Область техники, к которой относится изобретение
Настоящее изобретение относится к способам синтеза и промежуточным соединениям в синтезе макроциклического ингибитора протеазы вируса гепатита C (HCV).
Уровень техники
Вирус гепатита C (HCV) является основной причиной хронического гепатита, который может прогрессировать в фиброз печени, приводя к циррозу, конечной стадии заболевания печени, и к печеночно-клеточному раку (HCC), делая его основным показанием к пересадке печени. К недостаткам существующей в настоящее время терапии HCV, основу которой составляет (пегилированный) интерферон-альфа (IFN-α) в сочетании с рибавирином, относятся ограниченная эффективность, значительные побочные эффекты и плохая переносимость многими пациентами. Это ускорило поиск более эффективной, удобной и лучше переносимой терапии.
Репликации генома HCV способствуют несколько ферментов, в том числе серинпротеаза NS3 вируса HCV и ее связанный кофактор NS4A. Описаны различные соединения, которые ингибируют данный фермент. Описаны линейные и макроциклические ингибиторы серинпротеазы NS3 с центральным замещенным пролиновым остатком (WO 05/073195) и с центральным циклопентильным остатком (WO 05/073216). Среди них макроциклические производные привлекают своей выраженной активностью против HCV и лучшим фармакокинетическим профилем.
В заявке WO 2007/014926 описаны макроциклические циклопентильные и пролиновые производные, включая соединение (I), структура которого представлена далее. Соединение (I) является очень эффективным ингибитором серинпротеазы HCV и особенно привлекает своим благоприятным фармакокинетическим профилем. Благодаря своим свойствам данное соединение выбрано потенциальным кандидатом для исследования как лекарственное средство против гепатита C. Соответственно существует необходимость производства больших количеств данного активного ингредиента на основании способов, которые обеспечивают высокий выход продукта и высокую степень его чистоты. В заявке WO 2008/092955 описаны способы и промежуточные соединения для получения соединения (I:

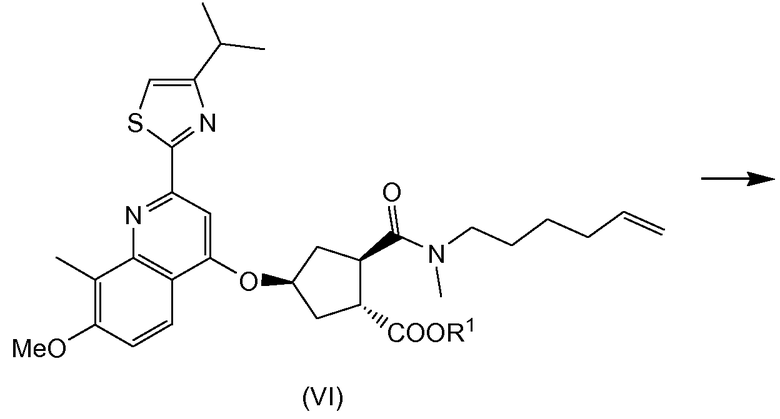
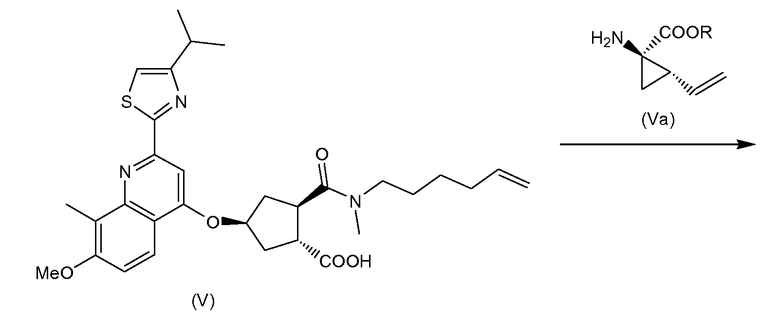
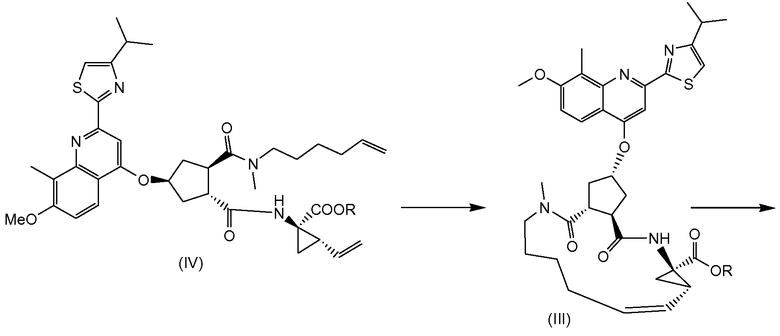
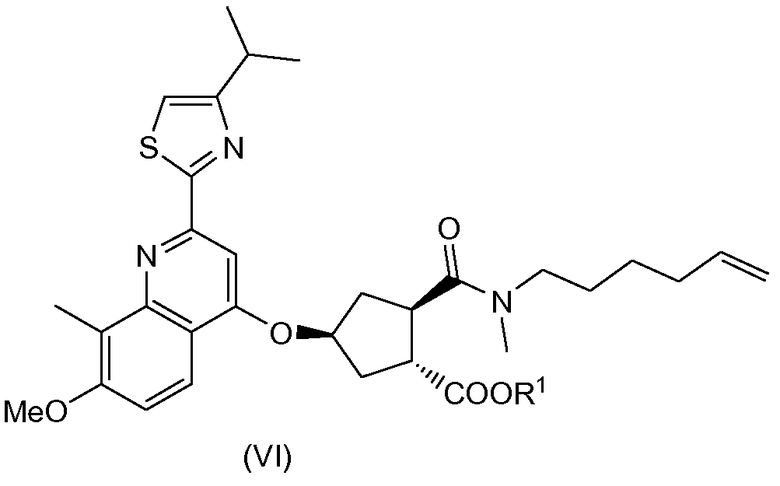
Соединение (I) можно получить исходя из промежуточного соединения (VI), из которого гидролизом сложноэфирной группы получают карбоновую кислоту (V), которая затем вступает в реакцию амидообразования с циклопропильной аминокислотой (Va). Полученное промежуточное соединение (IV) циклизуют по реакции олефинового обмена в присутствии подходящего металлического катализатора, например катализатора на основе илиденового производного рутения. Полученный макроциклический сложный эфир (III) затем гидролизуют в макроциклическую кислоту (II). Последняя вступает в реакцию амидообразования с сульфониламидом (V) с получением конечного продукта (I). Эти реакции описаны в приведенной ниже схеме. В данной и следующих схемах реакций или формулах индивидуальных соединений, R означает алкил C1-4, в частности R означает алкил C1-3, например R - алкил C1-2, или в одном варианте осуществления R - этил. R1 означает алкил C1-4, в частности R означает алкил C1-3, например R - алкил C1-2; или R1 - метил; или R1 - этил.

