Область техники, к которой относится изобретение
Изобретение относится к области биотехнологии и генетической инженерии, и может быть использовано при создании генно-терапевтических препаратов, предназначенных для использования в медицине, а именно в гепатологии для лечения фиброза печени различной степени тяжести.
Уровень техники
Из уровня техники известны лекарственные средства для лечения фиброза печени (Gene therapy by hepatocyte growth factor results in regression of experimental liver fibrosis RJGHC. - 2010. - Vol. 20. - No 4. - P. 22-28. N.A. Dzhoyashvili, N.I. Kalinina, I.B. Beloglazova, Z.I. Tsokolayeva, P.I. Makarevich, Yu.L. Perov, Ye.V. Parfenova, V.A. Tkachuk; Translational research on HGF: A phase I/II study of recombinant human HGF for the treatment of fulminant hepatic failure. Hepatol Res. 2008 Nov; 38 Suppl 1:S88-92. doi: 10.1111/j.1872-034X.2008.00432.X. Ido A, Moriuchi A, Marusawa H, Ikeda K, Numata M, Yamaji N, Setoyama H, Ida H, Oketani M, Chiba T, Tsubouchi H.; Urokinase-type plasminogen activator gene therapy in liver cirrhosis is mediated by collagens gene expression down-regulation and up-regulation of MMPs, HGF and VEGF. J Gene Med. 2006 Nov; 8(11): 1291-9 Bueno M, Salgado S, Beas-Zarate C, Armendariz-Borunda J.), в которых в качестве основного действующего вещества использованы плазмидные конструкции, содержащие по отдельности гены фактора роста гепатоцитов или урокиназы человека. Вышеупомянутые плазмидные генетические конструкции содержат белок-кодирующие участки ДНК соответствующих генов и при их введении в клетки млекопитающих в результате процессов транскрипции и трансляции обеспечивают синтез и последующую секрецию из клеток белков фактора роста гепатоцитов или урокиназы. Биологическая активность соответствующих белков обеспечивает поддержание функции и деления клеток печени-гепатоцитов, а также разрушение белков внеклеточного матрикса, откладывающихся в ткани при фиброзе. Способ лечения фиброза печени основан на многократном внутривенном введении указанных средств в количествах не более 3,75 мг/кг (для грызунов).
Однако для достижения терапевтического эффекта таких моногенных препаратов требутся многократное введение терапевтических плазмид, что удорожает лечение и делает его более продолжительным.
Наиболее близким, известным из уровня техники, является лекарственное средство и способ лечения экспериментального фиброза печени с помощью одновременного введения животным аденовирусов, несущих генетические конструкции, с которых происходит синтез белков - фактора роста гепатоцитов (HGF) и урокиназы (Treatment of experimental hepatic fibrosis by combinational delivery of urokinase-type plasminogen activator and hepatocyte growth factor genes. Lin Y, Xie WF, Chen YX, Zhang X, Zeng X, Qiang H, Chen WZ, Yang XJ, Han ZG, Zhang ZB. Liver Int. 2005 Aug; 25(4):796-807). Введение вирусных частиц с терапевтическими генами обеспечивает эффективное заражение клеток печени и секрецию ими соответствующих белков, биологическая активность которых приводит к уменьшению фибротического поражения ткани печени.
Несмотря на высокую эффективность данный подход имеет ряд ограничений и недостатков. Получение вирусных частиц существенно удорожает производство препарата и значительно повышает требования к обеспечению безопасности. Использование вирусных частиц на основе аденовируса не позволяет использовать данный способ лечения фиброза у пациентов, поскольку не может быть одобрен регламентирующими органами в связи с побочными эффектами, связанными с введением в организм частиц вируса.
Заявляемое решение основано на использовании препарата, включающего смесь невирусных плазмидных конструкций, содержащих гены HGF и урокиназы, которые при одновременном введении в клетки печени обеспечивают синтез и секрецию соответствующих белков, биологическая активность которых способствует излечиванию от фиброза. В то же время, создание заявляемого препарата существенно проще, чем получение вирусных частиц, поскольку производство невирусных конструкций не требует применения специальных мер безопасности, необходимых при работе с вирусами. Используемые невирусные конструкции удовлетворяют критериям безопасности, установленными регламентирующими органами для лечения заболеваний человека, и выгодно отличаются от моногенных препаратов на основе урокиназы и фактора роста гепатоцитов, поскольку эффективнее за счет использования синергического действия сразу двух биологически активных белков. Способ лечения с использованием заявляемого средства требует меньшего времени и количества введений препарата для достижения лучшего терапевтического эффекта по сравнению с существующими аналогами на основе моногенных препаратов.
Раскрытие изобретения
Задачей изобретения является создание высокоэффективного и безопасного генотерапевтического препарата, предназначенного для направленного лечения фиброза печени.
Техническим результатом, на достижение которого направлено заявленное изобретение, является обеспечение безопасного и упрощенного производства препарата, а также его безопасное использование в клинике, как для медицинских работников, так и для пациентов. Данный результат достигается за счет использования невирусных конструкций, содержащих терапевтические гены.
Для заявляемого препарата характерны следующие преимущества: простота получения в больших количествах, биологическая безопасность получения, высокая стабильность, сочетание сразу двух биологически активных компонентов. Для заявляемых способов применения лекарственного средства характерны простота применения, безопасность для медицинского персонала и пациентов, более высокая эффективность по сравнению с аналогами, сокращение числа вводимых доз, необходимых для достижения терапевтического эффекта.
Поставленная задача решается тем, что лекарственное средство для лечения фиброза печени в качестве активно действующего вещества содержит смесь из двух плазмидных конструкций, содержащих терапевтические гены HGF и урокиназы человека. Входящие в состав заявляемого средства плазмидные конструкции могут содержаться в следующем соотношении: pC4W-HGFopt (кодирует HGF) в интервале концентраций от 0,5 до 0,7 мг/мл; pVax1-UPAopt (кодирует урокиназу) от 0,3-0,5 мг/мл, причем суммарная концентрация ДНК должна составлять 1+/-0,01 мг/мл. Лекарственное средство может представлять собой жидкую субстанцию или лиофильно высушенный препарат. В одном из вариантов выполнения средства входящие в него плазмидные ДНК содержат следующие регуляторные элементы: для плазмиды pVax1-UPAopt промотор ранних генов цитомегаловируса (в скобках указано положение в нуклеотидной последовательности 137-724), старт ко дон трансляции в оптимальном окружении (759-770), оптимизированная кДНК урокиназы (765-2063), сигнал полиаденилирования мРНК гормона роста быка (3129-2354); для плазмиды pC4W-HGFopt промотор ранних генов цитомегаловируса (1-660), последовательность интрона бета-глобина (661-1323), старт кодон трансляции в оптимальном окружении (1342-1370), оптимизированная кДНК HGF (1371-3557), посттранскрипционный регуляторный элемент из вируса гепатита лесного сурка (3579-4170), синал полиаденилирования мРНК гормона роста быка (4181-4408). (Фиг. 1, 2, 25, 26). Эти регуляторные элементы обеспечивают высокий уровень продукции белков урокиназы и фактора роста гепатоцитов человека, при введении данных плазмидных ДНК в клетки млекопитающих, в том числе человека.
Поставленная задача решается тем, что способ получения заявляемого лекарственного средства включает получение бактериальных штаммов-продуцентов, несущих плазмиды с генами HGF и урокиназы человека, наращивание биомассы бактерий, несущих соответствующие плазмидные конструкции, выделение и очистка ДНК плазмидных конструкций с последующим смешением. Очистку проводят с достижением конечного содержания примесей на 1 мг: белки E.coli - не более 10 мкг (≤1%), РНК E.coli - не более 10 мкг (≤1%), геномная ДНК E.coli - не более 10 мкг (≤1%), бактериальные эндотоксины - не более 40 ЕЭ, и содержанием суперспиральной формы плазмидной ДНК - не менее 800 мкг (≥80%) смешением полученных водных растворов плазмидных конструкций в пропорциях 50-70% pC4W-HGFopt:30-50% pVax1-UPAopt с последующей лиофильной сушкой до остаточной влажности не более 0,005 мг на 1 мг. По итогам очистки нуклеотидную последовательность получаемых плазмидных конструкций подтверждают с помощью анализа длин фрагментов, получающихся при гидролизе специфическими ферментами - эндонуклеазами рестрикции, определяют уровень примесей, чистоту и концентрацию растворов, соответствующих плазмидных ДНК.
В одном из вариантов осуществления способа плазмидная конструкция pC4W-HGFopt имеет нуклеотидную последовательность гена HGF, представленную на фиг. 25, а плазмидная конструкция pVax1-UPAopt имеет нуклеотидную последовательность гена урокиназы, представленную на фиг. 26.
Поставленная задача решается также тем, что способ лечения фиброза печени включает введение заявляемого лекарственного средства в организм млекопитающего в фармацевтически приемлемом количестве. В одном из вариантов способа возможно 2-кратное с интервалом 2-3 дня внутривенное введение заявляемого средства в количестве, не превышающем 3,75 мг/кг для грызунов и 0,3125 мг/кг для человека. При использовании лиофильно высушенного средства, перед введением в организм млекопитающего его предварительно растворяют в изотоническом растворе.
Таким образом, лекарственное средство включает в себя смесь из двух невирусных плазмидных конструкций, одна из которых содержит терапевтический ген фактора роста гепатоцитов, вторая - ген урокиназы человека. Плазмидные конструкции обеспечивают высокий уровень секреции соответствующих биологически активных белков (в культуре клеток - в интервале от 5 до 1000 нг на 1 мл среды) при введении в клетки млекопитающих, в том числе человека.
Способ получения лекарственного средства включает следующие стадии: создание бактериальных штаммов продуцентов, содержащих невирусные плазмидные конструкции, кодирующие урокиназу и фактор роста гепатоцитов, получение бактериальной биомассы, содержащей вышеупомянутые плазмидные конструкции, выделение и очистку соответствующих плазмидных конструкций, получение смеси растворов плазмидных конструкций с суммарной концентрацией ДНК 1+/-0,2 мг/мл, содержащей определенные пропорции отдельных компонентов (в интервале концентраций от 0,3 до 0,70 мг/мл индивидуальных плазмид), получение лиофилизованных (высушенных) препаратов плазмидных конструкций.
Способ лечения фиброза печени включает одно или многократное, в зависимости от тяжести фиброза печени, введение заявленного препарата в виде водного изотонического раствора, который можно получить растворением лиофилизованного препарата, взятого в терапевтически приемлемом количестве (например, 3,75 мг/кг - для животных и 0,3125 мг/кг - для человека для внутривенного введения).
После введения в пораженную печень плазмидные конструкции экспрессируются в течение одной-двух недель, обеспечивая повышение продукции HGF и урокиназы. Биомишенями этих белковых факторов являются специфичные рецепторы на поверхности гепатоцитов, клеток Ито и макрофагов печени, белки c-met и uPAR. Факторы роста, продуцируемые в результате введения в пораженную печень плазмидных конструкций, взаимодействуют со специфичными биомишенями. Взаимодействие HGF со своей биомишенью приводит к активации каскадов внутриклеточной сигнализации, в результате чего происходят подавление апоптоза, стимуляция пролиферации гепатоцитов, а также предотвращение активации дифференцировки клеток Ито в про-фибротические миофибробласты, что обусловливает ускоренное восстановление количества функционально активных клеток печени. Взаимодействие урокиназы со своим рецептором на поверхности клеток-мишеней вызывает как активацию каскадов внутриклеточной сигнализации, так и запуск околоклеточного протеолиза. В результате происходит подавление активности трансформирующего фактора роста, подавление экспрессии генов коллагена 1α1, фибронектина, ингибитора активатора плазминогена 1 типа и тканевого ингибитора матриксных металлопротеиназ 1 типа, а также повышение продукции матриксной металлопротеиназы-13, что обусловливает непосредственное анти-фибротическое действие исследуемого лекарственного средства.
Кроме того, при изучении свойств урокиназы авторами настоящего изобретения было выявлено, что помимо тромболитической активности, урокиназа обладает способностью участвовать в регуляции ремоделирования органов и тканей, регуляции клеточной пролиферации и миграции, регуляции клеточной дифференцировки, а также в процессах регуляции регенерации. Обнаружено, что урокиназа принимает участие в регуляции активности множества факторов роста, как протеолитически активируя их, так и влияя на экспрессию их рецепторов.
Краткое описание чертежей
Изобретение поясняется чертежами.
На фиг. 1 и 2 представлены схемы плазмидных конструкций: на фиг. 1 - плазмидная конструкция, несущая UPA - ген урокиназы, на фиг. 2 - HGF - ген фактора роста гепатоцитов. Цифрами обозначены положения нуклеотидов.
На фиг. 3 представлен характерный спектр поглощения высокоочищенной плазмидной ДНК в диапазоне длин волн от 240 нм до 320 нм.
На фиг. 4 - анализ спектров поглощения растворов ДНК с разными разбавлениями.
На фиг. 5 - электрофоретический анализ продуктов гидролиза плазмидных ДНК рестриктазами.
На фиг. 6 - график зависимости длины пробега от логарифма размера фрагмента.
На фиг. 7 - результаты измерения пролиферативной активности культуры клеток под действием HGF, по оси абсцисс указано время, по оси ординат - значения среднего времени удвоения количества клеток.
На фиг. 8 - фотографии стеллатных клеток мыши, увеличение 40х, сразу после выделения (слева), на следующий день (справа), светятся вакуоли с ретинолом.
На фиг. 9-13 - результаты измерения относительных уровней мРНК белков внеклеточного матрикса стеллатных клеток, обработанных средой, содержащей урокиназу, секретируемую клетками HEK293, трансфицированными плахмидой pVax1-UPAopt. На фиг. 9 представлено изменение уровня мРНК альфа-гладкоклеточного актина (α-SMA) при добавлении кондиционированной среды клеток линии HEK. На фиг. 10 - изменение уровня мРНК трансформирующего фактора роста бета (TGF-β) при добавлении кондиционированной среды клеток линии HEK. На фиг. 11 - изменение уровня мРНК тканевого ингибитора матриксных металлопротеиназ (TIMP-1) при добавлении кондиционированной среды клеток линии HEK. На фиг. 12 - изменение уровня мРНК коллагена I типа при добавлении кондиционированной среды клеток линии HEK. Фиг. 13 - изменение уровня мРНК коллагена III типа при добавлении кондиционированной среды клеток линии HEK.
На фиг. 14 представлены фотографии гистологически окрашенных срезов ткани печени. Группа отрицательного контроля (сверху А и В). Мыши, которым в течение Недель 1-4 вводили 30% CCl4 на оливковом масле в дозе 1 мкл/1 г массы тела. В течение Недель 5-6 животные этой группы не получали никакого лечения. Пикросириус красный (слева А), гематоксилин-эозин (справа В), увеличение 10х. Группа терапии (снизу С и D). Недели 1-4 - индукция фиброза, Недели 5-6 - введение комбинации плазмид HGF и uPA 1 раз в неделю методом гидродинамического удара. Пикросириус красный (слева С), гематоксилин-эозин (справа D), увеличение 10х.
На фиг. 15 и фиг. 16 представлены фотографии гистологически окрашенных срезов печени мышей. Группа положительного контроля (препарата сравнения). Недели 1-4 - индукция фиброза, Недели 5-6 - введение 1 раз плазмиды HGF методом гидродинамического удара методом гидродинамического удара в дозе 3,75 мкг плазмидной ДНК на 1 г массы тела. Группа здоровых животных. Пикросириус красный (слева), гематоксилин-эозин (справа). Увеличение 10х.
На фиг. 17 - кинетическая кривая ПЦР.
На фиг. 18 - уровень мРНК компонентов внеклеточного матрикса в печени мышей.
На фиг. 19 - фотографии иммуногистохимически окрашенных срезов печени. Иммуногистохимичесское окрашивание срезов печени на коллаген I типа. Контрольная группа (без терапии) (А), группы терапии (В), группа терапии HGF (С), здоровый контроль (D).
На фиг. 20 - мммуногистохимическое окрашивание срезов печени на альфа-гладкоклеточный актин. Контрольная группа (без терапии) (А), группы терапии (В), группа терапии HGF (С), здоровый контроль (D).
На фиг. 21 - зависимость изменения поглощения при 340 нм от времени образца реакционной смеси для измерения активности АЛТ. По оси абсцисс - время в минутах, по оси ординат - значения оптической плотности при 340 нм.
На фиг. 22 - гистограммы средних значений активности АЛТ, измеренные в условных единицах активности на литр. Количество животных в каждой группе было больше или равно четырем. 1 - контрольные животные с индуцированным фиброзом печени; 2 - животные с индуцированным фиброзом, которые получали разрабатываемое лекарственное средство; 3 - животные с индуцированным фиброзом, которые получали препарат сравнения; 4 - контрольные животные без фиброза, инъецированные оливковым маслом; 5 - здоровые мыши, которые не получали никаких инъекций.
На фиг. 23 - зависимость изменения поглощения при 340 нм от времени образца реакционной смеси для измерения активности ACT. По оси абсцисс - время в минутах, по оси ординат - значения оптической плотности при 340 нм.
На фиг. 24. - гистограммы средних значений активности ACT, измеренные в условных единицах активности на литр. Количество животных в каждой группе было больше или равно четырем. 1 - контрольные животные с индуцированным фиброзом печени; 2 - животные с индуцированным фиброзом, которые получали разрабатываемое лекарственное средство; 3 - животные с индуцированным фиброзом, которые получали препарат сравнения; 4 - контрольные животные без фиброза, инъецированные оливковым маслом; 5 - здоровые мыши, которые не получали никаких инъекций.
На фиг. 25 представлена нуклеотидная последовательность гена HGF, представленная в плазмидной конструкции pC4W-HGFopt (с №1371 по №3557).
На фиг. 26 - нуклеотидная последовательность гена урокиназы, входящая в плазмидную конструкцию pVax1-UPAopt (с №765 по №2063).
На фиг. 27 - структурная формула плазмидной ДНК pC4W-HGFopt.
На фиг. 28 - структурная формула плазмидной ДНК pVax1-UPAopt.
Осуществление изобретения
Для создания заявленного лекарственного средства используют две различные невирусные плазмидные конструкции, несущие терапевтические гены урокиназы и фактора роста гепатоцитов. Для создания лекарственного средства можно использовать любые плазмидные конструкции, которые обеспечивают продукцию урокиназы и фактора роста гепатоцитов при введении в клетки млекопитающих. Данные конструкции должны содержать элементы ДНК, которые обеспечивают высокий уровень транскрипции и трансляции соответствующих генов, обеспечивая высокий уровень синтеза и секреции кодируемых ими белков урокиназы и фактора роста гепатоцитов в клетках млекопитающих. В изобретении может быть использована последовательность гена HGF, оптимизированная для получения больших количеств белка, которая представлена в материалах патента RU 2385936. Схемы соответствующих конструкций и их полная структура приведены на Фиг. 1, 2, 27 и 28.
Урокиназа - фактор, который расщепляет ряд белков внеклеточного матрикса, способствует миграции клеток и ремоделированию ткани, стимулирует ангиогенез. Для продуцирования uPA используют ДНК, содержащую природный ген урокиназы человека. Исходная последовательность гена была модифицирована таким образом, чтобы при ее включении в состав эукариотического вектора обеспечивался более высокий уровень экспрессии названного белка в клетках, трансфицированных этой генетической конструкцией. Фактор роста гепатоцитов (HGF) стимулирует регенерацию печеночной ткани, оказывает защитное действие на гепатоциты и другие клетки, предотвращая их апоптоз, а также оказывает антифиброзное действие, индуцируя синтез протеиназ внеклеточного матрикса. HGF стимулирует миграцию резидентных стволовых клеток сердца из мест их локализации в участки повреждения, в частности, при инфаркте миокарда - в зону инфаркта. Для продуцирования HGF используют ДНК, содержащую природный ген фактора роста гепатоцитов человека, однако при его использовании не удается получить высокий уровень экспрессии данного белка.
Заявляемый препарат относится к фармакологической группе биологических препаратов для генной терапии, гепатопротекторов. Введение препарата внутривенно, может стимулировать процесс восстановления поврежденной фиброзом печени, влияя на выживаемость гепатоцитов, способствуя разрушению отложений коллагена и других белков, замещающих при фиброзе паренхиму печени.
