Настоящая заявка испрашивает приоритет согласно §119(a) главы 35 Свода законов США на основании предварительной заявки на европейский патент №10195675.3, поданной 17 декабря 2010 г., содержание которой полностью включено в настоящее описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
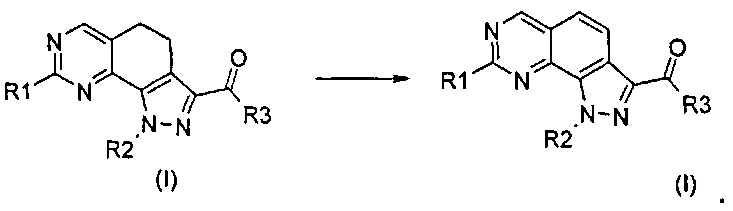
Настоящее изобретение относится к некоторым замещенным пиразоло[4,3-h]хиназолиновым соединениям, которые модулируют активность ингибиторов протеинкиназ и, в частности, Pim киназы (Pim-1, Pim-2 и/или Pim-3). Вследствие этого соединения согласно настоящему изобретению подходят для применения при лечении заболеваний, вызванных нарушением регуляции активности протеинкиназы. В настоящем изобретении также предложены способы получения указанных соединений, фармацевтические композиции, содержащие указанные соединения, и способы лечения заболеваний с применением фармацевтических композиций, содержащих указанные соединения.
УРОВЕНЬ ТЕХНИКИ
Нарушение функции протеинкиназ (PKs) является признаком многих заболеваний. Значительная часть онкогенов и протоонкогенов, вовлеченных в развитие злокачественных новообразований у человека, кодирует PKs. Повышенная активность PKs также наблюдается при многих доброкачественных заболеваниях, таких как доброкачественная гиперплазия предстательной железы, наследственный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток сосудов, обусловленная атеросклерозом, фиброз легких, артрит гломерулонефрит и послеоперационный стеноз и рестеноз.
PKs также принимают участие в воспалительных процессах и в размножении вирусов и паразитов. PKs также могут играть важную роль в патогенезе и развитии нейродегенеративных заболеваний.
В отношении общих сведений о нарушении работы или дерегуляции PKs см., например, Current Opinion in Chemical Biology 1999, 3, 459-465 и Carcinogenesis 2008, 29, 1087-1091.
Первоначально определенные как гены, активированные посредством провирусного мутагенеза в лимфоме модельной мыши, PIMs (PIM1, PIM2 и/или PIM-3 на всем протяжении настоящей заявки) представляют собой серин/треониновые протеинкиназы. PIM киназы слабо экспрессированы в нормальных тканях, но сверхэкспрессированы или даже мутированы в отдельных видах злокачественных новообразований у человека, включая лимфому, лейкемию, рак предстательной железы, поджелудочной железы и желудка [Shah с соавт., Eur. J. Cancer, 44, 2144-51, (2008)].
PIM киназы постоянно активны, и их активность способствует росту и выживанию клеток новообразований in vitro и in vivo посредством модификации растущего количества обычных, а также изоформ-специфичных субстратов, включая различные регуляторы клеточного цикла и медиаторы апоптоза. Похоже, что PIM1, но не PIM2, также служит медиатором хоуминга и миграции нормальных и злокачественных гемопоэтических клеток путем регуляции поверхностной экспрессии хемокинового рецептора [Brault с соавт., Haematologica, 95, 1004-1015 (2010)].
Существует все больше доказательств того, что PIM1 и PIM2 киназы могут быть вовлечены в медиацию онкогенного действия некоторых онкогенов, вызывающих острые миелогенные лейкозы (ОМЛ). В частности, онкогенная роль мутаций FLT3 (ITD и KD мутаций, присутствующих в 30% ОМЛ-ов) и/или транслокаций, вовлекающих ген MLL (происходящих в 20% ОМЛ-ов), (Kumar с соавт., J. Mol. Biol. 348, 183-193 (2005)). PIM1 более экспрессирован в FLT3-ITD трансформированных клетках ОМЛ, чем в клетках костного мозга ДТ. Данные свидетельствуют о том, что ингибирование PIM1, а также PIM2, может способствовать FLTS-ITD-зависимой гибели клеток ОМЛ. Интересно, что клетки, трансформированные посредством мутаций FLT3, что придает устойчивость к низкомолекулярным ингибиторам тирозинкиназы, по-прежнему были чувствительны к нокдауну PIM2 или PIM-1 и PIM-2 посредством РНК-интерференции (Kim с соавт. (2005) Blood 105: 1759-67).
Кроме того, сообщалось о сверхэкспрессии PIM2 и его связи с развитием различных злокачественных новообразований, которые происходят из линии B-клеток, таких как хронический лимфолейкоз (ХЛЛ), диффузная B-крупноклеточная лимфома (ДВКЛ), лимфома из клеток мантии (ЛКМ) или миелома (Cohen с соавт. (2004) Leuk. Lymphoma 94:51; Huttmann с соавт (2006) Leukemia 20 1774).
Интересно, что PIM и AKT/PKB, по-видимому, играют частично совпадающие роли в медиации роста и выживания гемопоэтических клеток, скорее всего, из-за перекрывания субстратов, подобных BAD, p21WAF1/CIP1, p27KIP1 или Cot/Tpl-2 [Choudhary с соавт., Mol Cell. 36 326-39 (2009)].
Было показано, что PIM киназы контролируют устойчивость к ингибированию mTOR (рапамицину), пролиферацию и выживание. Следовательно, комбинация низкомолекулярных ингибиторов, воздействующих на различные киназы, регулирующие выживаемость клеток, может иметь большое значение для создания мощной терапевтической платформы для лечения рака [Amaravadi R. с соавт. J. Clin. Invest., 2005, 115(10): 2618-24]. Синтез онкогенного белка через elF4E связывающий белок 1 (4Е-BP1), похоже, не зависит от mTOR и контролируется PIM-2. Указанные результаты показывают, что онкогенный комплекс elF4F, инициирующий трансляцию, может быть блокирован с применением низкомолекулярных ингибиторов PIM-2 [Tamburini J. с соавт., Blood, 2009, 114(8), 1718-27 и Brault L. с соавт., Haematologica, 2010, 95(6) 1004-1015].
Пиразолхиназолиновые производные, обладающие активностью, направленной на ингибирование киназы, также были описаны в WO 04/104007 на имя Pharmacia Italia S.P.A. Некоторые конкретные соединения вышеуказанной заявки WO 04/104007 исключены из настоящей общей формулы.
Несмотря на указанные достижения, в данной области по-прежнему существует необходимость в эффективных агентах для лечения указанных заболеваний.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении обнаружен новый класс замещенных пиразол[4,3-h]хиназолиновых соединений, обладающих более высокой активностью по сравнению с известными соединениями. Было обнаружено, что указанные соединения в неожиданно низкой концентрации способны препятствовать пролиферации опухолевых клеток человека, тем самым максимально увеличивая противоопухолевую эффективность при одновременном снижении риска возникновения побочных эффектов, связанных с введением более высоких количеств лекарственных средств.
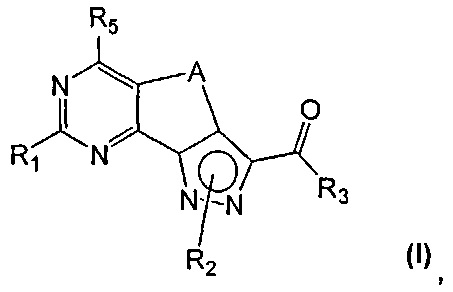
Новые соединения имеют структуру, представленную формулой (I)
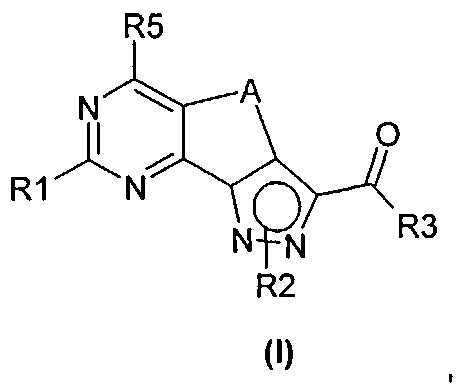
где
R1 представляет собой водород, CN, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила,
C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила или группы XR4, где
X представляет собой двухвалентный радикал, выбранный из O, S, SO, SO2 и NR6, где
R6 представляет собой необязательно замещенный линейный или разветвленный C1-C6алкил или R6 и R4 совместно с атомом азота, с которым они связаны, могут образовать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
R2 представляет собой водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, линейного или разветвленного C2-C6алкенила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R3 представляет собой группу, выбранную из NR'Rʺ и N(OH)R', где
R' и Rʺ независимо друг от друга представляют собой водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R4 представляет собой группу, выбранную из необязательно замещенного линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R5 представляет собой водород или NR'Rʺ, где
R' и Rʺ независимо друг от друга представляют собой водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или совместно с атомом азота, с которым они связаны, R' и Rʺ могут образовать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
A представляет собой двухвалентную группу, выбранную из -(CH2)2- и -CH=CH-, и
их фармацевтически приемлемые соли, за исключением:
1-метил-8-(пиперидин-1-ил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида
и 1-метил-8-(метилсульфонил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида.
В настоящем изобретении также предложены способы синтеза замещенных пиразоло[4,3-h]хиназолиновых соединений, представленных формулой (I), полученных способом, состоящим из стандартных синтетических трансформаций, и изомеров, таутомеров, гидратов, сольватов, комплексов, метаболитов, пролекарств, носителей, N-оксидов.
В настоящем изобретении также предложен способ лечения заболеваний, вызванных и/или связанных с нарушением регуляции активности протеинкиназ, в частности, человеческих PIM-1, PIM-2, PIM-3, Flt-3, c-Kit, MPS1 (TTK), представителей семейства PLK, протеинкиназы C в различных изоформах, Met, PAK-4, PAK-5, PERK, STLK-2, DDR-2, Aurora 1, Aurora 2, Bub-1, Chk1, Chk2, HER2, C-raf, B-raf raf1, Melk, PDK1, MEK1, MAPK, EGF-R, PDGF-R, FGF-R, IGF-R, PI3K, киназы weel, Src, Abl, Akt, MAPK, ILK, MK-2, IKK-2, Cdc7, Nek, семейства киназ Cdk/циклин, конкретнее PIM-1, PIM-2, PIM-3 человека, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества замещенного пиразоло[4,3-h]хиназолинового соединения, представленного формулой (I), определенной выше.
Способ согласно настоящему изобретению предназначен для лечения заболевания, вызванного и/или связанного с нарушением регуляции активности протеинкиназы, выбранного из группы, состоящей из злокачественного новообразования, клеточных пролиферативных нарушений и заболеваний и нарушений, вызванных иммунными клетками.
Другой способ согласно настоящему изобретению предназначен для лечения конкретных видов злокачественных новообразований, включая, но не ограничиваясь ими, карциному, такую как рак мочевого пузыря, молочной железы, толстой кишки, почек, печени, легкого, включая мелкоклеточный рак легкого, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы и кожи, включая сквамозную карциному; гемобластозы из клеток лимфоидной линии, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гемобластозы из клеток миелоидной линии, включая острый и хронический миелогенный лейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому нейробластому, глиому и шванномы; другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы, саркому Калоши и мезотелиому, и другие.
Другой способ согласно настоящему изобретению предназначен для лечения конкретных клеточных пролиферативных нарушений, таких как, например, доброкачественная гиперплазия предстательной железы, семейный аденоматоз толстой кишки, нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток сосудов, обусловленная атеросклерозом, фиброз легких, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
Другой способ согласно настоящему изобретению предназначен для лечения заболеваний и нарушений, вызванных иммунными клетками, таких как воспалительные и аутоиммунные заболевания, например, рассеянный склероз, системная красная волчанка, воспалительные заболевания кишечника (ВЗК), болезнь Крона, синдром раздраженного кишечника, панкреатит, неспецифический язвенный колит, дивертикулез, миастения, васкулит, псориаз, склеродермия, бронхиальная астма, аллергия, системный склероз, витилиго, артрит, такой как остеоартрит, ювенильный ревматоидный артрит, анкилозирующий спондилоартрит.
Кроме того, способ согласно настоящему изобретению также обеспечивает ангиогенез опухоли и ингибирование метастаз, а также лечение отторжения пересаженных органов и реакции «трансплантат против хозяина».
В настоящем изобретении также предложена фармацевтическая композиция, содержащая одно или более соединений формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель, носитель или разбавитель.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I), в комбинации с известными видами противоопухолевой терапии, такими как лучевая терапия или режим химиотерапии в комбинации с цитостатическими или цитотоксическими агентами, агентами, представляющими собой антибиотики, алкилирующими агентами, антиметаболитными агентами, гормональными агентами, иммунологическими агентами, агентами, представляющими собой интерфероны, ингибиторами циклооксигеназы (например, ингибиторами ЦОГ-2), ингибиторами матриксной металлопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, агентами против рецепторов факторов роста, агентами против HER, агентами против EGFR, агентами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути сигнальной трансдукции ras-raf, ингибиторами клеточного цикла, другими ингибиторами cdks, агентами, связывающими тубулин, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и т.п.
В настоящем изобретении также предложен способ ингибирования активности протеинкиназы PIM-1, PIM-2, PIM-3 in vitro, который включает приведение указанной киназы в контакт с эффективным количеством соединения формулы (I), определенной выше.
Дополнительно в настоящем изобретении предложен продукт или набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, определенную выше, или его фармацевтические композиции и один или более химиотерапевтических агентов, в качестве комбинированного лекарственного средства для одновременного, раздельного или последовательного применения в противоопухолевой терапии.
В другом аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, определенная выше, для применения в качестве лекарственного средства.
Кроме того, в настоящем изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, определенной выше, в производстве лекарственного средства с противоопухолевой активностью.
Наконец, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, определенная выше, для применения в способе лечения злокачественного новообразования.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фигуре 1 представлены превращения соединений I-VIII, включая получение соединения формулы (I), где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; a R2, R3, R4, R5, R', Rʺ и A определены выше.
На Фигуре 2 представлены уровни pBAD (s112), полученные посредством вестерн-блоттинга, который показывает дозозависимое снижение количества белка BAD (фосфо-BAD) в MV-4-11 клетках (линия лейкозных клеток человека, содержащих FLT3/ITD мутацию) при обработке соединением формулы (I), представленным соединением 85, по сравнению с клетками, обработанными ДМСО (контрольный первый ряд).
ПОДРОБНОЕ ОПИСАНИЕ ПРИМЕРНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Далее будут приведены подробные ссылки на некоторые варианты реализации настоящего изобретения, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Несмотря на то, что изобретение будет описано в сочетании с приведенными вариантами реализации, следует понимать, что указанные варианты не предназначены для ограничения настоящего изобретения. Напротив, изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, определенный его формулой. Специалист в данной области обнаружит множество способов и материалов, аналогичных или эквивалентных описанным в настоящей заявке, которые могут быть применены при практической реализации настоящего изобретения. Настоящее изобретение никоим образом не ограничивается описанными способами и материалами. В случае, если один или более включенных источников, патентов и аналогичных материалов отличаются от или противоречат указанной заявке, включая, но не ограничиваясь ими, термины, срок применения, описанные технологии и т.п., указанная заявка имеет преимущественную силу. Если не указано иное, все технические и научные термины, используемые в настоящей заявке, означают то же, что обычно понимает под ними специалист в области, к которой относится настоящее изобретение. Несмотря на то, что при практической реализации или испытании настоящего изобретения могут быть применены способы и материалы, аналогичные или эквивалентные описанным в настоящей заявке, подходящие способы и материалы описаны ниже. Все публикации, патентные заявки, патенты и другие источники, упомянутые в настоящей заявке, полностью включены в настоящее описание посредством ссылок. Номенклатура, используемая в настоящей заявке, основана на систематической номенклатуре IUPAC, если не указано иное.
Если не указано иное, когда речь идет о соединениях формулы (I) самих по себе, а также о любой фармацевтической композиции указанных соединений или о любом терапевтическом воздействии, включающем применение указанных соединений, настоящее изобретение включает все изомеры, таутомеры, гидраты, сольваты, комплексы, метаболиты, пролекарства, носители, N-оксиды и фармацевтически приемлемые соли соединений согласно настоящему изобретению.
Другими словами, в случае легкости получения из соединений формулы (I), определенной выше, изомеры, таутомеры, гидраты, сольваты, комплексы, метаболиты, пролекарства, носители и N-оксиды указанных соединений также являются объектом настоящего изобретения.
Метаболит соединения формулы (I) представляет собой любое соединение, в которое указанное соединение формулы (I) превращается in vivo, например, при введении млекопитающему, нуждающемуся в этом. Как правило, что, однако, не является ограничивающим примером, при введении соединения формулы (I) указанное производное может превратиться в различные соединения, включая, например, более растворимые производные, такие как гидроксилированные производные, которые легко выводятся из организма. Следовательно, в зависимости от пути, по которому таким образом происходит метаболизм, любое из указанных гидроксилированных производных можно рассматривать в качестве метаболита соединений формулы (I).
Пролекарства представляют собой любые соединения с ковалентными связями, которые высвобождают in vivo активное исходное лекарственное средство согласно формуле (I).
N-оксиды представляют собой соединения формулы (I), где азот и кислород связаны семиполярной связью.
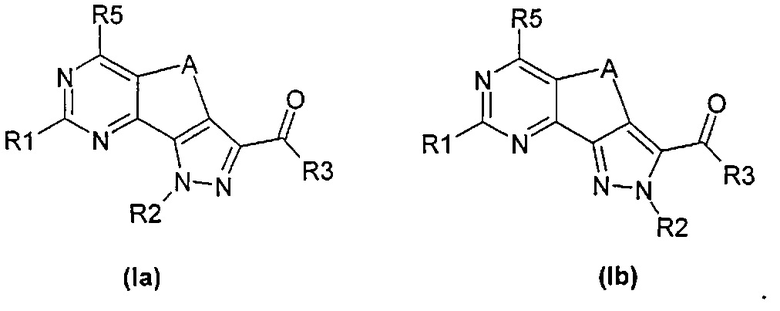
В формуле (I)
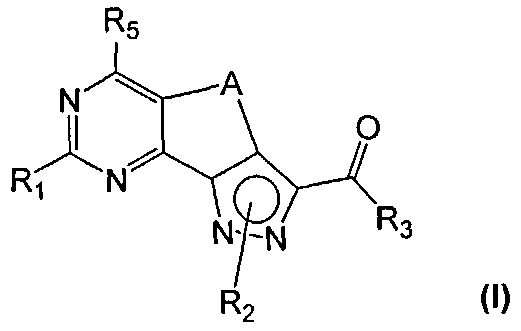
R2 может быть связан с любым из атомов азота пиразольного кольца согласно формулам (Ia) и (Ib)
Если в соединении согласно настоящему изобретению присутствует стереогенный центр или другая разновидность центра изомерии, в настоящую заявку включены все формы такого изомера или изомеров, включая энантиомеры и диастереомеры. Соединения, содержащие стереогенный центр, могут быть применены в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическая смесь может быть разделена с применением общеизвестных технологий, а отдельный энантиомер может быть применен сам по себе. В тех случаях, когда соединения содержат ненасыщенные двойные связи углерод-углерод, в объем настоящего изобретения входят как цис (Z), так и транс (E) изомеры.
В тех случаях, когда соединения могут существовать в таутомерных формах, будь то находящиеся в равновесии или преимущественно в одной форме, каждую форму рассматривают как включенную в настоящее изобретение.
Таким образом, если не предусмотрено иное, в случае если в соединениях формулы (I) R2 представляет собой водород, указана только одна из следующих таутомерных форм формулы (Ia') или (Ib'), оставшаяся форма также включена в объем настоящего изобретения:
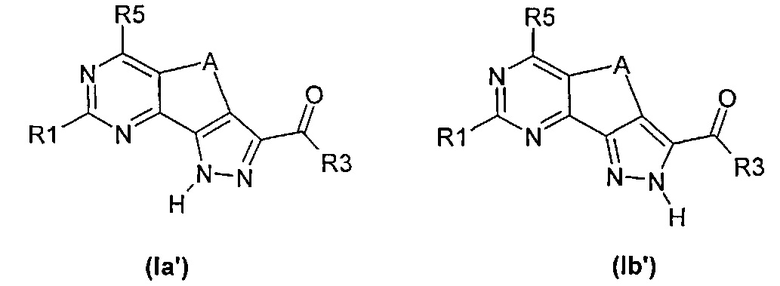
В тех случаях, когда соединения могут существовать в других таутомерных формах, таких как кето-енольные таутомеры, будь то находящиеся в равновесии или преимущественно в одной форме, каждую таутомерную форму рассматривают как включенную в настоящее изобретение.
Термин «арил» включает карбоциклические или гетероциклические углеводороды с кольцевыми фрагментами в количестве от 1 до 2, конденсированными или связанными друг с другом посредством простых связей, где по меньшей мере одно из колец является ароматическим; любой ароматический гетероциклический углеводород, если он присутствует, также именуется как гетероарильная группа, содержащая 5-6-членное кольцо с гетероатомами в количестве от 1 до 3, выбранными из N, O и S.
Примерами арильных групп согласно настоящему изобретению являются, например, фенил, бифенил, α- или β-нафтил, дигидронафтил, тиенил, бензотиенил, фурил, бензофуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, изоиндолил, пуринил, хинолил, изохинолил, дигидрохинолинил, хиноксалинил, бензодиоксолил, инданил, инденил, триазолил и т.п.
Термин «гетероциклил» (также известный как «гетероциклоалкил») означает 3-7-членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, где один или более атомов углерода заменены гетероатомами, такими как азот, кислород и сера. Неограничивающими примерами гетероциклильных групп являются, например, пиран, пирролидин, пирролин, имидазолин, имидазолидин, пиразолидин, пиразолин, тиазолин, тиазолидин, дигидрофуран, тетрагидрофуран, 1,3-диоксолан, пиперидин, пиперазин, морфолин и т.п.
Термин «C3-C7циклоалкил», охватывающий и C4-C7циклоалкил, означает, если не предусмотрено иное, 3-7-членное моноциклическое кольцо только из атомов углерода, которое может содержать одну или более двойных связей, но не имеет полностью сопряженной π-электронной системы. Неограничивающими примерами циклоалкильных групп являются циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексен, циклогексадиен, циклогептан, циклогептен, циклогептадиен.
Термин «линейный или разветвленный C1-C6алкил», охватывающий и C1-C4алкил, означает любую из групп, таких как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил и т.п.
Термин «линейный или разветвленный C2-C6алкенил» означает любую из групп, таких как, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил, 1-гексенил и т.п.
Согласно настоящему изобретению, если не предусмотрено иное, любая из вышеуказанных групп R1, R2, R4, R6, R' и Rʺ может быть необязательно замещена по любому из свободных положений одной или более группами, например, группами в количестве от 1 до 6, независимо выбранными из атома галогена, нитро, оксогрупп (=O), циано, C1-C6алкила, полифторированного алкила, полифторированного алкокси, алкенила, алкинила, гидроксиалкила, арила, арилалкила, гетероциклила, C3-C7циклоалкила, гидрокси, алкокси, арилокси, гетероциклилокси, метилендиокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси, гетероциклилкарбонилокси, алкилиденаминоокси, карбокси, алкоксикарбонила, арилоксикарбонила, циклоалкилоксикарбонила, гетероциклилоксикарбонила, амино, уреидо, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонила, алкиламинокарбонила, диалкиламинокарбонила, ариламинокарбонила, гетероциклиламинокарбонила, алкоксикарбониламино, гидроксиаминокарбонил алкоксиимино, алкилсульфониламино, арилсульфониламино, гетероциклилсульфониламино, формила, алкилкарбонила, арилкарбонила, циклоалкилкарбонила, гетероциклилкарбонила, алкилсульфонила, арилсульфонила, аминосульфонила, алкиламиносульфонила, диалкиламиносульфонила, ариламиносульфонила, гетероциклиламиносульфонила, арилтио, алкилтио, фосфоната и алкилфосфоната.
В свою очередь, в случае необходимости каждый из вышеуказанных заместителей может быть дополнительно замещен одной или более вышеуказанными группами.
В связи с этим, термин «атом галогена» означает атом фтора, хлора, брома или йода.
Термин «циано» означает остаток -CN.
Термин «нитро» означает группу -NO2.
Термины «алкенил» и «алкинил» означает любую из вышеуказанных линейных или разветвленных C2-C6 алкильных групп, дополнительно содержащую двойную или тройную связь соответственно. Неограничивающими примерами алкенильных или алкинильных групп согласно настоящему изобретению являются, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил, 1-гексенил, этинил, 2-пропинил, 4-пентинил и т.п.
Термин «полифторированный алкил» или «полифторированный алкокси» означает любую из вышеуказанных линейных или разветвленных C1-C6 алкильных или алкоксильных групп, которые замещены более чем одним атомом фтора, такую как, например, трифторметил, трифторэтил, 1,1,1,3,3,3-гексафторпропил, трифторметокси и т.п.
Термины «алкокси», «арилокси», «гетероциклилокси» и их производные означают любую из вышеуказанных C1-C6 алкильных, арильных или гетероциклильных групп, связанных с остальной молекулой через атом кислорода (-O-).
Из всего вышеизложенного специалисту понятно, что любая группа, наименование которой представляет собой составное наименование, такая как, например, ариламино, как обычно должна быть истолкована по частям, из которых она получена, например, по аминогруппе, которая дополнительно замещена арилом, где арил определен выше.
Аналогично любой из терминов, таких как, например, алкилтио, алкиламино, диалкиламино, алкоксикарбонил, алкоксикарбониламино, гетероциклилкарбонил, гетероциклилкарбониламино, циклоалкилоксикарбонил и т.п., включает группы, где алкильные, алкокси, арильные, C3-C7 циклоалкильные и гетероциклильные фрагменты определены выше.
Фармацевтически приемлемые соли соединений формулы (I) включают соли присоединения неорганических или органических кислот, например, азотной, соляной, бромоводородной, серной, хлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой, фумаровой, молочной, щавелевой, малоновой, яблочной, малеиновой, винной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изэтионовой и салициловой кислоты. Соль добавления кислоты соединений согласно настоящему изобретению предпочтительно выбрана из гидрохлорида или мезилата.
Фармацевтически приемлемые соли соединений формулы (I) также включают соли, полученные с применением неорганических или органических оснований, например, гидроксидов щелочных или щелочноземельных металлов, в частности, натрия, калия, кальция, аммония или магния, карбонатов или бикарбонатов, ациклических или циклических аминов, предпочтительно метиламина, этиламина, диэтиламина, триэтиламина, пиперидина и т.п.
Предпочтительные соединения формулы (I) представляют собой соединения, где
R1 представляет собой CN, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу XR4, где
X представляет собой двухвалентный радикал, выбранный из O, S, SO, SO2 и NR6, где
R6 представляет собой необязательно замещенный линейный или разветвленный C1-C6алкил, или R6 и R4 совместно с атомом азота, с которым они связаны, могут образовать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
R2 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, линейного или разветвленного C2-C6алкенила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила;
R5 представляет собой водород или NR'Rʺ, где
R' и Rʺ представляют собой водород,
и R3, R4 и A определены выше.
Другой предпочтительный класс соединений формулы (I) представляет собой соединения, где
R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу XR4, где
X представляет собой двухвалентный радикал, выбранный из O, S, SO, SO2 и NR6, где
R6 представляет собой необязательно замещенный линейный или разветвленный C1-C6алкил, или R6 и R4 совместно с атомом азота, с которым они связаны, могут образовать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
R2 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, гетероциклила и гетероциклилалкила;
R3 представляет собой группу, выбранную из NR'Rʺ и N(OH)R', где
R' и Rʺ представляют собой водород,
и R4, R5 и A определены выше.
Следующий предпочтительный класс соединений формулы (I) представляет собой соединения, где
R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, арила и гетероциклила, или группу XR4, где
X представляет собой двухвалентный радикал, выбранный из O, S и NR6, где
R6 представляет собой необязательно замещенный линейный или разветвленный C1-C6алкил, или R6 и R4 совместно с атомом азота, с которым они связаны, могут образовывать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
R3 представляет собой NR'Rʺ, где
R' и Rʺ представляют собой водород;
R4 представляет собой группу, выбранную из необязательно замещенного линейного или разветвленного C1-C6алкила, арила, арилалкила и гетероциклила;
R5 представляет собой водород,
и R2 и A определены выше.
Особенно предпочтительный класс соединений формулы (I) представляет собой соединения, где
R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила и арила, или группу XR4, где
X представляет собой двухвалентный радикал, выбранный из O, S и NR6, где
R6 представляет собой необязательно замещенный линейный или разветвленный C1-C6алкил, или R6 и R4 совместно с атомом азота, с которым они связаны, могут образовать 5-6-членную гетероарильную или гетероциклильную группу, необязательно содержащую один дополнительный гетероатом, выбранный из N, O и S;
R4 представляет собой группу, выбранную из необязательно замещенного линейного или разветвленного C1-C6алкила, арила и гетероциклила;
A представляет собой двухвалентную группу -CH=CH-,
и R2, R3 и R5 определены выше.
ОЦЕНКА БИОЛОГИЧЕСКОГО ДЕЙСТВИЯ
Определение активности соединения формулы (I) по отношению к киназе Pim может быть осуществлено с помощью ряда прямых и косвенных способов обнаружения. Некоторый иллюстративные соединения, описанные в настоящей заявке, анализировали на предмет определения активности связывания киназы Pim, включая изоформы Pim-1, Pim-2 и Pim-3 (Пример 901) и активности in vitro против опухолевых клеток (Пример 902). Некоторые иллюстративные соединения согласно настоящему изобретению имели значения показателя активности связывания Pim IC50 менее примерно 1 микромоль (мкмоль). Некоторые соединения согласно настоящему изобретению имели значения показатели активности против опухолевых клеток EC50 менее примерно 1 микромоль (мкмоль). Соединения формулы (I), имеющие Ki/IC50/EC50 менее 1 мкмоль в анализах, описанных в Примерах 901 и 902, могут терапевтически подходить для применения в качестве ингибиторов киназы Pim (Pim-1, Pim-2 и/или Pim-3).
Иллюстративные соединения формулы (I) в Таблице 1 получали, определяли и испытывали на ингибирование киназы Pim способами согласно настоящему изобретению, и указанные соединения имеют следующие структуры и соответствующие наименования (ChemBioDraw Ultra, Version 11.0, CambridgeSoft Corp., Кембридж, MA).
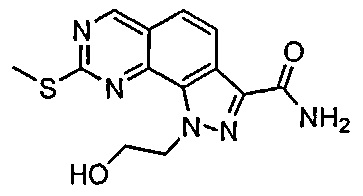
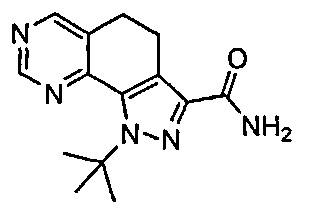
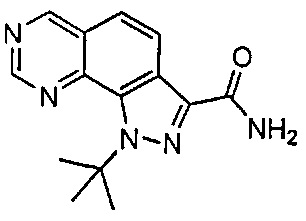
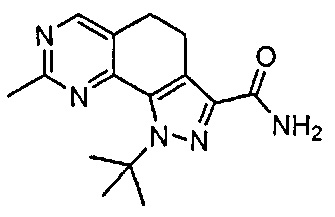
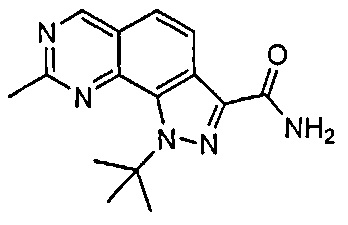
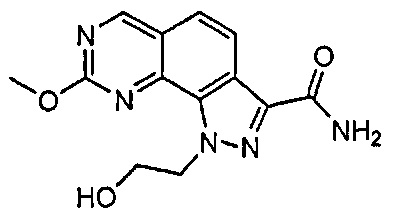
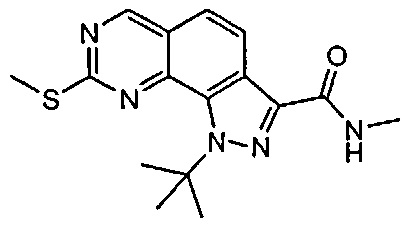
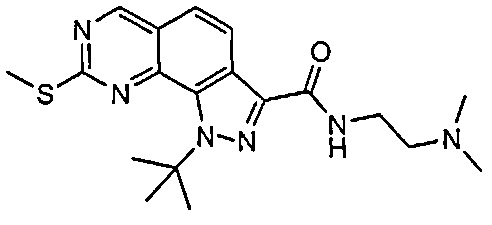
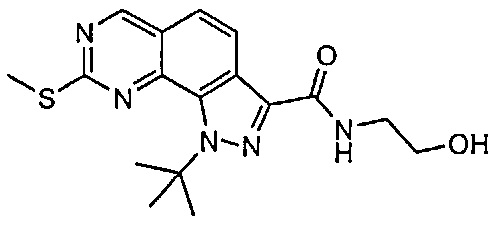
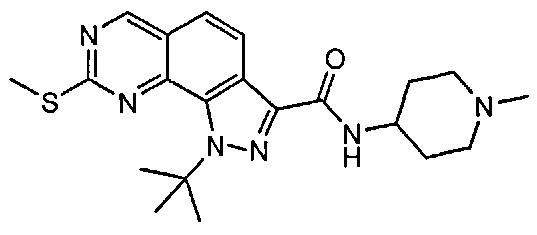
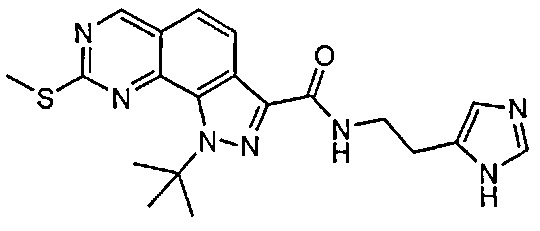
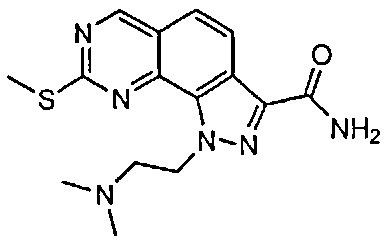
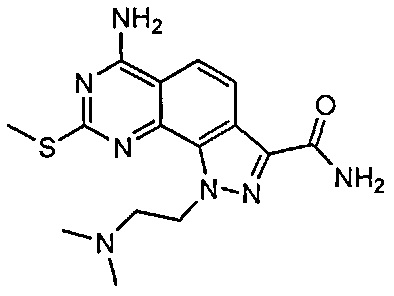
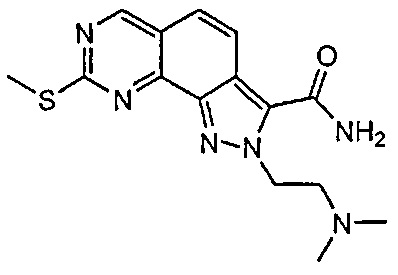
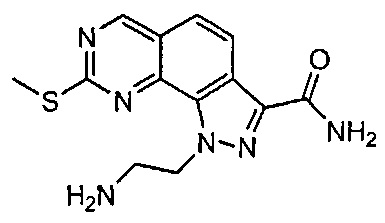
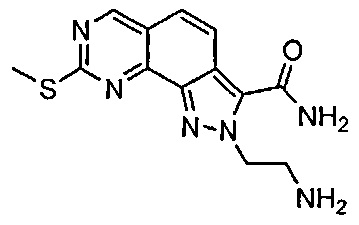
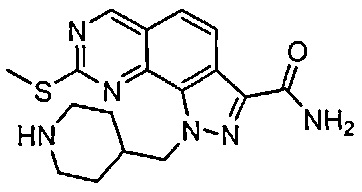
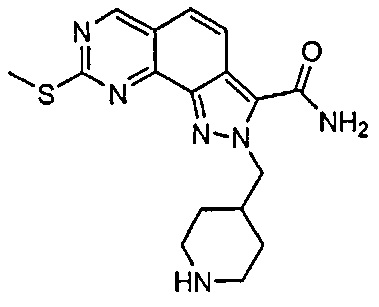

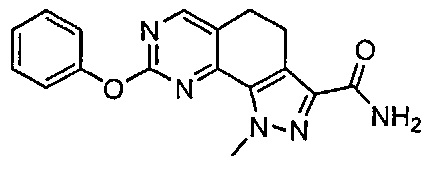
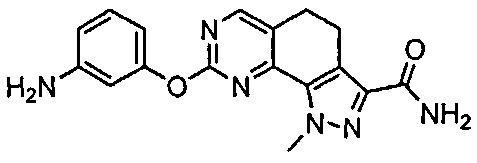
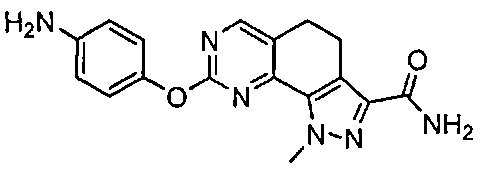
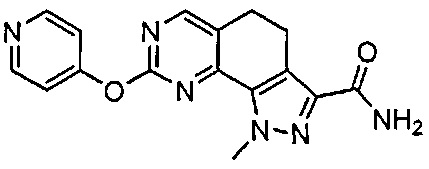
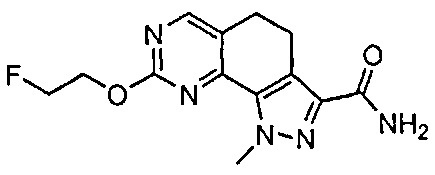
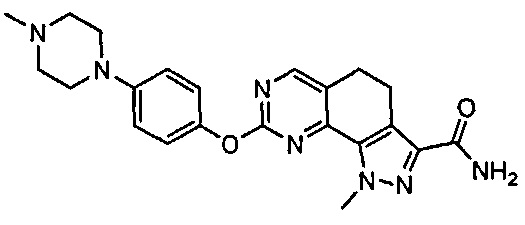
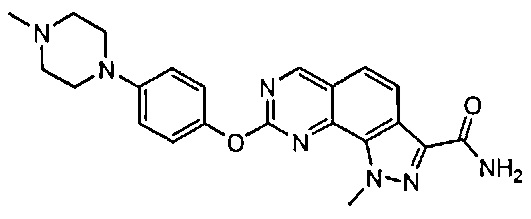
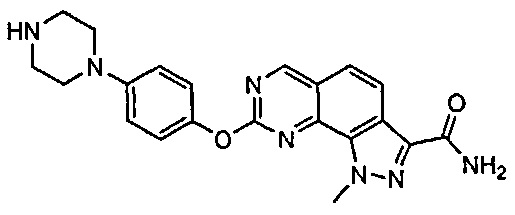
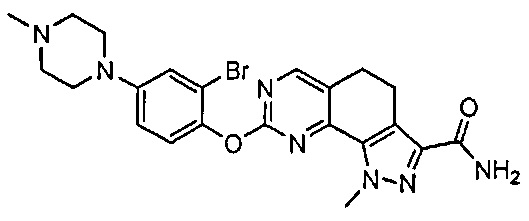
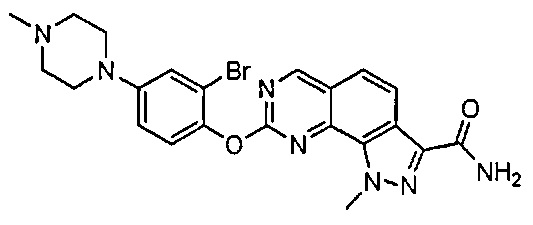
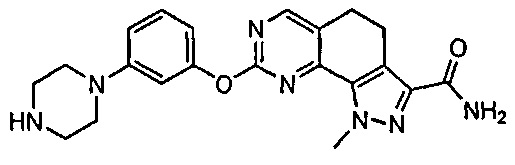
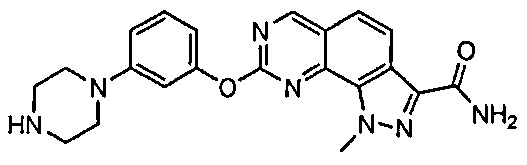
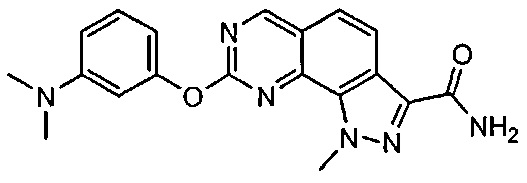

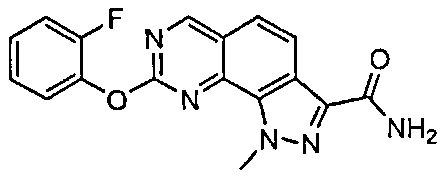
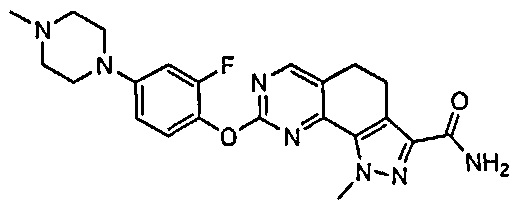
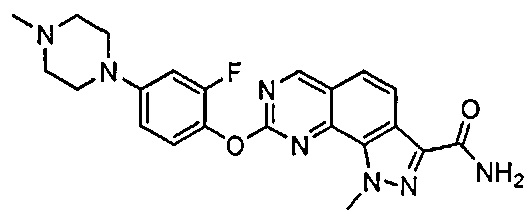

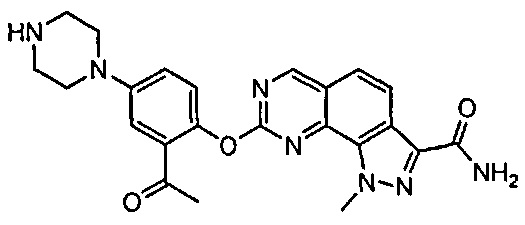
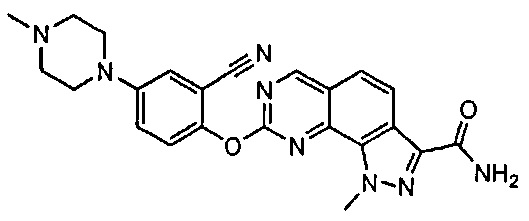
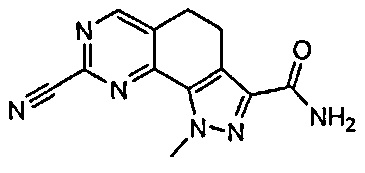
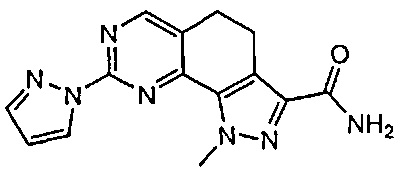

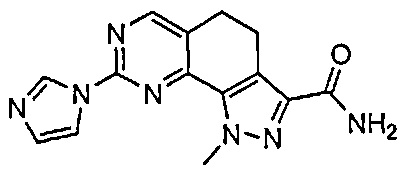
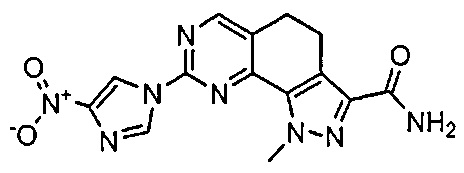
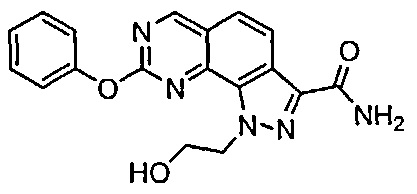
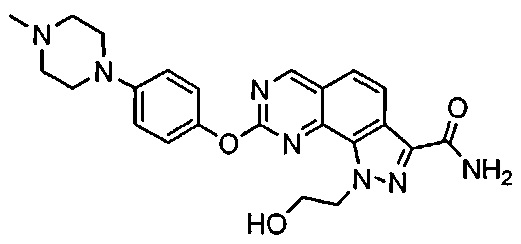
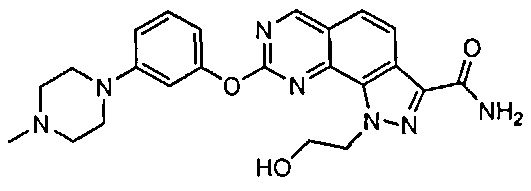
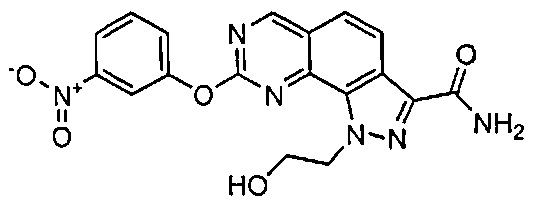
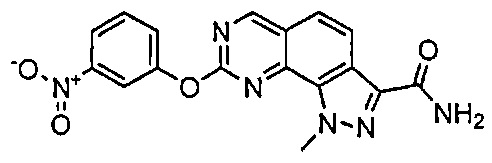
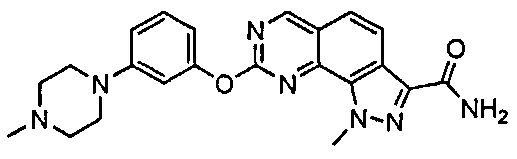
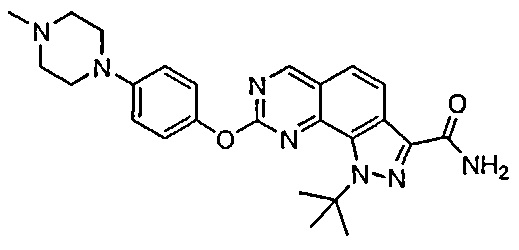
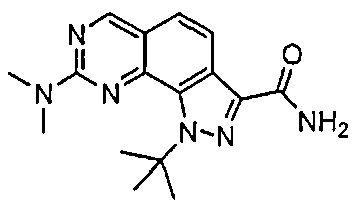
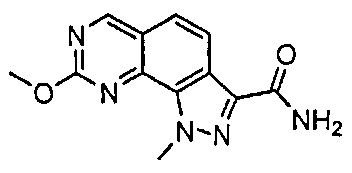
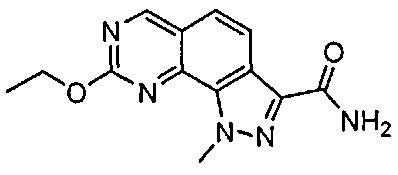
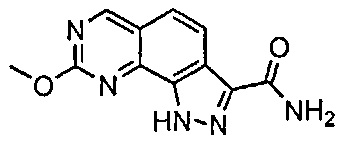
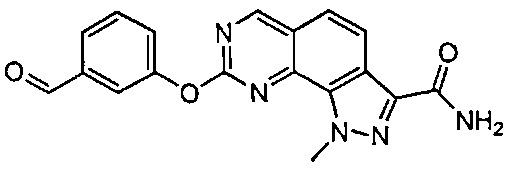
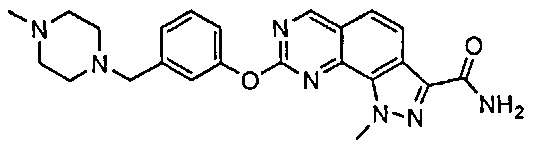
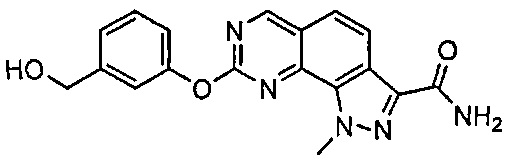
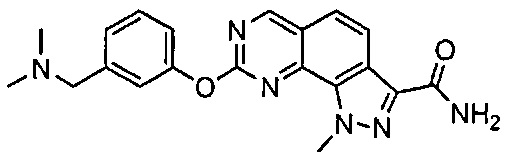
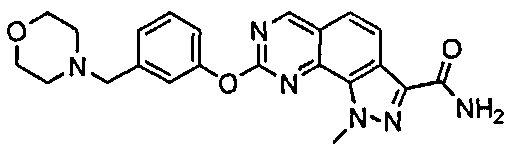
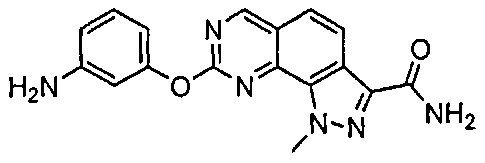
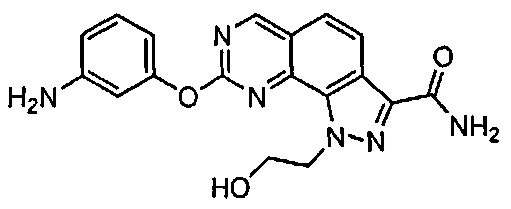
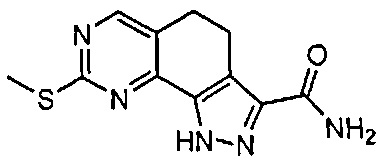
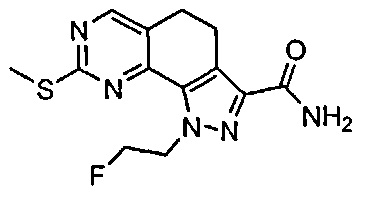

