Настоящее изобретение направлено на способ получения цеолитов и цеолитоподобных структур, допированных металлами. Конкретно, в описанном способе используют стадию сублимации с целью введение металла в каналы/полости цеолитного материала. Таким образом, в соответствии с такой сухой методикой исключено использование растворителя, что позволяет устранить определенные недостатки методик ионного обмена во влажном состоянии, пропитки или других способов добавления металлов, известных в данной области техники.
Допированные металлами цеолиты или цеолитоподобные структуры и их применение, конкретно, при каталитическом превращении оксидов азота, например, содержащихся в отходах или отработавших газах, известны в данной области техники. В допированные цеолиты и цеолитоподобные структуры введен, по меньшей мере, один металлический каталитически активный компонент. Каталитически активный металлический компонент, как правило, представляет собой переходный металл, конкретно, каталитически активный металл, например, медь или железо, и т.д. Такие допированные металлами цеолиты и цеолитоподобные структуры применяют, конкретно, либо в чистом виде, либо в качестве составляющего элемента покрытия каталитически активных структур.
Способ добавления/обмена металла представляет собой ключевую стадию превращения «белого» цеолита или цеолитоподобной структуры в активную форму для требуемого катализатора, например, предназначенного для облегчения селективного каталитического восстановления (СКВ) оксида азота и диоксида азота (NO и NO2 соответственно, которые далее в настоящем описании называют оксидами азота или NOx) с помощью мочевины/NH3 (или аналогичного восстановителя на основе азота) в выпускном тракте транспортного средства. Таким образом, получение цеолитов и цеолитоподобных структур, легированных металлом путем допирования/обмена, очень важно в академическом и коммерческом отношении, о чем свидетельствует большое количество патентов и общедоступных изданий на эту тему. Разнообразные способы получения допированных металлами цеолитов можно разделить на несколько классов.
Во-первых, существует «истинный» ионный обмен, включающий обработку щелочного, щелочноземельного или аммониевого цеолита/цеолитоподобной структуры в буферном растворе соли подходящего металла, возможно, при повышенной температуре, с целью замещения путем ионного обмена катиона (Na+, K+, 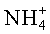
Во-вторых, введение металла можно осуществить путем пропитки в водной среде или в суспензии протонированной или аммониевой формы цеолита/цеолитоподобной структуры соответствующим предшественником с последующим прокаливанием при высокой температуре. Этот способ иногда также называют ионным обменом, но это, в строгом смысле, некорректно, поскольку ионный обмен происходит только в ходе прокаливания после разложения предшественника, что приводит к образованию подвижных ионов. Таким образом, этот способ может иметь меньшую эффективность, чем «истинный» ионный обмен, и может привести к наличию индукционной фазы при работе катализатора, поскольку материал мог провести недостаточное время при повышенной температуре для достижения эффективного замещения протонов в составе структуры ионами металлов. Примеры такого подхода описаны в патентах US 5908806, US 5116586, US 5270024, US 5271913, US 5516497, US 5776423, Lee и др. App Cat B Env 5 (1994) c.7-21 и Sueto и др., а также в J. Chem. Soc. Faraday Trans. 93 (4) 1997 c.659-664.
Металлические частицы можно также ввести непосредственно в каркасную структуру синтетическим способом, например, как это происходит при синтезе Cu-ALPO-34 (Me-ALPO означает допированный металлом алюмофосфат), Cu-APSO-34 (Me-APSO означает допированный металлом кремнийалюмофосфат) и других соответствующих систем Me-ALPO и Me-APSO, что описано в патенте EP 1142833 B1, Frache и др., в Cat Today 75 (2002) с.359-365 и Palella и др. в J Catal 217 (2003) с.100-106.
В качестве альтернативы, металл можно ввести в цеолит/цеолитоподобную структуру путем ионного обмена в твердом состоянии (ИОТ). Такое введение металла осуществляют по реакции в гомогенной смеси цеолита/цеолитоподобной структуры и подходящего летучего при высокой температуре предшественника, например, фторида, хлорида и других солей металлов при температуре от 400 до 800°C, в зависимости от конкретного предшественника. Примеры и более подробное описание такого способа приведены в патентах US 5434114, US 5545784, Beyer и др., Zeolites 8 (1988) с.79-81, Weckhuysen и др., J Catal 175 (1988) с.338-346, и Brandenberger и др. в Cat Rev 50 (4) (2008) с.492-531.
Модификацию металлом можно также осуществить с помощью химического осаждения паров (ХОП). В таком способе цеолит «дегазируют» при температуре более 500°C при пониженном давлении с целью удаления адсорбированных веществ, например, воды, а затем при комнатной температуре подвергают воздействию насыщенных паров летучего предшественника металла, который также обычно представляет собой летучий галогенид или фторуглеродную соль металла. Этому подходу посвящены патент US 6043177, статья Chen и Sachtler в Cat Today 42 (1998) с.73-83, а также статья Kuroda и др. в Chem Comm 22 (1997) с.2241-2242. Дополнительный и, в некотором смысле, родственный способ введения металла описан в WO 2008/009453 A2 (или EP 0955080 B1), в котором «дегазированный» аммониевый цеолит/цеолитоподобную структуру гомогенизировали совместно с каталитически активным металлом и прокаливали при температуре от 400 до 600°C в течение времени от 10 до 16 ч при пониженном давлении в присутствии азотсодержащего соединения, например, соли аммония. Заявлено, что, поскольку допирование протекает в качестве части ионно-обменной реакции в твердом состоянии в защитной атмосфере, например, в атмосфере NH3 или N2, такие анаэробные условия прокаливания обеспечивают относительно долговременную устойчивость допированного металлом цеолита. Аналогичный способ описан в патенте US 2010075834, в котором описан способ получения ионообменных цеолитов, включающий следующие стадии:
I) обеспечение сухой смеси
А) цеолита
Б) соединения каталитически активного металла,
II) тщательное измельчение смеси,
III) нагревание смеси в реакторе до определенной температуры,
IV) поддержание смеси при определенной температуре,
V) охлаждение до комнатной температуры и получение допированного металлом цеолита.
Особенность такого способа состоит в том, что внутреннее давление в реакторе в ходе нагревания поддерживают в диапазоне от 0 до 200 мбар. Соответственно, каталитически активный металл, предпочтительно, выбирают из группы, включающей Cu, Co, Rh, Pd, Ir, Pt, Ru, Fe, Ni, V. Каталитически активный металл применяют в форме соли, например, нитрата, сульфата, сульфита, гидроксида, нитрита и т.д., либо в форме комплексного соединения.
Однако, несмотря на такой большой объем работ, опубликованных на данный момент, сохраняется значительная потребность в отношении разработки простого, надежного и дешевого способа получения металлсодержащих цеолитов/цеолитоподобных структур. Таким образом, хотя формальный ионный обмен высокоэффективен для получения активных катализаторов с высокой дисперсностью металла, такой процесс включает большое количество стадий, некоторые из которых медленны и дороги для коммерческого применения, а также могут потребовать осуществления нескольких циклов для обеспечения оптимального содержания металла; например, в патенте US 6221324 описано получение Na-Y фожазита, допированного Cu/Ca путем ионного обмена, в этом способе каждая последующая стадия ионного обмена (на Ca, а затем на Cu) требует перемешивания суспензии соли и цеолита в течение 24 ч. Аналогично, патент US 7049261 обеспечивает способ получения катализатора Cu-ZSM5, содержащего 2,9 мас.% Ca, предполагающий трехкратное повторение стадии ионного обмена при перемешивании длительностью 24 ч с последующим фильтрованием, промыванием и сушкой при 110°C в течение 12 ч. Наконец, материал прокаливают в течение 5 ч при 500°C, что приводит к общей продолжительности синтеза, составляющей почти 5 дней. Следует также отметить, что ионный обмен в цеолитах может быть осложнен, поскольку некоторые вещества, например, соли железа, образуют большие гидратные оболочки, что затрудняет или даже препятствует переносу ионов железа на цеолит. Еще одна проблема заключается в большом объеме потока отходов процесса ионного обмена, содержащих смесь щелочных и щелочноземельных металлов, гидроксида аммония и нитратов, образовавшихся в ходе многократных стадий промывки, и эти отходы требуют дорогостоящей обработки перед тем, как их можно будет безопасно выпустить окружающую среду.
Некоторые из этих проблем облегчаются с применением способа пропитки/прокаливания, но, как указано выше, получаемые материалы обладают худшими характеристиками в свежем виде в силу пониженной эффективности начальной стадии ионного обмена. Кроме того, применение в промышленном масштабе требует осуществления такого процесса посредством суспензионного осаждения, в котором предшественник, например, Cu(NO3)2·3H2O (или его раствор) смешивают с суспензией, содержащей белый цеолит/цеолитоподобную структуру. Однако растворение предшественника приводит к образованию кислоты в суспензии, что приводит к необходимости добавления основания (то есть вещества с pH более 7) с целью нейтрализации кислоты до того, как цеолит/цеолитоподобная структура сможет быть повреждена химическим воздействием, а также с целью инициирования осаждения растворимых металлических структур, что отмечено выше. К выбору применяемого основания необходимо подходить с осторожностью, с целью ограничения комплексообразования с участием иона металла и основания, поскольку это может снизить способность иона проникать в структуру пор/каналов каркасной структуры. Более того, само основание может воздействовать/повреждать цеолитный материал, что создает дополнительную проблему при использовании традиционного влажного способа. Для устранения таких проблем можно попытаться применять органические основания, например, гидроксид тэтраэтиламмония (TEAH), что описано, например, в патенте US 5908806. Однако в ходе прокаливания такие органические вещества могут подвергнуться неполному сгоранию, то есть они скорее разлагаются в среде с ограниченным содержанием O2 с образованием вредных побочных продуктов, что, в свою очередь, снижает производительность процесса и требует осуществления дополнительных стадий в ходе прокаливания. В коммерческом масштабе это оказывало бы влияние на стоимость способа (дополнительные скрубберы и/или замедленный процесс прокаливания со сниженной загрузкой материала с целью снижения вредных выбросов). Кроме того, известно, что допированные металлами цеолиты или цеолитоподобные структуры, получаемые пропиткой/прокаливанием, могут иметь ограниченную устойчивость. Конкретно, наблюдается нарушение структуры и дезактивация при температуре более 800°C, вызванные дестабилизацией каркаса в результате кислотно-основных химических взаимодействий в суспензии. Такая дополнительная нестабильность ставит под вопрос коммерческое внедрение технологии СКВ, поскольку такие высокие температуры встречаются в ходе регенерации дизельных фильтров.
Последняя проблема возникает в ходе процессов ионного обмена и пропитки при введении в цеолиты или цеолитоподобные структуры каталитически активных компонентов, которые имеют различные устойчивые степени окисления, например, железа, ванадия или меди. Следовательно, в ходе ионного обмена в водной среде каталитически активные частицы могут быть дополнительно окислены до достижения более устойчивой с точки зрения термодинамики, но менее эффективной с точки зрения катализа степени окисления.
Использование цеолитоподобных структур с допированным каркасом, полученных путем прямого синтеза, устраняет указанные выше проблемы. Однако было обнаружено, что такие материалы представляют проблемы в отношении гидротермической устойчивости, что отражается в пониженной степени кристалличности и площади поверхности после состаривания, а также пониженной активности Cu в каркасе, по сравнению с Cu в традиционных ионообменных положениях (Frache и др. Cat Today 75 (2002) с.359-365 и Palella и др. J Catal 217 (2003) с.100-106). Более того, увеличенная сложность синтеза связана с проблемами в отношении воспроизводимости концентраций допирующего вещества и положения его атомов в каркасе, а оба этих фактора влияют на характеристики катализатора.
Проблемы, характерные для НОТ, ХОП и связанных способов, в широком смысле аналогичны, и их можно обобщить следующим образом. Во-первых, все эти способы связаны с выбросами опасных и, в некоторых случаях, едких и/или токсичных побочных продуктов, например, HF, HCl и т.д. С учетом того, что такие выбросы происходят при температурах от 400 до 800°C, потенциально в присутствии остатков влаги образуется опасная среда как для людей, так и для целостности структуры каркаса. Во-вторых, требуемые температуры могут превышать предельно допустимые для термический стабильности каркаса, что, по меньшей мере, приведет к спеканию (росту кристаллов), а это может отрицательно сказаться на характеристиках получаемого катализатора в результате увеличения среднего пути для свободной диффузии. В-третьих, может наблюдаться избыточное остаточное загрязнение цеолита/цеолитоподобной структуры щелочными или другими металлами. Такое загрязнение вызвано избыточным ионным обменом металла со слабокислотными центрами, связанными с дефектами каркаса. Такие загрязнения можно удалить путем промывания, но только с последующей обработкой щелочными материалами, например, NaOH, что может привести к дополнительному отрицательному воздействию на каркас. Кроме того, необходимость использования пониженного давления перед и/или в ходе допирования металлом создает технические и экономические сложности при увеличении масштаба процесса. Далее, такие процессы, как описанные выше, могут привести к невыгодному неоднородному распределению металла. Наконец, в случае допированных железом цеолитов, каталитически активные ионы Fe2+ при высоких температурах могут быть дополнительно окислены до неактивных ионов Fe3+.
Указано, что ключевой способ применения допированных металлами цеолитов/цеолитоподобных структур заключается в нейтрализации NOx путем селективного каталитического восстановления (СКВ) с использованием азотсодержащего восстановителя. Оксиды азота являются хорошо известными токсичными побочными продуктами работы двигателей внутреннего сгорания (ДВС), электрогенераторов, работающих на ископаемом топливе, а также промышленных процессов. NO образуется по реакциям свободных радикалов в процессе сжигания (см. И.Б. Зельдович, Acta Physico-chern. USSR, 21 (1946) 577), то есть:
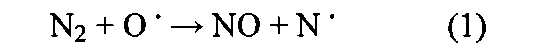
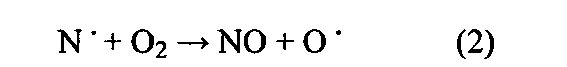
NOx токсичны для живых существ (P.E. Morrow J. Toxicol Environ Health 13(2-3), (1984), 205-27) и вносят вклад в действие ряда источников загрязнения, например, кислотных дождей, фотохимического смога и озона, которые отрицательно воздействуют на здоровье людей (M.V. Twigg, Applied Catalysis B, vol.70, (2007), 2). Таким образом, для регулирования выбросов NOx вводятся строгие ограничения, например, ЕВРО 5 и ЕВРО 6 [директива (EC) №715/2007 Европейского парламента, 20 июня 2007, Official Journal of the European Union L 171/1, Twigg, Applied Catalysis B, т.70, (2007), с.2-25 и R.M. Heck, R.J. Farrauto Applied Catalysis A т.221, (2001), c.43-457, а приведенные также в этих изданиях ссылки].
Регулирование выбросов NOx стехиометрическими бензиновыми двигателями осуществляют с помощью трехкомпонентных каталитических нейтрализаторов (см. SAE 2005-01-1111). Однако, трехкомпонентные катализаторы эффективны только при стехиометрических соотношениях воздух : топливо, и неэффективны для дизельных или других работающих при недостатке топлива, то есть при избытке кислорода, циклов сжигания, например, для двигателей с прямым впрыском топлива с обедненной смесью. Таким образом, преимущества дизельных двигателей в отношении ресурса, высокого крутящего момента при низких оборотах коленчатого вала и повышенная топливная экономичность/сниженные выбросы CO2 и УВ (углеводородов) также обеспечивают простор для достижений в отношении снижения выбросов NOx. Для удовлетворения требований к таким выбросам было разработано несколько технологий дообработки выхлопных газов, например, СКВ с использованием мочевины/NH3.
