Сущность изобретения
Настоящее изобретение относится к новым соединениям, которые могут использоваться для лечения группы расстройств и нарушений, связанных с амилоидными белками, в частности с глазными расстройствами, такими как глаукома или возрастная дегенерация желтого пятна (AMD), и заболеваний или состояний, связанных с амилоидоподобными белками. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим эти соединения, и к использованию этих соединений для производства лекарственных средств для лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками. Также описывается способ лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками.
Соединения по настоящему изобретению могут также использоваться при лечении или предотвращении глазных болезней, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, в частности, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, связанными с бета-амилоидными белками, таких как нейронная дегенерация. Указанные патологические нарушения могут происходить, например, в различных тканях глаза, таких как зрительный кортекс, приводя к кортикальной недостаточности зрения; как передняя камера и как зрительный нерв, приводя к глаукоме; как хрусталик, приводя к катаракте из-за осаждения бета-амилоидных белков; как стекловидное тело, приводя к глазным амилоидозам; как сетчатка, приводя к первичной дегенерации сетчатки и дегенерации желтого пятна, например, к возрастной дегенерации желтого пятна; как зрительный нерв, приводя к друзам зрительного нерва, к оптической нейропатии и к невриту зрительного нерва; и как роговая оболочка, приводя к решетчатой дистрофии.
Область техники, к которой относится изобретение
Множество возрастных заболеваний основываются на амилоидных или амилоидоподобных белках или связаны с ними и отличаются, в частности, возникновением внеклеточных отложений амилоидных или амилоидоподобных материалов, которые вносят вклад в патогенез, а также в развитие заболевания.
Эти нейродегенеративные заболевания включают как расстройства центральной нервной системы, так и расстройства периферической нервной системы, в частности, глазные расстройства.
Такие расстройства включают, но, не ограничиваясь этим, неврологические расстройства, такие как болезнь Альцгеймера (AD), заболевания или состояния, отличающиеся потерей способности к когнитивному запоминанию, такие, например, как умеренные когнитивные нарушения (MCI), деменция с тельцами Леви, синдром Дауна, наследственная церебральная геморрагия с амилоидозом голландского типа; гуамский комплекс паркинсонизма-деменции. Другие расстройства, которые основываются на амилоидоподобных белках или связаны с ними, представляют собой прогрессирующий супрануклеарный паралич, рассеянный склероз; болезнь Крейтцфельда-Якоба, болезнь Паркинсона, деменцию, связанную с ВИЧ, ALS (амиотрофический боковой склероз), миозит с тельцами включения (IBM), диабет взрослых; сенильный сердечный амилоидоз; эндокринные опухоли и другие заболевания, включая связанные с амилоидами глазные болезни, которые нацелены на различные ткани глаза, такие как зрительный кортекс, включая кортикальную недостаточность зрения; как передняя камера и зрительный нерв, включая глаукому; как хрусталик, включая катаракту из-за осаждения бета-амилоидных белков; стекловидное тело, включая глазные амилоидозы; как сетчатка, включая первичную дегенерацию сетчатки и дегенерацию желтого пятна, например, возрастную дегенерацию желтого пятна; как зрительный нерв, включая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и как роговая оболочка, включая решетчатую дистрофию.
Бета-амилоид (Aβ) является главным компонентом сенильных бляшек при болезни Альцгеймера (AD). Эти бляшки вызываются аномальным процессингом белка-предшественника амилоида (APP) и участвуют в нейропатии AD. Aβ также, как показано недавно, участвует в развитии глазных расстройств, таких как глаукома, посредством апоптоза ганглионарных клеток сетчатки (RGC). Связь между глаукомой и AD демонстрируется в нескольких исследованиях на пациентах с AD, демонстрирующих также потери RGC, связанные с типичными глаукоматозными изменениями, такими как оптическая нейропатия и ослабление зрительной функции.
Глаукома представляет собой группу заболеваний зрительного нерва, включающих потерю ганглионарных клеток сетчатки (RGC), при характерной картине оптической нейропатии. Глаукома часто, но не всегда, сопровождается повышением глазного давления, которое может представлять собой результат блокировки циркуляции водной жидкости или ее дренирования.
Хотя повышенное внутриглазное давление представляет собой значительный фактор риска для развития глаукомы, порог внутриглазного давления, который был бы определяющим для возникновения глаукомы, не может быть определен.
Повреждение может также вызываться плохим подводом крови к витальным волокнам зрительного нерва, слабостью структуры нерва, и/или проблемой со здоровьем самих нервных волокон.
Нелеченая глаукома приводит к необратимому повреждению зрительного нерва и возникающей в результате потере поля зрения, которая может развиться до слепоты.
RGC представляют собой нервные клетки, которые передают зрительные сигналы от глаза в мозг. Каспаза-3 и каспаза-8, два главных фермента в процессе апоптоза, активируются в этом процессе, приводя к апоптозу RGC. Каспаза-3 расщепляет белок-предшественник амилоида (APP) с получением нейротоксичных фрагментов, включая амилоид β. В отсутствие защитного воздействия APP, аккумуляция амилоида β в слое ганглионарных клеток сетчатки вызывает гибель RGC и необратимую потерю зрения.
Различные типы глаукомы классифицируются как открытоугольные глаукомы, если состояние является хроническим, или закрытоугольные глаукомы, если острая глаукома возникает внезапно. Глаукома обычно воздействует на оба глаза, но заболевание может развиваться в одном глазу быстрее, чем в другом.
Хроническая открытоугольная глаукома (COAG), также известная как первичная открытоугльная глаукома (POAG), представляет собой наиболее распространенный тип глаукомы. COAG вызывается микроскопической блокировкой трабекулярной сети, которая уменьшает дренирование оттока водной жидкости в венозный синус склеры, и повышает внутриглазное давление (IOP). POAG обычно воздействует на оба глаза и тесно связана с возрастом и положительной семейной историей. Частота ее появления увеличивается у пожилых людей, поскольку механизм дренирования жидкости из глаза может постепенно забиваться с возрастом. Повышение внутриглазного давления у субъектов, подверженных хронической открытоугольной глаукоме, не сопровождается никакими симптомами, пока не почувствуется потеря в центральной области зрения.
Остроугольная глаукома (AACG) или закрытоугольная глаукома представляет собой относительно редкий тип глаукомы, отличающийся внезапным увеличением внутриглазного давления до 35-80 мм рт. ст., приводя к острой боли и необратимой потере зрения. Внезапное повышение давления вызывается перекрыванием угла фильтрования и блокировкой дренажных каналов. Индивидуумы с узкими углами имеют повышенный риск внезапной закупорки угла. AACG обычно возникает в одном глазу, но существует риск для обоих глаз. Возраст, катаракта и псевдокапсулярная эксфолиация также являются факторами риска, поскольку они связаны с увеличением хрусталика и расширением или сужением угла. Внезапный приступ глаукомы может быть связан с острой глазной болью и головной болью, с воспалением глаз, тошнотойой и с помутнением зрения.
Смешанная глаукома или глаукома с сочетанным механизмом представляет собой смесь или сочетание открытоугольной и закрытоугольной глаукомы. Она возникает у пациентов с острой ACG, у которых угол открывается после лазерной иридотомии, но которые продолжают нуждаться в лекарственных средствах для контроля IOP, а также у пациентов с POAG или с псевдоэксфолиативной глаукомой, у которых постепенно развивается сужение угла.
Глаукома при нормальном давлении (NTG), известная также как глаукома при низком давлении (LTG), отличается прогрессирующим повреждением зрительного нерва и потерей периферического зрения, сходными с теми, которые наблюдаются при других типах глаукомы; однако внутриглазное давление находится в нормальном диапазоне, или оно даже ниже нормального.
Врожденная (младенческая) глаукома представляет собой относительно редкий наследуемый тип открытоугольной глаукомы. Недостаточное развитие области дренирования приводит к повышению давления в глазу, которое может приводить к потере зрения из-за повреждения зрительного нерва и к увеличению глаза. Ранняя диагностика и лечение являются критичными для сохранения зрения у младенцев и детей, подверженных этому заболеванию.
Вторичная глаукома может возникать в результате повреждения глаза, воспаления радужной оболочки глаза (а), диабета, или использования стероидов у индивидуумов, восприимчивых к стероидам. Вторичная глаукома может также быть связана с или закупоркой или блокировкой ретинальной вены.
Пигментарная глаукома отличается отсоединением гранул пигмента от радужной оболочки. Гранулы вызывают блокировку глазной системы дренирования, приводя к повышению внутриглазного давления и повреждению зрительного нерва.
Эксфолиативная глаукома (псевдокапсулярная эксфолиация) отличается отложением хлопьевидного материала на передней капсуле и в углу глаза. Аккумуляция хлопьевидного материала блокирует систему дренирования, и повышает глазное давление.
Диагноз глаукомы может быть поставлен с использованием различных исследований. определяет давление в глазу с помощью измерения тонуса или твердости его поверхности. Для этого исследования доступны несколько типов тонометров, наиболее распространенным является аппланационный тонометр. Пахиметрия определяет толщину роговой оболочки, которая, в свою очередь, является мерой внутриглазного давления. Гониоскопия дает возможность для исследования угла фильтрования и площади дренирования глаза. Гониоскопия может также определять, могут ли аномальные кровеносные сосуды блокировать дренирование водной жидкости из глаза. Офтальмоскопия делает возможным исследование зрительного нерва и может детектировать отпадение слоя нервных волокон или изменение диска зрительного нерва, или зазубривание этой структуры (образования в ней углубления), которое может вызываться повышением внутриглазного давления или отпадением аксонов. Гониоскопия является также полезной при оценке повреждения нерва из-за плохого протекания крови или повышенного внутриглазного давления. Исследования поля зрения субъективно картируют поле зрения, это может детектировать признаки глаукоматозного повреждения зрительного нерва. Они представляются с помощью конкретных картин потерь поля зрения. Глазная когерентная томография, объективная мера потерь слоя нервных волокон, осуществляется посредством наблюдения толщины слоя волокон зрительного нерва (которая изменяется при глаукоме) посредством разницы в прохождении света через поврежденную аксональную ткань.
Дегенерация желтого пятна представляет собой распространенное заболевание глаз, которое вызывает ухудшение состояния желтого пятна, которое представляет собой центральный участок сетчатки (ткани толщиной с лист бумаги на задней внутренней поверхности глаза, где светочувствительные клетки посылают зрительные сигналы в мозг). Острое, четкое, 'переднее' зрение вырабатывается с помощью желтого пятна. Повреждение желтого пятна приводит к развитию слепых пятен и к помутнению или нарушению зрения. Возрастная дегенерация желтого пятна (AMD) является главной причиной ухудшения зрения в Соединенных Штатах для людей в возрасте больше 65 и является главной причиной реальной слепоты среди Европейцев. Приблизительно 1,8 миллиона американцев в возрасте 40 и старше имеют развитую AMD, а еще 7,3 миллиона людей с промежуточной стадией AMD имеют значительный риск потери зрения. Правительство оценивает, что к 2020 году будет 2,9 миллиона людей с развитой AMD. Жертвы AMD часто бывают неприятно удивлены и озабочены, обнаруживая, насколько мало известно о причинах и лечении этого состояния с потерей зрения.
Имеются две формы дегенерации желтого пятна: сухая дегенерация желтого пятна и влажная дегенерация желтого пятна. Сухая форма, при которой клетки желтого пятна начинают медленно разрушаться, диагностируется у 85 процентов случаев дегенерации желтого пятна. Обычно сухая AMD воздействует на оба глаза, хотя и один глаз может потерять зрение, в то время как другой глаз остается нетронутым. Друзы, которые представляют собой желтые осаждения под сетчаткой, представляют собой распространенные ранние признаки болезни сухой AMD. Риск развития развитой сухой AMD или влажной AMD увеличивается, когда количество или размер друз увеличивается. Иногда бывает, что сухая AMD развивается и вызывает потерю зрения без перехода во влажную форму заболевания; однако бывает также, что сухая AMD на ранней стадии внезапно переходит во влажную форму.
Влажная форма, хотя она составляет только 15 процентов от всех случаев, дает 90 процентов слепоты, и считается развитой AMD (ранней или промежуточной стадии влажной AMD нет). Влажной AMD всегда предшествует сухая форма заболевания. Когда сухая форма ухудшается, у некоторых людей начинается аномальный рост кровеносных сосудов позади желтого пятна. Эти сосуды являются очень хрупкими, и будут иметь утечки жидкости и крови (отсюда 'влажная' дегенерация желтого пятна), вызывая быстрое повреждение желтого пятна.
Сухая форма AMD сначала часто вызывает легкое помутнение зрения. Центр зрения, в частности, может стать мутным и этот участок становится больше, когда болезнь прогрессирует. Симптомы могут и не замечаться, если поражается только один глаз. При влажной AMD, могут появляться прямые линии, волнистые, и может происходить быстрая потеря центрального зрения.
Диагностика дегенерации желтого пятна, как правило, включает исследование расширенного глаза, исследование остроты зрения и глазного дна с использованием процедуры, называемой исследованием глазного дна, чтобы помочь в диагностике AMD, и - если ожидается влажная AMD, может также осуществляться флуоресцеиновая ангиография. Если сухая AMD достигает развитых стадий, то современного лечения для предотвращения потери зрения не существует. Однако высокие дозы конкретных препаратов антиоксидантов и цинка могут отсрочить или предотвратить развитие промежуточной AMD до развитой стадии. Macugen® (пегаптаниб натрий, инъекция), лазерная фотокоагуляция и фотодинамическая терапия могут контролировать аномальный рост кровеносных сосудов и кровотечение в желтом пятне, что является полезным для некоторых людей, которые имеют влажную AMD; однако зрение, которое уже потеряно, не будет восстанавливаться с помощью этих методик. Если зрение уже потеряно, то имеются средства для людей с плохим зрением, которые могут помочь улучшить качество жизни.
Один из самых ранних признаков возрастной дегенерации желтого пятна (AMD) представляет собой аккумуляцию внеклеточных отложений, известных как друзы, между базальной отпадающей оболочкой пигментированного эпителия сетчатки (RPE) и мембраной Бруха (BM). Недавние исследования, осуществленные Anderson et al. подтвердили, что друзы содержат бета-амилоидный белок. (Experimental Eye Research 78 (2004) 243-256).
Прионы вызывают нейродегенеративные заболевания, такие как скрепи у овец, бычью губкообразную энцефалопатию у крупного рогатого скота и болезнь Крейтцфельдта-Якоба у людей. Единственный известный компонент частицы представляет собой изоформу белка, присущую скрепи, PrPSc. Хотя прионы размножаются, нет доказательств того, что они содержат нуклеиновую кислоту. PrPSc получается из неинфекционного клеточного белка PrPC с помощью посттрансляционного процесса, в течение которого PrPC подвергается выраженному конформационному изменению.
Белок PrPSc, присущий скрепи, играет критическую роль в нейронной дегенерации, и в течение развития заболевания подвергается трехстадийному преобразованию, следующим образом: PrPC (нормальная клеточная изоформа белка) - PrPSc: инфекционная форма (изоформа белка, присущая скрепи) - белок PrP27-30.
Такой каскад событий происходит в течение развития болезни Крейтцфельдта-Якоба (CJD), Куру, синдрома Герстманна-Страусслера-Шайнкера (GSS), летальной врожденной асомнии у людей, скрепи у овец и коз, энцефалопатии у норок и бычьей губкообразной энцефалопатии у крупного рогатого скота.
Клеточный нетоксичный белок (PrPC) представляет собой сиалогликопротеин с молекулярной массой от 33000 до 35000, который экспрессируется в основном в нейронах. При заболеваниях, указанных выше, PrPC преобразуется в измененную форму (PrPSc), которая отличается от своего нормального гомолога своей относительной стойкостью к дигестии с помощью протеаз. PrPSc аккумулируется в центральной нервной системе пораженных животных и индивидуумов, и его протеазоустойчивая сердцевина агрегирует вне клеток.
Амилоидоз не представляет собой отдельного заболевания, но скорее представляет собой разнообразную группу прогрессирующих болезненных процессов, отличающихся отложениями во внеклеточных тканях воскообразного крахмалоподобного белка, называемого амилоидным белком, который аккумулируется в одном или нескольких органах или системах организма. Когда амилоидные отложения увеличиваются в размерах, они начинают вмешиваться в нормальное функционирование органа или системы организма. Имеется, по меньшей мере, 15 различных типов амилоидоза. Главные формы представляют собой первичный амилоидоз без известных предшественников, вторичный амилоидоз, который следует после какого-нибудь другого состояния, и наследственный амилоидоз.
Вторичный амилоидоз возникают у людей, которые имеют хроническую инфекцию или воспалительное заболевание, такое как туберкулез, бактериальную инфекцию, называемую наследственной средиземноморской лихорадкой, костные инфекции (остеомиелит), ревматоидный артрит, воспаление тонкого кишечника (грануломатозный илеит), болезнь Ходжкина и проказу.
Друзы зрительного нерва представляют собой глобулярные конкреции белка и солей кальция, которые как считается, представляют собой секреции наследственно изменённых сосудистых структур, которые воздействуют на слой аксональных нервных волокон. Эта аккумуляция происходит в перипапиллярном слое нервных волокон и, как считается, повреждает слой нервных волокон либо непосредственно, посредством сжатия, либо опосредованно, прерывая сосудистое снабжение слоя нервных волокон. Она обычно становится видимой после первого десятилетия жизни пораженных индивидуумов. Чаще всего она происходит на обоих глазах, но может также происходить в одном глазу и может вызывать умеренную потерю периферического зрения в течение многих лет.
Оптическая нейропатия представляет собой заболевание, отличающееся повреждением зрительного нерва, вызываемым демиелинизацией, блокировкой поступления крови, недостаточностью питания или токсинами. Демиелинизирующие оптические нейропатии (смотри неврит зрительного нерва, ниже), как правило, вызываются фундаментальным процессом демиелинизации, таким как рассеянный склероз. Блокировка поступления крови, известная как ишемическая оптическая нейропатия, может приводить к гибели или дисфункции клеток зрительного нерва. Неартериальная ишемическая оптическая нейропатия обычно встречается у людей среднего возраста. Факторы риска включают высокое давление крови, диабет и атеросклероз. Артериальная ишемическая оптическая нейропатия обычно встречается у людей постарше после воспаления артерий (артериита), в частности, темпоральной артерии (темпоральный артериит). Потеря зрения может быть быстрой или развиваться постепенно в течение 2-7 дней, и повреждение может происходить на одном или обоих глазах. У людей с оптической нейропатией, вызываемой экспонированием токсина или питательной недостаточностью, обычно поражаются оба глаза.
Примерно 40% людей с неартериальной ишемической оптической нейропатией испытывают самопроизвольное улучшение со временем. Неартериальная ишемическая оптическая нейропатия лечится посредством контролирования давления крови, диабета и уровней холестерина. Артериальная ишемическая оптическая нейропатия лечится с помощью больших доз кортикостероидов для предотвращения потери зрения во втором глазу.
Неврит зрительного нерва связан с умеренной или острой потерей зрения в одном или обоих глазах и может вызываться системным процессом демиелинизации (смотри выше), вирусной инфекцией, вакцинацией, менингитом, сифилисом, рассеянным склерозом и внутриглазным воспалением (увеитом). Движение глаз может быть болезненным, и зрение может ухудшаться с повторением рецидивов. Диагностика включает исследования реакций зрачков и определение того, набух ли диск зрительного нерва. Магнитно-резонансная томография (MRI) может показать наличие рассеянного склероза или, редко, опухоли, которая давит на зрительный нерв, в этом случае зрение улучшается после того, как давление опухоли ослабляется. В большинстве случаев неврит зрительного нерва проходит сам в течение нескольких месяцев без лечения. В некоторых случаях, может быть необходимым лечение с помощью кортикостероидов, вводимых внутривенно.
Катаракта представляет собой помутнение, которое развивается в хрусталике глаза или в его оболочке. Катаракты, как правило, вызывают прогрессирующую потерю зрения, и могут вызывать слепоту, если остаются нелечеными. При моргагниевой катаракте, кортекс катаракты постепенно ожижается с образованием молочно-белой жидкости и может вызывать острое воспаление, если капсула хрусталика разрушается, и дает протечку. Если ее не лечить, катаракта может также вызвать факоморфическую глаукому. Катаракты могут быть врожденными по природе или вызываться генетическими факторами, старостью, долговременным ультрафиолетовым излучением, экспонированием для радиации, диабетом, ранением глаз или физической травмой.
Экстракапсулярная (ECCE) хирургия представляет собой наиболее эффективное лечение при лечении катаракты. При операции, хрусталик удаляется, но большая часть капсулы хрусталика остается интактной. Факоэмульсификация, малый надрез на одной стороне роговой оболочки, как правило, используется для разрушения хрусталика перед извлечением.
Глазной амилоидоз представляет собой наследственное расстройство, связанное с врожденной амилоидопатической полиневропатией (FAP) типа I, и отличающийся аномальными конъюктивальными сосудами сухой кератоконъюнктивит, аномалии зрачка и, в некоторых случаях, помутнение стекловидного тела и вторичную глаукому. FAP типа I связана с мутациями в транстиретине (TTR), тетрамерном белке плазмы (преальбумине), синтезируемом в печени, в пигментном эпителии сетчатки 2 и в сосудистой оболочке мозга. Различные мутации заставляют транстиретин полимеризоваться в виде складчатой структуры из амилоидных фибрил, приводя к наследственному амилоидозу. Наиболее часто встречающаяся мутация представляет собой TTR-met303, при которой метионин замещает валин в положени 30 в транстиретине.
FAP типа IV связана с решетчатой дистрофией роговой оболочки (LCD). Решетчатая дистрофия роговой оболочки представляет собой наследственный, первичный, обычно билатеральный амилоидоз роговой оболочки, отличающийся присутствием решетчатых преломляющих линий с двойным контуром в строме роговой оболочки. LCD типа I (Бибера-Хааба-Диммера) представляет собой аутосомальное доминантное, билатерально симметричное расстройство роговой оболочки, отличающееся присутствием многочисленных полупрозрачных тонких решетчатых линий с белыми точками и слабым помутнением в верхнем и среднем слоях центральной стромы. Симптомы возникают в течение первого или второго десятилетий жизни, вызывая прогрессирующую потерю зрения. Большинство пациентов нуждаются в трансплантации роговой оболочки к 40 годам жизни. LCD типа II связана с системным амилоидозом (синдромом Меритоя) и отличается присутствием толстых решетчатых линий в лимбе, центральной роговой оболочке и строме. Зрение не ухудшается до поздней старости. LCD типа III поражает людей среднего возраста, и отличается присутствием толстых решетчатых линий, которые простираются от лимба до лимба. LCD типа III A отличается аккумуляцией амилоидных отложений в строме и присутствием полос амилоидного белка между стромой и слоем Баумана, LCD типа III A отличается от LCD типа III присутствием эрозии роговой оболочки, возникновением белых пятен и аутосомальной доминантной наследственной структурой.
Лечения для глаукомы нет. Большинство видов лечения для глаукомы создаются для понижения и/или контроля внутриглазного давления (IOP), которое может повредить зрительный нерв, который передает зрительную информацию в мозг. Глазные капли от глаукомы часто являются первым выбором по сравнению с хирургическим лечением глаукомы, и могут быть очень эффективными при контроле IOP для предотвращения повреждения глаз. Медицинские препараты для лечения глаукомы классифицируются по их активным химическим соединениям, и могут быть перечислены в следующих далее категориях, (при этом лекарственные средства, одобренные в настоящее время, показаны в скобках):
- Бета-блокаторы (тимоптик, бетоптик, исталол, тимолол) работают посредством уменьшения продуцирования (водной) жидкости в глазу.
- Ингибиторы угольной ангидразы (трусопт, азопт, диамокс, наптазан, даранид) уменьшают скорость продуцирования внутриглазной жидкости.
- Альфа-адренергические агонисты (альфаган, альфаган-P, иопидин) также уменьшают продуцирование внутриглазной жидкости.
- Простагландины (ксалатан, лумиган, траватан Z, рескула) перенаправляют дренирование внутриглазной жидкости в другой путь в задней части глаза, уменьшая тем самым избыточное глазное давление.
- Парасимпатомиметики (карбахол, пилокарпин) работают посредством повышения оттока водной жидкости из глаза, увеличивая, таким образом, дренирование внутриглазной жидкости.
- Эпинефрин уменьшает скорость продуцирования внутриглазной жидкости, и увеличивает отток водной жидкости из глаза.
Наряду с медицинскими препаратами, предназначенными для контроля IOP, определенные исследовательские способы лечения глаукомы концентрируются на защите зрительного нерва. Лекарственное средство мемантин от болезни Альцгеймера в настоящее время исследуется при показаниях глаукомы в качестве нейропротектора. Однако рандомизированное клиническое исследование антагониста N-метил-d-аспартата (NMDA) мемантина при открытоугольной глаукоме не показывает значительной эффективности.
Дополнительные виды лечения глаукомы представляют собой лазерную хирургию, которая включает трабекулопластику, процедуру, которая помогает внутриглазной жидкости покидать глаз более эффективно. Согласно Glaucoma Foundation, примерно 80% пациентов реагируют на процедуру достаточно хорошо, чтобы отложить или исключить дополнительную операцию. Однако давление опять увеличивается в глазах у половины всех пациентов в пределах двух лет после лазерной хирургии, согласно National Eye Institute. Хирургическую операцию осуществляют, если медицинские препараты и начальное лазерное лечение оказываются безуспешными при понижении давления внутри глаз. Один из типов операции, трабекулэктомия, создает отверстие в стенке глаза, так что внутриглазная жидкость может удаляться. Однако примерно у одной трети пациентов после трабекулэктомии развивается катаракта в пределах пяти лет, согласно Glaucoma Foundation. Если трабекулэктомия не дает результата, дополнительные хирургические процедуры включают помещение дренажной трубки в глаз между роговицей и радужной оболочкой и использование лазерного или криогенного лечения для разрушения ткани в глазу, которая создает внутриглазную жидкость. Хирургическая операция может сохранить оставшееся зрение у пациента, но она не улучшает зрения. На самом деле зрение может ухудшаться после хирургической операции.
Современная терапия для лечения глаукомы старается замедлить развитие потери поля зрения посредством понижения и контроля внутриглазного давления. Как рассмотрено выше, это делается либо с помощью лекарственных средств, понижающих IOP, либо с помощью лазерной трабекулопластики. Долговременные исследования воздействий понижения IOP, как показано, являются эффективными при замедлении развития заболевания у некоторых пациентов. К сожалению, имеются пациенты, которые продолжают терять поле зрения, несмотря на понижение их IOP, или пациенты вообще не реагирующие на лекарственные средства, понижающие IOP. По этой причине, имеется необходимость в разработке новых видов лечения, нацеленных на параметр, иной, чем внутриглазное давление. Такая новая цель представляет собой нейропротекцию RGC.
Возрастная дегенерация желтого пятна (AMD) является главной причиной слепоты среди европейцев в возрасте 65. Хотя, в исследованиях дегенерации желтого пятна достигнут большой прогресс в последнее время, не существует видов лечения, которые предотвращают гибель нейронных клеток, которая происходит в ходе заболевания. Не существует также окончательных видов лечения для других глазных болезней, связанных с нейронной дегенерацией, связанной с бета-амилоидными белками, таких как кортикальная недостаточность зрения, друзы зрительного нерва, оптическая нейропатия, неврит зрительного нерва, глазной амилоидоз и решетчатая дистрофия.
Амилоидные отложения, как правило, содержат три компонента. Фибриллы амилоидных белков, которые составляют примерно 90% амилоидного материала, содержат один из нескольких различных типов белков. Эти белки способны складываться в так называемые "бета-складчатые" листовые фибриллы, уникальную конфигурацию белка, которая демонстрирует сайты связывания с конго красным, что приводит к возникновению уникальных свойств окрашивания амилоидного белка. В дополнение к этому, амилоидные отложения тесно связаны с компонентом (AP) амилоида P (пятиугольного), гликопротеина, родственного обычному амилоиду P сыворотки (SAP), и с сульфатированными гликозаминогликанами (GAG), сложными угледоводородами соединительной ткани.
Одна из разработок в направлении лечения расстройств и нарушений, связанных с амилоидным белком, или состояний, связанных с амилоидоподобными белками, таких как болезнь Альцгеймера и прионные заболевания, представляет собой конструирование молекул, которые связываются с аномальной конформацией β-листа Aβ и PrP, соответственно, тем самым предотвращая агрегацию этих молекул. Конформация β-листа пептидов отличается тем, что образуются водородные связи в виде регулярной структуры между соседними нитями аминокислот. Это система приводит к возникновению стабильной трехмерной структуры. Акцепторы H-связи (группа C=O) и доноры H-связи (группа NH) чередуются во встречающихся в природе пептидах в конформации β-листа с атомами, которые должны связываться, располагаясь примерно на одной линии. В каждой нити аминокислот, расстояния между соседними донорами H-связи и акцепторами H-связи попадают в конкретные пределы. В частности, расстояние между донором H-связи (группой NH) и акцептором H-связи (группой C=O) в одном аминокислотном остатке составляет от 3,5 до 4,0 Å. Расстояние между акцептором H-связи (группой C=O) одного аминокислотного остатка и донором H-связи (группой NH) следующего аминокислотного остатка, которые участвуют в связывании различных нитей, составляет от 2,6 до 2,9 Å. Другими словами, расстояния между соседними донорами H-связи и акцепторами H-связи в пределах одной аминокислотной нити изменяются в следующих пределах:
донор H-связи (аминокислота 1) - акцептор H-связи (аминокислота 1) = 3,5-4,0 Å;
акцептор H-связи (аминокислота 1) - донор H-связи 2 (аминокислота 2) = 2,6-2,9 Å.
Лиганды, которые конструируются для связывания β-листов, в идеале имеют порядок доноров H-связи и акцепторов H-связи, который является комплементарным к порядку доноров H-связи и акцепторов H-связи в аминокислотных нитях β-листа.
В WO 03/095429 и в Rzepecki et al., Synthesis (2003) 12, 1815-1826 описываются синтетические молекулы, которые, как сказано, связываются с β-конформацией Aβ или PrP, тем самым предотвращая их агрегацию. Для этой цели синтезированы определенные молекулы, содержащие два или более аминопиразольных остатка, связанных с помощью линкеров, содержащих карбонильную группу, например, “AmpOx” и “Тример ”.

“AmpOx”

“Тример”
Некоторые из молекул, описанных в WO 03/095429, как сказано, имеют ингибирующее воздействие на образование агрегатов Aβ в двух биофизических анализах. Согласно Rzepecki et al., Synthesis (2003) 12, 1815-1826 одна из молекул, описанных там, способна уменьшать агрегацию рекомбинантного PrPC в растворе. Однако физико-химические свойства, в этих исследованиях не изучались.
WO 2008/061795 описывает определенные гетероциклические соединения, которые являются пригодными для лечения заболеваний, связанных с амилоидными или амилоидоподобными белками.
Физико-химические свойства играют ключевую роль в преодолении гематоэнцефалического барьера нейротерапевтическими препаратами. Факторы, релевантные для успешного действия лекарственных средств для CNS, обсуждаются в (H. Pajouhesh и G. R. Lenz, NeuroRx®: J. Am. Soc. Exp. Neurother. (2005) Vol. 2, 541). Они включают коэффициент распределения между водой и н-октанолом (LogP), то есть, в основном, липофильность соединения. Некоторые соединения, описанные в WO 03/095429 и в Rzepecki et al., Synthesis (2003) 12, 1815-1826, имеют неблагоприятные вычисленные значения LogP и, по этой причине, как ожидается, они не преодолевают гематоэнцефалический барьер. В частности, “AmpOx” имеет вычисленное значение LogP ниже нуля.
Другие соединения, описанные в указанных выше документах, имеют свойства, которые делают их непригодными для введения пациенту из-за их вредных побочных воздействий. Например, “Тример” является мутагенным, канцерогенным и метаболически нестабильным.
Сущность изобретения
Целью настоящего изобретения является получение соединений, которые могут использоваться при лечении заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включая амилоидоз, но в особенности, глазных болезней, таких как глаукома. Соединения должны быть способны преодолевать гематоэнцефалический барьер. Кроме того, они должны быть фармацевтически приемлемыми, в частности, они не должны иметь мутагенных или канцерогенных свойств или быть метаболически нестабильными. Соединения должны иметь разумно высокую растворимость в воде, сохраняя при этом свою биологическую активность.
Другой целью настоящего изобретения является создание улучшенных возможностей для лечения субъектов, пораженных глазными болезнями, связанными с патологическими нарушениями/изменениями в тканях зрительной системы, в частности, связанными с патологическими нарушениями/изменениями в тканях зрительной системы, связанными с бета-амилоидными белками, такими, например, как нейронная дегенерация. Указанные патологические нарушения могут возникать, например, в различных тканях глаза, таких как зрительный кортекс, приводя к кортикальной недостаточности зрения; как передняя камера и как зрительный нерв, приводя к глаукоме; как хрусталик, приводя к катаракте из-за осаждения бета-амилоидных белков; как стекловидное тело, приводя к глазному амилоидозу; как сетчатка, приводя к первичной дегенерации сетчатки и дегенерации желтого пятна, например, к возрастной дегенерации желтого пятна; как зрительный нерв, приводя к возникновению друз зрительного нерва, оптической нейропатии и невриту зрительного нерва; и как роговая оболочка, приводя к решетчатой дистрофии.
Авторы настоящего изобретения неожиданно обнаружили, что эти цели могут быть достигнуты с помощью соединений общей формулы (I), как описано далее. Соответственно, настоящее изобретение относится к соединению общей формулы (I).
В другом аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей соединение общей формулы (I).
Еще один аспект настоящего изобретения относится к применению соединения общей формулы (I) для получения лекарственного средства для лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включая амилоидоз.
Также, в настоящем документе описывается способ лечения заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения общей формулы (I).
В предпочтительном варианте осуществления, заболевание или состояние представляет собой глазную болезнь или состояние. Более предпочтительно, заболевание представляет собой глаукому, еще более предпочтительно, заболевание выбрано из группы, состоящей из хронической (идеопатической) открытоугольной глаукомы, глаукомы с блокированием зрачка, глаукомы развития, глаукомы, связанной с другими глазными расстройствами, глаукомы, связанной с повышенным эписклеральным венозным давлением, глаукомы, связанной с воспалением и травмой, глаукомы после внутриглазной хирургической операции, глаукомы при повышенном давлении, глаукомы при нормальном давлении, острой закрытоугольной глаукомы, подострой закрытоугольной глаукомы, хронической закрытоугольной глаукомы, глаукомы с сочетанным механизмом, врожденной (младенческой) глаукомы, ювенильной глаукомы с аниридией, глаукомы, связанной с расстройствами эндотелия роговой оболочки, глаукомы, связанной с расстройствами радужной оболочки и ресничного тела, глаукомы, связанной с расстройствами хрусталика, глаукомы, связанной с расстройствами сетчатки, сосудистой оболочки глаза и стекловидного тела, глаукомы, связанной с отслоением сетчатки и витреоретинальными нарушениями, неоваскулярной глаукомы, пигментной глаукомы, синдрома эксфолиации, открытоугольной глаукомы, вызываемой проблемами с хрусталиком, глаукомы, связанной с утолщением и смещением хрусталика, глаукомы, связанной с кератитом, эписклеритом и склеритом, глаукомы, связанной с блокировкой реснитчатого тела (злокачественной), глаукомы при афакии и артифакии, при пролиферации клеток эпителия, волокон и эндотелия, глаукомы, связанной с хирургической операцией на роговице, и глаукомы, связанной с витреоретинальной хирургической операцией.
Еще один аспект настоящего изобретения относится к применению соединения общей формулы (I) для производства лекарственного средства для лечения или ослабления воздействий глазных болезней, связанных с патологическими нарушениями/изменениями в тканях зрительной системы.
Также, в настоящем документе описывается способ лечения или ослабления воздействий глазных болезней, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, включающий введение субъекту, нуждающемуся в таком лечении, эффективного количества соединения общей формулы (I).
Глазные болезни, связанные с патологическими нарушениями/изменениями в тканях зрительной системы, в частности, связанные с патологическими нарушениями/изменениями в тканях зрительной системы, связанных с бета-амилоидными белками, такими, например, как нейронная дегенерация. Указанные патологические нарушения могут происходить, например, в различных тканях глаза, таких как зрительный кортекс, приводя к кортикальной недостаточности зрения; как передняя камера и зрительный нерв, приводя к глаукоме; как хрусталик, приводя к катаракте из-за осаждения бета-амилоидных белков; как стекловидное тело, приводя к глазному амилоидозу; как сетчатка, приводя к первичной дегенерации сетчатки и дегенерации желтого пятна, например, возрастной дегенерации желтого пятна; как зрительный нерв, приводя к возникновению друз зрительного нерва, оптической нейропатии и невриту зрительного нерва; и как роговая оболочка, приводя к решетчатой дистрофии.
В одном из предпочтительных вариантов осуществления глазная болезнь или состояние выбраны из группы, состоящей из глаукомы, нейронной дегенерации, кортикальной недостаточности зрения, катаракты из-за осаждения бета-амилоидных белков, глазного амилоидоза, первичной дегенерации сетчатки, дегенерации желтого пятна, например, возрастной дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дистрофии.
В другом аспекте настоящее изобретение относится к смеси (такой как фармацевтическая композиция), содержащей соединение по настоящему изобретению и необязательно, по меньшей мере, одно дополнительное биологически активное соединение и/или фармацевтически приемлемый носитель и/или разбавитель и/или наполнитель. Дополнительное биологически активное вещество может представлять собой известное соединение, используемое при лечении заболеваний и расстройств, которые вызываются амилоидными или амилоидоподобными белками или связаны с ними.
В одном из вариантов осуществления дополнительное биологически активное соединение предпочтительно выбрано из группы, состоящей из бета-блокаторов, ингибиторов угольной ангидразы, альфа- или бета-адренергических агонистов, простагландинов, парасимпатомиметиков, ингибиторов холинэстераз, усилителей синтеза накопления или высвобождения ацетилхолина, агонистов постсинаптических рецепторов ацетилхолина, антагонистов рецепторов N-метил-D-аспартата глютамата, соединений, используемых при лечении амилоидозов, соединений против оксидативного стресса, антиапоптотических соединений, хелаторов металлов, ингибиторов репарации ДНК, таких как пирензепин и метаболиты, 3-амино-1-пропансульфоновой кислоты (3APS), 1,3-пропандисульфоната (1,3PDS), активаторов α-секретазы, ингибиторов β- и γ-секретазы, нейротрансмиттеров, расщепителей β-листов, аттрактантов клеточных компонентов, вычищающих/понижающих содержание бета-амилоидных белков, ингибиторов расщепленных бета-амилоидных белков с N-окончаниями, включая пироглютаматированный бета-амилоидный белок 3-42, противовоспалительных молекул или ингибиторов холинэстеразы (ChEI), таких как такрин, ривастигмин, донепезил и/или галантамин, агонистов M1, других лекарственных средств, включая любое лекарственное средство, модифицирующее амилоиды, и питательные добавки, антитела, вакцины.
В другом предпочтительном варианте осуществления дополнительное биологически активное соединение выбрано из группы, состоящей из тимоптика, бетоптика, исталола, тимолола, трусопта, азопта, диамокса, наптазана, даранида, альфагана, альфагана-p, иопидина, ксалатана, лумигана, траватана Z, рескулы, карбахола, пилокарпина, эпинефрина и мемантина.
В другом предпочтительном варианте осуществления, дополнительное биологически активное соединение представляет собой антитело, предпочтительно, моноклональное антитело, включая любое функционально эквивалентное антитело или его функциональные части. Предпочтительно, антитело, более предпочтительно, моноклональное антитело, может включать любое функционально эквивалентное антитело или его функциональные части, оно представляет собой антитело, которое связывает амилоид β. Предпочтительно, антитело, более предпочтительно, моноклональное антитело, которое может включать любое функционально эквивалентное антитело или его функциональные части, представляет собой антитело, это антитело, при совместном инкубировании с амилоидными мономерными и/или полимерными растворимыми амилоидными пептидами, например, с β-амилоидными мономерными пептидами, такими как Aβ мономерные пептиды 1-39; 1-40, 1-41 или 1-42, и/или с полимерным растворимым β-амилоидным пептидом, содержащим множество Aβ мономерных единиц, но в особенности, с Aβ1-42 мономерным и/или Aβ полимерным растворимым амилоидным пептидом, содержащим множество Aβ1-42 мономерных единиц, ингибирует агрегацию Aβ мономеров в виде высокомолекулярных полимерных фибрилл или нитей, и в дополнение к этому, при совместном инкубировании вместе с предварительно сформированными высокомолекулярными полимерными амилоидными фибриллами или нитями, сформированными посредством агрегации амилоидных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких, например, как Aβ мономерные пептиды 1-39; 1-40, 1-41 или 1-42, но в особенности, с Aβ1-42 мономерными пептидами, способно к дизагрегации предварительно сформированных полимерных фибрилл или нитей. В одном из вариантов осуществления, антитело может представлять собой химерное антитело или его функциональную часть, или гуманизированное антитело или его функциональную часть. В другом варианте осуществления, антитело может представлять собой моноклональное антитело, выбранное из группы антител, имеющих характерные свойства антитела, продуцируемого линией клеток гибридом:
a) FP 12H3, депозит от 01 декабря 2005 года и 09 декабря 2005 года, соответственно, как DSM ACC2752;
b) FP 12H3-C2, депозит от 01 декабря 2005 года и 09 декабря 2005 года, соответственно, как DSM ACC2750;
c) FP 12H3-G2, депозит от 01 декабря 2005 года и 09 декабря 2005 года, соответственно, как DSM ACC2751;
d) ET 7E3, депозит от 08 декабря 2005 года как DSM ACC2755; и
e) EJ 7H3, депозит от 08 декабря 2005 года как DSM ACC2756.
В другом варианте осуществления, антитело может представлять собой гуманизированное антитело, демонстрирующее легкую цепь и тяжелую цепь, как изображено в SEQ ID No. 2 и SEQ ID No. 4 Международной заявки № PCT/US2007/073504.
Еще в одном варианте осуществления антитело может представлять собой гуманизированное антитело, демонстрирующее вариабельную область легкой цепи и вариабельную область тяжелой цепи, как изображено в SEQ ID No. 1 и SEQ ID No. 3 Международной заявки № PCT/US2007/073504.
В другом варианте осуществления дополнительное биологически активное соединение может представлять собой антигенный пептидный фрагмент Aβ, состоящий из единственного или повторяющегося фрагмента секвенирования из множества смежных аминокислотных остатков из части Aβ пептида с N-окончанием, в частности, фрагмента секвенирования между 13 и 15 непрерывными аминокислотными остатками. Aβ антигенный пептидный фрагмент может представлять собой Aβ1-15 пептидный антиген, такой как пальмитоилированный Aβ1-15 пептидный антиген, модифицированный с помощью ковалентно присоединенных пальмитоильных остатков, в частности, между 2 и 4, более конкретно, на 4 остатках, на каждом конце пептида, разбавленного в липосоме.
Дополнительное биологически активное вещество или соединение может оказывать свое биологическое воздействие с помощью такого же или сходного механизма, как и соединение в соответствии с настоящим изобретением, или с помощью независимого механизма действия или с помощью множества родственных и/или независимых механизмов действия.
Во всех вариантах осуществления настоящего изобретения соединение по настоящему изобретению и/или дополнительное биологически активное соединение предпочтительно используется в терапевтически эффективном количестве.
Также описывается способ сбора данных для диагностики заболевания или состояния, связанного с амилоидными белками, в образце или у пациента, который включает:
(a) приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный белок, с соединением по настоящему изобретению;
(b) предоставление возможности соединению для связывания с амилоидным белком;
(c) детектирование соединения, связанного с белком; и
(d) необязательное коррелирование присутствия или отсутствия соединения, связывающегося с амилоидным белком с присутствием или отсутствием амилоидного белка в образце или в конкретной части тела или участке тела.
Другой вариант осуществления настоящего изобретения представляет собой способ определения степени присутствия амилоидогенных бляшек в ткани и/или телесной жидкости, включающей:
(a) получение образца, репрезентативного для ткани и/или телесной жидкости, которые нужно исследовать;
(b) исследование образца на присутствие амилоидного белка с помощью соединения по настоящему изобретению;
(c) определение количества соединения, связанного с амилоидным белком; и
(d) вычисление присутствия бляшек в ткани и/или телесной жидкости.
В предпочтительном варианте осуществления, определения на стадии(c) осуществляются так, что присутствие или отсутствие соединения, связывающегося с амилоидным белком, коррелирует с присутствием или отсутствием амилоидного белка.
Другой аспект относится к способу сбора данных для определения предрасположенности к заболеванию или состоянию, связанному с амилоидным белком, у пациента, включающему детектирование специфичного связывания соединения по настоящему изобретению с амилоидным белком в образце или in situ, который включает стадии:
(a) приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный белок, с соединением по настоящему изобретению, это соединение специфично связывается с амилоидным белком;
(b) предоставление возможности соединению для связывания с амилоидным белком, с образованием комплекса соединение/белок;
(c) детектирование образования комплекса соединение/белок;
(d) необязательное коррелирование присутствия или отсутствия комплекса соединение/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участке тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Еще один аспект настоящего изобретения представляет собой способ сбора данных для мониторинга минимального остаточного заболевания у пациента после лечения с помощью антитела или композиции вакцины, где способ включает:
(a) приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный белок, с соединением по настоящему изобретению, это соединение специфично связывается с амилоидным белком;
(b) предоставление возможности соединению для связывания с амилоидным белком с образованием комплекса соединение/белок;
(c) детектирование образование комплекса соединение/белок;
(d) необязательное коррелирование присутствия или отсутствия комплекса соединение/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участка тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Также описывается способ сбора данных для предсказания реакции у пациента, которого лечат с помощью антитела или композиции вакцины, который включает:
(a) приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный белок, с соединением по настоящему изобретению, это соединение специфично связывается с амилоидным белком;
(b) предоставление возможности соединению для связывания с амилоидным белком с образованием комплекса соединение/белок;
(c) детектирование образования комплекса соединение/белок;
(d) необязательное коррелирование присутствия или отсутствия комплекса соединение/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участке тела; и
(e) необязательное сравнение количества комплекса соединение/белок с нормальным контрольным значением.
Другой аспект настоящего изобретения представляет собой набор для исследования для детектирования и диагностики заболевания или состояния, связанного с амилоидным белком, содержащий соединение по настоящему изобретению. Предпочтительно, набор для исследования содержит контейнер, содержащий одно или несколько соединений в соответствии с настоящим изобретением, и инструкции для использования соединения для целей связывания с амилоидным белком с образованием комплекса соединение/белок и детектирования образования комплекса соединение/белок, так что присутствие или отсутствие комплекса соединение/белок коррелирует с присутствием или отсутствием амилоидного белка.
Определения
В пределах обозначений настоящей заявки применяются следующие определения:
"Алкил" относится к насыщенному органическому остатку, состоящему из атомов углерода и водорода. Примеры соответствующих алкильных групп имеют 1-6 атомов углерода, предпочтительно, 1-4 атома углерода, и включают метил, этил, пропил и бутил.
"Алкилен" относится к двухвалентной алкильной группе. Указанные выше комментарии по поводу "алкила" относятся по аналогии и к этому варианту осуществления.
"Циклоалкил" относится к циклическому органическому остатку, состоящему из атомов углерода и водорода. Примеры соответствующих алкильных групп имеют 5-10 атомов углерода, предпочтительно, 5 или 6 атомов углерода, и включают циклопентил и циклогексил.
"Гетероциклоалкил" относится к циклоалкильной группе, как определено выше, в которой один из атомов углерода заменен гетероатомом, который выбран, например, из N, O или S, или остатком, содержащим гетероатом (например, N, O и/или S). Примеры возможных гетероциклоалкильных групп включают пирролидин, тетрагидрофуран, пиперидин, и тому подобное.
"Алкенил" относится к органическому остатку, состоящему из атомов углерода и водорода, который включает, по меньшей мере, одну двойную связь. Примеры соответствующих алкенильных групп имеют 2-6 атомов углерода, предпочтительно, 2-4 атома углерода, и включают пропенил и бутенил.
"Алкинил" относится к органическому остатку, состоящему из атомов углерода и водорода, который включает, по меньшей мере, одну тройную связь. Примеры соответствующих алкинильных групп имеют 2-6 атомов углерода, предпочтительно, 2-4 атома углерода, и включают пропинил и бутинил.
"Арил" относится к ароматическому органическому остатку, состоящему из атомов углерода и водорода, который предпочтительно имеет 5-10 атомов углерода, более предпочтительно, 5 или 6 атомов углерода. Пример представляет собой фенильное кольцо.
"Гетероарил" относится к арильной группе, как определено выше, в которой один из атомов углерода заменен гетероатомом, который, например, выбран из N, O или S, или остатком, содержащим гетероатом (например, N, O и/или S). Примеры возможных гетероарильных групп включают пиридин, и тому подобное.
"Алкокси" относится к группе -O-алкил.
"Аминоалкилен" относится к группе -алкилен-NR14R15.
Если группа определяется как являющаяся "необязательно замещенной", она может иметь один или несколько заместителей, выбранных из Hal, C1-6 алкила или C1-6 алкокси.
"Hal" относится к F, Cl, Br, и I. Предпочтительный Hal представляет собой F и Cl, более предпочтительно, F.
Соединения по настоящему изобретению, имеющие один или несколько оптически активных атомов углерода, могут существовать как рацематы и рацемические смеси, диастереомерные смеси и индивидуальные диастереомеры, энантиомерные смеси и отдельные энантиомеры, таутомеры, атропизомеры и ротамеры. Все изомерные формы включены в настоящее изобретение. Соединения, описанные в настоящем изобретении, содержащие олефиновые двойные связи, включают E и Z геометрические изомеры. Также включенными в настоящее изобретение являются все солевые формы, полиморфы, гидраты и сольваты.
Предпочтительные определения, приведенные в разделе "Определения", относятся ко всем вариантам осуществления, описанным ниже, если не утверждается иного.
Подробное описание изобретения
В первом варианте осуществления настоящее изобретение относится к соединению общей формулы (I)

Пиридиновые кольца A, B и C являются независимо незамещенными или замещенными одним или несколькими заместителями, которые независимо выбраны из группы, состоящей из: C1-6-алкилен-C(=NR13)-NHR14, C1-6-алкилен-C(O)-NH-CN, C1-6-алкилен-C(O)-NR16-C1-6-алкилен-NR14R15, C1-6-алкилен-C(O)-NR14R15, C1-6-алкилен-C(O)-OR13, C1-6-алкилен-NR16-C(=NR13)-NR14R15, C1-6-алкилен-NR16-C(O)-NR14R15, C1-6-алкилен-NR16-C(O)-OR14, C1-6-алкилен-NR16-C(O)-R14, C1-6-алкилен-NR14R15, C1-6-алкилен-NR16-SO2-NR14R15, C1-6-алкилен-NR16-SO2R14, C(=NR13)-NHR14, C(O)-NH-CN, C(O)-NR16-C1-6-алкилен-NR14R15, C(O)-NR16-NR14R15, C(O)-NR14R15, C(O)-OH, C(O)-OR13, C(O)-R13, CHal3, CN, Hal, NO2, NR13-C(=NR13)-NR14R15, NR16-C(O)-NR14R15, NR16-C(O)-OR14, NR16-C(O)-R14, NR14R15, NR16-SO2-NR14R15, NR16-SO2R14, O-C1-6-алкилен-C(O)-NR14R15, O-C(O)-NR14R15, O-C(O)-R13, OR13, S(O)t-C1-6-алкилен-C(O)-NR14R15, S(O)t-C(O)-OR13, S(O)tR13, SO2-NR14R15, C1-6-алкила, C5-10-циклоалкила, C5-10-циклоалкил-C1-6-алкилена, 5-10-членного гетероциклоалкила, галогеналкила, имеющего 1-6 атомов углерода, 6-10-членного гетероциклоалкил-C1-6-алкилена, C2-6-алкенила, C2-6-алкинила, C5-10-арила, 5-10-членного гетероарила, C5-10-арил-C1-6-алкилена, 5-10-членного гетероарил-C1-6-алкилена, C1-6-алкокси-C1-6-алкилена и аминоалкилена, где алкиленовая группа имеет 1-6 атомов углерода, где алкил, циклоалкил, циклоалкилалкилен, гетероциклоалкилен, гетероциклоалкилалкилен, алкенил, алкинил, арил, гетероарил, арилалкилен, гетероарилалкилен, алкоксиалкилен и аминоалкилен могут быть необязательно замещенными. В предпочтительном варианте осуществления пиридиновые кольца A, B и C являются независимо незамещенными или замещенными одним или двумя заместителями. В предпочтительном варианте осуществления заместители независимо выбраны из группы, состоящей из: C1-6-алкила, галогеналкила, имеющего 1-6 атомов углерода, Hal или OR13, более предпочтительно, они независимо выбраны из группы, состоящей из: C1-6-алкила или OH. Наиболее предпочтительно, пиридиновые кольца A, B и C являются незамещенными.
L1 и L2 независимо выбраны из остатков, имеющих формулу (a) или (b)