Для получения больших количеств вышеупомянутых плазмидных конструкций, нанограммовые количества плазмидных ДНК (по отдельности) используют для трансформации коммерчески доступных штаммов кишечной палочки Esherichia coli, генотип которых позволяет использовать их для наработки больших количеств плазмидной ДНК. Трансформированные плазмидной ДНК бактерии размножают в приемлемой жидкой среде с необходимым антибиотиком для получения биомассы, содержащей плазмидную ДНК. Плазмидную ДНК выделяют и очищают, используя коммерчески доступные наборы или стандартные методы выделения плазмидной ДНК. Выделенные по отдельности плазмидные конструкции растворяют в бидистиллированной воде, доводя концентрацию ДНК в водном растворе до значения 1 мг/мл +/-0,2 мг/мл. Содержание примесей и состав плазмидных конструкций подтверждают доступными аналитическими методами, которые приняты для лабораторного анализа лекарственных средств, а также для определения качества и количества ДНК в растворе. Из полученных растворов индивидуальных плазмидных конструкций готовят смесь, содержащую водный раствор плазмиды pC4W-HGFopt (кодирует HGF человека) в пропорции от 30 до 70% и водный раствор плазмиды pVax1-UPAopt (кодирует урокиназу человека) в пропорции от 30 до 70%. В полученном таким образом препарате определяют количество примесей и соответствие препарата расчетной формуле, используя набор аналитических методов, принятых для анализа генотерапевтических конструкций, используемых в медицине. Для получения лиофилизованного препарата, водный раствор полученной смеси плазмид лиофильно высушивают. В результате получают сухой препарат с содержанием влаги не более 0,005 мг на 1 мг сухого вещества, который перед введением животным или пациентам растворяют в изотоническом водном растворе солей из расчета 1 мг на 1 мл раствора.
Для использования заявляемого лекарственного средства для лечения фиброза печени, лекарственное средство, если оно находится в сухом лиофилизованном состоянии, предварительно полностью растворяют в изотоническом стерильном солевом растворе в концентрации 1+/-0,01 мг/мл в дозах не более 3, 75 мг/кг для грызунов и не более 0,3125 мг/кг для человека. Введение препарата проводят двукратно с интервалом в 2-3 дня внутривенно. Эффективность лечения фиброза печени проводят гистологически и иммуногистохимически через 1-3 недели после введения последней дозы препарата, используя окрашивание срезов ткани печени с помощью гематоксилина и эозина и пикросириуса красного, а также с помощью специфических антител на коллаген по стандартным методикам с последующей визуализацией и документированием изображений. Уровень мРНК белков внеклеточного матрикса в образцах тканей печени, определяют используя метод обратной транскрипции-полимеразной цепной реакции с использованием в качестве матрицы образцов тотальной РНК, выделенной из тканей печени. Снижение уровня этих мРНК свидельствует об успешности лечения фиброза. Восстановление ткани печени также определяют, определяя уровень ферментов-метилтрансфераз ALT и AST в сыворотке крови с помощью стандартных диагностических наборов. Снижение уровней этих ферментов свидетельствует об успехе лечения. Для определения эффективности лечения фиброза сравнивают данные полученные в результате анализа состояния печени экспериментальных и контрольных здоровых мышей без фиброза печени.
Ниже представлены примеры реализации изобретения, которые не ограничивают настоящее изобретение, а лишь демонстрируют возможность его осуществления с достижением технического результата.
Пример 1. Получение штаммов продуцентов, необходимых для наработки лекарственного средства
Для трансформации и наработки плазмидной ДНК pC4W-HGFopt, которая содержит ген фактора роста гепатоцитов, используют штамм Е. coli XL1Blue-HGFopt, производный от штамма Е. coli XL1-Blue.
Для трансформации и наработки плазмидной ДНК pVAX-UPAopt, которая содержит ген урокиназы, используют штамм Е. coli XL1-Blue-UPAopt, производный от штамма Е. coli XL1-Blue.
Исходный штамм Е. coli XL1-Blue является музейным и хранится во Всероссийской Коллекции Промышленных Микроорганизмов. Полное видовое название исходного штамма Escherichia coli XL1-Blue.
Трансформированные штаммы Е. coli XL1-Blue-UPAopt и Е. coli XL1Blue-HGFopt отличаются от исходного штамма Е. coli XL1-Blue устойчивостью к антибиотику канамицину и ампициллину, соответственно, и обеспечиваемой введенной в состав штамма плазмидой pC4W-HGFopt или pVAX-UPAopt.
Генетические характеристики
Штамм бактерий E.coli XL1Blue-HGFopt имеет следующие генетические характеристики: endAl gyrA96(nalR) thi-1 recA1 relA1 lac glnV44 F'[::Tn10 proAB+lacIq Δ(lacZ)M15] hsdR17(rK- mK+), обладает устойчивостью к налидиксовой кислоте и тетрациклину и дополнительно содержит плазмиду pC4W-HGFopt в эписомном состоянии, придающую штамму устойчивость к ампициллину.
Штамм бактерий E.coli XL1Blue-UPAopt имеет следующие генетические характеристики: endA1 gyrA96(nalR) thi-1 recA1 relA1 lac glnV44 F'[::Tn10 proAB+lacIq Δ(lacZ)M15] hsdR17(rK- mK+), обладает устойчивостью к налидиксовой кислоте и тетрациклину и дополнительно содержит плазмиду pVAX-UPAopt в эписомном состоянии, придающую штамму устойчивость к канамицину.
Культурально-морфологические характеристики
Клетки мелкие, прямые, утолщенной палочковидной формы, грамотрицательные, неспороносные слабо подвижные, капсул и спор не образуют. Размер клеток около 4x1 мкм. Клетки хорошо растут на простых питательных средах. При росте на LB-агаре «BD» образуются круглые, гладкие, выпуклые, мутные, блестящие, серые колонии, с ровными краями. При росте в жидких средах (в среде LB) образуют интенсивную ровную муть.
Биологические характеристики
Аэроб. Температурный диапазон роста от плюс 4 до плюс 42°С при оптимуме pH 6,5-7,5. В качестве источника азота используют как минеральные соли в аммонийной и нитратной формах, так и органические соединения в виде аминокислот, пептона, триптона, дрожжевого экстракта и т.д. В качестве источника углерода используют аминокислоты, глицерин, углеводы.
Промышленные штаммы по предварительным оценкам не обладают патогенными, вирулентными, токсигенными свойствами. Исследование патогенных свойств штаммов будет проведено на последующих этапах разработки лекарственного препарата.
Способ получения штаммов-продуцентов
Штаммы-продуценты XL1Blue-HGFopt и XL1Blue-UPAopt для наработки плазмидных ДНК получали трансформацией исходного штамма Е. coli XL1-Blue плазмидой, соответственно, pC4W-HGFopt или pVAX-UPAopt, с последующим отбором рекомбинантных клонов на среде LB с ампициллином и канамицином, соответственно при 37°С.
Технология получения штаммов продуцентов трансформацией исходного штамма плазмидой не зависит в данном лабораторном процессе от типа плазмиды (pC4W-HGFopt или pVAX-UPAopt), поэтому в дальнейшем изложении не будет уточняться, какая именно плазмида используется в процессе, но подразумевается, что проводятся два идентичных процесса получения трансформированного штамма, содержащего одну из плазмид pC4W-HGFopt или pVAX-UPAopt. Остальные стадии технологического процесса до этапа смешивания двух субстанций и получения комбинированной субстанции так же проводятся одинаково для обоих трансформированных штаммов.
Для получения компетентных клеток бактерии исходного штамма Е. coli XL1-Blue высевали в стерильную пробирку емкостью 15 мл, содержащую 5 мл среды LB (1 л среды LB содержит 10 г триптона, 5 г дрожжевого экстракта, 10 г NaCl, pH среды доводят до 7,5 при помощи 1 М NaOH и стерилизуют автоклавированием). Пробирку инкубировали в течение ночи при 37°С в воздушном термостате. Культуру бактерий переносили в коническую стеклянную колбу емкостью 2 л, содержащую 400 мл среды LB и инкубировали в шейкере-инкубаторе при 37°С с покачиванием (300 об/мин) до того момента, когда оптическая плотность суспензии бактерий, измеренная при 600 нм, не составила 0,5. Далее суспензию клеток переносили в охлажденные до 4°С центрифужные стаканы емкостью 250 мл и центрифугировали в рефрижераторной центрифуге при 3000 g в течение 30 мин при 4°С. Осадок клеток суспендировали в 200 мл охлажденного до 4°С стерильного раствора 50 мМ CaCl2 и инкубировали суспензию при 0°С (во льду) в течение 30 мин, после чего суспензию клеток снова переносили в охлажденные до 4°С центрифужные стаканы и центрифугировали в рефрижераторной центрифуге при 3000 g в течение 30 мин при 4°С. Осадок клеток суспендировали в 40 мл охлажденного до 4°С стерильного раствора 50 мМ CaCl2, добавляли 6 мл стерильного глицерина, аликвотировали по 1 мл в ампулы для замораживания клеточных культур и замораживали в жидком азоте. Ампулы хранили при температуре минус 70°С.
Трансформацию компетентных клеток E.coli плазмидой проводили следующим образом. Ампулу с суспензией компетентных клеток, хранящуюся при минус 70°С, помещали в лед, дожидались размораживания, и 100 мкл суспензии переносили в стерильную пробирку емкостью 1,5 мл, находящуюся во льду. К суспензии бактерий добавляли 1 мкл стерильного раствора 1 мкг/мл плазмидной ДНК в буфере ТЕ (10 мМ Tris-HCl, 0,2 мМ EDTA) и инкубировали в течение 15 мин при 0°С (во льду). Далее пробирку помещали в водяную баню с температурой воды 43°С и инкубировали в течение 60 сек. После инкубации суспензию бактерий переносили на чашку Петри с микробиологическим агаром, приготовленным на среде LB и дополнительно содержащим 0,3 мг/мл ампициллина, и инкубировали в течение ночи при 37°С.
Колонии переносили в стерильные стеклянные колбы объемом 100 мл, содержащие 50 мл среды ТВ с 0,3 мг/мл ампициллина и инкубировали в шейкере-инкубаторе при 37°С с покачиванием (300 об/мин) в течение ночи для получения биомассы и измерения выхода плазмиды.
Пример 2. Культивирование бактерий и сбор биомассы для изготовления плазмидных ДНК pC4W-HGFopt и pVAX-UPAopt
Для получения биомассы для изготовления плазмидной ДНК pC4W-HGFopt (первого компонента субстанции) или pVAX-UPAopt (второго компонента субстанции), проводят следующие процедуры (шаги 1, 2 и 4 выполняют в ламинарном шкафу в стерильных условиях).
Размораживание и подращивание клеточных культур
1. Из колбы со средой с солями и амипициллином в стерильную стеклянную коническую колбу емкостью 250 мл переносят 50 мл среды при помощи серологической пипетки объемом 25 мл.
2. Из рабочего банка бактерий E.coli штамма-продуцента XL1Blue-HGFopt (в случае изготовления плазмидной ДНК pC4W-HGFopt) или штамма-продуцента XL1 Blue-UPAopt (в случае изготовления плазмидной ДНК pVAX-UPAopt), хранящегося при минус 70°С, размораживают одну ампулу и при помощи серологической пипетки объемом 1 мл переносят 1 мл культуры бактерий в колбу с 50 мл среды, приготовленную на предыдущем шаге.
3. Колбу помещают в шейкер-инкубатор и культивируют бактерии 24 ч при 37°С со скоростью качания 25 об/мин.
Выращивание культур в колбах и сбор биомассы
4. В 4 колбы объемом 2 л, содержащие по 1000 мл среды ТВ с солями и антибиотиком, вносят по 10 мл культуры бактерий, полученной на предыдущем шаге.
5. Колбы помещают в шейкер-инкубатор (Т-1) и культивируют бактерии 20 ч при 37°С со скоростью качания 250 об/мин.
6. По окончании культивирования из каждой колбы отбирают по 1 мл культуры бактерий при помощи серологической пипетки объемом 1 мл и измеряют оптическую плотность при длине волны 600 нм, которая должна быть не менее 3.
7. Культуру бактерий, полученную на шаге 5, переливают из 2 колб в 6 центрифужных стаканов объемом 470 мл, по 333 мл на стакан, уравновешивают стаканы попарно и центрифугируют при 6000 об/мин 30 мин на центрифуге (Ф-5) J-21C в роторе JA10 при 4°С.
8. Жидкость сливают, культуру бактерий из оставшихся 2 колб переливают в те же центрифужные стаканы, по 333 мл на стакан, уравновешивают стаканы попарно и центрифугируют при 6000 об/мин 30 мин на центрифуге (Ф-5) J-21C в роторе JA10 при 4°С.
9. Жидкость сливают, стаканы с осадком бактерий центрифугируют при 2000 об/мин 3 мин на центрифуге (Ф-5) J-21C в роторе JA10 при 4°С.
10. Остатки жидкости в стаканах убирают при помощи серологической пипетки объемом 5 мл.
11. Все осадки переносят при помощи шпателя из стаканов в предварительно взвешенную пробирку объемом 50 мл, взвешивают с целью определить вес биомассы и замораживают в морозильной камере при минус 70°С. Биомассу хранят не более 90 сут.
Пример 3. Характеристика лекарственного средства и подтверждение его подлинности
Лекарственное средство содержит следующие компоненты: ДНК (pC4W-HGFopt) 0,05 г, ДНК (pVAX-UPAopt) 0,05 г, натрия хлорид 0,9 г, вода для инъекций 100 мл. Структура соответствующих плазмид фиг. 1 и 2.
Описание. Субстанция представляет собой бесцветную прозрачную жидкость или лиофилизованный осадок, который перед внутривенным введением или для анализа необходимо растворить в изотоническом растворе.
Подлинность. Соответствует смеси плазмидных ДНК pC4W-HGFopt и pVAX-UP Aopt в весовой пропорции 1:1.
Определяют следующими методами.
1. Методом спектрофотометрии: в спектральном диапазоне 240 нм - 320 нм максимум поглощения должен быть единственным и находиться в пределах длин волн от 255 нм до 265 нм; отношение А260/А280 должно быть в пределах от 1,8 до 2,0; отношение А320/А260 не должно превышать 0,01. Примеры кривых поглощения растворов ДНК приведены на фиг. 3 и 4.
2. Методом рестрикционного анализа: при обработке рестриктазами Sail и BstEII размеры фрагментов должны составлять 715 п. н., 852 п. н, 1375 п. н., 2197 п. н. и 4658 п. н. (всего 5 фрагментов); при обработке рестриктазами BbsI и HindIII размеры фрагментов должны составлять 696 п. н., 1386 п. н., 1668 п. н., 2369 п. н. и 3820 п. н. (всего 5 фрагментов); при обработке рестриктазами HindIII и EcoRV размеры фрагментов должны составлять 554 п. н., 782 п. н, 886 п. н., 1532 п. н. и 5350 п. н. (всего 5 фрагментов). Количество наблюдаемых фрагментов для каждой обработки должно совпадать с указанным. Полученные в результате анализа значения размеров фрагментов не должны отличаться от указанных более чем на 20%. Отношение весового количества плазмидной ДНК pC4W-HGFopt к весовому количеству плазмидной ДНК pVAX-UPAopt должно быть в пределах от 0,8 до 1,2.
Прозрачность раствора. Раствор субстанции после размораживания должен быть прозрачным по сравнению с водой для инъекций.
Цветность раствора. Раствор субстанции после размораживания должен быть бесцветным по сравнению с водой для инъекций.
pH. pH раствора субстанции должен быть в пределах от 5,2 до 7,2. Определение проводят потенциометрическим методом.
Посторонние примеси
Содержание белков E.coli в субстанции должно быть не более 10 мкг на 1 мг ДНК плазмидной ДНК. Определение примеси белков E.coli методом иммуноферментного анализа.
Содержание РНК E.coli в субстанции должно быть не более 10 мкг на 1 мг плазмидной ДНК. Определение примеси РНК E.coli методом окрашивания SYBR Green II.
Содержание геномной ДНК E.coli в субстанции должно быть не более 50 мкг на 1 мг плазмидной ДНК. Определение примеси геномной ДНК E.coli методом полимеразной цепной реакции.
Содержание бактериальных эндотоксинов. Содержание бактериальных эндотоксинов в субстанции должно быть не более 93 ЕЭ/мл. Гель-тромб тест с лизатом амебоцитов мечехвоста Limulus polyphemus, количественный анализ методом В, по ОФС 42-0062-07 (ГФ XII, ч. 1, стр. 128-136).
Токсичность. Субстанция должна быть нетоксичной в дозе 0,1 мг плазмидной ДНК на мышь весом 19-21 г. Определение проводят на белых мышах. Тест-доза на одно животное 0,1 мг субстанции, разведенной стерильным 0,9%-ным раствором натрия хлорида для инъекций до конечного объема 0,5 мл. Субстанцию вводят в хвостовую вену. Подготовку животных и испытания осуществляют в соответствии с ОФС 42-0060-07 ГФ XII.
Стерильность. Субстанция должна быть стерильной. Испытание проводят методом прямого посева по ГФ XII, ч.1, ОФС 42-0066-07, стр. 155.
Количественное определение.
Концентрация нуклеиновой кислоты. Концентрация нуклеиновой кислоты в субстанции должна быть в пределах от 0,8 мг/мл до 1,2 мг/мл. Определение проводят методом спектрофотометрии.
Содержание хлорида натрия. Содержание натрия хлорида в субстанции должно быть в пределах от 0,85 г до 0,95 г на 100 мл. Определение проводят аргентометрическим титрованием по методу Фольгарда в модификации Колдуэлла и Мойера.
Содержание основного вещества. Содержание основного вещества в субстанции должно быть не менее 90%. Определение проводят методом электрофоретического разделения в агарозном геле.
Пример 4. Выделение и очистка плазмидных ДНК pC4W-HGFopt и pVAX-UPAopt
Выполняется одинаковым образом для плазмид pC4W-HGFopt и pVAX-UPAopt.
Приготовление растворов
Для проведения первой стадии очистки плазмидной ДНК готовят следующие стоковые растворы.
1. 18 М NaOH. Для приготовления 1 л раствора взвешивают на весах с точностью до 0,1 г 720 г гидроксида натрия, переносят в стеклянную мерную колбу емкостью 1000 мл, добавляют деионизованную воду и перемешивают на магнитной мешалке до полного растворения. Объем раствора доводят до 1000 мл деионизованной водой. Хранят в пластиковом флаконе емкостью 1000 мл при комнатной температуре не более 90 сут.
2. 1 М Трис-HCl, pH 8,0. Для приготовления 2 л раствора взвешивают на весах (КП-3) с точностью до 0,1 г 242,2 г трис(гидроксиметил)аминометана, переносят в стеклянную мерную колбу емкостью 2000 мл, добавляют 1600 мл очищенной воды и перемешивают на магнитной мешалке до полного растворения. Затем измеряют pH раствора и доводят pH до значения 8,0 добавлением концентрированной соляной кислоты (около 100 мл). После этого доводят объем раствора до 2000 мл деионизованной водой. Хранят в пластиковых флаконах емкостью 1000 мл при 4°С не более 90 сут.
3. 0,5 М EDTA, pH 8,0. Для приготовления 1 л раствора взвешивают на весах с точностью (КП-3) до 0,1 г 146,1 г этилендиаминтетрауксусной кислоты (EDTA), переносят в стеклянную мерную колбу емкостью 1000 мл, добавляют 800 мл деионизованной воды и перемешивают на магнитной мешалке. Затем измеряют pH раствора и доводят pH до значения 8,0 добавлением 18 М раствора NaOH. После полного растворения вещества доводят объем раствора до 1000 мл деионизованной водой. Хранят в пластиковом флаконе емкостью 1000 мл при 4°С не более 90 сут.
4. 3 М ацетат калия, pH 5,5. Для приготовления 2 л раствора взвешивают на весах с точностью до 0,1 г 588,9 г ацетата калия, переносят в стеклянную мерную колбу емкостью 2000 мл, добавляют 1000 мл деионизованной воды и перемешивают на магнитной мешалке до полного растворения. Затем измеряют pH раствора (КП-1) и доводят pH до значения 5,5 добавлением концентрированной уксусной кислоты (около 320 мл). После этого доводят объем раствора до 2000 мл деионизованной водой. Хранят в пластиковых флаконах емкостью 1000 мл при 4°С не более 90 сут.