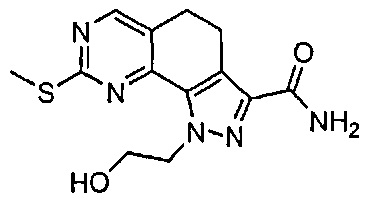
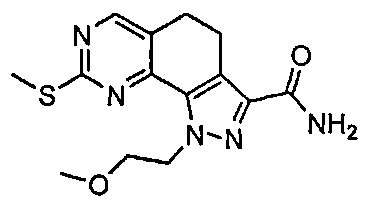
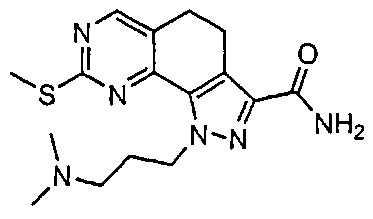
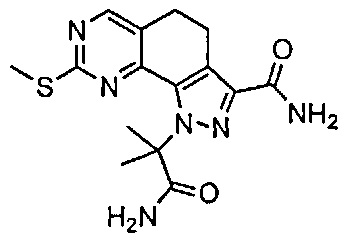
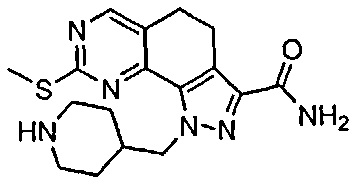
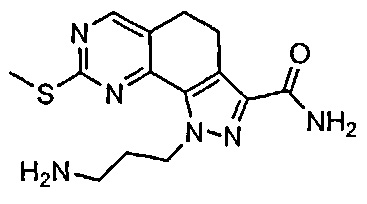
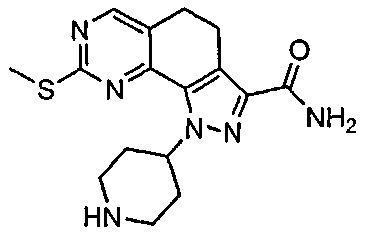
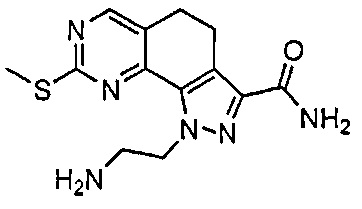
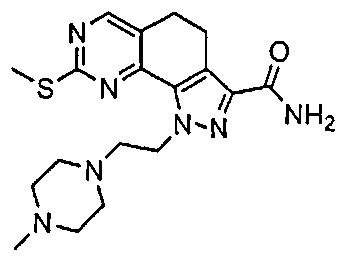
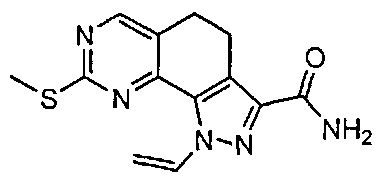


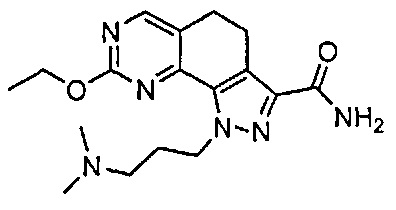
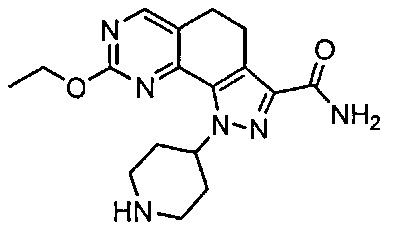

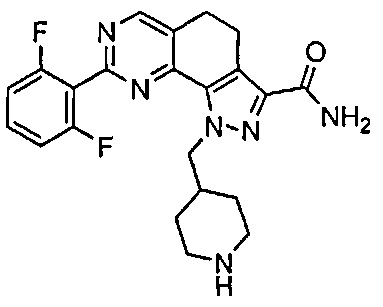
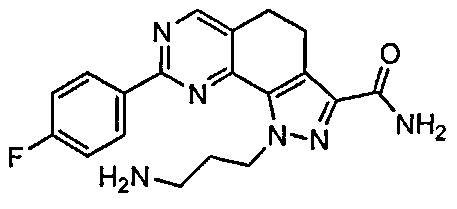
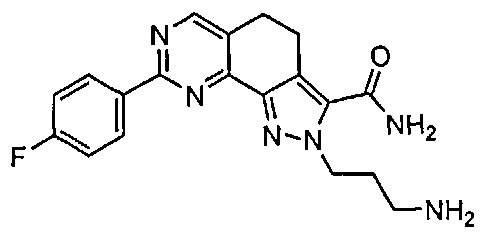
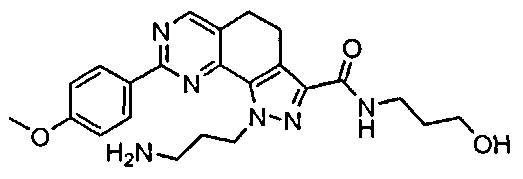
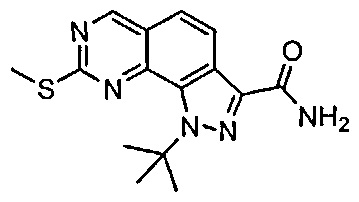
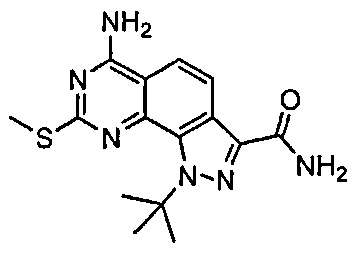
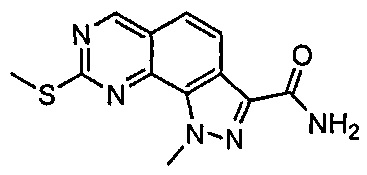

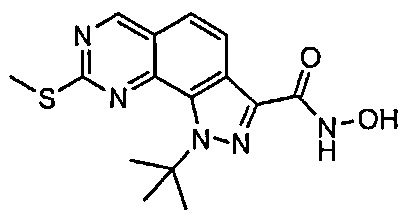
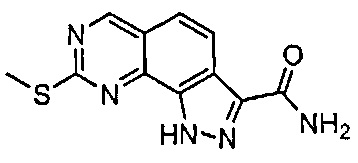
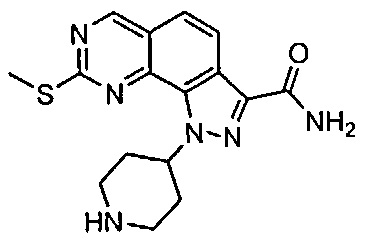
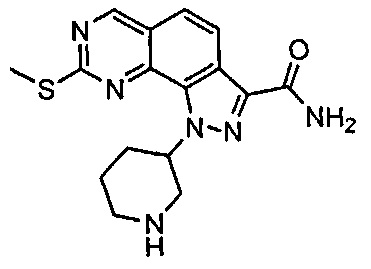
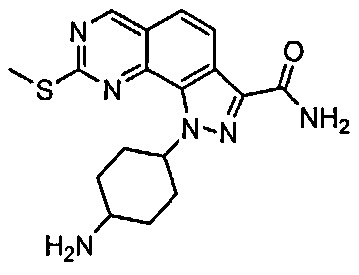
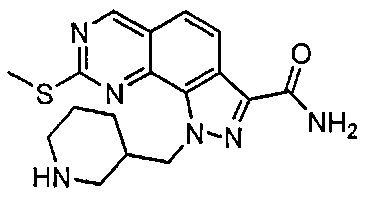
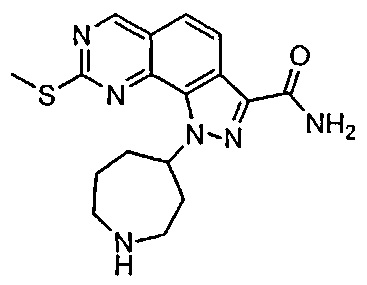
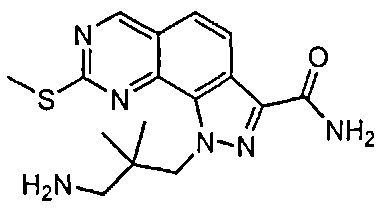
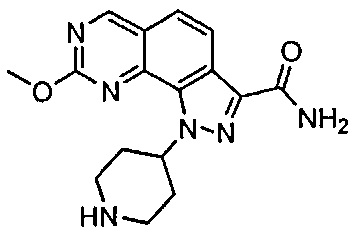
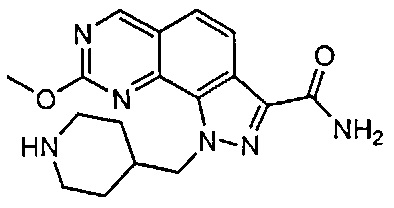
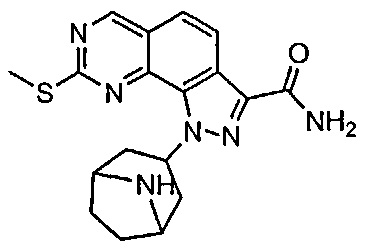
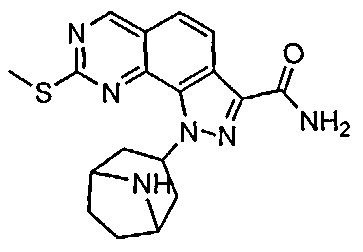
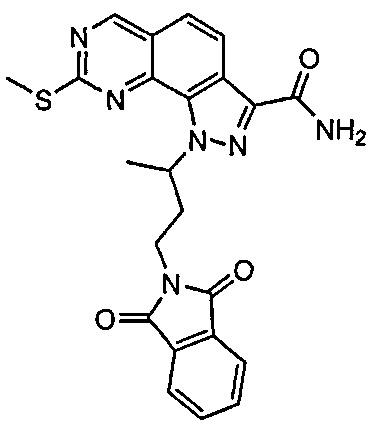
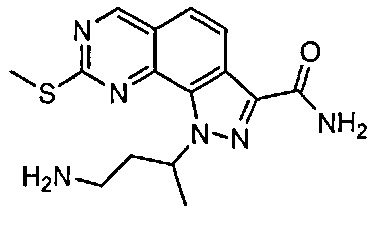

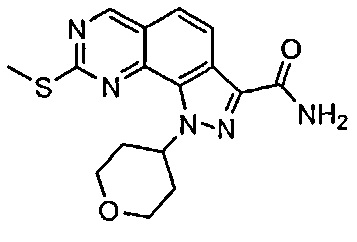
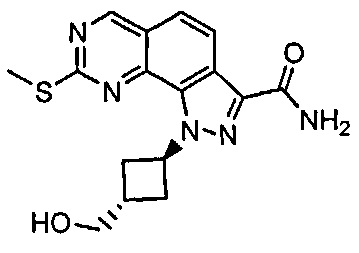
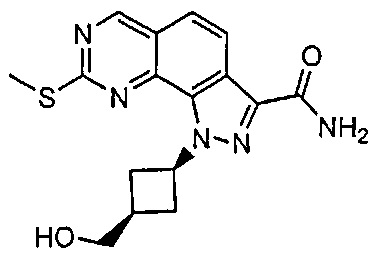
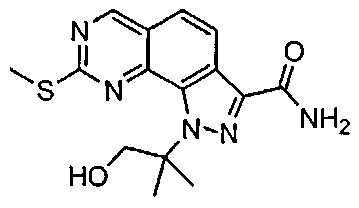

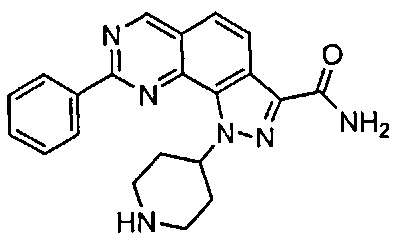
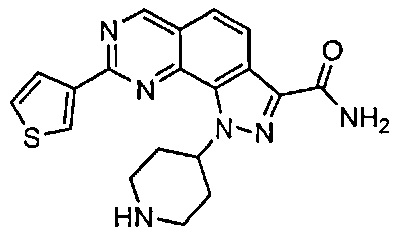
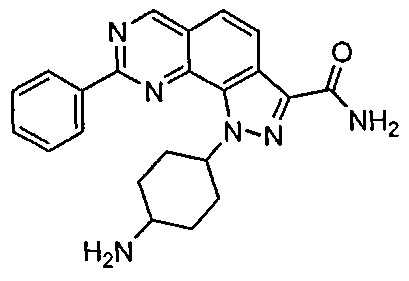

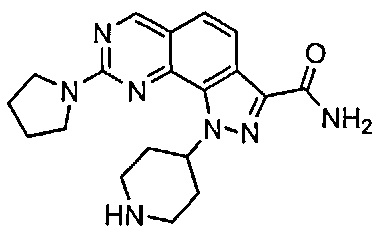
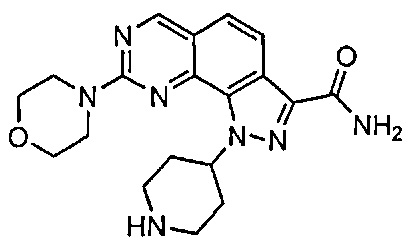
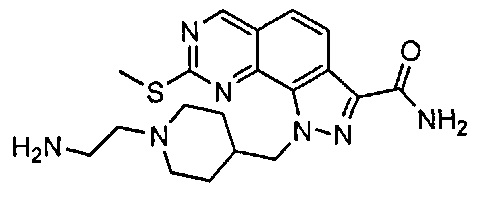

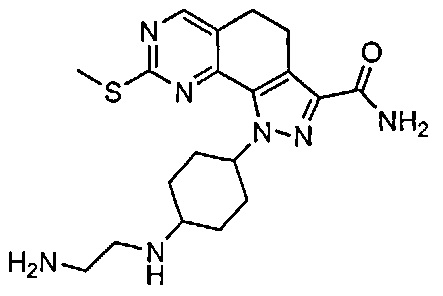
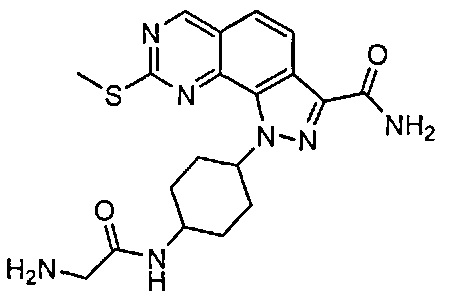
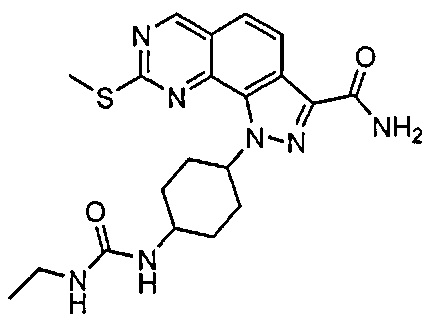
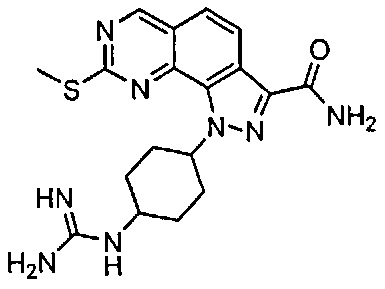
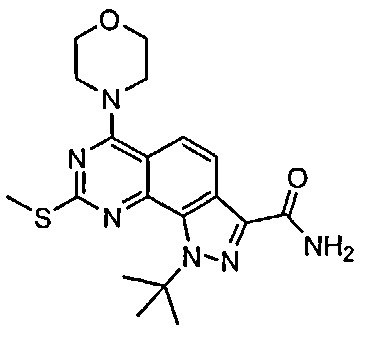
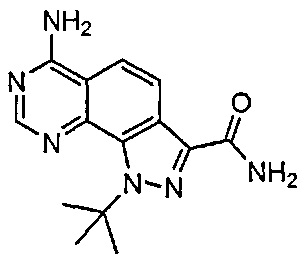
Для ссылки на любое конкретное соединение формулы (I) согласно настоящему изобретению, необязательно в виде фармацевтически приемлемой соли, см. экспериментальный раздел и формулу изобретения.
В настоящем изобретении также предложен способ получения соединения формулы (I), определенной выше, с применением путей реакции и схем синтеза, описанных ниже, технологий, доступных в данной области, и легкодоступных материалов. Получение некоторых вариантов реализации настоящего изобретения описано в следующих примерах, но специалистам в данной области будет понятно, что описанные способы получения могут быть легко приспособлены для получения других вариантов реализации настоящего изобретения. Например, синтез соединений согласно настоящему изобретению, не описанных в примерах, может быть выполнен путем изменений, очевидных специалистам в данной области, например путем подходящей защиты групп, мешающих протеканию реакции по желаемому пути, путем применения других подходящих реагентов, известных в данной области, или путем произведения стандартных изменений условий реакции. В качестве альтернативы другие реакции, упомянутые в настоящей заявке или известные в данной области, будут признаны приспосабливаемыми для получения других соединений согласно настоящему изобретению.
На Фигуре 1 представлено получение соединения формулы (I), где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; a R2, R3, R4, R5, R', Rʺ и A определены выше.
Всем специалистам в данной области будет понятно, что любое превращение, осуществляемое согласно указанным способам, может потребовать стандартных изменений, таких как, например, защита групп, мешающих протеканию реакции по желаемому пути, применение других подходящих реагентов, известных в данной области, или произведение стандартных изменений условий реакции.
Таким образом, способ согласно настоящему изобретению включает:
эт. 1) смешивание соединения формулы (II)
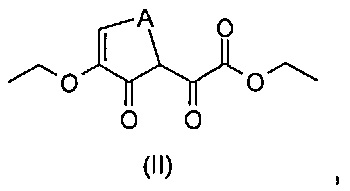
где A представляет собой -(CH2)2-, с производным гидразина формулы (III)
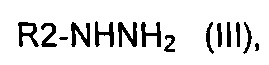
где R2 определен выше, в кислых условиях с получением соединения формулы (IV)
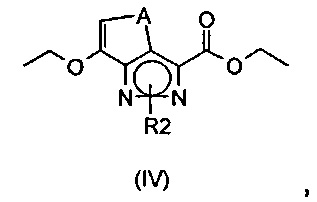
где A представляет собой -(CH2)2-, a R2 определен выше; или
эт. 1a) смешивание соединения формулы (IV), где R2 представляет собой водород, с соединением формулы (V)
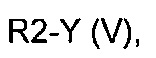
где R2 определен выше, но не водород, a Y представляет собой подходящую уходящую группу, такую как йод, бром, хлор или сульфонатную группу (например, -OS(O)2CF3, -OS(O)2CH3 или -OS(O)2PhMe), с получением соединения формулы (IV), где R2 определен выше, но не водород;
эт. 2) смешивание полученного соединения формулы (IV) с диалкилацеталем диметилформамида с получением соединения формулы (VI)
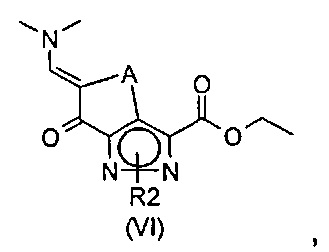
где A представляет собой -(CH2)2-, a R2 определен выше;
эт. 3) проведение реакции полученного соединения формулы (VI) согласно любому из альтернативных этапов (эт. 3a), (эт. 3b) или (эт. 3c):
эт. 3a) с изотиомочевиной формулы (VIIa) или ее солью:
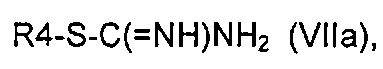
где R4 определен выше;
эт. 3b) с изомочевиной формулы (VIIb) или ее солью:

где R4 определен выше;
эт. 3c) с подходящим амидином формулы (IX) или его солью:

где R1 представляет собой водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или его солью
с получением соединения формулы (VIII)
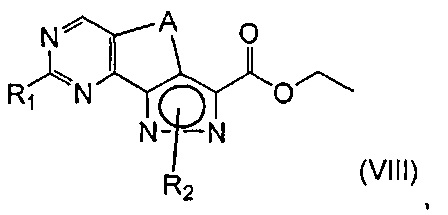
где R1 представляет собой соответственно SR4, OR4, водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, A представляет собой -(CH2)2-, a R2 и R4 определены выше;
эт. 4) смешивание полученного соединения формулы (VIII) с окисляющим агентом или в условиях дегидрирования в присутствии Pd или Pt катализатора с получением соединения формулы (VIII), где R1 представляет собой соответственно SR4, OR4, водород или необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, A представляет собой -CH=CH-, a R2 и R4 определены выше;
эт. 5) проведение реакции соединения формулы (VIII), полученного как описано на этапе эт. 3a), эт. 3b), эт. 3c) или эт. 4), где R2 представляет собой защитную группу, такую как трет-бутил или тритил, в кислых условиях, с получением соединения формулы (VIII), где R2 представляет собой водород, превращение полученного соединения формулы (VIII) в смесь двух соединений формулы (VIIIa) и формулы (VIIIb), где R2 определен выше, но не водород, посредством реакции с соединением формулы (V)

где Y определен выше, и R2 определен выше, но не водород:
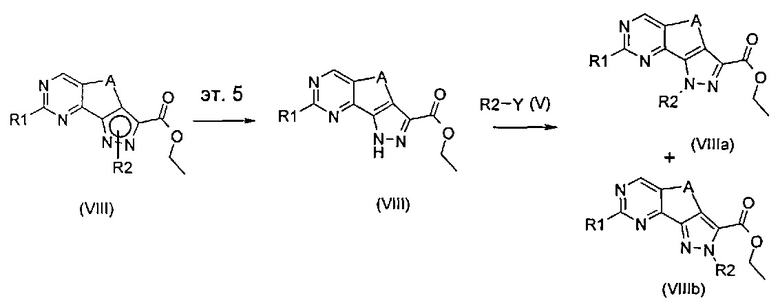
эт. 6) проведение реакции соединения формулы (VIII), полученного как описано на этапе эт. 3a), эт. 3b), эт. 3c), эт. 4) или эт. 5), согласно любому из альтернативных этапов эт. 6a), эт. 6b) или эт. 6c):
эт. 6a) с гидроксидом аммония или амином формулы (XII)

где R' и Rʺ определены выше, с получением соединения формулы (I)
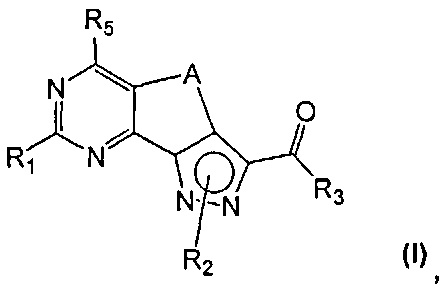
где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; R3 представляет собой NR'Rʺ, R5 представляет собой водород, a R2, R4 и A определены выше;
эт. 6b1) в условиях кислотного или щелочного гидролиза с получением соединения формулы (XI) или его соли:
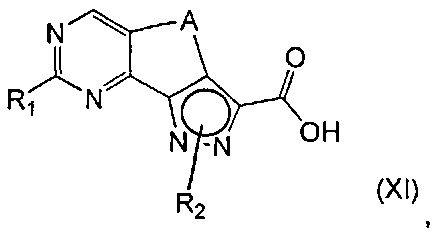
где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; a R2, R4 и A определены выше;
эт. 6b2) смешивание полученного соединения формулы (XI) или его соли с солью аммония или производным формулы (XII), или производным формулы (X)
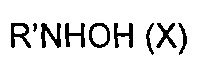
в щелочных условиях и в присутствии подходящего конденсирующего агента с получением соединения формулы (I)
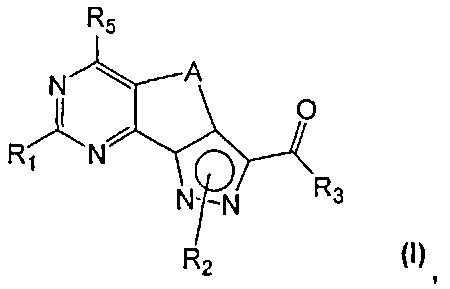
где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; R5 представляет собой водород, a R2, R4, R3 и A определены выше;
эт. 6c) с солью аммония или подходящим амином формулы (XII) в присутствии сильного основания с получением соединения формулы (I)
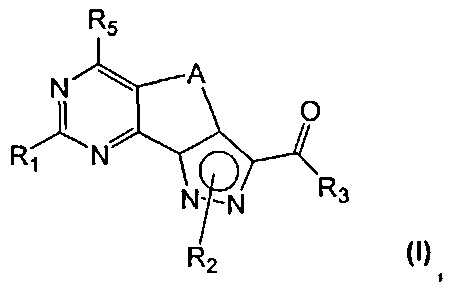
где R1 представляет собой водород, необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, или группу SR4 или OR4; R3 представляет собой NR'Rʺ, a R2, R4, R5, A, R' и Rʺ определены выше;
необязательно превращение соединение формулы (I) в другое соединение формулы (I) и при необходимости превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
В настоящем изобретении дополнительно предложен альтернативный способ получения соединения формулы (I), определенной выше, приведенный ниже на Схеме 2.
Схема 2

На вышеуказанной Схеме R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, R2 определен выше, но не водород, R3 и A определены выше; указанный альтернативный способ включает следующие этапы:
эт. 7) проведение реакции соединения формулы (XIII)
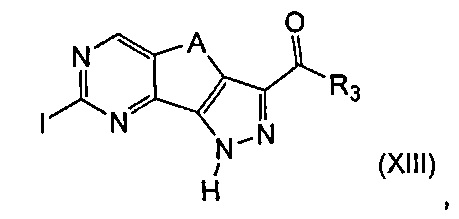
где R3 и A определены выше, с соединением формулы (V)

где Y определен выше, a R2 определен выше, но не водород;
эт. 8) проведение реакции полученного соединения формулы (XIV)
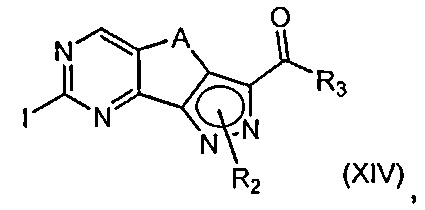
где R2 определен выше, но не водород, a R3 и A определены выше, с соединением формулы (XV)
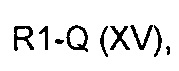
где R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, a Q представляет собой подходящую группу, такую как -B(OH)2, -B(OAlk)2, -Sn(Alk)4, -Al(Alk)3, ZnHal, MgHal или ZrCp2Hal,
причем указанное соединение может подвергаться образованию углеродной связи, катализируемому палладием, с получением соединения формулы (I)
где R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, R2 определен выше, но не водород, R3 и A определены выше, а R5 представляет собой водород;
необязательно превращение соединение формулы (I) в другое соединение формулы (I) и при необходимости превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
В настоящем изобретении дополнительно предложен альтернативный способ получения соединения формулы (VIII), определенной выше, приведенный ниже на Схеме 3.
Схема 3
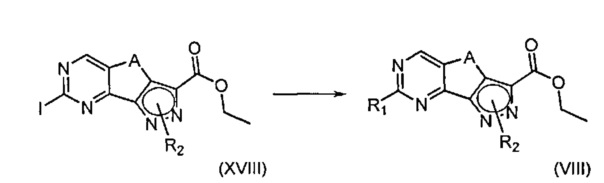
На вышеуказанной Схеме R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, R2 определен выше, но не водород, и A определен выше; указанный альтернативный способ включает следующий этап:
эт. 9) проведение реакции соединения формулы (XVIII)
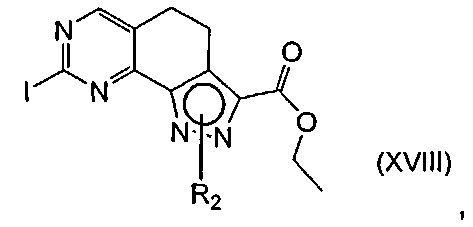
где R2 определен выше, но не водород, и A определен выше, с соединением формулы R1-Q (XV)
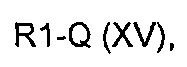
где R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, a Q определен выше,
причем указанное соединение может подвергаться образованию углеродной связи, катализируемому палладием, с получением соединения формулы (VIII).
Как определено выше, соединения формулы (I), которые получают способом, представляющим собой объект настоящего изобретения, беспрепятственно могут быть превращены в другие соединения формулы (I) путем выполнения операций согласно общеизвестным условиям синтеза, при этом существуют следующие примеры возможных превращений:
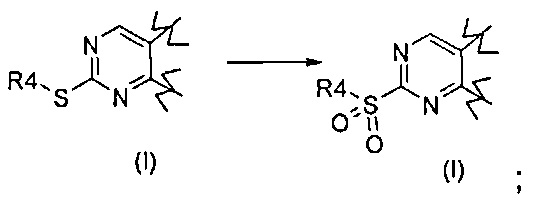
Превр. a) превращение соединения формулы (I), где R1 представляет собой группу, такую как R4-S-, где R4 определен выше, в соединение формулы (I), где R1 представляет собой группу R4-S(O)2-, в условиях окисления:
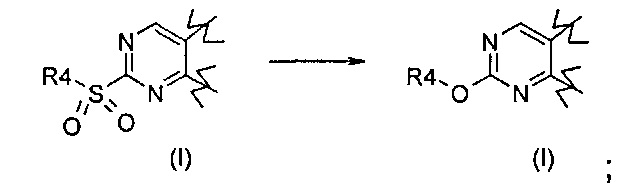
Превр. b) превращение соединения формулы (I), где R1 представляет собой группу, такую как R4-S(O)2-, где R4 определен выше, в соединение формулы (I), где R1 представляет собой группу R4-O-, путем проведения реакции сульфонильного производного с соединением формулы R4-OH (XVI):
Превр. c) превращение соединения формулы (I), где R1 представляет собой группу, такую как R4-S(O)2-, в соединение формулы (I), где R1 представляет собой группу R4-NR6, где R4 и R6 определены выше, путем проведения реакции сульфонильного производного с соединением формулы R4 R6-NH (XVII):
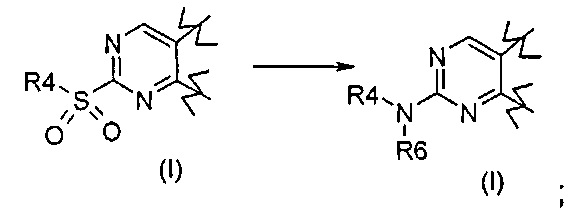
где R4 и R6 определены выше,
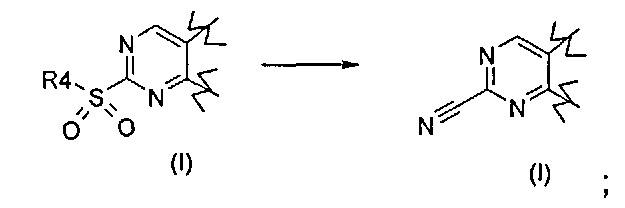
Превр. d) превращение соединения формулы (I), где R1 представляет собой группу, такую как R4-S(O)2-, в соединение формулы (I), где R1 представляет собой -CN, путем проведения реакции сульфонильного производного с цианидом натрия (NaCN) или цианидом калия (KCN):
Превр. e) превращение соединения формулы (I), где R1, R3 и A определены выше, a R2 представляет собой водород, в соединение формулы (I), где R2 определен выше, но не водород, посредством реакции с соединением формулы (V):
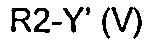
где Y' представляет собой OH или группу, которая необязательно в результате активации может служить подходящей уходящей группой, такую как йод, бром, хлор или сульфонатная группа (например, -OS(O)2CF3, -OS(O)2CH3 или -OS(O)2PhMe), a R2 определен выше, но не водород,
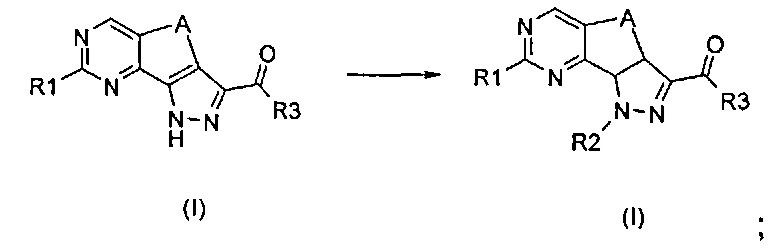
Превр. f) превращение соединения формулы (I), где R2 представляет собой галогенэтил, в соединение формулы (I), где R2 представляет собой винил, путем создания щелочных условий:
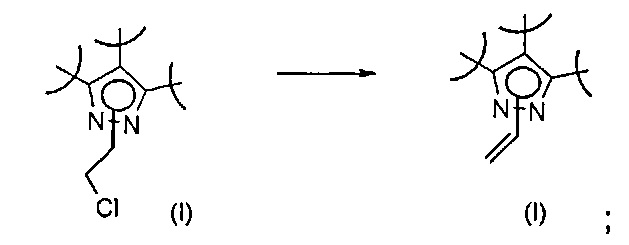
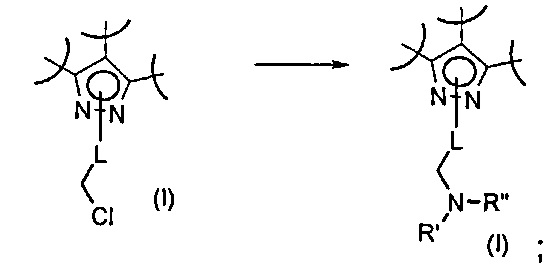
Превр. g) превращение соединения формулы (I), где R2 представляет собой группу формулы L-CH2Cl, где L представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, замещенную хлором, путем проведения реакции с соединением формулы R'RʺNH (XII) с получением соединения формулы (I), где R2 представляет собой группу L-CH2NR'Rʺ, a R' и Rʺ определены выше:
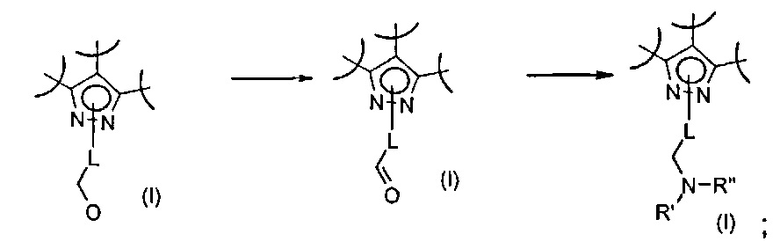
Превр. h) превращение соединения формулы (I), где R2 представляет собой группу формулы L-CH2OH, где L определен выше, в соединение формулы (I), где R2 представляет собой группу L-CH2NR'Rʺ, путем превращения группы CH2OH в CHO, а затем проведения реакции полученного альдегидного производного с соединением формулы R'RʺNH (XII) в присутствии подходящих восстанавливающих агентов:
Превр. i) превращение соединения формулы (I), где R2 представляет собой группу формулы L-COOалкил, где L определен выше, в соединение формулы (I), где R2 представляет собой L-CH2OH, путем проведения реакции с подходящим восстанавливающим реагентом:
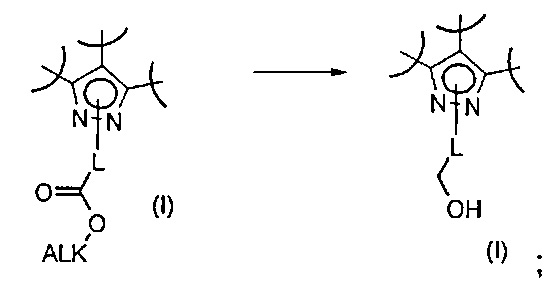
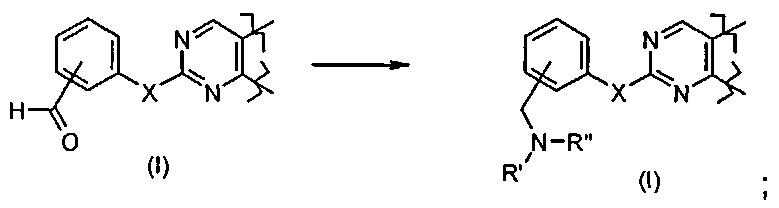
Превр. j) превращение соединения формулы (I), где X представляет собой O, и R4 представляет собой арил, то есть фенил, замещенный -CHO, в другое соединение формулы (I), где R4 представляет собой арил, то есть фенил, замещенный CH2NR'Rʺ, где R' и Rʺ определены выше, путем обработки амином формулы R'Rʺ-NH (XII) в присутствии подходящих восстанавливающих агентов:
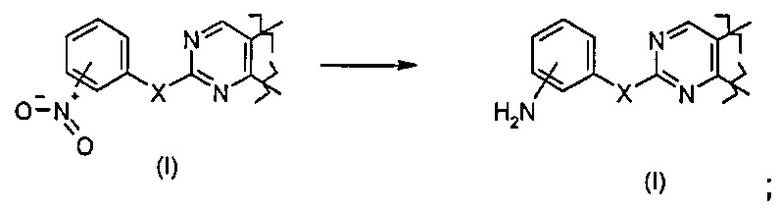
Превр. k) превращение соединения формулы (I), где X представляет собой O, и R4 представляет собой арил, то есть фенил, замещенный -NO2, в другое соединение формулы (I), где R4 представляет собой арил, то есть фенил, замещенный NH2, путем обработки подходящим восстанавливающим агентом:
Превр. l) превращение соединения формулы (I), где R2, R3 и A определены выше, a R1 представляет собой группу, такую как Me-S-, в соединение формулы (I), где R1 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, арила и гетероциклила, путем проведения реакции с соединением формулы R1-Q' (XV), где R1 определен выше, a Q' представляет собой подходящую группу, такую как -B(OH)2, -MgHal, -ZnHal,
причем указанное соединение может подвергаться образованию углеродной связи, катализируемому палладием:
Превр. m) превращение соединения формулы (I), где R1 и R2 определены выше, R5 представляет собой водород, и A представляет собой -CH=CH-, в соединение формулы (I), где R5 представляет собой NR'Rʺ, путем проведения реакции с солью аммония или подходящим амином формулы R'RʺNH (XII) в присутствии сильного основания;
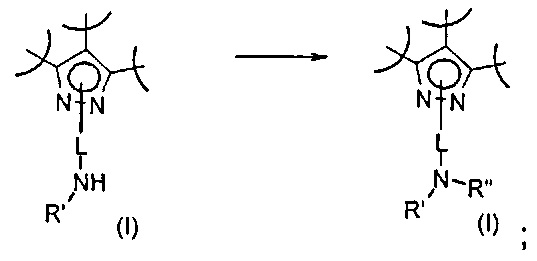
Превр. n) превращение соединения формулы (I), где R2 представляет собой группу формулы L-N(H)R', где L и R' определены выше, путем обработки соединением формулы Rʺ-CHO (XIX) в условиях получения соединения формулы (I), где R2 представляет собой группу формулы L-NR'Rʺ, где R' и Rʺ определены выше:
Превр. o) превращение соединения формулы (I), где R2 представляет собой группу формулы L-N(H)R', где L и R' определены выше, путем обработки соединением формулы R7-COW (XX), где R7 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, W представляет собой гидроксильную группу или галоген, в условиях получения соединения формулы (I), где R2 формулы L-N(R')COR7, где R' определен выше:
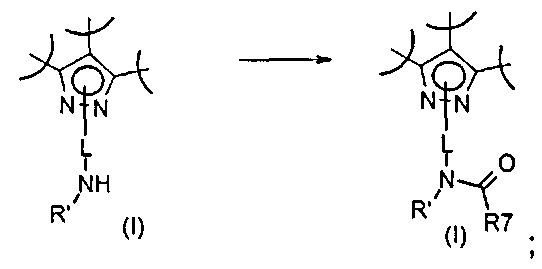
Превр. p) превращение соединения формулы (I), где R2 представляет собой группу формулы L-N(H)R' где L и R' определены выше, путем обработки соединением формулы R8-N=C=O (XXI), где R8 представляет собой необязательно замещенную группу, выбранную из линейного или разветвленного C1-C6алкила, C3-C7циклоалкила, циклоалкилалкила, арила, арилалкила, гетероциклила и гетероциклилалкила, в условиях получения соединения формулы (I), где R2 представляет собой группу формулы L-N(R')CONHR8, где R' определен выше:

