Настоящая заявка является частичным продолжением заявки 08/093225, поданной 16 июля 1993 г.
Настоящее изобретение относится к новым промежуточным соединениям и способу получения соединений, которые ингибируют протеазу, кодируемую вирусом иммунодефицита человека (ВИЧ); и, в частности, L-735524, или к их фармацевтически приемлемым солям. Эти соединения представляют ценность для предотвращения инфицирования HIV, для лечения инфекций, вызванных ВИЧ, и для лечения возникающего синдрома приобретенного иммунодефицита (СПИД).
Более конкретно, рассматриваемый способ включает реакцию амиденолята, полученного из амида, такого как N-(2(R)-гидрокси-1(S)-индалил)-3-фенилпропанамид, с активированным нерацемическим производным глицидола, таким как 2(S)-глицидилтозилат, с получением эпоксидного промежуточного соединения, которое является ключевым промежуточным соединением, необходимым для получения соединений, являющихся ингибиторами ВИЧ протеазы, включая L-735524. Предложен также усовершенствованный способ синтеза специфических диалкиламинов, используемых для получения ингибиторов ВИЧ протеазы.
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), представляет собой этиологический агент комплексного заболевания, которое включает прогрессирующее разрушение иммунной системы (синдром приобретенного иммунодефицита, СПИД) и вырождение центральной и периферической нервной системы. Этот вирус ранее был известен как LAV, HTLV, -III или ARV. Общей особенностью ретровирусной репликации является интенсивный пост-трансляционный процессинг предшественников полипротеинов за счет кодируемой вирусом протеазы с выработкой зрелых вирусных протеинов, требуемых для сборки и функционирования вирусов. Ингибирование такого процессинга предотвращает продуцирование обычно инфекционного вируса. Так например, авторы Kohl, N.E. et al. Proc. Nat' (Acad. Sci., 85, 4686 (1988) продемонстрировали, что генетическая инактивация ВИЧ кодируемой протеазы приводит к продуцированию незрелых, неинфекционных вирусных частиц. Эти результаты указывают на то, что ингибирование ВИЧ протеазы представляет ценный метод для лечения СПИД и предотвращения или лечения инфицирования ВИЧ.
Нуклеотидная последовательность ВИЧ демонстрирует наличие pol гена в открытой считывающей рамке (Rather, L. et. al., Nayure, 313, 277 (1985). Гомологичность аминокислотной последовательности представляет доказательство того, что pol последовательность кодирует обратную транскриптазу, эндонуклеазу и ВИЧ протеазу (Toh, H. et. al, EMBO J., 4, 1267 (1985); Power, M.D., et al. , Science, 231, 1567 (1986); Pearl, L.H. et al., Nature, 329, 351 (1987)). Конечные продукты-соединения, включая L-735524, который представлен в примере 4 далее, которые можно получить из промежуточных соединений согласно изобретению и по способу настоящего изобретения, являются ингибиторами ВИЧ протеазы, и раскрыты в EPO 541168, опубликованный 12 мая 1993 г.
Ранее синтез L-735524 и родственных соединений осуществляли с помощью 12-стадийного процесса, в котором использовали гидрокси-защищенный дигидро-5(S)-гидроксиметил-3-(2H)-фуранон, который алкилировался, способ включал замену спиртовой отщепляемой группы на алкилированном фураноне пиперидиновым фрагментом. Затем продукт присоединения гидролизовали с раскрытием фуранонового кольца в гидроксикислотный фрагмент, и эту кислоту, наконец, присоединяли к 2(R)-гидрокси-1(S)-аминоиндану. Этот процесс описан в EPO 541168. Крайняя громоздкость этой схемы (12 стадий) делает этот способ длительным и трудоемким, и требует использования множества дорогостоящих реагентов и дорогих исходных материалов. Способ, требующий меньше стадий и реагентов, обеспечил бы экономические и времясберегающие преимущества.
В EPO 541168 представлен также модифицированный способ получения L-735524 и родственных соединений, основанный на диастереоселективном алкилировании енолята, получаемого из N-(2(R)-гидрокси-1(S)-индан-N,O-изопропилиден-ил)-3-фенилпропанамида, в котором C3-C5 трехуглеродный фрагмент вводят в виде аллильной группы, а затем окисляют. С этим способом связаны некоторые следующие проблемы: (a) для введения трехуглеродного глицидильного фрагмента необходимы 4 стадии, (b) в этом способе используют высокотоксичный OsO4, и (c) на стадии дигидрооксилирования достигается низкая диастереоселективность. Таким образом, необходим способ, при котором трехуглеродный фрагмент вводился бы непосредственно в нужной хиральной окисленной форме.
Кроме того, синтез хирального пиперазинового промежуточного соединения осуществлялся из 2-пиразинкарбоновой кислоты по способу из 6 стадий и требовал использования дорогих реагентов, таких как BOC-ON и EDC (этилендихлорид). Таким образом, желателен был бы также и более короткий путь к пиперазиновому промежуточному производному, в котором не нужно было бы использовать дорогие реагенты.
Из литературы известны несколько примеров конденсации стабилизированных карбанионов с глицидолом и его производными (активированными или неактивированными); однако ни один из известных способов не дает новый эпоксид с хорошим выходом. См. , например, Hanson, R.M. Chem. Rev. 1991, 91, 437-475. В случае активированных глицидольных производных и нуклеофилов углерода это связано, главным образом, с ожидаемым и нежелательным "двойным" присоединением нуклеофила к эпоксидному продукту. Далее, ни в одном из известных примеров нет амидного фрагмента как карбанион стабилизирующей группы (амидонолят), и ни один из известных примеров не включает присоединение содержащей стабилизированный карбанион хиральности к хиральному, нерацемическому глицидольному производному (двойная диастереоселективность).
Была продемонстрирована конденсация стабилизированных карбанионов с активированными нерацемическими глицидольными производными, малонатный анион присоединяли как к нерацемическому эпихлоргидрину i, так и к нерацемическому глицидилтрифлату ii, с получением циклопропил-лактона iii. См., например, Pirrung, M. C. , et al., Helvetica Chimica Acta 1989, 72, 1301-1310, и Burgess, K. , et al. J. Org. Chem. 1992, 57, 5931-5936. В данном случае промежуточный эпоксид подвергается дальнейшей реакции, давая циклопропильную кольцевую систему. В случае i, начальная реакция с малонатным анионом проходит по эпоксидному концу /C3/, тогда как в случае ii, начальная реакция протекает по трифлатному C1 концу.

Родственным примером является реакция сульфон-стабилизированного карбаниона, полученного из V, с глицидилтозилатом iv с получением гидрокситозилатом vi. См. Baldwin J. E. et al., J. Chem. Soc., Chem. Commun. 1992, 1249-1251. В этом случае, хотя двойное присоединение карбаниона и не является основной проблемой, необходима дополнительная стадия для превращения промежуточного гидрокси-тозилата vi в целевой эпоксид vii.

Аналогично, из литературы неизвестно и не предполагается, что азотсодержащие нуклеофилы можно селективно присоединять к активированным производным глицидола, снимая при этом проблему двойного присоединения.
Известна также конденсация амидэнолятов, полученных из N-(2(R)-гидрокси-1(S)-индан-N, O-изопропилиден-ил)-3-фенилпропан-амида 1, с защищенными альфа-аминоэпоксидами viii с получением целевого гидроксиэтилендипептидного изостерного производного ix с высокой степенью стереоконтроля в отношении C2-(R)-стереоцентра. См., например, Askin, D., et al. J.Org.Chem. 1992, 57, 2771-2773 и патент США 5169952 Askin, D., et al. После гидролиза получают гидроксиэтилендипептидные (с удаленной защитой) изостерные ингибиторы.

Известно разделение 2-пиперазинкарбоновой кислоты с помощью (+)-CSA. См. , например, Felder, E et al., Helvetica Chim. Acta, 1960, 43, 888. Однако в литературе известны примеры разделения пиперазинамидов.
В настоящем изобретении предложен усовершенствованный способ получения ингибиторов ВИЧ протеазы по сравнению с известными ранее. Кроме того, он значительно короче, гораздо более диастереоселективен, приводит к более высоким выходам соединений, раскрытых в EPO 541168, и в частности L-735524, не требует использования таких токсических реагентов, как тетраоксид осмия, или таких исключительно дорогостоящих реагентов, как (S)-(+)-дигидро-5-(гидроксиметил)-2(3H)-фуранон.
Краткое содержание изобретения
Настоящее изобретение включает новые способы синтеза таких промежуточных эпоксидов, как 3, которые используют для синтеза ингибиторов ВИЧ протеазы. Изобретение включает также реакцию амидэнолята, такого как энолят, полученный из 1, с активированным нерацемическим производным глицидола, таким как 2(S)-глицидилтозилат 2, с получением эпоксидного продукта, такого как 3, в одну стадию и с высоким выходом. Результат этой реакции является неожиданным, так как предполагалось, что эпоксид 3 будет далее вступать в реакцию в условиях реакций присоединения, давая большое количество димерного продукта 3-a, и таким образом давая низкий выход продукта 3.

