Изобретение относится к новым химическим соединениям ряда карбазола, именно к производным 2,3,4,9-тетра- гидро-1Н-карбазол-1-уксусной кислоты общей формулы
TU
(I)
СН2СООН
г Rs
R б) R, RG
де a) R.J - бензил; этил; водород;
водород или 8-этил; 2-метил-2-пропенил; этил;
R R - водород или Rj-7-хлор; R J - 8-метил;
в)R - 1,1-пиметил-2 пропенил; Rg - этил;
R3 - водород;
R - 8-фтор или RJ - 7-хлор; R 4 - 8-метилi
г)R - 2-пропенил; RJ. - метил;
Rs - водород; К - водо- род или 8-этил, или RJ ч этил, R, - водород, R - водород, 8-метил, 6-, 7- или 8-этил, 8-пропияг 8-изопрогаш, 6-бутил, б или 8-фтор, 5-s 6-, 7- или 8-хлрр, 5-, 6-, 7- или 8-бром, 8-йод, или RJ - 8-метил, - 5-метил, 7-фтор,. 5- или 7- . хлорs 7-бром, или RS ™ 8-хлор,Е4-5-метил,5-, 6- или 7-хлор, или R3 - 8-бром, R4 5-метил, или RJ - 7-хлор, R4 - 5-хлор, или Ra - пропил, R3 - водород, R - 8- этил, или бензметанами новая соль соединения 1, где RI - 2-пропенил, RJJ и R -этил, RJ - РОД,
оторые проявляют болеутоляющее и ротивовоспалительное действие, а акже к метиловому эфиру 2,3,4,9- етрагидро-1Н-карбазол-1-уксусной ислоты общей формулы
(II)
з HR2 CH2COOCH3
5
0
5
0
5
0
5
0
5
где R15R2, RJ и R4 имеют указанные значения, который является промежуточным продуктом для синтеза соединений, iпроявляющих болеутоляющее и противовоспалительное действие.
Цель изобретения - поиск в ряду карбазола новых соединений, обладающих более высоким болеутоляющим и противовоспалительным действием.
Изобретение иллюстрируется следующими примерами.
П р и м е-р 1. Цис-1,8-диэтил- 2,3,4,9-тетрагидро-4-(2-пропенил)- 1Н-карбазол-1-уксусная кислота.
а)Получение 2-этилциклогексанона. 2-Зтилциклогексанол (1,6 моль,
I204 г, 226 мл) перемешивают в 3,2 л ацетона при 0ЙС и обрабатывают 8н« реактивом Джонса (приготовленного из 106,8 г Сг03э суспендированной в 92мл концентрированной серной кислоты и разбавленной 400 мл воды) до появления устойчивой оранжевой окраски ( ЧЗО мл) . Затем к реакционной смеси добавляют изопропанол, пока она сно- ва не окрашивается в зеленый цвет, и выливают ее в 2 л эфира. Образующийся продукт промывают 6 раз рассолом, порциями по 500 мл, высушивают над MgSO и отгоняют растворитель. После молекулярной перегонки (т. кип. 80-85йС при 25 мм рт„ ст.) получают 184 г (1,46 моль, выход 91% (2-этилциклогексанона в виде бесцветной маслянистой жидкости.
б)Получение метилового эфира 1- этил-2-оксоциклогексанонуксусной кислоты.
I
Гидрид калия (417 ммоль, 70 мл,-о
л/ 6М в минеральном масле) помещают в атмосфере азота в трехгорлую колбу, снабженную механической мешалкой, и трижды промывают петролейным эфиром (эта промывка не является обязательной) . Затем в колбу добавляют тетра- гидрофуран (200 мл, перегнанные из смеси натрия и Ph$,CO) , а затем медленно в течение примерно 15 мин раствор 2-этилциклогексанона (VIII) (50 г, 396 ммоль) в 200 мл тетрагид- рофурана. Через 1 мин в колбу добавляют 495 мл 1М EtjB в тетра идрофу- ране, а еще через 1 ч 594 ммоль (91 г, 56 мл) метилбромацетата. Суспензию желтого цвета перемешивают в течение 2,5 ч, выливают в 800 мл воды тщательно отделяют с помощью декантации от избытка КН и экстрагирую
4 раза петролейным эфиром порциями п 300 мл. Объединенные органические фазы высушивают над сульфатом натрия и концентрируют в вакууме. Полученный продукт перегоняют в колонке Burpo длиной 6 дюймов и собирают фракцию, кипящую при 107-118 С и
0,8 мм рт.ст. (два региоизомера со стадии алкопирования). Полученный продукт подвергают очистке с помощью флеш-хроматографии (колонка диаметром 4 дюйма, элюент: 7,5%-ный раствор этилацетата в петролейном эфире, высота слоя силикагеля 5,5 дюйма), получая в результате 35,33 г (178,2 ммоль, выход 45%) бесцветной маслянистой жидкости„ Целевым продуктом является продукт с меньшей
482915в
вижная фаза: 15%-ный раствор этилацетата в петролейном эфире) R 0,9.
