Настоящее изобретение относится к способу получения карбоновых кислот с этиленовой ненасыщенностью или их эфиров, в частности, α, β ненасыщенных карбоновых кислот или их эфиров, более конкретно акриловых кислот или их эфиров, таких как (алк)акриловые кислоты или алкил(алк)акрилаты, в частности (мет)акриловая кислота или алкил(мет)акрилаты, конденсацией карбоновой кислоты или сложных эфиров с формальдегидом или его источником, таким как диметоксиметан, в присутствии катализаторов, в частности, но не исключительно, к способу получения (мет)акриловой кислоты или ее алкилэфиров, например, метилметакрилата, конденсацией пропионовой кислоты или ее алкилэфиров с формальдегидом или его источником, таким как диметоксиметан, в присутствии такой каталитической системы. Изобретение, в частности, относится к получению метакриловой кислоты (МАК) и метилметакрилата (ММА).
Такие кислоты или их эфиры могут быть получены взаимодействием алкановой кислоты (или сложного эфира) формулы R3-CH2-COOR4, где R3 и R4 каждый представляет собой независимо подходящий заместитель, известный в области акриловых соединений, такой как водород или алкил-группа, особенно, низшая алкил-группа, имеющая, например, 1-4 углеродных атома, с подходящим источником метилена, таким как формальдегид. Таким образом, например, метакриловая кислота или ее алкилэфиры, в частности, метилметакрилат, могут быть получены каталитической реакцией пропионовой кислоты или соответствующего алкилэфира, например, метилпропионата, с формальдегидом в качестве источника метилена в соответствии с последовательностью реакций 1:
R3-CH2-COOR4+HCHO→R3-CH(CH2OH)-COOR4
или
R3-CH(CH2OH)-COOR4→R3-C(:CH2)-COOR4+Н2О
(Последовательность 1)
Примером последовательности реакций 1 является последовательность реакций 2:
CH3-CH2-COOR4+HCHO→CH3-CH(CH2OH)-COOR4
CH3-CH(CH2OH)-COOR4→CH3-C(:CH2)-COOR4+Н2О
(Последовательность 2)
Другой последовательностью реакций является последовательность, которая использует ацеталь:
R3-CH2-COOR4+R'OCH2OR”→R3-C(:CH2)-COOR4+R'OH+R”OH
(Последовательность 3)
Теоретическим примером последовательности реакций 3 является последовательность реакций 4, которая использует диметоксиметан:
CH3-CH2-COOR4+CH3OCH2OCH3→CH3-C(:CH2)-COOR4+2CH3OH
(Последовательность 4)
Использование диметоксиметана, таким образом, теоретически дает безводную систему, которая позволяет избежать трудности последующего отделения воды и/или последующего гидролиза продукта. Кроме того, использование диметоксиметана позволяет избежать использования свободного формальдегида, но тем не менее действует в общем смысле как источник формальдегида. Отсутствие воды и свободного формальдегида может значительно упростить выделение ММА из потока продукта.
Однако, на практике последовательность 4 является проблематичной, поскольку метанол дегидратируется до простого диметилового эфира и воды. Кроме того, диметоксиметан разлагается в каталитических условиях до простого диметилового эфира и формальдегида. Любая вода, образовавшаяся в указанных реакциях, может гидролизовать исходное сложноэфирное питание или дать его соответствующую кислоту, что может быть нежелательным.
US 4560790 описывает получение α, β ненасыщенных карбоновых кислот или их эфиров конденсацией метилаль-(диметоксиметана) с карбоновой кислотой или ее эфиром с использованием катализатора общей формулы М1/М2/Р/О, где М1 представляет собой металл группы IIIb, предпочтительно, алюминий, и М2 представляет собой металл группы IVb, предпочтительно, кремний.
Как указано выше, известный способ получения ММА представляет собой каталитическую конверсию метилпропионата (МЕП) до ММА с использованием формальдегида. Подходящим катализатором для этого является цезиевый катализатор на носителе, например, диоксиде кремния.
US 4118588 рассматривает получение метилметакрилата и метакриловой кислоты при взаимодействии пропионовой кислоты или метилпропионата с диметоксиметаном в присутствии катализаторов на основе фосфатов и/или силикатов магния, кальция, алюминия, циркония, тория и/или титана, и также в присутствии 0-0,5 моль воды на 1 моль ацеталя. Предпочтительными фосфатами являются алюминий, цирконий, торий и титан. Катализаторы обычно содержат оксидный модификатор для улучшения каталитической активности. Фосфат магния не является типичным примером, и фосфат кальция не является типичным примером в отдельности, но являются примером, когда предусматривается оксидный модификатор. Результаты плохо сравниваются с другими фосфатами, в частности, с фосфатом алюминия.
В работе Gupta et al., The Beilstein Lournal of Organic Chemistry, 2009, 5, № 68 рассматривается конденсация Кневенагеля между ароматическими альдегидами и малононитрилом, этилцианоацетатом или малоновой кислотой с гидроксиапатитом, нанесенным на карбонат цезия, в воде. Однако конденсация с малоновой кислотой дает декарбоксилирование.
Гидроксиапатит кальция существует в ряде кристаллических форм. Кроме того, рассматриваются аморфные предшественники гидроксиапатита с соотношениями кальций:фосфор, которые подобны таким соотношениям для кристаллических форм. Они могут превратиться в кристаллический гидроксиапатит либо при физической, либо при химической обработке. Кристаллические формы обычно делятся на два типа: стержни и пластины, но кристаллические наносферы являются также известными. Указанные три кристаллические формы хорошо описаны в научной литературе. Типичные природные стержнеподобные и пластиноподобные кристаллические формы гидроксиапатита рассматриваются во многих документах, например, в работах:

Гидроксиапатит в стержнеподобной кристаллической форме может иметь такие структуры, как бантоподобные или цветкоподобные структуры (Chemical Physics Letters, 2004, 396 (Liu)).
Условия получения различных кристаллических форм гидроксиапатита кальция также хорошо подтверждены документами

Кроме того, конверсия наносфер в стержнеподобные и листоподобные структуры рассмотрена в работе Tao et al., J. Phys. Chem. B, 2007, 111, 13410.
В частности, способы получения стержней гидроксиапатита хорошо описаны в литературе. Стержни гидроксиапатита были успешно синтезированы с использованием гидротермального способа (Zhang et al., Journal of Crystal Growth, 2007, 308, 133-140), влажного химического способа (Materials Chemistry and Physics, 2004, 86, 69-73 (Liu et al.)), пиролиза с ультразвуковым распылением (Materials Science and Engineering A, 2007, 449-451, 821-824 (An et al.)) и способа золь-гель (Particuology, 2009, 7, 466 (Padmanabhan et al.)).
Большая часть интереса к природным кристаллическим формам гидроксиапатита относится к их использованию или применению в исследовании биомедицинских применений благодаря их подобности костям человека. Несколько исследований морфологических эффектов относятся к промышленным каталитическим применениям гидроксиапатита.
Кристаллические сферы или наносферы или аморфные фосфаты кальция с соотношениями кальций:фосфор, подобными кристаллическим гидроксиапатитам в форме сфер и наносфер, также хорошо описаны в литературе и обычно являются предпочтительными у изготовителей (Tao et al., J. Phys. Chem. B, 2007, 111, 13410). Иногда кристаллическая сердцевина капсулируется аморфной оболочкой с созданием сфер. Однако аморфные сферы могут образовываться первоначально с последующей последовательной кристаллизацией, как рассмотрено в работе Kandori et al., Polyhedron, 2009, 28, 3036. Каталитические применения гидрокси-апатита являются известными, но не указывают на кристалличность или конкретные кристаллические формы, рассмотренные здесь. Благодаря широкой доступности наносферических аморфных предшественников или кристаллических форм гидроксиапатита можно предположить, что каталитические применения относятся к указанным общей аморфной или наносферической форме, если не указано иное.
Неожиданно, теперь найдено, что дисперсные фосфаты металлов конкретной кристаллической формы являются заметно селективными катализаторами для получения α, β карбоновых кислот с этиленовой ненасыщенностью или их эфиров конденсацией соответствующей кислоты или эфира с источником метилена, таким как формальдегид или диметоксиметан, обеспечивая высокую селективность и получение низшего простого диметилового эфира (ДМЭ). В частности, катализаторы являются особенно подходящими для получения эфиров α, β карбоновых кислот с этиленовой ненасыщенностью, поскольку они дают мало воды в таких реакциях.
Согласно первому аспекту настоящего изобретения предусматривается способ получения карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, предпочтительно, α, β карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, способ, который содержит стадии взаимодействия формальдегида или подходящего его источника с карбоновой кислотой или ее эфиром в присутствии катализатора и, необязательно, в присутствии спирта, где катализатор содержит кристаллы фосфата металла группы II, имеющие стержне- или иглоподобную морфологию, или подходящий их источник.
Подходящие примеры фосфатов в соответствии с настоящим изобретением включают в себя гидроксиапатит, пирофосфат, гидроксифосфат, РО4 2- фосфат и их смеси, более предпочтительно, гидроксиапатит, пирофосфат и их смеси.
Под термином «подходящий их источник» в отношении кристаллов фосфата понимается, что морфология может быть образована на месте из источника фосфата в условиях реакции. Поэтому один фосфат может действовать как источник другого фосфата. Например, пирофосфаты группы II могут образовать гидроксиапатиты группы II в условиях реакции, и, таким образом, пирофосфат является подходящим источником гидроксиапатита.
Под термином «подходящий его источник» в отношении формальдегида понимается, либо что свободный формальдегид может быть образован на месте из источника в условиях реакции, либо что источник может действовать как эквивалент свободного формальдегида в условиях реакции, например, он может образовать такое же реакционное промежуточное соединение, как формальдегид, так что имеет место эквивалентная реакция.
Ссылки на стержнеподобную морфологию кристаллов фосфатов металлов являются самопоясняющими для специалиста в данной области техники, но в сомнительном случае может быть указан кристалл с предпочтительным ростом в одном ключевом направлении (ось z) и значительно меньшим ростом во втором и третьем направлении (оси x и y). Более конкретно, стержнеподобный кристалл имеет длину, ширину и толщину. Ось z может быть определена как длина. Оси x и y могут быть определены взаимозаменяемо как ширина и толщина. Отношение толщины к ширине может быть неодинаковым. Альтернативно, отношение ширина:толщина может быть по существу равным, например, оно может составлять от 1:2 до 2:1, более типично, от 2:3 до 3:2, и, наиболее типично, от 3:4 до 4:3. В любом случае толщина и ширина являются меньше длины; где соотношение главных размеров длина (ось z): толщина и/или ширина (оси x и y) обычно является >2, более обычно >5, наиболее обычно >10.
Стержнеподобная форма, как определено в настоящем изобретении, предназначена охватывать любой кристалл, который имеет вышеуказанные размеры и поэтому имеет свойство кристалла или внешний вид, макроскопически или микроскопически, быть в форме удлиненного элемента со сходством со стержнем. Поэтому стержнеподобная форма охватывает любую из официальных форм кристалла, способную к особенности стержнеподобного кристалла, т.е. гексагональную, орторомбическую, тетрагональную, моноклиническую, триклиническую или кубическую. Предпочтительно, кристаллическая форма стержнеподобных кристаллов настоящего изобретения является гексагональной.
