Область техники
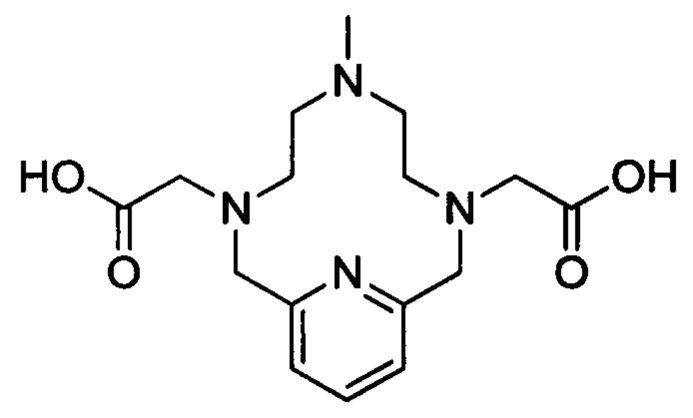
Настоящее изобретение относится к хелатным соединениям и их применению в качестве контрастных агентов в способах магнитно-резонансной томографии (МРТ).
Описание уровня техники
МРТ - это метод медицинской визуализации, в котором области тела визуализируют с помощью ядер выбранных атомов, особенно ядер водорода. Сигнал МРТ зависит от окружения визуализируемых ядер и их продольного и поперечного времен релаксации, Т1 и Т2. Таким образом, в случае, когда визуализируемым ядром является протон, интенсивность МРТ сигнала будет зависеть от таких факторов, как плотность протонов и химическое окружение протонов. Контрастные агенты можно использовать в МРТ для улучшения контраста визуализации. Они влияют на время релаксации T1, Т2 и/или Т2* и тем самым влияют на контрастность изображений.
Известно, что времена релаксации T1, Т2 и/или Т2* можно оптимизировать для хелатированных парамагнитных контрастных агентов путем структурной модификации. Особенно важным является присутствие и время удерживания молекулы воды, связанной с парамагнитным ионом, и корреляционное время вращения контрастного агента. Присутствие и время удерживания молекулы воды, связанной с парамагнитным ионом, можно регулировать путем выбора парамагнитного иона и хелатирующего фрагмента. Корреляционное время вращения можно регулировать путем изменения размера контрастного агента.
Хорошо известны некоторые типы контрастных агентов для применения в МРТ. Контрастные агенты для MP кровотока, в частности частицы суперпарамагнитного оксида железа, остаются внутри сосудистой системы в течение длительного времени. Было показано, что они чрезвычайно полезны для улучшения контрастности, например, в печени, а также для обнаружения аномалий проницаемости капилляров, таких как «подтекающие» стенки капилляров в опухолях, которые являются результатом ангиогенеза опухолей.
Растворимость парамагнитного хелатного соединения в воде также является важным фактором, когда их используют в качестве контрастных агентов для МРТ, поскольку их вводят пациентам в относительно больших дозах. Таким образом, в случае парамагнитного хелатного соединения с высокой растворимостью в воде требуется меньший объем введения, и, следовательно, легче вовдится пациенту и вызывает меньше дискомфорта. Водорастворимые парамагнитные хелатные соединения, то есть комплексные соединения, состоящие из хелатирующего агента и иона парамагнитного металла, хорошо известны - в частности, коммерчески доступные хелатные соединения гадолиния Omniscan™ (GE Healthcare), Dotarem™ (Guerbet), Gadavist™ (Bayer) и Magnevist™ (Bayer). Из-за низкой молекулярной массы они быстро распределяются во внеклеточном пространстве (то есть крови и интерстициальной ткани) после введения в сосудистую систему. Они также быстро выводятся из организма.
В некоторых публикациях описана работа, проведенная с целью разработки усовершенствованных парамагнитных хелатных соединений. Например, в US 8540966 раскрыты структуры следующей общей формулы:

где L является линкером, a R представляет собой Н или С2-70 аминополиольный фрагмент. В экспериментальных примерах из US 8540966 сравнивают некоторые из этих соединений с коммерчески доступными хелатными соединениями гадолиния, чтобы продемонстрировать аналогичный фармакокинетический профиль, но более высокую релаксивность.
В ЕР1931673 раскрыта структура следующей общей формулы:
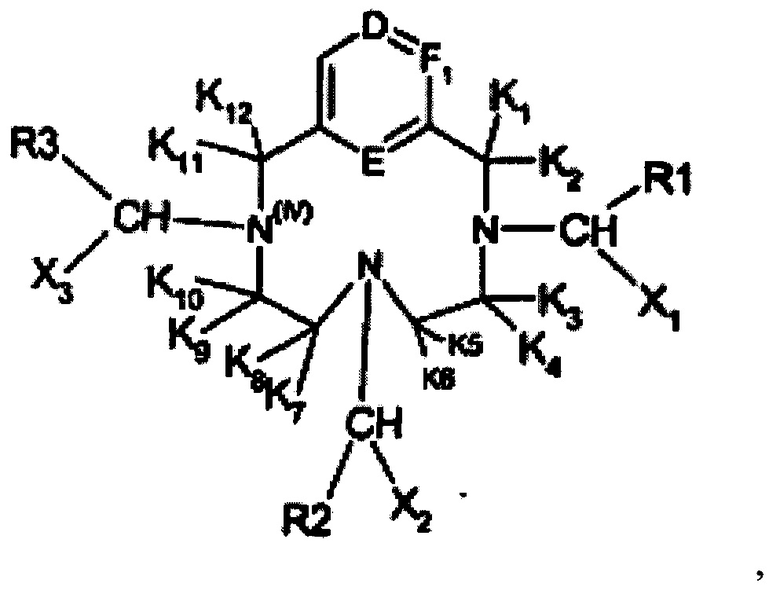
где каждый R в указанной выше структуре определен в ЕР 1931673 как координирующий лиганд, и каждый X содержит по меньшей мере одну C1-6 гидроксиалкильную группу. В ЕР 1931673 сделан акцент на свойствах релаксивности указанных соединений. В ЕР 1931673 указано, что указанные соединения могут комплексоваться с ионом парамагнитного металла, выбранного из Gd3+, Mn2+ и Fe3+, но в действительности фокус сделан на хелатные структуры, которые подходят для устойчивого комплексообразования с Gd3+, например, образования следующего содержащего гадолиний комплекса:

Все раскрытые комплексы являются гептадентатными, так как четыре азота и три карбоксильные группы координируются комплексообразующим ионом металла. Вредное воздействие гептадентатных хелатных соединений марганца было описано в WO 2011073371.
Ключевое свойство хелатных соединений для МРТ заключает для парамагнитного иона в его сохранении внутри структуры хелата как можно дольше. Парамагнитный ион, высвобождаемый из хелата in vivo, может оказывать влияние на биологические процессы и потенциально индуцирует токсичность. Способность хелата удерживать парамагнитный ион (в данной заявке называемая также стабильностью) также является свойством, которое можно регулировать путем синтеза структуры хелатирующего фрагмента. Наибольший интерес представляет кинетическая стабильность, определяемая как время полураспада, которая показывает степень инерции в отношении изменения химического окружения (то есть эндогенных ионов). В указанных выше публикациях не обсуждается инертность переметаллирования описанных в них соединений.
Как можно предположить на основе анализа коммерчески доступных агентов и акцента, сделанного в уровне техники, гадолиний является наиболее широко используемым ионом парамагнитного металла в хелатных соединениях для МРТ, что обусловлено его благоприятными характеристиками релаксивности. Стабильность парамагнитного иона внутри структуры хелата особенно важна для хелатов гадолиния, так как проблемы, связанные со свободным гадолинием и токсичностью, хорошо известны. Из-за этих проблем существует мотивация для поиска альтернативы гадолинию.
Ион марганца (II) является парамагнитной частицей с высоким спиновым числом и длительным временем релаксации электронов, причем потенциал контрастного агента с высокой релаксивностью на основе марганца (II) был указан в литературе ( Advances in Inorganic Chemistry, 2009, 61(09), 63-129). Было, однако, показано, что некоторые хелатные соединения марганца (II), разработанные к настоящему времени, являются значительно менее стабильными по сравнению с соответствующими хелатными соединениями гадолиния. Например, марганцевые хелаты DOTA (MnDOTA) в несколько сот раз менее стабильны по сравнению с соответствующим комплексом гадолиния (GdDOTA (
Advances in Inorganic Chemistry, 2009, 61(09), 63-129). Было, однако, показано, что некоторые хелатные соединения марганца (II), разработанные к настоящему времени, являются значительно менее стабильными по сравнению с соответствующими хелатными соединениями гадолиния. Например, марганцевые хелаты DOTA (MnDOTA) в несколько сот раз менее стабильны по сравнению с соответствующим комплексом гадолиния (GdDOTA ( , В; Inorganic Chemistry, 2012(12), 1975-1986).
, В; Inorganic Chemistry, 2012(12), 1975-1986).
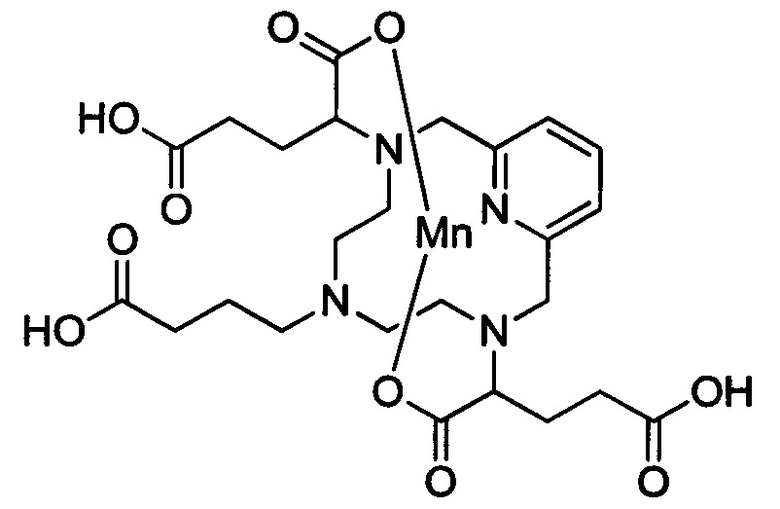
Работа, описанная в WO 2011073371, посвящена синтезу молекул, обеспечивающих высокую стабильность хелатного соединения и высокую релаксивность. Это делает указанные соединения очень подходящими для применения в качестве контрастных агентов для МРТ. Типичное соединение из WO 2011073371 имеет следующую структуру (в данной заявке называемую также "хелат Mn уровня техники"):

Тем не менее, все еще существуют возможности для других хелатных соединений марганца, имеющих улучшенную и долговременную кинетическую стабильность при сохранении подходящих свойств релаксации.
Сущность изобретения
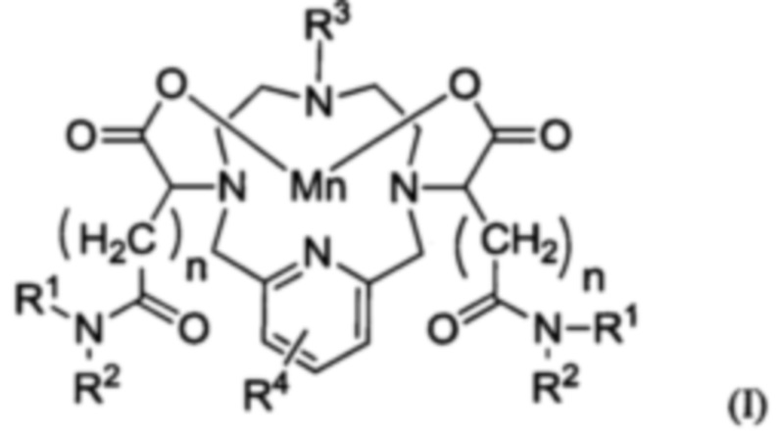
В одном аспекте настоящего изобретения предложено соединение формулы I:
или его соль или сольват, где:
каждый R1 независимо выбран из группы, включающей С1-20 гидроксиалкил, С1-6 алкил, С3-6 арил, необязательно замещенный одним или более заместителей, выбранных из галогена и -C(=O)-NH-C1-6 гидроксиалкила, или углеводного фрагмента;
каждый R2 независимо выбран из группы, включающей С1-20 гидроксиалкил, С1-6 алкил или водород;
R3 выбран из группы, включающей С1-3 алкил или -(CH2)m-C(=O)-NR5R6, где m является целым числом от 2 до 5, и R5 и R6 являются такими, как, соответственно, указано для R1 и R2;
R4 представляет от 0 до 3 заместителей, выбранных из группы, включающей гидрокси, C1-6 алкил и C1-6 гидроксиалкил; при этом
каждый n является целым числом от 0 до 4;
и при этом соединение формулы I содержит по меньшей мере две гидроксигруппы.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы I по изобретению, включающий:
(i) активацию карбоксильных групп соединения формулы II пептидным реагентом

(ii) конденсацию указанного активированного соединения формулы II аминной составляющей заместителя -NR1R2 с получением указанного соединения формулы I, в котором R1 и R2 являются такими, как указано в п. 1.
В другом аспекте настоящего изобретения предложен способ получения соединения формулы II, указанного в настоящей заявке, включающий алкилирование соединения формулы III:
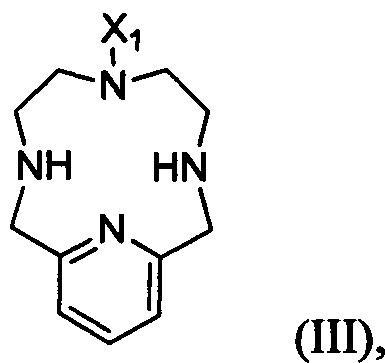
в котором X1 является метилом или -(СН2)3-СООН.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы I по изобретению совместно с биосовместимым носителем, в форме, приемлемой для введения млекопитающим.
В другом аспекте настоящего изобретения предложен способ, включающий:
(i) введение субъекту соединения формулы I по изобретению или фармацевтической композиции по изобретению;
(ii) регистрацию сигналов магнитного резонанса (MP), полученных от указанного субъекта или органов указанного субъекта, в которых распределено указанное соединение;
(iii) генерацию MP изображений и/или MP спектров на основе указанных зарегистрированных сигналов.
Соединения по настоящему изобретению продемонстрировали, что они обладают свойствами, которые указывают на возможность их применения в качестве контрастных агентов для МРТ.
Растворимость соединений по изобретению, определяемая как описано в Примере 13, демонстрирует, что они подходят для применения в качестве контрастных агентов для МРТ.
Измерения in vitro релаксивности для оценки эффективности соединений по изобретению (см. Пример 14) показали, что эти соединения способствуют увеличению и продольной, и поперечной скоростей релаксации (например, 1/T1 и 1/Т2, соответственно) молекул воды, координированных ионом металла.
Эксперименты для оценки кинетической инертности соединений по изобретению проводили в присутствии конкурирующих ионов металлов Cu2+ и Zn2+ в слабокислом растворе (см. Пример 15). Эти эксперименты показали, что соединения по настоящему изобретению обладают благоприятными свойствами по сравнению с уровнем техники.
Соединения по изобретению продемонстрировали улучшение кинетической инертности хелатных соединений на основе Mn (II) с более медленной диссоциацией по сравнению с соединением уровня техники.
Следовательно, в итоге, соединения по настоящему изобретению демонстрируют более выгодный баланс между эффективностью контрастного агента и улучшенной in vivo стабильностью, не демонстрируемую прежде хелатными соединениями Mn (II). Дополнительная in vivo стабильность является указанием на то, что соединения по изобретению могут оказаться привлекательной альтернативой соединениям Gd (III) в следующем поколении клинических контрастных агентов для медицинского МРТ.
Краткое описание чертежей
Фиг. 1 иллюстрирует кинетику диссоциации хелатов на основе Mn (II), протестированных методами из Примера 15.
Фиг. 2 и 3 показывают 1Н NMRD (англ. - Nuclear magnetic relaxation dispersion - дисперсия ядерно-магнитной релаксации) профили, записанные для хелатов на основе Mn (II), измеренные методом, описанным в Примере 14.
Фиг. 4 и 5 иллюстрируют результаты экспериментов переметаллирования, выполненных для хелатного соединения Mn уровня техники, как описано в Примере 15.
Фиг. 6 и 7 показывают сравнение результатов переметаллирования, полученных для соединения уровня техники и определенных соединений по изобретению, как описано в Примере 15.
Фиг. 8 показывает зависимость времени продольной релаксации (то есть, Т1) от времени для соединения по изобретению.
Фиг. 9 и 10 иллюстрируют превращение, вызванное конкуренцией Cu2+ с хелатными соединениями Mn по изобретению в сравнении с хелатным соединением Mn уровня техники.
Фиг. 11 иллюстрирует результаты, полученные для реакции переметаллирования с Zn2+ с хелатом Mn уровня техники, в сравнении с соединением по изобретению.
Подробное описание предпочтительных воплощений изобретения
Для более ясного и точного описания и понимания предмета предложенного изобретения ниже приведены определения и типичные воплощения для специфических понятий, используемых в описании и формуле изобретения настоящего изобретения. В данной заявке любой приведенный для иллюстрации пример специфического понятия следует воспринимать как неограничивающий пример.
Термины «содержащий» или «содержит» имеют в данной заявке свое обычное значение и предполагают, что агент или композиция должна иметь перечисленные существенные признаки или компоненты, но, кроме этого, могут присутствовать и другие. Термин «содержащий» в качестве предпочтительного подпонятия включает «по существу состоящий из», которое означает, что композиция имеет перечисленные компоненты, без присутствия других признаков или компонентов.
Термин «алкил», отдельно или в сочетании, обозначает алкильный радикал с прямой цепью или разветвленной цепью общей формулы CnH2n+1. Примеры таких радикалов включают метил, этил и изопропил.
Термин «гидроксил» относится к группе -ОН.
Термин «гидроксиалкил» относится к алкильной группе, указанной выше, содержащей гидроксильный заместитель, указанный выше.
Термин «арил» относится к функциональной группе или заместителю, содержащему ароматическое кольцо, обычно ароматическому углеводороду, примеры которого включают фенил и пиридил. В одном из воплощений арильные группы по настоящему изобретению являются ароматическими 6-членными кольцами, содержащими от 0 до 3 гетероатомов, выбранных из О, N и S.
Термин «галогено» или «галоген» обозначает заместитель, выбранный из фтора, хлора, брома или йода.
Термин «углеводный фрагмент» относится к производному альдегида или кетона многоатомного спирта и включает моносахаридные, дисахаридные и олигосахаридные остатки. Неограничивающие примеры включают фруктозные, глюкозные и сахарозные остатки.
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой С1-12 гидроксиалкил.
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой С3-6 гидроксиалкил.
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой С6 гидроксиалкил.
В одном из воплощений указанного соединения формулы I каждый R1 независимо выбран из группы, включающей:
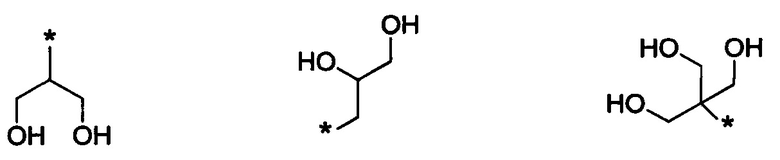

где в каждом случае звездочка обозначает место присоединения остатка соединения формулы I.
В одном из воплощений указанного соединения формулы I каждый R1 независимо выбран из группы, включающей:

где в каждом случае звездочка обозначает место присоединения остатка соединения формулы I.
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой С3-6 арил, замещенный одним или более заместителей, выбранных из галогена и -C(=O)-NH-C1-6 гидроксиалкила.
В одном из воплощений указанного соединения формулы I указанный С3-6 арил является фенилом.
В одном из воплощений указанного соединения формулы I указанный галоген является йодом.
В одном из воплощений указанного соединения формулы I -C(=O)-NH-C1-6 гидроксиалкил представляет собой -C(=O)-NH-CH2-C(OH)-CH2-C(OH).
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой:

где звездочка обозначает место присоединения остатка соединения формулы I.
В одном из воплощений указанного соединения формулы I каждый R1 представляет собой:
где звездочка обозначает место присоединения остатка соединения формулы I.
В одном из воплощений указанного соединения формулы I каждый R1 является одинаковым.
В одном из воплощений указанного соединения формулы I каждый R2 представляет собой С1-3 алкил.
В одном из воплощений указанного соединения формулы I каждый R2 представляет собой метил.
В одном из воплощений указанного соединения формулы I каждый R2 представляет собой водород.
В одном из воплощений указанного соединения формулы I каждый R2 представляет собой С1-20 гидроксиалкил.
В одном из воплощений указанного соединения формулы I каждый R2 представляет собой C1-6 гидроксиалкил. Когда каждый R2 представляет собой C1-6 гидроксиалкил, в одном из воплощений каждый R1 также представляет собой С1-6 гидроксиалкил, и в другом воплощении R2 и R1 являются одинаковыми.
В одном из воплощений указанного соединения формулы I каждый R2 является одинаковым.
В одном из воплощений указанного соединения формулы I каждый n является целым числом от 1 до 3.
В одном из воплощений указанного соединения формулы I n представляет собой 1.
В одном из воплощений указанного соединения формулы I n представляет собой 2.
В одном из воплощений указанного соединения формулы I каждый n представляет собой 3.
В одном из воплощений указанного соединения формулы I R3 представляет собой С1-3 алкил.
В одном из воплощений указанного соединения формулы I R3 представляет собой метил.
В одном из воплощений указанного соединения формулы I R3 представляет собой -(CH2)m-C(=O)-NR5R6, как указано в данной заявке.
В одном из воплощений указанного соединения формулы I R5 является таким, как указано для R1 в данной заявке.
В одном из воплощений указанного соединения формулы I R6 является таким, как указано для R2 в данной заявке.
В одном из воплощений указанного соединения формулы I m представляет собой 3.
В одном из воплощений указанного соединения формулы I n представляет собой 2.
В одном из воплощений указанного соединения формулы I R4 представляет О заместителей.
В одном из воплощений указанного соединения формулы I R4 представляет 1 или 2 гидроксигруппы.
В одном из воплощений указанного соединения формулы I R4 представляет 2 гидроксигруппы в мета-положениях пиридильного кольца.
В одном из воплощений указанное соединение формулы I содержит по меньшей мере 4 гидроксигруппы.
В одном из воплощений указанное соединение формулы I содержит от 4 до 15 гидроксигрупп.
В одном из воплощений указанное соединение формулы I содержит от 5 до 10 гидроксигрупп.
В одном из воплощений соединения по изобретению указанный Mn обогащен изотопом Mn, выбранным из группы, включающей 52Mn и 54Mn. В одном из воплощений указанный изотоп Mn представляет собой 54Mn.
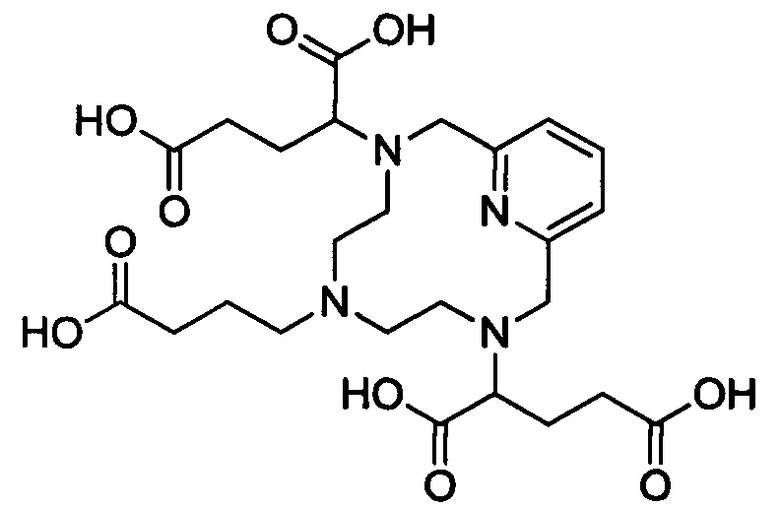
Неограничивающие примеры соединений формулы I представляют собой следующие соединения:





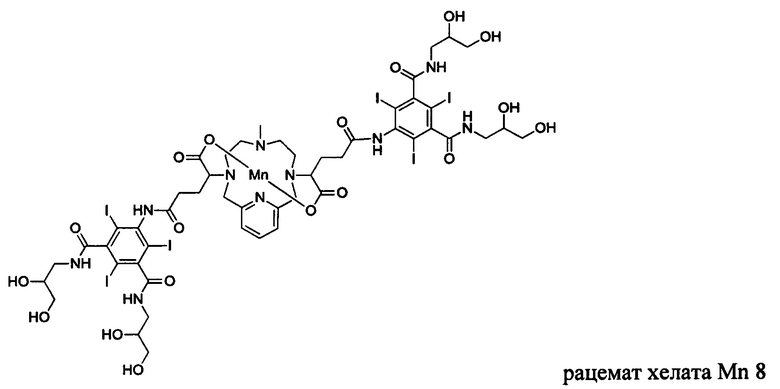
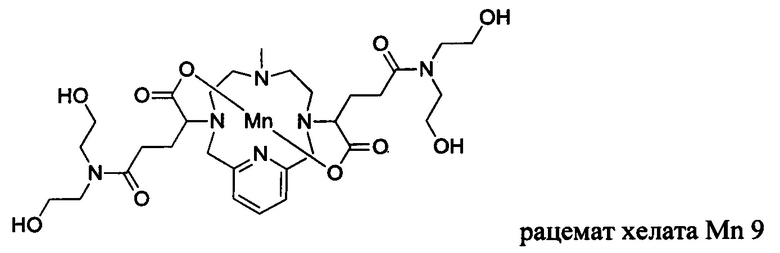
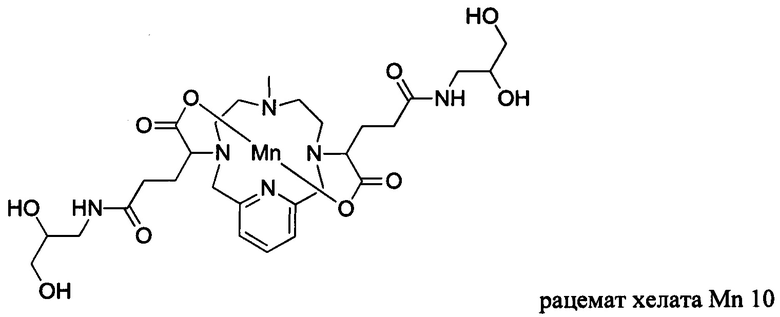

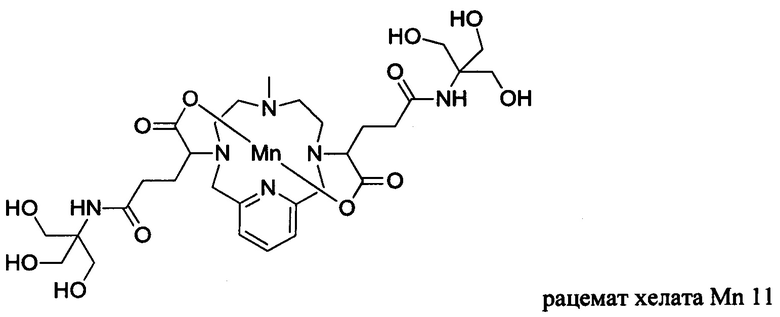
В соединениях формулы I атомы углерода, присоединенные к карбоксильным ответвлениям, являются стереоцентрами. Соединения формулы I по изобретению могут быть обеспечены в виде рацемической смеси или в виде энантиомерно обогащенной смеси, или рацемическую смесь можно разделить с помощью хорошо известных способов и отдельный энантиомер можно использовать отдельно. В одном из воплощений соединение формулы I является либо рацемической смесью, либо диастереомерно чистым. В одном из воплощений соединение формулы I является диастереомерно чистым.
Неограничивающие примеры диастереомерно чистых соединений формулы I представляют собой следующие соединения:



Гидрофильное получение производных с получением соединений по изобретению достигается через амидную связь с алифатическим линкером. Амид, который является некоординирующей присоединенной группой, находится слишком далеко от иона марганца и поэтому не будет координироваться. Точная длина некоординирующего линкера у R3 формулы I очень важна: если она слишком короткая (то есть если m=1 в формуле I), то есть риск того, что амидная группа координирует ион марганца, тем самым блокируя доступ молекуле воды, что значительно снижает релаксивность комплекса в целом. Длина некоординирующего линкера, присоединенного к карбоксиметильному ответвлению (координирдинационной группе), может быть короткой (то есть, если n=0 в формуле I), поскольку одна и та же «группа» не способна выполнять роль двух координационных групп (координационный угол будет слишком напряженным).
Соединения формулы I можно синтезировать несколькими методами синтеза, известными специалисту в данной области техники, из коммерчески доступных исходных материалов. Подходящие источники марганца для введения в хелатное соединение при получении соединений по настоящему изобретению включают соли карбоната (MnCO3), оксид (MnO), ацетат (Mn(ОАс)2), хлорид (MnCl2), гидроксид (Mn(ОН)2), оксалат (MnC2O4), формиат (Mn(НСО2)2) и нитрат (Mn(NO3)2. Следующие общие методики можно использовать и/или легко адаптировать для получения соединений формулы I:

Вкратце:
А: Тозилирование аминоэтанола дает азиридин (Carrillo, Arkivoc, 2007).
В: Азиридинирование аминобутановой кислоты (Каталог Sigma Aldrich 56-12-2). В одном из воплощений азиридинирование метиламина протекает в чистом ацетонитриле. В одном из воплощений для этой аминокислоты используют активацию амина основанием. Необязательно кислотная функциональная группа может быть защищена в виде сложного эфира.
С: Циклизация с 2,6-бис(хлорметил)пиридином (Каталог Sigma Aldrich 3099-28-3). В одном из воплощений эту стадию проводят в ацетонитриле с карбонатом калия в качестве основания.
D: Детозилирование при использовании, в одном из воплощений, концентрированной серной кислоты. В одном из воплощений эта стадия проходит количественно.
Е: Бромирование на основе способа, описанного в литературе (Henig, J.,  Engelmann, J., Gottschalk, S., & Mayer, ч. a. (2010). Inorganic Chemistry, 49(13), 6124-38).
Engelmann, J., Gottschalk, S., & Mayer, ч. a. (2010). Inorganic Chemistry, 49(13), 6124-38).
F: Алкилирование полиамина. В одном из воплощений эту стадию проводят в водном растворе. В другом воплощении, в котором вторичные галогениды реагируют медленно (первичные алкилгалогениды взаимодействуют хорошо), можно синтезировать бис-сложный эфир (Е) и перейти на органический растворитель для улучшения скорости реакции.
G: Комплексообразование при использовании MnCl2. Осаждение избытка Mn с помощью основания.
Н: Активация карбоксильных групп пептидными реагентами. В одном из воплощений эти реагент представляют собой EDCI и/или НОВТ (как описано в ЕР 2457914 В1). Конденсация с подходящим амином (например, меглумином).
Если соединение формулы I содержит в качестве R1 замещенный арил, такой как трийодированный фенил, соединения можно получить при использовании или адаптировании следующей схемы реакций:

Неограничивающий выбор соединений по изобретению был синтезирован, как описано ниже в Примерах 1-10 и соединение уровня техники было синтезировано, как описано в Примере 11. Эти соединения были охарактеризованы in vitro и/или in vivo, как описано в Примерах 12-15.
Подходящие способы для in vitro характеристики стабильности хелатов можно найти в литературе ( , J.-M. Journal of Magnetic Resonance Imaging: JMPT, 2009, 30(6), 1249-58 и Baranyai, Z. Chemistry - A European Journal, 2015, 21(12), 4789-4799). Другие подходящие способы включают in vitro исследования физиологических сред (то есть сыворотки или плазмы крови человека) для контроля инертности переметаллирования. Другой подходящий способ оценки инертности переметаллирования состоит в измерении удерживания ионов металлов in vivo после введения хелатированного металла. Известно, что неповрежденные хелаты обычно проявляют очень быструю кинетику клиренса.
, J.-M. Journal of Magnetic Resonance Imaging: JMPT, 2009, 30(6), 1249-58 и Baranyai, Z. Chemistry - A European Journal, 2015, 21(12), 4789-4799). Другие подходящие способы включают in vitro исследования физиологических сред (то есть сыворотки или плазмы крови человека) для контроля инертности переметаллирования. Другой подходящий способ оценки инертности переметаллирования состоит в измерении удерживания ионов металлов in vivo после введения хелатированного металла. Известно, что неповрежденные хелаты обычно проявляют очень быструю кинетику клиренса.
В одном аспекте по изобретению соединение формулы I обеспечивают в виде фармацевтической композиции.
«Фармацевтическая композиция» - это композиция, содержащая соединение по изобретению совместно с биосовместимым носителем в форме, подходящей для введения млекопитающему. «Биосовместимый носитель» - это текучая среда, предпочтительно жидкость, в которой соединение формулы I суспендируют или растворяют, так что полученная композиция является физиологически переносимой, то есть может вводить в тело млекопитающего без токсического эффекта или неприятного дискомфорта (что можно понимать как определение термина «подходящая для введения млекопитающему»).
Фармацевтическая композиция по изобретению подходит для применения в качестве контрастной среды для магнитного резонанса (MP) в магнитно-резонансной томографии (МРТ) организма человека и не относящегося к человеку животного.
В одном из воплощений фармацевтическая композиция по изобретению может содержать один или более фармацевтически приемлемых эксципиентов. Они предпочтительно не влияют на изготовление, хранение или применение конечной композиции.
Неограничивающие примеры подходящих фармацевтически приемлемых эксципиентов включают буферные агенты, стабилизаторы, антиоксиданты, регулирующие осмоляльность агенты, регулирующие рН агенты, дополнительный хелатирующий агент и нестабильные комплексы физиологически приемлемых ионов. Эти и другие подходящие эксципиенты хорошо известны специалистам в данной области техники и более подробно описаны, например, в WO 1990003804, ЕР 0463644-А, ЕР 0258616-А и US 5876695, содержание которых включено в данную заявку посредством ссылки. Фармацевтическая композиция по изобретению в одном из воплощений находится в форме, подходящей для парентерального введения, например путем инъекции. Фармацевтическая композиция по изобретению может, следовательно, быть составлена с помощью физиологически приемлемых эксципиентов для введения способом, полностью известным в данной области техники. Например, соединение формулы I необязательно при добавлении фармацевтически приемлемых эксципиентов, можно суспендировать или растворить в водной среде с получением раствора или суспензии, которую затем стерилизуют.
Неограничивающим примером подходящего буферного агента является гидрохлорид трометамина.
Термин "дополнительный хелатирующий агент" обозначав любое соединение, способное захватывать свободный парамагнитный ион (марганца), но не парамагнитный ион (марганца), удерживаемый внутри комплексов по настоящему изобретению, как описано в ЕР2988756А1. Хотя небольшие количества являются существенными для здоровья человека, избыточное воздействие свободных ионов марганца может приводить к нейродегенеративному расстройству, известному как "манганизм", которое имеет симптомы, напоминающие болезнь Паркинсона. Тем не менее, фундаментальная проблема, связанная с Mn, равно как и с другими металлами, в качестве контрастных агентов состоит в стабильности их хелатирования. Стабильность хелатирования является важным свойством, которое отражает потенциальное высвобождение свободных ионов металла in vivo. Известно, что существует корреляция между количеством дополнительного хелатирующего агента в композиции с парамагнитным хелатным соединением и количеством парамагнитного металла, распределенного в животных моделях (Sieber 2008 J Mag Res Imaging; 27(5): 955-62). Следовательно, в другом воплощении, количество дополнительного хелатирующего агента выбирают так, что он может выступать как поглотитель Mn для снижения или предотвращения высвобождения Mn из композиции после инъекции. Оптимальное количество свободного хелатирующего агента даст в результате фармацевтическую композицию, имеющую подходящие физико-химические свойства (то есть вязкость, растворимость и осмоляльность) и не имеющую токсикологических эффектов, таких как дефицит цинка в случае слишком большого количества хелатирующего агента. В US 5876695 описан, в частности, избыток линейного хелатирующего агента, в частности свободного DTP А, и этот неограничивающий пример дополнительного хелатирующего агента, подходящего для применения в фармацевтической композиции по настоящему изобретению. Этот принцип составления композиций применяют для таких продуктов, как Magnevist, Vasovist или Primovist. В WO 2009103744 описан аналогичный принцип составления композиций на основе добавления точного количества свободного хелатирующего агента так, чтобы иметь очень небольшой избыток указанного хелатного соединения и нулевую концентрацию свободного лантанида.
Физиологически приемлемый ион может в одном из воплощений быть выбран из физиологически приемлемых ионов, включая соли кальция и натрия, такие как хлорид кальция, аскорбат кальция, глюконат кальция или лактат кальция.
Парентерально вводимые формы должны быть стерильными и свободными от физиологически неприемлемых агентов и должны иметь низкую осмоляльность для сведения к минимуму раздражения или других негативных эффектов при введении и, следовательно, фармацевтическая композиция должна быть изотоничной или слабо гипертонической. Неограничивающие примеры подходящих носителей включают водные носители, при необходимости используемые для введения парентеральных растворов, такие как физиологический раствор для инъекций, физиологический раствор Рингера для инъекций, раствор декстрозы для инъекций, раствор декстрозы и хлорида натрия для инъекций, лактидный раствор Рингера для инъекций и другие растворы, такие как описаны в Фармацевтическом научном справочнике Ремингтона [Remington's Pharmaceutical Sciences], 22ое издание (2006 Lippincott Williams & Wilkins) и в Дополнении к Фмериканской фармакопее [The National Formulary 20] (https:/books.qooqle.com/books?id=03qixPEMwssC&q=THE+NATIONAL+FORMULARY&da=THE+NATIONAL+FORMULARY&hl=en&sa=X&ved=OCC8Q6AEwAGoVChMImfPHrdTavAIVJfNvChl RJw E).
В случае фармацевтической композиции по изобретению, предназначенной для парентерального введения, то есть путем инъекции содержащего ее препарата, дополнительно включает стадии, включающие удаление органического растворителя, добавление биосовместимого буфера и любых других необязательных ингредиентов, таких как эксципиенты или буферные агенты. В случае парентерального введения, стадии, гарантирующие, что фармацевтическая композиция является стерильной и апирогенной, тоже должны быть предприняты.
В другом воплощении, настоящее изобретение обеспечивает способ, включающий введение соединения формулы I, указанного в данной заявке, при генерировании MP изображений и/или MP спектров.
Способы введения и пациенты, рассматриваемые как подходящие в контексте настоящего изобретения, были описаны выше в данной заявке, в связи с фармацевтической композицией. Введение соединения формулы I предпочтительно проводят парентерально и наиболее предпочтительно внутривенно. Внутривенный путь введения представляет наиболее эффективный путь для доставки соединения в организм пациента. Кроме того, внутривенное введение не является существенным физиологическим вмешательством и не связано с существенным риском для здоровья. Соединение формулы I по изобретению предпочтительно вводят в виде фармацевтической композиции по изобретению, как указано выше. Способ по изобретению можно также рассматривать как включающий стадии (ii)-(iii), проводимые на субъекте, которому предварительно вводят соединение по изобретению. В одном из воплощений фармацевтическую композицию вводят в количестве, подходящем для улучшения контраста в способе MP визуализации (МРТ). Для более подробного описания способов МРТ читатель может обратиться к общим знаниям в данной области техники, например, как указано в Главе 27 "Контрастные агенты и магнитно-резонансная томография" ["Contrast Agents and Magnetic Resonance Imaging"] в книге "Магнитно-резонансная томография: физические и биологические принципы" ["Magnetic Resonance Imaging: Physical and Biological Principles"] (4oe издание 2015 Elsevier, Stewart Carlyle Bushong & Geoffrey Clarke, Eds.) или в "Контрастные агенты I: магнитно-резонансная томография " ["Contrast Agents I: Magnetic Resonance Imaging"] (2002 Springer-Verlang, Werner Krause, Ed.).
Способ по изобретению можно использовать для изучения биологического маркера или процесса в здоровых субъектах, или альтернативно у субъектов, для которых известно или предполагается патологическое состояние, связанное с аномальной экспрессией биологического маркера. Когда способ используют для визуализации пациента, для которого известно или предполагается патологическое состояние, он имеет преимущество в способе для диагностики указанного состояния.
Стадия способа по изобретению «детектирование» включает детектирование сигналов, испускаемых соединением формулы I, с помощью детектора, чувствительного к указанным сигналам. Стадию детектирования можно также рассматривать как накопление данных по сигналам.
Стадию способа по изобретению «генерирование» проводят с помощью компьютера, который применяет алгоритм реконструкции к накопленным данным сигналов с получением набора данных. Затем этот набор данных подвергают обработке для генерирования одного или более изображений и/или одного или более спектров, показывающих положение и/или количество сигналов.
"Субъектом" по изобретению может быть любой человек или животное. В одном из воплощений субъектом по изобретению является млекопитающее. В одном из воплощений указанный субъект представляет собой in vivo организм млекопитающего, не получающего лечения. В другом воплощении субъект по изобретению является человеком.
В представленном описании использованы примеры для раскрытия настоящего изобретения, в том числе наилучший вариант осуществления изобретения, и обеспечения специалисту в данной области техники знакомства с практической реализацией изобретения, включая получение и применение любых устройств или систем и осуществление любого из включенных способов. Объем притязаний по изобретению определяется формулой изобретения и может включать другие примеры, которые встречаются специалистам в данной области техники. Предполагается, что такого рода другие примеры входят в объем формулы изобретения, если они имеют структурные элементы, которые не отличаются от буквальной терминологии, используемой в формуле изобретения, или если они включают эквивалентные структурные элементы, которые несущественно отличаются от буквальной терминологии, используемой в формуле изобретения. Все патенты и патентные публикации, упомянутые в тексте, во всей полноте включены в данную заявку посредством ссылки, как если бы они были включены по отдельности.
Краткое описание примеров
В Примере 1 описан синтез хелата Mn 3.
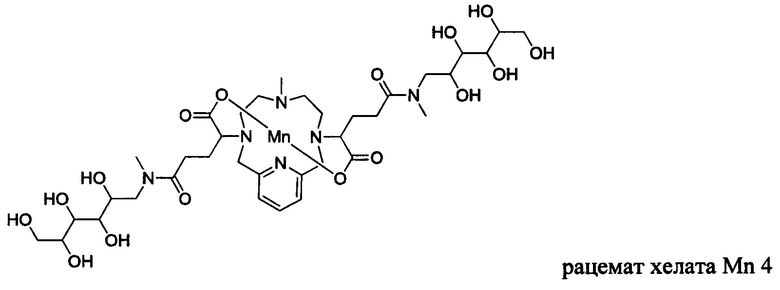
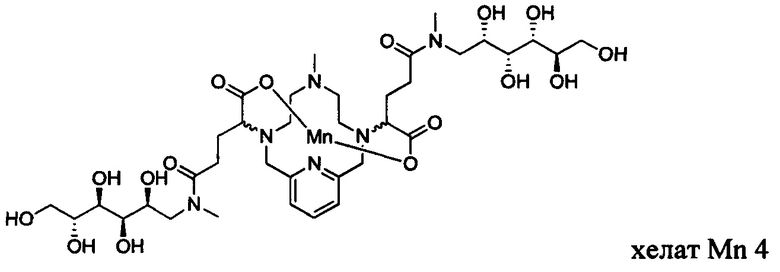
В Примере 2 описан синтез хелата Mn 4.
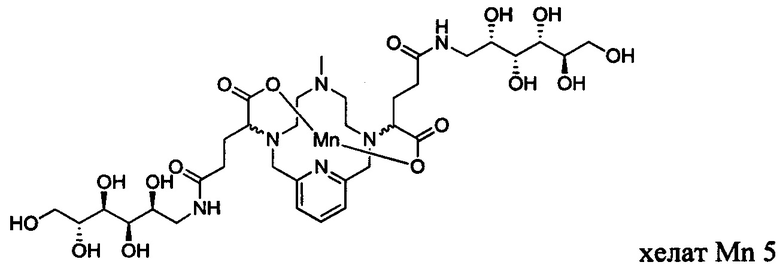
В Примере 3 описан синтез хелата Mn 5.
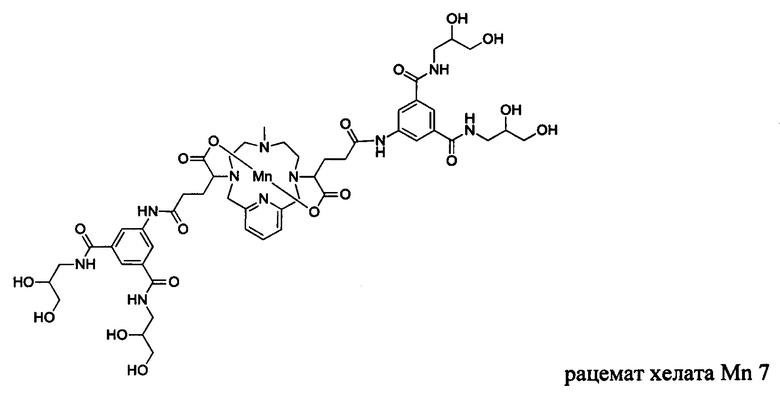
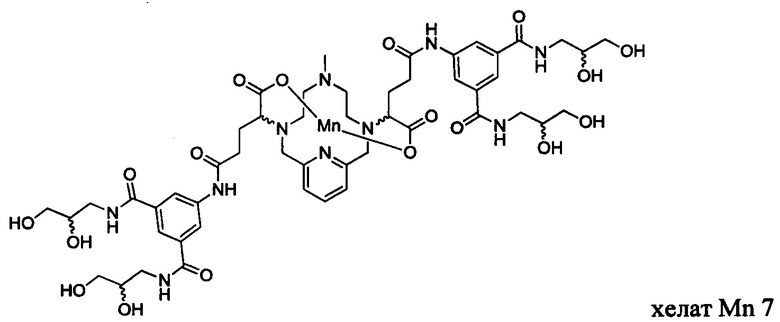
В Примере 4 описан синтез хелата Mn 7.
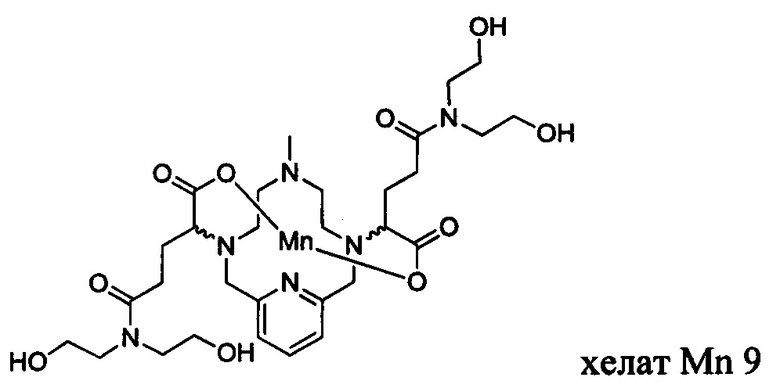
В Примере 5 описан синтез хелата Mn 9.
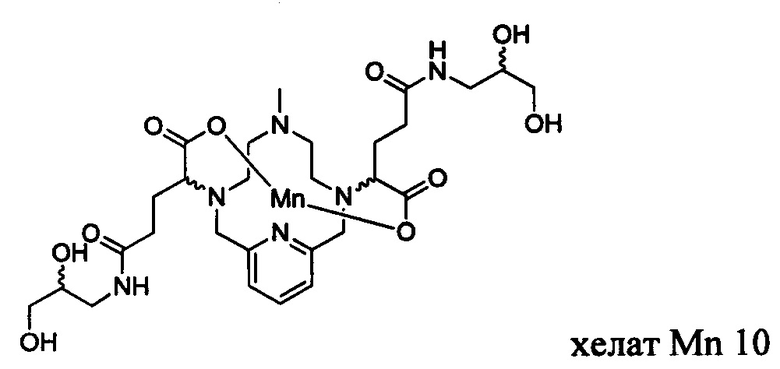
В Примере 6 описан синтез хелата Mn 10.
В Примере 7 описан синтез хелата Mn 11.
В Примере 8 описан синтез хелата Mn 5а.
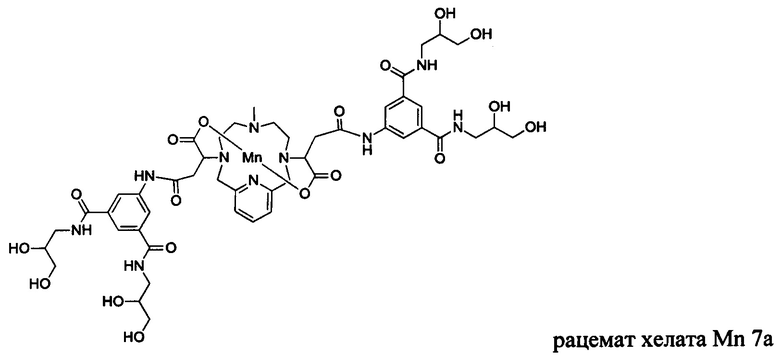
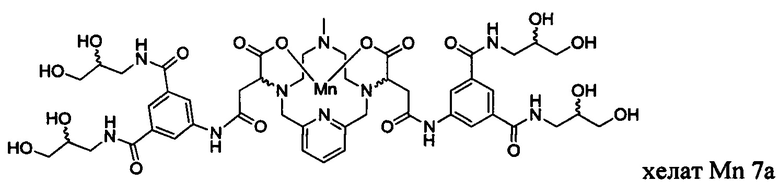
В Примере 9 описан синтез хелата Mn 7а.
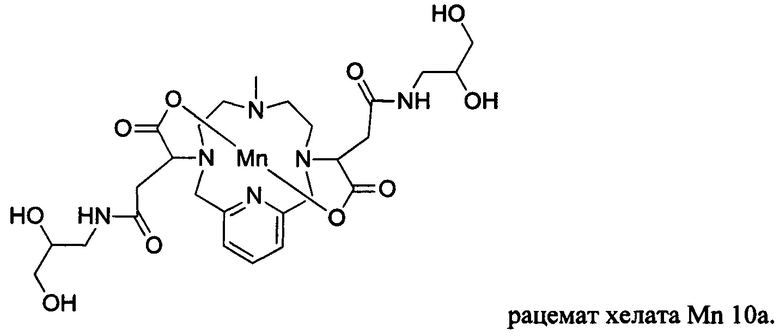
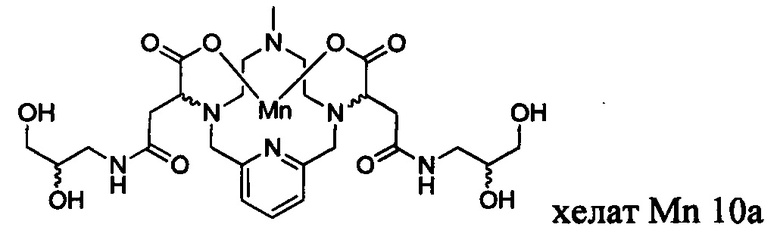
В Примере 10 описан синтез хелата Mn 10а.
В Примере 11 описан синтез хелата Mn уровня техники.
В Примере 12 описано in vivo исследование биораспределения 54Mn на крысах.
В Примере 13 описана характеристика растворимости соединений по изобретению в воде.
В Примере 14 описаны in vitro профили протонной релаксивности и дисперсии ядерной магнитной релаксации (NMRD) для соединений по изобретению.
В Примере 15 описаны эксперименты, проводимые для определения кинетики диссоциации соединений по изобретению.
Список сокращений, используемых в примерах
AcN ацетонитрил
ДМСО диметилсульфоксид
EDCI 1-этил-3-(3-диметиламинопропил)карбодиимид
EtOAc этилацетат
EtOH этанол
ч час(ы)
HOBt гидроксибензотриазол
МеОН метанол
ЯМР ядерный магнитный резонанс
NMRD дисперсия ядерной магнитной релаксации
Примеры
Пример I: Синтез хелата Mn 3
Пример 1(i): Синтез 4-метилбензолсулфоната 4-(бензилокси)-4-оксобутан-1-аммония

2л-овую 3-горлую колбу снабжали механической мешалкой, ловушкой Дина-Старка, обратным холодильником и впускным отверстием для азота. В колбу загружали 4-аминобутановую кислоту (41,522 г, 0,403 моль), n-толуолсульфоновую кислоту (91,912 г, 0,048 моль) и бензиловый спирт (201 мл). Полученный мутный раствор нагревали при кипении с обратным холодильником в течение 14 ч. В конце периода кипения в горячий реакционный раствор добавляли н-гептан (175 мл). Реакционной смеси давали охладиться до температуры окружающего воздуха. Полученные белые кристаллы выделяли с помощью фильтрования под вакуумом и перекристаллизовывали из смеси этилацетат/н-гептан 6:1 с получением 124,2 г (выход 84%) целевого продукта в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3, δ) 7,71 (5Н, д), 7,31 (5Н, м), 7,13 (2Н, д), 5,02 (2Н, с), 2,87 (2Н, м), 2,32 (5Н, м), 1,85 (2Н, м).
Пример 1(ii): Синтез бензил-4-(бис(2-(4-метилфенилсулъфонамидо)этил)-амино)бутаноата

В 2л-овый реактор с кожухом, снабженный 4-лопастной мешалкой в форме якоря, обратным холодильником и впускным отверстием для азота, загружали N-тозилазиридин (107,7 г, 0,546 моль) и безводный ацетонитрил (870 мл). Затем добавляли 4-метилбензолсулфонат 4-(бензилокси)-4-оксобутан-1-аммония (100 г, 0,274 моль) и безводный ацетонитрил (500 мл) с получением грязно-белой суспензии. Добавляли диизопропиламин (47,6 мл, 0,274 моль) и реакционную смесь перемешивали при 40°С в течение 16 ч. Затем реакционную смесь охлаждали до 22,5°С и перемешивали еще в течение 49 ч. Мутную белую суспензию отфильтровывали под вакуумом и прозрачный желтый фильтрат упаривали досуха. Неочищенный материал очищали методом хроматографии на силикагеле (от 50% гексанов в EtOAc до 10% гексанов в EtOAc; оба элюента содержали 1% триэтиламина) с получением 89,7 г (54%) целевого продукта в виде бесцветного масла. 1Н ЯМР (400 МГц, CD2Cl2, δ) 7,74 (4Н, д), 7,35 (9Н, м), 5,13 (2Н, м) 5,10 (2Н, с), 2,85 (4Н, м), 2,41 (10Н, м), 2,23 (4Н, м), 1,60 (2Н, м).
Пример 1(iii): Синтез защищенного циклического хелата с 3 ответвлениями
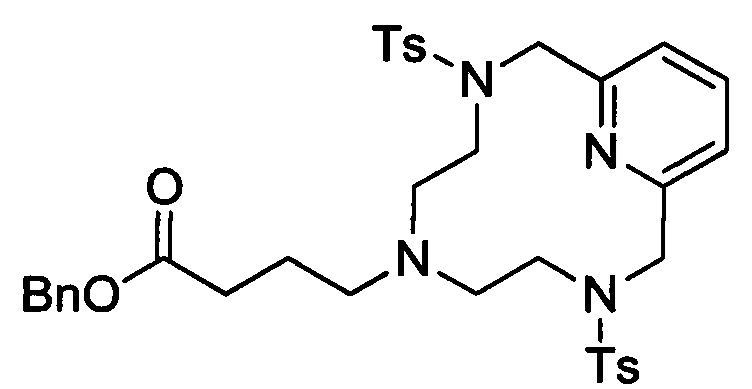
В 3л-овую 3-горлую круглодонную колбу, снабженную механической мешалкой, обратным холодильником и впускным отверстием для азота, загружали 8 стеклянных шариков диаметром 6 мм, бензил-4-(бис(2-(4-метилфенилсульфонамидо)этил)амино)-бутаноат (143,9 г, 0,245 моль) и 2,6-бис(хлорметил)пиридин (43,1 г, 0,245 ммоль). Добавляли безводный ацетонитрил (1,632 л), а затем безводный карбонат калия (135,5 г, 0,980 моль) и полученный раствор нагревали при 80°С в течение 47 ч. Затем полученную суспензию охлаждали до температуры окружающей среды и перемешивали еще в течение 65 ч. Добавляли безводный карбонат калия (67,0 г, 0,485 g) и реакционную смесь перемешивали при температуре окружающей среды в течение 27 ч. Нерастворимый карбонат калия удаляли с помощью фильтрования под вакуумом и прозрачный оранжевый фильтрат упаривали досуха. Неочищенный материал очищали методом хроматографии на силикагеле (от 100% CH2Cl2 до 10% МеОН в CH2Cl2; оба элюента содержали 1% триэтиламина) в получением 70,9 г (42%) целевого продукта в виде белой пены. 1H ЯМР (400 МГц, CD2Cl2, δ) 7,72 (5Н, м), 7,34 (9Н, м), 7,26 (2Н, д), 5,09 (2Н, с), 4,31 (4Н, с), 3,07 (4Н, м), 2,43 (6Н, с), 2,28 (8Н, м, 1,61 (2Н, м).
Пример 1(iv):
Синтез незащищенного циклического хелата с 3 ответвлениями
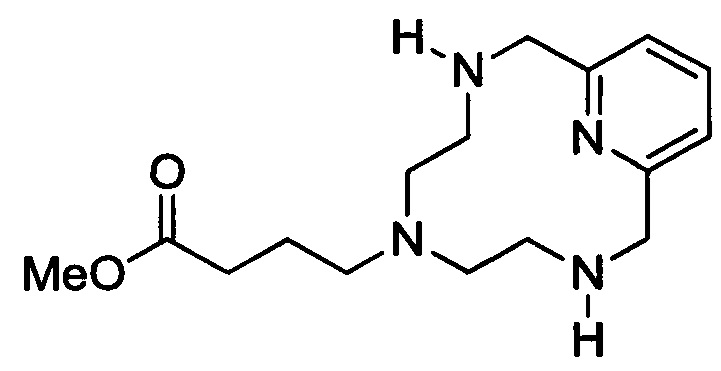
В 3л-овую 3-горлую круглодонную колбу, снабженную механической мешалкой, обратным холодильником и заглушкой, загружали защищенный циклический хелат с 3 ответвлениями (70,5 г, 92,3 ммоль) и концентрированную H2SO4 (282 мл) и нагревали при 100° в течение 19 ч. Полученный черный раствор охлаждали до температуры окружающей среды и доводили рН до 7 50%-ным масс. NaOH в воде. Добавляли МеОН (1 л) и твердые вещества удаляли путем фильтрования под вакуумом. Фильтрат концентрировали досуха, оставляя черный остаток, который перетирали с МеОН (500 мл) при 60°С в течение 1 ч. Нерастворимый материал удаляли методом фильтрования под вакуумом и фильтрат концентрировали досуха. Полученное тягучее окрашенное полутвердое вещество растворяли в МеОН (1 л) и рН доводили до примерно 1 концентрированной H2SO4 и перемешивали при температуре окружающей среды в течение 18 ч. Раствор затем нагревали до 60°С при перемешивании в течение 25 ч. Нерастворимый материал удаляли методом фильтрования под вакуумом и рН фильтрата доводили до 7 карбонатом калия. Нерастворенный карбонат калия удаляли методом фильтрования под вакуумом и фильтрат упаривали досуха. Полученное грязно-белое твердое вещество перетирали в безводном ацетонитриле (1 л) и нерастворимый материал удаляли методом фильтрования под вакуумом. Фильтрат упаривали досуха с получением 25,3 г (89,6%) целевого продукта (ESI: m/z=306 (М+Н+)) в виде белого твердого вещества.
Пример 1(v):
Синтез защищенного хелата Mn с 3 ответвлениями С5
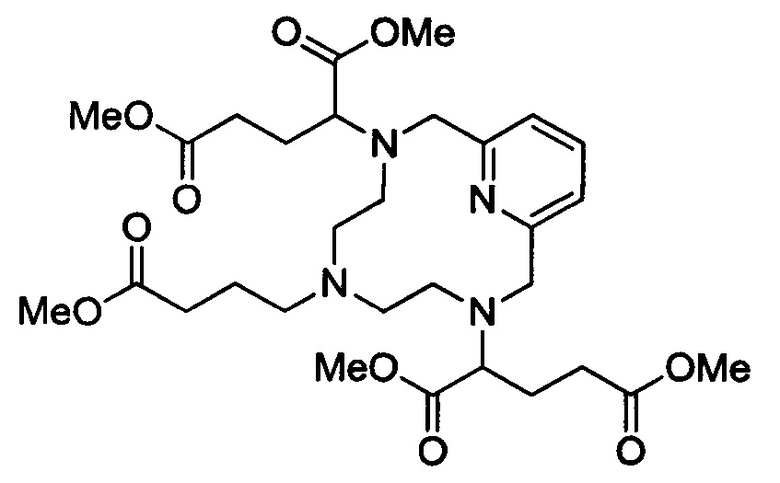
В 100 мл-овую круглодонную колбу, снабженную магнитной мешалкой и обратным холодильником, загружали незащищенный циклический хелат с 3 ответвлениями (2,776 г, 9,06 ммоль) и безводный ацетонитрил (60,4 мл). Затем добавляли триэтиламин (3,16 мл, 22,7 ммоль), а затем диметил-2-бромпентандиоат (4,982 г, 20,8 ммоль) и полученный раствор нагревали при 65°С в течение 20 ч. Добавляли вторую порцию диметил-2-бромпентандиоата (1,36 г, 5,7 ммоль) и продолжали нагревание в течение еще 23 ч. Растворитель удаляли под вакуумом и неочищенный материал очищали на C18 силикагеле (30% ацетонитрила в воде) с получением 2,883 г (51%) целевого продукта (ESI: m/z=623 (М+Н+)) в виде желтого масла.
Пример 1(vi): Синтез незащищенного хелата Mn с 3 ответвлениями С5
В 500 мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали защищенный хелат Mn с 3 ответвлениями С5 (14,010 г, 22,5 ммоль), растворенный в воде (225 мл), и добавляли 12,5 М NaOH (18,0 мл). Полученный раствор перемешивали при температуре окружающей среды в течение 18 ч. Затем рН доводили до 6 конц. HCl и удаляли растворитель под вакуумом. Неочищенный остаток очищали на C18 силикагеле (от 100% воды до 15% AcN в воде) с получением 12,43 г (100%) целевого продукта (ESI: m/z=553 (M+H+)) в виде желтого масла.
Пример 1(vii): Синтез хелата Mn с 3 ответвлениями С5
В 1 л-овую 3-горлую круглодонную колбу, снабженную магнитной мешалкой, загружали незащищенный хелат Mn с 3 ответвлениями С5 (12,43 г, 22,5 ммоль), тетрагидрат хлорида марганца (8,90 г, 45,1 ммоль) и воду (405 мл). Полученный раствор перемешивали при температуре окружающей среды в течение 12,5 ч. Затем рН доводили до 6 и реакционную смесь нагревали при 75°С в течение 7 ч. Раствор охлаждали до температуры окружающей среды и рН доводили до 8 насыщенным водным карбонатом натрия. Полученный белый осадок удаляли методом фильтрования под вакуумом и фильтрат упаривали досуха под вакуумом. Неочищенный остаток очищали на С18 силикагеле (100% воды) с получением 13,33 г (98%) целевого продукта (ESI: m/z=606 (М+Н+)) в виде желтого твердого вещества.
Пример 1(viii): Синтез хелата Mn 3

В 100 мл-овую 3-горлую колбу, снабженную магнитной мешалкой, загружали хелат Mn с 3 ответвлениями С5 (1,54 г, 2,5 ммоль) и разбавляли водой (26,8 мл). Добавляли N-метил-D-глюкамин (1,54 г, 7,9 ммоль), а затем EDCI-HCl (1, 64 г, 8,6 ммоль) и рН доводили до 6,4 1,0 М-ной HCl. Добавляли гидрат HOBt (0,140 г, 1,0 ммоль) и рН поддерживали при 6, пока перемешивали при температуре окружающей среды в течение 18 ч. Добавляли N-метил-D-глюкамин (0,77 г, 3,9 ммоль) и EDCI-HCl (0,82 г, 4,3 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 8 ч. Добавляли EDCI-HCl (0,42 г, 2,2 ммоль) и перемешивали при температуре окружающей среды в течение 17 ч. Затем весь растворитель удаляли под вакуумом, оставляя коричневой масло, которое очищали на C18 силикагеле (от 100% воды до 30% AcN в воде) с получением 1,6 г (56%) целевого продукта (ESI: m/z=1138 (М+Н+)).
Пример 2: Синтез хелата Mn 4
Пример 2(i): Синтез N,N'-((метилазандиил)бис(этан-2,1-диил))бис(4-метилбензолсульфонамида)
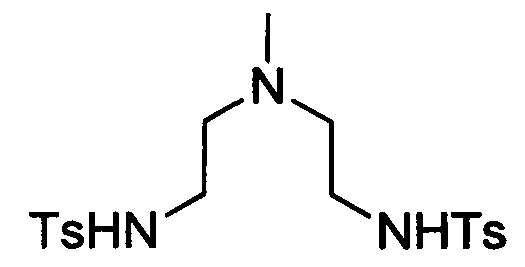
В 1 л-овую круглодонную колбу, снабженную магнитной мешалкой, загружали N-тозилазиридин (49 г, 248 ммоль) и AcN (450 мл). Добавляли 41%-ный водный метиламин (12 мл, 121 ммоль) и перемешивали при температуре окружающей среды в течение 36 ч. Добавляли вторую аликвоту N-тозилазиридина (1,7 г, 8,62 ммоль) и перемешивали при температуре окружающей среды в течение еще 48 ч. Растворитель удаляли под вакуумом и неочищенный остаток перекристаллизовывали из EtOH с получением 45 г (87%) целевого продукта в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6, δ) 7,68 (4Н, м), 7,36 (6Н, м), 2,75 (4Н, т), 2,38 (6Н, с), 2,22 (4Н, т), 1,93 (3Н, с).
Пример 2(ii): Синтез защищенного циклического хелата с 2 ответвлениями

В 12 л-овую 3-горлую круглодонную колбу, снабженную обратным холодильником и механической мешалкой, загружали N,N'-((метилазандиил)бис(этан-2,1-диил))бис(4-метилбензолсульфонамид) (93 г, 218,5 ммоль) и AcN (8,3 L). Добавляли 2,6-бис(хлорметил)пиридин (38,5 г, 218,5 ммоль) и полученный раствор нагревали при 80° в течение 16 ч. Реакционную смесь охлаждали до температуры окружающей среды и растворитель удаляли под вакуумом до момента начала кристаллизации. Полученные кристаллы собирали путем фильтрования под вакуумом с получением 86,9 г (75%) целевого продукта в виде белого твердого вещества (ESI: m/z=530 (М+Н+)).
Пример 2(iii): Синтез незащищенного циклического хелата с 2 ответвлениями
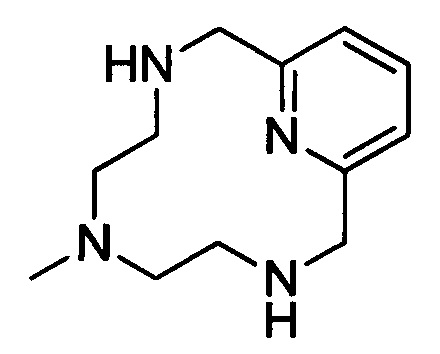
В 1 л-овую 3-горлую круглодонную колбу, снабженную механической мешалкой, загружали защищенный циклический хелат с 2 ответвлениями (150 г, 284 ммоль) и концентрированную серную кислоту (250 мл, 4,69 моль) и нагревали при 100°С в течение 15 ч. Раствор выливали на лед и рН доводили до 7,4 путем добавления 50%-ного масс. NaOH в воде, получая белое твердое вещество. Добавляли AcN (200 мл) и белое твердое вещество удаляли методом фильтрования под вакуумом. Фильтрат упаривали досуха с получением коричневой пены. Пену растворяли в воде (200 мл) и очищали на смоле Amberlite А26 в гидроксидной форме с получением 61 г (98%) целевого продукта в виде твердого вещества цвета дубовой коры. 1Н ЯМР (400 МГц, CD3CN, δ) 7,56 (1Н, м), 7,03 (2Н, м), 3,76 (4Н, с), 2,47 (4Н, м), 2,19 (3Н, с), 1,95 (4Н, с).
Пример 2(iv): Синтез защищенного хелата Mn с 2 ответвлениями С5
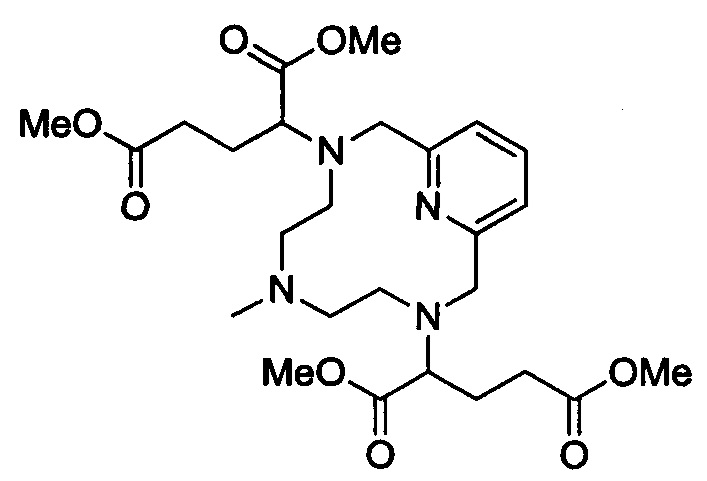
В 500 мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали незащищенный циклический хелат с 2 ответвлениями (20,0 г, 90,8 ммоль; полученный согласно Примеру 2(iii)) и AcN (160 мл). Добавляли диизопропилэтиламин (38,7 мл, 217 ммоль) и диметил-2-бромпентандиоат (47,7 г, 199,7 ммоль) и полученный раствор перемешивали при 65°С в течение 20 ч. Добавляли диизопропилэтиламин (9,75 мл, 54,6 ммоль) и диметил-2-бромпентандиоат (11,8 г, 49,4 ммоль) и полученный раствор перемешивали при 65°С в течение еще 19 ч. Растворитель удаляли под вакуумом, получая в остатке красное масло. Затем масло растворяли в воде (300 мл) и промывали EtOAc (300 мл). Затем слой EtOAc экстрагировали водой (2×50 мл) и объединяли с исходным водным слоем, удаляли воду под вакуумом, полученное в остатке красное масло применяли без дальнейшей очистки.
Пример 2(v): Синтез хелата Mn с 2 ответвлениями С5
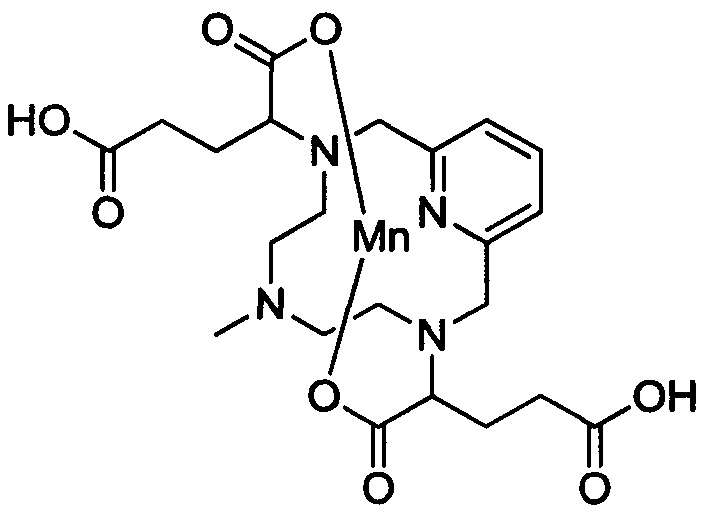
В 1 л-овую круглодонную колбу, снабженную магнитной мешалкой, загружали защищенный хелат Mn с 2 ответвлениями С5 (48,7 г, 90,8 ммоль) и воду (450 мл). Добавляли гидроксид натрия (29,1 г, 726 ммоль) и перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь промывали EtOAc (250 мл) и слои разделяли. Водный слой снова промывали EtOAc (2×100 мл) и водный слой собирали. В водный раствор добавляли тетрагидрат хлорида марганца (19,6 г, 99 ммоль). рН доводили до 7,1 6М-ным NaOH и перемешивали при температуре окружающей среды в течение 17 ч и затем при 90°С в течение 2,5 ч. После охлаждения до температуры окружающего воздуха, рН доводили до 10,1 50%-ным масс, водн NaOH и получали мелкий коричневый осадок. Осадок удаляли путем центрифугирования при 3000 об/мин в течение 20 мин и надосадочную жидкость собирали и упаривали досуха под вакуумом. Остаток растирали с МеОН (127 мл) при 40°С в течение 1,5 ч. Нерастворимое белое твердое вещество удаляли путем центрифугирования при 3000 об/мин в течение 30 мин. Надосадочную жидкость упаривали досуха под вакуумом с получением грязно-белого твердого вещества, которое очищали на C18 силикагеле (3% AcN в воде) с получением 36,8 г (75%) целевого продукта в виде грязно-белого твердого вещества (ESI: m/z=534 (М+Н+)).
Пример 2(vi): Синтез хелата Mn 4
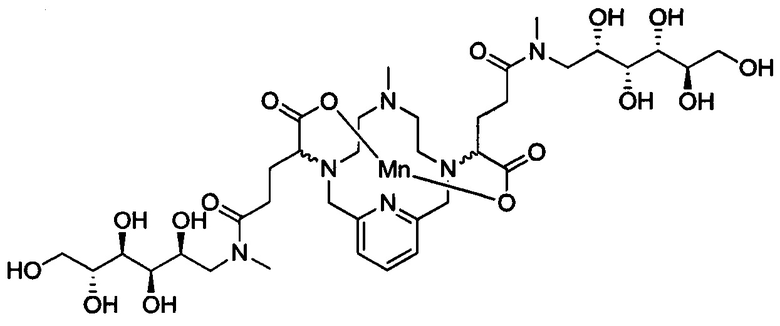
В 250 мл-овую 3-горлую круглодонную колбу, снабженную магнитной мешалкой, загружали хелат Mn с 2 ответвлениями С5 (4,40 г, 7,27 ммоль) и воду (76,5 мл). Добавляли N-метил-D-глюкамин (2,98 г, 15,3 ммоль), а затем EDCI-HCl (3,30 г, 17,2 ммоль) и HOBt гидрат (0,20 г, 1,47 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 7 ч. Добавляли EDCI-HCl (1,62 г, 8,45 ммоль) и рН поддерживали при 6, пока перемешивали в течение 20 ч при температуре окружающей среды. Добавляли N-метил-D-глюкамин (0,75 г, 3,84 ммоль) и EDCI-HCl (0,83 г, 4,32 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 3 дней. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на С18 силикагеле (от 100% воды до 20% AcN в воде) с получением 3,66 г (57%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=888 (М+Н+)).
Пример 3: Синтез хелата Mn 5
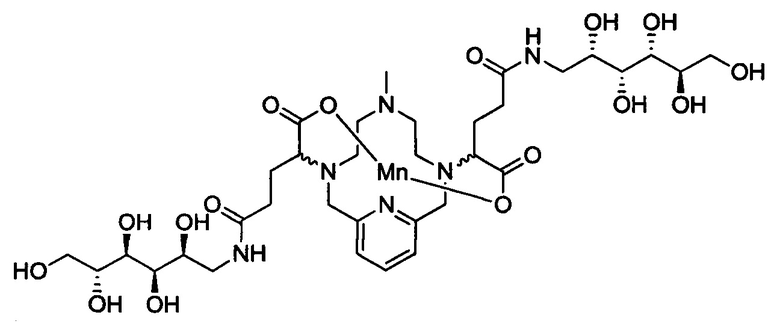
В 50 мл-овую 2-горлую колбу, снабженную магнитной мешалкой, загружали D-глюкамин (0,713 г, 3,94 ммоль) и воду (19,7 мл). рН полученного раствора доводили до 7,4 1,0 М-ной HCl и добавляли хелат Mn с 2 ответвлениями С5 (1,00 г, 1,87 ммоль; полученный согласно Примеру 2(v)), а затем EDCI-HCl (0,848 г, 4,42 ммоль) и гидрат HOBt (0,121 г, 0,787 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 8 ч. Добавляли D-глкжамин (0,359 г, 1,98 ммоль) и EDCI-HCl (0,433 г, 2,26 ммоль)) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 16 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на Cu силикагеле (от 100% воды до 20% AcN в воде) с получением 0,782 g (48%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=860 (М+Н+)).
Пример 4: Синтез хелата Mn 7

В 2-горлую колбу на 50 мл, снабженную магнитной мешалкой, загружали гидрохлорид 5-амино-N,N'-бис(2,3-дигидроксипропил)изофталамида (1,432 г, 3,94 ммоль) и воду (19,7 мл). рН полученного раствора доводили до 6 и добавляли хелат Mn с 2 ответвлениями С5 (1,005 г, 1,88 ммоль; полученный согласно Примеру 2(v)), а затем EDCI-HCl (0,858 г, 4,48 ммоль) и HOBt гидрат (0,108 г, 0,799 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 4,5 ч. добавляли EDCI-HCl (0,868 г, 4,53 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 16 ч. Добавляли EDCI-HCl (0,853 г, 4,44 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 7 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на Cu силикагеле (от 100% воды до 25% AcN в воде) с получением 0,600 г (28%) целевого продукта в виде слабо-желтого твердого вещества (ESI:m/z=1152(M+)).
Пример 5: Синтез хелата Mn 9
В 2-горлую колбу на 25 мл, снабженную магнитной мешалкой, загружали диэтаноламин (0,207 г, 1,97 ммоль) и воду (9,86 мл). рН полученного раствора доводили до 7 1,0 М-ной HCl и добавляли хелат Mn с 2 ответвлениями С5 (0,500 г, 0,956 ммоль; полученный согласно Примеру 2(v)), а после EDCI-HCl (0,445 г, 2,32 ммоль) и HOBt гидрат (0,045 г, 0,333 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 20 ч. Добавляли диэтаноламин (0,207 г, 1,97 ммоль) и EDCI-HCl (0,432 г, 2,25 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 8 ч. Добавляли диэтаноламин (0,207 г, 1,97 ммоль) и EDCI-HCl (0,448 г, 2,34 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 15,5 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на Cig силикагеле (от 100% воды до 30% AcN в воде) с получением 0,110 г (16%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=708 (М+Н+)).
Пример 6: Синтез хелата Mn 10

В 25 мл-овую 2-горлую колбу, снабженную магнитной мешалкой, загружали 3-аминопропан-1,2-диол (0,190 г, 2,03 ммоль) и воду (10,4 мл). рН полученного раствора доводили до 7 1,0 М-ной HCl и добавляли хелат Mn с 2ответвлениями С5 (0,603 г, 0,996 ммоль; полученный согласно Примеру 2(v)), а после EDCI-HCl (0,473 г, 2,47 ммоль) и HOBt гидрат (0,063 г, 0,466 ммоль). рН поддерживали на уровне 6, путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 7,5 ч. Добавляли 3-аминопропан-1,2-диол (0,095 г, 1,04 ммоль) и EDCI-HCl (0,453 г, 2,36 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 15,5 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на С18 силикагеле (от 100% воды до 20% AcN в воде) с получением 0,280 г (41%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=680 (М+Н+)).
Пример 7: Синтез хелата Mn 11

В 100 мл-овую 3-горлую круглодонную колбу, снабженную магнитной мешалкой, загружали основание Tris (0,632 г, 5,22 ммоль) и воду (26,0 мл). рН полученного раствора доводили до 7 1,0 М-ной HCl и добавляли хелат Mn с 2 ответвлениями С5 (1,500 г, 2,49 ммоль; полученный согласно Примеру 2(v)), а затем EDCI-HCl (1,141 г, 5,95 ммоль) и HOBt гидрат (0,160 г, 1,04 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 7,5 ч. Добавляли Tris (0,636 г, 5,25 ммоль) и EDCI-HCl (1,177 г, 6,14 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 17 ч. Добавляли Tris (0,624 г, 5,15 ммоль) и EDCI-HCl (1,133 г, 5,91 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 23 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на C18 силикагеле (от 100% воды до 20% AcN в воде) с получением 0,247 г (13%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=740 (М+Н+)).
Пример 8: Синтез хелата Mn 5а
Пример 8(i): Синтез защищенного хелата Mn с 2ответвлениями С4
В 250 мл-овую круглодонную колбу, снабженную механической мешалкой, загружали циклический хелат с 2 ответвлениями (19,85 г, 90,1 ммоль; полученный согласно Примеру 2(iii)), диметилмалеат (51,94 г, 360,4 ммоль), монтмориллонит K10 (36,0 г) и МеОН (36 мл). Полученную суспензию перемешивали при температуре окружающей среды в течение 26 ч. Нерастворимый материал удаляли путем фильтрования и прозрачный оранжевый фильтрат упаривали досуха. Остаток растворяли в EtOAc (200 мл) и экстрагировали водой (200 мл). Слои разделяли и водный слой упаривали досуха с получением 39,35 г (86%) в виде оранжевого твердого вещества (ESI: m/z=509 (М+Н+)), которое использовали без дальнейшей очистки.
Пример 8(H): Синтез хелата Mn с 2 ответвлениями С4
В 1 л-овую круглодонную колбу, снабженную магнитной мешалкой, загружали защищенный хелат Mn с 2 ответвлениями С4 (39,35 г, 77,2 ммоль), гидроксид натрия (24,71 г, 617 ммоль) и воду (500 мл). Полученный раствор перемешивали при 45°С в течение 4 ч. рН доводили до 7 концентрированной HCl и добавляли MnCl4-4Н2О (16,8 г, 84,9 ммоль). рН поддерживали при 7, пока перемешивали при 90°С в течение 2,5 ч перед охлаждением до температуры окружающей среды. рН доводили до 10,1 6,0 М-ным NaOH и полученный осадок удаляли путем центрифугирования при 3000 об/мин в течение 20 мин. Надосадочную жидкость собирали и упаривали досуха под вакуумом. Остаток растирали с МеОН (72 мл) при 40°С в течение 1,5 ч. Нерастворимое белое твердое вещество удаляли путем центрифугирования при 3000 об/мин в течение 30 мин. Надосадочную жидкость упаривали досуха под вакуумом с получением грязно-белого твердого вещества, которое очищали на Cu силикагеле (3% AcN в воде) с получением 25,8 г (66%) целевого продукта в виде грязно-белого твердого вещества (ESI: m/z=506 (М+Н+)).
Пример 8(iii): Синтез хелата Mn 5а

В 2-горлую 50-мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали хелат Mn с 2 ответвлениями С4 (0,667 г, 1,32 ммоль) и воду (13,0 мл). Добавляли глюкамин (0,505 г, 2,79 ммоль) и рН доводили до 7 1,0 М-ной HCl. Добавляли EDCI-HCl (0,599 г, 3,12 ммоль) и HOBt гидрат (0,036 г, 0,266 ммоль) и рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 7,5 ч. Добавляли вторую аликвоту EDCI-HCl (0,610 г, 3,18 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 16,5 ч. Добавляли третью аликвоту EDCI-HCl (0,610 г, 3,18 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 72 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на C18 силикагеле (от 100% воды до 30% AcN в воде) с получением 0,350 г (31%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=832 (М+Н+)).
Пример 9: Синтез хелата Mn 7а
В 3-горлую 100 мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали гидрохлорид 5-амино-N,N'-бис(2,3-дигидроксипропил)изофталамида (1,516 г, 4,17 ммоль) и воду (20 мл). рН полученного раствора доводили до 8 с помощью 1,0 М-ного NaOH и добавляли хелат Mn с 2 ответвлениями С4 (1,000 г, 1,98 ммоль; полученный согласно Примеру 8(H)), а затем EDCI-HCl (0,901 г, 4,70 ммоль). рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 6,5 ч. Добавляли вторую аликвоту EDCI-HCl (0,895 г, 4,67 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 16,5 ч. Добавляли третью аликвоту EDCI-HCl (0,417 г, 2,18 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 8 ч. Добавляли четвертую аликвоту EDCI-HCl (0,536 г, 2,80 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 17 ч. Мутную реакционную смесь отфильтровывали под вакуумом для удаления твердых веществ. Прозрачный желтый фильтрат упаривали досуха и неочищенный продукт очищали на C18 силикагеле (от 100% воды до 20% AcN в воде) с получением 0,494 г (22%) целевого продукта в виде слабо-желтого твердого вещества (ESI; m/z=1124 (М+)).
Пример 10: Синтез хелата Mn 10а

В 2-горлую 50 мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали хелат Mn с 2 ответвлениями С4 (0,667 г, 1,32 ммоль; полученный согласно Примеру 8(H)) и воду (13,0 мл). Добавляли 3-амино-1,2-пропандиол (0,253 г, 2,77 ммоль) и рН доводили до 7 1,0 М-ной HCl. Добавляли EDCI-HCl (0,599 г, 3,12 ммоль) и HOBt гидрат (0,036 г, 0,266 ммоль) и рН поддерживали на уровне 6 путем добавления по необходимости 1,0 М-ной HCl или 1,0 М-ного NaOH, пока перемешивали при температуре окружающей среды в течение 7 ч. Добавляли вторую аликвоту EDCI-HCl (0,610 г, 3,18 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 17,5 ч. Добавляли третью аликвоту EDCI-HCl (0,610 г, 3,18 ммоль) и рН поддерживали на уровне 6, пока перемешивали при температуре окружающей среды в течение 70 ч. Реакционный раствор упаривали досуха под вакуумом и неочищенный продукт очищали на Cu силикагеле (от 100% воды до 30% AcN в воде) с получением 0,160 г (19%) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=652 (М+Н+)).
Пример 11: Синтез хелата Mn уровня техники
Пример 11(i): Синтез защищенного хелата Mn с 0 ответвлениями

В 100 мл-овую 3-горлую круглодонную колбу, снабженную магнитной мешалкой и обратным холодильником, загружали защищенный циклический хелат с 2 ответвлениями (4,51 г, 8,53 ммоль; полученный согласно Примеру 2(H)) и концентрированную серную кислоту (18,0 мл) и нагревали при 100°С в течение 18 ч. Реакционную смесь охлаждали до температуры окружающей среды и помещали в ледяную баню перед доведением рН до 9,9 50%-ным водным NaOH. Полученную суспензию переносили в 250 мл-овую 3-горлую круглодонную колбу и добавляли безводный карбонат калия (11,78 г, 85,2 ммоль), а затем AcN (25 мл) и t-бутилбромацетат (6,64 г, 34,0 ммоль) и реакционную смесь нагревали при 70°С в течение 3 ч. Реакционную смесь охлаждали до температуры окружающей среды и твердые вещества удаляли путем фильтрования под вакуумом. Фильтрат экстрагировали AcN (3×50 мл) и органический слой упаривали досуха с получением темно-коричневого масла, которое очищали на С18 силикагеле (от 100% воды до 100% AcN в воде) с получением 1,28 г (3 3%) целевого продукта в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CD3CN, δ) 7,67 (1H, м), 7,12 (2Н, м), 5,14 (2Н, шир. с), 3,95 (4Н, м), 3,44 (4Н, м), 3,28 (6Н, м), 3,16 (2Н, м), 2,78 (3Н, с), 1,42 (18Н, с).
Пример 11(ii): Синтез незамещенного хелата Mn с 0 ответвлений
В 3-горлую 100 мл-овую круглодонную колбу, снабженную магнитной мешалкой и обратным холодильником, загружали защищенный хелат Mn с 0 ответвлений (1,28 г, 2,85 ммоль), AcN (8,4 мл) и ТГФ (21 мл). Добавляли 88%-ную водную муравьиную кислоту (29,1 мл, 556 ммоль) и полученный раствор нагревали при 65°С в течение 4 ч. Добавляли вторую аликвоту 88%-ной водной муравьиной кислоты (29,1 мл, 556 ммоль) и продолжали нагревание в течение еще 9 ч. Растворитель удаляли под вакуумом, получая в остатке желтое масло, которое применяли без дальнейшей очистки. 1Н ЯМР (400 МГц, CD3OD, δ) 7,74 (1H, м), 7,20 (2Н, м), 4,07 (4Н, м), 3,65 (4Н, м), 2,91 (3Н, с), 2,99 (4Н, м), 1,92 (4Н, м).
Пример 11(iii): Синтез хелата Mn уровня техники
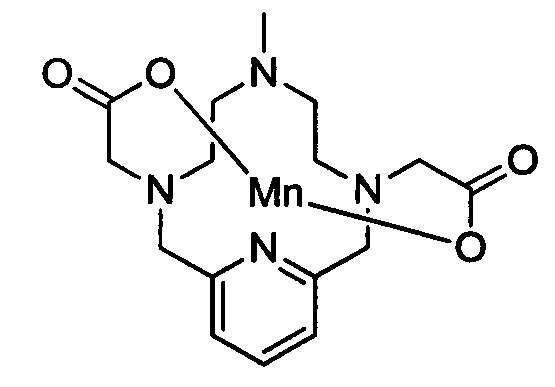
В 250 мл-овую круглодонную колбу, снабженную магнитной мешалкой, загружали незамещенный хелат Mn с 0 ответвлений (0,959 г, 2,85 ммоль) и тетагидрат хлорида марганца (II) (1,119 г, 5,65 ммоль). рН доводили по необходимости до 7,4 1,0 М-ным NaOH и 1,0 М-ной HCl и полученный раствор перемешивали при температуре окружающей среды в течение 15,5 ч. Затем рН доводили до 10 насыщенным водным карбонатом натрия и полученный грязно-белый осадок удаляли методом фильтрования под вакуумом. Фильтрат концентрировали досуха под вакуумом и очищали на C18 силикагеле (от 100% воды до 10% AcN в воде) с получением 0,511 г (46% за 2 стадии) целевого продукта в виде слабо-желтого твердого вещества (ESI: m/z=390 (М+)).
Пример 12: Исследование in vivo биораспределения 54Mn на крысах
Хелаты, меченые 54Mn, для исследования биораспределения были получены с использованием следующего способа. В стеклянный флакон объемом 3 мл, снабженный магнитной мешалкой, добавляли соответствующий марганецсодержащий хелат (1 мг) и 1,0 М формиат аммония, рН=4 (0,5 мл) (рН=5 для хелата Mn уровня техники). Затем добавляли 54MnCl2 в 1,0 М HCl (~500 мкКи) и полученный раствор нагревали при 40°С в течение 16 часов. Полученный раствор очищали с помощью препаративной ВЭЖХ для удаления нехелатированного Mn. Радиоактивную фракцию собирали и упаривали в вакууме досуха. Радиоактивный остаток растворяли в воде, содержащей нерадиоактивный хелат Mn (0,310 М), таким образом, что радиоактивность готовой смеси с дозировкой 0,620 ммоль Mn/кг в объеме пробы 2 мл/кг составляла ~ 30 мкКи.
Протокол эксперимента соответствовал Руководству по уходу и использованию лабораторных животных и был одобрен IACUC в General Electric Global Research (Niskayuna, NY). Самок крыс линии Sprague-Dawley (130-150 г; Charles River Laboratories; Massachusetts; USA) содержали в стандартных клетках, обеспечивавших свободный доступ к стандартному коммерческому корму и воде, при чередующемся 12-часовом цикле свет-темнота в помещениях с контролируемой температурой и влажностью. Перед инъекцией хелатов, меченых 54Mn, крыс анестезировали ингаляционным 3%-ным изофлураном (Piramal, NDC 66794-013-25; EZ-Anesthesia EZ700 Isoflurane Vaporizer, S/N 107). Место инъекции предварительно обрабатывали спиртовыми салфетками, и в хвостовую вену помещали временный катетер 27Ga (Surflo SROX2419V). 30 мкКи (0,74 МБк) меченого хелатом 54Mn в смеси с соответствующим нерадиоактивным хелатом на основе Mn (II), дозировали по 0,620 ммоль нерадиоактивного хелата Mn/кг в объеме пробы 2 мл/кг и скорости введения 1 мл/мин. После инъекции животных по одному размещали в клетках с проволочным дном, выстланных фильтровальной бумагой, до получения первой порции мочи. Затем крыс совместно содержали в клетках для длительного пребывания. Через 7 дней после инъекции животных умерщвляли погружением в СО2, а органы и ткани, представляющие интерес, извлекали и анализировали на радиоактивность с использованием гамма-счетчика Wizard 2480 (Perkin Elmer, Beaconsfield, UK).
Наивные крысы (Charles River Laboratories; Massachusetts; USA) получали однократную дозу 0,620 ммоль/кг (приблизительно 30 мкКи, 0,740 МБк) испытуемого образца через хвостовую вену путем инъекции. Кроме того, одной группе животных вводили физиологический раствор с соответствующей осмоляльностью (концентрированный NaCl для инъекций: часть АРР Pharmaceuticals # NDC 63323-187-30, лот №6008656, в смеси со стерильной водой для инъекций: часть Hospira # NDC 0409- 7990-09, лот №49-396-DKas) в качестве отрицательного контроля (Таблица 1). После умерщвления органы и ткани извлекали и анализировали на остаточную радиоактивность (т.е. содержание 54Mn в органах) через 7 дней после однократного введения.
Целью данного исследования являлась оценка распределения вещества в тканях у наивных крыс через 7 дней после однократной инъекции хелатов на основе Mn (II), меченых 54Mn (радиоактивный изотоп). Остаточную радиоактивность (например, % ID) измеряли в соответствующих собранных тканях (Таблица 2). Оценка уровня хелатов на основе Mn (II) показала, что содержание 54Mn находится в пределах уровня физиологической изменчивости марганца почти во всех образцах органов и тканей, и следовые количества присутствовали в выделительных органах. Обнаружение 54Mn в печени и почках ожидалось для всех оцениваемых соединений, поскольку эти органы являются частью основного пути экскреции для всех MP контрастных агентов. Хелаты на основе Mn (II) продемонстрировали пониженное распределение в сердце и мозге по сравнению с 54MnCl2 согласно литературным данным. Действительно, более низкие уровни (например, в пределах эндогенной изменчивости уровня) 54Mn, обнаруженные в мозге для хелата Mn 3 и хелата Mn 4, представляют большой интерес, поскольку мозг является одним из органов-мишеней токсичности для Mn (II). Удивительно, что более низкие уровни 54Mn в большеберцовой/малоберцовой и бедренной кости для хелатов Mn 3 и 4 также свидетельствуют об улучшенной стабильности этого класса соединений in vivo по сравнению с хелатом Mn предшествующего уровня техники, учитывая, что кости действуют как резервуар ионов свободных металлов (например, Mn (II)). Профили биораспределения трех хелатов на основе Mn (II) продемонстрировали, что стабильность парамагнитных комплексов in vivo является свойством, которое можно регулировать путем синтеза структуры хелатного фрагмента, принимая во внимание, что хелат Mn 3 и хелат Mn 4 проявляют улучшенную стабильность по сравнению с хелатом Mn предшествующего уровня техники.
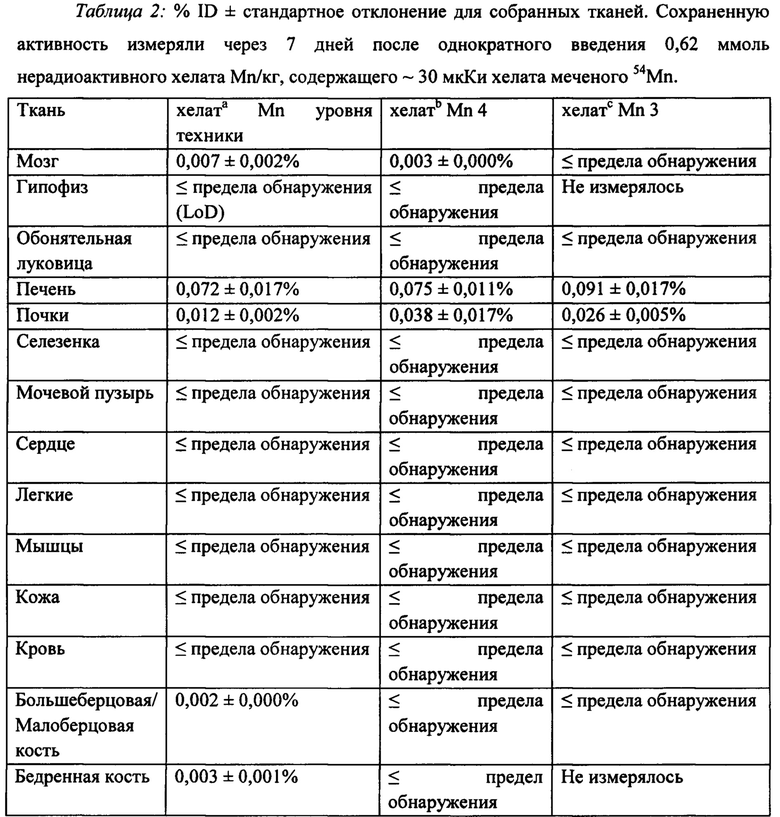
lLoD=0,002% ID, n=8; bLoD=0,002% ID, n=8; cLoD=0,005% ID, n=3
Пример 13: Растворимость в воде хелатов на основе Mn (II)
Растворимость хелатов на основе Mn (II) была подтверждена путем растворения очищенных хелатов на основе Mn (II) (чистота ~ 99%) в предписанном объеме растворителя (вода Millipore 18,2 MegaOhm производства компании Millipore BioCell Benchtop) для получения раствора заданного объема. Растворы визуально проверяли на гомогенность и, при необходимости, фильтровали через мембрану из ПТФЭ толщиной 0,45 мкм. Оценку проводили при 25°С.Конечные концентрации образца подтверждали путем определения конечной концентрации Mn (II) с помощью ИСП-МС (Spectro Arcos FHS12, S/N 10003910 или S/N 12006120).
Размер парамагнитного комплекса может влиять на растворимость в воде, следовательно, на распределение во внеклеточном пространстве и скорость выведения из организма. Адекватная растворимость в воде также требуется для поддержания низкого объема инъекции, чтобы облегчить введение контрастных веществ пациентам. С этой целью обычно выпускаются коммерчески доступные контрастные агенты Gd (III) с концентрацией 0,5 М. Растворимость хелатов на основе Mn (II) подтверждена в пределах стандартного диапазона растворимости (>0,5 М) независимо от увеличенного молекулярного размера для некоторых соединений (Таблица 3).

Пример 14: In vitro профили релаксивности протона и дисперсии ядерно-магнитной релаксации (NMRD) для хелатов на основе Mn(II)
Времена и продольной, и поперечной релаксации были измерены в сыворотке крови человека (BioreclamationIVT, Cat# HMSRM-M), чтобы продемонстрировать эффективность хелатов на основе Mn (II) в окружении, близком к физиологическому. Оценка релаксивности была проведена в диапазоне концентраций от 5 до 0 мМ Mn (II), при 40°С при использовании настольного ЯМР релаксометра Minispec Mq (Bruker Instruments, Rheinstetten, Germany) с рабочей частотой 60 MHz (1.4T) с последовательностью импульсов инверсия-восстановление. Продольная и поперечная релаксивности комплексов (например, r1 и r2 соответственно) вычисляли путем построения графика зависимости величины, обратной их временам релаксации, от концентрации марганца, как определено с помощью ИСП-МС (Spectro Arcos FHS12, S/N 10003910 или S/N 12006120) для каждого из хелатов на основе Mn (II). Зависимость T1 от приложенного В0 (то есть, магнитного поля) оценивалась через накопление профилей 1Н дисперсии ядерной магнитной релксации (NMRD). Предназначенный для этого релаксометр Stelar SMARTracer Fast Field Cycling с быстрым циклированием магнитного поля (0.01-10 МГц) и с ЯМР электромагнитом Bruker WP80, приспособленным к измерениям в переменном поле (20-80 MHz) и управляемым ПК-ЯМР консолью SMARTracer, был использован для записи профилей *Н NMRD в расширенном диапазоне частот Лармора (от 0,01 до 80 МГц). Температуру регулировали с помощью блока регулировки температуры VTC91 и поддерживали с помощью газового потока. Температуру определяли по предварительно откалиброванному термометру сопротивления из Pt. Релаксивность при сильном поле определяли с помощью ЯМР спектрометра Bruker AVANCE 400 МГц. Всего по 25 опорных точек на каждый NMRD профиль были зарегистрированы при каждой температуре, рН 6,9 и с концентрацией марганца в образцах 6,99, 6,78 и 4,45 мМ для хелата Mn уровня техники, хелата Mn 4 и хелата Mn 3, соответственно. Концентрация Mn (II) во всех образцах подтверждалась измерениями объемной магнитной восприимчивости (ОМВ). Релаксивность вычисляли путем вычитания скорости релаксации среды (дистиллированная вода) из скорости релаксации раствора комплекса Mn (II) при каждой величине поля и деления полученной разницы на концентрацию марганца, подтвержденную измерениями ОМВ.
Линейное приближение (коэффициент детерминации R2>0,99 для всех исследованных соединений) 1/T1 и I/T2 как функции от концентрации Mn дало величины r1 или r2, представленные в таблице 4 для сыворотки крови человека.
Представленные измеренные значения релаксивности всех хелатов на основе Mn (II) пригодны и находятся в пределах стандартной релаксивности (то есть, r1 и г2) имеющихся на рынке контрастных МРТ агентов (r1≥3 мМ-1с-1). Особый интерес представляет величина r1 (то есть, продольная релаксивность) хелатов на основе Mn (II), которая отражает способность хелатов формировать Т1 (или позитивный) контраст in vivo после iv введения. В свою очередь, величина r2 (то есть, поперечная релаксивность) отражает способность хелатов на основе Mn (II) формировать Т2 (или отрицательный) контраст после iv введения.
1Н NMRD профили были зарегистрированы для хелата Mn уровня техники и хелата Mn 4 (Фиг. 2 и Фиг. 3, соответственно). Оба оцениваемых хелата на основе Mn (II) показывают приемлемые 1Н NMRD профили с более низкими продольными релаксивностями, чем водный ион Mn (II), и только один разброс данных в профиле около 1 MHz. NMRD профиль хелата Mn 4 (Фиг. 3) показал небольшой перегиб r1 в частотном диапазоне от 60 до 80 МГц (~ 1,5 Т), который может оказаться перспективным для будущих применений клинической визуализации с улучшенной эффективностью хелатов на основе Mn (II) при клинически значимой величине магнитного поля. Этот высокопольный перегиб r1 является результатом небольшого уменьшения скорости вращений из-за большего молекулярного размера соединения.
Пример 15: Кинетика диссоциации хелатов на основе Mn (II) Кинетическую инертность оценивали при слабокислой рН благодаря низкой скорости диссоциации парамагнитного комплекса при рН физиологической среды и благодаря возможности гидролиза ионов металла при концентрациях, применяемых в экспериментах, и при значениях рН, близких к физиологическим.
Zn2+ обмен
Кинетику диссоциации хелатов на основе Mn (II) (концентрация 1 мМ) по изобретению оценивали через Zn2+ переметаллирование, регистрируя изменение во времени продольного времени релаксации (то есть, T1). Оценку проводили в ряде экспериментов, в том числе для различных концентраций (5, 10, 20, 40 эквивалентов Zn2+) конкурирующего иона металла с хелатом Mn уровня техники при 25°С и различных рН (рН: 5,1; 5,4; 5,7). Оценку кинетики диссоциации хелата Mn 4 (концентрация 1 мМ) проводили в присутствии 5 экв. Zn2+ при 25°С и рН 5,1 и 5,7. В обоих случаях оценки, реакционная смесь содержала 0,15 М NaCl и 50 мМ N-метилпиперазинового буфера (то есть, NMP). Изменения продольного времени релаксации контролировали при 1 МГц для хелата Mn уровня техники и при 0,01 МГц для хелата Mn 4. Действительно, высвобождение свободных ионов Mn2+, индуцированное Zn2+, приводит к снижению времени релаксации для обоих хелатов на основе Mn (II), оцененного во всех экспериментальных условиях. Частоты накопления для данного набора экспериментов были выбраны на основе разницы между наблюдаемой релаксивностью водного иона Mn (II) и хелатов на основе Mn (II) (например, хелата Mn уровня техники и хелата Mn 4), чтобы иметь достаточно различий между двумя типами химических веществ. Частоты были выбраны на основе профилей 1Н NMRD, как показано на Фиг. 1 для хелата Mn, уровня техники, хелата Mn 4 и MnCl2.
Cu2+ обмен
Оценку кинетики диссоциации хелата Mn уровня техники, хелата Mn 4 и хелата Mn 3 при концентрации 0,2 мМ проводили через Cu2+ переметаллирование, регистрируя изменение во времени поглощения в УФ/видимой области спектра в присутствии избытка конкурирующего иона металла (10 и 40 эквивалентов Cu2+) при 25°С и различных рН (рН: 5,0; 5,2; 5,4). Во всех случаях, реакционная смесь содержала 0,15 М NaCl и 50 мМ NMP. Длину волны 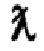 для накопления спектров в УФ/видимой области выбирали на основе разницы между наблюдаемым поглощением для хелатов на основе Mn (II) (например, хелата Mn уровня техники, хелата Mn 3 и хелата Mn 4) и вновь образованного Cu2+ - комплекса, чтобы иметь наилучшее различие между двумя типами химических веществ. Спектры в УФ/видимой части спектра накапливали при =300 нм на спктрометре Perkin-Elmer Lambda 19. Действительно, замещение иона Mn (II), индуцированное Cu2+, приводило к увеличению наблюдаемого поглощения при =300 нм для всех хелатов на основе Mn (II) во всех экспериментальных условиях. Данное увеличение происходило благодаря образованию Cu2+ - комплекса.
для накопления спектров в УФ/видимой области выбирали на основе разницы между наблюдаемым поглощением для хелатов на основе Mn (II) (например, хелата Mn уровня техники, хелата Mn 3 и хелата Mn 4) и вновь образованного Cu2+ - комплекса, чтобы иметь наилучшее различие между двумя типами химических веществ. Спектры в УФ/видимой части спектра накапливали при =300 нм на спктрометре Perkin-Elmer Lambda 19. Действительно, замещение иона Mn (II), индуцированное Cu2+, приводило к увеличению наблюдаемого поглощения при =300 нм для всех хелатов на основе Mn (II) во всех экспериментальных условиях. Данное увеличение происходило благодаря образованию Cu2+ - комплекса.
Результаты экспериментов по переметаллированию
Zn2+и Cu2+ переметаллирование изучали при использовании избытка обменивающегося иона металла для демонстрации кинетической инертности хелатов на основе Mn (II). Для любого из экспериментов по переметаллированию, проводимого с хелатом Mn уровня техники (например, при разных рН; разной концентрации Zn или Cu2+) было очевидно, что изменение скорости релаксации T1 (для Zn2+ - обмена; Фиг. 4) или изменение поглощения в УФ/видимой области спектра (для Cu2+ - обмена; Фиг. 5), вызванное конкурирующим ионом металла, представляло собой "одноступенчатый" процесс, наилучшим образом описываемый моно-экспоненциальной функцией. Действительно, хелат Mn уровня техники демонстрировал реакцию обмена, описываемую уравнением (1) псевдо-первого порядка (1) при скорости реакции, прямо пропорциональной суммарной концентрации хелатов на основе Mn (II). Скорость диссоциации, которую описывает kнабл., вычисляли по уравнению (1), где [MnL]cyм. является суммарной концентрацией хелатов на основе Mn (II) и t является временем наблюдения.
В отличие от хелата Mn уровня техники, для хелата Mn 4 и хелата Mn 3 кривые поглощения в УФ/видимой области спектра в зависимости от времени в реакции переметаллирования с Cu2+ не описывались с помощью моно-экспоненциальной функции, а демонстрировали двухступенчатую природу (то есть, двухстадийные механизмы процесса диссоциации). Двухступенчатая природа процесса Mn (II) - Cu2+ обмена была очевидной и подтверждалась для хелата Mn 3 и хелата Mn 4 при всех рН и оцениваемых концентрациях конкурирующего иона металла. Сравнение между кинетическими кривыми для хелата Mn уровня техники, хелата Mn 3 и хелата Mn 4, описывающими переметаллирование с Cu2+ при разных рН, представлено на Фиг. 6 и Фиг. 7. Профиль кривых ясно демонстрирует разный механизм диссоциации трех соединений при анализе в одинаковых экспериментальных условиях.
Для подтверждения двухступенчатой природы кривых кинетики диссоциации для хелата Mn 3 и хелата Mn 4 оценку реакции переметаллирования с Zn2+ проводили для хелата Mn 4. Кривые продольного времени релаксации (то есть, Ti) в зависимости от времени записывали при рН 5,1 и 5,7 (Фиг. 8) и они подтверждали двухступенчатое поведение кинетических кривых, то есть процесса диссоциации хелата Mn 4.
Хелат Mn 3 и хелат Mn 4 демонстрировали реакцию обмена, определяемую суммой двух независимых экспоненциальных функций. Эти двухступенчатые кинетические кривые можно хорошо описать математически с помощью биэкспоненциального уравнения (2), где А является наблюдаемым поглощением; b, A1 и А2 являются константами, специфичными для двухступенчатого процесса диссоциации для хелатов на основе Mn (II); kнабл1 и kнабл2 представляют константы скорости, описывающие двухступенчатую диссоциацию и t является временем наблюдения.

Скорости двухступенчатой диссоциации для хелата Mn 3 и хелата Mn 4, описываемой kнабл1 и kнабл2, и одноступенчатой диссоциации для хелата Mn уровня техники, описываемой kнабл, вычисляли по уравнению (2) и (1), соответственно, для разных экспериментальных условий, используемых в серии экспериментов.
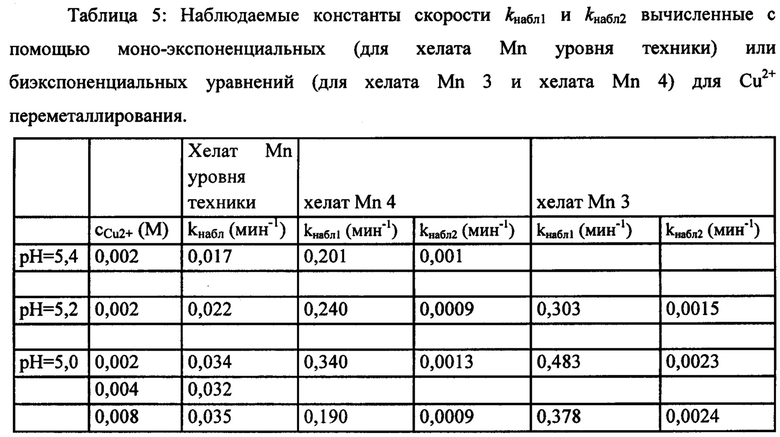
Из сравнения кинетических кривых и предварительной kнабл на качественном уровне очевидно, что хелат Mn 3 и хелат Mn 4 диссоциируют по другому механизму, описываемому биэкспоненциальной функцией, и с более медленной скоростью по сравнению с хелатом Mn уровня техники. Для количественного сравнения кинетической инертности этих хелатов на основе Mn (II), для реакции Cu2+ обмена было записано прямое сравнение развития поглощения в УФ/видимой области спектра при 300 нм. Прямое сравнение поглощения в УФ/видимой области спектра позволило количественно определить степень превращения хелатов на основе Mn (II) в хелаты на основе Cu2+ во время реакции переметаллирования, которую можно соотнести с кинетической инертностью комплекса Mn (II). "Степень превращения" рассчитывали в соответствии с уравнением (3), где At - это поглощение в момент времени t (или в конце реакции), А0 - поглощение в нулевой момент времени (или в начале накопления данных) и Аравн - поглощение в состоянии равновесия.
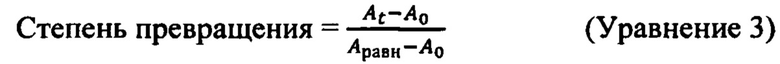
Прямое сравнение степени превращения, вычисленной для любого из экспериментов по переметаллированию, проводимого с хелатом Mn уровня техники, хелатом Mn 3 и хелатом Mn 4 (например, при различных рН, различных концентрациях Zn2+ или Cu2+), демонстрировало, что хелат Mn уровня техники достигает полной степени превращения за гораздо более короткое время, чем хелат Mn 3, а комплекс хелата Mn 4 диссоциирует еще медленнее. Степень превращения, обусловленная конкурирующим Cu2+, рассчитана и показана на Фиг. 9 и 10.
При том же математическом подходе сравнение степени превращения, обусловленной реакцией переметаллирования с Zn2+, проводили для хелата Mn уровня техники и хелата Mn 4 (Фиг. 11). Данная оценка ясно демонстрирует более высокую кинетическую инертность хелата Mn 4 в сравнении с хелатом Mn уровня техники.
Таким образом, хелат Mn 3 и хелат Mn 4 демонстрировали другой и более медленный механизм диссоциации в Cu2+ и Zn2+ переметаллировании по сравнению с хелатом Mn уровня техники. Неожиданно, улучшенная кинетическая инертность была продемонстрирована при прямом сравнении степеней превращения этих трех соединений, имеющихся во время реакций переметаллирования между MnL (то есть, комплексом Mn (II)) и Zn2+ или Cu2+ в одинаковых экспериментальных условиях. Все результаты кинетики диссоциации продемонстрировали явное улучшение общей кинетической инертности хелата Mn 3 и хелата Mn 4 по сравнению с хелатом Mn уровня техники, взятым в качестве образца сравнения. Действительно, хелат Mn 4 является наиболее кинетически инертным соединением по результатам всех экспериментальных условий. Можно предположить, что присутствие боковых цепей с -ОН группами может создавать вокруг меньшего по размеру металла Mn (II) защитную оболочку, успешно защищая Mn от диссоциации, тем самым внося вклад в повышении кинетической инертности этого класса хелатов на основе Mn (II).
| название | год | авторы | номер документа |
|---|---|---|---|
| Хелатные соединения марганца на основе тетраазабицикло-макроцикла, подходящие в качестве визуализирующих агентов для MRI | 2017 |
|
RU2769699C2 |
| ХЕЛАТЫ МАРГАНЦА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ КОНТРАСТНЫХ АГЕНТОВ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ (MPB) | 2010 |
|
RU2561718C2 |
| Новые марганецсодержащие наноструктуры | 2012 |
|
RU2638535C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2016 |
|
RU2743167C2 |
| Химические соединения для покрытия наноструктур | 2018 |
|
RU2757904C2 |
| МУЛЬТИМЕРНЫЕ КОНТРАСТНЫЕ АГЕНТЫ ДЛЯ МАГНИТНОГО РЕЗОНАНСА | 2006 |
|
RU2425831C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛОПОЛИАЗАМАКРОЦИКЛОФОСФОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2114115C1 |
| Контрастная композиция для магнитно-резонансной томографии и ангиографии на основе Mn-ДЦТА | 2020 |
|
RU2776412C2 |
| НОВЫЕ ХЕЛАТНЫЕ СОЕДИНЕНИЯ ГАДОЛИНИЯ С ВЫСОКОЙ РЕЛАКСИВНОСТЬЮ ДЛЯ ПРИМЕНЕНИЯ В МАГНИТНО-РЕЗОНАНСНОЙ ВИЗУАЛИЗАЦИИ | 2017 |
|
RU2755181C2 |
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛИЧЕСКИМИ ПОЛИАМИНОКИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ | 2000 |
|
RU2232763C2 |



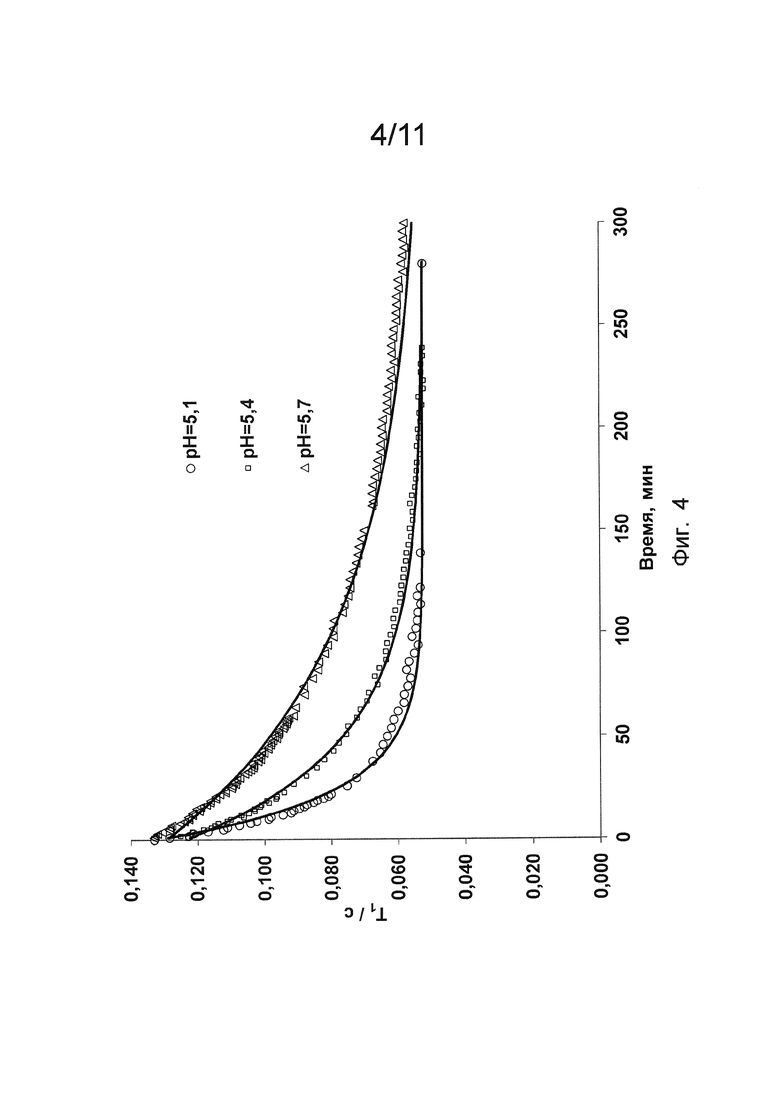

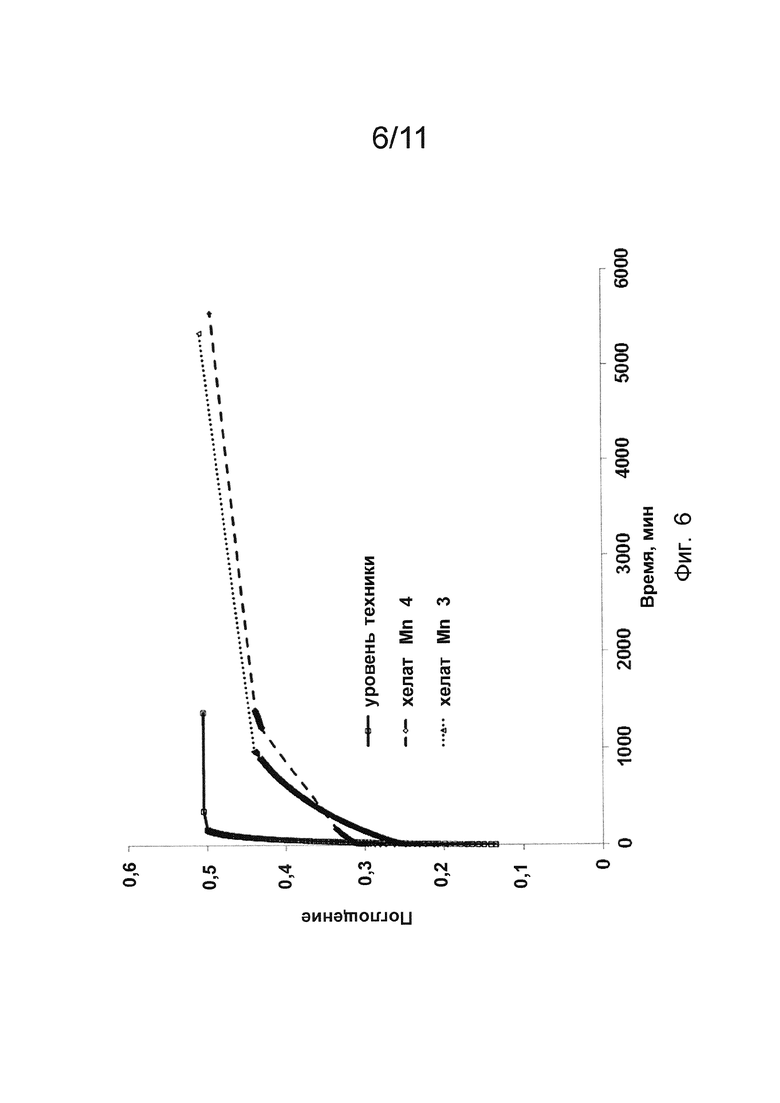

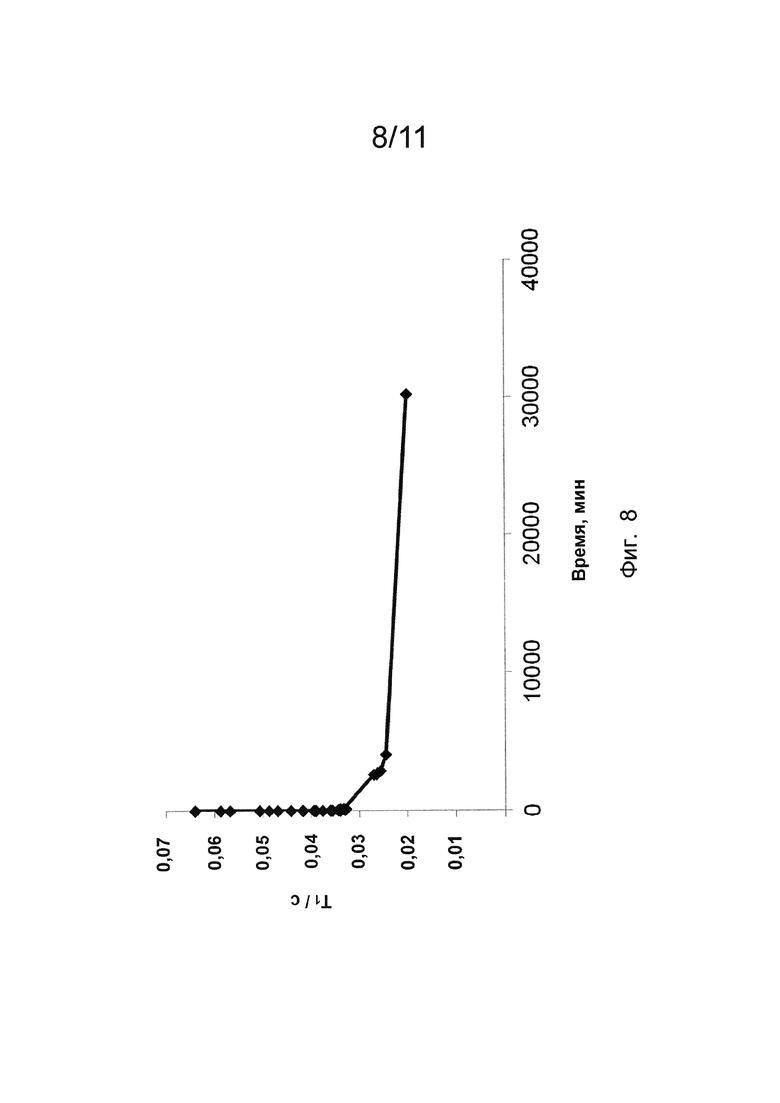


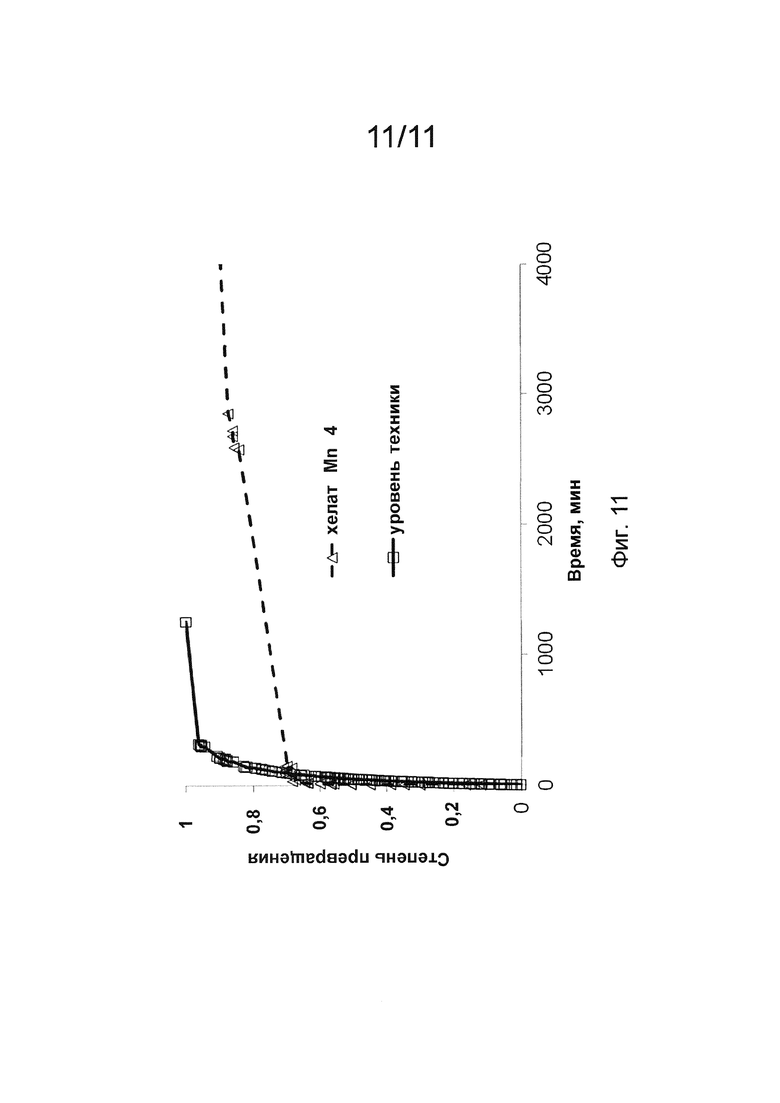
Настоящее изобретение относится к соединению формулы I:
 ,
,
в которой каждый R1 независимо выбран из группы, включающей С1-20 гидроксиалкил, C1-6 алкил, С3-6 арил, необязательно замещенный одним или более заместителями, выбранными из галогена и -C(=O)-NH-C1-6 гидроксиалкила; каждый R2 независимо выбран из группы, включающей C1-20 гидроксиалкил, C1-6 алкил или водород; R3 выбран из группы, включающей С1-3 алкил или -(CH2)m-C(=O)-NR5R6, где m является целым числом от 2 до 5, a R5 и R6 являются такими, как, соответственно, указано для R1 и R2; R4 отсутствует; каждый n является целым числом от 0 до 4; и при этом соединение формулы I содержит по меньшей мере две гидроксигруппы. Также предложены способ получения соединения формулы I, способ получения промежуточного соединения формулы II, фармацевтическая композиция и способ получения MP изображений и/или MP спектров. Предложенные соединения обладают улучшенной и долговременной кинетической стабильностью при сохранении подходящих свойств релаксации и могут применяться в качестве контрастных агентов в магнитно-резонансной томографии (МРТ). 5 н. и 39 з.п. ф-лы, 11 ил., 5 табл., 15 пр.
1. Соединение формулы I:

где:
каждый R1 независимо выбран из группы, включающей С1-20 гидроксиалкил, C1-6 алкил, С3-6 арил, необязательно замещенный одним или более заместителями, выбранными из галогена и -C(=O)-NH-C1-6 гидроксиалкила;
каждый R2 независимо выбран из группы, включающей C1-20 гидроксиалкил, C1-6 алкил или водород;
R3 выбран из группы, включающей С1-3 алкил или -(CH2)m-C(=O)-NR5R6, где m является целым числом от 2 до 5, a R5 и R6 являются такими, как, соответственно, указано для R1 и R2;
R4 отсутствует; и
каждый n является целым числом от 0 до 4;
и при этом соединение формулы I содержит по меньшей мере две гидроксигруппы.
2. Соединение по п. 1, в котором каждый R1 представляет собой С1-12 гидроксиалкил.
3. Соединение по любому из пп. 1 или 2, в котором каждый R1 представляет собой С3-6 гидроксиалкил.
4. Соединение по любому из пп. 1-3, в котором каждый R1 представляет собой С6 гидроксиалкил.
5. Соединение по любому из пп. 1-3, в котором каждый R1 независимо выбран из группы, содержащей:
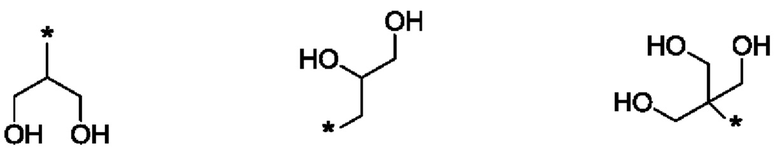
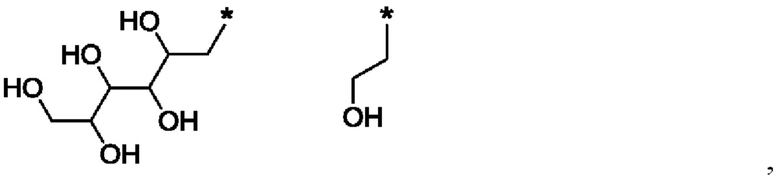
где в каждом случае звездочка обозначает место присоединения остатка соединения формулы I.
6. Соединение по п. 1, в котором каждый R1 представляет собой С3-6 арил, замещенный одним или более заместителями, выбранными из галогена и -C(=O)-NH-C1-6 гидроксиалкила.
7. Соединение по п. 6, в котором указанный С3-6 арил является фенилом.
8. Соединение по п. 6 или 7, в котором указанный галоген является йодом.
9. Соединение по любому из пп. 6-8, в котором указанный -C(=O)-NH-C1-6 гидроксиалкил представляет собой -C(=O)-NH-CH2-C(OH)-СН2-С(ОН).
10. Соединение по любому из пп. 6-9, в котором каждый R1 представляет собой:
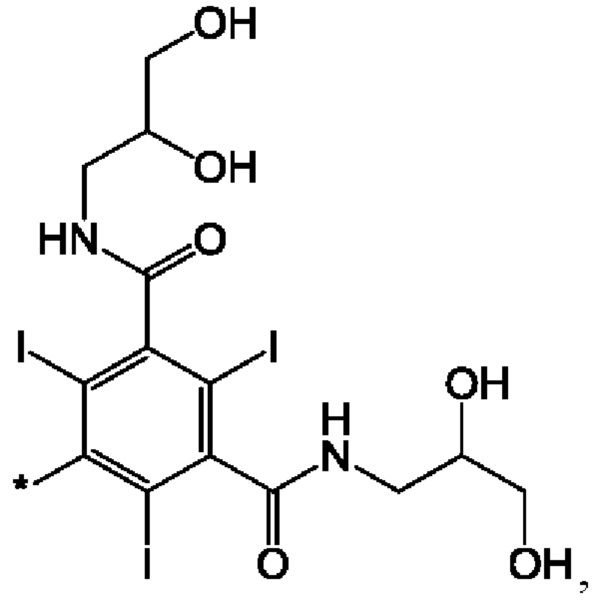
где звездочка обозначает место присоединения остатка соединения формулы I.
11. Соединение по любому из пп. 6-9, в котором каждый R1 представляет собой:
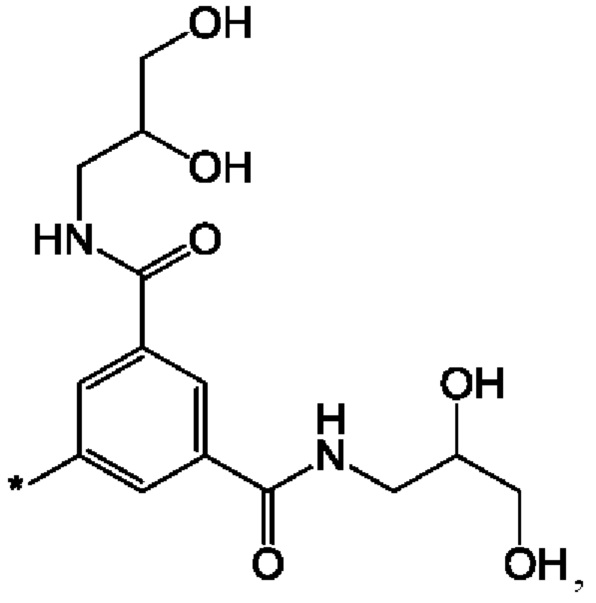
где звездочка обозначает место присоединения остатка соединения формулы I.
12. Соединение по любому из пп. 1-11, в котором каждый R1 является одинаковым.
13. Соединение по любому из пп. 1-12, в котором каждый R2 представляет собой C1-3 алкил.
14. Соединение по п. 13, в котором каждый R2 является метилом.
15. Соединение по любому из пп. 1-12, в котором каждый R2 является водородом.
16. Соединение по любому из пп. 1-12, в котором каждый R2 является С1-20 гидроксиалкилом.
17. Соединение по п. 16, в котором каждый R2 является C1-6 гидроксиалкилом.
18. Соединение по п. 17, в котором каждый R1 является С1-6 гидроксиалкилом.
19. Соединение по любому из пп. 1-18, в котором каждый R2 является одинаковым.
20. Соединение по любому из пп. 1-19, в котором каждый n является целым числом от 1 до 3.
21. Соединение по п. 20, в котором каждый n представляет собой 1.
22. Соединение по п. 20, в котором каждый n представляет собой 2.
23. Соединение по п. 20, в котором каждый n представляет собой 3.
24. Соединение по любому из пп. 1-23, в котором R3 представляет собой C1-3 алкил.
25. Соединение по п. 24, в котором R3 является метилом.
26. Соединение по любому из пп. 1-23, в котором R3 представляет собой -(CH2)m-C(=O)-NR5R6, как указано в п. 1.
27. Соединение по п. 26, в котором R5 является таким, как указано для R1 в любом из пп. 2-11.
28. Соединение по любому из пп. 26 или 27, в котором R6 является таким, как указано для R2 в любом из пп. 13-17.
29. Соединение по любому из пп. 26-28, в котором m представляет собой 3.
30. Соединение по п. 29, в котором n представляет собой 2.
31. Соединение по любому из пп. 1-30, которое содержит по меньшей мере 4 гидроксигруппы.
32. Соединение по п. 31, которое содержит 4-15 гидроксигрупп.
33. Соединение по п. 32, которое содержит 5-10 гидроксигрупп.
34. Соединение по любому из пп. 1-33, в котором указанный Mn обогащен изотопом Mn, выбранным из группы, включающей 52Mn и 54Mn.
35. Соединение по п. 1, которое выбрано из любого из следующих соединений:
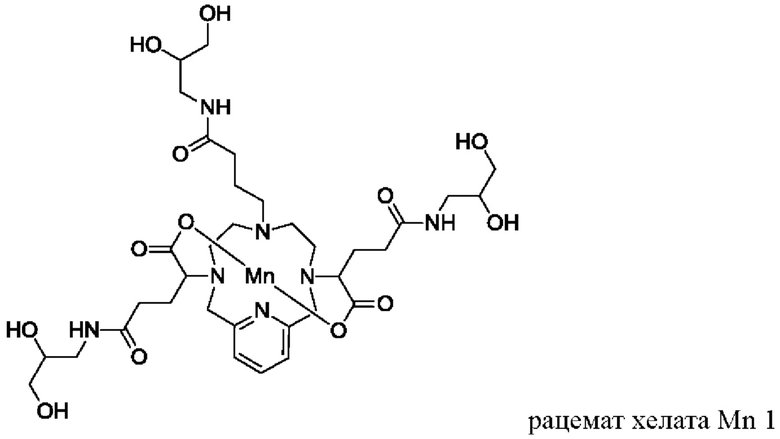
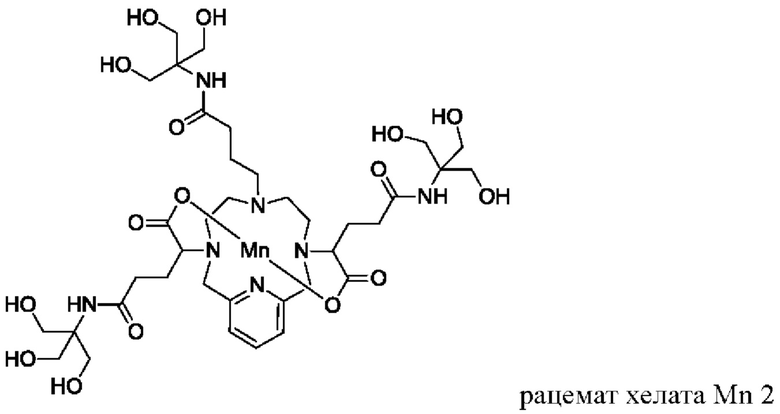

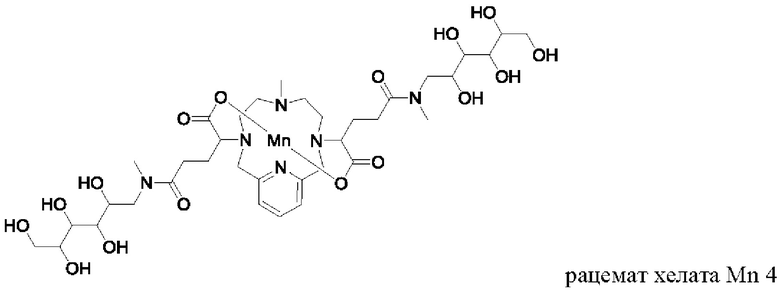
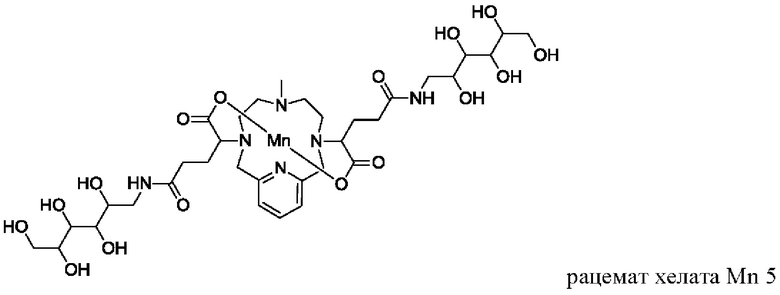


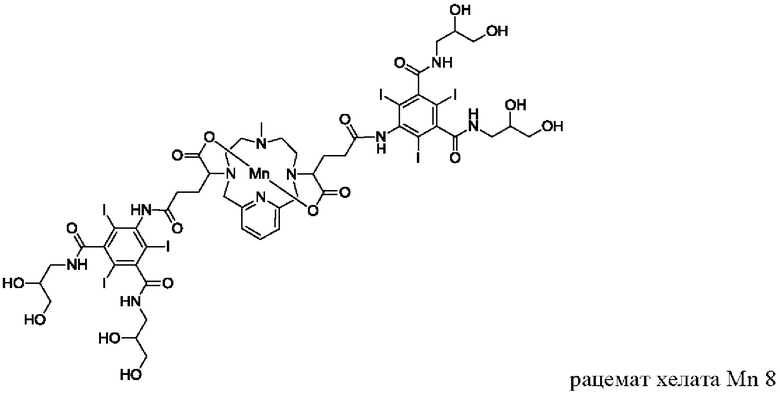
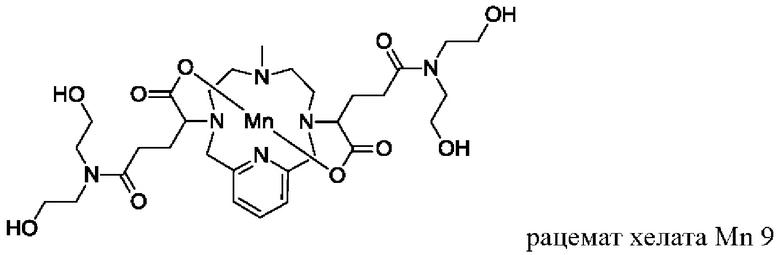


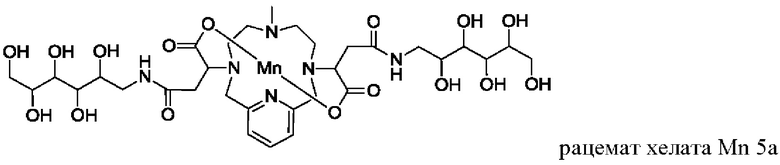
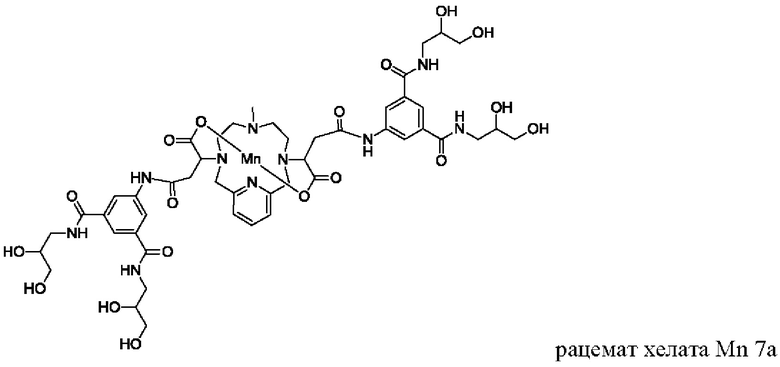

36. Соединение формулы I по любому из пп. 1-35, которое является диастереомерно чистым.
37. Соединение по п. 36, выбранное из любого из следующих соединений:
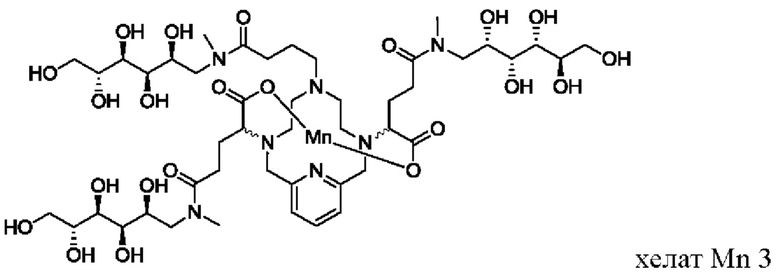
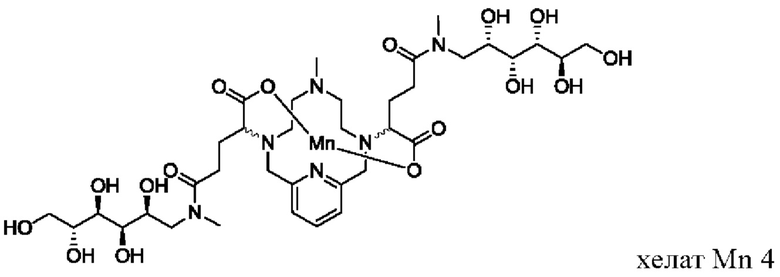
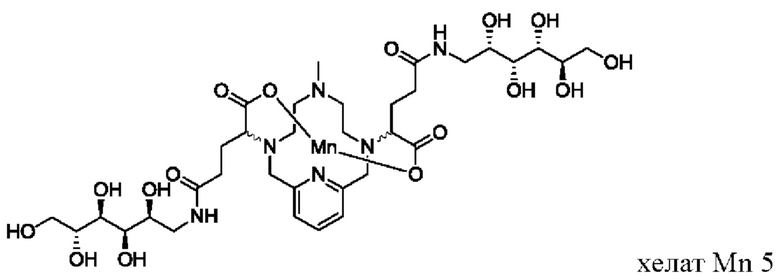





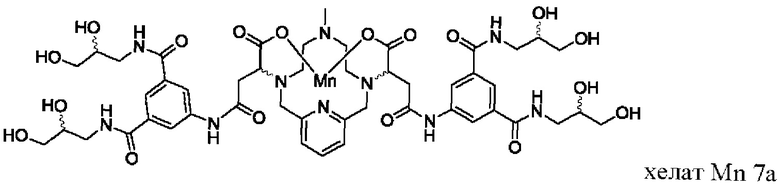
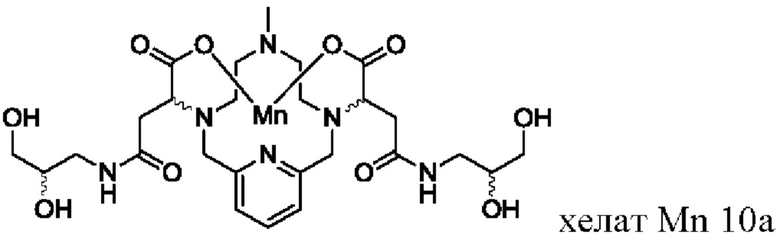
38. Способ получения соединения формулы I по любому из пп. 1-37, включающий:
(i) активацию карбоксильных групп соединения формулы II пептидным реагентом
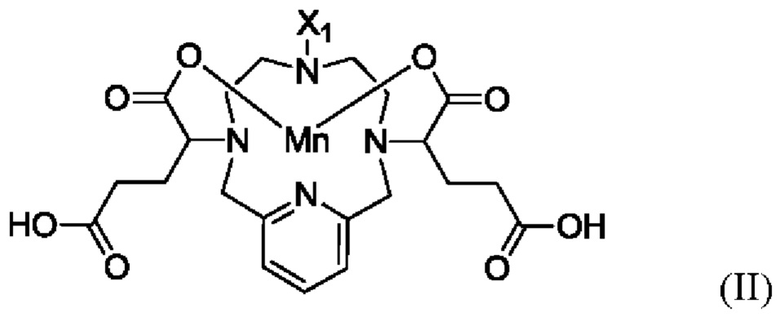
где X1 является метилом или -(СН2)3-СООН; затем
(ii) конденсацию указанного активированного соединения формулы II с аминной составляющей заместителя -NR1R2 с получением указанного соединения формулы I, в котором R1 и R2 являются такими, как указано в п. 1.
39. Способ по п. 38, в котором указанный пептидный реагент выбран из 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI) или гидроксибензотриазола (НОВТ).
40. Способ получения соединения формулы II, указанного в п. 38, включающий алкилирование соединения формулы III:
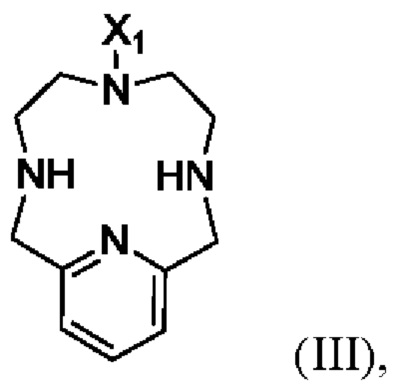
в котором X1 является метилом или -(CH2)3-СООН, с последующим комплексообразованием с использованием соединения Mn.
41. Фармацевтическая композиция для получения MP изображений и/или MP спектров, содержащая соединение формулы I по любому из пп. 1-37 совместно с биосовместимым носителем в форме, подходящей для введения млекопитающему.
42. Фармацевтическая композиция по п. 41, дополнительно содержащая один или более фармацевтически приемлемых эксципиентов.
43. Фармацевтическая композиция по п. 42, в которой указанные фармацевтически приемлемые эксципиенты выбраны из буферных агентов, стабилизаторов, антиоксидантов, регуляторов осмоляльности, регуляторов рН, дополнительного хелатирующего агента и слабых комплексов физиологически приемлемых ионов.
44. Способ получения MP изображений и/или MP спектров, включающий:
(i) введение субъекту соединения формулы I по любому из пп. 1-37 или фармацевтической композиции по любому из пп. 41-43;
(ii) детектирование сигналов магнитного резонанса (MP), полученных от указанного субъекта или органов указанного субъекта, в которых было распределено указанное соединение;
(iii) генерирование MP изображений и/или MP спектров на основе указанных детектированных сигналов.
| EP 1931673 A1, 18.06.2008 | |||
| WO 2011073371 A1, 23.06.2011 | |||
| WO 2006080022 A2, 03.08.2006 | |||
| КОМПЛЕКСЫ МЕТАЛЛОВ С БИЦИКЛИЧЕСКИМИ ПОЛИАМИНОКИСЛОТАМИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ | 2000 |
|
RU2232763C2 |