В свою очередь промежуточное соединение (VI) можно получить любым из способов, описанных в заявке WO 2008/092955, в частности исходя из гидроксициклопентилового бис-эфира (Xa), в том числе
(a) взаимодействием гидроксициклопентилового бис-эфира (Xa) и тиазолилзамещенного хинолинола (VII) в реакции образования простого эфира с получением хинолинилоксициклопентилового сложного бис-эфира (XII), в которой бензильноэфирную группу, которая находится в цис-положении к простой эфирной группе хинолинилоксициклопентилового сложного бис-эфира (XII), селективно отщепляют с образованием монокарбоновой кислоты (XI), которая затем вступает в реакцию амидообразования с алкенамином; таким образом, получают целевой конечный продукт (VI); или
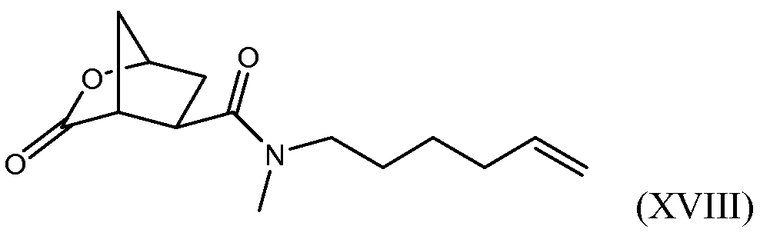
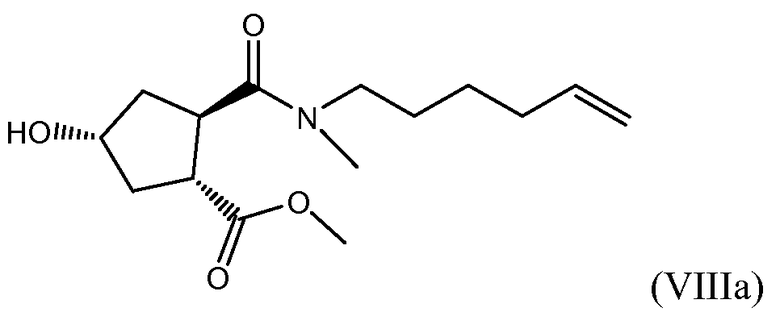
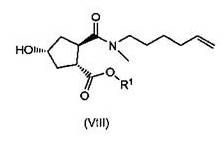
(b) селективной конверсией гидроксициклопентилового сложного бис-эфира (Xa) в монокарбоновую кислоту (IX), которая затем вступает в реакцию амидообразования с алкенамином; так получают гидроксициклопентиламид (VIII), который затем реагирует с тиазолилзамещенным хинолинолом (VII) и дает целевой конечный продукт (VI), как показано на следующей схеме реакции:
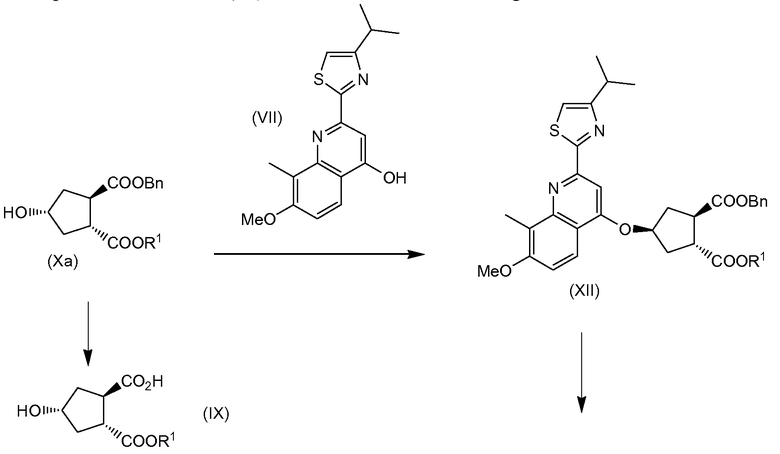
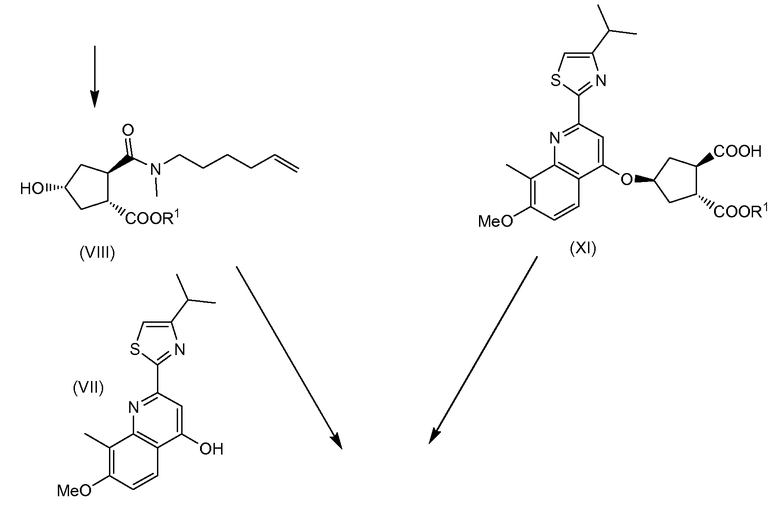
Каждый R1 в реакциях на данной схеме имеет значение, определенное выше, и предпочтительно R1 означает метил. Bn означает бензил.
Присутствие различных хиральных центров в соединении (I) и его предшественниках налагает определенные ограничения, потому что хиральная чистота важна для получения продукта, приемлемого для терапевтического применения. Промежуточное соединение (VI) имеет три хиральных центра, и получение правильной стереохимии всех трех центров налагает существенное ограничение на любые способы синтеза с целью получения данного соединения. Следовательно, способы получения (VI) должны давать продукты приемлемой хиральной чистоты без применения сложных процедур очистки, в которых теряются существенные количества нецелевых стереоизометрических форм.
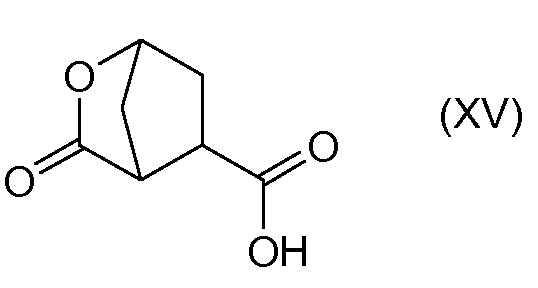
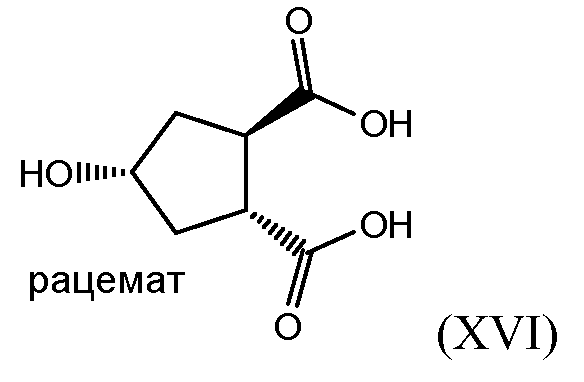
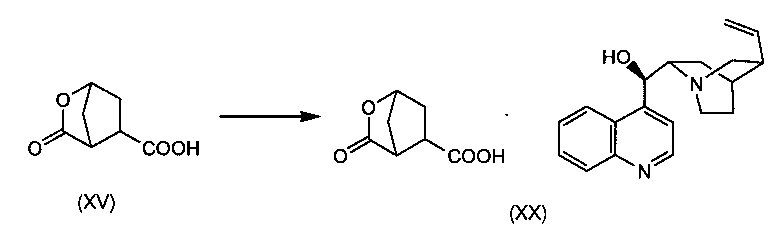
В заявке WO 2008/092955 описан способ синтеза промежуточного соединения (Xa) исходя из 4-оксоцикпопентил-1,2-бис-карбоновой кислоты (XVII) восстановлением кетогруппы в спирт с получением 4-гидроксициклопентил-1,2-бис-карбоновой кислоты (XVI), которую затем циклизуют в бициклический лактон (XV), при этом карбоксильную группу кислоты в бициклическом лактоне (XV) этерифицируют бензиловым спиртом с получением бензилового эфира лактона (XIV). В последнем лактоновый цикл раскрывают в реакции трансэтерификации в присутствии алканола C1-4 с получением гидроксициклопентилового бис-эфира (X), который затем разделяют на энантиомеры (Xa) и (Xb), как показано на следующей схеме реакции:

Каждый R1 в реакциях на данной схеме имеет значение, определенное выше, и предпочтительно R1 означает метил.
Недостаток приведенного выше способа состоит в том, что он включает разделение энантиомеров (X) хиральной колоночной хроматографией, и эту сложную процедуру трудно выполнять в крупномасштабном производстве.
