ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармацевтической композиции, включающей в качестве действующего ингредиента некоторые производные, образованные 1-β-D-арабинофуранозилцитозином (цитарабином) и насыщенными и мононенасыщенными жирными кислотами с длинной цепью. В частности, настоящее изобретение относится к фармацевтической композиции и способу получения такой композиции, подходящей для парентерального введения терапевтически эффективных доз указанных производных, с целью улучшить приверженность пациента терапии при лечении рака.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
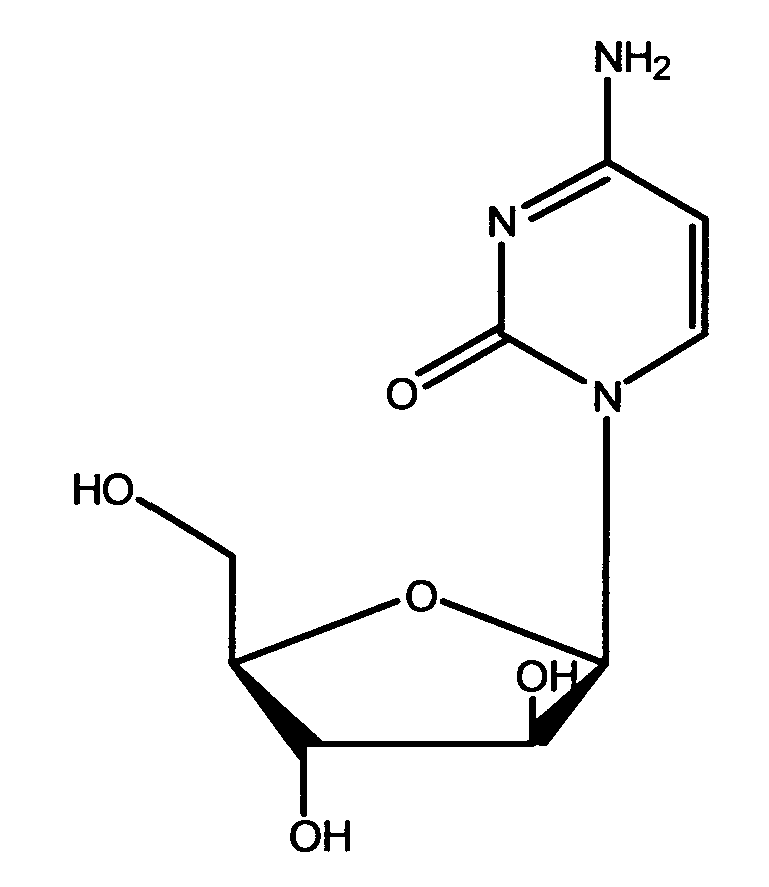
Цитарабин, известный также как Ara-C или Cytosar, давно известен в качестве химиотрапевтического средства для лечения острого миелоцитарного лейкоза. Цитарабин имеет формулу
Действующие ингредиенты фармацевтической композиции по настоящему изобретению включает производное цитарабина формулы (I)

где R1,R2 и R3 независимо выбраны из водорода и C18- и C20- насыщенных и мононенасыщенных ацильных групп, при условии, что все заместители R1, R2 и R3 не могут одновременно быть атомами водорода.
Цитарабин обладает ограниченной эффективностью против солидных опухолей (Frei et al., Cancer Res. 29 (1969), 1325-1332; Davis et al., Oncology, 29 (1974), 190-200; Cullinan et al., Cancer Treat. Rep. 61 (1977), 1725-1726) и даже при лечении лейкоза цитарабин находит лишь ограниченное применение из-за очень непродолжительного периода полужизни в биологической среде и высокой токсичности.
В целях преодоления этих недостатков, ряд авторов получили и протестировали пролекарственные производные цитарабина. Например, Hamamura и соавторы исследовали 3'-ацильное и 3',5'-диацильное производные цитарабина (J. Med. Chem. 19 (1976) №5, 667-674). Эти исследователи получили и протестировали многочисленные производные цитарабина с насыщенными или ненасыщенными сложноэфирными группами, содержащими от 2 до 22 атомов углерода, и обнаружили, что многие из этих соединений демонстрируют более высокую активность против L1210 лейкоза у мышей по сравнению с самим исходным нуклеозидом.
Хотя работы по сложноэфирным пролекарствам на основе цитарабина, включая 3'- и 5'-ацильные производные, продолжались (смотрите, например, Rubas et al., в Int. J. Cancer, 37, 1986, стр. 149-154, где протестированы липосомальные составы 5'-олеилцитарабина против L1210 лейкемии и меланомы B16) до сих пор подобные лекарства не стали доступными в клинической практике.
Основная причина того, почему цитарабин не применяется в лечении солидных опухолей, заключается в быстром клиренсе действующего соединения из раковых клеток и плазмы. По-видимому, невозможно достичь значительных внутриклеточных концентраций лекарственного средства в ткани новообразования, даже если интересующая опухоль восприимчива к цитарабину in vitro. Авторы изобретения показали ранее, что производные формулы (I) обладают увеличенным временем полужизни и меняют распределение препарата в тканях, что очень важно для терапевтического действия этих продуктов (WO 97/05154).
Развитие невосприимчивых раковых клеток является сложной проблемой в современной химиотерапии рака. Ранее было обнаружено, что одно из производных формулы (I), а именно элацитарабин (5'-O-(транс-9”-октадеценоил)-1-β-D-арабинофуранозилцитозин) демонстрирует такое же действие против невосприимчивых к цис-платину клеток (NHIK 3025/DDP) и невосприимчивых к MDR клеток (A549), как и против соответствующих восприимчивых клеточных линий. Это происходит потому, что сложноэфирные производные не являются субстратами клеточных механизмов выведения лекарственных препаратов, например, «насоса gp 120 MDR», ответственных за явление, проявляющееся в виде мультилекарственной невосприимчивости.
Тем не менее, включение терапевтически эффективного количества плохо растворимых производных формулы (I) в фармацевтическую композицию, подходящую для парентерального введения, представляет собой проблему. Для внутривенного введения указанных производных должна быть подобрана такая композиция эксципиентов, которая позволяет солюбилизировать эти производные. Производные цитарабина формулы (I) являются амфифильными, и обладают плохой растворимостью как в воде, так и в маслах. Это ограничивает выбор потенциальных эксципиентов, которые могут солюбилизировать такие производные. В качестве примера, элацитарабин обладает растворимостью <0,1 мкг/мл в деионизированной воде и <1 мкг/мл в фосфатном буфере с pH 7,4 при 25°C. Кроме того, предварительные исследования составов показали, что элацитарабин не обладает необходимой растворимостью в эмульсиях на основе соевого масла, что подтверждает низкую растворимость препарата в маслах.
