Область техники
Изобретение относится к персонализированной медицине для онкологических заболеваний, а именно к системе поддержки принятия клинических решений с использованием анализа активности внутриклеточных молекулярных путей, основанного на широкомасштабных мутационных профилях и данных по генной экспрессии.
Уровень техники
Онкологические заболевания характеризуются нарушением клеточного цикла, появлением бесконтрольно делящихся клеток, способных к быстрому росту и инвазии в прилежащие к опухоли зоны и метастазированию в отдаленные органы. Онкологические заболевания занимают лидирующие позиции среди причин смертности, как в России, так и в мире. Благодаря развитию современных подходов радио- и химиотерапии рака, а также ранней диагностики и совершенствованию техник хирургического удаления опухолей, в последние десятилетия удалось стабилизировать, а в некоторых случаях и снизить, показатели смертности от данного типа заболеваний.
На сегодняшний день разработаны и применяются в медицинской практике более 200 лекарственных препаратов для борьбы с раком и еще больше вариантов их комбинаций [https://www.cancer.gov/about-cancer/treatment/drugs]. Но по-прежнему остается открытым вопрос выбора терапии для конкретного пациента. Известно, что применение одного и того же препарата при одном и том же гисто-морфологическом типе опухоли у разных пациентов может иметь разный эффект: от полного регресса опухоли до ее активной прогрессии. Этот ответ невозможно предугадать лишь на основании истории болезни и гистологического типа опухоли, поэтому сегодня выбор терапии зачастую осуществляется вслепую из списка одобренных для данной нозологии препаратов, что часто ассоциировано с низкой эффективностью применения терапии и высокими показателями смертности от онкологических заболеваний.
Развитие современных наукоемких технологий массового параллельного секвенирования (или глубокого секвенирования) и микрочипового исследования транскриптомов позволили взглянуть на гистологически однотипные опухоли с новой стороны. Оказалось, что одинаковые гистолого-морфологические изменения тканей могут быть вызваны разными наборами мутаций, а также сопровождаться разными профилями экспрессии генов. Таким образом, в современном медицинском мире информация о гистолого-морфологическом типе опухоли постепенно отходит на второй план при выборе терапии, в то время как генетика опухоли конкретного пациента выходит на первый. Такой подход все чаще встречается в современной медицинской практике и имеет очевидные доказанные преимущества (Martel CL, Lara PN. Renal cell carcinoma: current status and future directions. Crit Rev Oncol Hematol. 2003 Feb;45(2):177-90).
23 мая 2017 года Управление по продуктам и лекарствам США медикаментов (FDA) впервые в истории одобрило в качестве показания к применению противоракового препарата Кейтруда (Пембролизумаб) наличие генетического маркера опухоли, а не локализацию или тип опухоли [https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm560167.html]. Персонализированная онкология по этой причине представляется будущим стандартом медицины. Поэтому актуальной задачей является создание биомедицинских платформ для интеллектуального подбора наиболее эффективной терапии и поиска прогностических опухолевых маркеров в случае конкретного пациента. Широкое внедрение подобных методик в будущем будет способствовать уменьшению частоты смертности от онкологических заболеваний.
В настоящее время существует ограниченное число платформ, использующих отдельные виды широкомасштабного генетического профилирования для консультирования докторов и пациентов. Примером является система Caris Molecular Intelligence (Russell et al. 2014; Green et al. 2014; Popovtzer et al. 2015; Vigneswaran et al. 2016). Использование платформы основано на анализе ограниченного спектра мутаций с ранее показанной клинической значимостью, а также на профилировании биообразцов пациентов на иммуногистохимической панели определения белковых онкомаркеров. Тем не менее, указанная система не использует результатов исследований полных спектров генной экспрессии, не производит анализ активации молекулярных путей и не предназначена для одновременной обработки мультиомиксных данных. Как следствие, возможности таких систем по использованию мультиомиксных данных для анализа внутриклеточных путей и для выработки клинически значимых рекомендаций существенным образом ограничены.
Уровень активности молекулярного пути в клетке зависит от концентраций входящих в него генных продуктов, участвующих в выполнении молекулярных функций, являющихся специализацией данного пути.
Для того, чтобы оценивать активацию молекулярных путей на основе данных по концентрации генных продуктов-участников этих путей ранее были разработаны такие методы, как TAPPA (Gao and Wang 2007), topology-based score (TB) (Ibrahim et al. 2012), Pathway-Express (PE) (Draghici et al. 2007), SPIA (Tarca et al. 2009), Oncofinder (Buzdin et al., 2013), IPANDA (Ozerov et al., 2016) и другие. Эти подходы используют различные формулы для расчета уровней активации каждого молекулярного пути, и их отличительными чертами является приведение вычисляемого уровня активации в зависимость от концентрации генных продуктов - участников пути, в исследуемом образце по сравнению с контрольными образцами.
Например, в патентной заявке US20170262578A1, описывается вычисление активации молекулярных путей (индекс Pathway Activation Strength, PAS) по формуле:
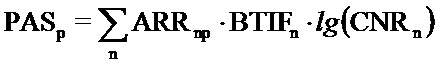
Здесь PASp - показатель уровня активации молекулярного пути p; ARRnp - роль генного продукта n в молекулярном пути p (принимает значения от -1 до 1 в зависимости от того, является ли генный продукт активатором или репрессором молекулярного пути, соответственно); BTIFn - это показатель того, является ли генный продукт n дифференциально экспрессируемым относительно контрольной группы образцов; lg - десятичный логарифм; case-to-normal ratio, CNRn - отношение уровня экспрессии гена n в исследуемом образце к усредненному уровню экспрессии в группе контрольных образцов. При этом данный алгоритм был предложен для обработки данных экспрессии матричной РНК (мРНК) и белковых профилей. Приложением вычисленных значений активации молекулярных путей может являться создание биомаркеров нового поколения, поскольку значения PAS могут маркировать различные физиологические и патологические состояния клеток, тканей и всего организма. При этом уровень генной экспрессии определяется непосредственно в самой патологической ткани пациента. Таковой в случае онкозаболеваний могут являться, например, свежие образцы биопсии или парафинизированные блоки опухолевой ткани.
В патентной заявке US20160224739 A1 предлагается использовать набор индексов активации молекулярных путей для предсказания ответа пациентов с раком молочной железы на различные виды химиотерапии. Для достижения этого результата, авторы заранее определяют справочные (референтные) значения индексов активации маркерных молекулярных путей для каждого из анализируемых видов химиотерапии, отдельно для группы пациентов, ответивших на нее (ответчиков) и для группы неответивших пациентов (неответчики). После этого, для исследуемого случая (например, блока парафинизированной опухолевой ткани) определяется уровней активации маркерных молекулярных путей и проводится анализ того, насколько эти уровни близки к значениям для референтных групп ответчиков и неответчиков. На основании сходства профилей индексов активации маркерных путей к той или иной группе, делается предсказание относительно ответа конкретного исследуемого пациента на данный вид химиотерапии.
С другой стороны, в заявке US20170193176A1, см. также статью Artemov et al., 2015, предлагается способ расчета относительной эффективности действия фармпрепаратов (Drug Score, DS) исходя из значений активации молекулярных путей. Для этого авторы предлагают формулу:
 ,
,
где DS - это показатель потенциальной эффективности таргетного препарата; d - это препарат, для которого производится анализ эффективности; PASp - показатель силы активации молекулярного пути p; AMCFp - показатель, учитывающий способность молекулярного пути p усиливать или ослаблять клеточные рост или гибель (при этом он принимает значения 1 и -1, соответственно); DTI - показатель, учитывающий способность препарата d ингибировать генный продукт t или нет (при этом он принимает значения 1 и 0, соответственно); NII- показатель, учитывающий вовлеченность или нет генного продукта t в молекулярный путь p (при этом он принимает значения 1 и 0, соответственно).
В отличие от системы Онкобокс, при оценке эффективности, этот метод не учитывает изменение эффективной концентрации молекулярных мишеней фармпрепаратов. Другое отличие - в том, что приведенный в заявке US20170193176A1, а также в заявке US20160132632A1 метод может анализировать только уровни экспрессии мРНК и белков, но не профили мутаций ДНК, распределение сайтов связывания транскрипционных факторов и микроРНК.
Для всех ранее опубликованных способов предсказания эффективности препаратов характерен ряд ограничений.
Во-первых, они не могут одновременно использовать широкий спектр результатов мультиомиксного профилирования генов: данные по генным мутациям, по профилю связывания транскрипционных факторов, по уровню экспрессии белков, мРНК и микроРНК. При этом ранее вообще никогда не публиковались способы формализованного предсказания эффективности большого числа препаратов разной специфичности исходя из профилей генных мутаций, экспрессии микроРНК и связывания транскрипционных факторов.
Во-вторых, ранее опубликованные методы для предсказания эффективности препаратов не использовали комбинирования уровней активации молекулярных путей и отдельной количественной меры уровней изменения (экспрессии или мутационной нагрузки) генов - непосредственных мишеней исследуемых препаратов, взятой вне контекста молекулярных путей.
В-третьих, ранее опубликованные подходы при анализе экспрессии мРНК и белка (протеомные профили) не решали проблему гармонизации данных в части объединения профилей генной экспрессии исследуемых образцов и набора нормальных (контрольных) образцов.
В-четвертых, при анализе уровней активации молекулярных путей, роль тех или иных генных продуктов в составе пути определялась при ручном курировании графа молекулярного пути. Это является источником операционных ошибок и ограничивает широкое применение таких технологий в силу невозможности эффективной ручной обработки сотен и тысяч молекулярных путей, включающих десятки и сотни генных продуктов.
В-пятых, в ранее опубликованных методах массового скрининга таргетных препаратов слабо проработаны различия в расчетных алгоритмах в зависимости от природы различных классов таргетных препаратов и от механизма их действия.
На сегодняшний день в клинической практике не существует эффективных способов прогнозирования эффективности действия существующих противоопухолевых препаратов для конкретного пациента, которые бы учитывали особенности молекулярного дисбаланса, образующегося при развитии конкретной опухоли. Как следствие, большинство пациентов получают стандартные препараты, выбор которых основан на клинических или морфологических параметрах, таких как стадия заболевания, размер опухоли, агрессивность развития заболевания и др., что зачастую приводит к тому, что пациенты не отвечают на терапию, и рост опухоли продолжается. Развитие персонализированного подхода к лечению раковых заболеваний исходя из молекулярных нарушений в организме пациента является актуальной задачей, и данное изобретение призвано расширить круг подходов, применяющихся для решения этой задачи.
Сущность изобретения
Задачей настоящего изобретения является создание эффективного, научно обоснованного подхода к персонализированной терапии для онкологических пациентов, то есть к выбору противоопухолевого препарата, наиболее подходящего для конкретного пациента и способного модулировать сигнальные пути таким образом, чтобы компенсировать патологические изменения в опухолевых тканях. Описанный в настоящем изобретении подход заключается в анализе изменений во внутриклеточных молекулярных путях: сигнальных, репарационных, метаболических, цитоскелетных и других, а также прогнозе клинической эффективности применения лекарственных средств для индивидуальных онкологических пациентов. Описанная в изобретении платформа Oncobox решает задачу перепозиционирования существующих лекарственных препаратов для новых показаний к применению, а также может решать задачу поиска молекулярных мишеней при разработке новых лекарственных препаратов направленного действия (таргетных препаратов).
В качестве исходных данных Oncobox использует результаты мультиомиксных исследований образцов клеток и тканей индивидуальных больных, а также лиц без медицинской патологии. В качестве биоматериала для исследования используются свежие, парафинизированные или законсервированные иным путем образцы ткани. Исследованиями, предоставляющими исходные данные для платформы Oncobox, может быть мультиомиксное генетическое профилирование: высокопроизводительное профилирование экспрессии генов на уровне мРНК с использованием гибридизации на микрочипах, глубокого секвенирования, ОТ-ПЦР в реальном времени или профилирование на уровне экспрессии белков с использованием технологий количественной протеомики, профилирование количественного спектра микроРНК-транскриптома биообразцов, а также глубокое секвенирование геномной ДНК или определение профилей связывания транскрипционных факторов с геномной ДНК.
На основе результатов мультиомиксного профилирования, платформа Oncobox вычисляет профили активации внутриклеточных сигнальных путей (сигналома), а затем оценивает потенциальную эффективность воздействия фармпрепаратов с известным спектром молекулярных мишеней на индивидуального больного.