г) Получение метилового эфира цис-1-этил-2-оксо-4-(2-проленил)- циклогексаноуксусной кислоты,
Исходный енон 6-карбометокси- метил-6-этил-2-циклогексан-1-он (X) (81,53 ммоль, 16,0 г) перемешивают в 82 мл сухого (перегнанного с ) при -78 С в атмосфере азота и добавляют к нему по каплям (122,30 ммоль (23,2 г) TiCl, , а затем
15 97,84 ммоль (11,18 г, 15,55 мл) аллил- триметилтилсилана. Через 1,5 ч реакцию прекращают, добавляя к реакционной смеси при -78 °С 50 мл МеОН, выливают ее в 200 мл воды и четырежды про10









| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2005 |
|
RU2382770C2 |
| ПРОИЗВОДНЫЕ 3-(ГЕТЕРОАРИЛАМИНО)-1,2,3,4-ТЕТРАГИДРО-9Н-КАРБАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2011 |
|
RU2562255C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2005 |
|
RU2430088C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2010 |
|
RU2525196C2 |
| Способ получения производных карбазола | 1979 |
|
SU900807A3 |
| ПРОИЗВОДНЫЕ 2,3,4,9-ТЕТРАГИДРО-1H-КАРБАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА CRTH2 | 2005 |
|
RU2404163C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ФИЗИОЛОГИЧЕСКИХ И/ИЛИ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ LHRH РЕЦЕПТОРОМ, ПОСРЕДСТВОМ ВЫШЕНАЗВАННЫХ ПРОИЗВОДНЫХ | 2008 |
|
RU2497806C9 |
| ПРОИЗВОДНЫЕ 1Н-ПИРИДО[3,4-В]ИНДОЛ-4-КАРБОКСАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2180904C2 |
| Способ получения производных простановой кислоты | 1973 |
|
SU648088A3 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2004 |
|
RU2318810C2 |
Изобретение касается замещенных гетероциклических веществ, в частности производных 2,3,4,9-тетрагидро-1H-карбазол-1-уксусной кислоты общей ф-лы @ , где а) R1-бензил
R2-C2H5
R3-H
R4-H, 8-C2H5
б) R1-2-CH3-2-пропенил
R2-C2H5
R3-R4-H
или R3-7CI
R4-8-CH3
в) R1-1,1-диметил-2-пропенил
R2-C2H5
R3-H
R4-8-F или R3-7CI
R4-8CH3
г) R1-2-пропенил
R2-CH3
R3-R4-H или R3-H и R4-8-CH3
6-, 7- или 8-C2H5-
8-C3H7
8-изо-C3H7
6-C4H9, 6- или 8-F
5-, 6-, 7- или 8-CI, 5-,6-, 7- или 8-BR, 8-иод
или R3-8-CH3
R4-5-CH3
7-F
5-или 7-CI
7-BR, или R3-8-CI
R4-5-CH3
5-, 6- или 7-CI
или R3-8-BR
R4-5-CH3, или R3-7-CI
R4-5-CI или R2-C3H7
R3-H
R4-8-C2H5 или бензметаминовая соль соединения при R1-2-пропенил
R2 и R4-C2H5
R3-H, проявляющих болеутолящее и противовоспалительное действие. Цель - создание новых промежуточных веществ, обеспечивающих получение новых более активных веществ указанного класса. Синтез промежуточных метиловых эфиров указанной кислоты где значения по "а-в" см. выше, "г" -R1-2-пропенил, R2-C2H5
R3-H, R4-H, 8-CH3, 6-, 7- или 8-C2H005
8-C3H7
8-изо-C3H7, 6-C4H9
6- или 8-F
5-, 6-,7- или 8- 5-,6-, 7- или 8-BR
8- иод или R3-8-CH3
R4-5-CH3
7-F
5- или 7-CI, 7-BR или R4-5-CH3 или R3-7-CI
R4-5-CI или R2-C3H7
R3-H
R4-8-C2H5
ведут, например, из метилового эфира цис-1-этил-2-оксо-4-(2-пропенил)циклогексануксусной кислоты и 2-этилфенилгидразина при кипячении в среде метанола. Затем полученный метиловый эфир цис-1,8-диэтил-2,3,4,9-тетрагидро-4-(2-пропенил)-1H- карбазол -1-уксусной кислоты кипятят с метанолом с получением чистоты целевого продукта 99,6%. Новые вещества оказывают противовосполительное и болеутолящее действие, лучше чем у известного аналога-4-пропил-1,8-диэтил-2,3,4,9-тетрагидро-1H-карбазол-1-уксусной кислоты, т.к. их эффективная доза составляет 0,01-0,02 мг/кг против 5 мг/кг. 2 с.п. ф-лы, 5 табл.
величиной ГЦ (из двух перекрывающихся 20 водят экстракцию эфиром порциями по
пятен на тонкослойной хромотографии) R 0,23 при использовании в каче- стве подвижной фазы смеси этилацетата и петролейного эфира. Около 5-10% 2,6-региоизомера (верхнее пятно) может быть вьщелено из полученного продукта.
в) Получение метилового зфира 1-ЭТИЛ-2-оксоциклогекс-3-енуксусной кислоты.
Исходный кетон, 2-карбометокси- метил-2-этилциклогексанон (141 ммоль, 28 г) перемешивают в 1,25 л этилацетата (высушенного с помощью молекулярного сита ЗА), после чего обрабатывают 169 ммоль (32,5 г) PhSeCl. Реакционную смесь перемешивают в атмосфере азота в течение 4 ч и обрабатывают 250 мл воды. Смесь энергично встряхивают в разделительной- воронке и органическую фазу возвращают в реакционную колбу, в которую добавляют 550 мл тетрагидрофурана и затем по каплям 35 мл 30%-ного водного раствора перекиси водорода. Реакционную смесь перемешивают в течение 1 ч и промывают вначале 500 мл воды, а затем 500 мл насыщенного водного раствора NajCQj. Полученный продукт высушивают над MgSO и концентрируют в вакууме. В результате флешхроматографии (колонка диаметром 4 дюйма, 20%-ный раствор этилацетата в петролейном эфире в качестве элю- ента, слой силикагеля высотой 5,5дюй- ма) получают 15,3 г (78,0 ммоль, выход 55%) продукта в виде бледно- желтой маслянистой жидкости. По данным тонкослойной хроматографии (под30
35
100 мл. После высушивания () и флеш-хроматографии (колонка длиной 95 мм, 15%-ный этилацетат в петролейном эфире в качестве элюента, сло 25 силикагеля толщиной 5,5 дюйма) получают 16,02 г (67,2 ммоль, выход 82% бесцветной маслянистой жидкости. Полученный продукт состоит в основном из одного диастереомера (при соотношении между диастереомерами не менее 9:1). Основным диастереомером является цис-изомер. По данным тонко слойной хроматографии с использованием в качестве подвижной фазы 154%ного этилацетата в петролейном эфире Ц 0,39.
д) Получение метилового эфира цис 1,8-диэтил-2,3,4,9-тетрагидро-4-(2- пропенил)-1Н-карбазол-1-уксусной ки- ,слоты.
Исходный кетон ,метиловый эфир цис- 1-этил-2-оксо-4-(2-пропенил)-цикло- гексануксусной кислоты (51,14 ммоль, 12,188 г) и 2-этилфенилгидра- зин (51,14 ммоль, 6,965 г) кипятят с обратным холодильником в 219 мл МеОН в течение 120 ч. Реакционную смесь затем охлаждают до 0°С, добавляют к ней 102 ммоль (8,0 г 7,3 мл) АсС1 и кипятят с обратным холодильником в течение еще 45 мин. После этого растворитель отгоняют и проводят фле хроматографию (колонка длиной 95 мм, 12%-ный этилацетат в петролейном эфи ре в качестве элюента, высота слоя силикагеля 5,5 дюйма), получая в результате 7,0 г (20,64 ммоль, выход 40%) маслянистой жидкости оранжевого цвета. По данным тонкослойной хро40
45
50
55
0 водят экстракцию эфиром порциями по
0
5
100 мл. После высушивания () и флеш-хроматографии (колонка длиной 95 мм, 15%-ный этилацетат в петролейном эфире в качестве элюента, слой 5 силикагеля толщиной 5,5 дюйма) получают 16,02 г (67,2 ммоль, выход 82%) бесцветной маслянистой жидкости. Полученный продукт состоит в основном из одного диастереомера (при соотношении между диастереомерами не менее 9:1). Основным диастереомером является цис-изомер. По данным тонкослойной хроматографии с использованием в качестве подвижной фазы 154% . ного этилацетата в петролейном эфире Ц 0,39.
