Цель изобретения:
Предлагается способ синтеза дротаверина, удовлетворяющего по качеству требованию Госфармакопеи, лишенный вышеуказанных недостатков с использованием доступных видов сырья, стандартного оборудования.
Цель достигается описываемым способом синтеза дротаверина. Ключевыми полупродуктами синтеза являются орто-диэтоксибензол (ОДЭБ) и диэтоксинитрил (ДЭН), в которых присутствуют необходимые «этокси» группы.
Стадия ТП-1: Получение о-диэтоксибензола (ОДЕБ) формулы 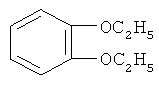 путем взаимодействия пирокатехина с диэтилсульфатом в щелочной среде при температуре 50-60ºС.
путем взаимодействия пирокатехина с диэтилсульфатом в щелочной среде при температуре 50-60ºС.
Стадия ТП-2: Получение диэтоксинитрила (ДЭН) формулы 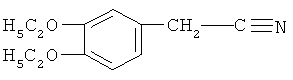 путем взаимодействия ОДЕБ с параформом в присутствии HCl и цианировании полученного диэтоксибензилхлорида (ДЭБХ) при температуре 80-85ºС.
путем взаимодействия ОДЕБ с параформом в присутствии HCl и цианировании полученного диэтоксибензилхлорида (ДЭБХ) при температуре 80-85ºС.
Далее по известной схеме, предложенной ЦХЛС-ВНИИХФИ:
Стадия ТП-3: Получение 3,4-диэтоксифенилуксусной кислоты (ДЭФУ кислоты) формулы 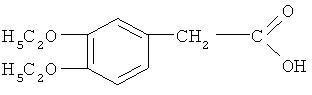 путем взаимодействия диэтоксинитрила со стадии ТП-2 с гидроксидом натрия и выделением диэтоксифенилуксусной кислоты соляной кислотой.
путем взаимодействия диэтоксинитрила со стадии ТП-2 с гидроксидом натрия и выделением диэтоксифенилуксусной кислоты соляной кислотой.
Стадия ТП-4: Получение диэтоксиамина (ДЭАмина) формулы 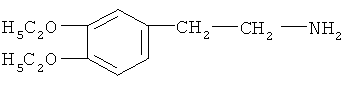 путем каталитического гидрирования диэтоксинитрила со стадии ТП-2, где в качестве катализатора используется никель - Ренея.
путем каталитического гидрирования диэтоксинитрила со стадии ТП-2, где в качестве катализатора используется никель - Ренея.
Стадия ТП-5: Получение этоксиамида (ЭАмида) формулы
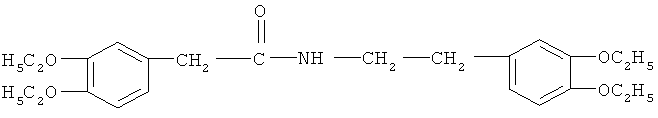 путем конденсации диэтоксифенилуксусной кислоты, полученной на стадии ТП-3 с диэтоксиамином, полученным на стадии ТП-4.
путем конденсации диэтоксифенилуксусной кислоты, полученной на стадии ТП-3 с диэтоксиамином, полученным на стадии ТП-4.
Стадия ТП-6: Получение технического дротаверина гидрохлорида формулы 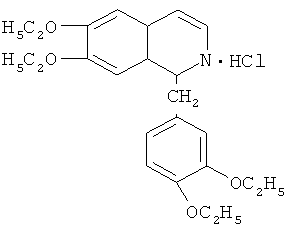 путем циклизации этоксиамида со стадии ТП-5 с помощью POCl3 и дегидрировании полученного вещества в присутствии катализатора никеля - Ренея.
путем циклизации этоксиамида со стадии ТП-5 с помощью POCl3 и дегидрировании полученного вещества в присутствии катализатора никеля - Ренея.
Стадия ТП-7: Получение фармакопейного дротаверина гидрохлорида перекристаллизацией технического дротаверина со стадии ТП-6 в среде дихлорэтана с подкислением хлористым водородом.
В качестве исходного сырья можно использовать пирокатехин и диэтоксисульфат, из которых синтезируется о-диэтоксибензол (или товарный о-диэтоксибензол, который производится и продается в частности Венгрией, Италией, …). В этом случае цикл производства дротаверина гидрохлорида сокращается на 120-130 часов (из 460-480), но увеличивается стоимость готового продукта, т.к. цена готового ОДЭБ значительно выше его себестоимости, в том случае, если его производишь сам.
Пример 1: В реактор загружается 600 л водопроводной воды и 300 кг (294,0 кг 100%) пирокатехина, реакционная масса нагревается до 50-60°C при работающей мешалке. Дается выдержка 2 часа и отбирается проба на полноту растворения пирокатехина. При положительной пробе в реактор загружается 886 кг диэтилсульфата, масса перемешивается в течение 0,5 часа, затем начинается прилив 800-820 л 45% раствора NaOH (pH реакционной массы должно быть 8-10 - розовое пятно на фенол-фталеиновой бумаге). После прилива щелочи дается выдержка 2 часа, еще раз проверяется pH среды. Из реакционной массы отгоняется «острым» паром полученный орто-диэтоксибензол. Процесс отгонкн контролируется через смотровое стекло. После окончания отгонки ОДЭБ отстаивается во флорентине в течение 2 часов, затем нижний слой (ОДЕБ) передается на сушку, которая проводится под вакуумом 0,1 ати при температуре 110-120°C. Через 24 часа сушки отбирается проба на содержание влаги в ОДЭБ. Сушка заканчивается при содержании влаги менее 0,5%. С операции получают 400-410 кг ОДЭБ с содержанием 99-99,5% (396 кг 100%), что составляет 89,2% от теории, считая на пирокатехин.
Пример 2: В реактор загружается 1200 л трихлорэтилена (товарного или регенерированного), 400 кг 99% о-диэтоксибензола (396 кг 100%), 105-120 кг параформа (марка A). Реакционная масса размешивается в течение 0,5 часа и в нее через барботер подается из газогерератора хлористый водород, температура реакционной массы должна быть 35-45°C. Хлористый водород подается в течение 4 часов, затем отбирается проба на процентное содержание в массе диэтоксибензилхлорида, которое должно быть 25-30%. При положительном анализе масса перемещается на делительную воронку, промывается водопроводной водой 2-3 раза, промытый раствор ДЭБХ еще раз проверяется на процентное содержание; нижний слой (раствор ДЭБХ в ТХЭ) передается на стадию цианирования, а верхний «кислый» слой сливается в «кислую» канализацию на станцию нейтрализации. Получают 1450-1500 л 25-30% раствора диэтоксибензилхлорида в трихлорэтилене.
В реактор-цианатор загружают 500 л водопроводной воды и 110-120 кг 89% (98,0 кг 100%; 5-10% избыток по отношению к ДЭБХ) натрия цианистого. Затем в реактор передают 1450-1500 л 25-30% раствора ДЭБХ, массу нагревают до температуры 80-85°C, при этой температуре дают выдержку в течение 4 часов, затем массу передают на делительную воронку для ее промывки водопроводной водой при температуре 45-50°C. Промытый раствор диэтоксинитрила в ТХЭ проверяют на отсутствие циан-ионов и передают на отгонку трихлорэтилена. Отгонку проводят при температуре 80-82°C и остаточном вакууме 0,1 ати. Массу передают на стадию вакуум-разгонки. Получают 480-520 кг технического диэтоксинитрила.
В вакуум-перегонный куб загружают 160-180 кг технического диэтоксинитрила (1/3 часть операции). Нагревают массу до 80-90°C, подают в реактор вакуум (сначала «общий» - 50-100 мм рт.ст, для отгонки остатков ТХЭ, затем «глубокий» - 5-15 мм рт.ст. для отгонки I-ой фракции). Отгонку основного количества ДЭН ведут при температуре 160-180°C и вакууме 5-15 мм рт.ст.. Первую фракцию собирают и накапливают, затем перегоняют, полученный ДЭН используют в производстве. Отогнанный трихлорэтилен и кубовый остаток передают на сжигание. С полной операции получают 190-220 кг 85-90% диэтоксинитрила (170 кг 100%), что составляет 34,8% от теории, считая на загруженный о-диэтоксибензол.
Далее диэтоксинитрил передается на стадию ТП-3 для производства 3,4-диэтоксифенилуксусной кислоты и стадию ТП-4 для производства диэтоксиамина.
Пример 3: В реактор-омылитель загружают 500 л водопроводной воды и 300 кг диэтоксинитрила (255,0 кг 100%). Нагревают реакционную массу до температуры 55-60ºС, дают выдержку в течение 0,5 часа и проверяют массу на полноту растворения. При положительном анализе приливают 70-75л 42-45% раствора NaOH, перемешивают массу в течение 1 часа и проверяют pH среды (должно быть 8-10, розовый цвет на фенолфталеиновой бумаге). Затем приливают 95-100 л 31% соляной кислоты, загружают 20 кг активированного угля, нагревают массу до температуры 95-100ºС. Дают выдержку в течение 0,5-1 часа при работающей мешалке, затем передавливают массу через друк-фильтр, заправленный диагональю и фильтрованной бумагой в реактор-кристаллизатор. Охлаждают массу до 50-55ºС, выделяют 30-31% соляной кислотой (рН 4-5, синее пятно на бумаге «конго») диэтоксифенилуксусную кислоту. Охлаждают массу 15-20ºС, дают выдержку для кристаллизации 2 часа. На центрифуге отжимают ДЭФУ кислоту от маточников, промывают ее водопроводной водой от кислоты. Отжимают. Выгружают и отбирают на анализ - содержание влаги в ДЭФУ кислоте. Получают 350-360 кг пасты диэтоксифенилуксусной кислоты с содержание влаги ~ 75% (260,0 100%), что составляет 93,3% от теории, считая на диэтоксинитрил.
Пример 4: В реактор загружают 300 л изопропилового спирта, 150 кг диэтоксинитрила 127,5 кг 100%). Нагревают реакционную массу до температуры 80-85ºС, дают выдержку для растворения ДЭН в течение 0,5-1 часа т передавливают ее в автоклав. Нагревают массу до температуры 80-85ºС; через барбатер подают в течение 0,5 часа газообразный аммиак. Затем загружают в автоклав 3,5-4,0 кг катализатора никель - Ренея в изопропиловом спирте. После этого подают в течение 3,5-4 часов водород под давлением до 60 ати. На насыщение массы водородом расходуется 5-6 баллонов. После того, как давление водорода стабилизируется, дают выдержку 1 час и передавливают реакционную массу через друк-фильтр, заправленный фильтровальным сукном в реактор, для отгонки спирта. Массу нагревают до температуры 85-90ºС и отгоняют изопропиловый спирт. Полученный технический диэтоксиамин 150-160 кг с содержанием 85-90% передают на стадию вакуум-разгонки.
В вакуум-перегонный куб загружают 150-160 кг техн. ДЭАмина, подают вакуум 15-20 мм рт.ст., нагревают массу до температуры 140-150ºС и перегоняют диэтоксиамин. Получают 125-130 кг 95% (122,0 кг 100%) диэтоксиамина, что составляет 93,8% от теории, считая на загруженный диэтоксинитрил.
Пример 5: В реактор загружают 150 л орто-ксилола, 85-87 кг (80,0 кг 100%) диэтоксиамина и 95-100 кг (74,2 кг 100%) пасты диэтоксифенилуксусной кислоты. Массу нагревают до температуры 85-90ºС. Холодильник работает как «прямой», отгоняет влагу из реакционной массы, процесс контролируют по смотровому стеклу. По окончании отгонки влаги, холодильник переключают на «обратный», массу нагревают до температуры 145-150ºС. при этой температуре дают выдержку 4 часа. Затем массу передавливают в кристаллизатор. Охлаждают ее до температуры 15-20ºС и дают выдержку для кристаллизации в течение 2-3 часов. Затем этоксиамид отфуговывают на центрифуге, промывают изопропиловым спиртом и передают на сушку.
Сушат этоксиамид при температуре 80-90ºС до содержания влаги менее 0,2%. С одной операции получают 124-125 кг этоксиамида с содержанием ~ 99% (123,0 кг 100%), что составляет 78,9% от теории, считая на загруженный диэтоксиамин.
Пример 6: В реактор загружают 250 л трихлорэтилена, 125 кг (123,0 кг 100%) этоксиамида, 49 кг хлорокиси фосфора. Нагревают массу до температуры 80-85ºС, холодильник работает как «прямой». По окончании отгонки влаги холодильник переключают на «обратный». Нагревают реакционную массу до кипения (температура 88-90ºС) и дают выдержку в течение 2 часов. Затем в реактор загружают 300 л орто-ксилола, перемешивают массу и выключают мешалку. Дают выдержку в течение 0,5-1 часа для экстракции образовавшегося вещества в о-ксилол. Затем сливают нижний слой - «кислый» трихлорэтилен (его после нейтрализации регенерируют), реакционную массу передавливают в реактор-дегидратор.
В дегидратор загружают 20-25 кг катализатора никель - Ренея, нагревают реакционную массу до температуры 145-150ºС, во время нагрева холодильник работает как «прямой». По окончании нагрева холодильник переключают на «обратный» и дают выдержку при 145-150ºС в течение 4 часов. По окончании выдержки массу передавливают через друк-фильтр, заправленный сукном, в кристаллизатор. Загружают в реакционную массу 40-45 л спирта, насыщенного хлористым водородом, содержание HCl ~ 30% (рН 3-5, синее пятно на бумаге «конго»). Охлаждают массу до температуры 5-10ºС, дают выдержку для кристаллизации хлоргидрата дротаверина. Отфуговывают его на центрифуге, промывают спиртом и передают на сушку.
Сушат хлоргидрат дротаверина при температуре 55-60ºС до содержания влаги менее 0,5%. С одной операции получают 95-96 кг технического хлоргидрата дротаверина (92,5 кг 100%), что составляет 72,1% от теории, считая на загруженный этоксиамид.
Пример 7: В реактор загружают 300 л этилового спирта и 100 кг технического дротаверина (97,0 кг 100%), нагревают до температуры 55-60ºС, дают выдержку для растворения в течение 0,5-1 часа. проверяют на полноту растворения и при положительном анализе передавливают реакционную массу через друк-фильтр, заправленный диагональю, бязью и фильтровальной бумагой в кристаллизатор. Реактор и друк-фильтр промывают 60 л этилового спирта, промывки присоединяют к маточному раствору, подкисляют реакционную массу спиртом, насыщенным хлористым водородом (pH 3-5, синее пятно на бумаге «конго»). Охлаждают массу до температуры 5-10ºС, дают выдержку для кристаллизации в течение 4 часов. Затем массу переносят на центрифугу, отжимают от маточников, промывают в 2 приема 75-80 л этанола. Проверяют дротаверин на полноту промывки, при положительном результате выгружают из центрифуги и передают на сушку.
Сушат дротаверин в вакуум-сушилке при температуре 55-60ºС при остаточном давлении 0,1-0,2 ати, в течение 2-3 часов. Затем отбирают анализ на содержание влаги (должно быть менее 0,5%). При положительном анализе хлоргидрат дротаверина выгружают из сушилки, взвешивают и отбирают пробу на полный анализ.
С одной операции получают 92-93 кг фармакопейного хлоргидрата дротаверина (мелкокристаллический порошок желто-зеленого цвета), что составляет 95,0% от технического хлоргидрата дротаверина, загруженного на перекристаллизацию.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДРОТАВЕРИНА ГИДРОХЛОРИДА | 2015 |
|
RU2661150C9 |
| СПОСОБ ПОЛУЧЕНИЯ ПАПАВЕРИНА ГИДРОХЛОРИДА | 2016 |
|
RU2647583C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3,4-ДИАЛКОКСИБЕНЗИЛЦИАНИДА | 1998 |
|
RU2149868C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-НИТРОБЕНЗОИЛХЛОРИДА | 2016 |
|
RU2617126C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛОВ АЛКОКСИФЕНИЛУКСУСНЫХ КИСЛОТ (ВАРИАНТЫ) | 1997 |
|
RU2133736C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-(6-МЕТИЛ-2,4-ДИОКСО-1,2,3,4-ТЕТРАГИДРО-5-ПИРИМИДИНСУЛЬФОН)-N'-ИЗОНИКОТИНОИЛГИДРАЗИДА | 2011 |
|
RU2458059C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВАЛИДОЛА | 2016 |
|
RU2626978C1 |
| Способ получения бентазона | 2023 |
|
RU2810483C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-(6-МЕТИЛ-2,4-ДИОКСО-1,2,3,4-ТЕТРАГИДРО-5-ПИРИМИДИНСУЛЬФОН)-N′-ИЗОНИКОТИНОИЛГИДРАЗИДА | 2010 |
|
RU2455301C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРЗАМЕЩЕННЫХ 4,4-ДИАМИНОБЕНЗАНИЛИДОВ | 2007 |
|
RU2385861C2 |
Изобретение относится к области органической химии, а именно к способу получения дротаверина гидрохлорида, заключающемуся в конденсации пирокатехина с диэтилсульфатом в щелочной среде с дальнейшей отгонкой полученного о-диэтоксибензола (ОДЭБ) «острым паром» [либо использовании для синтеза дротаверина гидрохлорида в виде исходного сырья товарного о-диэтоксибензола (ОДЭБ)]; последующем получении диэтоксинитрила (ДЭН) хлорметилированием ОДЭБ хлористым водородом и параформом в среде трихлорэтилена, дальнейшем цианировании полученного диэтоксибензилхлорида (ДЭБХ); последующем получении диэтоксифенилуксусной кислоты (ДЭФУ кислоты) и диэтоксиамина (ДЭАмина) из ДЭН; последующей конденсации ДЭФУ кислоты и ДЭАмина (получение этоксиамида); циклизации этоксиамида с помощью хлорокиси фосфора, дегидрировании полученного полупродукта; и последующей его перекристаллизации - получении фармакопейного дротаверина гидрохлорида. Технический результат: разработан новый способ получения дротавернина гидрохлорида, отличающийся использованием более дешевого простого оборудования, меньшими временными затратами. 3 з.п. ф-лы, 7 пр.
1. Способ получения дротаверина гидрохлорида:
конденсация пирокатехина с диэтилсульфатом в щелочной среде с дальнейшей отгонкой полученного о-диэтоксибензола (ОДЭБ) «острым паром» [либо использование для синтеза дротаверина гидрохлорида в виде исходного сырья товарного о-диэтоксибензола (ОДЭБ)];
последующее получение диэтоксинитрила (ДЭН) хлорметилированием ОДЭБ хлористым водородом и параформом в среде трихлорэтилена, дальнейшее цианирование, полученного диэтоксибензилхлорида (ДЭБХ);
последующее получение диэтоксифенилуксусной кислоты (ДЭФУ к-ты) и диэтоксиамина (ДЭАмина) из ДЭН;
последующая конденсация ДЭФУ кислоты и ДЭАмина (получение этоксиамида); циклизация этоксиамида с помощью хлорокиси фосфора, дегидрирование полученного полупродукта (получение технического дротаверина гидрохлорида);
и последующая его перекристаллизация (очистка) - получение фармакопейного дротаверина гидрохлорида
2. Способ по п.1 отличается от известных тем, что позволяет избежать «жестких» условий проведения операции омыления: температура 185ºС, парами хлористого водорода в среде пиридина.
3. Способ по п.1 позволяет использовать стандартное нержавстальное и эмалированное оборудование.
4. Способ по п.1 позволяет сократить число технологических стадий получения дротаверина гидрохлорида на одну (на две в случае использования в качестве исходного сырья ОДЕБ), тем самым сократить цикл производства на 120-130 часов из 460 часов.
| МАГНИТНАЯ ПОДВЕСКА | 0 |
|
SU234693A1 |
| Turks, Maris et al: "Practical synthesis of Drotaverine impurity standard", Rigas Tehniskas Universitates Zinatniskie Raksti, Serija 1: Materialzinatne un Lietiska Kimija, 23, pp.107-111, 2011 | |||

