Область техники
Настоящее изобретение относится к соединению циклопептида высокой чистоты и способу получения, а также относится к применению такого соединения циклопептида высокой степени чистоты.
Предшествующий уровень техники
Грибковая инфекция стала основной причиной высокой заболеваемости и смертности у иммунодефицитных больных. За последние 20 лет заболеваемость грибковой инфекцией значительно увеличилась. Люди с высоким уровнем риска заболеваний грибковыми инфекциями включают пациентов в критическом состоянии, хирургических пациентов и тех пациентов, которые страдают от ВИЧ-инфекции, лейкемии и других опухолей. Пациенты после трансплантации органов также подвергаются высокому риску грибковой инфекции.
Эхинокандины, как новый класс противогрибковых агентов, демонстрируют хорошие результаты в лечении инфекций, вызванных Candida или Aspergillus. Каспофунгин и Микафунгин являются представителями таких лекарственных средств. Эхинокандины угнетают грибок, подавляя образование 1,3-β гликозидной связи, и таким образом снижают вред человеческому телу и уменьшают побочные эффекты, оставаясь в то же время высокоэффективными. Таким образом, они более безопасны в использовании, чем традиционные противогрибковые агенты.
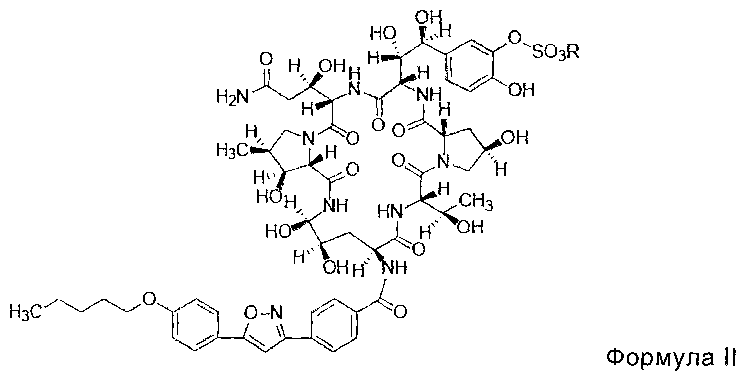
FK463 (натрия Микафунгин) является соединением формулы II (R является ионом натрия), которое разработано Japan Fujisawa Toyama Co., Ltd, Takaoka Plant под торговой маркой Mycamine, и в настоящее время продается в нескольких странах как противогрибковое средство для внутривенного введения. Его получают путем разрезания боковой цепи FR901379 как предшественника (соединение формулы III, R является ионом натрия или ионом водорода) с помощью фермента, формируя таким образом FR179642 (соединение формулы I, R является водородом или ионом натрия) (смотри патент США US 5376634, ЕР 0431350 и китайский патент CN 1161462C для конкретных способов), а затем химической модификации FR179642 (см патентную публикацию WO 9611210, WO 9857923, WO 2004014879 для конкретных способов получения и очистки).
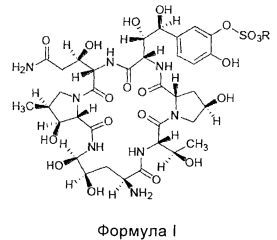
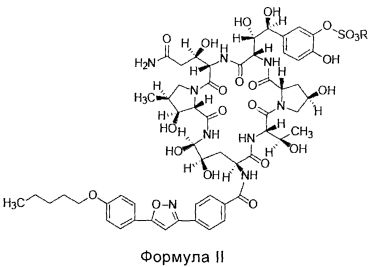
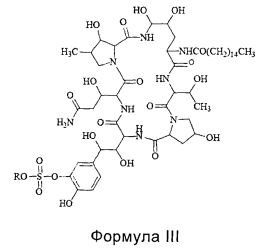
Конкретная схема показана следующим образом:

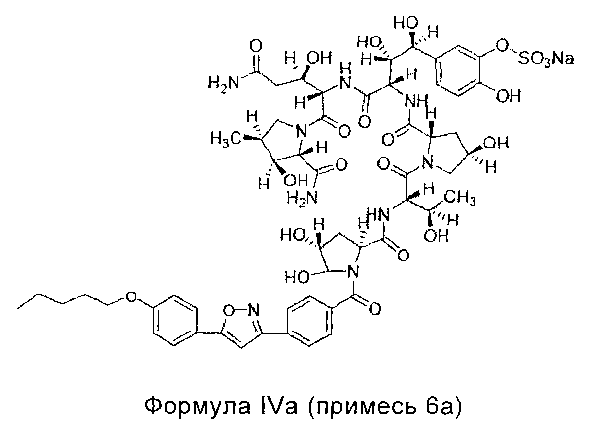
Тем не менее, авторы настоящего изобретения провели анализ существующей формулировки Микафунгина с помощью ВЭЖХ (Высокоэффективная жидкостная хроматография) и обнаружили, что в композиции содержатся примесь 6а, примесь 7а, примесь 8а, примесь 9а и примесь 10а. И изобретатели приготовили примесь 6а, примесь 7а, примесь 8а, примесь 9а и примесь 10а с помощью препаративной колонки в небольшом количестве и подтвердили структуру примеси с помощью МС (масс-спектрометрия) и 1H-NMR (ПМР - Протонный магнитный резонанс) как формулы IVa, Va, VIa, VIIa, VIIIa соответственно:
Примесь 6a, химическое название которой 5-[(1S,2S,3S)-4-[(1S,2R)-4-амино-1-[[(2S,3S,4S)-2-карбамил-3-гидрокси-4-метил-1-пирролил]карбонил]-2-гидрокси-4-оксобутил]амино]-3-[[[(2S,4R)-1-[(2S,3R)-2-[[[(2S,4R)-4,5-дигидрокси-1-[4-[5-[4-(пентилокси)фенил]-3-изоксазолил]бензоил]-2-пирролил]карбонил]амино]-3-гидроксибутирил]-4-гидрокси-2-пирролил]карбонил]амино]-1,2-дигидрокси-4-оксобутил]-2-гидроксифенил натрия сульфат
МС и ПМР данные примеси 6а перечислены ниже:
МС: 1314,5 [M+Na]+
ПМР (ДМСО-d6 (диметилсульфоксид)): 0,81-1,03 (6H, m), 1,12 (3H, d), 1,3-1,7 (4H, m), 1,7-2,1 (5H, m), 2,11-2,41 (3H, m), 2,52-2,62 (IH, m), 3,03-3,14 (IH, m), 3,62-4,65 (15H, m), 4,70-5,22 (10H, m), 5,24 (IH, d), 5,53 (IH, d), 6,53-6,71 (3H, m), 7,12-7,70 (7H, m), 7,82 (2H, d), 7,83-8,24 (5H, m), 8,61-9,11 (2H, m)
Примесь 7a, химическое название которой 5-[(1S,2S)-2-[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S)-3-[(R)-2-карбамоил-1-гидроксиэтил]-11,20,21,25-тетрагидрокси-15-[(R)-1-гидроксиэтил]-2,5,8,14,17,23-гексакарбонил-18-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензамид]-1,4,7,13,16,22-гексаазотрицикло[22.3.0.09,13]гептакозан-6-ил]-1,2-дигидроксиэтил]-2-гидроксифенил натрия сульфат:

МС и ПМР данные примеси 7а перечислены ниже:
MC: 1300,5 [M+Na]+
ПМР (ДМСО-d6): 0,87 (3H, t), 1,12 (3H, d), 1,42-1,65 (4H, m), 1,62-2,16 (6H, m), 2,11-2,43 (3H, m), 2,51-2,65 (IH, m), 3,04-3,11 (IH, m), 3,62-4,63 (15H, m), 4,71-5,23 (10H, m), 5,24 (IH, d), 5,69 (IH, d), 6,51-6,72 (3H, m), 7,11-7,74 (7H, m), 7,83 (2H, d), 7,84-8,11 (4H, m), 8,26 (IH, d), 8,61-9,12 (2H,m)
Примесь 8a, химическое наименование которой 5-[(1S,2S)-2-[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-3-[(R)-2-карбамоил-1-гидроксиэтил]-11,20,21,25-тетрагидрокси-15-[(R)-1-гидроксиметил]-26-метил-2,5,8,14,17,23-гексакарбонил-18-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензамид]-1,4,7,13,16,22-гексаазатрицикло[22.3.0.09,13]гептакозан-6-ил]-1,2-дигидроксиэтил]-2-гидроксифенил сульфат натрия
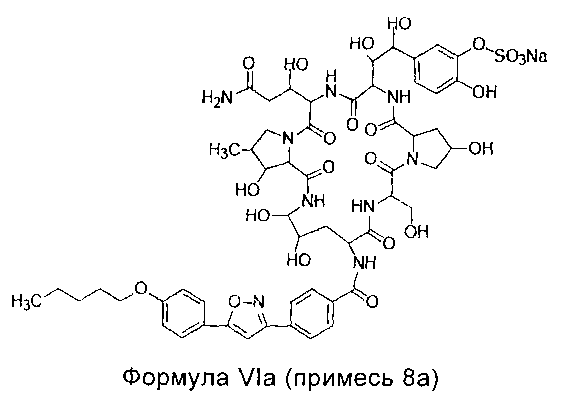
МС и ПМР данные примеси 8а перечислены ниже:
MC: 1300,4 [M+Na]+
ПМР (ДМСО-d6): 0,83-1,02 (6Н, m), 1,32-1,72 (4Н, m), 1,75-2,14 (6Н, m), 2,13-2,46 (3Н, m), 2,52-2,64 (IH, m), 3,01-3,13 (IH, m), 3,62-4,64 (15Н, m), 4,72-5,22 (10Н, m), 5,24 (IH, d), 5,53 (IH, d), 6,52-6,73 (3Н, m), 7,11-7,75 (7Н, m), 7,85 (2H, d), 7,85-8,17 (4Н, m), 8,27 (IH, d), 8,65-9,12 (2Н, m)
Примесь 9а, химическое название которой 5-[(1S,2S)-2-[(3S,6S,9S,11R,15S,18S,20R,21S,24S,25S,26S)-3-[(R)-2-карбамоил-1-гидроксиэтил]-11,20,21,25-тетрагидрокси-15-[(R)-1 -гидроксиэтил]-26-метил-2,5,8,14,17,23-18-гексакарбонил-18-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензамид]-1,4,7,13,16,22-гексаазатрицикло[22.3.0.09,13]гептакозан-6-ил]-1,2-дигидроксиэтил]-2-гидроксифенил сульфат натрия
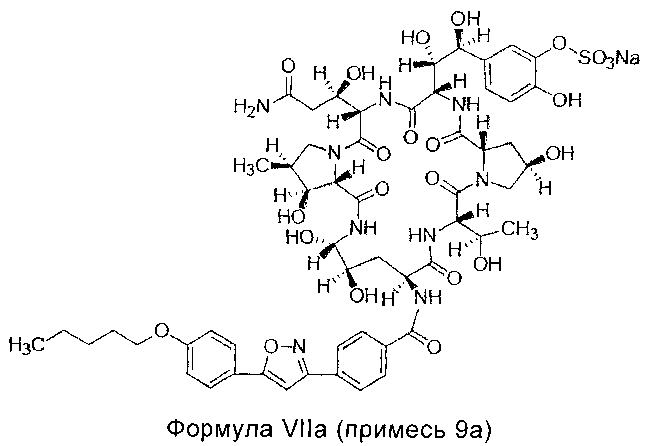
МС и ПМР данные примеси 9а перечислены ниже:
MC: 1314,4 [M+Na]+
ПМР (ДМСО-d6): 0,87-1,11 (6Н, t), 1,13 (3Н, d), 1,41-1,61 (4Н, m), 1,62-2,17 (6Н, m), 2,11-2,43 (3Н, m), 2,53-2,65 (IH, m), 3,05-3,11 (1Н, m), 3,62-4,63 (15Н, m), 4,71-5,23 (10Н, m), 5,26 (IH, d), 5,69 (IH, d), 6,53-6,72 (3Н, m), 7,11-7,72 (7Н, m), 7,81 (2Н, d), 7,86-8,11 (4Н, m), 8,28 (IH, d), 8,62-9,12 (2Н, m)
Примесь 10а, химическое название которой 5-[(1S,2S)-2-[(3S,6S,9S,11R,15S,18S,20R,21R,24S,25S,26S)-3-[(R)-2-карбамоил-1-гидроксиэтил]-11,21,25-тригидрокси-15-[(R)-1-гидроксиэтил]-26-метил-2,5,8,14,17,23-гексакарбонил-18-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензамид]-1,4,7,13,16,22-гексаазатрицикло[22.3.0.09,13]гептакозан-6-ил]-1,2-дигидроксиэтил]-2-гидроксифенил сульфат натрия
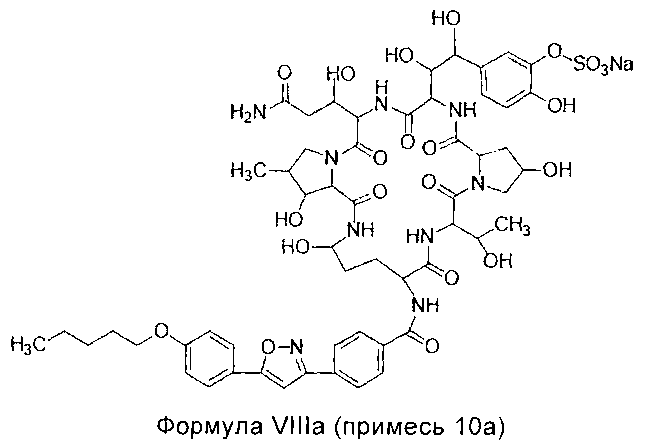
МС и ПМР данные примеси 9а перечислены ниже:
MC: 1298,4 [M+Na]+
ПМР (ДМСО-d6): 0,87-1,11 (6Н, t), 1,13 (3Н, d), 1,41-1,61 (4Н, m), 1,62-2,17 (8Н, m), 2,11-2,43 (3Н, m), 2,53-2,65 (IH, m), 3,05-3,11 (IH, m), 3,62-4,63 (14Н, m), 4,71-5,23 (10Н, m), 5,26 (IH, d), 5,69 (IH, d), 6,53-6,72 (3Н, m), 7,11-7,72 (7Н, m), 7,81 (2Н, d), 7,86-8,11 (4Н, m), 8,28 (IH, d), 8,62-9,12 (2Н, m)
В композиции количество примеси 6а больше чем 0,3%, общее количество примесей 7а и 8а больше чем 0,6%, количество примеси 9а больше чем 0,2%, количество примеси 10а больше чем 0,2%, а количество микафунгина всего лишь около 98,0%. Тем не менее, в соответствии с FDA (Food and Drug Administration - Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, США), API (active pharmaceutical ingredient - активные фармацевтические ингредиенты=действующее вещество) должно содержать примесей как можно меньше, чтобы применяться безопаснее. Например: по требованиям FDA, количество конкретной примеси должно поддерживаться ниже 0,1% (см подробнее в ICH Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, Q7A, Current Step 4 Version (November 10, 2000)).
WO03018615 описывает процесс кристаллизации Микафунгина натрия (например, соединение формулы II), который может создавать определенные эффекты очистки. Однако чистота коммерчески доступного препарата микафунгина натрия, полученного заявителем, Japan Fujisawa Pharmaceutical Company, из Микафунгина, полученного в результате этого процесса, составляет всего лишь около 98%, поэтому существует определенный клинический риск при применении препарата.
Как хорошо известно в данной области, чем выше чистота промежуточных лекарственных препаратов, тем выше чистота конечного лекарственного препарата, полученного с помощью химической модификации. Аналогично, чем выше чистота соединения формулы I в качестве промежуточного, тем выше чистота соединения формулы II (FK463), полученного путем химической модификации, после реакции. Если чистота соединения формулы II (FK463), полученного при химической модификации, является высокой, то давление на очистку соединения формулы II будет значительно освобождено, и конечный продукт, соединение формулы II (FK463), с высокой чистотой может быть получен с помощью простого процесса очистки. Из структуры структурных аналогов Микафунгина, таких как примесь 6, примесь 7, примесь 8, примесь 9, примесь 10, может быть определено, что вышеуказанные структурные аналоги, по существу, происходят из структурных аналогов соединения формулы I путем химической модификации реакции.