Химия СКВ включает сложный набор реакций разложения (реакция 3 для подачи мочевины) и окисления-восстановления (реакции с 4 по 9), включающих различные промежуточные продукты и реакции, которые формируют базу для академических и опытных исследований, таких как Fritz и Pitchon Арр Cat B 13 (1997) 1-25, Kondratenko и др. Арр Cat B 84 (2008) 497-504, Brüggemann и Keil J. Phys. Chem. C (2009), 113, 13930, SAE 2008-01-1184, SAE 2008-01-1323 и другие. Такие реакции обобщены уравнениями с 3 по 9. Уравнения с 4 по 6 подробно описывают желаемую химию СКВ. Однако могут протекать конкурентные процессы, например, паразитное окисление NH3 (реакции с 7 по 9). Это может привести к образованию N2, H2O и N2O, являющегося мощным парниковым газом (примерно в 300 раз сильнее, чем CO2), либо даже дополнительного количества NOx.
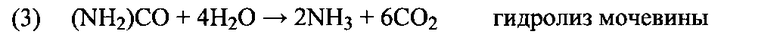

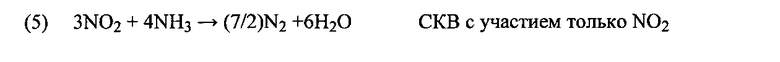




Основная реакция представлена уравнением (4). Однако было продемонстрировано, что при условиях практического применения реакция с участием смесей NO/NO2 в пропорции 50:50 приводит к наивысшей скорости превращения NOx (6) (E.S.J. Lox Handbook of Heterogeneous Catalysis 2e издание, с.2274-2345 и приведенные в ней ссылки). Все же, хотя, как известно, реакция между NH3 и NO2 (5) протекает, она не является преобладающей в кинетическом отношении. Таким образом, при повышении концентрации NO2 выше примерно 50% происходит сопутствующее снижение каталитической активности и скорости реакции (Grossale и др. J. Catal, 256 (2008) 312-322).
Следовательно, в данной области техники существует потребность в технологии, способной обеспечить высокоактивные и селективные катализаторы СКВ, легированные металлом путем допирования/ионного обмена, обладающие улучшенной гидротермической стабильностью и пониженной стоимостью. Это планируется осуществить путем разработки синтетического способа производства металлсодержащих цеолитов и цеолитоподобных структур. Такой способ должен обеспечить преимущества в отношении повышенной простоты и надежного пути изготовления, пониженного образования отходов, высокой дисперсности металла и пониженной стоимости процесса. Кроме того, он должен обеспечивать такие улучшения и одновременно обеспечивать материалы, имеющие широкий рабочий диапазон, стойкость к высокому содержанию NO2 и высокую стойкость к ядам на основе УВ и SOx, присутствующих в потоке отработавших газов, с целью обеспечения соответствия требованиям к современным многокомпонентным системам регулирования выбросов.
Цель настоящего изобретения заключается в разработке способа получения цеолитов и цеолитоподобных структур, легированных металлом путем допирования/ионного обмена, что обеспечит выгодные материалы для СКВ NOx, конкретно в отработавших газах двигателей внутреннего сгорания транспортных средств, работающих при условиях обедненной смеси. Кроме того, настоящее изобретение включает способ, обладающий преимуществами по сравнению с ранее описанными в данной области техники способами как с экологической, так и с экономической точек зрения.
Эти и другие цели, хорошо известные лицам, квалифицированным в данной области техники, достигаются путем применения способа получения допированных металлами цеолитов и цеолитоподобных структур, включающего следующие стадии:
I) обеспечение сухой гомогенной смеси цеолита или цеолитоподобной структуры с соединением - предшественником, включающим комплексное соединение, образованное переходным металлом, выбранным из группы, включающей V, Cr, Mn, Fe, Co, Ni, Cu, Nb, Mo, Ru, Rh, Pd, Ag и Ce, и лигандом, причем при разложении комплексного соединения при температуре от 100 до 500°C образуется металл или ион металла,
II) прокаливание смеси при достаточной температуре и в течение достаточного времени для осуществления сублимации в твердом состоянии металла или иона металла; и
III) получение допированного металлом цеолита или цеолитоподобной структуры
очень выгодным и неочевидным образом. В момент создания настоящего изобретения в данной области техники не предполагали, что стойкость материала и его каталитические свойства улучшаются по сравнению с ранее известными в данной области техники в такой степени, что либо применение меньшего количества материала по настоящему изобретению приводит к сравнимому эффекту, либо такое же количество материала приводит к лучшим результатам, а это, в свою очередь, приводит к снижению издержек при производстве в коммерческом масштабе.
Лица, квалифицированные в данной области техники, знают, какие цеолиты и цеолитоподобные структуры имеют значение при рассмотрении соответствующих структур каркаса, позволяющих осуществлять восстановление NOx. В связи с этим приведена конечная ссылка на определения и литературные источники, указанные выше. Для способа по настоящему изобретению, однако, некоторые цеолиты и цеолитоподобные структуры считаются предпочтительными. Их выбирают из группы, включающей одну из следующих структур или их смесь: цеолит типа фожазита, типа пентасила, чабазитный цеолит; или цеолитоподобную структуру, например, SAPO-34 или другие структуры на основе восьмичленных колец структурного типа CHA, и соответствующие структуры, например, типов AEI, AFT, AFX, DDR, ERI, ITE, ITW, KFI, LEV, LTA, PAU, RHO и UFI. Более предпочтительные структуры выбирают из группы, включающей тип пентасила, SAPO-34, конкретно, ZSM-5 и цеолит бета, а также цеолиты структурного типа чабазита. Наиболее предпочтителен чабазит/SAPO-34 и цеолит бета.
Было доказано, что переходные металлы, свободно вступающие в реакции окисления и восстановления, могут служить в качестве главных металлов, осуществляющих восстановление оксидов азота в соответствии с процессом СКВ. Выгодные металлы, обладающие такими свойствами, выбирают из группы, включающей металлы, называемые переходными, то есть 38 элементов групп с 3 по 12 Периодической таблицы элементов. В соответствии с настоящим изобретением, используют переходные металлы, выбранные из группы, включающей V, Cr, Mn, Fe, Co, Ni, Cu, Nb, Mo, Ru, Rh, Pd, Ag и Ce. Более предпочтительны металлы, выбранные из группы, включающей Fe, Cu и Ce. Наиболее предпочтительными в этом отношении металлами являются Cu и Fe.
С целью обеспечения возможности введения металла в каркас первичной структуры цеолита или цеолитоподобной структуры требуется применение предшественника металла с небольшой летучестью и подходящей температурой разложения, например, комплексного соединения, которое разлагается с получением металла или иона металла при температуре от 100 до 500°C, предпочтительно, от 200 до 450°C; оно может иметь структуру, соответствующую формуле (I):
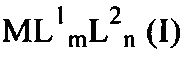 ,
,
в которой М представляет собой металл, выбранный из группы, указанной выше;
L1 может представлять собой карбонил, амин, алкил, алкокси, алкен, арен, фосфин или другой нейтральный координирующий лиганд, m может представлять собой число от 0 до 6, n может быть равно валентности M, а L2 выгодным образом включает дикетонат, кетоиминат или аналогичный член такого гомологического ряда, например, лиганд, соответствующий формуле (II):
 ,
,
в которой R1 и R2 независимо представляют собой алкил, замещенный акил, арил, замещенный арил, ацил и замещенный ацил.
Соединения-предшественники, включающие комплекс, образованный металлом и лигандом, включающим дикетонатную структуру, хорошо известны лицам, квалифицированным в данной области техники. Дополнительные подробности в отношении таких соединений и их получения можно найти в следующих источниках: Fernelius и Bryant Inorg Synth 5 (1957) 130-131, Hammond и др. Inorg Chem 2 (1963) 73-76, патенте WO 2004/056737 A1 и приведенных в них ссылках. Дополнительные лиганды, которые в составе комплекса способствуют образованию дикетонатной структуры, также были известны ранее в данной области техники, их примеры приведены Finn и др. в J Chem Soc (1938) 1254, Van Uitert и др. в J Am Chem Soc 75 (1953) 2736-2738, и David и др. в J Mol Struct 563-564 (2001) 573-578. Предпочтительные структуры с лигандами таких типов могут быть выбраны из группы, включающей R1 и R2 из формулы (II), представляющие собой алкилы. Более предпочтительные из таких лигандов выбирают из группы, включающей R1 и R2, представляющие собой метил или трет-бутил; наиболее предпочтителен ацетилацетонат (acac, R1 и R2 в формуле II представляют собой метальные группы).
Если используют соединения металлов с низкой валентностью, предпочтительными комплексами являются карбонильные, предпочтительно стабильные при комнатной температуре, с монооксидом углерода в качестве лиганда, с учетом их умеренной летучести и температур разложения. Синтез таких соединений хорошо известен, его обычно осуществляют путем восстановления соли металла в присутствии CO. Дополнительные подробности в отношении таких соединений и их получения можно найти в следующих изданиях: Abel Quart Rev 17 (1963) 133-159, Hieber Adv Organomet Chem 8 (1970) 1-28, Abel and Stone Quart Rev 24 (1970) 498-552, и Werner Angew Chem Int изд. 29 (1990) 1077.
Смесь цеолитов/цеолитоподобных структур с соединениями-предшественниками необходимо затем нагревать с целью увеличения мобилизирования металла в составе комплекса, чтобы он мог проникнуть путем диффузии в поры и каналы каркаса. Для этого нужно уделять внимание температуре: она должна быть достаточной для разложения предшественника и инициации и облегчения диффузии, и, в то же время, она не должна быть слишком высокой, что вызывает деградацию каркаса или избыточное спекание кристаллов цеолита/цеолитоподобной структуры. Таким образом, такое прокаливание, предпочтительно проводят при температуре выше 200°C. В особенно предпочтительном варианте смесь прокаливают при температуре от 200 до 650°C. В наиболее предпочтительном варианте используемая температура составляет от 350 до 450°C. Необходимо особо отметить, что данный процесс не зависит от пониженного давления или конкретных реакционных газов, и его можно осуществлять в атмосфере неподвижного или текущего газа, например, на воздухе или в атмосфере инертного газа, такого как N2, или в восстановительной атмосфере, включающей, например, примерно от 0,5 до 4% H2, без отрицательного воздействия на характеристики конечного катализатора.
Кроме того, необходимо отметить, что продолжительность прокаливания или нагревания должна находиться в подходящем диапазоне. Воздействие высокой температуры на смесь может, как правило, продолжаться до 12 часов. Предпочтительно, термическая обработка имеет продолжительность примерно от 1 до 5 ч. В особенно предпочтительном случае смесь подвергают воздействию высоких температур, как описано выше. Выгодным образом смесь подвергают воздействию температур примерно от 350 до 450°C в течение времени от 1 до 5 ч. Наиболее предпочтительно процесс осуществляют примерно при 350°C в течение времени от 90 до 150 минут.
С целью обеспечения сублимации/диффузии необходимого для катализа количества металла в порах, полостях и каналах цеолита и цеолитоподобной структуры смесь должна включать оба ингредиента в конкретных соотношениях. Таким образом, предпочтительно, чтобы смесь включала материал каркаса и соединение-предшественник в таких соотношениях, чтобы разложение предшественника приводило к обеспечению концентрации металла в цеолите/цеолитоподобной структуре, составляющей от примерно 0,01 до примерно 10 мас.% металла, предпочтительно, от 0,1 до 7,5 мас.%. Более предпочтительно, концентрация металла в цеолите/цеолитоподобной структуре должна составлять от примерно 1 до 4 мас.%. Наиболее предпочтительно, чтобы концентрация металла в цеолите/цеолитоподобной структуре должна составлять от примерно 1,5 до примерно 2,5 мас.%. Здесь необходимо отметить, что такое содержание металла несколько ниже, чем описанное в ранее изданной литературе, в которой указано, что повышенное содержание металла и требование к «избытку» металла защищает цеолит от гидротермического старения (WO 2010-054034 или WO 2008-106519 A1).
Второй предпочтительный вариант настоящего изобретения направлен на материал или смесь материалов, получаемую в соответствии со способом по настоящему изобретению, причем материал или смесь материалов при нанесении на носитель, как описано ниже, осуществляет катализ восстановления оксидов азота по реакции с азотсодержащим восстановителем даже при такой низкой температуре, как 100°C. Выражение «осуществляет катализ восстановления оксидов азота по реакции с азотсодержащим восстановителем даже при такой низкой температуре, как 100°C» следует понимать в том смысле, что восстановление протекает при 100°C в определенной степени. Предпочтительно, реакционная способность при 100°C, по сравнению с максимальной реакционной способностью материала или смеси материалов, составляет более 0,2%, более предпочтительно более 0,5%, наиболее предпочтительно более 1%.
В дополнительном аспекте настоящее изобретение направлено на катализатор, включающий материал или смесь материалов, полученные в соответствии со способом по настоящему изобретению, причем катализатор включает инертное термостойкое связующее, выбранное из группы, включающей оксид алюминия, оксид титана, нецеолитный оксид кремния-оксид алюминия, оксид кремния, оксид циркония и смеси перечисленного, нанесенные в виде покрытия на керамический монолит со сквозными каналами, пенообразный металлический субстрат или на субстрат с фильтрующими стенками. Предпочтительным образом, описанный выше катализатор получают способом, в котором материал или смесь материалов, описанные выше, и связующее наносят на отдельные зоны керамического монолита со сквозными каналами, пенообразного металлического субстрата или субстрата с фильтрующими стенками.
В еще одном дополнительном аспекте настоящее изобретение направлено на монолитный катализатор, полученный путем экструзии материала или смеси материалов в соответствии со способом по настоящему изобретению.
Другой аспект настоящего изобретения относится к применению катализатора или монолитного катализатора, описанных выше, в селективном каталитическом восстановлении оксидов азота. Выгодным образом использование такого катализатора или монолитного катализатора осуществляют так, что источник азотсодержащего восстановителя вводят с обеспечением эффективного отношения NH3:NOx (отношение α (альфа)) на входе в катализатор, составляющего от 0,5 до 2. Более того, применение катализатора или монолитного катализатора, описанных выше, предпочтительно, осуществляют при фиксируемом отношении NO:NO2 на входе в катализатор, составляющем от 1:0 до 1:3 по объему, предпочтительно, от 1:0,8 до 1:1,2, наиболее предпочтительно примерно 1:1.
Обычно материал или смесь материалов, получаемые в соответствии со способом по настоящему изобретению, имеют форму каталитического устройства, включающего корпус, расположенный вокруг субстрата, причем катализатор СКВ включает материал или смесь материалов и нанесен на субстрат. Кроме того, способ обработки отходящего газа бензинового ДВС (двигателя внутреннего сгорания), работающего при обедненных кислородом условиях, или ДВС с зажиганием от сжатия, или потока отработавших газов сжигания ископаемого топлива при обедненных кислородом условиях может включать введение указанного потока отработавших газов в такой катализатор СКВ и восстановление NOx в составе указанного потока отработавших газов до N2.
Материал или смесь материалов можно включить в состав путем соединения оксида алюминия, оксида кремния или другого подходящего связующего и, необязательно, других каталитически активных материалов, например, компонента для хранения кислорода (КХК) на основе церия, с получением смеси, сушки (активной или пассивной) и, необязательно, прокаливания смеси. Более конкретно, можно получить суспензию путем соединения материала по настоящему изобретению с оксидом алюминия или оксидом кремния и водой и, необязательно, с агентами для регулирования pH, например, неорганическими или органическими кислотами и основаниями и/или другими компонентами. Такую суспензию можно нанести в качестве реакционного покрытия на подходящий субстрат. Покрытый таким образом продукт можно высушить и подвергнуть термической обработке с целью закрепления реакционного покрытия на субстрате.
Такую суспензию, полученную описанным выше способом, можно высушить и подвергнуть термической обработке, например, при температурах примерно от 350 до примерно 1000°C, или, более конкретно, от примерно 400 до примерно 600°C, с получением конечного состава катализатора. В качестве альтернативы или в дополнение к этому суспензию можно нанести в качестве реакционного покрытия на субстрат, а затем подвергнуть термической обработке описанным выше способом, с целью регулирования площади поверхности и кристаллической природы носителя.