Предпочтительно, металл группы II фосфата изобретения может быть смесью металлов группы II, но, предпочтительно, выбран из Ca, Sr или Ва или их смесей, более предпочтительно, Ca или Sr, особенно, Са. Особенно предпочтительными катализаторами являются пирофосфат стронция, гидроксиапатит стронция, гидроксиапатит бария и гидроксиапатит кальция, которые имеют стержнеподобную морфологию в их кристаллической форме, более предпочтительными являются гидроксиапатит стронция, гидроксиапатит бария и гидроксиапатит кальция, наиболее предпочтительными являются гидроксиапатит стронция и гидроксиапатит кальция. Металл группы II магний более обычно используется в качестве легирующего металла с одним или более из Ca, Sr или Ва в фосфатах настоящего изобретения.
Предпочтительно, катализатор представляет собой, по меньшей мере, 50% мас./мас. фосфата металла, более предпочтительно, по меньшей мере, 70% мас./мас. фосфата металла, наиболее предпочтительно, по меньшей мере, 80% мас./мас. фосфата металла. Фосфат металла имеет значительную кристаллическую фракцию фосфата металла, но может также содержать аморфный материал. Известные кристаллические формы фосфатов металлов являются стержне-/иглоподобными, пластиноподобными или кристаллическими сферами. Авторами настоящего изобретения неожиданно было установлено, что кристаллические фосфаты металлов с, по меньшей мере, частью стержне-/иглоподобных кристаллов имеют неожиданно высокую селективность в настоящем изобретении.
Кристаллическая млорфология кристаллического фосфата металла может быть определена методами, известными специалистам в данной области техники, например, трансмиссионной электронной спектроскопией (ТЭМ) или сканирующей электронной спектроскопией (СЭМ) или по относительной интенсивности ДРЛ-пиков по сравнению с известными морфологическими вариантами кристаллических фосфатов металлов. Предпочтительно, стержне-/иглоподобные кристаллы являются в среднем численно доминирующей кристаллической формой в фосфате. Предпочтительно, стержне-/иглоподобные кристаллы являются в среднем численно доминирующей кристаллической формой по количеству средней поверхности ТЭМ-изображения, охватываемой в фосфате. Под доминирующей понимается, что кристаллическая форма является самой большой группой кристаллов. Однако, нет необходимости, чтобы стержне- или иглоподобная морфология была доминирующей кристаллической формой, чтобы изобретение было эффективным. Даже фосфат металла с меньшей частью кристаллов в стержне- или иглоподобной форме будет еще эффективным в качестве катализатора. Соответственно, кристаллы фосфата металла группы II, имеющие стержне- или иглоподобную морфологию, или подходящий их источник должны только присутствовать или стать присутствующими на уровне, который является эффективным для катализирования реакции с достаточной селективностью, такой как селективности, определенные ниже.
Предпочтительно, селективность реакции для карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, предпочтительно, продукта α, β карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, особенно, продукта (алк)акриловой кислоты или алкил(алк)акрилата, составляет, по меньшей мере, 40% моль, более предпочтительно, по меньшей мере, 60% моль, наиболее предпочтительно, по меньшей мере, 70% моль, особенно, по меньшей мере, 80 или 90% моль Обычные селективности, как определено выше, находятся в интервале 45-100% моль, более предпочтительно, 65-100% моль, наиболее предпочтительно, 75-100% моль, особенно, 85 или 90-100% моль % моль может быть определен газовой хроматографией. Селективность основана на % моль общего продукта, превращенного из исходной карбоновой кислоты или ее эфира. Например, если 100 г метилпропионата взаимодействует с получением 90 г метилпропионата и 10 г продукта, производного от пропионата, 9 г которого представляют собой метилметакрилат, тогда реакция имеет селективность к метилметакрилату 90%, которая может быть преобразована в селективность в % моль с использованием соответствующей молекулярной массы для определения молей метилпропионата, превращенного в продукт и молей полученного метилметакрилата и расчета % моль метилметакрилата. Аналогично, такой же анализ может быть выполнен для других компонентов, таких как метакриловая кислота. Подходящим устройством газовой хроматографии является газохроматограф Shimadzu GC GC2010, оборудованный колонкой RTX1701 (поставляемой фирмой Thames Restec UK Ltd) и пламяионизационным детектором ((ПИД)(FID)).
Композиции питания реактора и образцы конденсированного потока, выходящего из реактора, могут все анализироваться газовой хроматографией. Подходящим устройством является газохроматограф Shimadzu GC, указанный выше. Для каждого анализа итоговая хроматограмма может быть получена с использованием компьютерной программы Shimadzu's “GC Solutions” с получением площади пика для отдельных компонентов. Ответные ПИД-факторы для отдельных компонентов, полученные с использованием стандартов, применяются для преобразования площадей пиков сначала в % мас. и затем в % моль определяемого материала в образце.
Водосодержание продукта каталитической реакции может быть определено титрованием Карла-Фишера (Mettler Toledo DL38 с зондом DM143-SC, Hydranal Working Medium K and Composite K).
Предпочтительно, стержнеподобные кристаллы находятся в достаточно открытом размещении с обеспечением доступа к их поверхностям с осуществлением достаточного катализа. В затвердевшей массе кристаллов площадь поверхности стержнеподобных кристаллов, доступная для катализа, может быть снижена, таким образом, снижая, хотя не удаляя каталитическую эффективность. Соответственно, фосфатные кристаллы изобретения являются, предпочтительно, достаточно неагломерированными или незатвердевшими.
Предпочтительно, по меньшей мере, 10% моль/моль общего фосфата металла в катализаторе находится в кристаллической форме, более предпочтительно, по меньшей мере, 30% моль/моль, наиболее предпочтительно, по меньшей мере, 50% моль/моль Обычно аморфный материал (или фракция кристаллической фазы) может быть оценена на основе результатов ДРЛ по уравнению:
Xc=(1 - v112/300)/I300,
где I300 - интенсивность (300) дифракционного пика, и
v112/300 - интенсивность впадины между дифракционными пиками (112) и (300); Хс - степень кристалличности.
Обычно размер кристалла кристаллов фосфата металла по оси z находится в интервале 0,01-104 нм, более предпочтительно, 0,1-104 нм, наиболее предпочтительно, 0,1-103 нм, т.е. кристаллы изобретения представляют собой обычно нанокристаллы. В частности, стержни имеют обычно ширину 0,001-103 нм, более предпочтительно, ширину 0,01-103 нм, наиболее предпочтительно, ширину или толщину 0,1-103 нм и, предпочтительно, имеют соотношение габаритных размеров, определенное здесь. В предпочтительных вариантах кристаллы фосфатов металлов по осям z и х или y находятся в соответствующих интервалах 1-5000 нм и 0,1-500 нм, более предпочтительно, 5-1000 нм и 0,5-100 нм, наиболее предпочтительно, 10-500 нм и 1-50 нм. Соответственно, в данном контексте морфология кристаллов изобретения может быть названа наностержнями.
Преимущественно использование металлфосфатного катализатора в способе изобретения также дает в результате неожиданно низкие уровни простого диметилового эфира в потоке продукта, когда композиция формальдегидного компонента выпаренного питания реактора основана на формальдегиде или диметоксиметане.
Также было установлено, что катализатор настоящего изобретения имеет увеличенную эффективность, когда поверхностный слой кристаллов истощается ниже оптимального соотношения М:Р для гидроксиапатита, т.е. ниже 1,67. Было установлено, что соотношение М:Р поверхности кристалла в интервале 1,35-1,55 является особенно эффективным. Под соотношением поверхности здесь понимается соотношение, как определено рентгеновской фотоэлектронной спектроскопией ((РФС)(XPS)). Однако было также установлено, что использование низкого соотношения М:Р предшественника может дать конечные кристаллы с увеличенным соотношением М:Р поверхности выше объема. Блочное соотношение М:Р кристалла в интервале 1-1,3 может дать увеличенное соотношение М:Р поверхности, соответственно, выше, чем соотношение, найденное в объеме. Поэтому может быть, что увеличенная каталитическая эффективность найдена как результат благоприятного поверхностного размещения фосфата металла. Обычно соотношение М:Р поверхности, в частности, соотношение для Са:Р, находится в интервале 1,30-1,55. Это может быть структура гидроксиапатита металла, обедненная металлом.
Возможно, что особенно предпочтительный гидроксиапатит металла формулы M9(PO4)5OH(HPO4) является поэтому высоко каталитически активным с предпочтительным соотношение М:Р 1,5, где металлом является металл группы II, более предпочтительно, Ca, Sr или Ва, наиболее предпочтительно, Са или Sr, особенно, Са, или их смеси.
Общая формула гидроксиапатита металла ((ГАП)(НАР)) согласно настоящему изобретению может быть дана формулой I:
M10-x(PO4)6-x(OH)2-x(HPO4)x

в которой М представляет собой металл группы II, предпочтительно, Са, Sr или Ва или их смеси, более предпочтительно, Са или Sr или их смеси, и где x равен 0-1.
Общая формула пирофосфата металла согласно настоящему изобретению может быть дана как формула II:
M2P2O7
в которой М представляет собой металл группы II, предпочтительно, Са, Sr или Ва или их смеси, более предпочтительно, Са или Sr или их смеси.
Следует учесть, что мольное соотношение М:Р в чистом фосфате металла может варьироваться, например, около оптимального соотношения 5:3 для гидроксиапатита металла и 1:1 для пирофосфата металла. Гидроксиапатит металла обычно варьируется так, что является гидроксиапатитом с недостатком металла, тогда как пирофосфат металла может варьироваться так, что является обогащенным металлом. Мольное соотношение М:Р может варьироваться в интервале 0,8-1,8, но обычными интервалами М:Р поверхности являются 1,00-1,55, особенно, 1,10-1,50, более особенно, 1,20-1,50, как определено методом РФС, тогда как блочные мольные соотношения М:Р варьируются в интервале 0,8-1,8, более обычно, 1,00-1,70, как определено методом рентгеновской флуоресцентной спектрометрии ((РФлС)(XRF)). Подходящим прибором для определения поверхностных соотношений М:Р методом РФС (XPS) является рентгеновский фотоэлектронный спектрометр Kratos “Axis Ultra”. спектрометрии ((РФлС)(XRF)). Подходящим прибором для определения объемных соотношений М:Р методом РФлС (XRF) является прибор Oxford Instruments X-Supreme 8000, который основан на рентгеновских флуоресцентных измерениях рассеяния энергии (EDXRF).
Варьирование соотношений М:Р в конечных кристаллах может быть достигнуто варьированием соотношений М:Р предшественника и/или в случае мокрого способа получения рН раствора и/или температуры раствора.
Обычно получение стержне- или иглоподобной морфологии изобретения достигается соответствующими способами, известными специалисту в данной области техники, как уже установлено выше.