Превр. q) превращение соединения формулы (I), где R2 представляет собой группу формулы L-N(H)R', где L и R' определены выше, путем обработки соединением формулы R8-NHC(NH)G (XXII), где R8 определен выше, a G представляет собой подходящую уходящую группу, в условиях получения соединения формулы (I), где R2 представляет собой группу формулы L-N(R')NHCNHR8, где R' определен выше:
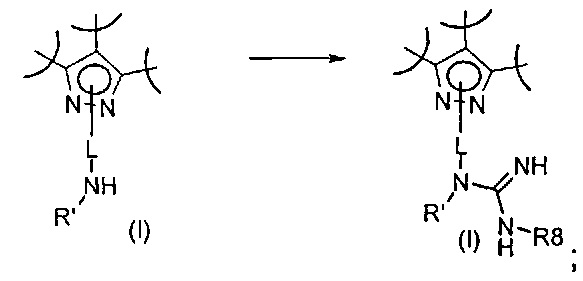
Превр. r) превращение соединения формулы (I), где R1, R2 и R3 определены выше, и A представляет собой двухвалентную группу, такую как CH2-CH2-, в соединение формулы (I), где A представляет собой группу -CH=CH-, путем обработки окисляющим агентом или в условиях дегидрирования в присутствии Pd или Pt катализатора:
Синтез соединения формулы (I) согласно способу синтеза, описанному ранее, может быть осуществлен поэтапно, при этом каждый промежуточный продукт перед проведением следующей реакции выделяют и очищают посредством стандартных технологий очистки, таких как, например, колоночная хроматография. В качестве альтернативы, два или более этапов последовательности синтеза могут быть выполнены так называемым «однореакторным» способом, известным в данной области, в результате чего выделяют и очищают только соединение, полученное в результате проведения двух или более этапов.
Согласно этапу (эт. 1) указанного способа проводят реакцию соединения формулы (II) с гидразином или производным гидразина формулы (III) в растворителе, таком как этанол, в присутствии уксусной кислоты, указанную реакцию проводят при температуре в диапазоне от комнатной температуры до 80°C, таким образом получают соединение формулы (IV).
Необязательно соединение формулы (IV), где R2 представляет собой водород, растворяют в подходящем растворителе, например, ацетонитриле, тетрагидрофуране, диметилформамиде или подобном растворителе, и к раствору добавляют подходящее основание, такое как гидрид натрия, или карбонат цезия. Затем добавляют соединение общей формулы R2Y (V) и смесь перемешивают в течение периода времени от примерно 2 часов до примерно 15 часов при температуре в диапазоне от примерно 20°C до примерно 80°C.
Согласно этапу (эт.2) указанного способа синтез енаминонового производного формулы (IV) осуществляют с применением диалкилацеталя N,N-диметилформамида, такого как, например, ди-трет-бутилацеталь диметилформамида, диэтилацеталь диметилформамида и т.п., в подходящем растворителе, таком как N,N-диметилформамид, N,N-диметилацетамид, толуол или другой подобный растворитель, при температуре в диапазоне от комнатной температуры до 100°C и в течение периода времени от 30 минут до примерно 24 часов.
Согласно одному из альтернативных этапов (эт. 3a), (эт. 3b) или (эт. 3c) указанного способа превращение соединения формулы (VI) в соединение формулы (VIII) осуществляют посредством применения изотиомочевины формулы (VIIa), или изомочевины формулы (VIIb), или подходящего амидина формулы (IX). Любую из вышеуказанных реакций проводят традиционными способами. Например, реакцию с метилизотиомочевиной или ее солью, такой как сульфат, проводят в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран, ацетонитрил и т.п., в присутствии основания, такого как ацетат калия, бикарбонат натрия, карбонат натрия или калия и т.п., при температуре в диапазоне от 50°C до 100°C и в течение периода времени от 2 часов до примерно 48 часов.
Согласно этапу (эт. 4) указанного способа соединение формулы (VIII) может подвергаться дегидрированию в присутствии необязательно нанесенного палладия или платины или 2,3-дихлор-5,6-дициано-1,4-бензохинона (ДДХ) так, чтобы получить соответствующее ароматическое производное формулы (VIII), то есть в подходящем растворителе, таком как толуол, 1,4-диоксан, хлорбензол, дихлорбензол, при температуре в диапазоне от 90°C до температуры обратной конденсации в течение периода времени от 2 часов до 8 часов.
Согласно этапу (эт. 5) указанного способа в отношении соединения формулы (VIII), где R2 представляет собой защитную группу, такую как трет-бутил или тритил, отщепление защитной группы может быть выполнено различными путями согласно традиционным способам. Предпочтительно проводят смешивание с соляной кислотой, трифторуксусной кислотой в присутствии подходящего растворителя, такого как дихлорметан и т.п., с получением соединения формулы (VIII), где R2 представляет собой водород.
Исходя из этого, проводят реакцию полученного соединения формулы (VIII) с соединением формулы R2Y (V), где R2 определен выше, но не водород, и Y определен выше, в присутствии основания, такого как карбонат калия или цезия, в подходящем растворителе, таком как ацетонитрил, N,N-диметилформамид. Последняя реакция может привести к получению смеси региоизомеров формулы (VIIIa) и (VIIIb), которые могут быть разделены известными способами, такими как хроматография на силикагеле или препаративная ВЭЖХ.
Согласно этапу (эт. 6a) указанного способа соединение формулы (VIII) превращают в соединение формулы (I) способами, хорошо известными в данной области, с превращением карбоксиэфирных групп (-COOEt) в карбоксамиды (-CONH2), N-замещенные карбоксамиды (-CONHR'), N,N-дизамещенные карбоксамиды (-CONR'Rʺ). Предпочтительно проводят реакцию с гидроксидом аммония в смеси метанола/N,N-диметилформамида при температуре в диапазоне от примерно 50°C до примерно 100°C.
Аналогичные рабочие условия применяют при получении N-замещенных карбоксамидов или N,N-дизамещенных карбоксамидов, при этом вместо аммиака или гидроксида аммония применяют подходящий первичный или вторичный амин.
Предпочтительно согласно этапу (эт. 6b1) указанного способа гидролиз соединения формулы (VIII) с получением соответствующей карбоновой кислоты соединения формулы (XI) проводят в кислых или щелочных условиях. Предпочтительно проводят реакцию с водными растворами щелочей, такими как водный раствор гидроксида лития, натрия или калия, в присутствии подходящего растворителя, такого как низший спирт, тетрагидрофуран, N,N-диметилформамид или их смеси; предпочтительно проводят реакцию с гидроксидом калия в смеси метанола/N,N-диметилформамида при температуре в диапазоне от примерно комнатной температуры до примерно 100°C. В соответствии с примененными рабочими условиями соединение формулы (XI) может быть получено в кислотной форме или, в качестве альтернативы, в форме соли.
Предпочтительно согласно этапу (эт. 6b2) указанного способа амидирование карбоновой кислоты формулы (XI) с получением соответствующего соединения формулы (I) проводят в присутствии хлорида аммония, или подходящего первичного или вторичного амина формулы R'RʺNH (XII), или замещенного гидроксиламинового производного формулы R'NHOH (X) в щелочных условиях, предпочтительно с применением N,N-диизопропил-N-этиламина или триэтиламина в подходящем растворителе, таком как дихлорметан, N,N-диметилформамид, тетрагидрофуран, 1,4-диоксан или N,N-диметилацетамид, в присутствии подходящего конденсирующего агента, например, дициклогексилкарбодиимида (ДЦК), 1-этил-3-(3'-диметиламинопропил)карбодиимида (ЭДК), 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазина (DHBT), тетрафторбората O-бензотриазолилтетраметилизоурония (TBTU), гексафторфосфата бензотриазол-1-илокситрипирролидинофосфония (PyBOP) или гексафторфосфата 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU). Указанную реакцию необязательно проводят в присутствии подходящего катализатора, такого как 4-диметиламинопиридин, или в присутствии дополнительного связывающего реагента, такого как N-гидроксибензотриазол. В качестве альтернативы, указанную реакцию также проводят, например, методом смешанных ангидридов с применением алкилхлорформиата, такого как этил, изопропил, бензилхлорформиат, в присутствии третичного амина, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин, в подходящем растворителе, таком как, например, толуол, дихлорметан, тетрагидрофуран, N,N-диметилформамид и т.п., при комнатной температуре.
Согласно этапу (эт. 6c) указанного способа карбоксиэфирная группа соединения формулы (VIII) может быть превращена в карбоксамид, или N-замещенные карбоксамиды, или N,N-дизамещенные карбоксамиды в щелочных условиях, таких как 1 н. бис-триметилсилиламид лития в ТГФ, с применением хлорида аммония или подходящего первичного или вторичного амина; предпочтительно реакцию проводят в тетрагидрофуране при температуре в диапазоне от 0°C до температуры обратной конденсации.
Примечательно, что при проведении указанной реакции в случае, если A представляет собой -CH=CH-, получали смесь желаемых продуктов формулы (I), где R3 представляет собой R'RʺN-, a R5 представляет собой водород или группу R'RʺN-. Затем указанные два производных выделяли из реакционной смеси традиционными способами, например, посредством хроматографии или препаративной ВЭЖХ.
Согласно этапу (эт. 7) указанного способа, проводят реакцию соединения формулы (XIII) с соединением формулы R2Y (V), где R2 определен выше, но не водород, и Y определен выше, в присутствии основания, такого как карбонат калия или цезия, в подходящем растворителе, таком как ацетонитрил, диметилформамид, с получением производных формулы (XIV).
Согласно этапу (эт. 8) указанного способа соединение формулы (XIV) можно превратить в соединение формулы (I) посредством применения любой из реакций кросс-сочетания, подходящих для образования связей углерод-углерод. Указанные реакции, которые хорошо известны в данной области, обеспечивают связь с подходящим металлоорганическим реагентом, таким как, например, борорганический (реакция Судзуки), оловоорганический (реакция Стилле), магнийорганический (реакция Кумада), цинкорганический, или алюминийорганический, или цирконийорганический реагент (реакция Негиши) и т.п. Предпочтительной реакцией является реакция Судзуки, в которой применяют подходящее производное арил или гетероарилбороновой кислоты в присутствии катализатора на основе палладия, такого как комплекс дихлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) с дихлорметаном (PdCl2(dppf)2⋅CH2Cl2), и основания, такого как карбонат натрия или цезия, в смеси растворителей, таких как диметоксиэтан и вода, при температуре в диапазоне от комнатной температуры до 80°C и в течение периода времени между 2 часами и целой ночью.
Согласно этапу (эт. 9) указанного способа соединение формулы (XVIII) можно превратить в соединение формулы (VIII) посредством применения любой из реакций кросс-сочетания, подходящих для образования связей углерод-углерод, таких как уже описанные на этапе 8.
Согласно превращению (превр. a) указанного способа, превращение тиогруппы в сульфонильную группу может быть достигнуто посредством проведения реакции с окисляющим агентом, хорошо известным специалистам в данной области, таким как, например, оксон в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, ацетон, необязательно в присутствии воды в качестве сорастворителя, или м-хлорпербензойной кислоты в присутствии подходящего растворителя, предпочтительно ДХМ, при комнатной температуре.
Согласно превращению (превр. b) указанного способа соединение формулы (I), где R4 определен выше, а X представляет собой -O-, можно легко получить путем проведения реакции соответствующего сульфонильного производного с производным формулы (XVI) R4-OH. Реакция может быть проведена в присутствии основания, такого как карбонат калия или натрия, гидроксид натрия или лития или другого подобного основания, в подходящем растворителе, таком как ацетонитрил, N,N-диметилформамид или диметилсульфоксид, и при температуре в диапазоне от комнатной температуры до примерно 100°C.
Примечательно, что при проведении указанной реакции с применением соединения формулы (I), где A представляет собой -CH2-CH2-, получали смесь желаемых продуктов формулы (I), где A представляет собой группу -CH2-CH2- или -CH=CH-. Затем указанные два производных выделяли из реакционной смеси традиционными способами, например, посредством хроматографии или препаративной ВЭЖХ.
Согласно превращению (превр. c) указанного способа сульфонильное производное формулы (I) обрабатывают подходящим нуклеофилом, таким как вторичный амин формулы R4R6NH, с получением соединения формулы (I), где R1 представляет собой R4R6N-. Указанную реакцию осуществляют с применением избытка указанного амина или, в качестве альтернативы, в подходящем растворителе, таком как, например, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, при температуре в диапазоне от комнатной температуры до примерно 100°C в течение периода времени от 2 часов до 24 часов.
Согласно превращению (превр. d) указанного способа превращение сульфонильного производного формулы (I) в соединение формулы (I), где R1 представляет собой -CN, может быть выполнено, например, с применением цианида натрия в подходящем растворителе, таком как ацетонитрил, N,N-диметилформамид или их смесь, при температуре в диапазоне от 20°C до температуры кипения в течение периода времени от 2 часов до 24 часов.
Согласно превращению (превр. e) указанного способа превращение соединения формулы (I) в другое соединение формулы (I) может быть выполнено с применением соединения формулы R2-Y' (V), где Y' представляет собой OH, в случае чего могут быть применены условия реакции Мицунобу, или Y представляет собой группу, которая необязательно после активации может служить уходящей группой, такую как атом галогена, тозилат, мезилат или трифлат.
В первом случае, то есть при применении схемы реакции Мицунобу, реакция может быть проведена с применением диалкилазодикарбоксилата, такого как диэтилазодикарбоксилат (ДЭАД), диизопропилазодикарбоксилат (ДИАД) или другого подобного соединения, в присутствии триалкилфосфина или триарилфосфина, предпочтительно трифенилфосфина, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, ацетонитрил. Если Y представляет собой галоген или группу, такую как тозилат, мезилат или трифлат или подобную группу, превращение может быть выполнено с применением подходящего основания, такого как, например, NaH, K2CO3, CS2CO3, DBU, KO-t-Bu и т.п., в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид и т.п. Указанные реакции могут быть проведены при температурах в диапазоне от 0°C до температуры обратной конденсации и в течение периода времени от 30 минут до примерно 48 часов.
Согласно превращению (превр. f) указанного способа соединение формулы (I), где R2 представляет собой галогенэтил, предпочтительно хлорэтил, обрабатывают основанием, предпочтительно 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU), при температуре в диапазоне от 20°C до 80°C, с получением соответствующего соединения формулы (I), где R2 представляет собой винил.
Согласно превращению (превр. g) указанного способа соединение формулы (I), где R2 представляет собой группу формулы L-CH2Cl, смешивают с подходящим нуклеофилом, таким как первичный или вторичный амин формулы (XII) NHR'Rʺ, в присутствии основания, такого как, например, карбонат калия, карбонат цезия, триэтиламин, DBU, в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил, N,N-диметилформамид и их смесь, при температуре в диапазоне от 50°C до 100°C и в течение подходящего периода времени, например, от 2 часов до 24 часов.
Согласно превращению (превр. h) указанного способа превращение соединения формулы (I), где R2 представляет собой группу L-CH2OH, в соединение формулы (I), где R2 представляет собой группу L-CH2NR'Rʺ, может быть выполнено несколькими способами, и рабочие условия точно установлены специалистами в данной области. Только в качестве примера далее описана двухэтапная последовательность, включающая сначала образование альдегида формулы (I), где R2 представляет собой группу L-CHO, который затем подвергают реакции с амином формулы (XII) NHR'Rʺ в условиях восстановительного аминирования. Соответственно, соединение формулы (I) с группой L-CH2OH сначала превращают в соответствующий альдегид путем обработки окисляющим агентом, таким как, например, 2-йодоксибензойная кислота (IBX), в подходящем растворителе, таком как этилацетат, тетрагидрофуран и т.п., при температуре в диапазоне от 50°C до температуры обратной конденсации в течение подходящего периода времени, например, от 30 минут до 4 часов. Затем полученный альдегид подвергают реакции с подходящим амином формулы (XII) NHR'Rʺ в присутствии восстанавливающего агента, такого как, например, цианоборгидрид натрия, триацетоксиборгидрид натрия или триацетоксиборгидрид тетраметиламмония, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид или их смеси, при температуре в диапазоне между 0°C и комнатной температурой в течение периода времени от 30 минут до 6 часов.
Согласно превращению (превр. i) указанного способа восстановление сложного эфира карбоной кислоты до соответствующего первичного спирта осуществляют с применением подходящего восстанавливающего агента, такого как, например, алюмогидрид лития, боргидрид лития, боргидрид натрия или другой подобный агент, в подходящем растворителе, таком как тетрагидрофуран, диэтиловый эфир, толуол, этанол и т.п., при температуре в диапазоне от 0°C до комнатной температуры в течение подходящего времени реакции, составляющего период между 30 минутами и 24 часами.
Согласно превращению (превр. j) указанного способа превращение альдегидного остатка в соответствующие алкиламиновые производные -CH2NR'Rʺ, где R' и Rʺ определены выше, может быть достигнуто посредством проведения реакции альдегидного производного с амином формулы (XII), определенной выше, в условиях восстановительного аминирования, предпочтительно с восстанавливающим агентом, таким как, например, цианоборгидрид натрия, триацетоксиборгидрид натрия или триацетоксиборгидрид тетраметиламмония, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид или их смеси, при температуре в диапазоне между 0°C и комнатной температурой в течение периода времени от 30 минут до 6 часов.
Согласно превращению (превр. k) указанного способа соединение формулы (I) с нитрогруппой превращали в соединение формулы (I) с аминогруппой в условиях восстановления, предпочтительно с восстанавливающим агентом, таким как, например, цинковая пыль, в присутствии хлорида аммония в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, этанол и вода или их смеси, при температуре в диапазоне между 50°C и температурой обратной конденсации в течение периода времени от 30 минут до 6 часов.
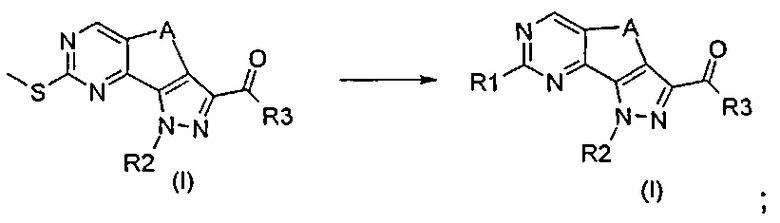
Согласно превращению (превр. l) указанного способа превращение соединения формулы (I), где R1 представляет собой Me-S-, в соединение формулы (I), где R1 представляет собой, например, арил или гетероарил, осуществляют посредством проведения реакции с подходящим металлооганическим реагентом, таким как, например, органобороновая кислота формулы R1-B(OH)2. Реакция представляет собой Pd-катализируемое Cu-опосредованное десульфитативное C-C кросс-сочетание, обычно называемое «реакция Либескинда-Срогла». Указанную реакцию осуществляют в присутствии подходящего источника палладия, такого как, например, тетракис(трифенилфосфино)палладий [Pd(PPh3)4] или другого подобного источника, карбоксилата меди(I) в качестве металлического кофактора, такого как тиофен-2-карбоксилат меди, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид, при температуре обратной конденсации в течение периода времени от 30 минут до 6 часов.
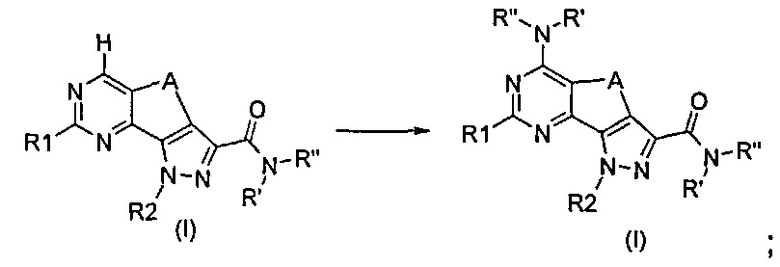
Согласно превращению (превр. m) указанного способа соединение формулы (I), где R5 представляет собой водород, можно превратить в соединение формулы (I), где R5 представляет собой R'RʺN-, в условиях применения сильного основания, такого как 1 н. бис-триметилсилиламид лития в ТГФ, с применением хлорида аммония или подходящего первичного или вторичного амина; предпочтительно реакцию проводят в тетрагидрофуране при температуре в диапазоне от 0°C до температуры обратной конденсации.
Согласно превращению (превр. n) указанного способа соединение формулы (I), в котором присутствует первичная или вторичная аминогруппа, такая как L-N(H)R', превращают в соответствующее производное вторичного или третичного амина. Предпочтительно реакцию проводят с альдегидом формулы RʺCHO (XIX) в условиях восстановительного аминирования, предпочтительно с применением восстанавливающего агента, такого как, например, цианоборгидрид натрия, триацетоксиборгидрид натрия или триацетоксиборгидрид тетраметиламмония, в подходящем растворителе, таком как тетрагидрофуран, 1,4-диоксан, N,N-диметилформамид или их смеси, при температуре в диапазоне между 0°C и комнатной температурой в течение периода времени от 30 минут до 6 часов.
Согласно превращению (превр. o) указанного способа соединение формулы (I), в котором присутствует первичная или вторичная аминогруппа, такая как L-N(H)R', превращают в соответствующее карбоксамидное производное путем проведения реакции с соединением формулы R7-COW (XX). Специалисту в данной области понятно, что указанную реакцию можно осуществить различными способами и в различных условиях, которые широко известны в данной области как применяемые для получения карбоксамидов. Например, если W представляет собой галоген, такой как хлорид, реакцию проводят в подходящем растворителе, таком как, например, дихлорметан, тетрагидрофуран, 1,4-диоксан, ацетонитрил или N,N-диметилформамид или другой подобный растворитель, при температуре в диапазоне от примерно -10°C до температуры обратной конденсации и в течение подходящего периода времени, например, от примерно 30 минут до примерно 96 часов. Реакцию проводят в присутствии подходящего акцептора протонов, такого как триэтиламин, N,N-диизопропилэтиламин или пиридин. Если W представляет собой гидроксильную группу, реакцию проводят в присутствии связывающего агента, такого как, например, тетрафторборат 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU), 1,3-дициклогексилкарбодиимид, 1,3-диизопропилкарбодиимид, 1-(3-диметиламинопропил)-3-этилкарбодиимид, N-циклогексилкарбодиимид-N'-пропилметилполистирол или N-циклогексилкарбодиимид-N'-метилполистирол, в подходящем растворителе, таком как, например, дихлорметан, тетрагидрофуран, 1,4-диоксан, ацетонитрил, N,N-диметилформамид, при температуре в диапазоне от примерно -10°C до температуры обратной конденсации и в течение подходящего периода времени, например, от примерно 30 минут до примерно 48 часов. Указанную реакцию необязательно проводят в присутствии подходящего катализатора, например, 4-диметиламинопиридина, или в присутствии дополнительного связывающего агента, такого как N-гидроксибензотриазол.
Согласно этапу (превр. p) указанного способа соединение формулы (I), в котором присутствует первичная или вторичная аминогруппа, такая как L-N(H)R', превращают в соответствующее производное мочевины путем проведения реакции с подходящим изоцианатом формулы R8-N=C=O (XXI) с получением соответствующего производного мочевины. Реакцию предпочтительно проводят в подходящем растворителе, таком как дихлорметан, тетрагидрофуран или другой подобный растворитель, при температуре в диапазоне от примерно 20°C до температуры обратной конденсации и в течение периода времени в диапазоне от примерно 30 минут до примерно 48 часов.
Согласно превращению (превр. q) указанного способа соединение формулы (I), в котором присутствует первичная или вторичная аминогруппа, такая как L-N(H)R', превращают в соответствующее гуанидиновое производное путем проведения реакции с соединением формулы R8NHC(NH)-G, где G представляет собой подходящую уходящую группу, такую как -S-Me, N-S(O)2CF3 или 1H-пиразолил. Например, указанная реакция может быть проведена в щелочных условиях, например, в присутствии триэтиламина или карбоната калия, в подходящем растворителе, таком как метанол, этанол, N,N-диметилформамид и их смеси. Предпочтительно реакцию проводят при температуре в диапазоне от комнатной температуры до примерно 80°C и в течение периода времени в диапазоне от примерно 30 минут до примерно 24 часов.
Согласно превращению (превр. r) указанного способа соединение формулы (I), где A представляет собой -(CH2)2-, может подвергаться дегидрированию в присутствии необязательно нанесенного палладия или платины или 2,3-дихлор-5,6-дициано-1,4-бензохинона (ДДХ) с получением соответствующего ароматического производного формулы (I) путем действия в подходящем растворителе, таком как толуол, 1,4-диоксан, хлорбензол, дихлорбензол, при температуре в диапазоне от 90°C до температуры кипения в течение периода времени в диапазоне от 2 часов до 8 часов.
Разумеется, любой из промежуточных продуктов, полученных при применении способов, описанных выше, в случае желания и необходимости можно превратить в другой промежуточный продукт аналогичным образом, то есть посредством любой из реакций превращения, описанных выше.
Исходя из всего вышеизложенного специалисту в данной области понятно, что любое соединение формулы (I), содержащее функциональную группу, которая может быть дополнительно дериватизирована до другой функциональной группы способами, хорошо известными в данной области, тем самым приводя к образованию других соединений формулы (I), предназначено для включения в объем настоящего изобретения.
Специалисту в данной области также понятно, что при необходимости реакционно-способные группы могут быть защищены, а затем удалены способами, хорошо известными в литературе, например, защитные группы в органическом синтезе.
Согласно любому варианту способа получения соединений формулы (I) исходные материалы и любые другие реактивы являются известными или легко получаемыми известными способами.
Соединение формулы (II) можно получить, как описано в WO 2004/104007.
Соединения формулы (XIII) и (XVIII) можно получить, как описано в WO 2008/074788.
Соединения формулы (III), (VIIa), (VIIb), (IX), (X), (XII), (XV), (XVII), (XIX), (XX), (XXI) и (XXII) являются коммерчески доступными или могут быть получены известными способами.
Соединения формулы (V) и (XVI) являются коммерчески доступными или могут быть получены известными способами, или могут быть получены, как описано ниже в экспериментальном разделе (от Получения O до Получения R).
Исходя из всего вышеизложенного специалисту в данной области понятно, что при получении соединений формулы (I) любым из вышеуказанных вариантов способа дополнительные функциональные группы в исходных материалах или промежуточных продуктах, наличие которых может привести к нежелательным побочным реакциям, необходимо должным образом защищать с применением традиционных технологий. Аналогично, превращение соединений с защищенными группами в свободные, незащищенные соединения может быть осуществлено известными способами.
Как будет легко понять, если соединения формулы (I), полученные способом, описанным выше, получают в виде смеси изомеров, ее разделение на отдельные изомеры формулы (I) с применением традиционных технологий входит в объем настоящего изобретения.
Традиционные технологии разделения рацемата включают, например, секционированную кристаллизацию производных, представляющих собой диастереомерные соли, или препаративную хиральную ВЭЖХ.
Соединения согласно настоящему изобретению могут быть введены в виде простых агентов или, в качестве альтернативы, в комбинации с известными видами противоопухолевой терапии, такими как лучевая терапия или режим химиотерапии в комбинации с цитостатическими или цитотоксическими агентами, агентами, представляющими собой антибиотики, алкилирующими агентами, антиметаболитными агентами, гормональными агентами, иммунологическими агентами, агентами, представляющими собой интерфероны, ингибиторами циклооксигеназы (например, ингибиторами ЦОГ-2), ингибиторами матриксной металлопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, агентами против рецепторов факторов роста, агентами против HER, агентами против EGFR, агентами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути сигнальной трансдукции ras-raf, ингибиторами клеточного цикла, другими ингибиторами cdks, агентами, связывающими тубулин, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и т.п.
В случае приготовления в виде препарата с фиксированными дозами препараты, представляющие собой такие комбинации, содержат соединения согласно настоящему изобретению в диапазоне доз, описанных ниже, и другой фармацевтически активный агент в диапазоне доз, соответствующем стандарту.
Если комбинационный состав является неприемлемым, соединения формулы (I) и известные противоопухолевые агенты могут быть применены последовательно.
Соединения формулы (I) согласно настоящему изобретению, подходящие для введения млекопитающему, например, людям, могут быть введены обычными путями, а уровень дозы зависит от возраста, массы, состояния пациента и пути введения.
Например, подходящая дозировка, установленная для перорального введения соединения формулы (I), может варьироваться от примерно 10 до примерно 500 мг на дозу, от 1 до 5 раз в сутки. Соединения согласно настоящему изобретению могут быть введены в различных дозированных формах, например, перорально в виде таблеток, капсул, таблеток, покрытых сахаром или пленочной оболочкой, жидких растворов или суспензий; ректально в виде суппозиториев; парентерально, например, внутримышечно или посредством внутривенной, и/или интратекальной, и/или интраспинальной инъекции или инфузии.
Настоящее изобретение также включает фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым наполнителем, который может представлять собой носитель или разбавитель.
Фармацевтические композиции, содержащие соединения согласно настоящему изобретению, обычно получают традиционными способами и вводят в подходящей фармацевтической форме. Например, твердые пероральные формы совместно с активным соединением могут содержать разбавители, например, лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; скользящие вещества, например, диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; связывающие агенты, например, крахмалы, гуммиарабик, желатин, метил целлюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; разрыхляющие агенты, например, крахмал, альгиновую кислоту, альгинаты или карбоксиметилкрахмал натрия; шипучие смеси; красители; подсластители; смачивающие агенты, такие как летицин, полисорбаты, лаурилсульфаты и, в общем, нетоксичные и фармакологически неактивные вещества, применяемые в фармацевтических составах. Указанные фармацевтические препараты могут быть произведены известным способом, например, посредством смешивания, гранулирования, таблетирования, покрытия сахаром или покрытия пленочной оболочкой.
Жидкие дисперсии для перорального введения могут представлять собой, например, сиропы, эмульсии и суспензии. Например, сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннит и сорбит.
Суспензии и эмульсии могут содержать в качестве носителей, например, натуральную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензия или растворы для внутримышечных инъекций совместно с активным соединением могут содержать фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль, и, при желании, подходящее количество лидокаина гидрохлорида.
Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду, или предпочтительно могут находиться в виде стерильных, водных, изотонических, солевых растворов, или могут содержать пропиленгликоль в качестве носителя.
Суппозитории совместно с активным соединением могут содержать фармацевтически приемлемый носитель, например, масло какао, полиэтиленгликоль, поверхностно-активное вещество полиоксиэтиленовый сложный эфир сорбита и жирной кислоты или лецитин.
Следующие примеры изложены далее с целью лучшей иллюстрации настоящего изобретения и не ставят каких-либо ограничений.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
ФАРМАКОЛОГИЯ
Соединения формулы (I) действуют в качестве ингибиторов протеинкиназы и, следовательно, подходят, например, для ограничения нерегулируемой пролиферации опухолевых клеток.
В терапии они могут быть применены в лечении различных опухолей, таких как определенные ранее, а также в лечении других нарушений пролиферации клеток и заболеваний и нарушений, вызванных иммунными клетками.
Ингибирующую активность ингибиторов PIM-1 и 2 и эффективность выбранных соединений определяли посредством анализов, описанных ниже.
Для ссылки на любое конкретное соединение формулы (I) согласно настоящему изобретению, необязательно в виде фармацевтически приемлемой соли см. экспериментальный раздел и формулу изобретения. В следующих примерах соединения согласно настоящему изобретению синтезировали с применением способов, описанных в настоящей заявке, или других способов, которые хорошо известны в данной области.
Сокращенные формы и аббревиатуры, используемые в настоящей заявке, имеют следующие значения:
Следующие примеры изложены далее с целью лучшей иллюстрации настоящего изобретения и не ставят каких-либо ограничений.
В настоящей заявке символы и обозначения, применяемые в способах, схемах и примерах, соответствуют тем, которые используются в современной научной литературе, например, Journal of the American Chemical Society или Journal of Biological Chemistry.
Если не указано иное, все материалы получали от частных поставщиков, наивысшей степени чистоты и применяли без дополнительной очистки. Безводный растворитель, такой как ДМФА, ТГФ, CH2Cl2 и толуол, получали от Aldrich Chemical Company. Все реакции с участием соединений, чувствительных к воздействию воздуха или влаги, проводили в атмосфере азота или аргона.
Биохимический анализ ингибиторов активности PIM-1 киназы
Ингибирующую активность предполагаемых ингибиторов киназы и эффективность выбранных соединений определяли с применением анализа транс-фосфорилирования.
Конкретные пептидные или белковые субстраты транс-фосфорилировали посредством конкретной ser-thr или tyr киназы указанных субстратов в присутствии АТФ, меченой 33P-γ-АТФ, и в присутствии оптимального буферного раствора и кофакторов.
В конце реакции фосфорилирования более чем 98% немеченого АТФ и радиоактивного АТФ захватывались избытком ионообменной смолы Dowex®; затем смола оседала на дно реакционного планшета под действием силы тяжести.
Надосадочную жидкость впоследствии извлекали и перемещали на счетную пластинку, а затем исследовали путем подсчета β-частиц.
Реагенты / условия анализа
Приготовление смолы Dowex®:
Отвешивали 500 г влажной смолы (SIGMA, полученная на заказ смола DOWEX® 1×8 200-400 меш, 2,5 кг) и разбавляли до 2 л 150 мМ формиатом натрия, pH 3,00.
Обеспечивали осаждение смолы (в течение нескольких часов), а затем удаляли надосадочную жидкость.
После трех промываний, как указано выше, в течение нескольких дней обеспечивали осаждение смолы и добавляли два объема (относительно объема смолы) 150 мМ буферного раствора формиата натрия.
Затем измеряли pH, который должен был составлять примерно 3,00.
Промытая смола стабильна более недели; запас смолы перед применением хранят при 4°C.
Буферный раствор для киназы (КБ):
Буферный раствор для анализа PIM-1 состоял из 50 мМ HEPES при pH 7,5 с 10 мМ MgCl2, 1 мМ ДТТ, 3 мкМ NaVO3 и 0,2 мг/мл БСА.
Полноразмерный PIM-1 человека экспрессировали и очищали, как описано в Bullock A.N. и др., J. Biol. Chem., 2005, 280, 41675-82.
Фермент демонстрировал линейную кинетику после этапа предварительной активации посредством автофосфорилирования в следующих условиях:
1,7 мкМ PIM1 выдерживали в течение 1 часа при 28°C КТ в присутствии 125 мкМ АТФ.
Условия анализа:
Концентрация АТФ: 200 мкМ.
33P-γ-АТФ: 6 нМ.
Концентрация фермента: 1 нМ.
Концентрация субстрата Aktide (регистрационный номер по реестру Химической реферативной службы 324029-01-8): 25 мкМ.
Автоматизированный анализ Dowex®:
Исследуемая смесь состояла из:
1) 3x ферментной смеси (приготовленной в буферном растворе для киназы 3X), 5 мкл/лунка;
2) 3x смеси субстрата и АТР (приготовленной в ddH2O), совместно с 33P-γ-АТФ, 5 мкл/лунка;
3) 3x исследуемого соединения (разбавленного ddH2O - 3% ДМСО), 5 мкл/лунка.
Разбавление соединений и схему анализа см. ниже.
Разбавление соединений
Для определения IC50 исследуемые соединения получали в виде 1 мМ раствора в 100% ДМСО и распределяли по 96-луночным планшетам: затем соединения высевали в первую колонку нового 96-луночного планшета (от A1 до G1), 100 мкл/лунка.
Для серийных разбавлений применяли автоматическую станцию (Biomek FX®, Beckman), производя разбавления 100% ДМСО в отношении 1:3 по линии от A1 до A10, аналогично разбавляли все соединения в колонке. Кроме того, готовили 4-5 копий из дочерних планшетов путем переформатирования 5 мкл раствора из планшетов указанной первой серии разбавления 100% ДМСО в 384-луночные планшеты с глубокими лунками: одну копию указанных планшетов серийного разбавления с исследуемыми соединениями размораживали в день проведения исследования, растворы разбавляли до рабочей концентрации (3-кратная конечная концентрация) с применением 162 мкл/лунка воды и применяли в анализах для определения IC50. В стандартном эксперименте наивысшая концентрация (3X) соединений, как правило, составляет 30 мкМ, тогда как наиболее низкая концентрация, как правило, составляет 1,5 нМ.
Каждый 384-луночный планшет обеспечивал по меньшей мере одну кривую стандартного ингибитора, представлявшего собой стауроспорин, и контрольные лунки (общая активность фермента по отношению к отсутствию ферментной активности) для оценки Z' и отношения сигнала к фону (S/B).
Схема анализа:
Готовили 384-луночные планшеты с V-образным дном (исследуемые планшеты) с 5 мкл соединения, разбавленного, как описано ранее, (3X), а затем помещали их в автоматическую станцию PlateTrak® 12 (Perkin Elmer; роботизированное устройство имело одну 384-конечную пипетирующую головку для начала анализа и одну 96-конечную головку для дозирования смолы) совместно с одной емкостью с ферментной смесью (3X) и одной емкостью со смесью АТФ (3X).
Данные анализировали посредством внутренне адаптированной версии пакета программ «Assay Explorer®», которая обеспечивала подбор сигмоидальных кривых, полученных в результате десяти разбавлений, для определения IC50 в программах вторичного анализа/подтверждения результата.
Способ анализа ингибирования киназы PIM-2: технология Dowex.
Буферный раствор для киназы (КБ):
Буферный раствор для анализа PIM-2 состоял из 50 мМ HEPES при pH 7,5 с 1 мМ MgCl2, 1 мМ ДТТ, 3 мкМ Na3VO4 и 0,2 мг/мл БСА.
Полноразмерный PIM-2 человека экспрессировали и очищали, как описано в Fedorov О. и др., (2007) PNAS 104, 51, 20523-28.
Условия анализа (конечные концентрации):
Концентрация фермента: 1,5 нМ.
Концентрация субстрата Aktide (регистрационный номер по реестру Химической реферативной службы 324029-01-8): 5 мкМ.
Концентрация АТФ: 4 мкМ.
Концентрация 33P-γ-АТФ: 1 нМ.
Автоматизированный анализ Dowex:
См. выше: процедура та же, что описана для PIM-1.
Анализ пролиферации клеток in vitro:
Клетки MV-4-11 (бифенотипический B миеломоноцитарный лейкоз) (1250 клетки/лунка) высевали в белые 384-луночные планшеты на полную среду (RPMI 1640 или EMEM плюс 10% фетальная бычья сыворотка) и обрабатывали соединениями, растворенными в 0,1% ДМСО, в течение 24 ч после посева. Указанные клетки инкубировали при 37°C и 5% CO2, и через 72 часа планшеты обрабатывали с применением анализа CellTiter-Glo® (Promega), следуя инструкции изготовителя. CellTiter-Glo® представляет собой гомогенный способ, основанный на определении количества присутствующей АТФ, индикатора метаболически активных клеток. Количество АТФ определяют с применением системы на основе люциферазы и D-люциферина, приводящих к испусканию света. Люминесцентный сигнал пропорционален количеству клеток, присутствующих в культуре.
Вкратце, 25 мкл/лунка раствора реагента добавляли в каждую лунку и через 5 минут показания со встряхиваемых микропланшетов снимали с помощью люминометра Envision® (PerkinElmer). Люминесцентный сигнал был пропорционален количеству клеток, присутствующих в культуре.
Ингибирующую активность оценивали, сравнивая обработанные данные с контрольными с применением программы Assay Explorer (MDL). IC50 вычисляли с применением сигмоидальной кривой интерполяции.
С учетом вышеизложенных анализов ингибирования, соединения формулы (I) согласно настоящему изобретению обладали высокой ингибирующей активностью по отношению к PIM-1, как правило, с IC50 намного ниже 1 мкМ и высокой ингибирующей активностью по отношению к PIM-2, как правило, с IC50 ниже 10 мкМ. Более того, соединения формулы (I) согласно настоящему изобретению демонстрируют высокую ингибирующую активность по отношению к пролиферации клеток MV-4-11, как правило, с IC50 в диапазоне от 0,010 до 2 мкМ.
В следующей Таблице A представлены экспериментальные данные для некоторых репрезентативных соединений формулы (I) согласно настоящему изобретению, подвергнутых испытанию с применением ферментов PIM-1 и 2 в конкретном анализе киназы in vitro, описанном выше (мкМ IC50).
Также в следующей Таблице A представлена ингибирующая активность некоторых наиболее близких по структуре соединений, известных из уровня техники, по отношению к PIM-1 и PIM-2.
Контрольное соединение 1 представляет собой 1-метил-8-(пиперидин-1-ил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид, указанное соединение имело код B76-X06-M00(C01)-D03; и контрольное соединение 2 представляет собой 1-метил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид, в патентной заявке WO 2004/104007, приведенной выше, указанное соединение имело код B67-X04-M00(C01-D03). Контрольные соединения 1 и 2 представляют собой первое и второе соединения, исключенные из объектов настоящего изобретения, соответственно.
Также в следующей Таблице A представлена антипролиферативная активность некоторых репрезентативных соединений согласно настоящему изобретению по отношению к клеткам MV-4-11 миеломоноцитарного лейкоза.
Неожиданно ингибирующая активность соединений согласно настоящему изобретению по отношению к PIM-1 и PIM-2 оказалась заметно выше, чем активность контрольных соединений.
Новые соединения согласно настоящему изобретению неожиданно обладают значительно более высокой ингибирующей активностью по отношению к PIM-1 и PIM-2, чем наиболее близкие по структуре соединения, известные из уровня техники, представляющие собой соединения согласно вышеуказанной WO 2004/104007, и, следовательно, при применении в терапии особенно полезны при лечении пролиферативных нарушений, вызванных измененной активностью киназ.
Также чтобы установить, что цитотоксичность соединений согласно настоящему изобретению была следствием ингибирования киназы PIM, было исследовано фосфорилирование проапоптотического белка BAD киназой PIM в присутствии или в отсутствии ингибиторов PIM в лейкозных клетках линии MV-4-11.
Было показано, что BAD, представляющий собой нижележащую мишень киназы PIM, был непосредственно фосфорилирован PIM-1, PIM-2 и PIM-3 по Ser112 (Pogacic V. и др. (2007) Cancer Research, 67(14); 6916-24).
Было обнаружено, что соединения согласно настоящему изобретению эффективно и дозозависимо ингибировали фосфорилирование BAD по Ser 112 в MV-4-11, тем самым подтвердив, что указанные соединения являются ингибиторами PIM. Результаты исследования представлены на Фигуре 2.
MV-4-11 клетки высевали в 6-луночные планшеты в концентрации 1,5×106 клеток/мл и обрабатывали PIM-ингибиторами в различных концентрациях в течение 3 часов. Клетки собирали, промывали буферным раствором PBS и лизировали в буфере для лизиса (2% SDS, 100 mM Трис при pH 7,5, 1:100 коктейля ингибиторов фосфатазы 1 (SIGMA) и 1:100 коктейля ингибиторов фосфатазы 2 (SIGMA) и ингибитор протеаз Complete (Roche). Концентрацию белка в лизатах определяли с применением ВСА Protein Assay Kit (Pierce). Равные количества белка наносили на трис-глициновый гель с градиентом концентрации полиакриламида 4-12% для проведения анализа SDS-PAGE. Затем белки переносили на PVDF мембраны (Millipore) для проведения вестерн-блоттинга. Мембраны исследовали с применением антител к фосфо-Bad (Ser112) (Cell Signaling Technologies).
ПРИМЕРЫ
В следующих примерах описано получение в результате синтеза некоторых соединений формулы (I) согласно настоящему изобретению.
Соединения согласно настоящему изобретению, полученные согласно следующим примерам, также были охарактеризованы данными анализа 1H ЯМР или ВЭЖХ/МС; данные ВЭЖХ/МС собирали любым из способов 1, 2, 3 и 4.
Способ ВЭЖХ/МС анализа 1
ВЭЖХ оборудование состояло из системы Waters Acquity™ UPLC, оснащенной детектором PDA 2996 Waters, и одноквадрупольного масс-спектрометра Micromass модель ZQ, оснащенного источником электрораспыления ионов (ИЭР). Управление измерительными приборами, сбор и обработку данных обеспечивали с помощью программного обеспечения Empower® и MassLynx® 4.0.
ВЭЖХ проводили при 45°C при скорости потока 0,8 мл/мин с применением колонки Waters Acquity® UPLC BEH C18 (2,1×50 мм), 1,7 микрон. Подвижная фаза A представляла собой буферный раствор, содержавший 0,1% муравьиной кислоты, с pH=3,3 с ацетонитрилом (98:2), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент составлял от 5 до 95% B в течение 2 минут, а затем поддерживали 95% B в течение 0,1 минуты. Объем вводимой пробы составлял 2 мкл. Масс-спектрометр работал в режиме определения положительно и отрицательно заряженных ионов, на капилляре создавали напряжение 3,5 кВ (ES+) и 28 B (ES-); температура источника ионизации составляла 120°C; напряжение на конусе составляло 14 B (ES+) и 2,8 кВ (ES-); устанавливали полное сканирование в диапазоне масс от 100 до 800 а.е.м.
Способ ВЭЖХ/МС анализа 2
ВЭЖХ/МС анализы проводили на масс-спектрометре типа ионная ловушка Finnigan MAT модель LCQ®, оснащенном источником ионов для ИЭР (электроспрея), масс-спектрометр непосредственно присоединяли к системе ВЭЖХ SSP4000 (Thermo Separation), оснащенной автосамплером Lc Pal (CTC Analytics) и детектором PDA UV6000LP.
ВЭЖХ проводили при 40°C при скорости потока 1,0 мл/мин с применением колонки Phenomenex Gemini C18, 3 мкм, 50×4,6 мм. Подвижная фаза A представляла собой 5 мМ ацетатный буферный раствор с pH 4,5 : ацетонитрил 95:5 (об.:об.), а подвижная фаза В представляла собой 5 мМ ацетатный буферный раствор с pH 4,5: ацетонитрил 5:95 (об.:об.), градиент составлял от 0 до 100% B в течение 7 минут, а затем поддерживали 100% B в течение 2 минут перед уравновешиванием. Полное время проведения жидкостной хроматографии составляло 10 минут. Объем вводимой пробы составлял 10 мкл.
Условия масс-спектроскопии: масс-спектрометр LCQ работал с интерфейсом, представлявшим собой электрораспылительную ионизацию (ИЭР), в режиме определения положительно и отрицательно заряженных ионов. Напряжение распылителя для ИЭР составляло 4,0 кВ, температура нагретого капилляра составляла 255°C, давление защитной азотной оболочки составляло 5,0 бар. Применяли режим полного сканирования (в диапазоне масс от 50 до 1000 а.е.м.).
Эксперименты MC/MC проводили автоматически, анализируя наиболее «интенсивной» ион каждого сканирования с помощью программного обеспечения Xcalibur®. Для фрагментации ионов-прекурсоров применяли 45% энергии столкновения.
Способ ВЭЖХ/МС анализа 3
ВЭЖХ оборудование состояло из системы Waters 2795 Alliance HT, оснащенной детектором PDA 2996 Waters, и одноквадрупольного масс-спектрометра Micromass модель ZQ, оснащенного источником электрораспыления ионов (ИЭР). Управление измерительными приборами, сбор и обработку данных обеспечивали с помощью программного обеспечения Empower® и MassLynx® 4.0.
ВЭЖХ проводили при 30°C при скорости потока 1,0 мл/мин с применением колонки Phenomenex C18 (4,6×50 мм), 3 микрона. Подвижная фаза A представляла собой буферный раствор, содержащий 5 мМ ацетата аммония, с pH=5,2 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент составлял от 10 до 90% B в течение 8 минут, а затем фаза B возрастала до 100% в течение 1,0 минуты. Объем вводимой пробы составлял 10 мкл. Масс-спектрометр работал в режиме определения положительно и отрицательно заряженных ионов, на капилляре создавали напряжение 3,5 кВ (ES+) и 28 B (ES-); температура источника составляла 120°C; напряжение на конусе составляло 14 B (ES+) и 2,8 кВ (ES-); устанавливали полное сканирование в диапазоне масс от 100 до 800 а.е.м.
Способ ВЭЖХ/МС анализа 4
ВЭЖХ оборудование состояло из системы Waters 2790 HPLC, оснащенной детектором PDA 996 Waters, и одноквадрупольного масс-спектрометра Micromass модель ZQ, оснащенного источником электрораспыления ионов (ИЭР). Управление измерительными приборами, сбор и обработку данных обеспечивали с помощью программного обеспечения Empower и MassLynx 4.0.
ВЭЖХ проводили при 25°C при скорости потока 1 мл/мин с применением колонки RP18 Waters X Terra (3,0×20 мм). Подвижная фаза A представляла собой буферный раствор, содержавший 0,05% гидроксида аммония, с pH=10 с ацетонитрилом (95:5), а подвижная фаза B представляла собой H2O/ацетонитрил (5:95); градиент составлял от 10 до 90% B в течение 4 минут, а затем поддерживали 90% B в течение 1 минуты. Объем вводимой пробы составлял 10 мкл. Масс-спектрометр работал в режиме определения положительно и отрицательно заряженных ионов, на капилляре создавали напряжение 2,5 кВ; температура источника составляла 120°C; напряжение на конусе составляло 10 B; устанавливали полное сканирование в диапазоне масс от 100 до 800 а.е.м.
Некоторые соединения формулы (I) согласно настоящему изобретению, полученные согласно следующим примерам, очищали посредством препаративной ВЭЖХ.
Рабочие условия определены ниже:
Способ препаративной ВЭЖХ/МС 1
ВЭЖХ оборудование состояло из системы Waters 2790 HPLC, оснащенной детектором PDA 996 Waters, и одноквадрупольного масс-спектрометра Micromass модель ZQ, оснащенного источником электрораспыления ионов (ИЭР). Управление измерительными приборами, сбор и обработку данных обеспечивали с помощью программного обеспечения Empower и MassLynx 4.0.
ВЭЖХ проводили при 25°C при скорости потока 20 мл/мин с применением колонки RP18 Waters X Terra, 10 микрон (19×250 мм). Подвижная фаза A представляла собой буферный раствор, содержавший 0,05% гидроксида аммония, с pH=10 с ацетонитрилом (95:5), а подвижная фаза B представляла собой ацетонитрил; градиент составлял от 10 до 90% B в течение 15 минут, а затем поддерживали 90% B в течение 3 минут. Объем вводимой пробы составлял 10 мкл.
Масс-спектрометр работал в режиме определения положительно и отрицательно заряженных ионов, на капилляре создавали напряжение 2,5 кВ; температура источника составляла 120°C; напряжение на конусе составляло 10 B; устанавливали полное сканирование в диапазоне масс от 100 до 800 а.е.м.
Способ препаративной ВЭЖХ/МС 2
ВЭЖХ оборудование состояло из системы Waters 2790 HPLC, оснащенной детектором PDA 996 Waters, и одноквадрупольного масс-спектрометра Micromass модель ZQ, оснащенного источником электрораспыления ионов (ИЭР). Управление измерительными приборами, сбор и обработку данных обеспечивали с помощью программного обеспечения Empower и MassLynx 4.0.
ВЭЖХ проводили при 25°C при скорости потока 20 мл/мин с применением колонки RP18 Waters X Terra, 10 микрон (19×250 мм). Подвижная фаза A представляла собой 0,1% трифторуксусной кислоты в воде/ацетонитрил (95:5), а подвижная фаза B представляла собой ацетонитрил; градиент составлял от 10 до 90% B в течение 15 минут, затем поддерживали 90% B в течение 3 минут. Объем вводимой пробы составлял 10 мкл.
Масс-спектрометр работал в режиме определения положительно и отрицательно заряженных ионов, на капилляре создавали напряжение 2,5 кВ; температура источника составляла 120°C; напряжение на конусе составляло 10 B; устанавливали полное сканирование в диапазоне масс от 100 до 800 а.е.м.
Точная МС
Данные точной массы в результате ИЭР(+) получали на приборе Waters Q-Tof Ultima®, непосредственно присоединенном к микроколоночному ВЭЖХ 1100 Agilent, как описано ранее (M. Colombo, F. Riccardi-Sirtori, V. Rizzo, Rapid Commun. Mass Spectrom., 2004, 18, 511-517).
1H-ЯМР спектрометрию проводили на приборе с одним отсеком Bruker AVANCE® 400 МГц с градиентами, оснащенном квадратурным ядерным датчиком QNP (датчиком со сменными вставками на 4 ядра - 1H, 13C, 19F и 31P) (ЯМР способ 1) или на Mercury VX 400, действовавшем при 400,45 МГц, оснащенном 5 мм датчиком двойного резонанса [1H(15N-31P) ID_PFG Varian].
Получение A
Этиловый эфир 1-трет-бутил-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты
[(IV), R2=трет-бутил, A=-(CH2)2-]
этап 1
К раствору 10 г (42,6 ммоль) этилового эфира (3-этокси-2-оксоциклогекс-3-ен-1-ил)(оксо)уксусной кислоты и 5 мл уксусной кислоты в абсолютном этаноле (150 мл) при комнатной температуре добавляли 6 г (48 ммоль) гидрохлорида трет-бутилгидразина. Указанную смесь перемешивали при 60°C в течение 3 часов. Летучие компоненты удаляли под вакуумом, остаток разбавляли ДХМ и промывали насыщенным водным раствором NaHCO3 и рассолом. Органическую фазу сушили сульфатом натрия, фильтровали и концентрировали. Необработанный материал очищали посредством колоночной хроматографии с применением силикагеля, элюируя этилацетатом и гексаном (1:2), с получением этилового эфира 1-трет-бутил-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты с выходом 90%. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,08 мин. 1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.)=4,18 (кв, J=6,83 Гц, 2H) 2,93-2,30 (3м, 6H) 1,58 (с, 9H), 1,16 (т, J=6,83 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C14H20N2O3 [M+H]+: 287,1366, полученное значение: 287,1356.
Следующее соединение было получено согласно аналогичному способу, но с применением гидразина:
Этиловый эфир 7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты [(IV), R2=H, A=-(CH2)2-]. 1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.)=14,39 (с, 1H), 4,27 (кв, J=7,11 Гц, 2H), 2,87 (т, J=6,10 Гц, 2H), 2,51 (м, 2H), 2,04 (м, 2H), 1,28 (т, J=7,07 Гц, 3H).
Получение B
Этиловый эфир 1-(4-метоксибензил)-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты [(IVa), R2=п-метоксибензил, A=-(CH2)2-] и этиловый эфир 2-(4-метоксибензил)-7-оксо-4,5,6,7-тетрагидро-2H-индазол-3-карбоновой кислоты [(IVb), R2=п-метоксибензил, A=-(CH2)2-].
этап 1a
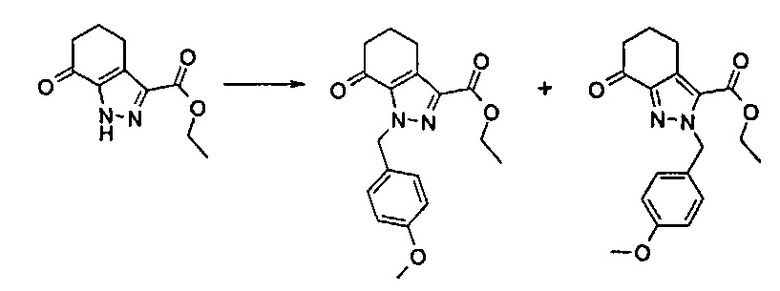
50 мг этилового эфира 7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты и 120 мг (0,37 ммоль) карбоната цезия растворяли в 1 мл безводного ДМФА, добавляли 42 мкл (0,288 ммоль) п-метоксибензилбромида и перемешивали при к.т. в течение ночи. Указанную смесь распределяли между H2O и ДХМ. Органический слой промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии (ДХМ/Ацетон 95/5) с получением двух региоизомеров:
этилового эфира 1-(4-метоксибензил)-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты с выходом 20%; 1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.)=7,10-7,30 (м, 2H), 6,75-6,96 (м, 2H), 5,64 (с, 2H), 4,29 (кв, J=7,16 Гц, 2H), 3,71 (с, 3H), 2,93 (т, J=6,10 Гц, 2H), 2,55 (дд, J=5,55, 7,26 Гц, 2H), 1,90-2,15 (м, 2H), 1,30 (т, J=7,14 Гц, 3H);
этилового эфира 2-(4-метоксибензил)-7-оксо-4,5,6,7-тетрагидро-2H-индазол-3-карбоновой кислоты с выходом 25%; 1H ЯМР (401 МГц, ДМСО-d6) δ = 7,12-7,32 (м, 2H), 6,76-6,97 (м, 2H), 5,70 (с, 2H), 4,32 (кв, J=7,12 Гц, 2H), 3,71 (с, 3H), 2,91 (т, J=6,10 Гц, 2H), 2,52-2,57 (м, 2H), 1,97-2,09 (м, 2H), 1,30 (т, J=7,08 Гц, 3H).
Получение C
Этиловый эфир (6E)-1-трет-бутил-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты [(VI), R2=трет-бутил, A=-(CH2)2-]
этап 2