где, по меньшей мере, один из L1 или L2 имеет формулу (b). Это обеспечивает то, что соединение, имеющее общую формулу (I), содержит 2,6-диаминопиридиновый остаток.
В формуле (a) R3 выбран из группы, состоящей из C(=NOR13)-R14, C(=NR13)-NR14R15, C(O)-C(=NR13)-NR14R15, C(O)-NR14R15, C(O)-OR13, R13, S(O)tNR14R15 и S(O)tR13. В предпочтительном варианте осуществления R3 представляет собой R13. В более предпочтительном варианте осуществления R3 выбран из группы, состоящей из водорода и C1-6-алкила. В еще более предпочтительном варианте осуществления R3 представляет собой водород.
R4, R5, R6, и R7 независимо выбраны из группы, состоящей из водорода, C1-6-алкилен-C(=NR13)NHR14, C1-6-алкилен-C(O)-NH-CN, C1-6-алкилен-C(O)-NR16-C1-6-алкилен-NR14R15, C1-6-алкилен-C(O)-NR16-NR14R15, C1-6-алкилен-C(O)-NR14R15, C1-6-алкилен-C(O)-OR13, C1-6-алкилен-NR16C(=NR13)NR14R15, C1-6-алкилен-NR16-C(O)-NR14R15, C1-6-алкилен-NR16-C(O)OR14, C1-6-алкилен-NR16-C(O)R14, C1-6-алкилен-NR14R15, C1-6-алкилен-NR16-SO2-NR14R15, C1-6-алкилен-NR16-SO2-R14, C(=NR13)NHR14, C(O)-NH-CN, C(O)-NR16-C1-6-алкилен-NR14R15, C(O)-NR16-NR14R15, C(O)-NR14R15, C(O)-OH, C(O)-OR16, CHal3, CN, CO-NR14R15, CO-R13, Hal, NO2, NR16C(=NR13)NR14R15, NR16-C(O)-NR14R15, NR16-C(O)-OR14, NR16-C(O)-R14, NR14R15, NR16-SO2-NR14R15, NR16-SO2-R13, O-C1-6-алкилен-C(O)-NR14R15, O-C(O)-NR14R15, OC(O)-R13, OR13, S(O)t-C1-6-алкилен-C(O)-NR14R15, S(O)t-C1-6-алкилен-C(O)-OR13, S(O)t-C(O)-NR14R15, S(O)t-C(O)-OR13, S(O)tR13, SO2-NR14R15, и SO2OR13. В предпочтительном варианте осуществления R4, R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и C1-6-алкила. В еще более предпочтительном варианте осуществления R4, R5, R6, и R7 представляют собой водород.
p равно 1 или 2. В предпочтительном варианте осуществления p равно 1.
В формуле (b) R12 выбран из группы, состоящей из C(=NOR13)-R14, C(=NR13)-NR14R15, C(O)-C(=NR13)-NR14R15, C(O)-NR14R15, C(O)-OR13, R13, S(O)tNR14R15, и S(O)tR13. В предпочтительном варианте осуществления R12 представляет собой R13. В более предпочтительном варианте осуществления R12 выбран из группы, состоящей из водорода и C1-6-алкила. В еще более предпочтительном варианте осуществления R12 представляет собой водород.
R8, R9, R10, и R11 независимо выбраны из группы, состоящей из водорода, C1-6-алкилен-C(=NR13)NHR14, C1-6-алкилен-C(O)-NH-CN, C1-6-алкилен-C(O)-NR16-C1-6-алкилен-NR14R15, C1-6-алкилен-C(O)-NR16-NR14R15, C1-6-алкилен-C(O)-NR14R15, C1-6-алкилен-C(O)-OR13, C1-6-алкилен-NR16C(=NR13)NR14R15, C1-6-алкилен-NR16-C(O)-NR14R15, C1-6-алкилен-NR16-C(O)OR14, C1-6-алкилен-NR16-C(O)R14, C1-6-алкилен-NR14R15, C1-6-алкилен-NR16-SO2-NR14R15, C1-6-алкилен-NR16-SO2-R14, C(=NR13)NHR14, C(O)-NH-CN, C(O)-NR16-C1-6-алкилен-NR14R15, C(O)-NR16-NR14R15, C(O)-NR14R15, C(O)-OH, C(O)-OR16, CHal3, CN, CO-NR14R15, CO-R13, Hal, NO2, NR16C(=NR13)NR14R15, NR16-C(O)-NR14R15, NR16-C(O)-OR14, NR16-C(O)-R14, NR14R15, NR16-SO2-NR14R15, NR16-SO2-R13, O-C1-6-алкилен-C(O)-NR14R15, O-C(O)-NR14R15, OC(O)-R13, OR13, S(O)t-C1-6-алкилен-C(O)-NR14R15, S(O)t-C1-6-алкилен-C(O)-OR13, S(O)t-C(O)-NR14R15, S(O)t-C(O)-OR13, S(O)tR13, SO2-NR14R15, и SO2OR13. В предпочтительном варианте осуществления R8, R9, R10 и R11 независимо выбраны из группы, состоящей из водорода и C1-6-алкила. В еще более предпочтительном варианте осуществления R8, R9, R10, и R11 представляют собой водород.
q равно 0, 1 или 2. В предпочтительном варианте осуществления q равно 1, поскольку эти соединения имеют улучшенную растворимость по сравнению с соединениями, в которых q равно 2.
t равно 1 или 2.
R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-6-алкила, C5-10-циклоалкила, C5-10-циклоалкил-C1-6-алкила, 5-10-членного гетероциклоалкила, галогеналкила, имеющего 1-6 атомов углерода, C5-10-гетероциклоалкил-C1-6-алкила, C2-6-алкинила, C5-10-арила, 5-10-членного гетероарила, C5-10-арил-C1-6-алкила, 5-10-членного гетероарил-C1-6-алкила или аминоалкила, где алкильная группа имеет 1-6 атомов углерода, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, галогеналкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил, который может необязательно быть замещенным, или R1 и R2, когда взятые вместе с атомом азота, к которому они присоединены, могут образовывать 3-8-членное кольцо, которое может необязательно содержать один или несколько дополнительных гетероатомов, выбранных из O, S или NR3, и где 3-8-членное кольцо может быть необязательно замещенным. В предпочтительном варианте осуществления R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-6-алкила, C5-10-циклоалкила и C5-10-арила. В более предпочтительном варианте осуществления R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-6-алкила и фенила. Наиболее предпочтительно, R1 и R2 независимо выбраны из группы, состоящей из водорода и C1-6-алкила. Еще более предпочтительно, R1 представляет собой водород и R2 представляет собой метил.
R16 независимо выбран из группы, состоящей из C(=NOR13)-R14, C(=NR13)-NR14R15, C(O)-C(=NR13)-NR14R15, C(O)-NR14R15, C(O)-OR13, R13, S(O)tNR14R15, и S(O)tR13. В предпочтительном варианте осуществления R16 представляет собой R13. В более предпочтительном варианте осуществления R16 выбран из группы, состоящей из водорода и C1-6-алкила. В еще более предпочтительном варианте осуществления R16 представляет собой водород.
R13 независимо выбран из группы, состоящей из водорода, C1-6-алкила, C5-10-циклоалкила, C5-10-циклоалкил-C1-6-алкила, 5-10-членного гетероциклоалкила, галогеналкила, имеющего 1-6 атомов углерода, 5-10-членного гетероциклоалкил-C1-6-алкила, C2-6-алкинила, C5-10-арила, 5-10-членного гетероарила, C5-10-арил-C1-6-алкила, 5-10-членного гетероарил-C1-6-алкила или аминоалкила, где алкильная группа имеет 1-6 атомов углерода, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, галогеналкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления R13 независимо выбран из группы, состоящей из водорода, C1-6-алкила и C5-10-арила. В более предпочтительном варианте осуществления R13 независимо выбран из водорода, C1-6-алкила и фенила, еще более предпочтительно, из водорода и C1-6-алкила.
R14 и R15 независимо выбраны из группы, состоящей из водорода, C1-6-алкила, C5-10-циклоалкила, C5-10-циклоалкил-C1-6-алкила, 5-10-членного гетероциклоалкила, галогеналкила, имеющего 1-6 атомов углерода, 5-10-членного гетероциклоалкил-C1-6-алкила, C2-6-алкинила, C5-10-арила, 5-10-членного гетероарила, C5-10-арил-C1-6-алкила, 5-10-членного гетероарил-C1-6-алкила или аминоалкила, где алкильная группа имеет 1-6 атомов углерода, где алкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, галогеналкил, гетероциклоалкилалкил, алкенил, алкинил, арил, гетероарил, арилалкил, гетероарилалкил и аминоалкил могут быть необязательно замещенными. В предпочтительном варианте осуществления R14 и R15 независимо выбраны из группы, состоящей из водорода, C1-6-алкила и C5-10-арила. В более предпочтительном варианте осуществления R14 и R15 независимо выбраны из водорода, C1-6-алкила и фенила, еще более предпочтительно из водорода и C1-6-алкила.
В случае, когда NR14R15 R14 и R15, взятые вместе с атомом азота, к которому они присоединены, могут образовывать 3-8-членное кольцо, которое может необязательно содержать один или несколько дополнительных гетероатомов, выбранных из O, S, или NR3, и где 3-8-членное кольцо может быть необязательно замещенным. В этом варианте осуществления 3-8-членное кольцо может представлять собой, например, пирролидин, пиррол, пиперидин или пиридин.
Предпочтительные соединения по настоящему изобретению представляют собой
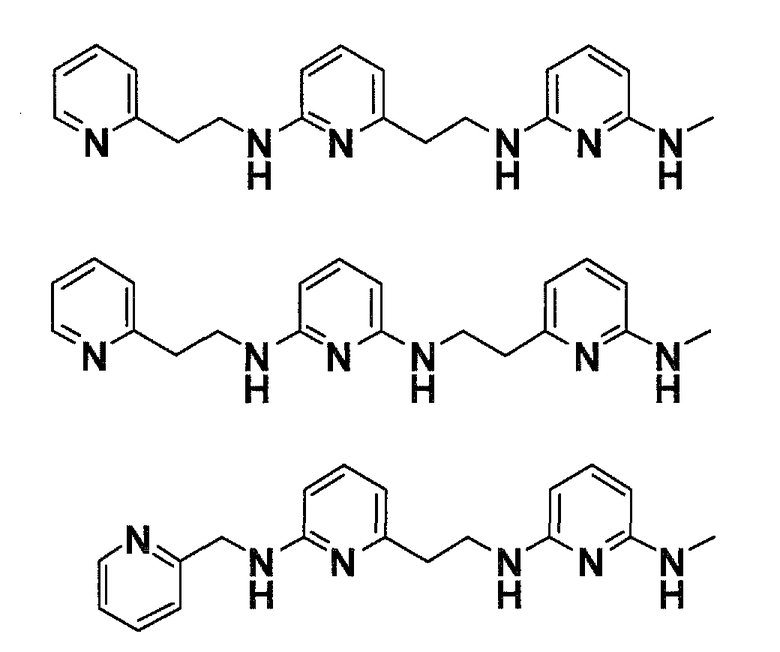
Соединения по настоящему изобретению, которые имеют общую формулу (I), имеют одновременно хорошую фармацевтическую активность и хорошую растворимость. Это связано с присутствием 3 пиридиновых колец и 2,6-диаминопиридиновой подструктуры.
Соединения по настоящему изобретению могут быть получены в соответствии с обычными способами, которые сходны, например, со способами, описанными в WO 2008/061795.
Соединения по настоящему изобретению могут синтезироваться с помощью общих способов, показанных на Схемах 1-8. Эти способы приводятся в качестве иллюстративных примеров, и не являются ограничивающими.
Общая схема синтеза для получения аминовых фрагментов, содержащих два пиридильных остатка с x = 1 или 2.
Схема 1

Коммерчески доступный 2-бром-6-метил-пиридин обрабатывают литийдиизопропиламином в соответствующем растворителе при -78°C для генерирования соответствующего аниона. Реакция аниона при -78°C с помощью диметилформамида и обработка реакционной смеси боргидридом натрия дает после очистки соответствующее гидроксильное производное с одной вытянутой в длину боковой цепью из атомов углерода. Защита гидроксильного остатка с помощью триизопропилсилилхлорида в соответствующем растворителе и использование соответствующего основания дает после очистки защищенный спирт. Реакция бром-заместителя защищенного спирта с соответствующим амином, с использованием условий аминирования Бухвальда (Pd-катализатор, лиганд, основание и растворитель) дает после очистки продукт связывания. Boc-защита аминового остатка достигается посредством нагревания исходного вещества вместе с ди-трет-бутил дикарбонатом, и последующей очистки. Силильную защитную группу удаляют с помощью тетра-н-бутиламмоний фторида с получением после очистки гидрокси производного. После активирования гидроксильного остатка с помощью метилсульфонилхлорида в соответствующем растворителе и использования соответствующего основания, промежуточное метилсульфонильное производное преобразуется в соответствующее азидное производное, посредством нагревания вместе с азидом натрия в соответствующем растворителе. Очистка дает желаемое азидное производное. Азидное производное обрабатывают трифенилфосфином, используя условия реакции Штаудингера, с получением соответствующего амина. Очистка дает желаемый аминовый фрагмент.
Общая схема синтеза для альтернативного получения аминового элемента структуры, содержащего два пиридила, соединенных C2-линкером.
Схема 2

Защита гидроксильного остатка коммерчески доступного (6-бромпиридин-2-ил)метанола с помощью триизопропилсилилхлорида в соответствующем растворителе и использование соответствующего основания дает после очистки защищенный спирт. Реакция бром-заместителя защищенного спирта с соответствующим амином, с использованием условий аминирования Бухвальда (Pd-катализатор, лиганд, основание и растворитель), дает продукт связывания после очистки. Boc-защита аминового остатка достигается посредством нагревания исходного вещества вместе с ди-трет-бутил дикарбонатом, и последующей очистки. Силил-защитную группу удаляют с помощью тетра-н-бутил аммоний фторида с получением после очистки гидрокси производного. После активирования гидроксильного остатка с помощью метилсульфонилхлорида в соответствующем растворителе и использования соответствующего основания, промежуточное метилсульфонильное производное преобразуется в соответствующее нитрильное производное посредством нагревания вместе с цианидом натрия в соответствующем растворителе. Очистка дает желаемое нитрильное производное. Обработка нитрильного производного с помощью никель(II)-хлорида и боргидрида натрия в соответствующем растворителе с последующей очисткой дает желаемый аминовый фрагмент.
Общая схема синтеза для получения аминового элемента структуры, содержащего два пиридильных остатка с помощью C3-линкеров
Схема 3
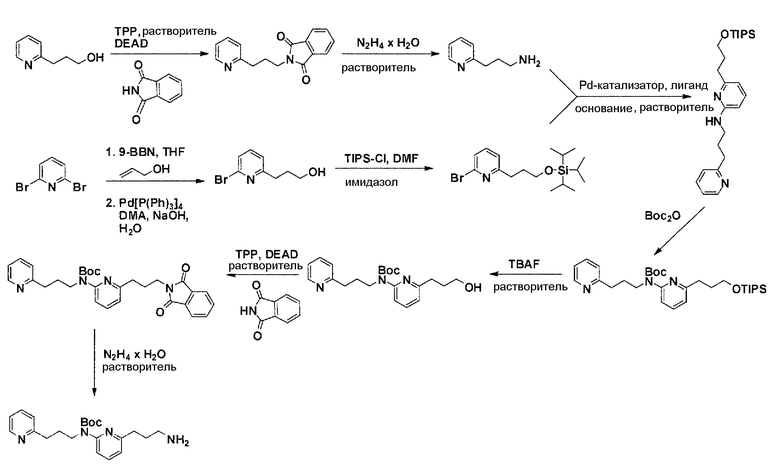
Коммерчески доступный 3-(пиридин-2-ил)пропан-1-ол преобразуется в соответствующее аминовое производное с помощью реакции Мицунобу, с использованием фталимида, с последующей обработкой очищенного промежуточного соединения с помощью гидразина гидрата в соответствующем растворителе. Очистка дает желаемый амин с C3-линкером. Коммерчески доступный 2,6-дибромпиридин получает возможность для взаимодействия с продуктом добавления аллилового спирта и 9-BBN в соответствующем растворителе, с использованием соответствующего Pd-катализатора в соответствующей смеси растворителей, с получением после очистки желаемого продукта алкилирования. Защита гидроксильного остатка с помощью триизопропилсилилхлорида в соответствующем растворителе и использование соответствующего основания дает после очистки защищенный спирт. Реакция бром-заместителя защищенного спирта с помощью соответствующего амина, с использованием условий аминирования Бухвальда (Pd-катализатор, лиганд, основание и растворитель), дает после очистки продукт связывания. Boc-защита аминового остатка достигается посредством нагревания исходного вещества вместе с ди-трет-бутилдикарбонатом и последующей очистки. Силил-защитную группу удаляют с помощью тетра-н-бутиламмоний фторида с получением гидрокси производного после очистки. Гидрокси производное преобразуют в соответствующее аминовое производное с помощью реакции Мицунобу, с использованием фталимида с последующей обработкой очищенного промежуточного соединения с помощью гидразина гидрата в соответствующем растворителе. Очистка дает желаемый аминовый фрагмент с C3-линкерами.
Общая схема синтеза для получения аминового элемента структуры, содержащего один пиридильный остаток
Схема 4

Коммерчески доступный 2-амино-6-метилпиридин нагревают вместе с ди-трет-бутилдикарбонатом с получением после очистки моно-Boc-защищенного производного. Boc-производное обрабатывают литийдиизопропиламином в соответствующем растворителе при -78°C для генерирования соответствующего аниона. Реакция аниона при -78°C с диметилформамидом и обработка реакционной смеси боргидридом натрия дают соответствующее гидроксильное производное с одной вытянутой в длину боковой цепью из атомов углерода после очистки. После активирования гидроксильного остатка с помощью метилсульфонилхлорида в соответствующем растворителе и использования соответствующего основания, промежуточное, метилсульфонильное производное преобразуется в соответствующее азидное производное, посредством нагревания вместе с азидом натрия в соответствующем растворителе. Очистка дает желаемое азидное производное. Моно-Boc-амино заместитель азидного производного обрабатывают гидридом натрия в соответствующем растворителе с последующим взаимодействием с метилйодидом с получением N-метилированного азидного производного после очистки. N-метилированное азидное производное обрабатывают трифенилфосфином, с использованием условий реакции Штаудингера, с получением соответствующего амина. Очистка дает желаемый аминовый фрагмент.
Общая схема синтеза для получения бромированного элемента структуры, содержащего один пиридильный остаток
Схема 5

Коммерчески доступный 2-амино-6-бром-пиридин обрабатывают ди-трет-бутилдикарбонатом, соответствующим основанием и 4-диметиламино пиридином в соответствующем растворителе с получением после очистки моно-Boc-производного. Обработка моно-Boc-производного гидридом натрия в соответствующем растворителе с последующим взаимодействием с метилйодидом, дает после очистки желаемый бромированный элемент структуры.
Общая схема синтеза для получения бромированного элемента структуры, содержащего два пиридильных остатка
Схема 6

Коммерчески доступный 2,6-дибромпиридин нагревают с соответствующим амином и соответствующим основанием в соответствующем растворителе с получением после очистки продукта моно-аминирования. Нагрев продукта аминирования вместе с ди-трет-бутилдикарбонатом дает после очистки желаемый бромированный элемент структуры.
Общая схема синтеза для получения соединений с x = 1, 2 или 3 и y = 1 или 2
Схема 7
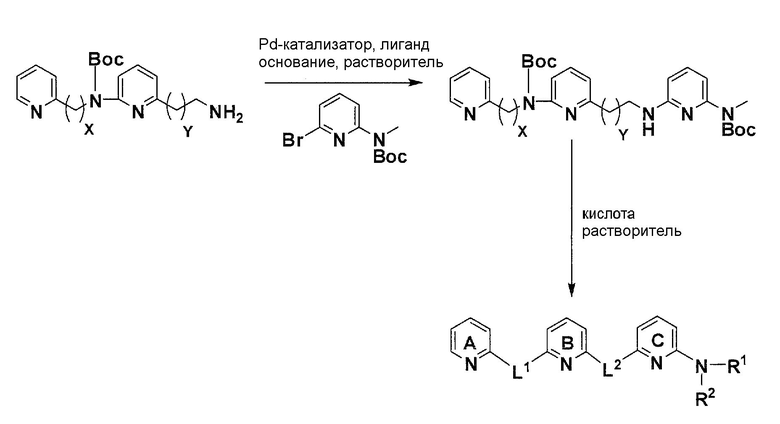
С использованием соответствующего амина и бромированного элемента структуры из описанной выше реакции аминирования, катализируемой Pd, с использованием условий Бухвальда (Pd-катализатор, лиганд, основание, растворитель), получают после очистки желаемый продукт аминирования. Отщепление Boc-защитных групп с помощью кислоты в соответствующем растворителе дает после лиофилизации желаемое конечное соединение.
Общая схема синтеза для получения соединений по настоящему изобретению
Схема 8

С использованием соответствующего амина и бромированного элемента структуры из описанной выше реакции аминирования, катализируемой Pd, с использованием условий Бухвальда (Pd-катализатор, лиганд, основание, растворитель), получают после очистки желаемый продукт аминирования. Расщепление Boc-защитных групп с помощью кислоты в соответствующем растворителе дает после лиофилизации желаемое конечное соединение.
Хотя является возможным введение соединений по настоящему изобретению по-отдельности, является предпочтительным приготовление их в виде фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Таким образом, настоящее изобретение также предусматривает фармацевтическую композицию, которая содержит терапевтически эффективное количество соединения формулы (I) в смеси с фармацевтически приемлемым наполнителем.
Фармацевтически приемлемые наполнители хорошо известны в области фармации, и описаны, например, в Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1991). Фармацевтический наполнитель может быть выбран в соответствии с предполагаемым способом введения и стандартной фармацевтической практикой. Наполнитель должен быть приемлемым в том смысле, что он не является вредным для того, кто его принимает.
Фармацевтически пригодные для использования наполнители, которые могут быть использованы при приготовлении фармацевтической композиции по настоящему изобретению, могут содержать, например, носители, связующие, разбавители, растворители, такие как одноатомные спирты, такие как этанол, изопропанол, и многоатомные спирты, такие как гликоли, и съедобные масла, такие как соевое масло, кокосовое масло, оливковое масло, сафлоровое масло, хлопковое масло, масляные сложные эфиры, такие как этилолеат, изопропилмиристат, связующие вещества, вспомогательные вещества, солюбилизаторы, загущающие агенты, стабилизаторы, разрыхлители, глиданты, смазывающие агенты, буферные агенты, эмульсификаторы, смачивающие агенты, суспендирующие агенты, подслащивающие агенты, красители, ароматизаторы, агенты для нанесения покрытий, консерванты, антиоксиданты, технологические добавки, модификаторы для доставки лекарственных средств и усилители, такие как фосфат кальция, стеарат магния, тальк, моносахариды, дисахариды, крахмал, желатин, целлюлоза, метилцеллюлоза, натрий карбоксиметилцеллюлоза, декстроза, гидроксипропил-ß-циклодекстрин, поливинилпирролидон, воски с низкими температурами плавления и ионообменные смолы.
Способы введения (доставки) соединений по настоящему изобретению включают, но, не ограничиваясь этим, один или несколько способов из: перорального (например, в виде таблетки, капсулы или в виде проглатываемого раствора), местного, мукозального (например, в виде назального спрея или аэрозоля для ингаляции), назального, парентерального (например, в форме инъекции), гастроинтестинального, интраспинального, внутрибрюшинного, внутримышечного, внутривенного, внутриматочного, внутриглазного, интрадермального, внутричерепного, внутритрахеального, интравагинального, интрацеребровентрикулярного, внутрицеребрального, подкожного, офтальмического (включая интравитреальное или интракамеральное), чрезкожного, ректального, буккального, эпидурального и сублингвального.
При офтальмическом введении, соединения могут вводиться, например, в форме глазных капель.
Например, соединения могут вводиться перорально в форме таблеток, капсул, суппозиториев, эликсиров, растворов или суспензий, которые могут содержать ароматизирующие или окрашивающие агенты, для применений с непосредственным, замедленным, модифицированным, задержанным по времени, импульсным или контролируемым высвобождением.
Таблетки могут содержать наполнители, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин, разрыхлители, такие как крахмал (предпочтительно, кукурузный, картофельный крахмал или крахмал тапиоки), натрий крахмал гликолят, натрий кроскармелоза, и определенные сложные силикаты, и связующие вещества для гранулирования, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза (HPC), сахароза, желатин и смола акации. В дополнение к этому, могут включаться смазывающие агенты, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Твердые композиции сходного типа могут также использоваться в качестве наполнителей в желатиновых капсулах. Предпочтительные наполнители в этом отношении включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров, агент может объединяться с различными подслащивающими или ароматизирующими агентами, подкрашивающими материалами или красителями, с эмульсифицирующими и/или суспендирующими агентами, и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин, и их сочетания.
Если соединения по настоящему изобретению вводятся парентерально, тогда примеры такого введения включают один или несколько способов из: внутривенного, внутриартериального, внутрибрюшинного, интратекального, интравентрикулярного, интрауретрального, интрастернального, внутричерепного, внутримышечного или подкожного способа введения соединения; и/или посредством использования методик вливания. Для парентерального введения, соединения лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например, достаточное количество солей или глюкозы, чтобы сделать раствор изотоничным с кровью. Водные растворы должны содержать соответствующие буферы (предпочтительно для поддержания pH от 3 до 9), если это необходимо. Приготовление соответствующих парентеральных препаратов при стерильных условиях легко осуществляется с помощью стандартных фармацевтических технологий, хорошо известных специалистам в данной области.
Как указано, соединения по настоящему изобретению могут вводиться интраназально или посредством ингаляции, и их удобно доставлять в форме ингалятора с сухим порошком или в форме аэрозольного спрея из контейнера высокого давления, насоса, распылителя или небулайзера с помощью использования соответствующего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, гидрофторалкана, такого как 1,1,1,2-тетрафторэтан (HFA134AT) или 1,1,1,2,3,3,3-гептафторпропан (HFA 227EA), диоксида углерода или другого соответствующего газа. В случае аэрозоля высокого давления, стандартная лекарственная форма может определяться посредством создания клапана для доставки отмеренного количества. Контейнер высокого давления, насос, распылитель или небулайзер может содержать раствор или суспензию активного соединения, например, с использованием смеси этанола и пропеллента в качестве растворителя, который может дополнительно содержать смазывающее вещество, например, сорбитантриолеат. Капсулы и картриджи (изготовленные, например, из желатина) для использования в ингаляторе или инсуффляторе могут приготавливаться так, чтобы они содержали порошкообразную смесь соединения и соответствующей порошкообразной основы, такой как лактоза или крахмал.
Альтернативно, соединения по настоящему изобретению могут вводиться в форме суппозитория или пессария, или они могут наноситься местным образом в форме геля, гидрогеля, примочки, раствора, крема, мази или мелкодисперсного порошка. Соединения по настоящему изобретению могут также вводиться дермально или трансдермально, например, посредством использования пластыря на коже.
Они могут также вводиться с помощью пульмонарного или ректального способа. Они могут также вводиться с помощью глазного способа. Для офтальмического использования, соединения могут приготавливаться в виде микронизированных суспензий в изотоническом, имеющем заданный pH, стерильном солевом растворе, или, предпочтительно, в виде растворов в изотоническом, имеющем заданный pH, стерильном солевом растворе, необязательно, в сочетании с консервантом, таким как бензилалконийхлорид. Альтернативно, они могут приготавливаться в виде мази, такой как петролят.
Для нанесения местным образом на кожу, соединения по настоящему изобретению могут приготавливаться в виде соответствующей мази, содержащей активное соединение, суспендированное или растворенное, например, в смеси с одним или несколькими веществами: минеральным маслом, жидким вазелином, белым вазелином, пропиленгликолем, эмульсифицирующим воском и водой. Альтернативно, они могут приготавливаться в виде соответствующих примочек или кремов, суспендированных или растворенных, например, в смеси с одним или несколькими из следующих веществ: минерального масла, сорбитана моностеарата, полиэтиленгликоля, жидкого парафина, полисорбата 60, воска сложных цетиловых эфиров, цетеарилового спирта, 2-октилдодеканола, бензилового спирта и воды.
Благодаря их высокой растворимости, соединения по настоящему изобретению являются особенно пригодными для способов введения, при которых соединения доставляются в жидкой среде. Примеры представляют собой глазные капли и другие растворы.
Как правило, врач определит реальную дозу, которая будет наиболее пригодной для использования для индивидуального субъекта. Конкретный уровень дозы и частота дозирования для любого конкретного индивидуума могут изменяться и будут зависеть от множества факторов, включая активность конкретного используемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выделения, сочетание лекарственных средств, тяжесть конкретного состояния и конкретного индивидуума, подвергающегося воздействию терапии.
Предлагаемая доза соединений в соответствии с настоящим изобретением для введения людям (с массой тела приблизительно 70 кг) составляет от 0,1 мг до 1 г, предпочтительно, от 1 мг до 500 мг активного ингредиента на стандартную лекарственную форму. Стандартная лекарственная форма может вводиться, например, 1-4 раза в день. Доза будет зависеть от способа введения. Будет понятно, что может быть необходимым осуществление рутинных изменений в дозировке в зависимости от возраста и массы пациента, а также от тяжести состояния, которое должно лечиться. Точная доза и способ ее введения, в конечном счете, будут определяться лечащим врачом или ветеринаром.
Соединения по настоящему изобретению могут быть также использованы в сочетании с другими терапевтическими агентами. Когда соединение по настоящему изобретению используется в сочетании со вторым терапевтическим агентом, активным против такого же заболевания, доза каждого соединения может отличаться от его дозы, когда соединение используется отдельно.
Сочетания, упоминаемые выше, могут удобно быть представлены для использования в форме фармацевтического препарата. Индивидуальные компоненты таких сочетаний могут быть введены либо последовательно, либо одновременно в виде раздельных или сочетанных фармацевтических препаратов с помощью любого обычного способа. Когда введение является последовательным, любое из соединений по настоящему изобретению или второго терапевтического агента может вводиться первым. Когда введение является одновременным, сочетание может быть введено либо в одной и той же, либо в иной фармацевтической композиции. Когда их объединяют в одном препарате, будет понятно, что оба соединения должны быть стабильными и совместимыми друг с другом и с другими компонентами препарата. Когда их вводят по-отдельности, они могут быть предусмотрены в виде любого удобного препарата, удобно, таким способом, как известно для таких соединений в данной области.
Фармацевтические композиции по настоящему изобретению могут быть получены способом, известным, таким образом, специалистам в данной области, как описано, например, в Remington's Pharmaceutical Sciences, 15th Ed., Mack Publishing Co., New Jersey (1991).
Заболевания, которые можно лечить с помощью соединений по настоящему изобретению, могут быть связаны с образованием аномальных белковых структур, в частности, аномальных структур β-листов. В контексте настоящего изобретения, аномальная белковая структура представляет собой белковую структуру, которая появляется, когда белок или пептид перестраивается из трехмерной структуры, которую он, как правило, принимает у здоровых индивидуумов, в другую трехмерную структуру, которая связана с патологическим состоянием. Подобным же образом, аномальная структура β-листа в контексте настоящего изобретения представляет собой структуру β-листа, которая появляется, когда белок или пептид перестраивается из трехмерной структуры, которую он, как правило, принимает у здорового индивидуума, в структуру β-листа, которая связана с патологическим состоянием.
В частности, в одном из вариантов осуществления заболевания, которое можно лечить с помощью соединений по настоящему изобретению, представляют собой заболевания или состояния, связанные с амилоидными или амилоидоподобными белками.
Это группа заболеваний и расстройств включает глазные болезни, связанные с амилоидными белками, которые нацелены на различные ткани глаза, такие как зрительный кортекс, включая кортикальную недостаточность зрения; как передняя камера и зрительный нерв, включая глаукому; как хрусталик, включая катаракту из-за осаждения бета-амилоидных белков; как стекловидное тело, включая глазные амилоидозы; как сетчатка, включая первичную дегенерацию сетчатки и дегенерацию желтого пятна, в частности, возрастную дегенерацию желтого пятна; как зрительный нерв, включая друзы зрительного нерва, оптическую нейропатию и неврит зрительного нерва; и как роговая оболочка, включая решетчатую дистрофию.
Способность соединения к ингибированию агрегации Aβ может определяться, например, с использованием флюоресцентной корреляционной спектроскопии, как описано в Rzepecki et al., J. Biol. Chem., 2004, 279(46), 47497-47505, или посредством использования спектрофлюоресцентного анализа, с использованием тиофлавина T.
В другом варианте осуществления соединения по настоящему изобретению могут быть использованы для лечения или ослабления воздействий глазных болезней, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, в частности, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, связанными с бета-амилоидными белками, такими как, например, нейронная дегенерация. Указанные патологические нарушения могут происходить, например, в различных тканях глаз, таких как зрительный кортекс, приводя к кортикальной недостаточности зрения; как передняя камера и как зрительный нерв, приводя к глаукоме; как хрусталик, приводя к катаракте из-за осаждения бета-амилоидных белков; как стекловидное тело, приводя к глазному амилоидозу; как сетчатка, приводя к первичной дегенерации сетчатки и дегенерации желтого пятна, например, к возрастной дегенерации желтого пятна; как зрительный нерв, приводя к друзам зрительного нерва, оптической нейропатии и невриту зрительного нерва; и как роговая оболочка, приводя к решетчатой дистрофии. Соединения по настоящему изобретению, как показано, являются особенно пригодными для лечения или предотвращения глаукомы.
Соединения в соответствии с настоящим изобретением могут также быть предусматрены в форме смеси, по меньшей мере, с одним дополнительным биологически активным соединением и/или фармацевтически приемлемым носителем и/или разбавителем и/или наполнителем. Соединение и/или дополнительное биологически активное соединение предпочтительно присутствует в терапевтически эффективном количестве.
Природа дополнительного биологически активного соединения будет зависеть от предполагаемого использования смеси. Дополнительное биологически активное вещество или соединение может оказывать свое биологическое воздействие с помощью такого же, как у соединения в соответствии с настоящим изобретением, или сходного механизма, или с помощью независимого механизма действия, или посредством разнообразных родственных и/или независимых механизмов действия.
Как правило, дополнительное биологически активное соединение может включать бета-блокаторы, ингибиторы угольной ангидразы, альфа- или бета-адренергические агонисты, простагландины, парасимпатомиметики, ингибиторы холинэстераз, усилители синтеза накопления или высвобождения ацетилхолина, агонисты постсинаптических рецепторов ацетилхолина или антагонисты N- рецепторов метил-D-аспартата глютамата. В частности, дополнительное биологически активное соединение может выбираться из группы, состоящей из соединения, используемого при лечении амилоидоза, соединений против оксидативного стресса, анти-апоптических соединений, хелаторов металлов, ингибиторов репарации ДНК, таких как пирензепин и метаболиты, 3-амино-1-пропансульфоновой кислоты (3APS), 1,3-пропандисульфоната (1,3PDS), активаторов α-секретазы, ингибиторов β- и γ-секретазы, нейротрансмиттера, расщепителей β-листа, аттрактантов клеточных компонентов, вычищающих/понижающих содержание бета-амилоидных белков, ингибиторов расщепленных бета-амилоидных белков с N-окончаниями, включая пироглютаматированный бета-амилоидный белок 3-42, противовоспалительных молекул или ингибиторов холинэстеразы (ChEI), таких как такрин, ривастигмин, донепезил и/или галантамин, агонистов M1, других лекарственных средств, включая любое лекарственное средство, модифицирующее амилоиды, и питательные добавки, антитела, включая любое функционально эквивалентное антитело или его функциональные части, Aβ антигенный пептидный фрагмент, состоящий из единственного или повторяющегося фрагмента секвенирования из множества смежных аминокислотных остатков из части Aβ пептида с N-окончанием.
В другом варианте осуществления, смеси в соответствии с настоящим изобретением могут содержать мемантин вместе с соединением по настоящему изобретению и, необязательно, фармацевтически приемлемый носитель и/или разбавитель и/или наполнитель.
Еще в одном варианте осуществления настоящего изобретения предусматрены смеси, которые содержат в качестве дополнительного биологически активного соединения агент для понижения внутриглазного давления вместе с соединением в соответствии с настоящим изобретением и, необязательно, фармацевтически приемлемый носитель и/или разбавитель и/или наполнитель.
В одном из предпочтительных вариантов осуществления дополнительное биологически активное соединение представляет собой антитело, включая любое функционально эквивалентное антитело или его функциональные части. Антитело может предпочтительно представлять собой моноклональное, химерное или гуманизированное антитело.
В другом аспекте настоящего изобретения, предусматрена смесь, содержащая, в дополнение к соединению по настоящему изобретению, антитело, включая его функциональные части, или, более конкретно, моноклональное антитело, включая его функциональные части, которые распознают амилоид β (Aβ) и связываются с ним, в частности, с нативной конформацией амилоида β, то есть, с амилоидными олигомерами и волокнами, но не с линейными амилоидными частицами.
В частности, указанные антитела способны ингибировать in vitro и in vivo агрегацию амлоидогенных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких, например, как Aβ мономерные пептиды 1-39; 1-40, 1-41, 1-42 или 1-43, но в особенности, Aβ1-42 мономерных пептидов, в виде высокомолекулярных полимерных амилоидных фибрилл или нитей. Посредством ингибирования агрегации амилоидогенных мономерных пептидов, эти антитела способны предотвращать или замедлять образование амилоидных бляшек, в частности, амилоидной формы (1-42), которая, как известно, становится нерастворимой посредством изменения вторичной конформации и которая составляет главную часть амилоидных бляшек в мозгу у заболевших животных или людей.
В другом аспекте настоящего изобретения, смесь содержит антитела, которые при совместном инкубировании вместе с предварительно сформированными высокомолекулярными полимерными амилоидными фибриллами или нитями, сформированными посредством агрегации амилоидных мономерных пептидов, в частности, β-амилоидных мономерных пептидов, таких, например, как Aβ мономерные пептиды 1-39; 1-40, 1-41, 1-42 или 1-43, но в особенности, Aβ1-42 мономерных пептидов, способны к дезагрегации указанных высокомолекулярных полимерных амилоидных фибрилл или нитей. Посредством дезагрегации амилоидогенных полимерных фибрилл или нитей, эти антитела способны предотвращать или замедлять образование амилоидных бляшек, что приводит к ослаблению симптомов, связанных с заболеванием, и к замедлению и обращению вспять его развития.
Еще в одном аспекте настоящего изобретения, смесь содержит антитело, но в особенности, моноклональное антитело или его функциональные части, это антитело является бифункциональным или биспецифичным в том смысле, что оно демонстрирует как свойство ингибирования агрегации, так и свойство дезагрегации, как определено в настоящем документе, выше, в частности, в сочетании с высоким уровнем конформационный чувствительности.
В одном из вариантов осуществления, смесь содержит антитело, которое распознает конформационный эпитоп, в частности, конформационный эпитоп, который присутствует в части амилоидного β пептида с N-окончанием, в частности, находящийся в следующей сердцевинной области части амилоидного β пептида с N-окончанием:
и связывается с ним.
В частности, это эпитоп, локализованный в области β-амилоидного белка между аминокислотными остатками 12 и 24, в частности, между остатками 14 и 23, более конкретно, между аминокислотными остатками 14 и 20, содержащий три различных сайта распознавания и связывания, эти остатки в основном участвуют в связывании β-амилоидного белка и располагаются в положении 16, 17, и в положении 19 и 20, и в положении 14, соответственно.
В конкретном варианте осуществления, смесь по настоящему изобретению содержит, в дополнение к соединению по настоящему изобретению, антитело, в частности, бифункциональное антитело, но в особенности, моноклональное антитело, в частности, бифункциональное моноклональное антитело, включая любое функционально эквивалентное антитело или его функциональные части, это антитело имеет характерные свойства антитела, продуцируемого линией клеток гибридом, выбранной из группы, состоящей из FP 12H3, FP 12H3-C2 и FP 12H3-G2, депозит от 01 декабря 2005 года и 09 декабря 2005 года, соответственно, как DSM ACC2752, DSM ACC 2750 и DSM ACC2751, соответственно, ET 7E3, депозит от 08 декабря 2005 года как DSM ACC2755 и EJ 7H3, депозит от 08 декабря 2005 года как DSM ACC2756.
Более конкретно, настоящее изобретение относится к антителу, включая любое функционально эквивалентное антитело или его функциональные части, продуцируемое линией клеток гибридом, выбранной из группы, состоящей из FP 12H3, FP 12H3-C2 и FP 12H3-G2, депозит от 01 декабря 2005 года и 09 декабря 2005 года, соответственно, как DSM ACC2752, DSM ACC 2750 и DSM ACC2751, соответственно, ET 7E3, депозит от 08 декабря 2005 года как DSM ACC2755, и EJ 7H3, депозит от 08 декабря 2005 года, как DSM ACC2756.
Указанные выше антитела описаны в опубликованной Международной заявке WO 2007/068412, которая включена в настоящий документ в качестве ссылки.
В дополнительном аспекте, антитело, которое содержится в смеси в соответствии с настоящим изобретением, представляет собой химерное антитело или его фрагмент, или гуманизированное антитело или его фрагмент. Эти и другие антитела, которые могут быть пригодными для использования в смесях в соответствии с настоящим изобретением, описаны, например, в Международной заявке PCT/US2007/073504, поданной 13 июля 2007 года.
Если антитело представляет собой гуманизированное антитело, оно предпочтительно демонстрирует легкую цепь и тяжелую цепь, как изображено в SEQ ID No. 2 и SEQ ID No. 4 Международной заявки № PCT/US2007/073504, или демонстрирует вариабельную область легкой цепи и вариабельную область тяжелой цепи, как изображено в SEQ ID No. 1 и SEQ ID No. 3 Международной заявки № PCT/US2007/073504. Эти последовательности также показаны в прилагаемом списке последовательностей.
Еще в одном аспекте настоящего изобретения, предусматрена смесь, которая содержит, в дополнение к соединению по настоящему изобретению, и как описано в настоящем документе выше, пептидный фрагмент из части Aβ пептида с N-окончанием, в частности, фрагмент Aβ пептида, состоящий из единственного или повторяющегося фрагмента секвенирования, в пределах между смежными аминокислотными остатками 13 и 15 из части Aβ пептида с N-окончанием, но, в частности, фрагмент Aβ пептида, состоящий из аминокислотных остатков, выбранных из группы, состоящей из остатков 1-15, 1-14 и 1-13 из части Aβ пептида с N-окончанием, более конкретно, из остатков 1-15, включая их функционально эквивалентные фрагменты, но в особенности, фрагмент Aβ пептида, как рассмотрено в настоящем документе, выше, присоединенный к частице носителю/вспомогательному веществу или инкорпорированный или разбавленный в нем, например в липосоме. Фрагмент пептида может содержаться в композиции вакцины. В частности, пептидный антиген модифицируется липофильным или гидрофобным остатком, что облегчает вставку в липидный бислой липосомного носителя/иммунного вспомогательного вещества, в частности, с помощью липофильного или гидрофобного остатка, который функционирует в качестве якоря для пептида в липосомном бислое, и который имеет такой размер, который приводит к позиционированию и стабилизации пептида вблизи поверхности липосомы.
В другом варианте осуществления настоящего изобретения, липофильный или гидрофобный остаток представляет собой жирную кислоту, триглицерид или фосфолипид, но в особенности, жирную кислоту, триглицерид или фосфолипид. В частности, гидрофобный остаток представляет собой пальмитиновую кислоту, и липосомный препарат может, в дополнение к этому, содержать вспомогательное вещество, такое, например, как липид A, алюминиевые квасцы, фосфат кальция, интерлейкин 1 и/или микрокапсулы полисахаридов и белков, но в особенности, детоксифицированный липид A, такой как монофосфорил- или дифосфориллипид A, или алюминиевые квасцы.
Эти и другие композиции, которые можно соответствующим образом использовать в смесях по настоящему изобретению, описаны, например, в опубликованной Международной заявке WO 2007/068411.
Диагностика заболевания или состояния, связанного с амилоидными белками, или предрасположенность к заболеванию или состоянию, связанному с амилоидным белком, у пациента может быть осуществлена посредством детектирования специфичного связывания соединения в соответствии с настоящим изобретением с амилоидным белком в образце или in situ, которое включает приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный антиген, с соединением по настоящему изобретению, которое связывает амилоидный белок, предоставляя возможность соединению по настоящему изобретению для связывания с амилоидным белком с образованием комплекса соединение/белок, детектирование образования комплекса соединение/белок и коррелирование присутствия или отсутствия комплекса соединение/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участке тела, необязательное сравнение количества указанного комплекса соединение/белок с нормальным контрольным значением, где увеличение в количестве указанного агрегата по сравнению с нормальным контрольным значением может указывать на то, что указанный пациент страдает от заболевания или состояния, связанного с амилоидными белками, или имеет риск его развития.
Мониторинг минимального остаточного заболевания у пациента после лечения с помощью соединения или смеси по настоящему изобретению, может осуществляться посредством детектирования специфичного связывания соединения по настоящему изобретению с амилоидным белком в образце или in situ, которое включает приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный антиген, с соединением по настоящему изобретению, которое связывается с амилоидным белком, предоставляя возможность соединению для связывания с амилоидным белком с образованием комплекса соединение/белок, детектирование образования комплекса соединение/белок и коррелирование присутствия или отсутствия комплекса соединение/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участке тела, необязательное сравнение количества указанного комплекса соединение/белок с нормальным контрольным значением, где увеличение количества указанного агрегата по сравнению с нормальным контрольным значением может указывать на то, что указанный пациент может по-прежнему страдать минимальным остаточным заболеванием.
Предсказание реакции пациента на лечение с помощью соединения или композиции или смеси по настоящему изобретению, может осуществляться посредством детектирования специфичного связывания соединения в соответствии с настоящим изобретением с амилоидным белком в образце или in situ, которое включает приведение в контакт образца или конкретной части тела или участка тела, содержащего, как ожидается, амилоидный белок, с соединением по настоящему изобретению, которое связывает амилоидный белок, предоставление возможности соединению для связывания с амилоидным белком с образованием комплекса соединение/белок, детектирование образования комплекса соединения/белок и коррелирование присутствия или отсутствия комплекса соединения/белок с присутствием или отсутствием амилоидного белка в образце или конкретной части тела или участке тела, необязательное сравнение количества указанного комплекса соединение/белок до и после лечения, где уменьшение количества указанного агрегата может указывать на то, что указанный пациент имеет высокий потенциал для положительной реакции на лечение.
Биологические образцы, которые могут быть использованы при диагностике заболевания или состояния, связанного с амилоидными белками, для диагностирования предрасположенности к заболеванию или состоянию, связанному с амилоидными белками, или для мониторинга минимального остаточного заболевания у пациента, или для предсказания реакции пациента на лечение с помощью соединения или композиции, или смеси в соответствии с настоящим изобретением, и как описано в настоящем документе, выше, представляют собой, например, жидкости, такие как сыворотка, плазма, слюна, желудочные выделения, слизь, цереброспинальная жидкость, лимфатическая жидкость, и тому подобное, или образцы тканей или клеток, полученные из организма, такие как нервная, мозговая, сердечная или сосудистая ткань. Для определения присутствия или отсутствия амилоидного белка в образце может использоваться любой иммунологический анализ, известный специалистам в данной области (смотри Harlow and Lane, Antibodies: A Laboratory Manual (Cold Spring Harbor Laboratory, New York, 1988, 555-612), такой, например, как анализы, которые используют опосредованные методы детектирования с использованием вторичных реагентов для детектирования, анализы ELISA и анализы с помощью иммунопреципитации и агглютинации. Подробное описание этих анализов приведено, например, в WO96/13590, Maertens and Stuyver, Zrein et al. (1998) и WO96/29605.
Для диагностики in situ, соединение или композиция, или смесь в соответствии с настоящим изобретением, и как описано в настоящем документе, выше, может быть введена в организм, который должен диагностироваться с помощью способов, известных в данной области, таких, например, как внутривенная, интраназальная, внутрибрюшинная, внутрицеребральная, внутриартериальная инъекция, так что может происходить специфичное связывание между соединением в соответствии с настоящим изобретением и амилоидным антигеном. Комплекс соединение/белок может детектироваться с помощью метки, прикрепленной к соединению.
Иммуннологические анализы, используемые в диагностических применениях или в применениях для диагностики предрасположенности к заболеванию или состоянию, связанному с амилоидным белком, или для мониторинга минимального остаточного заболевания у пациента, или для предсказания реакции пациента на лечение с помощью соединения или композиции, или смеси в соответствии с настоящим изобретением, и как описано в настоящем документе, выше, как правило, основаны на меченых антигенах, антителах или вторичных реагентах для детектирования. Эти белки или реагенты могут метиться соединениями, как правило, известными специалистам в данной области, включая ферменты, радиоактивные изотопы и флуоресцентные, люминесцентные и хромогенные вещества, включая окрашенные частицы, такие как коллоидное золото и латексные шарики. Среди них, радиоактивное мечение может быть использовано почти для всех типов анализов и с множеством вариантов. Метки, конъюгированные с ферментами, являются особенно полезными, когда радиоактивность должна быть исключена или когда требуются быстрые результаты. Флюорохромы, хотя и требуют дорогостоящего оборудования для их использования, обеспечивают очень чувствительный метод детектирования. Антитела, пригодные для этих анализов, включают моноклональные антитела, поликлональные антитела и аффинно очищенные поликлональные антитела.
Альтернативно, соединение по настоящему изобретению может метиться опосредованно, посредством взаимодействия с мечеными веществами, которые имеют сродство к иммуноглобулину, такими как белок A или G, или вторые антитела. Антитело может конъюгироваться со вторым веществом, и детектироваться с помощью меченого третьего вещества, имеющего сродство ко второму веществу, конъюгированному с антителом. Например, антитело может конъюгироваться с биотином, и конъюгат антитело-биотин детектируется с использованием меченого авидина или стрептавидина. Подобным же образом, антитело может конъюгироваться с гаптеном, и конъюгат антитело-гаптен детектируется с использованием меченого антитела анти-гаптен.
Специалисты в данной области будут знать эти и другие соответствующие метки, которые могут быть использованы в соответствии с настоящим изобретением. Связывание этих меток с антителами или их фрагментами может осуществляться с использованием стандартных методик, повсеместно известных специалистам в данной области. Типичные методики описаны Kennedy, J. H., et al., 1976 (Clin. Chim. Acta 70:1-31) и Schurs, A. H. W. M., et al. 1977 (Clin. Chim Acta 81:1-40). Методики связывания, рассмотренные в дальнейшем, представляют собой глютаральдегидный метод, периодатный метод, дималеимидный метод и другие, все они включаются в настоящий документ в качестве ссылок.
Современные иммунологические анализы используют метод двойного антитела для детектирования присутствия анализируемого вещества, где антитело метится опосредованно посредством способности взаимодействовать со вторым антителом, которое метится с помощью детектируемой метки. Второе антитело предпочтительно является таким, которое связывается с антителами животного, из которого получают моноклональное антитело. Другими словами, если моноклональное антитело представляет собой антитело мыши, тогда меченое второе антитело представляет собой антитело анти-мышь. Для моноклонального антитела, которое должно использоваться при анализе, описанном ниже, эта метка предпочтительно представляет собой шарик, покрытый антителом, в частности, магнитный шарик. Для поликлонального антитела, которое должно использоваться в иммунологическом анализе, описанном в настоящем документе, метка предпочтительно представляет собой детектируемую молекулу, например, радиоактивного, флуоресцентного или электрохемилюминесцентного вещества.
Альтернативная система с двойным антителом, часто упоминаемая как система быстрого формата, поскольку она адаптирована для быстрого определения присутствия анализируемого вещества, также может быть использована в рамках настоящего изобретения. Система требует высокого сродства между антителом и анализируемым веществом. В соответствии с одним из вариантов осуществления настоящего изобретения, присутствие амилоидного антигена определяется с использованием пары антител, каждое из которых является специфичным для амилоидного антигена. Одно из указанной пары антител упоминается в настоящем документе как "детектирующее антитело", а другое из указанной пары антител упоминается в настоящем документе как "антитело захвата". Моноклональное антитело может также использоваться либо как антитело захвата, либо как детектирующее антитело. Моноклональное антитело может также использоваться и как антитело захвата, и как детектирующее антитело, вместе, в одном анализе. Таким образом, один из вариантов осуществления настоящего изобретения использует сэндвич-метод с двойным антителом для детектирования амилоидного антигена в образце биологической жидкости. В этом методе, анализируемое вещество (амилоидный антиген) заключается между детектирующим антителом и антителом захвата, антитело захвата необратимо иммобилизируется на твердой подложке. Детектирующее антитело может содержать детектирующую метку для идентификации присутствия сэндвича антитело-анализируемое вещество и, таким образом, присутствия анализируемого вещества.
Иллюстративные вещества в твердой фазе включают, но, не ограничиваясь этим, микротитровальные планшеты, пробирки из полистирола, магнитные, пластиковые или стеклянные шарики и микроскопные стекла, которые хорошо известны в области радиоиммунологического анализа и ферментного иммуноанализа. Методы связывания антител с твердыми фазами также хорошо известны специалистам в данной области. Недавно, в качестве твердых подложек стали использоваться разнообразные пористые материалы, такие как нейлон, нитроцеллюлоза, ацетат целлюлозы, стекловолокна и другие пористые полимеры.
Осаждение бляшек в ткани и/или телесной жидкости (такой как слой ганглионарных клеток сетчатки животного, в частности, млекопитающего, но в особенности, человека, страдающего глазной болезнью, связанной с патологическими нарушениями/изменениями в тканях зрительной системы, в частности, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, связанными с бета-амилоидными белками) может быть вычислен с помощью методов, известных в данной области, таких, как описаны в Ding, J. -D. et al., "Targeting age-related macular degeneration with Alzheimer's disease based immunotherapies: Anti-amyloid-b antybody attenuates pathologies in an age-related macular degeneration mouse model", Vision Research (2007), doi:10,1016/j.visres.2007.07.025.
Соединение по настоящему изобретению может также быть включено в набор для исследований для детектирования амилоидного белка. Набор для исследований, как правило, содержит контейнер, содержащий одно или несколько соединений в соответствии с настоящим изобретением и инструкции для использования соединения для целей связывания с амилоидным белком с образованием комплекса соединение/белок и детектирования образования комплекса соединение/белок, так что присутствие или отсутствие комплекса соединение/белок коррелирует с присутствием или отсутствием амилоидного белка.
Термин "набор для исследований", как правило, относится к любому диагностическому набору, известному в данной области. Более конкретно, последний термин относится к диагностическому набору, как описано в Zrein et al. (1998).
ПРИМЕРЫ
Синтез соединений по настоящему изобретению, ингибирующих агрегацию Ab1-42, и анализ их биологической активности описан в следующих далее примерах, которые не рассматриваются как ограничивающие изобретение каким-либо образом.
Ингибирование агрегации Ab1-42 с помощью соединений по настоящему изобретению может быть измерено с использованием любого соответствующего анализа, известного в данной области. Описан стандартный анализ in vitro для измерения ингибирования агрегации.
Примеры приготовления
Все реагенты и растворители получают из коммерческих источников, и используют без дополнительной очистки. Протонные (1H) спектры регистрируют на ЯМР спектрометре на 400 МГц в дейтерированных растворителях. Масс-спектры (MS) регистрируют на спектрометре Finnigan MAT TSQ 7000. Хроматографию осуществляют с использованием силикагеля (Fluka: Silica gel 60, 0,063-0,2 мм), и соответствующие растворители являются такими, как указано в конкретных примерах. Тонкослойную хроматографию (TLC) осуществляют на пластинках с силикагелем, с УФ детектированием. Препаративную тонкослойную хроматографию (Prep-TLC) осуществляют на пластинках со слоем силикагеля толщиной 0,5 мм или 1 мм (Analtech: Uniplate, F254), и растворители указываются в конкретных примерах.
Пример приготовления 1 (соединение 5 и соединение 13):