Полученный катализатор включает цеолит/цеолитоподобную структуру, в которую путем ионного обмена введен металл по способу сублимации, описанному в настоящем описании. Катализатор может дополнительно включать инертное термостойкое связующее. Нанесенный катализатор можно затем нанести на субстрат. Субстрат может включать любой материал, предназначенный для применения в желаемой среде. Возможные материалы включают кордиерит, карбид кремния, металл, оксиды металлов (например, оксид алюминия и подобные), стекла и подобные, а также смеси, включающие, по меньшей мере, один из указанных материалов. Такие материалы могут находиться в форме набивки, экструдатов, фольги, перфорированного материала, матов, волокнистого материала, монолитов, например, сотовой структуры и подобных, монолитов с фильтрующими стенками (имеющих способность к отфильтровыванию дизельной сажи), других пористых структур, например, пористых стекол, губок, пены и подобных (в зависимости от конкретного устройства), а также комбинаций, включающих, по меньшей мере, один из указанных выше материалов и форм, например, в виде металлической фольги, губчатого оксида алюминия с открытыми порами, а также пористых стекол с ультранизкой степенью термического расширения. Более того, такие субстраты можно покрывать оксидами и/или гексаалюминатами, например, они могут представлять собой фольгу из нержавеющей стали, покрытую гексаалюминатным осадком. В качестве альтернативы, материал с допированной катионами решеткой можно экструдировать совместно с подходящими связующими и волокнами с получением монолита или монолитной структуры с проницаемыми стенками.
Хотя субстрат может иметь любой размер или геометрию, его размер или геометрию, предпочтительно, выбирают с целью оптимизации геометрической площади поверхности в соответствии с параметрами конкретного устройства для регулирования выбросов в отработавших газах. Обычно, субстрат имеет сотовую геометрию, причем каналы, проходящие через соты, имеют любую многогранную или круглую форму, причем предпочтительными являются, по существу, квадратная, треугольная, пентагональная, гексагональная, гептагональная или октагональная или аналогичная геометрия, вследствие простоты производства и повышенной площади поверхности.
После нанесения каталитически активного материала на субстрат последний можно расположить в корпусе с получением преобразователя (нейтрализатора). Корпус может иметь любую конструкцию и включать любой материал, подходящий для способа его применения. Подходящие материалы могут включать металлы, сплавы и подобные материалы, например, ферритную нержавеющую сталь (включая такие нержавеющие стали, как, например, стали 400 серии, например, SS-409, SS-439 и SS-441) и другие сплавы (например, включающие никель, хром, алюминий, иттрий и подобные металлы, позволяющие достичь повышенной устойчивости и/или стойкости к коррозии при рабочих температурах в окислительной или восстановительной атмосферах).
К одному или обоим концам корпуса могут быть коаксиально присоединены состоящие из таких же материалов, что и корпус, концевик (концевики), торцевая пластина (пластины), крышка (крышки) выпускного коллектора и подобные устройства с обеспечением герметичного соединения. Такие компоненты можно получать отдельно (например, литьем или подобными способами), или их можно получать вместе с корпусом с использованием таких способов, как, например, ротационная вытяжка или подобных.
Между корпусом и субстратом может находиться удерживающий материал. Удерживающий материал, который может иметь форму плиты, частиц или подобную, может представлять собой набухающий материал, например, материал, включающий вермикулитный компонент, то есть компонент, расширяющийся при нагревании, не набухающий материал, или их комбинацию. Такие материалы могут включать керамические материалы, например, керамические волокна, и другие материалы, например, органические и неорганические связующие и подобные, или комбинации, включающие, по меньшей мере, один из указанных выше материалов.
Таким образом, монолит, содержащий легированный металлом путем допирования/ионного обмена катализатор, вводят в поток отработавших газов двигателя, работающего при обедненной смеси. Это обеспечивает средство обработки указанного потока отработавших газов с целью понижения концентрации NOx путем пропускания указанного потока отработавших газов над указанным выше катализатором СКВ при суммарно окислительных условиях в присутствии вводимой в выхлопную систему мочевины или аммония, или другого азотсодержащего восстановителя, с целью облегчения каталитического превращения в безвредный с точки зрения окружающей среды газообразный азот.
На фиг.1 показаны данные моделирования газового испытания двух эталонных образцов 3% Cu-SAPO-34 A и B, полученных методом традиционной пропитки, описанным в примерах.
На фиг.2 показана модель газового испытания эталонных образцов A и B после двухчасового цикла состаривания при 780°C на воздухе.
На фиг.3 отражена активность образцов A и B после двухчасового цикла состаривания при 900°C на воздухе.
На фиг.4a приведено сравнение характеристик при термопрограммируемом восстановлении (ТПВ) с использованием H2 для свежих образцов 3% Cu-SAPO-34.
На фиг.4б показано H2 ТПВ образцов 3% Cu-SAPO-34 после состаривания при 780°C на воздухе.
На фиг.5a показано сравнение рентгеновских дифрактограмм свежих образцов 3% Cu-SAPO-34.
На фиг.5б обобщены рентгеновские дифрактограммы образцов по фиг.5а после состаривания на воздухе при 780°C.
На фиг.5в показаны рентгеновские дифрактограммы образцов по фиг.5а после состаривания на воздухе при 900°C.
На фиг.6а показана степень превращения NO для свежих образцов 3% Cu-SAPO-34, полученных с использованием медьсодержащих предшественников и способов, описанных в таблице 2.
На фиг.6б показана степень превращения NH3 для свежих образцов 3% Cu-SAPO34, полученных с использованием предшественников и способов, описанных в таблице 2.
На фиг.7а показана степень превращения NO для образцов по фиг.6а после цикла состаривания в течение 12 ч при 780°C на воздухе.
На фиг.7б показана степень превращения NH3 для материалов по фиг.6а после цикла состаривания в течение 12 ч при 780°C на воздухе.
На фиг.8а показана степень превращения NO для материалов, представленных на фиг.6а, после цикла состаривания в течение 2 ч при 900°C на воздухе.
На фиг.8б суммированы активности при превращении NH3 для материалов, представленных на фиг.6б, после цикла состаривания в течение 2 ч при 900°C на воздухе.
На фиг.9 показано сравнение активности двух свежих образцов на основе 3% Cu-SAPO-34, полученных стандартным способом (A) и путем сублимации (F), включающим способ с использованием Cu(acac)2 в качестве предшественника (F).
На фиг.10 показано сравнение активности двух образцов на основе 3% Cu-SAPO-34, полученных стандартным способом (A) и путем сублимации (F), состаренных при 780°C в течение 2 ч для (A) или в течение 12 ч для (F) соответственно.
На фиг.11 показано сравнение активности двух образцов на основе 3% Cu-SAPO-34, полученных стандартным способом (A) и путем сублимации (F), описанным в таблице 2, состаренных при 900°C в течение 2 ч на воздухе.
На фиг.12а показано влияние содержания меди на степень превращения NO серией свежих образцов на основе Cu-SAPO34, полученных путем сублимации при 500°C в атмосфере N2.
На фиг.12б показано влияние содержания меди на степень превращения NH3 серией свежих образцов на основе Cu-SAPO34, полученных путем сублимации при 500°C в атмосфере N2.
На фиг.13а показаны степени превращения NO для образцов по фиг.12а после цикла состаривания в течение 2 ч при температуре 780°C, на воздухе.
На фиг.13б показаны степени превращения NH3 для образцов по фиг.12б после цикла состаривания в течение 2 ч при температуре 780°C, на воздухе.
На фиг.14а показаны степени превращения NO для образцов по фиг.12а после цикла состаривания в течение 2 ч при температуре 900°C, на воздухе.
На фиг.14б показаны степени превращения NH3 для образцов по фиг.12б после цикла состаривания в течение 2 ч при температуре 900°C, на воздухе.
На фиг.15 показана активность свежих образцов на основе 2% Cu-SAPO34, полученных с помощью различных разновидностей способа сублимации, в ходе исследования активности оценивали влияние способа перемешивания.
На фиг.16 показаны характеристики образцов по фиг.12 после состаривания катализатора при 780°C на воздухе в течение 2 ч.
На фиг.17 показаны характеристики образцов по фиг.12 после состаривания катализатора при 900°C на воздухе в течение 2 ч.
На фиг.18 показано исследование влияния содержания меди при превращении NO в присутствии серии свежих образцов на основе Cu-SAPO34, полученных путем сублимации при 350°C на воздухе.
На фиг.19 приведены характеристики образцов по фиг.18 после состаривания в течение 2 ч при 780°C на воздухе.
На фиг.20 отражены характеристики образцов по фиг.18 после состаривания в течение 2 ч на воздухе при 900°C.
На фиг.21 показаны характеристики образцов по фиг.18 после состаривания катализатора при 950°C на воздухе в течение 2 ч.
На фиг.22 отражены данные в отношении степени превращения для образцов по фиг.18 после цикла гидротермического состаривания, включающего шестнадцатичасовое воздействие смеси воздуха и 10% пара при температуре 750°C.
На фиг.23 отражены характеристики образцов по фиг.18 после цикла гидротермического состаривания, включающего четырехчасовое воздействие смеси воздуха и 10% пара при температуре 900°C.
На фиг.24 показано сравнение данных по степени превращения NO образцов, содержащих 2,5 мас.% Fe на ZSM5 и цеолите бета, полученных путем влажной пропитки либо сублимационным способом, после небольшого состаривания перед началом испытаний.
На фиг.25 показано сравнение данных по степени превращения NO для образцов ZSM5 или цеолита бета, допированных 2 и 4 мас.% Cu, после небольшого состаривания перед началом испытаний.
Обозначения образцов
A: 3% Cu-SAPO34, полученный добавлением раствора Cu(NO3)2 к хорошо перемешанной суспензии SAPO34.
B: 3% Cu-SAPO34, полученный добавлением кристаллического Cu(NO3)2 к плохо перемешанной суспензии SAPO34.
C: 3% Cu-SAPO34, физическая смесь
D: 3% Cu-SAPO34, прокаленная физическая смесь
E: 3% Cu-SAPO34, полученный ионным обменом в водной среде
F: 3% Cu-SAPO34, полученный сублимацией ацетилацетоната меди
G: 3% Cu-SAPO34, полученный сублимацией оксалата меди (CuC2O4)
H: 3% Cu-SAPO34, полученный сублимацией ацетата меди (Cu(CH3COO)2)
I: 2% Cu-SAPO34, полученный сублимацией с перемешиванием с помощью смесителя для красок (см. основное описание)
J: 2% Cu-SAPO34, полученный сублимацией с перемешиванием с помощью кофемолки (см. основное описание)
K: 2% Cu-SAPO34, полученный сублимацией с перемешиванием с использованием верхнеприводной мешалки (см. основное описание)
Приведенный ниже набор данных включает широкое разнообразие примеров приготовления с различными содержанием металлов, предшественниками и условиями процесса, и является иллюстрацией гибкости способа допирования металлом и его применения для СКВ. Прямое сравнение с традиционными методиками синтеза (ионным обменом и пропиткой в суспензии/прокаливанием) приведено для иллюстрации преимуществ в отношении характеристик и стойкости материалов, получаемых способом по настоящему изобретению. Приведенные данные представляют собой серию примеров, выбранных из значительно более обширной работы, и они включают ссылки на измерения каталитических характеристик. Такие измерения осуществляли с использованием модели традиционного газофазного реактора с запорным потоком. В этих измерениях потоки газа, имитирующие поток отработавших газов ДВС с обедненной смесью, пропускали над и через просеянные частицы испытуемых образцов при различных условиях в отношении температуры; эффективность образца при восстановлении NOx определяли с помощью установленного в линии ИК-спектрометра с преобразованием Фурье. В таблице 1 подробно описаны все параметры эксперимента, использованные для получения приведенных в настоящем описании данных.
На фиг. с 1 по 3 обобщены характеристики производительности двух сравнительных эталонных образцов, содержащих 3% Cu-SAPO34 и обозначенных A и B соответственно, полученных традиционным способом пропитки/прокаливания (см. примеры). Эти данные отражают присущие этому способу недостатки и зависимость возможности синтеза «качественных» материалов от смешивания. Таким образом, образец B проявляет сниженную активность в свежем состоянии как в отношении активации NH3, так и в отношении последующего восстановления NOx. Образец A, полученный при хорошем смешивании, обеспечил на 20% более высокую степень превращения NOx на стадии «зажигания» реакции (T<200°C) и в диапазоне средних температур (200<Y<300°C). Тем не менее, степень превращения NO обоих этих образцов так и не превысила теоретического максимума в 90% при отношении сырья (α), составляющем 0,9. В самом деле, максимальная степень превращения для образца A составила примерно 80% при температурах от 210 до 270°C. Такая пониженная степень превращения NOx согласуется с ограниченной величиной примерно 90% степенью превращения NH3 в аналогичном температурном диапазоне, что свидетельствует о присущей данному образцу ограниченной активности. Такое ограничение степени превращения NH3 обоими образцами не наблюдается в диапазоне более высоких температур (более 300°C). Однако ни в одном из этих случаев повышение степени превращения NH3 не соответствует улучшению степени превращения NOx, и, конкретно, в случае образца A, наблюдается скорее уменьшение образования N2, то есть увеличение содержания NO и N2O, что объясняется повышенной скоростью паразитного окисления NH3 (см. реакции с 7 по 9 во введении). После состаривания в течение 2 ч на воздухе при 780°C образец A утратил активность в малой степени при пониженных температурах (менее 200°C), но проявил улучшенные степени превращения NO и NH3 при всех остальных температурах, что согласуется с мягким индукционным эффектом. Напротив, в случае образца B, низкотемпературные и пиковые степени превращения NO и NH3 оказались примерно на 20% выше, и образец проявлял более высокую эффективность утилизации NH3 при СКВ по сравнению со свежим состоянием. Такие эффекты напрямую связаны с явлением индукции. Такие данные согласуются с данными ТПВ, показанными на фиг.4а и 4б. Профиль образца A содержит два пика восстановления, один из пиков находится при температуре примерно 250°C, что соответствует восстановлению диспергированного CuO, а второй пик находится при температуре примерно 475°C, что свидетельствует о большей сложности восстановления молекул CuO, а это типично для оксида металла, стабилизированного/введенного ионным обменом в цеолит. В спектре образца B пик восстановления диспергированного CuO больше, а высокотемпературный пик примерно при 425°C меньше, что свидетельствует о присутствии в каркасе структур, введенных путем ионного обмена. После состаривания при 780°C на воздухе оба образца имеют аналогичные профили ТПВ, содержащие два пика. Это согласуется с высокотемпературным перемещением Cu на подвергнутые ионному обмену центры. Однако, такое перемещение Cu может также способствовать образованию объемных частиц CuO (тенорит), а такое вещество является активным катализатором окисления NH3 при более высоких температурах. Следовательно, неудивительно, что при температуре 400°C и выше образец B проявил пониженную селективность в отношении N2 в ходе превращения NO в результате повышенного образования NO и N2O, что также согласуется с увеличением паразитного окисления NH3. Повышенное неселективное окисление при использовании образца B также согласуется с данными ТПВ, показанными на фиг.4б; видно, что в профиле наблюдается дополнительный пик примерно при 275°C, который согласуется с восстановлением объемного CuO. После дальнейшего состаривания в течение 2 ч при 900°C на воздухе различия между двумя материалами с эквивалентным составом стали еще четче. Так, образец B полостью утратил активность и проявил только остаточную активность при паразитном окислении NH3 при температуре более 300°C. Напротив, образец A сохранил некоторую способность к СКВ, хотя и пониженную, с пиком степени превращения NO, составляющей примерно 35%. Такая катастрофическая дезактивация является поводом для беспокойства, поскольку пиковые температуры могут достигать или даже превышать 900°C, например, в ходе каталитической регенерации фильтра, особенно если двигатель переходит в режим холостого хода в середине цикла регенерации. Причины такой катастрофической дезактивации видны из рентгеновского дифракционного анализа (фиг.5а-5в). Для обоих свежих образцов характерны рефлексы, характерные для исходного каркаса типа SAPO-34. С помощью рентгеновской дифракции не обнаружено отчетливой фазы CuO или других фаз, связанных с разрушением каркаса SAPO-34 в ходе суспензионной обработки. Однако это не так после состаривания на воздухе при 780°C, когда образец B, полученный при «плохом» перемешивании, проявляет отчетливые рефлексы примерно при 21 и 35°, что согласуется с присутствием кристобалита, то есть фазы на основе минерального SiO2. Такая фаза может образовываться только при удалении Si из SAPO-34, то есть пропитка суспензией/прокаливание приводят к преждевременному коллапсу структуры каркаса. Такое разрушение очевидно для обоих образцов после состаривания при 900°C на воздухе (фиг.5в). Разрушение структуры сильнее в случае образца B, с учетом того, что при исследовании «хорошо» перемешанного образца A видны небольшие рефлексы, характерные для остатка фазы SAPO-34. Такие проблемы отражены в таблице 3, в которой приведены площади поверхности (БЭТ) образцов в свежем и состаренном состоянии. Видно, что образец B имеет сниженную удельную поверхность по БЭТ, и имеются свидетельства коллапса площади поверхности/структуры после состаривания при 780°C. Таким образом, из этих данных видно, что традиционный способ синтеза имеет большие недостатки в отношении как характеристик, так и стойкости.
С целью устранения таких недостатков была получена серия дополнительных сравнительных и испытательных порошков на основе Cu-SAPO34, что подробно описано в таблице 2 и в примерах.
Результаты исследований образцов в процессе СКВ приведены на фиг. с 6а по 8б. Величины активности этих образцов довольно сильно различаются. Свежие образцы E, F и H обеспечивают высокие степени превращения как NO, так и NH3, в то время как свежие образцы C, D и G даже при температуре более 350°C не проявляют увеличения склонности к неселективному окислению NH3 с соответствующим образованием NOx, и поэтому заметны отрицательные степени превращения NO. После состаривания при 780°C (12 ч, на воздухе) образцы E, F и H проявляют аналогичные или худшие характеристики, то есть наблюдалось отсутствие индукционного эффекта или преждевременное разрушение структуры, в то время как характеристики образцов C, D и G значительно улучшились, особенно характеристики образцов D и G, что отражает особенно сильный индукционный эффект. Дальнейшее состаривание при 900°C привело к снижению степени превращения NO и NH3 для всех образцов. Однако следует заметить, что снижение характеристик оказалось совсем небольшим по сравнению с образцами A и B, полученными традиционным способом. Таким образом, пиковые степени превращения NO составили от примерно 40 до примерно 70%, то есть они до двух раз выше, чем у наилучшего сравнительного порошка. Из этих данных очевидно, что разложение/сублимация Cu(acac)2 обеспечивает оптимальные характеристики катализатора в свежем и состаренном состоянии, при отсутствии индукционного периода или преждевременной дезактивации. Образец D (CuO, смешанный с SAPO34) также проявил высокую активность, но только после состаривания, что также согласуется с наличием индукционного эффекта, по аналогии с образцом B. Напротив, характеристики в свежем состоянии образца E (получен ионным обменом) конкурентоспособны по отношению к образцам, полученным путем сублимации, что отражает изначально оптимальное взаимодействие между Cu и каркасом. Однако этот образец плохо перенес состаривание, что объясняется спеканием несвязанных атомов Cu с образованием объемной фазы CuO, это приводит к сильному паразитному окислению NH3 с образованием NO, которое начинается при температуре примерно 250°C. Аналогично, образец H, полученный путем сублимации ацетата меди показал приемлемую активность в свежем состоянии, но стабильность этого порошка при состаривании оказалась недостаточной, поэтому дальнейшее его исследование не осуществляли.
Преимущества сублимационного способа по сравнению с суспензионной пропиткой/прокаливанием отмечены на фиг. с 9 по 11. Видно, что образец F в свежем состоянии проявляет более высокую активность при превращении NO и NH3. Он обеспечивает значительное улучшение характеристик «зажигания», степень превращения NO при 165°C примерно на 25% выше по сравнению со сравнительным образцом. Аналогично, характеристики в среднем температурном диапазоне оказались на 5-10% выше, а при температуре 350°C или более степень превращения NO оказалась одинаковой. Эти преимущества сохраняются после цикла состаривания при 780°C. В самом деле, несмотря на более жесткие условия состаривания (12 ч вместо 2 ч), образец F сохранил превосходные характеристики зажигания и обеспечил высокую степень превращения NO и NH3 в среднем температурном диапазоне. Улучшенная стойкость сублимированного образца дополнительно подтверждается данными, показанными на фиг.11. Таким образом, после цикла состаривания на воздухе при 900°C образец, полученный суспензионной пропиткой/прокаливанием, претерпел катастрофическую дезактивацию, для него пиковая степень превращения NO составила примерно 35%, в то время как образец, полученный сублимацией, обеспечил степень превращения, составляющую более 70%. Эти выгодные свойства отражаются на окислительно-восстановительных характеристиках образца F (фиг.4а и 4б). Так, профиль свежего образца F содержит два пика восстановления, причем высокотемпературный (500°C) пик окисления-восстановления относится к меди, стабилизированной в каркасе. При сравнении данных для свежего образца и состаренного при 780°C образца сдвига температуры данного пика не наблюдается, в отличие от образцов A и B. Это свидетельствует о том, что сублимационный синтез обеспечивает подходящее распределение меди в конкретном предпочтительном активном центре уже в свежем состоянии, в то время как традиционная методика синтеза требует дополнительной высокотемпературной обработки (индукции), то есть осуществления ионного обмена в твердом состоянии с целью достижения данного предпочтительного наиболее активного состояния. Данные, полученные с помощью рентгеновского дифракционного анализа (фиг. с 5а по 5в) согласуются с каталитическими характеристиками и данными ТПВ. Для свежего образца F характерны только рефлексы, связанные с SAPO34, хотя кристалличность этого образца выше, чем у образцов A и B (более сильные и отчетливые рефлексы). После состаривания при 780°C образец F сохранил высокую степень кристалличности, наблюдали лишь незначительные следы присутствия кристобалита (плечо при 21,5°). Дополнительное состаривание при 900°C, тем не менее, привело к разрушению структуры SAPO34, остались только ее следовые рефлексы, в целом дифрактограмма была, по существу, характерной для кристобалита. Такое разрушение структуры связывают с дестабилизацией дальнего порядка структуры SAPO34 в присутствии большого количества меди, поскольку на основании измерения активности можно заключить, что в образце сохраняется достаточное количество активных центров для обеспечения каталитической функции. Дополнительные тенденции в отношении дезактивации видны из данных о площади поверхности (таблица 3). Свежие образцы A и F имеют сравнимые удельные поверхности по БЭТ в свежем состоянии, примерно на 40 м2/г выше, чем у образца B, что согласуется с описанными проблемами. Однако, после состаривания при 780°C поверхность по БЭТ образца F снизилась не сильно, а у образцов A и B этот параметр сильно снизился, что согласуется с дестабилизацией каркаса, связанной со способом приготовления образцов, то есть сублимационный способ позволяет осуществлять допирование Cu с меньшим отрицательным воздействием на устойчивость каркаса при высоких температурах.
Эффективность ионного обмена металлов в сублимационном способе также позволяет снижать содержание допирующего металла с целью улучшения высокотемпературной устойчивости без снижения каталитической функции. Такая возможность показана на фигурах с 12а по 14б. На них показано, как влияет на состаривание изменение содержания Cu от 1 до 3 мас.%. Образцы готовили так, как описано в примерах, и во всех случаях хорошо перемешанные смеси предшественников прокаливали в токе N2. Кривые зависимости степени превращения NOx от температуры для свежих катализаторов четко свидетельствуют о преимуществах катализатора с содержанием меди не менее 2 мас.%, причем пиковая степень превращения NOx составляет примерно 85%. Аналогично отмечено почти количественное превращение NH3 в присутствии этих образцов при всех температурах ≥200°C. Сниженную низкотемпературную активность в превращении NOx при содержании меди 1 и 1,5% связывают с пониженной степенью превращения NH3, с пиковыми значениями на уровне примерно 88% и примерно 72% для образцов, содержащих 1,5% и 1% Cu соответственно. После состаривания при 780°C почти не наблюдается изменения характеристик образцов с повышенным содержанием Cu, пиковая степень превращения NOx по-прежнему близка к теоретическому максимуму при данном α. Напротив, образцы, содержащие 1 и 1,5% Cu, проявляют значительно улучшенные характеристики, и обеспечивают степень превращения NOx, составляющую 62 и 78% соответственно. Такое улучшение связывают с эффектом мобилизирования меди (индукция). Видно, что после цикла состаривания на воздухе при 900°C характеристики меняются. Таким образом, в присутствии образца, содержащего 3 мас.% Cu, наблюдается сниженная степень превращения NH3/NOx в результате протекания высокотемпературных реакций между медью и каркасом. После этого более жесткого цикла состаривания оптимальные характеристики отмечены при содержании меди 2 или 2,5%, также начинает проявлять конкурентоспособные характеристики образец, содержащий 1,5% меди. Однако основной итог данного испытания заключается в том, что новый способ синтеза обеспечивает улучшенную активность и устойчивость катализатора при пониженном содержании меди, причем эффективные материалы можно получать с помощью прокаливания в токе N2.
Преимущества, которые заключаются в достижении улучшенных характеристик при сниженных содержаниях металла, были подтверждены элементным анализом выборочных образцов на основании приведенных выше данных. Так, из таблицы 4 видно, что высокие характеристики достигаются при меньших содержаниях меди по сравнению с образцами, описанными ранее в данной области техники, например, в патентах WO 2010-054034 или WO 2008-106519 A1.
Как отмечено выше, особенное преимущество сублимации заключается в надежности способа синтеза. Это отражено на фиг. с 15 по 17, на которых показано влияние применения различных смешивающих устройств для получения гомогенной смеси соли с каркасным соединением. Эти данные подтверждают высокую активность образцов, допированных 2% Cu, полученных с помощью трех различных смешивающих устройств; во всех случаях наблюдалась лучшая степень превращения NH3 и NOx по сравнению с традиционными образцами, содержащими 3% меди. Более того, активности трех образцов различались на величину, не превышающую экспериментальной погрешности, и в свежем состоянии, и после циклов состаривания при 780°C и при 900°C. Здесь снова пиковая степень превращения NOx после наиболее жесткого цикла при 900°C составила примерно 80%, что является значительным улучшением по сравнению с традиционными способами. Таким образом, очевидно, что в способе по настоящему изобретению можно применять различные смешивающие устройства без отрицательного воздействия на свойства конечного катализатора.
Дополнительная демонстрация надежности способа приготовления и улучшенной термической стойкости образцов, полученных путем сублимации, проведена на фиг. с 18 по 21. Все образцы, представленные на этих чертежах, получали так, как описано в примерах, с конечным прокаливанием на воздухе в статических условиях. Полученные данные неудивительны; все образцы проявили активность и устойчивость, почти идентичные показанным на фиг. с 12а по 14б. Таким образом, степень превращения NOx в свежем состоянии превысила 85%, и образцы проявили высокую активность при низком паразитном окислении NH3/образовании NOx в широком температурном диапазоне. Характеристики после состаривания при 780°C изменились мало или вообще не изменились, за исключением улучшения, показанного для образцов, допированных 1 и 1,5% меди, в соответствии с фиг.12а/13а. Фиг.20 подтверждает недостаточную стойкость образца, содержащего 3% меди, после состаривания при 900°C на воздухе, что свидетельствует о начале протекания твердофазных реакций между допирующей медью и каркасом. Эти тенденции еще более очевидны после состаривания при 950°C на воздухе: образцы, содержащие 3 и 2,5% меди, почти полностью дезактивировались. Напротив, образец, содержащий 1,5% меди, сохранил высокую активность, пиковая степень превращения NOx составила почти 80%. Эти данные свидетельствуют о том, что гибкость сублимационного способа может позволить подбирать содержания допирующего металла в соответствии со способом применения, например, использовать цеолитоподобную структуру с пониженным содержанием меди для СКВ ДСФ (сажевый фильтр с покрытием для СКВ) в виде гомогенного слоя или зоны ДСФ. Использование зоны материала с пониженным содержанием меди, улучшенной стойкостью и высокой активностью после жесткого состаривания в ходе регенерации фильтра представляет особый интерес, с учетом пиковых температур в ходе регенерации ДСФ.
Дополнительные примеры стабильности, в данном случае в присутствии воздуха и пара (10%) при 750 и 900°C, показаны на фиг.22 и 23. На них проведено сравнение образцов, прокаленных на воздухе и в атмосфере N2, с различным содержанием меди. После состаривания в течение 16 ч при 750°C на воздухе в присутствии 10% пара все образцы сохранили отличную активность и обеспечили пиковую степень превращения NOx, составляющую примерно 80%. Разницы между образцами, прокаленными на воздухе, и образцами, прокаленными в атмосфере азота, с содержанием 2 или 2,5% Cu, не видно, замечено лишь небольшое снижение характеристик образца, содержащего 1,5% Cu. После прокаливания в течение 4 ч при 900°C в присутствии воздуха/пара наблюдается иная картина. В этом случае оба образца, содержащие 2,5% меди, проявили почти идентичные характеристики и претерпели катастрофическую дезактивацию. Аналогично, активность образцов, содержащих 2% меди, оказалась сравнимой, но очень плохой. Напротив, образец, содержащий 1,5% меди, сохранил приемлемые характеристики, пиковая степень превращения NOx составила примерно 57% после такого жесткого состаривания. Следует особо отметить, что эти данные отражают скорее фундаментальную неустойчивость в силу протекания твердофазных реакций при высокой температуре между медью и каркасом цеолитоподобной структуры, чем слабости нового способа синтеза, например, см. фиг.3 и 11. С гипотезой о сохранении лучшей активности при пониженном содержании меди также согласуются данные, представленные на фиг.14а/б и фиг.20.
Применение способа сублимации для введения частиц других металлов и для других структур каркаса показано на фиг.24 и 25 соответственно. Так, на фиг.24 приведена активность кратковременно состаренных промотированных железом цеолитов (2,5 мас.% Fe), конкретно, ZSM5 (SAR 23) и цеолита бета (SAR 38), полученных либо путем традиционной пропитки, либо путем сублимации. Все образцы оказались активными в процессе СКВ, но потребовалось использование более высоких температур, чем в случае использования образцов на основе меди, что согласуется с ранее описанными данными, например, в WO 2008/132452 A2; очевидно, что для каждой структуры каркаса активность значительно выше при использовании образцов, полученных сублимационным способом, а не путем традиционной пропитки, что является дополнительным подтверждением преимуществ нового способа. Более того, сублимационный способ можно также выгодным образом применять для получения других стандартных цеолитных систем, промотированных медью. Это следует из фиг.25, на котором показана активность кратковременно состаренных образцов ZSM5 и цеолита бета, промотированных 2 и 4 мас.% меди, полученных сублимацией Cu(acac)2 в соответствии со стандартным способом. Вновь, все образцы проявили высокую активность и обеспечили стехиометрическую или близкую к стехиометрической степень превращения NO с участием доступного NH3 в составе потока сырья.
Настоящее изобретение относится к разработке и применению улучшенного способа получения легированных металлом путем допирования/ионного обмена цеолитов/цеолитоподобных структур и их применению для удаления NOx, выработанных ДВС, с помощью селективного каталитического восстановления (СКВ) с использованием вводимого азотсодержащего восстановителя. Дополнительной особенностью данного способа является использование сухого, то есть безводного (или без использования другого растворителя) процесса, в котором ионы металла вводят в каркасный материал путем сублимации/разложения соответствующего предшественника металла, например, дикетоната, конкретных карбонильных комплексов или аналогичных структур в составе гомогенной смеси соединения-предшественника и цеолитов/цеолитоподобных структур. Еще одной дополнительной особенностью данного способа является его надежность, заключающаяся в том, что он не требует использования конкретной реакционной газообразной среды и пониженного давления. Он обеспечивает получение желаемого допированного металлом каркасного материала, который также является частью настоящего изобретения, без образования значимых количеств опасных или токсичных побочных продуктов.