5. 5 М ацетат калия. Для приготовления 1 л раствора взвешивают на весах с точностью до 0,1 г 490,8 г ацетата калия, переносят в стеклянную мерную колбу емкостью 1000 мл, добавляют 500 мл деионизованной воды и перемешивают на магнитной мешалке до полного растворения. После этого доводят объем раствора до 1000 мл деионизованной водой. Хранят в пластиковом флаконе емкостью 1000 мл при комнатной температуре не более 90 сут.
6. 20% SDS. Для приготовления 1 л раствора взвешивают на весах (КП-3) с точностью до 0,1 г 200 г додецилсульфата натрия (SDS), переносят в стеклянную мерную колбу емкостью 1000 мл, добавляют 600 мл деионизованной воды и перемешивают на магнитной мешалке до полного растворения. После этого доводят объем раствора до 1000 мл деионизованной водой.
Рабочие растворы
1. Раствор А (50 мМ Трис-HCl, pH 8.0, 10 мМ EDTA). Для приготовления 2 л раствора А в стеклянную мерную колбу емкостью 2000 мл добавляют 1800 мл деионизованной воды, 100 мл стокового раствора 1 М Трис-HCl, pH 8,0 и 40 мл стокового раствора 0,5 М EDTA, pH 8,0. После этого доводят объем раствора до 2000 мл деионизованной водой. Раствор В (140 мМ NaOH, 1% SDS). Для приготовления 2 л раствора В в стеклянную мерную колбу емкостью 2000 мл добавляют 1800 мл деионизованной воды, 15,6 мл стокового раствора 18 М NaOH и 100 мл стокового раствора 20% SDS. После этого доводят объем раствора до 2000 мл деионизованной водой. Таблица 5.16. Расход реактивов для приготовления раствора 140 мМ NaOH, 1% SDS (Раствор В).
2. Раствор 1 М ацетат калия, pH 5,5. Для приготовления 450 мл раствора в пластиковый флакон емкостью 500 мл добавляют 300 мл деионизованной воды и 150 мл стокового раствора 3 М ацетата калия, pH 5,5. Хранят при 4°С не более 90 сут.
3. Раствор С (50 мМ Трис-HCl, pH 8.0, 1 мМ EDTA). Для приготовления 1 л раствора С в стеклянную мерную колбу емкостью 1000 мл добавляют 800 мл деионизованной воды, 100 мл стокового раствора 1 М Трис-HCl, pH 8,0 и 4 мл стокового раствора 0,5 М EDTA, pH 8,0. После этого доводят объем раствора до 1000 мл деионизованной водой. Хранят в пластиковом флаконе емкостью 1000 мл при 4°С не более 90 сут.
4. Раствор D (13% PEG 8000, 1,6 М NaCl). Для приготовления 1 л раствора взвешивают на весах (КП-3) с точностью до 0,1 г 130 г полиэтиленгликоля (PEG) 8000 и 93,5 г хлорида натрия, переносят в стеклянную мерную колбу емкостью 1000 мл, добавляют 700 мл деионизованной воды и перемешивают на магнитной мешалке до полного растворения. После этого доводят объем раствора до 1000 мл деионизованной водой. Хранят в пластиковом флаконе емкостью 1000 мл при 4°С не более 90 сут.
5. Раствор ТЕ (10 мМ Трис-HCl, pH 8,0, 0,2 мМ EDTA). Для приготовления 500 мл раствора в пластиковый флакон емкостью 500 мл добавляют 495 мл деионизованной воды, 5 мл стокового раствора 1 М Трис-HCl, pH 8,0 и 0,2 мл стокового раствора 0,5 М EDTA. Хранят при 4°С не более 90 сут.
6. Раствор РНКазы А (10 мг/мл). Для приготовления раствора размораживают концентрат РНКазы А (100 мг/мл) из набора «EndoFree Plasmid Giga Kit» (Qiagen, США). В пластиковую пробирку вместимостью 1,5 мл при помощи ручных дозаторов переменного объема с наконечниками 100-1000 мкл и 20-200 мкл вносят 0,9 мл раствора С и 0,1 мл концентрата РНКазы А. Раствор перемешивают. Хранят при минус 20°С не более 90 сут.
Первую стадию очистки плазмидной ДНК выполняют следующим образом.
1. Подготавливают стеклянный стакан объемом 2000 мл, содержащий 1500 мл охлажденного до 4°С раствора А и пустой стеклянный стакан объемом 2000 мл. Пробирки с биомассой, хранящиеся при минус 70°С и содержащие в сумме от 80 г до 100 г биомассы, размораживают инкубированием 60 мин при комнатной температуре. В пробирки с биомассой добавляют раствор А из первого стакана и перемешивают биомассу стеклянной палочкой. По мере суспендирования биомассы суспензию бактерий переносят в пустой стакан при помощи ручного дозатора с наконечником 1-5 мл и добавляют в пробирки новые порции раствора А из первого стакана. После переноса всей биомассы во второй стакан туда выливают раствор А, оставшийся в первом стакане. Суспензию перемешивают на магнитной мешалке в течение 1 ч при 4°С. Затем суспензию разносят по 12 центрифужным стаканам объемом 470 мл, по 137 мл на стакан, при помощи серологической пипетки объемом 25 мл.
2. В каждый центрифужный стакан добавляют по 125 мл раствора В и перемешивают переворачиванием 10 раз. Стаканы инкубируют 5 мин при комнатной температуре.
3. В каждый стакан добавляют по 125 мл охлажденного до 4°С раствора 3 М ацетата калия, pH 5,5 и перемешивают переворачиванием 10 раз. Стаканы инкубируют 10 мин при комнатной температуре.
4. Первые 6 стаканов центрифугируют при 6000 об/мин 30 мин. на центрифуге J-21С (Ф-5) в роторе JA10 при 4°С (стаканы уравновешивают попарно при помощи раствора 1 М ацетата калия, pH 5,5).
5. Шаг 4 повторяют для оставшихся 6 центрифужных стаканов.
6. Жидкость из всех стаканов переливают в пластиковые флаконы емкостью 1000 мл и фильтруют через фильтры «QIAfilter Giga Cartridge» из набора «EndoFree Plasmid Giga Kit», в среднем по 900 мл жидкости на фильтр. Фильтрация происходит при подключении фильтра к водоструйному насосу при помощи силиконового шланга. Фильтрат собирают в пластиковые флаконы емкостью 1000 мл.
7. Жидкость, полученную на предыдущем шаге, разносят по 10 пластиковым флаконам емкостью 1000 мл, по 450 мл на флакон. В каждый флакон добавляют 360 мл изопропанола. Перемешивают взбалтыванием и инкубируют 1 ч или ночь при 4°С.
8. Жидкость в 3 флаконах взбалтывают, разносят по 6 центрифужным стаканам объемом 470 мл, по 405 мл на стакан и центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С (стаканы уравновешивают попарно).
9. Супернатант сливают, жидкость из следующих 3 флаконах взбалтывают, переливают в те же центрифужные стаканы и центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С (стаканы уравновешивают попарно).
10. Шаг 9 повторяют для следующих 3 флаконов.
11. Супернатант сливают, жидкость из последнего флакона переливают в те же центрифужные стаканы, по 135 мл на стакан, и центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С (стаканы уравновешивают попарно).
12. Супернатант сливают, стаканы с осадком центрифугируют при 2000 об/мин 3 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С. Остатки жидкости убирают при помощи серологической пипетки объемом 5 мл.
13. Осадок в каждом стакане суспендируют в 200 мл раствора С при помощи серологической пипетки объемом 25 мл, добавляют 200 мл раствора 5 М ацетата калия перемешивают взбалтыванием и инкубируют 1 ч при комнатной температуре.
14. Стаканы центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 20°С (стаканы уравновешивают попарно).
15. Супернатант из каждого стакана переливают в пластиковый флакон емкостью 1000 мл, добавляют 400 мл изопропанола, перемешивают взбалтыванием и инкубируют 1 ч или ночь при 4°С.
16. Жидкость в 3 флаконах взбалтывают, разносят по 6 центрифужным стаканам объемом 470 мл, по 400 мл на стакан, и центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С (стаканы уравновешивают попарно).
17. Супернатант сливают, жидкость из оставшихся 3 флаконов взбалтывают, переливают в те же центрифужные стаканы и центрифугируют при 6000 об/мин 30 мин на центрифуге J-21C в роторе JA10 при 4°С (стаканы уравновешивают попарно).
18. Супернатант сливают, стаканы с осадком центрифугируют при 2000 об/мин 3 мин на центрифуге J-21C (Ф-5) в роторе JA10 при 4°С. Остатки жидкости убирают при помощи серологической пипетки объемом 5 мл.
19. Осадок в каждом стакане суспендируют в 25 мл раствора С при помощи серологической пипетки объемом 25 мл и переносят в пластиковую пробирку объемом 50 мл. Затем при помощи ручного дозатора переменного объема с наконечником 20-200 мкл добавляют 25 мкл раствора РНКазы А и инкубируют 1,5 ч при 37°С.
20. По окончании инкубации в каждую пробирку добавляют по 25 мл раствора D и инкубируют 3 ч или ночь при комнатной температуре.
21. Пробирки центрифугируют при 4000 об/мин 40 мин на центрифуге B4i.
22. Супернатант сливают, пробирки с осадком центрифугируют при 3000 об/мин 3 мин. на центрифуге B4i. Остатки жидкости убирают ручным дозатором с наконечником 100-1000 мкл.
23. Осадок в каждой пробирке суспендируют в 12 мл раствора С при помощи серологической пипетки объемом 10 мл и пипетируют до полного растворения. Затем добавляют 12 мл раствора 5 М ацетата калия, перемешивают взбалтыванием, добавляют 24 мл изопропанола, перемешивают взбалтыванием и инкубируют 1 ч или ночь при минус 20°С.
24. Пробирки центрифугируют при 4000 об/мин 30 мин на центрифуге B4i.
25. Супернатант сливают, пробирки с осадком центрифугируют при 3000 об/мин 3 мин на центрифуге B4i. Остатки жидкости убирают ручным дозатором с наконечником 100-1000 мкл.
26. Осадок в каждой пробирке суспендируют в 7 мл раствора ТЕ при помощи серологической пипетки объемом 10 мл и пипетируют до полного растворения. Жидкость из всех пробирок собирают в одну пробирку объемом 50 мл.
27. Измеряют объем и оптическую плотность раствора (КП-4) плазмидной ДНК при 260 нм (OD260). Вычисляют концентрацию и количество ДНК. Для вычисления концентрации ДНК используют следующую формулу:
С=OD260⋅0,05 (мг/ мл), где С - концентрация ДНК.
Выход частично очищенной ДНК на данной стадии 120-240 мг.
Вторая стадия очистки плазмидной ДНК с помощью набора «EndoFree Plasmid Giga Kit» (Qiagen, США)
Процедура выполняется одинаковым образом для плазмид pC4W-HGFopt и pVAX-UPAopt.
Все буферные растворы и колонки (Ф-9) входят в состав набора «EndoFree Plasmid Giga Kit» и имеют названия, данные производителем. Исключения составляют реактивы, используемые на конечных стадиях очистки: изопропанол, этанол и стерильный раствор 0,9%-ного хлорида натрия.
Вторую стадию очистки плазмидной ДНК выполняют следующим образом (шаги 7-16 проводят в ламинарном шкафу в стерильных условиях).
1. Раствор плазмидной ДНК, полученный после проведения первой стадии очистки, размораживают инкубированием пробирки с раствором в пластиковом флаконе объемом 250 мл, содержащей 200 мл дистиллированной воды, 1 ч при комнатной температуре. Порцию раствора, содержащую 12 мг ДНК, переносят в пластиковый флакон емкостью 500 мл и добавляют буфер Р1 из набора до конечного объема 125 мл. Затем добавляют 125 мл раствора ТЕ, 125 мл буфера Р3 и перемешивают.
2. Шаг 1 проводят для других порций раствора плазмидной ДНК, полученного после проведения первой стадии очистки (суммарно от 10 до 20 порций). Дальнейшие шаги проводят параллельно для каждой порции раствора.
3. К порции раствора, полученной на предыдущем шаге, добавляют 30 мл охлажденного на льду буфера ER из набора, перемешивают и инкубируют на льду 30 мин.
4. Колонку "QIAGEN-tip 10000" (Ф-9) из набора уравновешивают 75 мл буфера QBT из набора.
5. Затем на колонку наносят порцию раствора, полученного на шаге 3, и дают возможность нанесенной жидкости пройти через колонку под действием силы тяжести.
6. На колонку наносят 600 мл буфера QC из набора и дают возможность нанесенной жидкости пройти через колонку под действием силы тяжести.
7. Плазмидную ДНК элюируют с колонки путем пропускания через нее 100 мл буфера QN из набора и дают возможность нанесенной жидкости пройти через колонку под действием силы тяжести. Элюат собирают в стерильный пластиковый флакон емкостью 250 мл.
8. Элюат с каждой колонки разносят по 4 стерильным пробиркам объемом 50 мл при помощи серологической пипетки на 10 мл, по 25 мл на пробирку. В каждую пробирку добавляют 20 мл изопропанола, перемешивают взбалтыванием и инкубируют 1 ч или ночь при минус 20°С.
9. Пробирки центрифугируют при 4000 об/мин 30 мин на центрифуге B4i (Ф-6).
10. Супернатант сливают, пробирки с осадком центрифугируют при 3000 об/мин 3 мин на центрифуге B4i. Остатки жидкости убирают при помощи серологической пипетки объемом 1 мл.
Осаждение ДНК этиловым спиртом, +4°С
11. Осадок в каждой пробирке суспендируют в 3 мл стерильного раствора 0,9%-ного хлорида натрия при помощи серологической пипетки объемом 10 мл и пипетируют до полного растворения. Жидкость из 4 пробирок собирают в одну пробирку объемом 50 мл, добавляют 30 мл этанола и инкубируют 1 ч или ночь при минус 20°С.
12. Пробирки центрифугируют при 4000 об/мин 30 мин на центрифуге B4i (Ф-6).
13. Супернатант сливают, пробирки с осадком центрифугируют при 3000 об/мин 3 мин на центрифуге B4i (Ф-6). Остатки жидкости убирают при помощи серологической пипетки объемом 1 мл.
Разведение осадка в растворе натрия хлорида 0,9%
14. Осадок в каждой пробирке суспендируют в 6 мл стерильного раствора 0,9%-ного хлорида натрия при помощи серологической пипетки объемом 10 мл и пипетируют до полного растворения. Жидкость из всех пробирок собирают в одну стерильную пластиковую пробирку емкостью 50 мл, перемешивают и измеряют объем и оптическую плотность раствора плазмидной ДНК при 260 нм. Вычисляют концентрацию и количество ДНК.
Выход очищенной плазмидной ДНК pC4W-HGFopt или pC4W-UPAopt на второй стадии очистки составляет от 50% до 85% или 60-200 мг в расчете на один цикл очистки.
Приготовление стерильного 0,9% раствора NaCl
После полного растворения натрия хлорида в воде раствор стерильно фильтруют через фильтр с размером пор 0,2 мкм.
Нормирование субстанций, содержащих плазмидную ДНК pC4W-HGFopt или pVAX-UPAopt, до концентрации 1 мг/мл
Концентрацию ДНК доводят до 1 мг/мл при помощи стерильного раствора 0,9%-ного хлорида натрия.
Для проведения предварительного контроля качества отбирают 5 мл раствора плазмидной ДНК в стерильную ампулу для замораживания.
Выход продукта составляет не менее 55 мл для раствора 1 мг/мл каждой из плазмидных ДНК pC4W-HGFopt и pC4W-UPAopt.
Пример 5. Смешивание в соотношении 1:1 субстанций, содержащих плазмидные ДНК pC4W-HGFopt и pC4W-UPAopt и лиофилизация
В стерильном пластиковом флаконе емкостью 250 мл в ламинарном шкафу в стерильных условиях смешивают равные объемы растворов, очищенных плазмидных ДНК pC4W-HGFopt и pVAX-UPAopt, полученных на предыдущей стадии и успешно прошедших предварительный контроль качества.
Полученная таким образом субстанция является стерильной и апирогенной. Субстанцию хранят в замороженном виде при минус 20°С.
Выход продукта на стадии составляет не менее 100 мл.
Для лиофилизации лекарственного средства раствор плазмидных ДНК помещают в подходящую емкость из стекла и лиофилизуют, используя лиофильную сушку и рекомендации производителей по высушиванию растворов, содержащих ДНК. После сушки флаконы с лиофилизатом плотно укупоривают и хранят в сухом прохладном месте.
Пример 6. Структурные и эмпирические формулы и молекулярная масса плазмид, входящих в лекарственное средство
Структурная формула плазмидной ДНК pC4W-HGFopt представлена на фиг. 1.
Структурная формула плазмидной ДНК pVax1-UPAopt представлена на фиг. 2.
Эмпирическая формула плазмидной ДНК pC4W-HGFopt:
C147717H185567N56673O90840P15140
Эмпирическая формула плазмидной ДНК pVax1-UPAopt:
C83784H105284N32316O51600P8600
Молекулярная масса плазмидной ДНК pC4W-HGFopt: 4677148,1 Да
Молекулярная масса плазмидной ДНК pVax1-UPAopt: 2657009,6 Да
Пример 7. Рестрикционный анализ смеси плазмидных ДНК
Этот анализ имеет целью подтвердить наличие определенных плазмидных ДНК, а именно pC4W-HGFopt и pVax1-UPAopt, в субстанции, а также проконтролировать равную представленность указанных плазмидных ДНК в смеси (весовая пропорция должна составлять 1:1). На фиг. 1 и 2 представлены функциональные карты плазмидных ДНК pC4W-HGFopt и pVax1-UPAopt. Показаны следующие функциональные элементы: оптимизированный ген фактора роста гепатоцитов HGFopt или оптимизированный ген ангиопоэтина 1 AGPopt, промотор ранних генов цитомегаловируса (CMV), интрон из гена β-глобина кролика, участок инициации трансляции, посттранскрипционный регуляторный элемент из вируса гепатита лесного сурка (WPRE), сигнал полиаденилирования мРНК из гена гормона роста быка, сигнал полиаденилирования мРНК ранних генов вируса SV40, ген устойчивости к ампициллину, ген устойчивости к канамицину, участок инициации репликации ColE1 (pUC ori). Указаны также координаты сайтов узнавания и расщепления плазмидных ДНК ферментами рестрикции HindIII, XbaI, EcoRV, PflMI, Acc65I, BbsI, BamHI и BstEII. Эти координаты получены на основании анализа нуклеотидных последовательностей плазмидных ДНК pC4W-HGFopt и pVax1-UPAopt (см. раздел 2.1.5. Структурная и эмпирическая формулы и молекулярная масса).
Для идентификации плазмидных ДНК применяют рестрикционный анализ, то есть расщепление ДНК определенными комбинациями ферментов рестрикции (рестриктазами), или секвенирование (определение нуклеотидной последовательности). В силу того, что субстанция содержит смесь двух различных плазмидных ДНК, непосредственное секвенирование субстанции невозможно.
После обработки рестриктазами фрагменты расщепления плазмидных ДНК разделяют при помощи электрофореза в агарозном геле в присутствии бромистого этидия, что позволяет определить их размеры, выраженные в числе пар нуклеотидов (п.н.). По наличию полного набора фрагментов расщепления, характерных для данной плазмидной ДНК, судят о ее присутствии в анализируемом образце. Проведенные расчеты показывают, что в случае смеси плазмидных ДНК pC4W-HGFopt и pVax1-UPAopt для подобного анализа оптимально использовать следующие комбинации рестриктаз.
1. HindIII и XbaI. Совместное расщепление этими рестриктазами должно приводить к появлению трех фрагментов с размерами 563 п. н., 1668 п. н. и 5339 п. н., характерных для плазмидной ДНК pC4W-HGFopt, и трех фрагментов с размерами 452 п. н., 929 п. н. и 2919 п. н, характерных для плазмидной ДНК pVax1-UPAopt.
2. HindIII и EcoRV. Совместное расщепление этими рестриктазами должно приводить к появлению четырех фрагментов с размерами 554 п. н., 782 п. н., 886 п. н. и 5348 п. н., характерных для плазмидной ДНК pC4W-HGFopt, и двух фрагментов с размерами 1354 п. н. и 2946 п. н., характерных для плазмидной ДНК pVax1-UPAopt.
3. PflMI и Acc65I. Совместное расщепление этими рестриктазами должно приводить к появлению двух фрагментов с размерами 2448 п. н. и 5122 п. н., характерных для плазмидной ДНК pC4W-HGFopt и двух фрагментов с размерами 974 п. н. и 3326 п. н., характерных для плазмидной ДНК pVax1-UPAopt.
4. BbsI и BamHI. Совместное расщепление этими рестриктазами должно приводить к появлению четырех фрагментов с размерами 651 п. н., 876 п. н., 2223 п. н. и 3820 п. н., характерных для плазмидной ДНК pC4W-HGFopt, и двух фрагментов с размерами 1589 п. н. и 2711 п. н., характерных для плазмидной ДНК pVax1-UPAopt.
Для контроля равной представленности двух плазмидных ДНК в смеси применяют денситометрирование цифровой фотографии продуктов электрофоретического разделения. Уровень яркости полосы на фотографии, соответствующей определенному фрагменту ДНК, пропорционален отношению размера данного фрагмента к полному размеру той плазмидной ДНК, продуктом расщепления которой он является, и также пропорционален исходному количеству этой плазмидной ДНК в образце. Поскольку размеры как самих плазмидных ДНК pC4W-HGFopt и pVax1-UPAopt, так и продуктов их расщепления вышеуказанными рестриктазами известны, весовое соотношение плазмидных ДНК в исходной смеси можно определить, измеряя и сравнивая уровни яркости полос, соответствующих какой-либо паре фрагментов ДНК, где один из фрагментов является продуктом расщепления pC4W-HGFopt, а другой - продуктом расщепления pVax1-UPAopt.
Приготовление растворов. Для приготовления растворов используют реактивы производства фирм Bio-Rad (США) и Sigma (США) или аналогичные.
1. 18 М раствор натрия гидроксида. 72 г натрия гидроксида помещают в мерную колбу вместимостью 100 мл, добавляют воду очищенную до метки и перемешивают на магнитной мешалке до полного растворения. Хранят в пластиковом флаконе вместимостью 250 мл при комнатной температуре. Срок годности 1 г.
2. 0,5 М раствор натрия эдетата. 168,1 г натрия эдетата помещают в мерную колбу вместимостью 1000 мл, добавляют около 800 мл воды очищенной и перемешивают с помощью магнитной мешалки. Измеряют pH полученного раствора и, при необходимости, доводят pH до значения 8,0 с помощью раствора 18 М натрия гидроксида, контролируют значение pH потенциометрически. Доводят объем раствора до метки водой. Хранят в пластиковом флаконе при температуре 4°С. Срок годности 1 г.
3. 1%-ный раствор бромистого этидия. 0,5 г бромистого этидия помещают в пробирку вместимостью 50 мл, добавляют воды очищенной до метки, перемешивают. Хранят в защищенном от света месте при комнатной температуре. Срок хранения 1 г.
4. 50-кратный концентрат буфера ТАЕ. 242,2 г трис(гидроксиметил) аминометана растворяют в 600 мл воды очищенной в мерной колбе вместимостью 1000 мл, добавляют 57,1 мл уксусной кислоты ледяной, 100 мл 0,5 М раствора натрия эдетата и доводят объем раствора водой очищенной до метки. Хранят в пластиковом флаконе при температуре 4°С. Срок годности 1 г.
5. Буфер ТАЕ с бромистым этидием (концентрация бромистого этидия 1 мкг/мл). В мерную колбу вместимостью 1000 мл вносят 20 мл 50-кратного концентрата буфера ТАЕ и доводят объем раствора водой очищенной до метки. Добавляют 100 мкл 1%-ного раствора бромистого этидия, перемешивают. Хранят в пластиковом флаконе при температуре 4°С. Срок годности 7 сут.
6. 5-кратный концентрат утяжелителя с краской-маркером. 25 г сахарозы и 0,05 г красителя Понсо С помещают в пробирку вместимостью 50 мл, добавляют 20 мл воды очищенной и 10 мл 0,5 М раствора натрия эдетата, перемешивают и доводят объем раствора водой очищенной до метки. Раствор разливают по 1 мл в пластиковые пробирки вместимостью 1,5 мл и хранят при температуре минус 20°С. Срок годности 3 г.
Обработка рестриктазами. Используют рестриктазы или их изошизомеры, 10-кратные реакционные буферы и маркеры размеров фрагментов ДНК фирмы Thermo Scientific (США) или аналогичные реактивы других производителей. Для проведения анализа готовят следующие реакционные смеси.
1. Пробирка №1. В коническую пробирку с крышкой из полипропилена для микропроб однократного применения вместимостью 0,5 мл вносят 16 мкл деионизованной воды, 2 мкл 10-кратного реакционного буфера «Y», 1 мкл образца субстанции, 0,5 мкл рестриктазы HindIII (10000 ед./мл) и 0,5 мкл рестриктазы XbaI (10000 ед./мл). После внесения всех компонентов смесь тщательно перемешивают.
2. Пробирка №2. Аналогичным образом в пробирку №2 вносят 16 мкл деионизованной воды, 2 мкл 10-кратного реакционного буфера «R», 1 мкл образца субстанции, 0,5 мкл рестриктазы HindIII (10000 ед./мл) и 0,5 мкл рестриктазы EcoRV (10000 ед./мл, торговое название изошизомера данной рестриктазы у фирмы Thermo Scientific - Eco32I). После внесения всех компонентов смесь тщательно перемешивают.
3. Пробирка №3. Аналогичным образом в пробирку №3 вносят 16 мкл деионизованной воды, 2 мкл 10-кратного реакционного буфера «R», 1 мкл образца субстанции, 0,5 мкл рестриктазы PflMI (10000 ед./мл, торговое название изошизомера данной рестриктазы у фирмы Thermo Scientific - Van91I) и 0,5 мкл рестриктазы Acc65I (10000 ед./мл). После внесения всех компонентов смесь тщательно перемешивают.
4. Пробирка №4. Аналогичным образом в пробирку №4 вносят 16 мкл деионизованной воды, 2 мкл 10-кратного реакционного буфера «О», 1 мкл образца субстанции, 0,5 мкл рестриктазы BbsI (10000 ед./мл, торговое название изошизомера данной рестриктазы у фирмы Thermo Scientific - BpiI) и 0,5 мкл рестриктазы BamHI (10000 ед./мл). После внесения всех компонентов смесь тщательно перемешивают.
Пробирки помещают в сухой воздушный термостат и инкубируют 16-20 ч при температуре 37°С. По окончании инкубации к пробиркам №1-4 добавляют пробирку «М» с маркерами размеров фрагментов ДНК, которую готовят внесением 16 мкл деионизованной воды, 2 мкл 10-кратного реакционного буфера «О» и 2 мкл препарата маркеров «GeneRuler DNA Ladder Mix (100-10,000 bp)». Далее проводят электрофорез в 0,8%-ном агарозном геле в присутствии бромистого этидия.
Подготовка агарозного геля. В качестве подложки (или емкости) для полимеризации агарозного геля используют крышку от иммунологического планшета (ТУ 64-2-278-79, или аналогичного) с размерами 12,6×8,5 см и высотой бортика 0,4 см. Подложку помещают на строго горизонтальную поверхность. Для формирования лунок в пластинке агарозного геля используют пластмассовую гребенку с 12 зубцами, высота каждого зубца 15 мм, ширина 8 мм, толщина 1 мм, расстояние между зубцами 2-3 мм. С помощью двух зажимов закрепляют гребенку так, чтобы все ее зубцы были «погружены» в емкость для полимеризации (т.е. концы зубцов гребенки находятся ниже бортиков подложки) на расстоянии 2 см от длинной стороны подложки и параллельно этой стороне. Расстояние от концов зубцов до дна подложки должно быть 1 мм.
В коническую стеклянную колбу вместимостью 250 мл вносят навеску 0,6 г агарозы (Bio-Rad, США) и добавляют 75 мл однократного буфера ТАЕ с бромистым этидием. Колбу помещают в микроволновую печь, разогревают до 80°С, перемешивают с использованием рукавиц до полного растворения агарозы, разогревают до кипения и помещают в стакан вместимостью 1 л со 100 мл дистиллированной воды для охлаждения. Колбу охлаждают 2-3 мин при помешивании, и расплав агарозы выливают в подложку, в которую «погружены» зубцы гребенки.
Агарозный гель полимеризуется в течение 40 минут при комнатной температуре. Важно, чтобы подложка с гелем в это время оставалась в покое, для этого на данном лабораторном столе не должны выполняться никакие другие экспериментальные процедуры. По истечении 40 минут гребенку осторожно извлекают из полученной пластинки агарозного геля. На этом этапе и при дальнейших операциях необходимо пользоваться тонкими латексными лабораторными перчатками. В тех местах, где в гель были погружены зубцы гребенки, остаются лунки правильной прямоугольной формы. Агарозный гель хрупок, и наличие жесткой подложки облегчает работу с ним, поэтому пластинку агарозного геля не извлекают из подложки.
Проведение электрофореза. Камеру для электрофореза Sub-Cell GT (Bio-Rad, США) или аналогичную помещают на горизонтальную поверхность лабораторного стола. В камеру наливают 1 л буфера ТАЕ с бромистым этидием. В буфер погружают и располагают горизонтально пластинку агарозного геля. Так как образцы ДНК должны мигрировать в агарозном геле по направлению от отрицательного электрода к положительному, правильным положением агарозной пластинки является такое, при котором лунки находятся ближе к отрицательному электроду.
В каждую пробирку с образцами №№1-4 и «М» вносят по 5 мкл 5-кратного концентрата утяжелителя с краской-маркером. Образцы из пробирок по очереди вносят в лунки агарозного геля. Для этого используют ручной дозатор (Gilson, Pipetman PI00, Франция или аналогичный) с установленным значением объема 25 мкл и одноразовые наконечники объемом до 200 мкл. Наконечник с набранным в него образцом опускают в центр лунки агарозного геля так, чтобы торец наконечника оказался приблизительно на середине высоты лунки. Затем плавно выдавливают образец, нажимая на поршень дозатора и не допуская активного перемешивания жидкости в лунке, что может привести к потере части образца или его затеканию в соседние лунки Образец погружается на дно лунки благодаря содержащейся в нем сахарозе, увеличивающей плотность жидкости. Процесс внесения образца в лунку геля контролируют визуально, поскольку краситель Понсо С придает образцу красный цвет.
Положительный электрод камеры для электрофореза соединяют с клеммой «+», а отрицательный электрод камеры - с клеммой «-» источника постоянного электрического тока. Электрофорез проводят в режиме стабилизации по напряжению при значении напряжения 80-100 V. При проведении электрофореза происходит тепловыделение, поэтому необходимо следить за температурой камеры и в случае нагрева выше 37°С снижать установленное значение напряжения. Электрофорез прекращают, когда фронт красителя Понсо С достигнет дальнего от лунок края агарозного геля.
Фотодокументирование. Пластинку агарозного геля извлекают из электрофорезной камеры и помещают гелем вниз на ультрафиолетовый трансиллюминатор с длиной волны излучения 254 нм (Helling, Германия) или аналогичный. На трансиллюминатор ставят светонепроницаемый ящик с цифровой камерой Кодак (Kodak DC290, Eastman Kodak, США) или аналогичный, включают ультрафиолетовое облучение и проводят фотографирование геля с оранжевым светофильтром в соответствии с руководством по эксплуатации. Фрагменты двухцепочечной ДНК, образовавшиеся в результате обработки рестриктазами, располагаются в геле в виде полос, параллельных длинным сторонам лунок. При облучении ультрафиолетовым светом фрагменты ДНК в комплексе с бромистым этидием флуоресцируют в оранжевой области спектра. Параметры съемки (экспозицию и контрастность) выбирают так, чтобы на снимке были заметны все видимые глазом светящиеся полосы в дорожке с препаратом маркеров (образец из пробирки «М»).
Обработка результатов. Определение размеров фрагментов. Цифровую фотографию распечатывают и измеряют длину пробега (расстояние от полосы до соответствующей лунки) для каждого фрагмента ДНК, регистрируемого на электрофореграмме в виде светлой полосы. Эти же измерения выполняют и для полос маркеров длин фрагментов ДНК. Затем строят кривую зависимости длины пробега от размера фрагмента ДНК, откладывая по оси абсцисс десятичные логарифмы выраженных в парах нуклеотидов (п.н.) размеров фрагментов маркеров, а по оси ординат - соответствующие длины пробега (в мм). Согласно прилагающемуся к препарату маркеров описанию фрагменты маркеров имеют следующие размеры в п. н. (в порядке убывания): 10000, 8000, 6000, 5000, 4000, 3500, 3000, 2500, 2000, 1500, 1200, 1000, 900, 800, 700, 600, 500, 400, 300, 200 и 100.
Пример фотографии агарозного геля приведен на фиг. 5.
Результаты измерений интерполируют полиномом третьей степени в соответствии с уравнением:
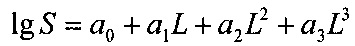 ,
,
где L - длина пробега, S - размер маркера, а0, а1, а2, а3 - коэффициенты полинома.
Эти коэффициенты определяют методом регрессионного анализа, используя программу CADRE Regression (CADRE Analytic, США) или аналогичную. Размеры фрагментов ДНК, образовавшихся после обработки субстанции рестриктазами, вычисляют при помощи полученного уравнения, подставляя туда соответствующие измеренные длины пробега. Допустимо также определять логарифмы размеров фрагментов непосредственно по графику, проводя через все построенные точки гладкую линию от руки. Количество наблюдаемых фрагментов в каждом случае должно совпадать с нормативным. Полученные в результате анализа значения размеров фрагментов не должны отличаться от нормативных более чем на 20%.
Обработка результатов. Денситометрирование. Цифровую фотографию пластинки агарозного геля обрабатывают с помощью программы Image Quant фирмы Molecular Dynamics (США), позволяющей определить относительное значение яркости каждой светящейся полосы в дорожке (в условных единицах). Значения яркости определяют для полос электрофореграммы, соответствующих фрагментам ДНК со следующими номинальными размерами:
1. при обработке рестриктазами HindIII и XbaI (пробирка №1):
- фрагмент 1668 п. н. из pC4W-HGFopt (значение яркости I1),
- фрагмент 929 п. н. из pVax1-UPAopt (значение яркости I2),
2. при обработке рестриктазами HindIII и EcoRV (пробирка №2):
- фрагмент 886 п. н. из pC4W-HGFopt (значение яркости I3),
- фрагмент 1354 п. н. из pVax1-UPAopt (значение яркости I4);
3. при обработке рестриктазами PflMI и Acc65I (пробирка №3):
- фрагмент 2448 п. н. из pC4W-HGFopt (значение яркости I5),
- фрагмент 974 п. н. из pVax1-UPAopt (значение яркости I6).
4. при обработке рестриктазами BbsI и BamHI (пробирка №4):
- фрагмент 2223 п. н. из pC4W-HGFopt (значение яркости I7),
- фрагмент 1589 п. н. из pVax1-UPAopt (значение яркости I8).
Отношение весового количества плазмидной ДНК pC4W-HGFopt к весовому количеству плазмидной ДНК pVax1-UPAopt в образце (величину R) вычисляют для четырех пар пиков по следующим формулам:
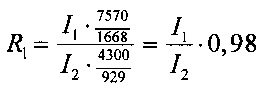
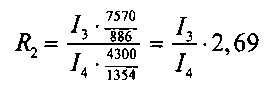
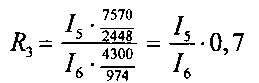
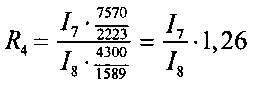
Далее вычисляют величину R как среднее значение R1, R2, R3, R4 и стандартную ошибку. Отношение весового количества плазмидной ДНК pC4W-HGFopt к весовому количеству плазмидной ДНК pVax1-UPAopt в образце (величина R) должно быть в пределах от 0,8 до 1,2.
Пример проведения рестрикционного анализа
Условия проведения анализа
1. Для анализа использовали рестриктазы HindIII, XbaI, Eco32I (изошизомер EcoRV), Van91I (изошизомер PflMI), Acc65I, BpiI (изошизомер BbsI), BamHI, Eco91I (изошизомер BstEII) (все no 10000 ед./мл), 10-кратные реакционные буферы «Y», «О», «R» и маркеры размеров фрагментов ДНК «GeneRuler DNA Ladder Mix (100-10,000 bp)» производства компании Thermo Scientific (США).
2. Время инкубации при обработке рестриктазами 20 часов, температура 37°С.
3. После завершения инкубации образцы подвергали электрофорезу в 0,8%-ном агарозном геле (агароза «Biotechnology Grade» производства компании Amresco, США) в буфере ТАЕ в присутствии бромистого этидия (1 мкг/мл).
4. По окончании электрофореза гель помещали на ультрафиолетовый трансиллюминатор с длиной волны излучения 254 нм и фотографировали с оранжевым светофильтром при помощи цифровой камеры Kodak DC290 производства Eastman Kodak, США. Электрофореграмма прилагается.
5. Цифровую фотографию распечатывали и измеряли длину пробега каждого фрагмента ДНК. Для маркеров размеров фрагментов ДНК строили график зависимости длины пробега от логарифма размера фрагмента, по построенному графику с использованием метода регрессионного анализа определяли размеры фрагментов анализируемых образцов. График, интерполяционное уравнение и таблица результатов определения размеров фрагментов прилагаются.
6. Цифровую фотографию подвергали денситометрированию с помощью программы Image Quant фирмы Molecular Dynamics (США), Определяли значения яркости (в условных единицах) для полос, соответствующих фрагментам 1668 п. н., 929 п. н (обработка HindIII+XbaI); 886 п. н., 1354 п. н. (обработка HindIII+Eco32I); 2448 п. н., 974 п. н (обработка Van91I+Acc65I); 2223 п. н., 1589 п. н (обработка BpiI+BamHI). По полученным данным вычисляли отношение весового количества плазмидной ДНК pC4W-HGFopt к весовому количеству плазмидной ДНК pVax1-UPAopt в образце (величина R) для 4 пар пиков. Вычисляли среднее значение величины R и стандартную ошибку. Таблица результатов денситометрирования прилагается.
Пример электрофоретического анализа приведен на фиг. 5.
На фиг. 6 приведен график зависимости длины пробега от логарифма размера фрагмента.
Результат регрессионного анализа
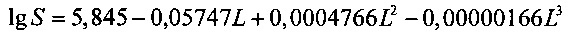
Нормативные требования: при обработке рестриктазами HindIII и XbaI размеры фрагментов должны составлять 452 п. н., 563 п. н., 929 п. н., 1668 п. н., 2919 п. н. и 5339 п. н. (всего 6 фрагментов); при обработке рестриктазами HindIII и EcoRV размеры фрагментов должны составлять 554 п. н., 782 п. н., 886 п. н., 1354 п. н., 2946 п. н. и 5348 п. н. (всего 6 фрагментов); при обработке рестриктазами PflMI и Acc65I размеры фрагментов должны составлять 974 п. н., 2448 п. н., 3326 п. н. и 5122 п. н. (всего 4 фрагмента); при обработке рестриктазами BbsI и BamHI размеры фрагментов должны составлять 651 п. н., 876 п. н., 1589 п. н., 2223 п. н., 2711 п. н. и 3820 п. н. (всего 6 фрагментов). Количество наблюдаемых фрагментов для каждой обработки должно совпадать с указанным. Полученные в результате анализа значения размеров фрагментов не должны отличаться от указанных более чем на 20%. Отношение весового количества плазмидной ДНК pC4W-HGFopt к весовому количеству плазмидной ДНК pVax1-UPAopt должно быть в пределах от 0,8 до 1,2.
Пример 8. Исследование эффективности лекарственного средства на основе невирусных конструкций, несущих гены фактора роста гепатоцитов и урокиназы in vitro на модели фиброза печени
Определение биологической активности лекарственного средства, на моделях in vitro
Определение биологической активности лекарственного средства на моделях in vitro осуществляют с помощью следующих экспериментов:
1) оценка влияние HGF на пролиферативную активность эндотелиальных клеток пупочной вены (HUVEC) с помощью МТТ-теста.
2) оценка влияния huPA (урокинзы) на продукцию компонентов внеклеточного матрикса звездчатыми клетками (клетками Ито).