Настоящее изобретение относится также к промежуточным эпоксидным соединениям и способам присоединения промежуточных эпоксидов к амиду формулы V, определенному далее, с образованием ВИЧ протеазных промежуточных и конечных продуктов.
В описании использованы следующие сокращения:
Обозначение - Защитная группа
BOC /Boc/ - трет-бутилоксикарбонил
CBz /Cbz/ - бензилоксикарбонил/карбобензокси/
TBS /TBDMS/ - трет-бутил-диметилсилил
Обозначение - активирующая группа
Ts или тозил или тозилат - пара-толуолсульфонил
Ns или нозил или нозилат - 3-нитробензолсульфонил
Tf или трифлил или трифлат - трифторметансульфонил
Ms или мезил или мезилат - метансульфонил
Обозначение - Реагент присоединения
BOP реагент - бензотриазол-1-илокситрис(диметиламино)-фосфонийгексафторфосфат
BOP-Cl - бис(2-оксо-3-оксазолидинил)фосфинивый хлорид
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлорид - Другие
BOC-ON - 2-(трет-бутилкарбонилоксиимино)-2-фенилацетонитрил
(BOC)2O (BOC2O или Boc2O) - ди-трет-бутилдикарбонат
n-Bu4N+F- - тетрабутиламмонийфторид
nBuLi (n-Buli) - н-бутиллитий
(S)-CSA - (1S)-(+)-10-камфорсульфоновая кислота
DIEA или DIPEA - диизопропилэтиламин
DMAP - диметиламинопиридил
DME - диметоксиэтан
DMF - диметилформамид
Et3N - триэтиламин
EtOAc - этилацетат
h - час/часы/
IPA - 2-пропанол
LDA - литийдиизопропиламид
L-PGA - (L)-пироглутаминовая кислота
TFA - трифторуксусная кислота
THF - тетрагидрофуран
TLC - Тонкослойная хроматография /ТСХ/
Подробное описание изобретения
В настоящем изобретении предложен новый способ получения промежуточных соединений формулы I и IV, которые используют при получении ингибиторов ВИЧ протеазы, и в частности соединений, раскрытых в EPO 541168. Способ получения промежуточных соединений формулы I: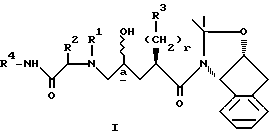
включает стадии:
/1/ взаимодействие или
a. глицидола формулы II
b. эпихлоргидрина формулы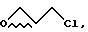
с амидом формулы III
в присутствии сильного основания, при низкой температуре, с получением аддукта IV
/2/ взаимодействие IV с амином формулы V с получением продукта I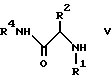
где стереоцентра соответствует либо R конфигурации, либо S конфигурации, либо является рецемическим;
X выбирают из группы, состоящей из -H, метансульфонила, трифторметансульфонила, пара-толуолсульфонила, бензолсульфонила и 3-нитробензолсульфонила;
r является целым числом от 0 до 5 /включительно/;
R1 и R2 независимо выбирают в каждом случае из группы, состоящей из 1/ водорода, 2/ -C1-4 алкила, незамещенного или замещенного одним или более из
а/ гидрокси,
b/ C1-3 алкокси,
c/ арила, незамещенного или замещенного одним или более из C1-4 алкила, гидрокси или арила,
d/ -W-арила или -W-бензила, где W представляет -O- или -S-,
e) 5-7-членной циклоалкильной группы, незамещенной или замещенной одним или более из:
i/ гидрокси, ii/ C1-3 алкокси, или iii/ арила,
f/ гетероцикла, незамещенного или замещенного одним или более из гидрокси, C1-4 алкила, C1-4 алкила, замещенного гидрокси или Boc,
g/ -NH-COOC1-3 алкила,
h/ -NH-CO-C1-3 алкила
i/ -NH-SO2C1-3 алкила,
j/ -COOR, или
k/ -//CH3/mO/n R, или
3/ арила, незамещенного или замещенного одним или более из группы:
a/ галоида,
b/ гидрокси,
c/ -NO2 или -N/R/2,
d/ C1-4 алкила,
e/ C1-3 алкокси, незамещенного или замещенного одним или более из -OH или C1-3 алкокси,
f/ -COOR,
g/ -CON/R/2,
h/ -CH2N/R/2,
i/ -CH2NHCOR,
j/ -CN,
k/ -CF3,
l/ -NHCOR,
арил C1-3 алкокси,
n/ арила,
o/ -NRSO2R,
p/ OP(O)(ORx)2, или
q) -R5, как определен ниже; или
R1 и R2 могут быть соединены вместе с азотом, к которому присоединен R1, и углеродом, к которому присоединен R2, с образованием 3-10 членной моноциклической или бициклической насыщенной кольцевой системы, которая состоит из азота, к которому присоединен R1, и 2 - 9 атомов углерода, например, такой как:
и которая не замещена или замещена одним или более из:
1/ гидрокси,
2/ C1-4 алкила, незамещенного или замещенного одним или более из: a/ галоида, b/ гидрокси, c/ C1-3 алкокси, d/ арила, e/ 5-7-членной циклоалкильной группы, незамещенной или замещенной одним или более из i/ гидрокси, ii/ C1-3 алкокси, или iii/ арила или f/ гетероцикла,
3/ C1-3 алкокси,
4/ -NH-COOC1-3алкила,
5/ -NH-CO-C1-3алкила,
7/ гетероцикла,
8/ -W-арила или
9/ -W-CO-арила, где W имеет указанные ранее значения; или
R1 и R2 могут быть соединены вместе с азотом, к которому присоединен R1, и углеродом, к которому присоединен R2 с образованием 3-10 членной моноциклической или бициклической насыщенной кольцевой системы, которая состоит из азота, к которому присоединен R1, 1 - 8 атомов углерода и одного или более незамещенного или замещенного гетероатома, выбранного из
1/ 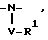
где V отсутствует или представляет -CO-Q- или -SO2-Q-,
R1 имеет указанные ранее значения для случая, когда R1 не зависит от и не присоединен к R2,
и когда Q отсутствует или представляет -O-, -N(R)- или гетероцикл, необязательно замещенный -C1-4 алкилом,
2/ 
3/  алкенила, незамещенного или замещенного арилом,
алкенила, незамещенного или замещенного арилом,
4/ 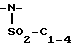 алкенила, незамещенного или замещенного арилом,
алкенила, незамещенного или замещенного арилом,
5/ -S(O)p-, где p представляет 0, 1 или 2, или
6/ -O-,
такой как, например,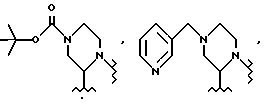
R3 выбирают из группы, состоящей из
1/ водорода,
2/ -C1-4 алкила,
3/ C5-C10 циклоалкила, необязательно замещенного гидроксигруппой,
4/ C6-C10 арила, незамещенного или замещенного одним или более из:
a/ галоида
b/ гидрокси
c/ -NO2 или -N(R)2,
d/ C1-4 алкила,
e/ C1-3 алкокси, незамещенный или замещенный одним или более из -OH или C1-3 алкокси,
f/ -COOR,
g/ -CON(R)2,
h/ -CH2N(R)2,
i/ -CH2NHCOHR,
j/ -CN,
k/ -CF3,
l/ -NHCOR,
m/ арил C1-3 алкокси,
n/ арила,
o/ -NRSO2R,
p/ -OP(O) (ORx)2, или
q/ -R5, как определен ниже, или
5/ моноциклического или бициклического гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, O и S, например, 2-приидила, 3-пиридила или 4-пиридила, и который незамещен или замещен R5 и необязательно одним или более из:
a/ галоида, b/ C1-4 алкила, или c/ C1-3 алкокси; m = 2, 3, 4 или 5; n = 0, 1, 2 или 3; R представляет водород или C1-4 алкил; Rx представляет H или арил; R4 представляет C1-5 алкил, разветвленный или неразветвленный; а
R5 представляет 1/ -W-(CH2)m-NR6R7, где W и m имеют указанные ранее значения, а R6 и R7 независимо выбирают в каждом случае из:
a/ водорода,
b/ C1-6 алкила, незамещенного или замещенного одним или более из i/ C1-3 алкокси, ii/ -OH, или iii/ -N/R/2,
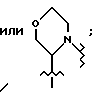
c/ ароматического гетероцикла незамещенного или замещенного одним или более из i/ C1-4 алкила, или ii/ -N/R/2, d/ или R6 и R7 соединены вместе с азотом, к которому они присоединены с образованием 5 - 7 членного гетероцикла, такого как морфолино, содержащего вплоть до двух дополнительных гетероатомов, выбранных из -N/R/, -O-, -S-, -S(O) или -S(O)2, гетероцикла, необязательно замещенного C1-4 алкилом,
2/ -(CH2)q-NR6R7, где q является целым числом от 1 до 5, а R6 и R7 имеют указанные ранее значения, за исключением того, что R6 или R7 не представляют H или незамещенный C1-6 алкил, или
3/ бензофурила, индолила, азациклоалкила, азабицикло C7-11 циклоалкила, или бензопиперидинила, незамещенного или замещенного C1-4 алкилом.
