Данная заявка заявляет приоритет по Предварительной патентной заявке США под номером 61/238113, поданной 28 августа 2009 года, которая включена в данный документ ссылкой во всей своей полноте.
1. ВВЕДЕНИЕ
Данное изобретение относится к ферментной заместительной терапии с увеличением дозы с помощью кислой сфингомиелиназы (ASM) для лечения субъектов-людей, имеющих недостаточность кислой сфингомиелиназы (ASMD), и, в частности, не неврологические проявления болезни Ниманна-Пика (NPD), и в определенных вариантах осуществления NPD типа B.
2. ПРЕДПОСЫЛКИ
Кислая сфингомиелиназа, E.C. 3.1.4.12, (ASM) представляет собой фермент - лизосомальную фосфодиэстеразу, которая гидролизует сфингомиелин, фосфолипидное запасное вещество, обнаруживаемое в мозге, печени, легких, селезенке и лимфатических узлах, на церамид и фосфорилхолин. Недостаточности в активности ASM приводят к неспособности организма разрушать сфингомиелин, вызывая образование лизосомальной болезни накопления, имеющей название болезнь Ниманна-Пика.
Болезнь Ниманна-Пика является врожденным аутосомным рецессивным нарушением накопления липидов, которое характеризуется чрезмерным накоплением сфингомиелина в лизосомах клеток, таких как макрофаги и нейроны, что нарушает нормальную клеточную функцию. Тип A болезни Ниманна-Пика представляет собой быстро прогрессирующее нейродегенеративное заболевание у новорожденных и обычно приводит к смерти в возрасте двух-трех лет. Тип B болезни Ниманна-Пика приводит к увеличению печени и селезенки и дыхательной недостаточности со смертельным исходом, который имеет место, как правило, в юности. Эти две формы болезни Ниманна-Пика, обе из которых связаны с недостаточностями ASM, в данном документе называются обобщенно болезнь Ниманна-Пика, или недостаточность ASM (ASMD). Другие типы болезни Ниманна-Пика, например тип C, не вовлекают мутации в гене ASM и не вносят напрямую вклад в функцию ASM. Природа биохимических и молекулярных дефектов, которые лежат в основе значительной клинической гетерогенности подтипов A и B, остается неизвестной. Хотя пациенты с обоими подтипами имеют остаточную активность ASM (приблизительно 1-10% от нормальной), биохимический анализ не может достоверно отличить два фенотипа. Более того, течение болезни NPD типа B является высоко вариабельным, и в настоящее время невозможно сопоставить тяжесть заболевания с уровнем остаточной активности ASM.
NPD чаще возникает среди особей ашкеназо-еврейского происхождения, чем в общей популяции. Как оценивается, частота возникновения заболевания типа A среди евреев ашкенази составляет приблизительно 1 на 40000, частота гена (q) приблизительно 1 на 200 и частота гетерозиготных носителей (2 pq) 1 на 100 (Goodman, 1979, в Genetic Disorders Among The Jewish People, John Hopkins Univ. Press, Baltimore, pp. 96-100). Частота возникновения гетерозиготного носителя типа B NPD в популяции евреев ашкенази является менее распространенной (Goodman, ранее). Объединенная частота гетерозиготных носителей для типов A и B NPD, как оценили, составляет приблизительно 1 на 70 среди особей ашкеназо-еврейского происхождения. Хотя ферментная диагностика пациентов, страдающих либо типом A, либо B NPD, может быть достоверно проведена (Spence and Callahan, ранее), определение ферментов у облигатных гетерозигот имеет доказанную проблематичность, особенно с применением лейкоцитов в качестве источника фермента. Вероятно, возникновение нейтральных сфингомиелиназ в нескольких источниках и (или) присутствие остаточной активности ASM, возникающей как результат мутантного аллеля, внесли вклад в неспособность достоверно различать носителей для любого подтипа заболевания. Даже применение культивированных фибробластов кожи, которые не экспрессируют нейтральную сфингомиелиназу, не давало однозначных результатов с гетерозиготами. В эпидемиологических исследованиях, проведенных в отдельных странах, объединенная частота возникновения болезни Ниманна-Пика A и B в некоторых странах в мире, как оценивается, находится в диапазоне от 1 на 167000 до 1 на 250000 новорожденных (Miekle et al., 1999 JAMA 281(3):249-254; Poorthuis et al., 1999 Hum Genet 105:151-156; Pinto et al., 2004 Euro. J. Hum. Gene. 12:87-92). Соотношение гетерозиготных носителей, как полагают, находится в диапазоне от 1 на 200 до 1 на 250 особей.
Ферментную заместительную терапию применяли для других лизосомальных болезней накопления. Ферментная заместительная терапия делает попытку восполнить недостаточную ферментативную активность с помощью экзогенно поставляемого фермента. В случае ферментной заместительной терапии для болезни Ниманна-Пика целью будет дать возможность пораженной особи обрабатывать сфингомиелин и избегать его накопления внутри лизосом. Чтобы быть эффективной, такая терапия изначально будет нуждаться в существенно большом количестве заместительного фермента для разрушения накопленного сфингомиелина, а также в непрерывном введении заместительного фермента во избежание дальнейшего накопления сфингомиелина.
3. КРАТКОЕ ОПИСАНИЕ
Данное изобретение относится к ферментной заместительной терапии с увеличением дозы для лечения субъектов-людей, имеющих ASMD - в частности, субъектов, имеющих не неврологические проявления NPD и, в конкретных вариантах осуществления, NPD типа B. Более конкретно, фермент, ASM, вводят таким пациентам в начальной низкой, нетоксичной дозе, которую затем увеличивают в последующих введениях. Самую высокую дозу ASM, которая переносится пациентом, затем можно использовать в качестве поддерживающей дозы. Альтернативно, в качестве поддерживающей дозы можно применять терапевтически эффективную дозу, которая меньше самой высокой переносимой дозы.
Данное изобретение основано, частично, на открытии, что дозы ASM, которые будут необходимы для устранения накопленного сфингомиелинового субстрата у субъектов-людей, т.е. пациентов с ASMD или пациентов с Ниманна-Пика, приводят к токсичным побочным эффектам (включая клинические признаки токсичности). Это особенно неожиданно у пациентов с менее тяжелой формой ASMD, NPD типа B, у которых есть недостаточность, но по крайней мере некоторая ферментная активность.
Более конкретно, лечение NPD будет требовать доз, достаточно высоких для достижения достаточного распределения фермента ASM в органах патологии (например, в частности, в печени, селезенке, легких, сердце, почке и мозге). Исследования в модели на мышах, нокаутных по ASM (ASKMO-мыши), показали, что основная часть введенной рекомбинантной ASM человека (rhASM) распределяется в печени и селезенке, где она уменьшает субстрат, но намного в меньшей степени в легком, сердце и мозге (Miranda et al. FASEB 2000, 14:1988; смотрите также фиг.9B He et al., 1999, Biochimica et Biophsyica Acta 1432: 251-264). В последующих исследованиях с помощью более высоких доз rhASM в ASMKO-мышиной модели субстрат уменьшался, и токсичность не наблюдалась при дозах ≤3,0 мг/кг; фактически, клинические симптомы токсичности не наблюдались вплоть до применения доз ≥10 мг/кг. Смотрите Dose Responsive Toxicological Findings Following Intravenous Administration of Recombinant Human Acid Sphingomyelinase (rhASM) to Acid Sphingomyelinase Knock-out (ASMKO) Mice. C. Nickerson, J. Murray, A. Vitsky, M. Hawes, S. Ryan, P. Ewing, B. Thurberg, L. Andrews. Dept Pharm/Tox, Pathology, Genzyme Corp., Framingham, MA., American Society of Human Genetics 2005; и Elevations of Pro-Inflammatory Cytokines and Decreases in Cardiovascular Hemodynamics Following Intravenous Administration of Recombinant Human Acid Sphingomyelinase (rhASM) to Acid Sphingomyelinase Knock-out (ASMKO) Mice. J. Murray, A.M. D'Angona, C. Nickerson, A. Vitsky, M. Hawes, S. Ryan, P. Ewing, B. Thurberg, L. Andrews. Dept. Pharmacology/Toxicology & Pathology, Genzyme Corp., Framingham, MA., Society of Toxicology 2006.
На основании этих данных ASKMO мы лечили субъектов-людей, имеющих не нейронопатическую ASMD, консервативной максимальной дозой 1,0 мг/кг rhASM, как описано в разделе 6, ниже. Достаточно неожиданно токсичность у субъектов-людей, включая появление связанных нежелательных явлений с клиническими симптомами, наблюдали при применении дозы до 0,3 мг/кг! Этот результат был особенно неожиданным, поскольку фермент ASM отсутствует в модели на нокаутных мышах, которая должна отражать более тяжелое состояние, чем у этих субъектов-людей, имеющих по крайней мере некоторую ферментативную активность и относительно легкую форму заболевания.
Не намереваясь быть связанным какой-либо теорией, токсические побочные эффекты, которые возникают при лечении ASM, могут являться результатом разрушения накопленного субстрата сфингомиелина у пациента с ASMD и высвобождения продукта, церамида, который является проапоптическим и вызывает провоспалительный цитокиновый ответ и гипербилирубинемию. Чтобы решить данную проблему, мы разработали режим, который обеспечивает безопасное введение высоких доз фермента ASM, необходимого для достижения достаточного распределения в органах патологии. В соответствии с этой схемой начальное лечение с ASM в очень низких дозах применяют для достижения медленного разрушения накопленного субстрата, которое сопровождается меньшими побочными эффектами. По мере исчерпания субстрата у субъекта (по мере «уменьшения по массе» запасного субстрата) дозу можно безопасно увеличивать.
В соответствии с этим протоколом вначале пациенту с заболеванием NPD вводят низкую нетоксичную дозу фермента ASM и с течением времени дозу увеличивают. По мере повышения дозы фермента ASM пациента можно подвергать мониторингу в отношении общей концентрации билирубина, продуцирования острофазовых агентов, продуцирования медиаторов воспаления и связанных нежелательных явлений. Введение низкой дозы ASM и увеличение дозы облегчает сокращение массы накопленного сфингомиелина. По мере сокращения массы сфингомиелина у пациента можно безопасно вводить более высокие дозы пациенту для обеспечения достаточного распределения фермента ASM в целевых органах (например, в печени, селезенке, легких, сердце, почке, мозге, костном мозге, скелете, суставах и т.д.). В определенных вариантах осуществления максимальную дозу, переносимую пациентом, можно использовать в качестве поддерживающей дозы. В некоторых вариантах осуществления, основываясь на состоянии пациента, поддерживающая доза может быть увеличена или уменьшена с течением времени.
В определенных вариантах осуществления лечение пациента подвергают мониторингу путем измерения уровней сфингомиелина в плазме, уровней церамида в плазме, продуцирования «острофазовых агентов» и медиаторов воспаления, которые являются мерой воспалительных реакций, концентраций билирубина (общей, прямой и непрямой) и (или) других биохимических маркеров для обеспечения стабильного ответа до увеличения дозы до следующего уровня. Эти маркеры включают, но без ограничений, C-реактивный белок (CRP) или CRP высокой чувствительности (hs-CRP), цитокины (например, IL-8, IL-6), кальцитонин и ферритин. В конкретных вариантах осуществления пациента можно подвергать мониторингу в отношении одного или нескольких связанных нежелательных явлений, которые могут включать, но без ограничений, системные симптомы (например, лихорадку, тошноту, рвоту, боль, миалгию) и желтуху.
Дозы менее 1 мг/кг являются предпочтительными для начала лечения. Начальную дозу последовательно увеличивают, пока не достигнут терапевтической дозы. Такое увеличение дозы можно применять для определения самой высокой переносимой дозы. Например, как только у пациента сокращается масса накопленного субстрата сфингомиелиназы, дозу можно далее увеличивать, пока не наблюдается токсичность. Поддерживающую дозу можно соответственно корректировать и можно постоянно и периодически повторно корректировать в зависимости от состояния пациента.
В конкретном варианте осуществления способ лечения субъекта-человека, имеющего недостаточность кислой сфингомиелиназы, включает: (a) схему для сокращения массы накопленного субстрата сфингомиелина у субъекта-человека, включающий: (i) введение начальной низкой нетоксичной дозы ASM субъекту-человеку; (ii) введение последовательно более высоких доз ASM субъекту-человеку и мониторинг субъекта в отношении одного или нескольких неблагоприятных побочных эффектов после каждой последующей дозы, которые определяются повышенным билирубином или связанным нежелательным явлением; и (b) поддерживающую схему, включающую введение дозы, равной или меньшей, чем самая высокая доза, переносимая субъектом, в качестве поддерживающей дозы для субъекта.
В другом конкретном варианте осуществления способ лечения субъекта-человека, имеющего недостаточность кислой сфингомиелиназы, включает введение rhASM по схеме с увеличением дозы со следующими последовательными дозами: 0,1 мг/кг; 0,3 мг/кг; 0,6 мг/кг; и 1,0 мг/кг, где каждую дозу rhASM вводят по меньшей мере дважды, и каждую дозу вводят с двухнедельными интервалами, и где пациента подвергают мониторингу в отношении токсических побочных эффектов перед повышением дозы до следующего уровня.
В другом конкретном варианте осуществления, описанном в данном документе, представлена кислая сфингомиелиназа (ASM) для применения в лечении недостаточности кислой сфингомиелиназы у субъекта-человека, подготовленная для введения: (a) в схеме для сокращения массы накопленного субстрата сфингомиелина, включающей: (i) введение начальной низкой нетоксичной дозы кислой сфингомиелиназы (ASM); (ii) введение последовательно более высоких доз ASM, и мониторинг субъекта в отношении одного или нескольких нежелательных побочных эффектов после каждой последующей дозы, которые определяют по повышенному билирубину или связанному нежелательному явлению; и (b) в поддерживающей схеме, включающей введение дозы, равной или меньшей, чем самая высокая доза, переносимая субъектом, в качестве поддерживающей дозы для субъекта.
В другом конкретном варианте осуществления, описанном в данном документе, представлена рекомбинантная ASM человека для применения в лечении недостаточности кислой сфингомиелиназы у субъекта-человека, подготовленная для введения в схеме с увеличением дозы со следующими последовательными дозами: 0,1 мг/кг; 0,3 мг/кг; 0,6 мг/кг; и 1,0 мг/кг, где каждую дозу вводят по меньшей мере дважды, и каждую дозу вводят с двухнедельными интервалами, и где субъекта подвергают мониторингу в отношении токсических побочных эффектов перед повышением дозы до следующего уровня.
3.1. ТЕРМИНОЛОГИЯ
Применяемые в данном документе выражения «приблизительно» и «примерно» применяются взаимозаменяемо в контексте данной величины для обозначения диапазона вокруг данной величины, где полученная величина является, по сути, такой же, как точно перечисленная величина. В конкретном варианте осуществления «приблизительно» означает в пределах 10%, 15%, 25% данной величины или диапазона.
Применяемое в данном документе выражение «пожилой человек» относится к человеку 65 лет или старше.
Применяемое в данном документе выражение «взрослый человек» относится к человеку, которому 18 лет или старше.
Применяемое в данном документе выражение «человек-ребенок» относится к человеку, которому от 1 года до 18 лет.
Применяемое в данном документе выражение «человек-младенец» относится к человеку от новорожденного до возраста 1 года.
Применяемое в данном документе выражение «человек-ребенок ясельного возраста» относится к человеку, которому от 1 года до 3 лет.
Применяемое в данном документе выражение «нежелательное явление» относится к «любому неблагоприятному медицинскому случаю у пациента или субъекта клинического испытания, которому ввели фармацевтический продукт», как определено в стандартной терминологии Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1. «Связанное нежелательное явление» представляет собой нежелательное явление, которое имеет причинную взаимосвязь с лечением.
Применяемое в данном документе выражение «поддерживающая доза (дозы)» и т.п. относится к дозировке, вводимой пациентам с ASMD для поддержания желательного терапевтического эффекта. В конкретных вариантах осуществления поддерживающая доза (дозы) поддерживает один, два, три, четыре или более из следующих желательных терапевтических эффектов: (i) снижение объема селезенки, что оценивается с помощью техник, известных в данном уровне техники, например МРТ (магнитно-резонансная терапия); (ii) снижение уровней сфингомиелина печени, что оценивается с помощью техник, известных в данном уровне техники, например биохимического анализа и (или) гистоморфометрического анализа образцов печени; (iii) увеличение способности переносить физическую нагрузку, что оценивается с помощью техник, известных в данном уровне техники, например максимальная рабочая нагрузка с помощью велоэргометрии, включая относительную расчетную максимальную рабочую нагрузку, пиковое потребление кислорода и продуцирование диоксида углерода; (iv) увеличение легочной функции, что оценивается с помощью техник, известных в данном уровне техники, например техник, описанных в American Thoracic Society, 1991, Am. Rev. Respir. Dis. 144: 1202-1218, такой как диффузионная емкость легких (DLCO), относительная расчетная форсированная жизненная емкость легких (FVC), которая измеряется с помощью, например, спирометрических техник, объем форсированного выдоха за 1 секунду (FEV1), который измеряется с помощью, например, спирометрических техник, и общая емкость легких; (v) уменьшение сфингомиелина бронхоальвеолярного лаважа (BAL); (vi) уменьшение объема печени, которое оценивается с помощью техник, известных в данном уровне техники, например МРТ: (vii) улучшение внешнего вида легкого, которое оценивается с помощью техник, известных в данном уровне техники, например изображения компьютерной томографии с высоким разрешением (CT) или рентгенографии грудной клетки; (viii) уменьшение концентрации сфингомиелина в печени, коже, плазме и сухой капле крови (DBS), которое измеряется с помощью, например, тандемной масс-спектрометрии; (ix) снижение или ослабление тяжести ASMD и (или) симптома, связанного с ней; (x) снижение длительности симптома, связанного с ASMD; (xi) профилактика рецидива симптома, связанного с ASMD; (xii) снижение частоты госпитализации субъекта; (vi) снижение длительности госпитализации; (xiii) увеличение выживания субъекта; (xiv) снижение смертности; (xv) уменьшение частоты госпитализации; (xvi) снижение числа симптомов, связанных с ASMD; (xvii) увеличение бессимптомного выживания пациентов с ASMD; (xviii) улучшение неврологической функции (например, психомоторной функции, социальной ответственности и т.д.); (xix) улучшение очищения легких, которое измеряется с помощью, например, количества клеток и профиля BAL; (xx) уменьшение уровней хитотриозидазы в сыворотке; (xxi) уменьшение уровней хемокина (c-c) motif ligand 18 (CCL18) в сыворотке; (xxii) улучшение липидного профиля (например, HDL [липопротеины высокой плотности, или ЛВП], LDL [липопротеины низкой плотности, или ЛНП], холестерина, триглицеридов и соотношения общий холестерин:HDL); и (xxiii) улучшенное качество жизни, которое оценивается с помощью, например, опросника. В определенных вариантах осуществления поддерживающая доза представляет собой самую высокую или максимальную дозу, переносимую пациентом.
В некоторых вариантах осуществления поддерживающая доза представляет собой дозу от 0,5 мг/кг до 1,5 мг/кг, от 0,75 мг/кг до 1,25 мг/кг, от 1 мг/кг до 2,5 мг/кг, от 1 мг/кг до 2,75 мг/кг, от 1,5 мг/кг до 2,5 мг/кг, от 1,5 мг/кг до 2,75 мг/кг, от 2 мг/кг до 2,5 мг/кг, от 2 мг/кг до 2,75 мг/кг, от 2,5 мг/кг до 2,75 мг/кг, от 2,5 мг/кг до 3 мг/кг, от 3 мг/кг до 4 мг/кг, от 3 мг/кг до 5 мг/кг, от 4 мг/кг до 5 мг/кг, от 2 мг/кг до 5 мг/кг или от 5 мг/кг до 10 мг/кг ASM. В определенных вариантах осуществления поддерживающая доза представляет собой дозу от 5 мг/кг до 15 мг/кг, от 10 мг/кг до 15 мг/кг, от 10 мг/кг до 20 мг/кг, от 15 мг/кг до 20 мг/кг, от 20 мг/кг до 30 мг/кг, от 25 мг/кг до 50 мг/кг, от 30 мг/кг до 40 мг/кг, от 30 мг/кг до 45 мг/кг или от 40 мг/кг до 50 мг/кг ASM. В некоторых вариантах осуществления поддерживающая доза составляет 0,75 мг/кг, 0,80 мг/кг, 0,85 мг/кг, 0,90 мг/кг, 0,95 мг/кг, 1 мг/кг, 1,1 мг/кг, 1,2 мг/кг, 1,25 мг/кг, 1,3 мг/кг, 1,4 мг/кг, 1,5 мг/кг, 1,75 мг/кг или 2 мг/кг ASM. В определенных вариантах осуществления поддерживающая доза составляет 2,5 мг/кг, 2,75 мг/кг, 3 мг/кг, 3,25 мг/кг, 3,5 мг/кг, 3,75 мг/кг, 4 мг/кг, 4,25 мг/кг, 4,5 мг/кг, 4,75 мг/кг, 5 мг/кг, 5,5 мг/кг, 6 мг/кг, 6,5 мг/кг, 7 мг/кг, 7,5 мг/кг, 8 мг/кг, 8,5 мг/кг, 9 мг/кг, 9,5 мг/кг или 10 мг/кг ASM. В некоторых вариантах осуществления поддерживающая доза составляет 11 мг/кг, 12 мг/кг,13 мг/кг, 14 мг/кг, 15 мг/кг, 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг или 50 мг/кг ASM. В некоторых вариантах осуществления поддерживающая доза составляет по меньшей мере 1 мг/кг, по меньшей мере 2 мг/кг, по меньшей мере 3 мг/кг, по меньшей мере 4 мг/кг, по меньшей мере 5 мг/кг, по меньшей мере 6 мг/кг, по меньшей мере 7 мг/кг, по меньшей мере 8 мг/кг ASM, при этом самая высокая доза составляет 10 мг/кг ASM. В определенных вариантах осуществления поддерживающая доза составляет по меньшей мере 10 мг/кг, по меньшей мере 15 мг/кг, по меньшей мере 20 мг/кг, по меньшей мере 25 мг/кг, по меньшей мере 30 мг/кг или по меньшей мере 35 мг/кг ASM, при этом самая высокая доза составляет 50 мг/кг.
Применяемое в данном документе выражение «нетоксичная доза (дозы)» и т.п. относится к дозировке, вводимой пациентам с ASMD, не приводя к одному, двум, трем или всем из следующего: (i) умеренное или тяжелое связанное нежелательное явление, которое определяется по клиническому симптому, который препятствует нормальному ежедневному функционированию и требует дополнительного мониторинга, вмешательства или лечения, или, патологическому лабораторному значению или процедурному результату клинической задачи, которая требует дальнейшего отслеживания, лечения или исследования. Смотрите, например, стандартную терминологию Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1; (ii) величина общего билирубина составляет больше 1,5 мг/дл, 1,75 мг/дл, 2,0 мг/дл, 2,1 мг/дл, 2,2 мг/дл, 2,3 мг/дл, 2,4 мг/дл, 2,5 мг/дл, 2,6 мг/дл, 2,7 мг/дл, 2,75 мг/дл, 2,8 мг/дл, 2,9 мг/дл, 3,0 мг/дл, 3,1 мг/дл, 3,2 мг/дл, 3,3 мг/дл, 3,4 мг/дл, 3,5 мг/дл, 3,6 мг/дл, 3,7 мг/дл, 3,8 мг/дл, 3,9 мг/дл или 4 мг/дл или в диапазоне от 2,1 мг/дл до 2,5 мг/дл, от 2,1 мг/дл до 3,0 мг/дл или от 2,1 мг/дл до 4 мг/дл, которая сохраняется в течение более 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM; (iii) концентрация церамида в плазме больше 8,2 мкг/дл, 8,3 мкг/дл, 8,4 мкг/дл, 8,5 мкг/дл, 8,75 мкг/дл, 9 мкг/дл, 9,5 мкг/дл, 10 мкг/дл, 11 мкг/дл, 12 мкг/дл, 13 мкг/дл, 14 мкг/дл, 15 мкг/дл, 16 мкг/дл, 17 мкг/дл, 18 мкг/дл, 19 мкг/дл, 20 мкг/дл, 25 мкг/дл, 30 мкг/дл, 35 мкг/дл, 40 мкг/дл, 45 мкг/дл, 50 мкг/дл, 55 мкг/дл, 60 мкг/дл, 65 мкг/дл, 70 мкг/дл, 75 мкг/дл или 80 мкг/дл или в диапазоне от 8,2 мкг/дл до 10 мкг/дл, от 8,5 мкг/дл до 10 мкг/дл, от 9 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 15 мкг/дл, от 10 мкг/дл до 20 мкг/дл, от 15 мкг/дл до 20 мкг/дл или от 20 мкг/дл до 30 мкг/дл через 6 часов, 8 часов, 10 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM; или (iv) острофазовый ответ/реакция. «Нетоксичная доза» ASM может изменяться в зависимости, например, от стабильности применяемого фермента, активности применяемого фермента и (или) пути введения фермента. Например, доза модифицированного фермента ASM с увеличенной активностью может быть ниже дозировки немодифицированной ASM. Специалист в данной области будет способен скорректировать дозу вводимого фермента на основании стабильности фермента, активности фермента и (или) пути введения фермента.
Острофазовая реакция представляет собой раннюю реакцию (как правило, например, в пределах 12-72 часа) после введения ASM, которая является характерной для воспалительного ответа. Острофазовый ответ можно оценить путем изменения концентрации острофазового агента (такого как, например, CRP/hs-CRP, ферритин, фибриноген, железо или трансферрин), изменения процентного соотношения нейтрофилов, изменения протромбинового времени или изменения частичного тромбопластинового времени. В конкретном варианте осуществления увеличение концентрации CRP/hs-CRP через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации CRP/hs-CRP у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрация CRP/hs-CRP в плазме, которая больше нормальной концентрации CRP/hs-CRP в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, может быть использована в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию CRP/hs-CRP в плазме больше чем примерно 8,1 мг/л, 8,2 мг/л, 8,3 мг/л, 8,4 мг/л, 8,5 мг/л, 9 мг/л, 9,5 мг/л, 10 мг/л, 11 мг/л или 12 мг/л или в диапазоне от 8,5 мг/л до 10 мг/л, или от 8,5 мг/дл до 12 мг/л, или от 10 мг/л до 12 мг/л через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно применять в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение в концентрации ферритина через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации ферритина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию ферритина в плазме, которая больше нормальной концентрации ферритина в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно применять в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию ферритина в плазме больше чем примерно 300 нг/мл, 325 нг/мл, 350 нг/мл, 375 нг/мл, 400 нг/мл, 425 нг/мл, 450 нг/мл, 475 нг/мл, 500 нг/мл, 525 нг/мл, 550 нг/мл, 575 нг/мл, 600 нг/мл, 625 нг/мл, 650 нг/мл, 675 нг/мл, 700 нг/мл, 725 нг/мл, 750 нг/мл, 775 нг/мл, 800 нг/мл, 850 нг/мл, 900 нг/мл, 950 нг/мл, 1000 нг/мл, 1050 нг/мл, 1100 нг/мл, 1150 нг/мл или 1200 нг/мл или в диапазоне от 600 нг/мл до 800 нг/мл, от 650 нг/мл до 850 нг/мл, от 600 нг/мл до 1000 нг/мл, от 600 нг/мл до 1200 нг/мл, от 800 нг/мл до 1000 нг/мл, от 900 нг/мл до 1000 нг/мл или от 1000 нг/мл до 1200 нг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно применять в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение в концентрации IL-8 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации IL-8 у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию IL-8 в плазме или сыворотке, которая больше чем нормальной концентрации IL-8 в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию IL-8 в плазме больше чем примерно 24 пг/мл, 50 пг/мл, 75 пг/мл, 100 пг/мл, 200 пг/мл, 300 пг/мл, 400 пг/мл, 500 пг/мл, 600 пг/мл, 700 пг/мл, 800 пг/мл или 900 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации IL-6 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации IL-6 у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию IL-6 в плазме или сыворотке, которая больше нормальной концентрации IL-6 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию IL-6 в плазме больше чем примерно 4,4 пг/мл, 6 пг/мл, 8 пг/мл, 10 пг/мл, 15 пг/мл, 20 пг/мл, 25 пг/мл или 30 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации кальцитонина в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации кальцитонина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию кальцитонина в плазме или сыворотке, которая больше нормальной концентрации кальцитонина в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию в плазме кальцитонина больше чем примерно 9,4 пг/мл, 20 пг/мл, 30 пг/мл, 40 пг/мл, 50 пг/мл, 75 пг/мл, 100 пг/мл, 150 пг/мл, 200 пг/мл или 250 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации фибриногена через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации фибриногена у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию фибриногена в плазме, которая больше нормальной концентрации фибриногена в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию в плазме фибриногена больше чем примерно 350 мг/дл, 375 мг/дл, 400 мг/дл, 425 мг/дл или 450 мг/дл или в диапазоне от 350 мг/дл до 400 мг/дл, от 350 мг/дл до 450 мг/дл или от 400 мг/дл до 450 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В одном варианте осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM по сравнению с относительным содержанием у пациента нейтрофилов от общего количества лейкоцитов до введения ASM можно использовать в качестве измерения острофазового ответа. В другом варианте осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов, которое больше нормального относительного содержания нейтрофилов от общей концентрации лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов, которое составляет 70%, 75%, 80%, 85%, 90%, 95% или больше, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа.
