Область техники
Данное изобретение относится к методам поиска биомаркеров заболеваний (биомедицинских состояний) и новых физиологически активных веществ на основе анализа данных высокопроизводительных биологических экспериментов на уровне клеточных сигнальных путей. Анализируемые данные могут включать данные генной экспрессии, метилирования ДНК, данные об индивидуальных генетических полиморфизмах, протеомные данные и др.
Уровень техники
Анализ биологических данных - трудоемкая задача, поскольку данные являются высокоразмерными, где количество измерений насчитывает десятки тысяч. Основным преимуществом алгоритмов анализа сигнальных путей является способность производить снижение размерности таких данных, при этом облегчая возможности их дальнейшей интерпретации и использования в моделях. Но несмотря на данное преимущество, алгоритмы остаются неидеальными в отношении извлечения набора значимых признаков. Под сигнальным путем обычно понимают последовательность молекул, посредством которых информация в клетке передается от клеточного рецептора внутрь клетки. Выделяют три основных подхода к анализу сигнальных путей [1, 2]: подход, основанный на анализе представленности групп генов Over-Representation Analysis or Enrichment Analysis (ORA), алгоритмы, основанные на оценке функциональных групп генов Functional Class Scoring (FCS), а также подход, основанный на топологии сетей взаимодействия групп генов (Pathway Topology (РТ)).
Метод, основанный на анализе представленности групп генов, использует долю генов в пути или в любой другой группе генов, которые дифференциально экспрессируются. Цель данного метода - получение списка наиболее релевантных путей, упорядоченных в соответствии с полученными для них значениями p-value. Основная гипотеза метода ORA заключается в том, что значимые пути могут быть определены по количеству генов, демонстрирующих дифференциальную экспрессию в пути при определенных условиях. Статистическая значимость между генами из путей и списком дифференциально экспрессирующихся генов определяется с помощью статистических тестов таких, как тест Фишера, тест на основе гипергеометрического распределения и т.д. Основным недостатком данного метода является то, что при его использовании не учитывается значимая биологическая информация о взаимной регуляции сигнальных путей и составляющих их генов, что впоследствии приводит к смещенным результатам.
Метод, основанный на оценке функциональных групп генов (FSC), предполагает, что важны не только сильные изменения экспрессии в индивидуальных генах, но и малые скоординированные изменения в функциональных наборах генов (т.е. самих путях). Все методы данной группы, за малым исключением, работают по следующей схеме:
1. Вычисляется статистика, показывающая значимость дифференциальной экспрессии гена (t-test, Z-score, ANOVA, Q-statistics и т.д.).
2. Вычисляется статистика, показывающая значимость активации сигнального пути (статистика Колмогорова-Смирнова, сумма, медиана или среднее значений статистик из п. 1 и т.д.).
3. Производится оценка значимости статистики, полученной в п. 2.
В отличие от метода ORA данный подход не использует строгое разделение генов на группы статистически значимых и незначимых, а использует все доступные молекулярные измерения для оценки активации сигнального пути. Однако недостатком данного метода, как и предыдущего метода, является то, что активация сигнального пути оценивается независимо от других путей, что неверно с биологической точки зрения, поскольку один ген может участвовать в нескольких процессах, образуя тем самым перекрытие путей. Также в данном методе часто значения дифференциальной экспрессии используются только для ранжирования генов и игнорируются в дальнейших расчетах, считая в дальнейшим их эквивалентными.
Третья группа алгоритмов, в отличии от предыдущих двух групп, использует не только информацию, относящуюся к списку генов в пути и корреляции между генами, а также основывается на топологии сетей взаимодействия генов или их продуктов в пути.
Алгоритм, на котором основывается изобретение, относится к данной группе методов. Поэтому в качестве ближайшего аналога следует рассмотреть алгоритм DART [3, 4].
Первичная информация о сигнальных путях представляется в этом алгоритме следующим образом. Пусть Р - сигнальный путь, состоящий из числа nu апрегулированных генов Pu в ответ на активацию пути и числа nd даунрегулированных генов Pd. Гены будем считать апрегулированными, если они имеют положительное значение 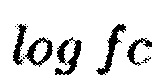 , и даунрегулированными, если наоборот. Значение рассчитывается следующим образом:
, и даунрегулированными, если наоборот. Значение рассчитывается следующим образом:
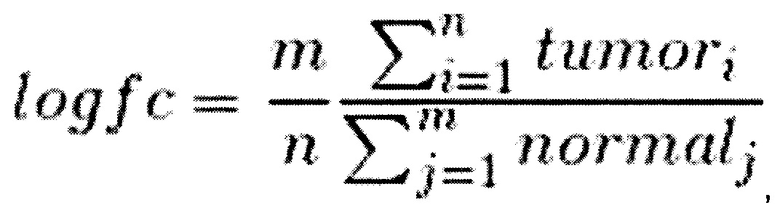
где tumori отвечает значению экспрессии i-го образца в экспериментальной группе, a normalj отвечает значению экспрессии j-го образца в контрольной группе.
На вход данный алгоритм принимает данные экспрессии генов X в виде таблицы, строки в котором отвечают числу генов G, а столбцы числу сэмплов ns. Входной датасет никак не связан с первичной информацией о сигнальных путях. Цель данного алгоритма - оценить значения активации сигнальных путей во всех образцах.
Первым шагом в данном алгоритме является оценка первичной информации в контексте входных данных. Данный шаг включает построение сети генной коэкспрессии на основе корреляции между значениями экспрессии всех генов, входящих в путь P. В данном алгоритме в качестве меры используется коэффициент корреляции Пирсона. Для проверки гипотезы о значимости полученных значений корреляции над коэффициентами выполняется преобразование Фишера (трансформация Фишера). После чего выводится матрица значений P-value для каждого значения. Далее производится оценка доли ложноположительных результатов с помощью пермутаций значений генной экспрессии. Парам генов со значением корреляции, прошедшим данный порог, впоследствии назначается ребро в строящейся сети.
Далее производится оценка значимости первичной информации о пути с помощью сравнения с полученной сетью коэкспрессии. Каждому ребру сети, полученной в п. 1 назначается бинарный вес (-1, 1) в зависимости от знака значения корреляции между парой генов. Также данной паре генов назначается бинарный знак -1, если поведение генов различно в ответ на активацию/ингибирование пути, и 1, если поведение одинаково. В дальнейшем ребро считается релевантным, если знаки в обеих операциях совпадают, нерелевантные ребра удаляются. Оценка релевантности данной сети выводится как доля релевантных ребер относительно общего числа ребер.
Метрика, оценивающая активацию сигнального пути выглядит следующим образом:
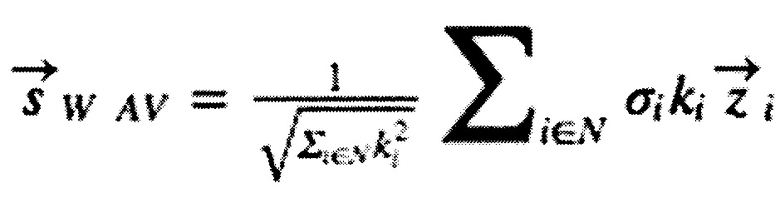 ,
,
где 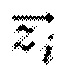 - нормализованная z-оценка профиля экспрессии генов, σi - знак активации в пути, ki - степень вершины, соответствующей гену i в коэкспрессионной сети.
- нормализованная z-оценка профиля экспрессии генов, σi - знак активации в пути, ki - степень вершины, соответствующей гену i в коэкспрессионной сети.
Сущность изобретения
Технической задачей, решаемой с помощью предлагаемого изобретения, является поиск биомаркеров заболеваний и физиологически активных веществ путем анализа омиксных данных на уровне сигнальных путей. Таким образом, в задачу входит осуществление расчетов, основанных на результатах экспериментально полученных данных по омиксным экспериментам (транскриптомика, протеомика, эпигенетика, геномика). Предварительно данные должны быть преобразованы в формат таблиц, содержащих оценки активации или ингибирования экспрессии генов, их продуктов, генетических или эпигенетических модификаций в исследуемом наборе образцов. Данные такого вида проецируются на схемы сигнальных путей с помощью расширенной версии алгоритма iPANDA (Ozerov IV, et al., "In silico Pathway Activation Network Decomposition Analysis (iPANDA) as a method for biomarker development", Nat Commun. 2016 Nov 16; 7:13427). Таким образом, должны быть получены оценки активации сигнальных путей в исследуемых образцах по сравнению с контролем, которые затем непосредственно используются для поиска биомаркеров и физиологически активных веществ.
Указанная задача решается путем применения способа оценки эффективности воздействия лекарственных кандидатов на физиологический процесс, выбранных из группы лекарственных кандидатов, состоящий по меньшей мере из следующих этапов: (а) получают данные по меньшей мере одного типа, собранные из полученных экспериментальным путем образцов, в которых протекает указанный физиологический процесс, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам метилирования геномной ДНК, (iv) полногеномные данные по мутациям в геномной ДНК, (v) данные по экспрессии малых некодирующих РНК, (vi) данные фенотипических скринингов с помощью системы CRISPR-CAS9, (vii) данные по фосфорилированию белков; (б) получают данные того же типа, что и данные, полученные на стадии (а), собранные из контрольных образцов, в которых не протекает указанный физиологический процесс; (в) определяют необходимость гармонизации данных, полученных на стадиях (а) и (б) и, при необходимости, осуществляют следующие операции гармонизации этих данных: препроцессинг, нормализацию и формирование фенотипических групп; (г) определяют набор генов, или их продуктов, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя данные, полученные на стадиях (а)-(в); (д) определяют набор сигнальных путей, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя набор генов, полученный на стадии (г); (е) определяют набор генов и сигнальных путей, статистически достоверно активированных или ингибированных после воздействия каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, на клетки или биологические ткани, в которых может протекать указанный физиологический процесс; (ж) определяют количественные показатели эффективности воздействия на указанный физиологический процесс для каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е), таким образом оценивая эффективность воздействия потенциальных лекарственных средств на указанный физиологический процесс.
В некоторых вариантах осуществления изобретения вышеуказанный способ оценки эффективности воздействия лекарственных кандидатов характеризуется тем, что (i) на стадиях (г) и (д) дополнительно проводят сравнение генов и сигнальных путей между различными состояниями, составляющими указанный физиологический процесс, и определяют набор генов и сигнальных путей, активированных или ингибированных в отдельных подгруппах образцов, соответствующих указанному физиологическому процессу, тем самым выделяя специфические по признакам болезни подгруппы пациентов для указанного физиологического процесса; и (ii) на стадии (ж) определяют количественные показатели эффективности воздействия в отдельных подгруппах образцов, соответствующих указанному физиологическому процессу, для каждого лекарственного кандидата, выбранного из группы потенциальных лекарственных веществ, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е).