10 г (37,8 ммоль) промежуточного продукта, представлявшего собой этиловый эфир 1-трет-бутил-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты, растворяли в 50 мл диметилацеталя N,N-диметилформамида и перемешивали при 110°C. Реакционную смесь перемешивали при указанной температуре в течение 16 часов. Реакционную смесь концентрировали, а затем распределяли между H2O и ДХМ. Органический слой промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения с количественным выходом. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,85 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 7,55 (с, 1H), 4,22-4,33 (м, 2H), 3,11 (с, 6H), 2,82 (с, 4H), 1,66 (с, 9H), 1,25-1,32 (м, 3H).
Получение D
Этиловый эфир 1-трет-бутил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=трет-бутил, A=-(CH2)2-]
этап 3a
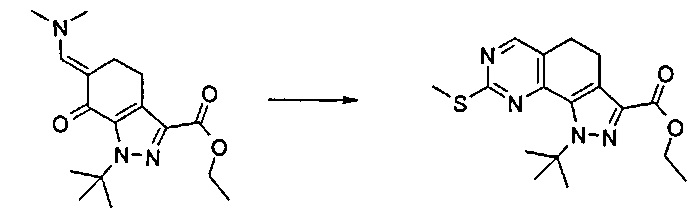
К раствору 9 г (28,17 ммоль) этилового эфира (6E)-1-трет-бутил-6-[(диметиламино)метилиден]-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты в 50 мл безводного ДМФА добавляли 9,2 г (112 ммоль) безводного ацетата калия и 23,52 г (84,51 ммоль) сульфата метилизотиомочевины. Реакционную смесь перемешивали при 100°C в течение 8 часов. Указанную смесь разбавляли этилацетатом, промывали H2O, сушили над Na2SO4, фильтровали и выпаривали. Необработанный материал очищали посредством хроматографии на силикагеле (этилацетат : гексан 1:3) с получением указанного в заголовке соединения (50%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 7,46 мин. 1H ЯМР (401 МГц, ДМСО-d6): δ = 8,58 (с, 1H), 4,22-4,38 (м, 2H), 2,95-3,01 (м, 2H), 2,80-2,86 (м, 2H), 2,57 (с, 2H), 1,76-1,83 (м, 9H). Значение МСВР (ИЭР), вычисленное для C17H22N4O2S [M+H]+: 347,1536, полученное значение: 347,1523.
Аналогичным способом были получены следующие соединения:
Этиловый эфир 8-(метилсульфанил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=тритил, A=-(CH2)2-]; 1H ЯМР (401 МГц, ДМСО-d6) δ = 7,19-7,35 (м, 10H), 6,92-7,01 (м, 6H), 4,22-4,31 (м, 2H), 3,04 (т, J=6,59 Гц, 1H), 2,55-2,60 (м, 2H), 2,41 (с, 3H), 1,22-1,30 (м, 4H);
Этиловый эфир 1-метил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=метил, A=-(CH2)2-]; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,55 (с, 1H), 4,33 (с, 3H), 4,29 (кв, J=7,08 Гц, 2H), 2,97-3,04 (м, J=1,10, 6,84 Гц, 2H), 2,88-2,95 (м, 2H), 2,56 (с, 3H), 1,31 (т, J=7,08 Гц, 3H);
Этиловый эфир 1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=2-гидроксиэтил, A=-(CH2)2-]; ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,27 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,55 (с, 1H), 4,87 (шир. с, 1H), 4,83 (т, J=6,04 Гц, 2H), 4,30 (кв, J=7,08 Гц, 2H), 3,82 (т, J=5,80 Гц, 2H), 2,97-3,06 (м, 2H), 2,85-2,95 (м, 2H), 2,55 (с, 3H), 1,32 (т, J=7,08 Гц, 3H); значение МСВР (ИЭР), вычисленное для C15H18N4O3S [M+H]+: 335,1172, полученное значение: 335,1175.
Получение E
Этиловый эфир 1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=H, R2=Me, A=-(CH2)2-]
этап 3c
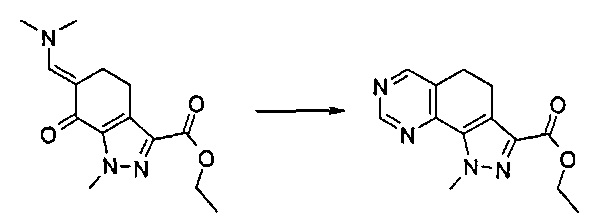
К раствору 0,5 г (1,80 ммоль) этилового эфира (6E)-6-[(диметиламино)метилиден]-1-метил-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты в 20 мл безводного ДМФА добавляли 746 мг (5,4 ммоль) карбоната калия и 510 мг (5,4 ммоль) ацетата формамидина. Реакционную смесь перемешивали при 100°C в течение 8 часов. Указанную смесь разбавляли этилацетатом, промывали H2O, сушили над Na2SO4, фильтровали и выпаривали. Остаток растирали в порошок с применением диэтилового эфира с получением 450 мг указанного в заголовке соединения (95%). 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,09 (с, 1H), 8,74 (с, 1H), 4,35 (с, 3H), 4,31 (кв, J=7,08 Гц, 2H), 2,98-3,05 (м, 4H), 1,33 (т, J=7,08 Гц, 3H).
Аналогичным способом были получены следующие соединения:
Этиловый эфир 1-трет-бутил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=H, R2=трет-бутил, A=-(CH2)2-]; ЖХ/МС (m/z): 301,1 [M+H]+, ВЭЖХ (254 нм) способом 2: Rt=5,89 мин; 1H ЯМР (500 МГц, ДМСО-d6) δ = 9,13 (с, 1H), 8,75 (с, 1H), 4,31 (кв, J=7,14 Гц, 2H), 2,95-3,06 (м, 2H), 1,81 (с, 9H), 2,85-2,95 (м, 2H), 1,31 (т, J=7,14 Гц, 3H); значение МСВР (ИЭР), вычисленное для C16H20N4O2 [M+H]+: 301,1659, полученное значение: 301,1663.
Следующие соединения были получены, как описано в получении E, с применением ацетамидина вместо формамидина:
Этиловый эфир 1,8-диметил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me, R2=Me, A=-(CH2)2-]; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,62 (с, 1H), 4,36 (с, 3H), 4,30 (кв, J=7,08 Гц, 2H), 2,91-3,05 (м, 4H), 2,65 (с, 3H), 1,32 (т, J=7,08 Гц, 3H);
Этиловый эфир 1-трет-бутил-8-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me, R2=трет-бутил, A=-(CH2)2-]; ЖХ/МС (m/z): 315,3 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt=6,52 мин.
Получение F
Этиловый эфир 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=трет-бутил, A=-CH=CH-]
этап 4
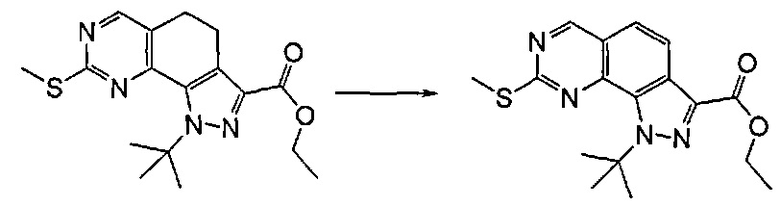
Раствор 1,5 г (4,35 ммоль) этилового эфира 1-трет-бутил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и 1,97 г (8,7 ммоль) DDQ в хлорбензоле нагревали с обратным холодильником в течение 4 часов. Летучие компоненты удаляли in vacuo, остаток растворяли в этилацетате и промывали насыщенным водным раствором NaHCO3. Органическую фазу сушили с применением Na2SO4, фильтровали и концентрировали. Необработанный материал очищали посредством колоночной хроматографии с применением силикагеля, элюируя этилацетатом и гексаном (1:4), с получением 1,19 г указанного в заголовке соединения (80%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 7,78 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,52 (с, 1H), 8,18-8,43 (м, 1H), 7,89 (д, J=8,79 Гц, 1H), 4,24-4,61 (м, 2H), 2,74 (с, 3H), 2,00 (с, 9H), 1,40 (т, J=7,08 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C17H20N4O2S [М+Н]+: 345,1380, полученное значение: 345,1371.
С применением тех же способов, что описаны в вышеприведенном примере, также были синтезированы следующие аналоги:
Этиловый эфир 1-метил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=метил, A=-CH=CH-]; 1H ЯМР (401 МГц, ДМСО-d6), сдвиг составлял 9,49 (с, 1H), 8,15 (д, J=8,79 Гц, 1H), 7,82 (д, J=8,79 Гц, 1H), 4,74 (с, 3H), 4,44 (кв, J=7,20 Гц, 2H), 2,73 (с, 3H), 1,40 (т, J=7,14 Гц, 3H);
Этиловый эфир 1-(2-гидроксиэтил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=2-гидроксиэтил, A=-CH=CH-]; ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,49 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,17 (д, J=8,67 Гц, 1H), 7,83 (д, J=8,79 Гц, 1H), 5,24 (т, J=5,92 Гц, 2H), 4,39-4,49 (м, 2H), 3,98 (т, J=5,80 Гц, 2H), 2,70 (с, 3H), 1,41 (т, J=7,14 Гц, 3H); значение МСВР (ИЭР), вычисленное для C15H16N4O3S [M+H]+: 333,1016, полученное значение: 333,1017;
Этиловый эфир 1-трет-бутил-8-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-, R2=трет-бутил, A=-CH=CH-]; ЖХ/МС (m/z): 313,2 [M+H]+, ВЭЖХ (254 нм) способом 2: Rt=6,79; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,62 (с, 1H), 8,33 (д, J=8,67 Гц, 1H), 7,92 (д, J=8,79 Гц, 1H), 4,39-4,50 (м, 2H), 2,92 (с, 3H), 2,02 (с, 9H), 1,41 (т, J=7,08 Гц, 3H); значение МСВР (ИЭР), вычисленное для C17H20N4O2 [M+H]+: 313,1659, полученное значение: 313,1653;
Этиловый эфир 1-трет-бутил-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=H, R2=трет-бутил, A=-CH=CH-]; ЖХ/МС (m/z): 299,1 [М+Н]+, ВЭЖХ (254 нм) способом 2: Rt=6,36 мин; 1H ЯМР (500 МГц, ДМСО-d6) δ = 9,75 (с, 1H), 9,56 (с, 1H), 8,44 (д, J=8,79 Гц, 1H), 7,98 (д, J=8,79 Гц, 1H), 4,46 (кв, J=7,14 Гц, 2H), 2,01 (с, 9H), 1,41 (т, J=7,00 Гц, 3H); значение МСВР (ИЭР), вычисленное для C16H18N4O2 [M+H]+: 299,1503, полученное значение: 299,1502.
Пример 1
1-(2-гидроксиэтил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 1)
этап 6a
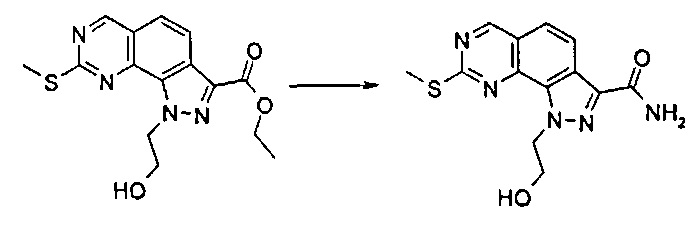
Суспензию 1,2 г (3,61 ммоль) этилового эфира 1-(2-гидроксиэтил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в 10 мл 7 н. NH3 в метаноле подвергали действию микроволнового излучения при 120°C в течение 4 часов. Летучие компоненты удаляли под вакуумом, остаток разбавляли диэтиловым эфиром и отфильтровывали твердое вещество с получением 0,6 г указанного в заголовке соединения (55%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,29 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,46 (с, 1H), 8,28 (д, J=8,67 Гц, 1H), 7,84 (с, 1H), 7,73 (д, J=8,79 Гц, 1H), 7,52 (шир. с, 1H), 5,21 (т, J=5,98 Гц, 2H), 4,82-4,96 (м, J=0,61 Гц, 1H), 3,94-4,07 (м, 2H), 2,70 (с, 3H). Значение МСВР (ИЭР), вычисленное для C13H13N5O2S [M+H]+: 304,0863, полученное значение: 304,0857.
С применением тех же способов, что описаны в вышеприведенном примере, также были синтезированы следующие аналоги:
1-трет-бутил-8-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-, R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 5), ЖХ/МС (m/z): 284,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt=5,95 мин;
1-трет-бутил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 3), ЖХ/МС (m/z): 270,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt=4,55 мин;
Пример 2
1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=H, R2=Me, R3=NH2, A=-(CH2)2-]
этап 6b1 и этап 6b2
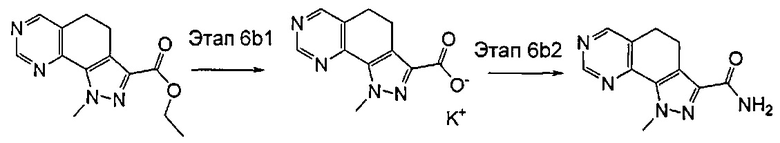
Этап a. Суспензию 440 мг (1,74 ммоль) этилового эфира 1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в этаноле (30 мл) и 300 мг (5,2 ммоль) гидроксида калия нагревали с обратным холодильником в течение 2 часов. Летучие компоненты удаляли под вакуумом, остаток разбавляли этанолом (3 мл) и отфильтровывали твердое вещество с получением 374 мг 1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксилата калия (80%).
Этап b. К суспензии 200 мг (0,74 ммоль) 1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксилата калия в ДМФА (10 мл) добавляли 286 мг (1,49 ммоль) EDCI, 1 мл (10 ммоль) ДИПЭА, 300 мг (1,98 ммоль) HOBt-NH4. Указанную смесь перемешивали при комнатной температуре в течение 18 часов, затем раствор разбавляли ДХМ и промывали насыщенным водным раствором NaHCO3. Органический слой сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством флэш-хроматографии, элюируя циклогексаном/EtOAc в соотношении 4/1, с получением 118 мг указанного в заголовке соединения (70%) в виде твердого вещества белого цвета. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,08 (с, 1H), 8,73 (с, 1H), 7,51 (шс, 2H), 4,33 (с, 3H), 3,05 (м, 2H), 2,97 (м, 2H).
С применением тех же способов, что описаны в вышеприведенном примере, также был синтезирован следующий аналог:
1-трет-бутил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=H, R2=трет-бутил, R3=NH2, A=-(CH2)2-] (соединение 2), ЖХ/МС (m/z): 272 [M+H]+, ВЭЖХ (254 нм) способом 2: Rt=4,28 мин. 1H ЯМР (500 МГц, ДМСО-d6) δ = 9,10 (с, 1H), 8,73 (с, 1H), 7,41 (шир. с, 1H), 7,31 (шир. с, 1H), 2,97-3,03 (м, 2H), 2,83-2,91 (м, 2H), 1,81 (с, 9H). Значение МСВР (ИЭР), вычисленное для C14H17N5O [M+H]+: 272,1506, полученное значение: 272,1514.
Пример 3
1-(2-гидроксиэтил)-8-метокси-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=R4=Me, X=O, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 6)
этап 3b, этап 4, этап 6a
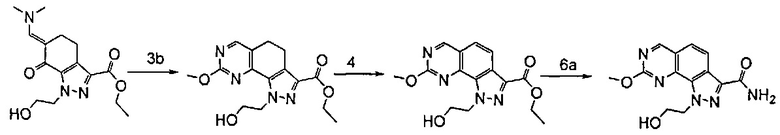
Получение этилового эфира 1-(2-гидроксиэтил)-8-метокси-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (этап 3b)
К раствору 15 г (49 ммоль) этилового эфира (6E)-6-[(диметиламино)метилиден]-1-(2-гидроксиэтил)-7-оксо-4,5,6,7-тетрагидро-1H-индазол-3-карбоновой кислоты в 50 мл безводного ДМФА добавляли 14,4 г (147 ммоль) безводного ацетата калия и 12,60 г (73,0 ммоль) сульфата метилизомочевины. Реакционную смесь перемешивали при 100°C в течение 16 часов. Указанную смесь разбавляли этилацетатом, промывали H2O, сушили над Na2SO4, фильтровали и выпаривали. Необработанный материал очищали на колонке (этилацетат : гексан 1:3) с получением 1,2 г указанного в заголовке соединения (10%). Значение МСВР (ИЭР), вычисленное для C15H18N4O4 [M+H]+: 319,3278, полученное значение: 319,3256.
Получение этилового эфира 1-(2-гидроксиэтил)-8-метокси-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (этап 4)
К суспензии 1,2 г (3,76 ммоль) этилового эфира 1-(2-гидроксиэтил)-8-метокси-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в толуоле (20 мл) добавляли 500 мг (2,2 ммоль) 4,5-дихлор-3,6-диоксоциклогекса-1,4-диен-1,2-дикарбонитрила. Указанную смесь подвергали действию микроволнового излучения при 100° в течение 3 часов в герметизированном сосуде. Летучие компоненты выпаривали, необработанный материал растворяли в этилацетате и разделяли с применением насыщенного раствора NaHCO3, органический слой концентрировали досуха. Остаток очищали посредством хроматографии (этилацетат/гексан 7/3) с получением 520 мг желаемого соединения (46%). Значение МСВР (ИЭР), вычисленное для C15H16N4O4 [M+H]+: 317,3119, полученное значение: 317,3126.
В ходе получения этилового эфира 1-(2-гидроксиэтил)-8-метокси-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты 250 мг (0,79 ммоль) указанного эфира растворяли в 7 М NH3 в метаноле (8 мл). Указанную смесь подвергали воздействию микроволнового излучения при 120° в течение 4 часов в герметизированном сосуде. Летучие компоненты выпаривали и необработанный материал очищали посредством хроматографии, элюируя ДХМ/MeOH 95/5, с получением 136 мг желаемого соединения (60%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,75 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,51 (с, 1H), 8,20 (д, J=8,67 Гц, 1H), 7,82 (шир. с, 1H), 7,74 (д, J=8,79 Гц, 1H), 7,51 (шир. с, 1H), 5,19 (т, J=5,92 Гц, 1H), 4,90 (т, J=6,29 Гц, 3H), 4,12 (с, 3H), 3,97-4,07 (м, J=5,00 Гц, 2H). Значение МСВР (ИЭР), вычисленное для C13H13N5O3 [M+H]+: 288,1091, полученное значение: 288,1086.
Получение G
1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксилат калия [(XI), R1=Me-S-, R2=трет-бутил, A=-CH=CH-]
этап 6b1
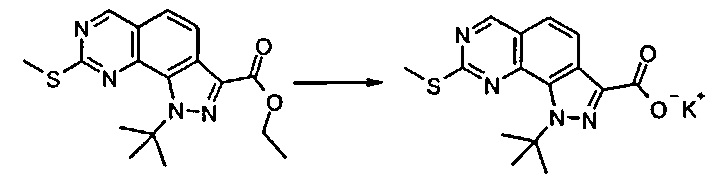
Этиловый эфир 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (300 мг 0,87 ммоль) суспендировали в 2 мл безводного этанола и обрабатывали 1,5 М раствором гидроксида калия в этаноле (5 мл, 7,5 ммоль) при комнатной температуре в течение 2 часов. Полученный осадок собирали посредством фильтрования с получением 215 мг указанного в заголовке соединения (70%) в виде твердого вещества грязно-белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,79 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,38 (с, 1H), 8,61 (д, J=8,54 Гц, 1H), 7,58 (д, J=8,67 Гц, 1H), 2,72 (с, 3H), 1,95 (с, 9H). Значение МСВР (ИЭР), вычисленное для C15H16N4O2S [M+H]+: 317,1067, полученное значение: 317,1052.
Пример 4
1-трет-бутил-N-метил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R'=Me, A=-CH=CH-] (соединение 7)
этап 6b2
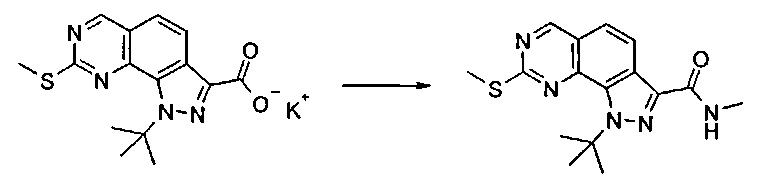
Суспензию 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксилата калия (50 мг, 0,142 ммоль) в безводном N,N-диметилформамиде (2 мл) обрабатывали гексафторфосфатом 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (HBTU) (82 мг, 0,255 ммоль) и гидрохлоридом метиламина (15 мг, 0,213 ммоль) в присутствии N,N-диизопропилэтиламина (100 мкл, 0,71 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разделяли между этилацетатом и водой, и органический слой сушили над Na2SO4, фильтровали и концентрировали. Необработанный продукт очищали посредством колоночной хроматографии с применением EtOAc/гексана 7/3 с получением 20 мг (45%) указанного в заголовке соединения в виде твердого вещества грязно-белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,89 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,42 (д, J=8,67 Гц, 1H), 8,32 (д, J=4,52 Гц, 1H), 7,80 (д, J=8,67 Гц, 1H), 2,87 (д, J=4,76 Гц, 3H), 2,74 (с, 3H), 1,98-2,04 (м, 9H). Значение МСВР (ИЭР), вычисленное для C16H19N5OS [M+H]+: 330,1383, полученное значение: 330,1383.
Действуя аналогичным способом, получали следующие соединения:
1-трет-бутил-N-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R'=2-(диметиламино)этил, A=-CH=CH-] (соединение 8), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,28 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,43 (д, J=8,67 Гц, 1H), 8,22 (т, J=5,86 Гц, 1H), 7,81 (д, J=8,67 Гц, 1H), 3,44 (кв, J=6,59 Гц, 2H), 2,74 (с, 3H), 2,46 (т, J=6,77 Гц, 2H), 2,21 (с, 6H), 2,01 (с, 9H); значение МСВР (ИЭР), вычисленное для C19H26N6OS [M+H]+: 387,1962, полученное значение: 387,1974;
1-трет-бутил-N-(2-гидроксиэтил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R'=2-гидроксиэтил, A=-CH=CH-] (соединение 9), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,07 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,43 (д, J=8,54 Гц, 1H), 8,22 (т, J=5,92 Гц, 1H), 7,81 (д, J=8,67 Гц, 1H), 4,80 (т, J=5,43 Гц, 1H), 3,58 (кв, J=6,02 Гц, 2H), 3,43 (кв, J=6,27 Гц, 2H), 2,74 (с, 3H), 2,01 (с, 9H); значение МСВР (ИЭР), вычисленное для C17H21N5O2S [M+H]+: 360,1489, полученное значение: 360,1495;
1-трет-бутил-N-(1-метилпиперидин-4-ил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R'=1-метилпиперидин-4-ил, A=-CH=CH-] (соединение 10), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,09 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,48 (с, 1H), 8,40 (д, J=8,67 Гц, 1H), 8,06 (д, J=8,06 Гц, 1H), 7,80 (д, J=8,79 Гц, 1H), 3,84 (дд, J=3,30, 7,57 Гц, 1H), 2,74-2,83 (м, 2H), 2,73 (с, 3H), 2,18 (с, 3H), 2,01 (с, 9H), 1,94-1,99 (м, 2H), 1,63-1,84 (м, 4H); значение МСВР (ИЭР), вычисленное для C21H28N6OS [M+H]+: 413,2118, полученное значение: 413,2112;
1-трет-бутил-N-[2-(1H-имидазол-5-ил)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R'=2-(1H-имидазол-5-ил)этил, A=-CH=CH-] (соединение 11), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,49 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,55 (д, J=2,93 Гц, 1H), 8,43 (д, J=8,67 Гц, 1H), 7,81 (д, J=8,67 Гц, 1H), 7,57 (д, J=0,98 Гц, 1H), 6,89 (с, 1H), 3,51-3,64 (м, 2H), 2,82 (т, J=7,38 Гц, 2H), 2,70-2,76 (м, 3H), 2,01 (с, 9H); значение МСВР (ИЭР), вычисленное для C20H23N7OS [M+H]+: 410,1758, полученное значение: 410,1751.
Получение H
Этиловый эфир 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=H, A=-CH=CH-]
этап 5

Этиловый эфир 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (800 мг, 2,32 ммоль) обрабатывали трифторуксусной кислотой (5 мл). Полученную смесь нагревали при 70°C и перемешивали в течение 4 часов. После удаления летучих компонентов in vacuo остаток растворяли в ДХМ, промывали насыщенным раствором NaHCO3, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (670 мг, >99%) в виде твердого вещества светло-желтого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,66 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,50 (с, 1H), 8,14 (д, J=8,67 Гц, 1H), 7,80 (д, J=8,79 Гц, 1H), 4,41-4,47 (м, 2H), 2,75 (с, 3H), 1,41 (т, J=7,14 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C13H12N4O2S [M+H]+: 289,0754, полученное значение: 289,0752.
Получение I
Этиловый эфир 1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIIIa), R1=Me-S-, R2=2-(диметиламино)этил, A=-CH=CH-]
и
этиловый эфир 2-[2-(диметиламино)этил]-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIIIb), R1=Me-S-, R2=2-(диметиламино)этил, A=-CH=CH-]
этап 5

К раствору этилового эфира 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (150 мг, 0,52 ммоль) в сухом диметилформамиде (4 мл) добавляли карбонат цезия (339 мг, 1,04 ммоль), с последующим добавлением 2-бром-N,N-диметилэтанамина (135 мг, 0,88 ммоль). Гетерогенную смесь перемешивали при комнатной температуре в течение 24 часов. Указанную смесь обрабатывали водой и подвергали экстракции EtOAc. Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством хроматографии на силикагеле, элюируя EtOAc/этанолом в соотношении 85/15, с получением этилового эфира 1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в виде основного изомера (60 мг, 32%), 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,51 (с, 1H), 8,16 (д, J=8,67 Гц, 1H), 7,83 (д, J=8,79 Гц, 1H), 5,29 (т, J=6,59 Гц, 1H), 4,32-4,49 (м, 2H), 2,94 (шир. с, 2H), 2,71 (с, 3H), 2,24 (шир. с, 6H), 1,41 (т, J=7,08 Гц, 3H); и
этилового эфира 2-[2-(диметиламино)этил]-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в виде минорного изомера (20 мг, 10%), 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,39 (с, 1H), 8,26 (д, J=8,78 Гц, 1H), 7,87 (д, J=8,78 Гц, 1H), 4,66 (т, J=6,57 Гц, 1H), 4,45 (кв, J=7,06 Гц, 2H), 2,71 (шир. с, 3H), 2,67 (с, 2H), 2,37 (шир. с, 6H), 1,46 (т, J=7,08 Гц, 3H).
Пример 5
1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-(диметиламино)этил, R3=NH2, A=-CH=CH-] (соединение 12)
этап 6c
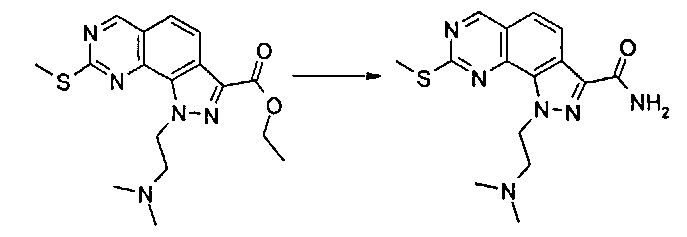
Этиловый эфир 1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (46 мг, 0,128 ммоль) суспендировали в 2 мл тетрагидрофурана. Добавляли хлорид аммония (20 мг, 0,384 ммоль) и 1 н. LiN(TMS)2 в ТГФ (0,8 мл, 0,8 ммоль). Указанную смесь перемешивали при комнатной температуре в течение 1 часа. Затем растворитель выпаривали досуха, остаток очищали посредством флэш-хроматографии на силикагеле (элюент представлял собой ДХМ/MeOH/NH4OH в соотношении 95/5/0,1) с получением 18 мг (42%) указанного в заголовке соединения и 2 мг побочного продукта, представлявшего собой 6-амино-1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,75 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,48 (с, 1H), 8,27 (д, J=8,67 Гц, 1H), 7,84 (шир. с, 1H), 7,74 (д, J=8,67 Гц, 1H), 7,54 (шир. с, 1H), 5,25 (т, J=6,71 Гц, 2H), 2,93 (шир. с, 2H), 2,71 (с, 3H), 2,23 (шир. с, 6H). Значение МСВР (ИЭР), вычисленное для C15H18N6OS [M+H]+: 331,1336, полученное значение: 331,1334.
6-амино-1-[2-(диметиламино)этил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-(диметиламино)этил, R3=NH2, R5=NH2, A=-CH=CH-] (соединение 13)
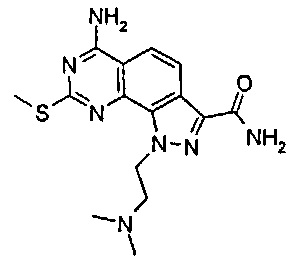
ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,97 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,01 (д, J=8,79 Гц, 1H), 7,88 (шир. с, 1H), 7,84 (д, J=8,91 Гц, 1H), 7,74 (шир. с, 1H), 7,45 (шир. с, 1H), 5,22 (т, J=5,61 Гц, 2H), 2,88 (шир. с, 2H), 2,58 (с, 3H), 2,15-2,29 (м, J=6,47 Гц, 6H). Значение МСВР (ИЭР), вычисленное для C15H19N7OS [M+H]+: 346,1445, полученное значение: 346,1439.
Пример 6
2-[2-(диметиламино)этил]-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-(диметиламино)этил, R3=NH2, A=-CH=CH-] (соединение 14)
этап 6a
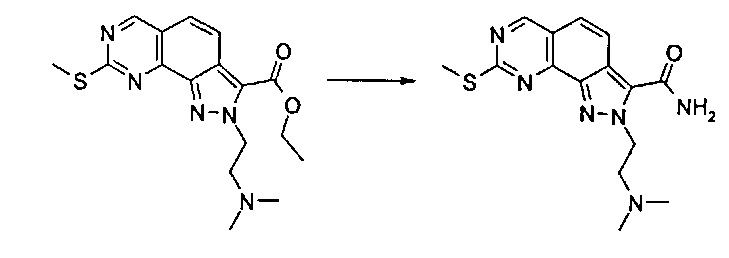
Суспензию 20 мг (0,055 ммоль) этилового эфира 2-[2-(диметиламино)этил]-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в 4 мл 7 н. NH3 в метаноле подвергали воздействию микроволнового излучения при 120°C в течение 4 часов. Летучие компоненты удаляли под вакуумом и остаток очищали посредством хроматографии на силикагеле (элюент представлял собой ДХМ/MeOH/NH3 в соотношении 97/3/1) с получением 2,6 мг указанного в заголовке соединения (15%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,13 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,34 (с, 1H), 8,34 (шир. с, 1H), 8,04 (шир. с, 1H), 7,80 (д, J=8,91 Гц, 1H), 7,59 (д, J=9,03 Гц, 1H), 4,89 (т, J=6,47 Гц, 2H), 2,80 (т, J=6,41 Гц, 2H), 2,69 (с, 3H), 2,18 (с, 6H). Значение МСВР (ИЭР), вычисленное для C15H18N6OS [M+H]+: 331,1336, полученное значение: 331,1331.
Пример 7
трет-Бутиловый эфир {2-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]этил}карбаминовой кислоты [(I), R1=Me-S-, R2=N1-2-[(трет-бутоксикарбонил)амино]этил, R3=NH2, A=-CH=CH-]
и
трет-бутиловый эфир {2-[3-карбамоил-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-2-ил]этил}карбаминовой кислоты [(I), R1=Me-S-, R2=N2-2-[(трет-бутоксикарбонил)амино]этил, R3=NH2, A=-CH=CH-]
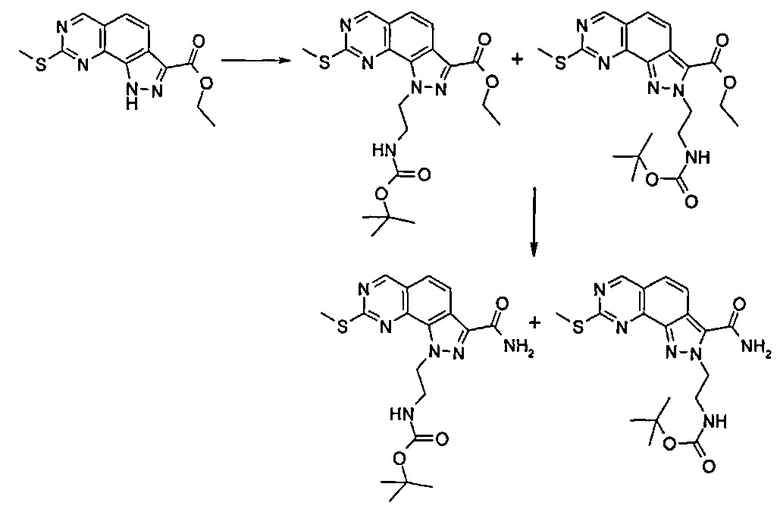
К раствору этилового эфира 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (200 мг, 0,69 ммоль) в сухом диметилформамиде (4 мл) добавляли карбонат цезия (337 мг, 1,03 ммоль) с последующим добавлением трет-бутилового эфира 2-бромэтилкарбаминовой кислоты (186 мг, 0,83 ммоль). Гетерогенную смесь перемешивали при комнатной температуре в течение 48 часов. Указанную смесь обрабатывали водой и подвергали экстракции EtOAc. Органический слой промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством хроматографии на силикагеле, элюируя EtOAc/этанолом в соотношении 85/15, с получением смеси двух неразделенных региоизомеров, представлявших собой этиловый эфир 1-{2-[(трет-бутоксикарбонил)амино]этил}-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и этиловый эфир 2-{2-[(трет-бутоксикарбонил)амино]этил}-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты, (282 мг, 95%).
Полученную смесь двух региоизомеров суспендировали в 10 мл 7 н. NH3 в метаноле и подвергали воздействию микроволнового излучения при 120°C в течение 5 часов. Летучие компоненты удаляли под вакуумом, остаток очищали посредством хроматографии на силикагеле (элюент представлял собой ДХМ/MeOH/NH3 в соотношении 97/3/1) с получением 145 мг трет-бутилового эфира {2-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]этил}карбаминовой кислоты в виде основного изомера (55%); ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,53 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,46 (с, 1H), 8,27 (д, J=8,79 Гц, 1H), 7,74 (с, 2H), 7,54 (шир. с, 1H), 6,91 (т, J=6,29 Гц, 1H), 5,21 (т, J=5,86 Гц, 2H), 3,58 (кв, J=6,14 Гц, 2H), 2,71 (с, 3H), 1,23 (с, 9H); значение МСВР (ИЭР), вычисленное для C18H22N6O3S [M+H]+: 403,1547, полученное значение: 403,1534; и
18 мг трет-бутилового эфира {2-[3-карбамоил-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-2-ил]этил}карбаминовой кислоты в виде минорного изомера (7%), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,08 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,33 (с, 1H), 8,08 (д, J=15,01 Гц, 2H), 7,84 (д, J=8,91 Гц, 1H), 7,60 (д, J=8,91 Гц, 1H), 6,95 (т, J=6,71 Гц, 1H), 4,82 (т, J=6,29 Гц, 2H), 3,49 (кв, J=6,51 Гц, 2H), 2,69 (с, 3H), 1,28 (с, 9H); значение МСВР (ИЭР), вычисленное для C18H22N6O3S [M+H]+: 403,1547, полученное значение: 403,1552.
При проведении синтеза согласно указанному способу получали следующее соединение:
трет-бутиловый эфир 4-{[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]метил}пиперидин-1-карбоновой кислоты [(I), R1=Me-S-, R2=трет-бутил 4-N1-метилпиперидин-1-карбоксилат, R3=NH2, A=-CH=CH-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,56 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,47 (с, 1H), 8,28 (д, J=8,79 Гц, 1H), 7,81 (с, 1H), 7,74 (д, J= 8,67 Гц, 1Н), 7,55 (с, 1H), 5,07 (д, J=7,57 Гц, 2H), 3,90 (д, J=12,08 Гц, 2H), 2,69-2,70 (м, 3H), 2,59-2,68 (м, J=2,01, 3,72 Гц, 2H), 2,22-2,41 (м, 1H), 1,41-1,51 (м, J=6,10 Гц, 2H), 1,38 (с, 9H), 1,22 (дкв, J=4,33, 12,31 Гц, 2H); значение МСВР (ИЭР), вычисленное для C22H28N6O3S [M+H]+: 479,1836, полученное значение: 479,1849.
Пример 8
Гидрохлорид 1-(2-аминоэтил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=N1-2-аминоэтил, R3=NH2, A=-CH=CH-] (соединение 15)
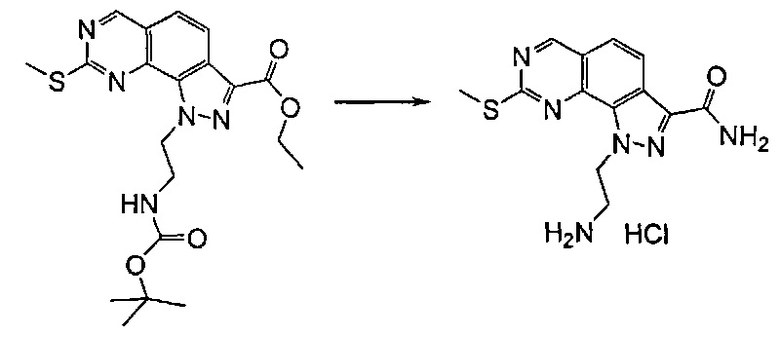
трет-Бутиловый эфир {2-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]этил}карбаминовой кислоты (40 мг, 0,1 ммоль) обрабатывали 1 мл 4 М HCl в 1,4-диоксане. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли in vacuo, полученный остаток растирали в порошок с применением диэтилового эфира, фильтровали и промывали Et2O и сушили in vacuo с получением 32 мг (97%) указанного в заголовке соединения в виде твердого вещества белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,59 мин. Соль HCl, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,50 (с, 1H), 8,29 (д, J=8,67 Гц, 1H), 8,12 (шир. с, 2H), 8,04 (шир. с, 1H), 7,78 (д, J=8,67 Гц, 1H), 7,65 (с, 1H), 5,32-5,58 (м, 2H), 3,57-3,72 (м, 2H), 2,72 (с, 3H). Значение МСВР (ИЭР), вычисленное для C13H14N6OS [M+H]+: 303,1023, полученное значение: 303,1021.
При проведении синтеза согласно указанному способу получали следующие соединения:
гидрохлорид 2-(2-аминоэтил)-8-(метилсульфанил)-2H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=N2-2-аминоэтил, R3=NH2, A=-CH=CH-] (соединение 16), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,33 мин; HCl соль, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,34-9,40 (м, 1H), 7,93-8,29 (м, 5H), 7,85-7,91 (м, 1H), 7,66 (д, J=9,03 Гц, 1H), 5,01 (т, J=6,16 Гц, 2H), 3,43-3,53 (м, 2H), 2,67-2,71 (м, 3H); значение МСВР (ИЭР), вычисленное для C13H14N6OS [M+H]+: 303,1023, полученное значение: 303,1017;
гидрохлорид 8-(метилсульфанил)-1-(пиперидин-4-илметил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=N1-(пиперидин-4-илметил), R3=NH2, A=-CH=CH-] (соединение 17), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,94 мин; соль HCl, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,49 (с, 1H), 8,62 (шир. с, 1H), 8,47 (д, J=10,25 Гц, 1H), 8,29 (д, J=8,67 Гц, 1H), 7,81 (с, 1H), 7,76 (д, J=8,67 Гц, 1H), 7,59 (с, 1H), 5,13 (д, J=7,32 Гц, 2H), 3,23 (д, J=12,21 Гц, 2H), 2,81 (кв, J=11,76 Гц, 2H), 2,74 (с, 3H), 2,36-2,48 (м, 1H), 1,43-1,68 (м, 4H); значение МСВР (ИЭР), вычисленное для C17H20N6OS [M+H]+: 357,1492, полученное значение: 357,149; и
гидрохлорид 8-(метилсульфанил)-2-(пиперидин-4-илметил)-2H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=N2-(пиперидин-4-илметил), R3=NH2, A=-CH=CH-] (соединение 18), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,7 мин; соль HCl, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,36 (с, 1H), 8,64 (д, J=10,37 Гц, 1H), 7,94-8,39 (м, 3H), 7,83 (д, J=9,03 Гц, 1H), 7,62 (д, J=9,03 Гц, 1H), 4,67-4,90 (м, 2H), 2,78-2,94 (м, 2H), 2,69 (с, 3H), 2,34-2,43 (м, 1H), 1,58-1,71 (м, J=12,45 Гц, 2H), 1,41-1,56 (м, 2H); значение МСВР (ИЭР), вычисленное для C17H20N6OS [M+H]+: 357,1492, полученное значение: 357,1481.
Пример 9
1-метил-8-[4-(пиперазин-1-ил)фенокси]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-(пиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 19)
Превращение b
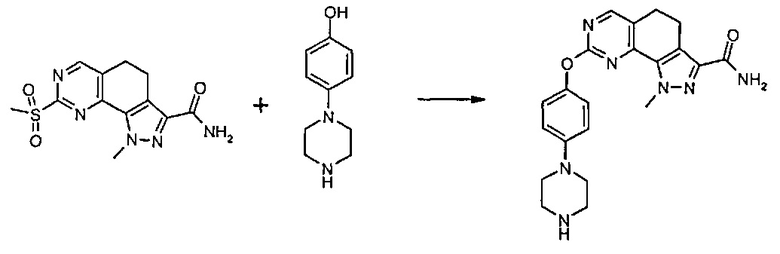
Проводили реакцию 154 мг (0,5 ммоль) 1-метил-8-(метилсульфонил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (полученного, как описано в WO 2004/104007 A1) и 107 мг (0,6 ммоль) 4-(пиперазин-1-ил)фенола в 5 мл безводного ДМФА в присутствии Cs2CO3 (0,487 г, 1,5 ммоль) при 70°C в течение 2 часов. После охлаждения реакционную смесь сушили под вакуумом, смешивали с ложкой диоксида кремния и элюировали посредством флэш-хроматографии (ДХМ/MeOH/7 н. NH3 в MeOH 9/1/0,4%) с получением желаемого продукта. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,73 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,68 (шир. с, 2H), 8,49 (с, 1H), 7,44 (с, 1H), 7,29 (с, 1H), 7,12-7,19 (м, 2H), 7,03-7,08 (м, 2H), 4,06 (с, 3H), 3,20-3,39 (м, 8H), 2,97-3,04 (м, 2H), 2,85-2,92 (м, 2H). Значение МСВР (ИЭР), вычисленное для C21H23N7O2 [M+H]+: 406,1986, полученное значение: 406,19825.
Действуя аналогичным способом, получали следующие соединения:
1-метил-8-фенокси-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 20), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,23 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,53 (с, 1H), 7,42-7,49 (м, 3H), 7,22-7,30 (м, 4H), 3,99 (с, 3H), 2,98-3,04 (м, 2H), 2,85-2,93 (м, 2H); значение МСВР (ИЭР), вычисленное для C17H15N5O2 [M+H]+: 322,1299, полученное значение: 322,1293;
8-(3-аминофенокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-аминофенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 21), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,43 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,51 (с, 1H), 7,45-7,51 (м, 1H), 7,26 (шир. с, 1H), 7,04 (т, J=7,99 Гц, 1H), 6,43 (ддд, J=0,85, 2,08, 8,06 Гц, 1H), 6,38 (т, J=2,14 Гц, 1H), 6,32 (ддд, J=0,79, 2,23, 7,96 Гц, 1H), 5,21 (шир. с, 2H), 4,06 (с, 3H), 2,98-3,04 (м, 2H), 2,85-2,92 (м, 2H); значение МСВР (ИЭР), вычисленное для C17H16N6O2 [M+H]+: 337,1408, полученное значение: 337,1411;
8-(4-аминофенокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-аминофенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 22), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,23 мин; 1H ЯМР (401 МГц, ДМСО-d6) сдвиг δ составлял 8,48 (с, 1H), 7,47 (с, 1H), 7,25 (шир. с, 1H), 6,84-6,94 (м, 2H), 6,54-6,63 (м, 2H), 4,99 (с, 2H), 4,04 (с, 3H), 2,95-3,04 (м, 2H), 2,83-2,91 (м, 2H); значение МСВР (ИЭР), вычисленное для C17H16N6O2 [М+Н]+: 337,1408, полученное значение: 337,1405;
1-метил-8-(пиридин-4-илокси)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=пиридин-4-илокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 23), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,68 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,81-8,86 (м, 2H), 8,79 (с, 1H), 7,53 (с, 1H), 7,32 (шир. с, 1H), 6,30-6,36 (м, 2H), 4,36 (с, 3H), 3,04-3,11 (м, 2H), 3,00 (шир. с, 2H); значение МСВР (ИЭР), вычисленное для C16H14N6O2 [M+H]+: 323,1251, полученное значение: 323,1238;
8-этокси-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=этил, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254) ВЭЖХ способ 2: Rt=4,47 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,48 (с, 1H), 7,45-7,48 (м, 1H), 7,26 (шир. с, 1H), 4,39 (кв, J=7,04 Гц, 2H), 4,29 (с, 3H), 2,95-3,03 (м, 2H), 2,81-2,90 (м, 2H), 1,36 (т, J=7,02 Гц, 3H); значение МСВР (ИЭР), вычисленное для C13H15N5O2 [M+H]+: 274,1299, полученное значение: 274,1296;
1-метил-8-(пропан-2-илокси)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=пропан-2-илокси, R2=метил, R3=NH2) A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,84 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,47 (с, 1H), 7,47 (шир. с, 1H), 7,26 (шир. с, 1H), 5,21 (квин, J=6,16 Гц, 1H), 4,28 (с, 3H), 2,96-3,03 (м, 2H), 2,80-2,89 (м, 2H), 1,35 (д, J=6,10 Гц, 6H); значение МСВР (ИЭР), вычисленное для C14H17N5O2 [M+H]+: 288,1455, полученное значение: 288,144;
1-метил-8-(2-оксопропокси)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-оксопропокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,81 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,42-8,50 (м, 1H), 7,49 (с, 1H), 7,27 (шир. с, 1H), 5,07 (с, 2H), 4,22 (с, 3H), 2,96-3,04 (м, 2H), 2,81-2,89 (м, 2H), 2,14 (с, 3H); значение МСВР (ИЭР), вычисленное для C14H15N5O3 [M+H]+: 302,1248, полученное значение: 302,1249; и
8-(2-фторэтокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-фторэтокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 24), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,23 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,51 (с, 1H), 7,41-7,59 (м, 1H), 7,28 (шир. с, 1H), 4,80-4,88 (м, 1H), 4,69-4,76 (м, 1H), 4,61-4,67 (м, 1H), 4,53-4,59 (м, 1H), 4,29 (с, 3H), 2,95-3,05 (м, 2H), 2,83-2,92 (м, 2H); значение МСВР (ИЭР), вычисленное для C13H14FN5O2 [M+H]+: 292,1205, полученное значение: 292,1209.
Пример 10
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 25)
и
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 26)
Превращение b
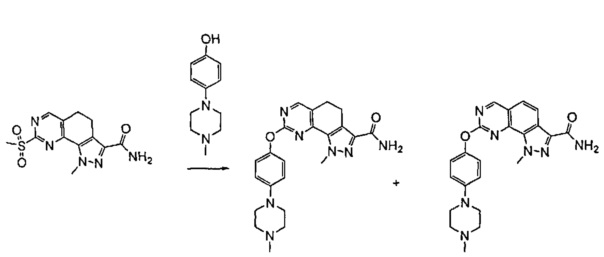
Проводили реакцию 400 мг (1,3 ммоль) 1-метил-8-(метилсульфонил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида и 360 мг (1,43 ммоль) 4-(4-метилпиперазин-1-ил)фенола в 25 мл безводного ДМФА в присутствии 717 мг (5,2 ммоль) K2CO3 при 70°C в течение 4 часов. После охлаждения реакционную смесь сушили под вакуумом, смешивали с ложкой диоксида кремния и элюировали посредством флэш-хроматографии (ДХМ/MeOH/7 н. NH3 в MeOH 9/1/0,4%) с получением смеси двух соединений в соотношении 1:1. Затем каждый продукт выделяли способом 2 препаративной ВЭЖХ с получением:
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,88 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,46 (с, 1H), 7,44 (с, 1H), 7,24 (с, 1H), 7,01-7,09 (м, 2H), 6,91-6,98 (м, 2H), 4,02 (с, 3H), 3,06-3,11 (м, 4H), 2,94-3,01 (м, 2H), 2,81-2,88 (м, 2H), 2,42-2,45 (м, 4H), 2,20 (с, 3H); значение МСВР (ИЭР), вычисленное для C22H25N7O2 [M+H]+: 420,2143, полученное значение: 420,2148; и
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,03 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,56 (с, 1H), 8,21 (д, J=8,67 Гц, 1H), 7,83 (с, 1H), 7,76 (д, J=8,91 Гц, 1H), 7,49 (с, 1H), 7,18-7,24 (м, 1H), 6,99-7,07 (м, 2H), 4,35 (с, 3H), 3,12-3,19 (м, 4H), 2,45-2,49 (м, 4H), 2,22-2,26 (м, 3H); значение МСВР (ИЭР), вычисленное для C22H23N7O2 [M+H]+: 418,1986, полученное значение: 418,1985.
Согласно аналогичной методике, но с применением подходящего исходного материала, получали следующие соединения:
1-метил-8-[4-(пиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 27), ЖХ/МС (254 нм) ВЭЖХ способом 2: Rt=3,91 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,57 (с, 1H), 8,70 (шир. с, 2H), 8,23 (д, J=8,67 Гц, 1H), 7,79 (шир. с, 1H), 7,77 (д, J=8,79 Гц, 2H), 7,52 (с, 1H), 7,25-7,31 (м, 2H), 7,09-7,14 (м, 2H), 4,36 (с, 3H), 3,25-3,40 (м, 8H); значение МСВР (ИЭР), вычисленное для C21H21N7O4 [M+H]+: 404,183, полученное значение: 404,1836;
8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-бром-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 28), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,35 мин; соль ТФУ, 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,52 (с, 1H), 7,57 (д, J=2,07 Гц, 1H), 7,47 (с, 1H), 7,28 (шир. с, 1H), 7,20-7,27 (м, 2H), 4,06 (с, 3H), 2,94-3,04 (м, 6H), 2,84-2,94 (м, 2H), 2,54 (шир. с, 4H), 2,26 (с, 3H); значение МСВР (ИЭР), вычисленное для C22H24BrN7O2 [M+H]+: 498,1248, полученное значение: 498,1239;
8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-бром-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 29), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,55 мин; соль ТФУ, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,59 (с, 1H), 8,24 (д, J=8,67 Гц, 1H), 7,83 (с, 1H), 7,78 (д, J=8,91 Гц, 1H), 7,73 (д, J=2,69 Гц, 1H), 7,51 (с, 1H), 7,35-7,42 (м, 1H), 7,26-7,32 (м, 1H), 4,36 (с, 2H), 2,95-3,08 (м, 2H), 2,54-2,61 (м, 2H), 2,29 (шир. с, 2H); значение МСВР (ИЭР), вычисленное для C22H22BrN7O2 [M+H]+: 496,1091, полученное значение: 496,1082;
1-метил-8-[3-(пиперазин-1-ил)фенокси]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(пиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-(CH2)2-] (соединение 30), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,9 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,51 (с, 1H), 7,47 (шир. с, 1H), 7,26-7,28 (м, 1H), 7,17-7,28 (м, 1H), 6,77-6,83 (м, J=1,22, 1,22, 8,42 Гц, 1H), 6,75 (т, J=2,26 Гц, 1H), 6,55-6,63 (м, 1H), 4,03 (с, 3H), 3,05 (дд, J=4,09, 5,92 Гц, 4H), 2,97-3,03 (м, 2H), 2,89 (с, 1H), 2,86-2,93 (м, 2H), 2,77-2,84 (м, J=4,09, 5,92 Гц, 4H); значение МСВР (ИЭР), вычисленное для C21H23N7O2 [M+H]+: 406,1986, полученное значение: 406,1980;
1-метил-8-[3-(пиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=3-(пиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 31), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,1 мин; соль ТФУ, 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,59 (с, 1H), 8,65 (шир. с, 2H), 8,24 (д, J=8,67 Гц, 1H), 7,79 (д, J=8,79 Гц, 2H), 7,51-7,54 (м, 1H), 7,38 (т, J=8,12 Гц, 1H), 7,01 (т, J=2,20 Гц, 1H), 6,96 (д, J=2,44 Гц, 1H), 6,86 (дд, J=1,59, 7,93 Гц, 1H), 4,34 (с, 3H), 3,36-3,41 (м, 4H), 3,22 (шир. с, 4H); значение МСВР (ИЭР), вычисленное для C21H21N7O2 [M+H]+: 404,183, полученное значение: 404,1835;
8-[3-(диметиламино)фенокси]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(диметиламино)фенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,53 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,51 (с, 1H), 7,47 (шир. с, 1H), 7,26 (шир. с, 1H), 7,22 (т, J=8,18 Гц, 1H), 6,62 (дд, J=2,08, 8,30 Гц, 1H), 6,57 (т, J=2,20 Гц, 1H), 6,51 (дд, J=1,77, 7,87 Гц, 1H), 4,05 (с, 3H), 2,98-3,04 (м, 2H), 2,85-2,94 (м, 8H); значение МСВР (ИЭР), вычисленное для C19H20N6O2 [M+H]+: 365,1721, полученное значение: 365,171;
8-[3-(диметиламино)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(диметиламино)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 32), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,8 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,57 (с, 1H), 8,22 (д, J=8,79 Гц, 1H), 7,83 (шир. с, 1H), 7,77 (д, J=8,91 Гц, 1H), 7,45-7,52 (м, 1H), 7,25-7,30 (м, 1H), 6,60-6,74 (м, 3H), 4,35 (с, 3H), 2,92 (с, 6H); значение МСВР (ИЭР), вычисленное для C19H18N6O2 [M+H]+: 363,1564, полученное значение: 363,1574;
8-(2-хлорфенокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-хлорфенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,55 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,55 (с, 1H), 7,62 (дд, J=0,92, 7,99 Гц, 1H), 7,48 (д, J=2,81 Гц, 1H), 7,40-7,46 (м, 2H), 7,33 (ддд, J=2,75, 6,26, 7,96 Гц, 1H), 7,27 (шир. с, 1H), 3,95 (с, 3H), 2,98-3,07 (м, 2H), 2,86-2,94 (м, J=7,93 Гц, 2H); значение МСВР (ИЭР), вычисленное для C17H14ClN5O2 [M+H]+: 356,0909, полученное значение: 356,0898;
8-(2-хлорфенокси)-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-хлорфенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 33), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,81 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,63 (с, 1H), 8,25 (д, J=8,79 Гц, 1H), 7,85 (шир. с, 1H), 7,80 (д, J=8,79 Гц, 1H), 7,68 (дд, J=1,46, 7,93 Гц, 1H), 7,55 (дт, J=1,53, 8,15 Гц, 1H), 7,46-7,53 (м, 2H), 7,34-7,43 (м, 1H), 4,23 (с, 3H); значение МСВР (ИЭР), вычисленное для C17H12ClN5O2 [M+H]+: 354,0753, полученное значение: 354,0746;
8-(2-фторфенокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-фторфенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,33 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,55 (с, 1H), 7,48 (шир. с, 1H), 7,28-7,46 (м, 4H), 7,26-7,28 (м, 1H), 3,97 (с, 3H), 2,97-3,05 (м, 2H), 2,87-2,94 (м, 2H); значение МСВР (ИЭР), вычисленное для C17H14FN5O2 [M+H]+: 340,1205, полученное значение: 340,1208;
8-(2-фторфенокси)-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-фторфенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 34), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,58 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,63 (с, 1H), 8,26 (д, J=8,79 Гц, 1H), 7,85 (с, 1H), 7,80 (д, J=8,79 Гц, 1H), 7,55 (дт, J=1,89, 7,90 Гц, 1H), 7,50 (шир. с, 1H), 7,44-7,49 (м, 1H), 7,38-7,44 (м, J=2,14, 4,64, 6,68, 6,68 Гц, 1H), 7,31-7,38 (м, 1H), 4,26 (с, 3H); значение МСВР (ИЭР), вычисленное для C17H12FN5O2 [M+H]+: 338,1048, полученное значение: 338,1037;
8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-фтор-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, А=-(CH2)2-] (соединение 35), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,06 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,65 (шир. с, 1Н), 8,50 (с, 1H), 7,45 (с, 1H), 7,23-7,34 (м, J=9,09, 9,09 Гц, 2Н), 7,07 (дд, J=2,81, 13,79 Гц, 1Н), 6,87 (дд, J=2,08, 9,03 Гц, 1Н), 4,07 (с, 3H), 3,78-3,98 (м, J=13,79 Гц, 2Н), 3,46-3,59 (м, 2Н), 3,08-3,23 (м, 2Н), 2,94-3,07 (м, 4Н), 2,84-2,94 (м, 5Н); значение МСВР (ИЭР), вычисленное для C22H24FN7O2 [M+H]+: 438,2049, полученное значение: 438,2039;
8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-фтор-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 36), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,25 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,68 (шир. с, 1Н), 9,56-9,61 (м, 1Н), 8,25 (д, J=8,67 Гц, 1Н), 7,79 (д, J=8,91 Гц, 2Н), 7,53 (с, 1Н), 7,41 (т, J=9,09 Гц, 1Н), 7,14 (дд, J=2,81, 13,79 Гц, 1Н), 6,93 (дд, J=2,26, 8,97 Гц, 1Н), 4,37 (с, 3H), 3,79-4,05 (м, J=8,79 Гц, 2Н), 3,47-3,61 (м, 2Н), 3,17 (шир. с, 2Н), 3,03 (шир. с, 2Н), 2,88 (с, 3H); значение МСВР (ИЭР), вычисленное для C22H22FN7O2 [M+H]+: 436,1892, полученное значение: 436,1875;
8-[2-ацетил-4-(4-метил пиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-ацетил-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, А=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,83 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,57 (шир. с, 1Н), 8,48 (с, 1Н), 7,44 (с, 1Н), 7,36 (д, J=2,93 Гц, 1Н), 7,27-7,32 (м, 2Н), 7,23-7,27 (м, 1Н), 4,03 (с, 3H), 3,84-3,96 (м, J=12,45 Гц, 2Н), 3,48-3,61 (м, J=11,23 Гц, 2Н), 2,98-3,06 (м, J=7,32 Гц, 4Н), 2,85-2,92 (м, 5Н), 2,45 (с, 3H); значение МСВР (ИЭР), вычисленное для C24H27N7O3 [M+H]+: 462,2248, полученное значение: 462,2229;
8-[2-ацетил-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-ацетил-4-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 37), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,99 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,60 (шир. с, 1Н), 9,57 (с, 1Н), 8,24 (д, J=8,79 Гц, 1Н), 7,78 (д, J=8,79 Гц, 2Н), 7,53 (с, 1Н), 7,42 (д, J=2,93 Гц, 1Н), 7,36-7,40 (м, 2Н), 4,32 (с, 3H), 3,88-4,04 (м, J=15,01 Гц, 2Н), 3,47-3,65 (м, J=11,47 Гц, 2Н), 2,96-3,12 (м, 2Н), 2,90 (шир. с, 3H), 2,46 (с, 3H); значение МСВР (ИЭР), вычисленное для C24H25N7O3 [M+H]+: 460,2092, полученное значение: 460,207;
8-[2-ацетил-4-(пиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-ацетил-4-(пиперазин-1-ил)фенокси)фенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,75 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 8,72 (д, J=5,37 Гц, 2Н), 8,49 (с, 1Н), 7,44 (с, 1Н), 7,35 (д, J=2,93 Гц, 1Н), 7,26-7,32 (м, 2Н), 7,22-7,26 (м, 1Н), 4,02 (с, 3H), 3,37-3,42 (м, 4Н), 3,28 (шир. с, 4Н), 2,98-3,04 (м, 2Н), 2,85-2,92 (м, 2Н), 2,41-2,46 (м, 3H); значение МСВР (ИЭР), вычисленное для C23H25N7O3 [M+H]+: 448,2092, полученное значение: 448,2071;
8-[2-ацетил-4-(пиперазин-1-ил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-ацетил-4-(пиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 38), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,9 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,57 (с, 1Н), 8,74 (шир. с, 2Н), 8,23 (д, J=8,67 Гц, 1Н), 7,78 (д, J=8,91 Гц, 2Н), 7,51-7,54 (м, 1Н), 7,42 (д, J=2,69 Гц, 1Н), 7,37-7,40 (м, 1Н), 7,32-7,37 (м, 1Н), 4,31 (с, 3H), 3,41-3,45 (м, 4Н), 3,30 (д, J=5,13 Гц, 4Н), 2,45-2,47 (м, 3H); значение МСВР (ИЭР), вычисленное для C23H23N7O3 [M+H]+: 446,1935, полученное значение: 446,1927;
8-[2-циано-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-циано-4-(4-метилпиперазин-1-ил, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,7 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ = 9,69 (шир. с, 1Н), 8,54 (с, 1Н), 7,53-7,56 (м, 1Н), 7,47 (шир. с, 1Н), 7,42-7,44 (м, 1Н), 7,31 (с, 1Н), 4,08 (с, 3H), 3,00-3,05 (м, 2Н), 2,89-2,95 (м, J=7,81 Гц, 2Н), 2,87 (с, 3H); значение МСВР (ИЭР), вычисленное для C23H24N8O2 [M+H]+: 445,2095, полученное значение: 445,2090; и
8-[2-циано-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=2-циано-4-(4-метилпиперазин-1-ил, R2=метил, R3=NH2, A=-CH=CH-] (соединение 39), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,1 мин; 1H ЯМР (401 МГц, ДМСО-d6) сдвиг составлял 9,71 (шир. с, 1Н), 9,63 (с, 1Н), 8,29 (д, J=8,79 Гц, 1Н), 7,75-7,87 (м, 2Н), 7,60 (д, J=2,93 Гц, 1Н), 7,55-7,59 (м, 1Н), 7,54 (шир. с, 1Н), 7,45-7,52 (м, 1Н), 4,37 (с, 3H), 3,89-4,05 (м, J=13,79 Гц, 2Н), 3,50-3,61 (м, J=14,28 Гц, 2Н), 3,19 (шир. с, 2Н), 3,03-3,13 (м, J=13,43 Гц, 2Н), 2,88 (с, 3H); значение МСВР (ИЭР), вычисленное для C23H22N8O2 [M+H]+: 443,1939, полученное значение: 443,1938.
Пример 11
8-(2-ацетилфенокси)-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид
и
8-[2-(2-гидроксифенил)-2-оксоэтил]-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид
Превращение b

Раствор 307 мг (1 ммоль) 1-метил-8-(метилсульфонил)-4,5-дигидро-1Н-пиразоло[4,3-h-хиназолин]-3-карбоксамида, 322 мг (1,5 ммоль) 2-гидроксиацетофенона, 975 мг (3,0 ммоль) карбоната цезия в примерно 15 мл NMP нагревали при 95°C в течение 1 часа. После исчезновения исходного материала реакционную смесь охлаждали и подвергали экстракции водой и этилацетатом. Образовавшийся осадок выделяли посредством фильтрования и очищали посредством препаративной обращенно-фазовой ВЭЖХ с получением двух соединений:
8-(2-ацетилфенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-ацетилфенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,0 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,52 (с, 1Н), 7,87 (дд, J=1,71, 7,81 Гц, 1Н), 7,67 (ддд, J=1,71, 7,45, 8,06 Гц, 1Н), 7,48 (шир. с, 1Н), 7,41 (дт, J=1,10, 7,57 Гц, 1Н), 7,35 (дд, J=0,85, 8,18 Гц, 1Н), 7,26 (шир. с, 1Н), 3,94 (с, 3H), 2,96-3,06 (м, 2Н), 2,89 (т, J=7,90 Гц, 2Н), 2,46 (с, 3H); значение МСВР (ИЭР), вычисленное для C19H17N5O3 [M+H]+: 364,1404, полученное значение: 364,1406; и
8-[2-(2-гидроксифенил)-2-оксоэтил]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2-ацетилфенокси, R2=метил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,12 мин; 1H ЯМР (500 МГц, ДМСО-d6) δ=11,62 (с, 1Н), 8,66 (с, 1Н), 7,92-7,97 (м, 1Н), 7,52 (с, 1Н), 7,48 (шир. с, 1Н), 7,27 (шир. с, 1Н), 6,98 (д, J=7,48 Гц, 1Н), 6,94-6,97 (м, 1Н), 4,70 (с, 2Н), 4,11 (с, 3H), 2,97-3,06 (м, 2Н), 2,89-2,95 (м, 2Н); значение МСВР (ИЭР), вычисленное для C19H17N5O3 [M+H]+: 363,3770, полученное значение: 363,3774.
Пример 12
8-циано-1-метил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=CN, R2=Me, R3=NH2, A=-(CH2)2-] (соединение 40)
Превращение d

136 мг (0,44 ммоль) 1-метил-8-(метилсульфонил)-4,5-дигидро-1Н-пиразоло[4,3-h-хиназолин]-3-карбоксамида растворяли в примерно 8 мл ДМФА и нагревали до 70°C в присутствии 116 мг (1,77 ммоль) цианида калия. Через 30 мин исходный материал исчезал, после чего реакционную смесь охлаждали и подвергали экстракции водой и этилацетатом. Высушенную над Na2SO4 и выпаренную органическую фазу суспендировали в абсолютном этаноле и перемешивали при 65°C в течение 2 часов. Посредством фильтрования выделяли 35 мг (31%) продукта в виде желтоватого порошка. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,67 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,89 (с, 1Н), 7,56 (шир. с, 1Н), 7,33 (шир. с, 1Н), 4,28 (с, 3H), 3,06 (с, 4Н). Значение МСВР (ИЭР), вычисленное для C12H10N6O [M+H]+: 255,0989, полученное значение: 255,0978.
Пример 13
1-метил-8-(1Н-пиразол-1-ил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=1Н-пиразол-1-ил, R2=Me, R3=NH2, A=-(CH2)2-] (соединение 41)
Превращение c

К раствору 100 мг (0,32 ммоль) 1-метил-8-(метилсульфонил)-4,5-дигидро-1Н-пиразоло[4,3-п-хиназолин]-3-карбоксамида в ДМСО (3 мл) добавляли 44 мг (0,65 ммоль) пиразола и 134,9 мг (0,976 ммоль) K2CO3. Указанную смесь перемешивали при 70°C в течение 4 часов. Реакционную смесь обрабатывали путем добавления воды, осадок отфильтровали и промывали водой. Твердые частицы промывали горячим этанолом и отфильтровали с получением 65 мг указанного в заголовке соединения (67%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,38 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,74 (с, 1Н), 8,69 (д, J=2,44 Гц, 1Н), 7,88 (д, J=0,85 Гц, 1Н), 7,51 (шир. с, 1Н), 7,31 (шир. с, 1Н), 6,62 (дд, J=1,59, 2,56 Гц, 1Н), 4,40 (с, 3H), 3,06 (д, J=6,96 Гц, 2Н), 2,97-3,02 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C14H13N7O [M+H]+: 296,1255, полученное значение: 296,1261.
Согласно аналогичной методике, но с применением подходящего исходного материала получали следующие соединения:
1-метил-8-[3-(трифторметил)-1H-пиразол-1-ил]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(трифторметил)-1H-пиразол-1-ил, R2=Me, R3=NH2, A=-(CH2)2-] (соединение 42), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,7 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,91 (дд, J=0,98, 2,69 Гц, 1Н), 8,82 (с, 1Н), 7,54 (шир. с, 1Н), 7,32 (шир. с, 1Н), 7,10 (д, J=2,56 Гц, 1Н), 4,38-4,40 (м, 3H), 2,96-3,15 (м, 4Н); значение МСВР (ИЭР), вычисленное для C15H12F3N7O [M+H]+: 364,1128, полученное значение: 364,1134;
8-(1Н-имидазол-1-ил)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=1Н-имидазол-1-ил, R2=Me, R3=NH2, A=-(CH2)2-] (соединение 43), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,33 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,74 (с, 1Н), 8,64 (т, J=0,98 Гц, 1Н), 7,99 (т, J=1,34 Гц, 1Н), 7,52 (шир. с, 1Н), 7,31 (шир. с, 1Н), 7,10-7,21 (м, 1Н), 4,34-4,38 (м, 3H), 3,03-3,10 (м, 2Н), 2,94-3,02 (м, 2Н); значение МСВР (ИЭР), вычисленное для C14H13N7O [M+H]+: 296,1255, полученное значение: 296,1249; и
1-метил-8-(4-нитро-1H-имидазол-1-ил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-нитро-1H-имидазол-1-ил, R2=Me, R3=NH2, A=-(CH2)2-] (соединение 44), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,82 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,93 (д, J=1,46 Гц, 1Н), 8,84 (с, 1Н), 8,81 (д, J=1,46 Гц, 1Н), 7,51-7,61 (м, 1Н), 7,20-7,41 (м, 1Н), 4,39 (с, 3H), 3,06 (дд, J=6,23, 12,94 Гц, 4Н); значение МСВР (ИЭР), вычисленное для C14H12N8O3 [M+H]+: 341,1105, полученное значение: 341,1103.
Пример 14
1-метил-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, X=SO2-, R2=Me, R3=NH2, A=-CH=CH-]
Превращение a

0,2 г (0,74 ммоль) 1-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид суспендировали в 10 мл ДХМ и подвергают реакции с 0,52 г (3,04 ммоль) М-ХПБК в течение 3 часов. Добавляли воду и NaHCO3 и твердые частицы отделяли, фильтровали и промывали насыщенным водным раствором NaHCO3 с получением 180 мг (80%) соединения белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,72 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,97 (с, 1Н), 8,60 (д, J=8,67 Гц, 1Н), 7,99 (д, J=8,79 Гц, 2Н), 7,63 (шир. с, 1Н), 4,72 (с, 3H), 3,61 (с, 3H). Значение МСВР (ИЭР), вычисленное для C12H11N5O3S [M+H]+: 306,0656, полученное значение: 306,0645.
Действуя аналогичным способом, получали следующие соединения:
1-(2-гидроксиэтил)-8-(метилсульфонил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, Х=SO2-, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,39; значение МСВР (ИЭР), вычисленное для C13H13N5O4S [M+H]+: 336,0761, полученное значение: 336,0776;
1-трет-бутил-8-(метилсульфонил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, X=SO2-, R2=трет-бутил, R3=NH2, A=-CH=CH-], ЖХ/МС (m/z): 348,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 4,59; и
8-(метилсульфонил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, X=SO2-, R2=H, R3=NH2, A=-CH=CH-], ЖХ/МС (m/z): 292,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 2,21.
Пример 15
1-(2-гидроксиэтил)-8-фенокси-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=фенокси, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 45)
Превращение b
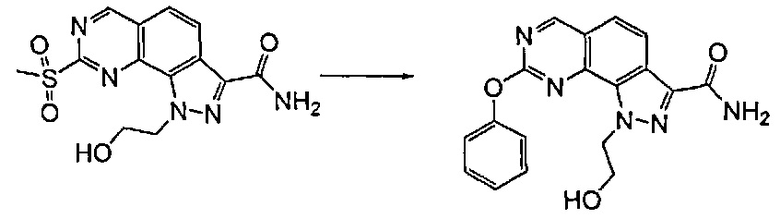
К раствору 100 мг (0,298 ммоль) 1-(2-гидроксиэтил)-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в N,N-диметилформамиде (5 мл) добавляли карбонат калия (160 мг, 1,2 ммоль) и фенол 35 мг (0,36 ммоль). Указанную смесь перемешивали при 70°C в течение 3 часов, а затем разделяли между EtOAc и H2O. Объединенные органические слои сушили над Na2SO4 и концентрировали in vacuo. Необработанный материал очищали посредством хроматографии (ДХМ/MeOH 9/1) с получением желаемого продукта (40 мг, 40%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,79 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,61 (с, 1Н), 8,25 (д, J=8,79 Гц, 1Н), 7,75-7,84 (м, J=8,79 Гц, 1Н), 7,45-7,57 (м, 3H), 7,31-7,40 (м, 3H), 4,69-4,75 (м, 2Н), 4,55-4,63 (м, 1Н), 3,59 (кв, J=5,61 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C18H15N5O3 [M+H]+: 350,1248, полученное значение: 350,1247.
С применением способов, аналогичных описанным в вышеприведенном примере, также были синтезированы следующие аналоги:
1-(2-гидроксиэтил)-8-[4-(4-метилпиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-(4-метилпиперазин-1-ил)фенокси, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 46), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,78 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,57 (с, 1Н), 8,23 (д, J=8,79 Гц, 1Н), 7,77 (д, J=8,91 Гц, 2Н), 7,46-7,53 (м, 1Н), 7,16-7,27 (м, 2Н), 6,98-7,10 (м, 2Н), 4,77 (т, J=5,19 Гц, 2Н), 4,55-4,63 (м, 1Н), 3,64 (кв, J=5,53 Гц, 2Н), 3,13-3,21 (м, 4Н), 2,52 (шир. с, 4Н), 2,26 (с, 3H); значение МСВР (ИЭР), вычисленное для C23H25N7O3 [M+H]+: 448,2092, полученное значение: 448,2082;
1-(2-гидроксиэтил)-8-[3-(4-метилпиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(4-метилпиперазин-1-ил)фенокси, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 47), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,92 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,59 (с, 1Н), 8,24 (д, J=8,79 Гц, 1Н), 7,79 (с, 1Н), 7,77 (с, 2Н), 7,48-7,50 (м, 1Н), 7,28-7,36 (м, 1Н), 6,84-6,93 (м, 2Н), 6,68-6,77 (м, 1Н), 4,70-4,78 (м, 2Н), 4,57 (т, J=5,68 Гц, 1Н), 3,62 (кв, J=5,53 Гц, 2Н), 3,15-3,22 (м, 4Н), 2,39-2,47 (м, 4Н), 2,21 (с, 3H); значение МСВР (ИЭР), вычисленное для C23H25N7O3 [M+H]+: 448,2092, полученное значение: 448,2086;
1-(2-гидроксиэтил)-8-(3-нитрофенокси)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-нитрофенокси, R2=2-гидроксиэтил, R3=NH2, A=-CH=CH-] (соединение 48), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,9 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,66 (с, 1Н), 8,30 (т, J=2,14 Гц, 1Н), 8,28 (д, J=8,79 Гц, 1Н), 8,20-8,24 (м, 1Н), 7,89-7,93 (м, 1Н), 7,80-7,86 (м, 2Н), 7,50-7,52 (м, 1Н), 4,69 (т, J=5,74 Гц, 1Н), 4,61-4,65 (м, 1Н), 3,57 (кв, J=5,45 Гц, 1Н); значение МСВР (ИЭР), вычисленное для C18H14N6O5 [M+H]+: 395,1099, полученное значение: 395,1098;
1-метил-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-нитрофенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 49); 1H ЯМР (401 МГц, ДМСО-d6) δ=9,65 (с, 1Н), 8,34 (т, J=2,14 Гц, 1Н), 8,27 (д, J=8,67 Гц, 1Н), 8,21 (ддд, J=0,98, 2,20, 8,18 Гц, 2Н), 7,91-7,95 (м, 2Н), 7,86 (шир. с, 1Н), 7,78-7,85 (м, 4Н), 7,51 (шир. с, 3H), 4,29 (с, 3H); значение МСВР (ИЭР), вычисленное для C17H12N6O4 [M+H]+: 365,0993, полученное значение: 365,0997;
1-метил-8-[3-(4-метил пиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=3-(4-метилпиперазин-1-ил)фенокси, R2=метил, R3=NH2, A=-CH=CH-] (соединение 50), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,24 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,58 (с, 1Н), 8,22 (д, J=8,67 Гц, 1Н), 7,83 (с, 1Н), 7,77 (д, J=8,79 Гц, 1Н), 7,49 (с, 1Н), 7,31 (т, J=8,18 Гц, 1Н), 6,90-6,93 (м, 1Н), 6,87 (дд, J=1,77, 8,36 Гц, 1Н), 6,71-6,77 (м, 1Н), 4,31-4,35 (м, 3H), 3,12-3,20 (м, 4Н), 2,44 (д, J=9,28 Гц, 4Н), 2,21 (с, 3H); значение МСВР (ИЭР), вычисленное для C22H23N7O2 [M+H]+: 418,1986, полученное значение: 418,1983; и
1-трет-бутил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-(4-метилпиперазин-1-ил)фенокси, R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 51), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,63 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,60 (с, 1Н), 8,34 (д, J=8,54 Гц, 1Н), 7,82 (д, J=8,79 Гц, 1Н), 7,67 (с, 1Н), 7,43-7,57 (м, 1Н), 7,12-7,20 (м, 2Н), 6,99-7,09 (м, 2Н), 3,09-3,21 (м, 4Н), 2,25 (с, 3H), 1,55 (с, 9Н); значение МСВР (ИЭР), вычисленное для C25H29N7O2 [M+H]+: 460,2456, полученное значение: 460,2448.
Пример 16
1-трет-бутил-8-(диметиламино)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=R4 и R6=Me, X=N, R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 52)
Превращение c

К раствору 10 мг (0,028 ммоль) 1-трет-бутил-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в N,N-диметилформамиде (2 мл) добавляли карбонат калия (160 мг, 0,112 ммоль) и 10 мкл мг (0,36 ммоль) 40% водного диметиламина. Указанную смесь перемешивали при комнатной температуре в течение 18 часов, а затем разбавляли H2O. Осадок отфильтровывали и промывали водой, после чего сушили in vacuo с получением 5 мг желаемого продукта (65%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,44 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,23 (с, 1Н), 8,05 (д, J=8,54 Гц, 1Н), 7,61 (шир. с, 1Н), 7,56 (д, J=8,67 Гц, 1Н), 7,41 (шир. с, 1Н), 2,00 (с, 9Н). Значение МСВР (ИЭР), вычисленное для C16H20N6O [M+H]+: 313,1772, полученное значение: 313,1777.
Пример 17
8-метокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, X=O, R2=метил, A=-CH=CH-] (соединение 53)
Превращение b
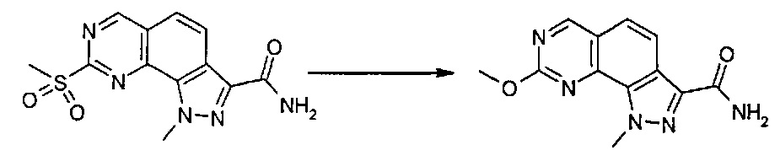
1-метил-8-(метилсульфонил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (30 мг 0,1 ммоль) растворяли в 3 мл метанола и добавляли карбонат калия (27 мг, 0,2 ммоль). Указанную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток растирали в порошок с применением диэтилового эфира, фильтровали и сушили с получением 20 мг указанного в заголовке продукта (80%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,28 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,51 (с, 1Н), 8,18 (д, J=8,67 Гц, 1Н), 7,83 (шир. с, 1Н), 7,73 (д, J=8,67 Гц, 1Н), 7,47-7,57 (м, 1Н), 4,70 (с, 3H), 4,14 (с, 3H). Значение МСВР (ИЭР), вычисленное для C12H11N5O2 [M+H]+: 258,0986, полученное значение: 258,0987.
Действуя аналогичным способом, получали следующие соединения:
8-этокси-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Et, X=O, R2=метил, A=-CH=CH-] (соединение 54), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,71 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,46-9,55 (м, 1Н), 8,17 (д, J=8,79 Гц, 1Н), 7,79-7,89 (м, 1Н), 7,72 (д, J=8,79 Гц, 1Н), 7,50 (шир. с, 1Н), 4,68 (с, 3H), 4,55-4,63 (м, 2Н), 1,46 (т, J=7,08 Гц, 3H); значение МСВР (ИЭР), вычисленное для C13H13N5O2 [M+H]+: 272,1142, полученное значение: 272,1149; и
8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Me, X=O, R2=H, A=-CH=CH-] (соединение 55), ЖХ/МС (m/z): 244,2 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 2,89 мин.
Пример 18
8-(3-формилфенокси)-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-формилфенил, X=O, R2=метил, A=-CH=CH-] (соединение 56)
Превращение b
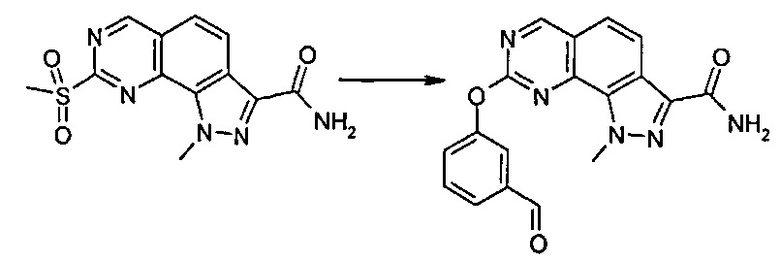
К раствору 120 мг (0,39 ммоль) 1-метил-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид в N,N-диметилформамиде (3 мл) добавляли карбонат калия (108 мг, 0,78 ммоль) и м-гидроксибензальдегид (70 мг, 0,58 ммоль). Указанную смесь перемешивали при 70°C в течение 4 часов, а затем разделяли между EtOAc и H2O. Объединенные органические слои сушили над Na2SO4 и концентрировали in vacuo с получением желаемого продукта (100 мг 74%). ЖХ/МС (m/z): 348,0 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 5,05 мин.
Пример 19
1-метил-8-{3-[(4-метиллиперазин-1-ил)метил]фенокси}-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-[(4-метилпиперазин-1-ил)метил]фенил, X=O, R2=метил, A=-CH=CH-] (соединение 57)
и
8-[3-(гидроксиметил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-гидроксиметилфенил, X=O, R2=метил, A=-CH=CH-] (соединение 58)
Превращение j
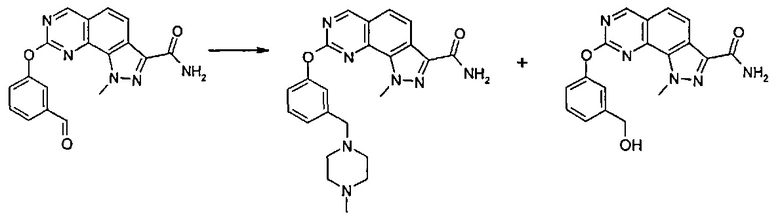
К раствору 30 мг (0,086 ммоль) 8-(3-формилфенокси)-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в N,N-диметилформамиде (2 мл) и уксусной кислоте (50 мкл) добавляли N-метилпиперазин (13 мкл, 0,13 ммоль) и цианоборгидрид натрия (6 мг, 0,26 ммоль). Указанный раствор перемешивали при к.т. в течение 3 часов, контролировали посредством ТСХ и ЖХ/МС (способ 1). Реакционную смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали на колонке (ДХМ/MeOH/NH4OH 95/5/0,1) с получением 1-метил-8-{3-[(4-метилпиперазин-1-ил)метил]фенокси}-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в качестве основного продукта (15 мг, 40%), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,14 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,60 (с, 1Н), 8,23 (д, J=8,79 Гц, 1Н), 7,83 (с, 1Н), 7,78 (д, J=8,79 Гц, 1Н), 7,49-7,50 (м, 1Н), 7,46 (т, J=7,81 Гц, 1Н), 7,26 (дт, J=1,71, 7,69 Гц, 3H), 4,27 (с, 3H), 3,53 (с, 2Н), 2,30-2,47 (м, 8Н), 2,17 (шир. с, 3H); значение МСВР (ИЭР), вычисленное для C23H25N7O2 [M+H]+: 432,2143, полученное значение: 432,2141; и
8-[3-(гидроксиметил)фенокси]-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в качестве минорного продукта (10 мг, 33%), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,54 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,60 (с, 1Н), 8,23 (д, J=8,79 Гц, 1Н), 7,83 (с, 1Н), 7,78 (д, J=8,79 Гц, 1Н), 7,49 (с, 1Н), 7,42-7,48 (м, 1Н), 7,30-7,33 (м, J=1,83 Гц, 1Н), 7,20-7,28 (м, 2Н), 5,27 (т, J=5,80 Гц, 1Н), 4,56 (д, J=5,74 Гц, 2Н), 4,30 (с, 3H); значение МСВР (ИЭР), вычисленное для C18H15N5O3 [M+H]+: 350,1248, полученное значение: 350,1253.
Действуя аналогичным способом, получали следующие соединения:
8-{3-[(диметиламино)метил]фенокси}-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-[(диметиламино)метил]фенил, X=O, R2=метил, A=-CH=CH-] (соединение 59), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,89 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,61 (с, 1Н), 8,23 (д, J=8,79 Гц, 1Н), 7,82 (с, 1Н), 7,78 (д, J=8,91 Гц, 1Н), 7,49-7,50 (м, 1Н), 7,46 (т, J=7,81 Гц, 1Н), 7,29 (д, J=1,46 Гц, 1Н), 7,21-7,28 (м, 2Н), 4,27 (с, 3H), 3,46 (с, 2Н), 2,18 (с, 6Н); значение МСВР (ИЭР), вычисленное для C20H20N6O2 [M+H]+: 377,1721, полученное значение: 377,1719; и
1-метил-8-[3-(морфолин-4-илметил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-(морфолин-4-илметил)фенил, X=O, R2=метил, A=-CH=CH-] (соединение 60), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,72 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,60 (с, 1Н), 8,23 (д, J=8,67 Гц, 1Н), 7,83 (с, 1Н), 7,78 (д, J=8,79 Гц, 1Н), 7,49-7,50 (м, 1Н), 7,46 (т, J=7,87 Гц, 1Н), 7,29-7,32 (м, 1Н), 7,23-7,29 (м, 2Н), 4,27 (с, 3H), 3,55-3,60 (м, 4Н), 3,53 (с, 2Н), 2,36-2,41 (м, 4Н); значение МСВР (ИЭР), вычисленное для C22H22N6O3 [M+H]+: 419,1826, полученное значение: 419,1815.
Пример 20
8-(3-аминофенокси)-1-метил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-аминофенил, X=O, R2=метил, A=-CH=CH-] (соединение 61)
Превращение k
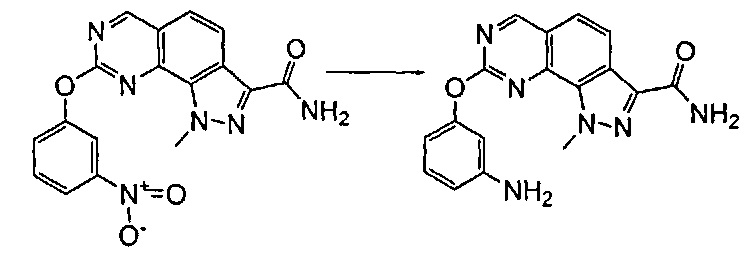
55 мг (0,15 ммоль) 1-метил-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида суспендировали в 1,4-диоксане (5 мл) и воде (1 мл), добавляли 43 мг (0,6 ммоль) цинковой пыли и 88 мг (1,5 ммоль) хлорида аммония. Указанную смесь перемешивали при 100°C в течение 2 часов. Летучие компоненты удаляли in vacuo, остаток растворяли в этилацетате и воде, органический слой подвергали экстракции и промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанное твердое вещество очищали посредством флэш-хроматографии на силикагеле (дихлорметан/метанол 95/5) с получением 10 мг (выход 20%) указанного в заголовке соединения. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,7 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,57 (с, 1Н), 8,22 (д, J=8,79 Гц, 1Н), 7,84 (с, 1Н), 7,77 (д, J=8,79 Гц, 1Н), 7,49 (с, 1Н), 7,11 (т, J=7,99 Гц, 1Н), 6,50-6,53 (м, 1Н), 6,41-6,50 (м, J=0,92, 2,23, 8,68, 8,68 Гц, 2Н), 5,26 (с, 2Н), 4,38 (с, 3H). Значение МСВР (ИЭР), вычисленное для C17H14N6O2 [M+H]+: 335,1251, полученное значение: 335,1258.
Действуя аналогичным способом, получали следующее соединение:
8-(3-аминофенокси)-1-(2-гидроксиэтил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=3-аминофенил, X=O, R2=2-гидроксиэтил, A=-CH=CH-] (соединение 62), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,26 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,58 (с, 1Н), 8,23 (д, J=8,79 Гц, 1Н), 7,74-7,83 (м, J=8,79 Гц, 1Н), 7,49 (с, 1Н), 7,12 (т, J=8,12 Гц, 1Н), 6,47-6,55 (м, 2Н), 6,40-6,47 (м, 1Н), 5,27 (с, 2Н), 4,81 (т, J=5,19 Гц, 2Н), 4,61 (т, J=5,68 Гц, 1Н), 3,69 (кв, J=5,57 Гц, 2Н); значение МСВР (ИЭР), вычисленное для C18H16N6O3 [M+H]+: 365,1357, полученное значение: 365,1363.
Получение J. Этиловый эфир 8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=Me-S-, R2=H]
этап 5
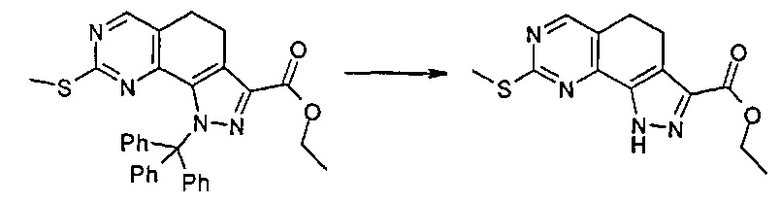
Этиловый эфир 8-(метилсульфанил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (1,0 г, 1,99 ммоль) в ДХМ (20 мл) обрабатывали трифторуксусной кислотой (5 мл). Полученную смесь перемешивали при к.т. в течение 1 часа и растворитель удаляли in vacuo. Остаток растирали в порошок с применением диэтилового эфира, фильтровали и сушили с получением 500 мг указанного в заголовке соединения (96%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,62 мин.
Пример 21
8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=H, R3=NH2, A=-(CH2)2-] (соединение 63)
этап 6a

Суспензию 0,5 г (1,91 ммоль) этилового эфира 8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в 5 мл 7 н. NH3 в метаноле выдерживали при 60°C в течение 72 часов. Летучие компоненты удаляли под вакуумом, остаток растирали в порошок с применением диэтилового эфира и отфильтровывали твердое вещество с получением 0,4 г указанного в заголовке соединения (80%). 1H ЯМР (401 МГц, ДМСО-d6) δ=14,13 (шир. с, 1Н), 8,52 (с, 1Н), 7,53 (шир. с, 1Н), 7,26 (шир. с, 1Н), 2,96-3,08 (м, J=7,45 Гц, 2Н), 2,83-2,94 (м, 2Н), 2,56 (с, 3H). Значение МСВР (ИЭР), вычисленное для C11H11N5OS [M+H]+: 262,0757, полученное значение: 262,07575.
Пример 22
1-(2-фторэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-фторэтил, R3=NH2, A=-(CH2)2-] (соединение 64)
Превращение e
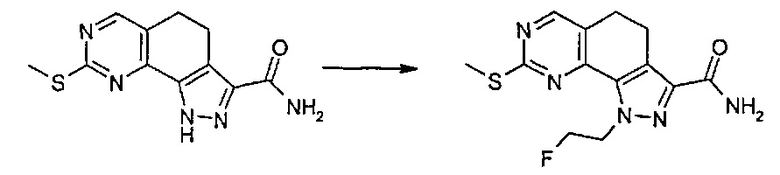
К раствору 8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (50 мг, 0,191 ммоль) в ацетонитриле добавляли 2-фтор-1-йодэтан (66,7 мг, 0,38 ммоль) и карбонат цезия (124 мг, 0,38 ммоль). Полученную смесь выдерживали при 60°C в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разделяли между этилацетатом и водой. Органические слои промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/EtOH 9/1) с получением указанного в заголовке соединения. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,82 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,52-8,58 (м, 1Н), 7,50 (шир. с, 1Н), 7,33 (шир. с, 1Н), 5,07-5,15 (м, J=4,88 Гц, 1Н), 5,03 (т, J=4,94 Гц, 1Н), 4,93-4,99 (м, 1Н), 4,85 (т, J=4,88 Гц, 1Н), 3,00-3,06 (м, J=7,69 Гц, 2Н), 2,85-2,93 (м, J=8,06 Гц, 2Н), 2,53 (с, 3H). Значение МСВР (ИЭР), вычисленное для C13H14FN5OS [M+H]+: 308,0976, полученное значение: 308,0976.
Действуя аналогичным способом, получали следующие соединения:
1-(2-хлорэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-хлорэтил, R3=NH2, A=-(CH2)2-] (соединение 65), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,25 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,55 (с, 1Н), 7,53 (шир. с, 1Н), 7,35 (шир. с, 1Н), 5,09 (т, J=6,23 Гц, 2Н), 4,11 (т, J=6,29 Гц, 2Н), 2,98-3,06 (м, 2Н), 2,86-2,92 (м, 2Н), 2,56 (с, 3H); значение МСВР (ИЭР), вычисленное для C13H14CIN5OS [M+H]+: 324,0681, полученное значение: 324,0676;
1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-гидроксиэтил, R3=NH2, -(CH2)2-] (соединение 66), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,1 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,53 (с, 1Н), 7,35-7,60 (м, 1Н), 7,28 (шир. с, 1Н), 4,84-4,90 (м, 1Н), 4,79 (т, J=6,16 Гц, 2Н), 3,79-3,90 (м, 2Н), 2,95-3,08 (м, 2Н), 2,82-2,94 (м, 2Н), 2,55 (с, 3H); значение МСВР (ИЭР), вычисленное для C13H15N5O2S [M+H]+: 306,1019, полученное значение: 306,1023;
1-(2-метоксиэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-метоксиэтил, R3=NH2, A=-(CH2)2-] (соединение 67), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,76 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,54 (с, 1Н), 7,47 (шир. с, 1Н), 7,30 (шир. с, 1Н), 4,90 (т, J=5,86 Гц, 2Н), 3,76-3,89 (м, 2Н), 3,23 (с, 3H), 2,98-3,06 (м, 2Н), 2,84-2,91 (м, 2Н), 2,54 (с, 3H); значение МСВР (ИЭР), вычисленное для C14H17N5O2S [M+H]+: 320,1176, полученное значение: 320,1187;
1-[3-(диметиламино)пропил]-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=3-(диметиламино)пропил, R3=NH2, A=-(CH2)2-] (соединение 68), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,69 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,53 (с, 1Н), 7,46 (шир. с, 1Н), 7,29 (шир. с, 1Н), 4,70-4,77 (м, 2Н), 2,97-3,05 (м, 2Н), 2,83-2,91 (м, 2Н), 2,54-2,58 (м, 3H), 2,22-2,31 (м, 2Н), 2,11 (с, 6Н), 1,92-2,03 (м, 2Н); значение МСВР (ИЭР), вычисленное для C16H22N6OS [M+H]+: 347,1649, полученное значение: 347,1638; и
1-(1-амино-2-метил-1-оксопропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=1-амино-2-метил-1-оксопропан-2-ил, R3=NH2, A=-(CH2)2-] (соединение 69), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,13 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,49 (с, 1Н), 7,42 (с, 1Н), 7,29 (с, 1Н), 6,96-7,14 (м, 1Н), 2,94-3,05 (м, 2Н), 2,75-2,85 (м, 2Н), 2,55 (с, 3H), 1,90 (с, 6Н); значение МСВР (ИЭР), вычисленное для C15H18N6O2S [M+H]+: 347,1285, полученное значение: 347,1282.
Пример 23
Гидрохлорид 8-(метилсульфанил)-1-(пиперидин-4-илметил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=пиперидин-4-илметил, R3=NH2, A=-(CH2)2-] (соединение 70)

К раствору 8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (50 мг 0,191 ммоль) в ацетонитриле добавляли трет-бутиловый эфир 4-(бромметил)пиперидин-1-карбоновой кислоты (105,71 мг, 0,38 ммоль) и карбонат цезия (124 мг, 0,38 ммоль). Полученную смесь выдерживали при 60°C в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разделяли между этилацетатом и водой. Органические слои промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/EtOH 9/1) с получением трет-бутилового эфира 4-{[3-карбамоил-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-1-ил]метил}пиперидин-1-карбоновой кислоты (30 мг, 0,65 ммоль). Затем указанное соединение обрабатывали 2 мл 4 М HCl в 1,4-диоксане. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли in vacuo, полученный остаток растирали в порошок с применением диэтилового эфира, фильтровали, промывали Et2O и сушили in vacuo с получением 32 мг указанного в заголовке соединения (97%) в виде твердого вещества белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,83 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,62 (д, J=10,01 Гц, 1Н), 8,55 (с, 1Н), 8,27-8,49 (м, J=8,42 Гц, 1Н), 7,44 (с, 1Н), 7,35 (с, 1Н), 4,63-4,78 (м, 2Н), 3,18-3,28 (м, J=6,10 Гц, 2Н), 2,99-3,07 (м, 2Н), 2,85-2,92 (м, J=7,93 Гц, 2Н), 2,74-2,85 (м, J=10,38 Гц, 2Н), 2,57 (с, 3H), 2,18-2,31 (м, 1Н), 1,56-1,70 (м, 2Н), 1,37-1,51 (м, J=3,72, 13,98 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C17H22N6OS [M+H]+: 359,1649, полученное значение: 359,1634.
Действуя аналогичным способом, получали следующие соединения:
гидрохлорид 1-(3-аминопропил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=3-аминопропил, R3=NH2, A=-(CH2)2-] (соединение 71), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,58 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,56 (с, 1Н), 7,79 (шир. с, 3H), 7,47-7,50 (м, 1Н), 7,38 (шир. с, 1Н), 4,77 (т, J=6,53 Гц, 2Н), 2,99-3,06 (м, 2Н), 2,86-2,93 (м, 2Н), 2,76-2,83 (м, 2Н), 2,54-2,59 (м, 3H), 2,17 (шир. с, 2Н); значение МСВР (ИЭР), вычисленное для C14H18N6OS [M+H]+: 319,1336, полученное значение: 319,1344;
8-(метилсульфанил)-1-(пиперидин-4-ил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=Me-S-, R2=пиперидин-4-ил, R3=NH2, A=-(CH2)2-] (соединение 72), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,65 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,58-8,65 (м, 1Н), 8,56 (с, 1Н), 8,46 (шир. с, 1Н), 7,41 (шир. с, 1Н), 7,38 (шир. с, 1Н), 5,52-5,73 (м, J=6,84, 6,84 Гц, 1Н), 3,46-3,62 (м, 2Н), 3,04-3,13 (м, J=6,84 Гц, 2Н), 2,97-3,04 (м, 2Н), 2,82-2,91 (м, J=8,18 Гц, 2Н), 2,58 (с, 3H), 2,19-2,32 (м, J=7,32 Гц, 4Н); значение МСВР (ИЭР), вычисленное для C16H20N6OS [M+H]+: 345,1492, полученное значение: 345; и
1-(2-аминоэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=Me-S-, R2=2-аминоэтил, R3=NH2, A=-(CH2)2-] (соединение 73), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,46 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,58 (с, 1Н), 7,92 (шир. с, 3H), 7,74 (с, 1Н), 7,43 (с, 1Н), 4,85-5,09 (м, 2Н), 3,42-3,57 (м, J=5,98 Гц, 2Н), 2,96-3,09 (м, 2Н), 2,80-2,94 (м, 2Н), 2,56 (с, 3H); значение МСВР (ИЭР), вычисленное для C13H16N6OS [M+H]+: 305,1179, полученное значение: 305,1191.
Пример 24
1-[2-(4-метилпиперазин-1-ил)этил]-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-(4-метилпиперазин-1-ил)этил, R3=NH2, A=-(CH2)2-] (соединение 74)
и
1-этенил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=этенил, R3=NH2, A=-(CH2)2-] (соединение 75)
Превращения f и g
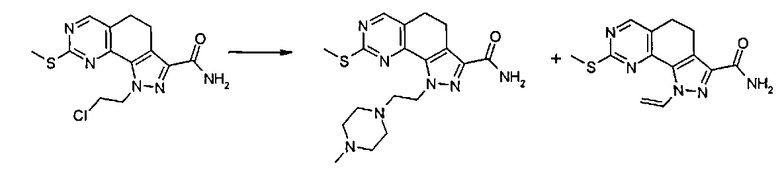
К раствору 1-(2-хлорэтил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (50 мг 0,15 ммоль) в метаноле (3 мл) добавляли N-метилпиперазин (62 мкл, 0,61 ммоль) и карбонат цезия (97 мг, 0,3 ммоль). Полученную смесь выдерживали при 60°C в течение 48 часов. Летучие компоненты удаляли под вакуумом, необработанное твердое вещество растворяли в этилацетате и воде, органический слой промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/EtOAc 8/2) с получением соединения, представлявшего собой 1-этенил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид, в виде твердого вещества желтого цвета (15 мг, выход 35%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,34 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,58 (с, 1Н), 8,35 (дд, J=8,73, 15,44 Гц, 1Н), 7,71 (с, 1Н), 7,46 (шир. с, 1Н), 5,96 (д, J=15,38 Гц, 1Н), 5,15 (д, J=8,67 Гц, 1Н), 3,00-3,09 (м, 2Н), 2,86-2,94 (м, 2Н), 2,56 (с, 3H). Значение МСВР (ИЭР), вычисленное для C13H13N5OS [M+H]+: 288,0914, полученное значение: 288,0913.
Сменив элюент на такой, как ДХМ/MeOH/NH4OH 9/1/0,1, собирали полярное соединение, представлявшее собой 1-(2-[N-метилпиперазин]этил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид, в виде твердого вещества желтого цвета (17 мг, выход 30%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,79 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,35 (с, 1Н), 7,46 (с, 1Н), 7,28 (с, 1Н), 4,82 (т, J=6,84 Гц, 2Н), 2,98-3,04 (м, 2Н), 2,88 (д, J=7,93 Гц, 2Н), 2,55 (с, 3H). Значение МСВР (ИЭР), вычисленное для C18H25N7OS [M+H]+: 388,1914, полученное значение: 388,1909.
Пример 25
1-{2-[4-(диметиламино)пиперидин-1-ил]этил}-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=2-[4-(диметиламино)пиперидин-1-ил]этил, R3=NH2, A=-(CH2)2-] (соединение 76)
Превращение h
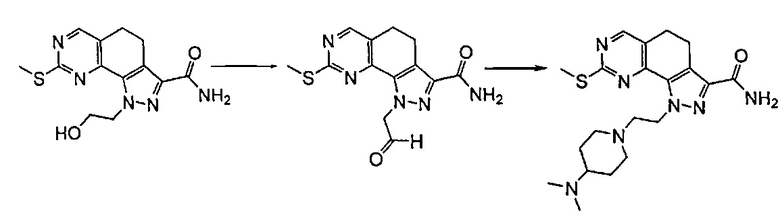
К раствору 1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (20 мг 0,077 ммоль) в этилацетате (2 мл), добавляли IBX (64 мг, 0,23 ммоль). Полученную смесь выдерживали при 80°C в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и летучие компоненты удаляли in vacuo, а альдегид в необработанном виде применяли в следующей реакции. Указанный альдегид растворяли в ТГФ/ДМФА (3/1 мл), после чего добавляли триацетоксиборгидрид натрия (49 мг, 0,233 ммоль) и 4-диметиламинопиперидин (20 мкл, 0,156 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 72 часов. Растворитель выпаривали и полученный необработанный материал очищали посредством хроматографии (ДХМ/MeOH/NH4OH 8/2/0,2) с получением 15 мг указанного в заголовке соединения (48%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,93 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,55 (с, 1Н), 7,46 (с, 1Н), 7,29 (с, 1Н), 4,81 (т, J=6,90 Гц, 2Н), 2,96-3,04 (м, 2Н), 2,79-2,93 (м, J=8,18 Гц, 5Н), 2,71 (т, J=6,59 Гц, 2Н), 2,56 (с, 3H), 2,14 (с, 6Н), 1,93-2,06 (м, J=16,11 Гц, 3H), 1,53-1,68 (м, J=14,16 Гц, 2Н), 1,04-1,20 (м, J=3,17 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C20H29N7OS [M+H]+: 416,2227, полученное значение: 416,2234.
Пример 26
Гидрохлорид 8-этокси-1-(пиперидин-4-илметил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R4=Et, X=O, R2=пиперидин-4-илметил, R3=NH2, A=-(CH2)2] (соединение 77)
Превращение e
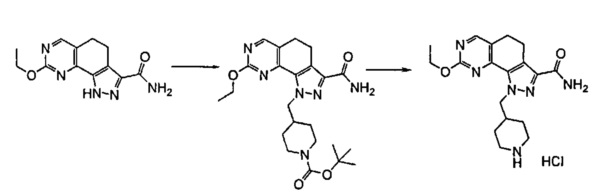
К раствору 8-этокси-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (40 мг, 0,154 ммоль) в ДМФА (3 мл) добавляли трет-бутиловый эфир 4-(бромметил)пиперидин-1-карбоновой кислоты (85 мг, 0,308 ммоль) и Cs2CO3 (121 мг, 0,370 ммоль). Указанную смесь нагревали до 70°C и перемешивали в течение 4 часов. Охлажденную смесь обрабатывали водой (10 мл) и подвергали экстракции этилацетатом. Объединенные органические фазы промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством флэш-хроматографии, элюируя ДХМ/EtOAc/EtOH в соотношении 7/2/1, с получением 50 мг (70%) желаемого продукта, представлявшего собой трет-бутиловый эфир 4-{[3-карбамоил-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил]метил}-пиперидин-1-карбоновой кислоты, в виде твердого вещества белого цвета. Последний продукт растворяли в ДХМ (2 мл) и добавляли 4 М HCl в 1,4-диоксане (2 мл). Указанный раствор перемешивали при к.т. в течение 1 часа и выпаривали досуха. Твердое вещество растирали в порошок с применением диэтилового эфира, фильтровали и сушили in vacuo с получением 40 мг желаемого продукта (95%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,65 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,65 (д, J=11,60 Гц, 1Н), 8,51 (с, 1Н), 8,31-8,46 (м, J=15,38 Гц, 1Н), 7,42 (шир. с, 1Н), 7,34 (с, 1Н), 4,69 (д, J=7,20 Гц, 1Н), 4,39 (кв, J=6,96 Гц, 1Н), 3,18-3,27 (м, 2Н), 2,97-3,04 (м, 2Н), 2,74-2,90 (м, J=7,93 Гц, 4Н), 2,18-2,32 (м, J=5,98 Гц, 1Н), 1,59-1,70 (м, J=11,84 Гц, 2Н), 1,40-1,53 (м, J=4,03 Гц, 2Н), 1,38 (т, J=7,02 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C18H24N6O2 [M+H]+: 357,2033, полученное значение: 357,2041.
Действуя аналогичным способом, получали следующие соединения:
1-[3-(диметиламино)пропил]-8-этокси-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Et, X=O, R2=3-(диметиламино)пропил, R3=NH2, A=-(CH2)2-] (соединение 78), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,63 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,49 (с, 1Н), 7,44 (с, 1Н), 7,29 (шир. с, 1Н), 4,67-4,78 (м, 2Н), 4,39 (кв, J=7,08 Гц, 2Н), 2,93-3,05 (м, 2Н), 2,81-2,93 (м, 2Н), 2,32-2,42 (м, J=1,92, 1,92, 3,72 Гц, 2Н), 2,19 (шир. с, 6Н), 1,90-2,07 (м, 2Н), 1,36 (т, J=7,02 Гц, 3H); значение МСВР (ИЭР), вычисленное для C17H24N6O2 [M+H]+: 345,2033, полученное значение: 345,203; и
гидрохлорид 8-этокси-1-(пиперидин-4-ил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R4=Et, X=O, R2=пиперидин-4-ил, R3=NH2, A=-(CH2)2-] (соединение 79), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,59 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,57-8,95 (м, 2Н), 8,52 (с, 1Н), 7,39 (шир. с, 2Н), 5,58 (квин, J=7,23 Гц, 1Н), 4,36-4,45 (м, 2Н), 2,96-3,02 (м, 3H), 2,82-2,89 (м, J=8,18 Гц, 2Н), 1,37 (т, J=7,02 Гц, 3H); значение МСВР (ИЭР), вычисленное для C17H22N6O2 [M+H]+: 343,1877, полученное значение: 343,1876.
Пример 27
8-этокси-1-[(1-метилпиперидин-4-ил)метил]-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R4=Et, X=O, R2=(1-метилпиперидин-4-ил)метил, R3=NH2, A=-(CH2)2-] (соединение 80)
Превращение n
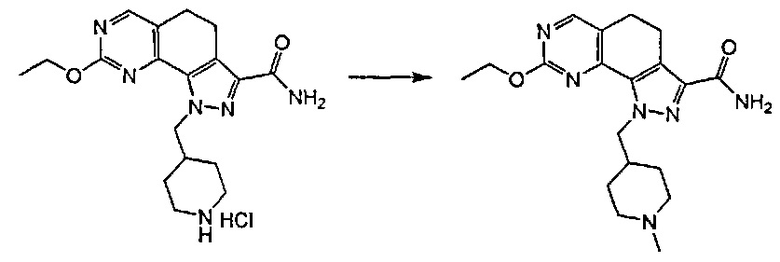
К раствору гидрохлорид 8-этокси-1-(пиперидин-4-илметил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (30 мг, 0,084 ммоль) в ДМФА (3 мл) добавляли 1 мл формальдегида (37% в воде, 12,3 ммоль) и триацетоксиборгидрид натрия (71 мг, 0,33 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Указанную смесь разбавляли водой и подвергали экстракции этилацетатом, органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали досуха. Необработанную смесь подвергали хроматографии на диоксиде кремния, элюируя ДХМ/EtOAc/EtOH в соотношении 7/2/1, с получением 20 мг желаемого продукта (64%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,7 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,49 (с, 1Н), 7,43 (с, 1Н), 7,30 (шир. с, 1Н), 4,65 (д, J=7,45 Гц, 1Н), 4,38 (кв, J=7,08 Гц, 2Н), 2,97-3,04 (м, 2Н), 2,83-2,90 (м, 2Н), 2,79 (шир. с, 1Н), 2,18 (шир. с, 2Н), 1,86-2,03 (м, 2Н), 1,44-1,53 (м, 2Н), 1,34-1,39 (м, 3H), 1,29 (д, J=4,03 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C19H26N6O2 [M+H]+: 371,219, полученное значение: 371,2191.
Получение K: трет-Бутиловый эфир 4-[(3-карбамоил-8-йод-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил)метил]пиперидин-1-карбоновой кислоты [(XIV)]
этап 7
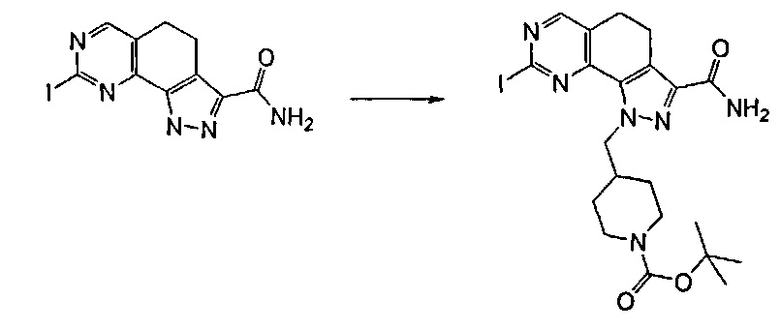
К раствору 50 мг (0,146 ммоль) 8-йод-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (полученного способом, описанным в WO 2008074788 A1) в ДМФА (3 мл) добавляли 61 мг (0,219 ммоль) трет-бутилового эфира 4-(бромметил)пиперидин-1-карбоновой кислоты и 95 мг CS2CO3 (0,293 ммоль). Указанную смесь нагревали до 70°C и перемешивали в течение 4 часов. Охлажденную смесь обрабатывали водой (10 мл) и подвергали экстракции этилацетатом. Органическую фазу промывали водой и рассолом, сушили над Na2SO4, и концентрировали. Необработанный материал очищали посредством флэш-хроматографии, элюируя ДХМ/EtOH в соотношении 9/1, с получением 63 мг (80%) желаемого продукта, указанного в заголовке, в виде твердого вещества белого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,51 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,46 (с, 1Н), 7,47 (с, 1Н), 7,32 (с, 1Н), 4,50-4,55 (м, 2Н), 3,84-3,96 (м, 2Н), 2,99-3,07 (м, 2Н), 2,84-2,95 (м, 2Н), 2,59-2,75 (м, 2Н), 2,07 (с, 1Н), 1,56 (шир. с, 2Н), 1,39 (с, 9Н), 1,08-1,22 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C21H27IN6O3 [M+H]+: 539,1262, полученное значение: 539,1274.
Действуя способом, аналогичным описанному выше, получали следующие соединения:
этиловый эфир 1-{3-[(трет-бутоксикарбонил)амино]пропил}-8-йод-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и этиловый эфир 2-{3-[(трет-бутоксикарбонил)амино]пропил}-8-йод-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты. ЖХ/МС (m/z): 528,0 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 6,43 мин.
Пример 28
Гидрохлорид 8-(2,6-дифторфенил)-1-(пиперидин-4-илметил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=2,6-дифторфенил, R2=пиперидин-4-илметил, R3=NH2, A=-(CH2)2-] (соединение 81)
этап 8
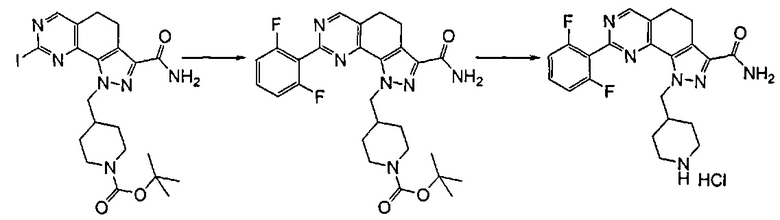
К раствору трет-бутилового эфира 4-[(3-карбамоил-8-йод-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил)метил]пиперидин-1-карбоновой кислоты (30 мг, 0,036 ммоль) в 3 мл 1,4-диоксана и 1 мл воды в атмосфере аргона последовательно добавляли 17,6 мг (0,11 ммоль) 2,6-дифторфенилбороновой кислоты, 13,7 мг (0,017 ммоль) комплекса 1,1'-бис(дифенилфосфино)ферроценпалладия с дихлорметаном и 54 мг (0,167 ммоль) карбоната цезия. Указанную смесь подвергали воздействию микроволнового излучения при 80° в течение 1 часа в герметизированном сосуде. Реакционную смесь фильтровали, пропуская через наполнитель celite, и растворитель выпаривали досуха. Затем необработанный материал разделяли между этилацетатом и водой, органический слой сушили над сульфатом натрия и растворитель удаляли in vacuo. После очистки посредством флэш-хроматографии на колонке, содержавшей силикагель, (ДХМ/EtOAc 7/3) получали 20 мг (70%) трет-бутилового эфира 4-{[3-карбамоил-8-(2,6-дифторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил]метил}пиперидин-1-карбоновой кислоты. Указанный продукт растворяли в ДХМ (2 мл) и добавляли 4 М HCl в 1,4-диоксане (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, растворитель выпаривали досуха. Твердое вещество обрабатывали этиловым эфиром и осадок собирали посредством фильтрования с получением 17 мг (85%) указанного в заголовке соединения в виде твердого вещества бледно-желтого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,14 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,89 (с, 1Н), 8,57 (д, J=9,76 Гц, 1Н), 8,30 (д, J=10,01 Гц, 1Н), 7,55-7,69 (м, 1Н), 7,45 (с, 1Н), 7,37 (с, 1Н), 7,23-7,33 (м, 2Н), 4,57-4,66 (м, 2Н), 3,16-3,26 (м, 2Н), 3,01-3,14 (м, 4Н), 2,69-2,85 (м, J=10,74 Гц, 2Н), 2,09-2,29 (м, J=7,20 Гц, 1Н), 1,56-1,70 (м, J=12,94 Гц, 2Н), 1,32-1,48 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C21H27IN6O3 [M+H]+: 425,1896, полученное значение: 425,1896.
Действуя способом, аналогичным описанному выше, получали следующее соединение:
8-(2,6-дифторфенил)-1-(2-фторэтил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=2,6-дифторфенил, R2=2-фторэтил, R3=NH2, A=-(CH2)2-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,31 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,89 (с, 1Н), 7,61 (tT, J=6,44, 8,45 Гц, 1Н), 7,53 (шир. С, 1Н), 7,35 (с, 1Н), 7,19-7,32 (м, 2Н), 4,96-5,11 (м, 2Н), 4,74-4,96 (м, 2Н), 2,98-3,18 (м, 4Н); значение МСВР (ИЭР), вычисленное для C18H14F3N5O [M+H]+: 374,1223, полученное значение: 374,1225.
Получение L: Этиловый эфир 8-(4-фторфенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=4-фторфенил, R2=тритил, A=-(CH2)2-]
этап 9

К раствору 500 мг (0,82 ммоль) этилового эфира 8-йод-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (полученного способом, описанным в WO 2008074788 A1) в толуоле (10 мл) добавляли 228 мг (1,63 ммоль) 4-фторбензолбороновой кислоты, 103 мг (2,46 ммоль) хлорида лития и 339,8 мг (2,46 ммоль) карбоната калия, растворенных в воде (0,5 мл). Реакционную смесь подвергали дегазации аргоном, затем добавляли 22 мг (0,041 ммоль, 5 мол.%) дихлорида бис(трифенилфосфин)палладия(II) и реакционную смесь выдерживали при 100°C в течение 60 мин. Результаты ЖХ/МС анализа свидетельствовали о полном превращении исходного материала в продукт. Реакционную смесь охлаждали до комнатной температуры, затем концентрировали in vacuo и объединяли с силикагелем. Необработанный продукт очищали посредством флэш-хроматографии, элюируя ДХМ/EtOH 10/0,5, с получением 380 мг желаемого продукта (80%). ЖХ/МС (m/z): 581,2 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 9,02.
Действуя способом, аналогичным описанному выше, получали следующие соединения:
этиловый эфир 8-(4-метоксифенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=4-метоксифенил, R2=тритил, A=-(CH2)2-], ЖХ/МС (m/z): 593,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 8,91 мин;
этиловый эфир 1-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=4-фторфенил, R2=3-(трет-бутоксикарбонил)аминопропил, A=-(CH2)2-]
и
этиловый эфир 2-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты [(VIII), R1=4-фторфенил, R2=3-(трет-бутоксикарбонил)аминопропил, A=-(CH2)2-], ЖХ/МС (m/z): 496,1 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 7,87 мин.
Получение M: 8-(4-фторфенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновая кислота [(XI), R1=4-фторфенил, R2=тритил, A=-(CH2)2-]
этап 6b1
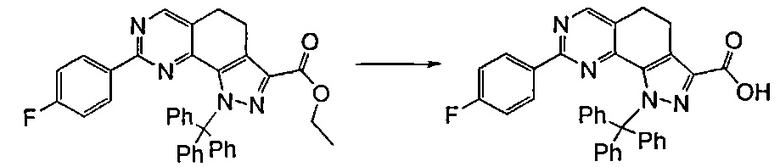
К раствору 380 мг (0,65 ммоль) этилового эфира 8-(4-фторфенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в этаноле (20 мл) добавляли 3 мл (6 ммоль) 2 н. водного раствора NaOH. Указанную смесь выдерживали при 80°C в течение 30 мин. После охлаждения до комнатной температуры летучие компоненты удаляли in vacuo. Остаток разбавляли водой и уксусной кислотой с pH=4, и образовывавшийся осадок отфильтровывали и промывали этанолом. Твердое вещество сушили в сушильной печи (40°C, в вакууме) в течение 2 часов с получением 360 мг продукта (99%). ЖХ/МС (m/z): 553,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 5,84.
Действуя способом, аналогичным описанному выше, получали следующие соединения:
8-(4-метоксифенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновую кислоту [(XI), R1=4-метоксифенил, R2=тритил, A=-(CH2)2-], ЖХ/МС (m/z): 565,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 6,03;
1-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновую кислоту [(XI), R1=4-фторфенил, R2=3-(трет-бутоксикарбонил)аминопропил, A=-(CH2)2-] и
2-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-2H-пиразоло[4,3-h]хиназолин-3-карбоновую кислоту [(XI), R1=4-фторфенил, R2=3-(трет-бутоксикарбонил)аминопропил, A=-(CH2)2-], ЖХ/МС (m/z): 468,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 4,27 мин (основной) и 4,55 мин (минорный).
Пример 29
8-(4-фторфенил)-N-(3-гидроксипропил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-фторфенил, R2=тритил, R'=3-гидроксипропил, A=-(CH2)2-]
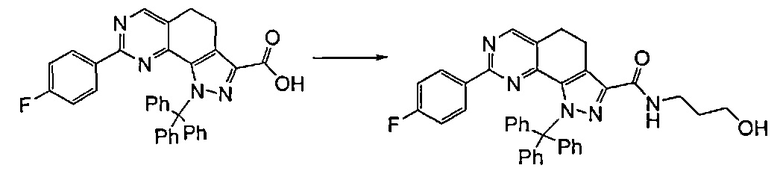
Проводили реакцию 358 мг (0,65 ммоль) 8-(4-фторфенил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в сухом ДМФА с 0,060 мл (0,78 ммоль) пропаноламина, 250 мг (0,78 ммоль) TBTU и 0,268 мл (1,5 ммоль) ДИПЭА при комнатной температуре в течение 3 часов. Реакционную смесь обрабатывали водой, насыщенным раствором NaHCO3 и подвергали экстракции этилацетатом. Органическую фазу, высушенную над Na2SO4, подвергнутую фильтрованию и выпариванию, очищали на диоксиде кремния с применением этилацетата/гексана в соотношении 7/3 с получением 300 мг (75%) чистого продукта. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 8,08 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,52 (с, 1Н), 7,34 (т, J=5,73 Гц, 1Н), 6,93-6,99 (м, 2Н), 4,52 (т, J=5,00 Гц, 1Н), 3,43-3,50 (м, 2Н), 3,25-3,32 (м, 2Н), 3,10 (д, J=8,05 Гц, 2Н), 2,78 (т, J=7,44 Гц, 2Н), 1,61 (квин, J=6,40 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C38H32FN5O2 [M+H]+: 610,2613, полученное 610,262.
Действуя способом, аналогичным описанному выше, получали следующее соединение:
N-(3-гидроксипропил)-8-(4-метоксифенил)-1-тритил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-метоксифенил, R2=тритил, R'=3-гидроксипропил, A=-(CH2)2-], ЖХ/МС (m/z): 622,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 7,07 мин.
Пример 30
8-(4-фторфенил)-N-(3-гидроксипропил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-фторфенил, R2=H, R'=3-гидроксипропил, A=-(CH2)2-]
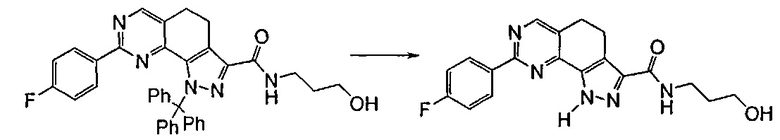
300 мг (0,5 ммоль) 8-(4-фторфенил)-N-(3-гидроксипропил)-1-тритил-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида растворяли в ДХМ и подвергали реакции с 2 мл трифторуксусной кислоты при комнатной температуре. Реакционную смесь выпаривали досуха и обрабатывали MeOH и насыщенным раствором NaHCO3 в течение 30 мин. Затем MeOH выпаривали, водную фазу подкисляли NaH2PO3 до pH=5,0 и подвергали экстракции ДХМ. Высушенный необработанный материал очищали на силикагеле с применением этилацетата. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,29 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=14,20 (с, 1Н), 8,77 (с, 1Н), 8,52-8,59 (м, 2Н), 8,23 (т, J=5,74 Гц, 1Н), 7,35-7,43 (м, 2Н), 4,51 (т, J=5,25 Гц, 2Н), 3,48 (кв, J=6,18 Гц, 2Н), 3,06-3,11 (м, 2Н), 3,00-3,06 (м, 2Н), 1,60-1,76 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C19H18FN5O2 [M+H]+: 368,1518, полученное значение: 368,153.
Действуя способом, аналогичным описанному выше, получали следующее соединение:
N-(3-гидроксипропил)-8-(4-метоксифенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=4-метоксифенил, R2=тритил, R'=3-гидроксипропил, A=-(CH2)2-], ЖХ/МС (m/z): 380,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 4,41 мин.
Пример 31
трет-Бутиловый эфир {3-[3-карбамоил-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил]пропил}карбаминовой кислоты [(I), R1=4-фторфенил, N1 R2=3-[(трет-бутоксикарбонил)амино]пропил, R3=NH2, A=-(CH2)2-]
и
трет-бутиловый эфир {3-[3-карбамоил-8-(4-фторфенил)-4,5-дигидро-2Н-пиразоло[4,3-h]хиназолин-2-ил]пропил}карбаминовой кислоты [(I), R1=4-фторфенил, N2 R2=3-[(трет-бутоксикарбонил)амино]пропил, R3=NH2, A=-(CH2)2-]
этап 6b2
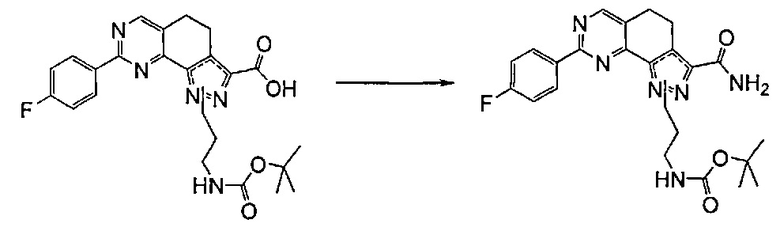
Проводили реакцию 358 мг (0,65 ммоль) 1-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и 2-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-2H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в сухом ДМФА (5 мл) с 147 мг (0,975 ммоль) HOBt*NH4, 250 мг (0,78 ммоль) TBTU и 0,268 мл (1,5 ммоль) ДИПЭА при комнатной температуре в течение 3 часов. Реакционную смесь обрабатывали водой, насыщенным раствором NaHCO3 и подвергали экстракции этилацетатом. Органическую фазу, высушенную над Na2SO4, подвергнутую фильтрованию и выпариванию, очищали на диоксиде кремния с применением этилацетата/гексана в соотношении 7/3 с получением 212 мг (70%) чистой смеси региоизомеров. ЖХ/МС (m/z): 467,2 [M+H]+и 467,2 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 5,62 и 6,12 мин.
Пример 32
1-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=4-фторфенил, N2 R2=3-аминопропил, R3=NH2, A=-(CH2)2-] (соединение 82)
и
2-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=4-фторфенил, N2 R2=3-аминопропил, R3=NH2, A=-(CH2)2-] (соединение 83)
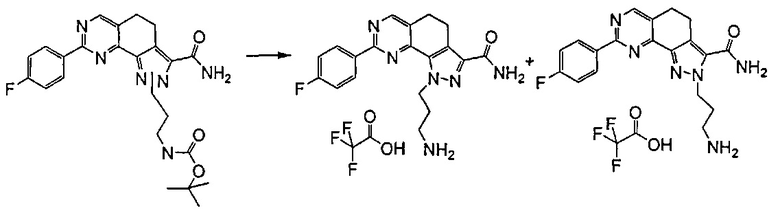
трет-Бутиловый эфир {3-[3-карбамоил-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-1-ил]пропил}карбаминовой кислоты и трет-бутиловый эфир {3-[3-карбамоил-8-(4-фторфенил)-4,5-дигидро-2Н-пиразоло[4,3-h]хиназолин-2-ил]пропил}карбаминовой кислоты суспендировали в ДХМ и подвергали реакции с 2 мл ТФУ при 60°C. Реакция завершалась через 90 мин, после чего реакционную смесь выпаривали. Необработанный материал очищали способом препаративной ВЭЖХ 2 и разделяли два региоизомера:
1-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат (основной), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,32 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,83 (с, 1Н), 8,43 (дд, J=5,68, 8,97 Гц, 2Н), 7,64 (шир. с, 3H), 7,48 (шир. с, 1Н), 7,40 (шир. с, 1Н), 7,38 (т, J=8,85 Гц, 2Н), 4,93 (т, J=6,47 Гц, 2Н), 3,05-3,13 (м, 2Н), 3,01 (т, J=7,20 Гц, 2Н), 2,82-2,91 (м, 2Н), 2,25 (квин, J=7,20 Гц, 2Н); значение МСВР (ИЭР), вычисленное для C19H19FN6O [M+H]+: 367,1677, полученное значение: 367,16785; и
2-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-2H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат (минорный), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,14 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,78 (с, 1Н), 8,46 (дд, J=5,68, 8,97 Гц, 2Н), 7,92 (шир. с, 1Н), 7,78 (шир. с, 1Н), 7,74 (шир. с, 3H), 7,37 (т, J=8,91 Гц, 2Н), 4,53 (т, J=6,84 Гц, 2Н), 2,93-3,05 (м, 4Н), 2,81-2,92 (м, 2Н), 2,16 (квин, J=7,26 Гц, 2Н); значение МСВР (ИЭР), вычисленное для C19H19FN6O [M+H]+: 367,1677, полученное значение: 367,16784.
Согласно указанной методике, но с применением подходящего замещенного производного, получали следующее соединение:
1-(3-аминопропил)-N-(3-гидроксипропил)-8-(4-метоксифенил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=4-метоксифенил, R2=3-аминопропил, R'=3-гидроксипропил, A=-(CH2)2-] (соединение 84), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло=3,52 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,79 (с, 1Н), 8,32-8,37 (м, 2Н), 8,11 (т, J=5,86 Гц, 1Н), 7,67 (шир. с, 3H), 7,06-7,14 (м, 2Н), 4,94 (т, J=6,53 Гц, 2Н), 3,86 (с, 3H), 3,49 (т, J=6,23 Гц, 2Н), 3,31 (шир. с, 2Н), 3,03-3,13 (м, 2Н), 3,00 (д, J=7,81 Гц, 2Н), 2,81-2,92 (м, 2Н), 2,26 (д, J=2,56 Гц, 2Н), 1,69 (квин, J=6,56 Гц, 2Н); вычисленная масса: 437,2296, полученная масса: 437,2291.
Получение N: трифторацетат 1-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и трифторацетат 2-(3-аминопропил)-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты
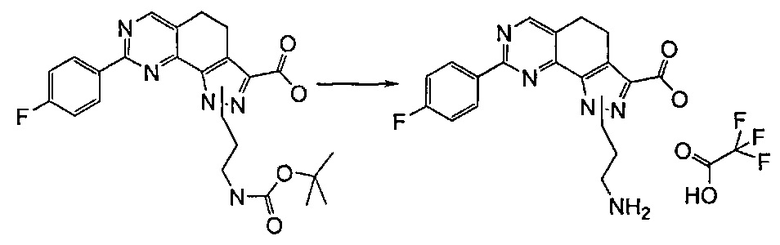
165 мг (0,35 ммоль) смеси 1-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты и 2-{3-[(трет-бутоксикарбонил)амино]пропил}-8-(4-фторфенил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты суспендировали в ДХМ и подвергали реакции с 2 мл ТФУ при 60°C. Реакция завершалась через 90 мин, после чего реакционную смесь выпаривали. Необработанный материал очищали способом препаративной ВЭЖХ 2 и обеспечивали в виде смеси двух региоизомеров. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,2 мин, то есть время удерживания для двух региоизомеров было одинаковым. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,83 (с, 1Н, минорный), 8,71 (с, 1Н, основной), 8,41-8,51 (м, 2Н), 8,34 (шир. с, 3H), 7,26-7,42 (м, 2Н), 4,96 (м, 2Н, минорный), 4,70-4,77 (м, 2Н, основной), 2,96-3,02 (м, 2Н), 2,88-2,95 (м, 2Н), 2,61 (т, J=5,92 Гц, 2Н), 2,11-2,21 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C19H18FN5O2 [M+H]+: 368,1518, полученное значение: 368,153.
Пример 33
1-трет-бутил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 85)
и
6-амино-1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R3=NH2, R5=NH2, A=-CH=CH-] (соединение 86)
этап 6c
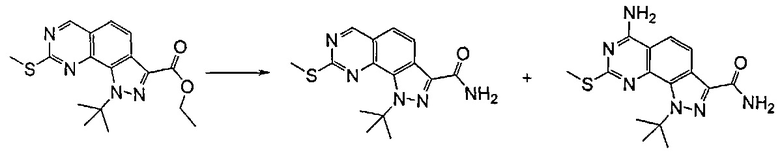
Этиловый эфир 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (170 мг, 0,494 ммоль) суспендировали в 4 мл безводного ТГФ и добавляли хлорид аммония (80 мг 1,48 ммоль). Реакционную смесь охлаждали до 0°C и добавляли 1 М LiN(TMS)2 в ТГФ (3 мл, 3 ммоль), после чего реакционную смесь перемешивали в течение 2 часов. Затем растворитель выпаривали досуха, остаток суспендировали в воде и фильтровали. Необработанный материал очищали посредством колоночной хроматографии на силикагеле, элюируя ДХМ/MeOH в соотношении 97/3, с получением 100 мг 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида (выход 65%) и 10 мг 6-амино-1-трет-бутил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (6%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,07 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,49 (с, 1Н), 8,42 (д, J=8,54 Гц, 1Н), 7,80 (д, J=8,79 Гц, 1Н), 7,75 (шир. с, 1Н), 7,53 (шир. с, 1Н), 2,74 (с, 3H), 2,00 (с, 9Н). Значение МСВР (ИЭР), вычисленное для C15H17N5OS [M+H]+: 316,1227, полученное значение: 316,1224.
6-амино-1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [R1=Me-S-, R2=трет-бутил, R3=NH2, R5=NH2, A=-CH=CH-], ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,08 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,16 (д, J=8,79 Гц, 1Н), 7,91 (д, J=8,79 Гц, 1Н), 7,84 (шир. с, 2Н), 7,62 (шир. с, 1Н), 7,42 (шир. с, 1Н), 2,61 (с, 3H), 1,98 (с, 9Н). Значение МСВР (ИЭР), вычисленное для C15H18N6OS [M+H]+: 331,1336, полученное значение: 331,1340.
Действуя аналогичным способом, получали следующие соединения:
1-метил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=Me, R3=NH2, A=-CH=CH-] (соединение 87), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,96 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,46 (с, 1Н), 8,26 (д, J=8,67 Гц, 1Н), 7,85 (шир. с, 1Н), 7,73 (д, J=8,79 Гц, 1Н), 7,52 (шир. с, 1Н), 4,71 (с, 3H), 2,73 (с, 3H); значение МСВР (ИЭР), вычисленное для C12H11N5OS [M+H]+: 274,0757, полученное значение: 274,0757;
6-амино-1-метил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=Me, R3=NH2, R5=NH2, A=-CH=CH-] (соединение 88), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,59; 1H ЯМР (401 МГц, ДМСО-d6) δ=8,00 (д, J=8,91 Гц, 1Н), 7,85 (шир. с, 1Н), 7,83 (д, J=8,91 Гц, 1Н), 7,74 (шир. с, 1Н), 7,42 (шир. с, 1Н), 4,65 (с, 3H), 2,59 (с, 3H); значение МСВР (ИЭР), вычисленное для C12H12N6OS [M+H]+: 289,0866, полученное значение: 289,0859;
1-трет-бутил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=H, R2=трет-бутил, R3=NH2, A=-CH=CH-] (соединение 3), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,63 мин; 1H ЯМР (500 МГц, ДМСО-d6) δ=9,71 (с, 1Н), 9,53 (с, 1Н), 8,55 (д, J=8,79 Гц, 1Н), 7,89 (д, J=8,78 Гц, 1Н), 7,80 (шир. с, 1Н), 7,58 (шир. с, 1Н), 2,01 (с, 9Н); значение МСВР (ИЭР), вычисленное для C14H15N5O [M+H]+: 270,1350, полученное 270,1357; и
6-амино-1-трет-бутил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=H, R2=трет-бутил, R3=NH2, R5=NH2, A=-CH=CH-] (соединение 122), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,29 мин; 1H ЯМР (500 МГц, ДМСО-d6) δ=8,65 (с, 1Н), 8,30 (д, J=9,06 Гц, 1Н), 8,12 (шир. с, 1Н), 8,02 (д, J=8,79 Гц, 1Н), 7,70 (шир. с, 1Н), 7,46-7,52 (м, 1Н), 1,98 (с, 9Н); значение МСВР (ИЭР), вычисленное для C14H16N6O [M+H]+: 285,1459, полученное значение: 285,1466.
Пример 34
1-трет-бутил-N-гидрокси-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R3=N(H)OH, A=-CH=CH-] (соединение 89)
этап 6b2
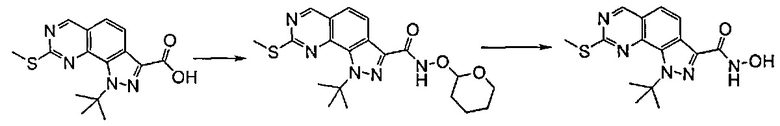
Проводили реакцию 40 мг (0,112 ммоль) 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты в сухом ДМФА (2 мл) с 25 мг (0,21 ммоль) O-(тетрагидро-2H-пиран-2-ил)гидроксиламина, 70 мг (0,21 ммоль) TBTU и 0,110 мл (1,19 ммоль) ДИПЭА при комнатной температуре в течение 3 часов. Реакционную смесь обрабатывали водой, насыщенным раствором NaHCO3 и подвергали экстракции этилацетатом. Органическую фазу, высушенную над Na2SO4, подвергнутую фильтрованию и выпариванию, очищали на диоксиде кремния с применением ДХМ/EtOAC в соотношении 7/3 с получением 23 мг (50%) 1-трет-бутил-8-(метилсульфанил)-N-(тетрагидро-2Н-пиран-2-илокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида. ЖХ/МС (m/z): 416,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 6,94 мин.
1-трет-бутил-8-(метилсульфанил)-N-(тетрагидро-2Н-пиран-2-илокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид растворяли в метаноле (2 мл) и добавляли 4 М HCl в диоксане (2 мл). Указанный раствор перемешивали при комнатной температуре в течение 2 часов, соответственно, летучие компоненты удаляли in vacuo. Необработанный материал сначала очищали с помощью силикагеля, элюируя ДХМ/MeOH в соотношении 6/1, с получением желаемого продукта, который подвергали очистке способом препаративной ВЭЖХ 1 с получением 5 мг указанного в заголовке продукта (30%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,95 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=11,16 (с, 1Н), 9,49 (с, 1Н), 9,15 (шир. с, 1Н), 8,33 (д, J=8,67 Гц, 1Н), 7,80 (д, J=8,67 Гц, 1Н), 2,73 (с, 3H), 1,99 (с, 9Н). Значение МСВР (ИЭР), вычисленное для C15H17N5O2S [M+H]+: 332,1176, полученное значение: 332,1185.
Пример 35
8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=H, R3=NH2, A=-CH=CH-] (соединение 90)
этап 6c
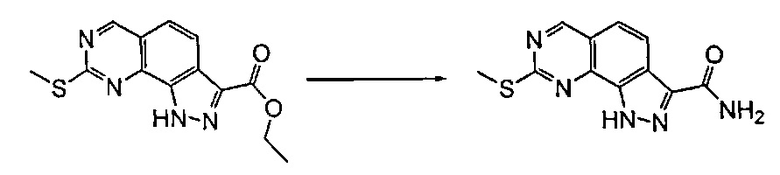
Этиловый эфир 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (210 мг 0,76 ммоль) суспендировали в 4 мл безводного ТГФ и добавляли хлорид аммония (120 мг, 2,28 ммоль). Реакционную смесь охлаждали до 0°C, добавляли 1 М LiN(TMS)2 в ТГФ (8 мл, 8,0 ммоль) и перемешивали в течение 2 часов. Затем растворитель выпаривали досуха, остаток поглощали ДХМ и промывали водой и рассолом. Органический слой сушили над Na2SO4, фильтровали и выпаривали под вакуумом. Необработанный материал очищали посредством колоночной хроматографии на силикагеле, элюируя ДХМ/MeOH в соотношении 9/1, с получением 180 мг указанного в заголовке соединения (выход 86%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,46 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,48 (с, 1Н), 8,24 (д, J=8,79 Гц, 1Н), 7,91 (шир. с, 1Н), 7,71 (д, J=8,67 Гц, 1Н), 7,49 (с, 1Н), 2,75 (с, 3H). Значение МСВР (ИЭР), вычисленное для C11H9N5OS [M+H]+: 260,0601, полученное значение: 260,0598.
Пример 36
трет-Бутиловый эфир 4-[3-карбамоил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты [(I), R1=Me-S-, R2=1-(трет-бутоксикарбонил)пиперидин-4-ил, R3=NH2, A=-CH=CH-]
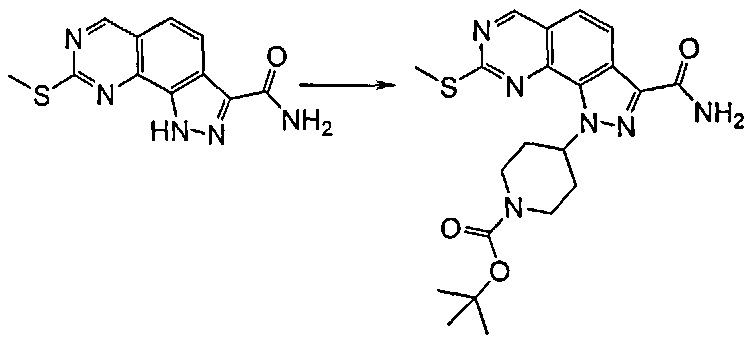
Проводили реакцию раствора 150 мг (0,58 ммоль) 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (5 мл), 205 мг (1,16 ммоль) трет-бутилового эфира 4-бромпиперидин-1-карбоновой кислоты и 377 мг (1,16 ммоль) карбоната цезия при 80°C в течение 18 часов. Указанную смесь охлаждали при к.т. и разделяли между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/EtOAc/EtOH в соотношении 6/4/0,5, с получением 200 мг желаемого соединения в виде твердого вещества белого цвета (78%). ЖХ/МС (m/z): 443,1 [M+H]+, ВЭЖХ (254 нм) способом 4: Rt составляло 2,53.
Действуя аналогичным способом, получали следующее соединение:
трет-бутиловый эфир 4-(3-карбамоил-8-метокси-1H-пиразоло[4,3-h]хиназолин-1-ил)пиперидин-1-карбоновой кислоты, ЖХ/МС (m/z): 427,2 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 5,61.
Пример 37
8-(метилсульфанил)-1-(пиперидин-4-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-) (соединение 91)
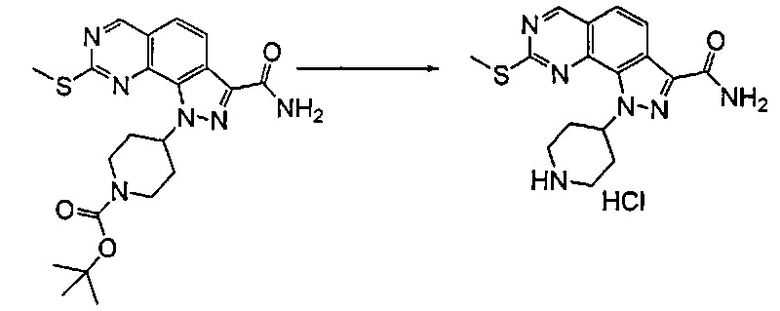
трет-Бутиловый эфир 4-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты растворяли в 1,4-диоксане (2 мл) и добавляли 4 М HCl в 1,4-диоксане (3 мл). Указанный раствор перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли in vacuo и остаток растирали в порошок с применением диэтилового эфира, фильтровали и сушили в сушильной печи (40°C) под вакуумом в течение 2 часов с получением 25 мг указанного в заголовке соединения (99%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,73 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,50 (с, 1Н), 8,85 (шир. с, 1Н), 8,64-8,78 (м, 1Н), 8,29 (д, J=8,67 Гц, 1Н), 7,73-7,82 (м, 2Н), 7,64 (с, 1Н), 6,16 (квин, J=7,05 Гц, 1Н), 3,52-3,61 (м, J=13,06 Гц, 2Н), 3,08-3,21 (м, 2Н), 2,75 (с, 3H), 2,37-2,47 (м, 4Н). Значение МСВР (ИЭР), вычисленное для C16H18N6OS [M+H]+: 343,1336, полученное значение: 343,1326.
Согласно аналогичной методике, но с применением подходящего исходного материала, получали следующие соединения:
8-(метилсульфанил)-1-(пиперидин-3-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=пиперидин-3-ил, R3=NH2, A=-CH=CH-] (соединение 92), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,86 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,41-9,59 (м, 1Н), 9,27 (шир. с, 1Н), 8,43 (шир. с, 1Н), 8,20-8,38 (м, 2Н), 7,75-7,84 (м, 1Н), 7,69 (с, 1Н), 6,30 (т, J=4,88 Гц, 1Н), 3,90 (д, J=12,33 Гц, 2Н), 3,78 (д, J=4,52 Гц, 2Н), 2,74 (шир. с, 3H), 2,27-2,42 (м, 3H), 2,14 (дд, J=5,61, 14,16 Гц, 2Н), 1,78 (д, J=5,37 Гц, 2Н); значение МСВР (ИЭР), вычисленное для C16H18N6OS [M+H]+: 343,1336, полученное значение: 343,1329;
1-(4-аминоциклогексил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=4-аминоциклогексил, R3=NH2, A=-CH=CH-] (соединение 93), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,05 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,48 (с, 1Н), 8,28 (д, J=8,67 Гц, 1Н), 8,00 (шир. с, 3H), 7,76 (д, J=8,79 Гц, 1Н), 7,72 (с, 1Н), 7,64 (с, 1Н), 6,18 (шир. с, 1Н), 2,69 (с, 3H), 2,25-2,37 (м, 2Н), 2,06-2,19 (м, 4Н), 1,79-1,95 (м, 2Н); значение МСВР (ИЭР), вычисленное для C17H20N6OS [M+H]+: 357,1492, полученное значение: 357,1491;
8-(метилсульфанил)-1-(пиперидин-3-илметил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=пиперидин-3-илметил, R3=NH2, A=-CH=CH-] (соединение 94), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,94 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=1Н 9,50 (с, 1Н), 8,63 (д, J=10,99 Гц, 1Н), 8,34-8,40 (м, 1Н), 8,30 (д, J=8,67 Гц, 1Н), 7,80 (с, 1Н), 7,78 (д, J=8,79 Гц, 1Н), 7,63 (с, 1Н), 5,01-5,19 (м, 2Н), 3,20 (д, J=13,79 Гц, 2Н), 3,02 (д, J=12,45 Гц, 1Н), 2,76-2,88 (м, 2Н), 2,74 (с, 3H), 2,55-2,66 (м, 1Н), 1,67-1,85 (м, 2Н), 1,31-1,64 (м, 2Н); значение МСВР (ИЭР), вычисленное для C17H20N6OS [M+H]+: 357,1492, полученное значение: 357,1489;
1-(азепан-4-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=азепан-4-ил, R3=NH2, A=-CH=CH-] (соединение 95), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,94 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,49 (м, 1Н), 8,87 (шир. с, 2Н), 8,28 (д, J=8,79 Гц, 1Н), 7,83 (с, 1Н), 7,76 (д, J=8,79 Гц, 1Н), 7,63 (с, 1Н), 6,05-6,59 (м, 1Н), 3,46-3,66 (м, 2Н), 3,10-3,24 (м, 1Н), 2,72 (с, 3H), 2,53-2,59 (м, 2Н), 2,39-2,46 (м, 1Н), 2,25-2,35 (м, 1Н), 2,04-2,17 (м, 1Н), 1,75-1,95 (м, 1Н); значение МСВР (ИЭР), вычисленное для C17H20N6OS [M+H]+: 357,1492, полученное значение: 357,1509;
1-(3-амино-2,2-диметилпропил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-S-, R2=3-амино-2,2-диметилпропил, R3=NH2, A=-CH=CH-] (соединение 96), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,08 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,50 (с, 1Н), 8,32 (д, J=8,67 Гц, 1Н), 7,99 (шир. с, 3H), 7,94 (с, 1Н), 7,78 (д, J=8,79 Гц, 1Н), 7,71 (с, 1Н), 5,29 (с, 2Н), 2,89 (кв, J=6,43 Гц, 1Н), 2,75 (с, 3H), 1,02 (с, 6Н); значение МСВР (ИЭР), вычисленное для C16H20N6OS [M+H]+: 345,1492, полученное значение: 345,1502;
8-метокси-1-(пиперидин-4-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-O-, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-] (соединение 97), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,43 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,55 (с, 1Н), 8,65-8,79 (м, 1Н), 8,68 (шир. с, 1Н), 8,45-8,58 (м, 1Н), 8,53 (шир. с, 1Н), 8,22 (д, J=8,67 Гц, 1Н), 7,79 (д, J=8,79 Гц, 1Н), 7,75 (с, 1Н), 7,62 (с, 1Н), 6,10 (квд, J=4,66, 9,67 Гц, 1Н), 4,16 (с, 3H), 3,51-3,65 (м, J=12,45 Гц, 1Н), 3,18-3,26 (м, 2Н), 2,37-2,46 (м, 4Н); значение МСВР (ИЭР), вычисленное для C16H18N6O2 [M+H]+: 327,1564, полученное значение: 327,15575; и
8-метокси-1-(пиперидин-4-илметил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид гидрохлорид [(I), R1=Me-O-, R2=пиперидин-4-илметил, R3=NH2, A=-CH=CH-] (соединение 98), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,54 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,53 (с, 1Н), 8,72-8,89 (м, 1Н), 8,57-8,70 (м, J=9,52 Гц, 1Н), 8,21 (д, J=8,67 Гц, 1Н), 7,80 (шир. с, 1Н), 7,77 (д, J=8,79 Гц, 1Н), 7,57 (с, 1Н), 5,09 (д, J=7,32 Гц, 2Н), 4,16 (с, 3H), 3,18-3,26 (м, J=12,69 Гц, 2Н), 2,72-2,89 (м, 2Н), 2,40-2,47 (м, 1Н), 1,50-1,68 (м, 4Н); значение МСВР (ИЭР), вычисленное для C17H20N6O2 [M+H]+: 341,1721, полученное значение: 341,1721.
Пример 38
1-[(3-экзо)-8-азабицикло[3.2.1]окт-3-ил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=8-азабицикло[3.2.1]окт-3-ил, R3=NH2, A=-CH=CH-] (соединение 99)
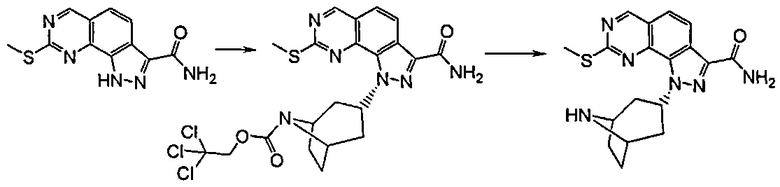
Проводили реакцию 50 мг (0,193 ммоль) 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (8 мл) с 200 мг (0,52 ммоль) 2,2,2-трихлорэтилового эфира 3-[(метилсульфонил)окси]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты и 140 мг (0,43 ммоль) карбоната цезия при 90°C в течение 4 часов. Указанную смесь охлаждали при к.т. и разделяли между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/EtOH в соотношении 9/1, с получением 73 мг (70%) 2,2,2-трихлорэтилового эфира 3-[3-карбамоил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-1-ил]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты в виде твердого вещества грязно-белого цвета. Указанное соединение (134 ммоль) растворяли в ТГФ (5 мл), после чего добавляли уксусную кислоту (5 мл) и 35 мг цинковой пыли (0,536 ммоль). Указанную смесь нагревали до 50°C и перемешивали в течение 18 часов. Летучие компоненты удаляли под вакуумом и остаток очищали посредством хроматографии, элюируя ДХМ/MeOH в соотношении 95/5. Необходимую дополнительную очистку осуществляли способом препаративной ВЭЖХ 1 с получением 25 мг указанного в заголовке соединения (50%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,08 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,50 (с, 1Н), 8,25 (д, J=8,67 Гц, 1Н), 7,62-7,79 (м, 3H), 7,52-7,60 (м, 1Н), 6,21-6,54 (м, 0H), 3,68 (шир. с, 2Н), 2,76 (с, 2Н), 2,22-2,36 (м, 2Н), 2,02 (шир. с, 2Н), 1,78-1,98 (м, 5Н); значение МСВР (ИЭР), вычисленное для C18H20N6OS [M+H]+: 369,1492, полученное значение: 369,15.
Действуя аналогичным способом, получали следующее соединение:
1-[(3-эндо)-8-азабицикло[3.2.1]окт-3-ил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=Me-S-, R2=8-азабицикло[3,2,1]окт-3-ил, R3=NH2, A=-CH=CH-] (соединение 100), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,0 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,49 (с, 1Н), 8,66 (шир. с, 1Н), 8,32 (д, J=8,79 Гц, 1Н), 7,85 (с, 1Н), 7,79 (д, J=8,79 Гц, 1Н), 7,63 (с, 1Н), 6,07 (т, J=8,12 Гц, 1Н), 4,07 (шир. с, 2Н), 2,81 (ддд, J=4,09, 8,03, 16,20 Гц, 2Н), 2,72 (с, 3H), 2,63 (д, J=16,36 Гц, 2Н), 2,05-2,19 (м, 2Н), 1,79-1,94 (м, 2Н); значение МСВР (ИЭР), вычисленное для C18H20N6OS [M+H]+: 369,1492, полученное значение: 369,1484.
Пример 39
1-[4-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)бутан-2-ил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=4-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)бутан-2-ил, R3=NH2, A=-CH=CH-] (соединение 101)
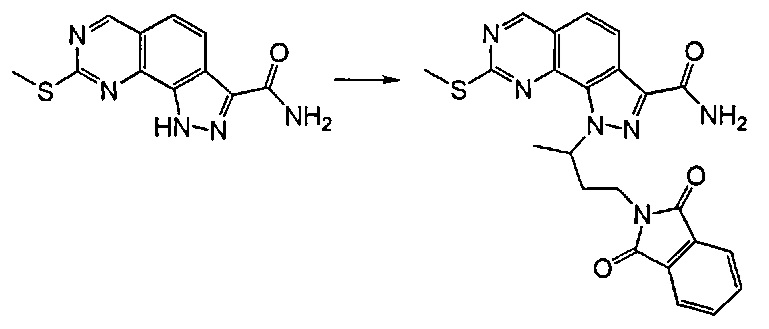
Проводили реакцию 50 мг (0,193 ммоль) 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (3 мл) с 108 мг (0,38 ммоль) 2-(4-бром-3-метилбутил)-1Н-изоиндол-1,3(2H)-диона и 132 мг (0,38 ммоль) карбоната цезия при 80°C в течение 3 часов. Указанную смесь охлаждали при к.т. и разделяли между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/EtOH в соотношении 9/1, с получением 57 мг (65%) желаемого соединения в виде твердого вещества бледно-желтого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,01 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,38 (с, 1Н), 8,21 (д, J=8,67 Гц, 1Н), 7,71-7,76 (м, 2Н), 7,69 (д, J=8,79 Гц, 1Н), 7,53-7,60 (м, 2Н), 6,29-6,45 (м, 1Н), 3,41-3,58 (м, 2Н), 2,72 (дт, J=5,25, 9,70 Гц, 1Н), 2,45 (д, J=7,57 Гц, 1Н), 2,41 (с, 3H), 1.59 (д, J=6,59 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C23H20N6O3S [M+H]+: 461,1391, полученное значение: 461,1412.
Пример 40
1-(4-аминобутан-2-ил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=4-аминобутан-2-ил, R3=NH2, A=-CH=CH-] (соединение 102)

К раствору 45 мг (0,097 ммоль) 1-[4-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)бутан-2-ил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида в ТГФ (5 мл) добавляли 1 М гидразин в ТГФ (5 мл). Указанную смесь перемешивали при 50°C в течение 3 часов. Указанную смесь охлаждали до к.т. и фильтровали, летучие компоненты удаляли под вакуумом, необработанный материал подвергали препаративной ВЭЖХ способом 1 с получением 20 мг указанного в заголовке продукта (60%). ЖХ/МС (254 нм) ВЭЖХ способом x: Rt составляло 3,98 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,50 (с, 1Н), 8,29 (д, J=8,67 Гц, 1Н), 7,81 (с, 1Н), 7,77 (д, J=8,79 Гц, 1Н), 7,62 (д, J=10,25 Гц, 2Н), 6,23 (шир. с, 1Н), 2,87 (тд, J=5,66, 11,50 Гц, 1Н), 2,71 (с, 3H), 2,61 (дт, J=6,41, 11,81 Гц, 1Н), 2,14-2,30 (м, 1Н), 1,66 (д, J=6,59 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C15H18N6OS [M+H]+: 331,1336, полученное значение: 331,134.
Пример 41
1-(1-азабицикло[2.2.2]окт-3-ил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=азабицикло[2.2.2]окт-3-ил, R3=NH2, A=-CH=CH-] (соединение 103)
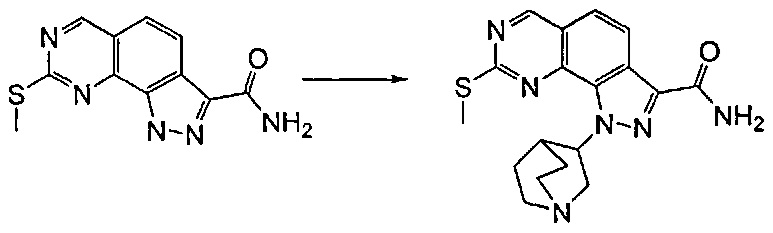
К раствору 64 мг (0,247 ммоль) 8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (3 мл) добавляли 300 мг (1,06 ммоль) 1-азабицикло[2.2.2]окт-3-илового эфира 4-метилбензолсульфоновой кислоты (полученного способом, описанным в Tetrahedron. Lett, 2000, 41, 271-274) и 55 мг (0,494 ммоль) трет-бутоксида калия. Указанную смесь нагревали до 100°C и перемешивали в течение 4 часов. Обеспечивали охлаждение реакционной смеси при комнатной температуре и разделяли указанную смесь между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/MeOH/NH4OH в соотношении 9/1/0,5, с получением 14 мг желаемого продукта (15%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,73 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,46 (с, 1Н), 8,17-8,41 (м, 1Н), 7,87-8,00 (м, 1Н), 7,68-7,80 (м, 1Н), 7,54 (с, 1Н), 5,94-6,38 (м, 1Н), 4,01 (дд, J=4,09, 13,98 Гц, 1Н), 3,37-3,48 (м, 1Н), 2,75-2,95 (м, 3H), 2,73 (с, 3H), 2,20-2,29 (м, 1Н), 1,80-1,95 (м, 1Н), 1,75 (ддд, J=4,76, 8,48, 13,12 Гц, 1Н), 1,34-1,47 (м, 1Н), 1,20-1,35 (м, 1Н). Значение МСВР (ИЭР), вычисленное для C18H20N6OS [M+H]+: 369,1492, полученное значение: 369,1508.
Пример 42
8-(метилсульфанил)-1-(тетрагидро-2Н-пиран-4-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=тетрагидро-2H-пиран-4-ил, R3=NH2, A=-CH=CH-] (соединение 104)
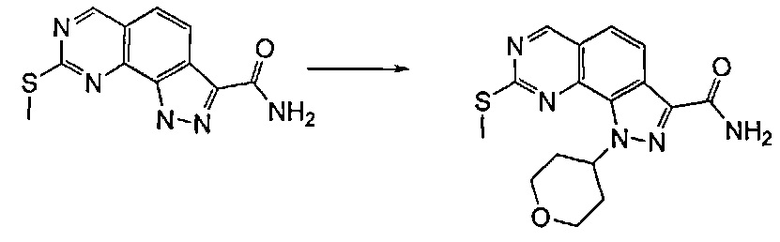
К раствору 30 мг (0,247 ммоль) 8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (3 мл) добавляли 62 мг (0,34 ммоль) тетрагидро-2H-пиран-4-илового эфира метансульфоновой кислоты (полученного согласно US 2003/6653489) и 87 мг (0,266 ммоль) карбоната цезия. Указанную смесь нагревали до 80°C и перемешивали в течение 4 часов. Обеспечивали охлаждение реакционной смеси при комнатной температуре и разделяли указанную смесь между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/MeOH/NH4OH в соотношении 9/1/0,5, с получением 32 мг желаемого продукта (38%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,41 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,47 (с, 1Н), 8,28 (д, J=8,67 Гц, 1Н), 7,84 (шир. с, 1Н), 7,75 (д, J=8,79 Гц, 1Н), 7,54 (с, 1Н), 6,25 (тт, J=4,32, 11,37 Гц, 1Н), 4,11 (дд, J=4,15, 11,47 Гц, 1Н), 3,48-3,65 (м, 1Н), 2,75 (с, 3H), 2,23-2,40 (м, 2Н), 2,13 (дд, J=2,38, 12,39 Гц, 2Н). Значение МСВР (ИЭР), вычисленное для C16H17N5O2S [M+H]+: 344,1176, полученное значение: 344,1166.
Пример 43
Метиловый эфир 3-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]циклобутанкарбоновой кислоты [(I), R1=Me-S-, R2=1-(метилоксикарбонил)циклобутан-3-ил, R3=NH2, A=-CH=CH-]
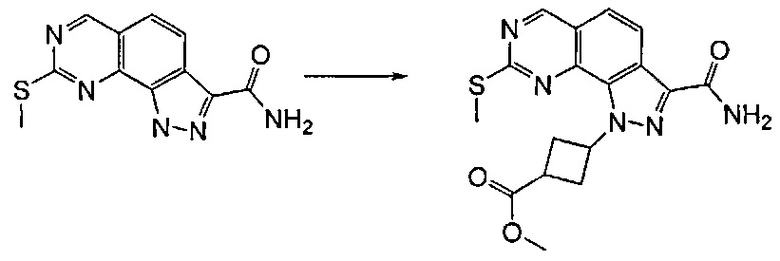
К раствору 100 мг (0,38 ммоль) 8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в ДМФА (5 мл) добавляли 200 мг (0,89 ммоль) метилового эфира 2-метил-4-[(метилсульфонил)окси]пентановой кислоты (полученного способом, описанным в WO 2009/71509 A1) и 191 мг (0,55 ммоль) карбоната цезия. Указанную смесь выдерживали при 90°C в течение 18 часов. Указанную смесь охлаждали при к.т. и разделяли между этилацетатом и водой. Органическую фазу промывали рассолом, сушили над Na2SO4 и выпаривали. Необработанный материал очищали посредством колоночной хроматографии, элюируя ДХМ/EtOH в соотношении 9/1, с получением 100 мг (70%) желаемого соединения в виде твердого вещества белого цвета. Указанный продукт содержал смесь цис- и транс-изомеров. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,55 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,44-9,51 (м, 1Н), 8,26 (дд, J=1,89, 8,73 Гц, 1Н), 7,87-8,02 (м, 1Н), 7,72-7,77 (м, 1Н), 7,58 (шир. с, 1Н), 6,24-6,93 (м, 1Н), 3,66-3,76 (м, 3H), 3,29 (с, 3H), 3,04-3,25 (м, 2Н), 2,79-3,02 (м, 3H). Значение МСВР (ИЭР), вычисленное для C17H17N5O3S [M+H]+: 372,1125, полученное значение: 372,1119.
Действуя аналогичным способом, получали следующее соединение:
метиловый эфир 2-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]-2-метилпропановой кислоты, ЖХ/МС (m/z): 360,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 4,75.
Пример 44
1-[транс-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=транс-3-(гидроксиметил)циклобутил, R3=NH2, A=-CH=CH-] (соединение 105)
и
1-[цис-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=цис-3-(гидроксиметил)циклобутил, R3=NH2, A=-CH=CH-] (соединение 106)
Превращение i
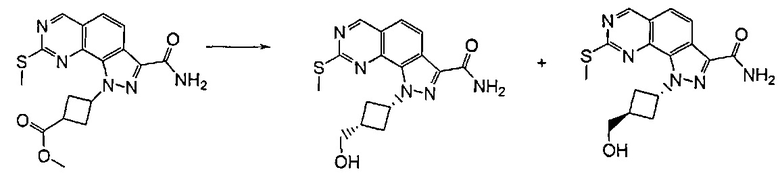
К раствору 70 мг (0,188 ммоль) метилового эфира 3-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]циклобутанкарбоновой кислоты (смеси цис- и транс-изомеров) в этаноле (5 мл) осторожно добавляли 30 мг (0,75 ммоль) боргидрида натрия. Указанную смесь перемешивали при комнатной температуре в течение 18 часов. После удаления растворителя остаток поглощали этилацетатом (20 мл), водой (10 мл) и насыщенным водным раствором NH4Cl (10 мл). Слои смешивали и разделяли. Органический экстракт промывали рассолом и сушили над Na2SO4, а затем фильтровали и концентрировали. Необработанный продукт очищали посредством колоночной хроматографии (ДХМ/EtOAc/EtOH 7/2/1) с получением продукта в виде твердого вещества грязно-белого цвета, который подвергали препаративной ВЭЖХ для разделения цис/транс-томеров с получением:
1-[цис-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 5,04 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,46 (с, 1Н), 8,26 (д, J=8,67 Гц, 1Н), 7,84 (с, 1Н), 7,73 (д, J=8,79 Гц, 1Н), 7,57 (с, 1Н), 6,39 (квин, J=8,33 Гц, 1Н), 4,55-4,67 (м, 1Н), 3,55 (д, J=6,10 Гц, 2Н), 2,73 (с, 3H), 2,57-2,65 (м, J=6,35 Гц, 1Н), 2,51-2,54 (м, 2Н), 2,31-2,40 (м, 1Н); значение МСВР (ИЭР), вычисленное для C16H17N5O2S [M+H]+: 344,1176, полученное значение: 344,1167; и
1-[транс-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,91 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,39-9,48 (м, 1Н), 8,14-8,34 (м, 1Н), 7,82-8,00 (м, 1Н), 7,73 (д, J=8,79 Гц, 1Н), 7,56 (с, 1Н), 6,64 (квин, J=7,78 Гц, 1Н), 4,71-4,97 (м, 1Н), 3,53-3,75 (м, 2Н), 2,82-2,97 (м, 2Н), 2,73-2,78 (м, 3H), 2,37-2,47 (м, 2Н); значение МСВР (ИЭР), вычисленное для C16H17N5O2S [M+H]+: 344,1176, полученное значение: 344,1174.
Действуя аналогичным способом, получали следующие соединения:
1-(1-гидрокси-2-метилпропан-2-ил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=1-гидрокси-2-метилпропан-2-ил, R3=NH2, A=-CH=CH-] (соединение 107), ЖХ/МС (m/z): 332,08 [M+H]+, ВЭЖХ (254 нм) способом 1: Rt составляло 1,14; и
1-(1-гидрокси-2-метилпропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=1-гидрокси-2-метилпропан-2-ил, R3=NH2, A=-(CH2)2-] (соединение 108), ЖХ/МС (m/z): 334,1 [M+H]+, ВЭЖХ (254 нм) способом 1: Rt составляло 0,63.
Пример 45
трет-Бутиловый эфир 4-(3-карбамоил-8-фенил-1H-пиразоло[4,3-h]хиназолин-1-ил)пиперидин-1-карбоновой кислоты [(I), R1=фенил, R2=1-(трет-бутоксикарбонил)пиперидин-4-ил, R3=NH2, A=-CH=CH-]
Превращение l
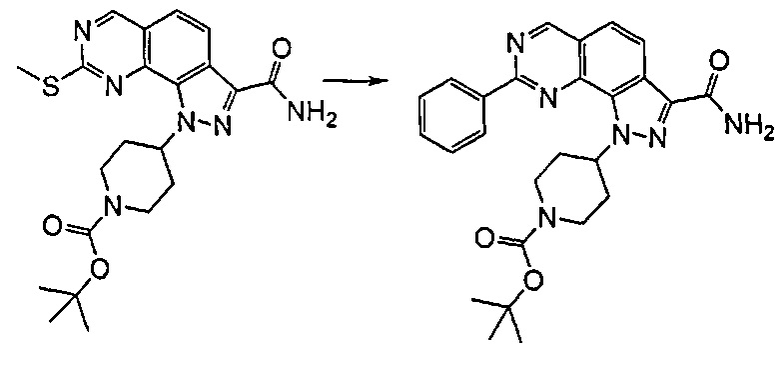
В сухой сосуд, подходящий для проведения микроволновых процессов, загружали 30 мг (0,068 ммоль) трет-бутилового эфира 4-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты, 3 мл сухого ТГФ, 16,5 мг (0,138 ммоль) фенилбороновой кислоты, 38,7 мг (0,203 ммоль) тиофенкарбоксилата меди и 7,8 мг (0,007 ммоль, 10 мол.%) Pd(PPh3)4. Затем указанную смесь выдерживали в микроволновом реакторе при 100°C в течение 60 мин. После охлаждения указанную смесь разбавляли этилацетатом и промывали 25% водным раствором аммиака. Водный слой снова подвергали экстракции этилацетатом, объединенные органические фазы сушили над Na2SO4 и остаток после выпаривания очищали посредством колоночной хроматографии на силикагеле (ДХМ/EtOAc/EtOH, 6/4/0,2) с получением 29 мг указанного в заголовке продукта (90%) в виде твердого вещества белого цвета. ЖХ/МС (m/z): 473,0 [M+H]+, ВЭЖХ (254 нм) способом 4: Rt составляло 2,95.
Пример 46
Гидрохлорид 8-фенил-1-(пиперидин-4-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=фенил, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-] (соединение 109)
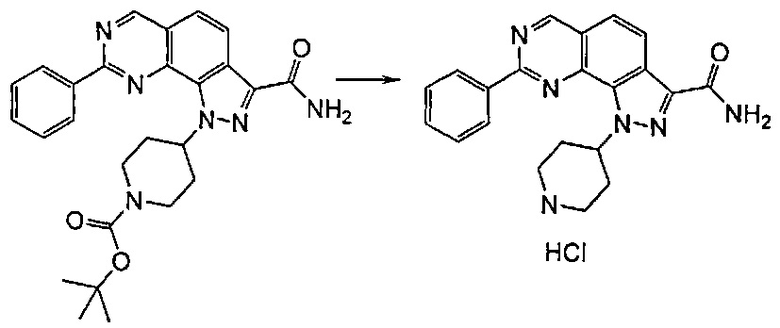
трет-Бутиловый эфир 4-(3-карбамоил-8-фенил-1H-пиразоло[4,3-h]хиназолин-1-ил)пиперидин-1-карбоновой кислоты растворяли в 1,4-диоксане (2 мл) и добавляли 4 М HCl в 1,4-диоксане (3 мл). Указанный раствор перемешивали при комнатной температуре в течение 1 часа. Летучие компоненты удаляли in vacuo и остаток растирали в порошок с применением диэтилового эфира, фильтровали и сушили в сушильной печи (40°C) под вакуумом в течение 2 часов с получением 27 мг указанного в заголовке соединения (99%). ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,47 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,82 (с, 1Н), 8,94 (шир. с, 1Н), 8,85 (шир. с, 1Н), 8,52-8,66 (м, 2Н), 8,41 (д, J=8,67 Гц, 1Н), 7,89 (д, J=8,67 Гц, 1Н), 7,84 (шир. с, 1Н), 7,57-7,73 (м, 4Н), 6,16-6,37 (м, 1Н), 3,54-3,67 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C21H20N6O [M+H]+: 373,1771, полученное 373,1783.
Действуя аналогичным способом, получали следующие соединения:
1-(пиперидин-4-ил)-8-(тиофен-3-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=тиофен-3-ил, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-] (соединение 110), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,32 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,74 (с, 1Н), 8,69 (шир. с, 1Н), 8,64 (дд, J=1,10, 3,05 Гц, 1Н), 8,59 (шир. с, 1Н), 8,37 (д, J=8,67 Гц, 1Н), 8,03 (дд, J=0,98, 5,00 Гц, 1Н), 7,85 (д, J=8,79 Гц, 1Н), 7,80 (с, 1Н), 7,77 (дд, J=3,05, 5,00 Гц, 1Н), 7,65 (с, 1Н), 6,11-6,43 (м, 1Н), 3,48-3,73 (м, J=12,94 Гц, 2Н), 3,35-3,42 (м, 2Н); значение МСВР (ИЭР), вычисленное для C19H18N6OS [M+H]+: 379,1336, полученное 379,1332;
гидрохлорид 1-(4-аминоциклогексил)-8-фенил-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=фенил, R2=4-аминоциклогексил, R3=NH2, A=-CH=CH-] (соединение 111), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,91 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,79 (с, 1Н), 8,59 (дд, J=1,89, 7,75 Гц, 2Н), 8,40 (д, J=8,67 Гц, 1Н), 8,04 (шир. с, 3H), 7,86 (д, J=8,79 Гц, 1Н), 7,73 (шир. с, 1Н), 7,69 (шир. с, 1Н), 7,56-7,67 (м, 3H), 6,32 (квин, J=4,61 Гц, 1Н), 2,10-2,46 (м, 6Н), 1,85-2,03 (м, J=4,52 Гц, 2Н); значение МСВР (ИЭР), вычисленное для C22H22N6O [M+H]+: 387,1928, полученное значение: 387,1929; и
1-(4-аминоциклогексил)-8-(тиофен-3-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид трифторацетат [(I), R1=тиофен-3-ил, R2=4-аминоциклогексил, R3=NH2, A=-CH=CH-] (соединение 112), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,73 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,71 (с, 1Н), 8,51-8,69 (м, 1Н), 8,36 (д, J=8,67 Гц, 1Н), 7,99 (д, J=5,13 Гц, 1Н), 7,89 (шир. с, 2Н), 7,83 (д, J=8,79 Гц, 1Н), 7,76 (дд, J=3,05, 5,00 Гц, 2Н), 7,58 (с, 1Н), 6,27 (квин, J=4,61 Гц, 1Н), 3,38-3,46 (м, J=4,64 Гц, 1Н), 2,31-2,42 (м, 2Н), 2,20-2,31 (м, 2Н), 2,04-2,18 (м, 2Н), 1,96 (дд, J=5,00, 13,06 Гц, 1Н); значение МСВР (ИЭР), вычисленное для C20H20N6OS [M+H]+: 393,1492, полученное значение: 393,1481.
Пример 47
трет-Бутиловый эфир 4-[3-карбамоил-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты [(I), R4=Me, X=SO2, R2=1-(трет-бутоксикарбонил)пиперидин-4-ил, R3=NH2, A=-CH=CH-]
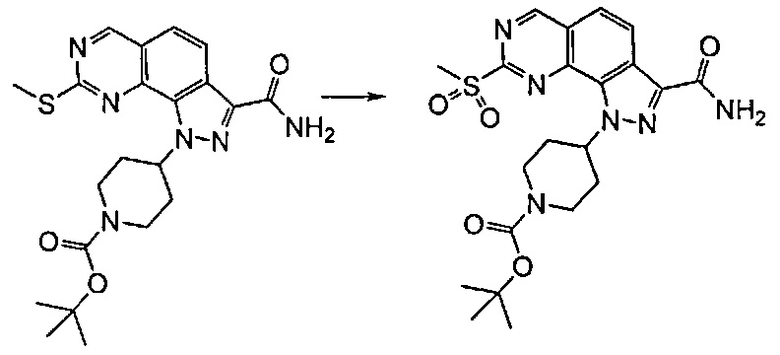
100 мг (0,225 ммоль) трет-бутилового эфира 4-[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты растворяли в 10 мл ДХМ. Добавляли 77,3 мг (0,448 ммоль) м-ХПБК (м-хлорпербензойной кислоты) при комнатной температуре и перемешивали при той же температуре в течение 60 мин. Добавляли 1 мл насыщенного водного раствора сульфита натрия и перемешивали в течение 30 мин. Реакционную смесь промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении с получением 100 мг (94%) указанного в заголовке соединения в виде твердого вещества желтого цвета. ЖХ/МС (m/z): 475,0 [M+H]+, ВЭЖХ (254 нм) способом 4: Rt составляло 2,01 мин.
Пример 48
Гидрохлорид 1-(пиперидин-4-ил)-8-(пирролидин-1-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=пирролидин-1-ил, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-] (соединение 113)
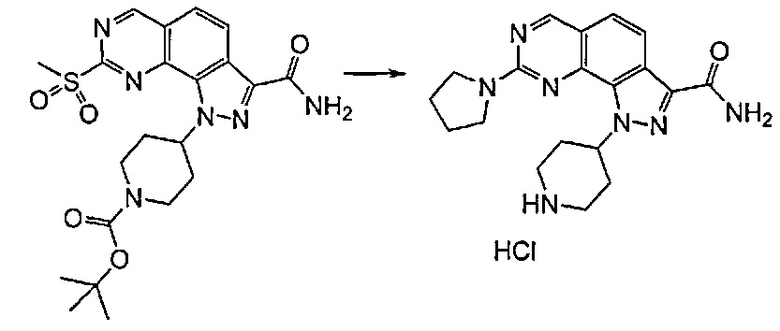
К раствору 30 мг (0,063 ммоль) трет-бутилового эфира 4-[3-карбамоил-8-(метилсульфонил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты в ТГФ (3 мл) добавляли 70 мкл (1 ммоль) пирролидина. Указанный раствор перемешивали при комнатной температуре в течение 1 часа и ход реакции контролировали посредством ЖХ/МС. Реакционную смесь охлаждали до комнатной температуры, отделенное твердое вещество фильтровали и промывали диэтиловым эфиром с получением 15 мг (50%) трет-бутилового эфира 4-[3-карбамоил-8-(пирролидин-1-ил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты. ЖХ/МС (m/z): 475,0 [M+H]+, ВЭЖХ (254 нм) способом 4: Rt составляло 2,01 мин.
15 мг (0,032 ммоль) трет-бутилового эфира 4-[3-карбамоил-8-(пирролидин-1-ил)-1H-пиразоло[4,3-h]хиназолин-1-ил]пиперидин-1-карбоновой кислоты растворяли в 1,4-диоксане (2 мл) и добавляли 3 мл (3 ммоль) 4 М HCl в 1,4-диоксане. Указанную смесь перемешивали при комнатной температуре в течение 1 ч. Летучие компоненты удаляли in vacuo и полученный остаток растирали в порошок с применением диэтилового эфира, фильтровали и сушили с получением 12 мг (93%) указанного в заголовке соединения. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 4,11 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,11-9,31 (м, 1Н), 8,89 (шир. с, 1Н), 8,57-8,82 (м, 1Н), 7,89 (д, J=8,67 Гц, 1Н), 7,64 (шир. с, 1Н), 7,41-7,58 (м, 2Н), 5,80-6,23 (м, 1Н), 3,69 (шир. с, 4Н), 3,50-3,62 (м, 2Н), 3,07-3,27 (м, 2Н), 2,35-2,45 (м, 4Н), 1,94-2,15 (м, 4Н). Значение МСВР (ИЭР), вычисленное для C19H23N7O [M+H]+: 366,2037, полученное значение: 366,2047.
Действуя аналогичным способом, получали следующее соединение:
гидрохлорид 8-(морфолин-4-ил)-1-(пиперидин-4-ил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=пирролидин-1-ил, R2=пиперидин-4-ил, R3=NH2, A=-CH=CH-] (соединение 114), ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,92 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,31 (с, 1Н), 8,82 (шир. с, 1Н), 8,73 (шир. с, 1Н), 7,98 (д, J=8,67 Гц, 1Н), 7,64-7,73 (м, 1Н), 7,51-7,62 (м, 2Н), 5,87-6,09 (м, 1Н), 3,85-3,91 (м, 4Н), 3,72-3,80 (м, 4Н), 3,54 (д, J=13,43 Гц, 2Н), 3,18 (д, J=5,86 Гц, 3H), 2,35-2,44 (м, 4Н); значение МСВР (ИЭР), вычисленное для C19H23N7O2 [M+H]+: 382,1986, полученное значение: 382, 2004.
Пример 49
Гидрохлорид 1-{[1-(2-аминоэтил)пиперидин-4-ил]метил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=1-(2-аминоэтил)пиперидин-4-илметил, R3=NH2) A=-CH=CH-] (соединение 115)
Превращение n
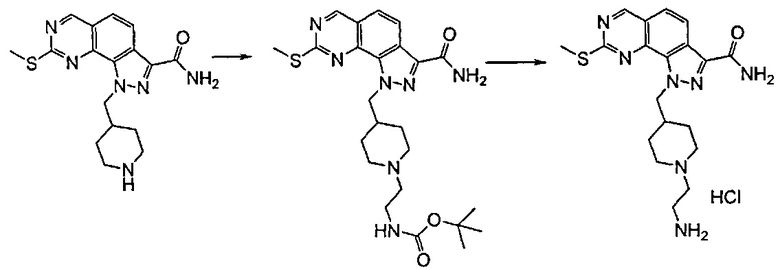
К раствору 26 мг (0,073 ммоль) 8-(метилсульфанил)-1-(пиперидин-4-илметил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в метаноле (1,5 мл) и ДМФА (1 мл) добавляли 13 мкл (0,219 ммоль) уксусной кислоты, 69,6 мг (0,438 ммоль) трет-бутилового эфира (2-оксоэтил)карбаминовой кислоты и 28 мг (0,438 ммоль) NaCNBH3. Указанную смесь перемешивали при к.т. в течение 18 часов, соответственно, летучие компоненты удаляли in vacuo. Остаток растворяли в этилацетате и разделяли с применением воды, органический слой промывали рассолом, сушили над Na2SO4 и концентрировали. Необработанный материал очищали посредством флэш-хроматографии (ДХМ/MeOH 95/5) с получением 27 мг трет-бутилового эфира [2-(4-{[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]метил}пиперидин-1-ил)этил]карбаминовой кислоты в виде твердого вещества белого цвета (75%). ЖХ/МС (254) ВЭЖХ способом 2: Rt=3,86 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,47 (с, 1Н), 8,28 (д, J=8,67 Гц, 1Н), 7,81 (с, 1Н), 7,74 (д, J=8,79 Гц, 1Н), 7,54 (шир. с, 1Н), 6,56 (шир. с, 1Н), 5,08 (д, J=7,32 Гц, 2Н), 2,93-3,05 (м, 2Н), 2,76-2,84 (м, J=10,25 Гц, 2Н), 2,71 (с, 3H), 2,27 (шир. с, 1Н), 2,14 (шир. с, 1Н), 1,75-1,94 (м, 2Н), 1,12-1,49 (м, 17Н). Значение МСВР (ИЭР), вычисленное для C24H33N7O3S [M+H]+: 500,2439, полученное значение: 500,2436.
Полученный трет-бутиловый эфир [2-(4-{[3-карбамоил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-1-ил]метил}пиперидин-1-ил)этил]карбаминовой кислоты растворяли в 2 мл 4 М HCl в 1,4-диоксане и перемешивали при к.т. в течение 2 часов. Затем летучие компоненты удаляли под вакуумом, остаток суспендировали с применением диэтилового эфира и фильтровали с получением 14 мг желаемого продукта в виде твердого вещества бледно-желтого цвета (90%). ЖХ/МС (254) ВЭЖХ способом 2: Rt=3,29 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=10,78 (шир. с, 1Н), 9,49 (с, 1Н), 8,30 (д, J=8,67 Гц, 1Н), 8,12-8,24 (м, 2Н), 7,86 (с, 1Н), 7,77 (д, J=8,79 Гц, 1Н), 7,58 (с, 1Н), 5,12 (д, J=7,32 Гц, 1Н), 3,47-3,54 (м, 2Н), 3,16-3,28 (м, 3H), 2,85-3,01 (м, 1Н), 2,76 (с, 2Н), 1,73-1,88 (м, 2Н), 1,60-1,71 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C19H25N7OS [M+H]+: 400,1914, полученное значение: 400,1918.
Действуя аналогичным способом, получали следующие соединения:
гидрохлорид 1-{4-[(2-аминоэтил)амино]циклогексил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=4-(2-аминоэтил)аминоциклогексил, R3=NH2, A=-CH=CH-] (соединение 116); значение МСВР (ИЭР), вычисленное для C19H25N7OS [M+H]+: 400,1914, полученное значение: 400,1908; и
гидрохлорид 1-{4-[(2-аминоэтил)амино]циклогексил}-8-(метилсульфанил)-4,5-дигидро-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=4-(2-аминоэтил)аминоциклогексил, R3=NH2, A=-(CH2)2-] (соединение 117); значение МСВР (ИЭР), вычисленное для C19H27N7OS [M+H]+: 402,2071, полученное значение: 402,2074.
Пример 50
Гидрохлорид 1-[4-(глициламино)циклогексил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида [(I), R1=Me-S-, R2=4-(глициламино)циклогексил, R3=NH2, A=-CH=CH-] (соединение 118)
Превращение o
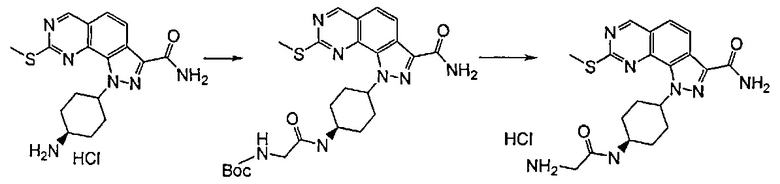
30 мг (0,084 ммоль) гидрохлорид 1-(4-аминоциклогексил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида растворяли в 2 мл диметилацетамида. К полученному раствору добавляли 21 мг (0,126 ммоль) N-(трет-бутоксикарбонил)глицина, 60 мкл (0,336 ммоль) ДИПЭА, 40 мг (0,126 ммоль) TBTU. Указанную смесь перемешивали при комнатной температуре в течение 18 часов, после чего раствор разделяли между этилацетатом и насыщенным водным раствором NaHCO3, органический слой промывали рассолом, сушили над Na2SO4 и выпаривали под вакуумом. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/MeOH/NH4OH 95/5/0,1) с получением 35 мг (81%) трет-бутилового эфира [2-({4-[3-карбамоил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-1-ил]циклогексил}амино)-2-оксоэтил]карбаминовой кислоты в виде твердого вещества белого цвета. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,47 (с, 1Н), 8,27 (д, J=8,67 Гц, 1Н), 7,78 (д, J=6,35 Гц, 1Н), 7,74 (д, J=8,67 Гц, 1Н), 7,64 (шир. с, 1Н), 7,55 (шир. с, 1Н), 6,86 (т, J=5,86 Гц, 1Н), 6,00 (шир. с, 1Н), 3,96 (д, J=2,56 Гц, 1Н), 3,62 (д, J=5,61 Гц, 2Н), 2,72 (с, 3H), 2,23-2,36 (м, 2Н), 1,91-2,11 (м, 4Н), 1,68-1,82 (м, 2Н), 1,38 (с, 9Н).
К раствору 20 мг (0,039 ммоль) трет-бутилового эфира [2-({4-[3-карбамоил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-1-ил]циклогексил}амино)-2-оксоэтил]карбаминовой кислоты в 1,4-диоксане (1 мл) добавляли 4 М HCl в 1,4-диоксане (2 мл 8 ммоль). Указанную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении, и необработанный материал разбавляли диэтиловым эфиром и фильтровали с получением 15 мг конечной солянокислой соли в виде твердого вещества желтого цвета (88%). ЖХ/МС (254) ВЭЖХ способом 2: Rt=3,3 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,48 (с, 1Н), 8,46 (д, J=6,96 Гц, 1Н), 8,27 (д, J=8,67 Гц, 1Н), 7,93-8,11 (м, 3H), 7,75 (д, J=8,79 Гц, 1Н), 7,67 (шир. с, 1Н), 7,51 (шир. с, 1Н), 5,83-6,10 (м, 1Н), 3,96-4,12 (м, 1Н), 3,56-3,71 (м, 2Н), 2,73 (с, 3H), 2,25-2,39 (м, 2Н), 2,04-2,15 (м, 2Н), 1,89-2,04 (м, 3H), 1,75-1,88 (м, 2Н). Значение МСВР (ИЭР), вычисленное для C19H23N7O2S [M+H]+: 414,1707, полученное значение: 414,1721.
Пример 51
1-{4-[(этилкарбамоил)амино]циклогексил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=4-[(этилкарбамоил)амино]циклогексил, R3=NH2, A=-CH=CH-] (соединение 119)
Превращение p
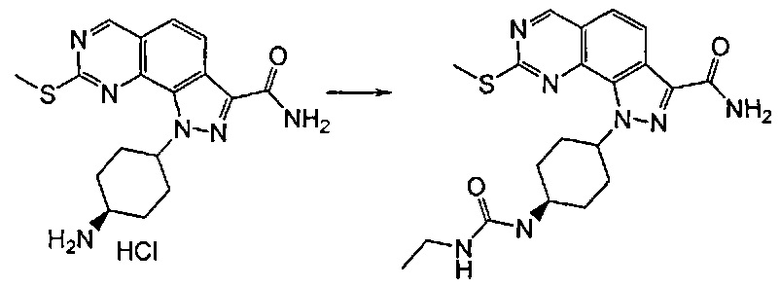
30 мг (0,084 ммоль) гидрохлорида 1-(4-аминоциклогексил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида растворяли в 2 мл диметилформамида. К полученному раствору добавляли 32 мкл (0,42 ммоль) этилизоцианата, 60 мкл (0,336 ммоль) ДИПЭА. Указанную смесь перемешивали при 100°C в течение 18 часов, после чего указанный раствор разделяли между этилацетатом и насыщенным водным раствором NaHCO3 и органический слой промывали рассолом, сушили над Na2SO4 и выпаривали под вакуумом. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/MeOH/NH4OH 95/5/1) с получением 4 мг (11%) 1-{4-[(этилкарбамоил)амино]циклогексил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида в виде твердого вещества белого цвета. ЖХ/МС (254) ВЭЖХ способом 2: Rt=4,38 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=9,47 (с, 1Н), 8,27 (д, J=8,67 Гц, 1Н), 7,74 (д, J=8,67 Гц, 1Н), 7,59 (с, 2Н), 6,06 (д, J=7,08 Гц, 1Н), 5,89-6,02 (м, 1Н), 5,81 (т, J=5,49 Гц, 1Н), 3,78-3,88 (м, 1Н), 3,00-3,07 (м, 2Н), 2,71-2,74 (м, 3H), 2,15-2,29 (м, 2Н), 1,99-2,11 (м, 3H), 1,90 (дд, J=3,54, 13,67 Гц, 2Н), 1,65-1,79 (м, 2Н), 1,01 (т, J=7,20 Гц, 3H). Значение МСВР (ИЭР), вычисленное для C20H25N7O2S [M+H]+: 428,1863, полученное значение: 428,1852.
Пример 52
1-(4-гуанидинциклогексил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=4-гуанидинциклогексил, R3=NH2, A=-CH=CH-] (соединение 120)
Превращение q
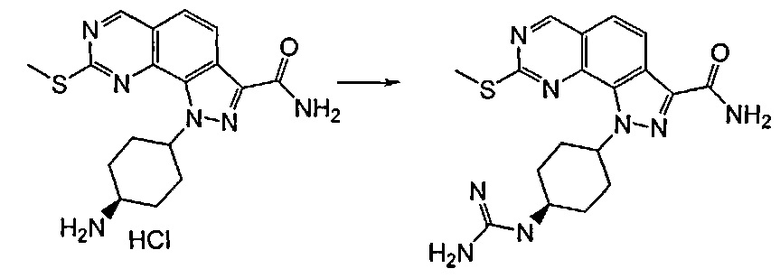
30 мг (0,084 ммоль) гидрохлорида 1-(4-аминоциклогексил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида растворяли в 2 мл диметилформамида. К полученному раствору добавляли 70 мг (0,34 ммоль) 3,5-диметил-1H-пиразол-1-карбоксамидина, 93 мкл (0,67 ммоль) ТЭА. Указанную смесь перемешивали при 100°C в течение 48 часов, раствор разделяли между этилацетатом и насыщенным водным раствором NaHCO3, органический слой промывали рассолом, сушили над Na2SO4 и выпаривали под вакуумом. Необработанный материал очищали посредством хроматографии на силикагеле (ДХМ/MeOH/NH4OH 95/5/1) с получением 6 мг (11%) 1-(4-гуанидинциклогексил)-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в виде твердого вещества желтого цвета. ЖХ/МС (m/z): 399,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 4,1 мин.
Пример 53
1-трет-бутил-8-(метилсульфанил)-6-(морфолин-4-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамид [(I), R1=Me-S-, R2=трет-бутил, R3=NH2, R5=морфолин-4-ил, A=-CH=CH-] (соединение 121)
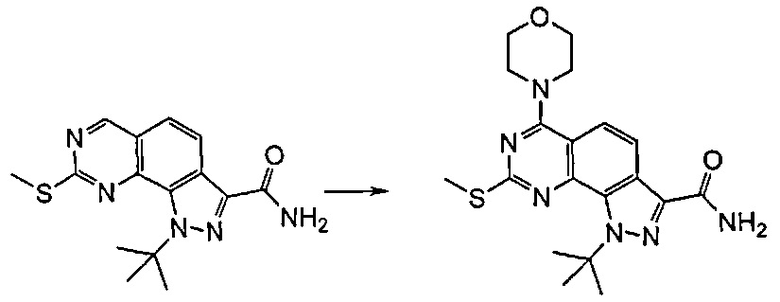
К раствору 50 мг (0,158 ммоль) 1-трет-бутил-8-(метилсульфанил)-1H-пиразоло[4,3-h]хиназолин-3-карбоксамида в безводном ТГФ (2 мл) добавляли 137 мкл (1,26 ммоль) морфолина, 1 М LiN(TMS)2 в ТГФ (4 мл, 4 ммоль). Указанную смесь перемешивали при к.т. в течение 48 часов, а затем разбавляли холодной водой и полученный осадок отфильтровывали. Необработанный материал подвергали ВЭЖХ/МС очистке способом 1 с получением 5 мг (8%) желаемого продукта. ЖХ/МС (m/z): 401,0 [M+H]+ ВЭЖХ (254 нм) способом 2: Rt=6,39 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,20 (д, J=8,91 Гц, 1Н), 7,67 (д, J=8,91 Гц, 2Н), 7,44 (с, 1Н), 3,75-3,82 (м, 4Н), 3,63-3,69 (м, 4Н), 2,64 (с, 3H), 1,97 (с, 9Н). Значение МСВР (ИЭР), вычисленное для C19H25N6O2S [M+H]+: 401,1754, полученное значение: 428,1757.
Получение O: 1-[2-гидрокси-5-(4-метилпиперазин-1-ил)фенил]этанон
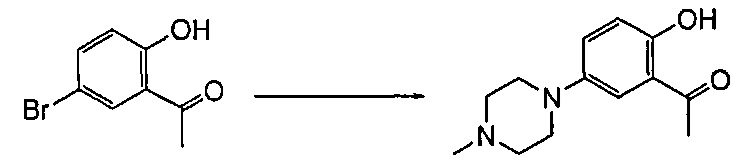
1,0 г (4,65 ммоль) 1-(5-бром-2-гидроксифенил)этанона в ТГФ (10 мл), 0,83 мл (7,45 ммоль) N-метилпиперазина, 16,2 мл (16,2 ммоль) LiHMDSA, 68 мг (0,074 ммоль) Pd2(dba)3 и 29,3 мг (0,074 ммоль) 2-дициклогексилфосфино-2'-N,N-диметиламинобифенила объединяли в атмосфере аргона в закрытом сосуде и перемешивали при 70°C в течение 40 минут. Летучие компоненты удаляли in vacuo, полученный остаток растворяли в воде и 1 н. HCl (pH=6) и разделяли с применением ДХМ. Органическую фазу промывали рассолом, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной флэш-хроматографии (ДХМ градиент до 10% MeOH/ДХМ) с получением 532 мг (51%) желаемого продукта в виде твердого вещества коричневого цвета. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 3,34 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=11,45 (с, 1Н), 7,21-7,28 (м, 2Н), 6,80-6,93 (м, 1Н), 2,98-3,10 (м, 4Н), 2,63 (с, 3H), 2,44-2,48 (м, 4Н), 2,23 (с, 3H). Значение МСВР (ИЭР), вычисленное для C13H18N2O2 [M+H]+: 235,1441, полученное значение: 235,1448.
Согласно аналогичной методике, но с применением подходящего исходного материала получали следующие соединения:
трет-бутиловый эфир 4-(3-ацетил-4-гидроксифенил)пиперазин-1-карбоновой кислоты, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 6,61 мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=11,49 (с, 1Н), 7,32 (д, J=2,93 Гц, 1Н), 7,25-7,30 (м, 1Н), 6,88 (д, J=8,91 Гц, 1Н), 3,41-3,52 (м, 4Н), 2,95-3,05 (м, 4Н), 2,64 (с, 3H), 1,42 (с, 9Н); значение МСВР (ИЭР), вычисленное для C17H24N2O4 [M+H]+: 321,1809, полученное значение: 321,1805;
2-гидрокси-5-(4-метилпиперазин-1-ил)бензонитрил, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=10,38 (шир. с, 1Н), 7,18 (дд, J=3,11, 9,09 Гц, 1Н), 7,08 (д, J=2,93 Гц, 1Н), 6,90 (д, J=9,15 Гц, 1Н); значение МСВР (ИЭР), вычисленное для C12H15N3O [M+H]+: 218,1288, полученное значение: 218,1287; и
2-фтор-4-(4-метилпиперазин-1-ил)фенол, ЖХ/МС (m/z): 211,1 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 4,00 мин.
Получение P: 4-(4-метилпиперазин-1-ил)фенол
2 г (11,24 ммоль) 4-(пиперазин-1-ил)фенола и 4,5 мл (37% раствор в воде, 56 ммоль) формальдегида суспендировали в смеси ТГФ/AcOH в соотношении 5/1 и перемешивали при комнатной температуре. Через 30 минут порциями добавляли 4,7 г (22,5 ммоль) триацетоксиборгидрида натрия. Реакционную смесь перемешивали в течение нескольких часов и концентрировали. Необработанный материал очищали на силикагеле с применением этилацетата/этанола/7 н. NH3 в метаноле в соотношении 8/2/0,2 с получением 2,3 г твердого вещества розового цвета в виде свободного основания. ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло 2,4 мин. 1H ЯМР (401 МГц, ДМСО-d6) δ=8,78 (с, 1Н), 6,71-6,84 (м, 2Н), 6,56-6,69 (м, 2Н), 2,85-3,03 (м, 4Н), 2,46 (шир. с, 4Н), 2,23 (с, 3H). Значение МСВР (ИЭР), вычисленное для C11H16N2O [M+H]+: 193,1336, полученное значение: 193,1328.
Действуя аналогичным способом, получали следующее соединение:
3-(4-метилпиперазин-1-ил)фенол, ЖХ/МС (254 нм) способом ВЭЖХ 2: Rt составляло мин; 1H ЯМР (401 МГц, ДМСО-d6) δ=9,07 (с, 1Н), 6,96 (т, J=8,12 Гц, 1Н), 6,34-6,38 (м, 1Н), 6,28 (т, J=2,32 Гц, 1Н), 6,19 (ддд, J=0,73, 2,20, 7,93 Гц, 1Н), 3,02-3,09 (м, 4Н), 2,41-2,46 (м, 4Н), 2,22 (с, 3H); значение МСВР (ИЭР), вычисленное для C11H16N2O [M+H]+: 193,1336, полученное 193,1329.
Получение Q
Этап a. Получение 4-(4-метилпиперазин-1-ил)фенилацетата
А смесь 1,080 мг (5,59 ммоль) 4-(4-метилпиперазин-1-ил)фенола и 1,72 мл (12,3 ммоль) ТЭА в ДХМ охлаждали в ледяной бане и по каплям добавляли 0,44 мл (6,15 ммоль) ацетилхлорида. После завершения реакции реакционную смесь фильтровали, органическую фазу немедленно подвергали экстракции водой, сушили над Na2SO4 и концентрировали с получением твердого вещества светло-розового цвета. ЖХ/МС (m/z): 235,4 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 1,89 мин.
Этап b. Получение 2-бром-4-(4-метилпиперазин-1-ил)фенилацетата
Раствор 247 мг (1,05 ммоль) 4-(4-метилпиперазин-1-ил)фенилацетата и 209 мг (1,1 ммоль) п-толуолсульфоновой кислоты в ацетонитриле охлаждали в ледяной бане и по каплям добавляли 0,056 мл (1,1 ммоль) брома. Реакционную смесь перемешивали при комнатной температуре до завершения реакции (3-4 часы), после чего разбавляли этилацетатом и подвергали экстракции водой и насыщенным раствором NaHCO3. Органическую фазу, высушенную над Na2SO4 и подвергнутую выпариванию, очищали посредством флэш-хроматографии (ДХМ/MeOH 85/15) с получением продукта в виде темно-коричневого масла. ВЭЖХ (254 нм) способом 2: Rt=4,05 мин. 1H ЯМР (400 МГц, ДМСО-d6) δ=(м.д.) 2,25 (с, 3H) 2,36 (с, 3H) 2,39 (м, 4H) 3,44 (м, 4H) 6,50 (дд, J=2,1,8,3 Гц, 1H) 7,03 (д, J=8,3 Гц, 1H) 7,21 (д, J=2,1 Гц, 1H). Вычисленная масса: 313,0546, полученная масса: 313,0538.
Этап c. Получение 2-бром-4-(4-метилпиперазин-1-ил)фенола
280 мг (1,03 ммоль) 2-бром-4-(4-метилпиперазин-1-ил)фенилацетата растворяли в метаноле и подвергали реакции с 2 мл 1 н. NaOH. Через два часа реакционную смесь подкисляли с применением ледяной уксусной кислоты, выпаривали и очищали на диоксиде кремния с применением ДХМ/MeOH в соотношении 9/1 и 0,4% 7 М NH3 в MeOH. ЖХ/МС (m/z): 273,2 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 2,96 мин.
Получение R: трет-бутиловый эфир 3-[(метилсульфонил)окси]азепан-1-карбоновой кислоты

Этап a. Получение трет-бутилового эфира 3-гидроксиазепан-1-карбоновой кислоты
Раствор 1,07 г (5 ммоль) трет-бутилового эфира 3-оксоазепан-1-карбоновой кислоты в 50 мл MeOH охлаждали до 0°C и добавляли 213 мг (6,1 ммоль) боргидрида натрия. Указанный раствор медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Летучие компоненты удаляли под вакуумом, остаток растворяли в этилацетате и разделяли с применением воды. Органический слой промывали рассолом, сушили с применением Na2SO4 и выпаривали. Необработанный материал очищали посредством флэш-хроматографии (EtOAc/гексан 3/7) с получением 0,75 г (75%) трет-бутилового эфира 3-гидроксиазепан-1-карбоновой кислоты в виде твердого вещества белого цвета. 1H ЯМР (401 МГц, ДМСО-d6) δ=4,34-4,65 (м, 1Н), 3,52-3,80 (м, 1Н), 3,05-3,25 (м, 2Н), 1,70-1,89 (м, 2Н), 1,58-1,69 (м, 1Н), 1,43-1,56 (м, 3H), 1,39 (с, 9Н).
Этап b. Получение трет-бутилового эфира 3-[(метилсульфонил)окси]азепан-1-карбоновой кислоты
К раствору 200 мг (0,93 ммоль) трет-бутилового эфира 3-гидроксиазепан-1-карбоновой кислоты в ДХМ (10 мл), охлажденному до 0°C, добавляли 190 мкл (1,4 ммоль) ТЭА и 114 мкл (1,4 ммоль) метансульфонилхлорида. Допускали нагревание указанного раствора до комнатной температуры и перемешивали указанный раствор в течение 2 часов. Смесь разбавляли насыщенным водным раствором NaHCO3, органический слой отделяли и промывали рассолом, сушили над Na2SO4 и концентрировали при пониженном давлении, полученное вязкое масло бледно-желтого цвета немедленно применяли на следующем этапе. ЖХ/МС (m/z): 294,0 [M+H]+, ВЭЖХ (254 нм) способом 3: Rt составляло 5,65 мин.
Согласно аналогичной методике, но с применением подходящего исходного материала получали следующее соединение:
2,2,2-трихлорэтиловый эфир (3-экзо)-3-[(метилсульфонил)окси]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты, ЖХ/МС (m/z): 381,1 [M+H]+ ВЭЖХ (254 нм) способом 3: Rt составляло 5,56 мин.
Пример 901 Активность связывания киназы Pim
Ферменты PIM-1, -2, и -3 получали в виде гибридных белков, экспрессированных в бактерии и очищенных посредством колоночной хроматографии IMAC (Sun X., Chiu J.F. и He Q.Y. (2005) Expert Rev. Proteomics, 2:649-657). Флуоресцентно меченый Pim-специфический пептидный субстрат был синтезирован на заказ American Peptide Company (Саннивейл, СА). Рабочий буферный раствор содержал 10 мМ HEPEC, pH 7,2, 10 мМ MgCl2, 0,01% Tween 20, 2 мМ ДТТ. Буферный раствор, останавливающий реакцию, содержал 190 мМ HEPEC, pH 7,2, 0,015% Brij-35, 0,2% покрывающего реагента 3 (Caliper Life Sciencec, Хопкинтон, MA), 20 мМ EDTA. Разделительный буферный раствор содержал 100 мМ HEPEC, pH 7,2, 0,015% Brij-35, 0,1% покрывающего реагента 3, 1:200 покрывающего реагента 8 (Caliper Life Sciences, Хопкинтон, MA), 10 мМ EDTA и 5% ДМСО.
Реакции PIM проводили в конечном объеме 10 мкл на лунку в 384-луночном планшете. Стандартная ферментативная реакция, инициированная посредством добавления 5 мкл 2X АТФ и исследуемого соединения к 5 мкл 2X фермента и FAM-пептида, содержала 20 пМ PIM1, 50 пМ PIM2 или 55 пМ PIM3, 1 мкМ FAM-пептида и 10 мкМ АТФ в рабочем буферном растворе. После 90 минут выдерживания при комнатной температуре реакцию фосфорилирования прекращали посредством добавления 10 мкл буферного раствора, останавливающего реакцию. Продукт и субстрат в каждой отдельной реакции разделяли на микрожидкостном чипе с 12 капиллярными трубками (Caliper Life Sciences, Хопкинтон, MA), приводимого в действие Caliper LC3000® (Caliper Life Sciences, Хопкинтон, MA). Разделение продукта и субстрата оптимизировали посредством выбора напряжений и давления с применением программного обеспечения CalipeR's Optimizer (Хопкинтон, MA). Применяли условия разделения, представлявшие собой напряжение на конце капиллярной трубки, к которому происходит перемещение, -500 В, напряжение на конце капиллярной трубки, от которого происходит перемещение, -2150 В и давление отбора -1,2 фунт/кв. дюйм. Продукт и флуорофор субстрата возбуждали при 488 нм и детектировали при 530 нм. Превращение субстрата вычисляли исходя из электрофореграммы с применением программного обеспечения HTS Well Analyzer (Caliper Life Sciences, Хопкинтон, MA). Вычисляли значения Ki для исследуемого соединения. См. Таблицу 1 для репрезентативного PIM1 LC3K Ki в области микромолярных концентраций иллюстративных соединений.
Пример 902 Анализы активности клеточной пролиферации in vitro
BaF3 представляет собой интерлейкин-3-зависимую линию про-B-клеток мыши, подходящую в качестве модельной системы для оценки активности и передачи сигнала по нисходящему пути киназы онкогенов («Ba/F3 cells and their use in kinase drug discovery», Warmuth M. и др. (январь 2007) Current Opinion in Oncology, Vol 19(1):55-60). Родительскую клеточную линию BaF3 получали из депозитария DSMZ. Далее получали BaF3 линии, трансфицированные PIM1 или PIM2. ИЛ-3 мыши приобретали у R&D Systems. G418 приобретали у Clontech. Среда для родительской клеточной линии BaF3 содержала RPMI, 10% ЭБС, 2 мМ L-глутамина, 2 нг/мл мИЛ-3. Среда для линий BaF3 PIM1 и 2 содержала RPMI, 10% ЭБС, 2 мМ L-глутамина, 250 мкг/мл. Среда для линии MM1.S (клетки множественной миеломы) содержала RPMI, 10% ЭБС, 2 мМ L-глутамина.
Родительские клетки, клетки BaF3 PIM1, клетки BaF3 PIM2 и клетки MM1.S (множественной миеломы) высевали в 384-луночный планшет в количестве 45 мкл/лунка по 2000 клеток/лунка, 5000 клеток/лунка, 5000 клеток/лунка и 10000 клеток/лунка соответственно. Исследуемое соединение добавляли в количестве 5 мкл/лунка. Клетки BaF3 (родительские и трансфицированные) выдерживали в течение ночи, тогда как клетки MM1.S выдерживали в течение 72 часов при 37°C, 5% CO2. Реагент CELL TITER GLO® (Promega) добавляли в количестве 50 мкл/лунка, планшеты выдерживали в течение 30 минут, их люминесценцию измеряли с помощью HT Analyzer. Далее вычисляли значения IC50/EC50 для исследуемого соединения.
Репрезентативные соединения согласно настоящему изобретению испытывали, как описано выше, и получали Ki/IC50/EC50, представленные ниже в Таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| Селективный ингибитор киназы JAK1 | 2020 |
|
RU2818002C2 |
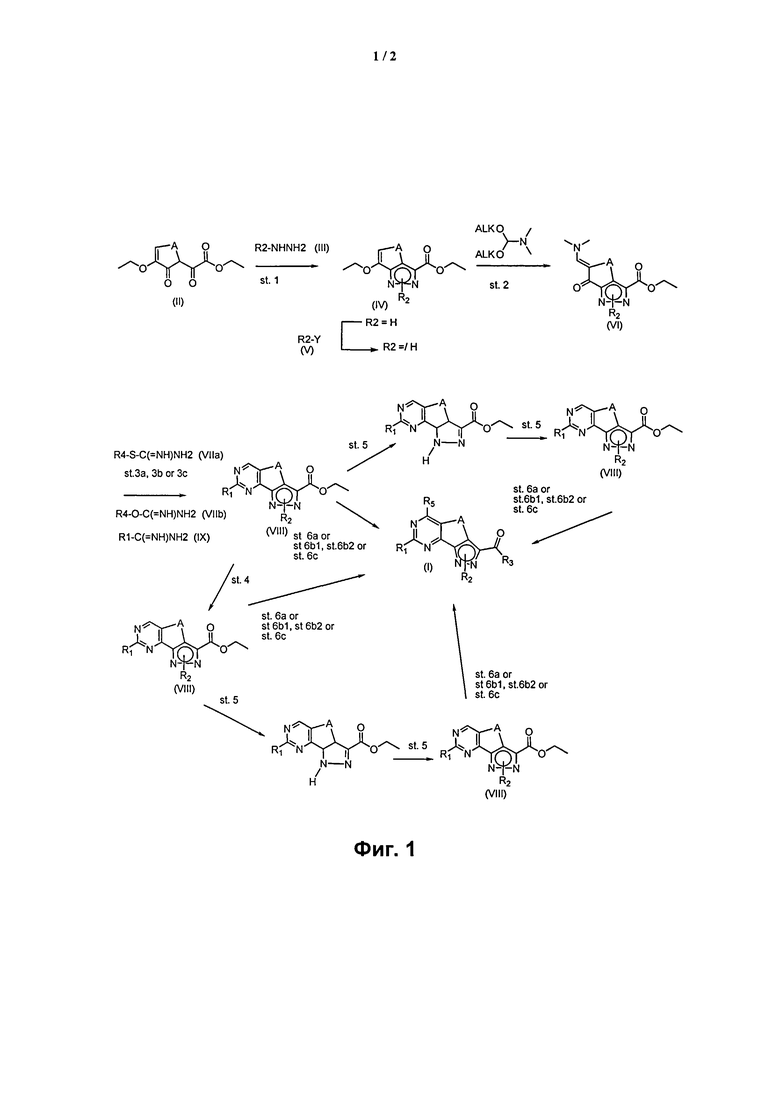

Настоящее изобретение относится к новым пиразоло[4,3-h]хиназолиновым соединениям, выбранным из группы, указанной ниже, и соответствующих общей формуле (I). Соединения обладают свойствами регуляции активности протеинкиназ PIM-1, PIM-2, PIM-3 и могут быть использованы для лечения заболеваний, вызванных и/или связанных с указанным нарушением, таких как рак, клеточные пролиферативные заболевания или нарушения, и заболевания и нарушения, связанные с иммунными клетками. Клеточное пролиферативное нарушение выбрано из группы, состоящей из доброкачественной гиперплазии предстательной железы, семейного аденоматоза толстой кишки, нейрофиброматоза, псориаза, пролиферации гладкомышечных клеток сосудов, обусловленной атеросклерозом, фиброза легких, артрита, гломерулонефрита и послеоперационного стеноза и рестеноза. Воспалительные и аутоиммунные заболевания выбраны из рассеянного склероза, системной красной волчанки, воспалительных заболеваний кишечника (ВЗК), болезни Крона, синдрома раздраженного кишечника, панкреатита, неспецифического язвенного колита, дивертикулеза, миастении, васкулита, псориаза, склеродермии, бронхиальной астмы, аллергии, системного склероза, витилиго, артрита, такого как остеоартрит, ювенильный ревматоидный артрит, анкилозирующий спондилоартрит. В формуле 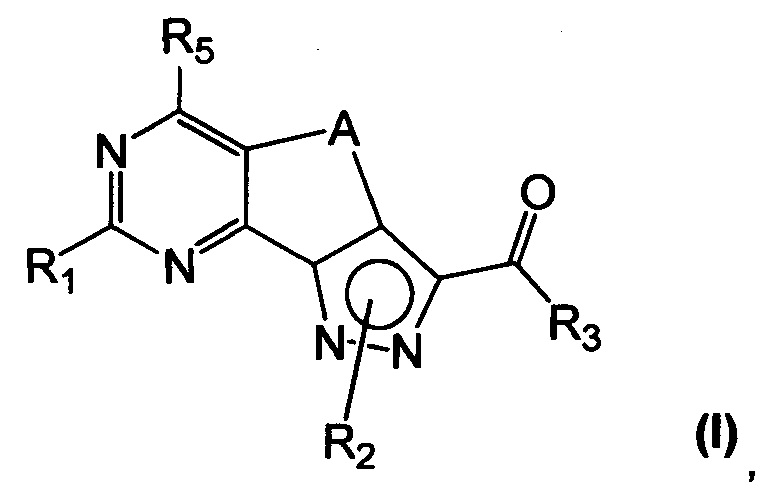 радикалы А, R1-R3, R5 имеют значения, соответствующие структуре соединений, которые выбраны из 1-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-(2-гидроксиэтил)-8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-(1-метилпиперидин-4-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-[2-(1Н-имидазол-5-ил)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,6-амино-1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,2-[2-(диметиламино)этил]-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-аминоэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,2-(2-аминоэтил)-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-1-(пиперидин-4-илметил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-2-(пиперидин-4-илметил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-фенокси-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(4-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-(пиридин-4-илокси)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(2-фторэтокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-метил-8-[3-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[3-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[3-(диметиламино)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(2-хлорфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-(2-фторфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[2-ацетил-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-ацетил-4-(пиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-циано-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-фенокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-метокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-этокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-формилфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-{3-[(4-метилпиперазин-1-ил)метил]фенокси}-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[3-(гидроксиметил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-{3-[(диметиламино)метил]фенокси}-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-метил-8-[3-(морфолин-4-илметил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-(2-гидроксиэтил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-фторэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-хлорэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-(2-метоксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-[3-(диметиламино)пропил]-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(1-амино-2-метил-1-оксопропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида и других соединений, указанных в п. 1 формулы изобретения. 7 н. и 8 з.п. ф-лы, 2 ил., 2 табл., 53 пр.
радикалы А, R1-R3, R5 имеют значения, соответствующие структуре соединений, которые выбраны из 1-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-(2-гидроксиэтил)-8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-(1-метилпиперидин-4-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-N-[2-(1Н-имидазол-5-ил)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,6-амино-1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,2-[2-(диметиламино)этил]-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-аминоэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,2-(2-аминоэтил)-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-1-(пиперидин-4-илметил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-2-(пиперидин-4-илметил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-фенокси-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(4-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-(пиридин-4-илокси)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(2-фторэтокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[4-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-метил-8-[3-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[3-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[3-(диметиламино)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(2-хлорфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-(2-фторфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[2-ацетил-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-ацетил-4-(пиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-[2-циано-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-фенокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-трет-бутил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-метокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-этокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-формилфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-метил-8-{3-[(4-метилпиперазин-1-ил)метил]фенокси}-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 8-[3-(гидроксиметил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-{3-[(диметиламино)метил]фенокси}-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-метил-8-[3-(морфолин-4-илметил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(3-аминофенокси)-1-(2-гидроксиэтил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-фторэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-хлорэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида, 1-(2-метоксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-[3-(диметиламино)пропил]-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,1-(1-амино-2-метил-1-оксопропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида и других соединений, указанных в п. 1 формулы изобретения. 7 н. и 8 з.п. ф-лы, 2 ил., 2 табл., 53 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль, выбранное из группы, состоящей из:
1-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-(2-гидроксиэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-(1-метилпиперидин-4-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-[2-(1Н-имидазол-5-ил)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
6-амино-1-[2-(диметиламино)этил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
2-[2-(диметиламино)этил]-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-аминоэтил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
2-(2-аминоэтил)-8-(метилсульфанил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-4-илметил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-2-(пиперидин-4-илметил)-2Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[4-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-фенокси-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(3-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(4-аминофенокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-(пиридин-4-илокси)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(2-фторэтокси)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[4-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-бром-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[3-(пиперазин-1-ил)фенокси]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[3-(пиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[3-(диметиламино)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(2-хлорфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(2-фторфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-фтор-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-ацетил-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-ацетил-4-(пиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[2-циано-4-(4-метилпиперазин-1-ил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-фенокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-(3-нитрофенокси)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[3-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-8-[4-(4-метилпиперазин-1-ил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-метокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-этокси-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-метокси-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(3-формилфенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-{3-[(4-метилпиперазин-1-ил)метил]фенокси}-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-[3-(гидроксиметил)фенокси]-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-{3-[(диметиламино)метил]фенокси}-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-[3-(морфолин-4-илметил)фенокси]-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(3-аминофенокси)-1-метил-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(3-аминофенокси)-1-(2-гидроксиэтил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-фторэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-хлорэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-гидроксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-метоксиэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[3-(диметиламино)пропил]-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(1-амино-2-метил-1-оксопропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-4-илметил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(3-аминопропил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-4-ил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(2-аминоэтил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[2-(4-метилпиперазин-1-ил)этил]-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-этенил-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-{2-[4-(диметиламино)пиперидин-1-ил]этил}-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-этокси-1-(пиперидин-4-илметил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[3-(диметиламино)пропил]-8-этокси-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-этокси-1-(пиперидин-4-ил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-этокси-1-[(1-метилпиперидин-4-ил)метил]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
6-амино-1-трет-бутил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
6-амино-1-метил-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-трет-бутил-N-гидрокси-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-4-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-3-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(4-аминоциклогексил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(пиперидин-3-илметил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(азепан-4-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(3-амино-2,2-диметилпропил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-метокси-1-(пиперидин-4-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-метокси-1-(пиперидин-4-илметил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[(3-экзо)-8-азабицикло[3.2.1]окт-3-ил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[(3-эндо)-8-азабицикло[3.2.1]окт-3-ил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[4-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)бутан-2-ил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(4-аминобутан-2-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(1-азабицикло[2.2.2]окт-3-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
8-(метилсульфанил)-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[транс-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[цис-3-(гидроксиметил)циклобутил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(1-гидрокси-2-метилпропан-2-ил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(1-гидрокси-2-метилпропан-2-ил)-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-{[1-(2-аминоэтил)пиперидин-4-ил]метил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-{4-[(2-аминоэтил)амино]циклогексил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-{4-[(2-аминоэтил)амино]циклогексил}-8-(метилсульфанил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-[4-(глициламино)циклогексил]-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-{4-[(этилкарбамоил)амино]циклогексил}-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида,
1-(4-карбамимидамидоциклогексил)-8-(метилсульфанил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида и
1-трет-бутил-8-(метилсульфанил)-6-(морфолин-4-ил)-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида.
2. Способ получения соединения по п. 1 формулы (I) или его фармацевтически приемлемых солей, за счет проведения этапа 6) приведения соединения формулы (VIII) во взаимодействие:
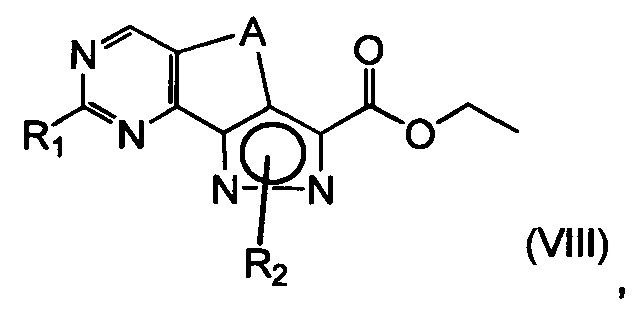
где R1, R2, R4 и А имеют значения, соответствующие структуре соединений по п. 1, согласно любому из альтернативных этапов 6а), 6b) или 6с):
этап 6а) с гидроксидом аммония или амином формулы (XII):

где R' и R'' имеют значения, соответствующие заместителям аминогруппы в соединениях по п. 1, с получением соединения формулы (I):
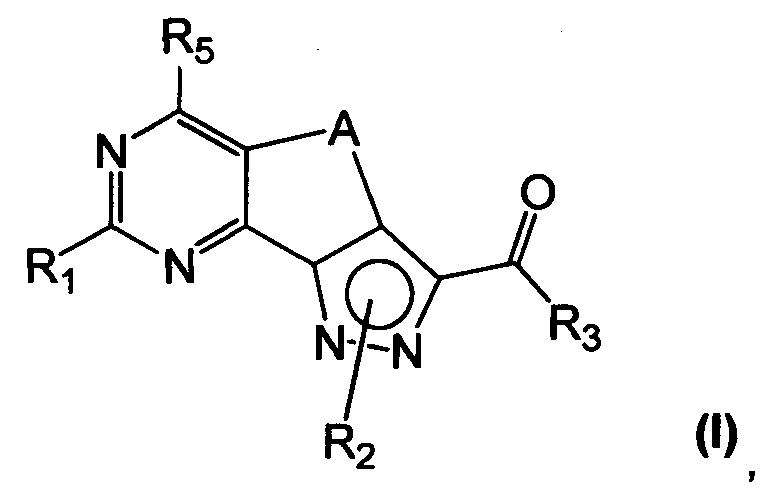
где R3 представляет собой NR'R'', R5 представляет собой водород, a R2, R4, A, R' и R'' определены выше;
этап 6b1) в условиях кислотного или щелочного гидролиза с получением соединения формулы (XI) или его соли:
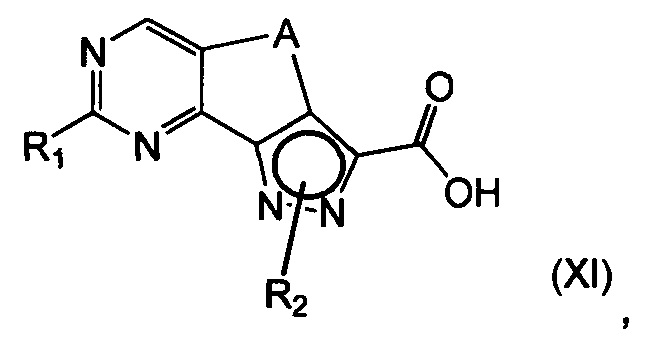
где R1, R2, R4 и А определены выше;
этап 6b2) смешивание полученного соединения формулы (XI) или его соли с солью аммония или производным формулы (XII), определенной выше, или производным формулы (X):

где R' определен выше, в щелочных условиях и в присутствии подходящего конденсирующего агента с получением соединения формулы (I):
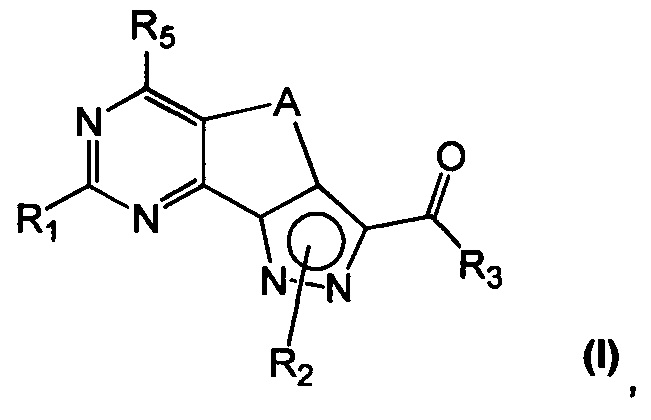
где R5 представляет собой водород, a R1, R2, R3, R4 и А определены выше;
этап 6с) с солью аммония в присутствии сильного основания с получением соединения формулы (I):
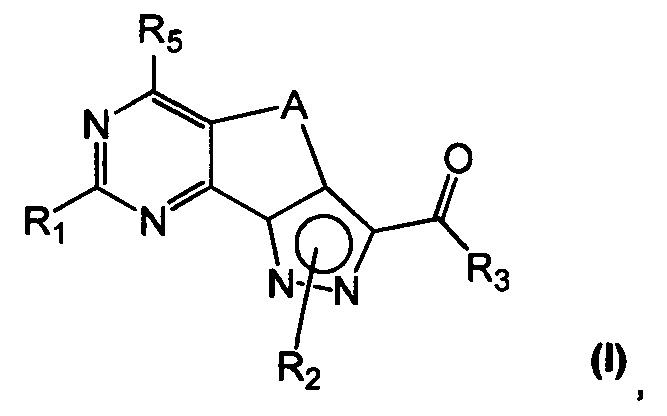
где R3 представляет собой NR'R'', a R1, R2, R4, R5, A, R' и R'' определены выше;
и необязательно, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
3. Способ по п. 2, отличающийся тем, что на этапе 6а) подходящий амин формулы (XII) применяют в присутствии сильного основания с получением соединения формулы (I):
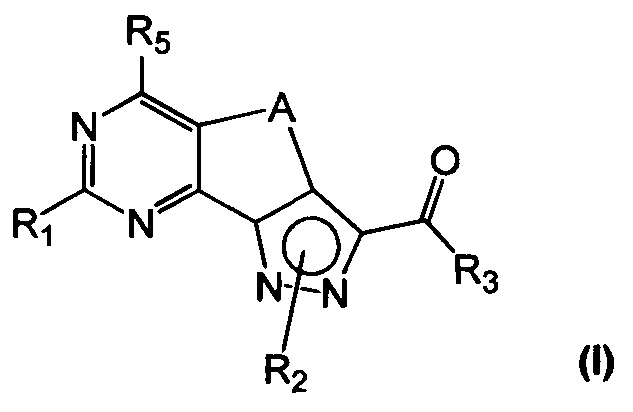
где R3 представляет собой NR'R'', a R1, R2, R4, R5, A, R' и R'' определены выше.
4. Способ получения соединения по п. 1 формулы (I) или его фармацевтически приемлемых солей, характеризующийся тем, что проводят этап 7) путем приведения во взаимодействие соединения формулы (XIII):
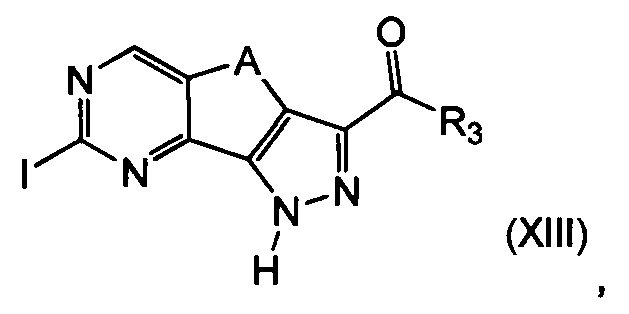
где R3 и А определены выше в п. 2, с соединением формулы (V):
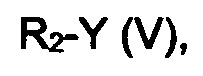
где Y представляет собой подходящую уходящую группу, такую как йод, бром, хлор или сульфонатная группа (например, -OS(O)2CF3, -OS(O)2СН3 или -OS(O)2PhMe), a R2 определен выше в п. 2, но не является водородом.
5. Способ получения соединения по п. 1 формулы (I), или его фармацевтически приемлемых солей, характеризующийся тем, что превращение проводят одним из следующих способов:
превращение е): превращение соединения формулы (I), где R1, R3 и А определены выше в п. 2, a R2 представляет собой водород, в соединение формулы (I), где R2 определен выше в п. 2, но не является водородом, путем проведения реакции с соединением формулы (V):
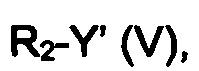
где Y' представляет собой ОН или группу, которая необязательно в результате активации может служить подходящей уходящей группой, такой как йод, бром, хлор или сульфонатная группа (например, -OS(O)2CF3, -OS(O)2СН3 или -OS(O)2PhMe), a R2 определен выше, но не является водородом;
превращение r): превращение соединения формулы (I), где R1, R2 и R3 определены выше, и А представляет собой двухвалентную группу, такую как -СН2-СН2-, в соединение формулы (I), где А представляет собой группу -СН=СН-, путем обработки окисляющим агентом или в условиях дегидрогенизации в присутствии Pd или Pt катализатора.
6. Соединение по п. 1 для применения в способе лечения заболевания, вызванного и/или связанного с нарушением регуляции активности протеинкиназ PIM-1, PIM-2, PIM-3, который включает введение эффективного количества указанного соединения.
7. Соединение для применения в способе лечения по п. 6, отличающееся тем, что указанное заболевание выбрано из группы, состоящей из рака, клеточных пролиферативных нарушений и заболеваний и нарушений, связанных с иммунными клетками.
8. Соединение для применения в способе лечения по п. 7, отличающееся тем, что рак выбран из группы, состоящей из карциномы, такой как рак мочевого пузыря, молочной железы, толстой кишки, почек, печени, легкого, включая мелкоклеточный рак легкого, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы и кожи, включая сквамозную карциному; гемобластозов из клеток лимфоидной линии, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому и лимфому Беркитта; гемобластозов из клеток миелоидной линии, включая острый и хронический миелогенный лейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухолей мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухолей центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; других опухолей, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы, саркому Капоши и мезотелиому.
9. Соединение для применения в способе лечения по п. 7, отличающееся тем, что клеточное пролиферативное нарушение выбрано из группы, состоящей из доброкачественной гиперплазии предстательной железы, семейного аденоматоза толстой кишки, нейрофиброматоза, псориаза, пролиферации гладкомышечных клеток сосудов, обусловленной атеросклерозом, фиброза легких, артрита, гломерулонефрита и послеоперационного стеноза и рестеноза.
10. Соединение для применения в способе лечения по п. 7, отличающееся тем, что воспалительные и аутоиммунные заболевания выбраны из рассеянного склероза, системной красной волчанки, воспалительных заболеваний кишечника (ВЗК), болезни Крона, синдрома раздраженного кишечника, панкреатита, неспецифического язвенного колита, дивертикулеза, миастении, васкулита, псориаза, склеродермии, бронхиальной астмы, аллергии, системного склероза, витилиго, артрита, такого как остеоартрит, ювенильный ревматоидный артрит, анкилозирующий спондилоартрит.
11. Способ in vitro ингибирования активности протеинкиназ PIM-1, PIM-2, PIM-3, включающий приведение в контакт указанного белка с эффективным количеством соединения по п. 1.
12. Фармацевтическая композиция для лечения заболеваний, вызванных и/или связанных с нарушением регуляции активности протеинкиназ PIM-1, PIM-2, PIM-3, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 1, и по меньшей мере один фармацевтически приемлемый наполнитель, носитель и/или разбавитель.
13. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, обладающее свойствами ингибитора протеинкиназ PIM-1, PIM-2 и PIM-3 для применения в качестве лекарственного средства.
14. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1 для применения в способе лечения рака, вызванного и/или связанного с нарушением регуляции активности протеинкиназ PIM-1, PIM-2 и PIM-3.
15. Применение соединения формулы (I) или его фармацевтически приемлемой соли по п. 1, обладающего свойствами ингибитора протеинкиназ PIM-1, PIM-2 и PIM-3 в производстве лекарственного средства с противораковой активностью.
| Maria Gabriella Brasca et al., Identification of N,1,4,4-tetramethyl-8-{ not 4-(4-methylpiperazin-1-yl)phenyl|amino} -4,5-dihydro-1H-pyrazolo not 4,3-h|quinazoline-3-carboxamide (PHA-848125), a potent, orally available cyclin dependent kinase inhibitor, Journal of Medicinal Chemistry, 2009, v | |||
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |
| Italo Beria et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Italo Beria,et al., Identification of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline Derivatives as a new class of orally and selective polo-like kinase 1 inhibitors, Journal of Medicinal Chemistry, 16.04.2010, v | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| Mauro Angiolini et al.,Structure-based optimization of potent PDK1 inhibitors, Bioorganic & Medicinal Chemistry Letters, 08.06.2010, v | |||
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Gabriella Traquandi, Identification of potent pyrazolo[4,3-h]quinazoline-3-carboxamides as multi-cyclin-dependent kinase inhibitor Journal of Medicinal Chemistry, 08.02.2010,v.53,no.5, p.2171-2187 | |||
| WO 2011012534 А1, 03.02.2011 | |||
| WO 2008074788 А1, 26.06.2008 | |||
| WO 2009156315 А,30.12.2009 | |||
| WO 2004104007 A1,02.12.2004 & EA 010904 B1 | |||
| WO 2004014352 А2,19.02.2004 | |||
| WO 03070706 А1, 28.08.2003 | |||
| WO 9828281 А1, 02.07.1998 | |||
| WO 9858926 А1, 30.12.1998 | |||
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |

