлаждали до 50°С со скоростью 9 к/ч.
Затем добавляли воду (410 мл) в течение 20 мин, смесь охлаждали до 20°С со скоростью 20°С/ч и перемешивали в течение 1 ч. Осажденные твердые вещества отсасывали, промывали водой (670 мл) и далее смесью воды (224 мл) и этанола (денатурированного толуолом, 283 мл). Сушили в вакуумном сушильном шкафу при 50°С в слабом потоке азота.
Выход: 86,3 г (82,9% от теоретического выхода)
Способ ВЭЖХ Е: минуты основного компонента: 12,7 мин
Анализ (ВЭЖХ мас. %): 99,9%
Чистота (площадь% ВЭЖХ): 99,7%
1Н ЯМР (400 МГц, [D6]DMSO): δ=8,72 (dd, J=2,7, 1,7 Гц, 1H), 8,28 (dd, J=8,3, 2,8 Гц, 1H), 7,87 (br s, 1H), 7,60 (br s, 1H), 7,34-7,40 (m, 1H), 7,12-7,26 (m, 3H), 5,87 (s, 2H). MS (ESI+): mlz=289 [M+H]+.
Пример 9
5-Фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-карбонитрил
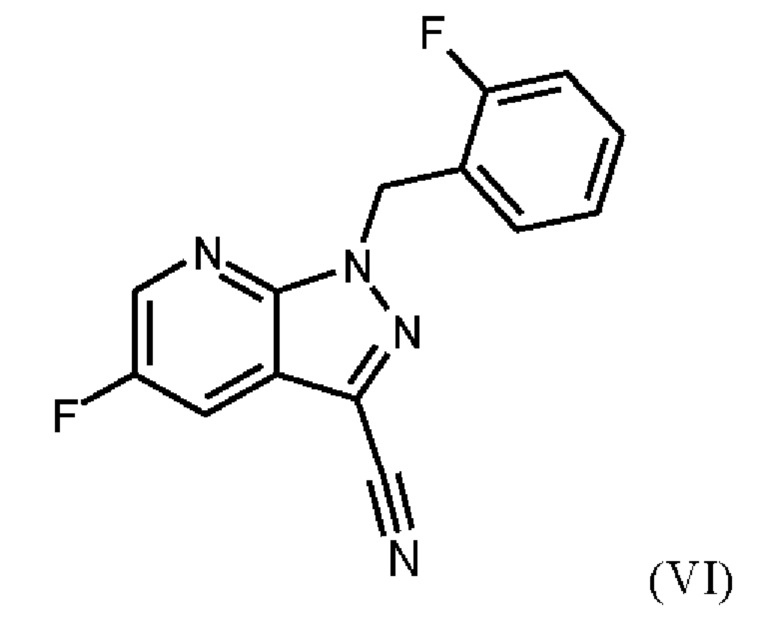
Соединение согласно Примеру 8 (99,9 мас.%, 80,0 г, 0,28 моль) нагревали до 103-107°С в сульфолане (187 мл) и ацетонитриле (39 мл). Медленно по каплям добавляли фосфорилхлорид (31,9 г, 0,21 моль) при перемешивании, капельную воронку промывали ацетонитрилом (13 мл) и затем смесь перемешивали при 107°С в течение 4 часов. Затем смесь охлаждали до 25°С и при соответствующем перемешивании добавляли ацетонитрил (13 мл), а затем воду (120 мл) с хорошей скоростью и при охлаждении, поддерживая внутреннюю температуру от 20°С до 30°С. Смесь перемешивали в течение 1 ч, нагревали до 50°С в течение 0,5 ч, перемешивали при этой температуре в течение 0,5 ч и охлаждали до 20°С в течение 1 ч. Затем раствор водн. аммиака (28%, 43,5 г) в воде (66,7 мл) добавляли по каплям в течение 1 часа и полученную смесь охлаждали до 5°С в течение 1 часа и перемешивали еще 0,5 часа. Осажденные твердые вещества собирали фильтрованием с отсасыванием, промывали водой (2 × 156 мл) и сушили при 50°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 71,9 (95,9% от теоретического выхода)
Способ ВЭЖХ F: минуты основного компонента: 15,3 мин
Анализ (ВЭЖХ мас. %): 100,1%
Чистота (площадь % ВЭЖХ): 99,4%
1Н ЯМР (400 МГц, [D6]DMSO): δ=8,87 (dd, J=2,6, 1,7 Гц, 1H), 8,52 (dd, J=8,1, 2,6 Гц, 1H), 7,17-7,42 (m, 4H), 5,87 (s, 2H). MS (ESI+): m/z=111 [M+H]+.
Пример 10
Гидрохлорид 5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-карбоксимидамида
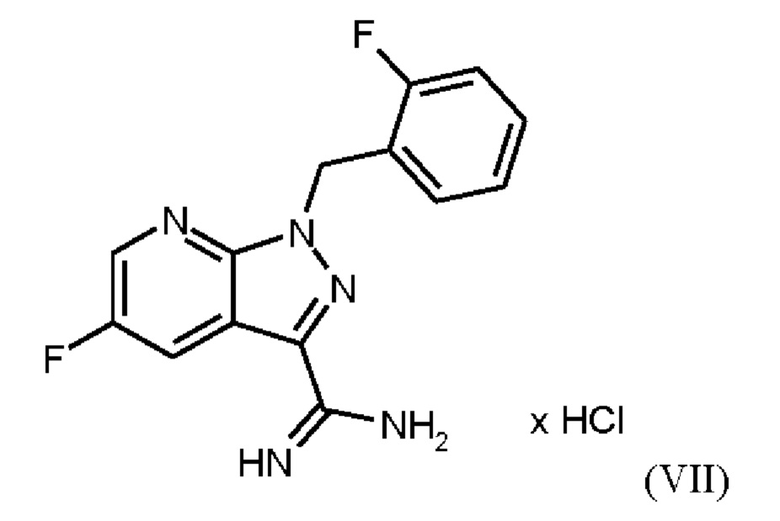
Соединение согласно Примеру 9 (98,8 мас. %, 80,0 г, 0,30 моль) суспендировали в метаноле (268 мл). Затем добавляли 30% метоксид натрия в метаноле (10,8 г, 0,06 моль) и смесь перемешивали при 22°С в течение 5 часов. Добавляли метанол (100 мл) и хлорид аммония (18,6 г, 0,35 моль), смесь нагревали до температуры кипения с обратным холодильником и перемешивали в течение 4,5 часов. Смесь охлаждали до 20°С, добавляли кизельгур (7,6 г) и перемешивали в течение 1 часа. Суспензию фильтровали и остаток на фильтре промывали метанолом (26 мл). Объединенные фильтраты концентрировали перегонкой при температуре рубашки 80°С, добавляли этилацетат (246 мл) и продолжали перегонку до получения около 100 мл дистиллята. Перегонку продолжали до достижения внутренней температуры 72°С при температуре рубашки 100°С. Тем временем непрерывно добавляли этилацетат (854 мл), чтобы поддерживать постоянный уровень наполнения. Смесь охлаждали до 20°С в течение 2 ч, добавляли 24 мл этанола, перемешивали 1 ч, суспензию фильтровали, остаток на фильтре промывали этилацетатом (157 мл) и сушили при 50°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 84,9 г (88,6% от теоретического выхода).
Способ ВЭЖХ D: минуты основного компонента: 13,8 мин
Анализ (ВЭЖХ мас. %): 98,6%
Чистота (площадь % ВЭЖХ): 99,9%
1Н ЯМР (400 МГц, [D6]DMSO): δ=9,35 (br s, 3Н), 8,86 (dd, J=2,5, 1,5 Гц, 1H), 8,48 (dd, J=8,8, 2,6 Гц, 1H), 7,36-7,43 (m, 1H), 7,29-7,35 (m, 1H), 7,22-7,28 (m, 1H), 7,15-7,20 (m, 1H), 5,90 (s, 2Н). MS (ESI+): m/z=288 [M+H]+.
Пример 11
2-[5-Фтор-1-(2-фторбензил)-1H-пиразоло[3,4-6]пиридин-3-ил]-5-[(Е)-фенилдиазенил]пиримидин-4,6-диамин
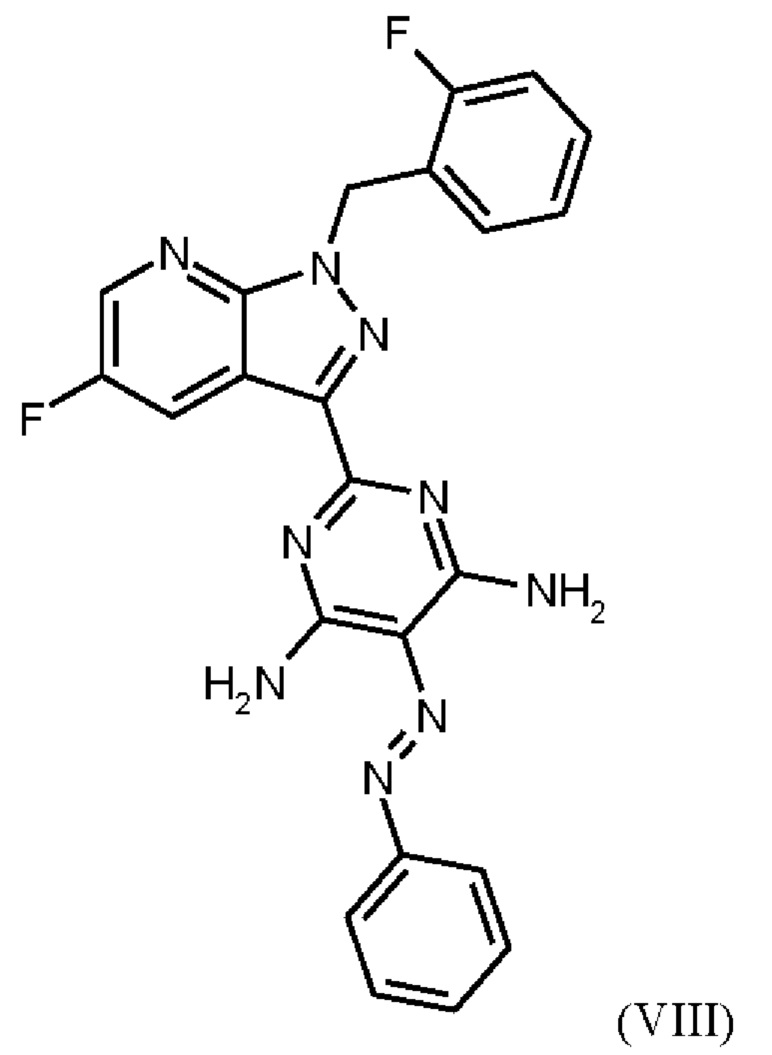
Конц. НСl (12,9 г, 130,9 ммоль) и воду (87,2 мл) добавляли по каплям при температуре от 0°С до 5°С к воде (87,2 мл) и анилину (6,0 г, 65,2 ммоль). Затем в течение 45 мин по каплям добавляли раствор нитрита натрия (4,6 г, 66,0 ммоль) в воде (11,1 мл) и смесь перемешивали при температуре от 0°С до 5°С в течение 15 мин. Затем при этой температуре по каплям в течение 45 мин добавляли раствор ацетата натрия (6,8 г, 82,6 ммоль) в воде (33,4 мл) и раствор малононитрила (4,4 г, 65,8 ммоль) в этаноле (11,7 г) добавляли по каплям в течение 1 часа. Капельную воронку промывали этанолом (68,5 мл) и смесь дополнительно перемешивали при температуре от 0°С до 5°С в течение 2 часов. Желтые твердые вещества собирали фильтрованием с отсасыванием, промывали водой (3×51 мл) и изопропанолом (3×26 мл) и сушили с отсасыванием. Еще влажный остаток растворяли в DMF (47,5 г) и триэтиламине (6,0 г, 59,4 ммоль), с получением раствора DMF [(Е)-фенилдиазенил]малононитрила (соединение (VIIIa) и триэтиламина (71,4 г). Соединение согласно Примеру 10 (97,7 мас. %, 14,0 г, 40,9 ммоль) суспендировали в DMF (25,7 г). Смесь нагревали до 100°С и раствор триэтиламина и [(E)-фенилдиазенил]малононитрила в DMF добавляли по каплям при этой температуре в течение 10 ч. Далее смесь перемешивали при 100°С в течение 12,5 ч. Затем охлаждали до 85°С, добавляли по каплям метанол (16,6 г) в течение 1 ч и полученную смесь охлаждали до 2°С в течение 5 ч и перемешивали в течение 1 ч. Твердые вещества собирали фильтрованием с отсасыванием, промывали DMF (5,5 г), метанолом (12 г), водой (76 г) и затем метанолом (12 г), сушили отсасыванием и затем сушили при 65°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 14,6 г (78,1% от теоретического выхода)
Способ ВЭЖХ С: минуты основного компонента: 18,6 мин
Анализ (ВЭЖХ мас. %): 98,6% Чистота (площадь % ВЭЖХ): 99,0%
1Н ЯМР (400 МГц, [D6]DMSO): δ=9,03 (dd, J=8,8, 2,8 Гц, 1H), 8,65-8,77 (m, 1H), 8,50 (br s, 2H), 8,02 (d, J=7,6 Гц, 2H), 7,86-7,98 (m, 2H), 7,44-7,57 (m, 2H), 7,32-7,44 (m, 2H), 7,11-7,31 (m, 3H), 5,84 (s, 2H). LC-MS (способ d): fR (мин)=1,15. MS (ESI+): m/z=458 [M+H]+.