Тем не менее, очень трудно отделить структурные аналоги соединения формулы I, особенно если принимать во внимание одновременно чистоту и выход при очистке. В CN91104847 описана очистка соединения формулы I, где YMC GEL ODS-AM 120 использовали в качестве наполнителя для основных целей очистки, и препаративную ВЭЖХ использовали для очистки соединения формулы I. Из формулы I видно, что соединение формулы I обладает сильной полярностью и хорошей гидрофильностью, и будет слабо удерживаться на МС GEL ODS-AM 120-наполнителе. Таким образом, хорошие эффекты очистки не могут быть получены с помощью препаративной ВЭЖХ с YMC GEL ODS-AM 120-наполнителем. Чистота соединения формулы I, полученного в процессе очистки CN 91104847, составляет всего 97,51%.
Кроме того, различные процессы очистки, обычно используемые в данной области техники, включая ионообменную смолу, макропористую абсорбционную смолу и обращенно-фазовую препаративную хроматографию, связанную с силикагелем С18, и нормальную препаративную хроматографию со сферическим силикагелем, используются изобретателями для очистки соединения формулы I с помощью хроматографии. Однако соединение формулы I не может быть получено с общей чистотой более 99,0%.
С помощью некоторых способов очистки, таких как кристаллизация и/или перекристаллизация, чистота соединения формулы I была улучшена изобретателями от примерно 97% до более чем 99,0% от общей чистоты (предпочтительно, более чем 99,8% от общей чистоты), а количество каждой единичной примеси составляет менее 0,25%. И выход при кристаллизации является высоким, что очень подходит для промышленного производства.
В WO 9611210, WO 03018615 и WO 2004014879 сообщались процесс синтеза и очистки для Микафунгина. В WO 9611210 описан способ разделения с помощью препаративной колонки, однако такой метод требует большого количества органического растворителя, что вызывает серьезное загрязнение окружающей среды, и его трудно масштабировать; а чистота полученного продукта невысока. В WO 03018615, WO 2004014879 очистку выполняют с помощью кристаллизации, однако примесь 6, примесь 7, примесь 8, примесь 9 и примесь 10 не могут быть эффективно удалены путем кристаллизации.
Таким образом, авторы настоящего изобретения желают получить соединение формулы I высокой чистоты, т.е. промежуточный препарат, путем некоторых способов очистки на стадии промежуточных препаратов. А затем соединение формулы II (FK463) высокой чистоты получают путем химической модификации с тем, чтобы получить Микафунгин высокой чистоты в соответствии с требованиями FDA.
Сущность изобретения
Одним объектом настоящего изобретения является создание вещества, такого как соединение формулы I высокой чистоты.
Другим объектом настоящего изобретения является создание способа получения вещества высокой чистоты (соединение формулы I).
Третьим объектом настоящего изобретения является обеспечение применения вещества высокой чистоты (соединение формулы I).
Четвертым объектом настоящего изобретения является обеспечение способа получения другого вещества высокой чистоты (соединение формулы II).
Пятым объектом настоящего изобретения является создание способа получения другого вещества высокой чистоты (соединение формулы II).
Соединение формулы I высокой чистоты
В настоящем изобретении предусмотрено соединение формулы I высокой чистоты, и степень чистоты составляет не менее 99,0%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламина.
В предпочтительном воплощении настоящего изобретения, чистота соединения формулы I составляет не менее 99,2%.
В предпочтительном воплощении настоящего изобретения, чистота соединения формулы I составляет не менее 99,5%.
В другом предпочтительном воплощении настоящего изобретения, чистота соединения формулы I составляет не менее 99,8%.
Чистоту соединения формулы I определяли методом ВЭЖХ.
Чистоту соединений формулы I и/или количество примесей рассчитывали следующим образом: площадь под кривой пика ВЭЖХ профиля для соединения формулы I и/или примеси делили на общую площадь под кривой профиля ВЭЖХ.
Метод ВЭЖХ перечислен ниже:
Колонка: АСЕ 3 AQ, 150×4,6 мм, 3 мкм
Подвижная фаза: А: 1000 мл воды, 10 мл метанола, 100 мкл трифторуксусной кислоты
В: 600 мл воды, 400 мл метанола, 100 мкл трифторуксусной кислоты
Скорость потока: 0,55 мл/мин
Температура колонки: 50°С
Градиент

Температура инжектора: 5°С
Длина волны детектирования: 225 нм
В другом предпочтительном воплощении настоящего изобретения, количество примеси А в соединении формулы I высокой чистоты составляет не более 0,25%.
В другом предпочтительном воплощении настоящего изобретения, относительное время удерживания (сокращенно RRT - relative retention time) примеси А при ВЭЖХ составляет около 0,45, то есть, 0,45±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество примеси А составляет не более 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примесей А составляет не более 0,05%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси А составляет не более 0,05%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси B в соединении формулы I высокой степени чистоты составляет не более 0,25%.
В другом предпочтительном воплощении настоящего изобретения, относительное время удерживания примеси В при ВЭЖХ составляет около 0,65, то есть 0,65±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество примеси В составляет не более 0,15%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси В составляет не более 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси В составляет от 0,03% до 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси В составляет не более 0,03%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси С в соединении формулы I высокой чистоты составляет не более 0,25%.
В другом предпочтительном воплощении настоящего изобретения, относительное время удерживания примеси С при ВЭЖХ составляет около 0,88, то есть 0,88±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество примеси С не больше чем 0,15%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси С не больше чем 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси С составляет от 0,02% до 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси С не более 0,02%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси D в соединении формулы I высокой степени чистоты составляет не более 0,20%.
В другом предпочтительном воплощении настоящего изобретения, относительное время удерживания примеси D при ВЭЖХ составляет около 1,08, то есть 1,08±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество примеси D составляет не более 0,15%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси D составляет не более 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси D составляет от 0,04% до 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси D составляет не более 0,04%.
В другом предпочтительном воплощении настоящего изобретения, относительное время удерживания примеси Е при ВЭЖХ составляет около 1,29, то есть 1,29±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество примеси Е составляет не более 0,10%.
В другом предпочтительном воплощении настоящего изобретения, количество примеси Е составляет не более 0,05%.
В следующем предпочтительном воплощении настоящего изобретения, количество примеси F в соединении формулы I высокой степени чистоты составляет не более 0,15%.
В следующем предпочтительном воплощении настоящего изобретения, относительное время удерживания примеси F при ВЭЖХ составляет около 1,92, то есть 1,92±0,02.
В следующем предпочтительном воплощении настоящего изобретения, количество примеси F составляет не более 0,10%.
В следующем предпочтительном воплощении настоящего изобретения, количество примеси F составляет не более 0,05%.
В другом предпочтительном воплощении настоящего изобретения, количество любых других соответствующих примесей в соединении формулы I высокой степени чистоты составляет не более чем 0,10%, и указанные другие соответствующие примеси относятся к примесям, отличным от примесей A-F, которые могут присутствовать.
В другом предпочтительном воплощении настоящего изобретения, количество любых других соответствующих примесей составляет не более 0,05%.
В другом предпочтительном воплощении настоящего изобретения, количество других соответствующих примесей составляет 0%.
Получение соединения формулы I высокой чистоты
После изучения авторы настоящего изобретения неожиданно обнаружили, что кристаллы с превосходной морфологией могут быть образованы из соединений формулы I путем растворения соединения в воде или смешанном растворе вода - смешиваемые с водой низшие спирты, и поддерживания раствора соединения формулы I около точки насыщения растворимости и поддержания значения рН раствора в заданном интервале. Такой процесс кристаллизации будет производить хорошие эффекты очистки, тем самым получая соединение формулы I высокой чистоты. Далее, поскольку соединение формулы I является циклопептидным соединением, пептидная связь, образуемая при конденсации аминокислоты, будет разрушена путем гидролиза в растворе при высокой температуре. Таким образом, процесс кристаллизации для соединения формулы I нужно контролировать при определенном температурном диапазоне, чтобы гарантировать, что циклопептид не будет деградировать при раскрытии кольца. Для способа получения по настоящему изобретению был выполнен большой объем работ интенсивных исследований по скринированию растворителей для кристаллизации, и было обнаружено, что кристаллы с превосходной морфологией могут быть образованы из соединений формулы I путем кристаллизации соединения в метаноле, этаноле, n-пропаноле, изопропаноле или смеси этих растворов, и процесс кристаллизации будет производить хорошие эффекты очистки. Тем не менее, при кристаллизации соединения в растворителе, таком как ацетон, ацетонитрил, этилацетат, соединение формулы I будет образовывать аморфный осадок, и эффекты очистки, такие как удаление примесей, не могут быть достигнуты за счет такого процесса осаждения.
Способ получения по настоящему изобретению включает в себя следующие этапы:
(a) растворение сырого соединения формулы I в воде или водном органическом растворителе (i) и контролирование рН раствора;
(b) получение соединения формулы I высокой чистоты путем снижения температуры и/или добавления органического растворителя (i).
На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), соотношение объемов органического растворителя (i) к воде в водном растворе органического растворителя (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно от 0,5 до 3,0.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), температуру понижают до температуры от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С и наиболее предпочтительно от 5 до 10°С.
В стадии (b), соотношение объемов органического растворителя (i) к раствору со стадии (а) составляет от 0,1 до 50, предпочтительно от 0,1 до 10 и наиболее предпочтительно от 1 до 5.
На стадии (а) и/или (b), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбранный из: метанола, этанола, n-пропанола, изопропанола или их смеси.
Соединение формулы I высокой чистоты, полученное на стадии (b), представляет собой кристаллы.
После стадии (b), могут быть следующие стадии:
(c) центрифугирование или фильтрование;
(d) удаление растворителя и большей части воды (сушка), так, чтобы получить соединения формулы I высокой чистоты.
Стадия (а)-(b) может быть повторена один или более раз для перекристаллизации, предпочтительно от 1 до 4 раз.
В одном воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в воде и контролирование рН раствора;
(b) получение кристаллов соединения формулы I путем снижения температуры раствора;
(c) центрифугирование или фильтрование;
(d) сушка, так, чтобы получить высокую чистоту соединения формулы I. На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, на основании общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), температуру снижают от -10 до 35°С, более предпочтительно от -5 до 30°С и наиболее предпочтительно от 5 до 10°С.
В другом воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в воде и контролирование рН раствора;
(b) добавление органического растворителя (i), таким образом, чтобы полностью осадить кристаллы соединения формулы I;
(c) центрифугирование или фильтрование;
(d) сушку кристаллов, так, чтобы получить высокую чистоту соединения формулы I.
На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 50 до 300 мг/мл соединения формулы I, на основе общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбирают из: метанола, этанола, n-пропанола, изопропанола или их смесей.
В стадии (b), соотношение объемов органического растворителя (i) к раствору со стадии (а) составляет от 0,1 до 50, предпочтительно от 0,1 до 10 и наиболее предпочтительно от 1 до 5.
В другом воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в воде, и контролирование рН раствора;
(b) снижение температуры раствора и добавление органического растворителя (i), с тем, чтобы полностью осадить кристаллы соединения формулы I;
(c) центрифугирование или фильтрование;
(d) сушка кристаллов, так, чтобы получить высокую чистоту соединения формулы I.
На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 50 до 300 мг/мл соединения формулы I, на основе общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбирают из: метанола, этанола, n-пропанола, изопропанола или их смесей.
На стадии (b), температуру понижают до температуры от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С и наиболее предпочтительно от 5 до 10°С.
На стадии (b), соотношение объемов органического растворителя (i) к раствору со стадии (а) составляет от 0,1 до 50, предпочтительно от 0,1 до 10 и наиболее предпочтительно от 1 до 5.
В другом воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в водном растворе органического растворителя (i) и контролирование рН раствора;
(b) снижение температуры раствора, с тем, чтобы осадить кристаллы соединения формулы I;
(c) центрифугирование или фильтрование;
(d) сушка кристаллов, так, чтобы получить высокую чистоту соединения формулы I.
На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), соотношение объемов органического растворителя (i) и воды в водном растворе органического растворителя (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, и наиболее предпочтительно от 0,5 до 3,0.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, на основе общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (а), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбирают из: метанола, этанола, n-пропанола, изопропанола, или их смесей.
На стадии (b) температуры понижают до температуры от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10°С.
В другом воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в водном растворе органического растворителя (i), и контролирование рН раствора;
(b) добавление органического растворителя (i), с тем, чтобы осадить кристаллы соединения формулы I;
(c) центрифугирование или фильтрование;
(d) сушка кристаллов, так, чтобы получить высокую чистоту соединения формулы I.
На стадии (а) температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а) соотношение объемов органического растворителя (i) и воды в водном растворе органического растворителя (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10 и наиболее предпочтительно от 0,5 до 3,0.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, на основе общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), соотношение объемов органического растворителя (i) и раствора со стадии (а) составляет от 0,1 до 10, предпочтительно от 1 до 5.
На стадии (а) и (b), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбирают из: метанола, этанола, n-пропанола, изопропанола или их смесей.
В другом воплощении настоящего изобретения, соединение формулы I высокой чистоты может быть получено с помощью следующих стадий:
(a) растворение соединения формулы I в водном растворе органического растворителя (i) и контролирование рН раствора;
(b) снижение температуры раствора и добавление органического растворителя (i), так, чтобы осадить кристаллы соединения формулы I;
(c) центрифугирование или фильтрование;
(d) сушка кристаллов, так, чтобы получить высокую чистоту соединения формулы I.
На стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), соотношение объемов органического растворителя (i) и воды в водном растворе органического растворителя (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, и наиболее предпочтительно от 0,5 до 3,0.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, на основе общего объема раствора.
На стадии (а), рН раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), температуру снижают до температуры от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С и наиболее предпочтительно от 5 to 10°С;
На стадии (b), соотношение объемов органического растворителя (i) и раствора со стадии (а) составляет от 0,1 до 50, предпочтительно от 0,1 до 10 и наиболее предпочтительно от 1 до 5.
На стадии (а) и (b), указанный органический растворитель (i) представляет собой С1-С4-низший спирт; предпочтительно выбирают из: метанола, этанола, n-пропанола, изопропанола, или их смесей.
Соединение формулы I, полученное с помощью способа, представленного в настоящем изобретении, обладает высокой чистотой; таким образом, его будет лучше использовать для получения соединения формулы II.
Соединение формулы II высокой чистоты
В одном аспекте, соединение формулы II высокой чистоты предусмотрено в настоящем изобретении, и его чистота по ВЭЖХ составляет не менее 98,80%; предпочтительно не менее 99,0%; более предпочтительно не менее 99,5%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламина.
Чистоту соединения формулы II определяют с помощью ВЭЖХ. Чистоту соединения формулы II и/или количество примесей рассчитывают следующим образом: площадь под кривой пика на профиле ВЭЖХ для соединения формулы II и/или примеси делят на общую площадь под кривой профиля ВЭЖХ.
Чистоту соединения формулы II в жидкой фазе определяют способом ВЭЖХ, и способ ВЭЖХ указан следующим образом:
ВЭЖХ Аналитическая колонка: YMC-ODS 250*4,6 мм, 5 мкм;
Подвижная фаза: ацетонитрил: фосфатный буфер = 70:45;
Программа элюирования: изократическая;
Скорость потока: 1,15 мл/мин;
Температура колонки: 35°С;
Длина волны детектирования: 210 нм;
Время прогона: 45 мин;
Растворитель: водный фосфатный буфер.
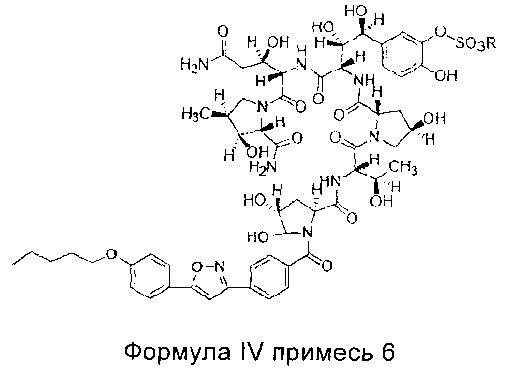
В одном предпочтительном воплощении настоящего изобретения количество примеси 6 (структура которой показана в формуле IV) в соединении формулы II высокой степени чистоты составляет менее 0,2%; предпочтительно менее 0,1%; более предпочтительно менее 0,05%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламина.
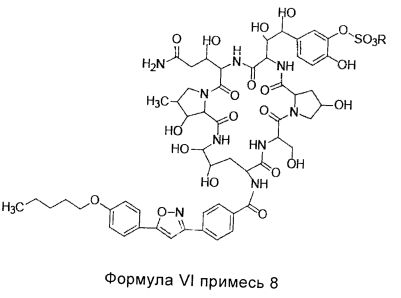
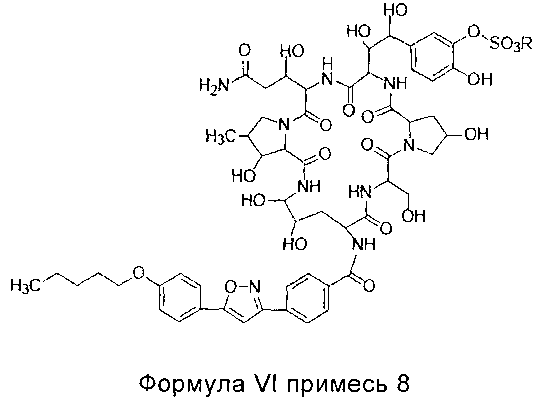
В одном предпочтительном воплощении настоящего изобретения, общее количество примеси 7 (структура которой показана в формуле V) и примеси 8 (структура которой показана в формуле VI) в соединении формулы II высокой степени чистоты составляет меньше 0,5%; предпочтительно менее 0,3%; более предпочтительно менее 0,1%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламин.

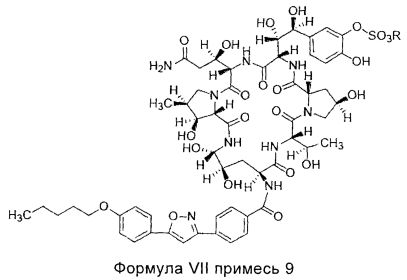
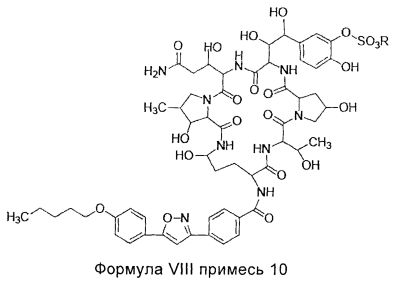
В другом предпочтительном воплощении настоящего изобретения количество примеси 9 (структура которой показана в формуле VII) в соединении формулы II высокой степени чистоты составляет менее 0,2%; предпочтительно менее 0,1%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламина.

В другом предпочтительном воплощении настоящего изобретения, количество примеси 10 (структура которой показана в формуле VIII) в соединении формулы II высокой степени чистоты, составляет менее 0,2%; предпочтительно менее 0,1%; где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; предпочтительно Н, ион натрия или ион диизопропилэтиламина.

В другом предпочтительном воплощении настоящего изобретения количество примеси 11 в соединении формулы II высокой степени чистоты составляет не более 0,15%; предпочтительно не более 0,1%; наиболее предпочтительно не более 0,05%. И относительное время удерживания примеси 11 при ВЭЖХ составляет около 0,96, т.е. 0,96±0,02.
В другом предпочтительном воплощении настоящего изобретения, количество любых других соответствующих примесей в соединении формулы II высокой степени чистоты составляет не более 0,02%; предпочтительно не более 0,01%; наиболее предпочтительно 0%; и указанные другие соответствующие примеси относятся к примесям, отличающимся от примесей 6-11, которые могут присутствовать.
Способ получения соединения формулы II высокой чистоты.
Способ получения соединения формулы II высокой чистоты, где циклопептидное соединение высокой чистоты по любому из пп. 1-11, используют в качестве сырья для получения соединения формулы II; где R представляет собой Н или ион диизопропилэтиламина или другие катионы, способные образовывать фармацевтически приемлемую соль.
Пути синтеза соединения были зарегистрированы в нескольких патентах, таких как WO 9611210, 9857923, 2604014879 и т.д.
Применение соединения формулы I высокой чистоты и композиций, его содержащих.
В настоящем изобретении представлено применение соединения формулы I высокой степени чистоты. В одном аспекте, оно может быть легко использовано для получения соединения формулы II высокой степени чистоты, где R представляет собой Н, диизопропилэтиламин или другие катионы, способные образовывать фармацевтически приемлемую соль.
В другом аспекте соединение формулы I высокой чистоты, представленное в данном изобретении, может быть непосредственно использовано для получения лекарственных препаратов для лечения грибковых инфекций. Также могут быть предусмотрены фармацевтическая композиция, содержащая соединение формулы I и фармацевтически приемлемый носитель.
Применение соединения формулы II высокой чистоты и композиций, его содержащих.
В настоящем изобретении представлено применение соединения формулы II высокой чистоты. Соединение формулы II высокой чистоты, представленное в настоящем изобретении, может быть непосредственно использовано для получения лекарственных препаратов для лечения грибковых инфекций. Также могут быть предусмотрены фармацевтическая композиция, содержащая соединение формулы II и фармацевтически приемлемый носитель.
В настоящем изобретении предложен способ получения фармацевтической композиции, содержащей соединение формулы II высокой чистоты:
Соединение формулы II высокой чистоты по настоящему изобретению смешивают с фармацевтически приемлемым носителем, так, чтобы получить фармацевтическую композицию, содержащую соединение формулы II высокой чистоты.
Как здесь указано, "соединение формулы I" или " формулы I соединение" можно использовать взаимозаменяемо, оба выражения относятся к соединению, имеющему следующую структурную формулу или его фармацевтически приемлемой соли:

где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль, предпочтительно Н, ион натрия или ион диизопропилэтиламина.
Предпочтительно, фармацевтически приемлемые соли включают: соли металлов, таких как соли щелочных металлов (такие как натриевая соль, калиевая соль), соли щелочноземельных металлов (такие как кальциевая соль, магниевая соль и т.п.), соли аммония, соли, образованные с органическими основаниями (например, соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, дициклогексиламиновая соль, N,N,-дибензилэтилендиаминовая соль, соль диизопропилэтиламина и т.д.), соли присоединения органической кислоты (такие как: формиат, ацетат, трифторацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), соли присоединения неорганической кислоты (например, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат и т.д.), соли, образованные с аминокислотой (например, аргинином, аспарагиновой кислотой, глутаминовой кислотой и т.д.), и подобные.
Как используется здесь, выражения "соединение формулы II" или "формулы II соединение" могут быть использованы взаимозаменяемо, оба выражения относятся к соединению, имеющему следующую структурную формулу, или его фармацевтически приемлемой соли:

где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль, предпочтительно Н, ион натрия или ион диизопропилэтиламина.
Предпочтительно фармацевтически приемлемые соли включают: соли металлов, такие как соли щелочных металлов (например, соль натрия, соль калия), соли щелочноземельных металлов (таких как соль кальция, соль магния и т.д.), соли аммония, соли, образованные с органическими основаниями (например, соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, соль дициклогексиламина, N, N, соль дибензилэтилендиамина, соль диизопропилэтиламина и т.д.), соли присоединения органической кислоты (такие как формиат, ацетат, трифторацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), соли присоединения неорганической кислоты (например, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат и т.д.), соли, образованные с аминокислотой (например, аргинином, аспарагиновой кислоты, глутаминовой кислоты и т.д.), и подобные.
Используемые здесь выражения «чистота соединения формулы I", "чистота соединения I" или "чистота по ВЭЖХ соединения I» могут быть использованы взаимозаменяемо, все они относятся к процентному соотношению измеренной площади пика соединения I и суммы площадей пиков всех пиков в условиях обнаружения при ВЭЖХ, предусмотренных настоящим изобретением (ВЭЖХ).
Используемые здесь выражения «чистота соединения формулы II", "чистота соединения II" или "чистота по ВЭЖХ соединения II" могут быть использованы взаимозаменяемо, все они относятся к процентному соотношению измеренной площади пика соединения II и суммы площадей пиков всех пиков в условиях обнаружения по ВЭЖХ, предусмотренных в соответствии с настоящим изобретением (ВЭЖХ).
Используемые здесь выражения «сырое соединение формулы I" или "неочищенное соединение I» могут быть использованы взаимозаменяемо, оба они относятся к смеси, где количество соединения I составляет меньше 98% в условиях обнаружения по ВЭЖХ, предусмотренных в настоящем изобретении. Сырое соединение I может быть получено с помощью способов, известных в данной области. Например, но не ограничиваясь этим, соединение формулы III (продукт, полученный при ферментации микроорганизмами) используют в качестве сырья для получения полусинтетического производного (например, соединения формулы I) через деацилирование с помощью деацилазы и сырое соединение I получают после выделения и очистки. Способ получения неочищенного соединения I можно смотреть, US 5376634, ЕР 0431350 и CN 1161462 C; и оно может быть получено с помощью коммерческих источников, таких как Fujisawa, Japan, но не ограничиваясь этим.
Используемый здесь термин "относительное время удерживания (RRT)" относится к отношению времени удерживания пика ко времени удерживания основного пика при определенных условиях ВЭЖХ. Например, при определенных условиях ВЭЖХ, если время удерживания основного пика составляет 1 минуту и время удерживания другого пика составляет 2 минуты, то относительное время удерживания (RRT) последнего составляет 2.
Используемый здесь термин "относительное время удерживания (RRT)" может колебаться в пределах указанного диапазона. Как хорошо известно в данной области, могут быть систематические ошибки при ВЭЖХ из-за адаптивности системы, приводящие к сдвинутому относительному времени удерживания (RRT). Как правило, предусмотрено, что интервал допустимых значений составляет ±0,02. Например, в соответствии со стандартами регистрации импортных лекарственных средств SFDA (State Food and Drug Administration = Государственное управление по контролю за продуктами и лекарствами) существует определенный интервал для относительного времени удерживания (RRT) для микафунгина натрия для инъекций и соответствующих примесей.
Используемое здесь выражение "С1-С4-низший спирт" относится к спиртам, число атомов углерода которых составляет от 1 до 4.
Используемые здесь "фармацевтически приемлемые соли" предпочтительно включают: соли металлов, такие как соли щелочных металлов (например, соль натрия, соль калия), соли щелочноземельных металлов (таких как соль кальция, соль магния и т.д.), соли аммония, соли, образованные с органическими основаниями (например, соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, соль дициклогексиламина, соль N,N,-дибензилэтилендиамина, соль диизопропилэтиламина и т.д.), соли присоединения органической кислоты (например, формиат, ацетат, трифторацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), соли присоединения неорганической кислоты (например, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат и т.д.), соли, образованные с аминокислотой (например, аргинином, аспарагиновой кислоты, глутаминовой кислоты, и т.д.), и тому подобное.
Как используется здесь, термин "фармацевтически приемлемый носитель" относится к носителю для введения терапевтического агента, включая различные наполнители и разбавители. Этот термин относится к таким носителям, которые сами по себе не являются необходимо активными ингредиентами и не будут производить чрезмерную токсичность при введении. Подходящие носители хорошо известны в данной области техники. В Remington′s Pharmaceutical Sciences (Mack Pub. Co., NJ 1991) можно найти всестороннее обсуждение фармацевтически приемлемых наполнителей. В композиции фармацевтически приемлемые носители могут включать жидкости, такие как вода, физиологический раствор, глицерин и этанол. Кроме того, вспомогательные вещества могут присутствовать в этих носителях, таких как разрыхлители, смачивающие агенты, эмульгаторы, рН-буферные вещества и тому подобное.
Преимущества данного изобретения в основном включают:
1. В настоящем изобретении чистота соединения формулы I была значительно улучшена, и примеси были значительно снижены, так, чтобы получить соединение формулы I высокой степени чистоты и решить технические проблемы, которые необходимо решить на предшествующем уровне.
2. Изобретатели выбрали конкретные условия получения путем многократных экспериментов, и были получены неожиданные технические эффекты, так что осуществляется способ получения для соединения формулы I высокой чистоты, и такой способ пригоден для крупномасштабного производства и с высоким выходом.
3. Новый способ получения соединения формулы II высокой степени чистоты обеспечивается в настоящем изобретении, где соединение формулы II может быть получено из соединения формулы I высокой степени чистоты. Давление при очистке соединения II будет значительно снижено, и конечный продукт, соединение формулы II высокой чистоты, может быть получено с помощью простого процесса очистки. Выход также значительно улучшен, тем самым достигнуты неожиданные технические эффекты.
Краткое описание чертежей
Фиг. 1 является ВЭЖХ профилем сырого соединения формулы I, где



Способ осуществления изобретения
Далее изобретение будет проиллюстрировано со ссылкой на следующие конкретные примеры. Следует понимать, что эти примеры предназначены только для иллюстрации изобретения, но не для ограничения объема данного изобретения. Для экспериментальных методов в следующих примерах без особых условий, они выполнены в обычных условиях или в соответствии с инструкциями производителя. Если не указано иное, все проценты, соотношения пропорций или части даны по массе.
Единицы в процентах вес/объем по изобретению хорошо известны специалистам в данной области техники, например, вес растворенного вещества в 100 мл раствора.
Если не указано иначе, все технические и научные термины, используемые здесь, имеют такое же значение, как обычно понимает специалист в данной области. Кроме того, любой процесс или материал, аналогичный или эквивалентный тем, которые описаны здесь, может быть использован в процессе по настоящему изобретению. Предпочтительные воплощения и материалы, описанные здесь, приведены только в качестве иллюстрации.
Чистоту соединения формулы I в жидкой фазе определяли способом ВЭЖХ и способ ВЭЖХ указан следующим образом:
Колонка: АСЕ 3 AQ, 150×4,6 мм, 3 мкм
Подвижная фаза: А: 1000 мл воды, 10 мл метанола, 100 мкл трифторуксусной кислоты
В: 600 мл воды, 400 мл метанола, 100 мкл
трифторуксусной кислоты
Скорость потока: 0,55 мл/мин
Температура колонки: 50°С
Градиент

Температура инъектора: 5°С
Длина волны детектирования: 225 нм
Чистоту соединения формулы II в жидкой фазе определяли способом
ВЭЖХ, и способ ВЭЖХ указан следующим образом:
ВЭЖХ Аналитическая колонка: YMC-ODS 250*4,6 мм, 5 мкм;
Подвижная фаза:ацетонитрил:фосфатный буфер = 70:45;
Программа элюирования: изократическая;
Скорость потока: 1,15 мл/мин;
Температура колонки: 35°С;
Длина волны детектирования: 210 нм;
Время прогона: 45 мин;
Растворитель: водный фосфатный буфер,
где относительное время удерживания соединения формулы II и соответствующих примесей составляет

Пример 1
Получение сырого соединения формулы I
76 г соединения формулы I в виде твердого порошка получали в соответствии со способом Примера 1 в патенте США U.S. Patent No. 5,376,634 и его количество определяли как 97,51% по данным ВЭЖХ (см. Фиг. 1 для ВЭЖХ-профиля)

Пример 2
Получение соединения формулы I высокой чистоты.
При 30°С 3,6 г сырого соединения I, полученного в Примере 1, растворяли в смешанном растворе, состоящем из 25 мл воды и 20 мл n-пропанола и перемешивали до полного растворения соединения I. рН доводили до 3,5 с использованием ледяной уксусной кислоты, и раствор постепенно охлаждали до 15°С,. Кристаллы соединения I осаждали и систему перемешивали в течение 5 часов при 15°С, так, чтобы кристаллы соединения I росли постепенно. По каплям добавляли 90 мл n-пропанола. После добавления полученную смесь перемешивали в течение 1 часа при 15°С. Кристаллы получали фильтрованием и сушили в вакууме с получением 3,5 г соединения I, чистоту которого определяли с помощью ВЭЖХ, как 99,00%. Количество основных соответствующих примесей приведено в Таблице 2.

Пример 3
Получение соединения формулы I высокой чистоты.
При 40°С 3,5 г соединения I, полученного в Примере 2 (чистота по ВЭЖХ которого была 99,00%), растворяли в смешанном растворе, состоящем из 19 мл воды и 16 мл n-пропанола и перемешивали до полного растворения соединения I. рН доводили до 2,0 с помощью ледяной уксусной кислоты, и раствор постепенно охлаждали до 15°С. Кристаллы соединения I осаждали и систему перемешивали в течение 5 часов при 15°С, так, что кристаллы соединения росли постепенно. По каплям добавляли 70 мл n-пропанола. После добавления полученную смесь перемешивали в течение 1 часа при 15°С. Кристаллы получали фильтрованием и сушили в вакууме с получением 3,4 г соединения I, чистоту которого определяли с помощью ВЭЖХ, как 99,23%. Количество основных соответствующих примесей приведено в таблице 3.
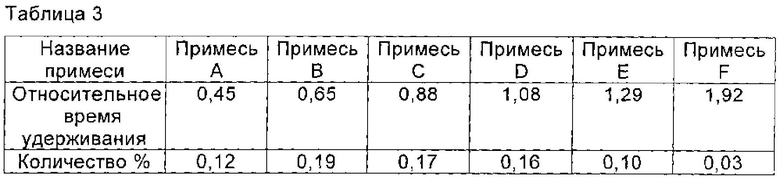
Пример 4
Получение соединения I высокой чистоты
При 40°С соединение I, полученное в Примере 3 (ВЭЖХ чистота которого была 99,23%), растворяли в смешанном растворе, рН которого доводили до 5,0 с помощью ледяной уксусной кислоты, состоящем из 8 мл воды и 7 мл n-пропанола, и перемешивали до полного растворения соединения I. Раствор охлаждали до 15°С. Кристаллы соединения I осаждали и систему перемешивали в течение 5 часов при 15°С, так что кристаллы соединения росли постепенно. По каплям добавляли 30 мл n-пропанола. После добавления полученную смесь перемешивали в течение 1 часа при 15°С. Кристаллы получали фильтрованием и сушили в вакууме с получением 3,3 г соединения I, чистоту которого определяли с помощью ВЭЖХ, как 99,57%. Количество основных соответствующих примесей приведено в таблице 4.
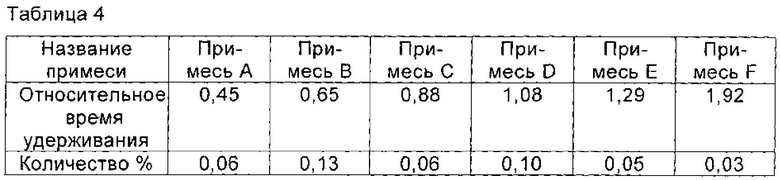
Пример 5
Получение соединения I высокой чистоты
При 40°С соединение I, полученное в Примере 4 (ВЭЖХ чистота которого была 99,57%) растворяли в смешанном растворе, рН которого доводили до 4,0 с помощью ледяной уксусной кислоты, состоящем из 8 мл воды и 7 мл n-пропанол, и перемешивали до полного растворения соединения I. Раствор охлаждали до 15°С. Кристаллы соединения I осаждали и систему перемешивали в течение 5 часов при 15°С, так что кристаллы соединения I росли постепенно. По каплям добавляли 30 мл n-пропанола. После добавления полученную смесь перемешивали в течение 1 часа при 15°С. Кристаллы получали фильтрованием и сушили в вакууме с получением 3,2 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 99,81%. Количество соответствующих примесей приведено в Таблице 5 и ВЭЖХ-профиле на Фиг. 2.
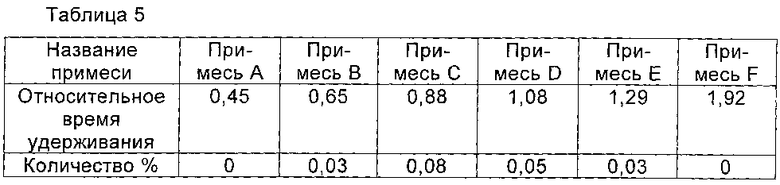
Пример 6
Получение соединения формулы I высокой чистоты
При 30°С 2,6 г соединения I, полученного в Примере 1, растворяли в 8 мл воды, рН доводили до 3,8 с помощью ледяной уксусной кислоты и перемешивали до полного растворения соединения I. По каплям медленно добавляли 10 мл n-пропанола. Кристаллы соединения I осаждали и систему перемешивали в течение 2 часов при 15°С. И затем 35 мл n-пропанола медленно добавляли по каплям. После добавления раствор, содержащий соединение I, медленно охлаждали до 15°С и перемешивали в течение еще 2 часов, для полного осаждения кристаллов соединения I. Влажный твердый осадок соединения I получали путем фильтрации. При 30°С 8 мл воды, рН которого доводили до 3,8 с помощью ледяной уксусной кислоты, использовали для растворения влажного твердого осадка соединения I и перемешивали до полного растворения соединения I. Раствор охлаждали до 15°С и медленно по каплям добавляли 10 мл n-пропанола для осаждения кристаллов соединения I. Систему перемешивали в течение еще 2 часов при 15°С. Медленно по каплям добавляли 35 мл n-пропанола. После добавления систему перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Влажный твердый осадок соединения I получали путем фильтрации. Описанную выше процедуру кристаллизации повторяли. После трехкратной перекристаллизации, соединение I получали путем фильтрации и сушили в вакууме с получением 2,4 г соединения I. Общий выход 92,3%, и чистоту соединения I определяли с помощью ВЭЖХ как 99,81%.
Пример 7
Получение соединения формулы I высокой чистоты
При 30°С 1,6 г соединения I, полученного в Примере 1, растворяли в смешанном растворе, состоящем из 80 мл воды и 80 мл этанола, и перемешивали до полного растворения соединения I. рН доводили до 2,8 с помощью ледяной уксусной кислоты, и раствор медленно охлаждали до -20°С. Раствор перемешивали при -20°С в течение 2 часов. Медленно по каплям добавляли 450 мл этанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,5 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 98,89%. Соответствующие примеси представлены в таблице 6.
Таблица 6

Пример 8
Получение соединения формулы I высокой чистоты
При 30°С 1,5 г соединения I, полученного в примере 7, растворяли в смешанном растворе, состоящем из 80 мл воды и 80 мл этанола, и перемешивали до полного растворения соединения I. рН доводили до 3,0 с помощью ледяной уксусной кислоты, и раствор медленно охлаждали до -20°С. Раствор перемешивали при -20°С в течение 2 часов. Медленно по каплям добавляли 450 мл этанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,4 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 99,36%. Соответствующие примеси представлены в таблице 7.
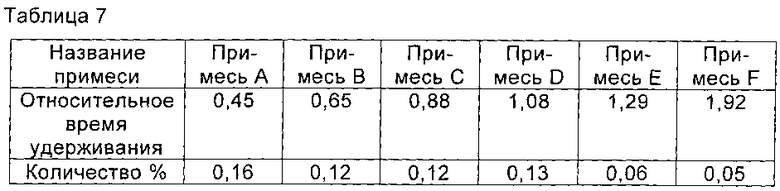
Пример 9
Получение соединения формулы I высокой чистоты
При 30°С 1,4 г соединения I, полученного в примере 8, растворяли в смешанном растворе, состоящем из 80 мл воды и 80 мл этанола, и перемешивали до полного растворения соединения I. рН доводили до 3,0 с помощью ледяной уксусной кислоты, и раствор медленно охлаждали до -20°С. Раствор перемешивали при -20°С в течение 2 часов. Медленно по каплям добавляли 450 мл этанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,3 г соединения I, чистоту которого определяли с помощью ВЭЖХ, как 99,68%. Соответствующие примеси представлены в таблице 8.

Пример 10
Получение соединения формулы I высокой чистоты
При 10°С 1,8 г соединения I, полученного в примере 1, растворяли в 10 мл воды, и перемешивали до полного растворения соединения I. рН доводили до 3,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 2°С. Раствор перемешивали при 2°С в течение 2 часов. Медленно по каплям добавляли 40 мл изопропанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,5 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 98,89%. Количество соответствующих примесей приведено в таблице 9.
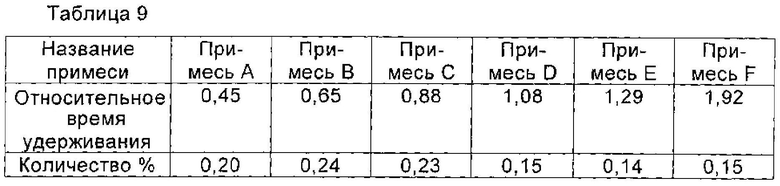
Пример 11
Получение соединения формулы I высокой чистоты
При 10°С 1,5 г соединения I, полученного в примере 10, растворяли в 10 мл воды, и перемешивали до полного растворения соединения I. рН доводили до 3,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 2°С. Раствор перемешивали при 2°С в течение 2 часов. Медленно по каплям добавляли 40 мл изопропанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,4 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 99,41%. Количество соответствующих примесей приведено в таблице 10.
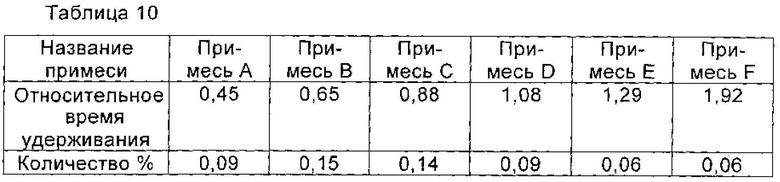
Пример 12
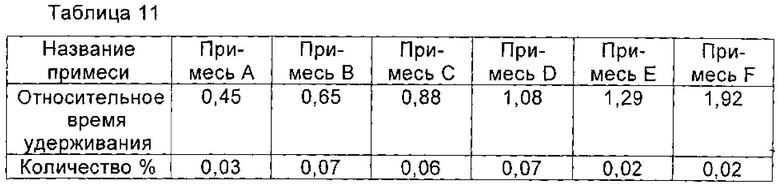
Получение соединения формулы I высокой чистоты.
При 10°С 1,4 г соединения I, полученного в примере 11, растворяли в 10 мл воды, и перемешивали до полного растворения соединения I. рН доводили до 3,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 2°С. Раствор перемешивали при 2°С в течение 2 часов. Медленно по каплям добавляли 40 мл изопропанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,3 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 99,73%. Количество соответствующих примесей приведено в таблице 11.
Пример 13
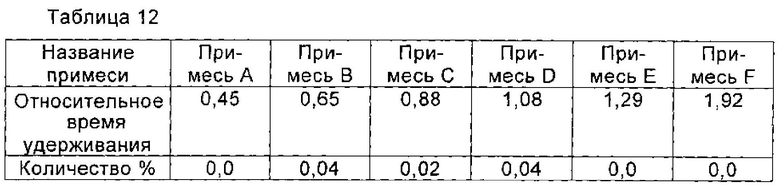
Получение соединения формулы I высокой чистоты
При 10°С 1,3 г соединения I, полученного в примере 12, растворяли в 10 мл воды и перемешивали до полного растворения соединения I. рН доводили до 3,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 2°С. Раствор перемешивали при 2°С в течение 2 часов. Медленно по каплям добавляли 40 мл изопропанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Кристаллы получали фильтрованием и сушили в вакууме с получением 1,2 г соединения I, чистоту которого определяли с помощью ВЭЖХ как 99,90%. Количество соответствующих примесей приведено в таблице 12.
Пример 14
Получение соединения формулы I высокой чистоты
При 20°С 2,0 г соединения I, полученного в примере 1, растворяли в смешанном растворе, состоящем из 5 мл воды и 15 мл метанола, и перемешивали до полного растворения соединения I. рН доводили до 4,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 10°С для осаждения кристаллов соединения I. Затем систему медленно охлаждали до -40°С и перемешивали при -40°С в течение еще 2 часов, для полного осаждения кристаллов соединения I. Медленно по каплям добавляли 80 мл метанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Влажное твердое соединение I получают путем фильтрации. При 20°С смесь раствора, содержащего 5 мл воды и 16 мл метанола, рН которого доводили до 4,5 с помощью ледяной уксусной кислоты, использовали для растворения влажного твердого соединения I и перемешивали в течение 30 минут до полного растворения соединения I. Раствор охлаждали до 10°С, и кристаллы соединения I выпадали в осадок. И затем систему медленно охлаждали до -40°С и перемешивали при -40°С в течение еще 2 часов. Медленно по каплям добавляли 80 мл метанола. После добавления систему перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Влажное твердое соединение I получали путем фильтрации. Описанную выше процедуру кристаллизацию повторяли. После трехкратной перекристаллизации соединение I получали путем фильтрации и сушили в вакууме с получением 1,5 г твердого соединения I. Общий выход составлял 89,0%, и чистоту соединения I определяли с помощью ВЭЖХ как 99,83%. Количество соответствующих примесей приведено в таблице 13.

Пример 15
Получение соединения формулы I высокой чистоты.
При 50°С 5,0 г соединения, полученного в примере 1, растворяли в 10 мл воды, и перемешивали до полного растворения соединения I. рН доводили до 5,0 с использованием ледяной уксусной кислоты и раствор охлаждали до 30°С для осаждения кристаллов соединения I. Затем систему медленно охлаждали до 2°С и перемешивали при 2°С в течение еще 10 часов. Влажное твердое соединение I получали путем фильтрации. При 50°С 9 мл воды, рН которой доводили до 5,0 с помощью ледяной уксусной кислоты, использовали для растворения влажного твердого соединения I и перемешивали в течение 30 мин до полного растворения соединения I. Раствор охлаждали до 30°С, и кристаллы соединения I выпадали в осадок. А затем систему медленно охлаждали до 2°С и перемешивали при 2°С в течение еще 10 часов. Влажное твердое соединение I получали путем фильтрации. Вышеуказанную процедуру кристаллизации повторяли. После трехкратной перекристаллизации соединение I получали путем фильтрации и сушили в вакууме с получением 3,0 г соединения I. Общий выход составлял 85,0%, и чистоту соединения определяли с помощью ВЭЖХ как 99,85%. Количество соответствующих примесей приведено в таблице 14.
Сравнительный пример 1
Влияние рН на получение соединения I высокой чистоты
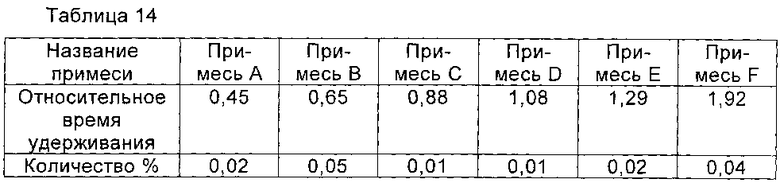
При 40°С 1,8 г соединения, полученного в Примере 1, растворяли в смешанном растворе, содержащем 8 мл воды и 6 мл изопропанола и перемешивали до полного растворения соединения I. рН доводили до 6,5 с помощью ледяной уксусной кислоты, и раствор охлаждали до 20°С для осаждения кристаллов соединения I. Систему перемешивали при 20°С в течение 2 часов. Медленно по каплям добавляли 35 мл изопропанола. После добавления раствор перемешивали в течение еще 2 часов для полного осаждения кристаллов соединения I. Влажное твердое соединение I получали путем фильтрации. При 40°С смешанный раствор, состоящий из 8 мл воды и 6 мл изопропанола, рН которого доводили до 6,5 с помощью ледяной уксусной кислоты, использовали для растворения влажного твердого соединения I и перемешивали до полного растворения соединения I. Раствор охлаждали до 20°С, и кристаллы соединения I выпадали в осадок. Систему перемешивали при 20°С в течение еще 2 часов. Медленно по каплям добавляли 35 мл изопропанола. После добавления раствор перемешивали еще в течение 2 часов для полного осаждения кристаллов соединения I. Влажное твердое соединение I получали путем фильтрации. Вышеуказанную процедуру кристаллизации повторяли. После четырехкратной перекристаллизации соединение I получали путем фильтрации и сушили в вакууме с получением 1,5 г соединения I. Общий выход 83,6%, и чистоту соединения определяли с помощью ВЭЖХ как 98,90%. Количество соответствующих примесей показано в Таблице 15.
Сравнительный Пример 2
Влияние растворителей на получение соединения I высокой чистоты
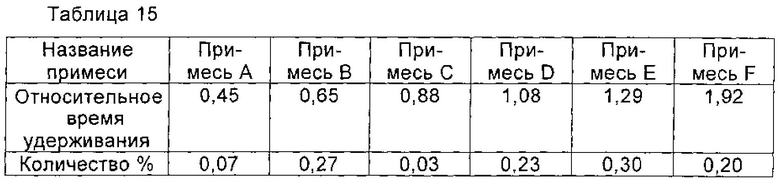
При 30°С 2,4 г соединения I, полученного в примере 1, растворяли в 7 мл воды, рН доводили до 3,8 с помощью ледяной уксусной кислоты, и полученную смесь перемешивали до полного растворения соединения I. Медленно добавляли 15 мл ацетонитрила и перемешивали в течение 2 часов, и твердые частицы осаждали. Микроструктуру твердых частиц наблюдали под микроскопом и обнаружили, что почти все твердые частицы были твердыми частицами неправильной формы. Соединение I получали путем фильтрации и сушили в вакууме, его чистоту определяли с помощью ВЭЖХ как 97,57%. Количество основных соответствующих примесей, которое почти не изменилось, приведено в таблице 16.

При 18°С 2,1 г соединения I, полученного в примере 1, растворяли в 7 мл воды, рН доводили до 3,8 с помощью ледяной уксусной кислоты, и полученную смесь перемешивали до полного растворения соединения I. Медленно добавляли 20 мл ацетона и перемешивали в течение 2 часов, и твердые частицы выпадали в осадок. Микроструктуру твердых частиц наблюдали под микроскопом и обнаружили, что почти все твердые частицы являлись твердыми частицами неправильной формы. Соединение I получали путем фильтрации и сушили в вакууме, его чистоту определяли с помощью ВЭЖХ, как 97,79%. Количество основных соответствующих примесей, которое почти не изменилось, приведено в таблице 17.
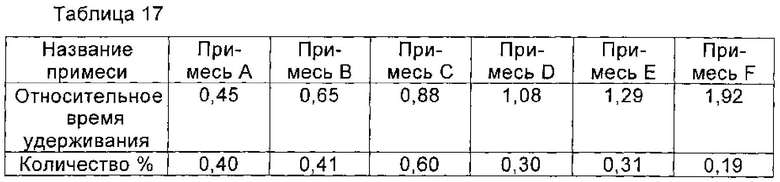
Пример 16
Получение соединения формулы II высокой чистоты из соединения формулы I высокой чистоты
Соединение формулы II синтезировали из соединения формулы I в соответствии со способом синтеза Микафунгина по WO 2004014879. Соединение формулы I, полученное в Примере 2 настоящей заявки (чистота по ВЭЖХ которого составляла 99,00% (1,00 г, 1,07 ммоль)), растворяли в 12 мл ДМФА (диметилформамид). Полученный раствор охлаждали до температуры ниже 0°С на ледяной бане. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), и температуру поддерживали при 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль), и реакцию нагревали до 2-6°С, и выдерживали в течение 4 часов. В конце реакции непосредственно в реакционную жидкость добавляли 60 мл этилацетата, перемешивали в течение еще 1 часа и фильтровали, чтобы получить Микафунгина диизопропилэтиламин. Соль растворяли в 30 мл ацетона и 30 мл этилацетата и фильтровали с помощью целлюлозного фильтра. Микафунгина диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. Чистоту Микафунгина диизопропилэтиламина определяли как 98,81% по данным ВЭЖХ, и выход составлял 90,6%. Хроматографические пики до 5 минут в профиле ВЭЖХ соответствуют растворителю и побочному продукту реакции, 1-гидроксибензотриазолу (НОВТ - hydroxybenzotriazole), который может быть легко удален с помощью процесса очистки, описанного в WO 2004014879. Количество основных соответствующих примесей приведено в таблице 18.

Пример 17
Получение соединения формулы II высокой чистоты из соединения формулы I высокой чистоты
Соединение формулы II синтезировали из соединения формулы I в соответствии со способом синтеза Микафунгина по WO 2004014879. Соединение формулы I, полученное в Примере 3 настоящей заявки (чистота по ВЭЖХ которого была 99,23% (1,00 г, 1,07 ммоль)) растворяли в 12 мл ДМФА. Полученный раствор охлаждали до температуры ниже 0°С на ледяной бане. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), и температуру поддерживали при 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль), и реакцию нагревали до 2-6°С, и выдерживали в течение 4 часов. В конце реакции непосредственно в реакционную жидкость добавляли 60 мл этилацетата, перемешивали в течение еще 1 часа и фильтровали, чтобы получить Микафунгина диизопропилэтиламин. Соль растворяли в 30 мл ацетона и 30 мл этилацетата и фильтровали с помощью целлюлозного фильтра. Микафунгин диизопропилэтиламин сушили в вакууме для удаления остаточного органического растворителя. Чистота Микафунгина диизопропилэтиламина была определена как 99,15% по данным ВЭЖХ, и выход составлял 91,6%. Хроматографические пики до 5 минут на профиле ВЭЖХ (см. Фиг. 3) соответствуют растворителю и побочному продукту реакции, 1-гидроксибензотриазолу (НОВТ), который может быть легко удален с помощью процесса очистки, описанного в WO 2004014879. Количество основных соответствующих примесей приведено в таблице 19.

Справочный пример 1
Получение примесей 6-10 в сыром соединении формулы II
23 г Микафунгина натрия получали из сырого соединения формулы I в качестве сырья, полученного в Примере 1 в соответствии со способом получения и очистки для Микафунгина по WO 2004014879.
Около 5 г Микафунгина натрия растворяли в 50 мл чистой воды, разделяли на 10 партий, и получали через препаративную колонку (Prep Nova-Pak®HR 7,8*300 мм). Для элюирования использовали смесь ацетонитрил:вода=70:45. Элюаты, относительное время удерживания которых составляли 0,71-0,74, 0,90-0,93, 1,08-1,10 и 1,11-1,13, собирали, концентрировали и сушили соответственно. Примеси, соответствующие вышеуказанным временам удерживания были определены как примесь 6, смеси примесей 7 и 8, примесь 9 и примесь 10, на основе МС and ПМР анализа. Их структуры приведены в формуле IV, формуле V, VI, формуле VII и формуле VIII соответственно.
Условия получения:
ВЭЖХ препаративной колонке: Prep Nova-Pak®HR 7,8*300 мм;
Подвижная фаза: ацетонитрил:вода=70:45;
Программа элюирования: изократическая;
Скорость потока: 4 мл/мин;
Температура колонки: 35°С;
Продолжительность (время прохода): 40 мин;
Разбавитель: вода.
Пример 18
Получение соединения формулы II высокой чистоты из соединения формулы I высокой чистоты и количества каждой отдельной примеси составляет менее 0,1%
Соединение формулы II синтезировали из соединения формулы I в соответствии со способом синтеза Микафунгин по WO 2004014879. Соединение формулы I, полученное в Примере 4 настоящей заявки (чистота по ВЭЖХ которого была 99,57% (1,00 г, 1,07 ммоль)), растворяли в 12 мл ДМФА. Полученный раствор охлаждали до температуры ниже 0°С на ледяной бане. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), и температуру поддерживали при 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль) и реакцию нагревали до 2-6°С, и выдерживали в течение 4 часов. В конце реакции непосредственно в реакционную жидкость добавляли 60 мл этилацетата, перемешивали в течение еще 1 часа, и фильтровали, чтобы получить Микафунгина диизопропилэтиламин. Соль растворяли в 30 мл ацетона и 30 мл этилацетата и фильтровали с помощью целлюлозного фильтра. Микафунгина диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. Чистота Микафунгина диизопропилэтиламин была определена как 99,49% по данным ВЭЖХ, и выход составил 95,1%. Количество основных соответствующих примесей приведено в таблице 20.

Пример 19
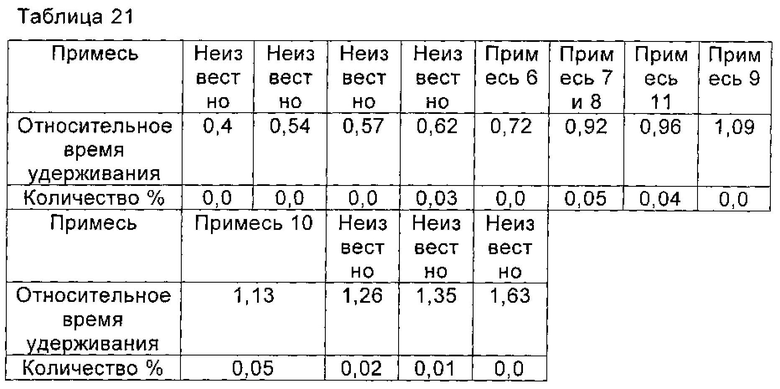
Получение соединения формулы II высокой чистоты с соединением формулы I высокой чистоты и количества каждой отдельной примеси составляет менее 0,1%
Соединение формулы II синтезировали из соединения формулы I в соответствии со способом синтеза Микафунгин по WO 2004014879. Соединение формулы I, полученное в Примере 5 настоящей заявки (чистота по ВЭЖХ которого была 99,81% (1,00 г, 1,07 ммоль)), растворяли в 12 мл ДМФА. Полученный раствор охлаждали до температуры ниже 0°С на ледяной бане. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль) и температуру поддерживали при 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль), и реакцию нагревали до 2-6°С, и выдерживали в течение 4 часов. В конце реакции непосредственно в реакционной жидкости добавляли 60 мл этилацетата, перемешивали в течение еще 1 часа и фильтровали, чтобы дать Микафунгина диизопропилэтиламин. Соль растворяли в 30 мл ацетона и 30 мл этилацетата и фильтровали с помощью целлюлозного фильтра. Микафунгина диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. Чистота Микафунгина диизопропилэтиламина была определена как 99,80% по данным ВЭЖХ, и выход составил 97,5%. Количество основных соответствующих примесей приведено в таблице 21.
Сравнительный пример 3
Получение соединения формулы II из сырого соединения формулы I
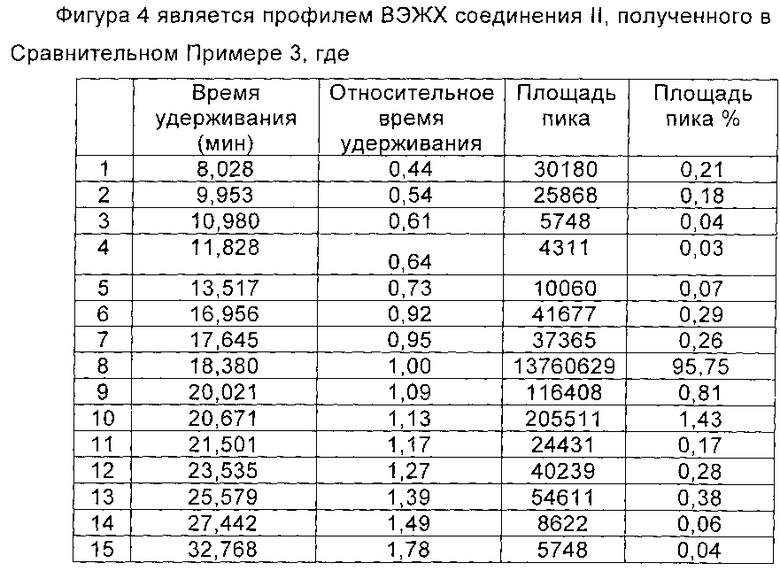
Соединение формулы II синтезировали из соединения формулы I в соответствии со способом синтеза Микафунгина в WO 2004014879. Соединение формулы I, полученное в примере 1 настоящей заявки (чистота по ВЭЖХ которого была 97,51% (10,7 ммоль)) растворяли в 12 мл ДМФА. Полученный раствор охлаждали до температуры ниже 0°С на ледяной бане. Добавляли диизопропилэтиламин (2,2 г, 16,7 ммоль), и температуру поддерживали при 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (5,3 г, 11,4 ммоль), и реакцию нагревали до 2-6°С, и выдерживали в течение 4 часов. В конце реакции непосредственно в реакционную жидкость добавляли 600 мл этилацетата, перемешивали в течение еще 1 часа и фильтровали, чтобы получить Микафунгина диизопропилэтиламин. Соль растворяли в 300 мл ацетона и 300 мл этилацетата и фильтровали с помощью целлюлозного фильтра. Микафунгина диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. Чистота Микафунгина диизопропилэтиламина была определена как 95,75% по данным ВЭЖХ, и мольный выход составил 75,2%. Хроматографические пики до 5 минут на профиле ВЭЖХ (см. Фиг. 4) соответствовали растворителю и побочному продукту реакции, 1-гидроксибензотриазолу (НОВТ), которые легко могут быть удалены с помощью процесса очистки, как приведено в WO 2004014879. Количество основных соответствующих примесей приведено в таблице 22.

Пример 20
Получение Микафунгина натрия высокой чистоты из Микафунгина диизопропилэтиламина высокой чистоты
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 по WO 2004014879.
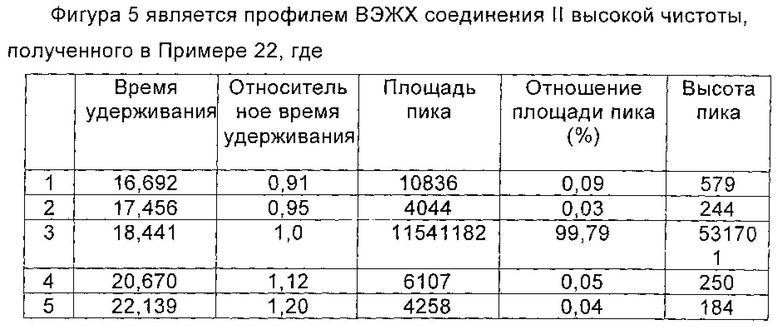
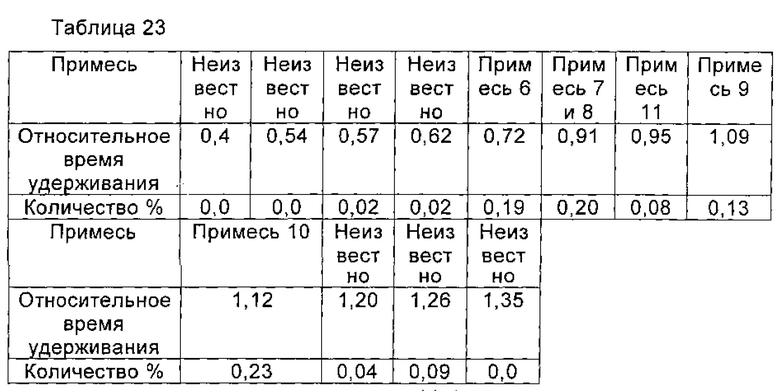
Микафунгина диизопропилэтиламин (0,97 ммоль), полученный в Примере 16 настоящей заявки, растворяли в 15 мл 75% водного метанола. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью 75% водного метанола до получения концентрации натрия Микафунгина менее 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 М NaOH. Собранную жидкость разбавляли чистой водой до концентрации метанола 35%. Полученный раствор наносили на 50 мл предварительно обработанной HP20ss макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 100 мл 35% водного метанола, а затем элюировали с помощью 200 мл 80% водного метанола. Элюат собирали, когда концентрация Микафунгина натрия составляла более 0,5 г/л, и сбор остановили, когда концентрация Микафунгина натрия была менее 0,5 г/л. Собирали все порции, включающие подходящую концентрацию Микафунгина натрия. Полученный Микафунгин натрия количественно анализировали с помощью ВЭЖХ (0,90 ммоль), и выход составил 93%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, с получением твердого вещества. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 99,00% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ) был полностью удален. Количество основных соответствующих примесей приведено в таблице 23.
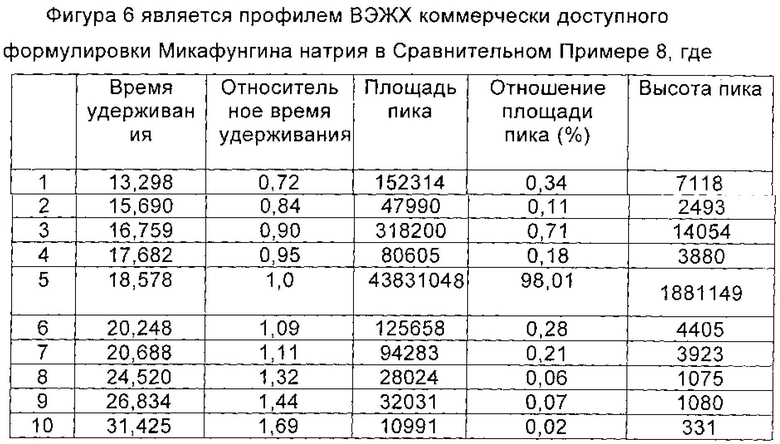
Сравнительный пример 4
Получение Микафунгина натрия из Микафунгина диизопропилэтиламина, полученного в Сравнительном Примере 3
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 по WO 2004014879.
Микафунгина диизопропилэтиламин (0,97 ммоль), полученный в Сравнительном Примере 3 настоящей заявки, растворяли в 15 мл 75% водного метанола. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью 75% водного метанола до получения концентрации Микафунгина натрия менее 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость разбавляли чистой водой, так чтобы концентрация метанола была 35%. Полученный раствор наносили на 50 мл предварительно обработанного HP20ss макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 100 мл 35% водного метанола, а затем элюировали с помощью 200 мл 80% водного метанола. Элюат собирали, когда концентрация натрия Микафунгин была более 0,5 г/л, и сбор прекращали, когда концентрация Микафунгина натрия была менее 0,5 г/л. Собирали все порции, содержащие подходящую концентрацию Микафунгина натрия. Полученный Микафунгина натрий количественно анализировали с помощью ВЭЖХ (0,88 ммоль), и выход составил 91%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, для получения твердого вещества. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 96,17% по данным ВЭЖХ, причем побочный продукт реакции, 1-гидроксибензотриазол (НОВТ) был полностью удален. Количество основных соответствующих примесей приведено в таблице 24.

Из Примера 20 и Сравнительного Примера 4 видно, что посредством простой процедуры очистки, Микафунгина натрий (чистота 98,99%) может быть получен из высокочистого Микафунгина диизопропилэтиламина, который получают из соединения формулы I высокой чистоты (чистота 99,0%). Однако, если соединение формулы I, чистота которого составляет 97,51%, полученное в соответствии с Примером 1 патента США US 5376634 использовали для получения Микафунгина диизопропилэтиламина высокой чистоты, то чистота Микафунгина натрия, полученного из Микафунгина диизопропилэтиламина, составляла всего 96,17%, Таким образом, Микафунгин натрия, полученный из соединения формулы I высокой чистоты, выше по чистоте.
Пример 21
Получение Микафунгина натрия высокой чистоты из Микафунгина диизопропилэтиламина высокой чистоты
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгина диизопропилэтиламин (0,98 ммоль), полученный в Примере 17 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью воды, пока концентрация натрия Микафунгин не была менее 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанной SP-207ss макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 120 мл 35% водного раствора этанола, а затем элюировали с помощью 150 мл 80% водного раствора этанола. Элюат собирали, когда концентрация Микафунгина натрия была более 0,5 г/л, и отбор останавливали, когда концентрация Микафунгина натрия была менее 0,5 г/л. Все порции, содержащие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгина натрий количественно анализировали с помощью ВЭЖХ (0,92 ммоль), и выход составил 93,9%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, для получения твердого вещества. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 99,40% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ) был полностью удален. Количество основных соответствующих примесей приведено в таблице 25.

Сравнительный Пример 5
Получение Микафунгина натрия из Микафунгина диизопропилэтиламина,полученного в Сравнительном Примере 3
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгин диизопропилэтиламин (0,98 ммоль), полученный в Сравнительном Примере 3 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью чистой воды до тех пор, пока концентрация Микафунгина натрия не была менее 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанной SP-207ss макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывают с помощью 120 мл 35% водного раствора этанола, а затем элюировали с помощью 150 мл 80% водного раствора этанола. Элюат собирали, когда концентрация Микафунгина натрия была более 0,5 г/л, и сбор прекращали, когда концентрация Микафунгина натрия была менее 0,5 г/л. Все порции, включающие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгина натрий количественно анализировали с помощью ВЭЖХ (0,84 ммоль), и выход составил 86,1%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, для получения твердого вещества. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 96,16% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ) был полностью удален. Количество основных соответствующих примесей приведено в таблице 26.

Из Примера 21 и Сравнительного Примера 5 видно, что посредством простой процедуры очистки Микафунгин натрия (чистота 99,40%) может быть получен из Микафунгина диизопропилэтиламина высокой чистоты, который получают из соединения формулы I высокой чистоты (чистота 99,23%). Однако, если соединение формулы I, чистота которого составляла 97,51%, полученное в соответствии с Примером 1 патента США US 5376634, использовали для подготовки Микафунгина диизопропилэтиламина высокой чистоты, то чистота Микафунгина натрия, полученного из Микафунгина диизопропилэтиламина, составит всего 96,16% с тем же процессом очистки. Таким образом, микафунгин натрия, полученный из соединения формулы I высокой чистоты, выше по чистоте.
Пример 22
Получение Микафунгина натрия высокой чистоты и количество каждой отдельной примеси составляет менее 0,1% от Микафунгина диизопропилэтиламина высокой чистоты
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгина диизопропилэтиламин (1,01 ммоль), полученный в Примере 18 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью воды, пока концентрация натрия Микафунгина не была менее 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанного SP-700 макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 100 мл 30% водного раствора ацетона, а затем элюировали с помощью 150 мл 80% водного раствора ацетона. Элюат собирали, когда концентрация Микафунгина натрия была больше 0,5 г/л, и сбор прекращали, когда концентрация Микафунгина натрия была меньше 0,5 г/л. Все порции, содержащие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгин натрия количественно анализировали с помощью ВЭЖХ (0,92 ммоль), и выход составил 91,1%. Порции, содержащие Микафунгин натрий, объединяли и отгоняли при пониженном давлении, в темноте, с получением твердого вещества. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 99,79% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ), был полностью удален. Количество основных соответствующих примесей приведено в таблице 27.
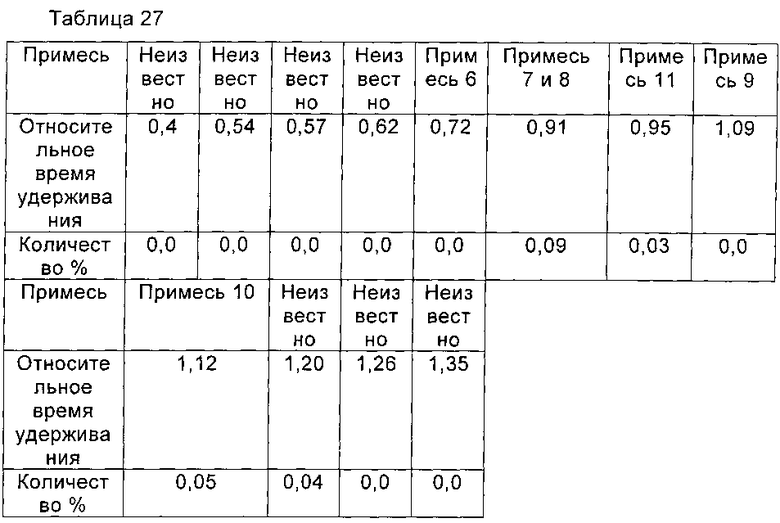
Сравнительный пример 6
Получение Микафунгина натрия из диизопропилэтиламина, полученного в Сравнительном Примере 3
Микафунгина натрий получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгин диизопропилэтиламин (1,01 ммоль), полученный в Сравнительном Примере 3 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 30 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью чистой воды до тех пор, пока концентрация натрия Микафунгин не была меньше 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанной SP-700 макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 100 мл 30% водного раствора ацетона, а затем элюировали с помощью 150 мл 80% водного раствора ацетона. Элюат собирали, когда концентрация Микафунгина натрия была больше 0,5 г/л, и сбор прекратили, когда концентрация Микафунгина натрия была меньше 0,5 г/л. Все порции, содержащие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгин натрия количественно анализировали с помощью ВЭЖХ (0,83 ммоль), и выход составил 82,1%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, получая твердое вещество. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 96,29% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ), был полностью удален. Количество основных соответствующих примесей приведено в таблице 28.
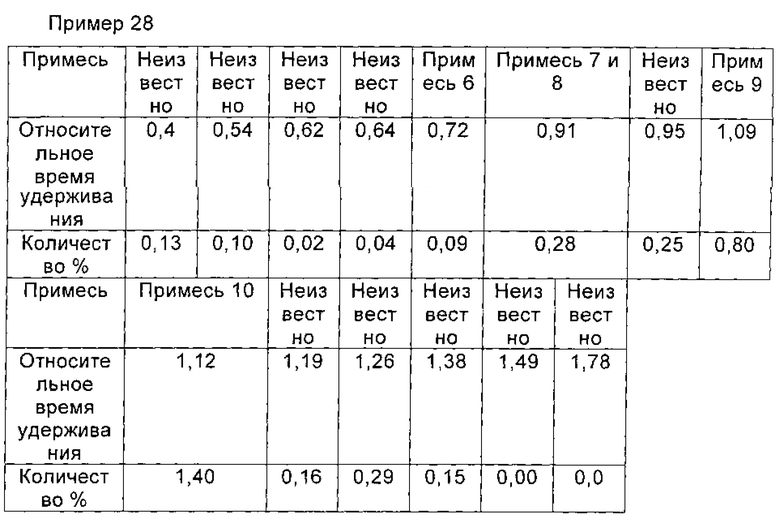
Из Примера 22 и Сравнительного Примера 6 видно, что посредством простой процедуры очистки Микафунгин натрия (чистота 99,79%) может быть получен из Микафунгина диизопропилэтиламина высокой чистоты, который получают из соединения формулы I высокой чистоты (чистота 99,57%). Однако, если соединение формулы I, чистота которого составляет 97,51%, полученное в соответствии с примером 1 патента США US 5376634, использовали для получения Микафунгина диизопропилэтиламина высокой чистоты, то чистота Микафунгина натрия, полученного из Микафунгина диизопропилэтиламина, составляла всего 96,29% при том же процессе очистки. Таким образом, микафунгин натрия, полученный из соединения формулы I высокой чистоты, выше по чистоте.
Пример 23
Получение Микафунгина натрия высокой чистоты из Микафунгина диизопропилэтиламина высокой чистоты, и количество каждой отдельной примеси составляет менее 0,1%
Микафунгин натрия получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгина диизопропилэтиламин (1,04 ммоль), полученный в Примере 19 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 25 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью воды до тех пор, пока концентрация Микафунгина натрия не была меньше 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанной HP2MG макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 200 мл 30% водного раствора этанола, а затем элюировали с помощью 200 мл 70% водного раствора этанола. Элюат собирали, когда концентрация Микафунгина натрия была больше 0,5 г/л, и сбор прекращали, когда концентрация Микафунгина натрия была меньше 0,5 г/л. Все порции, включающие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгин натрия количественно анализировали с помощью ВЭЖХ (0,92 ммоль), и выход составил 88,5%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, получая твердое вещество. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 99,88% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ) был полностью удален. Количество основных соответствующих примесей приведено в таблице 29.

Сравнительный пример 7
Получение Микафунгина натрия из диизопропилэтиламина, полученного в Сравнительном Примере 3
Микафунгин натрия получали из Микафунгина диизопропилэтиламина в соответствии со способом Примера 6 в WO 2004014879.
Микафунгина диизопропилэтиламин (1,04 ммоль), полученный в Сравнительном Примере 3 настоящей заявки, растворяли в 15 мл воды. Раствор, содержащий Микафунгина диизопропилэтиламин, загружали на 25 мл UBK510L ионообменной смолы. Нагруженную смолу элюировали с помощью чистой воды до тех пор, пока концентрация Микафунгин натрия не была меньше 1,0 г/л. Значение рН собранной жидкости доводили до 6,0 с помощью 0,1 моль/л NaOH. Собранную жидкость загружали на 40 мл предварительно обработанной HP2MG макропористой адсорбционной смолы для адсорбции. Нагруженную смолу промывали с помощью 200 мл 30% водного раствора этанола, а затем элюировали, используя 200 мл 70% водного раствора этанола. Элюат собирали, когда концентрация Микафунгина натрия была больше 0,5 г/л, и сбор прекращали, когда концентрация Микафунгина натрия была меньше 0,5 г/л. Все порции, содержащие подходящую концентрацию Микафунгина натрия, собирали. Полученный Микафунгина натрий количественно анализировали с помощью ВЭЖХ (0,86 ммоль), и выход составил 82,7%. Порции, содержащие Микафунгина натрий, объединяли и отгоняли при пониженном давлении, в темноте, получая твердое вещество. Твердое вещество сушили в вакууме, и чистота Микафунгина натрия была определена как 96,29% по данным ВЭЖХ, при этом побочный продукт реакции, 1-гидроксибензотриазол (НОВТ), был полностью удален. Количество основных соответствующих примесей приведено в таблице 30.
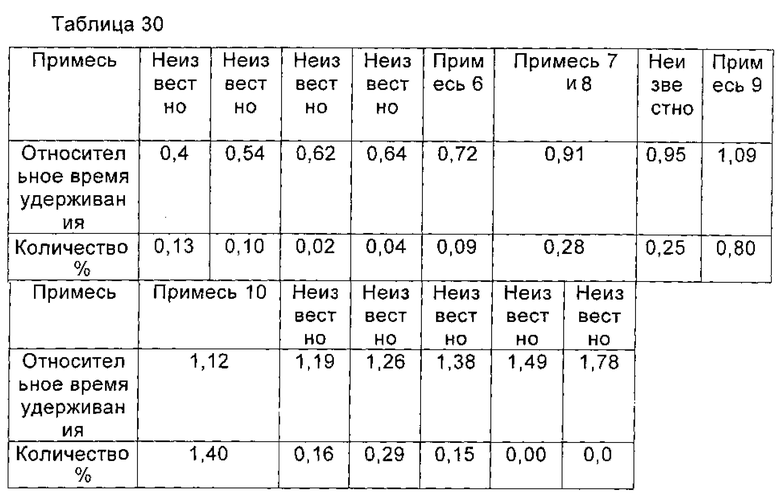
Из Примера 23 и Сравнительного Примера 7 видно, что посредством простой процедуры очистки Микафунгин натрия (чистота 99,88%) может быть получен из Микафунгина диизопропилэтиламина высокой чистоты, который получали из соединения формулы I высокой чистоты (чистота 99,81%). Однако, если соединение формулы I, чистота которого составляет 97,51%, полученное в соответствии с Примером 1 патента США US 5376634, использовали для получения Микафунгина диизопропилэтиламина, то чистота Микафунгина натрия, полученного из Микафунгина диизопропилэтиламина, является всего 96,29% при том же процессе очистки. Таким образом, микафунгин натрия, полученный из соединения формулы I высокой чистоты, выше по чистоте.
Сравнительный пример 8
Сравнение с коммерчески доступной композицией по содержанию примеси
Коммерчески доступные препараты: Наименование продукта: Микамин (MYCAMINE)
Общее название: Микафунгина натрий для инъекций
Производитель: Astellas Toyama Corporation Партия: 0901

Из таблицы 31 ясно, что чистота Микафунгина натрия, полученного в настоящем изобретении, значительно выше, чем у коммерчески доступного препарата, в то время как количество примеси 6, примеси 7, примеси 8, примеси 9 и примеси 10 значительно меньше, чем эти же количества в коммерчески доступном готовом препарате. На Фигуре 5 показан профиль ВЭЖХ для Микафунгина натрия, полученного в настоящем изобретении, и на Фигуре 6 - для коммерчески доступного препарата.

Лактозу растворяли в чистой воде (200 мл) при температуре ниже чем 50°С. После охлаждения ниже 20°С, в раствор лактозы добавляли соединение I высокой чистоты, полученное в Примере 3. Полученный раствор осторожно перемешивали, избегая пузырьков. Добавляли 2% водный раствор лимонной кислоты (0,95 мл), а затем в раствор добавляли 0,4% водный раствор NaOH (примерно 24 мл) для доведения рН до 5,5. И затем полученный раствор разбавляли чистой водой до получения заданного объема (250 мл). Полученный раствор разливали в 100 ампул (объем каждой составляет 10 мл) по 2,5 мл на каждую. Раствор в каждой ампуле лиофилизировали с использованием лиофилизатора в соответствии с обычными способами, так, чтобы получить лиофилизированные композиции, содержащие каждая по 25 мг соединения I.
Приведенные выше примеры являются лишь предпочтительными примерами для настоящего изобретения, и такие примеры не могут быть использованы для ограничения объема изобретения. Существенное техническое содержание в соответствии с настоящим изобретением широко определено в формуле изобретения. И любые субъекты или способы, совершенные другими, следует рассматривать как эквиваленты, и они подпадают под настоящие рамки, как определено в формуле изобретения, если указанные субъекты или способы такие же, как те, которые определены в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРАТ ЦИКЛОПЕПТИДНОГО СОЕДИНЕНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2594732C2 |
| КРИСТАЛЛ ЦИКЛОПЕПТИДА ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2607083C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ И ПРОИЗВОДНЫХ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2650687C1 |
| СПОСОБ ОЧИСТКИ ЦИКЛОЛИПОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2535489C1 |
| СПОСОБ ИЗГОТОВЛЕНИЯ ПРОТИВОГРИБКОВОГО АГЕНТА | 2012 |
|
RU2575237C2 |
| СПОСОБ ОЧИСТКИ ЦИКЛОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2538274C2 |
| СПОСОБ ОЧИСТКИ МИКАФУНГИНА | 2012 |
|
RU2583262C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В ПОЛУЧЕНИИ C5aR АНТАГОНИСТОВ | 2015 |
|
RU2712233C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 5-[2-[7-(ТРИФТОРМЕТИЛ)-5-[4-(ТРИФТОРМЕТИЛ)ФЕНИЛ]ПИРАЗОЛО[1,5-a]ПИРИМИДИН-3-ИЛ]ЭТИНИЛ]-2-ПИРИДИНАМИНА | 2012 |
|
RU2630700C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-БЕНЗИЛ-1-ФЕНЕТИЛ-ПИПЕРАЗИН-2,6-ДИОНА, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2014 |
|
RU2631323C2 |
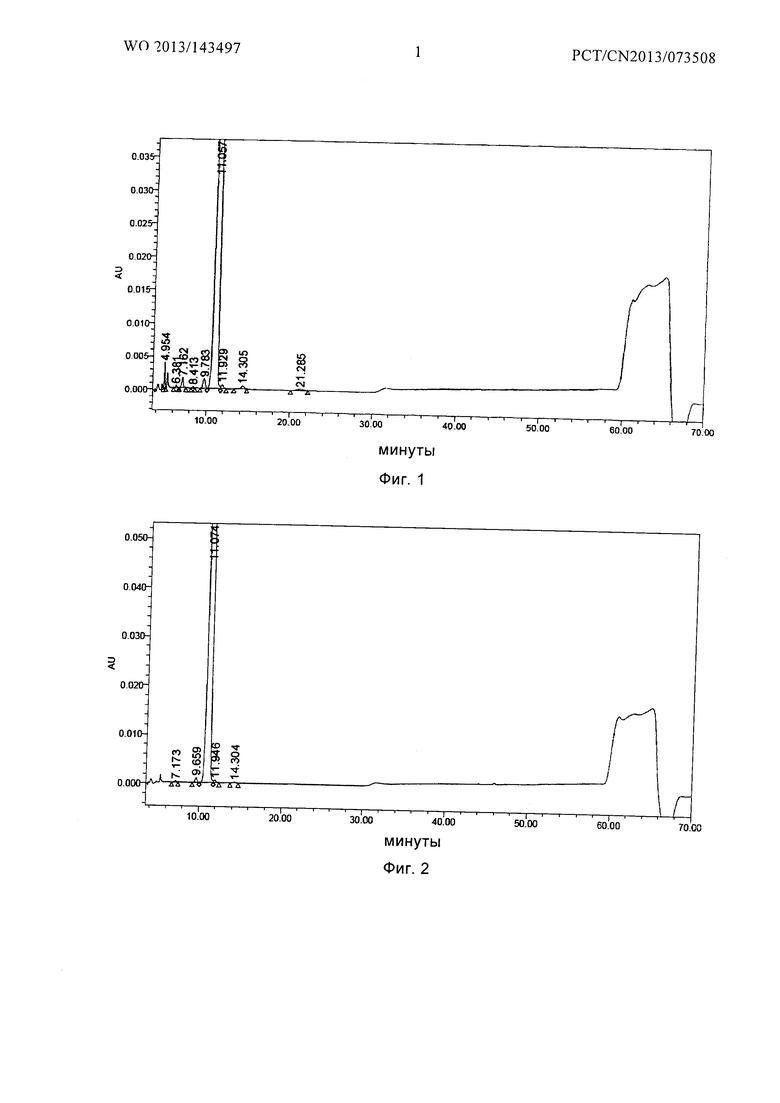


Изобретение относится к способу получения циклопептидного соединения формулы I высокой чистоты, где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль. Соласно способу (a) растворяют соединение формулы I в воде или водном органическом растворителе (i) и контролируют pH раствора; и (b) получают циклопептидное соединение формулы I высокой чистоты путем снижения температуры и/или добавления органического растворителя (i), где на стадии (a) pH раствора поддерживают на уровне от 2, 0 до 5,9; и на стадии (а) или (b) указанный органический растворитель (i) выбирают из С1-С4 низшего спирта. Способ позволяет получать соединение формулы I, чистота которого выше или равна 99,0%. 4 з.п. ф-лы, 6 ил., 31 табл., 24 пр.


1. Способ получения циклопептидного соединения формулы I высокой чистоты, в котором R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль, включающий следующие стадии:
(a) растворение сырого соединения формулы I в воде или водном органическом растворителе (i) и контролирование рН раствора;
(b) получение циклопептидного соединения высокой чистоты путем снижения температуры и/или добавления органического растворителя (i)
где на стадии (а) рН раствора поддерживают на уровне от 2,0 до 5,0; и
на стадии (а) и/или (b) указанный органический растворитель (i) выбирают из С1-С4 низшего спирта.
2. Способ получения по п. 1, где на стадии (а) рН раствора поддерживают на уровне от 3,5 до 4,5.
3. Способ получения по п. 1, где концентрация соединения формулы I в растворе на стадии (а) составляет от 10 до 500 мг/мл.
4. Способ получения по п. 1, где на стадии (а) и/или (b) указанный органический растворитель (i) выбирают из метанола, этанола, n-пропанола, изопропанола или их смесей.
5. Способ получения по п. 1, где стадия (а) - (b) может быть повторена один или несколько раз.
| WO 2004014879 A1, 19.02.2004 | |||
| СПОСОБ ОЧИСТКИ ЦИКЛОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2538274C2 |
| СПОСОБ ОЧИСТКИ ЦИКЛОЛИПОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2535489C1 |