Представленная далее схема 1 иллюстрирует этот процесс. Однако рассматриваемый способ не ограничен каким-либо конкретными заместителями, которые используют в схеме, которую используют для целей иллюстрации.

Промежуточные соединения формулы IV получают в результате реакции глицидола или его производного II и амида III в присутствии сильного основания. Сильное основание должно быть металлсодержащим основанием, и реакция может протекать в инертном безводном органическом растворителе, таком как, например, циклические или ациклические углеводороды, включая гексан, пентан, циклогексан и т.д. или без него. Подходящие сильные основания включают: LiN[(CH3)3Si] 2, KN[(CH3)3Si]2, NaN[(CH3)3Si]2, н-бутиллитий (н-BuLi), S-BuLi, трет-BuLi, трет-бутоксид калия, литийдиизопропиламид (LDA), литийизопропилциклогексиламид, литийпирролидид, литийтетраметилпиперидид, фениллитий, изопропилмагнийхлорид, изобутилмагнийхлорид и другие аналогичные сильные основания, известные специалистам. Предпочтительными сильными основаниями являются n-BuLi, S-BuLi,
LiN[(CH3)3Si]2 и LDA, причем наиболее предпочтительны n-BuLi и LiN[(CH3)3Si]2.
Предпочтительно использовать около 1-2 молярных эквивалентов сильного основания на 1 молярный эквивалент III, причем отношение около 1,15 : 1 молярных эквивалентов основания к III наиболее предпочтительно. Реакцию II и III можно осуществить, объединяя II и III в одном реакторе, а затем добавляя сильное основание, или это можно осуществить последовательно, то-есть вначале обрабатывая амин III основанием, а затем добавляя II. Сильное основание осуществляет замену водорода металлом в амиде III в положении альфа к карбонильной группе, с получением реакционноспособного металламидэнолята, который затем осуществляет раскрытие кольца эпоксида II в концевом положении, с получением продукта IV. Новый центр асимметрии создается в продукте изостера IV в положении 2.
Реакцию предпочтительно вести при низкой температуре, например, в интервале от около -82oC до 0oC. Для осуществления металлирования амида III температурный интервал поддерживают предпочтительно, от -82oC до -40oC, и наиболее предпочтительно, в интервале от -50oC до -45oC. Для осуществления взаимодействия между металлированным амидом и II с получением IV, температурный интервал поддерживают более предпочтительно от около -50oC до -10oC, и наиболее предпочтительно, от около -30 до -20oC в течение 4 - 5 часов
Предпочтительно, реакцию II с III ведут в эфирном растворителе. Эфирным растворителем является любой из растворителей, пригодных для использования на этой стадии реакции, включая, например, тетрагидрофуран, 1,2-диметоксиэтан, диэтиловый эфир и метил-трет-бутиловый эфир или их сочетания, причем предпочтителен тетрагидрофуран.
Соединение формулы I получают при взаимодействии соединения формулы IV с амином формулы V. Предпочтительно использовать от около 1 до около 3 молярных эквивалентов амина V на молярный эквивалент эпоксида IV, причем наиболее предпочтительно соотношение молярных эквивалентов V : IV равное около 1,05 : 1.
Эту реакцию можно вести в любом подходящем растворителе, например, выбранном из углеводородов, таком как толуол, таких простых эфирах как диэтиловый эфир, таком спирте как метанол, этанол или изопропанол, в таких нитрилах как ацетонитрил, и в таких сложных эфирах, как этилацетат, или в их комбинациях, причем предпочтительны спирты, а наиболее предпочтителен изопропанол. Температуру реакции можно поддерживать в интервале от комнатной до температуры кипения с обратным холодильником используемого растворителя, но предпочтительно вести реакцию при повышенных температурах, например, в интервале от 80oC до 90oC, и наиболее предпочтительно от около 83oC до 85oC.
Активированные глицидолы формулы II можно получить известными специалистам способами, такими как описанные, например, J. Klunder et. al, J. Org, Chem., 1989, 54 1295-1304 и в приведенных там ссылках.
Амиды формулы III можно получать в соответствии со стандартными процедурами, известными специалистам, такими как процедура примера 1, используя соответствующие исходные материалы.
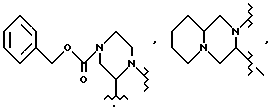
При необходимости в практике настоящего изобретения можно использовать такие защитные группы, как азотзащищающие группы. Например, азот в 4-положении 2-трет-бутилкарбоксамидпиперазина можно защитить такой группой, как BOC, CBZ, бензил, 4-метоксибензил, 2,4-диметоксибензил, трифторацетамид, триалкилсилил, или другими группами, известными специалистам.
Конечный продукт, ингибитор ВИЧ протеазы, получают из соединений формулы I с помощью удаления любых оставшихся защитных групп, в соответствии со способами удаления защитных групп, известными специалистам. Так, например, кетальные защитные группы можно удалить обработкой соединения I кислотой в присутствии метанола, или водной кислотой или 1 н HCl в ТГФ, с получением конечного продукта, ингибитора ВИЧ протеазы. Соединения формулы I можно далее заместить способами, известными специалистам.
В одном из вариантов настоящего изобретения стреоцентр a имеет S конфигурацию; X представляет пара-толуолсульфонил; r = 1; R1 и R2 соединены вместе с образованием циклической структуры, выбранной из группы, состоящей из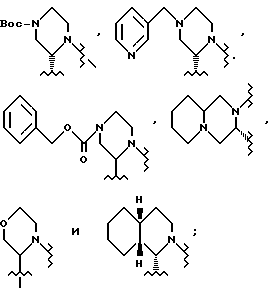
R3 выбирают из фенила,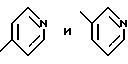
R4 представляет трет-бутил.
В этом варианте предпочтительным соединением формулы II является промежуточное соединение формулы IV-a:
Также в этом варианте предпочтительны соединения формулы I, которые являются промежуточными соединениями формул 2-a и 1-b: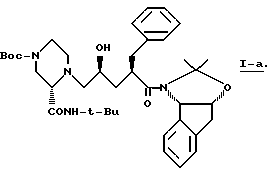
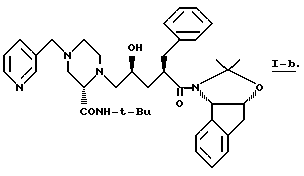
Соединение -1-b можно получить непосредственным присоединением 2(S)-трет-бутилкарбоксамид-4N-(метил-3-пиридил)-пиперазина к IV-a. Предпочтительно, конечный продукт L-735524 получают путем удаления защиты и пиколилирования 1-a, как указано в примерах 3-4.
Способы и промежуточные соединения настоящего изобретения пригодны для получения конечных продуктов, соединений, которые годятся для ингибирования ВИЧ протеазы, для предотвращения или лечения инфекции вирусом иммунодефицита человека (ВИЧ) и лечения сопутствующих патологических состояний, таких как СПИД. Лечение СПИД или предотвращение или лечение ВИЧ инфекции определяется как включающее /но не ограничивающееся этим/ лечение широкого круга состояний ВИЧ инфекций: СПИД, ARC (связанных со СПИД осложнений), как симптоматических, так и асимптоматических, а также случаев фактического или потенциального контакта с ВИЧ. Так например, конечные продукты, соединения, которые можно получить по способу настоящего изобретения, и промежуточные соединения настоящего изобретения пригодны для лечения ВИЧ инфекций после подозрений на возможное инфицирование, например, после переливания крови, трансплантации органов, замены биологических жидкостей, укусов насекомых, случайных уколов иглами или соприкосновения с кровью пациента во время операций.
Конечный продукт, ингибиторы ВИЧ протеазы, можно также использовать для препаратов и осуществления анализов скрининга на антивирусные соединения. Так например, конечные продукты можно использовать для выделения энзимных мутантов, которые являются прекрасными инструментами для скрининга при поиске более сильных антивирусных соединений. Кроме того, такие соединения можно использовать для установления или определения сайтов связывания других антивирусов с ВИЧ протеазой, например, при конкурирующем ингибировании. Такие конечные соединения, которые получены по способу настоящего изобретения, а также промежуточные соединения представляют собой промышленные и коммерческие продукты, предназначенные для продажи для этих целей.
Соединения-ингибиторы ВИЧ протеазы, которые можно получить из промежуточных соединений и в соответствии со способами настоящего изобретения, раскрыты EPO 541164. Ингибиторы ВИЧ протеазы можно вводить нуждающимся в таком лечении пациентам в фармацевтических композициях, содержащих фармацевтический носитель и терапевтически эффективное количество соединения или его фармацевтически приемлемой соли. В EPO 541164 раскрыты подходящие фармацевтические композиции, способы введения, формы солей и дозировки для этих соединений.
Соединения настоящего изобретения могут иметь асимметрические центры и имеют место в виде рацематов, рацемических смесей и в виде отдельных диастереоизомеров или энантиомеров, причем все изомерные формы входят в объем настоящего изобретения.
Если какая-либо переменная (например, арил, гетероцикл, R, R1, R2, n, X и т. д. ) встречается более одного раза в любом составляющем или в формулах I-V, его определение в каждом случае не зависит от его определений в каждом из других случаев. Кроме того, допустимы комбинации заместителей и/или переменных, только если такие комбинации приводят к стабильным соединениям.
В том смысле, как здесь использован, за исключением особых указаний, "алкил" включает как разветвленные, так и неразветвленные насыщенные алифатические углеводородные группы, содержащие определенное число атомов углерода (Me представляет метил, Et представляет этил, Pr-пропил, Bu-бутил; трет-Bu); "алкокси" представляет алкильную группу с указанным числом атомов углерода, присоединенную через кислородный мостик; "циклоалкил" включает насыщенные кольцевые группы, такие как циклопропил, циклобутил, циклопентил, циклогексил /Cyh/ и циклогептил. "Алкенил" включает углеводородные группы разветвленной или неразветвленной конфигурации с одной или более углерод-углеродных двойных связей, которые могут находиться в любой стабильной точке вдоль цепи, такие как этенил, пропенил, бутенил, пентенил и т.п. "Алкинил" включает углеводородные группы разветвленной или неразветвленной конфигурации с одной или более углерод-углеродной тройной связью, которые могут находиться в любой стабильной точке вдоль цепи, например, этинил, пропинил, бутинил, пентинил, и т.п. "Галоид" в том смысле, как здесь использован, представляет фтор, хлор, бром и иод. "Арил" в данном контексте означает фенил /Ph/ или нафтил.
Термины гетероцикл или гетероциклический в данном контексте, за исключением особых указаний, представляют стабильные 5-7 членные моно- или бициклические или стабильные 7-10 членные бициклические гетероциклические кольцевые системы, любое кольцо которых может быть насыщенным или ненасыщенным, и которые состоят из углеродных атомов и от одного до трех гетероатомов, выбранных из группы, состоящей из N, O и S, и в которых такие гетероатомы, как азот и сера могут быть необязательно окислены, а атом азота может быть необязательно кватернизован, и включают любую бициклическую группу, в которой любое из вышеопределенных гетероциалических колец сконденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, что приводит к созданию стабильной структуры. Примеры таких гетероциклических элементов включают пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролодинил, 2-оксоазепинил, азепинил, пиррол, 4-пиперидонил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолидинил, имидазолинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, тетерагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон и оксадиазолил. Морфолино представляет то же, что и морфолинил.
Представленные экспериментальные процедуры, использующие новый способ детально описаны далее. Эти процедуры являются лишь примерами, и их не следует рассматривать как ограничивающие новый способ настоящего изобретения.
Пример 1.
Получение амида 1: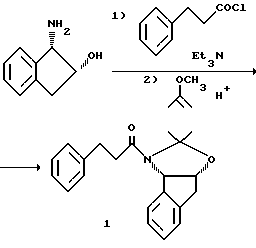
Раствор (-)-цис-1-аминоиндал-2-ола /884 г, 5,93 моля) в 17,8 л сухого ТГФ /KF = 55 мг/мл/ (KF означает титрование для воды Карла Фишера) и триэтиламина /868 мл, 6,22 моля/ в 50 мл круглодонной колбе, снабженной термопарным зондом, механической мешалкой, адаптором для ввода азота и барботером, охлаждают до 15oC. Затем в течение 75 минут добавляют 3-фенилпропионилхлорид /1000 г, 5,93 моля/, поддерживая температуру внутри реактора между 14 и 24oC за счет охладжающей бани со смесью лед-вода. После добавления смесь оставляют на 30 минут при 18-20oC, и по данным ВЭЖХ анализа следят за исчезновением (-)-цис-1-аминоиндан-2-ола.
За ходом реакции следят с помощью ВЭЖХ анализа: колонка 25 см Дюпон C8-PX, 60:40 ацетонитрил/10 мМ (KH2PO4/K2HPO4), 1,0 мл/мин, вводимый объем = 20 мл, детектирование = 200 нм, приготовление образца = 500 X разбавление. Приблизительные периоды удерживания:
Время удерживания /мин/ - идентичность
6,3 - цис-аминоинданол
Реакционную смесь обрабатывают пиридиний-п-толуолсульфонатом /241, 0,96 моля, 0,16 экв./ и перемешивают в течение 10 минут /pH смеси после разбавления 1 мл образца равным объемом воды находится между 4,3-4,6/. Затем добавляют 2-метоксипропен /1,27 л, 13,24 моля, 2,2 эквив/, и реакционную смесь нагревают до 38-40oC в течение 2 часов. Реакционную смесь охлаждают до 20oC и разделяют между этилацетатом /12 л/ и 5% водным NaHCO3 /10 л/, перемешивая смесь и выделяя соли. Этилацетатный экстракт промывают 5% водным NaHCO3 /10 л/ и водой /4 л/. Этилацетатный экстракт сушат перегонкой при атмосферном давлении, и растворитель меняют на циклогексан /полный объем примерно 30 л/. В конце перегонки и концентрирования /20% экстракционного объема этилацетата/ горячий циклогексановый раствор оставляют медленно остывать до 25oC для кристаллизации продукта. Полученную суспензию охлаждают далее до 10oC и оставляют на час. Продукт выделяют фильтрованием, и влажную лепешку промывают холодным /10oC/ циклогексаном /дважды по 899 мл/. Промывают лепешку в вакууме /26 дюймов = 6,6 мм рт.ст./ при 40oC до получения 1,65 кг ацетонида 1 (86,4%, 98% площади ВЭЖХ)
1H ЯМР (300,13 МгГц, CDCl3, основной ротамер) δ : 7,36-7,14 /м, 9H/, 5,03 /д, J = 4,4, 1H/, 4,66 /м, 1H/, 3,15 /м, 2H/, 3,06 /шир. с. 2H/, 2,97 /м, 2H/, 1,62 /c, 3H/, 1,37 /с, 3H/; 13C (75,5 МгГц, CDCl3, основной ротамер) δc/ 168,8, 140,9, 140,8, 140,4, 128,6, 128,5, 128,4, 127,1, 126,3, 125,8, 124,1, 96,5, 78,6, 65,9, 38,4, 36,2, 31,9, 26,5, 24,1.
Элементный анализ: Рассчитано для C21H23NO2: C 78,47; H 7,21; N 4,36. Найдено: C 78,65; H 7,24; N 4,40.
Пример 2.
Получение эпоксида 3:
a. тозилатный способ
Раствор ацетонида 1/1000 г, 3,11 моля/ и 2(S)-глицидилтозилата 2 /853 г, 3,74 моля, 1,2 экв. / в 15,6 л ТГФ /KF = 22 мг/мл/ в 50 л четырехгорлой круглодонной колбе, снабженной термопарой, механической мешалкой, капелькой воронкой и адаптором для ввода азота, дегазируют трижды за счет смены вакуума и продувки азотом, а затем охлаждают до -56oC. Затем в течение 2 часов добавляют литийгексаметилдисилазид (LiN[(CH3)3Si] 2) /2,6 л, 1,38 М, 1,15 экв. /, поддерживая температуру внутри реактора между -50 и -45oC. Реакционную смесь перемешивают при -45 до -40oC в течение 1 часа, а затем дают нагреться до -25oC в течение 1 часа. Эту смесь перемешивают при температуре от -25 до -22oC в течение 4 часов /или до тех пор, пока доля исходного ацетонида не составит 3% площади/.