Пример 11A
3-амино-3-({6-амино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]-5-[(Е)-фенилдиазенил]пиримидин-4-ил}амино)-2-[(Е)-фенилдиазенил]акрилонитрил
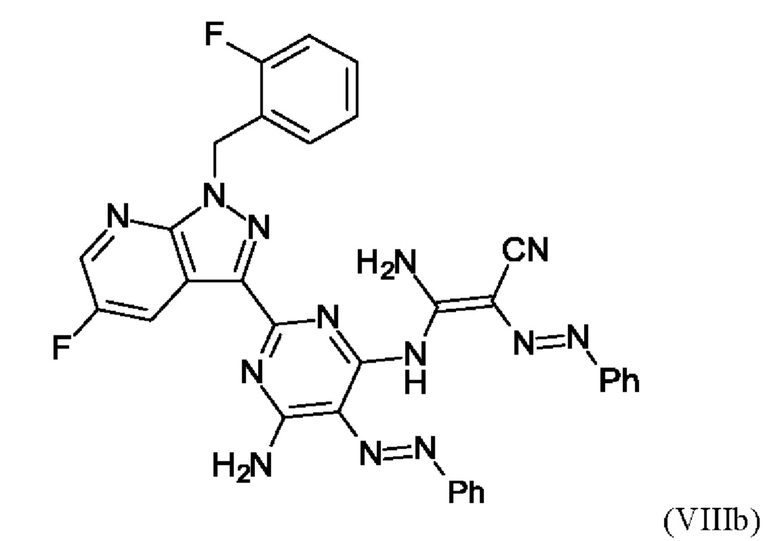
Соединение согласно Примеру 11 А получают как примесь соединения согласно Примеру 11 (VIII) реакцией двух молекул [(Е)-фенилдиазенил]малононитрила (соединение (VIIIa)) с соединением (VII) в присутствии триэтиламина. Если процесс проводят в соответствии с вышеописанными условиями, получают уровни примесей от 0,2 до 0,6 (площадь % ВЭЖХ). Эти уровни примеси полностью истощаются на последующих стадиях процесса, что приводит к высокочистому соединению формулы (I).
1Н ЯМР (500 МГц, DMF, 303К): δ=11,56-11,84 (т), 11,52 (br s), 11,01-11,28 (ь), 9,71 (br d,J=1,6 Гц), 9,65 (br d, J=1,6 Гц), 9,30 (br d, J=3,8 Гц), 9,22-9,29 (m), 9,10 (br d, J=8,2 Гц), 8,99-9,21 (m), 8,92-8,96 (m), 8,54 (br d, J=6,6 Гц), 8,15 (br d, J=7,6 Гц), 7,91-8,00 (m), 7,77 (br d, J=7,6 Гц), 7,69-7,75 (m), 7,62-7,65 (m), 7,58-7,63 (m), 7,51-7,56 (m), 7,46-7,52 (m), 7,44-7,49 (m), 7,41 (br t,J=7,7 Гц), 6,15 ppm (s)
13C ЯМР (126 МГц, DMF, 303K): δ=160,7, 158,6, 157,9, 156,9, 156,3, 156,0, 153,8, 152,8, 148,7, 140,0, 139,8, 131,2, 130,7, 129,7, 129,4, 127,3, 125,0, 123,8, 123,3, 122,4, 121,0, 117,6, 116,6, 115,7, 114,3, 96,1, 94,9, 45,1 ppm
Пример 12
2-[5-Фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-4,5,6-триамин
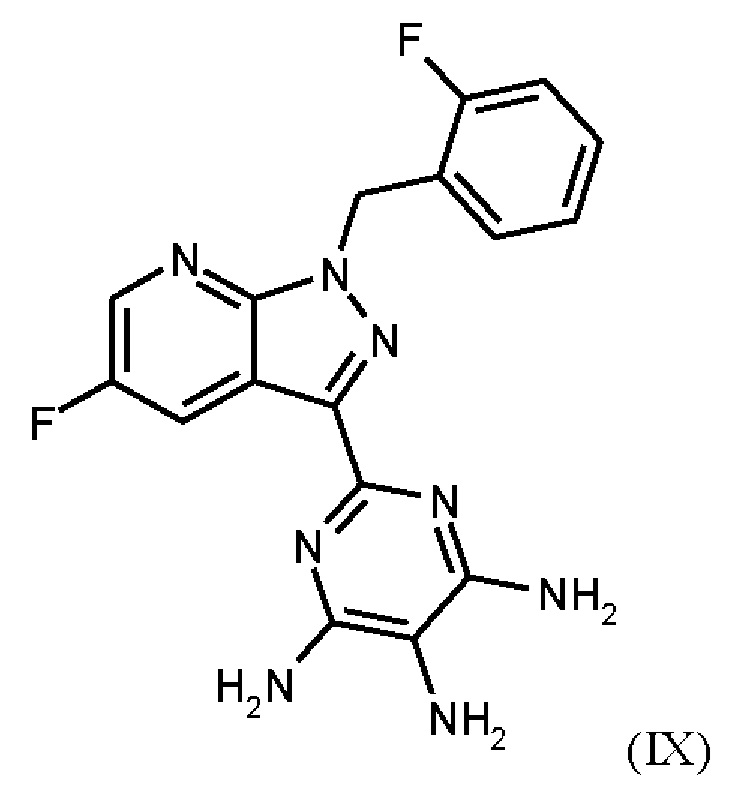
Соединение согласно Примеру 11 (97,7 мас. %, 100,0 г, 0,22 моль) сначала загружали в NMP (0,57 л), а затем добавляли 5% Pd/C (50% влажность, 2,2 г). Гидрирование проводили при 60°С и давлении водорода 60 бар при перемешивании в течение ночи. Смесь фильтровали и твердые вещества тщательно промывали NMP (48,4 мл). Фильтрат охлаждали до 20°С, затем добавляли воду (1,92 л) в течение 3 часов и смесь перемешивали в течение 1 часа. Твердые вещества собирали фильтрованием с отсасыванием, промывали водой (2 × 300 мл), сушили отсасыванием и затем сушили при 100°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 76,1 г (95,5% от теоретического выхода)
Способ ВЭЖХ В: минуты основного компонента: 10,6 мин
Анализ (ВЭЖХ мас. %): 98,6%
Чистота (площадь% ВЭЖХ): 98,6%
1H ЯМР (400 МГц, [D6]DMSO): δ=8,85 (dd, J=9,0, 2,9 Гц, 1H), 8,62 (dd, J=2,8, 1,7 Гц, 1Н), 7,32-7,39 (m, 1H), 7,10-7,26 (m, 3H), 5,86 (br s, 4H), 5,75 (s, 2H), 4,04 (br s, 2H). MS (ESI+): mlz=369 [M+H]+.
Примеры 13
Гидрохлорид метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b] пиридин-3-ил] пиримидин-5-ил}карбамата
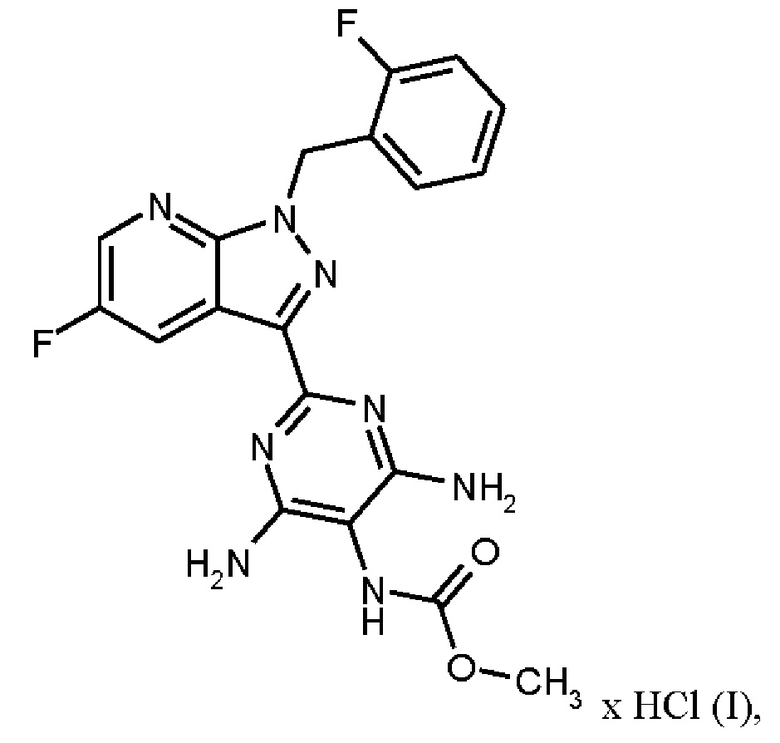
Пример 12 (96,7 мас. %, 250,0 г, 0,66 моль)в тетрагидрофуране (2,34 л) нагревали до 60°С и затем добавляли метилхлорформиат (72,6 г, 0,77 моль) в течение 15 минут. Смесь перемешивали при 60°С в течение 2 ч, твердые вещества собирали фильтрованием с отсасыванием при этой температуре и перемешивали с 1,54 л тетрагидрофурана при 55°С в течение 0,5 ч. Твердые вещества собирали фильтрацией с отсасыванием при этой температуре и снова перемешивали с 1,54 л тетрагидрофурана при 55°С в течение 0,5 часа. Твердые вещества собирали фильтрованием с отсасыванием при этой температуре, сушили с отсасыванием и затем сушили при 50°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 294,0 г (96,2% от теоретического выхода)
Способ ВЭЖХ А: минуты основного компонента: 9,4 мин
Анализ (ВЭЖХ мас. %): 98,49%
Чистота (площадь % ВЭЖХ): 99,14%
MS (ESIpos): m/z=427 (М+Н)+
1H ЯМР (600 МГц, [D6]DMSO): δ=13,3 (br s, 1H) 8,81 (m, 2Н), 8,41 (br s, 1H), 8,07 и 7,65 (2 br s, 4H), 7,40-7,13 (m, 4H), 5,90 (s, 2H), 3,66 (br s, 3H).
Пример 13A
N-{4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}формамид
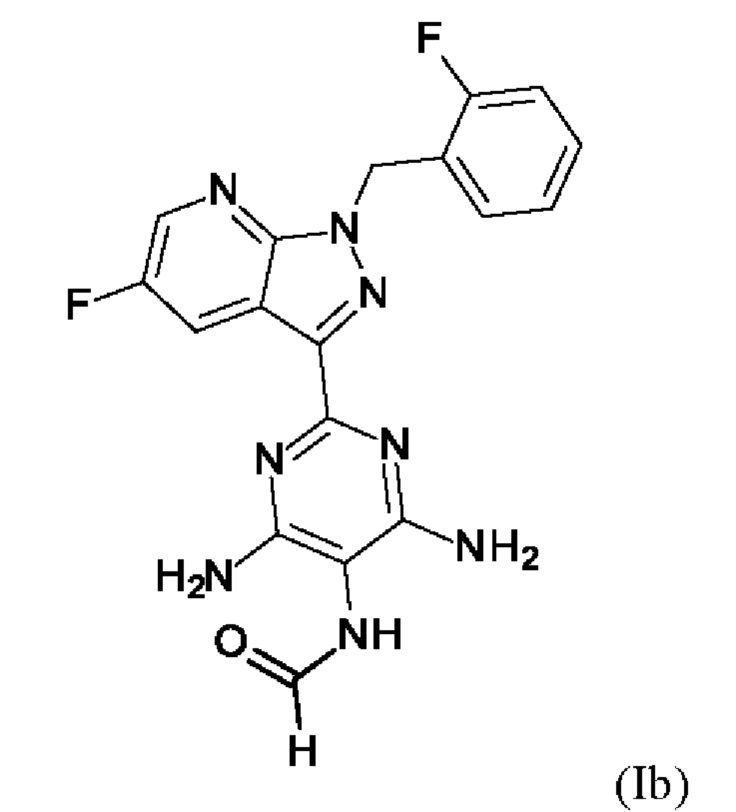
Соединение согласно Примеру 13А получают в виде примеси путем взаимодействия остаточного DMF с метилхлорформиатом и соединением формулы (IX), Пример 12, вместо гидрохлорида соединения формулы (I).
1Н ЯМР (600 МГц, [D6]DMSO): δ=8,89 (m, 1H), 8,85 (m, 1H), 8,66 (m, 1H), 8,12 (s, 1H), 7,38-7,34 (m, 1H) 7,24-7,13 (m, 3H), 6,41 (br s, 1H), 6,24 (br s, 3H), 5,79 (s, 2H).
