Изобретение относится к органической химии и касается способа получения спинохрома D (2,3,5,6,8-пентагидрокси-1,4-нафтохинона) - одного из основных пигментов, продуцируемых морскими ежами различных видов [1а-в]. Спинохром D был впервые обнаружен в иглах морского ежа Pseudocentrotus depressus японскими химиками, но они приписали ему неправильную молекулярную формулу [2]. Позже Томсон и сотр. установили правильную структуру этого пигмента, осуществив его полный синтез, и определили его свойства [3].
В основе структуры спинохрома D лежит циклическая система 5,8-дигидрокси-1,4-нафтохинона (нафтазарина). К настоящему времени около 30 таких пигментов, в которых в β-положениях циклического скелета содержатся гидрокси-, метокси-метильные, этильные, ацетильные и аминогруппы, выделено из морских ежей различных видов [4, 5]. Полигидрокси-1,4-нафтохиноны морских ежей, в том числе спинохром D, проявляют высокую антиоксидантную и антирадикальную активность и представляют новый класс природных антиоксидантов [4-11]. Спинохром D обладает также высокой антиалгальной [12, 13], антимикробной [14], антиаллергической [15] и гепатопротекторной [16] активностью.
На основе эхинохрома А (2,3,5,6,8-пентагидрокси-7-этил-1,4-нафтохинона) - одного из основных метаболитов морских ежей - созданы эффективные отечественные лекарственные препараты: Гистохром кардиологический® для лечения острого инфаркта миокарда и ишемической болезни сердца и Гистохром офтальмологический® для лечения пролиферативных и дегенеративных процессов, включая катаракту, и кровоизлияний различной природы в тканях глаза [17].
Интересно отметить, что икра и гонады морских ежей, содержащие полигидрокси-1,4-нафтохиноны, в том числе спинохром D, используются как высокоценимые деликатесные продукты питания в Японии, Китае и ряде стран Юго-Восточной Азии [4, 11].
Спинохром D представляет также ценность как удобный строительный блок для синтеза более сложных молекул природных полигидрокси-1,4-нафтохинонов, таких как эхинохром А, 2-ацетил-3,5,6,7,8-пентагидрокси-1,4-нафтохинон (спинохром С) [18] и 2,2'-(этан-1,1-диил)бис(3,5,6,7,8-пентагидрокси-1,4-нафтохинон) [19].
Возможности широкого практического использования полигидрокси-1,4-нафтохинонов морских ежей, в том числе спинохрома D, ограничиваются их малой доступностью. Так, содержание спинохрома D в морских ежах различных видов не превышает 0.001-0.003% от веса сухих ежей.
Возможны два пути получения нафтохиноидных пигментов морских ежей: либо извлечением из природных объектов, либо через полный органический синтез.
В отличие от таких пигментов морских ежей, как эхинохром А и спинохром Е (2,3,5,6,7,8-гексагидрокси-1,4-нафтохинон), технология получения спинохрома D из природного сырья в настоящее время не разработана. Таким образом, органический синтез остается единственным путем получения спинохрома D в препаративных количествах.
Известны несколько способов получения спинохрома D путем полного синтеза.
В первом из них (схема 1) [3] стартовым соединением стал 1,2,3-триметоксибензол (триметиловый эфир пирогаллола) 2, который в 4 стадии был превращен в кислоту 4.
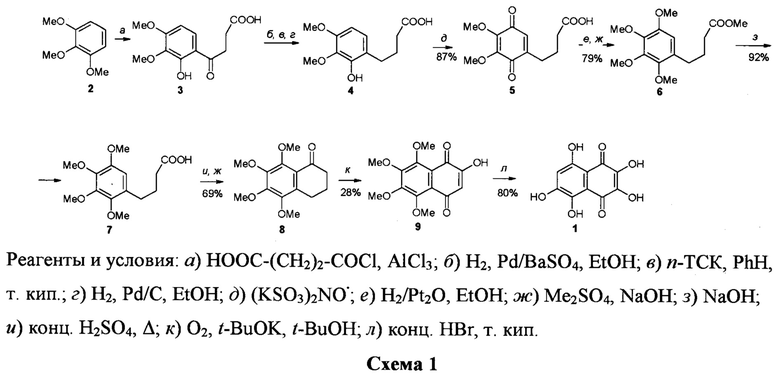
Последняя была превращена в 6 стадий в α-тетралон 8, окислением которого кислородом в сильноосновной среде был получен 2-гидрокси-5,6,7,8-тетраметокси-1,4-нафтохинон 9. Кислотный гидролиз тетраэфира 9 дал целевой спинохром D. Суммарный выход его на 12 стадий составил ~4.7%. Большое число стадий и низкий суммарный выход делают этот способ получения спинохрома D не имеющим практического значения.
Во втором синтезе нафтазариновый остов спинохрома D формировали через реакцию циклоацилирования 1,2-дигидрокси-3,4-диметоксибензола 11 монохлормалеиновым ангидридом 12 (схема 2) [20, 21].