Honda и др. (Tetrahedron Letters, vol. 22, no. 28, pp. 2679-2682, 1981) описали синтез (±)-брефельдина A с использованием следующих исходных веществ:
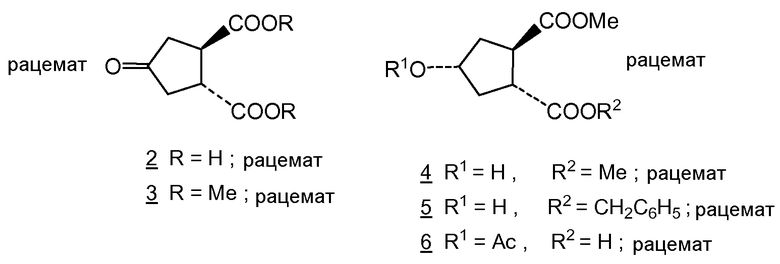
Honda и др. провели данный синтез исходя из dl-транс-4-оксоциклопентан-1,2-дикарбоновой кислоты 2, которую этерифицировали соответствующим метилэфиром 3 и восстанавливали на никеле Ренея в спирт 4. Частичным гидролизом 4 в монокарбоновую кислоту и бензилированием бензилбромидом получали преимущественно диастереоизомер 5, а именно диастереоизомер цис-положением гидроксильной и бензильноэфирной групп. Данный эфир в статье Honda и др. и соединение (X) представляют собой рацематы, но это взаимные диастереоизомеры, точнее эпимеры относительно атома углерода 4 при гидроксильной группе. Соединение (Xa) является одним из двух энантиомеров, получаемых разделением рацемического соединения (X). Другим энантиомером является соединение (Xb).
В заявке WO 2005/073195 описан синтез энантиомерически чистого бициклического лактона (8b) исходя из энантиомера 3,4-бис(метоксикарбонил)циклопентанона. Последний получали способом, который описали Rosenquist и др. (Acta Chemica Scandinavica 46 (1992) 1127-1129). Транс-изомер (3R,4R)-3,4-бис(метоксикарбонил)циклопентанона превращали в бициклический лактон (8b):
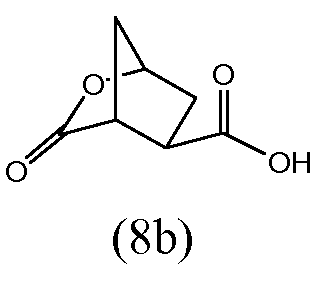
Кроме того, в заявке WO 2005/073195 описана дальнейшая модификация лактона (8b) в трет-бутиловый эфир при раскрытии лактонового цикла и реакции с надлежащим образом защищенными аминокислотами, например с этиловым эфиром (1R,2S)-1-амино-2-винилциклопропанкарбоновой кислоты, что в последнем примере дает:
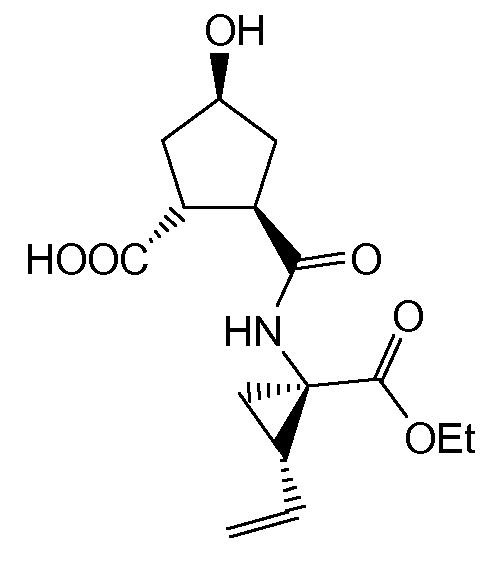
Синтез соединений (I) обязательно включает введение тиазолилзамещенного хинолинового остатка в циклопентильное кольцо посредством эфирной связи. Реакция Мицунобу представляет собой привлекательный способ получения ароматических алкилэфиров, в которых алкилэфир активируется и реагирует с фенолом. Кроме того, реакция Мицунобу, как правило, эффективнее, чем реакции O-арилирования, которые требуют дополнительные стадии синтеза. В этой мягкой реакции инвертируется стереохимия алкильной группы. В данной реакции образуются побочные продукты, например R'OOC-NH-NH-COOR', где R' - алкил C1-4, в частности этил или изопропил, другие азотсодержащие соединения и трифенилфосфиноксид, которые необходимо отделять от целевого конечного продукта.
Достоинством способов по настоящему изобретению является их применимость в крупномасштабном производстве. Исключаются сложные стадии очистки, в частности, с помощью хроматографии. В синтезе соединения (I) важно построение циклопентильной группы с правильной стереохимией ее трех хиральных центров.
Один из аспектов настоящего изобретения относится к способам получения промежуточных соединений (VIII) с высоким выходом и чистотой, особенно в отношении хиральной чистоты, которые пригодны для крупномасштабного промышленного применения.
Целью настоящего изобретения является предложение способов получения циклопентильных промежуточных соединений с правильной стереохимией, высоким выходом и чистой. В частности, настоящее изобретение относится к получению промежуточных соединений:
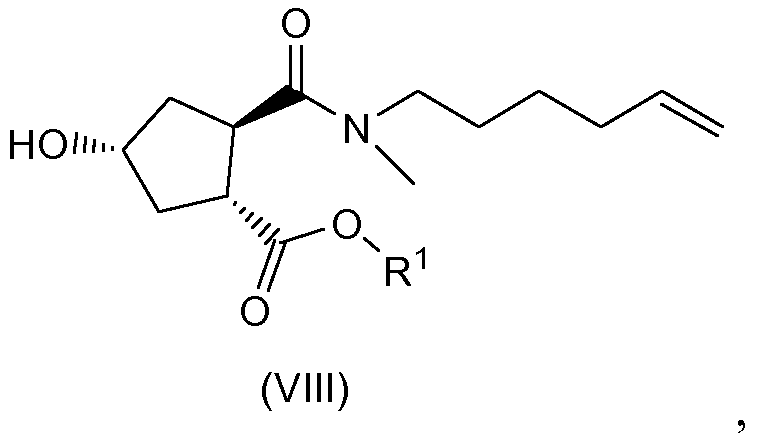
которые находят применение в получении соединения (I).