Применяемое в данном документе выражение «низкая, нетоксичная доза (дозы)» и т.п. в контексте начальной дозы или доз, вводимых субъекту, относится к дозировке, которая является первой дозой или дозами ASM, вводимыми субъекту для лечения ASMD, которая является нетоксичной. В определенных вариантах осуществления низкая, нетоксичная доза (дозы) представляет собой дозу от 0,001 мг/кг до 0,01 мг/кг, от 0,001 мг/кг до 0,01 мг/кг, от 0,001 мг/кг до 0,05 мг/кг, от 0,001 мг/кг до 0,1 мг/кг, от 0,001 мг/кг до 0,5 мг/кг, от 0,05 мг/кг до 0,275 мг/кг, от 0,075 мг/кг до 0,275 мг/кг, от 0,05 мг/кг до 0,2 мг/кг, от 0,075 мг/кг до 0,2 мг/кг, от 0,1 мг/кг до 0,275 мг/кг, от 0,1 мг/кг до 0,25 мг/кг, от 0,1 мг/кг до 1 мг/кг, от 0,5 мг/кг до 1 мг/кг, от 0,75 мг/кг до 1 мг/кг, от 0,1 мг/кг до 2 мг/кг, от 0,5 мг/кг до 2 мг/кг, от 0,75 мг/кг до 2 мг/кг, от 1 мг/кг до 2 мг/кг или от 1,25 мг/кг до 2 мг/кг, от 1,5 мг/кг до 2 мг/кг или от 1,75 мг/кг до 2 мг/кг ASM. В некоторых конкретных вариантах осуществления низкая, нетоксичная доза представляет собой дозу 0,001 мг/кг, 0,005 мг/кг, 0,0075 мг/кг, 0,01 мг/кг, 0,0125 мг/кг, 0,025 мг/кг, 0,05 мг/кг, 0,075 мг/кг, 0,1 мг/кг, 0,125 мг/кг, 0,15 мг/кг, 0,175 мг/кг, 0,2 мг/кг, 0,225 мг/кг, 0,25 мг/кг, 0,275 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,75 мг/кг, 0,8 мг/кг, 0,9 мг/кг или 1 мг/кг ASM.
Выражения «субъект» и «пациент» применяются в данном документе взаимозаменяемо для обозначения человека. В конкретном варианте осуществления человек диагностируется или был продиагностирован как имеющий ASMD.
Применяемое в данном документе выражение «терапевтически эффективный» в контексте введения дозы ASM субъекту относится к количеству ASM, которое приводит к благоприятному или терапевтическому эффекту. В конкретных вариантах осуществления выражение «терапевтически эффективный» в контексте введения дозы ASM субъекту относится к количеству ASM, которое является достаточным для достижения по меньшей мере одного, двух, трех, четырех или более из следующих эффектов: (i) уменьшение объема селезенки, которое оценивается с помощью техник, известных в данном уровне техники, например МРТ; (ii) снижение уровней сфингомиелина печени, которое оценивается с помощью техник, известных в данном уровне техники, например биохимический анализ и (или) гистоморфометрический анализ образцов печени; (iii) увеличение способности переносить физическую нагрузку, которое оценивается с помощью техник, известных в данном уровне техники, например максимальная рабочая нагрузка с помощью велоэргометрии, включая процентную расчетную максимальную рабочую нагрузку, пиковое потребление кислорода и продуцирование диоксида углерода; (iv) увеличение легочной функции, которое оценивается с помощью техник, известных в данном уровне техники, например, техник, описанных в American Thoracic Society, 1991, Am. Rev. Respir. Dis. 144: 1202-1218, таких как диффузионная емкость легких (DLCO), относительная расчетная форсированная жизненная емкость легких (FVC), которая измеряется с помощью, например, спирометрических техник, объем форсированного выдоха с течение 1 секунды (FEV1), который измеряется с помощью, например, спирометрических техник, и общая емкость легких; (v) уменьшение сфингомиелина бронхоальвеолярного лаважа (BAL); (vi) уменьшение объема печени, которое оценивается с помощью техник, известных в данном уровне техники, например МРТ: (vii) улучшение внешнего вида легкого, которое оценивается с помощью техник, известных в данном уровне техники, например изображения КТ высокого разрешения или рентгенографии грудной клетки; (viii) уменьшение концентрации сфингомиелина в коже, плазме и сухой капле крови (DBS), которое измеряется с помощью, например, тандемной масс-спектрометрии; (ix) снижение или облегчение тяжести ASMD и (или) симптома, связанного с ней; (x) снижение длительности симптома, связанного с ASMD; (xi) профилактика рецидива симптома, связанного с ASMD; (xii) снижение частоты госпитализации субъекта; (vi) снижение длительности госпитализации; (xiii) увеличение выживания субъекта; (xiv) снижение смертности; (xv) уменьшение частоты госпитализации; (xvi) снижение числа симптомов, связанных с ASMD; (xvii) увеличение бессимптомного выживания пациентов с ASMD; (xviii) улучшение неврологической функции (например, психомоторной функции, социальной ответственности и т.д.); (xix) улучшение очищения легких, что измеряется с помощью, например, количества клеток и профиля BAL; (xx) уменьшение уровней хитотриозидазы в сыворотке; (xxi) уменьшение уровней CCL18 в сыворотке; (xxii) улучшение липидного профиля (например, HDL, LDL, холестерина, триглицеридов и соотношения общий холестерин:HDL); и (xxiii) улучшенное качество жизни, которое оценивается с помощью, например, опросника.
Применяемые в данном документе выражения «терапии» и «терапия» могут относиться к любому протоколу (протоколам), способу (способам), композициям, составам и (или) средству (средствам), которые можно применять в лечении, тактике или облегчении ASMD или состояния или симптома, связанного с ней. В определенных вариантах осуществления выражения «терапии» и «терапия» относятся к биологической терапии, вспомогательной терапии и (или) другим терапиям, пригодным в лечении, тактике, профилактике или облегчении ASMD или состояния или симптома, связанного с ней. В вариантах осуществления выражение «терапия» относится к терапии, которая достигает одного, двух или более эффектов из следующих: (i) усиливает доставку ASM к местам патологии, (ii) усиливает активность ASM и (iii) усиливает стабильность ASM. В определенных вариантах осуществления выражение «терапия» относится к терапии, отличной от ASM. В конкретных вариантах осуществления «дополнительная терапия» и «дополнительные терапии» относятся к терапии, отличной от ASM.
Применяемое в данном документе выражение «токсический эффект (токсические эффекты)» и т.п. относится к одному, двум, трем или всем вариантам из следующих после введения дозы (доз) ASM: (i) умеренное или тяжелое связанное нежелательное явление, которое определяется по клиническому симптому, который препятствует нормальному ежедневному функционированию и требует дополнительного мониторинга, вмешательства или лечения, или патологическому лабораторному значению или процедурному результату клинической задачи, которые требуют дальнейшего мониторинга, лечения или исследования. Смотрите, например, стандартную терминологию Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1; (ii) величина общего билирубина больше 1,5 мг/дл, 1,75 мг/дл, 2,0 мг/дл, 2,1 мг/дл, 2,2 мг/дл, 2,3 мг/дл, 2,4 мг/дл, 2,5 мг/дл, 2,6 мг/дл, 2,7 мг/дл, 2,75 мг/дл, 2,8 мг/дл, 2,9 мг/дл, 3,0 мг/дл, 3,1 мг/дл, 3,2 мг/дл, 3,3 мг/дл, 3,4 мг/дл, 3,5 мг/дл, 3,6 мг/дл, 3,7 мг/дл, 3,8 мг/дл, 3,9 мг/дл или 4 мг/дл или в диапазоне от 2,1 мг/дл до 2,5 мг/дл, от 2,1 мг/дл до 3,0 мг/дл или от 2,1 мг/дл до 4 мг/дл, которая сохраняется в течение более 16 часов, 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM; (iii) концентрация церамида в плазме больше 8,2 мкг/дл, 8,3 мкг/дл, 8,4 мкг/дл, 8,5 мкг/дл, 8,75 мкг/дл, 9 мкг/дл, 9,5 мкг/дл, 10 мкг/дл, 11 мкг/дл, 12 мкг/дл, 13 мкг/дл, 14 мкг/дл, 15 мкг/дл, 16 мкг/дл, 17 мкг/дл, 18 мкг/дл, 19 мкг/дл, 20 мкг/дл, 25 мкг/дл, 30 мкг/дл, 35 мкг/дл, 40 мкг/дл, 45 мкг/дл, 50 мкг/дл, 55 мкг/дл, 60 мкг/дл, 65 мкг/дл, 70 мкг/дл, 75 мкг/дл или 80 мкг/дл или в диапазоне от 8,2 мкг/дл до 10 мкг/дл, от 8,5 мкг/дл до 10 мкг/дл, от 9 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 15 мкг/дл, от 10 мкг/дл до 20 мкг/дл, от 15 мкг/дл до 20 мкг/дл или от 20 мкг/дл до 30 мкг/дл через 6 часов, 8 часов, 10 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM; или (iv) острофазовый ответ.
Острофазовый ответ можно оценить по изменению концентрации острофазового агента (такого как, например, C-реактивный белок, ферритин, альбумин, IL-8, Il-6, кальцитонин, фибриноген, железо или трансферрин), изменению относительного содержания нейтрофилов, изменению протромбинового времени или изменению частичного тромбопластинового времени. В конкретном варианте осуществления увеличение концентрации CRP/hs-CRP через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации CRP/hs-CRP пациента до введения ASM можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию CRP/hs-CRP в плазме, которая больше нормальной концентрации CRP/hs-CRP в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию CRP/hs-CRP в плазме больше чем примерно 8,1 мг/дл, 8,2 мг/дл, 8,3 мг/дл, 8,4 мг/дл, 8,5 мг/дл, 8,6 мг/дл, 8,7 мг/дл, 8,8 мг/дл, 8,9 мг/дл 9 мг/дл, 9,5 мг/дл, 10 мг/дл, 11 мг/дл или 12 мг/дл или в диапазоне от 8,5 мг/дл до 10 мг/дл, или от 8,5 мг/дл до 12 мг/дл, или от 10 мг/дл до 12 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации ферритина через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации ферритина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию ферритина в плазме, которая больше нормальной концентрации ферритина в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию ферритина в плазме больше чем примерно 600 нг/мл, 625 нг/мл, 650 нг/мл, 675 нг/мл, 700 нг/мл, 725 нг/мл, 750 нг/мл, 775 нг/мл, 800 нг/мл, 850 нг/мл, 900 нг/мл, 950 нг/мл, 1000 нг/мл, 1050 нг/мл, 1100 нг/мл, 1150 нг/мл или 1200 нг/мл или в диапазоне от 600 нг/мл до 800 нг/мл, от 650 нг/мл до 850 нг/мл, от 600 нг/мл до 1000 нг/мл, от 600 нг/мл до 1200 нг/мл, от 800 нг/мл до 1000 нг/мл, от 900 нг/мл до 1000 нг/мл или от 1000 нг/мл до 1200 нг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации фибриногена через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации фибриногена у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию фибриногена в плазме, которая больше нормальной концентрации фибриногена в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию фибриногена в плазме больше чем примерно 350 мг/дл, 375 мг/дл, 400 мг/дл, 425 мг/дл или 450 мг/дл или в диапазоне от 350 мг/дл до 400 мг/дл, от 350 мг/дл до 450 мг/дл или от 400 мг/дл до 450 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления уменьшение концентрации альбумина через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации альбумина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления уменьшение концентрации альбумина на следующую величину 0,2, 0,4, 0,6, 1, 1,5, 2,0 г/дл от нормального диапазона 3,5-5,0 г/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления уменьшение концентрации ферритина через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации ферритина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления уменьшение концентрации ферритина на следующую величину 20, 40, 60, 80, 100, 120, 140, 160 мкг/дл от нормального диапазона 60-170 мкг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления уменьшение концентрации трансферрина через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации трансферрина у пациента до введения ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления уменьшение концентрации трансферрина на следующую величину 20, 40, 60, 80, 100, 120, 140, 160, 180 мг/дл от нормального диапазона 202-336 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В одном варианте осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, по сравнению с относительным содержанием нейтрофилов пациентов от общего количества лейкоцитов до введения ASM, можно использовать в качестве измерения острофазового ответа. В другом варианте осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов, которое больше нормального относительного содержания нейтрофилов от общей концентрации лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления увеличение относительного содержания нейтрофилов от общего количества лейкоцитов, которое составляет 70%, 75%, 80%, 85%, 90%, 95% или больше, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
4. КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1 является графическим представлением схемы протокола.
Фиг.2 представляет собой таблицу, показывающую демографию и исходные характеристики пациентов, включенных в описанный ниже протокол.
Фиг.3A, 3B и 3C представляют собой графики, отображающие уровни церамида в плазме (фиг.3A), и уровни сфингомиелина в плазме (фиг.3B), и уровень сфингомиелина в сухой капле крови (фиг.3C) с течением времени у различных пациентов, которым вводили различные дозы rhASM. Правые оси графиков отображают число пациентов и дозировку rhASM.
Фиг.4 представляет собой график, отображающий уровни общего билирубина, определенные с течением времени у различных пациентов, которым вводили различные дозы rhASM в ходе протокола. Правая ось графика отображает число пациентов и дозировку rhASM.
Фиг.5A-5G представляют собой графики, отображающие уровни CRP/hs-CRP (фиг.5A), процент нейтрофилов (фиг.5B), фибриногена (фиг.5C), ферритина (фиг.5D), IL-8 (фиг.5E), IL-6 (фиг.5F) и кальцитонина (фиг.5G), определенные с течением времени у различных пациентов, которым вводили различные дозы rhASM в ходе протокола. Правые оси графиков отображают число пациентов и дозировку rhASM.
Фиг.6 представляет собой таблицу возникающих в ходе лечения нежелательных явлений для четырех пациентов, каждые на различной дозе rhASM, где явления рассматриваются как связанные (возможно, вероятно или точно) с лечением.
Фиг.7 представляет собой диаграмму, показывающую период первичного лечения, который состоит из фаз с увеличением дозы и поддержанием дозы, протокола повторной дозы rhASM фазы 2 у пациентов с ASMD.
5. ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к ферментной заместительной терапии с увеличением дозы для лечения субъектов-людей, имеющих ASMD - особенно субъектов, имеющих не неврологические проявления NPD, и в определенных вариантах осуществления NPD типа B. Более конкретно, фермент, ASM, вводят таким пациентам в начальной низкой, нетоксичной дозе, которую затем повышают в последующих введениях. Самую высокую дозу ASM, переносимую пациентом, можно затем использовать в качестве поддерживающей дозы. Альтернативно, терапевтически эффективную дозу, которая меньше самой высокой переносимой дозы, можно использовать в качестве поддерживающей дозы.
Лечение NPD требует доз, достаточно высоких для достижения достаточного распределения фермента ASM в органах патологии (например, в селезенке, легких, сердце, почке и мозге). После внутривенного введения рекомбинантной ASM человека ASMKO-мышам основная часть активности ASM распределяется в печени с небольшими количествами ASM-ферментативной активности, обнаруживаемой в других органах патологии, таких как селезенка, сердце, почка и легкое (смотрите, например, фиг.9B He et al., 1999, Biochimia et Biophsyica Acta 1432: 251-264). Таким образом, очень высокие дозы будут необходимы для обеспечения распределения и доставки вводимого фермента в легкое, сердце и почку пациентов, страдающих от ASMD или болезни Ниманна-Пика.
Исследования в модели на мышах, нокаутных по ASM (ASKMO-мыши), показали, что основная часть вводимой rhASM распределяется в печени и селезенке, где она снижает субстрат, но намного в меньшей степени в легких, сердце и мозге (Miranda et al. FASEB 2000, 14:1988). В последующих исследованиях с применением более высоких доз rhASM в ASMKO-мышиной модели субстрат снижался, и токсичность не наблюдалась при дозах ≤3,0 мг/кг; фактически, клинические симптомы токсичности не наблюдались, пока не использовались дозы ≥10 мг/кг. Смотрите Dose Responsive Toxicological Findings Following Intravenous Administration of Recombinant Human Acid Sphingomyelinase (rhASM) to Acid Sphingomyelinase Knock-out (ASMKO) Mice, C. Nickerson, J. Murray, A. Vitsky, M. Hawes, S. Ryan, P. Ewing, B. Thurberg, L. Andrews. Dept Pharm/Tox, Pathology, Genzyme Corp., Framingham, MA., American Society of Human Genetics 2005; и Elevations of Pro-Inflammatory Cytokines and Decreases in Cardiovascular Hemodynamics Following Intravenous Administration of Recombinant Human Acid Sphingomyelinase (rhASM) to Acid Sphingomyelinase Knock-out (ASMKO) Mice, J. Murray, A.M. D'Angona, C. Nickerson, A. Vitsky, M. Hawes, S. Ryan, P. Ewing, B. Thurberg, L. Andrews. Dept. Pharmacology/Toxicology & Pathology, Genzyme Corp., Framingham, MA., Society of Toxicology 2006.
На основании этих данных ASKMO мы лечили субъектов-людей с не нейронопатической ASMD посредством консервативной максимальной дозы 1,0 мг/кг, как описано в разделе 6 ниже. Достаточно неожиданно, токсичность у субъектов-людей, включая возникновение нежелательных явлений с клиническими симптомами, наблюдалась с применением доз на уровне 0,3 мг/кг! Этот результат был особенно неожиданным, поскольку фермент ASM отсутствует в нокаутной мышиной модели, которая должна отражать более тяжелое состояние, чем субъекты-люди, у которых есть, по меньшей мере, некоторая ферментная активность и относительно легкое заболевание.
Не ограничиваясь какой-либо теорией, введение высоких доз ASM пациентам с NPD может привести к гидролизу больших количеств сфингомиелина в большие концентрации церамида, который может производить токсические побочные эффекты, наблюдаемые у этих пациентов с NPD. Фермент ASM гидролизует сфингомиелин, который является главным компонентом плазматической мембраны клеток (смотрите, например, Milaus et al., 2010 FEBS Letters 584: 1887-1894), в церамид и фосфохолин. Церамид, как известно, играет роль в клеточной смерти и, как известно, является проапоптическим агентом (смотрите, например, Smith and Schuchman, 2008, FASEB 22: 3419-3431).
Более того, в отличие от других лизосомальных ферментов, связанных с охарактеризованными лизосомальными болезнями накопления, ASM гидролизует сфингомиелин при нейтральном pH, обнаруживаемом в плазме, и кислом pH, обнаруженном в лизосоме (смотрите, например, Schissel et al., 1998. J. Biol. Chem. 273: 2738-2746). Способность фермента ASM функционировать в плазме может привести к гидролизу сфингомиелина, обнаруживаемого в липопротеинах и плазматической мембране клеток, что может увеличить количество продукта распада, церамида, который может вызвать токсические побочные эффекты, наблюдаемые у пациентов с NPD, которым вводят высокие дозы фермента ASM.
Для разрешения проблемы достижения достаточного распределения фермента ASM в органах патологии, при этом избегая или сводя к минимуму токсичность, связанную с введением высоких доз фермента, изобретатели разрабатывают схемы лечения, описанные в данном документе, в которых сначала вводят пациенту с NPD низкую, нетоксичную дозу фермента ASM и с течением времени увеличивают дозу. По мере увеличения дозы фермента ASM пациента можно подвергать мониторингу в отношении общей/прямой/непрямой концентраций билирубина, продуцирования острофазовых агентов, продуцирования медиаторов воспаления и связанных нежелательных явлений. Введение низкой дозы ASM и увеличение фермента облегчает сокращение массы накопленного сфингомиелина. Как только у пациента снижается масса накопленного сфингомиелина, пациенту можно безопасно вводить более высокие дозы фермента ASM для обеспечения достаточного распределения фермента ASM к целевым органам (например, печени, селезенке, легким, сердцу, почке, мозгу, костному мозгу, скелету, суставам и т.д.). В определенных вариантах осуществления максимальную дозу, переносимую пациентом, можно использовать в качестве поддерживающей дозы. Альтернативно, терапевтически эффективную дозу, которая меньше самой высокой переносимой дозы, можно использовать в качестве поддерживающей дозы. В некоторых вариантах осуществления, основываясь на состоянии пациента, поддерживающая доза может быть увеличена или уменьшена.
В определенных вариантах осуществления лечение пациента можно подвергать мониторингу путем измерения уровней сфингомиелина в плазме и уровней церамида в плазме, продуцирования «острофазовых агентов» и медиаторов воспаления, которые являются мерой воспалительных ответов, концентраций билирубина (общей, прямой или непрямой) и (или) других биохимических маркеров, для обеспечения стабильного ответа перед повышением дозы до следующего уровня. Эти маркеры включают, но без ограничений, CRP/hs-CRP, цитокины (например, IL-8, IL-6), кальцитонин и ферритин. В конкретных вариантах осуществления пациента можно подвергать мониторингу в отношении одного или нескольких связанных нежелательных явлений, которые могут включать, но без ограничений, системные симптомы (например, лихорадку, тошноту, рвоту, боль, миалгию и желтуху).
5.1. ПРОТОКОЛ УВЕЛИЧЕНИЯ ДОЗЫ
Описаны способы лечения ASMD, включающие введение одной или нескольких начальных, низких нетоксичных доз ASM субъекту для снижения количества сфингомиелина, который накопился у субъекта. После определенного периода времени доза ASM может быть увеличена, пока не будет достигнута самая высокая дозировка, переносимая субъектом, которая является терапевтически эффективной. После определения этой дозировки ее можно использовать в качестве поддерживающей дозы для лечения субъекта. Поддерживающая доза может вводиться каждую неделю, раз в две недели или каждый месяц субъекту для лечения ASMD. В некоторых вариантах осуществления субъекта, получающего поддерживающую дозу, подвергают мониторингу каждые 3 месяца, каждые 6 месяцев или каждый год в отношении одного, двух, трех или всех из следующих явлений: (i) связанные нежелательные явления; (ii) общие/прямые/непрямые концентрации билирубина; (iii) концентрация церамида в плазме; или (iv) острофазовый ответ. Если субъект испытывает связанное нежелательное явление умеренной интенсивности (например, связанное умеренное нежелательное явление), общая концентрация билирубина больше величины общего билирубина для человека без ASMD (например, здорового человека), концентрация церамида в плазме больше концентрации церамида в плазме человека без ASMD (например, здорового человека) или острофазовый ответ, то доза, вводимая субъекту, может быть оценена врачом или другим медицинским работником для определения того, нужно ли корректировать дозу.
В одном варианте осуществления способ лечения субъекта-человека, имеющего недостаточность кислой сфингомиелиназы, включает: (a) схему для сокращения массы накопленного субстрата сфингомиелина у субъекта-человека, включающую: (i) введение начальной низкой нетоксичной дозы ASM субъекту-человеку; (ii) введение последовательно более высоких доз ASM субъекту-человеку и мониторинг субъекта в отношении одного или нескольких нежелательных побочных эффектов после каждой последующей дозы, на которые указывают повышенный билирубин или связанное нежелательное явление; и (b) поддерживающую схему, включающую введение дозы, равной или меньшей, чем самая высокая переносимая доза субъектом, в качестве поддерживающей дозы субъекту. В определенных вариантах осуществления диапазоны начальной дозы составляют от 0,1 мг/кг до 0,5 мг/кг или от 0,1 мг/кг до 1 мг/кг ASM. В некоторых вариантах осуществления последовательно более высокие дозы вводят через одну, две, три или четыре недели после предыдущей дозы. В определенных вариантах осуществления последовательно более высокая доза примерно на 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, 1,2 мг/кг, 1,5 мг/кг, 1,75 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг или 5 мг/кг выше, чем предыдущая доза. В некоторых вариантах осуществления последовательно более высокая доза на от 0,1 до 0,5 мг/кг, от 0,1 мг/кг до 1 мг/кг, от 0,5 мг/кг до 1 мг/кг, от 0,5 мг/кг до 2 мг/кг, от 1 мг/кг до 2 мг/кг, от 2 мг/кг до 4 мг/кг или от 2 мг/кг до 5 мг/кг выше, чем предыдущая доза. В определенных вариантах осуществления самая высокая доза, переносимая субъектом, составляет от 1 мг/кг до 2,5 мг/кг. В некоторых вариантах осуществления самую высокую дозу вводят субъекту-человеку в качестве поддерживающей дозы.
В определенных вариантах осуществления способ лечения субъекта-человека, имеющего ASMD, включает: (a) сокращающее массу введение ASM для снижения количества сфингомиелина, который был накоплен у субъекта-человека, где сокращающее массу введение ASM включает: (i) введение низкой, нетоксичной дозы ASM субъекту-человеку; и (ii) введение последовательно более высоких доз ASM человеку, если субъект-человек не проявляет один или несколько нежелательных побочных эффектов, на которые указывает повышенная общая концентрация билирубина, повышенная концентрация церамида в плазме, продуцирование острофазовых агентов, продуцирование медиаторов воспаления или нежелательное явление (например, такое как определяемое с помощью стандартной терминологии Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1); и (b) поддерживающее введение ASM, где поддерживающее введение ASM включает повторное введение поддерживающей дозы ASM субъекту-человеку. В некоторых вариантах осуществления пациента подвергают мониторингу в течение периода времени после введения дозы ASM (например, 6 часов, 12 часов, 16 часов, 24 часа, 48 часов, 72 часа, каждую неделю или вплоть до следующей дозы), в отношении одного или нескольких нежелательных побочных эффектов или общего билирубина. В определенных вариантах осуществления поддерживающую дозу, которую вводят, можно корректировать во время курса лечения пациента. В некоторых вариантах осуществления поддерживающая доза, вводимая субъекту, является самой высокой дозой, переносимой субъектом.
В конкретном варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, начальной низкой, нетоксичной дозы ASM (например, дозы 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, от 0,1 мг/кг до 0,5 мг/кг или от 0,5 до 1 мг/кг ASM) и после определенного периода времени (например, 3 дня, 1 неделя, 2 недели или 3 недели) последовательное повышение дозы ASM, вводимой субъекту до тех пор, пока уровень активности ASM в одном или нескольких органах патологии не составит по меньшей мере 5%, 6%, 7%, 8%, 9%, 10%, 12%, 15%, 20%, 25%, 30%, 35%, 40%, 50%, 75%, 80%, 85%, 90%, 95% или более от активности ASM в соответствующем органе у субъекта (субъектов) без ASMD (например, здорового субъекта или популяции из 5, 10, 15, 20, 30, 40, 50, 75, 100, 150, 175 или более субъектов), что измеряется с помощью техник, известных в данном уровне техники, таких как, например, техника, описанная в He et al., 2003, Analytical Biochemistry 314: 116-120. В другом варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, дозы 0,1 мг/кг ASM и после определенного периода времени (например, 3 дня, 1 неделя, 2 недели или 3 недели) последовательное увеличение дозы ASM, вводимой субъекту до тех пор, пока уровень активности ASM в одном или нескольких из следующих органов патологии не составит от 5% до 10%, от 5% до 15%, от 5% до 20%, от 10% до 15%, от 10% до 20%, от 15% до 20%, от 15% до 25%, от 25% до 50%, от 50% до 75% или от 75% до 95% от нормальной активности ASM в соответствующем органе у субъекта (субъектов) без ASMD (например, здорового субъекта или популяции из 5, 10, 15, 20, 30, 40, 50, 75, 100, 150, 175 или более субъектов), что измеряется с помощью техник, известных в данном уровне техники, таких как, например, техника, описанная в He et al., 2003, Analytical Biochemistry 314: 116-120. В определенных вариантах осуществления дозу последовательно увеличивают, если общая концентрация билирубина меньше или равна 2,0 мг/дл или 2,1 мг/дл и субъект не испытывает умеренного или тяжелого связанного нежелательного явления. В конкретных вариантах осуществления активность ASM у нормальных здоровых субъектов, как оценивают, составляет примерно 20-40 единиц/мг белка для мозга, сердца, почки и печени на основании активности ASM в таких же органах у здоровых мышей с помощью анализа, описанного в Horinouchi et al., 1995, Nature Genetics 10: 288-293 (который включен путем ссылки в данный документ во всей своей полноте). В определенных вариантах осуществления активность ASM у нормальных здоровых субъектов, как оценивают, составляет примерно 15-25 единиц/мг белка для легкого и 10-15 единиц/мг белка для селезенки на основании активности ASM в таких же органах у здоровых мышей с помощью анализа, описанного в Horinouchi et al., 1995, Nature Genetics 10: 288-293. В определенных вариантах осуществления, как только активность ASM достигает нормального или определенного процента от нормального в одном или нескольких органах патологии, то субъекту можно вводить дозу, равную или меньше самой высокой дозы, переносимой субъектом, в качестве поддерживающей дозы. С течением времени поддерживающую дозу можно корректировать в зависимости от здоровья субъекта. В зависимости от состояний субъекта, поддерживающая доза может быть увеличена или уменьшена.
В одном варианте осуществления способ лечения ASMD включает: (a) введение человеку, нуждающемуся в этом, начальной низкой, нетоксичной дозы ASM; и (b) введение последовательно более высоких доз ASM, если человек не проявляет одного, двух, трех или четырех из следующих побочных эффектов после введения дозы ASM: (i) тяжелое связанное нежелательное явление, которое определяется с помощью, например, стандартной терминологии Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1; (ii) величина общего билирубина составляет больше 1,5 мг/дл, 1,75 мг/дл, 2,0 мг/дл, 2,1 мг/дл, 2,2 мг/дл, 2,3 мг/дл, 2,4 мг/дл, 2,5 мг/дл, 2,6 мг/дл, 2,7 мг/дл, 2,75 мг/дл, 2,8 мг/дл, 2,9 мг/дл, 3,0 мг/дл, 3,1 мг/дл, 3,2 мг/дл, 3,3 мг/дл, 3,4 мг/дл, 3,5 мг/дл, 3,6 мг/дл, 3,7 мг/дл, 3,8 мг/дл, 3,9 мг/дл или 4 мг/дл или в диапазоне от 2,1 мг/дл до 2,5 мг/дл, от 2,1 мг/дл до 3,0 мг/дл или от 2,1 мг/дл до 4 мг/дл, которая сохраняется в течение больше 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM; (iii) концентрация церамида в плазме больше 8,2 мкг/дл, 8,3 мкг/дл, 8,4 мкг/дл, 8,5 мкг/дл, 8,75 мкг/дл, 9 мкг/дл, 9,5 мкг/дл, 10 мкг/дл, 11 мкг/дл, 12 мкг/дл, 13 мкг/дл, 14 мкг/дл, 15 мкг/дл, 16 мкг/дл, 17 мкг/дл, 18 мкг/дл, 19 мкг/дл, 20 мкг/дл, 25 мкг/дл, 30 мкг/дл, 35 мкг/дл, 40 мкг/дл, 45 мкг/дл, 50 мкг/дл, 55 мкг/дл, 60 мкг/дл, 65 мкг/дл, 70 мкг/дл, 75 мкг/дл или 80 мкг/дл или в диапазоне от 8,2 мкг/дл до 10 мкг/дл, от 8,5 мкг/дл до 10 мкг/дл, от 9 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 15 мкг/дл, от 10 мкг/дл до 20 мкг/дл, от 15 мкг/дл до 20 мкг/дл или от 20 мкг/дл до 30 мкг/дл через 6 часов, 8 часов, 10 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM; или (iv) острофазовый ответ. В соответствии с этим вариантом осуществления человеку, нуждающемуся в этом, можно вводить последовательно более высокие дозы ASM до тех пор, пока человек не проявляет какой-либо один или несколько из пунктов (i)-(iv). В случае, когда человек проявляет любой один или несколько из пунктов (i)-(iv), то в зависимости от тяжести этого проявления такая же доза, которая привела к проявлениям по пунктам (i)- (iv), может быть повторена или доза может быть уменьшена до предыдущей дозы.
В другом варианте осуществления способ лечения ASMD включает: (a) введение человеку, нуждающемуся в этом, начальной дозы 0,1 мг/кг ASM; (b) мониторинг одного или нескольких из следующих явлений у человека после введения дозы ASM: (i) общая концентрация билирубина, (ii) проявление связанного нежелательного явления; (iii) острофазовый ответ; или (iv) концентрация церамида в плазме; (c) определение того, следует ли корректировать (например, увеличивать или уменьшать) или поддерживать дозу ASM на основании одного или нескольких из пунктов (i)-(iv); и (d) повторение этапов (b) и (c) после введения дозы ASM, определенной на предыдущем этапе (c). В другом варианте осуществления способ лечения ASMD включает: (a) введение человеку, нуждающемуся в этом, двух начальных доз 0,1 мг/кг ASM через 2-4 недели; (b) мониторинг одного или нескольких из следующих явлений у человека после введения каждой дозы ASM: (i) общая концентрация билирубина, (ii) проявление связанного нежелательного явления; (iii) острофазовый ответ; или (iv) концентрация церамида в плазме; (c) определение того, следует ли корректировать (например, увеличивать или уменьшать) или поддерживать дозу ASM на основании одного или нескольких из пунктов (i)-(iv); и (d) повторение этапов (b) и (c) после введения дозы ASM, определенной в предыдущем этапе (c). В соответствии с этими вариантами осуществления человеку можно вводить более высокую дозу ASM один или несколько раз через 2-4 недели, если начальная доза, которая составляет 0,1 мг/кг, или откорректированная доза, определенная на этапе (c), приводит к следующему: (i) общая концентрация билирубина меньше или равна 2,0 мг/дл до крайнего срока для введения другой дозы ASM; (ii) нет связанных нежелательных явлений или лишь легкие связанные нежелательные явления; (iii) церамид в плазме в пределах нормального диапазона через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 48 часов или 72 часа после введения последней дозы ASM; или (iv) нет острофазового ответа, или острофазовый ответ, который не является статистически достоверным. Однако дозу можно поддерживать или уменьшать, если начальная доза, которая составляет 0,1 мг/кг, или скорректированная доза, определенная на этапе (c), приводит к следующему: (i) общая концентрация билирубина 2,1 мг/дл или больше до крайнего срока для введения другой дозы ASM; (ii) связанное нежелательное явление; (iii) церамид в плазме выше нормального диапазона через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 48 часов или 72 часа после введения последней дозы ASM; или (iv) острофазовый ответ, который является статистически достоверным.
В другом варианте осуществления способ лечения ASMD включает: (A) введение человеку, нуждающемуся в этом, двух начальных доз 0,1 мг/кг через 2 недели; (B) мониторинг любого из следующих явлений: (i) общая концентрация билирубина, (ii) проявление связанного нежелательного явления, (iii) как (i), так и (ii) у человека после введения каждой начальной дозы ASM; (C) определение того, следует ли корректировать (например, увеличивать или уменьшать) или поддерживать дозу ASM на основании одного или нескольких из пунктов (i)-(iii), и (D) повторение этапов (B) и (C) после введения дозы ASM, определенной в предыдущем этапе (C), где (a) дозу, вводимую человеку, увеличивают, если общая концентрация билирубина меньше или равна 2,0 мг/дл, или у человека легкое связанное нежелательное явление; (b) человек продолжает получать текущую дозу 1-4 раза через 2-4 недели, если общая концентрация билирубина составляет от 2,1 мг/дл до 3,1 мг/дл, или у человека умеренное связанное нежелательное явление, и эту дозу поддерживают, если общая концентрация билирубина сохраняется больше 2,0 мг/дл после последней вводимой дозы; (c) дозу, вводимую человеку, уменьшают или более не вводят ASM, если общая концентрация билирубина больше 3 мг/дл, или у человека тяжелые связанные нежелательные явления.
В другом варианте осуществления способ лечения ASMD включает: (A) введение человеку, нуждающемуся в этом, двух начальных доз 0,3 мг/кг через 2 недели; (B) мониторинг любого из следующих явлений: (i) общая концентрация билирубина, (ii) проявление связанного нежелательного явления, (iii) как (i), так и (ii) у человека после введения каждой дозы ASM; (C) определение того, следует ли корректировать (например, увеличивать или уменьшать) или поддерживать дозу ASM на основании одного или нескольких из пунктов (i)-(iii), и (D) повторение этапов (B) и (C) после введения дозы ASM, определенной в предыдущем этапе (C), где (a) дозу, вводимую человеку, увеличивают, если общая концентрация билирубина меньше или равна 2,0 мг/дл, или у человека легкое связанное нежелательное явление; (b) человек продолжает получать текущую дозу 1-4 раза через 2-4 недели, если общая концентрация билирубина составляет от 2,1 мг/дл до 3,1 мг/дл, или у человека умеренное связанное нежелательное явление и эту дозу поддерживают, если общая концентрация билирубина остается больше 2,0 мг/дл после последней вводимой дозы; (c) дозу, вводимую человеку, уменьшают или больше не вводят ASM, если общая концентрация билирубина больше 3 мг/дл, или у человека тяжелые связанные нежелательные явления.
В конкретном варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, дозы 0,1 мг/кг ASM и после двух недель дозы 0,3 мг/кг ASM каждые две недели. В другом конкретном варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, дозы 0,1 мг/кг ASM, дозы 0,3 мг/кг ASM через две недели после введения 0,1 мг/кг дозы ASM и дозы 0,6 мг/кг ASM через две недели после введения дозы 0,3 мг/кг ASM. В другом конкретном варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, дозы 0,1 мг/кг ASM, дозы 0,3 мг/кг ASM через две недели после введения 0,1 мг/кг дозы ASM, дозы 0,6 мг/кг ASM через две недели после введения дозы 0,3 мг/кг ASM и дозы 1 мг/кг ASM через две недели после введения дозы 0,6 мг/кг ASM. В определенных вариантах осуществления каждая доза может быть повторена по меньшей мере два и предпочтительно от двух до четырех раз перед повышением дозы до следующего уровня. В соответствии с этими вариантами осуществления дозу повышают лишь в том случае, если величина общего билирубина равна или меньше 2,0 мг/дл и (или) субъект испытывает легкое связанное нежелательное явление. Дозу не повышают, если величина общего билирубина составляет от 2,1 до 3 мг/дл и (или) субъект испытывает умеренное связанное нежелательное явление. Дозу снижают до предыдущей переносимой дозы, если величина общего билирубина больше 3,0 мг/дл от общего билирубина и (или) субъект испытывает серьезное связанное нежелательное явление.
В конкретных вариантах осуществления пациента лечат от ASMD в соответствии с протоколом, описанным в разделе 8 или 9 и далее, ниже, или аналогичным протоколом.
В конкретном варианте осуществления способ лечения ASMD включает введение субъекту, нуждающемуся в этом, дозы 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг., 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, или от 0,1 мг/кг до 0,5 мг/кг, или от 0,1 мг/кг до 1 мг/кг ASM и после определенного периода времени (например, 3 дней, 1 недели, 2 недель, 3 недель или 4 недель) последовательное повышение дозы ASM, вводимой субъекту, если общая концентрация билирубина меньше или равна 2,1 мг/дл и субъект не испытывает умеренного или тяжелого связанного нежелательного явления. В некоторых вариантах осуществления дозу ASM последовательно увеличивают до тех пор, пока не будет достигнута максимальная или самая высокая доза, переносимая субъектом, которая является терапевтически эффективной. В определенных вариантах осуществления такую самую высокую или максимальную переносимую дозу вводят до тех пор, пока масса накопленного сфингомиелина в органах патологии не будет сокращена, после чего субъекту вводят поддерживающую дозу, которая ниже максимальной переносимой дозы и которая все еще является терапевтически эффективной. В некоторых вариантах осуществления поддерживающую дозу снижают с течением времени по мере улучшения состояния пациента.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD не вызывая одно, два или более тяжелых связанных нежелательных явлений или связанных нежелательных явлений, которые определяются с помощью, например, стандартной терминологии Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения общей концентрации билирубина относительно общей концентрации билирубина у пациента до введения ASM, где увеличение сохраняется в течение более двух дней, трех дней, пяти дней, одной недели, двух недель или трех недель. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая общей концентрации билирубина, которая больше нормальных общих концентраций билирубина, которая сохраняется в течение более 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM. Нормальная общая концентрация билирубина у человека без ASMD (например, здорового человека) составляет менее чем приблизительно 1,2 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая общей концентрации билирубина больше чем примерно 1,5 мг/дл, 1,75 мг/дл, 2,0 мг/дл, 2,1 мг/дл, 2,2 мг/дл, 2,3 мг/дл, 2,4 мг/дл, 2,5 мг/дл, 2,6 мг/дл, 2,7 мг/дл, 2,75 мг/дл, 2,8 мг/дл, 2,9 мг/дл, 3,0 мг/дл, 3,1 мг/дл, 3,2 мг/дл, 3,3 мг/дл, 3,4 мг/дл, 3,5 мг/дл, 3,6 мг/дл, 3,7 мг/дл, 3,8 мг/дл, 3,9 мг/дл или 4 мг/дл или в диапазоне от 2,1 мг/дл до 2,5 мг/дл, от 2,1 мг/дл до 3,0 мг/дл или от 2,1 мг/дл до 4 мг/дл, которая сохраняется в течение более 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза представляет собой самую высокую дозировку, которая является эффективной в лечении ASMD, не приводя к концентрации церамида в плазме, которая больше нормальной концентрации церамида в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация церамида в плазме у человека без ASMD (например, здорового человека) составляет примерно 1,5-8 мкг/дл. В определенных вариантах осуществления самая высокая переносимая доза является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации церамида в плазме больше чем примерно 8,2 мкг/дл, 8,3 мкг/дл, 8,4 мкг/дл, 8,5 мкг/дл, 8,75 мкг/дл, 9 мкг/дл, 9,5 мкг/дл, 10 мкг/дл, 11 мкг/дл, 12 мкг/дл, 13 мкг/дл, 14 мкг/дл, 15 мкг/дл, 16 мкг/дл, 17 мкг/дл, 18 мкг/дл, 19 мкг/дл, 20 мкг/дл, 25 мкг/дл, 30 мкг/дл, 35 мкг/дл, 40 мкг/дл, 45 мкг/дл, 50 мкг/дл, 55 мкг/дл, 60 мкг/дл, 65 мкг/дл, 70 мкг/дл, 75 мкг/дл или 80 мкг/дл или в диапазоне от 8,2 мкг/дл до 10 мкг/дл, от 8,5 мкг/дл до 10 мкг/дл, от 9 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 15 мкг/дл, от 10 мкг/дл до 20 мкг/дл, от 15 мкг/дл до 20 мкг/дл или от 20 мкг/дл до 30 мкг/дл через 6 часов, 8 часов, 10 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая острофазовый ответ. Острофазовый ответ можно оценить по изменению концентрации острофазового агента, изменению протромбинового времени, изменению частичного тромбопластинового времени или изменению относительного содержания нейтрофилов. Например, острофазовый ответ можно оценить по увеличению одного или нескольких из следующих факторов после введения ASM субъекту относительно этих факторов до введения ASM субъекту или относительно этих факторов у человека без ASMD (например, здорового человека): относительное содержание нейтрофилов, протромбиновое время (PT), частичное тромбопластиновое время (PTT), общая концентрация билирубина, концентрация C-реактивного белка (CRP/hs-CRP), амилоид A (SAA) сыворотки, компонент амилоид P сыворотки, ангиотензин-превращающий фермент (ACE), концентрация ферритина, концентрация IL-6, концентрация IL-8, концентрация кальцитонина, концентрация альбумина или концентрация фибриногена. Острофазовый ответ можно также оценить по уменьшению концентрации железа или концентрации альбумина после введения ASM субъекту относительно концентрации железа или концентрации альбумина у субъекта до введения ASM или относительно концентрации железа или концентрации альбумина у человека без ASMD (например, здорового человека).
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации CRP/hs-CRP через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации CRP/hs-CRP у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации CRP/hs-CRP в плазме, которая больше нормальной концентрации CRP/hs-CRP в плазме, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация CRP/hs-CRP в плазме у человека без ASMD (например, здорового человека) составляет менее чем примерно 8 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрации CRP/hs-CRP в плазме больше чем примерно 8,1 мг/дл, 8,2 мг/дл, 8,3 мг/дл, 8,4 мг/дл, 8,5 мг/дл, 8,6 мг/дл, 8,7 мг/дл, 8,8 мг/дл, 8,9 мг/дл 9 мг/дл, 9,5 мг/дл, 10 мг/дл, 11 мг/дл или 12 мг/дл или в диапазоне от 8,5 мг/дл до 10 мг/дл, или от 8,5 мг/дл до 12 мг/дл, или от 10 мг/дл до 12 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации ферритина через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации ферритина у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективнойв лечении ASMD, не приводя к концентрации ферритина в плазме, которая больше нормальной концентрации ферритина в плазме, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация ферритина в плазме у человека без ASMD (например, здорового человека) составляет 10-30 нг/мл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрации в плазме ферритина больше чем примерно 300 нг/мл, 325 нг/мл, 350 нг/мл, 375 нг/мл, 400 нг/мл, 425 нг/мл, 450 нг/мл, 475 нг/мл, 500 нг/мл, 525 нг/мл, 550 нг/мл, 575 нг/мл, 600 нг/мл, 625 нг/мл, 650 нг/мл, 675 нг/мл, 700 нг/мл, 725 нг/мл, 750 нг/мл, 775 нг/мл, 800 нг/мл, 850 нг/мл, 900 нг/мл, 950 нг/мл, 1000 нг/мл, 1050 нг/мл, 1100 нг/мл, 1150 нг/мл или 1200 нг/мл или в диапазоне от 600 нг/мл до 800 нг/мл, от 650 нг/мл до 850 нг/мл, от 600 нг/мл до 1000 нг/мл, от 600 нг/мл до 1200 нг/мл, от 800 нг/мл до 1000 нг/мл, от 900 нг/мл до 1000 нг/мл, или 1000 нг/мл, или от 1000 нг/мл до 1200 нг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, и ее можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации IL-8 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации IL-8 у пациента до введения ASM можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию IL-8 в плазме или сыворотке, которая больше нормальной концентрации IL-8 в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию IL-8 в плазме больше чем приблизительно 24 пг/мл, 50 пг/мл, 75 пг/мл, 100 пг/мл, 200 пг/мл, 300 пг/мл, 400 пг/мл, 500 пг/мл, 600 пг/мл, 700 пг/мл, 800 пг/мл или 900 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации IL-6 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации IL-6 у пациента до введения ASM можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию IL-6 в плазме или сыворотке, которая больше нормальной концентрации IL-6 в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию IL-6 в плазме больше чем примерно 4,4 пг/мл, 6 пг/мл, 8 пг/мл, 10 пг/мл, 15 пг/мл, 20 пг/мл, 25 пг/мл или 30 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления увеличение концентрации кальцитонина в плазме или сыворотке через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации кальцитонина у пациента до введения ASM можно использовать в качестве измерения острофазового ответа. В другом конкретном варианте осуществления концентрацию кальцитонина в плазме или сыворотке, которая больше нормальной концентрации кальцитонина в плазме через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, можно использовать в качестве измерения острофазового ответа. В определенных вариантах осуществления концентрацию кальцитонина в плазме больше чем примерно 9,4 пг/мл, 20 пг/мл, 30 пг/мл, 40 пг/мл, 50 пг/мл, 75 пг/мл, 100 пг/мл, 150 пг/мл, 200 пг/мл или 250 пг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM можно использовать в качестве измерения острофазового ответа.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, представляет собой самую высокую дозировку, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации фибриногена через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации фибриногена у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации фибриногена в плазме, которая больше нормальной концентрации фибриногена в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация фибриногена в плазме у человека без ASMD (например, здорового человека) составляет от 150 мг/дл до 300 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрации в плазме фибриногена больше чем примерно 350 мг/дл, 375 мг/дл, 400 мг/дл, 425 мг/дл или 450 мг/дл или в диапазоне от 350 мг/дл до 400 мг/дл, от 350 мг/дл до 450 мг/дл или от 400 мг/дл до 450 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения относительного содержания нейтрофилов от общего количества лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM по сравнению с относительным содержанием нейтрофилов пациентов от общего количества лейкоцитов до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к относительному содержанию нейтрофилов от общего количества лейкоцитов, которое больше нормального относительного содержания нейтрофилов от общей концентрации лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальное относительное содержание нейтрофилов от общего количества лейкоцитов у человека без ASMD (например, здорового человека) составляет от 45% до 60%. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения относительного содержания нейтрофилов от общего количества лейкоцитов, которое составляет 70%, 75%, 80%, 85%, 90%, 95% или больше через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения антител на вводимую ASM. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая реакции гиперчувствительности. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая синдром выброса цитокинов.
В определенных вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет от 1 мг/кг до 2,5 мг/кг, от 2 мг/кг до 3 мг/кг, от 3 мг/кг до 5 мг/кг, от 5 мг/кг до 10 мг/кг, от 10 мг/кг до 15 мг/кг, от 15 мг/кг до 20 мг/кг, от 15 мг/кг до 25 мг/кг, от 20 мг/кг до 30 мг/кг или от 25 мг/кг до 50 мг/кг. В некоторых вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет 1 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг, 5 мг/кг, 6 мг/кг, 7 мг/кг, 8 мг/кг, 9 мг/кг, 10 мг/кг, 11 мг/кг, 12 мг/кг, 13 мг/кг, 14 мг/кг или 15 мг/кг. В определенных вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг или 50 мг/кг.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая одного, двух или более тяжелых связанных нежелательных явлений или связанных нежелательных явлений, которые определяются с помощью, например, стандартной терминологии Clinical Data Interchange Standards Consortium Study Data Tabulation Model v.3.1.1. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения общей концентрация билирубина относительно общей концентрации билирубина у пациента до введения ASM, где увеличение сохраняется в течение более двух дней, трех дней, пяти дней, одной недели, двух недель или трех недель. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая общей концентрации билирубина, которая больше нормальных общих концентраций билирубина и которая сохраняется в течение более 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM. Нормальная общая концентрация билирубина у человека без ASMD (например, здорового человека) составляет менее чем примерно 1,2 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая общей концентрации билирубина, которая больше чем примерно 1,5 мг/дл, 1,75 мг/дл, 2,0 мг/дл, 2,1 мг/дл, 2,2 мг/дл, 2,3 мг/дл, 2,4 мг/дл, 2,5 мг/дл, 2,6 мг/дл, 2,7 мг/дл, 2,75 мг/дл, 2,8 мг/дл, 2,9 мг/дл, 3,0 мг/дл, 3,1 мг/дл, 3,2 мг/дл, 3,3 мг/дл, 3,4 мг/дл, 3,5 мг/дл, 3,6 мг/дл, 3,7 мг/дл, 3,8 мг/дл, 3,9 мг/дл или 4 мг/дл или в диапазоне от 2,1 мг/дл до 2,5 мг/дл, от 2,1 мг/дл до 3,0 мг/дл или от 2,1 мг/дл до 4 мг/дл, которая сохраняется в течение более 18 часов, 24 часов, 36 часов, 48 часов или 72 часов, 5 дней, одной недели, двух недель или трех недель после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации церамида в плазме, которая больше нормальной концентрации церамида в плазме, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация церамида в плазме у человека без ASMD (например, здорового человека) составляет примерно 1,5-8 мкг/дл. В определенных вариантах осуществления самая высокая переносимая доза является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации церамида в плазме больше чем примерно 8,2 мкг/дл, 8,3 мкг/дл, 8,4 мкг/дл, 8,5 мкг/дл, 8,75 мкг/дл, 9 мкг/дл, 9,5 мкг/дл, 10 мкг/дл, 11 мкг/дл, 12 мкг/дл, 13 мкг/дл, 14 мкг/дл, 15 мкг/дл, 16 мкг/дл, 17 мкг/дл, 18 мкг/дл, 19 мкг/дл, 20 мкг/дл, 25 мкг/дл, 30 мкг/дл, 35 мкг/дл, 40 мкг/дл, 45 мкг/дл, 50 мкг/дл, 55 мкг/дл, 60 мкг/дл, 65 мкг/дл, 70 мкг/дл, 75 мкг/дл или 80 мкг/дл или в диапазоне от 8,2 мкг/дл до 10 мкг/дл, от 8,5 мкг/дл до 10 мкг/дл, от 9 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 12 мкг/дл, от 10 мкг/дл до 15 мкг/дл, от 10 мкг/дл до 20 мкг/дл, от 15 мкг/дл до 20 мкг/дл или от 20 мкг/дл до 30 мкг/дл через 6 часов, 8 часов, 10 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая острофазовый ответ. Острофазовый ответ можно оценить по изменению концентрации острофазового агента, изменению протромбинового времени, изменению частичного тромбопластинового времени или изменению относительного содержания нейтрофилов. Например, острофазовый ответ можно оценить по увеличению одного или нескольких из следующих факторов после введения ASM субъекту относительно этих факторов до введения ASM субъекту или относительно этих факторов у человека без ASMD (например, здорового человека): относительное содержание нейтрофилов, протромбиновое время (PT), частичное тромбопластиновое время (PTT), общая концентрация билирубина, концентрация C-реактивного белка (CRP/hs-CRP), амилоид A (SAA) в сыворотке, компонент амилоид P в сыворотке, ангиотензин-превращающий фермент (ACE), концентрация ферритина, концентрация альбумина или концентрация фибриногена. Острофазовый ответ можно также оценить по уменьшению концентрации железа или концентрации альбумина после введения ASM субъекту относительно концентрации железа или концентрации альбумина у субъекта до введения ASM или относительно концентрации железа или концентрации альбумина у человека без ASMD (например, здорового человека).
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации CRP/hs-CRP через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM, относительно концентрации CRP/hs-CRP у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации CRP/hs-CRP в плазме, которая больше нормальной концентрации CRP/hs-CRP в плазме через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация CRP/hs-CRP в плазме у человека без ASMD (например, здорового человека) составляет менее чем примерно 8 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрацию CRP/hs-CRP в плазме больше чем примерно 8,1 мг/дл, 8,2 мг/дл, 8,3 мг/дл, 8,4 мг/дл, 8,5 мг/дл, 8,6 мг/дл, 8,7 мг/дл, 8,8 мг/дл, 8,9 мг/дл 9 мг/дл, 9,5 мг/дл, 10 мг/дл, 11 мг/дл или 12 мг/дл или в диапазоне от 8,5 мг/дл до 10 мг/дл, или от 8,5 мг/дл до 12 мг/дл, или от 10 мг/дл до 12 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации ферритина через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации ферритина у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации ферритина в плазме, которая больше нормальной концентрации ферритина в плазме, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация ферритина в плазме у человека без ASMD (например, здорового человека) составляет 10-30 нг/мл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрации ферритина в плазме больше чем примерно 600 нг/мл, 625 нг/мл, 650 нг/мл, 675 нг/мл, 700 нг/мл, 725 нг/мл, 750 нг/мл, 775 нг/мл, 800 нг/мл, 850 нг/мл, 900 нг/мл, 950 нг/мл, 1000 нг/мл, 1050 нг/мл, 1100 нг/мл, 1150 нг/мл или 1200 нг/мл или в диапазоне от 600 нг/мл до 800 нг/мл, от 650 нг/мл до 850 нг/мл, от 600 нг/мл до 1000 нг/мл, от 600 нг/мл до 1200 нг/мл, от 800 нг/мл до 1000 нг/мл, от 900 нг/мл до 1000 нг/мл или от 1000 нг/мл до 1200 нг/мл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения концентрации фибриногена через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM относительно концентрации фибриногена у пациента до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к концентрации фибриногена в плазме, которая больше нормальной концентрации фибриногена в плазме, через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальная концентрация фибриногена в плазме у человека без ASMD (например, здорового человека) составляет от 150 мг/дл до 300 мг/дл. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая концентрации фибриногена в плазме больше чем примерно 350 мг/дл, 375 мг/дл, 400 мг/дл, 425 мг/дл или 450 мг/дл или в диапазоне от 350 мг/дл до 400 мг/дл, от 350 мг/дл до 450 мг/дл или от 400 мг/дл до 450 мг/дл через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения относительного содержания нейтрофилов от общего количества лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM по сравнению с относительным содержанием нейтрофилов у пациента от общего количества лейкоцитов до введения ASM. В другом конкретном варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не приводя к относительному содержанию нейтрофилов от общего количества лейкоцитов, которое больше нормального относительного содержания нейтрофилов от общей концентрации лейкоцитов через 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM. Нормальное относительное содержание нейтрофилов от общего количества лейкоцитов у человека без ASMD (например, здорового человека) составляет от 45% до 60%. В определенных вариантах осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения относительного содержания нейтрофилов от общего количества лейкоцитов, которое составляет 70%, 75%, 80%, 85%, 90%, 95% или больше через 6 часов, 8 часов, 12 часов, 18 часов, 24 часа, 36 часов, 48 часов или 72 часа после введения дозы ASM.
В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая увеличения антител к вводимой ASM. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая реакции гиперчувствительности. В другом варианте осуществления самая высокая переносимая доза, которая является терапевтически эффективной, является самой высокой дозировкой, которая является эффективной в лечении ASMD, не вызывая синдром выброса цитокинов.
В определенных вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет от 1 мг/кг до 2,5 мг/кг, от 2 мг/кг до 3 мг/кг, от 3 мг/кг до 5 мг/кг, от 5 мг/кг до 10 мг/кг, от 10 мг/кг до 15 мг/кг, от 15 мг/кг до 20 мг/кг, от 15 мг/кг до 25 мг/кг, от 20 мг/кг до 30 мг/кг или от 25 мг/кг до 50 мг/кг. В некоторых вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет 1 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг, 5 мг/кг, 6 мг/кг, 7 мг/кг, 8 мг/кг, 9 мг/кг, 10 мг/кг, 11 мг/кг, 12 мг/кг, 13 мг/кг, 14 мг/кг или 15 мг/кг. В определенных вариантах осуществления самая высокая доза, переносимая субъектом, которая является терапевтически эффективной, составляет 20 мг/кг, 25 мг/кг, 30 мг/кг, 35 мг/кг, 40 мг/кг, 45 мг/кг или 50 мг/кг.
В определенных вариантах осуществления дозу ASM, описанную в данном документе, вводят субъекту каждые 3 дня, каждые 4 дня, каждые 5 дней, каждые 6 дней, каждую неделю, каждые 8 дней, каждые 9 дней, каждые 10 дней, каждые 11 дней, каждые 12 дней, каждые 13 дней, каждые 2 недели, каждые 3 недели, каждые 4 недели или каждые 5 недель. В некоторых вариантах осуществления дозу ASM, описанную в данном документе, вводят субъекту каждые 3-5 дней, каждые 3-7 дней, каждые 5-7 дней, каждые 5-10 дней, каждые 5-14 дней, каждые 7-14 дней, каждые 2-4 недели или каждые 3-5 недель. В определенных вариантах осуществления частоту введения дозы ASM изменяют по мере корректировки дозы. Например, дозу 0,3 мг/кг можно вводить каждую неделю или две недели, а дозу 1 мг/кг можно вводить каждые две недели или три недели.
В некоторых вариантах осуществления дозу ASM (например, поддерживающую дозу) вводят субъекту в течение периода 12 недель, 20 недель, 25 недель, 30 недель, 35 недель, 40 недель, 45 недель, 50 недель, 52 недель, 1 года, 1,5 лет, 2 лет, 2,5 лет, 3 лет, 3,5 лет, 4 лет или пока пациент не испытывает связанное нежелательное явление, величину общего билирубина больше величины билирубина для человека без ASMD (например, здорового человека), концентрацию церамида в плазме больше концентрации церамида в плазме человека без ASMD (например, здорового человека) или острофазовый ответ.
В определенных вариантах осуществления начальную низкую нетоксичную дозу ASM вводят субъекту каждые 3 дня, каждые 4 дня, каждые 5 дней, каждые 6 дней, каждую неделю, каждые 8 дней, каждые 9 дней, каждые 10 дней, каждые 11 дней, каждые 12 дней, каждые 13 дней, каждые 2 недели, каждые 3 недели, каждые 4 недели или каждые 5 недель. В некоторых вариантах осуществления дозу ASM, описанную в данном документе, вводят субъекту каждые 3-5 дней, каждые 3-7 дней, каждые 5-7 дней, каждые 5-10 дней, каждые 5-14 дней, каждые 7-14 дней, каждые 2-4 недели или каждые 3-5 недель. В определенных вариантах осуществления начальную низкую нетоксичную дозу ASM вводят в течение периода 4 недель, 6 недель, 8 недель, 12 недель, 14 недель или дольше.
В определенных вариантах осуществления последующее увеличение дозы ASM примерно на 0,1 мг/кг, 0,2 мг/кг, 0,3 мг/кг, 0,4 мг/кг, 0,5 мг/кг, 0,6 мг/кг, 0,7 мг/кг, 0,8 мг/кг, 0,9 мг/кг, 1 мг/кг, 1,2 мг/кг, 1,5 мг/кг, 1,75 мг/кг, 2 мг/кг, 3 мг/кг, 4 мг/кг или 5 мг/кг выше предыдущей дозы. В некоторых вариантах осуществления последующее увеличение дозы ASM на от 0,1 до 0,5 мг/кг, от 0,1 мг/кг до 1 мг/кг, от 0,5 мг/кг до 1 мг/кг, от 0,5 мг/кг до 2 мг/кг, от 1 мг/кг до 2 мг/кг, от 2 мг/кг до 4 мг/кг или от 2 мг/кг до 5 мг/кг выше предыдущей дозы.
Дозы ASM, описанные в данном документе, можно вводить любым путем, который является пригодным для достижения терапевтического эффекта. Конкретные пути введения доз ASM включают, но без ограничений, внутривенный, интравентрикулярный, внутрикожный, трансдермальный, подкожный, внутримышечный, интраназальный, ингаляцию, внутрилегочный, местный, трансмукозальный, интракраниальный, интратекальный, эпидуральный и интрасиновиальный. В одном варианте осуществления дозу ASM вводят систематически (например, парентерально) субъекту, нуждающемуся в этом. В другом варианте осуществления дозу ASM вводят местно субъекту, нуждающемуся в этом.
Доза фермента ASM, которая является эффективной в лечении соматических (не относящихся к центральной нервной системе) проявлений ASMD, не способна эффективно преодолевать гематоэнцефалический барьер. Таким образом, в конкретных вариантах осуществления ASM вводят пациенту интравентрикулярно или интратекально в мозг пациент с NPD. Смотрите, например, публикацию патентной заявки США № 2009/0130079 и 2009/0123451, которые включены в данный документ ссылкой во всей их полноте, для способов интравентрикулярной доставки ферментов, хранящихся в лизосомах, в мозг. В определенных вариантах осуществления ASM вводят пациенту интрацеребровентрикулярно. Смотрите, например, Dodge et al., 2009, Experimental Neurology 215: 349-357, которая включена в данный документ ссылкой во всей своей полноте, для способов интрацеребровентрикулярной инфузии ASM. В некоторых вариантах осуществления ASM вводят пациенту непрямыми интрапаренхимными инъекциями. Смотрите, например, Yang et al., 2007, Experimental Neurology 207: 258-266. В некоторых вариантах осуществления модифицированную форму ASM, которая направляет фермент в мозг, так как описано в разделе 5.2, ниже, вводят пациенту для лечения ASMD.
Только небольшой процент фермента ASM, вводимого пациенту, достигает легкого. Таким образом, в конкретных вариантах осуществления ASM вводят в легкие пациента. В определенных вариантах осуществления ASM вводят пациенту с помощью интраназального пути или ингаляции. Смотрите, например, Ziegler et al., 2009, Molecular Genetics and Metabolism 97: 35-42, которая включена в данный документ ссылкой во всей своей полноте, для информации, касающейся интраназального введения ASM. В некоторых вариантах осуществления пациенту для лечения ASMD вводят модифицированную форму ASM, которая направляет фермент в легкое, так как описано в разделе 5.2, ниже. В определенных вариантах осуществления ASM вводят пациенту, используя небулайзер. Доставка ASM в легкие может происходить с помощью внутрилегочной инъекции через бронхоскоп, дозирующий ингалятор или небулайзер.
В определенных вариантах осуществления фермент ASM вводят пациенту системно, а также вводят местно в конкретные органы, такие как мозг и легкое. В некоторых вариантах осуществления местное введение фермента ASM дополняется системным введением фермента. В конкретных вариантах осуществления фермент ASM вводят местно, например, в мозг или легкие, после того как была сокращена масса накопленного сфингомиелина у пациента с помощью системного введения (например, внутривенного введения).
В определенных вариантах осуществления пациента генотипируют перед введением ASM. В некоторых вариантах осуществления перед введением ASM определяют характер гликозилирования ASM, экспрессируемой пациентом. Введение ASM, которая является сходной/совместимой с эндогенно экспрессируемой пациентом, может снизить возможность иммуногенности.
В определенных вариантах осуществления активность эндогенно экспрессируемой ASM определяют до введения ASM. Активность эндогенно экспрессируемой ASM можно измерить в DBS и в культивируемых фибробластах с помощью техник, известных специалисту в данной области.
В конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают объем селезенки, что оценивается с помощью техник, известных в данном уровне техники, например МРТ. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают уровни сфингомиелина печени, что оценивается с помощью техник, известных в данном уровне техники, например биохимического анализа и (или) гистоморфометрического анализа образцов печени. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, увеличивают способность переносить физическую нагрузку, что оценивается с помощью техник, известных в данном уровне техники, например максимальной рабочей нагрузки с помощью велоэргометрии, включая процентную расчетную максимальную рабочую нагрузку, пиковое потребление кислорода и продуцирование диоксида углерода. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, увеличивают легочную функцию, что оценивается с помощью техник, известных в данном уровне техники, например DLCO, FVC, FEV и (или) TLC. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают сфингомиелин бронхоальвеолярного лаважа (BAL). В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают объем печени, что оценивается с помощью техник, известных в данном уровне техники, например МРТ. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, улучшают внешний вида легкого, что оценивается с помощью техник, известных в данном уровне техники, например изображение КТ высокого разрешения или рентгенография грудной клетки. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, улучшают очищение легких. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают концентрацию сфингомиелина в печени, коже, плазме и DBS. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают уровни хитотриозидазы в сыворотке. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают уровни CCL18 в сыворотке. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, улучшают липидный профиль у пациента (например, уменьшают холестерин). В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают или облегчают тяжесть ASMD и (или) симптом, связанный с ней. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают длительность симптома, связанного с ASMD. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, предупреждают рецидив симптома, связанного с ASMD. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают госпитализацию субъекта. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают длительность госпитализации. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, увеличивают выживание субъекта. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, снижают смертность субъекта. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают частоту госпитализации субъекта. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, уменьшают число симптомов, связанных с ASMD. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, увеличивают бессимптомное выживание пациентов с ASMD. В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, улучшают неврологическую функцию (например, психомоторную функцию, социальную ответственность и т.д.) субъекта.
В другом конкретном варианте осуществления способы лечения ASMD, приведенные в данном документе, улучшают качество жизни у пациента. В определенных конкретных вариантах осуществления способ лечения ASMD, приведенный в данном документе, улучшает качество жизни у пациента, что оценивается с помощью, например, Краткого опросника оценки утомляемости (BFI; Mendoza et al., 1999, Cancer 85(5): 1186-1196), Краткого опросника боли - краткая форма (BPI-SF; Cleeland et al., 1994, Ann Acad Med Singapore 23(2): 129-138), Анкеты оценки состояния здоровья с ASM, которая состоит из BFI, BPI-SF и Краткой формы 36 медицинского осмотра, Анкеты оценки хронических респираторных заболеваний самозаполняемого стандартизированного формата (CRQ-SAS; Schunemann et al., 2005, Eur. Respir. J. 25: 31-40) для оценки одышки и утомляемости и Измерения активности для терапии, следующей за интенсивной (AM-PAC), адаптированного для компьютерной обработки теста, который оценивает функциональные подвижности (например, передвижение, уход за собой и прикладное мышление).
Фермент - кислая сфингомиелиназа
ASM относится к любой форме фермента кислой сфингомиелиназы, которая сохраняет способность гидролизовать сфингомиелин до церамида и фосфорилхолина, что оценивается с помощью техник, известных специалисту в данной области, таких как описываемые в патентах США №№ 4039388, 4082781, 5686240 и 7563591 и международных публикациях №№ WO 2007/078806 и WO 2006/058385, которые включены в данный документ ссылкой в их полноте. В конкретном варианте осуществления фермент кислой сфингомиелиназы обладает способностью гидролизовать сфингомиелин до церамида и фосфорилхолина в анализе высокоэффективной жидкостной хроматографии на основе флюоресценции, описанном в He et al., 2003, Analytical Biochemistry 314: 116-120. В конкретном варианте осуществления кислая сфингомиелиназа обладает по меньшей мере 30%, 35%, 40%, 45%, 50%, 75%, 80%, 85%, 80%, 90%, 95% или 98% или от 30% до 50%, от 40% до 50%, от 50% до 75%, от 50% до 90%, от 75% до 80%, от 75% до 90%, от 75% до 95% или от 85% до 95% от активности ASM-1 (SEQ ID NO: 1, ниже), что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120.
В конкретном варианте осуществления ASM представляет собой ASM человека. Существуют различные изоформы ASM человека, получаемые в результате альтернативного сплайсинга. Одна из изоформ ASM человека, изоформа 1 ASM человека (которую иногда называют ASM-1), имеет аминокислотную последовательность, находящуюся под инвентарным номером UniProtKB/Swiss-Prot. P17405-1. Другая изоформа ASM человека, изоформа 2 человека (которую иногда называют ASM-2), имеет аминокислотную последовательность, находящуюся под инвентарным номером UniProtKB/Swiss-Prot. P17405-2. Третья изоформа ASM человека, изоформа 3 человека (которую иногда называют ASM-3), имеет аминокислотную последовательность, находящуюся под инвентарным номером UniProtKB/Swiss-Prot. P17405-3. Аминокислотная последовательность ASM-1, которая является наиболее распространенной изоформой, представлена ниже:






(SEQ ID NO:1). ASM-2 отличается от ASM-1 в том, что аминокислотные остатки 363-374 ASM-1 (т.е. IGGFYALSPYPG (SEQ ID NO:2)) замещены аминокислотами YLSSVETQEGKR (SEQ ID NO:3), и аминокислотные остатки 375-418 ASM-1 утрачиваются из ASM-2. ASM-3 отличается от ASM-1 в том, что аминокислотные остатки 363-418 ASM-1 утрачиваются из ASM-3. При условии что ASM-2 и ASM-3 обладают ферментативной активностью, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которые могут быть введены субъекту. В конкретном варианте осуществления при условии, что ASM-2 и ASM-3 обладают по меньшей мере 30%, 35%, 40%, 45%, 50%, 75%, 80%, 85%, 80%, 90%, 95% или 98% или от 30% до 50%, от 40% до 50%, от 50% до 75%, от 50% до 90%, от 75% до 80%, от 75% до 90%, от 75% до 95% или от 85% до 95% от активности ASM-1, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которые могут быть введены субъекту.
В конкретном варианте осуществления ASM человека имеет аминокислотную последовательность кислой сфингомиелиназы человека, раскрытую в SEQ ID NO:1, ранее, аминокислотную последовательность кислой сфингомиелиназы человека, раскрытую в патенте США № 6541218 (например, SEQ ID NO:2 в патенте США № 6541218), или аминокислотную последовательность, раскрытую на фиг.3 Schuchman et al., 1991, J. Biol. Chem. 266: 8531-8539, каждая из которых включена в данный документ ссылкой во всей своей полноте.
В конкретных вариантах осуществления ASM человека представляет собой процессированную зрелую форму. В других вариантах осуществления ASM человека представляет собой незрелую, непроцессированную форму. По отношению к ASM-1 незрелая форма составляет 629 аминокислот в длину и содержит сигнальный пептид, обнаруживаемый на аминокислотных остатках 1-46. Зрелая форма ASM-1 не содержит сигнального пептида и представляет собой от аминокислотного остатка 47 до аминокислотного остатка 629. ASM-1 человека содержит домен B-типа сапозина от аминокислотных остатков 85 до 169. По отношению к ASM-1 следующие аминокислотные остатки являются гликозилированными (в частности, N-связанными гликозилированными): 86, 175, 335, 395 и 520.
В дополнение к изоформам ASM человека существуют различные встречающиеся в природе варианты ASM человека. Например, были определены встречающиеся в природе варианты гена ASM человека с различными количествами единиц гексануклеотидных повторов, встречающихся в пределах участка гена, кодирующего предполагаемый сигнальный пептид ASM. Смотрите, например, Wan and Schuchman, 1995, Biochimica et Biophysica Acta 1270: 207-210 (которая включена в данный документ ссылкой во всей своей полноте), которая описывает определение пяти аллелей, соответствующих девяти, семи, шести, пяти и четырем гексануклеотидным повторам. Дополнительно, были определены встречающиеся в природе варианты ASM человека с отдельными аминокислотными изменениями. Смотрите, например, Schuchman et al., 1991, J. of Biol. Chem 266: 8531-8539 и Schuchman et al., 1991, Nucleic Acids Research 19(11): 3160, которые включены в данный документ ссылкой в их полноте, для информации, касающейся однонуклеотидных полиморфизмов. При условии что любой из встречающихся в природе вариантов ASM человека обладает ферментативной активностью, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которая может быть введена субъекту. В конкретном варианте осуществления при условии, что любой из встречающихся в природе вариантов ASM человека обладает по меньшей мере 30%, 35%, 40%, 45%, 50%, 75%, 80%, 85%, 80%, 90%, 95% или 98% или от 30% до 50%, от 40% до 50%, от 50% до 75%, от 50% до 90%, от 75% до 80%, от 75% до 90%, от 75% до 95% или от 85% до 95% от активности ASM-1, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которая может быть введена субъекту.
Были определены многие однонуклеотидные полиморфизмы (SNP) для гена, кодирующего ASM человека (смотрите, например, веб-сайт ncbi: ncbi.nlm.nih.gov/projects/SNP/ для примеров SNP). При условии что любой из SNP в гене кодирует ASM, которая обладает ферментативной активностью, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которая может быть введена субъекту. В конкретном варианте осуществления при условии, что любой из SNP в гене кодирует ASM, которая обладает по меньшей мере 30%, 35%, 40%, 45%, 50%, 75%, 80%, 85%, 80%, 90%, 95% или 98% или от 30% до 50%, от 40% до 50%, от 50% до 75%, от 50% до 90%, от 75% до 80%, от 75% до 90%, от 75% до 95% или от 85% до 95% активности ASM-1, что измеряется с помощью, например, анализа, описанного в He et al., 2003, Analytical Biochemistry 314: 116-120, их включают как ASM, которая может быть введена субъекту. В определенных вариантах осуществления ASM представляет собой модифицированную форму ASM человека. В конкретном варианте осуществления модифицированная форма ASM человека является формой, раскрытой в патенте США № 7527956, который включен в данный документ ссылкой во всей своей полноте. В другом варианте осуществления модифицированная форма ASM человека является формой, раскрытой в международной публикации № WO 2008/136451, которая включена в данный документ ссылкой во всей своей полноте. В некоторых конкретных вариантах осуществления ASM является модифицированной формой ASM человека, которая имеет одно, два или более из следующих свойств: (i) увеличенная направленность к местам патологии (например, легкому и (или) мозгу) относительно немодифицированной ASM человека, (ii) увеличенная стабильность относительно немодифицированной ASM человека и (iii) увеличенная активность относительно немодифицированной ASM человека. Техники, известные в данном уровне техники, могут использоваться для измерения стабильности, активности и направленности ASM. В конкретном варианте осуществления применяют техники, описанные в Gamacho et al., 2008, J. Pharmacol. Exp. Ther. 325: 400-408 (которая включена в данный документ ссылкой во всей своей полноте), для оценки направленности ASM. В другом конкретном варианте осуществления применяют техники, описанные в He et al., 1999, Biochimica et Biophysica Acta 1432: 251-264 или Dhami et al., 2001, Lab. Invest. 81: 987-999 (которые включены в данный документ ссылкой в их полноте), для оценки стабильности ASM. В другом конкретном варианте осуществления применяют техники, описанные в He et al., 2003, Analytical Biochemistry 314: 116-120 (которая включена в данный документ ссылкой во всей своей полноте) для оценки активности ASM.
В одном варианте осуществления ASM представляет модифицированную форму ASM человека, которая обладает повышенной ферментативной активностью относительно немодифицированной ASM человека, такой как ASM-1. Смотрите, например, Qiu et al., 2003, J. Biol. Chem. 278(35): 32744-32752 и патент США № 7527956 для мутантных форм рекомбинантной ASM человека с ферментативной активностью с более высокой активностью, чем у рекомбинантной ASM человека дикого типа (такая как, например, ASM-1). В некоторых вариантах осуществления ASM обладает повышенной ферментативной активностью относительно немодифицированной ASM человека (такой как, например, ASM-1) благодаря добавлению катионов цинка. Смотрите, например, Schissel et al., 1998, J. Biol. Chem 273: 18250-18259, которая включена в данный документ ссылкой во всей своей полноте, для обсуждения относительно роли цинка в активности ASM. В другом варианте осуществления ASM представляет собой модифицированную форму ASM человека, которая обладает повышенной аффинностью к природному рецептору ASM человека (например, манноза 6-фосфат или высокое содержание маннозы) относительно немодифицированной ASM человека (такой как, например, ASM-1). В конкретном варианте осуществления ASM конъюгирована с олигосахаридом, как описано в патенте США № 7001994 и международной публикации патентной заявки № WO 2010/075010 и публикации заявки на патент США № 2010/0173385 (которые включены в данный документ ссылкой в их полноте), для повышения направленности фермента к его природному рецептору. В другом варианте осуществления ASM представляет собой модифицированную форму ASM человека, которая связывается с альтернативным рецептором (например, молекулой внутриклеточной адгезии (ICAM)-1, что может увеличивать направленность к органам, таким как легкое), а не с природным рецептором для ASM. В варианте осуществления ASM представляет собой модифицированную форму ASM человека, которая обладает повышенной стабильностью относительно немодифицированной ASM человека (такой как, например, ASM-1).
В некоторых вариантах осуществления ASM конъюгирована или слита, непосредственно или опосредованно, с направляющей молекулой, такой как инсулиноподобный фактор роста (IGF)-I, IGF-II, лептин, гранулоцитоколониестимулирующий фактор (G-CSF) или гуманизированное антитело, которое связывается с рецептором инсулина человек (HIR). Смотрите, например, публикацию заявки на патент США № 2009/0029467 и патент США № 7560424 (который включен в данный документ ссылкой во всей своей полноте) для обсуждения применения IGF-I в качестве направляющей молекулы для ферментов, хранящихся в лизосомах, и патент США № 7396811 (который включен в данный документ ссылкой в его полноте) для обсуждения применения IGF-II в качестве направляющей молекулы для ферментов, хранящихся в лизосомах. Направляющие молекулы IGF-I и IGF-II могут облегчать направленность фермента ASM в лизосому. Для обсуждения применения гормонов, таких как G-CSF и лептин в качестве направляющих молекул смотрите, например, международную публикацию патентной заявки № WO 2007/091250 и публикацию заявки на патент США № 2010/0183577, которые включены в данный документ ссылкой в их полноте. Смотрите, например, публикацию заявки на патент США № 2004/0101904, которая включена в данный документ ссылкой во всей своей полноте, для обсуждения гуманизированного моноклонального антитела, которое связывается с рецептором инсулина человека (HIR), присоединенного к фармацевтическому средству, такому как фермент. Направляющие молекулы G-CSF, лептин и антитело, которое связывается с HIR, могут облегчать направленность фермента ASM в мозг.
В определенных вариантах осуществления ASM представляет собой модифицированную форму ASM (например, модифицированную форму ASM человека) с уменьшением активности относительно немодифицированной ASM (такой как немодифицированная ASM человека, например, ASM-1). В некоторых вариантах осуществления ASM представляет собой модифицированную форму ASM (например, модифицированную форму ASM человека) с увеличением направленности к местам патологии и уменьшением ферментативной активности относительно немодифицированной ASM (такой как немодифицированной ASM человека, например, ASM-1).
В определенных вариантах осуществления ASM представляет собой высокофосфорилированную форму ASM. Смотрите, например, публикацию заявки на патент США № 2002/0150981, которая включена в данный документ ссылкой, для обсуждения техник получения высокофосфорилированных форм лизосомальных ферментов.
Фермент ASM может быть получен любым способом, известным в данном уровне техники, включая без ограничения способы с рекомбинантными ДНК, клонирование кДНК (смотрите, например, Schuchman et al., 1991, J. Biol. Chem 266: 8531-8539 и патент США № 5773278, которые включены в данный документ ссылкой в их полноте для кДНК-клонов ASM человека), геномное клонирование, активацию генов (смотрите, например, патент США № 5641670, который включен в данный документ ссылкой во всей своей полноте для техники активации генов) или отобранные клеточные линии (например, клетки млекопитающих, дрожжей, прокариотов, насекомых (например, Sf 9, Sf21, Trichoplusia ni, Spodoptera frugiperda и Bombyx mori), и растительные клетки), которые продуцируют высокие уровни фермента ASM. ASM, а также способы получения ASM описаны, например, в патентах США №№ 5773278; 6541218; 5686240; и 7527956; каждый из которых включен в данный документ ссылкой во всей своей полноте.
В конкретном варианте осуществления ASM представляет собой рекомбинантно полученную ASM (например, рекомбинантно полученную ASM человека). Предпочтительными являются системы клеточной экспрессии, которые обладают клеточными механизмами и элементами для правильного процессинга, т.е. сигнальное отщепление, гликозилирование, фосфорилирование и белковая сортировка. Например, системы экспрессии клеток млекопитающих являются предпочтительными для экспрессии биологически активных ферментов, которые правильно сворачиваются и процессируются; при введении людям такие продукты экспрессии должны проявлять правильную направленность на ткань и не вызывать нежелательную иммунологическую реакцию.
В определенных вариантах осуществления ASM (например, ASM человека) получают с помощью сверхэкспрессии кДНК ASM в клетках млекопитающих. В конкретном варианте осуществления ASM человека получают с помощью сверхэкспрессии кДНК ASM человека в клетках яичника китайского хомяка (CHO). Смотрите, например, He et al., 1999, Biochimia et Biophsyica Acta 1432: 251-264 для способов сверхэкспрессии кДНК ASM человека в CHO-клетках и патент США № 6541218, каждый из которых включен ссылкой в данный документ во всей своей полноте. В определенных вариантах осуществления ASM человека получают с помощью сверхэкспрессии кДНК ASM человека в CHO-клетках, которые коэкспрессируют альфа-2,6-сиалитрансферазу. Смотрите, например, патент США № 5047335, включенный в данный документ ссылкой во всей своей полноте, который описывает CHO-клетки, сконструированные для экспрессии альфа 2,6-сиалитрансферазы.
В другом конкретном варианте осуществления клетки млекопитающего представляют собой клетки человека. Примеры клеток человека, которые можно применять для рекомбинантной экспрессии ASM (например, ASM человека), включают, без ограничений, клетки Crucell Per.C6, клетки HT 1080, клетки HeLa, клетки HEK 293, клетки 293T, клетки WI38, клетки HuT292, клетки LF 1043 (HEL), клетки MRC-5, клетки TMK-1, клетки BT483, клетки Hs578T, клетки HTB2, клетки HTB3, клетки HTB4, клетки BT 20, клетки T47D, клетки CRL7O3O, клетки HsS78Bst, клетки 721, клетки A2780, клетки A172, клетки A253, клетки COR-L23/R23, клетки COV-434, клетки DU145, клетки DuCaP, клетки EM2, клетки Saos-2, клетки U373, клетки WM39, клетки L132, клетки A-5489, клетки G-293, клетки G-401, клетки CAKI-1, клетки RD и клетки YAR. Другие иллюстративные клетки человека, которые можно применять для экспрессии ASM, включают такие линии клеток человека, которые перечислены в Базе данных иммунополиморфизма Европейской лаборатории молекулярной биологии и базе данных Национальных институтов здравоохранения США, а также те, которые коммерчески доступны из, например, Американской коллекции типовых культур, Манассас, Вирджиния, и Lifeline Cell Technology, Уолкерсвилль, Мэриленд. При применении клеточных линий человека для получения можно получить rhASM с помощью кДНК, гДНК для hASM или с помощью техник активации генов, таких как те, что описаны в патенте США № 5641670, который включен путем ссылки в данный документ во всей своей полноте.
Примеры других клеток млекопитающих, которые можно применять для получения ASM (например, ASM человека), включают, но без ограничений, клетки Vero, клетки VERY, клетки BHK, клетки COS, клетки MDCK или клетки 3T3. В определенных вариантах осуществления миеломные клетки применяют для получения ASM (например, ASM человека). Неограничивающие примеры миеломных клеток включают клетки NS0, клетки 45,6 TG1.7, клетки 9B5 клона AF-2, клетки 9B5 клона AF-2, клетки J558L, клетки MOPC 315, клетки MPC-11, клетки NCI-H929, клетки NP, клетки NS0/1, клетки P3 NS1 Ag4, клетки P3/NS1/1-Ag4-1, клетки P3U1, клетки P3X63Ag8, клетки P3X63Ag8.653, клетки P3X63Ag8U.1, клетки RPMI 8226, клетки Sp20-Ag14, клетки U266B1, клетки X63AG8.653, клетки Y3.Ag.1.2.3 и клетки YO.
В некоторых вариантах осуществления для экспрессии ASM применяют систему культуры клеток растений. Смотрите, например, патенты США №№ 5929304; 7504560; 6770799; 6551820; 6136320; 6034298; 5914935; 5612487; и 5484719, публикации заявок на патенты США №№ 2009/0208477, 2009/0082548, 2009/0053762, 2008/0038232, 2007/0275014 и 2006/0204487, и Shoji et al., 2008, Vaccine, 26(23):2930-2934, и D'Aoust et al., 2008, J. Plant Biotechnology, 6(9):930-940 (которые включены в данный документ ссылкой в их полноте) для клеток растений и способов получения белков с применением системы культуры клеток растений. В частности, заявки на патенты США №№ 2009/0208477, 2008/0038232 и 2006/0204487 описывают экспрессию и получение ферментативно активных с высоким содержанием маннозы лизосомальных ферментов с помощью корня трансгенных растений, в особенности клеток моркови. В конкретном варианте осуществления клетки моркови конструируют для экспрессии ASM. В определенных вариантах осуществления водоросли (например, Chlamydomonas reinhardtii) могут быть сконструированы для экспрессии ASM (смотрите, например, Rasala et al., 2010, Plant Biotechnology Journal (опубликованная онлайн 7 марта 2010 г., которая включена в данный документ ссылкой во всей своей полноте).
Тканевое и клеточное поглощение ASM опосредуется как остатками с высоким содержанием маннозы (например, у макрофагов), так и манноза-6-фосфатом (например, в печени). Таким образом, получение ASM с измененным содержанием гликана и (или) фосфорилированием может быть желательно для усиления распределения лекарственного средства. Например, ASM может быть получена с помощью клеток, имеющих мутацию, например, нокаутных в отношении по меньшей мере одной маннозидазы процессинга аппарата Гольджи. В одном варианте осуществления мутация снижает экспрессию гена, снижает уровни белка или активности или изменяет распределение или другие посттрансляционные модификации маннозидазы, например, процессинг углеводных цепей. В конкретном варианте осуществления мутация снижает уровень активности маннозидазы процессинга аппарата Гольджи. Мутация может быть в маннозидазе процессинга класса 1; маннозидазе процессинга класса 2; маннозидазе процессинга класса 1 и маннозидазе процессинга класса 2. Маннозидаза процессинга класса 1 включает: маннозидазу IA аппарата Гольджи; маннозидазу IB аппарата Гольджи; маннозидазу IC аппарата Гольджи. Маннозидаза процессинга класса 2 включает: маннозидазу II аппарата Гольджи. Смотрите, например, международную публикацию патентной заявки № WO 02/15927 и патент США № 7138262, которые включены в данный документ ссылкой в их полноте, для способов получения белка с высоким содержанием маннозы.
В определенных вариантах осуществления клетки, экспрессирующие ASM, культивируют в присутствии ингибитора маннозидазы, такого как антитело, кифуненсин, свайнсонин, манностатин, 6-дезокси-1,4-дидезокси-1,4-имино-D-маннитол (6-дезокси-DIM) или 6-дезокси-6-фтор-1,4-дидезокси-1,4-имино-D-маннитол (6-дезокси-6-фтор-DIM). Культивирование ASM в присутствии таких ингибиторов может привести к продуцированию ASM с высоким содержанием маннозы. В некоторых вариантах осуществления клетки, экспрессирующие ASM, включают последовательность нуклеиновой кислоты, такую как антисмысловая молекула или рибозим, которая может связываться с или инактивировать клеточную последовательность нуклеиновой кислоты маннозидазы, например мРНК, и ингибировать экспрессию белка. Смотрите, например, международную публикацию патентной заявки № WO 02/15927 и патент США № 7138262, которые включены в данный документ ссылкой в их полноте, для способов получения белка с высоким содержанием маннозы.
В определенных вариантах осуществления углеводные цепи рекомбинантно экспрессированного фермента ASM преобразуют с помощью последовательной обработки различными ферментами, такими как нейраминидаза, галактозидаза и β-N-ацетилглюкозаминидаза. Смотрите, например, патент США № 5549892, который включен в данный документ ссылкой во всей своей полноте, для способов преобразования углеводных цепей лизосомального фермента.
Поглощение ASM, опосредованное манноза-6-фосфатом (M6P), может быть усилено путем модификации ASM с получением высоко фосфорилированных маннозных остатков и M6P. Например, ASM можно модифицировать с помощью рекомбинантной технологии для введения дополнительного манноза-6-фосфата в ASM для усиления клеточного поглощения. Смотрите, например, Matsuoka et al., 2010 Mole. Ther, 18:1519-1526, которая включена в данный документ ссылкой во всей своей полноте. В других вариантах осуществления ASM можно объединить с высоко фосфорилированными олигосахаридными производными, содержащими манноза 6-фосфат (M6P). Смотрите, например, патент США № 7001994, публикацию заявки на патент США № US2010/0173385 и международную публикацию № WO2010/075010. В другом подходе можно применять систему дрожжевой культуры для экспрессии рекомбинантной ASM, которая содержит дополнительные высоко фосфорилированные остатки манноза-6-фосфата. Смотрите, например, Akeboshi et al., 2009 Glycobiology 19(9):1002-1009, которая включена путем ссылки в данный документ во всей своей полноте.
После продуцирования ASM ее можно выделить или очистить любым способом, известным в данном уровне техники, для выделения или очистки белка, например с помощью хроматографии (например, ионообменной, высокоэффективной жидкостной хроматографии, с помощью аффинности, в частности аффинности к ASM, с использованием белка A и сортирующей по размеру колоночной хроматографии), центрифугирования, дифференциальной растворимости или с помощью любой другой стандартной техники выделения или очистки белков. Там, где фермент ASM секретируется культивируемыми клетками, ASM можно легко извлечь из культуральной среды. Смотрите, например, He et al., 1999, Biochimia et Biophsyica Acta 1432: 251-264 для способов очистки ASM.
