Область техники
Настоящее изобретение относится к фармацевтической композиции, и, более конкретно, относится к фармацевтической композиции, содержащей гидрат соединения циклопептида, имеющий хорошую стабильность, а также к способу его получения и к его применению.
Предшествующий уровень техники
Грибковые инфекции стали основной причиной высокой заболеваемости и смертности у иммунодефицитных больных. На протяжении последних 20 лет заболеваемость грибковыми инфекциями значительно увеличилась. Группу риска составляют пациенты в тяжелом состоянии, пациенты после хирургических вмешательств, пациенты с ВИЧ-инфекцией, лейкемией и другими опухолями. Пациенты с трансплантированными органами, также подвержены высокому риску грибковой инфекции.
Эхинокандины, в качестве нового класса противогрибковых агентов, демонстрируют хорошие результаты в лечении инфекций, вызванных Candida или Aspergillus. Примерами таких лекарственных средств являются Каспофунгин и Микафунгин. Эхинокандины ингибируют грибок, подавляя образование 1,3-β гликозидной связи, таким образом, уменьшая вредное воздействие на человеческий организм и снижая побочные эффекты, при этом сохраняя высокую лекарственную эффективность. Таким образом, они более безопасны в использовании, чем традиционные противогрибковые агенты.
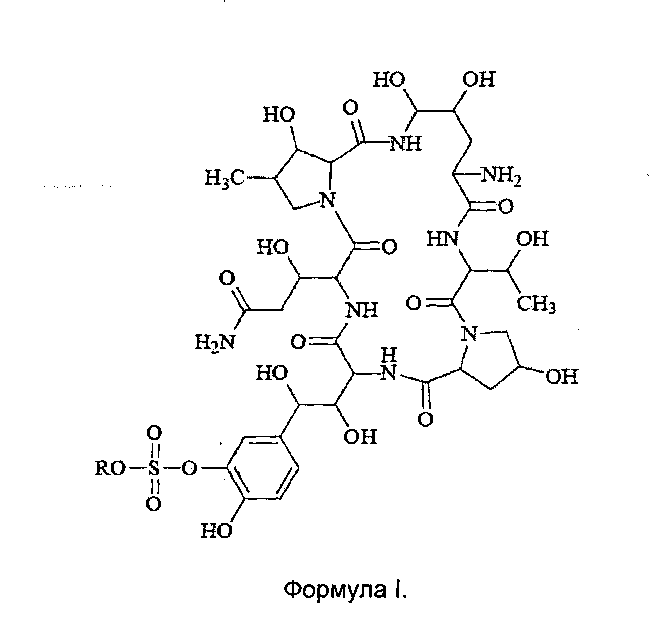
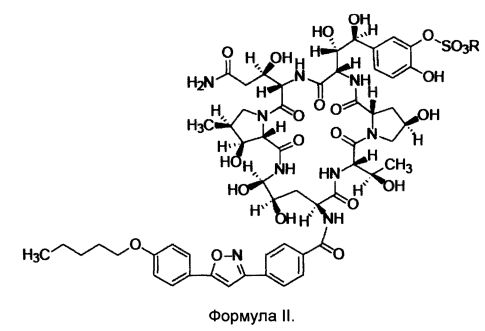
FK463 (Микафунгин натрия) представляет собой соединение формулы II (где R представляет собой ион натрия), разработанное Японским заводом Fujisawa Toyama Co., Ltd, в г. Такаока под торговой маркой Mycamine, и продаваемое в настоящее время в нескольких странах в качестве противогрибкового средства для внутривенного введения. Его получают путем разрезания боковой цепи предшественника FR 901379 (соединение формулы III, где R представляет собой ион натрия или ион водорода) с помощью фермента, с получением FR 179642 (соединение формулы I, где R представляет собой водород или ион натрия) (описание конкретных методов раскрыты в патенте США US5376634, ЕР 0431350 и патенте Китая CN1161462C), а затем химической модификации FR 179642 (описание конкретных подготовительных и очистных методов раскрыты в патентах WO 9611210, WO 9857923, WO 2004014879).
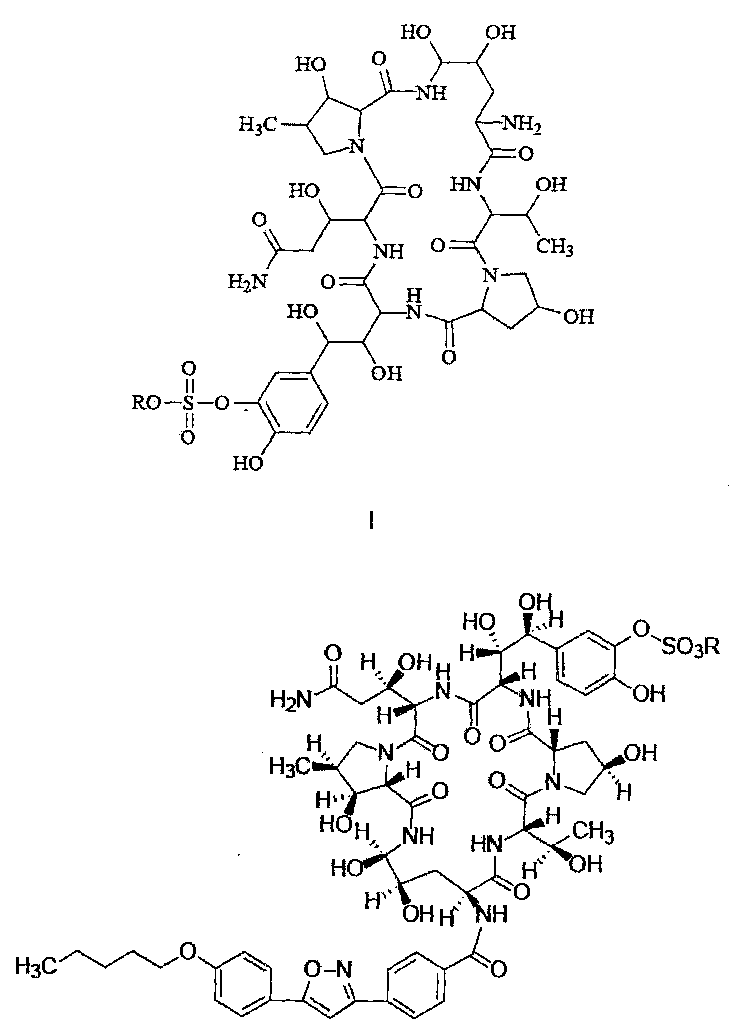
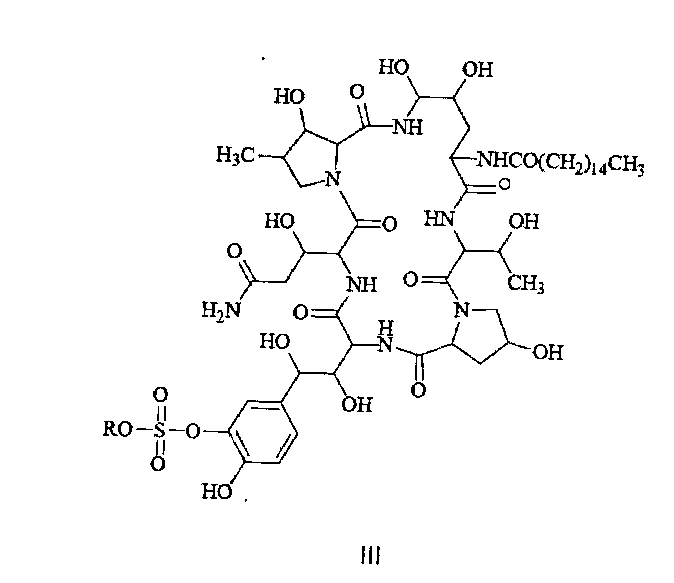
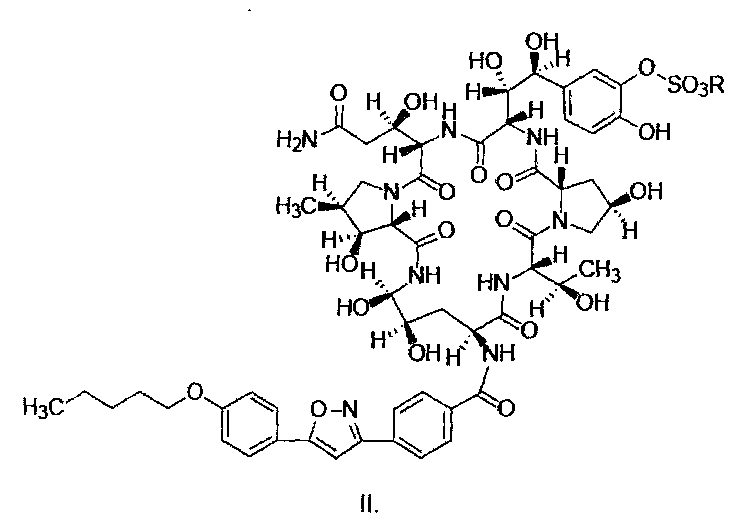
Конкретные схемы выглядят следующим образом:

Как хорошо известно, в данной области техники, стабильность лекарственного средства тесно связана с содержанием влаги. В литературе и книгах (например, "Фармацевтика"), обсуждающих стабильность лекарственных средств, говорится, что вода является средой для протекания химической реакции, и после того, как вода поглощается лекарственным средством в твердой форме, на поверхности лекарственного средства образуется жидкая пленка, в которой будет протекать гидролиз или реакция окислительного разложения. Присутствие следовых количеств воды может ускорить разложение нестабильных лекарственных средств. Содержание влаги в сырых лекарственных средствах, таких как ампициллин, следует поддерживать на относительно низком уровне, как правило, около 1%. Чем выше содержание влаги, тем быстрее идет разложение.
После обширных исследований авторы настоящего изобретения обнаружили, что содержание влаги в соединении формулы I имеет существенное влияние на стабильность соединения. Что еще более удивительно, авторы изобретения обнаружили, что высокое содержание влаги эффективно повышает стабильность соединения формулы I, вместо того, чтобы ускорять разложение соединения и снижать устойчивость соединения. Когда содержание влаги в соединении формулы I составляет менее 8%, стабильность соединения значительно снижена, как сказано выше. Изобретатели также обнаружили, что стабильность соединения формулы I меньше зависит от типов кристаллических форм, в то время как содержание влаги имеет решающее значение для стабильности соединения. Эти выводы являются неожиданными и были сформулированы изобретателями после проведения большого количества экспериментов.
Таким образом, в данной области техники существует насущная необходимость получения гидрата соединения формулы I, который обладает превосходной стабильностью и хорошо подходит для транспортировки и хранения.
Краткое описание изобретения
В одном аспекте, настоящее изобретение предлагает гидрат соединения формулы I, где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль, при этом содержание воды в гидрате более 8,0 масс. %, и R представляет собой, предпочтительно Н, ион натрия или диизопропилэтиламин ион;
В одном варианте осуществления изобретения, содержание воды составляет от 8 до 30 масс. %.
В дополнительном варианте осуществления изобретения, содержание воды составляет от 9,5 до 28,0 масс. %.
В другом варианте осуществления изобретения, гидрат получают посредством следующих стадий:
(a) растворение соединения формулы I в воде или водорастворимом органическом растворителе (I), и регулирование pH раствора, содержащего соединение формулы I;
(b) получение гидрата, включающего соединение формулы I, путем снижения температуры и/или добавления водорастворимого органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка гидрата, полученного на стадии (с) и регулирование содержания воды в массовых процентах в твердом веществе.
В дополнительном варианте осуществления изобретения упомянутый органический растворитель (i) выбран из С1-С4 низших спиртов.
В дополнительном варианте осуществления изобретения, низший спирт представляет собой один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
В дополнительном варианте осуществления, pH раствора, содержащего соединение формулы I, поддерживают на уровне от 2,0 до 5,0; в еще одном варианте осуществления, pH раствора, содержащего соединение формулы I, поддерживают на уровне от 3,5 до 4,5.
В дополнительном варианте осуществления изобретения, содержание воды в твердом веществе поддерживают на уровне более 8 масс. %.
В дополнительном варианте осуществления изобретения, содержание воды в твердом веществе поддерживают на уровне от 8 до 30 масс.%.
В дополнительном варианте осуществления изобретения, содержание воды в твердом веществе поддерживают на уровне от 9,5 до 28,0 масс. %.
В другом аспекте, гидрат соединения формулы I, получают с помощью настоящего изобретения, включающего следующие этапы:
(a) растворение соединения формулы I в воде или водном водорастворимом органическом растворителе (i), и регулирование pH раствора, содержащего соединение формулы I;
(b) получение гидрата, содержащего соединение формулы I, путем снижения температуры и/или добавления водорастворимого органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка гидрата, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате.
В дополнительном варианте осуществления упомянутый органический растворитель (i) выбран из С1-С4 низших спиртов.
В дополнительном варианте осуществления изобретения, низший спирт представляет собой один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
В дополнительном варианте осуществления изобретения, pH раствора, содержащего соединение формулы I, поддерживают на уровне от 2,0 до 5,0; в еще одном варианте осуществления, pH раствора, содержащего соединение формулы I, поддерживают на уровне от 3,5 до 4,5.
В дополнительном варианте осуществления изобретения, содержание воды в твердом веществе поддерживают на уровне более 8 масс. %.
В дополнительном варианте осуществления, содержание воды в твердом веществе поддерживают на уровне от 8 до 30 масс. %.
В дополнительном варианте осуществления изобретения, содержание воды в твердом веществе поддерживают на уровне от 9,5 до 28,0 масс. %.
В другом аспекте, изобретение предлагает применение гидрата для получения соединения формулы II по изобретению
В другом аспекте, изобретение предлагает применение гидрата для получения лекарственных средств для лечения грибковых инфекций
В другом аспекте, настоящее изобретение предлагает фармацевтическую композицию, содержащую вышеуказанный гидрат, и фармацевтически приемлемый носитель.
В другом аспекте, настоящее изобретение предлагает способ получения фармацевтической композиции, включающий смешивание гидрата с фармацевтически приемлемым носителем, в результате чего получают фармацевтическую композицию.
В другом аспекте, настоящее изобретение предлагает гидрат, полученный с помощью способов, приведенных выше.
Краткое описание графических материалов
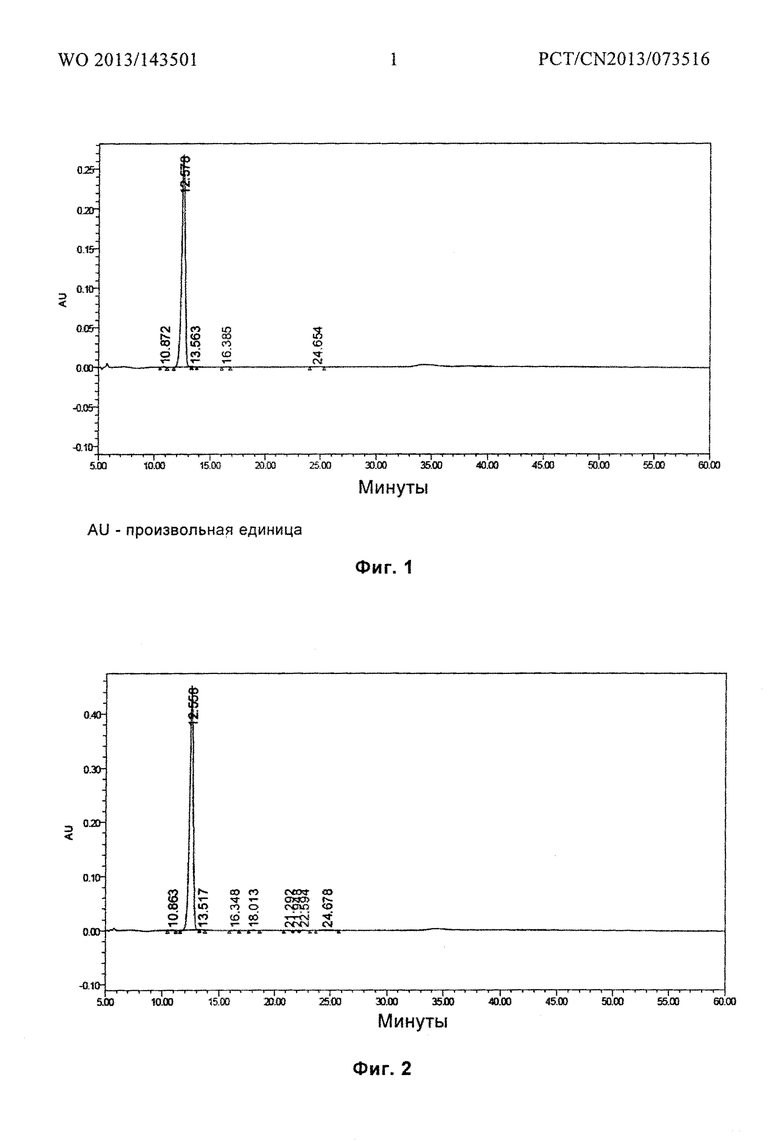
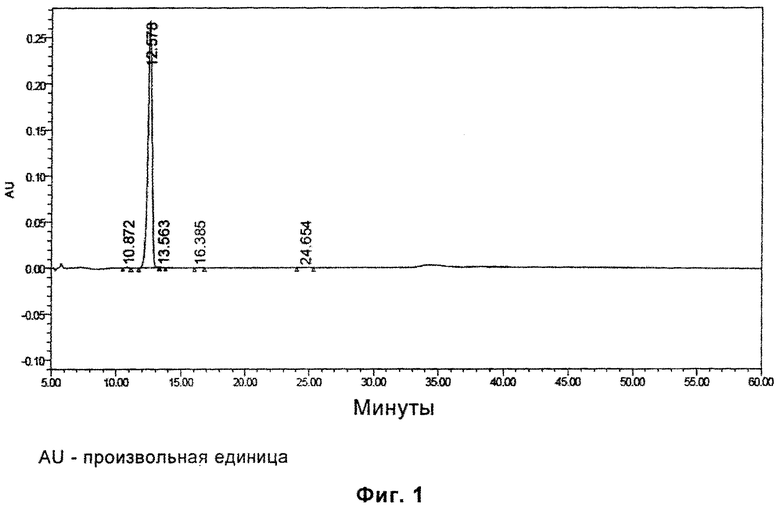
Фиг. 1 представляет паттерн ВЭЖХ (высокоэффективная жидкостная хроматография) для гидрата D, полученного в примере 2, после размещения при температуре 25°С в течение 6 месяцев.
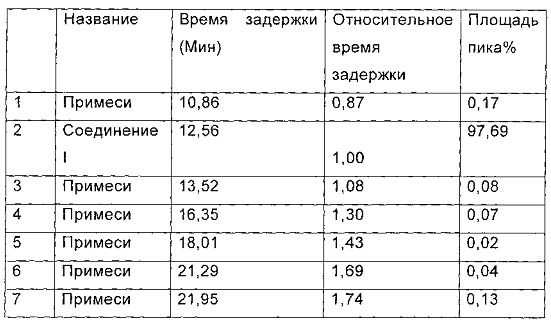
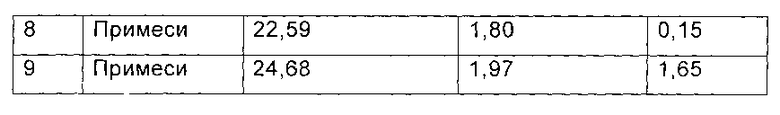
Фиг. 2 представляет паттерн ВЭЖХ для гидрата Y, полученного в примере 6, после размещения при температуре 25°С в течение 6 месяцев.
Фиг. 3 показывает количество примесей в образце для гидратов А, В, С, D и Е после размещения при температуре 25°С в течение 6 месяцев.
