Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям, которые могут применяться при лечении группы расстройств и нарушений, связанных с амилоидным белком, таких как болезнь Альцгеймера, и заболеваний или состояний, связанных с амилоидоподобными белками. Кроме того, настоящее изобретение относится к фармацевтическим композициям, включающим эти соединения, и к применению этих соединений для получения лекарственных средств для лечения или предотвращения заболеваний или состояний, связанных с амилоидными и/или амилоидоподобными белками. Кроме того, раскрывается способ лечения или предотвращения заболеваний или состояний, связанных с амилоидными и/или амилоидоподобными белками.
Соединения настоящего изобретения могут также применяться при лечении глазных заболеваний, связанных с патологическими нарушениями/изменениями в ткани визуальной системы, в частности, связанных с патологическими нарушениями/изменениями в ткани визуальной системы, имеющими отношение к бета-амилоиду, такими как нейрональная деградация. Указанные патологические нарушения могут возникать, например, в различных тканях глаза, таких как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталики, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, например, возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
Уровень техники
Многие заболевания в пожилом возрасте вызываются или связаны с амилоидными или амилоидоподобными белками, и они характеризуются, отчасти, образованием внеклеточных отложений амилоидного или амилоидоподобного вещества, которые влияют на патогенез, а также развитие заболевания. Эти заболевания включают, но этим не ограничивая, неврологические нарушения, такие как болезнь Альцгеймера (AD), заболевания или состояния, характеризующиеся потерей когнитивной способности памяти, такие как, например, умеренное когнитивное нарушение (MCI), деменция с тельцами Леви, синдром Дауна, врожденное внутримозговое кровоизлияние с амилоидозом (голландского типа); комплекс Гуама паркинсонизм-деменция. Другими заболеваниями, которые вызываются или связаны с амилоидными или амилоидоподобными белками, являются прогрессирующий супрануклеарный парез, множественный склероз; болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанная с ВИЧ деменция, ALS (боковой амиотрофический склероз), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте; старческий амилоидоз сердца; внутрисекреторные опухоли и другие заболевания, включающие связанные с амилоидом глазные заболевания, которые поражают различные ткани глаза, такие как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталик глаза, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, в частности, возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
Патогенез этих заболеваний может быть различным, но характерные отложения при этих заболеваниях часто содержат много общих для этих отложений молекулярных компонентов. В значительной степени, это может быть связано с локальной активацией провоспалительных проводящих путей, что приводит к одновременному отложению активированных компонентов комплемента, острофазовых агентов, иммунных модуляторов и других медиаторов воспаления.
Болезнь Альцгеймера (AD) представляет собой неврологическое нарушение, которое, как в основном считают, вызывается амилоидными бляшками, накоплением аномального отложения белков в мозге. Наиболее часто встречающийся тип амилоида, обнаруживаемый в мозге страдающих этим заболеванием людей, состоит в основном из Αβ волокон. Научно доказано, что увеличение и аккумуляция бета-амилоидного белка в бляшках приводит к гибели нервных клеток, что способствует развитию и прогрессированию болезни Альцгеймера. Гибель нервных клеток в стратегически важных областях головного мозга, в свою очередь, вызывает уменьшение количества нейротрансмиттеров и нарушение памяти. Белки, которые, главным образом, ответственны за образования бляшки, включают белок-предшественник амилоида (APP) и два пресенилина (пресенилин I и пресенилин II). Последующее расщепление белка-предшественника амилоида (APP), который конститутивно экспрессируется и катаболизируется в большинстве клеток, под действием ферментов β и γ секретазы приводит к высвобождению от 39 до 43 аминокислот Αβ пептида. Расщепление белков-предшественников амилоида, вероятно, повышает их предрасположенность к агрегации в бляшки. В частности, фрагмент Αβ(1-42) является именно тем фрагментом, который имеет высокую предрасположенность к образованию агрегатов, благодаря двум очень гидрофобным аминокислотным остаткам на его C-конце. Поэтому считают, что фрагмент Αβ(1-42) в основном участвует и содействует инициированию образования нейритических бляшек при болезни Альцгеймера и имеет, вследствие этого, высокое патологическое воздействие. Следовательно, существует необходимость в создании специфических молекул, действие которых направлено на предотвращение образования амилоидных бляшек.
Симптомы болезни Альцгеймера проявляются медленно, и сначала симптомом может являться только умеренная забывчивость. На этой стадии больные могут забывать недавние события, действия, имена знакомых людей или названия вещей и могут быть не способны решать простые математические задачи. По мере развития болезни симптомы становятся более заметными и достаточно серьезными для того, чтобы люди, страдающие болезнью Альцгеймера, и их близкие родственники начинали осознавать необходимость в обращении за медицинской помощью. Симптомы средней стадии болезни Альцгеймера включают забывание того, как выполняются простые задачи, такие как, например, гигиенический уход за телом, и возникновение проблем с речью, пониманием, чтением или письмом. Пациенты на более поздней стадии болезни Альцгеймера могут становиться беспокойными или агрессивными, могут уйти из дома и заблудиться, и, в конце концов, нуждаются в полном уходе.
В настоящее время единственным способом диагностирования болезни Альцгеймера является выявление бляшек и сплетений в ткани головного мозга при аутопсии после смерти больного. Поэтому, пока пациент еще жив, врачи могут только ставить диагноз "возможной" или "вероятной" болезни Альцгеймера. Используя современные методы, врачи могут достоверно диагностировать болезнь Альцгеймера до 90 процентов случаев с помощью различных способов диагностирования "возможной" болезни Альцгеймера. Врачи задают вопросы относительно общего состояния здоровья пациента, медицинских проблем в прошлом, и любых проблем, с которыми пациент сталкивается в процессе повседневной деятельности. Поведенческие тесты относительно памяти, способности решать задачи, внимания, способности к проведению вычислений, и способности выражать свои мысли позволяют получать информацию о когнитивном расстройстве, а медицинские исследования, такие как анализ крови, мочи или спинномозговой жидкости, и томограммы головного мозга могут давать некоторую дополнительную информацию.
Тактика лечения болезни Альцгеймера включает в себя медикаментозное и немедикаментозное лечение. В настоящее время лечение, направленное на изменение основного течения болезни (замедление или обратимость прогрессирования болезни), обычно не дает положительных результатов. Было показано, что лекарственные средства, которые восстанавливают дефицит (дефект) или неправильное функционирование химических мессенджеров нервных клеток (нейротрансмиттеров), в частности, ингибиторы холинэстеразы (ChEI), такие как такрин и ривастигмин, улучшают симптомы. Ингибиторы холинэстеразы препятствуют ферментативному расщеплению нейротрансмиттеров, что в результате увеличивает количество химических мессенджеров, способных передавать нервные сигналы в мозге.
В случае некоторых пациентов с ранней и средней стадией развития заболевания, лекарственные средства такрин (COGNEX®, Morris Plains, NJ), донепезил (ARICEPT®, Tokyo, JP), ривастигмин (EXELON®, East Hanover, NJ) или галантамин (REMINYL®, New Brunswick, NJ) могут способствовать предотвращению ухудшения некоторых симптомов в течение ограниченного периода времени. Еще одно лекарственное средство, мемантин (NAMENDA®, New York, NY), было опробовано для лечения умеренных и тяжелых случаев болезни Альцгеймера. Доступны также препараты, предназначенные для лечения психиатрических проявлений при болезни Альцгеймера. Кроме того, некоторые препараты позволяют контролировать поведенческие симптомы при болезни Альцгеймера, такие как бессонница, возбуждение, бред, страх и депрессия. Лечение этих симптомов часто позволяет пациентам чувствовать себя более комфортно и позволяет лицам, ухаживающим за пациентами, более легко осуществлять за ними уход. К сожалению, несмотря на существенные достижения при лечении, которые показали, что этот класс препаратов однозначно демонстрирует лучшие результаты, чем плацебо, заболевание продолжает прогрессировать, и средний результат воздействия этих препаратов на умственную деятельность всем еще остается весьма скромным. Многие из лекарственных средств, применяемых при медикаментозном лечении болезни Альцгеймера, такие как, например, ингибиторы холинэстеразы, кроме того, еще имеют побочные эффекты, которые включают желудочно-кишечное расстройство, печеночную токсичность и потерю массы тела.
Другие заболевания, которые вызываются или связаны с накоплением и отложением амилоидподобного белка, представляют собой умеренное когнитивное нарушение, деменцию с тельцами Леви (LBD), боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM) и дегенерацию желтого пятна, в частности, возрастную дегенерацию желтого пятна (AMD).
Умеренное когнитивное нарушение (MCI) является общим термином, обычно определяемое как незаметное, но поддающееся измерению нарушение памяти. Пациент с умеренным когнитивным нарушением испытывает проблемы с памятью в большей степени, чем обычно можно было бы ожидать в процессе старения, но не имеет других симптомов деменции, таких как помутнение сознания или нарушение мышления.
Деменция с тельцами Леви (LBD) представляет собой нейродегенеративное заболевание, которое может возникать у людей старше 65 лет, которое обычно вызывает симптомы когнитивного нарушения (нарушения мышления) и отклоняющихся от нормы изменений поведения. Симптомы могут включать когнитивное нарушение, неврологические признаки, расстройство сна и нарушение вегетативной регуляции. Когнитивное нарушение является в большинстве случаев признаком наличия деменции с тельцами Леви. Пациенты имеют периодически повторяющиеся случаи проявления спутанности сознания, которые постепенно становятся все более тяжелыми. Изменение когнитивной способности часто связано со степенью непостоянства внимания и внимательности. Когнитивное нарушение и изменения процесса мышления могут иметь продолжительность в течение нескольких минут, часов или дней.
Боковой амиотрофический склероз (ALS) характеризуется дегенерацией верхних и нижних двигательных нейронов. У ряда пациентов с боковым амиотрофическим склерозом, может иметь место деменция или афазия (ALS-D). Деменция обычно представляет собой лобно-височную деменцию (FTD), и во многих из этих случаев она характеризуется убиквитин-позитивными, тау-негативными включениями в нейронах зубчатой извилины медиальной и нижней поверхности полушария большого мозга и поверхностных слоев лобной и височной доли.
Миозит с включенными тельцами (IBM) представляет собой приводящее к инвалидности заболевание, обычно обнаруживаемое у людей старше 50 лет, при котором происходит воспаление мышечных волокон и начинается их атрофия - но при котором не повреждается головной мозг, и пациенты сохраняют полностью их умственные способности. Было обнаружено, что внутри мышечных клеток пациентов, страдающих этим самым распространенным прогрессирующим заболеванием мышц пожилых людей, при котором также повышается содержание β-амилоида, повышается содержание двух ферментов, участвующих в продуцировании β-амилоидного белка.
Дегенерация желтого пятна представляет собой общее глазное заболевание, которое вызывает разрушение желтого пятна, являющегося центральной областью сетчатки (тонкая как бумага ткань сзади глаза, откуда чувствительные к свету клетки посылают зрительные сигналы в головной мозг). Нормальный четкий находящийся впереди зрительный образ воспроизводится с помощью желтого пятна. Повреждение желтого пятна приводит к развитию слепых пятен и затуманенному или искаженному зрению. Возрастная дегенерация желтого пятна (AMD) является главной причиной нарушения зрения в США, и для людей старше 65 лет, живущих на Кавказе, она является основной причиной практической слепоты. Приблизительно 1,8 миллиона американцев в возрасте 40 лет и старше имеют запущенную форму дегенерации желтого пятна, и еще 7,3 миллиона человек с промежуточной стадией дегенерации желтого пятна подвержены существенному риску потери зрения. Согласно оценке правительства, к 2020 году запущенную форму дегенерации желтого пятна будут иметь 2,9 миллиона человек. Жертвы дегенерации желтого пятна удивлены и разочарованы тем, как мало известно о причинах и лечении этого состояния, приводящего к слепоте.
Существует две формы дегенерации желтого пятна: сухая дегенерация желтого пятна и влажная дегенерация желтого пятна. Сухая форма, при которой медленно начинают разрушаться клетки желтого тела, диагностируется в 85 процентах случаев дегенерации желтого пятна. Сухая дегенерация желтого пятна обычно поражает оба глаза, хотя в одном глазу может происходить потеря зрение, тогда как второй глаз остается неповрежденным. Друзы, которые представляют собой желтые отложения под сетчаткой, являются общими ранними признаками сухой дегенерации желтого пятна. Риск развития запущенной сухой дегенерации желтого пятна или влажной дегенерации желтого пятна повышается по мере увеличения числа или размера друз. Сухая дегенерация желтого пятна может переходить в запущенную стадию и вызывать потерю зрения без превращения во влажную форму заболевания; однако, существует возможность для ранней стадии сухой дегенерации желтого пятна неожиданно превращаться во влажную форму.
Влажная форма, несмотря на то, что она составляет только 15 процентов случаев, приводит в 90 процентах случаев к слепоте, и ее считают запущенной дегенерацией желтого пятна (при влажной дегенерации желтого пятна отсутствуют ранняя и промежуточная стадии). Влажной дегенерации желтого пятна всегда предшествует сухая форма заболевания. По мере того, как усугубляется сухая форма, у некоторых пациентов появляются аномальные кровеносные сосуды, растущие позади желтого пятна. Эти сосуды очень хрупкие, и из них происходит утечка жидкости и крови (отсюда и термин "влажная" дегенерация желтого пятна), что приводит к быстрому повреждению желтого пятна.
Сухая форма дегенерации желтого пятна в начальной стадии может часто приводить к слегка затуманенному зрению. Затем, в частности, центральная часть изображения может становиться размытой, и эта область увеличивается все больше и больше по мере прогрессирования заболевания. В случае поражения только одного глаза, симптомы могут быть незаметными. При влажной дегенерации желтого пятна, прямые линии могут представляться волнистыми, и может быстро возникнуть потеря центрального зрения.
Диагностирование дегенерации желтого пятна обычно включает расширенную проверку зрения, проверку остроты зрения, и осмотр задней части глаза с использованием процедуры, называемой исследованием глазного дна, для облегчения диагностирования дегенерации желтого пятна, и, если подозревается влажная дегенерация желтого пятна, то может быть также проведена ангиография с флуоресцеином. Если сухая дегенерация желтого пятна достигает запущенных стадий, то в настоящее время отсутствует лечение для предотвращения потери зрения. Однако специфическая высокая доза антиоксидантов и цинка может замедлить или предотвратить развитие промежуточной стадии дегенерация желтого пятна в запущенную стадию. Macugen® (инъекция пегаптаниба натрия), лазерная фотокоагуляция и фотодинамическая терапия позволяют контролировать аномальный рост кровеносных сосудов и кровотечение в желтом пятне, что оказывает положительное воздействие на некоторых пациентов, страдающих влажной дегенерацией желтого пятна; однако, если зрение уже потеряно, то с помощью этих методов его восстановить нельзя. Если зрение уже потеряно, то в этом случае могут быть использованы специальные устройства визуализации для слабого зрения, которые могут помочь улучшить качество жизни.
Одним из самых ранних признаков возрастной дегенерации желтого пятна (AMD) является накопление внеклеточных отложений, называемых друзами, между базальной пластинкой пигментного эпителия сетчатки (RPE) и оболочкой Бруха (BM). Недавние исследования, проведенные Андерсоном с соавторами (Anderson et al. Experimental Eye Research 78 (2004) 243-256), подтвердили тот факт, что друзы содержат бета-амилоид.
Прионы вызывают нейродегенеративные заболевания, такие как почесуха у овец, губчатая энцефалопатия крупного рогатого скота и болезнь Крейтцфельдта-Якоба у людей. Единственным известным компонентом приона является изоформа белка скрепи, PrPSc. Несмотря на то, что происходит рост числа прионов, тем не менее, доказательства того, что они содержат нуклеиновую кислоту, отсутствуют. PrPSc образуется из неинфекционного клеточного белка PrPC в результате посттрансляционного процесса, в котором PrPC подвергается глубокому конформационному изменению.
Белок скрепи PrPSc играет очень важную роль в нейронной дегенерации, и в процессе развития заболевания подвергается трехстадийной транзиции: PrPC (изоформа нормального клеточного белка) - PrPSc: инфекционная форма (изоформа белка скрепи) - белок PrP27-30.
Такой каскад превращений возникает в процессе развития болезни Крейтцфельдта-Якоба (CJD), нейровирусной инфекции куру, синдрома Герстмана-Штраусслера-Шейнкера (GSS), спорадической фатальной инсомнии у человека, почесухи у овец и коз, энцефалопатии у норок и губчатой энцефалопатии крупного рогатого скота.
Клеточный нетоксичный белок (PrPC) представляет собой сиалогликопротеин с молекулярной массой от 33000 до 35000, который экспрессирует в основном в нейронах. При упомянутых выше заболеваниях, PrPC превращается в измененную форму (PrPSc), которая отличается от ее нормального гомолога относительной устойчивостью к протеазному расщеплению. PrPSc накапливается в центральной нервной системе больных животных и людей, и его устойчивое к действию протеазы ядро образует внеклеточные агрегаты.
Амилоидоз не является единственной нозологической формой, а наоборот, он представляет собой разнообразную группу прогрессирующих заболеваний, характеризующихся внеклеточными отложениями в тканях воскообразного крахмалоподобного белка, называемого амилоидом, который накапливается в одном или более органах или системах организма. По мере того как образуются амилоидные отложения, они начинают отрицательно влиять на нормальную функцию органа или системы организма. Существуют по меньшей мере 15 различных типов амилоидоза. Основными формами являются первичный амилоидоз при отсутствии предшествующего известного заболевания, вторичный амилоидоз после некоторого другого состояния и наследственный амилоидоз.
Вторичный амилоидоз возникает у людей, которые страдают хронической инфекцией или воспалительным заболеванием, таким как туберкулез, бактериальная инфекция, называемая семейной средиземноморской лихорадкой, костные инфекции (остеомиелит), ревматоидный артрит, воспаление тонкого кишечника (гранулематозный илеит), болезнь Ходжкина и проказа.
Глаукома представляет собой группу заболеваний зрительного нерва, включающих потерю ганглиозных клеток сетчатки глаза (RGC) при характерной картине оптической нейропатии. Глаукома часто, но не всегда, сопровождается повышенным внутриглазным давлением, которое может быть результатом блокады водной циркуляции или водного оттока.
Несмотря на то, что повышенное внутриглазное давление является значимым фактором риска для развития глаукомы, тем не менее, не может быть определена пороговая величина внутриглазного давления, которая была бы определяющей для возникновения глаукомы.
Поражение может быть также связано с плохим кровоснабжением жизненно важных волокон зрительного нерва, недостаточностью в структуре нерва и/или проблемой состояния самих по себе нервных волокон.
Нелеченая глаукома приводит к необратимому повреждению зрительного нерва и в результате к дефекту поля зрения (скотоме), который может приводить к слепоте.
Ганглиозные клетки сетчатки глаза представляют собой нервные клетки, которые передают визуальные сигналы из глаза в головной мозг. В процессе, приводящем к апоптозу ганглиозных клеток сетчатки глаза, активируются два главных фермента апоптотического процесса, каспаза-3 и каспаза-8. Каспаза-3 расщепляет белок-предшественник амилоида (APP) с продуцированием нейротоксических фрагментов, включая β-амилоид. При отсутствии защитного действия белка-предшественника амилоида, накопление β-амилоида в слое ганглиозных клеток сетчатки глаза приводит к гибели ганглиозных клеток сетчатки глаза и необратимой потере зрения.
Различные типы глаукомы классифицируются как открытоугольные глаукомы, если состояние является хроническим, или как закрытоугольные глаукомы, если внезапно возникает острая глаукома. Глаукома обычно поражает оба глаза, но заболевание может прогрессировать более быстро в одном глазу, чем в другом.
Хроническая открытоугольная глаукома (COAG), также называемая первичной открытоугольной глаукомой (POAG), представляет собой самый распространенный тип глаукомы. Хроническая открытоугольная глаукома вызывается микроскопической блокадой в трабекулярной сети, что приводит к снижению водного оттока в венозном синусе склеры и росту внутриглазного давления (IOP). Первичная открытоугольная глаукома обычно поражает оба глаза и в значительной степени связана с возрастом и положительным семейным анамнезом. Частота ее возникновения повышается у пожилых людей, так как дренажная система глаз может с возрастом постепенно закупориваться. Повышение внутриглазного давления у субъектов, страдающих хронической открытоугольной глаукомой, не сопровождается никакими симптомами до тех пор, пока не проявится потеря центрального участка поля зрения.
Острая закрытоугольная глаукома (AACG) или закрытоугольная глаукома представляет собой относительно редкий тип глаукомы, характеризующийся внезапным повышением внутриглазного давления до 35-80 мм ртутного столба, приводящей к сильной боли и необратимой потери зрения. Внезапное повышение давления происходит в результате закупорки угла фильтрации и блокады дренажных каналов. Пациенты с узкими углами имеют повышенный риск внезапного закрытия угла. Острая закрытоугольная глаукома обычно возникает в одном глазу, но риск существует для обоих глаз. Возраст, катаракта и псевдокапсулярная эксфолиация также являются факторами риска, так как они связаны с разрастанием хрусталика глаза и сжиманием или сужением угла. Внезапный приступ глаукомы может сопровождаться сильной глазной болью и головной болью, воспалением глаза, тошнотой, рвотой и расплывчатым зрением.
Глаукома со смешанным или комбинированным механизмом представляет собой смесь или комбинацию открытоугольной и закрытоугольной глаукомы. Она поражает пациентов с острой закрытоугольной глаукомой, у которых открылся угол после лазерной иридотомии, но которые продолжают нуждаться в приеме медицинских препаратов для контроля внутриглазного давления, а также пациентов с первичной открытоугольной глаукомой или псевдоэксфолиативной глаукомой, у которых постепенно происходит сужение угла.
Глаукома с нормальным давлением (NTG), называемая также глаукомой с низким давлением (LTG), характеризуется прогрессирующим повреждением зрительного нерва и потерей периферического зрения, подобно тому, как это происходит при других типах глаукомы; однако, внутриглазное давление находится в интервале нормальных значений или даже ниже нормального.
Врожденная (инфантильная) глаукома является относительно редким наследственным типом открытоугольной глаукомы. Недостаточное развитие зоны дренирования вызывает повышенное давление в глазу, которое может привести к потери зрения в результате повреждения зрительного нерва и к разрастанию глаза. Ранний диагноз и лечение являются особо важными факторами для сохранения зрения у младенцев и детей, страдающих этим заболеванием.
Вторичная глаукома может быть результатом травмы глаза, воспаления радужной оболочки глаза (ирита), диабета, катаракты, или применения стероидов у восприимчивых к стероидам пациентов. Вторичная глаукома может быть также связана с отслоением сетчатки или окклюзией или закупоркой вены сетчатки глаза.
Пигментная глаукома характеризуется отслоением гранул пигмента от радужной оболочки глаза. Гранулы вызывают закупорку дренажной системы глаза, приводящей к повышенному внутриглазному давлению и повреждению зрительного нерва.
Эксфолиативная глаукома (псевдокапсулярная эксфолиация) характеризуется отложениями хлопьевидного вещества на передней капсуле и в углу глаза. Накопление хлопьевидного вещества блокирует дренажную систему и повышает глазное давление.
Диагностирование глаукомы может быть проведено с помощью различных тестов. С помощью глазной тонометрии определяют давление в глазу путем измерения тонуса или плотности его поверхности. Для этого теста могут быть использованы несколько типов тонометров, при этом самым распространенным является аппланационный тонометр. С помощью пахиметрии определяют толщину роговой оболочки глаза, на основе которой, в свою очередь, определяют внутриглазное давление. Гониоскопия позволяет обследовать угол фильтрации и дренажную зону глаза. Гониоскопия позволяет также определить, блокируют ли аномальные кровеносные сосуды отток водной жидкости из глаза. Офтальмоскопия позволяет обследовать зрительный нерв и может выявить спадание слоя нервных волокон или изменения в диске зрительного нерва, или вдавленность (коробление) этой структуры, которые могут быть вызваны повышенным внутриглазным давлением или аксональным выпадением. Гониоскопию также применяют при оценке повреждения нерва в результате недостаточного кровотока или повышенного внутриглазного давления. Исследование поля зрения позволяет субъективно отобразить поле зрения, которое может обнаруживать признаки глаукомного повреждения зрительного нерва. Оно изображается характерными образцами дефекта поля зрения. Оптическую когерентную томографию, объективный метод измерения потери слоя нервных волокон, проводят путем обследования толщины слоя волокон зрительного нерва (измененной при глаукоме) по разнице величины пропускания света через поврежденную аксональную ткань.
Друзы зрительного нерва представляют собой сферические конкременты белка и солей кальция, которые, как полагают, являются секрециями из врожденно измененных сосудистых структур, поражающие слой аксональных нервных волокон. Эти накопления возникают в слое перипапиллярных нервных волокон и, как полагают, повреждают слой нервных волокон, либо непосредственно в результате сжатия, либо косвенно вследствие нарушения кровоснабжения слоя нервных волокон. Они обычно становятся заметными после первых десяти лет жизни страдающих этим заболеванием пациентов. Наиболее часто они возникают в обоих глазах, но могут также поражать один глаз, и могут приводить к умеренной потере периферического зрения на протяжении многих лет.
Оптическая нейропатия представляет собой заболевание, характеризующееся повреждением зрительного нерва, вызванным разрушением миелинового слоя, закупоркой системы кровоснабжения, дефицитом питательных веществ, или токсинами. Демиелинизирующие оптические нейропатии (смотрите ниже неврит зрительного нерва) обычно вызываются лежащим в основе процессом разрушения миелинового слоя, таким как множественный склероз. Закупорка системы кровоснабжения, называемая ишемической оптической нейропатией, может приводить к гибели или нарушению функции клеток зрительного нерва. Несвязанная с артериитом ишемическая оптическая нейропатия обычно возникает у людей в зрелом возрасте. Факторы риска включают высокое кровяное давление, диабет и атеросклероз. Связанная с артериитом ишемическая оптическая нейропатия обычно возникает у пожилых людей после воспаления артерий (артериита), в частности, височной артерии (височного артериита). Потеря зрения может быть быстрой или развиваться постепенно на протяжении от 2 до 7 дней, и могут повреждаться один или оба глаза. У людей с оптической нейропатией, вызванной воздействием токсина или дефицитом питательных веществ, обычно поражаются оба глаза.
Около 40% людей с несвязанной с артериитом ишемической оптической нейропатией испытывают в течение времени спонтанное улучшение. Несвязанную с артериитом ишемическую оптическую нейропатию лечат путем контролирования кровяного давления, диабета и уровней холестерина. Связанную с артериитом ишемическую оптическую нейропатию лечат с помощью высоких доз кортикостероидов для предотвращения потери зрения во втором глазу.
Неврит зрительного нерва связан с умеренной или тяжелой потерей зрения в одном или обоих глазах и может вызываться системным процессом разрушения миелинового слоя (смотрите выше), вирусной инфекцией, вакцинацией, менингитом, сифилисом, множественным склерозом и внутриглазным воспалением (увеитом). Движение глаз может быть болезненным, и при повторных случаях может ухудшаться зрение. Диагностирование включает исследование реакций зрачков и определение наличия набухания диска зрительного нерва. Магнитно-резонансная томография (MRI) может выявить признаки множественного склероза или, в редких случаях, опухоль, давящую на зрительный нерв, в случае которой зрения улучшается, как только уменьшается давление опухоли. В большинстве случаев неврита зрительного нерва, состояние улучшается в течение нескольких месяцев без проведения лечения. В некоторых случаях, может быть необходимо лечение с помощью внутривенного введения кортикостероидов.
Катаракта представляет собой помутнение, которое развивается в хрусталике глаза или в его оболочке. Катаракты обычно приводят к прогрессирующей потере зрения и могут вызывать слепоту при отсутствии лечения. При морганиевой катаракте, кортикальный слой хрусталика постепенно разжижается с образованием молочно-белой жидкости, и это может вызывать тяжелое воспаление в случае перфорации мембраны хрусталика и истечений из нее. При отсутствии лечения, катаракта может также вызывать факоморфическую глаукому. Катаракты могут быть по своей природе врожденными или быть следствием генетических факторов, пожилого возраста, длительного воздействия ультрафиолетового излучения, воздействия радиации, диабета, повреждения глаза или физической травмы.
Экстракапсулярная (ECCE) хирургия является наиболее эффективным методом лечения катаракты. При хирургическом методе, хрусталик удаляют, но большую часть мембраны хрусталика оставляют нетронутой. Для разрушения хрусталика перед его извлечением обычно используют факоэмульсификацию, небольшой разрез на стороне роговой оболочки глаза.
Амилоидоз глаза представляет собой наследственное расстройство, связанное с семейной амилоидотической полинейропатией по типу I (FAP) и характеризующееся аномальными конъюнктивальными сосудами, сухим кератоконъюнктивитом, патологиями зрачка и, в некоторых случаях, помутнениями стекловидного тела и вторичной глаукомой. Семейная амилоидотическая полинейропатия по типу I (FAP) связана с мутациями в транстиретине (TTR), тетрамерном белке плазмы (преальбумине), синтезируемом в печени, пигментном эпителии сетчатки и хороидном сплетении мозга. Различные мутации вызывают полимеризацию транстиретина в складчатую структуру амилоидного волокна, приводя к наследственному амилоидозу. наиболее частой мутацией является TTR-met303, при которой метионин замещает валин в положение 30 в транстиретине.
Семейная амилоидотическая полинейропатия по типу IV связана с решетчатой дистрофией роговицы (LCD). Решетчатая дистрофия роговицы представляет собой наследственный первичный, обычно билатеральный, амилоидоз роговицы, характеризующийся присутствием линий с двойным контуром преломляющей решетки в строме роговой оболочки глаза. Решетчатая дистрофия роговицы по типу I (Бибера-Хааба-Диммера) представляет собой аутосомное доминантное билатерально симметричное нарушение роговицы, характеризующееся присутствием многочисленных пропускающих свет тонких линий решетки с белыми точками и слабым помутнением в поверхностных и средних слоях центральной стромы. Симптомы начинают проявляться в период первой или второй декады жизни, приводя к прогрессирующей потере зрения. Большинству пациентов к 40 годам требуется трансплантация роговицы. Решетчатая дистрофия роговицы по типу II связана с системным амилоидозом (синдромом Meretoja) и характеризуется присутствием толстых линий решетки в кайме, центральной роговой оболочке и строме. Не поражает зрение до наступления пожилого возраста. Решетчатая дистрофия роговицы по типу III поражает людей в зрелом возрасте и характеризуется присутствием толстых линий решетки, которые простираются от края до края. Решетчатая дистрофия роговицы по типу III A характеризуется накоплением амилоидных отложений в строме и присутствием лент амилоида между стромой и слоем Боумена. Решетчатая дистрофия роговицы по типу III A отличается от решетчатой дистрофии роговицы по типу III из-за присутствия изъязвлений роговицы, возникновения в склерах, и типа аутосомно-доминантного наследования.
Синдром Дауна (DS) или трисомия 21 представляет собой самое распространенное генетическое нарушение с частотой случаев приблизительно 1:700 рождений живого ребенка, и часто связано с различными врожденными аномалиями. Нарушение, которое вызывается присутствием лишней хромосомы 21, связано с ранними отложениями образующего бляшки бета-амилоидного белка и развитием болезни Альцгеймера в зрелом возрасте. Кроме того, многие люди с синдромом Дауна страдают от катаракты, начиная с самого детства, и многие страдают от врожденной глаукомы. Так как ген белка-предшественника амилоида, который расщепляется с образованием бета-амилоида, у людей расположен на длинном плече хросомы 21, сверхэкспрессия этого гена может приводить к повышенным уровням белка-предшественника амилоида и амилоидному отложению при синдроме Дауна.
Лекарства от глаукомы не существует. Медицинские препараты для лечения глаукомы включают средства, которые снижают продукцию внутриглазной жидкости в глазу, такие как бета-блокаторы (тимоптик, бетоптик), ингибиторы угольной ангидразы (трусопт, азопт), альфа-агонисты (альфаган, лопидин), и средства, которые изменяет направление дренажа внутриглазной жидкости на другой путь в задней части глаза, такие как простагландин (ксалатан). Лазерная хирургия включает трабекулопластику, процедуру, которая способствует более эффективному выводу внутриглазной жидкости из глаза. Согласно данным Фонда изучения глаукомы (Glaucoma Foundation), у приблизительно 80% пациентов этот метод дает довольно хорошие результаты, позволяющие отложить или исключить последующее хирургическое вмешательство. Однако, согласно данным Национального института глаза (National Eye Institute), после лазерной хирургии в течение двух лет у половины всех пациентов поднимается внутриглазное давление. Инцизионная хирургия проводится в том случае, если медикаментозное лечение и исходная лазерная терапия не позволяют успешно понизить внутриглазное давление. Один тип хирургического вмешательства, трабекулэктомия, создает отверстие в стенке глаза для обеспечения дренажа внутриглазной жидкости. Однако, согласно данным Фонда изучения глаукомы (Glaucoma Foundation), приблизительно у одной трети пациентов, подвергнутых трабекулэктомии, в течение пяти лет развивается катаракта. В случаях неудовлетворительных результатов при трабекулэктомии, дополнительные инцизионные методы включают размещение дренажной трубочки в глазу между роговой оболочкой глаза и радужной оболочкой глаза и использование лазера или замораживания для разрушения ткани в глазу, которая продуцирует внутриглазную жидкость. Хирургическое вмешательство может спасти оставшееся зрение у пациента, но оно не улучшает зрение. Зрение может даже ухудшаться после хирургического вмешательства.
Возрастная дегенерация желтого пятна (AMD) является главной причиной слепоты среди жителей Кавказа старше 65. Несмотря на большой прогресс, который достигнут за последнее время в изучении дегенерации желтого пятна, не существует лечения, которое предотвращает гибель нервных клеток, происходящую в процессе заболевания. Не существует также направленного лечения других глазных заболеваний, связанных с нейрональной деградацией, относящихся к бета-амилоиду, таких как кортикальные зрительные нарушения, друзы зрительного нерва, оптическая нейропатия, неврит зрительного нерва, амилоидная дистрофия роговицы глаза и решетчатая дегенерация роговицы.
Амилоидные отложения обычно содержат три компонента. Волокна амилоидного белка, которые составляют около 90% амилоидного вещества, включают один из нескольких различных типов белков. Эти белки способны складываться в так называемые волокнистые структуры "бета-складчатого" листа, уникальную конфигурацию белка, которая характеризуется участками связывания с красителем "конго красный", что позволяет получать специфическое окрашивание амилоидного белка. Кроме того, амилоидные отложения тесно связаны с компонентом амилоида P (пентагональным) (AP), гликопротеином, относящимся к сывороточному амилоиду P (SAP), и с сульфатированными гликоаминогликанами (GAG), сложными углеводами соединительной ткани.
Одним направлением исследования в области лечения болезни Альцгеймера и прионных заболеваний является дизайн молекул, которые связывают аномальную конформацию β-листов Αβ и PrP, соответственно, тем самым предотвращая агрегацию этих молекул. Конформация β-листа пептидов характеризуется тем, что в регулярной структуре образуются водородные связи между аминокислотами соседних цепочек. Эта конфигурация приводит к стабильной пространственной структуре. Акцепторы H-связи (группа C=O) и доноры H-связи (группа NH) чередуются в природных β-листах пептидов с атомами, с которыми они связываются, приблизительно на одной линии. Внутри каждой цепочки аминокислоты, расстояние между соседними донорами H-связи и акцепторами H-связи находятся внутри специфических интервалов. В частности, расстояние между донором H-связи (группой NH) и акцептором H-связи (группой C=O) внутри одного остатка аминокислоты составляет от 3,5 до 4,0 Å. Расстояние между акцептором H-связи (группой C=О) одного остатка аминокислоты и донора H-связи (группой NH) следующего остатка аминокислоты, участвующего в межцепочечном поперечном связывании составляет от 2,6 до 2,9 Å. Другими словами, расстояние между соседними донорами H-связи и акцепторами H-связи внутри одной цепочки аминокислоты изменяется в следующих интервалах:
донор H-связи (аминокислота 1) - акцептор H-связи (аминокислота 1) = 3,5-4,0 Å;
акцептор H-связи (аминокислота 1) - донор H-связи (аминокислота 2) = 2,6-2,9 Å.
Лиганды, которые предназначены для связывания β-листов, в идеале имеют порядок доноров H-связи и акцепторов H-связи, который является комплементарным в отношении порядка доноров H-связи и акцепторов H-связи в цепочках аминокислот β-листа.
В патентном документе WO 2007/002433 описаны конкретные производные пирроло[2,3-B]пиридина, которые заявлены в качестве подходящих ингибиторов протеинкиназы.
Задачей настоящего изобретения является создание соединений, которые могут быть использованы при лечении заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включая амилоидоз. Эти соединения должны быть способны преодолевать гематоэнцефалический барьер. Кроме того, они должны быть фармацевтически приемлемыми, в частности, они не должны обладать мутагенными или канцерогенными свойствами или быть метаболически нестабильными.
Кроме того, задачей изобретения является создание улучшенных вариантов лечения для субъектов, страдающих глазными заболеваниями, связанными с патологическими нарушениями/изменениями в ткани визуальной системы, в частности, связанными с патологическими нарушениями/изменениями в ткани визуальной системы, имеющими отношение к бета-амилоиду, такими как, например, нейрональная деградация. Указанные патологические нарушения могут возникать, например, в различных тканях глаза, таких как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталик, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, например, возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
Авторы настоящего изобретения неожиданно обнаружили, что эти задачи могут быть решены с помощью соединений общей формулы (I), описанных далее.
Сущность изобретения
Соответственно, настоящее изобретение относится к соединению общей формулы (I).
В дополнительном аспекте настоящее изобретение относится к фармацевтической композиции или радиофармацевтическому препарату, включающему соединение общей формулы (I).
Еще один аспект настоящего изобретения относится к применению соединения общей формулы (I) для получения лекарственного средства для лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включая амилоидоз.
Кроме того, в изобретении раскрывается способ лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения общей формулы (I).
Еще один аспект настоящего изобретения относится к применению соединения общей формулы (I) для получения лекарственного средства для лечения или облегчения поражающих воздействий глазных заболеваний, связанных с патологическими нарушениями/изменениями в тканях визуальной системы.
Кроме того, в изобретении раскрывается способ лечения или облегчения поражающих воздействий глазных заболеваний, связанных с патологическими нарушениями/изменениями в тканях визуальной системы, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения общей формулы (I).
Глазные заболевания, связанные с патологическими нарушениями/изменениями в тканях визуальной системы, связаны, в частности, с патологическими нарушениями/изменениями в ткани визуальной системы, имеющими отношение к бета-амилоиду, такими как, например, нейрональная деградация. Указанные патологические нарушения могут возникать, например, в различных тканях глаза, таких как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталик глаза, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, например возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
В дополнительном аспекте изобретение относится к смеси (такой как фармацевтическая композиция), включающей соединение согласно настоящему изобретению и, необязательно, по меньшей мере одно дополнительное биологически активное соединение и/или фармацевтически приемлемый носитель и/или разбавитель, и/или вспомогательное вещество. Дополнительное биологически активное вещество может представлять собой известное соединение, применяемое при медикаментозном лечении заболеваний и расстройств, которые вызываются или связаны с амилоидными или амилоидоподобными белками, включая амилоидоз, группы заболеваний и расстройств, связанных с амилоидным или амилоидоподобным белком, таким как Αβ белок, играющий активную роль в болезни Альцгеймера.
Дополнительное биологически активное вещество или соединение может оказывать свое биологическое действие по такому же или аналогичному механизму, что и соединение согласно изобретению, или по несвязанному с соединением механизму действия или по многочисленным связанным и/или несвязанным с соединением механизмам действия.
Кроме того, раскрывается способ сбора данных для диагностирования связанного с амилоидом заболевания или состояния в пробе или в организме пациента, который включает:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением согласно настоящему изобретению;
(b) связывание соединения с амилоидным белком;
(c) определение соединения, связанного с белком; и
(d) необязательное установление взаимосвязи между присутствием или отсутствием соединения, связывающего амилоидный белок, с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела.
Другим вариантом осуществления настоящего изобретения является способ определения степени отложения амилоидогенных бляшек в ткани и/или биологической жидкости организма, включающий:
(a) взятие пробы исследуемой ткани и/или биологической жидкости организма;
(b) испытание пробы на присутствие амилоидного белка с помощью соединения согласно настоящему изобретению;
(c) определение количества соединения, связанного с амилоидным белком; и
(d) расчет степени отложения бляшек в ткани и/или биологической жидкости организма.
Дополнительный аспект относится к способу сбора данных для выявления предрасположенности к связанному с амилоидом заболеванию или состоянию у пациента, включающий обнаружение специфического связывания соединения согласно настоящему изобретению с амилоидным белком в пробе или in situ, который включает стадии:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением согласно настоящему изобретению, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Еще одним аспектом настоящего изобретения является способ сбора данных с целью непрерывного контроля минимального остаточного заболевания у пациента после лечения с помощью композиции антитела или вакцины, где способ включает:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением согласно настоящему изобретению, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(е) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Кроме того, описан способ сбора данных с целью предсказания восприимчивости пациента к лечению с помощью композиции антитела или вакцины, который включает:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением согласно настоящему изобретению, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Дополнительным аспектом настоящего изобретения является набор для обнаружения и/или диагностирования связанного с амилоидом заболевания или состояния, включающий соединение согласно настоящему изобретению.
В еще одном аспекте настоящего изобретения соединение согласно настоящему изобретению применяют при ингибировании агрегации белка, в частности применяют при ингибировании агрегации Aβ1-42, агрегации тау-белка или агрегации альфа-синуклеина.
В одном аспекте изобретения раскрывается применение соединения согласно настоящему изобретению для получения лекарственного средства для (a) снижения отложения β-амилоидных бляшек и/или (b) ингибирования образования β-амилоидных бляшек, и/или (c) замедления увеличения отложения амилоидных бляшек в мозге пациента.
Дополнительный вариант осуществления изобретения предлагает способ (a) снижения отложения β-амилоидных бляшек и/или (b) ингибирования образования β-амилоидных бляшек, и/или (c) замедления увеличения отложения амилоидных бляшек в мозге пациента, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения согласно настоящему изобретению.
В еще одном варианте осуществления соединение согласно настоящему изобретению применяют при (a) снижении отложения β-амилоидных бляшек и/или (b) ингибировании образования β-амилоидных бляшек, и/или (c) замедлении увеличения отложения амилоидных бляшек в мозге пациента.
Согласно дополнительному аспекту, настоящее изобретение относится к применению соединения согласно настоящему изобретению для получения лекарственного средства для сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти.
Согласно еще одному аспекту, настоящее изобретение предлагает способ сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения согласно настоящему изобретению.
Еще один аспект настоящего изобретения относится к соединению согласно настоящему изобретению для применения при сохранении или повышении когнитивной способности памяти у субъекта, страдающего нарушением памяти.
Предпочтительные варианты осуществления изобретения приведены в зависимых пунктах формулы изобретения.
Индивидуальные соединения, которые раскрыты в патентном документе WO 2007/002433, не включаются в объем пунктов формулы изобретения, относящихся к соединениям или фармацевтической композиции. Эти соединения, например, приведены ниже:


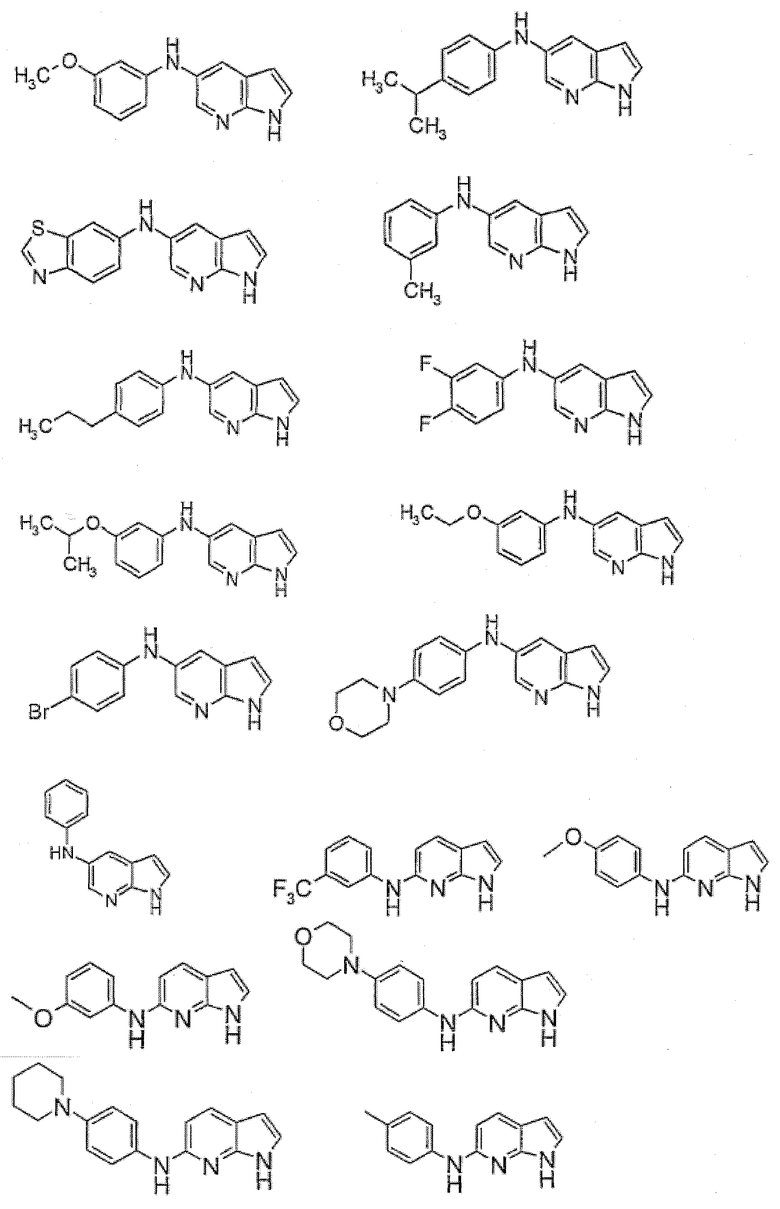


Эти соединения могут быть включены в объем других пунктов формулы изобретения или они могут быть исключены из объема других пунктов формулы изобретения. Соединения могут быть исключены либо в индивидуально, в комбинации двух или более, либо все соединения могут быть исключены из любых других пунктов формулы изобретения.
Краткое описание фигур
На фигуре 1 показаны количественные данные по кортикальной (a и b) и гиппокампальной (c и d) 6E10 иммунофлюоресценции.
На фигурах 2A и 2B, APPV171I приведены данные по исследованию трансгенных мышей, которые были подвергнуты обработке с помощью PBS (забуференного фосфатом физиологического раствора) и ACI-636.
На фигуре 3 показано разделения Boc-защищенного предшественника соединения 26 с помощью хиральной ВЭЖХ.
На фигуре 4 показан энантиомер, элюируемый первым после разделения.
На фигуре 5 показан энантиомер, элюируемый вторым после разделения.
На фигуре 6 показана рацемическая смесь (дигидрохлоридные соли) перед кристаллизацией.
На фигуре 7 показан энантиомер A (дигидрохлоридная соль) после кристаллизации, элюируемый первым.
На фигуре 8 показан энантиомер B (дигидрохлоридная соль) после кристаллизации, элюируемый вторым.
На фигуре 9 показан Boc-защищенный предшественник соединения 214b, полученный из диастереомера, элюируемого вторым.
На фигуре 10 показан Boc-защищенный предшественник соединения 214a, полученный из диастереомера, элюируемого первым.
На фигуре 11 приведены данные 1H-ЯМР элюируемого первым диастереомера (только область ароматики, примеси присутствует в интервале 7,1-7,3 м.д.).
На фигуре 12 приведены данные 1H-ЯМР элюируемого последним диастереомера (только область ароматики, примеси присутствует в интервале 7,1-7,3 м.д.).
Определения
В соответствии с сущностью настоящей заявки, применяются следующие определения:
"Алкил" относится к насыщенному органическому фрагменту, состоящему из атомов углерода и атомов водорода. Примеры подходящих алкильных групп имеют от 1 до 6 углеродных атомов, предпочтительно, от 1 до 4 углеродных атомов, и включают метил, этил, пропил и бутил.
"Циклоалкил" относится к циклическому органическому фрагменту, состоящему из атомов углерода и атомов водорода. Примеры подходящих циклоалкильных групп имеют от 5 до 10 углеродных атомов, предпочтительно, 5 или 6 углеродных атомов, и включают циклопентил и циклогексил.
"Гетероциклоалкил" относится к циклоалкильной группе, определенной выше, в которой по меньшей мере один из углеродных атомов был замещен на гетероатом, который выбирают, например, из N, O или S, или гетероатом- (например, N, O и/или S) содержащий фрагмент. Примеры возможных гетероциклоалкильных групп включают пирролидин, тетрагидрофуран, пиперидин и другие группы.
"Алкенил" относится к органическому фрагменту, состоящему из атомов углерода и атомов водорода, который включает по меньшей мере одну двойную связь. Примеры подходящих алкенильных групп имеют от 2 до 6 углеродных атомов, предпочтительно, от 2 до 4 углеродных атомов, и включают пропенил и бутенил.
"Алкинил" относится к органическому фрагменту, состоящему из атомов углерода и атомов водорода, который включает по меньшей мере одну тройную связь. Примеры подходящих алкинильных групп имеют от 2 до 6 углеродных атомов, предпочтительно, от 2 до 4 углеродных атомов, и включают пропинил и бутинил.
"Арил" относится к ароматическому органическому фрагменту, состоящему из атомов углерода и атомов водорода, который предпочтительно имеет от 5 до 10 углеродных атомов, более предпочтительно, 5 или 6 углеродных атомов. Примером является фенильное кольцо.
"Гетероарил" относится к арильной группе, определенной выше, в которой по меньшей мере один из углеродных атомов был замещен на гетероатом, который выбирают, например, из N, O или S, или гетероатом- (например, N, O и/или S) содержащий фрагмент. Примеры возможных гетероарильных групп включают пиридин и другие группы.
"Алкокси" относится к группе -O-алкил.
"Аминоалкил" относится к группе -алкил-NR1R2.
"Hal" или "галоген" относится к F, Cl, Br и I. Предпочтительным галогеном является F и Cl, более предпочтительным - F.
"Арилалкил" относится к группе арил-алкил-.
"Циклоалкилалкил" относится к группе циклоалкил-алкил-.
"Фторалкил" относится к алкильной группе, в которой один или более атомов водорода были замещены на атомы фтора.
"Галогеналкил" относится к алкильной группе, в которой один или более атомов водорода были замещены на атомы галогена.
"Гетероарилалкил" относится к группе гетероарил-алкил-.
"Гетероциклоалкилалкил" относится к группе гетероциклоалкил-алкил-.
"Гетероатомсодержащие фрагменты представляют собой фрагменты, которые содержат, например, N, O и/или S. Примеры таких фрагментов включают -C(O)-, -C(O)O- и -N(R50)-, в которых R50, для каждого случая присутствия, независимо выбирают из группы, состоящей из R20, S(O)tNR20R21, S(O)tR20, C(О)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 и C(O)NR20R21. Конкретные примеры включают -C(O)-, -C(O)O- и -N(R50)-, в которых R50, для каждого случая присутствия, независимо выбирают из группы, состоящей из H или C1-4 алкила.
Если группу определяют как "необязательно замещенную", она может иметь один или более заместителей, выбранных из галогена, C1-6 алкила, C1-6 алкокси, -SO2-алкила, -NH2, -NH(C1-6 алкил) или -N(C1-6 алкил)2.
Соединения настоящего изобретения, имеющие один или более оптически активных углеродов, могут существовать в виде рацематов и рацемических смесей, стереоизомеров (включая диастереомерные смеси и индивидуальные диастереомеры, энантиомерные смеси и индивидуальные энантиомеры, смеси конформационных изомеров и индивидуальные конформационные изомеры), таутомеров, атропоизомеров и поворотных изомеров. Все изомерные формы включаются в настоящее изобретение. Описанные в этом изобретении соединения, содержащие олефиновые двойные связи, включают E и Z геометрические изомеры. Кроме того, это изобретение включает все солевые формы, полиморфы, гидраты и сольваты.
На растворитель, содержащийся в сольватах, не накладывают конкретных ограничений, и он может быть любым фармацевтически приемлемым растворителем. Примеры включают воду и C1-4 спирты (такие как метанол или этанол).
Пациенты или субъекты, которые могут подвергаться лечению в настоящем изобретении, обычно представляют собой животных, в частности млекопитающих, более конкретно, людей.
Предпочтительные определения, приведенные в разделе "Определения", применяются ко всем описанным ниже вариантам осуществления, если не заявлено иначе.
Подробное описание изобретения
Настоящее изобретение относится к соединению формулы (I):
A-L1-B (I)
и всем его стереоизомерам, рацемическим смесям, фармацевтически приемлемым солям, гидратам, сольватам и полиморфам.
A выбирают из группы, состоящей из

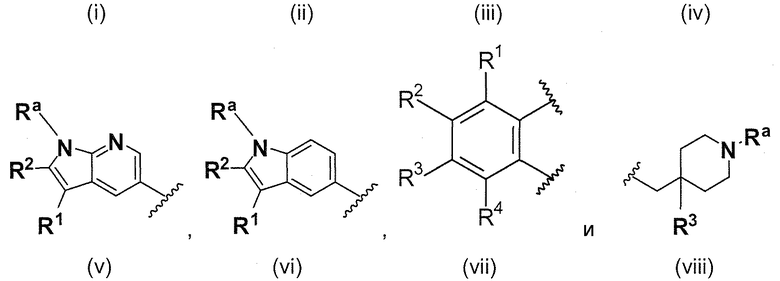 .
.
L1 направленно выбирают из группы, состоящей из
 .
.
Термин "направленно" означает, что связь, показанная слева в формуле (a), и две связи, показанные слева в формуле (b), связаны с A, в то время как связь, показанная справа в формуле (a), и связь, показанная слева в формуле (b), связаны с B. Для специалиста в этой области является очевидным, что формула (b) может только объединяться с формулой (vii) в качестве варианта A. В предпочтительном варианте осуществления, L1 представляет собой (a).
B выбирают из группы, состоящей из

R1, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R1, каждый независимо выбирают из водорода, галогена (такого как F), CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, -O-алкила, гетероциклоалкила (такого как  ,
,  или
или  ). Более предпочтительно. чтобы R1,
каждый независимо выбирали из водорода, F, CN, CF3, , и .
). Более предпочтительно. чтобы R1,
каждый независимо выбирали из водорода, F, CN, CF3, , и .
R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R2, каждый независимо выбирают из водорода, галогена (такого как F), CN, CF3,
CONR30R31, -N(R30)-C(O)-R31, -O-алкила, гетероциклоалкила (такого как , или ). В предпочтительном варианте осуществления, R2, каждый независимо выбирают из водорода, F, CN, CF3, CONR30R31, -O-алкила, , и .
R3, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R3, каждый независимо выбирают из водорода, галогена (такого как F), CN, CF3, -CONR30R31, -N(R30)-C(O)-R31,
-O-алкила, гетероциклоалкила (такого как , или ). Более предпочтительно, чтобы R3, каждый независимо выбирали из водорода, F, -O-алкила, CF3, , и .
R4, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R4, каждый независимо выбирают из водорода, галогена (такого как F), CN, CF3, -CONR30R31, -N(R30)-C(O)-R31, -O-алкила, гетероциклоалкила (такого как , или ). Более предпочтительно, чтобы R4 являлся водородом.
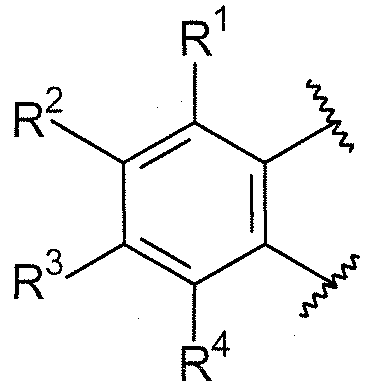
В дополнительном варианте осуществления, если любые из групп R1/R2/R3/R4 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5-8-членное кольцо, содержащее углеродные атомы и необязательно один или более гетероатомов, выбранных из O, S или N, или гетероатом- (N, O и/или S) содержащий фрагмент, где 5-8-членное кольцо может быть замещено с помощью NR20R21 или -O-алкила, где алкил может быть необязательно замещенным, или с помощью  . В одном варианте осуществления, R1 и R2, взятые вместе, могут образовывать 5-8-членное кольцо, содержащее углеродные атомы, такое как 6-членное карбоциклическое кольцо. Кольцо может быть насыщенным или включать одну или более двойных связей (включая ароматические кольца). Примерами групп, которые образуют кольцо, являются -(CH2)3- (то есть насыщенное 5-членное кольцо), -(CH2)4-, -O-(CH2)-O-. Эти кольца могут быть необязательно замещенными.
. В одном варианте осуществления, R1 и R2, взятые вместе, могут образовывать 5-8-членное кольцо, содержащее углеродные атомы, такое как 6-членное карбоциклическое кольцо. Кольцо может быть насыщенным или включать одну или более двойных связей (включая ароматические кольца). Примерами групп, которые образуют кольцо, являются -(CH2)3- (то есть насыщенное 5-членное кольцо), -(CH2)4-, -O-(CH2)-O-. Эти кольца могут быть необязательно замещенными.
Примеры необязательных заместителей 5-8-членного кольца включают -O-алкил, -O-алкилгалоген, -O-алкил-O-алкил, -NH-алкил-O-алкил и -алкил-SO2алкил.
Ra, каждый независимо выбирают из группы, состоящей из водорода, алкила, галогеналкила, S(O)tNR30R31, S(O)tR30, C(O)OR30, C(O)R30 и C(O)NR30R31. В предпочтительном варианте осуществления, Ra представляет собой водород или алкил.
Для каждого случая присутствия, Rb, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, Rb представляет собой водород, галоген (такой как F), CONR30R31 или алкил.
Для каждого случая присутствия, R20, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R20 представляет собой водород или алкил.
Для каждого случая присутствия, R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R21 представляет собой водород или алкил.
В одном варианте осуществления, R20 и R21, взятые вместе с атомом азота, к которому они присоединены, могут образовывать 3-8-членное кольцо, содержащее углеродные атомы и необязательно один или более дополнительных гетероатомов, выбранных из O, S или N, или гетероатом- (N, O и/или S) содержащий фрагмент, и где 3-8-членное кольцо может быть необязательно замещенным. Кольцо может быть насыщенным или включать одну или более двойных связей (включая ароматические кольца). В одном варианте осуществления кольцо является карбоциклическим, помимо атома азота, к которому R20 и R21 присоединены. В отличающемся варианте осуществления кольцо включает один дополнительный гетероатом (такой как N или O) помимо атома азота, к которому R20 и R21 присоединены.
Для каждого случая присутствия, R30, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R30 представляет собой водород или алкил.
Для каждого случая присутствия, R31, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления, R31 представляет собой водород или алкил.
Когда атом азота присутствует в кольце, образованном R1/R2/R3/R4, или кольце, образованном R20 и R21, он может находиться в любой подходящей форме. Возможные формы включают -
N(R50)- и -N=.
X, каждый независимо выбирают из группы, состоящей из CRb и N.
Y, каждый независимо выбирают из группы, состоящей из CRb и N.
t равняется 1 или 2.
p равняется 0, 1 или 2. В одном варианте осуществления, p равняется 0.
В предпочтительном варианте осуществления A выбирают из группы, состоящей из

L1 представляет собой

B выбирают из группы, состоящей из
 и
и

где:
R1, R2, R3, R4, Ra, R20, X и Y имеют такие же значения, как определенные выше;
V отсутствует или представляет собой NR20R21 ; и
z равняется 1 или 2.
В настоящем описании также предполагается любая комбинация упомянутых выше определений.
В одном варианте осуществления A имеет формулу (i)
 .
.
Предпочтительные варианты осуществления формулы (i) включают 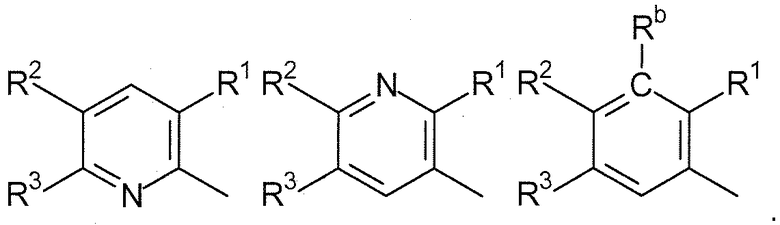 В одном предпочтительном варианте осуществления формула (i) представляет собой
В одном предпочтительном варианте осуществления формула (i) представляет собой 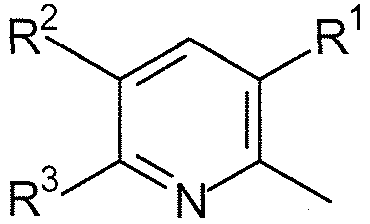 . В другом предпочтительном варианте осуществления формула (i) представляет собой
. В другом предпочтительном варианте осуществления формула (i) представляет собой  .
.
В этом варианте осуществления, R1, R2, R3 и Rb определены выше.
В одном варианте осуществления A имеет формулу (ii)

Предпочтительные варианты осуществления формулы (ii) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород или, предпочтительно, чтобы R1 и R2 вместе образовывали -(CH2)4-. В альтернативном варианте осуществления, R2 и Ra представляют собой водород, и R1 представляет собой , или , предпочтительно, чтобы R2 и Ra представляли собой водород, и R1 представлял собой или.
В одном варианте осуществления A имеет формулу (iii)

Предпочтительные варианты осуществления формулы (iii) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород или, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой , или . В одном предпочтительном варианте осуществления, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой или .
В одном варианте осуществления A имеет формулу (iv)
Предпочтительные варианты осуществления формулы (iv) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород или, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой , или . В одном предпочтительном варианте осуществления, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой или .
В одном варианте осуществления A имеет формулу (v)
 .
.
Предпочтительные варианты осуществления формулы (v) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород или, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой , или . В одном предпочтительном варианте осуществления, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой или .
В одном варианте осуществления A имеет формулу (vi)
 .
.
Предпочтительные варианты осуществления формулы (vi) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород или, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой , или . В одном предпочтительном варианте осуществления, предпочтительно, чтобы R2 представлял собой водород, и R1 представлял собой или .
В одном варианте осуществления A имеет формулу (vii)
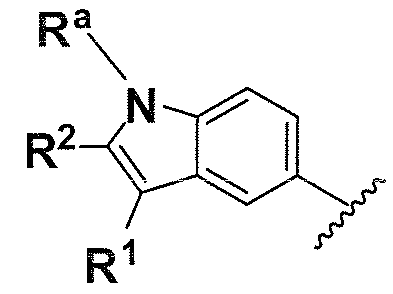
Предпочтительные варианты осуществления формулы (vii) включает те, в которых R1, R2, R3 и R4 представляют собой водород. Дополнительные предпочтительные варианты осуществления формулы (vii) включают

В одном варианте осуществления A имеет формулу (viii)

Предпочтительные варианты осуществления формулы (viii) включают те, в которых R3 представляет собой F, и Ra представляет собой водород, или в которых R3 и Ra представляют собой водород.
В одном варианте осуществления L1 имеет формулу (а)
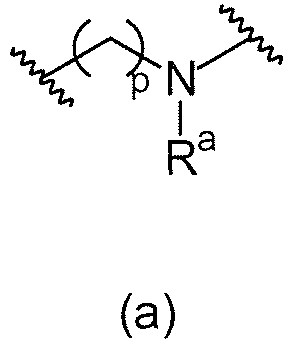 . В предпочтительном варианте осуществления p=0.
. В предпочтительном варианте осуществления p=0.
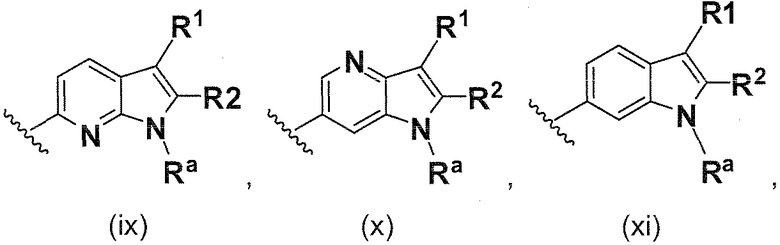
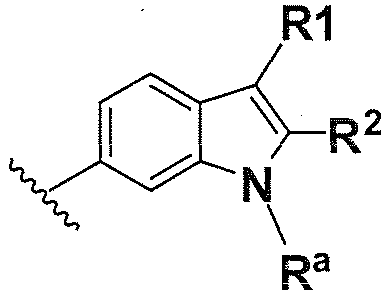
В одном варианте осуществления B имеет формулу (ix)
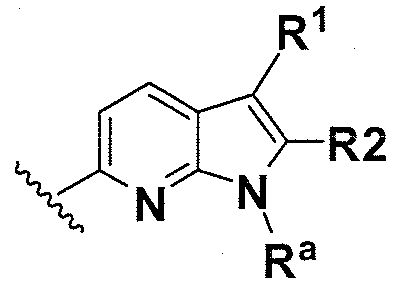
Предпочтительные варианты осуществления формулы (ix) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород. В дополнительных предпочтительных вариантах осуществления, R1 определен в общих определениях выше (примерами являются CN, -COO(C1-4 алкил), , или ; более предпочтительно, CN, , ) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
В одном варианте осуществления B имеет формулу (x)

Предпочтительные варианты осуществления формулы (x) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород. В дополнительных предпочтительных вариантах осуществления, R1 определен в общих определениях выше (примерами являются CN, -COO(C1-4 алкил), 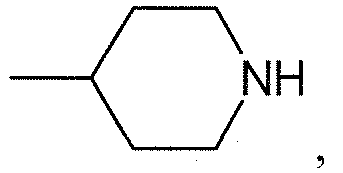
 ; более предпочтительно, CN,
; более предпочтительно, CN,  ) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
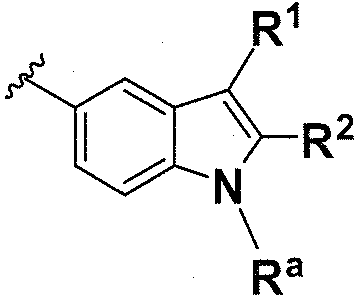
В одном варианте осуществления B имеет формулу (xi)
Предпочтительные варианты осуществления формулы (xi) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород. В дополнительных предпочтительных вариантах осуществления, R1 определен в общих определениях выше (примерами являются CN, -COO(C1-4 алкил),
; более предпочтительно, CN, ) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
В одном варианте осуществления B имеет формулу (xii)
Предпочтительные варианты осуществления формулы (xii) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород. В дополнительных предпочтительных вариантах осуществления, R1 определен в общих определениях выше (примерами являются CN, -COO(C1-4 алкил),
; более предпочтительно, CN, ) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
В одном варианте осуществления B имеет формулу (xiii)

Предпочтительные варианты осуществления формулы (xiii) включают те, в которых Ra представляет собой водород или C1-4 алкил. В дополнительных предпочтительных вариантах осуществления, R1 и R2 представляют собой водород. В дополнительных предпочтительных вариантах осуществления, R1 определен в общих определениях выше (примерами являются CN, -COO(C1-4 алкил),
; более предпочтительно, CN, ) и R2 представляет собой водород. В дополнительных предпочтительных вариантах осуществления, R1 и R2 могут образовывать кольцо, которое является необязательно замещенным. Примерами R1 и R2, которые образуют кольцо, являются -(CH2)3- и -(CH2)4-, которые могут быть необязательно замещенными.
В одном варианте осуществления предпочтительно, чтобы соединение настоящего изобретения имело формулу (I)
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой:

L1 представляет собой

B представляет собой

где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50 для каждого случая присутствия представляет собой R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N;
t равняется 1 или 2; и
z равняется 1 или 2.
В другом варианте осуществления предпочтительно, чтобы соединение настоящего изобретения имело формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой
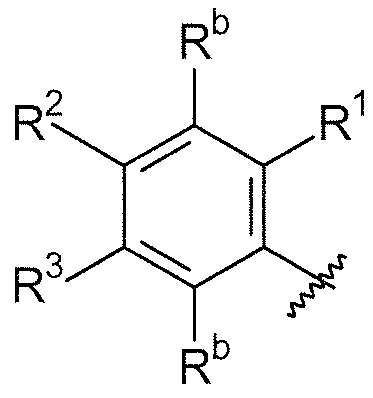
L1 представляет собой

B представляет собой
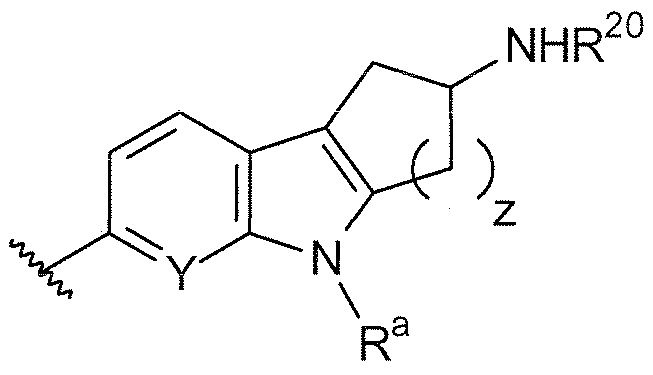
где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50, для каждого случая присутствия, представляет собой R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N;
t равняется 1 или 2; и
z равняется 1 или 2.
В дополнительном варианте осуществления предпочтительно, чтобы соединение настоящего изобретения имело формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой

L1 представляет собой
B представляет собой

где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50, для каждого случая присутствия, представляет собой R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N; и
t равняется 1 или 2.
В настоящем описании также предполагается любая комбинация упомянутых выше вариантов осуществления.
Предпочтительные соединения приведены в таблице 6.
Соединения настоящего изобретения могут быть синтезированы с помощью одного из общих методов, показанных на схемах 1-20. Эти методы приводятся только с целью иллюстрации и не являются ограничениями.
Общие схемы синтеза для получения 6-амино- или 6-бром-7-азаиндольных элементов структуры приведены далее.

Выпускаемый промышленностью 7-азаиндол или соответственно замещенное производное 7-азаиндола обрабатывали метахлорпербензойной кислотой в подходящем растворителе с получением N-оксидных/метахлорбензойной кислоты соответствующих солей. Обработка N-оксида/солей диметилсульфатом в подходящем растворителе с последующим взаимодействием промежуточного соединения с аммиаком в подходящем растворителе давала требуемые 6-амино-7-азаиндольные элементы структуры.
Обработка солей N-оксида основанием в подходящем растворителе давала после очистки N-оксид. Взаимодействие N-оксидов с гексаметилдисилазаном и бензоилбромидом в подходящем растворителе давало соответствующие N1-бензоил-защищенные 6-бром-7-азаиндольные производные. Гидролиз защитной группы с помощью гидроксида натрия в подходящем растворителе давал после очистки соответствующие 6-бром-7-азаиндольные производные. Защита N1-положения с помощью триизопропилсилильного фрагмента давала после очистки требуемые N1-защищенные 6-бром-7-азаиндольные производные.
Общая схема синтеза для получения 3-замещенного 6-бром-4-азаиндольного и 6-броминдольного элементов структуры при R=Boc, CH3, и X=CH, N.
Выпускаемый промышленностью 6-бром-4-индол (X=CH) обрабатывали гидридом натрия в подходящем растворителе, затем добавляли триизопропилсилилхлорид с получением после очистки N1-защищенных производных.
Выпускаемый промышленностью 6-бром-4-индол (X=CH) или 6-бром-4-азаиндол (X=N) обрабатывали с помощью N-Boc-пиперидин-4-она или N-метилпиперидин-4-она и соответствующим основанием в подходящем растворителе с получением после очистки продуктов конденсации. Гидрирование двойной связи с использованием подходящего катализатора и растворителя давало после очистки соответствующие продукты восстановления. Защита N1-положения с помощью триизопропилсилильного фрагмента давала после очистки требуемые 3-замещенные и N1-защищенные производные.
Общая схема синтеза для получения 3-замещенной 5-бром-7-азаиндольного и 5-бром-7-индольного элементов структуры при R=Boc, CH3.
Выпускаемый промышленностью 5-броминдол (X=CH) или 5-бром-7-азаиндол (X=N) обрабатывали с помощью N-Boc-пиперидин-4-она или N-метилпиперидин-4-она и соответствующего основания в подходящем растворителе с получением после очистки продуктов конденсации. Защита N1-положения с помощью триизопропилсилильного фрагмента давала соответствующие соединения. Гидрирование двойной связи с использованием соответствующего катализатора и растворителя давала после очистки требуемые N1-защищенные производные.
Общая схема синтеза для получения 3-замещенных 6-аминоиндольных элементов структуры этого изобретения при R=Boc, CH3.

Выпускаемый промышленностью 6-нитроиндол обрабатывали с помощью N-Boc-пиперидин-4-она или N-метилпиперидин-4-она и соответствующего основания в подходящем растворителе с получением после очистки продукта конденсации. Восстановление нитрогруппы и двойной связи с использованием соответствующего катализатора и растворителя давало после очистки требуемые 6-аминоиндольные производные. Защита N1-положения нитропроизводных от начальной конденсации с помощью триизопропилсилильного фрагмента давала соответствующие соединения, которые использовали для гидрирования без очистки. Восстановление нитрогруппы и двойной связи с использованием соответствующего катализатора и растворителя давало после очистки требуемые N1-TIPS-защищенные 6-аминоиндольные производные. Защита N1-положения нитропроизводного от начальной конденсации, содержащего N-Boc-защищенный пиперидиновый фрагмент, с помощью метильной группы давала после очистки требуемое соединение. Каталитическое восстановление нитрогруппы и двойной связи с использованием соответствующего катализатора и растворителя давала после очистки требуемое N1-метил-защищенное 6-аминоиндольное производное, где атом азота пиперидинового фрагмента защищен с помощью Boc-фрагмента.
Общая схема синтеза для получения трициклических 2-бром- или 2-аминотетрагидропиридо[2,3-b]индольных элементов структуры, когда R1 и R2, взятые вместе, образуют 5- или 6-членное кольцо, содержащее углеродные атомы.

Выпускаемый промышленностью 2,6-дибромпиридин обрабатывали гидратом гидразина в подходящем растворителе с получением моногидразинового производного. Конденсация с соответствующим циклическим кетоном в подходящем растворителе давала требуемые соединения гидразона. Термический синтез индола по Фишеру давал после очистки продукты трициклической циклизации. Реакция обмена бромпроизводного с аммиаком в присутствии оксида меди(I) давала после очистки требуемые трициклические 2-аминопиридо[2,3-b]индольные производные.
Общая схема синтеза для получения трициклических 2-бром-тетрагидропиридо[2,3-b]индольных (X=N, Y=Br, Z=H) или 6-бромтетрагидрокарбазольных (X=CH, Y=H, Z=Br) или 7-бром-тетрагидрокарбазольных (X=CH, Y=Br, Z=H) элементов структуры, когда R1 и R2, взятые вместе, образуют 6-членное кольцо, содержащее углеродные атомы, и 6-членное кольцо замещено с помощью NCH3Boc.
Общая схема синтеза для получения защищенных 4-амино- и 4-гидроксициклогексаноновых производных и незащищенных 4-гидроксициклогексаноновых производных.
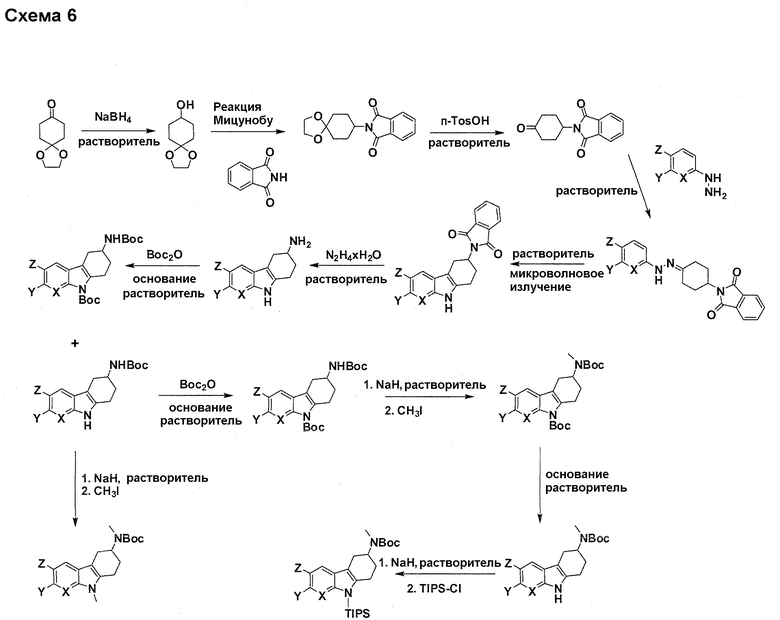
Выпускаемую промышленностью гидрохлоридную соль 4-амино-циклогексанола превращали в соответствующий фталимид-защищенный 4-аминоциклогексанол с использованитем фталимидного реагента и карбоната калия в соответствующем растворителе. Окисление спирта в кетон с помощью хлорхромата пиридиния в соответствующем растворителе давало после очистки фталимид-защищенный 4-аминоциклогексанон. Выпускаемый промышленностью моноэтиленацеталь 1,4-циклогександиона обрабатывали боргидридом натрия в соответствующем растворителе с получением соответствующего спирта. Обработка спирта фталимидом с использованием реакции Мицунобу давала после очистки требуемое соединение. Разложение ацеталей проводили путем кипячения исходного вещества с обратным холодильником со следовым количеством п-толуолсульфоновой кислоты в соответствующем смешанном растворителе с получением соответствующего фталимид-защищенного 4-аминоциклогексанона. Обработка продукта восстановления спирта бензоилхлоридом и затем разложение ацеталей с использованием следового количества п-толуолсульфоновой кислоты в соответствующем смешанном растворителе при кипячении с обратным холодильником давала соответствующий бензоил-защищенный 4-гидроксициклогексанон. Обработка продукта восстановления спирта с использованием следового количества п-толуолсульфоновой кислоты в соответствующем смешанном растворителе при кипячении с обратным холодильником давала соответствующий 4-гидроксициклогексанон.
Общая схема синтеза для получения трициклических 2-бром- тетрагидропиридо[2,3-b]индольных (X=N, Y=Br, Z=H) или 3-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=H, Z=Br), или 6-бромтетрагидрокарбазольных (X=CH, Y=H, Z=Br), или 7-бромтетрагидрокарбазольных (X=CH, Y=Br, Z=H) элементов структуры, когда R1 и R2, взятые вместе, образуют 6-членное кольцо, содержащее углероднын атомы, и 6-членное кольцо замещено с помощью NR20Boc.


Конденсация 2-бром-6-гидразинопиридина (X=N, Y=Br, Z=H) или 3-бром-6-гидразинопиридина (X=N, Y=H, Z=Br), или 4-бромфенилгидразина (X=CH, Y=H, Z=Br), или 3-бромфенилгидразина (X=CH, Y=Br, Z=H) с фталимид-защищенным 4-аминоциклогексаноновым производным в подходящем растворителе давала требуемые продукты конденсации гидразона. Синтез гидразона с помощью термического синтеза индола давал после очистки трициклические продукты циклизации. Фталимидную защитную группу удаляли путем обработки гидратом гидразина в соответствующем растворителе с получением первичных аминов. Обработка свободного амина с помощью Boc-ангидрида и подходящего основания в соответствующем растворителе давала после очистки моно-Boc и бис-Boc-защищенные производные. Обработка моно-Boc-производных гидридов натрия в подходящем растворителе и затем метилйодидом давала после очистки требуемые диметильные трициклические производные. Бис-Boc-защищенные соединения обрабатывали гидридом натрия в подходящем растворителе и затем добавляли подходящий алкилирующий агент с получением после очистки моноалкилированных продуктов при X=CH. В зависимости от реакционной способности алкилирующего агента, реакция идет при комнатной температуре или требует повышенных температур. Селективное разложение Boc-защитной группы, присоединенной к пиррольному кольцу, для X=N, достигалось путем добавления подходящего основания в соответствующем растворителе с получением требуемых соединений. Защита NH-фрагмента пиррольного кольца с помощью триизопропилсилильного фрагмента давала после очистки требуемые трициклические производные для X=N.
Общая схема синтеза для получения трициклических 2-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=Br, Z=H) или 3-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=H, Z=Br) элементов структуры, когда R1 и R2, взятые вместе, образуют 6-членное кольцо, содержащее углеродные атомы, и 6-членное кольцо замещено с помощью NR20Boc.

Нагревание выпускаемого промышленностью 2,6-дибромпиридина (X=N, Y=Br, Z=H) или 2,5-дибромпиридина (X=N, Y=H, Z=Br) с подходящим производным гидразина в соответствующем растворителе дает после очистки соответствующие 2-бром-6-гидразинопиридиновые или 3-бром-6-гидразинопиридиновые производные. Конденсация производных гидразина с выпускаемым промышленностью гидрохлоридом 2,2-диметилтриметиленкеталя 4-(метиламино)циклогексанона в подходящем растворителе в кислотных условиях, используемых в синтезе индола по Фишеру, дает продукты трициклической циклизации. Обработка основанием дает соответствующие продукты трициклической циклизации в виде свободного основания. Обработка свободного амина с помощью Boc-ангидрида и подходящего основания в соответствующем растворителе давала после очистки моно-Boc-защищенные производные.
Общая схема синтеза для получения трициклического 2-бром-6,9-диметилтетрагидропиридо[2,3-b]индола (X=N, Y=Br, Z=H), когда R1 и R2, взятые вместе, образуют 6-членное кольцо, содержащее углеродные атомы, и 6-членное кольцо замещено в этом же положении с помощью -CH3 и NR20Boc.


Выпускаемый промышленностью этиловый эфир 4-гидроксициклогексанкарбоновой кислоты обрабатывают триизопропилсилилхлоридом в подходящем растворителе с соответствующим основанием с получением силилзащищенного соединения. Гидролиз эфирного фрагмента с помощью основания в подходящем растворителе давал соответствующую карбоновую кислоту. Обработка карбоновой кислоты избытком диизопропиламина лития, полученного in situ путем реакции н-бутиллития и диизопропиламина, давала анион, реакцию прерывали путем добавления метилйодида с получением после очистки C-1 метилированной карбоновой кислоты. Карбоновую кислоту затем превращали в защищенный амин с помощью реакции перегруппировки Курциуса. Обработка изонатного промежуточного соединения реакции перегруппировки Курциуса соответствующим нуклеофильным реагентом, то есть трет-бутилатом калия, в подходящем растворителе давала после очистки требуемый защищенный амин. Обработка защищенного амина фторидом тетрабутиламмония в подходящем растворителе давала после очистки требуемые 4-гидроксил-производные. Окисление гидроксильного фрагмента с помощью хлорхромата пиридиния в подходящем растворителе давало после очистки соответствующее производное циклогексанона. Конденсация кетона с подходящим производным гидразина давала соединение гидразона. Соединение гидразона затем обрабатывали при условиях термического синтеза индола по Фишеру в подходящем растворителе с получением продукта циклизации, который непосредственно обрабатывали ди-трет-бутилдикарбонатом в подходящем растворителе. Реакция давала смесь моно- и бис-Boc-защищенных продуктов, которые разделяли с помощью хроматографии. Бис-Boc-производное превращали в моно-Boc-производное путем обработки основанием в подходящем растворителе. Моно-Boc-производное обрабатывали гидридом натрия в подходящем растворителе с получением соответствующей натриевой соли, которую обрабатывали алкилирующим агентом с получением после очистки требуемого продукта. В зависимости от реакционной способности алкилирующего агента реакция идет при комнатной температуре или требует повышенных температур.
Общая схема синтеза для получения трициклических 2-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=Br, Z=H) или 3-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=H, Z=Br), или 6-бромтетрагидрокарбазольных (X=CH, Y=H, Z=Br), или 7-бромтетрагидрокарбазольных (X=CH, Y=Br, Z=H) элементов структуры, когда R и R2, взятые вместе, образуют 6-членное кольцо, содержащее углеродные атомы, и 6-членное кольцо замещено с помощью OR20.

Бензоил-защищенный 4-гидроксициклогексанон обрабатывают подходящим производным гидразина в соответствующем растворителе с получением продукта конденсации. Производные гидрозона обрабатывают при условиях проведения термического синтеза индола по Фишеру при использовании микроволнового излучения с получением после очистки продуктов трициклической циклизации. Обработка трициклических продуктов гидридом натрия в подходящем растворителе и затем добавление метилйодида давала после очистки требуемые продукты. Разрушение эфирной защитной группы осуществлялось путем нагревания с основанием в подходящем растворителе с использованием микроволнового излучения с получением требуемых гидроксильных соединений. Алкилирование гидроксильной группы осуществляли с помощью подходящих алкилирующих агентов в соответствующем растворителе после активации гидроксильной группы в ее натриевую соль с использованием гидрида натрия. В зависимости от реакционной способности алкилирующего агента реакция идет при комнатной температуре или требует повышенных температур. После очистки получали требуемые продукты.
Общая схема синтеза для получения трициклических 2-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=Br, Z=H) или 3-бромтетрагидропиридо[2,3-b]индольных (X=N, Y=H, Z=Br), или 6-бромтетрагидрокарбазольных (X=CH, Y=H, Z=Br), или 7-бромтетрагидрокарбазольных (X=CH, Y=Br, Z=H) элементов структуры, когда R1 и R2, взятые вместе, образуют 6-членное кольцо, содержащее углеродные атомы, и 6-членное кольцо замещено с помощью OCH3.

Незащищенный 4-гидроксициклогексанон обрабатывают подходящим производным метилгидразина в соответствующем растворителе с получением продукта конденсации. Производные метилгидразона обрабатывают при условиях термического синтеза индола по Фишеру с использованием микроволнового излучения с получением после очистки продуктов трициклической циклизации. Обработка трициклических продуктов гидридом натрия в подходящем растворителе и затем добавление метилйодида давала после очистки требуемые продукты.
Общая схема синтеза для получения соединений этого изобретения.
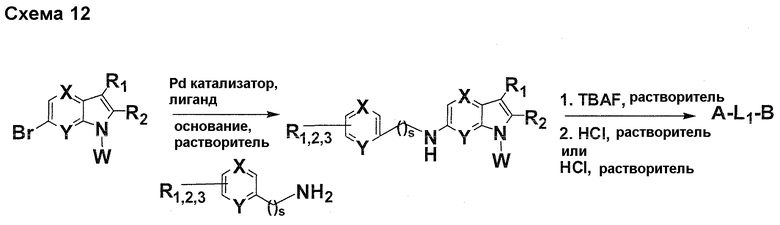
N-защищенные бромсодержащие элементы структуры при (X=CH, Y=N, Z=N, W=CH3 или TIPS) или (X=N, Y=CH, Z=CH, W=CH3 или TIPS), или (X, Z=CH, Y=N, W=CH3 или TIPS), или (X, Y, Z=CH, W=CH3 или Boc) подвергали взаимодействию с подходящими аминами или аминосодержащими элементами структуры при (X, Y=CH, s=0) или (X=N, Y=CH, s=0), или (X=CH, Y=N, s=0, 1, 2) с использованием условий реакции сочетания на Pd с соответствующим Pd-катализатором и соответствующим лигандом в подходящем растворителе с получением после очистки требуемых продуктов аминирования. Снятие защитных групп с N-TIPS-защищенных соединений с помощью фторида тетра-н-бутиламмония в подходящем растворителе давало после очистки требуемые соединения. Обработка N-незащищенных соединений хлористым водородом в подходящем растворителе давала конечные соединения. Для соединений, в которых W=CH3 или Boc, обработка хлористым водородом в подходящем растворителе давала конечные соединения.
Общая схема синтеза для получения соединений этого изобретения.

N-защищенные бромсодержащие элементы структуры (Y=CH, W=CH3, TIPS или Boc или Y=N, W=CH3 или TIPS) подвергали взаимодействию с подходящими аминами или аминсодержащими элементами структуры при (X, Z=CH) или (X=N, Z=CH), с использованием условий реакции сочетания на Pd с соответствующим Pd-катализатором и соответствующим лигандом в подходящем растворителе с получением после очистки требуемых продуктов аминирования. Снятие защитных групп с N-TIPS-защищенных соединений с помощью фторида тетра-н-бутиламмония в подходящем растворителе давало после очистки требуемые соединения. Обработка N-незащищенных соединений хлористым водородом в подходящем растворителе давала конечные соединения. Для соединений, в которых W=CH3 или Boc, обработка хлористым водородом в подходящем растворителе давала конечные соединения.
Общая схема синтеза для получения соединений этого изобретения.

N-защищенные бромсодержащие элементы структуры при (X=CH, Y=N, W=CH3 или TIPS) или (X=N, Y=CH, W=CH3 или TIPS), или (X, Y=CH, W=CH3 или TIPS) подвергали взаимодействию с подходящими аминсодержащими элементами структуры при (Z=CH, W=H или CH3, или TIPS) или (Z=N, W=H или CH3, или TIPS), с использованием условий реакции сочетания на Pd с соответствующим Pd-катализатором и соответствующим лигандом в подходящем растворителе с получением после очистки требуемых продуктов аминирования. Снятие защитных групп с N-TIPS-защищенных соединений с помощью фторида тетра-н-бутиламмония в подходящем растворителе давало после очистки требуемые соединения. Обработка N-незащищенных соединений хлористым водородом в подходящем растворителе давала конечные соединения. Для соединений, где (W=H или CH3), обработка хлористым водородом в подходящем растворителе давала конечные соединения.
Общая схема синтеза для получения соединений этого изобретения.

N-защищенные бромсодержащие элементы структуры при (X=CH, W=CH3 или TIPS) или (X=N, W=CH3 или TIPS) подвергали взаимодействию с подходящими аминосодержащими элементами структуры при (Z=CH, W=H или CH3, или TIPS) или (Z=N, W=H или CH3, или TIPS), с использованием условий реакции сочетания на Pd с соответствующим Pd-катализатором и соответствующим лигандом в подходящем растворителе с получением после очистки требуемых продуктов аминирования. Снятие защитных групп с N-TIPS-защищенных соединений с помощью фторида тетра-н-бутиламмония в подходящем растворителе давало после очистки требуемые соединения. Обработка N-незащищенных соединений хлористым водородом в подходящем растворителе давала конечные соединения. Для соединений, где (W=H или CH3), обработка хлористым водородом в подходящем растворителе давала конечные соединения.
Общая схема для получения 2-бромтетрагидропиридо[2,3-b]индольных элементов структуры, когда R1 и R2, взятые вместе, образуют (n+4)-членное кольцо, содержащее углеродные атомы.

Выпускаемый промышленностью 2,6-дибромпиридин обрабатывали производными гидразина (R=H, CH3) в подходящем растворителе с получением производного моногидразина. Конденсация с соответствующим циклическим кетоном в подходящем растворителе давала требуемые соединения гидразона. Термический синтез индола по Фишеру давал после очистки продукты трициклической циклизации.
Общая схема синтеза для получения трициклических 7-бромтетрагидропиридо[4,3-b]индольных (X=Br, Y=H) или 8-бромтетрагидропиридо[4,3-b]индольных (X=H, Y=Br) элементов структуры, когда R1 и R2, взятые вместе, образуют (n+4)-членное кольцо, содержащее углеродные атомы и NR20.
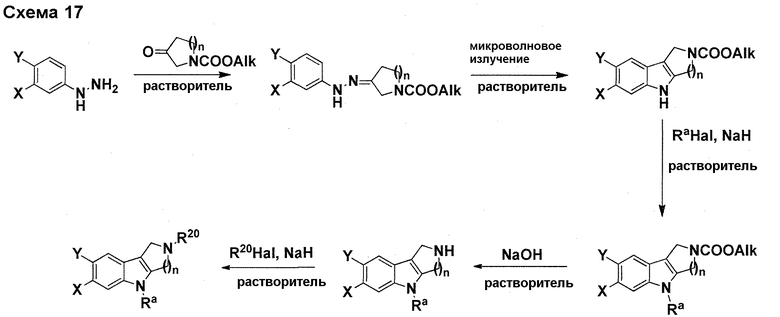
Выпускаемые промышленностью производные фенилгидразина обрабатывали соответствующим циклическим кетоном в подходящем растворителе с получением требуемых соединений гидразона. Синтез индола по Фишеру давал после очистки продукты трициклической циклизации. Обработка трициклических производных гидридом натрия в подходящем растворителе и затем соответствующим галогенированным алкилом давала соответствующий N-алкилированный продукт. Затем осуществляли снятие защиты с карбаматных производных путем добавления гидроксида натрия в соответствующем растворителе при кипячении с обратным холодильником с получением требуемых соединений. И наконец, дереватизировали алифатические NH-производные путем их обработки гидридом натрия и затем добавления соответствующего галогенированного алкила в подходящем растворителе с получением требуемых трициклических производных.
Общая схема синтеза для получения трициклических 2-бромтетрагидропирроло[2,3-b:4,5-c']дипиридиновых (X=N, Y=H, Z=Br), 3-бромтетрагидропирроло[2,3-b:4,5-c']дипиридиновых (X=N, Y=Br, Z=H), 7-бромтетрагидропиридо[4,3-6]индольных (X=CH, Y=H, Z= Br) или 8-бромтетрагидропиридо[4,3-b]индольных (X=CH, Y=Br, Z=H) элементов структуры, когда R1 и R2, взятые вместе, образуют (n+4)-членное кольцо, содержащее углеродные атомы и NR20.

Выпускаемые промышленностью производные гидразина обрабатывали соответствующим циклическим кетоном в присутствии подходящей кислоты в подходящем растворителе с получением после очистки требуемых соединений с помощью синтеза индола по Фишеру в кислой среде.
Общая схема синтеза для получения трициклических 2-бромтетрагидротиопирано[3',4':4,5]пирроло[2,3-b]пиридин-6,6-диоксидных (X=Br, Y=H) или 3-бромтетрагидротиопирано[3',4':4,5]пирроло[2,3-b]пиридин-6,6-диоксидных (X=H, Y=Br) элементов структуры, когда R1 и R2, взятые вместе, образуют (n+4)-членное кольцо, содержащее углеродные атомы и SO2.
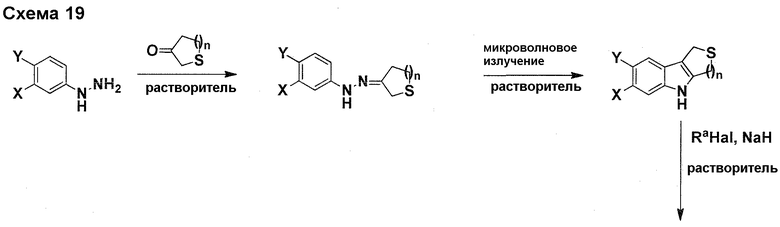
Выпускаемые промышленностью производные фенилгидразина обрабатывали соответствующим циклическим кетоном в подходящем растворителе с получением требуемых соединений гидразона. Синтез индола по Фишеру давал после очистки продукты трициклической циклизации. Обработка трициклических производных гидридом натрия в подходящем растворителе и затем соответствующим галогенированным алкилом давала соответствующий N-алкилированный продукт. Последующее окисление серосодержащих производных с использованием соответствующего окислителя давало требуемые сульфоновые соединения.
Общая схема синтеза для получения галогенированных алкилсульфонов.

Выпускаемые промышленностью производные метилтиоспиртов обрабатывали соответствующим окислителем в подходящем растворителе с получением требуемых гидроксисульфоновых производных. Соединения первичных спиртов превращали в соответствующие галогенпроизводные с помощью реакции Аппеля в подходящем растворителе.
Все реагенты и растворители приобретались у соответствующих фирм и использовались без дополнительной очистки. Спектры протонного (1H) ядерного магнитного резонанса регистрировали на ЯМР спектрометре Bruker EPX при 400 МГц в дейтерированных растворителях. Масс-спектры (MS) регистрировали на спектрометре Finnigan MAT TSQ 7000. Флэш-очистку проводили на системе очистки с помощью флэш-хроматографии Biotage Isolera One с использованием картриджей HP-Sil SNAP (фирмы Biotage), и градиент состава растворителя указывали в конкретных примерах. Тонкослойную хроматографию (ТСХ) проводили на пластинках с силикагелем, используя УФ-детектирование. Препаративную тонкослойную хроматографию (Prep-ТСХ) осуществляли на пластинках со слоем толщиной 0,5 мм или 1 мм силикагеля (Analtech: Uniplate, F254), и растворители указывали в конкретных примерах.
Несмотря на то, что возможно вводить соединения настоящего изобретения в чистом виде, предпочтительно приготавливать из них фармацевтическую композицию в соответствии с обычной фармацевтической практикой. Следовательно, изобретение также предлагает фармацевтическую композицию, которая включает терапевтически эффективное количество соединения формулы (I) в смеси с фармацевтически приемлемым вспомогательным веществом.
Фармацевтически приемлемые вспомогательные вещества хорошо известны в фармацевтике, и описаны, например, в монографии Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1991). Фармацевтическое вспомогательное вещество может быть выбрано с учетом предполагаемого способа введения и в соответствии с обычной фармацевтической практикой. Вспомогательное вещество должно быть приемлемым с точки зрения отсутствия его вредного воздействия на реципиента.
Фармацевтически подходящие вспомогательные вещества, которые могут быть использованы в составе фармацевтической композиции настоящего изобретения, могут включать, например, носители, среды, разбавители, растворители, такие как спирты, имеющие одну гидроксильную группу, такие как этанол, изопропанол, и многоатомные спирты, такие как гликоли, и пищевые масла, такие как соевое масло, кокосовое масло, оливковое масло, сафлоровое масло, хлопковое масло, эфиры жирных кислот, такие как этилолеат, изопропилмиристат, связующие, вспомогательные средства, солюбилизаторы, загустители, стабилизаторы, разрыхлители, глиданты, смазывающие вещества, буферные вещества, эмульгаторы, увлажняющие компоненты, суспендирующие вещества, подсластители, окрашивающие вещества, вещества, корригирующие вкус и запах, вещества для образования покрытий, консерванты, антиоксиданты, вспомогательные вещества, используемые в процессе производства, модификаторы и интенсификаторы доставки лекарственных средств, такие как фосфат кальция, стеарат магния, тальк, моносахариды, дисахариды, крахмал, желатин, целлюлоза, метилцеллюлоза, натрий-карбоксиметилцеллюлоза, декстроза, гидроксипропил-β-циклодекстрин, поливинилпирролидон, легкоплавкие воски и ионообменные смолы.
Способы введения (доставки) соединений изобретения включают, но этим не ограничивая, один или несколько из следующих способов, таких как пероральный (например, в виде таблетки, капсулы или в виде проглатываемого раствора), местный, мукозальный (например, в виде назального спрея или аэрозоля для ингаляции), назальный, парентеральный (например, в виде инъецируемой формы), гастроинтестинальный, интраспинальный, интраперитонеальный, внутримышечный, внутривенный, внутриматочный, внутриглазной, интрадермальный, интракраниальный, интратрахеальный, интравагинальный, интрацеребровентрикулярный, интрацеребральный, подкожный, офтальмический (включая введение в стекловидное тело или выстилающую ткань), трансдермальный, ректальный, буккальный, эпидуральный и сублингвальный.
Например, соединения могут быть введены перорально в форме таблеток, капсул, порошков, эликсиров, растворов или суспензий, которые могут содержать корригирующие вкус и запах вещества или окрашивающие вещества, для достижения мгновенного, замедленного, модифицированного, постоянного, периодического или контролируемого высвобождения лекарственного средства.
Таблетки могут содержать вспомогательные вещества, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновной фосфат кальция и глицин, разрыхлители, такие как крахмал (предпочтительно, кукурузный, картофельный или маниоковый крахмал), натрия крахмала гликолят, кроскармелоза натрия и конкретные комплексные силикаты, и связующие для грануляции, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза (HPC), сахароза, желатин и камедь. Кроме того, могут быть включены смазывающие вещества, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Твердые композиции аналогичного типа могут также быть использованы в качестве наполнителей в желатиновых капсулах. Предпочтительные вспомогательные вещества в данном случае включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров, лекарственное средство может быть объединено с различными подсластиталями или ароматизаторами, окрашивающим веществом или красителями, с эмульгаторами и/или суспендирующими веществами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин, и их комбинациями.
Если соединения изобретения вводят парентерально, то тогда примеры такого введения включают один или более из способов, таких как внутривенное, интраартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное или подкожное введение соединения; и/или с помощью методов инфузии. Для парентерального введения, соединения лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например, достаточные количества солей или глюкозы, для того чтобы сделать раствор изотоническим по отношении к крови. Водные растворы в случае необходимости должны быть соответствующим образом буферированы (предпочтительно до pH от 3 до 9). Приготовление подходящих парентеральных препаратов в стерильных условиях легко осуществимо с помощью стандартных фармацевтических методов, хорошо известных специалистам в этой области.
Как было указано, соединения настоящего изобретения могут быть введены интраназально или с путем ингаляции и удобно доставлены в форме ингалируемого сухого порошка или аэрозольной струи из контейнера под давлением, с помощью насоса, спрея или распылителя при использовании подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафиторэтана, гидрофторалкана, такого как 1,1,1,2-тетрафторэтан (HFA134AT) или 1,1,1,2,3,3,3-гептафторпропан (HFA 227EA), диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением, дозирующее устройство может быть создано путем установки клапана для доставки заданного количества. Контейнер под давлением, насос, спрей или распылитель могут содержать раствор или суспензию активного соединения, например, используя смесь этанола и пропеллента в качестве растворителя, который может дополнительно содержать смазывающее вещество, например, сорбитан триолеат. Для применения в ингаляторе или инсуффляторе могут быть приготовлены капсулы и картриджи (изготовленные, например, желатина), содержащие порошкообразную смесь соединения и подходящей порошкообразной основы, такой как лактоза или крахмал.
В качестве варианта, соединения настоящего изобретения могут быть введены в форме суппозитория или пессария, или они могут быть нанесены местно в форме геля, гидрогеля, лосьона, раствора, крема, мази или присыпки. Соединения настоящего изобретения могут также быть введены дермально или трансдермально, например, путем применения трансдермального пластыря.
Они могут также быть введены пульмонально или ректально. Они могут также быть введены окулярно. Для офтальмического применения, соединения могут быть приготовлены в виде тонко измельченных суспензий в изотоническом со скорректированной величиной pH стерильном физиологическом растворе, или, предпочтительно, в виде растворов в изотоническом со скорректированной величиной pH стерильном физиологическом растворе, необязательно, в комбинации с консервантом, таким как бензалкония хлорид. В качестве варианта, они могут быть приготовлены в мазе, такой как вазелин.
Для местного нанесения на кожу, соединения настоящего изобретения могут быть приготовлены в виде подходящей мази, содержащей активное соединение, суспендированное или растворенное, например, в смеси одного или более из следующих веществ: минерального масла, жидкого вазелина, медицинского вазелина, пропиленгликоля, эмульгирующего воска и воды. В качестве варианта, они могут быть приготовлены в виде подходящего лосьона или крема, будучи суспендированы или растворены, например, в смеси одного или более из следующих веществ: минерального масла, сорбитанмоностеарата, полиэтиленгликоля, жидкого парафина, полисорбата 60, цтиловых эфиров воска, цетеарилового спирта, 2-октилдодеканола, бензилового спирта и воды.
Обычно, врач определяет конкретную дозу, которая лучше всего подходит для конкретного субъекта. Конкретная величина дозы и частота дозирования для конкретного субъекта может изменяться и зависеть от ряда факторов, включающих активность применяемого конкретного соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость эксрекции, комбинацию лекарственных средств, тяжесть конкретного состояния и индивидуальную переносимость терапии.
Предполагаемая доза соединений согласно настоящему изобретению для введения человеку (приблизительно с массой тела 70 кг) составляет от 0,1 мг до 1 г, предпочтительно, от 1 мг до 500 мг активного ингредиента в разовой дозе. Разовая доза может быть введена, например, от 1 до 4 раз в сутки. Доза будет зависеть от способа введения. Следует иметь в виду, что может быть необходимым провести обычные корректировки дозы в зависимости от возраста и массы тела пациента, а также тяжести состояния, подвергаемого лечению. Точная величина дозы и способ ее введения будет определяться исключительно лечащим врачом или ветеринаром.
Соединения изобретения могут также применяться в комбинации с другими терапевтическими средствами. Когда соединение изобретения применяют в комбинации со вторым терапевтическим средством, активным против того же самого заболевания, доза каждого соединения может отличаться от той, которая используется в случае применения соединения отдельно.
Упомянутые выше комбинации могут быть удобно приготовлены для применения в форме фармацевтического препарата. Индивидуальные компоненты таких комбинаций могут быть введены либо последовательно, либо одновременно, в отдельных или комбинированных фармацевтических препаратах с помощью любого подходящего способа. При последовательном введении, первым может быть введено либо соединение изобретения, либо второе терапевтическое средство. При одновременном введении, комбинация может быть введена либо в одной и той же композиции, либо отдельными фармацевтическими композициями. При комбинации в одном и том же препарате, следует иметь в виду, что эти два соединения должны быть стабильными и совместимыми друг с другом и другими компонентами препарата. В случаи применения раздельных препаратов, они могут быть введены в любой удобной лекарственной форме удобными способами, которые известны в этой области для таких соединений.
Фармацевтические композиции изобретения могут быть приготовлены по существу известным для специалиста в этой области способом, описанным, например, в монографии Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1991).
Заболевания, которые могут подвергаться лечению с помощью соединений настоящего изобретения, могут быть связаны с образованием аномальных белковых структур, в частности, аномальных структур β-листов. В контексте настоящего изобретения, аномальная белковая структура представляет собой белковую структуру, которая возникает, когда происходит рефолдинг белка или пептида из пространственной структуры, которая обычно наблюдается у здоровых людей, в другую пространственную структуру, которая связана с патологическим состоянием. Аналогичным образом, аномальная структура β-листа в контексте настоящего изобретения представляет собой структуру β-листа, которая возникает, когда происходит рефолдинг белка или пептида из пространственной структуры, которая обычно наблюдается у здоровых людей, в структуру β-листа, которая связана с патологическим состоянием.
В частности, в одном варианте осуществления, заболевания, которые могут быть подвергнуты лечению с помощью соединений настоящего изобретения, представляют собой заболевания или состояния, связанные с амилоидными или амилоидоподобными белками.
Эта группа заболеваний и расстройств включает неврологические нарушения, такие как болезнь Альцгеймера (AD), заболевания или состояния, характеризующиеся потерей когнитивной способности памяти, такие как, например, умеренное когнитивное нарушение (MCI), деменция с тельцами Леви, синдром Дауна, врожденное внутримозговое кровоизлияние с амилоидозом (голландского типа); комплекс Гуама паркинсонизм-деменция. Другие заболевания, которые основаны или связаны с амилоидоподобными белками представляют собой прогрессирующий супрануклеарный парез, множественный склероз; болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, ALS (боковой амиотрофический склероз), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте; старческий амилоидоз сердца; внутрисекреторные опухоли, и другие заболевания, включая амилоид-связанные глазные заболевания, которые поражают различные ткани глаза, такие как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталик глаза, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, в частности возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
В предпочтительном варианте осуществления, соединения настоящего изобретения могут применяться для лечения болезни Альцгеймера, умеренного когнитивного нарушения (MCI), деменции с тельцами Леви (LBD), бокового амиотрофического склероза (ALS), миозита с включенными тельцами (IBM) и возрастной дегенерации желтого пятна (AMD). В конкретном предпочтительном варианте осуществления, соединения настоящего изобретения могут применяться для лечения болезни Альцгеймера.
Способность соединения ингибировать агрегацию Αβ может быть определена, например, с помощью флуоресцентной корреляционной спектроскопией, описанной в публикации Rzepecki et al., J. Biol. Chem., 2004, 279(46), 47497-47505 или с помощью спектро-флуоресцентного анализа с тиофлавином T.
В другом варианте осуществления, соединения настоящего изобретения могут применяться для лечения или облегчения поражающих воздействий глазных заболеваний, связанных с патологическими нарушениями/изменениями в тканях визуальной системы, в частности, связанных с патологическими нарушениеми/изменениями в ткани визуальной системы, имеющими отношение к бета-амилоиду, такими как, например, нейрональная деградация. Указанные патологические нарушения могут возникать, например, в различных тканях глаза, таких как зрительная зона коры головного мозга, вызывая кортикальные зрительные нарушения; передняя камера глаза и зрительный нерв, вызывая глаукому; хрусталик глаза, вызывая катаракту вследствие бета-амилоидного отложения; стекловидное тело, вызывая амилоидную дистрофию роговицы глаза; сетчатка, вызывая первичную дегенерацию сетчатки и дегенерацию желтого пятна, например возрастную дегенерацию желтого пятна; зрительный нерв, вызывая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и роговая оболочка глаза, вызывая решетчатую дегенерацию роговицы.
Соединения согласно настоящему изобретению могут также применяться в форме смеси по меньшей мере с одним дополнительным биологически активным соединением и/или фармацевтически приемлемым носителем и/или разбавителем и/или вспомогательным веществом. Предпочтительно, чтобы соединение и/или дополнительное биологически активное соединение присутствовали в терапевтически эффективном количестве.
Природа дополнительного биологически активного соединения зависит от предполагаемого применения смеси. Дополнительное биологически активное вещество или соединение может оказывать свое биологическое действие по такому же или аналогичному механизму, что и соединение согласно изобретению, или по несвязанному с соединением механизму действия или по многочисленным связанным и/или несвязанным с соединением механизмам действия.
Обычно, дополнительное биологически активное соединение может включать усилители нейронной передачи, психотерапевтические средства, ингибиторы ацетилхолинэстеразы, блокаторы кальциевых каналов, органические амины, бензодиазепиновые транквилизаторы, усилители синтеза, хранения и высвобождения ацетилхолина, агонисты постсинаптического рецептора ацетилхолина, ингибиторы моноаминоксидазы-A или -B, антагонисты рецептора N-метил-D-аспартата/глутамата, нестероидные противовоспалительные препараты, антиоксиданты и антагогнисты серотонинергического рецептора. В частности, дополнительное биологически активное соединение может быть выбрано из группы, состоящей из соединения, используемого при лечении амилоидоза, соединения против окислительного стресса, антиапоптотических соединений, соединений, образующих хелаты с металлами, ингибиторов репарации ДНК, таких как пирензепин и метаболиты, 3-амино-1-пропансульфоновой кислоты (3APS), 1,3-пропандисульфоната (1,3PDS), активаторов α-секретазы, ингибиторов β- и γ-секретазы, тау-белков, нейротрансмиттеров, разрушителей β-амилоидного белка, аттрактантов для очистки от бета амилоида/истощения клеточных компонентов, ингибиторов N-терминально усеченного бета амилоида, включая бета амилоид 3-42 в форме пироглутамата, противовоспалительных средств, или ингибиторов холинэстеразы (ChEI), таких как такрин, ривастигмин, донепезил и/или галантамин, агонистов М1, других лекарственных средств, включая любые лекарственные средства, модифицирующие амилоидные или тау-белки, и питательных добавок, антитела, включающего любое функционально эквивалентное антитело или его функциональные части, фрагмента Aβ антигенного пептида, состоящего из единственного или повторяющегося участка с множеством смежных аминокислотных остатков из N-терминальной части Aβ пептида.
В дополнительном варианте осуществления, смеси согласно изобретению могут включать ниацин или мемантин вместе с соединением согласно настоящему изобретению и, необязательно, фармацевтически приемлемый носитель и/или разбавитель, и/или вспомогательное вещество.
В еще одном варианте осуществления изобретения предлагаются смеси, которые включают в качестве дополнительного биологически активного соединения "атипичные нейролептики", такие как, например, клозапин, зипрасидон, рисперидон, арипипразол или оланзапин, для лечения позитивных и негативных психотических симптомов, включающих галлюцинации, бредовые идеи, нарушения мышления (проявляющиеся в заметной инкогерентности, нарушении планов, тангенциальности речи), и неправильное или дезорганизованное поведение, а также ангедонию, притупленный аффект, апатию, и социальную самоизоляцию, вместе с соединением согласно изобретению и, необязательно, фармацевтически приемлемым носителем и/или разбавителем, и/или вспомогательным веществом.
Другие соединения, которые могут быть удобно использованы в смесях в комбинации с соединением согласно настоящему изобретению, описаны, например, в патентном документе WO 2004/058258 (смотрите конкретно страницы 16 и 17), включая препараты целенаправленного терапевтического действия (страницы 36-39), алкансульфоновые кислоты и алканолсульфоновые кислоты (страницы 39-51), ингибиторы холинэстеразы (страницы 51-56), антагонисты рецептора N-метил-D-аспартата (страницы 56-58), эстрогены (страницы 58-59), нестероидные противовоспалительные средства (страницы 60 и 61), антиоксиданты (страницы 61 и 62), агонисты рецептора активатора пролиферации пероксисом (PPAR) (страницы 63-67), гипохолестеринемические средства (страницы 68 и 75), ингибиторы амилоида (страницы 75-77), ингибиторы образования амилоида (страницы 77-78), соединения, образующие хелаты с металлами (страницы 78-79), нейролептики и антидепрессанты (страницы 80-82), биологически активные добавки (страницы 83-89) и соединения, повышающие доступность биологические активных веществ в мозге (смотрите страницы 89-93) и пролекарства (страницы 93 и 94), содержание которого приводится в описании изобретения путем ссылки на него.
В одном предпочтительном варианте осуществления, дополнительное биологически активное соединение представляет собой антитело, включающее любое функционально эквивалентное антитело или его функциональные части. Предпочтительно, чтобы антитело могло представлять собой моноклональное, химерное или гуманизированное антитело.
В дополнительном аспекте изобретения предлагается смесь, включающая помимо соединения изобретения антитело, включая его функциональные части, или, более конкретно, моноклональное антитело, включая его функциональные части, которое распознает и связывает β-амилоид (Αβ), в частности, свойственную β-амилоиду конформацию, тот есть амилоидные олигомеры и волокна, а не линеаризованные амилоидные фрагменты.
В частности, указанные антитела способны ингибировать in vitro и in vivo агрегацию амилоидогенных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких как, например, Αβ мономерные пептиды 1-39; 1-40, 1-41, 1-42 или 1-43, и, особенно, Αβ1-42 мономерные пептиды, в высокомолекулярные полимерные амилодные волокна или нити. В результате ингибирования агрегации амилоидогенных мономерных пептидов, эти антитела способны предотвращать или замедлять образование амилоидных бляшек, в частности, амилоидной формы (1-42), которая, как известно, становится нерастворимой в результате изменения вторичной конформации и является основной частью амилоидных бляшек в мозге больных животных или людей.
В другом аспекте изобретения смесь включает антитела, которые при совместной инкубации с ранее образовавшимися высокомолекулярными полимерными амилоидными волокнами или нитями, образовавшимися в результате агрегации амилоидных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких, как например, Aβ мономерные пептиды 1-39; 1-40, 1-41, 1-42 или 1-43, но особенно Αβ1-42 мономерные пептиды, способны разрушать указанные высокомолекулярные полимерные амилоидные волокна или нити. В результате разрушения амилоидогенных полимерных волокон или нитей, эти антитела способны предотвращать или замедлять образование амилоидных бляшек, что приводит к облегчению симптомов, связанных с заболеванием, и отсрочке или обратному ходу его развития.
В еще одном аспекте изобретения смесь включает антитело, и, в частности, моноклональное антитело или его функциональные части, которое является бифункциональным или биспецифичным в том смысле, что оно обладает как свойством ингибирования агрегации, так и свойством разрушения, упомянутыми выше, в частности, вместе с высокой степенью конформационной чувствительности.
В одном варианте осуществления смесь включает антитело, которое распознает и связывает конформационный эпитоп, в частности, конформационный эпитоп, который присутствует в N-терминальной части β-амилоидного пептида, в частности, встроенного в следующую центральную зону N-терминальной части β-амилоидного пептида:

В частности, эпитоп был локализован в области β-амилоидного белка между аминокислотными остатками 12-24, в частности, между остатками 14-23, более конкретно, между аминокислотными остатками 14-20, включающий три сайта четкого распознавания и связывающих сайта, остатки которого в основном принимают участие в связывании β-амилоидного белка и расположены в положении 16, 17, и в положении 19 и 20, и в положении 14, соответственно.
В конкретном варианте осуществления смесь настоящего изобретения включает, помимо соединения изобретения, антитело, в частности, бифункциональное антитело, и, особенно, моноклональное антитело, в частности, бифункциональное моноклональное антитело, включающее любое функционально эквивалентное антитело или его функциональные части, где антитело имеет характерные свойства антитела, продуцированного линией клеток гибридомы, выбранных из группы, состоящей из FP 12H3, FP 12H3-C2 и FP 12H3-G2, депонированных в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2752, DSM ACC 2750 и DSM ACC2751, соответственно, ET 7E3, депонированных в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2755, и EJ 7H3 депонированных в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2756.
Более конкретно, изобретение относится к антителу, включающему любое функционально эквивалентное антитело или его функциональные части, продуцированному линией клеток гибридомы, выбранных из группы, состоящей из FP 12H3, FP 12H3-C2 и FP 12H3-G2, депонированных в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2752, DSM ACC 2750 и DSM ACC2751, соответственно, ET 7E3, депонированных в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2755, и EJ 7H3 депонированных в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2756.
Указанные выше антитела описаны в опубликованной Международной заявке WO 2007/068412, содержание которой приводится в описании настоящего изобретения путем ссылки на нее.
В дополнительном аспекте антитело, которое является составной частью смеси согласно изобретению, представляет собой химерное антитело или его фрагмент, или гуманизированное антитело или его фрагмент. Эти и другие антитела, которые удобно использовать в смесях согласно настоящему изобретению, описаны, например, в Международной заявке PCT/US2007/073504, зарегистрированной 13 июля 2007 года.
Если антитело представляет собой гуманизированное антитело, то, предпочтительно, чтобы оно имело легкую цепь и тяжелую цепь, которые показаны в последовательности SEQ ID NO: 2 и SEQ ID NO: 4 в Международной заявке № PCT/US2007/073504, или имеет вариабельную область легких цепей и вариабельную область тяжелых цепей, которые показаны в последовательности SEQ ID NO: 1 и SEQ ID NO: 3 в Международной заявке № PCT/US2007/073504. Эти последовательности также приведены в прилагаемом листинге последовательностей.
В еще одном аспекте изобретения предлагается смесь, которая включает, помимо описанного выше соединения согласно изобретению, пептидный фрагмент из N-терминальной части Αβ пептида, в частности, фрагмента Αβ пептида, состоящего из единственного или повторяющегося участка с множеством смежных аминокислотных остатков из N-терминальной части Aβ пептида, в частности, участка между 13 и 15 смежными аминокислотными остатками, и, в частности, фрагмента Αβ пептида, состоящего из аминокислотных остатков, выбранных из группы, состоящей из остатков 1-15, 1-14 и 1-13 из N-терминальной части Αβ пептида, более конкретно, из остатка 1-15, включая его функционально эквивалентные фрагменты, но, особенно, упомянутый здесь Αβ фрагмент, перед тем как его присоединить, или ввести или ресуспендировать в частице носителя/вспомогательного средства, такого как, например, липосома. Пептидный фрагмент может быть включен в композицию вакцины. В частности, пептидный антиген модифицируют с помощью липофильного или гидрофобного фрагмента, что облегчает его введение в липидные бислои липосомного носителя/иммуностимулятора, в частности, с помощью липофильного или гидрофобного фрагмента, который выполняет функцию якоря для пептида в липосомном бислое и имеет размер, который позволяет позиционировать и стабилизировать пептид в непосредственной близости от поверхности липосомы.
В дополнительном варианте осуществления изобретения липофильный или гидрофобный фрагмент представляет собой жирную кислоту, триглицерид или фосфолипид, и, в частности, жирную кислоту, триглицерид или фосфолипид. В частности, гидрофобный фрагмент представляет собой пальмитиновую кислоту, и липосомный препарат может, кроме того, содержать вспомогательное средство, такое как, например, липид A, алюминиевые квасцы, фосфат кальция, интерлейкин-1, и/или микрокапсулы полисахаридов и белков, и, в частности, детоксифицированный липид A, такой как монофосфорил или дифосфорил липида A, или алюминиевые квасцы.
Эти и другие композиции, которые удобно использовать в смесях настоящего изобретения, описаны, например, в опубликованной Международной заявке WO 2007/068411.
Диагностирование связанного с амилоидом заболевания или состояния или предрасположенности к связанному с амилоидом заболевания или состояния у пациента может осуществляться путем обнаружения специфического связывания соединения согласно изобретению с амилоидным белком в пробе или in situ, которое включает контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением изобретения, которое связывает амилоидный белок, связывание соединения с амилоидным белком с образованием комплекса соединение/белок, обнаружение образования комплекса соединение/белок и установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела, при необязательном сравнении количества указанного комплекса соединение/белок с нормальным контрольным значением, где увеличение количества указанного агрегата по сравнению с нормальным контрольным значением может указывать на то, что указанный пациент страдает от заболевания или состояния, связанного с амилоидом, или для него существует риск развития такого заболевания или состояния.
Мониторинг минимального остаточного заболевания у пациента после лечения с помощью соединения или смеси согласно изобретению может быть осуществлен путем обнаружения специфического связывания соединения согласно изобретению с амилоидным белком в пробе или in situ, которое включает контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением изобретения, которое связывает амилоидный белок, связывание соединения с амилоидным белком с образованием комплекса соединение/белок, обнаружение образования комплекса соединение/белок, и установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела, при необязательном сравнении количества указанного комплекса соединение/белок с нормальным контрольным значением, где увеличение количества указанного агрегата, по сравнению с нормальным контрольным значением, может указывать на то, что указанный пациент все еще страдает от минимального остаточного заболевания.
Предсказание восприимчивости пациента к лечению с помощью соединения или композиции или смеси согласно изобретению может быть осуществлено путем обнаружения специфического связывания соединения согласно изобретению с амилоидным белком в пробе или in situ, которое включает контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением согласно изобретению, которое связывает амилоидный белок, связывание соединения с амилоидным белком с образованием комплекса соединение/белок, обнаружение образования комплекса соединение/белок, и установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела, при необязательном сравнении количества указанного комплекса соединение/белок до и после начала лечения, где уменьшение количества указанного агрегата может указывать на то, что указанный пациент имеет высокую степень восприимчивости к лечению.
В дополнительном аспекте настоящего изобретения соединение формулы (I) может содержать радиоактивный изотоп (например, 125I, 124I, 123I или 18F). Эти соединения могут применяться для in vivo диагностирования или диагностической визуализации заболеваний, связанных с амилоидом, предпочтительно, болезни Альцгеймера, например, при таких методах, как однофотонная эмиссионная компьютерная томография или позитрон-эмиссионная томография (PET).
При использовании радиофармацевтических препаратов предпочтительно, чтобы соединение настоящего изобретения вводили в радиофармацевтический препарат, включающий соединение изобретения. "Радиофармацевтический препарат" определяется в настоящем изобретении как препарат, включающий соединение настоящего изобретения (такое как соединение формулы (I) или его соль) в форме, подходящей для введения млекопитающим, таким как люди. Предпочтительно, чтобы радиофармацевтический препарат дополнительно включал физиологически приемлемое вспомогательное вещество. Предпочтительно, чтобы введение осуществлялось путем инъекции препарата в виде водного раствора. Такой препарат может необязательно содержать дополнительные ингредиенты, такие как буферы; фармацевтически приемлемые солюбилизаторы (например, циклодекстрины или поверхностно-активные вещества, такие как Pluronic, Tween или фосфолипиды); фармацевтически приемлемые стабилизаторы или антиоксиданты (такие как аскорбиновая кислота, гентизиновая кислота или пара-аминобензойная кислота). Доза соединения настоящего изобретения может изменяться в зависимости от конкретного вводимого соединения, массы тела пациента и других переменных, что является очевидным для опытного лечащего врача. Обычно величина дозы будет находиться в интервале от 0,001 мкг/кг до 10 мкг/кг, предпочтительно 0,01 мкг/кг до 1,0 мкг/кг.
Биологические образцы, которые могут быть использованы при диагностировании заболевания или состояния, связанного с амилоидом, при диагностировании предрасположенности к заболеванию или состоянию, связанному с амилоидом, или для мониторинга минимального остаточного заболевания у пациента, или для предсказания восприимчивости пациента к лечению с помощью соединения или композиции или смеси согласно изобретению, и описанных в изобретении выше, представляют собой, например, жидкости, такие как сыворотка крови, плазма, слюна, секреция желудка, слизь, спинномозговая жидкость, лимфатическая жидкость, и другие подобные жидкости, или ткань или образцы клеток, полученные из организма, такие как нервная, мозговая, сердечная или сосудистая ткань. Для определения присутствия или отсутствия амилоидного белка в пробе, может быть использован любой иммунологический анализ, известный обычным специалистам в этой области (смотрите руководство Harlow and Lane, Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory, New York, 1988, 555 to 612), такой как, например, анализы, в которых применяются косвенные методы определения с использованием для обнаружения вторичных реагентов, твердофазный иммуноферментный анализ и методы иммунопреципитации и агглютинации. Подробное описание этих анализов приводится, например, в патентных документах WO96/13590 to Maertens and Stuyver, Zrein et al. (1998) и WO96/29605.
Для диагностирования in situ, соединение или композиция, или смесь согласно изобретению и описанные в изобретении выше могут быть введены в организм для диагностирования с помощью известных в этой области способов, таких как, например, внутривенная, интраназальная, интраперитонеальная, интрацеребральная, интраартериальный инъекция, для того чтобы могло произойти специфическое связывание соединения согласно изобретению и амилоидного антигена. Комплекс соединение/белок может быть обнаружен с помощью радиоактивной метки, которой помечено соединение.
Иммунологические анализы, применяемые при диагностировании заболевания или состояния, связанного с амилоидом, или при диагностировании предрасположенности к заболеванию или состоянию, связанному с амилоидом, или для мониторинга минимального остаточного заболевания у пациента, или для предсказания восприимчивости пациента к лечению с помощью соединения или композиции или смеси согласно изобретению, и описанных в изобретении выше, обычно основаны на меченых антигенах, антителах, или вторичных реагентах для обнаружения. Эти белки или реагенты могут быть помечены с помощью соединений, обычно известных специалистам в этой области, включающих ферменты, радиоизотопы и флуоресцентные, люминесцентные и хромогенные вещества, включающие окрашенные частицы, такие как коллоидное золото, и латексные микроносители. Из них, нанесение радиоактивной метки может быть использовано почти для всех типов анализа и в большинстве вариантов. Метки, конъюгированные с ферментом, особенно подходят для случая, когда должна быть исключена радиоактивность или когда необходимо быстро получить результаты. Флуорохромы, несмотря на то, что требует для их использования дорогого оборудования, тем не менее, обеспечивают очень чувствительные метод обнаружения. Антитела, применяемые в этих анализах, включают моноклональные антитела, поликлональные антитела и аффинно очищенные поликлональные антитела.
В качестве варианта, соединение изобретение может быть помечено косвенно в результате реакции с меченными веществами, которые имеют аффинность к иммуноглобулину, такими как белок A или G, или вторичные антитела. Антитело может быть конъюгировано со вторым веществом и обнаруживаться с меченым третьим веществом, имеющим аффинность ко второму веществу, конъюгированному с антителом. Например, антитело может быть конъюгировано с биотином, и конъюгат антитело-биотин может быть обнаружен с помощью меченого авидина или стрептавидина. Аналогично, антитело может быть конъюгировано с гаптеном, и конъюгат антитело-гаптен может быть обнаружен с помощью меченого антитела антигаптена.
Обычным специалистам в этой области хорошо известны эти и другие подходящие метки, которые могут быть использованы в соответствии с настоящим изобретением. Связывание этих меток с антителами или их фрагментами может быть осуществлено с помощью стандартных методов, хорошо известных обычным специалистам в этой области. Типичные методы описаны в публикациях Kennedy, J. H., et al., 1976, Clin. Chim. Acta 70: 1-31 и Schurs, A. H. W. M., et al. 1977, Clin. Chim Acta 81: 1-40. Методы пришивки, упомянутые в последней публикации, представляют собой глутаральдегидный метод, периодатный метод, дималеинимидный метод и другие, описание которых приводится в настоящем изобретении путем ссылки на публикацию.
В современных иммунологических анализах применяют метод двойных антител для обнаружения присутствия анализируемого вещества, где антитело метят косвенно в результате реакции со вторым антителом, которое уже было помечено поддающейся обнаружению меткой. Предпочтительно, чтобы второе антитело представляло собой антитело, которое связывает антитела животного, из которого получают моноклональное антитело. Другими словами, если моноклональное тело является мышиным антителом, то тогда меченое второе антитело представляет собой антимышиное антитело. Для моноклонального антитела, используемого в описанном ниже анализе, предпочтительно, чтобы эта метка представляла собой микроноситель с нанесенным на него антителом, в частности, магнитный микроноситель. Для поликлонального антитела, используемого в описанном в настоящем изобретении анализе, предпочтительно, чтобы метка представляла собой поддающуюся обнаружению молекулу, такую как радиоактивное, флуоресцентное или электрохемилюминесцентное вещество.
В объеме настоящего изобретения может быть также использована альтернативная система двойных антител, часто называемая системами быстрого формата, так как они предназначены для быстрых определений присутствия обнаруживаемого вещества. Система требует наличия высокой аффинности между антителом и обнаруживаемым веществом. Согласно одному варианту осуществления настоящего изобретения, присутствие амилоидного антигена определяют с помощью пары антител, каждая из которых специфична к амилоидному антигену. Одно антитело из указанной пары антител называют в описании изобретения "идентифицирующим антителом", а второе антитело из указанной пары антител называют в описании изобретения "иммобилизованным антителом". Моноклональное антитело может быть использовано либо в качестве иммобилизованного антитела, либо в качестве идентифицирующего антитела. Моноклональное антитело может быть также использовано и в качестве иммобилизованного, и в качестве идентифицирующего антитела, вместе в одном анализе. Таким образом, один вариант осуществления настоящего изобретения использует метод двойных антител для обнаружения амилоидного антигена в образце биологической жидкости. В этом методе, анализируемое вещество (амилоидный антиген) помещают между идентифицирующим антителом и иммобилизованным антителом, при этом иммобилизованное антитело необратимо иммобилизировано на твердом носителе. Идентифицирующее антитело должно содержать способную к обнаружению метку, для того чтобы обнаруживать присутствие сандвича антитело-анализируемое вещество и, таким образом, присутствие анализируемого вещества.
Примеры твердофазных веществ включают, но этим не ограничивая, панели микротитратора, пробирки из полистирола, магнитные полимерные или стеклянные гранулы и стекла, применение которых хорошо известны в радиоиммунологическом анализе и иммуноферментном анализе. Методы пришивки антител к твердым фазам также хорошо известны специалистам в этой области. Совсем недавно, в качестве твердых носителей был использован ряд пористых материалов, таких как нейлон, нитроцеллюлоза, ацетатцеллюлоза, стекловолокно и другие пористые полимеры.
Отложение бляшек в ткани и/или жидкости организма (такой как слой ганглиозных клеток сетчатки у животного, в частности, у млекопитающего, но, в особенности, у человека, страдающего глазным заболеванием, связанным с потологическими нарушениями/изменениями в тканях визуальной системы, в частности, связанным с патологическими нарушениями/изменениями ткани визуальной системы, имеющим отношение к бета амилоиду) может быть рассчитано известными в этой области методами, такими как метод, описанный в публикации Ding, J. et al., "Targeting age-related macular degeneration with Alzheimer's disease based immunotherapies: Anti-amyloid-b antibody attenuates pathologies in an age-related macular degeneration mouse model", Vision Research (2007), doi:10.1016/j.visres.2007.07.025.
Соединение согласно настоящему изобретению может также быть включено в диагностический набор для обнаружения амилоидного белка. Набор обычно включает контейнер, содержащий одно или более соединений согласно настоящему изобретению и инструкции по использованию соединения с целью связывания амилоидного белка с образованием комплекса соединение/белок и обнаружения образования комплекса соединение/белок, в соответствии с которыми присутствие или отсутствие комплекса соединение/белок взаимосвязано с присутствием или отсутствием амилоидного белка.
Термин "набор" относится в целом к любому известному в этой области диагностическому набору. Более конкретно, последний термин относится к диагностическому набору, описанному в публикации Zrein et al. (1998).
Ингибирование агрегации Αβ1-42 с помощью соединений настоящего изобретения может быть количественно определено при использовании любого известного в этой области подходящего метода анализа. Описан стандартный in vitro анализ для количественного определения ингибирования агрегации.
Синтез соединений изобретения, ингибирующих агрегацию Αβ1-42, и анализ их биологической активности описан в следующих примерах, которые ни в коем случае не следует рассматривать в качестве ограничений изобретения.
Примеры
Все реагенты и растворители приобретались у коммерческих фирм и использовались без дополнительной очистки. Протонные (1H) спектры ядерного магнитного резонанса регистрировали на ЯМР спектрометре при 400 МГц в дейтерированных растворителях. Масс-спектры (MS) регистрировали на спектрометре Finnigan MAT TSQ 7000. Хроматографию проводили с использованием силикагеля (Fluka: Silica gel 60, 0,063-0,2 мм) и подходящих растворителей, указанных в конкретных примерах. Флэш-очистку проводили на системе очистки с помощью флэш-хроматографии Biotage Isolera One с использованием картриджей HP-Sil SNAP (фирмы Biotage), и градиент состава растворителя указывали в конкретных примерах. Тонкослойную хроматографию (ТСХ) проводили на пластинках с силикагелем, используя УФ-детектирование. Препаративную тонкослойную хроматографию (Prep-ТСХ) осуществляли на пластинках со слоем толщиной 0,5 мм или 1 мм силикагеля (Analtech: Uniplate, F254), и растворители указывали в конкретных примерах.
Препаративный пример 1

Стадия А
Выпускаемый промышленностью 7-азаиндол (1,98 г, 16,8 ммоль) растворяли в смеси 1,2-диметоксиэтана и н-гептана (30 мл, 1:2), и смесь помещали в баню с холодной водой. Затем добавляли порциями при перемешивании м-хлорпероксибензойную кислоту (5,1 г, 19,5 ммоль, ~77%). После добавления половины количества м-хлорпероксибензойной кислоты образовывался осадок. После того, как добавление было закончено, смесь перемешивали при комнатной температуре в течение ночи. Осадок отфильтровывали, промывали с помощью смеси 1,2-диметоксиэтан/н-гептан (10 мл, 1:2) и сушили на воздухе с получением соли м-хлорпероксибензойной кислоты названного соединения в виде белого твердого вещества (4,41 г, 91%). Соль суспендировали в воде (45 мл), и добавляли водный раствор карбоната калия до тех пор, пока величина pH не достигала ~9. Растворители удаляли, и остаток очищали с помощью хроматографии на силикагеле, используя смесь дихлорметан/метанол (9/1) в качестве подвижной фазы, с получением названного соединения в виде серовато-белого твердого вещества (2 г, 91%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=6,60 (д, 1H), 7,07 (дд, 1H), 7,45 (д, 1H), 7,64 (д, 1H), 8,12 (д, 1H).
Стадия В
Названное соединение стадии А выше (1 г, 7,46 ммоль) растворяли в толуоле (150 мл). К этому раствору добавляли по каплям одновременно раствор гексаметилдисилазана (1,56 мл, 7,46 ммоль) в толуоле (75 мл) и раствор бензоилбромида (2,24 мл, 18,64 ммоль) в толуоле (75 мл). После того, как одновременное добавление было законченно, смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь промывали насыщенным раствором бикарбоната натрия (30 мл), солевым раствором (30 мл), и органическую фазу отделяли. Органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (10/90) с получением названного соединения в виде белого твердого вещества (1,48 г, 65%), которое непосредственно использовали на следующей стадии.
Стадия С
Названное соединение стадии В выше (1,48 г, 4,9 ммоль) растворяли в метаноле (90 мл), и добавляли 1M раствор гидроксида натрия. Смесь перемешивали при комнатной температуре в течение ночи и фильтровали для удаления нерастворимого материала. Фильтрат испаряли, и остаток суспендировали в дихлорметане (100 мл). Смесь подвергали воздействию ультразвука и затем перемешивали при комнатной температуре в течение 30 минут. Смесь фильтровали, и фильтрат испаряли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь дихлорметан/метанол (9/1) в качестве подвижной фазы, с получением названного соединения в виде белого твердого вещества (0,59 г, 60%).
1H-ЯМР (400 МГц, CDCl3): δ=6,53-6,56 (м, 1H), 7,26 (д, 1H), 7,39-7,42 (м, 1H), 7,84 (д, 1H), 10,5 (шир.с, 1H).
Стадия D
Названное соединение стадии С выше (0,59 г, 2,99 ммоль) растворяли в тетрагидрофуране (10 мл), и раствор охлаждали до 0°C. При 0°C добавляли порциями гидрид натрия (0,08 г, 3,3 ммоль). После того, как добавление было закончено, смесь перемешивали при комнатной температуре в течение 15 минут. Затем добавляли триизопропилсилилхлорид (0,4 мл, 3 ммоль), и смесь нагревали при ~85°C на песочной бане в течение 3 часов. Смесь разбавляли этилацетатом (50 мл) и солевым раствором (15 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (10/90), с получением названного соединения в виде бесцветного масла (0,53 г, 50%).
1H-ЯМР (400 МГц, CDCl3): δ=1,10-1,12 (м, 18H), 1,73-1,82 (м, 3H), 6,51 (д, 1H), 7,17 (д, 1H), 7,23 (д, 1H), 7,70 (д, 1H).
Стадия Е
Названное соединение стадии получения D выше (0,045 г, 0,127 ммоль) и выпускаемый промышленностью 2-(2-аминоэтил)пиридин (0,015 г, 0,127 ммоль) растворяли в толуоле (2 мл) и обрабатывали с помощью 2,2-бис-(дифенилфосфино)-1,1-нафталина (0,016 г, 0,025 ммоль) и трет-бутилата натрия (0,031 г, 0,33 ммоль). Затем реакционную смесь дегазировали путем барботирования аргона через реакционную смесь, затем добавляли хлороформеный комплекс трис(дибензилиденацетон)дипалладия (0,011 г, 0,0126 ммоль). Реакционный сосуд герметизировали, и смесь нагревали при ~115°C на песочной бане в течение 45 минут. Реакционную смесь разбавляли этилацетатом (20 мл), насыщенным раствором бикарбоната натрия (5 мл) и солевым раствором (5 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (20/80) для элюирования менее полярных побочных продуктов, затем смесь этилацетат/н-гептан (40/60), с получением названного соединения в виде бледно-оранжевого масла (0,04 г, 80%).
1H-ЯМР (400 МГц, CDCl3): δ=1,10-1,12 (м, 18H), 1,81-1,88 (м, 3H), 3,10 (т, 2H), 3,78 (т, 2H), 4,45-4,67 (шир.с, 1H), 6,20 (д, 1H), 6,37 (д, 1H), 6,94 (д, 1H), 7,10-7,14 (м, 2H), 7,56 (д, 1H), 7,58 (дт, 1H), 8,56 (д, 1H).
Препаративный пример 2

Стадия А
Названное соединение препаративного примера 1 стадии D (0,045 г, 0,127 ммоль) и выпускаемый промышленностью 2-(аминометил)пиридин (0,015 г, 0,127 ммоль) растворяли в толуоле (2 мл) и обрабатывали с помощью 2,2-бис-(дифенилфосфино)-1,1-нафталина (0,016 г, 0,025 ммоль) и трет-бутилата натрия (0,031 г, 0,33 ммоль). Затем реакционную смесь дегазировали путем барботирования аргона через реакционную смесь, затем добавляли хлороформенный комплекс трис(дибензилиденацетон)дипалладия (0,011 г, 0,0126 ммоль). Реакционный сосуд герметизировали, и смесь нагревали при ~115°C на песочной бане в течение 45 минут. Реакционную смесь разбавляли этилацетатом (20 мл), насыщенным раствором бикарбоната натрия (5 мл) и солевым раствором (5 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (20/80) для элюирования менее полярных побочных продуктов, затем смесь этилацетат/н-гептан (40/60), с получением названного соединения в виде бледно-оранжевого масла (0,044 г, 90%).
1H-ЯМР (400 МГц, CDCl3): δ=1,10-1,12 (м, 18H), 1,62-1,76 (м, 3H), 4,71 (с, 2H), 5,02-5,08 (шир.с, 1H), 6,33-6,37 (м, 2H), 6,92 (д, 1H), 7,10-7,14 (м, 1H), 7,30 (д, 1H), 7,56 (дт, 1H), 7,61 (д, 1H), 8,53 (д, 1H).
Препаративный пример 3

Стадия А
К раствору выпускаемого промышленностью 7-азаиндола (5 г, 42,3 ммоль) в диэтиловом эфире (350 мл) добавляли порциями м-хлорпербензойную кислоту (11 г, 63,4 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Осажденный продукт отфильтровывали и промывали диэтиловым эфиром (50 мл). Твердое вещество собирали и растворяли в смеси вода/ацетон (50 мл/10 мл) при нагревании. Смесь охлаждали до 5°C, и выкристаллизовывающийся продукт фильтровали и сушили на воздухе с получением названного соединения (11,7 г, 96%).
1H-ЯМР (400 МГц, CDCl3): δ=6,59 (д, 1H), 7,07 (дд, 1H), 7,46 (д, 1H), 7,66 (д, 1H), 8,14 (д, 1H), 12,4 (с, 1H).
Стадия В
К суспензии названного соединения стадии А выше (2 г, 6,92 ммоль) в сухом ацетонитриле (15 мл) добавляли диметилсульфат (0,885 г, 6,92 ммоль). Реакционную смесь нагревали при 70°C в течение 8 часов. Затем прозрачный раствор охлаждали до комнатной температуры. Раствор распределяли в три герметизированных пробирки и охлаждали до 0°C в атмосфере аргона. Затем в каждую пробирку добавляли 7M раствор аммиака в метаноле (5 мл). Герметизированные пробирки нагревали при 50-60°C в течение 48 часов. Растворитель удаляли, и остаток растворяли в этилацетате (200 мл), и органическую фазу промывали разбавленным раствором Na2CO3, водой и солевым раствором. Органическую фазу сушили над Na2SO4. Растворитель испаряли, и неочищенный продукт очищали с помощью хроматографии на диоксиде кремния, используя этилацетат, с получением названного соединения (0,5 г, 54%).
1H-ЯМР (400 МГц, CDCl3): δ=4,33 (м, 2H), 6,35 (дд, 1H), 6,38 (д, 1H), 6,99 (дд, 1H), 7,71 (д, 1H).
Препаративный пример 4

Стадия А
К раствору выпускаемого промышленностью 3-циано-7-азаиндола (2 г, 13,9 ммоль) в тетрагидрофуране (150 мл) добавляли м-хлорпербензойную кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Осадок отфильтровывали и сушили с получением названного соединения (1,89 г, 86%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,26 (д, 1H), 7,74 (д, 1H), 8,30 (д, 1H), 8,40 (с, 1H).
Стадия В
К суспензии названного соединения стадии А выше (1,89 г, 11,8 ммоль) в сухом ацетонитриле (15 мл) добавляли диметилсульфат (1,5 г, 11,8 ммоль), и реакционную смесь нагревали при 80°C в течение ночи. Прозрачный раствор охлаждали до комнатной температуры и переносили в три пробирки объемом 5 мл. Затем в каждую пробирку добавляли 7M раствор аммиака в метаноле (5 мл). Герметизированные пробирки нагревали при 70°C в течение 48 часов. Реакционную смесь концентрировали при пониженном давлении, и остаток выкристаллизовывали из этилацетата и н-гептана с получением названного соединения (0,9 г, 48%).
1H-ЯМР (400 МГц, CDCl3): δ=3,33 (шир.с, 2H), 6,00 (с, 1H), 6,42 (д, 1H), 7,64 (д, 1H), 7,83 (с, 1H).
Препаративный пример 5

Стадия А
К раствору выпускаемого промышленностью 6-броминдола (0,5 г, 2,55 ммоль) в тетрагидрофуране (20 мл) добавляли порциями гидрид натрия (0,095 г, 3,22 ммоль) при комнатной температуре. Суспензию перемешивали при комнатной температуре в течение 10 минут, и медленно добавляли триизопропилсилилхлорид (0,489 г, 2,55 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию останавливали с помощью воды, и растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (150 мл) и промывали водой, солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением остатка, который затем очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан 25/75) с получением названного соединения (0,890 г, 99%).
1H-ЯМР (400 МГц, CDCl3): δ=1,17 (д, 18H), 1,68-1,71 (м, 3H), 6,61 (д, 1H), 7,22-7,24 (м, 2H), 7,51 (д, 1H), 7,65 (с, 1H).
Препаративный пример 6
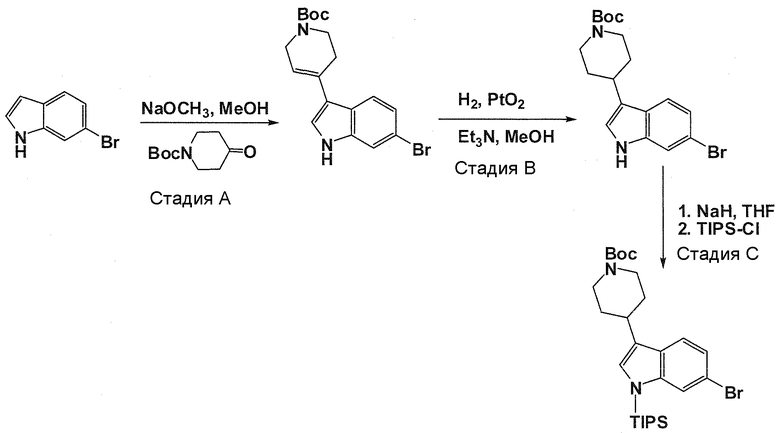
Стадия А
К раствору выпускаемого промышленностью 6-броминдола (2,5 г, 12,7 ммоль) в метаноле (15 мл) добавляли 25% раствор метоксида натрия в метаноле (2 мл), и реакционную смесь нагревали при 80°C в течение 2 дней. Затем реакционную смесь концентрировали до 1/3 ее начального объема и выливали в ледяную воду (100 мл) и экстрагировали этилацетатом (3×100 мл). Органическую фазу промывали водой, солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который затем очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан (20/80)), с получением названного соединения (3,5 г, 72%).
1H-ЯМР (400 МГц, CDCl3): δ=1,50 (с, 9H), 2,56 (м, 2H), 3,67 (т, 2H), 4,13 (д, 2H), 6,11 (с, 1H), 7,15 (д, 1H), 7,24 (д, 1H), 7,53 (д, 1H), 7,73 (д, 1H), 8,16 (с, 1H).
Стадия В
К раствору названного соединения стадии А (1,5 г, 3,97 ммоль) в метаноле (25 мл) добавляли триэтиламин (0,8 г, 7,94 ммоль), и смесь дегазировали. Затем, к реакционной смеси добавляли оксид платины(IV) (0,18 г, 0,79 ммоль), затем вакуумировали и заполняли газообразным водородом. Процедуру повторяли 2-3 раза, и реакционную смесь выдерживали в атмосфере водорода в течение ночи. Затем реакционную смесь фильтровали через слой целита. Фильтрат концентрировали и растворяли в этилацетате (200 мл), и органическую фазу промывали водой, солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который затем очищали на колонке с силикагелем, с получением названного соединения (0,95 г, 63%).
1H-ЯМР (400 МГц, CDCl3): δ=1,48 (с, 9H), 1,64 (м, 2H), 1,99-2,02 (м, 2H), 2,86-2,91 (м, 3H), 4,11-4,22 (м, 2H), 6,93 (д, 1H), 7,21 (дд, 1H), 7,48 (д, 1H), 7,51 (д, 1H), 8,09 (с, 1H).
Стадия С
К раствору названного соединения стадии В выше (0,7 г, 1,84 ммоль) в тетрагидрофуране (10 мл) добавляли гидрид натрия (0,088 г, 3,68 ммоль), и суспензию перемешивали в течение 10 минут. Затем добавляли триизопропилсилилхлорид (0,353 г, 1,84 ммоль). Реакционную смесь перемешивали в течение 30 минут. Реакционную смесь выливали в этилацетат (200 мл) и промывали водой, солевым раствором и сушили над Na2SO4. Растворители удаляли при пониженном давлении с получением неочищенного продукта, который затем очищали на колонке с силикагелем, используя смесь этилацетат/н-гептан (от 5/95 до 50/50), с получением названного соединения (0,9 г, 91%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,48 (с, 9H), 1,62-1,66 (м, 5H), 1,99-2,04 (м, 2H), 2,88-2,92 (м, 3H), 4,21-4,23 (м, 2H), 6,92 (с, 1H), 7,18 (дд, 1H), 7,45 (д, 1H), 7,58 (д, 1H).
Препаративный пример 7

Стадия А
К раствору выпускаемого промышленностью 6-бром-4-азаиндола (3 г, 15,3 ммоль) и 1-Boc-4-пиперидона (3,8 г, 19 ммоль) в метаноле (30 мл) добавляли 25% раствор метоксида натрия в метаноле (4 мл, 18,5 ммоль). Реакционную смесь затем перемешивали при 80°C в течение 3 часов. На данном этапе реакционную смесь охлаждали до комнатной температуры, выливали в ледяную воду (20 мл) и экстрагировали этилацетатом (350 мл). Органическую фазу промывали водой, и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, затем очищали на колонке с силикагелем с получением названного соединения (3 г, 52%).
1H-ЯМР (400 МГц, CDCl3): δ=1,60 (с, 9H), 2,55 (т, 1H), 2,66 (м, 2H), 3,81 (м, 2H), 4,27 (д, 2H), 7,22 (с, 1H), 7,44 (д, 1H), 7,91 (д, 1H), 8,63 (д, 1H), 8,97 (с, 1H).
Стадия В
К раствору названного соединения стадии А выше (0,45 г, 1,19 ммоль) в метаноле (15 мл) добавляли триэтиламин (54 мг, 0,53 ммоль), и смесь дегазировали. После добавления оксида платины(IV) (0,062 г, 0,185 ммоль), реакционную смесь вакуумировали и насыщали газообразным водородом (повторяли еще два раза). Реакционную смесь перемешивали в атмосфере водорода в течение 16 часов. Реакционную смесь фильтровали через целит, и фильтрат концентрировали. Остаток затем очищали на колонке с силикагелем, используя смесь этилацетат/н-гептан (10/90), с получением названного соединения (0,185 г, 42%).
1H-ЯМР (400 МГц, CDCl3): δ=1,50 (с, 9H), 1,69 (м, 2H), 2,15 (м, 2H), 2,97 (м, 2H), 3,23 (м, 1H), 4,22 (м, 2H), 7,18 (д, 1H), 7,82 (д, 1H), 8,26 (шир.с, 1H), 8,51 (д, 1H).
Стадия С
К раствору названного соединения стадии В выше (0,110 г, 0,28 ммоль) в тетрагидрофуране (5 мл) добавляли гидрид натрия (0,014 г, 0,56 ммоль). Суспензию перемешивали при комнатной температуре в течение 10 минут. После добавления триизопропилсилилхлорида (0,055 г, 0,28 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 30 минут. На этой стадии реакцию останавливали с помощью воды, выливали в этилацетат (150 мл), промывали водой и солевым раствором. Органическую фазу отделяли, сушили над Na2SO4, и растворитель испаряли при пониженном давлении. Неочищенный продукт очищали на колонке с силикагелем с получением названного соединения (0,105 г, 70%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,47 (с, 9H), 1,63-1,64 (м, 5H), 2,04-2,12 (м, 2H), 2,90-2,95 (м, 2H), 3,16-3,20 (м, 1H), 4,20-4,21 (м, 2H), 7,15 (с, 1H), 7,82 (д, 1H), 8,45 (д, 1H).
Препаративный пример 8

Стадия А
К раствору выпускаемого промышленностью 5-бром-7-азаиндола (3 г, 15,2 ммоль) и 1-Boc-4-пиперидона (4,2 г, 21,1 ммоль) в метаноле (25 мл) добавляли 25% раствор метоксида натрия в метаноле (4 мл, 18,5 ммоль). Затем реакционную смесь перемешивали при 80°C в течение 2 дней. Реакционную смесь выливали в этилацетат (350 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который затем выкристаллизовывали из смеси этилацетат/н-гептан, с получением названного соединения (4,2 г, 73%).
1H-ЯМР (400 МГц, CDCl3): δ=1,52 (с, 9H), 2,55 (с, 2H), 3,70 (т, 2H), 4,16 (м, 2H), 6,11 (с, 1H), 7,32 (с, 1H), 8,32 (д, 1H), 8,38 (д, 1H), 9,40 (шир.с, 1H).
Стадия В
К перемешиваемому раствору названного соединения стадии А выше (3,2 г, 8,4 ммоль) в тетрагидрофуране добавляли гидрид натрия (0,3 г, 12,6 ммоль), и суспензию перемешивали при комнатной температуре в течение 10 минут. Затем медленно добавляли триизопропилсилилхлорид (1,62 г, 8,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. На этой стадии реакцию останавливали с помощью воды и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4. Удаление растворителя давало неочищенный продукт, который очищали на колонке с силикагелем, с получением названного соединения (3,3 г, 75%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,52 (с, 9H), 1,83 (м, 2H), 2,56 (с, 2H), 3,51 (с, 2H), 3,71 (м, 2H), 4,16 (с, 2H), 7,04 (с, 1H), 7,26 (с,1H), 8,22 (с, 1H), 8,30 (дд, 1H).
Стадия С
К раствору названного соединения стадии В выше (1,63 г, 3,0 ммоль) в метаноле (20 мл) добавляли триэтиламин (0,6 г, 6,0 ммоль). Смесь дегазировали, и добавляли оксид платины(IV) (0,14 г, 0,6 ммоль). Затем колбу дважды вакуумировали и насыщали газообразным водородом. Реакционную смесь перемешивали в атмосфере водорода в течение 16 часов. Гетерогенную реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем, с получением названного соединения (0,240 г, 50%).
1H-ЯМР (400 МГц, CDCl3): δ=1,11 (д, 18H), 1,49 (с, 9H), 1,76-1,86 (м, 7H), 2,17-2,22 (м, 2H), 3,23 (м, 2H), 3,88-3,90 (м, 2H), 7,07 (с, 1H), 8,21 (д, 1H), 8,27 (д, 1H).
Препаративный пример 9

Стадия А
К раствору выпускаемого промышленностью 5-броминдола (3,5 г, 17,9 ммоль) в метаноле (50 мл) добавляли 1-Boc-4-пиперидон (5,1 г, 25,6 ммоль) и 25% раствор метоксида натрия в метаноле (8 мл, 37 ммоль), и реакционную смесь нагревали при 80°C в течение 2 дней. Реакционную смесь фильтровали. Твердое вещество промывали дважды этилацетатом и сушили под вакуумом с получением названного соединения (3 г). Фильтрат разбавляли этилацетатом (250 мл) и промывали водой, солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем, с получением дополнительного количества названного соединения (2,9 г). Суммарный выход составил 5,9 г, 86%.
1H-ЯМР (400 МГц, CDCl3): δ=1,52 (с, 9H), 2,55 (м, 2H), 3,69 (т, 2H), 4,16 (с, 2H), 6,12 (с, 1H), 7,19 (с, 1H), 7,26-7,33 (м, 2H), 8,01 (с, 1H), 8,29 (шир.с, 1H).
Стадия В
К дегазированному раствору названного соединения стадии А выше (1,8 г, 4,7 ммоль) в этилацетате (50 мл) добавляли оксид платины(IV) (0,2 г, 0,88 ммоль). Реакционную смесь вакуумировали и насыщали газообразным водородом. Процедуру повторяли дважды, и реакционную смесь выдерживали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную смесь фильтровали через целит, и фильтрат концентрировали. Неочищенный продукт очищали на колонке с силикагелем, используя смесь этилацетат/н-гептан (от 10/90 до 50/50), с получением названного соединения (0,75 г, 42%).
1H-ЯМР (400 МГц, CDCl3): δ=1,52 (с, 9H), 2,04 (м, 2H), 2,91 (м, 3H), 3,75 (м, 2H), 4,23-4,29 (м, 2H), 6,98 (с, 1H), 7,28 (м, 2H), 7,76 (с, 1H), 8,11 (с, 1H).
Стадия С
К раствору названного соединения стадии В выше (0,6 г, 1,58 ммоль) в тетрагидрофуране (20 мл) добавляли гидрид натрия (0,056 г, 2,37 ммоль), и суспензию перемешивали при комнатной температуре в течение 10 минут. Затем добавляли триизопропилсилилхлорид (0,34 г, 1,58 ммоль), и смесь перемешивали в течение 1 часа при комнатной температуре. Реакцию останавливали с помощью воды и концентрировали. Остаток растворяли в этилацетате (100 мл), промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем, используя смесь этилацетат/н-гептан (10/90), с получением названного соединения (0,6 г, 70%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,51 (с, 9H), 1,65-1,66 (м, 5H), 2,01-2,05 (м, 2H), 2,89-2,94 (м, 3H), 4,25-4,26 (м, 2H), 6,98 (с, 1H), 7,23 (дд, 1H), 7,34 (с, 1H), 7,36 (с, 1H), 7,72 (с, 1H).
Препаративный пример 10

Стадия А
К раствору названного соединения препаративного примера 9 стадии В (0,625 г, 1,64 ммоль) в THF (10 мл) добавляли NaH (0,059 г, 2,4 ммоль). Суспензию перемешивали в течение 5 минут. Затем медленно добавляли метилйодид (0,346 г, 2,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь выливали в этилацетат (150 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4. Растворитель концентрировали при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем (20-50%; от EtOAc до гептана) с получением названного соединения (0,485 г, 75%).
1H-ЯМР (400 МГц, CDCl3): δ=1,51 (с, 9H), 1,61-1,65 (м, 2H), 1,99-2,02 (м, 2H), 2,87-2,95 (м, 3H), 3,74 (с, 3H), 4,24-4,25 (м, 2H), 6,82 (с, 1H), 7,16 (д, 1H), 7,32 (д, 1H), 7,74 (с, 1H).
Препаративный пример 11

К раствору названного соединения (500 мг, 0,131 ммоль) в THF (10 мл) добавляли NaH (63 мг, 2,6 ммоль), и суспензию перемешивали в течение 5 минут. Добавляли метилйодид (185 мг, 0,131 ммоль), и реакционную смесь перемешивали в течение 30 минут. Затем реакцию останавливали с помощью воды и экстрагировали этилацетатом (50 мл ×3). Органическую фазу промывали солевым раствором и сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали на колонке с силикагелем (EtOAc:гептан; 20:30) с получением названного соединения (485 мг, 94%).
1H-ЯМР (400 МГц, CDCl3): δ=7,50-7,46 (м, 2H), 7,21 (дд, J=8,4, 1,7 Гц, 1H), 6,80 (с, 1H), 4,25 (с, 2H), 3,73 (с, 3H), 2,99-2,87 (м, 3H), 2,03-1,99 (м, 2H), 1,67-1,63 (м, 2H), 1,51 (с, 9H).
Препаративный пример 12

Стадия А
К раствору 5-броминдола (5 г, 25,3 ммоль) и 1-метилпиперидин-4-она (4,3 г, 38,2 ммоль) в метаноле (50 мл) добавляли (28%) NaOMe в метаноле (10 мл), и реакционную смесь нагревали при 90°C в течение 2 дней. Продукт осаждали, фильтровали и сушили под вакуумом с получением названного соединения (6,3 г, 85%).
1H-ЯМР (400 МГц, CDCl3): 2,28 (с, 3H), 2,55 (м, 2H), 3,04 (д, 2H), 3,34 (м, 2H), 6,07 (с, 1H), 7,21 (д, 1H), 7,35 (д, 1H), 7,45 (с, 1H), 7,92 (с, 1H).
Стадия В
К раствору названного соединения стадии А выше (0,5 г, 1,72 ммоль) в THF (100 мл) добавляли порциями гидрид натрия (0,1 г, 4,28 ммоль), и суспензию перемешивали при комнатной температуре в течение 10 минут. Затем медленно вводили триизопропилсилилхлорид (0,33 г, 1,72 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Реакцию останавливали с помощью воды, и растворитель удаляли. Остаток растворяли в этилацетате (150 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворители удаляли, и остаток очищали на колонке с силикагелем (метанол к EtOAc от 10% до 20%) с получением названного соединения (0,498 г, 65%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,67-1,69 (м, 3H), 2,45 (с, 3H), 2,64 (шир.с, 2H), 2,72 (т, 2H), 3,19 (с, 2H), 6,12 (с, 1H), 7,18 (с, 1H), 7,23 (д, 1H), 7,34 (д, 1H), 7,98 (с, 1H).
Стадия С
Раствор названного соединения стадии В выше (0,490 г, 1,09 ммоль) в этилацетате (50 мл) дегазировали, и добавляли оксид платины(IV) (0,150 г, 0,67 ммоль). Реакционную смесь вакуумировали и насыщали газообразным водородом. Процедуру повторяли 2-3 раза, и реакционную смесь перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали через слой целита. Фильтрат концентрировали и растворяли в этилацетате (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который затем очищали на колонке с силикагелем (MeOH к этилацетату от 5% до 12%) с получением названного соединения (0,15 г, 30%).
1H-ЯМР (400 МГц, CDCl3): δ=1,12 (д, 18H), 1,62-1,69 (м, 3H), 2,0-2,12 (м, 4H), 2,47 (т, 2H), 2,56 (с, 3H), 2,81-2,89 (м, 1H), 3,24 (д, 2H), 7,03 (с, 1H), 7,23 (д, 1H), 7,34 (д, 1H), 7,70 (с, 1H).
Препаративный пример 13

Стадия А
К раствору 6-броминдола (3 г, 15,3 ммоль) и 1-метилпиперидин-4-она (3,4 г, 30,6 ммоль) в метаноле (50 мл) добавляли (28%) NaOMe в метаноле (10 мл), и реакционную смесь нагревали при 90°C в течение 2 дней. Осажденный продукт отфильтровывали и сушили под вакуумом с получением названного соединения (4,1 г, 91%).
1H-ЯМР (400 МГц, CDCl3): δ=2,28 (с, 3H), 2,51-2,56 (м, 4H), 3,03 (с, 2H), 6,1 (с, 1H), 7,14 (д, 1H), 7,41 (с, 1H), 7,56 (с, 1H), 7,74 (д, 1H).
Стадия В
К раствору названного соединения стадии А выше (2 г, 6,8 ммоль) в смеси THF/диоксан (100 мл/10 мл) добавляли порциями гидрид натрия (0,25 г, 10,3 ммоль), и суспензию перемешивали при комнатной температуре в течение 10 минут. Затем медленно вводили триизопропилсилилхлорид (1,3 г, 6,8 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Реакцию останавливали с помощью воды, и растворитель удаляли, и остаток растворяли в этилацетате (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворители удаляли, и остаток очищали на колонке с силикагелем (метанол к EtOAc от 5% до 20%) с получением названного соединения (2,9 г, 95%).
1H-ЯМР (400 МГц, CDCl3): δ=1,12 (д, 18H), 1,61-1,69 (м, 3H), 2,41 (с, 3H), 2,61 (шир.с, 2H), 2,69 (т, 2H), 3,15 (д, 2H), 6,11 (с, 1H), 7,13 (с, 1H), 7,21 (д, 1H), 7,58 (с, 1H), 7,69 (д, 1H).
Стадия С
Раствор названного соединения стадии В выше (2,8 г, 6,2 ммоль) в этилацетате (50 мл) дегазировали, и добавляли оксид платины(IV) (0,18 г, 0,79 ммоль). Затем реакционную смесь вакуумировали и насыщали газообразным водородом. Процедуру повторяли 2-3 раза, и реакционную смесь перемешивали в атмосфере водорода в течение ночи. Затем реакционную смесь фильтровали через слой целита. Фильтрат концентрировали, и остаток растворяли в этилацетате (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением неочищенного продукта, который затем очищали на колонке с силикагелем (метанол к этилацетату от 5% до 12%) с получением названного соединения (2,3 г, 85%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,61-1,69 (м, 3H), 1,84-1,88 (м, 2H), 2,03-2,06 (м, 2H), 2,16 (т, 2H), 2,37 (с, 3H), 2,78 (тт, 1H), 2,99 (д, 2H), 6,97 (с, 1H), 7,19 (д, 1H), 7,47 (д, 1H), 7,59 (с, 1H).
Препаративный пример 14
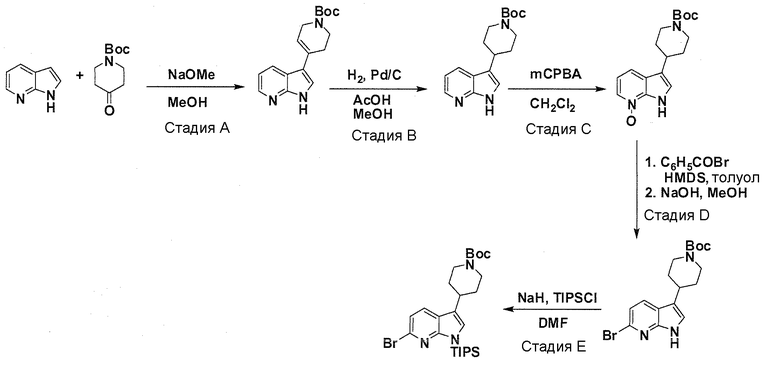
Стадия А
К раствору выпускаемого промышленностью 7-азаиндола (2 г, 16,8 ммоль) в метаноле (20 мл) добавляли 1-(трет-бутоксикарбонил)-4-пиперидон (6,75 г, 34 ммоль) и 25% метанольный раствор метоксида натрия (21,6 мл, 100 ммоль), и раствор кипятили с обратным холодильником в течение 2,5 часов. Реакционный раствор выливали в 50 мл ледяной воды, экстрагировали этилацетатом, и органический слой промывали водой, сушили и концентрировали. Полученный остаток выкристаллизовывали из смеси н-гексан/этилацетат с получением соединения (3,4 г, 67%).
1H-ЯМР (400 МГц, CDCl3): δ=1,46 (с, 9H), 2,56 (шир.с, 2H), 3,68 (т, 2H), 4,13 (д, 2H), 6,13 (с, 1H), 7,12 (дд, 1H), 7,34 (с, 1H), 8,20 (дд, 1H), 8,31 (дд, 1H), 11,28 (шир.с, 1H).
MS (ESI); m/z=299,94 (MH+).
Стадия В
К раствору названного соединения стадии А выше (1 г, 3,34 ммоль) в 65 мл метанола добавляли 1 мл уксусной кислоты, затем 500 мг 10% Pd/C. Смесь перемешивали в атмосфере водорода в течение 48 часов. Катализатор отфильтровывали через целит, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в этилацетате и промывали водным раствором NaHCO3. Органическую фазу испаряли с получением желтоватого соединения, которое очищали с помощью флэш-хроматографической системы Biotage (этилацетат/н-гептан: от 20 до 66%) с получением желтоватого соединения (0,92 г, 91%).
1H-ЯМР (400 МГц, CDCl3): δ=1,47 (с, 9H), 1,63-1,75 (м, 3H), 1,99-2,04 (м, 2H), 2,86-2,99 (м, 3H), 4,24 (шир.с, 2H), 7,07 (м, 2H), 7,95 (д, 1H), 8,32 (шир.с, 1H), 9,57 (шир.с, 1H).
MS (ESI); m/z=301,97 (MH+).
Стадия С
м-Хлорпербензойную кислоту (0,568 г, 1,69 ммоль) добавляли к холодному раствору (0°C) названного соединения стадии В выше (0,51 г, 1,69 ммоль) в 20 мл дихлорметана. Реакционную смесь подогревали до комнатной температуры и затем перемешивали в течение 12 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и затем концентрировали. Полученный остаток очищали с помощью флэш-хроматографической системы Biotage (метанол/дихлорметан: от 3 до 10%) с получением белого соединения (0,4 г, 74%).
1H-ЯМР (400 МГц, CDCl3): δ=1,47 (с, 9H), 1,61-1,65 (м, 2H), 1,96 (д, 2H), 2,86-2,90 (м, 3H), 4,21 (шир.с, 2H), 7,02 (т, 1H), 7,16 (с, 1H), 7,68 (д, 1H), 8,18 (д, 1H).
MS (ESI); m/z=317,96 (MH+).
Стадия D
К раствору названного соединения стадии С выше (0,38 г, 1,19 ммоль) в 24 мл толуола, добавляли одновременно гексаметилдисилазан (0,25 мл, 1,19 ммоль), растворенный в 12 мл толуола, и бензоилбромид (0,42 мл, 3,59 ммоль) в 14 мл толуола. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Остаток, полученный после испарения растворителя, растворяли в 10 мл метанола и обрабатывали с помощью 3,5 мл 1M раствора гидроксида натрия. После перемешивания в течение 2 часов, добавляли насыщенный водный раствор лимонной кислоты (10 мл). Водную фазу экстрагировали этилацетатом (100 мл). Органическую фазу промывали с помощью бикарбоната натрия и воды и затем сушили. Остаток, полученный после удаления растворителя, очищали с помощью флэш-хроматографической системы Biotage (метанол/дихлорметан: от 1 до 5%) с получением белого соединения (0,185 г, 40% за две стадии).
1H-ЯМР (400 МГц, CDCl3): δ=1,48 (с, 9H), 1,63-1,67 (м, 2H), 1,97 (д, 2H), 2,84-2,90 (м, 3H), 4,22 (д, 2H), 6,96 (с, 1H), 7,14 (д, 1H), 7,69 (д, 1H).
MS (ESI); m/z=381,85 (MH+).
Стадия Е
Названное соединение стадии D выше (0,1 г, 0,263 ммоль) растворяли в Ν,Ν'-диметилформамиде (6 мл), и раствор охлаждали до 0°C. При 0°C добавляли порциями 95% гидрид натрия (0,007 г, 0,289 ммоль). После того, как добавление было закончено, смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли триизопропилсилилхлорид (0,06 мл, 0,289 ммоль), и смесь перемешивали при комнатной температуре в течение 19 часов. Смесь разбавляли водой (50 мл). Водную фазу экстрагировали этилацетатом. Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя метанол/дихлорметан: от 1 до 5%, с получением названного соединения в виде белого твердого вещества (0,1 г, 71%).
1H-ЯМР (400 МГц, CDCl3): δ=1,10-1,11 (м, 18H), 1,66 (с, 9H), 1,67-1,63 (м, 2H), 1,80-1,73 (м, 3H), 1,97 (д, 2H), 2,90-2,84 (м, 3H), 4,22 (д, 2H), 6,96 (с, 1H), 7,14 (д, 1H), 7,69 (д, 1H).
MS (ESI); m/z=537,95 (MH+).
Препаративный пример 15

Стадия А
К раствору названного соединения препаративного примера 14 стадии D (1 г, 2,63 ммоль) в сухом тетрагидрофуране (25 мл) добавляли порциями при 0°C 60% гидрид натрия (0,111 г, 2,89 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли метилйодид (0,491 мл, 7,89 ммоль). После завершения реакции, которое контролировалось с помощью ТСХ на пластинке, раствор концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан от 15% до 40% с получением соединения в виде белого твердого вещества (0,984 г, 95%).
MS (ESI); m/z=394,48/396,48 (MH+).
Стадия В
К раствору названного соединения стадии А выше (0,688 г, 1,745 ммоль) в дихлорметане (50 мл) добавляли 2M хлористый водород в диэтиловом эфире (8,72 мл, 17,45 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали досуха с получением бежевого твердого вещества (0,654 г, 99%).
MS (ESI); m/z=294,60/296,60 (MH+).
Стадия С
К раствору названного соединения стадии В выше (0,654 г, 2,223 ммоль) в MeOH (50 мл) добавляли последовательно триэтиламин (0,781 мл, 5,56 ммоль), раствор формальдегида (0,196 мл, 2,445 ммоль) и триацетоксиборгидрид натрия (0,565 г, 2,67 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Смесь концентрировали досуха, и затем остаток разбавляли водой и этилацетатом. Экстракцию осуществляли с помощью 1M NaOH и солевого раствора. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха с получением ожидаемого соединения в виде желтоватого твердого вещества (0,411 г, 60%).
1H-ЯМР (400 МГц, CDCl3): δ=1,81 (т, J=11,6 Гц, 2H); 1,96 (д, J=11,6 Гц, 2H); 2,08 (т, J=11,2 Гц, 2H); 2,33 (с, 3H); 2,71 (т, J=11,2 Гц, 2H); 2,96 (д, J=9,6 Гц, 2H); 3,80 (с, 3H); 6,88 (с, 1H); 7,08-7,19 (м, 2H); 7,69-7,83 (м, 1H).
MS (ESI); m/z=308,59/310,59 (MH+).
Препаративный пример 16
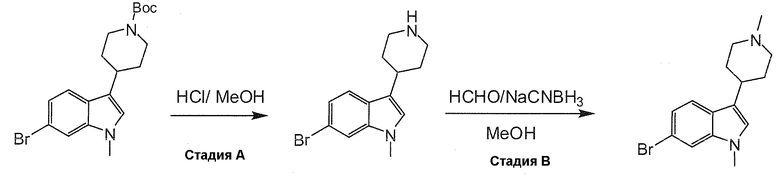
Стадия А
К раствору названного соединения (220 мг, 0,55 ммоль) в MeOH (3 мл) добавляли 3 н. HCl в MeOH (0,5 мл) и перемешивали реакционную смесь в течение ночи. Растворитель удаляли при пониженном давлении с получением названного соединения (180 мг, 98%).
Стадия В
К раствору соединения (220 мг, 0,75 ммоль) в MeOH (5 мл) добавляли NaCNBH3 (200 мг, 3,1 ммоль). Реакционную смесь перемешивали в течение ночи. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом MeOH/DCM (1/99→5/95), с получением названного соединения (92 мг, 30%).
1H-ЯМР (400 МГц, CDCl3): 7,46 (м, 2H), 7,20 (с, 1H), 6,89 (с, 1H), 3,72 (с, 3H), 3,48 (с, 3H), 2,94 (м, 1H), 2,68-2,63 (м, 3H), 2,21-2,04 (м, 5H).
MS (ESI): m/z=307,6 (M+H).
Препаративный пример 17

Стадия А
К раствору названного соединения препаративного примера 14 стадии D (0,500 г, 1,315 ммоль) в дихлорметане (10 мл) добавляли 2M хлористый водород в диэтиловом эфире (3,29 мл, 6,57 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции, суспензию концентрировали досуха с получением соединения в виде бежевого твердого вещества (0,443 г, 95%).
MS (ESI); m/z=280,53/282,52 (MH+).
Стадия В
К раствору названного соединения стадии А выше (0,443 г, 1,255 ммоль) и триэтиламина (0,353 мл, 2,509 ммоль) в метаноле (5 мл) добавляли раствор формальдегида (0,121 мл, 1,506 ммоль), затем триацетоксиборгидрид натрия (30,99 г, 1,882 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Смесь концентрировали досуха, и затем остаток разбавляли водой и этилацетатом. Экстракцию осуществляли с помощью 1M NaOH и солевого раствора. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха с получением ожидаемого соединения в виде белого твердого вещества (0,328 г, 89%).
MS (ESI); m/z=294,59/296,59 (MH+).
Стадия С
К раствору названного соединения стадии В выше (0,328 г, 1,115 ммоль) в сухом тетрагидрофуране (10 мл) добавляли порциями 60% гидрид натрия (0,045 г, 1,171 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли триизопропилсилилхлорид (0,255 мл, 1,204 ммоль). Реакционную смесь затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали досуха, затем остаток очищали флэш-хроматографией в смеси DCM/MeOH от 97:3 до 90:10 с получением ожидаемого соединения в виде белого аморфного твердого вещества (0,184 г, 37%).
1H-ЯМР (400 МГц, CDCl3): δ=1,00-1,23 (с, 18H); 1,69-1,87 (м, 3H); 1,95-2,12 (м, 4H); 2,25-2,40 (м, 2H); 2,47 (с, 3H); 2,70-2,89 (м, 1H); 3,08-3,23 (м, 2H); 6,99 (с, 1H); 7,08-7,17 (м, 1H); 7,67-7,79 (м, 1H).
MS (ESI); m/z=450,63/452,63 (MH+).
Препаративный пример 18

Стадия А
Выпускаемый промышленностью 6-нитроиндол (2,15 г, 13,27 ммоль) растворяли в метаноле (10 мл) и добавляли выпускаемый промышленностью N-Boc-4-пиперидон (3,93 г, 19,8 ммоль). После добавления 25% раствора метоксида натрия в метаноле (8,22 мл, 38 ммоль), смесь нагревали при ~100°C на песочной бане в течение ночи. Смесь разбавляли этилацетатом (150 мл) и промывали насыщенным раствором бикарбоната натрия (40 мл) и солевым раствором (40 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (40/60) для элюирования исходного реагента, затем смесь этилацетат/н-гептан (60/40), с получением названного соединения в виде желтого твердого вещества (2,56 г, 56%).
1H-ЯМР (400 МГц, ДМСО6): δ=1,39 (с, 9H), 2,48-2,51 (м, 2H), 3,54 (т, 2H), 4,00-4,04 (м, 2H), 6,16-6,19 (м, 1H), 7,84-7,90 (м, 2H), 7,98 (д, 1H), 8,31 (с, 1H), 11,8 (шир.с, 1H).
Стадия В
К раствору названного соединения стадии А выше (2,2 г, 6,3 ммоль) в тетрагидрофуране (50 мл) добавляли гидрид натрия (0,18 г, 7,56 ммоль). Черную суспензию перемешивали в течение 10 минут, и затем добавляли триизопропилсилилхлорид (1,24 г, 6,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. На этой стадии реакцию останавливали с помощью воды и концентрировали. Полученный остаток растворяли в EtOAc (300 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель испаряли при пониженном давлении с получением неочищенного продукта, который затем очищали на колонке с силикагелем с получением названного соединения (1,5 г, 46%).
1H-ЯМР (400 МГц, CDCl3): δ=1,17 (д,18H), 1,52 (с,9H), 1,73-1,79 (м, 3H), 2,59 (с, 2H), 3,72 (т, 2H), 4,17 (с, 2H), 6,16 (с, 1H), 7,46 (с, 1H), 7,88 (д, 1H), 8,06 (д, 1H), 8,47 (с, 1H).
MS (ESI) m/z: 500 (MH) 501 (M+2H).
Стадия С
К раствору названного соединения стадии В выше (1,5 г, 3,0 ммоль) в этилацетате (50 мл) добавляли 10% Pd/C. Смесь дегазировали под вакуумом и насыщали водородом. Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали, и растворитель испаряли. Остаток затем очищали с помощью хроматографии на диоксиде кремния, используя градиент этилацетат/н-гептан (20/80→50/50), с получением названного соединения в виде твердого вещества (0,51 г, 36%).
1H-ЯМР (400 МГц, CDCl3): δ=1,16 (д, 18H), 1,50 (с, 9H), 1,65-1,69 (м, 5H), 2,02-2,05 (м, 2H), 2,87-2,93 (м, 3H), 4,22-4,23 (м, 2H), 6,59 (д, 1H), 6,61 (д, 1H), 6,78 (с, 1H), 6,83 (с, 1H), 7,39 (д, 1Н).
Препаративный пример 19
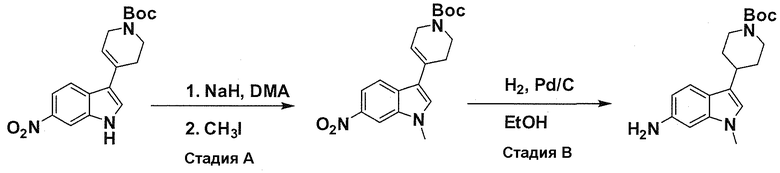
Стадия А
Названное соединение препаративного примера 17 стадии А (0,93 г, 2,71 ммоль) растворяли в N,N'-диметилацетамиде (10 мл), и смесь охлаждали до 0°C. При 0°C добавляли гидрид натрия (0,085 г, 3,45 ммоль), и смесь перемешивали при 0°C в течение 5 минут, и затем в течение 15 минут при комнатной температуре. После добавления метилйодида (0,175 мл, 2,72 ммоль), смесь перемешивали при комнатной температуре в течение 18 часов. Смесь разбавляли этилацетатом (100 мл) и водой (30 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворитель удаляли. Остаток очищали с помощью системы Biotage, используя градиент этилацетат/н-гептан (5/95→40/60), с получением названного соединения в виде желтого твердого вещества (0,87 г, 90%).
1H-ЯМР (400 МГц, CDCb): δ=1,54 (с, 9H), 2,55-2,60 (м, 2H), 3,70 (т, 2H), 3,90 (с, 3H), 4,14,4,17 (м, 2H), 6,15 (с, 1H), 7,30 (с, 1H), 7,90 (д, 1H), 8,04 (дд, 1H), 8,31 (д, 1H).
Стадия В
К раствору названного соединения стадии А выше (0,87 г, 2,44 ммоль) в этаноле (50 мл) добавляли катализатор 10% Pd/C (0,4 г). Смесь дегазировали под вакуумом и насыщали водородом. Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали, и растворитель испаряли. Остаток очищали с помощью системы Biotage, используя градиент этилацетат/н-гептан (5/95→100/0), с получением названного соединения в виде бледно-розовой пены (0,3 г, 38%).
1H-ЯМР (400 МГц, CDCl3): δ=1,52 (с, 9H), 1,58-1,66 (м, 3H), 2,00 (д, 2H), 2,85-2,91 (м, 3H), 3,64 (с, 3H), 4,15-4,27 (шир.с, 2H), 6,56-6,62 (3H), 7,40 (д, 1H).
Препаративный пример 20

Стадия А
Выпускаемый промышленностью 6-нитроиндол (2,15 г, 13,27 ммоль) растворяли в метаноле (15 мл), и добавляли выпускаемый промышленностью N-метил-4-пиперидон (2,19 мл, 19,8 ммоль). После добавления 25% раствора метоксида натрия в метаноле (8,22 мл, 38 ммоль), смесь нагревали при ~100°C на песочной бане в течение 30 часов. Смесь разбавляли водой (100 мл) и перемешивали при комнатной температуре в течение 10 минут. Осадок отфильтровывали, промывали с помощью метанол (25 мл) и сушили на воздухе с получением названного соединения в виде оранжевого твердого вещества (2,47 г, 72%).
1H-ЯМР (400 МГц, ДМСО6): δ=2,25 (с, 3H), 2,52-2,59 (м, 4H), 3,04 (с, 2H), 6,17 (с, 1H), 7,82 (с, 1H), 7,88 (д, 1H), 7,97 (д, 1H), 8,31 (с, 1H), 11,9 (шир.с, 1H).
Стадия В
К раствору названного соединения стадии А выше (1,1 г, 4,32 ммоль) в тетрагидрофуране (25 мл) добавляли гидрид натрия (0,136 г, 5,5 ммоль) при 0°C. Черную суспензию перемешивали в течение 5 минут при 0°C, и в течение 15 минут при комнатной температуре. Затем добавляли триизопропилсилилхлорид (0,58 мл, 4,35 ммоль). Реакционную смесь перемешивали при ~85°C на песочной бане в течение 2 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали насыщенным раствором бикарбоната натрия (25 мл) и солевым раствором (25 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением смеси названного соединения и исходного реагента. Эту смесь непосредственно использовали на следующей стадии.
Стадия С
К раствору смеси стадии В выше в этаноле (30 мл) добавляли катализатор 10% Pd/C (0,4 г). Смесь дегазировали под вакуумом и насыщали водородом. Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали, и растворитель испаряли. Остаток затем очищали с помощью хроматографии на диоксиде кремния, используя смесь дихлорметан/метанол (9/1), с получением смеси названного соединения вместе с соединением, у которого двойная связь не была восстановлена (0,22 г). Эту смесь снова гидрировали в этаноле (15 мл), используя описанный выше катализатор 10% Pd/C (0,11 г). Фильтрование раствора и испарение растворителя давали названное соединение в виде бесцветного стекла (0,2 г, 19% за 2 стадии).
1H-ЯМР (400 МГц, ДМСО6): δ=1,04-1,07 (м, 18H), 1,59-1,69 (м, 5H), 1,88 (д, 1H), 2,13-2,18 (м, 2H), 2,26 (с, 3H), 2,57-2,66 (м, 2H), 2,90 (д, 2H), 4,58-4,67 (шир.с, 2H), 6,38 (д, 1H), 6,69-6,72 (м, 2H), 7,17 (д, 1H).
Препаративный пример 21
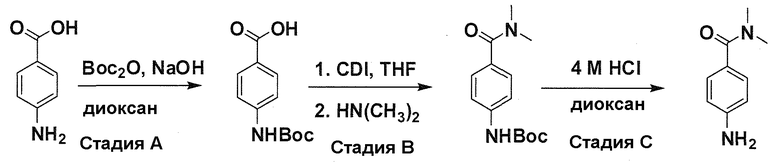
Стадия А
Выпускаемую промышленностью 4-аминобензойную кислоту (5 г, 36 ммоль) растворяли в 1M растворе гидроксида натрия (40 мл, 40 ммоль) и 1,4-диоксана (30 мл). После добавления ди-трет-бутилдикарбоната (7,85 г, 36 ммоль), смесь перемешивали при комнатной температуре в течение выходных дней. Диоксан удаляли под вакуумом, и остаток разбавляли водой (100 мл). Затем добавляли концентрированную хлористоводородную кислоту (~37%) до pH~3. Осадок отфильтровывали, промывали водой (100 мл) и сушили на воздухе с получением названного соединения в виде белого твердого вещества (6,3 г, 72%).
Стадия В
Названное соединение стадии А выше (0,43 г, 1,8 ммоль) суспендировали в тетрагидрофуране (10 мл) и обрабатывали с помощью карбонилдиимидазола (0,37 г, 2,26 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Затем к прозрачному раствору добавляли 2M раствор диметиламина в тетрагидрофуране (2,25 мл, 4,5 ммоль). Перемешивание продолжали в течение ночи, и растворители удаляли. Остаток растворяли в этилацетате (50 мл) и промывали с помощью 10% раствора лимонной кислоты в воде (15 мл) и солевого раствора (15 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением названного соединения в виде бесцветной пены (0,46 г, 96%).
1H-ЯМР (400 МГц, CDCl3): δ=1,54 (с, 9H), 3,00-3,12 (м, 6H), 6,63 (шир.с, 1H), 7,39-7,41 (м, 4H).
Стадия С
Названное соединение стадии В выше (0,46 г, 1,75 ммоль) растворяли в дихлорметане (2,2 мл) и обрабатывали с помощью 4M раствора хлористоводородной кислоты в 1,4-диоксане (2,2 мл, 8,8 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и разбавляли этилацетатом (20 мл). Затем добавляли насыщенный водный раствор карбоната натрия до pH~10. Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя этилацетат, с получением названного соединения в виде серовато-белого твердого вещества (0,16 г, 55%).
1H-ЯМР (400 МГц, CDCl3): δ=3,04 (с, 6H), 3,80 (шир.с, 2H), 6,67 (м, 2H), 7,31 (м, 2H).
Препаративный пример 22

Стадия А
Выпускаемую промышленностью 3-аминобензойную кислоту (5 г, 36 ммоль) растворяли в 1M растворе гидроксида натрия (40 мл, 40 ммоль) и 1,4-диоксана (30 мл). После добавления ди-трет-бутилдикарбоната (7,85 г, 36 ммоль), смесь перемешивали при комнатной температуре в течение выходных дней. Диоксан удаляли под вакуумом, и остаток разбавляли водой (100 мл). Затем добавляли концентрированную хлористоводородную кислоту (~37%) до pH~3. Осадок отфильтровывали, промывали водой (100 мл) и сушили на воздухе с получением названного соединения в виде белого твердого вещества (7,3 г, 84%).
Стадия В
Названное соединение стадии А выше (0,43 г, 1,8 ммоль) суспендировали в тетрагидрофуране (10 мл) и обрабатывали с помощью карбонилдиимидазола (0,37 г, 2,26 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. К прозрачному раствору затем добавляли 2M раствор диметиламина в тетрагидрофуране (2,25 мл, 4,5 ммоль). Перемешивание продолжали в течение ночи, и растворители удаляли. Остаток растворяли в этилацетате (50 мл) и промывали 10% раствором лимонной кислоты в воде (15 мл) и солевым раствором (15 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением названного соединения в виде бесцветной пены (0,36 г, 75%).
1H-ЯМР (400 МГц, CDCl3): δ=1,55 (с, 9H), 3,00 (с, 3H), 3,13 (с, 3H), 6,67 (шир.с, 1H), 7,08 (дт, 1H), 7,32 (т, 1H), 7,40-7,48 (м, 2H).
Стадия С
Названное соединение стадии В выше (0,36 г, 1,37 ммоль) растворяли в дихлорметане (2 мл) и обрабатывали с помощью 4M раствора хлористоводородной кислоты в 1,4-диоксане (2 мл, 8 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и разбавляли этилацетатом (20 мл). Затем добавляли насыщенный водный раствор карбоната натрия до pH~10. Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя этилацетат, с получением названного соединения в виде серовато-белого твердого вещества (0,08 г, 36%).
1H-ЯМР (400 МГц, CDCl3): δ=3,00 (с, 3H), 3,11 (с, 3H), 6,70-6,74 (м, 2H), 6,77 (дт, 1H), 7,18 (т, 1H).
Препаративный пример 23

Стадия А
К суспензии названного соединения препаративного примера 3 стадии А (2 г, 6,92 ммоль) в сухом ацетонитриле (15 мл) добавляли диметилсульфат (0,885 г, 6,92 ммоль). Реакционную смесь нагревали при 70°C в течение 8 часов. Затем прозрачный раствор охлаждали до комнатной температуры. Раствор распределяли в три герметизированных пробирки и охлаждали до 0°C в атмосфере аргона. Затем в каждую из герметизированных пробирок добавляли раствор трет-бутилового эфира 4-аминопиперидин-1-карбоновой кислоты (0,1 г) в метаноле (5 мл) и нагревали при 50-60°C в течение 2 дней. Растворитель удаляли, и остаток растворяли в этилацетате (200 мл). Органическую фазу промывали разбавленным раствором Na2CO3, водой, и солевым раствором и сушили над Na2SO4. Растворитель испаряли, и неочищенный продукт очищали на силикагеле (EtOAc) с получением названного соединения (0,130 г).
1H-ЯМР (400 МГц, CDCl3): δ=1,59 (с, 9H), 2,23 (м, 2H), 3,09 (м, 2H), 3,81 (м, 1H), 4,23 (м, 2H), 4,44 (д, 1H), 6,37 (д, 1H), 6,47 (д, 1H), 7,24 (д, 1H), 7,38 (с, 1H), 8,16 (д, 1H).
Препаративный пример 24

Стадия А
Выпускаемый промышленностью 2,6-дибромпиридин (4,12 г, 16,6 ммоль) суспендировали в этаноле (40 мл) и добавляли гидрат гидразина (10 мл, 97,6 ммоль) в воде (~50-60%). Смесь нагревали на песочной бане при ~115°C в течение 18 часов. Растворитель удаляли, и остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (60/40), с получением названного соединения в виде серовато-белого твердого вещества (3,05 г, 93%).
1H-ЯМР (400 МГц, CDCl3): δ=3,00-3,33 (шир.с, 2H), 6,00 (шир.с, 1H), 6,67 (д, 1H), 6,83 (д, 1H), 7,33 (т, 1H).
Стадия В
Названное соединение стадии А выше (0,84 г, 4,49 ммоль) растворяли в этаноле (16 мл) и воде (4 мл). После добавления циклогексанона (0,54 мл, 5,1 ммоль), смесь перемешивали при комнатной температуре в течение 1 часа. Осадок отфильтровывали, промывали с этанолом (5 мл) и сушили на воздухе с получением названного соединения в виде белого твердого вещества (0,88 мг, 73%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,50-1,64 (м, 6H), 2,20-2,23 (м, 2H), 2,39-2,41 (м, 2H), 6,80 (д, 1H), 7,00 (д, 1H), 7,42 (т, 1H), 9,83 (с, 1H).
Стадия С
Названное соединение стадии В выше (0,2 г, 0,75 ммоль) суспендировали в диэтиленгликоле (2 мл) и нагревали при 250°C в течение 30 минут, используя микроволновый реактор Initiator фирмы Biotage. Смесь разбавляли этилацетатом (40 мл) и водой (15 мл). Органическую фазу отделяли, промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью системы Biotage Isolera One system, используя градиент этилацетат/н-гептан (5/95→30/70), с получением названного соединения в виде серовато-белого твердого вещества (0,096 мг, 51%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,73-1,85 (м, 4H), 2,57 (т, 2H), 2,68 (т, 2H), 7,05 (д, 1H), 7,63 (д, 1H), 11,40 (с, 1H).
Стадия D
Названное соединение стадии С выше (0,06 г, 0,24 ммоль) суспендировали в этиленгликоле (2 мл) и 30% растворе гидроксида аммония (3 мл). После добавления окида меди(I) (0,005 г, 0,035 ммоль), смесь нагревали при 150°C в течение 45 минут, используя микроволновый реактор Initiator фирмы Biotage. Реакционную смесь разбавляли этилацетатом (30 мл) и смесью вода/гидроксид аммония (10 мл, 1/1). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители испаряли. Остаток очищали с помощью препаративной ТСХ, используя смесь дихлорметан/метанол (95/5), с получением названного соединения в виде коричневого твердого вещества (0,022 г, 50%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,67-1,78 (м, 4H), 2,46 (т, 2H), 2,55 (т, 2H), 5,26 (с, 2H), 6,12 (д, 1H), 7,34 (д, 1H), 10,30 (с, 1H).
Препаративный пример 25

Стадия А
К раствору названного соединения препаративного примера 23 стадии А (1 г, 5,37 ммоль) в этаноле (50 мл) добавляли циклопентанон (0,45 г, 5,37 ммоль), и реакционную смесь перемешивали в течение 3 часов при комнатной температуре. На этой стадии растворитель удаляли при пониженном давлении с получением названного соединения (1,36 г, количественно).
1H-ЯМР (400 МГц, CDCl3): δ=1,74-1,80 (м, 2H), 1,85-1,92 (м, 2H), 2,24 (т, 2H), 2,46 (т, 2H), 6,86 (д, 1H), 7,10 (д, 1H), 7,37 (т, 1H), 7,54 (с, 1H).
Стадия В
Раствор названного соединения стадии В выше (0,65 г, 2,55 ммоль) в диэтиленгликоле (11 мл) герметизировали в стеклянной пробирке для микроволнового синтеза (20 мл). Затем реакционную смесь нагревали при 250°C с помощью микроволнового излучения в течение 90 минут. Реакционную смесь охлаждали и выливали в этилацетат (120 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (дихлорметан), с получением названного соединения (0,120 г, 20%).
1H-ЯМР (400 МГц, CDCl3): δ=2,48-2,55 (м, 2H), 2,82 (т, 2H), 2,99 (т, 2H), 7,18 (д, 1H), 7,60 (д, 1H), 10,1 (с, 1H).
Стадия С
Названное соединение стадии С выше (0,1 г, 0,42 ммоль) растворяли в диэтиленгликоле (2 мл) и 25% растворе гидроксида аммония (3 мл), и добавляли оксид меди(I) (0,008 г, 0,058 ммоль). Затем реакционную смесь нагревали при 150°C, используя микроволновый реактор Initiator фирмы Biotage, в течение 90 минут. Реакционную смесь разбавляли дихлорметаном (200 мл), промывали водой и солевым раствором. Органическую фазу отделяли и сушили над Na2SO4. Остаток очищали на колонке с силикагелем (1/10, метанол/дихлорметан) с получением названного соединения (0,06 г, 83%).
1H-ЯМР (400 МГц, CDCl3): δ=2,46-2,49 (м, 2H), 2,78 (т, 2H), 2,87 (т, 2H), 6,36 (д, 1H), 7,55 (д, 1H), 8,35 (с, 1H).
Препаративный пример 26
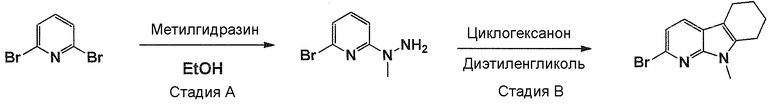
Стадия А
К суспензии 2,6-дибромпиридина (5 г, 21,11 ммоль) в этаноле (50 мл) добавляли метилгидразин (3,33 мл, 63,3 ммоль). Полученную смесь подогревали до 100°C в течение 20 часов. Реакционную смесь концентрировали досуха, и остаток очищали флэш-хроматографией (2×), используя смесь этилацетат/н-гептан (от 15% до 35%). Продукт получали в виде бледно-красноватой жидкости (2,1 г, 49%).
Стадия В
К смеси названного соединения стадии А выше (0,500 г, 2,475 ммоль) и циклогексанона (0,256 мл, 2,475 ммоль) добавляли этанол (соотношение: 1,000, объем: 10,00 мл). Полученный раствор перемешивали при комнатной температуре в течение 2 часов, затем растворитель удаляли под вакуумом. Масло разбавляли диэтиленгликолем (соотношение: 1,000, объем: 10 мл), и полученную смесь нагревали с помощью микроволнового излучения при 250°C в течение 35 минут. Темный раствор выливали в воду и фильтровали. Твердое вещество очищали флэш-хроматографией в смеси этилацетат/н-гептан (от 10% до 30%) с получением ожидаемого соединения в виде белого твердого вещества (0,307 г, 47%).
1H-ЯМР (400 МГц, CDCl3): δ=1,76-2,05 (м, 4H); 2,54-2,80 (м, 4H); 3,67 (с, 3H); 7,10 (д, J=8,0 Гц, 1H); 7,54 (д, J=8,0 Гц, 1H).
MS (ESI); m/z=265,69/267,69 (MH+).
Препаративный пример 27

Стадия А
К смеси названного соединения препаративного примера 25 стадии А (0,500 г, 2,475 ммоль) и циклопентанона (0,208 г, 2,475 ммоль) добавляли этанол (соотношение: 1,000, объем: 10,00 мл). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Затем растворитель удаляли под вакуумом. Масло разбавляли диэтиленгликолем (соотношение: 1,000, объем: 10,00 мл), и полученную смесь нагревали с помощью микроволнового излучения при 250°C в течение 35 минут.
Темный раствор выливали в воду и фильтровали. Твердое вещество очищали флэш-хроматографией в смеси этилацетат/н-гептан (от 10% до 30%) с получением ожидаемого соединения в виде желтоватого твердого вещества (0,264 г, 42%).
MS (ESI); m/z=251,66/253,67 (MH+).
Препаративный пример 28
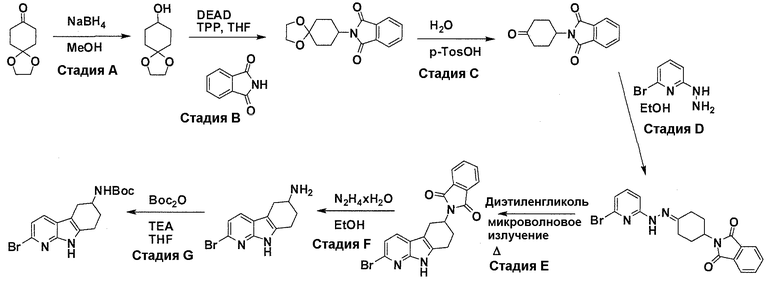
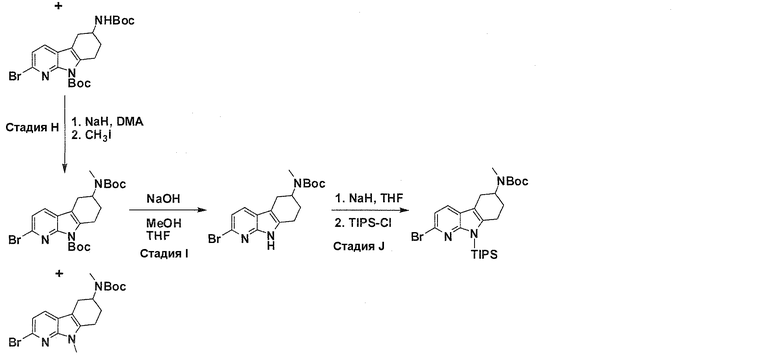
Стадия А
Выпускаемый промышленностью моноэтиленацеталь 1,4-циклогексадиона (5 г, 32 ммоль) растворяли в метаноле (65 мл), и смесь помещали в баню с холодной водой. Добавляли небольшими порциями боргидрид натрия (1,9 г, 50 ммоль) (реакция экзотермическая). После того, как добавление было закончено, смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли, и остаток растворяли в этилацетате (150 мл), воде (40 мл) и 1M NaOH (10 мл). Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (4×75 мл). объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением названного соединения в виде бесцветной жидкости (4,8 г, 94%).
1H-ЯМР (400 МГц, CDCl3): δ=1,53-1,70 (м, 4H), 1,78-1,95 (м, 4H), 3,77-3,83 (м, 1H), 3,94-3,97 (м, 4H).
Стадия В
Трифенилфосфин (12,57 г, 48,6 ммоль) и фталимид (3,56 г, 24,22 ммоль) растворяли в тетрагидрофуране (90 мл), и смесь охлаждали до 0°C. При 0°C добавляли раствор названного соединения стадии А выше (3,84 г, 24,2 ммоль) в тетрагидрофуране (90 мл), затем добавляли ~40% раствор диэтил азодикарбоксилата в толуоле (19,9 мл, 48,6 ммоль). Смесь перемешивали при 0°C в течение 10 минут, и затем при комнатной температуре в течение ночи. Растворители удаляли, и остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (20/80) в качестве подвижной фазы, с получением названного соединения в виде серовато-белого твердого вещества (3,5 г, 50%).
1H-ЯМР (400 МГц, CDCl3): δ=1,61-1,76 (м, 4H), 1,85-1,90 (м, 2H), 2,50-2,62 (м, 2H), 3,93-4,02 (м, 4H), 4,18 (тт, 1H), 7,66-7,71 (м, 2H), 7,80-7,83 (м, 2H).
Стадия С
Названное соединение стадии В выше (3,5 г, 12,1 ммоль) суспендировали в тетрагидрофуране (30 мл) и воде (20 мл). После добавления п-толуолсульфоновой кислоты (0,13 г, 0,67 ммоль), смесь нагревали при ~115°C на песочной бане в течение 2 часов. Смесь разбавляли этилацетатом (200 мл), и добавляли водный раствор бикарбоната натрия до pH~8-9. Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением названного соединения в виде серовато-белого твердого вещества (3 г, количественно).
1H-ЯМР (400 МГц, CDCl3): δ=2,17-2,24 (м, 2H), 2,60-2,71 (м, 4H), 2,80-2,90 (м, 2H), 4,78 (тт, 1H), 7,84-7,88 (м, 2H), 7,97-8,02 (м, 2H).
Стадия D
Названное соединение препаративного примера 23 стадии А (1,54 г, 8,23 ммоль) растворяли в этаноле (50 мл), и добавляли названное соединение стадии С выше (2 г, 8,23 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа до образования суспензии. Растворитель удаляли с получением названного соединения в виде серовато-белого твердого вещества (3,3 г, количественно).
1H-ЯМР (400 МГц, ДМСО-d6): δ=2,00-2,18 (м, 3H), 2,27-2,43 (м, 2H), 2,50-2,63 (м, 2H), 3,30-3,38 (м, 1H), 4,46 (тт, 1H), 7,00 (д, 1H), 7,18 (д, 1H), 7,62 (т, 1H), 7,93-8,00 (м, 4H), 10,1 (с, 1H).
Стадия Е
Названное соединение стадии D выше (1 г, 2,42 ммоль) суспендировали в диэтиленгликоле (10 мл) и нагревали при 250°C в течение 35 минут, используя микроволновый реактор Initiator фирмы Biotage. Реакционную смесь разбавляли этилацетатом (100 мл) и солевым раствором (20 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворитель удаляли. Описанную выше реакцию осуществляли еще 2 раза, и объединенный неочищенный продукт обрабатывали метанолом (5 мл). Осадок отфильтровывали, промывали с помощью метанола (5 мл) и сушили на воздухе с получением названного соединения в виде бежевого твердого вещества (1,57 г, 49%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=2,20-2,24 (м, 1H), 2,71-2,82 (м, 1H), 2,96-3,10 (м, 3H), 3,32-3,46 (м, 1H), 4,54-4,61 (м, 1H), 7,27 (д, 1H), 7,80 (д, 1H), 7,97-8,03 (м, 4H), 11,70 (с, 1H).
Стадия F
Названное соединение стадии Е выше (1,57 г, 3,96 ммоль) суспендировали в этаноле (50 мл) и обрабатывали с помощью 50-60% водного раствора гидрата гидразина (12 мл, 119 ммоль). Смесь перемешивали при комнатной температуре в течение ночи до образования прозрачного раствора. Растворители удаляли, и остаток обрабатывали дихлорметаном (150 мл). Смесь подвергали воздействию ультразвука в течение 5 минут и затем перемешивали при комнатной температуре в течение 30 минут. Осадок отфильтровывали и промывали дихлорметаном (15 мл). Объединенный фильтрат испаряли с получением названного соединения в виде бежевого твердого вещества (0,74 г, 70%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,53-1,62 (м, 1H), 1,90-1,95 (м, 1H), 2,20-2,28 (м, 1H), 2,69-2,73 (м, 2H), 2,80 (дд, 1H), 3,00-3,06 (м, 1H), 7,10 (д, 1H), 7,68 (д, 1H), 11,33-11,41 (шир.с, 1H).
Стадия G
Названное соединение стадии F выше (0,87 г, 3,28 ммоль) растворяли в тетрагидрофуране (60 мл) и обрабатывали с помощью триэтиламина (1,1 мл) и ди-трет-бутилдикарбоната (2,7 г, 12,4 ммоль). Смесь нагревали при ~40°C на песочной бане в течение ночи. Растворитель удаляли, и остаток обрабатывали диэтиловым эфиром (20 мл). Осадок отфильтровывали, и твердое вещество промывали с помощью диэтилового эфира (10 мл) с получением моно-Boc-защищенного промежуточного соединения в виде белого твердого вещества (0,86 г). Фильтрат испаряли с получением неочищенного названного соединения. Моно-Boc промежуточное соединение (0,86 г) растворяли в тетрагидрофуране (60 мл) и обрабатывали с триэтиламином (2,2 мл) и ди-трет-бутилдикарбонатом (5,4 г, 24,8 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, растворители удаляли, и неочищенный продукт объединяли с материалом из первой загрузки. Очистка неочищенного продукта на диоксиде кремния с использованием системы Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→30/75) давала названное соединение в виде белого твердого вещества/пены (1,16 г, 76%).
1H-ЯМР (400 МГц, CDCl3): δ=1,44 (с, 9H), 1,69 (с, 9H), 1,84-1,92 (м, 1H), 2,07-2,15 (м, 1H), 2,50 (дд, 1H), 3,00 (дд, 1H), 3,10 (т, 2H), 4,00-4,08 (м, 1H), 4,62-4,66 (м, 1H), 7,31 (д, 1H), 7,50 (д, 1H).
Моно-Boc промежуточное соединение: 1H-ЯМР (400 МГц, ДМСО-d6): δ=1,38 (с, 9H), 1,69-1,75 (м, 1Н), 1,93-2,00 (м, 1H), 2,41 (дд, 1H), 2,72-2,79 (м, 2H), 2,87 (дд, 1H), 3,66-3,72 (м, 1H), 7,00 (д, 1H), 7,11 (д, 1H), 7,70 (д, 1H), 11,47 (с, 1H).
Стадия H
Названное соединение стадии G выше (1,16 г, 2,5 ммоль) растворяли в Ν,Ν'-диметилацетамиде (8 мл), и смесь охлаждали до 0°C. При 0°C добавляли гидрид натрия (0,07 г, 3 ммоль), и смесь перемешивали при 0°C в течение 1 часа. Добавляли при 0°C метилйодид (0,2 мл, 3,32 ммоль), и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли этилацетатом (80 мл) и солевым раствором (20 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворитель удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния с использованием системы Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→15/85) с получением названного соединения в виде бесцветной пены (0,73 г, 61%), вместе с исходным реагентом (0,23 г, 20%) и соответствующим N1-метильным производным (0,053 г, 5%, белое твердое вещество).
1H-ЯМР (400 МГц, CDCl3): δ=1,46 (с, 9H), 1,69 (с, 9H), 1,91-2,04 (м, 2H), 2,62-2,77 (м, 2H), 2,84 (с, 3H), 3,00-3,10 (м, 1H), 3,22 (дд, 1H), 4,24-4,46 (шир.м, 1H), 7,31 (д, 1H), 7,50 (д, 1H).
N1-метильное производное: 1H-ЯМР (400 МГц, CDCl3): δ=1,46 (с, 9H), 2,00-2,10 (м, 2H), 2,70-2,90 (м, 7H), 3,70 (с, 3H), 4,21-4,50 (шир.м, 1H), 7,12 (д, 1H), 7,54 (д, 1H).
Стадия I
Названное соединение стадии H выше (0,88 г, 1,83 ммоль) растворяли в тетрагидрофуране (15 мл) и метаноле (15 мл). После добавления 1M раствора гидроксида натрия (15 мл, 15 ммоль), смесь перемешивали при комнатной температуре в течение ночи. Органические растворители удаляли, и остаток разбавляли водой (20 мл). Осадок отфильтровывали, промывали водой (5 мл) и сушили на воздухе с получением названного соединения в виде белого твердого вещества (0,67 г, 96%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,40 (с, 9H), 1,86-1,90 (м, 1H), 1,95-2,04 (м, 1H), 2,66-2,71 (м, 2H), 2,78 (с, 3H), 2,80-2,85 (м, 2H), 4,13-4,27 (шир.м, 1H), 7,18 (д, 1H), 7,75 (д, 1H), 11,55 (с, 1Н).
Стадия J
Названное соединение стадии I выше (0,38 г, 0,89 ммоль) суспендировали в тетрагидрофуране (5 мл), и смесь охлаждали до 0°C. При 0°C добавляли гидрид натрия (0,028 г, 1,15 ммоль), и смесь перемешивали при 0°C в течение 5 минут, и при комнатной температуре в течение 15 минут до получения прозрачного раствора. После добавления триизопропилсилилхлорида (0,12 мл, 0,9 ммоль), смесь нагревали при ~85°C на песочной бане в течение 1 часа. Смесь разбавляли этилацетатом (50 мл) и солевым раствором (15 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния с использованием системы Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→100/0) с получением названного соединения в виде бесцветного масла (0,27 г, 52%) вместе с исходным реагентом (0,14 г, 39%, белое твердое вещество).
1H-ЯМР (400 МГц, CDCl3): δ=1,12-1,16 (м, 18H), 1,51 (с, 9H), 1,81-1,90 (м, 3H), 1,98-2,03 (м, 2H), 2,72-2,80 (м, 2H), 2,85 (с, 3H), 2,97-3,08 (м, 2H), 4,25-4,57 (шир.м, 1H), 7,12 (д, 1H), 7,48 (д, 1H).
Препаративный пример 29

Стадия А
К раствору выпускаемого промышленностью 4-бромфенилгидразина (1,46 г, 6,5 ммоль) в этаноле (50 мл) добавляли раствор соединения препаративного примера 28 стадии C (1,6 г, 6,5 ммоль) в этаноле (15 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде твердого вещества (2,9 г, количественно).
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,85-7,86 (м, 4H), 7,31 (д, 8,4 Гц, 2H), 7,00 (д, 2H), 4,32-4,38 (м, 1H), 3,12-3,15 (м, 1H), 2,36-2,51 (м, 2,19-2,28 (м, 2H), 1,92-2,06 (м, 3H).
Стадия В
Раствор названного соединения стадии A (2,9 г, 7,0 ммоль) в диэтиленгликоле (45 мл) герметизировали в стеклянных пробирках для проведения микроволновых синтезов (20 мл). Затем реакционные пробирки нагревали при 250°C с помощью микроволнового излучения в течение 55 минут. Реакцию проводили три раза. Объединенную реакционную смесь собирали и растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (этилацетат/н-гептан 20%-40%), с получением названного соединения (2,1 г, 72%).
1H-ЯМР (400 МГц, CDCl3): δ=7,88-7,90 (м, 2H), 7,84 (шир.с, 1H), 7,75-7,78 (м, 2H), 7,53 (д, 1H); 7,22 (дд, 1H), 7,17 (д, 1H), 4,64-4,71 (м, 1H), 3,44-3,51 (м, 1H), 2,90-2,96 (м, 3H), 2,07 (м, 1H), 1,28 (шир.с, 1H).
Стадия С
К перемешиваемому раствору соединения стадии B (2 г, 5,0 ммоль) в THF (50 мл) добавляли NH2NH2∙H2O (500 мг 10 ммоль), и реакционную смесь перемешивали в течение ночи. Осадок фильтровали, и фильтрат концентрировали и использовали на следующей стадии без дополнительной очистки и идентификации.
Стадия D
К соединению стадии C (2,01 г) в THF (50 мл) добавляли (Boc)2O (2 г, 9,1 ммоль) и триэтиламин (1 г, 10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и неочищенный продукт выкристаллизовывали из смеси этилацетата и гептанов с получением моно-Boc-производного (1,15 г). Выкристаллизованное моно-Boc-производное растворяли в THF (50 мл) и добавляли избыток (Boc)2O (5 г, 22,9 ммоль) и триэтиламин (1,5 мл), и реакционную смесь перемешивали в течение 2 дней. Растворитель удаляли, и неочищенный продукт очищали хроматографически на колонке с силикагелем (этилацетат/н-гептан 10-20%) с получением названного соединения (1,35 г).
1H-ЯМР (400 МГц, CDCl3): δ=7,99 (д, 1H), 7,49 (д, 1H), 7,34 (дд, 1H), 4,68 (шир.с, 1H), 4,09 (шир.с, 1H), 3,11 (шир.с, 2H), 2,99 (дд, 1H), 2,49 (дд, 1H), 2,10-2,12 (м, 1H), 1,89-1,96 (м, 1H), 1,68 (с, 9H), 1,48 (с, 9H).
Стадия Е
Раствор названного соединения стадии D (370 мг, 0,79 ммоль) в DMA (7 мл) охлаждали до температуры ледяной бани, затем порциями добавляли NaH (40 мг, 1,59 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и охлаждали до температуры ледяной бани, и добавляли метилйодид (222 мг, 1,59 ммоль). Реакционную смесь доводили до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь растворяли в дихлорметане (250 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и остаток очищали колоночной хроматографией на силикагеле (от 0/100 до 15/85; смесь EtOAc/гептан) с получением названного соединения в виде белой пены (271 мг, 71%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,97 (д, 1H), 7,67 (с, 1H), 7,38 (д, 1H), 4,18-4,32 (м, 1H), 3,1-3,22 (м, 1H), 2,98-3,02 (м, 1H), 2,78 (с, 3H), 2,68-2,70 (м, 2H), 1,92-1,97 (м, 2H), 1,61 (с, 9H), 1,42 (с, 9H).
Препаративный пример 30
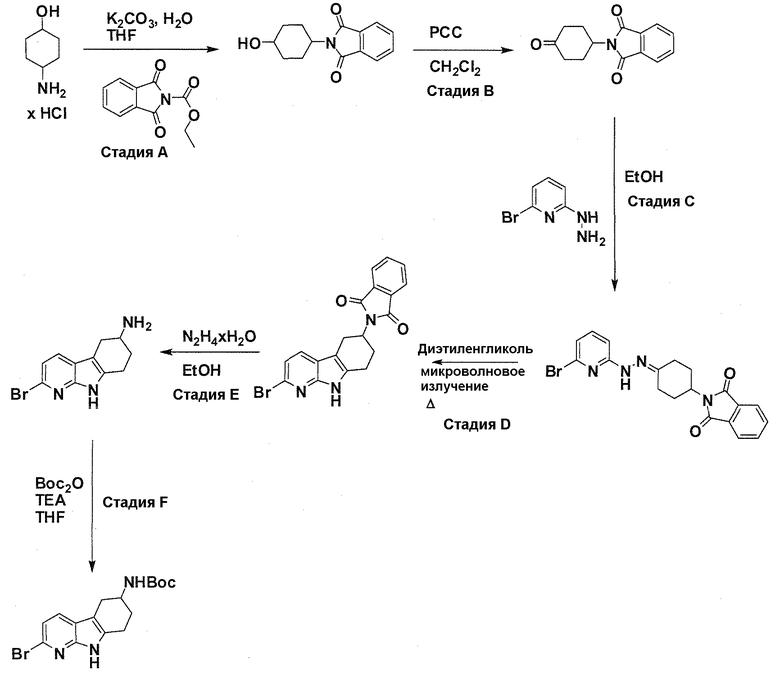
Стадия А
Выпускаемый промышленностью гидрохлоридная соль 4-аминоциклогексанола (25 г, 164 ммоль) растворяли в воде (350 мл). Затем добавляли карбонат калия (72 г, 328 ммоль), затем раствор выпускаемого промышленностью N-карбетоксифталимида в тетрагидрофуране (300 мл). Реакционную смесь затем интенсивно перемешивали при комнатной температуре в течение 3 дней. Тетрагидрофуран испаряли при пониженном давлении, и оставшуюся водную фазу экстрагировали дихлорметаном (2×300 мл) до тех пор, пока водная фаза не становилась прозрачной. Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворители испаряли при пониженном давлении с получением названного соединения в виде белого твердого вещества (28 г, 69%).
1H-ЯМР (400 МГц, CDCl3): δ=1,32-1,43 (м, 2H), 1,70-1,75 (м, 2H), 2,04-2,09 (м, 2H), 2,25-2,38 (м, 2H), 3,67-3,77 (м, 1H), 4,05-4,13 (м, 1H), 7,63-7,7,68 (м, 2H), 7,76-7,7,80 (м, 2H).
Стадия В
Названное соединение стадии А выше (28 г, 114 ммоль) растворяли в дихлорметане (990 мл), и добавляли порциями хлорохромат пиридиния (33,6 г, 157 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 8 часов. Затем добавляли порциями еще одну загрузку хлорохромата пиридиния (10,4 г, 48,6 ммоль), и перемешивание при комнатной температуре продолжали в течение 18 часов. Реакционную смесь фильтровали через слой целлита, и слой целита промывали дихлорметаном (400 мл). Объединенный фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (60/40) в качестве подвижной фазы, с получением названного соединения в виде белого твердого вещества (24,62 г, 88%)
1H-ЯМР (400 МГц, CDCl3): δ=2,17-2,24 (м, 2H), 2,60-2,71 (м, 4H), 2,80-2,90 (м, 2H), 4,78 (тт, 1H), 7,84-7,88 (м, 2H), 7,97-8,02 (м, 2H).
Стадия С
Названное соединение препаративного примера 23 стадии А (19 г, 101 ммоль) растворяли в этаноле (570 мл), и добавляли названное соединение стадии В выше (24,6 г, 101 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов до образования суспензии. Растворитель удаляли с получением названного соединения в виде серовато-белого твердого вещества (41,8 г, количественно).
1H-ЯМР (400 МГц, ДМСО-d6): δ=2,00-2,18 (м, 3H), 2,27-2,43 (м, 2H), 2,50-2,63 (м, 2H), 3,30-3,38 (м, 1H), 4,46 (тт, 1H), 7,00 (д, 1H), 7,18 (д, 1H), 7,62 (т, 1H), 7,93-8,00 (м, 4H), 10,1 (с, 1H).
Стадия D
Названное соединение стадии С выше (1,3 г, 3,15 ммоль) суспендировали в диэтиленгликоле (13 мл) и нагревали при 245°C в течение 35 минут, используя микроволновый реактор Initiator фирмы Biotage. Реакционную смесь разбавляли водой (90 мл), осадок отфильтровывали и сушили на воздухе. Эту реакционную последовательность повторяли до тех пор, пока названное соединение стадии С выше (41,8 г) полностью не расходовалось с получением названного соединения в виде серого твердого вещества (34,2 г, 85%). Неочищенный материал непосредственно использовали на следующей стадии.
Стадия Е
Названное соединение стадии D выше (34,2 г, 86,36 ммоль) суспендировали в этаноле (1080 мл) и обрабатывали с помощью 50-60% водного раствора гидрата гидразина (185 мл, 1990 ммоль). Смесь перемешивали при комнатной температуре в течение 2 дней. Осадок отделяли фильтрацией и промывали этанолом (150 мл). Объединенный фильтрат концентрировали до объема ~150 мл и экстрагировали дихлорметаном (2×450 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли с получением неочищенного названного соединения в виде темного твердого вещества (22,97 г, количественно).
Стадия F
Названное соединение стадии Е выше (22,97 г, 90,1 ммоль) растворяли в тетрагидрофуране (1000 мл) и обрабатывали с помощью триэтиламина (64 мл) и ди-трет-бутилдикарбоната (79 г, 367 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, и растворители удаляли при пониженном давлении. Остаток обрабатывали диэтиловым эфиром (600 мл) и перемешивали при комнатной температуре в течение 30 минут. Осадок отфильтровывали, промывали с помощью диэтилового эфира (250 мл) и сушили на воздухе с получением названного соединения в виде белого твердого вещества (11 г, 33%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,38 (с, 9H), 1,69-1,75 (м, 1h), 1,93-2,00 (м, 1H), 2,41 (дд, 1H), 2,72-2,79 (м, 2H), 2,87 (дд, 1H), 3,66-3,72 (м, 1H), 7,00 (д, 1H), 7,11 (д, 1H), 7,70 (д, 1H), 11,47 (с, 1H).
Препаративный пример 31

Стадия А
Названное соединение препаративного примера 30 стадии F (11 г, 30 ммоль) растворяли в Ν,Ν'-диметилацетамиде (140 мл), и смесь охлаждали до 0°C. Добавляли при 0°C гидрид натрия (2,2 г, 92 ммоль), и смесь перемешивали при 0°C в течение 2 часов. Затем добавляли метилйодид (9 мл, 145 ммоль), смесь перемешивали при 0°C в течение 5 минут. Удаляли ледяную баню, и через ~5 минут наблюдалось выделение тепла в результате экзотермической реакции. Реакционную смесь ненадолго помещали в водяную баню, и затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (700 мл), и осадок отфильтровывали. Твердый материал затем очищали с помощью хроматографии на диоксиде кремния, используя дихлорметан/ацетон, (98/2) с получением названного соединения в виде белого твердого вещества (10,6 г, 85%).
1H-ЯМР (400 МГц, CDCl3): δ=1,46 (с, 9H), 2,00-2,10 (м, 2H), 2,70-2,90 (м, 7H), 3,70 (с, 3H), 4,21-4,50 (шир.м, 1H), 7,12 (д, 1H), 7,54 (д, 1H).
Стадия В
Названное соединение стадии А выше (0,1 г, 0,253 ммоль) растворяли в дихлорметане (2 мл) и обрабатывали с помощью 2M раствора хлористого водорода (0,5 мл, 1 ммоль) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи. Затем к реакционной смеси добавляли триэтиламин (0,07 мл, 0,316 ммоль) и ацетилхлорид (0,316 мл, 1,034 ммоль). Перемешивание продолжали в течение еще 2 часов, затем смесь разбавляли дихлорметаном и промывали с помощью насыщенного водного раствора карбоната натрия. Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением остатка, который очищали с помощью системы флэш-хроматографической очистки Biotage (метанол/дихлорметан: 0→5%) с получением названного соединения в виде белого твердого вещества (0,065 г, 75% за 2 стадии).
1H-ЯМР (400 МГц, CDCl3): δ=1,99-2,03 (м, 1H), 2,12-2,19 (м, 4H), 2,67-2,73 (м, 1H), 2,81 (дд, 1H), 2,87-2,90 (м, 2H), 2,94-2,98 (м, 4H), 3,70 (д, 3H), 7,14 (дд, 1H), 7,53 (дд, 1H).
Препаративный пример 32

Стадия А
К суспензии выпускаемого промышленностью 2,6-дибромпиридина (10 г, 42,2 ммоль) в этаноле (50 мл) добавляли выпускаемый промышленностью метилгидразин (11,11 мл, 211 ммоль). Смесь нагревали при 80°C (температура реакционной смеси) в течение 48 часов. Реакционную смесь концентрировали досуха, и остаток очищали с помощью хроматографии на диоксиде кремния, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (15/75→35/65) с получением названного соединения в виде красноватого масла, которое становилось твердыи в результате стояния при комнатной температуре (7,6 г, 89%)
1H-ЯМР (400 МГц, CDCl3): δ=3,23 (с, 3H), 4,00 (шир.с, 2H), 6,70 (д, 1H), 6,82 (д, 1H), 7,27 (т, 1H).
Стадия В
Суспензию выпускаемого промышленностью гидрохлорида 4-(метиламино)циклогексанон-2,2-диметилтриметиленкеталя (3,7 г, 14,8 ммоль) и названного соединения стадии А выше (3 г, 14,8 ммоль) в диоксане (30 мл) помещали на ледяную баню. К перемешиваемой суспензии медленно добавляли концентрированную H2SO4 (3 мл). После завершения добавления H2SO4, реакционную смесь кипятили с обратным холодильником в течение 5 часов на песочной бане (~140°C). Реакционную смесь охлаждали до комнатной температуры, слой диоксана отбрасывали, и добавляли ледяную воду (20 мл). Смесь перемешивали до тех пор, пока не растворялся смолистый материал. Затем доводили pH реакционной смеси до pH=14 с помощью водного раствора NaOH. Водный слой подвергали экстракции дихлорметаном (200 мл), и органическую фазу промывали водой и солевым раствором. Органическую фазу отделяли, сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного названного соединения в виде серовато-белого твердого вещества (4,7 г, количественно).
Стадия С
К раствору неочищенного соединения стадии А выше (4,7 г) в тетрагидрофуране (50 мл) добавляли триэтиламин (5 мл) и ди-трет-бутилдикарбонат (10 г, 45,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, и остаток растворяли в дихлорметане (200 мл). Органическую фазу промывали водой, солевым раствором и сушили над Na2SO4. Органический растворитель удаляли при пониженном давлении, и остаток очищали на силикагеле, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (20/80→50/50) с получением названного соединения в виде белого твердого вещества (3,55 г, 61% за 2 стадии).
1H-ЯМР (400 МГц, CDCl3): δ=1,46 (с, 9H), 2,00-2,10 (м, 2H), 2,70-2,90 (м, 7H), 3,70 (с, 3H), 4,21-4,50 (шир.м, 1H), 7,12 (д, 1H), 7,54 (д, 1H).
Препаративный пример 33
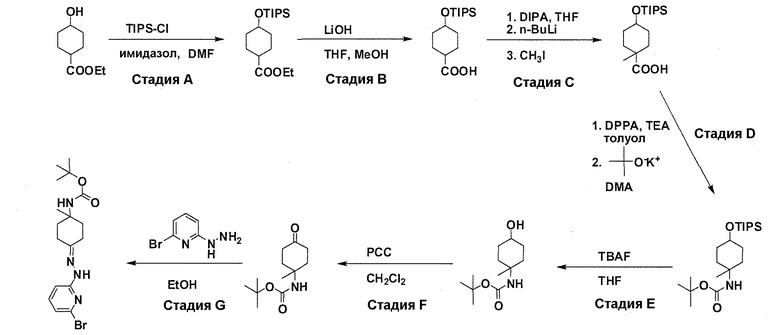

Стадия А
Выпускаемый промышленностью этиловый эфир 4-гидрокси-циклогексанкарбоновой кислоты (10,32 г, 59,92 ммоль) растворяли в N,N'-диметилформамиде (50 мл), и добавляли имидазол (8,16 г, 119,5 ммоль). После добавления триизопропилсилилхлорида (14,1 мл, 65,9 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли диэтиловым эфиром (150 мл) и промывали 1M раствором хлористоводородной кислоты (150 мл). Органическую фазу отделяли, и водную фазу экстрагировали диэтиловым эфиром (150 мл). Объединенную органическую фазу s промывали 1M раствором хлористоводородной кислоты (150 мл) и солевым раствором (150 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли с получением неочищенного названного соединения в виде бесцветной жидкости (18,7 г, 95%).
Стадия В
Неочищенное названное соединение стадии А выше (18,7 г, 65,17 ммоль) растворяли в тетрагидрофуране (90 мл) и метаноле (60 мл). После добавления гидрата гидроксида лития (5,47 г, 130,6 ммоль), реакционную смесь нагревали при ~70°C в течение 2 часов на песочной бане, и при комнатной температуре в течение ночи. Растворители удаляли, и остаток растворяли в воде (100 мл). Доводили pH реакционной смеси до pH~3-4 с помощью 1M раствора хлористоводородной кислоты, и водную фазу экстрагировали этилацетатом (2×200 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли с получением неочищенного названного соединения в виде бледно-желтого масла (16,65 г, 97%).
Стадия С
Диизопропиламин (9 мл, 65 ммоль) растворяли в тетрагидрофуране (100 мл), и смесь охлаждали до 0°C. Добавляли при 0°C 1,6M раствор н-бутиллития в н-гексане (38,5 мл, 61,6 ммоль), и реакционную смесь перемешивали при 0°C в течение 15 минут. Реакционную смесь затем охлаждали до -78°C, и добавляли по каплям раствор неочищенного названного соединения стадии В выше (8,3 г, 30,9 ммоль) в тетрагидрофуране (30 мл). После того, как добавление было закончено, охлаждающую баню удаляли, и реакционную смесь нагревали при ~60°C на песочной бане в течение 2 часов. Реакционную смесь снова охлаждали до -78°C, и добавляли метилйодид (2,1 мл, 34 ммоль). Реакционную смесь перемешивали при -78°C в течение 2 часов и затем подогревали до комнатной температуры в течение ночи. Реакционную смесь выливали в смесь диэтилового эфира (500 мл) и 1M раствора хлористоводородной кислоты (500 мл). Органическую фазу отделяли, и водную фазу экстрагировали диэтиловым эфиром (2×200 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан/метанол (15/84/1) в качестве подвижной фазы, с получением названного соединения в виде бледно-желтого масла (4,35 г, 50%), и извлекали исходный реагент в виде бледно-желтого масла (1,51 г, 18%).
1H-ЯМР (400 МГц, CDCl3): δ=1,03 (с, 18H), 1,17-1,24 (м, 7H), 1,43-1,52 (м, 2H), 1,64-1,71 (м, 1H), 1,77-1,84 (м, 2H), 2,18-2,23 (м, 2H), 3,65-3,73 (м, 1H),
извлекаемый исходный реагент:
1H-ЯМР (400 МГц, CDCl3): δ=1,03 (с, 18H), 1,20-1,80 (м, 7H), 1,94-2,05 (м, 4H), 2,28-2,33 (м, 1H), 3,62-3,68 (м, 1H).
Стадия D
Названное соединение стадии С выше (2,18 г, 6,92 ммоль) растворяли в толуоле (25 мл), и добавляли триэтиламин (1,08 мл, 7,7 ммоль). После добавления дифенилфосфорилазида (1,63 мл, 7,54 ммоль), реакционную смесь нагревали при ~115°C на песочной бане в течение 1 часа до тех пор, пока не заканчивалось выделение азота. Смесь охлаждали до 0°C и промывали насыщенным раствором бикарбоната натрия (15 мл), водой (15 мл) и солевым раствором (15 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители испаряли с получением неочищенного промежуточного соединения изоцианата. Неочищенное промежуточное соединение растворяли в Ν,Ν'-диметилацетамиде (10 мл), и добавляли порциями трет-бутоксид калия (0,8 г, 7,13 ммоль). После того, как добавление было закончено, реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом (80 мл) и водой (30 мл). Органическую фазу отделяли и промывали водой (20 мл) и солевым раствором (20 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (10/90) в качестве подвижной фазы, с получением названного соединения в виде бесцветного масла (1,57 г, 59%).
1H-ЯМР (400 МГц, CDC!3): δ=1,16 (с, 18H), 1,37-1,45 (м, 3H), 1,43 (с, 3H), 1,56 (с, 9H), 1,58-1,66 (м, 4H), 1,82-1,88 (м, 2H), 2,13-2,20 (м, 2H), 3,76-3,81 (м, 1H), 4,48 (шир.с, 1H).
Стадия Е
Названное соединение стадии D выше (1,45 г, 3,77 ммоль) растворяли в ацетонитриле (25 мл), и добавляли 1M раствор фторида тетрабутиламмония в тетрагидрофуране (14,8 мл, 14,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (80/20), с получением названного соединения в виде бесцветного масла (0,86 г, 99%).
1H-ЯМР (400 МГц, CDCl3): δ=1,42 (с, 3H), 1,54 (с, 9H), 1,57-1,70 (м, 3H), 1,85-1,93 (м, 3H), 2,19-2,24 (м, 2H), 3,70-3,73 (м, 1H), 4,45 (шир.с, 1H).
Стадия F
Названное соединение стадии Е выше (0,86 г, 3,76 ммоль) растворяли в дихлорметане (40 мл), и добавляли порциями хлорохромат пиридиния (1,18 г, 5,48 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, фильтровали через слой целлита, и целит промывали дихлорметаном (20 мл). Объединенный фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (60/40) в качестве подвижной фазы, с получением названного соединения в виде бесцветного масла (0,76 г, 89%)
1H-ЯМР (400 МГц, CDCl3): δ=1,42 (с, 3H), 1,45 (с, 9H), 1,77 (дт, 2H), 2,23-2,50 (м, 6H), 4,48 (шир.с, 1H).
Стадия G
Названное соединение препаративного примера 23 стадии А (0,63 г, 3,35 ммоль) растворяли в этаноле (20 мл), и добавляли названное соединение стадии F выше (0,76 г, 3,35 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа до тех пор, пока раствор не становился прозрачным. Растворитель удаляли с получением названного соединения в виде темно-желтого масла (1,33 г, количественно).
1H-ЯМР (400 МГц, CDCl3): δ=1,40 (с, 3H), 1,47 (с, 9H), 1,52-1,67 (м, 3H), 2,15-2,23 (м, 2H), 2,40-2,50 (м, 3H), 4,45 (шир.с, 1H), 6,90 (д, 1H), 7,15 (д, 1H), 7,40 (т, 1H), 7,85 (шир.с, 1H).
Стадия H
Названное соединение стадии G выше (0,7 г, 1,76 ммоль) растворяли в диэтиленгликоле (10 мл) и нагревали при 245°C в течение 60 минут, используя микроволновый реактор Initiator фирмы Biotage (давление: 10-11 бар). Реакционную смесь разбавляли водой (15 мл) и дихлорметаном (40 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением неочищенного названного соединения в виде темного масла. Эту процедуру повторяли еще один раз, и объединенный неочищенный материал непосредственно использовали на следующей стадии.
Стадия I
Неочищенное соединение стадии H выше растворяли в тетрагидрофуране (45 мл) и обрабатывали с помощью триэтиламина (1,7 мл) и ди-трет-бутилдикарбоната (2 г, 9,3 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, и растворители удаляли при пониженном давлении. Остаток очищали с помощью хроматографии на диоксиде кремния, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→30/70) с получением 3 различных фракций.
Фракция I: Бис-Boc-производное; желтое масло (0,15 г, 9%):
1H-ЯМР (400 МГц, CDCl3): δ=1,39 (с, 9H), 1,45 (с, 3H), 1,64 (с, 9H), 1,70-1,75 (м, 1H), 2,41-2,46 (м, 1H), 2,70 (д, 1H), 2,83 (д, 1H), 2,88-3,05 (м, 2H), 4,45 (шир.с, 1H), 7,27 (д, 1H), 7,48 (д, 1H).
Фракция II: извлеченное названное соединение стадии G; желтое масло (0,13 г, 9%).
Фракция III: Моно-Boc названное соединение; серовато-белое твердое вещество (0,26 г, 19%):
1H-ЯМР (400 МГц, CDCl3): δ=1,45 (с, 9H), 1,54 (с, 3H), 1,76-1,83 (м, 1H), 2,60-2,65 (м, 1H), 2,80-2,90 (м, 4H), 4,55 (с, 1H), 7,18 (д, 1H), 7,59 (д, 1H), 9,75 (шир.с, 1H).
Стадия J
Бис-Boc-производное стадии I выше (0,15 г, 0,32 ммоль) растворяли в тетрагидрофуране (2,6 мл) и метаноле (2,6 мл). После добавления 1M водного раствора гидроксида натрия (2,6 мл, 2,6 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи, и растворители испаряли. Остаток обрабатывали водой (20 мл), осадок отфильтровывали, промывали водой (5 мл) и сушили на воздухе с получением моно-Boc названного соединения стадии I в виде белого твердого вещества (0,09 г, 76%).
Стадия K
Моно-Boc названное соединение стадии I выше (0,35 г, 0,94 ммоль) растворяли в Ν,Ν'-диметилацетамиде (5 мл), и смесь охлаждали до 0°C. Добавляли при 0°C гидрид натрия (0,068 г, 2,8 ммоль), и смесь перемешивали при 0°C в течение 90 минут. Затем добавляли при 0°C метилйодид (0,27 мл, 4,45 ммоль), удаляли ледяную баню, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом (50 мл) и водой (20 мл). Органическую фазу отделяли, и водную фазу экстрагировали этилацетатом (20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя систему Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→30/70), с получением названного соединения в виде белого твердого вещества (0,31 г, 81%).
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 3H), 1,86-1,94 (м, 1H), 2,68-2-74 (м, 3H), 2,72 (с, 3H), 3,07-3,18 (м, 2H), 3,72 (с, 3H), 7,15 (д, 1H), 7,57 (д, 1H).
Препаративный пример 34

Стадия А
Названное соединение препаративного примера 28 стадии А (12 г, 75,9 ммоль) растворяли в дихлорметане (240 мл), и добавляли триэтиламин (14,5 мл). После добавления бензоилхлорида (12,7 г, 91,2 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли дихлорметаном (100 мл) и промывали водой (80 мл) и 10% водным раствором лимонной кислоты (2×80 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители испаряли при пониженном давлении с получением неочищенного названного соединения в виде масла.
Стадия В
Неочищенное названное соединение стадии А выше растворяли в тетрагидрофуране (85 мл) и воде (250 мл). Затем добавляли п-толуолсульфоновую кислоту (1 г, 5,8 ммоль), и смесь нагревали при ~115°C на песочной бане в течение 4 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (250 мл), и органическую фазу отделяли. Органическую фазу сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (20/80) в качестве подвижной фазы, с получением названного соединения в виде белого твердого вещества (10,7 г, 64% за 2 стадии).
1H-ЯМР (400 МГц, CDCl3): δ=2,27-2,45 (м, 4H), 2,52-2,61 (м, 2H), 2,74-2,84 (м, 2H), 5,55-5,60 (м, 1H), 7,60 (т, 2H), 7,73 (т, 1H), 8,21 (д, 2H).
Стадия С
Названное соединение препаративного примера 23 стадии А (3,69 г, 19,6 ммоль) растворяли в этаноле (85 мл), и добавляли названное соединение стадии В выше (4,28 г, 19,6 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, и растворители удаляли при пониженном давлении с получением названного соединения в виде желтой пены (7,6 г, количественно).
1H-ЯМР (400 МГц, CDCl3): δ=2,12-2,26 (м, 4H), 2,53-2,61 (м 2H), 2,68-2,84 (м, 2H), 7,02 (д, 1H), 7,30 (д, 1H), 7,55 (т, 1H), 7,57-7,62 (м, 2H), 7,71 (т, 1H), 8,17-8,22 (м, 2H).
Стадия D
Названное соединение стадии С выше (0,95 г, 2,45 ммоль) растворяли в диэтиленгликоле (9,5 мл) и нагревали при 245°C в течение 35 минут, используя микроволновый реактор Initiator фирмы Biotage. Реакционную смесь разбавляли водой (25 мл) и этилацетатом (100 мл). Органическую фазу отделяли, промывали солевым раствором (25 мл), сушили над Na2SO4, фильтровали, и растворители удаляли. Эту процедуру повторяли еще семь раз, и объединенный неочищенный материал очищали с помощью хроматографии на диоксиде кремния, используя дихлорметан/ацетон (98/2) в качестве подвижной фазы, с получением названного соединения в виде серовато-белого твердого вещества (3,4 г, 46%).
1H-ЯМР (400 МГц, CDCl3): δ=2,23-2,38 (м, 2H), 2,95-3,27 (м, 4H), 5,60 (5,57-5,61 (м, 1H), 7,22 (д, 1H), 7,44-7,47 (м, 2H), 7,58 (т, 1H), 7,65 (д, 1H), 8,06 (д, 2H), 9,88 (шир.с, 1H).
Стадия Е
Названное соединение стадии D выше (3,4 г, 9,19 ммоль) растворяли в Ν,Ν'-диметилацетамиде (40 мл), и раствор охлаждали до 0°C. Добавляли порциями при 0°C гидрид натрия (0,29 г, 11,95 ммоль). Смесь перемешивали при 0°C в течение 1 часа, и добавляли метилйодид (1,19 мл, 19,14 ммоль). Смесь перемешивали при 0°C в течение 5 минут, затем при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли этилацетатом (250 мл), 10% водным раствором лимонной кислоты (80 мл) и солевым раствором (50 мл). Органическую фазу отделяли, промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→30/70), с получением названного соединения в виде серовато-белого твердого вещества (2,42 г, 68%).
1H-ЯМР (400 МГц, CDCl3): δ=2,25-2,34 (м, 2H), 2,85-3,00 (м, 3H), 3,18 (дд, 1H), 3,75 (с, 3H), 5,52-5,57 (м, 1H), 7,15 (д, 1H), 7,42 (т, 2H), 7,53-7,57 (м, 2H), 8,00-8,04 (м, 2H).
Стадия F
Названное соединение стадии Е выше (0,33 г, 0,86 ммоль) растворяли в тетрагидрофуране (8 мл) и воде (8 мл). После добавления гидроксида калия (0,75 г, 13,4 ммоль), смесь нагревали при 140°C в течение 30 минут, используя микроволновый реактор Initiator фирмы Biotage. Реакционную смесь разбавляли этилацетатом (30 мл) и водой (10 мл). Органическую фазу отделяли, промывали солевым раствором (10 мл), сушили над Na2SO4, фильтровали, и растворители удаляли. Эту процедуру повторяли еще шесть раз с получением названного соединения в виде серовато-белого твердого вещества (1,74 г, 98%).
1H-ЯМР (400 МГц, CDCl3): δ=2,00-2,16 (м, 2H), 2,68 (дд, 1H), 2,75-2,80 (м, 1H), 2,88-2,93 (м, 1H), 3,06 (дд, 1H), 3,71 (с, 3H), 4,25-4,30 (м, 1H), 7,13 (д, 1H), 7,56 (д, 1H).
Стадия G
Названное соединение стадии F выше (0,2 г, 0,71 ммоль) растворяли в N,N'-диметилацетамиде (3 мл), и раствор охлаждали до 0°C. Добавляли при 0°C гидрид натрия (0,023 г, 0,92 ммоль). Смесь перемешивали при 0°C в течение 1 часа, и добавляли метилйодид (0,09 мл, 1,47 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли этилацетатом (30 мл), 10% водным раствором лимонной кислоты (8 мл) и солевым раствором (5 мл). Органическую фазу отделяли, промывали солевым раствором (5 мл), сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (5/95→40/60→100/0), с получением названного соединения в виде бледно-оранжевого масла, которое становилось твердым при стоянии при комнатной температуре (0,13 г, 63%).
1H-ЯМР (400 МГц, CDCl3): δ=2,05-2,19 (м, 2H), 2,70-2,78 (м, 2H), 2,86-2,93 (м, 1H), 3,30 (дд, 1H), 3,47 (с, 3H), 3,71 (с, 3H), 3,75-3,78 (м, 1H), 7,14 (д, 1H), 7,56 (д, 1H).
Препаративный пример 35

Стадия А
К раствору выпускаемого промышленностью 4-бромфенилгидразина (2,6 г, 11,6 ммоль) в этаноле (50 мл) добавляли производное кетона (2,54 г, 11,6 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде твердого вещества (4,5 г, количественно). Соединение использовали на следующей стадии без какой-либо очистки.
Стадия В
Раствор названного соединения стадии выше (4,4 г, 11,3 ммоль) в диэтиленгликоле (45 мл) герметизировали в трех различных пробирках для проведения микроволновых реакций (20 мл). Затем реакционные пробирки нагревали при 250°C с помощью микроволнового излучения в течение 55 минут. объединенную реакционную смесь собирали и растворяли в DCM (200 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (этилацетат/н-гептан 20%-30%), с получением названного соединения (1,77 г, 42%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 7,95 (д, 1H), 7,65 (т, 1H), 7,52 (дд, 1H), 7,24 (д, 1H), 7,12 (д, 1H), 5,46 (с, 1H), 3,34 (с, 3H), 3,13 (дд, 1H), 2,98-2,78 (м, 1H), 2,51 (с, 2H), 2,18 (д, 1H).
Стадия С
К перемешиваемому раствору соединения из приведенной выше стадии B (200 мг, 0,54 ммоль) в THF (50 мл) добавляли NaH (25 мг, 1,0 ммоль). Суспензию перемешивали в течение 10 минут. Затем добавляли раствор метилйодида (75 мг, 0,5 ммоль), и реакционную смесь перемешивали в течение 1 часа. Реакцию останавливали с помощью воды и экстрагировали этилацетатом (100 мл). Органическую фазу промывали солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный материал очищали на колонке с силикагелем (разбавитель: 10:90 EtOAc:гептан) с получением названного соединения (200 мг, 96%).
1H-ЯМР (400 МГц, CDCl3): δ=8,10-8,00 (м, 2H), 7,59-7,55 (м, 2H), 7,46-7,42 (м, 2H), 7,26-7,28 (м, 1H), 7,16 (д, 1H), 5,59-5,56 (м, 1H), 3,66 (с, 3H), 3,21 (дд, 1H), 3,01-2,92 (м, 3H), 2,33-2,27 (м, 2H).
Стадия D
К раствору соединения стадии C (200 мг, 0,52 ммоль) в THF:H2O (1:1, 8 мл) добавляли KOH (450 мг, 7,8 ммоль). Реакционную смесь нагревали при 140°C с помощью микроволнового излучения в течение 30 минут. Реакционную смесь экстрагировали этилацетатом (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель испаряли, и неочищенный материал очищали на колонке с силикагелем (разбавитель: 10:90 EtOAc:гептан) с получением названного соединения (137 мг, 69%).
1H-ЯМР (400 МГц, CDCl3): δ=7,56 (д, 1H), 7,23 (дд 1H), 7,10 (д, 1H), 4,48-4,07 (м, 1H), 3,60 (с, 3H), 3,05 (дд, 1H), 2,89 (дт, 1H), 2,83-2,61 (м, 3H), 2,20-1,96 (м, 2H).
Стадия Е
К раствору соединения с приведенной выше стадии D (130 мг, 0,46 ммоль) в THF (10 мл) добавляли NaH (22 мг, 0,92 ммоль). Суспензию перемешивали в течение 10 минут при комнатной температуре. Затем добавляли раствор метилйодида (129 мг, 0,92 ммоль), и реакционную смесь перемешивали в течение 1 часа. Затем реакцию осторожно останавливали водой и экстрагировали этилацетатом (150 мл). Органическую фазу промывали солевым раствором и сушили над Na2SO4. Растворитель удаляли и проводили очистку на колонке с силикагелем (элюент: 10:90 EtOAc:гептан) с получением названного соединения (115 мг, 85,8%).
1H-ЯМР (400 МГц, CDCl3) δ 7,57 (д, 1H), 7,21 (дд, 1H), 7,09 (д, 1H), 3,87-3,72 (м, 1H), 3,59 (с, 3H), 3,46 (с, 3H), 3,03 (дд, 1H), 2,90-2,83 (м, 1H), 2,77-2,68 (м, 2H), 2,19-2,12 (м, 1H), 2,19-1,98 (м, 1H).
Препаративный пример 36

Стадия А
К раствору выпускаемого промышленностью 3-бромфенилгидразина (2 г, 9,1 ммоль) в этаноле (50 мл) добавляли производное кетона (2 г, 9,1 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде твердого вещества (3,52 г, количественно). Продукт использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия В
Раствор названного соединения (3,5 г, 9,1 ммоль) в диэтиленгликоле (12 мл) герметизировали в стеклянной пробирке для проведения микроволновых реакций (20 мл). Затем реакционную смесь нагревали при 245°C с помощью микроволнового излучения в течение 50 минут. Реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем (этилацетат/н-гептан 10%-40%) с получением двух региоизомеров (1,17, 32%).
Региоизомер A (7-бром-2,3,4,9-тетрагидро-1H-карбазол-3-илбензоат) (670 мг):
1H-ЯМР (400 МГц, CDCl3): δ=8,05 (д, 1H), 7,83 (с, 1H), 7,57 (м, 1H), 7,47-7,42 (м, 2H), 7,32 (д, 1H), 7,21 (дд, 1H), 5,62-5,59 (м, 1H), 3,22 (дд, 1H), 3,10-2,76 (м, 3H), 2,56-2,07 (м, 2H).
Региоизомер B (5-бром-2,3,4,9-тетрагидро-1H-карбазол-3-илбензоат) (500 мг):
1H-ЯМР (400 МГц, CDCl3): δ=8,08 (д, 1H), 7,92 (с, 1H), 7,57 (м, 1H), 7,45 (т, 1H), 7,24 (т, 1H), 6,97 (т, 1H), 5,61 (м, 1H), 3,65 (дд, 1H), 3,38 (дд, 1H), 3,17-2,66 (м, 2H), 2,45-2,07 (м, 2H).
Стадия С
К перемешиваемому раствору региомера А соединения стадии 2 (450 мг, 1,2 ммоль) в THF (15 мл) добавляли NaH (58 мг, 2,4 ммоль). Суспензию перемешивали в течение 10 минут. Затем добавляли раствор метилйодида (338 мг, 2,4 ммоль), и реакционную смесь перемешивали в течение 2 часов. Реакцию останавливали с помощью воды и экстрагировали этилацетатом (200 мл). Органическую фазу промывали солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный материал очищали на колонке с силикагелем (этилацетат/н-гептан 10%-30%) с получением названного соединения (357 мг, 77,6%).
1H-ЯМР (400 МГц, CDCl3): δ=8,06-8,03 (м, 1H), 7,62-7,52 (м, 1H), 7,49-7,39 (м, 2H), 7,33 (д, 1H), 7,20 (дд, 1H), 5,76-5,41 (м, 1H), 3,65 (с, 3H), 3,23 (дд, 1H), 3,13-2,73 (м, 3H), 2,56-2,11 (м, 2H).
Стадия D
К раствору соединения стадии 3 (357 мг, 0,92 ммоль) в THF:H2O (1:1, 8 мл) добавляли KOH (450 мг, 7,8 ммоль). Реакционную смесь нагревали при 140°C с помощью микроволнового излучения в течение 30 минут. Реакционную смесь экстрагировали этилацетатом (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель испаряли, и неочищенный материал очищали на колонке с силикагелем (этилацетат/н-гептан 10%-40%) с получением названного соединения (160 мг, 62%).
1H-ЯМР (400 МГц, CDCl3) δ=7,42 (д, 1H), 7,32 (д, 1H), 7,19 (дд, 1H), 4,38-4,21 (м, 1H), 3,61 (с, 3H), 3,09 (дд, 1H), 2,82-2,70 (м, 3H), 2,16-1,96 (м, 2H).
Стадия Е
К раствору соединения стадии 4 (160 мг, 0,57 ммоль) в THF (5 мл) добавляли NaH (122 мг, 5,0 ммоль). Суспензию перемешивали в течение 10 минут при комнатной температуре. Затем добавляли раствор метилйодида (400 мг, 2,83 ммоль), и реакционную смесь перемешивали в течение 4 часов. Затем реакцию осторожно останавливали водой и экстрагировали этилацетатом (150 мл). Органическую фазу промывали солевым раствором и сушили над Na2SO4. Растворитель удаляли, и проводили очистку на колонке с силикагелем (ацетат/н-гептан 10%-40%) с получением названного соединения (120 мг, 71,8%).
1H-ЯМР (400 МГц, CDCl3): δ=7,40 (д, 1H), 7,32 (д, 1H), 7,18 (дд, 1H), 3,84-3,71 (м, 1H), 3,60 (с, 3H), 3,48 (с, 3H), 3,08 (дд, 1H), 2,87 (дт, 1H), 2,74-2,70 (м, 2H), 2,20-2,03 (м, 2H).
Препаративный пример 37

Стадия А
К раствору названного соединения (5 г, 31,6 ммоль) в THF (100 мл) добавляли NaH (1,2 г, 50 ммоль). Суспензию перемешивали в течение 10 минут при комнатной температуре. Добавляли медленно раствор метилйодида (5,5 г, 39 ммоль), и реакционную смесь перемешивали в течение 3 часов. Затем реакцию осторожно останавливали с помощью воды, и реакционную смесь концентрировали. Остаток растворяли в EtOAc (200 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (15/85→50/50), с получением названного соединения (4,3 г, 80%).
1H-ЯМР (400 МГц, CDCl3) δ: 3,95 (с, 3H), 3,35-3,29 (м, 5H), 1,87-1,80 (м, 4H), 1,75-1,68 (м, 2H), 1,60-1,52 (м, 2H).
Стадия В
К раствору названного соединения (6,4 г, 37,2 ммоль) в THF:H2O (1:1; 50 мл) добавляли п-толуолсульфоновую кислоту (640 мг, 3,86 ммоль). Реакционную смесь нагревали при 100°C в течение ночи.
Реакционную смесь экстрагировали этилацетатом (200 мл). Органическую фазу промывали водой, солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и остаток очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением названного соединения (4,2 г, 89%).
1H-ЯМР (400 МГц, CDCl3) δ: 3,64-3,62 (м, 1H), 3,42 (с, 3H), 2,62-2,54 (м, 2H), 2,31-2,26 (м, 2H), 2,14-2,06 (м, 2H), 1,99-1,92 (м, 2H).
Стадия С
К раствору 4-метоксициклогексанона (1,4 г, 10,8 ммоль) стадии B добавляли гидрохлорид(3-бромфенил)гидразина (2,4 г, 10,8 ммоль) в этаноле (50 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Затем растворитель удаляли при пониженном давлении с получением названного соединения (3,1 г, количественно). Продукт использовали на следующей стадии без какой-либо очистки.
Стадия D
Раствор названного соединения стадии D (3,1 г, 10,4 ммоль) в диэтиленгликоле (12 мл) герметизировали в стеклянной пробирке для микроволновых реакций (20 мл). Затем реакционную смесь нагревали при 250°C с помощью микроволнового излучения в течение 50 минут. Реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→30/70), с получением смеси даух региоизомеров (1,6 г, 53%).
Стадия Е
К раствору названного соединения с приведенной выше стадии D (1,4 г, 4,99 ммоль) в THF (30 мл) добавляли ди-трет-бутилдикарбонат (1,3 г, 5,9 ммоль) и каталитическое количество диметиламинопиридина (5 мг). Затем реакционную смесь перемешивали в течение 1 часа. Растворитель удаляли, неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (5/95→25/75), с получением двух региоизомеров. Быстро элюируемое соединение представляло собой региоизомер A (700 мг, 37%), а медленно элюируемое соединение представляло собой региоизомер B (1,1 г, 58%).
Региоизомер A: трет-бутил 5-бром-3-метокси-3,4дигидро-1H-карбазол-9(2H)-карбоксилат:
1H-ЯМР (400 МГц, CDCl3) δ: 8,15 (д, 1H), 7,34 (д, 1H), 7,06 (т, 1H), 3,77-3,71 (м, 1H), 3,48 (с, 3H), 3,47-3,42 (м, 1H), 3,18-3,11 (м, 2H), 3,05-2,97 (м, 1H), 2,10-1,95 (м, 2H), 1,67 (с, 9H).
Региоизомер B: трет-бутил 7-бром-3-метокси-3,4-дигидро-1H-карбазол-9(2H)-карбоксилат:
1H-ЯМР (400 МГц, CDCl3) δ=8,35 (д, 1H), 7,34 (дд, 1H), 7,23 (д, 1H), 3,78-3,73 (м, 1H), 3,47 (с, 3H), 3,19-3,12 (м, 1H) 3,00-2,95 (м, 2H), 2,69-2,63 (м, 1H), 2,15-2,09 (м, 1H), 2,05-1,96 (м, 1H), 1,68 (с, 9H).
Препаративный пример 38
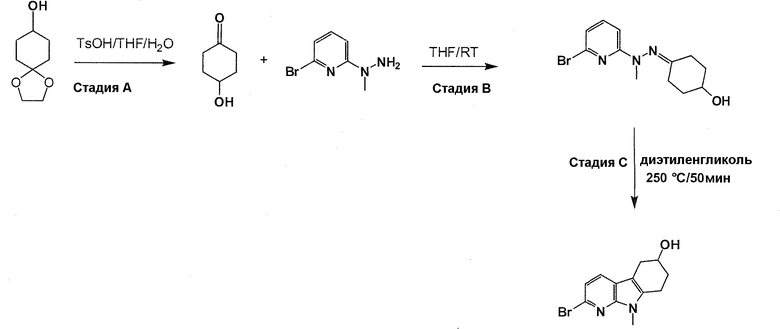
Стадия А
A раствор названного соединения (11 г, 70 ммоль) в THF:H2O (100 мл, 1:1) нагревали при 110°C в течение ночи. Реакционную смесь экстрагировали этилацетатом (250 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении с получением названного соединения (5,2 г, 65%).
Стадия В
К раствору 2-бром-6-(1-метилгидразинил)пиридина (5,2 г, 25,7 ммоль) в THF (50 мл) добавляли 4-гидроксициклогексанон (3,1 г, 27,1 ммоль). Реакционную смесь перемешивали в течение 1 часа. Растворитель удаляли при пониженном давлении с получением маслянистого соединения (7,6 г, количественно). Соединение использовали на следующей стадии без какой-либо очистки.
Стадия С
Раствор названного соединения стадии B (7,65 г, 25,7 ммоль) в диэтиленгликоле (45 мл) герметизировали в стеклянных пробирках для микроволновых реакций (20 мл). Реакционные пробирки нагревали при 250°C с помощью микроволнового излучения в течение 55 минут. Реакцию проводили с тремя загрузками. Объединенную реакционную смесь собирали и растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением неочищенного продукта, который очищали на колонке с силикагелем (этилацетат/н-гептан 20%-40%, с получением названного соединения (3,6 г, 50%).
1H-ЯМР (400 МГц, CDCl3) δ 7,58 (д, J=8,0 Гц, 1H), 7,16 (д, J=8,0 Гц, 1H), 4,29-4,28 (м, 1H), 3,69 (с, 3H), 3,08 (дд, J=15,4, 4,2 Гц, 1H), 2,93-2,85 (м, 1H), 2,80-2,66 (м, 2H), 2,17-1,99 (м, 3H).
Препаративный пример 39
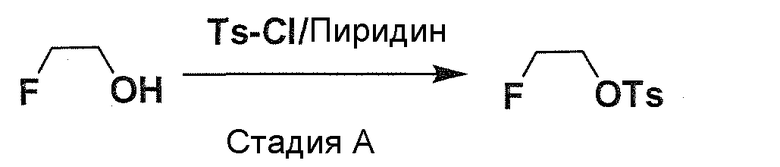
Стадия А
К раствору 2-фторэтанола (0,32 мл, 5 ммоль) в смеси пиридина и толуола (5 мл, 1:1) добавляли п-толуолсульфонилхлорид при 0°C. Реакционную смесь перемешивали в течение ночи. Реакционную смесь растворяли в EtOAc (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (5/95→30/70), с получением названного соединения в виде бесцветной жидкости (1,54 г, 67%).
1H-ЯМР (400 МГц, CDCl3) δ=7,85 (м, 2H), 7,40 (м, 2H), 4,65 (м, 1H), 4,55 (м, 1H), 4,35 (м, 1H), 4,25 (м, 1H), 2,48 (с, 3H).
Препаративный пример 40

Стадия А
К раствору названного соединения препаративного примера 38 стадии C (150 мг, 0,53 ммоль) в THF (15 мл) добавляли NaH (25 мг, 1,06 ммоль), и суспензию перемешивали в течение 5 минут. Затем добавляли раствор соединения (116 мг, 0,53 ммоль) стадии A. Реакционную смесь нагревали при 100°C в течение ночи. Реакционную смесь растворяли в EtOAc (100 мл), и промывали водой и солевым раствором и сушили над Na2SO4. Продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (10/90→20/80), с получением названного соединения (78 мг, 45%).
1H-ЯМР (400 МГц, CDCl3) δ 7,57 (д, 1H), 7,15 (д, 1H), 4,72-4,61 (м, 1H), 4,67 (т, 1H), 4,55 (т, 1H), 3,97-3,91 (м, 1H), 3,90 (т, 1H), 3,83 (т, H) 3,71 (с, 3H), 3,07 (дд, 1H), 2,93-2,88 (м, 1H), 2,80-2,72 (м, 2H), 2,22-2,20 (м, 1H), 2,14-2,05 (м, 1H).
Препаративный пример 41

Стадия А
К раствору названного соединения (5,6 г, 35,4 ммоль) в 2-йодпропане (28 мл) добавляли Ag2O (16 г, 69,2 ммоль), и реакционную смесь перемешивали в течение 4 дней. Затем реакционную смесь разбавляли диэтиловым эфиром и фильтровали, и фильтрат концентрировали при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (5/95→20/80), с получением названного соединения (2,6 г, 57% по прореагировавшему исходному реагенту).
1H-ЯМР (400 МГц, CDCl3) δ 3,95 (с, 4H), 3,80-3,59 (м, 1H), 3,48 (с, 1H), 1,81 (д, 4H), 1,76-1,61 (м, 3H), 1,56 (д, 2H), 1,15 (д, 6H).
Стадия В
К раствору названного соединения стадии A (3 г, 15,0 ммоль) в THF:H2O (20 мл, 1:1) добавляли п-толуолсульфоновую кислоту (0,5 г, 3 ммоль), и реакционную смесь нагревали при 100°C в течение ночи. Реакционную смесь экстрагировали с помощью EtOAc (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением названного соединения (2,1 г, 91%).
1H-ЯМР (400 МГц, CDCl3) δ 3,78 (д, 1H), 2,61 (с, 1H), 2,29 (с, 1H), 2,18-1,80 (м, 2H), 1,35-1,07 (м, 4H).
Стадия С
К раствору выпускаемого промышленностью 2-бром-6-(1-метилгидразинил)пиридина (620, 3,06 ммоль) в THF (5 мл) добавляли производное кетона стадии B (0,62 г, 3,06 ммоль). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде маслянистого материала (1 г, количественно). Продукт использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия D
Раствор названного соединения стадии C (1 г, 3,9 ммоль) в диэтиленгликоле (12 мл) герметизировали в стеклянной пробирке для микроволновых реакций (20 мл). Затем реакционную смесь нагревали при 245°C с помощью микроволнового излучения в течение 50 минут. Реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (10/90→50/50), с получением названного соединения (258 мг, 26%).
1H-ЯМР (400 МГц, CDCl3) δ 7,57 (д, 1H), 7,14 (д, 1H), 3,99-3,79 (м, 2H), 3,71 (с, 3H), 3,11-2,98 (м, 1H), 2,90 (дт, 1H), 2,79-2,72 (м, 1H), 2,69-2,62 (м, 1H), 2,20-2,11 (м, 1H), 2,08-1,93 (м, 1H), 1,23 (д, 3H), 1,22 (д, 3H).
Препаративный пример 42
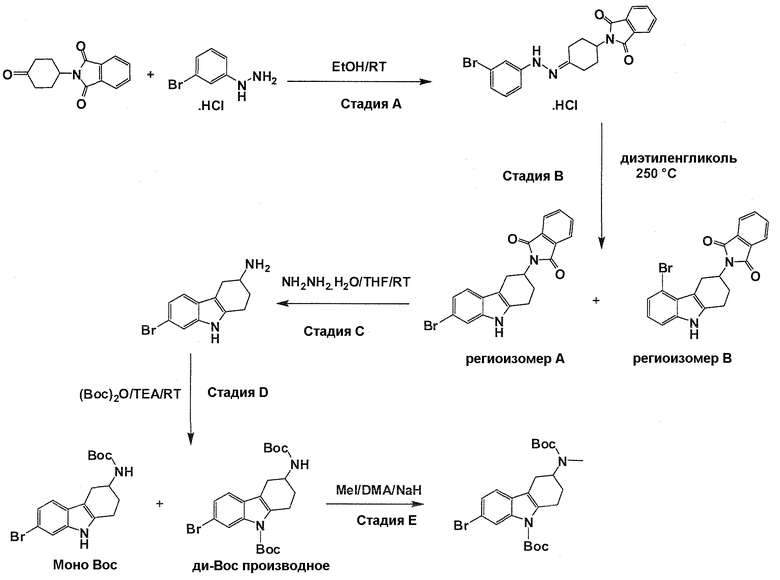
Стадия А
К раствору выпускаемого промышленностью 3-бромфенилгидразина (2,74 г, 12,2 ммоль) в этаноле (100 мл) добавляли производное кетона (3 г, 12,2 ммоль) в этаноле (50 мл). Реакционную смесь перемешивали в течение 5 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде твердого вещества (5 г, количественно). Продукт использовали на следующей стадии без какой-либо очистки.
Стадия В
Раствор названного соединения стадии A (5 г, 12,1 ммоль) в диэтиленгликоле (12 мл) герметизировали в стеклянной пробирке для микроволновых реакций (20 мл). Затем реакционную смесь нагревали при 245°C с помощью микроволнового излучения в течение 50 минут. Реакцию проводили для трех загрузок. Объединенную реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (10/90→55/45), с получением названных соединений в виде двух региоизомеров (3,3 г, 70%).
Региоизомер A (1,9 г) (2-(7-бром-2,3,4,9-тетрагидро-1H-карбазол-3-ил)изоиндолин-1,3-дион):
1H-ЯМР (400 МГц, CDCl3) δ 7,89 (м, 2H), 7,82-7,65 (м, 2H), 7,46 (с, 1H), 7,2-7,26 (м, 1H), 7,19 (д, 1H), 4,71-4,66 (м, 1H), 3,52-3,46 (м, 1H), 3,00-2,91 (м, 4H), 2,11-2,07 (м, 1H).
Региоизомер B (1,4 г) (2-(5-бром-2,3,4,9-тетрагидро-1H-карбазол-3-ил)изоиндолин-1,3-дион):
1H-ЯМР (400 МГц, CDCl3) δ 7,82 (дд, 2H), 7,70 (дд, 2H), 7,18 (д, 1H), 7,10 (д, 1H), 6,86 (т, 1H), 4,57 (м, 1H), 3,63-3,57 (м, 1H), 3,53-3,25 (м, 1H), 3,08-2,69 (м, 3H), 1,99 (д, 1H).
Стадия С
К перемешиваемому раствору региоизомера A соединения стадии B выше (980 мг, 2,48 ммоль) в THF (25 мл) добавляли NH2NH2 (2 мл). Реакционную смесь перемешивали в течение 2 дней. Твердое вещество отфильтровывали. Фильтрат концентрировали при пониженном давлении, и неочищенный продукт растворяли в DCM (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, неочищенный продукт использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия D
К неочищенному продукту с приведенной выше стадии (1,1 г) в THF (25 мл) добавляли (Boc)2O (4,5 г, 20,6 ммоль) и триэтиламин (2,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением быстро элюируемого соединения в виде ди-Boc-(650 мг) и медленно элюируемого соединения в виде моно-Boc-(830 мг) защищенного соединения.
Ди-Boc: (650 мг)
трет-бутил 7-бром-3-(трет-бутоксикарбониламино)-3,4-дигидро-1H-карбазол-9(2H)-карбоксилат:
1H-ЯМР (400 МГц, CDCl3) δ 8,35 (д, 1H), 7,35 (дд, 1H), 7,23 (д, 1H), 4,68 (с, 1H), 4,10 (с, 1H), 3,09-3,01 (м, 3H), 2,53 (дд, 1H), 2,13-2,00 (м, 1H), 2,03-1,75 (м, 1H), 1,69 (с, 9H), 1,48 (с, 9H).
Моно-Boc: (830 мг)
трет-бутил 7-бром-2,3,4,9-тетрагидро-1H-карбазол-3-илкарбамат:
1H-ЯМР (400 МГц, CDCl3) δ 7,45 (д, 1H), 7,30 (д, 3H), 7,20 (дд, 1H), 4,70 (с, 1H), 4,12 (с, 1H), 3,08 (дд, 1H), 2,85-2,81 (м, 3H), 2,57 (дд, 2H), 2,15-2,12 (м, 1H), 2,01-1,93 (м, 1H), 1,48 (с, 9H).
Стадия Е
Раствор ди-Boc-защищенного соединения с приведенной выше стадии (630 мг, 1,35 ммоль) в DMA (5 мл) охлаждали до температуры ледяной бани, и добавляли порциями NaH (65 мг, 2,7 ммоль). Суспензию перемешивали при комнатной температуре в течение 5 минут, и добавляли раствор метилйодида (380 мг, 2,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь растворяли в дихлорметане (250 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением названного соединения (435 мг, 67%).
1H-ЯМР (400 МГц, CDCl3) δ 8,34 (с, 1H), 7,35 (д, 1H), 7,23 (д, 1H), 4,50 (м, 1H), 3,28-3,24 (м, 1H), 3,08-3,0 (м, 1H), 2,87 (с, 3H), 2,75 (м, 2H), 1,98 (дд, 3H), 1,68 (с, 9H), 1,50 (с, 9H).
Препаративный пример 43
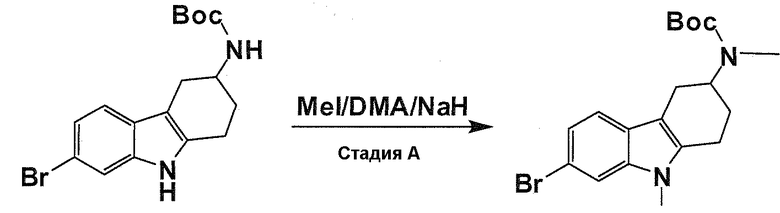
Стадия А
К раствору монозамещенного соединения препаративного примера 41 стадии D (750 мг, 2,05 ммоль) в DMA (5 мл) добавляли порциями NaH (65 мг, 2,7 ммоль). Суспензию перемешивали при комнатной температуре в течение 5 минут, и добавляли раствор метилйодида (380 мг, 2,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь растворяли в дихлорметане (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением названного соединения (455 мг, 56%).
1H-ЯМР (400 МГц, CDCl3) δ=7,40 (д, 1H), 7,31 (д, 1H), 7,18 (д, 1H), 4,50-4,33 (м, 1H), 3,59 (с, 3H), 2,88-2,74 (м, 7H), 2,08 (шир.с, 2H), 1,50 (с, 9H).
Препаративный пример 44
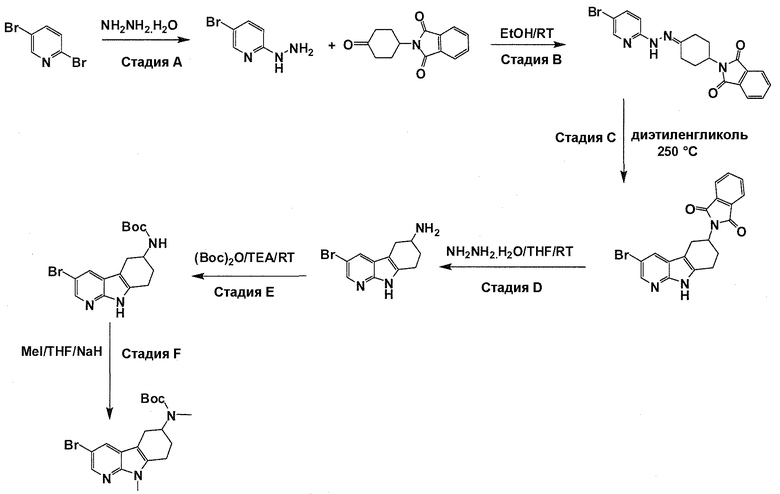
Стадия А
К раствору 2,5-дибромпиридина (15 г, 64 ммоль) добавляли NH2NH2∙H2O (20 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 дней. Растворитель удаляли при пониженном давлении, и неочищенный продукт растворяли в DCM (200 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→50/50), с получением названного соединения (5,1 г, 59% в расчете на прореагировавший исходный реагент).
1H-ЯМР (400 МГц, CDCl3) δ 8,16 (с, 1H), 7,56 (д, 1H), 6,69 (д, 1H), 6,04 (шир.с, 1H), 3,7 (шир.с, 2H).
Стадия В
К раствору названного соединения с приведенной выше стадии A (1,5 г, 6,7 ммоль) в этаноле (100 мл) добавляли производное кетона (1,6 г, 6,7 ммоль) в этаноле (50 мл). Реакционную смесь перемешивали в течение 5 часов при комнатной температуре. Растворитель удаляли с получением названного соединения в виде твердого вещества (3,1 г, количественно). Продукт использовали на следующей стадии без какой-либо очистки.
Стадия С
Раствор названного соединения с приведенной выше стадии B (3,7 г, 8,9 ммоль) в диэтиленгликоле (12 мл) герметизировали в стеклянной пробирке для микроволновых реакций (20 мл). Затем реакционную смесь нагревали при 245°C с помощью микроволнового излучения в течение 50 минут. Реакцию проводили для трех загрузок. Объединенную реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором. Органическую фазу сушили над Na2SO4, и растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→70/30), с получением названных соединений в виде двух региоизомеров (1 г, 28,5%). Продукт использовали на следующей стадии без какой-либо идентификации.
Стадия D
К перемешиваемому раствору соединения с приведенной выше стадии C (1,2 мг, 3,0 ммоль) в THF (50 мл) добавляли NH2NH2 (5 мл). Реакционную смесь перемешивали в течение 2 дней. Твердое вещество отфильтровывали. Фильтрат концентрировали при пониженном давлении, и неочищенный продукт растворяли в DCM (200 мл). Органическую фазу промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, неочищенный продукт (890 мг) использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия Е
К неочищенному продукту с приведенной выше стадии D (0,89 г) в THF (20 мл) добавляли (Boc)2O (2 г, 9,1 ммоль) и триэтиламин (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→60/40), с получением названного соединения (0,77 г, 24% суммарный выход за три стадии).
1H-ЯМР (400 МГц, CDCl3) δ 11,50 (с, 1H), 8,12 (д, 1H), 7,98 (д, 1H), 7,01 (д, 1H), 3,78-3,69 (м, 1H), 3,32 (с, 3H), 2,91-2,86 (м, 1H), 2,77-2,70 (м, 2H), 2,47-2,41 (м, 1H), 2,01-1,97 (м, 1H), 1,79-1,69 (м, 1H), 1,41 (с, 9H).
Стадия F
К раствору Boc-защищенного соединения с приведенной выше стадии E (700 мг, 1,9 ммоль) в DMA (10 мл) добавляли порциями NaH (180 мг, 7,5 ммоль). Суспензию перемешивали при комнатной температуре в течение 10 минут, и добавляли раствор метилйодида (1,2 г, 8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь растворяли в этилацетате (250 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (25/75→80/20), с получением названного соединения (435 мг, 67%).
1H-ЯМР (400 МГц, ДМСО) δ 8,20 (д, 1H), 8,06 (д, 1H), 4,26 (м, 1H), 3,65 (с, 3H), 3,00-2,88 (м, 2H), 2,79 (с, 3H), 2,73-2,70 (м, 2H), 2,04-1,99 (м, 2H), 1,42 (с, 9H).
Препаративный пример 45
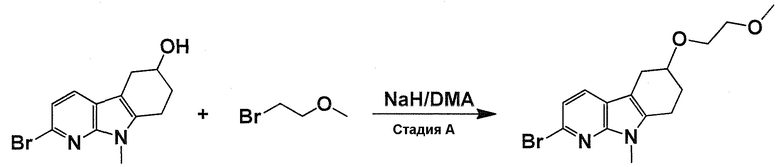
Стадия А
К раствору названного соединения (0,25 г, 0,88 ммоль) в DMA (5 мл) добавляли NaH (60 мг, 2,5 ммоль), и суспензию перемешивали в течение 5 минут. Затем добавляли раствор 1-бром-2-метоксиэтана (245 мг, 1,77 ммоль). Реакционную смесь перемешивали в течение 3 часов при 50°C на песочной бане. Реакционную смесь растворяли в EtOAc (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (20/80→80/20), с получением названного соединения (0,255 г, 85%).
1H-ЯМР (400 МГц, CDCl3) δ 7,56 (д, 1H), 7,14 (д, 1H), 3,91-3,85 (м, 1H), 3,77-3,74 (м, 2H), 3,70 (с, 3H), 3,59 (т, 2H), 3,42 (с, 3H), 3,07 (дд, 1H), 2,93-2,86 (м, 1H), 2,78-2,69 (м, 2H), 2,24-2,21 (м, 1H), 2,10-2,01 (м, 1H).
Препаративный пример 46

Стадия А
К раствору названного соединения препаративного примера 28 стадии G (400 мг, 0,858 ммоль) в DMA (5 мл) добавляли NaH (31 мг, 1,29 ммоль). Суспензию перемешивали в течение 5 минут при комнатной температуре, и добавляли 1-бром-2-метоксиэтан (120 мг, 0,869 ммоль). Реакционную смесь нагревали при 50°C в течение ночи. Реакционную смесь охлаждали, и растворяли в этилацетате (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (1/99→20/80), с получением названного соединения (150 мг, 41%).
1H-ЯМР (400 МГц, CDCl3) δ 7,50 (д, 1H), 7,10 (д, 1H), 4,69 (д, 1H), 4,38-4,14 (м, 2H), 4,04 (с, 1H), 3,65 (т, 2H), 3,24 (с, 3H), 3,01 (дд, 1H), 2,85-2,82 (м, 1H), 2,50 (дд, 1H), 2,19-2,06 (м, 1H), 1,95-1,86 (м, 1H), 1,42 (с, 9H).
Стадия В
К раствору моно-Boc-производного из приведенной выше стадии (80 мг, 0,188 ммоль) в DMA (2 мл) добавляли NaH (7 мг, 0,28 ммоль). К этой суспензии добавляли метилйодид (39 мг, 0,28 ммоль). Реакционную смесь перемешивали в течение 2 часов. Затем реакционную смесь растворяли в этилацетате (150 мл) и промывали водой и солевым раствором и сушили над Na2SO4. Растворитель удаляли, и неочищенный продукт очищали на колонке с силикагелем, используя систему очистки Biotage Isolera One с градиентом EtOAc/н-гептан (10/90→60/40), с получением названного соединения (70 мг, 85%).
1H-ЯМР (400 МГц, CDCl3) δ 7,53 (д, 1H), 7,13 (д, 1H), 4,49-4,24 (м, 4H), 3,70-3,67 (м, 2H), 3,27 (с, 3H), 3,27-2,70 (м, 6H), 2,04-2,03 (м, 2H), 1,48 (с, 9H).
Препаративный пример 47

Стадия А
К раствору LDA 1,8M (33,0 мл, 59,3 ммоль) в тетрагидрофуране (150 мл) при -75°C добавляли выпускаемый промышленностью 2-фторпиридин (4,25 мл, 49,4 ммоль). Смесь перемешивали в течение 4 часов при этой температуре. К полученной суспензии добавляли этилтрифторацетат (7,08 мл, 59,3 ммоль), при этом во время добавления внутренняя температура не должна подниматься выше -45°C. Реакционную смесь подогревали до комнатной температуры. Добавляли нитрометан (5,31 мл, 99 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Суспензию разбавляли этилацетатом (100 мл) и 50 мл 1,2M раствора HCl. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток суспендировали в дихлорметане и фильтровали с получением названного соединения в виде белых кристаллов (6,14 г). Маточный раствор концентрировали досуха и очищали флэш-хроматографией в смеси этилацетат/н-гептан (20/80-35/65) с получением дополнительного количества материала (4,45 г). суммарный выход составлял 10,59 г (84%).
Стадия В
Названное соединение стадии A выше (4,45 г, 17,51 ммоль) растворяли в этаноле (50 мл) и перемешивали в атмосфере водорода (0,141 г, 70,0 ммоль) в присутствии гидрат оксида платины(IV) (0,398 г, 1,751 ммоль). После расходования теоретического количества водорода, раствор фильтровали, и фильтрат кипятили с обратным холодильником в течение ночи. Затем растворитель удаляли при пониженном давлении. Названное соединение получали после хроматографической очистки с градиентом дихлорметан/метанол (от 98/2 до 9/1) с получением названного соединения (1,2 г, 34%).
MS (ESI); m/z=204,86 (MH+).
Стадия С
К раствору названного соединения стадии В выше (1,2 г, 5,88 ммоль) в тетрагидрофуране (15 мл) и пиридине (0,951 мл, 11,76 ммоль) добавляли тионилхлорид (0,858 мл, 11,76 ммоль) при 0°C. Полученную смесь перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь выливали в ледяную воду. Смесь нейтрализовывали (pH 6) с помощью насыщенного раствора карбоната натрия. Водный слой подвергали экстракции дихлорметаном. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаточный пиридин удаляли путем добавления толуола и концентрировали досуха 3 раза с получением названного соединения в виде коричневого твердого вещества (1,13 г, 100%).
MS (ESI); m/z=186,88 (MH+).
Стадия D
К раствору 3-хлорпербензойной кислоты (1,571 г, 9,11 ммоль) в тетрагидрофуране (50 мл) добавляли порциями при 0°C названное соединение стадии С выше (1,130 г, 6,07 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали досуха. Неочищенный продукт растирали в диэтиловом эфире и затем фильтровали (это повторяли несколько раз). Маточный раствор концентрировали досуха и очищали флэш-хроматографией, используя смесь дихлорметан/метанол, с получением названного соединения (1,37 г, 67%).
MS (ESI); m/z=202,84 (MH+).
Стадия Е
К раствору названного соединения стадии D выше (0,513 г, 1,430 ммоль) в сухом толуоле (30 мл) добавляли одновременно раствор гексаметилдисилазан (0,6 мл, 2,86 ммоль) в толуоле и раствор бензоилбромида (0,421 мл, 3,58 ммоль) в толуоле. Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали досуха. Остаток очищали флэш-хроматографией, используя смесь этилацетат/н-гептан (от 15/85 до 35/65), с получением названного соединения в виде белого твердого вещества (0,190 г, 36%).
Стадия F
К раствору названного соединения стадии Е выше (0,19 г, 0,515 ммоль) в метаноле (10 мл) добавляли 1M водный раствор гидроксида натрия (1,544 мл, 1,544 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 10 часов. Метанол затем удаляли при пониженном давлении. Остаток растворяли в этилацетате и насыщенном растворе бикарбоната натрия. Органический слой отделяли, сушили над Na2SO4 и концентрировали досуха с получением названного соединения в виде белого твердого вещества (0,122 г, 89%).
Стадия G
К раствору названного соединения стадии F выше (0,122 г, 0,460 ммоль) в сухом тетрагидрофуране (10 мл) добавляли порциями при 0°C 60% гидрид натрия (0,019 г, 0,483 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут, и затем добавляли триизопропилсилилхлорид (0,099 мл, 0,460 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали досуха. Остаток разбавляли этилацетатом. Экстракцию осуществляли с помощью насыщенного раствора бикарбоната и солевого раствора. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт очищали флэш-хроматографией, используя смесь этилацетат/н-гептан (от 15/85 до 40/60), с получением названного соединения в виде белого твердого вещества (0,188 г, 99%).
1H-ЯМР (400 МГц, CDCl3): δ=1,12 (д, 18H); 1,82 (h, 3H); 3,91 (с, 3H); 7,31 (д, 1H); 7,94 (с, 1H); 8,21 (д, 1Н).
13С-ЯМР Депт135 (400 МГц, CDCl3): δ=11,89; 17,99; 51,31; 121,62; 131,43; 137,75.
Препаративный пример 48

Стадия А
Смесь 0,6 мл ~70% раствора фтористого водорода в пиридине и дихлорметане (10 мл) охлаждали до -10°C, и добавляли по каплям раствор выпускаемого промышленностью эпоксида (2 г, 9,38 ммоль) в дихлорметане (10 мл). Затем смесь перемешивали при комнатной температуре в течение 4 часов, разбавляли дихлорметаном и промывали с помощью насыщенного водного раствора карбоната натрия. Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли с получением остатка, который очищали с помощью хроматографии на диоксиде кремния с градиентом этилацетат/н-гептан (20/80→50/50), с получением названного соединения в виде желтого масла (1,44 г, 66%).
1H-ЯМР (400 МГц, CDCl3): δ=1,44 (с, 9H), 1,47-1,63 (м, 2H), 1,85 (м, 2H), 3,08 (м, 2H), 3,56 (с, 1H), 3,61 (с, 1H), 3,92 (шир.с, 2H).
MS (ESI); m/z=233,98 (MH+).
Стадия В
Названное соединение стадии А выше (1,44 г, 6,17 ммоль) растворяли в пиридине (6 мл), и раствор охлаждали до 0°C. Добавляли п-толуолсульфонилхлорид (1,29 г, 6,79 ммоль), и смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь разбавляли дихлорметаном (250 мл) и промывали водой (50 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли. Остаток очищали, используя флэш-хроматографическую систему Biotage (этилацетат/н-гептан: 20/80→50/50), с получением названного соединения в виде желтого масла (2,2 г, 92%).
1H-ЯМР (400 МГц, CDCl3): δ=1,47 (с, 9H), 1,49-1,63 (м, 4H), 1,80 (м, 2H), 2,46 (с, 3H), 3,03 (м, 2H), 3,96 (шир.с, 2H), 7,36 (д, 2H), 7,79 (д, 2H).
Стадия С
Названное соединение стадии В выше (2,2 г, 5,68 ммоль) растворяли в Ν,Ν'-диметилформамиде (22 мл). Добавляли фталимид калия (1,052 г, 5,68 ммоль), и смесь нагревали при 150°C в течение 12 часов. После охлаждения до комнатной температуры, добавляли воду (100 мл), и смесь экстрагировали этилацетатом (3×250 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением названного соединения в виде белого твердого вещества (2,45 г), которое непосредственно использовали на следующей стадии.
MS (ESI); m/z=362,9 (MH+).
Стадия D
Названное соединение стадии С выше (2,45 г) суспендировали в этаноламине (6 мл). Смесь нагревали при 60°C в течение 2,5 часов. После охлаждения до комнатной температуры, добавляли воду (50 мл), и смесь экстрагировали этилацетатом (3×250 мл). Органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли. Остаток очищали, используя флэш-хроматографическую систему Biotage (метанол/дихлорметан: 20/80→50/50), с получением названного соединения в виде желтого масла (0,67 г, 46% за 3 стадии).
1H-ЯМР (400 МГц, CDCl3): δ=1,44 (с, 9H), 1,56 (м, 4H), 1,84 (м, 2H), 3,06 (м, 2H), 3,94 (шир.с, 2H).
MS (ESI): m/z=177,04 (M-t-Bu+H+), 217,96 (M-Me+H+), 233,01 (M+H+).
Препаративный пример 49
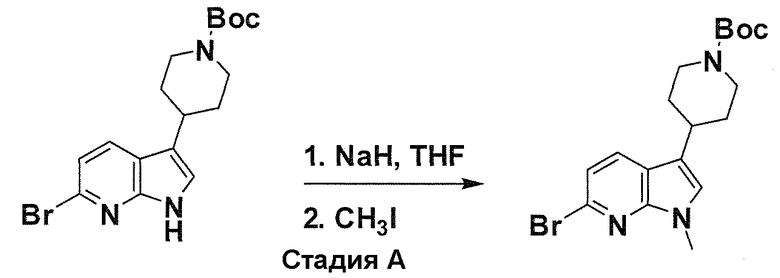
Стадия А
К раствору названного соединения препаративного примера 14, стадии D (1 г, 2,63 ммоль) в сухом тетрагидрофуране (25 мл) добавляли порциями при 0°C гидрид натрия (0,111 г, 2,89 ммоль, 60% в минеральном масле). Смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли метилйодид (0,491 мл, 7,89 ммоль). Раствор концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан (от 15% до 40%) с получением названного соединения в виде белого твердого вещества (0,98 г, 95%).
MS ESI: 394,48/396,48 (M+H).
Препаративный пример 50

Стадия А
1,2-диметоксиэтан (3 мл) добавляли к смеси названного соединения препаративного примера 3 (0,021 г, 0,156 ммоль), названного соединения препаративного примера 1, стадии D (0,050 г, 0,141 ммоль), хлороформенного комплекса трис(дибензилиденацетон)дипалладия (0,013 г, 0,014 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантена (Xantphos, 0,008 г, 0,014 ммоль) и карбоната цезия (0,09 г, 0,283 ммоль). Реакционную смесь дегазировали в течение 3 минут при комнатной температуре в атмосфере аргона и подвергали обработке ультразвуком. Колбу затем подогревали до 100°C в течение 1 часа. Реакционную смесь экстрагировали этилацетатом и солевым раствором. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали флэш-хроматографией, используя смесь этилацетат/н-гептан, с получением названного соединения в виде белого твердого вещества (0,07 г, 12%).
MS ESI: 406,06 (M+H).
Препаративный пример 51
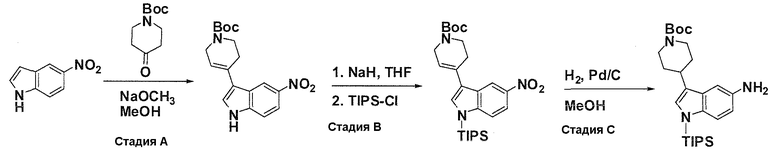
Стадия А
К раствору выпускаемого промышленностью 5-нитроиндола (3,4 г, 20,9 ммоль) и 1-Boc-4-пиперидона (6 г, 31,3 ммоль) в метаноле (50 мл) добавляли (28%) метоксид натрия в метаноле (10 мл), и реакционную смесь нагревали при 80°C в течение 2 дней. Осажденный продукт отфильтровывали и сушили с получением названного соединения в виде желтого твердого вещества (5,1 г, 72%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,42 (с, 9H), 3,2 (м, 2H), 3,56 (т, 2H), 4,0 (м, 2H), 6,17 (с, 1H), 7,54 (д, 1H), 7,69 (с, 1H), 8,00 (д, 1H), 8,69 (с, 1H).
Стадия В
К раствору названного соединения стадии А выше (4 г, 11,6 ммоль) в тетрагидрофуране (150 мл) добавляли порциями гидрид натрия (0,42 г, 17,4 ммоль). Окрашенную в темно-красный цвет суспензию перемешивали при комнатной температуре в течение 10 минут. Затем добавляли триизопропилсилилхлорид (2,23 г, 11,6 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали с помощью воды, и растворители удаляли. Остаток суспендировали в этилацетате (200 мл), и исходный реагент удаляли фильтрацией. Фильтрат концентрировали, и остаток очищали на колонке с силикагелем (этилацетат/н-гептан (20/80)→(60/40)) с получением названного соединения (2 г, 66%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,14 (д, 18H), 1,51 (с, 9H), 1,67-1,71 (м, 3H), 3,69-3,75 (м, 4H), 4,17 (с, 2H), 6,18 (с, 1H), 7,30 (с, 1H), 7,49 (д, 1H), 8,06 (д, 1H), 8,78 (с, 1H).
Стадия С
К раствору названного соединения стадии В выше (2 г, 4 ммоль) в этилацетате (150 мл) добавляли 10% Pd/C (0,5 г). Колбу вакуумировали и насыщали газообразным водородом. Затем реакционную смесь перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали и сушили с получением неочищенного продукта, который очищали на колонке с силикагелем, используя смесь этилацетат/н-гептан (20/80→50/50), с получением названного соединения (1,31 г, 69%).
1H-ЯМР (400 МГц, CDCl3): δ=1,14 (д, 18H), 1,51 (с, 9H), 1,61-1,69 (м, 5H), 2,02-2,05 (м, 2H), 2,89-2,91 (м, 3H), 4,23-4,24 (м, 2H), 6,68-6,70 (м, 1H), 6,92 (с, 1H), 6,99 (д, 1H), 7,30 (д, 1H).
Препаративный пример 52

Стадия А
К раствору названного соединения препаративного примера 33 стадии F (0,150 г, 0,534 ммоль) в сухом тетрагидрофуране (объем: 10 мл) добавляли порциями 60% гидрид натрия (0,022 г, 0,560 ммоль). Через 30 минут при комнатной температуре добавляли по каплям d3-йодметан (0,040 мл, 0,640 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан от 15% до 50% с получением ожидаемого соединения в виде желтоватого твердого вещества (0,95 г, 60%).
MS (ESI); m/z=298,65/300,68 (MH+).
Препаративный пример 53

Стадия А
К перемешиваемому этанолу (45 мл) добавляли этилкарбетокси-4-пиперидон (8,81 мл, 58,4 ммоль) и гидрохлорид 4-бромфенилгидразина (13,06 г, 58,4 ммоль). Полученную смесь подогревали до 140°C в течение 2 часов, затем перемешивали при комнатной температуре в течение ночи. Суспензию фильтровали, и осадок на фильтре промывали смесью EtOH/вода 1:1. Осадок сушили при 120°C в течение 1 часа с получением названного соединения в виде серовато-белого твердого вещества (12,75 г, 67%).
MS (ESI); m/z=323,54/325,54 (MH+).
Стадия В
К раствору названного соединения стадии A (2,5 г, 7,74 ммоль) в тетрагидрофуране (75 мл) добавляли порциями 60% гидрид натрия (0,311 г, 8,12 ммоль) при 0°C. Через 30 минут при комнатной температуре добавляли по каплям йодметан (0,532 мл, 8,51 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан (от 15 до 35%) с получением названного соединения в виде белого твердого вещества (2,52 г, 97%).
MS (ESI); m/z=337,66/339,61 (MH+).
Препаративный пример 54
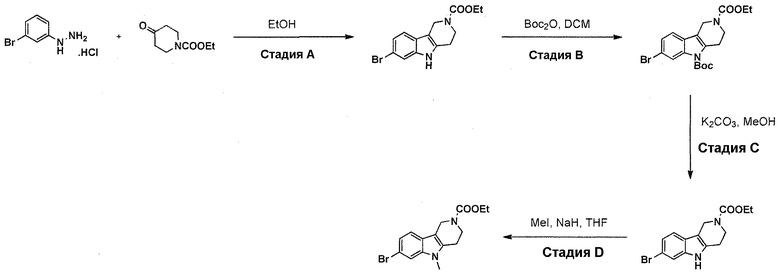
Стадия А
К перемешиваемому этанолу (45 мл) добавляли этилкарбетокси-4-пиперидон (4,41 мл, 29,2 ммоль) и гидрохлорид 3-бромфенилгидразина (6,53 г, 29,2 ммоль). Полученную смесь подогревали до 140°C в течение 12 часов, затем перемешивали при комнатной температуре в течение ночи. Суспензию фильтровали, и осадок на фильтре промывали смесью EtOH/вода 1:1 с получением названного продукта в виде смеси 2 изомеров (2,98 г, 32%).
Стадия В
К раствору названного соединения стадии A (2,979 г, 9,22 ммоль) в дихлорметане (150 мл) добавляли DMAP (0,056 г, 0,461 ммоль) и затем Boc2O (2,68 мл, 11,52 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали досуха. Остаток очищали флэш-хроматографией (2×), используя смесь этилацетат/н-гептан (от 10% до 20%) с получением названного соединения в виде белого твердого вещества (1,82 г, 47%).
1H-ЯМР (400 МГц, CDCl3): δ=1,30 (т, J=8,0 Гц, 3H); 1,66 (с, 9H); 3,08 (с, 2H); 3,79 (с, 2H); 4,20 (кв, J=8,0 Гц, 2H); 4,59 (с, 2H); 7,23 (д, J=8,0 Гц, 1H); 7,34 (дд, J1=1,6 Гц, J2=8,0 Гц, 1H); 8,36 (с, 1H).
Стадия С
К суспензии названного соединения стадии В (1 г, 2,362 ммоль) в метаноле (25 мл) добавляли карбонат калия (0,979 г, 7,09 ммоль). Полученную смесь кипятили с обратным холодильником в течение 3 часов. Реакционную смесь концентрировали досуха. Остаток экстрагировали этилацетатом и солевым раствором. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт очищали флэш-хроматографией во смесью этилацетат/н-гептан с получением названного соединения в виде белого твердого вещества (0,746 г, 98%).
MS (ESI); m/z=323,60/325,61 (MH+).
Стадия D
К раствору названного соединения стадии С (0,746 г, 2,308 ммоль) в сухом тетрагидрофуране (25 мл) добавляли порциями при 0°C 60% гидрид натрия (0,101 г, 2,424 ммоль). Через 30 минут перемешивания при комнатной температуре, добавляли по каплям йодметан (0,166 мл, 2,65 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 часов. Полученную смесь концентрировали досуха, и неочищенный продукт очищали флэш-хроматографией со смесью этилацетат/н-гептан с получением ожидаемого соединения в виде белого твердого вещества (0,657 г, 84%).
MS (ESI); m/z=337,66/339,56 (MH+).
Препаративный пример 55
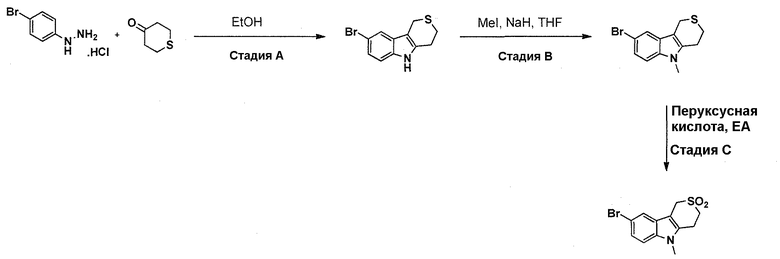
Стадия А
К смеси (4-бромфенил)гидразина, HCl (1 г, 4,47 ммоль) и дигодро-2H-тиопиран-4(3H)-она (0,520 г, 4,47 ммоль) добавляли абсолютный этанол (объем: 20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Полученную суспензию концентрировали досуха с получением соответствующего гидразона. Твердое вещество помещали в пробирки для проведения микроволновых реакций и суспендировали в этаноле (объем: 15 мл). Полученную суспензию нагревали с помощью микроволнового излучения до 125°C в течение 25 минут. Реакционную смесь добавляли по каплям в воду при интенсивном перемешивании. Полученную бежевую суспензию фильтровали фильтровали и затем сушили на воздухе в течение ночи. Неочищенный продукт выкристаллизовывали в этаноле. Оставшийся продукт в маточном растворе извлекали флэш-хроматографией со смесью DCM/MeOH от 2% до 5%. В результате получали названное соединение в виде белого твердого вещества (4,16 г, 87%).
1H-ЯМР (400 МГц, CDCl3): δ=3,02 (с, 4H); 3,82 (с, 2H); 7,16 (д, J=8,4 Гц, 1H); 7,24 (дд, J1=1,6 Гц, J2=8,4 Гц, 1H); 7,58 (д, J=2,0 Гц, 1H); 7,82 (с, 1H).
13C-ЯМР (400 МГц, CDCl3): δ=22,53; 25,17; 25,57; 111,89; 120,28; 124,42.
Стадия В
К раствору названного соединения стадии А (0,809 г, 3,02 ммоль) в сухом тетрагидрофуране (объем: 50 мл) добавляли при 0°C 60% гидрид натрия (0,139 г, 3,32 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли йодметан (0,283 мл, 4,53 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 12 часов. Смесь концентрировали досуха, и неочищенный продукт очищали флэш-хроматографией в смеси этилацетат/н-гептан 10-30% с получением названного соединения в виде желтого твердого вещества (0,811 г, 95%).
Стадия С
К раствору названного соединения стадии В (0,5 г, 1,772 ммоль) в этилацетате (объем: 4 мл) добавляли по каплям перуксусную кислоту (0,706 мл, 3,72 ммоль) при 0°C. Светло-желтую суспензию перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали досуха. Неочищенный продукт очищали флэш-хроматографией в смеси этилацетат/н-гептан от 40% до 65% с получением ожидаемого соединения в виде желтоватого твердого вещества (0,320 г, 57%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=3,31 (т, J=6,4 Гц, 2H); 3,52 (т, J=6,4 Гц, 2H); 3,66 (с, 3H); 4,46 (с, 2H); 7,26 (дд, J1=2,0 Гц, J2=8,8 Гц, 1H); 7,43 (д, J=8,8 Гц, 1H); 7,68 (д, J=2,0 Гц, 1H).
Препаративный пример 56

Стадия А
К смеси (3-бромфенил)гидразина, HCl (1 г, 4,47 ммоль) и дигидро-2H-тиопиран-4(3H)-она (0,520 г, 4,47 ммоль) добавляли абсолютный этанол (объем: 20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Полученную суспензию концентрировали досуха с получением соответствующего гидразона. Твердое вещество помещали в пробирки для проведения микроволновых реакций и суспендировали в этаноле (объем: 15 мл). Полученную суспензию нагревали с помощью микроволнового излучения до 125°C в течение 25 минут. Смесь добавляли по каплям в воду при интенсивном перемешивании. Проводили экстракцию в DCM в присутствии 1M HCl и воды. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт очищали флэш-хроматографией в смеси DCM/MeOH 95:5 с получением названного соединения в виде бежевого твердого вещества (0,328 г, 27%).
1H-ЯМР (400 МГц, CDCl3): δ=3,02 (с, 4H); 3,84 (с, 2H); 7,13-7,38 (м, 2H); 7,43 (с, 1H); 7,78 (с, 1H).
Стадия В
К раствору названного соединения стадии А (0,298 г, 1,111 ммоль) в сухом тетрагидрофуране (объем: 20 мл) добавляли при 0°C 60% гидрид натрия (0,0488 г, 1,167 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли йодметан (0,076 мл, 1,222 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 12 часов. Смесь концентрировали досуха, и неочищенный продукт очищали флэш-хроматографией в смеси этилацетат/н-гептан 10-30% с получением названного соединения в виде белого твердого вещества (0,310 г, 99%).
Стадия С
К раствору названного соединения стадии В (0,3 г, 1,063 ммоль) в этилацетате (соотношение: 1,000, объем: 20 мл) добавляли по каплям раствор перуксусной кислоты (0,403 мл, 2,126 ммоль) в этилацетате (соотношение: 1,000, объем: 20,00 мл) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь экстрагировали водой и насыщенным раствором Na2CO3. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан от 40% до 65% с получением ожидаемого соединения в виде белого твердого вещества (0,160 г, 48%).
1H-ЯМР (400 МГц, CDCl3): δ=3,35 (с, 4H); 3,62 (с, 3H); 4,33 (с, 2H); 7,21 (с, 2H); 7,42 (с, 1H).
Препаративный пример 57

Стадия А
К охлаждаемому льдом раствору 4-(метилтио)бутан-1-ола (2,5 г, 20,80 ммоль) в сухом дихлорметане (объем: 250 мл) добавляли порциями мета-хлорпероксибензойную кислоту (mCPBA; 9,32 г, 41,6 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Раствор концентрировали досуха. Остаток растворяли в смеси диэтилового эфира и воды. Бензойную кислоту экстрагировали в диэтиловый эфир. Водный слой концентрировали досуха. Остаток разбавляли с помощью DCM, и концентрированный раствор загружали в картридж Samplet для системы флэш-хроматографической очистки. Неочищенный продукт очищали флэш-хроматографией в смеси DCM/MeOH от 3 до 10% с получением названного соединения в виде бесцветного масла (2,95 г, 93%).
1H-ЯМР (400 МГц, CDCl3): δ=1,59-1,75 (м, 2H); 1,85-2,01 (м, 2H); 2,67 (с, 1H); 2,90 (с, 3H); 3,06 (дд, J1=J2=8,0 Гц, 2H); 3,64 (кв, J=5,6 Гц, 2H).
13C-ЯМР Депт135 (400 МГц, CDCl3): δ=22,17; 33,89; 43,46; 57,39; 64,50.
Стадия В
К смеси названного соединения стадии A (2,95 г, 19,38 ммоль), трифенилфосфина (7,62 г, 29,1 ммоль) и имидазола (1,979 г, 29,1 ммоль) в дихлорметане (объем: 250 мл) добавляли по каплям раствор йода (7,38 г, 29,1 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь концентрировали до половины объема, затем добавляли воду. Суспензию фильтровали, и проводили экстракцию раствором Na2SO3. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан от 40% до 60% с получением ожидаемого соединения в виде белого твердого вещества (1,2 г, 24%).
1H-ЯМР (400 МГц, CDCl3): δ=1,93-2,10 (м, 4H); 2,94 (с, 3H); 3,05 (т, J=7,6 Гц, 2H); 3,23 (т, J=6,2 Гц, 2H).
13C-ЯМР Депт135 (400 МГц, CDCl3): δ=4,84; 23,47; 31,56; 40,66 (CH3); 53,45.
Препаративный пример 58
Следуя методике, описанной в препаративном примере 54, за исключением того, что использовали пропил-производные, указанные на схеме реакции ниже, получали следующее соединение.

Выход: 72%
1H-ЯМР (400 МГц, CDCl3): δ=2,26-2,40 (м, 2H); 2,91 (с, 3H); 3,12 (т, J=7,6 Гц, 2H); 3,28 (т, J=6,8 Гц, 2H).
13C-ЯМР (400 МГц, CDCl3): δ=3,11; 26,00; 41,11 (CH3); 55,21.
Препаративный пример 59

Стадия А
К смеси дигидро-2H-тиопиран-4(3H)-она (5 г, 43,0 ммоль) в ацетоне (объем: 100 мл) добавляли оксон (52,9 г, 86 ммоль). Полученную смесь интенсивно перемешивали при комнатной температуре в течение 12 часов. Суспензию фильтровали, и твердое вещество промывали ацетоном. Органический слой концентрировали досуха с получением названного соединения в виде белого твердого вещества (5,83 г, 91%).
Стадия В
К смеси названного соединения стадии A (2 г, 13,50 ммоль) в метаноле (объем: 50 мл) добавляли при 0°C боргидрид натрия (0,511 г, 13,50 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Раствор концентрировали досуха. Остаток очищали флэш-хроматографией в смеси DCM/MeOH от 0% до 5% с получением названного соединения в виде белого твердого вещества (1,74 г, 86%).
13C-ЯМР Депт135 (400 МГц, CDCl3): δ=31,18; 46,74; 62,57.
Стадия С
К смеси названного соединения стадии B (1,0 г, 6,66 ммоль), трифенилфосфина (2,62 г, 9,99 ммоль) и имидазола (0,680 г, 9,99 ммоль) в дихлорметане (объем: 50 мл) добавляли по каплям раствор йода (2,53 г, 9,99 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 24 часов. Реакционную смесь фильтровали, затем концентрировали досуха. Остаток очищали флэш-хроматографией в смеси этилацетат/н-гептан от 40% до 75% с получением ожидаемого соединения в виде белого твердого вещества (0,684 г, 39%).
1H-ЯМР (400 МГц, CDCl3): δ=2,40-2,53 (м, 4H); 2,93-3,06 (м, 2H); 3,25-3,42 (м, 2H); 4,56-4,70 (м, 1H).
13C-ЯМР Депт135 (400 МГц, CDCl3): δ=24,74; 35,35; 50,40.
Препаративный пример 60

Стадия А
Высушенную в сушильном шкафу сосуд Шленка вакуумировали и заполняли аргоном. Процедуру повторяли 3-4 раза. Добавляли при комнатной температуре с помощью шприца диоксан (3 мл) и дегазировали путем барботирования через смесь аргона. Затем добавляли вместе 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил (XPhos, 0,054 г, 0,112 ммоль) и ацетат палладия(II) (0,009 г, 0,037 ммоль). Смесь нагревали при 110°C в течение 1 минуты. Реакционная смесь превращалась в прозрачный раствор красного цвета. Затем добавляли вместе в атмосфере аргона названное соединение препаративного примера 3 (0,050 г, 0,375 ммоль), выпускаемый промышленностью 6-бром-1-(триизопропилсилил)-1H-пирроло[3,2-b]пиридин (0,132 г, 0,375 ммоль) и трет-бутоксид натрия (0,12 г, 1,25 ммоль). Реакционную смесь нагревали при 110°C в течение 2 часов. Реакционную смесь разбавляли этилацетатом (150 мл). Органическую фазу промывали водой, солевым раствором, и сушили над Na2SO4. Растворитель удаляли, и остаток очищали с помощью хроматографии на диоксиде кремния, используя этилацетат, с получением названного соединения (0,045 г, 30%).
1H-ЯМР (400 МГц, CDCl3): δ=1,13 (д, 18H), 1,61-1,68 (м, 3H), 6,35 (дд, 1H), 6,60 (д, 1H), 6,80 (д, 1H), 6,94 (дд, 1H), 7,10 (шир.с, 1H), 7,39 (д, 1H), 7,73 (д, 1H), 8,20 (с, 1H), 8,54 (с, 1H), 9,27 (с, 2H).
Препаративные примеры 61-140
Следуя методике проведения реакции сочетания на Pd, описанной в препаративном примере 57, за исключением того, что использовались указанные в таблице 1 бромпроизводные и амины, были получены следующие соединения (см. в конце описания).
Препаративный пример 142

Стадия А
К раствору названного соединения препаративного примера 75 (0,120 г, 0,2 ммоль) в THF (1 мл) добавляли тетрабутиламмоний (0,052 г, 0,2 ммоль). Реакционную смесь перемешивали в течение 1 часа. Затем реакционную смесь концентрировали. Неочищенный продукт очищали на колонке с силикагелем (1:1, EtOAc:гептан) с получением названного соединения (0,08 г, 94%).
1H-ЯМР (400 МГц, CDCl3): δ=1,48 (с, 9H), 1,68-1,71 (м, 2H), 2,01-2,17 (м, 2H), 2,89-2,95 (м, 3H), 4,23-4,25 (м, 2H), 6,65 (м, 1H), 6,83-6,89 (м, 2H), 6,98-6,99 (м, 1H), 7,10 (с, 1H), 7,52 (д, 1H), 7,91 (с, 1H).
Стадия В
К раствору названного соединения стадии А выше (0,08 г, 0,187 ммоль) в THF (3 мл) добавляли гидрид натрия (0,007 г, 0,28 ммоль), затем добавляли метилйодид (0,026 г, 0,187 ммоль), и полученную реакционную смесь перемешивали в течение 1 часа. Затем реакцию останавливали с помощью воды и экстрагировали этилацетатом (3×50 мл). Органическую фазу промывали солевым раствором и сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали на колонке с силикагелем (EtOAc:гептан; 40:60) с получением названного соединения (0,025 г, 30%).
1H-ЯМР (400 МГц, CDCl3): δ=1,51 (с, 9H), 1,66-1,72 (м, 2H), 2,02-2,05 (м, 2H), 2,88-2,99 (м, 3H), 3,70 (с, 3H), 4,24-4,27 (м, 2H), 6,66 (д, 1H), 6,77 (с, 1H), 6,81-6,88 (м, 2H), 6,99-7,06 (м, 2H), 7,54 (д, 1H).
Препаративные примеры 143 и 144

Стадия А
К раствору названного соединения препаративного примера 69 (0,3 г, 0,323 ммоль) в THF (3 мл) добавляли фторид тетрабутиламмония (0,08 г, 0,323 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли, и остаток очищали на колонке с силикагелем (EtOAc-гептан; 20-100%) с получением названного соединения (0,127 г, 64%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,49 (с, 9H), 1,51 (с, 9H), 1,66 (м, 4H), 2,06 (м, 4H), 2,90 (м, 4H), 4,23 (м, 4H), 6,80 (д, 1H), 6,83 (дд, 1H), 6,94 (д, 1H), 6,96 (д, 1H), 7,07 (дд, 1H), 7,30 (д, 1H), 7,42 (д, 1H), 7,50 (д, 1H), 7,75 (с, 1H), 7,99 (с, 1H).
ESI MS (MH): 614.
Стадия В
К раствору названного соединения стадии А выше (0,05 г, 0,081 ммоль) в THF (5 мл) добавляли гидрид натрия (0,004 г, 0,16 ммоль), затем метилйодид (0,022 г, 0,15 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакцию останавливали каплей метанола, и растворители удаляли. Неочищенную смесь очищали на колонке с силикагелем с получением диметилированного названного соединения (0,021 г) и триметилированного названного соединения (0,010 г).
Диметилированное соединение 143, ESI MS (MH): 642 (M+H), 643 (M+2H), 644 (M+3H)
Триметилированное соединение 144, ESI MS (MH): 656 (M+H) 657 (M+2H), 658 (M+3H)
Препаративный пример 145

Стадия А
Трет-бутанол (2 мл) дегазировали путем обработки ультразвуком в течение 1 минуты при пропускании аргона через раствор. К дегазированному трет-бутанолу (1 мл) добавляли ацетат палладия(II) (0,003 г, 0,0127 ммоль) и 2-дициклогексилфосфино-2',4',6,-триизопропилбифенил (XPhos, 0,018 г, 0,038 ммоль). Эту смесь нагревали при ~100°C на песочной бане в течение 1 минуты для образования катализатора. К красноватому раствору катализатора затем добавляли раствор названного соединения препаративного примера 1, стадии D (0,045 г, 0,127 ммоль) и выпускаемый промышленностью 3,4-дифторанилин (0,019 г, 0,15 ммоль) в дегазированном трет-бутаноле (1 мл). После добавления карбонат калия (0,039 г, 0,28 ммоль), смесь нагревали на песочной бане при ~110°C в течение 3 часов. Смесь разбавляли этилацетатом (30 мл), водой (10 мл) и солевым раствором (10 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя смесь этилацетат/н-гептан (20/80), с получением названного соединения в виде серовато-белого твердого вещества (0,045 г, 87%).
1H-ЯМР (400 МГц, CDCl3): δ=1,12-1,16 (м, 18H), 1,80-1,90 (м, 3H), 6,22 (шир.с, 1H), 6,47 (д, 1H), 6,53 (д, 1H), 6,90-6,94 (м, 1H), 7,06 (т, 1H), 7,10 (д, 1H), 7,60-7,67 (м, 1H), 7,75 (д, 1H).
Препаративные примеры 146-256
Следуя методике проведения реакции сочетания на Pd, описанной в препаративном примере 145, за исключением того, что использовались указанные в таблице 2 бромпроизводные и амины, были получены следующие соединения (см. в конце описания).
Препаративный пример 258

Стадия А
К раствору названного соединения препаративного примера 66 (0,056 г, 0,091 ммоль) в THF (1 мл) добавляли фторид тетрабутиламмония (0,024 г, 0,091 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали, и остаток очищали на колонке с силикагелем (25%-100% EtOAc/гептаны: изократическая смесь) с получением названного соединения (0,026 г, 63%).
1H-ЯМР (400 МГц, CDCl3): δ=1,40 (с, 9H), 1,50 (м, 2H), 1,94 (м, 2H), 2,89 (м, 2H), 4,1 (м, 2H), 6,76 (д, 1H), 6,96 (д, 1H), 7,16 (дд, 1H), 7,43 (д, 1H), 7,94 (д, 1H), 8,96 (с, 1H), 10,5 (с, 1H), 12,2 (с, 1H).
Препаративный пример 259

Стадия А
Названное соединение препаративного примера 150 (0,049 г, 0,08 ммоль) растворяли в ацетонитриле (1 мл) и дихлорметане (1 мл) и обрабатывали с помощью 1M раствора фторида тетрабутиламмония (0,1 мл, 0,1 ммоль, 1,25 эквивалента на TIPS-группу) в тетрагидрофуране. Смесь перемешивали при комнатной температуре в течение 1 часа, и растворители удаляли. Остаток очищали с помощью хроматографии на диоксиде кремния, используя дихлорметан/ацетон (95/5), с получением названного соединения в виде бледно-желтого стекла (0,029 г, 82%).
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 3H), 1,94-2,08 (м, 2H), 2,70-2,95 (м, 7H), 4,25-4,56 (шир.м, 1H), 6,33 (шир.с, 1H), 6,56 (д, 1H), 6,92-7,03 (м, 1H), 7,06 (кв, 1H), 7,55-7,66 (м, 2H), 8,04 (шир.с, 1H).
Препаративный пример 260

Стадия А
К раствору названного соединения препаративного примера 82 (0,150 г, 0,3 ммоль) в THF (5 мл) добавляли нанесенный на полимер фторид (Aldrich 387789) (0,4 г). Смесь перемешивали в течение ночи, фильтровали, и растворитель испаряли. Раствор фильтровали и концентрировали, и остаток очищали на колонке с силикагелем (метанол к дихлорметану; от 5 до 15%) с получением названного соединения (0,075 г, 73%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,66-1,69 (м, 2H), 1,89-1,92 (м, 2H), 1,99-2,04 (м, 2H), 2,20 (с, 3H), 2,63-2,69 (м, 1H), 2,84-2,87 (м, 2H), 6,72-6,77 (м, 2H), 6,85-6,90 (м, 1H), 6,96 (с, 1H), 7,07 (с, 1H), 7,20 (abq, 1H), 7,44 (д, 1H), 8,09 (с, 1H), 10,5 (с, 1H).
Препаративные примеры 261-330
Следуя методике, аналогичной описанной в препаративных примерах 258(A), 259 (B), 260 (C), за исключением того, что использовались соединения из указанных в таблице 3 препаративных примеров, были получены следующие соединения (см. в конце описания).
Препаративный пример 331
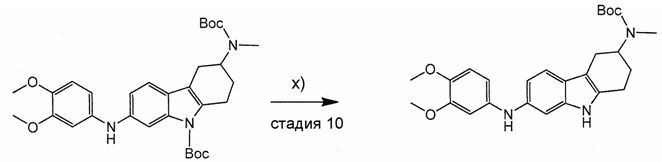
Препаративный пример 332

Стадия А
К раствору названного соединения препаративного примера 227 (0,127 г, 0,330 ммоль) в абсолютном этаноле (объем: 10 мл) добавляли гидроксид натрия (0,132 г, 3,30 ммоль). Полученную смесь нагревали с помощью микроволнового излучения до 180°C в течение 20 минут. Раствор концентрировали досуха. Продукт экстрагировали этилацетатом и солевым раствором. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали флэш-хроматографией в смеси DCM/MeOH с получением названного соединения в виде красноватого твердого вещества (0,070 г, 68%).
MS (ESI); m/z=314,71 (MH+).
Препаративные примеры 333-335
Следуя методике снятия защиты, описанной в препаративном примере 332, за исключением того, что использовались указанные в таблице 4 карбаматные производные, получали следующие соединения (см. в конце описания).
ПРИМЕР 1
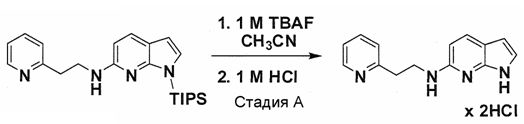
Стадия А
Названное соединение препаративного примера 1 (0,048 г, 0,125 ммоль) растворяли/суспендировали в ацетонитриле (1 мл) и обрабатывали с помощью 1M раствора фторида тетрабутиламмония (0,15 мл, 0,15 ммоль) в тетрагидрофуране. Смесь перемешивали при комнатной температуре в течение 1 часа, и растворители удаляли. Остаток очищали препаративной ТСХ, используя этилацетат, с получением свободного основания в виде оранжевого масла. Этот материал обрабатывали 1M водным раствором хлористого водорода (3 мл). Растворитель удаляли, используя лиофилизатор, с получением названного соединения в виде бледно-желтого твердого вещества (0,028 г, 73%).
1H-ЯМР (400 МГц, D2O): δ=3,32 (т, 2H), 3,78 (т, 2H), 6,41-6,44 (м, 2H), 7,02 (д, 1H), 7,79 (т, 1H), 7,87 (д, 1H), 8,00 (д, 1H), 8,38 (т, 1H), 8,53 (д, 1H).
MS (ESI); m/z=239,83 (MH+).
ПРИМЕРЫ 2-23
Следуя аналогичной методике, описанной в примере 1, за исключением того, что использовались указанные в таблице ниже соединения препаративных примеров. Для препаративных примеров без TIPS-защитной группы проводили только обработу водным раствором хлористого водорода. Для примеров 19-23 использовали нанесенный на полимер фтор для разрушения TIPS-защитных групп. Соединения всех примеров, приведенные в таблице 5, были получены в виде их HCl-солей, если не указано иначе (см. в конце описания).
ПРИМЕР 24
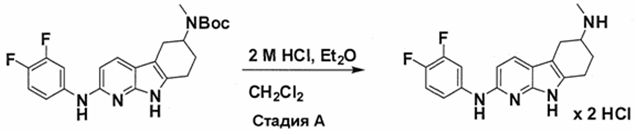
Стадия А
Названное соединение препаративного примера 259 (0,03 г, 0,07 ммоль) растворяли в дихлорметане (1 мл) и обрабатывали с помощью 2M раствора хлористого водорода (1 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали с помощью диэтилового эфира (5 мл) и затем сушили при пониженном давлении с получением названного соединения в виде серовато-белого твердого вещества (0,021 г, 75%).
1H-ЯМР (400 МГц, D2O): δ=1,93-2,05 (м, 1H), 2,20-2,59 (м, 1H), 2,65-2,80 (м, 6H), 3,10 (дд, 1H), 3,48-3,55 (м, 1H), 6,64 (д, 1H), 6,94-7,02 (м, 1H), 7,15-7,21 (м, 2H), 7,90 (д, 1H).
MS (ESI); m/z=328,95 (MH+).
ПРИМЕРЫ 25-203
Следуя аналогичной методике, описанной в примере 24, за исключением того, что использовались указанные в таблице ниже соединения препаративных примеров, получали следующие соединения. В случае образования осадка, растворители удаляли, и остаток растворяли в воде (5 мл). Испарение водного раствора с помощью лиофилизатора давало названные соединения. Соединения всех примеров, приведенные в таблице 6, были получены в виде их HCl-солей, если не указано иначе (см. в конце описания).
ПРИМЕР 204
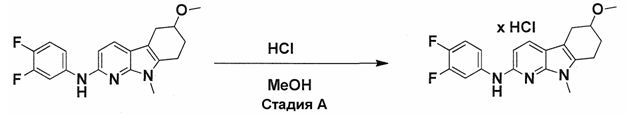
Стадия А
Названное соединение препаративного примера 203 (0,032 г, 0,093 ммоль) растворяли в метаноле (2 мл), и добавляли концентрированный водный раствор хлористого водорода. Прозрачный раствор замораживали в жидком азоте, и растворители испаряли с помощью лиофилизатора с получением названного соединения в виде серовато-белого твердого вещества (0,029 г, 81%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,79-1,87 (м, 1H), 2,02-2,08 (м, 1H), 2,46-2,50 (м, 1H), 2,63-2,77 (м, 2H), 2,88 (дд, 1H), 3,26 (с, 3H), 3,55 (с, 2H), 3,60-3,65 (м, 1H), 6,50 (д, 1H), 7,22-7,40 (м, 2H), 7,62 (д, 1H), 8,10 (ддд, 1H).
MS (ESI); m/z=344,77 (MH+).
ПРИМЕР 205

Использовали методику, аналогичную методике, описанной для примера 204, за исключением того, что использовалось названное соединение препаративного примера 204.
Выход: 76%
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,80-1,86 (м, 1H), 2,03-2,10 (м, 1H), 2,45-2,51 (м, 1H), 2,65-2,76 (м, 2H), 2,88 (дд, 1H), 3,30 (с, 3H), 3,55 (с, 3H), 3,61-3,66 (м, 1H), 5,90 (с, 2H), 6,46 (д, 1H), 6,81 (д, 1H), 7,04 (дд, 1Н), 7,57-7,61 (м, 2H).
MS (ESI); m/z=352,69 (MH+).
ПРИМЕР 206

Стадия А
К раствору названного соединения препаративного примера 332 (0,070 г, 0,223 ммоль) в метаноле (объем: 4 мл) добавляли раствор формальдегида (0,020 мл, 0,246 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем добавляли порциями триацетоксиборгидрид натрия (0,0568 г, 0,268 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Добавляли 1 мл 1,2M HCl в воде. После 5 минут перемешивания при комнатной температуре, раствор выливали в смесь воды, насыщенного Na2CO3 и солевого раствора. Экстракцию проводили этилацетатом 3 раза. Органические слои собирали, сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт очищали флэш-хроматографией в смеси DCM/MeOH от 98:2 до 90:10. Продукт растворяли в DCM, и медленно добавляли HCl в эфире для образования соответствующей HCl соли. Раствор концентрировали досуха, и твердое вещество сушили под вакуумом с получением ожидаемого соединения в виде фиолетового твердого вещества (0,028 г, 34%).
1H-ЯМР (400 МГц, MeOD): δ=3,07 (с, 3H); 3,14-3,23 (м, 2H); 3,47-3,60 (м, 1H); 3,66 (с, 3H); 3,77-3,91 (м, 1H); 4,28 (д, J=14,0 Гц, 1H); 4,63 (д, J=14,0 Гц, 1H); 6,70 (с, 1H); 6,79 (с, 1H); 6,94-7,10 (м, 2H); 7,23 (с, 1H); 7,35 (д, J=8,8 Гц, 1H).
MS (ESI); m/z= 328,71 (MH+).
ПРИМЕР 207

Стадия А
К смеси названного соединения препаративного примера 334 (0,036 г, 0,107 ммоль), названного соединения препаративного примера 54 (0,0281 мг, 0,107 ммоль) и карбоната калия (0,0297 мг, 0,215 ммоль) добавляли тетрагидрофуран (соотношение: 1,000, объем: 2 мл), затем ацетонитрил (соотношение: 1,000, объем: 2,000 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали досуха. Остаток очищали флэш-хроматографией в смеси DCM/MeOH от 0% до 10%. Продукт растворяли в DCM и добавляли HCl в эфире. Суспензию концентрировали досуха. Остаток растворяли в нескольких каплях MeOH, и добавляли этилацетат для инициирования осаждения. Суспензию концентрировали досуха с получением ожидаемого соединения в виде бежевого твердого вещества (0,020 г, 37%).
1H-ЯМР (400 МГц, ДМСО): δ=1,73-1,87 (м, 2H); 1,88-2,02 (м, 2H); 3,00 (с, 3H); 3,06-3,17 (м, 2H); 3,21 (т, J=7,6 Гц, 2H); 3,24-3,35 (м, 2H); 3,38-3,51 (м, 1H); 3,56 (с, 3H); 3,69-3,84 (м, 1H); 4,12-4,30 (м, 5H); 4,56 (д, J=13,6 Гц, 1H); 6,52-6,62 (м, 2H); 6,68-6,83 (м, 2H); 7,01 (с, 1H); 7,26-7,36 (м, 1H); 10,62 (с, NH).
MS (ESI); m/z=470,68 (MH+).
ПРИМЕРЫ 208-210
Названные соединения получали в соответствии с методикой, описанной в примере 207, используя указанное в таблице 7 йод-производное (см. в конце описания).
ПРИМЕРЫ 211-218
Если обрабатывать названные соединения препаративных примеров, описанные в примере 207, с помощью йод-производных, то можно получить требуемые соединения, указанные в таблице 8 (см. в конце описания).
ПРИМЕРЫ 219-222
Если обрабатывать названные соединения препаративных примеров с помощью винилсульфоновых производных согласно патентному документу WO2008/150799, то можно получить требуемые соединения, указанные в таблице 9 (см. в конце описания).
Соединения настоящего изобретения, имеющие один или более оптически активных углеродов, могут существовать в виде рацематов и рацемических смесей, диастереомерных смесей и индивидуальных диастереомеров, энатиомерных смесей и индивидуальных энантиомеров, таутомеров, атропоизомеров и ротамеров, при этом все изомерные формы входят в объем настоящего изобретения. Описанные в этом изобретении соединения, содержащие олефиновые двойные связи, включают как E, так и Z геометрические изомеры. В это изобретение также входят все солевые формы, полиморфы, гидраты и сольваты. Все упомянутые выше соединения входят в объем изобретения.
Определение ингибирования агрегации Aβ1-42
Способность примеров соединений изобретения ингибировать агрегацию Aβ1-42 пептида определяли с помощью спектрофлуоресцентного анализа с тиофлавином T (ThT-анализ), описанного ниже.
Приготовление пленки Aβ пептида
Лиофилизированный порошок AJ31-42 (Bachem) ресуспендировали в гексафторизопропаноле (HFIP) до 1 мМ. Раствор пептида подвергали воздействию ультразвука в течение 15 минут при комнатной температуре, перемешивали в течение ночи, и отбирали аликвоты в несиликонизированные микроцентрифужные пробирки. Гексафторизопропанол затем испаряли в потоке аргона. Полученную пленку пептида was сушили под вакуумом в течение 10 минут, хорошо герметизировали и хранили при -80°C до использования.
Определение ингибирования агрегации Aβ1-42
Для анализа ингибирования агрегации Aβ1-42, опосредованного синтетическими соединениями, синтетические соединения из примеров растворяли перед каждым экспериментом в безводном диметилсульфоксиде (ДМСО, Sigma-Aldrich) с достижением содержания 7,4 мМ. Пленку пептида Aβ1-42 растворяли в ДМСО с достижением содержания 400 мкМ. Раствор для анализа в PBS приготавливали в несиликонизированных инкубационных пробирках с достижением следующих концентраций: 330 мкМ синтетического соединения, 33 мкМ Aβ1-42, 10 мкМ тиофлавина T (ThT), и 12,8% ДМСО. Следовательно, окончательное мольное отношение синтетического соединения к Aβ1-42 составляло 10:1. Для измерения максимального значения относительной флуоресценции (RFU), приготавливали пробу для положительного контроля, не содержащую синтетического соединения. Приготавливали пробу для отрицательного контроля для каждого синтетического соединения, не содержащую Aβ1-42. Для установления воспроизводимости между независимыми экспериментами во всех анализах, в качестве контрольного соединения использовали триммер 3-аминопиразола (Rzepecki et. al. Synthesis 2003, 1815). Растворы затем инкубировали в течение 24 часов при 37°C и считывали спектрофлуоресценцию (в относительных единицах флуоресценции; RFU) шести параллельных проб в черных 384-луночных планшетах (Perkin-Elmer) на спектрофлуориметре FluoroCount фирмы Perkin-Elmer. Ингибирование агрегации выражают как средний% ингибирования ±1 стандартное отклонение (SD) в соответствии со следующим уравнением:

Были измерены значения для следующих примеров соединений, приведенных в таблице 10 (см. в конце описания):
Ингибирование с помощью синтетических соединений агрегации Aβ1-42 заранее сфомированных Aβ1-42 волокон. Результаты выражены как среднее значение +/- стандартное отклонение двух независимых экспериментов.
Ингибирование агрегации Aβ1-42 заранее сфомированных Aβ1-42 волокон не может быть определено для примеров 5 и 7 вследствие очень сильной автофлуоресценции.
Пример 211: Воздействие ACI-636 на образование бляшек у hAPPSL трансгенных мышей
Целью этого исследования являлась оценка свойств ACI636 для ингибирования образования бляшек и разрушения существующих бляшек.
211.1 Методы
Трансгенные (Tg) мыши, сверхэкспрессирующие человеческий амилоидный белок (hAPP), являются удобными моделями для изучения влияния лекарственных средств на продуцирование, разрушение и отложение амилоида. Для исследования использовали мышей, сверхэкспрессирующих 751 аминокислотную форму hAPP с мутацией London (V717I) и мутацией Swedish (KM670/671NL) под контролем мышиного промотора Thy-1 (hAPPSL мыши). hAPPSL мыши характеризуются возрастным увеличением β-амилоидных пептидов (Aβ) и образованием бляшек, состоящих из амилоидных отложений в раннем возрасте (начиная с 4-6 месяцев). Тяжесть патологии головного мозга коррелирует с увеличением возраста и расстройствами поведения. Начиная с возраста в 13-14 месяцев, самкам hAPPSL Tg мышей вводили один раз в сутки 10 мг/кг ACI-636 путем принудительного кормления, как это показано в таблице, приведенной в конце описания.
В конце периода лечения, животных умерщвляли и собирали образцы крови (плазмы) и мозга. Определяли воздействие ACI-636 на Aβ уровни (Aβ38, Aβ40, Aβ42) в четырех различных фракциях гомогената мозга (TBS, Triton X-100, SDS и FA) с помощью набора Aβ-kit фирмы Mesoscale Discovery. Кроме того, в образцах мозга иммуногистохимически определяли амилоидные отложения, обнаруживаемые с помощью 6E10 антител. Уровни Aβ оценивали путем сравнения с пептидным эталоном в виде пг/мг массы влажного мозга.
211.2 Результаты
В целом, животные, получающие ACI-636 в течение 14 дней, демонстрировали тенденцию к увеличению содержания растворимого Aβ (фракции TBS и Triton X-100), и пониженному содержанию нерастворимого Aβ (фракции SDS и FA), по сравнению с животными, которым вводили PBS. Эффект лечения животных с помощью ACI-636 по сравнению с плацебо (PBS) называют "различием, вызванным лечением". Количественное определение кортикальной (a и b) и гиппокампальной (c и d) 6E10 иммунофлуоресценции приведено на фигуре 1. Лечение самок мышей в течение 14 дней оказывает воздействие на число и суммарную площадь бляшек (примечание: минимальный размер объекта >150 мкм2). Сокращения: Плацебо (A); ACI-636 10 мг/кг/день (B).
По сравнению с плацебо, лечение с помощью ACI636 систематически снижает отложения бляшек у самок Tg мышей после 14 дней лечения.
211.3 Выводы
Животные, получавшие ACI636 в течение 14 дней, продемонстрировали тенденцию к увеличению содержания растворимого Aβ (фракции TBS и Triton X-100), и пониженному содержанию нерастворимого Aβ (фракции SDS и FA), по сравнению с животными, которым вводили плацебо. При введении в течение 14 дней, ACI636 селективно понижает отложение амилоида у самок hAPP751SL трансгенных мышей, как во внеклеточном отложении бляшек, так и суммарное отложение амилоида, включая внеклеточный амилоид. Эффект был более четко выраженным в коре головного мозга, чем в гиппокампе.
Пример 212: Воздействие ACI-636 на поведение APPV171I трансгенных мышей
Целью этого исследования являлось определение эффективности ACI-636 в экспериментальной модели на мышах болезни Альцгеймера. Влияние лечения на возможности памяти оценивали с помощью теста на узнавание пространства (SRT)
212.1 Методы
212.1.1 Тест на узнавание пространства (SRT)
Тест на узнавание нового объекта (ORT) представляет собой in vivo тест для исследования действия испытуемого вещества на память у грызунов (мышей, крыс). Этот тест проводили для оценки непространственной памяти у грызунов на основании их способности узнавать новый объект в остальной знакомой для них окружающей среде (Ennaceur, A. and Delacour, J. Behav. Brain Res. 1988, 31, 47-59). Тест дает информацию по кратковременной памяти или долговременной памяти, промнестическому или амнестическому действию испытуемого соединения и исследовательскому поведению, которое связано с вниманием. На стадии обучения в тесте, перед животным ставят два одинаковых объекта, размещенных на открытом месте, для того чтобы оно познакомилось с объектами; в фазе ретенции (сохранения информации) перед животным ставят два различных объекта, размещенных на открытом месте: один знакомый объект, использовавшийся в фазе обучения, и один новый объект. В фазе ретенции, измеряют время, потраченное на осматривание каждого из объектов. Осматривание объекта определяется как время, в течение которого голова животного была направлена в сторону и в пределах 2 сантиметров от объекта. Не страдающие амнезией животные должны тратить больше времени на осматривание нового объекта, чем на осматривание знакомого для них объекта. Для того чтобы усложнить задачу для животных, тест ORT модифицировали для включения компонента пространственной памяти (тест на узнавание пространства (SRT)), используя два одинаковых объекта. При постановке теста SRT, один объект остается в фазе ретенции на том же самом месте, на котором он находился ранее в фазе изучения, в то время как другой объект ставят в другом место. Из суммарно 24 самок APPV171I тренсгенных мышей (в возрасте 10 месяцев), 12 мышам вводили через зонд один раз в сутки 10 мг/кг ACI-636, и 12 мышам вводили через зонд один раз в сутки PBS. После 21 дня лечения, проводили тест SRT в сером полипропиленовом лабиринте (круговое открытое поле, диаметр 40 см, высота 32,4 см) с использованием двух различных объектов (1 кубик из конструктора LEGO (2,9 см × 5,8 см) и черная стеклянная бутылочка (5,3 см × 5,3 см)). Эксперимент осуществлялся следующим образом:
День 1:
- Привыкание к аппаратуре (10 минут) каждого животного в отсутствие объекта.
День 2:
- Стадия обучения: два одинаковых объекта (кубик из конструктора LEGO или черную стеклянную бутылочку) выставлялись для обозрения (10 минут) в одной стороне лабиринта.
- Тест на ретенцию (через 3 часа после обучения): один из одинаковых объектов перемещали в противоположную сторону лабиринта и выставляли для обозрения (10 минут).
212.2 Результаты
На фигурах 2A и 2B представлены данные анализа для APPV171I трансгенных мышей, которые были подвергнуты лечению с помощью PBS и ACI-636. Статистический анализ индекса узнаваемости (RI) для продолжительности и частоты (фигуры 2A и 2B) с использованием двустороннего критерия Стьюдента показал значимую разницу между APPV171I трансгенными мышами, которых подвергали лечению с помощью PBS и ACI-636 (p<0,05).
212.3 Выводы
В тесте на узнавание пространства (SRT), APPV171I трансгенные мыши, которые подвергались лечению с помощью ACI-636, затрачивали больше времени на ознакомление с перемещенным объектом, чем со знакомым для них объектом, по сравнению с APPV171I трансгенными мышами, которые подвергались лечению с помощью плацебо (PBS), что демонстрирует воздействие ACI-636 на ретенцию кратковременной памяти при проведении теста на узнавание пространства.
Пример 213: Получение соединения 213a и 213b (энантиомеров соединения 26): хиральное разделение
213.1 Цель и способ проведения исследования
Целью этого исследования являлось разделение и идентификация двух энантиомеров соединения 26.
213.2 Методы
Разделение Boc-защищенного предшественника соединения 26 гидрохлорида (N2-(3,4-дифторфенил)-N6,9-диметил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-2,6-диамина; рацемата) с помощью ВЭЖХ с использованием хиральной фазы
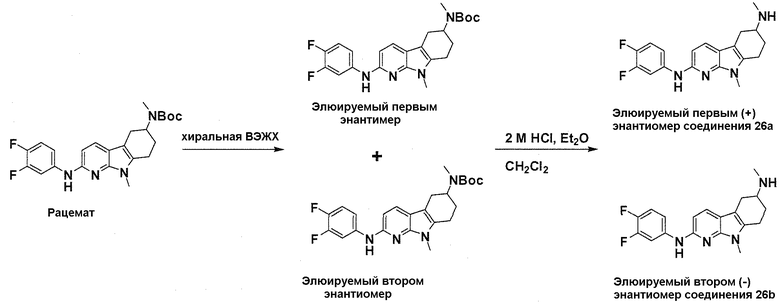
Разделяли энантиомеры Boc-защищенного предшественника соединения 26 (0,110 г, рацемат). Суммарно было получено 0,040 г элюируемого первым энантиомера и 0,033 г элюируемого вторым энантиомера. Согласно данным хиральной ВЭЖХ, каждый энантиомер содержал <1% другого энантиомера.
Условия проведения хиральной ВЭЖХ:
- chiralpak IA, 4,6×250 мм, 5 мкм;
- подвижная фаза: н-гептан/изопропанол (95:5);
- расход: 1 мл/мин;
- длина волны: 254 нм.
На фигуре 3 показано разделение Boc-защищенного предшественника соединения 26 с помощью хиральной ВЭЖХ:
Rt (элюируемый первым энантиомер): 22,7 мин;
Rt (элюируемый вторым энантиомер): 31,6 мин.
На фигуре 4 показан элюируемый первым энантиомер после разделения, а на фигуре 5 показан элюируемый вторым энантиомер после разделения.
213.3 Получение конечных соединений
Все реагенты и растворители приобретались у фирм-производителей и использовались без дополнительной очистки. Протонные (1H) спектры регистрировались на ЯМР спектрометре при 400 МГц в дейтерированных растворителях. Масс-спектры (MS) регистрировали на спектрометре Finnigan MAT TSQ 7000. Оптическое вращение энантиомеров определяли в метаноле на поляриметре JASCO при 25°C и длине волны 589 нм.
213.3.1 Получение соединения 213a
Элюируемый первым энантиомер после разделения с помощью ВЭЖХ (0,039 г, 0,088 ммоль) растворяли в дихлорметане (1,2 мл) и обрабатывали с помощью 2M раствора хлористого водорода (1,2 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали с помощью диэтилового эфира (5 мл) и сушили при пониженном давлении с получением названного соединения в виде серовато-белого вещества (0,029 г, 88%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,90-2,02 (м, 1H), 2,27-2,36 (м, 1H), 2,65-2,73 (м, 4H), 2,75-2,94 (м, 2H), 3,10-3,18 (м, 1H), 3,42-3,48 (м, 1H), 3,63 (с, 3H), 6,60 (д, 1H), 7,25-7,33 (м,1H), 7,36-7,42 (м, 1H), 7,70 (д, 1H), 8,14-8,20 (м, 1H).
MS (ESI); m/z=343,70 (MH+).
[α]25=+45,4°(c 0,066, MeOH).
213.3.2 Получение соединения 213b
Элюируемый вторым энантиомер после разделения с помощью ВЭЖХ (0,022 г, 0,05 ммоль) растворяли в дихлорметане (1 мл) и обрабатывали с помощью 2M раствора хлористого водорода (1 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали с помощью диэтилового эфира (5 мл) и сушили при пониженном давлении с получением названного соединения в виде серовато-белого вещества (0,013 г, 71%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,90-1,98 (м, 1H), 2,26-2,33 (м, 1H), 2,62-2,72 (м, 4H), 2,77-2,93 (м, 2H), 3,10-3,16 (м, 1H), 3,40-3,47 (м, 1H), 3,62 (с, 3H), 6,58 (д, 1H), 7,25-7,33 (м, 1H), 7,36-7,41 (м, 1H), 7,70 (д, 1H), 8,11-8,18 (м, 1H).
MS (ESI); m/z=343,68 (MH+).
[α]25=-37,5° (c 0,064, MeOH).
213.4 Выводы
Рацемический Boc-защищенный предшественник соединения 26 был успешно разделен на его два энантиомера. Разрушение защитной группы элюируемых первыми энантиомеров давало (+)-энантиомер соединения 213a, а элюируемый вторым энантиомер давал (-)-энантиомер соединения 213b.
Препаративный пример 336: получение индивидуальных энантиомеров трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата путем разделения диастереомеров
336.1 Цель и способ проведения исследования
Целью этого исследования являлась разработка способа получения индивидуальных энантиомеров трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)-карбамата путем разделения диастереомеров.
342.2 Методы
336.2.1 Принципы разделения энантиомеров
Рацемическое соединение подвергали взаимодействию с оптически активным реагентом L (производным аминокислоты) с получением смеси диастереомеров (RL и SL), которые разделяли с помощью хроматографии. Затем хиральный реагент удаляли путем обработки кислотой с получением энантиомерно обогащенного элемента структуры.
Схема синтеза для получения энантиомерно обогащенного трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата
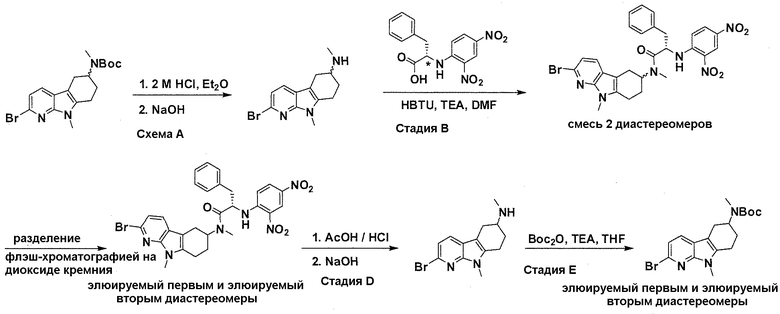
Получение хиральной вспомогательной (S)-2-((2,4-динитрофенил)амино)-3-фенилпропановой кислоты
К смеси L-фенилаланина (8,88 г, 53,7 ммоль) и 1-фтор-2,4-динитробензола (3,24 мл, 26,9 ммоль) добавляли триэтиламин (7,86 мл, 56,4 ммоль). Полученную смесь нагревали, используя микроволновый реактор Initiator фирмы Biotage, до 100°C в течение 4 минут. Твердое вещество разбавляли с помощью MeOH, и суспензию добавляли по каплям в дистиллированную воду. К суспензии добавляли дихлорметан, и органическую фазу отделяли. Органическую фазу затем экстрагировали 1,2 M раствором хлористого водорода и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали, и растворители испаряли. Остаток разбавляли дихлорметаном, и суспензию фильтровали. Осадок затем промывали небольшим количеством дихлорметана. Объединенные фильтраты испаряли, и остаток очищали флэш-хроматографией с градиентом дихлорметан/метанол (от 0% до 5%) с получением названного соединения.
MS ESI MeOH (Neg): 329,96 (M-H)/375,99 (M+FA-H)/661,13 (2M-H)/992,32 (3M-H).
Стадия А
К раствору рацемического трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата (0,9 г, 2,28 ммоль) в 20 мл дихлорметана, добавляли 11,34 мл (22,8 ммоль) 2M раствора хлористого водорода в диэтиловом эфире, и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. После разбавления дихлорметаном, раствор подщелачивали водным раствором гидроксида натрия до pH 13-14. Органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном (4×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением названного соединения в виде коричневатого масла.
Стадия В
Неочищенное названное соединение стадии А выше (2,28 ммоль) растворяли в N,N'-диметилформамиде (14 мл). Добавляли (S)-2-((2,4-динитрофенил)амино)-3-фенилпропановую кислоту (0,944 г, 2,85 ммоль), затем HBTU (1,08 г, 2,85 ммоль) и триэтиламин (0,68 мл, 5,01 ммоль). Раствор перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь разбавляли дихлорметаном (500 мл) и промывали водой (100 мл). Органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном (4×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением неочищенного продукта в виде оранжевого твердого вещества.
Стадия С
Разделение диастереомеров флэш-хроматографией на диоксиде кремния
Смесь диастереомеров стадии В выше разделяли с помощью флэш-хромаптографической системы Biotage Isolera One с получением 0,64 г элюируемого первым диастереомера и 0,7 г эолюируемого вторым диастереомера.
Использовали следующие условия:
Загрузка картриджа: 250 мг - 500 мг смеси, нанесенной на 100 г HP-Sil SNAP картриджа
Градиент растворителей: EtOAc/н-гептан: 30-50%
Хроматографическое разделение смеси диастереомеров проводили на силикагеле (расход: 50 мл/мин), используя градиент этилацетат/н-гептан (30/70→60/40).
1H-ЯМР данные элюируемого первым диастереомера (только область ароматики, примеси присутствуют в области 7,1-7,3 м.д.) приведены на фигуре 11.
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,07 (д, 1H), 7,35 (д, 1H), 7,51 (д, 1H), 7,71-7,76 (дд, 2H), 8,21-8,25 (м, 2H), 8,87-8,88 (м, 1H), 9,25-9,33 (дд, 2H).
1H-ЯМР данные элюируемого вторым диастереомера (только область ароматики, примеси присутствуют в области 7,1-7,3 м.д.) приведены на фигуре 12.
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,17 (д, 1H), 7,36 (д, 1H), 7,48 (д, 1H), 7,43 (д, 1H), 7,80 (д, 1H), 8,14 (дд, 1H), 8,23 (дд, 1H), 8,87 (дд, 1H), 9,32 (т, 2H).
Стадия D
Элюируемый вторым диастереоизомер (0,7 г, 1,15 ммоль) стадии С выше растворяли в 18 мл ледяной уксусной кислоты, и затем добавляли 18 мл концентрированной хлористоводородной кислоты (36-38%). Смесь нагревали при 120°C на песочной бане в течение 5 дней. Реакционную смесь охлаждали при комнатной температуре и затем разбавляли с помощью 400 мл дихлорметана и подщелачивали 1 M водным раствором гидроксида натрия до pH 13-14. Органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном (4×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали, и растворитель удаляли с получением неочищенного продукта в виде коричневатого масла.
Стадия E
Неочищенное соединение стадии D выше (1,15 ммоль) растворяли в тетрагидрофуране (16 мл) и обрабатывали с помощью триэтиламина (0,176 мл) и ди-трет-бутилдикарбоната (0,95 г, 12,4 ммоль). Смесь выдерживали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток очищали с помощью системы очистки Biotage (EtOAc/н-гептан: 10-40%) с получением Boc-защищенного трициклического элемента структуры, полученного из элюируемого вторым диастереомера в виде серовато-белого твердого вещества (0,22 г, 24% за 4 стадии).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,41 (с, 9H), 1,91-2,06 (м, 2H), 2,71 (м, 2H), 2,78 (с, 3H), 2,96-2,83 (м, 2H), 3,62 (с, 3H), 4,25 (шир.м, 1H), 7,17 (д, 1H), 7,78 (д, 1H).
Элюируемый первым диастереомер обрабатывали, как описано выше (стадия D и стадия Е) с получением трициклического элемента структуры из соответствующего элюируемого первым энантиомера.
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,41 (с, 9H), 1,91-2,06 (м, 2H), 2,71 (м, 2H), 2,78 (с, 3H), 2,96-2,83 (м, 2H), 3,62 (с, 3H), 4,25 (шир.м, 1H), 7,17 (д, 1H), 7,78 (д, 1H).
Препаративный пример 337: Получение индивидуальных энантиомеров трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата путем разделения диастереомерных солей
337.1 Цель и способ проведения исследования
Целью этого исследования являлась разработка способа разделения рацемического трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата через диастереомерные соли.
337.2 Методы
337.2.1 Принцип разделения энантиомеров
Рацемическое соединение в виде свободного основания подвергали взаимодействию с оптически активной кислотой с получением смеси диастереомерных солей (R+L1 -+S+L1 -). Кристаллизация диастереомерных солей в подходящем растворителе и удаление хирального вспомогательного вещества давали индивидуальные энантиомеры.
Схема синтеза для получения индивидуальных энантиомеров трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]-индол-6-ил)метил)карбамата

Стадия А
Раствор рацемического трет-бутил-(2-бром-9-метил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]индол-6-ил)метил)карбамата (12,48 г, 31,87 ммоль) в 300 мл дихлорметана охлаждали до 0°C. Затем добавляли при 0°C 150 мл (300 ммоль) 2M раствора хлористого водорода в диэтиловом эфире. После того, как добавление было закончено, ледяную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Осадок отфильтровывали, промывали диэтиловым эфиром (100 мл) и сушили на воздухе с получением дигидрохлоридной соли в виде серовато-белого твердого вещества (11,5 г, количественно). В снабженную магнитной мешалкой колбу объемом 250 мл, содержащую дигидрохлоридную соль (3,65 г, 10 ммоль) в виде суспензии в воде (37 мл), добавляли водный раствор гидроксида натрия (1,27 г, 32 ммоль NaOH в 22 мл воды) (время добавления 1 минута). Затем колбу, содержащую раствор гидроксида натрия, споласкивали дополнительным количеством воды (2×3 мл). полученную белую суспензию интенсивно перемешивали в течение 5 минут, затем добавляли дихлорметан (65 мл). Полученную смесь интенсивно перемешивали в течение 5 минут. Органическую фазу отделяли, и водную фазу экстрагировали 3×65 мл дихлорметаном. Объединенные органические фазы сушили над MgSO4 и испаряли под вакуумом при комнатной температуре досуха, получая 2,63 г (90%) названного соединения в виде желтого твердого вещества.
Разделение энантиомеров через диастереомерные соли:
Стадия В (реакция с (S)-(+)-миндальной кислотой с получением энантиомера B, Rt=26,1 мин)
В снабженную магнитной мешалкой колбу объемом 250 мл, содержащую названное соединение стадии А выше (5,46 г, 18,6 ммоль), растворенное в метаноле (30 мл), при 35°C добавляли (S)-(+)-миндальную кислоту (1,35 г, 8,9 ммоль), растворенную в метаноле (50 мл). Полученную смесь перемешивали при 35°C (внутренняя температура) в течение 24 часов и затем фильтровали через воронку с фильтром из спеченного стекла. Маточные растворы отделяли, и полученное белое твердое вещество промывали с помощью метанола при комнатной температуре (3×20 мл). Соль сушили под вакуумом с получением 2,55 г (31%) названного соединения (энантиомер B и (S)-(+)-миндальная кислота, ВЭЖХ 97% ee) в виде белого твердого вещества. Маточные растворы испаряли досуха, затем добавляли 0,5M водный раствор гидроксида натрия (20 мл). Водный раствор экстрагировали дихлорметаном (3×10 мл), объединенные органические фазы сушили над MgSO4 и испаряли под вакуумом при комнатной температуре с получением желтовато-коричневой пасты, соответствующей 7:3 (энантиомер A:энантиомер B) смеси энантиомеров (3,24 г, 59%).
(Взаимодействие маточных растворов с (R)-(-)-миндальной кислотой с получением энантиомера A, Rt=24,3 мин)
В снабженную магнитной мешалкой колбу объемом 100 мл, содержащую раствор 7:3 (энатиомер A:энантиомер B) смеси энантиомеров (3,24 г, 11 ммоль) в метаноле (30 мл), при 35°C добавляли (R)-(-)-миндальную кислоту (1,09 г, 7,2 ммоль), растворенную в метаноле (35 мл). Полученную смесь перемешивали при внутренней температуре 35°C в течение 24 часов. Кристаллическое твердое вещество отфильтровывали через воронку с фильтром из спеченного стекла, промывали метанолом при комнатной температуре (3×16 мл) и сушили под вакуумом с получением 3,08 г (63%) названного соединения (энантимер A и (R)-(-)-миндальная кислота, ВЭЖХ 97% ee) в виде белого твердого вещества.
Стадия С
Образование дигидрохлорида энантиомера A из соответствующего манделата
В снабженную магнитной мешалкой колбу объемом 250 мл, содержащую соль манделата энантиомера A (3,08 г, 6,9 ммоль), добавляли 1M раствор гидроксида натрия (30 мл) и дихлорметан (60 мл). После 5 минут интенсивного перемешивания, органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном (2×60 мл). Объединенные органические фазы промывали солевым раствором (30 мл), сушили над MgSO4 и испаряли досуха при комнатной температуре. Полученную коричнево-желтую пасту растворяли в 1M водном растворе хлористого водорода (50 мл), и растворитель испаряли досуха с получением белого твердого вещества. Когда это твердое вещество сушили под высоким вакуумом при 25°C до постоянной массы, оно плавилось, и его перекристаллизовывали с получением названного соединения в виде желтого твердого вещества (1,97 г, 78%, ВЭЖХ 97,8% ee).
Образование дигидрохлорида энантиомера B из соответствующего манделата
В снабженную магнитной мешалкой колбу объемом 100 мл, содержащую манделатную соль энантиомера B (2,1 г, 4,7 ммоль), добавляли 1M раствор гидроксида натрия (20 мл) и дихлорметан (40 мл). После 5 минут интенсивного перемешивания, органическую фазу отделяли, и водную фазу экстрагировали дихлорметаном (2×40 мл). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над MgSO4 и испаряли досуха при комнатной температуре. Полученную коричнево-желтую пасту растворяли в 1M водном растворе хлористого водорода (30 мл), и растворитель испаряли досуха с получением белого твердого вещества. Когда это твердое вещество сушили под высоким вакуумом при 25°C до постоянной массы, оно плавилось, и его перекристаллизовывали с получением названного соединения в виде желтого твердого вещества (1,57 г, 91%, ВЭЖХ 97,7% ee).
Условия хиральной ВЭЖХ:
Образец: 2 мг в расчете на основание/мл в смеси н-гептан/(этанол-0,2% DEA) (90:10).
Колонка: Chiracel AD-H.
Растворитель: н-гептан/(этанол-0,2% DEA) (90:10).
Объем вводимой пробы: 5 мкл.
На фигуре 6 приведена рацемическая смесь (дигидрохлоридные соли) перед кристаллизацией:
Rt (элюируемый первым энантиомер A): 24,3 мин,
Rt (элюируемый вторым энантиомер B): 26,1 мин.
На фигуре 7 приведен элюируемый первым энантиомер A (дигидрохлоридная соль) после кристаллизации.
На фигуре 8 приведен элюируемый вторым энантиомер B (дигидрохлоридная соль) после кристаллизации.
Стадия D
Boc-защита элюируемого первым энантиомера A
Дигидрохлорид 2-бром-N6,9-диметил-6,7,8,9-тетрагидро-5H-пиридо[2,3-b]-индол-6-амина (0,94 г 2,56 ммоль; энантиомер A) растворяли в тетрагидрофуране (50 мл) и обрабатывали с помощью триэтиламина (1,95 мл, 13,9 ммоль) и ди-трет-бутилдикарбонатом (1,8 г, 8,55 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток очищали, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (от 5/95 до 30/70), с получением названного соединения (энантиомер A) в виде серовато-белого твердого вещества (0,969 г, 96%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,41 (с, 9H), 1,91-2,06 (м, 2H), 2,71 (м, 2H), 2,78 (с, 3H), 2,96-2,83 (м, 2H), 3,62 (с, 3H), 4,25 (шир.м, 1H), 7,17 (д, 1H), 7,78 (д, 1H).
1H-ЯМР (400 МГц, CDCl3): δ=1,60 (с, 9H), 2,10-2,23 (м, 2H), 2,82-3,01 (м, 7H), 3,81 (с, 3H), 4,38-4,64 (шир.м, 1H), 7,25 (д, 1H), 7,67 (д, 1H).
Boc-защита элюируемого вторым энантиомера B
Элюируемый вторым энантиомер B обрабатывали,как описано выше (стадия С и стадия D), с получением соответствующего Boc-защищенного трициклического элемента структуры.
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,41 (с, 9H), 1,91-2,06 (м, 2H), 2,71 (м, 2H), 2,78 (с, 3H), 2,96-2,83 (м, 2H), 3,62 (с, 3H), 4,25 (шир.м, 1H), 7,17 (д, 1H), 7,78 (д, 1H).
337.3 Выводы
Энантиомерно чистые элементы структуры получали путем разделения рацемической смеси с помощью кристаллизации диастереомерных солей. Чистота энантиомерных элементов структуры составляла 97,8% ee для энантиомера A и 97,7% ee для энантиомера B.
Пример 214: Получение соединений 214a и 214b (энантиомеры соединения 26), используя названные соединения препаративного примера 336 (разделение диастереомеров)
214.1 Цель и способ проведения исследования
Целью этого исследования являлся синтез соединений 214a и 214b, двух энантиомеров соединения 26, используя энантиомерно обогащенные элементы структуры, полученные путем разделения диастереомеров.
214.2 Схема синтеза энантиомерно обогащенного сооединения 214b

Стадия А
Трет-бутанол (2 мл) добавляли к смеси 2-дициклогексил-фосфино-2',4',6'-триизопропилбифенила (XPhos, 0,036 г, 0,075 ммоль) и ацетата палладия(II) (0,0056 г, 0,025 ммоль). Смесь дегазировали путем обработки ультразвуком в течение 1 минуты при одновременном пропускании через раствор аргона. Эту смесь нагревали при ~100°C на песочной бане в течение 1 минуты для образования катализатора. К красноватому раствору катализатора затем добавляли названное соединение препаративного примера 336, полученное из элюируемого вторым диастереомера (0,1 г, 0,25 ммоль), выпускаемый промышленностью 3,4-дифторанилин (0,041 г, 0,31 ммоль) и карбонат калия (0,086 г, 0,625 ммоль). Смесь нагревали на песочной бане при ~110°C в течение 3 часов. Смесь разбавляли этилацетатом (100 мл) и водой (10 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали дважды, используя систему флэш-хроматографической очистки Biotage Isolera (MeOH/DCM: 0-1% затем EtOAc/н-гептан: 10-30%), с получением названного соединения в виде серовато-белого твердого вещества (0,087 г, 97%).
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 9H), 2,04 (м, 2H), 2,69-2,85 (м, 7H), 3,70 (с, 3H), 4,29-4,50 (шир.м, 1H), 6,51 (д, 1H), 7,00-7,02 (м, 1H), 7,05-7,12 (м, 1H), 7,61 (м, 2H).
MS (ESI); m/z=443,64 (MH+).
На фигуре 8 показан Boc-защищенный предшественник соединения 214b, полученный из элюируемого вторым энантиомера.
Стадия В
Названное соединение стадии А выше (0,08 г, 0,045 ммоль) растворяли в дихлорметане (2,0 мл) и обрабатывали с помощью 2M раствора хлористого водорода (2 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали с помощью диэтилового эфира (5 мл) и сушили при пониженном давлении с получением названного соединения в виде серовато-белого твердого вещества (0,068 г, 97%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,90 (м, 1H), 2,25-2,28 (м, 1H), 2,50-2,64 (м, 1H), 2,64 (с, 3H), 2,75-2,88 (м, 2H), 3,10 (дд, 1H), 3,40 (м, 1H), 3,57 (с, 3H), 6,54 (д, 1H), 7,20-7,27 (м, 1H), 7,31 (м, 1H), 7,65 (д, 1H), 8,10-8,05 (м, 1H).
214.3 Схема синтеза энантиомерно обогащенного соединения 214a

Для анантиомерно обогащенного элемента структуры, получаемого из элюируемого первым соединения препаративного примера 336, на стадии А применяли методику, аналогичную методике, описанной для синтеза соединения 214b, с получением Boc-защищенного предшественника соединения 214a.
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 9H), 2,04 (м, 2H), 2,69-2,85 (м, 7H), 3,70 (с, 3H), 4,29-4,50 (шир.м, 1H), 6,51 (д, 1H), 7,00-7,02 (м,1H), 7,05-7,12 (м, 1H), 7,61 (м, 2H).
MS (ESI): m/z=443,64 (MH+).
На фигуре 9 показан Boc-защищенный предшественник соединения 214a, получаемый из элюируемого первым диастереомера
Для анантиомерно обогащенного элемента структуры, получаемого из элюируемого первым диастереомера, на стадии В применяли методику, аналогичную методике, описанной для синтеза соединения 214b, с получением 214a.
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,90 (м, 1H), 2,25-2,28 (м, 1H), 2,50-2,64 (м, 1H), 2,64 (с, 3H), 2,75-2,88 (м, 2H), 3,10 (дд, 1H), 3,40 (м, 1H), 3,57 (с, 3H), 6,54 (д, 1H), 7,20-7,27 (м, 1H), 7,31 (м, 1H), 7,65 (д, 1H), 8,10-8,05 (м, 1H).
214.4 Выводы
Энантиомерно обогащенные соединения 214a и 214b получали с использованием элементов структуры, получаемых путем разделения диастереомеров. Элемент структуры, получаемый из элюируемого первым диастереомера, представлял собой энантиомерно обогащенное соединение (Boc-защищенное соединение 214a), содержащее 89% элюируемого первым энантиомера и 10,6% элюируемого вторым энантиомера. Элемент структуры, получаемый из элюируемого вторым диастереомера, представлял собой энантиомерно обогащенное соединение (Boc-защищенное соединение 214b), содержащее 28,5% элюируемого первым энантиомера и 69,4% элюируемого вторым энантиомера.
Пример 215: Получение соединений 215a и 215b (энантиомеров соединения 26), используя названные соединения препаративного примера 337 (разделение с помощью кристаллизации)
215.1 Цель и способ проведения исследования
Целью этого исследования являлся синтез соединений 215a и 215b, двух энантиомеров соединения 26, используя энантиомерно чистые элементы структуры, получаемые с помощью кристаллизации диастереомерных солей.
215.2 Схема синтеза энантиомерно чистого соединения 215b
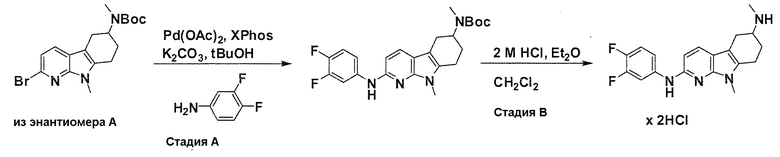
Стадия А
Трет-бутанол (13 мл) добавляли к смеси 2-дициклогексил-фосфино-2',4',6'-триизопропилбифенила (XPhos, 0,119 г, 0,25 ммоль) и ацетата палладия(II) (0,0198 г, 0,084 ммоль). Смесь дегазировали путем обработки ультразвуком в течение 1 минуты при одновременном пропускании через раствор аргона. Эту смесь нагревали при ~100°C на песочной бане в течение 1 минуты для образования катализатора. К красноватому раствору катализатора затем добавляли названное соединение препаративного примера 337, получаемое из элюируемого первым энантиомера A (0,33 г, 0,84 ммоль), выпускаемый промышленностью 3,4-дифторанилин (0,125 г, 0,99 ммоль), растворенный в 1 мл трет-бутанола, и карбонат калия (0,257 г, 1,85 ммоль). Смесь нагревали на песочной бане при ~110°C в течение 3 часов. Смесь разбавляли этилацетатом (250 мл) и водой (25 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (от 5/95 до 30/70), с получением названного соединения в виде серовато-белого твердого вещества (0,35 г, 95%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,42 (с, 9H), 1,92-2,04 (м, 2H), 2,67 (м, 2H), 2,78-2,92 (м, 7H), 3,61 (с, 3H), 4,08-4,34 (шир.м, 1H), 6,53 (д, 1H), 7,25-7,33 (м, 1H), 7,35-7,38 (м, 1H), 7,65 (д, 1H), 8,17 (дд, 1H), 9,16 (с, 1H).
1H-ЯМР (400 МГц, CDCl3): δ=1,58 (с, 9H), 2,10-2,18 (м, 2H), 2,82-3,00 (м, 7H), 3,77 (с, 3H), 4,34-4,63 (шир.м, 1H), 6,48 (шир.с, 1H), 6,60 (д, 1H), 7,09-7,12 (м, 1H), 7,13-7,22 (м, 1H), 7,67 (д, 1H), 7,85 (ддд, 1H).
MS (ESI); m/z=443,64 (MH+).
Стадия В
Названное соединение стадии А выше (0,35 г, 0,79 ммоль) растворяли в дихлорметане (8 мл). Раствор охлаждали при 0°C и затем обрабатывали 2M раствором хлористого водорода (8 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали диэтиловым эфиром (15 мл) и сушили с получением названного соединения в виде серовато-белого твердого вещества (0,32 г, 95%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,85-1,95 (м, 1H), 2,25-2,28 (м, 1H), 2,59-2,64 (м, 1H), 2,64 (с, 3H), 2,73-2,88 (м, 2H), 3,10 (дд, 1H), 3,40 (м, 1H), 3,57 (с, 3H), 6,54 (д, 1H), 7,20-7,27 (м, 1H), 7,32-7,34 (м, 1H), 7,65 (д, 1H), 8,10-8,05 (м, 1H).
[α]25 D=-54,0° (c 0,1, MeOH).
215.3 Схема синтеза энантиомерно чистого соединения 215a

Стадия А
Трет-бутанол (4 мл) добавляли к смеси 2-дициклогексил-фосфино-2',4',6'-триизопропилбифенил (XPhos, 0,031 г, 0,065 ммоль) и ацетата палладия(II) (0,0049 г, 0,022 ммоль). Смесь дегазировали путем обработки ультразвуком в течение 1 минуты при одновременном пропускании через раствор аргона. Эту смесь нагревали при ~100°C на песочной бане в течение 1 минуты для образования катализатора. К красноватому раствору катализатора затем добавляли названное соединение препаративного примера 337, получаемое из элюируемого вторым энантиомера B (0,086 г, 0,218 ммоль), выпускаемый промышленностью 3,4-дифторанилин (0,024 мл, 0,240 ммоль), растворенный в 1 мл трет-бутанола, и карбонат калия (0,060 г, 0,436 ммоль). Смесь нагревали на песочной бане при ~110°C в течение 3 часов. Смесь разбавляли этилацетатом (250 мл) и водой (25 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали, используя систему очистки Biotage Isolera One с градиентом этилацетат/н-гептан (от 10/90 до 30/70), с получением названного соединения в виде серовато-белого твердого вещества (0,095 г, 98%).
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 9H), 2,04 (м, 2H), 2,69-2,85 (м, 7H), 3,70 (с, 3H), 4,29-4,50 (шир.м, 1H), 6,51 (д, 1H), 7,00-7,02 (м,1H), 7,05-7,12 (м, 1H), 7,61 (м, 2H).
MS (ESI); m/z=443,68 (MH+).
Стадия В
Названное соединение стадии А выше (0,095 г, 0,21 ммоль) растворяли в дихлорметане (4 мл). Раствор охлаждали при 0°C и затем обрабатывали 2M раствором хлористого водорода (2 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали диэтиловым эфиром (10 мл) и сушили с получением названного соединения в виде серовато-белого твердого вещества (0,078 г, 87%).
1H-ЯМР (400 МГц, ДМСО-d6/D2O): δ=1,82-2,00 (м, 1H); 2,21-2,35 (м, 1H); 2,57-2,70 (м, 30 4H); 2,72-2,93 (м, 2H); 3,10 (дд, 1H); 3,34-3,48 (м, 1H); 3,58 (с, 3H); 6,56 (д, 1H); 7,25 (кв, 1H); 7,31-7,40 (м, 1H); 7,66 (д, 1H); 8,10 (дд, 1H).
MS (ESI); m/z=343,78 (M+H).
[α]25 D=+20,0° (c 0,1, MeOH).
215.4 Выводы
Энантиомерно чистые соединения 215a и 215b получали, используя энантиомерные элементы структуры, получаемые путем кристаллизации диастереомерных солей.
Пример 216: Получение соединений 216a и 216b (энантиомеров соединения 42), используя названные соединения препаративного примера 336 (разделение диастереомеров)
216.1 Цель и способ проведения исследования
Целью этого исследования являлся синтез соединений 216a и 216b, двух энантиомеров соединения 42, используя энантиомерно чистые элементы структуры, получаемые разделением диастереомеров.
216.2 Схема синтеза энантиомерно обогащенного соединения 216b
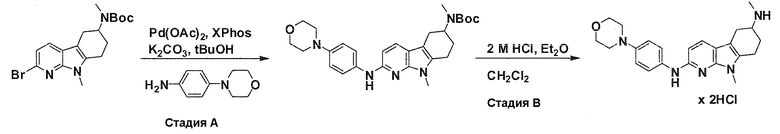
Стадия А
Трет-бутанол (2 мл) добавляли к смеси 2-дициклогексил-фосфино-2',4',6'-триизопропилбифенила (XPhos, 0,0108 г, 0,022 ммоль) и ацетата палладия(II) (0,0017 г, 0,0076 ммоль). Смесь дегазировали путем обработки ультразвуком в течение 1 минуты при одновременном пропускании через раствор аргона. Эту смесь нагревали при ~100°C на песочной бане в течение 1 минуты для образования катализатора. К красноватому раствору катализатора затем добавляли названное соединение препаративного примера 336, получаемое из элюируемого вторым диастереомера (0,03 г, 0,076 ммоль), выпускаемый промышленностью 4-морфолиноанилин (0,0169 г, 0,095 ммоль) и карбонат калия (0,0262 г, 0,19 ммоль). Смесь нагревали на песочной бане при ~110°C в течение 3 часов. Смесь разбавляли этилацетатом (100 мл) и водой (10 мл). Органическую фазу отделяли, сушили над Na2SO4, фильтровали, и растворители удаляли. Остаток очищали дважды, используя систему флэш-хроматографической очистки Biotage Isolera (MeOH/DCM: 0-1%, затем EtOAc/н-гептан: 10-30%), с получением названного соединения в виде серовато-белого твердого вещества (0,024 г, 64%).
1H-ЯМР (400 МГц, CDCl3): δ=1,49 (с, 9H), 2,02 (м, 2H), 2,76-2,86 (м, 6H), 3,02-3,32 (м, 2H), 3,63 (с, 3H), 3,88 (м, 4H), 4,31-4,52 (шир.м, 1H), 6,93 (шир.с, 1H), 7,17-7,19 (м, 1H), 7,24-7,27 (м, 1H), 7,52 (шир.с, 1H).
MS (ESI); m/z=492,72 (MH+).
Стадия В
Названное соединение стадии А выше (0,02 г, 0,040 ммоль) растворяли в дихлорметане (2,0 мл) и обрабатывали 2 M раствором хлористого водорода (0,5 мл) в диэтиловом эфире. Смесь перемешивали при комнатной температуре в течение ночи, и осадок отфильтровывали. Твердые вещества промывали диэтиловым эфиром (5 мл) и сушили при пониженном давлении с получением названного соединения в виде белого твердого вещества (0,016 г, 99%).
1H-ЯМР (400 МГц, D2O): δ=2,11 (м, 1H), 2,36 (м, 1H), 2,71-2,79 (м, 6H), 3,06-3,13 (м, 1H), 3,53 (м, 8H), 4,0 (м, 4H), 6,59 (с, 1H), 7,4 (м, 2H), 7,55 (м, 2H), 7,77 (с, 1H).
216.2 Схема синтеза энантиомерно обогащенного соединения 216a

Применяли методику, аналогичную методике для стадий А и B, описанной для синтеза соединения 216b, для энантиомерно обогащенного элемента структуры, получаемого из элюируемого первым соединения препаративного примера 336, с получением соединения 216a.
MS (ESI); m/z=391,99 (M+H)
216.4 Выводы
Энантиомерно обогащенные соединения 216a и 216b получали, используя элементы структуры, получаемые в результате разделения диастереомеров.
Пример 217: Получение соединений 217a и 217b (энантиомеров соединения 156), используя названные соединения препаративного примера 336 (разделение диастереомеров)
217.1 Цель и способ проведения исследования
Целью этого исследования являлся синтез соединений 217a и 217b, двух энантиомеров соединения 156, используя энантиомерно обогащенные элементы структуры, получаемые разделением диастереомеров.
217.2 Схема синтеза энантиомерно обогащенного соединения 217b

Стадия А
К смеси XPhos (0,0108 мг, 0,023 ммоль) и ацетата палладия(II) (0,0017 г, 0,008 ммоль) добавляли 4 мл дегазированного трет-бутанола (подвергнутого воздействию ультразвука в токе аргона). Полученный раствор подогревали до 80°C в течение 90 секунд (до тех пор, пока не появлялась темно-красная окраска раствора). Активированный катализатор переносили во вторую колбу, содержащую названное соединение препаративного примера 336, полученного из элюируемого вторым диастереомера выше (0,030 г, 0,076 ммоль), 2,3-дигидробензо[b][1,4]диоксин-6-амин (0,0115 г, 0,076 ммоль) и карбонат калия (0,021 г, 0,152 ммоль). Полученную смесь подогревали до 110°C в течение 3 часов. Реакционную смесь концентрировали досуха. Остаток очищали с помощью системы флэш-хроматографической очистки Biotage с градиентом EtOAc/н-гептан от 15% до 30% с получением названного соединения в виде бежевой пены (0,036 г, 100%).
MS (ESI); m/z=465,70 (M+H).
Стадия В
К раствору названного соединения стадии А выше (0,034 г, 0,073 ммоль) в 4 мл дихлорметана добавляли 2M раствор хлористого водорода в диэтиловом эфире (0,183 мл, 0,366 ммоль) и 0,050 мл метанола. Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции, смесь концентрировали досуха и добавляли несколько капель MeOH для растворения остатка. И наконец, добавляли EtOAc для осаждения продукта. Суспензию отфильтровывали и сушили под вакуумом в течение 2 часов, получая названное соединение в виде желтого твердого вещества (0,016 г, 50%).
1H-ЯМР (400 МГц, MeOD): δ=2,03-2,17 (м, 1H); 2,36-2,48 (м, 1H); 2,70-3,03 (м, 6H); 3,18-3,30 (м, 1H); 3,51-3,63 (м, 1H); 3,74 (с, 3H); 4,27 (с, 4H); 6,64 (д, J=8,0 Гц, 1H); 6,78-7,05 (м, 3H); 8,05 (д, J=8,0 Гц, 1H).
MS (ESI); m/z=365,75 (M+H)/406,79 (M+ACN+H).
217.3 Схема синтеза энантиомерно обогащенного соединения 217a
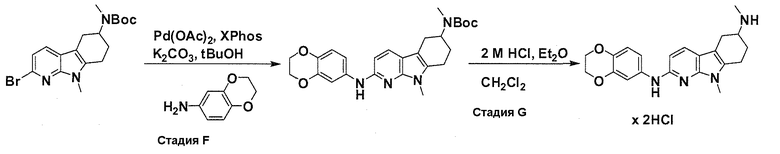
Применяли методику, аналогичную методике для стадии А, описанной для синтеза соединения 217b, для энантиомерно обогащенного элемента структуры, получаемого из элюируемого первым диастереомера препаративного примера 336, с получением Boc-защищенного предшественника соединения 217a.
MS (ESI); m/z=465,70 (M+H).
Стадия В
Применяли методику, аналогичную методике для стадии В, описанной для синтеза соединения 217b, для энантиомерно обогащенного элемента структуры, получаемого из элюируемого первым диастереомера, с получением соединения 217a (0,015 г, 42%).
1H-ЯМР (400 МГц, MeOD): δ=2,03-2,19 (м, 1H); 2,34-2,49 (м, 1H); 2,70-3,06 (м, 6H); 3,18-3,36 (м, 1H); 3,50-3,65 (м, 1H); 3,74 (с, 3H); 4,26 (с, 4H); 6,64 (д, J=8,0 Гц, 1H); 6,77-7,05 (м, 3H); 8,05 (д, J=8,0 Гц, 1H).
MS (ESI); m/z=365,74 (M+H)/406,77 (M+ACN+H).
217.4 Выводы
Энантиомерно обогащенные соединения 217a и 217b получали, используя элементы структуры, получаемые в результате разделения диастереомеров.
Пример 218: Получение соединений 218a и 218b (энантиомера соединения 156), используя названное соединение препаративного примера 337 (разделение кристаллизацией)
218.1 Цель и способ проведения исследования
Целью этого исследования являлся синтез соединений 218a и 218b, двух энантиомеров соединения 156, используя энантиомерно чистые элементы структуры, получаемые кристаллизациии диастереомерных солей.
218.2 Схема синтеза энантиомерно чистого соединения 218b
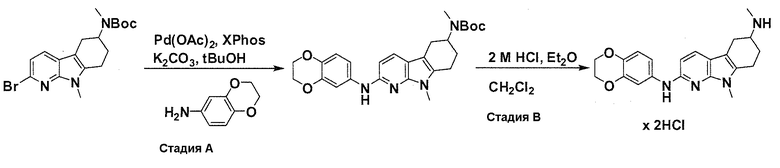
Стадия А
К смеси XPhos (0,0272 мг, 0,057 ммоль) и ацетата палладия(II) (0,0042 г, 0,019 ммоль) добавляли 4 мл дегазированного трет-бутанола (подвергнутого воздействию ультразвука в токе аргона). Полученный раствор подогревали до 80°C в течение 90 секунд (до тех пор, пока не появлялась темно-красная окраска раствора). Активированный катализатор переносили во вторую колбу, содержащую названное соединение препаративного примера 337, полученного из элюируемого первым энантиомера A (0,075 г, 0,190 ммоль), 2,3-дигидробензо[b][1,4]диоксин-6-амин (0,0288 мг, 0,190 ммоль) и карбонат калия (0,0526 мг, 0,380 ммоль). Полученную смесь подогревали до 110°C в течение 3 часов. Реакционную смесь концентрировали досуха. Остаток очищали с помощью системы флэш-хроматографической очистки Biotage с градиентом EtOAc/н-гептан от 15% до 35% с получением названного соединения в виде белого твердого вещества (0,083 г, 94%).
MS (ESI); m/z=465,70 (M+H).
Стадия В
К раствору названного соединения стадии А выше (0,082 г, 0,177 ммоль) в 4 мл дихлорметана добавляли 2M раствор хлористого водорода в диэтиловом эфире (0,183 мл, 0,366 ммоль) и 0,050 мл метанола. Полученную смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции, смесь концентрировали досуха и добавляли несколько капель MeOH для растворения остатка. И наконец, добавляли EtOAc для осаждения продукта. Суспензию отфильтровывали и сушили под вакуумом в течение 2 часов, получая названное соединение в виде желтоватого твердого вещества (0,074 г, 96%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,88-2,07 (м, 1H); 2,27-2,40 (м, 1H); 2,62 (т, J=5,2 Гц, 3H); 5 2,67-2,95 (м, 3H); 3,10 (дд, J1=5,2 Гц, J2=15,2 Гц, 1H); 3,33-3,48 (м, 1H); 3,60 (с, 3H); 4,15-4,27 (м, 4H); 6,53 (д, J=8,4 Гц, 1H); 6,75 (д, J=8,8 Гц, 1H); 7,06 (дд, J1=2,4 Гц, 8,4 Гц, 1H); 7,61 (с, 1H); 7,62 (д, J=10,0 Гц, 1H); 7,60 (д, J=2,4 Гц, 1H); 7,62 (д, J=8,4 Гц, 1H); 9,23 (с, 2H).
MS (ESI); m/z=365,75 (M+H)/406,79 (M+ACN+H).
218.3 Схема синтеза энантиомерно обогащенного соединения 218a

Применяли методику, аналогичную методике для стадии А, описанной для синтеза соединения 218b, для энантиомерно чистого элемента структуры, получаемого из элюируемого вторым энантиомера B препаративного примера 337, с получением Boc-защищенного предшественника соединения 218a.
MS (ESI); m/z=465,70 (M+H).
Стадия В
Применяли методику, аналогичную методике для стадии В, описанной для синтеза соединения 218b, для энантиомерно чистого элемента структуры, получаемого из элюируемого вторым энантиомера B, с получением соединения 218a (0,069 г, 85%) в виде светло-желтого твердого вещества.
1H-ЯМР (400 МГц, ДМСО-d6): δ=1,89-2,05 (м, 1H); 2,26-2,40 (м, 1H); 2,64 (т, J=5,2 Гц, 3H); 2,67-2,95 (м, 3H); 3,10 (дд, J1=4,4 Гц, J2=14,4 Гц, 1H); 3,33-3,48 (м, 1H); 3,60 (с, 3H); 4,13-4,27 (м, 4H, подводный пик); 6,53 (д, J=8,4 Гц, 1H); 6,76 (д, J=8,8 Гц, 1H); 7,05 (дд, J1=2,4 Гц, 8,4 Гц, 1H); 7,61 (с, 1H); 7,62 (д, J=10,0 Гц, 1H); 9,18 (с, 2H).
MS (ESI); m/z=365,74 (M+H)/406,77 (M+ACN+H).
Пример 219: Ингибирование агрегации бета-амилоидного (Aβ)1-42 пептида (спектрофлуоресцентный анализ с тиофлавином T (ThT) анализ)
Рацемические соединения и их энантиомеры подвергали испытанию на их способность ингибировать агрегацию амилоидного пептида с помощью ThT анализа. Для определения IC50, при проведении описанного выше ThT анализа использовали разбавления соединений до следующих значений: 330 мкМ, 82,50 мкМ, 20,63 мкМ, 5,16 мкМ, 1,29 мкМ, 0,32 мкМ и 0,08 мкМ

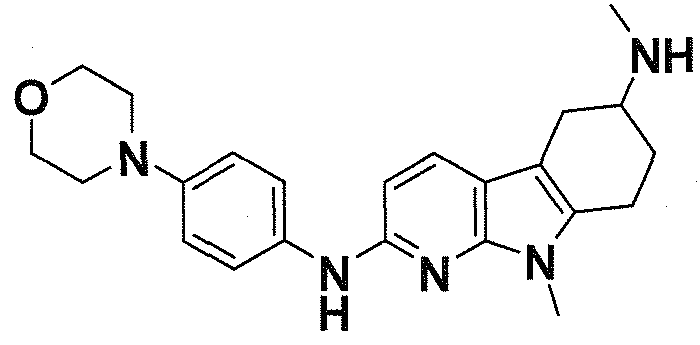

Пункты изобретения приводятся ниже:
1. Соединение формулы (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где
A выбирают из группы, состоящей из
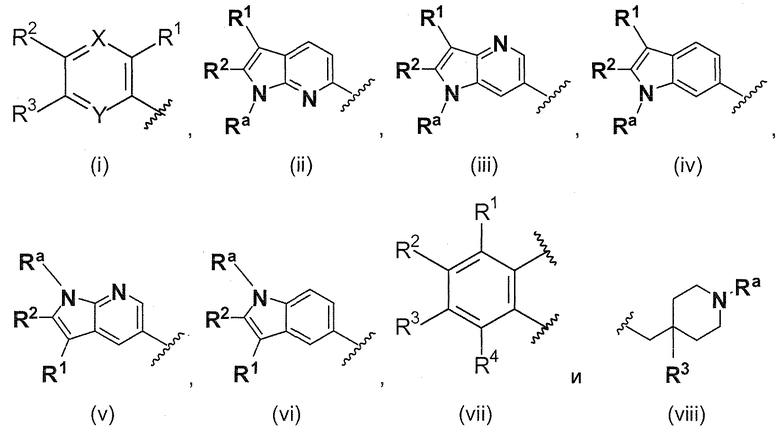
L1 направленно выбирают из группы, состоящей из

B выбирают из группы, состоящей из
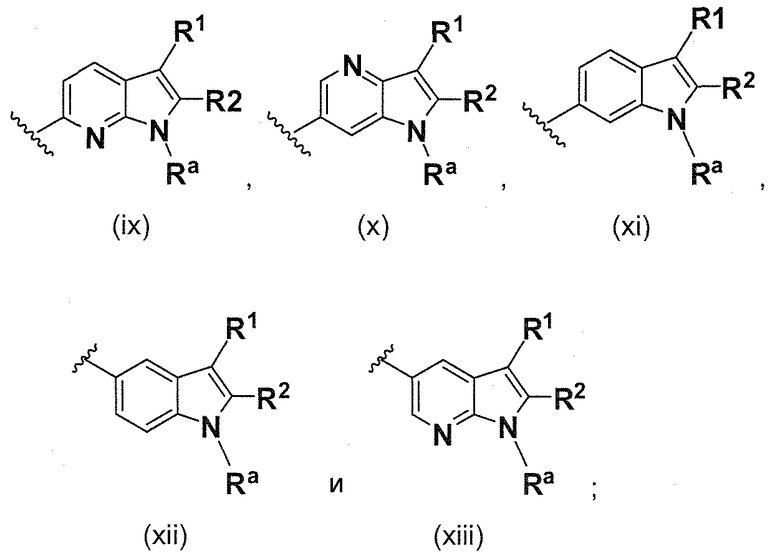
где
R1, R2, R3 и R4, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если любые из этих групп R1/R2/R3/R4 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5-8-членное кольцо, содержащее углеродные атомы и необязательно один или более гетероатомов, выбранных из O, S или N, или необязательно один или более гетероатом- (например, N, O и/илиr S) содержащих фрагментов, и где 5-8-членное кольцо может быть замещено с помощью NR20R21;
для каждого случая присутствия, Ra независимо выбирают из группы, состоящей из водорода, алкила, галогеналкила, S(O)tNR30R31, S(O)tR30, C(O)OR30, C(O)R30, и C(O)NR30R31;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными, или где R20 и R21, взятые вместе с азотом, к которому они присоединены, могут образовывать 3-8-членное кольцо, содержащее углеродные атомы и необязательно один или более дополнительных гетероатомов, выбранных из O, S или N, или необязательно один или более гетероатом- (например, N, O и/или S) содержащих фрагментов, и где 3-8-членное кольцо может быть необязательно замещенным;
X и Y, каждый независимо выбирают из группы, состоящей из CRb и N;
t равняется 1 или 2; и
p равняется 0, 1 или 2
при условии, что исключаются следующие соединения:


 .
.
2. Соединение по пункту 1, где
A выбирают из группы, состоящей из
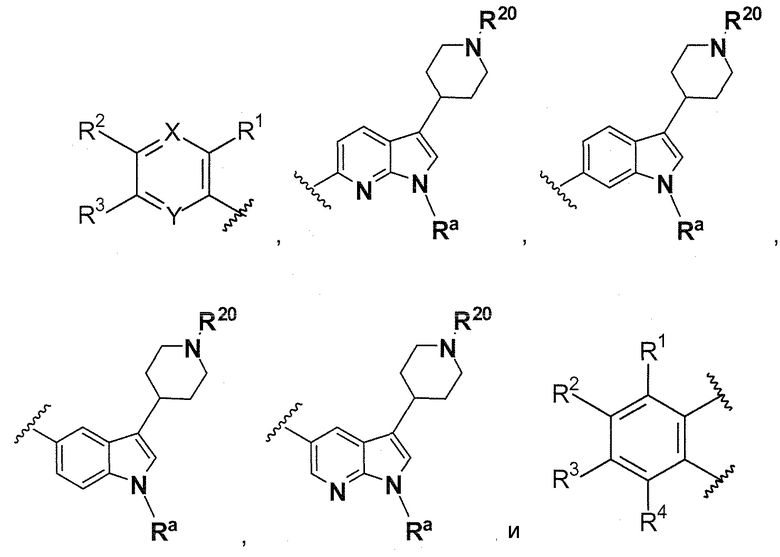
L1 представляет собой
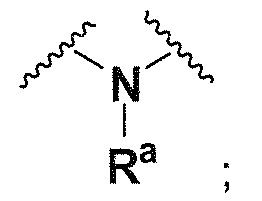
B выбирают из группы, состоящей из

где:
R1, R2, R3, R4, Ra, R20, X и Y имеют такие же значения, как в пункте 1;
V отсутствует или представляет собой NR20R21, и
z равняется 1 или 2.
3. Соединение по пункту 1 или 2, где A имеет формулу (i).
4. Соединение по любому из предшествующих пунктов, где L1 имеет формулу (a), и предпочтительно, чтобы p равнялось 0.
5. Соединение по любому из предшествующих пунктов, где R1, R2, R3 и R4, каждый независимо выбирают из водорода, галогена (таконо как F), CN, CF3, CONR30R31, -O-алкила, гетероциклоалкила (такого как 
 ).
).
6. Соединение по пункту 1, где формулу (vii) в A выбирают из  .
.
7. Соединение по любому из предшествующих пунктов, где Ra представляет собой водород или C1-4 алкил.
8. Соединение по пункту 1, где A имеет формулу (i)
 .
.
9. Соединение по пункту 1, где A имеет формулу (ii)
 .
.
10. Соединение по пункту 1, где A имеет формулу (iii)
 .
.
11. Соединение по пункту 1, где A имеет формулу (iv)
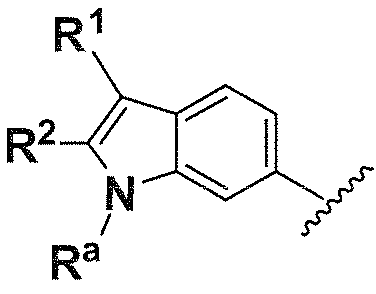 .
.
12. Соединение по пункту 1, где A имеет формулу (v)
 .
.
13. Соединение по пункту 1, где A имеет формулу (vi)
 .
.
14. Соединение по пункту 1, где A имеет формулу (vii)
 .
.
15. Соединение по пункту 1, где L1 имеет формулу (a)
 .
.
16. Соединение по пункту 1, где B имеет формулу (ix)
 .
.
17. Соединение по пункту 1, где B имеет формулу (x)
 .
.
18. Соединение по пункту 1, где B имеет формулу (xi)
 .
.
19. Соединение по пункту 1, где B имеет формулу (xii)
 .
.
20. Соединение по пункту 1, где B имеет формулу (xiii)
 .
.
21. Соединение по пункту 1, имеющее формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой:

L1 представляет собой:

B представляет собой:
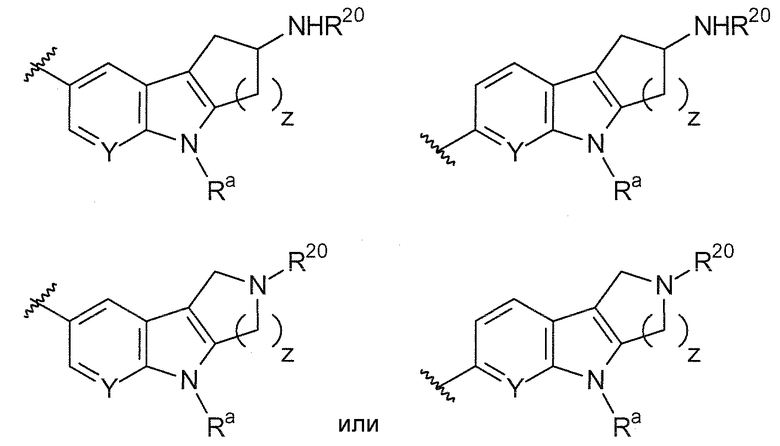
где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50 представляет собой для каждого случая присутствия R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N;
t равняется 1 или 2; и
z равняется 1 или 2.
22. Соединение по пункту 1, имеющее формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой:
L1 представляет собой:

B представляет собой:
 ,
,
где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50 представляет собой, для каждого случая присутствия, R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N;
t равняется 1 или 2; и
z равняется 1 или 2.
23. Соединение по пункту 1, имеющее формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой:
L1 представляет собой:
B представляет собой:
 ,
,
где
R1 и R2, каждый независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил и гетероарилалкил могут быть необязательно замещенными, или если R1 и R2 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из O, S или N, или гетероатом-содержащий фрагмент NR50;
R3 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия, Rb независимо выбирают из группы, состоящей из водорода, галогена, CN, CF3, CONR30R31, алкила, -O-алкила, -C(O)O-алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила и гетероарилалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
для каждого случая присутствия, R30, R31, R20 и R21, каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, циклоалкилалкила, гетероциклоалкила, фторалкила, гетероциклоалкилалкила, алкенила, алкинила, арила, гетероарила, арилалкила, гетероарилалкила и аминоалкила, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, фторалкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными;
R50 представляет собой, для каждого случая присутствия, R20, S(O)tNR20R21, S(O)tR20, C(O)OR20, C(O)R20C(=NRa)NR20R21, C(=NR20)NR21Ra, C(=NOR20)R21 или C(O)NR20R21;
Y, каждый представляет собой независимо CH или N; и
t равняется 1 или 2.
24. Соединение по пункту 1, имеющее формулу (I):
A-L1-B (I)
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли, гидраты, сольваты и полиморфы;
где A представляет собой:

L1 представляет собой:
и B представляет собой:

25. Соединение по любому из предшествующих пунктов, где соединение выбирают из группы, состоящей из
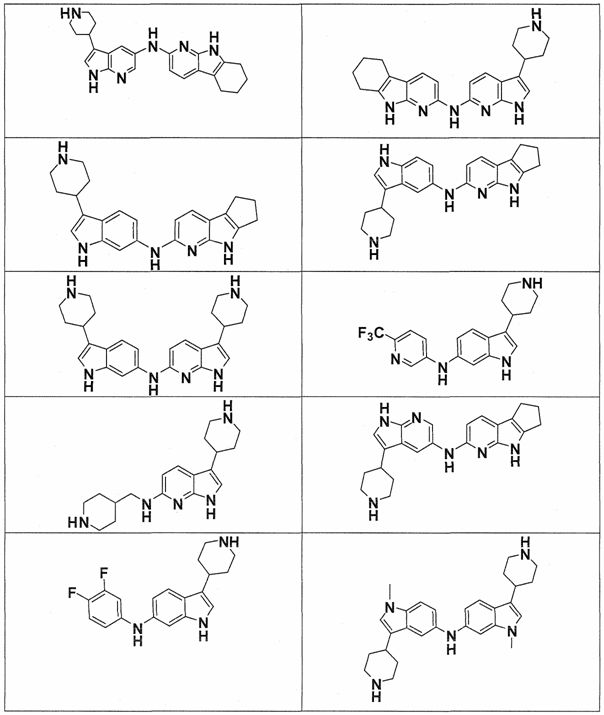

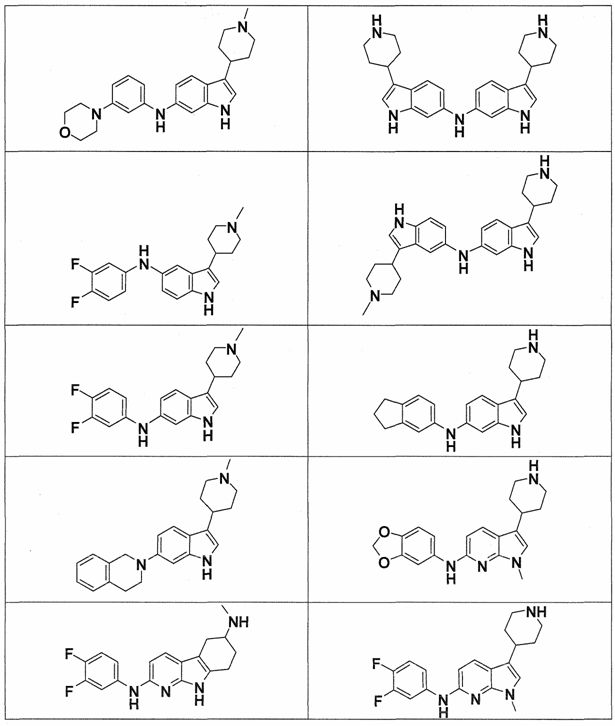
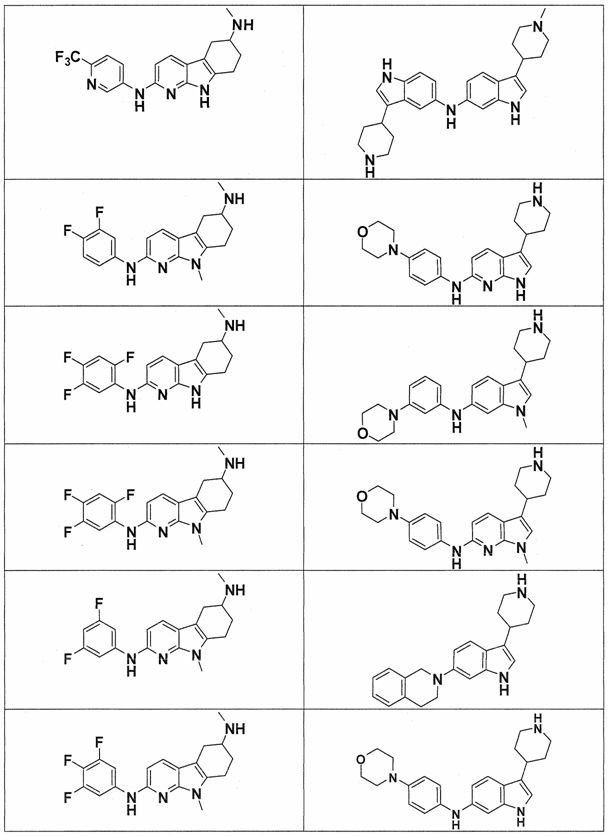

26. Соединение по любому одному из пунктов 1-25, включающее радиоактивный изотоп, где исключаются соединения, которые исключены в пункте 1.
27. Радиофармацевтический препарат, включающий соединение по любому одному из пунктов 1-25, содержащее радиоактивный изотоп, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
28. Фармацевтическая композиция, включающая соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1.
29. Фармацевтическая композиция по пункту 28, дополнительно включающая фармацевтически приемлемый носитель или вспомогательное вещество.
30. Применение соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для приготовления лекарственного средства для лечения или предотвращения заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком.
31. Применение по пункту 30, где заболевание представляет собой неврологическое нарушение.
32. Применение по пункту 31, где неврологическое нарушение представляет собой болезнь Альцгеймера (AD), деменцию с тельцами Леви (LBD), синдром Дауна, врожденное внутримозговое кровоизлияние с амилоидозом (голландского типа), комплекс Гуама паркинсонизм-деменция или умеренное когнитивное нарушение (MCI).
33. Применение по пункту 32, где неврологическое нарушение представляет собой болезнь Альцгеймера.
34. Применение по пункту 30, где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы.
35. Способ лечения или предотвращения заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
36. Способ по пункту 35, где заболевание представляет собой неврологическое нарушение.
37. Способ по пункту 36, где неврологическое нарушение представляет собой болезнь Альцгеймера (AD), деменцию с тельцами Леви (LBD), синдром Дауна, врожденное внутримозговое кровоизлияние с амилоидозом (голландского типа), комплекс Гуама паркинсонизм-деменция или умеренное когнитивное нарушение (MCI).
38. Способ по пункту 37, где неврологическое нарушение представляет собой болезнь Альцгеймера.
39. Способ по пункту 35, где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы).
40. Соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для применения при лечении или предотвращении заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком.
41. Соединение по пункту 40, где заболевание представляет собой неврологическое нарушение.
42. Соединение по пункту 41, где неврологическое нарушение представляет собой болезнь Альцгеймера (AD), деменцию с тельцами Леви (LBD), синдром Дауна, врожденное внутримозговое кровоизлияние с амилоидозом (голландского типа), комплекс Гуама паркинсонизм-деменция или умеренное когнитивное нарушение (MCI).
43. Соединение по пункту 42, где неврологическое нарушение представляет собой болезнь Альцгеймера.
44. Соединение по пункту 40, где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы.
45. Смесь, включающая соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, и необязательно по меньшей мере одно дополнительное биологически активное соединение и/или фармацевтически приемлемый носитель и/или разбавитель и/или вспомогательное вещество.
46. Смесь по пункту 45, где дополнительное биологически активное соединение представляет собой соединение, применяемое при лечении амилоидоза.
47. Смесь по пункту 45 или 46, где дополнительное биологически активное соединение выбирают из группы, состоящей из антител, вакцин, соединений против окислительного стресса, антиапоптотических соединений, соединений, образующих хелаты с металлами, ингибиторов репарации ДНК, таких как пирензепин и метаболиты, 3-амино-1-пропансульфоновой кислоты (3APS), 1,3-пропандисульфоната (1,3PDS), активаторов α-секретазы, ингибиторов β- и γ-секретазы, тау-белков, нейротрансмиттеров, разрушителей β-амилоидного белка, аттрактантов для очистки от бета амилоида/истощения клеточных компонентов, ингибиторов N-терминально усеченного бета амилоида, включая бета амилоид 3-42 в форме пироглутамата, противовоспалительных средств, или ингибиторов холинэстеразы (ChEI) таких как такрин, ривастигмин, донепезил и/или галантамин, агонистов М1 и других лекарственных средств, включая любые лекарственные средства, модифицирующие амилоидные или тау белки, и питательных добавок.
48. Смесь по пункту 47, где дополнительное биологически активное соединение представляет собой ингибитор холинэстеразы (ChEI).
49. Смесь по пункту 47, где дополнительное биологически активное соединение выбирают из группы, состоящей из такрина, ривастигмина, донепезила, галантамина, ниацина и мемантина.
50. Смесь по пункту 45, где дополнительное биологически активное соединение представляет собой антитело, в частности, моноклональное антитело, включающее любое функционально эквивалентное антитело или его функциональные части.
51. Смесь по пункту 50, где антитело, в частности, моноклональное антитело, включающее любое функционально эквивалентное антитело или его функциональные части, представляет собой антитело, которое связывает β-амилоид.
52. Смесь по пункту 50 или 51, где антитело, в частности, моноклональное антитело, включающее любое функционально эквивалентное антитело или его функциональные части, представляет собой антитело, которое при совместной инкубации с амилоидными мономерными и/или полимерными растворимыми амилоидными пептидами, в частности, с β-амилоидными мономерными пептидами, такими, как например, Aβ мономерные пептиды 1-39; 1-40, 1-41 или 1-42, и/или полимерным растворимым β-амилоидным пептидом, включающим множество Aβ мономерных звеньев, но особенно с Aβ мономерным и/или Aβ полимерным растворимым амилоидным пептидом, включающим множество Aβ1-42 мономерных звеньев, ингибирует агрегацию Aβ мономеров в высокомолекулярные полимерные волокна или нити и, кроме того, при совместной инкубации с ранее образовавшимися высокомолекулярными полимерными амилоидными волокнами или нитями, образовавшимися в результате агрегации амилоидных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких, как например, Aβ мономерные пептиды, способно разрушать агрегаты образовавшихся полимерных волокон или нитей.
53. Смесь по пункту 50, где антитело представляет собой химерное антитело или его функциональную часть, или гуманизированное антитело или его функциональную часть.
54. Смесь по пункту 50, где антитело представляет собой моноклональное антитело, выбранное из группы антител, имеющих характерные свойства антитела, продуцированного линией клеток гибридомы:
(a) FP 12H3, FP 12H3-C2 DSM ACC2752;
(b) FP 12H3-C2, депонированное в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2750;
(c) FP 12H3-G2, депонированное в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2751;
(d) ET 7E3, депонированное в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2755; и
(e) EJ 7H3, депонированное в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2756.
55. Смесь по пункту 50, где антитело представляет собой моноклональное антитело, выбранное из группы антител, продуцированных линией клеток гибридомы:
(a) FP 12H3, депонированное в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2752;
(b) FP 12H3-C2, депонированное в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2750;
(c) FP 12H3-G2, депонированное в немецкой коллекции микроорганизмов 1 декабря 2005 г. и 9 декабря 2005 г., соответственно, как DSM ACC2751;
(d) ET 7E3, депонированное в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2755; и
(e) EJ 7H3, депонированное в немецкой коллекции микроорганизмов 8 декабря 2005 г. как DSM ACC2756.
56. Смесь по пункту 50, где антитело представляет собой гуманизированное антитело, обладающее легкой цепью и тяжелой цепью, которые показаны в последовательности SEQ ID NO: 2 и SEQ ID NO: 4 в Международной заявке № PCT/US2007/073504.
57. Смесь по пункту 50, где антитело представляет собой гуманизированное антитело, обладающее вариабельной областью легких цепей и вариабельной областью тяжелых цепей, которые показаны в последовательности SEQ ID NO: 1 и SEQ ID NO: 3 в Международной заявке № PCT/US2007/073504.
58. Смесь по пункту 45, где дополнительное биологически активное соединение представляет собой фрагмент Aβ антигенного пептида, состоящий из единственного или повторяющегося участка с множеством смежных аминокислотных остатков из N-терминальной части Aβ пептида, в частности, участка между 13 и 15 смежными аминокислотными остатками.
59. Смесь по пункту 58, где фрагмент Aβ антигенного пептида представляет собой Aβ1-15 пептидный антиген.
60. Смесь по пункту 58, где Aβ1-15 пептидный антиген представляет собой пальмитоилированный Aβ1-15 пептидный антиген, модифицированный путем ковалентного присоединенения пальмитоильных остатков, в частности, от 2 до 4, более предпочтительно, 4 остатков, на каждом конце пептида, встроенного в липосому.
61. Смесь по любому одному из пунктов 45-60, где соединение и/или дополнительное биологически активное соединение присутствуют в терапевтически эффективном количестве.
62. Способ сбора данных для диагностирования связанного с амилоидом заболевания или состояния в пробе или в организме пациента, включающий:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются;
(b) связывание соединения с амилоидным белком;
(c) определение соединения, связанного с белком; и
(d) необязательное установление взаимосвязи между присутствием или отсутствием соединения, связывающего амилоидный белок, с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела.
63. Способ определения степени отложения амилоидогенных бляшек в ткани и/или биологической жидкости организма, включающий:
(a) взятие пробы исследуемой ткани и/или биологической жидкости организма;
(b) испытание пробы на присутствие амилоидного белка с помощью соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются;
(c) определение количества соединения, связанного с амилоидным белком; и
(d) расчет степени отложения бляшек в ткани и/или биологической жидкости организма.
64. Способ по пункту 63, где определение на стадии (с) проводят так, что присутствие или отсутствие соединения, связывающего амилоидный белок, взаимосвязано с присутствием или отсутствием амилоидного белка.
65. Способ сбора данных для выявления предрасположенности к связанному с амилоидом заболеванию или состоянию у пациента, включающий обнаружение специфического связывания соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, с амилоидным белком в пробе или in situ, который включает стадии:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
66. Способ сбора данных с целью непрерывного контроля минимального остаточного заболевания у пациента после лечения с помощью композиции антитела или вакцины, где способ включает:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
67. Способ сбора данных с целью предсказания восприимчивости пациента к лечению с помощью композиции антитела или вакцины, включающий:
(a) контактирование пробы или специфической части тела или области тела, относительно которых существует подозрение по поводу присутствия амилоидного белка, с соединением по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, в результате которого соединение специфически связывает амилоидный белок;
(b) связывание соединения с амилоидным белком с образованием комплекса соединение/белок;
(c) обнаружение образования комплекса соединение/белок;
(d) необязательное установление взаимосвязи между присутствием или отсутствием комплекса соединение/белок с присутствием или отсутствием амилоидного белка в пробе или специфической части тела или области тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
68. Диагностический набор для обнаружения и/или диагностирования связанных с амилоидом заболевания или состояния, включающий соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
69. Диагностический набор по пункту 68, включающий контейнер, содержащий одно или более соединений по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, и инструкции по использованию соединения с целью связывания амилоидного белка с образованием комплекса соединение/белок и обнаружения образования комплекса соединение/белок, в соответствии с которыми присутствие или отсутствие комплекса соединение/белок взаимосвязано с присутствием или отсутствием амилоидного белка.
70. Применение соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для получения лекарственного средства для лечения или предотвращения глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности, связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета амилоиду.
71. Применение по пункту 70, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например, возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
72. Способ лечения или предотвращения глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности, связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета амилоиду, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
73. Способ по пункту 72, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например, возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
74. Соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для применения при лечении или предотвращении глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности, связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета-амилоиду.
75. Соединение по пункту 74, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например, возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
76. Соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для применения при ингибировании агрегации белка, в частности, при ингибировании агрегации Aβ1-42, агрегации тау белка или агрегации альфа-синуклеина.
77. Применение соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для получения лекарственного средства для (a) снижения отложения β-амилоидных бляшек и/или (b) ингибирования образования β-амилоидных бляшек и/или (c) замедления увеличения отложения амилоидных бляшек в мозге пациента.
78. Способ (a) снижения отложения β-амилоидных бляшек и/или (b) ингибирования образования β-амилоидных бляшек и/или (c) замедления увеличения отложения амилоидных бляшек в мозге пациента, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
79. Соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для применения при (a) снижении отложения β-амилоидных бляшек и/или (b) ингибировании образования β-амилоидных бляшек и/или (c) замедлении увеличения отложения амилоидных бляшек в мозге пациента.
80. Применение по пункту 77, способ по пункту 78 или соединение по пункту 79, где снижение отложения β-амилоидных бляшек и/или ингибирование образования β-амилоидных бляшек и/или замедление увеличения отложения амилоидных бляшек приводит к уменьшению и/или облегчению воздействия заболевания или состояния, вызываемого или связанного с образованием и отложением β-амилоидных бляшек в мозге.
81. Применение соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для получения лекарственного средства для сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти.
82. Способ сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются.
83. Соединение по любому одному из пунктов 1-25, где исключаются соединения, которые исключены в пункте 1, или не исключаются, для применения при сохранении или повышении когнитивной способности памяти у субъекта, страдающего нарушением памяти.
84. Применение по пункту 30, способ по пункту 35 или соединение по пункту 40, где состояние, связанное с амилоидом, характеризуется утратой когнитивной способности памяти.
85. Применение, способ или соединение по пункту 84, где лечение состояния, связанного с амилоидом, характеризующееся утратой когнитивной способности памяти, приводит к улучшению сохранения когнитивной способности памяти.
86. Применение, способ или соединение по пункту 84, где лечение состояния, связанного с амилоидом, характеризующееся утратой когнитивной способности памяти, приводит к полному восстановлению когнитивной способности памяти.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2,6-ДИАМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДНЫМИ БЕЛКАМИ, ИЛИ ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ БОЛЕЗНЕЙ | 2010 |
|
RU2538208C2 |
| ИНГИБИТОРЫ ПРОДУЦИРОВАНИЯ / СЕКРЕЦИИ β-АМИЛОИДНОГО БЕЛКА | 2002 |
|
RU2304140C2 |
| ПИРИДИЛДИМЕТИЛСУЛЬФОНОВОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2404968C2 |
| НАФТИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ СМЯГЧЕНИЯ СИМПТОМОВ ПОСТКЛИМАКТЕРИЧЕСКОГО СИНДРОМА И ДРУГИХ СВЯЗАННЫХ С ЭСТРОГЕНОМ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, СПОСОБЫ ПОЛУЧЕНИЯ НАФТИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 1995 |
|
RU2165924C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2135484C1 |
| СОЕДИНЕНИЯ ДЛЯ ВИЗУАЛИЗАЦИИ АГРЕГАТОВ БЕЛКА ТАУ | 2017 |
|
RU2778739C2 |
| 6,7-МОДИФИЦИРОВАННЫЕ ПАКЛИТАКСЕЛЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1993 |
|
RU2125998C1 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА RIP-1 КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2800652C2 |
| ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2691105C1 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |

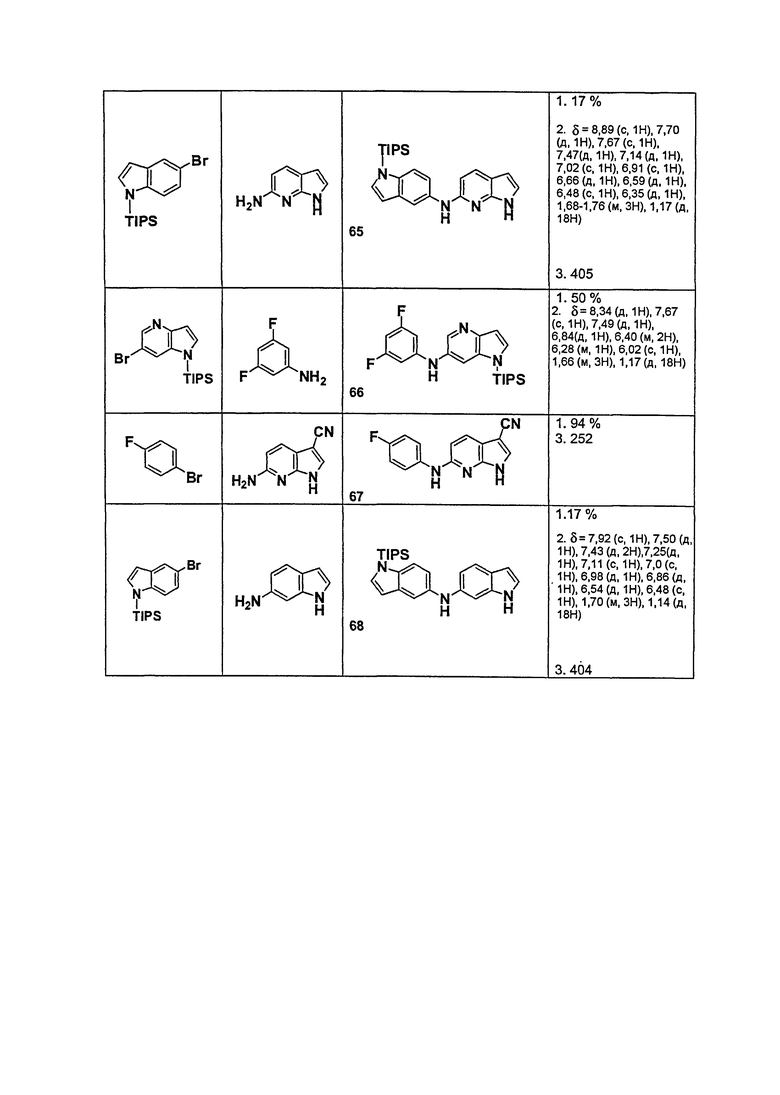


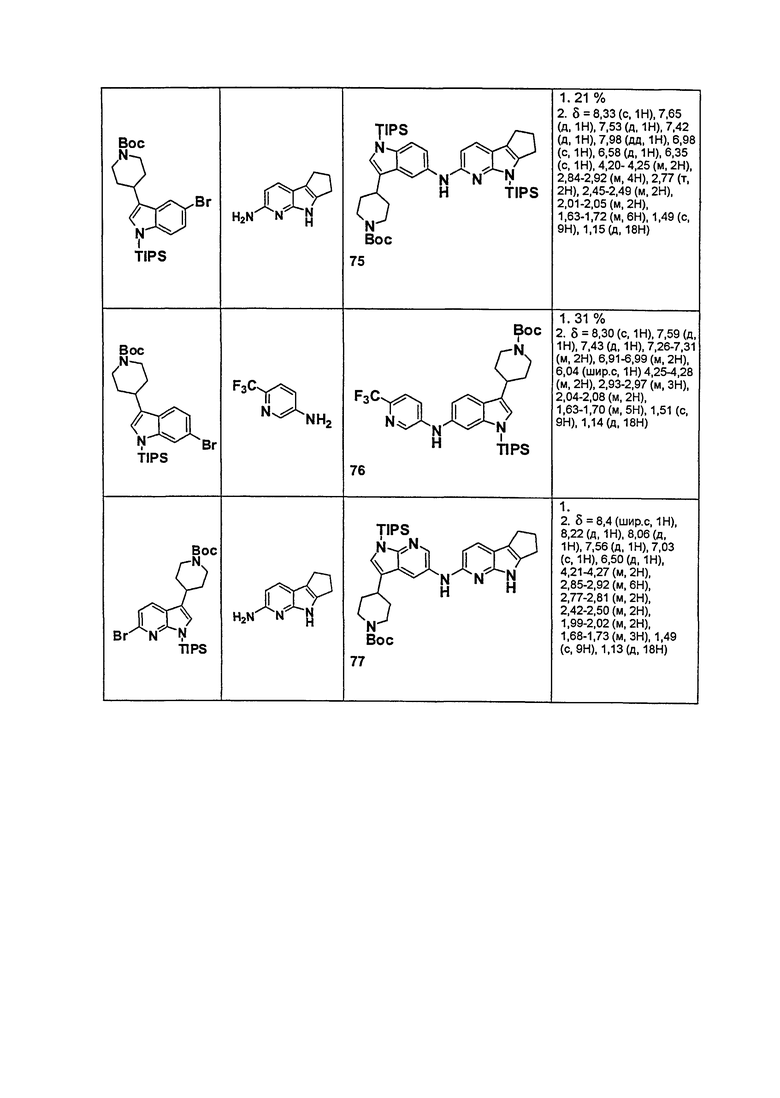



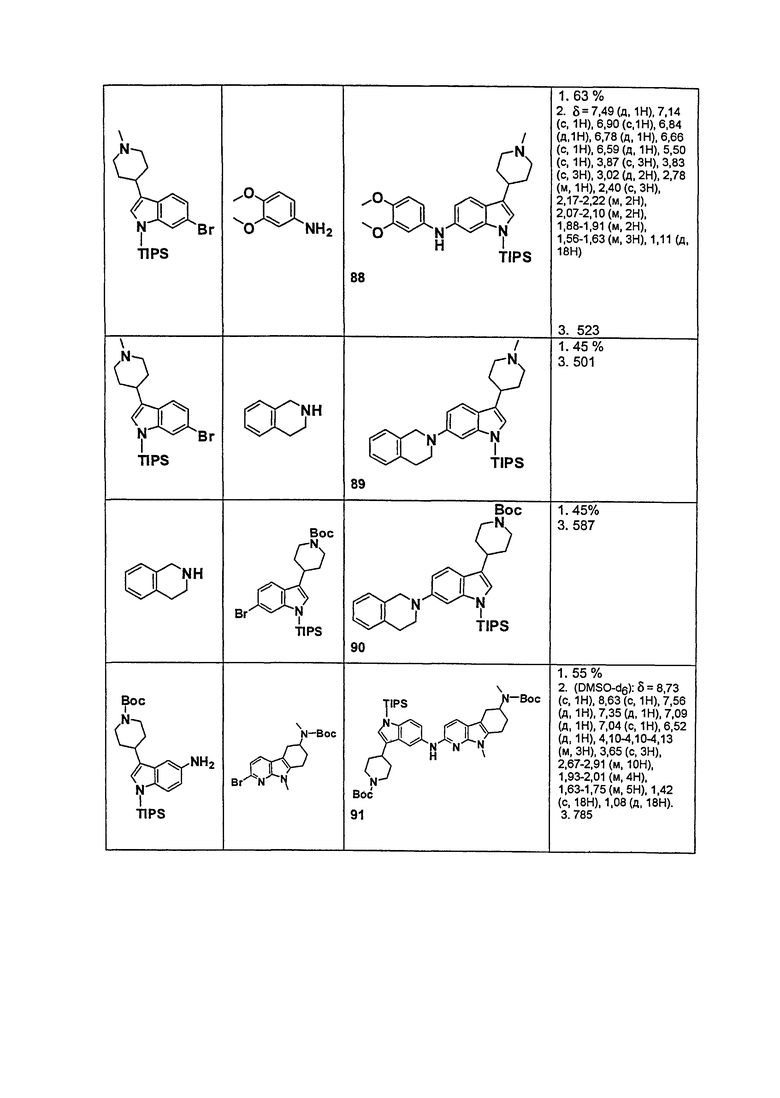



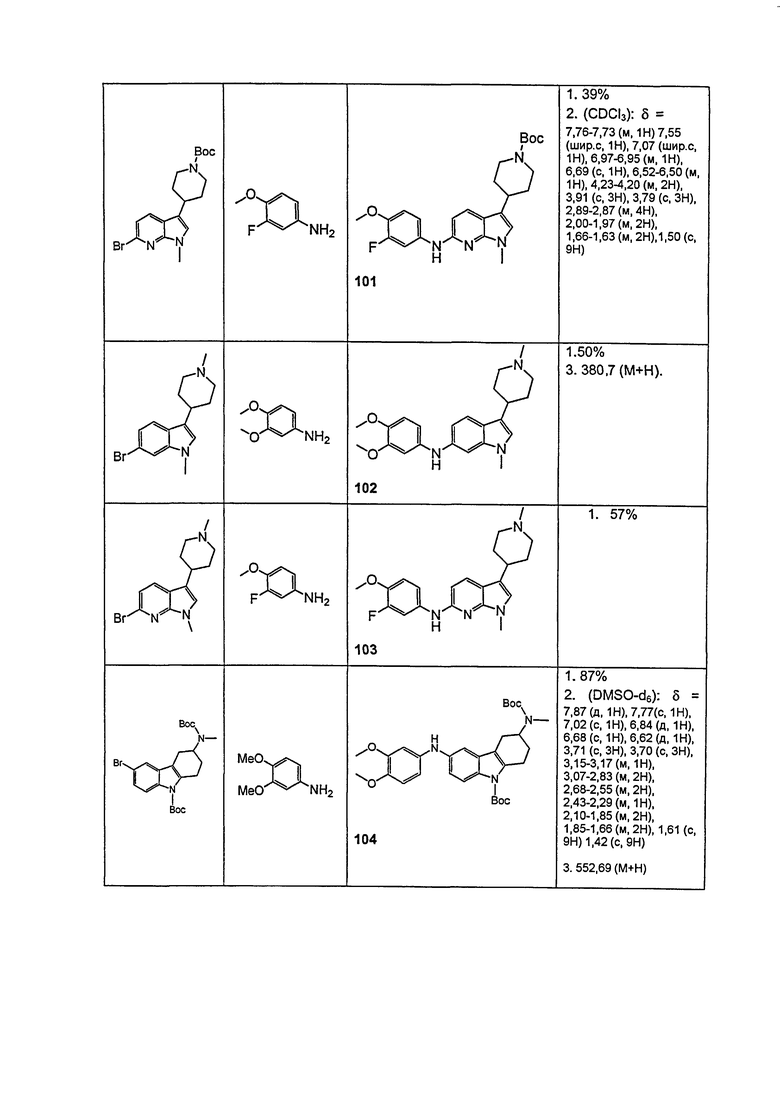



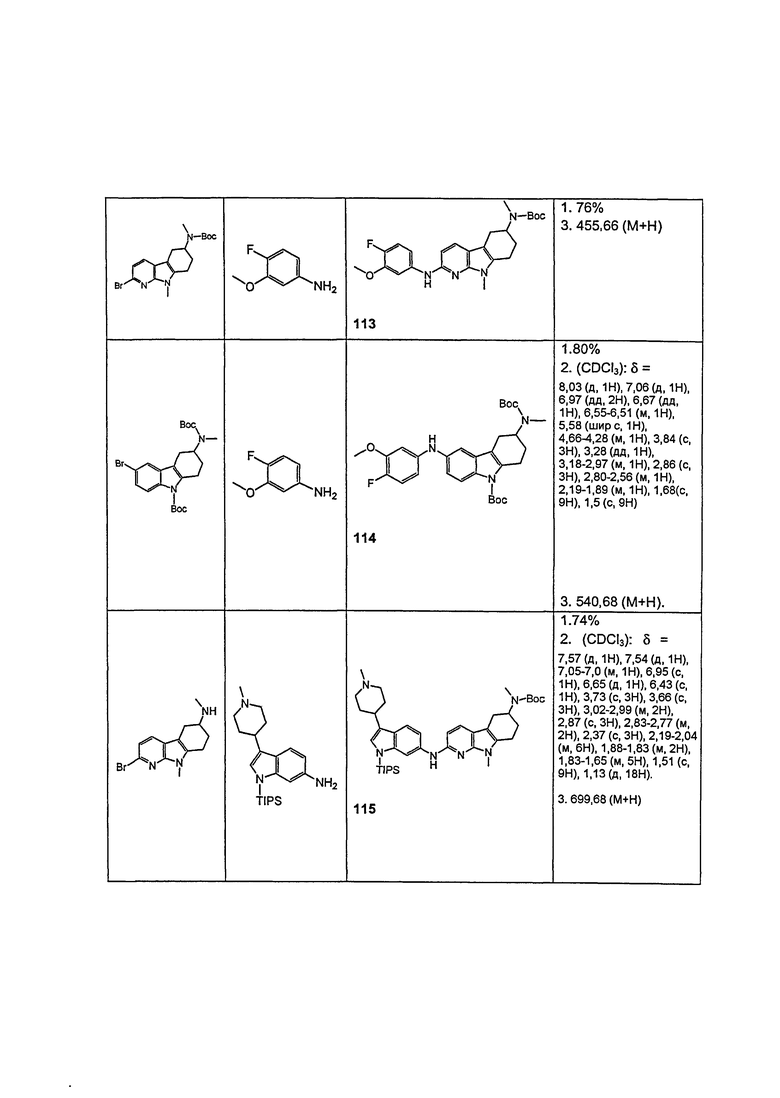







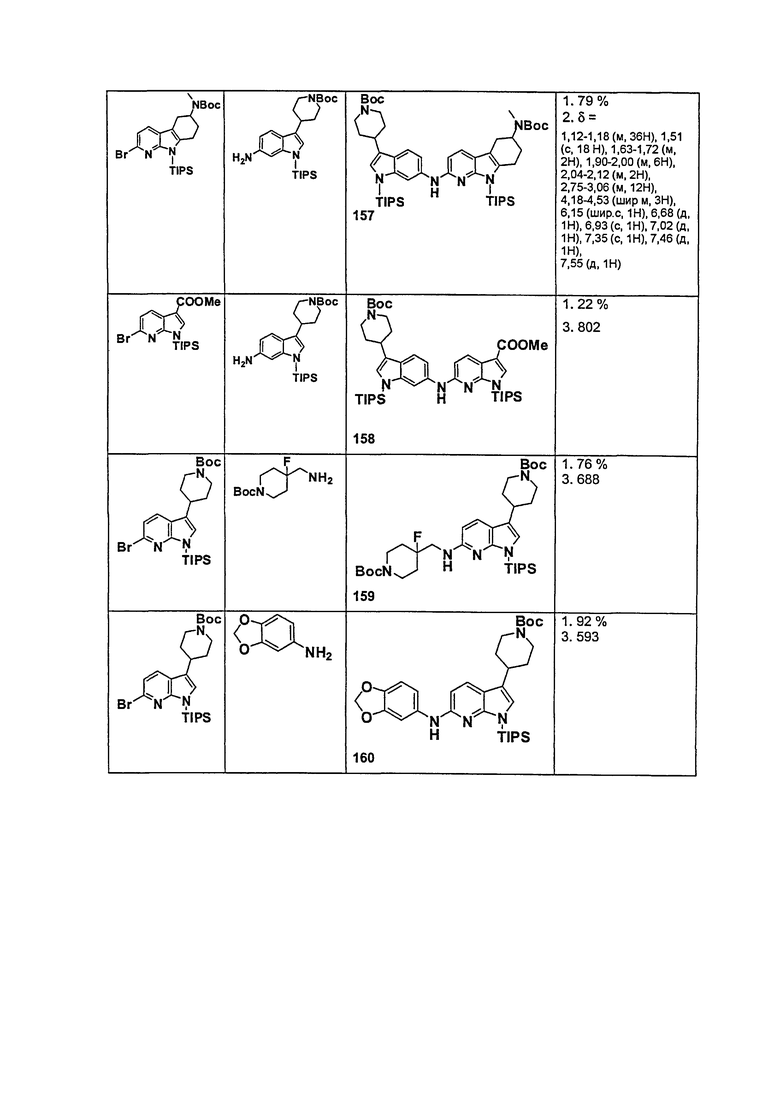








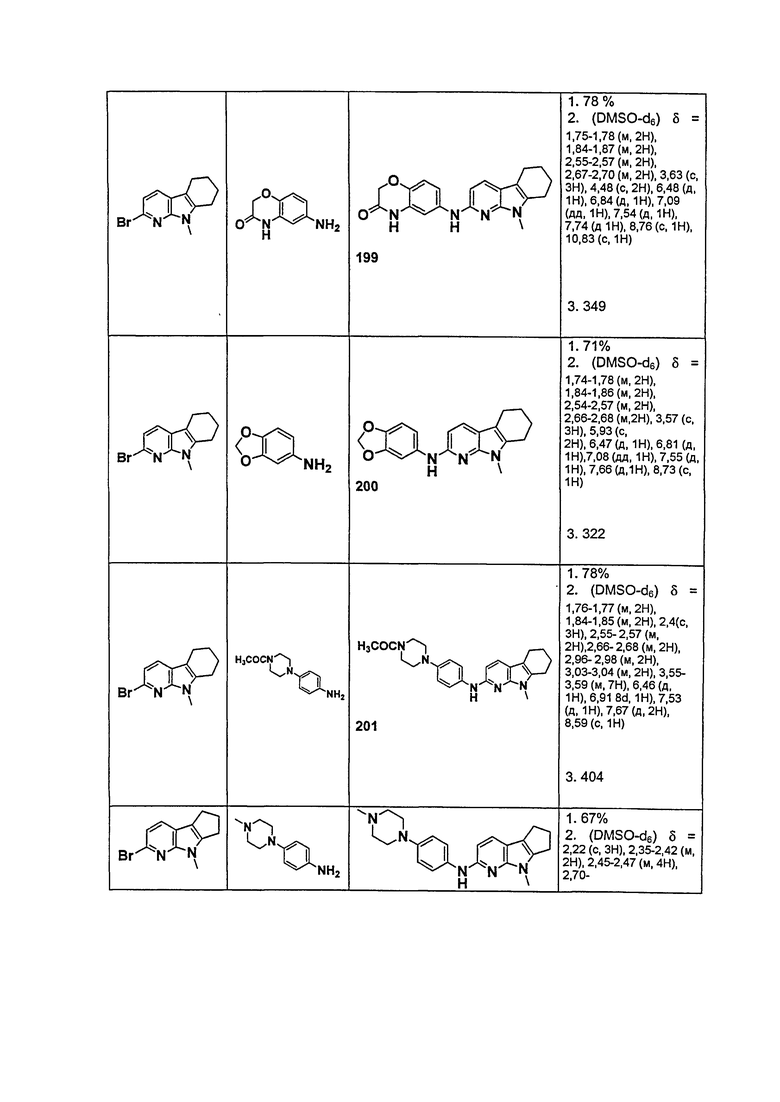
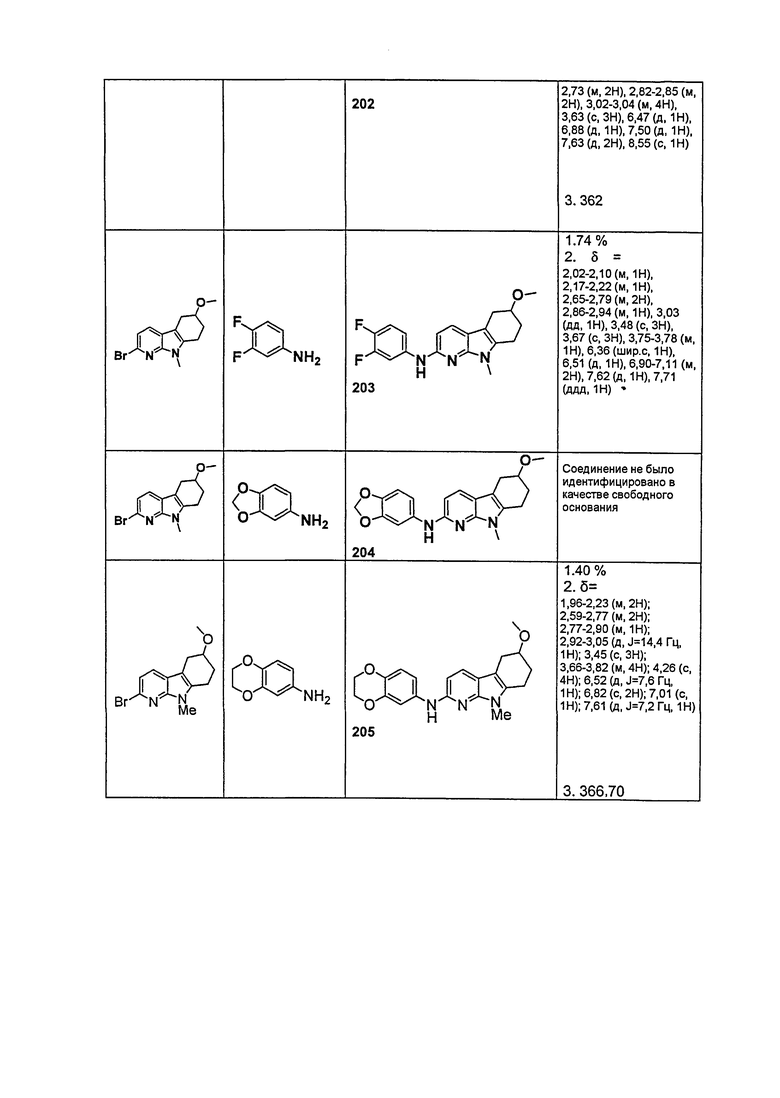







