Уровень техники изобретения
Полиморфное поведение лекарственных препаратов может иметь первостепенную важность в фармации и фармакологии. Полиморфы по определению представляют собой кристаллы той же самой молекулы, имеющие различные физические свойства как результат порядка молекул в кристаллической решетке. Различия в физических свойствах, демонстрируемые полиморфами, затрагивают такие фармацевтические параметры, как стабильность при хранении, сжимаемость и плотность (важные в составлении и производстве продукта), и скорости растворения (важный фактор в определении биодоступности). Различия в стабильности могут быть результатом изменений в реакционной способности (например, дифференцированное окисление, такое, что лекарственная форма изменяет окраску быстрее, когда состоит из одного полиморфа, чем когда состоит из другого полиморфа), или механические изменения (например, таблетки разрушаются при хранении, поскольку кинетически контролируемый полиморф превращается в термодинамически более стабильный полиморф) или обеих (например, таблетки одного полиморфа более чувствительны к разрушению при высокой влажности). В результате различий в растворимости/растворении в лучшем случае некоторые полиморфные превращения могут привести к потери эффективности или, к другой противоположности, токсичности. Кроме того, физические свойства кристалла могут быть важными в процессинге: например, один полиморф может с большей вероятностью образовывать сольваты или может оказаться трудным для фильтрования и промывки от примесей (то есть форма частицы, и распределение по размерам может отличать один полиморф от другого).
Каждое фармацевтическое соединение обладает оптимальной терапевтической концентрацией в крови и смертельной концентрацией. Биодоступность соединения определяет удельную дозу в лекарственной форме, необходимую, чтобы получить идеальный уровень в крови. Если лекарственный препарат может кристаллизоваться в виде двух или большего количества полиморфов, отличающихся по биодоступности, оптимальная доза будет зависеть от полиморфа, присутствующего в композиции. Некоторые лекарственные препараты показывают узкую границу между терапевтической и летальной концентрациями. Хлорамфеникол-3-пальмитат (САРР), например, является антибиотиком широкого спектра действия, который, как известно, кристаллизуется по меньшей мере в трех полиморфных формах и одной аморфной форме. Самая стабильная форма, А поступает в продажу. Различие в биологической активности между этим полиморфом и другой формой В в восемь раз, что создает возможность фатальных передозировок соединения, если непреднамеренно будет введена в виде формы В вследствие изменений во время процессинга и/или хранения. В связи с этим контролирующие органы, такие как Администрация США по контролю за пищевым продуктам и лекарственным веществам, начали осуществлять жесткий контроль над полиморфным содержанием активного компонента в твердых лекарственных формах. Как правило, для лекарственных препаратов, которые существуют в полиморфных формах, если что-нибудь кроме чистого, термодинамически преобладающего полиморфа должно поступить в продажу, контролирующий орган потребует контроля партии за партией. Таким образом, становится важным как по медицинским, так и по коммерческим причинам производить и продавать чистый препарат в виде его наиболее термодинамически стабильного полиморфа, по существу свободном от других кинетически контролируемых полиморфов.
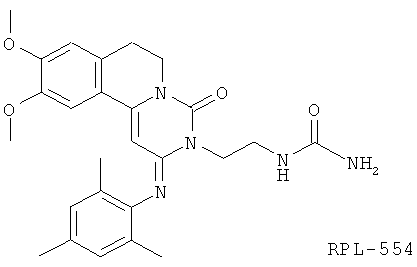
Патент США №6794391, 7378424 и 7105663, каждый из которых включен в настоящее изобретение посредством ссылки, раскрывает соединение RPL-554 (N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил] этил}мочевина).
Было бы полезно, обеспечивать композицию стабильного полиморфа RPL-554, который обладает преимуществом над менее стабильным полиморфами или аморфными формам, включая стабильность, сжимаемость, плотность, скорости растворения, увеличенную эффективность или отсутствие токсичности.
Краткое изложение сущности изобретения
Одним объектом настоящего изобретения является полиморфная форма N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил] этил} мочевины (RPL-554). В результате его уникальных свойств стабильности этот полиморф обеспечивает очень чистую N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил] этил}мочевину в ее наиболее термодинамически стабильной полиморфной форме.
Другим объектом настоящего изобретения является соединение N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил]этил} мочевину в форме кристаллического твердого тела, состоящего больше чем на 99 мас. % из N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил] этил} мочевины,
где по меньшей мере на 95% находится в полиморфной форме термодинамически стабильного полиморфа (I) N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил] этил} мочевины, указанного полиморфа (I) имеющего следующие структурные параметры, полученные посредством монокристального анализа:
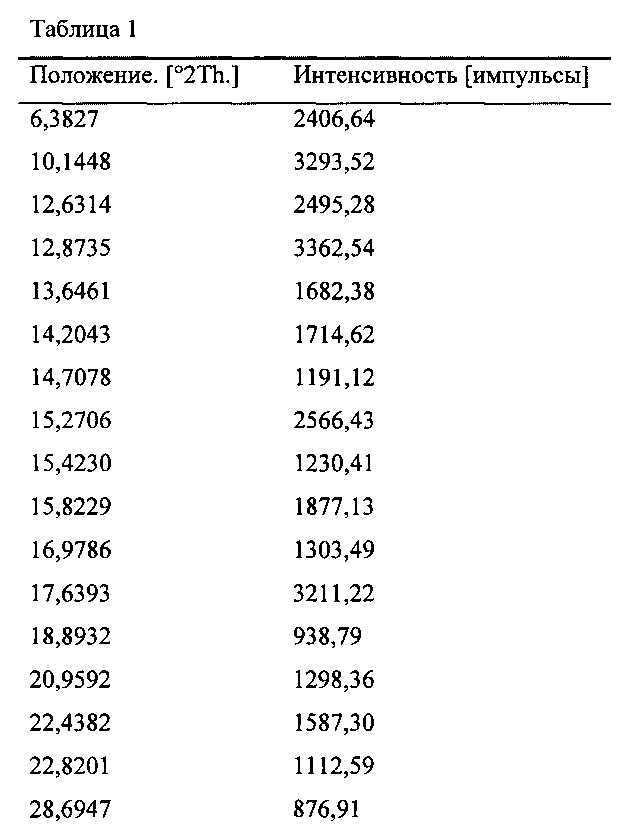
Другим объектом настоящего изобретения является кристаллический полиморф (I) N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1 -а]-изохинолин-3(4Н)-ил] этил} мочевины, имеющий рентгенограмму порошковой рентгеновской дифракции, включающей характеристические пики в единицах 29 при около 10,1° и около 12,9°.
В определенных примерах осуществления, указанная рентгенограмма порошковой рентгеновской дифракции дополнительно содержит характеристические пики в единицах 29, при около 15,3° и около 17,6°.
В другом варианте осуществления, указанная рентгенограмма порошковой рентгеновской дифракции содержит по меньшей мере 5 характеристических пиков, в единицах 2θ, выбранных из около 6,4° около 10,1°, около 12,6°, около 12,9°, около 13,6°, около 14,2°, около 14,7°, около 15,3°, около 15,4°, около 15,8°, около 17,0°, около 17,6°, около 18,9°, около 20,9° около 22,4°, около 22,8° и около 28,7°.
В определенных примерах осуществления полиморф имеет рентгенограмму порошковой рентгеновской дифракции по существу как показано на Фиг. 1.
В других примерах осуществления полиморф имеет кривую дифференциальной сканирующей калориметрии, показывая максимум около при 248°С.
В различных примерах осуществления полиморф имеет кривую дифференциальной сканирующей калориметрии по существу как показано на Фиг. 4.
В другом объекте изобретение обеспечивает твердую композицию, содержащую полиморф, как описано в настоящем изобретении.
В одном варианте осуществления изобретение обеспечивает композицию, которой по меньшей мере около 50 мас. % от общего количества N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил]этил} мочевины в указанной композиции присутствует в виде указанного полиморфа.
В других примерах осуществления изобретение обеспечивает композицию, в которой по меньшей мере около 70 мас. % от общего количества N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил]этил} мочевины в указанной композиции присутствует в виде указанного полиморфа.
В других примерах осуществления изобретение обеспечивает композицию, которой по меньшей мере около 90 мас. % от общего количества N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3(4Н)-ил] этил} мочевины в указанной композиции присутствует в виде указанного полиморфа.
В других примерах осуществления изобретение обеспечивает композицию, в которой по меньшей мере около 97 мас. % от общего количества N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил] этил} мочевины в указанной композиции присутствует в виде указанного полиморфа.
В другом варианте осуществления изобретение обеспечивает твердую композицию, содержащую полиморф, как описано выше, и фармацевтически приемлемый носитель.
В определенных примерах осуществления твердая композиция содержит полиморф и один или более дополнительных соединений.
В дополнительном варианте осуществления дополнительным соединением является известное терапевтическое средство.
В определенных примерах осуществления терапевтическое средство используется для лечения астмы, аллергической астмы, сенной лихорадки, аллергического ринита, бронхит, хронической обструктивной болезни легких (ХОБЛ), респираторного дистресс-синдрома взрослых (РДСВ), муковисцидоза, заболевания кожи, атопического дерматита, псориаза, воспаления глаз, мозговой ишемии или аутоиммунных заболеваний.
В других примерах осуществления изобретение обеспечивает твердую композицию в которой терапевтическое средство используется для лечения астмы или хронической обструктивной болезни легких.
В определенных аспектах изобретение обеспечивает способ получения полиморфа в соответствии с настоящим изобретением, включающий:
(a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил] этил)мочевины с растворителем для образования смеси, и
(b) нагревание при или выше температуры около 50°С в течение времени и в условиях, подходящих для образования указанного полиморфа.
В определенных аспектах изобретение обеспечивает способ получения полиморфа в соответствии с настоящим изобретением, включающий:
(a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил] этил}мочевины с растворителем для образования смеси,
(b) фильтрование указанной смеси,
(c) нагревание при или выше температуры около 55°С в течение времени и при условиях, подходящих для образования указанного полиморфа; и
(d) фильтрование и высушивание.
В определенных примерах осуществления растворителем является ДМСО, этанол, метанол, изопропиловый спирт, гексаны, пентан, этилацетат, дихлорметан или хлороформ. В дополнительном варианте осуществления растворителем является ДМСО или этанол.
В другом варианте осуществления, указанная смесь сохраняется при или выше температуры около 50°С в течение около 24-96 часов. В дополнительном варианте осуществления, указанная смесь сохраняется при или выше температуры около 50°С в течение около 72 часов. В других примерах осуществления указанная смесь сохраняется при или выше температуры около 55°С в течение около 24-96 часов. В различных примерах осуществления указанная смесь сохраняется при или выше температуры около 55°С в течение около 72 часов.
В других примерах осуществления указанная смесь высушивается в вакууме при температуре между 25 и 50°С. В дополнительном варианте осуществления указанная смесь высушивается в вакууме при 40°С.
В другом варианте осуществления изобретение предусматривает полиморф, как описано выше, полученный посредством способа, включающего
(a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил] этил} мочевины с растворителем для образования смеси, и
(b) нагревание при или выше температуры около 50°С в течение времени и при условиях, подходящих для образования указанного полиморф.
В другом варианте осуществления изобретение обеспечивает полиморф, как описано выше, полученный посредством способа, включающего
(a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3(4Н)-ил] этил} мочевины с растворителем для образования смеси,
(b) фильтрование указанной смеси,
(c) нагревание при или выше температуры около 55°С в течение времени и при условиях, подходящих для образования указанного полиморфа; и
(d) фильтрование и высушивание.
Другим объектом настоящего изобретения является способ лечения астмы, аллергической астмы, сенной лихорадки аллергического ринита, бронхита, хронической обструктивной болезни легких (ХОБЛ), респираторного дистресс-синдрома взрослых (РДСВ), муковисцидоза, заболевания кожи, атопического дерматита, псориаза, воспаления глаз, мозговой ишемии или аутоиммунных заболеваний у нуждающегося в этом млекопитающего, способ который включает введение указанному млекопитающему эффективного количества кристаллического полиморфа N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3 (4Н)-ил] этил}мочевины.
В одном варианте осуществления млекопитающим является человек.
Аморфная форма RPL554 является менее стабильной, так как при определенных условиях она может в той или иной степени превращаться в различные полиморфные формы, и это превращение может иметь место и при хранении. Кроме того, при крупномасштабном производстве аморфной формы, в соответствии с Современными правилами организации производства и контроля качества лекарственных средств (cGMP), к ее стабильности могут предъявляться еще более строгие требования, касающиеся чистоты, а также профиля примесей. Аморфная форма поэтому представляет собой проблемы в разработке воспроизводимого стандартного микронизированного сухого твердого порошка RPL554 для определенных применений композиции. Принимая во внимание, что кристаллическая полиморфная форма I может быть легко получена в большом масштабе согласно требованиям cGMP, чтобы обеспечить твердый RPL554 в качестве активного фармацевтического ингредиента (АФИ) в более систематическом и воспроизводимом процессе, процессы очистки и выделения для продукта кристаллической полиморфной формы I более приспособлены для крупномасштабного производства по cGMP. Вышеупомянутые преимущества приводят к обеспечению партий продукта, которые систематически соответствуют необходимым техническим требованиям cGMP. Как показано в одном аспекте настоящего изобретения, кристаллическая полиморфная форма I является наиболее термодинамически стабильной полиморфной формой RPL554, и также ожидается, что она будет иметь наиболее длительный срок годности при хранении. Это преимущество должно распространиться дополнительно на потенциальный срок годности любого коммерческого фармацевтического продукта RPL554, содержащего стабильную кристаллическую полиморфную форму I в качестве АФИ. Кроме того, кристаллическая полиморфная форма I более пригодна для разработки воспроизводимого стандартного микронизированного сухого твердого порошка RPL554 для определенных фармацевтических применений композиции.