За ходом реакции следят с помощью ВЭЖХ: 25 см • 4,6 нм Zorbax Silica колонка, 20% этилацетат в гексане, 2,0 мл/мин, вводимый объем 20 мл, детектирование на 254 нм, образец разбавлен в 100 раз. Приблизительные периоды удерживания:
время удержания - идентичность
5,5 - амид 1
6,5 - глицидилтозилат 2
13,5 - эпоксид 3
Реакционную смесь гасят деионизированной водой /6,7 л/ при -15oC, и разделяют этилацетатом /10 л/. Полученную смесь перемешивают, и слои разделяют. Этилацетатный экстракт промывают смесью 1% NaHCO3 /5 л/ и насыщенного NaCl /0,5 л/. Этилацетатный экстракт /28,3 л/ концентрируют вакуумной перегонкой /28 дюймов = 711 мм рт.ст./ и добавляют дополнительно этилацетат, чтобы завершить замену растворителя на этилацетат /конечный объем = 11,7 л/. Этилацетатный концентрат обрабатывают далее для замены растворителя на MeOH для кристаллизации продукта и концентрируют до конечного объема 3,2 л. Оставшийся этилацетатный растворитель удаляют, добавляя 10 л метанола и собирая 10 л дистиллята. Полученную суспензию перемешивают при 22oC в течение 1 часа, а затем охлаждают до 5oC и оставляют на 0,5 часа. Продукт выделяют фильтрованием, а влажную лепешку промывают холодным метанолом /2 • 250 мл/. Промытую лепешку сушат в вакууме /26 дюймов = 660 мм рт.ст./ при 25oC до получения 727 г эпоксида 3 /61,2%, 98,7% площадь основного эпоксида по данным ВЭЖХ/; 13С ЯРМ (300 МгГц, CDCl3) δ; 171,1, 140,6, 140,5, 139,6, 129,6, 128,8, 128,2, 137,2, 126,8, 125,6, 124,1, 96,8, 79,2, 65,8, 50,0, 48,0, 44,8, 39,2, 37,4, 36,2, 26,6, 24,1.
b. Способ с использованием эпихлоргидрина
/Получение эпоксида 3 /эпихлоргидринный способ/
Раствор ацетонида 1 /3,00 г, 9,33 ммоля/ в 47 мл высушенного на ситах ТГФ дегазируют азотом, и полученный раствор охлаждают до -78oC и обрабатывают 8,0 мл литийгексаметилдисилилазидного раствора /1,38М в ТГФ/. Полученный раствор оставляют при -79oC на 15 минут, затем прикапывают 2(S)-эпихлоргидрин /1,2 мл, 15,3 ммоля/, и полученной смеси дают нагреться до -25oC за 1 час, а затем оставляют на 1 час. Затем реакционную смесь снова охлаждают до -78oC и обрабатывают 3,0 мл литийгексаметилдисилазидного раствора, а затем 1,0 мл /S/-эпихлоргидрина. Реакционной смеси дают нагреться до -25oC и оставляют на 2 часа. Реакционную смесь гасят 20 мл насыщенного водного бикарбоната натрия и экстрагируют 120 мл EtOAc, и проводят обратную экстракцию 60 мл EtOAc. Объединенную органическую фазу промывают рассолом, сушат над сульфатом магния, фильтруют и концентрируют при пониженном давлении до получения 3,97 масла, которое очищают с помощью хроматографии на силикагеле /80 г SiO2 элюент 4:1 гексан/этилацетат/ до получения 2,9 г смеси целевого эпоксида 3 и промежуточного хлоргидрина. Порцию смеси /1,29 г/ растворяют в высушенном на ситах ТГФ /20 мл/ при 25oC и обрабатывают 1,73 г 25% /по весу/ раствору калий-трет-амилата, и полученную смесь выдерживают при 25oC в течение 1 часа. Реакционную смесь разделяют между этилацетатом и насыщенным бикарбонатом натрия. Органическую фазу промывают рассолом, сушат над сульфатом магния и концентрируют при пониженном давлении до масла. Это масло очищают хроматографически на силикагеле /80 г SiO2, элюент 4:1 гексан:этилацетат/ до получения 1,1 г /70% полный выход/ эпоксида 3/.
Пример 3.
Получение соединения 6
Суспензию 2(S)-трет-бутилкарбоксамид-4-N-Boc-пиперазина 4 /1950 г., 6,83 моля, более 99,5% ее/ /ее=этантиомерный избыток/ и эпоксида 3 /2456 г, 97,5: 2,5 смесь 4S/R эпоксидов, 6,51 моля/ в изопропаноле /2-пропанол, 18,6 л/ в 72-литровой круглодонной колбе, с четырьмя впускными отверстиями, снабженной механической мешалкой, обратным холодильником, паровой баней, покрытой тефлоном термопарой и вводом для азота, нагревают до кипения с обратным холодильником /внутренняя температура 84-85oC/. Спустя 40 минут получают гомогенный раствор. Полученную смесь кипятят с обратным холодильником в течение 28 часов.
Внутренняя температура при этом сохраняется 84-85oC. За ходом реакции следят с помощью ВЭЖХ /колонка 25 см Дюпон C8-RX, 60:40 ацетонитрил/10 мМ (KH2PO4/K2HPO4), 1,0 мл/мин, детектирование 220 нм, препарат образца = 2 мкл, реакционная смесь разбавлена до 1 мл ацетонитрилом/. Примерные периоды удерживания:
Время удерживания /мин/ - идентификация
4,8 - пиперазин 4
8,9 - эпоксид 3
15,2 - продукт присоединения 5
Через 28 часов оставшийся эпоксид 3 и продукт присоединения 5 /по данным ВЭЖХ анализа/ составляют 1,50% площади и 91 - 93% площади соответственно. Смесь охлаждают до 0 - 5oC и добавляют 10,9 л 6 н HCl, поддерживая температуру ниже 15oC. После завершения добавления смесь нагревают до 22oC. В это время отмечается выделение газа. Смесь выдерживают при 20 - 22oC в течение 6 часов.
За ходом реакции следят по данным ВЭЖХ: условия те же, что и ранее указанные. Приблизительные периоды удерживания:
время удерживания - идентификация
7,0 - цис-аминоинданол
11,9 - соединение 6
15,1 - продукт присоединения 5
Эту смесь охлаждают до 0oC и медленно добавляют 7,5 л 50% NaOH, для того, чтобы довести pH смеси до 11,6, причем температуру при этом поддерживают мене 25oC. Смесь разделяют между этилацетатом /40 л/ и водой /3 л/. Органическую фазу /60 л/ концентрируют при пониженном давлении /29 дюймов = 736 мм рт.ст./ и растворитель заменяют на ДМФ и концентрируют до конечного объема 10,5 л /KF = 1,8 мг/мл/. ВЭЖХ анализ продукта 6 в этилацетате дает 86,5%. Соединение 6, пенультимат, в ДМФ непосредственно используют на следующей стадии без дополнительной очистки. Для выделенного продукта 6: 13C ЯМР (75,4 МгГц, CDCl3) δ: 175,2, 170,5, 140,8, 140,5, 139,9, 129,1, 128,5, 127,9, 126,8, 126,5, 125,2, 124,2, 73,0, 66,0, 64,8, 62,2, 57,5, 49,5, 47,9, 46,4, 45,3, 39,6, 39,3, 38,2, 28,9.
Пример 4.

Получение L-735524-моногидрата
Раствор соединения 6 в ДМФ /105,5 л, KF = 10 мг/мл/ с предшествующей стадии смешивают с 8 л высушенного на ситах ДМФ /KF менее 30 мг/л и полученную смесь нагревают на паровой бане при давлении 30 дюймов = 762 мм рт. ст. для того, чтобы отогнать основную часть воды и/или остатки растворителей: изопропанола и этилацетата. Конечный объем концентрата составляет 13,5 л /KF = 1,8 мг/мл/; а затем добавляют тиэтиламин /2,86 л, 20,51 моля/ при температуре 25oC, а затем к раствору добавляют 3-пиколилхлоридгидрохлорид /96%, 1287 г 7,84 моля/. Полученную суспензию нагревают до 68oC.
За ходом реакции следят с помощью ВЭЖХ, используя условия, указанные на предшествующей стадии. Приблизительные времена удерживания:
Время удерживания /мин/ - идентификация
2,7 - ДМФ
4,2 - 3-пиколилхлорид
4,8 - L-735524
9,1 - соединение 6
Пример 5
Пиразин-2-трет-бутилкарбоксамид 9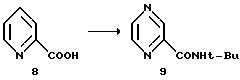
2-пиразинкарбоновая кислота /8/ - 3,35 кг /27 молей/
Оксалилхлорид - 3,46 кг /27,2 моля/
трет-бутиламин /KF = 460 мкг/мл/ - 9,36 д /89 молей/
EtOAc /KF = 56 мкг/мл/ - 9,36 л /89 молей/
ДМФ - 120 мл
1-пропанол - 30 л
Карбоновую кислоту 8 суспендируют в 27 л EtOAc и 120 мл ДМФ в 72-литровой трехгорлой колбе с механическим перемешиванием в атмосфере азота, и охлаждают суспензию до 2oC. Добавляют оксалилхлорид, поддерживая температуру между 5 и 8oC.
Добавление завершают за 5 часов. Во время экзотермического добавления выделяются CO и CO2. Образующаяся HCl преимущественно остается в растворе. Выпадает осадок, который, вероятно, представляет собой HCl соль пиразинхлорангидрида. Анализ образования хлорангидрида осуществляют, гася безводный образец реакционной смеси трет-бутиламином. В конце реакции остается менее 0,7% кислоты 8.
Анализ на завершение образования хлорангидрида важен, так как проведенная не до конца реакция приводит к образованию бис-трет-бутилоксамидной примеси.
За ходом реакции можно следить с помощью ВЭЖХ: колонка 25 см Дюпон Zorbax RXC8 при скорости потока 1 мл/мин и с детектированием при 250 нм; линейный градиент от 98% 0,1% водной H3PO4 и 2% CH3CN до 50% водной H3PO4 и 50% CH3CN за 30 минут. Периоды удерживания:
кислота 8 = 10,7 мин; амид 9 = 28,1 мин.
Реакционную смесь выдерживают при 5oC в течение 1 часа. Полученную суспензию охлаждают до 0oC, и трет-бутиламин добавляют с такой скоростью, чтобы температура внутри реактора оставалась ниже 20oC.
Добавление требует 6 часов, так как реакция крайне экзотермична. Небольшая часть образующегося трет-бутиламмонийгидрохлорида выпадает из реакционной смеси в виде хлопьеобразного белого твердого вещества.
Полученную смесь выдерживают при 18oC еще 30 минут. Выпавшую в осадок аммонийную соль удаляют фильтрованием. Фильтровальную лепешку промывают 12 л EtOAc. Объединенные органические фазы промывают 6 л 3% NaHCO3 и 2 х 2 л насыщенного водного NaCl. Органическую фазу обрабатывают 200 г Darco G60 угля и фильтруют через Solka Flok, и лепешку промывают 4 л EtOAc.
Обработка углем эффективно удаляет незначительную пурпурную окраску продукта.
Раствор 9 в EyOAc концентрируют при 10 мбар = 103 н/м2 до 25% начального объема. Добавляют 30 л 1-пропанола и перегонку продолжают до конечного объема 20 л.
В этот момент количество EtOAc было ниже пределов детектирования методом 1Н ЯМР /менее 1%/. Внутренняя температура при этой замене растворителя менее 30oC. Раствор соединения 3 в смеси 1-пропанол/EtOAc остается стабильным до рефлюкса при атмосферном давлении в течение нескольких дней.
При испарении аликвот получают твердый продукт желто-коричневого цвета с т. плавления 87-88oC. 13С ЯМР (75 МгГц, CDCl3, мд) 161,8, 146,8, 145,0, 143,8, 142,1, 51,0, 28,5.
Пример 6
рацемический 2-трет-бутил-карбоксамидпиперазин 10
Материалы:
Пиразин-2-трет-бутилкарбоксамид 9 2,41 кг /13,4 мол/ в 1-пропанольном растворе, 12 л, 20% Pd(OH)2C 16 вес.% вода 144 г.
Раствор пиразин-2-трет-бутилкарбоксамида 9 в 1-пропаноле помещают в автоклав емкостью 5 галлонов = 18,93 л. Добавляют катализатор и полученную смесь гидрируют при 65oC и давлении 40 пси = 2,8122 кг/см2 H2.
Через 24 часа реакционная смесь поглощает теоретически рассчитанное количество водорода, и по данным газовой хроматографии продукт 9 составляет менее 1%. Полученную смесь охлаждают, через нее продувают N2, и катализатор удаляют фильтрованием через Solka Flok. Катализатор промывают 2 л теплого 1-пропанола.
Было обнаружено, что использование теплого 1-пропанола при промывке фильтровальной лепешки улучшает фильтрацию и снижает потери продукта на фильтровальной лепешке.
За ходом реакции следят с помощью газовой хроматографии: Колонка 30 м Megabore, от 100 до 160oC при скорости 10oC/мин, выдерживают 5 минут, а затем при скорости 10oC/мин до 250oC; время удерживания : для 9 = 7,0 мин; для 10 = 9,4 мин. За ходом реакции можно также следить по данным ТСХ, используя растворитель EtOAc/MeOH /50:50/ и нингидрин в качестве проявляющего агента.
После выпаривания аликвот оказывается, что выход после амидирования и гидрирования составляет 88%, а концентрация 10 составляет 133 г/л.
После выпаривания аликвот получают продукт 10 в виде белого твердого вещества с т. плавления 150 - 151oC; 13С ЯМР (75 МгГц, D2O, мл) 173,5, 59,8, 52,0, 48,7, 45,0, 44,8, 28,7.
Пример 7.
Соль (S)-2-трет-бутил-карбоксиамидпиперазин-бис(S)-камфорсульфоновой кислоты (S)-II:
Материалы:
рацемический 2-трет-бутилкарбоксамидпиперазин 10 - 4,10 кг/22,12 моля/
в виде раствора в 1-пропаноле - в 25,5 кг растворит.
(S)-(+)-10-камфорсульфоновая кислота - 10,0 кг /43,2 моля/
1-пропанол - 12 л
Ацетонитрил - 39 л
Вода - 2,4 л
Раствор амина 10 в 1-пропаноле помещают в 100 литровую колбу с присоединением порционным концентратором. Раствор концентрируют при давлении 10 мбар = 103 н/м2 и при температуре менее 25oC до объема примерно 12 л.
В этот момент продукт осаждается из раствора, но при нагревании смеси до 50oC переходит обратно в раствор.
Анализ гомогенных аликвот указывает, что концентрация продукта 10 составляет 341 г/л. Концентрацию определяют по данным ВЭЖХ: колонка 25 см Дюпон Zorbax PXC8 при скорости потока 1,5 мл/мин и детектировании на 210 нм, изократно /98/3/ CH3CN/0,1% водн. H3PO4. Время удерживания продукта 10 : 2,5 мин.
Для получения прозрачного слегка коричневого раствора добавляют 39 л ацетонитрила и 2,4 л воды.
Содержание воды определяют с помощью KF титрования и интегрирование спектров 1Н ЯМР дает отношение CH3CN/1-пропанол/H2O = 26/8/1,6. Концентрация в растворе составляет 72,2 г/л.
(S)-10-камфорсульфоновую кислоту загружают за 30 минут четырьмя порциями при 20oC. Температура повышается до 40oC после добавления CSA. Через несколько минут образуется густой белый осадок. Белую суспензию нагревают до 76oC для растворения всей твердой части, а затем коричневатому раствору дают остыть до 21oC за 8 часов.
Продукт выпадает в осадок при 62oC. Его фильтруют без выдерживания при 21oC, и фильтровальную лепешку промывают 5 л CH3CN/1-пропанол/H2O 26/8/1,6 смесью растворителей. Все это сушат при 35oC в вакуумном термостате в атмосфере азота до получения 5,6 кг/39%/ продукта II в виде белого кристаллического твердого вещества с т. пл. 288 - 290oC /с разложением/. [α]
Показатель ее этого материала составляет 95% по данным следующего хирального ВЭЖХ анализа; аликвоту продукта II /33 мг/ суспендируют в 4 мл EtOH и 1 мл Et3N. Добавляют Boc2O /11 мг/ и реакционную смесь выдерживают в течение 1 часа. Растворитель полностью удаляют в вакууме, а остаток растворяют примерно в 1 мл EtOAc и фильтруют через пастеровскую пипетку с SiO2, используя в качестве элюента EtOAc. Фракции выпаренного продукта снова растворяют в гексанах в концентрации примерно 1 мг/мл. Энантиомеры разделяют на колонке Daicel Chiracell AS, используя систему растворителей гексан /JPA /97:3/ со скоростью 1 мл/мин, и детектируя на 228 нм.
Время удерживания : S антипод = 7,4 мин, R = 9,7 мин.
Пример 8.
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонилпиперазин 4, полученный из соли II.

Материалы
(S)-2-трет-бутилкарбоксамидпиперазин
Бис(S)-(+)-CSA соль 11,95% ее - 5,54 кг /8,53 моля/
Ди-трет-бутилкарбонат - 1,86 кг /8,53 моля/
Et3N - 5,95 л /42,6 моля/
EtOH строго 200 крепости - 55 л
EtOAc - 2 л
К (S)-CSA соли II в 100 литровой трехгорлой колбе с капельной воронкой в атмосфере азота добавляют EtOH, затем триэтиламин при 25oC. Твердая часть легко растворяется при добавлении Et3N•Boc2O растворяют в EtOAc и загружают в капельную воронку. Раствор Boc2O в EtOAc добавляют с такой скоростью, чтобы температура не повышала 25oC. Такое добавление занимает 3 часа. Реакционную смесь выдерживают в течение 1 часа после завершения добавления раствора Boc2O.
За ходом реакции можно следить по данным ВЭЖХ : 25 см Дюпон Zorbax RXC8 колонка, скорость потока 1 мл/мин и детектирование при 228 нм, изократная смесь (50/50) CH3CN/0,1 М KH2PO4 доводят pH до 6,8 с помощью NaOH. Время удерживания продукта 4 = 7,2 минуты. Хиральный анализ проводят, используя ту же систему, что и на предшествующей стадии. За ходом реакции можно также следить с помощью ТСХ, используя в качестве растворителя 100% EtOAc /Rf = 0,7/.
Затем раствор концентрируют примерно до 10 л при температуре внутри реактора ниже 20oC в концентраторе порционного типа при давлении 10 мбар = 103 н/м2. Растворитель заменяют, медленно доливая 20 л EtOAc, и затем снова концентрируя до 10 л. Реакционную смесь промывают 16 л. 5% водного раствора Na2CO3, дважды по 10 л деионизированной воды и дважды по 6 л насыщенного водного хлорида натрия. Объединенные водные промывки подвергают обратной экстракции 20 л EtOAc, и органическую фазу промывают дважды по 3 л воды и дважды по 4 л насыщенного водного хлорида натрия. Объединенные EtOAc экстракты концентрируют при давлении 140 мбар = 103 н/м2 и температуре внутри реактора менее 20oC в столитровом концентрате порционного типа до примерно 8 л. Замены растворителя циклогексаном достигают, медленно приливая примерно 20 л циклогексана, и снова концентрируя до примерно 8 л. К полученной суспензии добавляют 5 л циклогексана и 280 мл EtOAc, и полученную смесь нагревают до кипения с обратным холодильником, когда все переходит в раствор. Этот раствор охлаждают и при 58oC вводят 10 г затравки. Суспензию охлаждают до 22oC в течение 4 часов, и полученный продукт выделяют фильтрованием после того, как в течение часа выдерживают при 22oC. Фильтровальную лепешку промывают 1,8 л циклогексана и сушат в вакуумном термостате при 35oC в атмосфере азота до получения 1,87 кг (77%, более 99,8% площадки на хроматограмме ВЭЖХ, R-изомер ниже уровня детектирования) продукта 4 в виде слегка желтовато-коричневого цвета порошка. [α]
13ЯМР (75, МгГц, CDCL3, мл)170,1, 154,5, 79,8, 58,7, 50,6, 46,6, 43,6, 43,4, 28,6, 28,3.
Пример 9.
(S)-2-трет-бутил-карбоксамидпиперазин-бис-(L)пироглютаминовая кислота 12
Материалы
Рацемический 2-трет-бутилкарбоксамидпиперазин 10 /0,11 моля/ в 1-пропанольном растворе - 155 мл, анализ = 21,1 г L
L-пироглутаминовая кислота - 28 г /0,21 моля/
Вода - 5 мл
Раствор рацемического-2-трет-бутил-карбоксамидпиперазина 10 в 1-пропаноле загружают в 500 мл круглодонную колбу с холодильником, механической мешалкой и вводом для азота. Воду добавляют вместе с L-пироглютаминовой кислотой, и полученную суспензию кипятят с обратным холодильником. Гомогенный раствор желтого цвета охлаждают до 50oC, и вводят затравку - бис-(S)-PGA соли R амина /50 мг/. Сразу же образуется твердый продукт. Этот раствор охлаждают далее до 25oC и выдерживают в течение 16 часов. Твердую часть отфильтровывают при 22oC, и фильтровальную лепешку промывают 35 мл холодной смеси 1-пропанол/вода. Фильтровальную лепешку сушат при 35oC в вакуумном термостате, продуваемом азотом, до получения 23,74 г /48%/ (R)-2-трет-бутилкарбоксамидпиперазин-бис-(L)-пироглутаминовой кислоты. Величина ее материала составляет 98% по данным ВЭЖХ анализа на хиральность, описанного ранее. Маточные растворы желтого цвета содержит 22,6 г /46%/ соли (S)-2-трет-бутилкарбоксамидпиперазин бис(L)-пироглутаминовой кислоты 12, и величина ее составляет 95% по данным ВЭЖХ анализа на хиральность. Маточные растворы выпаривают и используют непосредственно на стадии защиты.
Пример 10.
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазин 4 из соли (S)-2-трет-бутилкарбоксамидпиперазин-бис(L)-пироглутаминовой кислоты 12
Материалы
соль (S)-2-трет-бутилкарбоксамидпиперазин-бис(L)-пираглутаминовой кислоты, 95% ее - 22,6 г /50,1 ммоля/
Ди-трет-бутилкарбонат - 11,1 г /50,1 ммоля/
Et3N - 35,5 мл /0,254 моля/
1-пропанол - 226 мл
EtOAc - 24 мл
К соли (S)-2-трет-бутилкарбоксамидпиперазин-бис/L/-пироглутаминовой кислоты в 500 мл трехгорлой колбе с капельной воронкой, в атмосфере азота добавляют 1-пропанол. При добавлении Et3N легко растворяется смолистое твердое вещество. В течение 2 часов при 22oC добавляют раствор Boc2O в EtOAc. Реакционную смесь выдерживают в течение 1 часа после добавления.
За ходом реакции можно следить по данным ВЭЖХ и ТСХ, используя тот же способ, что и для превращения 11 в 4.
Затем раствор концентрируют, и растворитель заменяют этилацетатом /200 мл/. Реакционную смесь промывают 50 мл 7% водного раствора Na2CO3, 2000 мл воды, сушат над сульфатом натрия и фильтруют. EtOAc раствор концентрируют, и растворитель заменяют циклогексаном /60 мл/. Добавляют EtOAc /1 мл/, и полученную смесь нагревают до кипения с обратным холодильником для растворения всей твердой части. Смесь охлаждают и вводят затравку /50 мг/ при 52oC. Суспензию охлаждают до 22oC в течение 2 часов, и продукт выделяют фильтрованием после выдерживания в течение 1 часа при 22oC. Фильтровальную лепешку промывают 8 мл циклогексана и сушат в вакуумном термостате при 35oC в атмосфере азота до получения 10,8 г /74%/ (более 99,9% площади хроматограммы ВЭЖХ, R-изомер ниже уровня детектирования) продукта 4 в виде грязно-белого порошка.
Пример 11.
Кинетическое выделение продукта 4 их (S/К)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонилпиперазина-13.
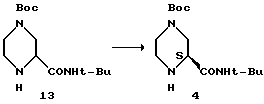
Материалы
Неочищенный (S/R)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонилпиперазин 13 - 1,40 г
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонилпиперазин 4 (более 99,5% ее) - 4 х 0,14 г
Метилциклогексан с 2% /объем/объем/EtOAc - 14 мл
Неочищенный смолистый продукт 13 растворяют в 14 мл смеси растворителей, нагревая до 90oC. Раствор оставляют охлаждаться, и с 10oC интервалами раствор засевают 0,14 г продукта 4 /более 99,5% ее/. При 55oC четвертая порция в 0,14 г затравки уже больше не растворяется, и при дальнейшем медленном охлаждении до комнатной температуры образуется белая кристаллическая масса. Реакционную смесь фильтруют, промывают 3 мл смеси растворителей метилциклогексан/EtOAc и сушат в вакуумном термостате в токе азота до получения 0,95 г белого твердого вещества. Определение энантиомерной чистоты на колонке Chiracell As дает 93% ее.
Пример 12.
Получение транс-3-(4-пиридил)акриловой кислоты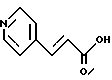
К раствору 4-пиридинкарбоксальдегида/ 36,7 мл, 0,384 моля/ и малоновой кислоты /40 г, 0,384 моля/ в 31 мл пиридина добавляют пиперидин /0,12 мл/, и полученную смесь нагревают до 100oC. Предостережение: выделяются большие объемы CO2. Через полчаса реакционную смесь охлаждают до комнатной температуры и раствор затвердевает. Его тщательно растирают с 240 мл воды и фильтруют, промывают дважды по 50 мл порциями воды. Твердую часть сушат в течение ночи при 42oC при давлении 10 мм рт.ст. до получения 37,1 г белого твердого вещества; т.пл. 295-297oC.
Пример 13.
Получение N-(2(R)-гидрокси-1(S)инданил)-транс-3-(4-пиридил)-акриламида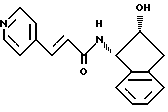
К суспензии транс-3-(4-пиридил)акриловой кислоты /10,0 г, 0,067 моля/ в 500 мл ТГФ добавляют триэтиламин /10,29 мл, 0,0738 моля/, и полученный раствор охлаждают до 0oC. Добавляют триметилацетилхлорид /8,68 мл, 0,0704 моля/, и реакционную смесь перемешивают в течение получаса. Добавляют через канюлю. 2(R)-гидрокси-1(S)-индан /10,0 г, 0,067 моля/, растворенный в 260 мл ТГФ. Спустя 2 часа реакционную смесь нагревают до комнатной температуры и перемешивают еще 15 часов. Растворитель удаляют в вакууме, а полученную твердую часть тщательно растирают с холодным этилацетатом /150 мл/ и фильтруют. Ее сушат в течение ночи при давлении 0,5 мм рт.ст. до получения 18,5 г твердого вещества белого цвета. Т.пл. 205-207oC.
Пример 14.
Получение N-(1,2-N,0-изопропилиден-2(R)-гидрокси-1(S)-инданил)- транс-3-(4-пиридил)акриламида
К суспензии N-(2(R)-гидрокси-1(S)-инданил(транс-3-(4-пиридил)-акриламида /18,5 г, 0,066 моля/ в 700 мл метиленхлорида добавляют диметоксипропан /49,0 мл, 0,402 моля/ а затем (+)-/камфорсульфоновую кислоту /46,8 г, 0,201 моля/. После 20 минут реакционная смесь становится гомогенной. Реакционную смесь перемешивают в течение 3 часов, промывают насыщенным бикарбонатом натрия /2 х 150 мл/. Водный слой экстрагируют метиленхлоридом /3 х 200 мл/, и объединенный органический слой сушат над сульфатом магния, фильтруют и концентрируют до масла. Очистка на хроматографической колонке с мгновенным испарением (100 х 150 мм колонка с силикагелем; градиентное элюирование 1:30: 69, 2: 30: 68, 3:30:67, 5:30:65 смесью MeOH : CHCl3, насыщенной NH3:CH2Cl2) приводит к получению 16,0 г белой пены (Rf = 0,46 в 5:30:65 MeOH : CHCl3 насыщенная NH3CH2Cl2/
Пример 15.
Получение N-(1,2-N, O-изопропилиден-2(R)гидрокси-1(S)инданил)- 3-(4-пиридил)пропиламида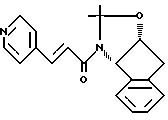
К N-(1,2-N, O-изопропилиден-2(R)-гидрокси-1(S)-инданил)-транс-3- (4-пиридил)акриламиду /16,0 г, 0,0499 моля/ растворенному в 200 мл этанола и 200 мл ТГФ, добавляют 14,0 г Pd(OH)2 на угле /20 вес.%/. Затем в колбу вводят H2 и реакционную смесь перемешивают в течение 9 часов. Раствор продувают аргоном, фильтруют через слой целита и промывают этанолом /100 мл/. Растворитель удаляют в вакууме, и полученный продукт очищают с помощью флешхроматографии (колонка с силикагелем 100 х 150 мм; градиентное элюирование 1:30:69, 2:30: 67, 5: 30: 65 смесь MeOH : CHCl3, насыщенная NH3:CH2Cl2), в результате чего получают 13,8 г белой пены (Rf = 0,5 в 5:30:65 MeOH : CHCl3, насыщенной NH3: CH2Cl2)
Пример 16
Получение N-(2(R)-гидрокси-1(S)-инданил)-транс-3-(3-пиридил)-ацетамида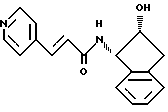
Используя практически тот же способ, что и для получения N-(2(R)-гидрокси-1(S)-инданил)-транс-3-(4-пиридил)акриламида, но заменяя соответствующие исходные материалы, получают указанное в заглавии соединение. Физические характеристики: т.пл. 119-120oC.
Элементный анализ: Рассчитано для C17H16N2O2 • 0,65H2O:
C 69,92; H 5,97; N 9,59. Найдено: C 69,94; H 5,74; N 9,84.
Пример 17.
Получение N-(1,2-N,O-изопропилиден-2(R)-гидрокси-1(S)-инданил)- транс-3-/3-пиридил/акриламида
Используя практически тот же способ, что и для получения N-(1,2-N,0-изопропилиден-2(R)-гидрокси-1(S)инданил)-транс-3-(4-пиридил) акриламида, но заменяя соответствующие исходные материалы, получают указанное в заглавии соединение. Физические характеристики: т.пл. 134 - 136. Элементный анализ: Рассчитано для C20H20N2O2 • 0,25 H2O: C 73,94; H 6,36; N 8,62. Найдено: C 73,95; H 6,18; N 8,70.
Пример 18.
Получение N-(1,2-N, O-изопропилиден-2(R)-гидрокси-1(S)-инданил)-3-(2-пиридил) пропиламида
Используя практически тот же способ, что и для получения N-(1,2-N,O-изопропилиден-2(R)-гидрокси-1(S)-инданил)-3-(4-пиридил) пропиламида, но заменяя соответствующие исходные материалы, получают указанное в заглавии соединение.
Хотя в изложенном описании изложены принципы настоящего изобретения, и примеры предложены в целях иллюстрации, следует учитывать, что практика настоящего изобретения охватывает все обычные варианты, изменения и модификации, попадающие в объем изобретения, сформулированный в формуле изобретения.
Изобретение относится к новым промежуточным соединениям и усовершенствованному способу получения соединений, которые ингибируют протеазу, кодируемую вирусом иммунодефицита человека (ВИЧ), и в частности L-735524, или к их фармацевтически приемлемым солям. Способ получения производных гетероциклических карбоновых кислот формулы I включает взаимодействие соединения формулы IV с амином формулы V, где стереоцентр а имеет или R-конфигурацию, или S-конфигурацию, или является рацемическим; r - целое число от 0 до 5 включительно; R1 и R2, взятые вместе с атомом азота, к которому присоединен R1, и атомом углерода, к которому присоединен R2, образуют 6-членную моноциклическую насыщенную кольцевую систему, состоящую из атома азота, к которому присоединен R1, из 4 атомов углерода и одного замещенного гетероатома 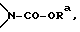 где Ra представляет алкил; R3 - фенил; R4 - C1 -C5 -алкил. Предложенный способ значительно короче, более диастереосективен, приводит к более высоким выходам целевых соединений. 4 с. и 16 з.п.ф-лы.
где Ra представляет алкил; R3 - фенил; R4 - C1 -C5 -алкил. Предложенный способ значительно короче, более диастереосективен, приводит к более высоким выходам целевых соединений. 4 с. и 16 з.п.ф-лы. 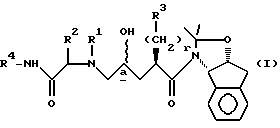

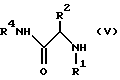

включающий взаимодействие соединения формулы IV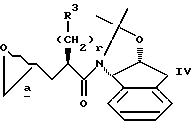
с амином формулы V
где стереоцентр  имеет или R-конфигурацию, или S-конфигурацию, или является рацемическим;
имеет или R-конфигурацию, или S-конфигурацию, или является рацемическим;
r представляет целое число от 0 до 5 включительно;
R1 и R2, взятые вместе с атомом азота, к которому присоединен R1, и атомом углерода, к которому присоединен R2, образуют 6-членную моноциклическую насыщенную кольцевую систему, состоящую из атома азота, к которому присоединен R1, из 4 атомов углерода и одного замещенного гетероатома
где Rа представляет незамещенный C1-4 алкил;
R3 представляет фенил;
R4 представляет C1-C5-алкил с прямой или разветвленной цепью.
а) соединения формулы II
или b) эпихлоргидрина структуры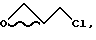
с амидом формулы III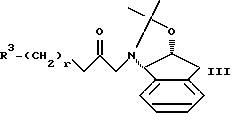
в присутствии сильного основания при низкой температуре, где X выбран из группы, состоящей из -H, метаносульфонила, трифторметансульфонила, п-толуолсульфонила, бензолсульфонила и 3-нитробензолсульфонила. имеет S-конфигурацию; r = 1; R1 и R2 соединены вместе с образованием циклической структуры
имеет S-конфигурацию; r = 1; R1 и R2 соединены вместе с образованием циклической структуры
R3 представляет фенил и R4 представляет трет-бутил.
8. Способ получения производных N-(1,2-N,O-изопропилиден-2(R)-гидрокси-1(S)-инданил)-3-(2-оксиранил)пропиламида формулы IV
где стереоцентр  имеет R-конфигурацию или S-конфигурацию или является рацемическим;
имеет R-конфигурацию или S-конфигурацию или является рацемическим;
r представляет целое число от 0 до 5 включительно;
R3 представляет фенил,
отличающийся тем, что осуществляют взаимодействие соединения формулы II
с амидом формулы III
где a, r и R3 имеют значения, определенные выше;
X выбран из группы, состоящей из -H, метансульфонила, трифторметансульфонила, п-толуолсульфонила, бензолсульфонила и 3-нитробензолсульфонила,
в присутствии сильного основания при низкой температуре. имеет S-конфигурацию; r = 1 и R3 представляет фенил.
имеет S-конфигурацию; r = 1 и R3 представляет фенил.
где стереоцентр  имеет R-конфигурацию или S-конфигурацию или является рацемическим;
имеет R-конфигурацию или S-конфигурацию или является рацемическим;
r представляет целое число от 0 до 5 включительно;
R3 представляет фенил. имеет S-конфигурацию; r = 1 и R3 представляет фенил.
имеет S-конфигурацию; r = 1 и R3 представляет фенил.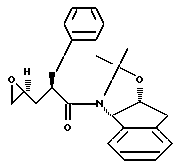
Приоритет по пунктам:
16.07.93 по пп.1 - 3 и 6 - 20;
26.01.94 по пп.4 и 5.
| Люминофоры в сине-фиолетовой области спектра | 1977 |
|
SU699001A1 |
| US 5169952 A, 1992 | |||
| Способ получения триалкил(органилтио) силанов или германов | 1974 |
|
SU480714A1 |
| EP 521686 A1, 07.01.93. | |||

