Область техники
Изобретение относится к области биотехнологии и персонализированной медицины. Объект заявки представляет собой способ идентификации активированных сигнальных путей (молекулярных механизмов) специфичных для развития определенного подтипа онкологических заболеваний у индивидуального пациента. Для этого используются данные о последовательности ДНК (экзом) и уровне экспрессии генов (транскриптом) для образцов нормальной ткани и ткани опухоли. В частности, изобретение позволяет генерировать персонализированные рекомендации для оптимизации схем противоопухолевой фармакотерапии и может быть использовано в медицине.
Уровень техники
Ключевой тенденцией в современном лечении онкологических заболеваний является то, что постановка диагноза, прогнозирование течения заболевания, оценки риска и ответа на терапию могут быть значительно улучшены путем стратификации пациентов на основании геномных, транскриптомных и эпигеномных характеристик опухоли. Под стратификацией подразумевается разделение пациентов на подгруппы объединенные общими активированными молекулярными механизмами, ответственными за развитие заболевания. При этом для каждой отдельной подгруппы пациентов со специфическим молекулярным механизмом онкогенеза может применяться специфическая лекарственная терапия. Такая персонализированная терапия может быть более эффективной по сравнению со стандартной схемой, т.к. учитывает специфику заболевания у отдельного пациента.
Особый интерес представляют методы, позволяющие при стратификации пациентов одновременно учесть различные типы данных, характеризующие молекулярные свойства опухоли, в частности - объединить данные о копийностях генов, имеющихся соматических мутациях и степени экспрессии соответствующих мРНК. Другим желательным свойством методов стратификации пациентов является интегрирование с существующим биологическим знанием, представленным в виде набора генных онтологий, сигнальных путей, а также сетей межгенных и межбелковых взаимодействий. Под интегрированием понимается учет и использование при поиске активированных молекулярных механизмов онкогенеза известной биологической информации хранящейся в различных базах данных, таких как Gene Onthology, KEGG, Wiki Pathways, DrugBank, Reactome, PharmGKB и подобных ресурсах. Данная область является относительно новой, но активно развивается в последнее время.
К настоящему моменту разработано несколько способов использования информации об уровне экспрессии и последовательностях генов для стратификации пациентов (Bennett В.D., Xiong Q., Mukherjee S. and Furey Т. S.A predictive framework for integrating disparate genomic data types using sample-specific gene set enrichment analysis and multi-task learning (2012) PLoS One 7, e44635; Ng S., Collisson E.A., Sokolov A., Goldstein Т., Gonzalez-Perez A., Lopez-Bigas N., Benz C., Haussler D, and Stuart J.M. PARADIGM-SHIFT predicts the function of mutations in multiple cancers using pathway impact analysis (2012) Bioinformatics 28, i640-i646; Xiong Q., Ancona N., Hauser E.R., Mukherjee S. and Furey T.S. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets (2012) Genome Res 22, 386-397). Однако ни в одном из указанных методов не сочетаются анализ активированных генных сетей с объединением экзомных и транскриптомных данных на уровне отдельного пациента.
Из остальных методов наиболее близок к патентуемому изобретению метод iCluster + (Мо Q., Wang S., Seshan V.Е., Olshen А.В., Schultz N., Sander С., Powers R.S., Ladanyi M. and Shen R. Pattern discovery and cancer gene identification in integrated cancer genomic data (2013) Proc Natl Acad Sci USA 110, 4245-4250). Авторами разработан единый математический аппарат, основанный на регрессионном и факторном анализе, и позволяющий объединить множество разнообразных типов данных. Так наличие мутации в каждом гене моделируется как бинарная переменная, что позволяет использовать метод логистической регрессии. Данные о копийностях генов трактуются в категориальной шкале, и для их анализа применяется мультиномиальная логит-модель. Наконец количество прочтений мРНК полученных методом RNASeq обрабатывается пуассоновской регрессией. Построенная модель оптимизируется путем отбора значимых переменных. Наибольший интерес представляют кластер-специфичные гены, характеризующие тот или иной молекулярный механизм. Однако для метода не разработана валидация построенной модели, большое число параметров значительно увеличивает риск чрезмерной подстройки модели под данные (переобучение), а найденным скрытым (латентным) переменным не дается какая-либо биологическая интерпретация. Также в методе не используется априорное биологическое знание (канонические сигнальные пути).