Пример 14
Метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамат в виде ди-диметилсульфоксид сольвата
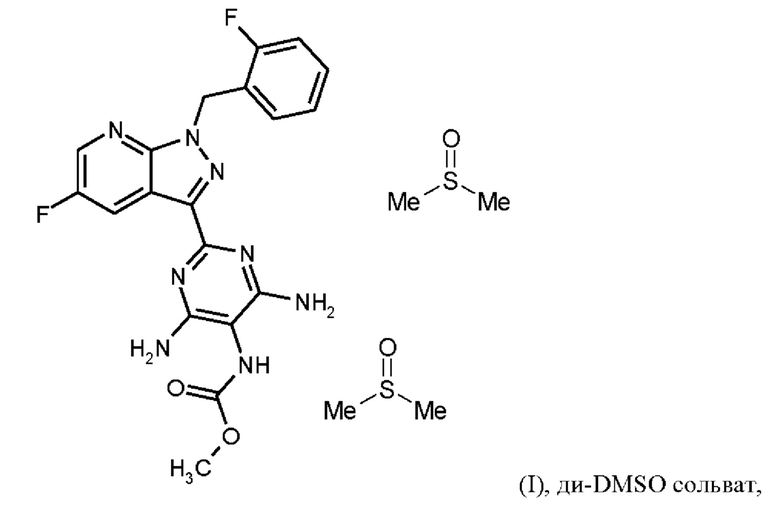
Соединение согласно Примеру 13 (280,0 г) (98,5 мас. %, 280,0 г, 0,60 моль) перемешивали в DMSO (635,8 мл) в течение 2 ч при 80°С. Три-н-бутиламин (140,0 г, 0,18 моль) и активированный углерод (16,8 г) добавляли и смесь перемешивали при 80°С в течение 15 мин. Суспензию фильтровали в горячем состоянии и остаток на фильтре промывали DMSO (173 мл), предварительно нагретым до 80°С. Объединенные фильтраты перемешивали при 60°С в течение 15 мин, охлаждали до 45°С в течение 1,5 ч, перемешивали при этой температуре в течение 0,5 ч и далее охлаждали до 20°С со скоростью 10 град/ч. В течение 1 ч добавляли этилацетат (1,98 л), нагревали до 45°С со скоростью 10 К/ч, перемешивали при этой температуре в течение 1 ч и снова охлаждали до 20°С со скоростью 10 К/ч. Перемешивали в течение ночи при 20°С, охлаждали до 20°С со скоростью 10 К/ч и перемешивали при этой температуре в течение 0,5 ч. Твердые вещества собирали фильтрованием с отсасыванием, промывали смесью DMSO (107,3 г) и этилацетата (536,7 г), сушили отсасыванием и затем сушили при 50°С в слабом потоке азота в вакуумном сушильном шкафу.
Выход: 271,9 г (77,7% от теоретического выхода)
Способ ВЭЖХ А: минуты основного компонента: 9,4 мин
Анализ (ВЭЖХ мас. %): 73,1%; 24,4% DMSO
Чистота (площадь % ВЭЖХ): 99,92%
1H ЯМР (400 МГц, [D6]DMSO): δ=8,89 (dd, J=9,0, 2,8 Гц, 1H), 8,66 (m, 1H), 7,99 и 7,67 (2 br s, 1H), 7,32-7,40 (m, 1H), 7,19-7,26 (m, 1H), 7,10-7,19 (m, 2Н), 6,22 (br s, 4Н), 5,79 (s, 2Н), 3,62 (br s, 3Н). LC-MS (cnoco6d): fR (мин)=0,79. MS (ESI+): m/z=427 [M+H]+.
Пример 15
Метил{4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил] пиримидин-5-ил}карбамат
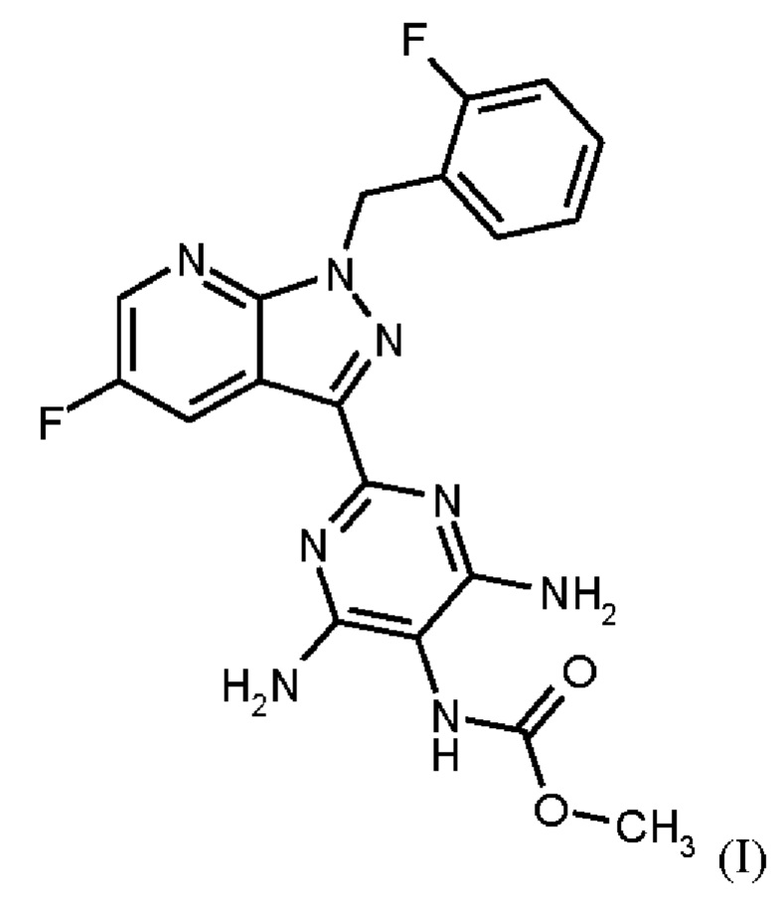
6,29 г метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в виде ди-DMSO сольвата (73,0% мас./мас. соединения формулы (I), 27,4% мас./мас. DMSO, (Пример 14)) суспендировали в 37,4 г DMSO и нагревали до 75°С. К полученному прозрачному раствору добавляли 15,7 г этанола и смесь перемешивали при 75°С в течение 15 мин. Раствор фильтровали, промывали 22,4 г DMSO. Фильтрат нагревали до 75°С и 53,4 г воды добавляли по каплям в течение 5 мин. Суспензию охлаждали до 20°С со скоростью 28 К/ч и добавляли 25,8 г изопропилацетата в течение 30 мин. Смесь перемешивали при 20°С еще 30 мин, и твердое вещество выделяли. Затем его промывали сначала 34,3 г этанола, а затем 34,8 г изопропилацетата. Влажный продукт сушили в течение ночи при 50°С при пониженном давлении в токе азота. Получали метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамат формулы (I) в кристаллической форме модификации I с очень высоким выходом и чистотой.
Выход: 4,24 г (92,6% от теоретического выхода)
Способ ВЭЖХ А: минуты основного компонента: 9,4 мин
Анализ (ВЭЖХ мас. %): 99,27%
Чистота (площадь % ВЭЖХ): 99,97%
MS (ESIpos): m/z=427 (М+Н)+
1H ЯМР (400 МГц, [D6]DMSO): δ=8,89 (dd, J=9,0, 2,8 Гц, 1H), 8,66 (m, 1H), 7,99 и 7,67 (2 br s, 1H), 7,32-7,40 (m, 1H), 7,19-7,26 (m, 1H), 7,10-7,19 (m, 2Н), 6,22 (br s, 4H), 5,79 (s, 2H), 3,62 (br s, 3Н).
LC-MS (способ d): 'R (мин)=0,79. MS (ESI+): m/z=427 [M+H]+.
Пример 16
Более высокая удельная производительность в центрифуге с выворачиваемым фильтром. Промышленный масштаб.
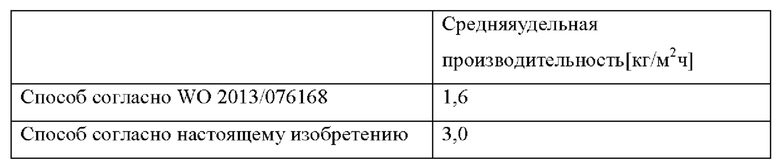
Эти данные показывают улучшенную способность к выделению материала, полученного способом согласно настоящему изобретению, по сравнению с материалом, полученным способом согласно WO 2013/076168.
Пример17
Улучшенная пропускная способность сита. Промышленный масштаб.

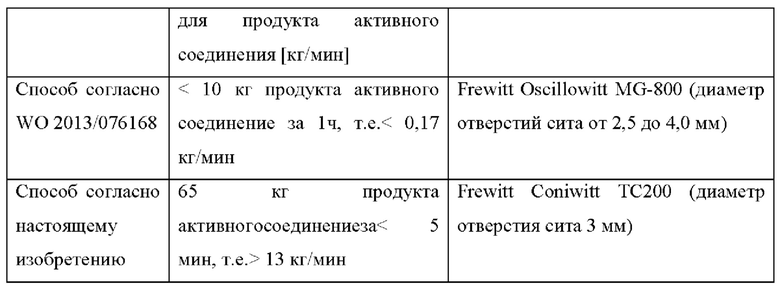
Наблюдается очень большая разница почти в 100 раз в пропускной способности сита активного продукта соединения (I) в кристаллической форме модификации I, полученного в соответствии со способом WO 2013/076168, по сравнению с продуктом активного соединения (I) в кристаллической форме модификации I, полученным способом согласно настоящему изобретению. Эта очень большая разница в пропускной способности сита обусловлена главным образом характеристиками материала продукта активного соединения и не может быть объяснена различными типами устройств.
Краткое описание чертежей
Фиг. 1: Соединение формулы (I) в кристаллической форме модификации I, полученное способом согласно WO 2013/076168, анализ с помощью сканирующей электронной микроскопии.
Фиг. 2: соединение формулы (I) в кристаллической форме модификации I, полученное согласно Примеру 15 согласно настоящему изобретению, анализ с помощью сканирующей электронной микроскопии.
Эти изображения показывают заметную разницу в структуре соединения формулы (I) в кристаллической форме модификации I в качестве активного соединения, что указывает на улучшенные свойства, среди прочего, в отношении способности к выделению продукта активного соединения, способности к выгрузке продукта активного соединения после выделения и сушки, а также транспортабельности, просеиваемости и микронизируемости продукта активного соединения, полученного способом согласно настоящему изобретению.

Изобретение относится к способу получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I, где рентгеновская дифрактограмма указанного соединения в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7. Согласно предлагаемому способу получают гидрохлорид соединения формулы (I) путем нагревания соединения формулы (IX) в тетрагидрофуране в качестве растворителя, добавления от 1,0 до 1,2 экв. метилхлорформиата, перемешивания в течение времени реакции от 1 до 10 ч и выделения гидрохлорида соединения формулы (I). Затем получают ди-DMSO сольват соединения формулы (I) путем растворения гидрохлорида соединения формулы (I) в DMSO, добавления три-н-бутиламина и активированного углерода, удаления активированного углерода, кристаллизации ди-DMSO сольвата путем охлаждения и добавления этилацетата, выделения ди-DMSO сольвата в кристаллической форме и промывки смесью DMSO и этилацетата. Затем получают соединение формулы (I) в кристаллической форме модификации I, где 1.1) ди-DMSO сольват соединения формулы (I) растворяют в DMSO и этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас.; 1.2) растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды; 1.3) образованную суспензию затем охлаждают до температуры от 5 до 50°С и 1.4) образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения добавлением изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0. Способ позволяет получать целевое соединение с высоким выходом, высокой степенью чистоты и улучшенными физическими характеристиками. Изобретение относится также к варианту способа получения соединения формулы (I) и способу получения гидрохлорида соединения формулы (I). 3 н. и 6 з.п. ф-лы, 2 ил., 17 пр.