Оценка влияния HGF на пролиферативную активность эндотелиальных клеток пупочной вены (HUVEC) с помощью МТТ-теста
МТТ-тест - широко используемый метод количественного определения клеточной выживаемости и пролиферации, основанный на расщеплении МТТ [3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид] (Sigma, USA) митохондриальным ферментом сукцинатдегидрогеназой и колориметрическом измерении образования продукта расщепления - формазана. Методика оптимизирована для использования спектрофотометра Multiscan Ascent (Labsystems).
Суспензию HUVEC в полной среде роста EGM-2 (Lonza, USA) высаживали по 100 мкл на 3 культуральных 96-луночный планшета с таким расчетом, чтобы на каждую экспериментальную точку приходилось по 3 лунки и конфлюент составил не более 50%.
Рассаживали клетки на 96-луночном планшете следующим образом:
1 - среда ЕВМ-2 (Endothelial Cell Basal Medium, Lonza, USA) с 2% фетальной бычьей сывороткой (HyClone, USA), без клеток (отрицательный контроль);
2 - среда ЕВМ-2 с 2% сывороткой, HUVEC (отрицательный контроль);
3 - среда ЕВМ-2 с 5% сывороткой (положительный контроль);
4 - среда ЕВМ-2 с 2% сывороткой с добавлением среды клеток HEK293 без трансфекции, HUVEC (отрицательный контроль);
5 - среда ЕВМ-2 с 2% сывороткой с добавлением среды, собранной с клеток HEK293, экспрессирующих вектор pc4W, без концентрирования среды (отрицательный контроль).
6 - среда ЕВМ-2 с 2% сывороткой с добавлением среды, собранной с клеток HEK293, экспрессирующих HGF, без концентрирования среды (опыт).
7 - среда ЕВМ-2 с 2% сывороткой с добавлением среды, собранной с клеток HEK293, экспрессирующих вектор pc4W, концентрирование среды (отрицательный контроль).
8 - среда ЕВМ-2 с 2% сывороткой с добавлением среды, собранной с клеток Далее клетки инкубировали при 37°С и 5% СО2 1-й планшет - в течение 24 часов,
2-й планшет - 48 часов, 3-й планшет - 72 часа. Готовили 10-кратный стоковый раствор МТТ на среде RPMI (Invitrogene/Gibco, USA) с концентрацией 5 мг/мл. В день эксперимента подготавливали рабочий раствор МТТ на среде RPMI (Invitrogene/Gibco, USA) с концентрацией 500 мкг/мл (разведение 1:10).
По окончании времени инкубации клетки промывали во всех планшетах теплой средой RPMI (Invitrogene/Gibco, USA) и наносили рабочий раствор МТТ (100 мкл на лунку) на период времени от 30 минут до 4 часов. Время инкубации зависит от типа клеток и конфлюентности и определяется по интенсивности выпадения в осадок пурпурных кристаллов формазана. Затем в лунки добавили по 100 мкл изопропанола/ 0.04 N HCl и ресуспендировали до полного растворения кристаллов формазана, при этом среда стала гомогенно-голубого цвета. Далее измеряли оптическую плотность сред на планшетном спектрофлуориметре при длине волны 570 нм и референсной длине волны 630 нм.
Пролиферативную активность оценивали, как зависимость оптической плотности от количества клеток, с помощью калибровочного графика, построенного с использованием серии разведений эндотелиальных клеток. Результаты измерений приведены на фиг. 7.
Полученные данные позволяют оценить время удвоения клеток. Расчет производили по следующей формуле:
Среднее время удвоения популяции Td:
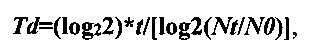
где t - время прироста популяции, Nt - количество клеток через время t, N0 -исходное количество клеток.
Результаты измерения пролиферативной активности культуры клеток под действием HGF приведены в таблицах 1 и 2, а также в виде графиков на фиг. 7.
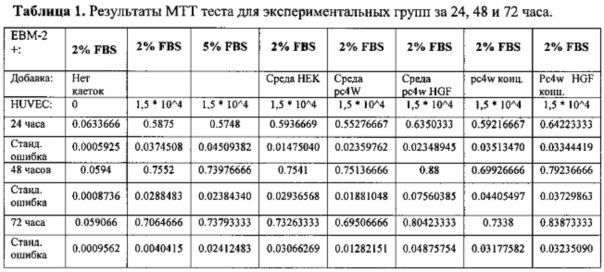
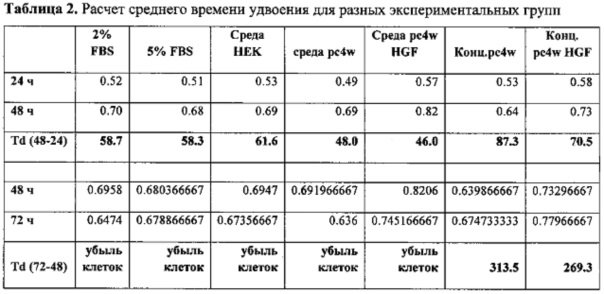
Выводы: максимальное время удвоения популяции (46 часов) было отмечено через 48 часов от начала эксперимента для клеток, которым добавляли кондиционированную среду НЕК, трансфецированных HGF. Через 72 часа от начала эксперимента максимальное среднее время удвоения популяции было отмечено для клеток, содержавшихся на среде с добавлением концентрированного супернатанта HEK, трансфецированных HGF.
Оценка влияния huPA (урокинзы человека) на продукцию компонентов внеклеточного матрикса звездчатыми клетками (клетками Ито) на пластике без покрытия при добавлении кондиционированной среды клеток HEK293, трансфецированных плазмидой с кДНК uPA человека.
Для выделения стеллатных клеток использовали 8 самцов линии Balb/C, массой 20-22 г. Мышей анестезировали путем интраперитонеального введения 500 мкл 8% хлоралгидрата. Далее проводили перфузию печени. Для этого после вскрытия грудной и брюшной полости, осуществляли канюляцию левого желудочка, для оттока жидкости вскрывали правое предсердие. Перфузию проводили со сменой растворов: 1) безкальциевый Хэнке с добавлением 0,5 мМ ЭДТА 10 минут со скоростью 1 мл/мин, 2) раствор проназы Е 1 мг/мл, приготовленный на Хэнксе с добавлением антибиотиков и HEPES до 0,2 мМ 10 минут со скоростью 1 мл/мин, 3) раствор коллагеназы 0,25 мг/мл, приготовленный на Хэнксе с добавлением антибиотиков и HEPES до 0,2 мМ 10 минут со скоростью 1 мл/мин.
Далее осуществлялось взятие печени. Печень в стерильных условиях измельчали с помощью ножниц и пропускали через 100 мкн ситечко. Далее гомогенизированный образец инкубировали в растворе ДНКазы I типа (NEB) на Хэнксе 10 минут при 37°С и помешивании.
На следующем этапе производили осаждение паренхиматозной фракции клеток с помощью центрифугирования в течение 5 минут при 4°С и 25 g 2 раза. При этом оба осадка утилизировали, а супернатант собирали и, объединив, откручивали при 400 g 10 минут, 4°С.
Осажденные клетки разводили в 2 мл DMEM/FT2 и наслаивали на 33% раствор Optiprep, чтобы сформировать градиент плотности, откручивали при 4С, 20 минут на 1400 g.
По окончании центрифугирования формировалось кольцо на разделе сред, содержащее преимущественно стеллатные клетки. Кольцо собирали в стерильную пробирку и 2 раза откручивали в растворе DMEM с добавлением бычьего сывороточного альбумина до 0,25%, антибиотика и HEPES до 20 мМ.
Затем клетки ресуспендировали в среду для культивирования (DMEM с высоким содержанием глюкозы, 10% фетальной бычьей сывороткой, антибиотиком) и высаживали на 48-луночные планшеты без покрытия.
На следующий день производили смену среды и постановку экспериментов. На фиг. 8 приведены фотографии выделенных стеллатных клеток мыши.
Восемь полученных образцов разделили следующим образом:
Образец 1 - контроль: стеллатные клетки в среде культивирования, для эксперимента на 24 часа
Образец 2 - стеллатные клетки с добавлением кондиционированной среды нетрансфецированых клеток HEK, для эксперимента на 24 часа.
Образец 3 - стеллатные клетки с добавлением кондиционированной среды клеток HEK, трансфецированных вектором pVax, не несущим вставки, для эксперимента на 24 часа.
Образец 4 - стеллатные клетки с добавлением кондиционированной среды клеток HEK, трансфецированных плазмидой pVax-uPa, для эксперимента на 24 часа
Образец 5 - контроль: стеллатные клетки в среде культивирования, для эксперимента на 48 часов.
Образец 6 - стеллатные клетки с добавлением кондиционированной среды нетрансфецированых клеток HEK, для эксперимента на 48 часов.
Образец 7 - стеллатные клетки с добавлением кондиционированной среды клеток HEK, трансфецированных вектором pVax, не несущим вставки, для эксперимента на 48 часов.
Образец 8 - стеллатные клетки с добавлением кондиционированной среды клеток HEK, трансфецированных плазмидой pVax-uPa, для эксперимента на 48 часов.
Материалы и методы
Тотальную РНК выделяли спустя 24 и 48 часов после добавления кондиционированных супернатантов. Выделение производили с помощью набора Trizol (Gibco/Life Technologies) в соответствие с приведенным протоколом. Количественная ПЦР-РВ производилась на амплификаторе «RotorGene 3000» (Corbett Reaserch) с использованием SYBR Green I-«Синтол» (Россия). Специфичность продуктов оценивалась с помощью проведения агарозного гель-электрофореза с содержанием агарозы 1%. Нормирование производили по бета-актину.
Результаты:
Добавление среды клеток линии HEK293 приводит к снижению уровня мРНК альфа-гладкоклеточного актина (α-SMA), трансформирующего фактора роста бета (TGF-β), тканевого ингибитора матриксных металлопротеиназ (TIMP-1), коллагенов I и III типов.
Результаты измерений представлены на фиг. 9-13.
Пример 9. Исследование эффективности влияния лекарственного средства на восстановление структуры печени при лечении экспериментального фиброза печени
Гистологическая оценка тканей печени мышей после индукции фиброза и введения терапевтических конструкций
Описание и методы
Гистологическую оценку фиброза производили с помощью окрашивания на гематоксилин и эозин и с помощью специфического окрашивания на коллаген. Для количественной оценки коллагеновых волокон проводилось окрашивание пикросириусом красным. Данный краситель обладает аффинностью к большинству типов коллагена, включая типы I и III.
Гистологическую картину срезов печени оценивали с помощью индекса гистологической активности (модифицированная шкала Кноделя), представленной в Таблице 3. Полуколичественный индекс гистологической активности, или модифицированная системы Кноделя (Ishak), представляет собой шкалу, предназначенную для оценки степени повреждения печени после внутрибрюшинного введения тетрахлорметана/приема этанола. Фибротические изменения оцениваются в баллах, максимальный балл - 6 - соответствует циррозу.
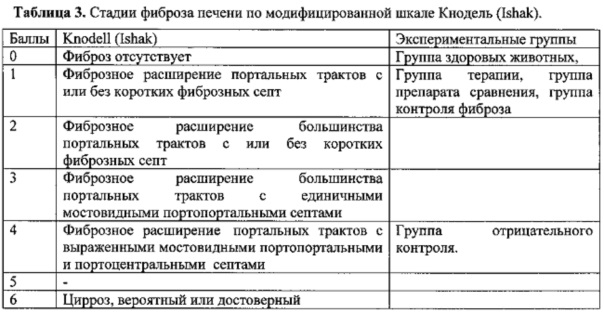
Эксперимент проводили на животных линии Balb/c, 22-25 г. Животные были разделены на 5 групп:
1) Группа отрицательного контроля (n=8): мыши, которым в течение Недель 1-4 вводили 30% CCl4 на оливковом масле в дозе 1 мкл/1 г массы тела. В течение Недель 5-6 животные этой группы не получали никакого лечения.
2) Группа терапии (n=8): мыши, которым в течение Недель 1-4 вводили 30% CCl4 на оливковом масле в дозе 1 мкл/1 г массы тела. В течение Недель 5-6 1 раз в неделю животные этой группы получали инъекции комбинации плазмид HGF и uPA методом гидродинамического удара в дозе 3,75 мкг плазмидной ДНК на 1 г массы тела.
3) Группа положительного контроля (препарата сравнения) (n=8): мыши, которым в течение Недель 1-4 вводили 30% CCl4 на оливковом масле в дозе 1 мкл/1 г массы тела. В течение Недель 5-6 1 раз в неделю животные этой группы получали инъекции плазмиды HGF методом гидродинамического удара в дозе 3,75 мкг плазмидной ДНК на 1 г массы тела.
4) Группа контроля развития фиброза (n=8): мыши, которым в течение Недель 1-4 вводили оливковое масло в дозе 1 мкл/1 г массы тела. В течение Недель 5-6 животные этой группы не получали никаких инъекций.
5) Группа здоровых животных (n=8)
Гидродинамический метод введения генетических конструкций в печень заключается во внутривенном введении большого объема раствора (примерно 8-10% от массы тела) за короткий промежуток времени (около 5-7 секунд).
Результаты представлены на фиг. 14-16.
Выводы: применение комбинации плазмид HGF и uPA (Геатофиброл) приводит к снижению выраженности фиброза с 4 до 1 балла по модифицированной шкале Кнодель (Ishak). Сопоставимые результаты получены для препарата сравнения.
Измерение уровня экспрессии генов с помощью полимеразной цепной реакции в реальном времени
Материалы и методы
Для изучения экспрессии выбранных генов использовался метод полимеразной цепной реакции в реальном времени (Real-Time PCR), выполненной на амплификаторе «RotorGene 3000» (Corbett Reaserch) (Австралия). Был использован набор реактивов для проведения ПЦР-РВ в присутствии интеркалирующего красителя SYBR Green I-«Евроген» (Россия).
Дизайн праймеров выполнялся с использованием программы DNASTAR.
Нуклеотидные последовательности праймеров приведены в таблице 4. Праймеры по предоставленным последовательностям были синтезированы фирмой ЗАО «Евроген» (Россия).


Метод ПЦР-РВ заключается в определении выхода продукта реакции после каждого цикла амплификации, построении по этим данным кинетической кривой ПЦР и определению относительной концентрации субстрата на основании анализа этой кривой.
Флуоресцентный краситель используется для детекции ПЦР-продукта и обеспечивает флуоресценцию, прямо пропорциональную количеству ПЦР-продукта-репортерную флуоресценцию.
Кинетическая кривая ПЦР в координатах «Уровень репортерной флуоресценции-цикл амплификации» имеет сигмоидную форму. На этой кривой можно выделить 3 стадии: инициации, когда ПЦР-продукта слишком мало для детекции; экспоненциальную стадию, отражающую экспоненциальную зависимость роста флуоресценции от цикла ПЦР; и плато, или стадию насыщения, на которой ампликоны уже не синтезируются.
Пороговый цикл, на котором достигается некоторый заданный уровень флуоресценции, называется threshold cycle, Ct. Его значение прямо пропорционально логарифму количества субстрата. Таким образом, ПЦР-РВ позволяет сравнивать количества субстрата при условии, что эффективность реакции и заданный уровень пороговой флуоресценции одинаковы для каждой из сравниваемых реакций.
Кинетическая кривая представлена на фиг. 17.
Количественную оценку проводят с использованием метода относительного количественного анализа (ΔΔCt). Для этого для каждого исследуемого гена (А) высчитывается разница в циклах ΔCt между исследуемым геном и геном домашнего хозяйства (house-keeping gene) HPD при выходе кривой в стадию экспоненциального роста. Пороговый уровень устанавливается вручную согласно рекомендациям к прибору и данным литературы. Далее высчитывается уровень экспрессии каждого гена, выраженный в числе копий. Для этого эффективность реакции принималась равной 2. Тогда в соответствии со значениями ΔCt высчитывалось число копий, равное 2-ΔCt. Затем статистически обсчитывалась разница значений уровня экспрессии между исследуемой и контрольной группами (2-ΔΔCt). Для каждой матрицы кДНК одновременно ставилась ПЦР-реакция в 2-х пробирках: с геном домашнего хозяйства и одним из исследуемых генов. Реакционная смесь включала в себя по 1 мкл каждого праймера (10рМ), 10 мкл реакционной смеси, 1 мкл кДНК и 12 мкл воды. Итого объем реакционной смеси составлял 27,5 мкл с учетом дополнительного объема в 10% на ошибку пипетирования. Реакционная смесь для PCR-RT (ЗАО «Синтол») содержит 2,5х ПЦР Буфер Б (KCl, трис-HCl (рН=8,8), 6,25 мМ MgCl2), Taq-полимеразу, дезоксинуклеозидфосфат, глицерол, Tween 20, интеркалирующий краситель SYBR Green I.
Условия ПЦР-РВ.
Денатурация при 95°С в течение 10 минут предшествовала 40 циклам амплификации. Каждый цикл состоял из 10 секунд денатурации при 95°С, 15 секунд отжига при 60°С и 20 секунд элонгации при 72°С. Затем проводилось плавление полученной двухцепочечной ДНК (каждые 5 секунд температура повышалась с 72°С до 95°С с шагом 1°С) для получения кривых плавления, отражающих специфичность проведенной PCR-реакции, то есть отсутствие дополнительных неспецифических продуктов реакции. Для анализа экспрессии гена hHGF в составе мышиной cDNA животных, получавших терапию hHGF и комбинацию hHGF и hPA, использовали полимеразную цепную реакцию (ПЦР). Для этого из образцов печеночной ткани выделяли тотальную РНК. Далее по указанному выше протоколу осуществляли синтез комплементарной ДНК. Предварительно проводили качественный и количественный анализ полученной РНК с помощью аналитической системы Experion (Automated Electrophoresis Station) и набора реактивов RNA StdSens Kit-«BioRad» (США). В реакцию обратной транскрипции брали по 10 мкг тотальной РНК, 4 мкл обратной транскриптазы M-MuLV Reverse Transcriptase (20u/ml), 2 мкл ингибитора рибонуклеаз (20u/ml), 4 мкл смеси нуклеотидов dNTP(10mM), 2 мкл праймера oligo(dT)18, 8 мкл буфера реакционного - «Fermentas» (Латвия). Дизайн праймеров выполнялся с использованием программы DNASTAR.
Реакционный состав ПЦР: верхняя смесь включала 10 мкл Red Mix, 1 мкл cDNA, 9 мкл деионизованной воды; нижняя смесь состояла из 1 мкл праймеров, 2 мкл dNTP и 2 мкл деионизованной воды. В качестве положительного контроля брали cDNA клеток линии HEK 293. Проба без cDNA и с мышиной cDNA животного, не получавшего инъекции указанных плазмидных конструкций, использовали в качестве отрицательного контроля. По окончании ПЦР ампликоны детектировались с помощью электрофореза в 1% агарозном геле. Результаты приведены на фиг. 18.
Введение заявляемого препарата в хвостовую вену методом гидродинамического удара приводит к снижению уровня мРНК компонентов внеклеточного матрикса (коллагена I, III, TGF-β, TIMP-1).
Статистически значимых различий уровней мРНК между группой мышей, получавших терапию заявляемым препаратом и группой, получавшей HGF, не отмечено.
Иммуногистохимический метод оценки эффективности влияния лекарственного средства на восстановление структуры печени.
Восстановление структуры печени после внесения плазмидных конструкций, содержащих кДНК генов HGF человека и uPA человека, оценивают с помощью иммунофлуоресцентного окрашивания криосрезов печени. Животных умерщвляют, выделяют печень, фиксируют ее 4% раствором формальдегида в течение 3 часов. Фиксированные таким образом образцы замораживают в жидком азоте в среде для замораживания Tissue-Tek (Sakura Finetek). Готовят срезы толщиной 6 мкм. Полученные срезы окрашивают первичными антителами к белкам внеклеточного матрикса. Визуализацию полученных образцов осуществляют с помощью микроскопа Leica6000, оборудованного CCD камерой (Leica Microsystems).
Протокол иммуногистохимического окрашивания
1. Срезы печени инкубировать 5 минут при комнатной температуре.
2. Погрузить срезы в охлажденный до -20°С ацетон. Фиксировать 10 минут.
3. Промыть 3×5 раз солевым изотоническим раствором (PBS).
4. Блокировать аутофлуоресценцию 0,05 М раствором ацетата аммония.