Если состав представляет собой систему, состоящую из частиц, существуют определенные требования к размеру частиц состава для внутривенного введения. Кроме того, парентеральные продукты должны быть стерильными и часто стерильное фильтрование является единственным подходящим способом для фармацевтических систем, состоящих из частиц. Это означает, что размер частиц этих составов должен быть менее 220 нм (0,22 мкм), что равно размеру пор стерильных фильтров. На практике для способа промышленного масштаба размер частиц должен быть существенно меньше, во избежание забивания фильтра.
Другая проблема заключается в том, что рекомендованная дневная доза при внутривенном введении элацитарабина в режиме монотерапии в настоящее время установлена на уровне 2000 мг/м2. Это значит, что для среднего пациента с поверхность тела 1,8 м2 суммарная дневная доза элацитарабина должна составлять 3600 мг. Это приводит к еще более сложным задачам: a) требуется увеличение концентрации лекарственного средства в составе для того, чтобы пациентам не пришлось осуществлять парентеральное введение слишком больших объемов жидкости, b) необходимо избегать применения антиоксидантов и консервантов, которые даже при добавлении в небольших количествах, будут поступать в организм в неприемлемо высоком общем количестве и c) необходимо ограничивать количество добавленных ПАВ и ко-солюбилизаторов по указанной выше причине.
Наконец, сложноэфирные производные формулы (I) склонны к гидролитическому расщеплению при физиологических значениях pH, скорость которого зависит от типа производного и буфера. Это выдвигает дополнительные требования как к составу, так и к параметрам производственного процесса. Как правило, предпочтительно, чтобы фармацевтический продукт был готов к употреблению. Для получения такого готового к употреблению продукта необходимо, чтобы указанные производные были защищены от гидролитического разрушения в водной среде парентерального состава в течение всего срока годности.
Настоящее изобретение позволяет решить все указанные выше проблемы.
КРАТКОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО МАТЕРИАЛА
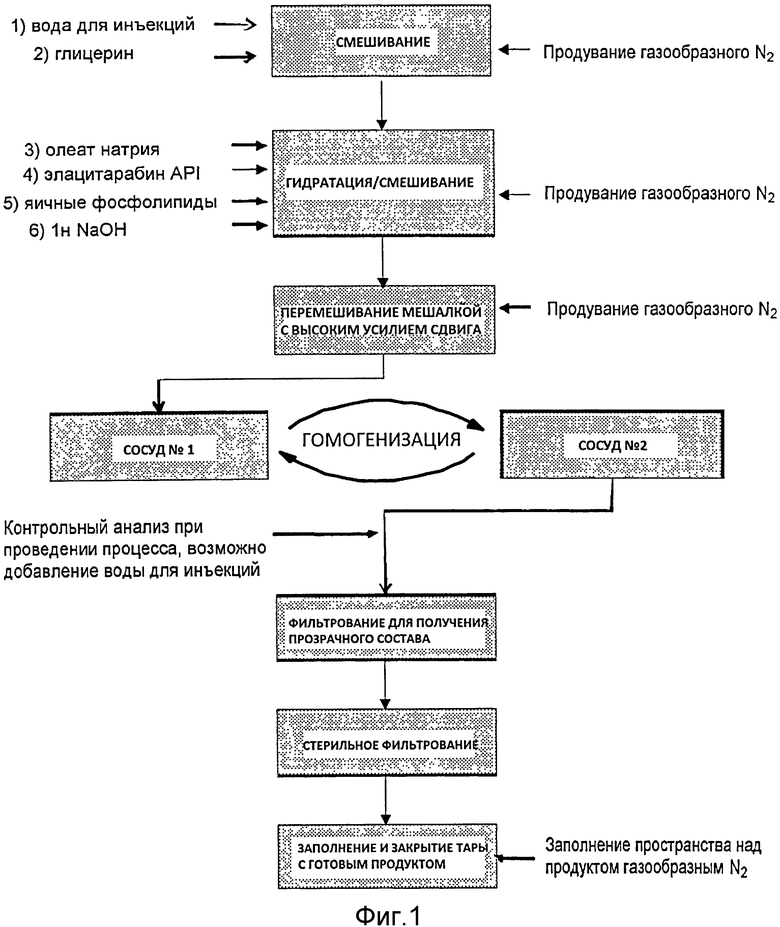
Фиг.1: Технологическая схема производства лекарственного препарата на основе элацитарабина.
Фиг.2: Концентрации в плазме элацитарабина, Ara-C и Ara-U (метаболита Ara-C, образующегося при деаминировании), средние значения для 61 пациента.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Основной целью настоящего изобретения являлась разработка фармацевтической композиции, подходящей для парентерального введения и включающей в качестве действующего ингредиента производные цитарабина формулы (I).
Далее, настоящее изобретение частично относится к парентеральным составам элацитарабина (5'-O-(транс-9”-октадеценоил)-1-β-D-арабинофуранозилцитозина), имеющего формулу
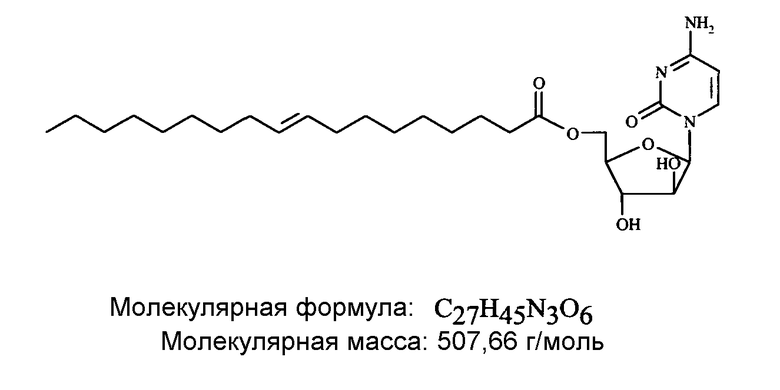
Кроме того, настоящее изобретение частично относится к составам элацитарабина, содержащим: элацитарабин (или его соль); солюбилизатор, включающий один или несколько фосфолипидов; ко-солюбилизатор, например, олеат натрия, а также изотонический агент, например, глицерин, в водной среде, предпочтительно при pH 6-8.
Далее, настоящее изобретение частично относится к способу получения составов элацитарабина на основе липидных наночастиц/липосом в водной среде.
Кроме того, настоящее изобретение частично относится к способу лечения расстройств, связанных с пролиферацией клеток, включающему введение субъекту, которому необходимо такое лечение, составов элацитарабина на основе липидных наночастиц, где указанный субъект страдает клеточным пролиферативным расстройством или имеется опасность развития такого расстройства. Из WO 97/05154 известно, что соединения формулы (I) применимы в лечении рака.
Заявители неожиданно обнаружили фармацевтическую композицию, подходящую для парентерального введения, и способ ее получения для производных цитарабина формулы (I), который позволяет изготовлять готовые к применению водные составы на основе фосфолипидных частиц с мольным соотношением лекарственного средства к липиду от 1:20 до 1:7, предпочтительно от 1:13 до 1:8, где указанные липидные частицы защищают указанное производное от гидролитического разрушения до цитарабина в течение 24 месяцев при хранении при 2-8°C в атмосфере азота. Кроме того, в способе по настоящему изобретению применяются природные фосфолипиды, полученные из яичного желтка, причем за счет включения в состав небольшого количества солей жирных кислот, эти фосфолипиды также защищены от гидролитического разрушения. Сформированные липидные наночастицы имеют гидродинамический диаметр <50 нм и могут быть легко подвергнуты стерильному фильтрованию. Кроме того, способ получения состава по настоящему изобретению способствует стабилизации более высоких концентраций лекарственного средства в указанных наночастицах и поддается масштабированию до промышленных объемов, подходящих для производства водных стерильных продуктов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Основной целью настоящего изобретения является разработка фармацевтической композиции на основе природных фосфолипидов, подходящей для парентерального введения и содержащей в качестве действующего средства производное цитарабина формулы (I), которая включает терапевтически эффективные дозы указанных производных и является такой же эффективной или более эффективной в лечении рака, чем коммерчески доступные продукты цитарабина.
Эта и другие цели настоящего изобретения достигаются с помощью фармацевтической композиции и способа ее получения, которые описаны в приложенной формуле изобретения.
Действующий фармацевтический ингредиент
Согласно одному из вариантов осуществления, в настоящем изобретении разработана фармацевтическая композиция, включающая в качестве действующего ингредиента производное цитарабина формулы (I)
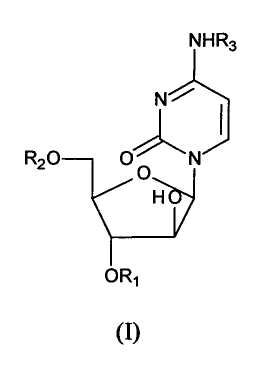
где R1, R2 и R3 независимо выбраны из водорода и C18- и C20- насыщенных и мононенасыщенных ацильных групп, при условии, что R1, R2 и R3 не могут одновременно являться атомами водорода или его фармацевтически приемлемую соль; где этот действующий ингредиент растворен или диспергирован в фосфолипидах.
Согласно предпочтительному варианту осуществления настоящего изобретения, фосфолипиды указанной фармацевтической композиции включают нейтрально заряженные фосфолипиды сами по себе, или в комбинации с другими фосфолипидами.
Цитарабин имеет четыре функциональные группы, которые могут образовывать производные, а именно 5'-, 3'- и 2'-гидроксильные группы и N4-аминогруппу. Реакционная способность 2'-гидроксильной группы в этом контексте является ограниченной, и рассматриваться не будет. Каждую из оставшихся групп можно селективно превратить в сложноэфирное или амидное производное, но, кроме того, можно получать дизамещенные производные (сложные диэфиры или эфирамиды) и тризамещенные производные. В случае ди- и тризамещенных производных ацильные заместители необязательно должны быть одинаковыми.
В данном случае предпочтительными для применения в качестве действующих ингредиентов фармацевтической композиции по настоящему изобретению являются моноацилзамещенные производные, т.е. соединения в которых, два заместителя из трех R1, R2 и R3, представляют собой атомы водорода. Особенно предпочтительно, чтобы монозамещение ацильной группой осуществлялось бы по положениям 3'-O или 5'-O остатка сахара, где положение 5'-O является наиболее предпочтительным.
Двойная связь мононенасыщенных ацильных групп может иметь либо цис-, либо транс-конфигурацию, хотя терапевтический эффект может различаться в зависимости от того, какая именно конфигурация применяется.
Положение двойной связи в мононенасыщенной ацильной группе также, по-видимому, оказывает влияние на активность. В данном случае заявители предпочитают применять сложные эфиры или амиды, ненасыщенная связь которых находится в положении ω-9. В ω-системе номенклатуры положение ω двойной связи мононенасыщенной жирной кислоты отсчитывают от концевой метильной группы так что, например, эйкозеновая кислота (C20:1 ω-9) включает 20 атомов углерода в цепи, и единственная двойная связь находится между 9 и 10 атомами углерода, считая от метильной концевой группы цепи. Заявители считают предпочтительным применение сложных эфиров, эфир-амидов и амидов, полученных из олеиновой кислоты (C18:1 ω-9, цис), элаидиновой кислоты (C18:1 ω-9, транс), эйкозеновой кислоты (кислот) (C20:1 ω-9, цис) и (C20:1 ω-9, транс), причем амиды и 5'-сложные эфиры в настоящем изобретении считаются наиболее предпочтительными производными.
Амид Ara-C(N4)-элаидиновой кислоты, сложный эфир Ara-C-5'-элаидиновой кислоты и сложный эфир Ara-C-3'-элаидиновой кислоты входят в число наиболее предпочтительных производных и, согласно предпочтительному варианту осуществления изобретения, элацитарабин (сложный эфир Ara-C-5'-элаидиновой кислоты или 5'-O-(транс-9”-октадеценоил)-1-β-D-арабинофуранозилцитозин) является действующим ингредиентом фармацевтической композиции по настоящему изобретению.
Производные формулы (I) получают по методикам известного уровня техники (для более подробного ознакомления смотрите, например, WO 97/05154).
Состав API
Ниже по тексту описана водная фармацевтическая композиция по настоящему изобретению. Как правило, эта водная композиция должна включать солюбилизатор API, ко-солюбилизатор, изотонический агент и регулятор pH.
Согласно предпочтительному варианту осуществления изобретения, фармацевтическая композиция включает элацитарабин, фосфатидилхолин, фосфатидилэтаноламин, сфингомиелин, лизофосфолипиды, природные липиды, жирные кислоты, олеат натрия, глицерин и воду. Особенно предпочтительный состав приведен ниже в таблице 1.