Из других методов можно упомянуть алгоритм PARADIGM (Ng S., Collisson E.A., Sokolov A., Goldstein Т., Gonzalez-Perez A., Lopes-Bigas N., Benz C., Haussler D. and Stuart J.M. PARADIGM-SHIFT predicts the function of mutations in multiple cancers using pathway impact analysis (2012) Bioinformatics 28, i640-i646; Vaske C.J., Benz S.C., Sanborn J.Z., Earl D., Szeto C., Zhy J., Haussler D. and Stuart J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM (2010) Bioinformatics 26, i237-245), который интегрирует данные о копийностях генов с транскриптомным анализом, используя обширную коллекцию сигнальных путей NCI PID (Schaefer С.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay Т. and Buetow K.H. PID: the Pathway Interaction Database (2009) Nucleic Acids Res 37, D674-679). Каждый сигнальный путь конвертируется в вероятностную модель (факторный граф), где моделируются регуляции транскрипции и трансляции. Аналогично методу максимального правдоподобия для каждого гена оценивается мера его активности при условии имеющихся данных. Полученная матрица активностей генов используется для проведения иерархического кластерного анализа пациентов. Найденные группы пациентов показывают достоверное различие в выживаемости. Однако в указанном методе не учитываются известные соматические мутации (экзомные данные), а для применения метода требуется значительная выборка пациентов, что ограничивает его применение на практике.
В большинстве других алгоритмов предложенных для стратификации пациентов (патенты ЕР 2297359 A1, US 20120252856 A1, 2473555 С2) предлагается использовать заранее зафиксированный набор генов либо входящих в общий сигнальный путь, либо отобранных в результате независимого поиска биомаркеров и не учитывающих известные белок-белковые взаимодействия. Это не позволяет идентифицировать случаи, когда у индивидуального пациента активирован другой регуляторный каскад, ответственный за развитие опухоли и не входящий в априорно заданный набор генов. Соответственно подход с фиксированным набором генов не может учесть протекание заболевания в каждом отдельном случае. Такие наборы генов представляют собой потенциальные многомерные биомаркеры, идентифицированные с помощью различных процедур статистического анализа транскриптомных данных, включая методы распознавания образов. Однако несмотря на проделанный объем работ в данной области, большинство найденных таким способом биомаркеров было невоспроизводимо в независимых исследованиях, а клиническая значимость была продемонстрирована для единичных наборов биомаркеров. Причинами слабой воспроизводимости результатов являются значительные статистические трудности при обработке данных характеризующимися десятками тысяч переменных (т.н. 'проклятие размерности') и наличие существенной биологической вариабельности между образцами.
Актуальность изобретения
Популяции клеток опухоли чрезвычайно гетерогенны как в морфологических, так и в функциональных аспектах, а соответствующие молекулярные механизмы ответственные за прогрессирование опухоли очень разнообразны. Поэтому одной из важнейших проблем современной онкологии является разработка персонализированного подхода к таргетной (целенаправленной) терапии опухолей, заключающегося в учете индивидуальных особенностей молекулярного профиля конкретного новообразования. Воздействие на специфические для опухоли нарушения в активности сигнальных каскадов все чаще рассматривается как основная причина эффективности ее ответа на ту или иную схему направленной терапии. Также важность персонифицированного подхода к выбору таргетной химиотерапии обусловлена высокой стоимостью и высокой токсичностью противоопухолевых препаратов. При этом ошибка в определении специфической терапии может выявиться только через несколько месяцев, когда прогрессирование опухоли вследствие неэффективной терапии приведет к необратимым последствиям. Резюмируя, можно заключить, что в области оптимизации схем лечения онкологических заболеваний как нигде справедлив подход "лечить больного, а не болезнь".
За последнее десятилетие существенный прогресс в области изучения молекулярных особенностей злокачественных клеток достигнут благодаря интенсивному развитию высокопроизводительных методов геномного секвенирования (Next-Generation Sequencing, NGS). Снижение стоимости технологий детектирования однонуклеотидных полиморфизмов (Single Nucleotide Polymorphism, SNP) и определения уровней экспрессии всей совокупности мРНК транскриптома (технология RNASeq) позволило проводить крупномасштабные международные проекты для получения информации о генетических профилях различных видов раковых заболеваний.
В то же время до сих пор не разработаны устоявшиеся алгоритмы и подходы по интеграции массивов различных типов NGS-данных с целью их биологического осмысления с точки зрения накопленных знаний о взаимодействии и регуляции генов и белков. Это сильно затрудняет клиническое применение результатов проведенных геномных и транскриптомных анализов. Для определения дерегулированных сигнальных каскадов и соответственно индивидуального подбора терапии необходимо разработать метод по наложению имеющихся результатов экзомного и транскриптомного анализа на заранее сформированные экспертом сигнальные каскады (pathways), биологические сети и генные сигнатуры.
Раскрытие изобретения
Разработанная методика представляет собой способ анализа данных о соматических мутациях и экспрессии генов для идентификации белков-мишеней в составе активированных сигнальных путей специфичных для развития определенного подтипа онкологических заболеваний у индивидуального пациента и генерации персонализированных рекомендаций для оптимизации схем противоопухолевой фармакотерапии.
Данные об экспрессии генов могут быть получены с помощью различных технологий. Наиболее распространены два подхода - метод биочипов (microarray) и метод RNASeq, представляющий собой секвенирование следующего поколения (NGS) и основанный на прямом прочтении последовательности нуклеотидов в молекулах дезоксирибонуклеиновых кислот.
