ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Приоритет настоящей заявки испрашивается на основании китайской патентной заявки №201510560190.7, поданной 2 сентября 2015 г. в патентное ведомство Китая, которая в порядке ссылки во всей полноте и во всех целях включена в настоящую заявку, как если бы она была конкретно и целиком изложена в ней.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к медицине, более точно, к соединениям, композициям, их получению и применению, при этом описанные соединения или композиции могут использоваться для ингибирования активности ксантиноксидазы и переносчика 1 уратов, а также для профилактики или лечения заболеваний, связанных с высоким содержанием мочевой кислоты в крови.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Мочевую кислоту, являющуюся конечным метаболитом пуриновых соединений в организме человека, в основном выводят почки, которые выделяют две трети ее общего количества. Накопление мочевой кислоты, вызванное ее сверхпродукцией или нарушениями экскреции, приводит к повышению ее содержания в крови, а затем к гиперурикемии. При рационе с нормальным содержанием пуринов гиперурикемией является уровень содержания мочевой кислоты в сыворотке крови натощак в различные дни выше 420 мкмоль/л у мужчин и выше 360 мкмоль/л у женщин. Причины гиперурикемии можно разделить на три типа: (1) увеличение продукции мочевой кислоты, (2) плохая экскреция мочевой кислоты, и (3) смешанный тип, при этом такая классификация применима для обнаружения причины гиперурикемии и назначения целевого лечения.
При перенасыщении крови мочевой кислотой урат натрия начинает образовывать кристаллы и осаждаться в синовиальной оболочке суставов, синовиальных сумок, хрящей или других тканей. Быстрые изменения уровней мочевой кислоты, высвобождение мельчайших кристаллов вследствие местного повреждения и изменений в покрытии кристаллов мочевой кислоты может вызывать повторяющуюся и пароксизмальную воспалительную реакцию, а затем подагру. Подагра относится, в частности, к острому артриту и хроническому отложению солей, преимущественно включая острые приступы артрита, образование подагрических узлов, отложение солей в сочетании с хроническим артритом, мочекислую нефропатию, мочекислые камни в мочевых путях и тяжелые симптомы, такие как поражение сустава и почечная недостаточность. Кроме того, подагре также сопутствует гипертензия, метаболический синдром, гиперлипидемия, диабет и резистентность к инсулину и другие заболевания (Terkeltaub RA. Clinical practice. Gout [J]. N Engl J Med. 2003, 349: 1647-1655)(Schlesinger N, Schumacher HR Jr. Gort: can management be improved [J]. Curr Opin Rheumatol. 2001, 13:240-244).
Гиперурикемия и подагра, которые угрожают здоровью человека, являются серьезным метаболическим заболеванием. Известно, что примерно у 5-12% пациентов с гиперурикемией впоследствии развивается подагра. Поскольку мочевая кислота является материальной основой гиперурикемии и подагры, снижение концентрации мочевой кислоты в крови может использоваться для профилактики или лечения гиперурикемии и подагры, а также снижения риска осложнений, связанных с гиперурикемией и подагрой.
В настоящее время существуют препараты двух типов, используемые для снижения уровня мочевой кислоты, один из которых используется для ингибирования продукции мочевой кислоты, а другой для увеличения экскреции мочевой кислоты.
Мочевая кислота образуется в результате потребления с пищей и эндогенного синтеза пурина, который в конечном итоге образуется при окислении ксантиноксидазы. Таким образом, ксантиноксидаза рассматривается в качестве важной мишени для лекарственных средств-ингибиторов продукции мочевой кислоты. Хотя сообщалось, что существующий препарат Лопурин, используемый для ингибирования продукции мочевой кислоты, эффективен при лечении гиперурикемии и различных заболеваний, вызываемых гиперурикемией, также отмечалось, что Лопурин имеет серьезные побочные эффекты, такие как синдром интоксикации, апластическая анемия, нарушение функции печени, эксфолиативный дерматит, синдром Стивенса-Джонсона и т.д. (Kazuhide Ogino и др., Nippon Rinsho {Japan Clinical), 2003, Vol. 61, Extra edition 1, pp. 197-201). Соответственно, требуется разработка препаратов с высокой эффективностью, низкой токсичностью и малыми побочными эффектов.