5. Для блокировки неспецифического связывания использовать 10% сыворотку/1% BSA в PBS животного, иммунизованного для создания вторичных антител.
6. Инкубировать в первичных антителах сутки при +4С.
7. Промыть в PBS 3×5 минут.
8. Инкубировать со вторичными антителами, приготовленными на 1% BSA, в течение 1 часа.
9. Промыть 3×5 минут в темноте.
10. Заключить с использованием водорастворимого заключающего агента (Immumount, FisherScientific).
11. Флуоресцентная микроскопия полученных срезов.
Фотографии окрашенных иммунофлуоресцентно срезов ткани печени представлены на фиг. 19-20.
Выводы: введение лекарственного средства на основе комбинации плазмид HGF и uPA приводит к снижению содержания компонентов внеклеточного матрикса (коллагена I типа, альфа-гладкоклеточного актина) в строме печени.
Влияние разрабатываемого лекарственного средства на уровень ферментов аланинаминотрансферазы и аспартатаминотрансферазы в сыворотке мышей с индуцированным фиброзом печени
Как уже было сказано ранее, для оценки эффективности разрабатываемого лекарственного средства in vivo мы разработали экспериментальную модель на мышах, которая позволяет индуцировать фиброз печени с помощью интраперитонеальных инъекций четыреххлористого углерода в оливковом масле. После последовательных инъекций у мышей, получающих четыреххлористый углерод, развивается фиброз печени, который можно детектировать с помощью гистологического окрашивания и путем оценки уровня маркерных молекул в образцах печени животных. Кроме того, повреждение печени при фиброзе приводит к гибели большого числа клеток, в том числе, гепатоцитов. Массированная гибель клеток сопровождается выбросом в кровь ферментов-аминотрансфераз, которые являются, таким образом, маркерами стресса и гибели клеток при повреждениях печени. Анализ активности аминотрансфераз, в частности, аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (ACT) является удобным методом оценки степени повреждения печени при фиброзе и циррозе, который широко используется в лабораторной диагностике в медицине и ветеринарии. Установлено, что значительное увеличение активности вышеупомянутых ферментов может быть индикатором патологии печени. Еще одним преимуществом данного метода является относительная доступность сыворотки крови животных, получение которой не требует специальных реактивов и процедур. Кроме того, существуют дешевые и коммерчески доступные наборы (в том числе отечественного производства) для рутинного определения уровней АЛТ и АЛТ в сыворотке или плазме крови.
Принцип метода.
В основе метода определения уровня ферментативной активности АЛТ и ACT лежит спектрофотометрическое измерение уровня NADH, количество которого в реакционной смеси, содержащей сыворотку, уменьшается под действием аминотрансфераз. Уменьшение концентрации NADH сопровождается уменьшением оптической плотности образца при длине волны 340 нм. Для измерения активности АЛТ и ACT мы использовали коммерческие наборы «Диаком-АЛТ» и «Диаком-АСТ», выпускаемых ЗАО «ДИАКОМ-ВНЦМДА» (Москва) в соответствии с рекомендациями и инструкциями производителя, которые приведены ниже (представлены части текста инструкций к соответствующим наборам, а также отсканированные инструкции к наборам). Для измерения оптической плотности при 340 нм использовали спектрофотометр Ultrospec 2100pro, оснащенный термостатируемыми отделением для кювет и подключенный к жидкостному термостату, а также пластиковые кюветы с длиной оптического пути 1 см. Для обработки результатов измерения использовали персональный компьютер, с предустановленным пакетом программ SWIFT II (Biochrom Ltd.), который позволяет измерять кинетику ферментативных реакций. Получение образцов сыворотки крови мышей.
Для получения образцов мышиной сыворотки кровь собирали с помощью пункции левого желудочка сердца. Для этого животных усыпляли с помощью ингаляции изофлурана в течение 1-1,5 минут, после чего проводили вскрытие грудной клетки, которое позволяет обнажить бьющееся сердце мыши. Для сбора крови использовали стандартные инсулиновые шприцы со съемными иглами. С одного животного удавалось получить до 1 мл крови, которую затем переносили в герметично закрывающиеся необработанные гепарином пробирки объемом 1,5 мл. Образцы крови в пробирках инкубировали в течение 5-15 минут при комнатной температуре для проведения коагуляции крови. Образовавшийся в результате коагуляции тромб осаждали с помощью центрифугирования пробирок в настольной центрифуге при комнатной температуре при 12000 об/мин в течение 5 минут. При этом содержимое пробирки разделялось на тромб и клеточный компоненты крови, которые оседали на дне, а также прозрачную сыворотку, которую собирали с помощью пипетки и переносили в чистую пробирку. Полученную сыворотку повторно центрифугировали, чтобы гарантировать полное отделение клеточных компонентов крови, после чего переносили супернатант в чистые пробирки и хранили при температуре -80С. Перед измерением активности аминотрансфераз образцы сыворотки размораживали на льду и хранили в течение измерения при температуре 0-4С.
Результаты измерения активности аланинаминотрансферазы.
Для измерения активности АЛТ в образцах сыворотки использовали рекомендации, представленные в соответствующей инструкции. Поскольку объем полученных образцов сыворотки был небольшим (не более 400 мкл для каждого животного), объем реакционной смеси был уменьшен ровно в 2 раза. Это позволило существенно снизить объем сыворотки, используемой для измерения, а также уменьшить объемы используемых реагентов, не влияя на результаты измерения. Таким образом, объем рабочего раствора для каждого измерения составлял 1 мл, а объем образца сыворотки 150 мкл. В случае, если объем сыворотки составлял менее 150 мкл непосредственно перед проведением реакции сыворотку разбавляли физиологическим раствором (0,14 М NaCl в стерильной воде).
Полученные в результате спектрофотометрических измерений активности АЛТ значения экспортировали в файлы формата.xls, которые позволяют анализировать данные в программе Excel (Microsoft).
Зависимость изменения оптической плотности от времени отображали в виде графиков, позволяющих оценить линейность снижения оптической плотности. Такой первичный анализ абсолютно необходим, поскольку вычисление активности из кинетических данных возможно только в случае линейной зависимости скорости катализируемой ферментом реакции от времени. Пример графика зависимости изменения оптической плотности образца от времени представлен на фиг. 21.
Как видно из графика, падение оптической плотности в представленном образце было линейно на протяжении всего времени измерения, поэтому для определения активности АЛТ из значения оптической плотности в начальный момент времени вычитали значение оптической плотности в конечный момент времени и делили полученную разность на 4 минуты (общее время измерения). Если зависимость поглощения образца от времени не была линейной, то такие образцы исключались из выборки результатов. Полученное значение умножали в соответствии с формулой на фактор пересчета, который был определен производителем набора заранее. Получившееся в результате значение после этого умножали на фактор разбавления, что, в итоге, позволило получить значения активности АЛТ в единицах активности на 1 литр объема.
В результате измерения активности АЛТ в образцах сыворотки мышей из контрольных групп, групп у которых был индуцирован фиброз, групп с индуцированным фиброзом, которым был введен препарат сравнения или разрабатываемое лекарственное средство были получены значения активности АЛТ. Анализ результатов позволил получить гистограммы, которые свидетельствуют о том, что введение разрабатываемого лекарственного средства мышам с индуцированным фиброзом печени позволяет снизить уровень активности АЛТ в сыворотке практически до уровня, наблюдаемого у здоровых животных. В то же время, достоверно установлено, что препарат сравнения в меньшей степени, по сравнению с заявляемым снижает уровень активности АЛТ. Схожие результаты по влиянию разрабатываемого лекарственного средства на уровень АЛТ в сыворотке крови мышей с индуцированным фиброзом печени были получены в результате проведения трех независимых экспериментов. Представленные на фиг. 22 гистограммы являются репрезентативным для всех трех экспериментов.