Отличие предлагаемого подхода от известных методов состоит в том, что идентификация активированных сигнальных путей проводится для каждого образца (пациента) индивидуально, т.е. набор потенциальных биомаркеров заранее не зафиксирован, а определяется 'на лету'. Также важной особенностью патентуемого подхода является ориентация на поиск именно регуляторных межгенных взаимодействий, а не набора дифференциально экспрессированных генов. Многие регуляторы входят в состав сигнальных путей и могут служить указанием на молекулярный механизм специфического подтипа заболевания. При этом уровень экспрессии самого регулятора часто меняется незначительно, что не позволяет выявить его при помощи стандартных методов поиска дифференциально экспрессируемых генов. Наконец в предлагаемом подходе предлагается способ интегрирования транскриптомных и экзомных данных путем проецирования их на известные сигнальные каскады. Для этого производится поиск сигнальных каскадов статистически достоверно обогащенных генами/белками, полученными в результате анализа данных по экспрессии и последовательностей ДНК. Изучение указанных сигнальных каскадов экспертом-биологом дает возможность построить модель развития заболевания для индивидуального пациента, а изучение состава найденных значимых сигнальных каскадов на предмет известных белков-мишеней дает возможность предложить персонализированную оптимальную противоопухолевую терапию.
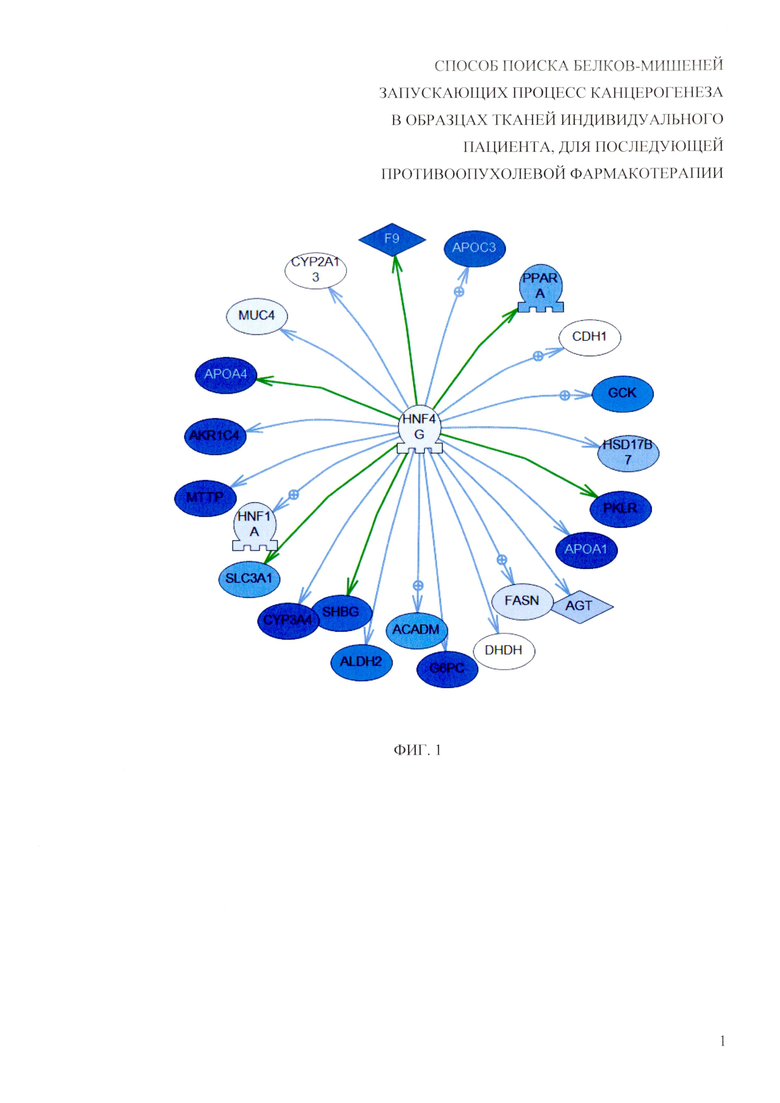
Для решения заявленной задачи были использованы методы поиска активированных генных регуляторных сетей по транскриптомным данным. Генная сеть в простейшем случае состоит из центрального регулятора (в частности, транскрипционного фактора) и генов, чья экспрессия зависит от указанного регулятора. Важным свойством генной регуляторной сети является то, что экспрессия всех генов скоординировано реагирует на изменение состояния, например, при сравнении опухоль - норма. Распределение логарифмов величин отношения экспрессии в опухоли по отношению к норме (log-ratio) для генов, контролируемых центральным регулятором, должно статистически достоверно отличаться от фонового распределения, построенного для всех генов, измеренных в эксперименте. Для поиска активированных генных регуляторных сетей предлагается использовать алгоритм SNEA, Subnetwork Enrichment Analysis (Sivachenko A.Y., Yuryev A., Daraselia N. and Mazo I. Molecular networks in microarray analysis (2007) J Bioinform Comput Biol 5, 429-456), однако возможны и другие алгоритмы поиска активированных генов-регуляторов, т.н. upstream regulators. Пример генной сети представлен на рисунке (фиг. 1), где цветом указан относительный уровень экспресии гена в опухоли по сравнению с нормой. В этом случае регулятор экспрессии - транскрипционный фактор HNF4G имеет пониженную экспрессию в опухоли, однако регулируемые им гены (в частности, АРОА1, PKLR, CYP3A4) показывают значительно более выраженное изменение в экспрессии. Таким образом, наблюдаемое изменение экспрессии указанных генов можно объяснить понижением экспрессии самого транскрипционного фактора - регулятора.
Поиск регуляторов, обуславливающих изменение экспрессии генов, играет важную роль при анализе молекулярных механизмов рака. Большинство онкогенов и опухолевых супрессоров как раз являются регуляторами генной экспрессии и играют ключевую роль в процессах туморогенеза, несмотря на то, что не являются наиболее дифференциально экспрессированными генами.
В качестве исходных данных о межгенной регуляции используется биологическая сеть (граф) ResNet, выпускаемая компанией Elsevier. Ее особенность заключается в том, что узлы графа - всевозможные классы молекулярно-биологических сущностей (белок, ген, малая молекула, функциональный класс, клеточный процесс, комплекс, т.д.) - связаны между собой различными типами ребер, включая регуляторные (регуляция транскрипции) и физические межгенные/межбелковые взаимодействия. Важно отметить, что сеть ResNet построена с помощью методик компьютерной обработки естественного языка (Natural Language Processing, NLP) и описывает более чем 1500000 взаимодействий полученных при анализе более чем 22000000 резюме и 880000 полных текстов статей по медико-биологической тематике. Поскольку в биологической сети Resnet представлены различные типы сущностей, то при поиске активированных регуляторных сетей при помощи алгоритма SNEA в качестве регулятора может выступать другая сущность, которая не измеряется в эксперименте, например группа генов/белков (функциональный класс), клеточный процесс и т.д.
Полученные транскриптомные данные анализируются на предмет выявления дифференциально экспрессированных генов, т.е. генов, которые статистически значимо изменили свою экспрессию в образце опухоли по сравнению с нормой. Для этого может использоваться любой алгоритм для анализа транскриптомных данных, такой, например, как edgeR (Dimont Е., Shi J., Kirchner R., Hide W., edgeRun: an R package for sensitive, functionally relevant differential expression discovery using an unconditional exact test. Bioinformatics. 2015 Aug l;31(15:2589-90. doi: 10.1093/bioinformatics/btv209. Epub 2015 Apr 21) или DESeq (Anders S., Huber W. Differential expression analysis for sequence count data. GenomeBiol. 2010; 11(10):R106. doi: 10.1186/gb-2010-11-10-r106. Epub 2010 Oct. 27).
Далее обрабатываются данные по соматическим мутациям. Анализ последовательности генома (экзома) пациента дает дополнительный уровень понимания функционирования молекулярных механизмов, ответственных за развитие опухоли. Для этого используется коллекция известных канонических сигнальных путей. Такая коллекция может быть собрана из открытых источников (базы данных KEGG http://www.genome.jp/kegg/, Reactome http://www.reactome.org/, WikiPathways http://www.wikipathways.org/index.php/WikiPathways, PID http://pid.nci.nih.gov/), так и дополнена сигнальными путями, специально созданными для этой цели экспертами-биологами. Каждый сигнальный путь представляет собой набор функционально родственных генов, связанных регуляторными соотношениями и/или белок-белковыми взаимодействиями. Среди такой коллекции известных канонических сигнальных путей отбираются сигнальные пути, которые достоверно обогащены рядом "важных" генов. К таким "важным" генам относятся ранее найденные регуляторы экспрессии, дифференциально экспрессированные гены, гены с идентифицированными соматическими мутациями (полученные, например, из базы данных COSMIC, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), гены-драйверы для которых известна из научной литературы их связь с онкологическими заболеваниями (Krishnan V.G. and Ng Р.С. Predicting cancer drivers: are we there yet? (2012) Genome Med 4, 88).
Для отбора сигнальных путей обогащенных "важными" генами используется известный из уровня техники статистический метод - точный тест Фишера (Rivals I., Personnaz L., Taing L. and Potier, M.C. Enrichment or depletion of a GO category within a class of genes: which test? (2007) Bioinformatics 23, 401-407), причем сигнальный путь считается достоверно обогащенным, если вычисленное значение уровня значимости не превышает величины 0,05.
Таким образом, определяются сигнальные пути, активированные у индивидуального пациента и играющие потенциально значимую роль в прогрессе заболевания. Эта информация затем может быть использована экспертом-биологом для построения персонализированной модели туморогенеза, а также использована для поиска лекарственных препаратов, мишенью которых являются найденные белки.
Таким образом, интеграция экзомных и транскриптомных данных производится на уровне канонических сигнальных путей, активированных у данного пациента.
Помимо поиска сигнальных путей, обогащенных регуляторами генной экспрессии, возможно проведение поиска сигнальных путей, на которых перепредставлены дифференциально экспрессирующиеся гены (также с помощью точного теста Фишера), которые являются потенциальными биомаркерами прогресса опухоли. Измеряя изменение экспрессии таких биомаркеров, а также взаимодействующих белков, возможно контролирование эффективности применяемой фармакотерапии.
Идентифицированные на предыдущем этапе сигнальные пути анализируются на предмет наличия в их составе белков-мишеней для лекарственных препаратов, предназначенных для противоопухолевой терапии, известных из литературных источников. Используя информацию о существующих лекарственных препаратах и их белках-мишенях, а также о составе найденных активированных канонических сигнальных путей, генерируются персонализированные рекомендации по оптимизации противоопухолевой фармакотерапии. Для этого проводится поиск в существующих базах данных (в частности, база DrugBank) информации о лекарственных препаратах и их мишенях на предмет присутствия белков, содержащихся в идентифицированных сигнальных путях, а именно - если белок-мишень для какого-либо лекарственного препарата, предназначенного для противоопухолевой терапии, входит в состав идентифицированного сигнального пути, то такой препарат рекомендуется пациенту для проведения химиотерапии.
Дополнительно возможен учет наличия у пациента полиморфизмов, для которых показана их связь с чувствительностью к определенному препарату. Для этого проводится поиск в существующих базах данных по фармакогеномике (в частности, база PharmGKB) на предмет присутствия аннотированных однонуклеотидных полимофизмов (SNP), для которых экспериментально установлена взаимосвязь между наличием данного SNP с и чувствительностью к терапии указанным фармакологическим препаратом.
Таким образом, удается объединить экспертное знание относительно известных межгенных и межбелковых регуляторных взаимодействиях (канонические сигнальные каскады) с индивидуальными особенностями развития опухоли как на уровне экзома (поиск регуляторов и сигнальных путей значимо обогащенных функциональными SNP), так и на уровне транскриптома (активированные генно-регуляторные сети определяются "на лету" отдельно для каждого пациента).
Найденные списки задействованных сигнальных путей, являющиеся результатом анализа индивидуального экзома и транскриптома, представляют значительный интерес при последующем экспертно-биологическом анализе. Отталкиваясь от найденных сигнальных путей и соответствующих межгенных и межбелковых регуляторных и других взаимодействий, эксперт-биолог может создать персонализированную модель развития онкологического заболевания в отдельном индивидууме и, соответственно, дать рекомендации относительно оптимальной противоопухолевой фармакотерапии.
Краткое описание чертежей
На фиг. 1 приведен пример активированной генной сети. Интенсивностью цвета закодировано изменение экспрессии в опухоли по сравнению с нормой. Незначительное изменение экспрессии регулятора (транскрипционного фактора HNF4G) приводит к существенному изменению экспрессии регулируемых им генов: АРОА1, PKLR, CYP3A4 и других генов.
Осуществление изобретения
Исходными данными являются набор транскриптомных данных и информация о соматических мутациях, представленная в виде набора однонуклеотидных полиморфизмов (SNP) или инделов. Экзомные и транскриптомные данные должны быть получены как для образца опухоли, так и для образца исходной (здоровой) ткани. Для экспертного анализа необходимо использование набора сигнальных каскадов (pathways), созданных специально для изучаемого онкологического заболевания и/или полученных из внешних источников. В качестве внешних источников сигнальных путей могут выступать базы KEGG, Reactome, Wikipathways и другие. Предпочтительно, чтобы наборы сигнальных путей были загружены в специализированный программный продукт, например Pathway Studio (Elsevier), IPA (Ingenuity Systems) или аналоги.
Продемонстрируем осуществление изобретения на примере.
Были получены образцы опухоли и здоровой ткани больного, страдающего от гепатоцеллюлярной карциномы невирусной этиологии. Было предложено провести анализ согласно данному описанию изобретения с целью подобрать максимально эффективную химиотерапию именно для указанного больного.
Биопсия была проведена во время операции по частичному хирургическому удалению опухоли. Доля содержания опухолевых клеток составила 80%. Парные образцы норма/опухоль были проанализированы методом секвенирования следующего поколения на секвенаторе Illumina HiSeq2000. Были получены как данные экзомного секвенирования, так и данные по экспрессии генов. Длина прочтения составила 100 нуклеотидов. Количество прочтений для ДНК в нормальной и опухолевой ткани составило 127243260 и 237545808 соответственно. Количество прочтений для РНК в нормальной и опухолевой ткани составило 83546978 и 107422142 соответственно.
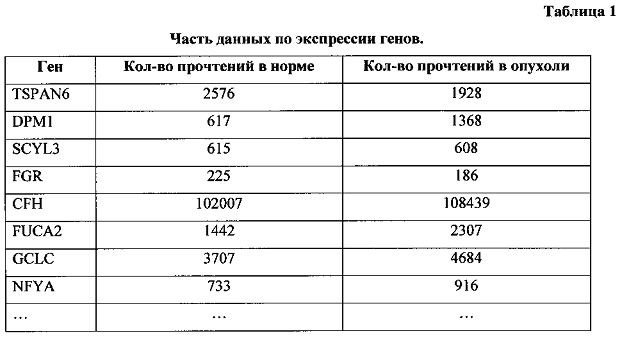
На первом этапе анализировались данные по экспрессии генов. В таблице 1 приведена часть полученных данных.