С другой стороны, около 90% гиперурикемии обусловлено снижением экскреции мочевой кислоты, при этом экскреция мочевой кислоты почками включает в основном четыре процесса: клубочковую фильтрацию, реабсорбцию в почечных канальцах и собирательных трубочках, секрецию в почечных канальцах и собирательных трубочках, а также реабсорбцию после секреции, при этом в каждом процессе участвует соответствующий белок, и, наконец, из организма выводится только 8-12% мочевой кислоты (Liu Ruoxia, Cang Luping, Wu Xinrong, Shangdong Medical Journal [J], 2002, 52(28)). Переносчик 1 уратов (URAT1) представляет собой трансмембранный белок, открытый Эномото и др., который находится на стороне щеточной каемки эпителиальных клеток проксимальных почечных канальцев и участвует в реабсорбции мочевой кислоты в проксимальных почечных канальцах. hURAT1, кодируемый геном SLC22A12 (содержащим 10 экзонов и 9 интронов) в хромосоме 11q13, содержит 555 аминокислотных остатков, 12 трансмембранных доменов, концевой домен -NH2 и концевой домен -СООН внутри клетки. Исследования показали, что у пациентов с почечной гиперурикемией произошла мутация гена SLC22A12 с потерей им способности кодировать зрелый белок URAT1, что говорит о том, что URAT1 является патогенным геном в отношении почечной гиперурикемии (Enomoto, Kimura Н, Chairoungdua А, и др. Molecular identification of a renal urate anion exchanger that regulates blood urate levels [J]. Nature, 2002, 417 (6887): 447-452), и что URAT1 важен для реабсорбции мочевой кислоты в почках и тесно связан с регулированием мочевой кислоты в крови. Таким образом, соединения, ингибирующие активность URAT1, могут использоваться для содействия экскреции, содержащейся в крови мочевой кислоты, а также для лечения или профилактики заболеваний, связанных с высоким уровнем мочевой кислоты в крови, включая гиперурикемию, подагру, отложение солей, подагрический артрит, заболевания почек, связанные с гиперурикемией, мочевые конкременты и т.п.
Сообщалось о том, что комбинация аллопуринола и средств, способствующих выведению мочевой кислоты, более эффективно снижает содержание мочевой кислоты в сыворотке крови, чем только аллопуринол (S Takahashi, Ann. Rheum. Dis., 2003, 62, 572-575). Таким образом, комбинация средства, способствующего выведению мочевой кислоты, и ингибитора продукции мочевой кислоты может обеспечивать недостижимый для монотерапии терапевтический эффект без соответствующих рисков, поскольку, например, монотерапия гиперурикемии средствами, способствующими выведению мочевой кислоты при плохом выведении мочевой кислоты может вызвать риск образования мочевых конкрементов, в то время как комбинация средства, способствующего выведению мочевой кислоты, и ингибитора продукции мочевой кислоты может обеспечивать лучший терапевтический эффект.
Препараты, ингибирующие как ксантиноксидазу, так и URAT1, обеспечивают лучший терапевтический эффект для пациентов и более удобны, чем комбинированные препараты. Они актуальный для лечения гиперурикемии, подагры и заболеваний, связанных с гиперурикемией.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В данном разделе вкратце изложены некоторые особенности настоящего изобретения, которыми оно не ограничено. Эти и другие особенности и варианты осуществления изобретения более подробно описаны далее. Существует более полное описание этих и других частей. Все ссылки, приведенные в настоящем описании, во всей их полноте в порядке ссылки включены в настоящую заявку. В случае различий между настоящим описанием и ссылками, преимущественную силу имеет настоящее описание.
В настоящем изобретении предложены соединения с антагонистической активностью в отношении как ксантиноксидазы, так и URAT1, которые могут использоваться при изготовлении лекарственного средства для профилактики или лечения заболеваний, связанные с высоким уровнем мочевой кислоты в крови, таких как гиперурикемия, подагра, отложение солей, подагрический артрит, заболевания почек, связанные с гиперурикемией и мочекаменной болезнью, и т.д. Соединения, согласно настоящему изобретению, обладают хорошей ингибирующей активностью в отношении как ксантиноксидазы, так и URAT1, а также отличными физико-химическими и фармакокинетическими свойствами.
В настоящем изобретении также предложен способ получения таких соединений и фармацевтических композиций, содержащих эти соединения, и способ применения этих соединений или композиций для лечения описанных выше заболеваний у млекопитающих, в особенности, у людей.