В других вариантах изобретения указанная задача решается путем использования системы для оценки эффективности воздействия лекарственных кандидатов на физиологический процесс, выбранных из группы лекарственных кандидатов, включающей:
- по меньшей мере один процессор;
- по меньшей мере одну память, которая содержит машиночитаемые инструкции, которые при их исполнении по меньшей мере одним процессором осуществляют оценку эффективности воздействия лекарственных кандидатов на физиологический процесс при помощи компьютерно-реализуемого способа, состоящего по меньшей мере из следующих этапов:
(а) получают данные по меньшей мере одного типа, собранные из полученных экспериментальным путем образцов, в которых протекает указанный физиологический процесс, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам метилирования геномной ДНК, (iv) полногеномные данные по мутациям в геномной ДНК, (v) данные по экспрессии малых некодирующих РНК, (vi) данные фенотипических скринингов с помощью системы CRISPR-CAS9, (vii) данные по фосфорилированию белков;
(б) получают данные того же типа, что и данные, полученные на стадии (а), собранные из контрольных образцов, в которых не протекает указанный физиологический процесс;
(в) определяют необходимость гармонизации данных, полученных на стадиях (а) и (б) и, при необходимости, осуществляют следующие операции гармонизации этих данных: препроцессинг, нормализацию и формирование фенотипических групп;
(г) определяют набор генов, или их продуктов, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя данные, полученные на стадиях (а)-(в);
(д) определяют набор сигнальных путей, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя набор генов, полученный на стадии (г);
(е) определяют набор генов и сигнальных путей, статистически достоверно активированных или ингибированных после воздействия каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, на клетки или биологические ткани, в которых может протекать указанный физиологический процесс;
(ж) определяют количественные показатели эффективности воздействия на указанный физиологический процесс для каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е), таким образом оценивая эффективность воздействия потенциальных лекарственных средств на указанный физиологический процесс.
При осуществлении изобретения достигается следующий технический результат: разработан новый, более эффективный способ оценки эффективности воздействия лекарственных кандидатов на физиологический процесс, использующий широкий спектр экспериментальных данных. Данный способ может быть автоматизирован, что предотвращает потенциальные ошибки, связанные с ручным расчетом, и позволяет учитывать изменения в сотнях и тысячах молекулярных путей, включающих десятки и сотни генных продуктов, характерных для конкретного пациента.
Краткое описание рисунков
Рис. 1. Пример ранжированного списка генов и сигнальных путей, биомаркеров заболевания. Гены (пути) отранжированы в соответствии с модулем уровня экспрессии (активации) таким образом, чтобы гены (пути), имеющие одинаковое направление активации в большинстве датасетов оказывались в верхней части списка. В примере использованы данные, соответствующие процессу мышечного старения. Топ 20 генов (путей) из списка отранжированы слева направо.
Рис. 2. Блок-схема создаваемого технического решения.
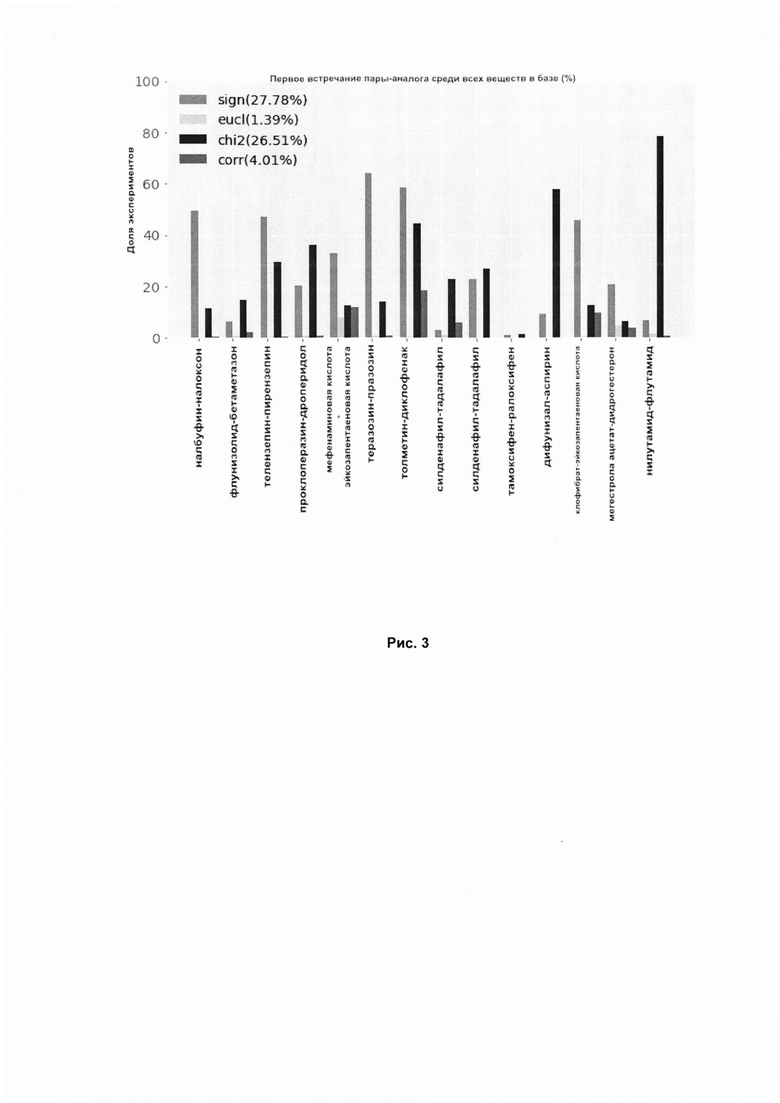
Рис. 3. Результаты ранжирования известных аналогов на основе исходных значений генной экспрессии. По оси X указаны известные аналоги, на которых проводилось тестирование, по оси Y отложен процент от числа всех веществ, в котором встречается аналогичное исследуемому вещество. Для каждого аналога приведены 4 метрики (перечислены слева направо): sign - количество сигнальных путей сонаправленных (активированных или ингибированных), eucl - евклидово расстояние, chi2 - тест Хи-квадрат, corr - корреляция Пирсона.
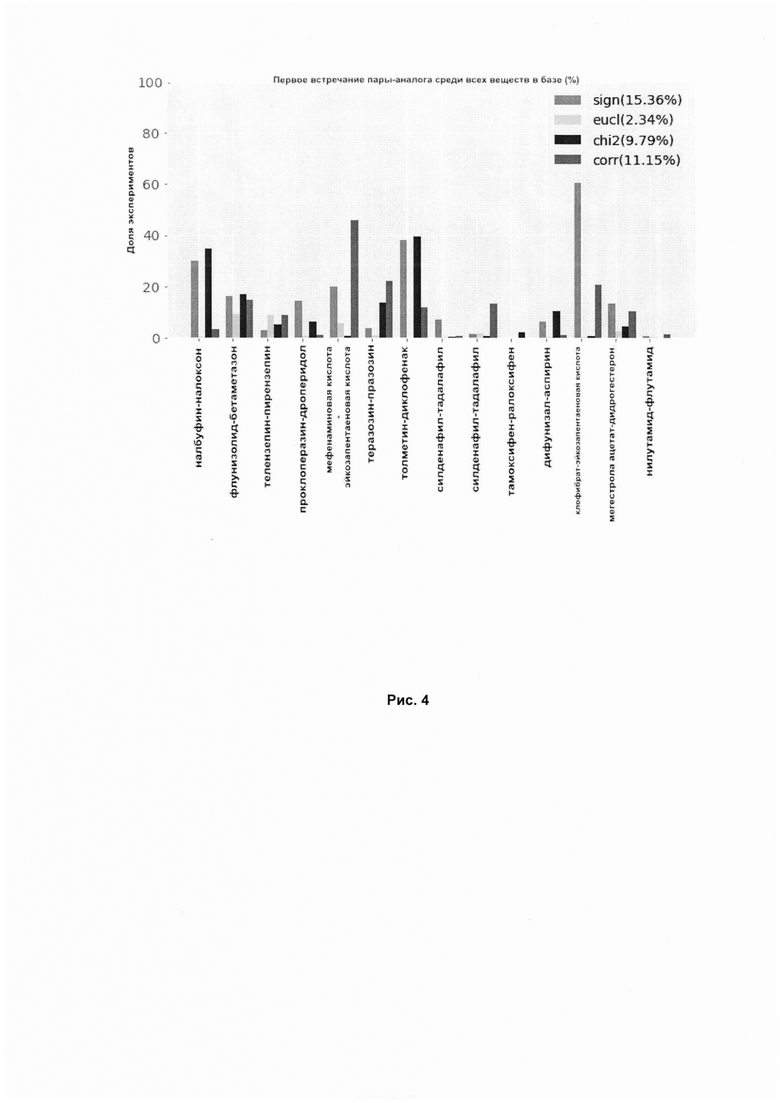
Рис. 4. Результаты ранжирования известных аналогов на основе значений активации сигнальных путей. По оси X указаны известные аналоги, на которых проводилось тестирование, по оси Y отложен процент от числа всех веществ, в котором встречается аналогичное исследуемому вещество. Для каждого аналога приведены 4 метрики (перечислены слева направо): sign - количество сигнальных путей сонаправленных (активированных или ингибированных), eucl - евклидово расстояние, chi2 - тест Хи-квадрат, corr - корреляция Пирсона.
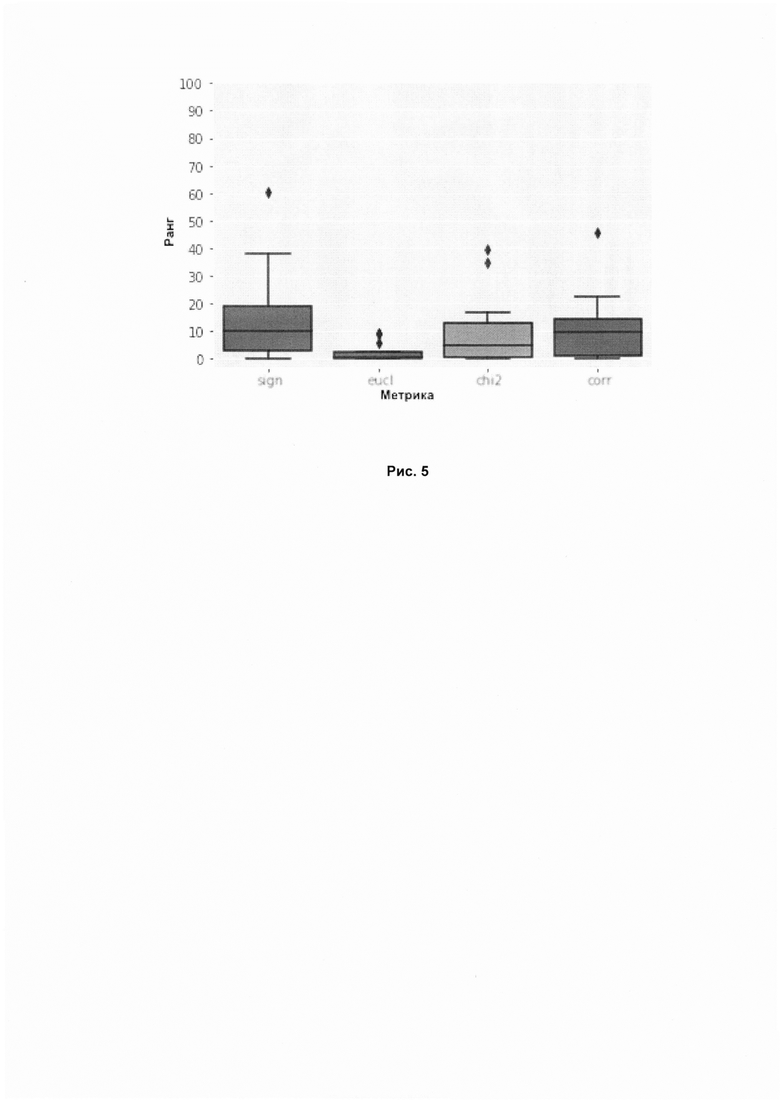
Рис. 5. Результаты ранжирования известных аналогов на основе значений активации сигнальных путей, оцененные с помощью различных метрик на заранее известных парах аналогов. График показывает ранг (позиция, нормированная на число элементов, среди которых производился поиск) аналога, найденного среди базы веществ, включающих различные временные метки и дозировки, относительно выбранного вещества, для которого производится поиск. По оси X указаны используемые метрики (перечислены слева направо): sign - количество сигнальных путей сонаправленных (активированных или ингибированных), eucl - евклидово расстояние, chi2 - тест Хи-квадрат, corr - корреляция Пирсона.