Исследование N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо [6,1-а]-изохинолин-3(4Н)-ил] этил} мочевины (RPL5 54), представленной в кристаллической форме (обозначаемой как Форма I), показало, что вещество было очень устойчива к изменениям и, что Форма I была в рамках выполненного исследования самой стабильной полиморфной формой известного вещества (см. экспериментальную часть).
Серия экспериментов была использована, чтобы обнаружить существование новых, чистых полиморфов соединения, причем использовалось: добавление антирастворителя; манипулирование с насыщенным раствором; созревание суспензии; стандартная перекристаллизация; исследования смешанных растворителей; и водная система кристаллизации.
Синтезировали аморфную фазу, и ее также использовали в некоторых исследованиях, в результате которых были сделаны аналогичные выводы о том, что Форма I является доминирующей, за исключением только нескольких экспериментов, в которых наблюдались изменения. В случаях, когда изменение происходило при синтезе небольших количеств, предпринимались попытки проводить процессы кристаллизации в большем масштабе, для того чтобы полностью охарактеризовать формы (чистые и смешанные) и определить, происходило ли это изменение вследствие сольватации или причин, не связанных с сольватацией. В большинстве случаев, вследствие сложного характера переменных параметров процесса, которые необходимо было повторить для осуществления таких кристаллизации и для общей стабильности систем, происходило образования Формы I. Главным исключением являлся ДМСО, в случае которого наблюдалась фаза с потенциально более высокой температурой плавления в виде смеси с Формой I, после цикла основного плавления с разложением вещества. Кроме того, было отмечено, что перед проведением процесса кристаллизации в большем масштабе, в случае ДМСО происходило образование двух других альтернативных фаз, каждая из которых отличались друг от друга, но, в конечном счете, представляли собой фазу, которая являлась наиболее вероятной менее стабильной формой и/или сольватом.
Предыдущие сообщения предполагали, что аморфное вещество могло быть получено при помощи дробления охлажденного расплава. Быстрое упаривание из дихлорметана обеспечило надежный метод, тогда как дробление охлажденного расплава просто приводило к разложению (аморфный по данным XRPD (порошковая рентгеновская дифракция), но<20% продукта по данным анализа ВЭЖХ).
Поскольку на первых порах не было известно, было ли вещество, полученное в результате собственного производства, аморфным или кристаллическим, исследования растворимости были повторены для обоих этих веществ, чтобы выступать в качестве руководящего принципа, с аморфной фазой, оказавшейся более растворимой.
В конечном счете, было показано, что Форма I в качестве термодинамически предпочтительной фазы и, кстати сказать, что манипуляция либо кристаллического, либо аморфного вещества, могла быть успешно достигнута в отношении измененного физического представления выделенных веществ, например, порошок против более кристаллической составляющей.
Из значения в течении прогрессии этого вещества было то, что применяемое давление приводило к образованию того, что, как предполагали, было аморфным веществом, как было показано посредством XRPD и ДСК. Тепловой профиль для этого вещества показал заметную экзотермическую кристаллизацию и последующий эндотермический пик плавления, который был очень близок к этому показателю Формы I. Это могло быть проверено, посредством повторения с большим количеством вещества, и выделением продукта после экзотермического события и проверкой посредством XRPD и ВЭЖХ.
Краткое описание чертежей
Фиг. 1: Сравнение результатов измерений RPL554 (сверху) и моделируемых по монокристальным данным (внизу).
Фиг.2: Сравнение расширенного диапазона два тета (5-13° 2θ).
Фиг. 3: Профиль просвечивающей XRPD для RPL554.
Фиг. 4: Кривые ДСК/ТГА (термогравиметрический анализ) для RPL554.
Фиг. 5: Сравнение XRPD добавлений антирастворителя ДМСО/гептан (CG1099)
Фиг. 6: Сравнение XRPD RPL554 и кристаллов, полученных в насыщаемом растворе ДМСО (JN376A4).
Фиг. 7: Наложение ДСК введения RPL554 и новой формы из насыщенного раствора ДМСО (JN376A4).
Фиг. 8. Избранные результаты созревания суспензии, показывающие Форму I для всех серий.
Фиг. 9. Профили XRPD для кристаллизации из водных смесей (эксперимент PF86).
Фиг. 10. Раздробленный охлажденный расплав Формы I RPL554 от 245°С (выполненный как PF84).
Фиг. 11. Профиль ДСК для раздробленного охлажденного RPL554 (от 245°С, PF84).
Фиг. 12. Профиль ДСК для размолотой Формы I RPL554.
Фиг. 13. Сравнение XRPD спрессованного образца и оригинального вещества RPL554 (CG1099 F).
Фиг.14. Сравнение ДСК аморфного вещества (полученного) посредством давления и RPL554 ДСК.
Фиг. 15. Сравнение ДСК RPL554 и партии аморфного RPL554 из дихлорметана (JN376).
Фиг. 16. Анализ XRPD RPL554 (CM CG1099) и аморфного вещества (JN376F). Фиг. 17. Сравнение XRPD для PF89A4 (ДМСО) с Формой I RPL554.
Фиг. 18. Комбинированные ДСК/ТГА для ДМСО масштабированной кристаллизации PF90.
Фиг. 19. Сравнение XRPD Формы I RPL554 с формой смешанной фазы ДМСО (PF90).
Фиг. 20. Получающаяся конформация для RPL554 на основе анализа дифракции рентгеновских лучей.
Фиг. 21. Образование водородных связей, вовлеченные в каркас RPL554. Фиг. 22. Мотив упаковки для RPL554.
Подробное описание изобретения
Исследование N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил]этил}мочевины (RPL554), представленной в кристаллической форме (обозначаемой как Форма I), показало, что вещество было очень устойчиво к изменениям и, что Форма I была в рамках выполненного исследования самой стабильной полиморфной формой известного вещества. Серия экспериментов была использована, чтобы проверить это, включая в рамках таких исследований использование аморфного вещества. Обе фазы после получения показали, что преобладала Форма I за исключением только нескольких экспериментов, где было индуцировано изменение. В случаях, где изменение было индуцировано в небольшом масштабе, были предприняты попытки для масштабирования кристаллизации для того, чтобы полностью охарактеризовать формы (чистую и смешанную), чтобы определить, произошло ли изменение вследствие сольватации или присутствия осадителя. В большинстве случаев, вследствие деликатного характера переменных процесса, требуемых для повторения такой кристаллизации, и общей стабильности систем, получается Форма I. Главным исключением был ДМСО, где наблюдался потенциал выше расплавленной фазы, как смесь с Формой I, после цикла главного плавления с разложением вещества.
В предыдущих сообщениях предполагалось, что аморфное вещество могло быть генерировано при помощи дробления охлажденного расплава. Быстрое упаривание из дихлорметана обеспечило надежный метод, тогда как дробление охлажденного расплава просто приводило к разложению (аморфный по данным XRPD, но <20% продукта по данным анализа ВЭЖХ).
Поскольку на первых порах не было известно, было ли вещество, полученное в результате собственного производства, аморфным или кристаллическим, исследования растворимости были повторены для обоих этих веществ, чтобы выступать в качестве руководящего принципа.
В конечном счете, было показано, что Форма I является термодинамически предпочтительной фазой и, что манипуляция либо с кристаллическим, либо с аморфным веществом могла быть успешно достигнута в отношении измененного физического представления выделенных веществ, например, порошок против более кристаллической составляющей. На этой основе 3 пробные кристаллизации были начаты с целью выращивания вещества, подходящего для монокристального анализа и для дополнения к индексации и уточнения элементарной ячейки, выполненных с полученными данными с порошком высокого качества.
Порошковая рентгеновская дифракция является идеальным инструментом для получения «отпечатков пальцев» фармацевтического продукта, представляющего интерес. Идеально, если порошковые данные касаются уникальной кристаллической формы с уникальным XRPD. Вследствие коллапса кристаллографической информации от 3D до 2D, типичный XRPD будет видеть наложение дифракционных пиков при высоких значениях двух тета. Вследствие этого факта, индексирование порошковых дифрактограмм (касающиеся данных параметров элементарной ячейки) остается трудным этапом, хотя обширный прогресс был достигнут за последние годы.
Данные, представленные в настоящем изобретении, потенциально касаются правильных параметров элементарной ячейки RPL554. Алгоритм в программном обеспечении предоставляет выходные параметры настройки элементарной ячейки с показателями достоверности. Чем выше эти значения, тем выше вероятность получить правильную элементарную ячейку.
Порошковая рентгеновская дифракция (XRPD) PANalytical X'Pert PRO: Дифрактограммы порошковой рентгеновской дифракции регистрировали на PANalytical diffractometer с использованием Сu Кα излучения (45 кВ, 40 мА), 9- 9 гониометра, фокусирующего зеркала, вертикальной щели (1/2"), щелей Соллера и как для падающего, так и расходящегося луча (4 мм) и детектора PIXcel. Программное обеспечение, используемое для сбора данных, было X'Pert Data Collector, version 2.2f, и данные были представлены использованием X'Pert Data Viewer, version 1.2d.
Образцы исследовали при внешних условиях и проанализировали посредством трансмиссионной XRPD с использованием фольги, используя порошок непосредственно после получения. Около 2-5 мг образца устанавливали на 96-ти позиционном держателе образца с подложкой из полиимидной пленки (Kapton, толщина 12,7 мкм) пленке. Данные собирали в диапазоне 3-40° 2θ с непрерывным сканированием (скорость 0,146° 2θ/с).
Дифференциальная сканирующая калориметрия (ДСК): Данные ДСК были получены на PerkinElmer Pyris 4000 DSC. Прибор калибровали по энергии и температуре с использованием сертифицированного индия. Заранее определенное количество в миллиграммах мг образца помещали в алюминиевую капсулу с точечными отверстиями и, как правило, нагревали при 20°C. min-1 от 30°С до 350°С, или варьировали, как это было необходимо для эксперимента. Для инструментального контроля и анализа данных использовали программное обеспечение Pyris Software v9.0-1.0174.
Термо-гравиометрический анализ (ТГА): Данные ТГА были получены на Pyris 1 TGA, оборудованным 20-позиционным автоматическим пробоотборником. Прибор был калиброван с использованием сертифицированного индия. Заранее определенное количество в миллиграммах образца загружали в предварительно взвешенную алюминиевую капсулу и нагревали при 40°С мин-1 от температуры окружающей среды до 400°С. Поток азотом при 20 мл мин-1 поддерживался над образцом. Для инструментального контроля и анализа данных использовали программное обеспечение Pyris Software v9.0-1.0174.
Гравиметрическая сорбция пара: Изотермы сорбции получали, используя Hiden Isochema moisture sorption analyser (модель IGAsorp), который управлялся посредством программного обеспечения IGAsorp Systems Software V6.50.48. Образец поддерживали при постоянной температуре (25°С) средствами инструментального контроля. Влажность контролировали, смешивая потоки сухого и влажного азота с общим потоком 250 мл мин-1. Прибор верифицировали по относительной влажности посредством измерения трех калиброванных солевых раствора Rotronic (10-50-88%). Изменение веса образца измеряли как функцию влажности с помощью микровесов (точность +/-0,005 мг). Определенное количество образца помещали в корзинку из нержавеющей стали тарированными ячейками при внешних условиях. Полный экспериментальный цикл состоял из двух сканирований (сорбция и десорбция) при постоянной температуре (25°С) и 10%-ых интервалах RH в 10-90%-ом диапазоне (90 минут для каждого уровня влажности). Этот тип эксперимента должен был продемонстрировать способность исследуемых образцов абсорбировать влагу (или не абсорбировать) в ряду ячеек в определенных диапазонов влажности.
Ядерный магнитный резонанс (ЯМР) 1Н: Спектры ЯМР регистрировали на приборе Broker 270 МГц, оборудованном автоматическим дозатором, и контролируемым посредством пульта управления DRX400. Автоматизированные эксперименты проводили с использованием программного обеспечения Delta NMR Processing & Control Software version 4.3. Образцы получали в d6-ДМСО, если не указано иное. Анализ осуществляли с использованием (ACD/ Specmanager 7.11).
Термодинамическая растворимость в воде, определяемая с помощью ВЭЖХ: Растворимость в воде определяли, диспергируя достаточное количество соединения в воде квалификации «для ВЭЖХ», чтобы получить максимальную конечную концентрацию >20 мг мл-1 родоначального соединения. Суспензию уравновешивали при 25°С в течение 24 часов. Суспензию затем фильтровали в ампулу ВЭЖХ. Фильтрат затем разбавляли в подходящее количество раз. Количественное определение осуществляли посредством ВЭЖХ относительно стандартного раствора с концентрацией приблизительно 0,5 мг мл-1 в смеси ацетонитрил: вода: (1:1). Инжектировали различные объемы стандарта, разбавленные и неразбавленные растворы образца. Растворимость вычисляли, используя площади пиков, определенные интегрированием пика, найденного при том же времени удерживания, как основной пик при инжекции стандарта.
Анализ осуществляли на приборе для жидкостной хроматографии Agilent 1100 series, оборудованным ультрафиолетовым детектором (DAD или VWD) @ 254 нм, и используя программной обеспечение Chemstation Rev. В.01.03 для обработки данных. Определение химической чистоты посредством ВЭЖХ.
Фармацевтические композиции
Фармацевтические композиции в соответствии с настоящим изобретением содержат терапевтически эффективное количество полиморфа в соответствии с настоящим изобретением, составленным вместе с одним или большим количеством фармацевтически приемлемых носителей. Как использующийся в настоящее изобретение, термин "фармацевтически приемлемый носитель" означает нетоксичный, инертный твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующее вещество или вспомогательное средство любого типа. Фармацевтические композиции в соответствии с настоящим изобретением могут вводиться людям и другим животным перорально, ректально, парентерально, интрацистернально, внутривагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально или как пероральный или назальный спрей. Другие способы введения лекарственного препарата включают ингаляцию лекарства из раствора или суспензии фармацевтических композиций в соответствии с настоящим изобретением в дозирующем ингаляторе (MDI) или небулайзере, или ингаляцию порошкообразного лекарственного препарата, содержащего терапевтически эффективное количество полиморфа в соответствии с настоящим изобретением, которое как правило, смешивается с вспомогательным веществом или фармацевтическими композициями в соответствии с настоящим изобретением из ингалятора сухого порошка (DPI).
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в фармакологии такие как, например, вода, спирт или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, полисорбат, диметилформамид, масла (в частности семян хлопчатника, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), моно- или диглицериды, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирной кислоты и сорбитана и смеси вышеперечисленного. Помимо инертных разбавителей, пероральные композиции могут также включать вспомогательные средства, такие как смачивающее средство, эмульсифицирующие и диспергирующие вещества, антиоксиданты, подсластители, вкусовые и ароматизирующие вещества. Жидкая лекарственная форма может также быть инкапсулирована в желатиновую капсулу, в которой соединение в соответствии с настоящим изобретением может быть растворено в фармацевтически приемлемом носителе, содержащем, например, один или более солюбилизирующих агентов (например, полисорбат 80 и моно и диглицериды), и другие подходящие вспомогательные вещества (например, антиоксиданты, такие как аскорбил пальмитат или подслащивающее или ароматизирующее средство).
Инъекционные формы, например, стерильные инъекционные водные или масляные суспензии могут быть составлены в соответствии с ранее известными техническими решениями с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов. Стерильная инъекционная форма может также быть стерильным инъекционным раствором, суспензией или эмульсией в нетоксичном, приемлемом для парентерального введения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут использоваться, присутствуют вода, раствор Рингера, U.S.Р. и изотонический раствор хлористого натрия. Кроме того, стерильные, нелетучие масла традиционно используются как растворитель или суспензионная среда. С этой целью может использоваться любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота используются в получении инъекционных лекарственных средств.
Чтобы продлить эффект лекарственного препарата, часто желательно замедлить абсорбцию лекарственного препарата при подкожной или внутримышечной инъекции. Это может быть достигнуто посредством использования жидкой суспензии кристаллического или аморфного вещества с плохой растворимостью в воде. Скорость абсорбции лекарственного препарата в этом случае зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, отсроченная абсорбция лекарственной формы, введенной парентерально, достигается посредством растворения или диспергирования лекарственного препарат в масляном носителе.
Лекарственные формы с немедленным высвобождением также рассмотрены настоящим изобретением.
Композициями для ректального или вагинального введения предпочтительно являются свечи, которые могут быть приготовлены, посредством смешения соединений в соответствии с настоящим изобретением с подходящими нераздражающими вспомогательными веществами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозитория, которые представляют собой твердые вещества при температуре окружающей среды, но жидкие при температуре тела, и поэтому расплавляются в прямой кишке или вагинальной полости и высвобождают полиморф активного соединения.
Твердые композиции подобного типа могут также использоваться в качестве наполнителей в мягких и твердых заполненных желатиновых капсул, используя такие вспомогательные вещества как лактозу или молочный сахар так же как высокомолекулярный полиэтиленгликоль и т.п.
Активные полиморфы могут также быть в микрокапсулированой форме с одним или более вспомогательными веществами, как отмечалось выше.
Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочкой, такими как энтеросолюбильное покрытие, покрытия контролируемого высвобождения и другие покрытия, известные в области технологии приготовления лекарственных средств. В таких твердых лекарственных формах активное соединение можно смешать по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включить в качестве обычной практики, дополнительные вещества кроме инертных разбавителей, например, скользящее при производстве таблеток и другие вспомогательные вещества для изготовления таблеток, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферизующие агенты.
Предпочтительно, полиморф в соответствии с настоящим изобретением формулируется в виде твердой дисперсии, где полиморф может быть распределен на молекулярном уровне в матрице, которая содержит фармацевтически приемлемый гидрофильный полимер. Матрица может также содержать фармацевтически приемлемое поверхностно-активное вещество. Подходящая технология для получения твердой дисперсии для составления полиморфа в соответствии с настоящим изобретением включает, но не ограничивается этим: экструзия расплава, распылительная сушка или упаривание растворителя.
Лекарственные формы для местного или трансдермального введения полиморфа в соответствии с настоящим изобретением включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, если это может требоваться. Офтальмологическая композиция, ушные капли, глазные мази, порошки и растворы также рассматриваются как входящие в объем настоящего изобретения.
Мази, пасты, кремы и гели могут содержать в дополнение к полиморфу в соответствии с настоящим изобретением, вспомогательные вещества, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакантовую камедь, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевую кислоту, тальк и оксид цинка или смеси вышеперечисленного.
Порошки и спреи могут содержать в дополнение к полиморфам в соответствии с настоящим изобретением вспомогательные вещества, такие как лактоза, тальк, кремниевую кислоту, гидроокись алюминия, силикаты кальция и полиамидный порошок или смеси этих веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды.
Трансдермальные пластыри имеют дополнительное преимущество в том, что обеспечивают контролируемую доставку полиморфа в организм. Такие лекарственные формы могут быть изготовлены растворением или диспергированием полиморфа в подходящей среде. Усилители абсорбции могут также использоваться для увеличения потока полиморфа через кожу. Скорость можно контролировать либо путем обеспечения мембраны с контролируемой скоростью, либо путем диспергирования соединения в матрице полимера или геле.
Биологическая активность
В другом объекте настоящее изобретение обеспечивает полиморфную композицию, содержащую полиморф в соответствии с настоящим изобретением и ветеринарно или фармацевтически приемлемый носитель или разбавитель. Предпочтительно композиция является фармацевтической композицией для медицины человека. В другом аспекте композиция в соответствии с настоящим изобретением является фармацевтической композицией для ветеринарии.
Полиморфные соединения в соответствии с настоящим изобретением являются ингибиторами PDE и таким образом обладают ценными фармакологическими свойствами, такими как бронходилатационная активность, как продемонстрировано с помощью ингибирования стимулируемого полем сокращения изолированной трахеи морской свинки, и противовоспалительную активность как проиллюстрировано в исследованиях мононуклеарных клеток человека, стимулируемых РНА (фитогемагглютинином). In vitro и in vivo данные указывают, что соединения имеют длительную продолжительность действия, как продемонстрировано их постоянными защитными эффектами против вызванного гистамином бронхоспазма у морской свинки при непосредственной ингаляции в легкие в виде сухого порошка. Настоящее изобретение поэтому также касается острого, хронического или профилактического лечения пациентов, страдающих от респираторных расстройств, включая, в частности, астму, аллергическую астму, сенную лихорадку, аллергический ринит, бронхит, хроническую обструктивную болезнь легких (ХОБЛ), респираторный дистресс-синдром взрослых (РДСВ) и муковисцидоз. Они могут также использоваться местно при кожных заболеваниях, таких как атопический дерматит и псориаз или при воспалении глаз или любой другой болезни, включая мозговую ишемию или аутоиммунные заболевания, при которых увеличение внутриклеточных концентраций цАМФ считают полезным.
Один или более полиморфов соединения могут присутствовать в ассоциации с одним или большим количеством нетоксичных фармацевтически и/или ветеринарно приемлемым носителем и/или разбавителем и/или вспомогательными лекарственными веществами и/или пропеллентами и при необходимости другими активными ингредиентами. Подходящие носители или разбавители известны специалистам в данной области техники (например, Handbook of Pharmaceutical Excipients (1994) 2nd Edition, Eds. A. Wade/PJ Weller, The Pharmaceutical Press, American Pharmaceutical Association).
Другим объектом настоящего изобретения является полиморф соединения для применения в медицине. Полиморфные соединения в соответствии с настоящим изобретением полезны в качестве ингибиторов изоферментов фосфодиэстеразы. Полиморфы соединений или композиций в соответствии с настоящим изобретением могут использоваться для предотвращения или лечения любого заболевания, в которых соединения или композиции являются полезными, но особенно для болезни, при которой желательно увеличение внутриклеточной концентрации цАМФ. Примеры болезней, против которых соединения являются полезными, включают респираторные заболевания, в частности, астму, бронхит, хроническую обструктивную болезнь легких (ХОБЛ), респираторный дистресс-синдром взрослых (РДСВ), аллергическую астму, сенную лихорадку, аллергический ринит и муковисцидоз. Они могут также использоваться местно при кожных заболеваниях кожи, таких как атопический дерматит или псориаз, воспаление глаз или любая другая болезнь, включая мозговую ишемию или аутоиммунные заболевания, при которых увеличение внутриклеточных концентраций цАМФ считается полезным.
Этот объект изобретения особенно относится к лечению людей, но также применим к общей ветеринарной промышленности, в особенности к домашним животным, таким как собаки и кошки и сельскохозяйственные животные, такие как лошади, свиньи, рогатый скот, овцы и т.д.
Уровни дозировки порядка от около 0,02 мг до около 200 мг для приема до трех раз ежедневно являются полезными для лечения вышеупомянутых состояний. Конкретнее, диапазон дозировки от около 0,2 мг до около 20 мг, принимаемые до трех раз ежедневно, является эффективным. Конкретный режим дозировки будет, однако, в конечном счете, определен лечащим врачом, и будут учитываться такие факторы как применяемое лечение, возраст, масса, серьезность симптомов и/или серьезность существующего лечения или лечения, которое будет применено, способ введения лекарственного препарата, побочные реакции и/или других противопоказания.
Лекарственное средство согласно этому объекту изобретения может быть дано пациенту вместе с другими активными компонентами, которые могут, например, быть другим соединением или полиморфом в соответствии с настоящим изобретением или другими соединениями. Примеры включают агонисты β2-адренорецептора, глюкокортикоидные стероиды для наружного применения, производные ксантина, антигистаминовые соединения, антагонисты лейкотриенов, ингибиторы синтеза лейкотриенов и/или комбинаций вышеперечисленного.
В соответствии с другим объектом настоящее изобретение обеспечивает применение полиморфа в соответствии с настоящим изобретением в изготовлении ингибитора изофермента типа III/IV фосфодиэстеразы (PDE3/4). Изобретение охватывает использование полиморфа в соответствии с настоящим изобретением в изготовлении бронходилатора и/или антиастматического лекарственного средства и/или лекарственного средства для предотвращения или лечения хронической обструктивной болезни легких (ХОБЛ).
Настоящее изобретение также касается способа лечения или профилактики болезни млекопитающего, у которого ожидалась бы польза от ингибитора изофермента фосфодиэстеразы и/или бронходилатора, способ, который включает введение указанному млекопитающему количества, эффективного, нетоксичного количества полиморфа в соответствии с настоящим изобретением. Настоящее изобретение охватывает способ лечения или предотвращения астмы и/или хронической обструктивной болезни легких (ХОБЛ) у млекопитающего.
Термин "субъект" как использующийся в настоящем изобретении относится к млекопитающему. Субъект поэтому относится к, например, собакам, кошкам, лошадям, коровам, свиньям, морским свинкам и т.п. Предпочтительно субъектом является человек. Когда субъектом является человек, субъект может быть либо пациентом, либо здоровым человеком.
Как использующийся в настоящем изобретении, термин "фармацевтически приемлемая соль" относится к тем солям соединений, образованных посредством способа в соответствии с настоящим изобретением, которые являются по результатам тщательной медицинской оценки подходящим для использования в контакте с тканями людей и низших животных без неспецифической токсичности, раздражения, аллергической реакции и т.п., и соразмерны с разумным отношением выгоды/риска. Фармацевтически приемлемые соли известны специалистам в данной области техники. Например, S. M. Berge, et al. описывает подробно фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19 (1977). Соли могут быть получены in situ во время окончательного выделения и очистки соединений в соответствии с настоящим изобретением, или специально посредством реакции свободной аминогруппы с подходящей органической кислотой. Примеры фармацевтически приемлемых солей включают, но не ограничиваются этим: нетоксичные соли, полученные посредством добавления кислоты, или соли аминогруппы, образованные с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота или с органическими кислотами, такими как уксусная кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота или при помощи других методов, используемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают, но не ограничиваются этим: адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецлсульфат, этансульфонат, формиат, фумарат, глюкогентоноат, глицерофосфат, глюконат, гемисульфат, гептоноат, гексаноат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2- нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалоат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и т.п. Типичные представители солей щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция или магния и т.п. Дополнительно фармацевтически приемлемые соли включают в соответствующих случаях, нетоксичные катионы аммония, четвертичного аммония и амина, образованные, с использованием противоионов, таких как галид, гидроксид, карбоксид, сульфат, фосфат, нитрат, алкилсульфонат, имеющий от 1 до 6 углеродных атомов, и арилзамещенного сульфонат.
Комбинации заместителей и переменных, предусматриваемых настоящим изобретением, являются только те, которые приводят к образованию стабильных соединений. Термин "стабильный", как использующийся в настоящем изобретении, относится к соединениям, которые обладают стабильностью, достаточной для изготовления, и которые поддерживает целостность соединения в течение достаточного периода времени, чтобы быть полезными для целей, описанных в настоящем изобретении (например, для терапевтического или профилактического введения субъекту).
Соединения, описанные в настоящем изобретении, содержат один или более асимметричных центров и таким образом дают начало энантиомерам, диастереомерам и другим стереоизомерным формам, которые могут быть определены в терминах абсолютной стереохимии, как (R)- или (S)- или как (D)-или (L)- для аминокислот. Настоящее изобретение подразумевает включение всех таких возможных изомеров, так же как их рацемических и оптически чистых форм. Оптические изомеры могут быть получены из их соответствующих оптически активных предшественников посредством методик, описанных выше, или разделением рацемических смесей. Разделение может быть осуществлено в присутствии агента для оптического расщепления, с помощью хроматографии или посредством многократной кристаллизации или некоторой комбинацией этих методик, которые известны специалистам в данной области техники. Более подробная информация относительно разделения энантиомеров может быть найдена в Jacques, et al., Enantiomers, Racemates, and Resolutions (John Wiley & Sons, 1981). Когда соединения, описанные в настоящем изобретении, содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не определено иначе, это означает, что соединения включают как Е, так и Z геометрические изомеры. Аналогично, все таутомерные формы также предназначены для включения. Конфигурация любой двойной связи углерод-углерод, появляющаяся в настоящем изобретении, выбирается для удобства только и не предназначена, чтобы определять особую конфигурацию, если в тексте не утверждается другое; таким образом двойная связь углерод-углерод, изображенная произвольно в настоящем изобретении в виде транс-, может быть цис-, транс- или смесью обоих в любом соотношении.
Синтезируемые соединения могут быть выделены из реакционной смеси и дополнительно очищены таким методом, как колоночная хроматография, жидкостная хроматография высокого давления или перекристаллизация. Как может быть оценено специалистом в данной области техники, дополнительные методы синтеза соединений формул в соответствии с настоящим изобретением будут очевидны для специалиста в данной области техники. Дополнительно, различные синтетические стадии могут быть осуществлены в альтернативной последовательности или порядке, давая ожидаемые соединения. Кроме того, растворители, температура, продолжительность реакции и т.д., очерченные в настоящем изобретении, даются только в иллюстративных целях, и специалист в данной области техники понимает, что вариации в условиях реакции могут произвести желаемые мостиковые макроциклические продукты в соответствии с настоящим изобретением. Методологии трансформаций синтетической химии и защитных групп (защита и удаление защиты), полезные в синтезе соединений, описанных в настоящем изобретении, известны специалисту в данной области техники и включают, например, такие как описано в R. Larock, Comprehensive Organic Transformations. VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed.. Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995), и последующих выпусках вышеперечисленного.
Соединения в соответствии с настоящим изобретением могут быть модифицированы, посредством присоединения различных функциональных групп с помощью любых синтетических средств, очерченных в настоящем изобретении, чтобы усилить селективные биологические свойства. Такие модификации известны специалисту в данной области техники и включают такие, которые увеличивают биологическое проникновение в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), увеличивают биодоступность, увеличивают растворимость, чтобы сделать возможным введение инъекции, изменить метаболизм и изменить уровень выделения.
Перечисление списка химических групп в любое определение переменной в настоящем изобретении включает определения этой переменной как любой единственной группы или комбинации перечисленных групп. Перечисление вариантов осуществления для переменной в настоящем изобретении включает такой вариант осуществления как любой единственный вариант осуществления или в сочетании с любыми другими вариантами осуществления или частями вышеперечисленного.
Примеры
Соединения и способы в соответствии с настоящим изобретением будут лучше поняты в связи со следующими примерами, которые предназначены как только иллюстрации, а не для ограничения объема изобретения. Различные изменения и модификации к раскрытым вариантам осуществлениям будут очевидны для специалиста в данной области техники, и такие изменения и модификации, включая без ограничения, такие, которые касаются химических структур, заместителей, производных, составов и/или способов изобретения, могут быть сделаны, не отступая от сущности изобретения и объема приложенной формулы изобретения.
Количество твердого RPL554 в форме кристаллического полиморфа (I) в приведенных примерах составляет более 95 мас. %.
Пример 1
Приблизительно 50 мг N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]-изохинолин-3(4Н)-ил]этил}мочевины (RPL554) растворяли в 0,5 мл ДМФА при 70°С. Прозрачному желтому раствору позволили охлаждаться очень медленно с продувкой азотом приблизительно до 30°С более чем 4 дня. Начальная партия вещества, которое имело кристаллическую природу, наблюдалась в растворе (около 0,4 мл). Этот процесс повторяли дважды и он дал вещество, подходящее для монокристального анализа в объемом жидкости около 0,2 мл. Образец передали для анализа, неизменного в пределах маточного раствора.
Монокристальные данные измеряли при низкой температуре (123 K) и при различной длине волны (0,71073  ). Чтобы обеспечить картину, которая дала бы прямое сравнение, была необходима ручная регулировка значений длины волны. После различных регулировок получали Фиг. 1, которая показывает наложение расщеплений между измеренными данными для RPL554 070638 посредством XRPD (вверху) и симулированной XRPD, полученной из монокристальных данных (Копия onyx2010a, внизу).
). Чтобы обеспечить картину, которая дала бы прямое сравнение, была необходима ручная регулировка значений длины волны. После различных регулировок получали Фиг. 1, которая показывает наложение расщеплений между измеренными данными для RPL554 070638 посредством XRPD (вверху) и симулированной XRPD, полученной из монокристальных данных (Копия onyx2010a, внизу).
Более внимательное рассмотрение (Фиг. 2) в двух диапазонах теты (5-13° 2θ) ясно демонстрирует общие черты между наблюдаемыми данными и симулированным порошковым образцом.
Алгоритм в программном обеспечении сравнивает в числовой форме наблюдаемые данные (RPL554 070638), измеренные трансмиссионной XRPD, и численные значения, полученные из монокристального анализа. Основные результаты, полученные из монокристального анализа, приведены ниже:

Программное обеспечение, применяемое для сравнения, считывает файл с измеренных (RPL554 070638) и запрашиваемых исходных параметров элементарной ячейки, извлеченных из монокристальных данных. Уточнение Pawley (алгоритм) затем возвращало численные значения, но программное обеспечение также иллюстрирует это численное значение посредством графического изображения результата. Численное значение для критерия адекватности между измеренными и теоретическими значениями для RPL554 составляло 2,9. Это значение отражает качество данных. Чем более низкое значение, тем будут лучше данные, и обычно значение меньшее 10 рассматривается как положительный хит.
Численные значения для RPL554 имели хорошее соответствие со значениями, полученными для монокристалла, измеренными при 123K. Таким образом, монокристальные данные дают превосходное согласование с данными по порошку, собранными для полученного самостоятельно RPL554, и поэтому является типичным представителем для кристаллографического профиля для известного термодинамически предпочтительного полиморфа (Форма I).
Пример 2. Исходное вещество
Образец 9,0 г ex-GMP сток-раствора RPL554 (070638-9) отбирали и анализировали посредством XRPD (Фиг. 3 и Таблица 1) и ДСК (Фиг. 4) для использования в качестве текущего набора данных сравнения. RPL554 (070638-9) получают путем выделения 2-бутанона в кристаллической форме с содержанием воды, равным 10 мас. %.
Другие исследования, такие как протонный ЯМР и ВЭЖХ на данном этапе не были важны для начала осуществления проекта как результаты, собранные предварительно. Выполнение исследований по растворимости вещества с использованием набора обычных органических веществ не было так же необходимым, поскольку предыдущие исследования четко показали поведение вещества. Начало исследования в присутствии других форм RPL554 являлось приоритетным.
Как известно, вещество является кристаллическим с началом плавления 246°С для единственного эндотермического пика, без последующей заметной кристаллизации или перехода, отличного от термического разложения. Начальная потеря веса приблизительно 4,2% отмечена с помощью ТГА, соответствующая эндотермическому пику плавления, который не связан с растворителем (вероятно, потеря аммиака перед полной фрагментацией). После получения исходных данных, были начаты эксперименты с веществом с целью впоследствии получить аморфное вещество. С этого момента вышеупомянутые данные будут упоминаться как Форма I.
Предполагается, что процесс получения RPL554 поставляет вещество в существующей форме и что суспензии из спирта, использованные для улучшения его чистоты, увеличивают его кристалличность, или что еще более важно, превращают аморфное вещество в кристаллическую форму.
Пример 3. Добавление антирастворителя
Добавление антирастворителя является известным методом получения аморфных веществ, новых полиморфов и также смешанных фаз. Принимая это во внимание, получали сток-раствор RLP554 в ДМСО. ДМСО выбрали, так как вещество имеет разумную растворимость в этом растворителе, который является также смешивающимся со множеством других растворителей.
Экспериментальные условия (CG1099):
300 мг RPL554 растворяли в горячем ДМСО (2 мл сток-раствора) и 0,15 мл этого раствора добавляли при энергичном перемешивании порциями к 12 холодным пробиркам, содержащим различные растворители (1,0 мл) при комнатной температуре. В почти всех случаях наблюдалось немедленное выпадение осадка.
Выделение осуществляли фильтрацией и высушиванием в вакууме при 40°С.
Результаты приведены ниже в Таблице 2:
Все образцы, которые возвращали твердое вещество после высушивания в вакууме при 45°C и анализа посредством XRPD, показали, что возвращалась Форма I. Заметными исключениями оказались диоксан, дихлорметан и гептан. Диоксан и дихлорметан давали растворы, которые оставили стоять и испариться, что в конечном счете приводило к Форме I.
Гептан оказался более интересным в том, что образовывал два слоя, оба окрашенные раствора, которые постепенно превращались в масло после интенсивного встряхивания и периодов отстаивания (слой ДМСО). После приблизительно 24 часов образовывались твердые частицы в опалесцирующем масле и поэтому смесь декантировали и высушивали в вакууме при 45°С. К сожалению, в то время как надеялись, что будет образовываться аморфное вещество, наблюдалась Форма I. Тем не менее, было решено, что повторение эксперимента было оправдано, но с меньшими затратами времени после образования преципитирующего масла. Это снова привело к образованию двух прозрачных слоев и постепенно приводило к преципитирующему слою ДМСО, который декантировали и анализировали как влажный, так и высушенный (CG1110Awet и CG1110A2 соответственно). Раствору в ДМСО также позволили испариться до твердого вещества, которое высушивали в вакууме (CG1110B2). Результаты продемонстрированы на Фиг. 5, показывающей, что все образцы были фактически идентичны начальному веществу (CG1099SM).
Пример 4. Насыщенные растворы
Насыщенные растворы безусловно являются лучшим методом получения чистых фаз новых полиморфов. Поэтому, известное количество Формы I RPL554 растворяли или диспергировали в течение долгого времени, чтобы получить насыщенный раствор при температуре, с последующей отделочной фильтрацией, чтобы удалить любую память формы (зародыши). Затем использовали контролируемое охлаждение, чтобы стимулировать кристаллизацию.
Экспериментальная часть (JN376):
24 Растворителя перемешивались при 45°С в присутствии 30 мг Формы I RPL554, чтобы предоставить начальную суспензию. Суспензии нагревали при 45°С в течение 4,5 часов перед горячей фильтрацией в предварительно нагретые пробирки, которые затем оставляли медленно испаряться перед высушиванием в вакууме при 45°С и анализом. Следует отметить, что образцы А и В (в Таблице 3) нагревали при 35°С в течение 24 часов под азотом, чтобы помочь испарению.
Условные обозначения: XT = кристаллический, нераств. = нерастворимое/очень мало растворимое вещество, следы порошка-недостаточный = недостаточно вещества, чтобы проанализировать
Из таблицы можно видеть, что во многих экспериментах не растворилось достаточное количество вещества, необходимое для анализа, и образуется или капля масла или порошок на дне пробирки после высушивания. Однако, несмотря на это много экспериментов дали вещество в порошковой форме, которая позволила провести XRPD-анализ. Самые успешные эксперименты, показавшие правильные кристаллы, выросшие из маточного раствора, были получены, и только один опыт показал какое-то соответствующее изменение формы (ДМСО).
Результаты для образца ДМСО показаны ниже (Фиг. 6 и 7), и в то время как вещество показывает некоторое сходство с Формой I, считается на основе XRPD и учета предпочтительной ориентации, что присутствует новый полиморф. Анализ ДСК вещества дает более ясный признак изменения формы с намного более низким началом главного эндотермического пика. Для ТГА и ЯМР-анализа было недостаточно вещества. По нашему мнению, оно является менее стабильной формой RPL554.
Пример 5. Созревания суспензии
24 х Созреваний суспензии осуществляли в попытке подвергнуть сжатию текущее кристаллическое твердое вещество в прохождении трансформации формы. Самый эффективный метод для такой работы должен использовать пролонгированные циклы нагревания/охлаждения, каким был подвергнута Форма I RPL554.
Экспериментальная часть (CG1100):
Около 20 мг вещества перемешивали в стеклянных пробирках примерно в 30 объемах растворителя и повторяли циклически между изотермическими периодами (8 часов) 20°С и 50°С. После 4 дней циклических повторов твердые частицы отфильтровывали и высушивали в вакууме при 45°С для анализа XRPD. Все результаты сравнивали с исходным веществом, RPL554 (Форма I); см. Таблицу 4.
Все образцы показали тот же самый профиль XRPD при сравнении с исходным веществом, демонстрируя, что Форма I RPL554 чрезвычайно устойчива к изменениям и очень вероятно является термодинамически самой стабильной формой соединения (Фиг. 8).
Пример 6. Смешанный растворитель для перекристаллизации
Поскольку профили растворимости могут заметно изменяться в присутствии систем смешанных растворителей, и из-за того, что эти тенденции могут быть предсказаны с большим трудом, планировали серию перекристаллизации с использованием множества систем. Это считали другим подходящим способом вызвать изменение формы, приводящее к открытию новых полиморфов.
Было отмечено во время исследований, что диоксан и кумен (отличные от обычно используемых ДМФА, ДМСО и тетралина), показали степень эффективности в растворении RPL554. Эти растворители сформировали основу нескольких начальных испытаний, которые брали растворы после отделочной фильтрации каждого растворителя и RPL554 и затем смешивали их горячими с серией известных антирастворителей различных типов, поддерживая температуру раствора, и затем позволяя медленно охладиться.
Экспериментальная часть (PF87):
Сток раствор с 100 мг RPL 554 получали, размешивая твердое вещество в 5 мл главного растворителя и оставляя нагреваться при 50°С, чтобы улучшить растворение. Добавляли дополнительный 1 мл (больше, если было необходимо) со-растворителя, чтобы улучшить растворение и сформировать конечную исходную смесь. Образцы оставляли перемешиваться при 50°С в течение 30 минут. К предварительно подогретой теплой пробирке (50°С) со сток-раствором добавили 2 мл антирастворителя, указанного в таблице; образцы оставили перемешиваться и упариваться. Образцы отфильтровывали и высушивали в течение ночи в вакуумном сушильном шкафу при 50°С перед анализом XRPD (см. Таблицу 5).
Все эксперименты, перечисленные в эксперименте PF87, приводили к Форме I, что дает дополнительное доказательство о термодинамической стабильности RPL554.
Пример 7. Исследования по кристаллизации в смешанных водных системах.
Серию кристаллизации в смешанных растворителях, в которых была водная основа, исследовали в эксперименте PF86.
Экспериментальная часть (PF86):
Горячий сток-раствор RPL554 (Форма I) в ДМСО (1 мл, 15 мг.мл-1) добавили к горячему раствору растворителя, смешанного с водой (1:1, об/об, 1 мл) как обозначено в Таблице 6 (50°С). Растворы оставили перемешиваться при температуре в течение 48 часов, охлаждали, фильтровали и затем высушивали в сушильном шкафу в течение ночи (40°С). Осуществляли анализ XRPD и сравнивали с исходным веществом.
Результаты для этого ряда экспериментов (Фиг. 9) снова показали, что Форма I была доминирующей.
Пример 8. Генерация аморфного вещества
В действительности существовало историческое экспериментальное доказательство образования аморфного вещества посредством нагревания Формы I RPL554 до 245°С и быстрого охлаждения расплава, осуществляли повторение этого метода (на основе ТГА) и получили такое же, прозрачное, желтое, стеклообразное вещество. Этот образец был впоследствии проанализирован с помощью XRPD и было показано, что он был аморфным (см. Фиг. 10).
Это веществом затем исследовали на приборе ДСК в попытке наблюдать стеклование и, если возможно, любые перекристаллизации. Результаты показаны на Фиг. 11.
Предполагаемое стеклование могло наблюдаться при приблизительно 120°С без наблюдаемой формальной перекристаллизации. В то время как проходил этот эксперимент, образец был представлен для анализа LC, поскольку стабильность при (повышенной) температуре, как было известно, была потенциальной проблемой (наблюдение ТГА). Хроматограмма показала присутствие многочисленных пиков с только 26% вещества, являющегося исходным веществом, RPL554. Этот результат показал, либо что необходимо осуществить измельчение охлажденного расплава с более чувствительными экспериментальными границами, либо что способ оказался несостоятельным. Эксперимент повторяли при более низкой температуре (240°С) в попытке избежать разложения и таким образом получить лучший профиль по показателю чистоты. Анализ LC (жидкостная хроматография) показал подобный профиль очень низкой чистоты и различные побочные продукты. Поэтому стало очевидно, что аморфное вещество не будет получено этим способом.
Затем было предпринято много других попыток получить аморфную фазу, начиная с физической манипуляции (размол, прикладываемое давление) и затем быстрое упаривание разбавленного раствора от растворителей, таких как дихлорметан и кумен. В этом эксперименте, измельченный образец (ступка и пестик), оказался очень электростатическим, и его невозможно было перенести на каптоновую пластинку для XRPD-анализа. Образец был, однако, успешно перенесен в капсулу для ДСК; наблюдаемая точка плавления оказалась ниже, чем у вещества сравнения, что указывало на возможность изменения формы. Повторение этого эксперимента потребуется для продолжения этого исследования, однако, аморфная фаза, не была получена таким подходом (рис. 12).
Прикладываемое давление (100 кН): 10 мг RPL554 оставили в системе пресс-шайбы на ночь, формируя тонкий и однородный диск толщиной меньше чем 0,1 мм. Диск осторожно размалывали и анализировали с помощью XRPD (Фиг. 13).
Сравнение XRPD прессованного вещества со стартовой Формой (используемая ссылка CG1099F), демонстрирует, что сжатие кристаллического вещества производит аморфную фазу. После этих наблюдений термический анализ (ДСК) осуществляли и доказали, что вещество был аморфным (Фиг. 14).
Переход из аморфного вещества показан посредством Tg при 75°С, которое затем повторно кристаллизуется при приблизительно 131°С прежде плавления при 242,6°С и 262,7°С последовательно. Первый эндотермический пик оказался немного ниже, чем у вещества сравнения, и мог потенциально быть связанной с другой формой (сравнения чистоты будут разумным для выполнения исследования перед признанием новой формы). Второе плавление могло также быть рассмотрено как отличная кристаллическая структура, хотя следует подчеркнуть, что она находится в районе разложения для этого вещества.
Быстрое упаривание растворителя: RPL554-070638, содержащийся в колбе, полностью растворяли в большом объеме дихлорметана при комнатной температуре (290 мл, 2,0 г). Растворитель затем быстро удаляли при нагревании, сохраняя RPL554-070638 полностью растворенным, избегая медленного осаждения исходного вещества и присутствия Формы I. Анализ ВЭЖХ показал высокую химическую чистоту и протонный ЯМР показал следы остаточного дихлорметана.
XRPD исходной партии показал аморфную фазу (JN376E), что было подтверждено посредством ДСК (Фиг. 15). Процесс нагревания индуцировал большой экзотермический пик, иллюстрирующий кристаллизацию при 152°С, с последующим эндотермическим пиком для ожидаемого плавления Формы I, хотя немного подавленным. Следует отметить тонкие различия между этой партией и аморфным веществом, полученным при применении давления (основное различие в температуре начального экзотермического пика и профиля после главного плавления).
Успешно выделив небольшое количество аморфного вещества, методику масштабировали до граммовых количеств вещества для использования в работе насыщенного раствора и суспензии. Эталонный профиль для этой партии (JN376F) показан на Фиг. 16.
Пример 9. Насыщенные растворы с аморфным веществом
Как указано, насыщенные растворы являются лучшим методом получения чистых фаз новых полиморфов. С этой целью брали аморфное вещество, и исследование осуществляли, используя насыщаемые растворы, как в JN376, с целью получения более высокой общей исходной концентрации.
Экспериментальная часть (PF89):
24 растворителя перемешивали при 45°C в присутствии 30 мг RPL554 (JN376F), чтобы получить исходную (более сконцентрированную) суспензию, чем с кристаллической фазой. Суспензии нагревали в течение 4,5 часов, чтобы увеличить растворимость. Каждую пробирку проверяли на полное растворение вещества. Если твердое вещество оставалось, добавляли дополнительный 1 мл известного растворителя пока не происходило полное растворение. Каждую пробирку оценивали перед горячей фильтрацией в предварительно нагретые пробирки, которые затем оставили медленно упариться. Обращаем внимание, что пробирки помещали под током азота, чтобы помочь испарению в течение 24 часов. Результаты этого эксперимента показаны ниже в таблице (Таблица 7):
Из таблицы можно заметить, что в некоторых экспериментах растворилось недостаточное количество вещества, чтобы провести анализ, или получались либо капелька масла, либо аморфной порошок на дне пробирки после высушивания. Однако, несмотря на это, во многих экспериментах были получены вещества в порошковой форме, что позволило провести XRPD-анализ. Наиболее успешные эксперименты, показавшие правильные кристаллы, выросшие из маточных растворов, были проведены, и только один образец показал какое-то соответствующее изменение формы (PF89A4, ДМСО).
Результаты для образцов ДМСО показаны ниже, и пока вещество демонстрирует некоторое подобие Формы I, считается на основе XRPD и учете предпочтительной ориентации, что новый полиморф присутствует. Анализ ДСК вещества дает более ясный признак изменения формы с намного более низким началом главного эндотермического пика. Оказалось недостаточно вещества для ТГА- и ЯМР-анализов. Этот результат масштабировали для дальнейшего исследования, хотя следует отметить, что это вещество не является тем же самым, но изменил профиль относительно RPL554 как JN376A4 (насыщенный раствор из ДМСО, Фиг. 6 и 7). Считалось, что это могло произойти вследствие относительного уровня сольватации.
Чтобы помочь пониманию сделанного анализа, и вследствие того, что высококачественная информация о моделировании была получена от предсказанных параметров элементарной ячейки (эксперимент индексации) для RPL554, новая фаза была сравнивалась с этими теоретическими данными.
Числовой анализ демонстрирует, что большая часть вещества представляет собой форму I с некоторыми пиками, которые не согласовывались. Это служит убедительным доказательством того, что образец был либо смесью двух форм, либо потенциально сольватированным продуктом. Из этих результатов не следовало полностью и непосредственно, что форма I обнаруживалась во всех случаях, где оставалось достаточно вещества для сбора, за исключением ДМСО, где были замечены незначительные изменения в профиле XRPD.
Пример 10. Автоматизированный эксперимент (расширенное исследование с антирастворителем)
Эксперимент был начат, чтобы расширить набор растворителей из тех, которые использовали в работе с добавлением антирастворителя и для осуществления такого смешивания растворителей при нагревании вместо разрушения охлаждением через использование антирастворителя (CG1099).
Экспериментальная часть (PF85):
Определенное количество RPL554 (1,5 г) и 20 мл ДМСО добавляли в пробирку и нагревали при 50°С, чтобы улучшить растворение. 200 мкл сток-раствора помещали в нагретую ячейку (50°С). 200 мкл перечисленных растворителей (таблица ниже) добавляли к ячейке и оставили перемешиваться на более чем 72 часов перед выделением. Образцы затем переносили на каптоновую пластинку для анализа XRPD. Результаты обрабатывали и сравнивали с исходным веществом RPL554 (Таблица 8). Там, где XRPD оказался недостаточным, чтобы дифференцировать от исходного вещества, осуществляли термический анализ, чтобы определить точки плавления (если вещество допускало).
Таким образом, из сведенных в таблицу результатов, особенно тех, на которые указывает ДСК собранных следов, можно видеть, что некоторые потенциально новые или смешанные фазы выделяются из более широкой библиотеки антирастворителей. В то время как диаграммы XRPD могут выглядеть первоначально отличающимися, вследствие интенсивности пиков, эффекты предпочтительной ориентации могут присутствовать, и тем самым может оказаться необходимым более глубокий анализ (предпочтенную ориентацию считали возможной вследствие визуального наблюдения за перенесенными кристаллами из автоматизированного эксперимента) (результаты не показаны). Такой анализ был также представлен посредством численного сравнения новых диаграмм XRPD с предсказанной диаграммой Формы I из индексирования. Идея состояла в том, чтобы получить столько же качественные данные о присутствии истинной новой фазы или же результатом фактически была смесь из исходных образцов.
Из параллельного эксперимента PF85 результаты обеспечили некоторую относительно интересную информацию о способности RPL554 демонстрировать различные кристаллические формы. Объединенные аналитические инструменты (XRPD/ДСК) показали, что по меньшей мере пять кристаллических форм потенциально существовали параллельно с Формой I RPL554, хотя было ощущение, что они были главным образом смесями. Эксперименты, которые дали наиболее вероятные доказательства новых фаз и были отобраны для масштабирования были (следует заметить, что другие системы, возможно, могли повторять эти результаты):
Численное сравнение вышеупомянутых 5 форм с расчетной диаграммой Формы I RPL554.
Данные, которые касаются потенциально новых полиморфов RPL554 и расчетного профиля Формы I (данные, не показаны). Для этого исследования было задано направление к определению параметров элементарной ячейки во время попыток индексировать Форму I RPL554. Алгоритм в программном обеспечении позволяет вводить параметры элементарной ячейки, в этом случае связанные с Формой I. Программное обеспечение затем вычисляет профиль, таким образом позволяет пользователю сравнить визуально с результатами измерений. Этот подход помогает подтвердить существование новой формы и/или новой формы, сосуществующей с Формой I (смесь). Почти во всех случаях, описанных выше, заметный результат состоит в том, что присутствовала Форма I и, главным образом, присутствовали смеси, а не новая, чистая фаза с потенциалом для D10, являющимся исключением. Все вышеперечисленные кандидаты были намечены для масштабирования.
Пример 11. Масштабирование потенциальных новых форм
Серия экспериментов по масштабированию была начата, чтобы получать больше вещества новых и/или смешанных форм. Таким образом, исследование могло быть осуществлено более детально, и пересечься экспериментальными суспензиями. Следует отметить, что такое масштабирование может быть нетривиальным, особенно там, где менее стабильные формы индуцировались в небольшом масштабе (технологические тонкости означают, что масштабирование может быть протестировано).
Экспериментальная часть (PF90 и JN386):
Сток-раствор RPL554 получали в горячем ДМСО. Раствор фильтровали отделочно и 1,75 мл (содержащие приблизительно 200 мг) добавляли к объему (4 мл) горячего растворителя (55°С). Образцы оставили перемешиваться и медленно упариваться в течение длительного времени, чтобы подражать условиям, которые производили потенциально новые полиморфы. После осаждения образцы отфильтровывали и высушивали в вакууме при 40°С перед анализом XRPD. Таблица, приведенная ниже (Таблица 9), суммирует эксперименты по масштабированию для RPL554.
С веществами в наличие анализ такой как ТГА был бы применим для определения, является ли сольватация причиной различий в профилях XRPD (например, смешанная фаза Формы 1 и сольват), или это происходит вследствие несольватированной смеси форм. В случае этилацетата, например, можно наблюдать, что масштабирование процесса дает Форму 1 и, таким образом, наиболее вероятно, что более заметные различия в начальном профиле XRPD происходило вследствие смеси Формы 1 и менее стабильной формы, которая превращалась в самый стабильный полиморф (более низкая т. пл. эндотермического пика, присутствующего в ДСК, вне фазы разложения, благоприятствует этому). Тот же самый комментарий верен для всех других растворителей кроме уксусной кислоты, которая давала смолу, и ДМСО, который давал смесь. В этом случае было доступно достаточно вещества, чтобы подтвердить присутствие Формы I и менее стабильной формы, которая со временем превращается в наиболее стабильную форму. Следует отметить, что она не идентична менее стабильным формам, идентифицированным из ДМСО в PF98A4 и JN376A4, показывая, что этот растворитель обеспечил ряд изменений главной формы, но что при масштабировании Форма I была все еще главным компонентом.
Анализ (Фиг. 18 и 19) связан с экспериментом масштабирования, вовлекающим ДМСО.
Данные XRPD снова ясно показывают присутствие пиков, которые не принадлежат Форме I. Как продемонстрировано в термическом анализе. Форма I присутствует в большинстве (образцов) (начало плавления при 246,1°С), в то время как сосуществует соединение с более высокой температурой плавления (начало при 261°С). Гравиметрический анализ демонстрирует присутствие свободного ДМСО в образце, который испаряется при продувании азотом в ТГА, при нагревании образца от комнатной температуры до 100°С. Это не может быть связано с включением растворителя в более высоко плавящееся соединение. Хотя весь свободный ДМСО, по всей видимости, исчезает после нагревания, кривая ТГА показывает медленную, но стабильную потерю веса между 100°С и 200°С, которая может быть связана с 'включенным' ДМСО, хотя без выделения чистой фазы, эти предположения трудно подтвердить. Протонный ЯМР, как и ожидалось, показывает присутствие ДМСО в образце.
В качестве заключения, эксперименты по масштабированию для RPL554, демонстрируют, что Форма I остается предпочтительной и также кинетически более предпочтительна. Вероятно, что большинство наблюдаемых смешанных фаз в конечном счете должно превратиться Форму I.
Пример 12. Растворимость в воде
Так как аморфное вещество и Форма I были в наличие, оба образца подвергли прямому исследованию растворимости в очищенной воде при 25°C. Этот эксперимент осуществляли, поскольку это было неизвестно, кристаллическое или аморфное вещество было первоначально выделено во время предыдущей самостоятельной синтетического эксперимента.
Экспериментальная часть:
Суспензию 20 мг кандидата перешивали в очищенной воде (1 мл, рН 7). Суспензию уравновешивали при 25°С в течение 24 часов. Суспензию затем фильтровали в ампулу для ВЭЖХ и фильтрат затем разбавляли в подходящее количество раз с определением количества с помощью ВЭЖХ относительно стандартного раствора.
Как ожидалось, доказанная аморфная фаза, хотя довольно малорастворимая, имела большую растворимость, чем Форма I в этом базовом исследовании. К сожалению, оба образца были слишком тонкодисперсны, чтобы быть эффективно отфильтрованными и выделенными для повторного анализа посредством XRPD, хотя известно, что Форма I получается из таких дисперсий.
Пример 13. Данные рентгеновского анализа: Релевантные параметры, связанные с монокристаллом RPL554
Данные рентгеновского анализа для RPL554 могут быть найдены в Таблицах 10-15 и Фиг. 20-22.
Содержание всех ссылок (включая литературные ссылки, выданные патенты, опубликованные заявки на патент и одновременно заявленные заявки на патент, находящиеся на рассмотрении) процитированные по тексту всей настоящей заявки тем самым явным образом включены полностью в настоящее изобретение посредством ссылок. Если иначе не определено, все технические и научные термины, использованные в настоящем изобретении, получают значение, общеизвестное специалисту в данной области техники.
Специалистам в данной области техники будет понятно или они будут в состоянии установить использование всего-навсего обычное экспериментирование, много эквивалентов конкретных вариантов осуществления изобретения, описанного в настоящее описании. Такие эквиваленты предназначены быть охваченными прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ЛЕЧЕНИЕ | 2015 |
|
RU2688191C2 |
| ПОЛИМОРФЫ ДОЦЕТАКСЕЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2437875C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 6-(1Н-ИМИДАЗОЛ-1-ИЛ)-2-ФЕНИЛХИНАЗОЛИНА И ЕГО СОЛЕЙ | 2010 |
|
RU2557547C2 |
| НОВЫЕ ПОЛИМОРФЫ И СОЛИ | 2011 |
|
RU2573384C2 |
| РАЗЛИЧНЫЕ ФОРМЫ 6-ХЛОР-2-ЭТИЛ-N-(4-(4(4-(ТРИФТОРМЕТОКСИ)ФЕНИЛ)ПИПЕРИДИН-1-ИЛ)БЕНЗИЛ)ИМИДАЗО[1,2-a]ПИРИДИН-3-КАРБОКСАМИДА | 2019 |
|
RU2826176C2 |
| ТВЕРДЫЕ ФОРМЫ (1,1-ДИОКСО-4-ТИОМОРФОЛИНИЛ)-[6-[[3-(4-ФТОРФЕНИЛ)-5-МЕТИЛ-4-ИЗОКСАЗОЛИЛ]МЕТОКСИ]-3-ПИРИДИНИЛ]-МЕТАНОНА | 2012 |
|
RU2618524C2 |
| КОМБИНАЦИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2014 |
|
RU2666624C2 |
| ТВЁРДЫЕ ФОРМЫ ИНГИБИТОРА КИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2013 |
|
RU2673077C2 |
| КЕТАМИН ПАМОАТ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2825336C2 |
| Фармацевтические композиции, содержащие RPL554 в HFA-134A для введения посредством ингаляции | 2019 |
|
RU2813959C2 |
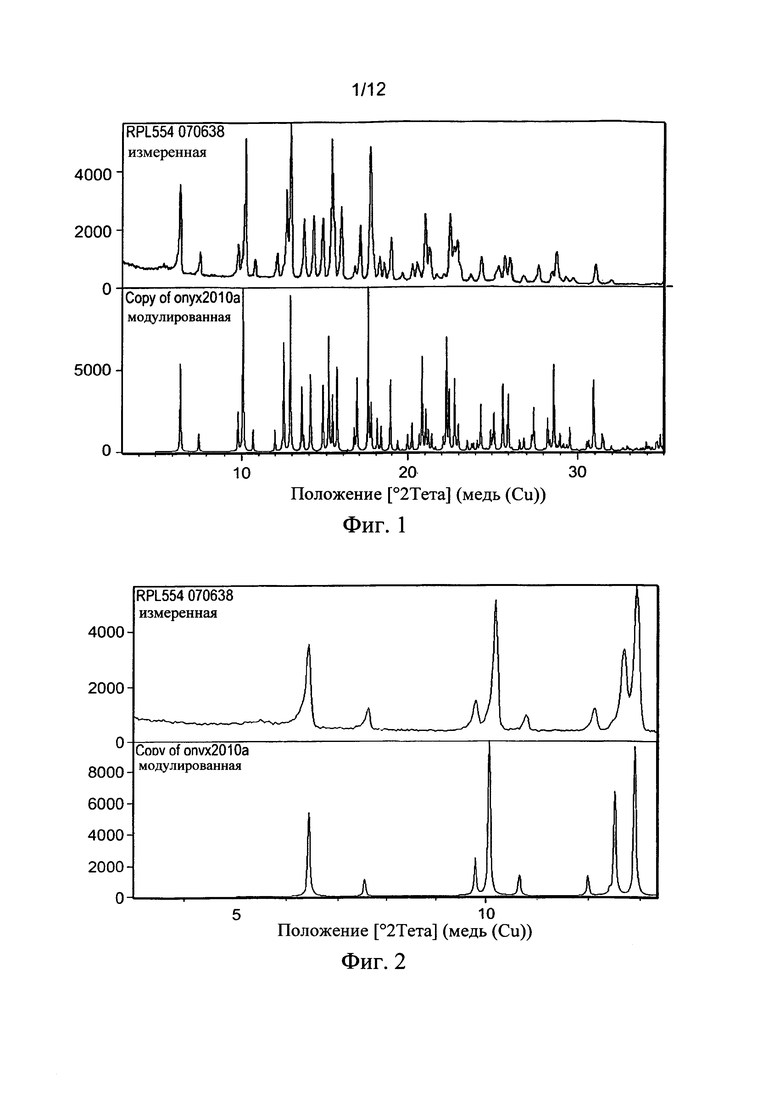
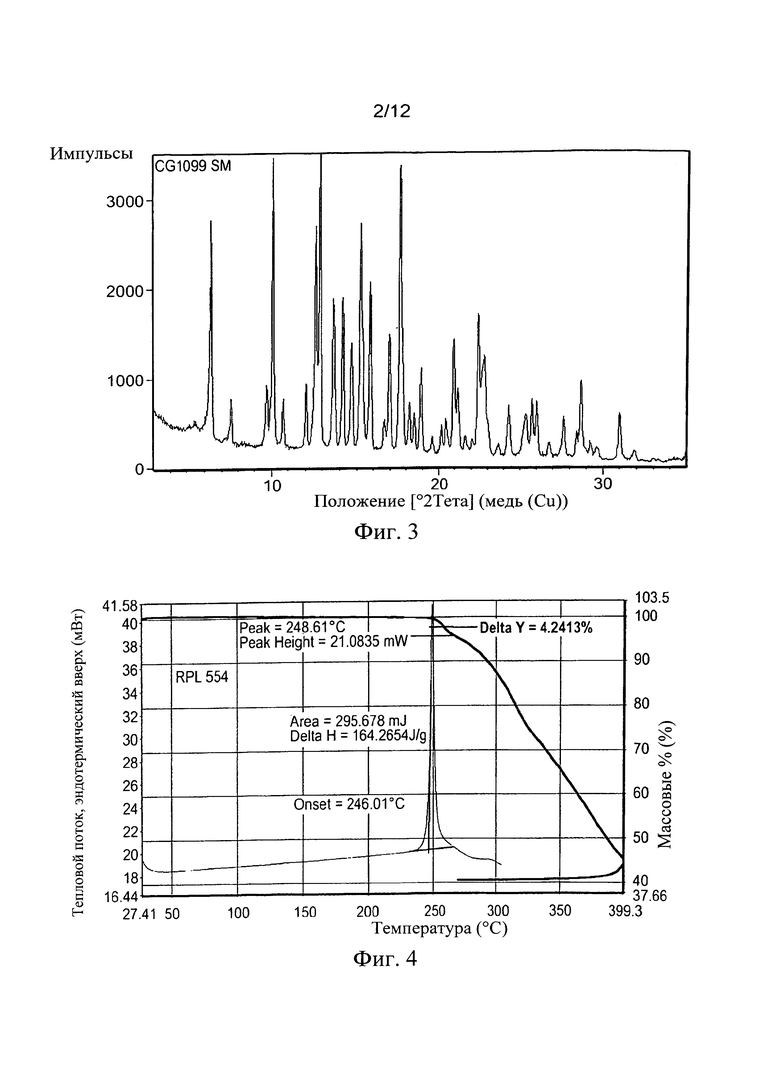
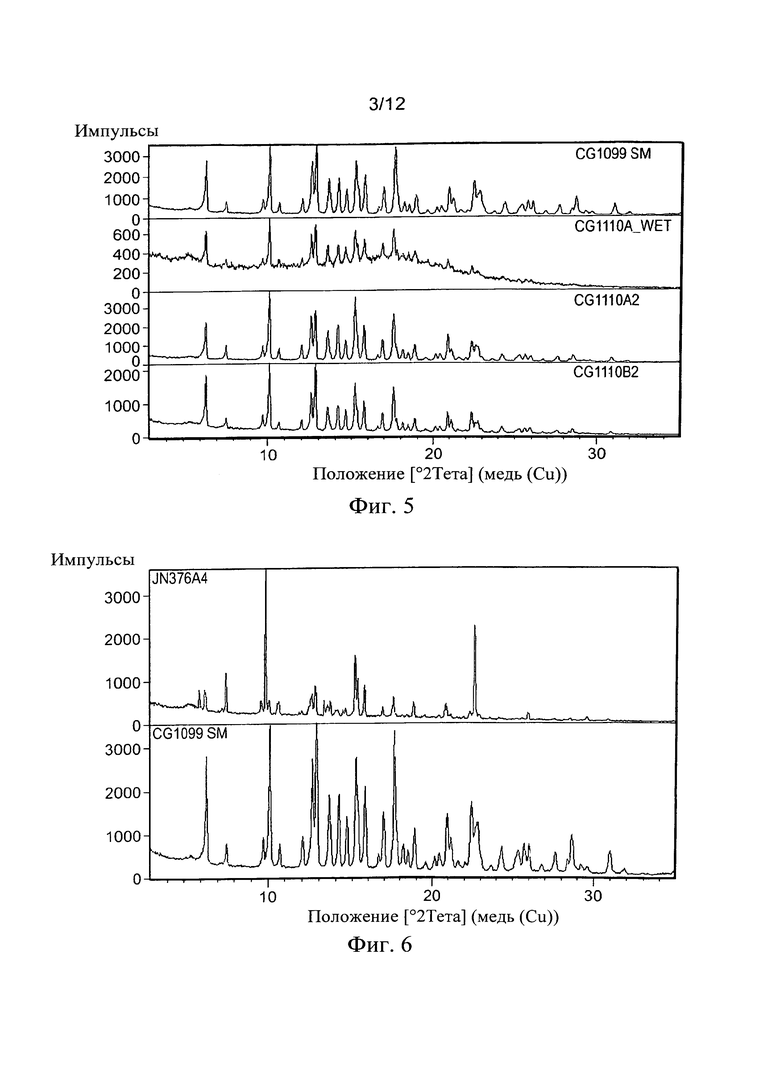
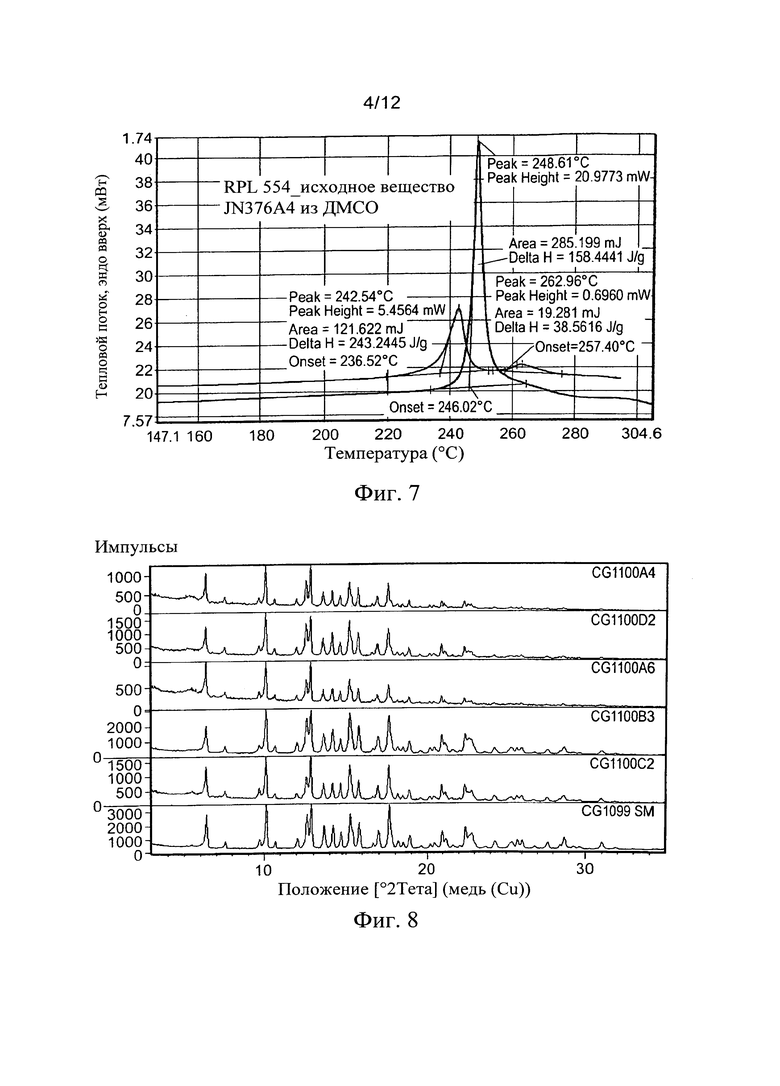

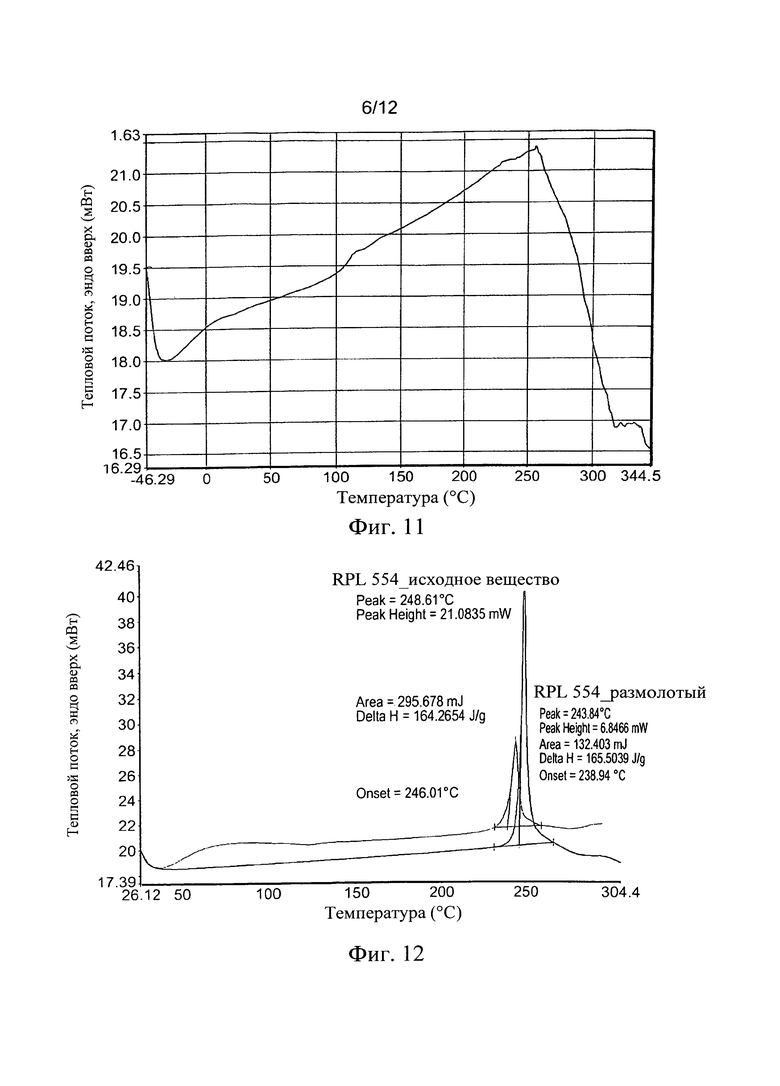
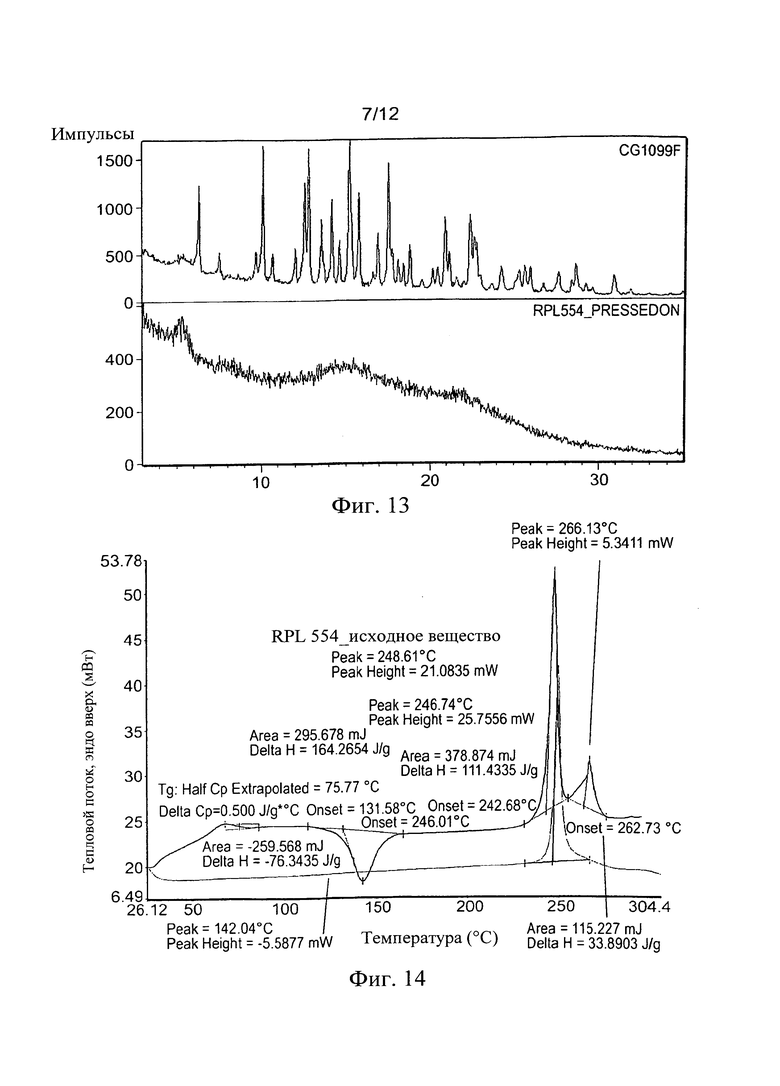
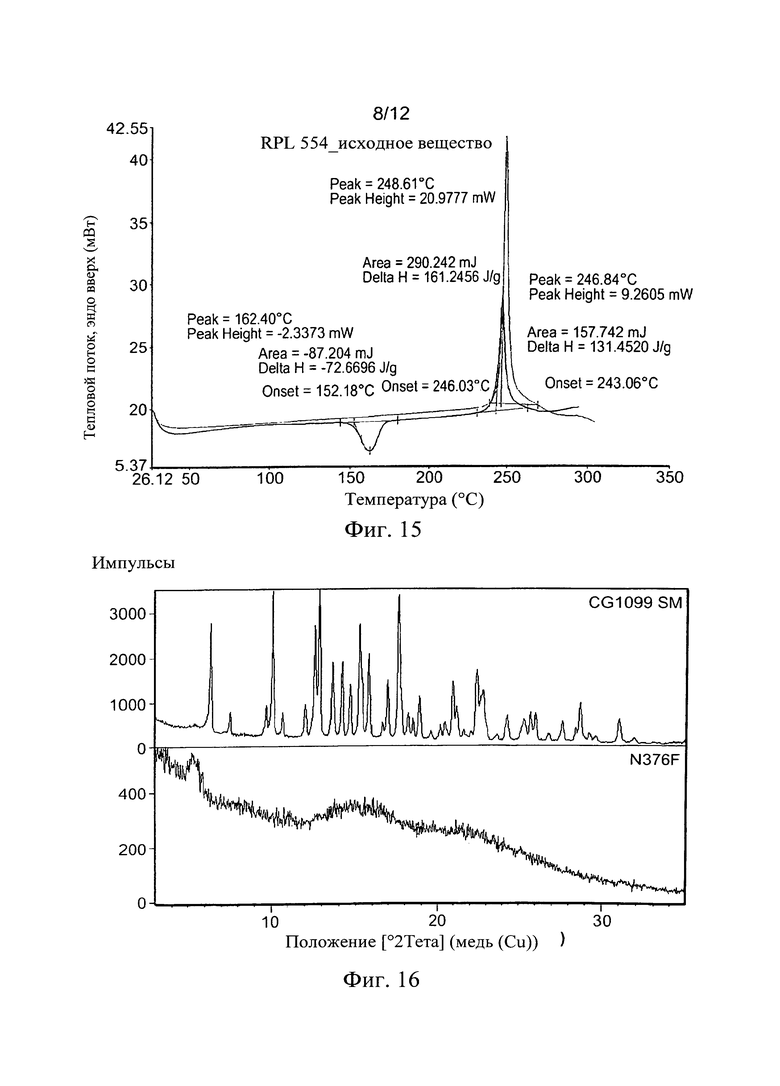
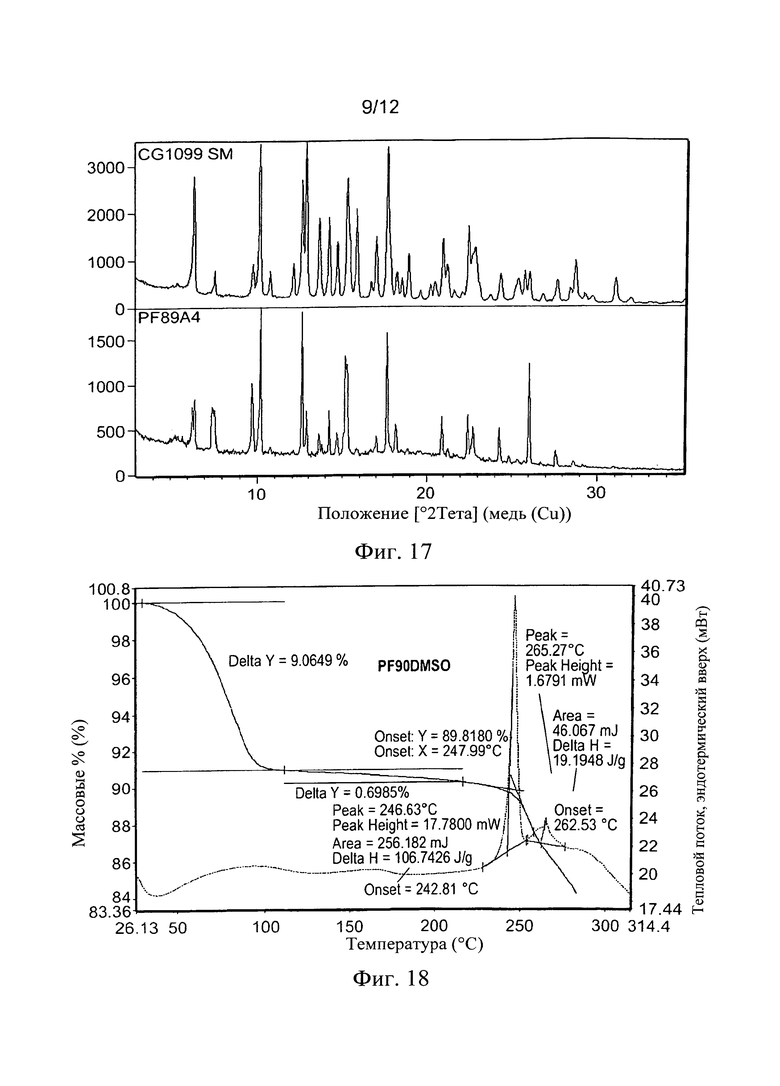
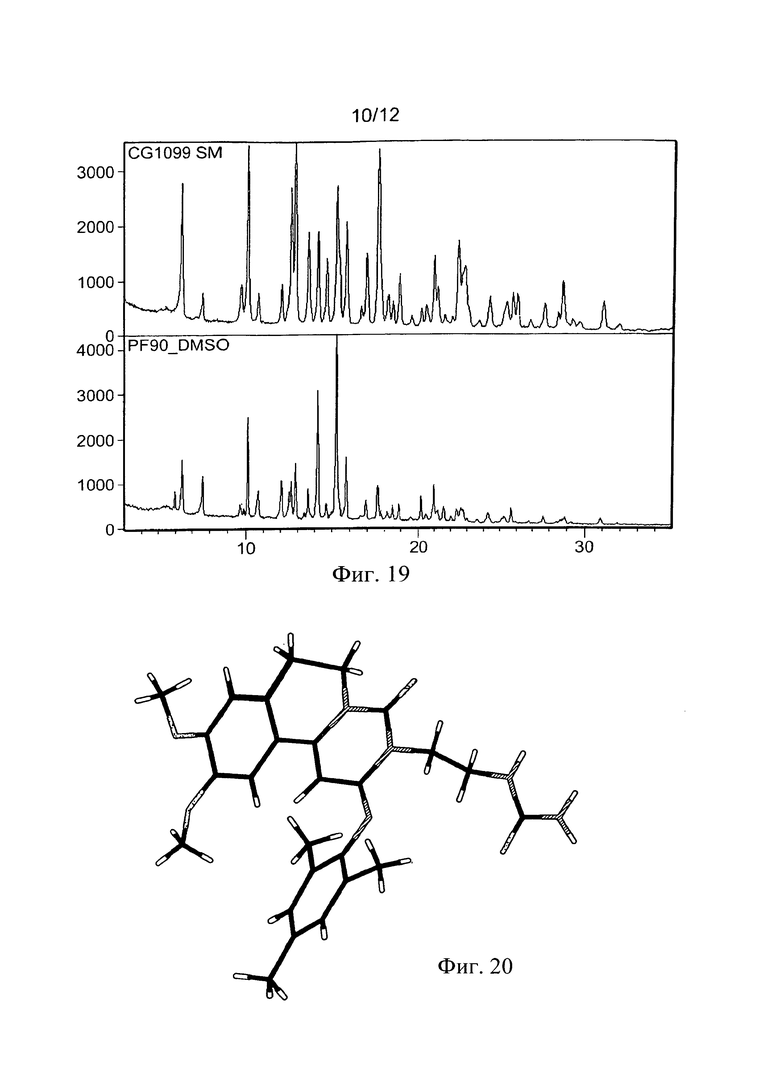
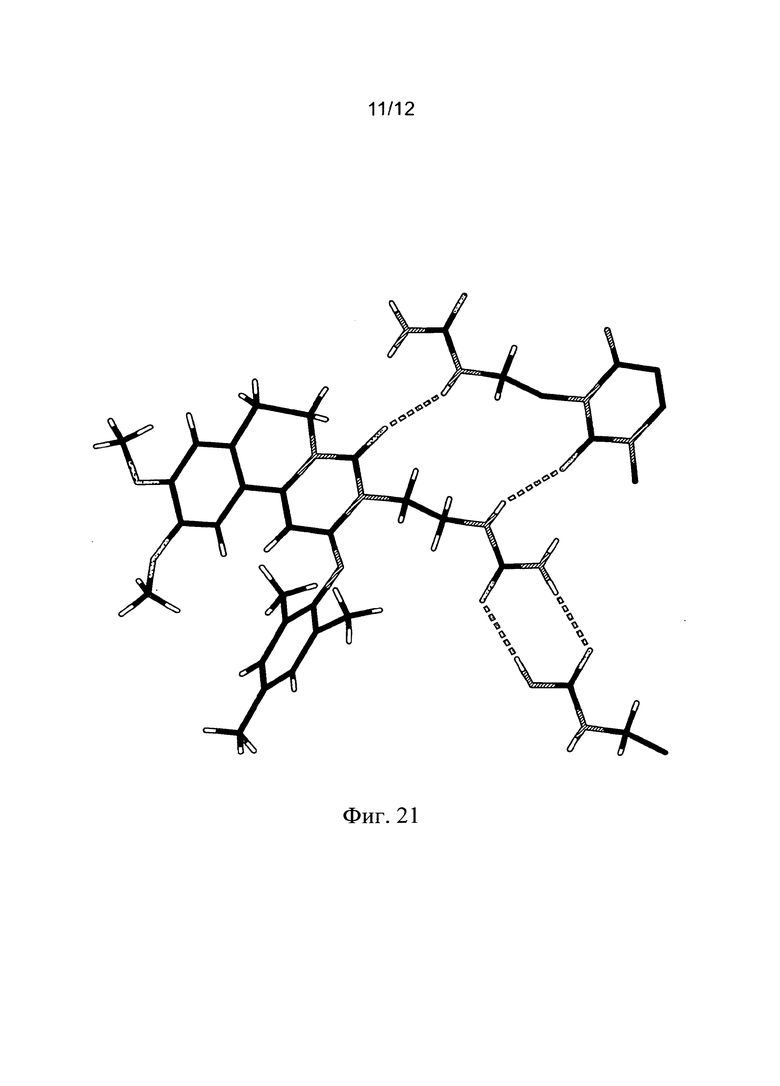

Настоящее изобретение относится к новому кристаллическому полиморфу N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины. Указанное соединение может быть использовано при лечении астмы, аллергической астмы, бронхита, хронической обструктивной болезни легких (ХОБЛ) или респираторного дистресс-синдрома. Кристаллическая структура указанного полиморфа имеет дифрактограмму порошковой рентгеновской дифракции, содержащую характеристические пики в единицах 2θ, при 10,1°, 12,9°, 15,3° и 17,6°, и следующие структурные параметры, полученные посредством монокристального анализа 
При этом по меньшей мере 95% полиморфа находится в термодинамически стабильной полиморфной форме. Изобретение также относится к способу получения указанного кристаллического полиморфа. Способ включает (a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины с растворителем с образованием смеси, (b) нагревание смеси при температуре в интервале 45-121°С в течение времени, подходящего для образования указанного полиморфа, и медленное охлаждение до температуры в интервале 30-40°С, при условиях, подходящих для образования указанного полиморфа; и (c) фильтрование и высушивание. Способ дополнительно может включать стадию фильтрации смеси после стадии (а). В качестве растворителя может быть использованы ДМСО, этанол, метанол, изопропиловый спирт, гексаны, пентан, этилацетат, дихлорметан или хлороформ. Указанная смесь сохраняется при или выше температуры около 50°С в течение около 24-96 часов. 4 н. и 15 з.п. ф-лы, 22 ил., 15 табл., 13 пр.
1. Кристаллический полиморф (I) N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины, имеющий дифрактограмму порошковой рентгеновской дифракции, содержащую характеристические пики в единицах 2θ, при 10,1°, 12,9°, 15,3° и 17,6°,
где полиморф имеет следующие структурные параметры, полученные посредством монокристального анализа:
2. Полиморф по п. 1, в котором указанная рентгенограмма порошковой рентгеновской дифракции содержит по меньшей мере 5 характеристических пиков в единицах 2θ, выбранных из 6,4°, 10,1°, 12,6°, 12,9°, 13,6°, 14,2°, 14,7°, 15,3°, 15,4°, 15,8°, 17,0°, 17,6°, 18,9°, 20,9°, 22,4°, 22,8° и 28,7°.
3. Кристаллический полиморф по п. 1 или 2, имеющий кривую дифференциальной сканирующей калориметрии, показывающую максимум около 248°С.
4. Твердая фармацевтическая композиция, проявляющая свойства PDE ингибитора и содержащая терапевтически эффективное количество полиморфа по п. 1 и фармацевтически приемлемый носитель.
5. Композиция по п. 4, в которой по меньшей мере 50 мас.% общей N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины в указанной композиции присутствует в виде указанного полиморфа.
6. Композиция по п. 4, в которой по меньшей мере 70 мас.% общей N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины в указанной композиции присутствует в виде указанного полиморфа.
7. Композиция по п. 4, в которой по меньшей мере 90 мас.% общей N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины в указанной композиции присутствует в виде указанного полиморфа.
8. Композиция по п. 4, в которой по меньшей мере 97 мас.% общей N-{2-[(2E)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины в указанной композиции присутствует в виде указанного полиморфа.
9. Способ получения кристаллического полиморфа по п. 1, включающий:
(a) объединение N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины с растворителем с образованием смеси,
(b) нагревание при температуре в интервале 45-121°С в течение времени, подходящего для образования указанного полиморфа, и медленное охлаждение до температуры в интервале 30-40°С, при условиях, подходящих для образования указанного полиморфа; и
(c) фильтрование и высушивание.
10. Способ по п. 9, дополнительно включающий стадию фильтрации смеси после стадии (а).
11. Способ по п. 9 или 10, в котором растворителем является ДМСО, этанол, метанол, изопропиловый спирт, гексаны, пентан, этилацетат, дихлорметан или хлороформ.
12. Способ по п. 11, в котором растворителем является ДМСО или этанол.
13. Способ по п. 9, в котором указанная смесь сохраняется при или выше температуры около 50°С в течение около 24-96 часов.
14. Способ по п. 13, в котором указанная смесь сохраняется при или выше температуры около 50°С в течение около 72 часов.
15. Способ по п. 9, в котором указанная смесь сохраняется при или выше температуры около 55°С в течение около 24-96 часов.
16. Способ по п. 13, в котором указанная смесь сохраняется при или выше температуры около 55°С в течение около 72 часов.
17. Способ по п. 9, в котором указанная смесь высушивается в вакууме при температуре от 25 до 50°С.
18. Способ по п. 17, в котором указанная смесь высушивается в вакууме при 40°С.
19. Способ лечения астмы, аллергической астмы, бронхита, хронической обструктивной болезни легких (ХОБЛ) или респираторного дистресс-синдрома взрослых (РДСВ), опосредованных PDE активностью, где способ содержит введение эффективного количества кристаллического полиморфа N-{2-[(2Е)-2-(мезитилимино)-9,10-диметокси-4-оксо-6,7-дигидро-2Н-пиримидо[6,1-а]изохинолин-3(4Н)-ил]этил}мочевины по п. 1.
| Устройство для автоматического останова уточно-перемоточных и тому подобных головок с двумя и более веретенами в головке | 1939 |
|
SU58308A1 |
| BASNI; LAL; et al.,TREQUINSIN,A POTENT NEW ANTIHYPERTENSIVE VASODILATATOR IN THE SERIES OF 2-(ARYLIMINO)-3-ALKYL-9,10-DIMETHOXY-3,4,6,7-TETRAHYDRO-2H-PYRIMIDO(6,1-A)ISOQUINOLINE-4-ONES, Journal of Medicinal Chemistry, 19840101, Vol:11, Nr:27, Page(s):1470 - 1480 | |||
| US 7378424 B2,27.05.2008 | |||
| MINO R.CAIRA, Crystalline | |||