Состав медицинского продукта элацитарабина
Fresenius-Kabi
1. Солюбилизатор
Фосфолипиды являются природными компонентами клеточных мембран, обладают высокой биосовместимостью и, как обнаружили заявители, применимы в составах элацитарабина, описанных в настоящей заявке. Фосфолипиды являются амфифильными молекулами, которые самопроизвольно образуют двухслойные структуры при контакте с водой и при дальнейшем разбавлении превращаются в микро- и наноузырьковые частицы, именуемые липосомами. Липосомы способны инкапсулировать молекулы лекарственного средства во внутренней водной части, окруженной двухслойной мембраной, или включать молекулы лекарственного средства в двухслойную структуру. Физико-химические свойства лекарственного соединения являются основным фактором, определяющим локализацию лекарственного средства в липосомальной частице. В зависимости от типа применяемых фосфолипидов и локализации лекарственного средства, липосомы могут действовать в качестве двух существенно различных систем доставки: 1) усовершенствованной системы доставки лекарств, способной обеспечивать отсроченное или регулируемое высвобождение, или 2) солюбилизатора/стабилизатора действующего вещества, приводящего к немедленному высвобождению состава, аналогично эмульсиям или суспензиям. Последний механизм является особенно актуальным в случае липофильных и амфифильных молекул, которые в основном локализованы в двухслойных структурах фосфолипидов и находятся близко к поверхности частиц.
Липофильные или амфифильные соединения могут встраиваться в двухслойную структуру вплоть до определенного молярного соотношения, не нарушая структуру липосом. Максимальная концентрация лекарственного средства в таком составе зависит от типа и концентрации фосфолипидов и физико-химических характеристик действующего соединения. Часто применяемое мольное соотношение лекарственного средства к фосфолипидам в таких составах составляет примерно 1:20.
Способность таких липосомальных структур захватывать лекарственное средство может быть увеличена с применением подходящего сорастворителя. Заявители неожиданно обнаружили, что в случае составов по настоящему изобретению, не только добавления <2,5% глицерина достаточно для стабилизации состава производного цитарабина при его содержании 8,8 мол.% относительно фосфолипидов, но, кроме того, добавление этого небольшого количества глицерина на соответствующей стадии производственного процесса должно стабилизировать состав при более высоком содержании лекарственного средства, равном 13 мол.% относительно количества фосфолипидов. Добавление глицерина на соответствующей стадии производства способствует также применению более низких температур в производственном процессе, что приводит к более низкой степени разрушения как действующего вещества, так и фосфолипидов. Поскольку указанное небольшое количество глицерина уже приводит к образованию слегка гипертонического продукта, важно не увеличивать содержание глицерина и, следовательно, осмолярность, еще больше.
Липосомы получают из природных или синтетических фосфолипидов, главным образом, фосфатидилхолина, который имеет нейтральный заряд при физиологических значениях pH. С целью стабилизации этих коллоидных частиц, в состав можно включать также небольшое количество отрицательно заряженного вещества. Электростатическое отталкивание, вызванное отрицательным зарядом частиц, создает эффективное препятствие для агрегации и образования более крупных частиц. Отрицательный заряд на частицах может возникать за счет любого отрицательно заряженного вещества, которое может быть включено в двухслойную структуру фосфолипида. Однако в литературе имеются данные, что добавление соли жирной кислоты, например, олеата натрия, в значительной степени способствует стабильности фосфолипидов, благодаря созданию благоприятного микроокружения (Werling et al., Eur. J.Pharm/Biopharm. 69 (2008) 1104-1113). Кинетика гидролиза фосфатидилхолина относится к псевдо 1-му порядку с минимальной скоростью в районе pH 6,5 (Grit et al., J. Pharm. Sci. 82 (1993) 362-366). В этой реакции образуются свободные жирные кислоты, которые снижают pH среды, и увеличивают величину отрицательного заряда на поверхности частиц. Werling и соавторы (смотрите ссылку выше) предположили, что добавление анионов жирных кислот к системе частиц на основе фосфолипидов, не только стабилизирует частицы за счет введения чистого отрицательного заряда, но также оказывает влияние на микроокружение поверхности, замедляет кинетику гидролиза и способствует общему улучшению физико-химической стабильности суспензии. Однако упомянутые авторы использовали мольное соотношение олеата натрия к яичным фосфолипидам 1:4, в то время, как в составах по настоящему изобретению мольное соотношение 1:23 неожиданно привело к аналогичному результату.
Элацитарабин является амфифильным соединением, которое в водной среде может быть солюбилизировано за счет связывания с двухслойными структурами, образованными очищенными яичными фосфолипидами. Очищенные яичные фосфолипиды являются смесью соединений. Основные (примерно 90%) фосфолипидные компоненты очищенных яичных фосфолипидов приведены в таблице 2. Кроме того, типовой профиль жирных кислот, их относительное количество и положение в молекуле яичного фосфатидилхолина (PC) показаны в таблице 3. Все фосфолипидные компоненты являются амфифильными соединениями, в определенной степени подобными элацитарабину (полярная «голова» - липофильный «хвост»). Эти амфифильные свойства используются в составах элацитарабина.
Основные компоненты очищенных яичных фосфолипидов
масса
2-LPC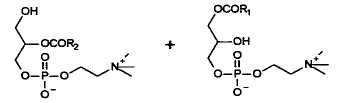

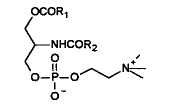
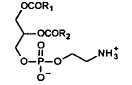
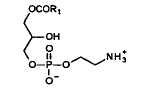
Положения различных жирных кислот во фракции PC яичных фосфолипидов
Концентрация очищенных яичных фосфолипидов была оптимизирована для включения целевого количества лекарственного вещества в двухслойную структуру фосфолипидов. Средняя молекулярная масса фосфолипидной фракции очищенных яичных фосфолипидов составляет 764 г/моль. Содержание липидов составляет приблизительно 91±1% мас./мас. Исходя из этого добавление 100 мг/мл очищенных яичных фосфолипидов к составу, показанному в таблице 1, эквивалентно содержанию липидов примерно 91±1% мг/мл. Соответственно, при концентрации липидов и API 91 мг/мл и 7,5 мг/мл соответственно, мольное отношение между липидами и API в составе элацитарабина составляет примерно 8:1.
Липидные частицы состава могут включать, не ограничиваясь этим, следующие фосфолипиды, которые действуют в качестве солюбилизаторов, компонентов, образующих двойные слои, или мицеллообразующих эксципиентов: фосфатидилхолин, фосфатидилэтаноламин, сфингомиелин, лизофосфолипиды, фосфатидилинозит, фосфатидилсерин, фосфатидилглицерин, фосфатидную кислоту, кардиолипин. Фосфолипиды могут присутствовать в любой форме, включая солевые или обессоленные, гидрированные или частично гидрированные, природные, полусинтетические или синтетические. Кроме того, возможно присоединение к фосфолипидам гидрофильных полимеров, например, полиэтиленгликоля (ПЭГ), чтобы избежать быстрого клиренса в результате деятельности ретикулоэндотелиальной системы (RES).
В предпочтительном варианте осуществления природные ненасыщенные фосфолипиды, полученные из куриных яиц, используются самостоятельно либо в виде комбинации.
В еще одном варианте осуществления настоящего изобретения природные яичные фосфолипиды включают цвиттерионный фосфолипид, например яичный фосфатидилхолин, который является нейтральным в диапазоне pH 6-8.
2. Ко-солюбилизатор
В одном из вариантов осуществления в композицию добавлен ко-солюбилизатор. В предпочтительном варианте осуществления, этот ко-солюбилизатор выбран из группы ПАВ. В более предпочтительном варианте осуществления этот ко-солюбилизатор играет также роль стабилизатора за счет введения отрицательного заряда. В еще более предпочтительном варианте осуществления выбрано анионное ПАВ, например, соль жирной кислоты. В еще более предпочтительном варианте осуществления этот ко-солюбилизатор также защищает фосфолипиды от гидролитического разрушения. В наиболее предпочтительном варианте осуществления упомянутый ко-солюбилизатор является олеатом натрия.
3. Изотонический агент
В одном из вариантов осуществления настоящего изобретения в фармацевтическую композицию включен изотонический агент. В более предпочтительном варианте осуществления изотонический агент выбран из следующего перечня: глицерин, пропиленгликоль, сахар, аминокислоты или белки, соли и их смеси.
В наиболее предпочтительном варианте осуществления упомянутый изотонический агент представляет собой глицерин. Глицерин в качестве сорастворителя добавляют для облегчения диспергирования частиц элацитарабина и включения лекарственного средства в липидные наночастицы. Количество добавленного глицерина находится в диапазоне от 0,1% до 30% масса/объем конечной фармацевтической композиции, более предпочтительно 1-10% масса/объем и, наиболее предпочтительно, 2-5% масса/объем от конечной фармацевтической композиции.
Количество изотонического агента может меняться в пределах от 1 до 50% конечной фармацевтической композиции, более предпочтительно от 5 до 15% и наиболее предпочтительно 7-10%. Все поддиапазоны в пределах от 1 до 50% включены в объем изобретения.
В другом варианте осуществления мольное отношение изотонического агента к общему количеству фосфолипидов составляет от 10:1 до 1:5, более предпочтительно от 5:1 до 1:1. Все поддиапазоны в пределах от 10:1 до 1:5 включены в объем изобретения.
4. Регулятор pH
В еще одном варианте осуществления, для регулирования pH и для усиления отрицательного заряда частиц к композиции добавляют основание. В настоящем описании основания включают химические соединения, которые акцептируют протоны. Примеры оснований включают, не ограничиваясь этим, гидроксиды металлов (например, гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния, гидроксид кальция, гидроксид бария и гидроксид стронция), карбонаты (например, карбонат лития, карбонат натрия, карбонат калия, карбонат магния, карбонат кальция, карбонат стронция и карбонат лантана), амины (например, аммиак) и их смеси.
В предпочтительном варианте осуществления это основание является гидроксидом натрия. Гидроксид натрия можно добавлять, например, в виде 0,1-10М раствора. Количество раствора подбирают таким образом, чтобы добиться pH конечной фармацевтической композиции 7. Все диапазоны концентраций, приводящие к конечному значению pH 6-8, входят в объем изобретения.
5. Изготовление состава по настоящему изобретению
Настоящее изобретение относится также к способу получения описанной выше фармацевтической композиции.
В одном из предпочтительных вариантов осуществления изобретения выбирают композицию эксципиентов, соотношение лекарственного средства к липидам и способ производства, которые благоприятствуют образованию двухслойной структуры липосом. В другом варианте осуществления выбирают такие параметры, которые благоприятствуют образованию мицеллярных наночастиц или комбинации мицелл и липосом.
В одном из вариантов осуществления диспергируемые в воде ингредиенты, например глицерин и олеат натрия, добавляют к нагретой воде, после чего добавляют гидроксид натрия и фосфолипиды. Добавляют производное формулы (I) и диспергируют до солюбилизации, используя миксер с высоким усилием сдвига. Затем массу продукта гомогенизируют в несколько циклов до достижения желаемого размера частиц, после чего осуществляют стерильное фильтрование и асептическое заполнение. В более предпочтительном варианте осуществления к нагретой воде добавляют глицерин и диспергируют в этой смеси производное формулы (I), используя миксер с высоким усилием сдвига. Добавляют остальные эксципиенты и перемешивают, после чего массу продукта гомогенизируют и осуществляют стерильное фильтрование, как указано выше.
На фиг.1 показана технологическая схема предпочтительного способа производства. Конкретно, на первой стадии воду для инъекций смешивают с глицерином и нагревают до целевой температуры 45°C. На второй стадии получения продукта постепенно добавляют олеат натрия, API элацитарабин, яичные фосфолипиды и NaOH и затем перемешивают, поддерживая температуру 45°C. На третьей стадии массу продукта перемешивают роторно-статорной мешалкой при 45°C в течение одного часа. На четвертой стадии продукт гомогенизируют при 25000 psi (фунтов/кв.дюйм) в течение не менее трех циклов для получения удовлетворительного размера частиц. Для контроля процесса измеряют средний размер частиц, профиль распределения размеров частиц и мутность после каждого цикла гомогенизации. Если мутность после цикла гомогенизации номер 3 составляет менее 600 NTU, состав считается готовым для следующей стадии способа. Если мутность превышает 600 NTU, осуществляют еще один цикл гомогенизации. При удовлетворительной мутности проводят анализ. Если результат анализа <102,0% коррекцию состава не проводят. Если результат ≥102,0%, добавляют рассчитанное количество воды для инъекций до достижения результата анализа 100%. Значение pH измеряют только для сведения. На пятой стадии осуществляют фильтрование для получения прозрачного состава, последовательно пропуская состав через фильтры 1,2 мкм и 0,45 мкм. Затем осуществляют стерильное фильтрование массы продукта через два стерильных 0,22 мкм фильтра. Фильтры проверяют на целостность и точку пузырька (давление, при котором начинают появляться пузырьки), используя воду для инъекций, и затем стерилизуют паром. Первый фильтр, который находится в непосредственном контакте с нестерильным продуктом, тестируют на целостность, используя массу продукта непосредственно перед стерильным фильтрованием. Оба фильтра тестируют на целостность и точку пузырька после фильтрования. На последней стадии способа продукт, прошедший стерильное фильтрование, непрерывным способом асептически разливают в стерилизованные, депирогенизированные стеклянные пузырьки. Наполненные пузырьки продувают азотом, и немедленно плотно закрывают стерильными резиновыми пробками и алюминиевыми колпачками.
Частицы готовой фармацевтической композиции подобны либо липосомам, т.е. представляют собой пузырьки, окруженные фосфолипидным двойным слоем, либо мицеллам, либо являются комбинацией частиц обоих видов. Размер частиц готовой фармацевтической композиции может находиться в пределах 5-45 нм, предпочтительно 9-25 нм, наиболее предпочтительно в диапазоне 10-20 нм при среднем размере частиц примерно 15 нм.
Фармацевтическая композиция по настоящему изобретению предпочтительно имеет жидкую форму, и может выпускаться в дозированных формах, например пузырьках, мешках для инфузии или подобных. Фармацевтической формой готовой композиции является суспензия или дисперсия либо липосом, либо мицелло-подобных наночастиц, либо комбинации частиц обоих типов.
Количество фосфолипидиной фазы в готовой фармацевтической композиции может меняться от примерно 0,1% до 50%, предпочтительно 1-15% и более предпочтительно 5-12%. В наиболее предпочтительном, но не ограничивающем варианте осуществления, количество фосфолипидной фазы составляет в готовой фармацевтической композиции 8-10%. Все поддиапазоны от 0,1% до 50% включены в объем настоящего изобретения.
Мольное отношение производного элацитарабина формулы (I) к общему количеству фосфолипидов в готовой фармацевтической композиции может находиться в пределах от 1:20 до 1:7. Наиболее предпочтительным диапазоном является от 1:13 до 1:8. Все поддиапазоны от 1:20 до 1:7 включены в объем изобретения. Предпочтительный вариант осуществления готового состава содержит от 5,0 мг/мл до 7,5 мг/мл элацитарабина, наиболее предпочтительно 7,5 мг/мл элацитарабина.
Мольное отношение яичного фосфатидилхолина к яичному фосфатидилэтаноламину в композиции может меняться в пределах от 1:1 до 99:1, предпочтительно от 2:1 до 80:1, причем наиболее предпочтительное отношение находится в диапазоне от 4:1 до 10:1. Все поддиапазоны соотношений от 1:1 до 99:1 включены в объем настоящего изобретения.
6. Дозировка
Термин «фармацевтически эффективное количество» в настоящем описании относится к количеству от примерно 0,001 до 10 граммов производного цитарабина формулы (I) или его фармацевтически приемлемой соли в день, более предпочтительно от примерно 10 мг до 6 граммов производного цитарабина формулы (I) или его фармацевтически приемлемой соли в день, в составе, содержащем 0,001-80% указанного производного или его соли и предназначенном для парентерального введения.
Фармацевтические композиции по настоящему изобретению применимы в лечении широкого спектра раковых заболеваний.
В случае солидных раковых опухолей, таких как рак яичников, немелкоклеточный рак легких, колоректальный рак и злокачественная меланома, предпочтительная схема внутривенного введения цитарабина включает введение 200 мг/м2 один раз в день в течение 5 дней, с 2-3-недельным перерывом между курсами введения.
В случае гематологических раковых заболеваний, например лейкозов, и конкретно включая острый миелоидный лейкоз, предпочтительно рекомендованная дневная доза для внутривенного введения элацитарабина, применяемого в виде единственного лекарственного средства или монотерапии, составляет 2000 мг/м2. Это означает, что для среднего пациента с площадью поверхности тела 1,8 м2, суммарная дневная доза элацитарабина должна составлять 3600 мг. В этом варианте осуществления предпочтительная схема внутривенного введения состава элацитарабина включает введение 2000 мг/м2 в день в виде непрерывной инфузии в течение 5 дней, с 2-3-недельным перерывом между курсами введения.
В случае гематологических раковых заболеваний, например лейкозов, и конкретно включая острый миелоидный лейкоз, предпочтительно рекомендованная дневная доза для внутривенного введения элацитарабина, применяемого в схеме комбинированной терапии (например, вместе с идарубицином), составляет 1000 мг/м2. Это означает, что для среднего пациента с площадью поверхности тела 1,8 м2, суммарная дневная доза элацитарабина должна составлять 1800 мг. В этом варианте осуществления предпочтительная схема внутривенного введения состава элацитарабина включает введение 1000 мг/м2 в день в виде непрерывной инфузии в течение 5 дней, с 2-3-недельным перерывом между курсами введения.
Далее по тексту, настоящее изобретение будет дополнительно объяснено с помощью примеров. Имеется в виду, что эти примеры являются только иллюстративными, и их не следует считать ограничивающими.
ПРИМЕРЫ
Пример 1:
Глицерин (2,22%) и олеат натрия (0,17%) добавляли к воде при 75°C и перемешивали. Добавляли 1М раствор гидроксида натрия до достижения значения pH 7-8. Добавляли 10% очищенных яичных фосфолипидов (PL90, Fresenius-Kabi) и диспергировали с помощью миксера с высоким усилием сдвига. Добавляли элацитарабин (0,5%) и перемешивали. Продукт гомогенизировали при 50°C в несколько циклов до достижения необходимого размера частиц. После этого продукт подвергали стерильному фильтрованию, разливали в стеклянные флаконы и герметично закрывали в атмосфере азота.
Сосуды хранили при 2-8°C, защищая от действия света, и следили за стабильностью препаратов данной партии в течение 24 месяцев. В течение этого периода исследования устойчивости препаратов наблюдали менее чем 2,5% уменьшение содержания элацитарабина.
Пример 2:
Готовили 2,22% мас./мас. раствор глицерина в воде и нагревали до 50°C. Добавляли элацитарабин до концентрации 0,75% мас./об. при энергичном перемешивании мешалкой с высоким усилием сдвига до получения тонкоизмельченной дисперсии. Одной порцией добавляли олеат натрия, гидроксид натрия и яичные фосфолипиды в тех же концентрациях, что и в примере 1, и тщательно перемешивали. Массу продукта гомогенизировали при 50°C в несколько циклов до достижения необходимого размера частиц, подвергали стерильному фильтрованию, разливали в стеклянные флаконы и герметично закрывали в атмосфере азота.
В отдельном эксперименте получали состав элацитарабина с содержанием действующего вещества 7,5 мг/мл согласно способу, приведенному ниже таблице 4:
Способ получения лекарственного продукта на основе элацитарабина и подробности изготовления препарата
Глицерин 3,330 кг
Проверка скорости перемешивания.
Гидратация и смешивание
API 1125г
Яичные фосфолипиды 15,0 кг
1н NaOH 112,5 г
Вода до 100%
Лопастная мешалка с целевой скоростью 300 об/мин и миксер с высоким усилием сдвига при 57Гц в течение 7±2 мин после добавления каждого исходного компонента.
Перемешивание мешалкой с высоким усилием сдвига
Миксер с высоким усилием сдвига при 57Гц в течение 60±5 мин.
Лопастная (пропеллерная) мешалка с целевой скоростью 400 об/мин в течение 5±2 мин после добавления воды.
Проверка массы.
Гомогенизация
Лопастная (пропеллерная) мешалка в подающей и приемной емкости с целевой скоростью 400 об/мин.
Проверка массы.
Фильтрование для получения прозрачного состава
Стерильное фильтрование
Заполнение и закрытие тары с готовым продуктом
Пример 3: Степень окисления и гидролиза фосфолипидов
Составы примера 1 тестировали на степень окисления и гидролиза фосфолипидов.
Для того чтобы избежать окисления жирных кислот, лекарственный продукт по настоящему изобретению производят в атмосфере азота с низким содержанием кислорода и флаконы продувают азотом перед запечатыванием.
Заявители провели эксперимент с целью определения и сравнения степени окисления жирных кислот в двух выбранных партиях лекарственного продукта, одна из которых имела возраст 16 месяцев с момента производства, а другая имела возраст 3 мес с момента производства и до начала программы исследования. Исследование этих партий производили параллельно, т.е. во всех экспериментах сравнивали один образец «свежей» партии и один образец «старой» партии. Для обоих образцов определяли пероксидное число, анизидиновое число, фосфолипидный профиль и общее содержание с помощью 31P-ЯМР, УФ-анализ проводили для определения соотношения сопряженных диенов и триенов, тест на содержание кислорода и малондиальдегидный анализ проводили для определения содержания циклических пероксидов.
Полученные результаты подтвердили, что степень окисления фосфолипидов является пренебрежимо малой.
Образцы описанных выше партий выдерживали также в атмосфере кислорода при 40°C, причем в этих условиях появилась возможность инициировать и измерить окисление. Кроме того, были получены партии плацебо, и впоследствии подвергнуты окислительному стрессу для подтверждения полученных выше результатов также и в отсутствии лекарственного соединения.
Результаты проведенных обширных исследований обеспечили достаточную гарантию того, что использование атмосферы азота и хранение лекарственного продукта при 2-8°C являются эффективными мерами для предотвращения окисления.
Другим важным путем разрушения фосфолипидов является гидролиз. Во время исследования стабильности продуктов осуществляли наблюдение за количеством лизофосфатидилхолина. Было показано, что в общей сложности менее 3,5 мол.% фосфатидилхолина подверглось гидролизу с образованием продуктов разложения во время изготовления и 24-месячного хранения при 2-8°C. Это подтверждает защитное действие олеата натрия на фосфолипиды.
Пример 4:
Проводили термический анализ состава, описанного в примере 1, по способу дифференциальной сканирующей калориметрии (DSC) для определения диапазона температур хранения и перевозки продукта. Было показано, что температура замерзания составляла -19,3°C, вероятно, из-за переохлаждения воды. Температура плавления составляла примерно -3,5°C. Эти данные позволили прийти к выводу, что перевозка и хранение при температуре 2-8°C не должны вызвать плавления или замерзания фосфолипидов и, следовательно, не должны оказать какого-либо отрицательного влияния на структуру частиц.
Пример 5:
Состав, описанный в примере 1, вводили 61 пациенту в рамках фазы II исследования монотерапии элацитарабином, в качестве второй резервной терапии для лечения острого миелогенного лейкоза (AML). Данные исследований продемонстрировали статистически значимое превосходство в эффективности у пациентов с рефракторным/рецидивирующим заболеванием с очень плохим прогнозом. Доля реакции пациентов составила 15%. Средний итоговый срок выживания составил 5,3 месяца, против 1,5 месяцев для исторических контрольных данных. Среднее выживание пациентов с терапевтическим эффектом составляло 13,5 месяцев. Доля выживших в течение 6 месяцев составляла 44%.
Побочные эффекты элацитарабина являются предсказуемыми и поддаются регулированию. Продукт хорошо переносился, также и пациентами пожилого возраста.
Фармакокинетические данные, представленные на фиг.2, показывают по крайней мере 10-кратное превышение концентрации в плазме для элацитарабина по сравнению с цитарабином (Ara-C).
Пример 6:
Заявители осуществили несколько попыток увеличить соотношение между лекарственным средством и фосфолипидами во внутривенных составах элацитарабина.
Первый состав элацитарабина, использованный в клинических испытаниях, имел концентрацию 10 мг/мл и точно такой же состав, который был описан в примере 1, но только с более высоким содержанием элацитарабина. Этот продукт претерпел осаждение через несколько месяцев после получения, и был отозван из клинических учреждений, в которых проводилось исследование. Проведенный впоследствии анализ супернатанта показал, что остаточное содержание лекарственного средства в липосомах составило 7-7,5 мг/мл.
Получали еще одну группу составов, чтобы исследовать эффект от других комбинаций фосфолипидов и от изменения способа приготовления состава, где измененный способ включал введение в композицию раствора действующего вещества в органическом растворителе. Первая серия экспериментов в общем виде представлена в таблице 5. Способ получения состоял из растворения фосфолипидов и элацитарабина в этаноле, после чего осуществлялось регулируемое введение полученного этанольного раствора в смесь глицерин/вода. Полученную массу продукта гомогенизировали, осуществляя до 7 циклов, и затем концентрировали до целевого объема с помощью фильтрования тангенциального потока (TFF) и тем же способом удаляли избыток этанола.
Составы яичный PC/яичный PG были дополнительно исследованы при более низких концентрациях лекарственного средства: 10, 8,5 и 7,5 мг/мл. Итоговое содержание действующего вещества составило соответственно 6,5, 6,8 и 5,7 мг/мл. Дальнейшее исследование показало, что соотношение между лекарственным средством и липидами резко падает при TFF фильтровании. Более важно, что способ введения раствора в этаноле, по-видимому, не дает возможности добиться более высокого соотношения между лекарственными средствами и липидами или какого-либо другого положительного эффекта для составов по настоящему изобретению по сравнению со значительно менее затратным исходным способом получения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОСОМАЛЬНЫЙ ПРЕПАРАТ ДЛЯ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ | 2017 |
|
RU2756755C2 |
| КОМПОЗИЦИИ АНТАГОНИСТОВ НЕЙРОКИНИНА-1 ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ | 2010 |
|
RU2642234C2 |
| ЛИПОСОМАЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ | 1995 |
|
RU2160099C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ УВЕЛИЧЕНИЯ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ АКТИВНОГО ВЕЩЕСТВА С ИСПОЛЬЗОВАНИЕМ ДАННОГО СОСТАВА | 1991 |
|
RU2104715C1 |
| ЛИПОСОМАЛЬНОЕ СРЕДСТВО НА ОСНОВЕ УБИХИНОЛА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2605616C1 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ | 2010 |
|
RU2476216C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2009 |
|
RU2522977C2 |
| ЖИРОВАЯ ЭМУЛЬСИЯ ДЛЯ ИНГАЛЯЦИИ | 1999 |
|
RU2212230C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2003 |
|
RU2361615C2 |
| ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ МАСЛА КРИЛЯ | 2012 |
|
RU2625760C2 |

Настоящее изобретение относится к парентеральным составам для введения некоторых производных, образованных 1-β-D-арабинофуранозилцитозином (цитарабином) и насыщенными и мононенасыщенными жирными кислотами с длинной цепью (элацитарабин). В частности, настоящее изобретение относится к парентеральной фармацевтической композиции, позволяющей вводить терапевтически эффективные дозы указанных производных, которая увеличивает приверженность пациентов терапии при лечении рака, а также способу ее получения. 2 н. и 13 з.п. ф-лы, 6 прим., 5 табл., 2 ил
1. Фармацевтическая композиция, включающая в качестве действующего ингредиента элацитарабина формулы (I)

или его фармацевтически приемлемую соль в качестве действующего ингредиента;
где указанный действующий ингредиент растворен или диспергирован в составе, содержащем солюбилизатор, включающий один или несколько фосфолипидов, ко-солюбилизатор, включающий ПАВ, и изотонический агент, представляющий собой глицерин,
где добавленное количество составляет 2-5% мас./об. от конечной фармацевтической композиции и, где размер частиц состава находится в пределах 5-45 нм.
2. Фармацевтическая композиция по п. 1, где солюбилизатор включает один или несколько фосфолипидов, которые выбраны из группы, состоящей из фосфатидилхолина, лизо-фосфатидилхолина 1, лизо-фосфатидилхолина 2, фосфатидилглицерина, фосфатидилэтаноламина, лизо-фосфатидилэтаноламина, фосфатидилинозита, фосфатидилсерина, фосфатидиновой кислоты, сфингомиелина и кардиолипина, каждый из которых может присутствовать в любой форме, включая солевые или обессоленные, гидрированные или частично гидрированные, природные, полусинтетические или синтетические.
3. Фармацевтическая композиция по п. 1, где фосфолипиды представляют собой природные ненасыщенные фосфолипиды, полученные из куриных яиц.
4. Фармацевтическая композиция по п. 1, где ко-солюбилизатором является олеат натрия.
5. Фармацевтическая композиция по п. 1, где pH состава находится в пределах от 6,0 до 8,0.
6. Фармацевтическая композиция по п. 1, где размер частиц состава находится в пределах 9-25 нм.
7. Фармацевтическая композиция по п. 1, где мольное отношение лекарственное средство:липид находится в пределах от 1:20 до 1:7.
8. Фармацевтическая композиция по п. 1, где мольное отношение лекарственное средство:липид находится в пределах от 1:13 до 1:8.
9. Фармацевтическая композиция по п. 1, где конечная концентрация элацитарабина в составе находится в пределах от 5,0 до 7,5 мг/мл.
10. Способ получения фармацевтической композиции по п. 1, включающий стадии:
a) смешивания воды с изотоническим агентом и нагревания полученной смеси при 45±5°C,
b) добавления к смеси стадии а) солюбилизатора, ко-солюбилизатора и действующего ингредиента и перемешивание с высоким усилием сдвига,
c) гомогенизации смеси стадии b) при действии на смесь высокого давления и
d) фильтрования полученного продукта.
11. Способ по п. 10, где перемешивание с высоким усилием сдвига на стадии b) осуществляют при 57 Гц в течение примерно 1 ч.
12. Способ по п. 10, где гомогенизацию при высоком давлении на стадии с) проводят под давлением примерно 25000 фунтов/кв. дюйм.
13. Фармацевтическая композиция по любому из пп. 1-9 для применения в способе лечения солидных опухолей, где указанный способ лечения включает введение указанной композиции в дозировке 200 мг/м2 один раз в день в течение 5 дней, с перерывом 2-3 недели между курсами введения.
14. Фармацевтическая композиция по любому из пп. 1-9 для применения в способе лечения гематологических опухолей, где указанный способ лечения включает введение указанной композиции в виде монотерапии в дозировке 2000 мг/м2 один раз в день в течение 5 дней, с перерывом 2-3 недели между курсами введения.
15. Фармацевтическая композиция по любому из пп. 1-9 для применения в способе лечения гематологических опухолей, где указанный способ лечения включает введение указанной композиции в схеме комбинированной терапии в дозировке 1000 мг/м2 один раз в день в течение 5 дней, с перерывом 2-3 недели между курсами введения.
| Schwendener R | |||
| A.; liposomes as carriers for lipophilic antitumor prodrugs | |||
| Incorporation characteristics and in vivo cytotoxic activity | |||
| Пневматический водоподъемный аппарат-двигатель | 1917 |
|
SU1986A1 |
| Аппарат для передачи фотографических изображений на расстояние | 1920 |
|
SU170A1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОТИВОРАКОВЫЙ АГЕНТ | 2000 |
|
RU2248969C2 |
| Счетная таблица | 1919 |
|
SU104A1 |
| EA 200501302, НГУЕН-ХУАН ЧО (СН), 28.04.2008 | |||
| СТР | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