Результаты измерения активности аспартатаминотрансферазы
Для измерения активности ACT в образцах сыворотки использовали рекомендации, представленные в соответствующей инструкции. Поскольку объем полученных образцов сыворотки был небольшим (не более 400 мкл для каждого животного), объем реакционной смеси был уменьшен ровно в 2 раза. Это позволило существенно снизить объем сыворотки, используемой для измерения, а также уменьшить объемы используемых реагентов, не влияя на результаты измерения. Таким образом, объем рабочего раствора для каждого измерения составлял 1 мл, а объем образца сыворотки 150 мкл. В случае, если объем сыворотки составлял менее 150 мкл непосредственно перед проведением реакции сыворотку разбавляли физиологическим раствором (0,14 М NaCl в стерильной воде).
Полученные в результате спектрофотометрических измерений активности ACT значения экспортировали в файлы формата.xls, которые позволяют анализировать данные в программе Excel (Microsoft).
Зависимость изменения оптической плотности от времени отображали в виде графиков, позволяющих оценить линейность снижения оптической плотности. Такой первичный анализ абсолютно необходим, поскольку вычисление активности из кинетических данных возможно только в случае линейной зависимости скорости катализируемой ферментом реакции от времени. Пример графика зависимости изменения оптической плотности образца от времени представлен на фиг. 23.
Как видно из графика, падение оптической плотности в представленном образце было линейно на протяжении всего времени измерения, поэтому для определения активности ACT из значения оптической плотности в начальный момент времени вычитали значение оптической плотности в конечный момент времени и делили полученную разность на 4 минуты (общее время измерения). Если зависимость поглощения образца от времени не была линейной, то такие образцы исключались из выборки результатов. Полученное значение умножали в соответствии с формулой на фактор пересчета, который был определен производителем набора заранее. Получившееся в результате значение после этого умножали на фактор разбавления, что, в итоге, позволило получить значения активности ACT в единицах активности на 1 литр объема.
В результате измерения активности ACT в образцах сыворотки мышей из контрольных групп, групп у которых был индуцирован фиброз, групп с индуцированным фиброзом, которым был введен препарат сравнения или разрабатываемое лекарственное средство были получены значения активности ACT. Анализ результатов позволил получить гистограммы, которые свидетельствуют о том, что введение разрабатываемого лекарственного средства мышам с индуцированным фиброзом печени позволяет снизить уровень активности ACT в сыворотке практически до уровня, наблюдаемого у здоровых животных. В то же время, достоверно установлено, что препарат сравнения в меньшей степени, по сравнению с заявляемым снижает уровень активности ACT. Схожие результаты по влиянию разрабатываемого лекарственного средства на уровень ACT в сыворотке крови мышей с индуцированным фиброзом печени были получены в результате проведения трех независимых экспериментов. Представленные на фиг. 24 гистограммы являются репрезентативным для всех трех экспериментов.
Таким образом, на основании данных по изменению активности сывороточных аминотрансфераз в мышах с индуцированным фиброзом печени под действием препарата сравнения и исследуемого лекарственного средства сделаны следующие заключения:
1) Препарат сравнения позволяет снизить уровень активности аминотрансфераз в сыворотке крови животных с индуцированным фиброзом печени по сравнению с контрольными животными;
2) Исследуемое лекарственное средство также снижает активность аминотрансфераз при введении животным с индуцированным фиброзом печени по сравнению с контрольными животными;
3) На основании полученных результатов можно утверждать, что снижение активности аминотрансфераз коррелирует с уровнем повреждения и эффективным восстановлением поврежденной фиброзом паренхимы печени;
4) Исследуемое лекарственное средство, судя по уровню сывороточных аминотрансфераз превосходит препарат сравнения по способности эффективно стимулировать восстановление поврежденной фиброзом печени.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СТИМУЛЯЦИИ АНГИОГЕНЕЗА В ИШЕМИНИЗИРОВАННЫХ ТКАНЯХ И КОМБИНИРОВАННОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ОСУЩЕСТВЛЕНИЯ СПОСОБА | 2015 |
|
RU2628706C2 |
| ГЕННО-ИНЖЕНЕРНАЯ КОНСТРУКЦИЯ ДЛЯ СТИМУЛЯЦИИ ПОСТТРАВМАТИЧЕСКОЙ РЕГЕНЕРАЦИИ ПЕРИФЕРИЧЕСКИХ НЕРВОВ | 2019 |
|
RU2719013C1 |
| ГЕН HGFopt ФАКТОРА РОСТА ГЕПАТОЦИТОВ | 2008 |
|
RU2385936C1 |
| ФРАГМЕНТ ДНК НС365, ПОЛИПЕПТИД, ШТАММ БАКТЕРИЙ ESCHERICHIA COLI - ПРОДУЦЕНТ ПОЛИПЕПТИДА, ОБЛАДАЮЩЕГО СПОСОБНОСТЬЮ СВЯЗЫВАТЬ АНТИТЕЛА К ВИРУСУ ГЕПАТИТА C | 1993 |
|
RU2041949C1 |
| ГЕН ММР1opt МЕТАЛЛОПРОТЕИНАЗЫ 1 | 2008 |
|
RU2378377C1 |
| ГЕННО-ИНЖЕНЕРНАЯ КОНСТРУКЦИЯ ДЛЯ СТИМУЛЯЦИИ АНГИОГЕНЕЗА | 2019 |
|
RU2737487C1 |
| Ген H5, кодирующий стабилизированную форму белка гемагглютинина вируса гриппа A (H5N8), рекомбинантная плазмидная ДНК pVAX-H5, содержащая ген H5 и обеспечивающая его экспрессию, и стабилизированная форма белка гемагглютинина вируса гриппа А (H5/N8), продуцируемая рекомбинантной плазмидной ДНК pVAX-H5 в организме животных и индуцирующая специфический иммунный ответ против высокопатогенного вируса гриппа A (H5N8) | 2024 |
|
RU2832515C1 |
| СПОСОБ СТИМУЛИРОВАНИЯ ВОССТАНОВЛЕНИЯ ПЕРИФЕРИЧЕСКОЙ ИННЕРВАЦИИ ТКАНЕЙ С ПОМОЩЬЮ ВЕКТОРНЫХ КОНСТРУКЦИЙ | 2011 |
|
RU2486918C1 |
| ГЕН VEGFopt ФАКТОРА РОСТА ЭНДОТЕЛИЯ СОСУДОВ ЧЕЛОВЕКА | 2008 |
|
RU2385937C1 |
| СРЕДСТВО ДЛЯ ОСУЩЕСТВЛЕНИЯ ТЕРАПЕВТИЧЕСКОГО АНГИОГЕНЕЗА И СПОСОБ ЕГО ОСУЩЕСТВЛЕНИЯ | 2009 |
|
RU2449799C2 |
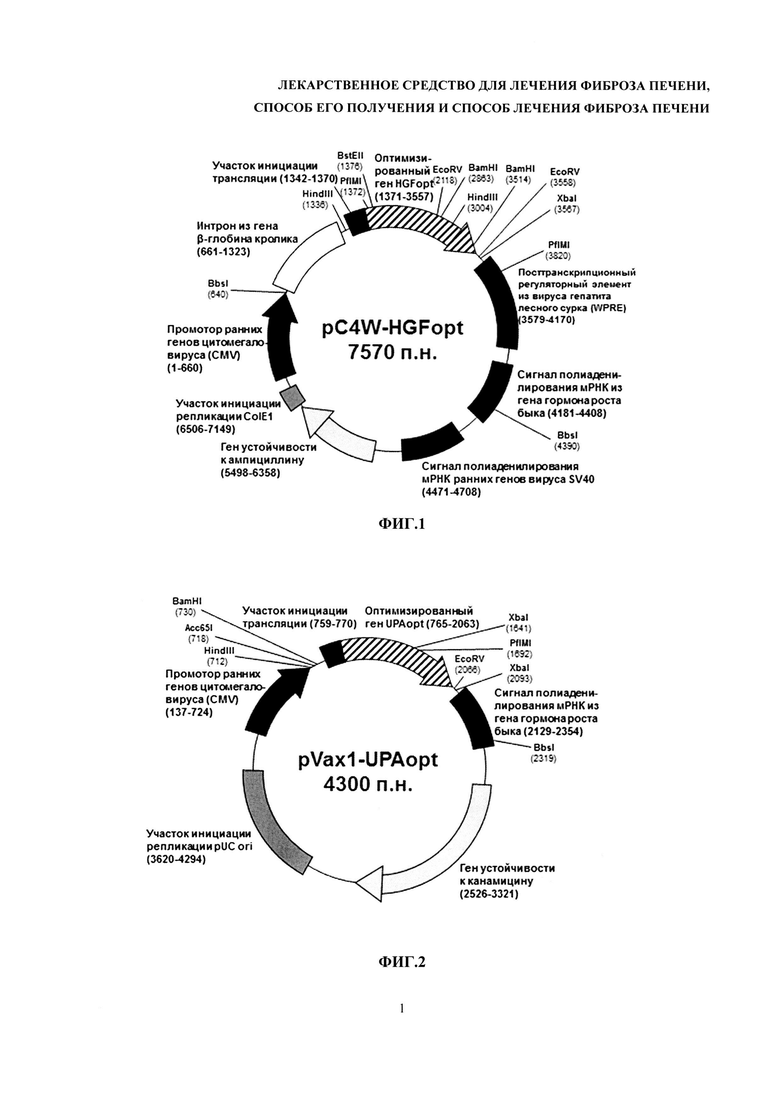
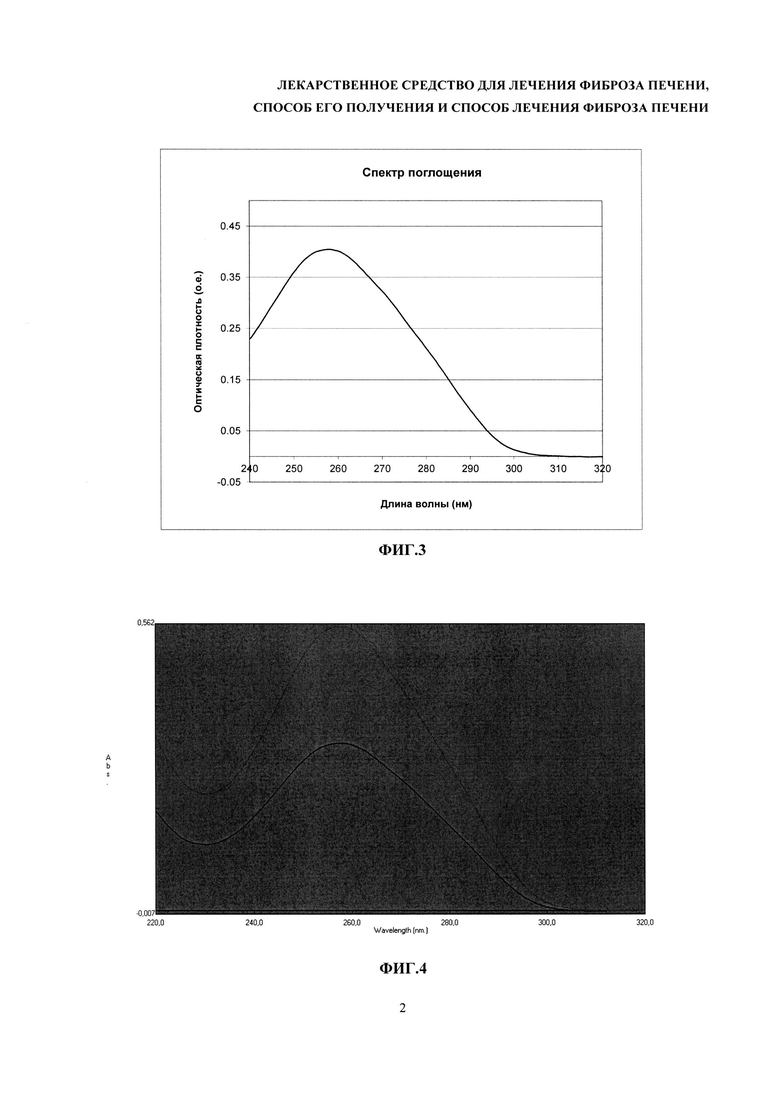
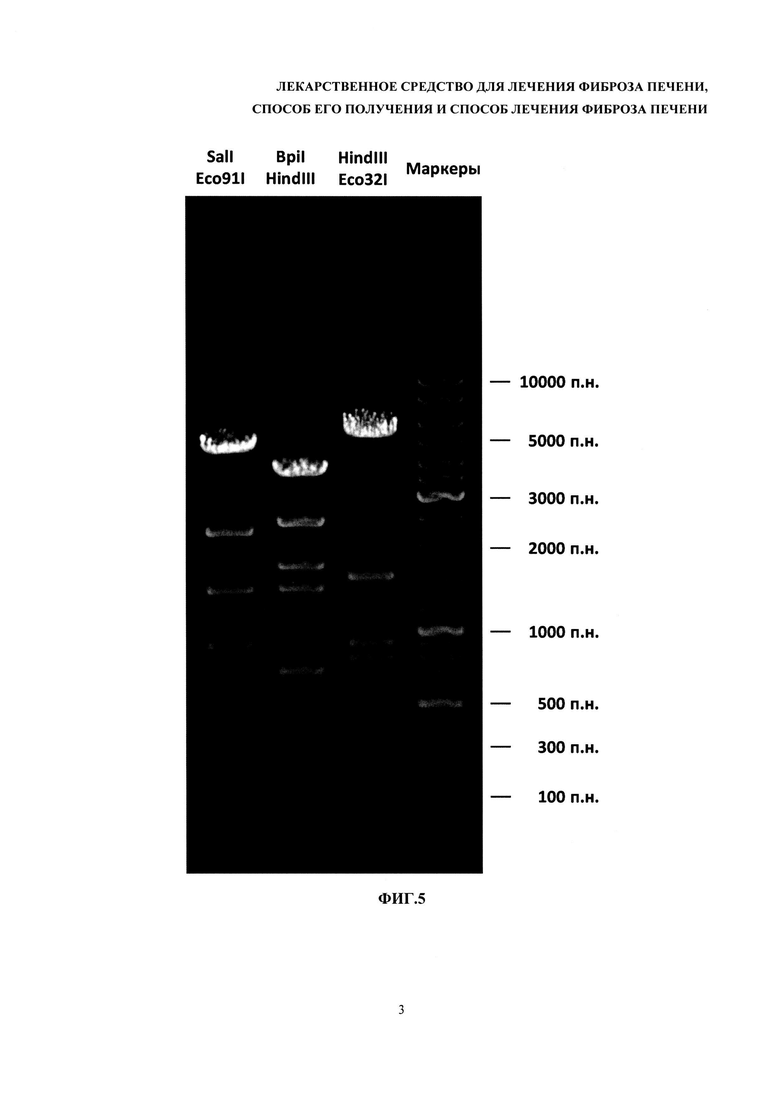
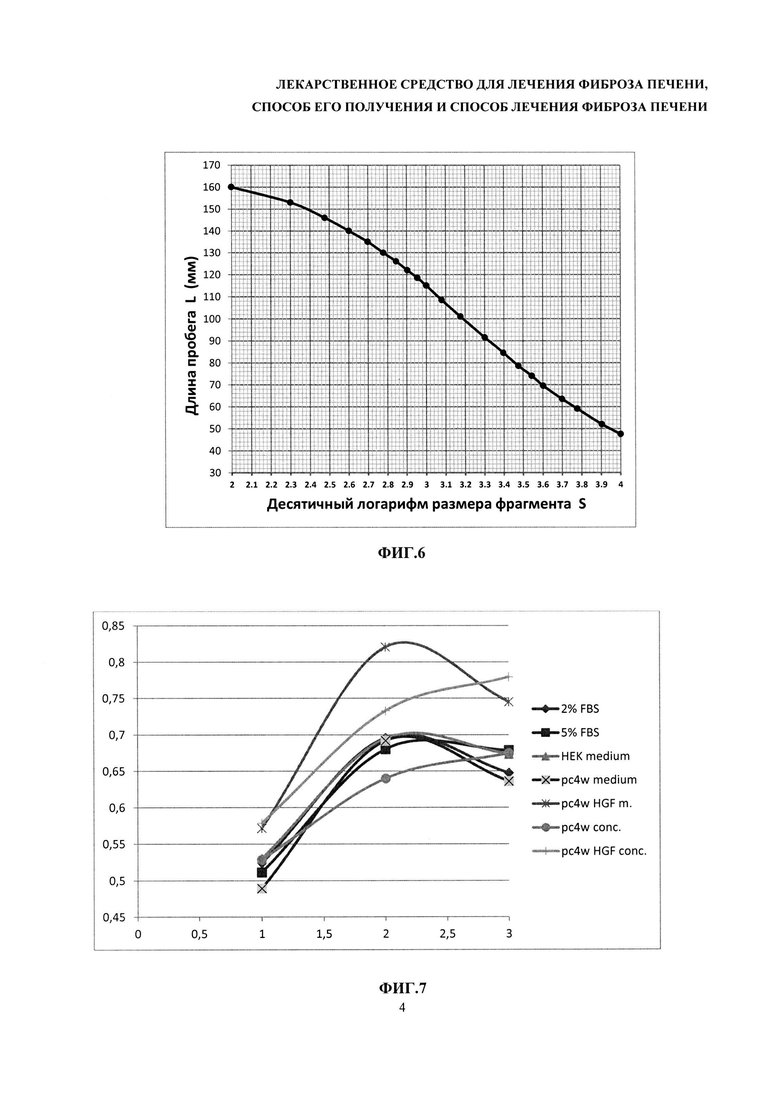
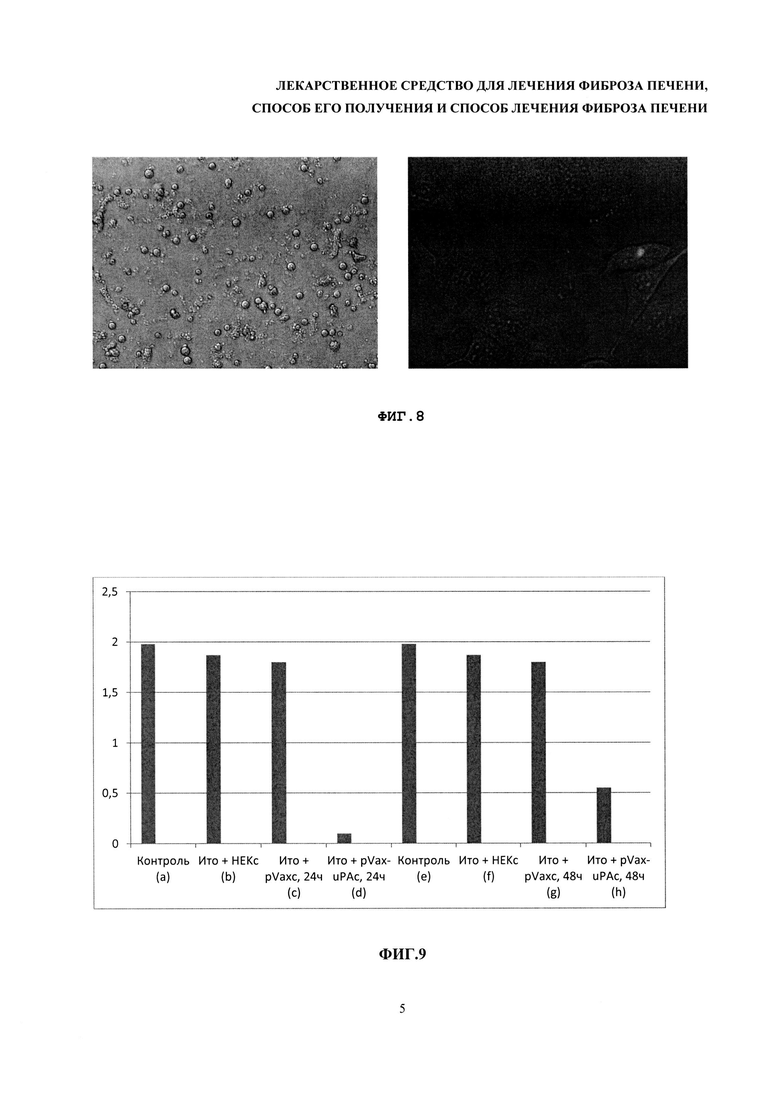
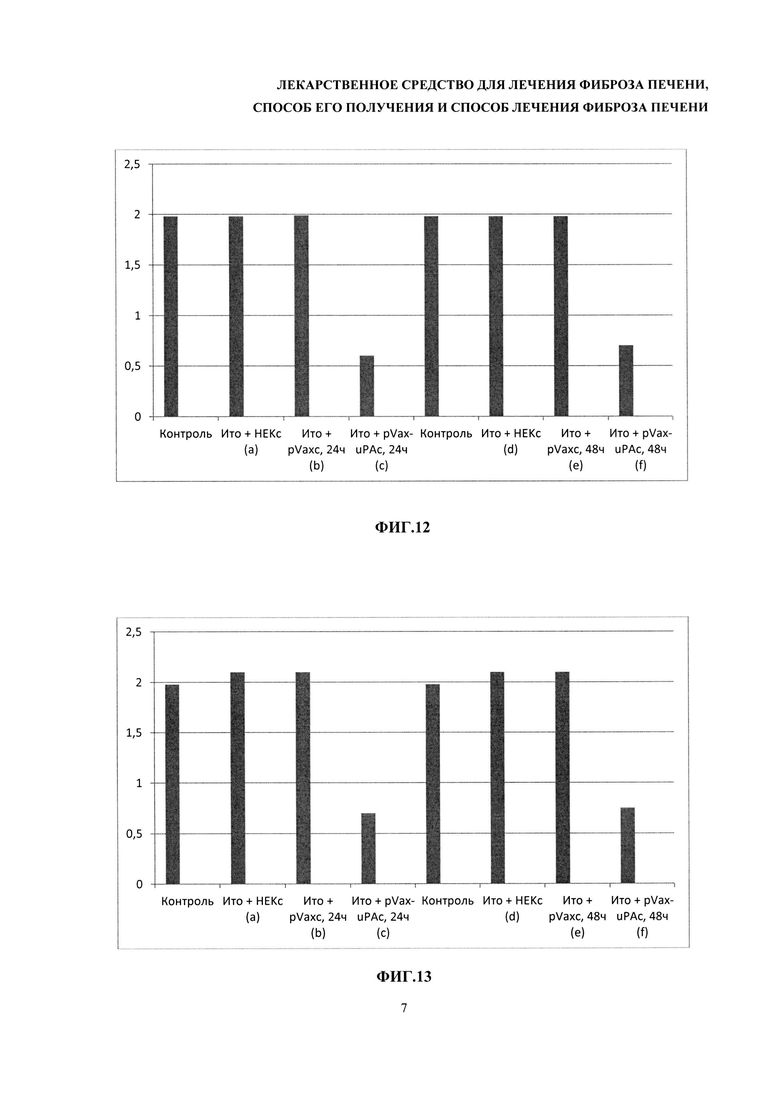
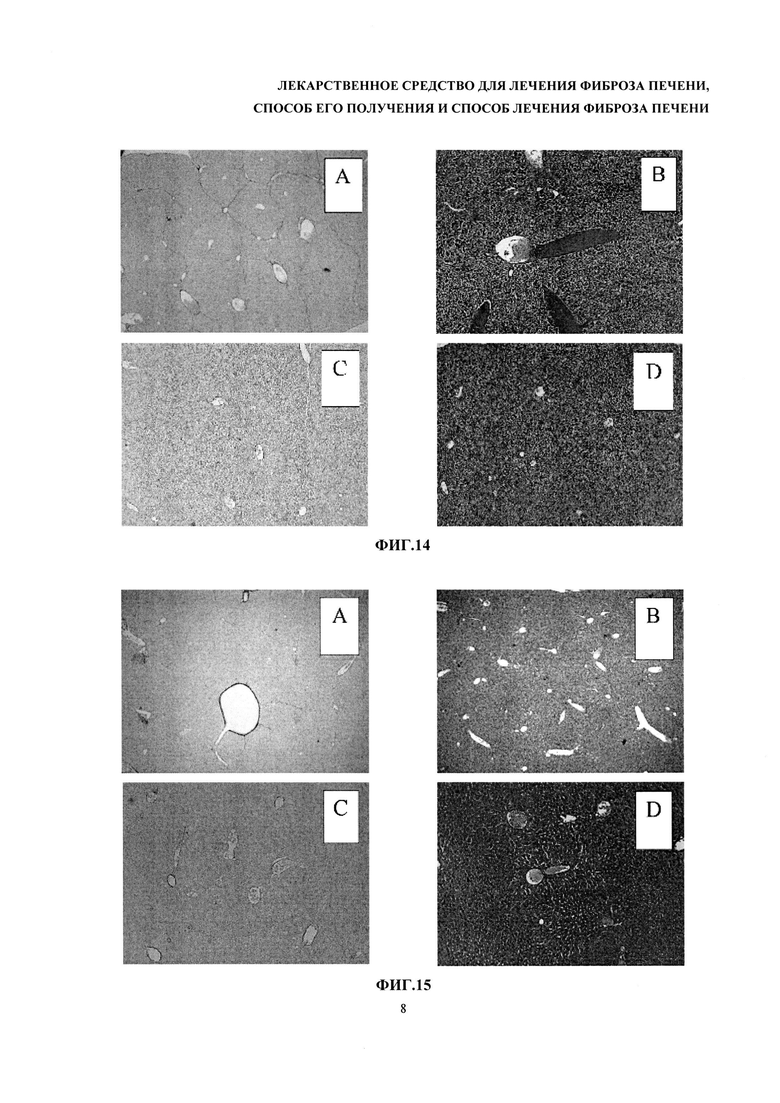
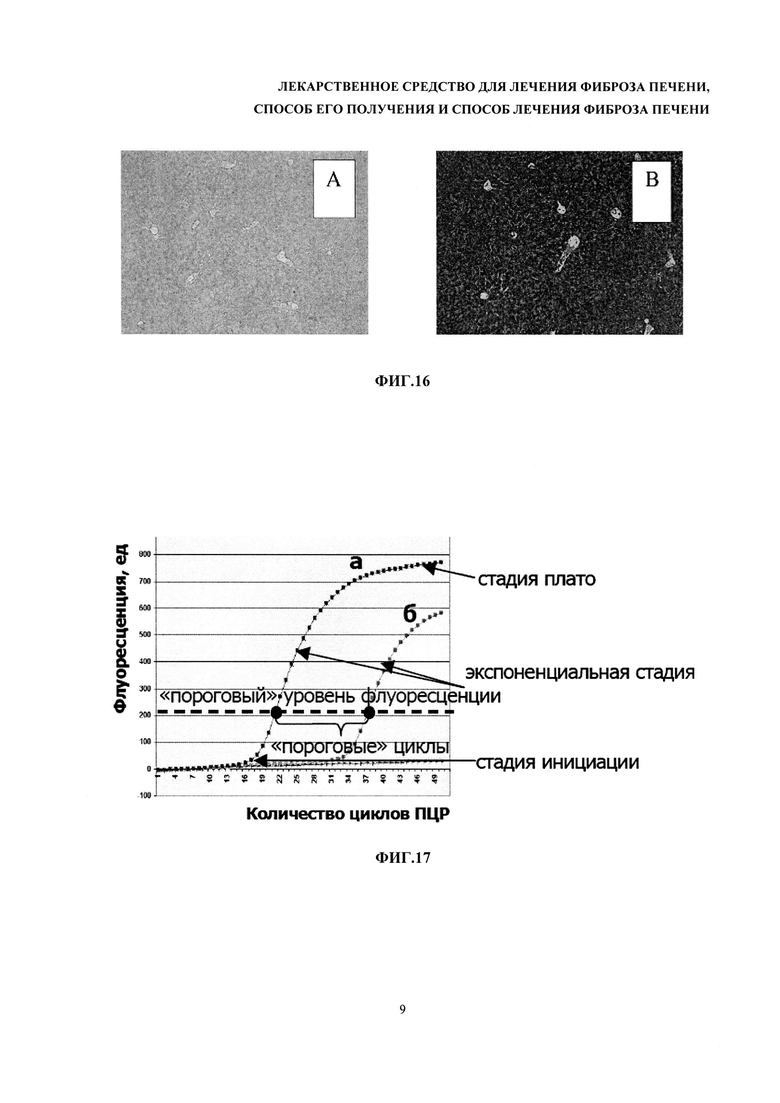
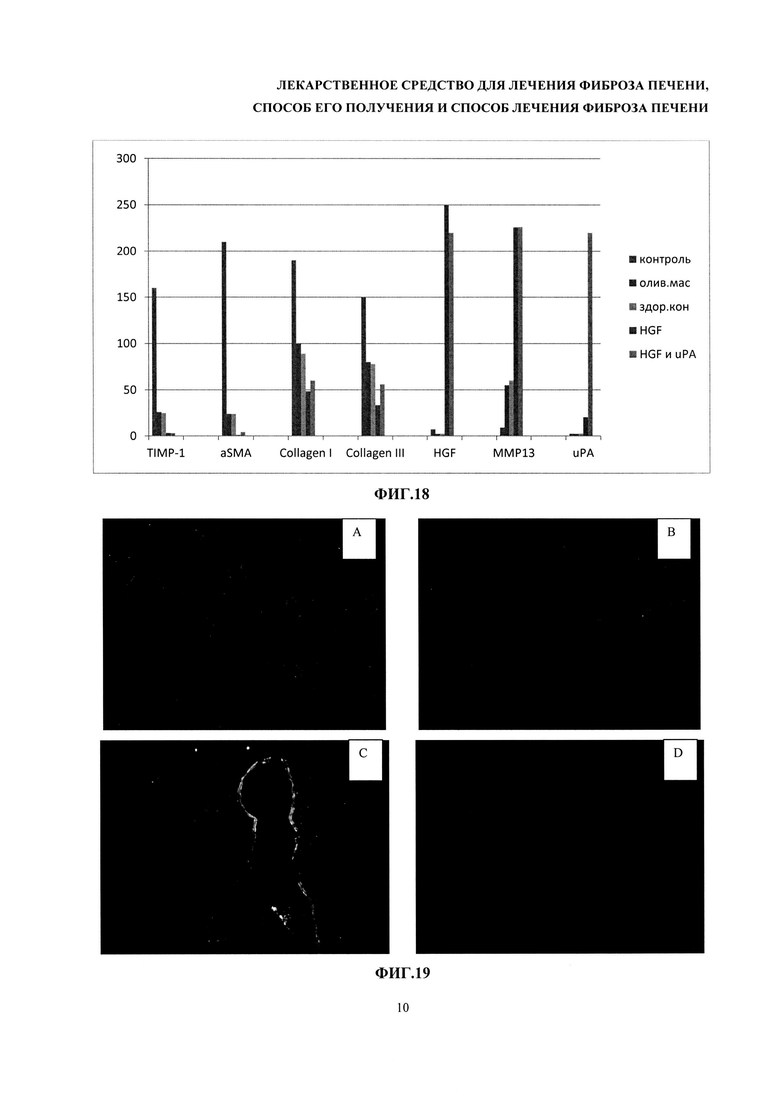
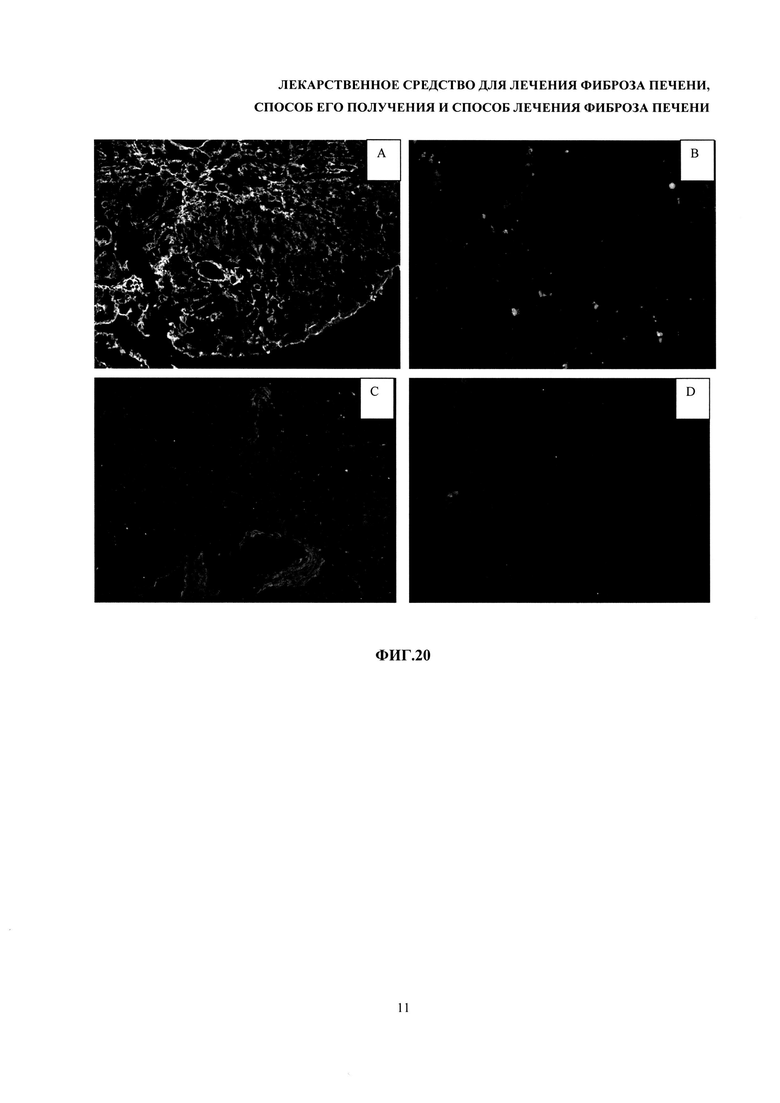
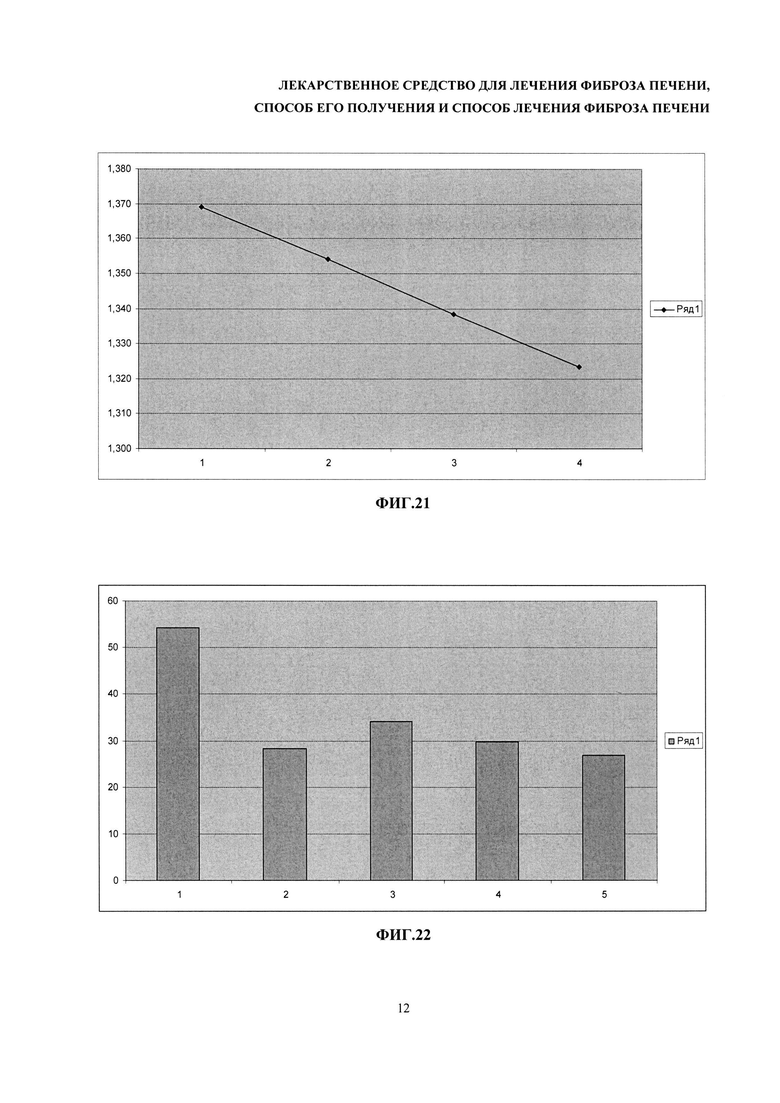
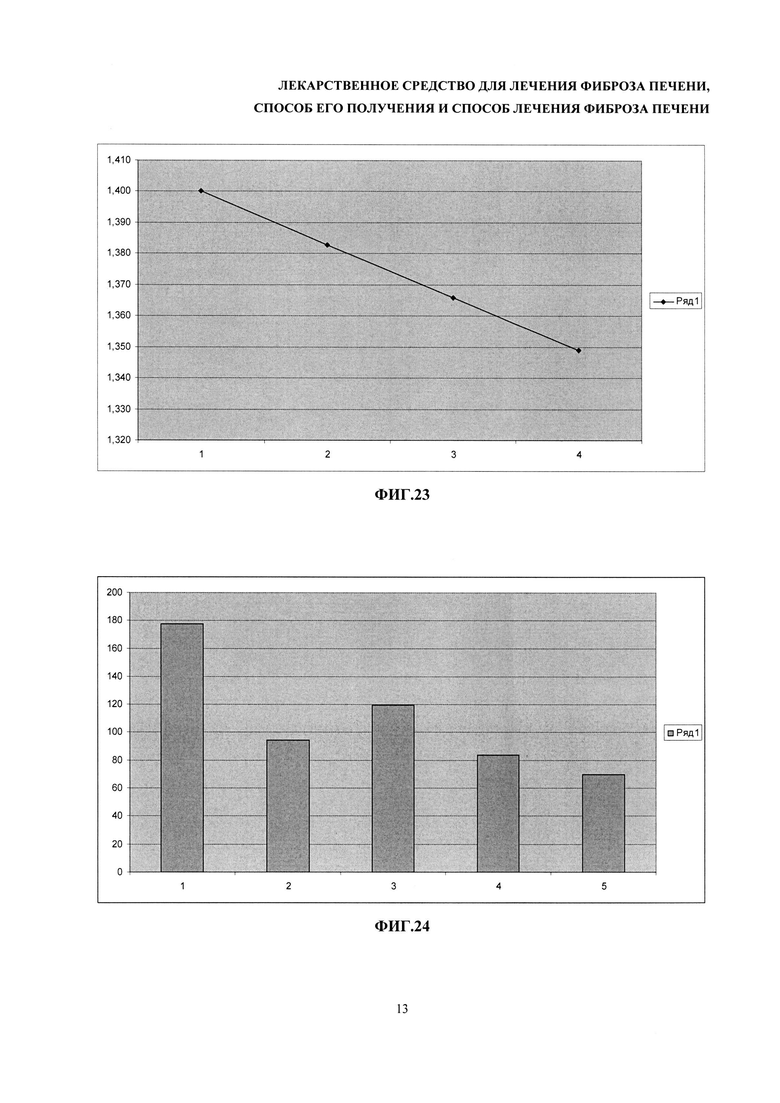








Изобретение относится к области биохимии, биотехнологии и генетической инженерии, в частности к лекарственному средству для лечения фиброза печени на основе смеси двух невирусных плазмидных конструкций. Первая невирусная плазмидная конструкция представляет собой pC4W-HGFopt и содержит ген, кодирующий фактор роста гепатоцитов человека. Вторая - pVax1-UPAopt и содержит ген, кодирующий урокиназу человека. В указанном лекарственном средстве плазмидные конструкции содержатся в следующих концентрациях: pC4W-HGFopt - от 0,5 до 0,7 мг/мл; pVax1-UPAopt - от 0,3 до 0,5 мг/мл, причем суммарная концентрация ДНК составляет 1±0,01 мг/мл. Настоящее изобретение раскрывает способ получения указанного лекарственного средства и способ лечения фиброза печени с использованием указанного лекарственного средства в фармацевтически приемлемом количестве. Настоящее изобретение позволяет получить лекарственное средство для лечения фиброза печени, обладающее повышенной эффективностью, являющееся безопасным и упрощенным в получении. 3 н. и 9 з.п. ф-лы, 28 ил., 4 табл., 9 пр.
1. Лекарственное средство для лечения фиброза печени на основе смеси двух невирусных плазмидных конструкций, содержащих терапевтические гены HGF и урокиназы человека, где первая невирусная плазмидная конструкция представляет собой pC4W-HGFopt и содержит ген, кодирующий фактор роста гепатоцитов человека, а вторая - pVax1-UPAopt и содержит ген, кодирующий урокиназу человека, при этом плазмидные конструкции содержатся в следующих концентрациях: pC4W-HGFopt - от 0,5 до 0,7 мг/мл; pVax1-UPAopt - от 0,3 до 0,5 мг/мл, причем суммарная концентрация ДНК составляет 1±0,01 мг/мл.
2. Средство по п. 1, характеризующееся тем, что оно представляет собой лифильно высушенный препарат.
3. Средство по п. 1, характеризующееся тем, что оно представляет собой жидкую субстанцию.
4. Средство по п. 1, характеризующееся тем, что оно содержит плазмидные ДНК, содержащие следующие регуляторные элементы: для плазмиды pVax1-UPAopt промотор ранних генов цитомегаловируса (с 137 по 724 н.п.), старт кодон трансляции в оптимальном окружении (759-770 н.п.), оптимизированная кДНК урокиназы (765-2063 н.п.), сигнал полиаденилирования мРНК гормона роста быка (3129-2354 н.п.); для плазмиды pC4W-HGFopt промотор ранних генов цитомегаловируса (1-660 н.п.), последовательность интрона бета-глобина (661-1323 н.п.), старт кодон трансляции в оптимальном окружении (1342-1370 н.п.), оптимизированная кДНК HGF (1371-3557 н.п.), посттранскрипционный регуляторный элемент из вируса гепатита лесного сурка (3579-4170 н.п.), сигнал полиаденилирования мРНК гормона роста быка (4181-4408 н.п.).
5. Способ получения лекарственного средства по п. 1, включающий получение двух бактериальных штаммов-продуцентов, первый из которых несет плазмиду pC4W-HGFopt, содержащую ген, кодирующий фактор роста гепатоцитов человека (HGF), а второй - плазмиду pVax1-UPAopt, содержащую ген, кодирующий урокиназу человека (UPA), наращивание биомассы указанных штаммов-продуцентов, несущих соответствующие плазмидные конструкции, выделение и очистка ДНК плазмидных конструкций с последующим смешением плазмиды pC4W-HGFopt в концентрации от 0,5 до 0,7 мг/мл и pVax1-UPAopt в концентрации от 0,3 до 0,5 мг/мл, причем суммарная концентрация ДНК составляет 1±0,01 мг/мл.
6. Способ по п. 5, характеризующийся тем, что очистку проводят с достижением конечного содержания примесей на 1 мг: белки E. coli - не более 10 мкг (≤1%), РНК E. coli - не более 10 мкг (≤1%), геномная ДНК E. coli - не более 10 мкг (≤1%), бактериальные эндотоксины - не более 40 ЕЭ, и содержанием суперспиральной формы плазмидной ДНК - не менее 800 мкг (≥80%) смешением полученных водных растворов плазмидных конструкций в пропорциях 50-70% pC4W-HGFopt:30-50% pVax1-UPAopt с последующей лиофильной сушкой до остаточной влажности не более 0,005 мг на 1 мг.
7. Способ по п. 6, характеризующийся тем, что по итогам очистки нуклеотидную последовательность получаемых плазмидных конструкций подтверждают с помощью анализа длин фрагментов, получающихся при гидролизе специфическими ферементами - эндонуклеазами рестрикции, определяют уровень примесей, чистоту и концентрацию растворов соответствующих плазмидных ДНК.
8. Способ по п. 6, характеризующийся тем, что плазмидная конструкция pC4W-HGFopt имеет нуклеотидную последовательность гена HGF:
ATGTGGGTGACCAAGCTGCTGCCAGCCCTGCTGCTGCAGCATGTCCTCCTGCATCTCCTCCTGCTCCCCATCGCCATCCCCTATGCAGAGGGACAAAGGAAAAGAAGAAATACAATTCATGAATTCAAAAAATCAGCAAAGACCACCCTGATCAAGATCGACCCCGCCCTGAAGATCAAGACCAAAAAAGTGAATACTGCAGACCAGTGTGCCAATAGATGTACCAGGAACAAGGGCCTGCCATTCACTTGCAAGGCTTTTGTTTTTGATAAAGCAAGAAAACAATGCCTCTGGTTCCCCTTCAATAGCATGTCAAGTGGAGTGAAAAAAGAATTTGGCCATGAATTTGACCTCTATGAAAACAAAGACTACATTAGAAACTGCATCATTGGTAAAGGACGCAGCTACAAGGGAACAGTATCTATCACTAAGAGTGGCATCAAATGTCAGCCCTGGAGTTCCATGATACCACACGAACACAGCTTTCTGCCTTCCAGCTACCGGGGCAAGGACCTGCAGGAGAACTACTGCCGGAACCCTCGAGGGGAAGAAGGGGGACCCTGGTGTTTCACAAGCAATCCAGAGGTACGCTACGAAGTCTGTGACATTCCTCAGTGTTCAGAAGTTGAATGCATGACCTGCAATGGGGAGAGTTATCGAGGTCTCATGGATCATACAGAATCAGGCAAGATTTGTCAGCGCTGGGATCATCAGACACCACACCGGCACAAATTCTTGCCTGAAAGATATCCCGACAAGGGCTTTGATGATAATTATTGCCGCAATCCCGATGGCCAGCCGAGGCCATGGTGCTATACTCTTGACCCTCACACCCGCTGGGAGTACTGTGCAATTAAAACATGCGCTGACAATACTATGAATGACACTGATGTTCCTTTGGAAACAACTGAATGCATCCAGGGCCAGGGCGAGGGCTACAGGGGCACTGTCAATACCATTTGGAATGGAATTCCATGTCAGCGTTGGGATTCTCAGTATCCTCACGAGCATGACATGACTCCTGAAAATTTCAAGTGCAAGGACCTGCGGGAGAACTACTGCCGGAATCCAGATGGGTCTGAATCACCCTGGTGTTTTACCACTGATCCAAACATCCGAGTTGGCTACTGCTCCCAAATTCCAAACTGTGATATGTCACATGGACAAGATTGTTATCGTGGGAATGGCAAAAATTATATGGGCAACCTGTCCCAGACCAGATCTGGCCTGACCTGTTCAATGTGGGACAAGAACATGGAGGACCTGCACCGGCACATCTTCTGGGAACCAGATGCAAGTAAGCTGAATGAGAATTACTGCCGAAATCCAGATGATGATGCTCATGGACCCTGGTGCTACACGGGAAATCCACTCATTCCTTGGGATTATTGCCCTATTTCTCGTTGTGAAGGTGATACCACACCTACCATCGTGAACCTGGACCACCCCGTGATCTCTTGCGCCAAGACCAAGCAGCTGCGGGTGGTGAACGGGATCCCAACACGAACAAACATAGGATGGATGGTTAGTCTGAGATACAGAAACAAGCACATCTGCGGCGGCTCCCTGATCAAGGAGAGTTGGGTTCTTACTGCACGACAGTGTTTCCCTTCTCGAGACTTGAAAGATTATGAAGCTTGGCTTGGAATTCATGATGTCCACGGAAGAGGAGATGAGAAATGCAAACAGGTTCTCAATGTTTCCCAGCTGGTATATGGCCCTGAAGGATCAGATCTGGTGCTGATGAAGCTGGCCAGGCCTGCTGTCCTGGATGATTTTGTGAGCACCATCGACCTGCCTAACTACGGATGCACAATTCCTGAAAAGACCAGTTGCAGTGTTTATGGCTGGGGCTACACTGGACTGATCAACTACGACGGCCTGCTGCGGGTGGCCCACCTGTACATCATGGGAAATGAGAAATGCAGCCAGCATCATCGAGGGAAGGTGACTCTGAATGAGTCTGAAATATGTGCTGGGGCTGAAAAGATTGGATCAGGACCATGTGAGGGGGATTATGGTGGCCCACTTGTTTGTGAGCAACATAAAATGAGAATGGTTCTTGGTGTCATTGTTCCTGGTCGTGGATGTGCCATTCCAAATCGTCCTGGTATTTTTGTCCGAGTAGCATATTATGCAAAATGGATCCACAAGATCATCCTGACCTACAAGGTGCCACAGTCCTGA
9. Способ по п. 5, характеризующийся тем, что плазмидная конструкция pVax1-UPAopt имеет нуклеотидную последовательность гена урокиназы:
ATGGGAAGAGCTCTGCTCGCTAGACTGCTACTTTGTGTGCTGGTGGTCTCTGATTCTAAAGGATCTAATGAGCTGCATCAGGTGCCTTCTAATTGCGACTGCCTGAATGGAGGCACATGCGTGTCTAACAAGTACTTCAGCAACATCCACTGGTGCAACTGTCCCAAGAAATTCGGAGGCCAGCATTGTGAGATTGACAAGTCCAAGACCTGCTACGAAGGGAATGGACATTTCTACAGAGGCAAAGCCTCTACTGACACCATGGGTAGACCATGTCTGCCTTGGAACTCTGCCACAGTGCTGCAGCAAACTTATCATGCTCATAGAAGCGATGCACTGCAGCTGGGACTGGGAAAACACAACTACTGCAGGAACCCTGATAACAGAAGGAGACCCTGGTGTTATGTGCAGGTCGGACTGAAACCACTGGTGCAGGAATGTATGGTGCATGATTGTGCTGATGGAAAGAAACCTTCTTCTCCACCTGAGGAACTGAAGTTCCAGTGTGGACAGAAAACCCTGAGACCCAGGTTCAAGATCATTGGAGGTGAGTTCACCACAATCGAGAACCAGCCTTGGTTTGCTGCCATTTATAGGAGACACAGGGGAGGCTCTGTGACATATGTGTGTGGAGGCTCTCTGATTTCTCCCTGTTGGGTGATTTCTGCTACTCACTGTTTCATCGACTATCCCAAGAAAGAGGACTACATCGTGTATCTGGGAAGGTCCAGACTGAATAGCAACACCCAGGGAGAAATGAAGTTCGAGGTGGAGAACCTGATCCTGCACAAGGACTACTCTGCTGATACACTGGCTCATCATAACGACATCGCACTGCTCAAAATCAGGAGCAAAGAGGGAAGATGTGCTCAGCCATCTAGAACTATCCAGACCATTTGCCTGCCTTCTATGTACAACGACCCACAGTTTGGCACATCTTGCGAAATCACAGGCTTTGGAAAGGAGAACTCCACCGACTATCTGTACCCTGAACAGCTGAAGATGACCGTGGTCAAGCTGATCAGCCACAGGGAATGCCAGCAACCTCATTACTATGGATCTGAGGTGACTACCAAGATGCTGTGTGCTGCAGATCCACAGTGGAAAACAGACTCCTGTCAGGGAGATTCTGGTGGACCTCTGGTGTGTTCTCTGCAGGGAAGAATGACTCTGACAGGAATTGTGTCTTGGGGAAGAGGATGTGCTCTGAAAGATAAGCCTGGAGTGTATACCAGAGTGTCTCACTTTCTTCCATGGATTAGGAGCCATACCAAGGAAGAGAATGGACTTGCTCTGTAA
10. Способ лечения фиброза печени, включающий введение средства по п. 1 в фармацевтически приемлемом количестве.
11. Способ по п. 10, характеризующийся тем, что осуществляют 2-кратное с интервалом 2-3 дня внутривенное введение в организм пациента в количестве, не превышающем 3,75 мг/кг для грызунов и 0,3125 мг/кг для человека.
12. Способ по п. 11, характеризующийся тем, что используют лиофильно высушенное средство, которое перед ведением в организм млекопитающего предварительно растворяют в изотоническом растворе.
| ДЖОЯШВИЛИ Н.А., и др., Генная терапия фактором роста гепатоцитов приводит к регрессии экспериментального фиброза печени, РЖГГК, 2010, No.4, с.22-28 | |||
| LIN Y., et al., Treatment of experimental hepatic fibrosis by combinational delivery of urokinase-type plasminogen activator and hepatocyte growth factor genes, Liver Int., 2005, 25(4), pp.796-807 | |||
| ГЕН HGFopt ФАКТОРА РОСТА ГЕПАТОЦИТОВ | 2008 |
|
RU2385936C1 |
| СПОСОБ СТИМУЛИРОВАНИЯ ВОССТАНОВЛЕНИЯ ПЕРИФЕРИЧЕСКОЙ ИННЕРВАЦИИ ТКАНЕЙ С ПОМОЩЬЮ ВЕКТОРНЫХ КОНСТРУКЦИЙ | 2011 |
|
RU2486918C1 |
| INVITROGEN, catalog no | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |

