Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям формулы (II), которые можно использовать для селективного выявления расстройств и аномалий, ассоциированных с агрегатами белка тау, таких как болезнь Альцгеймера (AD) и другие тауопатии, например, с использованием визуализации позитронно-эмиссионной томографией (ПЭТ). Настоящее изобретение также относится к промежуточным соединениям, которые можно использовать в получении таких визуализирующих агентов. Диагностические композиции, а также способы визуализации или диагностики с использованием вышеуказанных соединений и наборов, которые пригодны для получения радиофармацевтического препарата, также являются предметом настоящего изобретения.
Уровень техники
Болезнь Альцгеймера представляет неврологическое расстройство, которое, как полагается, вызвано амилоидными бляшками, внеклеточным накоплением аномальных отложений агрегатов бета-амилоида (Аβ) в головном мозге или в глазах. Другими важными нейропатологическими признаками при AD являются внутриклеточные нейрофибриллярные сплетения (NFT), которые возникают в результате агрегации гиперфосфорилированного белка тау (тубулин-ассоциированной единицы), фосфорилированного тау или патологического тау и его конформеров. AD разделяет эту патологию со многими нейродегенеративными тауопатиями, в частности, с определенными типами фронтотемпоральной деменции (FTD). В головном мозге пациентов с AD, таупатология (тауопатия) развивается позднее, чем амилоидная патологии, и это все еще является спорным вопросом, насколько белок Aβ является причиной развития AD, что составляет сущность гипотезы так называемого амилоидного каскада (Hardy et al., Science, 1992, 256, 184-185, и совсем недавно, Musiek et al., Nature Neurosciences, 2015, 18 (6), 800-806, «Three dimensions of the amyloid hypothesis: time, space and 'wingmen').
В настоящее время единственным определенным способом диагностики AD является идентификация бляшек и сплетений в ткани головного мозга гистологическим анализом материала, отобранного при биопсии или вскрытии после смерти человека. Помимо AD, белок тау играет важную роль в других (отличных от AD) нейродегенеративных заболеваниях. Такие тауопатии, отличные от AD, включают, например, надъядерный паралич (PSP), болезнь Пика (PiD) и кортикобазальную дегенерацию (CBD).
Следовательно, существует большой интерес к выявлению таупатологии in vivo. Визуализация белка тау с помощью ПЭТ позволяет по-новому взглянуть на отложение агрегатов белка тау в головном мозге человека и может позволить неинвазивно исследовать степень таупатологии, количественно оценить изменения в отложениях белка тау с течением времени, оценить их корреляцию с когнитивной функцией и анализировать эффективность терапии, направленной на белок тау. См. недавние обзоры Shah et al., J. Nucl. Med., 2014, 55 (6), 871-874: «Molecular Imaging Insights into Neurodegeneration: Focus on ТАУ PET Radiotracers», Jovalekic et al., EJNMMI Radiopharmacy and Chemistry, 2016, 1:11, «New protein deposition tracers in the pipeline», и Ariza et al., J. Med. Chem., 2015, 58 (11), 4365-82: «ТАУ PET Imaging: Past, Present and Future». Кроме того, недавно было опубликовано несколько патентных заявок, например: WO 2013/176698, WO 2009/102498, WO 2011/119565, US 8,932,557 B2 и US 8,691,187, B2 (Siemens Medical Solutions, Lilly), WO 2012/067863 и WO 2012/068072 (оба GE Healthcare) WO 2014/026881, WO 2014/177458, WO 2014/187762, WO 2015/044095, WO 2015/052105, WO 2015/173225 (Hoffmann-La Roche AG), WO 2015/188368 (Merck Sharp & Dohme) и WO 2016/124508 (UCB Biopharma SPRL), в которых заявлены новые соединения для визуализации белка тау.
Для достижения высокой селективности к мишени были использованы молекулярные зонды, которые распознают и связываются с патологической мишенью. Таким образом, селективность связывания с патологическим белком тау по сравнению с другими белковыми отложениями в головном мозге является основным требованием для визуализации белка тау. Для снижения интерференции фонового сигнала за счет неспецифического связывания с немишеневыми белками (например, связывания с Aβ или моноаминоксидазами), визуализирующие соединения должны связываться с высокой аффинностью с патологическим белком тау. Поскольку амилоидные или амилоидоподобные отложения, образованные из белков с различными первичными аминокислотными последовательностями, имеют общую четвертичную β-складчатую конформацию, то необходимы молекулярные зонды, которые могут дифференцировать такие структуры, чтобы избежать выявления других патологий (ложноположительных) и, следовательно, постановки ошибочного диагноза.
Сообщалось, что немишеневое связывание с моноаминоксидазой A или B является существенным ограничением для индикаторов белка тау, особенно T-807 и THK-5351 (Vermeiren C. et al., Alzheimers & Dementia. 2015; 11 (7) Supplement p1-2: «T807, a reported selective ТАУ tracer, binds with nanomolar affinity to monoamine oxidase A»; Ng, KP et al., Alzheimer's Research and Therapy 2017, 9:25: «Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain»). Немишеневое связывание с моноаминоксидазами A или B мешает интерпретации данных ПЭТ-визуализации с использованием T807 и THK5351 для белка тау. Присутствие моноаминоксидаз в нескольких областях головного мозга ограничивает интерпретацию результатов ПЭТ-визуализации с помощью этих индикаторов.
Помимо высокой селективности, способность связываться с различными изоформами белка тау также является важным аспектом для индикатора тау-белка. До сих пор большинство индикаторов демонстрируют связывание с белком тау при AD. Однако белок тау при AD представляет собой смесь двух изоформ, так называемых 3R-тау и 4R-тау. Другие тауопатии, отличные от AD, характеризуются преимущественным присутствием одной из этих изоформ. При болезни Пика (PiD) преимущественно присутствует изоформа 3R-тау, тогда как при прогрессирующем надъядерном параличе (PSP) и при кортикобазальной дегенерации (CBD) изоформа 4R-тау является существующей патологией.
Кроме того, молекулярные зонды также должны быть сконструированы таким образом, чтобы после введения они могли распространяться в организме и достигать своей мишени. Для визуализации агрегатов белка тау, ассоциированных с неврологическими расстройствами, такими как, например, болезнь Альцгеймера, необходимы визуализирующие агенты, которые могут проникать через гематоэнцефалический барьер и проходить в соответствующие области головного мозга. Для нацеливания на внутриклеточные агрегаты белка тау проницаемость клеток является дополнительным требованием к визуализирующим агентам. Еще одной предпосылкой для получения достаточного отношения сигнал/шум является быстрое вымывание соединений из нецелевых областей мозга (или другого органа-мишени). Кроме того, соединения не должны подвергаться дефторированию, поскольку поглощение костной тканью в черепе (за счет присутствия свободного фтора) будет вызывать значительное перетекание в головной мозг, что ограничивает применимость соединений (Chien D.T. et al., J. Alzheimers Dis., 2014; 38: 171-84).
Специфически раскрытое и наиболее усовершенствованное производное в WO 2013/176698 представляет собой 2,5-дизамещенное производное пиридина 18F-1 (см. также заявку на патент США № 8932557 B2).
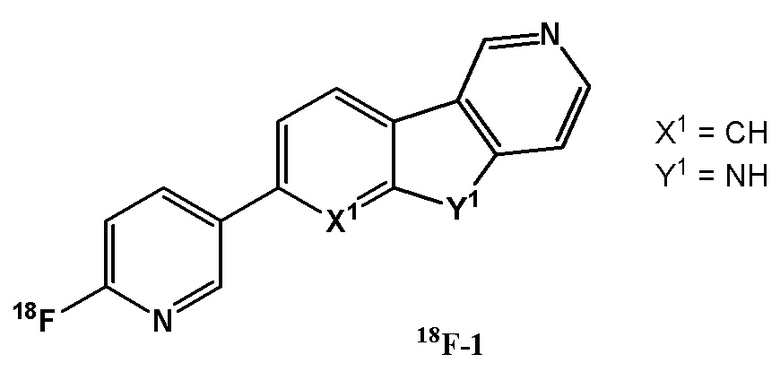
Соединение 18F-1 было исследовано в различных клинических испытаниях. Несмотря на то, что 18F-1, по-видимому, способно детектировать белок тау у пациентов с AD или амилоид-бета-позитивным легким когнитивным расстройством (MCI), сообщалось о различных его ограничениях.
Vermeiren и соавторы обнаружили, что соединение 18F-1 связывается с моноаминоксидазой A (MAO A) с KD, равной 1,5 нМ. Данные этих авторов однозначно демонстрируют, что соединение 18F-1 связывается с агрегатами белка тау и МАО-А с аналогичной высокой аффинностью. Полученные результаты побуждают с осторожностью относиться к интерпретации клинических данных для соединения 18F-1, поскольку МАО-А широко экспрессируется в большинстве областей головного мозга человека (Vermeiren et al., Alzheimers & Dementia. 2015; 11 (7) Supplement p1-2: T807- a reported selective ТАУ tracer, binds with nanomolar affinity to Monoamine oxidase A).
Сообщалось, что соединение 18F-1 имеет довольно сильный сигнал в областях базальных ганглиев мозга, например, в полосатом теле и черной субстанции, независимо от диагноза пациента. Сигнал 18F-1 в коре головного мозга не достигал «стабильного состояния» (промежуток времени, в течение которого отношение связывания в области-мишени к связыванию в контрольной ткани (т. е. мозжечке) было стабильным). Кроме того, кинетика 18F-1 в различных областях головного мозга была различной и не стабилизировалась в течение 150-мин периода сканирования (S. Baker, Human Amyloid Imaging Meeting, 2015).
Связывание соединения 18F-1 со срезами головного мозга человека с AD было продемонстрировано авторадиографией. Однако соединение 18F-1 показало ограничения в связывании со срезами головного мозга с патологиями тауопатий, отличных от AD: а) Lowe V.J. et al. An autoradiographic evaluation of AV-1451 ТАУ PET in dementia. Acta Neuropathologica Communications. 2016; 4:58; b) Marquie M. et al. Validating novel ТАУ Positron Emission Tomography Tracer [F-18]-AV-1451 (T807) on postmortem Brain Tissue. Annals of Neurology. 2015; 78:787; c) Gomez F. et al. Quantitative assessment of [18F]AV-1451 distribution in AD, PSP and PiD Post-Mortem Brain Tissue Sections relative to that of the anti-ТАУ antibody AT8. Journal of Nuclear Medicine. 2016; 57, S2: 348, d) Sander K. et al. Characterization of ТАУ positron emission tomography tracer AV1451 binding to postmortem tissue in Alzheimer's disease, primary TAYopathies, and other dementias. Alzheimers Dementia 2016, 12(11): 116-1124 e) Smith R. et al. Increased basal ganglia binding of 18F-AV-1451 in patients with progressive supranuclear palsy. Movement disorders 2016.
Также в клинике, 18F-1, по-видимому, имеет ограниченную ценность для детектирования белка тау у субъектов с PSP: а) Smith R. et al., ТАУ neuropathology correlates with FDG-PET, but nor with AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathologica 2017, 133:149-151; b) Smith R. et al. Increased basal ganglia binding of 18F-AV-1451 in patients with progressive supranuclear palsy. Movement disorders 2017, 32(1), 108-114.
Окончательные выводы по результатам этих исследований показывают, что T807/AV1451 не может быть надежным, чтобы отличить отдельных пациентов с PSP от контрольных здоровых субъектов. В основном это объясняется повышенным неспецифическим связыванием в структурах среднего мозга, таких как базальные ганглии. Поглощение, наблюдаемое в коре головного мозга и белом веществе, не отражало таупатологию при PSP.
Соединение 18F-2 раскрыто в WO 2015/052105.
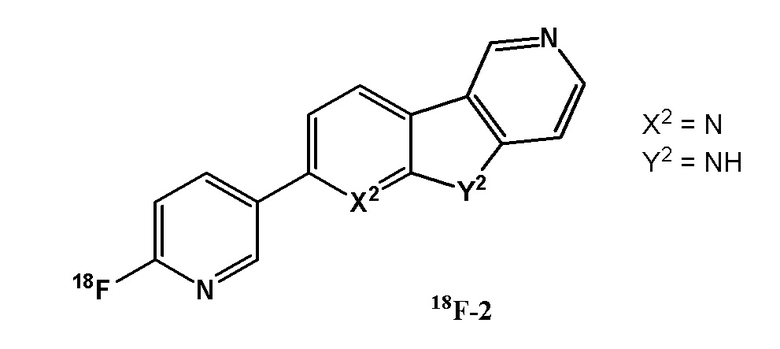
В WO 2015/052105 раскрывается только одно 18F-меченное соединение, и соответствующее производное, меченное тритием. Соединение содержит группу 2,5-дизамещенного пиридина (соединение 18F-2). В WO 2015/052105 не приводятся какие-либо данные о связывании с изоформами тау-белка при тауопатиях, отличных от AD, о связывании с MAO A (или иным образом о селективности к белку тау), поглощении в головном мозге, вымывании или удерживании в головном мозге здорового субъекта или каких-либо данные о дефторировании in vivo.
Было обнаружено, что 18F-2 не связывается с тканями головного мозга у пациентов с тауопатиями, отличными от AD, такими как болезнь Пика (PiD) и прогрессирующий надъядерный паралич (PSP) (Honer M. et al., In vitro binding of 3H-RO6958948, 3H-AV-1451, 3H-THK5351 and 3H-T808 to ТАУ aggregates in non-AD TAYopathies. Human Amyloid Imaging 2017, abstract 99).
Принимая во внимание вышеописанный уровень техники, целью настоящего изобретения было обеспечение соединения, которое обладает высокой аффинностью и селективностью по отношению к белку тау и, таким образом, подходит в качестве агента для ПЭТ-визуализации. Предпочтительно соединения по настоящему изобретению демонстрируют высокую аффинность к агрегатам белка тау, высокую селективность по отношению к патологическому белку тау по сравнению с другими мишенями в головном мозге и благоприятные фармакокинетические свойства без дефторирования. Требуемый агент для ПЭТ-визуализации белка тау должен связываться как с 3R-, так и с 4R-тау для нацеливания на тауопатии с AD и без AD, включая PiD, CBD и PSP.
Сущность изобретения
Следовательно, настоящее изобретение относится к следующим пунктам:
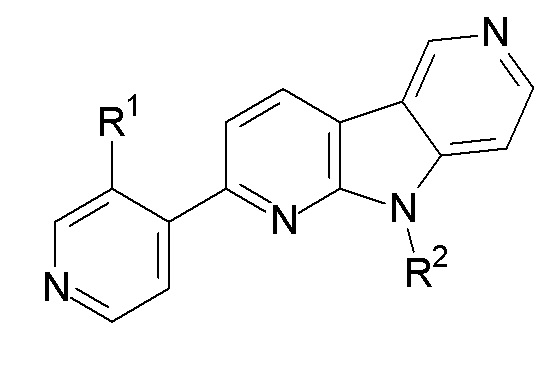
1. Соединение формулы (II):
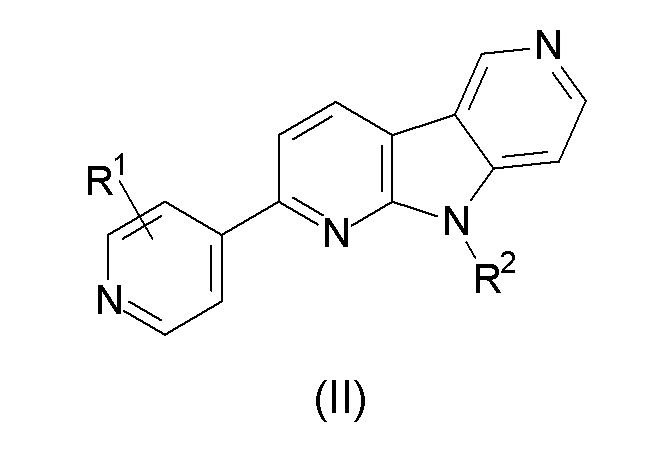
а также его фармацевтически приемлемые соли, гидраты, сольваты, пролекарства и полиморфы;
где:
R1 выбран из группы, состоящей из 18F, F и LG;
R2 представляет Н или PG;
PG представляет защитную группу;
LG представляет уходящую группу;
где любой Н формулы II может быть Н, 2Н или 3Н.
2. Соединение по п.1, которое представляет:
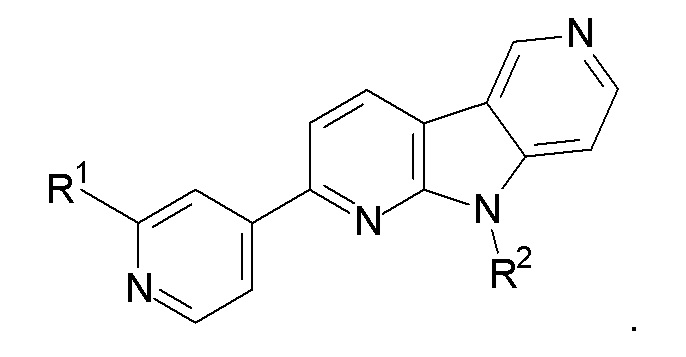
3. Соединение по п.1, которое представляет:
4. Соединение по пп.1, 2 или 3, где R1 представляет 18F, и R2 представляет Н.
5. Соединение по пп.1, 2 или 3, где R1 представляет F, и R2 представляет Н.
6. Соединение по пп.1, 2 или 3, где R1 представляет LG, и R2 представляет H или PG.
7. Соединение по пп.1, 2 или 3, где R1 представляет LG, и R2 представляет Н.
8. Соединение по пп.1, 2 или 3, где R1 представляет LG, и R2 представляет PG.
9. Соединение по пп.1, 2, 3, 6, 7 или 8, где LG представляет нитро, галоген или триметиламмоний.
10. Соединение по п.9, где LG представляет нитро или триметиламмоний.
11. Соединение по пп.1, 2, 3, 6, 8, 9 или 10, где PG представляет трет-бутилоксикарбонил (BOC), трифенилметил (тритил) или диметокситритил (DMT).
12. Соединение по п.11, где PG представляет трет-бутилоксикарбонил (BOC).
13. Соединение по пп.1, 2 или 3, где соединение является детектируемо меченым.
14. Соединение по п.13, где детектируемая метка выбрана из 2H, 3H и 18F.
15. Соединение по п.14, где детектируемой меткой является 18F.
16. Диагностическая композиция, содержащая соединение, как определено в любом из пп.4, 13, 14 или 15, и, необязательно, фармацевтически приемлемый носитель, разбавитель, адъювант или эксципиент.
17. Соединение, как определено в пп.4 или 15, для применения в диагностике.
18. Соединение, как определено в пп.4 или 15, для применения при визуализации агрегатов белка тау, в частности, для применения в позитронно-эмиссионной томографии для визуализации агрегатов белка тау.
19. Соединение, как определено в пп.4 или 15, для применения в диагностике расстройства, ассоциированного с агрегатами белка тау, или для применения в диагностике тауопатии, где, в частности, диагностика проводится с помощью позитронно-эмиссионной томографии.
20. Соединение для применения по п.19, где тауопатия представляет собой 3R-тауопатию.
21. Соединение для применения по п.19, где тауопатия представляет собой 4R-тауопатию.
22. Соединение для применения по п.19, где расстройство выбрано из болезни Альцгеймера (AD), семейной формы AD, болезни Крейцфельда-Якоба, деменции боксера, синдрома Дауна, болезни Герстмана-Штраусслера-Шейнкера, миозита с тельцами включения, церебральной амилоидной ангиопатии, связанной с прионными белками, черепно-мозговой травмы (TBI), бокового амиотрофического склероза, комплекса паркинсонизма-деменции Гуама, болезни двигательного нейрона с нейрофибриллярными сплетениями, отличной от болезни Гуама, болезни аргирофильных зерен, кортикобазальной дегенерации (CBD), диффузных нейрофибриллярных сплетений с кальцификацией, фронтотемпоральной деменции с паркинсонизмом, связанной с хромосомой 17, болезни Галлервордена-Шпатца, множественной системной атрофии, болезни Нимана-Пика типа C, паллидо-понто-нигральной дегенерации, болезни Пика (PiD), прогрессирующего субкортикального глиоза, прогрессирующего надъядерного паралича (PSP), подострого склерозирующего панэнцефалита, деменции, связанной только со сплетениями, постэнцефалитического паркинсонизма, миотонической дистрофии, тау-панэнцефалопатии, AD-подобных заболеваний с поражением астроцитов, некоторых прионных болезней (GSS с тау), мутаций в LRRK2, хронической травматической энцефалопатии, семейной британской деменции, семейной датской деменции, фронтотемпоральной лобарной дегенерации, гваделупского паркинсонизма, нейродегенерации с накоплением железа в мозге, SLC9A6-ассоциированной задержки умственного развития, таутопатии белого вещества с глобулярными глиальными включениями, синдрома травматического стресса, эпилепсии, деменции с тельцами Леви (LBD), наследственного церебрального кровоизлияния с амилоидозом (голландский тип), легкого когнитивного расстройства (MCI), рассеянного склероза, болезни Паркинсона, деменции, связанной с ВИЧ, диабета с началом во взрослом возрасте, старческого сердечного амилоидоза, эндокринных опухолей, глаукомы, амилоидоза глаза, первичной дегенерации сетчатки, макулярной дегенерации (например, возрастной макулярной дегенерации (AMD)), друза диска зрительного нерва, нейропатии зрительного нерва, неврита зрительного нерва и решетчатой дистрофии; предпочтительно болезни Альцгеймера.
23. Соединение для применения по п.22, где расстройство представляет болезнь Альцгеймера (AD).
24. Соединение для применения по п.22, где расстройством является болезнь Паркинсона или атипичный паркинсонизм.
25. Соединение для применения по п.22, где расстройство представляет прогрессирующий надъядерный паралич (PSP).
26. Соединение для применения по п.22, где расстройство представляет болезнь Пика (PiD).
27. Соединение для применения по любому из пп.18-26, где агрегаты белка тау визуализируются в головном мозге или в глазу, где предпочтительно детектируемая метка представляет 18F, и визуализация представляет позитронно-эмиссионную томографию.
28. Способ визуализации агрегатов белка тау, в частности, способ визуализации позитронно-эмиссионной томографией агрегатов белка тау, где эффективное количество соединения, как определено в пп. 4 или 15, вводят пациенту.
29. Способ диагностики расстройства, ассоциированного с агрегатами белка тау или тауопатии, где пациенту вводят эффективное количество соединения, как определено в пп. 4 или 15, где, в частности, диагностику проводят позитронно-эмиссионной томографией.
30. Способ по п.29, где тауопатия представляет собой 3R-тауопатию.
31. Способ по п.29, где тауопатия представляет собой 4R-тауопатию.
32. Соединение для применения по п.29, где расстройство выбрано из болезни Альцгеймера (AD), семейной формы AD, болезни Крейцфельда-Якоба, деменции боксера, синдрома Дауна, болезни Герстмана-Штраусслера-Шейнкера, миозита с тельцами включения, церебральной амилоидной ангиопатии, связанной с прионными белками, черепно-мозговой травмы (TBI), бокового амиотрофического склероза, комплекса паркинсонизма-деменции Гуама, болезни двигательного нейрона с нейрофибриллярными сплетениями, отличной от болезни Гуама, болезни аргирофильных зерен, кортикобазальной дегенерации (CBD), диффузных нейрофибриллярных сплетений с кальцификацией, фронтотемпоральной деменции с паркинсонизмом, связанной с хромосомой 17, болезни Галлервордена-Шпатца, множественной системной атрофии, болезни Нимана-Пика типа C, паллидо-понто-нигральной дегенерации, болезни Пика (PiD), прогрессирующего субкортикального глиоза, прогрессирующего надъядерного паралича (PSP), подострого склерозирующего панэнцефалита, деменции, связанной только со сплетениями, постэнцефалитического паркинсонизма, миотонической дистрофии, тау-панэнцефалопатии, AD-подобных заболеваний с поражением астроцитов, некоторых прионных болезней (GSS с тау), мутаций в LRRK2, хронической травматической энцефалопатии, семейной британской деменции, семейной датской деменции, фронтотемпоральной лобарной дегенерации, гваделупского паркинсонизма, нейродегенерации с накоплением железа в мозге, SLC9A6-ассоциированной задержки умственного развития, таутопатии белого вещества с глобулярными глиальными включениями, синдрома травматического стресса, эпилепсии, деменции с тельцами Леви (LBD), наследственного церебрального кровоизлияния с амилоидозом (голландский тип), легкого когнитивного расстройства (MCI), рассеянного склероза, болезни Паркинсона, деменции, связанной с ВИЧ, диабета с началом во взрослом возрасте, старческого сердечного амилоидоза, эндокринных опухолей, глаукомы, амилоидоза глаза, первичной дегенерации сетчатки, макулярной дегенерации (например, возрастной макулярной дегенерации (AMD)), друза диска зрительного нерва, нейропатии зрительного нерва, неврита зрительного нерва и решетчатой дистрофии; предпочтительно болезни Альцгеймера.
33. Способ по п.32, где расстройство представляет болезнь Альцгеймера (AD).
34. Способ по п.32, где расстройством является болезнь Паркинсона или атипичный паркинсонизм.
35. Способ по п.32, где расстройство представляет прогрессирующий надъядерный паралич (PSP).
36. Способ по п.32, где расстройство представляет болезнь Пика (PiD).
37. Способ по любому из пп.28-36, где агрегаты белка тау визуализируются в головном мозге или в глазу, где предпочтительно детектируемая метка представляет 18F, и визуализация представляет позитронно-эмиссионную томографию.
38. Применение соединения, как определено в пп.4 или 15, для производства диагностического агента для визуализации агрегатов белка тау, в частности, для визуализации агрегатов белка тау позитронно-эмиссионной томографией.
39. Применение соединения, как определено в пп.4 или 15, для производства диагностического агента для диагностики расстройства, ассоциированного с агрегатами белка тау, или для диагностики тауопатии, где, в частности, диагностика проводится позитронно-эмиссионной томографией.
40. Способ по п.39, где тауопатия представляет собой 3R-тауопатию.
41. Способ по п.39, где тауопатия представляет собой 4R-тауопатию.
42. Соединение для применения по п.39, где расстройство выбрано из болезни Альцгеймера (AD), семейной формы AD, болезни Крейцфельда-Якоба, деменции боксера, синдрома Дауна, болезни Герстмана-Штраусслера-Шейнкера, миозита с тельцами включения, церебральной амилоидной ангиопатии, связанной с прионными белками, черепно-мозговой травмы (TBI), бокового амиотрофического склероза, комплекса паркинсонизма-деменции Гуама, болезни двигательного нейрона с нейрофибриллярными сплетениями, отличной от болезни Гуама, болезни аргирофильных зерен, кортикобазальной дегенерации (CBD), диффузных нейрофибриллярных сплетений с кальцификацией, фронтотемпоральной деменции с паркинсонизмом, связанной с хромосомой 17, болезни Галлервордена-Шпатца, множественной системной атрофии, болезни Нимана-Пика типа C, паллидо-понто-нигральной дегенерации, болезни Пика (PiD), прогрессирующего субкортикального глиоза, прогрессирующего надъядерного паралича (PSP), подострого склерозирующего панэнцефалита, деменции, связанной только со сплетениями, постэнцефалитического паркинсонизма, миотонической дистрофии, тау-панэнцефалопатии, AD-подобных заболеваний с поражением астроцитов, некоторых прионных болезней (GSS с тау), мутаций в LRRK2, хронической травматической энцефалопатии, семейной британской деменции, семейной датской деменции, фронтотемпоральной лобарной дегенерации, гваделупского паркинсонизма, нейродегенерации с накоплением железа в мозге, SLC9A6-ассоциированной задержки умственного развития, таутопатии белого вещества с глобулярными глиальными включениями, синдрома травматического стресса, эпилепсии, деменции с тельцами Леви (LBD), наследственного церебрального кровоизлияния с амилоидозом (голландский тип), легкого когнитивного расстройства (MCI), рассеянного склероза, болезни Паркинсона, деменции, связанной с ВИЧ, диабета с началом во взрослом возрасте, старческого сердечного амилоидоза, эндокринных опухолей, глаукомы, амилоидоза глаза, первичной дегенерации сетчатки, макулярной дегенерации (например, возрастной макулярной дегенерации (AMD)), друза диска зрительного нерва, нейропатии зрительного нерва, неврита зрительного нерва и решетчатой дистрофии; предпочтительно болезни Альцгеймера.
43. Применение по п.42, где расстройство представляет болезнь Альцгеймера (AD).
44. Применение по п.42, где расстройством является болезнь Паркинсона или атипичный паркинсонизм.
45. Применение по п.42, где расстройство представляет прогрессирующий надъядерный паралич (PSP).
46. Применение по п.32, где расстройством представляет болезнь Пика (PiD).
47. Применение по любому из пп.38-46, где агрегаты белка тау визуализируются в головном мозге или в глазу, где предпочтительно детектируемая метка представляет 18F, и визуализация представляет позитронно-эмиссионную томографию.
48. Применение соединения по п.5 в качестве аналитического стандарта.
49. Применение соединения по п.5 в качестве инструмента для скрининга in vitro.
50. Способ получения соединения, как определено в п. 4, включающий взаимодействие соединения, как определено в п. 6, с [18F]фторирующим агентом, где способ дополнительно включает отщепление защитной группы PG, если она присутствует.
51. Способ по п.50, где [18F]фторирующий агент выбран из K18F, H18F, Cs18F, Na18F и тетра(C1-6 алкил)аммониевой соли 18F.
52. Способ получения диагностической композиции, как определено в п.16, включающий взаимодействие соединения, как определено в п.6, с [18F]фторирующим агентом, где способ дополнительно включает отщепление защитной группы PG, если она присутствует, и затем необязательно примешивание фармацевтически приемлемого носителя, разбавителя, адъюванта или эксципиента.
53. Набор для получения радиофармацевтического препарата, где указанный набор включает герметичный флакон, содержащий заранее определенное количество соединения, как определено в п.6.
54. Набор по п.53, который дополнительно содержит, по меньшей мере, один компонент, выбранный из реакционного растворителя, картриджа для твердофазной экстракции, реагента для отщепления защитной группы, растворителя для очистки, растворителя для формуляции и фармацевтически приемлемого носителя, разбавителя, адъюванта или эксципиента для формуляции.
55. Способ сбора данных для диагностики расстройства, ассоциированного с агрегатами белка тау в образце или у пациента, включающий:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, как определено в пп.13-15;
(b) обеспечение связывания соединения с агрегатом белка тау;
(с) детектирование соединения, связанного с агрегатом белка тау; и
(d) необязательно сопоставление наличия или отсутствия связывания соединения с агрегатом белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела.
56. Способ определения количества агрегата белка тау в ткани и/или жидкости организма, включающий:
(a) обеспечение образца, представляющего исследуемую ткань и/или биологическую жидкость;
(b) тестирование образца на присутствие агрегата белка тау с соединением, как определено в пп.13-15;
(с) определение количества соединения, связанного с агрегатом белка тау; и
(d) расчет количества агрегата белка тау в тканях и/или жидкости организма.
57. Способ сбора данных для определения предрасположенности к расстройству, ассоциированному с агрегатами белка тау, у пациента, включающий детектирование специфического связывания соединения, как определено в пп.13-15, с агрегатом белка тау в образце или in situ, который включает стадии:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, как определено в пп.13-15, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением.
58. Способ сбора данных для мониторинга остаточного проявления расстройства у пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, которого лечили лекарственным средством, где способ включает:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, как определено в пп.13-15, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением.
59. Способ сбора данных для прогнозирования реактивности пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, и получающего лечение лекарственным средством, включающий:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, как определено в пп.13-15, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением.
Понятно, что настоящее изобретение охватывает соединения формулы (II), в которых один или более соответствующих атомов заменены другим изотопом. Например, соединения формулы (II) включают соединения, в которых один или более атомов водорода заменены на тритий и/или один или более атомов водорода заменены на дейтерий.
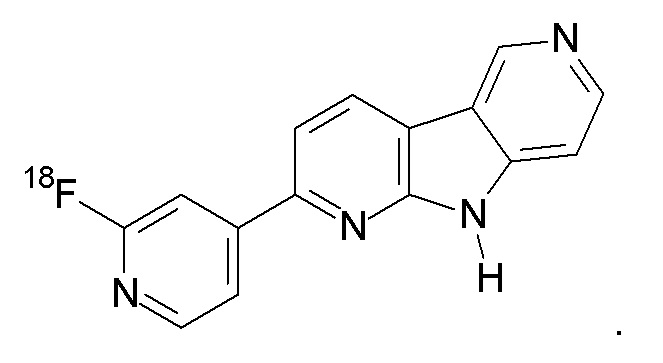
Заявители настоящего изобретения с удивлением обнаружили, что соединения формулы (II), в которой R1 представляет 18F или F, и R2 представляет H (соединения F-3a, F-3b, 18F-3a и 18F-3b соответственно), обладают существенно улучшенными свойствами по сравнению с соединениями 18F-1 или 18F-2 предшествующего уровня техники.
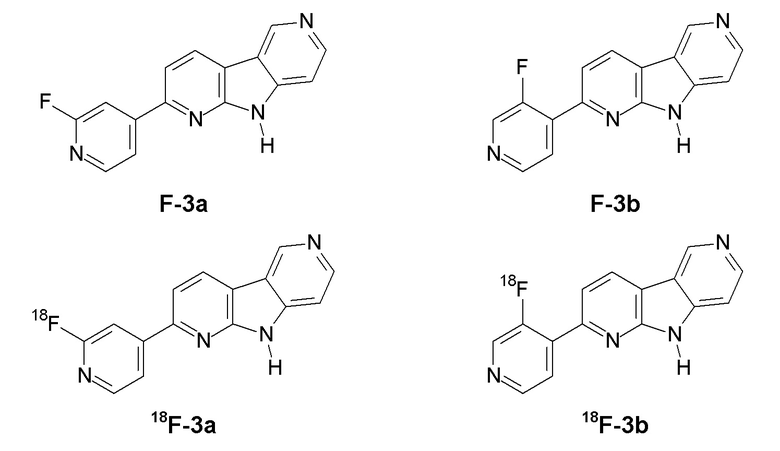
В данном документе F-3a и F-3b в совокупности будут относиться к «F-3», и 18F-3a и 18F-3b в совокупности будут относиться к «18F-3». Из них соединения F-3a и 18F-3a являются предпочтительными.
Краткое описание фигур
Фиг. 1: авторадиография срезов головного мозга субъекта с AD и HC с использованием соединения 18F-3a. В срезах головного мозга субъекта с AD было обнаружено сильное точечное окрашивание, которое можно было блокировать добавлением избытка соответствующего немеченого соединения. В срезах головного мозга здорового контрольного субъекта (HC) специфического сигнала не было видно.
Фиг. 2: кривые вымывания, показывающие клиренс активности из головного мозга у нормальных мышей для соединений 18F-1, 18F-2 и 18F-3a.
Фиг. 3: поглощение и вымывание в головном мозге 18F-3a у контрольного субъекта без деменции.
Фиг. 4: а) ПЭТ-изображение с использованием 18F-3a контрольного субъекта без деменции в аксиальной, сагиттальной и коронарной проекции, b) ПЭТ-изображение с использованием 18F-3a субъекта с AD в аксиальной, сагиттальной и корональной проекции.
Фиг. 5: ПЭТ-изображение с использованием 18F-3a субъекта с PSP в аксиальной, сагиттальной и коронарной проекции: а) на уровне черной субстанции, b) на уровне бледного шара.
Подробное описание изобретения
Настоящее изобретение относится к детектируемо меченым соединениям формулы (II):
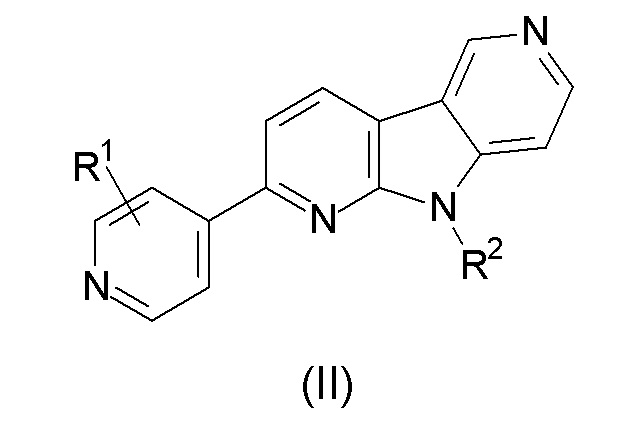
Предпочтительными соединениями по настоящему изобретению являются:
Более предпочтительными соединениями по настоящему изобретению являются:
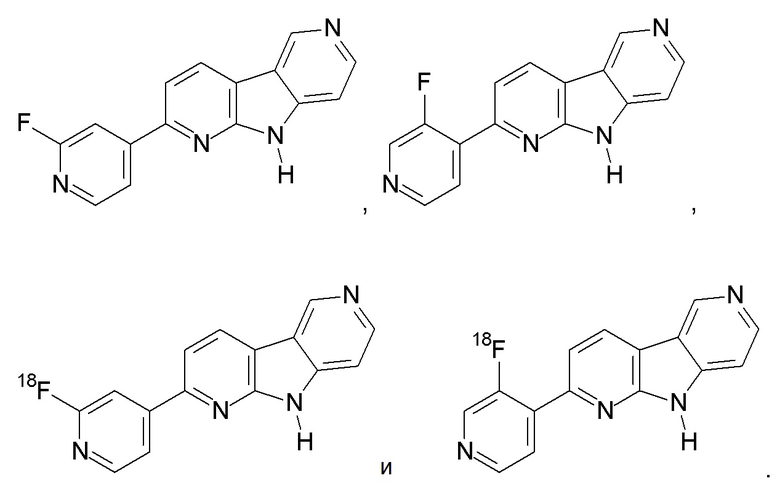
Еще более предпочтительными соединениями по настоящему изобретению являются:
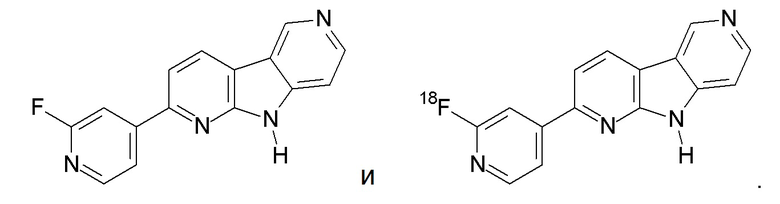
Еще более предпочтительным соединением по настоящему изобретению является:
Детектируемо меченые соединения по настоящему изобретению можно использовать для селективного выявления расстройств и аномалий, ассоциированных с агрегатами белка тау, таких как Альцгеймера и другие тауопатии, например, с использованием визуализации позитронно-эмиссионной томографией (ПЭТ). Настоящее изобретение также относится к промежуточным соединениям, которые можно использовать в получении таких визуализирующих агентов. Соединения по настоящему изобретению обладают высокой аффинностью к белку тау и связываются с изоформами белка тау, имеющими место как при болезни Альцгеймера (AD), так и при тауопатиях, отличных от AD, например, таких как прогрессирующий надъядерный паралич (PSP) и болезнь Пика (PiD). Поскольку они имеют низкую аффинность к бета-амилоиду и МАО А, то их можно использовать в качестве высокоселективных молекулярных зондов для связывания с патологическим белком тау и, таким образом, можно избежать выявления других патологий и постановки ошибочного диагноза.
18F-меченные соединения по настоящему изобретению также приводят к слабому сигналу в головном мозге здоровых субъектов, так что они могут уменьшить интерференцию фонового сигнала и, таким образом, обеспечить низкий предел обнаружения.
Благодаря хорошему поглощению тканью головного мозга, быстрому вымыванию из здорового мозга, низкой длительной задержке в здоровом мозге, а также отсутствию дефторирования in vivo 18F-меченные соединения по настоящему изобретению обеспечивают хорошее отношение сигнал/шум.
Кроме того, в соединения по настоящему изобретению можно легко вводить детектируемую метку, например, 18F, с высокими выходами.
Определения
Как здесь используется, термин «защитная группа» (PG) означает любую защитную группу, которая подходит для защиты аминогруппы во время предусмотренной химической реакции. Примеры подходящих защитных групп хорошо известны специалисту в данной области. Подходящие защитные группы обсуждаются, например, в руководстве Greene and Wuts, Protecting groups in Organic Synthesis, третье издание, стр. 494-653, которое включено в настоящий документ посредством ссылки. Защитные группы могут быть выбраны из карбаматов, амидов, имидов, N-алкиламинов, N-ариламинов, иминов, енаминов, боранов, N-P-защитных групп, N-сульфенила, N-сульфонила и N-силила. Конкретными предпочтительными примерами защитных групп (PG) являются карбобензилокси (Cbz), п-метоксибензилкарбонил (Moz или MeOZ), трет-бутилоксикарбонил (BOC), 9-флуоренилметилоксикарбонил (FMOC), бензил (Bn), п-метоксибензил (PMB), 3,4-диметоксибензил (DMPM), п-метоксифенил (PMP), трифенилметил (тритил), метоксифенилдифенилметил (MMT) или диметокситритил (DMT). Более предпочтительные примеры защитной группы PG включают трет-бутилоксикарбонил (BOC), диметокситритил (DMT) и трифенилметил (тритил). Еще одним предпочтительным примером защитной группы PG является трет-бутилоксикарбонил (ВОС).
Как здесь используется, термин «уходящая группа» (LG) означает любую уходящую группу и означает, что атом или группа атомов могут быть замещены другим атомом или группой атомов. Примеры приведены, например, в Synthesis (1982), p. 85-125, table 2, Carey and Sundberg, Organische Synthese, (1995), p. 279-281, table 5.8; или Netscher, Recent Res. Dev. Org. Chem., 2003, 7, 71-83, scheme 1, 2, 10 and 15 and others). (Coenen, Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions, (2006), in: Schubiger P.A., Friebe M., Lehmann L., (eds), PET-Chemistry - The Driving Force in Molecular Imaging. Springer, Berlin Heidelberg, pp.15-50, explicitly: scheme 4 pp. 25, scheme 5 pp 28, table 4 pp 30, Figure 7 pp 33). Предпочтительно «уходящая группа» (LG) представляет собой нитро, галоген или триметиламмоний. Более предпочтительно, «уходящая группа» (LG) представляет нитро или триметиламмоний. В одном предпочтительном варианте осуществления «уходящая группа» (LG) представляет нитро. В другом предпочтительном варианте осуществления «уходящая группа» (LG) представляет триметиламмоний.
Как здесь используется, термин «белок тау» относится к высокорастворимому белку, связывающему микротрубочки, в основном находящемуся в нейронах, и он включает основные 6 изоформ, расщепленные или усеченные формы и другие модифицированные формы, такие как возникающие в результате фосфорилирования, гликозилирования, гликирования, пролилизомеризации, нитрования, ацетилирования, полиаминирования, убиквитинирования, сумоилирования и окисления. Как здесь используется, патологический белок тау или агрегаты белка тау (нейрофибриллярные сплетения, NFT) относятся к нерастворимым агрегатам гиперфосфорилированного белка тау, содержащим спаренные спиральные нити и прямые нити. Их наличие является признаком AD и других заболеваний, известных как тауопатии.
Термин «полиморфы» относится к различным кристаллическим структурам соединений по настоящему изобретению. Они могут включать, не ограничивается этим, кристаллические структуры (и аморфные вещества) и все формы кристаллической решетки. Соли по настоящему изобретению могут быть кристаллическими и могут существовать в виде более чем одного полиморфа.
Сольваты, гидраты, а также безводные формы соединений по настоящему изобретению также охватываются изобретением. Растворитель, включенный в сольваты, особым образом не ограничивается и может представлять собой любой фармацевтически приемлемый растворитель. Примеры включают воду и С1-4 спирты (такие как метанол или этанол).
Как используется в дальнейшем в описании и в формуле изобретения, термин «пролекарство» означает любое ковалентно связанное соединение, которое высвобождает активный исходный фармацевтический препарат в результате биотрансформации in vivo. В монографии Goodman и Gilman («The Pharmacological Basis of Therapeutics, 8 ed, McGraw-Hill, Int. Ed. 1992, «Biotransformation of Drugs», p. 13-15), в общем описаны пролекарства, которая включена в настоящий документ посредством ссылки.
Как используется в дальнейшем в описании и в формуле изобретения, термин «фармацевтически приемлемая соль» относится к нетоксичным производным раскрытых соединений, где исходное соединение модифицируется посредством получения его солей неорганических и органических кислот. Неорганические кислоты включают, не ограничиваясь этим, кислоты, такие как карбоновая, соляная, азотная или серная кислота. Органические кислоты включают, не ограничиваясь этим, кислоты, такие как алифатические, циклоалифатические, ароматические, аралифатические, гетероциклические, карбоновые и сульфоновые кислоты. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Обычно такие соли можно получить взаимодействием форм этих соединений в виде свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в их смеси. Списки подходящих солей можно найти в руководстве Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p. 1445, раскрытие которого включено в настоящее описание посредством ссылки.
«Фармацевтически приемлемый» определяется как такие соединения, вещества, композиции и/или лекарственные формы, которые в рамках здравого медицинского заключения подходят для применения в контакте с тканями людей и животных без проявления чрезмерной токсичности, раздражения, аллергической реакции, или другой проблемы или осложнения, соразмерных с разумным соотношением польза/риск.
Пациентами или субъектами по настоящему изобретению обычно являются животные, особенно млекопитающие, более конкретно люди.
Ген тау содержит 16 экзонов, где основные изоформы белка тау кодируются 11 из них. Альтернативный сплайсинг экзона 10 генерирует изоформы белка тау с тремя (экзон 10 отсутствует) или четырьмя (экзон 10 присутствует) повторными доменами, известными как 3R- и 4R-тау соответственно (A. Andreadis et al., Biochemistry 31, (1992) 10626-10633; M. Tolnay et al., IUBMB Life, 55 (6): 299-305, 2003). При болезни Альцгеймера соотношение 3R- и 4R-изоформ сходно. В противоположность при некоторых тауопатиях преимущественно присутствует одна из двух изоформ. В настоящем документе термин «3R-тауопатия» относится к тауопатиям (таким как болезнь Пика (PiD)), при которых преимущественно присутствует 3R-изоформа. В настоящем документе термин «4R-тауопатия» относится к тауопатиям (таким как прогрессирующий надъядерный паралич (PSP) и кортикобазальная дегенерация (CBD)), при которых преимущественно присутствует 4R-изоформа.
Предпочтительные определения, приведенные в разделе «Определения», применимы ко всем вариантам осуществления, описанным здесь, если не указано иное.
Диагностические процедуры
Детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) особенно подходят для визуализации агрегатов белка тау. В отношение белка тау, то детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) способны связываться с различными типами агрегатов белка тау, такими как патологически агрегированный белок тау, гиперфосфорилированный белок тау, нейрофибриллярные сплетения, спаренные спиральные нити, прямые нити, нейротоксические растворимые олигомеры, полимеры и фибриллы.
За счет вышеуказанных характеристик связывания детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) подходят для применения в диагностике расстройств, ассоциированных с агрегатами белка тау. Детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) особенно подходят для визуализации отложений белка тау позитронно-эмиссионной томографией (ПЭТ). Обычно 18F-меченные соединения формулы (II) используют в виде детектируемо меченых соединений, если соединения предназначены для введения пациенту.
При визуализации агрегатов белка тау детектируемо меченое соединение формулы (II) (предпочтительно 18F-3, более конкретно 18F-3a) вводят и детектируют сигнал, исходящий от соединения, которое специфически связано с агрегатами белка тау. Специфическое связывание является результатом высокой аффинности связывания соединений формулы (II) с агрегатами белка тау.
В предпочтительном варианте осуществления детектируемо меченое соединение формулы (II) (предпочтительно 18F-3, более конкретно 18F-3a), используется для диагностики наличия тауопатии (предпочтительно болезни Альцгеймера). В данном способе детектируемо меченое соединение формулы (II) (предпочтительно 18F-3, более конкретно 18F-3a) вводят пациенту, для которого имеется подозрение на то, что он страдает тауопатией (предпочтительно болезнью Альцгеймера), или получают образец от такого пациента, и сигнал, исходящий от детектируемой метки, детектируется, предпочтительно, позитронно-эмиссионной томографией (PET).
Если какой-либо сигнал, исходящий от детектируемой метки, не детектируется, то способ по настоящему изобретению можно использовать для исключения тауопатии, что указывает на наличие неврологического расстройства, отличного от тауопатии.
В способах диагностики расстройства, ассоциированного с агрегатами белка тау, такого как болезнь Альцгеймера, или предрасположенности к нему у субъекта, способ включает:
а) введение млекопитающему диагностически эффективного количества детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a);
b) обеспечение возможности детектируемо меченому соединению по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) распространяться в интересующей ткани (такой как ткань головного мозга, глаза или жидкости организма, такие как спинномозговая жидкость (CSF)); и
c) визуализацию представляющей интерес ткани, где увеличение связывания детектируемо меченого соединения по настоящему изобретению (в частности 18F-3, более конкретно 18F-3a) с интересующей тканью по сравнению с нормальным контрольным уровнем связывания указывает на то, что субъект страдает или подвержен риску развития расстройства, ассоциированного с агрегатами белка тау.
Детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) можно использовать для визуализации агрегатов белка тау в любом образце или определенной части тела или области тела пациента, которые предположительно содержат агрегат белка тау. Детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно, 18F-3a) способны проходить через гематоэнцефалический барьер и проникать в глаз. Следовательно, они особенно подходят для визуализации агрегатов белка тау в головном мозге, в глазу (офтальмологическая и/или ретинальная визуализация), а также в жидкостях организма, таких как спинномозговая жидкость (CSF).
В диагностических применениях детектируемо меченые соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) предпочтительно вводят в диагностической композиции.
Диагностика расстройства, ассоциированного с белком тау, или предрасположенности к развитию расстройства, ассоциированного с белком тау, у пациента может быть достигнута детектированием специфического связывания детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) с агрегатами белка тау в образце или in situ, что включает:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с детектируемо меченым соединением по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a), которое связывается с агрегатом белка тау;
(b) обеспечение возможности детектируемо меченому соединению по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) связываться с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау (в дальнейшем сокращенно «комплекс соединение/агрегат белка»);
(c) детектирование образования комплекса соединение/белок;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/белок с наличием или отсутствием агрегатов белка тау в образце или определенной части тела или области; и
(e) необязательно сравнение количества соединения/белка с нормальным контрольным значением, где увеличение количества соединения/белка по сравнению с нормальным контрольным значением может указывать на то, что пациент страдает или имеет риск развития расстройства, ассоциированного с белком тау.
После того, как образец или определенная часть тела или область тела были приведены в контакт с детектируемо меченым соединением по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a), соединению обеспечивается возможность связываться с агрегатом белка тау. Количество времени, необходимое для связывания, будет зависеть от типа теста (например, in vitro или in vivo) и может быть определено специалистом в данной области путем рутинных экспериментов.
Затем соединение, связанное с агрегатом белка тау, может быть детектировано любым подходящим способом. Предпочтительным методом является позитронно-эмиссионная томография (ПЭТ).
Затем наличие или отсутствие соединения/белка необязательно сопоставляют с наличием или отсутствием агрегатов белка тау в образце или определенной части тела или области. Наконец, количество соединения/белка можно сравнить с нормальным контрольным значением, которое было определено в образце или определенной части тела или области тела здорового субъекта, где увеличение количества соединения/белка по сравнению с нормальным контрольным значением может указывать на то, что пациент страдает или подвержен риску развития расстройства, ассоциированного с белком тау.
Настоящее изобретение также относится к способу определения количества агрегата белка тау в ткани и/или жидкости организма. Данный способ включает стадии:
(a) обеспечение образца, репрезентативного для исследуемой ткани и/или биологической жидкости;
(b) тестирование образца на наличие агрегата белка тау с использованием детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно, 18F-3a);
(c) определение количества детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a), связанного с агрегатом белка тау; и
(d) расчет количества агрегата белка тау в ткани и/или жидкости организма.
Образец можно тестировать на наличие агрегата белка тау с использованием детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) приведением образца в контакт с детектируемо меченым соединением по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3а), обеспечением возможности детектируемо меченому соединению по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3а) связываться с агрегатом белка тау с образованием комплекса соединение/агрегат белка и детектированием образования комплекса соединение/белок, как пояснено выше.
Мониторинг минимального остаточного проявления расстройства у пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, который лечился лекарственным средством, с использованием детектируемо меченого соединения по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a), может быть достигнут посредством:
(а) приведения образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с детектируемо меченым соединением по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a);
(b) обеспечения возможности детектируемо меченому соединению по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) связываться с агрегатом белка тау с образованием комплекса соединение/агрегат белка;
(c) детектирования образования комплекса соединение/ агрегат белка;
(d) необязательно сопоставления наличия или отсутствия комплекса соединение/агрегат белка с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(e) необязательно сравнения количества соединения/агрегата белка с нормальным контрольным значением, где увеличение количества агрегата по сравнению с нормальным контрольным значением может указывать на то, что пациент все еще может страдать минимальным остаточным проявлением заболевания.
Каким образом проводить стадии (a)-(e) было уже пояснено выше.
Прогнозирование реактивности пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, и получающего лечение лекарственным средством, может быть достигнуто посредством:
(а) приведения образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с детектируемо меченым соединением по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a);
(b) обеспечения возможности детектируемо меченому соединению по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a) связываться с агрегатом белка тау с образованием комплекса соединение/агрегат белка;
(c) детектирования образования комплекса соединение/ агрегат белка;
(d) необязательно сопоставления наличия или отсутствия комплекса соединение/агрегат белка с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(e) необязательно сравнения количества соединения/агрегата белка с нормальным контрольным значением.
Каким образом проводить стадии (a)-(e) было уже пояснено выше.
В способе прогнозирования реактивности количество соединения/комплекса белка может быть необязательно сопоставлено на различные временные точки во время лечения, например, до и после начала лечения или на различные временные точки после начала лечения. Изменение, в частности, уменьшение количества соединения/комплекса белка, может указывать на то, что пациент обладает высокой способностью реагировать на соответствующее лечение.
Соединение по настоящему изобретению также может быть включено в тестовый набор в целях определения агрегата белка тау. Тестовый набор обычно включает контейнер, содержащий одно или более соединений по настоящему изобретению, и инструкции по применению соединения с целью связывания с агрегатом белка тау с образованием комплекса соединение/белок и детектирования образования комплекса соединение/белок так, что наличие или отсутствие комплекса соединение/белок коррелирует с наличием или отсутствием агрегатов белка тау.
Термин «тестовый набор» в общем относится к любому диагностическому набору, известному в данной области. Более конкретно, последний термин относится к диагностическому набору, как описано в публикации Zrein et al., Clin. Diagn. Lab. Immunol., 1998, 5, 45-49.
Диагностические композиции
«Диагностическая композиция» определяется в настоящем изобретении как композиция, содержащая детектируемо меченое соединение по настоящему изобретению (предпочтительно, 18F-меченное; в частности, 18F-3, более конкретно 18F-3a). Для применений in vivo диагностическая композиция должна быть в форме, подходящей для введения млекопитающим, таким как человек. Предпочтительно диагностическая композиция дополнительно содержит физиологически приемлемый носитель, разбавитель, адъювант или эксципиент. Введение пациенту предпочтительно осуществляется инъекцией композиции в виде водного раствора. Такая композиция может необязательно содержать дополнительные ингредиенты, такие как растворители, буферы; фармацевтически приемлемые солюбилизаторы; и фармацевтически приемлемые стабилизаторы или антиоксиданты.
Фармацевтически приемлемые эксципиенты хорошо известны в области фармации и описаны, например, в руководстве Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1975). Фармацевтический эксципиент может быть выбран с учетом предполагаемого пути введения и обычной фармацевтической практики. Эксципиент должен быть приемлемым в том смысле, что он не является токсичным для реципиента.
Фармацевтически пригодные эксципиенты, которые можно использовать в составе диагностической композиции по настоящему изобретению, могут включать, например, носители, наполнители, разбавители, растворители и пищевые масла, сложные эфиры масляных кислот, связующие вещества, адъюванты, солюбилизаторы, загустители, стабилизаторы, дезинтегранты, скользящие агенты, смазывающие агенты, буферные агенты, эмульгаторы, смачивающие агенты, суспендирующие агенты, подсластители, красители, ароматизаторы, покровные агенты, консерванты, антиоксиданты, технологические агенты, модификаторы и усилители доставки лекарственных препаратов.
Если детектируемо меченые соединения по настоящему изобретению (предпочтительно 18F-меченные, в частности, 18F-3, более конкретно 18F-3a) вводят парентерально, то примеры такого введения включают одно или более из: внутривенного, внутриартериального, внутрибрюшинного, интратекального, интравентрикулярного, интрауретрального, интрастернального, интракраниального, внутримышечного или подкожного введения соединений; и/или с использованием методов инфузии. Для парентерального введения соединения лучше всего использовать в форме стерильного водного раствора, который может содержать другие эксципиенты. Водные растворы должны быть подходящим образом забуферены (предпочтительно до рН от 3 до 9), если необходимо. Приготовление подходящих парентеральных составов в стерильных условиях легко осуществляется стандартными фармацевтическими методами, хорошо известными специалистам в данной области.
Доза детектируемо меченых соединений по настоящему изобретению (предпочтительно 18F-меченных, в частности, 18F-3, более конкретно 18F-3a) будет варьироваться в зависимости от конкретного соединения, которое следует вводить, массы тела пациента, размера и типа образца, и других переменных, которые будут очевидны для специалистов в данной области. Как правило, доза может предпочтительно находиться в диапазоне от 0,001 мкг/кг до 10 мкг/кг, предпочтительно от 0,01 мкг/кг до 1,0 мкг/кг. Радиоактивная доза может составлять, например, от 100 до 600 МБк, более предпочтительно от 150 до 450 МБк.
Диагностические композиции по настоящему изобретению могут быть получены известным специалистом способом, который описан, например, в руководстве Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1975).
Например, соединения по настоящему изобретению можно применять в липосомальной композиции, как описано в WO2016057812A1, которая содержит соединение формулы (II) в качестве лиганда для применения в селективном детектировании расстройств и аномалий, ассоциированных с агрегатами белка тау, посредством нерадиоактивной магнитно-резонансной визуализации (MRI).
В частности, в одном варианте осуществления заболевания или расстройства, которые могут быть выявлены и контролироваться детектируемо мечеными соединениями по настоящему изобретению (в частности, 18F-3, более конкретно 18F-3a), представляют заболевания или патологические состояния, ассоциированные с агрегатами белка тау.
Заболевания или патологические состояния, которые могут быть выявлены и контролироваться детектируемо мечеными соединениями по настоящему изобретению (в частности, 18F-3, более конкретно, 18F-3a), включают нейродегенеративные расстройства, такие как тауопатии. Примеры заболеваний и состояний, которые можно выявить и контролировать, вызваны или связаны с образованием нейрофибриллярных поражений. Это преобладающая патология головного мозга при тауопатии. Заболевания и патологические состояния включают гетерогенную группу нейродегенеративных заболеваний или патологических состояний, включая заболевания или патологические состояния, которые демонстрируют совместное наличие таупатологии и амилоидных патологий. Примеры заболеваний, включающих агрегаты белка тау, обычно входят в перечень тауопатий, и они включают, не ограничиваясь этим, болезнь Альцгеймера (AD), болезнь Крейцфельда-Якоба, деменцию боксера, синдром Дауна, болезнь Герстмана-Штраусслера-Шейнкера, миозит с тельцами включения, церебральную амилоидную ангиопатию, связанную с прионными белками, черепно-мозговую травму (TBI), боковой амиотрофической склероз, комплекс паркинсонизма-деменции Гуама, болезнь двигательного нейрона с нейрофибриллярными сплетениями, отличную от болезни Гуама, болезнь аргирофильных зерен, кортикобазальную дегенерацию (CBD), диффузные нейрофибриллярные сплетения с кальцификацией, фронтотемпоральную деменцию с паркинсонизмом, связанную с хромосомой 17, болезнь Галлервордена-Шпатца, множественную системную атрофию, болезнь Нимана-Пика типа C, паллидо-понто-нигральную дегенерацию, болезнь Пика (PiD), прогрессирующий субкортикальный глиоз, прогрессирующий надъядерный паралич (PSP), подострый склерозирующий панэнцефалит, деменцию, связанную только со сплетениями, постэнцефалитический паркинсонизм, миотоническую дистрофию, тау-панэнцефалопатию, AD-подобные заболевания с поражением астроцитов, некоторые прионные болезни (GSS с тау), мутации в LRRK2, хроническую травматическую энцефалопатию, семейную британскую деменцию, семейную датскую деменцию, фронтотемпоральную лобарную дегенерацию, гваделупский паркинсонизм, нейродегенерацию с накоплением железа в мозге, SLC9A6-ассоциированную задержку умственного развития, таутопатию белого вещества с глобулярными глиальными включениями, синдром травматического стресса, эпилепсию, деменцию с тельцами Леви (LBD), наследственное церебральное кровоизлияние с амилоидозом (голландский тип), легкое когнитивное расстройство (MCI), рассеянный склероз, болезнь Паркинсона, деменцию, связанную с ВИЧ, диабет с началом во взрослом возрасте, старческий сердечный амилоидоз, эндокринные опухоли, глаукому, амилоидоз глаза, первичную дегенерацию сетчатки, макулярную дегенерацию (например, возрастную макулярную дегенерацию (AMD)), друз диска зрительного нерва, нейропатию зрительного нерва, неврит зрительного нерва и решетчатую дистрофию. Предпочтительно заболевания и состояния, которые можно выявить и контролировать, включают болезнь Альцгеймера (AD), семейную форму AD, болезнь Крейцфельда-Якоба, деменцию боксера, синдром Дауна, болезнь Герстмана-Штраусслера-Шейнкера, миозит с тельцами включения, церебральную амилоидную ангиопатию, связанную с прионными белками, черепно-мозговую травму (TBI), боковой амиотрофический склероз, комплекс паркинсонизма-деменции Гуама, болезнь двигательного нейрона с нейрофибриллярными сплетениями, отличную от болезни Гуама, болезнь аргирофильных зерен, кортикобазальную дегенерацию (CBD), диффузные нейрофибриллярные сплетения с кальцификацией, фронтотемпоральную деменцию с паркинсонизмом, связанную с хромосомой 17, болезнь Галлервордена-Шпатца, множественную системную атрофию, болезнь Нимана-Пика типа C, паллидо-понто-нигральную дегенерацию, болезнь Пика (PiD), прогрессирующий субкортикальный глиоз, прогрессирующий надъядерный паралич (PSP), подострый склерозирующий панэнцефалит, деменцию, связанную только со сплетениями, постэнцефалитический паркинсонизм, миотоническую дистрофию, тау-панэнцефалопатию, AD-подобные заболевания с поражением астроцитов, некоторые прионные болезни (GSS с тау), мутации в LRRK2, хроническую травматическую энцефалопатию, семейную британскую деменцию, семейную датскую деменцию, фронтотемпоральную лобарную дегенерацию, гваделупский паркинсонизм, нейродегенерацию с накоплением железа в мозге, SLC9A6-ассоциированную задержку умственного развития и таутопатию белого вещества с глобулярными глиальными включениями, более предпочтительно болезнь Альцгеймера (AD), болезнь Крейцфельда-Якоба, деменцию боксера, боковой амиотрофический склероз, болезнь аргирофильных зерен, кортикобазальную дегенерацию (CBD), фронтотемпоральную деменцию с паркинсонизмом, связанную с хромосомой 17, болезнь Пика (PiD), прогрессирующий надъядерный паралич (PSP), деменцию, связанную только со сплетениями, комплекс паркинсонизма-деменции Гуама, болезнь Галлервордена-Шпатца и фронтотемпоральную лобарную дегенерацию. Предпочтительно заболевание или патологическое состояние представляет болезнь Альцгеймера.
Общий синтез 18F-меченных соединений по настоящему изобретению
Соединения, имеющие формулу (II), которые содержат метку 18F, можно получить взаимодействием соединения формулы (II), где R1 представляет LG, и R2 представляет H или PG, с 18F-фторирующим агентом, так что уходящая группа LG замещается на 18F. Получение включает отщепление защитной группы PG, если она присутствует.
Можно использовать любой подходящий 18F-фторирующий агент. Типичные примеры включают H18F, 18F-фториды щелочных или щелочноземельных металлов (например, K18F, Rb18F, Cs18F и Na18F). Необязательно, 18F-фторирующий агент можно использовать в сочетании с хелатирующим агентом, таким как криптанд (например: 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]гексакозан - Kryptofix®) или краун-эфир (например, 18-краун-6). Альтернативно, 18F-фторирующий агент может представлять тетраалкиламмониевую соль 18F или тетраалкилфосфониевую соль 18F; например, тетра(C1-6 алкил)аммониевую соль 18F или тетра(C1-6 алкил)фосфониевую соль 18F. Ее примеры включают [18F]фторид тетрабутиламмония и [18F]фторид тетрабутилфосфония. Предпочтительно 18F-фторирующий агент представляет K18F, H18F, Cs18F, Na18F или [18F]фторид тетрабутиламмония.
Реагенты, растворители и условия, которые можно использовать для 18F-фторирования, хорошо известны специалистам в данной области (L. Cai, S. Lu, V. Pike, Eur. J. Org. Chem., 2008, 2853-2873; J. Fluorine Chem., 27 (1985):177-191; Coenen, Fluorine-18 Labeling Methods: Features and Possibilities of Basic Reactions, (2006), in: Schubiger P.A., Friebe M., Lehmann L., (eds), PET-Chemistry - The Driving Force in Molecular Imaging. Springer, Berlin Heidelberg, pp.15-50). Предпочтительно растворители, используемые в 18F-фторировании, представляют ДМФА, ДМСО, ацетонитрил, DMA или их смеси, предпочтительно растворитель представляет ацетонитрил или ДМСО.
Если желательно, то соединение, имеющее формулу (II), может иметь R1, представляющий LG, и R2, представляющий PG, где защитная группа PG защищает амин во время реакции 18F-фторирования. Эта защитная группа для аминогруппы может быть впоследствии удалена. Способы удаления защитной группы для аминогруппы известны в данной области и включают, не ограничиваясь этим, кислотное расщепление.
Если желательно, то соединение формулы (II) может быть выделено и/или очищено дополнительно до применения. Соответствующие процедуры хорошо известны в данной области.
Предшественники соединений, имеющих формулу (II), в которой R1 представляет LG, и R2 представляет H или PG, могут быть предоставлены в наборе, который подходит для получения соединений формулы (II) взаимодействием с 18F-фторирующим агентом. В одном варианте осуществления набор включает герметичный флакон, содержащий заранее определенное количество предшественника соединения по настоящему изобретению. Например, набор может содержать от 1,5 до 75 мкмоль, предпочтительно от 7,5 до 50 мкмоль, более предпочтительно от 10 до 30 мкмоль предшественника соединения формулы (II) по настоящему изобретению. Необязательно, набор может содержать дополнительные компоненты, такие как реакционный растворитель, картридж для твердофазной экстракции, реагент для получения 18F-фторирующего агента, реагент для отщепления защитной группы, растворитель для очистки, растворитель для формуляции и фармацевтически приемлемый носитель, разбавитель, адъювант или эксципиент для формуляции.
Соединения по настоящему изобретению, в которых R1 представляет F, и R2 представляет H, можно использовать в качестве аналитического стандартного соединения или инструмента для скрининга in vitro.
Соединения по настоящему изобретению, в которых R1 представляет F, и R2 представляет H, можно использовать в качестве аналитического стандартного соединения для контроля качества и высвобождения соединения по настоящему изобретению, в котором R1 представляет 18F, и R2 представляет H.
Соединения по настоящему изобретению, в которых R1 представляет F, и R2 представляет H, можно использовать в качестве инструмента для скрининга in vitro для характеристики ткани с таупатологией и для тестирования соединений, нацеленных на патологию тау в такой ткани.
Настоящее изобретение иллюстрируется следующими примерами, которые не следует рассматривать в качестве ограничивающих изобретение.
Примеры
Все реагенты и растворители были получены из коммерческих источников и использовались без дополнительной очистки. Протонные (1Н) спектры снимали на ЯМР спектрометре Bruker DRX-400 МГц или на ЯМР спектрометре Bruker AV-400 МГц в дейтерированных растворителях. Масс-спектры (МС) снимали на масс-спектрометре Advion CMS. Хроматографию проводили с использованием силикагеля (Fluka: силикагель 60, 0,063-0,2 мм) и подходящих растворителей, указанных в конкретных примерах. Флэш-очистку проводили с помощью системы флэш-очистки Biotage Isolera One с использованием колонок HP-Sil (Biotage) или puriFlash (Interchim) и градиента растворителей, указанных в конкретных примерах. Тонкослойную хроматографию (ТСХ) проводили на пластинках с силикагелем с УФ-детектированием.
Несмотря на то, что некоторые из настоящих примеров не указывают на то, что соответствующие соединения были детектируемо мечеными, понятно, что соответствующие детектируемо меченые соединения могут быть легко получены, например, с использованием детектируемо меченых исходных веществ, таких как исходные вещества, содержащие атомы 3H.
Сокращенные обозначения
Препаративный пример А
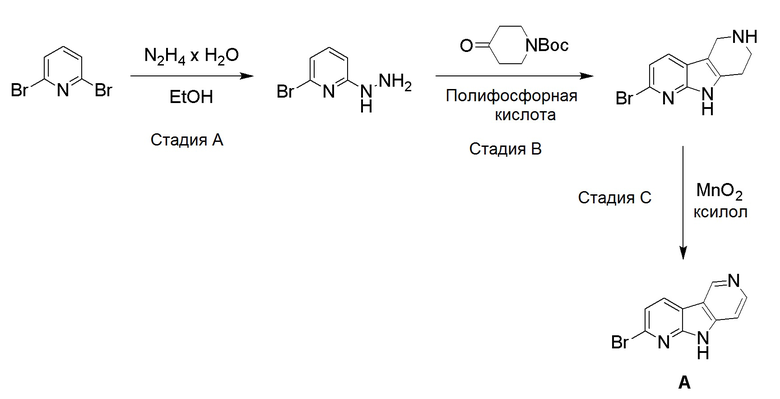
Стадия А
Коммерчески доступный 2,6-дибромпиридин (4,12 г, 16,6 ммоль) суспендировали в этаноле (40 мл) и добавляли гидразин гидрат (10 мл, 97,6 ммоль) в воде (~50-60%). Смесь нагревали на песочной бане при ~115°С в течение 18 ч. Растворитель удаляли и остаток очищали хроматографией на силикагеле, используя смесь этилацетат/н-гептан (60/40), с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (3,05 г, 93%).
1H-ЯМР (400 МГц, CDCl3): δ=7,33 (т, 1H), 6,83 (д, 1H), 6,67 (д, 1H), 6,00 (шир. с, 1H), 3,33-3,00 (шир. с, 2H).
Стадия В
Указанное в заголовке соединение со стадии A выше (10 г, 53,2 ммоль) и коммерчески доступный 1-Boc-4-пиперидон (10,6 г, 53,2 ммоль) добавляли в колбу емкостью 500 мл и перемешивали до получения гомогенной смеси. Затем добавляли полифосфорную кислоту (80 г, 115% на основе H3PO4) и смесь нагревали при ~160°C на песочной бане. При ~120°С защитная Вос-группа отщепляется, что приводит к вспениванию реакционной смеси. После полного отщепления Boc-группы пена разрушалась, и темную реакционную смесь перемешивали при ~160°C в течение 20 ч. Реакционной смеси давали охладиться до комнатной температуры и добавляли воду (400 мл). Реакционную смесь перемешивали/обрабатывали ультразвуком до тех пор, пока липкое вещество не растворялось. Затем реакционную смесь помещали на ледяную баню и рН раствора доводили до рН ~12 добавлением гранул твердого гидроксида натрия (экзотермическая реакция). Осадок собирали фильтрованием и промывали водой (400 мл) для удаления солей. Осадок растворяли в смеси дихлорметан/ метанол (9/1; 1500 мл) при обработке ультразвуком и промывали водой (2 × 400 мл) для удаления оставшихся солей и нерастворимого вещества. Органическую фазу высушили над Na2SO4, фильтровали и растворители удаляли при пониженном давлении. Темный остаток обрабатывали дихлорметаном (100 мл), обрабатывали ультразвуком в течение 5 мин и осадок собирали фильтрованием. Осадок промывали дихлорметаном (40 мл) и высушивали на воздухе с получением указанного в заголовке соединения в виде бежевого твердого вещества (3,5 г, 26%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=11,5 (шир. с, 1H), 7,72 (д, 1H), 7,15 (д, 1H), 3,86-3,82 (м, 2H), 3,06-3,00 (м, 2H), 2,71-2,65 (м, 2H).
Стадия С
Указанное в заголовке соединение со стадии B выше (1,75 г, 6,94 ммоль) суспендировали в ксилоле (380 мл) и добавляли оксид марганца (IV) (6,62 г, 76,9 ммоль). Затем реакционную смесь нагревали при ~160°С на песочной бане в течение 36 ч. Охлажденную реакционную смесь упаривали при пониженном давлении, остаток суспендировали в смеси дихлорметан/метанол (1/1; 400 мл) и перемешивали при комнатной температуре в течение 30 мин. Затем реакционную смесь фильтровали через бумажный фильтр для удаления оксида марганца (IV) и фильтр промывали метанолом (50 мл). Объединенные фильтраты упаривали при пониженном давлении и темный остаток очищали хроматографией на силикагеле (картридж HP-SIL 50 г) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/гептан (5/95-100/0) для удаления неполярных примесей, и затем смесь дихлорметан/метанол (9/1 → 4/1) с получением указанного в заголовке соединения в виде темно-желтого твердого вещества. Общий выход с 2 стадий составил 1,77 г (51%).
1H-ЯМР (400 МГц, ДМСО-d6): δ=12,52 (шир. с, 1H), 9,42 (с, 1H), 8,61 (д, 1H), 8,53 (д, 1H), 7,56-7,52 (м, 2H).
Препаративный пример В
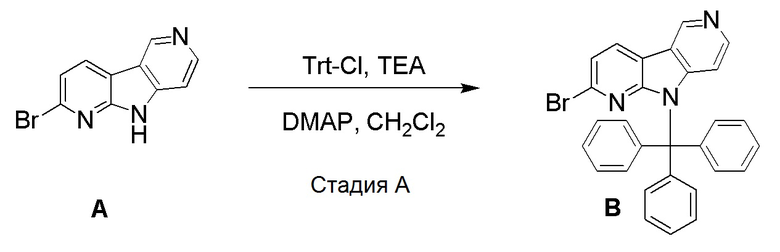
Стадия А
К суспензии указанного в заголовке соединения из препаративного примера А (0,776 г, 3,13 ммоль) в дихлорметане (65 мл) добавляли триэтиламин (1,86 мл, 13 ммоль) и тритилхлорид (2,63 г, 9,39 ммоль). После добавления 4-(диметиламино)пиридина (0,074 г, 0,608 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли дихлорметаном (150 мл) и водой (50 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на колонках HP-Sil SNAP (50 г) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения B в виде бледно-желтого твердого вещества (0,831 г, 54%). Непрореагировавшее исходное вещество выделяли промыванием колонки смесью этилацетат/метанол (90/10) с получением исходного вещества в виде не совсем белого твердого вещества (0,195 г, 25%).
1H-ЯМР (400 МГц, CDCl3) δ=9,22 (с, 1H), 8,23 (д, 1H), 8,13 (д, 1H), 7,48-7,42 (м, 7H), 7,33-7,22 (м, 12H), 6,41 (д, 1Н).
МС (ESI); m/z=490,03/491,96 [М+Н]+.
Препаративный пример С
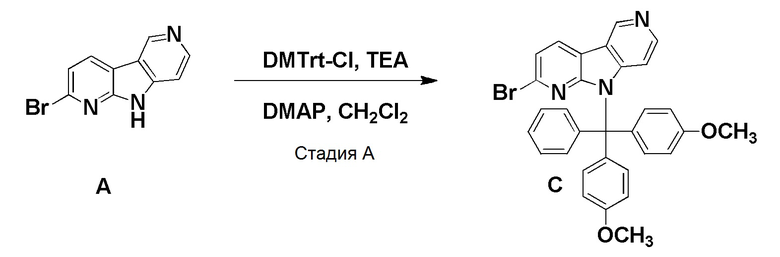
Стадия А
К суспензии указанного в заголовке соединения из препаративного примера A (0,482 г, 1,94 ммоль) в дихлорметане (40 мл) добавляли триэтиламин (1,15 мл, 8 ммоль) и 4,4'-(хлор (фенил)метилен)бис(метоксибензол); DMTrt-Cl) (1,963 г, 5,8 ммоль). После добавления 4-(диметиламино)пиридина (0,046 г, 0,377 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Реакционную смесь разбавляли дихлорметаном (100 мл) и водой (40 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на колонках HP-Sil SNAP (50 г) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения С в виде бледно-желтого твердого вещества (0,825 г, 72%).
1H-ЯМР (400 МГц, CDCl3) δ=9,23 (с, 1H), 8,23 (д, 1H), 8,13 (д, 1H), 7,39-7,31 (м, 6H), 7,29-7,25 (4H), 6,80 (д, 4Н), 6,41 (дд, 1Н), 3,81 (с, 6Н).
Пример 1 (ACI-2620)
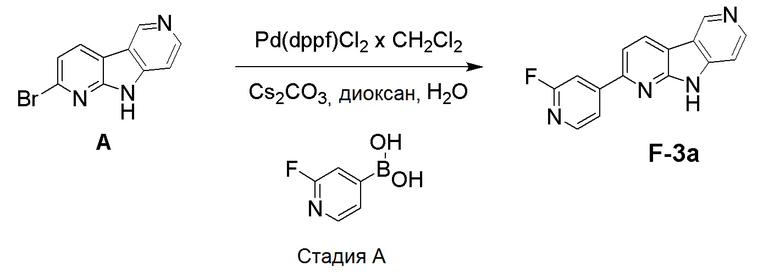
Стадия А
К смеси дегазированного 1,4-диоксана (4,3 мл) и воды (1 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0084 г, 0,01 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера А (0,05 г, 0,2 ммоль), (2-фторпиридин-4-ил)бороновой кислоты (0,035 г, 0,245 ммоль) и карбоната цезия (0,133 г, 0,41 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (60 мл) и водой (20 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (HP-SIL 25 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20), с получением указанного в заголовке соединения F-3a в виде не совсем белого твердого вещества (0,033 г, 63%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,50 (шир. с, 1H), 9,45 (с, 1H), 8,83 (д, 1H), 8,56-8,52 (м, 1H), 8,43-8,39 (м, 1H), 8,19-8,14 (м, 2H), 7,92 (с, 1H), 7,54-7,50 (м, 1H).
МС (ESI): m/z=265,04 [М+Н]+.
Пример 2 (ACI-2698)
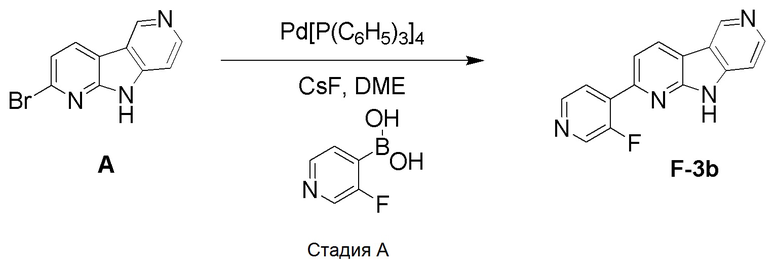
Стадия А
В пробирку для микроволнового реактора емкостью 5 мл добавляли указанное в заголовке соединение из препаративного примера A (0,05 г, 0,202 ммоль) и (3-фторпиридин-4-ил)бороновую кислоту (0,0398 г, 0,282 ммоль) в диметоксиэтане (соотношение: 2, объем: 1,334 мл) и метаноле (соотношение: 1, объем: 0,672 мл). Добавляли фторид цезия (0,0306 г, 0,202 ммоль) и полученную суспензию дегазировали в течение 5 мин аргоном. Затем добавляли тетракис(трифенилфосфин)палладий (0) (0,0419 г, 0,036 ммоль), пробирку герметично закрывали и реакционную смесь нагревали при 150°C в микроволновом реакторе Biotage Initiator в течение 30 мин (р=12 бар). Реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным раствором соли. Органический слой высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле (HP-SIL 10 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 80/20), с получением указанного в заголовке соединения F-3b в виде светло-коричневого твердого вещества (0,013 г, 24%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,46 (с, 1H), 8,82 (д, 1H), 8,77 (д, 1H), 8,63 (д, 1H), 8,56 (д, 1H), 8,09 (дд, 1Н), 7,91 (дд, 1Н), 7,54 (д, 1Н).
МС (ESI); m/z=265,16 [М+Н]+.
Пример 3 (ACI-2690)
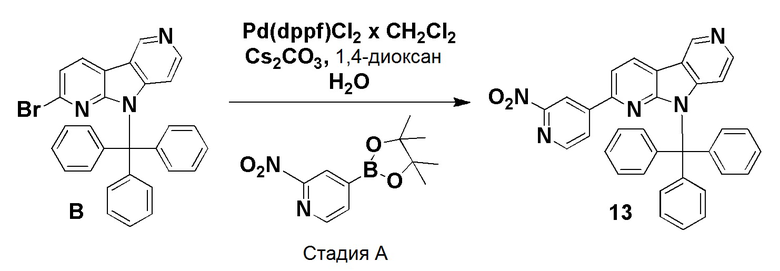
Стадия А
К смеси дегазированного 1,4-диоксана (4,3 мл) и воды (1 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис (дифенилфосфино)ферроцен]дихлорпалладий(II), комплекс с дихлорметаном (0,0084 г, 0,01 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,1 г, 0,2 ммоль), 2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,061 г, 0,245 ммоль) и карбоната цезия (0,133 г, 0,41 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (60 мл) и водой (20 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (колонка pufiFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения 13 в виде бледно-желтого твердого вещества (0,082 г, 75%).
1H-ЯМР (400 МГц, CDCl3) δ=9,32 (с, 1H); 8,56 (д, 1H), 8,48 (д, 1H), 8,33 (с, 1H); 8,30 (д, 1H), 7,85 (д, 1H), 7,69 (д, 1H), 7,58-7,54 (м, 5H), 7,32-7,25 (м, 10H), 6,48 (д, 1H).
МС (ESI): m/z=534,28 [М+Н]+.
Пример 4 (ACI-2756)
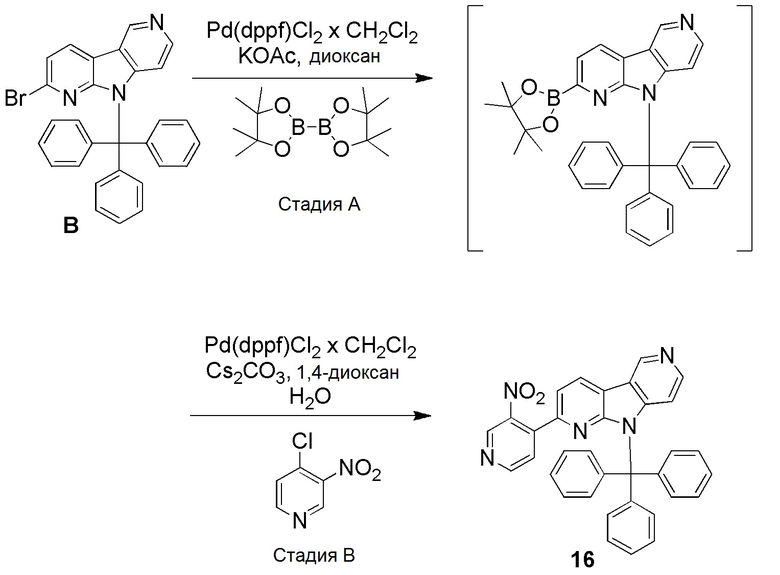
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A, описанное выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 4-хлор-3-нитропиридин (0,078 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (80 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения 16 в виде бледно-желтого твердого вещества (0,033 г, 15%).
1H-ЯМР (400 МГц, CDCl3) δ=9,30 (с, 1H), 9,02 (с, 1H), 8,68 (д, 1H), 8,42 (д, 1H), 8,26 (д, 1H), 7,49-7,45 (м, 5Н), 7,31 (д, 1Н), 7,27-7,22 (м, 10Н); 7,08 (д, 1Н), 6,44 (8Н, 1Н).
МС (ESI): m/z=533,59 [М+Н]+.
Пример 5 (нитро/Boc-предшественник) (ACI-2799)
Способ a: (ACI-2799-1)
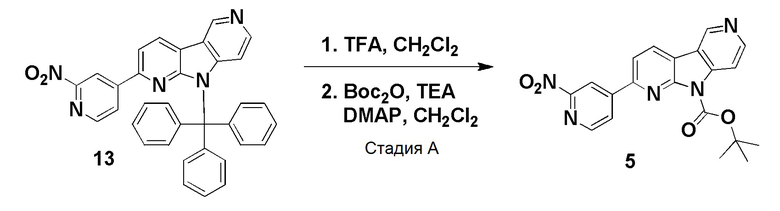
Стадия А
К раствору указанного в заголовке соединения из примера 3 (0,0396 г, 0,074 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч и добавляли метанол (2 мл). Растворители выпаривали в вакууме и остаток растворяли/суспендировали в метаноле (5 мл). Растворители выпаривали в вакууме и остаток снова растворяли/суспендировали в метаноле (5 мл). Растворители выпаривали в вакууме и остаток суспендировали в дихлорметане (2 мл). После добавления триэтиламина (1 мл, 7,2 ммоль), ди-трет-бутилдикарбоната (0,098 г, 0,43 ммоль) и 4-(диметиламино)пиридина (0,0018 г, 0,014 ммоль), реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли этилацетатом (50 мл) и водой (20 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), для элюирования неполярных побочных продуктов, и затем смесь этилацетат/метанол (95/5) с получением указанного в заголовке соединения 5 бледно-желтого цвета (0,0184 г, 63%).
1H-ЯМР (400 МГц, CDCl3) δ=9,36 (с, 1H), 9,15 (с, 1H), 8,82-8,76 (м, 2H), 8,57 (д, 1H), 8,45 (д, 1H), 8,36 (д, 1Н), 8,07 (д, 1Н), 1,87 (с, 9Н).
МС (ESI); m/z=391,82 [М+Н]+.
Способ b: (ACI-2799-2)
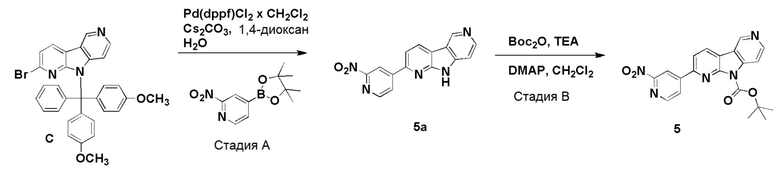
Стадия А
К смеси дегазированного 1,4-диоксана (2,2 мл) и воды (0,5 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0042 г, 0,005 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера С (0,055 г, 0,1 ммоль), 2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,0305 г, 0,12255 ммоль) и карбоната цезия (0,067 г, 0,205 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (20 мл), осадок собирали фильтрованием, промывали водой (10 мл) и метанолом (5 мл) и высушивали на воздухе с получением неочищенных указанных в заголовке соединений в виде серого твердого вещества (0,0277 г, 95%).
Стадия В
К суспензии неочищенного указанного в заголовке соединения со стадии A выше (0,0277 г, 0,095 ммоль) в дихлорметане (4 мл) добавляли триэтиламин (1 мл, 7,2 ммоль), ди-трет-бутилдикарбонат (0,2 г, 0,86 ммоль) и 4-(диметиламино)пиридин (0,0036 г, 0,028 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли этилацетатом (50 мл) и водой (20 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), для элюирования неполярных побочных продуктов, и затем смесь этилацетат/метанол (95/5) с получением указанного в заголовке соединения 5 в виде бледно-желтого твердого вещества (0,0261 г, 70%).
1H-ЯМР (400 МГц, CDCl3) δ=9,38 (с, 1H), 9,16 (с, 1H), 8,83-8,78 (м, 2H), 8,58 (д, 1H), 8,46 (д, 1H), 8,38 (д, 1Н), 8,09 (д, 1Н), 1,88 (с, 9Н).
МС (ESI); m/z=391,85 [М+Н]+; 291,74 [М+Н-Boc]+.
Пример 5а (нитро/Boc-предшественник) (ACI-2776)
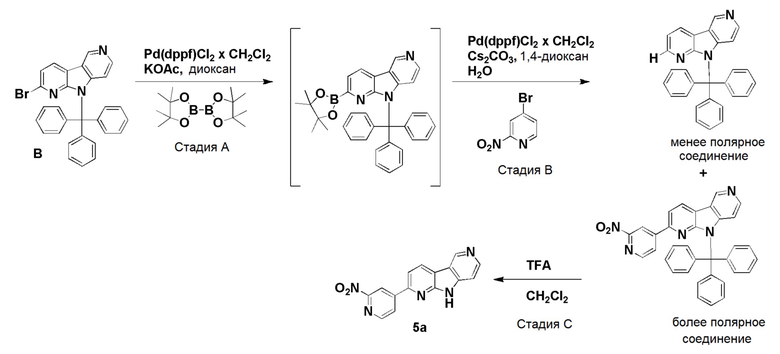
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 4-бром-2-нитропиридин (0,1 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной банев течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением более полярного указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,0437 г, 20%).
Более полярное указанное в заголовке соединение:
1H-ЯМР (400 МГц, CDCl3) δ=9,32 (с, 1H); 8,56 (д, 1H), 8,48 (д, 1H), 8,33 (с, 1H); 8,30 (д, 1H), 7,85 (д, 1H), 7,69 (д, 1H), 7,58-7,54 (м, 5H), 7,32-7,25 (м, 10H), 6,48 (д, 1H).
МС (ESI): m/z=534,28 [М+Н]+.
Менее полярный побочный продукт:
1H-ЯМР (400 МГц, CDCl3) δ=9,26 (с, 1H), 8,31 (дд, 1H), 8,23-8,19 (м, 2H), 7,52-7,46 (м, 5H), 7,28-7,22 (м, 10H), 7,14 (дд, 1H), 6,19 (д, 1H).
Стадия С
Указанное в заголовке соединение со стадии B выше (0,0437 г, 0,082 ммоль) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь разбавляли разведенным дихлорметаном (50 мл) и водой (20 мл). рН водной фазы доводили до рН ~12 добавлением 1 М водного раствора гидроксида натрия. Водный слой отбрасывали, и осадок в органическом слое собирали фильтрованием, промывали метанолом (10 мл) и высушивали на воздухе с получением указанного в заголовке соединения 5а в виде желтого твердого вещества (0,015 г, 63%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,75-12,5 (шир. с, 1H), 9,45-9,40 (шир. с, 1H), 9,10-9,05 (шир. с, 1H), 8,85-8,80 (шир. с, 2H), 8,68-8,63 (шир. с, 1H), 8,53-8,48 (шир. с, 1H), 8,27-8,22 (шир. с, 1H), 7,53-7,48 (шир. с, 1H).
МС (ESI): m/z=292,03 [М+Н]+.
Пример 6 (нитро/DMTr-предшественник) (ACI-2916)
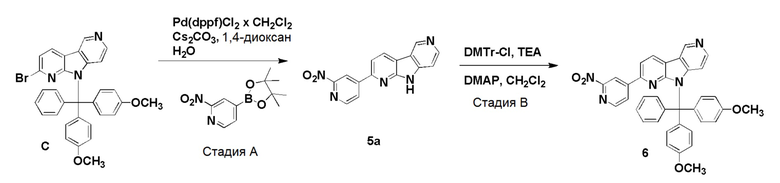
Стадия А
К смеси дегазированного 1,4-диоксана (2,2 мл) и воды (0,5 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0042 г, 0,005 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера С (0,055 г, 0,1 ммоль), 2-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,0305 г, 0,12525 ммоль) и карбоната цезия (0,067 г, 0,205 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (20 мл), осадок собирали фильтрованием, промывали водой (10 мл) и метанолом (5 мл) и высушивали на воздухе с получением неочищенных указанных в заголовке соединений в виде серого твердого вещества (0,0277 г, 95%).
Стадия В
К суспензии неочищенного указанного в заголовке соединения со стадии A выше (0,0277 г, 0,095 ммоль) в дихлорметане (4 мл), добавляли триэтиламин (1 мл, 7,2 ммоль), 4,4'-(хлор(фенил)метилен)бис(метоксибензол) (0,081 г, 0,29 ммоль) и 4-(диметиламино)пиридин (0,0036 г, 0,028 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, разбавляли этилацетатом (50 мл) и водой (20 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения 6 в виде бледно-желтого твердого вещества (0,0261 г, 44%).
1H-ЯМР (400 МГц, CDCl3) δ=9,32 (с, 1H), 8,58 (д, 1H), 8,50 (д, 1h), 8,36 (с, 1H), 8,30 (д, 1H), 7,85 (д, 1H), 7,74 (д, 1H), 7,52-7,42 (м, 6H), 7,27-7,23 (м, 4H), 6,80 (д, 4H), 6,49 (д, 1H), 3,78 (с, 6H).
Пример 7 (иод/Boc-предшественник) (ACI-3145)
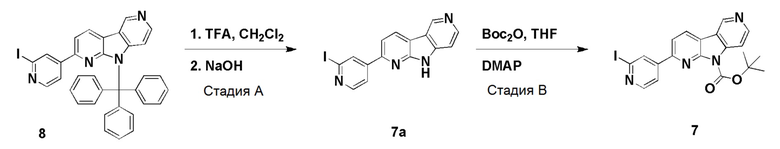
Стадия А
К раствору указанного в заголовке соединения из примера 8 (0,12 г, 0,195 ммоль) в дихлорметане (3,8 мл) добавляли трифторуксусную кислоту (3,01 мл, 39,1 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Растворители удаляли при пониженном давлении, остаток растворяли в 1 М водном гидроксиде натрия (20 мл) и экстрагировали дихлорметаном (3 × 50 мл). Органические фракции собирали, высушивали над Na2SO4 и очищали на колонках HP-Sil, используя градиент смеси дихлорметан/метанол (100/0 → 90/10), с получением указанного в заголовке соединения (0,025 г, 34%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,46 (с, 1H), 9,43 (д, 1H), 8,79 (д, 1H), 8,60 (д, 1H), 8,52 (дд, 2H), 8,20 (дд, 1Н), 8,14 (д, 1Н), 7,50 (м, 2Н).
МС (ESI): m/z=373,03 [М+Н]+.
Стадия В
К раствору указанного в заголовке соединения со стадии A выше (0,02 г, 0,067 ммоль) в тетрагидрофуране (5 мл) добавляли ди-трет-бутилдикарбонат (0,078 г, 0,336 ммоль) и 4- (диметиламино)пиридин (0,0082 г, 0,0672 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч и растворители удаляли в вакууме. Остаток очищали на колонках HP-Sil SNAP с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (100/0 → 50/50), с получением указанного в заголовке соединения 7 (0,018 г, 56%).
1H-ЯМР (400 МГц, CDCl3) δ=9,31 (д, 1H), 8,73 (д, 1H), 8,63 (д, 1H), 8,52-8,44 (м, 2H), 8,34 (дд, 1H), 7,99 (дд, 1H), 7,91 (д, 1H), 1,84 (с, 9H).
МС (ESI): m/z=473,03 [М+Н]+.
Пример 8 (иод/Tr-предшественник) (ACI-3143)
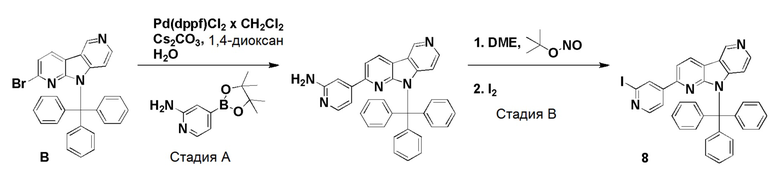
Стадия А
К смеси дегазированного 1,4-диоксана (30 мл) и воды (7 мл) в сухой пробирке для работы под давлением добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,043 г, 0,053 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,517 г, 1,054 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (0,278 г, 1,265 ммоль) и карбоната цезия (0,687 г, 2,109 ммоль). Затем реакционную смесь нагревали при 100°С в течение 4 ч. Растворители удаляли при пониженном давлении и остаток растворяли в этилацетате (40 мл) и 1 М водном растворе гидроксида натрия (40 мл). Фазы разделяли и органическую фазу промывали водой (2 × 50 мл). Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Растворители удаляли при пониженном давлении и остаток очищали на колонках HP-Sil SNAP (50 г), используя систему очистки Biotage Isolera One, применяя градиент смеси дихлорметан/метанол (100/0 → 90/10), с получением указанного в заголовке соединения (0,47 г, 89%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,39 (с, 1H), 8,68 (д, 1H), 8,23 (д, 1H), 7,92-7,75 (м, 2H), 7,67-7,51 (м, 6H), 7,38-7,18 (м, 9H), 6,65 (д, 1H), 6,59-6,44 (м, 2H), 5,72 (с, 2H).
Стадия В
К суспензии указанного в заголовке соединения со стадии A выше (0,47 г, 0,933 ммоль) в диметоксиэтане (40 мл) добавляли трет-бутилнитрит (0,134 мл, 1,12 ммоль) и йод (0,308 г, 1,213 ммоль). Реакционную смесь перемешивали при 50°С (внутренняя температура) в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли еще одну порцию трет-бутилнитрита (0,134 мл, 1,120 ммоль) и йода (0,2 г, 0,788 ммоль) и реакционную смесь перемешивали при 60°C (внутренняя температура) в течение 3 ч. Растворитель удаляли при пониженном давлении и остаток дважды очищали на колонках HP-Sil (100 г), используя градиент смеси дихлорметан/метанол (100/0 → 95/5) с медленным увеличением содержания метанола, со скоростью потока 15 мл/мин), с получением указанного в заголовке соединения 8 (0,12 г, 21%).
1H-ЯМР (400 МГц, CDCl3) δ=9,28 (с, 1H), 8,41 (д, 1H), 8,29 (дд, 2H), 7,76-7,67 (м, 2H), 7,62-7,52 (м, 6H), 7,38 (дд, 1H), 7,28 (м, 9H), 6,56 (д, 1H).
Пример 9 (хлор/Boc-предшественник) (ACI-2997)
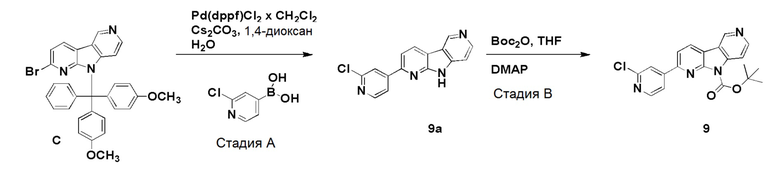
Стадия А
К смеси дегазированного 1,4-диоксана (30 мл) и воды (7 мл) в сухой пробирке для работы под давлением добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0225 г, 0,027 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера C (0,3 г, 0,545 ммоль), (2-хлорпиридин-4-ил)бороновой кислоты (0,103 г, 0,654 ммоль) и карбоната цезия (0,355 г, 1,09 ммоль). Затем реакционную смесь нагревали при 100°С в течение 4 ч. Растворители удаляли при пониженном давлении и остаток растворяли в этилацетате (40 мл) и воде (50 мл). Фазы разделяли и водную фазу снова экстрагировали этилацетатом (50 мл). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонках HP-Sil SNAP с использованием системы очистки Biotage Isolera One, применяя градиент смеси дихлорметан/метанол (100/0 → 90/10), с получением указанного в заголовке соединения (0,153 г, 26%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,48 (с, 1H), 9,45 (с, 1H), 8,83 (д, 1H), 8,57 (дд, 2H), 8,31-8,22 (м, 1H), 8,22-8,13 (м, 2Н), 7,53 (д, 1Н).
Стадия В
К суспензии указанного в заголовке соединения со стадии A выше (0,03 г, 0,107 ммоль) в тетрагидрофуране (5 мл) добавляли ди-трет-бутилдикарбонат (0,037 г, 0,16 ммоль) и 4- (диметиламино)пиридин (0,0065 г, 0,053 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч и растворители удаляли в вакууме. Остаток очищали на колонках HP-Sil SNAP с использованием системы очистки Biotage Isolera One, применяя градиент смеси дихлорметан/метанол (100/0 → 90/10), с получением указанного в заголовке соединения 9 (0,033 г, 81%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,51 (д, 1H), 8,87 (д, 1H), 8,71 (д, 1H), 8,59 (дд, 1H), 8,39 (д, 1H), 8,26 (дд, 1H), 8,22 (дд, 1H), 1,77 (с, 9H).
МС (ESI): m/z=280,81 [М+Н]+.
Пример 10 (трифлат/тритил) и пример 11 (трифалат/Boc-предшественник) (ACI-3538; ACI-3539)
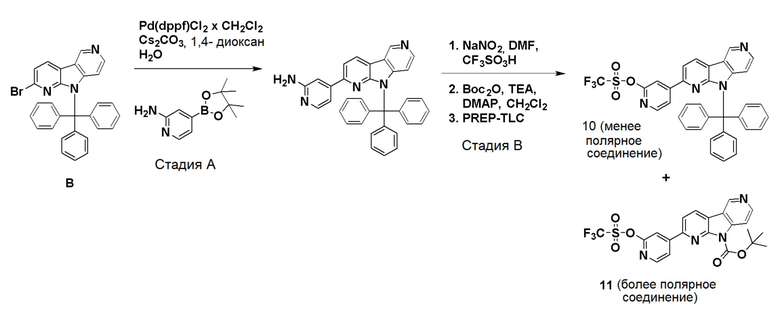
Стадия А
К смеси дегазированного 1,4-диоксана (8,7 мл) и воды (2 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера В (0,2 г, 0,4 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (0,110 г, 0,5 ммоль) и карбоната цезия (0,272 г, 0,84 ммоль). Затем реакционную смесь нагревали при ~120°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (40 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 97/3 → 95/5 → 95/5), с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (0,2 г, 97%).
1H-ЯМР (400 МГц, CDCl3) δ=9,26 (с, 1H); 8,40 (д, 1H), 8,30 (д, 1H), 8,01 (д, 1H), 7,73 (д, 1H), 7,65-7,59 (м, 5H), 7,31-7,24 (м, 10H), 6,98 (дд, 1H), 6,67 (д, 1H), 6,43 (д, 1H), 4,80 (шир.с, 2H).
Стадия В
Указанное в заголовке соединение со стадии A выше (0,2 г, 0,397 ммоль) суспендировали в N,N'-диметилформамиде (1,2 мл) и медленно добавляли трифторметансульфоновую кислоту (0,6 мл) при комнатной температуре (экзотермическая реакция). После добавления нитрита натрия (0,055 г, 0,795 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли дихлорметаном (40 мл) и водой (20 мл). Затем добавляли 2 М водный раствор гидроксида натрия до рН ~12. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли при пониженном давлении. Остаток суспендировали в дихлорметане (15 мл) и добавляли триэтиламин (2,7 мл) и ди-трет-бутилдикарбонат (0,621 г, 3,13 ммоль). После добавления 4-(диметиламино)пиридина (0,013 г, 0,1 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли этилацетатом (50 мл) и смесью насыщенный раствор соли/вода 1/1 (20 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (puriFlash 40 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанных в заголовке соединений 10 и 11. Смесь указанных в заголовке соединений разделяли препаративной ТСХ (20 × 20 см; 1000 мкм, Analtech), используя этилацетат в качестве подвижной фазы, с получением менее полярного указанного в заголовке соединения 10 в виде бледно-желтого твердого вещества (0,0357 г, 14%) и более полярного указанного в заголовке соединения 11 в виде бледно-желтого твердого вещества (0,0237 г, 12%).
Менее полярное указанное в заголовке соединение 10:
1H-ЯМР (400 МГц, CDCl3) δ=9,32 (с, 1H), 8,47 (д, 1H), 8,35 (д, 1H), 8,31 (д, 1H), 7,80 (д, 1H), 7,61-7,57 (м, 6H), 7,32-7,26 (м, 10H), 7,07 (с, 1H); 6,90 (д, 1H).
МС (ESI): m/z=637,25 [М+Н]+.
Более полярное указанное в заголовке соединение 11:
1H-ЯМР (400 МГц, CDCl3) δ=9,38 (с, 1H), 8,78 (д, 1H), 8,57-8,50 (м, 2H), 8,38 (д, 1H), 8,15 (д, 1H), 8,10 (с, 1Н), 8,00 (д, 1Н), 1,86 (с, 9Н).
МС (ESI): m/z=495,01 [М+Н]+.
Пример 12 (трифлат/NH-предшественник) (ACI-3545)
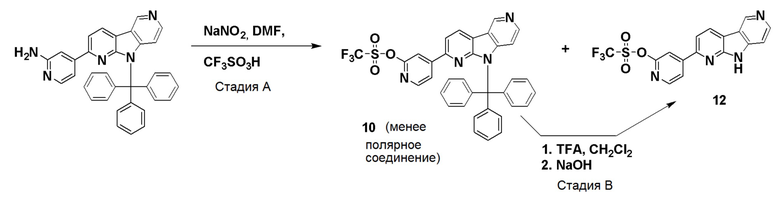
Стадия А
Указанное в заголовке соединение со стадии A примеров 10 и 11 (0,272 г, 0,54 ммоль) суспендировали в N,N'-диметилформамиде (1,8 мл). Реакционную смесь охлаждали до 0°С и медленно добавляли трифторметансульфоновую кислоту (0,9 мл) (экзотермическая реакция). Реакционной смеси давали подогреться до комнатной температуры, добавляли нитрит натрия (0,0825 г, 1,19 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли дихлорметаном (60 мл) и водой (25 мл); затем добавляли 2 М водный раствор гидроксида натрия до рН ~12. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (HP-SIL 25 г) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением менее полярного соединения 10 в виде бледно-желтого твердого вещества (0,0497 г, 14,5%). Затем градиент меняли на смесь дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20) с получением указанного в заголовке соединения 12 в виде серого твердого вещества (0,0425 г).
Стадия В
Менее полярное соединение 10 со стадии А выше (0,0497 г, 0,0078 ммоль), растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли дихлорметаном (30 мл) и водой (10 мл). рН водной фазы доводили до рН ~12 добавлением 2 М водного раствора гидроксида натрия. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (HP-SIL 10 г) с использованием системы очистки Biotage Isolera One, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20), с получением дополнительного количества указанного в заголовке соединения 12 в виде серого твердого вещества (0,0182 г) с общим выходом 0,0607 г (28,5%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,54 (шир. с, 1H), 9,46 (с, 1H), 8,87 (д, 1H), 8,64 (д, 1H), 8,56 (д, 1H), 8,42 (дд, 1H), 8,30 (д, 1H), 8,24 (д, 1H), 7,55 (дд, 1H).
МС (ESI): m/z=395,12 [М+Н]+.
Пример 14 (триметиламмоний/тритил-предшественник) (ACI-3591)
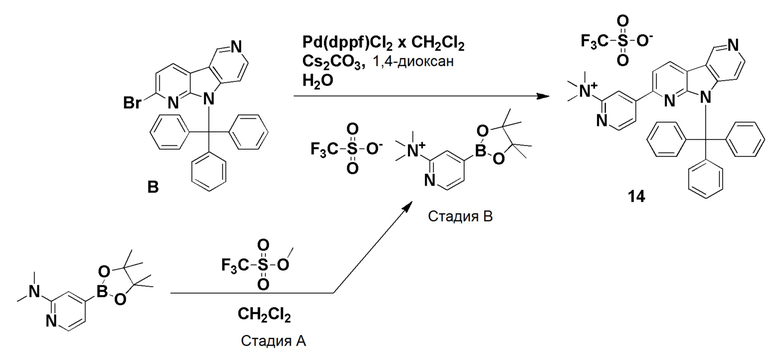
Стадия А
Коммерчески доступный N,N-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (0,25 г, 1 ммоль) растворяли в дихлорметане ( 5 мл). К полученному перемешиваемому раствору добавляли по каплям при комнатной температуре метилтрифторметансульфонат (0,124 мл, 1,1 ммоль). Раствор перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали для удаления дихлорметана и остаток высушивали в вакууме с получением желтой стеклообразной/пены, которую непосредственно использовали на следующей стадии.
Стадия В
К раствору дегазированного 1,4-диоксана (12 мл) и воды (3 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), указанное в заголовке соединение из препаративного примера B (0,4 г, 0,816 ммоль), неочищенное указанное в заголовке соединение со стадии A выше (~1 ммоль), и карбонат цезия (0,544 г, 1,68 ммоль). Реакционную смесь нагревали при ~120°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (150 мл) и водой (50 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (HP-Ultra 25 г) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), для элюирования непрореагировавшего исходного вещества и неполярных побочных продуктов. Затем градиент меняли на смесь дихлорметан/метанол (100/0 → 95/5 → 90/10) с получением производного диметиламина в виде бледно-желтого стеклообразного вещества (0,127 г, 29%; МС (ESI): m/z=532,27 [М+Н]+) и производного метиламина в виде серого твердого вещества (0,0547 г, 13%; МС (ESI): m/z=519,18 [М+Н]+). Градиент снова меняли на смесь дихлорметан/метанол (90/10 → 80/20) и выдерживали при (80/20) с получением указанного в заголовке соединения 14 в виде коричневого твердого вещества (0,104 г, 18%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,47 (с, 1H); 8,89 (д, 1H), 8,55 (д, 1H), 8-35-8,32 (м, 2H), 8,29 (д, 1H), 7,63-7,57 (м, 5H), 7,48 (д, 1H), 7,34-7,25 (м, 10H), 6,48 (д, 1H), 3,60 (с, 9H).
МС (ESI): m/z=546,26 [М+Н]+.
Пример 14а (триметиламмоний/NH-предшественник) (ACI-3613)
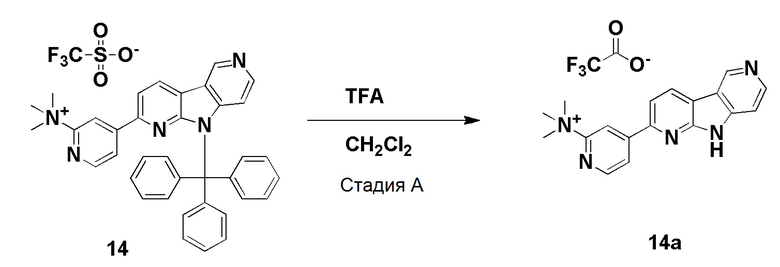
Стадия А
Указанное в заголовке соединение из примера 14 (0,199 г, 0,364 ммоль) суспендировали в дихлорметане (10 мл). После добавления трифторуксусной кислоты (10 мл) реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Растворители удаляли при пониженном давлении, остаток растворяли в метаноле (10 мл) и растворители удаляли при пониженном давлении. Обработку остатка метанолом повторяли еще два раза. Затем остаток суспендировали в дихлорметане (20 мл) и обрабатывали ультразвуком в течение ~5 мин. Осадок собирали фильтрованием, промывали дихлорметаном (10 мл) и высушивали на воздухе с получением указанного в заголовке соединения 14а в виде серого твердого вещества (0,127 г, 83%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=13,76 (шир.с, 1H), 9,84 (с, 1H); 8,12 (д, 1H), 8,89 (д, 1H), 8,80 (д, 1H), 8,75 (с, 1H), 8,54-8,50 (м, 2H), 8,04 (д, 1H), 3,72 (с, 9H).
МС (ESI): m/z=303,91 [М+Н]+.
Пример 15 (трифлат/DMTr-предшественник) (ACI-3546)
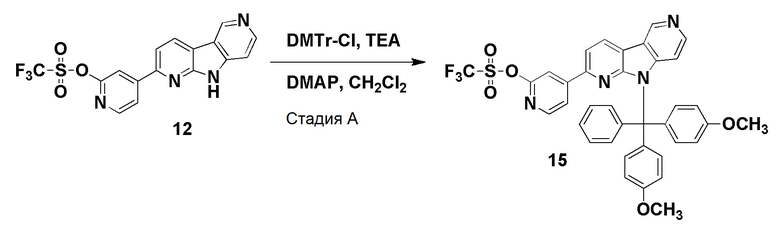
Стадия А
К суспензии указанного в заголовке соединения из примера 12 (0,0291 г, 0,0739 ммоль) в дихлорметане (3 мл) добавляли триэтиламин (0,046 мл, 0,926 ммоль), 4,4'-(хлор(фенил)метилен) бис(метоксибензол) (0,062 г, 0,222 ммоль) и 4-(диметиламино)пиридин (0,00175 г, 0,014 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, разбавляли этилацетатом (40 мл) и водой (15 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения в виде полутвердого вещества. Соединение обрабатывали н-гептаном (5 мл), обрабатывали ультразвуком в течение 5 мин и растворитель выпаривали при пониженном давлении с получением указанного в заголовке соединения 15 в виде не совсем белого твердого вещества (0,0348 г, 67%).
1H-ЯМР (400 МГц, CDCl3) δ=9,32 (с, 1H), 8,47 (д, 1H), 8,36-8,28 (м, 2H), 7,79 (д, 1H), 7,59-7,57 (м, 1H), 7,52-7,42 (м, 5H), 7,33-7,27 (м, 4H), 7,20-7,18 (м, 1H), 6,83-6,77 (м, 4H), 6,58 (д, 1H), 3,80 (с, 6H).
МС (ESI): m/z=697,28 [М+Н]+.
Пример 17 (мезилат/тритил-предшественник) (ACI-3540)
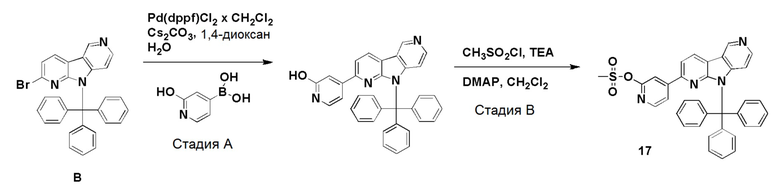
Стадия А
К смеси дегазированного 1,4-диоксана (13 мл) и воды (3 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,026 г, 0,03 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,3 г, 0,612 ммоль), (2-гидроксипиридин-4-ил)бороновой кислоты (0,104 г, 0,75 ммоль) и карбоната цезия (0,408 г, 1,15 ммоль). Затем реакционную смесь нагревали при ~120°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (120 мл) и водой (45 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением непрореагировавшего исходного соединения B в виде не совсем белого твердого вещества (0,1398 г, 47%). Затем градиент меняли на смесь дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20 → 80/20) с получением указанного в заголовке соединения в виде серого твердого вещества (0,0996 г, 32%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=11,52 (шир.с, 1H), 9,43 (с, 1H), 8,70 (д, 1H), 8,23 (д, 1H), 7,97 (д, 1H), 7,59-7,53 (м, 6H), 7,32-7,21 (м, 10H), 6,67 (д, 1H), 6,48 (д, 1H), 6,19 (дд, 1H).
МС (ESI): m/z=505,28 [М+Н]+.
Стадия В
К суспензии указанного в заголовке соединения со стадии A выше (0,080 г, 0,159 ммоль) в дихлорметане (5 мл) добавляли триэтиламин (0,2 мл, 1,431 ммоль), метансульфонилхлорид (0,0365 мл, 0,477 ммоль) и 4-(диметиламино)пиридин (0,0054 г, 0,023 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч, разбавляли этилацетатом (50 мл) и смесью воды/ насыщенного раствора соли (20 мл; 1/1). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения 17 в виде бледно-желтого твердого вещества (0,0438 г, 47%).
1H-ЯМР (400 МГц, CDCl3) δ=9,31 (с, 1H), 8,46 (д, 1H), 8,30-8,27 (м, 2H), 7,79 (д, 1H), 7,59-7,55 (м, 5H), 7,42 (дд, 1H), 7,32-7,26 (м, 10H), 7,08 (с, 1H), 6,65 (д, 1H).
МС (ESI): m/z=583,21 [М+Н]+.
Пример 17а (мезилат/NH-предшественник) (ACI-3572)
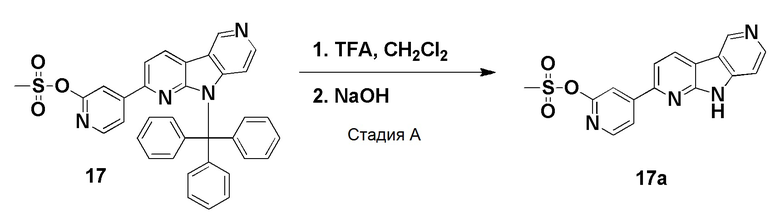
Стадия А
Указанное в заголовке соединение из примера 17 (0,0388 г, 0,0067 ммоль) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли дихлорметаном (30 мл) и водой (10 мл). рН водной фазы доводили до рН ~12 добавлением 2 М водного раствора гидроксида натрия. Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (HP-SIL 10 г) с использованием системы очистки Biotage Isolera One, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20), с получением указанного в заголовке соединения 17а в виде белого твердого вещества (0,0059 г, 26%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,51 (шир. с, 1H), 9,45 (с, 1H), 8,84 (д, 1H), 8,58-8,54 (м, 2H); 8,26 (дд, 1H), 8,20 (д, 1H); 8,05 (д, 1H), 7,54 (д, 1H), 3,68 (с, 3H).
МС (ESI): m/z=341,17 [М+Н]+.
Пример 18 (дейтерированное соединение)
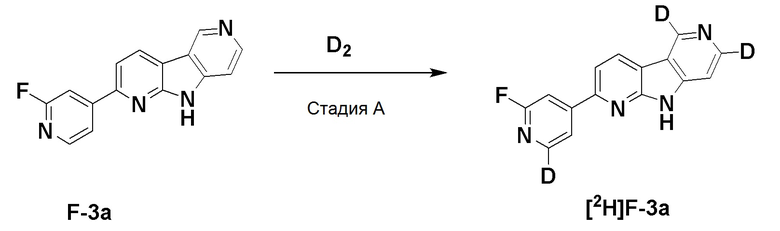
Стадия А
Указанное в заголовке соединение из примера 1 использовали в качестве исходного соединения для получения примера 18 ([2H]F-3a) путем прямого изотопного обмена водорода с черным родием.
МС (ESI): m/z=265 (45%) [М+Н]+; 266 (65%) [М+Н]+; 267 (100%) [М+Н]+; 268 (34%) [М+Н]+.
Пример 19 (тритированное соединение)
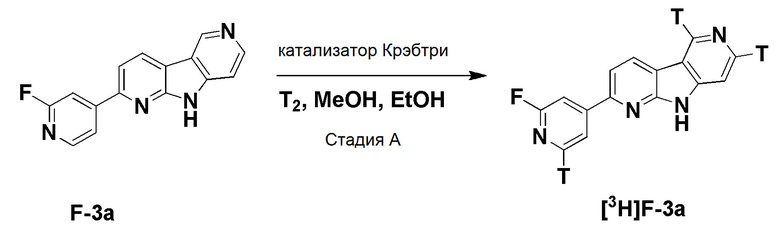
Стадия А
Указанное в заголовке соединение из примера 1 использовали в качестве исходного соединения для получения примера 19 ([3H]F-3a) посредством прямого обмена изотопами водорода с газообразным тритием (2,2 кюри/мл) с использованием катализатора Крэбтри в смеси метанол/этанол. После очистки с помощью ВЭЖХ (Phenomenex Prodigy ODS (2), 4,6 × 250 мм, 5 мкм; растворители A: вода с 0,1% ТФК; B: ацетонитрил; 0-20 мин, 0-100% B; выдерживали до 30 мин), [3H]F-3a получали проукт с радиохимической чистотой 98,7% и удельной активностью 24,6 кюри/ммоль).
МС (ESI): m/z=265 (100%) [М+Н]+; 267 (77,5%) [М+Н]+; 269 (41,3%) [М+Н]+; 271 (11,3%) [М+Н]+.
Сравнительный пример 2 (F-2) (ACI-2448)
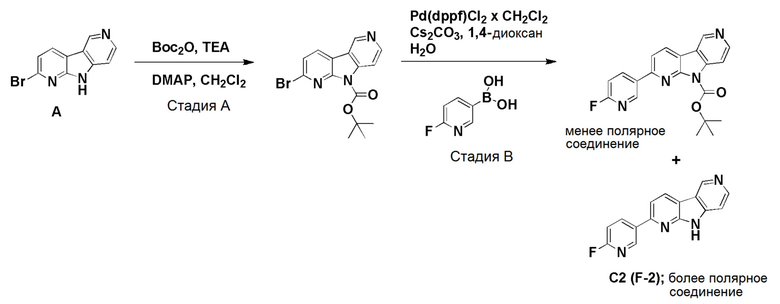
Стадия А
К суспензии указанного в заголовке соединения из препаративного примера А (0,430 г, 1,73 ммоль) в дихлорметане (25 мл) добавляли триэтиламин (1,93 мл, 13,89 ммоль) и ди-трет-бутилдикарбонат (2,27 г, 10,02 ммоль). После добавления 4- (диметиламино)пиридина (0,042 г, 0,34 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Растворители удаляли при пониженном давлении и остаток очищали на колонках HP-Sil SNAP (25 г) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (0,558 г, 92%).
1H-ЯМР (400 МГц, CDCl3) δ=9,28 (с, 1H), 8,73 (д, 1H), 8,22 (д, 2H), 7,59 8d, 1H), 1,80 (с, 9H).
Стадия В
К смеси дегазированного 1,4-диоксана (3 мл) и воды (0,7 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0058 г, 0,007 ммоль), с последующим добавлением указанного в заголовке соединения со стадии А выше (0,05 г, 0,143 ммоль), (6-фторпиридин-3-ил)бороновой кислоты (0,024 г, 0,17 ммоль) и карбоната цезия (0,092 г, 0,286 ммоль). Затем реакционную смесь нагревали при ~100°С на песочной бане в течение 4 ч. Реакционную смесь разбавляли этилацетатом (80 мл) и водой (35 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 12 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 98/2 → 95/5 → 90/10 → 80/20), с получением менее полярного Boc-защищенного соединения (0,0255 г, 49%) и более полярного сравнительного примера C2 (F-2) в виде не совсем белого твердого вещества (0,0116 г, 31%).
Более полярный сравнительный пример C2 (F-2):
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,40 (шир.с, 1H), 9,40 (с, 1H), 9,05 (с, 1H), 8,78-8,70 (м, 2H), 8,51 (д, 1H), 8,02 (д, 1H), 7,50 (д, 1H), 7,36 (дд, 1H).
МС (ESI): m/z=265,09 [М+Н]+.
Менее полярное Boc-защищенное соединение:
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,48 (с, 1H), 9,13 (д, 1H), 8,84-8,78 (м, 2H), 8,68 (д, 1H), 8,23 (д, 1H), 8,19 (д, 1H), 7,40 (дд, 1H), 1,75 с, 9H).
Синтез сравнительного примера C2 (F-2) был впервые описан в WO 2015/052105 (пример 1) с использованием другого способа синтеза.
Сравнительный пример 2 (F-2) предшественник (ACI-2449)
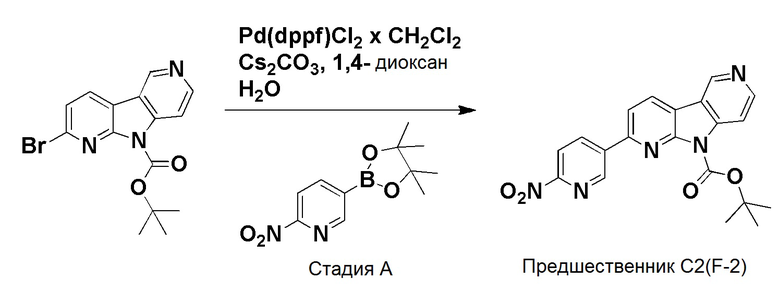
Стадия А
К смеси дегазированного 1,4-диоксана (3 мл) и воды (0,7 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0058 г, 0,007 ммоль), с последующим добавлением указанного заголовке соединения со стадии А сравнительного примера 2 (0,05 г, 0,143 ммоль), 2-нитро-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,0428 г, 0,17 ммоль) и карбоната цезия (0,092 г, 0,286 ммоль). Затем реакционную смесь нагревали при ~100°С на песочной бане в течение 4 ч. Реакционную смесь разбавляли этилацетатом (80 мл) и водой (35 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 12 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 98/2 → 95/5 → 90/10 → 80/20), с получением предшественника сравнительного примера С2 (F-2) в виде бледно-желтого твердого вещества (0,0173 г, 31%).
1H-ЯМР (400 МГц, CDCl3/CD3OD) δ=9,45 (д, 1H), 9,32 (с, 1H), 8,93 (дд, 1H), 8,68-8,64 (м, 2H), 8,46 (д, 1H), 8,35 (д, 1Н), 8,14 (д, 1Н), 1,82 (с, 9Н).
МС (ESI): m/z=392,13 [М+Н]+.
Синтез предшественника сравнительного примера С2 (F-2) был впервые описан в WO 2015/052105 (пример 3а) с использованием другого способа синтеза.
Сравнительный пример 5 (F-5) (ACI-2632)
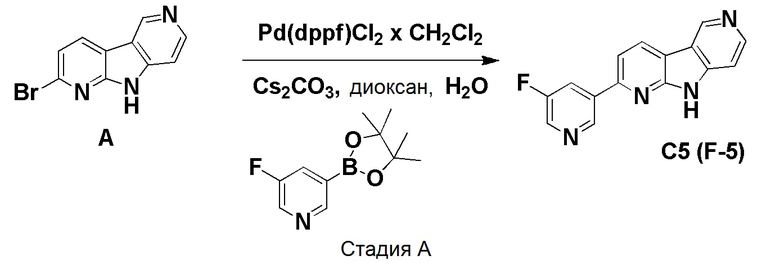
Стадия А
К смеси дегазированного 1,4-диоксана (4,3 мл) и воды (1 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0084 г, 0,01 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера А (0,05 г, 0,2 ммоль), 3-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,055 г, 0,246 ммоль) и карбоната цезия (0,133 г, 0,41 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (60 мл) и водой (20 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (HP-SIL 25 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20), с получением сравнительного примера C5 (F-5) в виде не совсем белого твердого вещества (0,022 г, 43%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,45 (шир.с, 1H), 9,45 (с, 1H), 9,31 (с, 1H), 8,80 (д, 1H), 8,67 (д, 1H). 8,53 (д, 1H), 8,46-8,40 (м, 1H), 8,11 (д, 1H), 7,52 (д, 1H).
МС (ESI): m/z=265,06 [М+Н]+.
Сравнительный пример 5 (F-5) предшественник (ACI-2719)
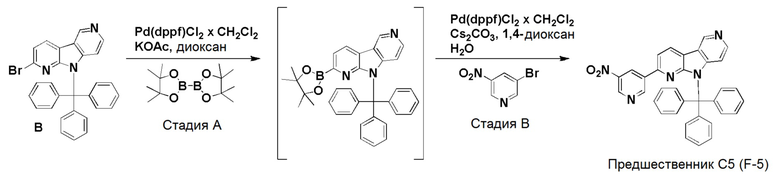
Стадия А
К смеси дегазированного 1,4-диоксана (4 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,1 г, 0,2 ммоль), бис(пинаколато)диборана (0,056 г, 0,22 ммоль) и ацетата калия (0,059 г, 0,6 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (4,3 мл) и воды (1 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль), 3-бром-5-нитропиридин (0,05 г, 0,245 ммоль) и карбонат цезия (0,133 г, 0,41 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч.
Реакционную смесь разбавляли этилацетатом (80 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C5 (F-5) в виде бледно-желтого твердого вещества (0,0144 г, 13%).
1H-ЯМР (400 МГц, CDCl3) δ=9,36 (д, 1H), 9,30 (с, 1H), 9,02 (д, 1H); 8,52-8,48 (м, 2H), 8,29 (д, 1H), 7,80 (д, 1H), 7,60-7,55 (м, 5H), 7,33-7,25 (м, 10H), 6,46 (д, 1H).
МС (ESI): m/z=533,67 [М+Н]+.
Сравнительный пример 6 (F-6) (ACI-2843)
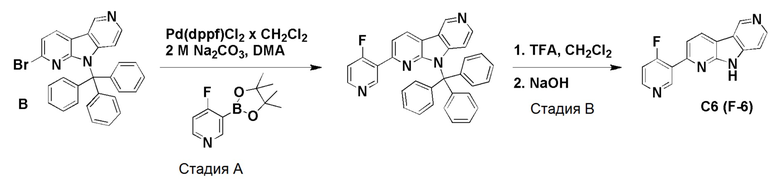
Стадия А
В пробирке для микроволнового реактора емкостью 20 мл растворяли указанное в заголовке соединение из препаративного примера B (0,2 г, 0,408 ммоль) и 4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (0,182 г, 0,816 ммоль) в N,N'-диметилацетамиде (5,10 мл). Добавляли карбонат натрия (0,816 мл, 1,631 ммоль) и полученный перемешивающийся раствор дегазировали в течение 5 мин. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном, и реакционную смесь нагревали до 110°С в течение 22 ч. Мониторинг ТСХ показал завершение реакции. Реакционную смесь разбавляли дихлорметаном, нерастворимые вещества отфильтровывали через целит, и фильтрат трижды промывали водой для удаления остаточных количеств N,N'-диметилацетамида. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Остаток очищали с использованием Biotage Isolera One (смесь дихлорметан/метанол от 100:0 до 90:10; колонка HP-Sil 25 г) с получением указанного в заголовке соединения (0,1036 г; 50%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,43 (с, 1H), 8,75 (д, 1H), 8,54 (дд, 1H), 8,26 (д, 1H), 8,17 (д, 1H), 7,83 (дд, 1H), 7,61-7,52 (м, 6H), 7,41 (дд, 1H), 7,35-7,20 (м, 9H), 6,46 (д, 1H).
МС [М+Н] +=507,43, 243,29.
Стадия В
В круглодонной колбе емкостью 25 мл растворяли указанное в заголовке соединение со стадии А выше (0,1 г, 0,199 ммоль), в дихлорметане (1 мл). Осторожно добавляли трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали в течение 18 ч при комнатной температуре. После охлаждения при 0°С реакционную смесь гасили до рН 10 с помощью 2 М раствора гидроксида натрия. Полученную суспензию фильтровали. Реакционную смесь промывали водой и насыщенным раствором соли. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Остаток очищали с использованием Biotage Isolera One (смесь дихлорметан/метанол от 100:0 до 90:10; колонка HP-Sil 10 г) с получением сравнительного примера C6 (F-6) (0,026 г; 47%).
1H-ЯМР (400 МГц, ДМСО-d6) δ 12,57 (с, 1H), 9,46 (с, 1H), 9,19 (д, 1H), 8,80 (д, 1H), 8,70 (с, 1H), 8,59-8,52 (м, 1Н), 7,81 (д, 1Н), 7,55 (д, 2Н).
МС [М+Н] +=265,29.
Сравнительный пример 6 (F-6) предшественник (ACI-2764)
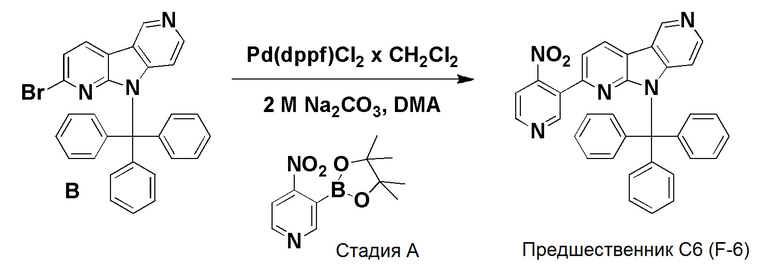
Стадия А
В пробирке для микроволнового реактора емкостью 20 мл растворяли указанное в заголовке соединение из препаративного примера B (0,2 г, 0,408 ммоль) и 4-нитро-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)пиридин (0,204 г, 0,816 ммоль) в N,N'-диметилацетамиде (5,10 мл). Добавляли карбонат натрия (0,816 мл, 1,631 ммоль) и полученный перемешивающийся раствор дегазировали в течение 5 мин. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль) и реакционную смесь нагревали до 110°С в течение 22 ч. Мониторинг ТСХ показал завершение реакции. Реакционную смесь разбавляли дихлорметаном, нерастворимые вещества отфильтровывали через целит, и фильтрат трижды промывали водой для удаления остаточных количеств N,N'-диметилацетамида. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Остаток очищали с использованием Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C6 (F-6) в виде бледно-желтого твердого вещества (0,056 г, 28%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,45 (с, 1H), 8,81 (д, 1H), 8,69 (д, 1H), 8,32-8,23 (м, 3H), 8,20 (д, 1H), 7,60 (дд, 6H), 7,36-7,22 (м, 9H), 6,52 (д, 1H), 5,76 (с, 1H).
МС (ESI): m/z=533,87 [М+Н]+.
Сравнительный пример 7 (F-7) (ACI-2731)
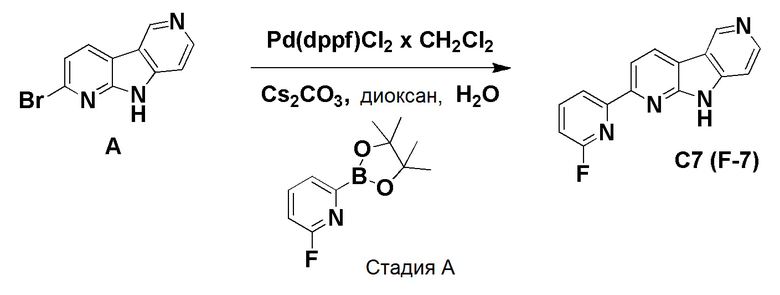
Стадия А
К смеси дегазированного 1,4-диоксана (4,3 мл) и воды (1 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,0084 г, 0,01 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера А (0,05 г, 0,2 ммоль), 2-фтор-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (0,055 г, 0,246 ммоль) и карбоната цезия (0,133 г, 0,41 ммоль). Затем реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (60 мл) и водой (20 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (HP-SIL 25 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10 → 80/20), с получением сравнительного примера C7 (F-7) в виде не совсем белого твердого вещества (0,033 г, 63%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,42 (с, 1H), 9,41 (с, 1H), 8,77 (д, 1H), 8,52 (д, 1H), 8,40 (дд, 1H), 8,27 (д, 1Н), 8,18 (кв, 1Н), 7,51 (д, 1Н), 7,26 (дд, 1Н).
МС (ESI): m/z=265,09 [М+Н]+.
Сравнительный пример 7 (F-7) предшественник (ACI-2778)
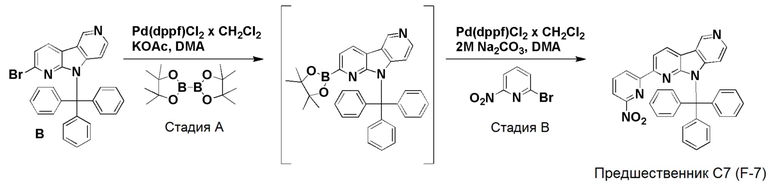
Стадия А
К смеси дегазированного N,N'-диметилацетамида (4 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,1 г, 0,2 ммоль), бис(пинаколато)диборана (0,056 г, 0,22 ммоль) и ацетата калия (0,059 г, 0,6 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
В пробирке для микроволнового реактора емкостью 20 мл растворяли неочищенное указанное в заголовке соединение со стадии A выше, 2-бром-6-нитропиридин (0,05 г, 0,245 ммоль) в N,N'-диметилацетамиде (5,10 мл). Добавляли карбонат натрия (0,408 мл, 0,816 ммоль) и полученный перемешивающий раствор дегазировали в течение 5 мин. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,017 г, 0,02 ммоль), и реакционную смесь нагревали до 110°С в течение 22 ч. Мониторинг ТСХ показал завершение реакции. Реакционную смесь разбавляли дихлорметаном, нерастворимые вещества отфильтровывали через целит, и фильтрат трижды промывали водой для удаления остаточных количеств N,N'-диметилацетамида. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Остаток очищали с использованием Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C7 (F-7) в виде бледно-желтого твердого вещества (0,0174 г, 16%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,43 (с, 1H), 9,38 (с, 1H), 8,81 (д, 1H), 8,60 (дд, 1H), 8,33 (д, 1H), 8,28- 8,24 (м, 2Н), 8,18 (д, 1Н), 8,10 (т, 1Н), 7,61 (д, 7Н), 7,47 (д, 4Н), 7,42 (д, 1Н), 7,28 (тт, 18Н), 6,58 (д, 1Н), 6,19 (д, 1Н).
МС (ESI): m/z=533,62 [М+Н]+.
Сравнительный пример 8 (F-8) (ACI-2876)
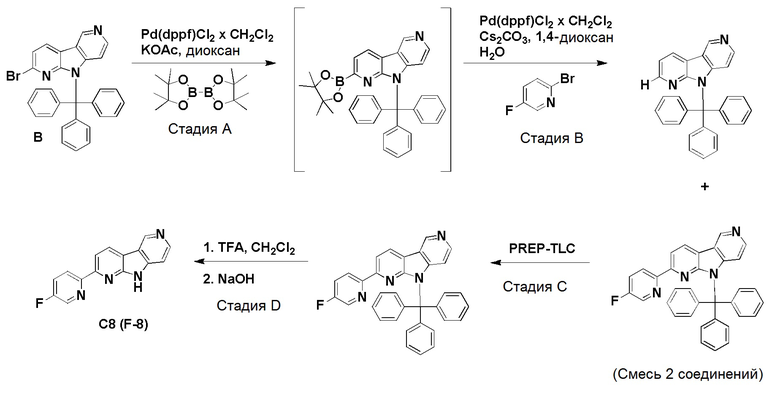
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-5-фторпиридин (0,086 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочного продукта (0,064 г).
Стадия С
Смесь указанного в заголовке соединения и побочного продукта со стадии B выше (0,064 г), очищали препаративной ТСХ с загрузкой ~0,03 г смеси на 1000 мкМ Analtech Uniplate (20 × 20 см) с использованием смеси дихлорметан/ацетон (90/10) в качестве подвижной фазы с получением более полярного указанного в заголовке соединения в виде не совсем белого твердого вещества (0,0385 г, 18,5% с 3 стадий).
1H-ЯМР (400 МГц, CDCl3) δ=9,26 (с, 1H), 8,45 (д, 1H), 8,38 (система AB, 2H), 8,25 (д, 1H), 7,62-7,58 (м, 5H), 7,30-7,18 (м, 12H), 6,56 (д, 1H).
Стадия D
Указанное в заголовке соединение со стадии C выше (0,0385 г, 0,076 ммоль) растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь разводили разведенным дихлорметаном (50 мл) и водой (20 мл). рН водной фазы доводили до рН ~12 добавлением 1 М водного раствора гидроксида натрия. Водный слой отделяли, экстрагировали дихлорметаном (25 мл), объединенный органический слой высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (колонка HP-SIL 10 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10), с получением сравнительного примера C8 (F-8) в виде белого твердого вещества (0,0079 г, 39,3%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,40 (шир.с, 1H), 9,40 (с, 1H), 8,77 (д, 1H), 8,72 (д, 1H), 8,55-8,50 (м, 2H), 8,35 (д, 1H), 7,95-7,90 (м, 1H), 7,51 (д, 1H).
МС (ESI): m/z=265,06 [М+Н]+.
Синтез сравнительного примера C8 (F-8) был впервые описан в WO2016/124508 (пример 18) с использованием другого способа синтеза.
Сравнительный пример 8 (F-8) предшественник (ACI-2877)
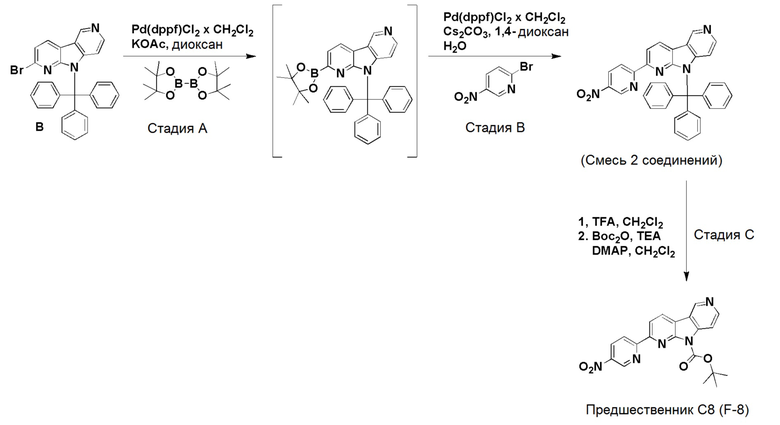
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-5-нитропиридин (0,1 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочного продукта (0,0788 г).
Стадия С
Смесь указанного в заголовке соединения со стадии B выше и побочного продукта (0,0788 г) растворяли в дихлорметане (10 мл) и добавляли трифторуксусную кислоту (2,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч и затем добавляли метанол (10 мл). Растворители выпаривали в вакууме и остаток суспендировали в метаноле (10 мл). Растворители снова выпаривали в вакууме и остаток суспендировали в дихлорметане (4 мл). После добавления триэтиламина (2 мл, 14,4 ммоль), ди-трет-бутилдикарбоната (0,2 г, 0,86 ммоль) и 4-(диметиламино)пиридина (0,0036 г, 0,028 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (40 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C8 (F-8) в виде бледно-желтого твердого вещества (0,0149 г, 25,7%).
1H-ЯМР (400 МГц, CDCl3) δ=9,55 (д, 1H), 9,36 (с, 1H), 8,88 (д, 1H), 8,77 (д, 1H), 8,72 (д, 1H), 8,65 (дд, 1H), 8,56 (д, 1H), 8,30 (д, 1H), 1,87 (с, 9H).
МС (ESI): m/z=391,93 [М+Н]+.
Сравнительный пример 9 (F-9) (ACI-2930)
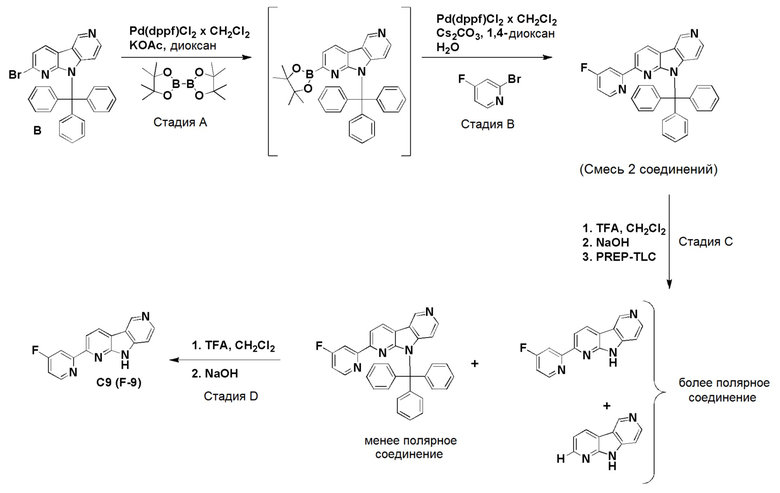
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-4-фторпиридин (0,086 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочного продукта (0,0489 г).
Стадия С
Смесь указанного в заголовке соединения и побочного продукта со стадии B выше (0,0489 г), растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь разбавляли разведенным дихлорметаном (50 мл) и водой (20 мл). рН водной фазы доводили до рН ~12 добавлением 1 М водного раствора гидроксида натрия. Водный слой отделяли, экстрагировали дихлорметаном (25 мл), объединенный органический слой высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток очищали препаративной ТСХ с загрузкой ~0,03 г смеси на 1000 мкМ Analtech Uniplate (20 × 20 см), используя смесь дихлорметан/метанол (90/10) в качестве подвижной фазы, с получением менее полярного указанного в заголовке соединения в виде не совсем белого твердого вещества (0,0145 г, 7% с 3 стадий) и более полярной смеси двух соединений.
1H-ЯМР (400 МГц, ДМСО-d6) δ=9,42 (с, 1H), 8,76 (д, 1H), 8,67 (дд, 1H), 8,35 (д, 1H), 8,27 (д, 1H), 7,67- 7,60 (м, 5H), 7,35-7,22 (м, 11H), 6,81 (дд, 1H), 6,60 (д, 1H).
Стадия D
Менее полярное указанное в заголовке соединение со стадии C выше (0,0145 г, 0,027 ммоль) растворяли в дихлорметане (3 мл) и добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли разведенным дихлорметаном (50 мл) и водой (20 мл). рН водной фазы доводили до рН ~12 добавлением 1 М водного раствора гидроксида натрия. Водный слой отделяли, экстрагировали дихлорметаном (25 мл), объединенный органический слой высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (HP-SIL 10 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10), с получением сравнительного примера C9 (F-9) в виде не совсем белого твердого вещества (0,0025 г, 33%).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,43 (шир.с, 1H), 9,45 (с, 1H), 8,82-8,77 (м, 2H), 8,54 (д, 1H), 8,44 (д, 1H), 8,22 (дд, 1H), 7,53 (д, 1H), 7,46-7,42 (м, 1H).
МС (ESI): m/z=264,63 [М+Н]+.
Сравнительный пример 9 (F-9) предшественник (ACI-2915)
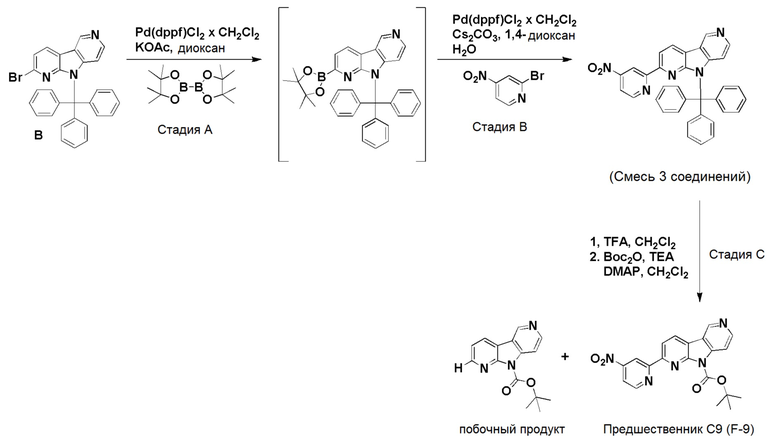
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-4-нитропиридин (0,1 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочных продуктов (0,076 г).
Стадия С
Смесь указанного в заголовке соединения со стадии B выше и побочных продуктов (0,076 г) растворяли в дихлорметане (10 мл) и добавляли трифторуксусную кислоту (2,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч и затем добавляли метанол (10 мл). Растворители выпаривали в вакууме и остаток суспендировали в метаноле (10 мл). Растворители снова выпаривали в вакууме и остаток суспендировали в дихлорметане (4 мл). После добавления триэтиламина (2 мл, 14,4 ммоль), ди-трет-бутилдикарбоната (0,2 г, 0,86 ммоль) и 4-(диметиламино)пиридина (0,0036 г, 0,028 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (40 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C9 (F-9) и побочного продукта в виде смеси ~1,1 (0,0231 г, бледно-желтое твердое вещество).
1H-ЯМР (400 МГц, CDCl3) δ=9,38 (д, 1H), 9,35 (д, 1H), 9,31 (с, 2H), 9,02 (д, 1H), 8,76-8,70 (м, 5H), 8,68 (д, 1H), 8,55 (д, 1H), 8,43-8,37 (м, 3H), 8,12 (дд, 1H), 8,07 (дд, 1H), 7,43 (д, 1H), 7,41 (д, 1H) 1,82 (с, 18Н).
МС (ESI): m/z=291,94 [MH-Boc указанного в заголовке соединения]+, 170,04 [MH+-Boc побочного продукта]+.
Сравнительный пример 10 (F-10) (ACI-2931)
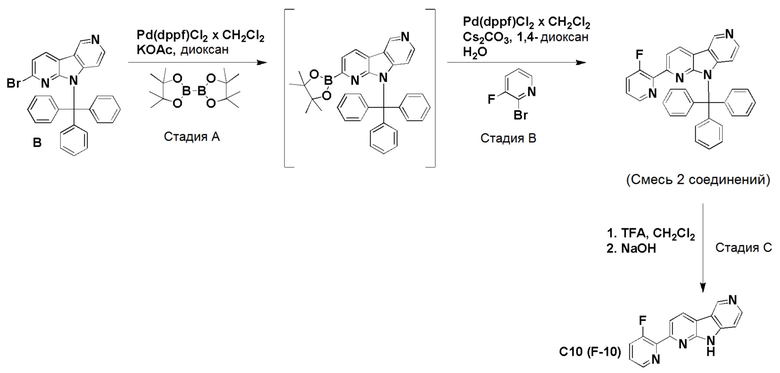
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной банев течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-3-фторпиридин (0,086 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочного продукта (0,0586 г).
Стадия В
Смесь указанного в заголовке соединения и побочного продукта со стадии B выше (0,0586 г), растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (1,8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь разбавляли разведенным дихлорметаном (50 мл) и водой (20 мл). рН водной фазы доводили до рН ~12 добавлением 1 М водного раствора гидроксида натрия. Водный слой отделяли, экстрагировали дихлорметаном (25 мл), объединенный органический слой высушивали над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (HP-SIL 10 г) с использованием системы Biotage Isolera, применяя градиент смеси дихлорметан/метанол (100/0 → 95/5 → 90/10), с получением сравнительного примера C10 (F-10) в виде не совсем белого твердого вещества (0,0067 г, 5,7% с 3 стадий).
1H-ЯМР (400 МГц, ДМСО-d6) δ=12,47 (шир.с, 1H), 9,45 (с, 1H), 8,80 (д, 1H), 8,63-8,61 (м, 1H), 8,55-8,53 (м, 1H), 8,00 (д, 1H), 7,94-7,88 (м, 1H), 7,63-7,58 (м, 1H), 7,52 (д, 1H).
МС (ESI): m/z=264,84 [М+Н]+.
Сравнительный пример 10 (F-10) предшественник (ACI-2941)
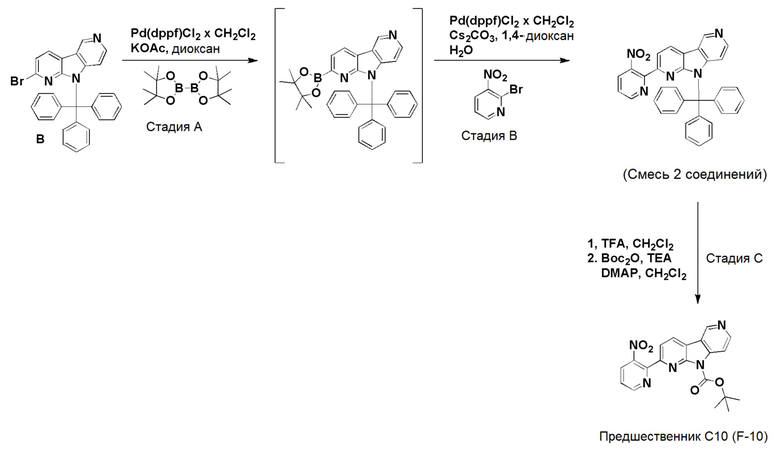
Стадия А
К смеси дегазированного 1,4-диоксана (8 мл) в пробирке для микроволнового реактора добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), с последующим добавлением указанного в заголовке соединения из препаративного примера B (0,2 г, 0,4 ммоль), бис(пинаколато)диборана (0,112 г, 0,44 ммоль) и ацетата калия (0,118 г, 1,2 ммоль). Затем реакционную смесь нагревали при ~95°С на песочной бане в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме с получением неочищенного указанного в заголовке соединения, которое непосредственно использовали на следующей стадии.
Стадия В
Неочищенное указанное в заголовке соединение со стадии A выше, растворяли в смеси дегазированного 1,4-диоксана (8,6 мл) и воды (2 мл) в пробирке для микроволнового реактора. Затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), комплекс с дихлорметаном (0,034 г, 0,04 ммоль), 2-бром-3-нитропиридин (0,1 г, 0,49 ммоль) и карбонат цезия (0,266 г, 0,82 ммоль) и реакционную смесь нагревали при ~115°С на песочной бане в течение 6 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (30 мл), органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители выпаривали в вакууме. Темный остаток очищали хроматографией на силикагеле (puriFlash 25 г, Interchim) с использованием системы Biotage Isolera, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением смеси указанного в заголовке соединения и побочного продукта (0,0538 г).
Стадия С
Смесь указанного в заголовке соединения со стадии B выше и побочного продукта (0,0538 г) растворяли в дихлорметане (4 мл) и добавляли трифторуксусную кислоту (2,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и затем добавляли метанол (10 мл). Растворители выпаривали в вакууме и остаток суспендировали в метаноле (10 мл). Растворители снова выпаривали в вакууме и остаток суспендировали в дихлорметане (4 мл). После добавления триэтиламина (2 мл, 14,4 ммоль), ди-трет-бутилдикарбоната (0,2 г, 0,86 ммоль) и 4-(диметиламино)пиридина (0,0036 г, 0,028 ммоль) реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (40 мл). Органическую фазу отделяли, высушивали над Na2SO4, фильтровали и растворители удаляли в вакууме. Остаток очищали на силикагеле (puriFlash 25 г, Interchim) с использованием системы очистки Biotage Isolera One, применяя градиент смеси этилацетат/н-гептан (5/95 → 100/0 → 100/0), с получением предшественника сравнительного примера C10 (F-10) в виде бледно-желтого твердого вещества (0,0194 г, 12,1% с 3 стадий).
1H-ЯМР (400 МГц, CDCl3) δ=9,35 (д, 1H), 8,90 (д, 1H), 8,73 (д, 1H), 8,58 (д, 1H), 8,24-8,17 (м, 3H), 7,57- 7,53 (м, 1Н), 1,73 (с, 9Н).
МС (ESI): m/z=391,92 [МН+], 291,90 [МН+-Boc].
Синтез 18F-меченных соединений
Общий способ 18F-фторирования A (прямое ароматическое 18F-фторирование)
[18F]фторид без добавления носителя (2-5 ГБк) улавливали на картридже для твердофазной экстракции Sep-Pak Accell Plus QMA light (Waters) и элюировали раствором K2CO3/Kryptofix® 2.2.2. Воду удаляли с использованием потока N2 при 120°С и выпаривали досуха с MeCN (3 × 1 мл). После этого раствор растворенного предшественника добавляли к высушенному комплексу K[18F]F-K222. Реакционный флакон закрывали и нагревали при обычном нагревании в течение 15 мин при 130°С. Затем реакционную смесь гасили водой и неочищенный продукт очищали с помощью полупрепаративной ВЭЖХ. Выделенный индикатор разводили водой (35 мл), улавливали на картридже C-18 Plus (Waters), промывали водой (5 мл), элюировали этанолом (1 мл) и формулировали в физиологическом растворе.
Общий способ 18F-фторирования В (прямое 18F-фторирование плюс снятие защиты)
Индикаторы синтезировали, начиная с [18F]фторида без добавления носителя (1-10 ГБк) прямым 18F-фторированием. Водный раствор [18F]фторида улавливали на картридже для твердофазной экстракции Sep-Pak Accell Plus QMA light (Waters) и элюировали раствором K2CO3/Kryptofix® 2.2.2. Воду удаляли с использованием потока N2 при 120°С и выпаривали с MeCN (3 × 1 мл) досуха. Затем соответствующий растворенный предшественник добавляли к высушенному комплексу K[18F]F-K222. Реакционный флакон закрывали и нагревали в течение 15 мин при 120-160°С (в нагревательном блоке). Для снятия защиты добавляли соляную кислоту, и смесь перемешивали в течение еще 10 мин при 110°С. После нейтрализации с использованием раствора гидроксида натрия, реакционную смесь гасили буфером на основе формиата аммония и улавливали на картридже C-18 Plus (Waters). Картридж промывали водой (5 мл), элюировали ацетонитрилом и затем неочищенный продукт очищали с помощью полупрепаративной ВЭЖХ. Выделенный индикатор разводили водой (25 мл), улавливали на картридже C-18 Plus (Waters), промывали водой (5 мл), элюировали этанолом (1 мл) и формулировали в физиологическом растворе.
Сравнительный пример 18F-1
18F-1 (680 МБк) синтезировали в соответствии с общим способом 18F-фторирования A, используя соответствующую молекулу нитропредшественника (M. Timothy et al., J. Labeled Comp. Radiopharm. (2013), 56 (14), 736-740) (2,8 мг, 7,1 мкмоль) в диметилсульфоксиде (0,6 мл).
Радиохимическую чистоту на уровне 100% определяли аналитической обращенно-фазовой ВЭЖХ (tR (RAD-индикатор)=3,19 мин). Идентичность 18F-1 подтверждали сравнением времени удерживания с нерадиоактивным стандартом F-1.
Сравнительный пример 18F-2
18F-2 (680 МБк) синтезировали в соответствии с общим способом 18F-фторирования A, используя предшественник сравнительного примера 2 (F-2) (WO 2015/052105) (3,4 мг, 8,7 мкмоль) в диметилсульфоксиде (0,6 мл). Радиохимическую чистоту на уровне 98% определяли аналитической обращенно-фазовой ВЭЖХ (tR (RAD-индикатор)=3,27 мин). Идентичность 18F-2 подтверждали сравнением времени удерживания с нерадиоактивным стандартом F-2.
Пример 18F-3а[18F]PI-2620
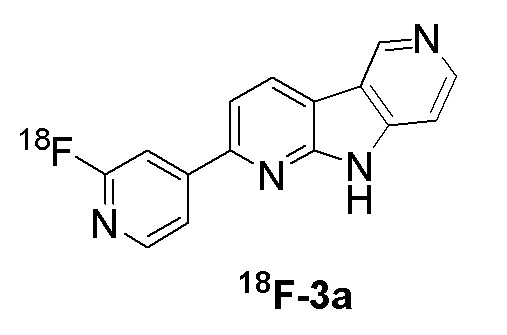
18F-3а (450 МБк) синтезировали в соответствии с общим способом 18F-фторирования В, используя молекулу предшественника соединения 13 (2,6 мг, 4,8 мкмоль) в диметилсульфоксиде (0,6 мл). Радиохимическую чистоту на уровне 100% определяли аналитической обращенно-фазовой ВЭЖХ (tR (RAD-индикатор)=3,31 мин). Идентичность 18F-3а подтверждали сравнением времени удерживания с нерадиоактивным стандартом F-3а.
Радиоактивно меченный пример 18F-3b и сравнительные примеры 18F-5, 18F-6, 18F-7, 18F-8, 18F-9, 18F-10 синтезировали в соответствии со способом B, исходя из соответствующих молекул-предшественников, как описано выше.
Определение связывания в гомогенатах головного мозга субъектов с AD и здоровых контролей
20 мкг гомогената головного мозга человека с болезнью Альцгеймера инкубировали с серией разведений каждого испытуемого соединения (от 1000 до 0,06 нМ) в присутствии 800 Бк 18F-меченного соединения, связывающегося с белком тау. Образцы встряхивали при 110 об/мин в течение 45 мин при 37°С. Затем образцы фильтровали через 96-луночные фильтрационные планшеты GF/B и дважды промывали 300 мкл аналитического буфера (PBS, содержащий 0,1% BSA и 2% ДМСО). Затем фильтрационные планшеты герметично закрывали и сверху помещали пленку для визуализации для планшетов Fuji (BAS-SR2025). Планшет для визуализации анализировали после экспозиции в течение ночи с использованием сканера Fuji Film BAS-5000. Неспецифический сигнал определяли с образцами, содержащими 18F-меченное стандартное соединение, связывающее белок тау, в присутствии аналитического буфера без субстрата головного мозга и конкурента. Специфическое связывание рассчитывали вычитанием неспецифического сигнала из сигнала анализированных образцов. Деблокированный 18F-меченный сигнал тау-связывающего соединения определяли как полное связывание. Значения IC50 рассчитывали с помощью Prism V6 (GraphPad) с установлением общего связывания на 100%.
Результаты:
Высокая аффинность к белку тау соединений F-1, F-2 и F-3a была установлена в конкурентном анализе с использованием гомогената головного мозга человека с AD. Значения IC50 для связывания белка тау на уровне <2 нМ были установлены для всех соединений.
Высокое отношение сигнал/шум между гомогенатом головного мозга субъектов с AD и гомогенатом головного мозга здоровых контролей получали с соединением 18F-3a с отношением, равным 6,7. Низкое отношение сигнал/шум между гомогенатом головного мозга субъектов с AD и гомогенатом головного мозга здоровых контролей, равное 1,3, было получено для соединения 18F-1.
Дополнительные данные были получены с использованием других тканей головного мозга человека.
Отношение сигнал/шум между гомогенатом головного мозга субъектов с AD и гомогенатом головного мозга здоровых контролей для соединения 18F-3a было определено как равное 14,0; 17,9 и 33,8 соответственно.
Достоверно более низкие значения были получены для соединения 18F-1, где эти отношения составляли только 1,7; 1,8 и 2,5.
Также соединение 18F-2 показало достоверно более низкие отношения между сигналом в гомогенатах головного мозга субъектов с AD и гомогенатах головного мозга здоровых контролей (3,3; 4,5; 6,9).
Авторадиография на срезах головного мозга человека
С использованием авторадиографии исследовали замороженные срезы человеческого головного мозга толщиной 18 мкм и срезы человеческого FFPE толщиной 6 мкм. Срезы головного мозга уравновешивали, по меньшей мере, в течение 1 ч в растворе 1×PBS перед использованием в эксперименте. Каждый срез мозга покрывали раствором 18F-меченного индикатора (200 Бк/мкл, 500 мкл) в 1×PBS. Для экспериментов с блокированием с соответствующим 19F-соединением избыток блокирующего соединения (10 мкМ) смешивали с 18F-соединением. Срезы головного мозга инкубировали с раствором индикатора в течение 1 ч при комнатной температуре, затем осушали и помещали в держатель для микроскопических препаратов. Затем микроскопические препараты промывали последовательно 1 × PBS в течение 1 мин; 70% EtOH в 1 × PBS в течение 2 мин; 30% EtOH в 1 × PBS в течение 2 мин; и 1 × PBS в течение 1 мин. Микроскопические препараты оставляли сушиться на воздухе в течение 30 мин перед тем, как поместить их в планшеты для визуализации Fuji для экспозиции в течение ночи. Планшеты для визуализации сканировали и измеряли сигнал с использованием программного обеспечения Fuji для получения авторадиографического изображения среза мозга.
Результаты:
Соединение 18F-3a тестировали в опытах с авторадиографией с использованием срезов головного мозга человека (с AD, PSP, PiD, HC). Используя срез головного мозга субъекта с AD, можно было обнаружить сильное точечное окрашивание, которое можно было блокировать добавлением избытка соответствующего немеченого соединения. В срезах здорового контроля (HC) специфический сигнал отсутствовал (фиг.1). Аналогичные результаты были получены для соединения 18F-3a на срезах головного мозга субъектов с PSP и PiD.
Определение аффинности связывания с бета-амилоидом в гомогенате головного мозга человека с AD
20 мкг гомогената головного мозга человека с болезнью Альцгеймера инкубировали с серией разведений каждого испытуемого соединения (от 1000 до 0,06 нМ) в присутствии 800 Бк 18F-меченного соединения, связывающегося с бета-амилоидом. Образцы встряхивали при 110 об/мин в течение 45 мин при 37°С. Затем образцы фильтровали через 96-луночные фильтрационные планшеты GF/B и дважды промывали 300 мкл аналитического буфера (PBS, содержащий 0,1% BSA и 2% ДМСО). Затем фильтрационные планшеты герметично закрывали и сверху помещали пленку для визуализации для планшетов Fuji (BAS-SR2025). Планшет для визуализации анализировали после экспозиции в течение ночи с использованием сканера Fuji Film BAS-5000. Неспецифический сигнал определяли с образцами, содержащими 18F-меченное соединение, связывающееся с бета-амилоидом, в присутствии аналитического буфера без субстрата головного мозга и конкурента. Специфическое связывание рассчитывали вычитанием неспецифического сигнала из сигнала анализированных образцов. Деблокированный 18F-меченный сигнал бета-амилоид-связывающего соединения определяли как полное связывание. Значения IC50 рассчитывали с помощью Prism V6 (GraphPad) с установлением общего связывания на 100%.
Результаты:
Низкая аффинность соединений F-1, F-2 и F-3a к бета-амилоиду было установлена в конкурентном анализе с использованием гомогената мозга человека с AD. Значения IC50 для связывания бета-амилоида на уровне <1 нМ были установлены для всех соединений.
Определение аффинности связывания с MAO A в гомогенате головного мозга HC
20 мкг гомогената головного мозга человека (без патологии AD) инкубировали с серией разведений каждого испытуемого соединения (от 1000 до 0,06 нМ) в присутствии 800 Бк 18F-меченного соединения, связывающегося с MAO-A. Образцы встряхивали при 110 об/мин в течение 45 мин при 37°С. Затем образцы фильтровали через 96-луночные фильтрационные планшеты GF/B и дважды промывали 300 мкл аналитического буфера (PBS, содержащий 0,1% BSA и 2% ДМСО). Затем фильтрационные планшеты герметично закрывали и сверху помещали пленку для визуализации для планшетов Fuji (BAS-SR2025). Планшет для визуализации анализировали после экспозиции в течение ночи с использованием сканера Fuji Film BAS-5000. Неспецифический сигнал определяли с образцами, содержащими 18F-меченный FEH в присутствии аналитического буфера без субстрата головного мозга и конкурента. Специфическое связывание рассчитывали вычитанием неспецифического сигнала из сигнала анализированных образцов. Деблокированный 18F-меченный сигнал FEH определяли как полное связывание. Значения IC50 рассчитывали с помощью Prism V6 (GraphPad) с установлением общего связывания на 100%.
Результаты:
В гомогенате головного мозга мышей соединение F-1 продемонстрировало высокую немишеневую аффинность к MAO A, равную 22 нМ, в конкурентном анализе 18F-FEH. Аффинность соединения F-2 была ниже и составляла 475 нМ, тогда как немишеневая аффинность к MAO A для соединения F-3a была еще ниже со значениями IC50 1400 нМ. При использовании гомогената мозга контрольного человека (здорового контроля) соединение F-1 показало высокую немишеневую аффинность к MAO A, равную 5 нМ, в конкурентном анализе FEH. Аффинность соединения F-2 была ниже и составляла 100 нМ, в то время как немишеневая аффинность к MAO A для соединения F-3a была еще ниже со значениями IC50 1100 нМ и 530 нМ соответственно.
Определение аффинности связывания с MAO В в гомогенате головного мозга HC
20 мкг гомогената головного мозга человека (без патологии AD) инкубировали с серией разведений каждого испытуемого соединения (от 1000 до 0,06 нМ) в присутствии 800 Бк 18F-меченного соединения, связывающегося с MAO-В. Образцы встряхивали при 110 об/мин в течение 45 мин при 37°С. Затем образцы фильтровали через 96-луночные фильтрационные планшеты GF/B и дважды промывали 300 мкл аналитического буфера (PBS, содержащий 0,1% BSA и 2% ДМСО). Затем фильтрационные планшеты герметично закрывали и сверху помещали пленку для визуализации для планшетов Fuji (BAS-SR2025). Планшет для визуализации анализировали после экспозиции в течение ночи с использованием сканера Fuji Film BAS-5000. Неспецифический сигнал определяли с образцами, содержащими 18F-меченный фтордепренил в присутствии аналитического буфера без субстрата головного мозга и конкурента. Специфическое связывание рассчитывали вычитанием неспецифического сигнала из сигнала анализированных образцов. Деблокированный 18F-меченный фтордепрениловый сигнал определяли как полное связывание. Значения IC50 рассчитывали с помощью Prism V6 (GraphPad) с установлением общего связывания на 100%.
Результаты:
В гомогенате мозга человека HC соединение F-1 продемонстрировало высокую немишеневую аффинность к МАО B, равную 170 нМ, в конкурентном анализе с 18F-меченным фтордепренилом. Аффинность соединения F-3 уменьшалась до значений > 1000 нМ.
Фармакокинетические исследования на здоровых мышах
Мышам NMRI (с массой тела в пределах 25-35 г) внутривенно вводили 18F-меченные соединения. Вводили 150 мкл 1×PBS раствора с 10%-15% EtOH или средой для разведения (57% воды для инъекций, 18% полиэтиленгликоля 400, 15% этанола, 10% воды), содержащей 18F-меченное соединение (2-10 МБк). Анестезию изофлураном проводили перед введением индикатора и поддерживали в течение периода получения изображения. ПЭТ-сканирование проводили с использованием ПЭТ/КТ-сканера для мелких животных SIEMENS INVEON (Siemens, Knoxville, TN). Получение данных ПЭТ начали непосредственно перед тем, как радиоактивную дозу вводили животному через хвостовую вену. Изображения получали в виде динамического сканирования в течение 60 мин.
Результаты:
Соединение 18F-1: максимальное поглощение: 5,3% ID/г, соотношение максимальное поглощения/30 мин: 6,8, удерживание в головном мозге на 60 мин: 0,8% ID/г, поглощение в костной ткани плечевого сустава на 60 мин: 4,0% ID/г.
Соединение 18F-2: максимальное поглощение: 5,7% ID/г, соотношение максимальное поглощения/30 мин: 10,9, удерживание в головном мозге на 60 мин: 0,6% ID/г, поглощение в костной ткани плечевого сустава на 60 мин: 6,2% ID/г.
Соединение 18F-3a: максимальное поглощение: 4,4% ID/г, соотношение максимальное поглощения/30 мин: 11,2, удерживание в головном мозге на 60 мин: 0,3% ID/г, поглощение в костной ткани плечевого сустава на 60 мин: не детектировали.
Максимальное поглощения в головном мозге было установлено на 100%, и были получены кривые вымывания для оценки клиренса активности из нормального мозга (фиг. 2).
Исследование с визуализацией человека
В клиническом испытании субъекты с AD или PSP, а также здоровые контроли без деменции (NDC) подвергались динамической ПЭТ-визуализации в течение более 3 ч после введения соединения 18F-3a 370 МБк болюсной инъекцией.
Результаты:
Первоначальные данные визуализации показывают сильное поглощение в головном мозге и быстрое вымывание из немишеневых областей. У NDC не наблюдали повышенного поглощения в сосудистом сплетении, базальных ганглиях, полосатом теле, мозжечковой миндалине, мозговых оболочках или других областях, отмеченных для других тау-агентов (фиг. 4а). 18F-3a показывает хорошее поглощение в головном мозге и быстрое вымывание из немишеневых областей головного мозга (см. фиг.3). При AD имело место очаговое асимметричное поглощение в височных, теменных и лобных долях (фиг. 4b). Наконец, у субъектов с PSP было очаговое повышенное поглощение в бледном шаре и черной субстанции (фиг. 5а и 5b).
Обобщенные данные по доклиническим характеристикам
(IC50 в гомогенате головного мозга человека с AD) b)
(< 2 нМ)a)
(< 2 нМ)a)
(< 2 нМ)a)
(определено авторадиографией на срезах головного мозга человека)c)
(IC50 в гомогенате головного мозга человека с AD)b)
> 1 мкМ
> 1 мкМ
> 1 мкМ
(IC50 в гомогенате головного мозга мыши)b)
(22 нМ)a)
(475 нМ)a)
(1400 нМ)a)
(IC50 в гомогенате HC)b)
(5 нМ)a)
(100 нМ)a)
(1100 нМ)a)
(IC50 в гомогенате HC)b
(170 нМ)
(> 1000 нМ)
(> 1000 нМ)
(отношение сигнал индикатора в гомогенате головного мозга человека с AD/сигнал в гомогенате головного мозга HC)c)
1,3a)
6,7a)
(дополнительные гомогенаты)
(отношение сигнал индикатора в 3 различных гомогенатах головного мозга человека с AD/сигнал в гомогенатах головного мозга HC)c)
1,7, 1,8, 2,5a)
3,3, 4,5, 6,9 a)
14,0, 17,9 33,8a)
(отношение сигнал индикатора в гомогенате головного мозга человека с AD/сигнал в гомогенате головного мозга мыши)c)
2,2a)
15,0a)
9,2a
(поглощение индикатора у здоровых мышей после в/в инъекции)c)
(5,3%ID/g)a)
(5,7%ID/g)a)
(4,4%ID/g)a)
(отношение поглощение индикатора в пике и на 30 мин у здоровых мышей)c)
(6,8)a)
(10,9)a)
(11,2)a)
(сигнал индикатора у здоровых мышей на 60 мин после в/в инъекции)c)
(0,8%ID/g)a)
(0,6%ID/g)a)
(0,3%ID/g)a)
(поглощение индикатора в костной ткани у здоровых мышей на 60 мин после в/в инъекции)c)
(4,0%ID/g) a)
(6,2%ID/g) a)
(< 0,5% ID/g)a), f)
- низкая, ○ средняя, + хорошая, ++ очень хорошая, +++ превосходная
а) собственные данные, см. экспериментальный раздел выше;
b) определено с нерадиоактивными фтор-19 производными F-1, F-2 и F-3a;
c) определено с радиоактивными фтор-18 производными 18F-1, 18F-2 и 18F-3a;
d) Marquie et al. 2015;
е) WO2015/052105;
f) дефторирование не обнаружено;
h) Honer et al., Human Amyloid Imaging Meeting 2017;
NA: отсутствует.
Как следует из таблицы 1, соединения 18F-1 и 18F-2 предшествующего уровня техники имеют ограничения, в частности, за счет:
низкого связывания с изоформами белка тау при тауопатиях, отличных от AD;
аффинности к МАО А и, следовательно, низкой селективности для белка тау;
отсутствия низкого сигнала в головном мозге здорового субъекта;
отсутствия быстрого вымывания из головного мозга здорового субъекта;
длительного удерживания в головном мозге здорового субъекта, и/или
дефторирования in vivo.
С другой стороны, соединение 18F-3a показывает:
специфическое связывание со срезами головного мозга человека с AD и тауопатиями, отличными от AD (примеры: сильный сигнал для PSP и PiD в отличие от сообщений для соединений 18F-1 и 18F-2);
более низкую аффинность к МАО А в цельном гомогенате головного мозга мыши (в 64 раза более высокая IC50, чем у соединения 18F-1, и в 3 раза более высокая IC50, чем у соединения 18F-2);
более низкую аффинность к МАО А в гомогенате головного мозга НС (в 220 раз более высокая IC50, чем у соединения 18F-1, и в 11 раз более высокая IC50, чем у соединения 18F-2);
более низкую аффинность к МАО В в гомогенате мозга HC (IC50> в 5 раз выше, чем у соединения 18F-1),
более высокое отношение сигнал/шум, определенное связыванием в гомогенате головного мозга человека с AD по сравнению с гомогенатом головного мозга HC (отношение в 5,2 раза выше, чем у соединения 18F-1);
более высокое отношение сигнал/шум, определенное связыванием в дополнительных гомогенатах головного мозга субъектов с AD и гомогенатом головного мозга HC (отношение в 8,2-13,5 раз выше, чем у соединения 18F-1, в 4,0-4,9 раз выше, чем у соединения 18F-2),
более высокое отношение сигнал/шум, определенное связыванием в гомогенате головного мозга субъекта с AD по сравнению с гомогенатом цельного мозга мыши (отношение в 4,2 раза выше, чем у соединения 18F-1);
более быстрое вымывание из здорового головного мозга (в 1,6 раза быстрее, чем у соединения 18F-1);
более низкое долгосрочное удерживание в головном мозге у здоровых мышей (в 2,7 раза ниже, чем у соединения 18F-1, и в 2 раза ниже, чем у соединения 18F-2);
отсутствие дефторирования у мышей (отсутствие поглощения в костной ткани в отличие от 4,0% ID/г для соединения 18F-1 и 6,2% ID/г для соединения 18F-2).
По меньшей мере, за счет его высокой аффинности к белку тау, более быстрого вымывания из головного мозга, более низкого долгосрочного удерживания в здоровом мозге и/или более низкой аффинности связывания с другими мишенями в головном мозге соединение 18F-3a обладает достоверно лучшими свойствами для определения и количественного определения отложений белка тау в головном мозге позитронно-эмиссионной томографией, чем соединения предшествующего уровня техники 18F-1 и 18F-2. В дополнение к выявлению и количественному определению отложений белка тау при AD, соединение 18F-3a может быть пригодным для клинической оценки тауопатий, отличных от AD.
Благоприятные доклинические характеристики 18F-3a были подтверждены у людей. 18F-3a показывает хорошее поглощение в головном мозге и быстрое вымывание из нецелевых областей головного мозга (см. фиг.3).
Характер поглощения, наблюдаемый у пациентов с AD и PSP, соответствовал предполагаемому характеру таупатологии (фиг. 4 и фиг. 5).
Удивительно, но 3a/18F-3a демонстрируют значительные преимущества по сравнению с его региоизомерами в отношении ключевых характеристик индикатора для ПЭТ-визуализации белка тау (таблица 2).
Аффинность связывания для тау была низкой для сравнительных примеров 6/18F-6 и 10/18F-10 и ниже для сравнительных примеров 5/18F-5, 7/18F-7 и 9/18F-9 (IC50 определена в гомогенате головного мозга человека с AD).
Низкая селективность к МАО А была установлена для сравнительных примеров 2/18F-2, 7/18F-7 и 8/18F.
Мечение радиоактивной меткой сравнительных примеров 18F-5, 18F-6, 18F-8, 18F-9 и 18F-10 было ниже (или не удавалось) в стандартных условиях.
Низкое поглощение в головном мозге мышей было обнаружено для сравнительного примера 18F-10.
Вымывание в головном мозге здоровых мышей было низким для сравнительных примеров 18F-5, 18F-7 и 18F-10.
Дефторирование у мышей было обнаружено для сравнительных примеров 18F-2, 18F-5, 18F-7 и 18F-10.