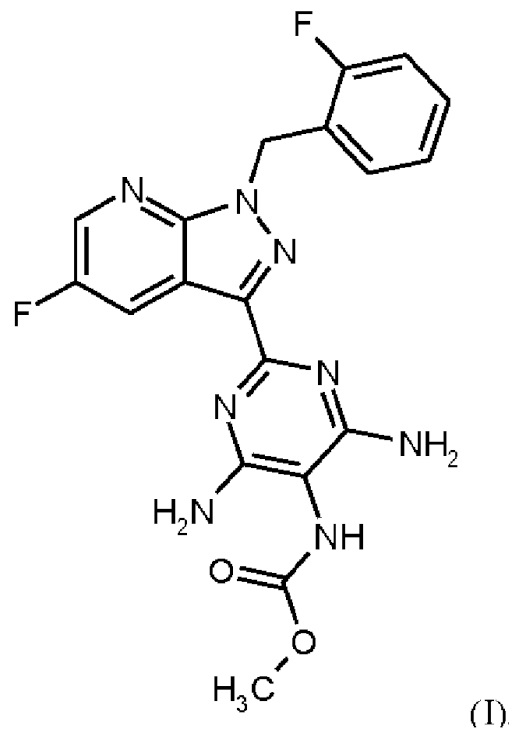
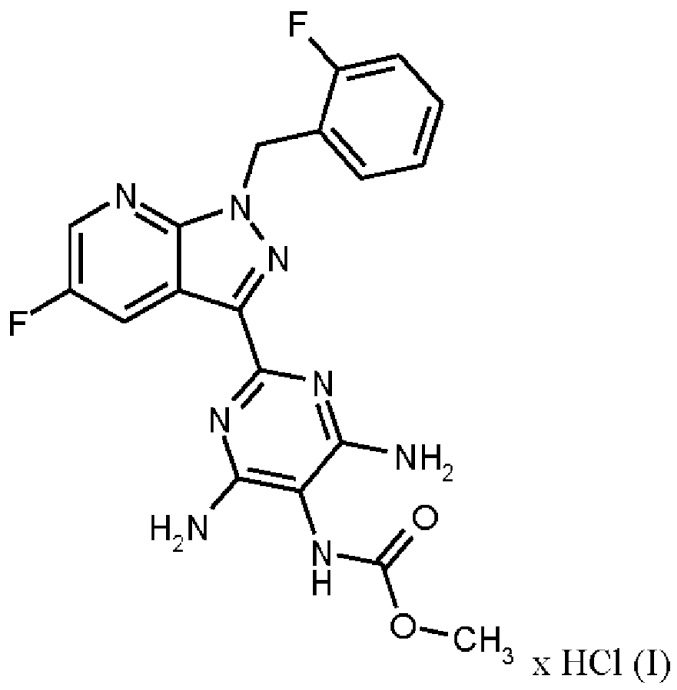
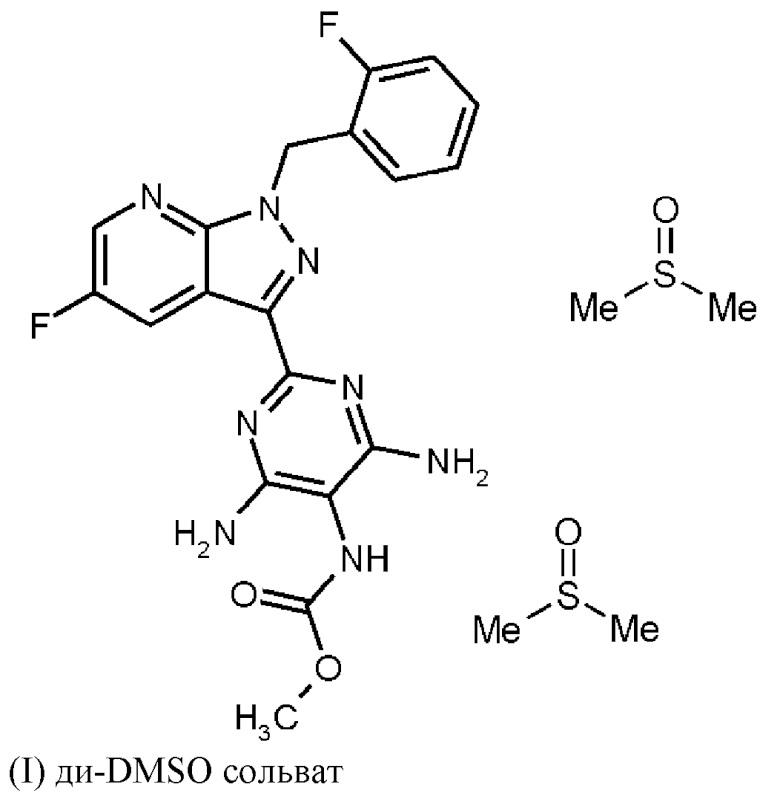
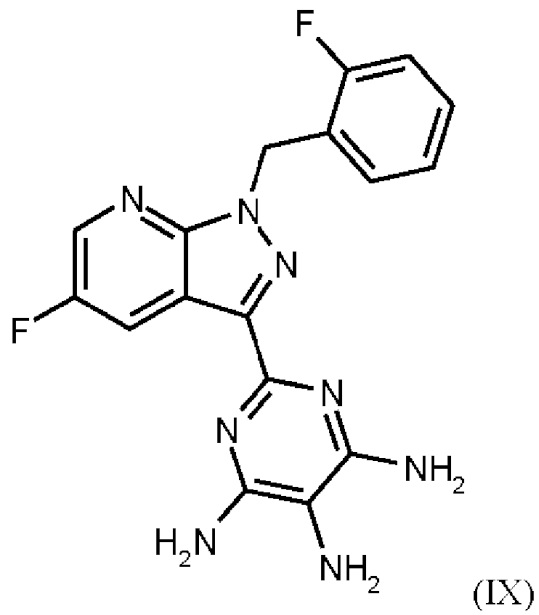
1. Способ получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I)
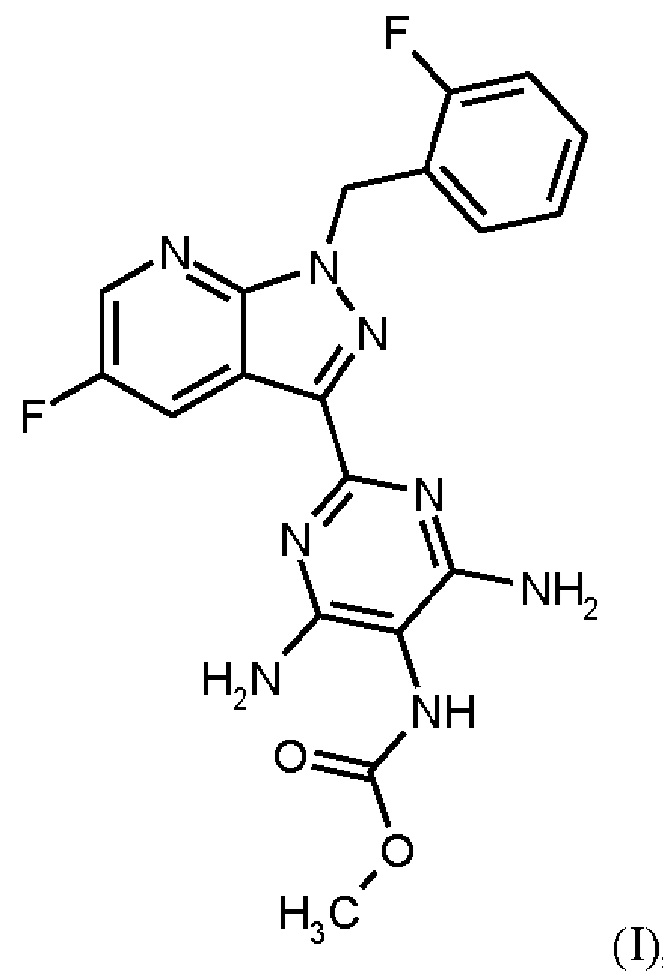
в кристаллической форме модификации I, где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7,
где гидрохлорид соединения формулы (I)
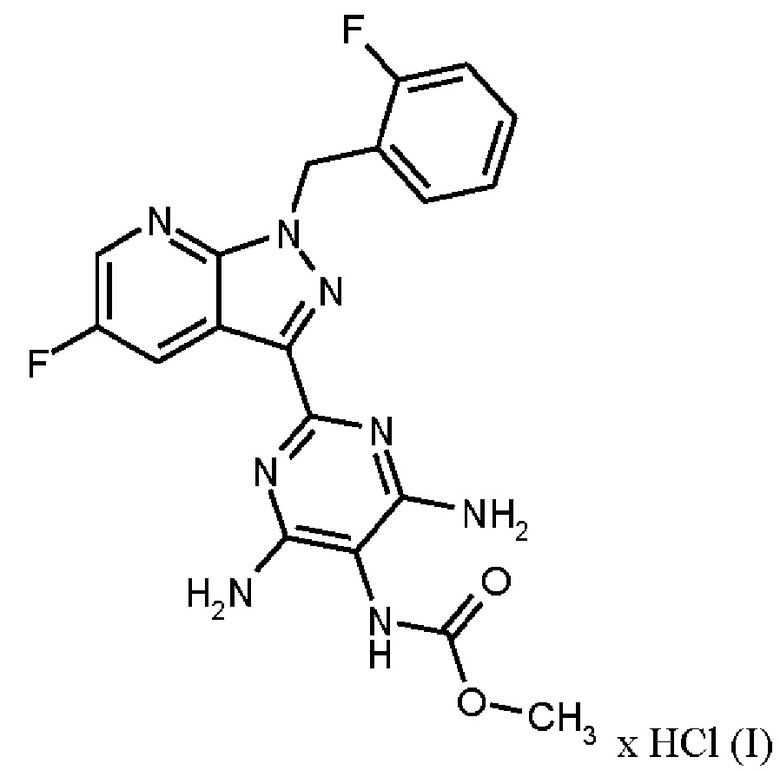
получают путем нагревания соединения формулы (IX)
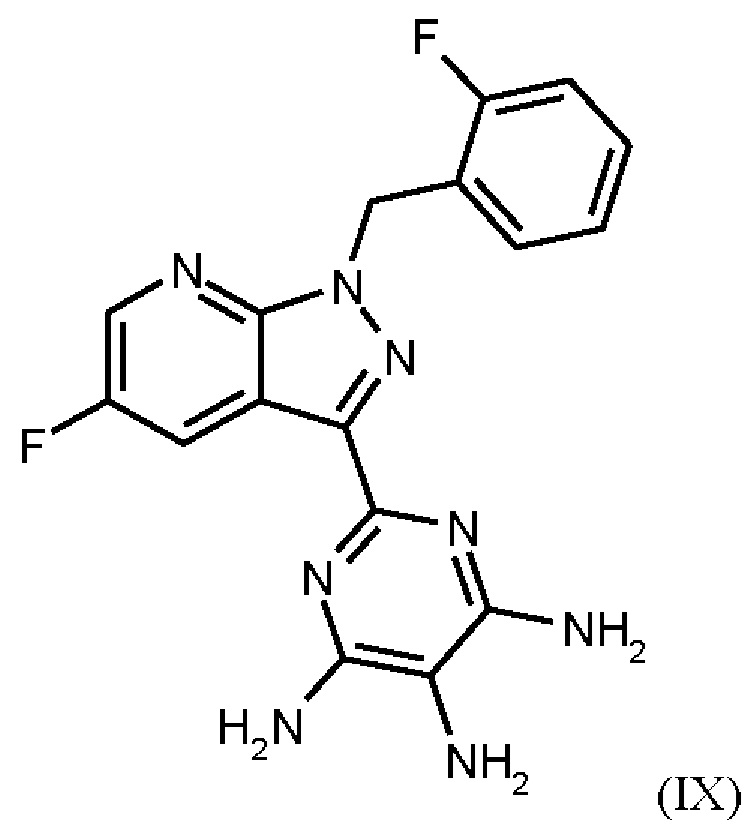
в тетрагидрофуране в качестве растворителя, добавления от 1,0 до 1,2 экв. метилхлорформиата, перемешивания в течение времени реакции от 1 до 10 ч и выделения гидрохлорида соединения формулы (I),
затем ди-DMSO сольват соединения формулы (I)
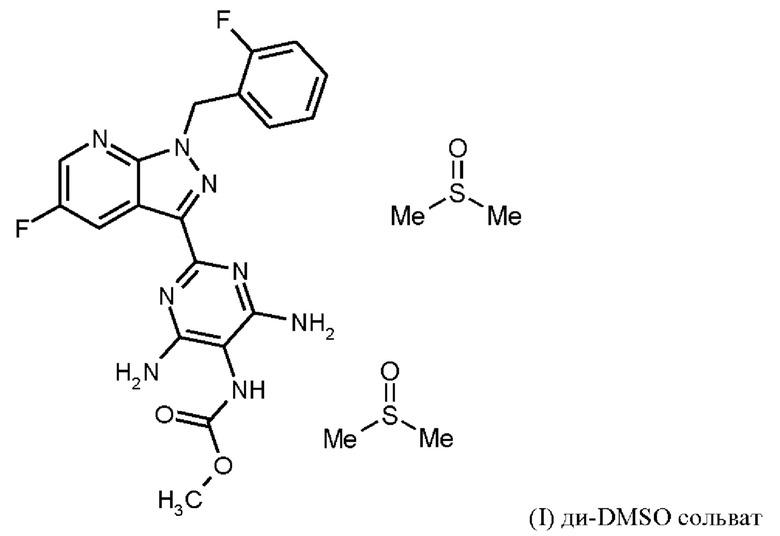
получают путем растворения гидрохлорида соединения формулы (I) в DMSO, добавления три-н-бутиламина и активированного углерода, удаления активированного углерода, кристаллизации ди-DMSO сольвата путем охлаждения и добавления этилацетата, выделения ди-DMSO сольвата в кристаллической форме и промывки смесью DMSO и этилацетата,
затем получают соединение формулы (I) в кристаллической форме модификации I, где
1.1) ди-DMSO сольват соединения формулы (I) растворяют в DMSO и этанол добавляют при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас.;
1.2) растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды;
1.3) образованную суспензию затем охлаждают до температуры от 5 до 50°С и
1.4) образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
2. Способ по п. 1, где соединение формулы (IX)
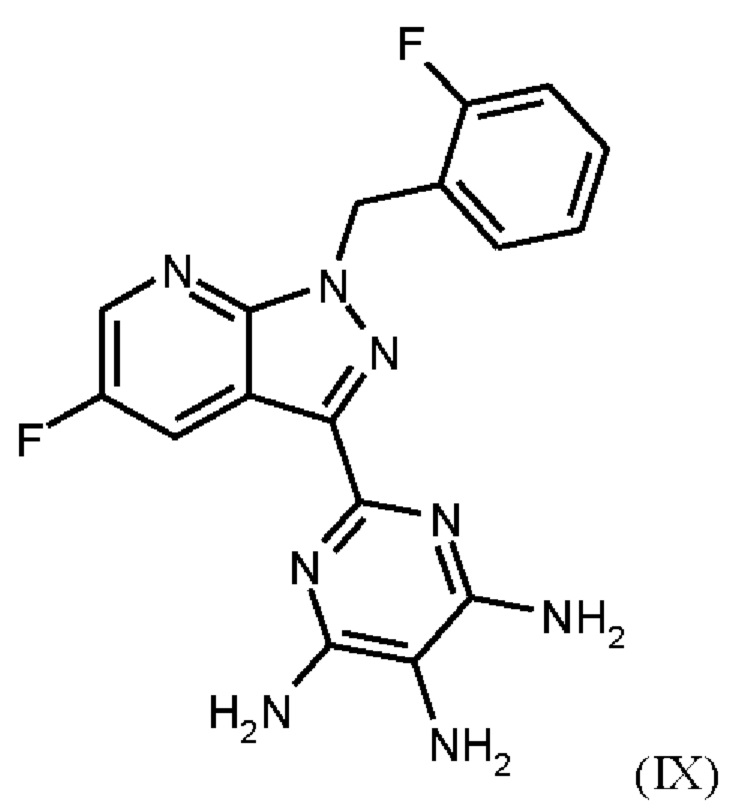
получают путем гидрирования соединения формулы (VIII)
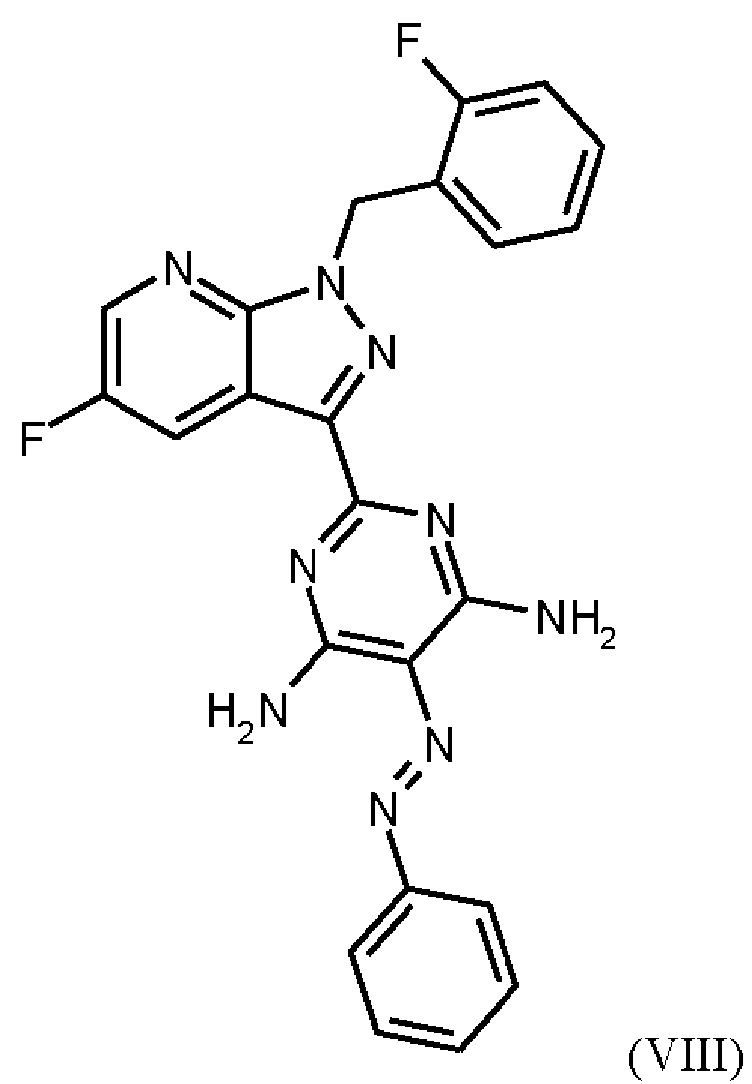
в NMP в качестве растворителя в присутствии водорода при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и выделения с получением соединения формулы (IX).