Указанная задача решается путем применения способа оценки клинической эффективности таргетных лекарственных средств для лечения пациента с пролиферативным или онкологическим заболеванием, выбранных из группы таргетных лекарственных средств, состоящего по меньшей мере из следующих этапов: (а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства, выбранного из указанной группы; (б) получают образец ткани пациента, имеющий пролиферативный фенотип; (в) получают данные по меньшей мере одного типа из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК;
(г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в);
(д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i) - (v), получаемых на стадии (д), совпадает с типом данных, соответственно (i) - (v), получаемых на стадии (в);
(е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д);
(ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i) - (v), используя данные, полученные на стадиях (в)-(е);
(з) определяют клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
В некоторых предпочтительных вариантах осуществления изобретения вышеуказанный способ оценки клинической эффективности таргетных лекарственных средств характеризуется тем, что данные, полученные из по меньшей мере одного контрольного образца ткани гармонизированы с данными, полученными на стадиях (в) и (д). В других вариантах способ характеризуется тем, что (i) на стадии (в) получают данные по меньшей мере двух типов; (ii) на стадии (ж) независимо рассчитывают количественный показатель эффективности по каждому типу данных; и (iii) оценивают клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств, усредняя рассчитанные для каждого типа данных количественные показатели эффективности. В особо предпочтительных вариантах на стадии (в) получают полногеномные гармонизированные данные по экспрессии белков, и полногеномные данные по мутациям в геномной ДНК.
В других вариантах изобретения указанная задача решается путем применения способа лечения пациента с пролиферативным или онкологическим заболеванием, состоящего по меньшей мере из следующих этапов: (а) получают информацию об имеющихся таргетных лекарственных средствах и формируют группу таргетных лекарственных средств; (б) оценивают клиническую эффективность таргетных лекарственных средств, выбранных из указанной группы таргетных лекарственных средств, согласно вышеуказанному способу оценки клинической эффективности таргетных лекарственных средств; (в) выбирают для лечения указанного пациента лекарственное средство, имеющее лучший, или один из лучших количественных показателей эффективности, определенных согласно вышеуказанному способу оценки клинической эффективности таргетных лекарственных средств.
В других вариантах изобретения указанная задача решается путем использования системы для оценки клинической эффективности таргетных лекарственных средств, выбранных из группы таргетных лекарственных средств, для пациента с пролиферативным или онкологическим заболеванием ткани, включающей:
- по меньшей мере, один процессор;
- по меньшей мере, одну память, которая содержит машиночитаемые инструкции, которые при их исполнении, по меньшей мере, одним процессором
- осуществляют оценку клинической эффективности указанных таргетных лекарственных средств, при помощи компьютерно-реализуемого способа, состоящего по меньшей мере из следующих этапов: (а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства, выбранного из указанной группы; (в) получают данные по меньшей мере одного типа из образца ткани пациента, имеющей пролиферативный фенотип, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК; (г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента; тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в); (д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i) - (v), получаемых на стадии (д), совпадает с типом данных, соответственно (i) - (v), получаемых на стадии (в); (е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента; тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д); (ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i) - (v), используя данные, полученные на стадиях (в)-(е); (з) оценивают клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
В других вариантах изобретения указанная задача решается путем применения способа определения наиболее эффективного лекарственного средства из группы таргетных лекарственных средств для пациента с пролиферативным или онкологическим заболеванием, состоящего по меньшей мере из следующих этапов: (а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства, выбранного из указанной группы; (б) получают образец ткани пациента, имеющий пролиферативный фенотип; (в) получают данные по меньшей мере одного типа из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК; (г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в); (д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i) - (v), получаемых на стадии (д), совпадает с типом данных, соответственно (i) - (v), получаемых на стадии (в); (е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д); (ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i) - (v), используя данные, полученные на стадиях (в)-(е);(з) определяют клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
При осуществлении изобретения достигается следующий технический результат: разработан новый, более эффективный способ оценки клинической эффективности таргетных лекарственных средств для индивидуального пациента с пролиферативным или онкологическим заболеванием ткани, использующий широкий спектр экспериментальных данных: данные по генным мутациям, по профилю связывания транскрипционных факторов, по уровню экспрессии белков (с учетом гармонизации), мРНК (с учетом гармонизации) и микроРНК, а также информацию по молекулярным мишеням таргетных лекарственных средств. Данный способ может быть автоматизирован, что предотвращает потенциальные ошибки, связанные с ручным расчетом, и позволяет учитывать изменения в сотнях и тысячах молекулярных путей, включающих десятки и сотни генных продуктов, характерных для конкретного пациента.
Краткое описание рисунков
Фиг. 1. Схема организации платформы Oncobox.
Фиг. 2. Схема алгоритма Shambhala для универсальной гармонизации экспрессионных профилей. Различные образцы (профили) (1,…, N) (см. левый нижний прямоугольник) добавляются по одному к вспомогательному (калибровочному) набору профилей P с помощью квантильной нормализации (Bolstad et al. 2003). Получившийся в результате этого набор профилей P1 подвергается затем конверсии в дефинитивную форму, характерную для набора профилей Q, с помощью кусочно-линейной гармонизации (Shabalin et al. 2008). Отличие данной конверсии от других опубликованных аналогов заключается в том, что 1) в процессе конверсии итеративным изменениям подвергается только набор профилей P1, в то время, как набор Q остается постоянным; 2) для кластеризации генов и образцов в наборах профилей P1 and Q используется сферическая (косинусоидальная), а не барицентрическая, как в методе XPN, мера близости (Shabalin et al. 2008). После данных процедур образец/профиль i = (1, …, N) считается гармонизированным.
Фиг. 3. Распределение белоккодирующих генов человека по значениям индексов GRES (обозначено на схеме как GRE).
Фиг. 4. Окрашивание гематоксилином и эозином указывает на умеренно дифференцированную внутрипеченочную холангиокарциному.
Фиг. 5. Клинический статус препаратов, отсортированных по значению индекса BES. Показатели клинической значимости (отложены по оси ординат): 1 - препарат одобрен для клинического применения при опухолях данной локализации; 0,85 - успешно прошел III фазу клинических испытаний; 0,7 - находится на III фазе клинических испытаний; 0,4 - успешно прошел II фазу; 0,3 - находится на II фазе клинических испытаний; 0,2 - успешно прошел I фазу; 0,1 - находится на I фазе клинических испытаний.
Фиг. 6. Приведение профилей генной экспрессии к универсальному виду с использованием метода Shambhala платформы Онкобокс. Показаны экспрессионные профили до и после гармонизации с помощью алгоритма Shambhala (верхний и нижний ряды панелей, соответственно). Показано распределение генов по уровням экспрессии (оси абсцисс). Исходные экспрессионные профили были получены для одного и того же биообразца (Stratagene Universal Human Reference RNA; UHRR Catalog #740000) с использованием разных экспериментальных платформ (слева направо): Illumina HiSeq 2000 (GPL11154), Illumina HumanHT-12 V4.0 expression beadchip (GPL10558), Affymetrix Human Gene 2.0 ST Array (GPL17930) и Affymetrix GeneChip PrimeView Human Gene Expression Array (GPL16043).
Фиг. 7. Индекс Жаккара для сравнения списков препаратов, которые попадали в верхнюю часть рейтинга, составленного по индексу эффективности DS1a, для исследуемых 11 нозологий (указаны заболевания по типу ткани).
Фиг. 8. Индекс Жаккара для сравнения списков препаратов, которые попадали в верхнюю часть рейтинга, составленного по индексу эффективности BES, для исследуемых 11 нозологий (указаны заболевания по типу ткани).
Фиг. 9. Зависимость рейтинга таргетных препаратов, рассчитанного системой Oncobox по транскриптомному профилю пациента с раком шейки матки из базы данных TCGA, от клинического рейтинга данных препаратов по базе clinicaltrials.gov (по состоянию на август 2017 года). Зеленым отмечены препараты, которые находились в верхней части рейтинга, красным - в нижней части.
Фиг. 10. Плотность распределения коэффициентов Анубиса, рассчитанных по индексу эффективности препаратов BES (верхний график) или DS1a (нижний график) для 306 пациентов с раком шейки матки (зеленым). Прозрачные столбцы гистограммы отражают распределение коэффициентов Анубиса при случайном перемешивании клинического статуса препаратов.
Подробное раскрытие изобретения
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Под «субъектом», или «пациентом», следует понимать человека (предпочтительно) или другое млекопитающее. Под «тканью» пациента или субъекта следует понимать значение, принятое в медицинской литературе - это система клеток и межклеточного вещества, объединённых общим происхождением, строением и выполняемыми функциями. В описании данного изобретения это может быть кровь, твердая ткань различного происхождения (например, эпителиальная, соединительная, нервная или мышечная) или часть любого органа пациента или субъекта.
Под образцом ткани пациента, имеющим пролиферативный фенотип, понимают образец опухолевой ткани, клетки которой обладают способностью к неконтролируемому делению в результате патологического изменения (обычно в результате изменения генетического аппарата клеток). Такой образец может быть частью доброкачественной или злокачественной опухоли.
Под пролиферативным или онкологическим заболеванием понимают заболевание, характеризуемое патологическим изменением генетического аппарата клеток, приводящее к неконтролируемому делению определенной популяции клеток. Примерами пролиферативных заболеваний могут служить миелопролиферативные заболевания, лимфопролиферативные заболевания, пролиферативные заболевания соединительной ткани и другие заболевания.
Под данными по экспрессии тотальной мРНК следует понимать данные, показывающие абсолютное или относительное количество всех или более 300 видов молекул мРНК в образце. Под полногеномными данными по сайтам связывания транскрипционных факторов следует понимать данные, показывающие области связывания заданного набора транскрипционных факторов с последовательностями ДНК в геноме субъекта. Этот набор определяется специалистом исходя из вовлеченности транскрипционных факторов в молекулярные пути, предположительно ответственные за заболевание. Аналогичным образом определяются полногеномные данные по экспрессии белков, полногеномные и полноэкзомные данные по мутациям в геномной ДНК, полногеномные данные по экспрессии микроРНК.
Под гармонизацией данных понимают приведение их к универсальному сравниваемому виду. При осуществлении данного изобретения необходимость гармонизации возникает при использовании данных по экспрессии тотальной мРНК или полногеномных данных по экспрессии белков, в случае, когда соответствующие данные контрольных образцов были получены независимо от данных пациента, в том числе, с использованием разных методов. В таком случае, для их гармонизации может быть использован предложенный авторами метод Shambhala, или другой метод, обеспечивающий возможность сравнения полногеномных экспрессионных данных, полученных на разных платформах. В случае, когда данные пациента и контрольных образцов получают параллельно с использованием единой методики, данные считаются гармонизированными без использования дополнительных алгоритмов. Аналогично, для полногеномных данных по мутациям в геномной ДНК, полногеномных данных по экспрессии микроРНК и полногеномных данных по сайтам связывания транскрипционных факторов необходимость в гармонизации отсутствует, и такие данные считаются гармонизированными без использования дополнительных алгоритмов.
Если не определено отдельно, технические и научные термины в данном описании имеют стандартные значения, общепринятые в научной и технической литературе.
Платформа Oncobox решает задачи прогноза клинической эффективности лекарственных средств для индивидуальных онкологических пациентов, а также задачу перепозиционирования существующих лекарственных препаратов и задачу поиска молекулярных мишеней при разработке новых лекарственных препаратов направленного действия (таргетных препаратов).
Задачи решаются при помощи технологии, основанной на молекулярном профилировании образцов патологической ткани, последующем анализе профилей активации молекулярных путей и вычислении сбалансированных индексов эффективности препаратов Balanced Efficiency Score (BES).
В качестве образцов патологической ткани могут браться образцы свежей, парафинизированной или законсервированной иным способом ткани. В предпочтительном варианте осуществления изобретения анализ ткани производится у пациента с пролиферативным или онкологическим заболеванием ткани. Также для патологической ткани предпочтительно проводить анализ достаточно гомогенного участка ткани пациента, имеющей пролиферативный фенотип. Для этого возможно проводить дополнительную очистку ткани, имеющей пролиферативный фенотип, от других окружающих ее тканей с помощью методов, известных специалистам.