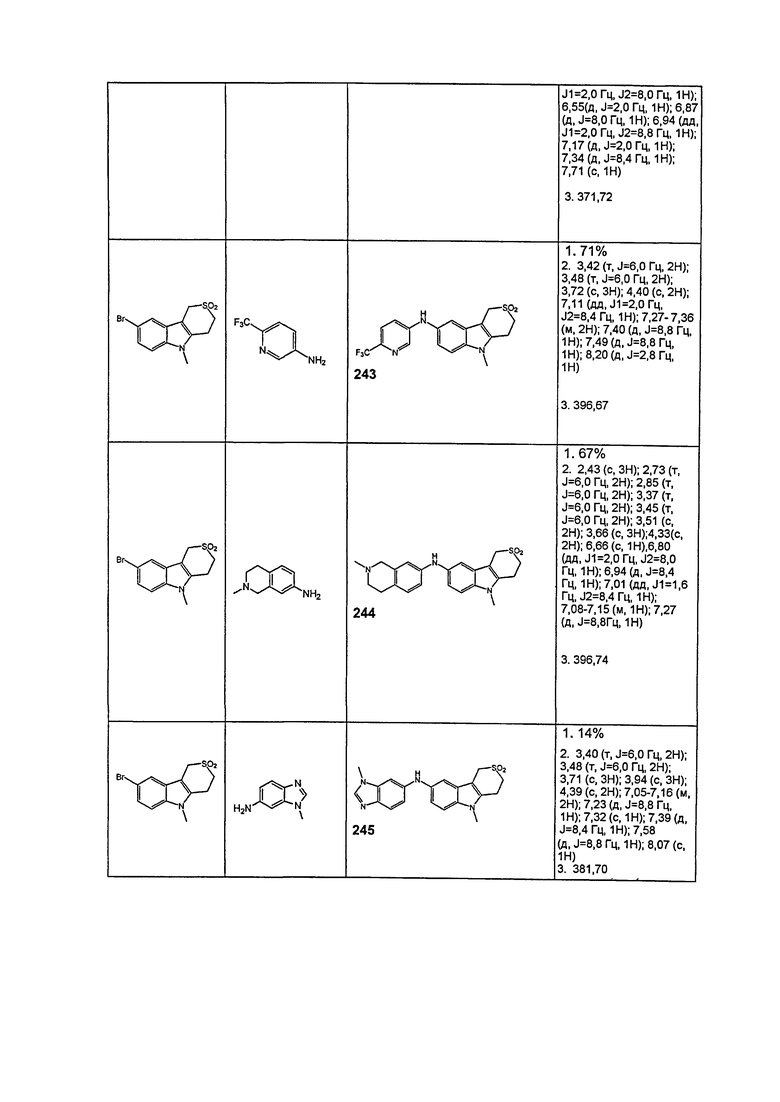


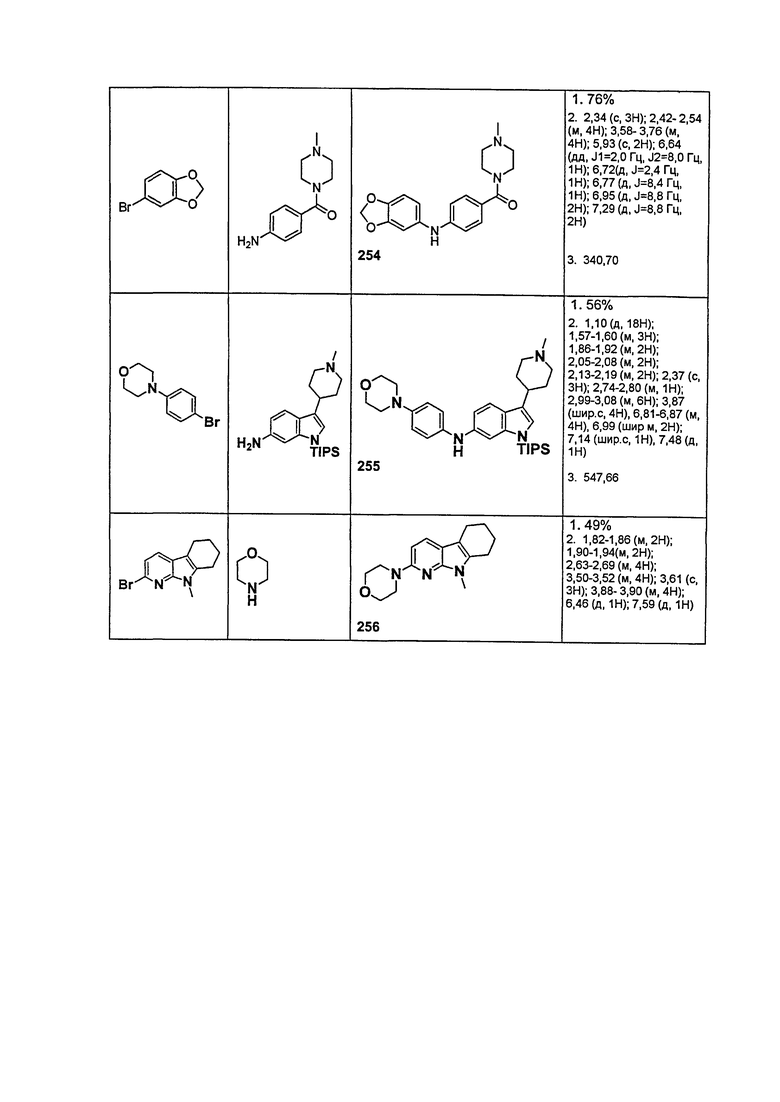


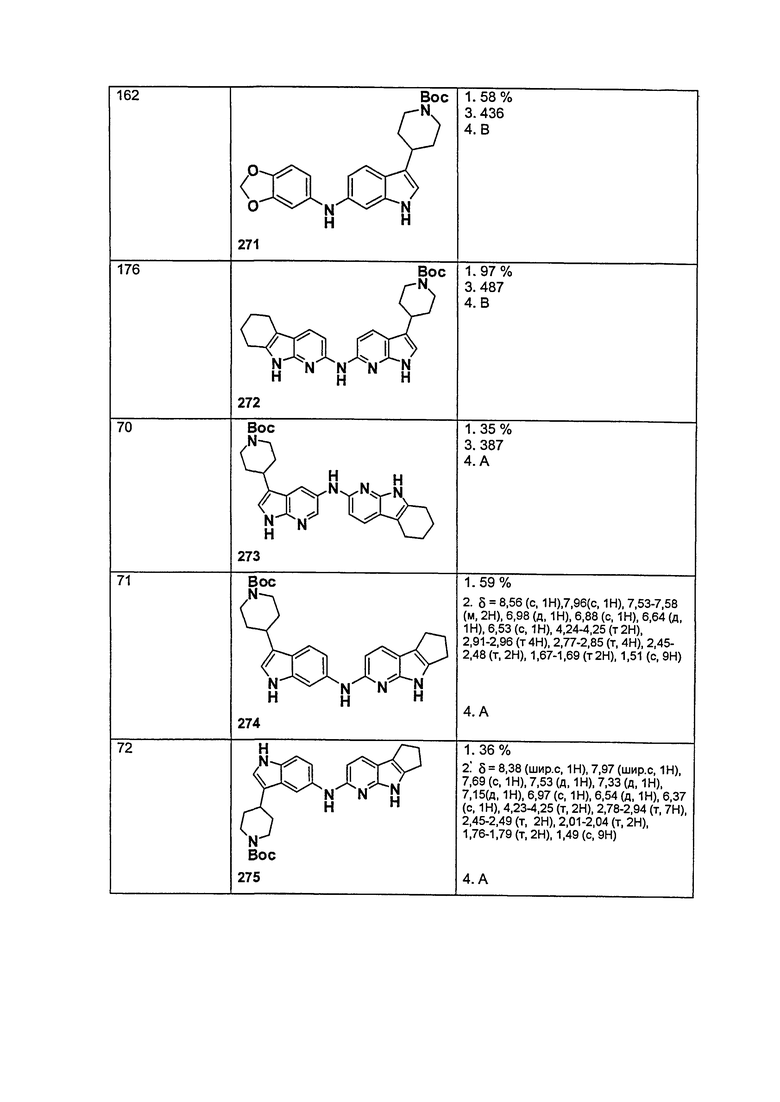

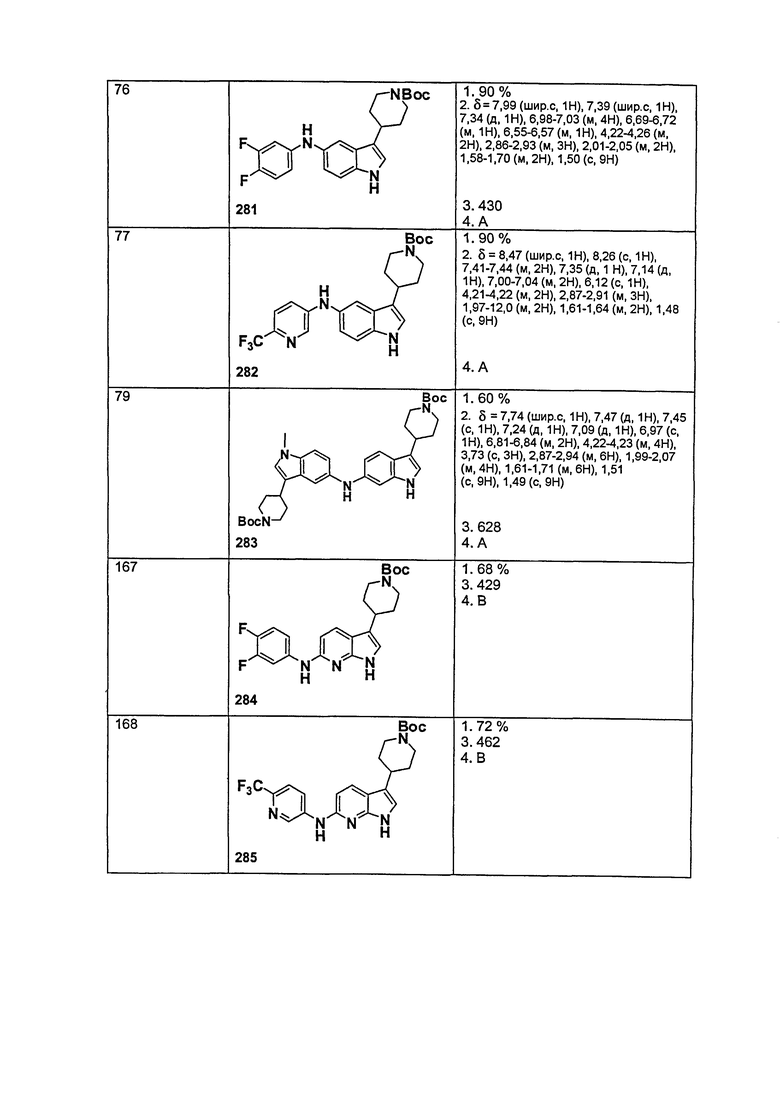

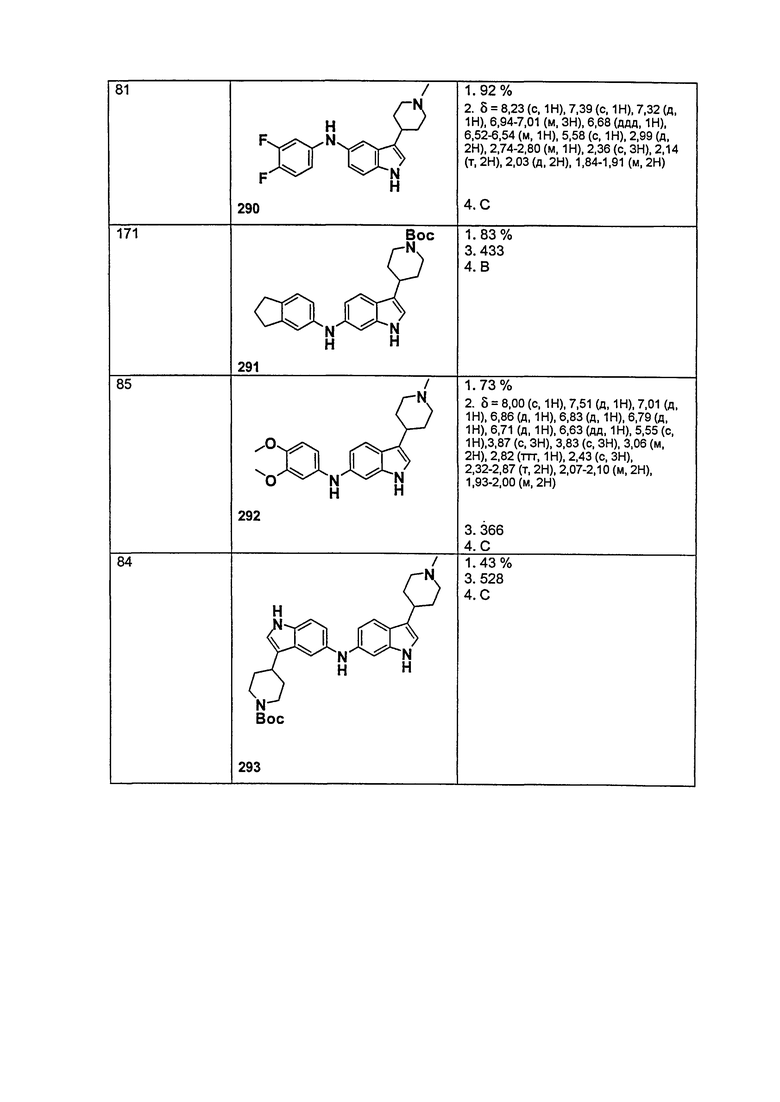

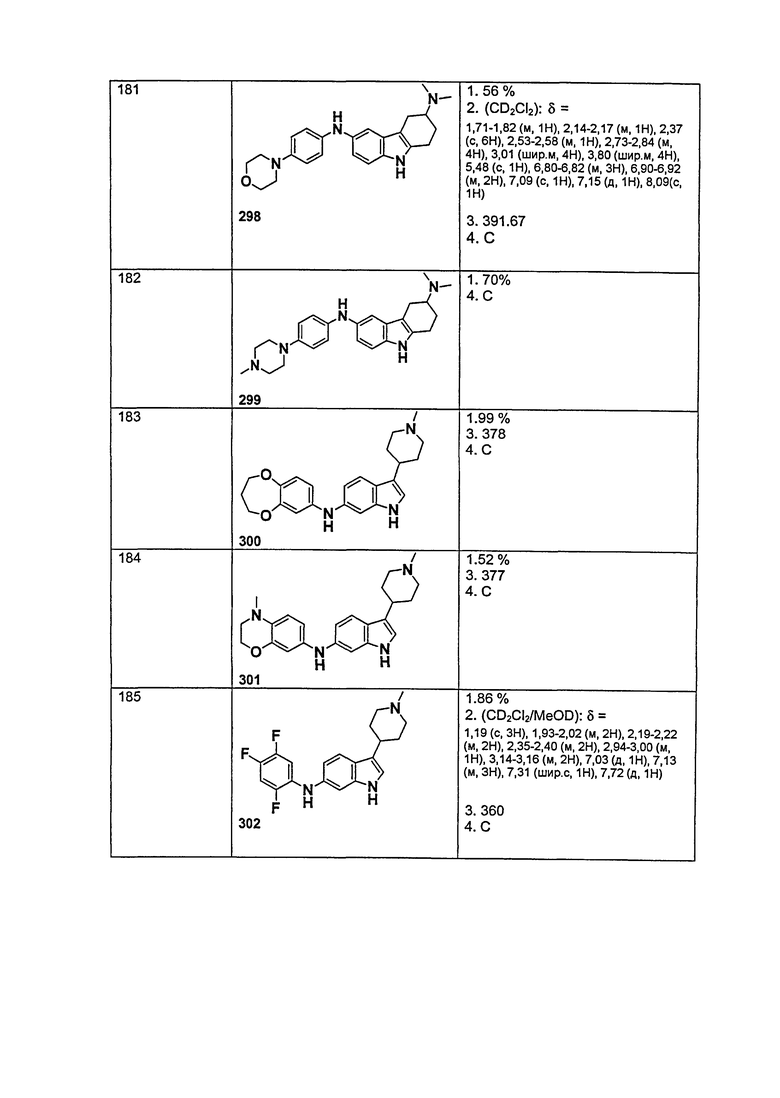




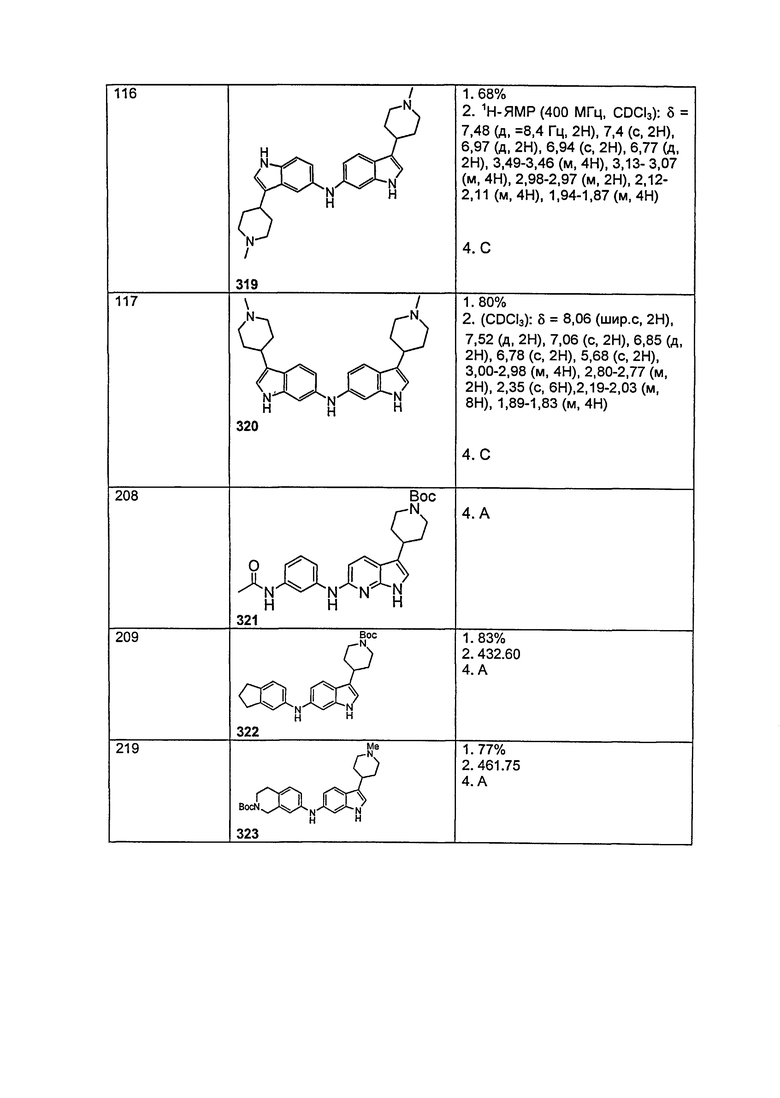

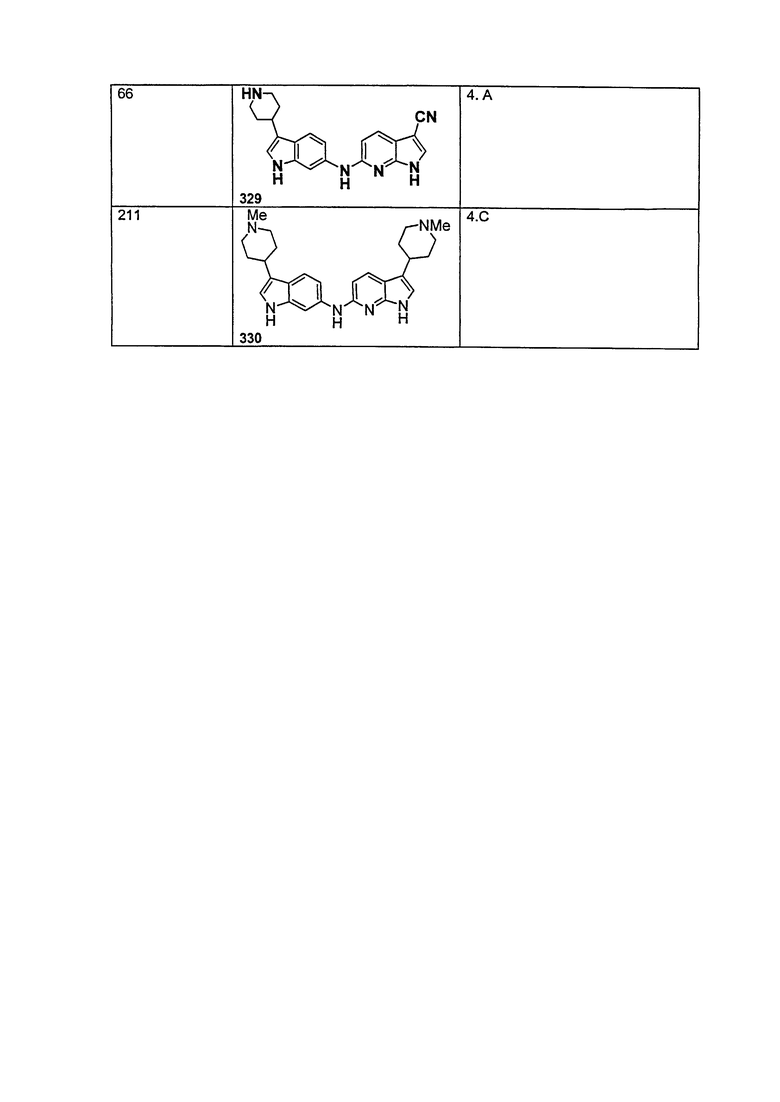
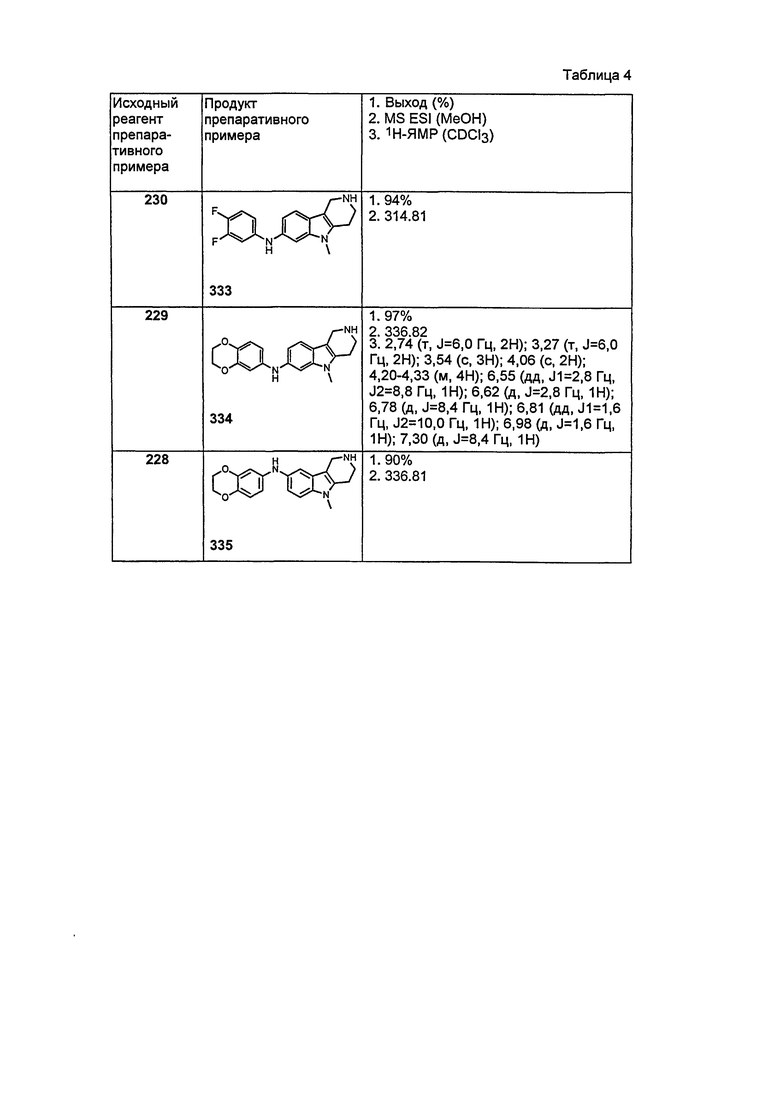

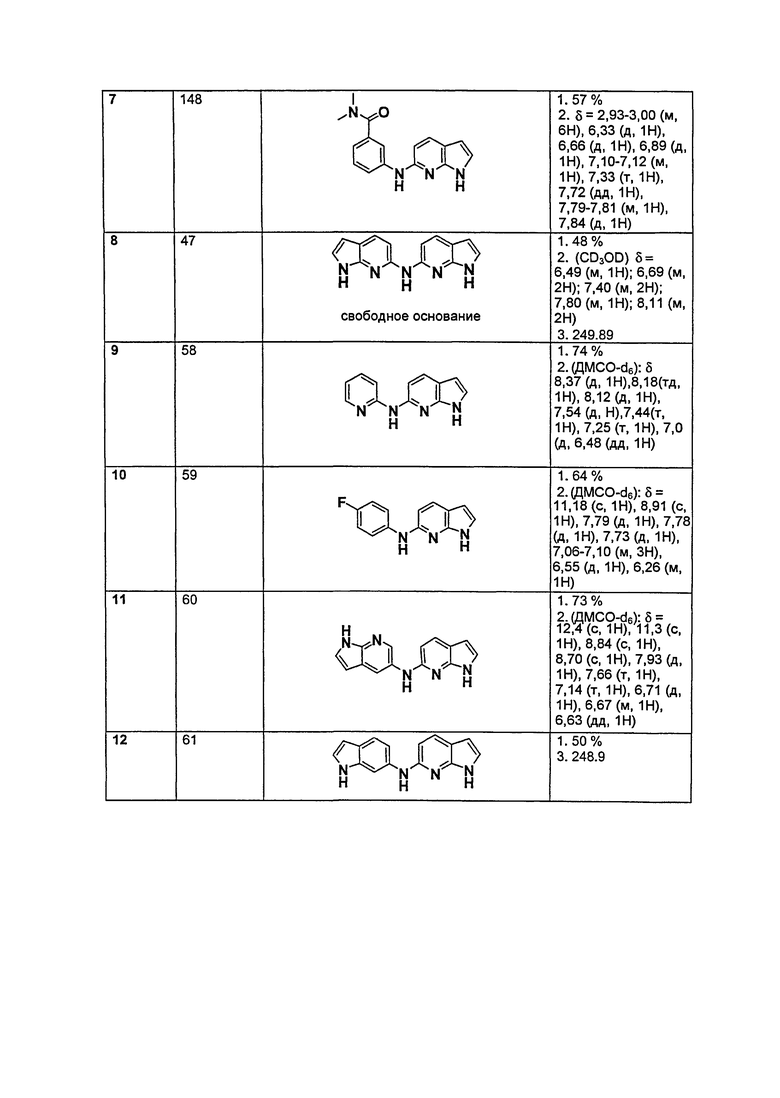



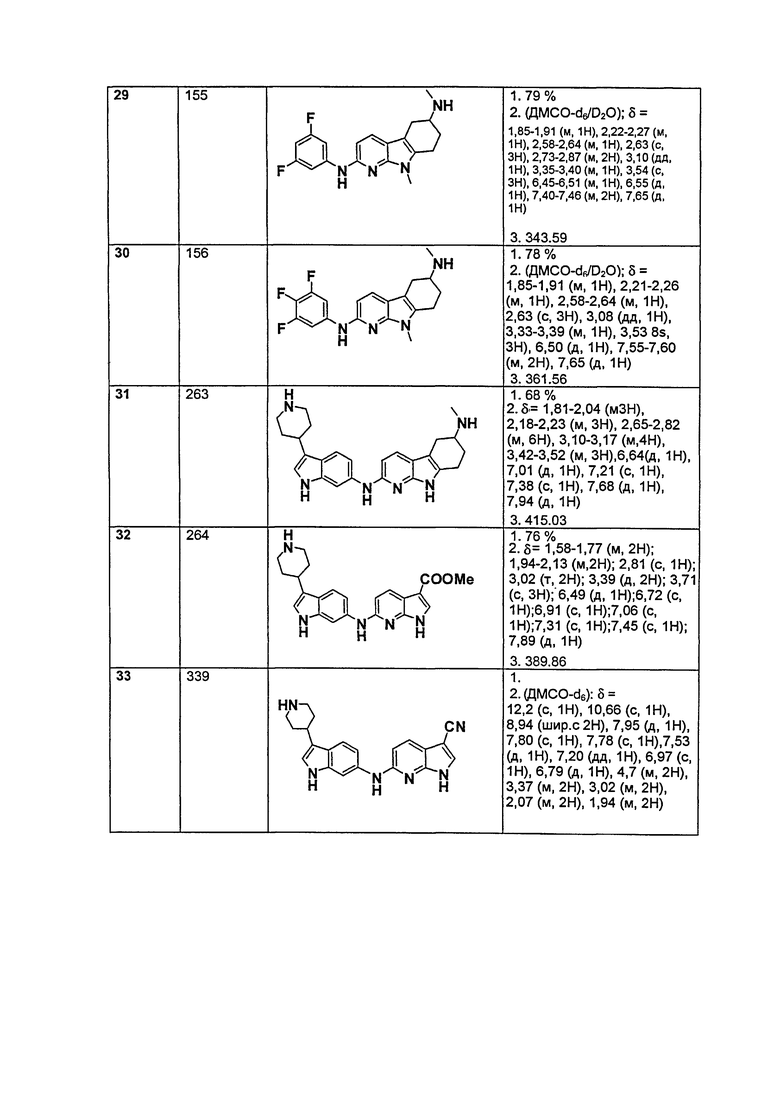
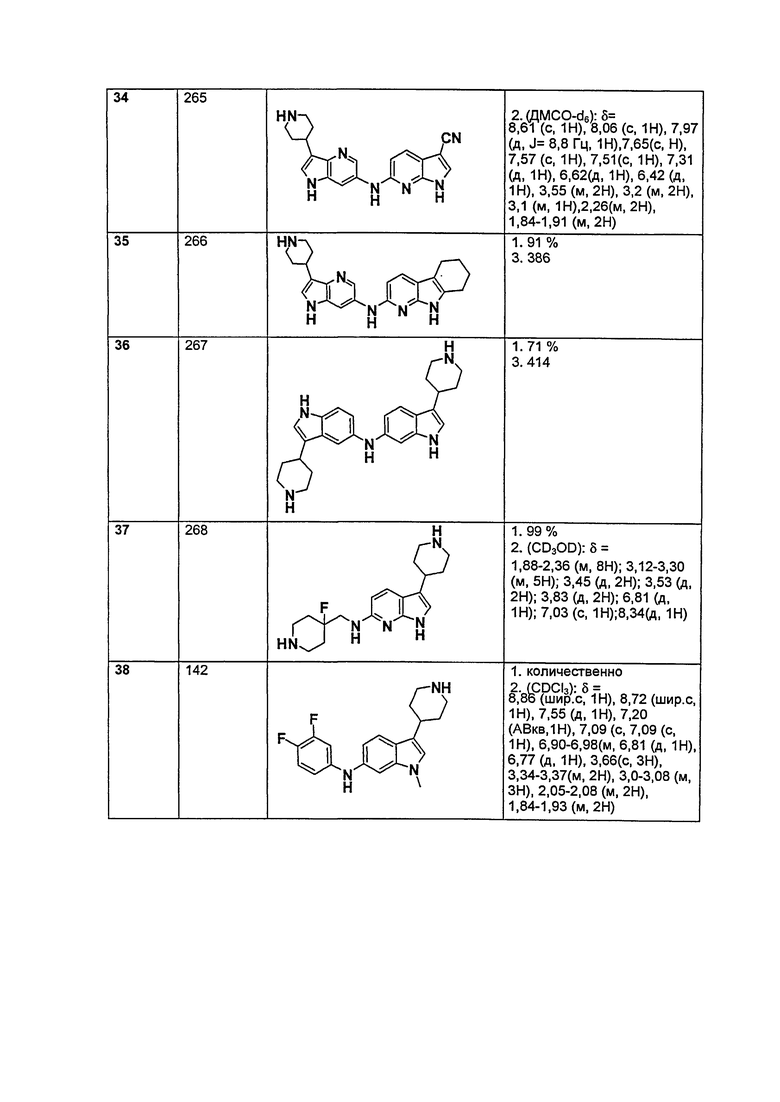

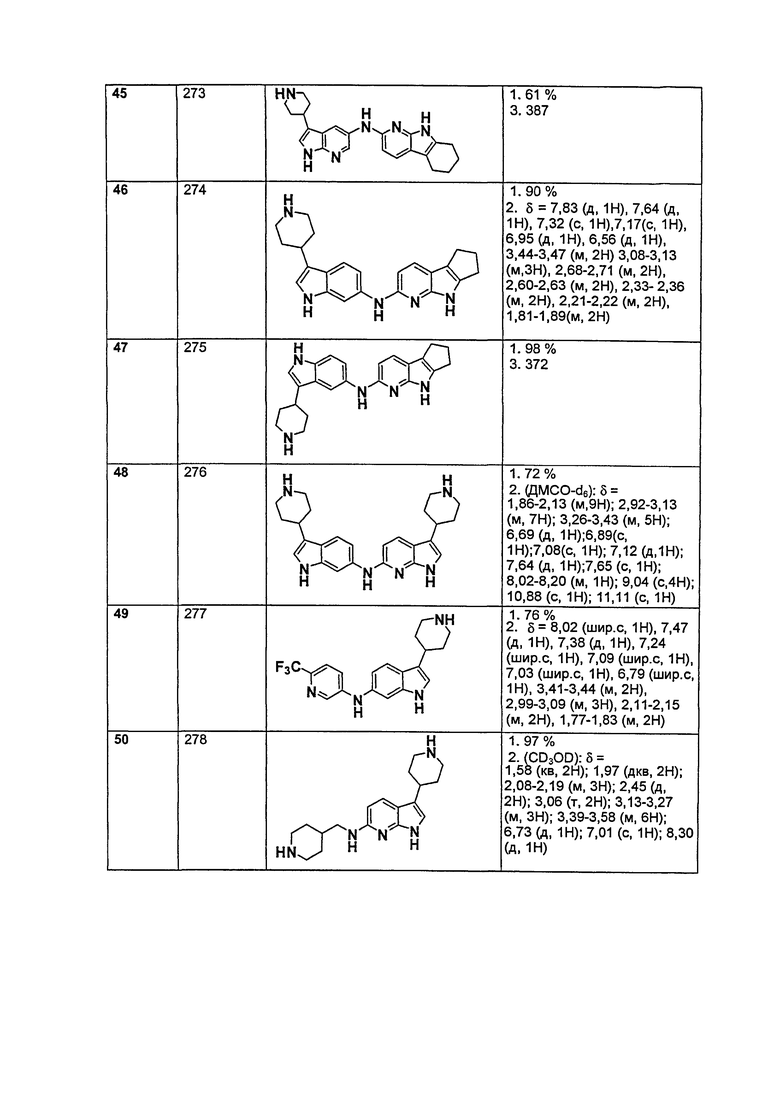

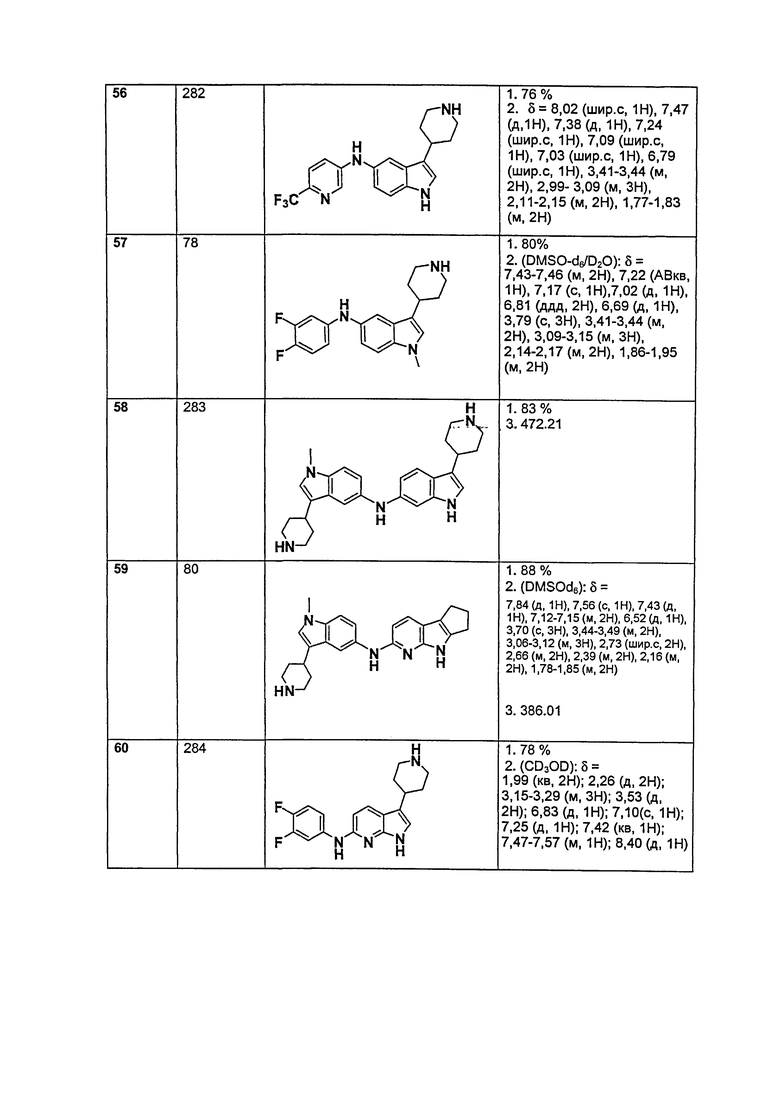



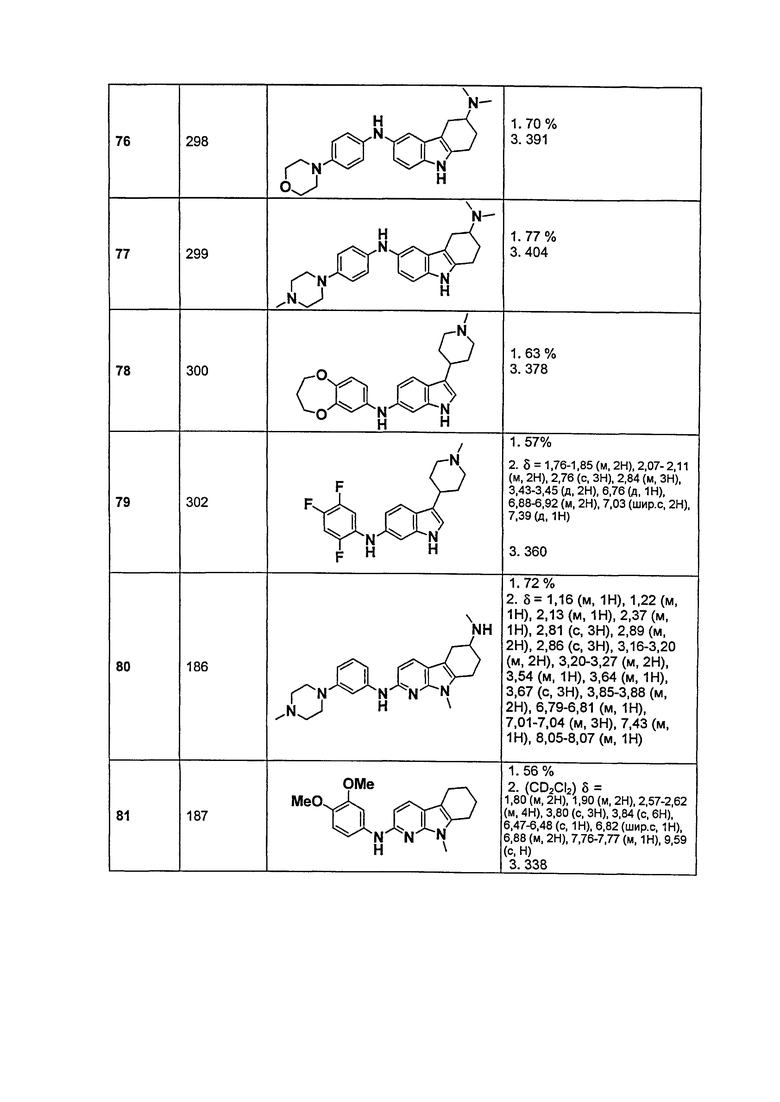


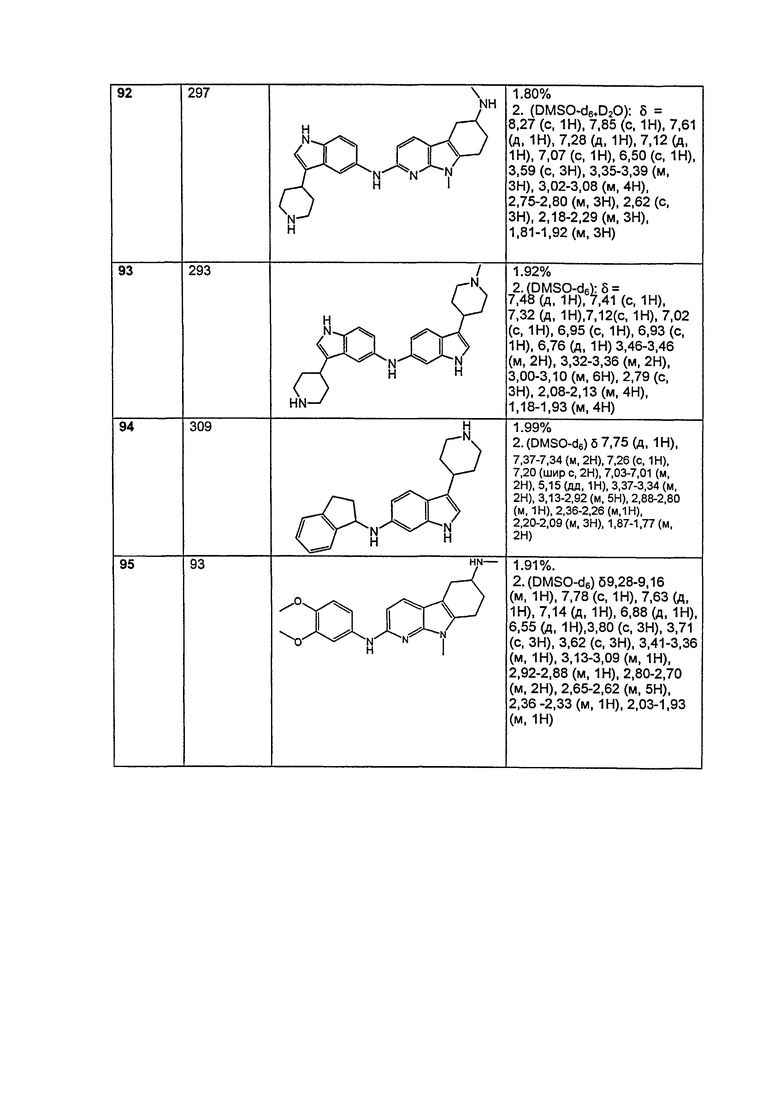






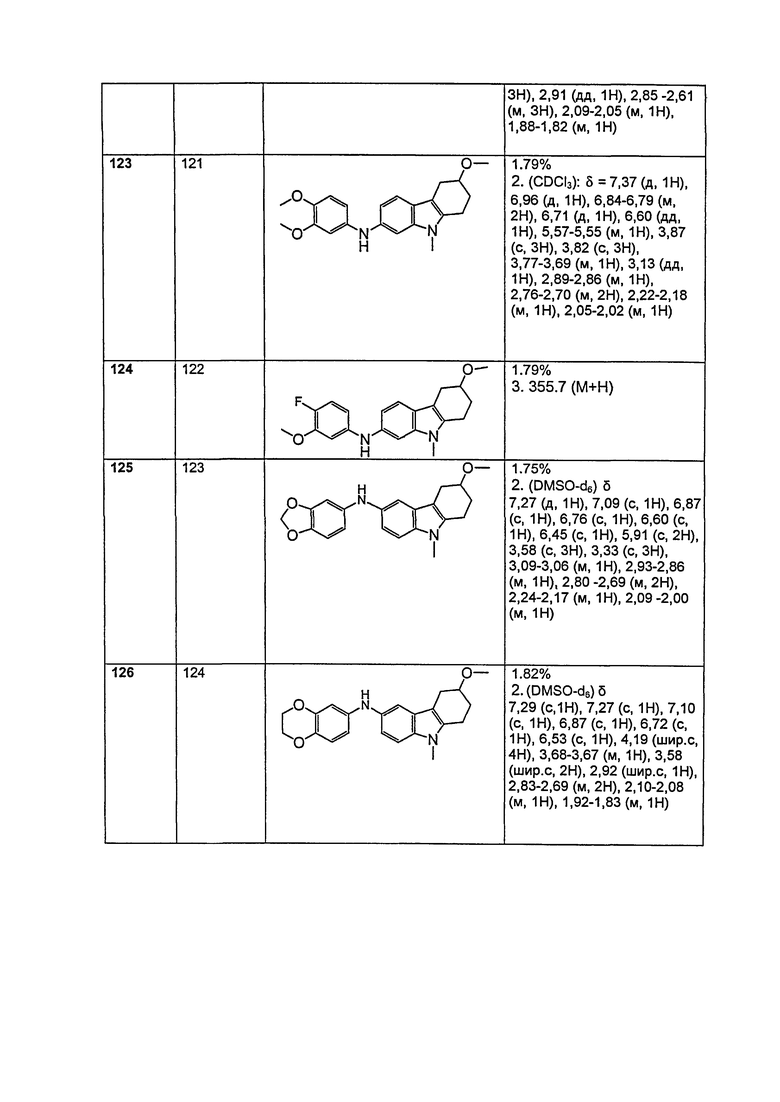
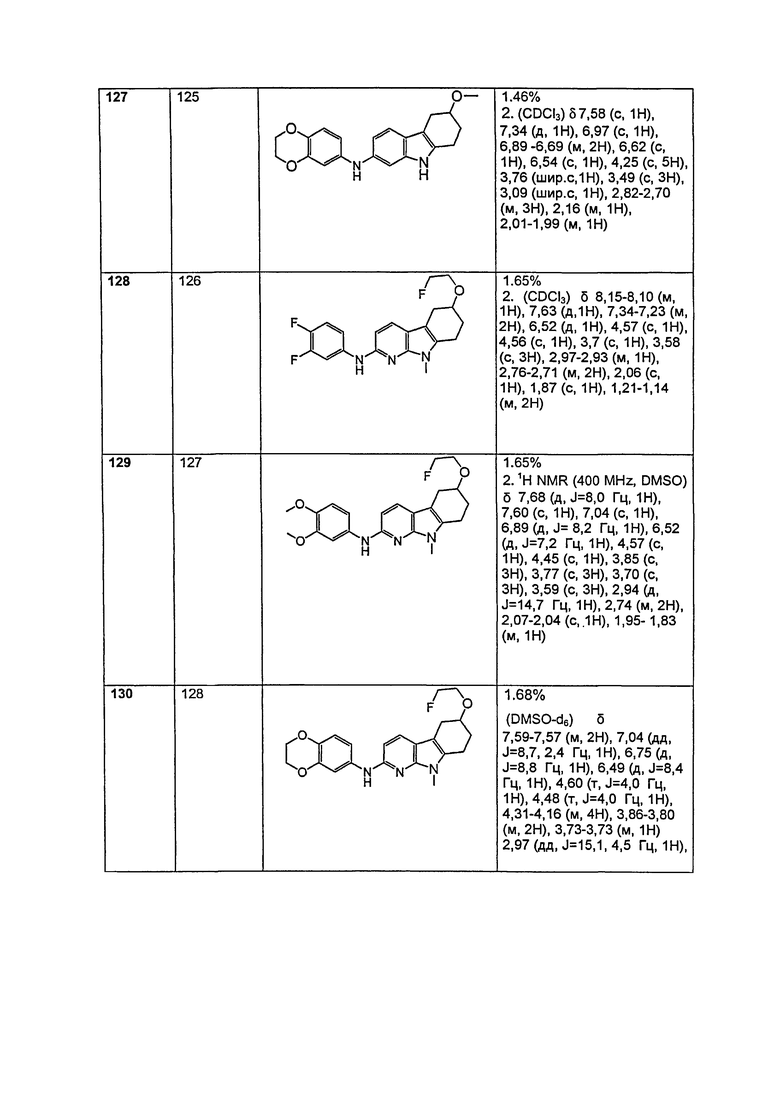
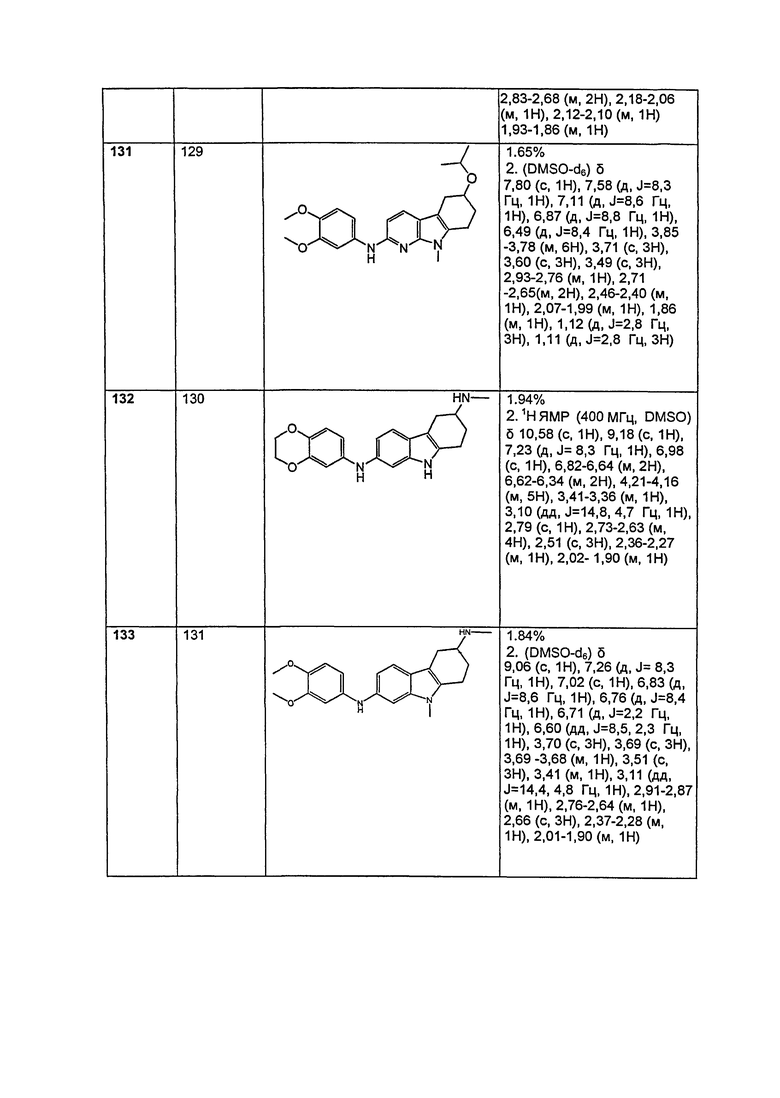
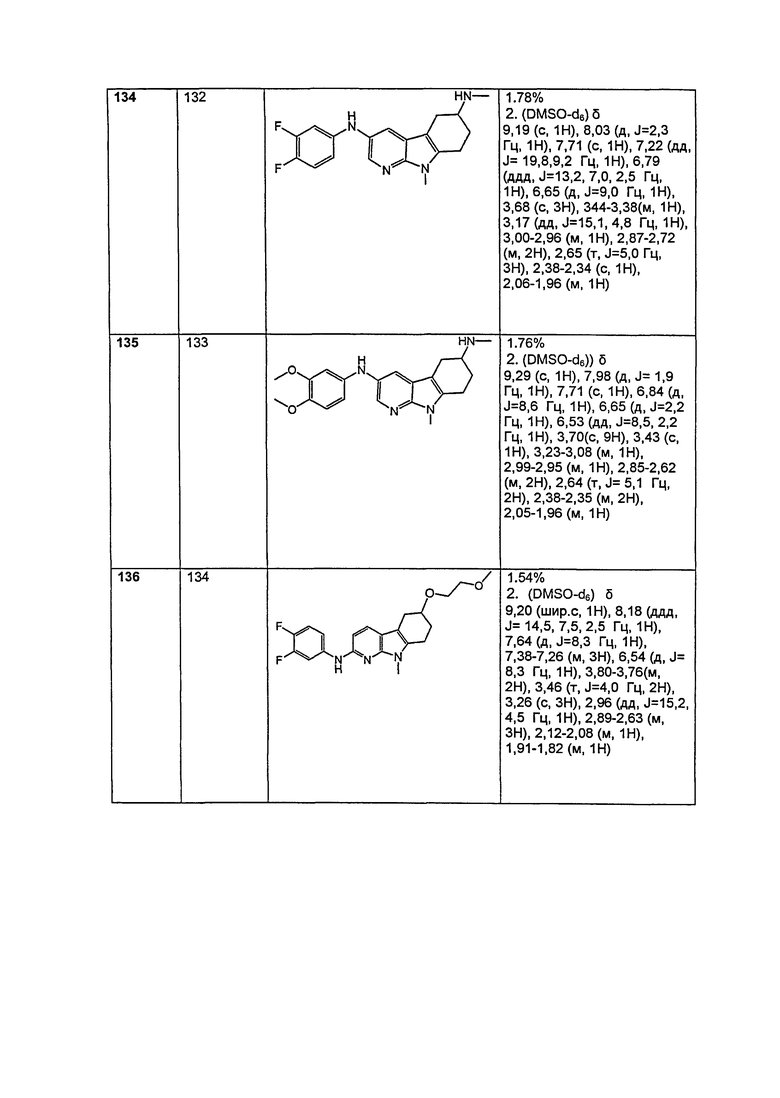


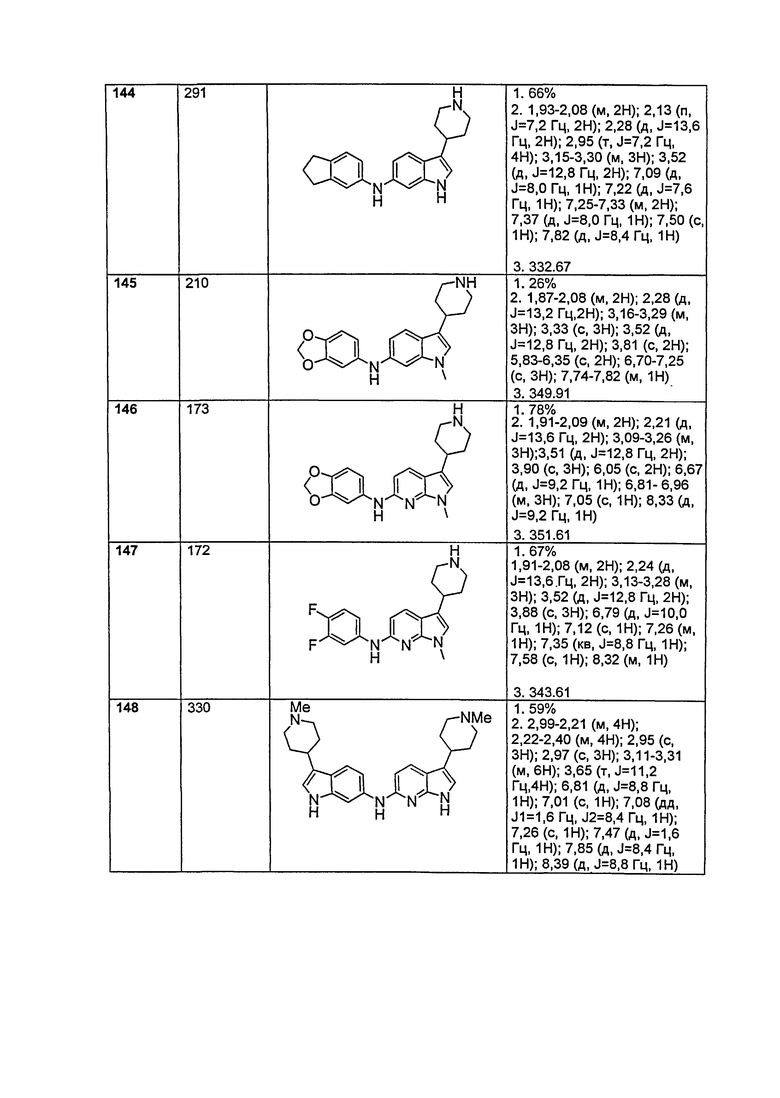

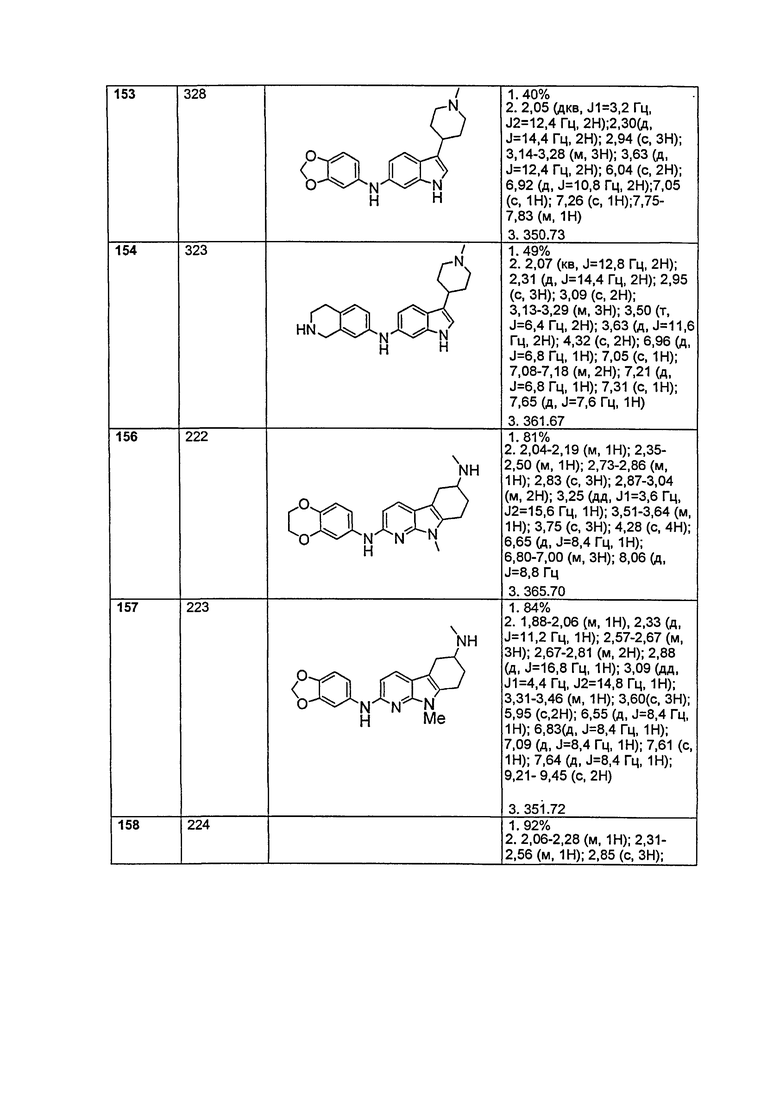
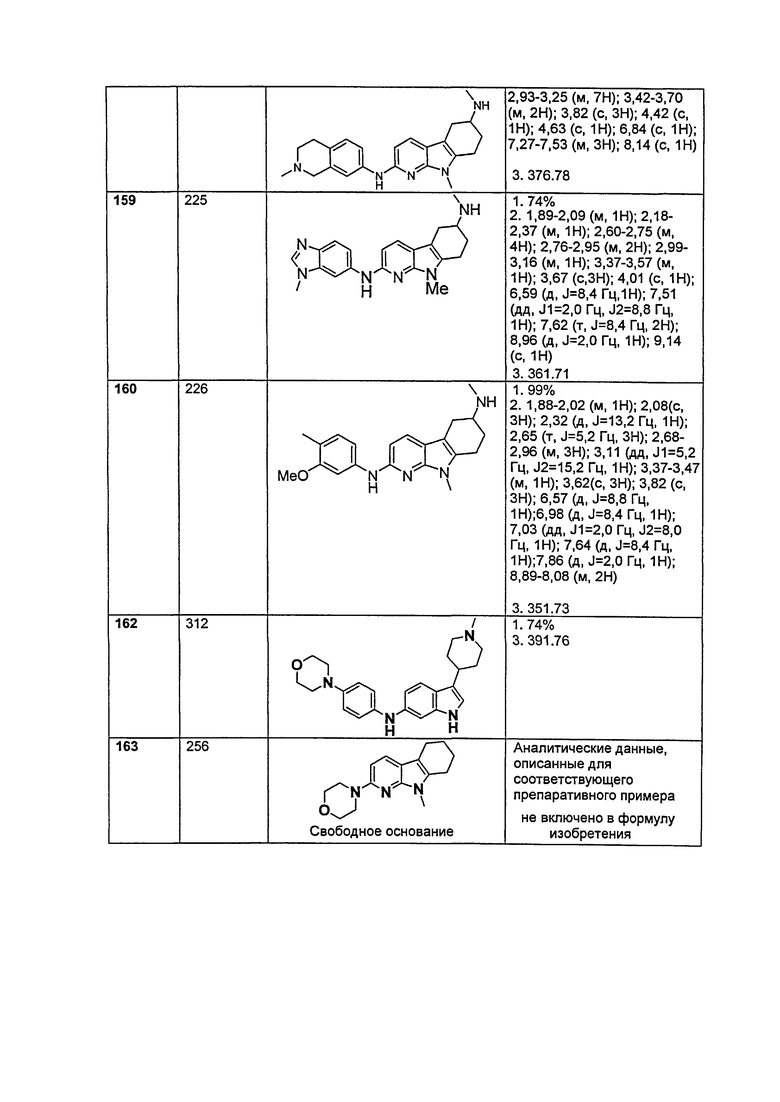




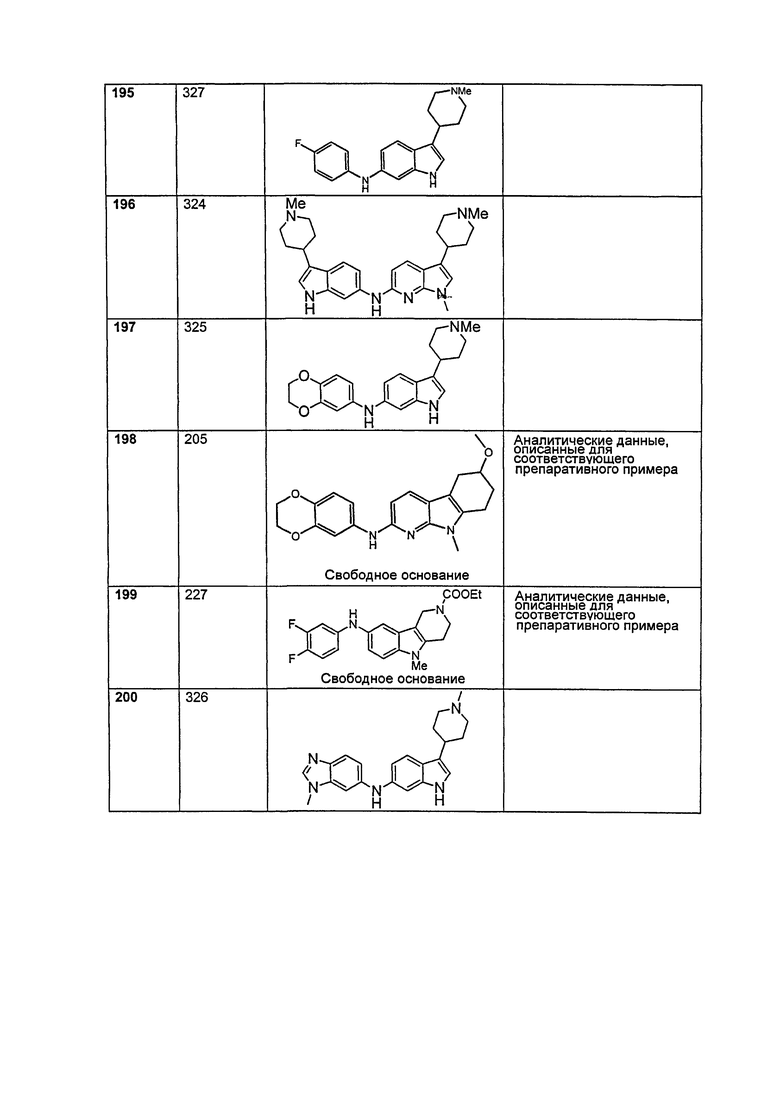
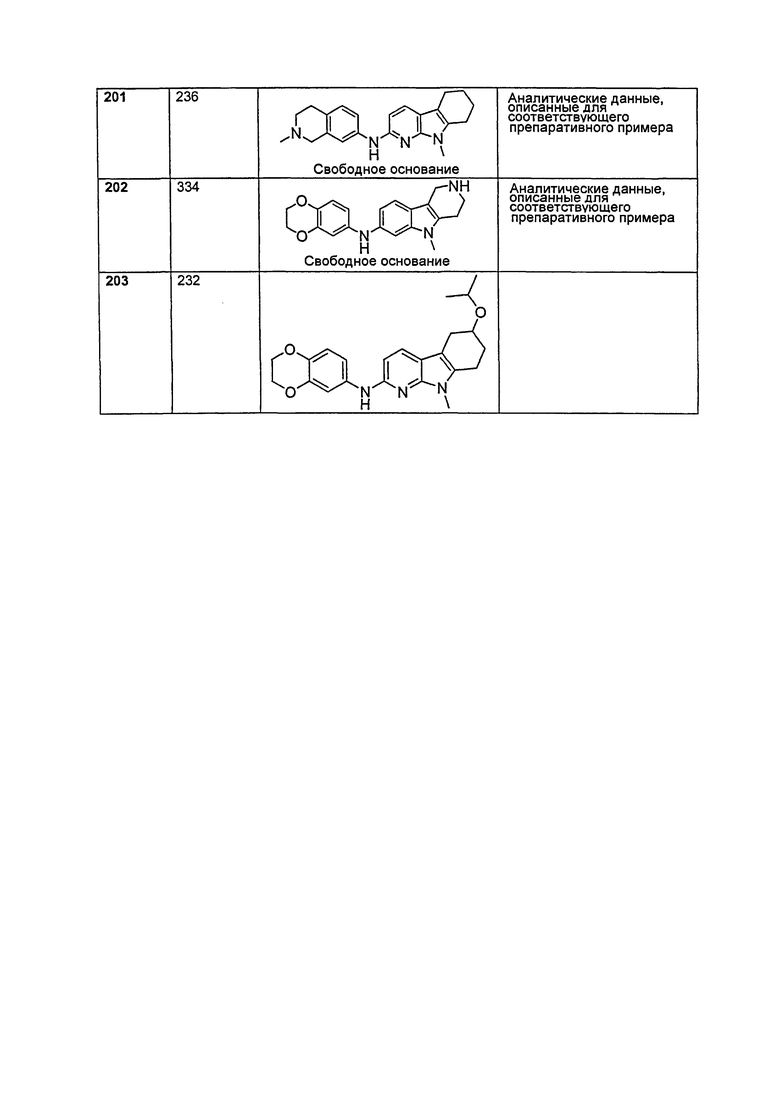