Предпочтительный способ получения для получения гидроксиапатитных и пирофосфатных стержне- или иглоподобных кристаллов согласно настоящему изобретению использует простой мокрый способ комбинирования нитрата металла группы II и диаммонийводородфосфата в качестве предшественников металла и фосфора, соответственно, в водном растворе с образованием осадка. Комбинирование нитрата и фосфата обычно имеет место между 20 и 115°С. Значение рН суспензии в процессе получения, предпочтительно, поддерживается в интервале 4,5-13. Непрерывное перемешивание может поддерживать продукт в суспензии. После вызревания продукт, предпочтительно, сушат и прокаливают при различных температурах в интервале от 300 до 700°C. Если присутствует более одного металла группы II, или если присутствуют другие металлы, водорастворимая соль металла (предпочтительно, нитрат) может раствориться в том же самом растворе в качестве первого нитрата группы II.
Другие предпочтительные способы включают в себя водную перекристаллизацию при образовании кристаллов на поверхности подложки в тех же условиях температуры и рН, как в простом мокром способе выше. Также возможным является нагревание предшественников катализатора в водяном паре (пропаривание), например, при 120°C с водным раствором аммиака при рН 10, или даже в условиях реакции 100-400°C. Возможные реагенты для пропаривания с водным раствором аммиака включают в себя широкий ряд соединений фосфата кальция, предпочтительно, со стехиометрическим соотношением Са:Р 1≤х≤1,5, таких как дикальцийфосфатдигидрат ((ДКФД)(DCPD)) или трикальцийфосфат ((ТКФ)(TCP)).
Еще другие технологии включают в себя термолиз в печи при <700°C. Для получения термолизом физическую смесь термически нестабильных соединений кальция и фосфора (например, нитрат кальция, гидроксид кальция, диаммонийводородфосфат, фосфорная кислота) нагревают в потоке воздуха при температурах до 700°C.
Кристаллическая форма гидроксиапатита ((ГАП)(НАР)) может быть определена методом ТЭМ или ДРЛ. Предпочтительно, она определяется ТЭМ-исследованием и, необязательно, подтверждается методом ДРЛ. Отсутствие или присутствие кристалличности, предпочтительно, определяется методом ДРЛ. Подходящим прибором для ДРЛ-анализа является диффрактометр Siemens Bruker D5000 Diffractometer D6. Подходящим прибором для ТЭМ-анализа является трансмиссионный электронный микроскоп Philips CM12.
Кристаллический ГАП имеет характерные ДРЛ-пики при 2 θ° 25,9 (002), 31,9 (211), 32,3 (112) и 33,0 (3000), все ± 0,2 2 θ°.
Согласно второму аспекту настоящего изобретения предусматривается каталитическая система, содержащая кристаллический металлфосфатный катализатор и носитель катализатора, где фосфат металла имеет стержне-/иглоподобную морфологию.
Преимущественно стержне-/иглоподобная морфология обеспечивает неожиданно высокую селективность в отношении к продукту кислоты с этиленовой ненасыщенностью или ее эфира в катализированной реакции согласно первому аспекту настоящего изобретения.
Стержне-/иглоподобная морфология кристалла фосфатов металлов является самопояснительной для специалиста в данной области техники, но в сомнительном случае может быть указан кристалл с предпочтительным ростом вдоль z-оси. Более конкретно, стержне-/иглоподобный кристалл имеет длину, ширину и толщину, у которого соотношение ширины и толщины находится в интервале от 1:2 до 2:1, более обычно, в интервале от 2:3 до 3:2 и, наиболее обычно, в интервале от 3:4 до 4:3. В любом случае толщина и ширина будут всегда намного меньше длины; у которого соотношение габаритных размеров длина (z-ось):толщина и/или ширина (оси х и y) составляет обычно >2, более обычно >3, наиболее обычно >5, особенно >10.
Необязательно, каталитические характеристики и/или уровень стержне-/иглоподобной морфологии могут быть модифицированы изменениями условий каталитического синтеза, таких как рН, температура, давление, соотношение М:Р, и путем легирования другими элементами, особенно, металлами.
Обычно значение рН каталитического синтеза может составлять, 4-13, более обычно, 4,5-12, наиболее обычно, 5-11,5, особенно, 6,5-11,5.
Температура раствора мокрого синтеза не является особенно критической и может составлять от 0 до 150°C, обычно от 10 до 130°C, более обычно от 20 до 125°C.
Давление реакции также не является критическим, и катализатор может быть получен при сниженном или высоком давлении. Обычно, однако, катализатор синтезируют при или около атмосферного давления.
Подходящие легирующие элементы могут присутствовать в катализаторе на уровне до 20% моль металла М. Подходящими легирующими катионами металла являются Cs, K, Rb, Na, Li, Zn, Ti, Si, Ln, Ce, Eu, Mg (если не используется как металл группы II), Ba (если не используется как металл группы II), Pb, Cd, Ag, Co, Cu, Ni и Zr. Предпочтительными легирующими веществами являются щелочные металлы группы I и щелочноземельные металлы группы II из приведенного выше перечня, более предпочтительно, металлы группы I, особенно, Cs.
Легирующие катионы могут заменять Са, Sr и/или Ва в приведенных выше формулах.
Подходящие легирующие анионы могут присутствовать на уровне до 20% моль фосфата. Подходящими легирующими анионами являются карбонат, хлорид и фторид. Они могут быть предложены для частичной замены металла группы II или фосфора или гидроксида здесь, как присуще.
Предпочтительно, реагент карбоновая кислота или ее эфир настоящего изобретения имеют формулу R3-CH2-COOR4, где R4 представляет собой либо водород, либо алкил- группу, и R3 представляет собой либо водород, алкил- либо арил-группу.
Формальдегид и его источники
Подходящим источником формальдегида может быть соединение формулы I:
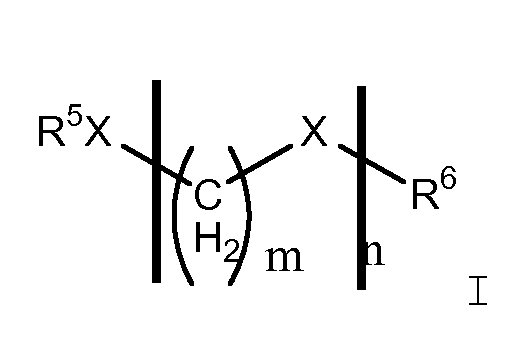
в которой R5 и R6 независимо выбраны из С1-С12 углеводородов или Н, Х представляет собой О, n представляет собой целое число от 1 до 100, и m равно 1.
Предпочтительно, R5 и R6 независимо выбраны из С1-С12 алкила, алкенила или арила, как определено здесь, или Н, более предпочтительно, С1-С10 алкила или Н, наиболее предпочтительно, С1-С6 алкила или Н, особенно, метила или Н. Предпочтительно, n представляет собой целое число от 1 до 10, более предпочтительно, от 1 до 5, особенно, 1-3.
Однако, могут быть использованы другие источники формальдегида, включая триоксан.
Поэтому подходящий источник формальдегида включает в себя любую равновесную композицию, которая может обеспечить источник формальдегида. Примеры источника формальдегида включают в себя (но не ограничиваясь этим) диметоксиметан, триоксан, полиоксиметилены R1-O-(CH2-O)i-R2, где R1 и/или R2 являются алкил-группами или водородом, i=1-100, пара-формальдегид, формалин (формальдегид, метанол, вода) и другие равновесные композиции, такие как смесь формальдегида, метанола и метилпропионата.
Обычно полиоксиметилены представляют собой высшие формали или полуформали формальдегида и метанола СН3-O-(CH2-O)i-СН3 («формаль-i») или СН3-O-(CH2-O)i-Н («полуформаль-i»), где i=1-100, предпочтительно, 1-5, особенно, 1-3, или другие полиоксиметилены с, по меньшей мере, одной неметильной концевой группой. Поэтому источником формальдегида также может быть полиоксиметилен формулы R31-O-(CH2-O)i-R32, где R31 и R32 могут быть одинаковыми или различными группами, и, по меньшей мере, один может быть выбран из С1-С10 алкил-группы, например, R31 представляет собой изобутил, и R32 представляет собой метил.
Предпочтительно, подходящий источник формальдегида выбран из диметоксиметана, высших полуформалей формальдегида и метанола СН3-O-(CH2-O)i-Н, где i=2, формалина или смеси, содержащей формальдегид, метанол и метилпропионат.
Особенно предпочтительно, что диметоксиметан может использоваться как источник формальдегида в настоящем изобретении. Преимущественно это обеспечивает возможность взаимодействия диметоксиметана с метилпропионатом с образованием ММА и метанола без получения воды. Это обеспечивает потенциально безводную систему, т.е. систему со сниженными требованиями к побочным реакциям с водой и отделению, чем при использовании других источников формальдегида, которые содержат или образуют воду. Кроме того, диметоксиметан является стабильным в отличие от других источников формальдегида, которые требуют воду и метанол, которые затем необходимо учитывать в последующих реакциях и отделении продукта. Другим преимуществом настоящего изобретения является низкий уровень разложения в настоящем изобретении диметоксиметана до простого диметилового эфира и формальдегида.
Предпочтительно, под термином «формалин» подразумевается смесь формальдегид:метанол:вода в соотношении (25-65% мас.):(0,01-25% мас.):(25-70% мас.). Более предпочтительно, под термином «формалин» подразумевается смесь формальдегид: метанол:вода в соотношении (30-60% мас.):(0,03-20% мас.):(35-60% мас.). Наиболее предпочтительно, под термином «формалин» подразумевается смесь формальдегид:метанол:вода в соотношении (35-55% мас.):(0,05-18% мас.):(42-53% мас.).
Предпочтительно, смесь, содержащая формальдегид, метанол и метилпропионат, содержит менее 5% мас. воды. Более предпочтительно, смесь, содержащая формальдегид, метанол и метилпропионат, содержит менее 1% мас. воды. Наиболее предпочтительно, смесь, содержащая формальдегид, метанол и метилпропионат, содержит 0,1-0,5% мас. воды.
Предпочтительно, кислота с этиленовой ненасыщенностью или ее эфир, получаемые способом настоящего изобретения, выбраны из метакриловой кислоты, акриловой кислоты, метилметакрилата, этилакрилата или бутилакрилата, более предпочтительно, им является сложный эфир с этиленовой ненасыщенностью, наиболее предпочтительно, метилметакрилат.
Способ настоящего изобретения является особенно подходящим для получения акриловой, алкакриловой, 2-бутеновой, циклогексеновой, малеиновой, итаконовой и фумаровой кислот и их алкилэфиров, а также метилензамещенных лактонов. Подходящими алкакриловыми кислотами и их эфирами являются (С0-8алк)акриловая кислота или алкил(С0-8алк)акрилаты, обычно от взаимодействия соответствующей алкановой кислоты или ее эфира с источником метилена, таким как формальдегид, в присутствии катализатора, предпочтительно, получение метакриловой кислоты или особенно метилметакрилата (ММА) из пропановой кислоты или метилпропионата, соответственно. Подходящие метилензамещенные лактоны включают в себя 2-метиленвалеролактон и 2-метиленбутиролактон из валеролактона и бутиролактона, соответственно.
Реакция настоящего изобретения может быть периодической или непрерывной реакцией.
Термин «алкил» при использовании здесь означает, если не определено иное, С1-С12 алкил и включает в себя группы; метил, этил, этенил, пропил, пропенил, бутил, бутенил, пентил, пентенил, гексил, гексенил и гептил, предпочтительно, алкил-группы выбраны из метила, этила, пропила, бутила, пентила и гексила, более предпочтительно, метила. Если не определено иное, алкил-группы могут, когда имеется достаточное число углеродных атомов, быть линейными или разветвленными, циклическими, ациклическими или частично циклическими/ациклическими, незамещенными, замещенными или оканчивающимися одним или более заместителей, выбранных из галогена, циано, нитро, -OR19, -OC(O)R20, -C(O)R21, -C(O)R22, -NR23R24 , -C(O)NR25R26, -SR29, -C(O)SR30, -C(S)NR27R28, незамещенного или замещенного арила или незамещенного или замещенного Het, где R19-R30 здесь, и обычно которые каждый независимо представляет собой водород, галоген, незамещенный или замещенный арил или незамещенный или замещенный алкил или в случае R21 - галоген, нитро, циано и амино, и/или прерываются одним или более атомов (предпочтительно, менее 4) ксилорода, серы, кремния или силано- или диалкилкремний-группами, или их смесями. Предпочтительно, алкил-группы являются незамещенными, предпочтительно, линейными и, предпочтительно, насыщенными.
Термин «алкенил» должен пониматься как «алкил», определенный выше, за исключением того, что, по меньшей мере, одна связь углерод-углерод здесь является ненасыщенной, и, соответственно, термин относится к С2-С12 алкенил-группам.
Термин «алк» или подобный должен (в отсутствие противоположной информации) приниматься в соответствии с вышеуказанным определением термина «алкил», за исключением того, что «С0 алк» означает незамещенный алкилом.
Термин «арил» при использовании здесь включает в себя пяти-десятизвенные, предпочтительно, пяти-восьмизвенные карбоциклические ароматические или псевдоароматические группы, такие как фенил, циклопентадиенил и инденил анионы и нафтил, которые группы могут быть незамещенными или замещенными одним или более заместителей, выбранных из незамещенных или замещенных арила, алкила (которая группа сама может быть незамещенной или замещенной или оканчиваться, как определено здесь), Het (которая группа сама может быть незамещенной или замещенной или оканчиваться, как определено здесь), галогена, циано, нитро, -OR19, -OC(O)R20, -C(O)R21, -C(O)R22, -NR23R24 , -C(O)NR25R26, -SR29, -C(O)SR30, -C(S)NR27R28, где R19-R30 каждый независимо представляет собой водород, незамещенный или замещенный арил или алкил (которая алкил-группа сама может быть незамещенной или замещенной или оканчиваться, как определено здесь), или в случае R21 - галоген, нитро, циано или амино.
Термин «галоген» при использовании здесь означает группу хлор, бром, иод или фтор, предпочтительно, хлор или фтор.
Термин “Het” при использовании здесь включает в себя от четырех- до двадцати-звенных, предпочтительно, четырех-десятизвенные кольцевые системы, которые кольца содержат один или более гетероатомов, выбранных из азота, кислорода, серы и их смесей, и которые кольца содержат нисколько, одну или более двойных связей или могут быть неароматическими, частично ароматическими или полностью ароматическими по характеру. Кольцевые системы могут быть моноциклическими, бициклическими или конденсированными. Каждая “Het” группа, идентифицированная здесь, может быть незамещенной или замещенной одним или более заместителей, выбранных из галогена, циано, нитро, оксо, алкила (которая алкил-группа сама может быть незамещенной или замещенной или оканчиваться, как определено здесь), -OR19, -OC(O)R20, -C(O)R21, -C(O)R22, -N(R23)R24 , -C(O)N(R25)R26, -SR29, -C(O)SR30, -C(S)N(R27)R28, где R19-R30 каждый независимо представляет собой водород, незамещенный или замещенный арил или алкил (которая алкил-группа сама может быть незамещенной или замещенной или оканчиваться, как определено здесь), или в случае R21 - галоген, нитро, амино или циано. Термин “Het”, таким образом, включает в себя такие группы, как необязательно замещенный азетидинил, пирролидинил, имидазолил, индолил, фуранил, оксазолил, изоксазолил, оксадиазолил, тиазолил, тиадиазолил, триазолил, оксатриазолил, тиатриазолил, пиридазинил, морфолинил, пиримидинил, пиразинил, хинолинил, изохинолинил, пиперидинил, пиразолил и пиперазинил. Замещение может быть по углеродному атому Het кольца или, когда соответствует, по одному или более гетероатомов.
“Het” группы могут быть также в форме оксида N.
Подходящие необязательные спирты в катализированной реакции настоящего изобретения могут быть выбраны из С1-С30 алканола, включая арил-спирты, которые могут быть, необязательно, замещены одним или более заместителей, выбранных из алкила, арила, Het, галогена, циано, нитро, -OR19, -OC(O)R20, -C(O)R21, -C(O)R22, -N(R23)R24 , -C(O)N(R25)R26, C(S)N(R27)R28, -SR29 или -C(O)SR30, как определено здесь. Высоко предпочтительными алканолами являются С1-С8 алканолы, такие как метанол, этанол, пропанол, изо-пропанол, изо-бутанол, трет-бутиловый спирт, фенол, н-бутанол и хлорокаприловый спирт, особенно, метанол. Хотя моноалканолы являются наиболее предпочтительными, также могут использоваться полиалканолы, предпочтительно, выбранные из ди-октаолов, таких как диолы, триолы, тетраолы и сахара. Обычно такие полиалканолы выбраны из 1,2-этандиола, 1,3-пропандиола, глицерина, 1,2,4-бутантриола, 2-(гидроксиметил)-1,3-прорандиола, 1,2,6-тригидроксигексана, пентаэритрита, 1,1,1-три(гидроксиметил)этана, наннозы, сорбозы, галактозы и других сахаров. Предпочтительные сахара включают в себя сахарозу, фруктозу и глюкозу. Особенно предпочтительными алканолами являются метанол и этанол. Наиболее предпочтительным алканолом является метанол. Количество спирта не является критическим. Обычно количества используются в избытке от количества этерифицируемого вещества. Таким образом, спирт может служить также как растворитель реакции, хотя, если требуется, могут также использоваться отдельные или дополнительные растворители.
Обычные условия температуры и давления в способе первого аспекта изобретения находятся в интервале от 100 до 400°C, более предпочтительно, 200-375°C, наиболее предпочтительно, 300-360°C; в интервале от 0,001 до 1 МПа, более предпочтительно, 0,03-0,5 МПа, наиболее предпочтительно, 0,03-0,3 МПа. Обычное время пребывания реагентов в присутствии катализатора составляет от 0,1 до 300 сек, более предпочтительно, 1-100 сек, наиболее предпочтительно, 2-30 сек, особенно, 3-20 сек.
Было установлено, что преимущественно использование катализатора настоящего изобретения дает заметно низкие уровни нежелательных побочных продуктов в реакции формальдегида или подходящего его источника с карбоновой кислотой или ее эфиром с получением карбоновой кислоты с этиленовой ненасыщенностью или ее эфира. В частности, заметно низкие уровни простого диметилового эфира (ДМЭ) получаются по сравнению с традиционными катализаторами, такими как фосфат алюминия. Кроме того, катализаторы обеспечивают превосходную селективность и активность.
Количество катализатора, используемого в способе настоящего изобретения не является обязательно критическим и определяется практическими вопросами способа, в котором он используется. Однако, количество катализатора обычно выбирается для осуществления оптимальной эффективности и выхода. Тем не менее специалист в данной области техники отметит, что минимальное количество катализатора должно быть достаточным для приведения примерно эффективной поверхности катализатора в контакт с реагентами во время контактирования. Кроме того, специалист в данной области техники заметит, что нереально установить верхний предел количества катализатора относительно реагентов, но что на практике это может регулироваться снова требуемым временем контактирования.
Относительное количество реагентов в способе изобретения может варьироваться в широких пределах, но обычно мольное отношение формальдегида или подходящего его источника к карбоновой кислоте или ее эфиру находится в интервале от 20:1 до 1:20, более предпочтительно, от 5:1 до 1:15. Наиболее предпочтительное соотношение зависит от формы формальдегида и способности катализатора высвобождать формальдегид из формальдегидных частиц. Так высоко реакционные формальдегидные вещества, у которых один или оба R31 и R32 в формуле R31-O-(CH2O)i-R32 представляют собой Н, требуют относительно низких соотношений, обычно, в данном случае мольное отношение формальдегида или подходящего его источника к карбоновой кислоте или ее эфиру находится в интервале от 1:1 до 1:9. Когда ни один из R31 и R32 не является Н, как, например, в СН3-СН2-ОСН3 или в триоксане, предпочтительными являются более высокие соотношения, обычно от 3:1 до 1:3.
Как указано выше, благодаря источнику формальдегида вода может также присутствовать в реакционной смеси. В зависимости от источника формальдегида может быть необходимо удалять часть или всю воду перед катализом. Поддержание более низких уровней воды, чем в источнике формальдегида, может быть предпочтительным для каталитической эффективности и/или последующей очистки продуктов. Предпочтительным является содержание воды в реакторе, по меньшей мере, 10% моль, более предпочтительно, менее 5% мол, наиболее предпочтительно, менее 2% моль
Мольное отношение спирта к кислоте или ее эфиру обычно находится в интервале от 20:1 до 1;20, предпочтительно, от 10:1 до 1:10, наиболее предпочтительно, от 5:1 до 1:5, например, 1:1. Однако, наиболее предпочтительное соотношение будет зависеть от количества воды, подаваемой к катализатору в реагентах, плюс количество, получаемое по реакции, так что предпочтительное мольное отношение спирта к общей воде в реакции будет составлять, по меньшей мере, 1:1 и, более предпочтительно, по меньшей мере, 3:1.
Реагенты могут подаваться в реактор независимо или после предварительного смешения, и способ реакции может быть непрерывным или периодическим. Предпочтительно, однако, используется непрерывный способ.
Обычно реакция имеет место в газовой фазе. Соответственно, подходящее конденсирующее оборудование обычно требуется для того, чтобы имела место конденсация потока продукта. Аналогично, может использоваться выпарной аппарат для доведения реагентов до температуры перед каталитическим слоем.
Предпочтительно, фосфат металла изобретения образует 50-100% мас. катализатора, более предпочтительно, 55-100% мас., наиболее предпочтительно. 60-100% мас., особенно, 70-100% мас., более особенно, 75-100% мас., наиболее особенно, 80-100% мас. Оставшуюся часть до 100% мас. катализатора составляют примеси, связующие или инертные материалы. Обычно фосфат металла образует примерно 80-90% мас. катализатора. Включенным в определение фосфата металла является фосфат с дефицитом металла, имеющий соотношения М:Р, определенные здесь.
Когда в настоящем изобретении используется связующее, оно может образовать до 50% мас. катализатора. Альтернативно связующее может использоваться вместе с носителем катализатора для связывания катализатора с носителем. В последнем случае связующее не образует часть катализатора как такового.
Подходящие связующие для катализатора настоящего изобретения являются известными специалистам в данной области техники. Неограничивающие примеры подходящих связующих включают в себя диоксид кремния (включая коллоидальный диоксид кремния), (диоксид кремния)-(оксид алюминия), такой как традиционный (диоксид кремния)-(оксид алюминия), оксид алюминия с покрытием диоксида кремния и диоксид кремния с покрытием оксида алюминия, и оксид алюминия, такой как (псевдо)боемит, гибсит, диоксид титана, оксид алюминия с покрытием диоксида титана, диоксид циркония, катионные глины и анионные глины, такие как сапонит, бентонит, каолин, сепиолит или гидротальцит, или их смеси. Предпочтительными связующими являются диоксид кремния, оксид алюминия и диоксид циркония или их смеси.
Частицы фосфата металла могут быть заделаны в связующее и наоборот. Обычно при использовании в качестве части катализатора связующее действует как адгезив для удержания частиц вместе. Предпочтительно, частицы гомогенно распределены в связующем или наоборот. Присутствие связующего обычно ведет к увеличению механической прочности конечного катализатора.
Обычная средняя площадь поверхности металлфосфатного катализатора находится в интервале 2-1000 м2г-1, более предпочтительно, 5-400 м2г-1, наиболее предпочтительно, 10-300 м2г-1, как определено многоточечным методом БЭТ с использованием анализатора площади поверхности и пористости Micrometritics TriStar 3000. Эталонным материалом, используемым для поверки характеристик прибора, является порошок углеродной сажи, поставляемый Micrometritics, с площадью поверхности 30,6 м2/г (±0,75 м2/г), номер части 004-16833-00.
Обычный средний размер частиц катализатора находится в интервале 1-10000 нм (10 мкм), более предпочтительно, 5-4000 нм (4 мкм), наиболее предпочтительно, 10-3000 нм (3 мкм), как измерено прибором Malvern Zetasiser Nano S c использованием светового рассеяния и с использованием стандартов НИНТ (NIST) (Национальный институт науки и техники).
Если материал является пористым, он является, предпочтительно, мезопористым со средним размером пор в интервале 2-50 нм. Размер пор может быть определен ртутной порометрией с использованием стандартов НИНТ.
Средний объем пор частиц катализатора может быть менее 0,01 см3/г, но обычно находится в интервале 0,01-5 см3/г, как измерено методом азотной адсорбции. Однако, микропористые катализаторы не являются наиболее предпочтительными, потому что они могут ингибировать движение реагентов через катализатор, и более предпочтительный объем пор находится в интервале 0,3-1,2 см3/г, как определено многоточечным методом БЭТ с использованием азотной адсорбции согласно ISO 12901-2:2006. Анализатор площади поверхности и пористости Micrometritics TriStar 3000 используется для определения объема пор, как в случае измерений площади поверхности, и используются те же стандарты.
В случае катализатора без носителя фосфат металла может использоваться непосредственно в форме частиц катализатора, либо движущихся свободно, либо вместе с подходящим связующим с созданием твердых тел желаемой формы и/или размера. Частицы могут быть любого подходящего размера и поэтому также в форме порошка, гранул или шариков либо с, либо без связующего. Обычно катализатор используется в форме неподвижного слоя и для этой цели может использоваться в отдельности или на носителе и в последнем случае может содержать подходящее каталитическое связующее для соединения с носителем.
Как указано выше, катализатор может использоваться на носителе. В данном случае металлфосфатный катализатор может образовать подходящее поверхностное покрытие на подходящем носителе для катализатора.
Для целей настоящего изобретения носитель не образует часть катализатора.
Фосфаты металла настоящего изобретения являются либо не нанесенными, либо нанесенными на подходящий носитель, например, оксид алюминия, диоксид кремния, нитрид кремния, карбид кремния, коллоидальный диоксид кремния, диоксид титана или фосфат алюминия.
Специалисту в данной области техники должно быть понятно, что катализатор изобретения может быть введен в носитель любым подходящим способом. Катализатор может быть фиксирован, предпочтительно, прокаливанием, на подходящем носителе после нанесения соединение на носитель с использованием подходящей соли в подходящем растворителе и последующей сушки носителя с покрытой поверхностью. Альтернативно, катализатор или подходящие солевые предшественники катализатора могут быть соосаждены с носителем или подходящими предшественниками носителя, такими как золь диоксида кремния из подходящего растворителя. Предпочтительно, используется оксидный носитель, более предпочтительно, оксидный носитель, как указано здесь.
Также можно использовать катализатор настоящего изобретения в смеси или предварительной смеси с другим катализатором согласно настоящему изобретению или иначе с или без подходящего связующего.
Обычно фосфат металла настоящего изобретения представляет собой нейтральную молекулу, и поэтому отрицательно заряженные фосфатные анионы, и, необязательно, гидроксид и любые другие неметаллы уравновешивают присутствующие положительно заряженные металлы.
Металлфосфатное соединение может быть нанесено на подходящий носитель, такой как диоксид кремния, нитрид кремния, карбид кремния, коллоидальный диоксид кремния, оксид алюминия, диоксид титана или фосфат алюминия. Носитель может быть или может не быть носителем, легированным металлом. Если носитель легирован щелочным металлом, легирующий агент щелочного металла может быть выбран из одного или более из цезия, калия, натрия или лития, предпочтительно, цезия или калия, более предпочтительно, цезия. Альтернативно, фосфат металла может быть сам легирован одним и более из вышеуказанных легирующих металлов.
Предпочтительно, когда используется отдельный носитель катализатора первого или второго аспекта, массовое соотношение катализатор:носитель находится в интервале от 10:1 до 1:50, более предпочтительно, от 1:1 до 1:20, наиболее предпочтительно, от 2:3 до 1:10.
Преимущественно селективность ненасыщенного сложного эфира увеличивается легирующими катионами, имеющими низкий заряд в радиальном отношении; так было установлено, что цезий является более селективным, чем литий. Поэтому предпочтительно, если используется, катионом легирующего металла является цезий, рубидий и/или калий, более предпочтительно, рубидий и/или цезий, наиболее предпочтительно, цезий.
Варианты изобретения теперь будут описаны со ссылкой на последующие неограничивающие примеры и фигуры и только путем иллюстрации, где:
на фигуре 1 показаны поверхностные и объемные соотношения М:Р для выбора образцов;
на фигуре 2 показано ТЭМ изображение кристаллов примера 1;
на фигуре 3 показано ТЭМ изображение сравнительного примера 4;
на фигуре 4 показано ТЭМ изображение кристаллов примера 3;
на фигуре 5 показано ТЭМ изображение кристаллов примера 6;
на фигуре 6 показано ТЭМ изображение кристаллов примера 8;
на фигуре 7 сравнивается морфология кристаллов методом ДРЛ для нескольких примеров и сравнительных примеров; и
на фигуре 8 показано ТЭМ изображение примера 11 на шкале 100 нм, показывающее присутствие наностержней.
Экспериментальная часть
Аналитические методы
Эксперименты с использованием метода ДРЛ
Образцы получают как сухие тонкопленочные образцы сухого спрессованного порошка, смонтированные на одиночных дисках кристаллов кремния. Используют следующие прибор и режимы.
Выходные данные находятся в форме дифрактограммы, показывающей интенсивность отражения (число сигналов в секунду) как функцию от угла 2θ. Идентификацию кристаллической фазы выполняют сравнением с эталонными ICDD (прежде JCPDS) дифрактограммами. Анализ интенсивности пиков или расширения пиков осуществляют с количественным определением морфологических параметров для кристаллической фазы.
Эксперименты методом РФлС (XRF)
Порошковые образцы измельчают и просеивают с получением размера частиц <100 мкм (меш). Приблизительно 1 г порошка слегка уплотняют в исходной чашке для образцов с тонкопленочным пропускающим основанием. Первичная чашка удерживается в приборе вторичной чашкой надежности также с тонкопленочным пропускающим основанием. Используют следующие прибор и условия.
(EDXRF)
(SDD)
Регистрируют интенсивности флуоресценции Са, Кα и Р Кα (число сигналов в секунду). Соотношение интенсивностей пиков было преобразовано с получением соотношения Са:Р для материала с использованием калибровочной шкалы, полученной от сигналов Са, Кα и Р Кα для стехиометрических эталонных материалов.
Эксперименты методом РФС (XPS)
Микрошпатель образца порошка помещают на кусок ленты без кремния, присоединенной к держателю образца прибора, и сыпучий порошок слабо разравнивают концом микрошпателя. Используют следующие прибор и режимы.
Созданную электронную спектроскопию для методов химического анализа (ESCA) используют для квалификации состава поверхности по элементарному атомному процентному содержанию. Глубина сигнала для оксидных материалов составляет приблизительно 3-5 нм, и предел определения составляет около 1 атом в 1000 (т.е. 0,1% ат. или 1000 ч./млн). Соотношения Са:Р сначала рассчитывают по экспериментальному атомному процентному содержанию и затем корректируют на присутствие поверхностных углеродистых частиц.
Эксперименты с методом ТЭМ
Образцы порошков материалов суспендируют в воде, и капли наносят на медные решетки, несущие Lacey углеродные пленочные подложки. После сушки их исследуют на ТЭМ Philips CM12 при ускоряющем напряжении 120 кВ.
Микрофотографии и электронные диффрактограммы собирают при соответствующих усилениях/длинах трубки. Выбранные участки анализируют с использованием связанной системы NORAN Vantage EDX. Наблюдаемый ряд морфологий, композиций и кристаллических частиц регистрируют как изображения. Используют следующие прибор и режимы.
Прибор Трансмиссионный электронный микроскоп Philips CM12
Ускоряющее напряжение 120 кВ
Группы экспериментов были выполнены по отношению к различным примерам изобретения и сравнительным примерам. Первая серия экспериментов была выполнена с использованием формальдегида в качестве потока питания, а вторая серия экспериментов была выполнена с использованием диметоксиметана в качестве потока питания. Анализ проводят с использованием газовой хроматографии, титрования формальдегида и с прибором Карла Фишера. Данные анализа используют для расчета выхода и селективности (ММА + МАК). Селективности в % моль по отношению к % моль (ММА + МАК) побочных продуктов диэтилкетона (ДЭК), простого диметилового эфира (ДМЭ) и толуола также представлены в виде таблицы ниже в результатах испытаний катализатора.
А. Формальдегидное питание

Пример 1
Пример получения 1
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 7 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4, растворенного в 50 мл деминерализованной воды при рН 7, добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 44 м2/г. Образец идентифицируют как гидроксиапатит кристаллического типа ДРЛ-анализом. Определяют наличие некоторого аморфного материала. ТЭМ подтверждает присутствие стержнеподобной кристаллической формы.
Испытание катализатора
3 г катализатора, как получено в примере получения 1, помещают в трубчатый реактор из нержавеющей стали, соединенный с выпарным аппаратом. Реактор нагревают до 350°C, а выпарной аппарат нагревают до 300°C. Смесь 56,2% моль метилпропионата, 33,7% моль метанола, 9,6% моль формальдегида и 0,5% моль воды пропускают через реактор с указанным временем контактирования. Конденсированную реакционную смесь анализируют газовой хроматографией с использованием Shimadzu GC, оборудованного колонкой DB1701 и пламяионизационным детектором. Для каждого анализа получаемую хроматограмму обрабатывают с использованием компьютерной программы Shimadzu c получением площадей пиков для отдельных компонентов. Факторы срабатывания ПИД для отдельных компонентов применяются для преобразования площадей пиков сначала в % мас. и затем в % моль определяемого материала в образце.
Селективность в отношении МАК или (МАК+ММА) рассчитывается по получаемому количеству компонента (выходное мольное содержание меньше мольного содержания питания) как процентное содержание мольного количества пропионата, превращенного в продукты.
Пример 2
Пример получения 2
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 9-10 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4, растворенного в 50 мл деминерализованной воды при рН 9-10, добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 9-10 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч.
Катализатор примера получения 2 испытывают, как описано в примере 1.
Пример 3
Пример получения 3
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 9-10 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4, растворенного в 50 мл деминерализованной воды при рН 9-10, добавляют по каплям к кипящему раствору нитрата кальция при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, затем фильтруют и промывают деминерализованной водой. После этого ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 9 м2/г. ДРЛ-анализом идентифицируют образец как монетит и пирофосфат. ТЭМ подтверждает присутствие пластино-, стержне-, листо- и сфероподобных кристаллических форм.
Катализатор испытывают, как описано в примере 1.
Пример 4
Пример получения 4
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в смеси 100 мл деминерализованной воды и 100 мл этанола. 7,9 г диаммонийводородфосфата (NH4)2HPO4, растворенного в 100 мл деминерализованной воды, добавляют по каплям к раствору нитрата кальция при температуре 25°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают до утра после завершения добавления по каплям, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 73 м2/г. ДРЛ-анализом идентифицируют образец как гидроксиапатит кристаллического типа. Определяют некоторое количество аморфного материала. ТЭМ подтверждает присутствие стержнеподобной кристаллической формы.
Катализатор испытывают, как описано в примере 1.
Пример 5
Пример получения 5
3 г катализатора, полученного как в примере получения 1, пропитывают 1% мас. цезия с использованием ацетата цезия в метаноле и испытывают, как описано в примере 1.
Сравнительный пример 1
Сравнительный пример получения 1
Катализатор синтезируют в соответствии со способом получения, рассмотренным в примере 4 патента US 4118588.
3 г диоксида титана TiO2 (номер 634662 Aldrich-каталога), 2,3 г фосфата алюминия (полученного как в сравнительном примере 2) и 0,75 г борной кислоты H3BO3 смешивают вместе. При введении 0,25 г мочевины в 5 мл деминерализованной воды получают пасту. Пасту сушат в течение 2 ч при 120°C и затем нагревают в течение 4 ч при 600°C.
Катализатор испытывают, как описано в примере 1. Наблюдается средняя селективность, но был определен высокий уровень ДМЭ.
Сравнительный пример 2
Сравнительный пример получения 2
37,5 г нонагидрата нитрата алюминия Al(NO3)3·9H2O и 13,2 г диаммонийводородфосфата (NH4)2HPO4 растворяют вместе в 160 мл деминерализованной воды, подкисленной азотной кислотой HNO3. Добавляют раствор гидроксида аммония до достижения рН 7. Образовавшийся гидрогель перемешивают в течение дополнительного 1 ч, после чего его фильтруют и промывают водой. Его сушат при 80°C до утра и затем прокаливают в воздушной среде при 600°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 181 м2/г.
Катализатор испытывают, как описано в примере 1. Наблюдается средняя селективность, но был определен высокий уровень ДМЭ.
Сравнительный пример 3
Используют коммерческий гидроксиапатит кальция от Aldrich с номером по каталогу 289396. ДРЛ-анализом подтверждается, что образец представляет собой гидроксиапатит кристаллического типа. Определяют некоторое содержание аморфного материала. ТЭМ показывает присутствие агломерированных неправильных сфероподобных частиц.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 1. Хотя селективность является средней, и уровень ДМЭ является низким, выход является очень низким, указывая на высокий уровень неактивности.
Сравнительный пример 4
Используют коммерческий гидроксиапатит кальция от Aldrich с номером по каталогу 677418.
ДРЛ-анализом подтверждается, что образец представляет собой гидроксиапатит кристаллического типа. ТЭМ показывает одинаковой формы наносферы обычно диаметра 50-100 нм (хотя с частью отдельных сфер диаметром 300-800 нм), с неочевидной любой несферической морфологией.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 1. Выход и селективность являются оба очень низкими.

Пример 6
Пример получения 6
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 11 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 11 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 96 м2/г. ДРЛ-анализом идентифицируют образец как гидроксиапатит кристаллического типа, хотя определяют некоторое количество аморфного материала. ТЭМ показывает высококристаллические наностержневые структуры, сгруппированные в пучки подобной ориентации.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 2.
Пример 7
Пример получения 7
14,2 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 7 гидроксидом аммония. 5,3 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 7 добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 64 м2/г. ДРЛ-анализом идентифицируют образец как гидроксиапатит кристаллического типа. Определяют некоторое количество аморфного материала.
Катализатор испытывают, как описано в примере 1.
Пример 8
Пример получения 8
14,2 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 7 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 7 добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. Площадь поверхности материала по методу БЭТ составляет 58 м2/г. ДРЛ-анализом идентифицируют главную фазу как гидроксиапатит кристаллического типа. Присутствует следовая фаза, подобная кальцийводородфосфату CaHPO4. Определяют некоторое количество аморфного материала. ТЭМ показывает присутствие стержне- и листоподобных кристаллических форм.
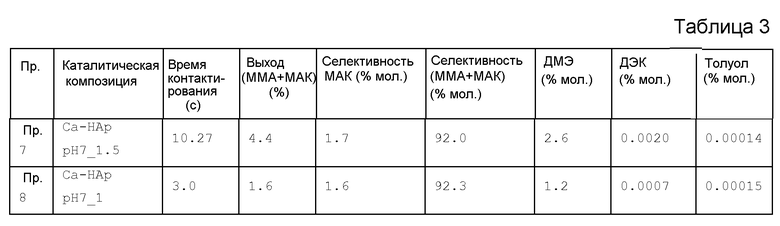
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 3.

Пример 9
Пример получения 9
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 7 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 7 добавляют по каплям к раствору нитрата кальция при температуре 25°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ТЭМ показывает короткие кристаллические наностержни длиной <100 нм и некоторое содержание аморфного материала. Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 4.
Пример 10
Пример получения 10
14,2 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 11 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 11 добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 11 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ДРЛ-анализом идентифицируют образец как гидроксиапатит кристаллического типа. Определяют некоторое количество аморфного материала. ТЭМ показывает плотно упакованные короткие кристаллические наностержни длиной <100 нм и диаметром примерно 10 нм. Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 4.
Пример 11
Пример получения 11
23,6 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 5 разбавленной водной азотной кислотой. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 5 добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 5 с помощью разбавленной водной азотной кислоты. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ТЭМ показывает крупные плоские структуры, ленто- или листоподобные, более 1 мкм в 2 их размерах. Края плоских структур являются разрушенными на параллельные наностержни с высоким соотношением габаритных размеров: более 100 нм в длину, но менее 20 нм в диаметре. ДРЛ-анализом идентифицируют образец как комбинацию фаз монетита СаНРО4 и пирофосфата Са2Р2О7, вероятно маскирующую лежащую ниже ГАП-фазу.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 4.
Пример 12
Пример получения 12
14,2 г тетрагидрата нитрата кальция Ca(NO3)2·4H2O растворяют в 100 мл деминерализованной воды, и рН корректируют до 5 разбавленной водной азотной кислотой. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 5 добавляют по каплям к раствору нитрата кальция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 5 с помощью разбавленной водной азотной кислоты. После этого суспензию фильтруют и промывают деминерализованной водой. Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ТЭМ показывает неоднородные частицы, преимущественно как листы, но также как стержни, окутанные аморфным материалом. ДРЛ-анализом идентифицируют присутствие пирофосфата Са2Р2О7. Также определяют наличие аморфного материала.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 4.

Пример 13
Пример получения 13
21,2 г нитрата стронция Sr(NO3)2 растворяют в 100 мл деминерализованной воды, и рН корректируют до 11 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 11 добавляют по каплям к раствору нитрата стронция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 11 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой.
Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ДРЛ-анализом идентифицируют образец как стронцийапатит кристаллического типа. ТЭМ-изображение показывает наностержни как единственную наблюдаемую морфологию, обычно длиной 100 нм и диаметром 20 нм.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 5.
Пример 14
Пример получения 14
19,0 г нитрата стронция Sr(NO3)2 растворяют в 100 мл деминерализованной воды, и рН корректируют до 11 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 11 добавляют по каплям к раствору нитрата стронция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 11 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой.
Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ДРЛ-анализом идентифицируют образец как стронцийапатит кристаллического типа. ТЭМ-изображение показывает плотно собранные в пучки наностержни обычно длиной 100 нм и диаметром 20 нм.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 5.
Пример 15
Пример получения 15
12,7 г нитрата стронция Sr(NO3)2 растворяют в 100 мл деминерализованной воды, и рН корректируют до 11 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 11 добавляют по каплям к раствору нитрата стронция при температуре 80°C при перемешивании. При добавлении фосфата к раствору нитрата образуется суспензия. Указанную маточную суспензию непрерывно перемешивают в течение 3 ч после завершения добавления по каплям, и рН поддерживается при 11 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой.
Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ДРЛ-анализом идентифицируют образец как стронцийапатит кристаллического типа. ТЭМ-изображение показывает пучки длинных наностержней, обычно длиной 100-500 нм и диаметром 10-20 нм.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 5.
В. Диметоксиметановое питание
Пример 16
Катализатор получают как в примере получения 1.
Испытание катализатора
3 г катализатора помещают в трубчатый реактор из нержавеющей стали, соединенный с выпарным аппаратом. Реактор нагревают до 350°C, а выпарной аппарат нагревают до 300°C. Смесь 70% моль метилпропионата и 30% мас. диметоксиметана пропускают через реактор. Конденсированную реакционную смесь анализируют газовым хроматографом, оборудованным CP-Sil 1701.
Пример 17
Катализатор получают как в примере получения 16.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 5
Катализатор получают как в сравнительном примере получения 1.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 6
Катализатор получают как в сравнительном примере получения 2.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 7
Сравнительный пример получения 7
3 г гидрата фосфата магния Mg3(PO4)2·xH2O (номер 344702 Aldrich-каталога) смешивают с 3 г фосфата алюминия (полученного как в сравнительном примере 2). Добавляют 5 мл деминерализованной воды с получением пасты. Пасту сушат в течение 2 ч при 120°C и затем нагревают в течение 4 ч при 600°C.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 8
Сравнительный пример получения 8
Катализатор синтезируют в соответствии со способом получения, рассмотренным в примере 3 патента US 4118588.
3 г диоксида титана TiO2 (номер 634662 Aldrich-каталога), 2,3 г фосфата кальция Са3(PO4)2 (номер 50552 Aldrich-каталога) и 0,75 г борной кислоты H3BO3 смешивают вместе. Добавляют 0,25 г мочевины в 5 мл деминерализованной воды с получением пасты. Пасту сушат в течение 12 ч при 120°C и затем нагревают в течение 3 ч при 580°C.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 9
Используют коммерческий гидроксиапатит кальция от Aldrich с номером каталога 289396.
ДРЛ-анализом подтверждают, что образец представляет собой гидроксиапатит кристаллического типа. ТЭМ показывает присутствие агломерированных неправильных сфероподобных частиц.
Определяют некоторое содержание аморфного материала.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 10
Используют коммерческий гидроксиапатит кальция от Aldrich с номером каталога 677418.
Площадь поверхности по методу БЭТ, определенная Aldrich, составляет 9,4 м2/г.
ДРЛ-анализом подтверждают, что образец представляет собой гидроксиапатит кристаллического типа. ТЭМ-анализ показывает присутствие сфероподобных кристаллов. Определяют некоторое содержание аморфного материала.
Катализатор испытывают, как описано в примере 16.
Сравнительный пример 11
Используют коммерческий Ca2P2O7 от Aldrich с номером каталога 693871.
Площадь поверхности по методу БЭТ, определенная Aldrich, составляет 12 м2/г.
ТЭМ показывает сфероподобные некристаллические частицы.
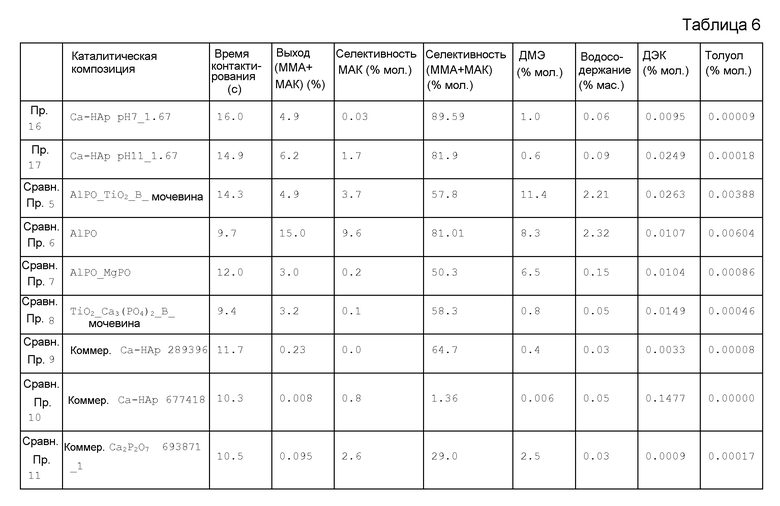
Катализатор испытывают, как описано в примере 16. Результаты представлены в таблице 6.

Пример 18
Катализатор получают как в примере получения 7.
Катализатор испытывают, как описано в примере 16. Результаты представлены в таблице 7.
Пример 19
Катализатор получают как в примере получения 8.
Катализатор испытывают, как описано в примере 16. Результаты представлены в таблице 7.
Пример 20
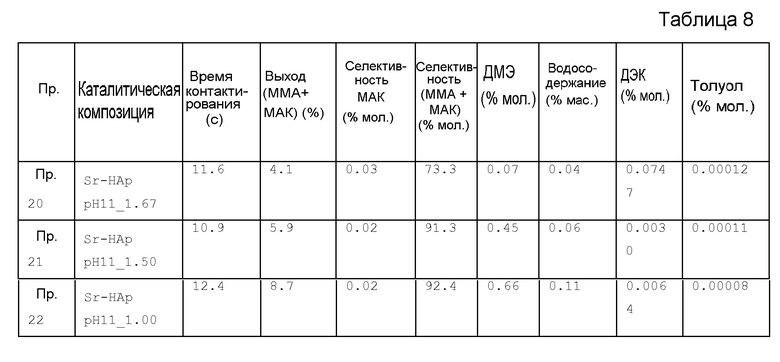
Катализатор примера получения 13 испытывают, как описано в примере 16. Результаты представлены в таблице 8.
Пример 21
Катализатор примера получения 14 испытывают, как описано в примере 16. Результаты представлены в таблице 8.
Пример 22
Катализатор примера получения 15 испытывают, как описано в примере 16. Результаты представлены в таблице 8.
Пример 23
Пример получения 23
21,2 г нитрата стронция Sr(NO3)2 растворяют в 100 мл деминерализованной воды, и рН корректируют до 7 гидроксидом аммония. 7,9 г диаммонийводородфосфата (NH4)2HPO4 в 50 мл деминерализованной воды при рН 7 добавляют по каплям к раствору нитрата стронция при температуре 80°C при перемешивании. Маточную суспензию непрерывно перемешивают в течение 3 ч, и рН поддерживается при 7 с помощью гидроксида аммония. После этого суспензию фильтруют и промывают деминерализованной водой.
Затем ее сушат до утра при 110°C и прокаливают в воздушной среде при 400°C в течение 1 ч. ДРЛ-анализом идентифицируют образец как пирофосфат стронция кристаллического типа. ТЭМ показывает крупные плоские структуры, лезвие- или листоподобные, обычно длиной 2-5 мкм и шириной 0,2-0,5 мкм. Плоские структуры окантованы пучками наностержневых структур с отдельными стержнями обычно диаметром 20 нм и длиной 200 нм.
Катализатор испытывают, как описано в примере 1. Результаты представлены в таблице 9.
Пример 24
Катализатор примера 23 испытывают с диметоксиметановым питанием, как описано в примере 16. Результаты представлены в таблице 9.

В таблице 10 показаны соотношения Са:Р синтеза различных примеров и сравнительных примеров, а также соотношения Са:Р в конечном кристалле (РФлС) и на поверхности кристалла (РФС). Сравнительный пример 12 представляет собой коммерческий пирофосфат в форме аморфных сфер, приобретенный от Aldrich с номером каталога 693871. Можно видеть, что идеальное соотношение гидроксиапатита 1,67 как объемного кристалла, так и поверхности кристалла является обедненным кальцием, но что поверхность является более обедненной. Однако при низких соотношениях М:Р синтеза, идеального для пирофосфатов 1:1, поверхность является богаче металлом, чем объем кристалла. Это предполагает образование предпочтительного поверхностного размещения на кристаллах. Поверхностное и объемное соотношения для ряда примеров представлены на фигуре 1. Можно видеть, что при высоких общих соотношениях поверхностное соотношение подавляется, и что при низких общих соотношениях поверхностное соотношение увеличивается.
Собирают ДРЛ данные по интенсивности пиков и сравнивают соотношения некоторых пиков для нескольких образцов. Результаты представлены на фигуре 7. Соотношение 002:211 для образцов изобретения может быть показателем сильного присутствия нано-стержней.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕН-НЕНАСЫЩЕННЫХ КАРБОНОВЫХ КИСЛОТ ИЛИ ИХ СЛОЖНЫХ ЭФИРОВ И КАТАЛИЗАТОР ДЛЯ ЭТОГО | 2013 |
|
RU2621687C2 |
| СИНТЕТИЧЕСКИЙ НАНОКРИСТАЛЛИЧЕСКИЙ ФОСФАТ КАЛЬЦИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2402482C2 |
| КОМПОЗИЦИЯ ДЛЯ УХОДА ЗА ПОЛОСТЬЮ РТА | 2013 |
|
RU2587054C1 |
| СОБСТВЕННО МАГНИТНЫЙ ГИДРОКСИАПАТИТ | 2011 |
|
RU2575566C2 |
| СОСТАВ ЗУБНОЙ ПАСТЫ | 2016 |
|
RU2686753C2 |
| КОЛЛАГЕНОВЫЙ МАТРИКС ИЛИ ГРАНУЛИРОВАННАЯ СМЕСЬ КОСТНОЗАМЕЩАЮЩЕГО МАТЕРИАЛА | 2020 |
|
RU2822395C2 |
| СРЕДСТВО ДЛЯ УХОДА ЗА ПОЛОСТЬЮ РТА И ПИЩЕВОЙ ПРОДУКТ ИЛИ НАПИТОК | 2016 |
|
RU2696495C2 |
| Способ получения композиционных биоматериалов хитозан/гидроксиапатит | 2020 |
|
RU2748799C1 |
| ИМПЛАНТАТЫ ДЛЯ ЗАМЕНЫ "НЕСУЩЕЙ НАГРУЗКУ" КОСТИ, ИМЕЮЩИЕ ИЕРАРХИЧЕСКИ ОРГАНИЗОВАННУЮ АРХИТЕКТУРУ, ПОЛУЧЕННЫЕ ПОСРЕДСТВОМ ПРЕВРАЩЕНИЯ РАСТИТЕЛЬНЫХ СТРУКТУР | 2011 |
|
RU2585958C2 |
| СПОСОБ ГИДРООКИСЛЕНИЯ ОЛЕФИНОВ ДО ОКСИДОВ ОЛЕФИНОВ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА НА ОСНОВЕ ОКИСЛЕННОГО ЗОЛОТА | 2000 |
|
RU2234369C2 |

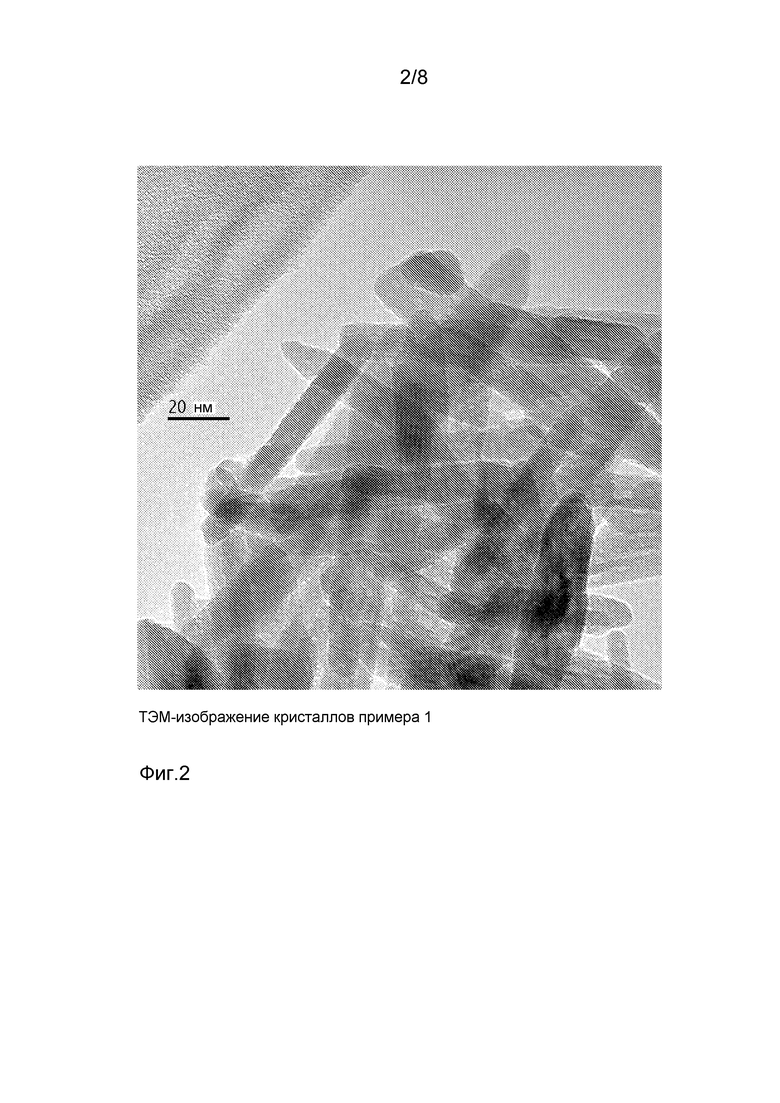



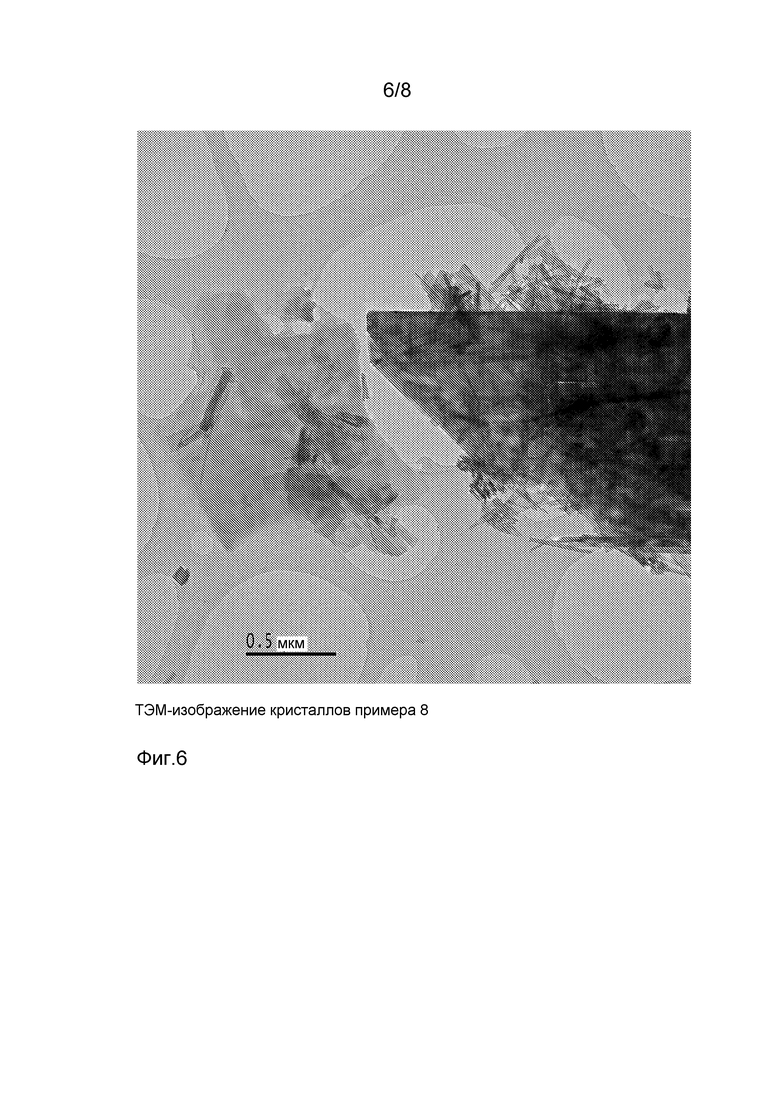

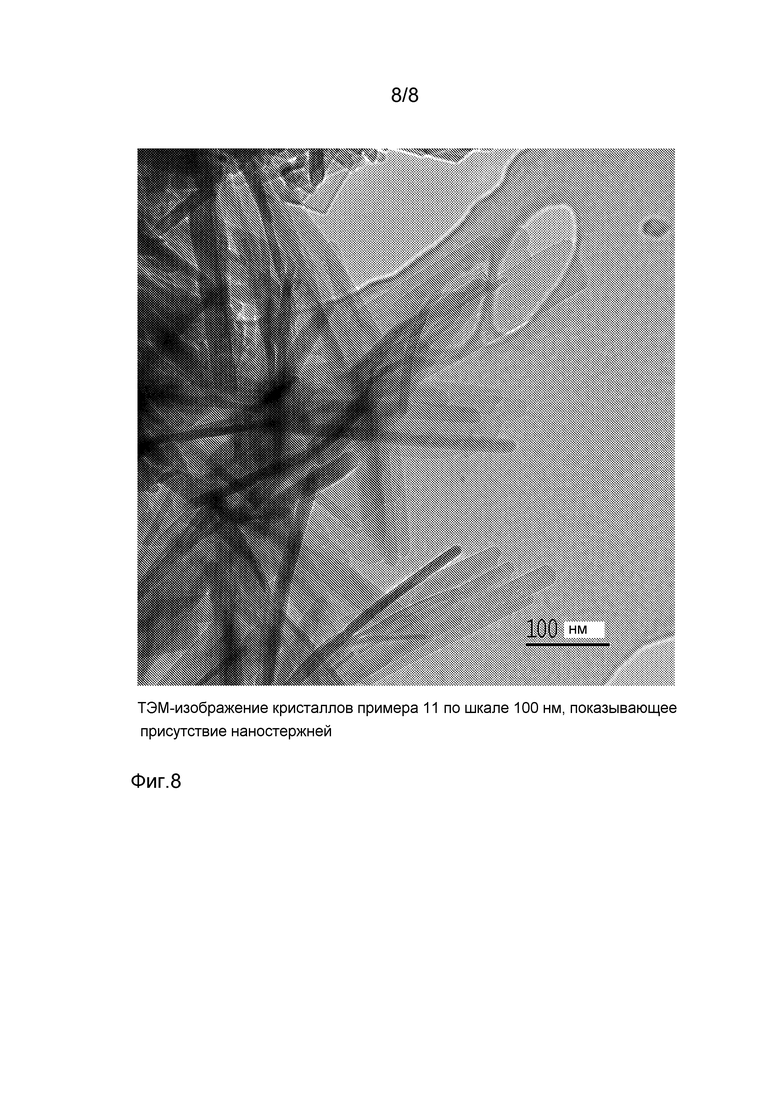
Изобретение относится к способу получения α, β карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, такой как метакриловая кислота или ее алкиловые эфиры, например, метилметакрилат. Способ включает стадии взаимодействия формальдегида или подходящего его источника с карбоновой кислотой или ее эфиром, например, пропионовой кислотой или ее алкиловыми эфирами, в присутствии катализатора и необязательно в присутствии спирта, выбранного из C1-C30 алканола, включая арил-спирты. Катализатор содержит кристаллы фосфата металла группы II, имеющие стержне- или иглоподобную морфологию. Фосфатом могут быть гидроксиапатит, пирофосфат, гидроксифосфат, РО4 2- фосфат или их смеси. Металл группы II может быть выбран из Ca, Sr, Ва или их смесей, например гидроксиапатит стронция и гидроксиапатит кальция. Также изобретение относится к каталитической системе, содержащей кристаллический металлфосфатный катализатор и носитель катализатора. Фосфат металла имеет стержне- или иглоподобную морфологию. Технический результат - высокая селективность продукта, 2 н. и 17 з.п. ф-лы, 10 табл., 24 пр.
1. Способ получения α, β карбоновой кислоты с этиленовой ненасыщенностью или ее эфира, включающий стадии взаимодействия формальдегида или его источника с карбоновой кислотой или ее эфиром в присутствии катализатора и необязательно в присутствии спирта, выбранного из C1-C30 алканола, включая арил-спирты, где катализатор содержит кристаллы фосфата металла группы II, имеющие стержне- или иглоподобную морфологию.
2. Способ по п. 1, в котором фосфат выбран из гидроксиапатита, пирофосфата, гидроксифосфата, PO4 2- фосфата и их смесей.
3. Способ по п. 1, в котором металл группы II фосфата выбран из Ca, Sr или Ва или их смесей.
4. Способ по пп. 1, 2 или 3, в котором катализаторы выбраны из гидроксиапатита стронция и гидроксиапатита кальция.
5. Способ по пп. 1, 2 или 3, в котором катализатор представляет собой, по меньшей мере, 50% мас./мас. фосфата металла.
6. Способ по пп. 1, 2 или 3, в котором селективность реакции к продукту α, β карбоновой кислоты с этиленовой ненасыщенностью или ее эфиру, особенно (алк)акриловой кислоты или алкил(алк)акрилата, составляет, по меньшей мере, 40% моль.
7. Способ по пп. 1, 2 или 3, в котором катализатор изобретения имеет свой поверхностный слой кристаллов, обедненный ниже соотношения (металл гидроксиапатита) : фосфат 1,67.
8. Способ по п. 7, в котором поверхностное соотношение металл : фосфат (М:Р) кристалла находится в интервале от 1,30 до 1,55.
9. Способ по пп. 1, 2 или 3, в котором реагент карбоновой кислоты или ее эфира настоящего изобретения имеет формулу R3-CH2-COOR4, где R4 представляет собой либо водород, либо алкил-группу, и R3 представляет собой либо водород, либо алкил- или арил-группу.
10. Способ по пп. 1, 2 или 3, в котором α, β карбоновая кислота с этиленовой ненасыщенностью или ее эфир, полученные способом изобретения, выбраны из акриловой, алкакриловой, 2-бутеновой, циклогексеновой, малеиновой, итаконовой и фумаровой кислот и их алкиловых эфиров, а также из метилензамещенных лактонов.
11. Способ по пп. 1, 2 или 3, в котором легирующие элементы присутствуют в катализаторе на уровне до 20% моль металла.
12. Способ по п. 11, в котором катионы легирующих металлов выбраны из Cs, K, Rb, Na, Li, Zn, Ti, Si, Ln, Ce, Eu, Mg (если не используется как металл группы II), Ва (если не используется как металл группы II), Pb, Cd, Ag, Co, Cu, Ni и Zr.
13. Способ по пп. 1, 2 или 3, в котором легирующие анионы присутствуют в катализаторе на уровне до 20% моль фосфата.
14. Способ по п. 13, в котором легирующие анионы выбраны из карбоната, хлорида и фторида.
15. Способ по пп. 1, 2 или 3, где катализатор нанесен на носитель, выбранный из оксида алюминия, диоксида кремния, нитрида кремния, карбида кремния, коллоидального диоксида кремния, диоксида титана или фосфата алюминия.
16. Способ по пп. 1, 2 или 3, в котором мольное соотношение М:Р составляет 0,8-1,8.
17. Способ по п. 2, в котором металл группы II фосфата выбирают из Ca, Sr или Ва, или их смеси.
18. Каталитическая система для взаимодействия формальдегида или его источника с карбоновой кислотой или ее эфиром, содержащая кристаллический фосфатный металла группы II катализатор и носитель катализатора, где фосфат металла группы II имеет стержне- или иглоподобную морфологию.
19. Каталитическая система по п. 18, где катализатор нанесен на носитель, выбранный из оксида алюминия, диоксида кремния, нитрида кремния, карбида кремния, коллоидального диоксида кремния, диоксида титана или фосфата алюминия.
| JP 7196314 A, 01.08.1995 | |||
| JP 10259012 A, 29.09.1998 | |||
| US 4118588 A, 03.10.1978 | |||
| Способ получения производных циклопропанкарбоновой кислоты в виде стереоизомеров или их смесей | 1981 |
|
SU1210661A3 |

