Область техники, к которой относится изобретение
Изобретение относится к способам синтеза и промежуточным продуктам синтеза макроциклического ингибитора протеазы вируса гепатита С (HCV).
Предпосылки создания изобретения
Вирус гепатита С (HCV) является основной причиной распространенного во всем мире хронического заболевания печени. После первоначальной острой инфекции у большинства инфицированных индивидуумов развивается хронический гепатит, так как HCV предпочтительно реплицируется в гепатоцитах, но не является непосредственно цитопатическим. Хронический гепатит может прогрессировать до фиброза печени, приводя к циррозу печени, конечной стадии заболевания печени, и НСС (печеночно-клеточному раку), являющемуся основной причиной трансплантаций печени. Это и большое количество пораженных болезнью пациентов делает HCV средоточием значительного медицинского исследования. Репликация генома HCV опосредуется большим количеством ферментов, среди которых находятся NS3-серинпротеаза HCV и связанный с ней кофактор, NS4A, который опосредует большое количество протеолитических расщеплений HCV-полипротеина, получающегося в результате генерации ферментов репликации HCV. Считают, что NS3-серинпротеаза является необходимой для вирусной репликации и представляет собой представляющую интерес мишень для нахождения лекарственного средства.
Современная анти-HCV-терапия базируется на (пэгилированном) интерфероне-альфа (IFN-б) в комбинации с рибавирином. Результатом этой терапии является не только ограниченная эффективность, так как только часть пациентов успешно излечивается, но и также в случае этой терапии сталкиваются со значительными побочными эффектами и плохой переносимостью препаратов множеством пациентов. Следовательно, существует потребность в более эффективной, пригодной и с улучшенной толерантностью терапии. Существует необходимость в дальнейших ингибиторах HCV, которые позволяют преодолеть недостатки современной HCV-терапии, такие, как побочные эффекты, ограниченная эффективность, возникновение резистентности, а также соответствующие неблагоприятные исходы.
Описаны различные агенты, ингибирующие NS3-серинпротеазу HCV и связанный с ней кофактор, NS4A. В Международной заявке WO-05/073195 раскрываются линейные и макроциклические ингибиторы NS3-серинпротеазы с замещенным центральным пролиновым остатком, а в случае Международной заявки WO-05/073216 - с центральным циклопентильным остатком. Среди них представляющими интерес являются макроциклические производные, которые проявляют явную активность против HCV и имеют хороший фармакокинетический профиль.
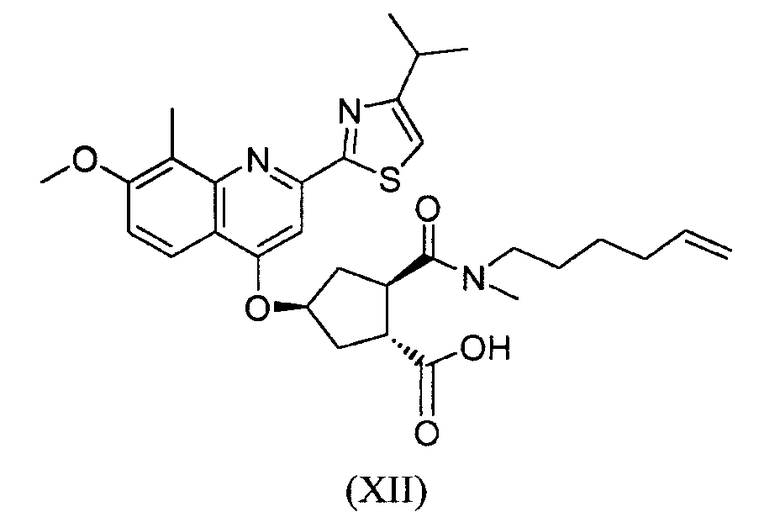
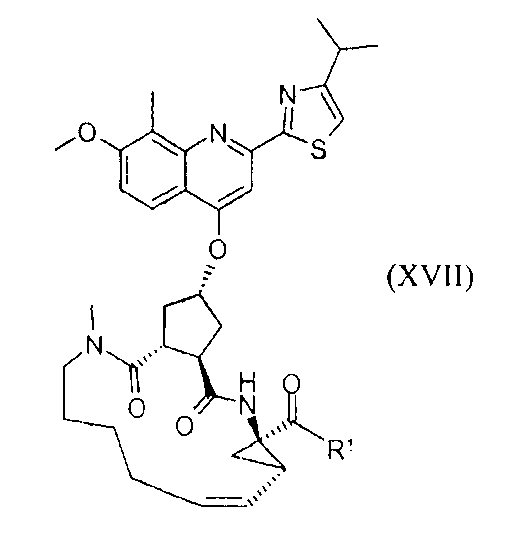
В настоящее время найдено, что конкретное макроциклическое соединение с центральным, замещенным хинолинилоксигруппой, циклопентильным остатком является особенно представляющим интерес с точки зрения эффективности, а также фармакокинетики. Оно представляет собой соединение формулы (XVII) со структурой, представленной ниже:

Соединение формулы (XVII) является очень эффективным ингибитором серинпротеазы вируса гепатита С (HCV) и описано в Международной заявке WO-2007/014926, опубликованной 8 февраля 2007 года. Вследствие его подходящих свойств, оно было выбрано в качестве потенциального «кандидата» для получения лекарственного средства против вируса гепатита С. Следовательно, существует необходимость получения больших количеств этого активного ингредиента на основе способов, которые обеспечивают получение продукта с высоким выходом и с высокой степенью чистоты.
Настоящее изобретение относится к способам получения соединения формулы (XVII) или его фармацевтически приемлемой соли, к получению промежуточных продуктов, используемых в этих способах, и к некоторым из этих промежуточных продуктов.
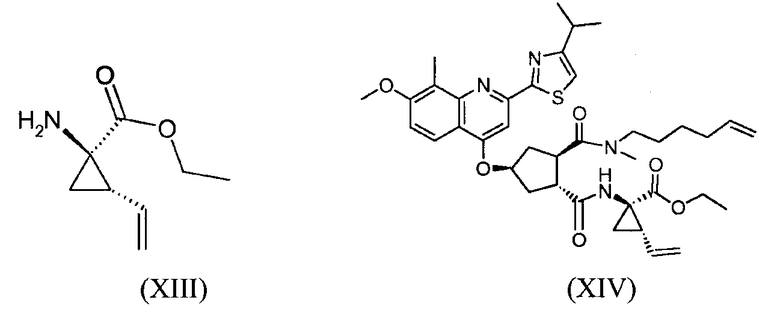
Соединение формулы (XVII) можно получать реакцией обмена, исходя из промежуточного продукта (XIV), который циклизуют для получения промежуточного продукта (XV), который затем гидролизуют до макроциклической кислоты (XVI). Последнюю связывают с сульфониламидом (XVII) посредством реакции образования амида, таким образом получая конечный продукт (XVII), как показано на следующей реакционной схеме:

Фармацевтически приемлемые солевые формы соединения формулы (XVII) можно получать путем введения во взаимодействие свободной формы этого соединения с кислотой или основанием.
В случае этой и последующих реакционных схем или представлениях индивидуальных соединений, например, в соединении (XIV), R означает С1-4-алкил, в особенности, R означает С1-3-алкил, более конкретно, R означает С1-2-алкил, или, в одном воплощении, R означает этил. Реакция превращения соединения (XV) в соединение (XVI) представляет собой реакцию гидролиза, которую предпочтительно проводят, используя основание, в водной среде, такой как смесь воды и растворимого в воде органического растворителя, такого, как тетрагидрофуран (ТГФ) или спирт, в особенности, спирт, из которого получают сложный эфир (XIV), или в смеси таких растворителей. Основание, которое используют, может представлять собой гидроксид щелочного металла, такой как, например, NaOH или KOH и, в особенности, он может представлять собой LiOH.
Промежуточный продукт (XIV) циклизуют реакцией обмена олефина в присутствии подходящего металлического катализатора, такого как, например, илиденовый катализатор на основе Ru, в особенности, необязательно замещенный акилиденовый или инденилиденовый катализатор, такой как бис(трициклогексилфосфин)-3-фенил-1Н-инден-1-илиденрутенийхлорид (Neolyst M1®) или бис(трициклогексилфосфин)[(фенилтио)метилен]-рутенийдихлорид. Другие катализаторы, которые можно использовать, представляют собой катализаторы первого и второго поколения Grubbs, т.е. бензилиденбис(трициклогексилфосфин)-дихлоррутений и (1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(фенилметилен)(трициклогексилфосфин)-рутений, соответственно. Особый интерес представляют катализаторы первого и второго поколения Hoveyda-Grubbs, которыми являются дихлор(о-изопропоксифенилметилен)-(трициклогексилфосфин)рутений(II) и 1,3-бис(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенил-метилен)рутений, соответственно. Реакции обмена можно проводить в подходящем растворителе, таком как, например, простой эфир, например, ТГФ, диоксан; галогенированные углеводороды, например, дихлорметан, CHCl3, 1,2-дихлорэтан и т.п., углеводороды, например, толуол. В предпочтительном воплощении, реакцию обмена проводят в толуоле.
Промежуточный продукт (XVI) может быть связан с циклопропилсульфонамидом реакцией образования амида, как например при использовании любого из способов образования амидной связи. В частности, соединение (XVI) можно обрабатывать с помощью связующего агента, например, N,N'-карбонилдиимидазола (CDI), N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (EEDQ), N-изобутилоксикарбонил-2-изобутилокси-1,2-дигидрохинолина (IIDQ), 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDCI) или бензотриазол-1-илокситриспирролидинофосфонийгексафторфосфата (коммерчески доступен как PyBOP®), в растворителе, таком как простой эфир, например, ТГФ, или галогенированный углеводород, например, дихлорметан, хлороформ, дихлорэтан, и вводить во взаимодействие с циклопропилсульфонамидом, предпочтительно, после взаимодействия соединения (XVI) со связующим агентом. Реакции соединения (XVI) с циклопропилсульфонамидом предпочтительно проводят в присутствии основания, например, триалкиламина, такого как триэтиламин или диизопропилэтиламин, или 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Промежуточный продукт (XVI) также можно превращать в активированную форму, например, в активированную форму, такую как галогенангидрид кислоты, в особенности, хлорангидрид или бромангидрид кислоты, или в активный сложный эфир, например, в кислоту, этерифицированную с помощью арилоксигруппы, такой, как фенокси, п-нитрофенокси, пентафторфенокси, трихлорфенокси, пентахлорфенокси и т.п.; или путем превращения макроциклической кислоты (XVI) в смешанный ангидрид.
Промежуточные продукты (XIV) являются исходными веществами для получения соединений формулы (XVII) и, следовательно, существует необходимость в разработке способов получения этих промежуточных продуктов при крупномасштабном производстве, т.е., в многотоннажном масштабе, или больше. Эти способы должны приводить к конечному продукту с высоким выходом и чистотой. В особенности, присутствие различных хиральных центров в молекуле вызывает особые проблемы, так как хиральная чистота является необходимой для получения продукта, который пригоден для терапевтического применения. Следовательно, способы получения соединения (XIV) должны приводить в результате к продуктам с пригодной хиральной чистотой, без применения обременяющих операций очистки с потерей значительных количеств нежелательных стереоизомерных форм.
Один из аспектов данного изобретения относится к способам получения промежуточных продуктов (XIV) с высоким выходом и чистотой, которые пригодны для крупномасштабного промышленного применения.
Данное изобретение также относится к промежуточным продуктам, которые пригодны для получения соединений формулы (XVII). Ряд таких промежуточных продуктов представляет собой:
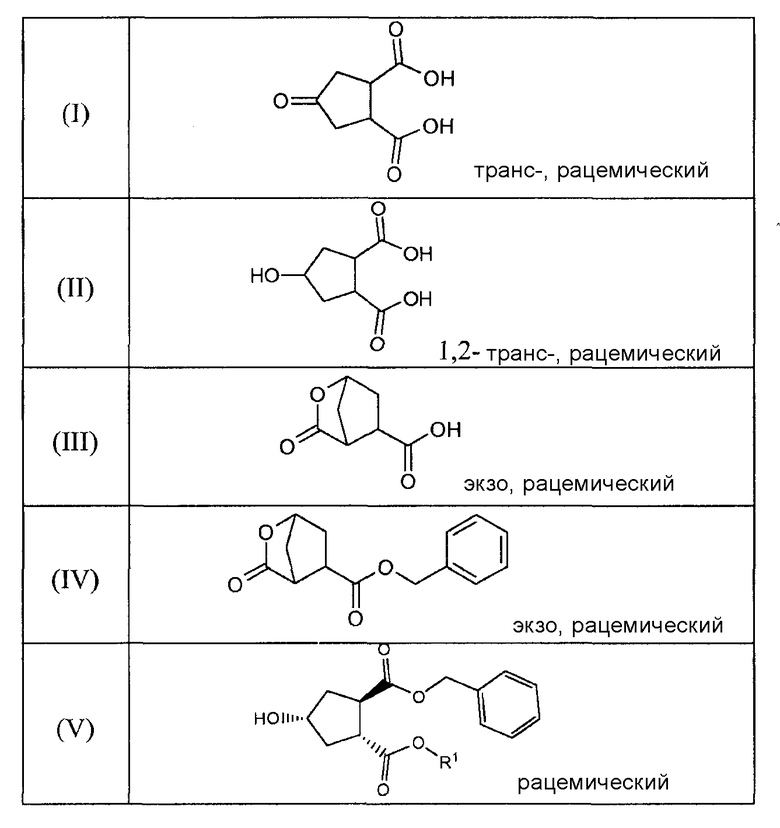
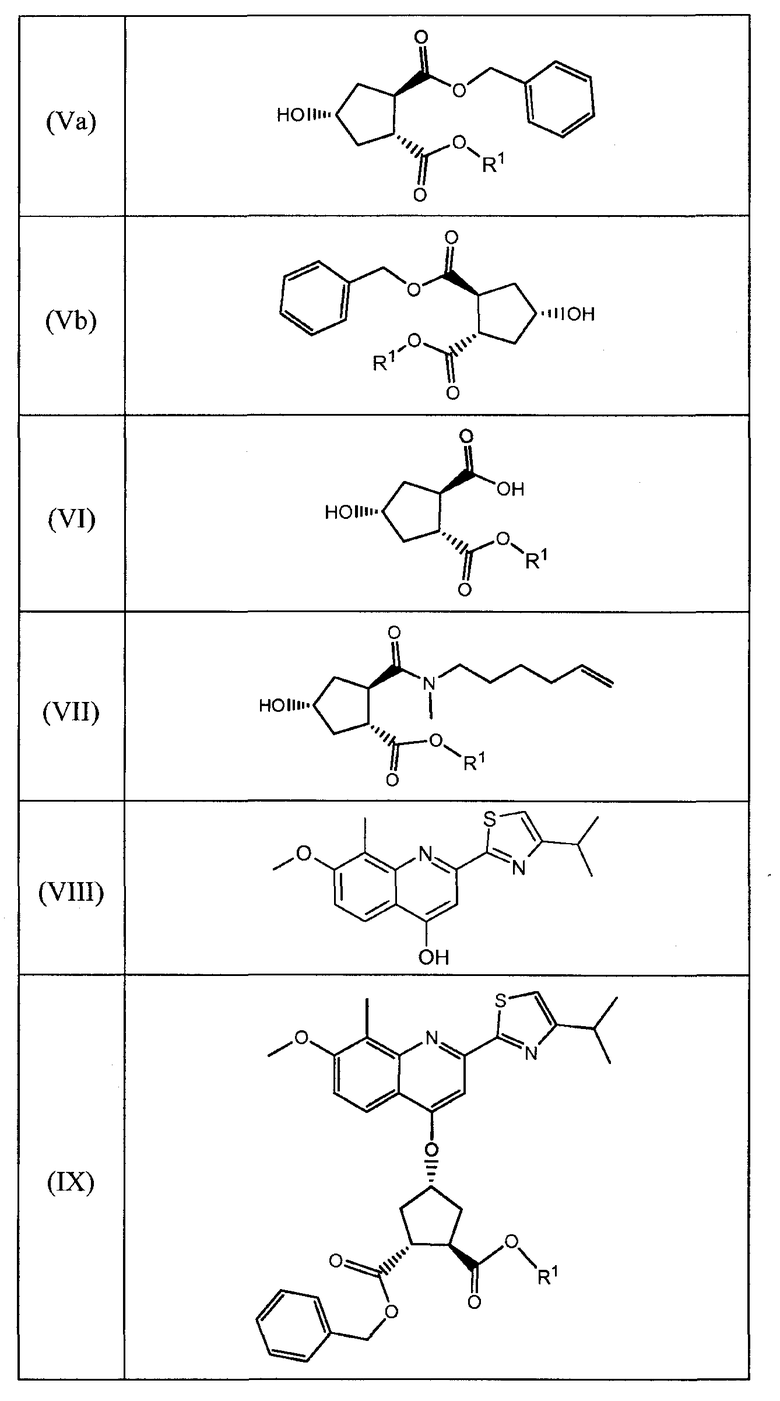


В соединениях, перечисленных в вышеприведенной таблице, R1 имеет значение, определенное ниже, и R имеет значение, как определено выше. В одном воплощении, R1 означает метил. В другом воплощении, R означает этил.
Honda и др., Tetrahedron Letters, том 22, № 28, сс.2679-2682, 1981, раскрывают синтез (±)-брефельдина А при использовании следующих исходных веществ:
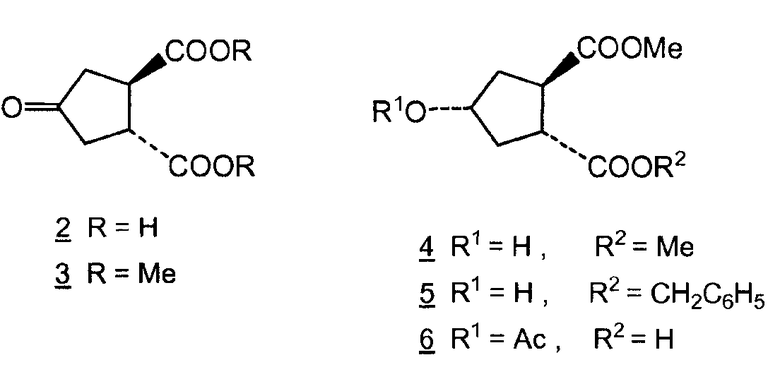
Синтез согласно Honda и др. осуществляют, исходя из dl-транс-4-оксоциклопентан-1,2-дикарбоновой кислоты (2), которую этерифицируют с получением соответствующего метилового эфира (3) и восстанавливают при использовании никеля Ренея до спирта (4). Частичный гидролиз продукта 4 и бензилирование приводят к получению преимущественно одного диастереоизомера сложного эфира 5, т.е. такого диастереоизомера, где гидроксильная и карбоксильная группы находятся в цис-положении. Последний сложный эфир 5, согласно Honda и др., и соединение (V) оба являются рацематами, но диастереоизомеры у каждого являются другими, более точно, эпимерами относительно атома углерода №4, несущего гидроксильную группу. Соединение (Va) представляет собой один из двух энантиомеров, получаемых разделением рацемического соединения (V). Другой энантиомер является соединением (Vb).
В Международной заявке WO-2005/073195 описывается синтез энантиомерночистого бициклического лактона (8b), исходя из энантиомера 3,4-бис(метоксикарбонил)циклопентанона. Последний получают, как описано Rosenquist и др. в Acta Chemica Scandinavica, 46, 1127-1129 (1992). Транс-изомер (3R,4R)-3,4-бис(метоксикарбонил)циклопентанона превращают в бициклический лактон (8b):

В Международной заявке WO-2005/073195, кроме того, далее, описывается модификация лактона (8b) до сложного трет-бутилового эфира, раскрытие лактона и связывание с соответствующим образом защищенными аминокислотами, например, с этиловым эфиром (1R,2S)-1-амино-2-винилциклопропанкарбоновой кислоты, где, в последнем случае, получают:

Получение соединений формулы (XVII) неизбежно включает введение тиазолилзамещенного хинолинового остатка в циклопентильное кольцо посредством простой эфирной связи. Реакция Mitsunobu представляет собой «привлекательный» реакционный путь для получения ароматических простых алкилэфиров, где простой алкиловый эфир активируют и вводят во взаимодействие с ароматическим спиртом. Кроме того, реакции по Mitsunobu вообще являются более эффективными, чем реакции О-арилирования, которые требуют дополнительных стадий синтеза. В случае протекающей в мягких условиях этой реакции инвертируется стереохимия алкильной части. Реакция приводит к увеличению побочных продуктов, таких как R'OOC-NH-NH-COOR', где R' означает С1-4-алкил и, в частности, этил или изопропил, другие азотсодержащие соединения и трифенилфосфиноксиды, которые необходимо отделять от желательного конечного продукта.
Способы согласно настоящему изобретению являются преимущественными, так как они пригодны для крупномасштабного производства. Уменьшено количество трудоемких стадий очистки, в особенности, путем хроматографии.
Кроме того, выбор защитных групп бензила (Bn) и С1-4-алкила, в особенности метила (Ме), в соединениях (V), (Va) и (Vb) позволяет осуществлять селективное манипулирование с этими соединениями. Сложный бензиловый эфир или сложный С1-4-алкиловый эфир (и, в частности, метиловый эфир) можно селективно расщеплять за счет использования различных реакционных условий для удаления бензильной группы или С1-4-алкильной группы, в частности, метильной группы. Кроме того, остаток сложного бензилового эфира в соединении (IV), (V) или (Va) вносит преимущество в том, что он позволяет осуществлять эффективное разделение соединений (IV), (V) или (Va) с помощью хиральной хроматографии и облегчает анализ и обнаружение этих соединений, так как бензильный остаток является УФ-активным.
Описание данного изобретения
В одном аспекте, настоящее изобретение относится к способу получения соединения формулы (XIV), исходя из промежуточного продукта (XI), который гидролизуют до кислоты (XII), которую, в свою очередь, связывают с эфиром циклопропиламинокислоты (XIII), получая желательный конечный продукт (XIV), как показано на следующей реакционной схеме:
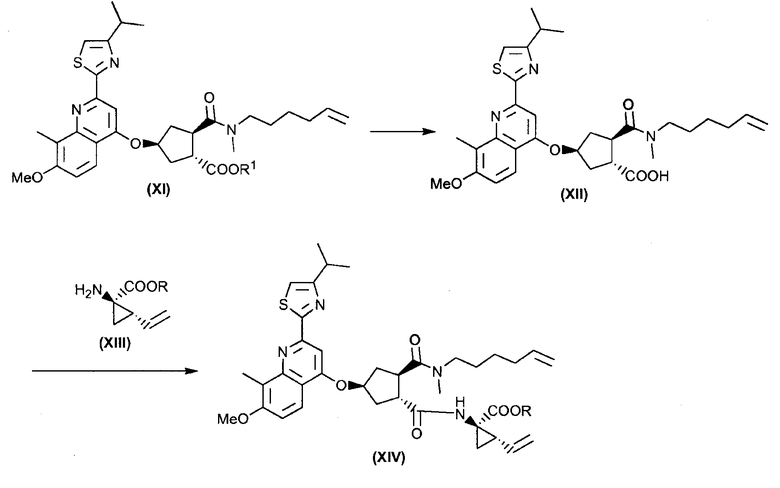
На вышеуказанной схеме, R имеет значение, как описано выше, т.е. С1-4-алкил, и R1, независимо от R, также означает С1-4-алкил. В одном воплощении, R означает этил. В другом воплощении, R1 означает метил. Представляет интерес способ, как показано выше, и промежуточные продукты формул (XI), (XIII) и (XIV), где R означает этил и R1 означает метил.
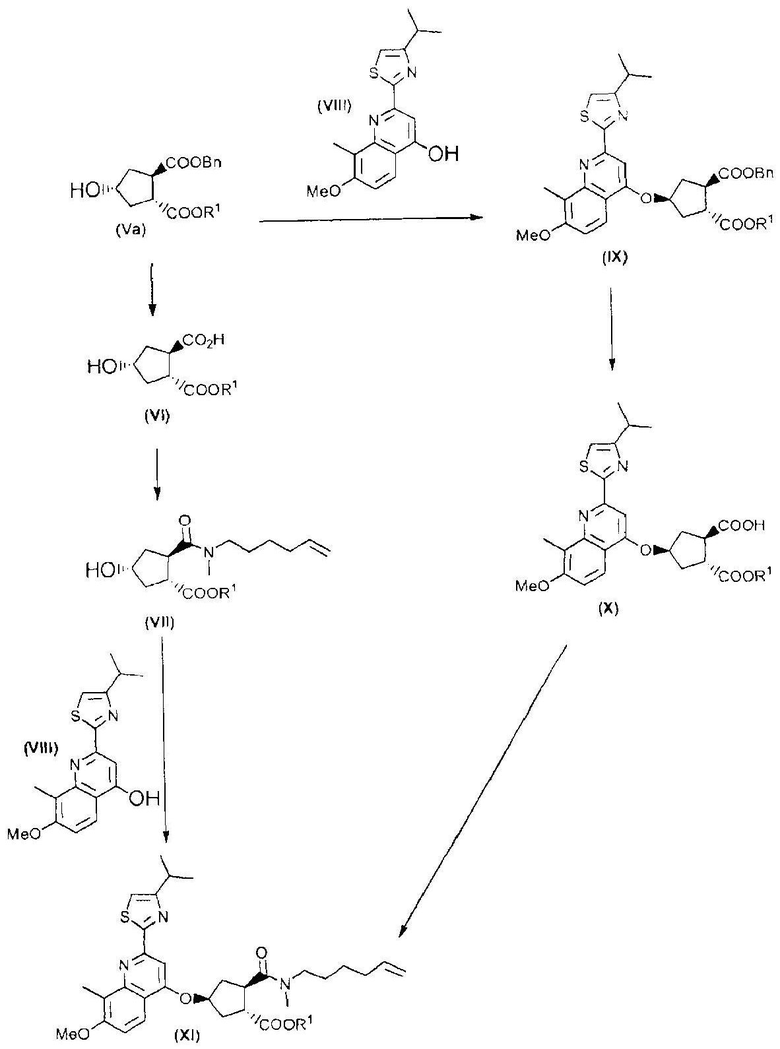
Промежуточный продукт (XI) является исходным веществом в вышеуказанном способе и его получение составляет дальнейший аспект данного изобретения. В соответствии с этим аспектом, изобретение относится к способу получения промежуточного продукта (XI), исходя из сложного гидроксициклопентилового бис-эфира формулы (Va), или
(а) путем введения во взаимодействие сложного гидроксициклопентилового бис-эфира формулы (Va) с тиазолилзамещенным хинолинолом (VIII) по реакции образования простого эфира, получая, таким образом, сложный хинолинилоксициклопентиловый бис-эфир формулы (IX), где сложноэфирную группу, которая находится в цис-положении по отношению к простой эфирной группе в сложном хинолинилоксициклопентиловом бис-эфире формулы (IX), селективно расщепляют до монокарбоновой кислоты (Х), которую, в свою очередь, связывают с алкениламином по реакции образования амида, получая, таким образом, желательный конечный продукт формулы (XI); или
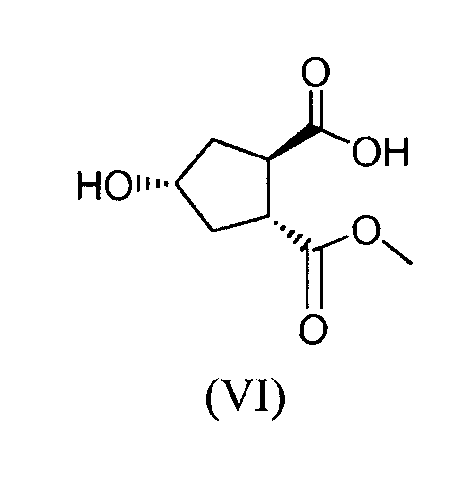
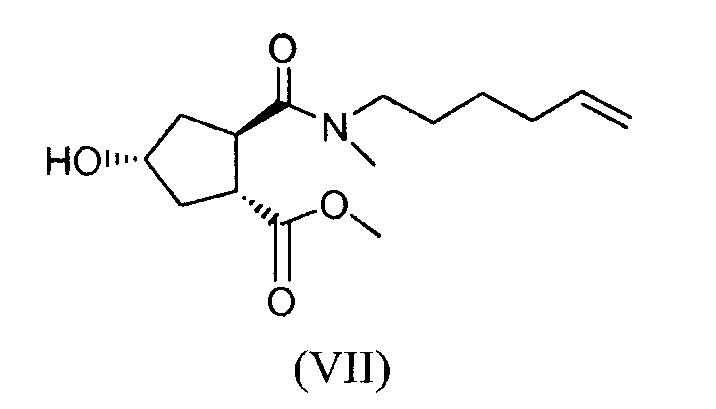
(b) путем селективного превращения сложного гидроксициклопентилового бис-эфира формулы (Va) в монокарбоновую кислоту (VI), которую, в свою очередь, связывают с алкениламином по реакции образования амида для получения гидроксициклопентиламида (VII), который, в свою очередь, вводят во взаимодействие с тиазолилзамещенным хинолинолом (VIII), получая, таким образом, желательный конечный продукт формулы (XI);
как показано на следующей реакционной схеме:
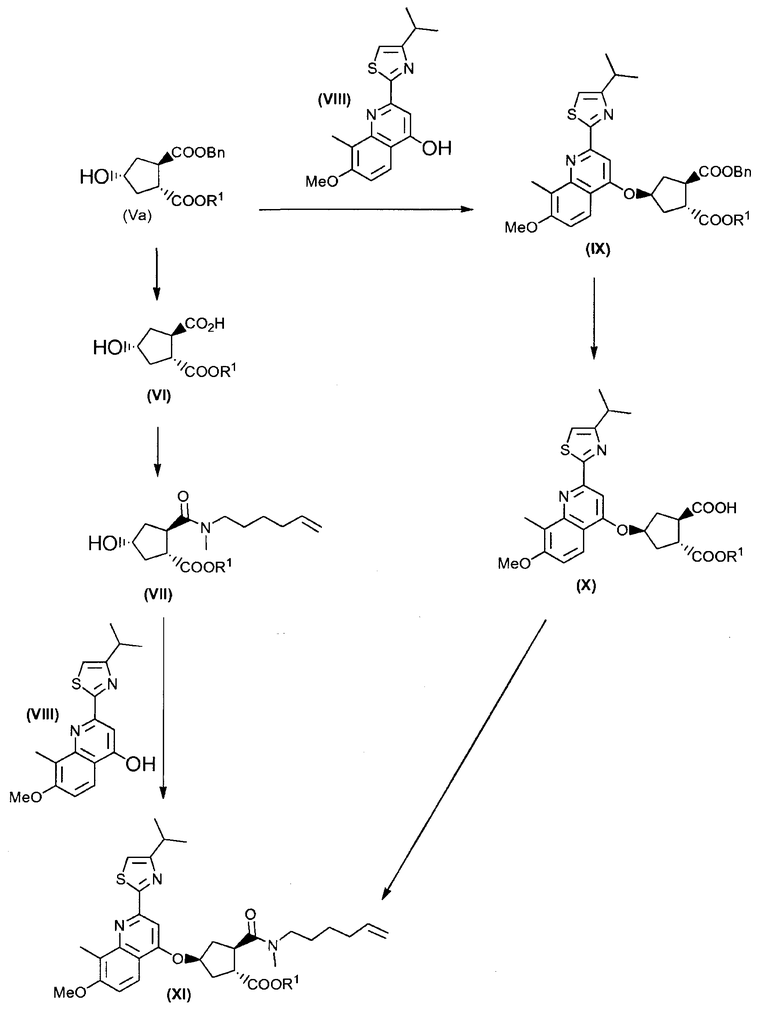
Каждый R1 в способах, представленных на вышеуказанной схеме, и, в частности, в промежуточных продуктах (Va), (VI), (VII), (IX), (X) и (XI), имеет значение, как описано выше, и, предпочтительно, R1 означает метил. Bn означает бензил.
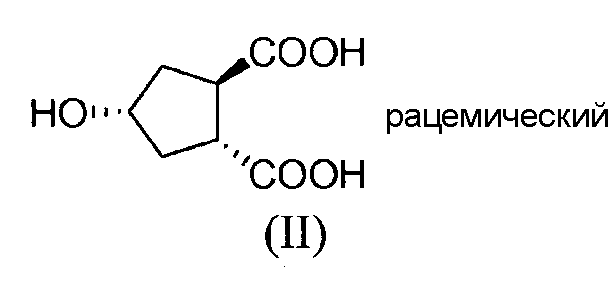
Промежуточный продукт (Va) является исходным веществом в вышеуказанном способе и его получение составляет дальнейший аспект данного изобретения. В соответствии с этим аспектом изобретение относится к способу получения промежуточного продукта (Va), исходя из 4-оксоциклопентил-1,2-бискарбоновой кислоты (I), путем восстановления кетогруппы до спирта, получая, таким образом, 4-гидроксициклопентил-1,2-бискарбоновую кислоту (II), которую, в свою очередь, циклизуют до бициклического лактона (III), где карбоксильную группу в бициклическом лактоне (III) этерифицируют с помощью бензилового спирта, получая, таким образом, бензиловый эфир лактона (IV), где лактон раскрывают и таким образом полученную карбоксильную группу этерифицируют с помощью С1-4-алканола, получая, таким образом, сложный гидроксициклопентиловый бис-эфир формулы (V), который, в свою очередь, разделяют на стереоизомеры (Vb) и (Va); как показано на следующей реакционной схеме:

Каждый R1 в способах, представленных на вышеуказанной схеме, и, в частности, в промежуточных продуктах (V), (Va) и (Vb), имеет значение, как описано выше, и, предпочтительно, R1 означает метил.
В одном воплощении, настоящее изобретение относится к применению соединений формул (I), (II), (III), (IV), (V), (Va), (Vb), (VI), (VII), (VIII), (IX), (X), (XI), (XII) или (XIV) в качестве промежуточных продуктов при получении соединения формулы (XVII) или его соли. Особый интерес представляют соединения формул (IX), (XI), (XII) и (XIV) и все промежуточные продукты, приводящие к образованию вышеуказанных соединений.
В другом воплощении, настоящее изобретение относится, по существу, к соединениям формул (II), (III), (IV), (V), (Va), (Vb), (VI), (VII), (IX), (X), (XI), (XII) или (XIV) и солям соединений формул (II), (III), (VI), (IX), (X) и (XII). Эти соединения могут быть в выделенной форме или в растворе. В особенности, соединения формул (VI), (IX), (X) или (XI) выделяют в твердой форме.
В одном воплощении, настоящее изобретение относится к способу получения соединения формулы (IX) или формулы (XI), где соединение формулы (Va), соответственно, формулы (VII), вводят во взаимодействие с соединением формулы (VIII) по реакции Mitsunobu. Эта реакция включает взаимодействие исходных веществ с азодикарбоксилатом формулы R'OOC-N=N-COOR', фосфином формулы R”3P, в инертном по отношению к реакции растворителе; где
R' означает этил или изопропил или трет-бутил;
R” означает, каждый, независимо, фенил, 2-пиридил, 3-пиридил или 4-пиридил.
Представленные в данном контексте реакции Mitsunobu представляют интерес в том, что они позволяют осуществлять пригодный способ получения соединений формул (XI) и (IX). Такое получение соединений формул (XI) и (IX), когда исходят из соединений (VII) и (Va), соответственно, включает инверсию стереохимии циклопентильного углерода, несущего гидроксильную группу или группу простого эфира.
Соединения формул (XI) и (IX) являются важными промежуточными продуктами, так как обнаружено, что оба соединения способны кристаллизоваться, в особенности, когда их смешивают со спиртовым растворителем, более конкретно, когда их смешивают с С1-4-алканолом. Кристаллизация соединений формул (XI) и (IX) представляет интерес в том, что она позволяет контролировать чистоту этих соединений, а также любых соединений, получаемых из них на последующих стадиях способа. В особенности, это дает возможность получать соединения формул (XI) и (IX) с более высокой энантиомерной чистотой.
Эта кристаллизация соединений формул (XI) и (IX) не только дает возможность удалять побочные продукты реакций Mitsunobu, по которым получают эти соединения, но также позволяет осуществлять последующее выделение соединений формул (XI) и (IX) из их соответствующих реакционных смесей простым путем. Это выделение без труда осуществляют путем замены растворителя, т.е. с помощью простого добавления спиртового растворителя к реакционной смеси, полученной по реакциям Mitsunobu, без проведения какой-либо дальнейшей манипуляции с реакционной смесью или любым ее компонентом.
Далее, так как соединения формул (XI) и (IX) нерастворимы в спиртовом растворителе, тогда как побочные продукты растворимы, это позволяет осуществлять непосредственную очистку соединений формул (XI) и (IX), выделяемых из реакционной смеси.
Способы, представленные в данном контексте, т.е. реакции Mitsunobu, с последующей заменой растворителя, обладают преимуществами в случае крупномасштабного производства. Другие способы выделения или очистки соединений формул (XI) и (IX), подобные хроматографии, гораздо меньше пригодны для крупномасштабного синтеза, требуют больше манипуляций и являются более дорогостоящими.
Как используется в вышеприведенном и нижеприводимом контексте, используют следующие определения, за исключением иначе указанного. Термин «галоген» является общим для фтора, хлора, брома и иода. Термин «С1-4-алкил» означает насыщенные углеводородные радикалы с линейной или разветвленной цепью, имеющие 1-4 атома углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил. Интерес представляют С1-4-алкильные радикалы без 2-метил-1-пропила. Термин «С1-3-алкил» является общим для метила, этила, 1-пропила и 2-пропила. Термин «С1-3-алкил» является общим для метила и этила. Термин «С1-4-алканол» относится к спирту, производному от С1-4-алкильной группы.
Фармацевтически приемлемые соли, которые может образовывать соединение формулы (XVII), являются фармацевтически приемлемыми аддитивными солями кислот или аддитивными солями оснований.
Аддитивные соли кислот, как таковые, получают путем введения во взаимодействие основной формы соединения формулы (XVII) с соответствующей кислотой, такой как, например, неорганическая кислота, такая как галогенводородная кислота, например, соляная или бромоводородная кислота, серная кислота, гемисерная кислота, азотная кислота, фосфорная кислота и подобные кислоты; или органическая кислота, такая как, например, уксусная кислота, аспарагиновая кислота, додецилсерная кислота, гептановая кислота, гексановая кислота, никотиновая кислота, пропановая кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, цикламеновая кислота, салициловая кислота, п-аминосалициловая кислота, памоевая кислота и подобные кислоты. Наоборот, формы аддитивной соли кислоты можно превращать в форму свободного основания путем обработки с помощью подходящего основания. Аддитивные соли оснований образуются путем обработки с помощью соответствующих органических и неорганических оснований. Соответствующие формы аддитивных солей оснований включают, например, соли аммония, соли щелочных и щелочноземельных металлов, например, соли лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, соли бензатина, N-метил-D-глюкамина, гидрабамина, и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Соединение формулы (XVII) имеет три хиральных центра и получение точной стереохимии в случае всех трех центров представляет собой важную задачу для любых способов синтеза, предназначенных для получения этого соединения. Для лучшего понимания стереохимии промежуточных продуктов, используемых в способах согласно настоящему изобретению, по причинам ясности приводятся следующие определения, хотя это известно в уровне техники.
Стереоизомерия представляет собой расположение атомов в молекулах, которые в отношении связности остаются теми же самыми, но их расположение в пространстве является различным в каждом изомере. Стереоизомеры можно разделять на две категории энантиомеров (зеркальные отображения) и диастереоизомеров (незеркальные отображения). Термин «энантиомер» относится к одной из пары молекул ненакладываемого зеркального отображения. Диастереомеры являются стереоизомерами, которые не являются энантиомерами или зеркальными отображениями друг друга. Диастереомеры могут иметь различные физические свойства и различную реакционную способность. Термин «рацемический» или «рацемат» относится к смеси равных количеств энантиомеров хиральной молекулы. Термин «эпимер» относится к стереоизомеру, который имеет другую конфигурацию только в случае одного из нескольких стереогенных центров. Таким образом, стереоизомеры различаются только по конфигурации в случае одного атома.
Общепринятая в настоящее время конвенция по номенклатуре стереохимических соединений является следующей:
- Если соединение изображено без стереосвязей, как изображено соединение формулы (III), тогда подразумевают, что соединение является рацемическим или конфигурация стереогенного(ых) центра(ов) не определена.
- Если соединение изображено со стереосвязями и, кроме того, указан один из дескрипторов «(±)», «rel» или «rac» в отношении химической структуры, тогда подразумевают, что соединение является рацемическим и стереохимия является относительной.
- Если соединение изображено со стереосвязями, но ни один из дескрипторов «(±)», «rel» или «rac» не указан в отношении структуры, тогда подразумевают, что соединение является оптически чистым соединением, иначе говоря, стереохимия является абсолютной.
Например, в ссылке Honda и др. обозначение «(±)», используют в заголовке статьи, подразумевая, что описывается рацемический синтез с рацемическими промежуточными продуктами. Однако вышеуказанной конвенции можно необязательно следовать во всех публикациях.
В одном воплощении, настоящее изобретение относится к применению соединения, выбираемого из соединений (I)-(XIV), перечисленных в вышеприведенной таблице, в качестве промежуточных продуктов для получения соединения формулы (XVII) или его солей.
В другом воплощении, настоящее изобретение относится к соединению формул (II), (III), (IV), (V), (Va), (Vb), (VI), (VII), (IX), (X), (XI), (XII) или (XIV) или к солям соединений формул (II), (III), (VI), (Х) и (XII), как указано в вышеприведенной таблице. Соединение может быть доступно в выделенной форме или в растворе.
Термин «выделенная форма», «выделенный», или любой его эквивалент, относится к твердому или жидкому состоянию, в котором соединение находится в чистой форме, т.е., по существу, свободно от других компонентов.
В другом воплощении, настоящее изобретение относится к соединению формул (VI), (IX), (X) или (XI), где соединение находится в твердой форме. Термин «твердая форма» включает как кристаллические, так и аморфные твердые формы, или любые их смеси.
Соединение формулы (I)
Соединение формулы (I) является коммерчески доступным или его можно получать в соответствии с методикой, описанной в примере 1.
Стадия (I) → (II)
Эта стадия относится к восстановлению кетогруппы в соединении формулы (I) до соответствующего спирта в соединении формулы (II). Последнее получают путем введения во взаимодействие соединения формулы (I) с водородом, в присутствии катализатора, необязательно в присутствии основания. Катализатор можно выбирать из катализаторов на основе благородных металлов, таких как родий-на-угле, родий-на-оксиде алюминия, платина-на-угле или платина-на-оксиде алюминия. Основание можно выбирать из гидроксида щелочного металла, в частности, гидроксида натрия, оксида алюминия или три-С1-4-алкиламина, такого как триэтиламин. По завершении взаимодействия, можно добавлять кислоту для превращения образовавшейся солевой формы обратно в свободную кислоту. Это может быть неорганическая кислота, такая как галогенводородная кислота, например, HCl, или серная кислота.
Стадия (II) → (III)
Эта стадия приводит к образованию лактона. Соединение формулы (III) или его соль получают путем введения во взаимодействие соединения формулы (II) или его соли с С1-4-алкилхлорформиатом формулы ClCOOR2, где R2 означает С1-4-алкил, в частности, метил, этил, пропил, изопропил, н-бутил или изобутил, и неорганическим основанием. В одном воплощении, можно выделять соединение формулы (III). Органическое основание может быть третичным амином, таким как три-С1-4-алкиламин, например, триэтиламин.
Стадия (III) → (IV)
Эта стадия приводит к образованию сложного бензилового эфира. Соединение формулы (IV) получают путем введения во взаимодействие соединения формулы (III) с бензиловым спиртом; исходное вещество (III) и бензиловый спирт могут взаимодействовать в присутствии агента образования сложного эфира, например, связующего агента. Или кислоту (III) можно превращать в активированную форму, как например путем введения во взаимодействие с хлорформиатом, в частности, с С1-4-алкилхлорформиатом формулы ClCOOR2, где R2 имеет значение, указанное выше, в присутствии органического основания.
Связующий агент можно выбирать из карбодиимидов, таких как EDCI, N,N'-дициклогексилкарбодиимид (DCC) или диизопропилкарбодиимид, или N,N'-карбонилдиимидазола (CDI), вместе с или без 4-диметиламинопиридина (DMAP), или этил/изопропил/изобутилхлорформиата. Органическое основание может быть третичным амином, таким как три-С1-4-алкиламин, например, триэтиламин.
Стадия (IV) → (V)
Стереоизомерную смесь (V) соединения формулы (Va) и соединения формулы (Vb) получают путем переэтерификации лактона в С1-4-алкиловый эфир. Применяемым растворителем предпочтительно является спирт, от которого происходит образующийся сложный эфир, т.е., если должен быть получен метиловый эфир, то реакцию проводят в метаноле. Это взаимодействие осуществляют в присутствии кислотного катализатора и с избытком спирта в качестве растворителя, как при проведении реакции в отношении образования сложного эфира. Кислотный катализатор может быть в виде неорганической кислоты, например, HCl, или в виде органической кислоты, такой как метансульфоновая кислота, или можно использовать кислые смолы, такие как Amberlyst 15™ (A15), которые можно без труда удалять путем фильтрации.
Стадия (V) → (Va) + (Vb)
Соединение формулы (Va) получают путем отделения его от соединения формулы (Vb), из смеси изомеров (Va) и (Vb), в частности, рацемической смеси соединений (Va) и (Vb). Это энантиомерное разделение можно осуществлять с помощью хроматографии на колонке с хиральной фазой или путем хиральной жидкостной хроматографии. Это включает использование хиральной стационарной фазы, например, стационарной фазы на основе полиамилозы или полицеллюлозы, такой как Chiralpak AD™.
Стадия (Va) → (VI)
Эта стадия включает расщепление сложного бензилового эфира до свободной кислоты. Соединение формулы (VI) получают из соединения формулы (Va) удалением бензиловой группы с помощью гидрирования. Это можно осуществлять путем использования водорода в присутствии катализатора. Катализатор можно выбирать из палладия-на-угле или соли палладия или гидроксида, как например ацетат палладия, хлорид палладия, гидроксид палладия или гидроксид палладия-на-угле.
Эту реакцию можно проводить в подходящем растворителе, который можно выбирать из простого эфира, в частности, из простого эфира, такого как метил-трет-бутиловый эфир (МВТЕ), или циклического простого эфира, такого как тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран (МеТГФ); кетона, такого как ацетон, метилизобутилкетон; спирта, такого как С1-4-алканол, например, метанол, этанол, пропанол; диполярного апротонного растворителя, такого как диметилформамид (ДМФА), диметилацетамид (DMA); углеводорода, такого как толуол; или любой смеси таких растворителей. Реакционную смесь, содержащую соединение (Va), катализатор и растворитель, можно перемешивать в атмосфере водорода. Соединение формулы (VI) можно кристаллизовать удалением путем фильтрации катализатора из реакционной смеси, промывки и осуществления необходимых замен растворителя, например, путем замены части или всего растворителя простым эфиром, таким как МТВЕ, затем, далее, необязательно введения затравки при использовании затравочных кристаллов соединения формулы (VI), при необязательном охлаждении до температуры в диапазоне от примерно -15°С до примерно 5°С. Если желательно, соединение формулы (VI) можно получать в растворе, свободном от катализатора, и можно затем использовать в растворенной форме при получении соединения формулы (VII).
Другие возможные способы дебензилирования, которые можно альтернативно применять на этой стадии, можно осуществлять путем гидрирования с переносом, используя смесь муравьиная кислота - триэтиламин, формиат натрия или калия, или путем гидросилилирования, используя, например, Et3SiH (TES-H), PhSiH3, Ph2SiH2, поли(метилгидросилоксан) (PMHS) или (RO)3SiH.
Стадия (VI) → (VII)
Соединение формулы (VII) получают из соединения формулы (VI) и N-метилгекс-5-ениламина (NMHA, также называемого как N-метил-5-гексен-1-амин) путем реакции образования амида. Последний может быть любым из таковых, обычно используемых в случае пептидного синтеза. Может быть использован связующий агент или кислота может быть активирована путем превращения ее в смешанный ангидрид или активный эфир. Связующие агенты, которые можно использовать, можно выбирать из EEDQ, N,N,N',N'-тетраметил-О-(7-азабензотриазол-1-ил)уронийгексафторфосфата (HATU), IIDQ, бензотриазол-1-илокситриспирролидинофосфоний-гексафторфосфата (коммерчески доступный как PyBOP®), DCC, EDCI или 1,3-диизопропилкарбодиимида. Можно добавлять катализатор, например, 1-гидроксибензотриазол (HOBt). Это взаимодействие обычно проводят в присутствии основания, в частности, амина в качестве основания, такого как третичный амин, например, триэтиламин, N-метилморфолин, N,N-диизопропилэтиламин (последний также указан выше как основание Хенига, DIPEA или DIEA). При реакциях образования амида предпочтительно избегают использования веществ, таких как О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (HATU) или О-(1Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU), потому что существует риск взрывов в случае крупномасштабного производства.
Реакцию связывания обычно проводят в инертном по отношению к реакции растворителе, который можно выбирать из простого эфира, в частности, циклического простого эфира, такого как ТГФ или МеТГФ, диполярного апротонного растворителя, такого как ДМФА, гексаметилфосфорамид (НМРТ), DMA, ацетонитрил; углеводорода, такого как толуол; галогенированного углеводорода, такого как дихлорметан, хлороформ, 1,2-дихлорэтан; спирта, такого как С1-4-алканол, например, метанол, этанол, пропанол; воды; или любой смеси таких растворителей.
Реакции связывания обычно проводят с необязательным перемешиванием, при температуре в диапазоне от примерно -20°С до температуры кипения с обратным холодильником реакционной смеси.
Стадия (VII) + (VIII) → (XI)
Эта реакционная стадия включает образование простой эфирной связи между циклопентильным остатком в соединении (VII) и хинолинильным остатком в соединении (VIII). Эта простая эфирная связь предпочтительно образуется посредством реакции Mitsunobu. Согласно этой реакции соединение формулы (VII) вводят во взаимодействие с соединением формулы (VIII) в присутствии азодикарбоксилата формулы R'OOC-N=N-COOR', фосфина формулы R”3P и органического растворителя; где каждый R' представляет собой, независимо, С1-4-алкил, в частности, этил, изопропил или трет-бутил; и каждый R” представляет собой, независимо, фенил, 2-пиридил, 3-пиридил или 4-пиридил. Предпочтительными являются диэтилазодикарбоксилат (DEAD) или диизопропилазодикарбоксилат (DIAD), в присутствии трифенилфосфина.
Органический растворитель можно выбирать из галогенированного углеводорода, такого как дихлорметан, простого эфира, в частности, циклического простого эфира, такого как тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран (МеТГФ), сложного эфира, такого как этилацетат, изопропилацетат, ароматического углеводорода, такого как толуол, или любой смеси таких растворителей.
Необязательно, начальную реакционную смесь, содержащую соединение формулы (VII), соединение формулы (VIII), фосфин формулы R”3P и органический растворитель, можно частично выпаривать, для того, чтобы удалить следы воды и/или спиртов. Эту начальную реакционную смесь можно охлаждать до температуры приблизительно 0°С, перед добавлением азодикарбоксилата формулы R'OOC-N=N-COOR'. Необязательно, воду можно добавлять после введения азодикарбоксилата, для того, чтобы удалить избыток последнего реагента. Любые нерастворимые побочные продукты, получаемые при взаимодействии, можно удалять путем фильтрации.
Выделение соединения формулы (XI)
Соединение формулы (XI) выделяют из реакционной смеси путем замены органического растворителя, полностью или частично, растворителем, выбираемым из С1-4-алканола, как, например, 1-бутанол, гептана, диизопропилового эфира или любой их смеси. Эту замену растворителя можно осуществлять дистилляцией или удалением растворителей, первоначально присутствующих в реакционной смеси, полностью или частично, и путем добавления после этого одного или более из вышеуказанных растворителей. Изменение температуры реакционной смеси также способствует осуществлению кристаллизации. Замену растворителей можно повторять также многократно и также при использовании множества растворителей, как желательно.
Выделение соединения формулы (XI) можно осуществлять путем фильтрации и высушивания. Кристаллизацию соединения формулы (XI) можно также улучшать путем затравки фильтрата или реакционной смеси с помощью затравочных кристаллов того же самого соединения.
Стадия (XI) → (XII)
На этой стадии, сложноэфирную группу -COOR1 в соединении формулы (XI) расщепляют до соответствующей кислоты. Можно использовать основание в водной среде. Основания, которые можно использовать, включают гидроксиды щелочных металлов, такие как гидроксид натрия или гидроксид калия, и, в особенности, гидроксид лития. Водной средой может быть вода или вода, смешанная с растворимым в воде органическим растворителем, таким как спирт, в частности, С1-4-алканол, например, метанол или этанол; простой эфир, в частности, циклический простой эфир, такой как ТГФ, МеТГФ, или любая смесь таких растворителей. Получают свободную кислоту формулы (XII) или ее соль щелочного металла, например, ее соль лития, натрия или калия.
В одном воплощении, гидроксид лития, гидроксид натрия или гидроксид калия вначале смешивают с растворителем перед добавлением соединения формулы (XI). Смесь, содержащую соединение формулы (XI), гидроксид лития, гидроксид натрия или гидроксид калия и растворитель, можно перемешивать при комнатной температуре. Если желательно, соединение формулы (XII), или его соль лития, натрия или калия, не выделяют, а используют в растворе для последующего превращения соединения формулы (XII) в соединение формулы (XIV).
Стадия (XII) → (XIV)
На этой стадии, соединение формулы (XII) или его соль, далее, вводят во взаимодействие с соединением формулы (XIII) или его солью согласно реакции образования амида, получая соединение формулы (XIV). Можно использовать те же самые реакционные условия, как описано выше в отношении стадии (VI) → (VII).
В одном воплощении, агент связывания амида выбирают из IIDQ, EDCI, DCC или 1,3-диизопропилкарбодиимида и, в частности, EEDQ в простом эфире, таком как ТГФ, в частности, в водном ТГФ. Реакционную смесь, указанную выше, можно перемешивать при комнатной температуре.
Стадия (Va) + (VIII) → (IX)
На этой стадии получают соединение формулы (IX), где соединение формулы (Va) вводят во взаимодействие с соединением формулы (VIII) до образования простой эфирной связи. Можно использовать реакцию Mitsunobu, как описано в случае стадии (VII) + (VIII) → (XI). Соединение (IX) можно кристаллизовать путем добавления С1-4-алканола, как, например, метанол, этанол, 1-пропанол или 1-бутанол. В одном воплощении, по окончании реакции Mitsunobu добавляют воду и побочные продукты удаляют фильтрацией. Фильтрат и любые промывочные жидкости изолятов можно концентрировать досуха или почти досуха и затем перекристаллизовывать из С1-4-алканола, например, метанола.
Выделение соединения формулы (IX)
Соединение формулы (IX), далее, можно выделять из реакционной смеси путем замены растворителя, полностью или частично, на растворитель, выбираемый из С1-4-спирта, гептана, диизопропилового эфира или любой их смеси. Эту замену растворителя можно осуществлять путем дистилляции или удаления растворителей, первоначально присутствующих в реакционной смеси, и после этого добавления растворителя, выбираемого из С1-4-алканола, гептана, диизопропилового эфира или любой их смеси. Изменение температуры реакционной смеси также способствует осуществлению замены растворителя. Эту замену растворителей можно повторять также многократно и также при использовании множества растворителей, как желательно.
Необязательно, выделение соединения формулы (XI) можно осуществлять путем фильтрации и высушивания. Получение соединения формулы (XI) можно также улучшать путем затравки фильтрата или реакционной смеси с помощью затравочных кристаллов того же самого соединения.
Стадия (IX) → (X)
Эта стадия включает расщепление сложного бензилового эфира в (IX) до получения соединения формулы (Х). Можно применять те же самые методики, описанные выше в отношении стадии (Va) → (VI).
Стадия (X) → (XI)
Соединение формулы (XI) можно получать путем введения во взаимодействие соединения формулы (Х) с NMHA и агентом образования амида, используя те же самые методики, как описанные выше в отношении стадии (VI) → (VII). Соединение (XI) можно очищать, как описано выше, или путем перекристаллизации из углеводорода, такого как гексан, гептан или октан, как описано выше. В одном воплощении, соединение (Х) и NMHA подвергают реакции связывания, реакционную смесь подкисляют, например, с помощью водного раствора HCl, и экстрагируют с помощью углеводорода, например, с помощью толуола. Углеводородный растворитель удаляют и остаток перекристаллизуют из гептана.
Примеры
Следующие примеры приводятся для иллюстрации настоящего изобретения и не ограничивают его объема охраны.
Пример 1
Получение соединения формулы (I)
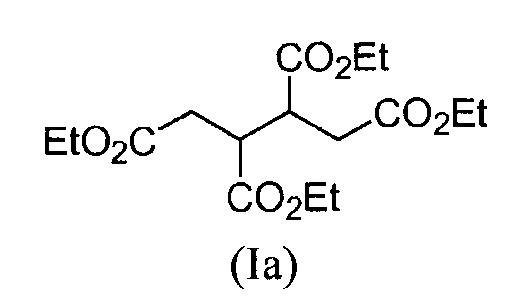
К раствору 1,2,3,4-бутантетракарбоновой кислоты (0,99 моль, 234,16 г) в этаноле (750 мл) и толуоле (500 мл) добавляют серную кислоту (0,56 моль, 30,05 мл, 55,30 г), в виде одной порции. Эту смесь кипятят с обратным холодильником в течение 4,5 часов. Растворитель удаляют путем дистилляции до тех пор, пока внутренняя температура не достигнет 110°С. Добавляют толуол (500 мл), в виде одной порции, и смесь кипятят с обратным холодильником при удалении воды путем азеотропной перегонки, используя ловушку Дина-Старка. После охлаждения до температуры 80°С, добавляют этанол (500 мл) и смесь кипятят с обратным холодильником в течение 16 часов. Растворитель удаляют путем дистилляции до тех пор, пока внутренняя температура не достигнет 105°С. Добавляют толуол (500 мл), в виде одной порции, и смесь кипятят с обратным холодильником в течение 1 часа при азеотропном удалении воды. Смесь охлаждают до температуры 22°С, добавляют 50 мл воды и смесь перемешивают в течение нескольких минут. Два слоя разделяют и толуольную фазу промывают с помощью 50 мл воды. Объединенные толуольные слои промывают с помощью водного раствора карбоната натрия (15 %масс./масс.) (375,00 мл). Толуольную фазу выпаривают досуха, получая масло желтого цвета, которое также сушат в вакууме, при температуре 50°С, в течение ночи, получая 315,7 г (91% отдельный выход) соединения формулы (Ia), в виде окрашенного в желтый цвет масла.
Н-ЯМР (CDCl3 - 400 МГц), δ м.д. 1,23-1,28 (м, 12Н), 2,38-2,43 (м, 2Н), 2,74-2,81 (м, 2Н), 3,28-3,31 (м, 2Н), 4,11-4,17 (м, 2Н).
ИК-спектр (пленка): 2970, 2940, 2900, 1730, 1525, 1500, 1375, 1340.
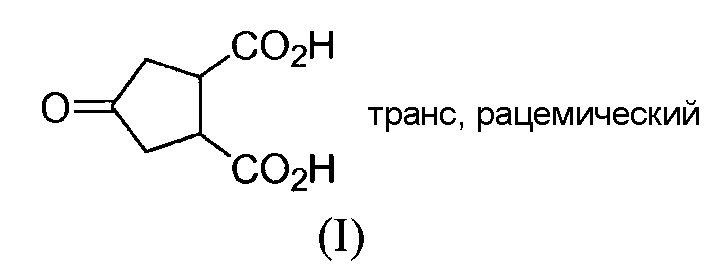
К раствору тетраэтилового эфира 1,2,3,4-бутантетракарбоновой кислоты (1,00 моль, 346,38 г) в метаноле (320,00 мл), при температуре 22°С, добавляют раствор метоксида натрия (30%-ный раствор в метаноле) (2,90 моль, 522,23 г). Смесь перемешивают при комнатной температуре в течение 16 часов. По каплям, в течение 15 минут, добавляют раствор концентрированной соляной кислоты (3,49 моль, 300,00 мл, 348,90 г) и воду (16,65 моль, 300,00 мл, 300 г). После добавления второй порции соляной кислоты (1,86 моль, 160,00 мл, 186,08 г), смесь нагревают при температуре кипения с обратным холодильником и растворитель отгоняют до тех пор, пока внутренняя температура не достигнет 100°С. Смесь кипятят с обратным холодильником в течение 16 часов. Затем смесь охлаждают до температуры 90°С. Добавляют 1 г активированного угля и смесь оставляют для дальнейшего охлаждения, при перемешивании, до температуры 50°С. Реакционную смесь отфильтровывают через целит. Фильтрат (900 мл) вносят в колбу емкостью 2000 мл. 400 мл Растворителя отгоняют при атмосферном давлении. При перемешивании, смесь оставляют охлаждаться до комнатной температуры. Твердые вещества отфильтровывают и промывают с помощью 100 мл воды. Продукт сушат в вакууме, при температуре 50°С, получая 156,6 г (выход 91% после выделения) соединения формулы (I) в виде твердого вещества белого цвета.
Н-ЯМР (ДМСО-d6 - 400 МГц), δ м.д. 2,23-2,38 (м, 2Н), 2,48-2,54 (м, 2Н), 3,20-3,27 (м, 2Н), 12,60 (уш.с, 2Н).
С-ЯМР (ДМСО-d6 - 100 МГц), д м.д.: 40,70, 43,17, 174,27, 213,45.
ИК-спектр (пленка), см-1: 3450, 3050, 2900, 1750, 1720, 1480, 1280, 1250, 1220, 1180, 1150.
Пример 2
Получение соединения формулы (II)
К суспензии 32,7 г (0,19 моль) соединения формулы (I) в 237,5 мл воды, в атмосфере азота, добавляют 1,0 мл (0,019 моль) водного раствора NaOH (50 %масс./масс.). Смесь нагревают до температуры 60°С и добавляют 2,5 г Rh/C (5 %масс./масс.). Реакционную колбу продувают водородом и перемешивают в атмосфере водорода до тех пор, пока не достигнут полной конверсии. Затем, нагретую реакционную смесь отфильтровывают через целит. Фильтровальный осадок дважды промывают с помощью 10 мл воды и фильтрат вносят в реакционную колбу емкостью 500 мл. Затем добавляют 60 мл 4-метил-2-пентанона и воду удаляют путем азеотропной перегонки до тех пор, пока внутренняя температура не достигнет 110°С. Смесь охлаждают до температуры 50°С. Добавляют 88 мл ацетона и 0,51 мл серной кислоты (95%-ная). Смесь перемешивают при температуре 22°С в течение 16 часов. Твердые вещества отфильтровывают и дважды промывают с помощью 10 мл ацетона. Эти вещества затем высушивают в вакууме при температуре 50°С, получая 21,84 г (выход 66% после выделения) соединения формулы (II) в виде твердого вещества белого цвета.
Н-ЯМР (D2O-d6 - 400 МГц), δ м.д., 1,80-1,86 (м, 1Н), 1,94-2,04 (м, 2Н), 2,33-2,40 (м, 1Н), 3,13 (кв, J=8,5 Гц, 1Н), 3,30 (кв, J=8,5 Гц, 1Н), 4,29-4,40 (м, 1Н).
С-ЯМР (D2O - 100 МГц), д м.д.: 37,61, 38,09, 44,92, 45,30, 71,67, 178,64, 178,92.
Пример 3
Получение соединения формулы (III)

К суспензии соединения формулы (II) (50 г, 0,29 моль) в 860 мл ТГФ добавляют триэтиламин (42,02 мл, 0,30 моль). Смесь перемешивают при комнатной температуре вплоть до полного растворения всего твердого вещества. Затем реакционную смесь охлаждают до температуры 0-5°С. По каплям добавляют этилхлорформиат (32,72 г, 0,30 моль) и смесь перемешивают в течение следующего часа при температуре 0-5°С. Реакционную смесь нагревают до температуры 22°С и перемешивают в течение следующих 5 часов. Потом реакционную смесь отфильтровывают через целит и твердые вещества промывают с помощью 25 мл ТГФ. Фильтрат выпаривают досуха. К остатку добавляют 50 мл этилацетата и смесь перемешивают при температуре 22°С в течение 15 минут. Твердые вещества отфильтровывают и промывают с помощью 10 мл холодного этилацетата, получая 21,67 г (выход 48% после выделения) соединения формулы (III) в виде твердого вещества белого цвета.
Н-ЯМР (CDCl3 - 400 МГц), δ м.д., 1,96 (д, J=10,8 Гц, 1Н), 2,27-2,29 (м, 3Н), 3,05 (т, J=6,8 Гц, 1Н), 3,23 (с, 1Н), 5,03 (с, 1Н).
С-ЯМР (CDCl3 - 100 МГц): 32,91, 37,87, 39,90, 45,24, 80,25, 167,33, 175,54.
Пример 4
Получение соединения формулы (IV)

600 мг (3,84 ммоль) Соединения формулы (III) (рацемическое), 0,418 мл (4,03 ммоль) бензилового спирта, 23,5 мг (0,19 ммоль) DMAP и 810 мг (4,23 ммоль) EDCI суспендируют в 38 мл этилацетата. Суспензию перемешивают в течение ночи при комнатной температуре. Потом добавляют 38 мл воды, полученную двухфазную смесь перемешивают при комнатной температуре в течение нескольких минут, затем оставляют декантироваться и водный слой удаляют. Органический слой сушат над сульфатом магния, отфильтровывают и концентрируют в вакууме, получая 993 мг сырого соединения формулы (IV) в виде почти бесцветного масла.
993 мг Сырого соединения формулы (IV) очищают с помощью флэш-хроматографии на силикагеле, используя смесь диизопропиловый эфир - гексан (3:1) в качестве элюента. Получают 834 мг (выход 88% после выделения) очищенного соединения формулы (IV) в виде бесцветного масла.
Физические данные: GC-MS (газовая хроматография - масс-спектрометрия): m/z = 246 (М+).
Н-ЯМР (600 МГц, CDCl3), δ м.д., 1,94 (д, J=10,95 Гц, 1Н), 2,18 (д, J=10,95 Гц, 1Н), 2,22-2,27 (м, 2Н), 2,95-3,00 (м, 1Н), 3,17 (с, 1Н), 4,97 (с, 1Н), 5,18 (с, 2Н), 7,33-7,43 (м, 5Н).
С-ЯМР (CDCl3 - 125 МГц), д м.д.: 33,31, 38,01, 39,77, 45,81, 67,31, 80,43, 128,27, 128,58, 128,72, 135,33, 172,37, 176,27.
350 г (1,79 моль) Соединения формулы (II) (рацемическое) и 262,1 мл (1,88 моль) триэтиламина суспендируют в 5,37 л ТГФ. Смесь охлаждают до температуры 0-5°С и добавляют 179,8 мл (1,88 моль) этилхлорформиата в течение 1-2 часов. Смесь перемешивают при температуре 0-5°С в течение 2 часов, затем - в течение ночи при комнатной температуре. Потом реакционную смесь отфильтровывают и твердые вещества дважды промывают с помощью 179 мл ТГФ. Растворители отгоняют из фильтрата и добавляют 4,48 л этилацетата для дистилляции остатка. К полученной смеси добавляют 194,6 мл (1,88 моль) бензилового спирта, 377,6 г (1,97 моль) EDCI и 10,9 г (89,5 моль) DMAP. Полученную суспензию перемешивают в течение ночи при комнатной температуре. Потом добавляют 1,79 л воды и два слоя разделяют. Органический слой сушат над сульфатом магния, отфильтровывают и концентрируют досуха в вакууме. Получают 435 г (выход 99%) сырого соединения формулы (IV) в виде масла желтого цвета.
Пример 5
Получение соединения формулы (V)

600 г (2,44 моль) Соединения формулы (IV) (рацемическое) растворяют в 10 л метанола. Добавляют 122 г смолы Amberlyst 15 и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смолу Amberlyst 15 отфильтровывают и фильтрат концентрируют в вакууме, получая 638 г (выход 94%) сырого соединения формулы (V).
300 г (1,22 моль) Соединения формулы (IV) (рацемическое) растворяют в 6 л метанола, добавляют 5 мл (0,08 моль) метансульфоновой кислоты и реакционную смесь перемешивают при комнатной температуре до тех пор, пока не исчезнет исходное вещество (приблизительно 2-3 часа). Затем добавляют 90 г карбоната натрия, предварительно растворенного в 0,9 л воды, и смесь концентрируют в вакууме. Остаток распределяют между 1,2 л этилацетата и 0,6 л воды. Органический слой сушат над сульфатом натрия, отфильтровывают и концентрируют в вакууме. Получают 300 г (89% выход) сырого соединения формулы (V) в виде масла.
1,75 г Сырого соединения формулы (V) очищают с помощью ВЭЖХ на силикагеле, получая 0,55 г очищенного соединения формулы (V).
GC-MS: m/z = 278 (М+). Н-ЯМР (400 МГц, CDCl3), δ м.д.,1,90-2,02 (м, 2Н), 2,07-2,17 (м, 1Н), 2,24 (ддд, 1Н), 3,14 (с, 1Н), 3,17-3,26 (м, 1Н), 3,47 (кв, 1Н), 3,65 (с, 3Н), 4,32-4,39 (м, 1Н), 5,13 (уш.с, 2Н), 7,26-7,39 (м, 5Н).
С-ЯМР (CDCl3 - 100 МГц), д м.д.: 38,50, 39,63, 45,21, 45,33, 52,23, 66,60, 72,56, 127,95, 128,20, 128,53, 135,85, 174,53, 175,72.
Пример 6
Получение соединений формул (Va) и (Vb)

749 г Сырого соединения формулы (V) (рацемическое) элюируют через Chiralpak AD, используя смесь гептан/метанол/этанол (70:15:15) в качестве элюента, получая 369 г соединения формулы (Va) и 57 г соединения формулы (Vb).
Анализы GC, GC-MS и ЯМР идентичны таковым очищенного соединения формулы (V) (рацемическое).
Пример 7
Получение соединения формулы (VI)
13,92 г (50 ммоль) Соединения формулы (Va) и 2,66 г (2,5 ммоль) сухого, 10 %масс./масс., Pd/C суспендируют в 250 мл ТГФ и суспензию перемешивают в атмосфере водорода. Катализатор отфильтровывают и промывают с помощью нескольких мл ТГФ. Фильтрат концентрируют в вакууме, получая 10,80 г (114%, «сырой» выход) сырого соединения формулы (VI), в виде масла, которое отверждается при стоянии.
ЖХ-MC: m/z = 189,1 (М+Н+).
Н-ЯМР (400 МГц, ДМСО-d6), δ м.д., 1,65-1,73 (м, 1Н), 1,74-1,81 (м, 1Н), 1,83-1,92 (м, 1Н), 2,19 (ддд, J=13,28, 10,01, 5,41 Гц, 1Н), 2,93-3,06 (м, 1Н), 3,13-3,25 (м, 1Н), 3,60 (с, 3Н), 4,08-4,22 (м, 1Н), 4,68 (с, 1Н), 12,27 (с, 1Н).
С-ЯМР (100 МГц, ДМСО-d6), д м.д.: 38,38, 39,11, 44,31, 44,45, 51,60, 70,50, 174,43, 175,69.
123,9 г (445 ммоль) Соединения формулы (Va) и 4,74 г (4,45 ммоль) сухого, 10 %масс./масс., Pd/C суспендируют в 668 мл ТГФ. Суспензию перемешивают в атмосфере водорода. Катализатор отфильтровывают и промывают с помощью 228 мл ТГФ. Фильтрат концентрируют в вакууме и остаток повторно суспендируют в 445 мл горячего гептана. Суспензию оставляют охлаждаться до комнатной температуры и выделяют, после фильтрации и высушивания, 81,25 г (выход 97%) соединения формулы (VI) в виде кристаллического вещества белого цвета.
11,13 г (40 ммоль) Соединения формулы (Va) и 0,85 г (0,8 ммоль) сухого, 10 %масс./масс., Pd/C суспендируют в 60 мл ТГФ. Суспензию перемешивают в атмосфере водорода. Катализатор отфильтровывают и 3 раза промывают с помощью 15 мл ТГФ. Фильтрат концентрируют в вакууме и остаток повторно суспендируют в 445 мл горячего этилацетата. Суспензию оставляют охлаждаться до комнатной температуры и выделяют, после фильтрации и высушивания, 5,12 г (выход 68%) соединения формулы (VI) в виде кристаллического вещества белого цвета.
20,87 г (75 ммоль) Соединения формулы (Va) и 3,19 г (0,75 ммоль) влажного, 5 %масс./масс., Pd/C суспендируют в 113 мл ТГФ. Суспензию интенсивно перемешивают в течение ночи в атмосфере водорода. Катализатор отфильтровывают и промывают с помощью 19 мл ТГФ и из фильтрата отгоняют 75 мл ТГФ. Затем добавляют 38 мл толуола и отгоняют 63 мл растворителя. Наконец, добавляют 101 мл МТВЕ. В почти прозрачный раствор вносят затравку кристаллического соединения формулы (VI). Суспензию охлаждают до температуры -5°С и перемешивают в течение ночи при температуре -5°С. Выделяют, после фильтрации и высушивания, 9,29 г (выход 66%) соединения формулы (VI) в виде кристаллического вещества белого цвета.
6,47 г (23,3 ммоль) Соединения формулы (Va) и 1,24 г (1,16 ммоль) сухого, 10 %масс./масс., Pd/C суспендируют в 23 мл ДМФА. Суспензию интенсивно перемешивают в течение ночи в атмосфере водорода. Катализатор отфильтровывают и промывают с помощью нескольких мл ДМФА. Фильтрат доводят до объема 50 мл с помощью ДМФА и полученный раствор используют на следующей стадии (амид связывают с NMHA, чтобы получить соединение формулы (VII)).
Пример 8
Получение соединения формулы (VII)
303 мг (2,67 ммоль) NMHA, 1,22 мл (6,99 ммоль) основания Хенига и 1,06 г (2,79 ммоль) HATU добавляют к раствору соединения формулы (VI) в ДМФА (2,33 ммоль в 5 мл). Смесь перемешивают в течение 2 часов при комнатной температуре, затем ДМФА и другие летучие вещества удаляют в вакууме. Остаток повторно растворяют в смеси МеТГФ-вода (5 мл каждого). Добавляют 1,5 мл концентрированной HCl, слои разделяют и водный слой экстрагируют с помощью 5 мл МеТГФ. Органические слои объединяют, сушат над карбонатом калия и сульфатом магния, отфильтровывают и фильтрат концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии на силикагеле, используя этилацетат в качестве элюента. Получают 373 мг (выход 44%) соединения формулы (VII) в виде масла светло-желтого цвета. Анализы ЯМР и GC-MS показывают следующее соединение в виде примеси:
GC-MS: m/z = 283 (М+).
Н-ЯМР (600 МГц, ДМСО-d6), δ м.д., 1,23-1,31 (м, 1Н), 1,33-1,40 (м, 1Н), 1,40-1,48 (м, 1Н), 1,51-1,63 (м, 2Н), 1,66-1,76 (м, 1Н), 1,80-1,91 (м, 1Н), 2,02 (кв, J=7,18 Гц, 1Н), 2,07 (кв, J=7,18 Гц, 1Н), 2,15-2,24 (м, 1Н), 2,80 и 2,99 (2с - ротамеры, 3Н), 3,13-3,22 (м, 1Н), 3,25-3,33 и 3,33-3,41 (2м - ротамеры, 2Н), 3,49 (кв, J=8,43 Гц, 1Н), 3,57 (с, 3Н), 4,16 (с, 1Н), 4,71 (т, 1Н), 4,91-5,09 (м, 2Н), 5,73-5,87 (м, 1Н).
С-ЯМР (150 МГц, ДМСО-d6), д м.д. (смесь ротамеров): 25,23 и 25,28, 26,00 и 27,61, 32,83 и 32,87, 33,20 и 34,62, 28,24 и 38,25, 39,33 и 40,13, 41,27 и 41,69, 44,92 и 45,11, 46,62, 48,67, 51,49, 70,77 и 70,81, 114,74 и 114,98, 138,38 и 138,60, 172,99 и 173,05, 174,65 и 174,68.
9,41 г (50 ммоль) Соединения формулы (VI), 5,94 г (52,5 ммоль) NMHA и 10,54 г (55 ммоль) EDCI суспендируют в 89 мл ТГФ. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют 100 мл МеТГФ и смесь последовательно промывают с помощью 50 мл воды, 50 мл 0,5 М водного раствора HCl, 50 мл 0,5 М водного раствора NaOH и 50 мл воды, сушат над сульфатом магния, отфильтровывают и концентрируют в вакууме. Получают 7,54 г (выход 53%) сырого соединения формулы (VII) в виде масла желтого цвета.
3,94 г Сырого соединения формулы (VII) очищают с помощью флэш-хроматографии на силикагеле, используя этилацетат в качестве элюента, получая 1,92 г очищенного соединения формулы (VII). Анализ GC показывает чистоту > 95%.
14,11 г (75 ммоль) Соединения формулы (VI), 8,91 г (78,8 ммоль) NMHA и 20,40 г (82,3 ммоль) EEDQ растворяют в 75 мл ТГФ. Реакционную смесь перемешивают в течение ночи при кипячении с обратным холодильником. Добавляют 1,27 г (11,3 ммоль) NMHA и 2,78 г (11,3 ммоль) EEDQ и кипятят с обратным холодильником в течение всей ночи. 63 мл Растворителя отгоняют и добавляют 75 мл ксилола, 75 мл воды и 13,5 мл концентрированной HCl. Слои разделяют и органический слой промывают с помощью 37,5 мл воды и концентрируют в вакууме, получая 22,47 г (106% «сырой» выход) сырого соединения формулы (VII) в виде масла светло-оранжевого цвета и это масло используют на следующей стадии, после того, как оно было получено. Анализ LC показывает, что главной примесью является некоторое количество остаточного ксилола.
1,0 г (5,3 ммоль) Соединения формулы (VI) и 1,2 г (5,8 ммоль) EDCI суспендируют в 10 мл дихлорметана. Суспензию перемешивают при комнатной температуре вплоть до получения раствора (приблизительно 15 минут). Затем добавляют 0,63 г (5,6 ммоль) NMHA и 6 мг (0,05 ммоль) DMAP и реакционную смесь перемешивают в течение ночи при комнатной температуре. Потом добавляют 10 мл этилацетата и смесь промывают с помощью 2 М водного раствора HCl, затем насыщенным солевым раствором. Органический слой концентрируют в вакууме, получая 0,94 г (выход 63%) сырого соединения формулы (VII) в виде масла желтого цвета. Анализ ЯМР показывает чистый продукт.
300 мг (1,6 ммоль) Соединения формулы (VI) и 180 мг (1,6 ммоль) NMHA растворяют в 5 мл ацетонитрила. Затем добавляют 0,6 г (1,6 ммоль) HBTU и 0,8 мл (4,8 ммоль) DIPEA и смесь перемешивают в течение 2 часов при комнатной температуре. Потом добавляют 20 мл этилацетата и смесь промывают с помощью 2 М водного раствора HCl, затем насыщенным солевым раствором. Органический слой концентрируют в вакууме. Получают 1,07 г (выход 51%) сырого соединения формулы (VII) в виде масла коричневого цвета.
40,0 г (0,21 моль) Соединения формулы (VI), 28 г (0,24 моль) NMHA и 63 г (0,26 моль) EEDQ растворяют в 400 мл ТГФ. Смесь перемешивают при температуре кипения с обратным холодильником вплоть до полного завершения реакции, затем разбавляют с помощью 400 мл МТВЕ и последовательно промывают 2 раза по 100 мл 1 М водного раствора HCl, 100 мл 1 М водного раствора NaOH и 50 мл насыщенного солевого раствора. Органический слой концентрируют в вакууме, получая 80 г сырого соединения формулы (VII), используемого на следующей стадии, после того, как оно было получено.
Пример 9
Получение соединения формулы (IX)

1,7 г (5,6 ммоль) Соединения формулы (Va) растворяют в 25 мл толуола. Этот раствор концентрируют в вакууме досуха для того, чтобы удалить следовые количества воды или остаточный спиртовой растворитель. К остатку добавляют 1,57 г (5 ммоль) соединения формулы (VIII), 1,77 г (6,75 ммоль) трифенилфосфина и 25 мл ТГФ. Смесь охлаждают до температуры 0°С и по каплям добавляют 1,24 мл (6,75 ммоль) диизопропилазодикарбоксилата (DIAD). Реакционную смесь перемешивают в течение 4 часов при температуре 0°С, затем в течение ночи при комнатной температуре. Для уничтожения избытка реагента Mitsunobu добавляют 0,5 мл (0,5 ммоль) 1 М водного раствора NaOH. Потом смесь концентрируют в вакууме досуха и остаток элюируют при использовании силикагеля с помощью этилацетата в качестве элюента. Предварительно очищенный продукт суспендируют в 10 мл кипящего метанола и суспензию охлаждают до комнатной температуры, перемешивают в течение ночи при комнатной температуре, затем в течение 1 часа при температуре 0°С. После фильтрации суспензии и высушивания, получают 2,05 г (выход 71%) очищенного соединения формулы (IX) в виде порошка белого цвета.
Физические данные: температура плавления: 125,1°С; [б]D: -9,1°; ЖХ-МС: m/z = 575 ([М+Н+]);
Н-ЯМР (400 МГц, ДМСО-d6), δ м.д., 1,33 (д, J=6,80 Гц, 6Н), 2,21-2,34 (м, 2Н), 2,41 (дд, 1Н), 2,58 (с, 3Н), 2,59-2,66 (м, 1Н), 3,14 (гепт, 1Н), 3,38 (ддд, 1Н), 3,55 (дт, J=10,58, 7,93 Гц, 1Н), 3,64 (с, 3Н), 3,95 (с, 3Н), 4,98 и 5,06 (AB, J=12,34 Гц, 2Н), 5,37 (с, 1Н), 7,14-7,19 (м, 2Н), 7,22-7,28 (м, 3Н), 7,38 (д, J=9,32 Гц, 1Н), 7,45 (д, J=3,78 Гц, 2Н), 7,88 (д, J=9,32 Гц, 1Н).
С-ЯМР (100 МГц, ДМСО-d6), д м.д.: 9,77, 22,30, 30,40, 35,10, 35,93, 44,66, 45,02, 51,90, 56,08, 66,00, 78,70, 95,35, 112,83, 115,50, 116,07, 120,07, 120,25, 127,60, 127,91, 128,25, 135,80, 147,86, 151,17, 157,96, 160,31, 164,27, 168,64, 173,12, 173,86.
20,75 г (74,5 ммоль) Соединения формулы (Va), 26,25 г (71 ммоль) соединения формулы (VIII) и 28 г (110 ммоль) трифенилфосфина растворяют в 391 мл толуола. Отгоняют 50 мл толуола для того, чтобы удалить любые следовые количества воды или остаточный спиртовой растворитель. Смесь охлаждают до температуры 0°С и по каплям добавляют 21,58 г (110 ммоль) DIAD. Реакционную смесь перемешивают в течение 2 часов при температуре 0°С. Для уничтожения избытка реагента Mitsunobu добавляют 7,1 мл воды. Смесь перемешивают при комнатной температуре в течение 10 минут, затем побочные продукты реакции Mitsunobu отфильтровывают и промывают с помощью 25 мл толуола. Фильтрат и промывочные слои концентрируют в вакууме и горячий маслянистый остаток разбавляют с помощью 355 мл метанола. Смесь охлаждают до температуры 0°С, затем перемешивают в течение ночи при температуре 0°С. Получают, после фильтрации суспензии и высушивания, 30,6 г (выход 75%) соединения формулы (IX) в виде порошка белого цвета.
Пример 10
Получение соединения формулы (Х)

1,50 г (2,61 ммоль) Соединения формулы (IX) и 1,25 мл (7,83 ммоль) триэтилсилана растворяют в 2,6 мл ТГФ. Добавляют 29,3 мг (0,13 ммоль) ацетата палладия и смесь перемешивают в течение ночи при кипячении с обратным холодильником. Смесь охлаждают до комнатной температуры. Затем добавляют 150 мг Norit A Supra, 150 мг дикалита и 0,2 мл 1 н. водного раствора HCl и смесь кипятят с обратным холодильником в течение 2-3 часов. Смесь отфильтровывают в горячем состоянии и фильтрат концентрируют в вакууме досуха. Остаток суспендируют в 2 мл кипящего метанола. Суспензию охлаждают до температуры 0°С и затем перемешивают в течение 1-2 часов. Получают, после фильтрации и высушивания, 800 мг (выход 63%) соединения формулы (Х) в виде порошка белого цвета.
Физические данные: температура плавления: 161,3°С; ЖХ-МС: m/z = 483 ([М-Н]-);
Н-ЯМР (400 МГц, ДМСО-d6), δ м.д., 1,34 (д, J=7,05 Гц, 6Н), 2,19-2,30 (м, 2Н), 2,38 (дд, J=13,85, 7,81 Гц, 1Н), 2,58 (с, 3Н), 2,60 (ддд, J=9,88, 4,85, 4,66 Гц, 1Н), 3,15 (гепт, 1Н), 3,23 (ддд, 1Н), 3,49 (ддд, J=10,70, 7,93, 7,81 Гц, 1Н), 3,67 (с, 3Н), 3,96 (с, 3Н), 5,34 (с, 1Н), 7,40 (д, J=9,32 Гц, 1Н), 7,44 (д, J=0,76 Гц, 1Н), 7,46 (с, 1Н), 7,96 (д, J=9,06 Гц, 1Н), 12,47 (с, 1Н).
С-ЯМР (100 МГц, ДМСО-d6), д м.д.: 9,77, 22,30, 30,40, 35,35, 36,06, 44,57, 45,09, 51,84, 56,05, 78,72, 95,30, 112,77, 115,44, 116,12, 120,07, 120,36, 147,83, 151,18, 157,94, 160,42, 164,25, 168,65, 174,11, 174,74.
4,48 г (7,79 ммоль) Соединения формулы (IX), 3,74 мл (23,37 ммоль) триэтилсилана и 87 мг (0,39 ммоль) ацетата палладия растворяют в 8 мл МеТГФ. Смесь перемешивают в течение ночи при кипячении с обратным холодильником. Смесь охлаждают до температуры 60°С. Добавляют 220 мг Norit A Supra, 220 мг дикалита, 0,52 мл воды и 0,6 мл концентрированной HCl и смесь кипятят с обратным холодильником в течение 1-2 часов. Смесь охлаждают до температуры 50-55°С, отфильтровывают и твердые вещества промывают с помощью 8 мл МеТГФ. Фильтрат и промывные жидкости объединяют и полученный раствор (15,4 г раствора - 25 %масс./масс. соединения формулы (Х)) используют на следующей стадии.
Пример 11
Получение соединения формулы (XI)
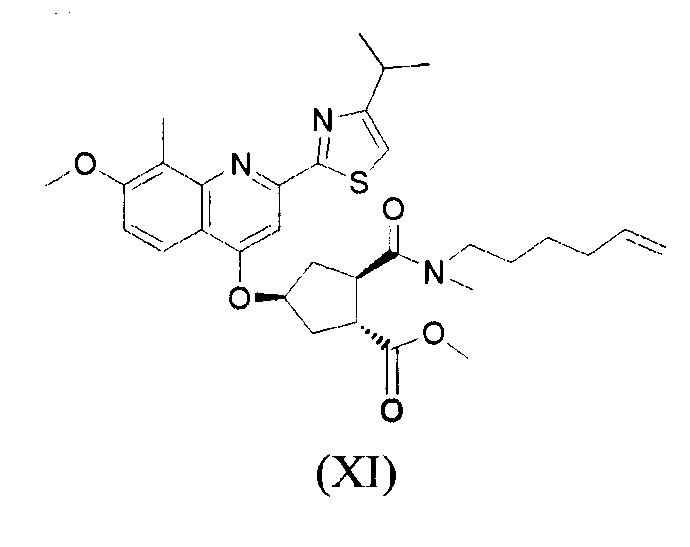
11,49 г (34,58 ммоль) Соединения формулы (VIII) и 9,52 г (36,30 ммоль) трифенилфосфина добавляют к раствору соединения формулы (VII) в толуоле (34,58 ммоль соединения формулы (VII) на 184,86 г раствора). 64 мл Растворителя отгоняют для того, чтобы удалить следовые количества воды и/или спиртов, затем смесь охлаждают до температуры 0°С. Добавляют 7,2 мл (36,30 ммоль) DIAD. Смесь перемешивают в течение 2 часов при температуре 0°С. После анализа, вводят дополнительное количество трифенилфосфина (0,9 г, 3,46 ммоль) и DIAD (0,68 мл, 3,46 ммоль) при температуре 0°С и смесь перемешивают далее при температуре 0°С в течение 1 часа, затем оставляют нагреваться до комнатной температуры в течение 15 часов, при перемешивании. Смесь затем охлаждают и перемешивают еще в течение 1-2 часов при температуре 0°С, после чего выпавшее в осадок твердое вещество отфильтровывают и промывают с помощью 17 мл толуола (твердое вещество состоит, главным образом, из трифенилфосфиноксида, 18 г, сырая масса). Из фильтрата отгоняют 140 мл растворителя, дистиллируют и добавляют 103,7 мл н-бутанола. Дистилляцию продолжают и отгоняют 88 мл растворителей, затем смесь охлаждают до температуры 80°С и добавляют 103,7 мл изопропанола и 1,73 г дикалита. Смесь отфильтровывают в горячем состоянии. Фильтрат охлаждают до температуры 30°С, вносят затравку при использовании соединения формулы (XI), охлаждают и перемешивают при температуре 0°С в течение 56 часов. Смесь отфильтровывают, фильтровальный осадок промывают с помощью 10,4 мл холодного изопропанола и продукт высушивают при температуре 70°С в вакууме. Выход: 15,00 г (71%).
Физические данные: температура плавления: 130,7°С; [б]D: -12,6°; ЖХ-МС: m/z = 580 ([М+Н]+).
Н-ЯМР (400 МГц, ДМСО-d6, смесь ротамеров), δ м.д., 1,19 (м, 2Н - один ротамер), 1,34 (д, J=6,80 Гц, 6Н), 1,36-1,43 (м, 4Н - один ротамер), 1,46-1,61 (м, 2Н - один ротамер), 1,76-1,87 (м, 1Н), 1,92 (кв, J=6,80 гц, 2Н - один ротамер) и 2,07 (кв, J=6,88 Гц, 2Н - один ротамер), 2,20-2,40 (м, 2Н), 2,58 (с, 3Н), 2,71-2,78 (м, 1Н), 2,79 (с, 3Н - один ротамер) и 2,98 (с, 3Н - один ротамер), 3,14 (гепт, J=6,75 Гц, 1Н), 3,21-3,52 (м, 4Н), 3,62 (с, 3Н), 3,62-3,70 (м, 1Н), 3,96 (с, 3Н), 4,86 (дд, J=10,20, 0,88 Гц, 1Н - один ротамер), 4,92 (дд, J=18,13, 1,51 Гц, 1Н - один ротамер), 4,96 (дд, J=11,08, 1,00 Гц, 1Н - один ротамер), 5,03 (дд, J=17,12, 1,51 Гц, 1Н - один ротамер), 5,33 (с, 1Н), 5,68 (ддт, J=17,12, 10,32, 6,74 Гц, 1Н - один ротамер) и 5,81 (ддт, J=17,00, 10,20, 6,68 Гц, 1Н - один ротамер), 7,40-7,48 (м, 3Н), 8,03 (д, J=8,81 Гц, 1Н - один ротамер) и 8,04 (д, J=9,06 Гц, 1Н - один ротамер).
С-ЯМР (100 МГц, ДМСО-d6, смесь ротамеров), д м.д.: 9,77, 22,26, 22,80, 25,14, 25,23, 25,90, 27,56, 30,40, 32,75, 32,86, 33,12, 34,61, 35,68, 35,73, 36,21, 36,76, 42,20, 42,61, 44,92, 45,19, 46,67, 48,58, 51,62, 51,65, 56,06, 78,23, 78,30, 95,35, 95,39, 112,77, 112,86, 114,61, 114,89, 115,47, 116,13, 116,17, 119,99, 120,03, 120,50, 120,56, 138,41, 138,49, 147,83, 147,85, 151,19, 151,20, 157,98, 158,00, 160,55, 160,58, 164,22, 168,64, 168,67, 171,93, 173,95, 174,10.
730 мг (1,51 ммоль) Соединения формулы (Х) и 213 мг (1,88 ммоль) NMHA растворяют в смеси ТГФ-МеТГФ (3 мл + 3 мл). Смесь нагревают до температуры 50°С. Затем добавляют 465 мг (1,88 ммоль) EEDQ и смесь перемешивают в течение ночи при температуре 50°С. Добавляют 43 мг (0,38 ммоль) NMHA и 93 мг (0,38 ммоль) EEDQ и смесь перемешивают в течение 2 дней при температуре 50°С. Смесь концентрируют в вакууме, затем повторно растворяют в 3 мл МеТГФ и последовательно промывают с помощью 6 мл 1 н. водного раствора HCl, 3 мл воды и 1,5 мл насыщенного солевого раствора. Органический слой сушат над сульфатом магния. Твердое вещество отфильтровывают и фильтрат концентрируют досуха в вакууме. Получают 0,95 г сырого соединения формулы (XI) в виде не совсем белого цвета твердого вещества.
Сырое твердое вещество повторно суспендируют в 6 мл кипящего гептана. Суспензию охлаждают до комнатной температуры и затем перемешивают в течение ночи при комнатной температуре. Получают, после фильтрации и высушивания, 710 мг (81% выход) очищенного соединения формулы (XI) в виде порошка белого цвета.
1,23 г (10,9 ммоль) NMHA и 2,69 г (10,9 ммоль) EEDQ добавляют к 15,4 г 25 %масс./масс. соединения формулы (Х), растворенного в МеТГФ (7,8 ммоль соединения формулы (Х)). Эту смесь перемешивают в течение ночи при температуре 50°С, затем охлаждают до комнатной температуры. После этого добавляют 28 мл воды, 3,1 мл концентрированной HCl и 10 мл толуола, полученные слои разделяют и органический слой промывают с помощью 16 мл воды. Органический слой обрабатывают с помощью 2,38 г основного оксида алюминия, 1,94 г дикалита и 1,01 г Norit A Supra и отфильтровывают. Фильтрат концентрируют в вакууме и остаток суспендируют в 51 мл гептана. Суспензию перемешивают в течение ночи при комнатной температуре, затем 2 часа при температуре 0°С. Получают, после фильтрации и высушивания, 2,25 г (выход 50%) соединения формулы (XI).
Пример 12
Получение соединения формулы (XII)
600 мг (1,03 ммоль) Соединения формулы (XI) растворяют в 4,1 мл ТГФ, затем добавляют 45,6 мг (1,1 ммоль) LiOH·Н2О, предварительно растворенного в 1 мл воды. Полученную смесь перемешивают в течение 2-3 часов при комнатной температуре. Анализы путем LC и ЖХ-МС показывают почти полную конверсию соединения формулы (XI) в соединение формулы (XII). Этот раствор, после его получения, используют на следующей стадии.
Физические данные: ЖХ-МС: m/z = 564 ([М-Н]-).
Пример 13
Получение соединения формулы (XIV)
1,14 ммоль Соединения формулы (XIII) и 294 мг (1,19 ммоль) EEDQ добавляют к 1,03 ммоль литиевой соли соединения формулы (XII) в виде раствора в смеси ТГФ-вода (4,1 мл + 1 мл). Смесь перемешивают в течение ночи при комнатной температуре. Затем добавляют 2,1 мл толуола и 1,55 мл 1 н. водного раствора HCl. Два слоя разделяют и органический слой последовательно промывают с помощью 0,52 мл воды, 1,55 мл 1 н. водного раствора NaOH, 0,52 мл воды и 0,52 мл насыщенного солевого раствора. Органический слой затем сушат над сульфатом натрия. Твердые вещества отфильтровывают и фильтрат концентрируют досуха в вакууме. Получают 698 мг (выход 96%) сырого соединения формулы (XIV) в виде стекловидного соединения. Анализы путем ЯМР и LC показывают > 90% чистоты.
Физические данные: ЖХ-МС: m/z = 703 ([М+Н]+).
Н-ЯМР (400 МГц, ДМСО-d6 - смесь ротамеров), δ м.д. 0,87 (т, J=7,30 Гц, 1Н), 1,06-1,19 (м, 3Н), 1,19-1,31 (м, 2Н), 1,33 (д, J=6,80 Гц, 6Н), 1,35-1,45 (м, 2Н), 1,46-1,66 (м, 2Н), 1,84-2,00 (м, 2Н), 2,00-2,18 (м, 3Н), 2,25-2,36 (м, 1Н), 2,58 (с, 3Н), 2,64-2,77 (м, 1Н), 2,80 (с, 3Н - один ротамер), 3,00 (с, 3Н - один ротамер), 3,08 - 3,30 (м, 2Н), 3,34 - 3,52 (м, 3Н), 3,97 (с, 3Н), 3,98-4,12 (м, 2Н), 4,82-5,13 (м, 3Н), 5,18-5,37 (м, 2Н), 5,55-5,86 (м, 2Н), 7,39-7,50 (м, 3Н), 8,06 (т, J=8,94 Гц, 1Н), 8,59 (с, 1Н - один ротамер), 8,73 (с, 1Н - один ротамер).
С-ЯМР (100 МГц, ДМСО-d6, смесь ротамеров), д м.д.: 9,79, 13,80, 13,96, 13,99, 14,12, 14,53, 18,59, 22,21, 22,30, 25,25, 25,32, 26,02, 27,64, 30,40, 32,07, 32,14, 32,81, 33,19, 34,64, 34,68, 36,31, 36,76, 36,95, 36,98, 42,28, 46,01, 46,43, 46,74, 48,74, 56,10, 60,33, 60,51, 60,59, 78,73, 78,80, 95,34, 95,38, 112,67, 112,76, 114,67, 114,88, 115,47, 116,20, 116,23, 117,34, 120,03, 120,05, 120,59, 120,62, 134,13, 134,18, 138,41, 138,49, 147,85, 151,22, 157,98, 158,0, 160,75, 164,25, 168,66, 168,69, 169,82, 169,85, 172,48, 172,51, 173,66, 173,83.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ ИНГИБИТОРОВ ПРОТЕАЗЫ HCV | 2011 |
|
RU2628081C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ГИДРОКСИАЛКИЛЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ 1-ФЕНИЛ-1,2,4-ТРИАЗОЛА | 2017 |
|
RU2742885C2 |
| НИТРООКСИПРОИЗВОДНЫЕ ЛОЗАРТАНА, ВАЛСАРТАНА, КАНДЕСАРТАНА, ТЕЛМИСАРТАНА, ЭПРОСАРТАНА И ОЛМЕСАРТАНА В КАЧЕСТВЕ БЛОКАТОРОВ РЕЦЕПТОРОВ АНГИОТЕНЗИНА II ДЛЯ ЛЕЧЕНИЯ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 2004 |
|
RU2374240C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БИЦИКЛО [3.1.0]ГЕКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЭТОЙ ЦЕЛИ | 2004 |
|
RU2388747C2 |
| 6-Хлор-3-(фенил-d5)-инден-1-он и его применение | 2013 |
|
RU2681221C2 |
| (22ζ)-6b-МЕТОКСИ-3a,5-ЦИКЛО-5a-ХОЛЕСТАН-24-ОН-22-ОЛ В КАЧЕСТВЕ ПОЛУПРОДУКТА В СИНТЕЗЕ (22R,23R)-3b-АЦЕТОКСИ-22,23-ИЗОПРОПИЛИДЕНДИОКСИ-24-МЕТИЛХОЛЕСТ-5-ЕНА | 1991 |
|
RU2024540C1 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 1999 |
|
RU2246941C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭПОТИЛОНА, НОВЫЕ ПРОИЗВОДНЫЕ ЭПОТИЛОНА, А ТАКЖЕ НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ РЕАЛИЗАЦИИ СПОСОБА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2404985C2 |
| ФОТОХРОМНЫЕ ОКСАЗИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРОИЗВОДСТВА | 2002 |
|
RU2295521C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ-4 | 2003 |
|
RU2323938C2 |
Настоящее изобретение относится к способам синтеза и промежуточным продуктам соединения формулы (XVII) и его солей. 6 н. и 26 з.п. ф-лы, 13 пр.
1. Способ получения соединения формулы (XIV), исходя из промежуточного продукта (XI), который гидролизуют до кислоты (XII), которую, в свою очередь, связывают с эфиром циклопропиламинокислоты (XIII), получая желательный конечный продукт (XIV), как показано на следующей реакционной схеме:

где R означает С1-4-алкил и R1, независимо от R, также означает С1-4-алкил.
2. Способ по п.1, где соединение формулы (XI) вводят во взаимодействие с гидроксидом щелочного металла, получая соединение формулы (XII) или его соль щелочного металла.
3. Способ по п.1, где соединение формулы (XII) или его соль, далее, вводят во взаимодействие с соединением формулы (XIII) или его солью и со связывающим амид агентом, получая соединение формулы (XIV).
4. Способ получения соединения формулы (XI) или его соли, исходя из сложного гидроксициклопентилового бис-эфира формулы (Va), или:
(a) путем введения во взаимодействие сложного гидроксициклопентилового бис-эфира формулы (Va) с тиазолилзамещенным хинолинолом (VIII) по реакции образования простого эфира, получая, таким образом, сложный хинолинилоксициклопентиловый бис-эфир формулы (IX), где сложноэфирную группу, которая находится в цис-положении по отношению к простой эфирной группе в сложном хинолинилоксициклопентиловом бис-эфире формулы (IX), селективно расщепляют до монокарбоновой кислоты (X), которую, в свою очередь, связывают с алкениламином по реакции образования амида, получая, таким образом, желательный конечный продукт формулы (XI); или
(b) путем селективного превращения сложного гидроксициклопентилового бис-эфира формулы (Va) в монокарбоновую кислоту (VI), которую, в свою очередь, связывают с алкениламином по реакции образования амида, получая гидроксициклопентиламид (VII), который, в свою очередь, вводят во взаимодействие с тиазолилзамещенным хинолинолом (VIII), получая, таким образом, желательный конечный продукт формулы (XI);
как показано на следующей реакционной схеме,
где R1 означает С1-4-алкил:
5. Способ получения промежуточного продукта (Va), исходя из 4-оксоциклопентил-1,2-бискарбоновой кислоты (I), путем восстановления кетогруппы до спирта, получая, таким образом, 4-гидроксициклопентил-1,2-бискарбоновую кислоту (II), которую, в свою очередь, циклизуют до бициклического лактона (III), где группу карбоновой кислоты в бициклическом лактоне (III) этерифицируют с помощью бензилового спирта, получая, таким образом, бензиловый эфир лактона (IV), где лактон раскрывают и таким образом полученную группу карбоновой кислоты этерифицируют с помощью С1-4-алканола, получая, таким образом, сложный гидроксициклопентиловый бис-эфир формулы (V), который, в свою очередь, разделяют на стереоизомеры (Vb) и (Va); как показано на следующей реакционной схеме, где R1 означает С1-4-алкил:
6. Способ по п.1, где соединение (XI) получают в соответствии со способом по п.4.
7. Способ по п.4 или 6, где соединение (Va) получают в соответствии со способом по п.5.
8. Способ получения соединения формулы (XIV) или формулы (XI) по любому из пп.4 или 6, где соединение формулы (IX) или формулы (XI) получают путем введения во взаимодействие соединения формулы (Va), соответственно, формулы (VII), с соединением формулы (VIII), в присутствии азодикарбоксилата формулы R'OOC-N=N-COOR', фосфина формулы R''3P и органического растворителя; где
R' означает этил или изопропил или трет-бутил;
R'' означает, каждый, независимо, фенил, 2-пиридил, 3-пиридил или 4-пиридил.
9. Способ получения соединения формулы (XIV) или формулы (XI) по любому из пп.4 или 6, где соединение формулы (VII) или формулы (XI) получают путем введения во взаимодействие соединения формулы (VI), соответственно, соединения формулы (X), с N-метилгекс-5-ениламином (NMHA) и агентом связывания амида, в инертном по отношению к реакции растворителе.
10. Способ по п.9, где связывающий амид агент выбирают из EEDQ, IIDQ, EDCI, DCC или 1,3-диизопропилкарбодиимида.
11. Способ получения соединения формулы (XIV) или формулы (XI) по любому из пп.4 или 6, где соединение формулы (VI) или соединение формулы (X) получают путем введения во взаимодействие соединения формулы (Va), соответственно, соединения формулы (IX), с восстановителем, в инертном по отношению к реакции растворителе.
12. Способ по п.11, где восстановителем является водород в присутствии металлического катализатора, или где восстановителем является муравьиная кислота или ее соль, смесь муравьиной кислоты и ее соли, триэтилсилан, трет-бутилдиметилсилан, фенилсилан или поли(метилгидросилоксан), необязательно в присутствии основания.
13. Способ по п.12, где восстановителем является водород в присутствии металлического катализатора.
14. Способ по п.13, где катализатор выбирают из палладия-на-угле, гидроксида палладия-на-угле, ацетата палладия или хлорида палладия.
15. Способ по любому из пп.12-14, где основанием является три- С1-4-алкиламин.
16. Способ по любому из пп.11-14, где органический растворитель выбирают из ТГФ, МеТГФ, уксусной кислоты, толуола или любой их смеси.
17. Способ по п.5, где соединение формулы (Va) получают путем разделения смеси (V) с помощью хирального разделения.
18. Способ по п.17, где хиральное разделение представляет собой хиральную колоночную хроматографию.
19. Способ по любому из пп.5 или 6, где соединение формулы (V) получают путем введения во взаимодействие соединения формулы (IV) с С1-4-алканолом и кислотным катализатором.
20. Способ по любому из пп.5 или 6, где соединение формулы (IV) получают путем введения во взаимодействие соединения формулы (III) с бензиловым спиртом, в присутствии связующего агента или в присутствии С1-4-алкилхлорформиата формулы ClCOOR'', где R'' означает С1-4-алкил, такой как метил, этил, 1-пропил, 1-бутил, 2-бутил, и органического основания.
21. Способ по п.20, где связующий агент выбирают из EDCI, DCC и диизопропилкарбодиимида.
22. Способ по любому из пп.5 или 6, где соединение формулы (III) или его соль получают путем введения во взаимодействие соединения формулы (II) или его соли с С1-4-алкилхлорформиатом формулы ClCOOR'', где R'' имеет значение, как описано в п.20, в присутствии органического основания.
23. Способ по любому из пп.5 или 6, где соединение формулы (II) получают путем взаимодействия соединения формулы (I) с водородом, в присутствии металлического катализатора, необязательно в присутствии основания.
24. Способ по п.23, где катализатор выбирают из родия-на-угле, родия-на-оксиде алюминия, платины-на-угле или платины-на-оксиде алюминия.
25. Способ по п.22, где основание выбирают из гидроксида натрия, оксида алюминия и три-С1-4-алкиламина.
26. Способ по любому из пп.1, 2, 3, 4, 5, 6, 10, 12, 13, 14, 17, 18, 21, 24 или 25, где R1 означает метил.
27. Способ по любому из пп.1, 2, 3, 4, 5, 6, 10, 12, 13, 14, 17, 18, 21, 24 или 25, где R означает этил.
28. Применение соединения, выбираемого из:



где R и R1, каждый, независимо, означает С1-4-алкил,
в качестве промежуточного продукта при получении соединения формулы (XVII):

или его фармацевтически приемлемой соли.
29. Применение по п.28, где R означает этил и R1 означает метил.
30. Соединение формулы (VI)
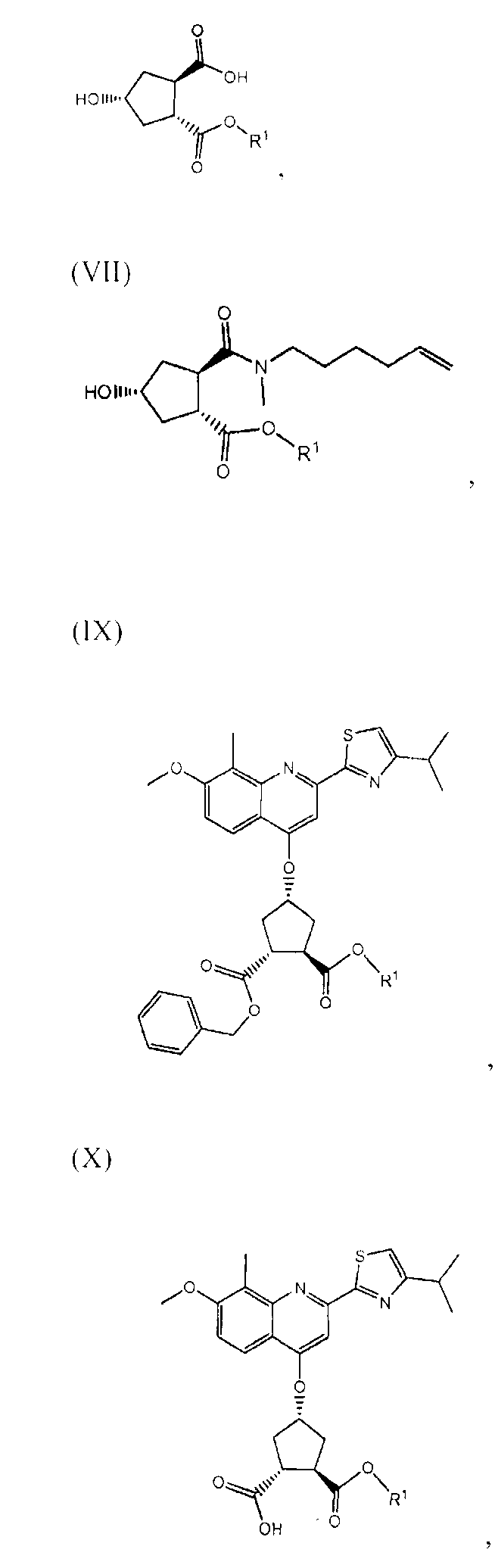
или
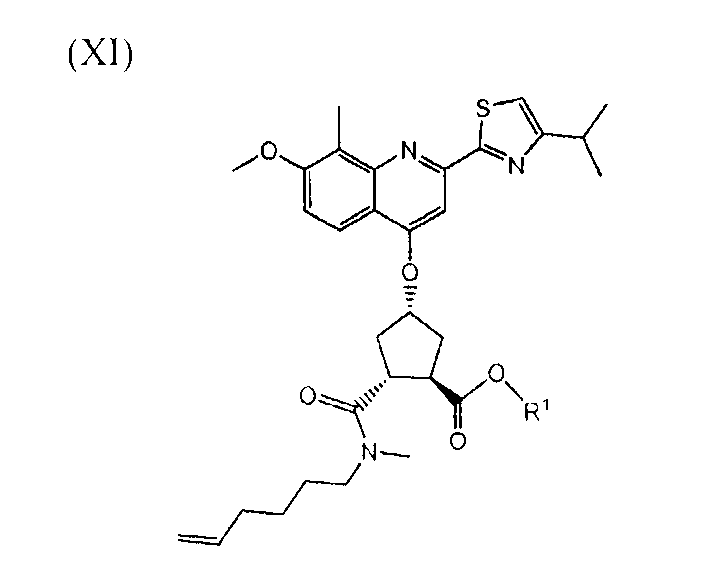
где R1 означает С1-4-алкил.
31. Соединение по п.30, где R1 означает метил.
32. Способ получения соединения формулы (XVII), включающий реакцию промежуточного продукта (XIV), который циклизуют для получения продукта (XV), который затем гидролизуют до макроциклической кислоты (XVI);
макроциклическую кислоту (XVI) связывают с сульфониламидом посредством реакции образования амида, таким образом, получая конечный продукт (XVII), как показано на следующей реакционной схеме:
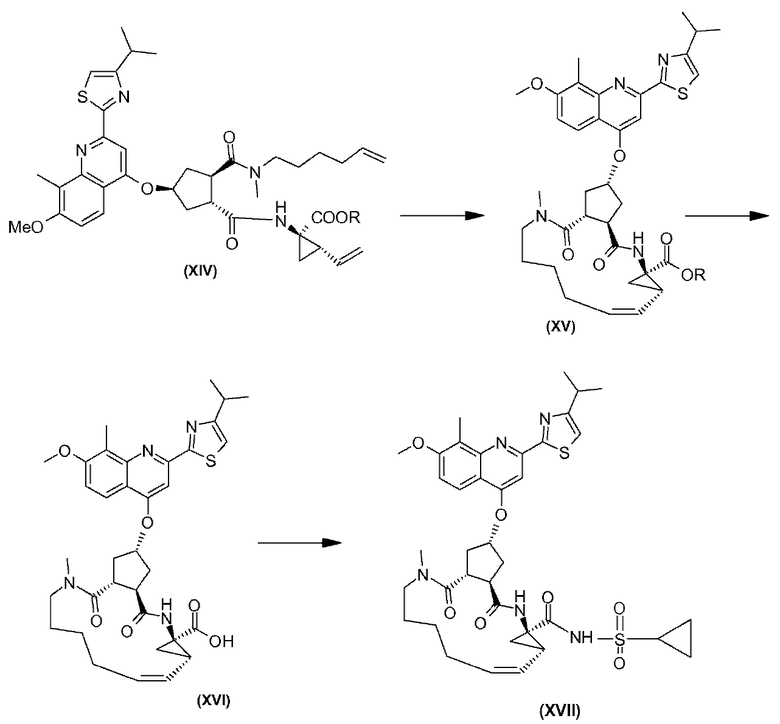
и где соединение (XIV) получают по способу по любому одному из пп.1-3 и 6-27.
| Устройство для определения количества поступившей в бак и отпущенной из бака жидкости | 1928 |
|
SU14188A1 |
| WO 2005073195 А2, 11.08.2005 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| BARTLETT PAUL A ET AL: "Total synthesis of brefeldin A" JOURNAL OF THE AMERICAN CHEMICAL | |||