д) Получение метилового эфира цис- 1,8-диэтил-2,3,4,9-тетрагидро-4-(2- пропенил)-1Н-карбазол-1-уксусной ки- ,слоты.
Исходный кетон ,метиловый эфир цис- 1-этил-2-оксо-4-(2-пропенил)-цикло- гексануксусной кислоты (51,14 ммоль, 12,188 г) и 2-этилфенилгидра- зин (51,14 ммоль, 6,965 г) кипятят с обратным холодильником в 219 мл МеОН в течение 120 ч. Реакционную смесь затем охлаждают до 0°С, добавляют к ней 102 ммоль (8,0 г 7,3 мл) АсС1 и кипятят с обратным холодильником в течение еще 45 мин. После этого растворитель отгоняют и проводят флеш- хроматографию (колонка длиной 95 мм, 12%-ный этилацетат в петролейном эфире в качестве элюента, высота слоя силикагеля 5,5 дюйма), получая в результате 7,0 г (20,64 ммоль, выход 40%) маслянистой жидкости оранжевого цвета. По данным тонкослойной хро0
5
0
5
матографии с использованием в качестве подвижной фазы 15%-ного этилацета та в петролейном эфире Rj 0,54.
е) Получение цис-1,8-диэтил- 2,3,4,9-тетрагидро-4-(2-пропенил)- 1Н-карбазол 1-уксусной кислоты.
Исходный эфир, метиловый эфир ци.с 1,8-диэтил-2,3,4,9-тетрагидро-4- (2-пропенил)-1Н-карбазол-1-уксусной кислоты (20,64 ммоль, 7,007 г) и КдСОз (24,77 ммоль, 3,423 г) кипятят с обратным холодильником в 165 мл MeOHj содержащей 21 мл воды, в атмосфере азота в течение 27,5 ч. Большую часть МеОН отгоняют в вакууме, а остток растворяют в 50 мл воды. Раствор подкисляют до рН 311 водным раствором НС1 и четырежды проводят экстракцию эфиром порциями по 50 мл„ Объединенные органические фаз высушивают над MgSOij и концентрируют. В результате флеш-хроматографии (колонка длиной 75 мм, 40%-ный этила- цетат в петролейном эфире в качестве элюента, высота слоя силикагеля 5,5 дюйма) получают 5,0152 г- (15,41 ммоль, выход 75%) маслянистой жидкости желтого цвета. По данным жидкостной хроматографии полученный продукт состоит в основном из одного изомера (при соотношении между дка стереомерами, как минимум, 9:1). Основным диастереомером является цис- изомер. 3 г полученного продукта пе- рекристаллизовывают из 20 мл смеси петролейного эфира и бензола в соотношении 2:1, получая в результате 1,3 г игольчатых кристаллов грязно- белого цвета (т.пл. 121-124°С). По данным жидкостной хроматографии перекристаллизованного материала степень чистоты его составляла 99,3%, После второй перекристаллизации степень чистоты составляла 99,6%.
ЯМР (CDClj(тетраметплсилан): 0,85 (т, ЗН, J 8, CHttCH9); 1,33 (т, J 8, 3H,ArCHjCH,)j 1,6-2,5 (м, 6Н, СЦвСН,, цикл СНг); 2,5-3,4 (м, 711, CHS CHfcCOO, , С ССНа)
& s
4,9-5,3 (м, 2Н, ); 5,6-6,3 (м, 1Н, )| 7,0-7,6 (м, ЗН, арома- тнч.), 8,98 (широкий с, 1Н, NH) .
ИК (КБг): 3600-3100 (ОН), 3400 (NH), 1710 (С - 0),
Пример 2. 1,8-Диэтил- 2,3,4,, 9-тетрагидро-4- (фенилметил) - 1Н-карбазол-1-уксусная кислота
п г 0 5 0
5
5
0
5
а)Получение метилового эфира 1-этил-2-оксо-4-(фенилметил)цикло- гексануксусной кислоты.
Исходный енон, метиловьй эфир 1-этил-2-оксоциклогекс-3-енуксусной кислоты, полученный по примеру 1, стадия в,(56,26 ммоль, 11,04 г), MeeS (11,25 мл) и 5,626 ммоль (1,15 г) CuBr. MeeS перемешивают в 165,5 мл тетрагидрофурана при -40°С в атмосфере азота и добавляют к смеси по каплям 56,26 ммоль (28,1 мл 2М раствора в тетрагидрофуране) PhCH/jMgCl. Реакцию затем прекращают, добавляя к реакционной смеси 150 мл 1М НС1 и четырежды проводят экстракцию петролейным эфиром, порциями по 100 мл. Объединенные органические фазы высушивают над и концентрируют. В результате флеш- хроматографки (колонка высотой 95мм, 12%-ный этилацетат в петролейном эфире в качестве элюента) получают 11,20 г (38,84 ммоль, выход 69%) смеси изомеров в виде маслянистой жидкости желтого цвета.
б)Получение метилового эфира 1,8- дизтил-2,3,4,9-тетрагидро-4-(фенил- метил)-1Н-карбазол -1-уксусной кислоты.
Смесь изомеров кетона, полученную на стадии а, (74,56 ммоль, 17,775 г) и 2-этилфенилгидразин (74,56 ммоль, 10,156 г) кипятят с обратным холодильником в 320 мл МеОН в атмосфере азота в течение 112 ч. Реакционную смесь затем охлаждают, добавляют к ней 112 ммоль (8,78 г, 8 мл) AcCl и кипятят в течение еще 45 мин. Реакционную смесь затем концентрируют в вакууме и подвергают флеш-хроматографии (колонка высотой 95 мм, 12%-ный этилацетат в петролейном эфире в качестве элюента, высота слоя силикагеля 5,5 дюйма), в результате чего получают 8,922 г (22,9 ммоль, выход 31%) смеси изомеров в виде маслянистой жидкости желтого цвета.
в)Получение 1, 8-д.иэтил-2,3,4,9- тeтpaгидpo-4-(фeнилмeтил)-1H-кapбa- зол-1-уксусной кислоты.