Сравнение региоизомеров
тау
[нМ]a,b
MAO A [нМ]a,b,d
(ACI-2620)(ACI-2620)
(< 5 нМ)
(> 1000 нМ)
(4,4-5,9%ID/г)
(11,2-16,6)
(< 0,5% ID/г) f
(ACI-2698)(ACI-2698)
(< 5 нМ)
(765 нМ)
(4,0%ID/г)
(19,4)
(ACI-2448) (ACI-2448)
(< 5 нМ)
(475 нМ)
(5,7%ID/г)
(10,9)
(6,2%ID/г)
(ACI-2632) (ACI-2632)
(6,3 нМ)
(> 1000 нМ)
(5,3%ID/г)
(9,6)
(2,9%ID/г)
(ACI-2843) (ACI-2843)
(43 нМ)
(> 1000 нМ)
(8,2%ID/г)
(14,5)
(4,8%ID/г)
(ACI-2731) (ACI-2731)
(7,1 нМ)
(268 нМ))
(8,5%ID/г)
(7,0)
(11,1%ID/г)
(ACI-2876) (ACI-2876)
(< 5 нМ)
(75 нМ)
(ACI-2930) (ACI-2930)
(8,4 нМ)
(> 1000 нМ)
(ACI-2931) (ACI-2931)
(103 нМ)
(> 1000 нМ)
(2,1%ID/г)
(3,6)
(6,2%ID/г)
- низкая, ○ средняя, + хорошая, ++ очень хорошая, +++ превосходная
а) собственные данные, см. экспериментальный раздел выше;
b) определено с нерадиоактивными фтор-19 производными F-1, F-2 и F-3a;
c) определено с радиоактивными фтор-18 производными 18F-1, 18F-2 и 18F-3a;
d) IC50 в гомогенатах головного мозга мыши;
f) дефторирование не обнаружено;
NA: отсутствует.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ДИАГНОСТИКИ И ЛЕЧЕНИЯ | 2017 |
|
RU2766139C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДНЫМИ ИЛИ АМИЛОИДОПОДОБНЫМИ БЕЛКАМИ | 2011 |
|
RU2603008C2 |
| ПЕНТАЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2754557C1 |
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2788916C2 |
| ПИРРОЛО[2,3-С]ПИРИДИНЫ В КАЧЕСТВЕ ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ ДЛЯ НЕЙРОФИБРИЛЛЯРНЫХ КЛУБКОВ | 2015 |
|
RU2695373C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА, ВИЗУАЛИЗИРУЮЩИЙ БЕЛОК ТАУ | 2011 |
|
RU2627694C2 |
| ИМИДАЗО[1,2-А]ПИРИДИН-7-АМИНЫ В КАЧЕСТВЕ СРЕДСТВ ВИЗУАЛИЗАЦИИ | 2014 |
|
RU2671506C2 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВОМ СВЯЗЫВАТЬ АМИЛОИД, И СПОСОБ ДЕТЕКЦИИ ОТЛОЖЕНИЙ АМИЛОИДА У МЛЕКОПИТАЮЩЕГО | 2004 |
|
RU2440995C2 |
| ПРОНИЦАЕМЫЕ ИНГИБИТОРЫ ГЛИКОЗИДАЗЫ И ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2707292C2 |
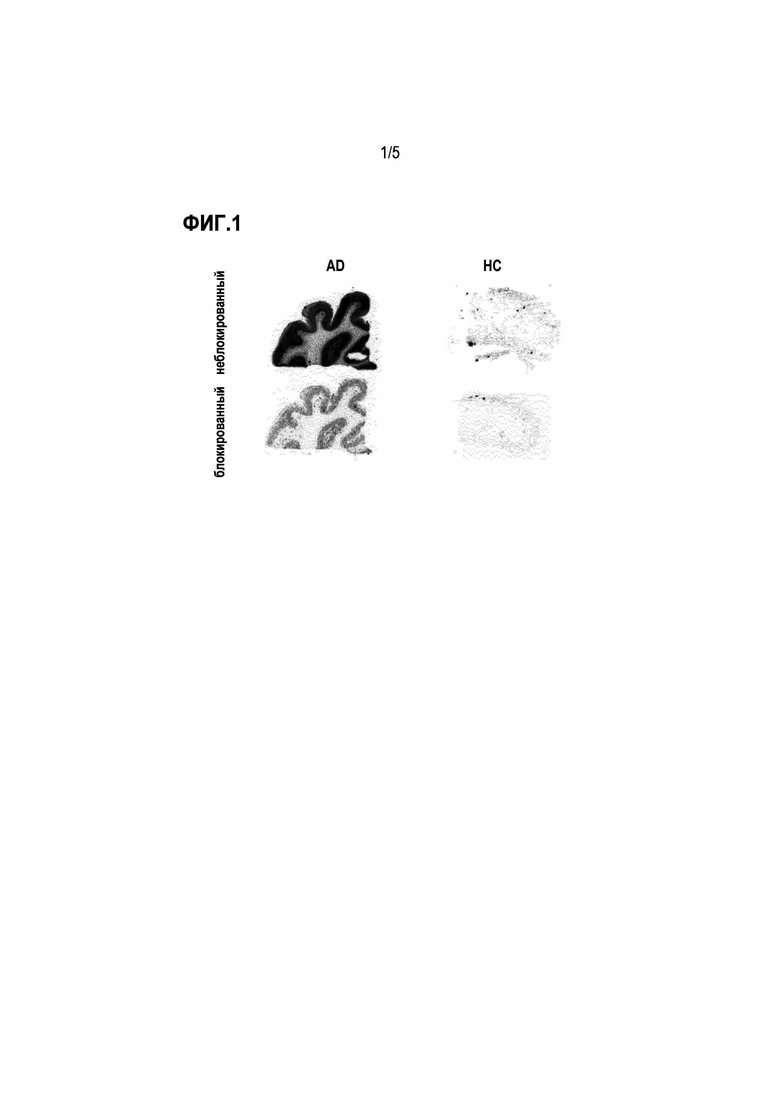
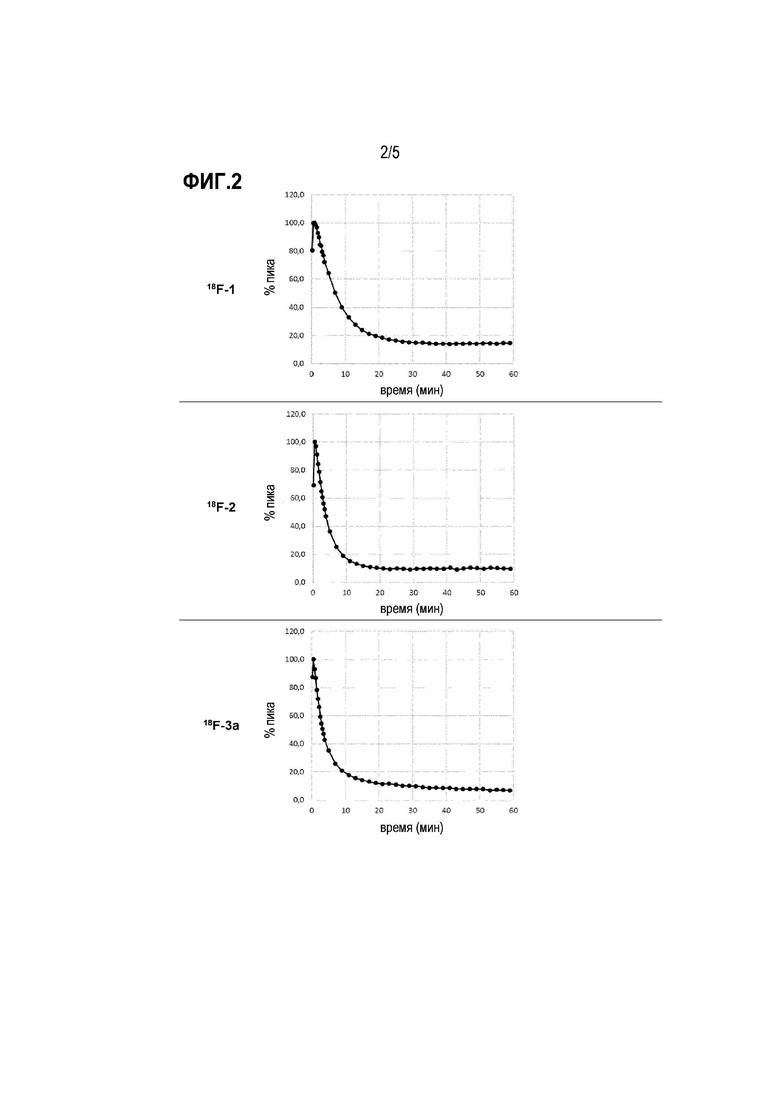
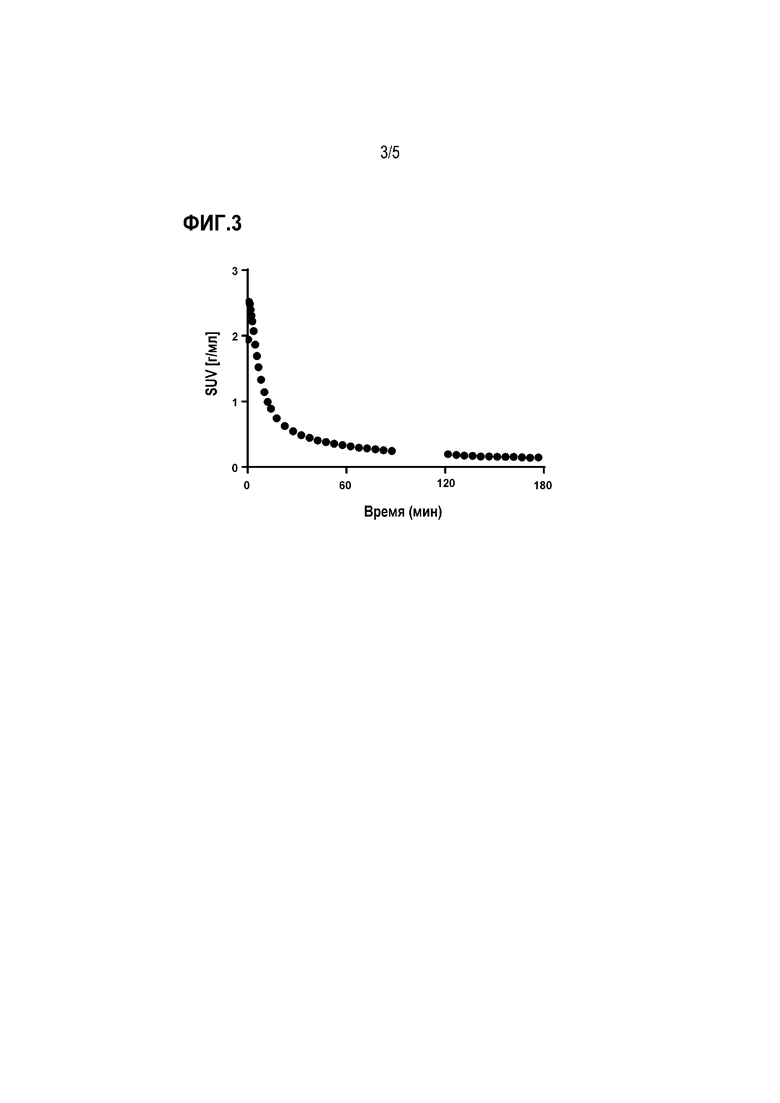
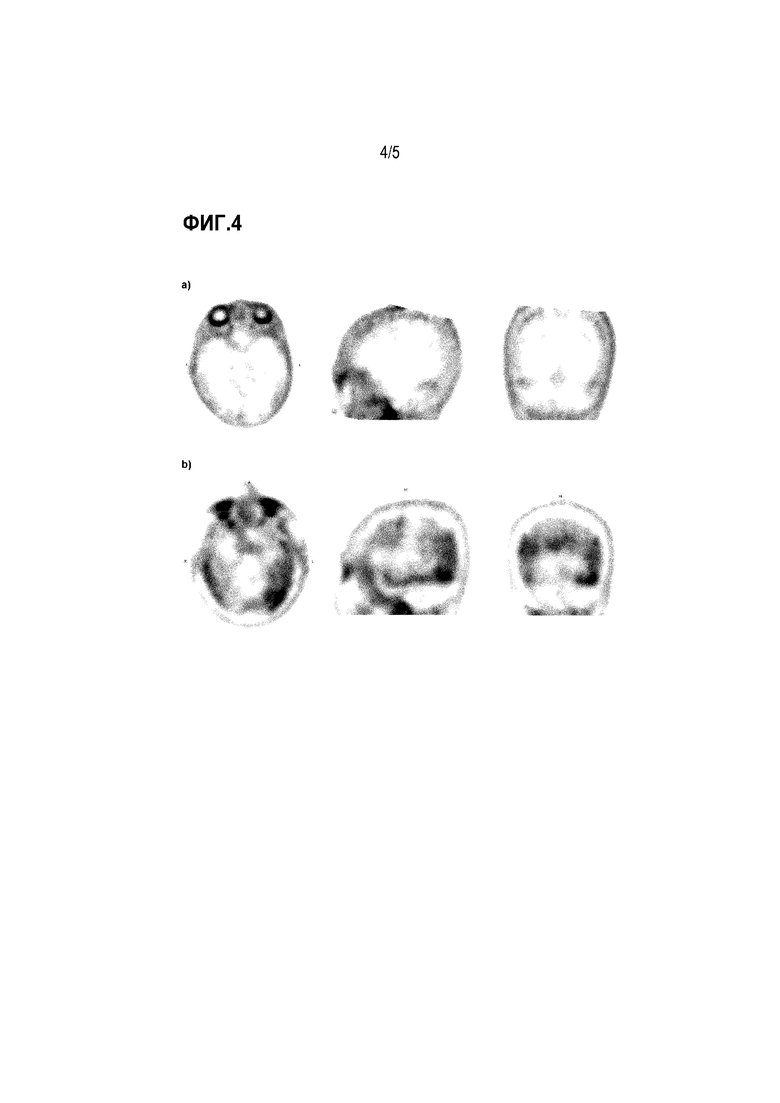
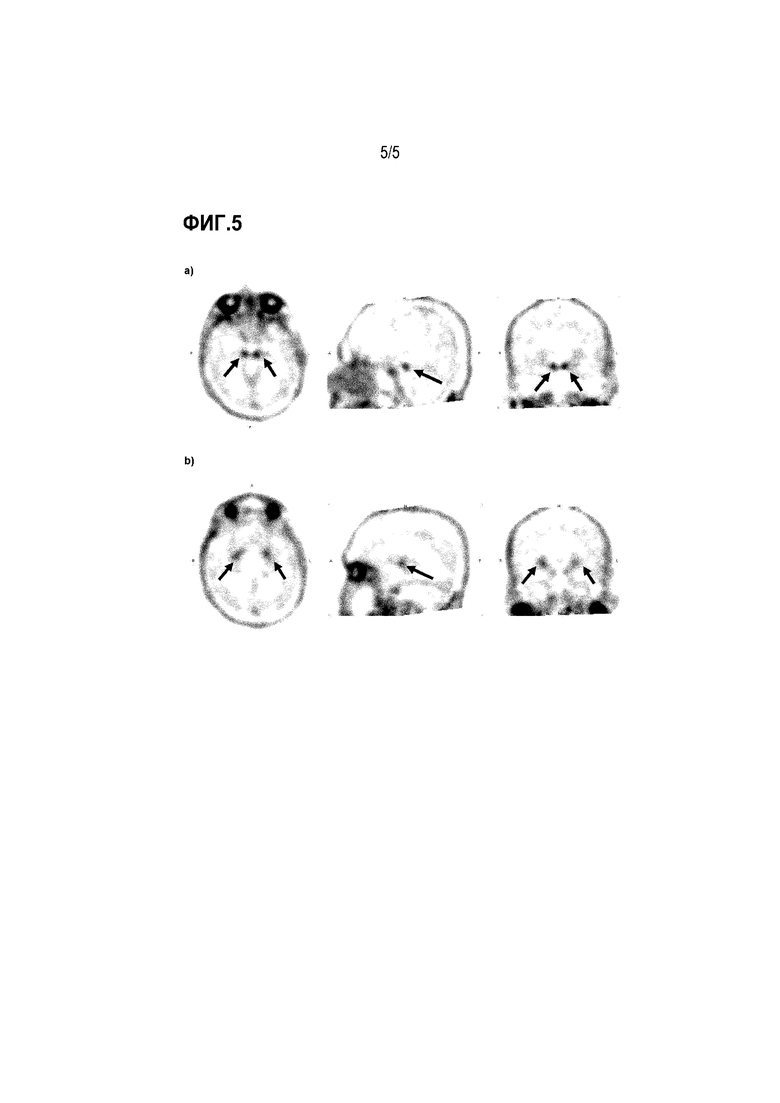
Изобретение относится к соединению формулы (II), где R1 представляет 18F; R2 представляет Н. Также изобретение относится к промежуточному соединению формулы (II), значения радикалов которые указаны в формуле изобретения, применению немеченного соединения формулы (II) в качестве аналитического стандарта, а также к способу получения меченого соединения формулы (II), набору для получения радиофармацевтического препарата, способу сбора данных для диагностики расстройства, ассоциированного с агрегатами белка тау, для определения предрасположенности к расстройству, ассоциированному с агрегатами белка тау, для мониторинга остаточного проявления расстройства у пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, и для прогнозирования реактивности пациента, страдающего расстройством, ассоциированным с агрегатами белка тау. Технический результат: получены гетероциклические соединения, которые можно использовать в селективном выявлении расстройств и аномалий, ассоциированных с агрегатами белка тау, таких как болезнь Альцгеймера и другие тауопатии, с использованием позитронно-эмиссионной томографии (ПЭТ). 10 н. и 11 з.п. ф-лы, 5 ил., 2 табл., 34 пр.
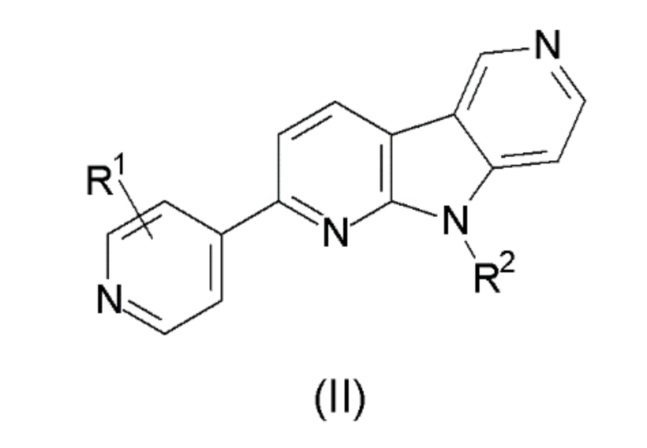
1. Соединение формулы (II)
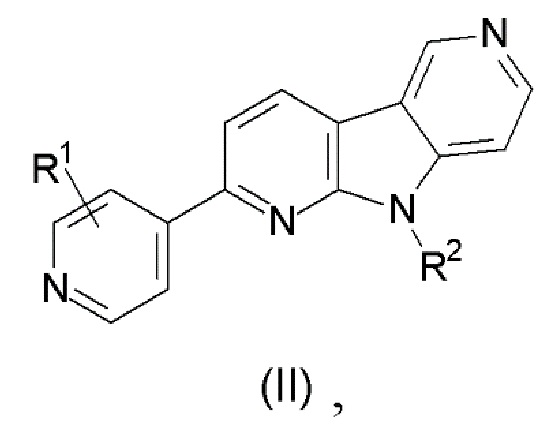
где R1 представляет 18F;
R2 представляет Н.
2. Соединение по п. 1, которое представляет
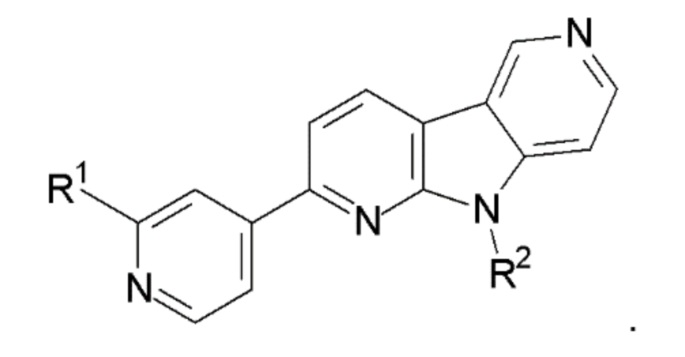
3. Соединение по п. 1, которое представляет
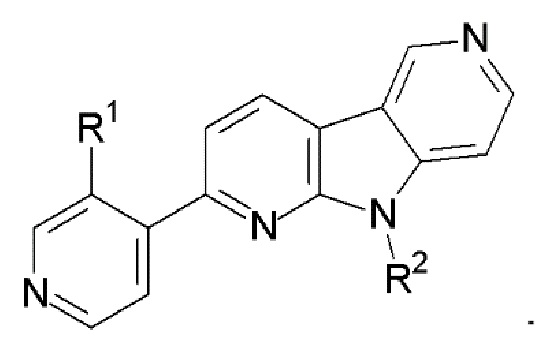
4. Соединение формулы (II)
где R1 представляет F;
R2 представляет Н.
5. Соединение по п. 4, которое представляет
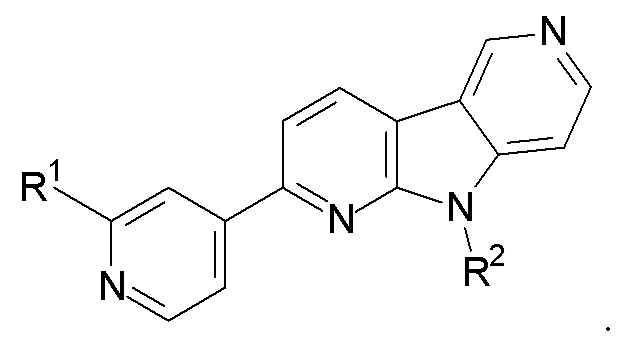
6. Соединение по п. 4, которое представляет
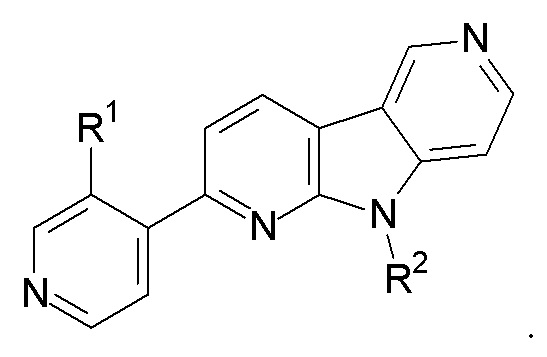
7. Соединение формулы (II)
где R1 представляет LG, где LG представляет уходящую группу, выбранную из нитро, галогена или триметиламмония;
R2 представляет Н или PG;
PG представляет защитную группу, такую как трет-бутилоксикарбонил (BOC), трифенилметил (тритил) или диметокситритил (DMT).
8. Соединение по п. 7, которое представляет
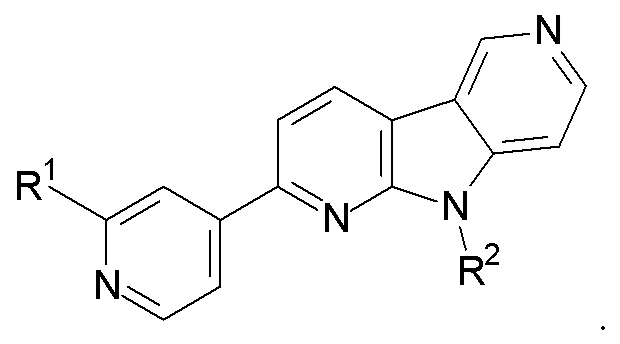
9. Соединение по п. 7, которое представляет
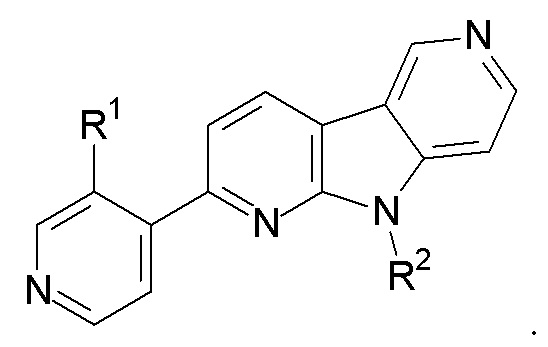
10. Соединение по п. 7, 8 или 9, где R2 представляет H.
11. Соединение по п. 7, 8 или 9, где R2 представляет PG.
12. Соединение по любому из пп. 7, 8 или 9, где PG представляет трет-бутилоксикарбонил (BOC).
13. Соединение по п. 1 для применения в диагностике расстройства, ассоциированного с агрегатами белка тау, где расстройство выбрано из болезни Альцгеймера (AD), прогрессирующего надъядерного паралича (PSP), кортикобазальной дегенерации (CBD), фронтотемпоральной лобарной дегенерации (FTLD) или болезни Пика (PiD) и где, в частности, диагностика проводится с помощью позитронно-эмиссионной томографии.
14. Соединение для применения по п. 13, где агрегаты белка тау визуализируются в головном мозге или в глазу, где визуализация предпочтительно представляет позитронно-эмиссионную томографию.
15. Применение соединения по п. 4 в качестве аналитического стандарта или в качестве инструмента для скрининга in vitro.
16. Способ получения соединения, где это соединение определено в п. 1, включающий взаимодействие соединения, где это соединение определено в п. 7, 8 или 9, где R2 представляет PG, с [18F] фторирующим агентом и отщепление защитной группы PG.
17. Набор для получения радиофармацевтического препарата, где указанный набор включает герметичный флакон, содержащий заранее определенное количество соединения, которое определено в пп. 7, 8 или 9, где R2 представляет собой PG.
18. Способ сбора данных для диагностики расстройства, ассоциированного с агрегатами белка тау в образце или у пациента, включающий:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, которое определено в пп. 1-3;
(б) обеспечение связывания соединения с агрегатом белка тау;
(с) детектирование соединения, связанного с агрегатом белка тау; и
(d) необязательно сопоставление наличия или отсутствия связывания соединения с агрегатом белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела,
где расстройство выбрано из болезни Альцгеймера (AD), прогрессирующего надъядерного паралича (PSP), кортикобазальной дегенерации (CBD), фронтотемпоральной лобарной дегенерации (FTLD) или болезни Пика (PiD).
19. Способ сбора данных для определения предрасположенности к расстройству, ассоциированному с агрегатами белка тау у пациента, включающий детектирование специфического связывания соединения, которое определено в пп. 1-3, с агрегатом белка тау в образце или in situ, который включает стадии:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, которое определено в пп. 1-3, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением;
где расстройство выбрано из болезни Альцгеймера (AD), прогрессирующего надъядерного паралича (PSP), кортикобазальной дегенерации (CBD), фронтотемпоральной лобарной дегенерации (FTLD) или болезни Пика (PiD).
20. Способ сбора данных для мониторинга остаточного проявления расстройства у пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, которого лечили лекарственным средством, где способ включает:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, которое определено в пп. 1-3, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением;
где расстройство выбрано из болезни Альцгеймера (AD), прогрессирующего надъядерного паралича (PSP), кортикобазальной дегенерации (CBD), фронтотемпоральной лобарной дегенерации (FTLD) или болезни Пика (PiD).
21. Способ сбора данных для прогнозирования реактивности пациента, страдающего расстройством, ассоциированным с агрегатами белка тау, и получающего лечение лекарственным средством, включающий:
(а) приведение образца или определенной части тела или области тела, предположительно содержащих агрегат белка тау, в контакт с соединением, которое определено в пп.1-3, где это соединение специфически связывается с агрегатом белка тау;
(b) обеспечение связывания соединения с агрегатом белка тау с образованием комплекса соединение/агрегат белка тау;
(с) детектирование образования комплекса соединение/агрегат белка тау;
(d) необязательно сопоставление наличия или отсутствия комплекса соединение/агрегат белка тау с наличием или отсутствием агрегата белка тау в образце или определенной части тела или области тела; и
(е) необязательно сравнение количества соединения/агрегата белка тау с нормальным контрольным значением;
где расстройство выбрано из болезни Альцгеймера (AD), прогрессирующего надъядерного паралича (PSP), кортикобазальной дегенерации (CBD), фронтотемпоральной лобарной дегенерации (FTLD) или болезни Пика (PiD).
| WO 2015052105 A1, 16.04.2015 | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |