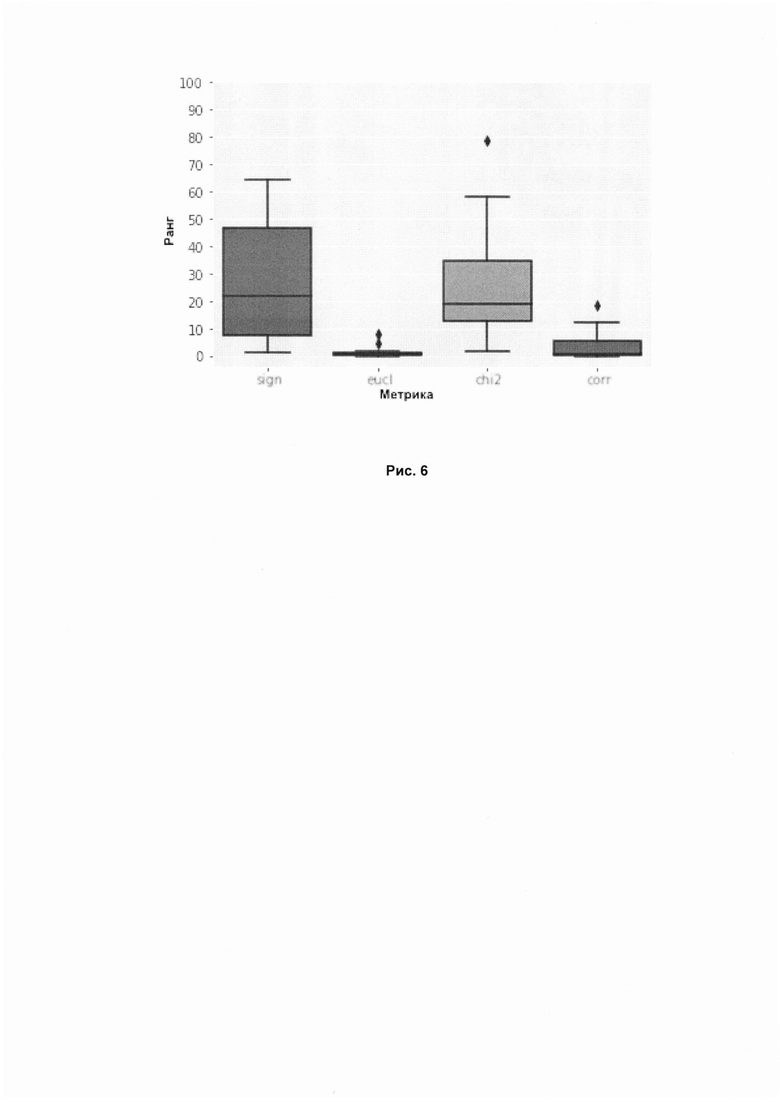
Рис. 6. Результаты ранжирования известных аналогов на основе значений дифференциальной экспрессии генов, оцененные с помощью различных метрик на заранее известных парах аналогов. График показывает ранг (позиция, нормированная на число элементов, среди которых производился поиск) аналога, найденного среди базы веществ, включающих различные временные метки и дозировки, относительно выбранного вещества, для которого производится поиск. По оси X указаны используемые метрики (перечислены слева направо): sign - количество сигнальных путей сонаправленных (активированных или ингибированных), eucl - евклидово расстояние, chi2 - тест Хи-квадрат, corr - корреляция Пирсона.
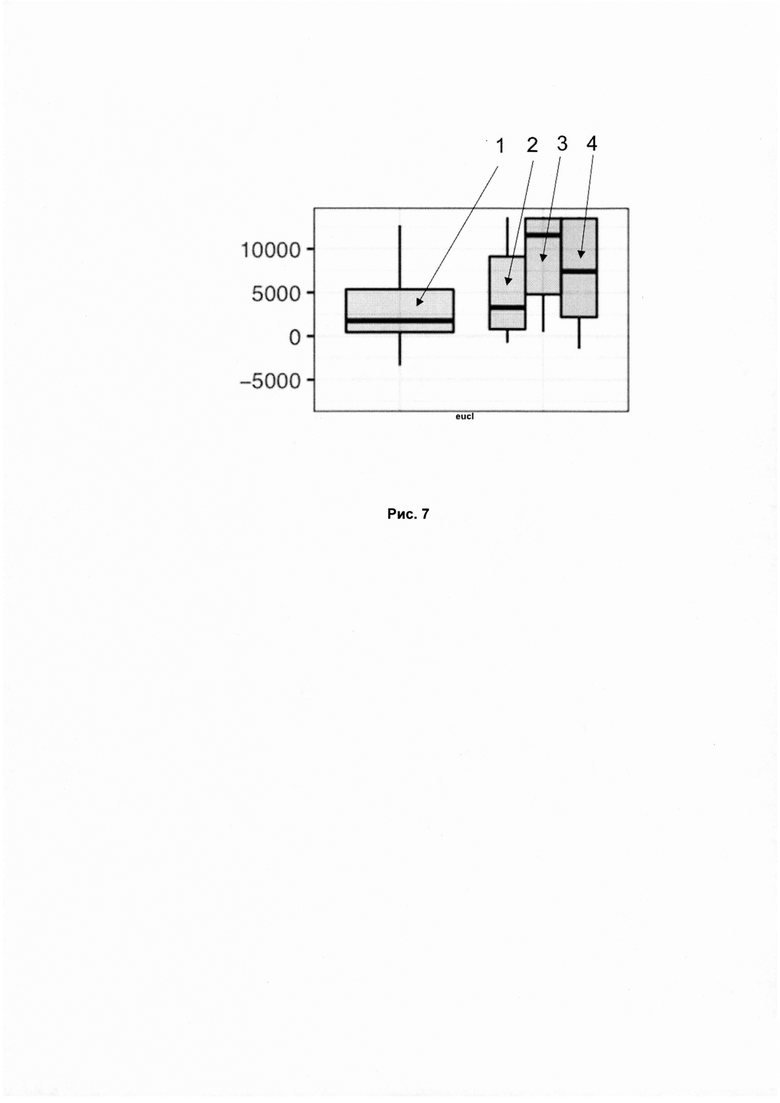
Рис. 7. Результаты ранжирования физиологически активных веществ для выбранных болезней для соответствующей евклидовой метрики (eucl - евклидово расстояние). Графики 3 и 4 соответствуют физиологически активным веществам, одобренным к использованию хотя бы для одной из болезней, график 3 - веществам, для которых не ведется новых клинических испытаний, график 4 - веществам, для которых ведутся испытания для других болезней, график 2 представляет препараты, клинические испытания для которых были приостановлены, график 1 представляет вещества без аннотации. По оси ординат отложены безразмерные значения евклидового расстояния между векторами активации сигнальных путей в образце, соответствующем заболеванию, и образце, соответствующем действию вещества. По оси X указана используемая метрика eucl - евклидово расстояние.
Подробное раскрытие изобретения
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из». Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе. Общая схема работы решения представлена на рисунке 2.
Результатом применения метода к омиксным данным по конкретному заболеванию (состоянию) является список активированных или ингибированныхгенов (их продуктов) и сигнальных путей, представляющих собой набор потенциальных биомаркеров данного заболевания (см. пример на Рис. 1). Вторым результатом использования метода является таблица с ранжированным списком физиологически активных веществ, в верхней части которого находятся вещества, с наибольшей вероятностью обладающие активностью против исследуемого заболевания (состояния) (см. пример в Таблице 1).
Таблица 1. Пример ранжированного списка физиологически активных веществ. В примере приведен список из 15 веществ, получивших максимальную оценку при ранжировании по схожести с подписью ДНК-повреждающих агентов с помощью предлагаемого технического решения.
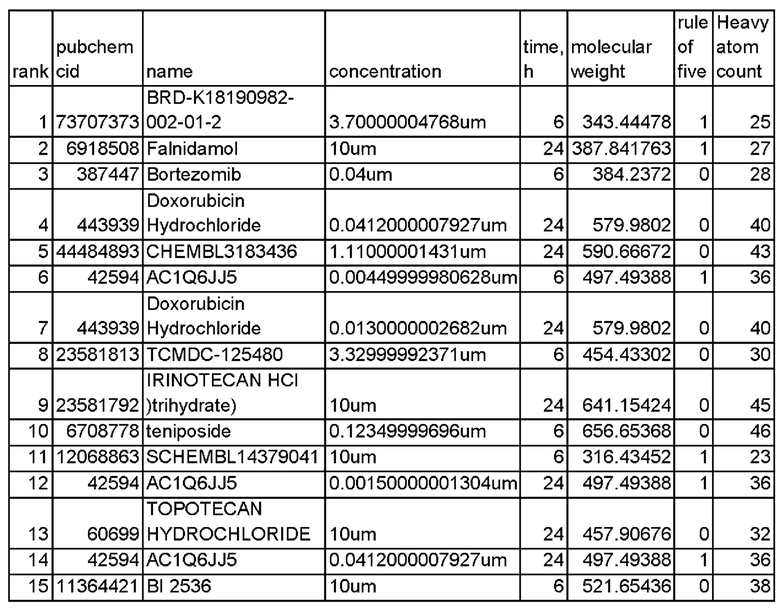
На Рис. 1 приведен список биомаркеров мышечного старения, найденных с помощью предлагаемого технического решения. Для 17 из 20 первых обнаруженных генов и 20 из 20 первых обнаруженных сигнальных путей имеются литературные данные, подтверждающие их связь с исследуемым процессом (мышечным старением). В таблице 1 приведен список веществ, вызывающих повреждения ДНК, полученный с помощью предлагаемого технического решения. 6 из 15 первых веществ являются известными ДНК-повреждающими агентами, в то время как еще 7 имеют структурное сходство с известными ДНК-повреждающими агентами. Таким образом, из приведенных на Рис. 1 и в Таблице 1 примеров следует, что технический результат совпадает с решаемой технической задачей.