3. Способ по п. 2, где соединение формулы (VIII)
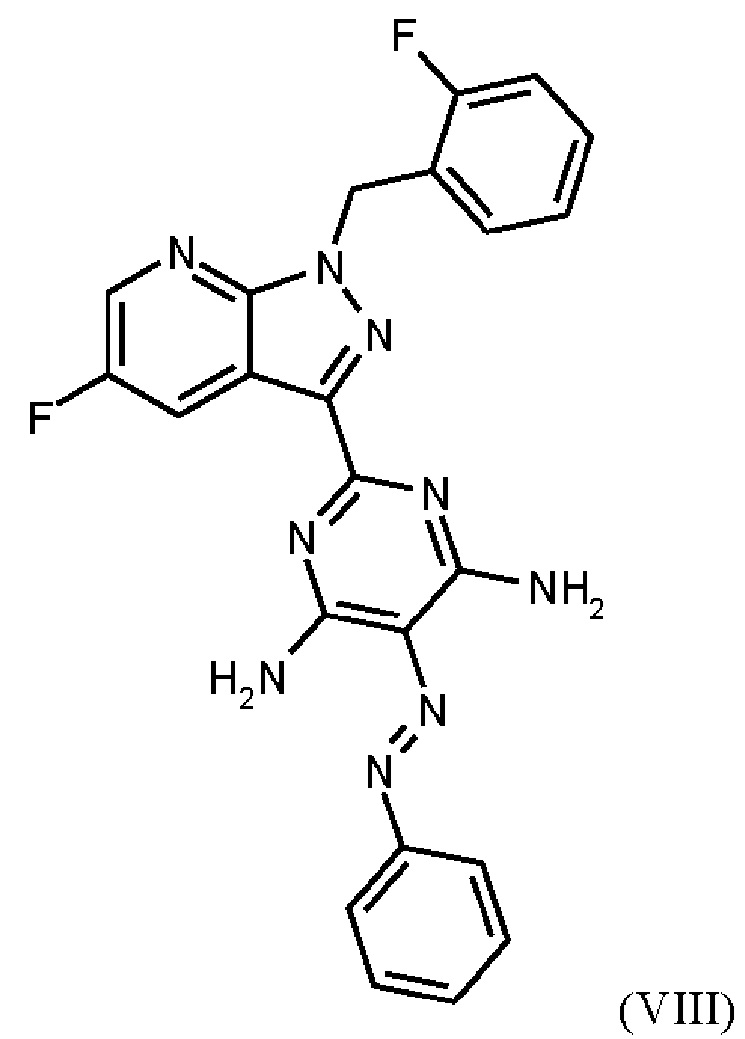
получают путем начального получения соединения формулы (VIIIa)
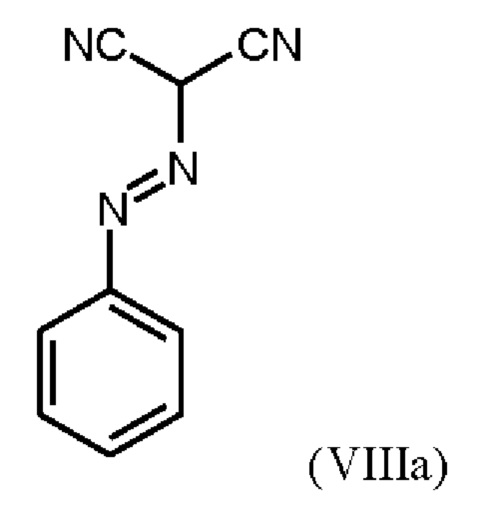
путем добавления концентрированной соляной кислоты в воде к анилину в воде, затем последовательного добавления раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII)
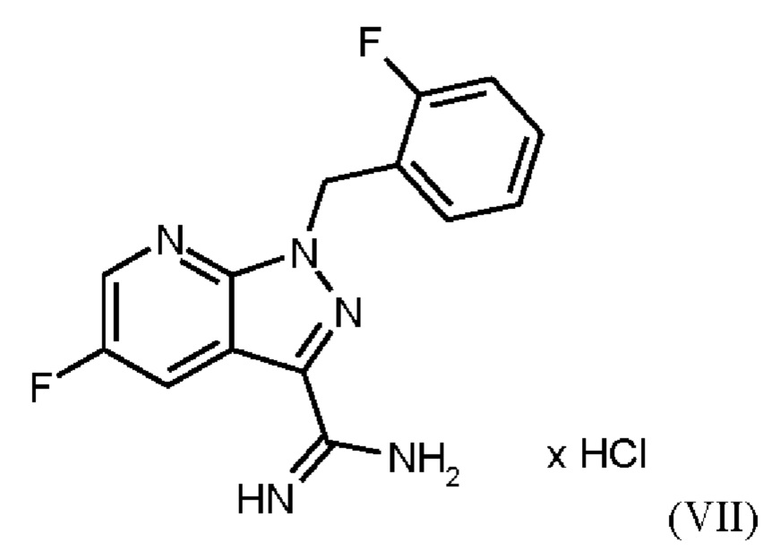
в DMF, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), добавления метанола и выделения соединения формулы (VIII).
4. Способ по п. 3, где соединение формулы (VII)
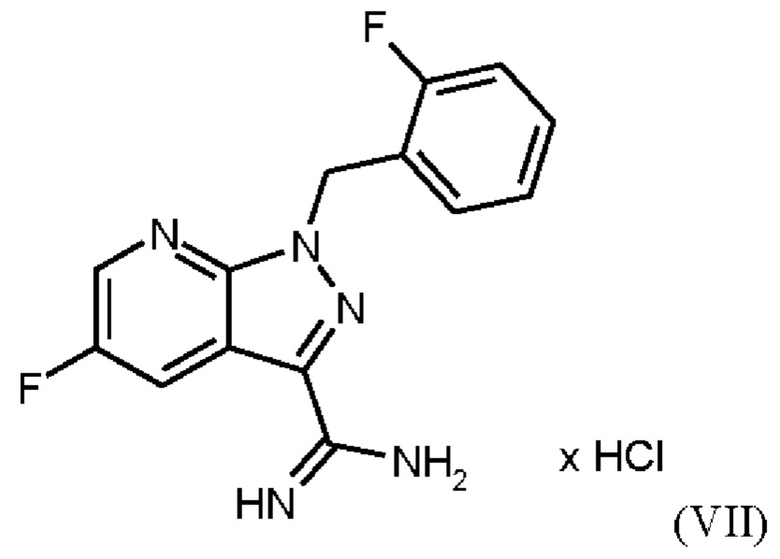
получают путем суспендирования соединения формулы (VI)
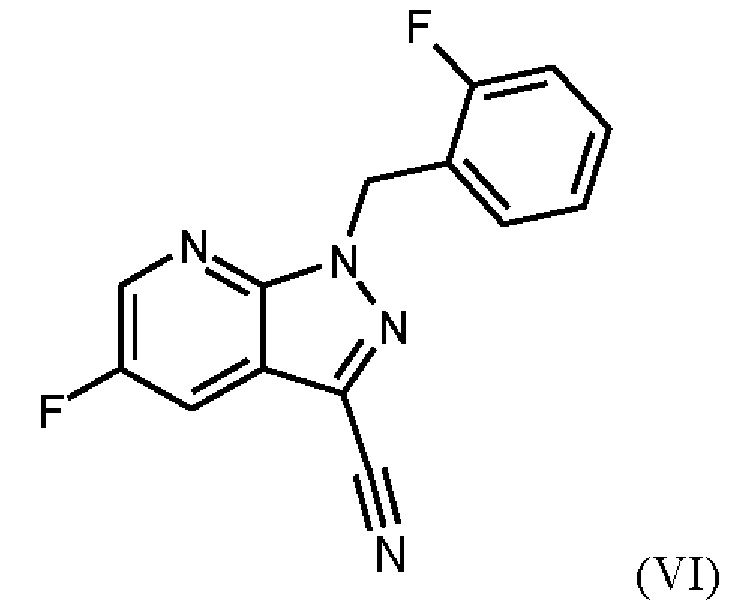
в метаноле, добавления метоксида натрия в метаноле, добавления метанола и хлорида аммония, фильтрации с использованием вспомогательного фильтра, концентрирования и добавления этилацетата, добавления этанола и выделения с получением соединения формулы (VII).
5. Способ по п. 4, где соединение формулы (VI)
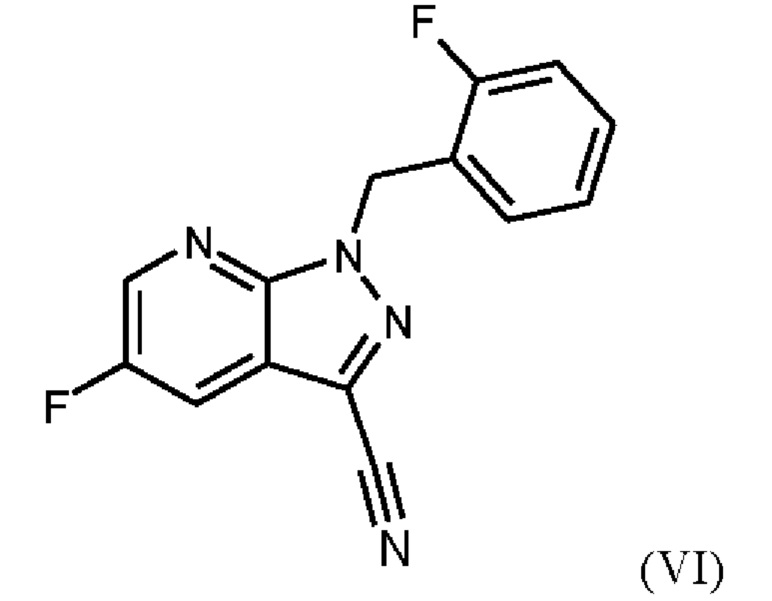
получают путем сначала получения соединения формулы (V)
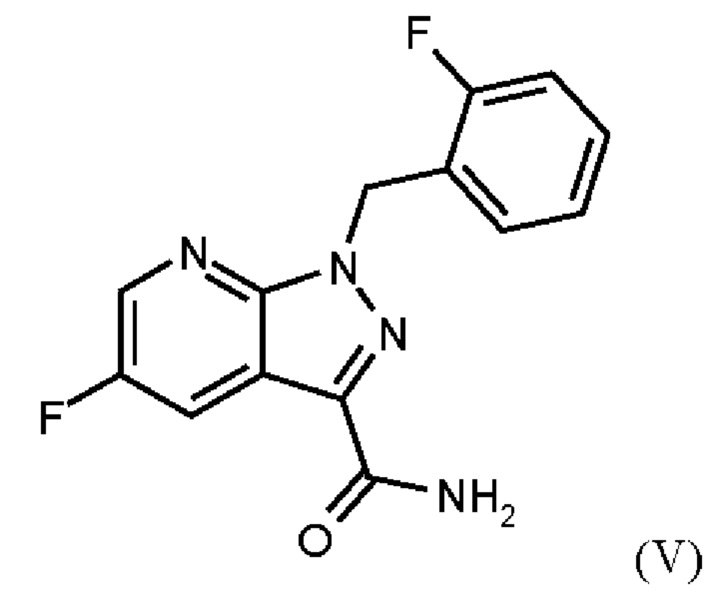
путем начальной загрузки соединения формулы (II)
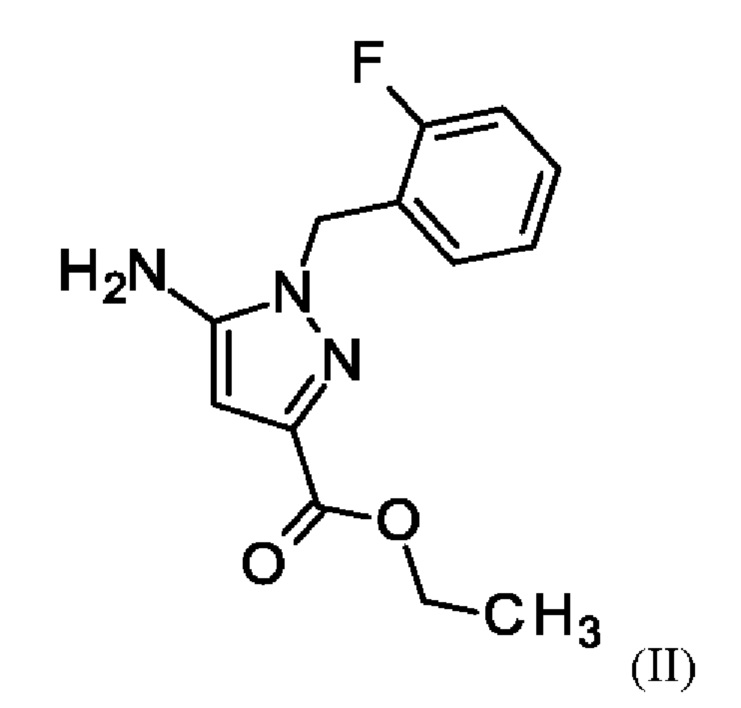
и хлорида лития в этаноле, загрузки соединения формулы (III)
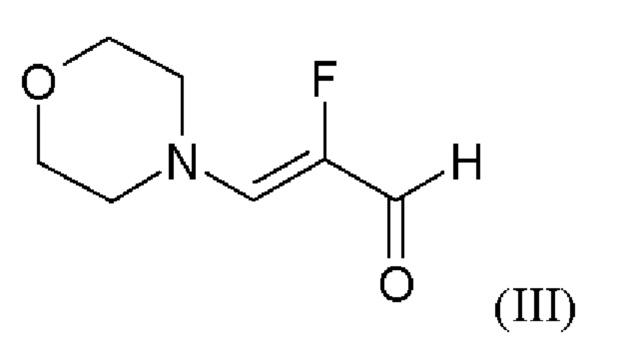
и хлортриметилсилана и нагревания с получением соединения формулы (IV)
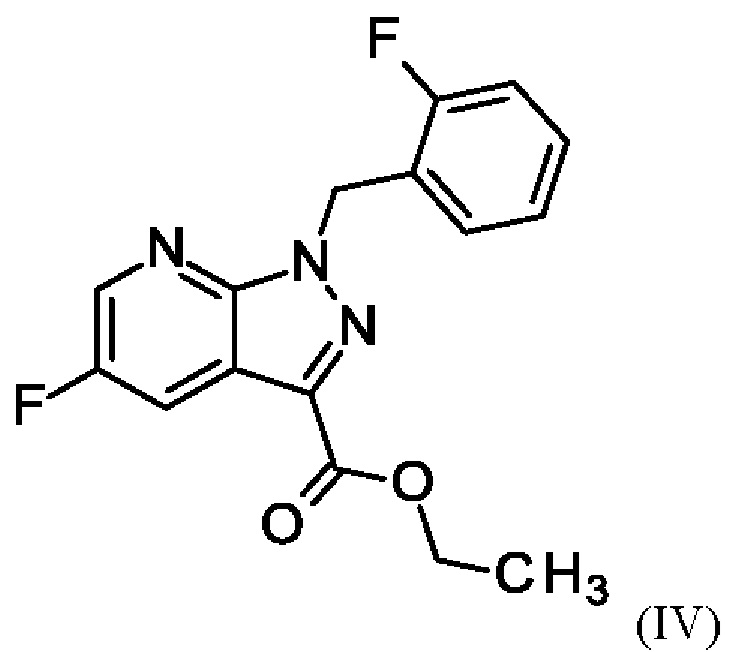
или альтернативно, изменяя порядок загрузки любого из исходных веществ,
добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений, при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V)
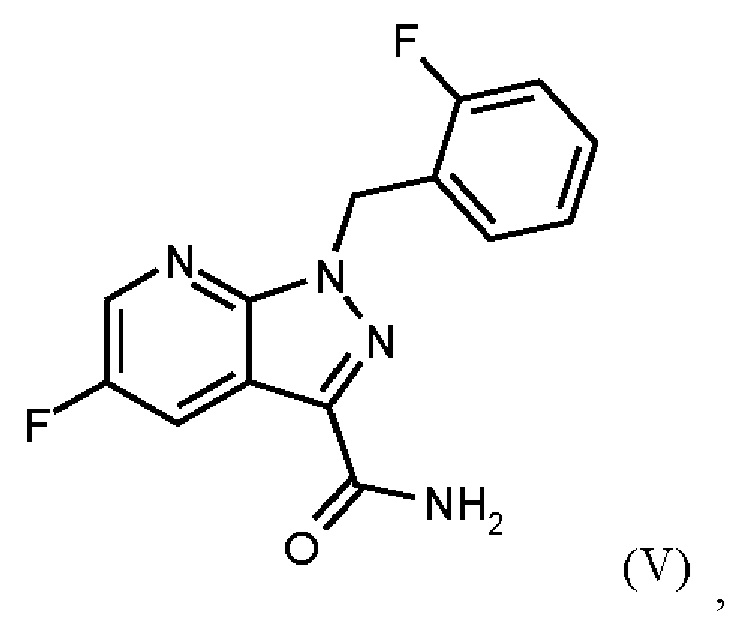
затем дегидратации соединения формулы (V) путем нагревания в сульфолане, ацетонитриле и фосфорилхлориде, добавления ацетонитрила и воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20 до 50°С, добавления аммиака в воде и выделения с получением соединения формулы (VI).
6. Способ по любому из пп. 1-5, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством % площади по ВЭЖХ) или более.
7. Способ получения гидрохлорида соединения формулы (I)
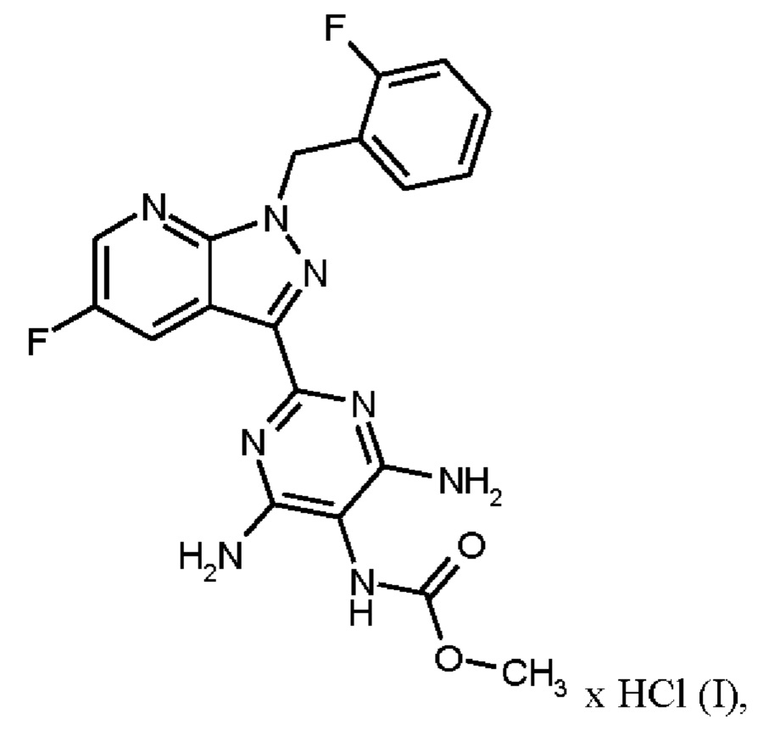
где соединение формулы (IX)
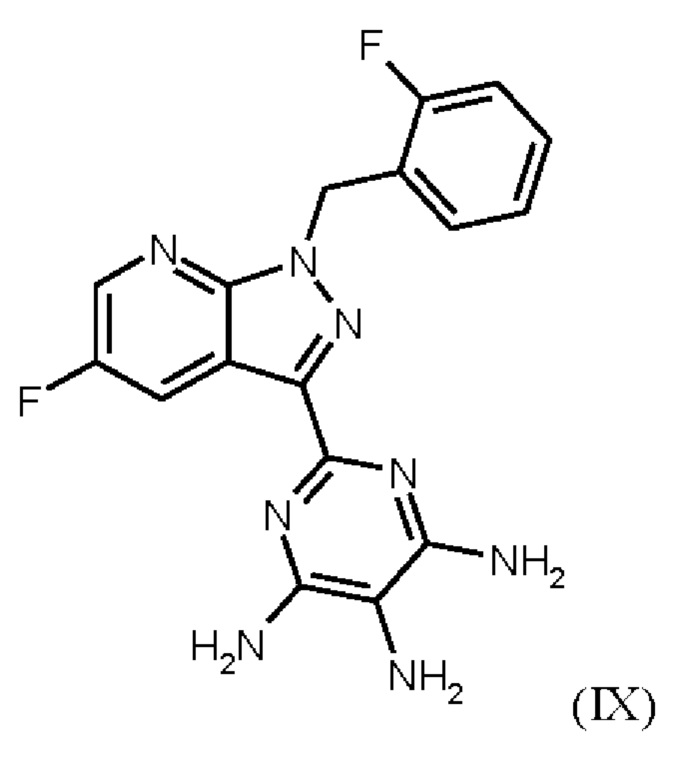
нагревают в тетрагидрофуране в качестве растворителя, добавляют от 1,0 до 1,2 экв. метилхлорформиата, перемешивают в течение времени реакции от 1 до 10 ч, выделяют гидрохлорид соединения формулы (I).
8. Способ получения метил {4,6-диамино-2-[5-фтор-1-(2-фторбензил)-1Н-пиразоло[3,4-b]пиридин-3-ил]пиримидин-5-ил}карбамата формулы (I) в кристаллической форме модификации I по п. 1
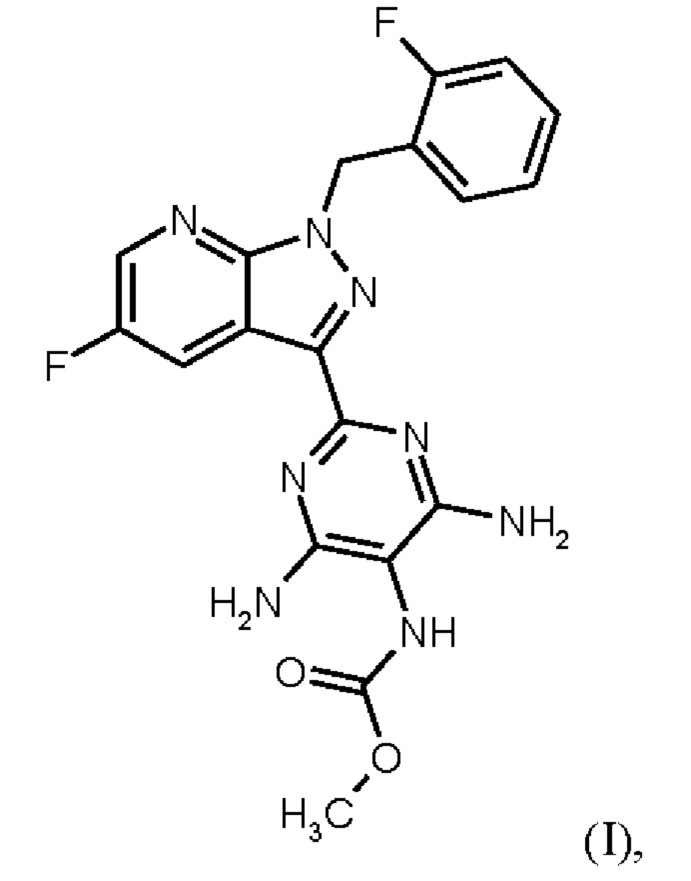
где рентгеновская дифрактограмма соединения формулы (I) в модификации I показывает максимумы пика угла 2-тета при 5,9, 6,9, 22,7 и
где соединение формулы (VI)
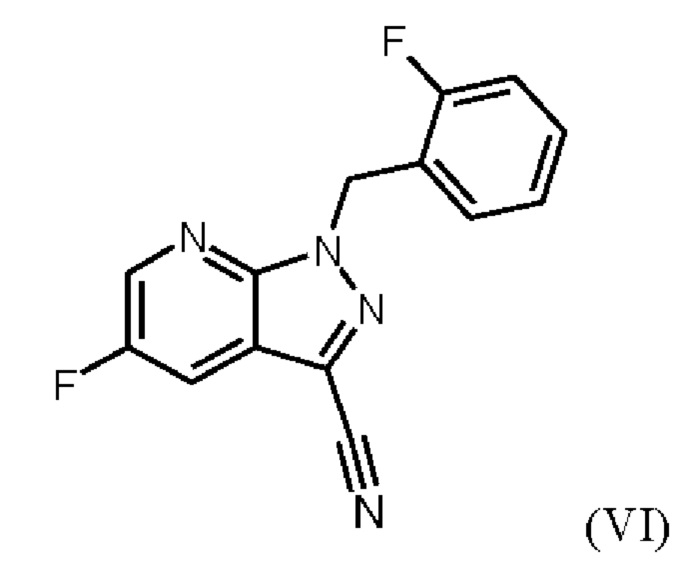
получают путем сначала получения соединения формулы (V)
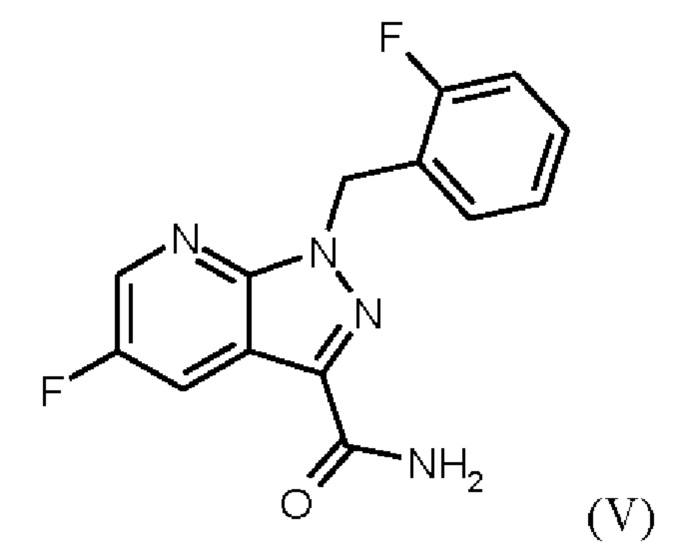
путем начальной загрузки соединения формулы (II)
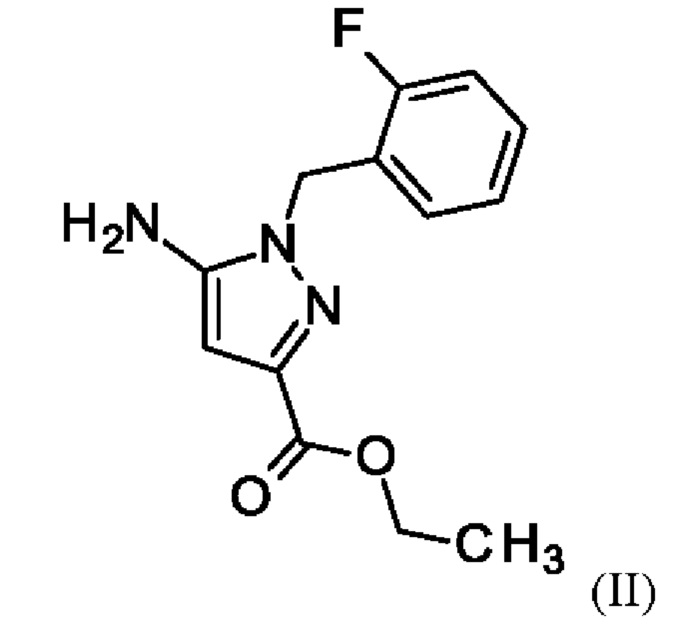
и хлорида лития в этаноле, загрузки соединения формулы (III)
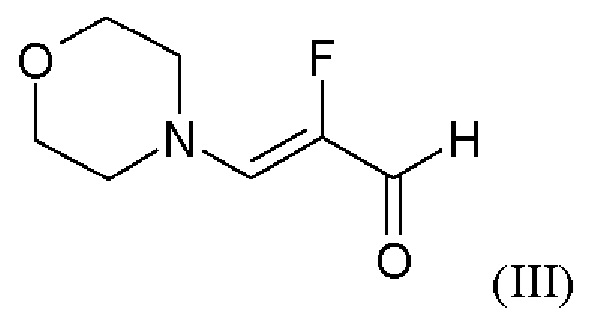
и хлортриметилсилана и нагревания с получением соединения формулы (IV)
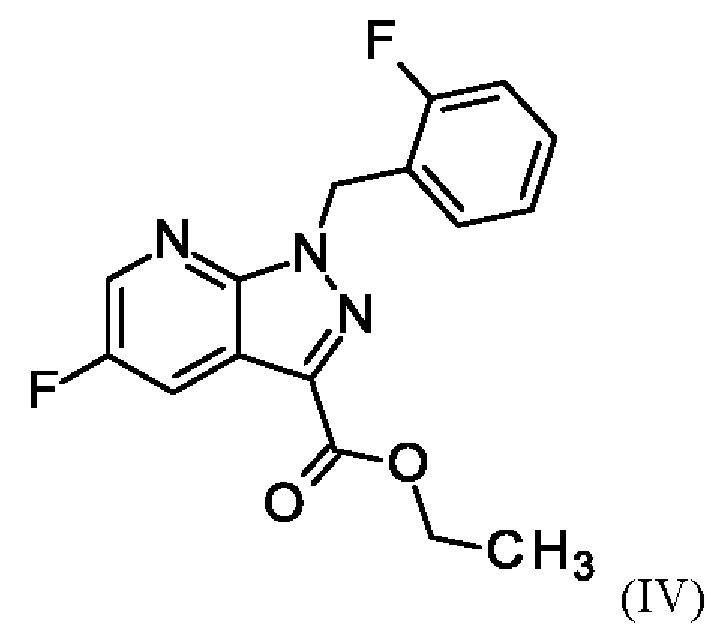
или альтернативно, изменяя порядок загрузки любого из исходных веществ,
добавления формамида и метоксида натрия в метаноле, отгонки низкокипящих соединений при доливке отогнанного объема формамидом, охлаждения, добавления воды, выделения твердых веществ, промывки и сушки с получением соединения формулы (V)
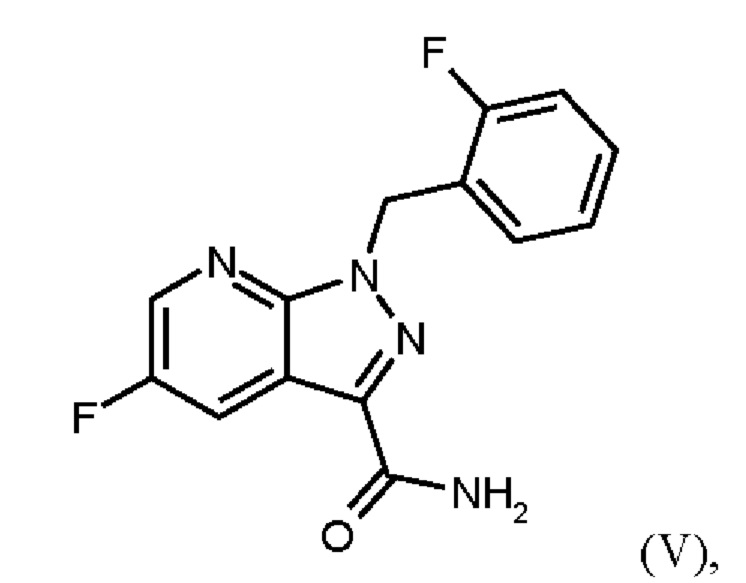
затем дегидратации соединения формулы (V) путем нагревания в сульфолане, ацетонитриле и фосфорилхлориде, добавления ацетонитрила и воды при соответствующей агитации и при хорошей скорости и при охлаждении, сохраняя внутреннюю температуру от 20 до 50°С, добавления аммиака в воде и выделения с получением соединения формулы (VI); затем соединение формулы (VII)
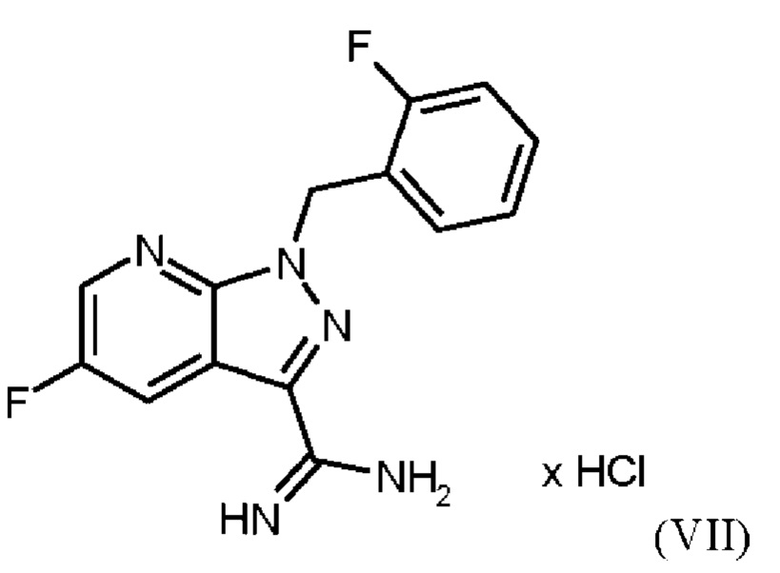
получают путем суспендирования соединения формулы (VI)
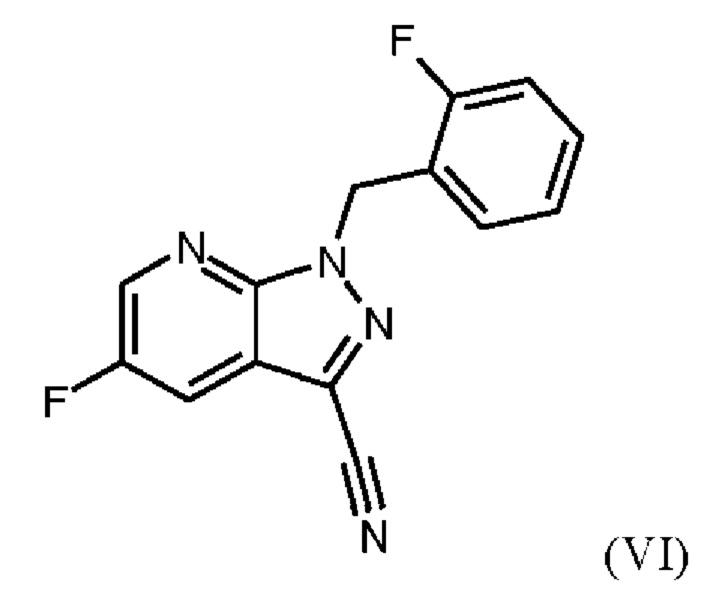
в метаноле, добавления метоксида натрия в метаноле, добавления метанола и хлорида аммония, фильтрации с использованием вспомогательного фильтра, концентрирования и добавления этилацетата, добавления этанола и выделения с получением соединения формулы (VII);
затем соединение формулы (VIII)
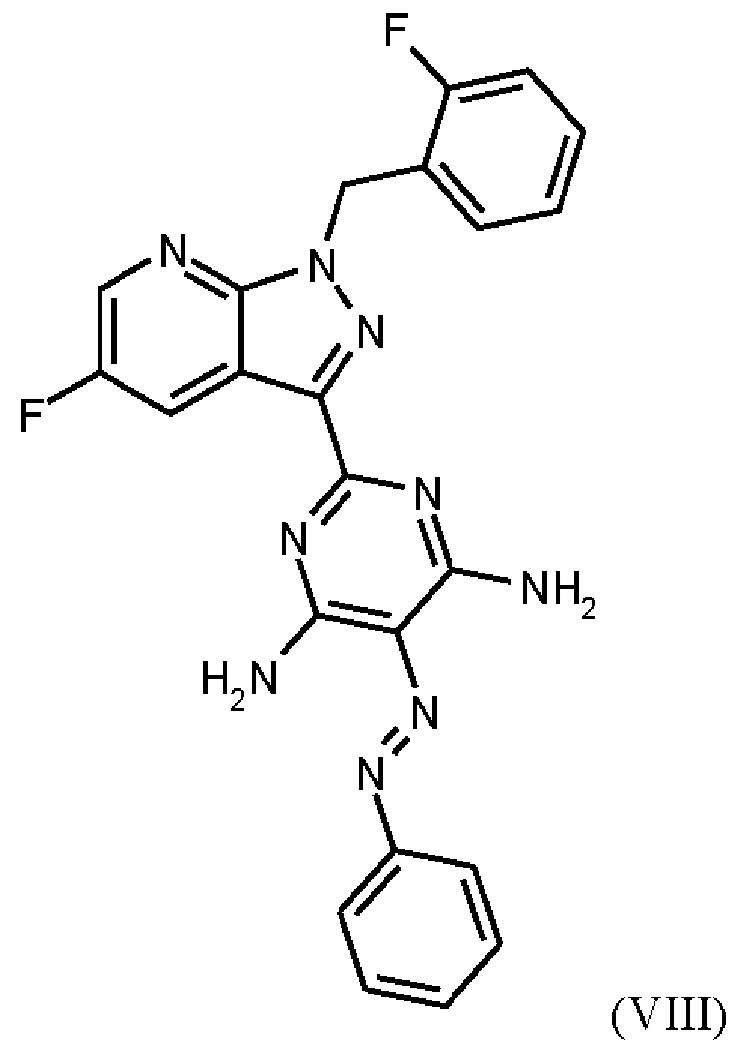
получают путем начального получения соединения формулы (VIIIa)
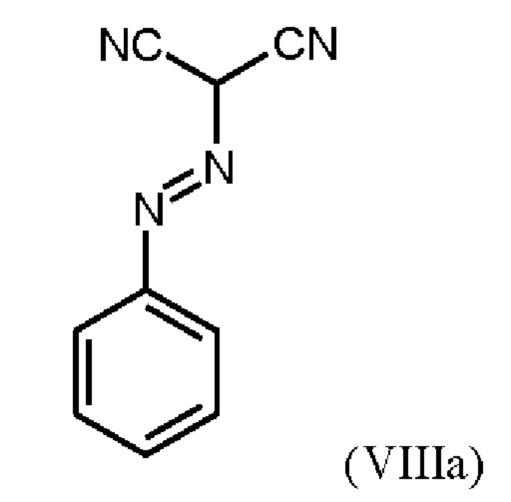
путем добавления концентрированной соляной кислоты в воде к анилину в воде, затем последовательного добавления раствора нитрита натрия в воде, раствора ацетата натрия в воде и раствора малононитрила в этаноле, выделения твердых веществ и промывки водой и изопропанолом с получением соединения формулы (VIIIa);