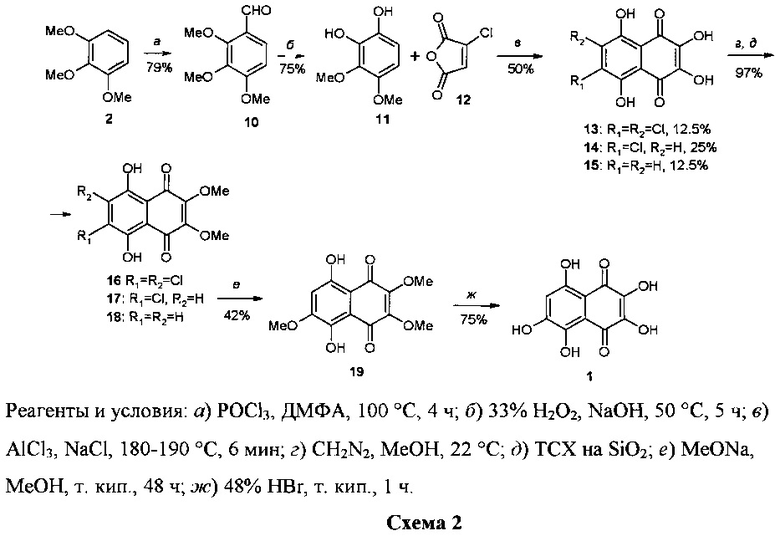
Исходный субстрат 11 получали в 2 стадии из триметилового эфира пирогаллола 2. Эта реакция циклоацилирования дает смесь трех продуктов 13-15 в мольном соотношении 1:2:1. Выход целевого продукта 14 не превышал при этом 25%. Авторы предположили, что побочные продукты 13 и 15 образуются в результате диспропорционирования двух молекул соединения 14. Полученную смесь соединений 13-15 обрабатывали без разделения метанольным раствором диазометана и образовавшуюся смесь диэфиров 16 - 18 разделяли методом ТСХ на пластинах с SiO2. В полученном продукте 17 осуществляли нуклеофильное замещение атома хлора на метоксигруппу действием большого избытка насыщенного раствора метилата натрия в метаноле при кипячении в течение 48 ч. Гидролиз триэфира 19 при кипячении в конц. HBr привел к образованию целевого спинохрома D 1. Суммарный выход его на 7 стадий составил 4.5%. Этот синтез также не имеет препаративного значения, поскольку содержит стадию конверсии 17→19, сильно ограничивающую его возможности. Эта конверсия требует применения огромного избытка насыщенного раствора MeONa в МеОН (на 30 мг (0.107 ммоль) субстрата 17 требуется применение 300 мл (1172 ммоль) насыщенного раствора MeONa в МеОН). При этом добиться полной конверсии субстрата 17 в триэфир 19 не удалось.
Недавно авторами осуществлен синтез спинохрома D, значительно отличающийся от вышеописанных (схема 3) [22].
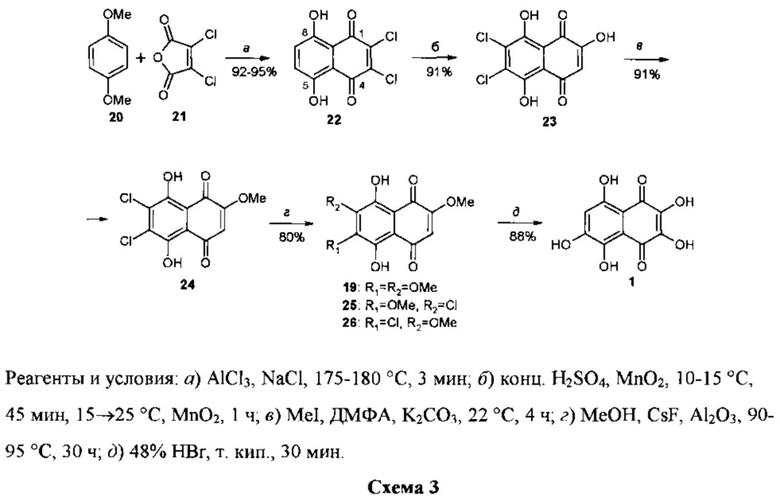
Стартовым соединением в этом подходе к синтезу спинохрома D явился известный 2,3-дихлорнафтазарин 22, легко получаемый циклоацилированием товарного 1,4-диметоксибензола 20 (диметилового эфира гидрохинона) дихлормалеиновым ангидридом 21 [23]. В отличие от условий, описанных в работе [23], циклоацилирование проводилось при 175-180°C в течение 3 мин, а не при 170°C (1-2 мин). Частично была изменена также методика обработки реакционной массы. Все это позволило получать стабильно высокие выходы продукта 22 (92-95%). Окисление соединения 22 товарным диоксидом марганца в растворе в конц. серной кислоте дало 2-гидрокси-6,7-дихлорнафтазарин 23 с выходом 91%.
Попытки нуклеофильного замещения обоих атомов хлора в 23 на метоксигруппы под действием разработанной авторами ранее системы реагентов MeOH-CsF-Al2O3 при 90-95°C в течение 30-40 ч [24] не увенчались успехом. При этом наблюдалось замещение лишь одного из атомов хлора в положении 6 либо в положении 7 с образованием смеси 2-гидрокси-6-метокси-7-хлор- и 2-гидрокси-7-метокси-6-хлорнафтазаринов. Этот результат привел к необходимости защиты β-OH-группы перед нуклеофильным замещением атомов хлора, поскольку ионизация этой группы в условиях реакции оказывала ингибирующее действие на процесс замещения из-за сильного электронодонорного эффекта алкоксидной группы.
С этой целью авторами был разработан способ защиты β-OH-группы в субстрате 23 в виде O-метилового эфира действием йодистого метила в растворе в диметилформамиде в присутствии поташа (K2CO3). Выход эфира 24 составил при этом 91%.
Взаимодействие эфира 24 с системой реагентов MeOH-CsF-Al2O3 при 90-95°C дало триэфир спинохрома D 19 с выходом 80% при полной конверсии субстрата. Из смеси продуктов реакции методом колоночной хроматографии на SiO2 выделены также небольшие количества продуктов неполного замещения атомов хлора 25 (8% от теор.) и 26 (2%). Кипячение раствора триэфира 19 в 48%-ной бромистоводородной кислоте дало целевой спинохром D 1 с выходом 88%.
Таким образом, разработанный 5-стадийный способ синтеза спинохрома D 20→22→23→24→19→1 позволяет получать его из коммерчески доступных продуктов с суммарным выходом 54%. С учетом того, что 2,3-дихлорнафтазарин 22 является сейчас коммерчески доступным (Aldrich), суммарный выход спинохрома D в 4-стадийной конверсии 22→23→24→19→1 повышается до 58%.
Из трех описанных выше способов синтеза спинохрома D последний наиболее близок к заявляемому по строению промежуточных продуктов синтеза и по сути выполняемых операций, поэтому он выбран в качестве способа-прототипа.
Недостатки способа-прототипа заключаются в следующем:
1) Использование на стадии конверсии 2,3-дихлорнафтазарина 22 в 2-гидрокси-6,7-дихлорнафтазарин 23 большого объема агрессивной конц. серной кислоты. В этой реакции окисления на 3.0 г субстрата 22 используется 120 мл конц. H2SO4. По окончании этой реакции данный объем кислоты разбавляют 600 мл воды при охлаждении и продукт реакции извлекают путем экстракции большим объемом этилацетата (400 мл). Остающийся кислый маточный раствор (~22-26%-ный раствор серной кислоты) требует применения процедуры нейтрализации для обеспечения его безопасности.
2) Полученный в реакции окисления 2-гидрокси-6,7-дихлорнафтазарин 23 не может быть использован прямо для полного замещения атомов хлора на метоксигруппы. Предварительно необходимо защитить 2-гидроксигруппу в виде метилового эфира действием йодистого метила в растворе в диметилформамиде в присутствии поташа. При этом наряду с целевым продуктом O-метилирования 24 образуется небольшое количество продукта C-алкилирования (4%) - 2,5,8-тригидрокси-3-метил-6,7-дихлор-1,4-нафтохинона, который отделяется от продукта 24 хроматографией на колонке с SiO2.
Задачей изобретения явилась разработка способа получения спинохрома D, свободного от недостатков способа-прототипа.
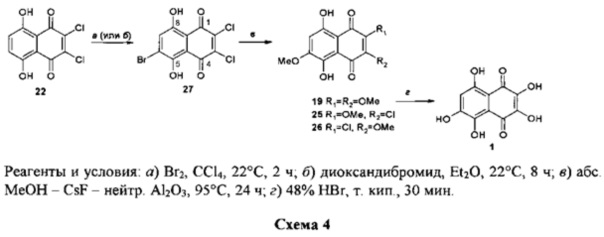
Задача решена способом получения спинохрома D, предусматривающим использование в качестве исходного соединения 2,3-дихлорнафтазарина, в котором согласно изобретению 2,3-дихлорнафтазарин бромируют под действием брома в четыреххлористом углероде или диоксандибромида в диэтиловом эфире, затем в образовавшемся 6-бром-2,3-дихлорнафтазарине замещают все галоидные атомы на метоксигруппы действием метанола, активированного фторидом цезия на поверхности оксида алюминия с получением 2,3,6-триметоксинафтазарина, который гидролизуют в целевой продукт кипячением в концентрированной бромистоводородной кислоте в течение 30 мин.
Суммарный выход спинохрома D составляет от 66-69% на три стадии 22→27→19→1.
Заявляемый способ представлен на схеме 4.
В данном способе получения спинохрома D используется стратегия, основанная на применении в качестве ключевого полупродукта синтеза триметилового эфира спинохрома D 19 тригалогензамещенного нафтазарина. Ранее эта стратегия в направленном синтезе спинохрома D не применялась.
Известен случай использования в качестве субстрата для получения триэфира 19 2,3,6-трихлорнафтазарина 29 [24] (схема 5).
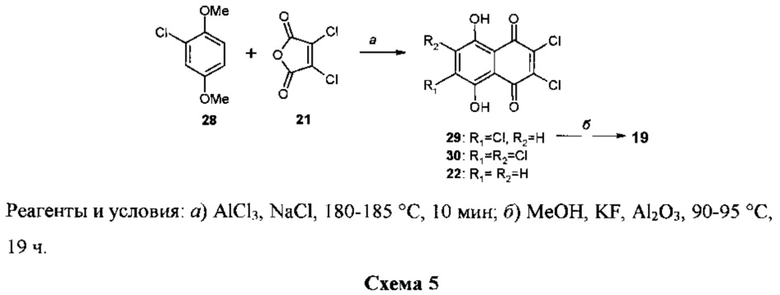
Соединение 29 получали ранее путем циклоацилирования диметилового эфира хлоргидрохинона 28 дихлормалеиновым ангидридом 21 в расплаве AlCl3-NaCl при 175-180°C в течение 3 мин [25]. Выход 29 составил 56%, а единственным побочным продуктом реакции явился дихлорнафтазарин 22 (9.5%). При использовании в качестве субстрата не диэфира 28, а свободного хлоргидрохинона и при проведении реакции в более жестких условиях (215-220°C, 5 мин) выход продукта 29 не превышал 39% [26]. О каких-либо побочных продуктах реакции в этой работе не сообщалось.
В результате проведенного авторами недавно исследования показано, что максимальный выход соединения 29 (61%) может быть достигнут при проведении этой реакции при 180-185°C в течение 10 мин [27]. Наряду с триэфиром 29 продуктами реакции являются также трихлорнафтазарин 30 (3%) и 2,3-дихлорнафтазарин 22 (10%). Эти побочные продукты образуются в результате диспропорционирования некоторой части исходного диэфира 28 на 2,3-дихлоргидрохинон и гидрохинон. Взаимодействие последних с дихлормалеиновым ангидридом 21 и приводит к появлению в реакционной смеси таких побочных продуктов, как 30 и 22. Необходимость хроматографической очистки 2,3,6-трихлорнафтазарина 29 от примесных соединений 30 и 22 заметно снижает возможности его использования в качестве субстрата для получения триметилового эфира спинохрома D 19. Конверсия субстрата 29 в триэфир 19 проводилась ранее действием системы реагентов MeOH-KF-Al2O3 при 90-95°C в течение 19 ч [24]. Выход триэфира 19 составил при этом 44%. О побочных продуктах реакции нуклеофильного замещения атомов хлора на метоксигруппы в субстрате 29 в работе [24] не сообщалось. Эта реакция была проведена авторами в иных условиях, когда в качестве катализатора использовался не KF, a CsF. Нагревание смеси субстрата 29 с системой реагентов MeOH-CsF-Al2O3 при 90-95°C в течение 32 ч дало смесь продуктов, из которой хроматографией на колонке с SiO2 были выделены триэфир 19 (выход 69%) и диэфиры 25 (8%) и 28 (11%). Конверсия субстрата 29 составила при этом 100%.
В заявляемом способе получения спинохрома D в качестве исходного соединения используют 2,3-дихлорнафтазарин 22, который может быть легко получен с выходом 95% путем циклоацилирования товарного диметилового эфира гидрохинона 20 дихлормалеиновым ангидридом 21 в условиях, описанных в способе-прототипе [22].
В настоящее время 2,3-дихлорнафтазарин 22 является коммерчески доступным продуктом (каталог фирмы «Aldrich» 2012-2014 года, стр. 923).
Бромирование субстрата 22 бромом в растворе в CCl4 или свежеприготовленным диоксандибромидом в растворе в диэтиловом эфире дает 6-бром-2,3-дихлорнафтазарин 27 с выходом 91-95%, тогда как 2,3,6-трихлорнафтазарин 29 может быть получен с максимальным выходом 61%, причем только в результате хроматографической очистки смеси продуктов реакции циклоацилирования субстрата 28 ангидридом 21.
Взаимодействие 6-бром-2,3-дихлорнафтазарина 27 с системой реагентов МеОН-CsF-Al2CO3 при 90-95°C в течение 24 ч приводит к образованию смеси продуктов, из которой хроматографией на колонке с SiO2 были выделены триэфир 19 (выход 82%) и диэфиры 25 (4%) и 26 (6%). Конверсия субстрата была количественной.
В способе-прототипе взаимодействие 2-метокси-6,7-дихлорнафтазарина 24 с системой реагентов MeOH-CsF-Al2O3 при 90-95°C протекало с полной конверсией субстрата в течение 30 ч. В результате хроматографического разделения смеси продуктов реакции были выделены триэфир 19 (выход 80%) и диэфиры 25 (8%) и 26 (2%).
По выходу целевого триэфира 19 эти процедуры сопоставимы, но в заявляемом способе полная конверсия субстрата в смесь продуктов 19, 25 и 26 достигается быстрее, чем в способе-прототипе (24 и 30 ч соответственно), что не является заведомо очевидным.
Конверсия триэфира 19 в целевой спинохром D осуществлялась путем кипячения его раствора в 48%-ной бромистоводородной кислоте в течение 30 мин. Выход спинохрома D на этой стадии составил 88%.
Разработанный способ получения спинохрома D лишен недостатков способа-прототипа.
Новизна предлагаемого метода синтеза спинохрома D заключается в применении нового подхода, в котором ключевую роль играет 6-бром-2,3-дихлорнафтазарин, ранее не применявшийся в синтезе спинохрома D и других пигментов морских ежей. Выше показано, что его использование в качестве субстрата на ключевой стадии нуклеофильного замещения галоидных атомов на метоксигруппы дает сопоставимый результат с 2-метокси-2,3-дихлорнафтазарином в качестве субстрата, используемом в способе-прототипе, но получение этого субстрата из 2,3-дихлорнафтазарина является гораздо более сложным и менее эффективным, чем конверсия 22 в субстрат 27.
Технический результат, обеспечиваемый изобретением, заключается в упрощении способа получения спинохрома D на стадии конверсии 2,3-дихлорнафтазарина в триметиловый эфир спинохрома D, в уменьшении числа стадий с четырех, как в способе-прототипе, до трех и в увеличении суммарного выхода целевого продукта с 58%, как в способе-прототипе, до 66-69%.
Сведения, подтверждающие возможность осуществления изобретения
Пример 1. Бромирование 2,3-дихлорнафтазарина 22 бромом в растворе в CCl4. Использованный в данном эксперименте 2,3-дихлорнафтазарин 22 был получен циклоацилированием 3.95 г (0.028 моль) диметилового эфира гидрохинона 20 9.5 г (0.057 моль) дихлормалеинового ангидрида 21 в условиях, описанных в способе-прототипе [22]. Выход составил 7.04 г (95% от теор.).
К перемешиваемому раствору 2,3-дихлорнафтазарина 22 (2.59 г, 10.0 ммоль) в 100 мл CCl4 прибавляют по каплям в течение 5 мин раствор брома (0.55 мл, 11.0 ммоль) в 15 мл CCl4 и реакционную смесь перемешивают при комнатной температуре в течение 2 ч, облучая светом УФ-лампы. Растворитель удаляют при пониженном давлении, остаток растворяют в 40 мл этанола и раствор кипятят в течение 5 мин. Смесь охлаждают до комнатной температуры и выдерживают при 5°C в течение ночи. Осадок отделяют на фильтре, высушивают и получают 0.308 г (91%) 6-бром-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона 27 в виде бордовых игл, т.пл. 197-199°C. Rf: 0.49 (н-гексан-бензол, 1:4 (по объему)). ИК-спектр (CDCl3, ν, см-1): 3450-2200 (α-ОН), 2965, 2925, 2870, 2848, 1625 (С=С), 1604 (С=O, С=С), 1567 (С=С), 1433, 1399, 1309, 1281, 1270, 1218, 1185, 1138, 1016. Спектр ЯМР 1Н (300 МГц, CDCl3, δ, м.д.): 7.68 (с, 1Н, Н-7), 12.28 (с, 1H, С(8)-ОН), 12.86 (с, 1H, С(5)-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д): 110.0 (С-8а), 110.3 (С-4а), 127.9 (С-7), 134.2 (С-6), 141.4 (С-3), 142.4 (С-2), 159.9 (С-5), 162.4 (С-8), 174.7 (С-1), 175.4 (С-4). Масс-спектр (ЭУ, 70 эВ, m/z, Iотн (%)): 337/339/341/343 [М+Н]+ (11), 336/338/340/342 [М]+ (100), 302/304/306 [М+Н-Cl]+ (7), 301/303/305 [М-Cl]+ (32), 266/268 [М-2Cl]+ (3), 258/260/262 [М+Н-Br]+ (8), 273/275/277 [М-Cl-СО]+ (12), 257/259/261 [M-Br]+ (45), 222/224 [М-Br-Cl]+ (4). Найдено (%): С, 35.38; Н, 0.81; Br, 23.80; Cl, 21.11. C10H3BrCl2O4. Вычислено (%): С, 35.54; Н, 0.89; Br, 23.64; Cl, 20.98.
Пример 2. Бромирование 2,3-дихлорнафтазарина 22 диоксандибромидом в растворе в диэтиловом эфире. Раствор 2,3-дихлорнафтазарина 22 (2.59 г, 10.0 ммоль) и свежеприготовленного диоксандибромида (2.48 г, 10.0 ммоль) [28] в 100 мл диэтилового эфира перемешивают при комнатной температуре в течение 8 ч. Реакционную смесь промывают 10%-ным раствором Na2S2O3⋅5H2O (3×25 мл), затем водой (3×25 мл) и высушивают над Na2SO4. Удаление растворителя при пониженном давлении дает 0.322 г (95%) 6-бром-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона 27, полностью идентичного описанному выше.
Пример 3. Нуклеофильное замещение галоидных атомов на метоксигруппы в 6-бром-5,8-дигидрокси-2,3-дихлор-1,4-нафтохиноне 27. Смесь хорошо высушенного субстрата 27 (1.69 г, 5.0 ммоль), безвод. CsF (3.80 г, 25.0 ммоль), активного нейтрального Al2O3 (4.46 г, 43.75 ммоль, 150 меш фирмы «Aldrich») и абс. МеОН (200 мл) перемешивают в герметичном сосуде при 90-95°C в течение 24 ч. После охлаждения до комнатной температуры адсорбент отделяют фильтрованием и последовательно промывают ацетоном (100 мл) и 10%-ной HCl (3 мл). Объединенные фильтраты упаривают при пониженном давлении, остаток разбавляют водой (200 мл) и экстрагируют CHCl3 (5×50 мл). Органический экстракт промывают последовательно водой (3×20 мл) и насыщ. раствором NaCl (20 мл), высушивают над безвод. Na2SO4 и концентрируют досуха при пониженном давлении. Остаток хроматографируют на колонке с SiO2 (G-60, 2.5×80 см), заполненной н-гексаном. Элюция смесью гексан-ацетон, 12:1 (по объему) дает 5,8-дигидрокси-2,7-диметокси-6-хлор-1,4-нафтохинон 26 (0.086 г, 6%) в виде красных пластинок, т.пл. 199-201°C. Rf: 0.42 (н-гексан-ацетон, 2:1 (по объему)). ИК-спектр (CDCl3, ν, см-1): 3400-2300 (α-ОН), 2940, 2926, 2852, 1607 (С=С), 1602 (С=O, С=С), 1573 (С=С), 1559 (C=С), 1472, 1445, 1409, 1307, 1271, 1160, 1127. Спектр ЯМР 1Н (300 МГц, CDCl3, δ, м.д.): 3.97 (с, 3H, С(2)-ОМе), 4.18 (с, 3H, С(7)-ОМе), 6.35 (с, 1Н, Н-3), 12.68 (с, 1H, С(8)-ОН), 13.26 (с, 1Н, С(5)-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 57.1 (С(2)-ОМе), 61.8 (С(7)-ОМе), 105.7 (С-4а), 109.1 (С-3), 110.8 (С-8а), 128.1 (С-6), 154.5 (С-7), 159.9 (С-2), 164.1 (С-8), 171.5 (С-4), 172.7 (С-5), 177.7 (С-1). Масс-спектр (ЭУ, 70 эВ, m/z, Iотн (%)): 284/286 [М]+ (100), 269/271 [М-СН3]+ (13), 266/268 [М-H2O]+ (18), 254/256 [М-СН2О]+ (10), 241/243 [М-СН3-СО]+ (15), 238/240 [М-H2O-СО]+ (14). Найдено (%): С, 50.73; Н, 3.15; Cl, 12.58. C12H9ClO6. Вычислено (%): С, 50.63; Н, 3.19; Сl, 12.45.
Элюция смесью гексан-ацетон, 10:1 (по объему) дает 5,8-дигидрокси-2,6-диметокси-7-хлор-1,4-нафтохинон 25 (0.057 г, 4%) в виде красных пластинок, т.пл. 195-197°C. Rf: 0.41 (н-гексан-ацетон, 2:1 (по объему)). ИК-спектр (CDCl3, ν, см-1): 3400-2250 (α-ОН), 2937, 2921, 2850, 1611 (С=C), 1604 (С=O, С=С), 1579, 1562, 1477, 1458, 1439, 1412, 1300, 1273, 1163, 1125. Спектр ЯМР 1Н (300 МГц, CDCl3, δ, м.д.): 3.97 (с, 3H, С(2)-ОМе), 4.26 (с, 3H, С(6)-ОМе), 6.34 (с, 1H, Н-3), 13.02 (с, 1Н, С(8)-ОН), 13.09 (с, 1Н, С(5)-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д.): 57.1 (С(2)-ОМе), 62.1 (С(6)-ОМе, 107.8 (С-4а), 108.4 (С-3), 108.7 (С-8а), 125.0 (С-7), 156.4 (С-6), 160.3 (С-2), 163.6 (С-5), 167.3 (С-4), 168.7 (С-8), 177.5 (С-1). Масс-спектр (ЭУ, 70 эВ, m/z, Iотн (%)): 284/286 [М]+ (100), 269/271 [М-СН3]+ (6), 266/268 [М-H2O]+ (36), 255/257 [М-СНО]+ (26), 241/243 [М-СН3-СО]+ (18), 213/215 [М-СН3-2СО]+ (17). Найдено (%): С, 50.76; Н, 3.11; Cl, 12.55. C12H9ClO6. Вычислено (%): С, 50.63; Н, 3.19; Сl, 12.45.
Элюция смесью н-гексан-ацетон, 5:1 (по объему) дает 5,8-дигидрокси-2,3,6-триметокси-1,4-нафтохинон 19 (1.148 г, 82%) в виде красно-коричневых игл, т.пл. 169-171°C (лит. [20, 21] 161-162°C; [29] 176-177°C). Rf: 0.40 (н-гексан-ацетон, 2:1 (по объему)). ИК-спектр (CDCl3, ν, см-1): 3400-2240 (α-ОН), 2944, 2928, 2849, 1610 (С=С), 1604 (С=O, С=С), 1575 (С=С), 1555 (С=С), 1451, 1408, 1366, 1292, 1210, 1157, 1086. Спектр ЯМР 1Н (300 МГц, CDCl3, δ, м.д.): 3.96 (с, 3H, С(6)-ОМе), 4.07 (с, 3H, С(3)-ОМе), 4.15 (с, 3H, С(2)-ОМе), 6.41 (с, 1H, Н-7), 12.93 (с, 1Н, С(5)-ОН), 13.04 (с, 1H, С(8)-ОН). Спектр ЯМР 13С (75 МГц, CDCl3, δ, м.д): 56.7 (С(6)-ОМе), 61.6 (С(3)-ОМе), 61.7 (С(2)-ОМе), 105.0 (С-8а), 107.5 (С-7), 109.4 (С-4а), 146.9 (С-3), 149.5 (С-2), 159.2 (С-6), 161.4 (С-5), 170.2 (С-8), 172.3 (С-1), 174.7 (С-4). Масс-спектр (ЭУ, 70 эВ, m/z, Iотн (%)): 281 [М+Н]+ (15), 280 [М]+ (100), 266 [М+Н-СН3]+ (6), 265 [М-СН3]+ (44), 263 [М+Н-H2O]+ (4), 262 [М-H2O]+ (16), 251 [М-СНО]+ (13), 247 [М-СН3-H2O]+ (11), 237 [М-СН3-СО]+ (11), 234 [М-H2O-СО]+ (22), 219 [М-H2O-СО-СН3]+ (14).
Пример 4. Конверсия триметилового эфира спинохрома D 19 в спинохром D 1. Смесь триэфира 19 (1.12 г, 4.0 ммоль) и конц. бромистоводородной кислоты (48%, 150 мл) кипятят в течение 30 мин, охлаждают до комнатной температуры, разбавляют водой (200 мл) и выдерживают при 5°C в течение ночи. Осадок отфильтровывают, промывают водой (4×10 мл) и высушивают. Получено 0.838 г (88%) 2,3,5,6,8-пентагидрокси-1,4-нафтохинона 1 (спинохрома D) в виде красно-коричневых игл, возгоняется без плавления при 282-288°C (лит. [3] возгоняется при 285-290°C; [20, 21] возгоняется при 280-290°C). Rf: 0.30 (н-гексан-ацетон, 2:1 (по объему)). Спектр ЯМР 1Н (300 МГц, DMSO-d6, δ, м.д): 6.48 (с, 1H, Н-3), 10.10 (уш.с, 1Н, С(7)-ОН), 10.41 (уш.с, 1H, С(6)-ОН), 11.46 (уш.с, 1Н, С(2)-ОН), 12.65 (с, 1H, С(5)-ОН), 12.70 (уш.с, 1H, С(8)-ОН). В растворе спинохрома D в DMSO-d6 доминирует таутомерная форма с 2,6,7-расположением β-OH-групп. Спектр ЯМР 13С (75 МГц, DMSO-d6, δ, м.д.): 102.0 (С-8а), 108.4 (С-4а), 109.3 (С-3), 140.1 (С-7), 142.3 (С-6), 151.1 (С-2), 157.1 (С-8), 161.0 (С-5), 179.1 (С-4), 181.5 (С-1). Масс-спектр (ЭУ, 70 эВ, m/z, Iотн. (%)): 238 [М]+ (100), 210 [М-СО]+ (65), 181 [М-СО-СНО]+ (14), 168 [М-СО-СН2СО]+ (15), 153 (8), 149 (9), 125 (11), 111 (17), 109 (10), 97 (21), 95 (16), 85 (18), 83 (21), 81 (15), 71 (29), 69 (24), 67 (10), 57 (37), 55 (30), 45 (32), 44 (29), 42 (18).
Источники информации
1. (a) Thomson R.H. Naturally occurring quinones, 2nd Ed, 1971, Academic Press, London, New York; (6) 3rd Ed, 1987, Chapman & Hall, London, New York; (в) 4th Ed, 1997, Chapman & Hall; London, New York.
2. Kuroda С., Ohshima H. // Proc. Imp. Acad. (Tokyo), 1940, V. 16, P. 214-219.
3. Anderson H.A, Smith J., Thomson R.H. // J. Chem. Soc, 1965, P. 2141-2144.
4. Zhou D.Y., Qin L, Zhu B.W., Wang X.D., Tan H., Yang J.F., Li D.M., Dong X.P., Wu H.T., Sun L.M., Li X.L., Murata Y. // Food Chemistry, 2011, V. 129, P. 1591-1597.
5. Utkina N.K., Pokhilo N.D. // Nat. Prod. Commun, 2012, V. 7, P. 901-904.
6. Богуславская Л.В., Храпова Н.Г., Максимов О.Б. // Изв. АН СССР. Сер. хим., 1985, №7, С. 1471-1476.
7. Лебедев А.В., Левицкая Е.Л., Тихонова Е.В., Иванова M.B. // Биохимия, 2001, Т. 66, С. 885-893.
8. Hatate Н., Murata Н., Наmа Y., Tanaka R., Suzuki N. // Fisheries Science, 2002, V. 68, P. 1641-1642.
9. Lebedev A.V., Ivanova M.V., Levitsky D.O. // Life Sciences, 2005, V. 76, P. 863-875.
10. Lebedev A.V., Ivanova M.V., Levitsky D.O. // Hemoglobin, 2008, V. 32, P. 165-179.
11. Kuwahara R., Hatate H., Yuki Т., Murata H, Tanaka R., Hama Y. // LWT - Food Science and Technology, 2009, V. 42, P. 1296-1300.
12. Service M., Wardlaw А.С.// Comp. Biochem. Physiol., 1984, V. 79B, P. 161-165.
13. Sime A.A.T. Biocidal compositions comprising polyhydroxynaphthoquinones. // GB Patent 2159056 (1985); Chem. Abstr., 1986, V. 104, 83795.
14. Стехова С.И., Шенцова E.Б., Кольцова Е.А, Кулеш Н.И. // Антибиотики и химиотерапия, 1988, Т. 33, С. 831-833.
15. Pozharitskaya O.N., Shikov A.N., Makarova M.N., Ivanova S.A, Kosman V.M., Makarov V.G., Bazgier V., Berka K., Otyepka M., Ulrichova J. // Planta Medica, 2013, V. 79, P. 1698-1704.
16. Артюков А.А., Попов A.M., Цыбульский А.В., Кривошапко О.H., Полякова Н.В. // Биомед. химия, 2012, Т. 58, С. 281-290.
17. Мищенко Н.П., Федореев С.А., Багирова В.Л. // Хим.-фарм. журн., 2003, Т. 37, №1, С. 49-53.
18. Chang С.W.J, Moore R.Е., Scheuer P.J. // J. Am. Chem. Soc, 1964, V. 86, P. 2959-2961.
19. Пелагеев Д.Н., Панченко М.Н., Похило Н.Д., Ануфриев В.Ф. // Изв. АН, Сер. хим., 2010, С. 1439-1443.
20. Singh I., Moore R.E., Chang C.W.J, Scheuer P.J. // J. Am. Chem.. Soc., 1965, V. 87, P. 4023-4024.
21. Singh I., Moore R.E., Chang C.W.J., Ogata R.Т., Scheuer P.J. // Tetrahedron, 1968, V. 24, P. 2969-2978.
22. Баланева H.H., Шестак О.П., Ануфриев В.Ф., Новиков В.Л. Синтез спинохрома D, метаболита различных видов морских ежей // Химия природ. соединений, 2016, №2, С 187-191.
23. Huot R., Brassard P. // Can. J. Chem., 1974, V. 52, P. 838-842.
24. Anufriev V.Ph., Novikov V.L. // Tetrahedron Lett., 1995, V. 36, P. 2515-2518.
25. Ануфриев В.Ф., Баланева Н.Н., Чижова А.Я., Новиков В.Л.. Еляков Г.Б. Ацилирование резорцинов, п-замещенных фенолов, гидрохинонов, их ацетатов и метиловых эфиров дихлормалеиновым ангидридом. // Владивосток, 1988, 23 с.; деп. в ВИНИТИ 07.06.88, 6275-В 88, РЖХим., 1988, Т. 19, №23 (1), 23Ж154.
26. Masuda K. Preparation of 2.3.6-trichloronaphthazarin and preparation of antitumor agents. // Japan Patent JP 07215906 A, Aug 15, 1995; Chem. Abstr., 1995, V. 124, 86608.
27. Новиков В.Л., Баланева Н.Н., Шестак О.П., Ануфриев В.Ф., Глазунов В.П. // Изв. АН. Сер. хим., 2016, №4, С. 993-1003.
28. Яновская Л.А., Терентьев А.П., Беленький Л.Н. // Журн. общ. химии, 1952, Т. 22, С. 1594-1601.
29. Masuda K., Funayama S., Komiyama K., Umezawa I. // J. Nat. Prod., 1987, V. 50, P. 958-960.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СПИНОХРОМА Е | 2014 |
|
RU2561280C1 |
| Способ получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) и промежуточные соединения, используемые в этом способе | 2016 |
|
RU2632668C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТАХИНОНОВ | 1990 |
|
RU2022959C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6,7-ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТОХИНОНОВ (СПИНАЗАРИНОВ) | 2012 |
|
RU2478607C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,5,8-ТРИГИДРОКСИ-6,7-ДИХЛОР-3-ЭТИЛ-1,4 НАФТОХИНОНА | 2001 |
|
RU2193550C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,6-7-ТРИМЕТОКСИ-3-ЭТИЛ-1,4-НАФТОХИНОНА | 2005 |
|
RU2277083C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,7-ТРИОКСИЮГЛОНА (СПИНОХРОМА В) | 2014 |
|
RU2568604C1 |
| СПОСОБ ПОЛУЧЕНИЯ СПИНОХРОМА А И БЕЛКА МОРСКИХ ЕЖЕЙ, ВЗАИМОДЕЙСТВУЮЩЕГО С ПОЛИГИДРОКСИНАФТОХИНОНОМ | 2008 |
|
RU2362573C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3,6,7-ТЕТРАГИДРОКСИНАФТАЗАРИНА | 2009 |
|
RU2411939C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,3,6-ТРИМЕТОКСИ-7-ЭТИЛ-1,4-НАФТОХИНОНА | 2001 |
|
RU2203265C2 |
Изобретение относится к способу получения спинохрома D (2,3,5,6,8-пентагидрокси-1,4-нафтохинона) - одного из нафтохиноидных пигментов морских ежей, обладающего высокой антиоксидантной и антирадикальной активностью и перспективного для использования в кардиологии и офтальмологии. Способ заключается в том, что 2,3-дихлорнафтазарин бромируют под действием брома в четыреххлористом углероде или диоксандибромида в диэтиловом эфире с получением 6-бром-2,3-дихлорнафтазарина, в котором замещают галоидные атомы на метоксигруппы действием метанола, активированного фторидом цезия на поверхности оксида алюминия, с получением 2,3,6-триметоксинафтазарина, который гидролизуют в целевой продукт кипячением в концентрированной бромистоводородной кислоте в течение 30 мин. Предлагаемый способ позволяет получить целевой продукт с высоким выходом при использовании простой и безопасной технологии. 4 пр.
Способ получения спинохрома D (2,3,5,6,8-пентагидрокси-1,4-нафтохинона), предусматривающий использование в качестве исходного соединения 2,3-дихлорнафтазарина, отличающийся тем, что 2,3-дихлорнафтазарин бромируют под действием брома в четыреххлористом углероде или диоксандибромида в диэтиловом эфире с получением 6-бром-2,3-дихлорнафтазарина, в котором замещают галоидные атомы на метоксигруппы действием метанола, активированного фторидом цезия на поверхности оксида алюминия, с получением 2,3,6-триметоксинафтазарина, который гидролизуют в целевой продукт кипячением в концентрированной бромистоводородной кислоте в течение 30 мин.
| Баланева Н.Н | |||
| и др | |||
| Синтез спинохрома D, метаболита различных видов морских ежей | |||
| Химия природных соединений, 2016, N 2, 187-191 | |||
| I.Singh et al, Spinochrome synthesis | |||
| Tetrahedron, 1968, 24(7), 2969-2978 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТАХИНОНОВ | 1990 |
|
RU2022959C1 |
| US 20130102650 A1, 25.04.2013 | |||
| Новиков В.Л | |||
| Основные направления исследований и достижения в области органического синтеза природных соединений в ТИБОХ ДВО РАН | |||
| Вестник ДВО РАН, 2014, N 1, 22-52. | |||