Метод разработки биомаркеров заболеваний и физиологически активных веществ на основе расширенной версии алгоритма iPANDA включает следующие основные шаги:
1) Подготовка мультиомиксных данных: препроцессинг полученных образцов, нормализация, разделение фенотипических групп, аннотация.
2) Анализ активированных или ингибированныхгенов (их продуктов)
3) Анализ активации сигнальных и метаболических путей на основе расширенной версии алгоритма iPANDA
4) Сбор баз данных болезней и лекарств
5) Поиск биологически активных веществ и биомаркеров
Входные данные
Метод разработки биомаркеров заболеваний и физиологически активных веществ на основе расширенной версии алгоритма iPANDA производит вычисления на основе данных, в которых протекает исследуемый физиологический процесс (т.е. исследуемый процесс, протекающий в организме, на клеточном уровне, например: заболевание, процесс старения и другие функциональные состояния), при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам метилирования геномной ДНК, (iv) полногеномные данные по мутациям в геномной ДНК; (v) данные по экспрессии малых некодирующих РНК; (vi) данные фенотипических скринингов с помощью системы CRISPR-CAS9; (vii) данные по фосфорилированию белков.
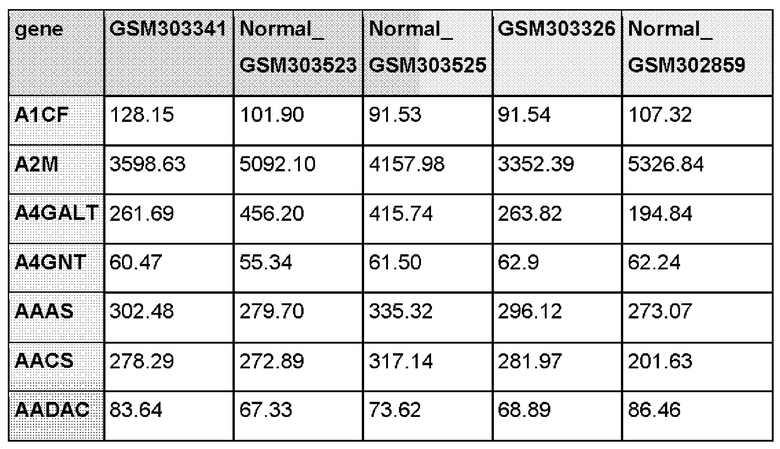
Входная информация представляют собой таблицу значений для данных, полученных с профилей генной экспрессии либо таблицу посчитанных значений 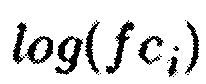 для данных генной экспрессии или иных типов данных, перечисленных выше. Строки этой таблицы представляют значения для генов, участвующих в эксперименте, и именуются уникальными именами (например, в системе gene IDs). Разработанный метод не имеет ограничений на число генов, участвующих в эксперименте. В первом случае значения в столбцах представляют значения генной экспрессии, полученные с конкретных образцов, участвующих в эксперименте, и именуются уникальными именами, отвечающими названию образца. Помимо образцов, участвующих в эксперименте, в таблице с входными данными должны присутствовать столбцы со значениями экспрессии, которые подразумевают "нормальное" состояние образцов вне эксперимента. Обычно в качестве "нормальных" рассматривают условия, относительно которых будет производиться сравнение значений, полученных в результате эксперимента. Названия таких образцов должны начинаться с приставки
для данных генной экспрессии или иных типов данных, перечисленных выше. Строки этой таблицы представляют значения для генов, участвующих в эксперименте, и именуются уникальными именами (например, в системе gene IDs). Разработанный метод не имеет ограничений на число генов, участвующих в эксперименте. В первом случае значения в столбцах представляют значения генной экспрессии, полученные с конкретных образцов, участвующих в эксперименте, и именуются уникальными именами, отвечающими названию образца. Помимо образцов, участвующих в эксперименте, в таблице с входными данными должны присутствовать столбцы со значениями экспрессии, которые подразумевают "нормальное" состояние образцов вне эксперимента. Обычно в качестве "нормальных" рассматривают условия, относительно которых будет производиться сравнение значений, полученных в результате эксперимента. Названия таких образцов должны начинаться с приставки  за которой следует уникальный идентификатор столбца. Каждый входной файл должен включать, как минимум, один столбец, отвечающий эксперименту, и, как минимум, один столбец, отвечающий "нормальным" условиям. В ином случае значения в столбцах есть посчитанные значения , отвечающие конкретным образцам. Расширение входных данных может быть любым, также как и разделитель данных внутри таблицы. В Таблице 2 приведен пример таблицы с входными данными.
за которой следует уникальный идентификатор столбца. Каждый входной файл должен включать, как минимум, один столбец, отвечающий эксперименту, и, как минимум, один столбец, отвечающий "нормальным" условиям. В ином случае значения в столбцах есть посчитанные значения , отвечающие конкретным образцам. Расширение входных данных может быть любым, также как и разделитель данных внутри таблицы. В Таблице 2 приведен пример таблицы с входными данными.
Таблица 2: Пример таблицы с входными данными.
Препроцессинг, нормализация и формирование фенотипических групп
Разработанный метод использует данные экспрессии генов (транскриптомика), их продуктов (протеомика), генетических (геномика) или эпигенетических (эпигенетика) модификаций в исследуемом наборе образцов и другие омиксные данные, упомянутые в разделе Входные данные. Поскольку входные данные могут быть получены из гетерогенных источников, шаг подготовки данных включает следующие операции гармонизации данных: препроцессинг, нормализацию и формирование фенотипических групп. Такой подход впоследствии позволяет комбинировать данные из множественных источников в интегрированные, последовательные и однозначные информационные продукты. Также поскольку входные данные могут быть получены с помощью различного экспериментального оборудования, для их пре процессии га и приведения к единому формату могут быть использованы различные алгоритмы, соответствующие входным данным.
Транскриптомные данные
Для обработки исходных данных с платформ Affymetrix используется разработанный программный конвейер на основе алгоритма fRMA (McCall, Jaffee, and Irizarry 2012). Использование данного подхода позволяет проводить обработку одиночных образцов или небольших групп образцов, без существенных потерь информации из-за шага нормализации, по сравнению с другими методами.
Обработка одноканальных чипов Agilent осуществляется с помощью программного пакета limma (Smyth, n.d.). Нормализация осуществлялась методом RMA (Brown 2004 - укажите журнал и номер). Каждый образец в результате обработки был преобразован в вектор нормализованных значений интенсивности, присвоенных генам, аннотированным согласно номенклатуре HGNC.
Обработка микрочиповых данных Illumina была произведена с помощью разработанного программного конвейера на основе программного пакета lumi (Du, Kibbe, and Lin 2008). Для обработки данных RNA-seq был использован программный конвейер r-make (http://physiology.med.cornell.edu/faculty/mason/lab/r-make/index.html).
Протеомные данные
Протеомные данные были получены из базы данных PRIDE, которая содержит 2251 набора данных масс-спектроскопии для Homo sapiens. Препроцессинг данных производился на основе шагов, предложенных в Trans-proteomic pipeline (ТРР). Первый этап подготовки данных - определить соответствие спектра пептида и его последовательности с помощью программы Tandem. Статистическая проверка качества результата этой программы осуществляется с помощью метода PeptideProphet, задающего вероятность каждому установленному соответствию. Анализ относительного количества белков и пептидов в пробе производился с помощью инструмента ASAPRatio, результатом которого является численная оценка количества белков в пробе, измеряемая в ppm (particles per million). Для статистической валидации полученных значений мы повторно использовали ProteinProphet. Данные каждой серии были квантильно нормализованы, а соответствие названий белков и генов (Gene Symbol) были получены из базы данных UniProt.
Для других типов данных могут быть использованы соответствующие рекомендованные производителями экспериментального оборудования или принятые сообществом методы анализа данных, вследствии чего они могут быть соотносены с экспрессией генов или их продуктов и использоваться в качестве входных данных расширенной версии алгоритма iPANDA.
После обработки данных, образцы могут быть сгруппированы в фенотипические группы на основе мета-информации (болезнь, ткань, возраст, вес, пол, расовая принадлежность). Для каждой исследуемой группы формируется группа контрольных здоровых образцов, с которыми производится сравнение на следующих этапах анализа.
Анализ активированных или ингибированныхгенов (их продуктов)
Анализ активированных или ингибированныхгенов или из продуктов проводится на основе оценки изменения в уровне генной экспрессии, экспрессии белка, наличия индивидуальных полиморфизмов или эпигенетических паттернов, например, локусов метилирования, в исследуемых образцах по сравнению с образцами нормальной ткани. В случае анализа экспрессии генов и белков такой оценкой может служить отношение экспрессии в исследуемых образцах по сравнению с усредненным уровнем экспрессии в соответствующих образцах нормальной ткани в логарифмическом масштабе 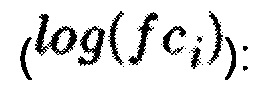
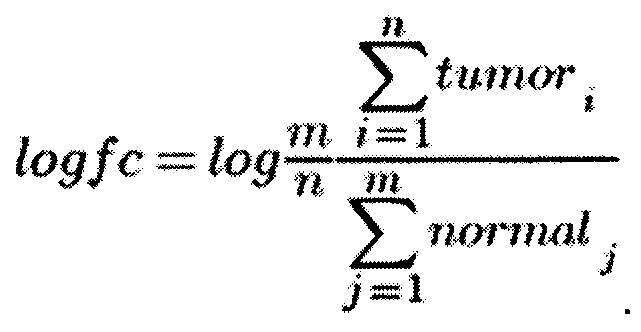
В случае дифференциальной экспрессии генов получены с помощью пакета limma (https://bioconductor.org/packages/release/bioc/html/limma.html) или DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
Пакет limma предназначен для анализа данных экспрессии генов, полученных с микрочипов или с помощью технологии РНК-секвенирования. Метод limma использует линейные модели для анализа эксперимента, что позволяет в целом оценить взаимодействие между образцами, а также выявить корреляции между выборками, которые могут присутствовать из-за повторения метода измерений. Пакет limma считает дисперсию для каждого гена, а также попарные корреляции между генами. Далее для каждого гена проводится тест Стьюдента и вычисляются значения p-value, а также скорректированные значения p-value. На основе полученных значений определяется значимость генов.
Пакет DESeq2 предназначен для анализа данных РНК-секвенирования. Данный метод также использует обобщенные линейные модели для анализа дифференциальной экспрессии генов. DESeq2 предполагает, что входные имеют негативное биномиальное распределение (также известное как гамма-Пуассоновское распределение) со средним значением μij и дисперсией σi. Среднее значение в данном случае вводится как количество прочтений qij, пропорциональное фрагментам ДНК для конкретного гена в конкретном образце, умноженное на коэффициент нормализации 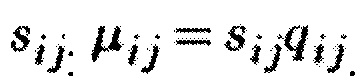 Обобщенная линейная модель строится следующим образом:
Обобщенная линейная модель строится следующим образом: 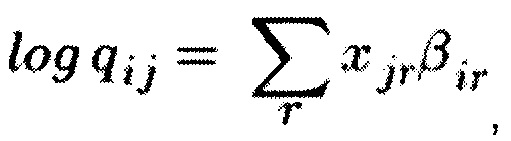 где xjr - матрица, построенная на основе входных значений, и представляющая дизайн эксперимента, βir - коэффициенты. В случае сравнения двух групп образцов (например, образцов, отвечающих эксперименту и контрольных образцов) коэффициенты обобщенной линейной модели показывают силу экспрессии гена. После построения модели на основе полученных коэффициентов βir строится гипотеза о том, что коэффициенты модели отличны от нуля. Для проверки данной гипотезы используется тест Вальда. На основе p-value, полученных после проведения теста, а также скорректированных p-value, вычисляется значимость генов.
где xjr - матрица, построенная на основе входных значений, и представляющая дизайн эксперимента, βir - коэффициенты. В случае сравнения двух групп образцов (например, образцов, отвечающих эксперименту и контрольных образцов) коэффициенты обобщенной линейной модели показывают силу экспрессии гена. После построения модели на основе полученных коэффициентов βir строится гипотеза о том, что коэффициенты модели отличны от нуля. Для проверки данной гипотезы используется тест Вальда. На основе p-value, полученных после проведения теста, а также скорректированных p-value, вычисляется значимость генов.
В случае других видов экспериментальных данных для получения оценок активации или ингибированиямогут быть использованы рекомендованные и принятые сообществом подходы для соответствующего типа данных.
Анализ активации сигнальных и метаболических путей на основе расширенной версии алгоритма iPANDA
Модуль iPANDA предназначен для эффективного анализа транскриптомных и других омиксных данных на уровне сигнальных путей клеточного метаболизма. В результате множество данных, полученных на уровне отдельных генов (белков), могут быть преобразованы в биологически значимые характеристики - уровень активации сигнальных путей в образцах клеток. Преобразованные таким образом данные могут быть использованы как сами по себе в задачах поиска новых биологически активных веществ, предсказания уровня чувствительности пациентов к определенному виду терапии, так и в качестве метода понижения шума и снижения размерности в исходных данных для дальнейшей тренировки моделей на основе различных техник машинного обучения, включая глубокие нейронные сети (ГНС). Метод iPANDA представляет собой устойчивый алгоритм анализа высокоразмерных биологических данных. Алгоритм производит оценку активации сигнального пути на основе анализа статистических и топологических весов генов, входящих в группу скоординированно экспрессирующихся генов. Поиск биомаркеров и физиологически активных веществ осуществляется на основе анализа совместно возбужденных и ингибированных сигнальных каскадов.
Оценка активации сигнальных путей
Метод iPANDA предназначен для анализа биологических данных на основе сгруппированных в модули генов. Известно, что экспрессия многих генов может изменяться согласованно за счет регуляции экспрессии одними и теми же транскрипционными факторами. Поэтому каждый модуль представляет собой не только набор генов, которые показывают значимую коэкспрессию, т.е. гены, содержащиеся в модуле экспрессируют вместе при определенных условиях, но и содержат гены, которое регулируются одними и теми же факторами транскрипции (процесс формирования генных модулей описан в главе Создание генных модулей). Поскольку данные могут включать гены, значения экспрессии которых не показывает значимой корреляции с другими генами, а также регуляция экспрессии которых не соотносится с регуляцией экспрессии остальных генов, то такие гены не объединяются в модули, а представляют из себя уникальные объекты. Исходя из этого функция оценки активации сигнального пути Р состоит из двух слагаемых. Первое слагаемое соответствует вкладу генов, не входящих в модули, второе учитывает гены, организующие модули. Конечная функция расчета значений активации сигнальных путей выглядит следующим образом:
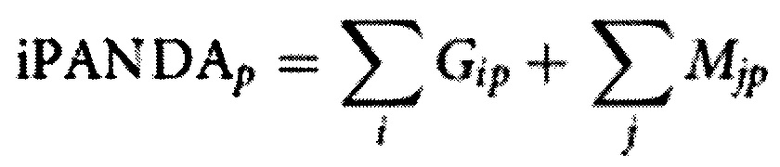 ,
,
где Gip - слагаемое, представляющее вклад индивидуальных генов, и 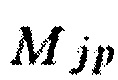 - слагаемое, представляющее вклад генных модулей. Данные слагаемые рассчитываются следующим образом:
- слагаемое, представляющее вклад генных модулей. Данные слагаемые рассчитываются следующим образом:
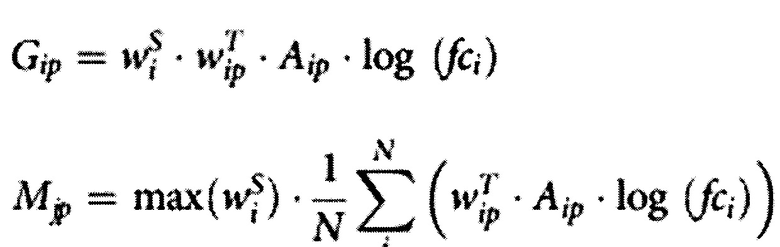 ,
,
где 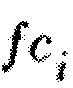 - величина, показывающая уровень изменчивости экспрессии для гена i в конкретном образце по сравнению со средним значением уровня экспрессии в контрольной группе. Поскольку предполагается, что уровни экспрессии генов имеют логарифмически нормальное распределение, в данных формулах с целью упрощения выражения рассматривается логарифм значений . Тем самым произведение логарифмов преобразуется в сумму. Каждый одиночный ген или генный модуль i имеет активирующий или ингибирующий эффект на сигнальный путь р в клетке. В соответствии с этим каждому из них при расчете уровня активации пути присваивается коэффициент Aip клеточного действия. Aip представляет дискретный коэффициент, показывающий влияние гена i на путь p. Данная величина имеет значение +1, если продукт гена положительно влияет на активацию пути p, и -1, если он несет отрицательный вклад. Члены
- величина, показывающая уровень изменчивости экспрессии для гена i в конкретном образце по сравнению со средним значением уровня экспрессии в контрольной группе. Поскольку предполагается, что уровни экспрессии генов имеют логарифмически нормальное распределение, в данных формулах с целью упрощения выражения рассматривается логарифм значений . Тем самым произведение логарифмов преобразуется в сумму. Каждый одиночный ген или генный модуль i имеет активирующий или ингибирующий эффект на сигнальный путь р в клетке. В соответствии с этим каждому из них при расчете уровня активации пути присваивается коэффициент Aip клеточного действия. Aip представляет дискретный коэффициент, показывающий влияние гена i на путь p. Данная величина имеет значение +1, если продукт гена положительно влияет на активацию пути p, и -1, если он несет отрицательный вклад. Члены 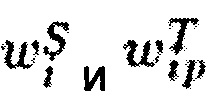 - статистические и топологические веса гена i, лежащие в диапазоне значений от 0 до 1. Описание данных коэффициентов приводится в разделе Топологические и статистические веса. Поскольку значения log
- статистические и топологические веса гена i, лежащие в диапазоне значений от 0 до 1. Описание данных коэффициентов приводится в разделе Топологические и статистические веса. Поскольку значения log 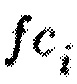 и
и 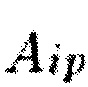 могут быть как положительными, так и отрицательными, значения активации пути iPANDA также могут иметь разные знаки. Положительные и отрицательные значения iPANDA соответствуют активации и ингибированию сигнального пути соответственно. Статистическая значимость сигнального пути считается с оценивается с помощью точного статистического теста Фишера:
могут быть как положительными, так и отрицательными, значения активации пути iPANDA также могут иметь разные знаки. Положительные и отрицательные значения iPANDA соответствуют активации и ингибированию сигнального пути соответственно. Статистическая значимость сигнального пути считается с оценивается с помощью точного статистического теста Фишера:
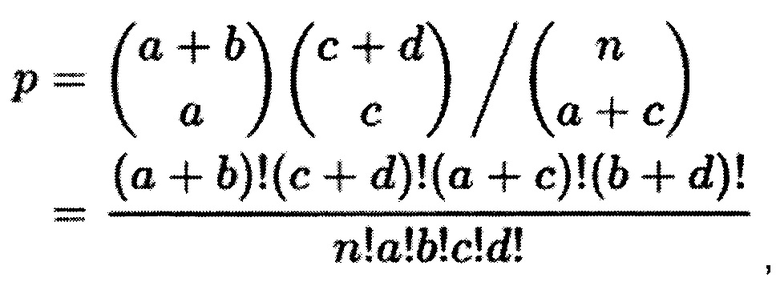
где данные формулы - данные таблицы, построенной на основе полученных данных об активации сигнальных путей.
Создание генных модулей
Поскольку известно, что экспрессия многих генов может изменяться согласовано в ходе клеточного метаболизма, то группы генов, демонстрирующих высокую корреляцию в уровне экспрессии, следует объединять в генные модули. Алгоритм iPANDA выбирает не только гены, которые экспрессирую одновременно, а также учитывает регуляцию экспрессии генов одними и теми же факторами транскрипции. Поэтому гены собираются в модули на основе попарной коэкспрессии и информации, полученной из базы данных транскрипционных факторов млекопитающих (Zheng G, et al., ITFP: an integrated platform of mammalian transcription factors. Bio informatics. 2008 Oct 15; 24(20):2416-7).
Коэкспрессионная матрица представляет собой матрицу парных корреляций между значениями экспрессии генов. Основным расширением алгоритма iPANDA является создание тканеспецифичных модулей коэкспрессии, поскольку поиск в базе данных биологически активных веществ ведется среди различных клеточных линий. Каждый модуль собирается на основе контрольных образцов определенного типа ткани, то корреляционная матрица рассчитывается для каждой ткани отдельно. В качестве метрики, показывающей коэкспрессию, используется коэффициент корреляции Пирсона:
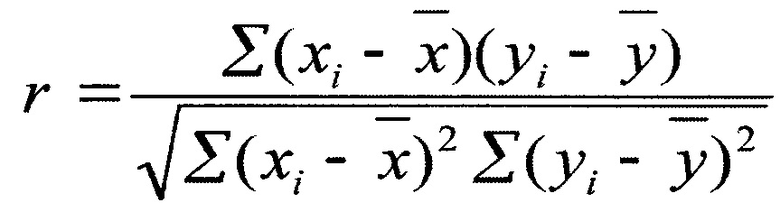
Генные модули образуются на основе полученных данных коэкспрессии с помощью алгоритма иерархической кластеризации. Кластеризация выполняется на основе матрицы расстояний между значениями корреляции. Расстояния рассчитываются следующим образом:
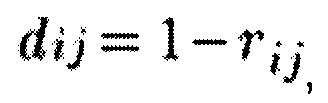
где rij - корреляция между уровнями экспрессии генов i и j. В качестве критерия связности используются методы average и complete. Модули рассчитываются для каждого из критериев отдельно. Для дальнейших расчетов были выбраны кластеры со средним уровнем попарной корреляции между уровнями экспрессии генов, входящих в кластер, не менее 0,3.