Платформа Oncobox эффективна для анализа большого спектра широкомасштабных молекулярных данных: данных по профилям мутаций в генах, по экспрессии мРНК генов, по экспрессии молекул микроРНК, специфических к соответствующим генным продуктам, по количественным протеомным профилям, а также по частоте встречаемости сайтов связывания транскрипционных факторов в генных регуляторных областях.
Первой решаемой технической задачей (1) является анализ изменений внутриклеточных молекулярных путей исходя из перечисленных видов данных. При этом к измеряемым внутриклеточным путям относятся сигнальные, репарационных, метаболические, цитоскелетные и другие молекулярные пути.
При этом первая техническая задача разделяется на ряд подзадач:
1.1. Разработка баз данных молекулярных путей и определение функциональной роли включенных в них генных продуктов
1.2. Разработка алгоритмов анализа активации молекулярных путей для данных по мутациям ДНК, экспрессии мРНК, белков, микроРНК, а также по профилям связывания транскрипционных факторов.
Второй решаемой технической задачей (2) является персонализированное прогнозирование клинической эффективности фармпрепаратов для индивидуальных пациентов, в том числе онкологических.
Вторая техническая задача разделяется на следующие подзадачи:
2.1. Разработка баз данных молекулярных мишеней фармпрепаратов.
2.2. Разработка алгоритмов персонализированного прогнозирования клинической эффективности фармпрепаратов на основе уровней активации молекулярных путей и других молекулярно-статистических данных.
Схема организации платформы Oncobox.
Общая схема работы платформы Oncobox приведена на Фиг.1.
Платформа Oncobox имеет ряд существенных преимуществ относительно иных опубликованных аналогов.
Во-первых, в отличие от других систем, она может одновременно использовать беспрецедентно широкий спектр результатов мультиомиксного профилирования генов: данные по генным мутациям, по профилю связывания транскрипционных факторов, по уровню экспрессии белков, мРНК и микроРНК. При этом в рамках платформы Oncobox впервые вводятся способы формализованного предсказания эффективности таргетных препаратов разной специфичности исходя из профилей генных мутаций, экспрессии микроРНК и связывания транскрипционных факторов. Возможность одновременного использования разных видов молекулярных данных дает платформе Oncobox уникальное преимущество проверки правильности выбора препаратов альтернативными методами. Впервые появляется возможность проведения единообразного сравнения геномных, транскриптомных либо эпигенетических данных, доступных для данного пациента. При этом итоговая рекомендация включает консенсусные препараты, рекомендованные с использованием наибольшего числа из доступных видов анализа.
Во-вторых, в отличие от других методов, платформа Oncobox использует комбинирование уровней активации молекулярных путей и специальной количественной меры изменения (экспрессии или мутационной нагрузки) генов - непосредственных мишеней препаратов. Это позволяет получать результаты расчета относительной эффективности действия фармпрепаратов более высокого качества, чем в ранее опубликованных методах, например, чем в заявке US20170193176A1, а также в статье Artemov et al., 2015. Сопоставление групп данных, полученных с помощью платформы Oncobox и ранее опубликованных подходов, приведено в Примере 8.
В-третьих, в рамках платформы Oncobox впервые вводится требование к унификации (гармонизации) данных в части объединения профилей генной экспрессии исследуемых образцов и наборов нормальных (контрольных) образцов перед анализом и предлагается оригинальный инструмент для осуществления этой задачи.
В-четвертых, в рамках платформы Oncobox впервые вводится автоматическая аннотация молекулярных путей. Предлагается оригинальный инструмент для определения роли каждого генного продукта в составе каждого молекулярного пути при анализе их уровней активации. Это одновременно снижает число операционных ошибок и повышает производительность платформы Oncobox до анализа любого количества молекулярных путей, включающих любое количество генных продуктов.
Другим важным отличием системы Oncobox является оригинальный способ расчета сбалансированного индекса эффективности для каждого препарата Balanced Efficiency Score (BES). На основании значений этого индекса, производится моделирование эффективности препаратов. BES рассчитывается с использованием единого алгоритма, включающего два базовых слагаемых: Drug Efficiency Score MP, DESMP (отражает вклад молекулярных путей) u Drug Efficiency Score TG, DESTG (отражает вклад конкретных молекулярных мишеней препаратов), при этом для различных типов препаратов используются разные весовые коэффициенты для DESMP u DESTG, варьирующие от -1 до 1.5. Впервые вводится суммирование слагаемых: DESMP u DESTG, разделение препаратов на классы и весовые коэффициенты в формуле для DESMP u DESTG в зависимости от этих классов. Разделение препаратов на классы осуществляется в соответствии с их известным механизмом действия и молекулярной специфичностью.
Предлагаемый метод может быть также использован для перепозиционирования существующих препаратов и для поиска новых мишеней при разработке новых фармпрепаратов.
Анализ внутриклеточных молекулярных путей
Количественный анализ молекулярных путей в исследуемом биообразце является первым этапом работы платформы Oncobox. Для этого система Oncobox использует единый базовый алгоритм анализа молекулярных путей, для которого ранее была показана способность минимизировать экспериментальные ошибки измерения (Aliper et al. 2017). Для каждого из типов анализируемых данных (мутации ДНК, профили экспрессии мРНК, белков, микроРНК, профиль связывания транскрипционных факторов), система Oncobox использует специфические модификации исходного алгоритма.
Анализ проводится на основе базы данных молекулярных путей с автоматически курируемыми функциональными ролями генных продуктов - участников каждого пути. В рамках системы Онкобокс, выделяют пять видов функциональных ролей для генных продуктов: активатор пути, репрессор, скорее активатор, скорее репрессор, а также генные продукты с невыясненной или противоречивой ролью.
Автоматическое курирование функциональной роли генных продуктов из базы молекулярных путей является одной из оригинальных особенностей платформы Oncobox. Оно устроено следующим образом.
Алгоритм расстановки коэффициентов, отражающих роль генных продуктов в активации молекулярного пути.
Алгоритм присвоения генным продуктам коэффициентов, отражающих роль генных продуктов в активации молекулярного пути (activator/repressor role, ARR) основан на анализе графа белок-белковых взаимодействий для каждого отдельного пути. Данный граф может быть построен вручную или с использование опубликованных баз данных молекулярных путей, таких как KEGG, biocarta, Reactome и др. В вершинах такого графа находятся гены, а наличие ребра между двумя вершинами свидетельствует о наличии белок-белкового взаимодействия между соотвествующими генными продуктами. Каждое ребро данного графа направлено, а также имеет параметр, указывающий на характер белок-белкового взаимодействия: «активация» или «ингибирование». Для корректной расстановки коэффициентов ARR данный граф должен быть связным, при этом достаточным условием является слабая связность.
Если граф белок-белковых взаимодействий для заданного молекулярного пути соответствует вышеуказанным критериям, коэффициенты ARR могут быть автоматически выставлены генным продуктам, которые входят в состав данного пути. Для этого авторы предлагают следующий рекурсивный алгоритм:
1) Инициализация: выявляется первая вершина, которая будет считаться центральным узлом графа. Для этого для каждой вершины V рассчитывается два параметра: N и M. N - количество вершин, которые могут быть достигнуты при движении из вершины V, M - количетво вершин, из которых можно достичь вершины V. Центральной вершиной будем считать такую вершину V, для которой параметр N+M будет максимальным. Центральной вершине присваивается ARR = 1. С данной вершины начинаем рекурсивно присваивать ARR другим вершинам
2) Рекурсия R: для каждой вершины V находим все вершины Pi, для которых в графе есть ребро Pi -> V или V -> Pi. Каждое ребро может быть рассмотрено в ходе рекурсии только один раз. В противном случае рекурсия будет вечной в случае появления циклов в графе. В том случае, если ребро имеет параметр «активация», то вершине Pi присваивается временный ARRtemp = 1. Если ребро имеет параметр «ингибирование», то вершине Pi присваивается временный ARRtemp = -1. В том случае, если при обходе графа вершина Pi ранее не встречалась, то присваиваем вершине Pi ARR = ARRtemp. В том случае, если при обходе графа вершина Pi ранее встречалась и присвоенный ранее ARR равен ARRtemp, то присваиваем вершине Pi ARR = ARRtemp. В том случае, если при обходе графа вершина Pi ранее встречалась и присвоенный ранее ARR не равен ARRtemp, то присваиваем вершине Pi ARR по правилу разрешения конфликтов. Правило разрешения конфликтов: в случае, если при обходе графа встречается ген, у которого уже указан ARR, но, согласно вышеизложенным правилам, данному гену требуется присвоить ARR, отличный от указанного, «конфликты» разрешаются по следующим правилам: 1) если знаки двух коэффициентов ARR различны, то результирующий ARR = 0; 2) если ARR отличаются на 0,5 и один из них положительный, то результирующий ARR = 0.5; 3) если ARR отличаются на 0,5 и один из них отрицательный, то результирующий ARR = -0.5. Затем для каждой вершины Pi, для которой модуль |ARR| равен 1, вызывается рекурсия R.
3) В результате алгоритм присвоит ARR вершинам графа. Данные коэффициенты могут быть использованы для расчета уровня активации молекулярных путей по вышеуказанным формулам.
Таким образом, генным продуктам, входящим в базу молекулярных путей, приписываются значения коэффициента ARR, отражающего функциональную значимость генного продукта в данном молекулярном пути.
При создании базы молекулярных путей, могут использоваться как опубликованные каталоги молекулярных путей, так и заданные пользователем самостоятельно. К опубликованным каталогам молекулярных путей относятся коллекции данных BioCarta, KEGG, NCI, Reactome и Pathway Central (Buzdin et al., 2017). В них собрана информация о 3125 молекулярных путях, совместно покрывающих около 11 000 белоккодирующих генов человека. Для использования в системе Oncobox, база данных для каждого молекулярного пути должна содержать следующую информацию:
1) Уникальные идентификаторы для всех генов, продукты которых включенных в молекулярные пути,
2) Роль каждого соответствующего генного продукта в функционировании конкретного молекулярного пути: роль активатора, репрессора, нейтральная роль, либо роли промежуточного активатора либо репрессора.
Базовый алгоритм анализа активации молекулярных путей основан на принятии следующих основных принципов.
Во-первых, граф молекулярных взаимодействий в каждом пути полагают в виде двух параллельных цепочек событий, где одна приводит к активации, а другая - к ингибированию молекулярного пути.
Во-вторых, уровень экспрессии каждого из генных продуктов - участников пути в состоянии низкой активности пути полагают меньшим, чем в состоянии высокой активности пути. Это положение принимается на основе данных о том, что в состоянии низкой активности пути было показано глубоконенасыщенное по концентрации состояние каждого из белков-переносчиков сигнала (Kuzmina and Borisov 2011; Aliper et al. 2017).
Важно отметить, что хотя базовый алгоритм включает понятие «экспрессии» гена (то есть при обычном толковании это уровни мРНК и белка), к нему могут сводиться также и другие измеряемые молекулярные характеристики:
- экспрессия микроРНК (влияет на экспрессию генов через ингибирование мРНК-мишеней),
- связывание транскрипционных факторов (регулирует экспрессию генов на уровне транскрипции),
- мутации геномной ДНК (влияют на экспрессию генов дикого типа путем появления мутантных аллелей). Авторами используется термин "приведенная экспрессия гена", охватывающий расчет экспрессии гена по вышеуказанным типам молекулярных данных.
Система Oncobox рассматривает все генные продукты-участники молекулярных путей в каждом отдельном пути как имеющие потенциально равные возможности вызывать активацию или ингибирование этого пути. Для анализа уровня активации молекулярных путей, в системе Oncobox используется следующая основная формула:
PALp = ∑n NIInp ⋅ ARRnp ⋅ ln CNRn / ∑n |ARRn|,
где PALp (Pathway Activation Level p) - это уровень активации молекулярного пути p; CNRn (case-to-normal ratio) - это отношение приведенных уровней экспрессии гена, кодирующего белок n, в исследуемом образце к норме (среднему значению у контрольной группы); ln - натуральный логарифм; NIInp - показатель принадлежности генного продукта n к пути p, принимает значения, равные единице для генных продуктов, входящих в путь и равные нулю для генных продуктов, не входящих в путь; дискретная величина ARRnp (activator/repressor role) определяется для гена n в пути p следующим образом и помещается в базу молекулярных путей:

Важным отличием базового алгоритма от ранее опубликованных (см. например Buzdin et al., Front Genet 2014) является то, что в системе Oncobox уровень активации молекулярного пути нормализуется на число генов-участников соответствующего пути с известной функциональной ролью, что отражается параметром |ARRn|.
В зависимости от типа получаемых молекулярных данных, в составе базового алгоритма по-разному рассчитывается параметр ln CNRn, то есть логарифм отношения приведенной экспрессии гена n в исследуемом образце к приведенной экспрессии гена n в контрольном образце. Ниже приведены варианты расчета параметра ln CNRn.
(1) - ln CNRn для данных по мутациям в геномной ДНК.
В каждом образце, для анализируемого гена n рассчитывается параметр MR (mutation rate):
MRn= 1000 ⋅ ∑ Nmut(n) / Lcds(n),
где Nmut(n) - это число выявленных мутаций в белоккодирующей части гена n в исследуемом образце; Lcds(n) - это длина белоккодирующей части гена n в парах нуклеотидов.
В свою очередь, параметр ln CNRn рассчитывается по формуле:
ln CNRn = ln CNR(MRn),
где CNR(MRn) - это отношение параметра MR в исследуемом образце к среднему значению MR в контрольной группе, для гена n.
(2) - ln CNRn для данных по сайтам связывания транскрипционных факторов.
В данном приложении, для каждого гена определяется консенсусная точка начала транскрипции. Для каждой точки начала транскрипции, определяется окрестность, составляющая по умолчанию участок от 5 т.п.н. выше точки начала транскрипции до 5 т.п.н. ниже точки транскрипции для соответствующего гена n. В данной окрестности, ведется подсчет картированных сайтов связывания транскрипционных факторов. Затем для каждого гена производится расчет параметра GRES (Gene Record Enrichment Score):
GRESn = m ⋅ TESn / ∑mi=1 TESi ,
где GRESn - это значение GRES для гена n; m - общее число исследуемых генов для данного образца; TESn - это количество картированных сайтов связывания транскрипционных факторов в окрестностях гена n; i - индекс, соответствующий идентификатору гена; сумма TESi по числу генов m - это общее число картированных сайтов связывания транскрипционных факторов в окрестностях всех исследуемых генов. Для каждого гена, значения GRES позволяют измерять уровень обогащения сайтами связывания транскрипционных факторов. Например, GRES =1 обозначает средний уровень обогащенности среди всех генов; GRES >1 обозначает уровень обогащения выше среднего по всем генам; GRES <1, напротив, обозначает то, что ген обеднен сайтами связывания транскрипционных факторов относительно всех генов.
Наконец, параметр ln CNRn рассчитывается по формуле:
ln CNRn = ln CNR(GRESn),
где CNR(GRESn) - это отношение параметра GRES в исследуемом образце к среднему значению GRES в контрольной группе, для гена n.
(3)- ln CNRn для данных по экспрессии мРНК
Важной особенностью системы Oncobox является то, что перед проведением расчетов активации молекулярных путей, для профилей мРНК проводится оригинальная совместная нормализация исследуемых образцов с группами соответствующих нормальных образцов.
Информация, содержащаяся в базах данных профилей генной экспрессии, представляют собой результаты исследований, полученных разными методами, включая микрочиповую гибридизацию мРНК, высокопроизводительное секвенирование и т.д. Каждый из этих методов реализован с использованием различных экспериментальных платформ от разных производителей. Находящиеся в открытом доступе такие хранилища данных содержат результаты для более чем 2 млн образцов, полученные в более чем 70 тыс. экспериментов (Cancer Genome Atlas Research Network 2008; https://www.ncbi.nlm.nih.gov/geo/).
Результаты количественного профилирования экспрессии генов, как правило, являются несравнимыми между собой (Demetrashvili et al. 2010). Для достижения удовлетворительного уровня однородности сравниваемых экспрессионных данных, в рамках системы Oncobox используется оригинальный метод гармонизации профилей генной экспрессии Shambhala, подходящий для унификации результатов, полученных как на одинаковых, так и на разных экспериментальных платформах.
Среди ранее опубликованных, могут быть отмечены методы гармонизации DWD (distance-weighted discrimination) (Huang et al. 2012), XPN (cross-platform normalization) (Shabalin et al. 2008) и PLIDA (platform-independent latent Dirichlet allocation) (Deshwar and Morris 2014), которые осуществляют глубокую реструктуризацию профилей генной экспрессии. Как правило, методы гармонизации экспрессионных профилей предусматривают поиск сходно экспрессирующихся на обеих платформах кластеров генов и белков, а затем производят пошаговое сближение экспрессионных профилей, полученных на двух разных экспериментальных платформах, в пределах каждого кластера. Тем не менее, все ранее опубликованные методы рассчитаны на гармонизацию максимально только двух наборов профилей экспрессии, полученных на разных экспериментальных платформах, причем число этих профилей в каждом из наборов, как правило, ограничено пороговым значением в сотню образцов. Это не позволяет применять данную группу методов для масштабного анализа массивов экспрессионных данных, включающих сотни и тысячи образцов.
В рамках системы Oncobox для этой цели используется разработанный нами оригинальный метод Shambhala, позволяющий проводить гармонизацию результатов профилирования генной экспрессии, полученных как на одинаковых, так и на разных платформах, который приводит их к универсальному сравниваемому виду. Метод Shambhala позволяет проводить гармонизацию для любого количества сравниваемых образцов, полученных на любом количестве экспериментальных платформ.
Метод Shambhala использует кластеризацию генов и образов с помощью стохастических генетических алгоритмов, а также кусочно-линейное итеративное сближение профилей генной экспрессии. Алгоритм Shambhala включает следующие особенности (Фиг. 2):
1. Осуществляется приведение (конверсия) гармонизируемого набора профилей (P1) к распределению уровней экспрессии, аналогичному эталонному (дефинитивному) набор профилей (Q). В качестве эталонного (дефинитивного) набора данных использован набор из ста экспрессионных профилей проекта Genotype Tissue Expression (GTEx) (GTEx Consortium 2013). Набор экспрессионных профилей GTEx (GSE45878) получен с помощью микрочиповой гибридизации мРНК на установке Affymetrix Human Gene 1.1 ST (GPL16977). В процессе конверсии, набор данных P1 итеративно приближается к набору профилей Q, который остается постоянным. Результатом такой конверсии будет являться набор экспрессионных профилей P2, который представляет из себя набор профилей P1, аналогичный тому, как если бы он был профилирован на той же платформе, что и эталонный (дефинитивный) набор профилей Q.
2. Для обеспечения большей устойчивости (меньших выбросов, обусловленных стохастической природой алгоритма кластеризации генов, входящего в алгоритм итеративной гармонизации), используется косинусоидальная метрика близости наборов вместо используемой в других методах (например, в методе XPN, Shabalin et al. 2008) евклидовой барицентрической (Krishna, 1999; Hornik, 2012).
3. Для обеспечения устойчивой конверсии в форму эталонного набора профилей Q всех образцов, депонированных в базах экспрессионных данных, каждый из конвертируемых профилей (i = 1,…N), по одному подвергается конверсии в составе вышеупомянутого набора профилей P1. Для формирования набора P1, каждый из экспрессионных профилей i (i = 1,…N) сначала квантильно нормализуется (Bolstad et al. 2003b) c заданным промежуточным (вспомогательным) набором профилей (P) для обеспечения единообразного масштаба калибровки по значениям экспрессий перед конверсией в форму набора профилей Q. Наконец, получившийся в результате такой конверсии набор P2 содержит гармонизированный профиль генной экспрессии i (i = 1,…N), см. Фиг. 2.
После гармонизации методом Shambhala, производят прямое сравнение экспрессионных профилей. Активность молекулярных путей вычисляют согласно указанной выше основной формуле, где в качестве ln CNRn берется натуральный логарифм от отношения гармонизированного уровня экспрессии гена n в исследуемом образце к норме (среднему значению у контрольной группы).
(4) - ln CNRn для данных количественной протеомики
Для анализа активации молекулярных путей на уровне данных белковой экспрессии, в системе Oncobox на первом этапе для гармонизации профилей сравниваемых образцов и группы норм, используется метод Shambhala, аналогично предыдущему приложению.
После этого рассчитывается активность молекулярных путей, где в качестве ln CNRn берется натуральный логарифм от отношения гармонизированного уровня экспрессии белка n в исследуемом образце к норме (среднему значению у контрольной группы).
(5) - ln CNRn для данных профилей микроРНК
Применение данного метода расчетов основано на использовании базы данных генных продуктов - мишеней микро РНК. Для использования в системе Oncobox, база данных для каждой включенной микроРНК должна содержать следующую информацию:
1) уникальное название и/или идентификатор микроРНК,
2) список уникальных идентификаторов генов молекулярных мишеней данной микроРНК.
В системе Oncobox, учет влияния микроРНК на приведенный уровень генной экспрессии осуществляется исходя из положения об ингибировании молекулами микроРНК своих мРНК-мишеней. Поэтому возрастание уровня микроРНК приводит к снижению приведенного уровня экспрессии соответствующей мРНК-мишени, и наоборот. При этом каждый генный продукт может иметь несколько регуляторных микроРНК, а каждая микроРНК - несколько генов-мишеней.
При этом ln CNRn вычисляется по следующей формуле:
ln CNRn = - ∑j ln miCNRi ⋅ miIIi, n,
где n - анализируемый в настоящий момент генный продукт, j - вся совокупность анализируемых системой микроРНК, i - анализируемая в настоящий момент микроРНК. Булева переменная microRNA involvement index (miIIi,n) указывает, является ли анализируемый генный продукт n мишенью для микроРНК i. При этом miIIi,n принимает значение, равное 1, когда анализируемый генный продукт n является мишенью для микроРНК i, и значение, равное 0 - когда не является. Величина miCNRi - это отношение количественно измеряемого уровня экспрессии микроРНК i в исследуемом образце к таковому среднему значению в контрольной группе. Отрицательный коэффициент перед знаком суммы отражает ингибирующую роль микроРНК для соответствующего генного продукта - мишени.
Для оптимального проведения оценки клинической эффективности таргетных лекарственных средств предпочтительно совмещать анализ различных типов молекулярных данных, полученных из патологической ткани пациента. Одной из возможных комбинаций является получение данных по изменению экспрессии белков (в молекулярных путях, а также белков-мишеней) и данных по накопленным в ткани мутациям. Эти два типа данных дополняют друг от друга и являются предпочтительными для проведения способа оценки клинической эффективности таргетных лекарственных средств, при условии достаточного качества этих данных. Остальные типы данных (связывание транскрипционных факторов, уровни микроРНК и мРНК) в основном влияют на уровень экспрессии белков в молекулярных путях, а также экспрессии белков-мишеней и действуют опосредованно. Данные этих типов могут быть использованы при осуществлении способа оценки клинической эффективности таргетных лекарственных средств в случаях, когда два предпочтительных типа данных недоступны или недостаточного качества.
Для получения информации из контрольных образцов здоровых субъектов можно использовать публично доступные данные, известные для конкретной ткани (например, данные экспрессии), так и, предпочтительно, экспериментальные данные, полученные из контрольных образцов здоровых субъектов на том же оборудовании, что и данные из исследуемого образца пациента. В последнем случае их можно получать параллельно с анализом образцов патологической ткани конкретного пациента. Для повышения точности оценки при выборе контрольных образцов желательно использовать образцы здоровых субъектов, имеющих как можно более схожие характеристики с пациентом, например, такие характеристики как пол, возраст и т.д. Минимальным является использование одного контрольного образца для одного образца пациента. Для повышения точности оценки рекомендуется использование от трех до двадцати контрольных образцов, что позволяет эффективно усреднять возможные отклонения, существующие в индивидуальных данных. Под усреднением следует понимать использование среднего арифметического значения усредняемых величин. В некоторых вариантах изобретения для усреднения используют среднее геометрическое значение усредняемых величин.
В предпочтительных вариантах осуществления изобретения необходимо получение полногеномных данных из образца пациента и контрольного образца. Однако осуществление изобретения возможно и с данными меньшего покрытия. Достаточными можно признать данные, оценивающие заданные параметры (уровни экспрессии микроРНК, белка или мРНК, профиль связывания транскрипционных факторов) для не менее 80% всех генных продуктов, входящих в состав отобранных и помещенных в базу молекулярных путей, в состав которых входят молекулярные мишени исследуемых препаратов. При этом крайне желательно получение данных для всех генных продуктов, которые являются известными молекулярными мишенями исследуемых препаратов. Минимально необходимый набор количественно охарактеризованных генных продуктов зависит (i) от количества и состава списка исследуемых препаратов и (ii) от набора молекулярных путей, которые добавлены пользователем технологии Онкобокс в референсную базу данных.
Для каждого таргетного лекарственного средства получают данные по меньшей мере от одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента. Под образцом ткани, не имеющим пролиферативного фенотипа, следует понимать образец ткани, взятый либо от "здорового" субъекта, не имеющего такого же онкологического заболевания, как и рассматриваемый пациент, либо взятый от рассматриваемого пациента, но из места, не пораженного онкологическим заболеванием.
Для осуществления изобретения возможно использование геномных данных неоптимального качества. Например, для данных экспрессии мРНК и микроРНК могут быть использованы любые типы входящих экспрессионных данных, способные обеспечить однозначное определение уровня экспрессии каждого генного продукта, который необходимо проанализировать, а также детекцию как минимум 1000-кратных различий в уровне экспрессии между отдельными транскриптами. Для мутационных профилей геномной ДНК могут быть использованы любые типы входящих данных геномного или экзомного секвенирования, полностью покрывающие кодирующие области генов, которые необходимо проанализировать, со средним по крайней мере 100-кратным уровнем покрытия. Для сайтов связывания транскрипционных факторов: количество анализируемых картированных сайтов связывания должно быть не менее, чем 10-кратное количество генов, которые необходимо проанализировать.
Расчет сбалансированного индекса эффективности для таргетных противоопухолевых препаратов.
К таргетным фармпрепаратам (лекарственным препаратам) относят препараты с известными молекулярными мишенями. В описании данного изобретения термин "таргетный препарат" ограничен препаратами определенных 16ти классов, или типов, приведенных в Таблице 1. Эти классы покрывают основные известные на сегодняшний день таргетные препараты, использующиеся в клинике. Препаратов под номерами 8, 9, 10, 14, 15 в Таблице 1 представляют собой препараты на основе иммуноглобулинов (антител), в то время как препараты других типов в Таблице 1 - это низкомолекулярные химические соединения (малые молекулы).
При формировании базы молекулярных мишеней для каждого таргетного лекарственного средства, в качестве источника может быть использована информация производителей фармпрепарата, а также научные публикации в специализированных журналах фармакологического, биохимического или биомедицинского профиля. Для использования в системе Oncobox, база данных для каждого включенного препарата должна содержать следующую информацию:
1. уникальное название и/или идентификатор препарата,
2. список уникальных идентификаторов генов - молекулярных мишеней данного препарата,
3. тип препарата по механизму действия (согласно Таблице 1).
Оценка системы Oncobox основывается на моделировании способности препаратов блокировать патологические изменения в молекулярных путях и одновременно блокировать генные продукты с патологически повышенными уровнями приведенной экспрессии. В отличие от известных аналогов, платформа Oncobox в качестве меры эффективности таргетных препаратов использует оригинальный параметр Balanced Efficiency Score (BES) для каждого препарата. При этом при расчете BES одновременно используются данные по активности молекулярных путей в исследуемом образце и данные по приведенной экспрессии генных продуктов - мишеней конкретного препарата. Значение BES вычисляется согласно формуле:
BESd = a⋅DESMPd + b⋅DESTGd,
где d - анализируемый таргетный препарат; a и b - весовые коэффициенты, варьирующие от -1 до 1.5 в зависимости от типа таргетного препарата d; DESMPd (Drug Efficiency Score for Molecular Pathways) - индекс активности препарата d, рассчитанный исходя из уровней активности молекулярных путей, содержащих его молекулярные мишени; DESTGd (Drug Efficiency Score for Target Genes) - индекс активности таргетного препарата d, рассчитанный исходя из уровней экспрессии индивидуальных генных продуктов - его молекулярных мишеней.
- Для вычисления значения DESMP используется следующая формула:
DESMPd = ∑t DTId, t⋅∑p PALp⋅AMCFp⋅NIIt,p,
где d – уникальный идентификатор таргетного препарата; t - уникальный идентификатор генного продукта-мишени препарата d; p - уникальный идентификатор сигнального пути; PALp - уровень активации молекулярного пути p; дискретная величина AMCF (activation-to-mitosis conversion factor) определяется так:
AMCF=1, когда активация молекулярного пути способствует выживанию, росту и делению клетки;
AMCF=0, когда нет данных относительно того, способствует ли активация молекулярного пути выживанию, росту и делению клетки, или когда такие данные, имеющиеся в распоряжении исследователя, противоречивы;
AMCF= -1, когда активация молекулярного пути препятствует выживанию, росту и делению клетки.
Дискретная величина DTI (drug-target index) определяется так:

Дискретная величина NII (node involvement index) определяется так:

- Для вычисления значения DESTG используется следующая формула:
DESTGd = ∑t DTId, ⋅∑p ln CNRt⋅ARRt, p⋅AMCFp⋅NIIt,p,
где d - уникальный идентификатор таргетного препарата; t - уникальный идентификатор генного продукта-мишени препарата d; p - уникальный идентификатор сигнального пути; CNRn (case-to-normal ratio) - отношение приведенных уровней экспрессии гена, кодирующего белок t, в исследуемом образце к норме (среднему значению у контрольной группы); ln - натуральный логарифм; определения DTId, t, AMCFp и NII аналогичны приведенным выше; дискретная величина ARRt, p (activator/repressor role) определяется для генного продукта t в пути p следующим образом и помещается в базу молекулярных путей:

При расчете значения сбалансированной эффективности BES для препарата d, используются весовые коэффициенты a и b, различающиеся в зависимости от природы препарата. Значения коэффициентов приведены в Таблице 1.
Для низкомолекулярных ингибиторов тирозинкиназ (нибов), оба весовых коэффициента равны 0.5, что отражает равную значимость активации таргетных молекулярных путей и уровня экспрессии таргетных генов в исследуемом образце патологической ткани. Это связано со способностью нибов блокировать свои молекулярные мишени и тем самым ингибировать их активность, а также тем самым модулировать прохождение сигнала по связанным с ними молекулярным путям. Для гормонов, оба весовых коэффициента принимают значение -0.5, это связано с тем, что они активируют, а не ингибируют свои молекулярные мишени и оказывают соответствующее воздействие на свои таргетные молекулярные пути. Для антигормонов, коэффициенты вновь принимают значение 0.5, что связано с ингибирующим механизмом их воздействия на свои молекулярные мишени, на продукцию гормонов или их молекулярную сигнализацию, а также на соответствующие таргетные молекулярные пути. Для ретиноидов, оба коэффициента равны -0.5, поскольку этот тип препаратов связывается с рецепторами ретиноевой кислоты и активирует ряд зависимых от них молекулярных путей. Для рапалогов (аналогов рапамицина), оба коэффициента равны 0.5, поскольку они обладают ингибирующим механизмом воздействия при связывании со своими молекулярными мишенями, а также на сигнализацию соответствующих молекулярных путей. Для мибов (ингибиторов протеасомы), оба коэффициента равны 0.5, поскольку эти препараты обладают ингибирующим механизмом воздействия при связывании со своими молекулярными мишенями, а также на сигнализацию молекулярных путей, включающих активность протеасомы. Для блокаторов VEGF, a=0 и b=1, поскольку данный класс препаратов непосредственно блокирует молекулы VEGF в кровотоке, не связываясь с мишенями внутри или на поверхности клетки и, таким образом, не оказывая прямого влияния на внутриклеточную сигнализацию. Для моноклональных антител, связывающихся с молекулярными мишенями на поверхности клетки (мабов), a=0 и b=1, поскольку основной механизм их действия заключается в активации иммунного цитотоксического ответа на клетки, содержащие связанные молекулы мабов на своей поверхности, и не включает модуляцию прохождения сигнала по молекулярным путям. Препараты киллермабы состоят из антител, связывающихся с молекулярными мишенями на поверхности клетки, связанными с цитотоксическими агентами. Связываясь с поверхностью клетки, киллермабы убивают ее, тем самым обеспечивая терапевтический эффект, не связанный с активацией молекулярных путей внутри клетки. Для них a=0 и b=1.5; в данном случае повышенное значение коэффициента b отражает собственную высокую цитотоксическую активность препаратов. Для препаратов-блокаторов de novo полимеризации тубулина, a=0 и b=1; это отражает неопределенную функцию большинства таргетируемых путей для выживания и пролиферации клетки, а также непосредственное ингибирующее воздействие данного класса препаратов при связывании с со своими молекулярными мишенями. Аналогичные коэффициенты заданы и для препаратов-ингибиторов гистондеацетилаз, по тем же причинам. В случае препаратов ДНК-алкилирующих агентов, a=0 и b= -1, что отражает неопределенную функцию большинства таргетируемых путей для выживания и пролиферации клетки, а также непосредственное ингибирование на эффект от препаратов данного класса от белков-участников систем репарации, таргетирующих алкилированную ДНК (отражено коэффициентом b= -1). Для иммунотерапевтических препаратов, оба коэффициента равны 0.5, в силу зависимости их воздействия от наличия как их непосредственных молекулярных мишеней, так и от активации молекулярных путей, связанных с лимфоцитарной инфильтрацией опухоли. Аналогично, препараты - блокаторы поли-ADP рибозы ингибируют репарацию ДНК и зависят как от наличия непосредственной молекулярной мишени, так и от активности таргетируемых молекулярных путей. Это отражено тем, что оба коэффициента приравниваются к 0.5.
Таблица 1. Значения весовых коэффициентов a и b для 16 классов таргетных противоопухолевых препаратов.
b = 0.5
b = 0.5
b = -0.5
b = 0.5
b = 0.5
b = 0.5
b = 0.5
b = 1
b = 1
b = 1.5
b = 1
b = 1
b = -1
b = 0.5
b = 0.5
b = 0.5
Система Oncobox позволяет количественно оценивать эффективность противоопухолевых препаратов, относящихся к 16 различным классам (Таблица 1). Разделение препаратов на классы осуществляется в соответствии с их известным механизмом действия и молекулярной специфичностью. Затем вычисляется сбалансированная величина Balanced Efficiency Score (BES), по-разному для разных классов противоопухолевых препаратов (Таблица 1). Затем на основании значений BES формируется персонализированный рейтинг таргетных противораковых препаратов для исследованного биообразца, например, взятого от онкобольного пациента, при этом к использованию могут быть рекомендованы препараты, обладающие положительным значением величины BES (BES >0).
Таким образом, в настоящем изобретении предложен способ оценки клинической эффективности таргетных лекарственных средств для лечения пациента с пролиферативным или онкологическим заболеванием, выбранных из группы таргетных лекарственных средств, состоящий по меньшей мере из следующих этапов: (а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства, выбранного из указанной группы; (б) получают образец ткани пациента, имеющий пролиферативный фенотип; (в) получают данные по меньшей мере одного типа из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК; (г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в); (д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i) - (v), получаемых на стадии (д), совпадает с типом данных, соответственно (i) - (v), получаемых на стадии (в); (е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д); (ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i) - (v), используя данные, полученные на стадиях (в)-(е);(з) определяют клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
Количественный показатель эффективности лекарственного средства (BES) для каждого из типов данных (i) - (v) определяют при помощи сложения двух частей (DESMPd и DESTGd), которые определяют из данных по активности молекулярных путей в исследуемом образце и данных по приведенной экспрессии генных продуктов -молекулярных мишеней препарата d), с учетом весовых коэффициентов (a и b), зависящих от типа лекарственного средства, и раскрытых в Таблице 1. При вычислении обеих частей (DESMPd и DESTGd) данные, полученные из образца пациента, нормируются на соответствующие данные того же типа, полученные из по меньшей мере одного контрольного образца.
В предпочтительных вариантах изобретения количественный показатель эффективности лекарственного средства d для каждого из типов данных (i) - (v) определяют при помощи формулы:
BESd = a ∙ ( ∑t DTId, t ∙ ∑p PALp ∙AMCFp ∙ NIIt,p) + b ∙(∑t DTId, t ∙ ∑p ln CNRt ∙ ARRt, p ∙ AMCFp ∙ NIIt,p).
При наличии нескольких типов данных от пациента определяют несколько количественных показателей эффективности для каждого из типов данных (i) – (v), после чего для оценки клинической эффективности таргетного лекарственного средства используют усредненный показатель эффективности.
Способ оценки клинической эффективности таргетных лекарственных средств может быть осуществлен с использованием вычислительного устройства, содержащего такие компоненты, как: один или более процессоров, по меньшей мере одну память, а также, предпочтительно, интерфейсы ввода/вывода, средство ввода/вывода, средство сетевого взаимодействия и другие компоненты. Процессор устройства выполняет основные вычислительные операции, необходимые для функционирования модулей исполняющего команды устройства. Процессор исполняет необходимые машиночитаемые команды, содержащиеся в оперативной памяти. Под памятью следует понимать любой накопитель информации, способный хранить необходимую программную логику, обеспечивающую требуемый функционал. Средство хранения данных может выполняться в виде HDD, SSD дисков, рейд массива, флэш-памяти, оптических накопителей информации (CD, DVD, MD, Blue-Ray дисков) и т.п. Выбор интерфейсов зависит от конкретного исполнения вычислительного устройства, которое может представлять собой персональный компьютер, мейнфрейм, серверный кластер, тонкий клиент, смартфон, кассовый аппарат и т.п. В качестве средств ввода/вывода данных может использоваться: клавиатура, джойстик, дисплей (сенсорный дисплей), проектор, тачпад, манипулятор мышь, трекбол, световое перо, динамики, микрофон и т.п.
Приложения системы Oncobox для анализа уровней активации молекулярных путей, для создания рейтингов эффективности препаратов, для поиска новых биомаркеров, для поиска молекулярных мишеней и перепозиционирования существующих фармацевтических препаратов проиллюстрированы ниже примерами.
Нижеследующие примеры работы системы приведены в целях раскрытия характеристик настоящего изобретения и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения.
Пример 1. Расчет индекса активации молекулярных путей исходя из данных по концентрации эпигенетических маркеров.
Полногеномные профили связывания 225 белков-транскрипционных факторов (ТФ) человека, полученные разными лабораториями в ходе экспериментов по иммунопреципитации хроматина (ChIP-seq) для клеточной линии К562 (эритролейкемия, иммортализованная клеточная линия), были загружены из базы данных ENCODE (https://www.encodeproject.org/chip-seq/transcription_factor/). Профили представляли собой нормированную на контроль интенсивность связывания ТФ в формате bedGraph (https://genome.ucsc.edu/goldenpath/help/bedgraph.html). В соответствии с протоколом обработки данных ChIP-seq референсная сборка человеческого генома hg19 была проиндексирована алгоритмом Бурроу-Уиллера с помощью программы BWA (https://www.encodeproject.org/pipelines/ENCPL220NBH/). Слияние fastq-файлов с сырыми данными, выравнивание на референсный геном и фильтрация проводились программами BWA, Samtools, Picard, Bedtools и Phantompeakqualtools (https://www.encodeproject.org) . Профили нормированного на контроль уровня связывания ТФ были получины программой Macs (https://www.encodeproject.org/pipelines/ENCPL138KID/). Эти профили были картированы на окрестности длиной 5 тысяч пар нуклеотидов для белок-кодирующих генов человека (координаты были загружены с USCS Browser, https://genome.ucsc.edu/cgi-bin/hgs, таблица RefGenes ). Для каждого гена были рассчитаны индексы GRES (Фиг. 3) и CNR(GRES) и далее для каждого молекулярного пути - индекс PAL.
Таким образом, с помощью системы Oncobox были найдены группы генов и молекулярные пути, активированные в опухолевой клеточной линии К562. Были охарактеризованы наиболее сильно активированные процессы: синтез белков, репликация и репарация ДНК, поддержание структуры ядра и хроматина, везикулярный транспорт и цитоскелет. В обоих случаях была обнаружена активация путей врожденной иммунной системы, что закономерно для клеточной линии миелоидного ряда.
Пример 2. Расчет рейтинга активности онкопрепаратов для индивидуальной опухоли исходя из активации молекулярных путей на основе данных по экспрессии мРНК.
Был произведен расчёт рейтинга потенциально эффективных противоопухолевых таргетных препаратов для пациента 72 лет с гистологически подтвержденной умеренно дифференцированной внутрипеченочной холангиокарциномой (Фиг. 4). Пациент был диагностирован в октябре 2015 года со следующими симптомами: умеренная потеря веса, боль в правом подреберье, потеря аппетита и астения, с показателем индекса Карнофского 70%. Результаты магнитно-резонансной томографии (МРТ) во время диагностики подтверждали диагноз. Опухоль не удалялась хирургическим путем из-за продвинутой стадии, нескольких внутрипеченочных образований и метастазов в легкие.
Сначала данный пациент получал лечение, считающееся лучшей клинической практикой: четыре курса химиотерапии (2 курса гемцитабина в сочетании с капецитабином и последующие 2 курса гемцитабина в сочетании с цисплатином) были проведены до мая 2016 года. Лечение было неэффективным, и опухоль увеличивалась в размерах согласно данным МРТ; в левой и правой долях появились дополнительные метастатические узлы с распространением на желчный проток и в желчный пузырь. Индекс шкалы Карнофского снизился до 60%. Пациент не отвечал на лечение, и для него был проведен расширенный молекулярный анализ опухоли с помощью системы Oncobox для определения альтернативных вариантов лечения.
Для этого сначала была выделена тотальная РНК из опухолевого образца, которая была использована для измерения уровня экспрессии 2163 генов на оборудовании CustomArray Inc. (США) с помощью метода микрочиповой гибридизации. В список 2163 генов входили гены из основных сигнальных путей человека, ассоциированных с онкопаталогиями, а также мишени всех таргетных противоопухолевых препаратов. В качестве образцов нормальной ткани, необходимых для расчётов вышеуказанных индексов, были использованы образцы печени без признаков патологических изменений, полученные от здоровых доноров. При помощи алгоритма Oncobox был составлен рейтинг таргетных препаратов согласно значениям индекса BES (Таблица 2).
Таблица 2. Рейтинг потенциально наиболее эффективных препаратов для пациента с холангиокарциномой согласно результатам платформы Oncobox.
Согласно результатам анализа, в мае 2016 года пациенту был назначен таргетный препарат Сорафениб, который является ингибитором тирозинкиназ. При МРТ-анализе в октябре 2016 года был обнаружен умеренный рост опухоли, что соответствует стабилизации болезни. Однако после лечения Сорафенибом пациент перестал жаловаться на боль в правом подреберье. МРТ, проведенная в январе 2017 г., выявила прогрессирование роста опухоли и дополнительные узлы в левом легком. Таким образом, время до прогрессирования составило около 6 месяцев. Кроме того, появились следующие побочные эффекты: покраснение, отек, боль на ладонях рук и подошвах ног. Поэтому было принято решение изменить лечение, для чего был выбран препарат Пазопаниб, другой тирозинкиназный ингибитор, рекомендованный на основе рейтинга, рассчитанного системой Oncobox. Лечение Пазопанибом началось в январе 2017 года. Контрольная МРТ в июле 2017 года выявила умеренный рост опухоли. При этом смена режима лечения привела к устранению побочных эффектов Сорафениба и общему улучшению качества жизни. По состоянию на октябрь 2017 года, пациент жив и физически активен, а индекс по шкале Карнофского составляет 100%.
Данный клинический случай свидетельствует о том, что персонализированное назначение тирозинкиназных ингибиторов при помощи системы Oncobox может быть эффективным с точки зрения общей выживаемости и качества жизни пациента при метастатической холангиокарциноме.
Пример 3. Расчет рейтинга активности онкопрепаратов для колоректального рака исходя из данных по мутациям геномной ДНК.
Рейтинг активности таргетных препаратов был составлен для 105 препаратов на основании полногеномных данных 1441 случаев колоректального рака при помощи платформы Oncobox. Данные были получены из базы данных COSMIC v76 (The Catalogue of Somatic Mutations In Cancer) (Forbes et al. 2008) и содержали информацию о 1 165 882 мутациях в составе 19897 генов.
Верхние позиции полученного рейтинга лекарственных препаратов представлены в Таблице 3 (указаны 9 препаратов с максимальными значениями Balanced Efficiency Score (BES)).
Таблица 3. Список препаратов, получивших наиболее высокие позиции рейтинга потенциальной эффективности исходя из значений MDS.
Пример 4. Моделирование новых молекулярных мишеней для опухолевых препаратов с помощью данных по мутациям ДНК.
Для оценки мутационного профиля первичных злокачественных опухолей печени были использованы данные полногеномного секвенирования из базы данных COSMIC v76 (The Catalogue of Somatic Mutations In Cancer) (Forbes et al. 2008), содержащие записи о 852964 мутациях 19491 генов 1654 опухолевых образцов. Для всех генов были определены нормализованные частоты мутаций (NMR). Далее были интегрированы в единую базу данные молекулярных путей, полученные из крупнейших международных баз данных: Reactome [doi: 10.1093/nar/gkt1102], NCI Pathway Interaction Database [doi: 10.1093/nar/gkn653], Kyoto Encyclopedia of Genes and Genomes [doi: 10.1093/nar/gks1239] и HumanCyc [(www.humancyc.org)]. Из 3125 путей в целях адекватности статистического анализа были отобраны молекулярные пути, содержащие более 10 генов. В результате дальнейшая работа велась с 1753 путями, содержащими 8755 генов, которые были рассмотрены в качестве потенциальных молекулярных мишеней для новых онкопрепаратов. Для всех молекулярных путей были определены величины PAL. В качестве спектра потенциальных таргетных препаратов были приняты 8755 условных препаратов, каждый из которых имел качестве мишени по одному гену из базы данных путей. Для всех генных продуктов - потенциальных мишеней были посчитаны значения индекса Balanced Efficiency Score с использованием платформы Онкобокс.
В результате были выявлены потенциальные гены-мишени для разработки новых терапевтических агентов. Примерами могут служить следующие потенциальные гены-мишени (Таблица 4).
Таблица 4. Рейтинги по индексу Balanced Efficiency Score для генов - потенциальных мишеней таргетной терапии, рассчитанные для злокачественных опухолей печени с помощью системы Oncobox.
Пример 5. Перепозиционирование известных лекарственных препаратов для злокачественных опухолей на основе данных по мутациям геномной ДНК.
При расчете рейтинга Balanced Efficiency Score для 105 препаратов с помощью системы Онкобокс были использованы полногеномные данные из базы данных COSMIC v76 (The Catalogue of Somatic Mutations In Cancer) (Forbes et al. 2008), содержащие записи о 852964 мутациях 19491 генов 1654 образцов первичных злокачественных опухолей печени. Максимальный мутационный рейтинг (первые 10 позиций) получили препараты: Regorafenib, Idelalisib, Masitinib, Thalidomide, Sorafenib, Tivantinib, Nintedanib (BIBF 1120), Crizotinib, Foretinib, Flavopiridol (Alvociclib).
Из них одобрен к применению при данном виде онкозаболеваний Regorafenib, успешно прошли третью фазу клинических испытаний Tivantinib и Nintedanib (BIBF 1120), а для препарата Flavopiridol (Alvociclib) завершена вторая фаза клинических испытаний. Остальные препараты, набравшие максимальный рейтинг Balanced Efficiency Score в данный момент не используются при опухолях печени и при отсутствии клинических испытаний, проводимых ранее, могут быть рекомендованы для рассмотрения применения при данной нозологии. Клинический статус всех препаратов, отсортированный согласно рейтингу Balanced Efficiency Score, приведен на Фиг. 5.
Пример 6. Гармонизация профилей генной экспрессии, полученных на разных экспериментальных платформах, с помощью системы Онкобокс.
Экспрессионные профили для одних и тех же образцов мРНК человека были получены в рамках проекта SEQC с использованием разных экспериментальных платформ и опубликованы в публичных базах данных (Su at al, 2014, SEQC/MAQC-III Consortium, 2014). Были взяты транскрипционные профили для коммерчески доступного образца мРНК человека Stratagene Universal Human Reference RNA (UHRR Catalog #740000) с использованием экспериментальных платформ микрочиповой гибридизации и глубокого секвенирования: Illumina HiSeq 2000 (GPL11154), Illumina HumanHT-12 V4.0 expression beadchip (GPL10558), Affymetrix Human Gene 2.0 ST Array (GPL17930), Affymetrix GeneChip PrimeView Human Gene Expression Array (GPL16043).
