Область изобретения
Настоящее изобретение относится к улучшенному способу приготовления Микафунгина или его солей.
Уровень техники
Микафунгин - это эхинокандин с антигрибковой активностью, представленный формулой (I):
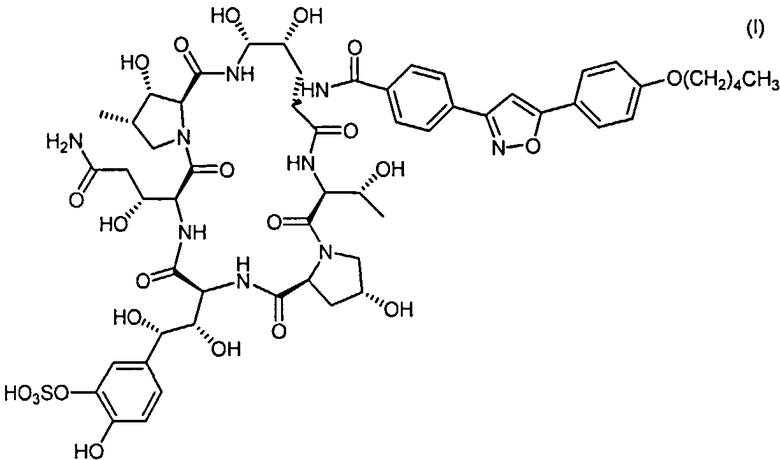
Микафунгин также известен как Пневмокандин А0, 1-[(4R,5R)-4,5-дигидрокси-N2-[4-[5-[4-(пентилокси) фенил]-3-изоксазолил]бензоил]-L-орнитин]-4-[(4S)-4-гидрокси-4-[4-гидрокси-3-(сульфокси)фенил]-L-треонин]. Микафунгин натрия, кроме того, известен как FK-463. Присвоенные регистрационные номера в химической реферативной службе 235114-32-6 для Микафунгина и 208538-73-2 для Микафунгина натрия.
Антигрибковая активность Микафунгина обусловлена его способностью ингибировать 1,3-β-D-глюкан синтазу и, таким образом приводить к лизису клеток грибка. Таким образом, Микафунгин полезен при лечении различных инфекций; в частности инфекций, вызванных штаммами например: Aspergillus, Cryptococcus, Candida, Mucor, Actinomyces, Histoplasma, Dermatophyte, Malassezia и Fusarium. Микафунгин является активным ингредиентом в одобренных лекарствах Mycamine® и Funguard®, которые используются в лечении и профилактике инфекций, вызванных кандидами.
Микафунгин является вторым одобренным антигрибковым агентом из группы эхинокандинов и в настоящее время используется во всем мире в хемотерапии опасных для жизни грибковых инфекций.
Микафунгин и его приготовление раскрыто в патенте США номер 6,107,458 выданного Fujisawa Pharmaceutical Co., Ltd. Способ приготовления Микафунгина также раскрыт в Главном Тезисе, "Процесс Разработки Микафунгина, Новый Липопептидный Противогрибковый Агент" Охигаши и др. В Journal of Synthetic Organic Chemistry, Япония, том 64, Номер 12, Декабрь 2006. Согласно способам, раскрытым в уровне техники, Микафунгин может быть получен из FR-901379, натурального продукта, выделенного из грибка Coleophoma empetri F-11899. С помощью ферментативного дезацилирования FR-901379, с последующим амидным присоединением 4-[5-(4-пентилокси)фенил)изоксазол-3-ил]бензойной кислоты, Микафунгин может быть получен.
Способ приготовления Микафунгина также раскрыт Фромтлингом и другими. Пептидное ядро, полученное после дезацилирования FR-901379, согласно Фромтлингу и другим, снова ацилировано с помощью активированной боковой цепи, 1-[4-[5-(4-пентилокси)фенил1)изоксазол-3-ил]бензоил]бензотриазол 3-оксида.
Различные способы производства Микафунгина натрия также раскрыты Хашимото и другие, The Journal of Antibiotics (2009) 62, 27-35.
Улучшенное изготовление Микафунгина описано в US 7199248. Данный способ содержит стадию добавления изолированной боковой цепи Микафунгина, например 1-[4-[5-(4-пентилокси)фенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазола, к дезацилированному пептидному ядру Микафунгина.
Охигаши и другие, Исследование Органических Процессов и Развитие 2005, том 9, страницы 179-184, раскрыли оптимизированный промышленный процесс приготовления Микафунгина, который также включал выделение активированной боковой цепи Микафунгина.
Все способы приготовления Микафунгина, раскрытые на данный момент, как было упомянуто выше, обладают общей особенностью - все известные методы предполагают выделение активированной боковой цепи Микафунгина перед реакцией с пептидным ядром Микафунгина, т.е. FR-901379. Таким образом, все известные способы осуществляются с помощью изолированной формы боковой цепи Микафунгина.
Цель настоящего изобретения - предоставить улучшенный и более промышленно эффективный способ приготовления Микафунгина
Краткое изложение изобретения
Цель настоящего изобретения, с одной стороны достигнута, используя способ, который включает в себя активацию боковой цепи Микафунгина и связывание вышеуказанной боковой цепи Микафунгина с пептидным ядром Микафунгина в однореакторном синтезе. То есть, способ настоящего изобретения включает в себя ацилирование пептидного ядра Микафунгина без изолирования боковой цепи Микафунгина из ее реакционной смеси. С другой стороны, изобретение включает в себя активацию и связывание боковой цепи Микафунгина с пептидным ядром Микафунгина в одной реакционной смеси. Таким образом, предложен способ, где стадия изолирования активированной боковой цепи Микафунгина является излишней.
Связывание кислотного соединения в виде боковой цепи Микафунгина с пептидным ядром, в виде ядра Микафунгина, может в итоге привести к различным нежелательным побочным продуктам. Ко всему прочему, упоминание произведено Охигаши и др., 2006, выше, обсуждая, в том числе, необходимость в подавлении побочных реакций во время ацилирования, в связи с наличием большого числа функциональных групп в ядре Микафунгина. Охигаши и др. 2006 более того, выяснили, что существует необходимость в оптимальных условиях очистки интермедиатов в синтезе. Несмотря на информацию из уровня техники, авторы настоящего изобретения неожиданно обнаружили, что боковая цепь Микафунгина может быть активирована и связана напрямую с ядром Микафунгина, без необходимости дополнительной стадии выделения активированной боковой цепи, где успешное связывание было достигнуто с минимальным образованием примесей. Способ настоящего изобретения обеспечивает преимущества в сравнении со способами из уровня техники, предусматривающими выделение боковой цепи Микафунгина. Например, однореакторный способ более эффективен с промышленной точки зрения в связи с укороченным временем синтеза, благодаря исключению стадии выделения. В дополнение, с улучшенным использованием боковой цепи Микафунгина, не будет наблюдаться никакой потери продукта вследствие стадии изоляции/очищения, приводя, в итоге, к всеобщему лучшему выходу.
Более конкретно, настоящее изобретение предоставляет однореакторный способ для производства Микафунгина или его соли, содержащий следующую последовательность стадий:
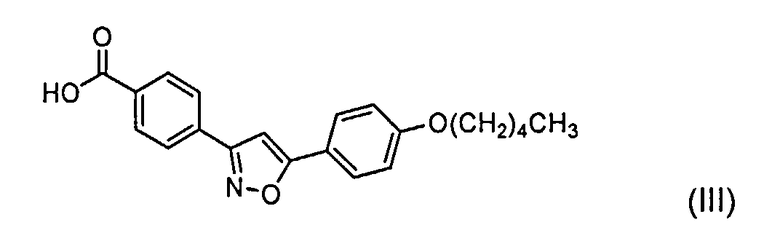
а) смешивание соединения с формулой (III)
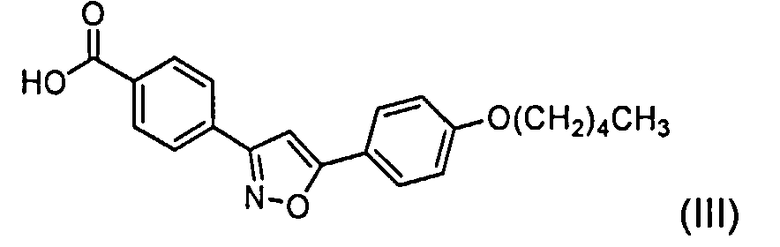
или его соли и связывающей добавки, выбранной из группы, состоящей из гидроксибензотриазола и этил-2-циано-2-(гидроксиимино)ацетата в растворителе;
б) добавление связывающего реагента к смеси, полученной на стадии а), где указанный связывающий реагент представляет собой карбодиимид;
в) добавление основания и соединения с формулой (II)
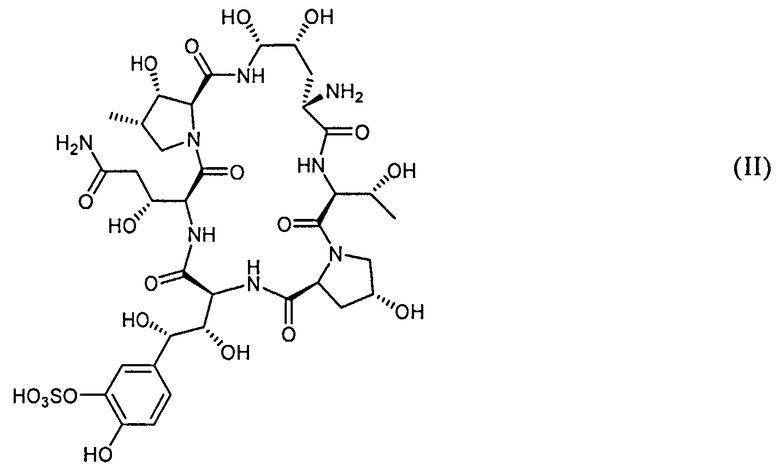
или его соли к смеси, полученной на стадии б).
Согласно одному из воплощений настоящего изобретения, связывающая добавка представляет собой 1-гидрокси-бензотриазол.
Согласно другому воплощению изобретения, связывающая добавка представляет собой 1-гидрокси-7-азобензотриазол.
Согласно еще одному воплощению изобретения, связывающая добавка представляет собой этил-2-циано(гидроксиимино)ацетат.
Согласно другому воплощению изобретения, связывающий реагент представляет собой
1-этил-3-(3-диметиламинопропил) карбодиимид (ЭДК) или его соль, предпочтительно хлороводородную соль ЭДК.
Согласно другому воплощению изобретения, растворитель, используемый на стадии а), указанной выше, - ДМФ.
Согласно другому воплощению изобретения, основание, используемое на стадии в) настоящего изобретения, - ДИПЭА.
Согласно еще одному воплощению изобретения, основание добавляется в смесь, полученную на стадии б), перед добавлением соединения с формулой II.
Согласно еще одному воплощению изобретения, основание добавляется в смесь, полученную на стадии б), после добавления соединения с формулой II.
Согласно другому воплощению изобретения, соль Микафунгина, полученная на стадии в), осаждается.
Согласно другому воплощению изобретения, соль Микафунгина, полученная на стадии в), осаждается, используя антирастворитель.
Согласно другому воплощению изобретения, соль Микафунгина, полученная на стадии в), осаждается, используя этилацетат.
Согласно еще одному воплощению изобретения, соль Микафунгина, полученная на стадии в), осаждается после остановки реакции добавлением метанола и ацетона.
Краткое описание фигур
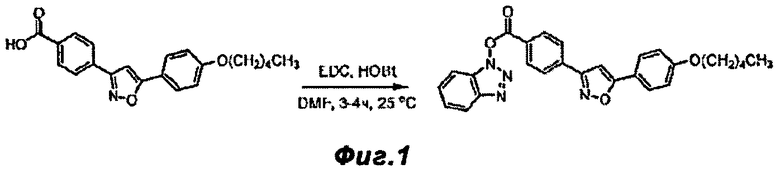
Фигура 1 - схема реакции, показывающая реакцию, описанную в Примере 1, т.е. отдельные реакции боковой цепи Микафунгина и присоединение связывающей добавки, представленной HOBt.
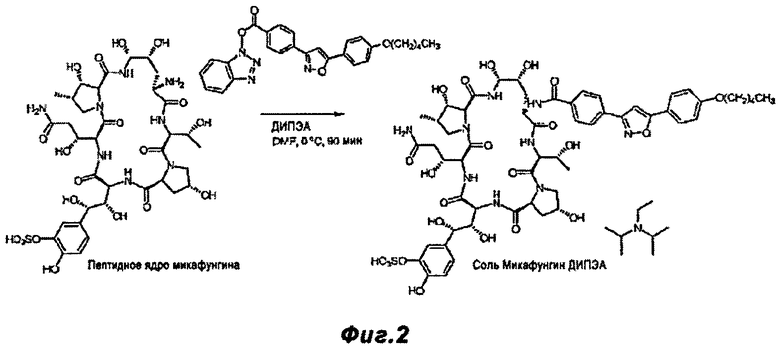
Фигура 2 - схема реакции, показывающая реакцию, описанную в примере 2, т.е. отдельную реакцию пептидного ядра Микафунгина и продукта реакции, показанного на Фигуре 1.
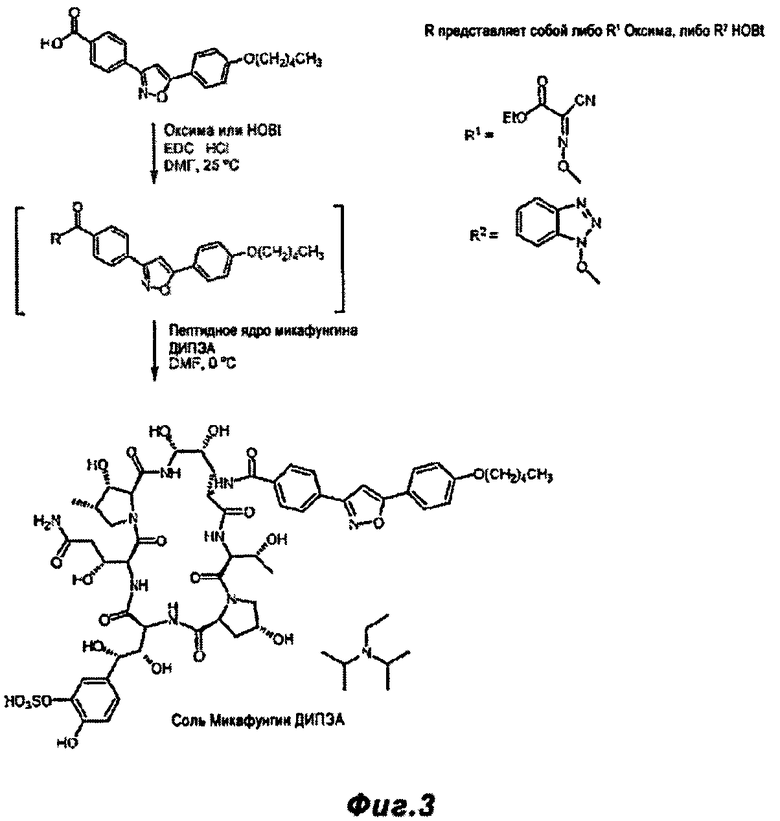
Фигура 3 - реакционная схема, показывающая реакции описанные в Примере 3 и Примере 4, т.е. где активация боковой цепи Микафунгина и реакция с пептидным ядром Микафунгина осуществлены по однореакторному способу, согласно настоящему изобретению.
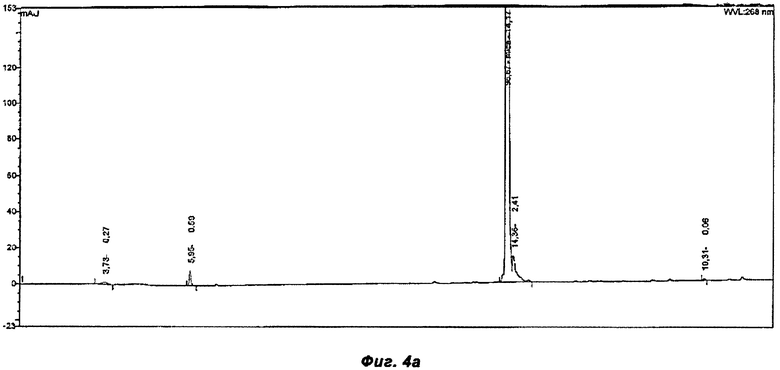
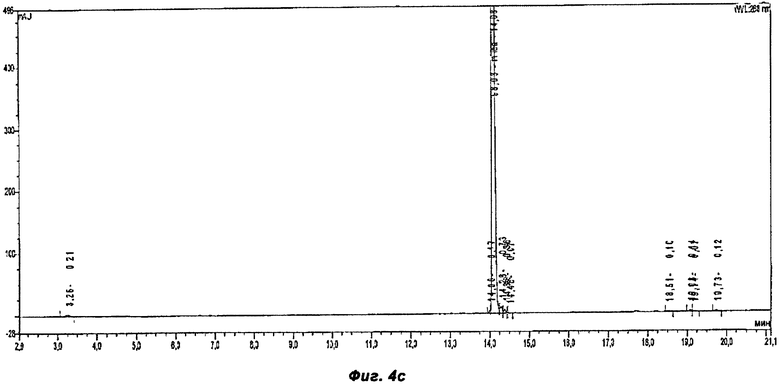
Фигура 4а показывает хроматограмму продукта постадийного способа, раскрытого в Примере 2.