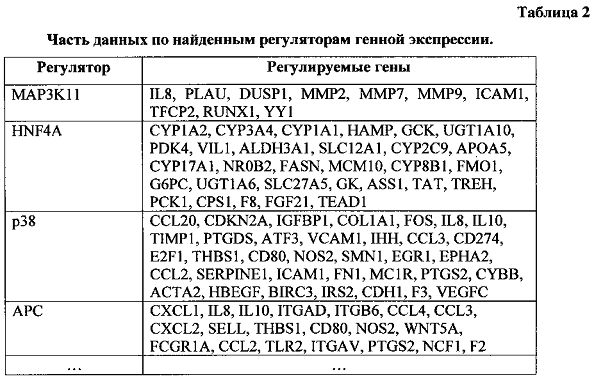
Полученные транскриптомные данные были использованы для поиска активированных генных регуляторных сетей с помощью алгоритма SNEA, Subnetwork Enrichment Analysis (Sivachenko et al., 2007). Всего было идентифицировано 23 регулятора. На следующей таблице приведена часть найденных генно-регуляторных сетей, состоящих из центрального регулятора и регулируемых им генов (табл. 2).
Далее с помощью алгоритма DEseq были проанализированы транскриптомные данные на предмет выявления дифференциально экспрессированных генов. Для этого использовали алгоритм DESeq (Anders&Huber, 2010). Всего было выявлено 314 таких генов, включая гены TGFA, SRC, IGF1, FOS, TGFB1, NRG1, CDK1, AREG, ID1, SPP1, АРОА1, SMAD3, MKI67, SMAD7.
Были проанализированы данные по экзомному секвенированию. Было выявлено 39 соматических несинонимических мутаций, из которых 36 мутаций представляли собой замены, две мутации являлись инсерциями и одна мутация представляла собой делецию.
Среди выявленных мутаций были предсказаны потенциальные гены-драйверы онкогенеза согласно алгоритму CHASM (Carter et al., 2009). Всего было предсказано три гена: ROR2, RPS6KA5 и NRAS.
Была создана коллекция канонических сигнальных путей, которая включала в себя данные из открытых баз данных KEGG http://www.genome.jp/kegg, Reactome http://www.reactome.org, WikiPathways http://www.wikipathways.org и PID http://pid.nci.nih.gov. Дополнительно был создан сигнальный путь, отражающий особенности функционирования EGFR-каскада в гепатоцеллюлярной карциноме. Всего в коллекцию было включено 314 сигнальных каскада.
Среди сигнальных каскадов из коллекции был проведен поиск сигнальных путей, значимо обогащенных несколькими категориями "важных" генов, а именно:
1) идентифицированными регуляторами генной экспрессии с помощью алгоритма SNEA;
2) генами с идентифицированными однонуклеотидными полиморфизмами и/или вставками/заменами, т.е. несинонимичными мутациями;
3) генами, показывающими дифференциальную экспрессию между опухолевой и здоровой тканью;
4) генами-"драйверами" опухолевого процесса согласно предсказанию веб-сервера CHASM (Carter Н., Chen S., Isik L., Tyekucheva S., Velculescu V.E., Kinzler K.W., Vogelstein В. and Karchin R. Cancer-specific high-throughput annotation of somatic mutation: computational prediction of driver missense mutations (2009) Cancer Res 69, 6660-6667).
Оценка значимости обогащения сигнального пути проводилась с помощью точного теста Фишера (Rivals et al., 2007), причем сигнальный путь считался значимо обогащенным, если вычисленное значение уровня значимости не превышает величины 0,05.
Приведем пример вычисления значимости обогащения сигнального пути генами, в которых были обнаружены соматические мутации. Всего таких генов было выявлено 39. Рассмотрим сигнальный каскад BDNF signaling pathway [source:Wikipathways], в который входит 140 генов. Составляется таблица, где определяется мера перекрывания сигнального каскада и генов с соматическими мутациями (табл. 3). Например, только один ген (RPS6KA5) одновременно входит в сигнальный каскад BDNF signaling pathway [source:Wikipathways] и мутирован в образце ткани пациента, в то время как среди остальных 139 генов каскада мутаций выявлено не было.

Далее согласно точному теста Фишера вычисляется уровень значимости p по формуле
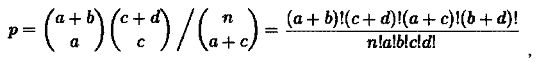
где n=a+b+c+d - общее количество генов. В данном случае значение p равно 0,238. Эта величина меньше общепринятого порога 0,05, а следовательно, данный сигнальный путь не будет считаться значимо обогащенным.