Полученные экспрессионные профили исходно существенно различались распределением генов по различным уровням экспрессии (Фиг. 6, верхние панели). После применения метода Shambhala в рамках платформы Онкобокс, экспрессионные профили были приведены к универсальному виду (Фиг. 6, нижние панели).
Пример 7. Комбинирование мутационного и экспрессионного способа рейтингования таргетных препаратов для пациента с раком головы и шеи на платформе Онкобокс.
Для взрослого онкобольного с раком головы и шеи 4 стадии заболевания с помощью платформы Онкобокс осуществлялся выбор следующей линии химиотерапии. Для этого у пациента была взята биопсия опухолевой ткани, была профилирована экспрессия мРНК и проведено полноэкзомное секвенирование ДНК на оборудовании Illumina HiSeq 2000. В качестве образца нормальной ткани был использован образец миндалины, полученный от здорового пациента, для которого профиль генной экспрессии был получен на том же оборудовании.
По профилям экспрессии мРНК в опухолевом и нормальном образцах, для 128 таргетных препаратов были рассчитаны величины Balanced drug Efficiency Score (BES). В таблице 5 приведены препараты с наибольшим значением индекса BES по данным экспрессии мРНК.
Таблица 5. Рейтинг таргетных препаратов с наибольшим значением BES для пациента с раком головы и шеи, согласно результатам профилирования экспрессии мРНК.
В параллель, для того же пациента индекс BES был рассчитан альтернативным способом, исходя из данных полноэкзомного секвенирования образца опухолевой ткани. Всего у данного пациента были найдены соматические мутации в 13 генах. Рейтинг таргетных препаратов согласно значениям BES, подсчитанным по данным мутаций геномной ДНК, приведен в Таблице 6.
Таблица 6. Рейтинг таргетных препаратов с наибольшим значением BES для пациента с раком головы и шеи, согласно результатам профилирования мутаций геномной ДНК.
Затем был составлен список тех препаратов, которые одновременно имели максимальные значения BES по данным мутаций геномной ДНК и экспрессии мРНК в опухолевой ткани (Таблица 7). Выяснилось, что многие такие препараты завершили третью фазу клинических испытаний или рекомендованы FDA (США) для применения при раке головы и шеи. Это свидетельствует в пользу эффективности и надежности комбинирования рейтингов таргетных препаратов, рассчитанных на экспрессионных и мутационных данных. По итогам исследования, в качестве следующей линии терапии для пациента был рекомендован препарат цетуксимаб. Возможность комбинирования рейтингов таргетных препаратов по итогам исследований профилей мРНК и геномной ДНК является отличительной особенностью платформы Онкобокс.
Таблица 7. Медицинский статус таргетных препаратов для лечения опухолей головы и шеи, которые одновременно имеют максимальный рейтинг BES по результатам профилирования экспрессии мРНК и мутаций геномной ДНК в опухолевой ткани пациента с раком головы и шеи.
(статус клин. испытаний)
Пример 8. Сравнение экспрессионного способа рейтингования таргетных препаратов на платформе Онкобокс с ранее опубликованными подходами.
Ниже приведена оценка сбалансированных индексов эффективности таргетных препаратов, рассчитанных при помощи способа по настоящему изобретению (используя предложенный индекс BES) в сравнении с ранее описанным в заявке US20170193176A1 способом, использующим для расчета только значения активации молекулярных путей (индекс Drug Score, далее обозначенный как DS1a).
Расчёт индексов эффективности таргетных препаратов был произведен по транскриптомным профилям онкопациентов из публичной базы данных TCGA (The Cancer Genome Atlas), после чего было произведено сравнение индексов эффективности с клиническим статусом таргетных препаратов.
База данных The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) содержит мутационные и транскриптомные профили онкопациентов с разными нозологиями. С помощью системы Oncobox авторы рассчитали сбалансированные индексы эффективности таргетных препаратов (BES) для пациентов 11 нозологий (Таблица 8). Для этих же пациентов были также рассчитаны индексы эффективности таргетных препаратов DS1a согласно методу, ранее опубликованному в заявке US20170193176A1, а также в статье Artemov et al., 2015.
Таблица 8. Количество проанализированных транскриптомных профилей пациентов из базы данных TCGA по нозологиям.
На первом этапе были проанализированы таргетные препараты, которые попадают в верхнюю часть рейтинга по версии отдельно по коэффициенту BES и отдельно по коэффициенту DS1a. Для этого вышеуказанные коэффициенты были рассчитаны для каждого отдельного транскриптомного профиля. Всего были проанализированы коэффициенты для 128 таргетных препаратов. Затем были найдены препараты, которые попадают в топ 10% рейтинга (топ-13 препаратов) для большей части пациентов. Получившиеся списки сравнили между разными нозологиями при помощи коэффициента Жаккара. Полученный график по парному сравнению всех нозологий для DS1a приведен на Фиг. 7. Оказалось, что топ-13 препаратов по версии коэффициента эффективности DS1a является одинаковым для разных нозологий (при этом конкретные позиции в рейтинге могут не совпадать). Это говорит о том, что DS1a не подходит для персонализированного назначения таргетной терапии онкопациентам, т.к. в соответствии с этим рейтингом большей части пациентов предполагается назначить одинаковые препараты.
Аналогичный анализ был проведен для рейтинга таргетных препаратов BES. В этом случае, напротив, топ-13 препаратов был вариабелен между нозологиями (Фиг. 8). Действительно, эффективность таргетных препаратов чаще всего варьирует между разными нозологиями. Таким образом, рейтинг BES, с одной стороны, в большей степени отражает клинические данные, и, с другой стороны, подходит для персонализированного назначения терапии онкопациентам.
Затем авторы оценили степень того, насколько рейтинг таргетных препаратов, рассчитанный системой Oncobox, соотносится с клиническим статусом данных препаратов. Для этого была проанализирована база данных clinicaltrials.gov, которая содержит в себе информацию о большей части клинических испытаний, в которых изучалась эффективность таргетных препаратов. В качестве примера были рассмотрены стадии клинических испытаний всех 128 таргетных препаратов для рака шейки матки. Затем, в зависимости от того, на какой клинической стадии находился препарат, ему проставлялся коэффициент от 0 до 1: 1 - если препарат одобрен для рака шейки матки, 0.85 - завершена третья фаза клинических испытаний, 0.7 - третья фаза клинических испытаний проходит в данный момент, 0.4 - вторая фаза клинических испытаний, 0.3 - первая фаза, 0 - данные по клиническим испытаниям отсутствуют. Исходя из этих данных, для каждого отдельно взятого пациента можно рассчитать, насколько персонализированный рейтинг соотносится с клиническим статусом препарата. Если в верхней половине рейтинга преобладают препараты, которые прошли начальные фазы клинических испытаний, или даже рекомендованы для данной нозологии, и при этом в нижней половине рейтинга таких препаратов меньше, значит данному пациенту скорее будет назначен препарат, успешно прошедший клинические испытания.
Для того, чтобы количественно оценить данный эффект, авторы предлагают использовать формулу:
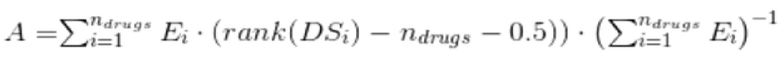 ,
,
где Ei - клинический коэффициент для препарата под номером i; DS - коэффициент эффективности данного таргетного препарата, например, BES или DS1a; ndrugs - количество таргетных препаратов (в нашем случае равно 128).
Полученный коэффициент А, названный нами коэффициентом Анубиса, даёт оценку того, насколько хорошо рассчитанные индексы эффективности препаратов соотносятся с их клиническим статусом. На Фиг. 9 приведен график зависимости рейтинга препарата по коэффициенту BES от клинического статуса препарата на примере одного пациента с раком шейки матки. Из графика видно, что препараты, которые находились на более поздних стадиях клинических испытаний находились скорее в верхней половине рейтинга. Коэффициент Анубиса, рассчитанный по вышеуказанной формуле, для данного пациента получился равным 14.83.
Авторы рассчитали коэффициент Анубиса для всех пациентов с раком шейки матки используя коэффициент эффективности препаратов BES и DS1a. Плотность распределения полученных значений коэффициента Анубиса приведена на Фиг. 10. Из графика видно, что в среднем коэффициенты Анубиса были выше в случае BES, чем в случае DS1a. Это говорит о том, что рейтинг препаратов по коэффициенту BES в большей степени соответствует клиническому статусу препаратов, чем для DS1a. Таким образом, коэффициент BES имеет явное преимущество перед DS1a, т.к. в верхней части рейтинга, построенного по BES, чаще содержатся препараты, успешно показавшие себя в клинических испытаниях.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ создания таргетной панели для исследования геномных регионов для выявления терапевтических биомаркеров ингибиторов иммунных контрольных точек (ИКТ) | 2023 |
|
RU2818360C1 |
| СПОСОБ ПОИСКА БЕЛКОВ-МИШЕНЕЙ, ЗАПУСКАЮЩИХ ПРОЦЕСС КАНЦЕРОГЕНЕЗА, В ОБРАЗЦАХ ТКАНЕЙ ИНДИВИДУАЛЬНОГО ПАЦИЕНТА, ДЛЯ ПОСЛЕДУЮЩЕЙ ПРОТИВООПУХОЛЕВОЙ ФАРМАКОТЕРАПИИ | 2013 |
|
RU2577107C2 |
| ПРОТИВООПУХОЛЕВЫЙ ИНДИВИДУАЛЬНЫЙ ПРОТЕОМ-ОСНОВАННЫЙ ТАРГЕТНЫЙ КЛЕТОЧНЫЙ ПРЕПАРАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ЭТОГО ПРЕПАРАТА ДЛЯ ТЕРАПИИ РАКА И ДРУГИХ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2012 |
|
RU2535972C2 |
| Метод разработки биомаркеров заболеваний и физиологически активных веществ на основе расширенной версии алгоритма iPANDA | 2018 |
|
RU2703534C1 |
| Способ дифференциальной диагностики рака яичников, кистозных образований яичника и рака тела матки | 2024 |
|
RU2836527C1 |
| Способ диагностики меланомы с помощью экзосомальных микроРНК | 2020 |
|
RU2769927C2 |
| Способ прогнозирования эволюции церебральных глиом | 2020 |
|
RU2740194C1 |
| СПОСОБ ДИАГНОСТИКИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2013 |
|
RU2547583C2 |
| СПОСОБ ДИАГНОСТИКИ МОЛЕКУЛЯРНОГО ФЕНОТИПА ПАЦИЕНТА, СТРАДАЮЩЕГО ЗАБОЛЕВАНИЕМ, СОПРОВОЖДАЮЩИМСЯ ХРОНИЧЕСКИМИ ВОСПАЛЕНИЯМИ | 2013 |
|
RU2672595C2 |
| ИНСЕРЦИЯ ТАРГЕТНОГО ГЕНА ДЛЯ УЛУЧШЕННОЙ КЛЕТОЧНОЙ ИММУНОТЕРАПИИ | 2017 |
|
RU2824204C2 |



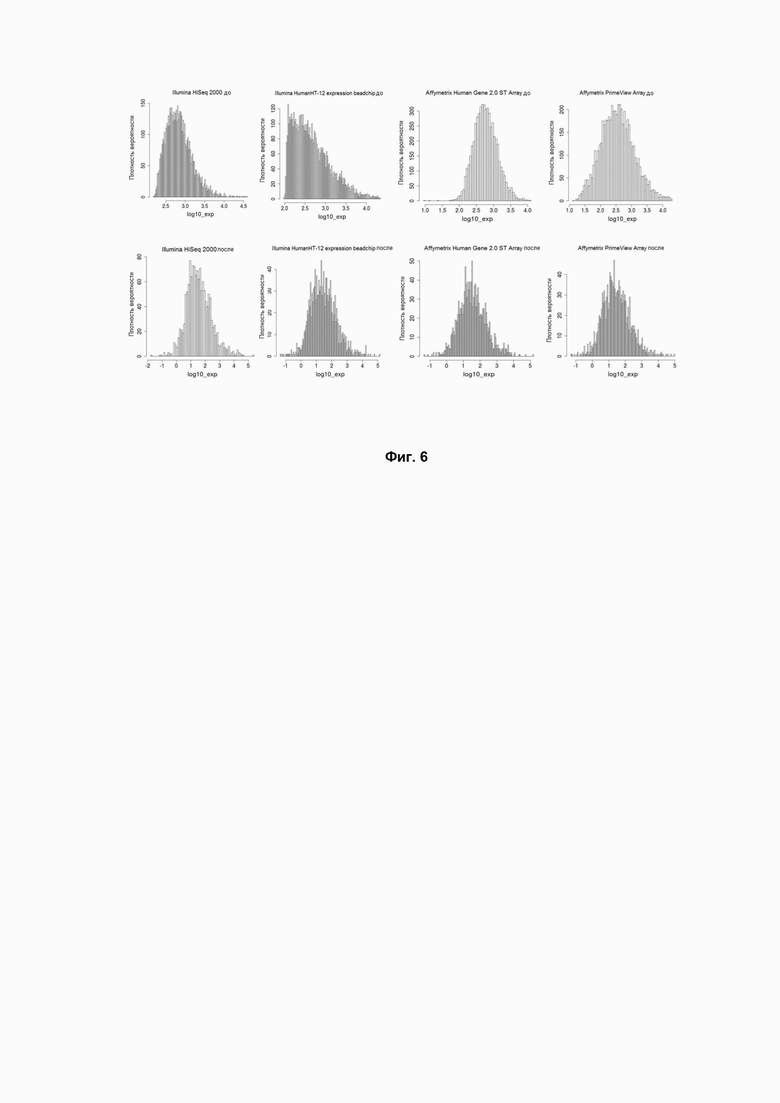
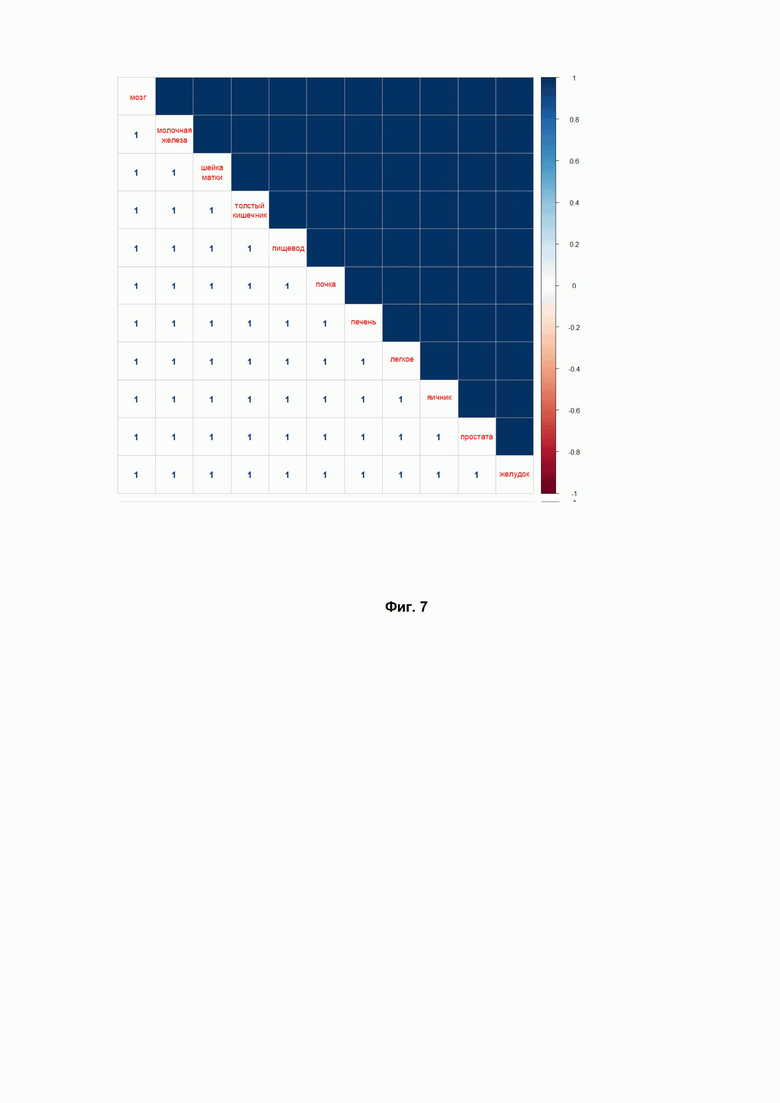
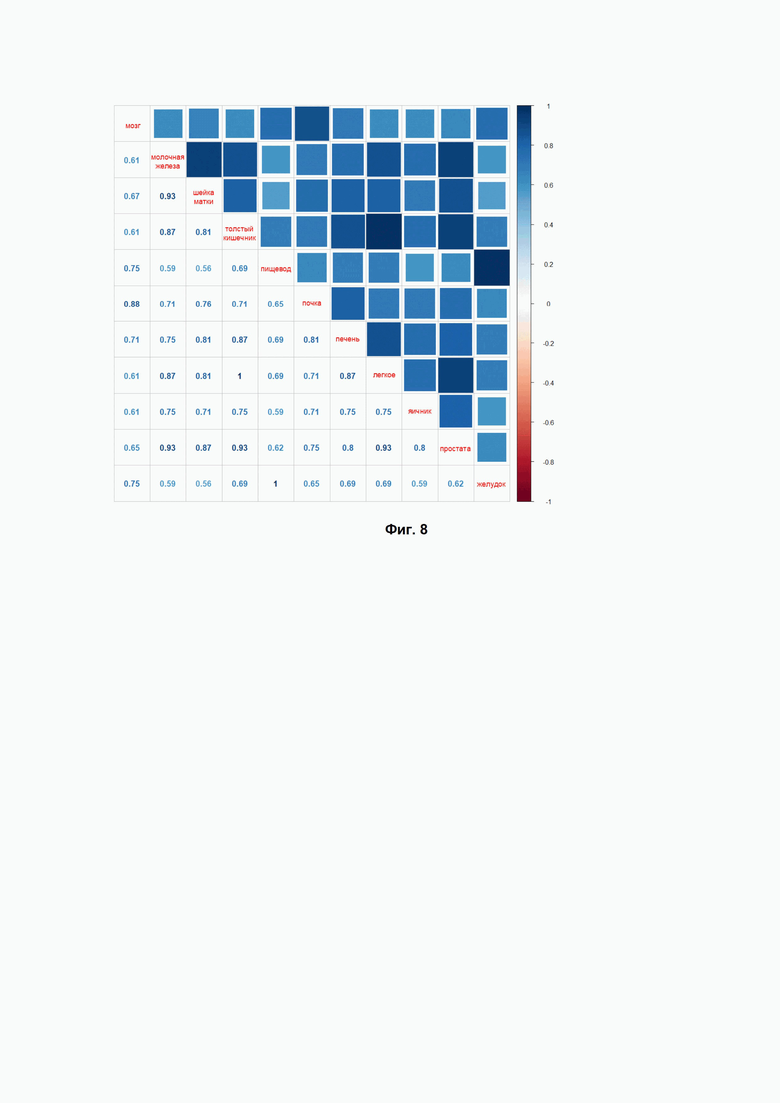


Предложенная группа изобретений относится к области медицины. Предложен способ оценки клинической эффективности таргетных лекарственных средств для лечения пациента с пролиферативным или онкологическим заболеванием, в котором получают данные по меньшей мере одного типа из образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК. Предложен способ лечения пациента с пролиферативным или онкологическим заболеванием и система для оценки клинической эффективности таргетных лекарственных средств, выбранных из группы таргетных лекарственных средств, для пациента с пролиферативным или онкологическим заболеванием ткани. Предложенная группа изобретений позволяет эффективно оценивать клиническую эффективность существующих таргетных лекарственных средств для индивидуального пациента с пролиферативным или онкологическим заболеванием. 3 н. и 3 з.п. ф-лы, 10 ил., 8 табл., 8 пр.
1. Способ оценки клинической эффективности таргетных лекарственных средств для лечения пациента с пролиферативным или онкологическим заболеванием, выбранных из группы таргетных лекарственных средств, состоящий по меньшей мере из следующих этапов:
(а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства;
(б) получают образец ткани пациента, имеющий пролиферативный фенотип;
(в) получают данные по меньшей мере одного типа из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК;
(г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в);
(д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i)–(v), получаемых на стадии (д), совпадает с типом данных, соответственно (i)–(v), получаемых на стадии (в);
(е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента, и тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д);
(ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i)–(v), используя только данные, полученные на стадиях (в)-(е);
(з) определяют клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
2. Способ по п. 1, в котором данные, полученные из по меньшей мере одного контрольного образца ткани, гармонизированы с данными, полученными на стадиях (в) и (д).
3. Способ по п. 2, в котором
(i) на стадии (в) получают данные по меньшей мере двух типов;
(ii) на стадии (ж) независимо рассчитывают количественный показатель эффективности по каждому типу данных; и
(iii) оценивают клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств, усредняя рассчитанные для каждого типа данных количественные показатели эффективности.
4. Способ по п. 3, в котором на стадии (в) получают полногеномные данные по экспрессии белков и полногеномные данные по мутациям в геномной ДНК.
5. Способ лечения пациента с пролиферативным или онкологическим заболеванием, состоящий по меньшей мере из следующих этапов:
(а) получают информацию об имеющихся таргетных лекарственных средствах и формируют группу таргетных лекарственных средств;
(б) оценивают клиническую эффективность таргетных лекарственных средств, выбранных из указанной группы таргетных лекарственных средств, согласно способу по п. 1;
(в) выбирают для лечения указанного пациента лекарственное средство, имеющее лучший, или один из лучших количественных показателей эффективности, определенных согласно способу по п. 1.
6. Система для оценки клинической эффективности таргетных лекарственных средств, выбранных из группы таргетных лекарственных средств, для пациента с пролиферативным или онкологическим заболеванием ткани, включающая:
- по меньшей мере один процессор;
- по меньшей мере одну память, которая содержит машиночитаемые инструкции, выполненные с возможностью осуществления по меньшей мере одним процессором компьютерно-реализуемого способа оценки клинической эффективности таргетных лекарственных средств, состоящего из по меньшей мере следующих этапов:
(а) получают информацию о молекулярных мишенях для каждого таргетного лекарственного средства, выбранного из указанной группы;
(в) получают данные по меньшей мере одного типа из образца ткани пациента, имеющей пролиферативный фенотип, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии тотальной мРНК, (ii) полногеномные данные по экспрессии белков, (iii) полногеномные данные по сайтам связывания транскрипционных факторов, (iv) полногеномные данные по мутациям в геномной ДНК, (v) полногеномные данные по экспрессии микроРНК;
(г) получают данные из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента; тип данных, получаемых из контрольного образца, совпадает с типом данных, полученных на стадии (в);
(д) получают данные по меньшей мере одного типа о молекулярных мишенях для каждого таргетного лекарственного средства из указанного образца, при этом тип данных выбирается из следующего списка: (i) данные по экспрессии мРНК молекулярных мишеней, (ii) данные по экспрессии молекулярных мишеней, (iii) данные по мутациям в генах молекулярных мишеней; (iv) данные по сайтам связывания транскрипционных факторов в генах молекулярных мишеней, (v) данные по экспрессии микроРНК, влияющих на экспрессию генов молекулярных мишеней, при этом каждый из типов данных (i)–(v), получаемых на стадии (д), совпадает с типом данных, соответственно (i)–(v), получаемых на стадии (в);
(е) получают данные о молекулярных мишенях для каждого таргетного лекарственного средства из по меньшей мере одного контрольного образца ткани, не имеющего пролиферативного фенотипа, при этом контрольный образец получают из ткани того же типа, что и указанная ткань пациента; тип данных из контрольного образца совпадает с типом данных, полученных на стадии (д);
(ж) определяют количественные показатели эффективности лекарственного средства для каждого из типов данных (i)–(v), используя только данные, полученные на стадиях (в)-(е);
(з) определяют клиническую эффективность для каждого таргетного лекарственного средства из группы таргетных лекарственных средств с помощью усреднения количественных показателей эффективности, определенных на стадии (ж).
| US 2016132632 A1, 12.05.2016 | |||
| US 2017193176 A1, 06.07.2017 | |||
| ARTEMOV A | |||
| et al | |||
| A method for predicting target drug efficiency in cancer based on the analysis of signaling pathway activation | |||
| Oncotarget | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Способ ослабления светочувствительности бромо-серебряных фотографических пластинок или фильмов | 1928 |
|
SU21945A1 |