Стадия A
Коммерчески доступный 2 амино-6-пиколин (10,8 г, 100 ммоль) обрабатывают раствором ди-трет-бутил дикарбоната (26,2 г, 120 ммоль) в дихлорметане (100 мл). Растворитель удаляют в вакууме, и остаток нагревают при ~70°C на песчаной бане в течение ночи. Смесь разбавляют этилацетатом (150 мл), и органическую фазу промывают 10% раствором лимонной кислоты (70 мл), насыщенным раствором бикарбоната натрия (70 мл) и насыщенным соляным раствором (70 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (10/90), для элюирования избытка реагента, затем, с использованием смеси этилацетат/петролейный эфир (20/80), с получением желаемого соединения в виде бесцветного масла, которое становится белым твердым продуктом при стоянии при комнатной температуре (19 г, 91%).
1H-ЯМР (400 МГц, CDCl3): д = 1,53 (с, 9H), 2,44 (с, 3H), 6,82 (д, 1H), 7,27 (ушир.с, 1H), 7,55 (т, 1H), 7,72 (д, 1H)
Стадия B
Гидрид натрия (0,84 г, 35 ммоль) суспендируют в N,N'-диметилформамиде (50 мл), и смесь охлаждают до 0°C. При 0°C раствор указанного в заглавии соединения из Стадии A, выше (6 г, 28,8 ммоль) в N,N'-диметилформамиде (20 мл) добавляют в течение периода 5 минут. После завершения добавления, реакционную смесь перемешивают при 0°C в течение 15 минут, а затем 60 минут при комнатной температуре. Затем добавляют метилйодид (2,39 мл, 38,5 ммоль) одной порцией, и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют этилацетатом (150 мл) и 10% раствором лимонной кислоты (150 мл). Органическую фазу отделяют и водную фазу экстрагируют этилацетатом (2×100 мл). Объединенную органическую фазу промывают 10% раствором лимонной кислоты (80 мл), насыщенным раствором бикарбоната натрия (80 мл) и насыщенным соляным раствором (80 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (10/90) с получением желаемого соединения в виде бледно-желтого масла (4,75 г, 74%).
1H-ЯМР (400 МГц, CDCl3): д = 1,53 (с, 9H), 2,48 (с, 3H), 3,38 (с, 3H), 6,87 (д, 1H), 7,40 (д, 1H), 7,52 (т, 1H)
Стадия C
Раствор LDA приготавливают посредством добавления 2 M раствора н-бутиллития (12 мл, 24 ммоль) при 0°C к перемешиваемому раствору N,N'-диизопропиламина (4 мл, 28,8 ммоль) в тетрагидрофуране (60 мл). Смесь перемешивают при 0°C в течение 1 часа, а затем охлаждают до -78°C. При -78°C раствор указанного в заглавии соединения из Стадии B, выше (2,13 г, 9,6 ммоль), в тетрагидрофуране (15 мл) добавляют в течение периода 5 минут. Смесь перемешивают при -78°C в течение 45 минут и позволяют ей нагреться до -50°C. Затем смесь охлаждают до -78°C и добавляют N,N'-диметилформамид (0,76 мл, 10,3 ммоль). После 15 минут при -78°C, добавляют метанол (8,4 мл) и уксусную кислоту (0,59 мл, 12,8 ммоль). Затем добавляют боргидрид натрия (0,34 г, 9,4 ммоль) при -78°C, и смесь перемешивают в течение ночи, и дают ей возможность для достижения комнатной температуры. Реакционную смесь разбавляют этилацетатом (80 мл) и промывают 10% раствором лимонной кислоты (50 мл) и насыщенным соляным раствором (50 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (20/80) для элюирования исходного вещества (0,7 г, извлечение 35%), затем, с использованием смеси этилацетат/петролейный эфир (60/40), с получением указанного в заглавии соединения в виде бледно-оранжевого масла (1,08 г, 44%).
1H-ЯМР (400 МГц, CDCl3): д = 1,56 (с, 9H), 2,98 (т, 2H), 3,37 (с, 3H), 4,05 (т, 2H), 6,88 (д, 1H), 7,53-7,60 (м, 2H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (1,07 г, 4,27 ммоль), растворяют в дихлорметане (10 мл), и добавляют триэтиламин (1,32 мл, 9,4 ммоль). После добавления метансульфонилхлорида (0,66 мл, 8,5 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют дихлорметаном (50 мл) и промывают 10% раствором лимонной кислоты (20 мл), насыщенным раствором бикарбоната натрия (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (50/50), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,93 г, 65%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 2,90 (т, 3H), 3,15 (т, 3H), 3,40 (с, 3H), 4,68 (т, 2H), 6,90 (т, 1H), 7,53-7,60 (м, 2H)
Стадия E
Указанное в заглавии соединение из Стадии D, выше (0,93 г, 2,8 ммоль), растворяют в N,N'-диметилацетамиде (10 мл), и добавляют азид натрия (0,91 г, 14 ммоль). Смесь нагревают на песчаной бане при ~75°C в течение 16 часов. Смесь разбавляют этилацетатом (80 мл) и 10% раствором лимонной кислоты (30 мл). Органическую фазу отделяют, промывают насыщенным раствором бикарбоната натрия (25 мл) и насыщенным соляным раствором (25 мл). Органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/петролейный эфир (20/80), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,69 г, 89%).
1H-ЯМР (400 МГц, CDCl3): д = 1,55 (с, 9H), 3,00 (т, 3H), 3,40 (с, 3H), 3,71 (т, 2H), 6,88-6,92 (м, 1H), 7,53-7,60 (м, 2H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,69 г, 2,5 ммоль), растворяют в тетрагидрофуране (20 мл), и добавляют трифенилфосфин (0,79 г, 3 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов, и добавляют воду (10 мл). Перемешивание продолжают в течение 5 часов и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5) для элюирования неполярных побочных продуктов, затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 7 M аммиака в метаноле (10 мл на 500 мл), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,52 г, 82%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 1,68 (ушир.с, 2H), 2,88 (т, 2H), 3,11 (т, 2H), 3,40 (с, 3H), 6,88 (д, 1H), 7,50 (д, 1H), 7,53 (т, 1H)
Стадия G
Раствор LDA приготавливают посредством добавления 1,6 M раствора н-бутиллития в гексане (51 мл, 81,2 ммоль) при 0°C к перемешиваемому раствору N,N'-диизопропиламина (13,5 мл, 97,4 ммоль) в тетрагидрофуране (60 мл). Смесь перемешивают при 0°C в течение 15 мин, а затем добавляют при -78°C к раствору коммерчески доступного 2-бром-6-метил-пиридина (5 г, 29,1 ммоль) в тетрагидрофуране (90 мл). Смесь перемешивают при -78°C в течение 25 минут, а затем добавляют N,N'-диметилформамид (7,9 мл, 107 ммоль). После 30 минут при -78°C, добавляют метанол (80 мл) и уксусную кислоту (6,1 мл, 132 ммоль). Затем добавляют боргидрид натрия (1,1 г, 28 ммоль) при -78°C, и смесь перемешивают в течение ночи и дают ей возможность для достижения комнатной температуры. Реакционную смесь разбавляют этилацетатом (150 мл) и промывают 10% раствором лимонной кислоты (80 мл) и насыщенным соляным раствором (80 мл). Органическую фазу отделяют, и водную фазу экстрагируют этилацетатом (2×150 мл). Объединенную органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5), с получением указанного в заглавии соединения в виде бледно-желтого масла (5 г, 85%).
1H-ЯМР (400 МГц, CDCl3): д = 3,01 (т, 2H), 3,09 (т, 1H), 4,02 (кв., 2H), 7,16 (д, 1H), 7,34 (д, 1H), 7,43 (т, 1H)
Стадия H
Указанное в заглавии соединение из Стадии G, выше (5 г, 24,75 ммоль), растворяют в N,N'-диметилформамиде (100 мл), и добавляют имидазол (4,84 г, 74,25 ммоль). После добавления хлортриизопропилсилана (7,92 мл, 37,1 ммоль), смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь разбавляют простым диэтиловым эфиром (300 мл) и промывают 10% раствором лимонной кислоты (3×40 мл) и насыщенным соляным раствором (100 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (5/95), с получением указанного в заглавии соединения в виде бесцветной жидкости (7,36 г, 83%).
1H-ЯМР (400 МГц, CDCl3): д = 0,92-1,13 (м, 21H), 3,00 (т, 2H), 4,08 (кв., 2H), 7,22 (д, 1H), 7,33 (д, 1H), 7,45 (т, 1H)
Стадия I
Указанное в заглавии соединение из Стадии H, выше (0,21 г, 0,6 ммоль) и указанное в заглавии соединение из Стадии F, выше (0,16 г, 0,63 ммоль), растворяют в толуоле (11 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,082 г, 0,12 ммоль) и трет-бутилатом натрия (0,16 г, 1,63 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,054 г, 0,06 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~80-85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (60 мл) и водой (20 мл). Органическую фазу промывают насыщенным раствором бикарбоната натрия (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием смеси этилацетат/н-гептан (20/80), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,26 г, 78%).
1H-ЯМР (400 МГц, CDCl3): д = 0,92-1,13 (м, 21H), 1,52 (с, 9H), 2,83 (т, 2H), 3,03 (т, 2H), 3,42 (с, 3H), 3,66 (кв., 2H), 4,02 (т, 2H), 4,95 (ушир.с, 1H), 6,26 (д, 1H), 6,49 (д, 1H), 6,88 (дд, 1H), 7,31 (т, 1H), 7,54-7,57 (м, 2H)
Стадия J
Указанное в заглавии соединение из Стадии I, выше (0,26 г, 0,53 ммоль), растворяют в тетрагидрофуране (1 мл), и добавляют ди-трет-бутил дикарбонат (0,17 г, 0,84 ммоль). После добавления 4-диметиламинопиридина (0,006 г, 0,05 ммоль), смесь нагревают на песчаной бане при ~65°C в течение ночи. Добавляют другую загрузку ди-трет-бутил дикарбоната (0,17 г, 0,84 ммоль) и 4-диметиламинопиридина (0,006 г, 0,05 ммоль), и продолжают нагрев при ~75°C в течение 12 часов. Затем оставшийся растворитель удаляют, и после добавления ди-трет-бутил дикарбоната (0,17 г, 0,84 ммоль), нагрев продолжают при ~75°C в течение ночи. Остаток очищают с помощью хроматографии на силикагеле с использованием н-гептана для элюирования избытка смеси ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (10/90), с получением указанного в заглавии соединения в виде бесцветного масла (0,27 г, 80%).
1H-ЯМР (400 МГц, CDCl3): д = 0,92-1,13 (м, 21H), 1,47 (с, 9H), 1,52 (с, 9H), 2,97 (т, 2H), 3,09 (т, 2H), 3,38 (с, 3H), 4,06 (т, 2H), 4,31 (т, 2H), 6,83-6,87 (м, 1H), 6,93 (д, 1H), 7,35 (д, 1H), 7,47-7,52 (м, 3H)
Стадия K
Указанное в заглавии соединение из Стадии J, выше (0,27 г, 0,428 ммоль), растворяют в ацетонитриле (5 мл), и добавляют 1 M раствор тетрабутиламмония фторида (2,14 мл, 2,14 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение выходных, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бесцветного масла (0,19 г, 92%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,53 (с, 9H), 2,98 (т, 2H), 3,07 (т, 2H), 3,3,5 (с, 3H), 3,78 (ушир.с, 1H), 3,98-4,04 (м, 2H), 4,28 (т, 2H), 6,86-6,88 (м, 2H), 7,39 (д, 1H), 7,45-7,57 (м, 3H)
Стадия L
Указанное в заглавии соединение из Стадии K, выше (0,19 г, 0,396 ммоль), растворяют в дихлорметане (2 мл), и добавляют триэтиламин (0,12 мл, 0,9 ммоль). После добавления метансульфонилхлорида (0,06 мл, 0,8 ммоль), смесь перемешивают при комнатной температуре в течение 1 часа. Смесь концентрируют, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (50/50), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,2 г, 90%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,53 (с, 9H), 2,90 (с, 3H), 3,09 (т, 2H), 3,15 (т, 2H), 3,37 (с, 3H), 4,31 (т, 2H), 4,68 (т, 2H), 6,86 (д, 1H), 6,91 (д, 1H), 7,47-7,59 (м, 4H)
Стадия M
Указанное в заглавии соединение из Стадии L, выше (0,2 г, 0,36 ммоль), растворяют в N,N'-диметилацетамиде (1,3 мл), и добавляют азид натрия (0,12 г, 1,8 ммоль). Смесь нагревают при ~75°C на песчаной бане в течение ночи. Смесь разбавляют этилацетатом (25 мл) и 10% лимонной кислотой (10 мл). Органическую фазу отделяют, промывают насыщенным соляным раствором (10 мл), сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (20/80), с получением указанного в заглавии соединения в виде бесцветного масла (0,16 г, 92%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,53 (с, 9H), 3,01 (т, 2H), 3,10 (т, 2H), 3,37 (с, 3H), 3,73 (т, 2H), 4,32 (т, 2H), 6,84 (дд, 1H), 6,90 (д, 1H), 7,46-7,55 (м, 4H)
Стадия N
Указанное в заглавии соединение из Стадии M, выше (0,16 г, 0,33 ммоль), растворяют в тетрагидрофуране (4 мл), и добавляют трифенилфосфин (0,1 г, 0,39 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 часов, а затем добавляют воду (2 мл). Перемешивание продолжают в течение 14 часов, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5), затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл), с получением указанного в заглавии соединения в виде бесцветного масла (0,13 г, 85%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,53 (с, 9H), 2,90 (т, 1H), 3,08 (т, 2H), 3,14 (т, 2H), 3,37 (с, 3H), 4,32 (т, 2H), 6,85-6,89 (м, 2H), 7,39 (д, 1H), 7,44-7,55 (м, 3H)
Стадия O
Указанное в заглавии соединение из Стадии N, выше (0,13 г, 0,28 ммоль), и 2-бромпиридин (0,043 г, 0,27 ммоль) растворяют в толуоле (4,8 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,034 г, 0,059 ммоль) и трет-бутилатом натрия (0,082 г, 0,853 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладия хлороформа (0,025 г, 0,027 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~80-85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (30 мл) и водой (10 мл). Органическую фазу промывают насыщенным раствором бикарбоната натрия (10 мл) и насыщенным соляным раствором (10 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием смеси этилацетат/н-гептан (60/40) для элюирования смеси продуктов реакции. Менее полярный продукт отделяют от более полярного продукта с помощью пластин для препаративной TLC (Analtech, 0,5 мм) с использованием смеси этилацетат/н-гептан (70/30) в качестве подвижной фазы, с получением указанных в заглавии соединений.
Менее полярный продукт: (0,029 г, бледно-желтое масло, 16%)
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 1,51 (с, 9H), 3,19 (т, 2H), 3,32 (с, 3H), 4,30 (т, 2H), 4,59 (т, 2H), 6,81-6,88 (м, 4H), 7,07-7,09 (м, 2H), 7,34 (д, 1H), 7,43-7,51 (м, 5H), 8,30-8,32 (м, 2H)
Более полярный продукт: (0,035 г, бледно-желтое масло, 22%)
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 18H), 3,03 (т, 2H), 3,10 (т, 2H), 3,32 (с, 3H), 3,68-3,74 (м, 2H), 4,32 (т, 2H), 5,00 (ушир.с, 1H), 6,34 (д, 1H), 6,51-6,56 (м, 1H), 6,82-6,89 (м, 2H), 7,31-7,53 (м, 5 H), 8,08 (ушир.с, 1H)
Стадия P
Более полярный продукт из Стадии O, выше (0,035 г, 0,06 ммоль), растворяют в хлороформе (1,1 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (1,1 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (2 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают с получением указанного в заглавии соединения в виде оранжевого стекла (0,026 г, 90%).
1H-ЯМР (400 МГц, D2O): д = 2,92 (с, 3H), 3,03-3,09 (м, 4H), 3,68-3,76 (м, 4H), 6,63 (д, 1H), 6,73 (д, 1H), 6,79-6,87 (м, 3H), 6,92 (д, 1H), 7,68-7,85 (м, 4H)
(MS (ESI); m/z = 349,52 (MH+)
Стадия Q
Менее полярный продукт из Стадии O, выше (0,029 г, 0,046 ммоль), растворяют в хлороформе (0,8 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (0,8 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (2 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде оранжевого стекла (0,024 г, 97%).
1H-ЯМР (400 МГц, D2O): д = 2,91 (с, 3H), 3,00 (т, 2H), 3,21 (т, 2H), 3,63 (т, 2H), 4,49 (т, 2H), 6,60-6,64 (м, 2H), 6,70 (д, 1H), 6,81 (д, 1H), 7,26 (т, 2H), 7,38 (д, 2H), 7,62 (т, 1H), 7,70 (т, 1H), 7,99-8,04 (м, 2H), 8,13 (д, 2H)
MS (ESI); m/z = 426,42 (MH+)
Пример приготовления 2 (соединение 1):
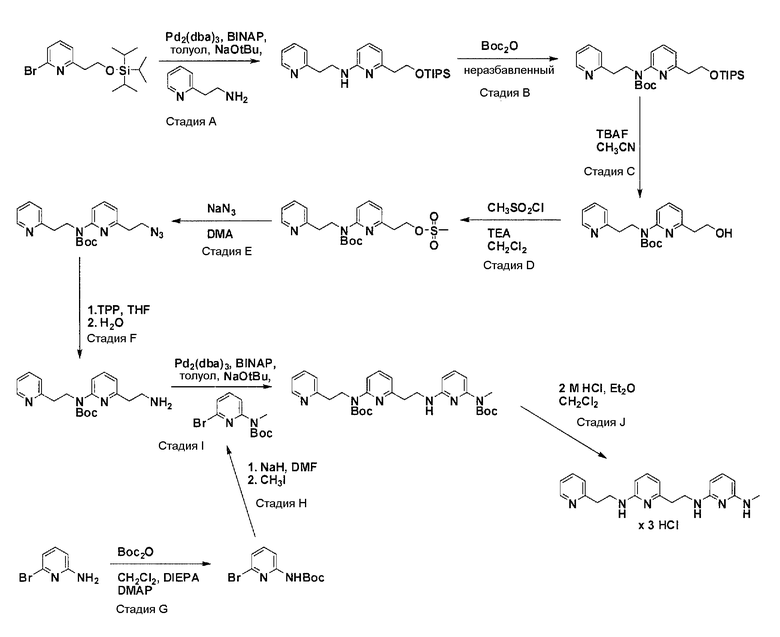
Стадия A
Указанное в заглавии соединение из Примера 1, Стадия H (0,9 г, 2,51 ммоль), и коммерчески доступный 2(2-аминоэтил)-пиридин (0,34 г, 2,78 ммоль), растворяют в толуоле (45 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,33 г, 0,48 ммоль) и трет-бутилатом натрия (0,63 г, 6,6 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением трис(дибензилиденацетон)дипалладия (0,22 г, 0,024 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~80-85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (150 мл), водой (30 мл) и насыщенным соляным раствором (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, затем с использованием этилацетата, с получением указанного в заглавии соединения в виде темно-желтого масла. Три дополнительных опыта дают в целом 3,4 г (84%).
1H-ЯМР (400 МГц, CDCl3): д = 0,98-1,10 (м, 21H), 2,85 (т, 2H), 3,10 (т, 2H), 3,68 (кв., 2H), 4,03 (т, 2H), 4,81 (ушир.с, 1H), 6,24 (д, 1H), 6,50 (д, 1H), 7,13-7,18 (м, 2H), 7,32 (т, 1H), 7,60 (т, 1H), 8,58 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (1,7 г, 4,26 ммоль), растворяют в дихлорметане (30 мл), и добавляют ди-трет-бутил дикарбонат (4,75 г, 21,3 ммоль). Растворитель удаляют, и маслянистый остаток нагревают на песчаной бане при ~75°C в течение 18-36 часов до тех пор, пока TLC не покажет расходование исходных материалов. Затем смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (10/90) для удаления избытка ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бледно-желтого масла. Два опыта дают в целом 3,6 г (86%).
1H-ЯМР (400 МГц, CDCl3): д = 0,98-1,10 (м, 21H), 1,42 (с, 9H), 2,92 (т, 2H), 3,11 (т, 2H), 4,01 (т, 2H), 4,30 (т, 2H), 6,90 (д, 1H), 7,07-7,10 (м, 1H), 7,12 (д, 1H), 7,36 (д, 1H), 7,46-7,56 (м, 2H), 8,49 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (3,9 г, 7,81 ммоль), растворяют в ацетонитриле (100 мл) и обрабатывают 1 M раствором тетрабутиламмония фторида (30 мл, 30 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде бледно-желтого масла (2,57 г, 95%).
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 2,98 (т, 2H), 3,11 (т, 2H), 4,04 (т, 2H), 4,30 (т, 2H), 6,85 (д, 1H), 7,06-7,10 (м, 1H), 7,16 (д, 1H), 7,38 (д, 1H), 7,48-7,57 (м, 2H), 8,46 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (2,57 г, 7,49 ммоль), растворяют в дихлорметане (45 мл), и добавляют триэтиламин (2,33 мл, 16,9 ммоль). Смесь охлаждают до 0°C, и добавляют метансульфонилхлорид (1,17 мл, 15 ммоль). После завершения добавления, смесь перемешивают при 0°C в течение 5 мин, а затем при комнатной температуре в течение 1 часа. Растворители выпаривают, и остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата с получением указанного в заглавии соединения в виде бледно-желтого масла (3,1 г, 98%).
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 2,91 (с, 3H), 3,11-3,17 (м, 4H), 4,30 (т, 2H), 4,66 (т, 2H), 6,89 (д, 1H), 7,08-7,11 (м, 1H), 7,15 (д, 1H), 7,46 (д, 1H), 7,53 (т, 1H), 7,56 (дт, 1H), 8,49 (д, 1H)
Стадия E
Указанное в заглавии соединение из Стадии D, выше (1,57 г, 3,73 ммоль), растворяют в N,N'-диметилацетамиде (17,5 мл), и добавляют азид натрия (1,22 г, 18,6 ммоль). Смесь нагревают при ~75°C на песчаной бане в течение ночи. Смесь разбавляют этилацетатом (150 мл) и водой (40 мл). Органическую фазу отделяют, промывают насыщенным соляным раствором (40 мл), сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бесцветного масла. Два опыта дают в целом 2,4 г, 87%.
1H-ЯМР (400 МГц, CDCl3): д = 1,43 (с, 9H), 2,97 (т, 2H), 3,12 (т, 2H), 3,69 (т, 2H), 4,31 (т, 2H), 6,87 (д, 1H), 7,07-7,10 (м, 1H), 7,12 (д, 1H), 7,46 (д, 1H), 7,51-7,58 (м, 2H), 8,50 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (2,4 г, 6,52 ммоль), растворяют в тетрагидрофуране (60 мл), и добавляют трифенилфосфин (2,15 г, 8,17 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 часов, а затем добавляют воду (30 мл). Перемешивание продолжают в течение 16 часов, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5) для удаления неполярных побочных продуктов, затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл), с получением указанного в заглавии соединения в виде бледно-желтого масла (2,1 г, 95%).
1H-ЯМР (400 МГц, CDCl3): д = 1,43 (с, 9H), 2,83 (т, 2H), 3,06-3,14 (м, 4H), 4,31 (т, 2H), 6,84 (д, 1H), 7,05-7,09 (м, 1H), 7,12 (д, 1H), 7,37 (д, 1H), 7,48 (т, 1H), 7,52 (дт, 1H), 8,47 (д, 1H)
Стадия G
Коммерчески доступный 2-амино-6-бром-пиридин (4,25 г, 24,6 ммоль) растворяют в дихлорметане (50 мл) и N,N'-диизопропилэтиламине (5,25 мл, 30,7 ммоль), и добавляют 4-диметиламинопиридин (0,15 г, 1,23 ммоль). После добавления раствора ди-трет-бутил дикарбоната (5,9 г, 27 ммоль) в дихлорметане (15 мл), смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют дихлорметаном (100 мл) и промывают 10% лимонной кислотой (50 мл) и насыщенным соляным раствором (50 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (5/95), с получением указанного в заглавии соединения в виде белого твердого продукта (2,15 г, 32%). Промывка колонки смесью этилацетат/н-гептан (10/90) дает соответствующее бис-Boc-производное в виде белого твердого продукта (1,95 г, 21%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 7,13 (д, 1H), 7,27 (ушир.с, 1H), 7,51 (т, 1H), 7,90 (д, 1H)
Бис-Boc производное:
1H-ЯМР (400 МГц, CDCl3): д = 1,48 (с, 18H), 7,27 (д, 1H), 7,40 (д, 1H), 7,60 (т, 1H)
Стадия H
Гидрид натрия (0,18 г, 7,38 ммоль) суспендируют в N,N'-диметилацетамиде (10 мл), и смесь охлаждают до 0°C. При 0°C раствор указанного в заглавии соединения из Стадии G, выше (1,66 г, 6,1 ммоль) в N,N'-диметилацетамиде (5 мл) добавляют в течение периода 5 минут. После завершения добавления, реакционную смесь перемешивают при 0°C в течение 5 минут, а затем 60 минут при комнатной температуре. Затем добавляют одной порцией метилйодид (0,5 мл, 8,12 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют этилацетатом (100 мл) и промывают 10% раствором лимонной кислоты (30 мл), насыщенным раствором бикарбоната натрия (30 мл) и насыщенным соляным раствором (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (5/95), с получением желаемого соединения в виде бесцветной жидкости (1,54 г, 90%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 3,40 (с, 3H), 7,17 (д, 1H), 7,47 (т, 1H), 7,74 (д, 1H)
Стадия I
Указанные в заглавии соединения из Стадии F (0,525 г, 1,54 ммоль) и Стадии H (0,423 г, 1,53 ммоль), выше, растворяют в толуоле (24 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,186 г, 0,3 ммоль) и трет-бутилатом натрия (0,383 г, 3,98 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением трис(дибензилиденацетон)дипалладия (0,133 г, 0,15 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (100 мл), водой (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, затем, с использованием смеси этилацетат/н-гептан (60/40) для элюирования желаемого соединения. Сырое указанное в заглавии соединение от четырех опытов дополнительно очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-оранжевого масла (3 г, 89%).
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 9H), 1,52 (с, 9H), 3,02 (т, 2H), 3,18 (т, 2H), 3,32 (с, 3H), 3,70-3,74 (м, 2H), 4,38 (т, 2H), 5,20 (ушир.с, 1H), 6,13 (д, 1H), 6,83-6,88 (м, 2H), 7,06-7,09 (м, 1H), 7,13 (д, 1H), 7,31 (т, 1H), 7,40 (д, 1H), 7,50 (т, 1H), 7,53 (дт, 1H), 8,48 (д, 1H)
Стадия J
Указанное в заглавии соединение из Стадии I, выше (3 г, 5,47 ммоль), растворяют в дихлорметане (50 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (50 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Остаток растворяют в воде (30 мл) и фильтруют через 0,2-мкм картридж фильтра (10 мл на картридж). Три картриджа промывают водой (5 мл). Объединенный фильтрат собирают, и растворитель выпаривают с использованием устройства для сушки вымораживанием, с получением указанного в заглавии соединения в виде бледно-оранжевой пены (2,2 г, 88%).
1H-ЯМР (400 МГц, D2O): д = 2,79 (с, 3H), 3,02 (т, 2H), 3,33 (т, 2H), 3,57 (т, 2H), 3,81 (т, 2H), 5,81 (д, 1H), 5,92 (д, 1H), 6,71 (д, 1H), 6,81 (д, 1H), 7,50 (т, 1H), 7,78 (т, 1H), 7,83-7,91 (м, 2H), 8,42 (т, 1H), 8,59 (д, 1H)
MS (ESI); m/z = 349,42 (MH+)
Пример приготовления 3 (альтернативная схема синтеза для соединения 1):

Стадия A
Коммерчески доступный (6-бромпиридин-2-ил)метанол (1 г, 5,3 ммоль) растворяют в N,N'-диметилформамиде (20 мл), и добавляют имидазол (0,97 г, 14,85 ммоль). После добавления триизопропилсилилхлорида (1,58 мл, 7,42 ммоль), смесь перемешивают при комнатной температуре в течение выходных. Смесь разбавляют простым диэтиловым эфиром (80 мл) и промывают 10% раствором лимонной кислоты (25 мл) и насыщенным соляным раствором (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (5/95). Фракции, содержащие защищенный спирт, собирают, и растворители выпаривают, с получением бесцветной жидкости (1,7 г, 92%). Защищенный спирт (0,85 г, 2,47 ммоль) и коммерчески доступный 2-(2-аминоэтил)пиридин (0,35 г, 2,78 ммоль), растворяют в толуоле (45 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,33 г, 0,48 ммоль) и трет-бутилатом натрия (0,63 г, 6,625 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением трис(дибензилиденацетон)дипалладия (0,22 г, 0,24 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~80-85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (1000 мл), водой (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием этилацетата, с получением указанного в заглавии соединения в виде коричневого масла. Два опыта дают в целом 1,6 г (84%).
1H-ЯМР (400 МГц, CDCl3): д = 1,06-1,08 (м, 18 H), 1,13-1,24 (м, 3H), 3,07 (т, 2H), 3,64-3,69 (м, 2H), 4,72 (с, 2H), 4,81 (ушир.с, 1H), 6,28 (д, 1H), 6,86 (д, 1H), 7,12-7,18 (м, 2H), 7,43 (т, 1H), 7,59 (дт, 1H), 8,56 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (1,6 г, 4,15 ммоль), растворяют в дихлорметане (30 мл), и добавляют ди-трет-бутил дикарбонат (4,75 г, 21,3 ммоль). Растворитель удаляют, и маслянистый остаток нагревают на песчаной бане при ~ 75°C в течение 24 часов до тех пор, пока TLC не покажет расходование исходных материалов. Затем смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (10/90) для удаления избытка ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бледно-желтого масла (1,75 г, 86%).
1H-ЯМР (400 МГц, CDCl3): д = 1,06-1,08 (м, 18 H), 1,13-1,24 (м, 3H), 1,44 (с, 9H), 3,10 (т, 2H), 4,31 (т, 2H), 4,80 (с, 2H), 7,05-7,08 (м, 1H), 7,12 (д, 1H), 7,25-7,33 (м, 2H), 7,53 (дт, 1H), 7,60 (т, 1H), 8,48 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (1,75 г, 3,6 ммоль), растворяют в ацетонитриле (45 мл) и обрабатывают 1 M раствором тетрабутиламмония фторида (13 мл, 13 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде бледно-оранжевого масла (1,16 г, 97%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 3,18 (т, 2H), 4,31 (т, 2H), 4,40-4,52 (ушир.с, 1H), 4,70 (с, 2H), 6,87-6,90 (м, 1H), 7,08-7,16 (м, 2H), 7,54-7,62 (м, 3H), 8,55 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (0,33 г, 1 ммоль), растворяют в дихлорметане (5 мл), и добавляют триэтиламин (0,28 мл, 2 ммоль). Смесь охлаждают до 0°C, и добавляют метансульфонилхлорид (0,13 мл, 1,7 ммоль). После завершения добавления, смесь перемешивают при 0°C в течение 5 мин, а затем при комнатной температуре в течение 1 часа. Растворители выпаривают, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,31 г, 75%).
1H-ЯМР (400 МГц, CDCl3): д = 1,46 (с, 9H), 3,07 (с, 3H), 3,11 (т, 2H), 4,33 (т, 2H), 5,22 (с, 2H), 7,07-7,16 (м, 3H), 7,53-7,66 (м, 3H), 8,48 (д, 1H)
Стадия E
Указанное в заглавии соединение из Стадии D, выше (0,3 г, 0,75 ммоль), растворяют в этаноле (17,5 мл), и добавляют цианид калия (0,24 г, 3,75 ммоль). Смесь нагревают при ~85°C на песчаной бане в течение 1 часа. Растворитель удаляют, и остаток растворяют с помощью этилацетата (30 мл) и воды (5 мл). Органическую фазу отделяют, промывают насыщенным соляным раствором (5 мл), сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,118 г, 46%).
1H-ЯМР (400 МГц, CDCl3): д = 1,47 (с, 9H), 3,17 (т, 2H), 3,83 (с, 2H), 4,34 (т, 2H), 7,04-7,11 (м, 2H), 7,18 (д, 1H), 7,56 (дт, 1H), 7,60-7,63 (м, 2H), 8,49 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,118 г, 0,35 ммоль), растворяют в сухом этаноле (1,2 мл), и добавляют никель(II)-хлорид (0,045 г, 0,35 ммоль). К реакционной смеси добавляют порциями боргидрид натрия (0,04 г, 1,05 ммоль) (реакция экзотермическая). После завершения добавления, черную реакционную смесь перемешивают при комнатной температуре в течение 2 часов до тех пор, пока не израсходуются все исходные материалы. Черную реакционную смесь фильтруют через слой целита, и этот слой промывают этанолом (25 мл). Бледно-желтый фильтрат выпаривают и остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (9/1) для удаления неполярных побочных продуктов, затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл), с получением указанного в заглавии соединения в виде желтого масла (0,084 г, 70%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 3,03 (т, 2H), 3,16 (т, 2H), 3,30 (т, 2H), 3,12-3,48 (ушир.с, 2H), 4,32 (т, 2H), 6,88 (д, 1H), 7,07-7,12 (м, 1H), 7,17 (д, 1H), 7,42 (д, 1H), 7,53 (т, 1H), 7,58 (дт, 1H), 8,57 (д, 1H)
MS (ESI); m/z = 342,98 (MH+)
Стадия G
Указанные в заглавии соединения из Стадии F, выше (0,084 г, 0,245 ммоль), и из Препаративного примера 2, Стадия I (0,068 г, 0,245 ммоль), выше, растворяют в толуоле (4 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,03 г, 0,048 ммоль) и трет-бутилатом натрия (0,062 г, 0,64 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением трис(дибензилиденацетон)дипалладия (0,021 г, 0,024 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (30 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием этилацетата для элюирования желаемого соединения. Сырое указанное в заглавии соединение дополнительно очищают с помощью PREP-TLC с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,04 г, 29%).
1H-ЯМР (400 МГц, CDCl3): д = 1,48 (с, 9H), 1,50 (с, 9H), 3,02 (т, 2H), 3,16 (т, 2H), 3,31 (с, 3H), 3,70 (т, 2H), 4,38 (т, 2H), 5,17-5,27 (ушир.с, 1H), 6,11 (д, 1H), 6,81-6,86 (м, 2H), 7,06-7,09 (м, 1H), 7,13 (д, 1H), 7,29 (т, 1H), 7,38 (д, 1H), 7,48 (т, 1H), 7,52 (дт, 1H), 8,48 (д, 1H)
Стадия H
Указанное в заглавии соединение из Стадии G, выше (0,075 г, 0,137 ммоль), растворяют в дихлорметане (2 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Остаток растворяют в воде (5 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают с использованием устройства для сушки вымораживанием, с получением указанного в заглавии соединения в виде бледно-оранжевой пены (0,045 г, 72%).
1H-ЯМР (400 МГц, D2O): д = 2,79 (с, 3H), 3,02 (т, 2H), 3,33 (т, 2H), 3,57 (т, 2H), 3,81 (т, 2H), 5,81 (д, 1H), 5,92 (д, 1H), 6,71 (д, 1H), 6,81 (д, 1H), 7,50 (т, 1H), 7,78 (т, 1H), 7,83-7,91 (м, 2H), 8,42 (т, 1H), 8,59 (д, 1H)
MS (ESI); m/z = 349,42 (MH+)
Пример приготовления 4 (соединение 2)
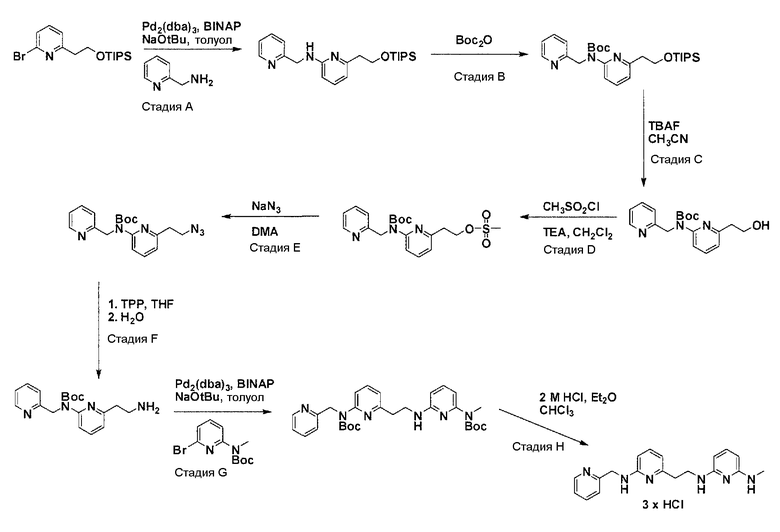
Стадия A
Указанное в заглавии соединение из Примера 1, Стадия H (0,35 г, 0,95 ммоль), и коммерчески доступный 2-аминометилпиридин (0,108 г, 1 ммоль) растворяют в толуоле (17 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,13 г, 0,19 ммоль) и трет-бутилатом натрия (0,245 г, 2,58 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладия хлороформа (0,086 г, 0,095 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~80-85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (40 мл), водой (10 мл) и насыщенным соляным раствором (10 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием этилацетата для элюирования продукта. Сырой материал снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (80/20), с получением указанного в заглавии соединения в виде бледно-оранжевого масла (0,28 г, 75%).
1H-ЯМР (400 МГц, CDCl3): д = 0,98-1,10 (м, 21H), 2,88 (т, 2H), 4,02 (т, 2H), 4,65 (д, 2H), 5,51 (ушир.с, 1H), 6,28 (д, 1H), 6,53 (д, 1H), 7,16-7,20 (м, 1H), 7,30-7,38 (м, 2H), 7,63 (дт, 1H), 8,58 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (0,28 г, 0,73 ммоль), растворяют в дихлорметане (5 мл), и добавляют ди-трет-бутил дикарбонат (0,8 г, 3,65 ммоль). Растворитель удаляют, и маслянистый остаток нагревают на песчаной бане при ~75°C в течение 3 дней. Затем смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (10/90) для удаления избытка ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,32 г, 91%).
1H-ЯМР (400 МГц, CDCl3): д = 0,85-1,10 (м, 21H), 1,38 (с, 9H), 2,88 (т, 2H), 3,90 (т, 2H), 5,31 (с, 2H), 6,91 (д, 1H), 7,12-7,17 (м, 1H), 7,28 (д, 1H), 7,54-7,62 (м, 2H), 7,67 (д, 1H), 8,54 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (0,32 г, 0,66 ммоль), растворяют в ацетонитриле (8 мл) и обрабатывают 1 M раствором тетрабутиламмония фторида (3,3 мл, 3,3 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде бледно-оранжевого масла (0,18 г, 84%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 2,91 (т, 2H), 3,69 (ушир.с, 1H), 3,88 (т, 2H), 5,27 (с, 2H), 6,88 (д, 1H), 7,15-7,20 (м, 1H), 7,30 (д, 1H), 7,59 (т, 1H), 7,63-7,67 (м, 2H), 8,54 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (0,18 г, 0,56 ммоль), растворяют в дихлорметане (3 мл), и добавляют триэтиламин (0,17 мл, 1,26 ммоль). Смесь охлаждают до 0°C, и добавляют метансульфонилхлорид (0,09 мл, 1,12 ммоль). Затем реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Затем смесь помещают в колонку с силикагелем, уравновешенную с помощью этилацетата. Колонку проявляют этилацетатом, с получением указанного в заглавии соединения в виде бледно-желтого масла (0,22 г, 96%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 2,82 (с, 3H), 3,07 (т, 2H), 4,43 (т, 2H), 5,30 (с, 2H), 6,91 (д, 1H), 7,18-7,23 (м, 1H), 7,29 (д, 1H), 7,61 (т, 1H), 7,69 (дт, 1H), 7,78 (д, 1H), 8,57 (д, 1H)
Стадия E
Указанное в заглавии соединение из Стадии D, выше (0,22 г, 0,54 ммоль), растворяют в N,N'-диметилацетамиде (2,1 мл), и добавляют азид натрия (0,18 г, 2,7 ммоль). Смесь нагревают при ~75°C на песчаной бане в течение ночи. Смесь разбавляют этилацетатом (25 мл) и водой (10 мл). Органическую фазу отделяют, промывают насыщенным соляным раствором (10 мл), сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бесцветного масла (0,15 г, 78%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 2,88 (т, 2H), 3,49 (т, 2H), 5,30 (с, 2H), 6,90 (д, 1H), 7,13-7,18 (м, 1H), 7,22 (д, 1H), 7,59-7,66 (м, 2H), 7,77 (д, 1H), 8,55 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,15 г, 0,41 ммоль), растворяют в тетрагидрофуране (4 мл), и добавляют трифенилфосфин (0,13 г, 0,5 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, а затем добавляют воду (2 мл). Перемешивание продолжают в течение выходных, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5) затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл), с получением указанного в заглавии соединения в виде бесцветного масла (0,135 г, 98%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 2,79 (т, 2H), 2,94 (т, 2H), 5,30 (с, 2H), 6,88 (д, 1H), 7,12-7,16 (м, 1H), 7,26 (д, 1H), 7,58 (т, 1H), 7,61 (дт, 1H), 7,70 (д, 1H), 8,54 (д, 1H)
Стадия G
Указанное в заглавии соединение из Стадии Приготовления F, выше (0,135 г, 0,41 ммоль), и указанное в заглавии соединение из Препаративного Примера 2, Стадия H (0,085 г, 0,4 ммоль), растворяют в толуоле (7,2 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,05 г, 0,08 ммоль) и трет-бутилатом натрия (0,125 г, 1,25 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,037 г, 0,04 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (40 мл), насыщенным раствором бикарбоната натрия (10 мл) и насыщенным соляным раствором (10 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5) для элюирования менее полярных побочных продуктов, затем, с использованием смеси дихлорметан/метанол (9/1) для элюирования смеси 2 соединений, согласно данным TLC (дихлорметан/метанол (9/1)). Растворители удаляют, и сырую смесь (87 мг, 46% в целом) непосредственно используют на следующей стадии.
Стадия E
Указанные в заглавии соединения из Стадии B, выше (0,06 г, 0,183 ммоль), и Стадии D, выше (0,048 г, 0,167 ммоль), растворяют в толуоле (3 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,021 г, 0,034 ммоль) и трет-бутилатом натрия (0,049 г, 0,51 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,015 г, 0,0167 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярных примесей, затем, с использованием смеси этилацетат/н-гептан (70/30) для элюирования желаемого соединения. Для удаления оставшихся примесей, сырой продукт дополнительно очищают с помощью Prep-TLC (Analtech, 0,5 мм) с использованием смеси дихлорметан/метанол (95/5) в качестве подвижной фазы, с получением указанного в заглавии соединения в виде темно-желтого масла (0,023 г, 25%).
1H-ЯМР (400 МГц, CDCl3): д = 1,40 (с, 9H), 1,50 (с, 9H), 2,92 (т, 2H), 3,30 (с, 3H), 3,50-3,54 (м, 2H), 4,69 (ушир.с, 1H), 5,31 (с, 2H), 5,92 (д, 1H), 6,82-6,88 (м, 2H), 7,11-7,14 (м, 1H), 7,27-7,30 (м, 2H), 7,55-7,62 (м, 2H), 7,70 (д, 1H), 8,53 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,023 г, 0,04 ммоль), растворяют в хлороформе (0,8 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (0,8 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (2 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают с получением указанного в заглавии соединения в виде темно-желтого стекла (0,016 г, 84%).
1H-ЯМР (400 МГц, D2O): д = 2,79 (с, 3H), 3,07 (т, 2H), 3,58 (т, 2H), 5,00 (с, 2H), 5,81 (д, 1H), 5,92 (д, 1H), 6,82 (д, 1H), 6,89 (д, 1H), 7,51 (т, 1H), 7,83-7,91 (м, 3H), 8,43 (т, 1H), 8,63 (д, 1H)
MS (ESI); m/z = 335,44 (MH+)
Пример приготовления 5 (соединение 6):

Стадия A
Коммерчески доступный 2-(2-аминоэтил)пиридин-2-ил (0,037 г, 0,3 ммоль) и указанное в заглавии соединение из Препаративного Примера 2, Стадия H (0,072 г, 0,25 ммоль), растворяют в толуоле (4,5 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,031 г, 0,05 ммоль) и трет-бутилатом натрия (0,075 г, 0,78 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,023 г, 0,025 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, затем, с использованием смеси этилацетат/н-гептан (80/20) для элюирования желаемого соединения. Для удаления оставшихся примесей, сырой продукт дополнительно очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (80/20), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,07 г, 85%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 3,14 (т, 2H), 3,36 (с, 3H), 3,66-3,76 (м, 2H), 4,81 (ушир.с, 1H), 6,11 (д, 1H), 6,88 (д, 1H), 7,13-7,19 (м, 2H), 7,36 (т, 1H), 7,61 (дт, 1H), 8,56 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (0,062 г, 0,189 ммоль), растворяют в хлороформе (2,75 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (2,75 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (5 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде темно-желтого стекла (0,047 г, 84%).
1H-ЯМР (400 МГц, D2O): д = 2,78 (с, 3H), 3,28 (т, 2H), 3,67 (т, 2H), 5,80 (д, 1H), 5,91 (д, 1H), 7,50 (т, 1H), 7,82 (т, 1H), 7,86 (д, 1H), 8,40 (т, 1H), 8,56 (д, 1H)
MS (ESI); m/z = 229,30 (MH+)
Пример приготовления 6 (соединение 7):

Стадия A
Указанное в заглавии соединение из Примера 1, Стадия F (0,075 г, 0,3 ммоль), и указанное в заглавии соединение из Примера 2, Стадия H (0,072 г, 0,25 ммоль), растворяют в толуоле (4,5 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,031 г, 0,05 ммоль) и трет-бутилатом натрия (0,075 г, 0,78 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,023 г, 0,025 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси вместе с желаемым соединением (0,098 г). Продукт дополнительно очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,09 г, 78%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,54 (с, 9H), 3,04 (т, 2H), 3,33 (с, 3H), 3,44 (с, 3H), 3,64-3,70 (м, 2H), 5,02 (ушир.с, 1H), 6,12 (д, 1H), 6,86-6,90 (м, 2H), 7,37 (т, 1H), 7,53-7,57 (м, 2H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (0,09 г, 0,196 ммоль), растворяют в хлороформе (2,9 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (2,9 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (5 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают с получением указанного в заглавии соединения в виде темно-желтого стекла (0,057 г, 80%).
1H-ЯМР (400 МГц, D2O): д = 2,78 (с, 3H), 2,89 (с, 3H), 2,98 (т, 2H), 3,57 (т, 2H), 5,80 (д, 1H), 5,90 (д, 1H), 6,63 (д, 1H), 6,77 (д, 1H), 7,50 (т, 1H), 7,68 (т, 1H)
MS (ESI); m/z = 258,28 (MH+)
Пример приготовления 7 (соединение 4):
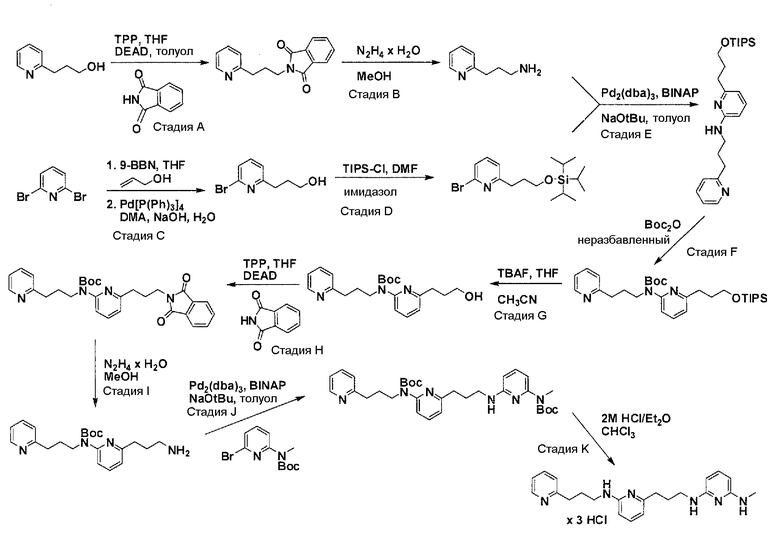
Стадия A
Трифенилфосфин (3,8 г, 14,4 ммоль) и фталимид (1,08 г, 7,4 ммоль), растворяют в тетрагидрофуране (20 мл), и смесь охлаждают до 0°C. При 0°C смесь коммерчески доступного 2-пиридин-1-пропанола (1 г, 7,4 ммоль) в тетрагидрофуране (10 мл) и 40% раствора диэтилазодикарбоксилата в толуоле (6 мл, 14,4 ммоль) добавляют в течение периода 5 мин. Смесь перемешивают в течение ночи, и дают ей возможность для достижения комнатной температуры. Растворители удаляют, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде красного масла (1,9 г, 98%).
1H-ЯМР (400 МГц, CDCl3): д = 2,13-2,21 (м, 2H), 2,87 (т, 2H), 3,80 (т, 2H), 7,07-7,11 (м, 1H), 7,20 (д, 1H), 7,58 (дт, 1H), 7,69-7,72 (м, 2H), 7,83-7,86 (м, 2H), 8,50 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (1,9 г, 7 ммоль), растворяют в метаноле (50 мл) и обрабатывают 50% раствором гидразина в воде (1,4 мл, 14 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Твердый продукт обрабатывают дихлорметаном (100 мл) и обрабатывают ультразвуком в течение 5 мин с получением суспензии, которую перемешивают при комнатной температуре в течение 30 мин. Смесь фильтруют, и осадок промывают 30 мл дихлорметана. Фильтрат концентрируют, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (9/1) для элюирования окрашенной примеси, затем, с использованием смеси дихлорметан/метанол (1:1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл, с получением указанного в заглавии соединения в виде коричневой жидкости (0,69 г, 70%).
1H-ЯМР (400 МГц, CDCl3): д = 1,43 (ушир.с, 2H), 1,85-1,92 (м, 2H), 2,74 (т, 2H), 2,83 (т, 2H), 7,08-7,12 (м, 1H), 7,16 (д, 1H), 7,59 (дт, 1H), 8,52 (д, 1H)
Стадия C
Аллиловый спирт (0,087 мл, 1,5 ммоль) растворяют в тетрагидрофуране (3 мл) и смесь охлаждают до 0°C. При 0°C добавляют 0,4 M раствор 9-борабицикло[3,3,1]нонана в гексане (11,25 мл, 4,5 ммоль), и перемешивание при 0°C продолжают в течение 15 мин. Затем смесь перемешивают при комнатной температуре в течение 4 часов, и растворители выпаривают. Остаток растворяют в тетрагидрофуране (8 мл), и добавляют 3 M водный раствор гидроксида натрия (2 мл, 6 ммоль). После добавления раствора коммерчески доступного 2,6-дибром-пиридина (0,46 г, 1,95 ммоль) в N,N'-диметилацетамиде (10 мл), смесь обрабатывают ультразвуком в течение 5 мин, при этом барботируют поток аргона через смесь. Затем добавляют тетракис(трифенилфосфин)палладий(0) (0,17 г, 0,156 ммоль), и смесь нагревают при ~95°C на песчаной бане в течение 90 мин. Смесь разбавляют этилацетатом (50 мл) и промывают 10% лимонной кислотой (20 мл) и насыщенным соляным раствором (15 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (30/70) для элюирования окрашенной примеси затем, с использованием смеси этилацетат/н-гептан (60/40), с получением сырого продукта. Объединенные сырые продукты от этого и двух дополнительных опытов дополнительно очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (50/50), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,48 г, 49%).
1H-ЯМР (400 МГц, CDCl3): д = 1,97-2,03 (м, 2H), 2,92 (т, 2H), 3,68-3,73 (м, 2H), 7,12 (д, 1H), 7,34 (д, 1H), 7,42 (т, 1H)
Примечание: 1H-ЯМР показывает присутствие малых количеств продуктов разложения 9-BBN, но материал является достаточно чистым для использования на следующей стадии.
Стадия D
Указанное в заглавии соединение из Стадии C, выше (0,48 г, 2,23 ммоль), растворяют в N,N'-диметилформамиде (10 мл), и добавляют имидазол (0,3 г, 4,46 ммоль). После добавления хлортриизопропилсилана (0,43 г, 2,23 ммоль), смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь разбавляют этилацетатом (60 мл) и промывают 10% раствором лимонной кислоты (3×15 мл) и насыщенным соляным раствором (15 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (5/95), с получением указанного в заглавии соединения в виде бесцветной жидкости (0,66 г, 79%).
1H-ЯМР (400 МГц, CDCl3): ( = 1,05-1,14 (м, 21H), 1,94-2,01 (м, 2H), 2,88 (т, 2H), 3,72 (т, 2H), 7,14 (д, 1H), 7,31 (д, 1H), 7,45 (т, 1H)
Стадия E
Указанные в заглавии соединения из Стадии B (0,136 г, 1 ммоль) и из Стадии D (0,35 г, 0,95 ммоль), выше, растворяют в толуоле (17 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,13 г, 0,19 ммоль) и трет-бутилатом натрия (0,25 г, 2,58 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,086 г, 0,095 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~85°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (80 мл), водой (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, смеси этилацетат/н-гептан (80/20) для элюирования желаемого соединения. Сырой продукт снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (80/20), с получением указанного в заглавии соединения в виде темно-желтого масла (0,34 г, 83%).
1H-ЯМР (400 МГц, CDCl3): д = 1,05-1,14 (м, 21H), 1,92-1,97 (м, 2H), 2,05-2,12 (м, 2H), 2,68 (т, 2H), 2,91 (т, 2H), 3,27-3,33 (м, 2H), 3,74 (т, 2H), 4,60-4,63 (ушир.с, 1H), 6,18 (д, 1H), 6,46 (д, 1H), 7,10-7,13 (м, 1H), 7,17 (д, 1H), 7,32 (т, 1H), 7,60 (дт, 1H), 8,52 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,34 г, 0,79 ммоль), растворяют в дихлорметане (5 мл), и добавляют ди-трет-бутил дикарбонат (0,86 г, 3,95 ммоль). Растворитель удаляют, и маслянистый остаток нагревают на песчаной бане при ~75°C в течение 3 дней до тех пор, пока все исходные материалы не будут израсходованы согласно данным TLC. Затем смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (10/90) для элюирования избытка ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде оранжевого масла (0,38 г, 92%).
1H-ЯМР (400 МГц, CDCl3): д = 1,05-1,14 (м, 21H), 1,49 (с, 9H), 1,99-1,96 (м, 2H), 2,05-2,12 (м, 2H), 2,74-2,84 (м, 4H), 3,72 (т, 2H), 4,01 (т, 2H), 6,88 (д, 1H), 7,07-7,10 (м, 1H), 7,12 (д, 1H), 7,39 (д, 1H), 7,52 (т, 1H), 7,58 (дт, 1H), 8,51 (д, 1H)
Стадия G
Указанное в заглавии соединение из Стадии F, выше (0,38 г, 0,72 ммоль), растворяют в ацетонитриле (8 мл), и добавляют 1 M раствор тетрабутиламмония фторида (3,6 мл, 3,6 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде бледно-желтого масла (0,25 г, 94%).
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 2,00-2,11 (м, 4H), 2,81-2,89 (м, 4H), 3,68 (т, 2H), 4,02 (т, 2H), 6,88 (д, 1H), 7,08-7,12 (м, 1H), 7,16 (д, 1H), 7,41 (д, 1H), 7,52 (т, 1H), 7,59 (т, 1H), 8,47 (д, 1H)
Стадия H
Трифенилфосфин (0,35 г, 1,36 ммоль) и фталимид (0,1 г, 0,68 ммоль), растворяют в тетрагидрофуране (3 мл), и смесь охлаждают до 0°C. При 0°C добавляют смесь указанного в заглавии соединения из Стадии G, выше (0,25 г, 0,68 ммоль), в тетрагидрофуране (2 мл) и 40% раствор диэтилазодикарбоксилата в толуоле (0,55 мл, 1,36 ммоль) в течение периода 5 мин. Смесь перемешивают в течение ночи, и дают ей возможность для достижения комнатной температуры. Растворители удаляют, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде оранжевого масла (382 мг).
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 2,00-2,17 (м, 4H), 2,77 (т, 2H), 2,81 (т, 2H), 3,76 (т, 2H), 4,03 (т, 2H), 6,88 (д, 1H), 7,04-7,08 (м, 1H), 7,14 (д, 1H), 7,38 (д, 1H), 7,48 (т, 1H), 7,55 (т, 1H), 7,69-7,72 (м, 2H), 7,82-7,86 (м, 2H), 8,48 (д, 1H)
Примечание: 1H-ЯМР показывает присутствие малых количеств диэтилгидразин-1,2-дикарбоксилата, но материал является достаточно чистым для использования на следующей стадии.
Стадия I
Указанное в заглавии соединение из Стадии H, выше (0,34 г, 0,68 ммоль), растворяют в метаноле (7 мл) и обрабатывают 50% раствором гидразина в воде (0,14 мл, 1,36 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют. Твердый продукт обрабатывают дихлорметаном (20 мл) и обрабатывают ультразвуком в течение 5 мин с получением суспензии, которую перемешивают при комнатной температуре в течение 30 мин. Смесь фильтруют, и осадок промывают 10 мл дихлорметана. Фильтрат концентрируют, и остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (9/1) для элюирования окрашенной примеси затем, с использованием смеси дихлорметан/метанол (1:1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл, с получением указанного в заглавии соединения в виде бледно-желтого масла (0,16 г, 62%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 1,86 (кв., 2H), 2,08 (кв., 2H), 2,70-2,76 (м, 4H), 2,83 (т, 2H), 4,04 (т, 2H), 6,85 (д, 1H), 7,08-7,11 (м, 1H), 7,14 (д, 1H), 7,40 (д, 1H), 7,52 (т, 1H), 7,57 (т, 1H), 8,51 (д, 1H)
Стадия J
Указанное в заглавии соединение из Стадии I, выше (0,075 г, 0,2 ммоль), и указанное в заглавии соединение из Примера 2, Стадия H (0,055 г, 0,193 ммоль), растворяют в толуоле (3,1 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,024 г, 0,039 ммоль) и трет-бутилатом натрия (0,05 г, 0,52 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,017 г, 0,019 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, затем, с использованием смеси этилацетат/н-гептан (60/40) для элюирования желаемого соединения. Сырой продукт снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,094 г, 89%).
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 9H), 1,52 (с, 9H), 2,00-2,12 (м, 4H), 2,78-2,84 (м, 4H), 3,27-3,33 (м, 5H), 4,02 (т, 2H), 4,70 (ушир.с, 1H), 6,08 (д, 1H), 6,86 (д, 2H), 7,08-7,13 (м, 2H), 7,34 (т, 1H), 7,40 (д, 1H), 7,48-7,57 (м, 2H), 8,50 (д, 1H)
Стадия K
Указанное в заглавии соединение из Стадии J, выше (0,094 г, 0,16 ммоль), растворяют в хлороформе (2,3 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (2,3 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (5 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде оранжевого стекла (0,06 г, 76%).
1H-ЯМР (400 МГц, D2O): д = 1,97 (кв., 2H), 2,06 (кв., 2H), 2,73-2,79 (м, 5H), 3,07 (т, 2H), 3,25 (т, 2H), 3,36 (т, 2H), 5,82-5,87 (м, 2H), 6,66 (д, 1H), 6,73 (д, 1H), 7,50 (т, 1H), 7,72 (т, 1H), 7,78 (т, 1H), 7,83 (д, 1H), 8,39 (т, 1H), 8,51 (д, 1H)
MS (ESI); m/z = 377,17 (MH+)
Пример приготовления 8 (соединение 3)

Стадия A
Коммерчески доступный 2,6-дибромпиридин (0,5 г, 2,1 ммоль), растворяют в N,N`-диметилацетамиде (5 мл), и добавляют коммерчески доступный 2-пиридилэтиламин (0,26 г, 2,1 ммоль). После добавления бикарбоната калия (0,23 г, 2,3 ммоль), смесь нагревают при ~110°C на песчаной бане в течение 5 часов. Смесь разбавляют этилацетатом (80 мл) и промывают водой 83×20 мл. Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде оранжевого масла (0,13 г, 21%).
1H-ЯМР (400 МГц, CDCl3): д = 3,07 (т, 2H), 3,68 (м, 2H), 5,17 (ушир.с, 1H), 6,30 (д, 1H), 6,70 (д, 1H), 7,10-7,21 (м, 3 H), 7,58 (т, 1H), 8,52 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (0,13 г, 0,46 ммоль), растворяют в дихлорметане (3 мл), и добавляют ди-трет-бутил дикарбонат (0,5 г, 2,32 ммоль). Растворитель удаляют, и маслянистый остаток нагревают на песчаной бане при ~75°C в течение 3 дней. Затем смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (20/80) для элюирования избытка ди-трет-бутилдикарбоната, затем, с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,095 г, 53%).
1H-ЯМР (400 МГц, CDCl3): д = 1,49 (с, 9H), 3,18 (т, 2H), 4,38 (т, 2H), 7,11-7,14 (м, 1H), 7,18 (д, 1H), 7,23 (д, 1H), 7,48 (т, 1H), 7,59-7,67 (м, 2H), 8,52 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (0,09 г, 0,238 ммоль), и указанное в заглавии соединение из Примера 1, Стадия F (0,07 г, 0,27 ммоль), растворяют в толуоле (4,25 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,032 г, 0,048 ммоль) и трет-бутилатом натрия (0,061 г, 0,65 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,021 г, 0,024 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5) для элюирования неполярной примеси, затем, с использованием смеси этилацетат/н-гептан (60/40) для элюирования желаемого соединения. Сырой продукт снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,12 г, 94%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 1,53 (с, 9H), 3,04 (т, 2H), 3,18 (т, 2H), 3,43 (с, 3H), 3,68-3,73 (м, 2H), 4,30 (т, 2H), 5,00 (ушир.с, 1H), 6,14 (д, 1H), 6,83 (д, 1H), 6,86-6,89 (м, 1H), 7,09-7,11 (м, 1H), 7,16 (д, 1H), 7,38 (т, 1H), 7,53-7,58 (м, 3H), 8,51 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (0,12 г, 0,22 ммоль), растворяют в хлороформе (3,1 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (3,1 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (5 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде оранжевого стекла (0,074 г, 74%).
1H-ЯМР (400 МГц, D2O): д = 2,90 (с, 3H), 3,00 (т, 2H), 3,31 (т, 2H), 3,58 (т, 2H), 3,69 (т, 2H), 5,88-5,93 (м, 2H), 6,61-6,65 (м, 1H), 6,79 (д, 1H), 7,50-7,73 (м, 1H), 7,70 (т, 1H), 7,84-7,91 (м, 2H), 8,42 (т, 1H), 8,58 (д, 1H)
MS (ESI); m/z = 349,49 (MH+)
Пример приготовления 9 (соединение 9):
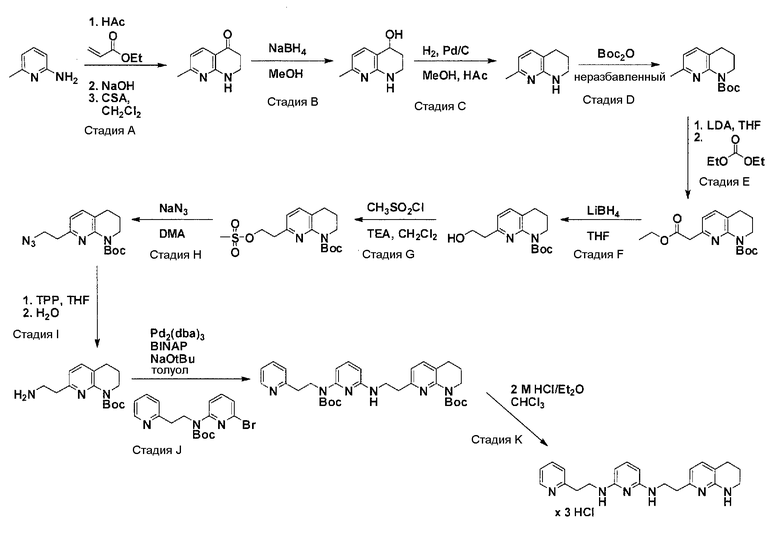
Стадия A
К коммерчески доступному 2-амино-6-метилпиридину (25,46 г, 235 ммоль) добавляют этилакрилат (26 мл, 239 ммоль) и уксусную кислоту (6 мл, 105 ммоль). Эту смесь нагревают при ~150°C на песчаной бане в течение 50 часов. Смесь охлаждают до комнатной температуры, и добавляют 6 Н гидроксид натрия (120 мл, 720 ммоль). Затем смесь нагревают при ~120°C на песчаной бане в течение 1 часа. Смесь охлаждают до комнатной температуры, и добавляют концентрированную хлористоводородную кислоту до тех пор, пока pH не достигнет приблизительно 4-5, при охлаждении на льду. Образуется полимерный преципитат, и смесь фильтруют. Фильтрат выпаривают, и остаток обрабатывают метанолом (100 мл). Полученную суспензию перемешивают при комнатной температуре в течение 30 минут и фильтруют. Преципитат промывают метанолом (30 мл), и объединенные фильтраты выпаривают, с получением коричневатой липкой массы. Этот сырой материал растворяют в дихлорметане (400 мл), и раствор помещают на ледяную баню. Затем добавляют по каплям хлорсульфоновую кислоту (162 мл, 2430 ммоль). После завершения добавления, смесь перемешивают при комнатной температуре в течение 2 часов. Затем смесь помещают обратно на ледяную баню, и осторожно добавляют воду (800 мл). После завершения добавления, кислотный раствор делают щелочным, доводя pH примерно до ~6 посредством добавления гидроксида натрия. Затем добавляют карбонат натрия для доведения pH до ~10-11. Образуется преципитат, и смесь экстрагируют этилацетатом (3×400 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (75/25) для элюирования неполярной примеси, затем, с помощью этилацетата, с получением указанного в заглавии соединения в виде желтого твердого продукта (12,2 г, 32%).
1H-ЯМР (400 МГц, CDCl3): д = 2,40 (с, 3H), 2,69 (т, 2H), 3,58-3,62 (м, 2H), 5,31 (ушир.с, 1H), 6,58 (д, 1H), 7,97 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (7 г, 43,2 ммоль) суспендируют в метаноле (170 мл), и добавляют порциями боргидрид натрия (2,94 г, 77,7 ммоль). После завершения добавления, смесь перемешивают при комнатной температуре в течение 1 часов до превращения в прозрачный раствор. Затем добавляют уксусную кислоту (21 мл), и растворители удаляют. Остаток растворяют в воде (250 мл), и водную фазу промывают дихлорметаном (2×100 мл). Водную фазу делают щелочной (pH~10) посредством добавления карбоната натрия и экстрагируют этилацетатом (6×150 мл). Объединенную органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют, с получением указанного в заглавии соединения в виде беловатого твердого продукта (6,39 г, 90%).
1H-ЯМР (400 МГц, CDCl3): д = 1,82-1,90 (м, 1H), 1,95-2,01 (м, 1H), 2,30 (с, 3H), 3,10 (ушир.с, 1H), 3,31-3,36 (м, 1H), 3,48 (дт, 1H), 4,72 (т, 1H), 5,32 (ушир.с, 1H), 6,38 (д, 1H), 7,32 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (6,39 г, 38,9 ммоль), растворяют в метаноле (130 мл) и уксусной кислоте (65 мл). После добавления катализатора, 10% палладия на угле (1,6г), смесь гидрируют в течение 3 дней. Смесь фильтруют, катализатор промывают метанолом (50 мл), и объединенные фильтраты выпаривают. Ацетатную соль растворяют в воде (200 мл) и pH доводят до pH~10 посредством добавления карбоната натрия. Водную фазу экстрагируют дихлорметаном (3×150 мл). Объединенную органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют с получением свободного амина в виде белого твердого продукта (4,42 г, 76%).
1H-ЯМР (400 МГц, CDCl3): д = 1,86-1,92 (м, 2H), 2,30 (с, 3H), 2,68 (т, 2H), 3,37-3,41 (м, 2H), 4,80 (ушир.с, 1H), 6,34 (д, 1H), 7,03 (д, 1H)
Стадия D
К указанному в заглавии соединению из Стадии C, выше (7,42 г, 50,13 ммоль), добавляют раствор ди-трет-бутилдикарбоната (33,7 г, 150,4 ммоль) в дихлорметане (100 мл). Растворитель удаляют, и маслянистый остаток нагревают при ~75°C на песчаной бане в течение 18 часов. Реакционную смесь очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (10/90) для удаления избытка ди-трет-бутил дикарбоната, затем, с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде белого твердого продукта (11,2 г, 90%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 1,87-1,93 (м, 2H), 2,46 (с, 3H), 2,70 (т, 2H), 3,72 (т, 2H), 6,80 (д, 1H), 7,24 (д, 1H)
Стадия E
Раствор LDA приготавливают посредством добавления 1,6 M раствора н-бутиллития (39,24 мл, 62,82 ммоль) при 0°C к перемешиваемому раствору N,N'-диизопропиламина (9,9 мл, 75,4 ммоль) в тетрагидрофуране (45 мл). Смесь перемешивают при 0°C в течение 15 мин. Затем при -78°C к раствору указанного в заглавии соединения из Стадии D, выше (5,6 г, 22,56 ммоль), добавляют по каплям раствор LDA и диэтилкарбонат (10,08 мл, 83,1 ммоль) в тетрагидрофуране (72 мл). Смесь перемешивают при -78°C в течение 40 минут. Реакция гасится посредством добавления раствора насыщенного хлорида аммония (100 мл) при -8°C. Смеси дают возможность для достижения комнатной температуры и разбавляют этилацетатом (200 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (50/50), с получением указанного в заглавии соединения в виде желтого масла, которое становится твердым при стоянии при комнатной температуре (7 г, 96%).
1H-ЯМР (400 МГц, CDCl3): д = 1,25 (т, 3H), 1,51 (с, 9H), 1,88-1,93 (м, 2H), 2,72 (т, 2H), 3,71-3,75 (м, 4H), 4,13 (кв., 2H), 6,96 (д, 1H), 7,34 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (2 г, 6,25 ммоль), растворяют в тетрагидрофуране (40 мл), и добавляют порциями боргидрид лития (0,18 г, 8,14 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем добавляют воду (25 мл), и смесь перемешивают при комнатной температуре в течение 10 мин. После добавления этилацетата (150 мл), органическую фазу отделяют, и водную фазу экстрагируют этилацетатом (50 мл). Объединенную органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием этилацетата, с получением указанного в заглавии соединения в виде бесцветного масла (1,48 г, 85%), затем, с использованием смеси дихлорметан/метанол (4/1), с получением продукта снятия N-Boc защиты в виде желтого масла (0,13 г, 11%).
Указанное в заглавии соединение:
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,88-1,93 (м, 2H), 2,70 (т, 2H), 2,92 (т, 2H), 3,77 (т, 2H), 3,98 (т, 2H), 5,53 (ушир.с, 1H), 6,75 (д, 1H), 7,30 (д, 1H)
продукт снятия N-Boc защиты:
1H-ЯМР (400 МГц, CDCl3): д = 1,87-1,93 (м, 2H), 2,03 (с, 1H), 2,68 (т, 2H), 2,80 (т, 2H), 3,40 (т, 2H), 3,89 (т, 2H), 6,32 (д, 1H), 6,48 (ушир.с, 1H), 7,10 (д, 1H)
Стадия G
Указанное в заглавии соединение из Стадии F, выше (822 мг, 2,95 ммоль), растворяют в дихлорметане (15 мл), и добавляют триэтиламин (0,9 мл, 6,5 ммоль). После добавления метансульфонилхлорида (0,46 мл, 5,87 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Смесь разбавляют дихлорметаном (50 мл) и промывают 10% раствором лимонной кислоты (20 мл), насыщенным раствором бикарбоната натрия (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (80/20), с получением указанного в заглавии соединения в виде бесцветного масла (0,83 г, 78%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,88-1,93 (м, 2H), 2,72 (т, 2H), 2,92 (с, 3H), 3,12 (т, 2H), 3,74 (т, 2H), 4,65 (т, 2H), 6,86 (д, 1H), 7,32 (д, 1H)
Стадия H
Указанное в заглавии соединение из Стадии G, выше (0,83 г, 2,22 ммоль), растворяют в N,N'-диметилацетамиде (5,5 мл), и добавляют азид натрия (0,76 г, 11,65 ммоль). Смесь нагревают на песчаной бане при ~75°C в течение 16 часов. Смесь разбавляют этилацетатом (55 мл) и 10% раствором лимонной кислоты (15 мл). Органическую фазу отделяют, промывают насыщенным раствором бикарбоната натрия (15 мл) и насыщенным соляным раствором (15 мл). Органическую фазу сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде бледно-желтого масла (0,6 г, 84%).
1H-ЯМР (400 МГц, CDCl3): д = 1,51 (с, 9H), 1,88-1,93 (м, 2H), 2,72 (т, 2H), 2,98 (т, 2H), 3,69 (т, 2H), 3,74 (т, 2H), 6,84 (д, 1H), 7,32 (д, 1H)
Стадия I
Указанное в заглавии соединение из Стадии H, выше (0,6 г, 1,98 ммоль), растворяют в тетрагидрофуране (8 мл), и добавляют трифенилфосфин (0,63 г, 2,38 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 часов, а затем добавляют воду (4 мл). Перемешивание продолжают в течение ночи, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5), затем, с использованием смеси дихлорметан/метанол (1/1), содержащей 10 мл 7 M аммиака в метаноле на 500 мл, с получением указанного в заглавии соединения в виде бледно-желтого масла (0,5 г, 91%).
1H-ЯМР (400 МГц, CDCl3): д = 1,52 (с, 9H), 1,66 (ушир.с, 2H), 1,89-1,96 (м, 2H), 2,72 (т, 2H), 2,87 (т, 2H), 3,11 (т, 2H), 3,78 (т, 2H), 6,80 (д, 1H), 7,25 (д, 1H)
Стадия J
Указанное в заглавии соединение из Примера 8, Стадия B (0,090 г, 0,238 ммоль), и указанное в заглавии соединение из Стадии I, выше (0,075 г, 0,27 ммоль), растворяют в толуоле (4,25 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,032 г, 0,048 ммоль) и трет-бутилатом натрия (0,061 г, 0,65 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,021 г, 0,024 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~110°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (60/40), с получением сырого соединения, указанного в заглавии. Сырой материал снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (80/20), с получением указанного в заглавии соединения в виде желтого масла (0,58 г, 42%).
1H-ЯМР (400 МГц, CDCl3): д = 1,40 (с, 9H), 1,52 (с, 9H), 1,86-1,92 (м, 2H), 2,67-2,71 (м, 2H), 2,93-2,99 (м, 2H), 3,10-3,17 (м, 2H), 3,68-3,73 (м, 2H), 3,73-3,80 (м, 2H), 4,23-4,27 (м, 2H), 6,40 (д, 1H), 6,68 (д, 1H), 6,80 (д, 1H), 7,06-7,09 (м, 1H), 7,12 (д, 1H), 7,23-7,29 (м, 2H), 7,50-7,56 (м, 1H), 8,50-8,53 (м, 1H)
Стадия K
Указанное в заглавии соединение из Стадии J, выше (0,058 г, 0,1 ммоль), растворяют в хлороформе (1,6 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (1,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (4 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде темно-желтого стекла (0,042 г, 86%).
1H-ЯМР (400 МГц, D2O): д = 1,71-1,78 (м, 2H), 2,57-2,61 (м, 2H), 2,79-2,84 (м, 2H), 3,22-3,30 (м, 4H), 3,47-3,50 (м, 2H), 3,59-3,63 (м, 2H), 5,80 (д, 1H), 6,44 (д, 1H), 7,33 (д, 1H), 7,42-7,83 (м, 4H), 8,35 (т, 1H), 8,51 (д, 1H)
MS (ESI); m/z = 375,29 (MH+)
Пример приготовления 10 (соединение 10):
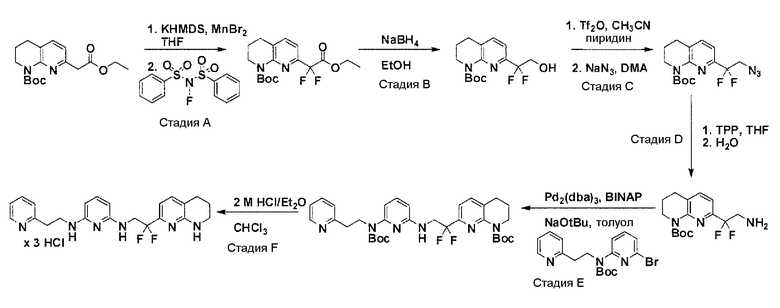
Стадия A
Калий бис(триметилсилил)амид (5,99 г, 30 ммоль), растворяют в тетрагидрофуране (90 мл), и раствор охлаждают до -78°C. При -78°C добавляют одной порцией указанное в заглавии соединение из Примера 9, Стадия E (3,2 г, 10 ммоль), и смесь перемешивают при -78°C в течение 45 минут. Марганец(II)бромид (4,3 г, 20 ммоль) добавляют одной порцией, и перемешивание продолжают при -78°C в течение 30 минут. Затем добавляют одной порцией N-фторбензолсульфонимид (8,9 г, 28,2 ммоль) при -78°C. Смесь перемешивают при -78°C в течение 30 минут и позволяют ей нагреться до комнатной температуры в течение ночи. Смесь разбавляют насыщенным раствором бикарбоната натрия (250 мл) и этилацетатом (300 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде бледно-оранжевого масла (2,15 г, 60%).
1H-ЯМР (400 МГц, CDCl3): д = 1,30 (м, 3H), 1,50 (с, 9H), 1,88-1,96 (м, 2H), 2,77-2,82 (м, 2H), 3,73-3,78 (м, 2H), 4,30-4,36 (м, 2H), 7,31-7,34 (м ,1H), 7,48-7,51 (м, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (2,15 г, 6 ммоль), растворяют в этаноле (8 мл), и смесь охлаждают до 0°C. Затем добавляют порциями боргидрид натрия (0,23 г, 6 ммоль) в течение периода 10 минут. После завершения добавления, смесь перемешивают в течение ночи, и дают ей возможность для достижения комнатной температуры. Смесь разбавляют этилацетатом (80 мл), водой (10 мл) и 10% раствором лимонной кислоты (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде беловатого твердого продукта (1,08 г, 57%).
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 9H), 1,92-1,98 (м, 2H), 2,76-2,80 (м, 2H), 3,77-3,82 (м, 2H), 4,14 (т, 2H), 5,28 (ушир.с, 1H), 7,30-7,38 (м, 1H), 7,52-7,56 (м, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше ((1,08 г, 3,44 ммоль), растворяют в ацетонитриле (6 мл), и добавляют пиридин (0,36 мл, 5,4 ммоль). Смесь охлаждают до 0°C, и добавляют по каплям ангидрид трифторметансульфоновой кислоты (0,63 мл, 3,77 ммоль). После завершения добавления, смесь перемешивают при 0°C в течение 30 минут. Смесь разбавляют простым диэтиловым эфиром (100 мл) и промывают 10% лимонной кислотой (10 мл) и насыщенным соляным раствором (10 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют с получением сырого трифлата в виде оранжевого масла. Сырой трифлат растворяют в N,N'-диметилацетамиде (7,5 мл), и добавляют азид натрия (1,1 г, 17,2 ммоль). Смесь нагревают при ~75°C на песчаной бане в течение 3 часов. Смесь разбавляют простым диэтиловым эфиром (100 мл) и промывают водой (20 мл) и насыщенным соляным раствором (20 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бледно-желтого масла, которое становится твердым при стоянии при комнатной температуре (0,87 г, 742%).
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 9H), 1,92-1,98 (м, 2H), 2,79 (т, 2H), 3,78 (т, 2H), 3,97 (т, 2H), 7,36 (д, 1H), 7,51 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (0,87 г, 2,58 ммоль), растворяют в тетрагидрофуране (10 мл), и добавляют трифенилфосфин (0,81 г, 3,1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 часов, а затем добавляют воду (10 мл). Перемешивание продолжают в течение ночи, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (98/2), затем, с использованием смеси дихлорметан/метанол (95/5), с получением сырого соединения, указанного в заглавии. Сырой материал снова очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (99/1), затем, с использованием смеси дихлорметан/метанол (9/1), с получением указанного в заглавии соединения в виде желтой жидкости, которая становится твердой/воскообразной при стоянии при комнатной температуре (0,79 г, 98%). Указанное в заглавии соединение содержит микроскопические количества трифенилфосфиноксида.
1H-ЯМР (400 МГц, CDCl3): д = 1,50 (с, 9H), 1,70 (ушир.с, 2H), 1,90-1,97 (м, 2H), 2,74-2,79 (м, 2H), 3,40 (т, 2H), 3,74-3,79 (м, 2H), 7,31 (д, 1H), 7,48 (д, 1H)
Стадия E
Указанное в заглавии соединение из Примера 8, Стадия B (0,09 г, 0,238 ммоль), и указанное в заглавии соединение из Стадии D, выше (0,085 г, 0,27 ммоль), растворяют в толуоле (4,25 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,032 г, 0,048 ммоль) и трет-бутилатом натрия (0,061 г, 0,65 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,021 г, 0,024 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~115°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (40/60), с получением указанного в заглавии соединения в виде темно-желтого воска (0,62 г, 43%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 9H), 1,57 (с, 9H), 1,92-1,98 (м, 2H), 2,76-2,80 (м, 2H), 3,13-3,18 (м, 2H), 3,77-3,82 (м, 2H), 4,22-4,32 (м, 4H), 6,38 (д, 1H), 6,52 (ушир.с, 1H), 6,82 (д, 1H), 7,06-7,09 (м, 1H), 7,17-7,20 (м, 1H), 7,26-7,31 (м, 2H), 7,47-7,56 (м, 2H), 8,51 (м, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,06 г, 0,1 ммоль), растворяют в хлороформе (1,6 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (1,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (4 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают с получением указанного в заглавии соединения в виде темно-желтого стекла (0,039 г, 74%).
1H-ЯМР (400 МГц, D2O): д = 1,73-1,81 (м, 2H), 2,67-2,71 (м, 2H), 3,23-3,28 (м, 2H), 3,32-3,38 (м, 2H), 3,62-3,67 (м, 2H), 3,97 (т, 2H), 5,90-5,93 (м, 1H), 6,80-6,83 (м, 1H), 7,44-7,83 (м, 5H), 8,38 (т, 1H), 8,50-8,53 (м, 1H)
MS (ESI); m/z = 411,45 (MH+)
Пример приготовления 11 (соединение 12):

Стадия A
Медный порошок (4,5 г, 70,8 ммоль) суспендируют в N,N'-диметилформамиде (22,5 мл) и 2-бромпиридине (4,5 г, 28,5 ммоль), и добавляют 2-бром-2,2-дифторацетат (6 г, 29,6 ммоль). Смесь нагревают при ~72°C на песчаной бане в течение ночи. Смесь разбавляют этилацетатом (60 мл), и добавляют раствор калия дигидрогенфосфата (8,58 г, 63 ммоль) в воде (50 мл). Смесь перемешивают при комнатной температуре в течение 30 минут, и фильтруют. Преципитат промывают этилацетатом (30 мл), и органическую фазу отделяют от фильтрата. Органическую фазу промывают водой (2×20 мл), сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (20/80), с получением указанного в заглавии соединения в виде желтого масла (3,17 г, 55%).
1H-ЯМР (400 МГц, CDCl3): д = 1,33 (т, 3H), 4,38 (кв., 2H), 7,40-7,45 (м, 1H), 7,72 (д, 1H), 7,86 (т, 1H), 8,66 (д, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (3,17 г, 15,8 ммоль), растворяют в этаноле (18 мл), и колбу помещают на водяную баню. Затем добавляют порциями боргидрид натрия (0,6 г, 16 ммоль) в течение периода 10 минут. После завершения добавления, смесь перемешивают при комнатной температуре в течение 90 минут. Смесь разбавляют этилацетатом (60 мл), и 10% раствор лимонной кислоты добавляют до тех пор, пока в смеси не прекратится пенообразование. Добавляют дополнительную воду (25 мл), и органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (95/5), с получением указанного в заглавии соединения в виде беловатого твердого продукта (2 г, 79%).
1H-ЯМР (400 МГц, CDCl3): д = 3,52 (ушир.с, 1H), 4,23 (т, 2H), 7,40-7,45 (м, 1H), 7,72 (д, 1H), 7,87 (т, 1H), 8,61 (д, 1H)
Стадия C
Указанное в заглавии соединение из Стадии B, выше (1,8 г, 11,3 ммоль), растворяют в ацетонитриле (18 мл), и добавляют пиридин (1,18 мл, 17,8 ммоль). Смесь охлаждают до 0°C, и добавляют по каплям ангидрид трифторметансульфоновой кислоты (2,09 мл, 12,4 ммоль). После завершения добавления, смесь перемешивают при 0°C в течение 30 минут. Смесь разбавляют простым диэтиловым эфиром (200 мл) и промывают 10% лимонной кислотой (60 мл) и насыщенным соляным раствором (60 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют с получением сырого трифлата в виде коричневого масла. Сырой трифлат растворяют в N,N'-диметилацетамиде (25 мл), и добавляют азид натрия (3,69 г, 56,7 ммоль). Смесь нагревают при ~75 °C на песчаной бане в течение 3 часов. Смесь разбавляют простым диэтиловым эфиром (200 мл) и промывают 10% лимонной кислотой (60 мл) и насыщенным соляным раствором (60 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде бесцветной жидкости (1,29 г, 62%).
1H-ЯМР (400 МГц, CDCl3): д = 4,02 (т, 2H), 7,39-7,45 (м, 1H), 7,72 (д, 1H), 7,86 (т, 1H), 8,68 (д, 1H)
Стадия D
Указанное в заглавии соединение из Стадии C, выше (1,4 г, 7,6 ммоль), растворяют в тетрагидрофуране (30 мл), и добавляют трифенилфосфин (2,4 г, 9,1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 48 часов, а затем добавляют воду (15 мл). Перемешивание продолжают в течение ночи, и растворители удаляют в вакууме. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/метанол (98/2), затем, с использованием смеси дихлорметан/метанол (95/5), с получением указанного в заглавии соединения в виде бледно-желтой жидкости (1,05 г, 87%).
1H-ЯМР (400 МГц, CDCl3): д = 1,42 (с, 2H), 3,42 (т, 2H), 7,35-7,40 (м, 1H), 7,68 (д, 1H), 7,82 (т, 1H), 8,65 (д, 1H)
Стадия E
Коммерчески доступный 2,6-дибромпиридин (0,5 г, 2,1 ммоль), растворяют в N,N`-диметилацетамиде (5 мл), и добавляют указанное в заглавии соединение из Стадии D, выше (0,31 г, 2,1 ммоль). После добавления бикарбоната калия (0,23 г, 2,3 ммоль), смесь нагревают при ~145°C на песчаной бане в течение 8 часов. Смесь разбавляют этилацетатом (100 мл) и промывают водой (30 мл) и насыщенным соляным раствором (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (30/70), с получением указанного в заглавии соединения в виде оранжевого масла (0,13 г, 20%).
1H-ЯМР (400 МГц, CDCl3): д = 4,20 (дт, 2H), 5,08 (ушир.с, 1H), 6,42 (д, 1H), 6,71 (д, 1H), 7,20 (д, 1H), 7,35-7,39 (м, 1H), 7,66 (д, 1H), 7,79 (т, 1H), 8,63 (д, 1H)
Стадия F
Указанное в заглавии соединение из Стадии E, выше (0,12 г, 0,39 ммоль) и указанное в заглавии соединение из Примера 10 Стадия D (0,14 г, 0,44 ммоль), растворяют в толуоле (7 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,053 г, 0,078 ммоль) и трет-бутилатом натрия (0,1 г, 1,06 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,035 г, 0,039 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~115°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси дихлорметан/ацетон (95/5), с получением сырого указанного в заглавии соединения. Сырой материал снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (50/50), с получением указанного в заглавии соединения в виде темно-желтого масла (0,62 г, 29%).
1H-ЯМР (400 МГц, CDCl3): д = 1,56 (с, 9H), 1,90-1,97 (м, 2H), 2,74-2,80 (м, 2H), 3,78-3,82 (м, 2H), 4,13-4,27 (м, 4H), 4,50 (ушир.с, 1H), 5,68-5,70 (м, 1H), 5,92-5,98 (м, 2H), 7,10 (т, 1H), 7,26-7,32 (м, 2H), 7,45-7,50 (м, 1H), 7,63-7,68 (м, 1H), 7,73-7,79 (м, 1H), 8,63-8,67 (м, 1H)
Стадия G
Указанное в заглавии соединение из Стадии F, выше (0,06 г, 0,11 ммоль), растворяют в хлороформе (1,6 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (1,6 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (4 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде темно-желтого стекла (0,048 г, 77%).
1H-ЯМР (400 МГц, D2O): д = 1,71-1,78 (м, 2H), 2,63-2,69 (м, 2H), 3,30-3,36 (м, 2H), 3,90-4,02 (м, 4H), 5,81-8,48 (м, 9H)
MS (ESI); m/z = 447,38 (MH+)
Пример приготовления 12 (соединение 11)
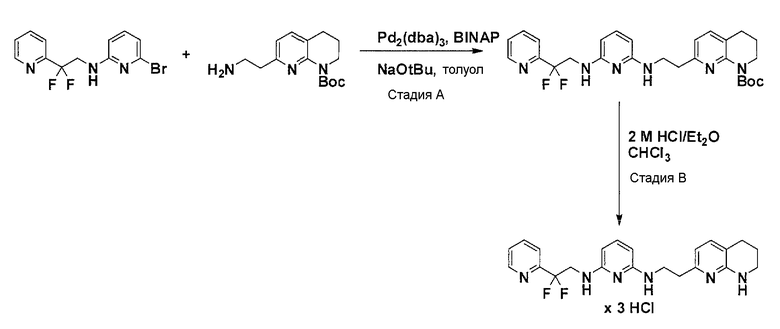
Стадия A
Указанное в заглавии соединение из Примера 11, Стадия E (0,13 г, 0,42 ммоль), и указанное в заглавии соединение из Примера 9, Стадия I (0,13 г, 0,48 ммоль), растворяют в толуоле (7,6 мл) и обрабатывают 2,2-бис-(дифенилфосфино)-1,1-нафталином (0,058 г, 0,084 ммоль) и трет-бутилатом натрия (0,11 г, 1,15 ммоль). Затем реакционную смесь дегазируют посредством барботирования аргона через реакционную смесь с последующим добавлением комплекса трис(дибензилиденацетон)дипалладий хлороформ (0,038 г, 0,042 ммоль). Реакционную емкость герметизируют, и смесь нагревают при ~115°C на песчаной бане в течение 45 минут. Реакционную смесь разбавляют этилацетатом (20 мл), водой (5 мл) и насыщенным соляным раствором (5 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют, и растворители удаляют. Остаток очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (70/30), с получением сырого указанного в заглавии соединения. Сырой материал снова очищают с помощью хроматографии на силикагеле с использованием смеси этилацетат/н-гептан (70/30), с получением указанного в заглавии соединения в виде серой пены (0,07 г, 32%).
1H-ЯМР (400 МГц, CDCl3): д = 1,54 (с, 9H), 1,86-2,04 (м, 2H), 2,66-2,74 (м, 2H), 2,93-3,08 (м, 2H), 3,48-3,66 (м, 2H), 3,72-3,80 (м, 2H), 4,18 (т, 2H), 4,5 (ушир.с, 1H), 5,65-5,98 (м, 2H), 6,21-6,40 (м, 1H), 6,76-6,83 (м, 1H), 7,10 (т, 1H), 7,25-7,36 (м, 2H), 7,61-7,68 (м, 1H), 7,72-7,77 (м, 1H), 8,60-8,64 (м, 1H)
Стадия B
Указанное в заглавии соединение из Стадии A, выше (0,07 г, 0,14 ммоль), растворяют в хлороформе (2 мл) и обрабатывают 2 M раствором хлористого водорода в простом диэтиловом эфире (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, и растворители удаляют с использованием шприца. Твердый материал растворяют в воде (4 мл) и фильтруют через 0,2-мкм картридж фильтра. Фильтрат собирают, и растворитель выпаривают, с получением указанного в заглавии соединения в виде темно-желтого стекла (0,063 г, 88%).
1H-ЯМР (400 МГц, D2O): д = 1,63-1,75 (м, 2H), 2,44-2,59 (м, 2H), 2,73-2,85 (м, 2H), 3,18-3,28 (м, 2H), 3,41-3,49 (м, 2H), 3,93 (т, 2H), 5,73-5,80 (м, 1H), 6,38-6,43 (м, 1H), 7,25-7,50 (м, 4 H), 7,61-7,65 (м, 1H), 7,85-7,90 (м, 1H), 8,42-8,48 (м, 1H)
MS (ESI); m/z = 411,45 (MH+)
Пример приготовления 13 (соединение 8):
Соединение 8 получают, как описано в WO2008/061795.
Экспериментальные результаты
Способ измерения растворимости:
1. Материалы, реагенты и оборудование
Планшетный шейкер, центрифуга (Eppendorf, радиус 8 см), ВЭЖХ (Dionex P580), колонка: Agilent Zorbax Eclipse XDB-C18 с быстрым разрешением (4,6×50 мм, 3,5 мМ, Agilent), бесцветные микропробирки 1,5 мл (Eppendorf, 1,5 мл), микропипетки 100-1000 мл, микропипетки 10-100 мл, фосфатный буфер Дульбекко, ДМСО, формиат аммония, муравьиная кислота 98-100%, UP-H2O (H2O согласно Фармакопее США), ацетонитрил, с качеством для ВЭЖХ, метанол, GR анализ, мембранный фильтр из PVDF.
2. Способ
2.1 Приготовление PBS 5x (хранят при 4°C) и PBS 1x для анализа
PBS 5x
Растворяют все содержание соли PBS (D-5652-10L) в 2 л UP-H2O.
PBS 1x
Перед анализом, разбавляют 5 кратно PBS 5x с получением 30 мл PBS 1x и фильтруют раствор с использованием шприца и любой гидрофильной мембраны, например, мембраны из PVDF.
2.2 Приготовление растворителя для ВЭЖХ (хранят при RT (комнатной температуре))
Растворитель A: 13,3 мМ формиата аммония/6,5 мМ муравьиной кислоты/UP-вода
Растворяют 820 ± 1 мг формиата аммония и 245 мкл муравьиной кислоты в 1000 мл UP-H2O.
Растворитель B: 6,0 мМ формиата аммония/2,9 мМ муравьиной кислоты/ 90% ацетонитрила/10% UP-воды
Растворяют 378±1 мг формиата аммония и 110 мкл муравьиной кислоты в 900 мл ацетонитрила и 100 мл UP-H2O.
2.3 Приготовление исходного раствора соединения
Растворяют соединение в ДМСО при концентрации 25 мМ (минимум 50 мл).
2.4 Получение стандартной кривой
Приготавливают 15 мл смеси метанол/H2O (6/4).
Приготавливают 5 стандартных калибровочных растворов: 250 мкМ, 200 мкМ, 50 мкМ, 12,5 мкМ и 3,13 мкМ в метаноле/H2O (6/4).
Получают препараты для каждой стандартной концентрации в микропробирках 1,5.
Переносят непосредственно 250-300 мкл из каждой микропробирки во флакон для ВЭЖХ.
Осуществляют ВЭЖХ (из микропробирок №6-№1), используя следующие условия:
Колонка C18, 0,7 мл/мин, 20°C, УФ детектирование на 254 нм, объем инжектирования: 20 мкл; и один из следующих градиентов:
Градиент для очень полярных/гидрофильных соединений
Градиент для менее полярных/липофильных соединений
2.5. Приготовление образца для исследования растворимости в воде
Приготавливают образцы соединения, по три для каждого опыта
Осторожно перемешивают его (350 об/мин) в течение 24 часов при комнатной температуре.
По прохождении времени инкубирования, центрифугируют его при 2500 g (5500 об/мин) в течение 30 мин.
Отбирают 200 мкл супернатанта для анализа с помощью ВЭЖХ, с использованием таких же условий, как описано в 2.4.
2.6. Обработка данных
Интегрируют площадь каждого стандартного точечного пика при 254 нм.
Определяют стандартную кривую для соединения посредством построения графика площади как функции от теоретической концентрации. Устанавливают уравнение стандартной кривой на основе линейной регрессии (с пересечением при 0, R2 ≥ 0,90).

Вычисляют среднюю площадь для каждой тройки образцов, приготовленных в водной фазе.
Концентрацию соединения в супернатанте определяют с помощью следующей формулы:
Растворимость соединения определяют с помощью следующей формулы:
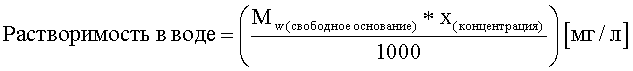
Пример: Ингибирование агрегации пептида (Ab)1-42 амилоидного бета белка (анализ ThT)
Ряд малых молекул исследуют на их способность ингибировать агрегацию пептида (Ab) 1-42 амилоидного бета-белка с использованием анализа спектра флюоресценции с тиофлавином T.
Приготовление пленки пептида Ab
Лиофилизированный порошок Ab1-42 (Bachem) разбавляют в гексафторизопропаноле (HFIP) до 1 мМ. Раствор пептида обрабатывают ультразвуком в течение 15 мин при комнатной температуре, перемешивают в течение ночи, и аликвоты помещают в пробирки для микроцентрифуги, не покрытые силиконом. Затем HFIP испаряют в потоке аргона. Полученную пептидную пленку сушат в вакууме в течение 10 мин, плотно герметизируют, и хранят при -80°C до использования.
Ингибирование агрегации Ab1-42
Для анализа ингибирования агрегации Ab1-42, опосредуемой малыми молекулами, малые молекулы растворяют перед каждым экспериментом в безводном диметилсульфоксиде (ДМСО, Sigma-Aldrich) с получением концентрации 7,4 мМ. Пептидную пленку Ab1-42 растворяют в ДМСО с получением концентрации 400 мкМ. Раствор для анализа в буфере PBS приготавливают в пробирках для инкубирования, не покрытых силиконом, с получением следующих концентраций: 330 мМ малых молекул, 33 мМ Ab1-42, 10 мкМ тиофлавина T (ThT) и 12,8% ДМСО. Следовательно, конечное молярное отношение малых молекул к Ab1-42 составляет 10:1. Приготавливают положительный контроль без малых молекул для измерения максимума RFU. Отрицательный контроль без Ab1-42 приготавливают для каждого вида малых молекул. Тример 3-аминопиразола (Trimer) исследуют во всех анализах для обеспечения воспроизводимости в независимых экспериментах. Растворы инкубируют в течение 24 часов при 37°C, и регистрируют спектры флюоресценции (относительные единицы флюоресценции; RFU) на шести одинаковых образцах в черных 384-луночных планшетах для анализов (Perkin-Elmer) на спектрофлуориметре Perkin-Elmer FlouroCount. Ингибирование агрегации выражается как средний % ингибирования или ±1 стандартное отклонение (SD) в соответствии со следующим уравнением:
Пороговый критерий для выбора функциональных молекул определяют при способности 50% ингибирования. Молекулы, показывающие способность к ингибированию выше 70%, считаются очень сильными кандидатами.
Для определения IC50, используют следующие разбавления соединений при анализе ThT, описанном выше:
330 мкМ, 82,50 мкМ, 20,63 мкМ, 5,16 мкМ, 1,29 мкМ, 0,32 мкМ и 0,08 мкМ
Пример: Воздействие соединения по настоящему изобретению в модели крыс хронической глазной гипертонии/глаукомы
Модель крысы для хронической глазной гипертонии (OHT)/глаукомы получают посредством инъекции гипертонического солевого раствора в эписклеральные вены одного глаза крыс Dark Agouti. 18 крыс получают интравитреальную инъекцию, содержащую объем 5 мкл соединения 1 (ACI-260) при концентрации 74 мг/л в глаз с OHT. 18 крыс служат в качестве отрицательного контроля и получают 5 мкл солевого раствора, и 18 крыс служат в качестве положительного контроля и получают 6 мкл конго красного (1,46 мг/мл). По 6 животных/в каждый момент времени подвергают эвтаназии через 3, 8 и 16 недель после лечения.
Внутриглазное давление (IOP) измеряют с использованием Tonopen каждые 4 недели после дозирования и перед умерщвлением (в пределах 3 дней). За 5-7 дней перед умерщвлением, животные получают интрацеребральную инъекцию из 5 мкл 4% FluoroGold, чтобы пометить ганглионарные клетки сетчатки (RGC). Для количественного определения жизнеспособных RGC, изображения обрабатывают с использованием специальной системы программного обеспечения для анализа изображения, и жизнеспособные RGC выражают как количество клеток на квадратный миллиметр.
Через 3 недели, ни у одного животного не наблюдается уменьшения внутриглазного давления. Однако количество жизнеспособных RGC значительно увеличивается в группе, леченной с помощью соединения 1 (p<0,001), и в группе, которой вводили конго красный (p<0,001), по сравнению с контрольной группой. Через 8 и 16 недель, эти результаты подтверждаются, и это говорит о том, что соединение 1 является нейропротектором. Результаты показаны на Фигуре 1.
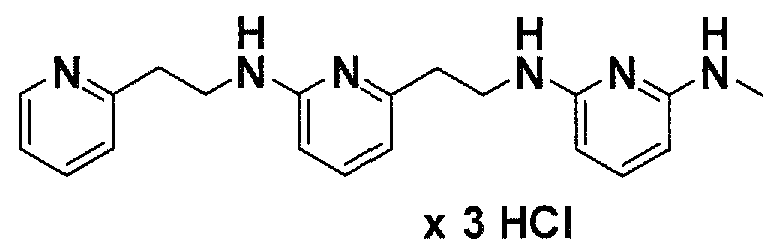
298 мг/л

71 мг/л
38 мг/л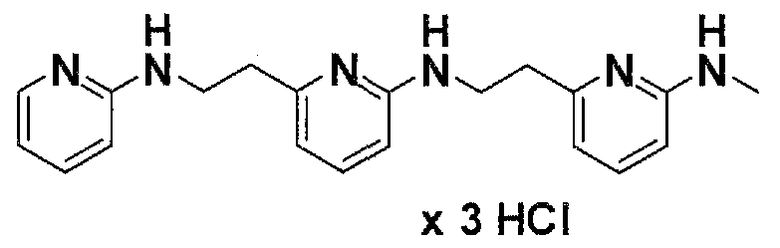

Как можно увидеть посредством сравнения результатов, полученных с помощью соединений в соответствии с настоящим изобретением (соединения 1, 2, 3 и 4), более длинный линкер уменьшает растворимость.
Соединение 5 (не в соответствии с настоящим изобретением) не имеет 2,6-диаминопиридинового остатка. Как можно увидеть, биологическая активность значительно уменьшается.


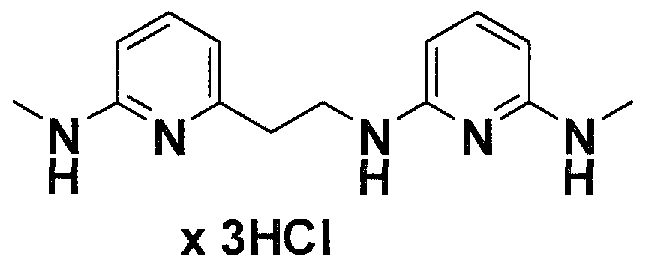
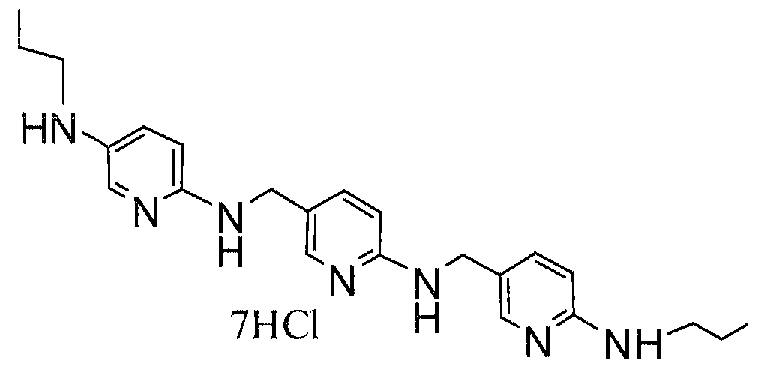
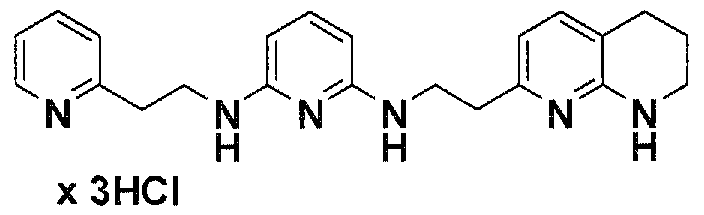
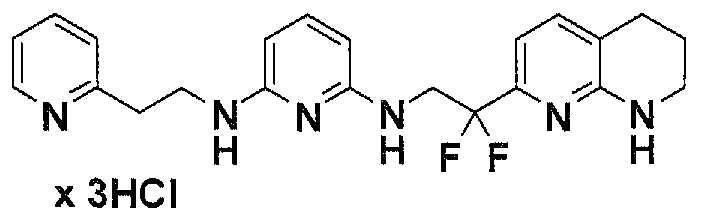

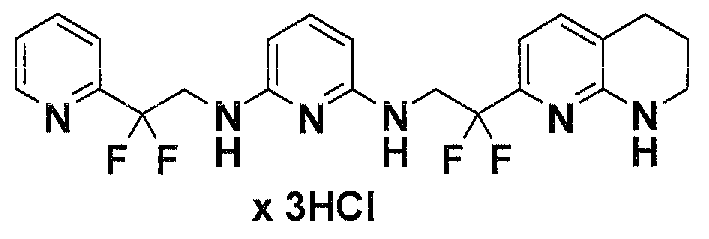
Соединение 6 (не в соответствии с настоящим изобретением) имеет только два пиридиновых кольца. Соединение 7 (не в соответствии с настоящим изобретением) имеет только два пиридиновых кольца. Оба этих соединения имеют значительно пониженную активность по сравнению с соединением 1 (в соответствии с настоящим изобретением).
Растворимость соединения 8 (не в соответствии с настоящим изобретением), которое имеет 2,5-диаминопиридиновый остаток, значительно хуже, чем у соединения 1 (в соответствии с настоящим изобретением), которое имеет 2,6-диаминопиридиновый остаток.
Соединения 9, 10 и 11 имеют активность, сравнимую с соединением 1, но их растворимость хуже. Только для соединения 12, как растворимость, так и активность хуже, по сравнению с соединением 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДНЫМИ ИЛИ АМИЛОИДОПОДОБНЫМИ БЕЛКАМИ | 2011 |
|
RU2603008C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДОМ ИЛИ АМИЛОИДПОДОБНЫМИ БЕЛКАМИ | 2007 |
|
RU2469026C2 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2135484C1 |
| КАРБАЗОЛСОДЕРЖАЩИЕ СУЛЬФОНАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ КРИПТОХРОМА | 2013 |
|
RU2654484C1 |
| ПРОИЗВОДНЫЕ МОРФОЛИНОПУРИНА, ОБЛАДАЮЩИЕ PI3K И/ИЛИ mTOR ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2009 |
|
RU2490269C2 |
| СОЕДИНЕНИЯ ДЛЯ ВИЗУАЛИЗАЦИИ АГРЕГАТОВ БЕЛКА ТАУ | 2017 |
|
RU2778739C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ | 1995 |
|
RU2136666C1 |
| СОЕДИНЕНИЕ ТРИАЗИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНСКИХ ЦЕЛЯХ | 2015 |
|
RU2692789C2 |
| ПРОИЗВОДНЫЕ АМИДОВ И КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АСАТ-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 1996 |
|
RU2128165C1 |





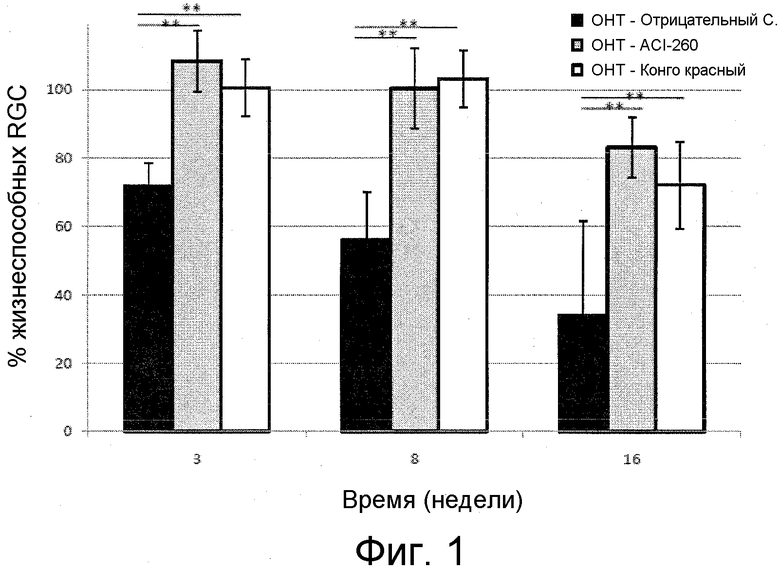
Изобретение относится к соединениям формулы (I)
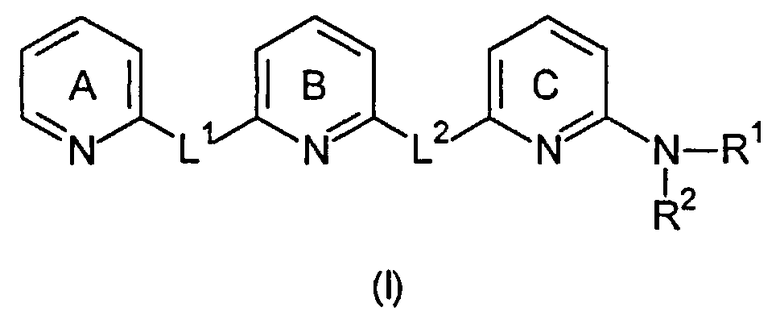
где пиридиновые кольца А, В и С являются независимо незамещенными или замещенными одним или несколькими заместителями, которые независимо выбраны из группы, состоящей из: C1-6-алкила, галогеналкила, имеющего 1-6 атомов углерода, Hal или OR13; L1 и L2 независимо выбраны из остатков, имеющих формулу (а) или (b)
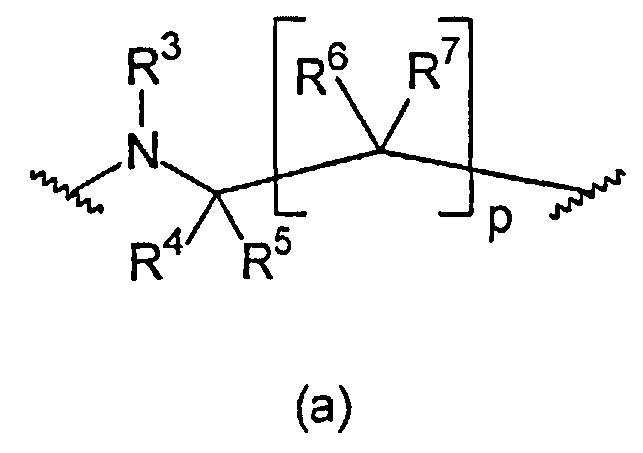

где, по меньшей мере, один из L1 или L2 имеет формулу (b); R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-6-алкила и фенила; R3 выбран из водорода и C1-6-алкила; R4, R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и C1-6-алкила; R8, R9, R10 и R11 независимо выбраны из группы, состоящей из водорода и C1-6-алкила; R12 выбран из группы, состоящей из водорода и C1-6-алкила; R13 независимо выбран из группы, состоящей из водорода, C1-6-алкила и фенила; р равно 1 или 2; q равно 0, 1 или 2 и Hal выбран из группы, состоящей из F, Cl, Br, и I, которые могут быть использованы при лечении группы расстройств и нарушений, связанных с амилоидным белком. 4 н. и 13 з.п.ф-лы, 1 ил., 6 таб., 13 пр.
1. Соединение, имеющее общую формулу (I)
где
пиридиновые кольца А, В и С являются независимо незамещенными или замещенными одним или несколькими заместителями, которые независимо выбраны из группы, состоящей из: C1-6-алкила, галогеналкила, имеющего 1-6 атомов углерода, Hal или OR13;
L1 и L2 независимо выбраны из остатков, имеющих формулу (а) или (b)
где, по меньшей мере, один из L1 или L2 имеет формулу (b);
R1 и R2 независимо выбраны из группы, состоящей из водорода, C1-6-алкила и фенила;
R3 выбран из водорода и C1-6-алкила;
R4, R5, R6 и R7 независимо выбраны из группы, состоящей из водорода и C1-6-алкила;
R8, R9, R10 и R11 независимо выбраны из группы, состоящей из водорода и C1-6-алкила;
R12 выбран из группы, состоящей из водорода и C1-6-алкила;
R13 независимо выбран из группы, состоящей из водорода, C1-6-алкила и фенила;
р равно 1 или 2;
q равно 0, 1 или 2 и
Hal выбран из группы, состоящей из F, Cl, Br, и I;
или его фармацевтически приемлемые соли.
2. Соединение по п. 1, где пиридиновые кольца А, В и С являются независимо незамещенными или замещенными одной или несколькими алкильными группами или атомами галогена.
3. Соединение по п. 1, где пиридиновые кольца А, В и С являются независимо незамещенными или замещенными одной или несколькими группами, независимо выбранными из C1-6 алкила или ОН.
4. Соединение по п. 1, где пиридиновые кольца А, В и С являются незамещенными.
5. Соединение по любому из пп. 1-4, где R1 и R2 независимо выбраны из водорода и алкила.
6. Соединение по п. 1, где q равно 2.
7. Соединение по п. 1, где соединение выбрано из


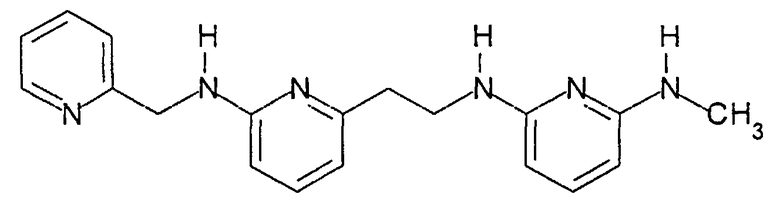
или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция для лечения группы расстройств и нарушений, связанных с амилоидными белками, заболеваний или состояний, связанных с амилоидными или амилоидоподобными белками, и глазных болезней, связанных с патологическими нарушениями/изменениями в тканях зрительной системы, содержащая соединение по любому из пп. 1-7.
9. Фармацевтическая композиция по п.8, дополнительно содержащая фармацевтически приемлемый носитель или наполнитель.
10. Применение соединения по любому из пп. 1-7 для получения лекарственного средства для лечения или предотвращения заболевания или состояния глаз, выбранного из группы, состоящей из глаукомы, нейронной дегенерации, кортикальной недостаточности зрения, катаракты из-за осаждения бета-амилоидных белков, глазного амилоидоза, первичной дегенерации сетчатки, дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дистрофии.
11. Соединение по п. 1 для лечения или предотвращения заболевания или состояния глаз, выбранного из группы, состоящей из глаукомы, нейронной дегенерации, кортикальной недостаточности зрения, катаракты из-за осаждения бета-амилоидных белков, глазного амилоидоза, первичной дегенерации сетчатки, дегенерации желтого пятна, друз зрительного нерва, оптической нейропатии, неврита зрительного нерва и решетчатой дистрофии.
12. Применение по п. 10, где заболевание представляет собой глаукому.
13. Соединение по п. 11, где заболевание представляет собой глаукому.
14. Применение по п. 12, где заболевание представляет собой хроническую (идиопатическую) открытоугольную глаукому, глаукому с блокированием зрачка, глаукому развития, глаукому, связанную с другими глазными расстройствами, глаукому, связанную с повышенным эписклеральным венозным давлением, глаукому, связанную с воспалением и травмой, глаукому после внутриглазной хирургической операции, глаукому при повышенном давлении, глаукому при нормальном давлении, острую закрытоугольную глаукому, подострую закрытоугольную глаукому, хроническую закрытоугольную глаукому, глаукому с сочетанным механизмом, врожденную (младенческую) глаукому, ювенильную глаукому с аниридией, глаукому, связанную с расстройствами эндотелия роговой оболочки, глаукому, связанную с расстройствами радужной оболочки и ресничного тела, глаукому, связанную с расстройствами хрусталика, глаукому, связанную с расстройствами сетчатки, сосудистой оболочки глаза и стекловидного тела, глаукому, связанную с отслоением сетчатки и витреоретинальными нарушениями, неоваскулярную глаукому, пигментную глаукому, синдром эксфолиации, открытоугольную глаукому, вызываемую проблемами с хрусталиком, глаукому, связанную с утолщением и смещением хрусталика, глаукому, связанную с кератитом, эписклеритом и склеритом, глаукому, связанную с блокировкой реснитчатого тела (злокачественную), глаукому при афакии и артифакии, и при пролиферации клеток эпителия, волокон и эндотелия, глаукому, связанную с хирургической операцией на роговице, и глаукому, связанную с витреоретинальной хирургической операцией.
15. Соединение по п. 13, где заболевание представляет собой хроническую (идиопатическую) открытоугольную глаукому, глаукому с блокированием зрачка, глаукому развития, глаукому, связанную с другими глазными расстройствами, глаукому, связанную с повышенным эписклеральным венозным давлением, глаукому, связанную с воспалением и травмой, глаукому после внутриглазной хирургической операции, глаукому при повышенном давлении, глаукому при нормальном давлении, острую закрытоугольную глаукому, подострую закрытоугольную глаукому, хроническую закрытоугольную глаукому, глаукому с сочетанным механизмом, врожденную (младенческую) глаукому, ювенильную глаукому с аниридией, глаукому, связанную с расстройствами эндотелия роговой оболочки, глаукому, связанную с расстройствами радужной оболочки и ресничного тела, глаукому, связанную с расстройствами хрусталика, глаукому, связанную с расстройствами сетчатки, сосудистой оболочки глаза и стекловидного тела, глаукому, связанную с отслоением сетчатки и витреоретинальными нарушениями, неоваскулярную глаукому, пигментную глаукому, синдром эксфолиации, открытоугольную глаукому, вызываемую проблемами с хрусталиком, глаукому, связанную с утолщением и смещением хрусталика, глаукому, связанную с кератитом, эписклеритом и склеритом, глаукому, связанную с блокировкой реснитчатого тела (злокачественную), глаукому при афакии и артифакии, и при пролиферации клеток эпителия, волокон и эндотелия, глаукому, связанную с хирургической операцией на роговице, и глаукому, связанную с витреоретинальной хирургической операцией.
16. Смесь, содержащая соединение по любому из пп. 1-7 и необязательно, по меньшей мере, одно дополнительное биологически активное соединение и/или фармацевтически приемлемый носитель и/или разбавитель и/или наполнитель.
17. Смесь по п. 16, в которой дополнительное биологически активное соединение представляет собой соединение, используемое при лечении амилоидоза и/или внутриглазного давления.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ЛОГАРИФМИЧЕСКИЙ АРИФМОМЕТР | 1928 |
|
SU12452A1 |