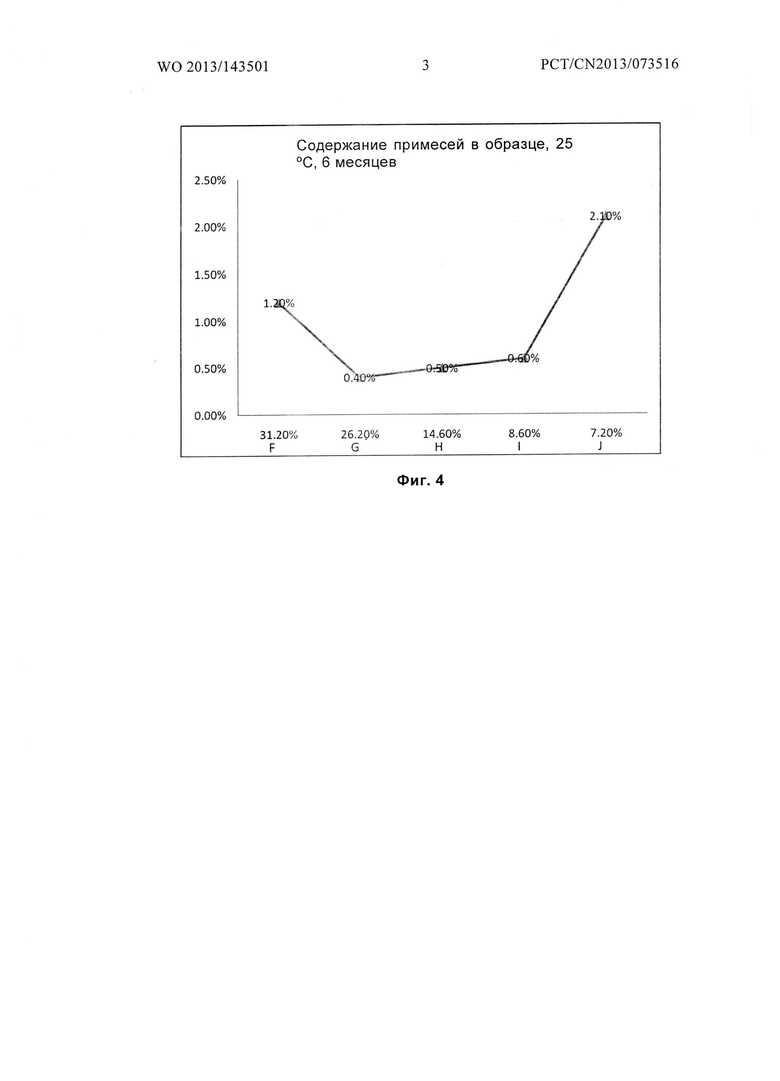
Фиг. 4 показывает количество примесей в образце для гидратов F, G, Н, I и J после размещения при температуре 25°С в течение 6 месяцев.
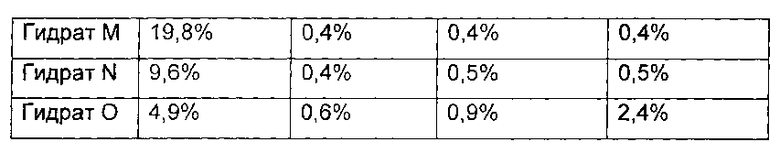
Фиг. 5 показывает количество примесей в образце для гидратов K, L, М, N и О после размещения при температуре 25°С в течение 6 месяцев.
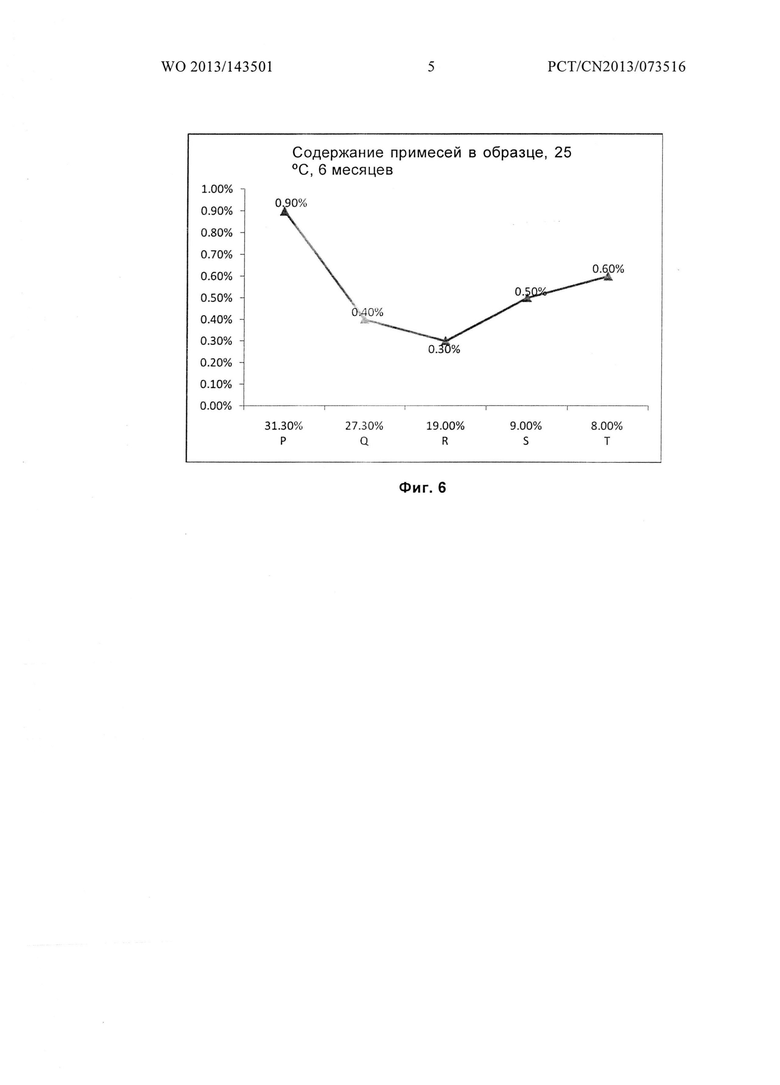
Фиг.6 показывает количество примесей в образце для гидратов Р, Q, R, S и Т после размещения при температуре 25°С в течение 6 месяцев.
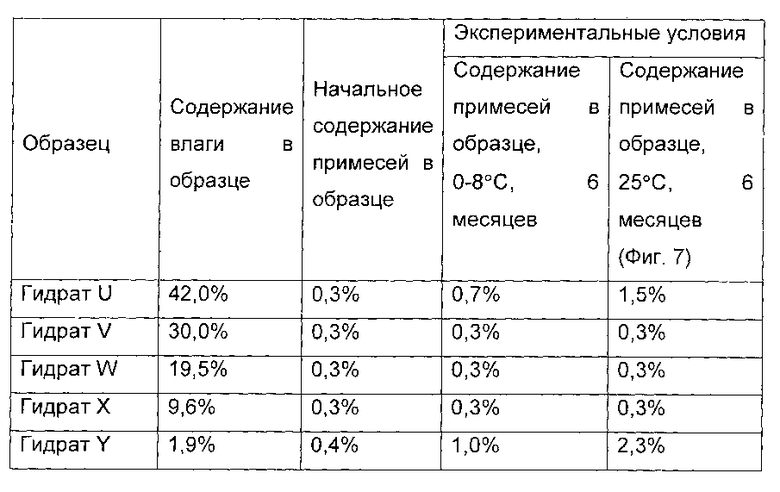
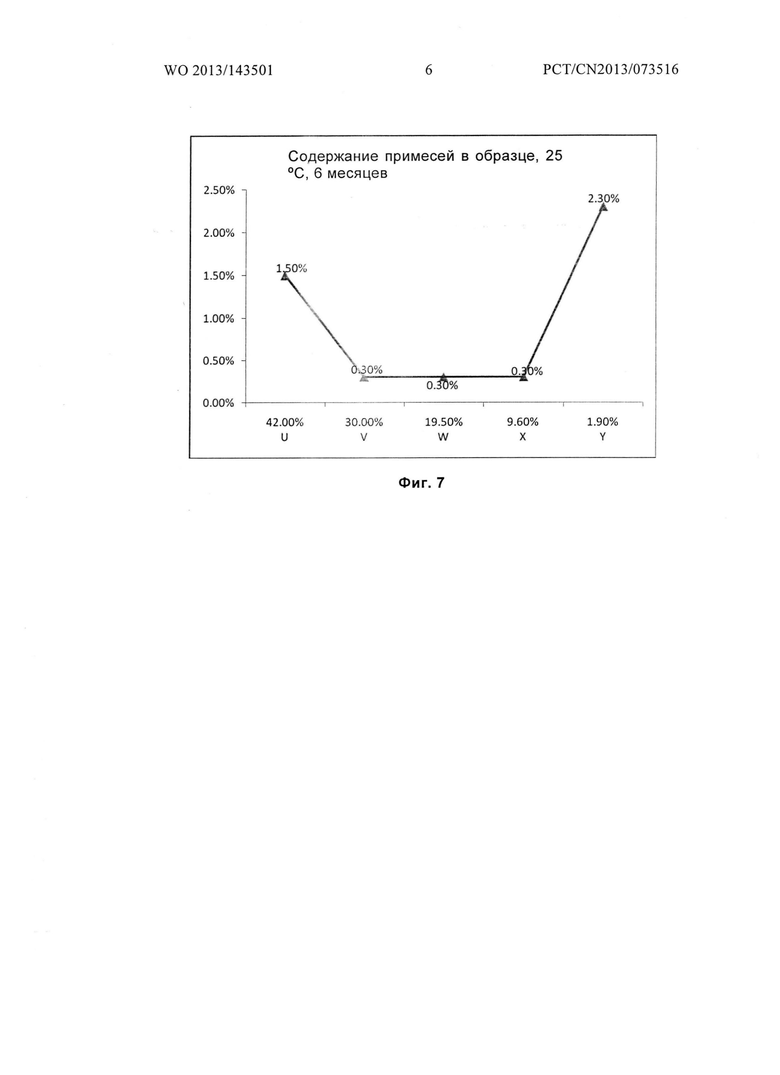
Фиг. 7 показывает количество примесей в образце для гидратов U, V, W, X и Y после размещения при температуре 25°С в течение 6 месяцев.
Режим выполнения изобретения
После обширных исследований авторы настоящего изобретения обнаружили, что гидраты соединений формулы I могут быть получены путем растворения соединения в воде или в смеси водорастворимых низших спиртов, поддержания растворимости раствора, содержащего соединение формулы I, в области насыщения, и поддержания значения pH раствора в обозначенном диапазоне.
Еще более важно то, что гидрат, образованный из соединения формулы I, содержит воду, и авторы настоящего изобретения после проведения обширных исследований обнаружили, что содержание влаги в гидрате соединения формулы I будет иметь существенное влияние на стабильность гидрата.
Для способа получения по настоящему изобретению, был проведен большой объем исследований на скрининговых растворителях для кристаллизации, и было обнаружено, что кристаллы с превосходной морфологией могут быть образованы из соединения формулы I путем кристаллизации соединения в метаноле, этаноле, н-пропаноле, изопропаноле или их смеси, и соединение формулы I с превосходной стабильностью может быть получено путем поддержания уровня влаги в пределах определенного диапазона.
При кристаллизации соединения в таком растворителе, как ацетон, ацетонитрил, этилацетат, из соединения формулы I будет сформирован аморфный осадок с низкой стабильностью, и, это является причиной разницы в стабильности между аморфными твердыми телами и кристаллическими веществами. Тем не менее, даже в случае аморфного твердого вещества, если содержание влаги в нем поддерживают в пределах определенного диапазона, твердое вещество будет обладать большей стабильностью по сравнению с твердым веществом с другим содержанием влаги. pH является еще одним ключевым параметром для получения кристаллов с улучшенной стабильностью из соединения формулы I; вне границ определенного диапазона pH, вещество перейдет в аморфную форму.
Определения
Термин "эффективное количество" так как он использован в данном описании, относится к носителям для введения терапевтического агента, в том числе к различным наполнителям и разбавителям. Этот термин относится к носителям, которые сами по себе не обязательно являются активными ингредиентами, и не будут чрезмерно токсичными при введении.
Подходящие носители хорошо известны специалистам в данной области техники. В "Remington′s Pharmaceutical Sciences" (Mack Pub. Co., NJ 1991), можно найти подробное обсуждение фармацевтически приемлемых носителей. В композиции, фармацевтически приемлемые носители могут включать в себя жидкости, такие как вода, физиологический раствор, глицерин и этанол. Кроме того, с этими носителями могут присутствовать вспомогательные вещества, такие как дезинтеграторы, смачивающие агенты, эмульгаторы, pH-буферные вещества и так далее.
Фармацевтическая композиция может быть приготовлена в виде различных лекарственных форм в зависимости от способа введения. Лекарственные формы вводят следующими способами: пероральное введение, ингаляционное распыление, ректальное, назальное, буккальное, местное, парентеральное введение, такими способами как подкожная, внутривенная, внутримышечная, внутрибрюшинная, интратекальная, интравентрикулярная, надчревная и внутричерепная инъекции или инфузии, либо посредством эксплантатов резервуара.
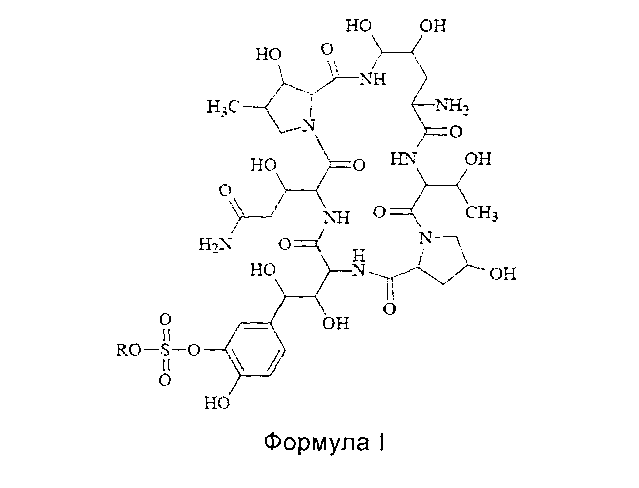
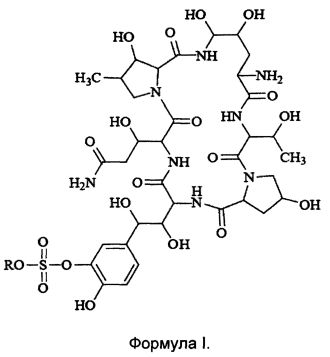
Термины "соединение формулы I" или "соединение формулы I" так, как они используются здесь, могут быть использованы взаимозаменяемо, оба из них относятся к соединению, имеющему следующую структурную формулу или к его фармацевтически приемлемой соли:
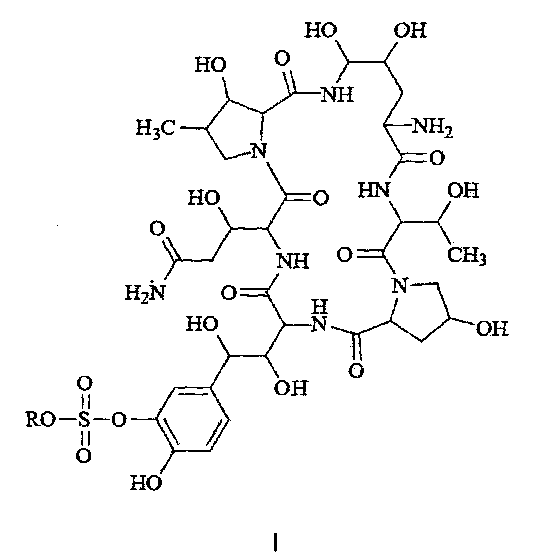
где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль.
Предпочтительно, фармацевтически приемлемые соли включают: соли металлов, такие как соли щелочных металлов (например, соль натрия, соль калия), соли щелочноземельных металлов (таких как соли кальция, соли магния и т.п.), соли аммония, соли, образованные с органическими основаниями (например, соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, соль дициклогексиламина, N, N, дибензилэтилендиамин соль, соль диизопропилэтиламина и т.д.), органические кислотно-аддитивные соли (такие как формиат, ацетат, трифторацетат, малеат, тартрат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), аддитивные соли неорганической кислоты (например, гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат и т.д.), соли, образованные с аминокислотой (например, аргинин, аспарагиновую кислоту, глутаминовую кислоту, и т.п.), и тому подобные. Предпочтительно R представляет собой Н, ион натрия или ион диизопропилэтиламина.
Соединение формулы I может быть получено с помощью традиционных в данной области способов, например, но, не ограничиваясь им, способ получения этого соединения был описан в патентной заявке WO 9611210; в качестве альтернативы, соединение может быть также получено из коммерческих источников, таких как, но, не ограничиваясь им, Fujisawa, Япония.
Термин "С1-С4 низший спирт" так, как он используется здесь, относится к спиртам, количество атомов углерода в которых составляет от 1 до 4.
Все свойства данного изобретения, упомянутые выше или в приведенных ниже примерах, могут быть, необязательно, объединены. Все свойства, раскрытые в данном описании, могут быть использованы в любой комбинации. Любое альтернативное свойство, служащее той же, эквивалентной или аналогичной цели может заменить любое свойство, раскрытое в этом описании. Поэтому, если не указано иное, описанные свойства, представляют собой только общие примеры эквивалентных или аналогичных свойств.
Получение гидратов соединения формулы I
После обширных исследований авторы настоящего изобретения обнаружили, что устойчивые гидраты соединений формулы I могут быть получены путем растворения соединения в воде или в смеси водорастворимых органических растворителей, поддержания растворимости раствора, содержащего соединение формулы I, близкой к насыщению, поддержания значения pH раствора в указанном диапазоне и изменения некоторых факторов, таких как температура кристаллизации, молярная концентрация, скорость охлаждения или скорость перемешивания и время кристаллизации, а затем вакуумной сушки.
На основании приведенных выше результатов, было сделано настоящее изобретение.
В настоящем изобретении, представлен стабильный гидрат соединения формулы I, в котором содержание воды в гидрате составляет более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В настоящем изобретении предложен способ получения гидрата соединения формулы I, включающий следующие стадии:
(a) растворение соединения формулы I в воде или водном органическом растворителе (I), и поддержание pH раствора;
(b) получение гидрата соединения формулы I путем снижения температуры и / или добавлени органического растворителя (I);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате.
При этом,
На стадии (а) температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а) соотношение объемов органического растворителя (i) и воды, в водном органическом растворителе (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно от 0,5 до 3,0.
На стадии (а) раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а) pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b) температуру понижают от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10°С.
На стадии (b) соотношение объема органического растворителя (i) к раствору со стадии (а) составляет от 0,1 до 10, предпочтительно от 1 до 5.
На стадии (а) и/или (b), указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно, один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола, и изопропанола.
В стадии (d) содержание воды в твердом веществе поддерживается на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
После полной кристаллизации, гидраты могут быть разделены путем фильтрования, декантации растворителя и тому подобное, предпочтительно, путем фильтрования. Гидраты могут быть промыты водой и, наконец, высушены в вакууме, с получением гидратов соединения формулы I.
В одном варианте осуществления настоящего изобретения, гидрат соединения формулы I получают путем следующих стадий:
(a) растворение соединения формулы I в воде, и поддержание pH раствора;
(b) получение гидрата соединения формулы I путем снижения температуры;
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а), pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b) температуру понижают от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10.
На стадии (d), содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В другом варианте осуществления настоящего изобретения, гидрат соединения формулы I получают посредством следующих стадий:
(a) растворение соединения формулы I в воде, и регулирование pH раствора;
(b) получение гидрата соединения формулы I путем добавления органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 50 до 300 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а), pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола, и изопропанола.
На стадии (b), соотношение объема органического растворителя (i) к раствору со стадии (а) составляет от 0,1 до 10, и предпочтительно от 1 до 5.
На стадии (d), содержание воды в твердом веществе поддерживается на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В другом варианте осуществления настоящего изобретения, гидрат соединения формулы I получают посредством следующих этапов:
(a) растворение соединения формулы I в воде, и регулирование pH раствора;
(b) получение гидрата соединения формулы I путем снижения температуры и добавления органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а), раствор содержит от 10 до 500 мг/мл, предпочтительно от 50 до 300 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а), pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b), указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола, и изопропанола.
На стадии (b) температуру понижают от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10°С.
На стадии (b) соотношение объема органического растворителя (I) к раствору со стадии (а) составляет от 0,1 до 10, и предпочтительно от 1 до 5.
На стадии (d) содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В другом варианте осуществления настоящего изобретения, гидрат соединения формулы I получают посредством следующих шагов:
(a) растворение соединения формулы I в водном органическом растворителе (I), и регулирование pH раствора;
(b) получение гидрата соединения формулы I путем снижения температуры;
(в) получение гидрата центрифугированием или фильтрованием;
(г) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а), температура для растворения составляет от 10 до 50°С, предпочтительно, от 20 до 40°С.
На стадии (а) соотношение объема органического растворителя (i) и воды, в водном органическом растворителе (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно от 0,5 до 3,0
На стадии (а) раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а) pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (а), указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола, и изопропанола.
На стадии (b), температуру понижают от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10°С.
На стадии (d), содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В другом варианте осуществления настоящего изобретения, гидрат соединения формулы I получают посредством следующих этапов:
(a) растворение соединения формулы I в водном органическом растворителе (i), и регулирование pH раствора;
(b) получение гидрата соединения формулы I путем добавления органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а) температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а) соотношение объема органического растворителя(0 и воды, в водном органическом растворителе (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно от 0,5 до 3,0.
На стадии (а) раствор содержит от 10 до 500 мг/мл, предпочтительно, от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а) pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b) соотношение объема органического растворителя (i) к раствору на стадии (а) составляет от 0,1 до 10, и, предпочтительно от 1 до 5.
На стадии (d) содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
На стадии (а) и (b) указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно, один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
В стадии (d) содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
В другом варианте осуществления настоящего изобретения, гидрат соединения формулы I получают посредством следующих этапов:
(a) растворение соединения формулы I в водном органическом растворителе (i), и поддержание pH раствора;
(b) получение гидрата соединения формулы I путем снижения температуры и добавления органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка твердого вещества, полученного на стадии (с) и регулирование содержания воды в массовых процентах в гидрате;
при этом,
на стадии (а) температура для растворения составляет от 10 до 50°С, предпочтительно от 20 до 40°С.
На стадии (а) соотношение объема органического растворителя (i) и воды, в водном органическом растворителе (i) составляет от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно, от 0,5 до 3,0.
На стадии (а) раствор содержит от 10 до 500 мг/мл, предпочтительно от 100 до 400 мг/мл соединения формулы I, в расчете на общий объем раствора на стадии (а).
На стадии (а), pH раствора поддерживают на уровне от 2,0 до 5,0, предпочтительно от 3,5 до 4,5.
На стадии (b) температуру понижают от -40 до 35°С, предпочтительно от -10 до 35°С, более предпочтительно от -5 до 30°С, и наиболее предпочтительно от 5 до 10°С.
На стадии (b) соотношение объем органического растворителя (i) к раствору на стадии (а) составляет от 0,1 до 10, и предпочтительно от 1 до 5.
На стадии (а) и (b), указанный органический растворитель (i) представляет собой С1-С4 низший спирт; предпочтительно один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
На стадии (d) содержание воды в твердом веществе поддерживают на уровне более 8,0 масс. %; предпочтительно от 8,0 до 30 масс. %; наиболее предпочтительно от 9,5 до 28 масс. %.
Идентификация и свойства
После того, как гидрат соединения формулы I был получен, его свойства были изучены авторами изобретения с использованием различных методов и инструментов.
В одном варианте осуществления настоящего изобретения для обнаружения массового процента воды в композиции, использовали традиционные в данной области техники методы детекции. Например, для обнаружения содержания влаги использовали метод Карла Фишера (KF).
В другом варианте осуществления настоящего изобретения, чистоту образца, полученного способом по изобретению, определяли с помощью высокоэффективной жидкостной хроматографии и изучали стабильность образца. Условия проведения ВЭЖХ перечисляются в следующем порядке:
ВЭЖХ: Waters 1525-717-2498
Хроматографическая колонка: АСЕ 3 AQ, 150×4,6 мм, 3 мкм Подвижная фаза: А: 1000 мл воды, 10 мл метанола, 100 мкл трифторуксусной кислоты
В: 600 мл воды, 400 мл метанола, 100 мкл трифторуксусной кислоты
(Используемые реагенты для ВЭЖХ, приобретены у TEDIA Company, Inc)
Скорость потока: 0,55 мл/мин
Температура колонки: 50°С
Градиент:

Температура инжектора: 5°С
Длина волны детектирования: 225 нм
Исследование устойчивости гидрата соединения формулы I
Гидрат соединения формулы I по изобретению является стабильным, подходит для промышленного производства, для транспортировки и хранения. С помощью теста на стабильность, авторы изобретения определили, что гидрат соединения формулы I, полученный способом по изобретению, имеет хорошую стабильность. Гидрат можно длительно хранить при 25°, что решает проблему его транспортировки (APIs).
После интенсивных исследований авторы настоящего изобретения дополнительно обнаружили, что лекарственная стабильность гидрата соединения формулы I тесно связана с содержанием влаги. При содержании влаги более 8,0 %, гидрат обладает хорошей стабильностью и его можно хранить при температуре 25°С в течение длительного срока.
При содержании влаги менее 8,0 %, продукт можно длительно хранить при температуре от 0 до 8°С с небольшим разложением продукта. Однако, при длительном хранении продукта при 25°С, разложение продукта будет значительным.
Применения
Настоящее изобретение предлагает применение гидрата соединения формулы I. В одном аспекте, он может быть применен для получения соединения формулы II. В литературе, например в патентах WO 9611210, WO 03018615 и WO 2004014879 описаны процессы синтеза.
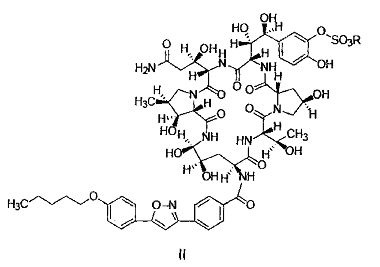
На основании приведенных выше результатов, настоящее изобретение дополнительно предоставляет фармацевтическую композицию, включающую гидрат соединения формулы I и фармацевтически приемлемый носитель.
Преимущества изобретения в основном включают в себя:
1. Путем повторных экспериментов авторы настоящего изобретения выбрали конкретные условия получения, и обнаружили неожиданные технические эффекты, так что представленный способ получения высоко-стабильного гидрата соединения формулы I пригоден для крупномасштабного производства.
2. Гидрат соединения формулы I обладает превосходной стабильностью, и значительно превосходит соединение формулы I, содержание воды в котором составляет менее 8,0%, а также соединение формулы I, полученное в предшествующем уровне техники.
3. Осуществление способа по изобретению является простым, и полученный гидрат с высокой стабильностью подходит для транспортировки и хранения, что снижает стоимость производства и приводит к непредсказуемым техническим эффектам.
Далее изобретение будет проиллюстрировано со ссылкой на следующие конкретные примеры. Следует понимать, что эти примеры предназначены только для иллюстрации изобретения, но не для ограничения объема изобретения.
Экспериментальные методы в следующих примерах выполнены в обычных условиях или в соответствии с инструкциями производителя. Если не указано иное, все проценты, соотношения пропорции или части даны по массе.
Единицы процентов вес/объем в изобретении, хорошо известны специалистам в данной области, например, вес растворенного вещества в 100 мл раствора.
Если не указано иное, все технические и научные термины, используемые здесь, имеют традиционное значение для понимания специалистом в данной области техники. Кроме того, любой процесс или материал, сходный или эквивалентный тем, которые описаны здесь, могут быть использованы в способе по настоящему изобретению. Предпочтительные варианты и материалы, описанные здесь, приведены лишь в качестве иллюстраций.
Пример 1
Получение соединения I
153 г соединения формулы I в твердом порошке было получено в соответствии со способом, описанным в примере 1 в патенте США № 5376634. Методом Карла Фишера было определено, что содержание влаги в соединении формулы I составляет 3,4%. Для изучения стабильности было взято 2,0 г полученного выше образца. Исследование проводили следующим образом: образец помещали в закрытую посуду и при температуре от 0 до 8°С на срок 6 месяцев, и при температуре 25°С на срок 6 месяцев, соответственно; и затем анализировали количество примесей. Первоначально, количество примесей в соединении формулы I составляло 2,4%; после инкубирования при температуре от 0 до 8°С в течение 6 месяцев, количество примесей в образце составило 3,0%; и после инкубирования при температуре 25°С в течение 6 месяцев, количество примесей в пробе составило 4,9%.
Пример 2
Получение гидратов А, В, С, D, и Е, содержащих соединение формулы I
При 50°С, 7,0 г твердого порошка соединения формулы I, полученного в примере 1, растворяли в смешанном растворе, состоящем из 10 мл воды и 8 мл н-пропанола, и перемешивали в течение 30 минут, до полного растворения соединения формулы I. pH доводили до 3,5 с помощью ледяной уксусной кислоты. Раствор охлаждали до 25°С и гидраты соединений формулы I осаждали.
Систему перемешивали в течение 5 часов при 25°С, так что гидрат соединения формулы I постепенно вырос. И затем медленно по каплям добавляли 36 мл н-пропанола и полученную систему перемешивали при 25°С в течение 2 часов.
Гидрат соединения формулы I получали фильтрованием. Гидрат соединения формулы I сушили под вакуумом при температуре от 20°С до 25°С в течение 1 часа, 1,0 г гидрата был отобран и назван гидрат А соединения формулы I, а содержание воды в гидрате А составило 29,5 масс. %.
Оставшийся образец дополнительно сушили в течение 0,5 ч. 1,0 г гидрата был отобран и назван гидрат В соединения формулы I, а содержание воды в гидрате В составило 27,1 масс. %. Оставшийся образец дополнительно сушили в течение 3 часов. 1,0 г гидрата был отобран и назван гидрат С соединения формулы I, а содержание воды в гидрате С составило 12,5 масс. %.
Оставшийся образец дополнительно сушили в течение 1,5 часов. 1,0 г гидрата был отобран и назван гидрат D соединения формулы I, а содержание воды в гидрате D составило 9,5 масс. %. P2O5 был помещен в вакуумную печь, а оставшийся образец дополнительно сушили в течение 2 часов. 1,0 г гидрата был отобран и назван гидрат Е соединения формулы I, а содержание воды в гидрате D составило 6,1 масс. %.
Полученные выше образцы были исследованы на устойчивость следующим образом: гидрат А, гидрат В, гидрат С, гидрат D, гидрат Е помещали в герметичные контейнеры на 6 месяцев при температуре от 0 до 8°С, и на 6 месяцев при температуре 25°С, соответственно; а затем анализировали количества примесей.
Характеристики паттерна ВЭЖХ (Фиг. 1) для гидрата D после инкубации при температуре 25°С в течение 6 месяцев, показаны в следующей таблице:
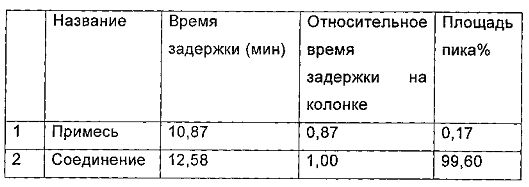
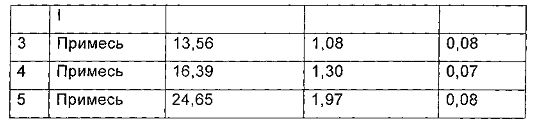

Пример 3
Получение гидратов F, G, Н, I, и J, содержащих соединение формулы I
При температуре 30°С, 16 г соединения формулы I, полученного в примере 1, растворяли в 90 мл воды и перемешивали в течение 2 часов до полного растворения соединения формулы I. pH доводили до 2,0 с помощью ледяной уксусной кислоты. Медленно по каплям добавляли 610 мл этанола, и гидраты соединений формулы I осаждали. Раствор охлаждали до 11°С и перемешивали при 11°С в течение 2 часов. Гидрат соединения формулы I получали фильтрованием.
Гидрат соединения формулы I высушивали под вакуумом при температуре от 20°С до 25°С в течение 0,5 часа. 1,0 г гидрата был отобран и назван гидратом F соединения формулы I, а содержание воды в гидрате F составило 31,2 масс. %. Оставшийся образец дополнительно сушили в течение 0,5 ч. 1,0 г гидрата был отобран и назван гидратом G соединения формулы I, а содержание воды в гидрате G составило 26,2 масс. %.
Оставшийся образец дополнительно сушили в течение 2 часов. 1,0 г гидрата был отобран и назван гидрат Н соединения формулы I, а содержание воды в гидрате Н составило 14,6 масс. %. Оставшийся образец дополнительно сушили в течение 2 часов. 1,0 г гидрата был взят и назван гидрат I соединения формулы I, а содержание воды в гидрате I составило 8,6 масс. %. P2O5 был помещен в вакуумную печь, а оставшийся образец дополнительно сушили в течение 1 часа. 1,0 г гидрата был отобран и назван гидрат J соединения формулы I, а содержание воды в гидрате J составило 7,2 масс. %.
Показатели стабильности приведены в следующей таблице:

Пример 4
Получение гидратов K, L, М, N и О, содержащих соединение формулы I
При 28°С, 18 г соединения формулы I, полученного в примере 1, растворяли в смешанном растворе, состоящем из 50 мл воды и 50 мл изопропанола, и перемешивали в течение 1 часа до полного растворения соединения формулы I. pH доводили до 3,6 с помощью ледяной уксусной кислоты. Раствор охлаждали до 17°С и гидраты соединений формулы I осаждали.
Систему дополнительно охлаждали до -10°С и перемешивали в течение более 2 часов. Гидрат соединения формулы I получали фильтрованием. Гидрат соединения формулы I сушили под вакуумом при температуре от 20°С до 25°С в течение 0,5 часа. 1,0 г гидрата был отобран и назван гидрат К соединения формулы I, а содержание воды в гидрате К составило 29,5 масс. %. Оставшийся образец дополнительно сушили в течение 0,5 ч. 1,0 г гидрата был отобран и назван гидрат L соединения формулы I, а содержание воды в гидрате L составило 27,5 масс. %.
Оставшийся образец дополнительно сушили в течение 3 часов. 1,0 г гидрата был отобран и назван гидрат М соединения формулы I, а содержание воды в гидрате М составило 19,8 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран и назван гидрат N соединения формулы I, а содержание воды в гидрате N составило 9,6 масс. %. Р2О5 помещали в вакуумную печь, а оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран и назван гидрат О соединения формулы I, а содержание воды в гидрате О составило 4,9 масс. %.
Показатели стабильности приведены в следующей таблице:

Пример 5
Подготовка гидратов Р, Q, R, S, и Т, содержащих соединение формулы I
При 25°С, 10,0 г соединения формулы I, полученного в примере 1, растворяли в смешанном растворе, состоящем из 40 мл воды и 64 мл метанола и перемешивали в течение 2 часов до полного растворения соединения формулы I. pH доводили до 3,5 с помощью ледяной уксусной кислоты. Медленно по каплям добавляли 300 мл метанола, и гидраты соединений формулы I осаждали. Гидрат соединения формулы I получали фильтрованием. Гидрат соединения формулы I сушили под вакуумом при температуре от 20°С до 25°С (перемешивали в течение 2 часов) в течение 0,5 часа.
1,0 г гидрата был отобран и назван гидрат Р соединения формулы I, а содержание воды в гидрате Р составило 31,3 масс. %. Оставшийся образец дополнительно сушили в течение 0,5 ч. 1,0 г гидрата был отобран и назван гидрат Q соединения формулы I, а содержание воды в гидрате Q составило 27,3 масс. %.
Оставшийся образец дополнительно сушили в течение 3 часов. 1,0 г гидрата был отобран и назван гидрат R соединения формулы I, а содержание воды в гидрате R составило 19,0 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран и назван гидрат S соединения формулы I, а содержание воды в гидрате S составило 9,0 масс. %. Р2О5 был помещен в вакуумную печь, а остальные образцы дополнительно сушили в течение 1 часа. 1,0 г гидрата был отобран и назван гидрат Т соединения формулы I, а содержание воды в гидрате Т составило 8 масс. %.
Показатели стабильности приведены в следующей таблице:


Пример 6
Получение гидратов U, V, W, X, и Y, содержащих соединение формулы I
При температуре 40°С, 15 г соединение формулы I, полученное в примере 1, растворяли в 50 мл воды, перемешивали до полного растворения соединения формулы I. pH доводили до 4,0 с помощью ледяной уксусной кислоты.
Раствор охлаждали до 22°С и гидраты соединений формулы I осаждали. Систему дополнительно охлаждали до 5°С и перемешивали в течение 10 часов при 5°С. Гидрат соединения формулы I получали фильтрованием. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа. 1,0 г гидрата был отобран, и назван гидрат U соединения формулы I, а содержание воды в гидрате U составило 42,0 масс. %. Остальные образцы дополнительно сушили в течение 2 часов. 1,0 г гидрата был отобран, и назван гидрат V соединения формулы I, а содержание воды в гидрате V составило 30,0 масс. %.
Оставшийся образец дополнительно сушили в течение 2 часов. 1.0 г гидрата был отобран, и назван гидрат W соединения формулы I, а содержание воды в гидрате W составило 19,5 масс. %.
P2O5 помещали в вакуумную печь, а оставшийся образец дополнительно сушили в течение 2 часов. 1,0 г гидрата был отобран, и назван гидрат X соединения формулы I, а содержание воды в гидрате X составило 9,6 масс. %.
Показатели стабильности приведены в следующей таблице:
Данные паттерна ВЭЖХ (Фиг. 2) для гидрата Y, помещенного при температуре 25°С на срок 6 месяцев, приведены в следующей таблице:
Пример 7
Получение гидратов а, b, и с содержащих соединение формулы I
При температуре 40°С, 15 г соединения формулы I, полученного в примере 1, растворяли в 50 мл воды, и перемешивали до полного растворения соединения формулы I. pH доводили до 5,0 с помощью ледяной уксусной кислоты. Раствор охлаждали до 22°С и гидрат соединения формулы I осаждали. Медленно добавляли 150 мл этанола и перемешивали в течение 2 часов. Гидрат соединения формулы I получали фильтрованием. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа.
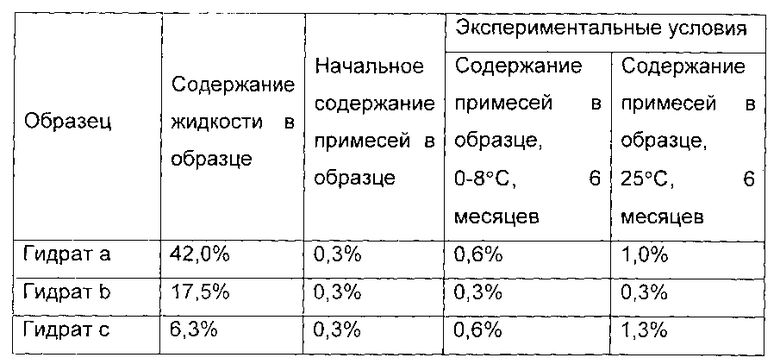
1.0 г гидрата был отобран и назван гидрат а соединения формулы I, а содержание воды в гидрате а составило 42,0 масс. %. Оставшийся образец дополнительно сушили в течение 3,5 часов. 1,0 г гидрата был отобран и назван гидрат b соединения формулы I, а содержание воды в гидрате b составило 17,5 масс. %. Р2О5 помещали в вакуумную печь, а остальные образцы дополнительно сушили в течение 3 часов. 1,0 г гидрата был отобран, и назван гидрат с соединения формулы I, а содержание воды в гидрате с составило 6,3 масс. %.
Показатели стабильности показаны в следующей таблице:
Пример 8
Получение гидратов d, е, и f, содержащих соединение формулы I
При температуре 20°С, 12 г соединения формулы I, полученного в примере 1, растворяли в 40 мл воды, и перемешивали до полного растворения соединения формулы I. pH доводили до 4,5 с помощью ледяной уксусной кислоты. Медленно добавляли 180 мл н-пропанола и перемешивали в течение 2 часов, и гидраты соединений формулы I осаждали. Гидрат соединения формулы I получали фильтрованием. Систему перемешивали в течение более чем 2 часов. Гидрат соединения формулы I получали фильтрованием.
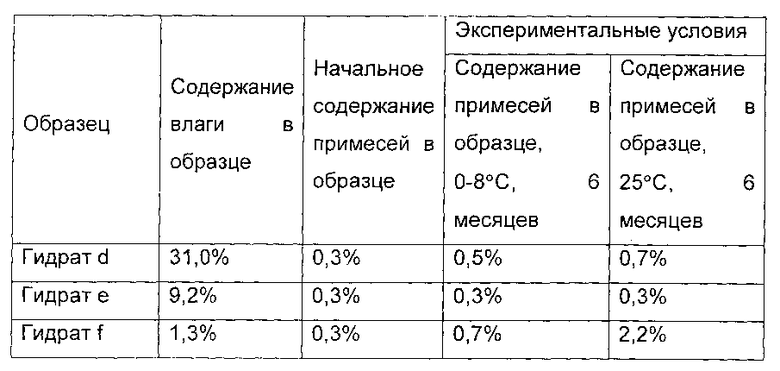
Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа. 1,0 г гидрата был отобран, и назван гидрат d соединения формулы I, а содержание воды в гидрате d составило 31,0 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат е соединения формулы I, а содержание воды в гидрате составило 9,2 масс. %. Р2О5 помещали в вакуумную печь, а остальные образцы дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат f соединения формулы I, и содержание воды в гидрате f составило 1,3 масс. %.
Показатели стабильности приведены в следующей таблице:
Из приведенных выше примеров можно заключить, что гидраты соединений формулы I, содержание влаги в которых составляет от 8,0 до 30 масс. %, будут обладать превосходной стабильностью. Если содержание влаги в гидрате соединения формулы I, более 30% или менее 8,0%, их стабильность будет значительно ниже.
Пример 9
Получение гидратов g, h, и l, содержащих соединение формулы I (действие pH)
В 30°С, 12 г соединения I, полученного в примере 1, растворяли в 60 мл воды, и перемешивали до полного растворения соединения. pH доводили до 1,8 с помощью ледяной уксусной кислоты. 200 мл этанола медленно добавляли, и соединение I выпадало в осадок.
Систему перемешивали в течение еще 1 часа, и фильтровали. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа. 1,0 г гидрата был отобран и назван гидрат g соединения формулы I, а содержание воды в гидрате г составило 36,0 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат h соединения формулы I, а содержание воды в гидрате h составило 14,5 масс. %.
P2O5 помещали в вакуумную печь, а остальные образцы дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат I соединения формулы I, а содержание воды в гидрате I составило 6,3 масс. %.
Показатели стабильности приведены в следующей таблице:

Пример 10
Получение гидратов j, k, и I, содержащих соединение формулы I (действие pH)
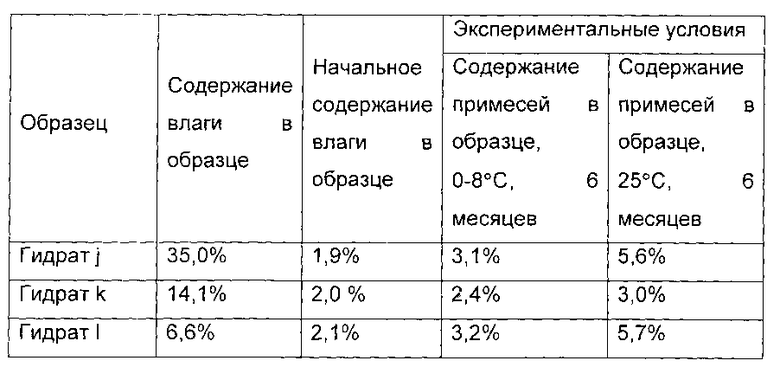
В 30°С, 12 г соединения I, полученного в примере 1, растворяли в 60 мл воды, и перемешивали до полного растворения соединения. pH доводили до 5,4 с помощью ледяной уксусной кислоты. 200 мл этанола медленно добавляли, и соединение формулы I осаждали. Систему перемешивали в течение еще 1 часа и фильтровали. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа. 1,0 г гидрата был отобран, и назван гидрат j соединения формулы I, а содержание воды в гидрате] составило 35,0 масс. %.
Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат к соединения формулы I, а содержание воды в гидрате к составило 14,1 масс. %. P2O5 был помещен в вакуумную печь, а остальные образцы дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат I соединения формулы I, а содержание воды в гидрате I составило 6,6 масс. %.
Показатели стабильности приведены в следующей таблице:
Пример 11
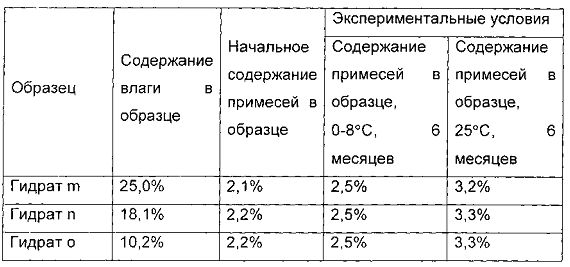
Получение гидратов m, n, и о, содержащих соединение формулы I (действие растворителя)
При температуре 20°С, 4,8 г соединения I, полученного в примере 1, растворяли в 14 мл воды. pH доводили до 4,0 с помощью ледяной уксусной кислоты. Полученную систему перемешивали в течение 2 часов до полного растворения соединения I.
Медленно добавляли 35 мл ацетонитрила и перемешивали в течение 2 часов, и твердое вещество осаждали. Систему перемешивали в течение еще 2 часов, и фильтровали. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа.
1,0 г гидрата был отобран, и назван гидрат m соединения формулы I, а содержание воды в гидрате m составило 25,0 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат n соединения формулы I, а содержание воды в гидрате n составило 18,1 масс. %. P2O5 был помещен в вакуумную печь, а оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат о соединения формулы I, а содержание воды в гидрате о составило 10,2 масс. %.
Показатели стабильности приведены в следующей таблице:
Пример 12
Получение гидратов р, q и r, содержащих соединение формулы I (действие растворителя)
При температуре 18°С, 4,2 г соединения I, полученного в примере 1, растворяли в 14 мл воды. pH доводили до 4,0 с помощью ледяной уксусной кислоты. Полученную систему перемешивали в течение 1 часа до полного растворения соединения I. 40 мл ацетона медленно добавляли и перемешивали в течение 2 часов, и твердое вещество осаждали.
Систему перемешивали в течение еще 2 часов, и фильтровали. Гидрат соединения формулы I сушили в вакууме при температуре от 20°С до 25°С в течение 1 часа. 1,0 г гидрата был отобран, и назван гидрат р соединения формулы I, а содержание воды в гидрате р составило 23,8 масс. %. Оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат q соединения формулы I, а содержание воды в гидрате q составило 15,6 масс. %. P2O5 помещали в вакуумную печь, а оставшийся образец дополнительно сушили в течение 4 часов. 1,0 г гидрата был отобран, и назван гидрат г соединения формулы I, а содержание воды в гидрате г составило 7,6 масс. %.
Показатели стабильности приведены в следующей таблице:
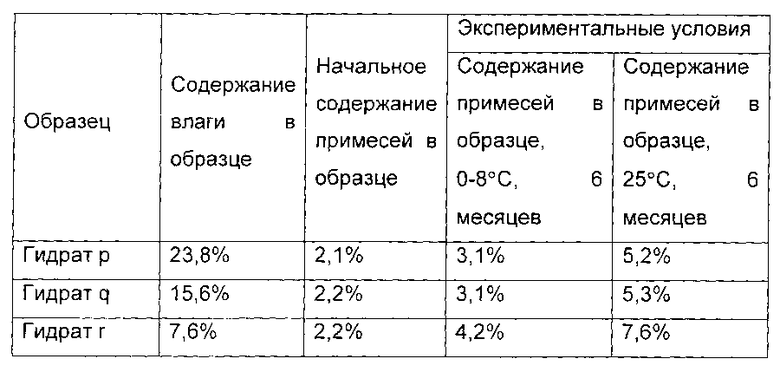
Из приведенных выше примеров можно заключить, что pH и растворитель, будут существенно влиять на стабильность гидрата. Если не поддерживать рН на уровне от 2,0 до 5,0, и использовать растворитель, отличный от тех, что использованы в настоящем изобретении, стабильность гидрата будет существенно ниже. Тем не менее, даже в указанных выше условиях, гидрат соединения формулы I, содержание влаги в котором составляет от 8,0 до 30 масс. %, будет иметь более высокую стабильность по сравнению с гидратом соединения формулы I, содержание влаги в котором более 30 масс. % или менее 8,0 масс. %.
Пример 13
Получение соединения формулы II
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидрат соединения формулы I, полученный в примере 2 настоящей заявки (1,07 ммоль, 1,00 г) растворяли в 12 мл ДМФА (М,1Ч-Диметилформамид). Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), и поддерживали температуру 0°С. МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль) медленно добавляли и нагревали реакционную смесь до 2-6°С, и выдерживали в течение 4 часов.
60 мл этилацетата добавляли непосредственно в реакционную смесь в конце реакции, перемешивали в течение еще 1 часа, и фильтровали, с получением микафунгин диизопропилэтиламина. Соль растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота микафунгин диизопропилэтиламина составляля 99,35% и выход составил 91,9%.
Пример 14
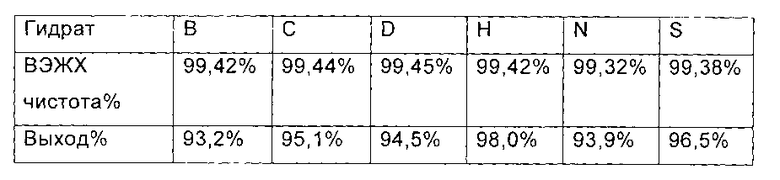
Получение соединения формулы II из гидратов В, С, D, Н, N, S соединения формулы I
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидраты В, С, D, Н, N и S соединения формулы I, полученного в примере 2, примере 3, в примере 4, примере 5 настоящего изобретения (1,07 ммоль, 1,00 г) растворяли в 12 мл ДМФА, соответственно. Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), соответственно, и поддерживали температуру 0°С.
Медленно добавляли МКС-8
(1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бен зотриазол) (0.53 г, 1.14 ммоль) и реакционную смесь нагревали до 2-6°С, и выдерживали в течение 4 часов. 60 мл этилацетата добавляли непосредственно в каждую реакционную жидкость в конце реакции, перемешивали в течение еще 1 часа, и фильтровали с получением Микафунгин диизопропилэтиламина. Соль, полученную выше, растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота и выход Микафунгин диизопропилэтиламина, приведены в следующей таблице:
Сравнительный пример 1
Получение соединения формулы II из гидрата соединения формулы I с содержанием влаги менее 8%
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидраты Е, J, О, Y, С и F соединения формулы I, полученного в примере 2, примере 3, примере 4, примере 6 и примере 8 настоящего изобретения (1,07 ммоль, 1,00 г) растворяли в 12 мл ДМФ, соответственно. Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом.
Добавляли диизопропилэтиламин (0.22 г, 1.67 ммоль), соответственно, и температуру поддерживали на уровне 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил) изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль), реакционную смесь нагревали до 2-6°С и выдерживали в течение 4 часов. 60 мл этилацетата добавляли в каждую реакционную смесь по окончании реакции, перемешивали в течение еще 1 часа и фильтровали с получением Микафунгин диизопропилэтиламина.
Соль растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота и выход Микафунгин диизопропилэтиламина, приведены в следующей таблице:
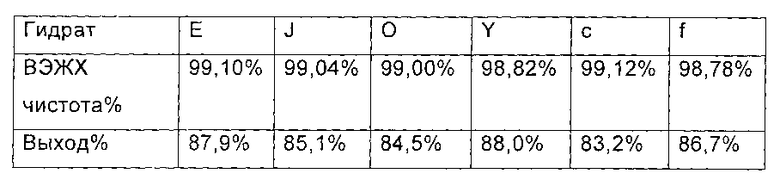
На основании приведенных выше сравнительных примеров, можно сделать вывод, что ВЭЖХ чистота и выход соединения формулы II снижались, когда был использован гидрат соединения формулы I, содержащий менее 8% влаги.
Сравнительный пример 2
Получение соединения формулы II из гидрата соединения формулы I с содержанием влаги более 30%
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидраты F, Р, U, a, d и g соединения формулы I, полученного в примере 3, примере 5, примере 6, примере 7, примере 8 и примере 9 настоящего изобретения (1,07 ммоль, 1,00 г) растворяли в 12 мл ДМФ, соответственно. Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом.
Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), соответственно, и поддерживали температуру на уровне 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил) изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль) и реакционную смесь нагревали до 2-6°С, и выдерживали в течение 4 часов. 60 мл этилацетата добавляли непосредственно в каждую реакционную смесь в конце реакции, перемешивали в течение еще 1 часа, и фильтровали с получением Микафунгин диизопропилэтиламина. Соль растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота и выход Микафунгин диизопропилэтиламина, приведены в следующей таблице:
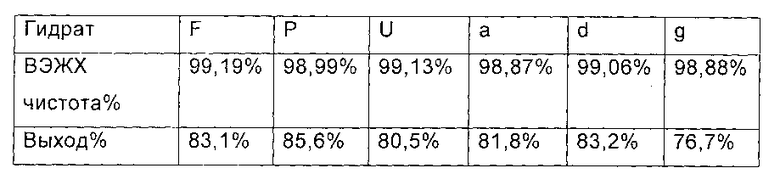
Из приведенных выше сравнительных примеров можно заключить, что ВЭЖХ чистота и выход соединения формулы II снижались, когда был использован гидрат соединения формулы I, содержащий более 30% влаги.
Сравнительный пример 3
Получение соединения формулы II из гидрата соединения формулы I из примера 1
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидрат соединения формулы I, полученного в примере 1 (1.07 ммоль, 1.00 г) растворяли в 12 мл ДМФА, соответственно. Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом. Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль) соответственно, и поддерживали температуру на уровне 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил)изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль) и реакционную смесь нагревали до 2-6°С, и выдерживали в течение 4 часов.
60 мл этилацетата добавляли непосредственно в реакционную смесь в конце реакции, перемешивали в течение еще 1 часа, и фильтровали с получением Микафунгин диизопропилэтиламина. Соль растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота Микафунгин диизопропилэтиламина составила 95,7%, а выход 75,2%.
Из приведенных выше сравнительных примеров, можно заключить, ВЭЖХ чистота и выход соединения формулы II значительно снижались, когда использовали гидрат соединения формулы I из примера 1.
Пример 15
Получение соединения формулы II из гидрата соединения формулы I
Соединение формулы II синтезировали из соединения формулы I способом, использованным для синтеза Микафунгина в патенте WO 2004014879.
Гидраты h, i, j, k, q и r соединения формулы I, полученного в примере 9, примере 10 и примере 12 настоящей заявки (1,07 ммоль, 1,00 г) растворяли в 12 мл ДМФА, соответственно. Полученный раствор охлаждали до температуры ниже 0°С в бане со льдом.
Добавляли диизопропилэтиламин (0,22 г, 1,67 ммоль), соответственно, и поддерживали температуру на уровне 0°С. Медленно добавляли МКС-8 (1-[4-[5-(4-пентилоксифенил) изоксазол-3-ил]бензоилокси]-1Н-1,2,3-бензотриазол) (0,53 г, 1,14 ммоль) и реакционную смесь нагревали до 2-6°С, и выдерживали в течение 4 часов.
60 мл этилацетата добавляли непосредственно в каждую реакционную жидкость в конце реакции, перемешивали в течение еще 1 часа, и фильтровали с получением Микафунгин диизопропилэтиламина. Соль растворяли в 30 мл ацетона и 30 мл этилацетата, крахмалили и фильтровали. Микафунгин диизопропилэтиламин сушили в вакууме, чтобы удалить остаточный органический растворитель. ВЭЖХ чистота и выход Микафунгин диизопропилэтиламина приведены в следующей таблице:
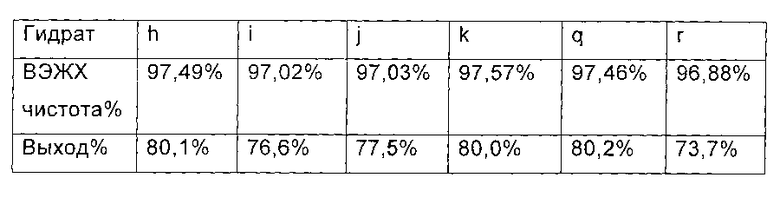
Из приведенных выше сравнительных примеров можно заключить, что ВЭЖХ чистота и выход соединения формулы II значительно снижались, когда использовали гидрата соединения формулы I. Однако, по сравнению с гидратом соединения формулы I , содержание влаги в котором находилось вне границ интервала от 8.0 до 30%, ВЭЖХ чистота и выход соединения формулы II, полученного из гидрата соединения формулы I, содержание влаги, в котором поддерживалось в пределах от 8,0% до 30 %, были лучше.
Пример 16
Получение фармацевтической композиции

20 г лактозы растворяли в чистой воде (200 мл) при температуре ниже 50°С. После охлаждения ниже 20°С, в раствор лактозы добавляли 2,5 г гидрата В, содержащего соединение формулы I, полученное в примере 2.
Полученный раствор осторожно перемешивали, чтобы избежать пузырьков. Добавляли 2% водный раствор лимонной кислоты (0,95 мл), а затем в раствор добавляли 0,4% водный NaOH (примерно 24 мл) для доведения pH до 5,5.
И затем полученный раствор разбавляли чистой водой до получения заданного объема (250 мл). Полученный раствор разливали в 100 ампул (объем которых составляет 10 мл), по 2,5 мл в каждую. Раствор в каждой ампуле лиофилизировали традиционным способом с использованием лиофилизатора с получением лиофилизированных композиций, каждая из которых содержала 25 мг гидрата, содержащего соединение формулы I.
Приведенные выше примеры являются лишь предпочтительными примерами осуществления настоящего изобретения, не могут быть использованы для ограничения объема изобретения. Существенное техническое содержание изобретения определено в широком смысле в формуле изобретения. Любые компоненты или способы, использованные другими, следует рассматривать как эквиваленты, попадающие в объем изобретения, как определено в формуле изобретения, если указанные объекты или способы являются теми же, как те, которые определены в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛОПЕПТИДНОЕ СОЕДИНЕНИЕ ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2603345C2 |
| КРИСТАЛЛ ЦИКЛОПЕПТИДА ВЫСОКОЙ ЧИСТОТЫ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2607083C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ И ПРОИЗВОДНЫХ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2650687C1 |
| СПОСОБ ОЧИСТКИ ЦИКЛОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2538274C2 |
| СПОСОБ ОЧИСТКИ ЦИКЛОЛИПОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2535489C1 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2790017C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИ-5-[2-(4-(ТРИФТОРМЕТИЛФЕНИЛ)ЭТИЛАМИНО)]БЕНЗОЙНОЙ КИСЛОТЫ | 2021 |
|
RU2836445C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 5-[2-[7-(ТРИФТОРМЕТИЛ)-5-[4-(ТРИФТОРМЕТИЛ)ФЕНИЛ]ПИРАЗОЛО[1,5-a]ПИРИМИДИН-3-ИЛ]ЭТИНИЛ]-2-ПИРИДИНАМИНА | 2012 |
|
RU2630700C2 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В ПОЛУЧЕНИИ C5aR АНТАГОНИСТОВ | 2015 |
|
RU2712233C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРАЗИКВАНТЕЛА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2671202C1 |


Изобретение относится к гидрату соединения формулы I, где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль, при этом содержание воды в гидрате от 8 до 30 мас.%. Гидрат обладает высокой стабильностью. Также описаны способ получения гидрата и его применение. 6 н. и 5 з.п. ф-лы, 7 ил., 16 пр.
1. Гидрат соединения формулы I, где R представляет собой Н или катион, способный образовывать фармацевтически приемлемую соль; при этом содержание воды в гидрате составляет от 8 до 30 мас.%
2. Гидрат по п. 1, где содержание воды составляет от 9,5 до 28,0 мас.%.
3. Гидрат по п. 1 или 2, получаемый посредством следующих стадий:
(a) растворение соединения формулы I в воде или водорастворимом органическом растворителе (i), и регулирование pH раствора, содержащего соединение формулы I;
(b) получение гидрата, содержащего соединение формулы I, путем снижения температуры и/или добавления водорастворимого органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка гидрата, полученного на стадии (с), и регулирование содержания воды в массовых процентах в твердом веществе в указанном диапазоне.
4. Гидрат по п. 3, где указанный органический растворитель (i) выбран из С1-С4 низших спиртов.
5. Гидрат по п. 4, в котором низший спирт представляет собой один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
6. Способ получения гидрата соединения формулы I по п. 1, включающий следующие этапы:
(a) растворение соединения формулы I в воде или водорастворимом органическом растворителе (i), и регулирование pH раствора, содержащего соединение формулы I;
(b) получение гидрата, содержащего соединение формулы I, путем снижения температуры и/или добавления водорастворимого органического растворителя (i);
(c) получение гидрата центрифугированием или фильтрованием;
(d) вакуумная сушка гидрата, полученного на стадии (с), и регулирование содержания воды в массовых процентах в твердом веществе в указанном диапазоне; где указанный органический растворитель (i) выбран из С1-С4 низших спиртов.
7. Способ получения по п. 6, где низший спирт представляет собой один или несколько спиртов, выбранных из группы, состоящей из метанола, этанола, н-пропанола и изопропанола.
8. Применение гидрата по любому из пп. 1-5 для получения соединения формулы II
9. Применение гидрата по любому из пп. 1-5 для получения лекарственных средств, для лечения грибковых инфекций.
10. Фармацевтическая композиция для лечения грибковых инфекций, содержащая гидрат по любому из пп. 1-5 и фармацевтически приемлемый носитель.
11. Способ получения фармацевтической композиции по п. 10, включающий смешивание гидрата по любому из пп. 1-5 с фармацевтически приемлемым носителем, в результате чего получают фармацевтическую композицию по п. 10.
| ЦИКЛОПЕПТИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2108342C1 |
| СПОСОБ ОЧИСТКИ ЦИКЛОЛИПОПЕПТИДНЫХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕЙ | 2011 |
|
RU2535489C1 |

