Область техники
Настоящее изобретение относится к реакции аминирования, в частности к катализатору для получения органического амина каталитическим аминированием, его получению и применению.
Уровень техники
Амины являются очень важными промышленными органическими соединениями и широко применяются в различных областях, например, в качестве растворителей, медицинских промежуточных продуктов, смоляного сырья, текстильных добавок, инсектицидов, стабилизаторов каучука, резистов, а также в очистке и в переработке пластмасс. Три основных способа получения аминов представляют собой гидроаминирование карбонильных соединений, гидроаминирование спиртов и гидрирование нитрилов.
Гидроаминирование карбонильных соединений представляет собой, например, реакцию ацетона, водорода и аммиака с образованием изопропиламина. Гидроаминирование спиртов включает, например, гидроаминирование этанола аммиаком в присутствии водорода с образованием этиламина, гидроаминирование изопропанола аммиаком в присутствии водорода с образованием изопропиламина, гидроаминирование бутанола аммиаком в присутствии водорода с образованием бутиламина, и гидроаминирование гександиола аммиаком в присутствии водорода с образованием гександиамина и т.д. Гидрированием нитрила является, например, гидрирование ацетонитрила до этиламина и гидрирование адипонитрила до гександиамина.
Патент США № US 4409399 описывает способ получения жирных аминов. Применяемый катализатор состоит из (1) оксида или гидроксида меди, (2) оксида или гидроксида никеля и (3) оксида или гидроксида металла группы IIA.
Заявка на патент Китая № CN 102658162 A описывает катализатор для синтеза этиленамина и способ получения этиленамина. Катализатор состоит из трех частей, а именно основного активного компонента, вспомогательного агента и аминированного носителя, при этом основной активный компонент является одним или несколькими компонентами, выбранными из Ni и Со, и составляет 1-40% от общей массы катализатора, и вспомогательный агент представляет собой один или несколько агентов, выбранных из группы, состоящей из Fe, Cu, Ru, Re, K, Zn и В и их оксидов, и составляет 0,1-20% от общей массы катализатора; аминированный носитель получают аминированием одного или нескольких носителей, выбранных из группы, состоящей из SiO2 и Al2O3.
Однако каталитическую активность, конверсию сырья, селективность по продукту и каталитическую стабильность существующих катализаторов для реакции аминирования все еще необходимо улучшать.
Сущность настоящего изобретения
Цель настоящего изобретения заключается в обеспечении катализатора, пригодного для получения органических аминов каталитическим аминированием, его получения и применения, при этом катализатор показывает улучшенные характеристики при применении в реакции аминирования, такие как по меньшей мере одну характеристику из улучшенной каталитической активности, улучшенной конверсии реакции, улучшенной селективности продукта и улучшенной стабильность катализатора.
Для достижения вышеуказанной цели в одном аспекте настоящее изобретение обеспечивает катализатор, пригодный для получения органических аминов путем каталитического аминирования, содержащий неорганический пористый носитель, содержащий алюминий и/или кремний, и активный металлический компонент, нанесенный на носитель, причем активный металлический компонент содержит по меньшей мере один металл, выбранный из металлов группы VIII и группы IB, где носитель имеет адсорбционную емкость по аммиаку от 0,25 до 0,65 ммоль/г, как измерено с помощью теста NH3-TPD.
Предпочтительно, носитель содержит матрикс, содержащий первый компонент носителя и необязательно второй компонент носителя, и примесный элемент, где первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита и диоксида титана, и примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, за исключением натрия и хлора, металлический элемент по меньшей мере является элементом, выбранным из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантанидов; неметаллический элемент по меньшей мере выбирают из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA.
Предпочтительно, катализатор дополнительно содержит металлический промотор, нанесенный на носитель, и металлический промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и ряда лантаноидов.
В другом аспекте, обеспечивают способ получения катализатора настоящего изобретения, причем способ включает стадии:
1) обеспечение неорганического пористого носителя, содержащего алюминий и/или кремний, который имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом;
2) загрузки активного металлического компонента и необязательно металлического промотора на носитель; и
3) проведения термической обработки и необязательно восстановительной обработки материала, полученного на стадии 2) для получения катализатора.
В еще другом аспекте, настоящее изобретение обеспечивает способ получения органических аминов, включающий:
контактирование сырья для аминирования, реагента для аминирования и катализатора согласно настоящему изобретению для реакции аминирования в присутствии водорода для получения органического амина, где сырье для аминирования выбирают из группы, состоящей из спиртов, кетонов, спиртаминов, альдегидов и их комбинации; и реагент для аминирования выбирают из группы, состоящей из аммиака, первичных аминов, вторичных аминов и их комбинаций.
Катализатор настоящего изобретения демонстрирует улучшенные характеристики, в частности улучшенную каталитическую
активность, конверсию реакции, селективность продукта и/или стабильность катализатора, при применении для получения органических аминов каталитическим аминированием.
Другие характеристики и преимущества настоящего изобретения будут описаны более подробно в подробном описание в настоящем изобретении ниже.
Подробное описание настоящего изобретения
Настоящее изобретение далее будет подробно описано со ссылкой на его конкретные варианты осуществления. Следует отметить, что конкретные варианты осуществления настоящего изобретения представлены только в целях иллюстрации и никоим образом не предназначены для ограничения.
Любое конкретное числовое значение, включая конечные точки числового диапазона, описанного в контексте настоящего изобретения, не ограничивается его точным значением, но должно интерпретироваться для дальнейшего включения всех значений, близких к указанному точному значению, например, всех значений в пределах ±5% указанного точного значения. Более того, в отношении любого числового диапазона, описанного в настоящем изобретении, могут быть сделаны произвольные комбинации между конечными точками диапазона, между каждой конечной точкой и любым конкретным значением в пределах диапазона или между любыми двумя конкретными значениями в пределах диапазона, чтобы обеспечить один или несколько новых числовых диапазонов, где указанный новый числовой диапазон(ы) также следует считать специально описанным в настоящей заявке.
Если не указано иначе, термины, применяемые в настоящем изобретении, имеют то же значение, которое обычно понятно специалисту в данной области техники; и если термины определены в настоящем изобретении и их определения отличаются от общепринятого понимания в данной области техники, определение, данное в настоящем изобретении, имеет преимущественную силу.
В настоящей заявке, адсорбционную емкость по аммиаку носителя и катализатора измеряют NH3-TPD тестом, где адсорбционную емкость по аммиаку выражают как измеренное количество десорбированного аммиака.
В настоящей заявке, удельная площадь поверхности, объем пор и долю пор, имеющих различные диаметры пор носителя, измеряют способом адсорбции-десорбции азота согласно GB/T6609.35-2009.
В настоящей заявке, выражение "С2-20" обозначает содержащий 2-20 атомов углерода, например, содержащий 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 атомов углерода. Аналогично, выражение "С1-12" обозначает содержащий 1-12 атомов углерода.
В настоящей заявке, размеры зерен активного металлического компонента и металлического промотора измеряют
рентгеноструктурным анализом.
В настоящей заявке, если не указано иначе, данные давления являются манометрическими.
В контексте настоящего изобретения, в дополнение к предметам изобретения, которые указаны явно, любой предмет или предметы изобретения, которые не упомянуты, считаются такими же, как те, которые являются известными в данной области техники, без каких-либо изменений. Кроме того, любой из вариантов осуществления, описанных в настоящем изобретении, может быть свободно объединен с другим одним или несколькими вариантами осуществления, описанными в настоящем изобретении, и полученные таким образом технические решения или идеи рассматриваются как часть исходного раскрытия или исходного описания настоящего изобретения, и не должны рассматриваться как новый предмет изобретения, который не был раскрыт или не ожидался в настоящем изобретении, если только специалисту в данной области техники не ясно, что такая комбинация является явно неадекватной.
Все патентные и непатентные документы, цитируемые в настоящем изобретении, включая, но не ограничиваясь, учебники и журнальные статьи, включены в настоящее изобретение полностью посредством ссылки.
Как описано выше, в первом аспекте, настоящее изобретение обеспечивает катализатор, пригодный для получения органических аминов каталитическим аминированием, содержащий неорганический пористый носитель, содержащий алюминий и/или кремний, и активный металлический компонент, содержащий по меньшей мере один металл, выбранный из металлов группы VIII и группы IB, где носитель имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом.
Согласно настоящему изобретению, металл группы VIII может представлять собой, например, кобальт, никель или палладий, и металл группы IB может представлять собой, например, медь. В предпочтительном варианте осуществления металл в составе активного металлического компонента выбирают из кобальта, никеля, палладия, меди или их комбинации, более предпочтительно выбирают из кобальта, никеля или их комбинации.
В катализаторе настоящего изобретения, металл группы IB, такой как медь, можно применять отдельно в качестве активного металлического компонента, и в данном случае его обычно применяют в относительно большем количестве; его также можно применять в сочетании с металлом группы VIII, и в данном случае его обычно применяют в относительно меньшем количестве. При применении в сочетании с металлом группы VIII, таким как кобальт, никель и палладий, металл группы IB обычно упоминается в настоящем изобретении как металлический промотор.
В предпочтительном варианте осуществления, носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г, как измерено NH3-TPD тестом.
В предпочтительном варианте осуществления, носитель содержит матрикс, содержащий первый компонент носителя и необязательно второй компонент носителя, и примесный элемент, где первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита и диоксида титана, и примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, исключая натрий и хлор, металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантанидов, предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана; неметаллический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, и предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
В следующем предпочтительном варианте осуществления, примесный элемент в носителе получен от катионов металлов и ионов кислотных радикалов, но не включает ион натрия или хлорид-ион; катион металла представляет собой по меньшей мере катион металла, выбранный из группы, состоящей из катионов металлов группы IA, ионов металлов группы IIA, ионов металлов группы VA и ионов металлов ряда лантанидов, предпочтительно по меньшей мере катион металла, выбранный из группы, состоящей из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария и иона лантана; ион кислотного радикала представляет собой по меньшей мере один ион кислотного радикала, выбранный из неметаллических ионов кислотных радикалов, предпочтительно по меньшей мере неметаллический ион кислотных радикалов, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
В предпочтительном варианте осуществления, носитель имеет по меньшей мере одну из следующих характеристик:
носитель имеет адсорбционную способность по углекислому газу 0,05-0,4 ммоль/г, предпочтительно 0,05-0,3 ммоль/г, более предпочтительно 0,06-0,2 ммоль/г, и где носитель имеет адсорбционную способность по углекислому газу в описанном выше диапазоне, это благоприятствует повышению селективности катализатора по продукту и уменьшению образования побочных продуктов;
примесный элемент присутствует в носителе в количестве 0,03-6 масс. %, предпочтительно 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, относительно общего веса матрикса;
носитель имеет удельную площадь поверхности 120-240 м2/г, предпочтительно 120-210 м2/г и более предпочтительно 125-200 м2/г;
носитель имеет объем пор - 0,45-1,2 мл/г, предпочтительно 0,45-1,1 мл/г и более предпочтительно 0,5-1 мл/г;
доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%, предпочтительно большей чем или равной 70%, более предпочтительно 70-90%, и доля объема пор, имеющих диаметр пор меньше чем 7 нм к объему пор носителя, предпочтительно составляет 0-10%, например 0-8%; где носитель имеет описанное выше распределение диаметра пор, это благоприятствует повышению поверхностной диффузии катализатора и повышению активности и селективности продукта катализатора;
матрикс носителя содержит комбинацию оксида алюминия и диоксида титана при весовом соотношении 1,5-5:1, предпочтительно 2-4,5:1; и
содержание оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 75 масс. % или более, более предпочтительно 80-100 масс. %, исходя из общего веса матрикса.
В предпочтительном варианте осуществления катализатора настоящего изобретения, активный металлический компонент присутствует в количестве 5-46 г, предпочтительно 10-42 г, например, 13-40 г, на 100 г матрикса.
В предпочтительном варианте осуществления, катализатор дополнительно содержит металлический промотор, нанесенный на носитель, и металлический промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и металлов ряда лантаноидов, предпочтительно содержит по меньшей мере один металл, выбранный из группы, состоящей из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се. Более предпочтительно, металлический промотор присутствует в количестве 0-10 г, предпочтительно 0,1-10 г, более предпочтительно 0,5-8 г, на 100 г матрикса.
В некоторых более предпочтительных вариантах осуществления, металлический промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, где весовое отношение металла группы VIIB к металлу группы IB, рассчитывается как металлический элемент, составляет 0,05-15:1, предпочтительно 0,1-12:1; или металлический промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB, где весовое отношение металла группы VIIB к металлу группы IIB составляет 0,2-20:1, предпочтительно 0,3-6:1; или металлический промотор содержит комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB при весовом соотношении металла группы VIB к металлу группы IB и к металлу группы IIB 0,1-10:0,1-10:1, предпочтительно 0,2-8:0,2-8:1. Особенно предпочтительно, металл группы VIIB выбирают из марганца и/или рения, металл группы IB представляет собой по меньшей мере металл, выбранный из группы, состоящей из меди, серебра и золота, металл группы IIB выбирают из цинка, и металл группы VIB выбирают из молибдена и/или вольфрама.
Согласно настоящему изобретению, носитель для катализатора можно получить способами, известными в данной области техники, пригодными для получения носителей, обладающих приведенными выше свойствами, на которые нет конкретных ограничений в настоящей заявке. Предпочтительно, носитель можно получить способом, включающим следующие стадии: подвергание смеси, содержащей химический элемент и матрикс или его предшественник, формованию, сушке и кальцинированию последовательно для получения носителя, где матрикс содержит первый компонент носителя и необязательно второй компонент носителя, причем первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, и второй компонент носителя выбирают из диатомита, белого порошка титана или их комбинации. Молекулярным ситом может быть, например, молекулярное сито ZSM-5 или ZSM-11. При применении предшественника матрикса предшественником оксида алюминия может быть псевдобемит, и предшественником оксида кремния может быть кремниевая кислота, ортокремниевая кислота или силикагель.
В указанном выше способе получения носителя предшественником матрикса является псевдобемит. Псевдобемит можно получить по меньшей мере одним из способов карбонизации, способом алюминийорганического гидролиза, способом с применением сульфата алюминия и азотнокислотным способом. Удельная площадь поверхности псевдобемита предпочтительно составляет 250-410 м2/г, более предпочтительно 260-400 м2/г, и более предпочтительно 260-380 м2/г, такая как 250-330 м2/г или 265-410 м2/г; псевдобемит предпочтительно имеет объем пор 0,7-1,3 мл/г, более предпочтительно 0,7-1,2 мл/г, более предпочтительно 0,8-1,2 мл/г, например, 0,8-1,3 мл/г или 0,78-1,2 мл/г. Катализатор с лучшими характеристиками можно получить, применяя псевдобемит с определенной структурой пор.
В указанном выше способе получения носителя, где исходный материал, обеспечивающий предшественник матрикса, уже содержит требуемое количество примесного элемента, формование может быть просто выполнено с применением данного исходного материала, и где исходный материал, обеспечивающий предшественник матрикса, не содержит примесный элемент или содержание примесного элемента является низким (недостаточным), можно осуществлять введение дополнительного примесного элемента.
В указанном выше способе получения носителя, примесный элемент можно обеспечивать с применением модификатора носителя, содержащего по меньшей мере одно соединение, способное образовывать катион (исключая ион натрия) и/или анион (исключая хлорид ион), где катион представляет собой по меньшей мере катион, выбранный из группы, состоящей из катионов группы IA, ионов металлов группы IIA, ионов металлов группы VA, ионов металлов ряда лантанидов, предпочтительно по меньшей мере катион, выбранный из группы, состоящей из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария и иона лантана; анион представляет собой по меньшей мере анион, выбранный из группы, состоящей из неметаллического иона кислотного радикала, по меньшей мере анион, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
Предпочтительно, модификатор носителя представляет собой по меньшей мере модификатор носителя, выбранный из группы, состоящей из борной кислоты, бората никеля, бората кобальта, бората калия, плавиковой кислоты, фторида калия, фторида кобальта, фторида никеля, фосфорной кислоты, фосфата алюминия, фосфата трикалия, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, нитрата висмута, нитрата калия, сульфата калия, карбоната калия, нитрата магния, сульфата магния, основного карбоната магния, нитрата кальция, основного карбоната кальция, нитрата стронция, фосфата стронция, сульфата стронция, нитрата бария, нитрата лантана и селеновой кислоты.
В указанном выше способе получения носителя, способ формования можно выбрать из замешивания, раскатывания, вальцевания и подобных.
В указанном выше способе получения носителя, модификатор носителя применяют в таком количестве, что примесный элемент присутствует в количестве 0,03-6 масс. %, предпочтительно 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, например 0,08-1 масс. % (например, может составлять 0,08 масс. %, 0,1 масс. %, 0,2 масс. %, 0,3 масс. %, 0,4 масс. %, 0,45 масс. %, 0,5 масс. %, 0,55 масс. %, 0,6 масс. %, 0,7 масс. %, 0,8 масс. %, 0,85 масс. %, 0,9 масс. %, 0,95 масс. %, 1 масс. %, или величину между любыми двумя из них), относительно общего веса матрикса. Специалист в данной области может определить количество исходного материала (например, модификатора носителя) для компонента на основе количества указанного компонента в конечном носителе, и поэтому количества некоторых исходных материалов не приводят в настоящем изобретении.
В указанном выше способе получения носителя, условия сушки могут включать: температуру 80-150°С (например, 80°С, 85°С, 90°С, 95°С, 100°С, 110°С, 115°С, 120°С, 125°С, 130°С, 140°С, 150°С, или величину между любыми двумя из них), такую как 100-150°С, 80-120°С или 100-120°С, продолжительность сушки 6-20 ч (например, 6 ч, 7 ч, 7,5 ч, 8 ч, 8,5 ч, 9 ч, 9,5 ч, 10 ч, 10,5 ч, 10 ч, 11 ч, 11,5 ч, 12 ч, 12,5 ч, 13 ч, 14 ч, 14,5 ч, 15 ч, 15,5 ч, 16 ч, 17 ч, 18 ч, 19 ч, 20 ч, или величину между любыми двумя из них), такую как 10-20 ч, 5-15 ч или 8-12 ч.
В указанном выше способе получения носителя, условия кальцинирования могут включать: температуру 500-1100°С (например, 600°С, 650°С, 680°С, 700°С, 750°С, 800°С, 850°С, 900°С, 950°С, 990°С, 1000°С, 1050°С, 1100°С или величину между любыми двумя из них), такую как б00-1100°С, 550-1050°С, 530-1000°С или 550-1000°С, и продолжительность кальцинирования 2-20 ч (например, 2 ч, 2,5 ч, 3 ч, 3,5 ч, 4 ч, 4,5 ч, 5 ч, 5,5 ч, 6 ч, 7 ч, 8 ч, 9 ч, 9,5 ч, 10 ч, 10,5 ч, 11 ч, 12 ч, 15 ч, 18 ч, 20 ч или величину между любыми двумя из них), такую как 4-20 ч, 4-10 ч или 5-8 ч.
Катализатор настоящего изобретения можно применять после восстановления, например, его можно восстановить с помощью водородосодержащего газа при 350-500°С, предпочтительно 350-450°С, такой как 400-450°С. Водородсодержащий газ может быть чистым газообразным водородом или газообразным водородом, разбавленным инертным газом, таким как смесь азота и водорода. Температуру восстановления постепенно повышают в процессе восстановления, причем повышение температуры происходит не слишком быстро, например, не более 20°С в час. Продолжительность восстановления можно определить, отслеживая образование Н2О в системе восстановления, то есть, когда в системе восстановления не образуется новая Н2О, восстановление прекращают, и специалист в данной области техники может соответственно выбрать продолжительность восстановления, для которого подробное описание в настоящем изобретении опущено для краткости, например, продолжительность восстановления может составлять 2-5 часов при самой высокой температуре. Восстановление можно проводить непосредственно в реакторе с последующей каталитической реакцией. Также возможно проводить восстановление в отдельном реакторе, также называемое внереакторным восстановлением, и пассивирование можно проводить после восстановления газовой смесью, содержащей кислород, перед выгрузкой катализатора из реактора, причем температура пассивирования составляет, например, от 10 до 60°С и, в частности, от 20 до 40°С. Катализатор, прошедший внереакторное восстановление и пассивирование, можно активировать перед применением, применяя водород или смесь водорода и азота при температуре, например, 150-250°С, предпочтительно 170-240°С, такой как 170-200°С.
Продолжительность активации можно определить, отслеживая образование Н2О в системе активации, то есть, когда в системе активации не образуется новая Н2О, активацию прекращают, и специалист в данной области техники может соответственно выбрать продолжительность активации, для которой подробное описание в настоящем изобретении опущено для краткости. Например, продолжительность активации при самой высокой температуре может составлять, например, от 1 до 5 часов, предпочтительно от 2 до 3 часов, или катализатор можно применять без активации, в зависимости от того, в какой степени активный металлический компонент и металлический промотор катализатора окислены.
Во втором аспекте, обеспечивают способ получения катализатора настоящего изобретения, включающего стадии:
1) обеспечения неорганического пористого носителя, содержащего алюминий и/или кремний, который имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом;
2) загрузки активного металлического компонента и необязательно металлического промотора на носитель; и
3) проведения термической обработки и необязательно восстановительной обработки материала, полученного на стадии 2), для получения катализатора,
В предпочтительном варианте осуществления, носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г, как измерено NH3-TPD тестом.
В предпочтительном варианте осуществления, указанное "обеспечение неорганического пористого носителя, содержащего алюминий и/или кремний" стадии 1) включает применение смеси, содержащей примесный элемент и матрикс или ее предшественник, в формовании, сушке и прокаливании последовательно с получением носителя, где матрикс содержит первый компонент носителя и необязательно второй компонент носителя, первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита, порошка титановых белил или их комбинации, предпочтительно первый компонент носителя представляет собой оксид алюминия, предшественник первого компонента носителя представляет собой псевдобемит, имеющий удельную площадь поверхности 250-410 м2/г, предпочтительно 260-400 м2/г, более предпочтительно 260-380 м2/г и объем пор 0,7-1,3 мл/г, предпочтительно 0,7-1,2 мл/г, более предпочтительно 0,8-1,2 мл/г; примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, исключая натрий и хлор, и металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантаноидов, предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана; неметаллический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, и предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
В следующем предпочтительном варианте осуществления, примесный элемент обеспечивают, применяя модификатор носителя, и примесный элемент и модификатор носителя можно выбрать, как описано выше, в первом аспекте, подробное описание которых опущено в настоящем изобретении для краткости. Еще более предпочтительно, модификатор носителя применяют в таком количестве, что полученный в результате носитель имеет содержание примесного элемента 0,03-6 масс. %, предпочтительно 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, например 0,08-1 масс. % (например, 0,08 масс. %, 0,1 масс. %, 0,2 масс. %, 0,3 масс. %, 0,4 масс. %, 0,45 масс. %, 0,5 масс. %, 0,55 масс. %, 0,6 масс. %, 0,7 масс. %, 0,8 масс. %, 0,85 масс. %, 0,9 масс. %, 0,95 масс. %, 1 масс. %, или величину между любыми двумя из них), относительно общего веса матрикса.
В следующем предпочтительном варианте осуществления, способ для формования можно выбрать из замешивания, раскатывания, вальцевания и подобных.
В следующем предпочтительном варианте осуществления, условия сушки стадии 1) включают: температуру-80-150°С, и продолжительность сушки 6-20 ч; и/или условия кальцинирования включают: температуру 500-1100°С, и продолжительность кальцинирования 2-20 ч.
В следующем предпочтительном варианте осуществления, стадия 1) имеет характеристики, как описано выше для способа получения носителя в первом аспекте, подробное описание которых опущено в настоящем изобретении для краткости.
В предпочтительном варианте осуществления, указанная загрузка стадии 2) включает пропитку указанного носителя раствором, содержащим предшественник указанного активного металлического компонента и необязательно предшественник указанного металлического промотора, при этом предпочтительно раствор для пропитки имеет рН в пределах 3,5-5,5. Регулирование рН раствора для пропитки в указанном выше диапазоне может дополнительно улучшить диспергируемость активного металлического компонента.
Согласно настоящему изобретению, пропитку осуществляют погружением носителя в подходящий раствор, содержащий предшественники указанного активного металлического компонента и металлического промотора, так что предшественник загружают на носитель адсорбцией. Способ пропитки можно классифицировать как сухая пропитка, влажная пропитка, множественная пропитка, смешанная пропитка, аэрозольная пропитка и подобные. Сухая пропитка и мокрая пропитка соответственно означают пропитку носителя, находящегося в сухом состоянии или предварительно смоченного водой, предшественником активного металлического компонента.
Множественная пропитка означает многократную пропитку смешанным раствором предшественника(ов) одного или более компонентов или множественную пропитку разными предшественниками, и при множественной пропитке сушку и прокаливание необходимо проводить после каждого раза пропитки для «закрепления» нанесенных компонентов. Смешанная пропитка означает совместное пропитывание предшественниками активного металлического компонента и металлического промотора, при котором не происходит реакции преципитации между данными двумя предшественниками. Пропитка распылением означает распыление раствора для пропитки на непрерывно вращающийся носитель с помощью пульверизатора таким образом, чтобы раствор для пропитки как раз заполнил объем пор носитель до насыщения. Данные способы пропитки могут быть соответствующим образом выбраны в соответствии с реальными условиями получения катализатора настоящего изобретения.
В предпочтительном варианте осуществления, предшественники активного металлического компонента и металлического промотора представляют собой растворимые соли соответствующих металлов, такие как нитраты, формиаты, оксалаты, лактаты и подобные. Растворителем, применяемым для образования раствора соли металла для пропитки носителя, является вода, хотя можно применять и некоторые органические растворители, такие как этанол. Пропитку носителя раствором соли металла можно осуществлять в любой требуемой последовательности или непрерывно несколькими растворами, содержащими одну или более солей металлов. Все стадии пропитки или одну стадию пропитки можно выполнять в несколько этапов, и также может варьироваться порядок пропитки. Концентрацию раствора подбирают таким образом, чтобы на носитель наносили требуемое количество металла.
Согласно настоящему изобретению, носитель, загруженный активным металлическим компонентом и необязательно металлическим промотором, подвергают термической обработке на стадии 3), причем указанная термическая обработка предпочтительно включает кальцинирование или комбинацию сушки и кальцинирования. Например, термическая обработка может включать сушку пропитанного носителя при 80-150°С, более предпочтительно 80-120°С. Продолжительность сушки можно соответствующим образом выбрать в зависимости от температуры сушки, количества материала, подлежащего сушке, оборудования для сушки и подобных, и может составлять, например, от 6 до 20 часов, например, 8 часов, при условии, что содержание влаги после сушки не влияет на последующее кальцинирование. Далее сушка может сопровождаться кальцинированием при 150-500°С для удаления из соли кристаллизационной воды или разложения соли на оксиды, происходящим при 300-500°С в течение 1-6 часов. В случае нескольких пропиток предпочтительно проводить сушку и кальцинирование после каждой пропитки.
В настоящей заявке, загрузка активного металлического компонента и металлического промотора не оказывает существенного влияния на микроструктуру катализатора, поэтому полученный катализатор имеет структуру пор, аналогичную структуре пор носителя.
В третьем аспекте, настоящее изобретение обеспечивает носитель, который представляет собой неорганический пористый материал, содержащий алюминий и/или кремний, где носитель имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом.
В предпочтительном варианте осуществления, носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г, как измерено NH3-TPD тестом.
В предпочтительном варианте осуществления, носитель содержит матрикс, содержащий первый компонент носителя и необязательно второй компонент носителя, и примесный элемент, где первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита и диоксида титана, и примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, исключая натрий и хлор, металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантаноидов, предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана; неметаллический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, и предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
В следующем предпочтительном варианте осуществления, примесный элемент в носителе получают из катионов металла и ионов кислотного радикала, но не включает ион натрия или ион хлорида; катион металла представляет собой по меньшей мере катион, выбранный из группы, состоящей из катионов металлов группы IA, ионов металлов группы IIA, ионов металлов группы VA и ионов металлов ряда лантанидов, предпочтительно по меньшей мере катион, выбранный из группы, состоящей из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария и иона лантана; ион кислотного радикала представляет собой по меньшей мере ион, выбран из неметаллических ионов кислотного радикала, предпочтительно по меньшей мере ион, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
В предпочтительном варианте осуществления, носитель имеет по меньшей мере одну из следующих характеристик:
носитель имеет адсорбционную способность по углекислому газу 0,05-0,4 ммоль/г, предпочтительно 0,05-0,3 ммоль/г, более предпочтительно 0,06-0,2 ммоль/г,
примесный элемент присутствует в носителе в количестве 0,03-6 масс. %, предпочтительно 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, относительно общего веса матрикса,
носитель имеет удельную площадь поверхности 120-240 м2/г, предпочтительно 120-210 м2/г, и более предпочтительно 125-200 м2/г;
носитель имеет объем пор 0,45-1,2 мл/г, предпочтительно 0,45-1,1 мл/г и более предпочтительно 0,5-1 мл/г;
доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%, предпочтительно 70% или более, более предпочтительно 70-90%, и предпочтительно доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-10%, например, 0-8%;
матрикс носителя содержит комбинацию оксида алюминия и диоксида титана при весовом соотношении 1,5-5:1, предпочтительно 2-4,5:1; и
содержание оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 75 масс. % или более, более предпочтительно 80-100 масс. %, исходя из общего веса матрикса.
В четвертом аспекте, настоящее изобретение обеспечивает применение катализатора согласно настоящему изобретению или носителя согласно настоящему изобретению для получения органических аминов каталитическим аминированием.
В пятом аспекте, настоящее изобретение обеспечивает способ получения органических аминов, включающий: в присутствии водорода, приведение в контакт исходного материала для аминирования и реагента для аминирования с катализатором согласно настоящему изобретению для реакции аминирования с получением органического амина.
В предпочтительном варианте осуществления, исходный материал для аминирования выбирают из группы, состоящей из спиртов, кетонов, аминоспиртов, альдегидов и их комбинаций, более предпочтительно выбирают из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов, С2-20 альдегидов и их смесей. Более предпочтительно, исходный материал для аминирования выбирают из группы, состоящей из этанола, ацетальдегида, н-пропанола, пропионового альдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканала, гексадеканола, гексадеканала, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенетилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6- гександиаля, 1,8-октандиола, 1,8-октандиаля, 1,12 -додекандиола, 1,12-додекандиальдегида, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и их смесей.
В настоящей заявке, реагент для аминирования относится к реагенту, способному обеспечивать аминогруппу и/или группа амина. Предпочтительно, реагент для аминирования выбирают из группы, состоящей из аммиака, первичных аминов, вторичных аминов и их комбинаций, более предпочтительно выбирают из группы, состоящей из аммиака, С1-12 первичных аминов, С2-12 вторичных аминов и их смесей, таких как по меньшей мере один из алкиламина, циклоалкиламина, и аралкиламина, и даже более предпочтительно С1-4 алкиламина. Особенно предпочтительно, реагент для аминирования выбирают из групп, состоящих из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина, диэтиламина и их смесей.
В предпочтительном варианте осуществления, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, предпочтительно 1-5:2-33:1, более предпочтительно 1-5:2-30:1, температуру 105-220°С, предпочтительно 110-220°С, более предпочтительно 130-200°С, давление 0,7-25 МПа, предпочтительно 0,8-25 МПа, более предпочтительно 1-15 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч);
В некоторых предпочтительных вариантах осуществления, исходный материал для аминирования представляет собой одноатомный спирт и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, предпочтительно 1-4:2-9:1, более предпочтительно 1-4:2-8:1, температуру 130-210°С, предпочтительно 130-200°С, давление 1-4 МПа, предпочтительно 1-3,5 МПа, более предпочтительно 1-2,5 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления, исходный материал для аминирования представляет собой кетон или альдегид и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, предпочтительно 1-4:2-5:1, температуру 105-180°С, предпочтительно 110-180°С, более предпочтительно 110-170°С, давление 0,7-3,5 МПа, предпочтительно 0,7-2,5 МПа, более предпочтительно 0,8-2,5 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления, исходный материал для аминирования представляет собой аминоспирт и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, предпочтительно 1-4:3-20:1, температуру 130-200°С, предпочтительно 135-200°С, давление 1-18 МПа, предпочтительно 1-15 МПа, более предпочтительно 1-11 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления, исходный материал для аминирования представляет собой двухатомный спирт и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 1-4:3-33:1, температуру 130-220°С, предпочтительно 130-210°С, давление 1-15 МПа, предпочтительно 4-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
В некоторых предпочтительных вариантах осуществления, исходный материал для аминирования представляет собой смесь 1,6-гександиола, циклогексимида и 6-амино-1-гексанола, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 1-4:3-33:1, более предпочтительно 1-4:3-30:1, температуру 130-220°С, предпочтительно 130-210°С, давление 1-25 МПа, предпочтительно 2-25 МПа, более предпочтительно 4-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч).
Первый тип вариантов осуществления
В первом типе вариантов осуществления настоящего изобретения, обеспечивают катализатор, выполняющий функцию катализа гидроаминирования спиртов с получением органических аминов, причем катализатор содержит носитель и активный металлический компонент и необязательно металлический промотор, нанесенный на носитель, характеризующийся тем, что носитель представляет собой по меньшей мере носитель, выбранный из группы, состоящей из допированного оксида алюминия, допированного оксида кремния, допированных молекулярных сит и допированных алюмосиликатов; носитель имеет адсорбционную емкость по аммиаку 0,25-0,6 ммоль/г, и носитель имеет адсорбционную способность по углекислому газу 0,05-0,3 ммоль/г; активный металлический компонент представляет собой кобальт и/или никель.
Катализатор первого типа вариантов осуществления настоящего изобретения обладает высокой каталитической активностью и высокой селективностью при применении в реакции гидроаминирования. Например, при применении для гидроаминирования этанола катализатор обладает высокой реакционной активностью и высокой селективностью образования этиламина при меньшем образовании метилэтиламина, метилдиэтиламина, этил-н-пропиламина и этил-втор-бутиламина. При прмиенении катализатора для гидроаминирования 1,6-гександиола образование тяжелых компонентов и других примесей является небольшим, а селективность образующегося гександиамина является высокой. После длительного периода оценки срока службы установлено, что катализатор первого типа вариантов осуществления настоящего изобретения обладает стабильной каталитической активностью, за счет регулирования кислотности и щелочности катализатора в соответствующем диапазоне улучшаются адсорбционно-десорбционные характеристики катализатора, таким образом, способствуя диффузии реакционной системы, ускорению скорости реакции, снижению отложения углерода и замедлению закупорка порового канала.
Предпочтительно, носитель имеет адсорбционную емкость по аммиаку 0,3-0,5 ммоль/г, более предпочтительно 0,3-0,42 ммоль/г.
Предпочтительно, носитель имеет адсорбционную способность по углекислому газу 0,06-0,2 ммоль/г, более предпочтительно 0,06-0,17 ммоль/г.
Предпочтительно, носитель содержит матрикс, который представляет собой по меньшей мере матрикс, выбранный из группы, состоящей из оксида алюминия, оксид кремния, молекулярных сит и алюмосиликатов, и примесный элемент, содержащий металлический элемент и неметаллический элемент. Предпочтительно, весовое отношение металлического элемента к неметаллическому элементу может составлять 1:0,05-50, более предпочтительно 1:0,2-8.
Более предпочтительно, металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA, и металлических элементов ряда лантаноидов, и более предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана.
Более предпочтительно, неметаллический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, и более предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
Более предпочтительно, примесный элемент в носителе получен из катионов металлов и ионов кислотных радикалов, но не включает ионы натрия или хлорид ионы. Поскольку примесный элемент вводят при получении носителя, примесный элемент в основном присутствует в объемной фазе носителя. Более предпочтительно, катион металла может быть по меньшей мере катионом, выбранным из группы, состоящей из катионов металлов группы IA, ионов металлов группы IIA, ионов металлов группы VA и ионов металлов ряда лантанидов, более предпочтительно по меньшей мере катион, выбранный из группы, состоящей из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария и иона лантана; ион кислотного радикала может представлять собой по меньшей мере ион, выбранный из неметаллических ионов кислотного радикала, более предпочтительно по меньшей мере ион, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
Предпочтительно, примесный элемент присутствует в носителе в количестве 0,03-2 масс. %, более предпочтительно 0,08-1 масс. %, относительно общего веса матрикса.
Предпочтительно, носитель имеет удельную площадь поверхности 120-240 м2/г.
Предпочтительно, носитель имеет объем пор 0,5-1 мл/г.
Согласно первому типу вариантов осуществления настоящего изобретения, активный металлический компонент может присутствовать в количестве 5-42 г, предпочтительно 10-35 г, более предпочтительно 10-30 г, на 100 г матрикса.
Согласно первому типу вариантов осуществления настоящего изобретения, катализатор может дополнительно содержать металлический промотор для лучшего проявления характеристик катализатора, для оптимизации доли продуктов реакции и для уменьшения нежелательных побочных реакций. Металлический промотор может представлять собой по меньшей мере металлический промотор, выбранный из группы, состоящей из группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантанидов, предпочтительно по меньшей мере металлический промотор из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се. Предпочтительно, металлический промотор может увеличиться в количестве от 0 до 10 г, примерно от 0,5 до 6 г, на 100 г матрицы.
В первом типе вариантов осуществления настоящего изобретения, обеспечивают способ получения органических аминов, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором, как описано выше, в присутствии водорода для проведения реакции аминирования.
Согласно первому типу вариантов осуществления настоящего изобретения, исходный материал для аминирования или реагент для аминирования можно выбрать, как описано выше, подробное описание которых опущено в настоящем изобретении для краткости.
Согласно первому типу вариантов осуществления настоящего изобретения, условия аминирования могут включать: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, температуру 130-200°С, давление 1-15 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, предпочтительно 2-3:4-6:1, температуру 130-200°С, предпочтительно 160-180°С, давление 1-4 МПа, предпочтительно 1-2 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч), предпочтительно 0,4-0,6 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой кетон или альдегид, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, температуру 110-180°С, давление 1-3,5 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой аминоспирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-20:1, предпочтительно 2-3:10-15:1, температуру 135-200°С, предпочтительно 170-190°С, давление 1-11 МПа, предпочтительно 8-10 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч), предпочтительно 0,4-0,6 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой двухатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 2-3:10-15:1, температуру 130-210°С, предпочтительно 180-190°С, давление 1-15 МПа, предпочтительно 8-10 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч), предпочтительно 0,4-0,6 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола (назваемого аминогексанолом для краткости), и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 3-4:10-20:1, температуру 130-210°С, предпочтительно 180-200°С, давление 1-15 МПа, предпочтительно 5-10 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч), предпочтительно 0,4-0,6 м3/(м3⋅ч).
Второй тип вариантов осуществления
Во втором типе вариантов осуществления настоящего изобретения, обеспечивают катализатор, обладающий функцией получения аминов каталитическим аминированием, содержащий носитель и активный металлический компонент и металлический промотор, нанесенные на носитель, где активный металлический компонент представляет собой кобальт и/или никель, металлический промотор представляет собой комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, и носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г.
Предпочтительно, носитель имеет адсорбционную емкость по аммиаку 0,3-0,56 ммоль/г, более предпочтительно 0,35-0,45 ммоль/г.
Катализатор второго типа вариантов осуществления настоящего изобретения содержит конкретный металлический промотор, обладает высокой каталитической активностью и в то же время обладает высокой селективностью
Предпочтительно, носитель содержит матрикс, содержащий оксида алюминия и необязательно дополнительный носитель, выбранный из оксида кремния и/или молекулярных сит, и примесный элемент. Более предпочтительно, примесный элемент присутствует в носителе в количестве 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, относительно веса матрикса.
Более предпочтительно, носитель в основном состоит из (допированного) оксида алюминия, и может дополнительно содержать (допированный) оксид кремния или подобные, тем самым дополнительно улучшая структуру пор и структурную стабильность катализатора. Особенно предпочтительно, содержание оксида алюминия в носителе составляет 75 масс. % или более, более предпочтительно 80-100 масс. %, исходя из общего веса матрикса.
Предпочтительно, примесный элемент в носителе представляет собой неметаллический элемент, предпочтительно по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена. Более предпочтительно, примесный элемент вводят при получении носителя в виде по меньшей мере элемента, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
Предпочтительно, доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя составляет 70-90%.
Предпочтительно, доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-8%.
Предпочтительно, доля объема пор, имеющих диаметр пор более чем 27 нм, к объему пор носителя составляет 15-30%.
Предпочтительно, носитель имеет удельную площадь поверхности 125-200 м2/г.
Предпочтительно, носитель имеет объем пор 0,45-1,1 мл/г.
Предпочтительно, активный металлический компонент может присутствовать в количестве 8-45 г (например, может быть любым из 8, 9, 15, 18, 20, 23, 25, 26, 30, 35, 36, 38, 40, 42, 45 или величиной между любыми двумя из них), на 100 г матрикса.
Предпочтительно, металлический промотор может присутствовать в количестве 0,1-10 г (например, может быть любым из 0,1, 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 8,5, 8,7, 9, 10 или величиной между любыми двумя из них), на 100 г матрикса.
Согласно второму типу вариантов осуществления настоящего изобретения, катализатор дополнительно содержит металлический промотор для лучшего проявления характеристик катализатора, оптимизации пропорции продуктов реакции и уменьшения нежелательных побочных реакций. Металлический промотор представляет собой смесь по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, где весовое отношение металла группы VIIB к металлу группы IB в металлическом промоторе предпочтительно составляет 0,05-15:1, более предпочтительно 0,1-12:1, еще более предпочтительно 0,5-2:1. Предпочтительно, металл группы VIIB выбирают из марганца и/или рения. Предпочтительно, металл группы IB представляет собой по меньшей мере металл, выбранный из группы, состоящей из меди, серебра и золота. Изобретатели настоящего изобретения обнаружили, что катализатор, обладающий лучшим каталитическим эффектом, можно получить при применении предпочтительной комбинации металлического промотора.
Согласно второму типу вариантов осуществления настоящего изобретения, применение носителя со специфической пористой структурой и адсорбционной емкостью по аммиаку может обеспечить катализатор, проявляющий высокую каталитическую активность и высокую селективность при применении для гидроаминирования спиртов; и получение небольшого количества побочных продуктов, включая метилэтиламин, метилдиэтиламин, этил-н-пропиламин, этил-втор-бутиламин и подобные, при применении для гидроаминирования этанола. При применении катализатора для гидроаминирования 1,6-гександиола образование тяжелых компонентов и других примесей является небольшим. После длительной оценки срока службы установлено, что катализатор имеет стабильные каталитические характеристики, и за счет регулирования кислотности (особенно адсорбционной емкости по аммиаку) катализатора в соответствующем диапазоне улучшают адсорбционно-десорбционные характеристики катализатора, и в сочетании с поровым каналом, имеющим особую структуру, это способствует диффузии реакционной системы, ускоряет скорость реакции, снижает отложение углерода и замедляет закупорку порового канала.
Во втором типе вариантов осуществления настоящего изобретения, обеспечивают способ получения органических аминов, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором, как описано выше, в присутствии водорода для проведения реакции аминирования.
Согласно второму типу вариантов осуществления настоящего изобретения, исходный материал для аминирования или реагент для аминирования можно выбрать, как описано выше, подробное описание которых опущено в настоящем изобретении для краткости.
Согласно второму типу вариантов осуществления настоящего изобретения, условия аминирования могут включать: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-33:1, температуру 110-220°С, давление 0,8-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч).
Предпочтительно, когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, температуру 130-210°С, давление 1-3,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0, 8 м3/(м3⋅ч).
Предпочтительно, когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-5:1, температуру 110-170°С, давление 0,8-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч).
Предпочтительно, когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, температуру 130-200°С, давление 1-18 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-33:1, температуру 130-220°С, давление 4-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
Третий тип вариантов осуществления
В третьем типе вариантов осуществления настоящего изобретения, обеспечивают катализатор, способный катализировать гидроаминирование спиртов, содержащий носитель и металлический активный компонент и металлический промотор, нанесенный на носитель, где металлический активный компонент представляет собой кобальт и/или никель, металлический промотор представляет собой комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB, и носитель имеет адсорбционную емкость по аммиаку 0,3-0,7 ммоль/г.
Катализатор третьего типа вариантов осуществления настоящего изобретения содержит специальный металлического промотора, обладает высокой каталитической активностью и в то же время обладает высокой селективностью и дает мало побочных продуктов.
Предпочтительно, носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г и адсорбционную способность по углекислому газу 0,05-0,3 ммоль/г.
Предпочтительно, носитель содержит матрикс и примесный элемент, матрикс содержит носитель на основе оксида алюминия и необязательно дополнительный носитель, который представляет собой по меньшей мере носитель, выбранный из группы, состоящей из оксида кремния, молекулярных сит и диатомита. Более предпочтительно, примесный элемент присутствует в носителе в количестве 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, относительно общего веса матрикса.
Более предпочтительно, носитель в основном состоит из (допированного) носителя на основе оксида алюминия, и может дополнительно содержать (допированный) оксид кремния и подобный, тем самым дополнительно улучшая диффузивность канала и стабильность пористой структуры катализатора. Особенно предпочтительно, содержание носителя на основе оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 80-100 масс. %, исходя из общего веса матрикса.
Предпочтительно, примесный элемент в носителе выбирают из металлического элемента и неметаллического элемента, и не включает натрий или хлор. Весовое отношение металлического элемента к неметаллическому элементу предпочтительно составляет 1:0,1-40. Примесный элемент присутствует в предшественнике носителя или добавляют при получении носителя, так что примесный элемент в основном присутствует в объемной фазе носителя.
Более предпочтительно, металлический элемент может представлять собой по меньшей мере металлический элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантаноидов, и более предпочтительно по меньшей мере металлический элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана.
Более предпочтительно, неметаллический элемент можно получить из по меньшей мере одного неметаллического иона кислотного радикала, более предпочтительно получить из по меньшей мере неметаллического иона кислотного радикала, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Неметаллический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена.
Предпочтительно, доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%, предпочтительно 70-90%, более предпочтительно 70-75%. Более предпочтительно, доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-10%, предпочтительно 5-8%. Более предпочтительно, доля объема пор пор, имеющих диаметр пор более чем 27 нм, к объему пор носителя составляет 10%-30%, предпочтительно 20%-30%.
Предпочтительно, носитель имеет удельную площадь поверхности 120-205 м2/г.
Предпочтительно, носитель имеет объем пор 0,45-1,2 мл/г.
Предпочтительно, носитель имеет адсорбционную способность по углекислому газу 0,05-0,4 ммоль/г.
Предпочтительно, металлический активный компонент может присутствовать в количестве 14-46 г (например, может составлять 14, 15, 20, 25, 30, 32, 35, 38, 40, 42, 45, 46 или величину между любыми двумя из них), на 100 г матрикса.
Предпочтительно, металлический промотор может присутствовать в количестве 0,1-10 г (например, может составлять 0,1, 0,5, 1, 1,2, 1,5, 1,8, 2, 3, 4, 5, 6, 7, 7,2, 7,5, 7,8, 8, 9, 10 или величину между любыми двумя из них), на 100 г матрикса.
Согласно второму типу вариантов осуществления настоящего изобретения, катализатор дополнительно содержит металлический промотор, как описано выше, для лучшего проявления характеристик катализатора, оптимизации пропорции продуктов реакции и уменьшения нежелательных побочных реакций. Весовое отношение металла группы VIIB-группы IIB в металлическом промоторе предпочтительно составляет 0,2-20:1, более предпочтительно 0,3-6:1, более предпочтительно 1-5:1. Предпочтительно, металл группы VIIB выбирают из марганца и/или рения. Предпочтительно, металл группы IIB выбирают из цинка.
Согласно третьему типу вариантов осуществления настоящего изобретения, применение носителя, имеющего специфическую структур пор, адсорбционную емкость по аммиаку и адсорбционную способность по углекислому газу, может обеспечить проявление катализатором высокой каталитической активности, и в то же время, проявление высокой селективности, при применении для гидроаминирования спиртов; и при применении катализатора для гидроаминирования н-пропанола образование других примесей является незначительным. При применении катализатора для гидроаминирования 1,6-гександиола образование тяжелых компонентов и других примесей является небольшим. После длительного периода оценки срока службы установлено, что катализатор имеет стабильные каталитические характеристики, и за счет регулирования кислотности и щелочности катализатора в соответствующем диапазоне, характеристики адсорбции-десорбции промежуточного продукта реакции на поверхности катализатора улучшается, так что диффузия реакционной системы улучшается, скорость реакции увеличивается, отложение углерода и закупорка порового канала уменьшаются, и срок службы катализатора эффективно продлевается.
В третьем типе вариантов осуществления настоящего изобретения, обеспечивают способ получения органических аминов, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором, как описано выше, в присутствии водорода для проведения реакции аминирования.
Согласно третьему типу вариантов осуществления настоящего изобретения, исходный материал для аминирования или реагент для аминирования можно выбрать, как описано выше, подробное описание которых опущено в настоящем изобретении для краткости.
Согласно третьему типу вариантов осуществления настоящего изобретения, условия аминирования могут включать: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-30:1, температуру 110-220°С, давление 0,8-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-9:1, температуру 130-200°С, давление 1-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой кетон или альдегид, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, температуру 110-180°С, давление 0,8-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой аминоспирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-20:1, температуру 130-200°С, давление 1-15 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, циклогексимида и 6-амино-1-гексанола или двухатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-30:1, температуру 130-220°С, давление 1-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Четвертый тип вариантов осуществления
В четвертом типе вариантов осуществления настоящего изобретения, обеспечивают катализатор, обладающий функцией катализа аминирования спиртов, содержащий носитель, и активный металлический компонент и металлический промотор, нанесенные на носитель, где активный металлический компонент представляет собой кобальт и/или никель, металлический промотор представляет собой комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB, и носитель имеет адсорбционную емкость по аммиаку 0,25-0,6 ммоль/г.
Катализатор четвертого типа вариантов осуществления настоящего изобретения содержит специальный металлического промотора, обладает высокой каталитической активностью, и в то же время, обладает высокой селективностью и дает мало побочных продуктов.
Предпочтительно, носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г.
Предпочтительно, носитель содержит матрикс и примесный элемент, где матрикс содержит носителя на основе оксида алюминия и дополнительный носитель, и указанный дополнительный носитель выбирают из оксида кремния и/или молекулярных сит.
Предпочтительно, предшественник оксида кремния и/или предшественник молекулярных сит и подобный добавляют при получении предшественника оксида алюминия, применяемого в носителе, и диффузивность и стабильность пористой структуры катализатора можно дополнительно сильно улучшить после получения носителя. Предпочтительно, содержание носителя на основе оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 80-97 масс. %, исходя из общего веса матрикса.
Предпочтительно, примесный элемент присутствует в носителе в количестве 0,05-5 масс. %, предпочтительно 0,08-3 масс. %, относительно общего веса матрикса.
Предпочтительно, примесный элемент вводят по меньшей мере в виде, выбранном из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона. Примесный элемент предпочтительно представляет собой по меньшей мере элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена. Согласно четвертому типу вариантов осуществления настоящего изобретения, неметаллический элемент вводят в процессе получения предшественника носителя, так что примесный элемент в основном присутствует в объемной фазе носителя, а не присоединен к его поверхности.
Предпочтительно, доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%, предпочтительно 70-90%. Более предпочтительно, доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-8%, предпочтительно 0-5%.
Предпочтительно, носитель имеет удельную площадь поверхности 120-210 м2/г.
Предпочтительно, носитель имеет объем пор 0,45-1,1 мл/г.
Предпочтительно, активный металлический компонент может присутствовать в количестве 10-46 г, предпочтительно 18-38 г, на 100 г матрикса.
Предпочтительно, металлический промотор может присутствовать в количестве 0,1-10 г, предпочтительно 0,5-6g г, на 100 г матрикса.
Согласно четвертому типу вариантов осуществления настоящего изобретения, катализатор дополнительно содержит металлический промотор, как описано выше, для лучшего проявления характеристик катализатора, оптимизации пропорции продуктов реакции и уменьшения нежелательных побочных реакций. Весовое отношение металла VIB группы, металла IB группы и металла группы IIB в металлическом промоторе предпочтительно составляет 0,1-10:0,1-10:1, более предпочтительно 0,2-8:0,2-8:1, более предпочтительно 0,5-4:0,5-6:1. Предпочтительно, металл группы VIB выбирают из молибдена и/или вольфрама. Предпочтительно, металл группы IB представляет собой по меньшей мере металл, выбранный из группы, состоящей из меди, серебра и золота. Предпочтительно, металл группы IIB выбирают из цинка.
Согласно четвертому типу вариантов осуществления настоящего изобретения, применение носителя, имеющего специфическую пористую структуру и адсорбционная емкость по аммиаку, может обеспечить катализатор, обладающий высокой каталитической активностью, и в то же время обладающий высокой селективностью при применении для гидроаминирования спиртов; и при применении катализатора для гидроаминирования н-пропанола образование других примесей является незначительным. При применении катализатора для гидроаминирования 1,6-гександиола образование тяжелых компонентов и других примесей является меньшим. После длительного периода оценки срока службы установлено, что катализатор обладает стабильной каталитической активностью, и за счет модификации носителя улучшаются адсорбционно-десорбционные характеристики катализатора, улучшается диффузия реакционной системы, увеличивается скорость реакции, отложение углерода снижается и замедляется закупорка порового канала.
В четвертом типе вариантов осуществления настоящего изобретения, обеспечивают способ получения органических аминов, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором, как описано выше, в присутствии водорода для проведения реакции аминирования.
Согласно четвертому типу вариантов осуществления настоящего изобретения, исходный материал для аминирования или реагент для аминирования можно выбрать, как описано выше, подробное описание которых опущено в настоящем изобретении для краткости.
Согласно четвертому типу вариантов осуществления настоящего изобретения, условия аминирования могут включать: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, температуру 105-220°С, давление 0,7-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 М3/(М3⋅Ч).
Предпочтительно, исходный материал для аминирования представляет собой одноатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-8:1, температуру 130-200°С, давление 1-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой кетон или альдегид, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-5:1, температуру 105-180°С, давление 0,7-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой аминоспирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, температуру 130-200°С, давление 5-18 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
Предпочтительно, исходный материал для аминирования представляет собой смесь 1,6-гександиола, циклогексимида и 6-амино-1-гексанола или двухатомный спирт, и условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, температуру 130-220°С, давление 2-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Пятый тип вариантов осуществления
В пятом типе вариантов осуществления настоящего изобретения, обеспечивают содержащий титан катализатор, обладающий функцией аминирования, содержащий носитель и активный металлический компонент и необязательно металлический промотор, нанесенный на носитель, где носитель содержит матрикс и необязательно примесный элемент, где матрикс содержит оксид алюминия и диоксид титана при весовом соотношении 1,5-5:1, активный металлический компонент представляет собой кобальт и/или никель, и катализатор имеет адсорбционную емкость по аммиаку 0,2-0,7 ммоль/г, предпочтительно 0,3-0,4 ммоль/г.
Катализатор пятого типа вариантов осуществления настоящего изобретения обладает высокой каталитической активностью, и в то же время, обладает высокой селективностью, при применении для гидроаминирования спиртов. В частности, когда катализатор применяют для гидроаминирования 1,6-гександиола, образование тяжелых компонентов и других примесей является меньшим, и селективность к гександиамину является высокой. После длительного периода оценки срока службы было обнаружено, что катализатор имеет стабильную каталитическую производительность, скорость реакции увеличивается, отложение углерода уменьшается, и закупорка порового канала замедляется.
Диоксид титана имеет сильную кислотность, содержит как В-кислоту, так и L-кислоту, и обеспечивает сильное взаимодействие с диспергированными на нем компонентами, в отличие от оксида алюминия. Композитный носитель на основе оксида алюминия и диоксида титана оказывает различное каталитическое действие на разные реакции. Авторы настоящего изобретения, путем тщательного исследования и тонкой дифференциации, обнаружили, что включение соответствующего количества диоксида титана в псевдобемит при получении носителя может особенно эффективно улучшить каталитическую эффективность полученного катализатора.
Предпочтительно, весовое отношение оксида алюминия к диоксиду титану составляет 2-4,5:1. При применении данного конкретного соотношения каталитические характеристики полученного катализатора могут быть дополнительно улучшены.
Предпочтительно, активный металлический компонент может присутствовать в количестве 13-40 г, предпочтительно 20-36 г, на 100 г матрикса.
Предпочтительно, металлический промотор может присутствовать в количестве 0-10 г, предпочтительно 2,5-8 г, на 100 г матрикса.
Согласно пятому типу вариантов осуществления настоящего изобретения, содержащий титан катализатор может дополнительно содержать металлический промотор для лучшего проявления характеристик катализатора, оптимизации пропорции продуктов реакции и уменьшения нежелательных побочных реакций. Металлический промотор может представлять собой по меньшей мере металлический промотор, выбранный из группы, состоящей из элементов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантаноидов, предпочтительно по меньшей мере металлический промотор из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се, и более предпочтительно по меньшей мере металлический промотор из Mo, Mn, Re, Cu, Ag, Zn и La.
Предпочтительно, содержащий титан катализатор имеет адсорбционную способность по углекислому газу 0,07-0,2 ммоль/г.
Предпочтительно, содержащий титан катализатор имеет удельную площадь поверхности 130-180 м2/г.
Предпочтительно, содержащий титан катализатор имеет объем пор 0,55-0,75 мл/г.
Предпочтительно, в содержащем титан катализаторе, доля объема пор, имеющих радиус пор меньше чем 4 нм к суммарному объему пор, является предпочтительно меньшей чем 20% (например, 3%, 5%, 7%, 8,5%, 10,5%, 11,5%, 12,5%, 15,5%, 18,5%, 19%, 19,5% или величину между любыми двумя из них), доля объема пор, имеющих радиус пор больше чем 10 нм, к суммарному объему пор является предпочтительно меньшей чем 15% (например, 3%, 5%, 7%, 8,5%, 9%, 10,5%, 11,5%, 12,5%, 13,5%, 14%, 14,5% или величину между любыми двумя из них), и доля объема пор, имеющих радиус пор 4-10 нм, к суммарному объему пор является большей чем или равной 65% (например, 66%, 70% 72%, 73%, 75%, 76,5%, 77%, 77,5%, 78,5%, 79%, 80% или величину между любыми двумя из них).
Предпочтительно, носитель содержащего титан катализатора допируют серой в количестве 0,1-0,5 г, более предпочтительно 0,15-0,4 г, на 100 г матрикса.
В пятом типе вариантов осуществления настоящего изобретения, загрузка активного металлического компонента и металлического промотора не оказывает существенного влияния на адсорбционную емкость по аммиаку, адсорбционную способность по углекислому газу, удельную площадь поверхности, характеристики структуры пор и подобные катализатора и, таким образом, соответствующие свойства носителя катализатора являются аналогичными описанным выше (например, различия в пределах ±5%), подробное описание которых опущено в настоящем изобретении для краткости.
Предпочтительно, содержащий титан катализатор может быть в виде полосы, листа, клевера или зубчатого сферы.
Катализаторы согласно пятому типу вариантов осуществления настоящего изобретения можно получить загрузкой, сначала обеспечивая носитель, содержащий матрикс и необязательно примесный элемент, причем матрикс содержит оксид алюминия и диоксид титана при весовом соотношении 2-5:1, и затем загрузкой активного металлического компонента и необязательно металлического промотора на носитель.
В пятом типе вариантов осуществления настоящего изобретения, обеспечивают способ получения содержащего титан катализатора, обладающего функцией аминирования, включающий:
1) применение псевдобемита и диоксида титана в замешивании, формовании и кальцинировании для получения носителя, где псевдобемит и диоксид титана применяют в таком количестве, что весовое отношение оксида алюминия к диоксиду титана в полученном носителе составляет 1,5-5:1;
2) загрузку активного металлического компонента и необязательно металлического промотора на полученный носитель, где активный металлический компонент содержит кобальт и/или никель.
Предпочтительно, псевдобемит и диоксид титана применяют в таком количестве, что весовое отношение оксида алюминия к диоксиду титана в полученном носителе составляет 2-4,5:1.
Предпочтительно, псевдобемит получают по меньшей мере одним из способа карбонизации, способа алюминийорганического гидролиза, способа с применением сульфата алюминия и способа с применением азотной кислоты. Органический алюминий предпочтительно представляет собой С1-С10 органический алюминий, предпочтительно изопропоксид алюминия.
Предпочтительно, диоксид титана обеспечивают порошком титановых белил, который получают способом осаждения и/или золь-гель способом, и исходный материал, способный обеспечить титан, применяемый в способе, может представлять собой органический источник титана (такой как органический титанат) и/или неорганический источник титана (такой как неорганическая соль титана). Неорганический источник титана может представлять собой один или более из TiCl4, Ti(SO4)2, TiOSO4, TiOCl2, гидроксид титана, соль нитрата титана, соль фосфата титана и подобные, и органический источник титана может представлять собой один или более из алкоксидов титана жирные спирты и органотитанаты. Органотитанат представляет собой органотитанат, имеющий формулу M4TiO4, где М представляет собой алкильную группу, содержащую от 1 до 4 атомов, где четыре М могут быть одинаковыми или разными, предпочтительно органотитанат представляет собой один или более, выбранный из изопропилтитаната, н-пропилтитаната, тетрабутилтитаната и тетраэтилтитаната. Конкретными примерами исходного материала, способного обеспечить титан, могут быть, но не ограничиваются: TiOCl2, тетрахлорид титана, сульфат титана, титанилсульфат, тетрапропилтитанат (включая различные изомеры тетрапропилтитаната, такие как тетраизопропилтитанат и тетра-н-пропилтитанат), тетрабутилтитанат (включая различные изомеры тетрабутилтитаната, такие как тетра-н-бутилтитанат) и тетраэтилтитанат.
Предпочтительно, содержание сульфатного радикала в порошке титановых белил составляет 0,2-3 масс. %. Порошок титановых белил более предпочтительно представляет собой порошок титановых белил, полученный способом гидролиза аммиаком сульфата титана или оксисульфата титана, и применяя данный порошок титановых белил, можно получить катализатор, содержащий носитель с более желательной структурой пористого канала и с лучшими каталитическими характеристиками.
Предпочтительно, активный металлический компонент применяют в таком количестве, что активный металлический компонент присутствует в количестве 13-40 г (например, 13 г, 15 г, 18 г, 20 г, 24 г, 25 г, 26 г, 27 г, 28 г, 30 г, 33 г, 35 г, 36 г, 37 г, 38 г, 40 г или величину между любыми двумя из них), на 100 г носителя.
Предпочтительно, для лучшего проявления характеристик катализатора, оптимизации пропорции продуктов реакции и уменьшения нежелательных побочных реакций, металлический промотор можно также загружать на носитель. Металлический промотор применяют в таком количестве, что металлический промотор присутствует в количестве 0-10 г (например, 1 г, 2 г, 2,5 г, 3 г, 4 г, 5 г, 6 г, 7 г, 8 г, 8,5 г, 9 г, 10 г, или величину между любыми двумя из них), на 100 г носителя.
Предпочтительно, металлический промотор может представлять собой меньшей мере металлический промотор, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и металлов ряда лантаноидов, предпочтительно по меньшей мере металлический промотор из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се, более предпочтительно по меньшей мере металлический промотор из Mo, Mn, Re, Cu, Ag, Zn и La.
Предпочтительно, на стадии 1) нет специальных требований к условиям замешивания, формования и кальцинирования. Для получения носителя, имеющего подходящую прочность, замешивание осуществляют в присутствии кислоты (в частности по меньшей мере одной из неорганической кислоты, такой как азотная кислота, серная кислота и фтороводородная кислота, и органической кислоты, такой как муравьиная кислота, уксусная кислота, лимонная кислота) в количестве 2-10 г на 100 г порошка. Температура кальцинирования на стадии 1) может составлять 550-1100°С, предпочтительно 800-1050°С. Продолжительность кальцинирования на стадии 1) может составлять 2-6 ч.
Предпочтительно, на стадии 2) загрузку можно осуществлять пропиткой, то есть, носитель можно пропитывать раствором, содержащим предшественник активного металлического компонента и необязательно предшественник металлического промотора, и затем сушить и кальцинировать.
Предпочтительно, способ может дополнительно включать стадию восстановления продукта, полученного на стадии 2) для получения катализатора с активным металлическим компонентом и восстанавливаемым металлическим промотором, присутствующим в восстановленном состоянии. Восстановление можно проводить водородосодержащим газом при 350-500°С, предпочтительно при 350-450°С. Водородсодержащий газ может быть чистым газообразным водородом или газообразным водородом, разбавленным инертным газом, таким как смесь азота и водорода. Температуру восстановления постепенно увеличивают при восстановлении, и повышение температуры предпочтительно не является слишком быстрым, например, не более чем на 20°С в час. Продолжительность восстановления можно определить, отслеживая образование Н2О в системе для восстановления, то есть когда в системе восстановления не образуется новая Н2О, восстановление прекращают, и специалист в данной области техники может соответствующим образом выбрать продолжительность восстановления, подробное описание которого опущено в настоящем изобретении для краткости, например, продолжительность восстановления может составлять 2-5 часов при самой высокой температуре.
Восстановление можно осуществлять непосредственно в реакторе с последующей каталитической реакцией. Также возможно проводить восстановление в отдельном реакторе, называемое также внереакторным восстановлением, и пассивирование можно осуществлять после восстановления газовой смесью, содержащей кислород, до выгрузки катализатора из реактора, при этом температура пассивирования составляет, например, от 10 до 60°С и в частности от 20 до 40°С. Катализатор, прошедший внереакторное восстановление и пассивирование, может быть активирован перед применением с применением водорода или смеси водорода и азота, например, при от 150°С до 250°С, предпочтительно от 170°С до 200°С.
Продолжительность активации можно определить наблюдением за образованием Н2О в системе активации, то есть, когда в системе активации не образуется новая Н2О, активацию прекращают, и специалист в данной области может соответствующим образом выбрать продолжительность активации, подробное описание которых опущено в настоящем изобретении для краткости. Например, продолжительность активации при самой высокой температуре может составлять, например, от 1 до 5 часов, предпочтительно от 2 до 3 часов, или катализатор можно применять без активации, в зависимости от степени окисления активного металлического компонента и металлического промотора катализатора.
В пятом типе вариантов осуществления настоящего изобретения, обеспечивают способ получения органических аминов, включающий: осуществление контакта аммиака и/или органического амина со спиртом в присутствии содержащего титан катализатора, как описано выше, для проведения реакции аминирования.
Предпочтительно, спирт может представлять собой по меньшей мере спирт, выбранный из группы, состоящей из замещенных или незамещенных одноатомных спиртов, содержащих 2-20 атомов углерода, и замещенных или незамещенных двухатомных спиртов, содержащих 2-2 0 атомов углерода (замещающая группа может представлять собой амино группу), например, по меньшей мере спирт из этанола, н-пропанола, изопропанола, н-бутанола, изобутанола, 2-этилгексанола, октанола, додеканола, гексадеканола, циклопентанола, циклогексанола, бензилового спирта, фенетилового спирта, этиленгликоля, 1,3-пропандиола, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,6-гександиола, 1,8-октандиола, 1,8-октандиаля, 1,12-додекандиола, этаноламина (2-аминоэтанола), 6-амино-1-гексанола (называемого для краткости аминогексанолом) и подобных.
Предпочтительно, органический амин может представлять собой первичный амин и/или вторичный амин. Например, можно применять алкиламин, содержащий от 1 до 4 атомов углерода, например, по меньшей мере один из монометиламина, диметиламина, метилэтиламина, моноэтиламина, диэтиламина и подобных. Реакцию аминирования предпочтительно проводят с применением избытка аммиака или амина, например, аммиак или органический амин можно применять в количестве от 1 до 50 молей, предпочтительно от 3 до 20 молей, более предпочтительно от 3 до 15 молей на моль гидроксильных групп в спирте. Реакцию аминирования спирта осуществляют в присутствии водорода, т.е. в гидроусловиях. Водород можно применять в количестве от 1 до 10 моль на моль гидроксильных групп в спирте. Парциальное давление водорода составляет, например, от 0,05 до 2 МПа. Температура реакции аминирования может составлять 100-300°С. Давление реакции аминирования может быть от 0,1 до 25 МПа, предпочтительно от 1 до 15 МПа. Часовая объемная скорость жидкости спирта может быть от 0,1-0, 8 м3 (м3⋅ч).
Например, реакция аминирования представляет собой, например, гидроаминирование этанола для получения этиламина, гидроаминирование н-пропанола для получения н-пропиламина, гидроаминирование изопропанола для получения изопропиламина, гидроаминирование н-бутанола для получения н-бутиламина, гидроаминирование этиленгликоля для получения этилендиамина, гидроаминирование этаноламина для получения этиленгликоля, гидроаминирование 1,3-пропандиола для получения 1,3-пропандиамина, гидроаминирование 1,4-бутандиола для получения 1,4-бутандиамина, гидроаминирование 1,6-гександиола для получения 1,6-гексадиамина, или гидроаминирование 1,12-додекандиола для получения 1,12-додекандиамина.
Согласно пятому типу вариантов осуществления настоящего изобретения, реакцию аминирования можно проводить периодически или непрерывно. Реакция аминирования может быть газофазной реакцией или жидкофазной реакцией в слое со струйным течением жидкости.
Также обеспечивают в настоящем изобретении некоторые конкретные варианты осуществления для проведения реакций аминирования, применяя катализатор согласно пятому типу вариантов осуществления настоящего изобретения.
Например, настоящее изобретение обеспечивает способ получения 1,6-гександиамина из 1,6-гександиола, применяя катализатор согласно пятому типу вариантов осуществления настоящего изобретения, включающий проведение реакции гидроаминирования с 1,6-гександиолом, аммиаком и водородом при температуре 130-200°С и давлении реакции 1-11 МПа, часовой объемной скорости жидкости подачи 1,6-гександиола 0,1-0,8 м3/(м3⋅ч), и молярном отношении водорода к аммиаку и к гександиолу 1-4:3-20:1, для получения гександиамина, 6-амино-1-гексанола и гексаметиленимина.
Настоящее изобретение обеспечивает способ получения 1,6-гександиамина из 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, применяя катализатор согласно пятому типу вариантов осуществления, включающий проведение реакции гидроаминирования смесью 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, аммиака и водорода при температуре реакции 130-200°С, давлении реакции 1-11 МПа, часовой объемной скорости жидкости смеси 0,1-0,8 м3/(м3⋅ч) и молярном отношении водорода к аммиаку и к смеси 1-4:3-20:1, для получения реакционного раствора, содержащего 1,б-гександиамин, 6-амино-1-гексанол и циклогексимид.
В особенно предпочтительных вариантах осуществления, настоящее изобретение описывает следующие технические решения:
А1, катализатор, выполняющий функцию катализа гидроаминирования спирта для получения органического амина, который содержит носитель, активный металлический компонент и необязательный металлический промотор, где активный металлический компонент и необязательный металлический промотор нанесены на носитель, и носитель является по меньшей мере носителем, выбранным из группы, состоящей из допированного оксида алюминия, допированного оксида кремния, допированных молекулярных сит и допированных алюмосиликатов; носитель имеет адсорбционную емкость по аммиаку 0,25-0,6 ммоль/г, и адсорбционную способность по углекислому газу 0,05-0,3 ммоль/г; и активный металлический компонент представляет собой кобальт и/или никель.
А2, катализатор по пункту А1, где носитель имеет адсорбционную емкость по аммиаку 0,3-0,5 ммоль/г;
и/или носитель имеет адсорбционную способность по углекислому газу 0,06-0,2 ммоль/г;
и/или, содержание допированного элемента в носителе составляет 0,03-2 масс. %, предпочтительно 0,08-1 масс. %, относительно общего веса элементов, отличных от примесных элементов в носителе;
и/или примесный элемент в носителе содержит металлический элемент и неметаллический элемент, где металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантаноидов, предпочтительно по меньшей мере металлический элемент, выбранный из группы, состоящей из кальция, магния, калия, висмута, стронция, бария и лантана; неметаллический элемент представляет собой по меньшей мере неметаллический элемент, выбранный из группы, состоящей из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA и неметаллических элементов группы VIIA, предпочтительно по меньшей мере неметаллический элемент, выбранный из группы, состоящей из бора, фтора, фосфора, серы и селена; предпочтительно, примесный элемент в носителе получен из катионов металлов и ионов кислотных радикалов, но не включает ион натрия или хлорид ион; катион металла представляет собой по меньшей мере катион металла, выбранный из группы, состоящей из катиона металлов группы IA, ионов металлов группы IIA, ионов металлов группы VA и ионов металлов ряда лантаноидов, предпочтительно по меньшей мере катион металла, выбранный из группы, состоящей из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария и иона лантана; ион кислотного радикала представляет собой по меньшей мере ион кислотного радикала, выбранный из группы, состоящей из неметаллических ионов кислотных радикалов, предпочтительно по меньшей мере ион кислотного радикала, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или носитель имеет удельную площадь поверхности 120-240 м2/г;
и/или носитель имеет объем пор 0,5-1 мл/г;
и/или содержание активного металлического компонента составляет 5-42 г, предпочтительно 10-35 г, на 100 г матрикса.
A3, катализатор по пункту А1 или А2, где носитель получают способом, включающим стадии: последовательного формования, сушки и кальцинирования смеси, содержащей источник примесного элемента и носителя, где источник носителя представляет собой по меньшей мере источник носителя, выбранный из группы, состоящей из псевдобемита, оксида кремния, молекулярных сит и алюмосиликатов.
А4, катализатор по пункту A3, где примесный элемент обеспечивают модификатором носителя, предпочтительно модификатор носителя представляет собой по меньшей мере одно из соединений, способных обеспечивать катион или анион, где катион представляет собой по меньшей мере катион, выбранный из группы, состоящей из катионов группы IA, ионов металлов группы IIA, ионов металлов группы VA и ионов металлов лантанидов, предпочтительно по меньшей мере катион, выбранный из группы, состоящей из ионов кальция, ионов магния, ионов калия, ионов висмута, ионов стронция, ионов бария и ионов лантана;
и/или анион представляет собой по меньшей мере анион, выбранный из группы, состоящей из неметаллических ионов кислотных радикалов, предпочтительно по меньшей мере анион, выбранный из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона.
А5, катализатор по пункту А4, где модификатор носителя представляет собой по меньшей мере модификатор носителя, выбранный из группы, состоящей из борной кислоты, бората никеля, бората кобальта, бората калия, фтороводородной кислоты, фторида калия, фторида кобальта, фторида никеля, фосфорной кислоты, фосфата алюминия, фосфата трикалия, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, нитрата висмута, нитрата калия, сульфата калия, карбоната калия, нитрата магния, сульфата магния, основного карбоната магния, нитрата кальция, основного карбоната кальция, нитрата стронция, фосфата стронция, сульфата стронция, нитрата бария, нитрата лантана и селеновой кислоты.
Предпочтительно, псевдобемит имеет удельную площадь поверхности 250-330 м2/г и объем пор 0,8-1,3 мл/г.
А6, катализатор по любому из пунктов А3-А5, где условия сушки включают: температуру 80-150°С, и продолжительность 6-20 ч;
и/или условия кальцинирования включают: температуру 600-1100°С и продолжительность 2-20 ч.
А7, способ получения катализатора по любому из пунктов А1-А6, включающий: загрузку активного металлического компонента и необязательно металлического промотора на носитель.
А8, носитель, как определено по любому из пунктов А1-А6.
А9, применение катализатора по любому из пунктов А1-А6, или способ по пункту А7, или носитель по пункту А8, в аминировании для получения органических аминов.
А10, способ получения органического амина, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором по любому из пунктов А1-А6 в присутствии водорода для проведения реакции аминирования;
Или альтернативно, способ включает: просеивание катализатора, содержащего носитель, как определено по любому из пунктов А1-А6, и осуществление контакта исходного материала для аминирования и реагента для аминирования с просеянным катализатором в присутствии водорода для проведения реакции аминирования.
А11, способ по пункту А10, где условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, температуру 130-200°С, давление 1-15 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере материал, выбранный из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере один из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканала, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, 1,12-додекандиола, 1,12-додекандиальдегида, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, ацетона, этиленгликоля и 1,3-пропандиола;
и/или реагент для аминирования представляет собой по меньшей мере реагент для аминирования, выбранный из группы, состоящей из аммиака, С1-12 первичного амина и С2-12 вторичного амина, предпочтительно по меньшей мере один из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
А12, способ по пункту А11, где:
когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, температуру 130-200°С, давление 1-4 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, температуру 110-180°С, давление 1-3,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-20:1, температуру 135-200°С, давление 1-11 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-35:1, температуру 130-210°С, давление 1-15 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-35:1, температуру 130-210°С, давление 1-15 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
B1, катализатор, обладающий способностью катализировать аминирование для получения амина, содержащий носитель, и активный металлический компонент и металлический промотор, нанесенные на носитель, где активный металлический компонент представляет собой кобальт и/или никель; и металлический промотор представляет собой комбинацию по меньшей мере один металл группы VIIB и по меньшей мере один металл группы IB.
B2, катализатор по пункту В1, где носитель содержит носитель на основе оксида алюминия, примесный элемент и необязательно дополнительный носитель, выбранный из оксида кремния и/или молекулярных сит; носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г; доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем или равной 70%.
B3, катализатор по пункту В1 или В2, где содержание носителя на основе оксида алюминия в носителе составляет 75 масс. % или более, предпочтительно 80-100 масс. %, исходя из общего количества носителя на основе оксида алюминия и указанного дополнительного носителя;
и/или содержание примесного элемента составляет 0,05-6 масс. %, предпочтительно 0,08-4 масс. %, относительно веса матрикса;
и/или примесный элемент в носителе представляет собой неметаллический элемент, и его предпочтительно вводят при получении носителя в виде по меньшей мере иона, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или носитель имеет адсорбционную емкость по аммиаку 0,3-0,56 ммоль/г;
и/или доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя составляет 70-90%, и доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-8%;
и/или носитель имеет удельную площадь поверхности 125-200 м2/г;
и/или носитель имеет объем пор 0,45-1,1 мл/г;
и/или активный металлический компонент присутствует в количестве 8-45 г, на 100 г матрикса;
и/или содержание металлического промотора составляет 0,1-10 г, на 100 г матрикса;
и/или весовое отношение металла группы VIIB к металлу группы IB в металлическом промоторе составляет 0,05-15:1, предпочтительно 0,1-12:1;
и/или, металл группы VIIB выбирают из марганца и/или рения;
и/или, металл группы IB представляет собой по меньшей мере металл, выбранный из группы, состоящей из меди, серебра и золота.
В4 катализатор по любому из пунктов В1-В3, где носитель получают способом, включающим: последовательное формование, сушку и кальцинирование смеси, содержащей предшественник оксида алюминия, примесный элемент и необязательно предшественник дополнительного носителя, где указанный дополнительный предшественник носителя выбирают из предшественника оксида кремния и/или предшественника молекулярных сит, примесный элемент обеспечивают модификатором носителя, и модификатор носителя представляет собой по меньшей мере одну неорганическую кислоту.
B5, катализатор по пункту В4, где неорганическая кислота представляет собой по меньшей мере неорганическую кислоту, выбранную из неорганических кислот, содержащих неметаллические ионы кислотных радикалов, предпочтительно по меньшей мере неорганическую кислоту, выбранную из борной кислоты, фтороводородной кислоты, фосфорной кислоты, серной кислоты и селеновой кислоты.
B6, катализатор по пункту В4 или В5, где предшественник оксида алюминия представляет собой псевдобемит, псевдобемит имеет удельную площадь поверхности 260-380 м2/г и объем пор 0,78-1,2 мл/г.
B7, катализатор по любому из пунктов В4-В6, где условия сушки включают: температуру 80-150°С и продолжительность 6-20 ч;
и/или условия кальцинирования включают: температуру 550-1050°С и продолжительность 2-20 ч.
B8, способ получения катализатор по любому из пунктов В1-В7, включающий: загрузку активного металлического компонента и металлического промотора на носитель.
B9, носитель, как определено по любому из пунктов В2-В7.
B10, применение катализатора по любому из пунктов В1-В7, или способ по пункту В8, или носитель по пункту В9, в аминировании для получения органических аминов.
B11, способ получения органического амина, включающий следующие стадии: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором по любому из пунктов В1-В7 в присутствии водорода для прведения реакции аминирования;
или альтернативно, способ включает: просеивание катализатора, содержащего носитель, как определено по любому из пунктов В1-В7, и осуществление контакта исходного материала для аминирования и реагента для аминирования просеянным катализатором в присутствии водорода для проведения реакции аминирования.
B12, способ по пункту В11, где условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-33:1, температуру 110-220°С, давление 0,8-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере исходный материал, выбранный из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере один из этанола, из ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или реагент для аминирования представляет собой по меньшей мере реагент, выбранный из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичны аминов, предпочтительно по меньшей мере один из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
В13, способ по пункту В12, где:
когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, температуру 130-210°С, давление 1-3,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-5:1, температуру 110-170°С, давление 0,8-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, температуру 130-200°С, давление 1-18 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-33:1, температуру 130-220°С, давление 4-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч).
С1, катализатор, обладающий способностью катализировать гидроаминирование спирта, содержащий носитель, и металлический активный компонент и металлический промотор, нанесенный на носитель, где металлический активный компонент представляет собой кобальт и/или никель; металлический промотор представляет собой комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB.
С2, катализатор по пункту С1, где носитель содержит носитель на основе оксида алюминия, примесный элемент и необязательно дополнительный носитель, который представляет собой по меньшей мере дополнительный носитель, выбранный из группы, состоящей из оксида кремния, молекулярных сит и диатомита; носитель имеет адсорбционную емкость по аммиаку 0,3-0,7 ммоль/г и адсорбционную способность по углекислому газу 0,05-0,4 ммоль/г; долю объема пор, имеющих диаметр пор в диапазоне 7-27 нм к объему пор носителя, больше чем 65%.
С3, катализатор по пункту С1 или С2, где содержание носителя на основе оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 80-100 масс. %, исходя из общего количества носителя на основе оксида алюминия и указанного дополнительного носителя;
и/или примесный элемент присутствует в носителе в количестве 0,05-6 масс. %, предпочтительно 0,08-4 масс. %, относительно общего веса матрикса;
и/или примесный элемент в носителе выбирают из металлических элементов и неметаллических элементов и не включает натрий или хлор; металлический элемент представляет собой по меньшей мере элемент, выбранный из группы, состоящей из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA и металлических элементов ряда лантаноидов, предпочтительно по меньшей мере один из ионов кальция, магния, калия, висмута, стронция, бария и лантана; неметаллические элементы получены из по меньшей мере одного из неметаллических ионов кислотных радикалов, предпочтительно по меньшей мере из одного, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г и адсорбционную способность по углекислому газу 0,05-0,3 ммоль/г;
и/или доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя составляет 70-90%, и доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-10%;
и/или носитель имеет удельную площадь поверхности 120-205 м2/г;
и/или носитель имеет объем пор 0,45-1,2 мл/г;
и/или содержание металлического активного компонента составляет 14-46 г, на 100 г матрикса;
и/или содержание металлического промотора составляет 0,1-10 г, на 100 г матрикса;
и/или весовое отношение металла группы VIIB к металлу группы IIB в металлическом промоторе составляет 0,2-20:1, предпочтительно 0,3-6:1;
и/или, металл группы VIIB выбирают из марганца и/или рения;
и/или металл группы IIB выбирают из цинка.
С4, катализатор по любому из пунктов С1-С3, где носитель получают способом, включающим стадии: последовательного формования, сушки и кальцинирования смеси, содержащей примесный элемент, предшественник оксида алюминия и необязательно предшественник дополнительного носителя, где указанный предшественник дополнительного носителя представляет собой по меньшей мере предшественник дополнительного носителя, выбранный из группы, состоящей из предшественника оксида кремния, предшественника молекулярных сит и предшественника диатомита.
С5, катализатор по пункту С4, где примесный элемент обеспечивают модификатором носителя, который представляет собой по меньшей мере модификатор носителя, выбранный из группы, состоящей из борной кислоты, бората никеля, бората кобальта, бората калия, фтороводородной кислоты, фторида калия, фторида кобальта, фторида никеля, фосфорной кислоты, фосфата алюминия, фосфата трикалия, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, нитрата висмута, нитрата калия, сульфата калия, карбоната калия, нитрата магния, сульфата магния, основного карбоната магния, нитрата кальция, основного карбоната кальция, нитрата стронция, фосфата стронция, сульфата стронция, нитрата бария, нитрата лантана и селеновой кислоты.
С6, катализатор по пункту С4 или С5, где предшественник оксида алюминия представляет собой псевдобемит, псевдобемит имеет удельную площадь поверхности 265-410 м2/г и объем пор 0,7-1,2 мл/г.
С7, катализатор по любому из пунктов С4-С6, где условия сушки включают: температуру 80-150°С и продолжительность 6-20 ч;
и/или условия кальцинирования включают: температуру 500-1100°С и продолжительность 2-20 ч.
С8, способ получения катализатор по любому из пунктов С1-С7, включающий: загрузку металлического активного компонента и металлического промотора на носитель.
С9, носитель, как определено по любому из пунктов С2-С7.
С10, применение катализатора по любому из пунктов С1-С7, или способ по пункту С8, или носитель по пункту С9, в аминировании для получения органических аминов.
С11, способ получения органических аминов, включающий следующие стадии: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором по любому из пунктов С1-С7 в присутствии водорода для проведения реакции аминирования;
или альтернативно, способ включает: просеивание катализатора, содержащего носитель, как определено по любому из пунктов С1-С7, и осуществление контакта исходного материала для аминирования и реагента для аминирования с просеянным катализатором в присутствии водорода для проведения реакции аминирования.
С12, способ по пункту С11, где условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-30:1, температуру 110-220°С, давление 0,8-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере исходный материал, выбранный из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере один из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутилальдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или реагент для аминирования представляет собой по меньшей мере реагент для аминирования, выбранный из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере один из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
С13, способ по пункту С12, где, когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-9:1, температуру 130-200°С, давление 1-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, температуру 110-180°С, давление 0,8-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-20:1, температуру 130-200°С, давление 1-15 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-30:1, температуру 130-220°С, давление 1-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
D1, катализатор, обладающий функцией катализа аминирования спиртов, содержащий носитель, и активный металлический компонент и металлический промотор, нанесенные на носитель, где активный металлический компонент представляет собой кобальт и/или никель; металлический промотор представляет собой комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB.
D2, Катализатор по пункту D1, где носитель содержит носитель на основе оксида алюминия, примесный элемент и дополнительный носитель, выбранный из оксида кремния и/или молекулярных сит; носитель имеет адсорбционную емкость по аммиаку 0,25-0,6 ммоль/г; доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%.
D3, катализатор по пункту D1 или D2, где содержание носителя на основе оксида алюминия в носителе составляет70 масс. % или более, предпочтительно 80-97 масс. %, исходя из общего количества носителя на основе оксида алюминия, и указанный дополнительный носитель;
и/или содержание примесного элемента составляет 0,05-5 масс. %, предпочтительно 0,08-3 масс. %, исходя из массы носителя;
и/или примесный элемент в носителе представляет собой неметаллический элемент, предпочтительно введенный в виде по меньшей мере примесного элемента, выбранного из группы, состоящей из борат-иона, фторид-иона, фосфат-иона, сульфат-иона и селенат-иона;
и/или носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г;
и/или доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя составляет 70-90%, и доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-8%;
и/или носитель имеет удельную площадь поверхности 120-210 м2/г;
и/или носитель имеет объем пор 0,45-1,1 мл/г;
и/или активный металлический компонент присутствует в количестве 10-46 г, предпочтительно 18-38 г, на 100 г носителя;
и/или содержание металлического промотора составляет 0,1-10 г, предпочтительно 0,5-6 г, относительно 100 г носителя;
и/или металлический промотор содержит металл группы VIB, металл группы IB и металл группы IIB при весовом соотношении 0,1-10:0,1-10:1, предпочтительно 0,2-8:0,2-8:1;
и/или металл группы VIB выбирают из молибдена и/или вольфрама;
и/или, металл группы IB представляет собой по меньшей мере металл группы IB, выбранный из группы, состоящей из меди, серебра и золота;
и/или металл группы IIB выбирают из цинка.
D4, катализатор по любому из пунктов D1-D3, где носитель получают способом, включающим стадии: последовательного формования, сушки и кальцинирования смеси, содержащей предшественник оксида алюминия, примесный элемент и предшественник дополнительного носителя, где указанный предшественник дополнительного носителя выбирают из предшественника оксида кремния и/или предшественника молекулярных сит.
D5, катализатор по пункту D4, где примесный элемент обеспечивают по меньшей мере одной из борной кислоты, фтороводородной кислоты, фосфорной кислоты, серной кислоты и селеновой кислоты.
D6, катализатор по пункту D4 или D5, где предшественник оксида алюминия представляет собой псевдобемит, псевдобемит имеет удельную площадь поверхности 260-400 м2/г и объем пор 0,8-1,2 мл/г.
D7, катализатор по любому из пунктов D4-D6, где условия сушки включают: температуру 80-150°С и продолжительность 6-20 ч;
и/или условия кальцинирования включают: температуру 500-1100°С и продолжительность 2-20 ч.
D8, способ получения катализатор по любому из пунктов D1-D7, включающий: загрузку активного металлического компонента и металлического промотора на носитель.
D9, носитель, как определено по любому из пунктов D2-D7.
D10, применение катализатора по любому из пунктов D1-D7, или способ по пункту D8, или носитель по пункту D9, в аминировании для получения органические амины.
D11, способ получения органических аминов, включающий следующие стадии: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором, как определено по любому из пунктов D1-D7, в присутствии водорода для проведения реакции аминирования.
D12, способ по пункту D11, где условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, температуру 105-220°С, давление 0,7-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч);
и/или исходный материал для аминирования представляет собой по меньшей мере исходный материал, выбранный из группы, состоящей из С2-20 спиртов, С3-20 кетонов, С2-20 аминоспиртов и С2-20 альдегидов, предпочтительно по меньшей мере один из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутилальдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола и 1,12-додекандиола;
и/или реагент для аминирования представляет собой по меньшей мере реагент для аминирования, выбранный из группы, состоящей из аммиака, С1-12 первичных аминов и С1-12 вторичных аминов, предпочтительно по меньшей мере один из аммиака, монометиламина, диметиламина, метилэтиламина, моноэтиламина и диэтиламина.
D13, способ по пункту D12, где, когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-8:1, температуру 130-200°С, давление 1-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0, 8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-5:1, температуру 105-180°С, давление 0,7-2,5 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, температуру 130-200°С, давление 5-18 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
или когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола или двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-35:1, температуру 130-220°С, давление 2-25 МПа и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/(м3⋅ч).
Е1, содержащий титан катализатор, обладающий функцией аминирования, содержащий носитель, и активный металлический компонент и необязательно металлический промотор, нанесенные на носитель, и катализатор содержит оксид алюминия и диоксид титана при весовом соотношении 1,5-5:1, и активный металлический компонент представляет собой кобальт и/или никель.
Е2, содержащий титан катализатор по пункту Е1, где весовое отношение оксида алюминия к диоксиду титана составляет 2-4,5:1;
и/или активный металлический компонент присутствует в количестве 13-40 г, предпочтительно 20-36 г, на 100 г носителя;
и/или содержание металлического промотора составляет 0-10 г, предпочтительно 2,5-8 г, относительно 100 г носителя;
и/или металлический промотор представляет собой по меньшей мере металлический промотор, выбранный из группы, состоящей из элементов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантаноидов, предпочтительно по меньшей мере один из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се.
Е4, содержащий титан катализатор по любому из пунктов Е1-Е3, где содержащий титан катализатор имеет адсорбционную емкость по аммиаку 0,3-0,4 ммоль/г, адсорбционную способность по углекислому газу 0,07-0,2 ммоль/г, удельную площадь поверхности 130-180 м2/г, объем пор 0,55-0,75 мл/г, долю объема пор, имеющих радиус пор меньше чем 4 нм, к суммарному объему пор меньше чем 20%, и долю объема пор, имеющих радиус пор больше чем 10 нм, к суммарному объему пор меньше чем 15%;
и/или носитель допирован серой в количестве 0,1-0,5 г, предпочтительно 0,15-0,4 г, на 100 г оксида алюминия и диоксида титана.
Е5, способ получения содержащего титан катализатора, обладающего функцией аминирования, включающий:
(1) применение псевдобемита и порошка титановых белил в замешивании, формовании и кальцинировании для получения носителя, где псевдобемит и порошок титановых белил применяют в таком количестве, что весовое отношение оксида алюминия к диоксиду титана в полученном носителе составляет 2-5:1;
(2) загрузку активного металлического компонента и необязательно металлического промотора на полученный носитель, где активный металлический компонент содержит кобальт и/или никель.
Е6, способ по пункту Е4, где псевдобемит и порошок титановых белил применяют в таком количестве, что весовое отношение оксида алюминия к диоксиду титана в полученном носителе составляет 2-4,5:1;
и/или, псевдобемит получают по меньшей мере одним из способа карбонизации, способом алюмоорганического гидролиза, способа с применением сульфата алюминия и способа с применением азотной кислоты, предпочтительно способом карбонизации или способом с применением сульфата алюминия;
и/или порошок титановых белил получают способом осаждения и/или золь-гель способом, и предпочтительно порошок титановых белил получают, применяя титанилсульфата в качестве исходного соединения.
Е7, способ по пункту Е5, где содержание сульфата в порошке титановых белил составляет 0,2-3 масс. %.
Е8, способ по любому из пунктов Е4-Е6, где активный металлический компонент применяют в таком количестве, что активный металлический компонент присутствует в количестве 13-40 г, предпочтительно 20-36 г, на 100 г носителя;
и/или количество металлического промотора является таким, что металлический промотор присутствует в количестве 0-10 г, предпочтительно 2,5-8 г, на 100 г носителя;
и/или металлический промотор представляет собой по меньшей мере металлический промотор, выбранный из группы, состоящей из элементов группы VIB, группы VIIB, группы IB, группы IIB и элементов ряда лантаноидов, предпочтительно по меньшей мере один из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се.
Е8, содержащий титан катализатор, полученный способом по любому из пунктов Е4-Е7.
Е9, применение содержащего титан катализатора по любому из пунктов Е1-Е3 или Е8, или способ по любому из пунктов Е4-Е7, в аминировании спирта.
Е10, способ получения органического амина, включающий: осуществление контакт аммиака и/или органического амина со спиртом в присутствии содержащего титан катализатора по любому из пунктов Е1-Е3 или Е8 для проведения реакции аминирования;
или альтернативно, содержащий титан катализатор получают способом по любому из пунктов Е4-Е7, и когда аммиак и/или органический амин контактирует со спиртом в присутствии полученного в результате содержащего титан катализатора для реакции аминирования.
Примеры
Настоящее изобретение будет дополнительно проиллюстрировано со ссылкой на следующие примеры, но настоящее изобретение ими не ограничивается.
В следующих примерах и сравнительных примерах, если не указано иное, применяемые реагенты и исходные материалы являются коммерчески доступными продуктами, которые являются химически чистыми.
Приборы для тестирования и способы, применяемые в следующих примерах, являются следующими: 1) NH3-TPD тест
Приборы для тестирования: Autochem 2920 полностью автоматический прибор для химической адсорбции (автоматическая система определения характеристик катализатора), изготовленный MICROMERITICS, USA;
Условия испытания: приблизительно 0,1 г образца точно взвешивали, помещали в пробирку, нагревали до температуры 600°С со скоростью 10°С/мин при продувке газообразным гелием, выдерживали 1 ч и охлаждали до температуры 120°С; газ заменяли на смешанный газ 10% NH3-He, адсорбировали в течение 60 минут, затем газ снова заменяли продувкой газообразным гелием в течение 1 часа, подсчет начинали после того, как базовая линия стабилизировалась; и полученный продукт нагревали до температуры 600°С со скоростью 10°С/мин, выдерживали 30 мин, регистрацию прекращали, и эксперимент заканчивали. Интегральный расчет проводили по площади пика, получая степень десорбции NH3, и степень десорбции применяли для характеристики адсорбционной емкости по аммиаку образца.
2) CO2-TPD тест
Приборы для тестирования: Autochem 2920 полностью автоматический прибор для химической адсорбции (автоматическая система определения характеристик катализатора), изготовленный MICROMERITICS, USA;
Условия испытания: приблизительно 0,1 г образца точно взвешивали, помещали в пробирку, нагревали до температуры 600°С со скоростью 10°С/мин при продувке газообразным гелием, выдерживали 1 ч и охлаждали до температуры 120°С; газ заменяли на смешанный газ 10% NH3-He, адсорбировали в течение 60 минут, затем газ снова заменяли продувкой газообразным гелием в течение 1 часа, подсчет начинали после того, как базовая линия стабилизировалась; и полученный продукт нагревали до температуры 600°С со скоростью 10°С/мин, выдерживали 30 мин, регистрацию прекращали, и эксперимент заканчивали. Интегральный расчет проводили по площади пика, получая степень десорбции СО2, и степень десорбции применяли для характеристики СО2 адсорбционной емкости образца.
3) BET тест
Приборы для тестирования: ASAP2420 полностью автоматический физико-химический анализатор адсорбции (автоматический анализатор микропор и хемосорбции), изготовленный MICROMERITICS, USA;
Условия испытания: газ для испытания: N2 (99,999% чистота); условия дегазации: нагрев до 350°С со скоростью 10°С/мин и вакуумирование в течение 4 ч; условия анализа: проведение полного анализа на изотерме мезопор для получения удельной площади поверхности и объема пор.
4) Рентгеноструктурный анализ
Приборы для тестирования: рентгеновский дифрактометр Empyrean производства PANalytical B.V. с анодной мишенью Cu мишенью и детектором Pixcel 3D;
Условия испытаний: напряжение трубки 40 кВ, ток трубки 40 мА, щель расходимости 1/4°, щель антирасходимости 1/2°, высота приемной щели 7,5 мм, скорость сканирования 0,013°/шаг, диапазон сканирования 5-90°.
Размер зерен активного металлического компонента и металлического промотора, если они присутствуют, получали расчетным путем с применением уравнения Шеррера.
Серия примеров I
Первый тип вариантов осуществления настоящего изобретения более подробно проиллюстрирован ниже со ссылкой на примеры серии примеров I. В следующих примерах серии примеров I применяемый порошок псевдобемита имел содержание (Al2O3) в пересчете на сухую массу 72 масс. %.
Пример I-1
Тетрагидрат нитрата кальция (аналитически чистый), азотная кислота и разбавленный кислый водный раствор фосфорной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 298 m2/г и объемом пор 1,21 мл/г) при замешивании, и после замешивания полученную в результате смесь формовали в виде полосок с диаметром 5 мм, нарезали на длину 4 мм, сушили при 100°С в течение 12 ч, и затем кальцинировали при 720°С в течение 8 часов для получения требуемого носителя, где количество тетрагидрата нитрата кальция (аналитически чистый) составляло 2,95 г, количество азотной кислоты составляло 6,5 г, и количество фосфорной кислоты составляло 0,63 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) растворяли в воде, получая 182 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением два раза, сушили при 120°С в течение 8 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-I-1.
Пример I-2
Водный раствор нитрата калия (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор борной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 286 м2/г и объемом пор 0,88 мл/г) при замешивании, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 120°С в течение 8 часов, и затем кальцинировали при 690°С в течение 10 ч для получения требуемого носителя, где количество нитрата калия (аналитически чистый) составляло 0,26 г, количество азотной кислоты составляло 5,2 г, и количество борной кислоты составляло 4,57 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
151,7 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) растворяли в воде, получая 168 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 390°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 часов, получая катализатор A-I-2.
Пример I-3
Водный раствор гексагидрата нитрата магния (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор серной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 310 м2/г и объемом пор 0,92 мл/г) при замешивании, формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 15 ч, и затем кальцинировали при 780°С в течение 10 ч для получения требуемого носителя, где количество гексагидрата нитрата магния (аналитически чистый) составляло 0,84 г, количество азотной кислоты составляло 6,1 г, и количество серной кислоты составляло 1,22 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
50,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 32,6 г 50 масс. % водного раствора нитрата магния растворяли в воде, получая 156 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов получая катализатор A-I-3.
Пример I-4
Водный раствор пентагидрата нитрата висмута (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор фосфорной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 321 м2/г и объемом пор 0,93 мл/г) при замешивании, формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 80°С в течение 20 ч, и затем кальцинировали при 660°С в течение 15 ч для получения требуемого носителя, где количество пентагидрата нитрата висмута (аналитически чистый) составляло 1,86 г, количество азотной кислоты составляло 6,5 г, и количество фосфорной кислоты составляло 1,9 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
75,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 50,6 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) растворяли в воде, получая 166 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-I-4.
Пример I-5
Водный раствор нитрата бария (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор борной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 275 м2/г и объемом пор 0,85 мл/г) при замешивании, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 150°С в течение 6 ч, и кальцинировали при 810°С в течение 5 ч для получения требуемого носителя, где количество нитрата бария (аналитически чистый) составляло 0,19 г, количество азотной кислоты составляло 6,5 г, и количество борной кислоты составляло 2,29 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
126,4 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) и 2,9 г перрената аммония (с чистотой 99%) растворяли в воде, получая 160 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 390°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 часов, получая катализатор A-I-5.
Пример I-6
Водный раствор нитрата цезия (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор серной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 269 м2/г и объемом пор 0,86 мл/г) при замешивании, формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение 8 часов, и затем кальцинировали при 830°С в течение 4 ч для получения требуемого носителя, где количество нитрата цезия составляло 0,03 г, количество азотной кислоты составляло 6,2 г, и количество серной кислоты составляло 0,09 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
201,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) растворяли в воде, получая 156 мл раствора; 7,4 г тетрагидрата молибдата аммония (аналитически чистый) растворяли в воде, получая 78 мл раствора; раствор нитрата кобальта загружали на 100 г носителя, полученного пропиткой распылением в два раза; и затем раствор молибдата аммония загружали на носитель пропиткой распылением один раз, сушили в течение 4 часов при 120°С после пропитки распылением, затем кальцинировали в течение 4 часов при 400°С, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 часов при 430°С, получая катализатор A-I-6.
Пример I-7
Водный раствор гексагидрата нитрата лантана (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор серной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 259 м2/г и объемом пор 0,88 мл/г) при замешивании, после замешивания полученную в результате смесь формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 10 ч, и затем кальцинировали при 980°С в течение 5 ч для получения требуемого носителя, где количество гексагидрата нитрата лантана (аналитически чистый) составляло 0,47 г, количество азотной кислоты составляло 5 г, и количество серной кислоты составляло 0,61 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
100,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98) и 14,1 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 176 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов получая катализатор A-I-7.
Пример I-8
Водный раствор гексагидрата нитрата лантана (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор фтороводородной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 291 м2/г и объемом пор 0,93 мл/г) при замешивании, формовали в зубчатые сферы диаметром 4 мм, сушили при 90°С в течение 12 ч, и затем кальцинировали при 900°С в течение 3 ч для получения требуемого носителя, где количество гексагидрата нитрата лантана (аналитически чистый) составляло 0,62 г, количество азотной кислоты составляло 5,5 г, и количество фтороводородной кислоты составляло 0,05 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
126 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 25,3 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) растворяли в воде, получая 177 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением 3 раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-I-8.
Пример I-9
Водный раствор тетрагидрата нитрата кальция (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор фтороводородной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 312 м2/г и объемом пор 1,02 мл/г) при замешивании, экструдировали в гранулы клеверной формы толщиной 4 мм, сушили при 100°С в течение 8 часов, и затем кальцинировали при 930°С в течение 3 ч для получения требуемого носителя, где количество тетрагидрата нитрата кальция (аналитически чистый) составляло 3,54 г, количество азотной кислоты составляло 6,5 г, и количество фтороводородной кислоты составляло 0,13 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 1,3 г нитрата серебра (аналитически чистый) растворяли в воде, получая 188 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением 3 раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 390°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 часов получая катализатор A-I-9.
Пример I-10
Глинопорошок оксида кремния (с удельной площадью поверхности 385 м2/г и объемом пор 0,95 мл/г) применяли в качестве исходного материала, формовали шаровым прокатыванием с разбавленным кислым водным раствором, содержащим нитрат магния, азотную кислоту и серную кислоту до зубчатых сфер диаметром 4 мм, сушили при 80°С в течение 15 часов, и затем кальцинировали при 750°С в течение 8 часов для получения требуемого носителя, где количество гексагидрата нитрата магния (аналитически чистый) составляло 8,44д г, количество азотной кислоты составляло 6,1 г, и количество серной кислоты составляло 4,28 г, на 100 г глинопорошка оксида кремния, рассчитанного как SiO2.
Остальная часть способа была такой же, как описано в примере 3, получая катализатора A-I-10.
Пример I-11
Водный раствор нитрата калия (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор фосфорной кислоты последовательно добавляли к порошку псевдобемита (с удельной площадью поверхности 261 м2/г и объемом пор 0,83 мл/г) при замешивании, формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 10 ч, и затем кальцинировали при 1030°С в течение 5 ч для получения требуемого носителя, где количество нитрата калия (аналитически чистый) составляло 1,29 г, количество азотной кислоты составляло 6,3 г, и количество фосфорной кислоты составляло 4,9 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3.
Остальная часть способа была такой же, как описано в примере 3, получая катализатора A-I-11.
Пример I-12
ZSM-5 порошок (с удельной площадью поверхности 354 м2/г, объемом пор 0,57 мл/г, кристалличностью 97,5%, SiO2/Al2O3=61) замешивали, водный раствор тетрагидрата нитрата кальция (аналитически чистый), азотную кислоту и разбавленный кислый водный раствор фосфорной кислоты последовательно добавляли к нему при замешивании, полученную в результате смесь формовали в зубчатые сферы диаметром 3,0 мм, сушили при 120°С в течение 10 часов, и затем кальцинировали при 920°С в течение 5 часов для получения требуемого носителя, где количество тетрагидрата нитрата кальция (аналитически чистый) составляло 1,56 г, количество азотной кислоты составлялло 8,2 г, и количество фосфорной кислоты составляло 2,9 г, на 100 г ZSM-5 порошка.
Остальная часть способа была такой же, как описано в примере 7, получая катализатор A-I-12.
Пример 1-13
Получение носителя была таким же, как описано в примере 1-72,56 г хлорида палладия (с чистотой 99,5%) и 38,2 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 171 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом постепенным повышением температуры при скорости 20°С/ч, и наконец восстанавливали при 230°С в течение 3 часов, получая катализатор A-I-13. Сравнительный пример 1-1
Катализатор получали, как описано в примере 5, за исключением того, что применяемое количество борной кислоты составляло 3,43 г, применяемое количество нитрата бария (аналитически чистый) составляло 4,76 г, и применяемое количество азотной кислоты составляло 6,5 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3. Полученный катализатор обозначали как D-I-1.
Сравнительный пример 1-2
Катализатор получали, как описано в примере 3, за исключением того, что только разбавленный кислый водный раствор азотной кислоты добавляли при замешивании, где применяемое количество азотной кислоты составляло 6,1 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3. Полученный катализатор обозначали как D-I-2.
Сравнительный пример I-3
Катализатор получали, как описано в примере 3, за исключением того, что азотную кислоту и разбавленный кислый водный раствор фосфорной кислоты добавляли при замешивании, где применяемое количество фосфорной кислоты составляло 15,82 г, и применяемое количество азотной кислоты составляло 6,2 г, на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3. Полученный катализатор обозначали как D-I-3.
Пример испытаний I-1
Элементный состав носителей и катализаторов анализировали плазменно-эмиссионной спектроскопией, содержание элемента (иона), отличного от носителя, выражали в массе элемента в расчете на 100 г матрикса, т.е. носителя, рассчитанного на основе непримесного элемента (например, рассчитанного на основе Al2O3, когда псевдобемит применяли как источник носителя); полученные выше носители были охарактеризованы способами NH3-TPD, CO2-TPD, ВЕТ-адсорбцей- десорбцией азота, и результаты представлены в таблице 1-1.
Таблица 1-1 Свойства носителей примеров и сравнительных примеров
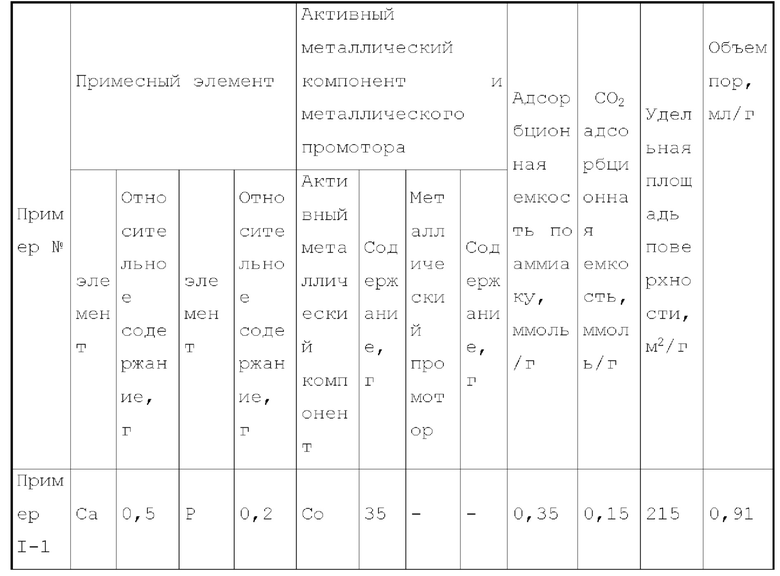
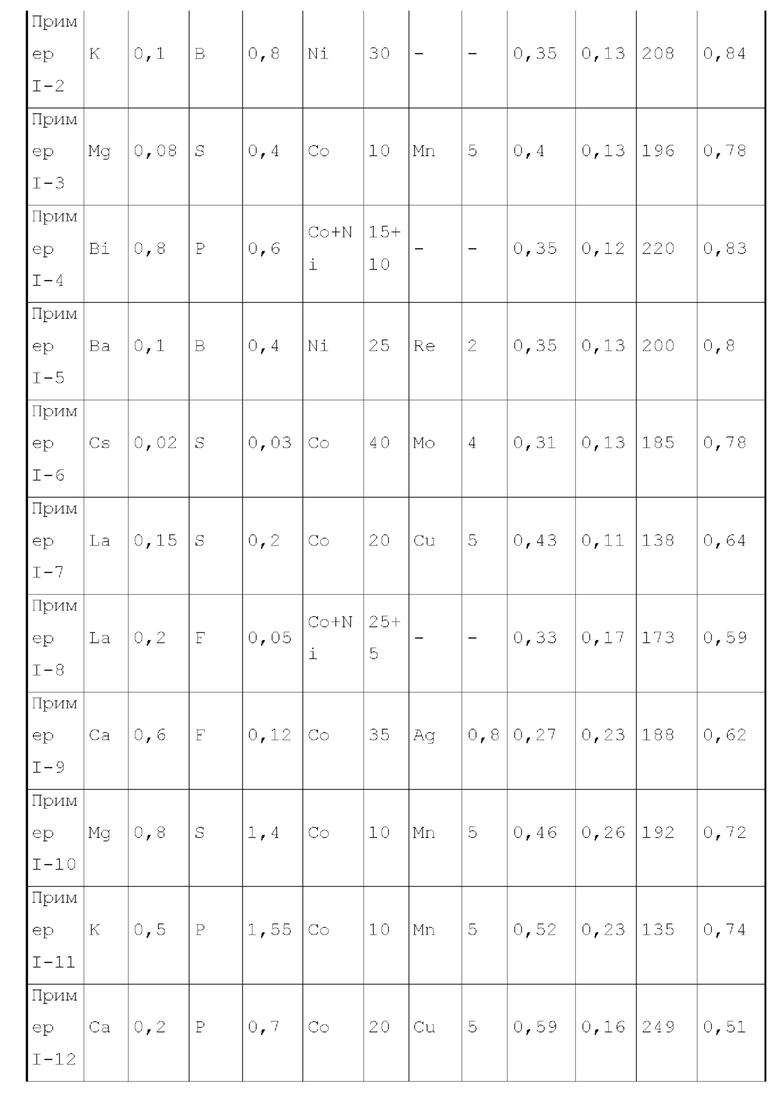
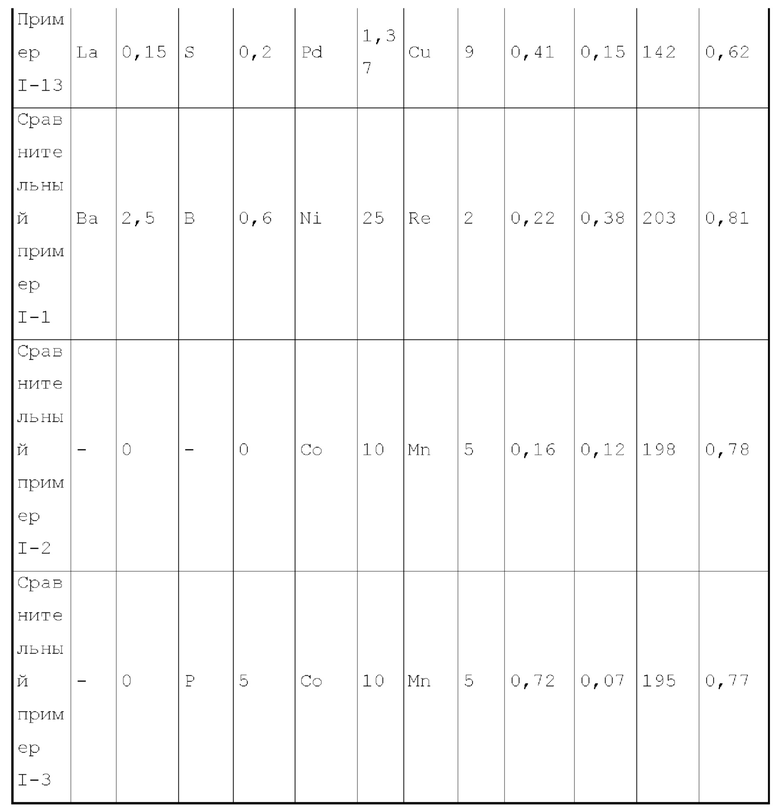
Пример испытаний 1-2
Данный пример испытаний иллюстрирует способ получения 1,6-гександиамина гидроаминированием 1,6-гександиола, применяя катализаторы первого типа вариантов осуществления настоящего изобретения.
100 мл катализаторов, полученных в приведенных выше примерах, соответственно отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 165°С, давление системы повышали до 8,8 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 100°С и затем направляли в верхний конец реактора, нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к 1,6-гександиолу составляло 3:14:1, и объемная скорость жидкой фазы 1,6-гександиола составляла 0,42 ч-1, каталитическую реакцию аминирования проводили в реакторе, при температуре реакции 185°С и давлении реакции 8,8 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (а именно после 360 ч реакции). Результаты анализа показаны в таблице 1-2.
Анализ образца проводился способом газовой хроматографии и калибровали с применением поправочного коэффициента полученного стандартного образца;
Конверсию и селективность рассчитывали, исходя из молярного содержания каждого компонента в реакционном растворе.
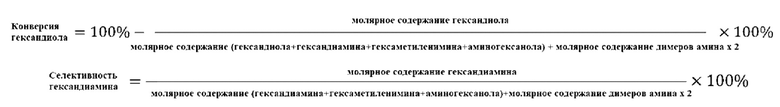
Селективность по гексаметиленимину рассчитывали путем изменения числителя в приведенном выше уравнении для расчета селективности гександиамина на молярное содержание гексаметиленимина, селективность по аминогексанолу рассчитывали путем изменения числителя в приведенном выше уравнении для расчета селективности гександиамина на молярное содержание аминогексанола и т.д., и селективность к «другим» рассчитывали путем замены числителя в приведенном выше уравнении для расчета селективности гександиамина на молярное содержание димеров амина ×2, димер амина относится к димеру 1,6-гександиамина (т.е. бис(гексаметилен)триамину, также известному как N-(6-аминогексил)-1,6-гександиамин) и димеру 1,6-гександиамина с гексаметиленимином (т.е. N-(6-аминогексил)гексаметиленимину).
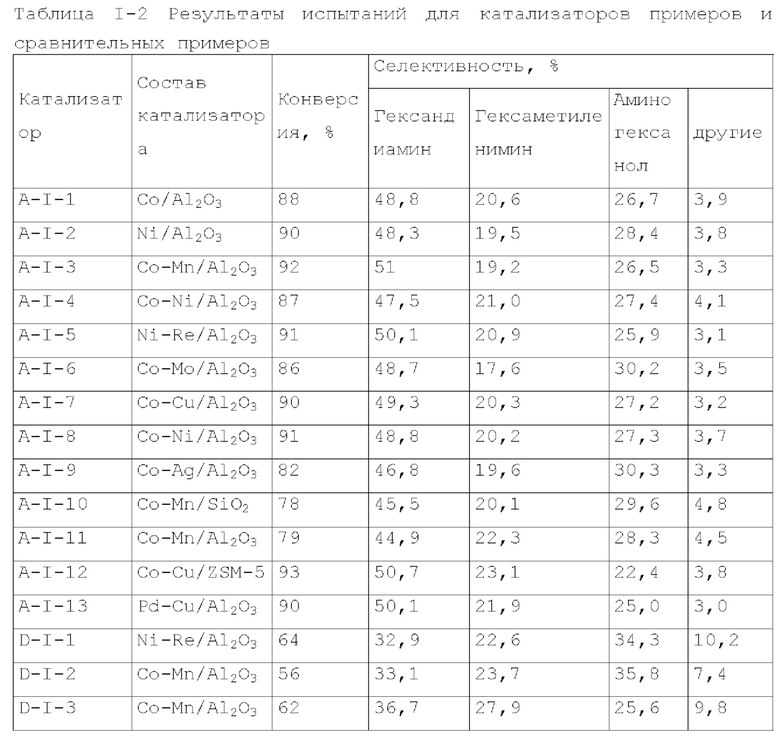
Как видно из данных таблицы 1-2, катализатор настоящего изобретения имеет более высокую конверсию и более высокую активность, чем катализаторы сравнения, что свидетельствует о том, что катализатор настоящего изобретения обеспечивает более высокую скорость реакции.
Все катализаторы испытывали в течение 360 часов, а затем выгружали для характеристики, и установлено, что катализаторы D-I-1, D-I-2 и D-I-3, полученные в сравнительных примерах, имеют явно сниженную удельную площадь поверхности и объем пор, а их количества нагара соответственно составляет 11 масс. %, 9,1 масс. % и 8,9 масс. %, в то время как катализаторы, полученные в примерах настоящего изобретения, не имеют явного изменения удельной площади поверхности и объема пор, а количество нагара у них меньше 2 масс. %.
Кроме того, когда катализаторы A-I-1-A-I-11 подвергали каталитической реакции в течение 1000 часов, отбирали пробу реакционного раствора и анализировали, и конверсия и селективность явно не изменились по сравнению с 360 часами, а именно величина снижения конверсии была не более чем 2%, величина снижения селективности была не более чем 1%, тогда как конверсия и селективность катализаторов D-I-1 to D-I-3 явно снижались по сравнению с 360 часами, когда катализаторы D-I-1-D-I-3 подвергали каталитической реакции в течение 1000 часов, и конверсия соответственно снижалась до 31%, 28% и 35%. Селективность гександиамина снижалась до 25%, 23% и 26%, соответственно.
Пример испытаний I-3
Данный пример испытаний иллюстрирует способ получения этиламина гидроаминированием этанола, применяя катализатор согласно первому типу вариантов осуществления настоящего изобретения
100 мл катализатора A-I-3, полученного в примере I-3, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 160°С, давление системы повышали до 1,75 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 110°С, затем направляли в верхний конец реактора, этанол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, молярное отношение водорода к аммиаку и к этанолу составляло 2: 5:1, объемная скорость жидкой фазы этанола составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 175°С и давлении реакции 1,75 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний 1-2), результаты анализа показаны в таблице 1-3.
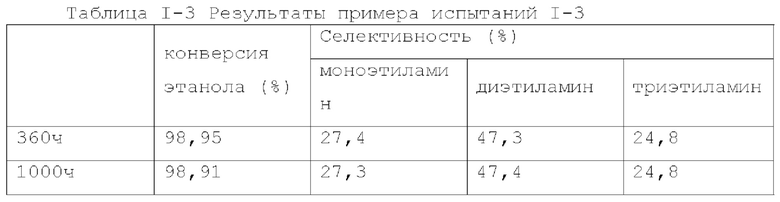
Пример испытаний 1-4
Данный пример испытаний иллюстрирует способ получения этилендиамина гидроаминированием этаноламина, применяя катализатор согласно первому типу вариантов осуществления настоящего изобретения
100 мл катализатора A-I-3, полученного в примере I-3, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 180°С, давление системы повышали до 9,6 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 100°С и направляется в верхний конец реактора, затем этаноламин подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, молярное отношение водорода к аммиаку и к этаноламину составляло 3:12:1, объемная скорость жидкой фазы этаноламина составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 180°С и давлении реакции 9,6 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний 1-2), результаты анализа показаны в таблице 1-4.
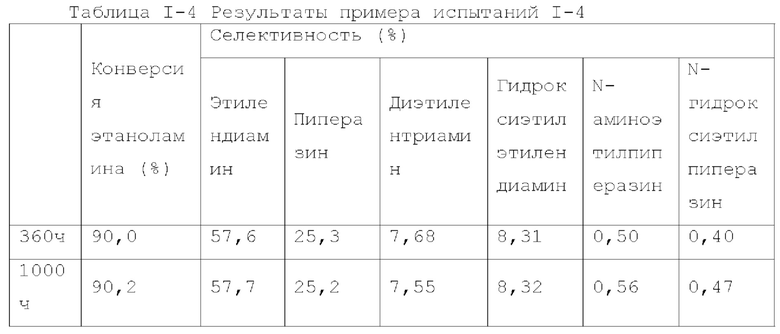
Пример испытаний I-5
Данный пример испытаний иллюстрирует способ получения гександиамина из смеси 1,6-гександиола, гексаметиленимина и аминогексанола, применяя катализатор согласно первому типу вариантов осуществления настоящего изобретения.
100 мл катализатора A-I-3, полученного в примере I-3, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 170°С, давление системы повышали до 8,2 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 110°С и затем направляли в верхний конец реактора, раствор смеси 53 масс. % 1,6-гександиола, 30 масс. % гексаметиленимина и 17 масс. % 6-амино-1-гексанола подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, молярное отношение водорода к аммиаку и к общему количеству трех веществ в смешанном растворе составляло 4:15:1, объемная скорость жидкой фазы смешанного раствора составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 190°С и давлении реакции 8,2 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний 1-2). Результаты анализа показаны в таблице 1-5.
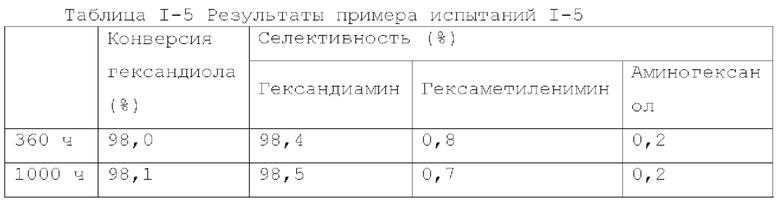
Серия примеров II
Второй тип вариантов осуществления настоящего изобретения далее подробно проиллюстрирован ниже со ссылкой на примеры из серии примеров II. В следующих примерах серии примеров II, порошок псевдобемита получали способом с применением сульфата алюминия и он содержал (Al2O3) на сухое вещество 72 масс. %; золь оксида кремния получали у Qingdao Ocean Chemical Co., Ltd, под торговым названием JN-40.
Пример II-1
Порошок псевдобемита (с удельной площадью поверхности 346 м2/г, объемом пор 1,13 мл/г, содержанием фосфора 0,22 г, на 100 г порошка, рассчитанного как Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 15 об. % азотной кислоты, формовали в полоски с диаметром 5 мм, сушили при 120°С в течение 12 часов, и затем кальцинировали при 800°С в течение 5 часов, получая носитель. Параметры носителя показаны в таблице II-1.
186,5 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 19,5 г 50 масс. % водного раствора нитрата магния и 14,13 г тригидрата нитрата меди растворяли в воде, получая 132 мл раствор, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 2 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 480°С в течение 2 ч, получая катализатор A-II-1.
Пример II-2
Порошок псевдобемита (с удельной площадью поверхности 333 м2/г, объемом пор 1,04 мл/г, содержанием бора 0,53 г, на 100 г порошка, рассчитанного как Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 10 об. % азотной кислоты, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 150°С в течение 6 часов, и затем кальцинировали при 820°С в течение 4 часов, получая носитель. Параметры носителя показаны в таблице II-1.
151,7 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), 13,0 г 50 масс. % водного раствора нитрата магния и 8,48 г тригидрата нитрата меди растворяли в воде, получая 148 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов. 1,12 г перрената аммония растворяли в воде, получая 78 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, и затем кальцинировали при 370°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 3 ч, получая катализатор A-II-2.
Пример II-3
Порошок псевдобемита (с удельной площадью поверхности 369 м2/г, объемом пор 0,99 мл/г, содержанием серы 2,88 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 15 об. % азотной кислоты, формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 20 ч, и затем кальцинировали при 840°С в течение 5 ч, получая носитель. Параметры носителя показаны в таблице II-1.
45,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 1,57 г нитрата серебра растворяли в воде, получая 102 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 1,50 г перрената аммония растворяли в воде, получая 70 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 390°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 420°С в течение 4 ч, получая катализатор A-II-3.
Пример II-4
Порошок псевдобемита (с удельной площадью поверхности 375 м2/г, объемом пор 1,15 мл/г, содержанием фтора 0,08 г, на 100 г порошка, рассчитанного как Al2O3) и ZSM-5 (имеющийся в продаже, полученный Nankai University, SiO2/Al2O3=45 (молярное отношение)) равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 12 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 8 часов, и затем кальцинировали при 810°С в течение 4 ч, получая носитель. Параметры носителя показаны в таблице II-1.
126,0 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 0,79 г нитрата серебра растворяли в воде, получая 192 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 8,70 г перрената аммония растворяли в воде, получая 81 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 3 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 420°С в течение 4 ч, получая катализатор A-II-4.
Пример II-5
Порошок псевдобемита (с удельной площадью поверхности 298 м2/г, объемом пор 0,85 мл/г, содержанием фосфора 0,18 г, на 100 г порошка, рассчитанного как Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 15 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 2,5 мм, сушили при 110°С в течение 14 ч, и затем кальцинировали при 890°С в течение 4 ч, получая носитель. Параметры носителя показаны в таблице II-1.
75,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 13,0 г 50 масс. % водного раствора нитрата магния и 3,15 г нитрата серебра растворяли в воде, получая 112 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 380°С в течение 4 часов. 2,90 г перрената аммония растворяли в воде, получая 70 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 2 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 3 ч, получая катализатор A-II-5.
Пример II-6
Порошок псевдобемита (с удельной площадью поверхности 348 м2/г, объемом пор 1,09 мл/г, и содержанием бора 3,66 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 15 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 810°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
126,4 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), 26,1 г 50 масс. % водного раствора нитрата магния и 1,57 г нитрата серебра растворяли в воде, получая 146 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 380°С в течение 4 часов. 4,40 г перрената аммония растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 350°С в течение 6 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 ч, получая катализатор А-II-6.
Пример II-7
Порошок псевдобемита (с удельной площадью поверхности 374 м2/г, объемом пор 1,18 мл/г, содержанием серы 0,81 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 10 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 750°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
201,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 11,30 г тригидрата нитрата меди и 9,80 г 50 масс. % водного раствора нитрата магния растворяли в воде, получая 132 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 4,70 г перрената аммония растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 2 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 460°С в течение 2 ч, получая катализатор A-II-7.
Пример II-8
Порошок псевдобемита (с удельной площадью поверхности 355 m2/г, объемом пор 1,05 мл/г, и содержанием серы 0,88 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 10 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 810°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
100,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 14,30 г тригидрата нитрата меди и 1,57 г нитрата серебра растворяли в воде, получая 116 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 360°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 5 ч при 420°С, получая катализатор A-II-8.
Пример II-9
Порошок псевдобемита (с удельной площадью поверхности 380 м2/г, объемом пор 1,01 мл/г, содержанием фтора 0,82 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 15 об. % азотной кислоты, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 810°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 13,0 г 50 масс. % водного раствора нитрата магния и 1,57 г нитрата серебра растворяли в воде, получая 154 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 1,50 г перрената аммония растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 380°С в течение 3 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 3 ч, получая катализатор A-II-9.
Пример II-10
Порошок псевдобемита (с удельной площадью поверхности 328 м2/г, объемом пор 0,97 мл/г, содержанием серы 0,95 г, на 100 г порошка, рассчитанного как Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 810°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
226,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 26,1 г 50 масс. % водного раствора нитрата магния и 0,79 г нитрата серебра растворяли в воде, получая 162 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 0,7 г перрената аммония растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, и затем кальцинировали при 420°С в течение 2 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 4 ч, получая катализатор A-II-10.
Пример II-11
Порошок псевдобемита (с удельной площадью поверхности 304 м2/г, объемом пор 1,06 мл/г, и содержанием фосфора 4,6 г, на 100 г порошка, рассчитанного как Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в цилиндрические стержни диаметром 4 мм, сушили при 120°С в течение 7 ч, и затем кальцинировали при 810°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
141,1 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 13,0 г 50 масс. % магния нитрата водный раствор и 0,79 г нитрата серебра растворяли в воде, получая 152 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 2,90 г перрената аммония растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 460°С в течение 2 ч, получая катализатор A-II-11.
Пример II-12
Катализатор получали, как описано в примере II-3, за исключением того, что применяемый порошок псевдобемита не содержал примесных элементов и имел удельная площадь поверхности 298 м2/г и объем пор 1,08 мл/г, и полученный катализатор обозначали как A-II-12.
Сравнительный пример II-1
Порошок псевдобемита (с удельной площадью поверхности 376 м2/г, объемом пор 1,01 мл/г и без серы) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 10 об. % азотной кислоты, и формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение 5 ч, и затем кальцинировали при 860°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
Остальная часть способа была такой же, как описано в примере 8, получая катализатор D-II-1.
Сравнительный пример II-2
Порошок псевдобемита (с удельной площадью поверхности 358 м2/г, объемом пор 1,15 мл/г и без серы) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 10 об. % азотной кислоты, и формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 12 ч, и затем кальцинировали при 890°С в течение 6 ч, получая носитель. Параметры носителя показаны в таблице II-1.
226,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 0,79 г нитрата серебра растворяли в воде, получая 162 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 4 ч, получая катализатор D-II-2.
Пример испытаний II-1
Элементный состав носителя и катализаторов анализировали с помощью плазменно-эмиссионной спектроскопии, где содержания примесного элемента, активного металлического компонента и металлического промотора выражены по массе в расчете на 100 г матрикса; полученные выше носители были охарактеризованы NH3-TPD, BET способом адсорбции-десорбции азота, и результаты приведены в таблице II-1.
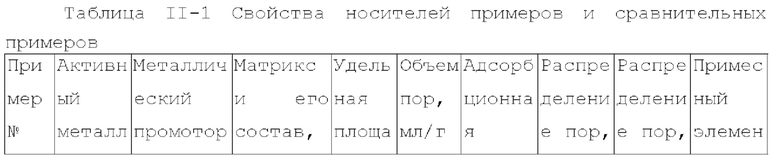
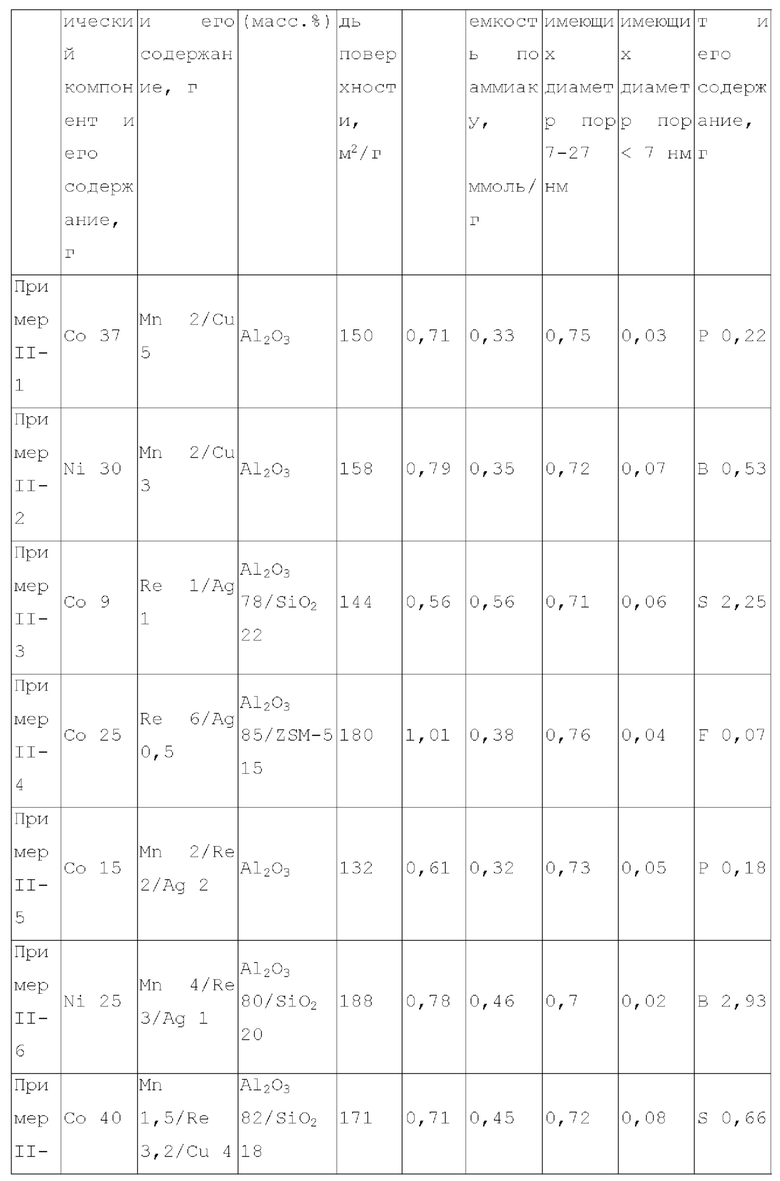
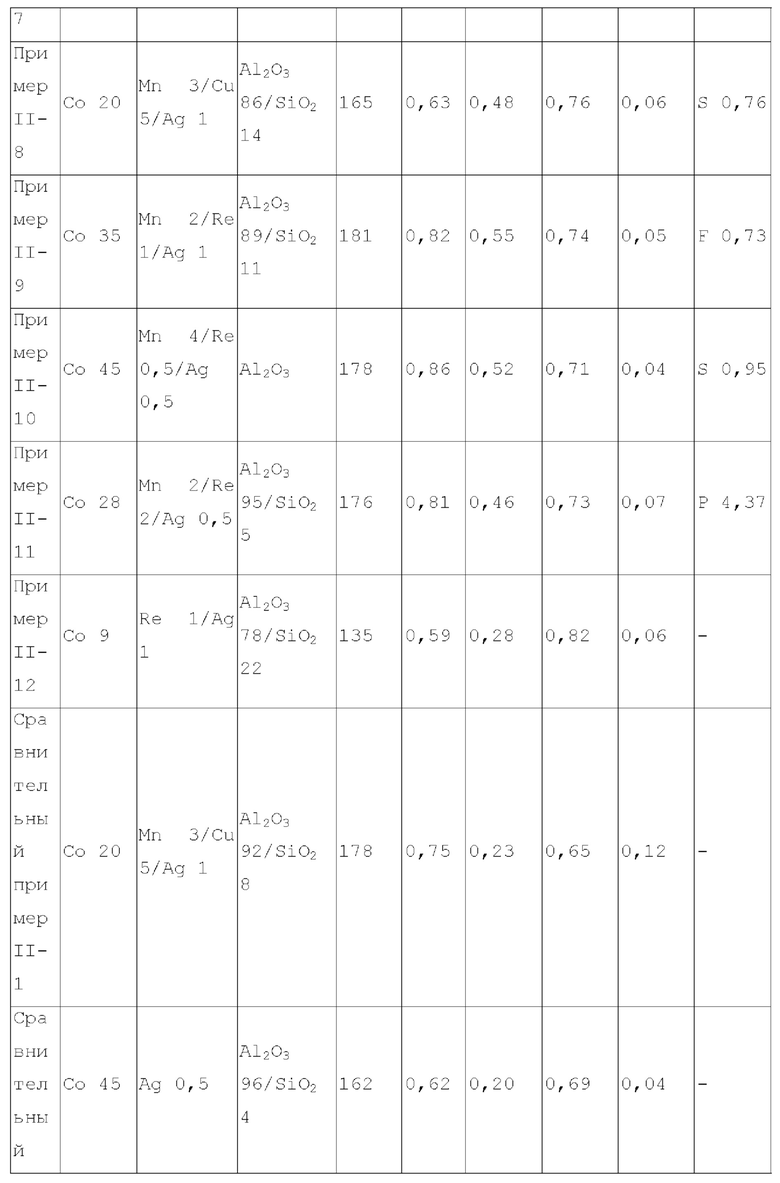

Пример испытаний II-2
Данный пример испытаний иллюстрирует способ получения 1,6-гександиамина гидроаминированием 1,6-гександиола, применяя катализатор второго типа вариантов осуществления настоящего изобретения.
100 мл катализаторов, полученных в примерах и сравнительных примерах, соответственно отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 9,5 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 155°С и затем направляли в верхний конец реактора, затем нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к 1,6-гександиолу составляло 3:8:1, объемная скорость жидкой фазы 1,6-гександиола составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 200°С и давлении реакции 11 МПа, и после того, как реакция стала стабильной (а именно после 500 ч реакции), реакционный раствор отбирали и анализировали. Результаты анализа показаны в таблице II-2.
Анализ образца проводился способом газовой хроматографии и калибровали с применением поправочного коэффициента полученного стандартного образца.
Конверсию и селективность рассчитывали, исходя из молярного содержания каждого компонента в реакционном растворе, и способы расчета были такими же, как описано в примере испытаний 1-2.

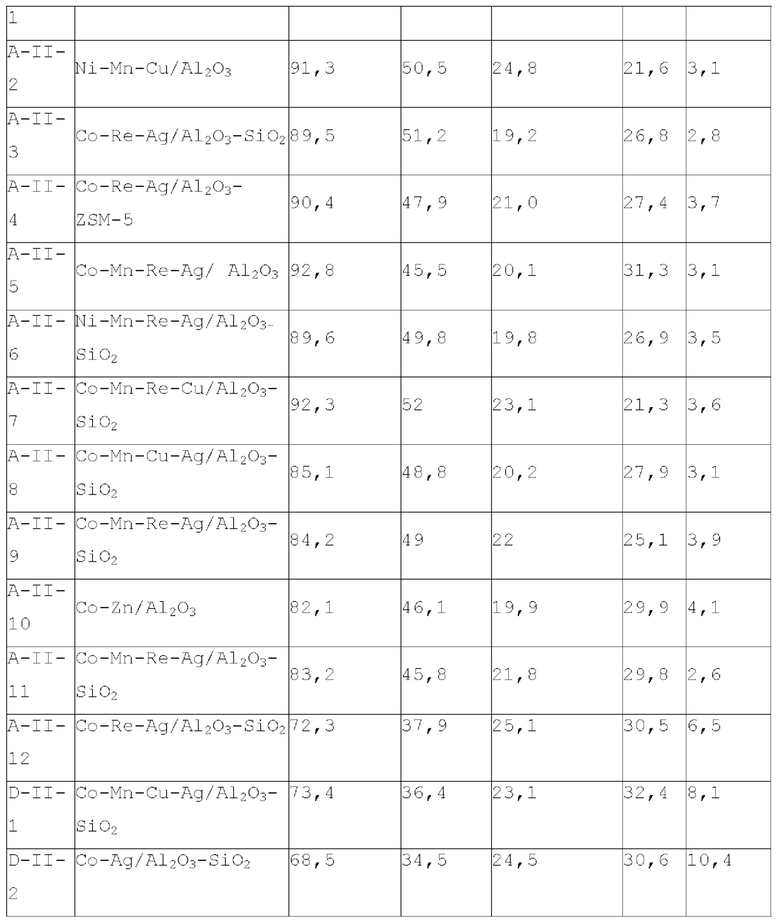
Как видно из данных таблицы II-2, катализатор настоящего изобретения показывает более высокую конверсию и более высокую селективность гександиамина, что свидетельствует об улучшении каталитической активности по сравнению с катализатором, выпадающим из настоящего изобретения, и более низкую селективность по отношению к «другим», т.е. аминам с 12 или более атомов углерода, что указывает на то, что катализатор настоящего изобретения имеет более низкую степень нагара и более высокую стабильность.
После длительного испытания на стабильность в течение 500 ч, проведенного соответственно на катализаторах A-II-3 и D-II-2, соответственно были проведены BET и XRD испытания на катализаторах до и после применения, результаты которых приведены в таблице II-3. Результаты показывают, что удельная площадь поверхности, объем пор и размер зерен активного металла катализатора A-II-3 практически не изменяются, в то время как удельная площадь поверхности и объем пор катализатора D-II-2 уменьшаются в определенной степени, и размер зерен активного металла явно увеличен по сравнению с катализаторами до реакции, что свидетельствует о том, что катализатор настоящего изобретения обладает лучшей стабильностью и меньшим нагарообразованием.
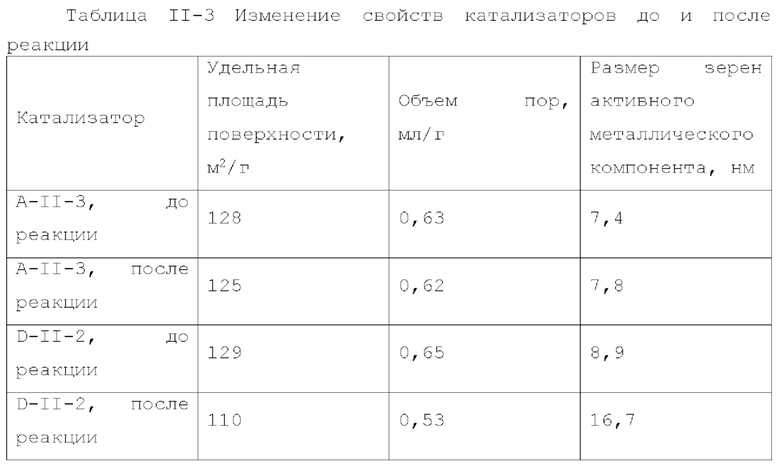
Пример испытаний II-3
100 мл катализатора A-II-3, полученного в примере II-3, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 240°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 1,8 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 160°С и затем направляли в верхний конец реактора, этанол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, молярное отношение водорода к аммиаку и к этанолу составляло 3:5:1, объемная скорость жидкой фазы этанола составляла 0,6 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 185°С и давлении реакции 2,8 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали. Результаты анализа показаны в таблице II-4.
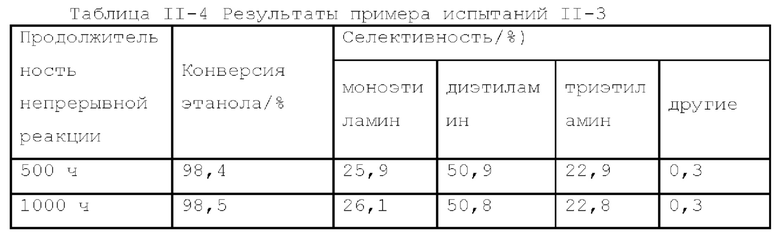
Пример испытаний II-4
100 мл катализатора A-II-5, полученного в примере II-5, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 240°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 8 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 150°С и направляется в верхний конец реактора, этаноламин подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к этаноламину составляло 3:8:1, объемная скорость жидкой фазы этаноламина составляла 0,6 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 195°С и давлении реакции 9,5 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний II-2). Результаты анализа показаны в таблице II-5.
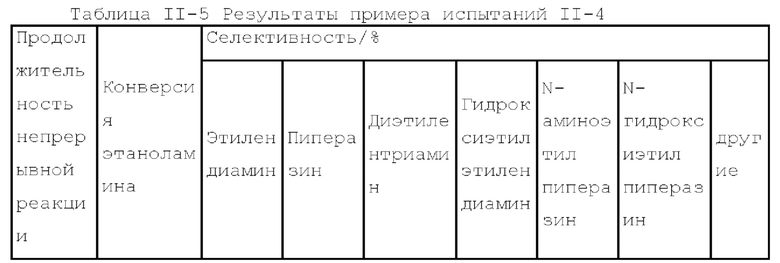
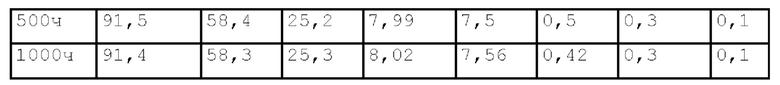
Серия примеров III
Третий тип вариантов осуществления настоящего изобретения далее подробно проиллюстрирован со ссылкой на примеры из серии примеров III. В следующих примерах серии примеров III, порошок псевдобемита получали способом с применением сульфата алюминия, и он содержал (Al2O3) на сухое вещество 72 масс. %; золь оксида кремния получали у Qingdao Ocean Chemical Co., Ltd, под торговым названием JN-40.
Пример III-1
Порошок псевдобемита (с удельной площадью поверхности 375 м2/г, объемом пор 0,99 мл/г, содержанием бора 3,89 г, на 100д Al2O3) замешивали с разбавленным кислым водным раствором (содержащим нитрат кальция в отмеренном количестве, с содержанием кальция 0,11 г, на 100 г Al2O3), и формовали в полоски с диаметром 5 мм, сушили при 120°С в течение 12 часов, и затем кальцинировали при 800°С в течение 10 часов, получая носитель. Параметры носителя показаны в таблице III-1.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 26,1 г 50 масс. % водного раствора нитрата магния и 6,83 г нитрата цинка растворяли в воде, получая 138 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С получая катализатор А-III-1.
Пример III-2
Порошок псевдобемита (с удельной площадью поверхности 405 м2/г, объемом пор 1,06 мл/г, содержанием серы 0,79 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат магния в отмеренном количестве, с содержанием магния 0,78 г, на 100 г Al2O3), экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 120°С в течение 15 часов, и затем кальцинировали при 840°С в течение 4 часов, получая носитель. Параметры носителя показаны в таблице III-1.
161,8 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), 39,1 г 50 масс. % водного раствора нитрата магния и 6,83 г нитрата цинка растворяли в воде, получая 146 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 5 часов после каждого раза пропитки распылением, и затем кальцинировали при 390°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 ч, получая катализатор А-III-2.
Пример III-3
Порошок псевдобемита (с удельной площадью поверхности 386 м2/г, объемом пор 1,09 мл/г, содержанием серы 0,84 г, на 100 г Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащим нитрат калия в отмеренном количестве, с содержанием калия 2,1 г, на 100 г Al2O3), формовали в зубчатые сферы диаметром 4 мм, сушили при 100°С в течение 20 часов, и затем кальцинировали при 850°С в течение 6 часов, получая носитель. Параметры носителя показаны в таблице III-1.
50,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 13,0 г 50 масс. % водного раствора нитрата магния и 6,83 г раствора нитрата цинка растворяли в воде, получая 104 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С получая катализатор A-III-3.
Пример III-4
Порошок псевдобемита (с удельной площадью поверхности 398 м2/г, объемом пор 0,99 мл/г, содержанием фтора 0,85 г, на 100 г Al2O3) и ZSM-5 (имеющийся в продаже, полученный Nankai University, SiO2/Al2O3=45 (молярное отношение)) равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат висмута в отмеренном количестве, с содержанием висмута 1,8 г на 100 г применяемого порошкового псевдобемита, рассчитанного как Al2O3), формовали в зубчатые сферы диаметром 4 мм, сушили при 140°С в течение 10 ч, и затем кальцинировали при 750°С в течение 10 ч, получая носитель. Параметры носителя показаны в таблице III-1.
100,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 3,3 г 50 масс. % водного раствора нитрата магния и 4,55д г раствора нитрата цинка растворяли в воде, получая 188 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-III-4.
Пример III-5
Порошок псевдобемита (с удельной площадью поверхности 375 м2/г, объемом пор 1,15 мл/г, содержанием фосфора 0,33 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат бария в измеренном количестве, с содержанием бария 1,7 г, на 100 г Al2O3), формовали в зубчатые сферы диаметром 4 мм, сушили при 150°С в течение 6 часов, и затем кальцинировали при 950°С в течение 3 часов, получая носитель. Параметры носителя показаны в таблице III-1.
75,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 4,55д г раствора нитрата цинка растворяли в воде, получая 104 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 380°С в течение 4 часов. 2,9 г перрената аммония растворяли в воде, получая 45 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, и затем кальцинировали при 380°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 30°С/ч, и наконец восстанавливали при 450°С в течение 4 ч, получая катализатор A-III-5.
Пример III-6
Порошок псевдобемита (с удельной площадью поверхности 320 м2/г, объемом пор 1,03 мл/г, содержанием бора 0,24 г, на 100 г Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат кальция в отмеренном количестве, с содержанием кальция 1,20 г, на 100 г Al2O3), формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 110°С в течение 16 ч, и затем кальцинировали при 880°С в течение 5 ч, получая носитель. Параметры носителя показаны в таблице III-1.
126,4 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) и 22,75 г нитрата цинка растворяли в воде, получая 150 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 8 часов и затем кальцинировали при 360°С в течение 6 часов после каждого раза пропитки распылением. 4,30 г перрената аммония растворяли в воде, получая 67 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-III-6.
Пример III-7
Порошок псевдобемита (с удельной площадью поверхности 355 м2/г, объемом пор 0,99 мл/г, содержанием серы 2,65 г, на 100 г Al2O3) и золь оксида кремния равномерно смешивали, замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат магния в измеренном количестве, с содержанием магния 1,4 г, на 100 г Al2O3), формовали в полоски с диаметром 5 мм, сушили при 120°С в течение 15 часов, и затем кальцинировали при 550°С в течение 20 часов, получая носитель. Параметры носителя показаны в таблице III-1.
201,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 2,28 г нитрата цинка растворяли в воде, получая 136 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов после каждого раза пропитки распылением. 1,40 г перрената аммония растворяли в воде, получая 62 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 480°С в течение 2 ч, получая катализатор A-III-7.
Пример III-8
Порошок псевдобемита (с удельной площадью поверхности 275 м2/г, объемом пор 0,99 мл/г, содержанием фтора 0,08 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую кальция нитрата в отмеренном количестве, с содержанием кальция 0,80 г, на 100 г Al2O3), формовали в полоски с диаметром 5 мм, сушили при 150°С в течение 8 часов, и затем кальцинировали при 800°С в течение 8 часов, получая носитель. Параметры носителя показаны в таблице III-1.
100,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 13,65 г нитрата цинка растворяли в воде, получая 122 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов после каждого раза пропитки распылением. 1,40 г перрената аммония растворяли в воде, получая 55 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 3 ч, получая катализатор A-III-8.
Пример III-9
Порошок псевдобемита (с удельной площадью поверхности 379 м2/г, объемом пор 0,99 мл/г, содержанием фосфора 0,2 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат кальция в отмеренном количестве, с содержанием кальция 1,30 г, на 100 г Al2O3), формовали в цилиндрические стержни диаметром 5 мм, сушили при 100°С в течение 18 часов, и затем кальцинировали при 880°С в течение 4 часов, получая носитель. Параметры носителя показаны в таблице III-1.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 19,5 г 50 масс. % водного раствора нитрата магния и 19,36 г нитрата цинка растворяли в воде, получая 152 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 380°С в течение 4 часов. 1,44 г перрената аммония растворяли в воде, получая 70 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 420°С в течение 3 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 6 ч при 420°С, получая катализатор A-III-9. Пример III-10
Порошок псевдобемита (с удельной площадью поверхности 392 м2/г, объемом пор 0,99 мл/г, содержанием серы 0,97 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат кальция в отмеренном количестве, с содержанием кальция 0,05 г, на 100 г Al2O3), формовали в полоски с диаметром 5 мм, сушили при 80°С в течение 20 часов, и затем кальцинировали при 700°С в течение 10 часов, получая носитель. Параметры носителя показаны в таблице III-1.
226,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 26,1 г 50 масс. % нитрата магния и 2,28 г нитрата цинка растворяли в воде, получая 162 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 6 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 2,88 г перрената аммония растворяли в воде, получая 75 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 4 ч при 440°С, получая катализатор A-III-10.
Пример III-11
Порошок псевдобемита (с удельной площадью поверхности 315 м2/г, объемом пор 0,99 мл/г, содержанием фосфора 4,64 г, на 100 г Al2O3) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту (содержащую нитрат кальция в отмеренном количестве, с содержанием кальция 1,25 г, на 100 г Al2O3), формовали в клеверы диаметром 3 мм, сушили при 120°С в течение 8 часов, и затем кальцинировали при 750°С в течение 8 часов, получая носитель. Параметры носителя показаны в таблице III-1.
141,1 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 32,6 г 50 масс. % водного раствора нитрата магния и 2,28 г нитрата цинка растворяли в воде, получая 154 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов. 5,76 г перрената аммония растворяли в воде, получая 71 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов и затем кальцинировали при 390°С в течение 6 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 450°С в течение 3 ч, получая катализатор A-III-11.
Пример III-12
Катализатор A-III-12 получали, как описано в примере III-6, за исключением того, что применяемый порошок псевдобемита составляло не содержал примесных элементов и имел удельную площадь поверхности 366 м2/г и объем пор 1,05 мл/г.
Сравнительный пример III-1
Катализатор получали, как описано в примере III-6, за исключением того, что применяемый порошок псевдобемита не содержал примесных элементов и имел удельную площадь поверхности 325 м2/г и объем пор 0,95 мл/г, и нитрат цинка заменяли 15,2 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), получая катализатор D-III-1.
Сравнительный пример III-2
Катализатор получали, как описано в примере III-9, за исключением того, что применяемый порошок псевдобемита не содержал примесных элементов и имел удельную площадь поверхности 349 м2/г и объем пор 1,13 мл/г, и нитрат магния и перренат аммония заменяли на 20,1 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), получая катализатор D-III-3.
Сравнительный пример III-3
Катализатор получали, как описано в примере III-2, за исключением того, что порошок псевдобемита (с удельной площадью поверхности 396 м2/г, объемом пор 1,21 мл/г, содержанием серы 4,25 г, на 100 г Al2O3) применяли, и нитрат цинка заменяли на 5,79 г нонагидрата нитрата железа, получая катализатор D-III-3.
Пример испытаний III-1
Элементный состав носителей и катализаторов анализировали с помощью плазменно-эмиссионной спектроскопии, где содержания основного элемента, активного металлического компонента и металлического промотора выражены по массе в расчете на 100 г матрикса; полученные выше носители были охарактеризованы NH3-TPD, C02-TPD, BET способом сорбции-десорбции азота, и результаты представлены в таблице III-1.
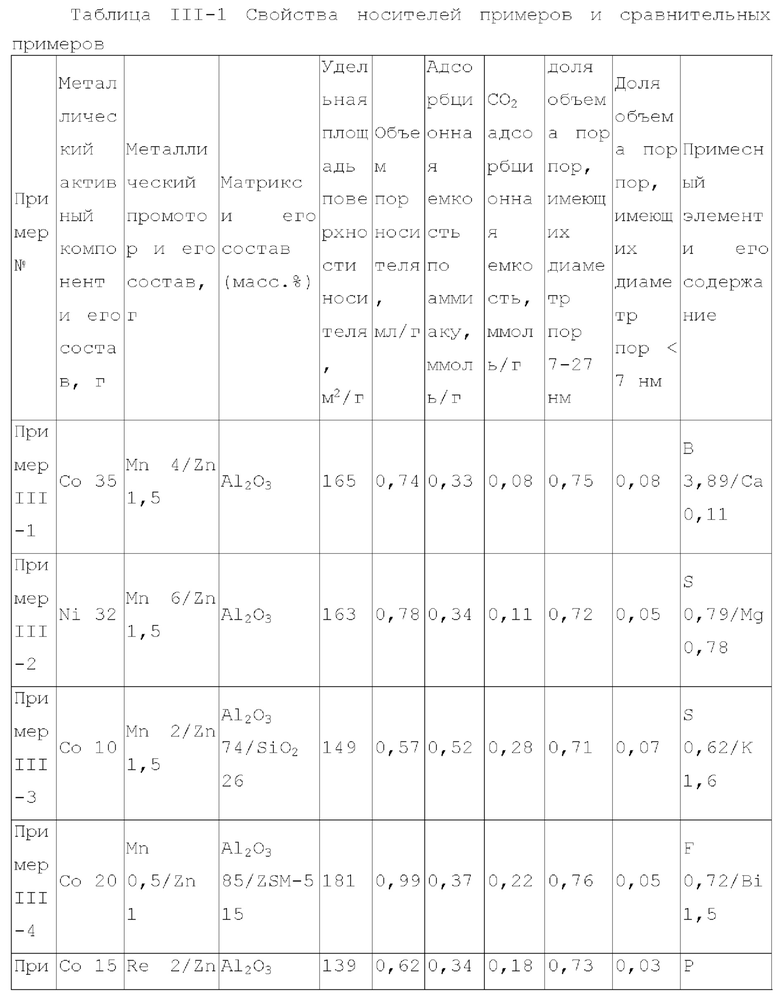
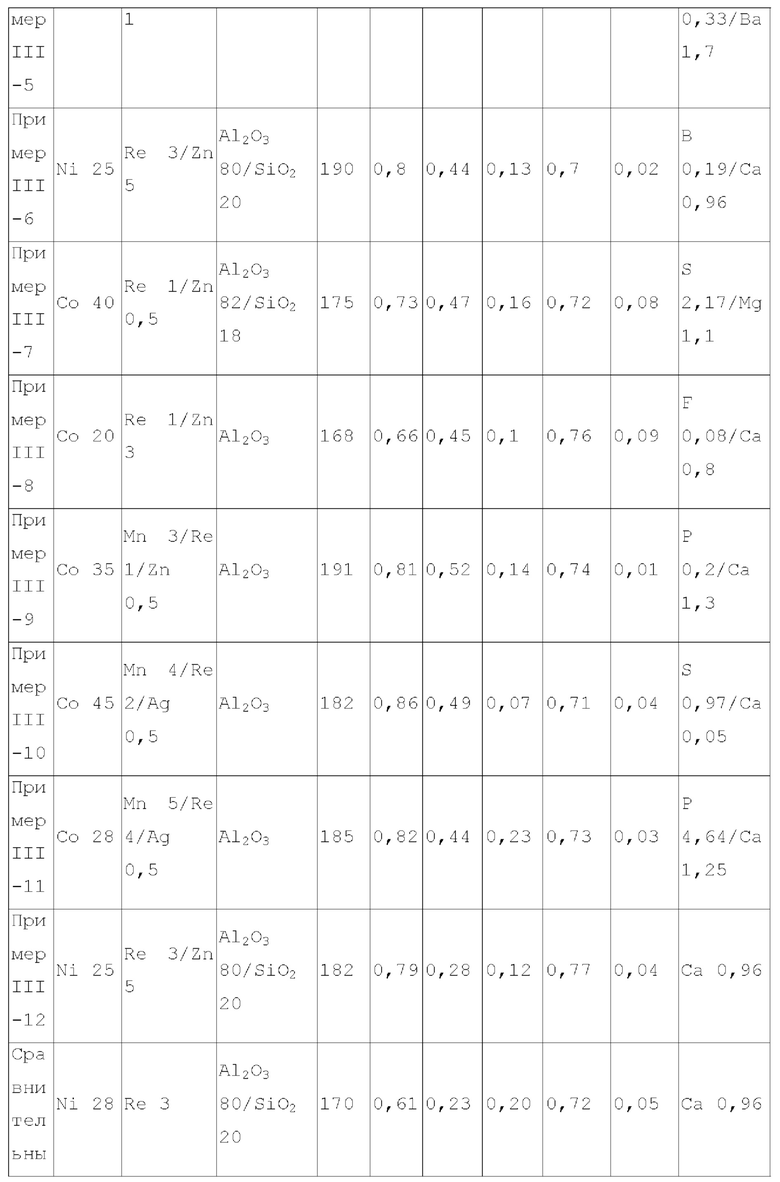
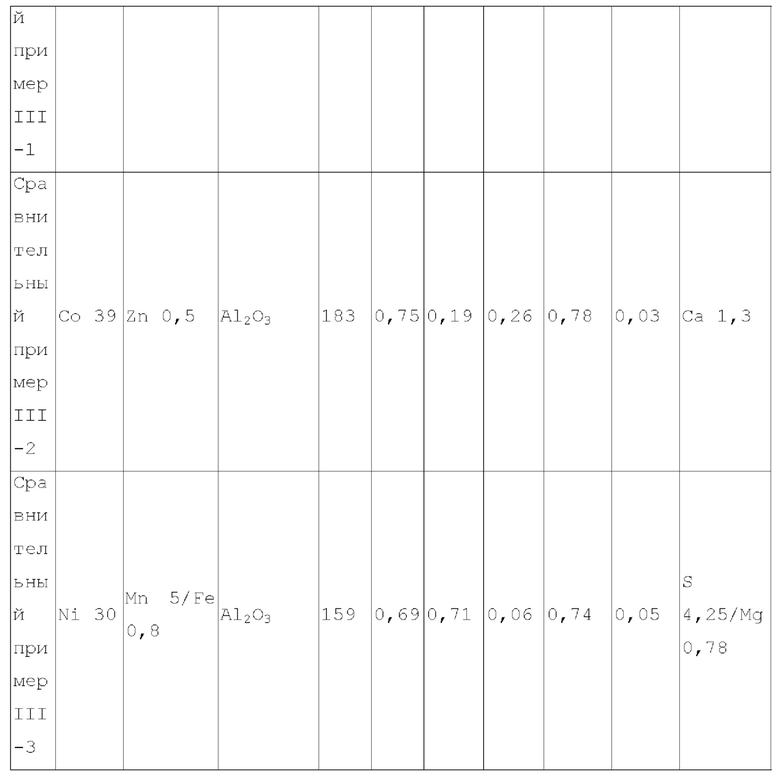
Пример испытаний III-2
Данный пример испытаний иллюстрирует способ получения 1,6-гександиамина гидроаминированием 1,6-гександиола, применяя катализатор третьего типа вариантов осуществления настоящего изобретения.
100 мл катализаторов, полученных в примерах и сравнительных примерах, соответственно отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 12 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 160°С и направляли в верхний конец реактора, затем нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к1,6-гександиолу составляло 3:8:1, и объемная скорость жидкой фазы 1,6-гександиола составляла 0,6 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 205°С и давлении реакции 13 МПа, и после того, как реакция стала стабильной (а именно после 10 ч реакции), реакционный раствор отбирали и анализировали. Результаты анализа показаны в таблице III-2.
Анализ образца проводили способом газовой хроматографии и калибровали с применением поправочного коэффициента полученного стандартного образца.
Конверсию и селективность рассчитывали, исходя из молярного содержания каждого компонента в реакционном растворе, и способы расчета были такими же, как описано в примере испытаний 1-2.
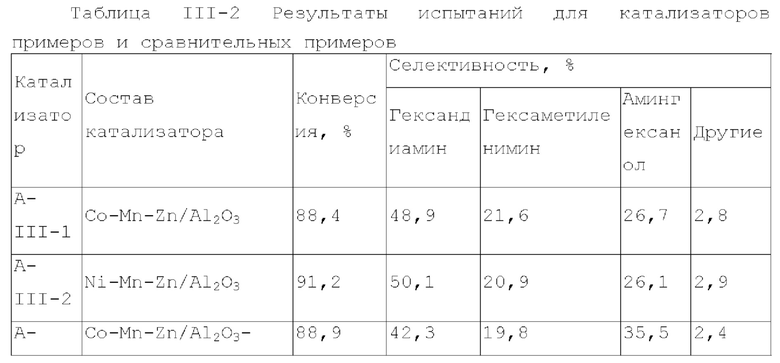
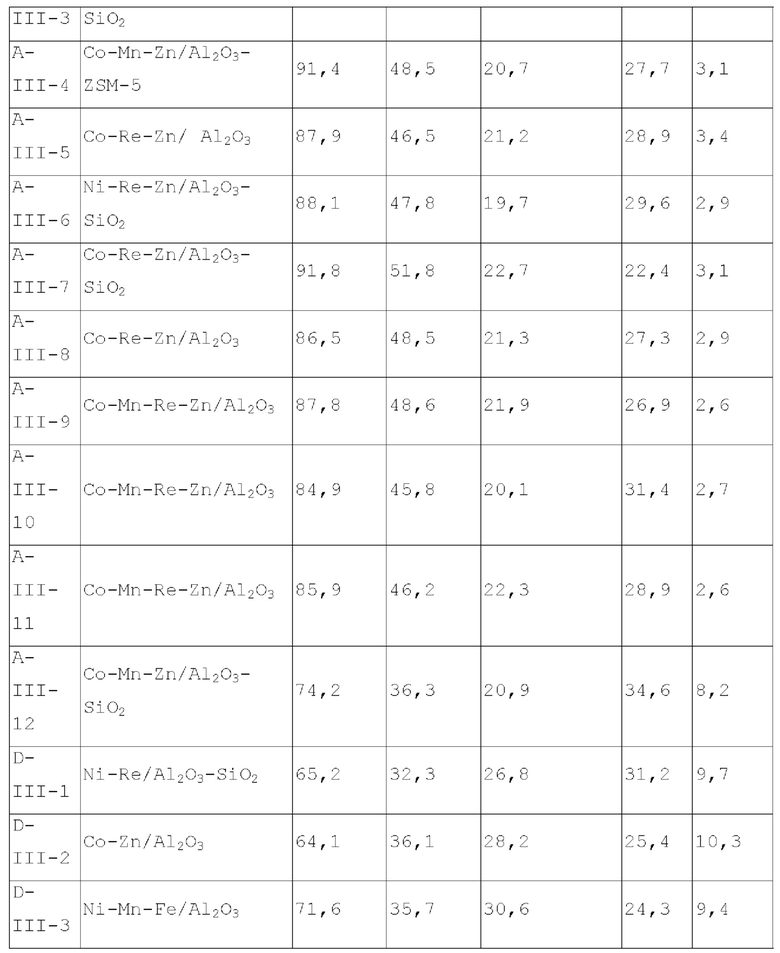
Катализаторы подвергали длительному тесту на стабильность в течение 500 ч в тех же условиях, и BET и XRD испытания проводили на катализаторах до и после применения. Результаты показывают, что удельная площадь поверхности и объемы пор катализаторов А-III-1-A-III-11 практически не изменились (т.е. восстановление составило не более 2%) до и после применения, в то время как удельная площадь поверхности и объем пор катализаторов D-III-1-D-III-3 значительно снижаются (оба более чем на 8%) после применения, что свидетельствует о том, что катализатор настоящего изобретения обладает большей стабильностью и меньшим меньшим образованием нагара.
Дальнейшее обнаружение показало, что катализатор A-III-2 по-прежнему показывает снижение конверсии не более чем на 5% после 1000 ч применения, и величина снижения удельной площади поверхности и объема пор составляет не более 5%. Видно, что катализатор настоящего изобретения имеет более длительный срок службы.
Пример испытаний III-3
Данный пример испытаний иллюстрирует способ получения н-пропиламина гидроаминированием н-пропанола, применяя катализатор третьего типа вариантов осуществления настоящего изобретения.
100 мл катализатора A-III-2, полученного в примере III-3, отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 172°С, давление системы повышали до 1,5 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 150°С и затем направляли в верхний конец реактора, н-пропанол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, молярное отношение водорода к аммиаку и к н-пропанолу составляло 3:5:1, объемная скорость жидкой фазы пропанола составляла 0,75 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 180°С и давлении реакции 2 МПа, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний III-2). Результаты анализа показаны в таблице III-3.
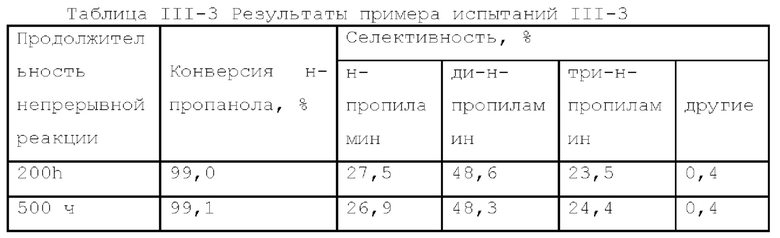
Серия примеров IV
Четвертый тип вариантов осуществления настоящего изобретения дополнительно подробно проиллюстрирован ниже со ссылкой на примеры серию примеров IV. В последующих примерах серии примеров IV применяемый порошок псевдобемита имел содержание (Al2O3) на сухое вещество 72 масс. %; золь оксида кремния приобретали у Qingdao Ocean Chemical Co., Ltd под торговым названием JN-40.
Пример IV-1
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 380 м2/г и объемом пор 1,02 мл/г, порошок псевдобемита, содержащий примесный элемент серы, с содержанием S 2,15 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, растворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника в кальцинированном носителе, составляла 4% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 120°С в течение 8 часов, и кальцинировали при 650°С в течение 5 ч для получения требуемого носителя.
186,5 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 6,83 г гексагидрата нитрата цинка (аналитически чистый) и 5,65 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 148 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением и затем кальцинировали при 400°С в течение 4 часов. 1,8 г тетрагидрата молибдата аммония (аналитически чистый) затем растворяли в воде, получая 74 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор А-IV-1.
Пример IV-2
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 375 м2/г и объемом пор 0,98 мл/г, порошок псевдобемита, содержащий примесный элемент бора, с содержанием В 0,53 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника в кальцинированном носителе, составляла 11% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 100°С в течение 12 ч, и кальцинировали при 590°С в течение 8 часов для получения требуемого носителя.
151,7 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), 6,83 г гексагидрата нитрата цинка (аналитически чистый) и 5,65 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 156 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. Затем, 3,7 г тетрагидрата молибдата аммония растворяли в воде, получая 78 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-IV-2.
Пример IV-3
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 380 м2/г и объемом пор 1,02 мл/г, порошок псевдобемита, содержащий примесный элемент серы, с содержанием S 2,15 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественник в кальцинированном носителе, составляла 4% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 120°С в течение 8 часов, и кальцинировали при 650°С в течение 5 ч для получения требуемого носителя.
55,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 6,83 г гексагидрата нитрата цинка (аналитически чистый) и 5,65 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 144 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 1,8 г тетрагидрата молибдата аммония растворяли в воде, получая 72 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-IV-3.
Пример IV-4
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 340 м2/г и объемом пор 1,13 мл/г, порошок псевдобемита, содержащий примесный элемент фосфора, с содержанием Р 0,18 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, предшественник молекулярных сит ZSM-5 (ZSM-5 порошок, доступный Catalyst Plant of Nankai University, SiO2/Al2O3=45 (молярное отношение), то же самое ниже) добавляли в начале, так что масса Al2O3, полученного из порошка псевдобемита в кальцинированном носителе, составляла 85% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в зубчатые сферы диаметром 4 мм, сушили при 80°С в течение 20 ч, и затем кальцинировали при 530°С в течение 6 ч для получения требуемого носителя.
126 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 4,55 г нитрата цинка и 0,79 г нитрата серебра (аналитически чистый) растворяли в воде, получая 210 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 3,7 г тетрагидрата молибдата аммония (аналитически чистый) затем растворяли в воде, получая 105 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-IV-4.
ример IV-5
Носитель получали, как описано в примере IV-4, за исключением того, что предшественник молекулярных сит ZSM-5 добавляли в начале при получении порошка псевдобемита, так что Al2O3, полученный из порошка псевдобемита в кальцинированном носителе, составлял 85% общей массы носителя.
126 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 9,1 г гексагидрата нитрата цинка (аналитически чистый) и 1,57 г нитрата серебра (аналитически чистый) растворяли в воде, получая 208 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением и затем кальцинировали при 400°С в течение 4 часов. 7,4 г тетрагидрата молибдата аммония (аналитически чистый) затем растворяли в воде, получая 104 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор А-IV-5.
Пример IV-6
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 341 м2/г и объемом пор 1,11 мл/г, порошок псевдобемита, содержащий примесный элемент фосфора, с содержанием Р 4,2 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, предшественник молекулярных сит ZSM-5 добавляли в начале, так что масса Al2O3, полученного из порошка псевдобемита в кальцинированном носителе, составляла 85% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в зубчатые сферы диаметром 4 мм, сушили при 80°С в течение 20 ч, и затем кальцинировали при 530°С в течение 6 ч для получения требуемого носителя.
Остальная часть способа была такой же, как описано в примере IV-4, получая катализатор A-IV-6.
Пример IV-7
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 288 м2/г и объемом пор 0,93 мл/г, порошок псевдобемита, содержащий примесный элемент серы, с содержанием S 0,88 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника, в кальцинированном носителе составляла 8% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в клеверы диаметром 4 мм, сушили при 100°С в течение 8 ч, и затем кальцинировали в течение 4 ч при 850°С для получения требуемого носителя.
201,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 2,28 г гексагидрата нитрата цинка (аналитически чистый) и 8,48 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 146 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением и затем кальцинировали при 400°С в течение 4 часов. Затем, 2,7 г метавольфрамата аммония (аналитически чистый) растворяли в воде, получая 73 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор А-IV-7.
Пример IV-8
Применяли носитель, полученный в примере IV-7.
100,8 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 2,28 г гексагидрата нитрата цинка (аналитически чистый) и 8,48 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 150 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 2,7 г метавольфрамата аммония (аналитически чистый) затем растворяли в воде, получая 75 мл раствора, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор А-IV-8.
Пример IV-9
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 281 м2/г и объемом пор 0,87 мл/г, порошок псевдобемита, содержащий примесный элемент фтора, с содержанием F 0,82 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника, в кальцинированном носителе составляла 21% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, экструдировали в гранулы клеверной формы толщиной 4 мм, сушили при 90°С в течение 18 ч, и кальцинировали при 770°С в течение 9 ч для получения требуемого носителя.
176,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 2,28 г гексагидрата нитрата цинка (аналитически чистый) и 11,3 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 130 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением и затем кальцинировали при 400°С в течение 4 часов. 2,0 г метавольфрамата аммония (аналитически чистый) затем растворяли в воде до раствора 65 мл, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор А-IV-9.
Пример IV-10
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 274 м2/г и объемом пор 0,85 мл/г, порошок псевдобемита, содержащий примесный элемент серы, с содержанием S 0,95 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника, в кальцинированном носителе составляла 25% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в гранулы клеверной формы диаметром 3,5 мм, сушили при 150°С в течение 6 ч, и затем кальцинировали в течение 6 ч при 930°С для получения требуемого носителя.
227,5 г гексагидрата нитрата никеля (технический тип, с чистотой 98%), 2,28 г гексагидрата нитрата цинка (аналитически чистый) и 1,10 г нитрата серебра (аналитически чистый) растворяли в воде, получая 156 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением 3 раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов. 5,4 г метавольфрамата аммония (аналитически чистый) затем растворяли в 52 мл воды, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-IV-10.
Пример IV-11
Порошок псевдобемита, полученный способом с применением сульфата алюминия (с удельной площадью поверхности 265 м2/г и объемом пор 0,81 мл/г, порошок псевдобемита, содержащий примесный элемент фосфора, с содержанием Р 3,1 г, на 100 г порошка псевдобемита, рассчитанного как Al2O3; в получении порошка псевдобемита, водорастворимое стекло (т.е. водный раствор силиката натрия), применяемое как SiO2 предшественник, добавляли в начале, так что масса SiO2, полученного из SiO2 предшественника, в кальцинированном носителе составляла 28% общей массы носителя) замешивали с разбавленным кислым водным раствором, содержащим 5 об. % азотную кислоту, формовали в гранулы клеверной формы диаметром 3 мм, сушили при 100°С в течение 8 часов, и кальцинировали при 1020°С в течение 5 часов для получения требуемого носителя.
141,1 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%), 2,28 г гексагидрата нитрата цинка (аналитически чистый) и 2,20 г нитрата серебра (аналитически чистый) растворяли в воде, получая 144 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением 3 раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, и затем кальцинировали при 400°С в течение 4 часов. 5,4 г метавольфрамата аммония (аналитически чистый) затем растворяли в 48 мл воды, раствор загружали на промежуточный продукт, полученный выше пропиткой распылением, сушили при 120°С в течение 4 часов, затем кальцинировали при 400°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 ч при 430°С, получая катализатор A-IV-11.
Пример IV-12
Катализатор получали, как описано в примере IV-1, за исключением того, что применяемый порошок псевдобемита имел содержание примесного элемента Р 1,33 г на 100 г порошка псевдобемита, рассчитанного как Al2O3, удельную площадь поверхности 375 м2/г и объем пор 0,98 мл/г, получая катализатор A-IV-12.
Сравнительный пример IV-1
Катализатор получали, как описано в примере IV-2, за исключением того, что применяемый порошок псевдобемита не содержал примесный элемент и имел удельную площадь поверхности 391 м2/г и объем пор 1,06 мл/г, и гексагидрат нитрата цинка отсутствовал в растворе, применяемом для пропитки распылением, получая катализатор D-IV-1.
Сравнительный пример IV-2
Катализатор получали, как описано в примере IV-8, за исключением того, что применяемый порошок псевдобемита не содержал примесный элемент и имел удельную площадь поверхности 349 м2/г и объем пор 1,17 мл/г, и тригидрат нитрата меди отсутствовал в растворе, применяемом для пропитки распылением, получая катализатор D-IV-2.
Сравнительный пример IV-3
Катализатор получали, как описано в примере IV-2, за исключением того, что применяемый порошок псевдобемита не содержал примесный элемент и имел удельную площадь поверхности 391 м2/г и объем пор 1,06 мл/г, и что гексагидрат нитрата цинка заменяли на 16,03 г гексагидрата нитрата магния (аналитически чистый), получая катализатор D-IV-3. Сравнительный пример IV-4
Катализатор получали, как описано в примере IV-2, за исключением того, что применяемый порошок псевдобемита не содержал примесный элемент и имел удельную площадь поверхности 391 м2/г и объем пор 1,06 мл/г, молибдат аммония не добавляли, и гексагидрат нитрата цинка заменяли на 8,84 г тетрагидрата нитрата кальция, получая катализатор D-IV-4.
Пример испытаний IV-1
Элементный состав носителей и катализаторов анализировали с помощью плазменно-эмиссионной спектроскопии, где содержание примесного элемента выражали по массе на 100 г матрикса, и содержание активного металлического компонента и металлического промотора выражали по массе на 100 г матрикса; полученные выше носители были охарактеризованы NH3-TPD, BET способами сорбции-десорбции азота, и результаты приведены в таблице IV-1.
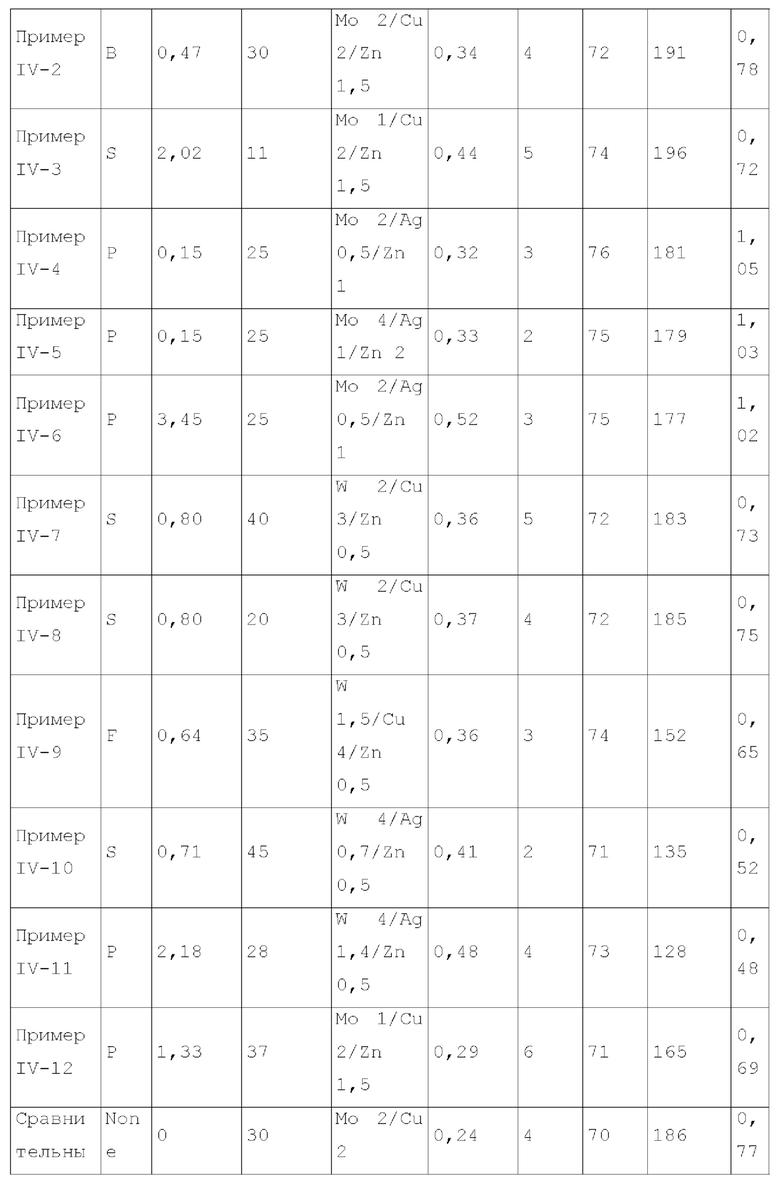
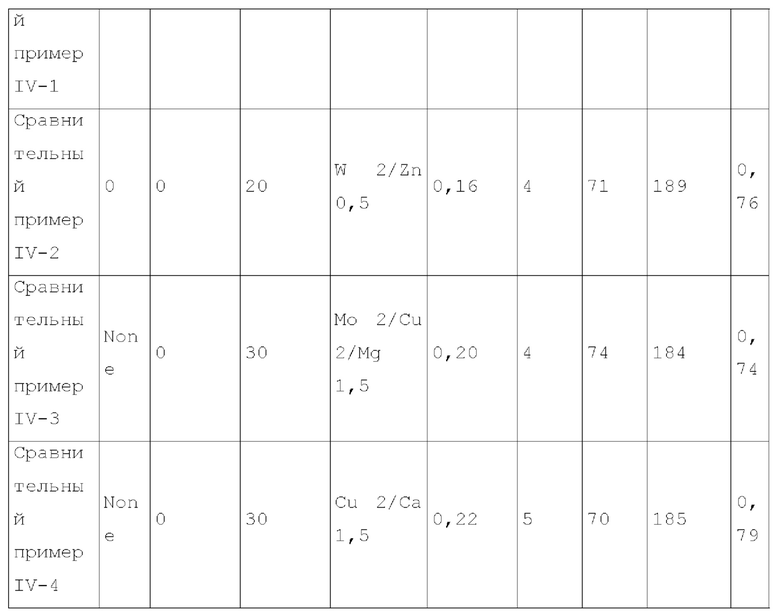
Пример испытаний IV-2
Данный пример испытаний иллюстрирует способ получения 1,6-гександиамина гидроаминированием 1,6-гександиола, применяя катализатор четвертого типа вариантов осуществления настоящего изобретения.
100 мл катализаторов, полученных в примерах и сравнительных примерах, соответственно отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 165°С, давление системы повышали до 11 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 125°С и направляется в верхний конец реактора, затем нагретый и расплавленный 1,6-гександиол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к 1,6-гександиолу составляло 2:12:1, объемная скорость жидкой фазы 1,6-гександиола составляла 0,35 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 170°С и давлении реакции 11 МПа, и после реакции в течение 20 ч, реакционный раствор отбирали и анализировали. Результаты анализа показаны в таблице IV-2.
Анализ образца проводился способом газовой хроматографии и калибровался с применением поправочного коэффициента полученного стандартного образца.
Конверсию и селективность рассчитывали, исходя из молярного содержания каждого компонента в реакционном растворе, и способы расчета были такими же, как описано в примере испытаний 1-2.
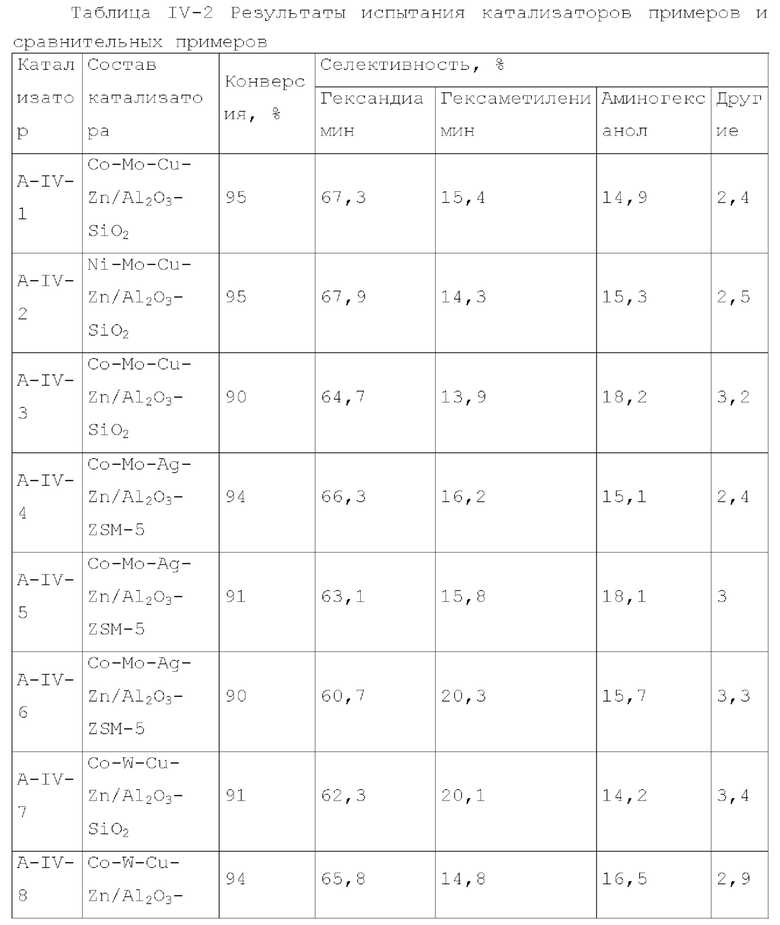
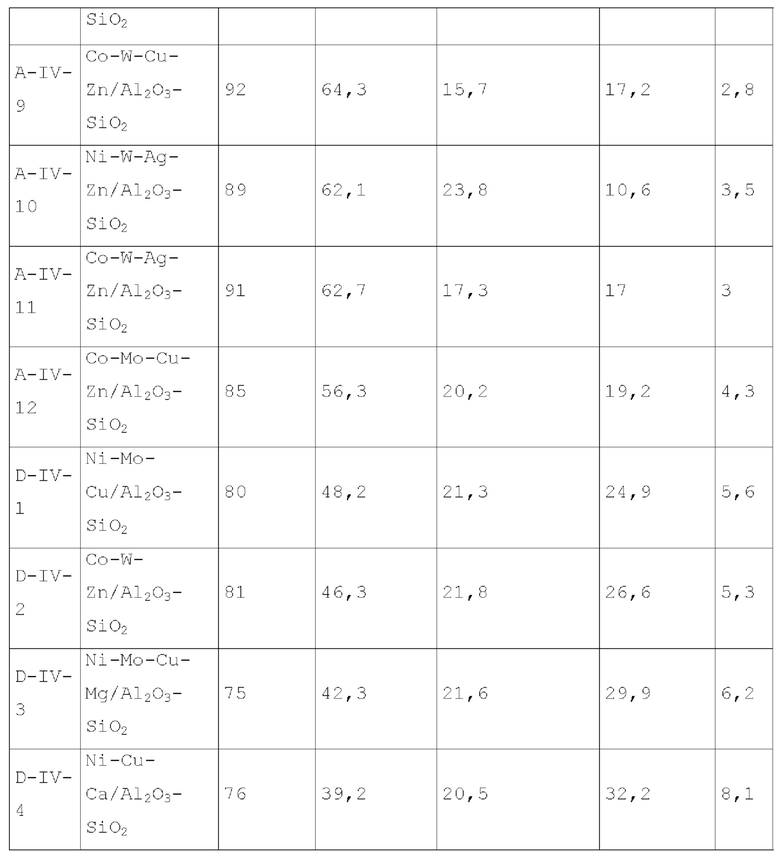
Пример испытаний IV-3
Данный пример испытаний иллюстрирует способ получения н-пропиламина гидроаминированием н-пропанола, применяя катализатор четвертого типа вариантов осуществления настоящего изобретения.
100 мл катализатора A-IV-3, полученного в примере IV-3, отмеряли, загружали в реактор с неподвижным слоем и активировали водородом при 220°С в течение 2 часов, затем охлаждали до 140°С, давление системы повышали до 2 МПа, применяя водород, аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 110°С и затем направляли в верхний конец реактора, н-пропанол подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к н-пропанолу составляло 3:6:1, объемная скорость жидкой фазы пропанола составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе, и после того, как реакция стала стабильной, реакционный раствор отбирали и анализировали (условия анализа, и способы расчета конверсии и селективности являются аналогичными способам примера испытаний IV-2). Результаты анализа показаны в таблице IV-3.

Пример испытаний IV-4
Катализаторы A-IV-1, A-IV-4, D-IV-1, D-IV-2 и D-IV-3 загружали в реакторы с неподвижным слоем, соответственно, в тех же условиях, что и в примере испытаний IV-2, с той лишь разницей, что время реакции увеличивали, и испытание проводили в течение 400 часов. Анализ и сравнение проводили на реакционном растворе после начальной стабилизации реакции, то есть при времени реакции 20 часов (условия анализа, способы расчета конверсии и селективности являются такими же, как в примере испытаний IV-2), и раствор реакции после 400 часов реакции (условия анализа, и способы расчета конверсии и селективности являются такими же, как в примере испытаний IV-2). Результаты анализа приведены в таблице IV-4.
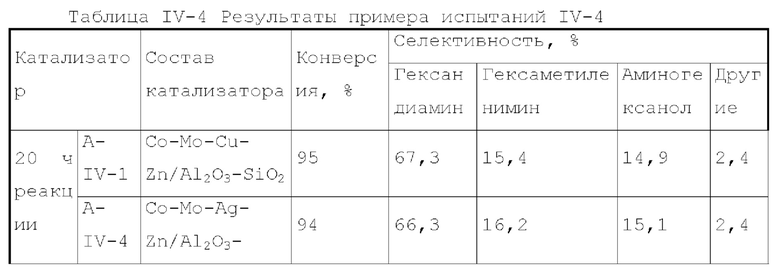
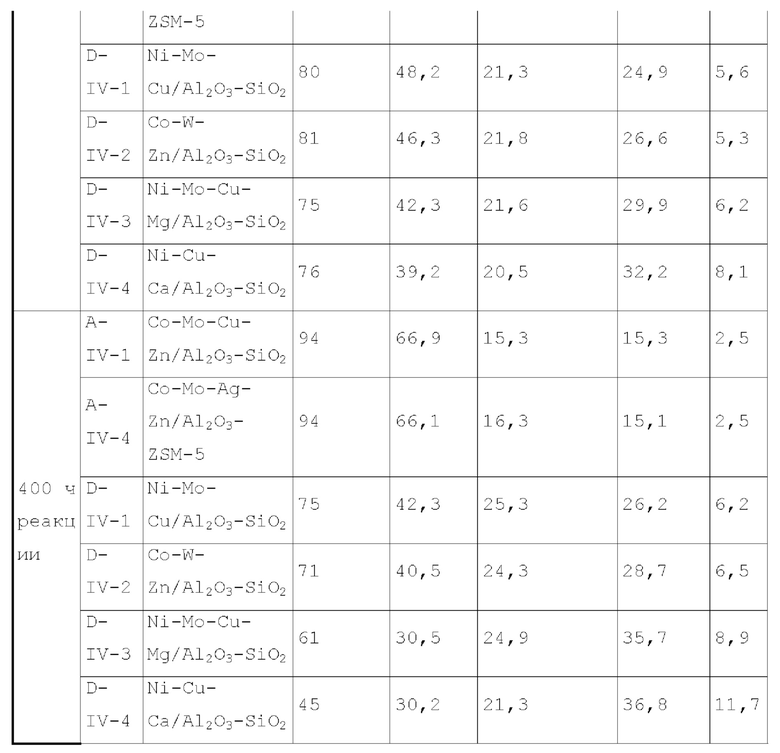
Как видно из вышеприведенной таблицы, после испытаний катализаторов A-IV-1 и A-IV-4 в течение 400 часов активность и селективность катализаторов существенно не снижались (а именно величина снижения меньше чем 2%), при этом активность и селективность катализаторов D-IV-1-D-IV-4 значительно снижались, и образуется больше побочных продуктов, что указывает на то, что катализатор настоящего изобретения обладает более высокой стабильностью.
Серия примеров V
Пятый тип вариантов осуществления настоящего изобретения далее подробно проиллюстрирован ниже со ссылкой на примеры серии примеров V. В следующих примерах серий примеров V применяемый порошок псевдобемита имел содержание (Al2O3) на сухое вещество 70 масс. %; порошок титановых белил получали гидролизом титанилсульфата аммиаком, с содержанием сульфата 2,5 масс. % и содержанием TiO2 95 масс. %, и он был коммерчески доступен от Shandong Dongjia Chemical Company, под торговым названием SA-100.
Пример V-1
820 г порошка псевдобемита (полученного способом с применением азотной кислоты, с удельной площадью поверхности 380 м2/г и объемом пор 0,96 мл/г) и 300 г порошка титановых белила однородно смешивали в смесителе, замешивали с 790 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты, формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 120°С в течение ночи и кальцинировали при 800°С в течение 4 часов для получения требуемого носителя.
126 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) растворяли в воде, получая 150 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-V-1.
Пример V-2
780 г порошка псевдобемита (полученного способом карбонизации, с удельной площадью поверхности 405 м2/г и объемом пор 1,01 мл/г) и 220 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 775 мл разбавленного кислого водного раствора, содержащего 7 об. % уксусную кислоту, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 120°С в течение ночи, и кальцинировали при 850°С в течение 3 часов для получения требуемого носителя.
140,4 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) растворяли в воде, получая 144 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 390°С в течение 4 часов, и затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение 3 часов, получая катализатор A-V-2.
Пример V-3
820 г порошка псевдобемита (полученного способом с применением сульфата алюминия, с удельной площадью поверхности 380 м2/г и объемом пор 1,26 мл/г) и 180 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 785 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты, формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение ночи и затем кальцинировали при 820°С в течение 3 часов для получения требуемого носителя.
134,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 43,4 г 50 масс. % раствора нитрата магния растворяли в воде, получая 155 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-V-3.
Пример V-4
1000 г порошка псевдобемита (полученного способом с применением азотной кислоты, с удельной площадью поверхности 380 м2/г и объемом пор 0,96 мл/г) и 200 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 785 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты, формовали в полоски с диаметром 5 мм, нарезали на длину 4 мм, сушили при 120°С в течение ночи, и затем кальцинировали при 800°С в течение 4 часов для получения требуемого носителя.
129,2 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 4,0 г нитрата серебра (аналитически чистый) растворяли в воде, получая 162 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-V-4.
Пример V-5
1500 г порошка псевдобемита (полученного способом карбонизации, с удельной площадью поверхности 405 м2/г и объемом пор 1,01 мл/г) и 250 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 777 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты, экструдировали в гранулы клеверной формы толщиной 3 мм, сушили при 120°С в течение ночи, и затем кальцинировали в течение 3,5 часов при 820°С для получения требуемого носителя.
132,6 г гексагидрата нитрата никеля (технический тип, с чистотой 98%) и 7,7 г перрената аммония растворяли в воде, получая 140 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 390°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 440°С в течение
3 часов, получая катализатор A-V-5.
Пример V-6
950 г порошка псевдобемита (полученного способом с применением сульфата алюминия, с удельной площадью поверхности 380 м2/г и объемом пор 0,96 мл/г) и 320 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 767 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты и 2 об. % серной кислоты, формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение ночи, и затем кальцинировали при 820°С в течение 5 ч для получения требуемого носителя.
182,6 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) растворяли в воде, получая 170 мл раствора, 16 г тетрагидрата молибдата аммония (аналитически чистый) растворяли в воде, получая 138 мл раствора, раствор нитрата кобальта загружали на 100 г носителя, полученного пропиткой распылением в два раза, затем раствор молибдата аммония загружали на носитель, полученный пропиткой распылением один раз, затем сушили в течение 4 часов при 120°С и кальцинировали в течение 4 часов при 400°С, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали в течение 3 часов при 430°С, получая катализатор A-V-6.
Пример V-7
1000 г порошка псевдобемита (полученный способом с применением сульфата алюминия, с удельной площадью поверхности 380 м2/г и объемом пор 0,96 мл/г) и 150 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 770 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты и 2 об. % серной кислоты, формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение ночи, и кальцинировали при 820°С в течение 3,5 часов для получения требуемого носителя.
177,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 15,9 г тригидрата нитрата меди (аналитически чистый) растворяли в воде, получая 140 мл раствора, раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-V-7.
Пример V-8
780 г порошка псевдобемита (полученного способом с применением сульфата алюминия, с удельной площадью поверхности 380 м2/г и объемом пор 0,96 мл/г) и 220 г порошка титановых белил равномерно смешивали в смесителе, замешивали с 782 мл разбавленного кислого водного раствора, содержащего 5 об. % азотной кислоты, 2 об. % фтороводородной кислоты и 0,5 масс. % лантана нитрата, формовали в зубчатые сферы диаметром 4 мм, сушили при 120°С в течение ночи, и кальцинировали в течение 4 часов при 820°С для получения требуемого носителя.
134,4 г гексагидрата нитрата кобальта (технический тип, с чистотой 98%) и 30,3 г гексагидрата нитрата цинка (аналитически чистый) растворяли в воде, получая 160 мл раствора, и раствор загружали на 100 г носителя, полученного пропиткой распылением в два раза, сушили при 120°С в течение 4 часов после каждого раза пропитки распылением, затем кальцинировали при 400°С в течение 4 часов, затем восстанавливали водородом, постепенно повышая температуру при скорости 20°С/ч, и наконец восстанавливали при 430°С в течение 3 часов, получая катализатор A-V-8.
Сравнительный пример V-1
Катализатор получали, как описано в примере V-3, за исключением того, что применяемое количество порошка псевдобемита составляло 500 г, и применяемое количество порошка титановых белил составляло 500 г, и 12,8 г KNO3 добавляли при замешивании, получая катализатор D-V-1.
Сравнительный пример V-2
Катализатор получали, как описано в примере V-2, за исключением того, что применяемое количество порошка псевдобемита составляло 900 г, и применяемое количество порошка титановых белил составляло 100 г, получая катализатор D-V-2.
Пример испытаний V-1
Элементный состав катализатора был проанализирован с помощью плазменно-эмиссионной спектроскопии, и результаты показали, что вся сера в порошке титановых белил сохраняется в носителе, и содержание других элементов является практически таким же, как и количество подаваемого сырья; катализаторы, полученные выше, были охарактеризованы NH3-TPD, C02-TPD, BET способам сорбции-десорбции азота, и результаты приведены в таблице V-1.
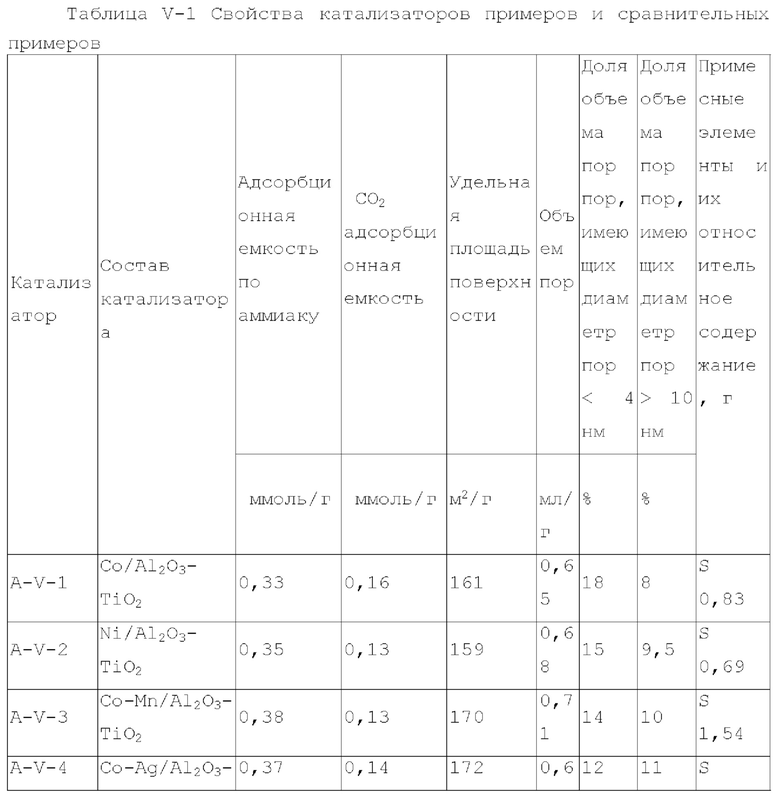
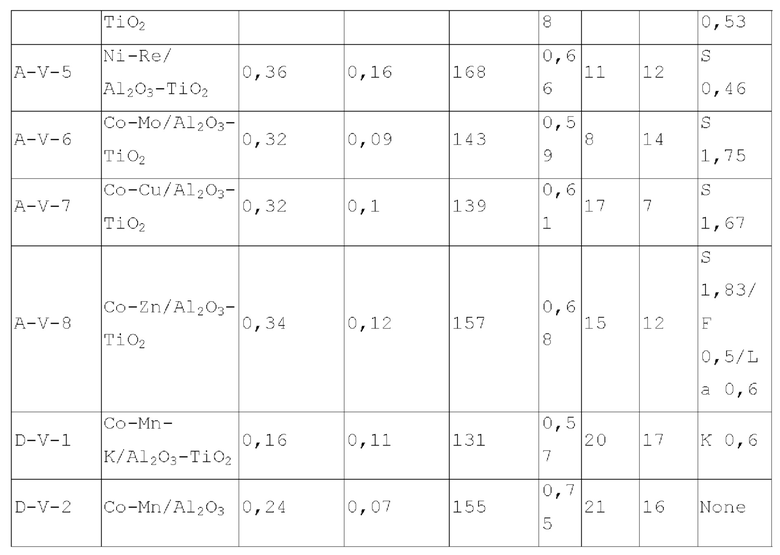
Пример испытаний V-2
100 мл полученных катализаторов A-V-1-A-V-8 и полученных катализаторов D-V-1 и D-V-2 соответственно отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 12,5 МПа, применяя водород, аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 150°С и подавали в реактор, нагретый и расплавленный 1,6-гександиол подавали в реактор дозируемым насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к 1,6-гександиолу составляло 3:12:1, и объемная скорость жидкой фазы 1,6-гександиола составляла 0,4 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 175°С и давлении реакции 12,5 МПа, и реакционный раствор отбирали и анализировали после стабилизации условий реакции в течение 2 часов. Результаты анализа показаны в таблице V-2.
Анализ образца проводился способом газовой хроматографии и калибровался с применением поправочного коэффициента полученного стандартного образца.
Конверсию и селективность рассчитывали, исходя из молярного содержания каждого компонента в реакционном растворе, и способы расчета были такими же, как описано в примере испытаний 1-2.
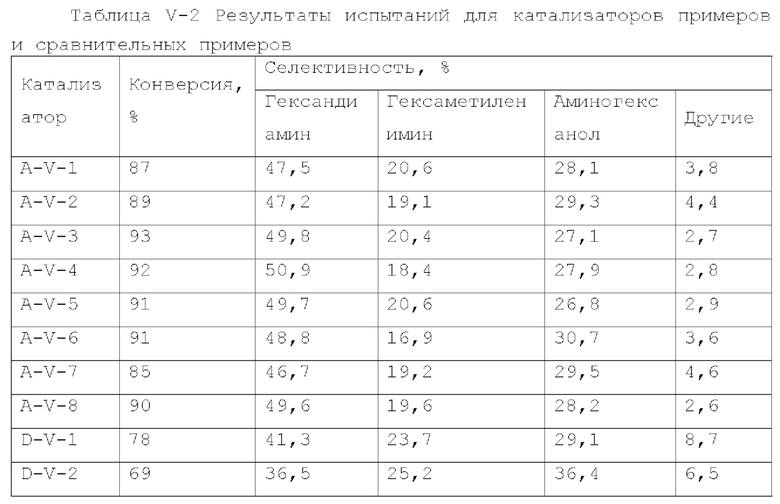
Как видно из данных таблицы выше, содержание «других», т.е. примесей, катализаторов D-V-1 и D-V-2 является значительно большим, и конверсия данных катализаторов является значительно большей, по сравнению с другими катализаторами.
После 196 часов оценки каждого катализатора катализатор выгружали для характеризации. Видно, что удельная площадь поверхности и объем пор катализаторов А-В-1-А-В-8 практически не изменились, и явного образования нагара не наблюдали, в то время как удельная площадь поверхности катализаторов D-V-1 и D-V-2 соответственно уменьшилась на 7% и 9%, объем пор соответственно уменьшился на 7%, и их степень образование нагара составляет соответственно 5,6 масс. % и 6,7 масс. %, что свидетельствует о том, что катализатор настоящего изобретения обладает более стабильными каталитическими характеристиками и может ускорять скорость реакции, уменьшать образование нагара и замедлять закупорку порового канала.
Пример испытаний V-3
100 мл полученных катализаторов отмеряли, загружали в реактор с неподвижным слоем, активировали водородом при 220°С в течение 2 часов, затем охлаждали до 168°С, давление системы повышали до 8 МПа, применяя водород, затем аммиак дозировали в реакционную систему с помощью дозирующего насоса, предварительно нагревали до 150°С и затем направляли в верхний конец реактора, смешанный раствор гександиола, гексаметиленимина и 6-амино-1-гексанола (50 масс. % гександиола, 32 масс. % гексаметиленимина и 18 масс. % 6-амино-1-гексанола) подавали в верхний конец реактора дозирующим насосом, водород стабильно вводили через массовый расходомер газа, где молярное отношение водорода к аммиаку и к смешанному раствору составляло 3:18:1, объемная скорость жидкой фазы смешанного раствора составляла 0,5 ч-1, каталитическую реакцию аминирования проводили в реакторе при температуре реакции 185°С и давлении реакции 8 МПа, и после стабилизации условий реакции в течение 2 часов, реакционный раствор отбирали и анализировали (условия анализа и способы расчета конверсии и селективности являются такими же, как в примере испытаний V-2). Результаты анализа показаны в таблице V-3.
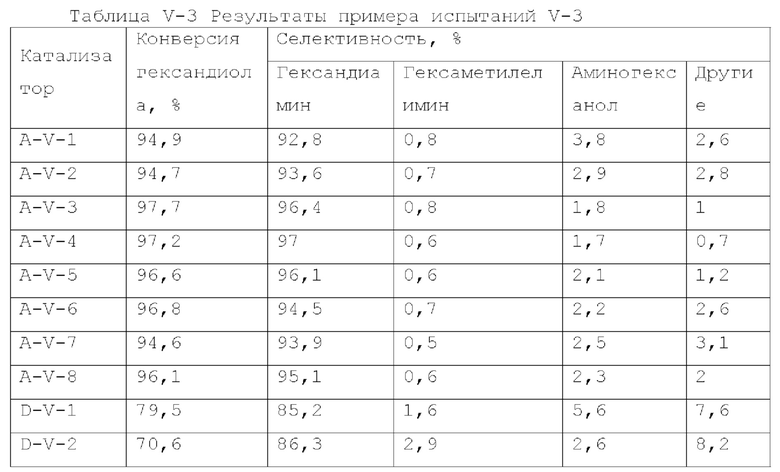
Как видно из данных таблицы выше, катализатор настоящего изобретения способен обеспечить более высокую конверсию гександиола и более высокую селективность гександиамина по сравнению с катализаторами D-V-1 и D-V-2.
Настоящее изобретение подробно проиллюстрирован выше со ссылкой на предпочтительные варианты осуществления, но это не предназначено для ограничения данными вариантами осуществления. В соответствии с изобретательским замыслом настоящего изобретения могут быть выполнены различные модификации, и данные модификации должны быть в пределах объема настоящего изобретения.
Следует отметить, что различные технические признаки, описанные в приведенных выше вариантах осуществления, можно комбинировать любым подходящим образом без противоречия, и во избежание ненужного повторения различные возможные комбинации не описывают в настоящем изобретении, но данные комбинации также должны находиться в рамках объема настоящего изобретения.
Кроме того, различные варианты осуществления настоящего изобретения могут быть произвольно объединены до тех пор, пока это сочетание не выходит за пределы сущности настоящего изобретения, и данные объединенные варианты осуществления следует рассматривать как описание настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОР АМИНИРОВАНИЯ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2837051C1 |
| Катализатор для обессеривания жидких нефтепродуктов, его получение и применение | 2017 |
|
RU2745372C2 |
| Материал-носитель из оксида алюминия и способ его получения, катализатор гидрирования и способ гидрирования остаточного масла | 2018 |
|
RU2753336C1 |
| КАТАЛИЗАТОР ГИДРИРОВАНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2021 |
|
RU2836692C1 |
| КОМПОЗИЦИЯ, СПОСОБНАЯ СНИЖАТЬ ВЫБРОСЫ CO И NOX, ЕЕ СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ И СПОСОБ КАТАЛИТИЧЕСКОГО КРЕКИНГА В ПСЕВДООЖИЖЕННОМ СЛОЕ | 2018 |
|
RU2772281C2 |
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ДЛЯ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2832734C1 |
| КАТАЛИЗАТОР РИФОРМИНГА НАФТЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2015 |
|
RU2693018C2 |
| СФЕРОПОДОБНЫЙ СУПЕРМАКРОПОРИСТЫЙ МЕЗОПОРИСТЫЙ МАТЕРИАЛ И СОДЕРЖАЩИЙ ЕГО ПОЛИОЛЕФИНОВЫЙ КАТАЛИЗАТОР | 2020 |
|
RU2829734C1 |
| Катализатор, способ его получения, применение и способ извлечения серы | 2017 |
|
RU2670606C9 |
| КАТАЛИЗАТОР КАТАЛИТИЧЕСКОГО КРЕКИНГА И ЕГО ПОЛУЧЕНИЕ | 2018 |
|
RU2755891C2 |
Изобретение относится к реакции аминирования, в частности к катализатору для получения органического амина каталитическим аминированием, его получению и применению. Катализатор, пригодный для получения органических аминов каталитическим аминированием, содержит неорганический пористый носитель, содержащий алюминий и/или кремний, и активный металлический компонент, нанесенный на носитель, где активный металлический компонент выбран из кобальта, никеля или их комбинации, где носитель имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено тестом NH3-TPD. Способ получения указанного катализатора включает стадии: 1) обеспечения неорганического пористого носителя, содержащего алюминий и/или кремний, который имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом; 2) загрузки активного металлического компонента на носитель и 3) проведения термической обработки материала, полученного на стадии 2) с получением катализатора, причем термическая обработка включает кальцинирование или комбинацию сушки и кальцинирования. Также описан способ получения органического амина. Технический результат - получение катализатора, демонстрирующего улучшенные характеристики, в частности улучшенную каталитическую активность, конверсию реакции, селективность продукта и/или стабильность катализатора, при его применении для получения органических аминов каталитическим аминированием. 3 н. и 12 з.п. ф-лы, 19 табл., 76 пр.
1. Катализатор, пригодный для получения органических аминов каталитическим аминированием, содержащий неорганический пористый носитель, содержащий алюминий и/или кремний, и активный металлический компонент, нанесенный на носитель, где активный металлический компонент выбран из кобальта, никеля или их комбинации, где носитель имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено тестом NH3-TPD.
2. Катализатор по п. 1, где носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г, как измерено тестом NH3-TPD.
3. Катализатор по п. 1, где носитель содержит матрикс и примесный элемент, причем матрикс содержит первый компонент носителя и необязательно второй компонент носителя, где первый компонент носителя выбирают из оксида алюминия, оксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита и диоксида титана, и
примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, и он не включает натрий или хлор, где металлический элемент выбирают из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA, металлических элементов ряда лантаноидов или их комбинации, и предпочтительно выбирают из кальция, магния, калия, висмута, стронция, бария, лантана или их комбинации; неметаллический элемент выбирают из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA, неметаллических элементов группы VIIA или их комбинации, предпочтительно выбирают из бора, фтора, фосфора, серы, селена или их комбинации;
предпочтительно, примесный элемент в носителе получен из катионов металлов и/или ионов кислотных радикалов, но не включает ион натрия или хлорид ион; катион металла выбирают из катионов металлов группы IA, ионов металлов группы IIA, ионов металлов группы VA, ионов металлов ряда лантаноидов или их комбинации, предпочтительно выбирают из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария, иона лантана или их комбинации; ион кислотного радикала выбирают из неметаллических ионов кислотных радикалов, предпочтительно выбирают из борат-иона, фторид-иона, фосфат-иона, сульфат-иона, селенат-иона или их комбинации.
4. Катализатор по п. 3, где носитель имеет по меньшей мере одну из следующих характеристик:
носитель имеет адсорбционную способность по углекислому газу 0,05-0,4 ммоль/г, предпочтительно 0,05-0,3 ммоль/г, более предпочтительно 0,06-0,2 ммоль/г;
примесный элемент присутствует в носителе в количестве 0,03-6 масс. %, предпочтительно 0,05-6 масс. %, более предпочтительно 0,08-4 масс. %, относительно общего веса матрикса;
носитель имеет удельную площадь поверхности 120-240 м2/г, предпочтительно 120-210 м2/г и более предпочтительно 125-200 м2/г;
носитель имеет объем пор 0,45-1,2 мл/г, предпочтительно 0,45-1,1 мл/г и более предпочтительно 0,5-1 мл/г;
доля объема пор, имеющих диаметр пор в диапазоне 7-27 нм, к объему пор носителя является большей чем 65%, предпочтительно 70% или более, более предпочтительно 70-90%, и предпочтительно доля объема пор, имеющих диаметр пор меньше чем 7 нм, к объему пор носителя составляет 0-10%, например 0-8%;
матрикс в носителе содержит комбинацию оксида алюминия и диоксида титана при весовом соотношении 1,5-5:1, предпочтительно 2-4,5:1; и
содержание оксида алюминия в носителе составляет 70 масс. % или более, предпочтительно 75 масс. % или более, более предпочтительно 80-100 масс. %, исходя из общего веса матрикса.
5. Катализатор по любому из пп. 1-4, где активный металлический компонент присутствует в количестве 5-46 г, предпочтительно 10-42 г, например 13-40 г, на 100 г матрикса.
6. Катализатор по любому из пп. 1-5, дополнительно содержащий металлический промотор, нанесенный на носитель, и металлический промотор содержит по меньшей мере один металл, выбранный из группы, состоящей из металлов группы VIB, группы VIIB, группы IB, группы IIB и металлов ряда лантаноидов, предпочтительно по меньшей мере один металл, выбранный из Cr, Mo, W, Mn, Re, Cu, Ag, Au, Zn, La и Се;
предпочтительно, металлический промотор присутствует в количестве 0-10 г, предпочтительно 0,1-10 г, более предпочтительно 0,5-8 г, на 100 г матрикса.
7. Катализатор по п. 6, где:
металлический промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IB, где весовое отношение металла группы VIIB к металлу группы IB, в расчете на металлический элемент, составляет 0,05-15:1, предпочтительно 0,1-12:1; или
металлический промотор содержит комбинацию по меньшей мере одного металла группы VIIB и по меньшей мере одного металла группы IIB, где весовое отношение металла группы VIIB к металлу группы IIB, в расчете на металлический элемент, составляет 0,2-20:1, предпочтительно 0,3-6:1; или
металлический промотор содержит комбинацию по меньшей мере одного металла группы VIB, по меньшей мере одного металла группы IB и по меньшей мере одного металла группы IIB, где весовое отношение металла группы VIB к металлу группы IB и к металлу группы IIB, в расчете на металлический элемент, составляет 0,1-10:0,1-10:1, предпочтительно 0,2-8:0,2-8:1,
предпочтительно, металл группы VIIB выбирают из марганца, рения или их комбинации, металл группы IB выбирают из меди, серебра, золота или их комбинации, металл группы IIB представляет собой цинк, и металл группы VIB выбирают из молибдена, вольфрама или их комбинации.
8. Способ получения катализатора по любому из пп. 1-7, включающий стадии:
1) обеспечения неорганического пористого носителя, содержащего алюминий и/или кремний, который имеет адсорбционную емкость по аммиаку 0,25-0,65 ммоль/г, как измерено NH3-TPD тестом;
2) загрузки активного металлического компонента на носитель; и
3) проведения термической обработки материала, полученного на стадии 2) с получением катализатора,
причем термическая обработка включает кальцинирование или комбинацию сушки и кальцинирования.
9. Способ по п. 8, где носитель имеет адсорбционную емкость по аммиаку 0,3-0,6 ммоль/г, как измерено тестом NH3-TPD, или
стадия 2) включает загрузку на носитель активного металлического компонента и дополнительно металлического промотора; или
стадия 3) включает проведение термической обработки и дополнительно восстановительной обработки материала, полученного на стадии 2).
10. Способ по п. 8, где указанное "обеспечение неорганического пористого носителя, содержащего алюминий и/или кремний" на стадии 1) включает применение смеси, содержащей примесный элемент и матрикс или его предшественник, в последовательно осуществляемых формовании, сушке и кальцинировании, с получением носителя,
где матрикс содержит первый компонент носителя и необязательно второй компонент носителя, причем первый компонент носителя выбирают из оксида алюминия, диоксида кремния, молекулярных сит, алюмосиликатов или их комбинаций, второй компонент носителя выбирают из диатомита, порошка титановых белил или их комбинации, предпочтительно, первый компонент носителя представляет собой оксид алюминия, и предшественник первого компонента носителя представляет собой псевдобемит, имеющий удельную площадь поверхности 250-410 м2/г, предпочтительно 260-400 м2/г, более предпочтительно 260-380 м2/г, и объем пор 0,7-1,3 мл/г, предпочтительно 0,7-1,2 мл/г, более предпочтительно 0,8-1,2 мл/г;
примесный элемент выбирают из металлического элемента, неметаллического элемента или их комбинации, и он не включает натрий или хлор, где металлический элемент выбирают из металлических элементов группы IA, металлических элементов группы IIA, металлических элементов группы VA, металлических элементов ряда лантаноидов или их комбинации, и предпочтительно выбирают из кальция, магния, калия, висмута, стронция, бария, лантана или их комбинации; неметаллический элемент выбирают из неметаллических элементов группы IIIA, неметаллических элементов группы VA, неметаллических элементов группы VIA, неметаллических элементов группы VIIA или их комбинации, предпочтительно выбирают из бора, фтора, фосфора, серы, селена или их комбинации,
предпочтительно, условия сушки на стадии 1) включают: температуру 80-150°С и продолжительность сушки 6-20 ч; и
предпочтительно, условия кальцинирования стадии 1) включают: температуру 500-1100°С и продолжительность кальцинирования 2-20 ч.
11. Способ по п. 9, где примесный элемент обеспечивают, применяя модификатор носителя, содержащий по меньшей мере одно соединение, способное обеспечивать катион и/или анион, где катион выбирают из катионов группы IA, ионов металлов группы IIA, ионов металлов группы VA, ионов металлов ряда лантаноидов или их комбинации, предпочтительно из иона кальция, иона магния, иона калия, иона висмута, иона стронция, иона бария, иона лантана или их комбинации;
анион выбирают из неметаллических ионов кислотных радикалов, предпочтительно выбирают из борат-иона, фторид-иона, фосфат-иона, сульфат-иона, селенат-иона или их комбинации;
предпочтительно, модификатор носителя выбирают из борной кислоты, бората никеля, бората кобальта, бората калия, фтороводородной кислоты, фторида калия, фторида кобальта, фторида никеля, фосфорной кислоты, фосфата алюминия, фосфата трикалия, дигидрофосфата калия, гидрофосфата калия, фосфата магния, фосфата кальция, серной кислоты, сульфата кобальта, сульфата никеля, сульфата алюминия, сульфата кальция, нитрата висмута, нитрата калия, сульфата калия, карбоната калия, нитрата магния, сульфата магния, основного карбоната магния, нитрата кальция, основного карбоната кальция, нитрата стронция, фосфата стронция, сульфата стронция, нитрата бария, нитрата лантана, селеновой кислоты или их комбинации.
12. Способ по любому из пп. 8-11, где загрузка на стадии 2) включает пропитку носителя раствором, содержащим предшественник активного металлического компонента и необязательно предшественник металлического промотора.
13. Способ получения органического амина, включающий: осуществление контакта исходного материала для аминирования и реагента для аминирования с катализатором по любому из пп. 1-7 в присутствии водорода для проведения реакции аминирования с получением органического амина,
где исходный материал для аминирования выбирают из этанола, ацетальдегида, н-пропанола, пропиональдегида, изопропанола, н-бутанола, бутиральдегида, изобутанола, изобутиральдегида, 2-этилгексанола, 2-этилгексальдегида, октанола, октаналя, додеканола, додеканаля, гексадеканола, гексадеканаля, циклопентанола, циклогексанола, циклооктанола, циклододеканола, бензилового спирта, бензальдегида, фенэтилового спирта, фенилацетальдегида, 1,4-бутандиола, 1,4-бутандиаля, 1,5-пентандиола, 1,5-глутаральдегида, 1,6-гександиола, 1,6-гександиаля, 1,8-октандиола, 1,8-октандиаля, 1,12-додекандиола, 1,12-додекандиальдегида, этаноламина, пропаноламина, изопропаноламина, 6-аминогексанола, диэтаноламина, диизопропаноламина, диметилэтаноламина, ацетона, этиленгликоля, 1,3-пропандиола или их комбинации;
реагент для аминирования выбирают из аммиака, С1-12 первичных аминов, С2-12 вторичных аминов или их комбинации.
14. Способ по п. 13, где условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-5:2-35:1, предпочтительно 1-5:2-33:1, более предпочтительно 1-5:2-30:1, температуру 105-220°С, предпочтительно 110-220°С, более предпочтительно 130-200°С, давление 0,7-25 МПа, предпочтительно 0,8-25 МПа, более предпочтительно 1-15 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-1 м3/(м3⋅ч).
15. Способ по п. 14, где:
когда исходный материал для аминирования представляет собой одноатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-10:1, предпочтительно 1-4:2-9:1, более предпочтительно 1-4:2-8:1, температуру 130-210°С, предпочтительно 130-200°С, давление 1-4 МПа, предпочтительно 1-3,5 МПа, более предпочтительно 1-2,5 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-0,8 м3/ (м3⋅ч);
когда исходный материал для аминирования представляет собой кетон или альдегид, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:2-6:1, предпочтительно 1-4:2-5:1, температуру 105-180°С, предпочтительно 110-180°С, более предпочтительно 110-170°С, давление 0,7-3,5 МПа, предпочтительно 0,7-2,5 МПа, более предпочтительно 0,8-2,5 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,1-1 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/(м3⋅ч);
когда исходный материал для аминирования представляет собой аминоспирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 1-4:3-25:1, предпочтительно 1-4:3-20:1, температуру 130-200°С, предпочтительно 135-200°С, давление 1-18 МПа, предпочтительно 1-15 МПа, более предпочтительно 1-11 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч);
когда исходный материал для аминирования представляет собой двухатомный спирт, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 1-4:3-33:1, температуру 130-220°С, предпочтительно 130-210°С, давление 1-15 МПа, предпочтительно 4-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/ (м3⋅ч), или
когда исходный материал для аминирования представляет собой смесь 1,6-гександиола, гексаметиленимина и 6-амино-1-гексанола, условия аминирования включают: молярное отношение водорода к реагенту для аминирования и к исходному материалу для аминирования 0,3-4:3-45:1, предпочтительно 1-4:3-35:1, более предпочтительно 1-4:3-33:1, более предпочтительно 1-4:3-30:1, температуру 130-220°С, предпочтительно 130-210°С, давление 1-25 МПа, предпочтительно 2-25 МПа, более предпочтительно 4-25 МПа, и объемную скорость жидкой фазы исходного материала для аминирования 0,06-0,8 м3/(м3⋅ч), предпочтительно 0,1-0,8 м3/ (м3⋅ч).
| Модификатор для флотации несуль-фидНыХ Руд | 1979 |
|
SU839574A1 |
| Собиратель для флотационного извле-чЕНия глиНиСТыХ шлАМОВ из КАлийСОдЕР-жАщиХ Руд | 1979 |
|
SU839575A1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОВ ИЗ ГЛИЦЕРИНА | 2008 |
|
RU2480449C2 |
| СПОСОБ АМИНИРОВАНИЯ | 1998 |
|
RU2215734C2 |
| УСОВЕРШЕНСТВОВАННЫЕ ЦЕОЛИТЫ И МОЛЕКУЛЯРНЫЕ СИТА И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2278818C2 |
| ИЗВЛЕЧЕНИЕ АММИАКА ИЗ ПРОДУВОЧНОГО ГАЗА | 2003 |
|
RU2314255C2 |
| Способ улавливания аммиака | 1990 |
|
SU1787505A1 |