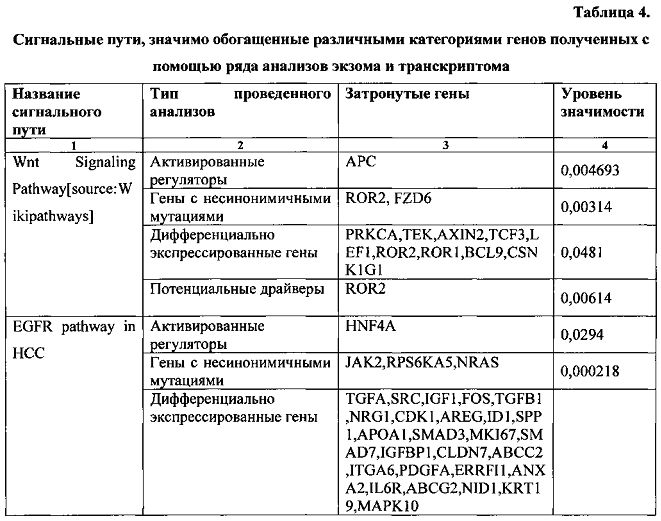
Ниже приведены (табл. 4) результаты поиска значимо обогащенных сигнальных путей.
14

Как следует из таблицы, в исследуемом пациенте активны сигнальные пути WNT signaling, EGFR signaling, и р38-МАРК signalling. Был проведен поиск в базах данных DrugBank и PharmGKB на предмет того, существуют ли известные белки-мишени, которые входят в состав указанных сигнальных путей. В таблице 5 приведены лекарственные препараты и их белки-мишени, входящие в состав указанных сигнальных путей.
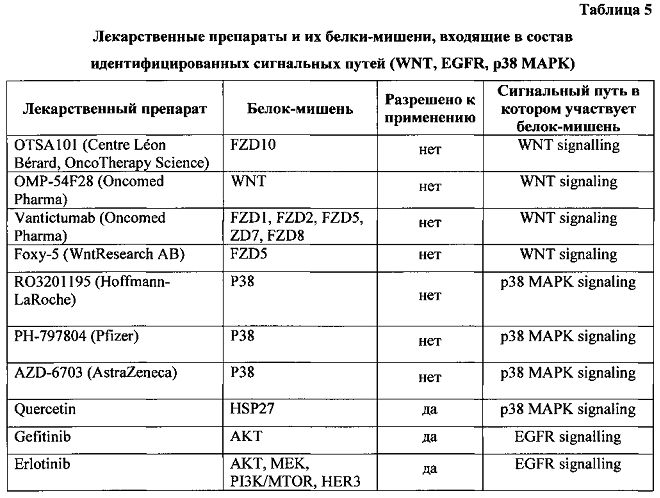
На сегодняшний момент лекарственные препараты, ингибирующие сигнальный каскад WNT (например, Vantictumab, OTSA101 и OMP-54F28), пока находятся в первой фазе клинических испытаний (Blagodatski et al., 2014). В то же время для сигнального каскада EGFR signalling существует ряд активно использующихся в клинике препаратов, а именно такие лекарственные средства как гефитиниб и эрлотиниб (Dienstmann R., De Dosso S., Felip E. and Tabernero J., Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol Oncol. 2012 Feb; 6(1): 15-26. doi:10.1016/j.molonc.2011.11.009. Epub 2011 Dec 6). Перспективно также применение кверцетина, как действующего на сигнальный каскад р38 MAPK (Chen S.F., Nieh S., Jao S.W., Liu C.L., Wu С.H., Chang C.Y. and Lin Y.S. Quercetin suppresses drug-resistant spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer cells (2012) PLoS One 7, e49275), но в то же время не являющегося препаратом выбора для лечения онкологических заболеваний.
Список найденных мутаций был проанализирован на наличие полиморфизмов, для которых известна их связь с чувствительностью к какому-либо препарату. Была выявлена мутация в гене FAS (1377G/A), для которой была показана повышенная чувствительность клеток опухоли к препарату (Ma F., Liao Y., Zi-Ping W., Xu В. Single nucleotide polymorphisms in FAS and FASL and sensitivity to gefitinib in patients with advanced non-smal cell lung cancer, J Clin Oncol 28, 2010 (suppl; abstr e13529)). Также была обнаружена герминативная однонуклеотидная замена в гене DPYD (C29R) которая ассоциирована с пониженной чувствительностью к 5-фторурацилу (Offer S.М., Wegner N.J., Fossum С., Wang K., Diasio R.B. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity, Cancer Res. 2013 Mar 15; 73(6):1958-68. doi: 10.1158/0008-5472. CAN-12-3858. Epub 2013 Jan 17).
Итого были сформированы рекомендации по персонализированной противоопухолевой терапии на основе анализа данных о последовательностях и экспрессии генов для индивидуального пациента.
Химиотерапевтом было одобрено применение гефитиниба как препарата, ингибирующего EGFR-signalling каскад, а также вследствие наличия у пациента полиморфизма, обуславливающего повышенную чувствительность к данному лекарству. В результате лечения гефитинибом у пациента удалось добиться стойкой ремиссии заболевания. Таким образом, была подтверждена эффективность патентуемого метода по идентификации белков-мишеней, запускающих процесс канцерогенеза у индивидуального пациента, для последующей противоопухолевой фармакотерапии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВООПУХОЛЕВЫЙ ИНДИВИДУАЛЬНЫЙ ПРОТЕОМ-ОСНОВАННЫЙ ТАРГЕТНЫЙ КЛЕТОЧНЫЙ ПРЕПАРАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ЭТОГО ПРЕПАРАТА ДЛЯ ТЕРАПИИ РАКА И ДРУГИХ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2012 |
|
RU2535972C2 |
| ПЛАТФОРМА АНАЛИЗА ГЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ ONCOBOX | 2018 |
|
RU2741703C1 |
| Способ получения фармацевтического агента для иммунотерапии рака | 2016 |
|
RU2719033C2 |
| Метод разработки биомаркеров заболеваний и физиологически активных веществ на основе расширенной версии алгоритма iPANDA | 2018 |
|
RU2703534C1 |
| ПРОТИВООПУХОЛЕВЫЙ ТАРГЕТНЫЙ КЛЕТОЧНЫЙ ПРОДУКТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2757812C2 |
| Способ выявления первично-рефрактерной формы множественной миеломы в дебюте заболевания | 2020 |
|
RU2749612C1 |
| ИНДИВИДУАЛИЗИРОВАННЫЕ ПРОТИВООПУХОЛЕВЫЕ ВАКЦИНЫ | 2012 |
|
RU2670745C9 |
| ИНДИВИДУАЛИЗИРОВАННЫЕ ПРОТИВООПУХОЛЕВЫЕ ВАКЦИНЫ | 2012 |
|
RU2779946C2 |
| ПРОТИВООПУХОЛЕВАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2728748C2 |
| ХИМЕРНЫЕ АНТИГЕННЫЕ РЕЦЕПТОРЫ С МУТИРОВАННЫМИ КОСТИМУЛЯТОРНЫМИ ДОМЕНАМИ CD28 | 2018 |
|
RU2800922C2 |
Изобретение относится к области биотехнологии, а именно к способу поиска белков-мишеней, запускающих процесс канцерогенеза, в образцах тканей индивидуального пациента, для последующей противоопухолевой фармакотерапии. Определяют методом секвенирования транскриптомные данные об уровнях экспрессии генов в образцах. Получают набор активированных генно-регуляторных сетей, состоящих из центрального гена регулятора и регулируемых им генов. Определяют из транскриптомных данных дифференциально экспрессированные гены. Определяют методами секвенирования изменения в последовательностях ДНК, представленных в виде набора однонуклеотидных полиморфизмов в образцах. Определяют среди найденных генетических вариантов гены-драйверы. Проводят поиск канонических сигнальных путей, значимо обогащенных ранее найденными центральными генами регуляторами экспрессии, дифференциально экспрессированными генами, генами с идентифицированными однонуклеотидными полиморфизмами и инделами, генами-драйверами. Проводят поиск методом перебора в составе идентифицированных сигнальных путей существующих белков-мишеней для лекарственных препаратов, предназначенных для противоопухолевой терапии. Предложенное изобретение позволяет с высокой эффективностью найти белки-мишени, запускающие процесс канцерогенеза, в образцах тканей индивидуального пациента. 1 ил., 5 табл.
Способ поиска белков-мишеней, запускающих процесс канцерогенеза, в образцах тканей индивидуального пациента, для последующей противоопухолевой фармакотерапии, включающий следующие стадии:
а) определение методом секвенирования следующего поколения транскриптомных данных об уровнях экспрессии генов в образцах, полученных из ткани опухоли и здоровой ткани индивидуального пациента с последующей обработкой полученных данных с помощью алгоритма Subnetwork Enrichment Analysis и получением набора активированных генно-регуляторных сетей, состоящих из центрального гена регулятора и регулируемых им генов;
б) определение из транскриптомных данных дифференциально экспрессированных генов с помощью алгоритма DEseq;
в) определение методами секвенирования изменений в последовательностях ДНК, представленных в виде набора однонуклеотидных полиморфизмов и инделов в образцах, полученных из ткани опухоли и здоровой ткани индивидуального пациента;
г) определение среди найденных генетических вариантов генов-драйверов с помощью алгоритма Cancer-specific High-throughput Annotation of Somatic Mutations;
д) поиск канонических сигнальных путей, значимо обогащенных ранее найденными центральными генами регуляторами экспрессии, дифференциально экспрессированными генами, генами с идентифицированными однонуклеотидными полиморфизмами и инделами, генами-драйверами, для отбора сигнальных путей используют точный тест Фишера;
е) поиск методом перебора в составе идентифицированных сигнальных путей существующих белков-мишеней для лекарственных препаратов, предназначенных для противоопухолевой терапии, причем из найденных белков-мишеней выбирают белки с учетом имеющихся у пациента полиморфизмов, для которых установлена их связь с чувствительностью к определенному препарату.