Описание изобретения
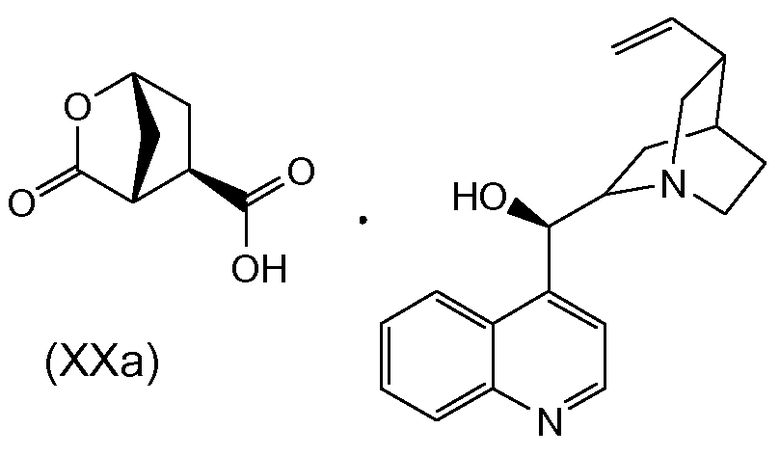
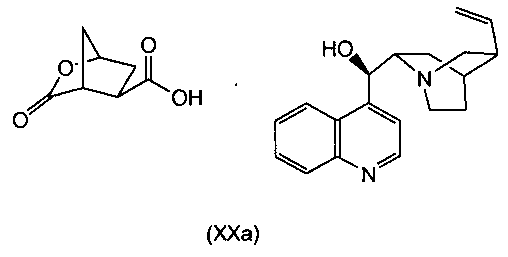
В одном аспекте настоящее изобретение относится к способу получения соединения (VIII) исходя из цинхонидиновой соли (XXa), которая реагирует с N-метил-гексенамином (NMHA) (XIX) в реакции амидообразования с получением амида бициклического лактона (XVIII), в котором раскрывают лактонную группу с получением целевого продукта (VIII). Данные реакции проиллюстрированы на приведенной ниже схеме, в которой R1 имеет определенное выше значение:
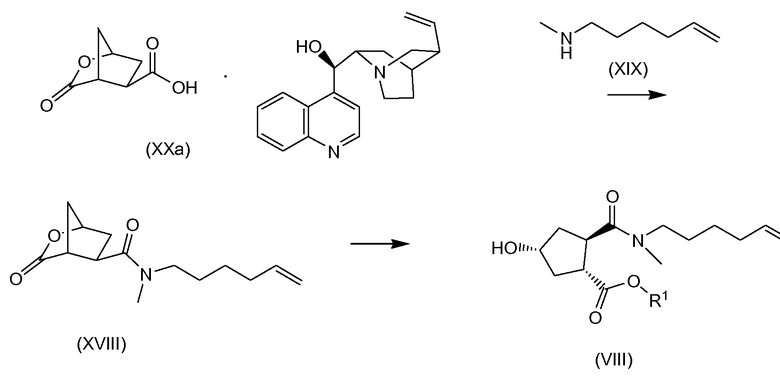
Другой аспект настоящего изобретения относится к получению цинхонидиновой соли (XXa) при разделении смеси диастереоизомерных солей (XX) селективной кристаллизацией (XXa). Соль (XX) получают, в свою очередь, синтезом цинхонидиновой соли рацемической бициклической лактонкарбоновой кислоты (XV), как показано на следующей схеме реакции:


Следующий аспект настоящего изобретения относится к цинхонидиновой соли:

Данную соль используют как промежуточное соединение в получении промежуточного соединения (VIII) и, следовательно, в получении ингибитора HCV (I).
Преимуществом способа синтеза по настоящему изобретению является получение правильной стереохимии циклопентильной группы без применения хиральной хроматографии. Показана селективная кристаллизация цинхонидиновой соли (XXa) с высокой хиральной чистотой.
Реакция цинхонидиновой соли (XXa) с NMHA (XIX) представляет собой реакцию амидообразования, в которой исходные вещества реагируют со связывающим амиды соединением в инертном для данной реакции растворителе в присутствии основания. К пригодным для использования растворителям относятся галогенированные углеводороды, например, дихлорметан (DCM) или хлороформ, простые эфиры, например тетрагидрофуран (THF) или 2-метилтетрагидрофуран (MeTHF), спирты, например метанол или этанол, углеводородные растворители, например толуол или ксилол, полярные апротонные растворители, например диметилформамид (DMF), диметиламин (DMA), ацетонитрил или их смеси. Предпочтительны дихлорметан, MeTHF, метанол, этанол, толуол или их смеси. К связывающим амиды соединениям относятся, например, N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ), N-изопропоксикарбонил-2-изопропокси-1,2-дигидрохинолин (IIDQ), в частности его гидрохлорид, N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)уронийгексафторфосфат (HATU), бензотриазол-1-ил-окси-трис-пирролидинофосфонийгексафторфосфат (товарное наименование PyBOP®), 1,1'-карбонилдиимидазол (CDI), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDI или EDCI), а также его гидрохлорид, дициклогексилкарбодиимид (DCC), или 1,3-диизопропилкарбодиимид, O-бензотриазол-N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU) и т.д. Можно добавить катализатор, например 1-гидроксибензотриазол (HOBt) или 4-диметиламинопиридин (DMAP). Реакцию обычно проводят в присутствии основания, в частности амина, в том числе третичного амина, например триэтиламина, N-метилморфолина, N,N-диизопропилэтиламина (последний также называют основанием Хюнига, DIPEA или DIEA). Предпочтительно не использовать основание. В одном варианте осуществления реакцию проводят в DCM или MeTHF с EEDQ, возможно, с добавлением метанола в конце реакции, при нагревании реакционной смеси с обратным холодильником.
В альтернативном варианте осуществления соль (XXa) можно расщепить на цинхонидин и бициклический лактон, и последний может реагировать с NMHA в реакции амидообразования, как описано выше. Обнаружено, что саму цинхонидиновую соль (XXa) можно использовать в реакции амидообразования, после чего цинхонидин можно легко удалить при приготовлении реакционной смеси, в частности, обрабатывая последнюю кислотой, например HCl, и смывая побочные продукты с водными фазами.
Лактонную группу в полученном амиде бициклического лактона (XVIII) раскрывают по реакции трансэтерификации со спиртом, который может также служить растворителем, в частности с алканолом C1-4, например метанолом или этанолом, в присутствии кислоты. В качестве кислоты можно использовать сильную органическую кислоту, например метансульфокислоту. Можно добавлять растворители, в частности простой эфир, например THF или MeTHF, или углеводородные растворители, например, толуол или ксилол. В реакции трансэтерификации образуется сложный эфир используемого спирта, например метиловый эфир, если реакцию проводят в метаноле.
В свою очередь, цинхонидиновую соль (XX) можно получить, обрабатывая рацемическую бициклическую лактонкарбоновую кислоту (XV) цинхонидином. Как правило, рацемическую соль (XX) не выделяют, а оставляют в растворе, из которого кристаллизуется целевой изомер (XXa). В одном варианте осуществления суспензию цинхонидина добавляют к раствору (XV) при слегка повышенной температуре, а затем дают смеси остыть для кристаллизации целевой соли (XXa). Дальнейшую очистку можно проводить перекристаллизацией. К пригодным растворителям для растворения (XV) относятся сложноэфирные растворители, например этилацетат, в то время как к пригодным растворителям для суспензии цинхонидина относится ацетонитрил. В одном варианте осуществления солеобразование осуществляют при температуре около 50-70°C, в частности около 60°C, и смеси дают остыть почти до комнатной температуры, например до температуры около 20-25°C, например, около 22°C. Дальнейшую очистку можно осуществлять перекристаллизацией из соответствующего растворителя или смеси растворителей, в частности из спирта, например алканола C1-4, в том числе изопропанола, или повторным суспендированием в растворителе или смеси растворителей, например, в смеси этанол/вода с содержанием компонентов, например, 95/5 масс.%.
Обнаружение возможности выделения соли (XXa) кристаллизацией открывает изящный способ получения бициклического лактона высокой энантиомерной чистоты. Перекристаллизация или повторное суспендирование обеспечивает дальнейшую очистку данной соли. Можно использовать (XXa) как исходное вещество в дальнейшем синтезе промежуточных соединений (XVIII) и (VIII), как описано выше. Последнее затем можно конвертировать в промежуточное соединение (VI), важный компонент в получении соединения (I).
Рацемическую бициклическую лактонкарбоновую кислоту (XV) получают, как описано в заявке WO 2008/092955 и показано выше на схеме, иллюстрирующей получение (Xa) и (Xb). В частности, (XV) получают восстановлением кетоциклопентандикарбоновой кислоты (XVII) в соответствующую гидроксидикарбоновую кислоту (XVI), которую затем конвертируют в (XV) лактонообразованием. Для восстановления кетонной группы в гидроксильную в (XVIII) можно использовать водород в присутствии катализатора на основе благородного металла, например родия на угле (Rh/C) или никеля Ренея в инертном для реакции растворителе, например в воде. Полученную гидроксициклопентандикарбоновую кислоту (XVI) можно конвертировать в соль, например в соль третичного амина, в том числе в соль триэтиламина.
Циклизацию через образование лактона (XVII) можно осуществить реакцией с хлорформиатом, например с этил- или метилхлорформиатом. Для этой реакции используют инертный для реакции растворитель, в том числе кетон, например ацетон, или простой эфир, например THF или MeTHF, или ацетонитрил. Можно добавлять основание, в том числе третичный амин, например триэтиламин:

В одном варианте осуществления настоящее изобретение относится к применению соединений (XX) или (XXa) как промежуточных соединений для получения соединения (I) или его солей.
В другом варианте осуществления, настоящее изобретение относится к самим соединениям (XX) или (XXa). Данные соединения можно выделять в чистом виде или использовать в растворе. В частности, соединения (XX) или (XXa) выделяют в виде твердых веществ.
Дальнейшую переработку соединений (VIII) в конечные продукты (I) проводят, как указано выше в схемах реакций и, в частности, как описано в заявке WO 2008/092955. В эту дальнейшую переработку входит реакция Мицунобу, которая включает стереохимическую инверсию циклопентильного атома углерода при гидроксильной группе.
Промежуточное соединение (VI) можно кристаллизовать, в частности, из смеси со спиртовым растворителем, в том числе из смеси с алканолом C1-4. Кристаллизация промежуточного соединения (VI) позволяет контролировать чистоту данного соединения, а также любых полученных из него соединений на последующих стадиях процесса. В частности, данное свойство позволяет получать промежуточное соединение (VI) более высокой энантиомерной чистоты.
Эта кристаллизация промежуточного соединения (VI) позволяет не только удалить побочные продукты реакции Мицунобу, в которой получают данные соединения, но также затем легко выделить промежуточное соединение (VI) из реакционной смеси. Такое выделение осуществляют простой заменой растворителя, в частности добавлением спиртового растворителя в реакционную смесь, полученную в реакции Мицунобу, без необходимости какого-либо дальнейшего воздействия на реакционную смесь или какой-либо ее компонент. Далее, поскольку промежуточное соединение (VI) нерастворимо в спирте, в отличие от побочных продуктов, это позволяет немедленно выделять промежуточное соединение (VI) из реакционной смеси.
Термины, используемые в тексте настоящей заявки, имеют следующие определения, если не указано иное. Термин «галоген» означает «фтор», «хлор», «бром» и «йод». Термин «алкил C1-4» означает линейные или разветвленные насыщенные углеводородные радикалы, содержащие от 1 до 4 атомов углерода, в том числе, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-l-пропил, 2-метил-2-пропил. Термин «алкил C1-3» объединяет метил, этил, 1-пропил, и 2-пропил. Термин «C1-2» объединяет метил и этил. Термин «алканол C1-4» означает спирт, содержащий алкильную группу C1-4.
Общепринятое условие представления стереохимических соединений, которое также применяется к настоящей заявке, заключается в следующем:
- Соединение, представленное без стереосвязей, например соединение (XV), является рацематом, или конфигурация стереогенного центра (центров) не определена.
- Соединение, представленное со стереосвязями и одним из обозначений «(±)», «относительный» или «рацемический», является рацематом, имеющим относительную стереохимию.
- Соединение, представленное со стереосвязями, но без описаний «(±)»,«относительный» или «рацемический», является нерацемическим (скалемическим) или энантиометрически обогащенным, т.е. имеющим абсолютную стереохимию.
Например, в названии своей статьи Honda и др. использовали обозначение «(±)», показав, что в ней описан синтез рацемата из рацемических промежуточных соединений. Однако приведенное выше условие не обязательно соблюдают во всех публикациях.
Хиральную чистоту выражают как энантиомерное отношение (ER). Для солей значение ER означает отношение двух энантиомеров в диастереоизомерной смеси. См., например, промежуточное соединение (XV).
В определенных вариантах осуществления термин «около» при использовании в отношении численной величины может пропускаться, что означает точную величину. В других вариантах осуществления данный термин может означать численную величину, к которой он относится, с точностью ±10%, ±5% или ±1%.
ПРИМЕРЫ
Следующие примеры, предназначенные для иллюстрации настоящего изобретения, не следует истолковывать как ограничение объема настоящего изобретения.
ПРИМЕР 1
К суспензии 32,7 г (0,19 моль) промежуточного соединения (XVII) (рацемат) в 237,5 мл воды в атмосфере азота добавляли 1,0 мл (0,019 моль) 50 масс.% водного раствора NaOH. Смесь нагревали до 60°C и добавляли 2,5 г Rh/C (5 масс.%). Реакционную колбу продували водородом и продолжали перемешивание в атмосфере водорода до достижения полной конверсии. Теплую реакционную смесь фильтровали через целит (диатомит). Осадок на фильтре дважды промывали 10 мл воды. Добавляли триэтиламин (55,61 мл, 0,40 моль) и 80% объема растворителя отгоняли при давлении 30 мбар. Реакционную колбу присоединяли к ловушке Дина-Старка, наполненной 2-метилтетрагидрофураном. К реакционной смеси добавляли 2-метилтетрагидрофуран (100 мл) и нагревали с обратным холодильником в течение 4 часов для удаления остаточной воды. При атмосферном давлении отгоняли 80% объема растворителя. Смесь охлаждали до 50°C и добавляли ацетон (380 мл). Смесь дополнительно охлаждали до 22°C и снова добавляли ацетон (760 мл) добавляли. Полученную суспензию охлаждали в атмосфере азота до -5°C и добавляли триэтиламин (27,8 мл, 20,24 г, 0,2 моль). Затем по каплям добавляли этилхлорформиат (22,68 г, 0,21 моль) и перемешивали смесь при 0°C в течение 3 часов. Реакционную смесь нагревали до 22°C и перемешивали еще 12 часов, затем фильтровали через дикалит и промывали твердую фазу ацетоном (100 мл). Полученный раствор (XV) в ацетоне использовали в следующем примере для получения соответствующей цинхонидиновой соли.
ПРИМЕР 2: получение цинхонидиновой соли (XXa)
Способ 1
Приблизительно 80% объема растворителя отгоняли при атмосферном давлении. Добавляли этилацетат (190 мл) и промывали органический раствор водным раствором HCl (2 M, 114 мл), получая раствор (XV) в этилацетате. Раствор (XV) в этилацетате добавляли к суспензии цинхонидина (55,94 г, 0,19 моль) в ацетонитриле (760 мл) при 60°C. Полученную смесь перемешивали при 60°C в течение 10 минут и затем охлаждали до 22°C и фильтровали. Твердую фазу перекристаллизовывали из изопропанола (1500 мл) и получали после высушивания 24,8 г (выход 29%) белого твердого вещества. Хиральная чистота (ER): 89/11.
Спектр ЯМР 1H (DMSO-d6, 400 МГц, δ м.д.): 1,45-1,86 (м, 6H), 1,93-2,19 (м, 3H), 2,32 (ш.с, 1H), 2,56-2,80 (м, 2H), 2,90-3,07 (м, 2H), 3,12-3,29 (м, 1H), 3,30-3,52 (м, 1H), 4,93-5,03 (м, 3H), 5,52 (д, J=5,6 Гц, 1H), 5,80-5,89 (м, 1H), 7,5 (д, J=4,2 Гц, 1H), 7,6 (т, J=5,6 Гц, 1H), 8,0 (д, J=9,3 Гц, 1H), 8,3 (д, J=8,1 Гц, 1H), 8,8 (д, J=4,6 Гц, 1H).
Способ 2
Приблизительно 80% объема растворителя отгоняли при атмосферном давлении. Добавляли этилацетат (522 мл) и отгоняли приблизительно 50% объема растворителя. Остаток охлаждали до 22°C и добавляли этилацетат (180 мл). Полученную суспензию фильтровали и добавляли фильтрат к суспензии цинхонидина (55,94 г, 0,19 моль) в ацетонитриле (760 мл). Данную смесь нагревали до 60°C, перемешивали в течение 10 минут, затем охлаждали до 22°C и фильтровали. Твердую фазу перекристаллизовывали из изопропанола (1500 мл) и получали после высушивания 24,8 г (выход 29%) белого твердого вещества. Хиральная чистота (ER): 90/10.
Способ 3
По методике способа 2, но с заменой суспензии цинхонидина (55,94 г, 0,19 моль) в ацетонитриле (760 мл) суспензией цинхонидина (55,94 г, 0,19 моль) в изопропаноле (325 мл) и этаноле (325 мл) получали 24,8 г (29%) белого твердого вещества. Хиральная чистота (ER): 92/8.
Химическую чистоту (XXa), а также значение ER можно повысить перекристаллизацией или повторным суспендированием солей, как описано в следующих трех методиках.
Растворяли 12 г неочищенного (XXa) (химическая чистота: титрование кислотой 96,2%, титрование основанием 102,2%; хиральная чистота (ER): 78,7/21,3) в 500 мл 2-пропанола при нагревании с обратным холодильником. Смеси давали медленно остыть. Если не начиналась самопроизвольная кристаллизация, в смесь вносили затравочные кристаллы (XXa) при 40°C, затем перемешивали при данной температуре в течение 2 часов. После охлаждения до комнатной температуры смесь продолжали перемешивать в течение 2 часов, фильтровали, промывали 50 мл 2-пропанола и после высушивания в вакууме при 50°C получали 5,51 г белого продукта. Химическая чистота: титрование кислотой 99.6%, титрование основанием 98,4%; хиральная чистота (ER): 88,1/11,9.
Смесь 5,3 г (XXa) с ER=87,0/13,0 и 0,5 г (XXa) с ER=90,6/9,4 растворяли при нагревании с обратным холодильником в 160 мл этанола, содержащем 5 масс.% воды. Прозрачному раствору давали медленно остыть. Если не начиналась самопроизвольная кристаллизация, в смесь вносили затравочные кристаллы (XXa) при 45°C. После охлаждения до комнатной температуры смесь продолжали перемешивать в течение 14 часов, фильтровали и промывали 10 мл этанола, содержащего 5 масс.% воды, и после высушивания в вакууме при 50°C получали 4,21 г белого продукта. Хиральная чистота (ER): 96,5/3,5.
Смесь 25 г (XXa) с ER=87,0/13,0 и 2,5 г (XXa) с ER=90,6/9,4 нагревали с обратным холодильником в 160 мл этанола, содержащего 5 масс.% воды. После одночасового нагревания с обратным холодильником суспензии давали остыть до комнатной температуры в течение 2 часов и после этого перемешивали в течение 14 часов. Затем смесь фильтровали, промывали 15 мл этанола, содержащего 5 масс.% воды, и после высушивания в вакууме при 50°C получали 22,96 г белого продукта. Хиральная чистота (ER): 97,6/2,4.
ПРИМЕР 3: получение (XVI) и его солей с триэтиламином (XVIa)
(a) Растворяли 344 мг (2 ммоль) (XVII) и 725 мг (4 ммоль) пентагидрата гидроксида тетраметиламмония в смеси 2,5 мл метанола и 2,5 мл MeTHF. Раствор перемешивали ночь при комнатной температуре в атмосфере водорода в присутствии 82 мг катализатора (влажный родий на активированном угле). Катализатор отфильтровывали и разбавляли фильтрат метанолом до конечного объема, составляющего 100 мл. Анализ методом жидкостной хроматографии показал образование 46% (XVI) в виде его двухзамещенной соли тетраметиламмония, в то время как по-прежнему присутствовало 41% (XVII) в виде его двухзамещенной соли тетраметиламмония.
(b) К 400 г водного раствора, содержащего 6,6 масс.% (XVI), добавляли 44,4 мл триэтиламина. В вакууме отгоняли 330 г растворителя, затем маслянистому остатку давали остыть до 50°C и добавляли 51,5 мл ацетона, получая суспензию. Данную суспензию охлаждали до комнатной температуры и добавляли еще 155 мл ацетона. Суспензию охлаждали до 5°C и перемешивали ночь при данной температуре. Твердую фазу отфильтровывали, промывали холодным ацетоном и сушили при 70°C в вакууме, получая 15,05 г комплекса (XVI) с различными количествами триэтиламина (XVIa) в виде белого кристаллического порошка. Выход: 37%. Например, можно получить комплекс (XVI), содержащий 1/3 или 2 молекулы триэтиламина6

где x составляет от 1/3 до 3, например x=1/3; x=2.
Очистка (XVIa)
(a) Суспендировали 2,00 г неочищенного (XVIa) в 10,4 мл ацетона и нагревали суспензию с обратным холодильником, затем ей давали остыть до комнатной температуры. Твердую фазу отфильтровывали, промывали ацетоном, сушили при 50°C в вакууме и получали 210 мг чистого (XVIa) в виде белого порошка. Выход: 35%.
(b) Суспендировали 2,00 г неочищенного (XVIa) в 10,4 мл бутанола и нагревали суспензию с обратным холодильником, затем ей давали остыть до комнатной температуры. Твердую фазу отфильтровывали, промывали ацетоном, сушили при 50°C в вакууме и получали 190 мг очищенного (XVIa) в виде белого порошка. Выход: 14%.
ПРИМЕР 4: получение (XVIII)
(a) Суспендировали 14,18 г (31,5 ммоль) (XXa) (ER=90/10), 3,92 г (34,6 ммоль) NMHA и 8,56 г (34,6 ммоль) EEDQ в 157 мл DCM и полученную суспензию нагревали ночь с обратным холодильником. Добавляли 47,2 мл метанола и продолжали нагревание с обратным холодильником еще сутки. Затем реакционную смесь концентрировали в вакууме и распределяли остаток между 47 мл толуола и 79 мл водного раствора 1 М HCl. Органический слой последовательно промывали 31,5 мл воды, 31,5 мл водного раствора 1 М NaOH и 31,5 мл воды, затем концентрировали в вакууме и получали 11,93 г неочищенного (XVIII) (ER=90/10), который использовали без очистки на следующей стадии.
(b) Суспендировали 2,50 г (5,55 ммоль) (XXa), 691 мг (6,10 ммоль) NMHA и 1,51 г (6,10 ммоль) EEDQ в 28 мл THF и нагревали суспензию с обратным холодильником в течение 2 суток. Добавляли 22 мл толуола и отгоняли 28 мл растворителя. После охлаждения до 50-60°C добавляли 19,4 мл водного раствора 1 M HCl и разделяли два слоя. Органический слой промывали 5,6 мл воды, затем концентрировали в вакууме, очищали остаток флэш-хроматографией и получали 910 мг (XVIII). Выход: 65%. Спектр ЯМР 1H (CDCl3, 600 МГц, δ м.д.) показал присутствие двух ротамеров в соотношении 55/45): 1,26-1,38 (м, 2H), 1,43-1,60 (м, 2 H), 2,01 (м, 2 H), 2,07-2,21 (м, 4 H), 2,86 (с, 3H - минорный ротамер), 2,89-2,97 (м, 2 H), 2,97 (с, 3H - основной ротамер), 3,21 (ддд, 1H - минорный ротамер, J=14,7, 9,1, 5,8 Гц), 3,29 (м, 1H -минорный ротамер), 3,31 (т, 2H, J=7,6 Гц - основной ротамер) 4,87-4,93 (м, 2H) 4,96 (д, 1H, J=16,2 Гц) 5,71 (м, 1H). Спектр ЯМР 13C (CDCl3, 600 МГц, δ м.д.) показал присутствие двух ротамеров: основной ротамер 25,86, 26,39, 33,24, 33,94, 35,17, 37,43, 37,97, 45,71, 47,95, 80,67, 114,71, 138,26, 170,91, 177,33; минорный ротамер 25,7, 27,72, 33,14, 33,69, 34,29, 36,72, 38,02, 46,19, 49,61, 80,64, 115,22, 137,73, 171,17, 177,28.
(c) Суспендировали 17,85 г (39,6 ммоль) (XXa) (ER=97,6/2,4), 4,71 г (41,6 ммоль) NMHA и 10,78 г (43,6 ммоль) EEDQ в 198 мл MeTHF. Суспензию нагревали с обратным холодильником в течение 2 суток, затем охлаждали до комнатной температуры. Твердую фазу (содержавшую, главным образом, цинхонидин) отфильтровывали и промывали толуолом. К объединенному фильтрату добавляли 40 мл воды и 7,14 мл концентрированной HCl. Разделяли два полученных слоя, органический слой промывали 20 мл воды, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали хроматографически на силикагеле (элюент: этилацетат/гептан в соотношении 65/35) и получали 9.35 г (XVIII) в виде масла. Выход: 68%.
ПРИМЕР 5: получение (VIIIa) как промежуточное соединение (VIII), в котором R 1 - метил
(a) Растворяли 1,05 г (4,2 ммоль) (XVIII) в 25 мл метанола. Добавляли 0,014 мл (0,2 ммоль) метансульфокислоты и перемешивали реакционную смесь в течение трех суток при комнатной температуре. Летучие вещества отгоняли в вакууме и повторно растворяли остаток в смеси, содержащей по 15 мл толуола и водного раствора 0,33 M NaOH. Слои разделяли, органический слой сушили над сульфатом магния, концентрировали в вакууме и получали 330 мг неочищенного (VIIIa) в виде масла (выход: 28%).
(b) Суспендировали 20,0 г (44,4 ммоль) (XXa), 5,53 г (48,8 ммоль) NMHA и 12,08 г (48,8 ммоль) EEDQ в 222 мл метанола. Смесь нагревали с обратным холодильником в течение 24 часов, затем добавляли 178 мл толуола. Отгоняли 250 мл растворителей и полученную суспензию охлаждали до 30°C. Добавляли 155 мл водного раствора 1 М HCl и разделяли два слоя. Водный слой дважды экстрагировали 44 мл толуола. Объединенные органические слои сушили над сульфатом магния, фильтровали и получали 122,78 г раствора, содержащего 4,1 масс.% (VIIIa) в толуоле. Выход: 40%.
(c) В вакууме концентрировали 18,42 г раствора, содержащего 4,1 масс.% (VIIIa) в толуоле, остаток очищали флэш-хроматографией (элюент: этилацетат/DCM в соотношении 15/85) и получали 680 мг химически чистого (VIIIa).
(d) Суспендировали 44,06 г (97,8 ммоль) (XXa) (ER=92,4/7,6), 12,18 г (107,6 ммоль) NMHA и 26,60 г (107,6 ммоль) EEDQ в 490 мл метанола. Смесь нагревали ночь с обратным холодильником, затем добавляли 391 мл толуола и отгоняли 750 мл растворителей. К остатку добавляли 156 мл воды и 30,8 мл концентрированной HCl. Разделяли два полученных слоя и экстрагировали водный слой 98 мл толуола, затем 98 мл MeTHF. Объединенные органические слои сушили над сульфатом магния, фильтровали и получали 384 г раствора, содержащего 3,6 масс.% (VIIIa) в смеси MeTHF/толуол. Выход: 50%.
(e) Суспендировали 19 г (42,2 ммоль) (XXa) (ER=93,4/6,6), 5,01 г (44,3 ммоль) NMHA и 11,46 г (46,4 ммоль) EEDQ в 210 мл THF. Суспензию нагревали ночь с обратным холодильником, затем охлаждали до комнатной температуры. Твердую фазу (главным образом, цинхонидин) отфильтровывали и промывали 84 мл толуола. К объединенным фильтратам добавляли 42 мл воды и 7,6 мл концентрированной HCl. Разделяли два слоя, органический слой промывали 21 мл воды, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток растворяли в 84 мл метанола, добавляли 0,14 мл метансульфокислоты, раствор перемешивали ночь при комнатной температуре, затем нагревали с обратным холодильником в течение 24 часов, прежде чем охлаждали до комнатной температуры. Добавляли 223 мг карбоната натрия и перемешивали смесь в течение 1 часа при комнатной температуре. Добавляли 295 мл толуола, отгоняли 160 мл растворителей и получали 184,9 г раствора, содержащего 5,3 масс.% (VIIIa) в толуоле. Выход: 82%.
(f) Суспендировали 19 г (42,2 ммоль) (XXa) (ER=93,4/6,6), 5,34 г (47,2 ммоль) NMHA и 12,51 г (50,5 ммоль) EEDQ в 210 мл толуола. Суспензию нагревали с обратным холодильником в течение 2 суток, затем охлаждали до комнатной температуры. Количественный анализ на месте показал выход 80% (XVIII). Твердую фазу (главным образом, цинхонидин) отфильтровывали и промывали 42 мл толуола. К объединенным фильтратам добавляли 42 мл воды и 7,6 мл концентрированной HCl. Разделяли два слоя, органический слой промывали 21 мл воды, затем 21 мл соляного раствора и концентрировали отгонкой 206 мл растворителей. К концентрату добавляли 84 мл метанола и 0,14 мл метансульфокислоты. Полученный раствор перемешивали ночь при комнатной температуре. Добавляли 223 мг карбоната натрия добавляли и продолжали перемешивать еще 1-2 часа. Добавляли 295 мл толуола добавляли и отфильтровывали полученную твердую фазу. Отгоняли 183 мл растворителей и получали 180,5 г раствора, содержащего 2,7 масс.% (VIIIa) в толуоле. Общий выход (VIIIa): 41%.
ПРИМЕР 6: получение (VIa) как промежуточное соединение (VI), где R 1 - метил

(a) Суспендировали 20,0 г (44,4 ммоль) (XXa) (ER=90,1/9,9), 5,53 г (48,8 ммоль) NMHA и 12,08 г (48,8 ммоль) EEDQ в 222 мл метанола. Смесь нагревали с обратным холодильником в течение 24 часов, затем добавляли 178 мл толуола. Отгоняли 250 мл растворителей и полученную суспензию охлаждали до 30°C. Добавляли 155 мл водного раствора 1 M HCl добавляли и разделяли два слоя. Водный слой дважды экстрагировали 44 мл толуола. Объединенные органические слои сушили над сульфатом магния, фильтровали и получали 122,78 г раствора, содержащего 4,1 масс.% (VIIIa) в толуоле. К 98,22 г данного раствора добавляли 11,17 г (35,5 ммоль) (VII) и 9,78 г (37,3 ммоль) трифенилфосфина и охлаждали смесь до 0°C. По каплям добавляли 7,4 мл (37,3 мл) DIAD, затем полученную реакционную смесь перемешивали при 0°C в течение 2 часов, при этом образовывался осадок. Добавляли 0,1 мл уксусной кислоты и отфильтровывали осадок. Фильтрат концентрировали в вакууме и растворяли остаток в 71 мл кипящего изопропанола. Раствор охлаждали до 0°C и оставляли для кристаллизации (VIa). Твердую фазу отфильтровывали, промывали холодным изопропанолом, сушили в вакууме и получали 6,32 г (VIa) (ER=97,2/2,8). Выход из (XXa): 31%.
(b) К 382,8 г раствора, содержащего 3,6 масс.% (48,6 ммоль) (VIIIa) в смеси MeTHF/толуол, добавляли 18,53 г (48,6 ммоль) (VII) и 19,7 г (75,4 ммоль) трифенилфосфина. Отгоняли 118 г растворителей и полученный остаток охлаждали до 0°C. По каплям добавляли 14,9 мл (75,4 ммоль) DIAD и перемешивали реакционную смесь в течение 2 часов при 0°C. Полученный твердый осадок (содержащий, главным образом, трифенилфосфиноксид) отфильтровывали и промывали холодным толуолом. Из объединенных фильтратов отгоняли 140 г растворителей, затем добавляли 97 мл 1-бутанола и отгоняли 77 г растворителей. Смесь охлаждали до 80°C и добавляли 97 мл изопропанола и 2,43 г дикалита. После перемешивания в течение нескольких минут при нагревании с обратным холодильником горячую смесь фильтровали и полученный фильтрат охлаждали до 40°C. Добавляли 14 мг (VIa) в качестве затравочного материала и охлаждали смесь до 0°C. После ночного перемешивания при 0°C добавляли 48 мл изопропанола и продолжали перемешивание при 0°C в течение 2 часов. (VIa) выделяли фильтрованием, промывали 9,7 мл холодного изопропанола и сушили при 70°C в вакууме. Получали первую порцию, содержащую 8,77 г (VIa) (выход: 28%). Маточные растворы концентрировали в вакууме, остаток очищали флэш-хроматографией на силикагеле и получали вторую порцию, содержащую 12,1 г (VIa) (выход: 43%).
(c) К 58,9 г (8,3 ммоль) раствора, содержащего 4 масс.% (VIIIa) в толуоле, добавляли 2,86 г (9 ммоль) (VII) и 2,29 г (10,2 ммоль) трифенилфосфина. Суспензию сушили отгонкой 27 мл растворителя, затем охлаждали до 0°C. По каплям добавляли 8,7 мл (10,2 ммоль) DIAD и перемешивали реакционную смесь в течение 1-2 часов при 0°C. Твердую фазу отфильтровывали и промывали 4,2 мл толуола. Из объединенных фильтратов отгоняли 27 растворителя. Добавляли 25 мл 1-бутанола и отгоняли 25 мл растворителей. Остаток охлаждали до 80°C, добавляли 25 мл изопропанола и 415 мг дикалита, суспензию нагревали с обратным холодильником и подвергали горячему фильтрованию. Фильтрат охлаждали до 30°C и добавляли 2,4 мг (VIa) в качестве затравочного материала. Суспензию охлаждали до 0°C и перемешивали ночь при этой температуре. (VIa) отфильтровывали, промывали 2,5 мл холодного изопропанола, сушили в вакууме и получали 24,3 г белого порошка. Выход: 80%.


Изобретение относится к цинхонидиновой соли формулы (XXa), к способу ее получения, применению в качестве промежуточного соединения при получении соединения (VIII) и к способу получения соединения (VIII) из соединения формулы (XXa). Технический результат - новое промежуточное соединение формулы (XXa) для синтеза макроциклического ингибитора протеазы вируса гепатита С (HCV). 4 н. и 21 з.п. ф-лы, 6 пр.
1. Способ получения соединения (VIII) исходя из цинхонидиновой соли (ХХа), которая вступает с N-метил-гексенамином (NMHA) (XIX) в реакцию амидообразования с получением амида бициклического лактона (XVIII), в котором лактонную группу раскрывают с получением целевого продукта (VIII), как показано ниже на схеме, где R1 представляет собой C1-4 алкил:

2. Способ по п. 1, в котором R1 является метилом.
3. Способ по п. 1, в котором реакцию амидообразования проводят в присутствии связывающего амиды реагента в инертном для реакции растворителе.
4. Способ по п. 3, в котором реакцию проводят в присутствии основания.
5. Способ по п. 2, в котором реакцию амидообразования проводят в присутствии связывающего амиды реагента в инертном для реакции растворителе.
6. Способ по п. 5, в котором реакцию проводят в присутствии основания.
7. Способ по любому из пп. 3, 4, 5 или 6, в котором растворитель содержит галогенированные углеводороды, простые эфиры, спирты, углеводородные растворители, диполярные апротонные растворители или их смеси.
8. Способ по п. 7, в котором галогенированные углеводороды представляют собой дихлорметан (DCM) или хлороформ, и/или простые эфиры представляют собой тетрагидрофуран (THF) или 2-метилтетрагидрофуран (MeTHF), и/или спирты представляют собой метанол или этанол, и/или углеводородные растворители представляют собой толуол или ксилол, и/или диполярные апротонные растворители представляют собой DMF, DMA или ацетонитрил.
9. Способ по любому из пп. 3, 4, 5 или 6, в котором связующее амиды вещество содержит:
N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ), N-изопропокси-карбонил-2-изопропокси-1,2-дигидрохинолин (IIDQ), N,N,N′,N′-тетраметил-О-(7-азабензотриазол-1-ил)уронийгексафторфосфат (HATU), бензотриазол-1-ил-окси-трис-пирролидино-фосфонийгексафторфосфат, CDI, 1-этил-3-(3-диметиламинопропил) карбодиимид (EDCI) или его гидрохлорид, дициклогексилкарбодиимид (DCC), 1,3-диизопропилкарбодиимид, или О-бензотриазол-N,N,N′,N′ - тетраметил-уронийгексафторфосфат (HBTU).
10. Способ по п. 9, в котором связующее амиды вещество используют в присутствии катализатора.
11. Способ по п. 10, в котором катализатор является 1-гидроксибензотриазолом (HOBt) или 4-диметиламинопиридином (DMAP).
12. Способ по п. 4 или 6, в котором в качестве основания используют третичный амин.
13. Способ по п. 12, в котором третичный амин представляет собой триэтиламин, N-метилморфолин, N,N-диизопропилэтиламин.
14. Способ получения цинхонидиновой соли (ХХа), которую получают кристаллизацией из рацемической соли (XX):

15. Способ по п. 14, в котором рацемическую соль (XX) получают реакцией бициклической лактонкарбоновой кислоты (XV) с цинхонидином:
16. Способ по п. 15, в котором суспензию цинхонидина добавляют к раствору (XV) при температуре от около 50°С до около 70°С и затем дают смеси остыть с кристаллизацией при этом целевого продукта (ХХа).
17. Способ по п. 15, в котором (XV) растворяют в растворителе, выбранном из сложных эфиров, а растворители для суспензии цинхонидина представляют собой ацетонитрил.
18. Способ по п. 17, в котором растворитель, выбранный из сложных эфиров, представляет собой этилацетат.
19. Способ по п. 16 или 17, в котором солеобразование проводят при температуре от около 50°С до около 70°С, в частности около 60°С, и смеси дают остыть почти до комнатной температуры, например до температуры от около 20°С до около 25°С.
20. Способ по п. 19, в котором солеобразование проводят при температуре около 60°С, и смеси дают остыть до комнатной температуры в интервале температуры от около 20°С до около 25°С.
21. Способ по п. 16 или 17, в котором соль подвергают дальнейшей очистке перекристаллизацией из подходящего растворителя или смеси растворителей, или повторным суспендированием в растворителе или смеси растворителей.
22. Способ по п. 21, в котором растворителем для перекристаллизации служит С1-4 алканол, или растворителем или смесью растворителей для повторного суспендирования служит смесь этанола и воды.
23. Способ по п. 22, в котором С1-4 алканол представляет собой изопропанол, и/или растворитель или смесь растворителей для повторного суспендирования представляет собой смесь этанола и воды в соотношении воды к этанолу 5/95 масс. %.
24. Цинхонидиновая соль формулы:
25. Применение цинхонидиновой соли (ХХа), определенной в п. 24, в качестве промежуточного соединения при получении промежуточного соединения (VIII).
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| СПОСОБ ПОЛУЧЕНИЯ R(+)-1,2,3,6-ТЕТРАГИДРО-4-ФЕНИЛ-1-[(3-ФЕНИЛ-3-ЦИКЛОГЕКСЕН-1-ИЛ)МЕТИЛ]ПИРИД ИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2144026C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3,3-ДИАРИЛПРОПИЛАМИНОВ (ВАРИАНТЫ) И СОЕДИНЕНИЯ (ВАРИАНТЫ) | 2001 |
|
RU2270188C2 |