Преимущества и особенности настоящего изобретения включают перечисленные ниже:
а) Простота. Способ включает смешивание двух сухих порошков с последующей высокотемпературной обработкой. Отсутствует необходимость в сложных смешивающих установках или системах для обработки суспензий. Для сухого способа отсутствуют требования к фильтрованию суспензий, промывке или сушке. Более того, способ невосприимчив к атмосфере или давлению в реакторе в ходе прокаливания. Это является преимуществом по сравнению с ранее описанными способами, поскольку ни защитный, ни восстановительный газ применять не нужно.
б) Затраты. Простота синтеза, даже не связанная с оборудованием и способом, описанными в пункте а), позволяет снизить затраты на материалы. Дополнительное снижение затрат связано с отсутствием необходимости использования оборудования для отслеживания pH, температуры и других параметров суспензии.
в) Время. Получение конечного порошка можно осуществить в течение 2 ч, в отличие от многосуточных временных затрат традиционных процессов ионного обмена во влажном состоянии, или многочасовых временных затрат на суспензионную пропитку/прокаливание (продолжительное смешивание для обеспечения гомогенности, ограничение влияния экзотермического эффекта при смачивании цеолита/цеолитоподобной структуры на химию суспензии, и так далее).
г) Уменьшенное влияние на окружающую среду. В отличие от ранее описанных в данной области техники процессов, в способе по настоящему изобретению образование побочных продуктов ограничивается стехиометрическими количествами CO2, образующегося в ходе разложения лигандов предшественника. Отсутствует образование больших количеств водных отходов, что происходит при ионном обмене, или образование потенциально токсичных выбросов, например, газообразных HF или HCl, как в случае ИОТС, или образование азотсодержащих соединений (органических аминов или оксидов азота), отмеченное для способа суспензионной пропитки/прокаливания (после сжигания NH3 или азоторганических оснований, используемых для регулирования pH суспензии/осаждения металла). Более того, с учетом стехиометрической природы синтеза, для получения катализатора не требуется избытка материалов или дополнительных химикатов, что снижает воздействие на окружающую среду до минимума.
д) Более надежный и гибкий способ введения допирующего вещества. Для определения количества вводимого вещества требуется простой расчет потери при сжигании материала предшественника. Отсутствие каких-либо дополнительных химических веществ или процессов снижает суммарные допуски до абсолютного минимума.
е) Выгодные характеристики. В отличие от традиционного способа суспензионной пропитки/прокаливания, сублимационный способ позволяет вводить металл непосредственно в конкретный активный ионно-обменный центр цеолита/цеолитоподобной структуры. Таким образом, индукционного периода не наблюдается. Кроме того, с учетом увеличенной эффективности допирования металлом при сублимационном способе, отсутствует необходимость в «перегрузке» цеолита/цеолитоподобной структуры с целью обеспечения «полного» допирования металлом, необходимого для оптимальных характеристик. Это обеспечивает улучшение селективности катализатора, то есть снижение паразитного окисления NH3, что может быть связано с образованием каталитически активной фазы, например, CuO (тенорит), не сплошной или заметной для традиционного метода рентгеновской дифракции. Во-вторых, достигается улучшенная стойкость/устойчивость к старению металлсодержащего каркасного материала, поскольку пониженные пределы содержания металла ограничивают протекание высокотемпературных (>750°C) твердофазных реакций между допирующим веществом и каркасом, а они являются основной причиной разрушения фазы и образования каталитически неактивных новых фаз в ходе/после состаривания. Наконец, в сухом способе сублимации отсутствует необходимость в модификаторах pH или реологии суспензии, например, HNO3 или TEAH. Использование кислотных или основных модификаторов обоих таких классов затруднительно, поскольку такие вещества могут реагировать с цеолитом или цеолитоподобной структурой и удалять атомы из каркаса, что дестабилизирует его структуру. Такие повреждения невозможно обнаружить в свежих порошках, но известно, что они отрицательно воздействуют на высокотемпературную стойкость конечных продуктов синтеза.
Определения
Необходимо отметить, что выражения «первый», «второй» и подобные, использованные в настоящем описании, не означают какого-либо порядка приоритета, а используются только для отделения одного элемента от другого, а использование формы единственного числа не является ограничением количества, а использовано только для обозначения наличия, по меньшей мере, одного из указанных элементов. Более того, все описанные в настоящем описании диапазоны являются охватывающими и совместимыми, например, диапазоны «до примерно 25 массовых процентов (мас.%), предпочтительно, от примерно 5 до примерно 20 мас.%, более предпочтительно, от примерно 10 до примерно 15 мас.%» включает все конечные и промежуточные значения диапазонов, например, «от примерно 5 до примерно 25 мас.%, от примерно 5 до примерно 15 мас.%» и так далее.
Цеолит: Цеолиты представляют собой микропористые кристаллические алюмосиликатные материалы, особенностью которых является хорошо упорядоченная трехмерная структура с однородными структурами пор/каналов/полостей размером от 3 до 10 Ангстрем (в зависимости от типа каркаса), а также способность вступать в ионный обмен, что делает возможным диспергирование каталитически активных катионов в структуре цеолитов.
Цеолитоподобная структура: Цеолитоподобные структуры представляют собой структурные изотипы/изоморфы цеолитов, но, вместо структуры каркаса, являющейся производным связанных тетраэдров оксида кремния и оксида алюминия, их основой является, например, алюмофосфат (ALPO), кремнийалюмофосфат (SAPO), металлалюмофосфат (Me-ALPO) или металлкремнийалюмофосфат (MeAPSO).
Цеолитный материал представляет собой материал на основе структурных форм, характерных для цеолитов или цеолитоподобных структур.
Под выражением «дикетонатные структурные лиганды» подразумевают лиганд, то есть ион или молекулу, способные связываться с центральным атомом с образованием координационного комплекса, имеющего два набора химических функциональностей, проявляющих кето-енольные формы. Так, кето, то есть кетоновые/альдегидные (углеводород, содержащий карбонил или C=O) - енольные (ненасыщенные спиртовые, то есть C=C-OH) формы относятся к органической химии. Ключевой особенностью кето-енольных систем является свойство, называемое таутомерией, что означает химическое равновесие между кето-формой и енольной формой, включая возможность взаимного перехода одной формы в другую путем переноса протонов и сдвига электронов, образующих связи.
Описанные выше катализатор и способ, а также другие особенности изобретения, будут оценены и понятны лицам, квалифицированным в данной области техники на основании приведенного ниже подробного описания, чертежей и формулы настоящего изобретения.
Примеры
Сравнительный образец A:
150 г H+ формы SAPO34 при постоянном перемешивании добавляли к 480 мл деионизированной воды с получением суспензии, содержащей 24% твердых веществ. Такое добавление вызывало экзотермический эффект в суспензии, в связи с чем данную стадию осуществляли с особой осторожностью. Суспензию непрерывно перемешивали в течение не менее чем 30 минут с целью рассеивания тепла экзотермической реакции, протекающей при смачивании цеолитоподобной структуры. Далее при интенсивном перемешивании в 30 г деионизированной воды растворяли 17,1 г кристаллов Cu(NO3)3·3H2O. Полученный таким образом раствор добавляли по каплям в воронку, образующуюся при перемешивании суспензии, в течение 15 минут. С целью облегчения щелочного осаждения (pH>7) меди, полученную суспензию с pH, составляющим примерно 4, обрабатывали путем добавления по каплям 48 г раствора гидроксида тетраэтиламмония (35 мас.% TEAH) в соответствии со способом по патенту US 5908806, с целью достижения конечного pH, составляющего от 7 до 8. Образец перемешивали в течение еще 60 минут, после чего сушили в течение 20 ч при 65°C на воздухе, а затем прокаливали 2 ч при 500°C на воздухе. Полученный порошок просеивали, как указано в таблице 1, и испытывали без дальнейшей модификации.
Сравнительный образец B:
150 г H+ формы SAPO34 при постоянном перемешивании добавляли к 240 мл деионизированной воды с получением суспензии, содержащей 38,5% твердых веществ. Такое добавление вызвало нагревание суспензии, в связи с чем данной стадии уделяли особую осторожность, суспензию непрерывно перемешивали в течение не менее чем 30 минут с целью рассеивания тепла. Далее в воронку, образующуюся при перемешивании суспензии, добавляли 17,1 г кристаллов Cu(NO3)3·3H2O в течение 1 минуты с получением суспензии с pH, составляющим 4. Вновь, суспензию осаждали путем добавления по каплям 48 г раствора гидроксида тетраэтиламмония (35 мас.% TEAH), с целью достижения конечного pH, составляющего от 7 до 8. Полученная таким образом суспензия оказалась крайне вязкой, перемешивание было затруднено. Образец перемешивали в течение еще 60 минут, после чего сушили в течение 20 ч при 65°C на воздухе, а затем прокаливали 2 ч при 500°C на воздухе. Полученный порошок просеивали, как указано в таблице 1, и испытывали без дальнейшей модификации.
Методика осаждения примерно 100 г 3 мас.% Cu на порошок SAPO34 путем сублимации/смешивания в твердом состоянии (образец F):
12,2 г ацетилацетоната меди (24,4 мас.% меди, поставлен Aldrich) слегка перемешивали с 100 г SAPO34 в герметичной пластиковой бутылке на 250 мл. Затем добавляли 10 г шариков Y-стабилизированного ZrO2 (диаметром 5 мм). Бутылку закрывали и помещали во встряхивающее устройство для красок (Aldrich Model RM 500, 0,55 КВт) и гомогенизировали ее содержимое в течение 5 минут с помощью вибрации. Бутылку удаляли из встряхивающего устройства, и смесь пропускали через грубое сито с целью удаления шариков. Наконец, смешанные порошки переносили в сосуд для прокаливания и нагревали на воздухе в статических условиях (или, в качестве альтернативы, в токе азота) до 350°C (скорость нагрева 5°C/мин) в течение 2 ч с получением активного порошкообразного катализатора, просеянного и испытанного в соответствии с таблицей 1.
Методика получения порошка SAPO34, содержащего 3 мас.% Cu, путем смешивания в твердом состоянии с использованием CuO в качестве предшественника меди (образец C):
Данный материал готовили в соответствии с описанной выше методикой, за исключением того, что смесь включала 0,194 г CuO (содержащего 79,88 мас.% Cu) и 5 г H-SAPO34, и материал не прокаливали после получения гомогенизированной смеси.
Методика получения порошка SAPO34, содержащего 3 мас.% Cu, путем смешивания в твердом состоянии и прокаливания с использованием CuO в качестве предшественника меди (образец D):
Данный материал готовили в соответствии с описанной выше методикой, за исключением того, что смесь включала 0,194 г CuO (содержащего 79,88 мас.% Cu) и 5 г H-SAPO34. После смешивания бутылку извлекали из встряхивающего устройства, и измельченную среду пропускали через грубое сито с целью удаления шариков. Наконец, смешанные порошки переносили в сосуд для прокаливания и нагревали на воздухе в статическом состоянии до 350°C (скорость нагрева 5°C/мин) в течение 2 ч с получением активного порошкообразного катализатора.
Сравнительный пример E: получение 3% Cu-SAPO34 путем ионного обмена в водной среде:
150 г H+ формы SAPO34 при постоянном перемешивании добавляли к 200 мл (то есть 200 г) деионизированной воды с получением суспензии, содержащей 42,8% твердых веществ. Вновь, на данной стадии уделяли особое внимание рассеянию тепла, выделяющегося вследствие экзотермического эффекта, связанного со смачиванием порошка. Суспензию дополнительно непрерывно перемешивали в течение не менее чем 30 минут с целью рассеяния тепла. Далее, при интенсивном перемешивании в 400 г деионизированной воды растворяли 14,0 г ацетата меди(II) (содержащего 32 мас.% Cu). Полученный таким образом раствор добавляли по каплям в воронку, образующуюся при перемешивании суспензии цеолитоподобной структуры, в течение 15 минут. Полученную смесь перемешивали в течение ночи, после чего сушили в течение 10 ч при 110°C на воздухе, а затем прокаливали 2 ч при 500°C на воздухе. Полученный порошок просеивали, как указано в таблице 1, и испытывали без дальнейшей модификации.
Методика получения примерно 100 г порошков SAPO34, содержащих 3 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием оксалата меди в качестве предшественника меди (сравнительный образец G):
Образец готовили описанным выше способом, за исключением того, что смесь включала 7,5 г оксалата меди(II) (содержащего 39,8 мас.% Cu) и 100 г H-SAPO34.
Методика получения примерно 100 г порошков SAPO34, содержащих 3 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетата меди в качестве предшественника меди (сравнительный образец H):
Образец готовили описанным выше способом, за исключением того, что смесь включала 9,3 г ацетата меди(II) (содержащего 32 мас.% Cu) и 100 г H-SAPO34.
Методика получения примерно 100 г порошков SAPO34, содержащих 2,5 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника:
Образцы готовили описанным выше способом, за исключением того, что смесь включала 10,1 г ацетилацетоната меди(II) и 100 г H-SAPO34.
Методика получения примерно 100 г порошков SAPO34, содержащих 2 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника:
Образцы готовили описанным выше способом, за исключением того, что смесь включала 8,07 г ацетилацетоната меди(II) и 100 г H-SAPO34.
Методика получения примерно 100 г порошков SAPO34, содержащих 1.5 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника:
Образцы готовили описанным выше способом, за исключением того, что смесь включала 6,0 г ацетилацетоната меди(II) и 100 г H-SAPO34.
Методика получения примерно 100 г порошков SAPO34, содержащих 1 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника:
Образцы готовили описанным выше способом, за исключением того, что смесь включала 4,0 г ацетилацетоната меди(II) и 100 г H-SAPO34.
Получение примерно 100 г порошков SAPO34, содержащих 2 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника и кофемолки в качестве смесителя (образец J):
Данный материал готовили описанным выше способом, за исключением того, что смесь тщательно гомогенизировали с использованием герметичной кофемолки (универсальное измельчающее устройство IKA M20) в течение 5 минут перед прокаливанием.
Методика получения примерно 100 г порошков SAPO34, содержащих 2 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника и верхнеприводной мешалки в качестве смесителя (образец K):
Образец готовили описанным выше способом, за исключением того, что смесь перед прокаливанием тщательно гомогенизировали в открытом стакане с использованием верхнеприводного смешивающего устройства с высоким сдвиговым усилием (HiTEC-Zang модель MiscoPakt@-mini-35) в течение 5 минут.
Методика получения примерно 120 г порошка ZSM5, содержащего 2,5 мас.% Fe, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната железа в качестве предшественника Fe:
Образцы готовили в соответствии со способом, описанным выше в отношении Cu/SAPO34, за исключением того, что смесь включала 15,8 г ацетилацетоната железа(III) и 109 г ZSM5.
Методика получения примерно 120 г порошка цеолита бета, содержащего 2.5 мас.% Fe, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната железа в качестве предшественника Fe:
Образцы готовили в соответствии со способом, описанным выше в отношении Cu/SAPO34, за исключением того, что смесь включала 15,8 г ацетилацетоната железа(III) и 114 г цеолита бета.
Методика получения сравнительного образца Fe(NO3)3
218 г 
Сравнительный образец цеолита бета (2,5 мас.% Fe) из Fe(NO3)3:
Образец готовили описанным выше способом, за исключением того, что применяли 
Методика получения примерно 120 г порошков ZSM5, содержащих 2 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника Cu:
Образцы готовили в соответствии со способом, описанным выше в отношении Cu/SAPO34, за исключением того, что смесь включала 8,25 г ацетилацетоната меди(II) и 115 г цеолита ZSM5.
Методика получения примерно 120 г порошка цеолита бета, содержащего 2 мас.% Cu, путем смешивания в твердом состоянии/сублимации с использованием ацетилацетоната меди в качестве предшественника Cu:
Образцы готовили в соответствии со способом, описанным выше в отношении Cu/SAPO34, за исключением того, что смесь включала 8,25 г ацетилацетоната меди(II) и 115 г цеолита бета.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИЗАТОРЫ СКВ: ПЕРЕХОДНЫЙ МЕТАЛЛ/ЦЕОЛИТ | 2008 |
|
RU2506989C2 |
| ЦЕОЛИТНЫЕ КАТАЛИЗАТОРЫ, СОДЕРЖАЩИЕ МЕТАЛЛЫ | 2013 |
|
RU2634899C2 |
| АЛЮМОСИЛИКАТНЫЙ ЦЕОЛИТ, СОДЕРЖАЩИЙ ПЕРЕХОДНЫЙ МЕТАЛЛ | 2009 |
|
RU2535706C2 |
| СПОСОБ ПОЛУЧЕНИЯ И АКТИВАЦИИ ПОЛИМЕТАЛЛИЧЕСКИХ ЦЕОЛИТНЫХ КАТАЛИЗАТОРОВ, СОСТАВ И ПРИМЕНЕНИЕ КАТАЛИЗАТОРА ДЛЯ РАЗЛОЖЕНИЯ NO | 2002 |
|
RU2297278C2 |
| Способ приготовления медьсодержащих цеолитов и их применение | 2020 |
|
RU2736265C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАЛЛООБМЕННЫХ ЦЕОЛИТОВ ПОСРЕДСТВОМ ИОННОГО ОБМЕНА В ТВЁРДОМ СОСТОЯНИИ ПРИ НИЗКИХ ТЕМПЕРАТУРАХ | 2014 |
|
RU2674152C2 |
| СИНТЕТИЧЕСКИЙ ЦЕОЛИТ, СОДЕРЖАЩИЙ КАТАЛИТИЧЕСКИЙ МЕТАЛЛ | 2017 |
|
RU2740186C2 |
| Ce-Zr-R-O-КАТАЛИЗАТОРЫ, ПРЕДМЕТЫ, ВКЛЮЧАЮЩИЕ Ce-Zr-R-O-КАТАЛИЗАТОРЫ, И СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ Ce-Zr-R-O-КАТАЛИЗАТОРОВ | 2007 |
|
RU2444405C2 |
| СМЕШАННЫЕ КАТАЛИТИЧЕСКИЕ КОМПОЗИЦИИ МЕТАЛЛ-МЕЛКОПОРИСТОЕ МОЛЕКУЛЯРНОЕ СИТО С 8-ЧЛЕННЫМИ КОЛЬЦАМИ, КАТАЛИТИЧЕСКИЕ УСТРОЙСТВА, СИСТЕМЫ И СПОСОБЫ | 2013 |
|
RU2717953C2 |
| ЦЕОЛИТНЫЕ МАТЕРИАЛЫ ТИПА СНА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПРИМЕНЕНИЕМ ЦИКЛОАЛКИЛАММОНИЕВЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2627399C2 |
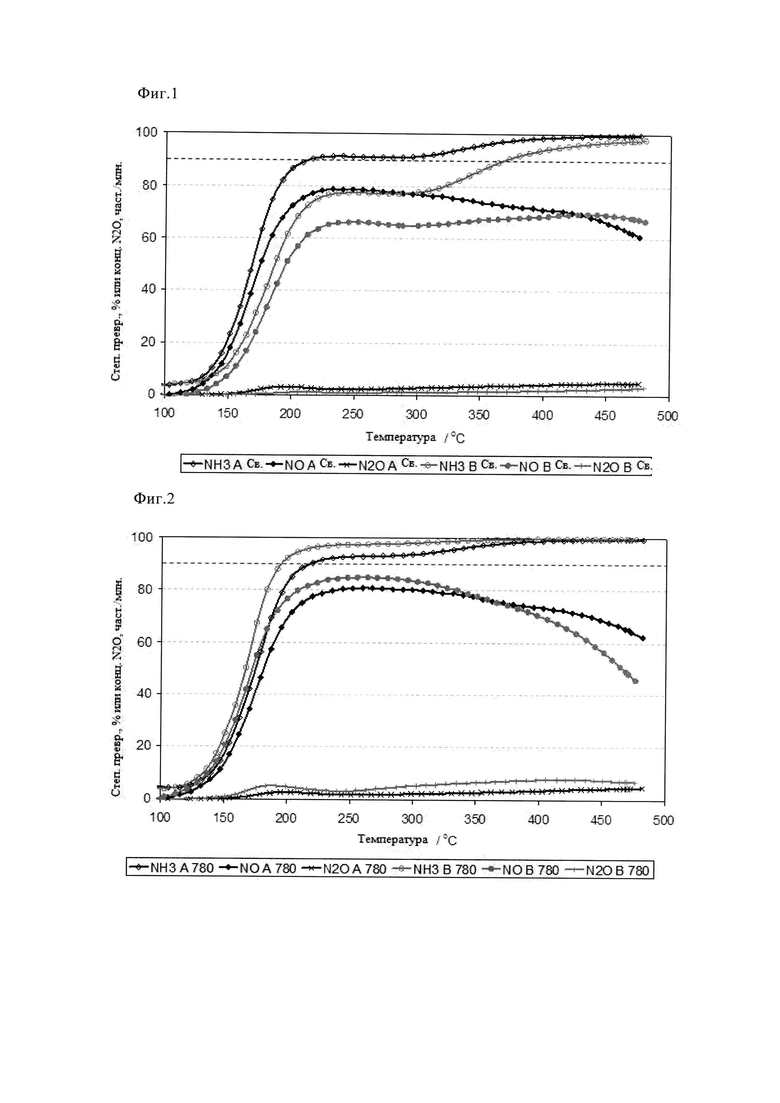

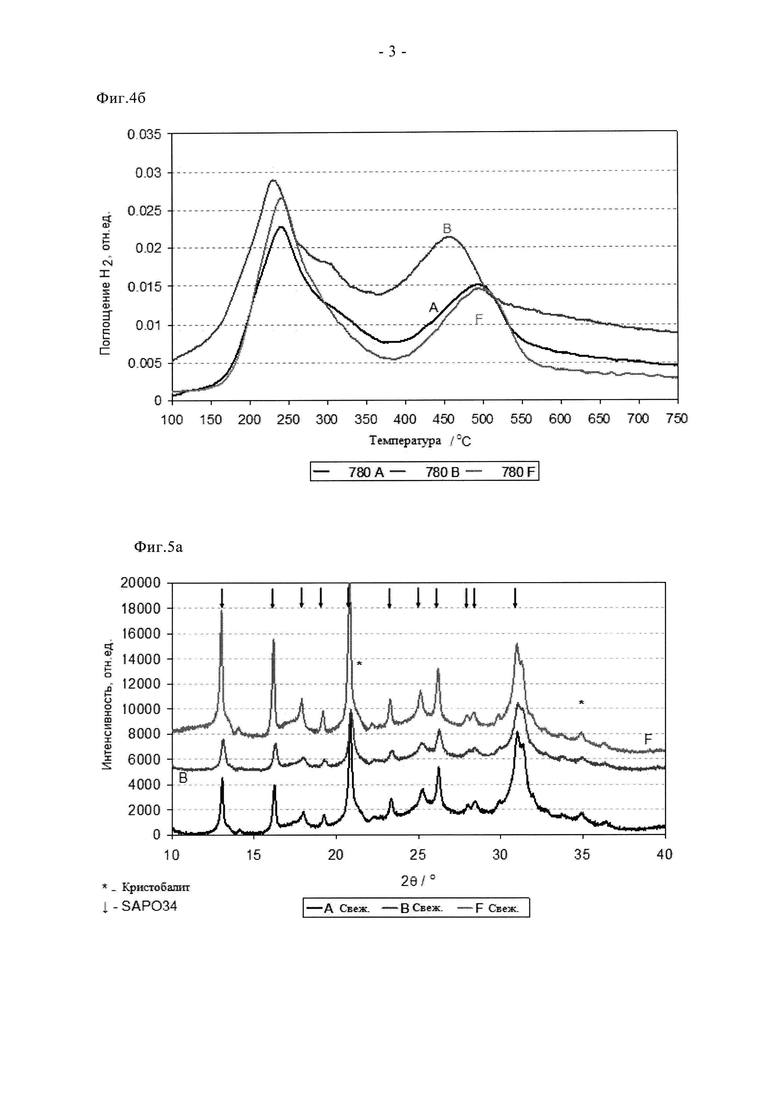
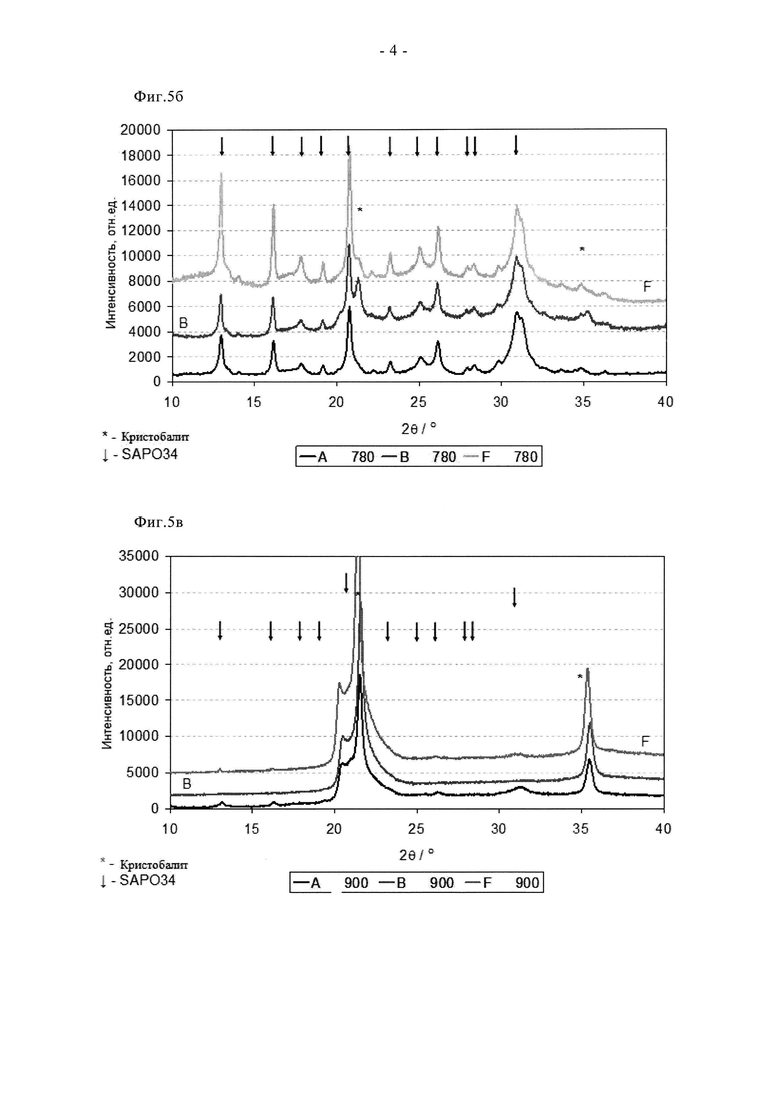
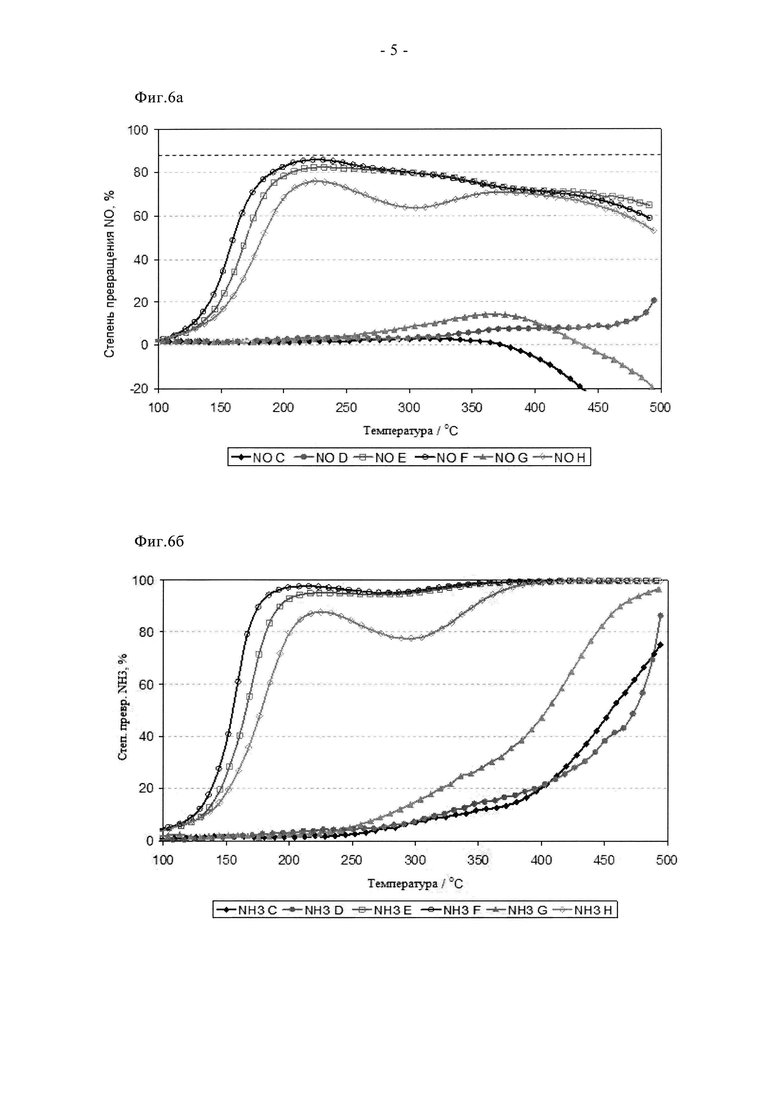

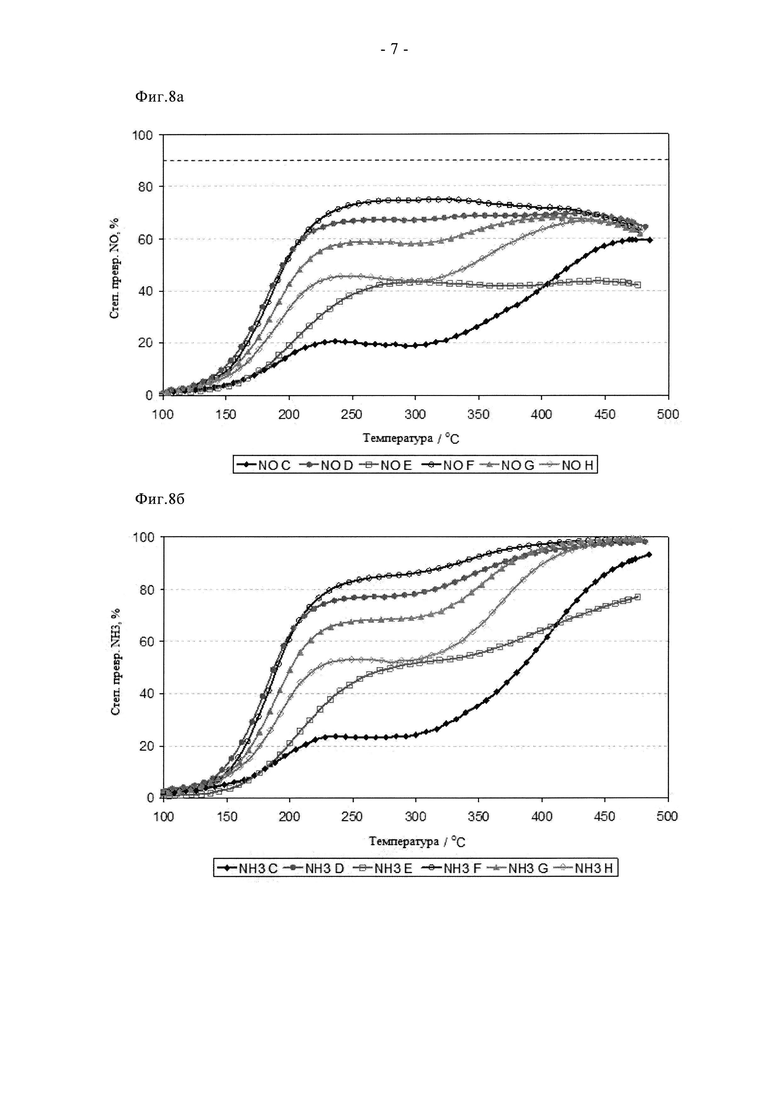
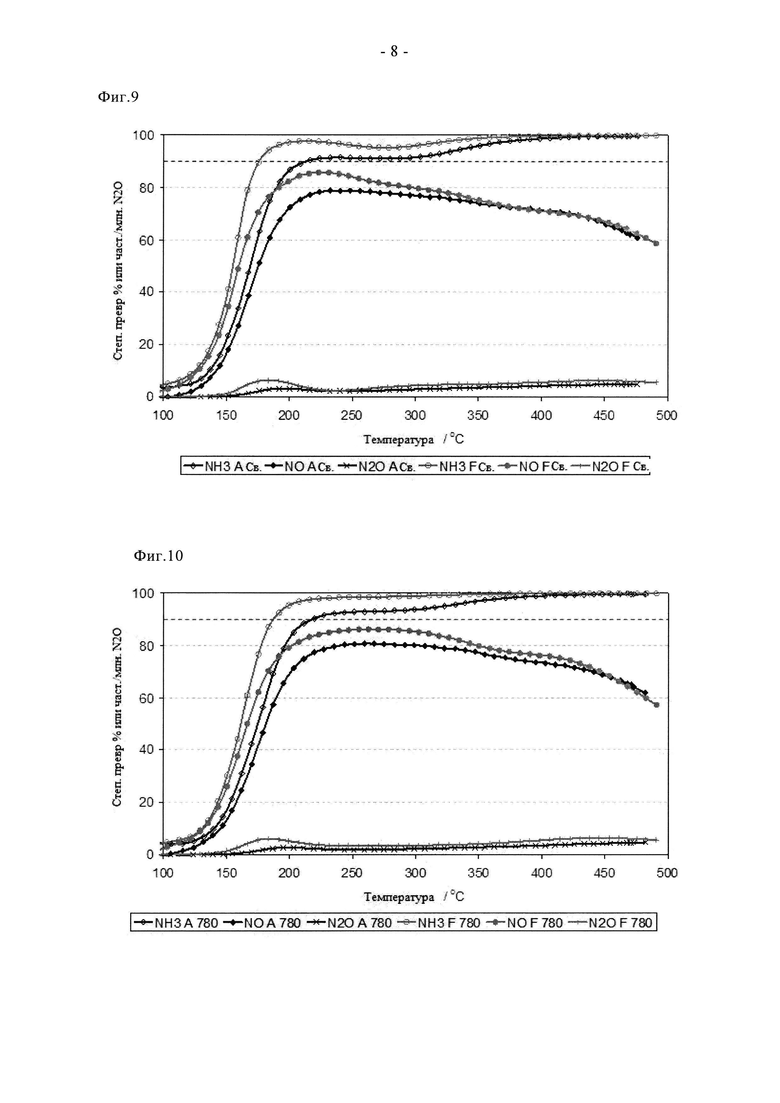
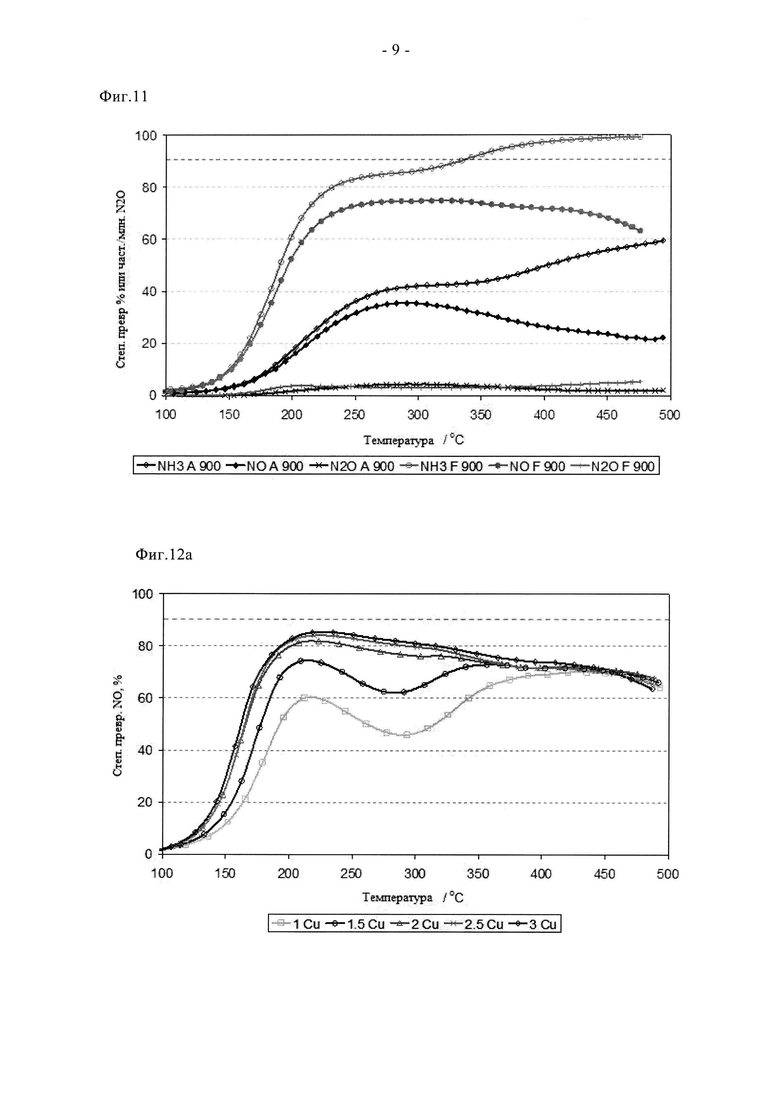
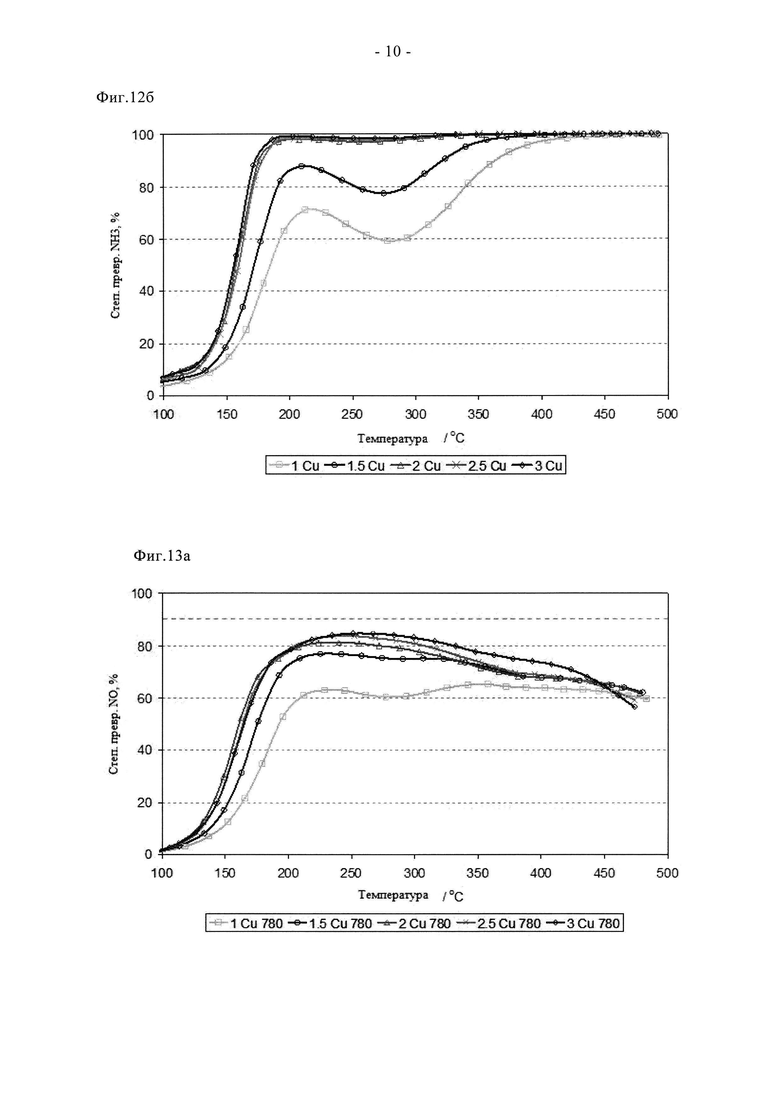
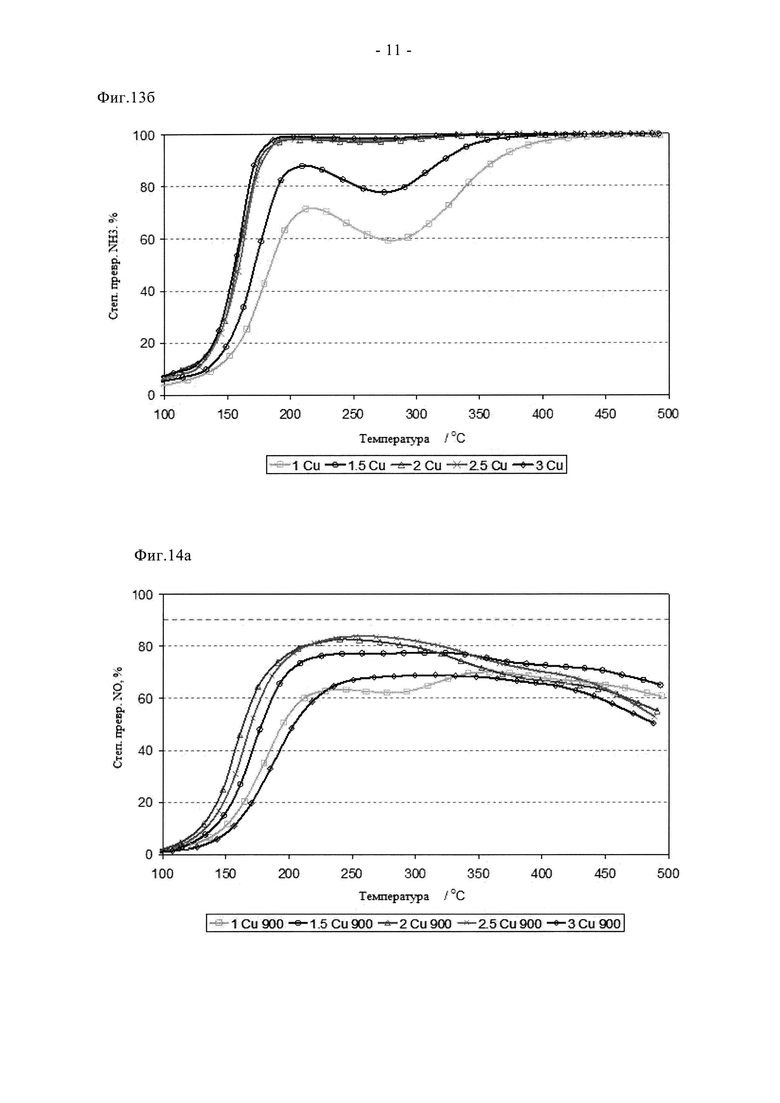

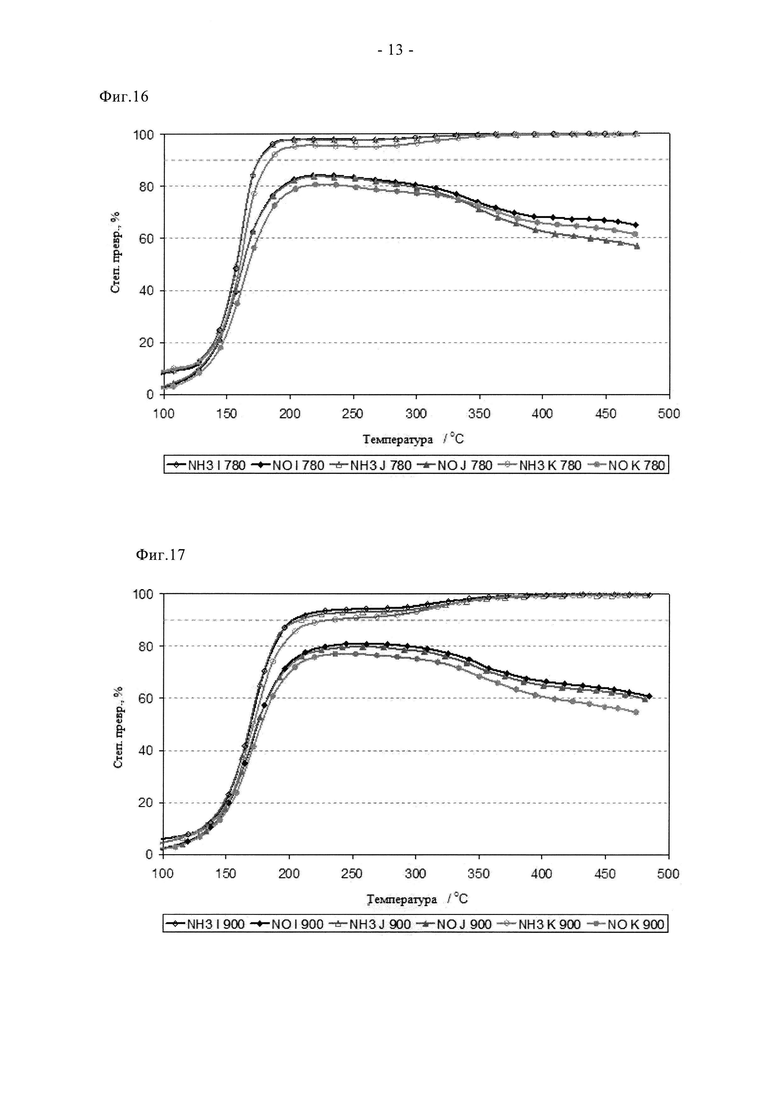
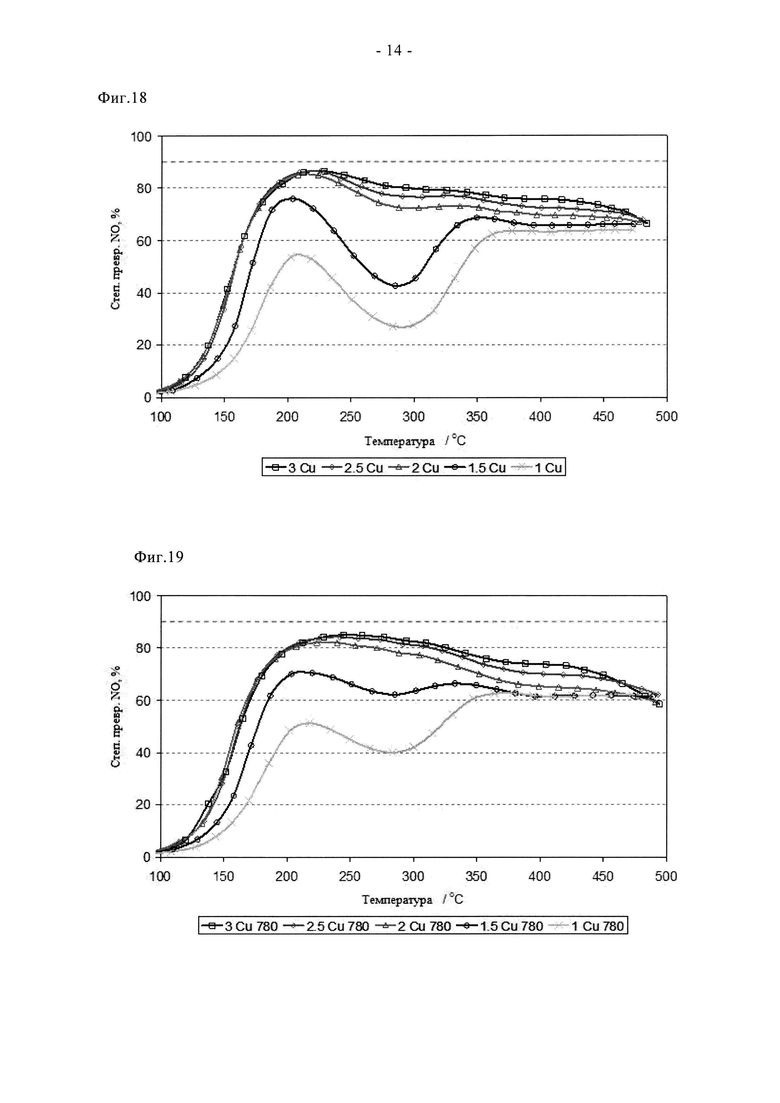

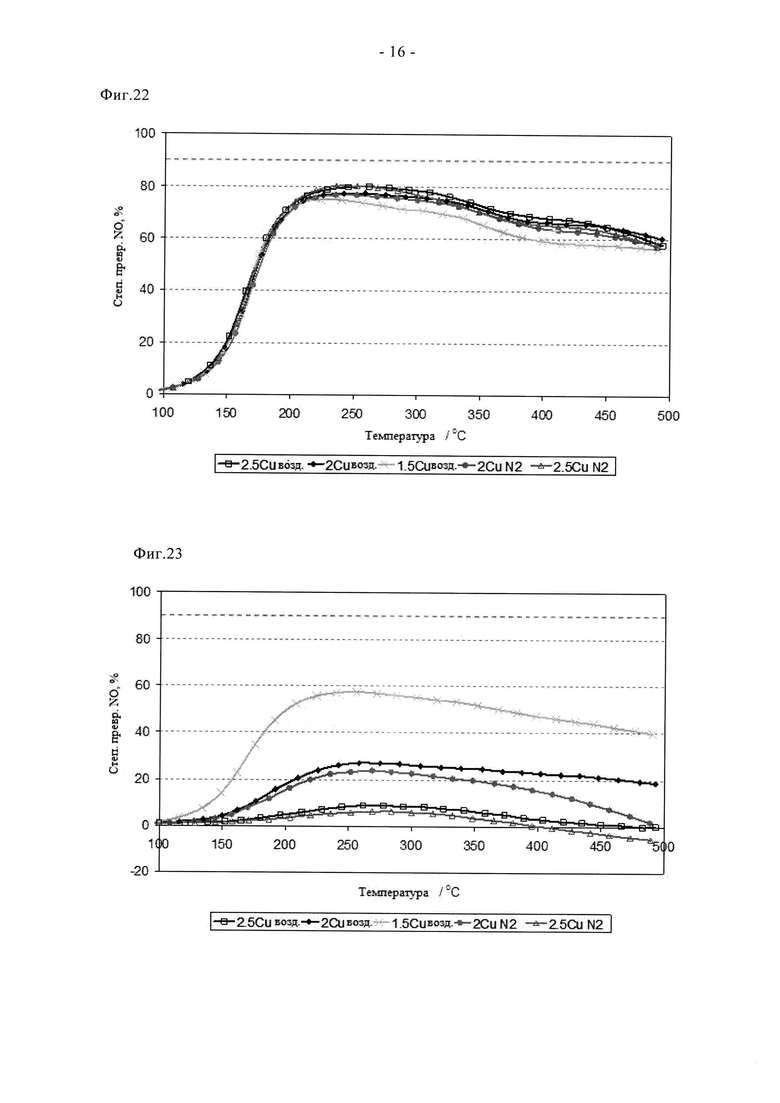
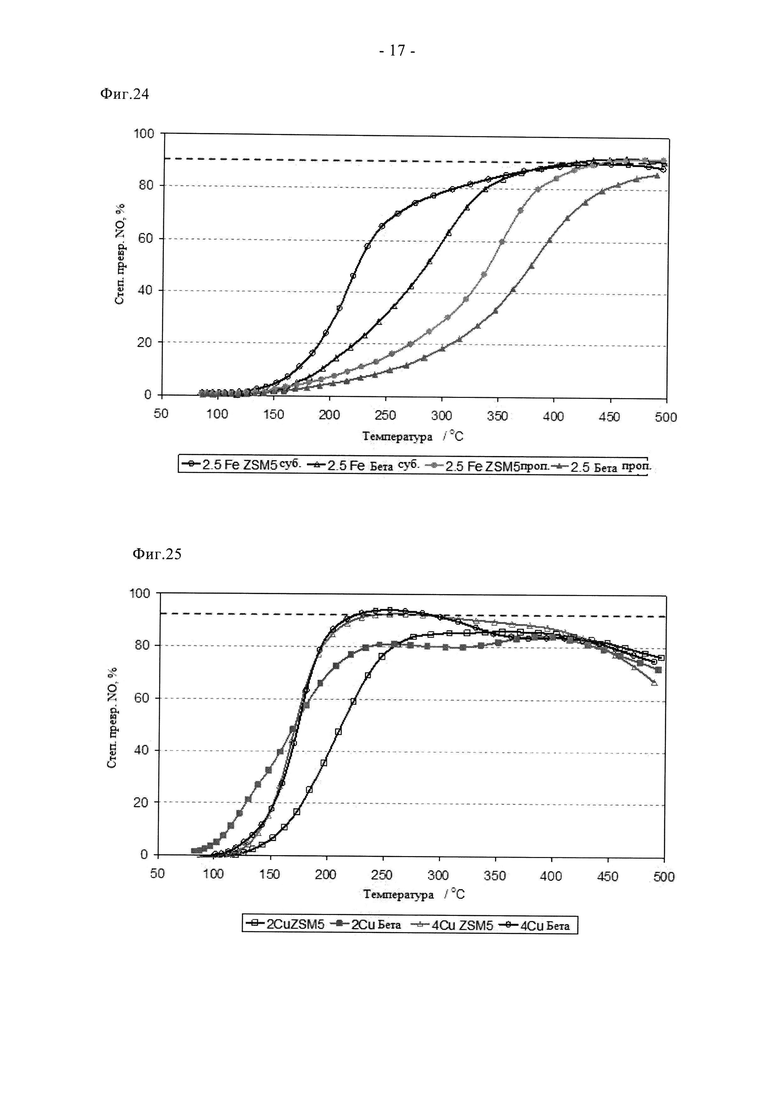
Изобретение относится к катализатору для восстановления оксидов азота, включающему материал или смесь материалов. Катализатор включает инертное термостойкое связующее, выбранное из группы, включающей оксид алюминия, оксид титана, не цеолитный оксид кремния-оксид алюминия, оксид кремния, оксид циркония и смеси перечисленного, нанесен на керамический монолит со сквозными каналами, пенообразный металлический субстрат или на субстрат с фильтрующими стенками. При этом материал или смесь материалов получены способом получения допированных металлами цеолитов и цеолитоподобных структур. Способ включает следующие стадии: I) обеспечение сухой гомогенной смеси цеолита или цеолитоподобной структуры с одним или более соединениями-предшественниками, включающими комплексное соединение, образованное переходным металлом и лигандом, соответствующим структурной формуле (I): ML1 mL2 n (I), в которой М представляет собой металл, выбранный из группы, включающей V, Cr, Mn, Fe, Со, Ni, Cu, Nb, Mo, Ru, Rh, Pd, Ag и Се, L1 представляет собой карбонил, амин, алкил, алкокси, алкен, арен, фосфин или другой нейтральный координирующий лиганд, m представляет собой число от 0 до 6, n представляет собой число, равное валентности М, и L2 представляет собой дикетонат или родственный член данного гомологического ряда, например лиганд, соответствующий формуле (II):