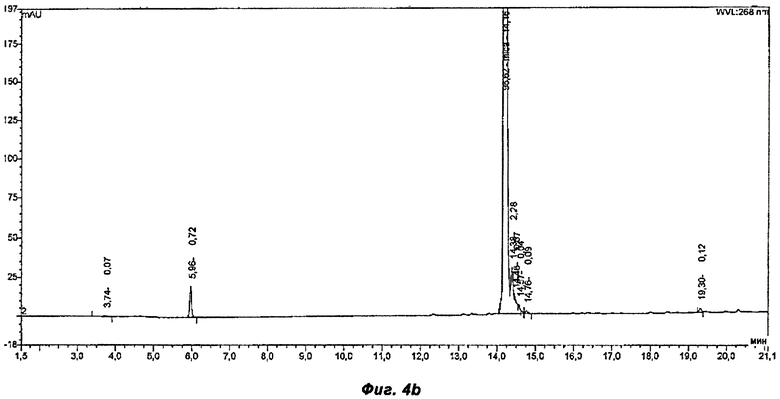
Фигура 4б показывает хроматограмму продукта однореакторного способа, раскрытого в Примере 3.
Фигура 4в показывает хроматограмму продукта однореакторного способа, раскрытого в Примере 4.
Детальное описание изобретения
Согласно настоящему изобретению, Микафунгин представляет собой любое соединения, включающее структуру
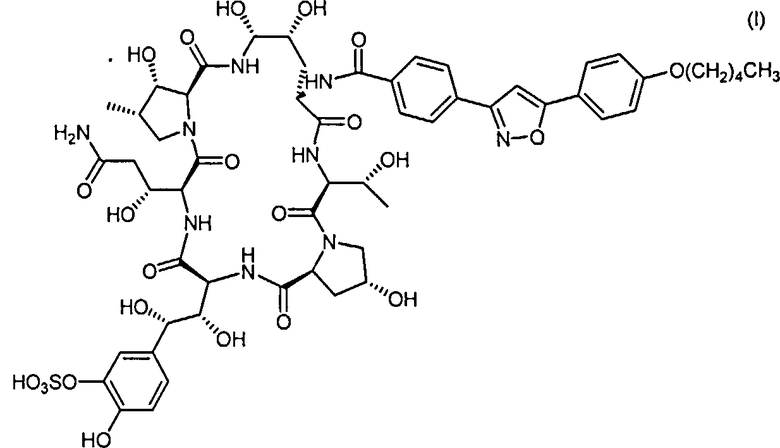
или его соли. Выражение "его соли" предназначено охватить любые соли Микафунгина, которые могут быть полезны с целью приготовления и/или очистки Микафунгина или любых других фармацевтически приемлемых солей Микафунгина, полезных в качестве активного ингредиента в медицинских препаратах. В данном отношении, неограниченный список солей Микофунгина - это натриевая соль, калиевая соль, диизопропилэтиламиновая (ДИПЭА) соль и другие.
Согласно настоящему изобретению, Микафунгин готовится в однореакторном синтезе, сначала а) смешивая боковую цепь Микафунгина со связывающей добавкой, б) затем добавлением связывающего реагента к смеси стадии а), и в) в конце добавляя к этой смеси основание и соединение формулы II изображенного раннее, например пептидное ядро Микафунгина.
Боковая цепь Микафунгина в кислотной форме представляет собой вещество с химическим названием 4-[5-(4-пентилокси)фенил]изоксазол-3-ил]бензойная кислота. Регистрационный номер в химической реферативной службе - 179162-55-1 и название, присвоенное Фуджисавой - FR-195752. Это соединение, которое здесь также называется как боковая цепь Микафунгина, в кислотной форме, может быть представлено формулой III
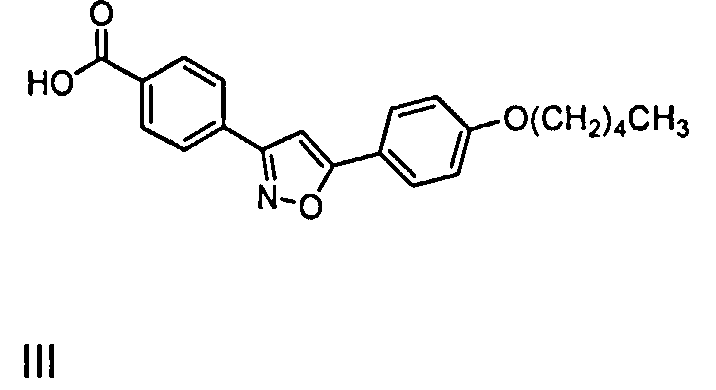
Пептидное ядро Микафунгина представлено формулой II
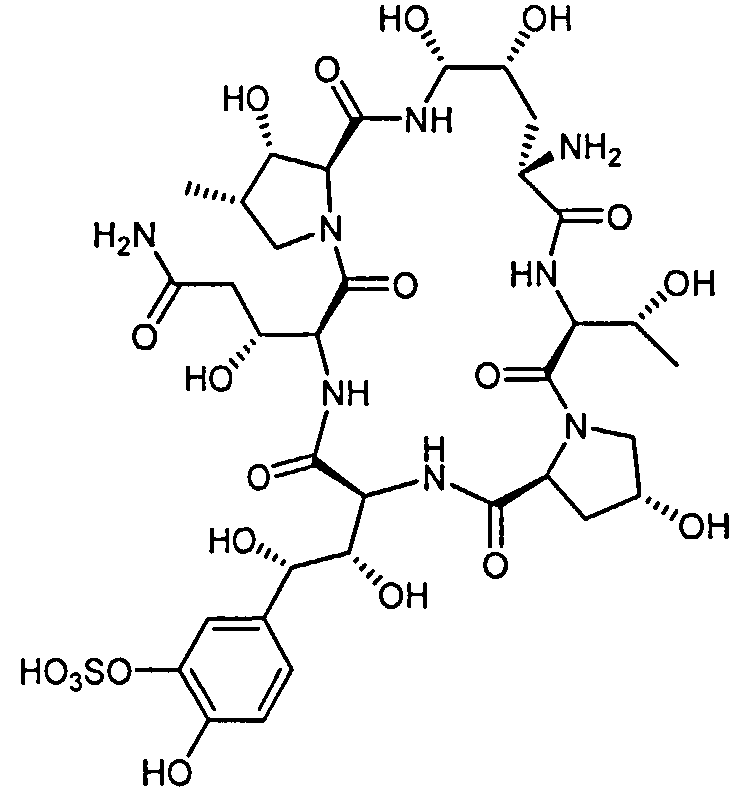
Термин "пептидное ядро Микафунгина", использованный здесь, предназначен включить в себя также соли вещества с формулой II. Например, натриевая соль вышеуказанного соединения, также известного как FR-179642 (Фромтлинг и другие в Drugs of the Future, 1998, том 23, номер 12, страницы 1273-1278).
В Микафунгине боковая цепь Микафунгина соединяется с пептидным ядром Микафунгина амидной связью. Различные способы образования амидной связи, то есть реакции карбоновой кислоты и амина, раскрыты в уровне техники и используются в синтезе пептидов. В большинстве случаев формирование амидной связи требует использования активации карбоновой кислоты с помощью связывающего реагента и связывающей добавки, согласно Маделейн М. Жули и Кеннет М. Лэссен, в Arkivoc, 2010 (viii), 189-250 и Эрик Валер и Марк Бредли, 2009, Chem. Soc. Rev., 38, 606-631. Также существуют примеры, где достаточно простые пептидные структуры приготовлены с помощью формирования амидной связи по однореакторному способу согласно Пю и другие, 2009, Organic Process Research & Development, 13, 321-314, где описан способ формирования пептидов в водно-этаноловой смеси.
Тем не менее, нигде в уровне техники однореакторный способ сочетания карбоновой кислоты и аминогруппы для сложной молекулы, такой как пептидное ядро микафунгина, не был предложен.
В данном контексте, термин "Пептидное ядро Микафунгина" или "ядро Микафунгина" - соединение, получаемое ферментативным дезацилированием остатка пальмитоиловой кислоты из FR-901379, показанной в формуле II, описанной выше. Фуджисава дал название FR-179642 для пептидного ядра Микафунгина и название PR-133303 для натриевой соли пептидного ядра Микафунгина. Приписанный регистрационный номер для данного вещества в химической реферативной службе 168110-44-9. В данном контексте, пептидное ядро Микафунгина предназначено включить в себя это вещество; так же как и соли данного вещества, например натриевую соль FR-133303.
Первая стадия в однореакторном способе, согласно настоящему изобретению - смешивание боковой цепи Микафунгина и связывающей добавки. После указанного смешивания, связывающий реагент добавляют к смеси, что приводит к активации боковой цепи Микафунгина и реакции с добавкой.
Термин "связывающая добавка" как здесь использовано, относится к любому веществу, которое усиливает реактивность активированной боковой цепи Микафунгина и упрощает связывание с первичным амином пептидного ядра Микафунгина. Преимущество использования связывающей добавки состоит в том, что она уменьшает образование побочных продуктов.
Существует огромный выбор связывающих добавок (смотри Chem. Soc. Rev., 2009, том 38, страницы 606-631 Валер и Бредли). Термин гидроксбензотриолы предназначен включить в себя гидроксибензотриазолы, гидроксиазабензотриазолы и их замещенные производные. Например, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, 6-хлор-1-гидроксибензотриазол и т.д.
Согласно способу настоящего изобретения, 1-гидроксибензотриол (HOBt) и этил 2-циано-2-(гидроксиимино)ацетат (Oxyma Pure®, CAS №3849-21-6, далее Оксима) показали свою полезность. Применение указанных связывающих добавок приводит к низкому образованию побочных продуктов и высоким выходам желаемого Микафунгина.
Термин "связывающие реагенты" используемый согласно настоящему изобретению - любое вещество, способное активировать карбоновую кислоту боковой цепи Микафунгина, в присутствии связывающей добавки, и, таким образом, облегчать ее реакцию с амином структуры ядра Микафунгина.
В качестве связывающего реагента, согласно настоящему изобретению, могут быть использованы производные карбодиимида, представленные следующей формулой: Ra-N=C=N-Rb, где Ra и Rb одинаковые или разные, и каждая независимо представлена алифатической, гетероалифатической, карбоциклической или гетероциклической группой, все указанные группы возможно могут быть замещены. Согласно одному аспекту настоящего изобретения, 1-этил-3-(3-диметиламинопропил) карбодиимид (ЭДК) используется как связывающий реагент. Согласно предпочтительному аспекту настоящего способа, хлороводородная соль ЭДК используется как связывающий реагент.
Из уровня техники известно, что карбодиимиды могут быть использованы как связывающие реагенты, смотри Валер и Бредли, Chem. Soc. Rev Июнь 2008. Отрицательно заряженный кислород боковой цепи Микафунгина предпочтительно ведет себя как нуклеофил, атакуя центральный углерод диимидовых групп. Способ настоящего изобретения показан ниже, используя Оксиму в качестве связывающей добавки и ЭДК в качестве связывающего реагента, смотри схему 1.
Подходящие растворители для однореакторной реакции включают полярные апротонные органические растворители. Неограниченный список подходящих растворителей включает диметилформамид (ДМФ), диметилацетамид (ДМА), диметилсульфоксид (ДМФО) и т.д., а также их смеси.
Подходящие основания для однореакторной реакции включают органические или неорганические основания, способные протонировать аминогруппу пептидного ядра Микафунгина. Неограниченный список подходящих оснований включает ДИПЭА, NaHCO3, Na2CO3 и т.д.
Подходящие температуры для активации боковой цепи Микафунгина находятся в диапазоне от 0°C до 40°C, предпочтительно 20°C-30°C.
Подходящие температуры для связывания боковой цепи Микафунгина с пептидным ядром Микафунгина находятся в диапазоне от -5°С до 10°С, предпочтительно 0°C. Также, предпочтительно достигать указанных температур до добавления основания.
Подходящее полное время реакции для однореакторного способа составляет примерно от 4 ч до 20 ч. Предпочтительно, 2-4 ч для активации боковой цепи Микафунгина и от 90 мин до 2 ч для связывания активированного Микафунгина с пептидным ядром Микафунгина.
Важно, чтобы связывающий реагент был добавлен после смешивания связывающей добавки и боковой цепи Микафунгина.
Схема реакции 1 ниже демонстрирует активацию боковой цепи Микафунгина и реакцию активированной боковой цепи Микафунгина с пептидным ядром Микафунгина, происходящие в однореакторном процессе настоящего изобретения.
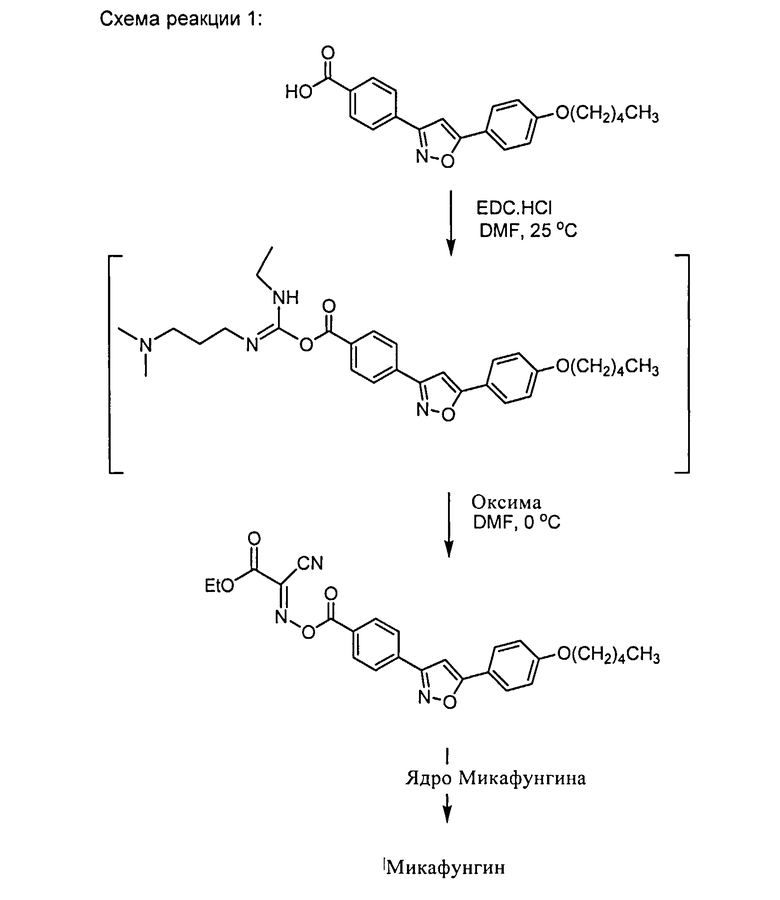
После связывания активированной боковой цепи Микафунгина с пептидным ядром Микафунгина, полученный продукт может быть осажден в виде соли Микафунгина. Термин "соль Микафунгина" может, в данном контексте, относиться к любой фармацевтически активной соли или соли, полезной для дальнейшей очистки Микафунгина. В последнем случае, солью Микафунгина может, к примеру, быть соль N,N-диизопропил этиламина (ДИПЭА). Любая фармацевтически приемлемая соль Микафунгина может затем быть приготовлена, используя Микафунгин, приготовленный согласно настоящему изобретению. Полезные фармацевтически приемлемые соли могут быть выбраны из группы, состоящей из калиевых и натриевых солей.
Микафунгин, приготовленный согласно настоящему изобретению может возможно дальше быть очищен, используя способы хорошо известные квалифицированному специалисту. Согласно одному воплощению настоящего изобретения, продукт из стадии в) настоящего изобретения трансформируют в соль ДИПЭА Микафунгина в последствие переводят в фармацевтически приемлемую соль Микафунгина, такую как натриевая соль Микафунгина и далее очищается хроматографией.
Микафунгин, приготовленный согласно настоящему изобретению, может быть использован в производстве фармацевтических композиций, полезных для лечения и предупреждение инфекционных заболеваний млекопитающих, включая человека и животных. Указанные фармацевтические композиции могут быть приготовлены, используя стандартные методики, широко известные в уровне техники. Фармацевтическая композиция может быть также подвержена обычным фармацевтическим процедурам, таким как стерилизация.
Например, фармацевтическая композиция может быть приготовлена в виде дозированной единицы, содержащей заданное количество очищенного Микафунгина, одного или в комбинации с другим активным ингредиентом, и вместе с фармацевтически приемлемыми эксципиентами. Термин "фармацевтически приемлемый эксципиент" относится к одному или нескольким препаратам, подходящим для осуществления или улучшения доставки Микафунгина, содержащегося в фармацевтической композиции, пациентам, которым он необходим. "Фармацевтически приемлемый эксципиент" может быть представлен наличием вспомогательных средств, таких как: консерванты, стабилизаторы, смачивающие реагенты, эмульгаторы, буферы и подобные. Лактоза - пример подходящего стабилизатора для Микафунгина, особенно в лиофильной форме. Квалифицированному специалисту известны различные фармацевтические эксципиенты подходящие для приготовления фармацевтических препаратов, содержащих противогрибковые соединения, такие как Микафунгин. Композиция согласно настоящему изобретению может быть приготовлена для парентерального введения, например внутривенного введения.
Пациенту, которому необходима композиция, согласно настоящему изобретению может быть введена подходящая доза Микафунгина. Подходящая суточная доза для человека или млекопитающего может широко разниться в зависимости от состояния пациента и других факторов. Суточная доза может быть определена квалифицированным специалистом, используя стандартные методы, и которые широко используются в лечении и предупреждении инфекций при введении Mycamine®.
Квалифицированный специалист из последующих примеров уяснит множество преимуществ настоящего изобретения. Эксперименты и результаты ниже должны пониматься в качестве неограничивающих примеров.
Пример 1: Приготовление активированной боковой цепи Микафунгина
FR-195752 (10.0 г) и HOBt (5.2 г) (включающий 12% воды) были растворены в DMF (142 мл) и перемешены в течение 5 минут. EDC HCl (6.6 г) было добавлено к суспензии. Реакционная смесь перемешивалась в течение 4 часов при 25°C. Реакционную смесь влили в 426 мл ацетонитрила (ACN) и перемешивали 18 часов при 25°C. Белую суспензию отфильтровали и сушили на фильтре 2 часа. Выделенное количество продукта составило 12.6 г (95%) с чистотой по ВЭЖХ: 98.0%.
Пример 2: Связывание активированной боковой цепи Микафунгина с пептидным ядром Микафунгина
FR-179642 (10.0 г) растворили в безводном ДМФ (200 мл) перемешиванием при 25°C в течение 10 мин. Смесь была охлаждена до 0°C. Добавили боковую цепь Микафунгина (4.50 г) и активированную кислоту, описанную в примере 1, с последующим добавлением ДИПЭА (2.25 мл). Все вещества растворились спустя 5 минут, и смесь перемешивали 90 минут при 0°C. Добавили смесь метанола (50 мл) и ацетона (100 мл) и температуру повысили до 10°C. Смесь перемешивали при данной температуре в течение 60 минут. Этилацетат (1000 мл) медленно добавили в течение 2.5 ч. Полученную суспензию перемешивали 15 ч, и продукт собрали фильтрацией под давлением, используя AIdrich 1000 мл стеклянный фильтропресс, снабженный крупнозернистым керамическим фильтрующим диском. Фильтрат промыли этилацетатом (1500 мл) и высушили на фильтре в течение 15 минут. Последующая сушка в эксикаторе при 25°C в течение 3 ч дала 12.8 г (86%) белого твердого вещества.
Пример 3: Активирование и сочетание боковой цепи Микафунгина с пептидным ядром Микафунгина
FR-195752 (394 мг) и HOBt гидрат (197 мг) растворили в ДМФ (15 мл) при 25°C. Добавили ЭДК HCI (166 мг) и смесь перемешивали при 25°C в течение 4 ч. Смесь охладили до 0°C, и добавили ДИПЭА (0.223 мл), с последующим добавлением FR-179642 (1.00 г). Смесь перемешивали 90 минут при 0°C. Смесь метанола (2.5 мл) и ацетона (5 мл) добавили, и смесь нагрели до 10°C и перемешивали 60 минут. Этилацетат (100 мл) добавили в течение 30 минут, и полученную суспензию перемешивали в течение 16 ч при 10°C. Твердый продукт собрали фильтрацией, промыли этилацетатом (150 мл) и высушили на фильтре 15 мин. Последующая просушка в эксикаторе при 25°C в течение 2 ч дала 1.47 г (98% выход) белого твердого вещества. Чистота по ВЭЖХ 96.6%
Пример 4: Активация и связывание боковой цепи Микафунгина с пептидным ядром Микафунгина
FR-195752 (2.06 г,) и Оксима (834 мг) были растворены в ДМФ (60 мл) при 25°C. ЭДК HCl (1.07 г) был добавлен, и смесь перемешивали при 25°C в течение 2 ч. Реакционная смесь превратилась в ярко-желтый раствор. Желтый раствор был охлажден до 0°C, и ДИПЭА (1.1 мл) был добавлен, с последующим добавлением FR-179642 (5.00 г). Смесь перемешивали 90 минут при 0°C. Смесь метанола (15 мл) и ацетона (30 мл) была добавлена, и смесь подогрели до 10°C и перемешивали 60 минут. Этилацетат (300 мл) был добавлен в течение 30 минут, и результирующая смесь перемешивалась 20 ч при 10°C. Твердый продукт был собран фильтрацией, промыт этилацетатом (100 мл) и высушен на фильтре в течение 15 минут. Дополнительная сушка в эксикаторе при 25°C в течение 2 ч дала: 6.68 г (90% выход) серо-белого твердого вещества.
Пример 5: Сравнение хроматограмм из Примеров 2-4
ВЭЖХ хроматограммы неочищенных продуктов из примеров 2-4 были проанализированы с помощью ВЭЖХ (Обратно-фазная хроматография) Таблица 1 показывает результаты ВЭЖХ хроматограмм, сравнивая 3 разных способа (смотри Фиг.4а-4в). Пример 2 представляет собой постадийный способ и примеры 3 и 4 - однореакторные способы, используя HOBt и Оксиму как связывающие добавки соответственно. Оксима не представлен в хроматограмме в неочищенном продукте. В реакционной смеси Оксима появляется на 8.5 минуте.
Способы 3 и 4 более эффективны, так как используется меньше растворителя и сокращено время реакции, в связи с тем, что исключено выделение активированной кислоты. Массовые выходы существенно выше в однореакторных способах, а именно 98% и 95% в примерах 3 и 4 соответственно, в сравнении с только 82% в ныне известных способах. Как было показано в примерах, однореакторный способ, согласно настоящему заявлению, дает производство Микафунгина той же чистоты, что и в ныне известных постадийных способах, но с повышенным выходом и примерно одинаковой чистотой неочищенных продуктов, как было показано на Фиг.4а-в.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ МИКАФУНГИНА | 2012 |
|
RU2583262C2 |
| ЦИКЛОПЕПТИДНОЕ СОЕДИНЕНИЕ ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2603345C2 |
| АНАЛОГ ДОЛОСТАТИНА | 1993 |
|
RU2132334C1 |
| ПЕПТИДИЛ-ДИАЦИЛГЛИЦЕРИДЫ | 2009 |
|
RU2545347C2 |
| НОВЫЕ СПОСОБЫ ПОЛУЧЕНИЯ БАРУСИБАНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2726414C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| КРИСТАЛЛ ЦИКЛОПЕПТИДА ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2607083C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| СПОСОБ ЖИДКОФАЗНОГО СИНТЕЗА H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 2015 |
|
RU2694051C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАСПОФУНГИНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2489442C2 |
Изобретение относится к однореакторному способу приготовления Микафунгина. Способ включает в себя ацилирование пептидного ядра Микафунгина без изолирования активированной боковой цепи Микафунгина из ее реакционной смеси. Однореакторный способ получения позволяет укоротить время синтеза и повысить выход. 13 з.п. ф-лы, 4 ил., 1 табл., 5 пр.
1. Однореакторный способ для производства Микафунгина или его соли, включающий следующую последовательность стадий:
а) смешивание соединения с формулой (III)
или его соли и связывающей добавки, выбранной из группы, состоящей из гидроксибензотриазолов и этил-2-циано-2-(гидроксиимино)ацетата в растворителе;
б) добавление связывающего реагента к смеси, полученной на стадии а), где указанный связывающий реагент представляет собой карбодиимид;
в) добавление основания и соединения с формулой (II)
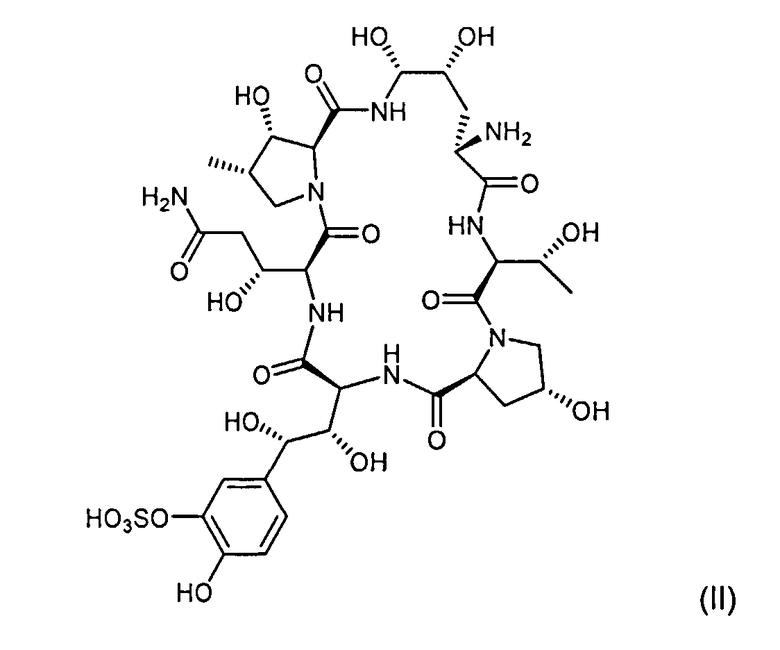
или его соли к смеси, полученной на стадии б).
2. Способ по п.1, где связывающая добавка представляет собой 1-гидрокси-бензотриазол.
3. Способ по п.1, где связывающая добавка представляет собой 1-гидрокси-7-азобензотриазол.
4. Способ по п.1, где связывающая добавка представляет собой этил-2-циано(гидроксиимино)ацетат.
5. Способ по любому из пп.1-4, где связывающий реагент представляет собой 1-этил-3-(3-диметиламинопропил) карбодиимид (ЭДК) или его соль.
6. Способ по п.5, где связывающий реагент представляет собой хлороводородную соль ЭДК.
7. Способ по п.1, где растворитель, использованный на стадии а), представляет собой ДМФ.
8. Способ по п.1, где основание, использованное на стадии в), представляет собой диизопропилэтиламин.
9. Способ по п.1, где основание добавляется к смеси, полученной на стадии б), перед добавлением соединения с формулой II.
10. Способ по п.1, где основание, добавляется к смеси, полученной на стадии б), после добавления соединения с формулой II.
11. Способ по п.1, где соль Микафунгина, полученная на стадии в), осаждается.
12. Способ по п.11, где соль Микафунгина, полученная на стадии в), осаждается с использованием антирастворителя.
13. Способ по п.12, где антирастворитель представляет собой этилацетат.
14. Способ по п.11, где соль Микафунгина, полученная на стадии в), осаждается после остановки реакции добавлением метанола и ацетона.
| ПОЛИПЕПТИДНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2165423C2 |
| US 2005227914 A1, 13.10.2005 | |||
| YANGWEI JOHN PU ET AL: "A Practical Method for Functionalized Peptide or Amide Bond Formation in Aqueous-Ethanol Media with EDC as Activator +", ORGANIC PROCESS RESEARCH & DEVELOPMENT, 2009, vol | |||
| Насос | 1917 |
|
SU13A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

