Изобретение относится к органической химии, конкретно к способу получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D), который может найти применение в медицине и косметологии. Изобретение также касается промежуточных соединений, используемых в этом способе.
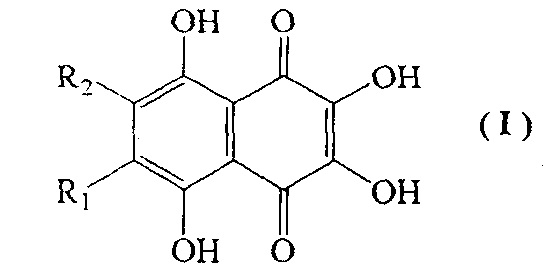
Полигидроксилированные производные нафтазарина (5,8-дигидрокси-1,4-нафтохинона) - природные соединения общей формулы (I), выделяемые из растений, грибов, микроорганизмов и морских животных, образуют группу соединений с практически полезными свойствами [Thomson R.Н. Naturally occurring quinones. London-New York: Chapman and Hall, 1987. 3rd ed. 732 p.; Blackie Academic and Professional, London - New York, 1997, 4th ed., 746 р.]. Гидроксинафтазарины проявляют выраженные антигрибковые и антимикробные свойства [GB 2159056 А, 1984].
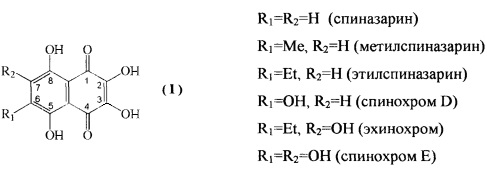
Специфической особенностью производных нафтазарина является легкость таутомерных переходов между хиноидной и бензеноидной частями нафтазаринового ядра, которая обуславливает реакционную спрособность атомов (групп), присоединенных к различным кольцам нафтазаринового ядра, и появление у них антиокислительной активности [Изв. АН СССР, сер. хим. 1985, №7, С. 1471-1476]. Описано применение метилспиназарина в качестве средства для лечения нервных расстройств и снижения кровяного давления [JP 7399389, 1977]. Предложено использовать полигидроксинафтохиноны, а также их хлор- и алкоксипроизводные в качестве средств для окраски человеческих волос [GB 2110723А, 1982, GB 2119411 А, 1983]. Примером биологически-активных полигидроксинафтазаринов является эхинохром - наиболее доступный пигмент морских ежей, соединение формулы (I), с R1=Et, R2=OH, на основе которого создан лекарственный препарат «Гистохром™», применяемый для лечений ишемической болезни сердца, инфаркта миокарда, травм и ожогов глаз [Хим. - Фарм. журн. 2003. Т. 37. №1. С. 48].
Установлено, что эхинохром эффективно защищает кардиомиоциты от побочных токсических эффектов противоопухолевых лекарственных препаратов, вызывающих повышенную продукцию активных форм кислорода и снижение митохондриального мембранного потенциала в клетках сердца [Marine Drugs 2014. V. 12. С. 2922-2936]. Эхинохром увеличивает массу митохондрий клеток и процесс окислительного фосфорилирования [Marine Drugs 2014. V. 12. С. 4602-4615], что может быть клинически полезным при лечении различных митохондриальных дисфункций. Как ингибитор ацетилхолинэстеразы, эхинохром перспективен в качестве средства для лечения нервно-мышечных расстройств, болезней Альцгеймера и Паркинсона [Marine Drugs 2014. V. 12. С. 3560-3573].
Высокая эффективность эхинохрома и разнообразные направления его использования в клинической практике делают перспективным поиск в ряду полигидроксинафтазаринов новых антиоксидантов - аналогов эхинохрома.
Наиболее близким к эхинохрому по строению хиноидного ядра, содержащего пять β-гидроксильных групп, отвечающих за антиоксидантные свойства и биологическую активность, является спинохром D, соединение формулы I (R1=OH, R2=Н). Наличие свободного положения в бензеноидной части позволяет присоединять разнообразные заместители и делает спинохром D привлекательным субстратом для синтеза новых аналогов эхинохрома с полезными свойствами.
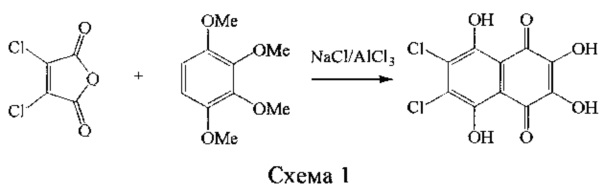
Известны способы получения 2,3-дигидрокси-6,7-замещенных нафтазаринов, в которых исходят из метоксипроизводных 1,2,3,4-тетрагидроксибензола и производных малеинового ангидрида (схема 1) [Tetrahedron 1968, V. 24, Р. 2969-2978; Austr. J. Chem. 1987, V. 40, Р. 119-1120]
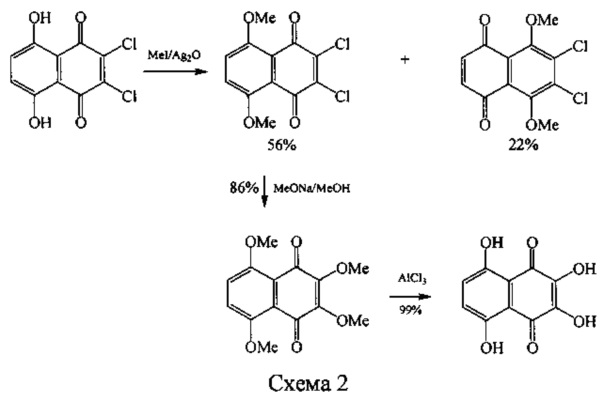
В другом варианте исходят из доступного 2,3-дихлорнафтазарина, при этом для облегчения замещения атомов хлора и блокирования таутомерных переходов в нафтазариновом ядре применяют предварительное метилирование α-гидроксильных групп нафтазарина (схема 2) [Can. J Chem. 1974, V. 54. Р. 838-842]
Однако в упомянутых методах получения спиназаринов использование труднодоступных метоксипроизводных 1,2,3,4-тетрагидроксибензола, ограниченность ассортимента доступных замещенных производных малеинового ангидрида (схема 1) либо многостадийность синтеза (схема 2) не позволяют получать спиназарины с различными заместителями R1 и R2 (формула I).
Среди известных способов получения 6,7-замещенных 2,3-дигидрокси-нафтазаринов предпочтительными, с точки зрения промышленного применения, являются способы, которые включают прямое замещение атомов хлора в доступных 6,7-замещенных 2,3-дихлорнафтазаринах на алкоксильные радикалы с последующим деалкилированием полученных эфиров под действием бромоводородной кислоты или безводного хлорида алюминия [RU 2022959, 15.11.1994; Tetrah. Lett. 1995, V. 36. P. 2515-2518].
В качестве прототипа нами выбран способ получения спинохрома D, описанный в патенте [RU 2022959, 15.11.1994] и представленный на схеме 3.
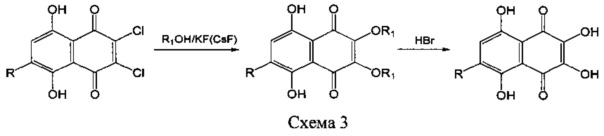
Согласно этому способу спинохром D получают замещением атомов хлора на алкоксигруппы в 2,3-дихлорнафтазаринах (схема 3), где R=OMe, Cl, ОН, под действием системы реагентов: метилцеллозольв (этилцеллозольв) - KF(CsF) - оксид алюминия, при температуре кипения растворителя с последующим О-дезалкилированием образующихся простых эфиров метилцеллозольва (этилцеллозольва) - 2,3-ди-О-алкокси-и 2,3,6-три-О-алкоксизамещенных производных нафтазарина под действием концентрированной бромоводородной кислоты при повышенной температуре.