Фермент ASM может быть составлен для любого пути введения (например, инфузии, подкожного, внутримышечного, интратекального, интравентрикулярного, интраназального, ингаляции или внутрикожного). Фермент ASM может поставляться в лиофилизированной форме, которую восстанавливают перед применением, например в стерильном солевом растворе (например, 0,9% хлориде натрия) или стерильной воде. Альтернативно, фермент ASM может поставляться в водной форме. В определенных вариантах осуществления ASM вводят субъекту в составе, содержащем цинк. В некоторых вариантах осуществления фермент ASM вводят пациенту с помощью инфузии, используя, например, шприцевой насос или инфузионный мешок с насосом.
В определенных вариантах осуществления ASM вводят субъекту в носителе, таком как липосомы или поликатионный носитель. Смотрите, например, патент США № 5716614, который включен в данный документ ссылкой во всей своей полноте, в отношении носителей, которые можно применять для введения ASM. В некоторых вариантах осуществления ASM вводят субъекту в нацеленных на ICAM-1 наноносителях. Смотрите, например, Muro et al., 2006, Mol. Ther. 13(1): 135-141, которая включена в данный документ ссылкой, для доставки ASM с помощью ICAM-1-нацеленных наноносителей.
5.2. ПОПУЛЯЦИИ ПАЦИЕНТОВ
В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который имеет или который диагностируется как имеющий одну или несколько мутаций в гене, кодирующем кислую сфингомиелиназу. В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который имеет или который диагностируется как имеющий болезнь Ниманна-Пика (NPD). В одном варианте осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человеком, который имеет или который диагностируется как имеющий NPD типа A. В другом конкретном варианте осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который имеет или который диагностируется как имеющий NPD типа B.
В определенных вариантах осуществления субъект, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, имеет одну или несколько мутаций в гене SMPD1. В некоторых вариантах осуществления мутация представляет собой миссенс-мутацию. В других вариантах осуществления мутация представляет собой делецию, которая приводит к делеции одного, двух или более аминокислотных остатков. В конкретных вариантах осуществления субъект, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, имеет одну или несколько мутаций.
Смотрите, например, Simonaro et al., 2002, Am. J. Hum. Genet. 71: 1413-1419, которая включена в данный документ ссылкой, в отношении мутаций в гене кислой сфингомиелиназы (обозначенный SMPD1).
В определенных вариантах осуществления субъект, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, эндогенно экспрессирует ASM с 2-5%, 5-10%, 5-15%, 5-20%, 5-30% или 20-30% активностью нормальной ASM человека, например, ASM-1. В некоторых вариантах осуществления субъект, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, эндогенно экспрессирует ASM с менее чем 30%, 25%, 20%, 15%, 10%, 5%, 4%, 3%, 2% или 1% активностью нормальной ASM человека, например, ASM-1. Смотрите, например, патенты США №№ 4039388, 4082781, 5686240 и 7563591 и международные публикации №№ WO 2007/078806 и WO 2006/058385, которые включены в данный документ ссылкой в их полноте, в отношении техник, которые можно применять для измерения активности ASM. В конкретном варианте осуществления для измерения активности ASM применяют анализ высокоэффективной жидкостной хроматографией на основе флюоресценции, описанный в He et al., 2003, Analytical Biochemistry 314: 116-120 (которая включается в данный документ ссылкой).
В определенных вариантах осуществления субъект, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, проявляет один или несколько симптомов NPD. Симптомы NPD включают, без ограничений, вздутия живота, гепатомегалию, спленомегалию, гепатоспленомегалию, нейтропению, заболевание легкого, лимфоаденопатию, наличие гистохимически отличительных пенистых клеток NPD, анемию (например, микроцитарную анемию), тромбоцитопению, рецидивирующую рвоту, хронический запор, расстройство роста (например, уменьшенный линейный рост и вес тела), задержку полового созревания, рецидивирующий кровоподтек, рецидивирующее кровотечение, атерогенный липидный профиль (высокое содержание холестерина, триглицеридов, LDL и низкое содержание HDL), боль (головная боль, боль в спине, конечностях, животе), утомляемость, чувство быстрого насыщения, низкую выносливость, остеопению, неврологические проявления и трудности дыхания (например, интерстициальное заболевание легкого, диспноэ). Неврологическое проявление NPD включает синдром вишневой косточки, гипотонию, мышечную слабость, психомоторное торможение, спастичность, социальную невосприимчивость, раздражительность и пароксизмы.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является субъект-человек, который проявляет два или более клинических признаков, присущих не невропатической NPD. В конкретных вариантах осуществления субъектом, который получает лечение от ASM в соответствии со способами, приведенными в данном документе, является субъект-человек, который проявляет два или более клинический признаков, присущих не невропатической NPD: тромбоцитопению, анемию, нейтропению, гепатомегалию, спленомегалию и заболевание легкого. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является субъект-человек, который проявляет два или более клинических признаков, присущих невропатической NPD.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с объемом селезенки в два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать или двенадцать раз больше, чем объем селезенки здорового человека, что оценивается с помощью техник, известных в данном уровне техники, таких как, например, магнитно-резонансная томография (МРТ). В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с объемом селезенки, который в восемь раз больше, чем объем селезенки здорового человека, что оценивается с помощью техник, известных в данном уровне техники, таких как, например, МРТ. В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с объемом селезенки в восемь-двенадцать, девять-двенадцать, десять-двенадцать или двенадцать-четырнадцать раз больше, чем объем селезенки здорового человека, что оценивается с помощью техник, известных в данном уровне техники, таких как, например, МРТ. В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с объемом селезенки в ≥8 раз больше нормы (MN) (т.е. 1,6% веса тела), что оценивается с помощью техник, известных в данном уровне техники, таких как, например, МРТ.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с диффузионной емкостью легких (DLCO) от 20% до 80%, от 25% до 80%, от 30% до 80%, от 40% до 80%, от 50% до 80% или от 60% до 80% от расчетной DLCO здорового человека. В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек с DLCO от 20% до 90%, от 25% до 90%, от 30% до 90%, от 40% до 90%, от 50% до 90%, от 60% до 90% или от 70% до 90% от расчетной DLCO здорового человека. DLCO измеряет скорость диффузии ограниченного процессами диффузии газа (например, моноксид углерода) за минуту через альвеолокапиллярную мембрану. DLCO может быть рассчитана путем сравнения количества моноксида углерода, которое выдыхается после известного количества вдыхаемого моноксида углерода. Для оценки DLCO можно применять техники, известные в данном уровне техники.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-младенец. В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, которому 2-3 месяца, 2-6 месяцев, 3-6 месяцев, 4-6 месяцев, 5-8 месяцев или 6-9 месяцев. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-ребенок. В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, которому 1-3 года, 2-3 года, 3-5 лет, 4-5 лет, 5-7 лет или 6-9 лет.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является взрослый человек. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, описанными в данном документе, является пожилой человек. В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, которому 10-18 лет, 10-20 лет, 12-20 лет, 15-20 лет, 20-25 лет, 21-25 лет, 21-30 лет, 25-30 лет, 30-35 лет, 35-40 лет, 40-45 лет, 45-50 лет, 50-55 лет, 55-60 лет, 60-65 лет, 65-70 лет, 70-75 лет, 75-80 лет, 80-85 лет, 85-90 лет, 90-95 лет или 95-100 лет.
В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-женщина. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-мужчина. В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-женщина, которая не является беременной или кормящей грудью. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек-женщина, которая является беременной, или станет беременной, или кормит грудью.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, у которого не было трансплантации жизненно-важного органа (например, трансплантации печени или костного мозга). В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, у которого была трансплантация жизненно-важного органа (например, трансплантация печени или костного мозга). В некоторых вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, у которого не было общей или частичной спленэктомии. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, у которого была общая или частичная спленэктомия.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который не имеет или у которого не было диагностировано одно или несколько из следующих заболеваний: активный гепатит B, активный гепатит C, инфекция вируса иммунодефицита человека (ВИЧ), цирроз или серьезное кардиологическое заболевание (например, умеренная или тяжелая легочная гипертензия, или дисфункция клапанов, или фракция выброса левого желудочка по эхокардиографии менее 50%, менее 40%, менее 30% или менее 20%). В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который имеет или у которого было диагностировано одно или несколько из следующих заболеваний: активный гепатит B, активный гепатит C, инфекция вируса иммунодефицита человека (ВИЧ), цирроз или значительное кардиологическое заболевание (например, умеренная или тяжелая легочная гипертензия, или дисфункция клапанов, или фракция выброса левого желудочка по эхокардиографии менее 50%, менее 40%, менее 30% или менее 20%).
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который не имеет одного или нескольких из следующих: международное нормализованное отношение (INR) составляет более 1,25, 1,5, 1,75 или 2, количество тромбоцитов менее 60×103 на мкл, аланинаминотрансфераза (ALT) более 250 МЕ/Л, аспартатаминотрансфераза более 250 МЕ/л, или общий билирубин более 1,5 мг/дл, 1,75 мг/дл или 2 мг/дл. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который имеет одно или несколько из следующих: международное нормализованное отношение (INR) составляет более 1,25, 1,5, 1,75 или 2, количество тромбоцитов менее 60×103 на мкл, аланинаминотрансфераза (ALT) более 250 МЕ/Л, аспартатаминотрансфераза более 250 МЕ/Л, или общий билирубин более 1,5 мг/дл, 1,75 мг/дл или 2 мг/дл.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который не получает один или несколько из следующих медикаментов: хлорпромазин, имипрамин или дезипрамин. В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который получает один или несколько из следующих медикаментов: хлорпромазин, имипрамин или дезипрамин.
В определенных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который не получает растительные добавки или медикаменты, которые могут вызвать или пролонгировать кровотечение (например, антикоагулянты, ибупрофен, аспирин, чесночные добавки, гинкго и женьшень) или имеют потенциальную гепатотоксичность (например, ингибиторы 3-гидрокси-3-метилглутарил [HMG]-CoA редуктазы, эритромицин, вальпроевая кислота, антидепрессанты, кава и эхинацея). В других вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который получает одну или несколько из следующих растительных добавок или медикаментов: антикоагулянты, ибупрофен, аспирин, чесночные добавки, гинкго, женьшень, ингибиторы 3-гидрокси-3-метилглутарил [HMG]-CoA редуктазы, эритромицин, вальпроевая кислота, антидепрессанты, кава и эхинацея.
В конкретных вариантах осуществления субъектом, который получает лечение от ASMD в соответствии со способами, приведенными в данном документе, является человек, который соответствует одному, двум или более, или всем критериям для субъектов в рабочих примерах в разделе 6, 8 и 9 и далее.
5.3. МОНИТОРИНГ ЛЕЧЕНИЯ
В соответствии со способом, обеспеченным в данном документе, ряд параметров (например, факторов или маркеров) можно подвергать мониторингу до, во время и (или) после введения дозы ASM. В определенных вариантах осуществления физическое исследование проводят до введения ASM и, если необходимо или рекомендовано, во время курса лечения с ASM. Физическое исследование может включать следующие оценки: общий внешний вид, кожа, голова, уши, глаза, нос и горло (HEENT), лимфатические узлы, сердце, легкие, живот, конечности/суставы, неврологический, умственный статус и рефлексы. В определенных вариантах осуществления основные показатели жизнедеятельности, непрерывную частоту сердечных сокращений, частоту дыхания, температуру и насыщение кислородом можно оценить до, во время и (или) после введения дозы ASM. Частоты сердечных сокращений можно подвергать мониторингу непрерывно с помощью телеметрии, начиная, например, с 6 часов до введения дозы ASM до 72 часов после введения дозы ASM.
В конкретных вариантах осуществления клинический анализ крови с подсчетом форменных элементов, азотом мочевины крови (BUN), бикарбонатом, креатинином, глюкозой, мочевой кислотой, кальцием, фосфатом, альбумином, общим белком, натрием, калием, хлоридом, лактатдегидрогеназой, креатинкиназой, креатинкиназой с MB фракцией и анализом мочи (включая цвет, внешний вид, удельный вес, pH, белок, глюкозу, кетоны, билирубин, гемоглобин, и микроскопию мочи, если назначено) могут быть проведены в определенные моменты времени до, во время и (или) после введения дозы ASM. В определенных вариантах осуществления печеночные пробы, исследования коагуляции и (или) липидный профиль натощак могут быть проведены до, во время и (или) после введения дозы ASM. Пробы функций печени могут включать оценку концентраций аланинаминотрансферазы (ALT), аспартатаминотрансферазы (AST), щелочной фосфатазы (AP), гамма-глутамилтрансферазы (GGT) и общего и прямого билирубина. Исследования коагуляции могут включать оценку протромбинового времени (PT), частичного тромбопластинового времени (PTT), международного нормализованного отношения (INR), концентрации D-димера и концентрации фибриногена. Оценки липидного профиля натощак могут включать оценки общего холестерина (TC), липопротеина низкой плотности (LDL), липопротеина высокой плотности (HDL), липопротеина очень низкой плотности (VLDL) и триглицеридов.
В определенных вариантах осуществления биопсию кожи проводят до, во время и (или) после введения дозы ASM. В некоторых вариантах осуществления биопсию печени проводят до, во время и (или) после введения дозы ASM. Уровни сфингомиелина в биопсиях кожи и печени можно оценить посредством гистологического анализа с помощью MetaMorph.
В определенных вариантах осуществления тесты легочной функции проводят до, во время и (или) после введения дозы ASM. Калибровка оборудования для тестирования легочной функции и протоколы проведения теста могут быть стандартизированы согласно руководствам Американского общества торакальной медицины (ATS) (ATS, 1991, Am Rev Respir Dis 144: 1202-1218). В определенных вариантах осуществления относительную расчетную форсированную жизненную емкость легких (FVC) оценивают до, во время и после введения дозы ASM. FVC представляет собой общий объем воздуха, выдыхаемого во время маневра форсированного выдоха. FVC можно измерить с помощью стандартных спирометрических техник.
В определенных вариантах осуществления объем форсированного выдоха за 1 секунду (FEV1) может быть проведен до, во время и после введения дозы ASM. FEV1 - это объем воздуха, выбрасываемого во время первой секунды FVC. FEV1 может быть измерен с помощью стандартных спирометрических техник.
В определенных вариантах осуществления общую емкость легких (TLC) можно оценить до, во время и после введения дозы ASM. TLC - это общий объем воздуха в легких после максимального усилия на вдохе. TLC можно измерить с помощью общей плетизмографии.
В определенных вариантах осуществления DLCO можно оценить до, во время и после введения дозы ASM. DLCO измеряет скорость диффузии ограниченного процессами диффузии газа (моноксида углерода, CO) в минуту через альвеолокапиллярную мембрану. DLCO может быть рассчитана путем сравнения количества моноксида углерода (CO), выдыхаемого после известного количества вдохнутого CO. Гелий, который не диффундирует через альвеолокапиллярную мембрану, может быть включен как изотоп с вдыхаемым CO для контроля захвата воздуха.
В определенных вариантах осуществления рентгенография грудной клетки (задне-передняя и боковая) может быть получена до, во время и (или) после введения дозы ASM. В определенных вариантах осуществления брюшную МРТ получают до, во время и (или) после введения дозы ASM.
В определенных вариантах осуществления образец крови забирают до, во время и (или) после введения дозы ASM, для оценки биомаркеров, концентрации билирубина и относительного содержания нейтрофилов от общего количества лейкоцитов. В конкретном варианте осуществления концентрацию одного или нескольких из следующих биомаркеров оценивают с помощью техник, известных специалисту в данной области: концентрация CRP/hs-CRP, концентрация сфингомиелина, концентрация железа, концентрация ферритина, концентрация кальцитонина, концентрация альбумина, SAA, компонент амилоида P сыворотки, ACE, CCL18, хитотриозидаза, трансферрин, концентрация фибриногена и концентрация сфингомиелина в плазме, концентрация церамида в плазме. В определенных вариантах осуществления концентрацию одного или нескольких из следующих цитокинов оценивают с помощью техник, известных специалисту в данной области: IL-6 и IL-8.
В определенных вариантах осуществления концентрацию антител против ASM (например, IgG к рекомбинантной ASM человека и (или) IgE к рекомбинантной ASM человека) оценивают до, во время и (или) после введения дозы ASM. В некоторых вариантах осуществления концентрацию одного, двух или более факторов комплемента оценивают до, во время и (или) после введения дозы ASM. В определенных вариантах осуществления концентрацию сывороточной триптазы оценивают до, во время и (или) после введения дозы ASM. В определенных вариантах осуществления тест кожи для определения IgE-опосредованных реакций к ASM оценивают до, во время и (или) после введения дозы ASM.
В определенных вариантах осуществления фармакокинетический профиль для ASM измеряют до, во время и (или) после введения дозы ASM. В некоторых вариантах осуществления количество клеток BAL измеряют до, во время и (или) после введения дозы ASM. Смотрите раздел 8 и далее, ниже, для других параметров, которые могут быть оценены до, во время и (или) после введения дозы ASM.
В определенных вариантах осуществления описываемый в данном документе фактор или маркер оценивают за 1 неделю, 72 часа, 48 часов, 24 часа, 12 часов, 6 часов, 4 часа, 2 часа, 1 час, 30 минут или 15 минут до введения дозы ASM. В некоторых вариантах осуществления описываемый в данном документе фактор или маркер оценивают во время введения дозы ASM. Например, описываемый в данном документе фактор или маркер оценивают во время введения дозы ASM. В определенных вариантах осуществления описываемый в данном документе фактор или маркер оценивают через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов, 16 часов, 18 часов, 24 часа, 36 часов, 48 часов, 72 часа, 4 дня, 5 дней, 6 дней, 1 неделю, две недели, три недели, четыре недели, один месяц, два месяца, три месяца или дольше после введения дозы ASM. В определенных вариантах осуществления описываемый в данном документе фактор или маркер оценивают через несколько часов или недель после определенного числа доз ASM. Например, описываемый в данном документе фактор или маркер можно оценивать через каждые четыре недели, месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев или 6 месяцев после определенного числа доз ASM. В определенных вариантах осуществления фактор или маркер оценивают каждые 4 недели, месяц, 2 месяца, 3 месяца, 4 месяца 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев или 12 месяцев. В некоторых вариантах осуществления фактор или маркер оценивают каждый месяц - 2 месяца, каждый месяц - 4 месяца, каждые 2 месяца - 4 месяца, каждые 2 месяца - 4 месяца, каждые 3 месяца - 4 месяца, каждые 2 месяца - 5 месяцев, каждые 3 месяца - 5 месяцев, каждые 4 месяца - 5 месяцев, каждые 2 месяца - 6 месяцев, каждые 3 месяца - 6 месяцев, каждые 4 месяца - 6 месяцев или каждые 5 месяцев - 6 месяцев. В определенных вариантах осуществления фактор или маркер оценивают каждые 6 месяцев - 8 месяцев, каждые 6 месяцев - 12 месяцев, каждые 8 месяцев - 12 месяцев, каждые 9 месяцев - 12 месяцев или каждые 10 месяцев - 12 месяцев. В некоторых вариантах осуществления один или несколько описываемых в данном документе факторов или маркеров не оценивают до, во время и (или) после введения дозы ASM.
В определенных вариантах осуществления результаты из оценки одного или нескольких факторов или маркеров указывают на то, что дозировку ASM следует откорректировать.
5.4. БИОЛОГИЧЕСКИЕ ОБРАЗЦЫ
В соответствии со способами, описанными в данном документе, биологический образец подвергают одному или нескольким этапам предварительной обработки до обнаружения и (или) измерения клеточной популяции, фактора или маркера (например, биомаркера). В определенных вариантах осуществления биологическую жидкость предварительно обрабатывают путем центрифугирования, фильтрации, осаждения, диализа или хроматографии или путем комбинации таких этапов предобработки. В других вариантах осуществления образец ткани предварительно обрабатывают с помощью замораживания, химической фиксации, погружения в парафин, дегидратации, пермеабилизации или гомогенизирования с последующим центрифугированием, фильтрацией, осаждением, диализом или хроматографией или с помощью комбинации таких этапов предобработки. В определенных вариантах осуществления образец предварительно обрабатывают путем удаления клеток определенного типа из образца или удаления инородных веществ из образца до определения числа или количества конкретного клеточного типа (клеточных типов) в образце согласно способам, описанным в данном документе.
Биологический образец может быть образцом ткани, биологической жидкостью, выделением или любым другим образцом от человека-субъекта. В некоторых вариантах осуществления биологический образец представляет собой образец крови или костного мозга. В определенных вариантах осуществления биологический образец представляет собой образец ткани (например, биопсия печени, кожи или легкого). В некоторых вариантах осуществления биологический образец представляет собой биологическую жидкость, такую как моча. В определенных вариантах осуществления биологический образец представляет собой мокроту или образец назального выделения. В некоторых вариантах осуществления биологический образец представляет собой мазок ротовой полости.
Техники, известные в данном уровне техники, могут использоваться для оценки присутствия, числа, количества или относительного содержания определенного типа (типов) клеток, присутствующих в биологическом образце. Например, клетки могут быть отсортированы с использованием клеточного сортера с активацией флюоресценции (FACS). Сортировка клеток с активацией флюоресценции (FACS) представляет собой известный способ для разделения частиц, включая клетки на основании свойств флюоресценции частиц. Смотрите, например, Kamarch, 1987, Methods Enzymol 151:150-165. Возбуждение лазером флюоресцентных молекул в отдельных частицах приводит к небольшому электрическому заряду, позволяющему провести электромагнитное разделение положительных и отрицательных частиц из смеси. Антитело или лиганд, используемые для обнаружения антигенной детерминанты, присутствующей на клеточной поверхности конкретных клеток, метят флюорохромом, таким как FITC или фикоэритрин. Клетки инкубируют с флюоресцентно меченым антителом или лигандом в течение периода времени, достаточного для того, чтобы обеспечить связывание меченого антитела или лиганда с клетками. Клетки пропускают через клеточный сортер, обеспечивая отделение клеток интереса от других клеток. Отсортированные на FACS частицы могут непосредственно распределяться в отдельные лунки микротитровальных планшетов для облегчения разделения.
Магнитные гранулы могут также быть использованы для разделения клеток. Например, клетки могут быть отсортированы, используя технику сортировки клеток с активацией магнитным полем (MACS), способ для разделения частиц на основании их способности связываться с магнитными гранулами (0,5-100 мкм диаметра). Разнообразие пригодных модификаций может быть осуществлено на магнитных микросферах, включая ковалентную добавку к антителу, которая, в частности, распознает молекулу на поверхности клетка - твердая фаза или гаптен. Затем применяется магнитное поле для физической манипуляции выбранными гранулами. В конкретном варианте осуществления антитела к маркеру поверхности клетки крови соединяются с магнитными гранулами. Гранулы затем смешиваются с культурой клеток крови для обеспечения связывания. Клетки затем пропускают через магнитное поле для отделения клеток, имеющих маркеры поверхности клеток крови интереса. Эти клетки затем могут быть выделены.
В некоторых вариантах осуществления поверхность культуральной чашки может быть покрыта антителами и используется для отделения клеток с помощью способа, называемого пэннинг. Отдельные чашки могут быть покрыты антителом, специфическим для конкретных клеток. Клетки могут быть добавлены вначале в чашку, покрытую специфическими для клеток крови антителами интереса. После тщательной промывки клетки, которые остаются связанными с чашкой, будут клетками, которые экспрессируют клеточные маркеры интереса. Примеры антигенных детерминант или маркеров клеточной поверхности включают, но без ограничений, CD2 для T лимфоцитов и клеток-натуральных киллеров, CD3 для T лимфоцитов, CD11a для лейкоцитов, CD28 для T лимфоцитов, CD19 для B лимфоцитов, CD20 для B лимфоцитов, CD21 для B лимфоцитов, CD22 для B лимфоцитов, CD23 для B лимфоцитов, CD29 для лейкоцитов, CD14 для моноцитов. CD41 для тромбоцитов, CD61 для тромбоцитов, CD66 для гранулоцитов, CD67 для гранулоцитов и CD68 для моноцитов и макрофагов.
Присутствие, концентрацию или количество маркера (включая биомаркер) или фактора можно оценить, используя техники, известные в данном уровне техники. Присутствие, концентрацию или количество маркера (включая биомаркер) или фактора могут быть измерены на уровне белка и (или) уровне РНК, используя техники, известные специалисту в данной области. На уровне белка, иммуноанализы, такие как ELISA и иммунопреципитация и вестерн-блоттинг анализы могут использоваться для измерения присутствия, концентрации или количества маркера (включая биомаркер) или фактора. Кроме того, FACS может использоваться для измерения присутствия, концентрации или количества маркера (включая биомаркер) или фактор. На уровне РНК, РВ-ПЦР (полимеразная цепная реакция в реальном времени) и нозерн-блоттинг анализы могут использоваться для измерения присутствия, концентрации или количества маркера (включая биомаркер) или фактора.
5.5. СОВМЕСТНЫЕ ТЕРАПИИ
В некоторых вариантах осуществления способ лечения ASMD включает введение ASM в комбинации с одной или несколькими дополнительными терапиями. Применяемое в данном документе выражение «в комбинации» относится, в контексте введения ASM, к введению дозы ASM до, совместно с или после введения одной или нескольких дополнительных терапий (например, средств) для применения в лечении ASMD. Применение выражения «в комбинации» не ограничивает порядок, в котором ASM и одну или несколько дополнительных терапий вводят субъекту. В конкретных вариантах осуществления интервалы времени между введением дозы ASM и введением одной или нескольких дополнительных терапий могут быть следующими: приблизительно 1-5 минут, 1-30 минут, 30 минут - 60 минут, 1 час, 1-2 часа, 2-6 часов, 2-12 часов, 12-24 часа, 1-2 дня, 2 дня, 3 дня, 4 дня, 5 дней, 6 дней, 7 дней, 1 неделя, 2 недели, 3 недели, 4 недели, 5 недель, 6 недель, 7 недель, 8 недель, 9 недель, 10 недель, 15 недель, 20 недель, 26 недель, 52 недели, 11-15 недель, 15-20 недель, 20-30 недель, 30-40 недель, 40-50 недель, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 1 год, 2 года или любой период времени между ними. В определенных вариантах осуществления дозу ASM и одну или несколько дополнительных терапий вводят менее чем через 1 день, 1 неделю, 2 недели, 3 недели, 4 недели, один месяц, 2 месяца, 3 месяца, 6 месяцев, 1 год, 2 года или 5 лет.
В некоторых вариантах осуществления комбинированные терапии, обеспеченные в данном документе, включают введение дозы ASM каждые 2-4 недели, и введение одной или нескольких дополнительных терапий один раз в неделю, один раз каждые 2 недели, один раз каждые 3 недели, один раз каждые 4 недели, один раз каждый месяц, один раз каждые 2 месяца (например, приблизительно 8 недель), один раз каждые 3 месяца (например, приблизительно 12 недель) или один раз каждые 4 месяца (например, приблизительно 16 недель). В определенных вариантах осуществления ASM и одну или несколько дополнительных терапий вводят циклически субъекту. Циклическая терапия включает введение ASM в течение периода времени, с последующим введением одной или нескольких дополнительных терапий в течение периода времени, и повторение этого последовательного введения. В определенных вариантах осуществления циклическая терапия может также включать период покоя, когда ASM или дополнительную терапию не вводят в течение периода времени (например, 2 дня, 3 дня, 4 дня, 5 дней, 6 дней, 7 дней, 1 неделю, 2 недели, 3 недели, 4 недели, 5 недель, 10 недель, 20 недель, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 12 месяцев, 2 года или 3 года). В одном варианте осуществления число вводимых циклов составляет от 1 до 12 циклов, от 2 до 10 циклов или от 2 до 8 циклов.
В некоторых вариантах осуществления способы для лечения ASMD, обеспеченные в данном документе, содержат введение ASM в качестве отдельного средства в течение периода времени до введения ASM в комбинации с дополнительной терапии. В определенных вариантах осуществления способы для лечения ASMD, обеспеченные в данном документе, содержат введение дополнительной терапии отдельно в течение периода времени до введения ASM в комбинации с дополнительной терапией.
В некоторых вариантах осуществления введение ASM и одной или нескольких дополнительных терапий в соответствии со способами, представленными в данном документе, имеют аддитивный эффект относительно введения ASM или указанной одной или нескольких дополнительных терапий отдельно. В некоторых вариантах осуществления введение ASM и одной или нескольких дополнительных терапий в соответствии со способами, представленными в данном документе, имеют синергический эффект относительно введения ASM или указанной одной или нескольких дополнительных терапий отдельно.
Применяемое в данном документе выражение «синергический» относится к эффекту введения дозы ASM в комбинации с одной или несколькими дополнительными терапиями (например, агентами), где комбинация является более эффективной, чем аддитивные эффекты любых двух или более монотерапий (например, агентов). В конкретном варианте осуществления синергический эффект комбинированной терапии позволяет применение более низких дозировок (например, субоптимальные дозы) ASM или дополнительной терапии и (или) менее частое введение ASM или дополнительной терапии субъекту. В определенных вариантах осуществления способность использовать более низкие дозировки ASM или дополнительной терапии и (или) вводить ASM или указанную дополнительную терапию менее часто снижает токсичность, связанную с введением ASM или указанной дополнительной терапии, соответственно, субъекту без снижения эффективности ASM или указанной дополнительной терапии, соответственно, в лечении ASMD. В некоторых вариантах осуществления синергический эффект приводит к улучшенной эффективности ASM и каждой из указанных дополнительных терапий в лечении ASMD. В некоторых вариантах осуществления синергический эффект комбинации ASM и одной или нескольких дополнительных терапий избегает или снижает неблагоприятные или нежелательные побочные эффекты, связанные с применением любой монотерапии.
В конкретных вариантах осуществления одну или несколько дополнительных терапий вводят в комбинации с ASM субъектам для снижения или облегчения одного или нескольких из следующих явлений, что может быть связано с введением конкретной дозы ASM: (i) связанное нежелательное явление, (ii) величина общего билирубина больше, чем величина билирубина для человека без ASMD (например, здорового человека), (iii) концентрация церамида в плазме больше, чем концентрация церамида в плазме человека без ASMD (например, здорового человека), или (iv) острофазовый ответ. В конкретных вариантах осуществления одну или несколько дополнительных терапий вводят в комбинации с ASM субъектам для увеличения легочной функции при минимизации одного или нескольких из следующих явлений, которые могут быть связаны с введением конкретной дозы ASM: (i) связанное нежелательное явление, (ii) величина общего билирубина больше, чем величина билирубина для человека без ASMD (например, здорового человека), (iii) концентрация церамида в плазме больше, чем концентрация церамида в плазме человека без ASMD (например, здорового человека), или (iv) острофазовый ответ.
В определенных вариантах осуществления одну или несколько дополнительных терапий вводят в комбинации с ASM субъекту для контроля или ослабления симптомов, связанных с ASMD. В некоторых вариантах осуществления одна или несколько дополнительных терапий, которые вводят в комбинации с ASM субъекту, представляют собой обезболивающие. Специфические примеры терапий, которые могут быть введены в комбинации с ASM, включают, без ограничений, следующие: N-ацетил-L-цистеин (NAC), S-аденозил-L-метионин (SAM), антитело интерлейкин (IL)-6, антитело IL-6 рецептора, дексаметазон, L-Nil, (селективный ингибитор индуцибельной NOS), L-NAME (селективный ингибитор NOS), основной фактор роста фибробластов (b-FGF), имипрамин (ингибитор сфингомиелиназы), D609 (ингибитор сфингомиелиназы) и N-олеоилэтаноламин (NOE; ингибитор церамида).
В определенных вариантах осуществления шапероны, такие как шапероны-малые молекулы, вводят в комбинации с ASM. Смотрите, например, патент США № 7750050 и международные публикации №№ WO 2004/045574 и WO 2010/015816, которые включены в данный документ ссылкой, для средств (например, малых молекул), которые могут быть введены в комбинации с ASM субъекту. В некоторых вариантах осуществления шаперон (например, шаперон-малая молекула) делает одно, два или все из следующего: увеличивает направленность ASM к местам патологии, стабилизирует активность ASM и усиливает активность ASM.
В некоторых вариантах осуществления глюкокортикостероид, такой как дексаметазон, вводят в комбинации с ASM. Смотрите, например, патент США № 7658916, который включен в данный документ ссылкой во всей своей полноте, для средств (например, глюкокортикостероидов), которые могут быть введены в комбинации с ASM.
В некоторых вариантах осуществления молекулу, уменьшающую субстрат, вводят в комбинации с ASM субъекту. В конкретных вариантах осуществления молекулу (например, малую молекулу), которая либо уменьшает количество сфингомиелина, снижает скорость синтеза сфингомиелина, либо и уменьшает и снижает, вводят в комбинации с ASM субъекту. Смотрите, например, Li et al. 2007, Biochim Biophys Act 1771(9):1186-1194 (которая включена в данный документ ссылкой во всей своей полноте) для ингибиторов сфингомиелинсинтазы, таких как трициклодекан-9-ил-ксантогенат и малые интерферирующие РНК сфингомиелинсинтазы.
В некоторых вариантах осуществления терапию, которая снижает потенциальную иммуногенность ASM, вводят в комбинации с ASM субъекту. В определенных вариантах осуществления бенедрил вводят в комбинации с ASM субъекту.
Комбинация ASM и одной или нескольких дополнительных терапий может быть введена субъекту в одной фармацевтической композиции. Альтернативно, ASM и одна или несколько дополнительных терапий могут быть введены одновременно субъекту в отдельных фармацевтических композициях. ASM и одна или несколько дополнительных терапий могут быть введены последовательно субъекту в отдельных фармацевтических композициях. ASM и одна или несколько дополнительных терапий могут также быть введены субъекту одинаковыми или различными путями введения.
5.6. ФАРМАЦЕВТИЧЕСКИЕ ПРОДУКТЫ
В одном аспекте, описанном в данном документе, представлены готовые упакованные и меченные фармацевтические продукты. В одном варианте осуществления фармацевтический продукт содержит стандартную лекарственную форму ASM в соответствующем сосуде или контейнере (например, стеклянной ампуле или другом контейнере, который является герметически закрытым). В некоторых вариантах осуществления стандартная лекарственная форма представляет собой лиофилизированную форму ASM, и при этих условиях фармацевтический продукт может содержать второй контейнер со стерильным солевым раствором или стерильной водой для восстановления лиофилизированной формы ASM. В других вариантах осуществления стандартная лекарственная форма представляет собой водную форму ASM, которая не нуждается в восстановлении перед введением субъекту. В конкретных вариантах осуществления стандартная лекарственная форма ASM содержит ASM в количестве, достаточном для введения низкой, нетоксичной дозы фермента субъекту. В определенных вариантах осуществления стандартная лекарственная форма является подходящей для выбранного пути введения ASM субъекту. В конкретном варианте осуществления стандартная лекарственная форма является подходящей для внутривенной доставки субъекту.
В одном варианте осуществления фармацевтический продукт содержит стандартную лекарственную форму ASM в соответствующем сосуде или контейнере (например, стеклянной ампуле или другом контейнере, который является герметически закрытым) и инфузионном насосе или шприцевом насосе для введения ASM субъекту.
Как и в случае с любым фармацевтическим продуктом упаковочный материал и контейнер разрабатываются, чтобы защищать стабильность продукта во время хранения и транспортировки. Дополнительно, фармацевтический продукт включает инструкции для применения или другой информационный материал, который советует врачу, младшему медицинскому персоналу или пациенту, как соответствующе лечить ASMD. Другими словами, фармацевтический продукт включает средства по инструкции, назначающие или предлагающие схему дозирования, включая, без ограничения, фактические дозы (например, протокол с увеличением дозы), процедуры отслеживания и другую информацию о мониторинге. В определенных вариантах осуществления фармацевтический продукт включает средства для генотипирования пациента (например, ПЦР праймеры для SMPD1 гена).
6. ПРИМЕР 1: ИССЛЕДОВАНИЕ НА ЧЕЛОВЕКЕ ФЕРМЕНТНОЙ ЗАМЕСТИТЕЛЬНОЙ ТЕРАПИИ РЕКОМБИНАНТНОЙ КИСЛОЙ СФИНГОМИЕЛИНАЗОЙ (rhASM) У ВЗРОСЛЫХ ЛЮДЕЙ С НЕДОСТАТОЧНОСТЬЮ ASM
6.1. ВВЕДЕНИЕ
ASMD - это аутосомное рецессивное, лизосомальное нарушение накопления, которое происходит, когда сфингомиелин является недоступным для того, чтобы нормально катаболизироваться до церамида и фосфорилхолина. Следовательно, сфингомиелин аккумулируется внутри клеток в первую очередь ретикулоэндотелиальной системы, приводя к гепатоспленомегалии, анемии, тромбоцитопении и интерстициальное заболевание легкого. Также часто обнаруживается замедление роста и атерогенный липидный профиль. Пациенты, которые имеют от небольшой до отсутствия остаточной активности ASM, проявляют наиболее тяжелые симптомы с началом в младенчестве, задержкой в развитии, нейродегенерацией и смертью к 3 годам (болезнь Ниманна-Пика типа A, NPD A). Пациенты с более высокими количествами остаточной активности ASM имеют различные возрасты начала, гетерогенные проявления и соматические симптомы, от небольшого до отсутствия неврологического поражения и, как правило, выживанием до взрослого возраста (NPD B). В настоящее время отсутствует лечение для пациентов с ASMD.
Ферментная заместительная терапия (ERT) была успешно использована для лечения некоторых лизосомальных расстройств накопления, включая заболевание Гоше, мукополисахаридоз типов I, II, и VI, заболевание Фабри и заболевание Помпе. Рекомбинантные лизосомальные ферменты человека вводятся внутривенно и поглощаются клетками посредством рецептор-опосредованного эндоцитоза для последующей направленности в лизосомы. Правильность принципа лечения ASMD было продемонстрировано лабораторией Dr. Edward Schuchman (Медицинский центр Mount Sinai) в ASM нокаутной мышиной модели (ASMKO), где внутривенные инъекции рекомбинантной ASM человека (rhASM) эффективно снижали уровни сфингомиелина в печени и селезенке, и в меньшей степени в легком (Miranda, et al., FASEB 2000; 14:1988). Однако уровни сфингомиелина не были снижены в мозге из-за неспособности rhASM преодолевать гематоэнцефалический барьер. Из-за того факта, что ASMKO мышь не имеет остаточной активности ASM или белка и развивается быстрое и тяжелое неврологическое заболевание, это животное рассматривалось как наиболее соответствующая модель NPD - тип A (смотрите, Buccinna et al., 2009, J. Neurochem. 109:105-115).
Дополнительные исследования подтвердили, что дозы rhASM раз в две недели снижали уровни сфингомиелина у ASMKO мышей зависимым от дозы образом (0,3-3 мг/кг). Уровни, при которых не наблюдались нежелательные эффекты (NOAEL) для однократной и повторной дозировки, были определены как 0,3 и 3 мг/кг, соответственно, у ASMKO мышей. Последующие попытки сократить уровни сфингомиелина в легком с более высокими дозами rhASM привели к неожиданной токсичности. При дозах ≥10 мг/кг ASMKO мыши, но не нормальные животные, проявляли воспаление печени, адреналовую геморрагию, сердечно-сосудистый шок и смерть в сопровождении сильно повышенных уровней цитокина, подтверждающих синдром выброса цитокинов. Токсичность и повышение цитокинов, наблюдаемые с высокими дозами rhASM, должны быть облегчены или предупреждены с помощью предварительного лечения ASMKO мышей некоторыми более низкими дозами rhASM, подтверждая, что скорость и количество распада сфингомиелина играет ключевую роль.
У ASMKO мыши, модели для NPD - типа A, токсичность не наблюдалась после однократных доз ≤0,3 мг/кг rhASM и повторных дозы ≤3,0 мг/кг rhASM; тяжелая токсичность не наблюдалась, пока не вводили однократные дозы ≥10 мг/кг rhASM. Следовательно, консервативная начальная доза 0,03 мг/кг rhASM была выбрана для лечения однократной дозой субъектов-людей для обеспечения 10-кратного предела безопасности по отношению к NOAEL однократной дозы (0,3 мг/кг), наблюдаемому у ASMKO мышей. Максимальная доза 1,0 мг/кг rhASM была выбрана для лечения однократной дозой субъектов-людей для обеспечения 10-кратного предела безопасности по отношению к дозе, при которой серьезная токсичность наблюдалась у ASMKO мыши (10 мг/кг). При выполнении протокола, изложенного ниже, токсичность у субъектов-людей неожиданно наблюдалась с дозой до 0,3 мг/кг rhASM.
6.2. МАТЕРИАЛЫ И СПОСОБЫ
6.2.1. СХЕМА ПРОТОКОЛА ДЛЯ ЧЕЛОВЕКА
Этот протокол представлял собой одноцентровое исследование фазы 1 с однократной дозой, с увеличением дозы. Главными целями исследования были оценить безопасность и фармакокинетику однократных доз rhASM у взрослых людей с не нейронопатической ASMD (Ниманна-Пика B). Однократные дозы 0,03, 0,1, 0,3, 0,6 и 1,0 мг/кг rhASM были введены инфузией последовательно когорте доз. Исходная схема исследования требовала минимум 15 пациентов (5 когорт по 3 пациента каждая). Из-за сложностей с включением пациентов, протокол был изменен так, чтобы первые 2 когорты включали 3 пациента каждая и последние 3 когорты включали 2 пациента каждая. Независимый комитет отслеживания данных наблюдал за проведением исследования, и все процедуры протокола были подтверждены IRB. Фиг.1 отображает поток пациентов протокола.
6.2.2. ПАЦИЕНТЫ
Чтобы соответствовать протоколу, пациенты должны были иметь возраст 18-65 лет и иметь недостаточную ASM ферментную активность, объем селезенки ≥2× от нормального, AST и ALT≤250 МЕ/л, билирубин ≤3,6 мг/дл, INR≤1,5, DLCO>30% расчетной, и тромбоциты ≥60000/мл. Пациенты исключались, если они имели цирроз (по биопсии печени), существенное заболевание сердца, общую спленэктомию или принимали медикаменты или растительные добавки, которые были потенциально гепатотоксическими, стимулировали кровотечение или ингибировали rhASM.
Было включено общее количество 13 пациентов, и 11 пациентам вводили инфузией rhASM. rhASM была получена путем сверхэкспрессии ASM кДНК в клетках яичника китайского хомяка. Средний возраст пациентов, которые подвергались инфузии, составлял 30,8 лет, все были представителями белой европеоидной расы (не испанцами/не латиноамериканцами), и средний объем селезенки составлял в 10,8 больше от нормального. Один имел частичную спленэктомию; остальные пациенты имели интактные селезенки в начале протокола. Фиг.2 отображает демографию и исходные характеристики пациентов в этом протоколе.
Как только скрининг был завершен и было подтверждено соответствие требованиям, пациенты были приняты в кардиологическое отделение (CCU) на ночь для исходной телеметрии и проведения инфузии следующим утром с rhASM. Пациенты были подвергнуты мониторингу в течение 72 часов после введения дозы на телеметрии (24 часов в CCU и 48 часов в Центре общих клинических исследований). На 14 день пациентов возвращали и оставляли на ночь и на 28 день оставляли для амбулаторного посещения.
Далее представлены наборы медицинских оценок, которые были получены в ходе выполнения протокола:
- Физическое исследование - дни 0, 1, 2, 14 и 28
- Химические исследования, гематология и анализ мочи - перед инфузией, затем 24, 48, 72 часа; дни 14, 28
- Исследования функции печени - перед инфузией, затем q12 часов по 72 часа, дни 14, 28, альдостерон, кортизол, дельта-4-андростенедион - перед инфузией, затем q12 часов по 72 часа
- Исследование стимуляции ACTH - скрининг, день 14 телеметрия - непрерывная по 72 часа ЭКГ, эхокардиограмма - перед инфузией, в конце инфузии и после инфузии на 1, 2, 6, 12 и 4 часа, день 14
- Сердечные биомаркеры (BNP, сердечный тропонин-I, CPK-MB) - перед инфузией, затем 2, 6, 12, 24 часа
- Уровни сфингомиелина, церамида в плазме - перед инфузией, затем 24, 48, 72 часа; дни 14 и 28
- Исследование легочной функции и CXR - скрининг, день 14
- Объем печени и селезенки с помощью МРТ - скрининг, день 14
- Биопсии печени и кожи - скрининг, день 14
- Биомаркеры - предварительная доза, дни 14, 28
- Исследование анти-rhASM IgG - перед инфузией, день 28
- Фармакокинетика - в пределах 30 мин перед инфузией, 15 мин после начала инфузии, в конце инфузии и со следующими временными интервалами после инфузии: 15 мин, 30 мин, 45 мин; 1 час, 2, 3, 4, 6, 8, 12, 18, 24, 48, 72 часа.
- Цитокины (IL-1a, IL-1b, IL-6, G-CSF, GM-CSF, MIP-1a, TNF-a) - фармакокинетические временные точки (выше) плюс день 14
6.3. РЕЗУЛЬТАТЫ
Как изображено на фиг.3A и 3B, уровни церамида плазмы и уровни сфингомиелина плазмы, соответственно, были определены при нескольких временных точках. Уровни церамида плазмы показали дозозависимое повышение к 6 часам и достигли пика в 18-72 часа. Уровни сфингомиелина плазмы были нормальными в исходном состоянии и не показали существенного изменения с течением времени. У пациента № 12112, который получил самую высокую дозу (1 мг/кг), уровень сфингомиелина плазмы вырос после введения дозы и достиг пика в 72 часа.
Фиг.4 изображает уровни общего билирубина, определенные за несколько временных точек в ходе протокола. Общий билирубин показал связанное с дозой возрастание за 24 часа и достиг пика в 48-60 часов. Самый высокий общий билирубин был 4,7 мг/дл у пациента № 12112, который получил самую высокую дозу (1 мг/кг). Существовали пропорциональные увеличения прямого и непрямого билирубина. Увеличения не наблюдались в ALT, AST или щелочной фосфатазе. Было легкое увеличение в GGT по 72 часа (не показано) у двух пациентов, которые получили самую высокую (1 мг/кг, пациент № 12112) и самую низкую (0,03 мг/кг, пациент № 10503) дозы. Уровни гемоглобина и гематокрита были стабильными в течение протокола, указывая на то, что гемолиз не был ответственным за повышенные уровни билирубина.
Фиг.5A-5G отображают уровни маркеров острофазового ответа (воспаления), CRP/hs-CRP, % нейтрофилов, фибриноген, ферритин, IL-8, IL-6 и кальцитонин, соответственно, определенные в определенные временные точки для нескольких пациентов. CRP/hs-CRP показал временное связанное с дозой повышение за 24 часа, с пиком в 48-72 часа, и возвратился в норму на день 14. Другие острофазовые агенты также показали увеличения (% нейтрофилов, фибриноген, ферритин, PT и PTT) и уменьшения (железо, альбумин). Определенные биомаркеры воспаления, например цитокины IL-6 и IL-8, и кальцитонин, также показали существенное повышение, которое было дозозависимым и достигало пика к 48 часам после инфузии. Среди этих медиаторов воспаления самые большие изменения в нисходящем порядке произошли у IL-8 (пик 33,8× верхней границы нормы), кальцитонина (пик 33,4× верхней границы нормы), CRP (пик 9,8× верхней границы нормы) и ферритина (3,9× верхней границы нормы). Не было изменения в количестве тромбоцитов или уровне продуктов деградации фибрина (не показано). Повышения лабораторных острофазовых агентов коррелировало с системными клиническими симптомами лихорадки, тошноты, рвоты, головной боли, боли и миалгий у некоторых пациентов.
Фиг.6 представляет собой схему лечения возникших после начала лечения нежелательных явлений для четырех пациентов каждая на различную дозу rhASM, где явления рассматривались как связанные (возможно, вероятно или точно) с лечением. Как представлено на схеме, пациент № 11509, который получал дозу 0,3 мг/кг rhASM, проявлял среднетяжелые нежелательные явления, начиная на день 2.
В отношении связанных нежелательных явлений, о которых сообщалось в протоколе, не наблюдалось значительных сердечно-сосудистых изменений с помощью телеметрии, ЭКГ, эхокардиограммы или биомаркеров (BNP, сердечный тропонин-I, CPK-MB) или адреналовой гормональной дисфункции. Один пациент (№ 12112, 1,0 мг/кг когорта) имел повышенный уровень кортизола утром через 24 часа после введения дозы, что, вероятнее всего, представляло собой нормальный физиологический стрессовый ответ на некоторые происходящие умеренные связанные нежелательные явления. Дополнительно, четырех из шести пациентов, получающих ≥0,3 мг/кг rhASM, испытывали общим количеством 19 клинических и лабораторных связанных нежелательных явлений, оцененных как связанные с лекарственным средством. Интенсивность связанных нежелательных явлений была в диапазоне от легкой до тяжелой, но большинство были умеренными и не нуждались в каком-либо вмешательстве. Они включали следующее: лихорадка (n=2), боль [миалгия; боль в животе, голени и бедре] (n=2), тошнота (n=2), иктеричность склер и уробилиноген мочи (n=1), утомляемость (n=1), рвота (n=1), лимфацитарный инфильтрат/гепатоклеточная деградация по биопсии печени (n=1), острофазовая реакция (n=4), повышенный билирубин (n=2) и увеличенный D-димер фибрина (n=1). Начало клинических симптомов начиналось через 12 часов после инфузии и прекращалось за 72 часа, кроме боли в бедре у одного пациента, которая началась через 72 часа. На день 14 биопсия печени у одного пациента (0,6 мг/кг rhASM, пациент № 12313) показала два новых очага лимфоцитарных инфильтратов: один был очень маленьким (0,1 мм в диаметре), а другой был средним (0,5 мм в диаметре), и что было связано с гепатоклеточной деградацией. Этот протокол был остановлен, когда первый пациент в когорте 5 (1 мг/кг rhASM, пациент № 12112) испытал ограниченную дозой токсичность гипербилирубинемии (пик 4,7 мг/дл).
Результаты этого показали неожиданное и отсроченное начало связанных с дозой клинических и лабораторных нежелательных явлений у людей, страдающих от ASMD при дозах rhASM 0,3 мг/кг и более высоких. Дополнительно, два основных наблюдения лабораторной безопасности были произведены в виду результатов этого протокола. Один касающийся гипербилирубинемии, где пропорционально дозе рост прямого и непрямого билирубина наблюдался без существенных маркеров повреждения печени (AST, ALT, AP) или случая гемолиза (гемоглобин и гематокрит). Два пациента имели слабо повышенный GGT в 48-72 часа, и один пациент имел два новых очага в печени лимфоцитарных инфильтратов, один из которых был связан с гепатоклеточной деградацией.
Другое наблюдение касалось острофазового ответа, т.е. воспаления. Результаты показали увеличение следующего: CRP/hs-CRP, % нейтрофилов, ферритина, IL-6, IL-8, кальцитонина, фибриногена, PT, PTT и уменьшение железа и альбумина в ответ на повышение дозировок rhASM. Системные клинические симптомы лихорадки, тошноты, рвоты, боли и миалгии были, вероятнее всего, связаны с острофазовым ответом. Результаты не показали доказательства синдрома выброса цитокинов, связанного с сердечнососудистыми изменениями, и доказательства адреналовой гормональной дисфункции.
6.4. ВЫВОДЫ
Описанный выше протокол представляет собой первый опыт с ферментной заместительной терапией у взрослых людей-пациентов с ASMD. При биоактивных дозах у людей rhASM не вызывала синдром выброса цитокинов, связанный с сердечно-сосудистыми изменениями, или вызывала адреналовую гормональную дисфункцию. Основные наблюдения по безопасности были связанными с дозой гипербилирубинемией и острофазовым ответом. Оба нежелательных явления были, вероятно, связаны с разрушением сфингомиелина до церамида и фосфилхолина, но точные молекулярные механизмы являются не полностью понятными. Некоторые биомаркеры безопасности были определены, включая билирубин, церамид, CRP/hs-CRP, 1L-8, 1L-6, кальцитонин и ферритин. Дополнительно, на основании обнаруженных фактов гипербилирубинемии максимальная переносимая начальная доза rhASM составила 0,6 мг/кг.
Более того, факт возникновения нежелательных явлений с клиническими симптомами у пациентов, которые наблюдались при дозе до 0,3 мг/кг, был неожиданным, учитывая, что NOAEL («уровень, при котором не наблюдаются нежелательные эффекты») у ASMKO мышей составлял 0,3 мг/кг. Причем клинические симптомы токсичности у ASMKO мышей не наблюдались, пока не использовали дозы больше чем или равные 10 мг/кг.
7. ПРИМЕР 2. ПРОТОКОЛ ТОКСИЧНОСТИ ВНУТРИВЕННОЙ ИНЪЕКЦИИ ПОВТОРНОЙ дозЫ ПОСЛЕ ФАЗЫ СОКРАЩЕНИЯ МАССЫ СФИНГОМИЕЛИНА У НОКАУТНЫХ ПО КИСЛОЙ СФИНГОМИЕЛИНАЗЕ МЫШЕЙ
Этот пример описывает исследование потенциальной токсичности повторного внутривенного введения рекомбинантной кислой сфингомиелиназы человека (rhASM) после фазы сокращения массы сфингломиелина у нокаутных по кислой сфингомиелиназе (ASMKO) мышей. Установив, что токсические побочные эффекты становились видимыми для наблюдения при начальных дозах 10 мг/кг rhASM у ASMKO мышей, последующее исследование было разработано, чтобы определить, будет ли введение растущих доз рекомбинантной кислой сфингомиелиназы человека (rhASM) у ASMKO мышей достаточно сокращать массу накопленного сфингомиелина так, чтобы более высокая доза rhASM могла быть введена ASMKO мышам для направленности в места патологии, такие как легкое и мозг, с минимальной или не наблюдаемой токсичностью.
JK ASMKO мышам вводили 3 мг/кг rhASM на дни исследования (SD) 1, 3, 5, 7 (фаза сокращения массы сфингомиелина). Начиная с SD 9 и продолжая раз в две недели в течение 13 недель (7 доз) мыши получали дозы лечения 3, 10 или 30 мг/кг rhASM (фаза лечения).
rhASM внутривенно вводили посредством болюсной инъекции в латеральную хвостовую вену самцу и самке ASMKO мышей. Группы 1-3 получали 3 мг/кг rhASM в ходе фазы сокращения массы сфингомиелина (SD 1, 3, 5 и 7). Группы 1, 2 и 3 получали 3, 10 и 30 мг/кг rhASM, соответственно, в ходе фазы лечения (SD 9, 23, 37, 51, 65, 79 и 93). У одной подгруппы мышей в каждой группе (первые 2 мыши/пол в группе 1 и первые 4 мыши/пол в группах 2-3) брали кровь перед исследованием, а также через 5 минут и 4 часов после доз 1, 4, и 7 фазы лечения для анализа на токсикокинетику (TK) (5 минут) и уровни церамида (4 часа). У другой подгруппы мышей в каждой группе (последние 2 мыши/пол в группе 1 и последние 4 мыши/пол в группах 2-3) брали кровь перед исследованием, а также через 4 и 24 часа после доз 1, 4 и 7 фазы лечения для анализа многоаналитных профилей грызунов (4 часа) и уровней белка острой фазы/пробы функции печени (24 часа). Смотрите схему исследования в таблице 2, ниже.
Из-за вероятности анафилактических реакций, начинающихся с первой дозой фазы лечения и каждой дозой после этого, каждая мышь находилась под строгим наблюдением в отношении признаков анафилаксии (например, беспокойство, жевание, потирание морды, крапивница, эдема, летаргия и неопрятная шерсть) после введения испытуемого препарата. Мыши получали дифенилгидрамин (DPH), начиная перед первой дозой фазы лечения и каждой дозой после этого.
DPH при концентрации 5 мг/мл был получен путем разведения 50 мг/мл коммерчески доступного маточного раствора стерильным 0,9% солевым раствором. DPH вводили интраперитонеально (IP) в дозе 20 мг/кг (4 мл/кг) на основании самого последнего веса тела, за 10-20 минут до введения дозы rhASM для профилактики возможных анафилактических реакций. Если любое животное демонстрировало реакцию гиперчувствительности после введения исследуемого препарата, несмотря на предварительное лечение DPH, то вторую дозу DPH вводили IP в дозе 10 мг/кг (2 мл/кг).
Всех мышей умертвили посредством асфиксии CO2 на SD 100. После эвтаназии кровь собирали для анализа на клиническую патологию и иммуногенность через пункцию сердца. Вскрытие проводили на всех мышах после сбора крови. Все ткани консервировали в 10% нейтрального забуференного формалина (NBF) с последующим гистопатологическим анализом. Кусочек печени, селезенки, почки и легкого консервировали в 2% глютаральдегида/2% параформальдегида для анализа сфингомиелиновой нагрузки. Кусочек печени, селезенки, почки и легкого также собирали и хранили при ≤-70°C для возможного будущего анализа.
После вскрытия все оставшиеся ткани, включая тушку, помещали в 10% NBF.
Схема исследования
Наблюдения при жизни
Первый день исследования рассматривался как SD1 (первый день введения дозы). Животным измеряли вес тела один раз каждую неделю во время курса исследования на SD-1 и по понедельникам каждой недели после этого. Наблюдения за клетками проводили один раз в день с понедельника по пятницу. Любые отклонения и наблюдения нормы записывали. Клинические наблюдения после введения дозы/присвоения баллов проводили непосредственно до, через 10-20 и 50-70 минут после каждого введения дозы. Любые отклонения и наблюдения нормы записывали. Лечащий ветеринар и (или) директор исследования проводил консультацию в случае нежелательных реакций, и соответствующие действия предпринимались на основании их рекомендаций.
Эвтаназия
Если была нежелательная реакция, которая поражала здоровье и благополучие животного, то животному проводили эвтаназию посредством асфиксии CO2, вскрывая через грудную, брюшную и краниальную полости и помещая в 10% нейтрального забуференного формалина (NBF) для возможного дальнейшего анализа. Если животное было обнаружено мертвым, то целую тушку консервировали в 10% NBF для возможного дальнейшего анализа. В конце исследования всех выживших животных подвергали эвтаназии с помощью асфиксии CO2.
Сбор образцов
Анализ дозы: Приблизительно пятьсот микролитров (500 мкл) исследуемого препарата из всех уровней доз собирали в пределах 1-10 минут после состава на каждый день дозировки и хранили на сухом льду до переноса в морозильную камеру, установленную для поддержания температуры ≤-70°C. Анализируемые образцы доз переносили на SD 1, 3, 5, 7, 9, 23, 37, 51, 65, 79 и 93 и хранили при ≤-70°C до анализа. Анализируемые образцы доз измеряли посредством анализа A280.
Сбор крови: Для сбора крови для анализа TK, многоаналитных профилей грызунов и уровней белка острой фазы/пробы функции печени мышам вводили анестезию со смесью изофлурана и кислорода. Сборы крови проходили на SD -1, 9, 51 и 93 (перед испытанием и на 1-ю, 4-ю и 7-ю дозы фазы лечения, соответственно) согласно таблицам сбора крови (смотрите таблицы 4 и 5, ниже) и следующему тексту.
Токсикокинетика rhASM: У первых 2 мышей/пол в группе 1 и первых 4 мышей/пол в группах 2-3 брали образец крови для анализа максимальных rhASM уровней перед исследованием (SD -1) и через 5 минут после 1-й, 4-й и 7-й доз фазы лечения. Кровь из всех животных собирали из ретро-орбитального сплетения у мышей без сознания. Приблизительно 60 мкл цельной крови собирали в гематокритные пробирки и давали свернуться при комнатной температуре в течение по меньшей мере 1 минуты. Сыворотку получали из этих образцов путем центрифугирования в течение 5 минут при 10000 оборотов в минуту (RPM). После центрифугирования сыворотку собирали и хранили на сухом льду до переноса в морозильную камеру, выставленную для поддержания температуры ≤-70°C. После переноса все образцы хранили при ≤-70°C до анализа. TK образцы переносили на SD 1, 9, 51 и 93. TK образцы измеряли с помощью фермент-связанного иммуносорбентного анализа (ELISA).
Анализ уровней церамида: У первых 2 мышей/пол в группе 1 и первых 4 мышей/пол в группах 2-3 брали образец крови для анализа уровней церамида перед исследованием (SD -1) и через 4 часа после 1-й, 4-й и 7-й доз фазы лечения. Кровь из всех животных собирали из ретро-орбитального сплетения у мышей без сознания. Приблизительно 240 мкл цельной крови собирали в пробирки с EDTA калия и помещали на качалку Nutator в течение до 30 минут для предотвращения образования сгустка. Плазму получали из этих образцы с помощью центрифугирования в течение 5 минут при 10000 об/мин. После центрифугирования плазму собирали и хранили на сухом льду до переноса в морозильную камеру, установленную для поддержания температуры ≤-70°C. Как только перенесли, все образцы хранили при ≤-70°C до анализа с помощью масс-спектрометрии. Образцы церамида переносили не позже, чем SD 94. Образцы церамида измеряли с помощью масс-спектрометрии.
Анализ многоаналитных профилей грызунов (MAP): У последних 2 мышей/пол в группе 1 и последних 4 мышей/пол в группах 2-3 брали образец крови для анализа многоаналитных профилей грызунов перед исследованием (SD -1) и через 4 часа после 1-й, 4-й и 7-й доз фазы лечения. Кровь из всех животных собирали из ретро-орбитального сплетения у мышей без сознания. Приблизительно 150 мкл цельной крови собирали в пробирках препаративной центрифуги для сыворотки и давали образоваться сгустку в течение по меньшей мере 30 минут. Сыворотку получали из этих образцов с помощью центрифугирования в течение 5 минут при 10000 об/мин. После центрифугирования сыворотку собирали и хранили на сухом льду до переноса в морозильную камеру, установленную для поддержания температуры ≤-70°C. Как только перенесли, все образцы хранили при ≤-70°C до транспортировки не позднее чем на SD 94 для анализа многоаналитных профилей грызунов. Образцы транспортировали в Rules Based Medicine на сухом льду. Таблица многоаналитных профилей грызунов (смотрите таблицу 3, ниже) перечисляет аналиты, которые были измерены.
Многоаналитные профили грызунов
Анализ уровней белка острой фазы/пробы на функцию печени: У последних 2 мышей/пол в группе 1 и последних 4 мышей/пол в группах 2-3 брали образец крови для анализа уровней белка острой фазы (сывороточный амилоид-A и сывороточный амилоид-P) и функции печени (билирубин и аланин аминотрансфераза (ALT)) перед исследованием (SD -1) и через 24 часа после 1-й, 4-й и 7-й доз фазы лечения. Кровь из всех животных собирали из ретро-орбитального сплетения у мышей без сознания. Приблизительно 150 мкл цельной крови собирали в пробирки препаративной центрифуги для сыворотки и давали образоваться сгустку в течение по меньшей мере 30 минут. Сыворотку получали из этих образцов с помощью центрифугирования в течение 5 минут при 10000 об/мин. После центрифугирования сыворотку собирали и хранили на сухом льду до переноса в морозильную камеру, установленную для поддержания температуры ≤-70°C. Как только перенесли, все образцы хранили при ≤-70°C до транспортировки не позднее чем на SD 94 для анализа белка острой фазы/пробы на функцию печени. Образцы транспортировали в Analytics на сухом льду.
Таблица сбора крови перед исследованием (SD -1)
Таблица сбора крови на SD 9, 51 и 93
Вскрытие
Конечный сбор крови для анализа клинической патологии и иммуногенности: После эвтаназии всем животным делали сердечную пункцию, проводимую для сбора цельной крови (приблизительно ≥500 мкл) из всех основных животных исследования для анализа клинической патологии и иммуногенности. Приблизительно 150 мкл цельной крови помещали в пробирки с EDTA калия, и аккуратно переворачивали для анализа гематологических параметров. После аккуратного переворачивания все образцы хранили при комнатной температуре, раскачивая на качалке Nutator, пока образцы не сохраняли при 2-10°C до анализа. Оставшуюся кровь (из забора через сердечную пункцию после удаления 150 мкл для гематологического анализа) помещали в пробирку препаративной центрифуги для сыворотки, давали образоваться сгустку при комнатной температуре в течение по меньшей мере 30 минут, откручивали на центрифуге в течение 5 минут при 10000 об/мин и собирали сыворотку. Приблизительно 30 мкл сыворотки помещали в пробирку Эппендорфа для анализа иммуногенности, тогда как оставшуюся сыворотку помещали в пробирку Эппендорфа для клинического химического анализа. Образцы на иммуногенность хранили на сухом льду, пока их не сохраняли в морозильной камере, установленной для поддержания температуры ≤-70°C. Образцы на иммуногенность измеряли с помощью ELISA. Образцы для клинического химического анализа помещали на сухой лед, пока все образцы не сохраняли при ≤-20°C до анализа. В таблице гематологических аналитов и клинических химических аналитов (смотрите таблицу 6, ниже) перечисляются аналиты, которые были измерены.
Таблица гематологических и клинических химических аналитов
Результаты из анализа гематологии и анализов клинической химии из всех животных были интерпретированы патоморфологом, получившим профессиональную сертификацию.
Сбор ткани: После эвтаназии на SD 100 все животные подвергались вскрытию для сбора тканей. Вскрытие включало изучение внешних признаков тушек, внешних отверстий тела, брюшной и грудной полостей, органов и тканей. Макроскопические наблюдения записывались. В таблице сбора тканей (смотрите таблицу 7, ниже) перечисляются ткани, которые были собраны в 10% NBF. Макроскопические повреждения также собирали и хранили в 10% NBF. Оставшиеся тушки консервировали в 10% NBF.
Таблица сбора тканей
Выделенные жирным шрифтом образцы будут взвешены
Массы органов: Во время вскрытия вышеперечисленные выделенные жирным шрифтом органы были взвешены; парные органы были взвешены вместе. Массы записывали в форму масс органов. Процентные соотношения массы органа к массе тела и массы органа к массе мозга рассчитывали на основании масс тела, взятых до вскрытия.
Гистопатология: Образцы от всех животных, а также макроскопических повреждений от любого животного в исследовании передавали на гистологию. Законсервированные ткани, перечисленные выше, из каждого животного погружали в парафин. Все ткани разрезали, окрашивали гематоксилином и эозином и исследовали микроскопически. Все срезы тканей исследовали микроскопически для оценки гистологических изменений патоморфологом, получившим профессиональную сертификацию.
Образцы печени, селезенки, почки и легкого из всех животных собирали и фиксировали в 2% глютаральдегиде/2% параформальдегиде для анализа сфингомиелиновой нагрузки. Затем эти ткани фиксировали в дихромате калия/тетроксиде осмия и погружали в Epon. Срезы по одному микрону окрашивали дубильной кислотой и толуидином синим для исследования с помощью световой микроскопии. Сфингомиелиновую нагрузку в каждом образце оценивали количественно с помощью компьютерной морфометрии с программным обеспечением MetaMorph патоморфологом, получившим профессиональную сертификацию.
Образцы печени, селезенки, почки и легкого от всех животных собирали, замораживали в жидком азоте и хранили замороженными при ≤-70°C для возможного будущего анализа.
Анализ образцов
Анализ дозы измеряли посредством A280 анализа и токсикокинетики, а образцы на иммуногенность были анализированы с помощью ELISA.
Анализ на церамид измеряли посредством масс-спектрометрии.
Гематологические образцы и образцы для клинической патологии измеряли с использованием гематологического анализатора Sysmex XTV 2000 IV и клинического химического анализатора Radnox Daytona, соответственно.
Образцы тканей обрабатывались, а гистопатологический анализ интерпретировался патоморфологом, получившим профессиональную сертификацию согласно SOP. Результаты анализа клинической патологии интерпретировались патоморфологом, получившим профессиональную сертификацию согласно SOP.
8. ПРИМЕР 3. ФЕРМЕНТНАЯ ЗАМЕСТИТЕЛЬНАЯ ТЕРАПИЯ С РАСТУЩЕЙ дозОЙ РЕКОМБИНАНТНОЙ КИСЛОЙ СФИНГОМИЕЛИНАЗОЙ ЧЕЛОВЕКА (rhASM)
8.1. ВВЕДЕНИЕ
Протокол, описанный ниже, использует увеличение дозы у одного пациента как факультативное средство для достижения более высоких повторных доз rhASM. Оценивают безопасность, эффективность и фармакокинетику (PK) rhASM инфузий в дозах 0,3, 0,6 и 1,0 мг/кг, вводимых каждые 2 недели (q2w) в течение 40 недель, а также долгосрочную безопасность и эффективность rhASM инфузий.
8.2. МАТЕРИАЛЫ и СПОСОБЫ
8.2.1.СХЕМА ПРОТОКОЛА
12 пациентов с NP-типа B, подлежащих лечению, принадлежат к любому полу. Пациенты будут получать по меньшей мере 1 дозу 0,1 мг/кг rhASM изначально. Пациентам, которые переносят 0,1 мг/кг rhASM инфузию, затем вводят растущие дозы 0,3 мг/кг, 0,6 мг/кг и 1 мг/кг каждые 2 недели, по мере переносимости. Пациентов, которые переносят 1,0 мг/кг, затем стратифицируют по объему селезенки (в <12 раз или ≥12 раз больше нормального) и рандомизируют для получения одной из 2 целевых доз: 1,0 мг/кг rhASM или 3,0 мг/кг; пациентам, рандомизированным в группу 3,0 мг/кг, будут повышать до 2,0 мг/кг и затем до 3,0 мг/кг, по мере переносимости. Пациентов, которые не переносят 0,1 мг/кг дозу rhASM, заменяют. Пациентов, которые не могут переносить увеличение до более высоких доз, будут оставлять на максимальной переносимой ими дозе в течение 26-недельного периода поддерживающей дозы.
У всех пациентов дозы будут повышены от 0,1 мг/кг до их целевой дозы. Корректировки дозировки во время периода увеличения будут происходить с интервалами каждые 2 недели, пока не будет достигнута целевая доза (при переносимости). После начальной инфузии (0,1 мг/кг rhASM), а также последующих доз последствия для пациентов, т.е. возникновение нежелательного явления (AE)/тяжесть и сывороточный билирубин, hsCRP и другие белки острой фазы, и церамид, будут рассмотрены. Следующие критерии будут определять следующую дозу, вводимую во время периода увеличения дозы:
1. величина общего билирубина ≤2,0 мг/дл или легкое AE → повышать до следующей дозы (если необходимо);
2. величина общего билирубина =2,1-3,0 мг/дл или умеренное AE → нет увеличения дозировки; повторить текущую дозу;
3. величина общего билирубина >3,0 мг/дл) или тяжелое AE → уменьшение дозы до предыдущей вводимой/переносимой.
Любое серьезное нежелательное явление (SAE), которое, по оценкам, связано с rhASM, значение билирубина, которое не уменьшается до уровня <2,0 мг/дл до следующей запланированной дозы, или любое AE, которое становится проблемой, касающейся безопасности rhASM при вводимой дозе, может быть рассмотрено как ограничивающая дозу токсичность (DLT). Если пациент испытывает DLT, то последующие дозы для пациента должны быть временно приостановлены. Пациентам могут провести пробу с повторным введением дозы для получения дозы, которая приводит к DLT, и при переносимости лечение будет протекать так, как планировалось изначально.
Если пациент не может переносить 2 дозы по 0,1 мг/кг (т.е. начальная инфузия и одна доза для пробы с повторным введением), они не будут получать лечение с растущими дозами, не смогут продолжать лечение и будут заменены в исследовании. Пациенты, которые переносят 0,3 мг/кг дозу, будут получать начальную инфузию rhASM (0,1 мг/кг) и после 2 недель - 0,3 мг/кг rhASM (при условии, что соблюдаются критерии увеличения дозы после дозы 0,1 мг/кг). Последующие дозы rhASM будут вводиться во время периода увеличения дозы q2w в течение максимум 18 недель. Если пациенту успешно увеличили дозу до 0,3 мг/кг и он, следовательно, соответствует критериям № 2 или № 3 (выше), то пациент может быть подвергнут пробе с повторным введением дозы дважды. Если проба с повторным введением дозы является неудачной (т.е. целевая доза не может быть достигнута), то пациент будет продолжать получать rhASM в дозе 0,1 мг/кг в течение оставшегося 40-недельного периода лечения.
Пациенты, подлежащие лечению с режимом дозы 0,6 мг/кг, будут получать начальную инфузию rhASM (0,1 мг/кг) с последующей 1 дозой 0,3 мг/кг rhASM, с последующей дозой 0,6 мг/кг rhASM q2w в оставшийся 40-недельный период лечения (при условии что предыдущие инфузии rhASM 0,1 мг/кг и 0,3 мг/кг хорошо переносятся). Пациенты, подлежащие лечению с режимом 1,0 мг/кг, будут получать начальную инфузию rhASM (0,1 мг/кг) с последующей 1 дозой 0,3 мг/кг rhASM, с последующей 1 дозой 0,6 мг/кг rhASM, с последующей 1,0 мг/кг rhASM q2w в оставшийся 40-недельный период лечения (при условии, что предыдущие rhASM инфузии 0,1 мг/кг, 0,3 мг/кг и 0,6 мг/кг хорошо переносятся). Если пациенту успешно повысили дозу от 0,3 до 0,6 мг/кг или от 0,6 до 1,0 мг/кг и он, следовательно, соответствует критериям № 2 или № 3 (выше), пациент может быть подвергнут пробе с повторным введением дважды. Если проба с повторным введением дозы является неудачной (т.е. целевая доза не может быть достигнута), то пациент будет продолжать получать более низкую переносимую дозу в течение оставшегося периода 40-недельного лечения.
После фазы 40-недельного лечения пациентам может быть позволено продолжать лечение на уровне их поддерживающей дозы.
8.2.2. ПАЦИЕНТЫ
Каждый пациент должен соответствовать следующим критериям включения, чтобы получать лечение в соответствии с этими режимами:
1. документированная ASM недостаточность, соответствующая болезни Ниманна-Пика (N-P);
2. диффузионная емкость легких (DLCO) >20% и ≤80% от расчетного нормального значения;
3. объем селезенки ≥8 раз от нормального (≥1,6% веса тела);
4. пациенты-женщины с детородным потенциалом должны иметь негативный сывороточный тест на беременность для β-hCG и быть согласны применять медицинский приемлемый противозачаточный метод в течение длительности протокола.
8.2.3. ЛЕЧЕНИЕ
Пациенты будут получать однократную дозу 0,1 мг/кг rhASM IV в течение приблизительно 35-минутного периода времени. Руководства для введения приведены в таблице 8, ниже. Пациентам, которые переносят начальную 0,1 мг/кг rhASM инфузию, будут увеличивать дозу до целевой дозы лечения (rhASM 0,3, 0,6 или 1,0 мг/кг). Все пациенты будут получать дозы, повышенные от 0,1 мг/кг до их целевой дозы. Корректировки дозировки во время периода увеличения будут происходить с интервалами каждые 2 недели, пока не будет достигнута целевая доза.
Введение rhASM
Этап 2: 60 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 50 мл/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 100 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 50 мл/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 100 мл/час за 20 мин (+/-5 мин), если нет IAR
Этап 4: 167 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (+/-5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 30 мл/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 60 мл/час за 20 мин (+/-5 мин), если нет IAR
Этап 4: 100 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (+/-5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (+/-5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Каждая степень спленомегалии пациента от исходного уровня может быть записана в разах от нормы (× нормы) в качестве индикатора тяжести заболевания. Нормальный объем селезенки равен 0,2% веса тела.
При каждом посещении пациенты должны оцениваться в отношении новых или продолжающихся нежелательных явлений (AE).
Клинические предельные значения могут быть измерены как % изменение от исходного уровня к 26 неделе периода поддерживающей дозы. Предельное значение первичной эффективности - это уменьшение объема селезенки, что измеряется с помощью МРТ. Предельные значения вторичной эффективности включают следующее: уменьшение уровня сфингомиелина печени; увеличение способности переносить физическую нагрузку, что определяется с помощью % расчетной максимальной рабочей нагрузки с помощью велоэргометрии; увеличенную легочную функцию как % расчетной DLCO; увеличенное очищение легких, что может быть определено по количеству клеток и профилю бронхоальвеолярного лаважа (BAL), уровни сфингомиелина, церамида, цитокина и хитотриозидазы. Предельные значения третичной эффективности могут включать следующее: уменьшение объема печени, что измеряется с помощью МРТ; увеличенную легочную функцию, что может быть определено по % расчетной FVC, FEV1, TLC; улучшенный внешний вид легкого, определенный по изображению КТ высокого разрешения, рентгенографии грудной клетки; улучшенный липидный профиль, что определяется по уровням ЛВП, ЛНП, общего холестерина, триглицеридов и соотношению общего холестерина к ЛВП холестерину; улучшенное количество тромбоцитов; гемоглобин; уменьшенные уровни сфингомиелина в коже, плазме, DBS; и улучшение других биомаркеров, таких как CCL18, ACE.
9. ПРИМЕР 4: ФЕРМЕНТНАЯ ЗАМЕСТИТЕЛЬНАЯ ТЕРАПИЯ С РАСТУЩЕЙ дозОЙ РЕКОМБИНАНТНОЙ КИСЛОЙ СФИНГОМИЕЛИНАЗЫ (rhASM) ЧЕЛОВЕКА
9.1 ВВЕДЕНИЕ
В протоколе, описанном ниже, используется повторная доза, сравнение доз для оценки безопасности, эффективности и фармакокинетики рекомбинантной кислой сфингомиелиназы человека (rhASM) у взрослых пациентов с ASMD. Цели протокола включают: (i) оценку безопасности увеличения дозы rhASM; (ii) оценку безопасности, эффективности и фармакокинетики максимальной переносимой или рандомизированной дозы rhASM, вводимой внутривенно каждые 2 недели в течение 26 недель; и (iii) оценку долговременной безопасности и эффективности rhASM инфузий, вводимых внутривенно каждые 2 недели от недели 26 до завершения протокола (длительностью по меньшей мере 182 недели).
9.2 МАТЕРИАЛЫ и СПОСОБЫ
9.2.1. ПАЦИЕНТЫ
Каждый пациент должен соответствовать следующим критериям включения, чтобы получать лечение в соответствии с этими режимами:
1. документированная недостаточность кислой сфингомиелиназы (ASM), соответствующая болезни Ниманна-Пика (NPD).
2. два или более характерных клинических признака, включая тромбоцитопению, анемию, нейтропению, гепатомегалию, спленомегалию и легочное заболевание, соответствующие не нейронопатической NPD.
3. диффузионная емкость легких моноксида углерода (DLCO)>20% и ≤80% от расчетного нормального значения;
4. объем селезенки ≥8 раз больше нормального (MN) (т.е. ≥1,6% веса тела); частичная спленэктомия может разрешаться, если проведена ≥1 год назад от скрининга/исходного значения, а остаточный объем селезенки составляет ≥8 MN;
5. пациенты-женщины с детородным потенциалом должны иметь негативный сывороточный тест на беременность для β-hCG и быть согласны применять медицински приемлемый противозачаточный метод в течение длительности протокола.
9.2.2. СХЕМА ПРОТОКОЛА
Пациенты, которые переносят rhASM при 0,1 мг/кг, могут быть включены в протокол. Для каждого пациента участие в протоколе может состоять из 3 периодов:
1. скрининг/исходный уровень (от -60 до -1 дней);
2. период первичного лечения (приблизительно 32 до 46 недель).
Фаза увеличения дозы (УД) (первичное УД - приблизительно 6-16 недель).
- При безопасно растущей от 0,1 до 0,3 до 0,6 до 1,0 мг/кг rhASM пациенты могут быть стратифицированы по объему селезенки (<12 и ≥12 MN) и рандомизированы либо в непрерывную дозировку 1,0 мг/кг, либо непрерывную с увеличением дозы через 2,0 и 3,0 мг/кг, а затем получают 3,0 мг/кг rhASM или максимальную переносимую дозу (вторичное УД - 4 недели) остальное время протокола.
- Нерандомизированные пациенты могут оставаться на максимальной переносимой ими дозе (<1,0 мг/кг rhASM).
Фаза поддерживающей дозы (ПД) (26 недель на максимальной переносимой или рандомизированной дозе).
3. Период долговременного лечения (по меньшей мере 182 недели при максимальной переносимой или рандомизированной дозе)
Оценки скрининга/исходного уровня могут быть завершены по меньшей мере за 24 часа до начальной инфузии rhASM. Включение в протокол обусловлено тем, что пациент способен перенести 2 дозы rhASM по 0,1 мг/кг. Изначально все соответствующие критериям пациенты могут получать однократную дозу 0,1 мг/кг rhASM в День 1 увеличения дозы (УД). Пациенты, которые не способны переносить эту дозу, будут подвергнуты пробе с повторным введением дозы через 2 недели. Пациенты, которые не могут переносить 2 дозы 0,1 мг/кг rhASM (т.е. начальная доза и доза пробы с повторным введением дозы), не могут продолжать исследование и заменяются, чтобы обеспечить включение приблизительно 12 пациентов (которые могут переносить rhASM при дозе 0,1 мг/кг).
Дозы rhASM могут повышаться каждые 2 недели от 0,1 до 0,3 до 0,6 до 1,0 мг/кг во время фазы первичного УД. Во время фазы первичного УД пациенты могут получать rhASM дозы, безопасно повышенные каждые 2 недели от 0,1 до 1,0 мг/кг (как показано на фиг.7).
Во время фаз первичного и вторичного УД следующие критерии увеличения дозы могут определять следующую дозу rhASM, которая подлежит введению:
1. Если общий билирубин (самое высокое значение до следующей запланированной дозы rhASM, и включает взятие крови перед инфузией) составляет ≤2,0 мг/дл, или отсутствие нежелательного явления/легкое связанное нежелательное явление (AE) → Повышать до следующей дозы.
2. Если общий билирубин составляет от >2,0 до <3,0 мг/дл, или умеренное связанное AE → Повторить эту же дозу.
- Если общий билирубин остается >2,0 мг/дл вплоть до следующей дозы → Временно остановить дальнейшее введение доз пациенту (потенциальная ограничивающая дозу токсичность [DLT]).
3. Если общий билирубин составляет ≥3,0 мг/дл, или тяжелое связанное AE → Уменьшить дозу.
- Если общий билирубин остается >2,0 мг/дл вплоть до следующей дозы → Временно остановить дальнейшее введение доз пациенту (потенциальная DLT).
4. Пациенты могут быть подвергнуты пробе с повторным введением дозы (на конкретном уровне дозы) столько раз, сколько необходимо, только в фазе первичного УД (проба с повторным назначением препарата не разрешается в фазе вторичного УД) до того, как пациентов рандомизируют или оставят на их максимальной переносимой дозе, к концу 16-недельной фазы первичного УД.
5. Включенные пациенты (т.е. те, которые могут переносить rhASM при дозе 0,1 мг/кг) могут быть заменены, если им не повышают дозу до 1,0 мг/кг и рандомизируют, или не могут переносить их целевую рандомизированную дозу (т.е. 1,0 или 3,0 мг/кг rhASM). Эти пациенты могут продолжать находиться в протоколе с максимальной переносимой ими дозой.
Если инфузия пропущена, то пациент может получить такую же дозу, как предыдущая инфузия. Если превышен промежуток между посещениями ±5 дней, то эта инфузия не может быть сделана, и пациент должен получить свою следующую инфузию в запланированное время для этой инфузии.
Для целей этого протокола, если любой из следующих критериев соблюдается, то результаты могут рассматриваться как указывающие на потенциальную ограничивающую дозу токсичность (DLT) для rhASM при данной дозе:
- любое серьезное нежелательное явление (SAE), подтвержденное врачом как связанное с rhASM, или
- общий билирубин остается >2,0 мг/дл вплоть до следующей дозы, или
- любое нежелательное явление (AE) которое, с точки зрения врача, создает проблему, касающуюся безопасности rhASM при вводимой дозе.
Тяжесть связанного нежелательного явления будет оцениваться врачом. Легкое связанное нежелательное явление является обычно временным и может требовать лишь минимального лечения или терапевтического вмешательства. Умеренное связанное нежелательное явление обычно облегчается с помощью дополнительного специфического терапевтического вмешательства. Это явление препятствует обычной жизнедеятельности, вызывая дискомфорт, но не имеет существенного или постоянного риска нанесения вреда субъекту. Тяжелое нежелательное явление нарушает обычную жизнедеятельность или существенно влияет на клинический статус, или может требовать интенсивного терапевтического вмешательства.
Все пациенты, которые получали повышенные дозы и безопасно переносили 1,0 мг/кг rhASM к 16 неделе, могут быть разделены на страты по объему селезенки (<12 и ≥12 MN) и рандомизированы в соотношении 1:1 по любому из следующего:
- продолжают получение 1,0 мг/кг rhASM (или максимальной переносимой дозы) каждые 2 недели в течение 26 недель; или
- продолжают увеличение дозы (УД) через 2,0 и 3,0 мг/кг, а затем получают 3,0 мг/кг rhASM (или максимальную переносимую дозу) каждые 2 недели в течение 26 недель.
Во время фазы вторичного УД пациенты, рандомизированные в 3,0 мг/кг группу, могут иметь только 4 недели на повышение их rhASM дозы до 3,0 мг/кг до начала 26-недельной фазы ПД при дозе 3,0 мг/кг (или максимальной переносимой дозе; смотрите фиг.7).
Фаза первичного УД может находиться в диапазоне от приблизительно 6 до 16 недель по длительности для каждого пациента. Это может зависеть от индивидуальных ограничений переносимости пациента, если они есть, с которыми встречается пациент во время увеличения дозы rhASM. Во время фазы первичного УД каждый пациент может получать дозу rhASM, повышаемую каждые 2 недели от 0,1 до 1,0 мг/кг (как показано на фиг.7).
Фаза первичного УД может завершаться, когда пациент получает и переносит дозу 1,0 мг/кг rhASM или достигает 16 недели, что случается даже раньше. Если ни один из критериев, ведущих к пробе с повторным введением дозы или уменьшению дозы, не соблюдается, то пациент может получить однократную инфузию каждой дозы (т.е. всего 4 инфузии rhASM), что дает в результате фазу первичного УД из 6 недель. Однако если критерии, ведущие к пробе с повторным введением дозы или уменьшению дозы, соблюдаются, то длительность фазы первичного УД может быть дольше, чем 6 недель, но не будет превышать 16 недель.
Пациенты, которые получали повышенные дозы и безопасно переносили 1,0 мг/кг к 16 неделе, могут быть разделены на 1 из 2 групп целевых доз (т.е. 1,0 или 3,0 мг/кг rhASM).
Пациенты, рандомизированные в группу, которая получает 3,0 мг/кг rhASM, могут входить в фазу вторичного УД (смотрите фиг.7), состоящую из 2 инфузий: первая инфузия (т.е. 2,0 мг/кг) будет через 2 недели после первой переносимой дозы 1,0 мг/кг, а вторая инфузия (т.е. 3,0 мг/кг или максимальная переносимая доза) будет через 4 недели после первой переносимой дозы 1,0 мг/кг. Следовательно, фаза вторичного УД может быть завершена через 4 недели после первой переносимой дозы 1,0 мг/кг. Не допускается проба с повторным введением дозы. Если пациент не переносит первую дозу 2,0 мг/кг, то вторая rhASM доза может быть уменьшена до максимальной переносимой дозы (т.е. 1,0 мг/кг) или временно приостановлена в случае потенциальной лимитирующей дозу токсичности (DLT).
Пациенты могут продолжать получать rhASM при максимальной переносимой или рандомизированной дозе, вводимой IV каждые 2 недели во время 26-недельной фазы ПД периода первичного лечения. Это может включать как рандомизированных, так и не рандомизированных пациентов.
Как указано на фиг.7, пациенты в группе с 1,0 мг/кг дозой могут начинать фазу ПД при их первой дозе 1,0 мг/кг после рандомизации, тогда как пациенты в группе с 3,0 мг/кг могут начинать фазу ПД при их третьей дозе rhASM после рандомизации. Последующие rhASM дозы могут быть введены IV каждые 2 недели в течение 26 недель в фазе ПД для всех рандомизированных пациентов.
Нерандомизированные пациенты могут начинать фазу ПД при их первой дозе rhASM через 2 недели после их 16-недельной фазы УД. Эти пациенты могут продолжать на их максимальной переносимой дозе rhASM (<1,0 мг/кг) каждые 2 недели в течение в общем 26 недель.
Если рандомизированный пациент испытывает умеренное/тяжелое связанное AE (т.е. связанную с инфузией реакцию [IAR]) во время инфузии в фазе ПД, то пациенту могут прекратить rhASM инфузию и начать заново (т.е. прервать) или уменьшить скорость инфузии. При условии того, что потенциальная DLT не возникает, пациент может продолжать на своей дозе rhASM в течение оставшейся части фазы ПД.
После того как оценки всего 26-недельного протокола завершены, все пациенты (включая нерандомизированных пациентов) могут входить в период долговременного лечения и продолжать лечение rhASM (при дозе rhASM, которая вводилась во время фазы ПД) IV каждые 2 недели в течение по меньшей мере 182 недель [3,5 года] по длительности или до прекращения протокола. Период долговременного лечения включает посещение для завершения протокола (или исключения пациента/досрочного завершения терапии) и телефонный звонок для наблюдения за безопасностью (30-37 дней после последней дозы rhASM). Индивидуальные дозы rhASM пациентов могут быть откорректированы в период долговременного лечения на основании результатов анализов периода первичного лечения. Любое необходимое увеличение дозы может происходить так, как описано ранее.
9.3. ЛЕЧЕНИЕ
Пациенты будут получать внутривенные инфузии rhASM при дозах 0,1, 0,3, 0,6 и 1,0 мг/кг каждые 2 недели во время фазы первичного УД. Пациенты, рандомизированные в группу с 3,0 мг/кг rhASM, будут получать 2,0 мг/кг до получения 3,0 мг/кг (или максимальной переносимой дозы) на 2 недели позже во время фазы вторичного УД. Во время 26-недельной фазы ПД пациентам будут вводить 1,0 или 3,0 мг/кг (или максимальную переносимую дозу) rhASM каждые 2 недели после фазы УД. Во время периода долговременного лечения rhASM доза, которая вводилась во время фазы ПД, будет вводиться IV каждые 2 недели.
Пациенты будут получать каждую внутривенную инфузию rhASM в течение периода приблизительно 35-135 минут (мин) в зависимости от дозы (смотрите таблицу 9 ниже). Лечение по протоколу (rhASM) будет вводиться инфузией с использованием стандартного программируемого инфузионного насоса и 0,22 микронного с низким связыванием белка встроенного фильтра. Для расчета дозы в мг/кг применяют действительный вес для пациентов с индексом массы тела (BMI) ≤30 (вес в кг)/(рост в метрах); для пациентов с BMI>30, рассчитывают вес, соответствующий BMI 30, используя рост пациента.
Введение rhASM
Этап 2: 60 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 50 мл/час за 20 мин (±5 мин), если нет IAR
Этап 3: 100 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 50 мл/час за 20 мин (±5 мин), если нет IAR
Этап 3: 100 мл/час за 20 мин (±5 мин), если нет IAR
Этап 4: 167 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 30 мл/час за 20 мин (±5 мин), если нет IAR
Этап 3: 60 мл/час за 20 мин (±5 мин), если нет IAR
Этап 4: 100 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 30 мл/час за 20 мин (±5 мин), если нет IAR
Этап 3: 60 мл/час за 20 мин (±5 мин), если нет IAR
Этап 4: 100 мл/час за 20 мин (±5 мин), если нет IAR
Этап 5: 200 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 5: 2,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Этап 2: 30 мл/час за 20 мин (±5 мин), если нет IAR
Этап 3: 60 мл/час за 20 мин (±5 мин), если нет IAR
Этап 4: 100 мл/час за 20 мин (±5 мин), если нет IAR
Этап 5: 200 мл/час за 20 мин (±5 мин), если нет IAR
Этап 6: 300 мл/час для оставшейся части инфузии, если нет IAR
Этап 2: 0,3 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 3: 0,6 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 4: 1,0 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 5: 2,0 мг/кг/час за 20 мин (±5 мин), если нет IAR
Этап 6: 3,0 мг/кг/час для оставшейся части инфузии, если нет IAR
Клинические предельные значения могут быть измерены как процентное (%) изменение от исходного уровня в объеме селезенки, увеличенном по сравнению с нормальным (как измерено с помощью магнитно-резонансной томографии), через 26 недель после получения максимальной переносимой или рандомизированной дозы rhASM. Вторичные предельные значения эффективности включают объем печени (как измерено с помощью МРТ); легочный снимок (с помощью изображения компьютерной томографии с высоким разрешением и рентгенографии грудной клетки); испытания легочной функции, включая процентную расчетную диффузионную емкость легких по моноксиду углерода, процентную расчетную форсированную жизненную емкость легких, объем форсированного выдоха за 1 секунду и общую емкость легких; способность переносить физическую нагрузку с помощью велоэргометрии, включая процентную расчетную максимальную рабочую нагрузку, пиковое потребление кислорода и продукцию диоксида углерода; общую врачебную оценку изменения; эффективность биомаркеров, таких как сывороточная хитотриозидаза, CCL18 и ACE; и гематологические параметры, такие как количество тромбоцитов, уровень гемоглобина. Оценки эффективности могут также включать липиды натощак, исследования коагуляции, биомаркеры, связанные с заболеванием, измерения общего состояния здоровья и фотографии пациента.
Оценки безопасности могут быть проведены до, во время и (или) после введения дозы. Оценки безопасности, которые могут быть проведены, включают следующее: клиническое обследование с неврологической оценкой, основные показатели жизнедеятельности (например, систолическое и диастолическое кровяное давление, температура, частота сердечных сокращений, частота дыхания и насыщение кислородом), ЭКГ, УЗИ с допплерометрическим исследованием, химический состав крови (например, натрий, калий, кальций, хлорид, азот мочевины крови, креатинин, мочевая кислота, ALT, аспартат-аминотрансфераза, общий билирубин, лактат-дегидрогеназа, щелочной фосфат, общий белок, альбумин, глюкоза, холестерин и т.д.), гематология (например, клинический анализ крови), анализ мочи, биомаркерный анализ на биомаркеры, связанные с раком, гормоны, цитокины (например, IL-8 и IL-6), риск сердечнососудистых заболеваний, острофазовые агенты и другие клеточные процессы, сывороточная концентрация анти-rhASM IgG, сывороточная концентрация анти-rhASM IgE, активность триптазы сыворотки и исследование кожи на гиперчувствительность. Кроме того, оценки фармакокинетики и фармакодинамики могут быть проведены до, во время и (или) после введения дозы. Например, уровни сфингомиелина можно оценить в печени и коже с помощью биопсии, а также в плазме и DBS с помощью тандемной масс-спектрометрии.
Все ссылки, процитированные в данном документе, включены ссылкой во всей их полноте и для всех целей в той же степени, как если бы каждая отдельная публикация или патент или патентная заявка были конкретно и отдельно указаны как включенные ссылкой в их полноте для всех целей.
Данное изобретение не ограничено в объеме специфическими вариантами осуществления, описанными в данном документе. Безусловно, различные модификации данного изобретения в дополнение к тем, что были описаны, будут очевидны для специалистов данной области техники из вышеизложенного описания изобретения и сопровождающих фигур. Такие модификации, как подразумевается, попадают в объем приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕРМЕНТНАЯ ЗАМЕСТИТЕЛЬНАЯ ТЕРАПИЯ С УВЕЛИЧЕНИЕМ ДОЗЫ ДЛЯ ЛЕЧЕНИЯ НЕДОСТАТОЧНОСТИ КИСЛОЙ СФИНГОМИЕЛИНАЗЫ | 2020 |
|
RU2833674C2 |
| ФЕРМЕНТНАЯ ЗАМЕСТИТЕЛЬНАЯ ТЕРАПИЯ С УВЕЛИЧЕНИЕМ ДОЗЫ ДЛЯ ЛЕЧЕНИЯ НЕДОСТАТОЧНОСТИ КИСЛОЙ СФИНГОМИЕЛИНАЗЫ | 2010 |
|
RU2731616C2 |
| Лечение патологических состояний кости у пациентов с дефицитом кислой сфингомиелиназы | 2018 |
|
RU2795570C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ НЕДОСТАТОЧНОСТИ КИСЛОЙ СФИНГОМИЕЛИНАЗЫ | 2019 |
|
RU2826120C2 |
| КОМПОЗИЦИИ ФОРБОЛОВЫХ ЭФИРОВ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ УМЕНЬШЕНИЯ ПРОДОЛЖИТЕЛЬНОСТИ ЦИТОПЕНИИ | 2015 |
|
RU2730998C2 |
| АНТИТЕЛО К РЕЦЕПТОРУ IL-6 ДЛЯ ЛЕЧЕНИЯ ЮВЕНИЛЬНОГО ИДИОПАТИЧЕСКОГО АРТРИТА | 2020 |
|
RU2822089C2 |
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2020 |
|
RU2828027C2 |
| СПОСОБЫ СНИЖЕНИЯ ЧИСЛА БАЗОФИЛОВ | 2014 |
|
RU2618455C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АГОНИСТОВ FXR | 2017 |
|
RU2737324C2 |
| ЛЕЧЕНИЕ КОРОНАВИРУСНОЙ ИНФЕКЦИИ | 2021 |
|
RU2837859C1 |



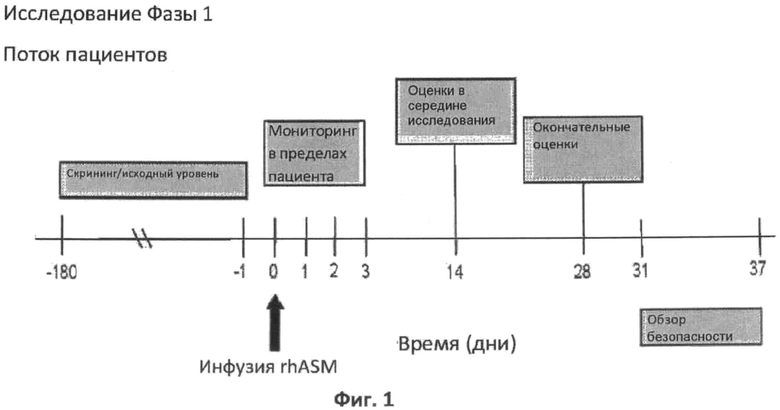
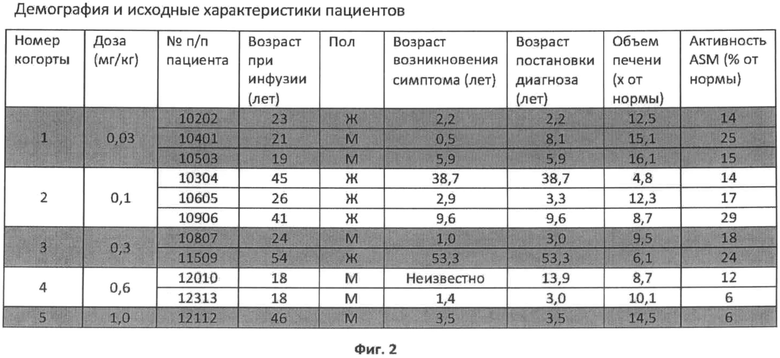
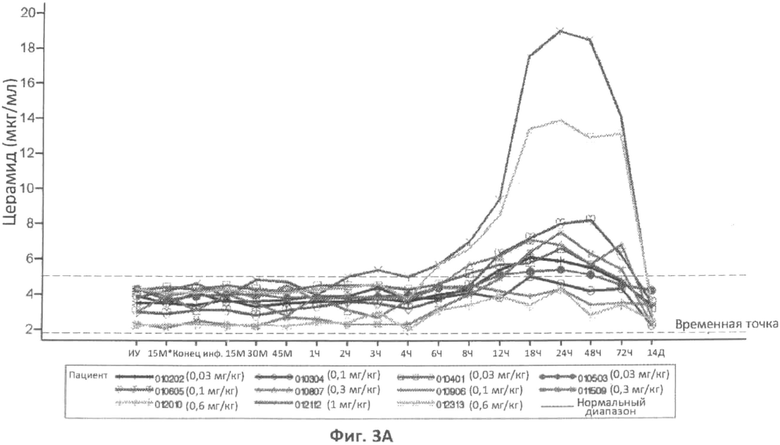
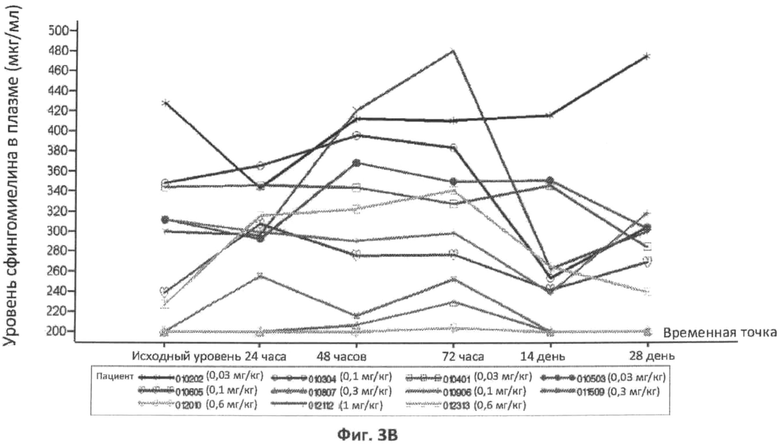
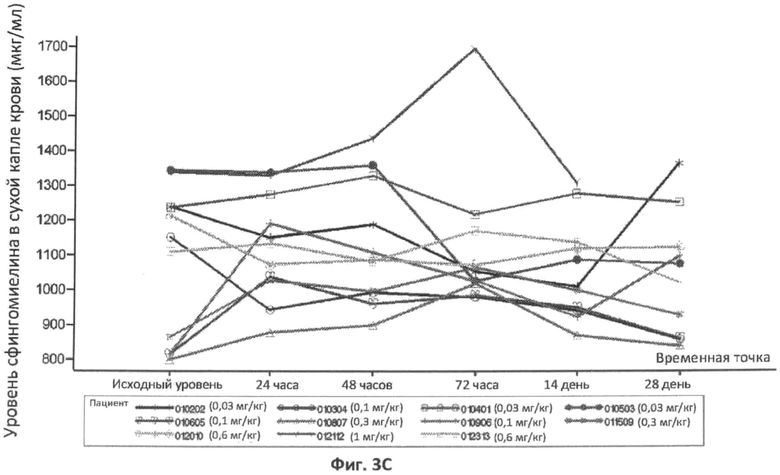
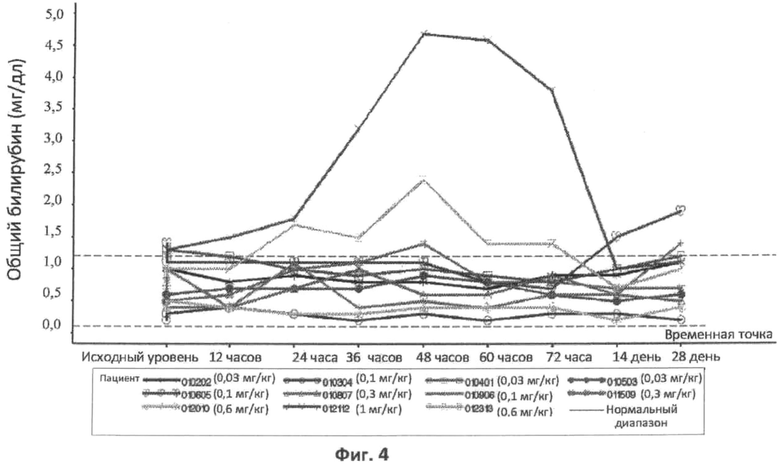
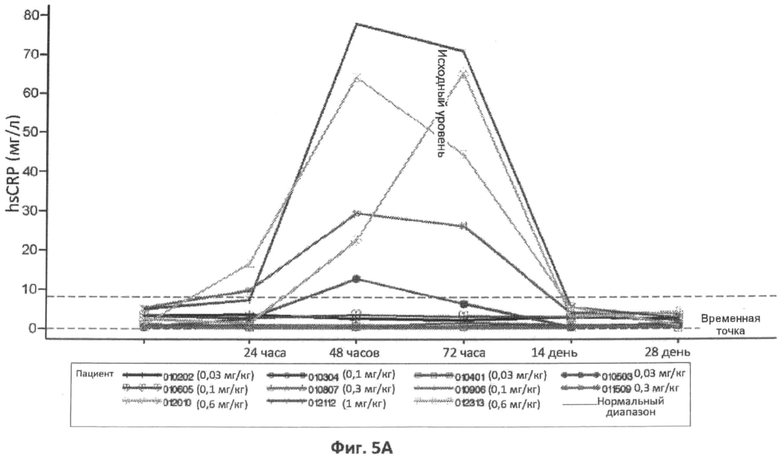
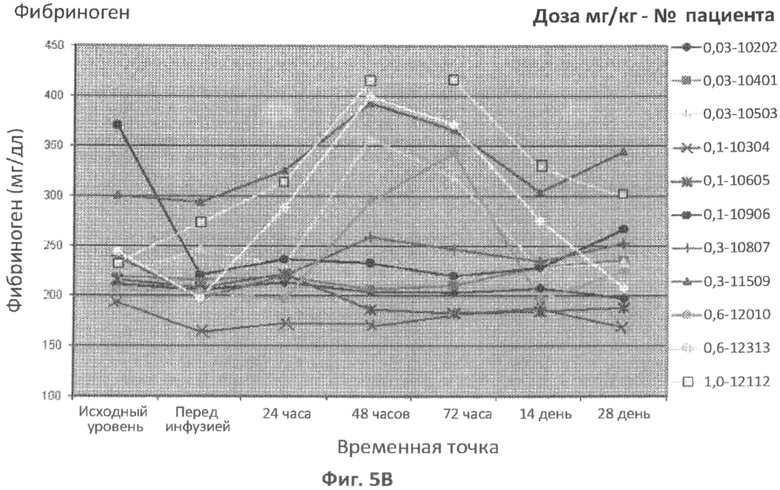
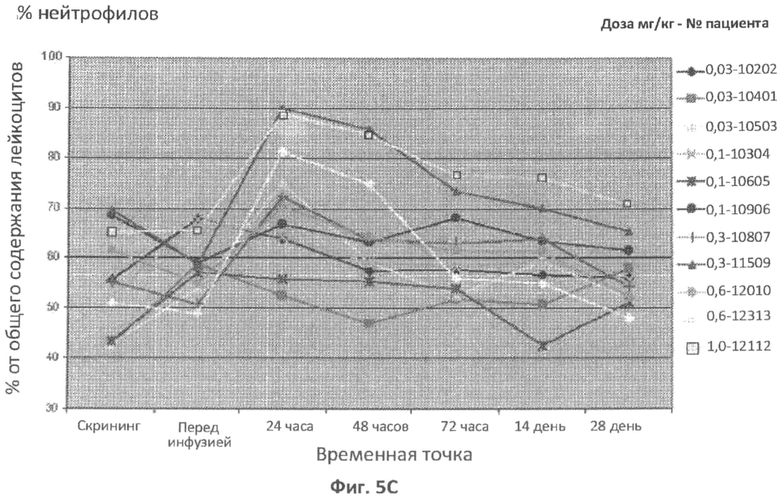

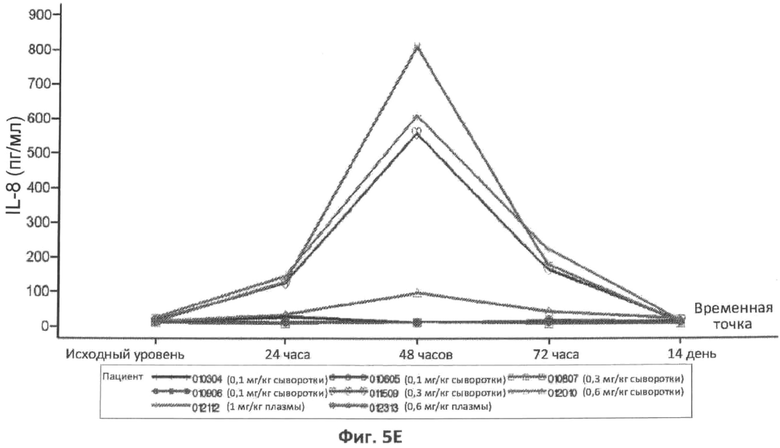
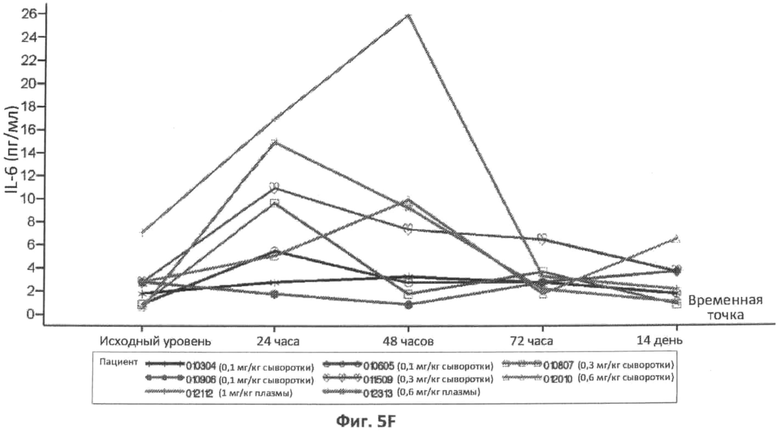
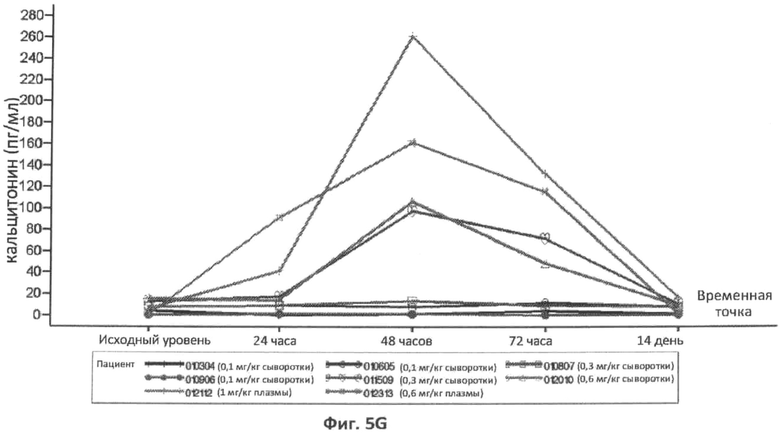
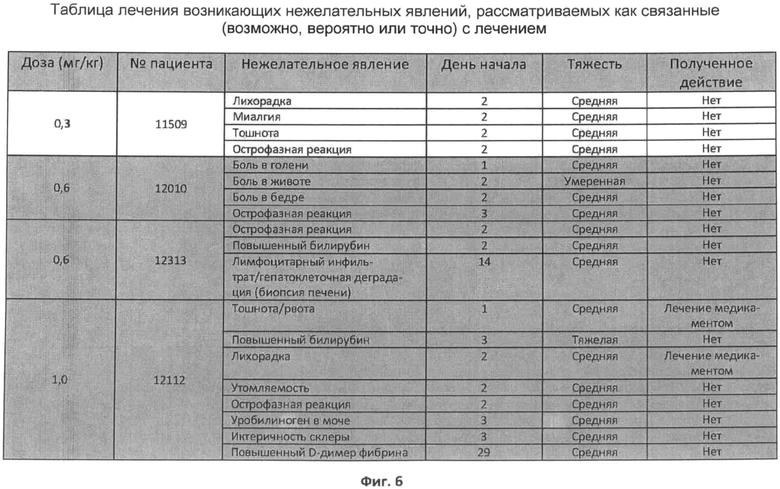
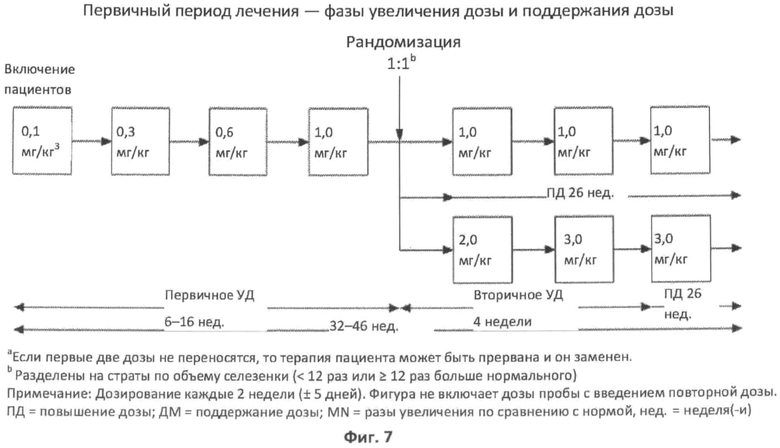
Группа изобретений относится к медицине и предназначена для лечения субъектов-людей, имеющих недостаточность кислой сфингомиелиназы. Лечение осуществляют с введения начальной низкой нетоксичной дозы кислой сфингомиелиназы (ASM). Затем последовательно вводят более высокие дозы ASM. Выполняют мониторинг в отношении одного или нескольких нежелательных побочных эффектов после каждой последующей дозы, на которые указывают повышенные концентрация общего билирубина, концентрация церамида в плазме, концентрация С-реактивного белка (CRP) в плазме или связанное нежелательное явление. Затем применяют поддерживающую схему, включающую введение дозы ASM, равной или меньшей, чем самая высокая доза, переносимая субъектом, в качестве поддерживающей дозы субъекту. В другом воплощении изобретения лечение включает введение кислой сфингомиелиназы (ASM) по схеме с увеличением дозы в следующих последовательных дозах: 0,1 мг/кг, 0,3 мг/кг и 0,6 мг/кг. Использование группы изобретений обеспечивает безопасное введение высоких доз фермента ASM, что сопровождается меньшими побочными эффектами. 2 н. и 33 з.п. ф-лы, 15 ил., 9 табл., 4 пр.
1. Способ лечения субъекта-человека, имеющего недостаточность кислой сфингомиелиназы, включающий:
(a) схему для сокращения массы накопленного субстрата сфингомиелина, включающую:
(i) введение начальной низкой нетоксичной дозы кислой сфингомиелиназы (ASM);
(ii) введение последовательно более высоких доз ASM и мониторинг субъекта в отношении одного или нескольких нежелательных побочных эффектов после каждой последующей дозы, на которые указывают повышенная концентрация общего билирубина, повышенная концентрация церамида в плазме, повышенная концентрация С-реактивного белка (CRP) в плазме или связанное нежелательное явление; и
(b) поддерживающую схему, включающую введение дозы ASM, равной или меньшей, чем самая высокая доза, переносимая субъектом, в качестве поддерживающей дозы субъекту.
2. Способ по п. 1, в котором ASM представляет собой rhASM.
3. Способ по п. 2, в котором диапазон начальной дозы составляет от 0,05 мг/кг до 0,275 мг/кг rhASM.
4. Способ по п. 3, в котором начальная доза составляет 0,1 мг/кг rhASM.
5. Способ по любому из пп. 1-4, в котором последовательно более высокие дозы вводят через одну, две, три или четыре недели после предыдущей дозы.
6. Способ по п. 5, в котором последовательно более высокая доза примерно на 0,1-1,0 мг/кг выше предыдущей дозы или примерно на 0,1-0,5 мг/кг выше предыдущей дозы.
7. Способ по любому из пп. 1-4, в котором самая высокая доза, переносимая субъектом-человеком, составляет от 1 мг/кг до 3 мг/кг.
8. Способ по любому из пп. 1-4, в котором самую высокую переносимую дозу вводят субъекту-человеку в качестве поддерживающей дозы.
9. Способ по п. 7, в котором самую высокую переносимую дозу вводят субъекту-человеку в качестве поддерживающей дозы.
10. Способ по любому из пп. 1-4, в котором поддерживающая доза представляет собой терапевтически эффективную дозу, которая меньше, чем самая высокая переносимая доза.
11. Способ по любому из пп. 1-4, который дополнительно включает мониторинг субъекта во время поддерживающей схемы в отношении одного или нескольких нежелательных побочных эффектов, на которые указывают повышенный билирубин или связанное нежелательное явление; и корректировку поддерживающей дозы.
12. Способ по п. 9, в котором поддерживающую дозу вводят субъекту каждые две-четыре недели.
13. Способ по п. 10, в котором поддерживающую дозу вводят субъекту каждые две-четыре недели.
14. Способ по любому из пп. 1-4, где человек имеет объем селезенки, в ≥8 раз превышающий нормальный.
15. Способ по любому из пп. 1-4, в котором дозы вводят внутривенно, внутрикожно, подкожно или внутримышечно.
16. Способ по любому из пп. 1-4, в котором недостаточностью кислой сфингомиелиназы является болезнь Ниманна-Пика (NPD) типа А.
17. Способ по любому из пп. 1-4, в котором недостаточностью кислой сфингомиелиназы является NPD типа В.
18. Способ по любому из пп. 1-4, в котором субъект-человек имеет миссенс-мутацию в гене, кодирующем кислую сфингомиелиназу.
19. Способ по п. 18, в котором мутация представляет собой L302P, H421Y или R496L.
20. Способ по любому из пп. 1-4, в котором субъект-человек имеет мутацию в гене, кодирующем кислую сфингомиелиназу, и мутация представляет собой ΔR608.
21. Способ лечения субъекта-человека, имеющего недостаточность кислой сфингомиелиназы, включающий введение кислой сфингомиелиназы (ASM) по схеме с увеличением дозы в следующих последовательных дозах:
(a) 0,1 мг/кг,
(b) 0,3 мг/кг и
(c) 0,6 мг/кг,
где: (i) каждую дозу вводят по меньшей мере дважды, и каждую дозу вводят с двухнедельными интервалами, и (ii) где субъекта подвергают мониторингу в отношении токсических побочных эффектов перед повышением дозы до следующего уровня.
22. Способ по п. 21, в котором ASM представляет собой рекомбинантную человеческую кислую сфингомиелиназу (rhASM).
23. Способ по п. 21, в котором последующая более высокая доза ASM составляет 1 мг/кг.
24. Способ по п. 23, в котором последующая более высокая доза ASM составляет 2 мг/кг.
25. Способ по п. 24, в котором последующая более высокая доза ASM составляет 3 мг/кг.
26. Способ по любому из пп. 21-25, в котором дозы вводят внутривенно, внутрикожно, подкожно или внутримышечно.
27. Способ по любому из пп. 21-25, в котором недостаточностью кислой сфингомиелиназы является болезнь Ниманна-Пика (NPD) типа А.
28. Способ по любому из пп. 21-25, в котором недостаточностью кислой сфингомиелиназы является NPD типа В.
29. Способ по любому из пп. 21-25, в котором субъект-человек имеет миссенс-мутацию в гене, кодирующем кислую сфингомиелиназу.
30. Способ по п. 29, в котором мутация представляет собой L302P, H421Y или R496L.
31. Способ по любому из пп. 21-25, в котором субъект-человек имеет мутацию в гене, кодирующем кислую сфингомиелиназу, и мутация представляет собой ΔR608.
32. Способ по любому из пп. 21-25, где человек имеет объем селезенки, в ≥8 раз превышающий нормальный.
33. Способ по п. 1, где указанная повышенная концентрация общего билирубина представляет собой концентрацию общего билирубина выше 1,5 мг/дл.
34. Способ по п. 1, где указанная повышенная концентрация церамида в плазме представляет собой концентрацию церамида в плазме выше 10 мкг/мл.
35. Способ по п. 1, где указанная повышенная концентрация CRP в плазме представляет собой концентрацию CRP в плазме выше 12 мг/л.
| US 2005048047 A1, 03.03.2005 | |||
| US 2007280925 A1, 06.12.2007 | |||
| PATAT AA | |||
| Designing and interpreting the results of first-time-to-man studies | |||
| Dialogues Clin Neurosci., 2000 Sep, 2(3), p.203-12 | |||
| SIMONARE CM et al | |||
| The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations | |||
| Am J Hum Genet., 2002 Dec, 71(6), p.1413-9 | |||
| WO 2007084737 A2, 26.07.2007. |