Смесь изомеров эфира (22,9 ммоль, 8,922 г), полученную на стадии б, и К4С03 (27,48 ммоль, 3,798 г) кипятят с обратным холодильником в 183 мл и 23 мл воды в атмосфере азота в течение 26,5 ч. Большую часть МеОН отгоняют в вакууме, а остаток растворяют в 50 мл воды. Полученный раствор подки
сляют до рН/ 1 ЗМ водным раствором НС1, четырежды проводят экстракцию эфиром, порциями по 50 мл, высушивают органическую фазу над MgSO и концентрируют ее, В результате флеш- хроматографии (колонка высотой 75 мм, 50%-ный этилацетат в петролейном эфире в качестве элюента, толщиной слоя силикагеля 5,5 дюйма) получают 6,7712 г (18,03 ммоль, выход 79%) маслянистой жидкости желтого цвета. С помощью обратнофазной (С j) хроматографии выделяют примерно по 1 г каждого из изомеров, которые пере- кристаллизовывают из смеси петролей- ного эфира и бензола в соотношении примерно 2:1. В результате получают белые кристаллы. Оба изомера высушивают в вакууме (72°С, силикагеля в качестве осушителя) в течение 8 ч. Первый изомер, вымываемый при обратнофазной хроматографии (смесь СН3С1 и НаО в соотношении 60:40 с добавкой 0,001 М КН2РО,(), обозначенный как изомер А, имеет температуру плавления 185-186 С. Второй вымываемый изомер, обозначенный как изомер В, имеет т.пл. 181-184°С.
Изомер А: ЯМР (CDC13 (тетраметил- силан): 0,9 (т, ЗН, J 9, СНаСНэ); 1,36 (т, ЗН; J 9, ЛгСН0СН3); 7,5- 2,3 (м, 7Н, цикл. CHg. и СИ); 2,6- 3,6 (м, PhCHrj, ArCHrj, ); 7,07,6 (м, 8Н, ароматич.); 8,9 (широкий с, 1Н, NH).
ИК (КВг): 3440 (NH); ЗбОО-ЗООО(ОН) 3060 (СН ароматич.); 3000-2880 (СН алифатич.); 1710 ().
Изомер В: Я11Р (CDClj/тетраметилси- лан): 0,88 (т, J 9, ЗН, СНаСК3); 1,36 т, J 9, ЗН, АгСМ,СК4); 1,6- 2,2 (м, 7Н, цикл. СИ, и СН); 2,7- 3,5 (м, 6Н, PhCHqi, ArCIU, CHa,COO); 6,9-7,6 (м, ароматич.); 9,0 (широкий, с, 1Н, NH).
ИК (КВг): 3600-2500 (СН); 3420 (NH); 1700 ().
П р и м е р 3. Цис-1-этил-2,3,4,9- тетрагидро-4-(2-пропенил)-1Н-карбазол 1-уксусная кислота.
а) Получение метилового эфира цис-1-этил-2,3,4,9-тетрагидро-4-(2- пропенил)-1Н-карбазол-1-уксусной кислоты.
5 г (20,98 ммоль) метилового эфира цис-1-этил-2-оксо-4-(2-пропенил)цикло гексануксусной кислоты, полученного по примеру 1, стадия г, и 2,27 г
10
- ,. 25
.
.„
0
5
5
5
(21 ммоль, 2,07 мл) 2-фенилгидразина кипятят с обратным холодильником в атмосфере азота в 21 мл толуола для удаления воды (азеотропная перегонка) . Через 24 ч толуол отгоняют в вакууме, а остаток растворяют в 15 мл уксусной кислоты. К полученному раствору добавляют 27,27 ммоль (3,87 г, 3,35 мл) бортрифторэтерата и кипятят смесь с обратным холодильником в атмосфере азота в течение 20 минут. Затем ее выливают в 40 мл воды и четырежды проводят экстракцию эфиром порциями по 40 мл. Объединенные органические фазы дважды промывают насыщенным водным раствором бикарбоната натрия порциями по 20 мл и высушивают над сульфатом магния. После флеш- хроматограиши (колонка высотой 75 мм, 7%-ный этилацетат в петролейном эфире в качестве элюента, высота слоя силикагеля 5,5 дюйма) получают 3,04 г (9,81 ммоль, выход 47%) маслянистой жидкости оранжевого цвета. Rr 0,65 при использовании в качестве подвижной фазы 15%-ного этилацетата в петролейном эфире.
ИК (чистый) 3490 (NH); 2850-3050 (СН); 1715 ().
ЯМР (CDClj/тетраметилсилан, 200 мГц): 0,836 (т, J 7,5, ЗН СНс.СК,); 1,258 (т, J 7,15, ЗН, АгСНгСНэ); 1,6-2,4; 2,6-2,8 (м, 8К, CLUCH}, CHQCHcC, .е); 2,664 (5, 2Н, СНвСООМе); 3,05 (м, ,СН) ; 3,703 (с, ЗН, ОСНг); 5,0-5,1 (м, 2Н, C-CHi); 5,8-6,0 (м, 1Н, ); 7,0-7,6 (м, 4Н, ароматич.); 9,3 (широкий с, 1Н, NH). I
б) Получение цис-1-этил-2,3,4,9- тетрагидро-4-(2-пропенил)-1Н-карба- зол-1-уксусной кислоты.
Раствор исходного эфира, метилового эфира цис-1-этил-2,3,4,9- тетрагидро-4-(2-пропенил)-1Н-карбазол- 1-уксусной кислоты (9,71 ммоль, 3,01 г) и карбонат калия (14,57 ммоль, 2,01 г) кипятят с обратным холодильт ником в атмосфере азота в 78 мл метанола и 9,7 мл воды в течение 24 ч. После этого большую часть метанола отгоняют в вакууме, а остаток суспендируют в 15 мл воды. Суспензию подкисляют до рН 1 1М НС1 и четырежды проводят экстракцию эфиром порциями по 60 мл. Объединенные эфирные фазы высушивают над сульфатом магния и концентрируют. После флеш-хромато
1114
графин (.колонка диаметром 50 мм, 25%-ный этилацетат в петролейном эфире в качестве элюента, высота слоя силикагеля 5,5 дюйма) получают 2,72 г (9,19 ммоль, выход 95%) продукта в виде маслянистой жидкости.. Кристаллизация его из смеси петролейного эфира и бензола в соотношении 85:15 дает 2,2 г грязно-белого порошко- образного продукта. Высушенный при 78 °С над пятиокисью фосфора в течение 7 ч он имеет т. пл. 103-105 C.
Рассчитано, %: С 76,74; Н 7,79; N 4,71.
Найдено, %: С 76,84; I 7,70; N 4,73.
ИК (КВг): 3420 (NH); 3600-3000 (ОН); 2860-3100 (-СН); 1715 (),
В примерах 4-7, 11, 10, 12-19, 21, 24, 25-30, 32, 34,- 36, 37 к 39 метиловый эфир цис-1-этил-2-оксо- 4-(2-пропенил)-циклогексануксусной кислоты (полученный по способу и соответствии с примером 1, стадия г) подвергался взаимодействию с соответствующим арилгидразином при кипячении с обратным холодильником в среде толуола в течение 12-100 часов с образованием в результате гидра- зона, после чего полученный продукт концентрировали в вакууме и растворяли в уксусной кислоте. После добавления к раствору эфирата трех- фтористого бора и кипячения смеси с обратным холодильником в течение 5- 260 мин получали соединения формулы (41), которые затем подвергали гидролизу с помощью гидроокиси натрия в среде водного раствора этанола с образованием в результате соединений формулы (1).
В случае примеров 8 и 11 метиловы эфир 1--метил-2-оксо 4-(2 пропенил)- циклогексануксусной кислоты (полученный по способу в соответствии с примером 1, стадия г, с той разницей что вместо 2-этилциклогексанона в качестве исходного материала использовали 2-метилциклогексанон в соответствии с примером 1, стадия б) подвер гали взаимодействию с соответствующи гидразином с последующим гидролизом,
В случае примера 20 процесс проводили так, как в примерах 8 и 11, н в качестве исходного материала вмест 2-метилциклогексанона использовался 2-прогпшциклогексанон.
В случае примеров 22 и 33 метиловый эфир 4-(1,1-диметил 2-пропе
5
0
5
9
5 0
5 0
5
0
нил)-1-э тил-2-оксоциклогексануксус- ной кислоты (полученный по способу в соответствии с примером 1, стадия г, с той разницей, что вместо аплилтри- метилсилана использовался (З-метил-2- бутенил)-трибутилин подвергали взаимодействию с соответствующим гидразином с последующим гидролизом.
В случае примеров 23 и 38 процесс проводили так, как в примерах 22 и 33, с той разницей, что вместо (3- метил-2-бутенил)-трибутилина использовали (2-метил-2-пропенил) трибу- тилин.
В случае примера 25 метиловый эфир 1-этил-2-оксо-4-(фенил-метил)- циклогексануксусиой кислоты (полученный по способу в соответствии с примером 2, стадия а) подвергали взаимодействию с фенилгидразином с последующим гидролизом
Пример 40. Бензметанаминовая соль (Ш-цис)-1,8-диэтил-2,3,4,9- тётрагидро-4-(2-пропенил)-1Н-карба- зол-1-уксусной кислоты.
а) Получение 2,3-диметокси-стрих- нидин-10-оновой соли (1:1) (1К-цис)- 1,8-ДИЭТИЛ-2,3,4,9-тетрагидро-4-(2- пропенил)-1И-карбазол-1-уксусной кислоты,
6 ммоль (1,947 г) цис-1, 8-диэтил- 2,3,4,9-тетрагидр0-4-(2-пропенил)- 1Н-карбазол-1-уксусной кислоты (полученной по способу в соответствии с примером 1, стадия е) и 6,0 ммоль (2,583 г) 2,3-диметокси-стрихни- дин-10-он дигидрата (дигидрат бру- цина) растворяют в 25 мл горячего этанола. К полученному прозрачному раствору добавляют 6,25 мл воды и оставляют его на ночь при комнатной температуре. Выпадающие белые кристаллы соли отфильтровывают и промывают 5 мл смеси этанола и воды в соотношении 1:1. Маточный раствор используют для выделения второго энан- тиомера в примере 45 на стадии а. Полученную соль (1,94 г) снова помещают в горячий этанол (14 мл) и добавляют по каплям 3,5 мл воды, не давая раствору остывать. Затем раствор оставляют на ночь при комнатной температуре. Выпадающие в результате кристаллы (1,73 г) отфильтровывают, промывают 5 мл смеси этанола и воды в соотношении 1:1 и высушивают в течение ночи в вакууме Выход полученного соединения составляет 80% от теоретического; т. пл. 128-130 00
б) Бензметанаминовая соль (1:1) (1R-цис)-1,8-ДИЭТИЛ-2,3,4,9-тетра- гидро-4-(2-пропенил)- 1Н-карбазол- 1-уксусной кислоты.
Исходную 2,3-диметокси-стрихнидин 10-оновую соль (1:1) (1К-ция)-1,8- диэтил-2,3,4,9-тетрагидро-4-(2-про- пенил)-1Н-карбазол-1-уксусной кислот (1,73 г) суспендируют в 100 мл эфира и добавляют к суспензии при перемешивании 1М соляную кислоту. После разделения органической соли промывают насыщенным водным раствором хлористого натрия, высушивают его над сульфатом натрия, фильтруют и упаривают получая бесцветную маслянистую жидкость (765 мг, выход 99%). Полученны продукт растворяют в 5 мл эфира и добавляют к раствору раствор бензмета- намина (255 мг) в 2 мл эфира. В результате образуется прозрачный раствор. По истечении 2 ч при комнатной температуре из него выпадает осадок кристаллического продукта. Раствор помещают на ночь в холодильник. После этого кристаллы отфильтровывают, промывают небольшим количеством эфира и высушивают в высоком вакууме при комнатной температуре, В результате получают 715 мг целевого соединения (т. пло 133,5-134°С (У)ъ -93 ). Хиральная чистота полученного продукта, определенная с помощью высокоскоростной жидкостной колоночной хроматографии на метиловом эфире свободной кислоты на хиральной хроматографической колонке, равнялас 99,9%. Абсолютную и относительную конфигурацию определяли с помощью рентгенографической кристаллографии.
Рассчитано, %: С 77,74; Н 8,39; N 6,48.
Найдено, %: С 77,58; Н 8,16; N 6,51.
ИК (КВг): 3340 (NH): СООН (широкая); 3080-2850 (СН); 640 ().
ЯМР (CDC13 , 200 мГц): 0,85 (т, ЗН, J 7,5); 1,32 (т, ЗН, J 7,6); 1,6-2,3 (м,7Н); 2,6 (м,2Н); 2,7 (м, 1Ю; 2,80 (q, 2H, J 7,6); 3 (м, 1Н); 3,86 (с, 2Н); 4,3 (широкий с); 5,5,2 (м, 2Н); 5,8-6 (м, 1Н), 6,9-7,1 (м, 2Н); 7,2-7,4 (м, 6Н), 9,9 (широкий с, 1Н).
П р им е р 41. Бензметанаминовая соль (1:1) (15-цис)-1„8-диэтил- 2,3,4,9-тетрагидро-4-(2-пропенил)- 1Н-карбазол-1-уксусной кислоты.
0
5
0
5
0
5
0
5
0
5
а)Получение 2,3-диметокси-стрих- нидин-10-оновой соли (1:1) (15-цис)- 1,8-диэтил-2,3,4,9-тетрагидро-4(2-пропенил)-1Н-карбазол-1-уксусной кислоты.
Маточный раствор после первой кристаллизации в примере 44, на стадии а насыщают водой (5 мл) и помещают на ночь в холодильник, Выпадающие кристаллы отфильтровывают и промывают смесью этанола и воды (10 мл) в соотношении 1:1, В результате получают обогащенную смесь соли бру- цинад которую растворяют в 10 мл горячего этанола. К раствору добавляют 1 мл воды и вносят в него затравку продукта, полученного в примере 44 . на стадии я. Раствор с затравкой оставляют на ночь при комнатной температуре. Выпадающие кристаллы (385 мг) отфильтровывают, а маточный раствор насыщают водой (30 мл). Выпадающий осадок (1,5 г) растворяют в 8 мл горячего этанола, добавляют к раствору 3 мл воды и оставляют его на ночь при комнатной температуре. Выпадающие кристаллы отфильтровывают и добавляют к фильтру 5 мл воды. При кристаллизации, которая протекает в течение ночи (раствор оставляют в холодильнике) , образуется белое кристаллическое соединение С 1,1 г). После двойной перекристаллизации из горячего этанола (3 мл) и воды (1,5 мл) при комнатной температуре получают соль бруцина (600 мг), выход 28%). Хиральная степень чистоты, определенная с помощью высокоскоростной жидкостной колоночной хроматографии на метиловом эфире свободной кислоты, равна.99,9%.
б)Бензметанаминовая соль (1:1)
(18-цис)-1,8-диэтил-2,3,4,9-тетрагид- ро-4-(2-пропенил)-1Н)-карбазол-1-уксусной кислоты.
Соль бруцина (600 мг), полученную на стадии а, суспендируют в эфире и добавляют к суспензии Ш соляную кислоту. После разделения органический слон промывают насыщенным водным раствором хлористого натрия, высушивают его над сульфатом натрия, фильтруют и упаривают, получая в результате бесцветную маслянистую жидкость (265 мг, выход 99%). Полученный продукт растворяют в 1,5 мл эфира и добавляют к раствору бензметанамина (88 мг) в 0,5 мл эфира. При стоянии в течение 2 ч при комнатной температуре, а затем в течение ночи в холодильнике из раствора выпадает осад кристаллического продукта. Кристаллы отфильтровывают, промывают небольшим количеством эфира и высушивают в вакууме при комнатной температуре. В результате получают 248 мг целевого соединения (выход 70%, т. пл. 133- 134°С, (oOjj +91,5° хиральная сте- пень чистоты, определенная с помощью высокоскоростной жидкостной колоночной хроматографии на метиловом эфире свободной кислоты, равна 99,9%.
Рассчитано, %: С 77,74; И 8,39; N 6,48.
Найдено, %: С 77,75; Н 8,29; N 6,49.
ИК (КВг): 3340 (NH); СООИ (широкая) ; 3080-2850 (СН); 1640 ().
ЯМР (CDC13, 200 мГц): 0,85 (т, ЗН, J 7,5); 1,3 (т, ЗН, J - 7,6); 1,6-2,3 (м, 7Н); 2,5 (м, 2Н) 2,65 (м, 1Н); 2,8 (q, 2H, J 7,6); 3 (м, 111), 3,85 (с, 2И); 4,63 (широ- кая с); 5-5,2 (м, 2Н) ; 5,8-6 (м, 1Н); 6,9-7,1 (м, 211); 7,2-7,4 (м, 6Н); 10 (широкий с, 1Н).
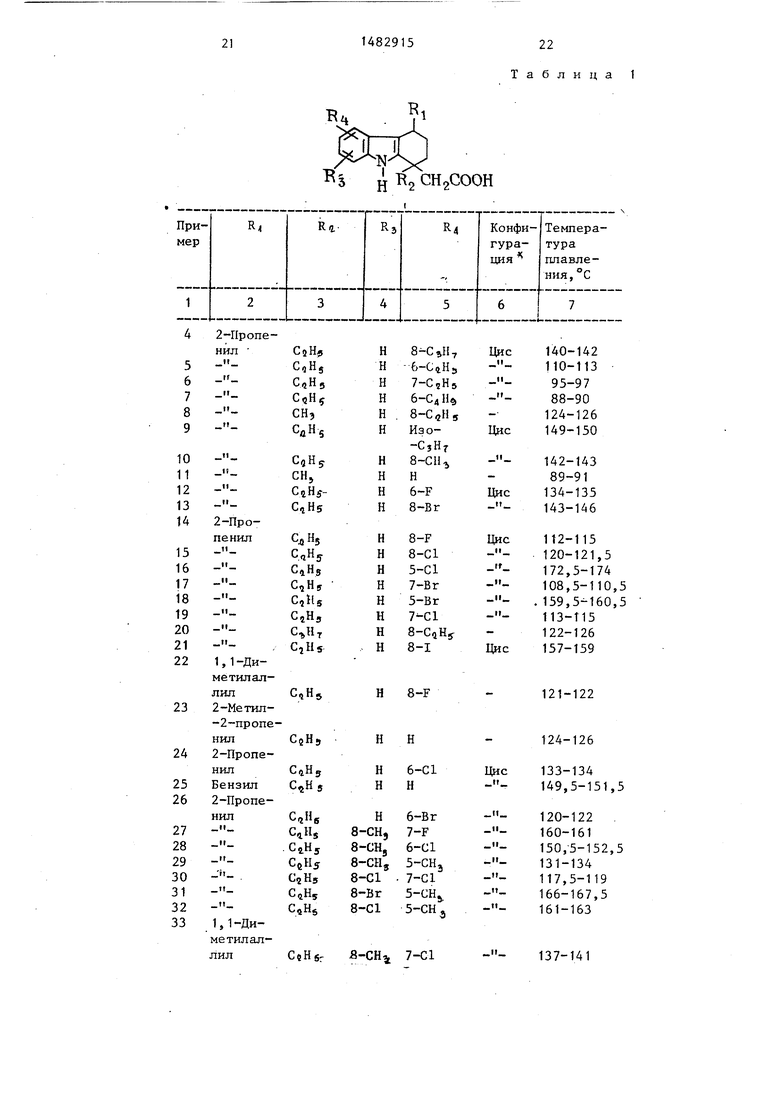
Температуры плавления соединений (I) приведены в табл. 1.
Противовоспалительное действие соединений (I) иллюстрируется с помощью стандартных фармакологических тестов, например теста, именуемого предупреждение отечности. Назна- чение этого теста состоит в определении способности испытуемых лекарст оказывать противовоспалительное действие на крысах. Результаты этого теста используются для первичного от бора лекарств, оказывающих противовоспалительное действие.
В качестве подопытных животных использовались самцы крыс Sprague Dawley весом 180-200 г. За 18 ч до проведения испытаний животных переставали кормить, но давали им вдоволь воды.
Полный адырант Фрейнда готовили путем суспендирования 5 мг мертвых высушенных микробактерий butyricum (Difeo) в 1 мл жидкого парафина. Испытуемые соединения растворяли в дистиллированной воде или суспендировали в дистиллированной воде с добавкой нескольких капель Твин 80 в зависимости от их растворимости Для первичного отбора все препараты вводили перорально после промывания
желудка в количестве 0,5 мл/100 г веса при дозе 100 мг/кг. Каждая группа состояла из 10 животных.
Животным вводили внутрикожно в левую заднюю лапу 0,1 мл полного адъю анта Фрейнда. Испытуемое соединение или индиферентный носитель вводили непосредственно перед адьювантом и через 24 и 48 ч (через 0,1 и 2 дня) после адъюванта. Объем задней лапы, в которую проводилась инъекция,измеряли с помощью плетизмометра (Buxco Electronic Inc.) перед введением адъюванта и через 24 ч после последнего введения испытуемого препарата (через три дня). Разница между последним и первоначально измеренным объемом задней лапы представляет собой объем отечности. В качестве контрольного лекарства использовали фе- нилбутазон (вводился перорально в количестве 50 мг/кг).
Рассчитывали объем отечности (в мл + SEM) для каждой группы поДопыт- ных животных и вычисляли процент защиты (в результате действия лекарства) по формуле
(c-t) 100 % защиты 1
где с - объем отечности для контрольных животных (которым не вводилось лекарство), t - объем отечности для животных, которым вводилось испытуемое лекарство.
Другой тест, проводившийся для определения полезности соединений в соответствии с настоящим изобретением, назывался Влияние лекарств на пишущее движение у мышей, индуцированное фенилхиноном.
Назначение этого теста заключается в определении способности испытуемых лекарств тормозить чувствительный к боли ответ у мышей, кото7 рым вводился химический раздражитель Этот тест используется для первичного отбора болеутоляющих лекарств, действующих как на периферическую, так и на центральную системы.
В качестве подопытных животных использовались самцы швейцарских белых мышей весом 15-25 г. За 18 ч до начала испытаний животных не кормили, но давали им вдоволь воды.
Испытуемые лекарства растворяли или суспендировали в зависимости от
их растворимоет.i в 0,5%-ной метил- целлюлозе или 0,5%-ном растворе Твин 80. Лекарства вводили путем промывания желудка в количестве С, 5 мл/кг. Для первичного отбора вводимая доза всех лекарств составляла 200 мг/кг. Каждая группа подопытных животных состояла из 10 мышей.
ТруППаМ ЖИВОТНЫХ, СОСТОЯВШИМ ИЗ
5 мышей, вводили испытуемое соединение или контрольный индиферентный носитель. Через 60 мин после этого животным вводили внутрибрюшинно 0,02%-ный раствор фенилхинона (PBQ, 2-фенил-1, 4-бензохинон) в количеств 0,3 мл на 2Д г веса и помещали их в индивидуальные боксы для наблюдения. При этом подсчитывалось количество пишущих движений и конвульсивных абдоминальных движений, совершаемых каждой мышью в течение последующих 15 мин. Опыт повторялся с другой группой из 5 мышей и рассчитывалось количество движений, приходящееся на каждую мышь для группы из 10 мышей.
Сравнивались опыты с контрольными мышами и мышами, которым вводилось лекарство, и по результатам опытов рассчитывался процент защиты по формуле
(c-t) IOO % защиты )
где с - количество пишущих движений у мышей из контрольной группы,
t - количество пишущих движений у мышей, которым вводи- лось лекарство.
Кроме того, для определения полезности предлагаемых соединений проводился тест под названием Тест на давление на лапу на крысах. Назна- чение этого теста состоит в оценке способности лекарств, оказывающих действие на периферическую и центральную систему, тормозит реакцию на боль, вызываемую при воздействии на воспаленную лапу.
В качестве подопытных животных использовались самцы крыс Spraque Dawiey весом 180-200 г. Ночью, перед введением лекарства, животным не давали есть.
Полный адъювант Фрейнда (ПАФ) готовили путем суспендирования мертвых и высушенных микробактерий butiri-
|п
20 5
0
5
0
.с
0
5
cum (Difeo) 1 мл жидкого парафина. Испытуемые соединения растворяли или суспендировали в зависимости от их растворимости в дистиллированной воде с добавкой нескольких капель. Tween 80. Приготовленные препараты вводили путем промывания желудка в количестве 0,5 мл на 100 г веса. Каждая группа состояла из 10 животных.
Каждая группа подопытных животных состояла из 10 крыс. Аппаратура представляет собой устройство, создающее усилие, возрастающее с постоянной скоро стьк. Это усилие непрерывно контролируется с помощью стрелочного устройства, перемещающегося вдоль линейной шкалы, и измеряется в граммах. Воспалительная реакция возбуждалась у крыс путем введения им внут- рикожно в левую заднюю лапу 0,1 мл адъюванта Фрейнда. Испытуемое соединение или индиферентный носитель вводили через 24 часа после адъгаван- та. Перед введением лекарства производили отбор крыс по чувствительности их лап. В опытах использовали только крыс, которые реагировали на болевой порог ниже 10 г (усилие, приложенное к воспаленной лапе). Болевой порог (писк, издаваемый при ощущении боли) определяли через 1 и 3 ч на воспаленной и больной лапе у животных из контрольной группы и у животных, которым вводилось лекарство. При оценке действия лекарства на болевой порог использовалось большее из двух полученных значений.
Принималось, что вводимое соединение оказывает действие, если определенный болевой порог был в полтора раза выше, чем у животных из контрольной группы. В каждой группе определялось количество животных, у которых наблюдалось обезболивающее действие вводимых соединений.
После этого определялась величина EDgo (доза, при которой эффект болеутоления наблюдается у 50% животных) при использовании по меньшей мере 3 доз.