затем нагревания соединения формулы (VII)
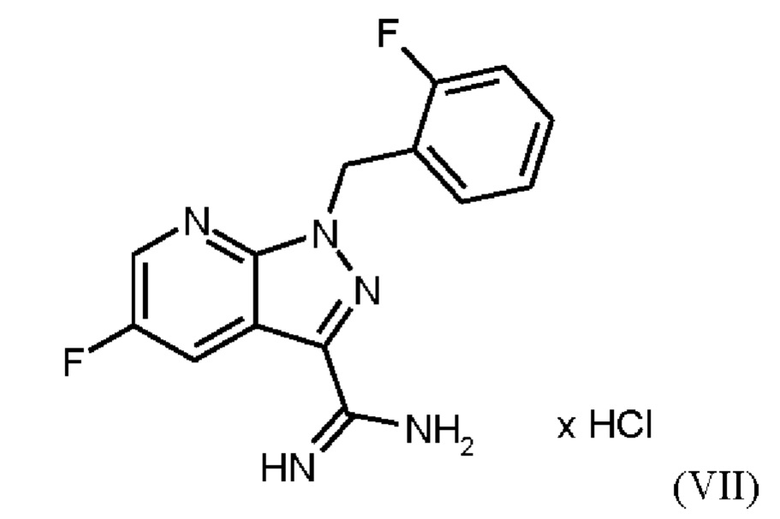
в DMF, добавления соединения формулы (VIIIa), растворенного в DMF, и от 1,2 до 1,7 экв. триэтиламина по отношению к соединению формулы (VII), добавления метанола и выделения соединения формулы (VIII);
затем соединение формулы (IX)
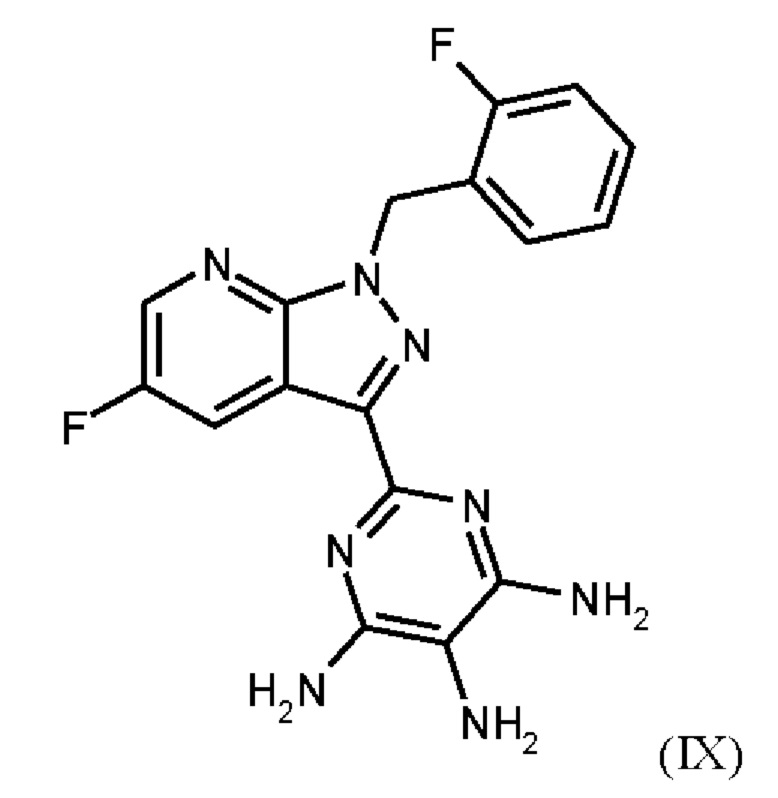
получают путем гидрирования соединения формулы (VIII)
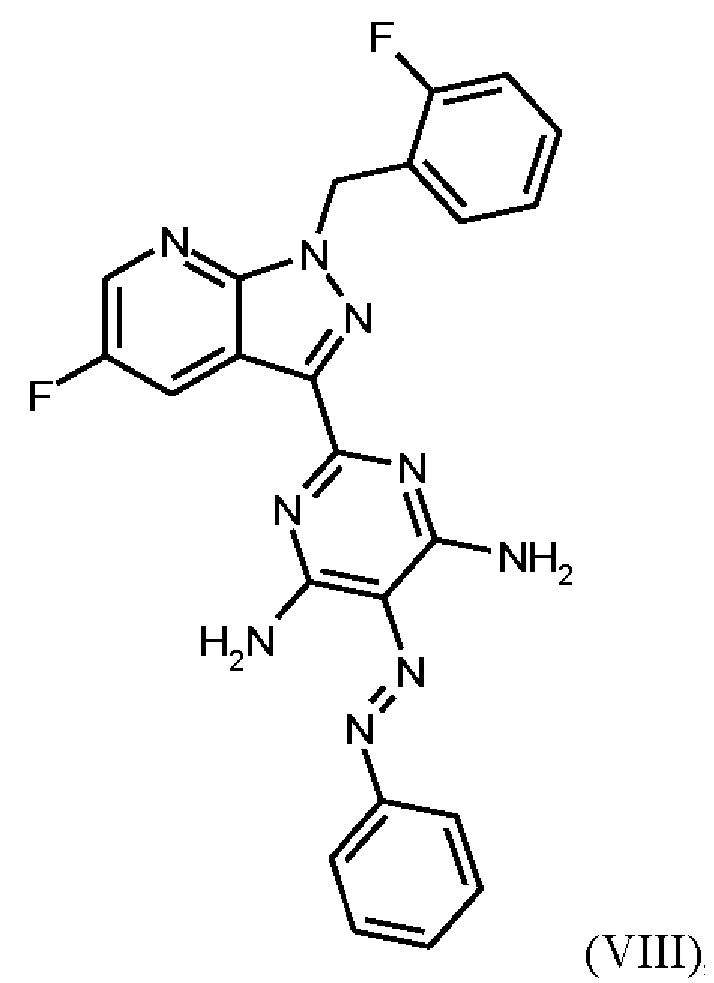
в NMP в качестве растворителя в присутствии водорода при катализе катализатором, выбранным из группы, состоящей из палладия на активированном углероде, платины на углероде, гидроксида палладия и никеля Ренея, кристаллизации путем добавления воды и выделения с получением соединения формулы (IX);
затем гидрохлорид соединения формулы (I)
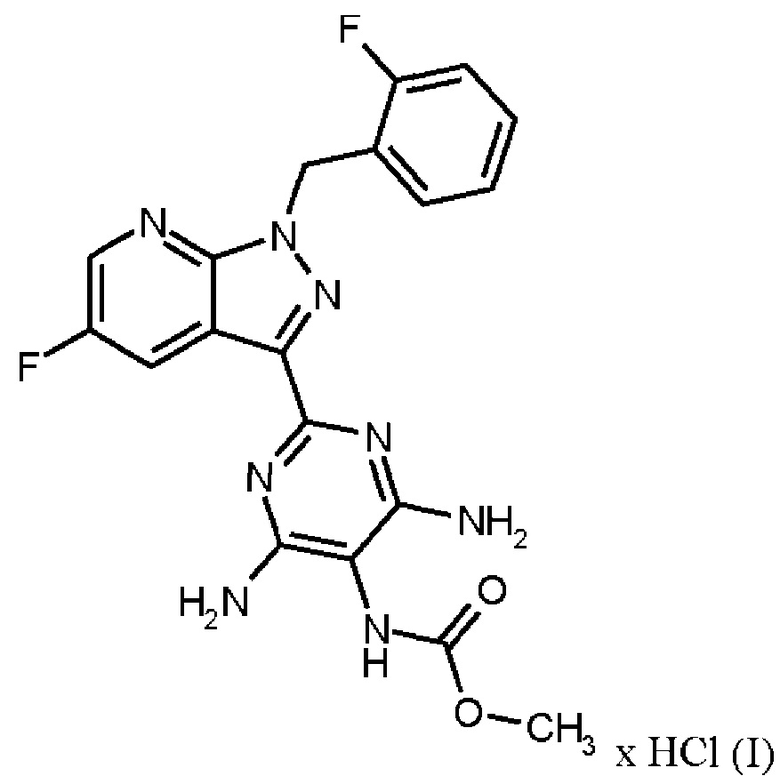
получают путем нагревания соединения формулы (IX)
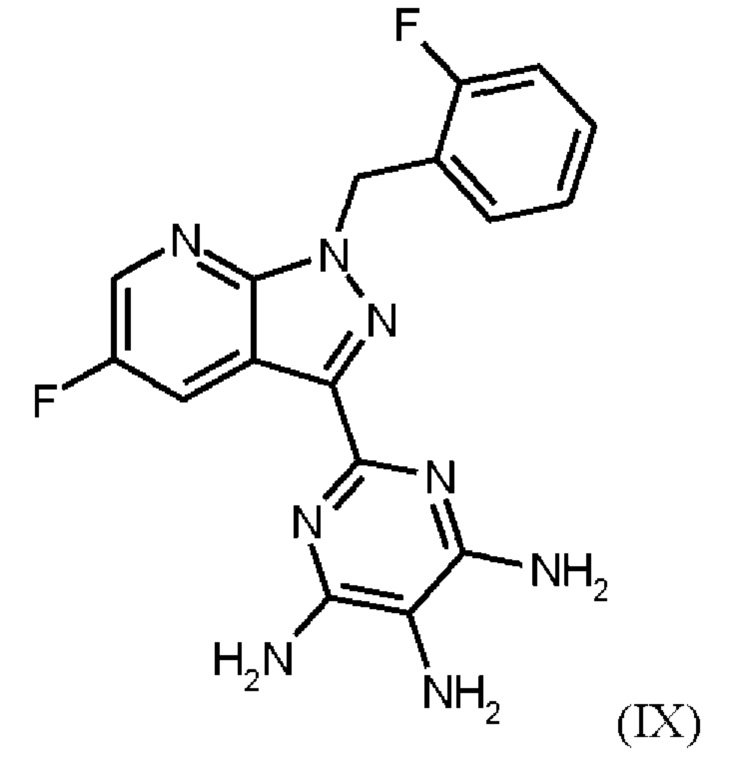
в тетрагидрофуране в качестве растворителя, добавления от 1,0 до 1,2 экв. метилхлорформиата, перемешивания в течение времени реакции от 1 до 10 ч и выделения гидрохлорида соединения формулы (I),
затем ди-DMSO сольват соединения формулы (I)
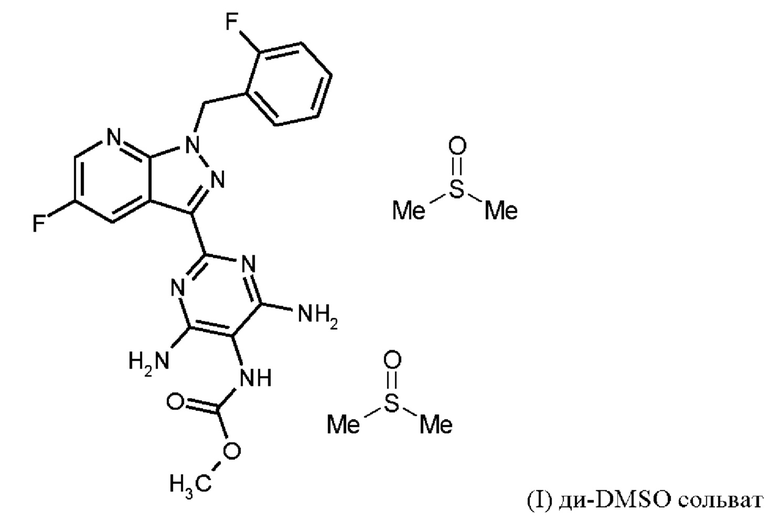
получают путем растворения гидрохлорида соединения формулы (I) в DMSO, добавления три-н-бутиламина и активированного углерода, удаления активированного углерода, охлаждения и добавления этилацетата, выделения ди-DMSO сольвата в кристаллической форме, промывки смесью DMSO и этилацетата,
наконец, получают соединение формулы (I) в кристаллической форме модификации I, где
1.1) ди-DMSO сольват соединения формулы (I) растворяют в DMSO и этаноле при соотношении DMSO и этанола от 2:1 до 6:1 мас./мас.;
1.2) растворенное соединение формулы (I) затем кристаллизуют из раствора путем добавления воды;
1.3) образованную суспензию затем охлаждают до температуры от 5 до 50°С и
1.4) образованные кристаллы на стадии 1.2 затем агломерируют с получением продукта активного соединения путем добавления изопропилацетата, где соотношение массы изопропилацетата и суммы массы соединения формулы (I) плюс массы этанола составляет от 0,3 до 2,0.
9. Способ по п. 8, где соединение формулы (I) в кристаллической форме модификации I получают с чистотой 99,90% (как измерено посредством % площади по ВЭЖХ) или более.
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Автоматические весы для сыпучих тел | 1931 |
|
SU31632A1 |