Недостатками упомянутого способа получения спинохрома D являются: использование в качестве стартового соединения малорастворимого 6-метокси-2,3-дихлорнафтазарина, который получают из 6-гидрокси-2,3-дихлорнафтазарина путем его метилирования токсичным диазометаном, применение дорогостоящих фторидов щелочных металлов, проведение замещения в сухих высококипящих растворителях (метил- или этилцеллозольва); необходимость в тщательном высушивании реагентов и аппаратуры, проведение процесса в атмосфере азота при 125-135°С (температуре кипения растворителей); проведение деалкилирования под действием кипящей концентрированной бромоводородной кислоты; недостаточно высокий выход продукта.
Так, для 6-метокси-2,3-дихлорнафтазарина выход 2,3,6-триалкоксинафтазарина составил на стадии замещения 82%, а для 6-гидрокси-2,3-дихлорнафтазарина выход 2,3-диалкоксинафтазарина составил 45%. Последующая стадия исчерпывающего О-дезалкилирования под действием кипящей концентрированной бромоводородной кислоты протекала с выходами 48-62%, и суммарный выход спинохрома D на две стадии получения по этому способу составил 28-39%. Следует отметить, что использование на завершающей стадии О-дезалкилирования бромистоводородной кислоты приводит к загрязнению целевого спинохрома D токсичными галогеннафтазаринами, неизбежно образующимися в ходе конкурентной реакции замещения алкоксильных радикалов на атомы брома.
Технический результат, обеспечиваемый предлагаемым изобретением, заключается в получении 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) без примесей токсичных галогеннафтазаринов с более высоким выходом (40-51%), чем в способе прототипе (28-39%), что стало возможным в результате использования новых хорошо растворимых промежуточных соединений, позволяющих проводить процесс в мягких условиях, эффективно удалять защитную алкоксигруппу на завершающей стадии и получать спинохром D и новые промежуточные соединения с использованием доступных и дешевых реагентов.
Настоящее изобретение относится к способу получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D), в котором исходят из доступного соединения формулы II
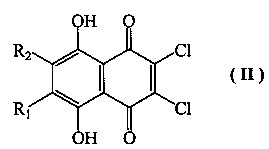
где R1=OH и R2=H,
причем способ включает следующие стадии:
a) взаимодействие соединения формулы II с первичными неразветвленными спиртами С3-С5 при катализе серной или метансульфокислотой, с удалением образующейся при этом воды азеотропной отгонкой спирта С3-С5, с получением 6-алкоксизамещенных соединений формулы II, где R1=н-пропил-О- и R2=H, R1=н-бутил-О- и R2=H, R1=н-амил-О- и R2=Н;
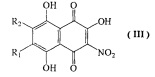
b) взаимодействие 6-алкоксизамещенных соединений формулы II, где R1 и R2 имеют указанные выше значения с 4-, 6 кратным мольным избытком азотистокислого натрия в смеси ацетон-этанол при температуре кипения с обратным холодильником, с получением соединений формулы III в виде смеси изомеров
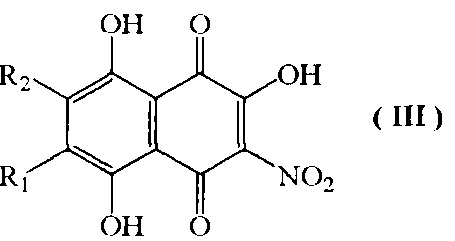
где R1=H, a R2=н-пропил-О-, н-бутил-О-, н-амил-О-;
R2=H, а R1=н-пропил-О-, н-бутил-О-, н-амил-О-;
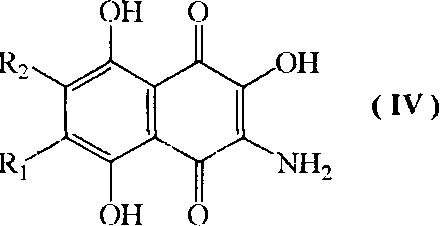
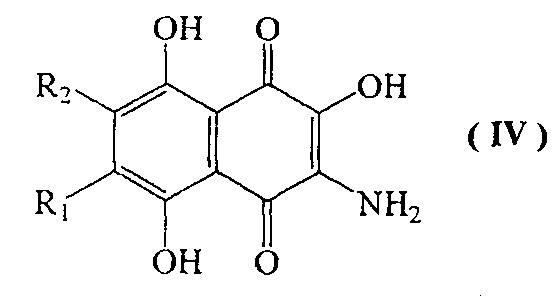
с) взаимодействие соединения формулы III, где R1 и R2 имеют указанные выше значения, с восстановителем с образованием соединения формулы (IV)
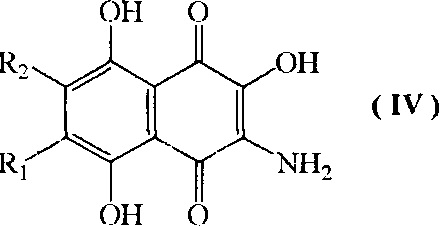
где R1=H, a R2=н-пропил-О-, н-бутил-О-, н-амил-О-;
R2=H, a R1=н-пропил-О-, н-бутил-О-, н-амил-О-;
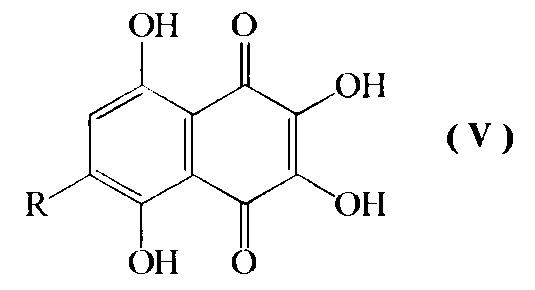
d) превращение соединения формулы IV путем кислотно-катализируемого гидролиза в смеси диметилсульфоксид-муравьиная кислота-серная кислота-вода при температуре кипения с обратным холодильником в 6-алкоксипроизводное спинохрома D формулы V
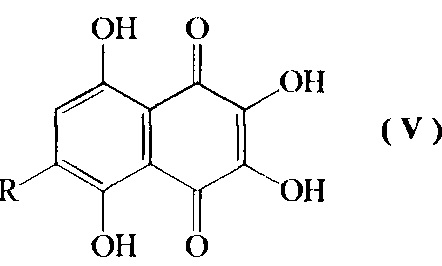
где R=н-пропил-О-, н-бутил-О-, н-амил-О-;
е) превращение соединения V в целевой продукт - спинохром D - путем кислотного гидролиза при температуре кипения смеси муравьиная кислота-метансульфокислота-вода с обратным холодильником.
Промежуточные соединения II, III, IV являются новыми и получены авторами впервые.
Изобретение относится также к промежуточным соединениям формулы V
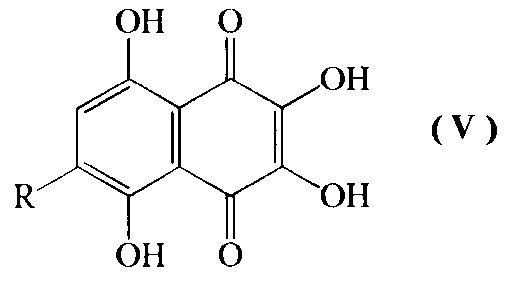
где R=н-пропил-O-, н-бутил-О-, н-амил-О-.
Промежуточные соединения формулы V в доступной патентной и научно-технической литературе не обнаружены.
Известна реакция 6,7-замещенных 2,3-дихлорнафтазаринов формулы II, где R1 и R2 представляют собой одновременно Н, Me, Cl или R1=H, a R2=Me, Et, трет-Bu, Cl, ОМе, OEt; R1=Me, a R2=Cl, ОМе, OEt, ОН; R1=Et, a R2=Cl, ОМе, ОН с нитритом натрия с получением соответствующих соединений формулы III, где R1 и R2 имеют вышеупомянутые значения [RU 2437870, 20.07.2011]. Однако данный способ не охватывает получение спинохрома D, что обусловлено специфическими свойствами исходного дихлорнафтазарина формулы II, где R1=OH и R2=H. Данный хинон может быть вовлечен во взаимодействие с NaNO2 только после защиты β-гидроксильной группы 6-гидрокси-2,3-дихлорнафтазарина путем его превращения в 6-метоксипроизводное нафтазарина формулы II, где R1=OMe и R2=H.
Вследствие специфических особенностей строения β-гидроксинафтазарины, не имеющие заместителей в соседнем положении, не алкилируются галоидными алкилами, алкилсульфатами и ортоэфирами [Synth Commun 1997, V. 27, 119-126]. Единственным известным на данный момент эффективным препаративным способом защиты β-гидроксидихлорнафтазарина формулы II, где R1=OH и R2=H, является метилирование токсичным диазометаном.
Предложенное в настоящей заявке алкилирование β-гидроксидихлорнафтазарина первичными неразветвленными спиртами С3-С5 при катализе неорганическими/органическими кислотами с удалением образующейся при этом воды азеотропной отгонкой алканола С3-С5 или улавливанием воды осушителем в аппарате Сокслета протекает с высокими выходами, дает хорошо растворимые промежуточные соединения, которые могут быть легко очищены кристаллизацией и хроматографическими методами. Последующее восстановление нитрогруппы во вновь полученных 2-гидрокси-3-нитронафтазаринах позволяет быстро и с высокими выходами синтезировать новую группу аминопроизводных нафтазарина формулы IV, которые обладают более высокой растворимостью в сравнении с известным аминогидроксинафтазарином формулы IV, где R1=OMe, R2=H, что существенно упрощает экстракцию, хроматографическое выделение и кристаллизацию продуктов.
Для замещения обоих атомов хлора, находящихся во втором и третьем положениях нафтазаринового ядра соединений общей формулы II, на стадии b) в качестве реакционной среды предпочтительно использовать смесь этанола и ацетона, предпочтительно при температуре кипения с обратным холодильником.
На стадии с) соединение формулы III восстанавливают под действием подходящего восстановителя, например Na2S2O4 или Na2S, в соединение формулы IV. На стадии d) соединение формулы IV превращают в соединение формулы V путем кипячения в смесях водных растворов минеральных и органических кислот.
Для ускорения конверсии соединения IV в соединение V в реакционную среду добавляют органический растворитель, не снижающий температуру кипения реакционной массы, например диметилсульфоксид. Конверсию предпочтительно проводить в смеси муравьиная кислота-вода-диметилсульфоксид-серная кислота при температуре кипения с обратным холодильником.
На стадии е) гидролиз соединения формулы V, проводимый в смеси муравьиная кислота-метансульфокислота-вода при температуре кипения с обратным холодильником, дает спинохром D - целевое соединение формулы I, в котором R1=OH, a R2=H.
Изобретение иллюстрируется примерами конкретного выполнения.
ПРИМЕР 1. Получение 5,8-дигидрокси-6-н-пропокси-2,3-дихлор-1,4-нафтохинона
Смесь 274 мг (1.00 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 50 мл н-пропанола и 0.10 мл 90% серной кислоты осторожно кипятят 20 ч с отгонкой ~10 мл н-пропанола до полной конверсии исходного хинона. Реакционную смесь упаривают при пониженном давлении до маслообразного состояния, остаток растворяют в 30 мл толуола, промывают водой 3×10 мл, органический слой упаривают без осушки, остаток растворяют при нагревании в 5 мл хлороформа, добавляют 10 мл этанола и выдерживают 2 ч при -18°С. Выпавшие кристаллы отделяют, промывают холодным этанолом, высушивают в вакууме и получают 242 мг (89%) соединения формулы II, в котором R1=н-пропил-O, a R2=Н (Rƒ0.86, гексан-бензол-ацетон, 2:1:1). Красные иглы, т. пл. 202-204°С (хлороформ-этанол).
ИК-спектр (CHCl3, v/см-1): 2972, 2942, 2883, 1615, 1600, 1558, 1455, 1404, 1352, 1303, 1271, 1248, 1191. Спектр ЯМР 1Н (500 МГц, CDCl3, δ, м.д.): 1.09 (т, J=7.4 Гц, 3Н, -СН2СН3), 1.95 (м, 2Н, -СН2СН3), 4.03 (т, J=6.6 Гц, 2Н, -ОСН2СН2), 6.24 (с, 1Н, ArH), 12.78 (с, 1H, α-ОН), 13.29 (с, 1H, α-ОН). Спектр ЯМР 13С (125 МГц, ДМСО-d6, δ, м.д): 10.24, 21.60, 70.79, 102.33, 106.73, 108.97, 140.56, 151.19, 157.11, 160.77 (2), 179.58, 181.48. Масс-спектр высокого разрешения, m/z 314.9830 [М-Н]-. Вычислено для C13H9Cl2O5 314.9833.
ПРИМЕР 2. Получение 6-н-бутокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона
Смесь 274 мг (1.00 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 30 мл н-бутанола и 0.50 мл 90% серной кислоты осторожно кипятят 8.5 ч с отгонкой 5 мл н-бутанола до полной конверсии исходного хинона. В реакционную смесь добавляют 15 мл толуола для растворения выпавшего осадка, промывают водой 2×10 мл, органический слой упаривают досуха без осушки в вакууме, остаток растворяют при нагревании в 5 мл хлороформа, добавляют 10 мл этанола и выдерживают 2 ч при -18°С, выпавшие кристаллы отделяют, промывают холодным этанолом и высушивают в вакууме. Получают 273 мг (83%) соединения формулы II, в котором R1=н-бутил-О, а R2=H (Rƒ0.88, гексан-бензол-ацетон, 2:1:1). Красные призмы, т. пл. 178-180°С (хлороформ-этанол).
ИК-спектр (CHCl3, v/см-1): 2963, 2940, 2877, 1615, 1599, 1558, 1459, 1405, 1351, 1303, 1271, 1250, 1182, 1118 см-1. Спектр ЯМР 1Н (700 МГц, CDCl3, δ, м.д.): 1.01 (т, J=7.4 Гц, 3Н, -СН2СН3), 1.53 (м, 2Н, -CH2CH3), 1.90 (м, 2Н, -ОСН2СН2), 4.07 (т, J=6.6 Гц, 2Н, -ОСН2СН2), 6.24 (с, 1H, ArH), 12.78 (с, 1Н, α-ОН), 13.29 (с, 1Н, α-ОН). Спектр ЯМР 13С (176 МГц, CDCl3, δ, м.д.): 13.64, 19.08, 30.24, 70.20, 108.53, 109.92, 110.19, 132.83, 135.36, 156.30, 157.62, 160.16, 178.67, 184.75. Масс-спектр высокого разрешения, m/z 328.9979 [М-Н]-. Вычислено для C14H11Cl2O5 328.9989.
ПРИМЕР 3. Получение 6-н-бутокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона
Смесь 274 мг (1.00 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 30 мл н-бутанола и 2.30 г сульфокатионита КУ-2 (Н+) помещают в нижнюю часть аппарата Сокслета, в верхнюю часть аппарата помещают 4.5 г высушенных молекулярных сит 4А и кипятят 28 ч, смолу отфильтровывают, промывают 30 мл горячего бутанола. Объединенный бутанольный экстракт упаривают при пониженном давлении, остаток растворяют в 6 мл горячего хлороформа и высаживают продукт добавлением 15 мл этанола. Выпавшие кристаллы промывают холодным этанолом и получают 285 мг (86%) соединения формулы II, в котором R1=н-бутил-О, a R2=H (Rƒ0.88, гексан-бензол-ацетон 2:1:1). Красные призмы, т. пл. 178-180°С (хлороформ-этанол).
ПРИМЕР 4. Получение 6-н-бутокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона
Смесь 274 мг (1.00 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 30 мл н-бутанола и 0.30 мл (4.62 ммоль) метансульфокислоты помещают в нижнюю часть аппарата Сокслета, в верхнюю часть загружают 5.5 г высушенных молекулярных сит 4А и кипятят 35 ч, реакционную смесь упаривают до маслообразного состояния, остаток растворяют в 30 мл толуола и промывают 3×15 мл воды. Толуольный слой упаривают досуха, остаток растворяют в 6 мл горячего хлороформа, добавляют 15 мл этанола и выдерживают 3 ч при -18°С. Выпавшие кристаллы отфильтровывают, промывают холодным этанолом, высушивают в вакууме и получают 250 мг (73%) соединения формулы II, в котором R1=н-бутил-О, a R2=H, с т. пл. 178-180°С (хлороформ-этанол).
ПРИМЕР 5. Получение 6-н-бутокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона
Смесь 820 мг (3.00 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 30 мл н-бутанола и 0.50 мл 90% серной кислоты помещают в нижнюю часть аппарата Сокслета, в верхнюю часть загружают 5.5 г высушенных молекулярных сит 4А и осторожно кипятят 9.5 ч, реакционную смесь упаривают до маслообразного состояния, остаток растворяют в 30 мл толуола и промывают 3×15 мл воды. Толуольный слой упаривают досуха, остаток растворяют в 12 мл горячего хлороформа, добавляют 15 мл этанола и выдерживают 3 ч при -18°С. Выпавшие кристаллы отфильтровывают, промывают холодным этанолом, высушивают и получают 846 мг (85%) соединения формулы II, в котором R1=н-бутил-О, a R2=Н, с т. пл. 178-180°С (хлороформ-этанол).
ПРИМЕР 6. Получение 6-н-амилокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинона
Смесь 2000 мг (7.33 ммоль) 5,6,8-тригидрокси-2,3-дихлор-1,4-нафтохинона, 100 мл н-амилового спирта и 0.80 мл 90% серной кислоты осторожно кипятят 6 ч с отгонкой 15 мл н-амилового спирта до полной конверсии исходного хинона. В реакционную смесь добавляют 60 мл толуола, переносят в делительную воронку, органический слой промывают водой (3×180 мл), упаривают н-амиловый спирт азеотропной отгонкой с водой, добавляют толуол и упаривают экстракт досуха. Остаток растворяют при нагревании в 15 мл хлороформа, добавляют 20 мл этанола и выдерживают 2 ч при -18°С, выпавшие кристаллы отделяют, промывают холодным этанолом и высушивают в вакууме. Получают 2027 мг (80%) соединения формулы II, в котором R1=н-амил-О, а R2=Н (Rƒ 0.90, гексан-бензол-ацетон, 2:1:1). Красные призмы, т. пл. 166-168°С (хлороформ-этанол).
ИК-спектр (CHCl3, v/см-1): 3054, 2960, 2875, 2361, 2342, 16015, 1600, 1558, 1458, 1404, 1304, 1272, 1250, 1204, 1192, 1118. Спектр ЯМР 1Н (500 МГц, CDCl3, δ, м.д.): 0.95 т (J=7.4 Гц, 3Н, -СН2СН3), 1.41 м (2Н, -СН2СН3), 1.48 м (2Н, ОСН2СН2СН2), 1.92 м (2Н, -ОСН2СН2), 4.06 т (J=6.7 Гц, 2Н, ОСН2СН2), 6.23 с (1Н, ArH), 12.79 с (1H, α-ОН), 13.29 с (1Н, α-ОН). Спектр ЯМР 13С (125 МГц, CDCl3, δ, м.д.): 13.87, 22.28, 27.91, 27.96, 70.50, 108.53, 109.93, 110.19, 132.82, 135.35, 156.28, 157.60, 160.15, 179.69, 184.77. Масс-спектр высокого разрешения, m/z 343.0133 [М-Н]-. Вычислено для C15H11Cl2O5 343.0146.
ПРИМЕР 7. Получение смеси 2,5,8-тригидрокси-3-нитро-6-н-пропокси-1,4-нафтохинона и 2,5,8-тригидрокси-3-нитро-7-н-пропокси-1,4-нафтохинона
5,8-Дигидрокси-6-н-пропокси-2,3-дихлор-1,4-нафтохинон 662 мг (2.0 ммоль), растворяют в смеси 15 мл ацетона и 15 мл этанола, добавляют 554 мг (8.0 ммоль) нитрита натрия и кипятят с перемешиванием 1 ч. Реакционную смесь упаривают досуха, добавляют ~3 мл концентрированной соляной кислоты, отфильтровывают неорганические соли, промывают осадок ацетоном, фильтрат упаривают досуха с толуолом, из остатка препаративной ТСХ на силикагеле в системе растворителей гексан-бензол-ацетон, 3:1:1, выделяют основную полосу красного цвета и получают 499 мг (77%) смеси 2,5,8-тригидрокси-3-нитро-7-н-пропокси-1,4-нафтохинона и 2,5,8-тригидрокси-3-нитро-6-н-пропокси-1,4-нафтохинона (Rƒ 0.52, гексан-бензол-ацетон, 2:1:1). Красный аморфный порошок.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 2973, 2941, 2883, 1754, 1632, 1595,1480, 1462, 1418, 1368, 1335, 1290, 1160. Спектр ЯМР 1Н (500 МГц, CDCl3, δ, ТМС) основного изомера (вычленен из спектра смеси): 1.10 (т, 3Н, -СН2СН3, J=7.1 Гц), 1.96 (м, 2Н, -ОСН2СН2СН3), 4.11 (т, 2Н, -ОСН2СН2СН3, J=6.7 Гц), 6.51 (с, 1H, ArH), 12.36 (с, 1Н, α-ОН), 13.18 (с, 1Н, α-ОН). Спектр ЯМР 1Н минорного изомера 1.09 (т, 3Н, -СН2СН3, J=7.5 Гц), 1.96 (м, 2Н, ОСН2СН2СН3), 4.07 (т, 2Н, ОСН2СН2СН3, J=6.7 Гц), 6.54 (с, 1H, ArH), 12.16 (с, 1H, α-ОН), 13.24 (с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 308.0410 [М-Н]-. Вычислено для C13H10NO8 308.0412.
ПРИМЕР 8. Получение смеси 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона
6-н-Бутокси-5,8-дигидрокси-2,3-дихлор-1,4-нафтохинон 250 мг (0.75 ммоль), растворяют в смеси 15 мл ацетона и 15 мл этанола, добавляют 312 мг (4.5 ммоль) нитрита натрия и кипятят при перемешивании 1 ч. Реакционную смесь упаривают при пониженном давлении, добавляют 1.5 мл концентрированной соляной кислоты, отфильтровывают неорганические соли, фильтрат обрабатывают в условиях, описанных в примере 7, и получают 214 мг (89%) смеси 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона (Rf=0.56, гексан:бензол:ацетон=2:1:1). Красный аморфный порошок.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 3492, 3347, 3100, 2964, 2877, 1755, 1661, 1630, 1610, 1595, 1545, 1480, 1460,1418, 1368, 1332, 1294, 1137, 1069. Спектр ЯМР 1Н (700 МГц, ДМСО-d6, δ, ТМС) основного изомера (вычленен из спектра смеси): 0.94 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.44 (м, 2Н, -СН2СН3), 1.74 (м, 2Н, -ОСН2СН2-), 4.11 (т, 2Н, -ОСН2СН2-, J=6.4 Гц), 6.67 (с, 1Н, ArH), 12.97 (уш.с, 1Н, α-ОН), 14.39 (уш.с, 1Н, α-ОН). Спектр ЯМР 1Н минорного изомера 0.94 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.44 (м, 2Н, -СН2СН3), 1.74 (м, 2Н, -ОСН2СН2-), 4.07 (т, 2Н, -ОСН2СН2-, J=6.4 Гц), 6.81 (с, 1H, ArH), 12.47 (уш.с, 1Н, α-ОН), 14.76 (уш.с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 322.0565 [М-Н]-. Вычислено для C14H12NO8 322.0568.
ПРИМЕР 9. Получение смеси 6-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона
5,8-Дигидрокси-6-н-амилокси-2,3-дихлор-1,4-нафтохинон 500 мг (1.45 ммоль), растворяют в смеси 25 мл ацетона и 25 мл этанола, добавляют 604 мг (8.75 ммоль) нитрита натрия и кипятят при перемешивании 1.0 ч. Реакционную смесь упаривают при пониженном давлении, добавляют 2 мл концентрированной соляной кислоты, 15 мл ацетона, отфильтровывают неорганические соли, фильтрат обрабатывают в условиях, описанных в примере 7, и получают 393 мг (80%) смеси 6-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона (Rƒ0.58, гексан-бензол-ацетон, 2:1:1). Красный аморфный порошок.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 3502, 3348, 3056, 2960, 2936, 2875, 1672, 1611, 1545, 1482, 1544, 1481, 1460, 1420, 1295, 1192, 1138, 1067. Спектр ЯМР 1Н (500 МГц, CDCl3, δ, ТМС) основного изомера (вычленен из спектра смеси): 0.95 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.41 (м, 2Н, -СН2СН3), 1.48 (м, 2Н, СН2СН2СН3), 1.93 (м, 2Н, -ОСН2СН2-), 4.13 (т, 2Н, -ОСН2СН2-, J=6.4 Гц), 6.51 (с, 1Н, ArH), 12.37 (с, 1Н, α-ОН), 13.17 (с, 1Н, α-ОН). Спектр ЯМР 1Н минорного изомера 0.95 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.41 (м, 2Н, -СН2СН3), 1.48 (м, 2Н, СН2СН2СН3), 1.93 (м, 2Н, -ОСН2СН2-), 4.10 (т, 2Н, -ОСН2СН2-, J=6.4 Гц), 6.54 (с, 1H, ArH), 12.17 (с, 1Н, α-ОН), 13.23 (с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 336.0721 [М-Н]-. Вычислено для C15H14NO8 336.0725.
ПРИМЕР 10. Получение смеси 3-амино-2,5,8-тригидрокси-6-н-пропокси-1,4-нафтохинона и 3-амино-2,5,8-тригидрокси-7-н-пропокси-1,4-нафтохинона
Смесь 2,5,8-тригидрокси-3-нитро-6-н-пропокси-1,4-нафтохинона и 2,5,8-тригидрокси-3-нитро-7-н-пропокси-1,4-нафтохинона 172 мг (0.55 ммоль), растворяют в смеси 13 мл н-бутанола и 15 мл этилацетата, прибавляют 13 мл воды и при энергичном перемешивании за 1 ч порциями вносят раствор 468 мг (2.28 ммоль) 85% дитионита натрия. Реакционную смесь перемешивают дополнительно 0.5 ч, затем переносят в делительную воронку. Органический слой промывают водой (3×10 мл), упаривают с добавлением толуола и получают 153 мг (98%) смеси 3-амино-2,5,8-тригидрокси-6-н-пропокси-1,4-нафтохинона и 3-амино-2,5,8-тригидрокси-7-н-пропокси-1,4-нафтохинона (Rƒ0.52, гексан-бензол-ацетон, 2:1:1). Черный кристаллический порошок.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 3511, 3450, 3399, 2971, 2938, 2881, 1712, 1664, 1632, 1599, 1563, 1480, 1461, 1407, 1337, 1311, 1279. Спектр ЯМР 1Н (500 МГц, ДМСО-d6, δ, ТМС) основного изомера (вычленен из спектра смеси): 0.98 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.76 (м, 2Н, -ОСН2СН2-), 4.04 (т, 2Н, -ОСН2СН2-, J=6.5 Гц), 6.00 (уш.с, 2Н, NH2), 6.67 (с, 1H, ArH), 9.80 (уш.с, 1H, β-ОН), 12.66 (с, 1Н, α-ОН), 13.00 (с, 1Н, α-ОН). Спектр ЯМР 1Н минорного изомера 0.98 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.76 (м, 2Н, -ОСН2СН2-), 4.05 (т, 2Н, -ОСН2СН2-, J=6.6 Гц), 6.42 (уш.с, 2Н, NH2), 6.60 (с, 1Н, ArH), 9.52 (уш.с, 1H, β-ОН), 12.64 (с, 1Н, α-ОН), 13.40 (с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 278.0668 [М-Н]-. Вычислено для C13H11NO6 278.0670.
ПРИМЕР 11. Получение смеси 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона
Смесь 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 196 мг (0.60 ммоль) растворяют в смеси 15 мл н-бутанола и 10 мл этилацетата, прибавляют 10 мл воды и при энергичном перемешивании за 3 ч порциями вносят раствор 500 мг (2.44 ммоль) 85% дитионита натрия в 12 мл воды. После прибавления дитионита реакционную смесь перемешивают 20 мин, переносят в делительную воронку, органический слой отделяют, промывают водой (3×10 мл), упаривают с добавлением толуола и получают 151 мг (81%) смеси изомеров 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона (Rƒ 0.56, гексан-бензол-ацетон, 2:1:1). Черный кристаллический порошок.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 3509, 3443, 3399, 3156, 2964, 2937, 2876,1719, 1664, 1633, 1598, 1480,1461,1409, 1337, 1302, 1280, 1188, 1145, 1064.
Спектр ЯМР 1Н (700 МГц, ДМСО-d6, δ, ТМС) основного изомера (вычленен из спектра смеси): 0.94 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.44 (м, 2Н, -CH2CH3), 1.74 (м, 2Н, -ОСН2СН2-), 4.08 (т, 2Н, -ОСН2СН2-, J=6.5 Гц), 5.99 (уш.с, 2Н, NH2), 6.68 (с, 1Н, ArH), 9.80 (уш.с, 1Н, β-ОН), 12.64 (с, 1Н, α-ОН), 13.00 (с, 1Н, α-ОН). Спектр ЯМР 1Н минорного изомера 0.94 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.44 (м, 2Н, -СН2СН3), 1.74 (м, 2Н, -ОСН2СН2-), 4.10 (т, 2Н, -ОСН2СН2-, J=6.5 Гц), 6.43 (уш.с, 2Н. NH2), 6.61 (с, 1H, ArH), 9.50 (уш.с, 1Н, β-ОН), 12.64 (с, 1Н, α-ОН), 13.40 (с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 292.0832 [М-Н]-. Вычислено для C14H14NO6 292.0827.
ПРИМЕР 12. Получение смеси 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона
Смесь 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 291 мг (0.90 ммоль) растворяют в смеси 20 мл этилацетата и 10 мл этанола, прибавляют 10 мл воды и при энергичном перемешивании за 6 ч порциями вносят раствор 742 мг (3.62 ммоль) 85% дитионита натрия в 15 мл воды. После прибавления дитионита натрия реакционную смесь перемешивают 30 мин, переносят в делительную воронку, органический слой отделяют, промывают водой (3×10 мл), упаривают с добавлением толуола и получают 257 мг (97%) смеси изомеров 3-амино-2,5,8-тригидрокси-6-н-бутокси-1,4-нафтохинона и 3-амино-2,5,8-тригидрокси-7-н-бутокси-1,4-нафтохинона (Rƒ 0.56, гексан-бензол-ацетон, 2:1:1) Черный кристаллический порошок.
ПРИМЕР 13. Получение смеси 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона
Смесь 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 148 мг (0.46 ммоль) растворяют в 20 мл этилацетата, прибавляют 20 мл воды и при энергичном перемешивании за 3 ч порциями вносят раствор 377 мг (1.84 ммоль) 85% дитионита натрия в 12 мл воды. После прибавления дитионита натрия реакционную смесь перемешивают 20 ч, переносят в делительную воронку, органический слой отделяют, промывают водой (3×10 мл), упаривают с добавлением толуола и получают 111 мг (82%) смеси изомеров 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона (Rf=0.56, гексан-бензол-ацетон, 2:1:1). Черный кристаллический порошок. Соотношение изомеров ~5:1.
ПРИМЕР 14. Получение смеси 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона
К смеси 6-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-бутокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 374 мг (1.16 ммоль) прибавляют 40 мл воды и при энергичном перемешивании за 20 мин порциями вносят раствор 1504 мг (11.6 ммоль) 60% моногидрата сульфида натрия в 10 мл воды. После прибавления раствора сульфида натрия реакционную смесь перемешивают 8 час, подкисляют 2 N HCl до рН 7-8, отфильтровывают выпавший черный осадок, промывают 30 мл воды, сушат и получают 306 мг (90%) смеси изомеров 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона (Rƒ 0.56, гексан-бензол-ацетон, 2:1:1) Черный кристаллический порошок. Соотношение изомеров ~5:1.
ПРИМЕР 15. Получение смеси 6-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона и 7-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона
Смесь 6-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 229 мг (0.68 ммоль) растворяют в 30 мл этилацетата и 10 мл этанола, прибавляют 50 мл воды и при энергичном перемешивании за 1 ч порциями вносят раствор 560 мг (2.73 ммоль) 85% дитионита натрия в 10 мл воды. После прибавления дитионита натрия реакционную смесь перемешивают 20 мин, переносят в делительную воронку, приливают 30 мл толуола, органический слой отделяют, промывают водой (2×15 мл), упаривают с добавлением толуола и получают 194 мг (93%) смеси изомеров 6-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона и 7-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона (Rƒ 0.58, гексан-бензол-ацетон, 2:1:1). Черный кристаллический порошок. Соотношение изомеров ~5:1.
ИК-спектр смеси изомеров (CHCl3, v/см-1): 3510, 3446, 3399, 2960, 2936, 2875, 2361, 2342, 1665, 1633, 1599, 1564, 1479, 1460, 1408, 1337, 1305, 12679, 1189, 1145. Спектр ЯМР 1Н (500 МГц, CDCl3, δ, ТМС) основного изомера (вычленен из спектра смеси): 0.94 (т, 3Н, -СН2СН3, J=7.2 Гц), 1.43 м (4Н, СН2СН2СН3), 1.90 м (2Н, -ОСН2СН2) 4.06 т (J=6.6 Гц, 2Н, -ОСН2СН2-), 4.69 (уш.с, 2Н, NH2), 6.46 (с, 1Н, ArH), 6.54 (уш.с, 1Н, β-ОН), 12.23 (с, 1Н, α-ОН), 12.71 (с, 1H, α-ОН). Спектр ЯМР 1Н минорного изомера 0.95 (т, 3Н, -СН2СН3, J=7.0 Гц), 1.43 (м, 4Н, СН2СН2СН3), 1.90 (м, 2Н, -ОСН2СН2), 4.07 (т, 2Н, -ОСН2СН2-, J=6.6 Гц), 4.89 (уш.с, 2Н, NH2), 6.41 (с, 1H, ArH), 6.50 (с, 1H, β-ОН), 12.53 (с, 1H, α-ОН), 12.62 (с, 1Н, α-ОН). Соотношение изомеров ~5:1. Масс-спектр высокого разрешения смеси изомеров, m/z 306.0990 [М-Н]-. Вычислено для C15H16NO6 306.0983.
ПРИМЕР 16. Получение смеси 6-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона и 7-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона
К смеси 6-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона и 7-н-амилокси-2,5,8-тригидрокси-3-нитро-1,4-нафтохинона 530 мг (1.57 ммоль) прибавляют 56 мл воды и при энергичном перемешивании за 20 мин порциями вносят раствор 2042 мг (15.7 ммоль) 60% моногидрата сульфида натрия в 15 мл воды. После прибавления раствора сульфида натрия реакционную смесь перемешивают 8 ч, подкисляют 2 N HCl до рН 7-8 и получают 440 мг (91%) смеси изомеров 6-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона и 7-н-амилокси-3-амино-2,5,8-тригидрокси-1,4-нафтохинона (Rƒ0.58, гексан-бензол-ацетон, 2:1:1). Черный кристаллический порошок. Соотношение изомеров ~5:1.
ПРИМЕР 17. Получение 2,3,5,8-тетрагидрокси-6-н-пропокси-1,4-нафтохинона
Смесь 3-амино-2,5,8-тригидрокси-6-н-пропокси-1,4-нафтохинона и 3-амино-2,5,8-тригидрокси-7-н-пропокси-1,4-нафтохинона 147 мг (0.52 ммоль) растворяют в смеси 8 мл 85% муравьиной кислоты, 0.20 мл диметилсульфоксида, 0.5 мл 25% серной кислоты и перемешивают при температуре кипения с обратным холодильником в течение 35 мин. Реакционную смесь охлаждают до комнатной температуры, смывают в делительную воронку 30 мл этилацетата, разбавляют 30 мл холодной воды, органический слой промывают водой (3×10 мл), 10 мл насыщенного раствора хлорида натрия, сушат над сульфатом натрия и упаривают на роторном испарителе с добавлением толуола. Из остатка препаративной ТСХ на силикагеле в системе гексан-бензол-ацетон, 2:1:1 выделяют 86 мг (68%) 2,3,5,8-тетрагидрокси-6-н-пропокси-1,4-нафтохинона (Rƒ 0.46, гексан-бензол-ацетон, 2:1:1). Красные кристаллы, т. пл. 211-213°С (гексан-ацетон).
ИК-спектр (CHCl3, v/см-1): 3430, 2971, 2941, 2883, 1691, 1636, 1592, 1483, 1461, 1411, 1349, 1303, 1278, 1240, 1193, 1115, 1061, 1014. Спектр ЯМР 1Н (500 МГц, ДМСО-d6, δ, ТМС): 0.98 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.77 (м, 2Н, -СН2СН3), 4.13 (т, 2Н, OCH2CH2, J=6.2 Гц), 6.71 (с, 1Н, ArH), 10.23 (уш.с, 1H, β-ОН). 10.37 (уш.с, 1Н, β-ОН), 12.73 (с, 2Н, α-ОН). Спектр ЯМР 13С (125 МГц, ДМСО-d6, δ, ТМС): 10.24, 21.60, 70.80, 102.33, 106.73, 108.97, 140.57, 142.40, 151.19, 157.12, 160.70, 179.58, 181.48. Масс-спектр высокого разрешения, m/z 279.0511 [М-Н]-. Вычислено для С13Н11О7 279.0510.
ПРИМЕР 18. Получение 2,3,5,8-тетрагидрокси-6-н-бутокси-1,4-нафтохинона
Смесь 3-амино-6-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-бутокси-2,5,8-тригидрокси-1,4-нафтохинона 220 мг (0.75 ммоль) растворяют в смеси 12 мл 85% муравьиной кислоты, 0.40 мл диметилсульфоксида, 1 мл 25% серной кислоты и перемешивают при температуре кипения с обратным холодильником в течение 0.5 ч. Реакционную смесь охлаждают, переносят в делительную воронку, разбавляют 40 мл воды и экстрагируют 2×25 мл этилацетата. Объединенный органический слой промывают 2×15 мл воды, высушивают сульфатом натрия, растворитель удаляют в вакууме с добавлением толуола, из остатка кристаллизацией из смеси ацетон-гексан получают 209 мг (93%) 6-н-бутокси-2,3,5,8-тетрагидрокси-1,4-нафтохинона (Rƒ 0.48, гексан-бензол-ацетон, 2:1:1). Красные кристаллы, т. пл. 190-193°С (гексан-ацетон).
ИК-спектр (CHCl3, v/см-1): 3431, 2964, 2940, 2877, 1710, 1691, 1637, 1592, 1483, 1460, 1412, 1356, 1302, 1279, 1240,1192, 1115, 1067. Спектр ЯМР 1Н (700 МГц, ДМСО-d6, δ, ТМС): 0.94 (т, 3Н, -СН2СН3, J=7.4 Гц), 1.44 (м, 2Н, -СН2СН3), 1.74 (м, 2Н, -ОСН2СН2), 4.11 (т, 2Н, -ОСН2СН2, J=6.5 Гц), 6.72 (с, 1H, ArH), 10.23 (уш.с, 1Н, β-ОН), 10.42 (уш.с, 1Н, β -ОН), 12.72 (с, 1Н, α-ОН), 12.76 (с, 1H, α-ОН). Спектр ЯМР 13С (176 МГц, ДМСО-d6, δ, ТМС): 13.59, 18.62, 30.20, 69.14, 102.33, 106.78, 108.95, 140.56, 142.23, 151.32, 157.15, 160.87, 179.45, 181.41. Масс-спектр высокого разрешения, m/z 293.0662 [М-Н]-. Вычислено для C14H13O7 293.0667.
ПРИМЕР 19. Получение 6-н-амилокси-2,3,5,8-тетрагидрокси-1,4-нафтохинона
Смесь 3-амино-6-н-амилокси-2,5,8-тригидрокси-1,4-нафтохинона и 3-амино-7-н-амилокси-2,5,8-тригидрокси-1,4-нафтохинона 400 мг (1.30 ммоль) растворяют в смеси 20 мл 85% муравьиной кислоты, 0.6 мл диметилсульфоксида, 2 мл 25% серной кислоты и кипятят с перемешиванием с обратным холодильником в течение 35 мин. Реакционную смесь охлаждают, переносят в делительную воронку, разбавляют 60 мл воды и экстрагируют этилацетатом (2×50 мл). Объединенный органический слой промывают водой (2×40 мл), высушивают сульфатом натрия, растворитель удаляют в вакууме с добавлением толуола, из остатка препаративной ТСХ на силикагеле в системе растворителей гексан-бензол-ацетон, 2:1:1, выделяют 316 мг (79%) 6-н-амилокси-2,3,5,8-тетрагидрокси-1,4-нафтохинона (Rƒ 0.50, гексан-бензол-ацетон, 2:1:1). Красные кристаллы, т. пл. 165-168°С (гексан-ацетон).
ИК-спектр (CHCl3, v/см1): 3429, 2960, 2936, 2875, 2865, 1691, 1636, 1592, 1483, 1460, 1411, 1379, 1348, 1303, 1277. Спектр ЯМР 1Н (500 МГц, ДМСО-d6, δ, ТМС): 0.90 (т, 3Н, -СН2СН3, J=7.1 Гц), 1.37 (м, 4Н, -CH2CH2CH3), 1.76 (м, 2Н, -ОСН2СН2-), 4.10 (т, 2Н, -ОСН2СН2-, J=6.5 Гц), 6.71 (с, 1Н, ArH), 10.21 (с, 1Н, β-ОН), 10.47 (с, 1H, β-ОН), 12.72 (с, 1H, α-ОН), 12.76 (с, 1H, α-ОН). Спектр ЯМР 13С (125 МГц, ДМСО-d6, δ, ТМС): 13.92, 21.82, 27.58, 27.88, 69.42, 102.32, 106.75, 108.96,140.57, 142.24, 151.22, 157.13, 160.79, 179.55, 181.51. Масс-спектр высокого разрешения, m/z 307.0828 [М-Н]-. Вычислено для С15Н15О7 307.0823.
ПРИМЕР 20. Получение 2,3,5.6,8-пентагидрокси-1,4-нафтохинона (спинохрома D)
2,3,5,8-Тетрагидрокси-6-н-пропокси-1,4-нафтохинон 140 мг (0.50 ммоль) растворяют в смеси 6.5 мл 98% муравьиной кислоты и 0.90 мл метансульфокислоты и кипятят 4 ч с обратным холодильником. Реакционную смесь разбавляют 30 мл воды, переносят в делительную воронку и экстрагируют 3×20 мл этилацетатом, объединенный органический слой промывают 2×30 мл воды, сушат над сульфатом натрия, упаривают из остатка кристаллизацией из смеси ацетон-гексан, получают 102 мг (86%) 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D), красные кристаллы, возгоняются без плавления при температуре выше 285°С.
Спектр ЯМР 1Н (700 МГц, ацетон-d6, δ, ТМС): 6.55 (с, 1H, ArH), 8.77 (уш.с, 1H, β-ОН), 8.98 (уш.с, 1H, β-ОН), 9.90 (уш.с, 1Н, β-ОН), 12.34 (с, 1Н, α-ОН), 12.49 (уш.с, 1Н, α-ОН). Спектр ЯМР 13С (176 МГц, ацетон-d6, δ, ТМС): 103.23, 109.59, 110.17, 139.94, 141.87, 151.09, 157.34, 162.17, 180.45, 182.81.
ПРИМЕР 21. Получение 2,3,5.6,8-пентагидрокси-1,4-нафтохинона (спинохрома D)
2,3,5,8-Тетрагидрокси-6-н-бутокси-1,4-нафтохинон 88 мг (0.30 ммоль) растворяют в смеси 5 мл мл 98% муравьиной кислоты и 0.50 мл метансульфокислоты и перемешивают 4 ч при температуре кипения с обратным холодильником. Реакционную смесь разбавляют 25 мл воды и выдерживают при +5°С 2 ч, выпавшие кристаллы отделяют, промывают водой, холодным этанолом, высушивают в вакууме и получают 66 мг (92%) 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) (Rƒ0.30, гексан-бензол-ацетон, 2:1:1). Красные кристаллы, возгоняются без плавления выше 285°С (ацетон).
ПРИМЕР 22. Получение 2,3,5.6,8-пентагидрокси-1,4-нафтохинона (спинохрома D)
6-н-Амилокси-2,3,5,8-тетрагидрокси-1,4-нафтохинон 155 мг (0.50 ммоль) растворяют в смеси 6.5 мл 98% муравьиной кислоты и 0.90 мл метансульфокислоты и перемешивают 4 ч при температуре кипения с обратным холодильником. Реакционную смесь разбавляют 30 мл воды, экстрагируют этилацетатом (3×25 мл), объединенный органический слой промывают водой (2×30 мл), сушат над сульфатом натрия, упаривают и кристаллизацией остатка из ацетона получают 93 мг (78%) 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 6,7-ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТОХИНОНОВ (СПИНАЗАРИНОВ) | 2012 |
|
RU2478607C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТАХИНОНОВ | 1990 |
|
RU2022959C1 |
| Способ получения спинохрома D | 2016 |
|
RU2612265C1 |
| Флуоресцентно-меченые дезоксиуридинтрифосфаты | 2016 |
|
RU2637310C1 |
| Дезоксиуридинтрифосфаты, связанные с цианиновыми красителями сульфамидоалкильными линкерами, для использования в ПЦР | 2017 |
|
RU2667070C1 |
| ФОСФОРОАМИДАТНЫЕ ПРОИЗВОДНЫЕ 5-ФТОР-2'-ДЕЗОКСИУРИДИНА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2614406C2 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ 1,4-НАФТОХИНОНА, ОБЛАДАЮЩИЕ ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РАКОВЫМ КЛЕТКАМ ЧЕЛОВЕКА В КУЛЬТУРЕ | 2010 |
|
RU2443678C1 |
| СРЕДСТВО, СТИМУЛИРУЮЩЕЕ АПОПТОЗ КЛЕТОК ЛЕЙКЕМИИ ЧЕЛОВЕКА (ВАРИАНТЫ) | 2007 |
|
RU2372919C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5,8-ДИГИДРОКСИ-2,6-7-ТРИМЕТОКСИ-3-ЭТИЛ-1,4-НАФТОХИНОНА | 2005 |
|
RU2277083C1 |
| ЗАМЕЩЕННЫЕ 2-[2-(3-ОКСОМОРФОЛИН-4-ИЛ)ЭТИЛТИО]БЕНЗИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ АНКСИОЛИТИЧЕСКОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2373202C2 |
Изобретение относится к способу получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) формулы I, где R1=OH и R2=H, который применяют в медицине и косметологии, а также к новым промежуточным соединениям формулы V, где R=н-пропил-О-, н-бутил-О-, н-амил-О-. Способ включает взаимодействие соединения формулы II, где R1=OH и R2=Н, с первичными неразветвленными спиртами С3-С5 при катализе серной или метансульфокислотой, с удалением образующейся при этом воды азеотропной отгонкой спирта С3-С5, с получением соединений формулы II, где R1=н-пропил-О- и R2=H, R1=н-бутил-О- и R2=H, R1=н-амил-О- и R2=H, с последующим взаимодействием полученных 6-алкоксизамещенных соединений формулы II с 4-, 6-кратным мольным избытком азотистокислого натрия в смеси ацетон-этанол при температуре кипения с обратным холодильником, с получением соединений формулы III в виде смеси изомеров, где R1=H, a R2=н-пропил-О-, н-бутил-О-, н-амил-О- и R2=H, а R1=н-пропил-О-, н-бутил-О-, н-амил-О-. Полученные соединения формулы III восстанавливают под действием дитионита натрия или сульфида натрия с образованием соединения формулы IV, где R1 и R2 определены выше, с последующим превращением соединения формулы IV путем кислотно-катализируемого гидролиза в смеси диметилсульфоксид-муравьиная кислота-серная кислота-вода при температуре кипения с обратным холодильником в соединение формулы V, где R=н-пропил-O-, н-бутил-О-, н-амил-О-, которое путем кислотно-катализируемого гидролиза в смеси муравьиная кислота-метансульфокислота-вода при температуре кипения с обратным холодильником превращают в целевое соединение - спинохром D. Предлагаемый способ позволяет получить целевой продукт с выходом 40-51%. 2 н.п. ф-лы, 22 пр.
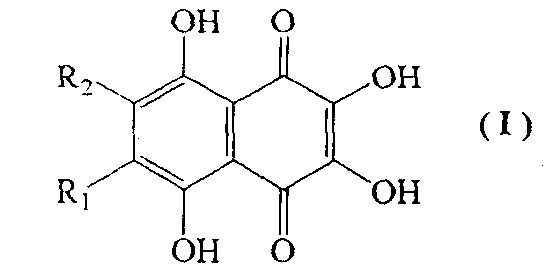
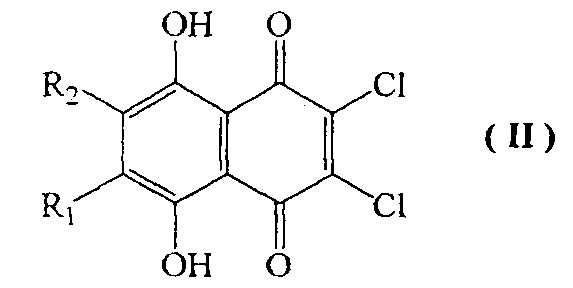
1. Способ получения 2,3,5,6,8-пентагидрокси-1,4-нафтохинона (спинохрома D) формулы I
где R1=OH и R2=H,
включающий взаимодействие соединения формулы II
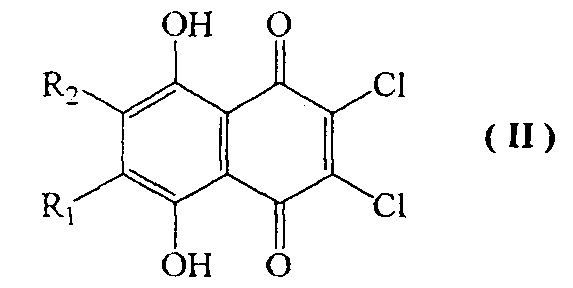
где R1=OH и R2=H,
с первичными неразветвленными спиртами С3-С5 при катализе серной или метансульфокислотой, с удалением образующейся при этом воды азеотропной отгонкой спирта С3-С5, с получением соединений формулы II, где R1=н-пропил-О- и R2=H, R1=н-бутил-О- и R2=H, R1=н-амил-О- и R2=H, с последующим взаимодействием 6-алкоксизамещенных соединений формулы II, где R1 и R2 имеют указанные выше значения с 4-, 6-кратным мольным избытком азотистокислого натрия в смеси ацетон-этанол при температуре кипения с обратным холодильником, с получением соединений формулы III в виде смеси изомеров
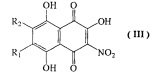
где R1=H, a R2=н-пропил-О-, н-бутил-О-, н-амил-О- и R2=H, а R1=н-пропил-О-, н-бутил-О-, н-амил-О-;
с последующим восстановлением соединения формулы III под действием дитионита натрия или сульфида натрия с образованием соединения формулы IV, где R1 и R2 определены выше,
с последующим превращением соединения формулы IV путем кислотно-катализируемого гидролиза в смеси диметилсульфоксид-муравьиная кислота-серная кислота-вода при температуре кипения с обратным холодильником в соединение формулы V
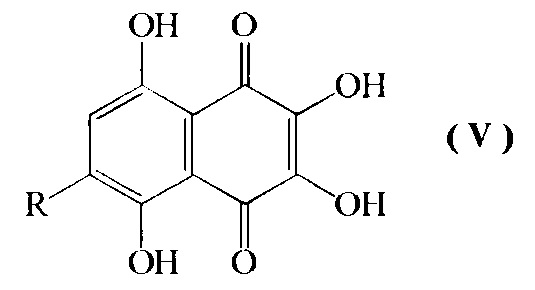
где R=н-пропил-O-, н-бутил-О-, н-амил-О-,
с последующим превращением соединения формулы V путем кислотно-катализируемого гидролиза в смеси муравьиная кислота-метансульфокислота-вода при температуре кипения с обратным холодильником в целевое соединение - спинохром D.
2. Промежуточные соединения формулы V
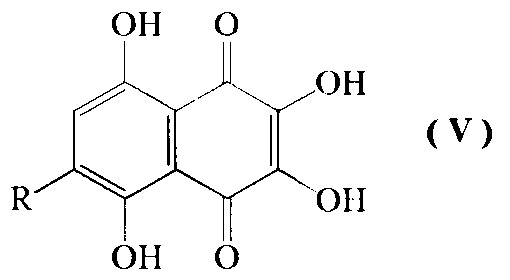
где R=н-пропил-О-, н-бутил-О-, н-амил-О-.
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТАХИНОНОВ | 1990 |
|
RU2022959C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6,7-ЗАМЕЩЕННЫХ 2,3,5,8-ТЕТРАГИДРОКСИ-1,4-НАФТОХИНОНОВ (СПИНАЗАРИНОВ) | 2012 |
|
RU2478607C1 |
| H | |||
| A | |||
| Anderson et al., 382 | |||
| Naturally occurring quinones | |||
| Part VI | |||
| Spinochrome D | |||
| Journal of the Chemical Society, 1965, 2141-2144. | |||

