Перекрестная ссылка на родственные заявки
Данная заявка заявляет приоритет предварительной заявки США 61/236477, поданной 24 августа 2009. Содержание этой заявки введено в данное описание посредством ссылки.
Область техники
Настоящее изобретение относится к способу получения соединений и их солей, полезных в качестве средств, стимулирующих нейрогенез. Более подробно изобретение направлено на способ получения дизамещенных пиперазинов с бензильным фрагментом и фрагментом никотиновой кислоты.
Предшествующий уровень техники
Патент США 7560553, введенный в данное описание посредством ссылки, описывает различные соединения, включая класс соединений, получение которых описано здесь, в качестве средств, стимулирующих нейрогенез. Таким образом, соединения, получаемые в соответствии со способом изобретения, полезны в лечении различных состояний, которые выигрывают от промотирования нейрогенеза посредством пролиферации/дифференциации гипокампальных мультипотентных стволовых клеток-предшественников и предшественников нейронов. Такие состояния включают болезнь Альцгеймера, умеренное когнитивное нарушение, деменцию, паралич, черепно-мозговую травму, повреждение спинного мозга, шизофрению и т.п. Способ получения, предоставляемый данным изобретением, избегает использования учетных препаратов, таких как бензилпиперазин.
Раскрытие изобретения
Способ по изобретению использует либо пиперазин, защищенный по одному из азотов в кольце, либо селективную реакцию только по одному из азотов в кольце, и замещенную никотиновую кислоту в качестве исходных веществ и завершается получением дизамещенного пиперазина, содержащего бензильный заместитель у одного из азотов в кольце. Получение может дополнительно включать превращение этого дизамещенного пиперазина в подходящую соль. Таким образом, в одном аспекте, изобретение направлено на способ получения соединения формулы:

в которой
R1 представляет собой алкил;
R2 является H или алкилом;
каждый R3 и R4 является независимо алкилом, алкенилом, галогеном, арилом, гетероарилом, арилалкилом, гетероарилалкилом, NR2, SR или OR, где R означает алкил или арил;
n означает 0, 1 или 2;
m означает 0, 1, 2 или 3;
в котором способ включает реагирование соединение формулы
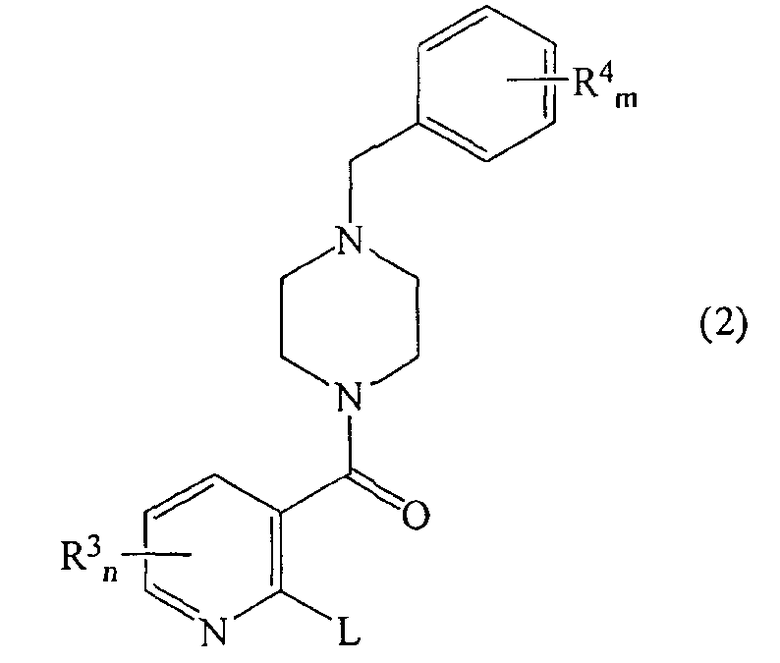
в которой R3, R4, m и n являются такими как определено для формулы (1) и L представляет собой уходящую группу,
с соединением формулы
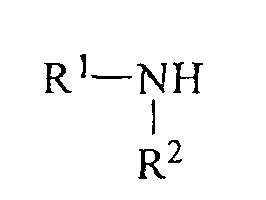
в которой R1 и R2 являются такими, как определено для формулы (1).
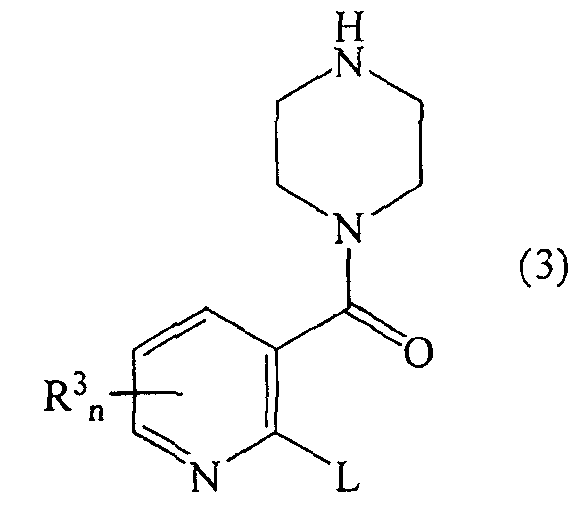
Соединение формулы (2) можно получить по реакции соединения формулы
в которой R3 и n являются такими, как определено для формулы (1) и L представляет собой уходящую группу,
с соединением формулы
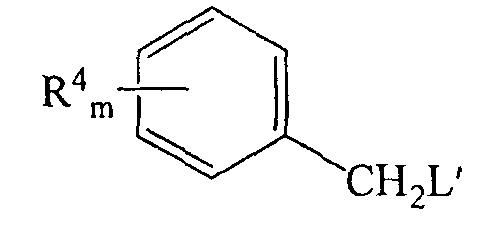
в которой R4 и m являются такими, как определено для формулы (1), и L' представляет собой уходящую группу,
или с соединением формулы

до образования имина, с последующим восстановлением вышеупомянутого имина.
В свою очередь, соединение формулы (3) можно получить по реакции соединения формулы
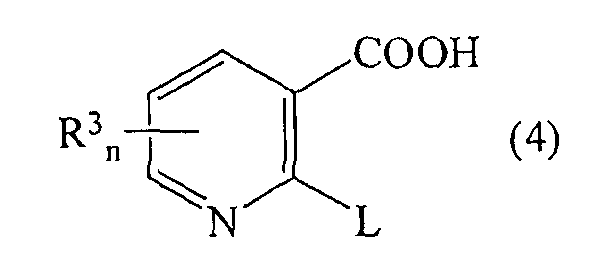
в котором R3 и n являются такими, как определено для формулы (1) и L представляет собой уходящую группу,
с соединением формулы
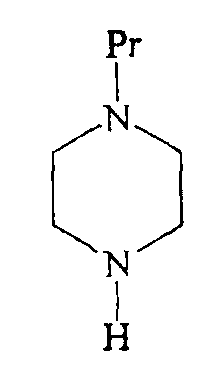
в которой Pr представляет собой защитную группу,
с последующим удалением защитной группы или путем селективного связывания с одним из азотов при использовании незащищенного пиперазина. Реакцию, в которой используют защитную группу, можно провести либо при контактировании соединения формулы (4) с защищенным пиперазином, в присутствии агента, способствующего образованию пептидной связи, или путем превращения соединения формулы (4) в соответствующий бензоил галогенид и присоединения защищенного пиперазина в присутствии слабого основания.
Соединение формулы (1) можно также превратить в подходящую кислотно-аддитивную соль, такую как сульфатную, фосфатную, гидрогалогенидную, цитратную, фумаратную, тозилатную или безилатную соль. Могут образовываться как моно-, так и бисоли.
Краткое описание чертежей
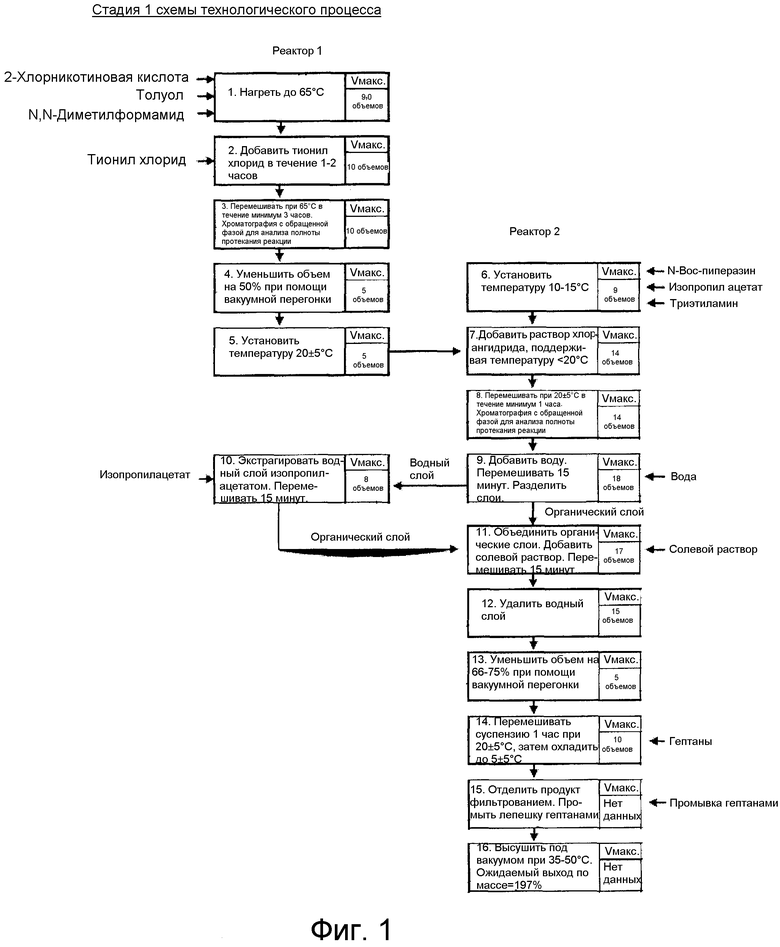
Фиг. 1 показывает оптимальный процесс для стадии 1В схемы 1.
Фиг. 2 показывает оптимальный процесс для стадии 2 схемы 1.
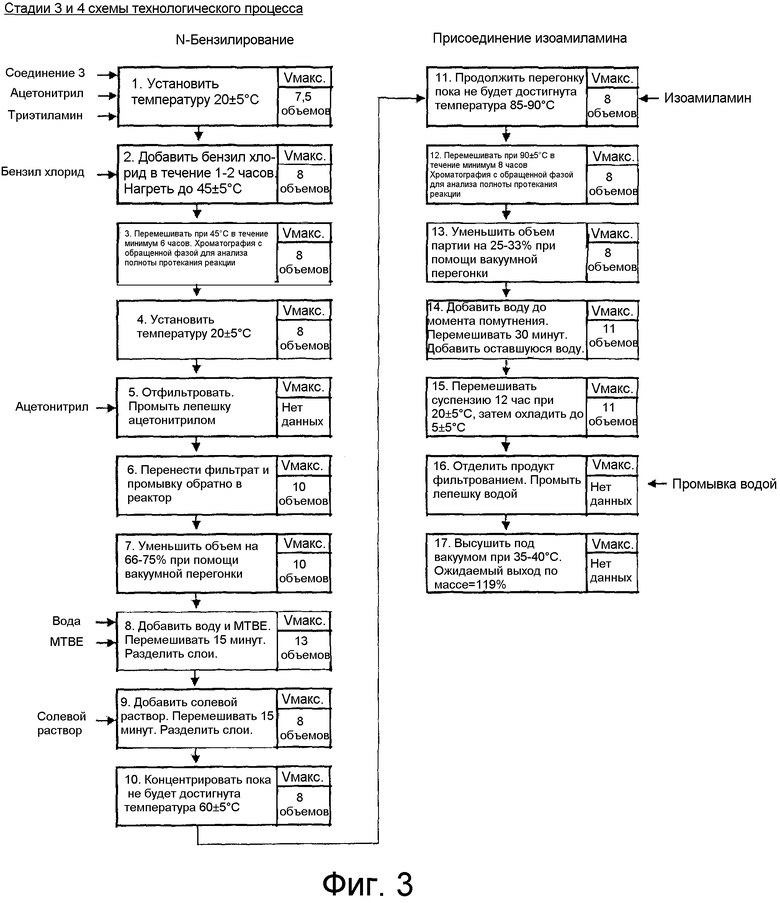
Фиг. 3 показывает оптимальный процесс для стадий 3 и 4 схемы 1.
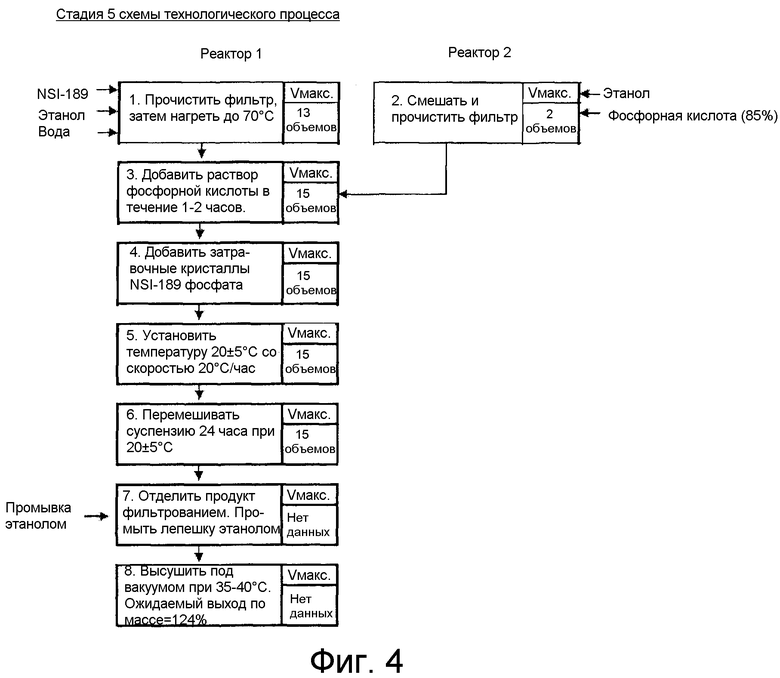
Фиг. 4 показывает оптимальный процесс для стадии 5 схемы 1.
Способы осуществления изобретения
Соединение формулы (1) и его соли и, в частности, соединение формулы 1E и его соли, как было показано, обладают активностью в отношении стимуляции нейрогенеза, как описано в вышеупомянутом патенте США 7560553. Настоящее изобретение описывает улучшенный способ получения этих соединений, как проиллюстрировано ниже в примерах 1-5.
В более общем смысле, получение этих соединений включает следующие стадии.

Как показано на схеме 1, необязательно замещенная никотиновая кислота, содержащая уходящую группу в положении 2, реагирует с наполовину защищенным пиперазином в присутствии агента, способствующего образованию пептидной связи, в присутствии слабого основания и в соответствующем растворителе. Обычно реакцию проводят в условиях окружающей среды для получения защищенной формы соединения формулы (3), с которого затем снимают защиту в кислоте в гидрофильном растворителе при слегка повышенных температурах. Получившийся в результате продукт формулы (3) реагирует с необязательно замещенным бензилом, содержащим уходящую группу в метиленовой части, в присутствии слабого основания и подходящего растворителя, также при повышенных температурах, для получения соединения формулы (2), которое не требует выделения, но реагирует с первичным или вторичным амином при повышенных температурах и в соответствующем растворителе до получения соединения формулы (1). Соединение формулы (1) может затем реагировать с 1 или 2 молями кислоты до получения кислотно-аддитивной соли. Если стадию 3 проводят путем замены бензил-L' на бензальдегид, образуется имин, который затем восстанавливают до амина, используя боргидрид натрия, цианборгидрид натрия, триацетоксиборгидрид натрия или боргидрид лития в практически любом органическом растворителе.
Обычно температура, при которой проводят стадию 1А, лежит между 20°C и 30°C; обычно основания включают триэтиламин или другие третичные амины и избыток малополярного непротонного растворителя, такого как бутил ацетат или изопропил ацетат. Стадию 2 обычно проводят при температуре между 50°C и 60°C, используя сильную кислоту, такую как HCl или серную кислоту в присутствии спиртового растворителя. Стадии 3 и 4 проводят при температуре между 45°C и 60°C для стадии 3 и между 80°C и 90°C для стадии 4. Стадию 3 проводят, используя слабое основание, такое как триэтиламин, и апротонный растворитель, такой как ацетонитрил или ДМСО. Стадию 4 также проводят в присутствии апротонного растворителя.
Стадию 5 проводят в условиях, зависящих от природы кислоты; либо один, либо два эквивалента кислоты можно использовать для получения подходящей соли.
В качестве альтернативы стадии 1A, соединение формулы (3 Pr) можно получить, используя стадию 1B, избегая применения дорогого реагента, способствующего образованию белковой связи.
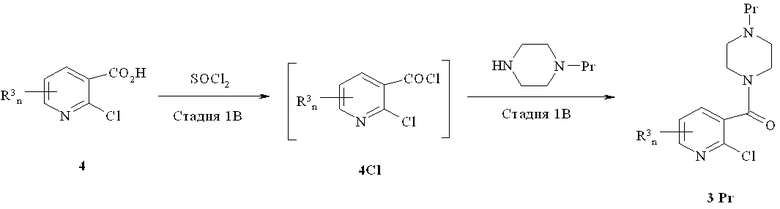
Стадию 1В обычно проводят при температуре между 60°C и 70°C обычно в присутствии основания, такого как третичный амин, в избытке малополярного непротонного растворителя, такого как бутил ацетат или изопропил ацетат. Таким образом, стадию 1B проводят в условиях, аналогичных тем, которые используют на стадии 1A, за исключением того, что никотиновую кислоту превращают в ацилгалогенид в присутствии SOCl2.
Оставшаяся часть схемы может оставаться той же самой, хотя можно улучшить выход посредством небольшого понижения температуры, при которой проводят стадию 3.
Как отмечено выше, оба R1 и R2 могут быть алкилами, а также включены алкильные заместители из тех, которые необязательно присутствуют в фрагментах никотиновой кислоты и бензила; кроме того, заместителями могут быть NR2, SR, OR, где R означает алкил. Заместители R3 и R4 могут также быть алкенилами.
Используемые здесь термины «алкил» и «алкенил» включают прямоцепочечные, с разветвленной цепью и циклические одновалентные углеводородные радикалы и их комбинации, которые содержат только C и H, если не являются замещенными. Примеры включают метил, этил, изопропил, изобутил, циклогексил, циклопентилэтил, 2-пропенил, 3-бутенил и т.п. Общее число атомов углерода в каждой такой группе иногда описывают, например, так что если группа содержит до 10 атомов углерода, это можно представить как 1-10C или как C1-C10 или C1-10. Вообще предпочтительно, чтобы один из R1 и R2 представлял собой H, а другой алкил, с максимум 10 или 8 атомов углерода, и R3 и R4, если они изображают алкил или алкенил, обычно содержат максимум 8 или 6 атомов углерода.
Обычно алкильные и алкенильные заместители в изобретении содержат 1-10C(алкил) или 2-10C(алкенил). Предпочтительно они содержат 1-8C(алкил) или 2-8C(алкенил). Иногда они содержат 1-4C (алкил) или 2-4C(алкенил). Отдельно взятая группа может включать более чем одну двойную связь; такие группы включены в определение термина «алкенил».
Алкильные и алкенильные группы могут быть незамещенными или замещенными до такой степени, до которой такое замещение химически целесообразно с точки зрения получения и свойств конечного продукта. Незамещенные формы являются предпочтительными.
Как дополнительно отмечено выше, R3 и R4 могут также представлять собой арил или гетероарил.
Используемый здесь «арил» относится к моноциклическому или конденсированному бициклическому фрагменту, обладающему свойствами ароматичности; примеры включают фенил и нафтил. Аналогично, «гетероарил» относится к таким моноциклическим или бициклическим кольцевым системам, которые содержат в качестве членов кольца один или несколько гетероатомов, выбираемых из O, S и N. Включения гетероатома допускают ароматичность в 5-ти членных кольцах, а также в 6-ти членных кольцах. Обычные гетероароматические системы включают моноциклические C5-C6 ароматические группы, такие как пиридил, пиримидил, пиразинил, пиридазинил, тиенил, фуранил, пирролил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил и имидазолил и конденсированные бициклические фрагменты, образованные при конденсации одной из этих моноциклических групп с фенильным кольцом или с любыми гетероароматическими моноциклическими группами, до образования C8-C10 бициклической группы, такой как индолил, бензимидазолил, индазолил, бензотриазолил, изохинолил, хинолил, бензотиазолил, бензофуранил, пиразолопиридил, хиназолинил, хиноксалинил, циннолинил и т.п. Любая моноциклическая или конденсированная кольцевая бициклическая система, которая обладает свойствами ароматичности в терминах распределения электронной плотности по кольцевой системе, включена в это определение. Оно также включает бициклические группы, в которых, по меньшей мере, кольцо, которое непосредственно присоединено к оставшейся молекуле, обладает свойствами ароматичности. Обычно кольцевые системы содержат 5-12 атомов членов кольца. Предпочтительно моноциклические гетероарилы содержат 5-6 кольцевых членов, и бициклические гетероарилы содержат 8-10 кольцевых членов.
Аналогично, «арилалкил» и «гетероарилалкил» относятся к ароматическим и гетероароматическим кольцевым системам, которые связаны с местом их присоединения посредством связывающей группы, такой как алкилен, включая насыщенные или ненасыщенные, циклические или ациклические линкеры, которые необязательно содержат один или несколько гетероатомов, выбираемых из O и S. Обычно линкером является C1-C8 алкильный или C1-C8 гетероалкильный линкер. Арилалкильная группа может, например, представлять собой фенильное кольцо и C1-C4 алкилен, где алкильные или гетероалкильные группы могут необязательно замыкаться в цикл, образуя кольцо, такое как циклопропан, диоксолан или оксациклопентан.
Используемый здесь термин «алкилен» относится к двухвалентной углеводородной группе; поскольку она является двухвалентной, она может связывать вместе две другие группы. Обычно она упоминается как -(CH2)n-, где n равно 1-8 и предпочтительно n равно 1-4, при этом, там, где это специально отмечено, алкилен может также быть замещенным другими группами и может иметь другие длины, и открытые валентности не обязательно должны находиться на противоположных концах цепи. Таким образом, -CH(Me)- и -C(Me)2- можно также отнести к алкиленам, также как и циклическую группу, такую как циклопропан-1,1-диил.
Арильная, гетероарильная, арилалкильная и гетероарилалкильная группы могут быть незамещенными или замещенными до такой степени, до которой такое замещение химически целесообразно с точки зрения получения и свойств конечного продукта. Незамещенные формы являются предпочтительными.
Используемый здесь термин «галоген» включает фтор, хлор, бром и йод заместители. Хлор и бром заместители являются особенно предпочтительными.
Подходящие уходящие группы для L и L' включают галогено группы, такие как хлор, йод или бром группы, тозилаты (OTs), и трифлаты (OTf). Другие подходящие уходящие группы включают мезилаты (OMs) и брозилаты (OBr).
Реагенты, способствующие образованию пептидной связи, включают О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурониум тетрафторборат (TBTU), а также 1-этил-3-(3-диметиламинопропил)-карбодиимид гидрохлорид (EDC), N-гидроксибензотриазол (HOBt), карбонил диимидазол (CDI), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурониум гексафторфосфат (HATU), Ν,Ν' дициклогексилкарбодиимид (DCC) и N-гидроксисукцинимид (NHS).
Подходящие средства для защиты групп включают 9-фторэтилметилкарбамат (Fmoc) и t-бутилкарбамат (Boc), а также TBDMS, TMS, TES, TIPS, TBDPS, бензоил и карбаматы или амиды в целом.
Эти списки не являются исчерпывающими и многие подходящие уходящие группы, защитные группы и реагенты, способствующие образованию пептидной связи, известны в данной области и могут быть коммерчески доступны.
Все эти реакции можно проводить в органических растворителях или в водно-органических растворителях, таких как тетрагидрофуран (THF), диметилформамид (DMF), метиленхлорид, MTBE, все алканы, NMP, DMA, EtOAc, и кроме тех, которые используют для стадии (2), за исключением спиртовых растворителей.
Предпочтительные варианты осуществления включают те, в которых R2 представляет собой H и R1 является этилом, пропилом, бутилом или амилом, включая их изоформы. Дополнительными предпочтительными формами являются те, в которых m и/или n равны 0 или 1, предпочтительно 0. Предпочтительной уходящей группой является галогеногруппа, предпочтительно хлоргруппа.
Следующие примеры предназначены для того, чтобы проиллюстрировать изобретение, а не для того, чтобы ограничить его.
Примеры 1-5 подробно описывают следующую последовательность реакций.

Пример 1
Получение защищенного 3E (Стадия 1A)
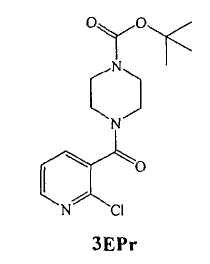
A. Хлорникотиновую кислоту (5,0 г) (4 эквив.) (4Е) загружали в круглодонную колбу, за которой следовала загрузка ацетонитрила (безводного, 40 мл) и TBTU (1,4 эквив.). К получившемуся в результате раствору добавляли триэтиламин (2,0 эквив.) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Boc-пиперазин (1,4 эквив.) добавляли порциями, поддерживая температуру внутри колбы <20°C. Реакцию медленно нагревали до 40°C и судили о полноте протекания по ВЭЖХ анализу после четырех часов.
Реакционную смесь гасили насыщенным раствором NaHCO3 (40 мл) и экстрагировали изопропил ацетатом (IPAc) (2×40 мл). Органические слои объединяли и промывали 50% солевым раствором (40 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали до одной четвертой от исходного объема. Получившееся в результате масло превращали в густую суспензию при перемешивании.
Добавляли простой метилтретбутиловый эфир (MTBE, 100 мл), и получившуюся в результате суспензию охлаждали на бане из воды со льдом и перемешивали в течение одного часа. Твердые вещества собирали фильтрованием через фильтровальную бумагу Whatman® № 1, и лепешку на фильтре промывали холодным MTBE (20 мл). Твердое вещество сушили в вакуумной печи при температуре окружающей среды до получения 6,7 г (выход 65%) 3EPr в виде светло-коричневого твердого вещества.
Вышеупомянутую реакцию повторяли в идентичных условиях и в том же масштабе, получая в результате 6,5 г (выход 65%) 3EPr.
B. Методику из пункта A повторяли, используя 1 г вместо 5 г хлорникотиновой кислоты и соответствующие количества других реагирующих веществ и используя этил ацетат или изопропил ацетат в качестве растворителей. Выходы составили:
• Этил ацетат: 50% с чистотой по ВЭЖХ 95,5% (AUC @ 226 нм).
• Изопропил ацетат: 80% с чистотой по ВЭЖХ 97,8% (AUC @ 226 нм).
C. В модифицированной методике из пункта A, хлорникотиновую кислоту (5,0 г) загружали в круглодонную колбу, за которой следовала загрузка ацетонитрила (марки ЧДА, 40 мл) и триэтиламина (2,0 эквив.). К получившемуся в результате раствору добавляли TBTU (1,4 эквив.) и смесь перемешивали при температуре окружающей среды в течение 30 минут. Boc-пиперазин (1,4 эквив.) добавляли порциями, поддерживая температуру внутри колбы <20°C. Реакцию перемешивали при температуре окружающей среды в течение выходных, и судили о полноте протекания по ВЭЖХ анализу после 50 часов. Реакционную смесь гасили насыщенным раствором NaHCO3 (40 мл) и экстрагировали IPAc (2×40 мл). Органические слои объединяли и промывали 50% солевым раствором (40 мл).
Органический слой высушивали над Na2SO4, фильтровали и концентрировали до одной четвертой от исходного объема.
К получившемуся в результате маслу добавляли MTBE (100 мл), и получившуюся в результате суспензию перемешивали при температуре окружающей среды в течение 5,5 часов и в течение еще двух часов на бане из воды со льдом. Твердые вещества собирали фильтрованием через фильтровальную бумагу Whatman® № 1, и лепешку на фильтре промывали холодным MTBE (20 мл). Продукт сушили в вакуумной печи при температуре окружающей среды до получения 6,3 г (выход 61%) 3EPr в виде светло-коричневого твердого вещества. Чистота по ВЭЖХ составила >99,9% (AUC @ 226 нм).
D. Реакцию из пункта С масштабировали до 10 г и приводили к завершению через 16 часов. IPAc экстракт, полученный после водной обработки, проводимой тем же самым способом, как описано выше, делили на две равные части. Каждую часть уменьшали до 20 г (≈1:1 IPAc/продукт по массе) при пониженном давлении.
Часть 1: К получившейся в результате суспензии добавляли MTBE (100 мл). Получившуюся в результате суспензию перемешивали при температуре окружающей среды в течение 16 часов и в течение еще двух часов на бане из воды со льдом. Твердые вещества собирали фильтрованием через фильтровальную бумагу Whatman® № 1, и лепешку на фильтре промывали холодным MTBE (20 мл). Продукт сушили в вакуумной печи при температуре окружающей среды до получения 6,8 г (выход 66%) 4EPr в виде светло-коричневого твердого вещества. Чистота по ВЭЖХ составила >99,9% (AUC @ 226 нм).
Часть 2: Процесс для части 2 был таким же, как и для части 1, но использовали гептаны в качестве антирастворителя, получая в результате 8,2 г (выход 80%) 4EPr в виде светло-коричневого твердого вещества. Чистота по ВЭЖХ составила >99,9% (AUC @ 226 нм).
Пример 2
Получение 3EPr через стадию 1B
A. Хлорникотиновую кислоту (5,0 г, 31,7 ммоль) загружали в круглодонную колбу, за которой следовала загрузка толуола (безводный, 40 мл) и DMF (120 мкл, 0,05 эквив.). Получившуюся в результате суспензию нагревали до 55°C затем по каплям в течение 5 минут добавляли тионил хлорид (4,6 мл, 2,0 эквив.). Суспензию перемешивали при 55°C в течение трех часов, в ходе которых наблюдали выделение газа, и смесь становилась гомогенной. Отбирали образец и гасили его в метаноле, содержащем триэтиламин, для того, чтобы получить метиловый эфир для ВЭЖХ анализа. ВЭЖХ анализ показал, что превращение кислоты в хлорангидрид завершено. Колбу снабжали аппаратом для дистилляции и нагревали с обратным холодильником. Удаляли приблизительно 20 мл растворителя, затем раствор охлаждали до температуры окружающей среды. В отдельную колбу загружали N-Boc-пиперазин (7,1 г, 1,2 эквив.), ацетонитрил (30 мл, 6 объемов) и триэтиламин (13,3 мл, 3,0 эквив.). Отмечали слабую эндотермичность. Затем добавляли подготовленный раствор хлорангидрида кислоты со скоростью, при которой внутренняя температура поддерживалась ниже 35°C. Получившуюся в результате суспензию перемешивали в течение одного часа при температуре окружающей среды. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь гасили насыщенным раствором NaHCO3 (20 мл), и водные слои экстрагировали изопропил ацетатом (20 мл). Органические слои объединяли и промывали водой (10 мл). ВЭЖХ анализ воды показал некоторую потерю продукта в водном слое. Органические слои концентрировали до приблизительно двух объемов и затем добавляли гептаны (50 мл) для того чтобы вызвать преципитацию. Полученную в результате суспензию перемешивали при температуре окружающей среды в течение 30 минут, охлаждали до 0-5°C в течение одного часа, фильтровали и промывали гептанами. Влажную лепешку затем сушили в течение ночи под вакуумом до получения 9,85 г 3EPr [MDM-W-1(14), выход 95%, 99,8% по ВЭЖХ] в виде светло-желтого твердого вещества.
B. Методику из пункта A этого примера проводили, используя 1,2 эквивалента тионил хлорида и 1,1 эквивалент N-Boc-пиперазина. Реакцию 2-хлорникотиновой кислоты с тионил хлоридом проводили при 65°C для лучшего контроля выделения газа. Реакцию промежуточного соединения хлорангидрида кислоты и N-Boc-пиперазина проводили в IPAc вместо ацетонитрила, для того чтобы помочь предотвратить преципитацию бикарбоната натрия в ходе гашения. Реакция дала 3EPr в виде не совсем белого твердого вещества [MDM-W-5(8), 9,83 г, выход 95%, >99,9% по ВЭЖХ].
C. Гашение и обработка реакции водным раствором бикарбоната натрия может приводить к образованию эмульсии, которая требует времени на разделение. Переход на гашение водой снимает эту проблему при малом масштабе; однако, по мере увеличения масштаба, продолжает сохраняться значительный слой в виде лохмотьев. Слой в виде лохмотьев можно растворить путем слабого нагревания двухфазной смеси до 30-35°C.
Пример 3
Снятие защиты (Стадия 2)
A. 1 г 3EPr, полученного в примере 1 или в примере 2 обрабатывали 2 эквивалентами 5-6 н. HCl в 2-пропаноле при 50°C. Реакция, как было обнаружено, прошла за 6 часов.
B. Способ из пункта A повторяли с 6,7 г 3EPr. К раствору 3EPr (6,65 г) в 2-пропаноле (5 объемов) добавляли 5-6 н. HCl в 2-пропаноле (2 эквив.). Реакцию нагревали до 40°C и судили о полноте протекания по ВЭЖХ анализу после четырех часов. За это время образовалась суспензия белого цвета.
Реакцию охлаждали до температуры окружающей среды, и твердые вещества отделяли фильтрованием через фильтровальную бумагу Whatman® № 1. Лепешку на фильтре промывали 2-пропанолом (20 мл). Твердое вещество сушили под высоким вакуумом до получения 4,63 г (выход 86%) 3E·HCl в виде твердого вещества белого цвета. 1H ЯМР соответствовал данной структуре, и чистота по ВЭЖХ составляла >99,9% (AUC @ 226 нм).
C. Процесс из пункта A повторяли, используя 11,5 г 3EPr. К раствору 3EPr (11,5 г) в IPA (70 мл, 6 объемов) добавляли 5-6 н. HCl в IPA (2 эквив.). Реакцию нагревали до 50°C, и судили о полноте протекания по ВЭЖХ анализу после девяти часов. За это время образовалась суспензия белого цвета.
Реакцию охлаждали до температуры окружающей среды, и твердые вещества отделяли фильтрованием через фильтровальную бумагу Whatman® № 1. Лепешку на фильтре промывали IPA (2×15 мл). Твердое вещество сушили под высоким вакуумом до получения 9,01 г (выход 97%) 3E·HCl в виде твердого вещества белого цвета. 1H ЯМР соответствовал данной структуре, и чистота по ВЭЖХ составляла >99,9% (AUC @ 226 нм).
В каждом из предыдущих случаев добавление кислоты в 2-пропаноле можно проводить при более высоких температурах, например, при 55°C или 60°C. Это лучше контролирует выделение газа.
D. Соединение 3EPr (9,0 г, 27,6 ммоль) загружали в круглодонную колбу, за которым следовал 2-пропанол (5 объемов). Суспензию нагревали до 55°C, в ходе чего суспензия становилась гомогенной, и добавляли по каплям 5-6 н. HCl в 2-пропаноле (2 эквив.). Реакционную смесь перемешивали при 55°C в течение четырех часов, в ходе которых формировалась густая суспензия. ВЭЖХ анализ показал завершение реакции. Получившуюся в результате суспензию охлаждали до температуры окружающей среды и фильтровали, промывая 2-пропанолом (2 объема). Влажную лепешку сушили под вакуумом при температуре окружающей среды до получения 3E [MDM-W-11(3), 6,9 г, выход 96%, чистота по ВЭЖХ >99,9%].
E. Реакцию из пункта D масштабировали в шестикратном размере и оценивали по калориметрии реакции (RC1, Mettler-Toledo). Прибор учета расхода газа конструировали и калибровали так, чтобы быть уверенными в точном измерении выделения газа. Соединение 3EPr (56,6 г, 174 ммоль) суспендировали в 2-пропаноле (300 мл), и суспензию нагревали до 55°C, в ходе чего смесь становилась гомогенной. При помощи насоса добавляли хлористоводородную кислоту (1 эквив.) в 2-пропаноле (3,8 M) с линейной скоростью за 30 минут, в ходе чего отмечали отхождение газа и начало преципитации. Реакцию затем оставляли перемешиваться в течение 30 минут перед добавлением хлористоводородной кислоты (1 эквив.) с той же скоростью. Получившуюся в результате суспензию перемешивали в течение четырех часов при 55°C. Суспензию охлаждали до температуры окружающей среды и фильтровали, промывая 2-пропанолом, до получения 44,0 г твердого вещества светло-желтого цвета после высушивания в течение выходных при температуре окружающей среды под вакуумом [MDM-W-56(1), выход 97%, чистота по ВЭЖХ >99,9%]. Наблюдали очень слабый эндотермический температурный профиль, дающий энтальпию реакции -57,8 кДж/моль и изменение адиабатической температуры в -9,6 K. Скорость выделения газа была слабой. Интегрирование кривой расхода массы дало выделение газа в ходе эксперимента в 3,9 л. Кривая расхода массы показала, что скорость выделения газа замедлялась практически сразу после прекращения добавления HCl, давая возможность предположить, что выделение газа целесообразно считать доз-контролируемым.
Пример 4
Превращение в 1E (Стадии 3 и 4)
A. Неочищенный препарат 3E, полученный в примере 3, подвергали комплексообразованию с TFA и реакции с бензальдегидом, и продукт очищали колоночной хроматографией (2-6% метанол/DCM). Фракции, содержащие продукт, собирали, и растворитель удаляли при пониженном давлении до получения соединения 2E в виде густого масла. 1Н ЯМР соответствовал приписываемой структуре. Поскольку 2E представляло собой масло, 3E превращали в соединение 1Е двух стадийным телескопическим способом.
B. К 0,6 г препарата 3E·HCl в 2-пропаноле добавляли триэтиламин (2 эквив.), за которым следовал бензилхлорид (1,2 эквив.). Получившуюся в результате суспензию нагревали до 50°C, в ходе чего она превращалась в прозрачный раствор. За реакцией следили по ВЭЖХ и судили о завершении через три часа.
Реакционную смесь охлаждали до температуры окружающей среды, и твердые вещества (TEA·HCl соль) отфильтровывали. К фильтрату добавляли изоамиламин (10 эквив.), и получившийся в результате раствор нагревали до 75°C. За реакцией следили по ВЭЖХ и обнаружили, что она подверглась только 36% конверсии за 48 часов.
C. Методику из пункта B осуществляли с 3,5 г 3E·HCl в ацетонитриле (20 мл). Реакцию проводили с 1,0 эквивалентом бензилхлорида в присутствии триэтиламина (3,0 эквив.). О полноте протекания реакции судили по данным ВЭЖХ анализа после перемешивания при 50°C в течение 4,5 часов. Реакционную смесь охлаждали до температуры окружающей среды, и твердую фазу отфильтровывали. Фильтрат прокачивали насосом до сухости. Остаток растворяли в изоамиламине (20 мл) и нагревали до 90°C. О полноте протекания реакции судили по данным ВЭЖХ анализа спустя 24 часа. Реакционную смесь охлаждали до температуры окружающей среды и уменьшали объем растворителя до достижения массы остатка в 9,5 г. К этому добавляли гептаны (30 мл), что приводило к образованию светло-коричневой суспензии. Это перемешивали при температуре окружающей среды в течение одного часа и еще часа на бане из воды со льдом. Твердую фазу собирали фильтрованием через фильтровальную бумагу Whatman® № 1, и лепешку на фильтре промывали холодной водой (2×20 мл). Продукт сушили в вакуумной печи, получая в результате 3,98 г (выход 70%) соединения 1E с чистотой по ВЭЖХ >99,9% (AUC @ 226 нм).
D. Альтернативно, к 8,0 г образца 3E·HCl в ацетонитриле (48 мл, 6 объемов) добавляли триэтиламин (2,5 эквив.), за которым следовал бензилхлорид (1,05 эквив.). Получившуюся в результате суспензию нагревали до 50°C, в ходе чего она превращалась в прозрачный раствор. Реакцию контролировали ВЭЖХ, и о полноте протекания судили через 3,5 часа (3,3% непрореагировавшего 3E·HCl). Реакционную смесь охлаждали до температуры окружающей среды, и твердую фазу (TEA·HCl соль) отфильтровывали.
Фильтрат выпаривали до достижения массы раствора 18 г (≈1:1 ацетонитрил/продукт по массе). К этому добавляли изоамиламин (≈4:1 изоамиламин/ацетонитрил, 10 эквив. изоамиламина), и получившийся в результате раствор нагревали до 85°C. О полноте протекания реакции судили по данным ВЭЖХ анализа спустя 19 часов (3,0% непрореагировавшего 2E). Реакционную смесь охлаждали до температуры окружающей среды, и растворитель удаляли при пониженном давлении до установления массы раствора 22 г (1 г растворителя на грамм 1E). При охлаждении получали влажное твердое вещество и его истирали в порошок с гептанами (6 г на грамм 1Е). Суспензию перемешивали при температуре окружающей среды в течение 16 часов, и твердую фазу собирали фильтрованием через фильтровальную бумагу Whatman® № 1, и лепешку на фильтре промывали гептанами (20 мл), затем водой (2×20 мл). Продукт сушили в вакуумной печи при температуре окружающей среды до получения 7,78 г (выход 69% за две стадии) 1E в виде твердого вещества светло-коричневого цвета. Чистота по ВЭЖХ составила >99,9% (AUC @ 226 нм).
E. Стадии 3 и 4 проводили в масштабе 6 г, следуя методике, изложенной выше. Соединение 3E (6,0 г, 22,9 ммоль) суспендировали в ацетонитриле (30 мл) и добавляли триэтиламин (9,6 мл, 3 эквив.), с последующим добавлением бензилхлорида (2,8 мл, 1,05 эквив.). Реакцию нагревали при 50°C в течение 24 часов. Анализ ВЭЖХ, проведенный через 20 часов и еще раз через 24 часа, показал отсутствие дальнейшего прогресса (10,4% 3E остались непрореагировавшими), и реакцию охлаждали до температуры окружающей среды и фильтровали для удаления солей аммония. Раствор затем концентрировали под вакуумом до приблизительно двух объемов, получая концентрированный раствор неочищенного 2E (чистота неочищенного продукта 80%). Затем добавляли изоамиламин (26 мл, 10 эквив.), и реакцию нагревали с обратным холодильником (81°C) в течение 24 часов. Анализ ВЭЖХ, проведенный через 20 часов и еще раз через 24 часа, показал отсутствие дальнейшего прогресса (чистота неочищенного продукта составила 73%), и реакцию охлаждали до температуры окружающей среды и концентрировали при пониженном давлении до приблизительно 4 объемов. Затем добавляли гептаны (35 мл), и получившуюся в результате суспензию перемешивали в течение выходных. Мелкозернистую суспензию отфильтровывали и промывали водой, после чего твердая фаза растворилась, не оставив ничего на воронке фильтра. Двухфазный фильтрат экстрагировали IPA и затем концентрировали до состояния масла. Масло растворяли в IPA (30 мл); медленно добавляли воду (36 мл) до тех пор, пока раствор не становился слегка мутным, и затем добавляли небольшое количество соединения 1E [DSJ-F-20(15)] для того чтобы вызвать кристаллизацию. Получившуюся в результате суспензию фильтровали, промывая водой, и сушили в течение ночи под вакуумом, получая 5,46 г соединения 1Е [MDM-W-26(8), выход 65%, чистота 99,9% по данным ВЭЖХ, 98,6 вес.% по данным 1Η ЯМР].
F. N-бензилирование осуществляли при температурах в интервале от 25°C до 75°C для того чтобы определить оптимальную температуру для реакции и ее термическую устойчивость. Скорость реакции повышалась с температурой, но все приближалось к общей конечной точке 95-96% степени конверсии после 20 часов, вне зависимости от температуры. ВЭЖХ анализ показал небольшую разницу в чистоте неочищенных продуктов, но происходило заметное изменение цвета при температуре выше 45°C, доводящее реакционный раствор до светло-оранжевого состояния. Температуру реакции 45°C считали оптимальной в терминах скорости реакции и сведения к минимуму изменения цвета и расслаивания осадка.
G. Способ можно улучшить путем увеличения количества бензил хлорида до 1,1-1,15 эквивалентов и путем небольшого понижения температуры реакции до 45°C для того, чтобы уменьшить обесцвечивание. Обработку водой после N-бензилирования вводили для удаления примесей, возникающих в результате реакции N-бензилирования до реакции образования 1E. Выделение продукта проводили путем прямой кристаллизации из реакционной смеси (изоамиламина) путем добавления воды в качестве антирастворителя. Потери продукта в фильтрате обычно составляли менее 7%. 1E выделяли в виде твердого вещества белого цвета с выходом приблизительно 80% с очень высокой чистотой.
Пример 5
Полное осуществление схемы 1
A. 50 г препарата 2-хлорникотиновой кислоты обрабатывали N-Boc-пиперазином (1,2 эквив.) в присутствии триэтиламина (2 эквив.) и TBTU (1,4 эквив.). Реакцию проводили в IPAc (300 мл, 6 объемов) и при температуре окружающей среды. О полноте протекания реакции судили по ВЭЖХ анализу спустя 12 часов. После фильтрования и обработки водой IPAc эктракт уменьшали до 180 г под вакуумом (~1:1 IPAc/продукт по массе).
К получившейся в результате суспензии добавляли гептаны (~1:1 IPAc/продукт по массе). Суспензию, которая получилась в результате этого, перемешивали при температуре окружающей среды в течение 16 часов и еще два часа на бане из воды со льдом. Продукт собирали фильтрованием через фильтровальную бумагу Whatman® №, и лепешку на фильтре промывали гептанами (2×25 мл). Продукт сушили в вакуумной печи при температуре окружающей среды до получения 78,53 г (выход 76%) соединения 3EPr в виде твердого вещества коричневого цвета. Чистота по ВЭЖХ составила 98,9% (AUC @ 226 нм).
B. 73,53 г соединения 3EPr, полученного по пункту A, подвергали реакции снятия защитной группы Boc- в присутствии 2 эквивалентов 5-6 н HCl в IPA. Реакцию проводили при 50°C в IPA (6 объемов). О полноте протекания реакции судили по данным ВЭЖХ анализа спустя семь часов. Реакционную смесь охлаждали до температуры окружающей среды и фильтровали через фильтровальную бумагу Whatman® № 1. Лепешку на фильтре промывали IPA (2×50 мл) и сушили при высоком вакууме до получения 56,31 г (выход 95%) 3E·HCl в виде твердого вещества коричневого цвета. Чистота по ВЭЖХ составила >99,9%.
C. 54,0 г препарата 3E·HCl затем обрабатывали бензилхлоридом (1,05 эквив.) в присутствии триэтиламина (3 эквив.). Реакцию проводили при 50°C в ацетонитриле (6 объемов). О полноте протекания реакции судили по данным ВЭЖХ анализа спустя восемь часов. Реакционную смесь охлаждали до температуры окружающей среды, и твердые вещества отфильтровывали через фильтровальную бумагу Whatman® № 1. Лепешку на фильтре промывали ацетонитрилом (2×25 мл). Растворитель удаляли при пониженном давлении до получения массы раствора 110 г (≈1:1 ацетонитрил/продукт по массе).
К нему добавляли изоамиламин (220 г) до получения соотношения изоамиламин/ацетонитрил 4:1. Получившийся в результате раствор нагревали до 85°C, и о полноте протекания реакции судили по данным ВЭЖХ анализа спустя 22 часа. Реакционную смесь охлаждали до температуры окружающей среды и удаляли растворитель при пониженном давлении до получения массы раствора 150 г. К получившейся в результате смеси добавляли гептаны (6 объемов). Суспензию перемешивали при температуре окружающей среды в течение 16 часов, и твердые вещества собирали фильтрованием через фильтровальную бумагу Whatman® №, и лепешку на фильтре промывали гептанами (250 мл×2), а затем водой (250 мл×2). Продукт сушили в вакуумной печи при температуре окружающей среды до получения 60,66 г (выход 80% за две стадии) 1E в виде твердого вещества светло-коричневого цвета. Чистота по данным ВЭХЖ составила >99,9% (AUC @ 226 нм).
Пример 6
Получение фосфатной соли
A. 22 литровую трехгорлую круглодонную колбу снабжали капельной воронкой, дефлегматором, термопарой, и в колбонагреватель помещали верхнеприводную мешалку. В колбу загружали этанол (7,9 л, Pharmco lot # 0802062), затем деионизированную воду (420 мл). Следующим в реактор загружали 1E (700 г, 2,1 моль), и получившуюся в результате смесь нагревали до 75°C. 1 M раствор H3PO4 в этаноле (4,5 л, 4,5 моль, 2,1 эквив.) загружали быстрым потоком за период времени 30 мин. Получившуюся в результате смесь перемешивали в течение 15 мин, и добавляли 1Ε·Η3ΡО4 (0,5 г) в качестве затравки для перекристаллизации. Получившийся в результате прозрачный раствор охлаждали до температуры окружающей среды со скоростью 20°C/ч.
Охлажденную суспензию перемешивали при температуре окружающей среды в течение 11 ч и фильтровали через фильтровальную бумагу Whatman® № 1. Для содействия переносу и для промывания лепешки на фильтре использовали этанол (2,8 л×2). Продукт сушили под вакуумом до постоянной массы при 25°C до получения 1Ε·Η3Ρ04 в виде твердого вещества белого цвета (751 г, выход 62%). ВЭЖХ анализ показал чистоту >99,9% (AUC @ 226 нм) и 1H ЯМР соответствовал данной структуре.
B. Соединение 1E (4,9 г, 13,3 ммоль) растворяли в 5% смеси воды в этаноле при 75°C и затем добавляли 1M фосфорную кислоту в этаноле (2,1 эквив.) и затем добавляли 1М раствор фосфорной кислоты в этаноле (2,1 эквив.). Получившийся в результате раствор охлаждали до температуры окружающей среды со скоростью 20°C/ч, в ходе чего быстро образовывался преципитат. Смесь повторно нагревали для растворения преципитата и затем в систему вносили затравку 1E фосфата и охлаждали как описано выше. Получившуюся в результате суспензию перемешивали в течение ночи при температуре окружающей среды и затем фильтровали, промывая этанолом, до получения 4,9 г 1E фосфата (выход 79%, >99,9% по ВЭЖХ) в виде твердого вещества белого цвета. Результаты показали, что внесение затравки является существенным для формирования кристаллов соответствующей формы.
C. Четыре реакции, приводящие к образованию фосфатной соли, проводили в масштабе 10 г при следующих условиях:
MDM-W-126: 1,25 эквивалентов H3PO4, 12 объемов EtOH
MDM-W-128: 1,25 эквивалентов H3PO4, 12 объемов 5% воды в EtOH
MDM-W-130: 1,0 эквивалент H3PO4, 12 объемов
MDM-W-131: 1,0 эквивалент H3PO4, 12 объемов 5% воды в EtOH
Каждую реакцию нагревали до 70°C, вносили затравку 1E фосфата [0,1 вес.%, DAJ-F-40(2)] и охлаждали до 20°C со скоростью 20°C/час. Получившуюся в результате густую суспензию перемешивали в течение ночи, фильтровали (промывая EtOH) и сушили до постоянной массы. Результаты этих реакций представлены в таблице 1. Вообще, суспензии, получаемые в реакциях, проводимых с использованием 5% воды в EtOH, были более удобны в обращении.
Скрининг образования фосфатной соли
Физические свойства монофосфатной соли
Растворимость в воде составила >36 мг/мл в условиях температуры окружающей среды, и соль является кристаллической по данным XRPD анализа.
DSC анализ показал один эндотермический процесс при 179°C, который соответствовал плавлению.
Анализ влагопоглощения показал, что вещество является умеренно гигроскопичным, адсорбируя 4,4 вес.% воды при относительной влажности 60% и 11,2 вес.% при относительной влажности 90%.
Анализ по непосредственным составляющим показал отношение формулы (1) к противоиону от 1:1,6 до 1:2,3 в различных партиях соли.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОЧИСТКА 2-НИТРО-4-МЕТИЛСУЛЬФОНИЛБЕНЗОЙНОЙ КИСЛОТЫ | 2002 |
|
RU2287521C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2125561C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 5-[2-[7-(ТРИФТОРМЕТИЛ)-5-[4-(ТРИФТОРМЕТИЛ)ФЕНИЛ]ПИРАЗОЛО[1,5-a]ПИРИМИДИН-3-ИЛ]ЭТИНИЛ]-2-ПИРИДИНАМИНА | 2012 |
|
RU2630700C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-(3-(4-(1-АМИНОЦИКЛОБУТИЛ)ФЕНИЛ)-5-ФЕНИЛ-3H-ИМИДАЗО[4,5-B]ПИРИДИН-2-ИЛ)ПИРИДИН-2-АМИНА | 2015 |
|
RU2677478C2 |
| СОЛЬ (СОЛИ) ДИМЕТИЛАМИДА 7-ЦИКЛОПЕНТИЛ-2-(5-ПИПЕРАЗИН-1-ИЛ-ПИРИДИН-2-ИЛАМИНО)-7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-6-КАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2631243C2 |
| СИНТЕЗ N-(4-ФТОРБЕНЗИЛ)-N-(1-МЕТИЛПИПЕРИДИН-4-ИЛ)-N'-(4-(2-МЕТИЛПРОПИЛОКСИ)ФЕНИЛМЕТИЛ)КАРБАМИДА, А ТАКЖЕ ЕГО ТАРТРАТА И КРИСТАЛЛИЧЕСКИХ ФОРМ | 2005 |
|
RU2417986C2 |
| Имидазопирролопиразиновые производные, полезные для лечения заболеваний, вызванных аномальной активностью протеинкиназ Jak1, Jak3 или Syk | 2010 |
|
RU2711869C2 |
| СОЛЬ ОМЕКАМТИВА МЕКАРБИЛА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2663663C2 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ ЧИСТОГО ИЛИ ОБОГАЩЕННОГО ПИПЕРАЗИН-2- ТРЕТ.БУТИЛКАРБОКСАМИДНОГО СУБСТРАТА И РАЦЕМИЧЕСКИЙ 2-ТРЕТ-БУТИЛКАРБОКСАМИД-4-(3- ПИКОЛИЛ)ПИПЕРАЗИН | 1995 |
|
RU2135482C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3S-3-АМИНО-3-ПИРИДИЛПРОПИОНОВОЙ КИСЛОТЫ И ЕЕ ПРОИЗВОДНЫХ И ПРОМЕЖУТОЧНОЕ ВЕЩЕСТВО | 2000 |
|
RU2228929C2 |

Изобретение относится к области органической химии, а именно к способу получения производного пиперазина формулы (1), где R1 представляет собой C1-С5алкил; R2 является Н; n составляет 0; m составляет 0; включающий взаимодействие соединения формулы (2) с соединением формулы 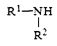 , где R1 представляет собой С1-С5алкил; R2 является Н; в присутствии апротонного растворителя при температуре между 80°C и 90°C. Технический результат: разработан новый способ получения соединения формулы (1), которое может быть полезно в качестве средства, стимулирующего нейрогенез. 5 з.п. ф-лы, 4 ил., 1 табл., 6 пр.
, где R1 представляет собой С1-С5алкил; R2 является Н; в присутствии апротонного растворителя при температуре между 80°C и 90°C. Технический результат: разработан новый способ получения соединения формулы (1), которое может быть полезно в качестве средства, стимулирующего нейрогенез. 5 з.п. ф-лы, 4 ил., 1 табл., 6 пр.

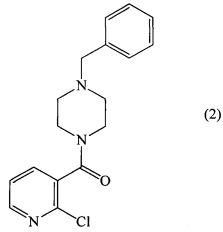
1. Способ получения соединения формулы:
в которой
R1 представляет собой C1-С5алкил;
R2 является Н;
n составляет 0;
m составляет 0;
включающий взаимодействие соединения формулы
с соединением формулы
где
R1 представляет собой С1-С5алкил;
R2 является Н;
в присутствии апротонного растворителя при температуре между 80°C и 90°C.
2. Способ по п.1, где соединение формулы (2) получают взаимодействием соединения формулы
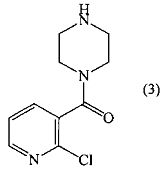
с соединением формулы
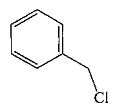
в присутствии слабого основания и апротонного растворителя при температуре между 45°C и 60°C.
3. Способ по п.2, где соединение формулы (3) получают взаимодействием соединения формулы
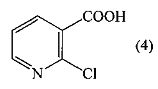
с соединением формулы
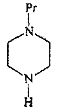
в которой Pr представляет собой защитную группу, в присутствии агента, способствующего образованию пептидной связи в присутствии слабого основания и малополярного непротонного растворителя при температуре между 20°C и 30°C, с последующим удалением защитной группы с использованием сильной кислоты в присутствии спиртового растворителя при температуре между 50°C и 60°C.
4. Способ по п.3, где Pr представляет собой Boc или Fmoc.
5. Способ по п.1, дополнительно включающий превращение соединения формулы (1) в фармацевтически приемлемую соль.
6. Способ по п.5, где соль является фосфатной солью.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

