Область техники, к которой относится изобретение
Изобретение относится к хелатным соединениям и к их применению в качестве контрастирующих агентов в магнитно-резонансных процедурах. Описание предшествующего уровня техники
Магнитно-резонансная визуализация (MRI) представляет собой метод медицинской визуализации, при котором области тела визуализируют с использованием ядер определенных атомов, в частности ядер атомов водорода. Сигнал MRI зависит от среды, окружающей используемые для визуализации ядра, и времен их продольной и поперечной релаксации, Т1 и Т2. Так, в том случае, когда используемым для визуализации ядром является протон, интенсивность сигнала MRI будет зависеть от таких факторов, как протонная плотность и химическое окружение протонов. При MRI для улучшения контрастности изображений можно использовать контрастирующие агенты. Они работают, воздействуя на время релаксации T1, Т2 и/или Т2*, и ввиду этого влияют на контрастность изображений.
Известно, что для хелатных парамагнитных контрастирующих агентов можно провести оптимизацию времен релаксации T1, Т2 и/или Т2* путем структурной модификации. Особенно важное значение имеет присутствие и время удерживания молекулы воды, связанной с парамагнитным ионом, и время корреляции вращательного движения контрастирующего агента. Присутствие и время удерживания молекулы воды, связанной с парамагнитным ионом, можно изменять путем выбора парамагнитного иона и хелатирующей группировки. Время корреляции вращательного движения можно изменять, варьируя размер контрастирующего агента.
Также известно, что парамагнитный ион может затрагивать биологические пути и оказывать токсическое действие, поэтому необходимо, чтобы парамагнитный ион как можно дольше удерживался в составе хелата. Способность хелата удерживать парамагнитный ион, в дальнейшем именуемая как стабильность, также является свойством, которое можно изменять путем конструирования структуры группировки хелатного соединения. Особый интерес представляет кинетическая стабильность, измеряемая как диссоциационный период полувыведения, который показывает степень инертности по отношению к измененному химическому окружению (т.е. эндогенным ионам).
Известно несколько типов контрастирующих агентов, которые используют при MRI. MR-контрастирующие агенты для пулов крови, например частицы суперпарамагнитного оксида железа, удерживаются в сосудистой системе в течение длительного периода времени. Они оказались чрезвычайно полезными для усиления контрастности, например, в печени, но также и для выявления нарушений проницаемости капилляров, например, «утечки» из стенок капилляров в опухолях, являющейся результатом опухолевого ангиогенеза.
Растворимость парамагнитных хелатов в воде также является важным фактором при их использовании в качестве контрастирующих агентов для MRI, поскольку их вводят пациентам в относительно больших дозах. В случае парамагнитного хелата с высокой растворимостью в воде требуется меньший объем для инъекции, следовательно его легче вводить пациенту, и это вызывает меньший дискомфорт. Водорастворимые парамагнитные хелаты, т.е. комплексы хелатора и иона парамагнитного металла, хорошо известны, например имеющиеся в продаже хелаты на основе гадолиния Omniscan™ (GE Healthcare), Dotarem™ (Guerbet), Gadavist™ (Bayer) и Magnevist™ (Bayer). Благодаря своей низкой молекулярной массы они быстро распределяются по внеклеточному пространству (т.е. по крови и интерстициальной ткани) при введении в сосудистую систему. Они также относительно быстро выводятся из организма.
В US 8540966 содержится информация относительно следующей обобщенной структуры:

где L представляет собой линкер, и R представляет собой Н или С2-70аминополиольную группировку. В экспериментальных примерах в US 8540966 приводится сравнение некоторых имеющихся в продаже хелатов на основе гадолиния с хелатами на основе гадолиния по изобретению с целью демонстрации наличия фармакокинетического профиля, аналогичного профилю у имеющихся в продаже хелатов на основе гадолиния, но с более высокой релаксационной способностью. В US 8540966 не описывается какого-либо влияния аминополиольных группировок на инертность в отношении трансметаллирования.
В ЕР 1931673 содержится информация относительно следующей обобщенной структуры:

Каждый R в приведенной выше структуре определяется в ЕР 1931673 как координирующий лиганд, а каждый X содержит по меньшей мере одну С1-6гидроксиалкильную группу. В ЕР 1931673 подчеркиваются релаксационные свойства соединений, которые там раскрыты. В содержащейся в ЕР 1931673 информации отмечается, что приведенное выше соединение может образовывать комплекс с ионом парамагнитного металла, выбранным из Gd3+, Mn2+ и Fe3+, но в действительности основное внимание уделяется хелатным структурам, которые подходят для образования стабильного комплекса с Gd3+, например таким, как приведенный ниже гадолиний-содержащий комплекс:

Все приведенные в качестве примера комплексы являются гептадентатами, поскольку входящий в комплекс ион металла координируют четыре атома азота и три группы карбоновой кислоты. Вредное воздействие гептадентатных хелатов на основе марганца описано в WO 2011073371. В ЕР 1931673 не описывается какого-либо влияния гидроксиалкильных группировок на инертность в отношении трансметаллирования.
Как можно понять на основании применения имеющихся в продаже агентов и главной идеи документов предшествующего уровня техники, ион гадолиния является самым широко используемым ионом парамагнитного металла для используемых при MRI хелатов.
Ион марганца(II) представляет собой разновидность парамагнитного иона с высоким спиновым числом и длительным временем электронной релаксации, и в литературе были сообщения о потенциале контрастирующего агента с высокой релаксационной способностью на основе марганца(II) (Toth, Е; Advances in Inorganic Chemistry, 2009, 61(09), 63-129). Однако хелаты на основе марганца(II) оказались намного менее стабильными по сравнению с соответствующими хелатами на основе гадолиния. Например, хелат на основе марганца и DOTA (1,4,7,10-тетраазациклододекан-1,4,7,10-тетрауксусная кислота) (MnDOTA) в несколько сотен раз менее стабилен по сравнению с соответствующим гадолиниевым комплексом (GdDOTA) (Drahos, В; Inorganic Chemistry, 2012(12), 1975-1986).
Таким образом, важной проблемой, требующей разрешения, является проблема получения новых хелатов на основе марганца, демонстрирующих высокую стабильность при сохранении эффективных релаксационных свойств.
Некоторые стабильные хелаты на основе марганца описаны в WO 2011073371. Было продемонстрировано, что молекулярное конструирование, описанное в этой заявке, способствует достижению высокой стабильности хелатов и их высокой релаксационной способности. Это делает данные соединения весьма подходящими для применения в качестве контрастирующих агентов при MRI. Типичное соединение из WO 2011073371 имеет следующую структуру:

Тем не менее, все еще существуют возможности для дальнейших улучшений в плане стабильности и релаксационной способности. Краткое описание сущности изобретения
Согласно одному из аспектов настоящее изобретение относится к соединению формулы I или его соли либо сольвату:

где:
X представляет собой О или S;
Y представляет собой О, S или Q-R3, где Q представляет собой N или СН, и R3 выбран из группы, содержащей С1-20гидроксиалкил, С1-6алкил или атом водорода;
Z представляет собой O-L-R4, S-L-R4 или Q-R3-(L-R4), где Q и R3 являются такими, как определено для Y, и L представляет собой необязательный линкер, выбранный из группы, содержащей С1-6алкиленовый, С1-6гидроксиалкиленовый и полиэтиленгликолевый (PEG) линкер, R4 выбран из группы, содержащей С1-6алкил-R5, С3-6арил-R5, гидрокси, -О-С1-3алкил-R5, сульфонил, 5-6-членное гетероциклическое кольцо, углеводную группировку, хелатную группировку, аминокислотную группировку, где R5 представляет собой один или более чем один необязательный заместитель, выбранный из гидрокси, амино, оксо, галогена, С1-3алкила, сульфонамида или -С(=O)-NH-С1-6гидроксиалкила; или Z сам образует часть углеводной группировки или 5-6-членного гетероциклического кольца;
R1 представляет собой С1-3алкил или -(CH2)m-Y1-C(=X1)-Z1, где X1, Y1 и Z1 являются такими, как определено для X, Y и Z, a m представляет собой целое число от 2 до 5;
R2 представляет собой 0-3 заместителя, независимо выбранных из группы, содержащей гидрокси, галоген, амино, амидо, С1-6алкил и С1-6гидроксиалкил; и
каждое n представляет собой целое число от 0 до 15;
при этом соединение формулы I содержит по меньшей мере две группы гидрокси; и
при условии, что если Y представляет собой Q-R3, где Q представляет собой СН, то Z не является Q-R3-(L-R4), где Q представляет собой N.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы I по изобретению вместе с биосовместимым носителем в форме, подходящей для введения млекопитающему.
Согласно другому аспекту настоящее изобретение относится к способу, включающему:
(1) введение субъекту соединения формулы I по изобретению или фармацевтической композиции по изобретению;
(2) обнаружение сигналов магнитного резонанса (MR) от указанного субъекта или частей указанного субъекта, в которых распределено указанное соединение;
(3) формирование MR изображений и/или MR спектров на основании указанных обнаруженных сигналов.
Согласно другому аспекту настоящего изобретения предложено соединение формулы I по изобретению для применения в способе по изобретению.
Хелаты на основе марганца(П) по настоящему изобретению являются кинетически стабильными, демонстрируют оптимальную кинетику обмена молекул воды и могут быть использованы в качестве контрастирующих агентов при MRI.
Несмотря на то, что соединения, описанные в WO 2011073371, были охарактеризованы как очень стабильные (т.е. проявляющие высокий уровень инертности в отношении трансметаллирования), авторам настоящего изобретения неожиданным образом удалось успешно получить хелаты на основе марганца, обладающие еще более высокой стабильностью. Эта удивительная инертность в отношении трансметаллирования достигается за счет встраивания хелата на основе марганца в гидрофильный экранирующий слой. Гидрофильный экранирующий слой конструируют посредством присоединения гидрофильных «плеч» к хелатирующим карбоксильным функциональным группам. Несмотря на то, что полезные эффекты полигидроксилированных хелатов в отношении релаксационных свойств известны из предшествующего уровня техники, неожиданное влияние на кинетику диссоциации, продемонстрированное в настоящем изобретении, не было тем не менее описано ранее.
Подробное описание предпочтительных воплощений
Чтобы более ясно и лаконично описать и выделить предмет заявленного изобретения, ниже приведены определения и типичные воплощения для конкретных терминов, использованных в настоящем описании и формуле изобретения. Любой иллюстративный пример конкретных терминов в данном описании следует рассматривать как неограничивающий пример.
Термины «содержащий» или «содержит» имеют свое традиционное значение во всей этой заявке и подразумевают, что агент или композиция должен (должна) иметь перечисленные существенные признаки или компоненты, но и что помимо них могут присутствовать другие признаки или компоненты. Термин «содержащий» включает в себя в качестве предпочтительного подмножества термин «состоящий по существу из», что означает, что композиция имеет перечисленные компоненты без наличия других признаков или компонентов.
Термин «соль» по изобретению включает в себя физиологически приемлемые соли присоединения кислот, такие как соли, происходящие из неогранических кислот, например, соляной, бромистоводородной, фосфорной, метафосфорной, азотной и серной кислот, и соли, происходящие из органических кислот, например, винной, трифторуксусной, лимонной, яблочной, молочной, фумаровой, бензойной, гликолевой, глюконовой, янтарной, метансульфоновой и пара-толуолсульфоновой кислот.
Подходящий «сольват» по изобретению выбран из этанола, воды, физиологического раствора, физиологического буфера и гликоля.
Термин «алкил», по отдельности или в комбинации, означает алкильный радикал с прямой или разветвленной цепью, имеющий общую формулу CnH2n+1 Примеры таких радикалов включают метил, этил и изопропил.
Термин «гидроксил» относится к группе -ОН.
Термин «гидроксиалкил» относится к алкильной группе, определенной выше, содержащей гидроксильный заместитель, определенный выше.
Термин «арил» относится к функциональной группе или заместителю, происходящей(ему) из ароматического кольца, обычно ароматического углеводорода, примеры которой(ого) включают фенил и пиридил. В одном из воплощений арильные группы по настоящему изобретению происходят из ароматических 6-членных колец с 0-3 гетероатомами, выбранными из О, N и S.
Термин «атом галогена» или «галоген» означает заместитель, выбранный из атомов фтора, хлора, брома или йода.
Термин «углеводная группировка» относится к альдегидному или кетоновому производному многоатомного спирта и включает в себя остатки моносахарида, дисахарида и олигосахарида. Неограничивающие примеры включают остатки фруктозы, глюкозы и сахарозы.
Термин «алкилен» относится к бивалентной группе -(СН2)х-, где х представляет собой целое число от 1 до 6. Термин «гидроксиалкилен» относится к алкилену, содержащему один или более заместителей гидрокси.
ПЭГ линкер
Термин «гидрокси» относится к группе -ОН.
Термин «сульфонил» относится к группе -SO2.
Термин «аминокислотная группировка» относится к группировке из 1-3 аминокислот.
Термин «хелатная группировка» относится к заместителю, который представляет собой металло-хелат, при этом термин «металло-хелат» относится к координационному комплексу, в котором ион металла связан с окружающим массивом молекул или анионов, содержащихся в хелатном соединении.
«Хелатирующее соединение» определяется в настоящем описании как органическое соединение, способное образовывать координационные связи с ионом парамагнитного металла посредством двух или более донорных атомов. В типичном хелатном соединении, подходящем для настоящего изобретения, 2-6, и предпочтительно 2-4, донорных атома для металла расположены таким образом, что в результате получаются 5- или 6-членные кольца (благодаря наличию некоординирующего остова, состоящего либо из атомов углерода, либо некоординирующих гетероатомов, связывающего донорные атомы для металла). Примеры подходящих типов донорных атомов, когда ион металла представляет собой ион парамагнитного металла, включают амины, тиолы, амиды, оксимы и фосфины. В одном из воплощений ионом металла является ион марганца.
Термин «амино» относится к группе -NR'R", где R' и R" независимо представляют собой атом водорода или алкил.
Термин «амидо» относится к группе -C(=O)NR'R", где R' и R" являются такими, как определено для термина «амино».
Термин «оксо» относится к группе =O.
Термин «сульфонамид» относится к группе -SO2-NH2.
В одном из воплощений соединения по настоящему изобретению X представляет собой О, Y представляет собой Q-R3, где Q представляет собой N, и Z представляет собой Q-R3-(L-R4), где Q представляет собой N.
В одном из воплощений соединения по настоящему изобретению X представляет собой S, Y представляет собой Q-R3, где Q представляет собой N, и Z представляет собой Q-R3-(L-R4), где Q представляет собой N.
В одном из воплощений соединения по настоящему изобретению X представляет собой О, либо Y представляет собой О, либо Z представляет собой O-L-R4, и если Y не представляет собой О, то он представляет собой Q-R3, где Q представляет собой N, и если Z не представляет собой O-L-R4, то он представляет собой Q-R3-(L-R4), где Q представляет собой N.
В одном из воплощений соединения по настоящему изобретению X представляет собой S, либо Y представляет собой О, либо Z представляет собой O-L-R4, и если Y не представляет собой О, то он представляет собой Q-R3, где Q представляет собой N, и если Z не представляет собой О, то он представляет собой Q-R3-(L-R4), где Q представляет собой N.
В одном из воплощений соединения по настоящему изобретению X представляет собой О, Y представляет собой Q-R3, где Q представляет собой N, и Z представляет собой Q-R3-(L-R4), где Q представляет собой СН.
В одном из воплощений соединения по настоящему изобретению X представляет собой О, либо Y представляет собой О, либо Z представляет собой O-L-R4, и если Y не представляет собой О, то он представляет собой Q-R3, где Q представляет собой СН, и если Z не представляет собой О, то он представляет собой Q-R3-(L-R4), где Q представляет собой СН.
В одном из воплощений соединения по настоящему изобретению каждая группа -L-R4 представляет собой С1-12гидроксиалкил.
В одном из воплощений соединения по настоящему изобретению каждая группа -L-R4 представляет собой С3-6гидроксиалкил.
В одном из воплощений соединения по настоящему изобретению каждая группа -L-R4 представляет собой С6гидроксиалкил.
В одном из воплощений соединения по настоящему изобретению каждая группа -L-R4 независимо выбрана из группы, содержащей:


где в каждом случае звездочка означает место присоединения к остальной части соединения формулы I.
В одном из воплощений соединения по настоящему изобретению каждый R4 независимо выбран из группы, содержащей:

где в каждом случае звездочка означает место присоединения к остальной части соединения формулы I.
В одном из воплощений соединения по настоящему изобретению каждый R4 представляет собой С3-6арил-R5, где R5 выбран из галогена и группы -C(=O)-NH-C1-6гидроксиалкила.
В одном из воплощений соединения по настоящему изобретению указанный галоген представляет собой атом йода.
В одном из воплощений соединения по настоящему изобретению указанный С3-6арил представляет собой фенил.
В одном из воплощений соединения по настоящему изобретению каждый R4 представляет собой трийодированный фенил.
В одном из воплощений соединения по настоящему изобретению каждый R4 представляет собой:
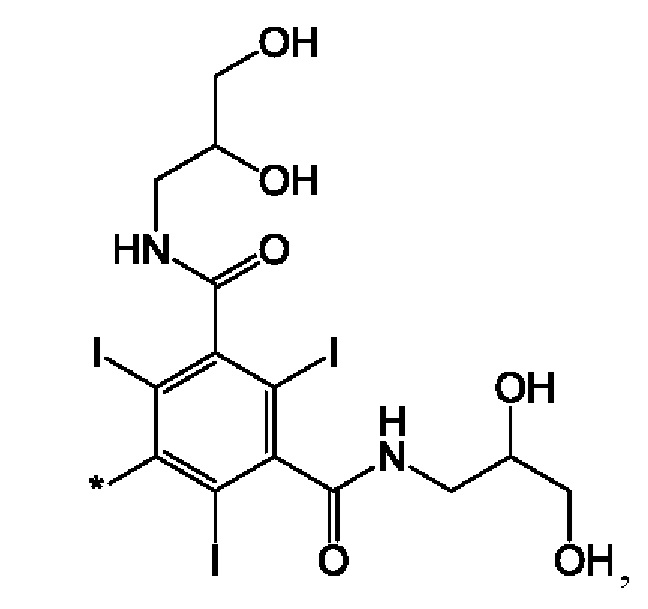
где звездочка означает место присоединения к остальной части соединения формулы I. В одном из воплощений соединения по настоящему изобретению каждый R4 представляет собой одну и ту же группу.
В одном из воплощений соединения по настоящему изобретению каждый R3 независимо выбран из группы, содержащей С1-3алкил или атом водорода.
В одном из воплощений соединения по настоящему изобретению каждый R3 представляет собой С1-3алкил.
В одном из воплощений соединения по настоящему изобретению каждый R3 представляет собой метил.
В одном из воплощений соединения по настоящему изобретению каждый R3 представляет собой атом водорода.
В одном из воплощений соединения по настоящему изобретению каждый R3 представляет собой одну и ту же группу.
В одном из воплощений соединения по настоящему изобретению каждое n представляет собой целое число от 1 до 6.
В одном из воплощений соединения по настоящему изобретению каждое n равно 2.
В одном из воплощений соединения по настоящему изобретению m равно 3.
В одном из воплощений соединения по настоящему изобретению R1 представляет собой С1-3алкил.
В одном из воплощений соединения по настоящему изобретению R1 представляет собой метальную группу.
В одном из воплощений соединения по настоящему изобретению R1 представляет собой -(CH2)m-Y-C(=X)-Z, где X, Y, Z и m являются такими, как определено в настоящем описании.
В одном из воплощений соединения по настоящему изобретению R2 представляет собой 0 заместителей.
В одном из воплощений соединения по настоящему изобретению R2 представляет собой 2 группы гидрокси.
В одном из воплощений соединения по настоящему изобретению указанные гидроксильные группы находятся в пиридильном кольце в мета-положениях.
В одном из воплощений соединение по настоящему изобретению содержит по меньшей мере 4 группы гидрокси.
В одном из воплощений соединение по настоящему изобретению содержит 4-15 групп гидрокси.
В одном из воплощений соединение по настоящему изобретению содержит 5-10 групп гидрокси.
В одном из воплощений соединения по настоящему изобретению указанный Mn представляет собой обогащенный изотоп Mn, выбранный из группы, содержащей 52Mn и 54Mn.
Неограничивающие примеры конкретных соединений формулы I по изобретению включают следующие соединения:




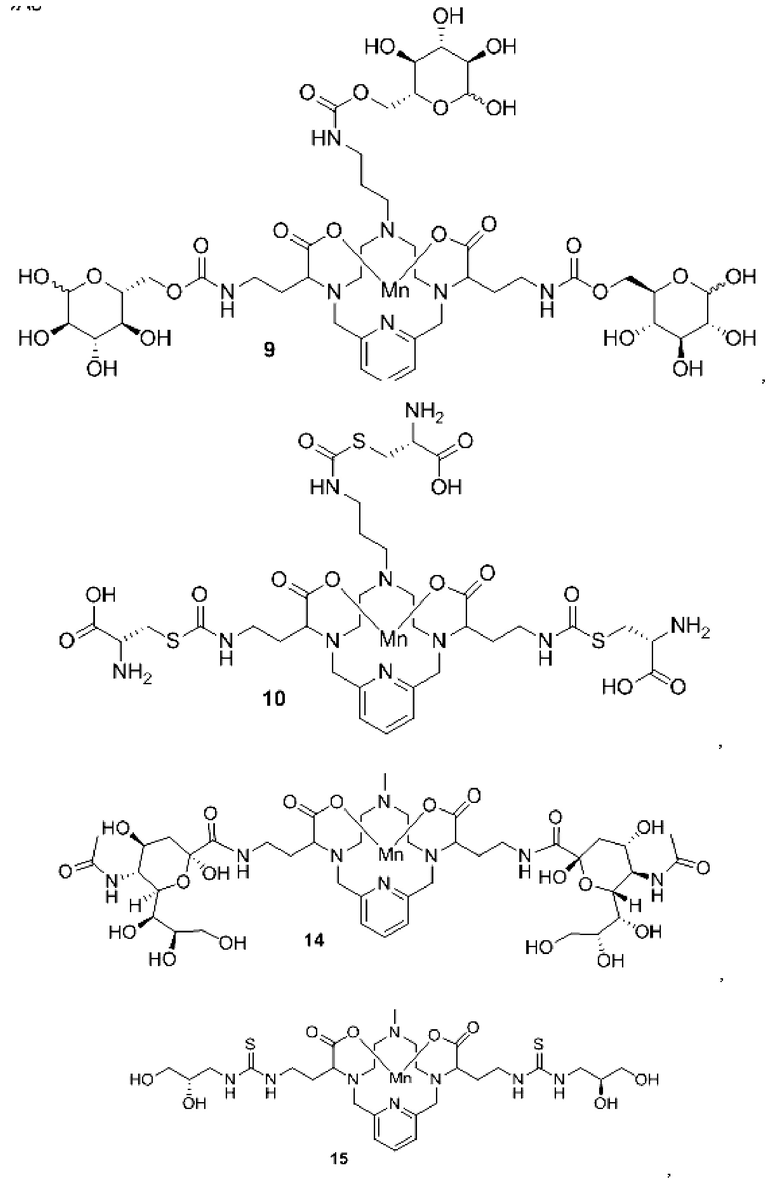
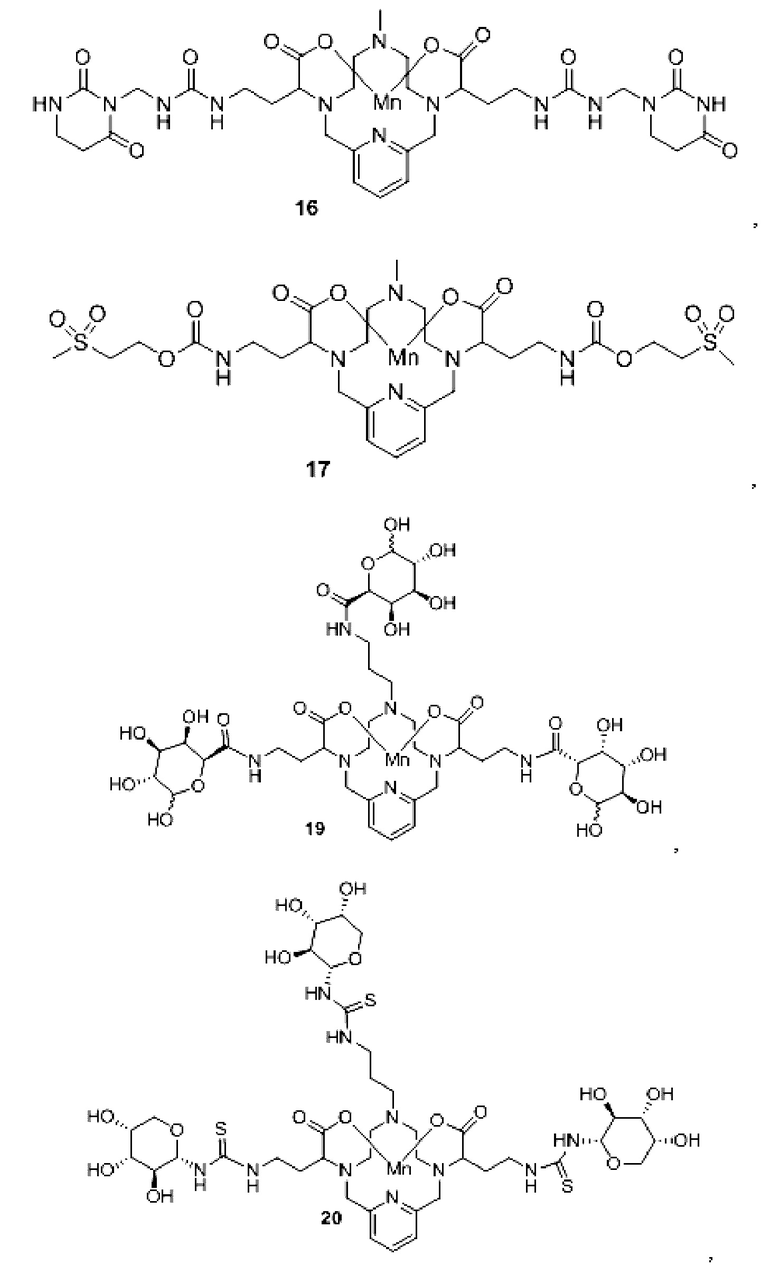





В соединениях формулы I атомы углерода, присоединенные к карбоксилатным «плечам», представляют собой стереоцентры. Соединения формулы I по изобретению могут быть представлены в виде рацемической смеси или в виде энантиомерно обогащенной смеси, либо рацемическую смесь можно разделить с использованием хорошо известных методов, а индивидуальный энантиомер можно использовать отдельно. В одном из воплощений соединение формулы I либо представляет собой рацемическую смесь, либо является диастереомерно чистым.
Получение гидрофильных производных соединений по изобретению достигается с использованием некоординирующей присоединяющей группы, выбранной из амида, мочевины, тиомочевины, карбамата, тиокарбамата или дитиокарбамата. Некоординирующая присоединяющая группа располагается слишком далеко от иона марганца, чтобы участвовать в значительной степени в образовании координационных связей с ионом марганца, что тем самым обеспечивает непосредственно образование координационных связей между ионом марганца и молекулами воды и, как следствие, наблюдаемую контрастность. Длина некоординирующего линкера в формуле I играет очень важную роль, если он слишком короткий (например, если в формуле I m=1), то существует риск, что он будет координировать ион марганца, тем самым блокируя доступ молекулы воды, что резко снизит общую релаксационную способность комплекса. Длина некоординирующего линкера, присоединенного к карбоксиметильному «плечу» (координирующей группе), может быть небольшой (т.е. когда в формуле I n=0), поскольку одно и то же «плечо» не может содействовать двум координирующим группам (координационный угол будет слишком деформированным).
Соединения формулы I могут быть синтезированы с использованием нескольких путей синтеза, известных специалисту в данной области техники, из имеющихся в продаже исходных веществ. Подходящие источники марганца для инкорпорирования в хелаты при получении соединений по настоящему изобретению включают соли, представляющие собой карбонат (MnCO3), оксид (MnO), ацетат (Mn(ОАс)2), хлорид (MnCl2), гидроксид (Mn(ОН)2), оксалат (MnC2O4), формиат (Mn (HCO2)2) и нитрат (Mn(NO3)2.
Для получения соединений формулы I можно использовать и/или легко адаптировать следующую обобщенную методику:
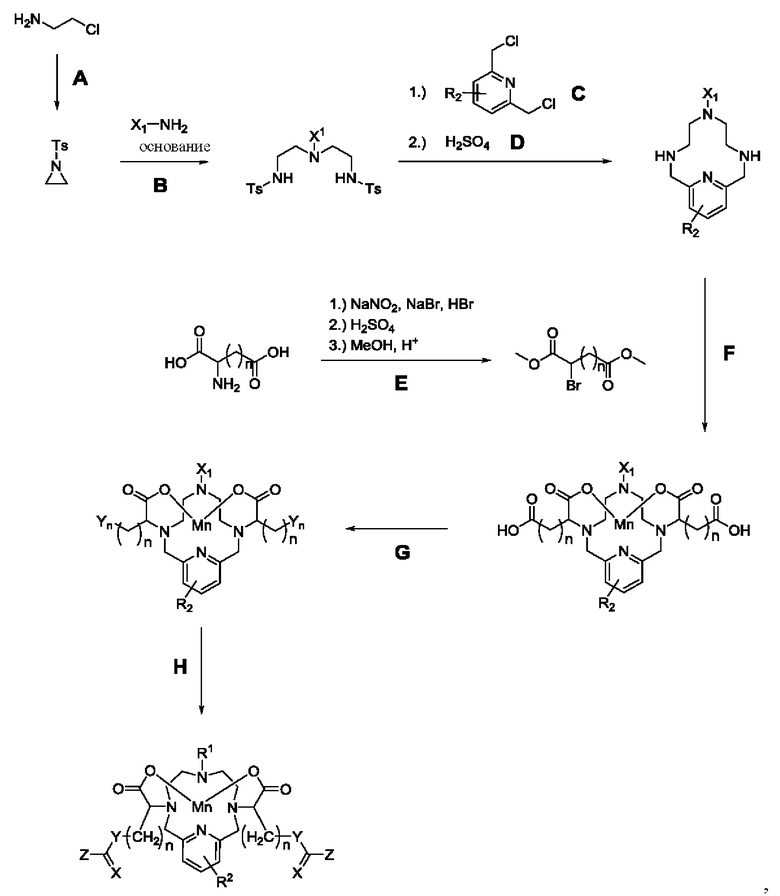 где R2, X, Y и Z являются такими, как определено где-либо еще в настоящем описании. X1 представляет собой СН3 или СН2СН2СН2СООН. Yn определен по-разному на приведенной ниже стадии Н.
где R2, X, Y и Z являются такими, как определено где-либо еще в настоящем описании. X1 представляет собой СН3 или СН2СН2СН2СООН. Yn определен по-разному на приведенной ниже стадии Н.
Кратко:
А: в результате тозилирования аминоэтанола получают азиридин (Carrillo, Arkivoc, 2007);
В: азиридинирование аминобутановой кислоты (номер 56-12-2 по каталогу Sigma-Aldrich). В одном из воплощений азиридинирование метиламина происходит в неразбавленном ацетонитриле. В одном из воплощений для этой аминокислоты с целью активации аминогруппы используют какое-нибудь основание. Возможно, что кислотная функциональная группа может быть защищена в виде сложного эфира;
С: циклизация с 2,6-бис(хлорметил)-пиридином (номер 3099-28-3 по каталогу Sigma-Aldrich). В одном из воплощений эту стадию проводят в ацетонитриле с карбонатом калия в качестве основания;
D: детозилирование с использованием в одном из воплощений концентрированной серной кислоты. В одном из воплощений эту стадию проводят количественно;
Е: бромирование, основанное на описанном в литературе методе (Henig, J., Toth, Е., Engelmann, J., Gottschalk, S., & Mayer, H. a. (2010), Inorganic Chemistry, 49(13), 6124-38);
F: алкилирование полиамина/введение металла, гидролиз сложного эфира. В одном из воплощений эту стадию проводят в водном растворе. В тех воплощениях, где реакция с вторичными галогенидами протекает медленно, возможен синтез сложного бис-эфира (Е) и перенесение в органический растворитель для увеличения скорости алкилирования. После проведения алкилирования сложные эфиры можно подвергнуть гидролизу в присутствии основания и затем, после перехода к нейтральному значению рН, можно ввести ион Mn(II), используя MnCl2, а избыток Mn можно осадить с применением основания;
G: активация карбоксилата с образованием Yn (где n представляет собой различные группы Y, Y1-5, которые определены в данном описании для соединения формулы I);
Н: реакции сочетания различных активированных соединений с нуклеофилами и электрофилами:
карбоксилаты можно активировать до изоцианата с использованием дифенилфосфорилазида (Oilman, J. W. and Otonari, Y. A. (1993) Synthetic Communications, 23(3), 335-341), получая активированное промежуточное соединение Y1, которое далее может быть приведено во взаимодействие на стадии Н с аминами ([а] Ranjan, A. et al. (2014), Organic Letters, 16(21), 5788-5791. [b] Petersen, T. P. et al. (2013), 19(28), 9343-9350), спиртами ([a] Amamoto, Y. et al. (2007), Journal of the American Chemical Society, 129(43), 13298, [b] Kim, I. et al. (2004), Journal of Medicinal Chemistry, 47(8), 2110-2122) или тиолами ([a] Peters, K. (2001), Journal of Enzyme Inhibition, 16(4), 339-350, [b] Safavi-Sohi, R. (2016), ACS Applied Materials & Interfaces, 8(35), 22808-22818) с получением мочевин, карбаматов и S-тиокарбаматов, соответственно;
помимо этого изоцианат Y1 можно подвергнуть расщеплению путем обработки основанием, получая амин Y2 (Chavboun, I. et al. (2015), Journal of Natural Products, 78(5), 1026-1036);
аминеодержащие производные Y2 можно привести во взаимодействие с различными кар бо новыми кислотами, изоцианатами, изотиоцианатами и хлорформиатами, получая амиды, мочевины, тиомочевины и карбаматы, соответственно;
альтернативно, амин Y2 можно преобразовать в активированный тиоизоцианат Y3 путем обработки в различных условиях, включая, но не ограничиваясь этим, обработку тионилдиимидазолом в DMF (диметилформамиде). После этого можно провести реакцию сочетания активированных электрофилов тиоизоцианатов с аминами, спиртами или тиолами, получая тиомочевины, О-тиокарбаматы и дитиокарбаматы, соответственно;
помимо этого, имеют место некоторые случаи, когда Yn может быть активирован в виде спирта (см. синтез диола 36) с получением спиртов (Y4), которые могут взаимодействовать с изоцианатами, изотиоцианатами с получением карбаматов и О-тиокарбаматов, соответственно.
В дополнение к этому имеют место случаи, когда Yn может быть активирован в виде нуклеофильного тиола (Y5), что показано для синтеза дитиола 40. Последующее взаимодействие тиолов с изоцианатами или изотиоцианатами будет приводить к образованию S-тиокарбаматов и дитиокарбаматов, соответственно.
На стадии Н для добавления гетероцикла  можно использовать соединение следующей формулы:
можно использовать соединение следующей формулы:

Полученные соединения формулы I имеют структуру формулы Ia:

Если соединение формулы I содержит в качестве R4 замещенный арил, такой как трехйодистый фенил, то для получения таких соединений можно использовать или адаптировать следующую реакционную схему (где X, Y, Z, R2 и n являются такими, как определено в настоящем описании, а Y2 и Z2 представляют собой функциональные группы, подходящие для обеспечения возможности осуществления реакции конъюгирования:
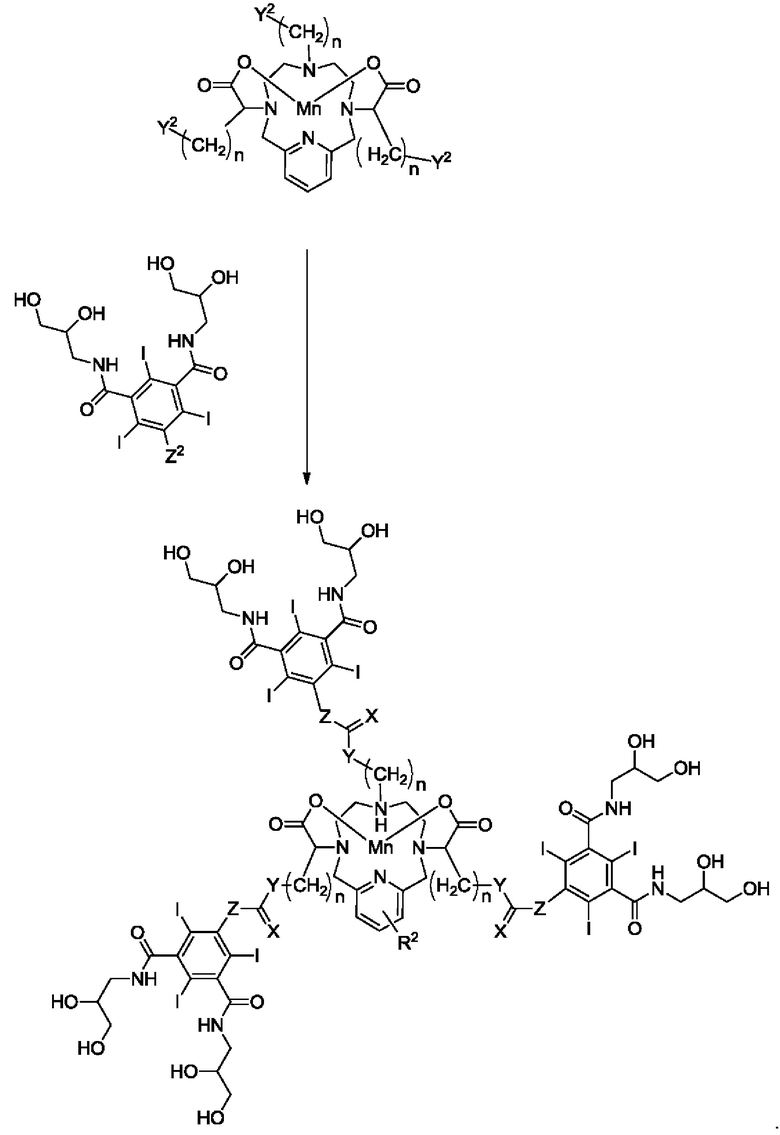
В приведенных ниже экспериментальных примерах синтез конкретных соединений проводят либо в соответствии с реакционными схемами, например, описанными выше, либо в соответствии с альтернативными реакционными схемами. Эти альтернативные реакционные схемы также могут быть легко адаптированы специалистом в данной области техники для получения других соединений формулы I.
Обратная амидная химическая структура хелатных соединений на основе Mn по настоящему изобретению отличает их от похожих хелатных соединений на основе Mn из предшествующего уровня техники. Полагают, что соединения по настоящему изобретению обладают повышенной стабильностью с точки зрения удержания Mn, а также в отношении метаболизма in vivo.
Методы, подходящие для определения стабильности хелатов in vitro, можно найти в литературных источниках (Idee, J.-M. Journal of Magnetic Resonance Imaging: JMRI, 2009, 30(6), 1249-58 и Baranyai, Z. Chemistry - A European Journal, 2015, 21(12), 4789-4799). Другие подходящие методы включают исследования in vitro физиологических сред (т.е. сыворотки или плазмы крови человека) для мониторинга инертности в отношении трансметаллирования. Другой подходящий метод оценки инертности в отношении трансметаллирования может заключаться в измерении удержания ионов металлов in vivo после введения хелатированного металла. Известно, что для интактных хелатов обычно характерна кинетика очень быстрого выведения.
Согласно одному из аспектов изобретения соединение формулы I предложено в составе фармацевтической композиции. «Фармацевтическая композиция» представляет собой композицию, содержащую соединение по изобретению вместе с биосовместимым носителем, в форме, подходящей для введения млекопитающему. «Биосовместимый носитель» представляет собой жидкость, в частности жидкость, в которой соединение формулы I суспендируется или растворяется, в результате чего полученная композиция является физиологически переносимой, т.е. может быть введена в организм млекопитающего без проявления токсичности или без вызывания чрезмерного дискомфорта (что можно понимать как определение термина «подходящая для введения млекопитающему»).
Фармацевтическая композиция по изобретению подходит для применения в качестве магнитно-резонансной (MR) контрастирующей среды при магнитно-резонансной визуализации (MRI) организма человека и не являющегося человеком животного.
В одном из воплощений фармацевтическая композиция по изобретению может содержать один или более чем один фармацевтически приемлемый эксципиент. Они, соответственно не влияют на приготовление, хранение или применение конечной композиции.
Неограничивающие примеры подходящих фармацевтически приемлемых эксципиентов включают буферные агенты, стабилизаторы, антиоксиданты, регулирующие осмоляльность агенты, регулирующие рН агенты, избыточное хелатирующее соединение и слабые комплексы физиологически переносимых ионов. Эти и другие подходящие эксципиенты будут хорошо известны специалистам в данной области техники и дополнительно описаны, например, в WO 1990003804, ЕР 0463644-А, ЕР 0258616-А и US 5876695, содержание которых включено в данное описание посредством ссылки. Фармацевтическая композиция по изобретению в одном из воплощений представлена в форме, подходящей для парентерального введения, например, в форме инъекции. Ввиду этого фармацевтическая композиция по изобретению может быть приготовлена для введения с использованием физиологически приемлемых эксципиентов способом, в полном объеме известным специалисту в данной области техники. Например, соединение формулы I, возможно с добавлением фармацевтически приемлемых эксципиентов, можно суспендировать или растворять в водной среде, а полученный раствор или суспензию затем подвергать стерилизации.
Неограничивающим примером подходящего буферного агента является гидрохлорид трометамина.
Термин «избыточное хелатирующее соединение» определяется как любое соединение, способное захватывать свободный парамагнитный ион (марганца), но не парамагнитный ион (марганца), удерживаемый в комплексах по данному изобретению, как описано в ЕР 2988756 А1. Поскольку для поддержания здоровья человека необходимы небольшие количества, передозировка свободными ионами марганца может приводить к нейродегенеративному расстройству, известному как «отравление марганцем» с симптомами, напоминающими болезнь Паркинсона. Однако, фундаментальная проблема применения Мn, а также других металлов в качестве контрастирующих агентов, заключается в стабильности их хелатов. Стабильность хелатов является важным свойством, которое отражает возможное высвобождение свободных ионов металла in vivo. Известно, что в животных моделях существует корреляция между количеством избыточного хелатирующего соединения в парамагнитной хелатной композиции и количеством осажденного парамагнитного металла (Sieber, 2008, J. Mag. Res. Imaging, 27(5): 955-62). Ввиду этого, в другом воплощении выбирают такое количество избыточного хелатирующего соединения, что оно могло действовать как поглотитель Mn, чтобы уменьшать или предотвращать высвобождение Mn из композиции после инъекции. Оптимальное количество свободного хелатирующего соединения будет приводить к получению фармацевтической композиции, имеющей подходящие физико-химические свойства (т.е. вязкость, растворимость и осмоляльность) и позволяющей избежать токсических эффектов, таких как истощение по цинку в случае слишком большого содержания свободного хелатирующего соединения. В US 5876695 описывается, в частности, избыток линейного хелата, в частности, свободной DTPA (диэтилентриаминпентауксусная кислота), и это является неограничивающим примером избыточного хелатирующего соединения, подходящего для применения в фармацевтической композиции по настоящему изобретению. Данная стратегия в отношении композиций используется для таких продуктов, как Magnevist™, Vasovist™ или Primovist. В WO 2009103744 описывается аналогичная стратегия в отношении композиций, основанная на добавлении точного количества свободного хелата с тем, чтобы иметь очень небольшой избыток указанного хелата и нулевую концентрацию свободного лантанида.
Физиологически переносимый ион в одном из воплощений может быть выбран из физиологически переносимых ионов, которые включают ионы солей кальция или натрия, такие как хлорид кальция, аскорбат кальция, глюконат кальция или лактат кальция.
Парентерально вводимые формы должны быть стерильными, не содержать физиологически неприемлемых агентов и должны иметь низкую осмоляльность, чтобы свести к минимуму раздражение или другие неблагоприятные эффекты после введения, и поэтому фармацевтическая композиция должна быть изотонической или слегка гипертонической. Неограничивающие примеры подходящих носителей включают водные носители, обычно используемые для введения парентеральных растворов, такие как раствор хлорида натрия для инъекций, раствор Рингера для инъекций, раствор декстрозы для инъекций, раствор декстрозы и хлорида натрия для инъекций, лактатный раствор Рингера для инъекций и другие растворы, например, описанные в Remington's Pharmaceutical Sciences, 22ое издание (2006, Lippincott Williams & Wilkins) и Национальном формуляре: (https://books.google.com/books?id=O3qixPEMwssC&q=THE+NATIONAL+FORMULARY&dq=THE+NATIONAL+FORMULARY&hl=en&sa=X&ved=0CC8Q6AEwAGoVChMImfPHrdTqyAIVJfNyCh1RJw_E).
Что касается фармацевтической композиции по изобретению, подлежащей введению парентерально, т.е. путем инъекции, то при ее приготовлении дополнительно используют стадии, включающие удаление органического растворителя, добавление биосовместимого буфера и любых возможных дополнительных ингредиентов, таких как эксципиенты или буферы. Для парентерального введения также необходимо предпринимать шаги для обеспечения стерильности и апирогенности фармацевтической композиции.
В другом воплощении настоящего изобретения предложен способ, включающий введение соединения формулы I, определенного в данном описании, для формирования MR изображений и/или MR спектров.
Способы введения и субъекты, рассматриваемые как подходящие в контексте настоящего изобретения, описаны в данной заявке выше применительно к фармацевтической композиции. Введение соединения формулы I предпочтительно выполняют парентерально и наиболее предпочтительно внутривенно. Внутривенный путь представляет собой наиболее эффективный путь доставки соединения по всему организму субъекта. Кроме того, внутривенное введение не представляет существенного физического вмешательства или существенного риска для здоровья. Соединение формулы I по изобретению предпочтительно вводят в составе фармацевтической композиции по изобретению, как определено выше. Способ по изобретению также можно понимать как способ, включающий стадии (2)-(3), выполняемые для субъекта, которому соединение по изобретению было введено предварительно. В одном из воплощений фармацевтическую композицию вводят в количестве, подходящем для усиления контрастности в методе MR визуализации (MRI). Для более подробной информации о методах MRI читатель отсылается к обычным общеизвестным сведениям в данной области техники, которые, например, приведены в главе 27 «Контрастирующие агенты и магнитно-резонансная визуализация» ("Contrast Agents and Magnetic Resonance Imaging") в книге «Магнитно-резонансная визуализация: физические и биологические принципы» ("Magnetic Resonance Imaging: Physical and Biological Principles") (4oe издание, 2015, Elsevier, изд. Stewart Carlyle Bushong & Geoffrey Clarke) или в книге «Контрастирующие агенты I: магнитно-резонансная визуализация» ("Contrast Agent I: Magnetic Resonance Imaging") (2002, Springer-Verlang, изд. Werner Krause).
Способ по изобретению можно использовать для изучения наличия биологического маркера или процесса у здоровых субъектов или, альтернативно, у субъектов, которые, как известно, имеют патологическое состояние, ассоциированное с аномальной экспрессией биологического маркера, или у которых подозревают наличие такого состояния. Если данный способ используют для получения изображений от субъекта, который, как известно, имеет патологическое состояние или у которого подозревают наличие такого состояния, то он находит применение в способе диагностики указанного состояния.
Стадия «обнаружения» в способе по изобретению включает обнаружение сигналов, испускаемых соединением формулы I, с использованием детектора, чувствительного к указанным сигналам. Эту стадию обнаружения также можно понимать как получение данных о сигналах.
Стадию «формирования» в способе по изобретению осуществляют посредством компьютера, применяя к собранным данным о сигналах алгоритм реконструкции с получением набора данных. Затем работают с этим набором данных, формируя одно или более изображений и/или один или более спектров, демонстрирующих местоположение источника сигналов и/или их количество.
«Субъектом» в контексте изобретения может быть любой человек или животное. В одном из воплощений субъектом по изобретению является млекопитающее. В одном из воплощений указанным субъектом является интактный организм млекопитающего in vivo. В другом воплощении субъектом по изобретению является человек.
В данном письменном описании соединения из примеров используют для раскрытия изобретения, включая наилучший вариант осуществления изобретения, а также для предоставления любому специалисту в данной области техники возможности использовать данное изобретение на практике, включая создание и использование любых устройств или систем и осуществление любых предусмотренных способов. Патентоспособный объем изобретения определяется формулой изобретения и может включать другие типичные соединения, которые встретятся специалистам в данной области техники. Подразумевается, что такие другие типичные соединения включены в объем формулы изобретения, если они содержат структурные элементы, которые не отличаются от буквальной терминологии формулы изобретения, или если они включают эквивалентные структурные элементы с несущественными различиями от буквальной терминологии формулы изобретения. Все патенты и патентные заявки, упомянутые в данном тексте, тем самым включены посредством ссылки во всей своей полноте, как если бы они были включены по отдельности. Краткое описание примеров
В приведенных ниже примерах описываются способы, посредством которых может быть получен неограниченный набор соединений по настоящему изобретению. Соединения 3-5, 8-10, 14-17, 19-22, 24-26, 28, 30, 32, 34, 37, 38, 41, 42 и 51 представляют собой примеры соединений по настоящему изобретению. Для любых соединений из примеров, которые являются примерами соединений с возможным использованием, могут потребоваться корректировки с учетом вариаций растворимости реагентов и реакционной способности, включая применение альтернативных растворителей, различных продолжительностей реакции, температур или концентраций, либо альтернативных, но эквивалентных систем реагентов. Любые такие корректировки считаются рутинными для специалиста в данной области техники.
Синтез бисизоцианата 12
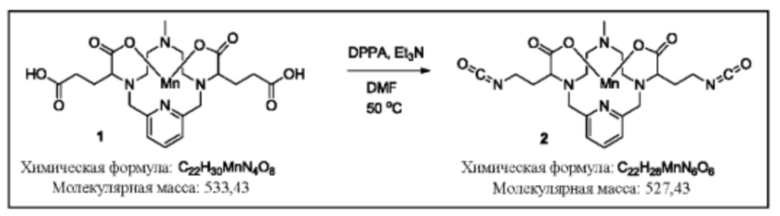
Исходное вещество 1 суспендируют в дегазированном диметилформамиде. Добавляют триэтиламин (2 эквивалента), затем дифенилфосфорилазид (2 эквивалента). Реакционную смесь перемешивают с нагреванием при 50°С, пока данные аналитической хроматографии не укажут на израсходование исходного вещества. Для завершения реакции может потребоваться применение дополнительных эквивалентов триэтиламина и дифенилфосфорилазида. После завершения реакции изоцианат 2 используют непосредственно в последующих реакциях в виде раствора в диметилформамиде (DMF).
Синтез конъюгата 3
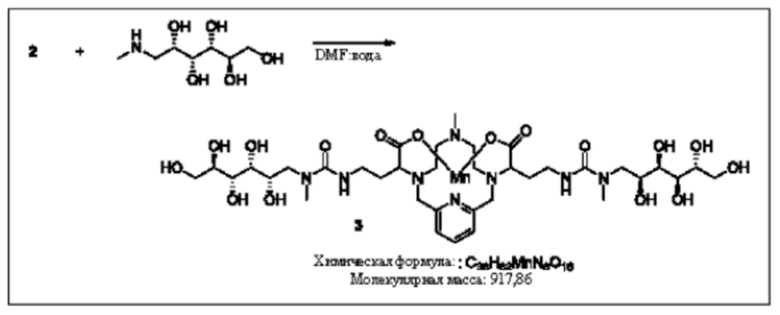
Раствор N-метил-D-глюкамина в воде (2 эквивалента) добавляют к изоцианату 2 в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка N-метил-D-глюкамина. Продукт 3 очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикателе и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 4

Раствор 2-(2-гидроксиэтокси)этилацетата в DMF (2 эквивалента) добавляют к раствору изоцианата 2 в DMF. К реакционной смеси добавляют каталитическое количество дилаурата дибутилолова и смесь нагревают до 70°С для завершения реакции; чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. По завершении реакции растворитель удаляют при пониженном давлении, остаток суспендируют в растворе метанола, содержащем каталитическое количество мет оксида натрия. Реакцию оставляют протекать, пока не будет наблюдаться полного гидролиза ацетатных защитных групп. По завершении реакции реакционную смесь концентрируют при пониженном давлении и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикат еле и элюируя смесями вода-ацетонитрил или вода-метанол
Синтез конъюгата 5

Раствор гидрохлорида 2-(диэтиламино)этантиола (2 эквивалента) и триэтиламина (2 эквивалента) в дегазированном DMF добавляют к раствору изоцианата 2 в DMF. Реакционную смесь оставляют перемешиваться при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции растворитель удаляют при пониженном давлении и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез трисизоцианата 7

Исходное вещество 6 суспендируют в дегазированном диметилформамиде. Добавляют триэтиламин (3 эквивалента), затем дифенилфосфорилазид (3 эквивалента). Реакционную смесь перемешивают с нагреванием при 50°С, пока данные аналитической хроматографии не укажут на израсходование исходного вещества. Для завершения реакции может потребоваться применение дополнительных эквивалентов триэтиламина и дифенилфосфорилазида. По завершении реакции изоцианат 7 используют непосредственно в последующих реакциях в виде раствора в DMF.
Синтез конъюгата 8

Раствор 5-амино-N,N'-бис(2,3-дигидроксипропил)изофталамида в DMF (2 эквивалента) добавляют к изоцианату 7 в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка 5-амино-N,N'-бис(2,3-дигидроксипропил)изофталамида. Продукт 8 очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 9

Синтез конъюгата 9. Раствор 1,2,3,4-тетраацетата β-D-глюкопиранозы в DMF (3 эквивалента) добавляют к раствору изоцианата 7 в DMF. К реакционной смеси добавляют каталитическое количество дилаурата дибутилолова и смесь нагревают до 70°С для завершения реакции; чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. Для завершения реакции могут потребоваться дополнительные порции 1,2,3,4-тетраацетата β-D-глюкопиранозы. По завершении реакции растворитель удаляют при пониженном давлении, остаток суспендируют в растворе метанола, содержащем каталитическое количество метоксида натрия. Реакцию оставляют протекать, пока не будет наблюдаться полного гидролиза ацетатных защитных групп. По завершении реакции реакционную смесь концентрируют при пониженном давлении и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 10
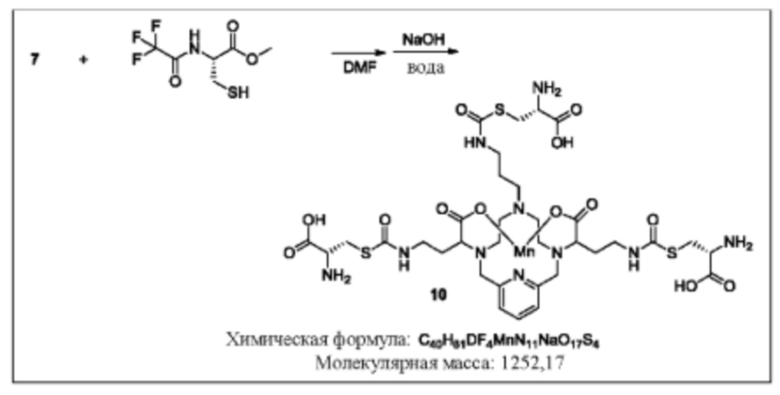
Раствор метилового эфира N-трифторацетил-L-цистеина (3 эквивалента) в дегазированном DMF добавляют к раствору изоцианата 7 в DMF. Реакционную смесь оставляют перемешиваться при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции растворитель удаляют при пониженном давлении, и остаток обрабатывают раствором гидроксида натрия в воде для удаления метилового эфира и трифторацетамидных защитных групп. Значение рН реакционной смеси доводят до 7 путем добавления водного раствора HCl и реакционную смесь непосредственно очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез функционализированного амином хелата 11

Бис-изоцианат 2 растворяют в безводном метаноле при комнатной температуре. После этого добавляют раствор NaOH в метаноле, чтобы гидролизовать полученный бис-метилкарбамат до требуемого амина 11. За протеканием реакции следят, используя аналитическую хроматографию. По завершении значение рН реакционной среды доводят до 7, и растворитель удаляют при пониженном давлении. При необходимости выделенный остаток можно очистить с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле и элюирования смесями вода-ацетонитрил или вода-метанол, получая чистое соединение 11.
Альтернативный синтез функционализированного амином хелата 11

Пирамин 12 и метил-2-бром-4-(1,3-диоксоизоиндолин-2-ил)бутаноат (2 эквивалента) растворяют в ацетонитриле и добавляют триэтиламин (2,2 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции в сторону образования соединения 13 следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и полученный остаток очищают с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле и элюирования смесями вода-ацетонитрил или вода-метанол, получая соединение 13. Соединение 13 суспендируют в 6 М водном растворе HCl и нагревают с перемешиванием до 100°С в течение 24 ч. По окончании этого промежутка времени реакционную смесь фильтруют для удаления нерастворившихся твердых веществ, фильтрат собирают и его рН доводят до 7,4, используя 50%-й (масс/масс) раствор гидроксида натрия. Как только рН реакционной смеси установится на уровне 7,4, к реакционной смеси добавляют MnCl2×Н2О (приблизительно 2,4 эквивалента) и смесь перемешивают при комнатной температуре. К реакционной смеси дополнительно добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12, добавляя водный раствор NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления водного раствора HCl. Требуемое соединение 11 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 14

Аминопирамин 13 растворяют в воде и добавляют N-ацетилнейраминовую кислоту (2,1 экв.) и EDCI-HCl (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида) (2,1 экв.). Значение рН доводят до 6,5, используя HCl, и затем добавляют HOBt (1-гидроксибензотриазол) (0,4 экв.). Значение рН полученного раствора поддерживают приблизительно равным 6 при одновременном перемешивании в течение 16 ч. Затем добавляют дополнительное количество EDCI-HCl (1,1 экв.), рН реакционной смеси поддерживают приблизительно равным 6 и перемешивают в течение еще 16 ч. Растворитель удаляют в вакууме и требуемое соединение 14 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 15
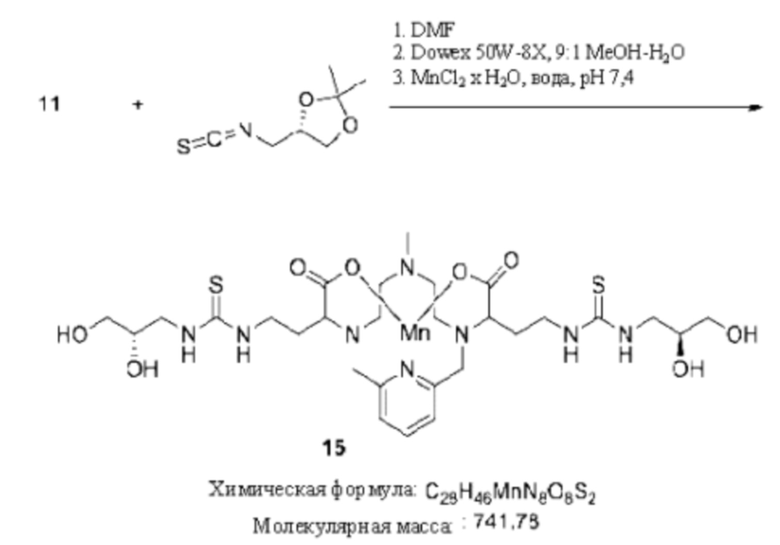
Раствор (S)-4-(изотиоцианатометил)-2,2-диметил-1,3-диоксолана в DMF (2 эквивалента) добавляют к диамину 11 в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка (S)-4-(изотиоцианатометил)-2,2-диметил-1,3-диоксолана. По завершении реакции реакционную смесь концентрируют при пониженном давлении, остаток переносят в смесь 9:1 МеОН-Н2О и добавляют смолу Dowex 50W-8X. Эту гетерогенную смесь перемешивают при комнатной температуре и оценку протекания реакции осуществляют посредством аналитической хроматографии. По завершении реакции смолу удаляют фильтрованием, и фильтрат концентрируют при пониженном давлении. Остаток переносят в воду и рН смеси доводят до 7,4. Добавляют дополнительное количество MnCl2×Н2О (1 эквивалент) и смесь перемешивают при комнатной температуре. К реакционной смеси дополнительно добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12, добавляя водный раствор NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления водного раствора HCl. Требуемое соединение 15 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 16
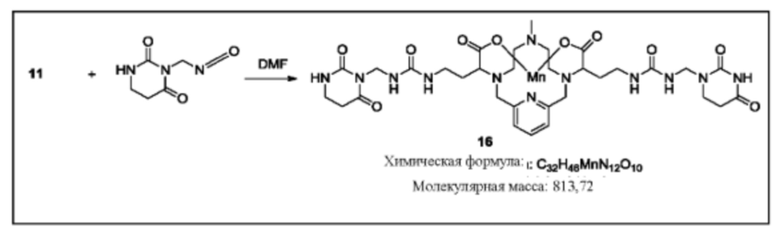
Раствор соединения 11 в дегазированном DMF добавляют к 3-(изоцианатометил)дигидропиримидин-2,4(1Н,3Н)-диону (2 эквивалента) в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции необходимо добавление избытка 3-(изоцианатометил)дигидропиримидин-2,4(1Н,3Н)-диона. Продукт 16 очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 17

Раствор 2-(метилсульфонил)этил-хлорформиата в DMF (2 эквивалента) добавляют к раствору соединения 11 и триэтиламина (2,5 эквивалента) в DMF при 0°С. Раствор оставляют нагреваться до комнатной температуры в течение ночи. Чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. Для завершения реакции может потребоваться добавление дополнительных порций 2-(метилсульфонил)этил-хлорформиата. По завершении реакции растворитель удаляют при пониженном давлении, остаток очищают с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле и элюирования смесями вода-ацетонитрил или вода-метанол, получая указанное в заголовке соединение 17.
Синтез функционализированного амином хелата 18

Трисизоцианат 7 растворяют в безводном метаноле при комнатной температуре. После этого добавляют раствор NaOH в метаноле, чтобы осуществить гидролиз полученного трис-метил-карбамата. За протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции значение рН реакционной смеси доводят до 7 и растворитель удаляют при пониженном давлении. При необходимости выделенный остаток может быть очищен с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле и элюирования смесями вода-ацетонитрил или вода-метанол, получая чистое соединение 18.
Синтез конъюгата 19
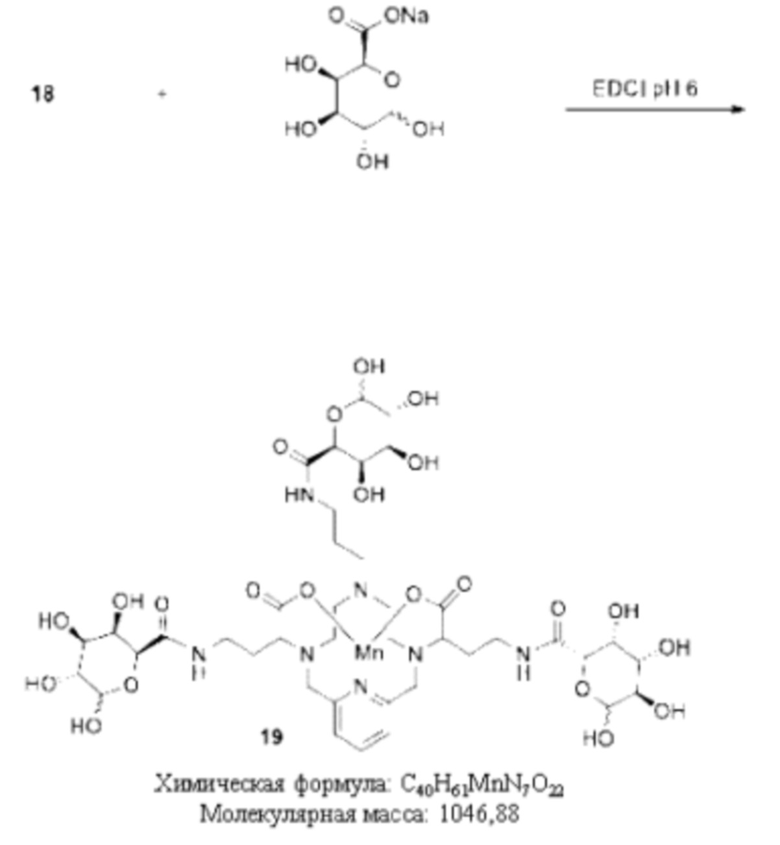
Аминопирамин 18 растворяют в воде и добавляют натриевую соль D-галактуроновой кислоты (3,1 экв.) и EDCI-HCl (3,1 экв.). Значение рН доводят до 6,5, используя HCl, и затем добавляют HOBt (0,6 экв.). Значение рН полученного раствора поддерживают приблизительно равным 6 при одновременном перемешивании в течение 16 ч. Затем добавляют дополнительное количество EDCI-HCl (1,6 экв.), рН реакционной смеси поддерживают приблизительно равным 6 и перемешивают в течение еще 16 ч. Растворитель удаляют в вакууме и требуемое соединение 19 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 20

Раствор 2,3,4-три-О-ацетил-α-D-арабинопиранозилизотиоцианата в дегазированном DMF (3,1 эквивалента) добавляют к триамину 18 в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка изотиоцианата. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и остаток переносят в раствор метанола, содержащий каталитическое количество метилата натрия. Реакцию оставляют протекать, пока не будет наблюдаться полного гидролиза ацетатных защитных групп. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 21

Раствор соединения 18 в дегазированном DMF добавляют к 3-(изоцианатометил)тетрагидротиофен-1,1-диоксиду (3 эквивалента) в DMF. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре и за протеканием реакции следят, используя методы аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка изотиоцианата. 3-(изоцианатометил)тетрагидротиофен-1,1-диоксида. Продукт 21 очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 22

Раствор (2,2-диметил-1,3-диоксолан-4-ил)метил-хлорформиата в DMF (3 эквивалента) добавляют к раствору соединения 18 и триэтиламина (3,5 эквивалента) в DMF при 0°С. Раствор оставляют нагреваться до комнатной температуры в течение 16 ч. Чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. Для завершения реакции может потребоваться добавление дополнительных порций (2,2-диметил-1,3-диоксолан-4-ил)метил-хлорформиата. По завершении реакции растворитель удаляют при пониженном давлении, остаток переносят в смесь 9:1 МеОН-H2O и добавляют смолу Dowex 50W-8X. Эту гетерогенную смесь перемешивают при комнатной температуре и оценку протекания реакции осуществляют посредством аналитической хроматографии. По завершении реакции смолу удаляют фильтрованием, и фильтрат концентрируют при пониженном давлении. Остаток переносят в воду и рН смеси доводят до 7,4. Добавляют дополнительное количество MnCl2×Н2О (1 эквивалент) и смесь перемешивают при комнатной температуре. К реакционной смеси дополнительно добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12, добавляя водный раствор NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления водного раствора HCl. Требуемое соединение 22 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез бис-изотиоцианата 23
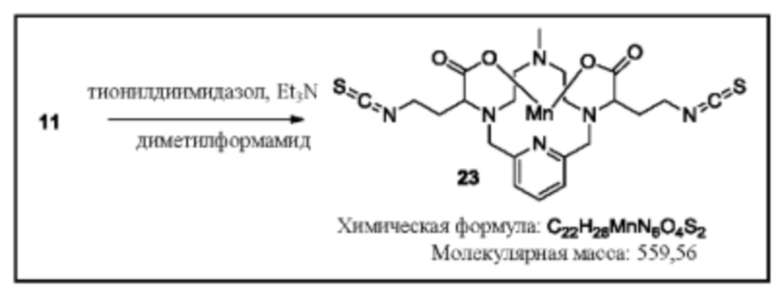
Исходное вещество 11 суспендируют в дегазированном диметилформамиде. Добавляют триэтиламин (2 эквивалента), затем тионилдиимидазол (2 эквивалента). Реакционную смесь перемешивают с нагреванием при 80°С, пока данные аналитической хроматографии не укажут на израсходование исходного вещества. Для завершения реакции может потребоваться применение дополнительных эквивалентов триэтиламина и тионилдиимидазола. По завершении реакции бис-тиоизоцианат 23 используют непосредственно в последующих реакциях в виде раствора в DMF.
Синтез бис-тиомочевины 24

К раствору соединения 23 в DMF при комнатной температуре добавляют 2,2'-азандиилдиэтанол (2,1 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции следят посредством аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка 2,2'-азандиилдиэтанола. По завершении реакции растворитель удаляют при пониженном давлении, остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 24.
Синтез конъюгата 25

Монометиловый эфир олигополиэтиленгликоля (в котором n находится в диапазоне от 0 до 150 и который может представлять собой отдельное химическое соединение или смесь химических соединений, состоящих из соединений с различными значениями «n») растворяют в DMF при 0°С и обрабатывают NaH, взятым в субстехиометрическом количестве. Смесь перемешивают при 0°С в течение 1 ч и затем к смеси добавляют раствор соединения 23 в DMF. Смесь продолжают перемешивать, медленно нагревая до комнатной температуры в течение нескольких часов. За протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции растворитель удаляют при пониженном давлении, остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 25.
Синтез конъюгата 26

Раствор соединения 23 в DMF добавляют к раствору гомоцистеина HCl в воде. Значение рН раствора доводят до 6,5, смесь оставляют для взаимодействия и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют, получая неочищенный остаток, который далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 26.
Синтез трис-изотиоцианата 27
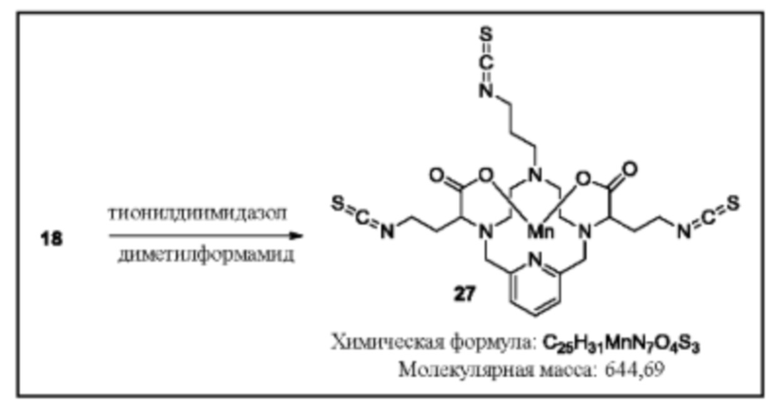
Исходное вещество 18 суспендируют в дегазированном диметилформамиде. Добавляют триэтиламин (3 эквивалента), затем тионилдиимидазол (3 эквивалента). Реакционную смесь перемешивают с нагреванием при 80°С, пока данные аналитической хроматографии не укажут на израсходование исходного вещества. Для завершения реакции может потребоваться применение дополнительных эквивалентов триэтиламина и тионилдиимидазола. По завершении реакции трис-тиоизоцианат 23 используют непосредственно в последующих реакциях в виде раствора в DMF.
Синтез конъюгата 28
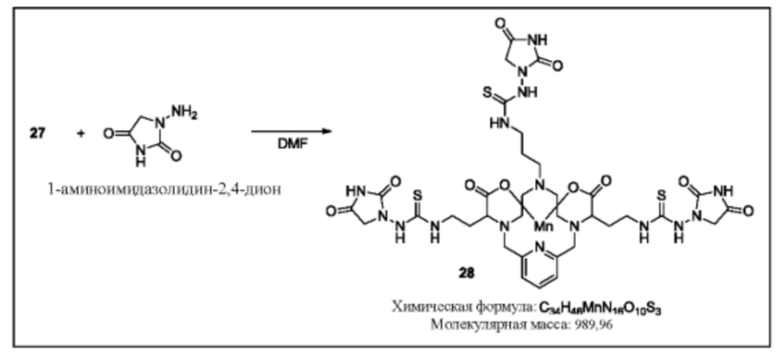
К раствору соединения 27 в DMF при комнатной температуре добавляют 1-аминоимидазолидин-2,4-дион (3,1 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции следят посредством аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка 1-аминоимидазолидин-2,4-диона. По завершении реакции растворитель удаляют при пониженном давлении, остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 28.
Синтез партнера сочетания 32а
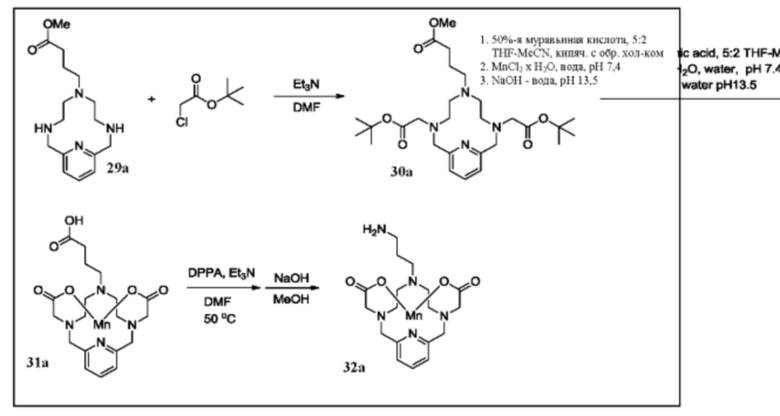
Исходное вещество 29а растворяют в DMF и добавляют триэтиламин (3 эквивалента), затем трет-бутилхлорацетат.
Синтез конъюгата 30
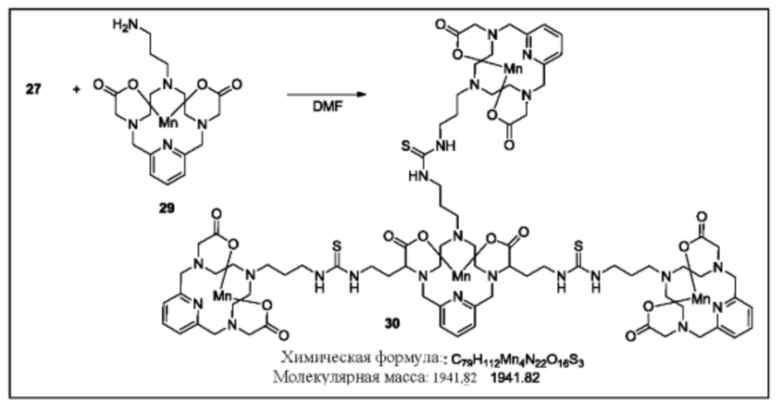
К раствору соединения 27 в DMF при комнатной температуре добавляют соединение 29 (3,1 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции следят посредством аналитической хроматографии. Для завершения реакции может быть необходимо добавление избытка соединения 29. По завершении реакции растворитель удаляют при пониженном давлении, остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 30.
Синтез конъюгата 32

Раствор соединения 31 в DMF (3 эквивалента) добавляют к раствору изотиоцианата 27 в DMF, содержащему 4-диметиламинопиридин (0,3 экв.) и дилаурат дибутилолова (0,03 экв.). Для завершения реакции смесь нагревают до 70°С; чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. Для завершения реакции могут потребоваться дополнительные порции соединения 31. По завершении реакции растворитель удаляют при пониженном давлении, остаток суспендируют в растворе метанола, содержащем каталитическое количество метоксида натрия. Реакцию оставляют протекать, пока не будет наблюдаться полного гидролиза ацетатных защитных групп. По завершении реакции реакционную смесь концентрируют при пониженном давлении и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 34

Раствор соединения 27 в DMF добавляют к раствору соединения 33 в воде. Значение рН раствора доводят до 6,5, смесь оставляют для взаимодействия и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют, получая неочищенный остаток, который далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 34.
Синтез диола 36
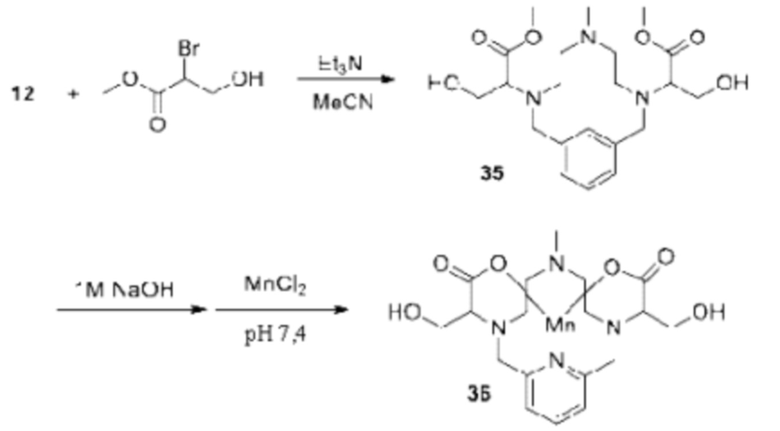
Пирамин 12 и метил-2-бром-3-гидроксипропаноат (2 эквивалента) растворяют в ацетонитриле и добавляют триэтиламин (2,2 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции в сторону образования соединения 35 следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и полученный остаток очищают с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле с элюированием смесями вода-ацетонитрил или вода-метанол, получая соединение 35. Соединение 35 растворяют в воде и добавляют твердый NaOH (4 эквивалента). Реакционную смесь перемешивают при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции в реакционную смесь помещают датчик рН и значение рН реакционной смеси доводят до 7,4, используя водный раствор HCl. После достижения желаемого значения рН к реакционной смеси добавляют MnCl2×Н2О (приблизительно 1,1 эквивалента) и смесь перемешивают при комнатной температуре. К этой реакционной смеси добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12, добавляя водный раствор NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления водного раствора HCl. Требуемое соединение 36 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 37
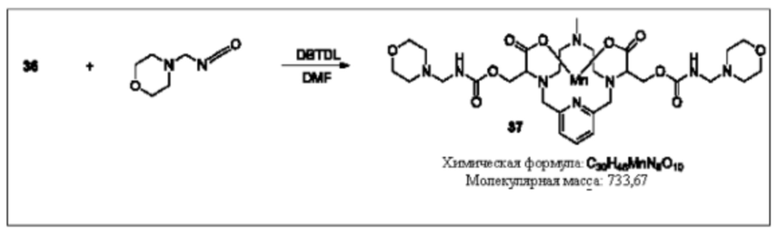
Раствор соединения 36 в DMF добавляют к раствору 4-(изоцианатометил)морфалина (2,1 эквивалента) в DMF. К реакционной смеси добавляют каталитическое количество дилаурата дибутилолова и для завершения реакции смесь нагревают до 70°С; чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. При необходимости к реакционной смеси добавляют дополнительные количества 4-(изоцианатометил)морфолина. По завершении реакции реакционную смесь концентрируют при пониженном давлении, остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением соединения 37.
Синтез конъюгата 38

Раствор 4-изотиоцианатобензолсульфонамида в DMF (2,1 эквивалента) добавляют к раствору соединения 36 в DMF, содержащему 4-диметиламинопиридин (0,3 экв.) и дилаурат дибутилолова (0,03 экв.). Для завершения реакции смесь нагревают до 70°С, чтобы убедиться в протекании реакции, используют методики аналитической хроматографии. Для завершения реакции могут потребоваться дополнительные порции 4-изотиоцианатобензолсульфонамида. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез дитиола 40

Пирамин 12 и метил-2-(тозилокси)-4-(тритилтио)бутаноат (2 эквивалента) растворяют в ацетонитриле и добавляют триэтиламин (2,2 эквивалента). Реакционную смесь продолжают перемешивать при комнатной температуре и за протеканием реакции в сторону образования соединения 39 следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют при пониженном давлении, и полученный остаток очищают с использованием обращенно-фазовой хроматографии на С-18 функционализированном силикагеле с элюированием смесями вода-ацетонитрил или вода-метанол, получая соединение 39. Защищенное соединение 39 растворяют в 2 М водном растворе HCl, который тщательно дегазируют в атмосфере азота перед применением. Реакционную смесь нагревают в атмосфере инертного газа, способствуя гидролизу метилового эфира и расщеплению тритилового тиоэфира. За протеканием этой реакции следят посредством аналитической хроматографии, по завершении реакции значение рН реакционной смеси доводят до 7,4 путем добавления дегазированного 2 М раствора NaOH. После достижения желаемого значения рН к реакционной смеси добавляют MnCl2×H2O (приблизительно 1,0 эквивалента) и смесь перемешивают при комнатной температуре. К этой реакционной смеси добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12, добавляя водный раствор NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления дегазированного водного раствора HCl. Требуемое соединение 40 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 41

Раствор 2-(изоцианатометокси)уксусной кислоты (2 эквивалента) и триэтиламина (2 эквивалента) в дегазированном DMF добавляют к раствору соединения 40 в дегазированном DMF. Реакционную смесь оставляют перемешиваться при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции растворитель удаляют при пониженном давлении, и остаток очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез конъюгата 42

4-(2-Изотиоцианатоэтил)морфолин (2,1 эквивалента) добавляют к раствору соединения 40 в воде. Значение рН раствора доводят до 7,2, смесь оставляют для взаимодействия и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют, получая неочищенный остаток, который далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 42.
Альтернативный синтез соединения 41


Промежуточное соединение 39 растворяют в дихлорметане и добавляют к раствору трифторуксусной кислоты, триизопропилсилана и воды (95:2,5:2,5 об/об/об). Реакционную смесь оставляют для взаимодействия при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции реакционную смесь концентрируют, получая неочищенный остаток, который далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол, с получением указанного в заголовке соединения 43. К раствору соединения 43 в дегазированном DMF добавляют раствор 2-(изоцианатометокси)уксусной кислоты (2 эквивалента) и триэтиламина (2 эквивалента) в дегазированном DMF. Реакционную смесь оставляют перемешиваться при комнатной температуре и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции растворитель удаляют при пониженном давлении, и остаток обрабатывают 1 М раствором NaOH в воде. Для наблюдения за гидролизом метиловых эфиров используют аналитическую хроматографию. По завершении реакции значение рН реакционной смеси доводят до 7,4 путем добавления 2 М водного раствора HCl, к реакционной смеси добавляют MnCl2×Н2О (приблизительно 1,0 эквивалента) и смесь перемешивают при комнатной температуре. К этой реакционной смеси добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12 путем добавления водного раствора NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления дегазированного водного раствора HCl. Требуемое соединение 41 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Альтернативный синтез соединения 42

4-(2-Изотиоцианатоэтил)морфолин (2,1 эквивалента) добавляют к раствору соединения 43 в воде. Значение рН раствора доводят до 7,2, смесь оставляют для взаимодействия и за протеканием реакции следят, используя аналитическую хроматографию. По завершении реакции рН реакционной смеси повышают до 13,5 путем добавления 2,0 М водного раствора NaOH. За омылением метиловых эфиров следят, используя аналитическую хроматографию. По завершении реакции рН реакционной смеси снижают до 7,4 путем добавления 2 М раствора HCl, к реакционной смеси добавляют MnCl2×Н2О (приблизительно 1,0 эквивалента) и смесь перемешивают при комнатной температуре. К реакционной смеси добавляют раствор NaOH для поддержания рН реакционной смеси в диапазоне от 7,0 до 7,4. За динамикой включения марганца следят посредством аналитической хроматографии и по завершении реакции рН реакционной смеси доводят до 12 путем добавления водного раствора NaOH. Реакционную смесь продолжают перемешивать в течение 1 ч при комнатной температуре, затем реакционную смесь фильтруют для удаления твердых веществ, фильтрат собирают и его рН доводят до 7,4 путем добавления дегазированного водного раствора HCl. Требуемое соединение 41 далее очищают, используя обращенно-фазовую хроматографию на С-18 функционализированном силикагеле и элюируя смесями вода-ацетонитрил или вода-метанол.
Синтез соединения 51

2-Бром-6-(2,2,2-трифторацетамидо)гексановая кислота (44). N-ε-Трифторацетамидо-L-лизин (25 г; 103,2 ммоль) и бромид натрия (37,2 г; 361,2 ммоль) добавляли в 3-горлый снабженный рубашкой реакционный сосуд емкостью 500 мл, оснащенный внутренней термопарой, механической мешалкой и насыпной воронкой. После этого твердые вещества растворяли в 69 мл воды и водном растворе HBr (20,9 мл; 8,9 М). Насыпную воронку удаляли и к реакционному сосуду присоединяли капельную воронку, имеющую входное отверстие для азота, в которую был загружен нитрит натрия (12,8 г; 185,8 ммоль), предварительно растворенный в 16,5 мл воды. Выходящие в ходе реакции газы пропускали через раствор сульфита натрия, после чего выпускали в вытяжной шкаф. Внутреннюю температуру реакционной смеси уменьшали до значения меньше 0°С и затем к реакционной смеси медленно добавляли раствор нитрита натрия с такой скоростью, чтобы внутренняя температура реакционной смеси не превышала 3°С. По завершении добавления нитрита натрия реакционная смесь была почти бесцветной. Капельную воронку, которая содержала раствор нитрита натрия, удаляли и к реакционному сосуду присоединяли вторую капельную воронку, в которую предварительно была загружена концентрированная серная кислота (5,5 мл). Серную кислоту добавляли к реакционной смеси с такой скоростью, чтобы внутренняя температура реакционной смеси не превышала 5°С. Реакционная смесь становилась коричневой, и из нее в процессе этого добавления выделялся бром. После добавления серной кислоты капельную воронку, в которой содержалась серная кислота, изымали и выполняли активную продувку N2 для удаления выделившегося брома из реакционной смеси и пространства над жидкой фазой. После барботирования в течение 20 минут при комнатной температуре реакционная смесь становилась значительно более светлой. Активную продувку N2 прекращали и проводили распределение реакционной смеси, используя 80 мл метил-трет-бутилового эфира (МТВЕ). Смесь быстро перемешивали в течение пяти минут и затем выполняли разделение фаз. Окрашенный желто-коричневым цветом органический слой собирали, а водный слой экстрагировали двумя дополнительными порциями МТВЕ по 70 мл. Объединенные органические слои промывали, используя несколько порций раствора Na2SO3, до почти бесцветного состояния и затем последовательно промывали рассолом (100 мл), сушили (MgSO4), фильтровали и концентрировали при пониженном давлении, получая масло бледно-желтой окраски. Выделенное вещество сушили под высоким вакуумом в течение ночи и после этого отбирали образец для аналитического определения характеристик (по данным UPLC-MS (сочетание сверхэффективной жидкостной хроматографии и масс-спектрометрии) продемонстрировано более чем 80%-ное превращение в соединение 44 (m/z 306)). Выделенное вещество взвешивали (28,1 г) и остаток затем переносили на следующую стадию без дополнительной очистки.
Метил-2-бром-6-(2,2,2-трифторацетамидо)гексаноат (45). Соединение 44 (28,1 г) растворяли в метаноле (350 мл) и добавляли n-толуолсульфоновую кислоту (0,35 г, 1,8 ммоль). Смесь нагревали при 65°С в атмосфере азота в течение ночи. По прошествии этого времени нагревание останавливали и реакционную смесь оставляли охлаждаться до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении, получая желтое масло, которое далее очищали флэш-хроматографией на силикагеле (колонка 330 г, 200 мл/мин, элюирование смесями этилацетата в гексанах). Использовали следующую программу для градиента: начальные условия: 5% этилацетата в гексанах, удерживание: 5% этилацетата в гексанах в течение прохождения 1 объема колонки, затем выполнение линейного градиента: до 50% этилацетата в гексанах в течение прохождения 12 объемов колонки, окончательно удерживание: 50% этилацетата в гексанах в течение прохождения еще 2 объемов колонки. Мониторинг элюатов с колонки проводили по изменению сигнала в ультрафиолетовом (УФ) диапазоне при 230 нм и 254 нм и с использованием испарительного светорассеяния. Желаемый продукт элюировали с колонки в диапазоне от 6,4 до 9 объемов колонки. Выделенный продукт представлял собой бесцветное масло (20,3 г; 63 ммоль); [α]D20,0=-31,10±0,01 (с=10,07 г/100 мл в МеОН), ESI-MS (MS с электрораспылительной ионизацией) 322, 320 m/z; 1H ЯМР (ядерный магнитный резонанс) (CD3CN, 599,19 МГц) δ 7.58 (br. s., 1Н), δ 4.35 (t., 1Н), δ 3.73 (s., 3Н), δ 3.25 (dd., 2Н), δ 2.08-2.02 (m., 1Н), δ 1.99-1.93 (m., 1Н), δ 1.61-1.53 (m., 2Н), δ 1.51-1.44 (m., 1Н), δ 1.40-1.33 (m., 1Н); 13С ЯМР (CD3CN, 150,83 МГц) δ 171.18, 157.83 (q., J=37,0 Гц), 117.17 (q., J=286,7 Гц), 53.57, 46.94, 40.03, 35.09, 28.59 и 25.01; 19F ЯМР (CD3CN, 564,32 МГц) δ -76.67.

Соединение (47). В колбу емкостью 100 мл, содержащую метилпирамин 46 (5 г; 21,7 ммоль), добавляли 45 мл безводного MeCN, затем 9,3 мл (52,1 ммоль) диизопропилэтиламина. После перемешивания в течение 5 минут к реакционной смеси добавляли соединение 45 (15,3 г, 74,8 ммоль). Реакционный сосуд помещали в масляную баню на 18 ч, установленную на 65°С. По прошествии этого времени к реакционной смеси добавляли дополнительно 1,9 мл (10,8 ммоль) диизопропилэтиламина и 3 г (9,4 ммоль) соединения 45. Реакционную смесь оставляли перемешиваться в масляной бане при 65°С в течение еще 24 ч. После отведенного промежутка времени реакционную смесь охлаждали до температуры окружающей среды и далее концентрировали при пониженном давлении, получая красный маслянистый остаток. Выделенный остаток распределяли между этилацетатом и водой и проводили разделение фаз. Органический слой промывали двумя дополнительными порциями воды, и затем объединенные водные промывки экстрагировали повторно двумя дополнительными порциями этилацетата. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, получая красное масло, которое вспенивалось под вакуумом. Выделенное масло растирали с двумя порциями диэтилового эфира по 40 мл каждая в течение 10 минут при температуре окружающей среды, и затем эфирный слой декантировали и отбрасывали. Остаток промывали 50 мл воды (нагретой в течение 10 мин в водяной бане при 60°С). Водную суспензию оставляли охлаждаться до температуры меньше 35°С и затем смесь экстрагировали двумя порциями диэтилового эфира по 15 мл каждая. Эфирные экстракты отбрасывали. Водный слой разбавляли порцией рассола эквивалентного объема и полностью экстрагировали этилацетатом (процедуры экстракции продолжали до тех пор, пока по данным высокоэффективной жидкостной хроматографии (HPLC) (5×30 мл) не было получено практически никакого сигнала, указывающего на наличие продукта в водном слое). Объединенные этилацетатные экстракты сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, получая почти бесцветную пену (14,3 г; 20,5 ммоль; 94%), ESI-MS 699,70 (m/z).
Соединение (48). В круглодонную колбу емкостью 50 мл, снабженную магнитным стержнем для перемешивания, добавляли соединение 47 (2,018 г; 2,89 ммоль) в H2O (9 мл). Добавляли KOH (1,302 г; 23,2 ммоль) и полученный раствор перемешивали при температуре окружающей среды в течение 1,5 ч. HPLC-MS анализ показал, что реакция прошла полностью (ESI-MS: 479 (М+Н+) (m/z)). Значение рН реакционного раствора доводили до 10,1, используя конц. HCl, и этот раствор использовали без дополнительной очистки.
Соединение (49). MnCl2⋅4H2O (0,638 г; 3,22 ммоль) добавляли к раствору с рН 10,1, описанному в разделе о синтезе соединения 5, рН снижали до 5,8 и доводили до 7,0, используя 3,4 М водный раствор KOH. Затем полученный раствор затем нагревали до 90°С в течение 3 ч, после чего охлаждали до температуры окружающей среды. Затем рН повышали до 10,0, используя KOH, и полученный мутный коричневый раствор перемешивали при температуре окружающей среды в течение 14,5 ч. После фильтрования через PTFE (политетрафторэтиленовый) фильтр (0,45 мкм) получали слегка мутный фильтрат, который концентрировали досуха в вакууме. Полученный коричневый остаток растирали с МеОН (10 мл), получая белое твердое вещество в прозрачном коричневом растворе. Твердое вещество удаляли фильтрованием через PTFE фильтр (0,45 мкм) и прозрачный коричневый фильтрат концентрировали досуха в вакууме. Полученный коричневый остаток растворяли в Н2О (5 мл) и очищали на С18 диоксиде кремния (от 5% AcN (ацетонитрил) в воде до 10% AcN в воде), получая 1,41 г (92%) желаемого продукта в виде бледно-желтого твердого вещества. ESI-MS: 532 (М+Н+) (m/z).
Соединение (51). 20% (масс/масс) водный раствор соединения 50 (1,651 мл; 3,11 ммоль) добавляли в 2-горлую колбу емкостью 25 мл, снабженную магнитным стержнем для перемешивания и датчиком рН. Значение рН раствора глицериновой кислоты подводили от 2,2 до 7,2, используя 3,4 М раствор KOH. Добавляли соединение 49 (0,701 г; 1,38 ммоль), затем EDCI-HCl (0,634 г; 3,31 ммоль) и гидроксибензотриазола гидрат (0,028 г; 0,183 ммоль). Значение рН полученного раствора поддерживали в диапазоне от 6 до 7 путем добавления 6 М раствора HCl и/или 3,4 М раствора KOH, в зависимости от ситуации, при одновременном перемешивании при температуре окружающей среды в течение 4,5 ч. Добавляли EDCI-HCl (0,530 г; 2,76 ммоль) и полученный раствор перемешивали при температуре окружающей среды в течение еще 15,5 ч. Весь растворитель удаляли в вакууме, получая золотисто-желтое масло, которое очищали на С18 диоксиде кремния (от 2% AcN в воде до 20% AcN в воде), получая 0,31 г (32%) желаемого продукта в виде беловатого твердого вещества. ESI-MS: 708 (М+Н+) (m/z).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛИЧЕСКИМИ ПОЛИАМИНОКИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ | 2000 |
|
RU2232763C2 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| ГЕТЕРОБИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ГЕТЕРОБИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 1993 |
|
RU2128656C1 |
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |
| АЛКИЛСУЛЬФОНАМИДХИНОЛИНЫ С АФФИННОСТЬЮ К РЕЦЕПТОРАМ NK-3 | 2006 |
|
RU2421447C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ FGFR4, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ, СОДЕРЖАЩИЕ ИХ, И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2827699C1 |
| СПОСОБ ОБРАБОТКИ ФТОРИДА | 2008 |
|
RU2475448C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ TRK | 2011 |
|
RU2735545C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В ЛЕЧЕНИИ РАССТРОЙСТВ, АССОЦИИРОВАННЫХ С ВАНИЛОИДНЫМ РЕЦЕПТОРОМ TRPV1 | 2007 |
|
RU2458055C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ TRK | 2011 |
|
RU2594742C2 |
Изобретение относится к соединению формулы I:
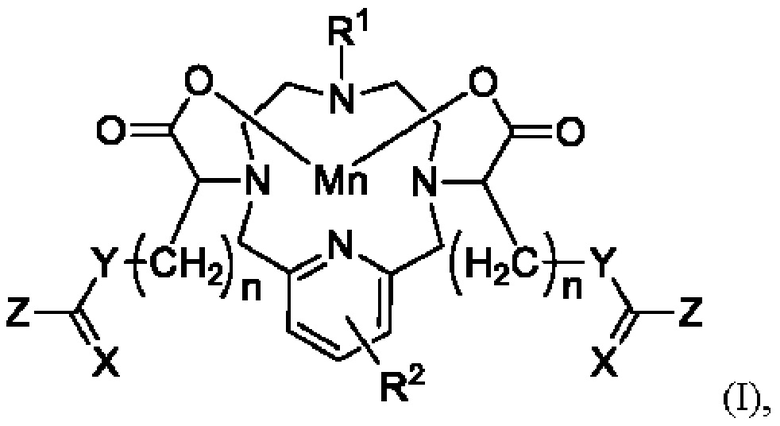 в которой X представляет собой О; Y представляет собой Q-R3, где Q представляет собой N, и R3 выбран из группы, содержащей C1-6алкил или атом водорода; Z представляет собой Q-R3-(L-R4), где Q представляет собой N или СН, и R3 представляет собой С1-20гидроксиалкил, C1-6алкил или атом водорода, и L представляет собой необязательный линкер, выбранный из группы, содержащей C1-6алкиленовый, C1-6гидроксиалкиленовый и полиэтиленгликолевый (PEG) линкер, R4 выбран из группы, содержащей C1-6алкил-R5, С3-6арил-R5, гидрокси, -О-С1-3алкил-R5, сульфонил, 5-6-членное гетероциклическое кольцо, выбранное из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина, углеводную группировку, выбранную из моносахаридных и дисахаридных остатков, хелатную группировку
в которой X представляет собой О; Y представляет собой Q-R3, где Q представляет собой N, и R3 выбран из группы, содержащей C1-6алкил или атом водорода; Z представляет собой Q-R3-(L-R4), где Q представляет собой N или СН, и R3 представляет собой С1-20гидроксиалкил, C1-6алкил или атом водорода, и L представляет собой необязательный линкер, выбранный из группы, содержащей C1-6алкиленовый, C1-6гидроксиалкиленовый и полиэтиленгликолевый (PEG) линкер, R4 выбран из группы, содержащей C1-6алкил-R5, С3-6арил-R5, гидрокси, -О-С1-3алкил-R5, сульфонил, 5-6-членное гетероциклическое кольцо, выбранное из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина, углеводную группировку, выбранную из моносахаридных и дисахаридных остатков, хелатную группировку  аминокислотную группировку, при этом R5 представляет собой один или более чем один необязательный заместитель, выбранный из гидрокси, амино, оксо, сульфонамида или -С(=O)-NH-С1-6гидроксиалкила; или Z сам по себе образует часть углеводной группировки, выбранной из моносахаридных и дисахаридных остатков, или 5-6-членного гетероциклического кольца, выбранного из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина; R1 представляет собой С1-3алкил; R2 отсутствует; и каждое n представляет собой целое число от 1 до 6; при этом соединение формулы I содержит по меньшей мере две группы гидрокси. Также предложены фармацевтическая композиция, способ формирования MR изображений и/или MR спектров с применением соединения формулы I. Предложенные соединения формулы (I) представляют собой полиаза-макроцикл с боковыми «плечами» в виде остатков карбоновой кислоты, обеспечивающими образование комплекса с марганцем в качестве металла ядра, и подходят для применения в качестве контрастирующих агентов при магнитно-резонансной визуализации (MRI). 4 н. и 14 з.п. ф-лы, 26 пр.
аминокислотную группировку, при этом R5 представляет собой один или более чем один необязательный заместитель, выбранный из гидрокси, амино, оксо, сульфонамида или -С(=O)-NH-С1-6гидроксиалкила; или Z сам по себе образует часть углеводной группировки, выбранной из моносахаридных и дисахаридных остатков, или 5-6-членного гетероциклического кольца, выбранного из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина; R1 представляет собой С1-3алкил; R2 отсутствует; и каждое n представляет собой целое число от 1 до 6; при этом соединение формулы I содержит по меньшей мере две группы гидрокси. Также предложены фармацевтическая композиция, способ формирования MR изображений и/или MR спектров с применением соединения формулы I. Предложенные соединения формулы (I) представляют собой полиаза-макроцикл с боковыми «плечами» в виде остатков карбоновой кислоты, обеспечивающими образование комплекса с марганцем в качестве металла ядра, и подходят для применения в качестве контрастирующих агентов при магнитно-резонансной визуализации (MRI). 4 н. и 14 з.п. ф-лы, 26 пр.
1. Соединение формулы I:

где:
X представляет собой О;
Y представляет собой Q-R3, где Q представляет собой N, и R3 выбран из группы, содержащей C1-6алкил или атом водорода;
Z представляет собой Q-R3-(L-R4), где Q представляет собой N или СН и R3 представляет собой С1-20гидроксиалкил, C1-6алкил или атом водорода, и L представляет собой необязательный линкер, выбранный из группы, содержащей C1-6алкиленовый, C1-6гидроксиалкиленовый и полиэтиленгликолевый (PEG) линкер, R4 выбран из группы, содержащей C1-6алкил-R5, С3-6арил-R5, гидрокси, -О-С1-3алкил-R5, сульфонил, 5-6-членное гетероциклическое кольцо, выбранное из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина, углеводную группировку, выбранную из моносахаридных и дисахаридных остатков, хелатную группировку  аминокислотную группировку, при этом R5 представляет собой один или более чем один необязательный заместитель, выбранный из гидрокси, амино, оксо, сульфонамида или -С(=O)-NH-С1-6гидроксиалкила; или Z сам по себе образует часть углеводной группировки, выбранной из моносахаридных и дисахаридных остатков, или 5-6-членного гетероциклического кольца, выбранного из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина;
аминокислотную группировку, при этом R5 представляет собой один или более чем один необязательный заместитель, выбранный из гидрокси, амино, оксо, сульфонамида или -С(=O)-NH-С1-6гидроксиалкила; или Z сам по себе образует часть углеводной группировки, выбранной из моносахаридных и дисахаридных остатков, или 5-6-членного гетероциклического кольца, выбранного из остатка N-ацетилнейраминовой кислоты, дигидропиримидин-2,4(1Н,3Н)-диона, тетрагидротиофен-1,1-диоксида, имидазолидин-2,4-диона и морфолина;
R1 представляет собой С1-3алкил;
R2 отсутствует; и
каждое n представляет собой целое число от 1 до 6;
при этом соединение формулы I содержит по меньшей мере две группы гидрокси.
2. Соединение по п. 1, где X представляет собой О, Y представляет собой Q-R3, где Q представляет собой N, и Z представляет собой Q-R3-(L-R4), где Q представляет собой N.
3. Соединение по п. 1, где X представляет собой О, Y представляет собой Q-R3, где Q представляет собой N, и Z представляет собой Q-R3-(L-R4), где Q представляет собой СН.
4. Соединение по любому из пп. 1-3, где каждая группа -L-R4 представляет собой С1-12гидроксиалкил.
5. Соединение по любому из пп. 1-4, где каждая группа -L-R4 независимо выбрана из группы, содержащей:

где в каждом случае звездочка означает место присоединения к остальной части соединения формулы I.
6. Соединение по любому из пп. 1-5, где каждый R4 представляет собой одну и ту же группу.
7. Соединение по любому из пп. 1-6, где каждый R3 независимо выбран из группы, содержащей C1-3алкил или атом водорода.
8. Соединение по п. 7, где каждый R3 представляет собой С1-3алкил.
9. Соединение по п. 8, где каждый R3 представляет собой атом водорода.
10. Соединение по любому из пп. 1-8, где каждый R3 представляет собой одну и ту же группу.
11. Соединение по любому из пп. 1-10, которое содержит по меньшей мере 4 группы гидрокси.
12. Соединение по п. 1, выбранное из следующих соединений:



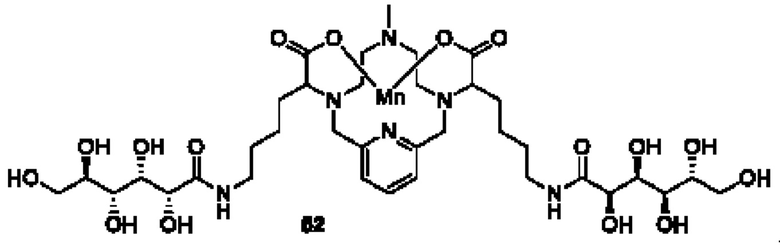
13. Соединение формулы I по любому из пп. 1-12, которое либо представляет собой рацемическую смесь, либо является диастереомерно чистым.
14. Фармацевтическая композиция для формирования MR изображений и/или MR спектров, содержащая соединение формулы I по любому из пп. 1-13 вместе с биосовместимым носителем, в форме, подходящей для введения млекопитающему.
15. Фармацевтическая композиция по п. 14, дополнительно содержащая один или более чем один фармацевтически приемлемый эксципиент.
16. Фармацевтическая композиция по п. 15, где указанные фармацевтически приемлемые эксципиенты выбраны из буферных агентов, стабилизаторов, антиоксидантов, регулирующих осмоляльность агентов, регулирующих рН агентов, избыточного хелатирующего соединения и слабых комплексов физиологически переносимых ионов.
17. Способ формирования MR изображений и/или MR спектров, включающий:
(1) введение субъекту соединения формулы I по любому из пп. 1-13 или фармацевтической композиции по любому из пп. 14-16;
(2) обнаружение сигналов магнитного резонанса (MR) от указанного субъекта или частей указанного субъекта, в которых распределено указанное соединение;
(3) формирование MR изображений и/или MR спектров на основании указанных обнаруженных сигналов.
18. Применение соединения формулы I по любому из пп. 1-13 в способе по п. 17.
| WO 2007042506 A1, 19.04.2007 | |||
| PAN D | |||
| et al, Manganese-based MRI contrast agent: past, present, and future, Tetrahedron, 2011, v | |||
| Приспособление для получения кинематографических стерео снимков | 1919 |
|
SU67A1 |
| Приспособление для плетения проволочного каркаса для железобетонных пустотелых камней | 1920 |
|
SU44A1 |
| Вертикальный водотрубный паровой котел | 1927 |
|
SU8431A1 |
| GALE E.M | |||
| et al, Manganese Alternative to Gadolinium for MRI Contrast, J | |||
| Am | |||
| Chem., 2015, v | |||
| Способ приготовления строительного изолирующего материала | 1923 |
|
SU137A1 |
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |
| Металлическая сцепка для приводных ремней | 1929 |
|
SU15548A1 |
| WEN J | |||
| et al, A mononuclear Mn2+ complex based on a novel tris-(ethyl | |||