Впоследствии все полученные группы модулей, соответствующие критериям связывания, и модули, собранные на основе информации о факторах транскрипции, объединяются в единый модуль в ходе рекурсивной процедуры на основе попарного пересечения при условии, что совместное пересечение составляет более 70%.
Такой подход позволяет присваивать топологические коэффициенты не отдельным генам, а кластерам согласовано экспрессирующихся генов, входящих в модули. Гены, не вошедшие ни в один модуль, здесь и далее будут называться одиночными генами. Также как и для одиночных генов, в ходе анализа образца для каждого генного модуля рассчитывается соответствующий уровень экспрессии, отнесенный к нормальному уровню 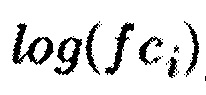
Топологические и статистические веса
Топологический вес 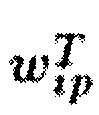 для каждого гена i в пути p вычисляются путем декомпозиции карты сигнального пути. Для оценки топологического веса рассчитываются все возможные обходы по генной сети, которая представляет из себя направленный граф, основанный на взаимодействии генов в сигнальном каскаде. В качестве вершин графа рассматриваются гены или генные модули, а в качестве ребер - биохимические взаимодействия. Вершины, которые не имеют входных ребер являются стартовыми точками обхода, а вершины, которые не имеют выходных ребер, являются конечными точками. Предполагается, что граф не имеет циклов. Число обходов Nip пути р, которые включают ген i, рассчитываются для каждого гена.
для каждого гена i в пути p вычисляются путем декомпозиции карты сигнального пути. Для оценки топологического веса рассчитываются все возможные обходы по генной сети, которая представляет из себя направленный граф, основанный на взаимодействии генов в сигнальном каскаде. В качестве вершин графа рассматриваются гены или генные модули, а в качестве ребер - биохимические взаимодействия. Вершины, которые не имеют входных ребер являются стартовыми точками обхода, а вершины, которые не имеют выходных ребер, являются конечными точками. Предполагается, что граф не имеет циклов. Число обходов Nip пути р, которые включают ген i, рассчитываются для каждого гена.
Расчет весов производится на основе следующей формулы:
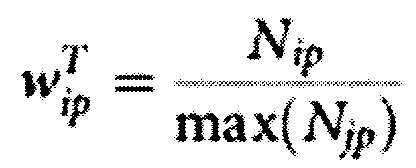 .
.
Статистические веса зависят от значений p-value, которые рассчитываются для значений генной экспрессии в конкретном образце с помощью библиотеки для анализа дифференциальной экспрессии генов limma. Для фильтрации ложной активности генов, не показывающих различий между группами, обычно используется метод резкого порогового значения p-value. Однако такой метод часто приводит к нестабильным результатам, из-за чего впоследствии оценки активации сигнальных путей получаются смещенными. Поэтому в разработанном алгоритме используется метод плавной отсечки, реализованный с помощью следующей функции:
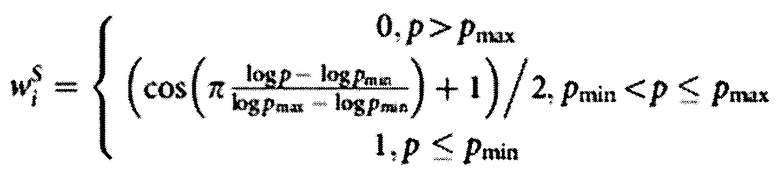 ,
,
где pmin и pmax наименьшее и наибольшее значения группового теста для генов.
Сбор и препроцессинг баз данных болезней и лекарств
Для сбора базы омиксных данных могут быть использованы такие базы омиксных данных как: BioProject (http://www.ncbi.nlm.nih.gov/bioprojecty), Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), ArrayExpress (https://www.ebi.ac.uk/arrayexpress/), PRIDE (https://www.ebi.ac.uk/pride/archive/), DDBJ (http://www.ddbj.nig.ac.jp/), ENA (http://www.ebi.ac.uk/ena), ENCODE (https://genome.ucsc.edu/ENCODE/), Human Protein Atlas (http://www.proteinatlas.org/), Proteomics DB (http://www.proteomicsdb.org/) и другие источники. Для каждого образца, где это возможно, могут быть определены болезнь, ткань, возраст, вес, пол, расовая принадлежность и другие параметры. Все образцы, относящиеся к заболеваниям должны быть аннотированы согласно онтологии Disease Ontology (http://disease-ontology.org/) или другой, а к тканям - согласно онтологии BRENDA Tissue Ontology (http://www.brenda-enzymes.org/ontology.php?ontology_id=3) или другой.
Поиск биомаркеров и физиологически активных веществ
Разработанный метод позволяет производить поиск биомаркеров и физиологически активных веществ на основе полученных значений активации сигнальных путей. В качестве входных данных используется информация об влиянии интересующего вещества на профиль активации сигнальных путей в данном виде клеток, ткани или заболевании. Список наиболее активированных или ингибированныхгенов и сигнальных путей представляет собой список потенциальных биомаркеров. Для поиска физиологически активных веществ исследуемый профиль активации сигнальных путей должен быть сравнен с профилями активации сигнальных путей других веществ из базы для того же вида клеток (ткани) с использованием метрики схожести. В качестве метрики схожести векторов активации сигнальных путей были использованы следующие варианты: sign, eucl, chi2 - тест Хи-квадрат, corr.
Метрика sign показывает количество сигнальных путей сонаправленных (активированных или ингибированных) в исследуемом профиле активации сигнальных путей и профиле активации сигнальных путей другого веществ из базы для того же вида клеток (ткани):
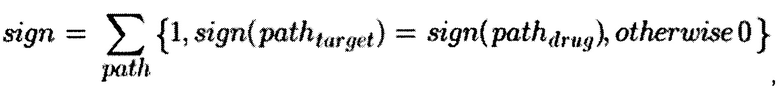
где  - значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы. Данная метрика показывает насколько близки исследуемые профили на молекулярном уровне. На примере лекарств значение данной метрики может интерпретироваться следующим образом: чем больше схожих биологических процессов протекает в профилях веществ, тем вероятнее, что данные вещества воздействуют на одни и те же потенциальные мишени (например, молекулы).
- значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы. Данная метрика показывает насколько близки исследуемые профили на молекулярном уровне. На примере лекарств значение данной метрики может интерпретироваться следующим образом: чем больше схожих биологических процессов протекает в профилях веществ, тем вероятнее, что данные вещества воздействуют на одни и те же потенциальные мишени (например, молекулы).
Метрика eucl представляем меру Евклидова расстояния в пространстве значений активации и ингибирования сигнальных путей, показывая близость между профилями в данном многомерном пространстве:
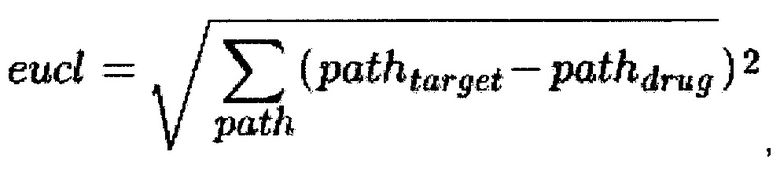
где 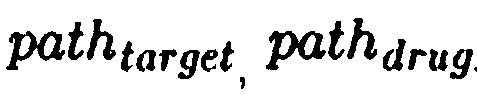 - значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы. Чем ниже значение eucl - тем ближе в пространстве сигнальных путей находятся вещества, тем более схожи они между собой.
- значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы. Чем ниже значение eucl - тем ближе в пространстве сигнальных путей находятся вещества, тем более схожи они между собой.
Метрика chi2 представляет статистический тест Хи-квадрат, который показывает значимость взаимосвязи между значениями активации сигнальных путей в исследуемом профиле и профилями из базы веществ. Данный тест выполняется на основе построения таблиц сопряженности активации и ингибирования сигнальных путей в исследуемых профилях. Оценка данного теста отражает меру схожести исследуемых профилей, отвечая на вопрос о статистически значимых различиях между количеством активированных и ингибированных сигнальных путей в профилях.
Метрика corr показывает корреляцию Пирсона между значениями активации сигнальных путей, отражая линейную зависимость между исследуемыми значениями величин:

где 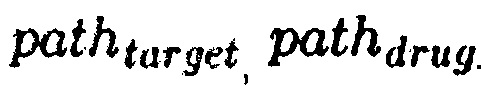 - значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы,
- значения активации сигнального пути path в исследуемом профиле активации и профиле вещества из базы, 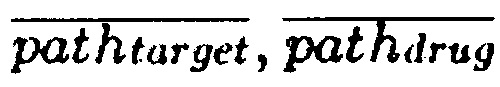 - средний значения оценок сигнальный путей.
- средний значения оценок сигнальный путей.
Выходные данные представляются в виде ранжированного списка веществ, обладающих активностью против исследуемого заболевания или схожего с исследуемым веществом. Вещества ранжируются в соответствии с оценкой схожести веществ для интересующей метрики. Аналогичный метод может применяться для перепозиционирования препаратов. В этом случае происходит расчет схожести профиля активации сигнальных путей для референсного вещества с профилями заболеваний из базы, а на выходе получается ранжированный список заболеваний, в верхней части которого оказываются заболевания, против которых с наибольшей вероятностью активно исследуемое вещество.
Созданное техническое решение предназначено для поиска биомаркеров и биологически активных веществ на основе анализа оценок активации сигнальных путей, полученных с помощью расширенной версии алгоритма iPANDA. Результатом работы такого технического решения является список веществ, которые являются схожими по химическим и биологическим свойствам с интересующим нас веществом, а также список биомаркеров исследуемого заболевания (на уровне отдельных генов и на уровне сигнальных путей).
Нижеследующие примеры осуществления способа приведены в целях раскрытия характеристик настоящего изобретения и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения.
Пример 1. Метод поиска биологически активных веществ в базе LINCS.
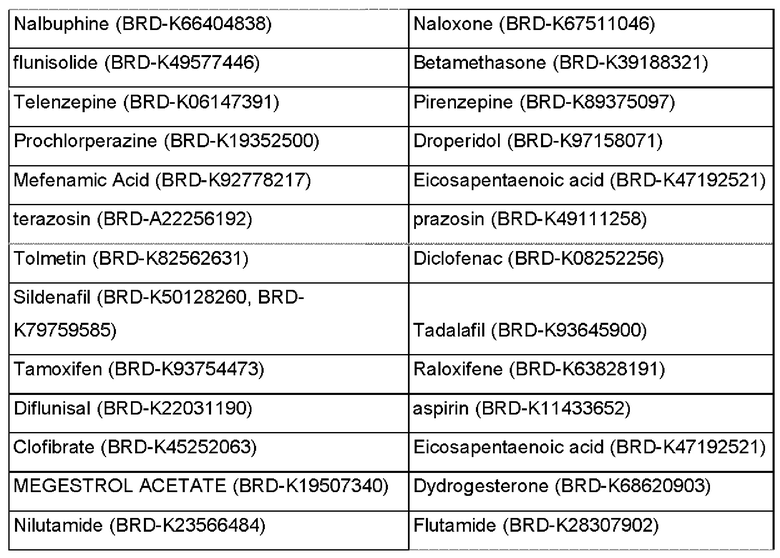
Эффективность разработанного технического решения была продемонстрирована на парах известных аналогичных химических веществ, для которых экспериментально показан сходный механизм действия. В качестве аналогичных веществ рассматривались пары приведенные в Таблице 3.
Таблица 3: Пары аналогичных химических веществ.
Исходные данные для тестирования брались из базы данных LINCS (http://www.lincsproject.org/) и представляли собой данные генной экспрессии в клеточных линиях после воздействия исследуемых веществ. Для каждого вещества, действовавшего в определенной концентрации и в течение определенного времени инкубации, входные данные включали образцы, полученные при воздействии данного вещества, а также набор контрольных (интактных) образцов для той же клеточной линии. На основе исходных данных такого вида были получены оценки относительной экспрессии генов для каждого вещества в формате отношения экспрессии в образцах с добавлением вещества к экспрессии генов в интактных образцах 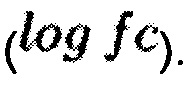 Кроме того, с помощью расширенной версии алгоритма iPANDA были рассчитаны оценки активации сигнальных путей для каждого из веществ, временных и концентрационных точек. При этом была использована внутренняя база данных сигнальных путей, включающая 2303 уникальных пути. Полученные данные по экспрессии генов и активации/ингибировании сигнальных путей использовались в качестве входных в разработанном техническом решении.
Кроме того, с помощью расширенной версии алгоритма iPANDA были рассчитаны оценки активации сигнальных путей для каждого из веществ, временных и концентрационных точек. При этом была использована внутренняя база данных сигнальных путей, включающая 2303 уникальных пути. Полученные данные по экспрессии генов и активации/ингибировании сигнальных путей использовались в качестве входных в разработанном техническом решении.
В ходе тестирования вектор значений активации сигнальных путей для одного из веществ в каждой паре выбирался в качестве референсного, а затем рассчитывалась схожесть векторов активации сигнальных путей для всех остальных веществ из использованной базы данных с референсным. Список всех веществ из базы ранжировался в соответствии с полученными значениями схожести. Ранг второго вещества в паре показывал точность предлагаемого технического решения. При этом данные, использованные для ранжирования, включали векторы активации сигнальных путей для одних и тех же веществ с различными временами инкубации и концентрациями внутри одной клеточной линии. Расчеты для различных клеточных линий проводились независимо друг от друга. Схожесть векторов активации сигнальных путей (экспрессии генов) измерялась на основе следующих метрик: sign -число значений с совпадающим знаком, eucl - Евклидово расстояния, chi2 - тест Хи-квадрат, corr - корреляция Пирсона. На Рисунках 3 и 4 представлены результаты ранжирования веществ из базы относительно референсных веществ на основе генной экспрессии и значений активации сигнальных путей соответственно.
Графики показывает результаты поиска для приведенных выше пар в сравнении с аналогичным способом поиска схожих веществ, но на основе генной экспрессии. Точки на графике - ранг (позиция, нормированная на число элементов, среди которых производился поиск) аналога, который оказался ближе всего по своим биологическим свойствам к веществу, для которого осуществлялся поиск. Рисунки 5 и 6 показывают усредненную оценку по всем веществам для используемых метрик.
Евклидова метрика показала наилучший результат для поиска аналогичных веществ в базе LINCS на основе значений активации/ингибирования сигнальных путей. Также можно заметить, что медиана у всех метрик находится ниже отметки 10 у всех метрик, кроме метрики sign, что говорит о том, что аналогичное вещество встречается среди первых 10%, и ниже отметки 15 у метрики sign, что говорит о том, что аналогичное вещество встречается среди первых 15% процентов выборки для поиска. Появление выбросов, один из которых можно наблюдать на графике, может объясняться зашумленностью данных, среди которых производится поиск.
Пример 2. Поиск веществ, обладающих терапевтическими свойствами против определенных заболеваний.
Поиск веществ, обладающих терапевтическими свойствами против определенных заболеваний осуществлялся на основе данных по клиническим испытаниям, которые были получены в результате объединения двух баз данных, включающих одобренные к использованию препараты и вещества, не прошедшие клинические испытания:
1. Drug Repurposing Hub by Broad Institute (https://clue.io/repurposing)
2. Данные из статьи об эффективности препаратов с точки зрения терапевтических механизмов (Shih HP, Zhang X, Aronov AM. Nat Rev Drug Discov. 2018 Jan; 17(1):78).
Из базы Drug Repurposing Hub были отобраны 8011 пар болезнь-лекарство, которые запущены на рынок и одобрены к использованию. Соответственно из базы, прилагающейся к статье, были отобраны преимущественно препараты, не прошедшие клинические испытания или отозванные с рынка. Данных пар болезнь-препарат было получено 6105. Собранные данные были сопоставлены сданными транскриптомных ответов различных клеточных линий на химический препарат из базы LINCS L1000. Также независимо из баз экспрессионных данных GEO и Array Express были получены данные о 212 датасетах, отвечающих 53 болезням. Собранные данные были препроцессированны, нормализованы и аннотированны на уровне генов. Впоследствии был применен метод iPANDA для получения оценок активации сигнальных путей. Затем для каждой из пар болезнь-препарат были посчитаны оценки схожести векторов активации сигнальных путей. Данные оценки были получены с помощью евклидовой метрики между противоположно направленными путями, нормированной на стандартное отклонение для оценок каждого сигнального пути в отдельности, рассчитанное по совокупности образцов для болезни и препарата. На рис. 7 представлены результаты применения метода для поиска физиологически активных веществ, отвечающих заболеваниям.
Таким образом, из рисунка 7 следует, что рассчитанная схожесть векторов активации сигнальных путей в парах одобренное для лечения вещество - соответствующая болезнь достоверно выше, чем в случайных парах вещество болезнь. Следовательно, применение предлагаемого технического решения для ранжирования физиологически активных веществ в среднем дает более высокий ранг набору веществ, достоверно имеющих терапевтическую активность против исследуемого заболевания по сравнению с набором случайных веществ.
Пример 3. Метод поиска биомаркеров заболеваний.
Данное техническое решение было применено для поиска биомаркеров ряда заболеваний, описанных ниже. При этом для всех описанных случаев с помощью применения метода удалось найти подтвержденные данными литературы биомаркеры исследованных заболеваний. В качестве входных данных для поиска биомаркеров были использованы данные из публично-доступных репозиториев омиксных данных TCGA (https://cancergenome.nih.gov/) и GEO (https://www.ncbi.nlm.nih.gov/geo/).
1. DCLK1 и другие новые биомаркеры рака головы и шеи С помощью предлагаемого технического решения было проведено исследование экспрессии гена DCLK1 и содержащих этот ген сигнальных путей при различных локализациях рака головы и шеи на основе данных из базы TCGA (https://cancergenome.nih.gov/). В соответствии с литературными данными (https://www.ncbi.nlm.nih.gov/pubmed/24384857), было показано, что экспрессия DCLK1 понижена в раковых клетках по сравнению со здоровыми. Были найдены дифференциально активированные сигнальные пути, связанные с геном DCLK1.
Было показано, что ген DCLK1 экспрессируется в фибробластах (Becht Е, et al., Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016 Oct 20; 17(1):218), а не в опухолевых клетках. Также был найден список генов, специфичных для конкретной опухоли, которые могут являться потенциальными прогностическими маркерами рака головы и шеи. Была показана связь экспрессии гена DCLK1 с семейством сигнальных путей NOTCH.
2. Исследование различных локализаций рака головы и шеи
С помощью алгоритма анализа сигнальных путей iPANDA было проведено исследование различных локализаций рака головы и шеи. Была показана схожесть различных типов рака головы и шеи на уровне генов и сигнальных путей. Была подтверждена роль МАР-киназного пути в данном типе рака, что соотноситься с литературными данными (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3946852/). Также была показана высокая активность STAT3-пути у пациентов, инфицированных вирусом папилломы человека (https://www.ncbi.nlm.nih.gov/pubmed/16503733).
3. Изучение роли внутриклеточных сигнальных систем в этиологии болезни alopecia areata путем сравнительного анализа транскриптома в норме и при патологии.
Были проанализированы транскриптомы здоровых и больных алопецией людей и выявлены гены, индуцирующие развитие заболевания. Определены сигнальные пути, наиболее и наименее активированные при alopecia areata. Полученные результаты соотносятся с литературными данными. В частности была показана активация сигнального пути Chemokine (https://link.springer.com/chapter/10.3920/978-90-8686-728-8 10), каскада JAK-STAT (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4646834/) и гиперэкспрессия цитокинов (https://www.ncbi.nlm.nih.gov/pubmed/23797621).
4. Изучении роли сигнальных путей при заболеваниях витилиго и мелазма
С помощью алгоритма анализа сигнальных путей iPANDA были найдены наиболее активированные сигнальные пути при данных заболеваниях. В полном соответствии с литературными данными (https://www.ncbi.nlm.nih.gov/pubmed/19580478) ими оказались Oxidative_phosphorylation_Main_Pathway и Ribosome_Main_Pathway. Данные, полученные после обработки iPANDA являются релевантными и подтверждаются другими методами.
5. Исследование неалкогольной жировой болезни печени (НАЖБП)
В результате данного исследования с помощью алгоритма iPANDA был найден ряд генов, дифференциально экспрессированных в образцах пациентов с НАЖБП. Большая часть генов совпадает с уже описанными ранее маркерами, например: AKR1B10 (Ryaboshapkina М, Hammar М. Human hepatic gene expression signature of non-alcoholic fatty liver disease progression, a meta-analysis. Sci Rep. 2017 Sep 27; 7(1):12361), MMP9 (https://www.ncbi.nlm.nih.gov/pubmed/19604544). S100AB (Liu X, et al., PLoS One. 2015 May 19; 10(5):e0127352), JUN (https://www.ncbi.nlm.nih.gov/pubmed/24492282). Также были найдены новые, ранее не ассоциированные с НАЖБП таргеты - MGAM и NCAM2
6. Исследование экспрессии генов и активации сигнальных путей при алкогольной болезни печени.
При проведении данного исследования с помощью алгоритма iPANDA были найдены специфичные для алкогольной болезни печени сигнальные пути Epac-Rap1 signaling и Wnt/b-catehine. Также было показано, что нарушение взаимодействия между белками Ерас и Rap1 и ингибирование белка Rap1 уменьшает вероятность возникновения фиброза печени. Эти результаты полностью соответствуют литературным данным по исследованиям на мышах (Sien-Sing Yang, Alcoholic Liver Disease in the Asian-Pacific Region with High Prevalence of Chronic Viral Hepatitis, Journal of Medical Ultrasound, Volume 24, Issue 3, September 2016, Pages 93-100).
7. Молекулярные механизмы влияния ультрафиолетового излучения
В результате сравнительного исследования образцов, подвергшихся облучению ультрафиолетом и контрольных с помощью алгоритма iPANDA были найдены наиболее дифференциально экспрессированные гены. Среди нах SP8, MLANA, TYR, DCT, TRIM63. Также были найдены сигнальные пути, преобладающие при ультрафиолетовом излучении: Bile Salt and Organic anion SLC transporters, c-Kit Receptor Signaling, BRCA1 Pathway, Notch Signaling (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4102648/, https://www.ncbi.nlm.nih.gov/pubmed/9679765).
8. Исследование экспрессии генов и активации сигнальных путей при боковом амиотрофическом склерозе (БАС).
Алгоритм iPANDA был применен для изучения этиологии заболевания.
Был проведен сравнительный анализ транскриптомов контрольной группы и пациентов и определены дифференциально экспрессированные гены-кандидаты, вовлеченные в процесс развития БАС. Исследование выявило повышенную экспрессию генов ARSB, CDH13, DDX18, GNPTAB и MTMR6, а также пониженную экспрессию генов B4GALT5, NMRK1, BAIAP2, DERL2 и CYP17A1. Наиболее активированные сигнальные пути в патогенезе БАС:
• ARSB, GNPTAB - KEGG_Lysosome_Main_Pathway
• B4GALT5 - KEGG_Mucin_type_O_Glycan_biosynthesis
• NMRK1 - KEGG_Nicotinate_and_nicotinamide_metabolism
• CYP17A1 - KEGG_Ovarian_steroidogenesis_Main_Pathway
9. Исследование механизмов остеоартрита
При проведении данного исследования с помощью алгоритма iPANDA были обнаружены дифференциально экспрессированные гены и сигнальные пути, характерные для остеоартрита. В основном гены и пути связаны с воспалительными процессами. Наиболее характерными генами являются TNF, TRAF1, NFKB2, FOS, а также интерлейкины- факторы воспаления - IL17B и IL2. Результаты исследования соотносятня с ранее известными литературными данными (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4997945/).
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные случаи приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть, понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЛАТФОРМА АНАЛИЗА ГЕНЕТИЧЕСКОЙ ИНФОРМАЦИИ ONCOBOX | 2018 |
|
RU2741703C1 |
| МЕТОД ПОИСКА ТЕРАПЕВТИЧЕСКИ ЗНАЧИМЫХ МОЛЕКУЛЯРНЫХ МИШЕНЕЙ ДЛЯ ЗАБОЛЕВАНИЙ ПУТЕМ ПРИМЕНЕНИЯ МЕТОДОВ МАШИННОГО ОБУЧЕНИЯ К КОМБИНИРОВАННЫМ ДАННЫМ, ВКЛЮЧАЮЩИМ ГРАФЫ СИГНАЛЬНЫХ ПУТЕЙ, ОМИКСНЫЕ И ТЕКСТОВЫЕ ТИПЫ ДАННЫХ | 2022 |
|
RU2798897C1 |
| СПОСОБ ПОИСКА БЕЛКОВ-МИШЕНЕЙ, ЗАПУСКАЮЩИХ ПРОЦЕСС КАНЦЕРОГЕНЕЗА, В ОБРАЗЦАХ ТКАНЕЙ ИНДИВИДУАЛЬНОГО ПАЦИЕНТА, ДЛЯ ПОСЛЕДУЮЩЕЙ ПРОТИВООПУХОЛЕВОЙ ФАРМАКОТЕРАПИИ | 2013 |
|
RU2577107C2 |
| ОТБОР БОЛЬНЫХ РАКОМ ДЛЯ ВВЕДЕНИЯ ИНГИБИТОРОВ СИГНАЛЬНОГО ПУТИ Wnt НА ОСНОВАНИИ СТАТУСА МУТАЦИИ RNF43 | 2013 |
|
RU2636000C2 |
| Способ создания таргетной панели для исследования геномных регионов для выявления терапевтических биомаркеров ингибиторов иммунных контрольных точек (ИКТ) | 2023 |
|
RU2818360C1 |
| СИСТЕМА ДЛЯ МОДЕЛИРОВАНИЯ МОЛЕКУЛЯРНЫХ ВЗАИМОДЕЙСТВИЙ, ПРИНИМАЮЩИХ УЧАСТИЕ В ВОСПАЛИТЕЛЬНОМ ПРОЦЕССЕ | 2020 |
|
RU2804998C2 |
| БИОЛОГИЧЕСКИЕ МАРКЕРЫ ДЛЯ ИДЕНТИФИКАЦИИ ПАЦИЕНТОВ ДЛЯ ЛЕЧЕНИЯ АНТАГОНИСТАМИ VEGF | 2013 |
|
RU2659173C2 |
| БИОМАРКЕРЫ ДЛЯ ТЕРАПИИ НА ОСНОВЕ ИНГИБИТОРА HEDGEHOG | 2012 |
|
RU2602185C2 |
| СПОСОБ ПРИМЕНЕНИЯ AXL В КАЧЕСТВЕ МАРКЕРА ЭПИТЕЛИАЛЬНО-МЕЗЕНХИМАЛЬНОГО ПЕРЕХОДА | 2010 |
|
RU2586493C2 |
| МикроРНК-124 В КАЧЕСТВЕ БИОМАРКЕРА | 2014 |
|
RU2687366C2 |


Предложенная группа изобретений относится к области медицины. Предложены способ и система для оценки эффективности воздействия лекарственных кандидатов, выбранных из группы лекарственных кандидатов, на физиологический процесс. Способ включает получение данных из образцов, в которых протекает указанный физиологический процесс, и из контрольных образцов, в которых не протекает указанный физиологический процесс, определение набора генов или их продуктов и сигнальных путей, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, определение набора генов и сигнальных путей после воздействия каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, на клетки или биологические ткани, в которых может протекать указанный физиологический процесс, и определение показателей эффективности воздействия на указанный физиологический процесс для каждого лекарственного кандидата. Предложенная группа изобретений обеспечивает создание более эффективного способа оценки эффективности воздействия лекарственных кандидатов на физиологический процесс. 2 н. и 1 з.п. ф-лы, 7 ил., 3 табл., 3 пр.
1. Способ оценки эффективности воздействия лекарственных кандидатов на физиологический процесс, выбранных из группы лекарственных кандидатов, состоящий по меньшей мере из следующих этапов:
(а) получают данные по меньшей мере одного типа, собранные из полученных экспериментальным путем образцов, в которых протекает указанный физиологический процесс, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам метилирования геномной ДНК, (iv) полногеномные данные по мутациям в геномной ДНК, (v) данные по экспрессии малых некодирующих РНК, (vi) данные фенотипических скринингов с помощью системы CRISPR-CAS9, (vii) данные по фосфорилированию белков;
(б) получают данные того же типа, что и данные, полученные на стадии (а), собранные из контрольных образцов, в которых не протекает указанный физиологический процесс;
(в) определяют необходимость гармонизации данных, полученных на стадиях (а) и (б) и, при необходимости, осуществляют следующие операции гармонизации этих данных: препроцессинг, нормализацию и формирование фенотипических групп;
(г) определяют набор генов, или их продуктов, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя данные, полученные на стадиях (а)-(в);
(д) определяют набор сигнальных путей, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя набор генов, полученный на стадии (г);
(е) определяют набор генов и сигнальных путей, статистически достоверно активированных или ингибированных после воздействия каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, на клетки или биологические ткани, в которых может протекать указанный физиологический процесс;
(ж) определяют количественные показатели эффективности воздействия на указанный физиологический процесс для каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е), таким образом оценивая эффективность воздействия потенциальных лекарственных средств на указанный физиологический процесс.
2. Способ по п. 1, характеризующийся тем, что
(i) на стадиях (г) и (д) дополнительно проводят сравнение генов и сигнальных путей между различными состояниями, составляющими указанный физиологический процесс, и определяют набор генов и сигнальных путей, активированных или ингибированных в отдельных подгруппах образцов, соответствующих указанному физиологическому процессу, тем самым выделяя специфические по признакам болезни подгруппы пациентов для указанного физиологического процесса;
(ii) на стадии (ж) определяют количественные показатели эффективности воздействия в отдельных подгруппах образцов, соответствующих указанному физиологическому процессу, для каждого лекарственного кандидата, выбранного из группы потенциальных лекарственных веществ, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е).
3. Система для оценки эффективности воздействия лекарственных кандидатов на физиологический процесс, выбранных из группы лекарственных кандидатов, включающая:
- по меньшей мере один процессор;
- по меньшей мере одну память, которая содержит машиночитаемые инструкции, которые при их исполнении по меньшей мере одним процессором осуществляют оценку эффективности воздействия лекарственных кандидатов на физиологический процесс при помощи компьютерно-реализуемого способа, состоящего по меньшей мере из следующих этапов:
(а) получают данные по меньшей мере одного типа, собранные из полученных экспериментальным путем образцов, в которых протекает указанный физиологический процесс, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам метилирования геномной ДНК, (iv) полногеномные данные по мутациям в геномной ДНК, (v) данные по экспрессии малых некодирующих РНК, (vi) данные фенотипических скринингов с помощью системы CRISPR-CAS9, (vii) данные по фосфорилированию белков;
(б) получают данные того же типа, что и данные, полученные на стадии (а), собранные из контрольных образцов, в которых не протекает указанный физиологический процесс;
(в) определяют необходимость гармонизации данных, полученных на стадиях (а) и (б) и, при необходимости, осуществляют следующие операции гармонизации этих данных: препроцессинг, нормализацию и формирование фенотипических групп;
(г) определяют набор генов, или их продуктов, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя данные, полученные на стадиях (а)-(в);
(д) определяют набор сигнальных путей, статистически достоверно активированных или ингибированных в образцах, в которых протекает указанный физиологический процесс, по сравнению с контрольными образцами, в которых не протекает указанный физиологический процесс, используя набор генов, полученный на стадии (г);
(е) определяют набор генов и сигнальных путей, статистически достоверно активированных или ингибированных после воздействия каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, на клетки или биологические ткани, в которых может протекать указанный физиологический процесс;
(ж) определяют количественные показатели эффективности воздействия на указанный физиологический процесс для каждого лекарственного кандидата, выбранного из группы лекарственных кандидатов, сравнивая наборы генов и сигнальных путей, определенных на стадиях (г)-(е), таким образом оценивая эффективность воздействия потенциальных лекарственных средств на указанный физиологический процесс.
| OZEROV I.V | |||
| et al | |||
| In silico Pathway Activation Network Decomposition Analisis (iPANDA) as a method for biomarker development | |||
| Nat Commun | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| WO 2012104764 A2, 09.08.2012 | |||
| TRAYNARD P | |||
| et al | |||
| Logic Modeling in Quantitative Systems Pharmacology | |||
| CPT Pharmacometrics Syst Pharmacol | |||
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| НЕМУДРЫЙ А.А | |||
| и др | |||
| Системы редактирования геномов TALEN и CRISPR-Cas инструменты открытий | |||
| Acta Naturae (русскоязычная версия) | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |