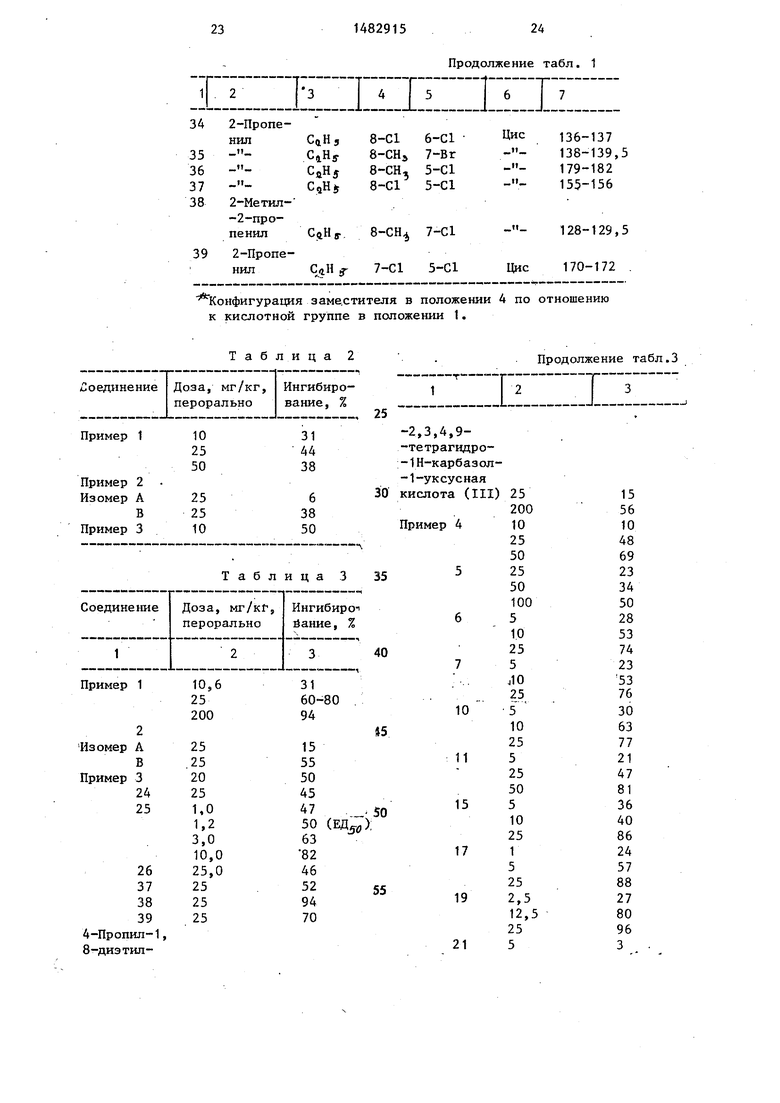
Данные биологических испытаний представлены в табл. 2-5.
Соединение (I) при испытуемых дозах не проявляли признаков токсичности.
Таким образом, соединения (I) обладают высокой болеутоляющей и противовоспалительной активностью, пре-
восходящей известную 4-пропил-1,8- -диэтил-2,3,4,9-тетрагидро-1Н-карба- зол-1-уксусной кислоты (III)..
Формула изобретения
В
з н R2 CH-POOH
де a) Е.Ц - бензилу R - этил1, RS - водород; R 4 - водород или 8-этил;
б)R - 2-метил 2-пропенил; E.J - этил;
RS R/) - водород или
7-хлор1, R/1 - 8-метил,
в)R,, - 1,1-диметил-2-пропенил; RQ - этил,
RJ т водород}
R4 - 8-фтор или R3 - 7-хлор, R - 8-метил;
г)Ry - 2-пропенил, Rg - метил,
R3R.
водород.
R4 водород ИЛИ 8-ЭТИЛ, ИЛИ RjJ
этил, RJ - водород, R/S водород, 8-метил, 6-s 7- или 8-этил, 8-пропил, 8-изопропил, 6-бутил, 6- или 8-фтор, 5-, 6-, 7- или 8-хлор, 5-, 6-, 7- или 8-бром, 8-иод или Ry - 8-метил, - 5-метил, 7-фтор, 5 или 7-хлор, 7-бром, или RJ - хлор, R4 - 5-метил, 5-, 6-, или 7-хлор, или R, - 8-бром, R4 - 5-метил, или Ry - 7-хлор, R$ - 5-хлор, или Rg - пропил, RJ - водород, R/J - 8-этил, или бензмв танаминовая соль соединения (I) , где R( - 2-пропенил, RJ. и R - этил, R3 - водород,
роявляющие болеутоляющее и противо
оспалительное действие.
10
кд н кг сн2соосн3
5
0
5
0
5
0
5
0
5
где a) R - бензил; Ru - этил; R3 - водород; R4 - водород или 8-этил;
б)R - 2-метил-2-пропенил; Rt - этил;
Rj R о, - водород или Ra 7-хлор;R4 8-метил,
в)R - 1, 1-диметил-2-пропенил, R4 - этил,
R - водород, R4 - 8-фторJ или RS 7-хлор; R - 8-метил,
г)Rf - 2-пропенил; - метил;
R - водород,
RJI - водород, 8-метил, 6-,
7- или 8-этил, 8-пропил, 8-изопропил, 6-бутил,
6-или 8-фтор, 5-, 6-,
7-или 8-хлор, 5-, 6-, 7- или 8-бром, 8-иод или RJ - 8-метил, R - - 5-метил, 7-фтор, 5- или 7-хлор, 7-бром, или RJ - 8-хлор, RJ - 5-метил, 5-, 6- или 7-хлор, или RS - 8-бром, R л - 5-метил, или RJ - 7-хлор, RH - 5-хлор, или RJ. - пропил, R, - водород,
&л - 8-этил,
как промежуточный продукт для синтеза производных 2,3,4,9-тетра-гидро- -1Н-карабазол-1-уксусной кислоты, проявляющих болеутоляющее и противовоспалительное действие.
Приоритет по признакам: 22.04.85 при R - 2-пропенил или бензил, R$. - этил, Rg - водород, R# п водород или 8-этил,
#3 н 2 СН2СООН
2-ПропеТаблица 1
Продолжение табл. 1
2S Продолжение табл.3
-1482915
26 Продолжение табл.3
Таблица 4
27148291528
Продолжение табл.4
Примечание. Приведенные, значения представляют собой процент торможения при дозах, мг/кг, величины которых даны в скобках, или величину ЕД5в, мг/кг.
Таблица 5
| Патент ША Р 4057559, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