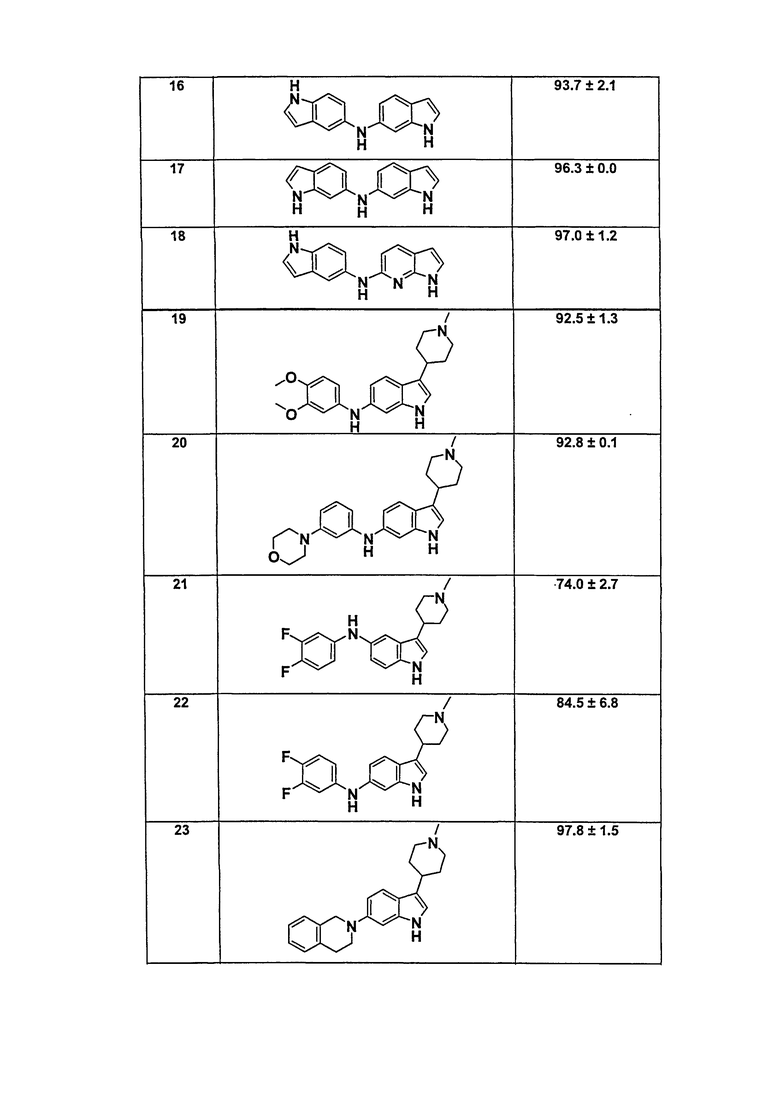

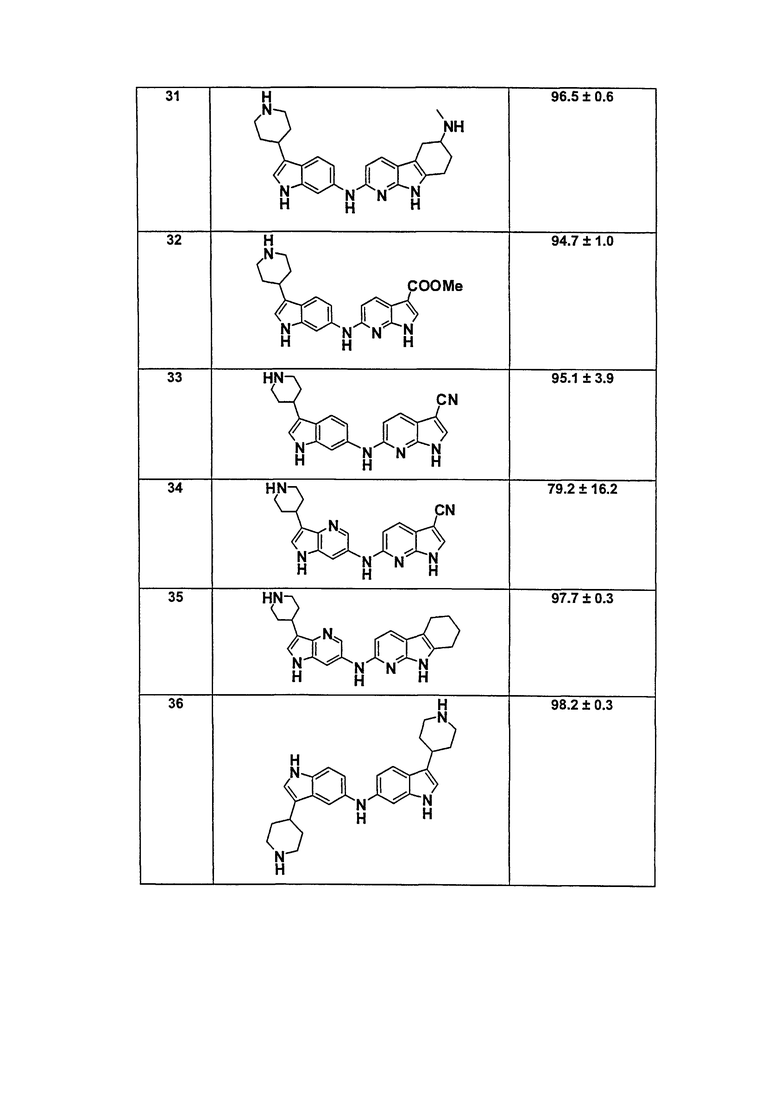
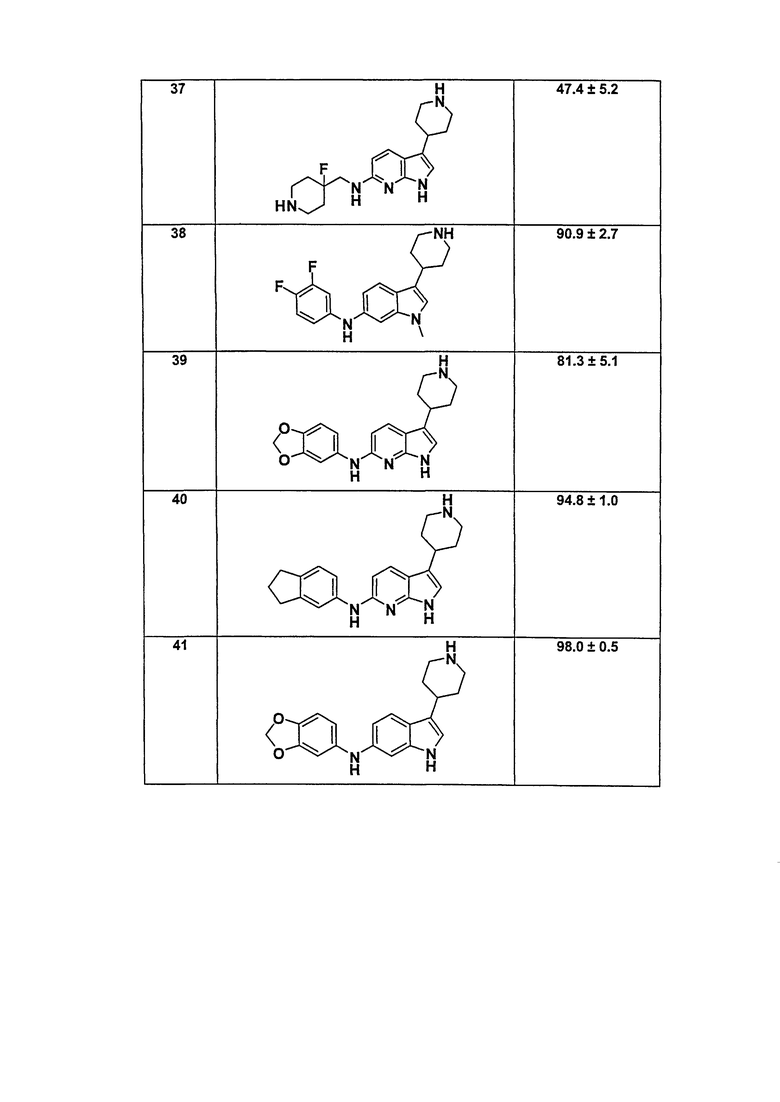
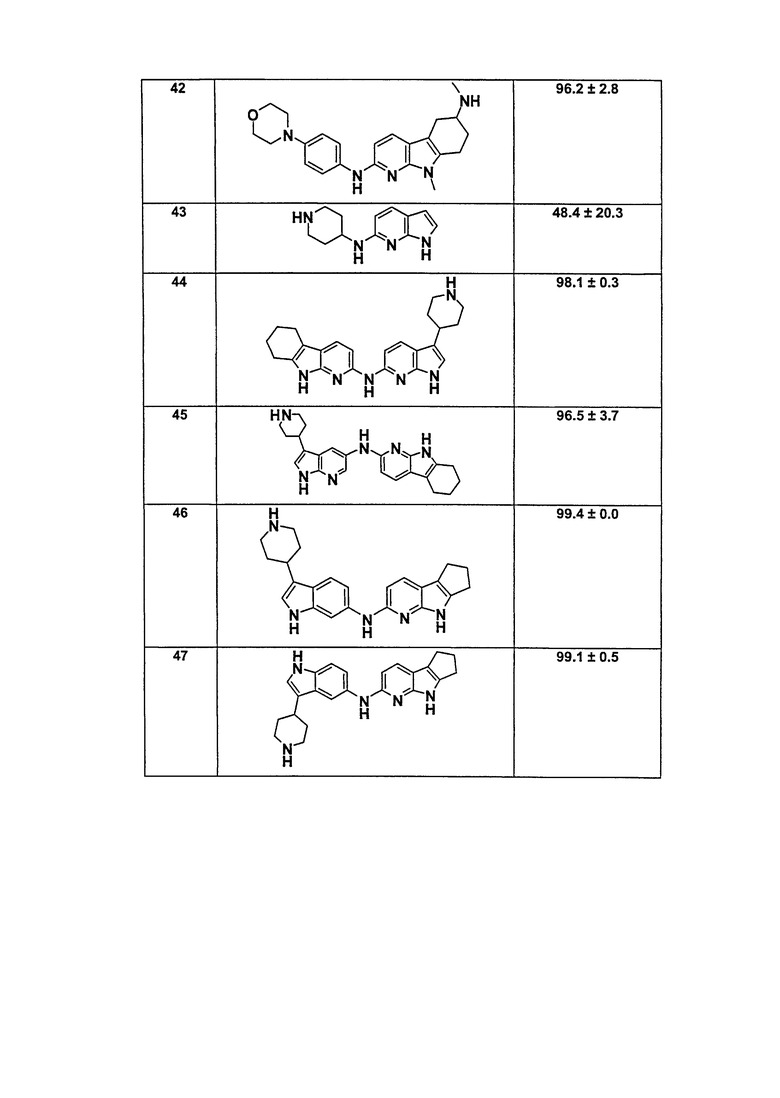
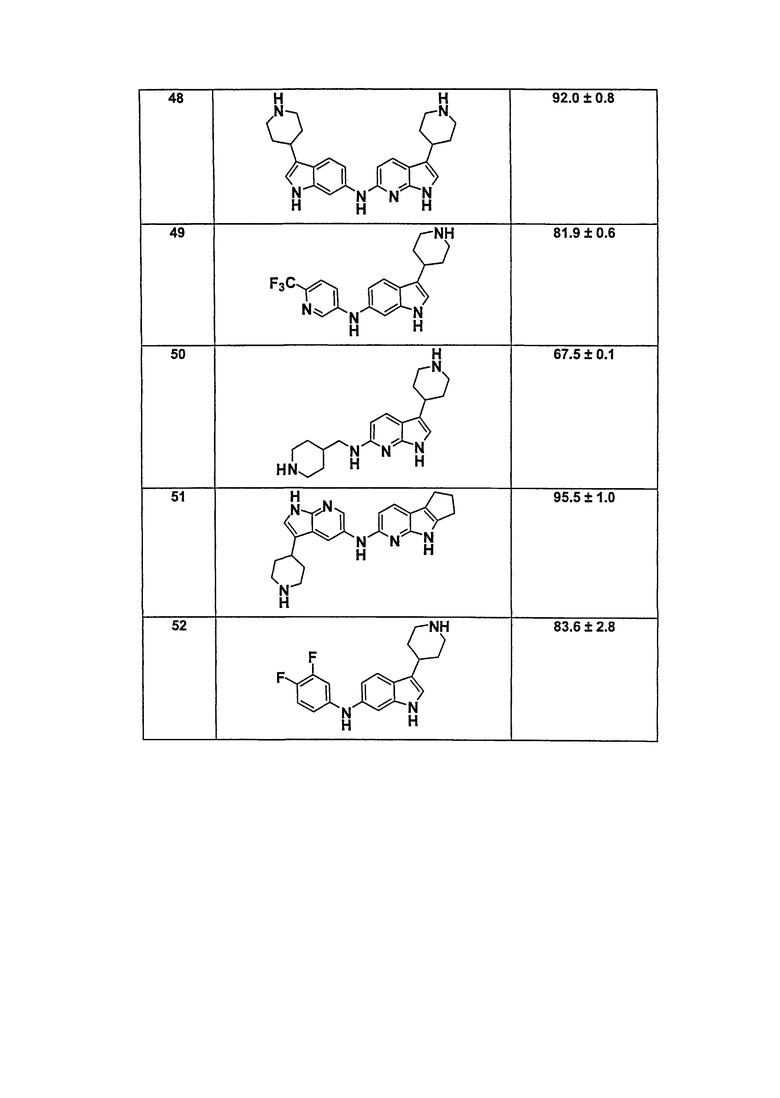
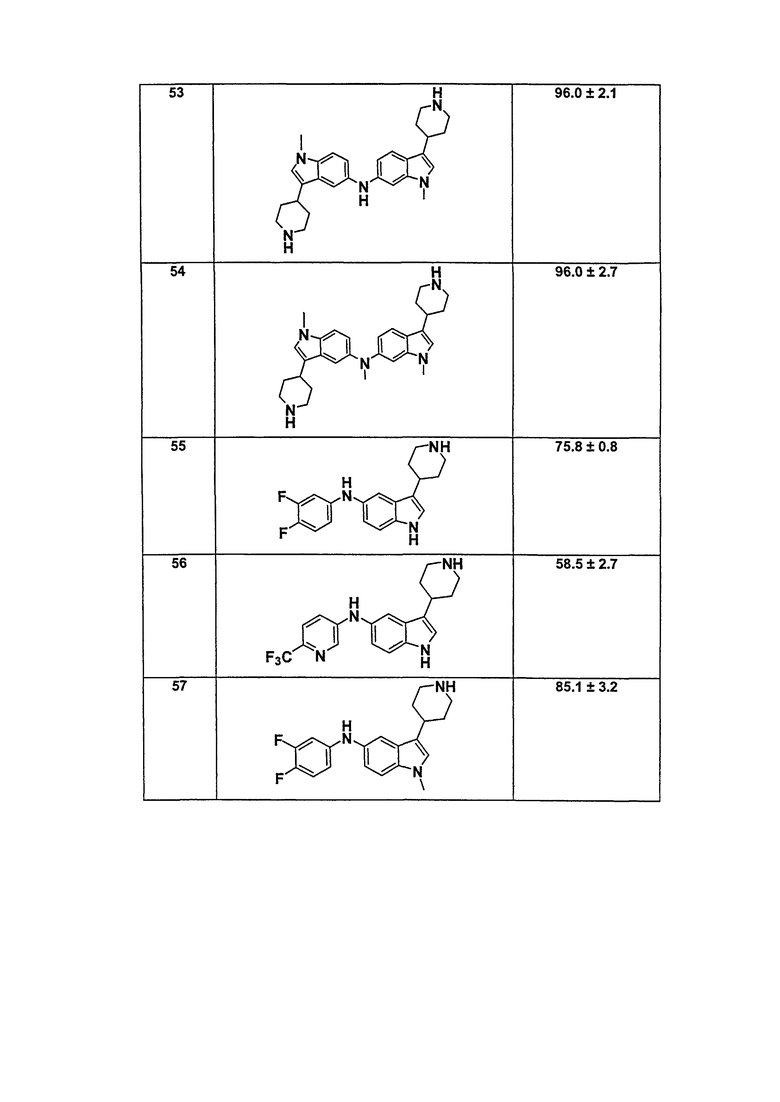
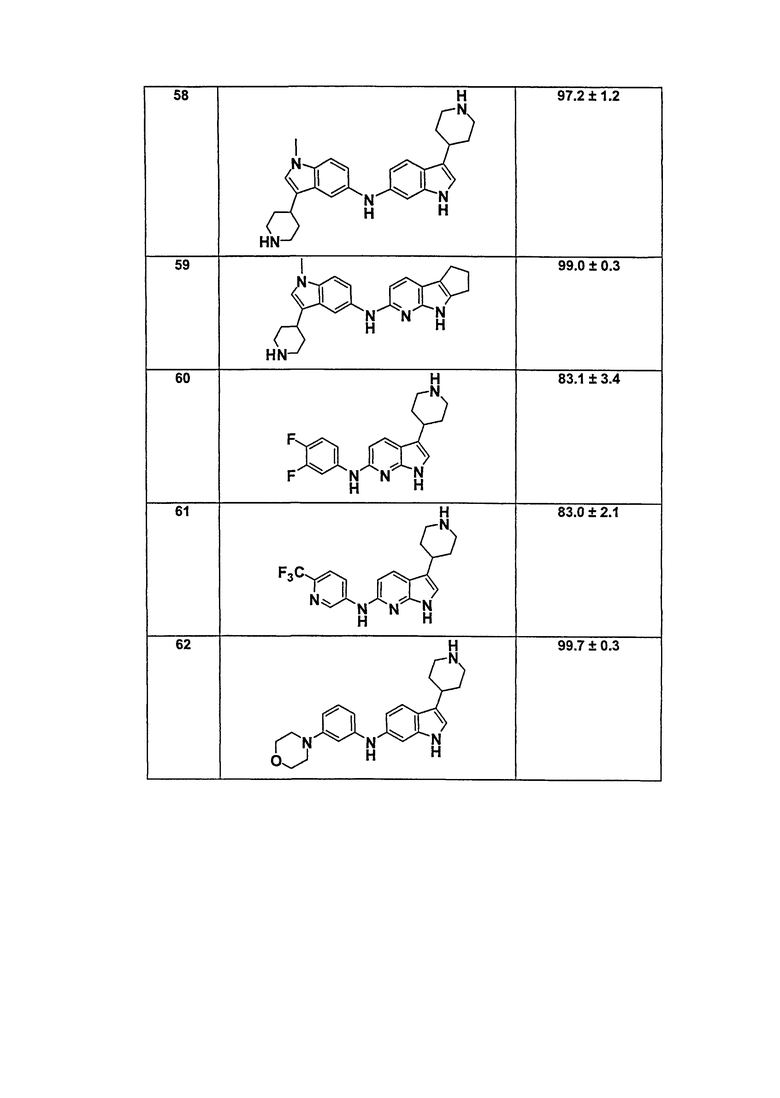


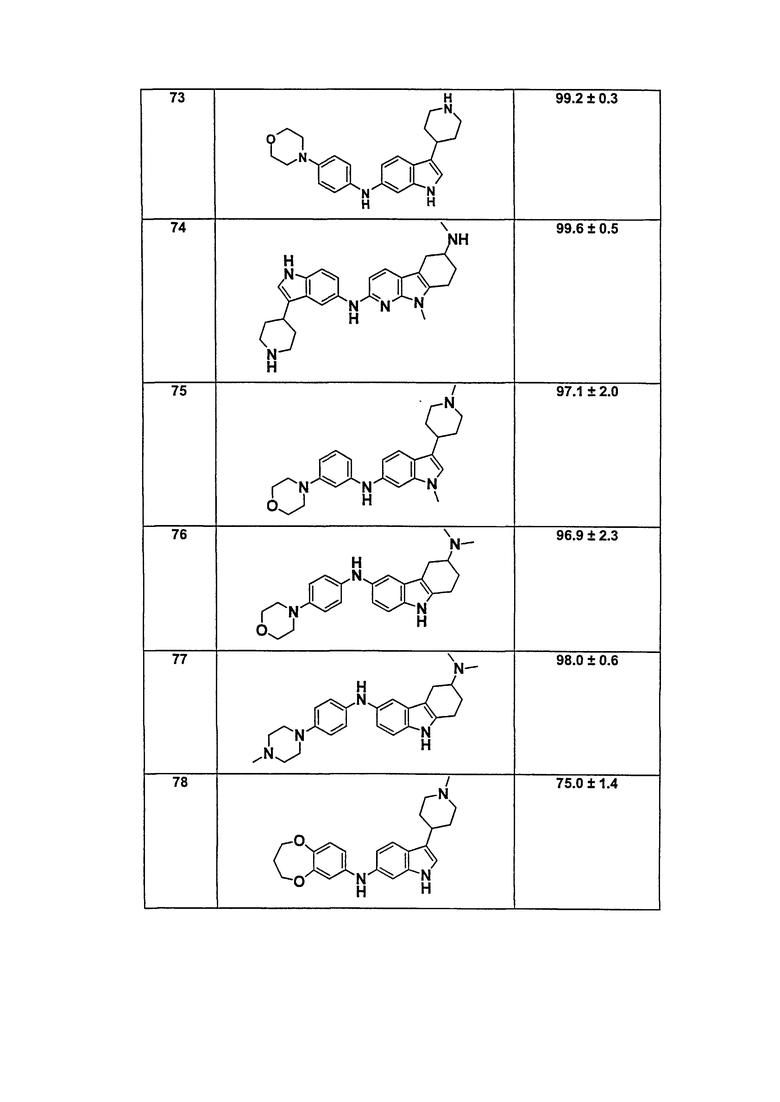
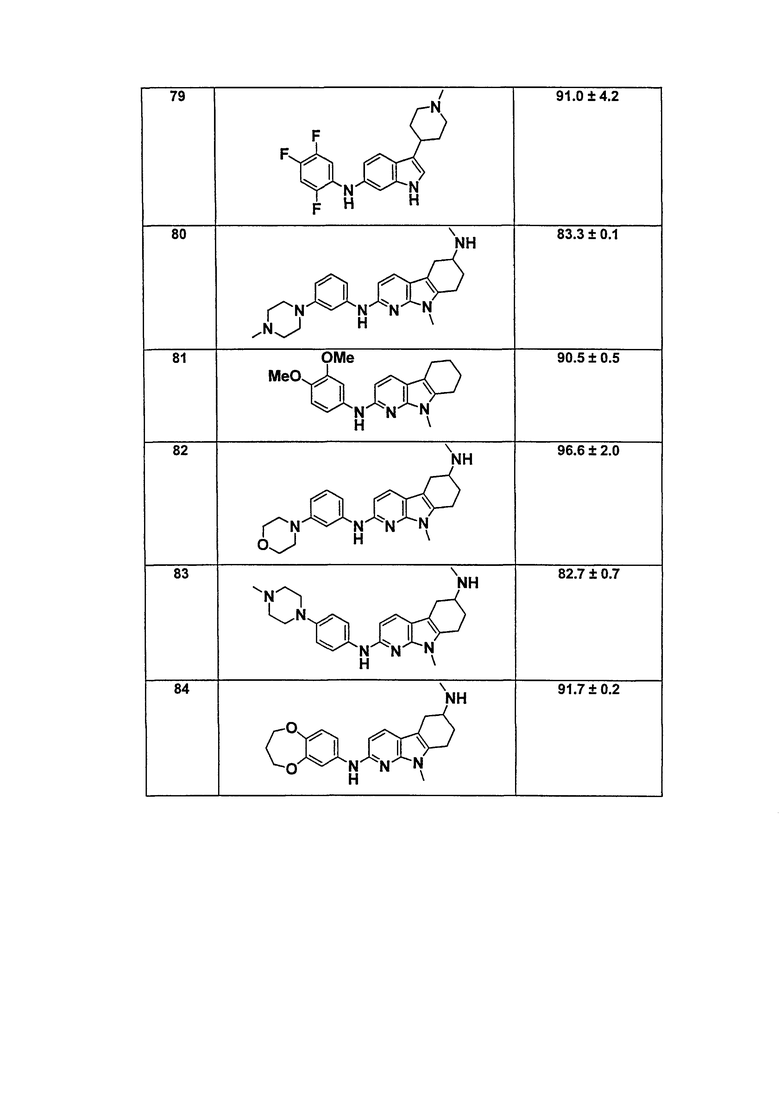





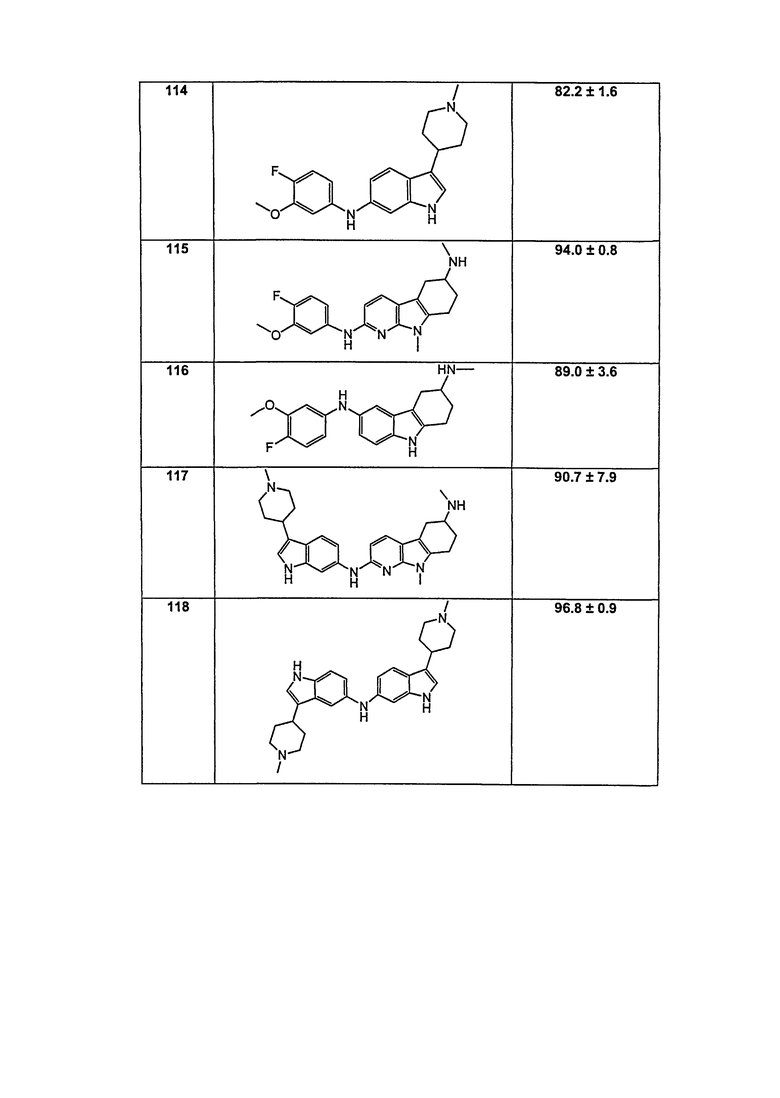


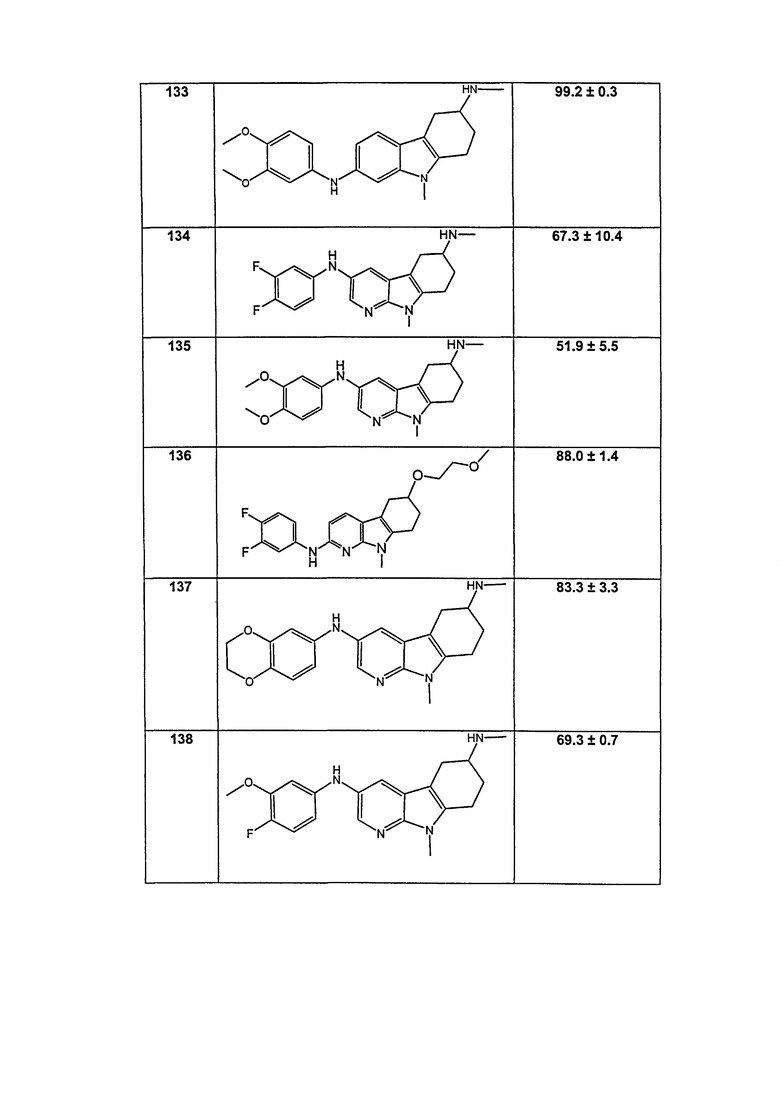



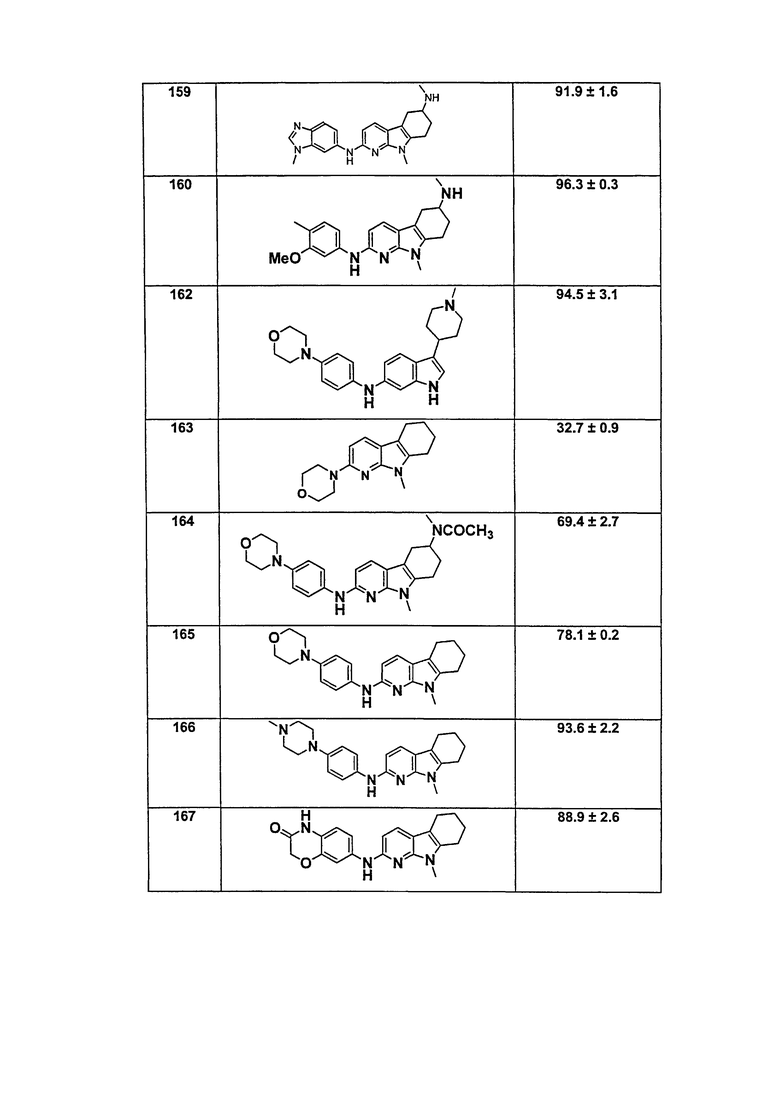











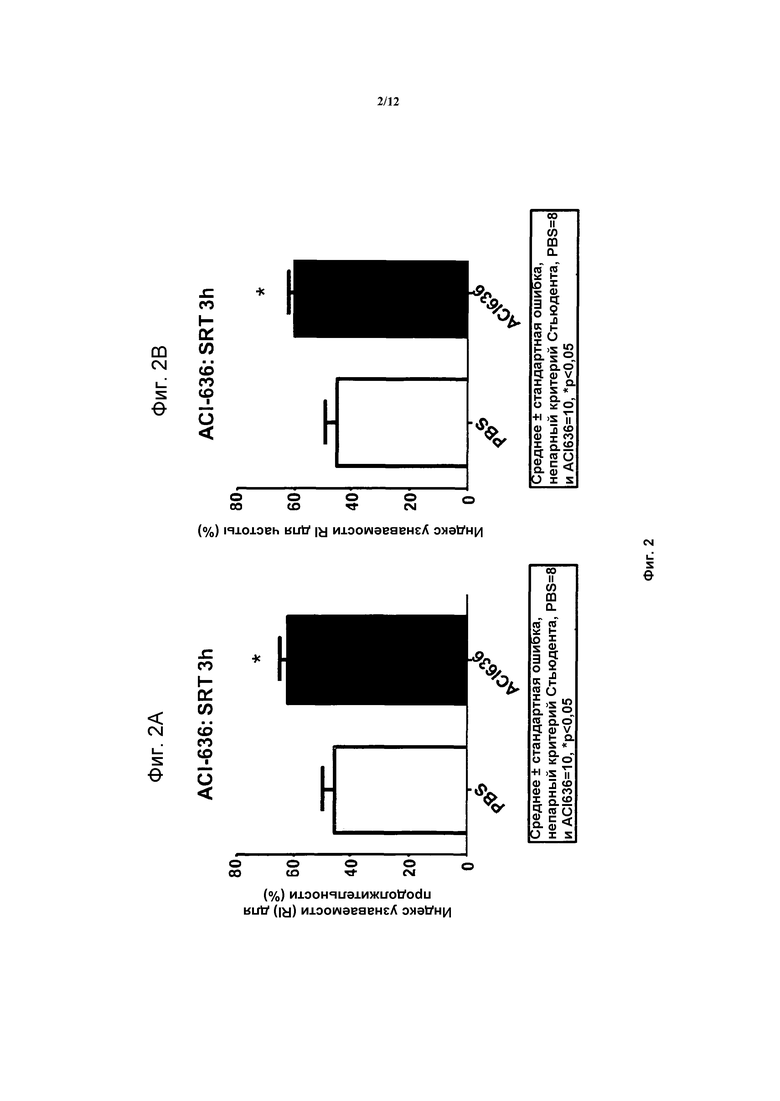







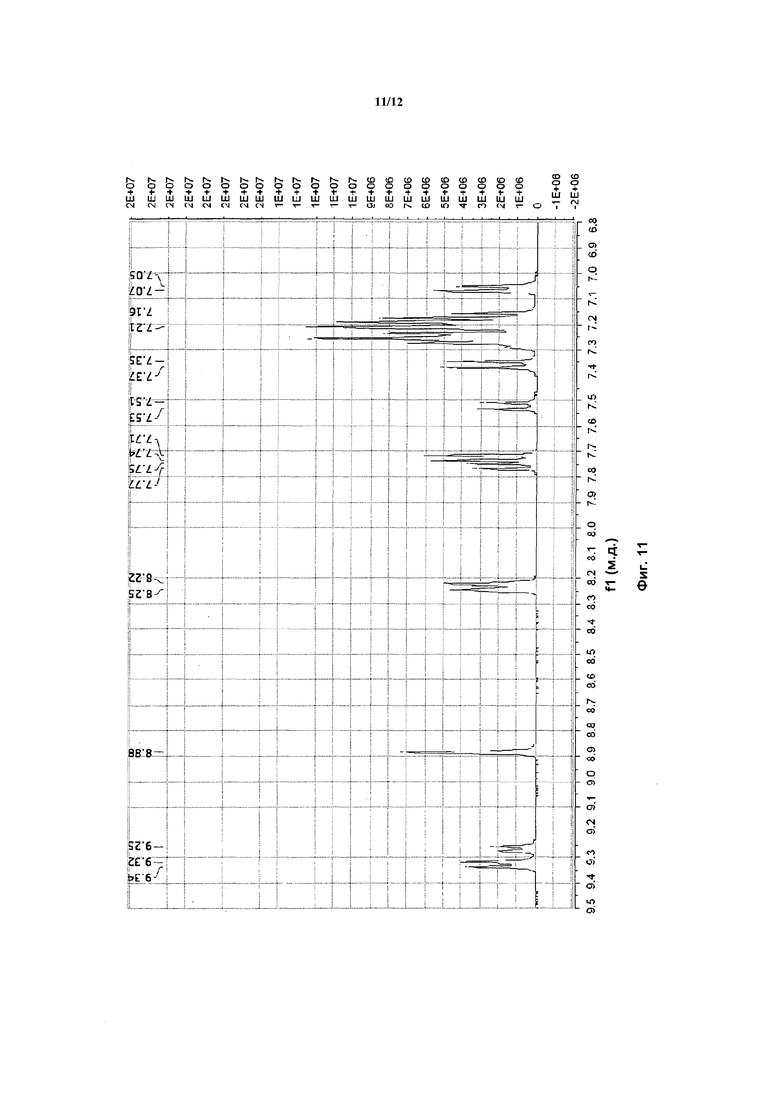
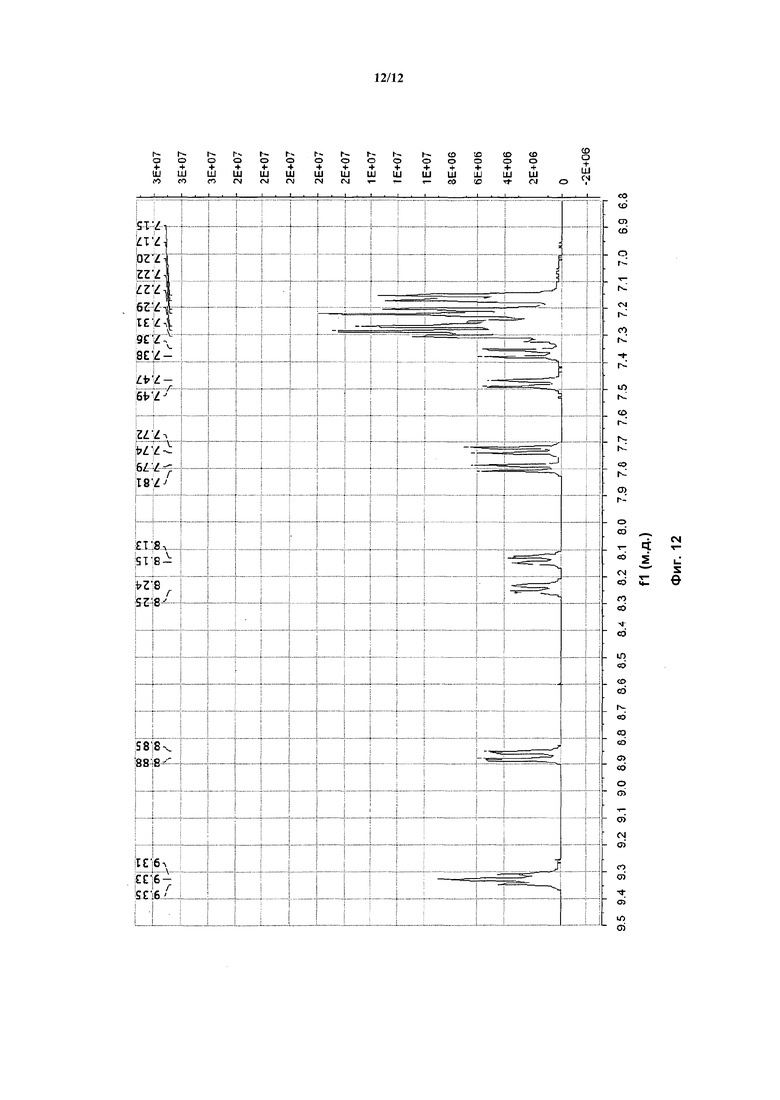
Изобретение относится к соединениям формулы (I), обладающим свойствами ингибитора агрегации Aβ1-42, фармацевтической композиции на их основе, их применению, способу лечения с их использованием, набору для обнаружения или диагностирования заболеваний, способу снижения отложения β-амилоидных бляшек и способу сохранения или повышения когнитивной способности. Соединения могут применяться при лечении группы расстройств и нарушений, связанных с амилоидным белком, таких как болезнь Альцгеймера, и заболеваний или состояний, связанных с амилоидоподобными белками, а также при лечении глазных заболеваний, связанных с патологическими нарушениями/изменениями в ткани визуальной системы. В общей формуле (I) А выбирают из группы, состоящей из (i), (ii), (iii), (iv), (v), (vi) и (viii), L1 выбирают из группы, состоящей из (a), В выбирают из группы, состоящей из (ix), (x), (xi), (xii), (xiii), где R1, R2 и R3 выбирают из группы водорода, галогена, CN, -CONR30R31, -N(R30)-С(О)-R31, алкила, -O-алкила, -С(О)О-алкила, 6-членного гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, фторалкила, где гетероциклоалкил может быть необязательно замещенным ацетильной или метильной группой, или, если любые из этих групп R1/R2/R3 являются смежными, они могут быть взяты вместе и образовывать 5-8-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из О, S или N, и где 5-8-членное кольцо может быть замещено NR20R21 или -O-алкилом, где алкил может быть замещен F, метокси или SO2Me; Ra выбирают из водорода, алкила; Rb выбирают из водорода, галогена, CONR30R31; R30, R31, R20 и R21 выбирают из водорода, алкила; X и Y выбирают из CRb и N; p равняется 0, 1 или 2. 11 н. и 25 з.п. ф-лы, 12 ил., 10 табл., 219 пр.

 ,
, 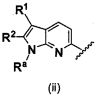 ,
, 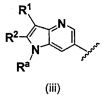 ,
, 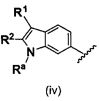 ,
, 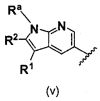 ,
,  , и
, и 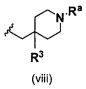 ,
, ,
,  ,
,  ,
,  и
и  ,
, 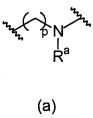
1. Соединение формулы (I):
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли:
где
А выбирают из группы, состоящей из
, , ,
, и ,
L1 направленно выбирают из группы, состоящей из
,
В выбирают из группы, состоящей из
, , ,
и ,
где
R1, R2 и R3, каждый независимо, выбирают из группы, состоящей из водорода, галогена, CN, -CONR30R31, -N(R30)-С(О)-R31, алкила, -O-алкила, -С(О)О-алкила, 6-членного гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, фторалкила, где гетероциклоалкил может быть необязательно замещенным ацетильной или метильной группой,
или, если любые из этих групп R1/R2/R3 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5-8-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из О, S или N, и где 5-8-членное кольцо может быть замещено с помощью NR20R21 или -O-алкила, где алкил может быть необязательно замещенными F, метокси или SO2Me,
для каждого случая присутствия Ra независимо выбирают из группы, состоящей из водорода, алкила;
для каждого случая присутствия Rb независимо выбирают из группы, состоящей из водорода, галогена, CONR30R31;
для каждого случая присутствия R30, R31, R20 и R21, каждый независимо, выбирают из группы, состоящей из водорода, алкила;
X и Y, каждый независимо, выбирают из группы, состоящей из CRb и N;
p равняется 0, 1 или 2;
при условии, что исключаются следующие соединения:






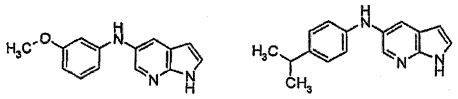


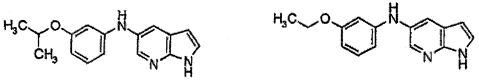









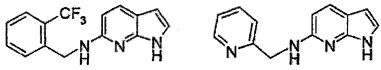

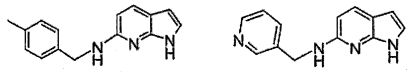



 и
и 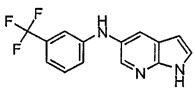
2. Соединение по п. 1, где
А выбирают из группы, состоящей из
 ,
, 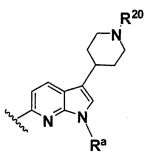 ,
, 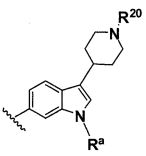 ,
,
 ,
,  ,
,
L1 представляет собой
 ,
,
В выбирают из группы, состоящей из
 ,
,  ,
,  и
и
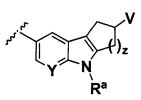 ,;
,;
где:
R1, R2, R3, Ra, R20, X и Y имеют такие же значения, как в п. 1;
V отсутствует или представляет собой NR20R21, и
z равняется 1 или 2.
3. Соединение по п. 1, где p равно 0.
4. Соединение по п. 1, где R1, R2 и R3, каждый независимо, выбирают из водорода, галогена (такого, как F), CN, CONR30R31, -O-алкила, гетероциклоалкила (такого, как 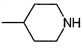 ,
,  или
или  ).
).
5. Соединение по п. 1, где Ra представляет собой водород или C1-4 алкил.
6. Соединение по п. 1, имеющее формулу (I):
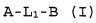
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли:
где А представляет собой:
 ,
,
L1 представляет собой:
 ,
,
В представляет собой:


 или
или  ,
,
где
R2 и R3, каждый независимо, выбирают из группы, состоящей из водорода, галогена, CN, CONR30R31, алкила, 6-членного гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, фторалкила, где гетероциклоалкил может быть необязательно замещенным ацетильной или метильной группой,
или, если R2 и R3 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из О, S или N;
R1 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия Rb независимо выбирают из группы, состоящей из водорода, галогена, CONR30R31;
для каждого случая присутствия R30, R31 и R20, каждый независимо, выбирают из группы, состоящей из водорода, алкила;
Y, каждый, представляет собой независимо СН или N;
z равняется 1 или 2.
7. Соединение по п. 1, имеющее формулу (I):

и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли:
где А представляет собой:
 ,
,
L1 представляет собой:
 ,
,
В представляет собой:
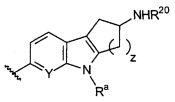 ,
,
где
R2 и R3, каждый независимо, выбирают из группы, состоящей из водорода, галогена, CN, CONR30R31, алкила, 6-членного гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, фторалкила, где гетероциклоалкил может быть необязательно замещенным ацетильной или метильной группой,
или, если R2 и R3 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из О, S или N;
R1 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия Rb независимо выбирают из группы, состоящей из водорода, галогена, CONR30R31;
для каждого случая присутствия R30, R31 и R20, каждый независимо, выбирают из группы, состоящей из водорода, алкила;
Y, каждый, представляет собой независимо СН или N;
z равняется 1 или 2.
8. Соединение по п. 1, имеющее формулу (I):
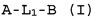
и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли:
где А представляет собой:
 ,
,
L1 представляет собой:
 ,
,
В представляет собой:
 ,
,
где
R2 и R3, каждый независимо, выбирают из группы, состоящей из водорода, галогена, CN, CONR30R31, алкила, 6-членного гетероциклоалкила с 1-2 гетероатомами, выбранными из N и О, фторалкила, где гетероциклоалкил может быть необязательно замещенным ацетилной или метильной группой,
или, если R2 и R3 являются смежными, они могут быть необязательно взяты вместе и могут образовывать 5- или 6-членное кольцо, содержащее углеродные атомы и необязательно один или два гетероатома, выбранных из О, S или N;
R1 представляет собой водород или галоген;
Ra представляет собой водород или алкил;
для каждого случая присутствия Rb независимо выбирают из группы, состоящей из водорода, галогена, CONR30R31;
для каждого случая присутствия R30, R31 и R20, каждый независимо, выбирают из группы, состоящей из водорода, алкила;
Y, каждый, представляет собой независимо СН или N.
9. Соединение по п. 1, имеющее формулу (I):

и все его стереоизомеры, рацемические смеси, фармацевтически приемлемые соли:
где А представляет собой:
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
,  или
или  ,
,
L1 представляет собой
 ,
,
и
В представляет собой
 или
или 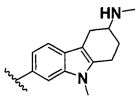 .
.
10. Соединение по п. 1, где соединение выбирают из группы, состоящей из



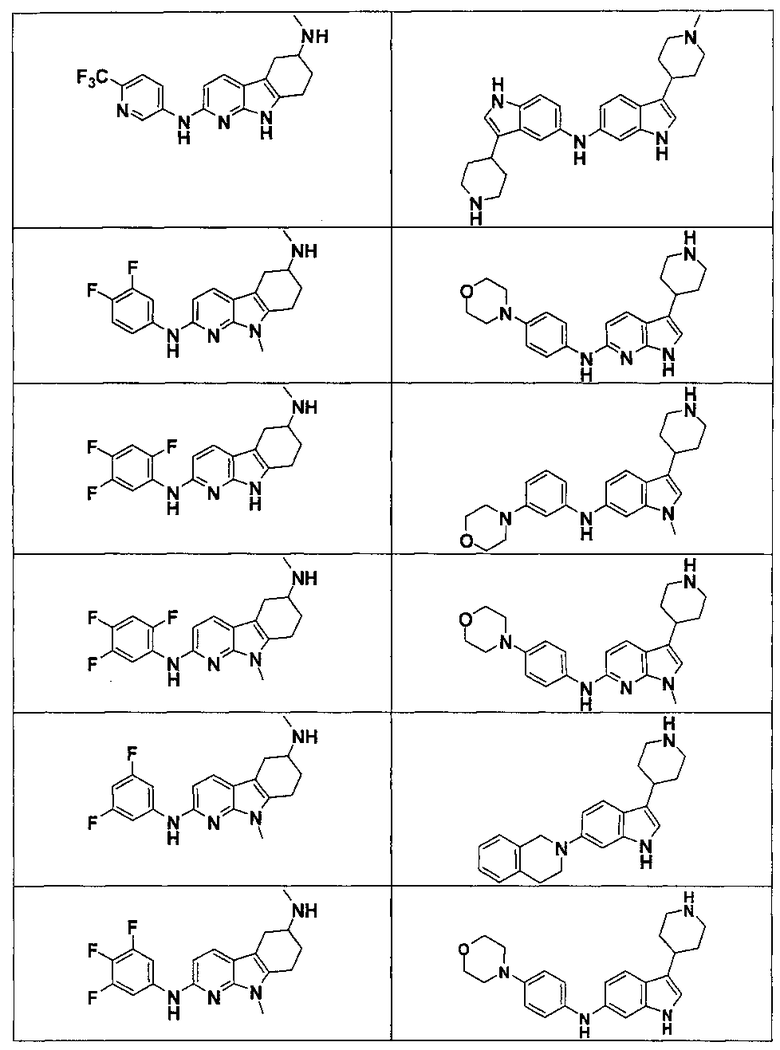

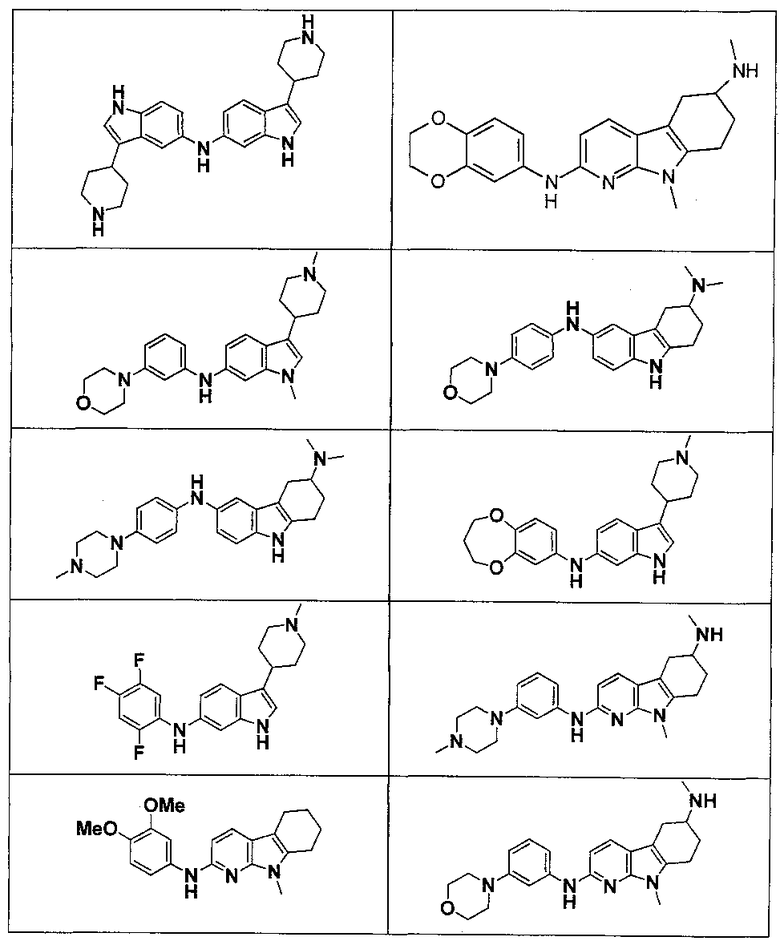
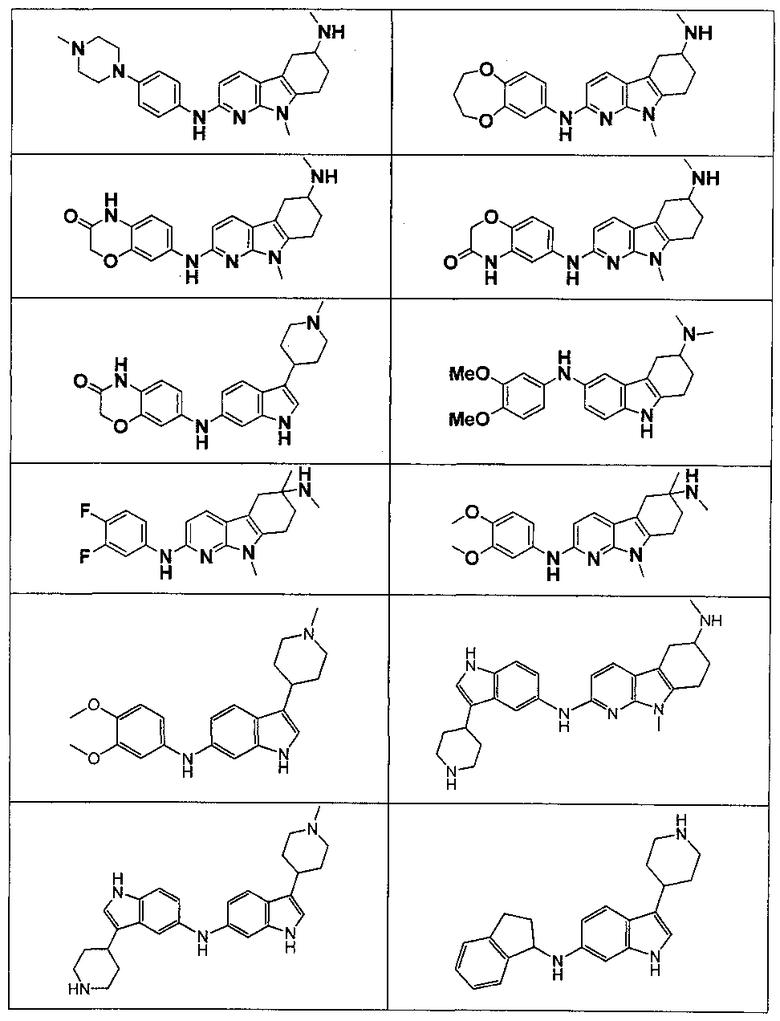






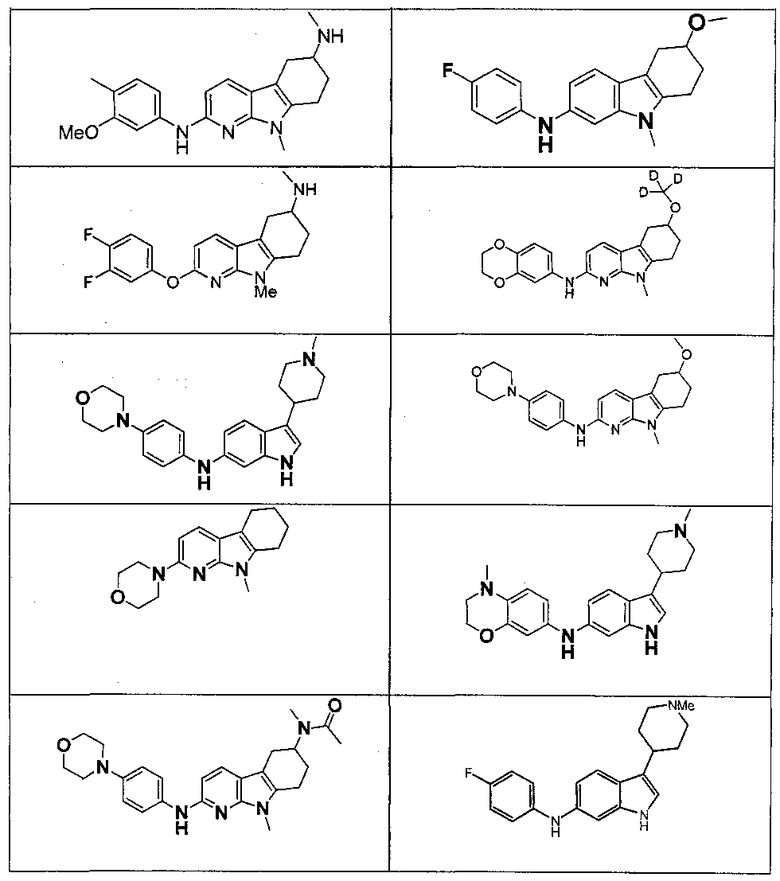
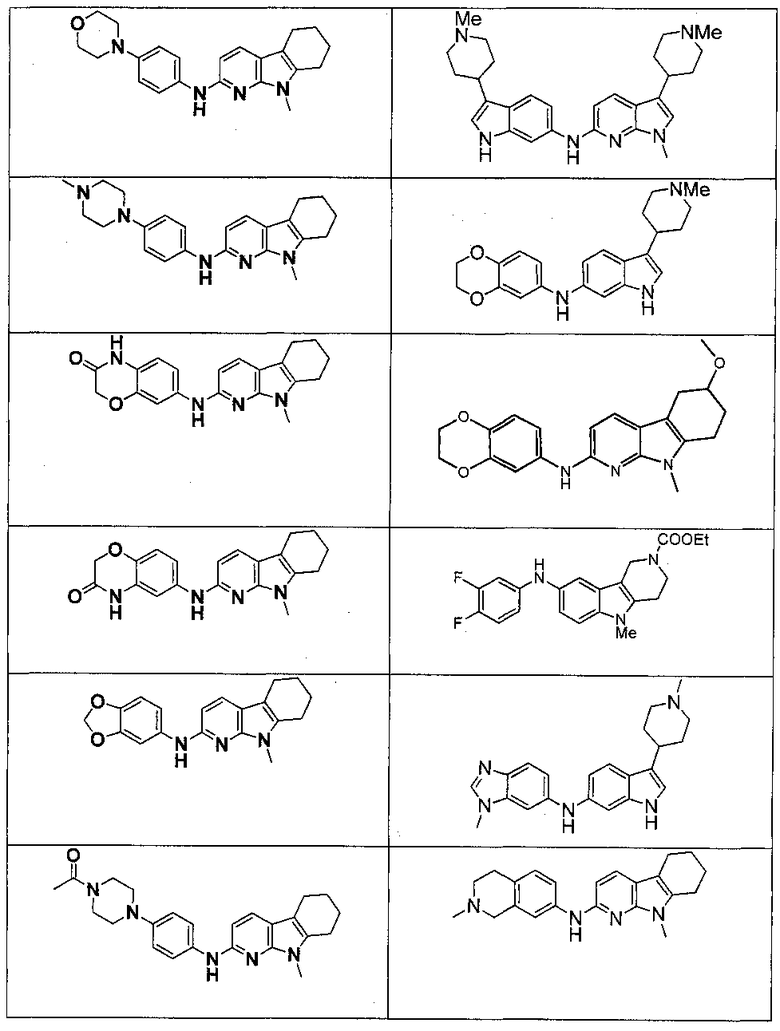
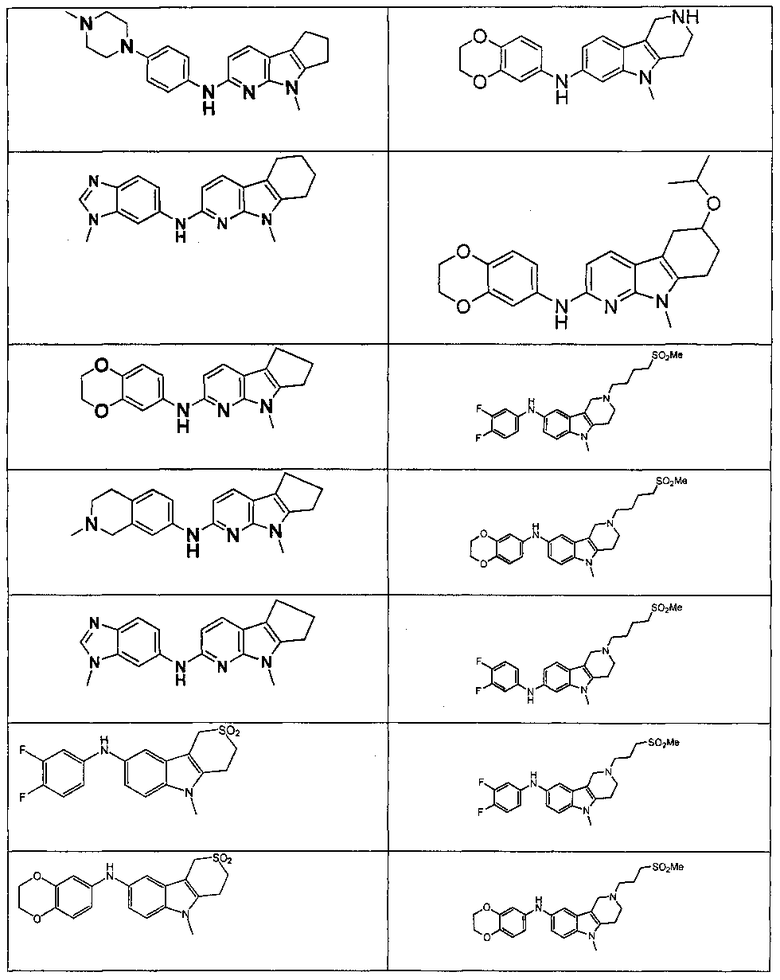


11. Фармацевтическая композиция, ингибирующая агрегацию Aβ1-42, включающая соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, где фармацевтическая композиция дополнительно включает фармацевтически приемлемый носитель или вспомогательное вещество.
12. Применение соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются, для получения лекарственного средства для лечения или предотвращения заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком.
13. Применение по п. 12, где заболевание представляет собой неврологическое нарушение, выбранное из группы, состоящей из болезни Альцгеймера (AD), деменции с тельцами Леви (LBD), синдрома Дауна, врожденного внутримозгового кровоизлияния с амилоидозом (голландского типа), комплекса Гуама паркинсонизм-деменция или умеренного когнитивного нарушения (MCI), или где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую, как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы.
14. Применение по п. 13, где неврологическое нарушение представляет собой болезнь Альцгеймера.
15. Способ лечения или предотвращения заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются.
16. Способ по п. 15, где заболевание представляет собой неврологическое нарушение, выбранное из группы, состоящей из болезни Альцгеймера (AD), деменции с тельцами Леви (LBD), синдрома Дауна, врожденного внутримозгового кровоизлияния с амилоидозом (голландского типа), комплекса Гуама паркинсонизм-деменция или умеренного когнитивного нарушения (MCI), где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую, как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы.
17. Способ по п. 16, где неврологическое нарушение представляет собой болезнь Альцгеймера.
18. Соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, для применения при лечении или предотвращении заболевания или состояния, связанного с амилоидным и/или амилоидоподобным белком.
19. Соединение по п. 18, где заболевание представляет собой неврологическое нарушение, выбранное из группы, состоящей из болезни Альцгеймера (AD), деменции с тельцами Леви (LBD), синдрома Дауна, врожденного внутримозгового кровоизлияния с амилоидозом (голландского типа), комплекса Гуама паркинсонизм-деменция или умеренного когнитивного нарушения (MCI), или где заболевание представляет собой прогрессирующий супрануклеарный парез, множественный склероз, миозит с включенными тельцами (IBM), болезнь Крейтцфельда-Якоба, болезнь Паркинсона, связанную с ВИЧ деменцию, боковой амиотрофический склероз (ALS), миозит с включенными тельцами (IBM), заболевание диабетом в зрелом возрасте, старческий амилоидоз сердца, внутрисекреторные опухоли, глаукому, амилоидную дистрофию роговицы глаза, первичную дегенерацию сетчатки, дегенерацию желтого пятна (такую, как возрастная дегенерация желтого пятна (AMD)), друзы зрительного нерва, оптическую нейропатию, неврит зрительного нерва или решетчатую дегенерацию роговицы.
20. Соединение по п. 19, где неврологическое нарушение представляет собой болезнь Альцгеймера.
21. Набор для обнаружения и/или диагностирования связанных с амилоидом заболеваний или состояний, включающий
контейнер, содержащий одно или несколько соединений по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются, и
инструкцию по использованию соединения с целью связывания амилоидного белка с образованием комплекса соединение/белок и обнаружения образования комплекса соединение/белок, в соответствии с которыми присутствие или отсутствие комплекса соединение/белок взаимосвязано с присутствием или отсутствием амилоидного белка.
22. Применение соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются, для получения лекарственного средства для лечения или предотвращения глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета-амилоиду.
23. Применение по п. 22, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
24. Способ лечения или предотвращения глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета-амилоиду, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются.
25. Способ по п. 24, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
26. Соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, для применения при лечении или предотвращении глазного заболевания или состояния, связанного с патологическим нарушением/изменением ткани визуальной системы, в частности связанного с патологическим нарушением/изменением ткани визуальной системы, имеющим отношение к бета-амилоиду.
27. Соединение по п. 26, где глазное заболевание или состояние выбирают из группы, состоящей из нейрональной деградации, кортикальных зрительных нарушений, глаукомы, катаракты вследствие бета-амилоидного отложения, амилоидной дистрофии роговицы глаза, первичной дегенерации сетчатки, дегенерации желтого пятна, например возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дегенерации роговицы.
28. Соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, для применения при ингибировании агрегации белка, в частности, при ингибировании агрегации Aβ1-42, агрегации тау-белка или агрегации альфа-синуклеина.
29. Применение соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются, для получения лекарственного средства для (а) снижения отложения β-амилоидных бляшек, и/или (b) ингибирования образования β-амилоидных бляшек, и/или (с) замедления увеличения отложения амилоидных бляшек в мозге пациента.
30. Способ (а) снижения отложения β-амилоидных бляшек, и/или (b) ингибирования образования β-амилоидных бляшек, и/или (с) замедления увеличения отложения амилоидных бляшек в мозге пациента, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются.
31. Соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, для применения при (а) снижении отложения β-амилоидных бляшек, и/или (b) ингибировании образования β-амилоидных бляшек, и/или (с) замедлении увеличения отложения амилоидных бляшек в мозге пациента.
32. Применение по п. 29, где снижение отложения β-амилоидных бляшек, и/или ингибирование образования β-амилоидных бляшек, и/или замедление увеличения отложения амилоидных бляшек приводит к уменьшению и/или облегчению воздействия заболевания или состояния, вызываемого или связанного с образованием и отложением β-амилоидных бляшек в мозге.
33. Применение соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются, для получения лекарственного средства для сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти.
34. Способ сохранения или повышения когнитивной способности памяти у субъекта, страдающего нарушением памяти, включающий введение субъекту, которому необходимо такое лечение, эффективного количества соединения по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, или не исключаются.
35. Соединение по любому из пп. 1-10, где исключаются соединения, которые исключены в п. 1, для применения при сохранении или повышении когнитивной способности памяти у субъекта, страдающего нарушением памяти.
36. Применение по п. 12, где состояние, связанное с амилоидом, характеризуется утратой когнитивной способности памяти и лечение состояния, связанного с амилоидом, приводит к улучшению сохранения когнитивной способности памяти или к полному восстановлению когнитивной способности памяти.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| УСТРОЙСТВО ДЛЯ ПРЕДОХРАНЕНИЯ ОБУВИ ОТ СКОЛЬЖЕНИЯ | 1999 |
|
RU2149573C1 |