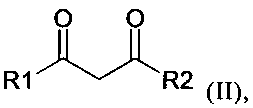
где R1 и R2 независимо представляют собой алкил, замещенный акил, арил, замещенный арил, ацил и замещенный ацил; II) прокаливание смеси без применения пониженного давления при достаточной температуре и в течение достаточного времени для обеспечения мобилизирования и разложения соединения-предшественника; и III) получение допированного металлом цеолита или цеолитоподобной структуры. Также предложены монолитный катализатор для восстановления оксидов азота и применение катализаторов. Изобретение позволяет получить катализатор, не применяя растворители, и обладающего преимуществами по сравнению с известными способами как с экологической, так и с экономической точек зрения. 3 н. и 9 з.п. ф-лы, 34 ил, 4 табл., 1 пр.
1. Катализатор для восстановления оксидов азота, включающий материал или смесь материалов, причем указанный катализатор включает инертное термостойкое связующее, выбранное из группы, включающей оксид алюминия, оксид титана, не цеолитный оксид кремния-оксид алюминия, оксид кремния, оксид циркония и смеси перечисленного, нанесенный на керамический монолит со сквозными каналами, пенообразный металлический субстрат или на субстрат с фильтрующими стенками, причем материал или смесь материалов получены способом получения допированных металлами цеолитов и цеолитоподобных структур, включающим следующие стадии:
I) обеспечение сухой гомогенной смеси цеолита или цеолитоподобной структуры с одним или более соединениями-предшественниками, включающими комплексное соединение, образованное переходным металлом и лигандом, соответствующим структурной формуле (I):
ML1 mL2 n (I),
в которой
М представляет собой металл, выбранный из группы, включающей V, Cr, Mn, Fe, Со, Ni, Cu, Nb, Mo, Ru, Rh, Pd, Ag и Се, и
L1 представляет собой карбонил, амин, алкил, алкокси, алкен, арен, фосфин или другой нейтральный координирующий лиганд,
m представляет собой число от 0 до 6,
n представляет собой число, равное валентности М, и
L2 представляет собой дикетонат или родственный член данного гомологического ряда, например лиганд, соответствующий формуле (II):
в которой:
R1 и R2 независимо представляют собой алкил, замещенный акил, арил, замещенный арил, ацил и замещенный ацил; и
II) прокаливание смеси без применения пониженного давления при достаточной температуре и в течение достаточного времени для обеспечения мобилизирования и разложения соединения-предшественника; и
III) получение допированного металлом цеолита или цеолитоподобной структуры.
2. Катализатор по п.1, в котором материал или смесь материалов, а также связующее нанесены на отдельные зоны керамического монолита со сквозными каналами, пенообразного металлического субстрата или субстрата с фильтрующими стенками.
3. Монолитный катализатор для восстановления оксидов азота, полученный путем экструзии материала или смеси материалов, которые получены способом получения допированных металлами цеолитов и цеолитоподобных структур, включающим следующие стадии:
I) обеспечение сухой гомогенной смеси цеолита или цеолитоподобной структуры с одним или более соединениями-предшественниками, включающими комплексное соединение, образованное переходным металлом и лигандом, соответствующим структурной формуле (I):
ML1 mL2 n (I),
в которой
М представляет собой металл, выбранный из группы, включающей V, Cr, Mn, Fe, Со, Ni, Cu, Nb, Mo, Ru, Rh, Pd, Ag и Се, и
L1 представляет собой карбонил, амин, алкил, алкокси, алкен, арен, фосфин или другой нейтральный координирующий лиганд,
m представляет собой число от 0 до 6,
n представляет собой число, равное валентности М, и
L2 представляет собой дикетонат или родственный член данного гомологического ряда, например лиганд, соответствующий формуле (II):
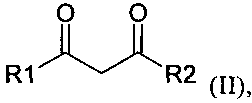
в которой:
R1 и R2 независимо представляют собой алкил, замещенный акил, арил, замещенный арил, ацил и замещенный ацил; и
II) прокаливание смеси без применения пониженного давления при достаточной температуре и в течение достаточного времени для обеспечения мобилизирования и разложения соединения-предшественника; и
III) получение допированного металлом цеолита или цеолитоподобной структуры.
4. Катализатор по п.1, 2 или 3, в котором цеолиты или цеолитоподобные структуры выбирают из группы, включающей одну из следующих структур или их смесь: цеолит типа фожазита, типа пентасила, чабазитный цеолит или цеолитоподобную структуру, например SAPO-34 или другие структуры на основе восьмичленных колец структурного типа СНА и соответствующие структуры, например, типов AEI, AFT, AFX, DDR, ERI, ITE, ITW, KFI, LEV, LTA, PAU, RHO и UFI.
5. Катализатор по п.1, 2 или 3, в котором металл выбирают из группы, включающей Fe, Cu, Со, Ag и Се.
6. Катализатор по п.1, 2 или 3, в котором лиганд комплексного соединения выбирают из одного из или смеси веществ, выбранных из группы, включающей дикетонатную структуру и карбонилсодержащие молекулы.
7. Катализатор по п.1 или 2, в котором смесь прокаливают при температуре от не менее чем 200 до 650°С.
8. Катализатор по п.7, в котором смесь прокаливают при температуре от 350 до 450°С в течение времени от 1 до 5 ч.
9. Катализатор по п.1, 2 или 3, в котором смесь включает цеолитный или цеолитоподобный материал и соединение-предшественник, с целью обеспечения последующего содержания допирующего металла, составляющего от 0,01 до 10 мас. % металла.
10. Применение катализатора или монолитного катализатора по одному или более из пп. с 1 по 9 для селективного каталитического восстановления оксидов азота.
11. Применение катализатора или монолитного катализатора по п.10, причем вводят источник азотсодержащего восстановителя с целью обеспечения эффективного отношения NH3:NOx (отношения α) на входе в катализатор, составляющего от 0,5 до 2.
12. Применение катализатора или монолитного катализатора по одному или более из пп. 10 и/или 11, причем отношение NO:NO2, регистрируемое на входе в катализатор, составляет от 1:0 до 1:3 по объему.
| Устройство для подачи нити | 1978 |
|
SU749781A1 |
| et al, Characterization of intrazeolitic Fe3+ prepared by chemical vapor deposition of [(C5H5)Fe(CO)2]2 inside NaY and FSM-16 zeolites and their catalytic activities towards phenol hydroxylation, MATERIALS RESEARCH BULLETIN, 2003, v | |||
| Способ сужения чугунных изделий | 1922 |
|
SU38A1 |
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| WO 2008009453 A2, 24.01.2008 | |||
| US 2009196812 A1, 06.08.2009 | |||
| ПОЛУЧЕНИЕ НАНЕСЕННЫХ КАТАЛИЗАТОРОВ НА ОСНОВЕ МЕТАЛЛ/ОКСИД МЕТАЛЛА ПУТЕМ ПРЕДШЕСТВУЮЩЕЙ ХИМИЧЕСКОЙ НАНОМЕТАЛЛУРГИИ В ОПРЕДЕЛЕННЫХ РЕАКЦИОННЫХ ПРОСТРАНСТВАХ ПОРИСТЫХ НОСИТЕЛЕЙ С ПОМОЩЬЮ МЕТАЛЛОРГАНИЧЕСКИХ И/ИЛИ НЕОРГАНИЧЕСКИХ ПРЕДШЕСТВЕННИКОВ И МЕТАЛЛСОДЕРЖАЩИХ ВОССТАНОВИТЕЛЕЙ | 2005 |
|
RU2380155C2 |
| Катализатор для очистки выхлопных газов от окислов азота | 1974 |
|
SU652868A3 |