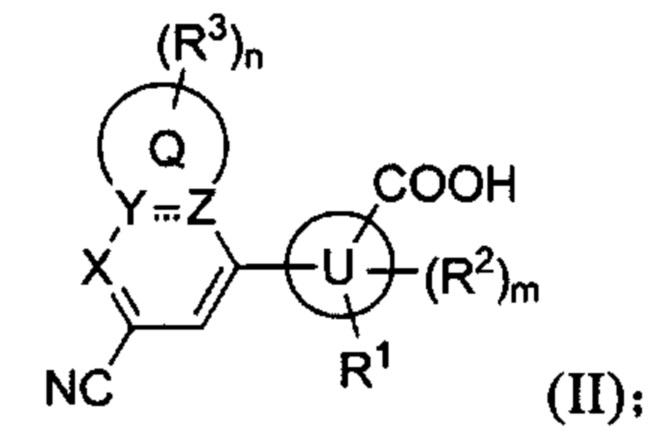
В частности, согласно одной из особенностей изобретения, предложено соединение, имеющее Формулу (I), или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль или пролекарство,
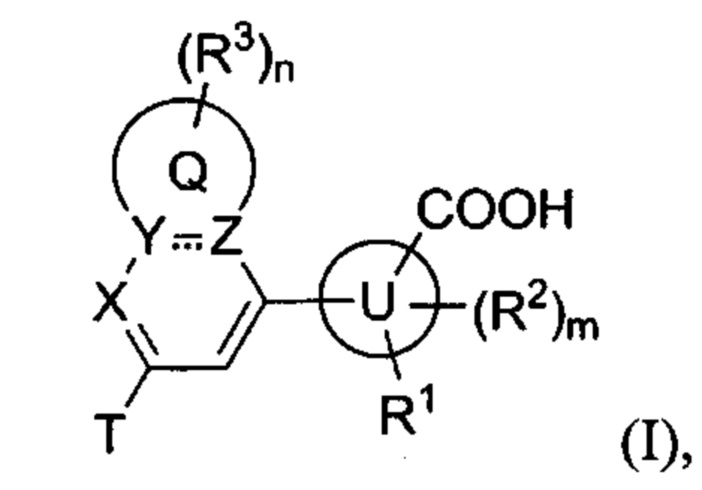
где:
U представляет собой фенил или 5- или 6-членный гетероарил;
каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, С1-6 алкил, С2-6 алкенил, С2-6алкинил, C1-6галогеналкил, С1-6алкокси, C1-6галогеналкокси, C1-6алкиламино, C1-6галоалкиламино, от 3- до 8-членный циклоалкил или 3- до 8-членный гетероциклил, при этом каждый из C1-6-алкила, С2-6-алкенила, С2-6-алкинила, C1-6-галогеналкила, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членного циклоалкила или от 3- до 8-членного гетероциклила независимо и необязательно замещен 1, 2, 3, 4 или 5 заместителями, выбранными из ОН, оксо (=О), NH2, NO2 или CN;
Т представляет собой Н, D, F, Cl, Br, NO2, CN или CF3;
X представляет собой CR4 или N;
R4 представляет собой Н, D, галоген, C1-6-алкил, C1-6-галогеналкил, C1-6-алкокси, C1-6-алкиламино или C1-6-галогеналкокси;
каждый из Y и Z независимо представляет собой С, СН или N;
 представляет собой одинарную или двойную связь;
представляет собой одинарную или двойную связь;
Q представляет собой фен, С4-7 карбоцикл, 4-7-членный гетероцикл или 5-или 6-членное гетероароматическое кольцо;
каждый R3 независимо представляет собой Н, D, галоген, оксо (=О), ОН, NH2, NO2, CN, C1-6-алкил, С2-6-алкенил, С2-6-алкинил, C1-6-галогеналкил, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членный циклоалкил, 3- до 8-членный гетероциклил, от 5- до 10-членный гетероарил, фенил, нафтил или G, при этом каждый из C1-6-алкила, С2-6-алкенила, С2-6-алкинила, C1-6-галогеналкила, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членного циклоалкила, от 3- до 8-членного гетероциклила, от 5- до 10-членного гетероарила независимо и необязательно замещен 1, 2, 3, 4 или 5 заместителями, выбранными из ОН, оксо (=О), NH2, NO2, CN или G;
G представляет собой замещенный C1-6 алифатический углеводород, где каждая из метиленовых групп C1-6 алифатического углеводорода необязательно и независимо замещена J;
J представляет собой -NH-, -S-, -О-, -С(=O)-, -C(=O)NH-, -SO-, -SO2-, -NHC(=O)-, -С(=O)O-, -SO2NH- или -NHC(=O)NH-;
m равно 0, 1, 2 или 3; и
n равно 0, 1, 2, 3 или 4;
при условии, что:
1. когда Т представляет собой F, Cl, Br или CF3, R1 представляет собой ОН;
2. когда Т представляет собой Н, 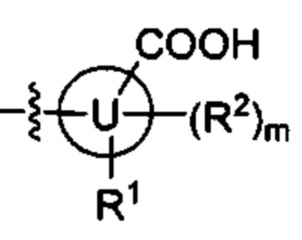 представляет собой
представляет собой
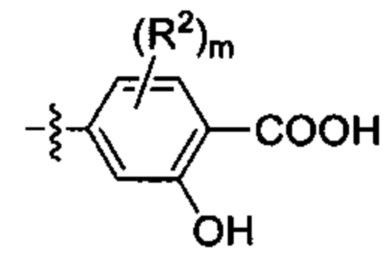 , a Q не означает фен;
, a Q не означает фен;
3. когда Т представляет собой NO2, R1 не означает Н.
В некоторых вариантах осуществления предложенным соединением, имеющим Формулу (I), является соединение, имеющее Формулу (II), или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль и пролекарство,
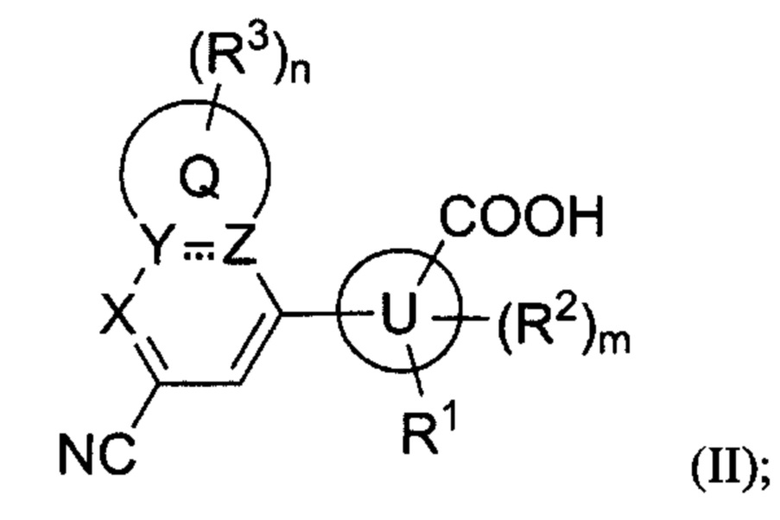
где Q, U, X, Y, Z, R1, каждый R2, каждый R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления U представляет собой фенил, пиридинил, пиримидинил, пиразинил, пиридазинил, 1,3,5-триазинил, пирролил, фуранил, тиазолил, тиенил, оксазолил или изоксазолил.
В других вариантах осуществления U представляет собой фенил 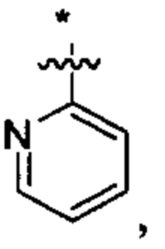
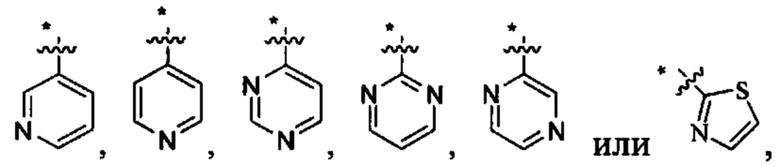 при этом "*" относится к положению кольца, присоединенного к
при этом "*" относится к положению кольца, присоединенного к 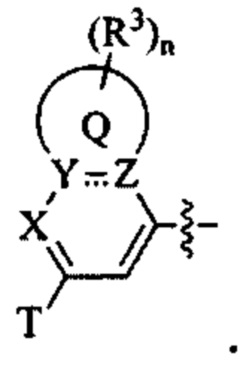
В некоторых вариантах осуществления предложенным соединением, имеющим Формулу (I), является соединение с Формулой (III), или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль и пролекарство,
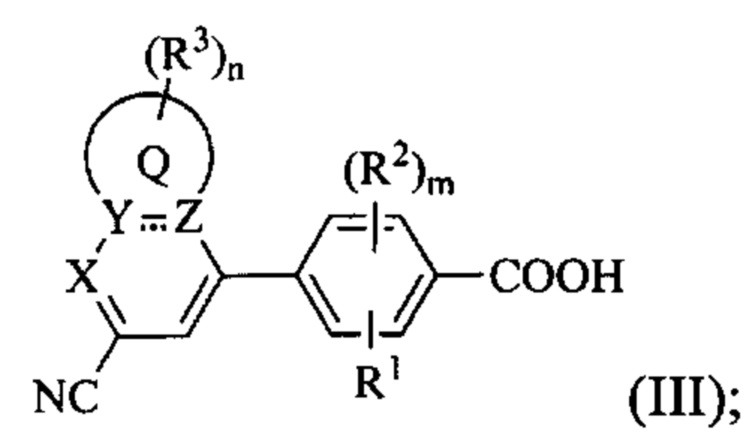
где Q, X, Y, Z, R1, каждый R2, каждый R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления изобретения каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, С1-4-алкил, С2-4-алкенил, С2-4-алкинил, C1-4-галогеналкил, C1-4-алкокси, С1-4-галогеналкокси, С1-4-алкиламино, C1-4-галоалкиламино, от 3- до 6-членный циклоалкил или от 3- до 6-членный гетероциклил, при этом каждый из С1-4-алкила, С2-4-алкенила, С2-4-алкинил, C1-4-галогеналкила, С1-4-алкокси, С1-4-галогеналкокси, C1-4-алкиламино, C1-4-галоалкиламино, от 3- до 6-членного циклоалкила или от 3- до 6-членного гетероциклила независимо и необязательно замещен 1, 2 или 3 заместителями, выбранными из ОН, оксо (=O), NH2, NO2 или CN.
В других вариантах осуществления изобретения каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, метил, этил, изопропил, бутил, гидроксиметил, гидроксиэтил, аминометил, дифторметил, трифторметил, метокси, этокси, изопропокси, трет-бутокси, n-бутокси, метиламино, этиламино, дифторметокси, трифторметокси, ацетил, ацетокси, ацетиламино, циклопропил, циклобутил, циклопентил, циклогексил, оксиранил, пирролидинил или тетрагидрофуранил.
В некоторых вариантах осуществления изобретения каждый R3 независимо представляет собой Н, D, галоген, оксо (=О), ОН, NH2, NO2, CN, метил, этил, изопропил, дифторметил, трифторметил, метокси, этокси, изопропокси, дифторметокси, трифторметокси, формил, карбокси, формамидо, ацетил, карбамоил, пропилсульфонамидо, циклопропил, циклобутил, имидазолил, пиразолил, тиазолил, оксазолил, пиридил, пиримидинил, хинолил, индолил, фенил или нафтил.
В некоторых вариантах осуществления изобретения каждый R4 представляет собой Н, D, галоген, метил, этил, изопропил, трет-бутил, n-бутил, дифторметил, трифторметил, метокси, этокси, трет-бутокси, метиламино, дифторметокси или трифторметокси.
В некоторых вариантах осуществления  представляет собой
представляет собой 
В некоторых вариантах осуществления 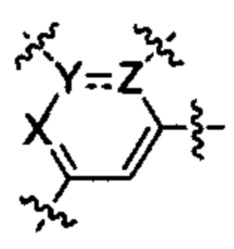 представляет собой
представляет собой 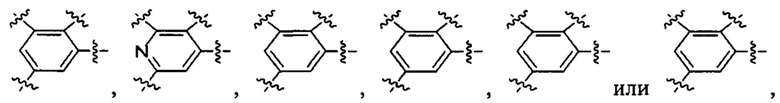 при этом *1 относится к положению, присоединенному к U-кольцу.
при этом *1 относится к положению, присоединенному к U-кольцу.
Согласно другой особенности изобретения, предложена фармацевтическая композиция, содержащая описанное соединение.
В некоторых вариантах осуществления предложена фармацевтическая композиция, дополнительно содержащая фармацевтически приемлемый эксципиент, носитель, адъювант, растворитель или их сочетание.
В других вариантах осуществления изобретения, предложена фармацевтическая композиция, дополнительно содержащая лекарственное средство для профилактики или лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, при этом действующее вещество лекарственного средства отличается от соединения согласно настоящему изобретению, а лекарственное средство представляет собой колхицин, нестероидное противовоспалительное средство, глюкокортикоид, средство против мочевой кислоты, средство выведения мочевой кислоты, средство подщелачивания мочи или их сочетание.
Согласно другой особенности изобретения, предложено применение описанного соединения или фармацевтической композиции при изготовлении лекарственного средства для профилактики или лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью у субъекта.
Согласно другой особенности изобретения, предложен способ профилактики или лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью у субъекта, включающий введение субъекту терапевтически эффективного количества описанного соединения или фармацевтической композиции.
Согласно другой особенности изобретения, предложено описанное соединение или фармацевтическая композиция для применения при профилактике или лечении гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью у субъекта.
Согласно другой особенности изобретения, предложено применение описанного соединения или фармацевтической композиции при изготовлении лекарственного средства для снижения уровня мочевой кислоты в крови.
Согласно другой особенности изобретения, предложен способ снижения уровня мочевой кислоты в крови у субъекта, включающий введение субъекту терапевтически эффективного количества описанного соединения или фармацевтической композиции.
Согласно другой особенности изобретения, предложено описанное соединение или фармацевтическая композиция для применения при снижении уровня мочевой кислоты в крови.
Согласно другой особенности изобретения, предложено применение описанного соединения или фармацевтической композиции при изготовлении лекарственного средства для ингибирования как ксантиноксидазы, так и переносчика 1 уратов.
Согласно другой особенности изобретения, предложен способ ингибирования ксантиноксидазы и переносчика 1 уратов у субъекта, включающий введение субъекту терапевтически эффективного количества описанного соединения или фармацевтической композиции.
Согласно другой особенности изобретения, предложено описанное соединение или фармацевтическая композиция для применения при ингибировании ксантиноксидазы и переносчика 1 уратов.
Согласно другой особенности изобретения, предложен способ получения, отделения или очистки соединения Формулы (I).
Биологические испытания показывают, что соединения согласно настоящему изобретению могут применяться в качестве эффективных ингибиторов ксантиноксидазы и переносчика 1 уратов.
Любой описанный вариант осуществления может быть объединен с другими вариантами осуществления при условии, что они не противоречат друг другу, даже если варианты осуществления описаны применительно к различным особенностям настоящего изобретения. Кроме того, любой технический признак одного варианта осуществления может применяться к соответствующему техническому признаку других вариантов осуществления изобретения при условии, что они не противоречат друг другу, даже если варианты осуществления описаны применительно к различным особенностям настоящего изобретения.
В данном разделе кратко изложены некоторые особенности, описанные в настоящем изобретении, без намерения ограничить настоящее изобретение. Эти и другие особенности и варианты осуществления изобретения более подробно описаны далее.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ОПРЕДЕЛЕНИЯ И ОБЩАЯ ТЕРМИНОЛОГИЯ
Теперь приведено подробное описание некоторых вариантов осуществления изобретения, примеры которых проиллюстрированы прилагаемыми структурными формулами. Подразумевается, что изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в его объем, определяемый формулой изобретения. Специалистам в данной области техники известные многочисленные способы и материалы, аналогичные или эквивалентные описанным в настоящем изобретении, которые могут применяться при осуществлении настоящего изобретения. Настоящее изобретение никоим образом не ограничено описанными в нем способами и материалами. Если один или несколько включенных в порядке ссылки источников, патентов и аналогичных материалов отличается от настоящей заявки или противоречит ей, включая без ограничения определяемые термины, использование терминов, описанные методы и т.п., предпочтение отдается настоящей заявке.
Следует также учесть, что определенные признаки изобретения, которые для ясности описаны в контексте нескольких отдельных вариантов осуществления, также могут использоваться в комбинации в одном из вариантов осуществления. И наоборот, различные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут также использоваться отдельно или в любой применимой субкомбинации.
Если не указано иное, все используемые технические и научные термины имеют такое же значение, в котором их обычно используют специалисты в той области техники, к которой относится настоящее изобретение. Все упоминаемые патенты и публикации, во всей полноте в порядке ссылки включены в настоящую заявку.
Если не указано иное, в настоящей заявке применяются следующие определения. В целях настоящего изобретения химические элементы идентифицируют в соответствии с периодической таблицей элементов, версия CAS и Справочником по химии и физике, 75-е издание, 1994 г. Кроме того, общие принципы органической химии описаны в "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999 и "March's Advanced Organic Chemistry" авт. Michael B. Smith и Jerry March, John Wiley & Sons, New York: 2007, все содержание которых в порядке ссылки включено в настоящую заявку.
Подразумевается, что используемые неопределенные и определенные артикли означают "по меньшей мере один" или "один или несколько", если не указано иное или это явно не противоречит контексту. Таким образом, используемые артикли служат для обозначения одного или нескольких (т.е. по меньшей мере, одного) грамматических подлежащих. В качестве примера, "компонент" представляет собой один или несколько компонентов, и, таким образом, при реализации описанных вариантов осуществления возможно, предусмотрено и может применяться или использоваться несколько компонентов.
Используемый термин "субъект" относится к животному. Как правило, животным является млекопитающее. Субъект также относится, например, к приматам (например, людям мужского или женского пола), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т.п. В некоторых вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек.
Используемый термин "пациент" относится к человеку (включая взрослых и детей) или другому животному. В некоторых вариантах осуществления "пациент" относится к человеку.
Термин "включает" является открытым и включает раскрытое содержание, но не исключают другое содержание.
Термин "стереоизомер" относится к соединениям, которые имеют идентичное химическое строение, но отличаются друг от друга расположением атомов или групп в пространстве. Стереоизомеры включают энантиомер, диастереомеры, конформер (ротамер), геометрический (цис/транс) изомер, атропоизомер и т.д.
Термин "хиральный" относится к молекулам, которые обладают свойством не накладываться на зеркальное отображение, а термин "ахиральный" относится к молекулам, способным накладываться на их зеркальное отображение.
Термин "энантиомеры" относится к двум стереоизомерам соединения, которые не являются способными накладываться зеркальными отображениями друг друга.
Термин "диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, молекулы которого не являются зеркальными отображениями друг друга. Диастереомеры обладают различными физическими свойствами, например, точками плавления, точками кипения, спектральными свойствма и биологической активностью. Смесь диастереомеров можно разделяться методами анализа с высоким разрешением, такими как электрофорез и хроматография, например, ВЭЖХ.
Используемые стереохимические определения и условные обозначения обычно соответствуют S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. и Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994.
Многие органические соединения существуют в оптически активных формах, т.е., обладают способностью вращать плоскость линейно поляризованного света. При описании оптически активного соединения префиксы D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(-в). Префиксы d и  или (+) и (-) используются для обозначения знака вращения линейно поляризованного света соединением, при этом (-) или
или (+) и (-) используются для обозначения знака вращения линейно поляризованного света соединением, при этом (-) или  означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Конкретный стереоизомер может быть обозначаться как энантиомер, а смесь таких стереоизомеров называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называют рацемической смесью или рацематом, что может иметь место при отсутствии стереоселективности или стереоспецифичности в химической реакции.
означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Конкретный стереоизомер может быть обозначаться как энантиомер, а смесь таких стереоизомеров называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называют рацемической смесью или рацематом, что может иметь место при отсутствии стереоселективности или стереоспецифичности в химической реакции.
Любой асимметричный атом (например, углерода или т.п.) описанного соединения(-й) может присутствовать в рацемической или энантиомерно обогащенной, например, (R)-, (S)- или (R, S)-конфигурации. В некоторых вариантах осуществления изобретения каждый асимметричный атом имеет энантиомерный избыток, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70%, по меньшей мере, 80%, по меньшей мере, 90%, по меньшей мере, 95%, или, по меньшей мере, 99% в (R)- или (S)-конфигурации.
В зависимости от выбора исходных веществ и процедур соединения могут присутствовать в форме одного из возможных стереоизомеров или в виде их смесей, таких как рацематы и смеси диастереоизомеров, в зависимости от числа асимметрических атомов углерода. Оптически активные (R)- и (S)-изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или разделены с использованием традиционных методик. Если соединение содержит двойную связь, заместителем может являться Е- или Z-конфигурация. Если соединение содержит двузамещенный циклоалкил, заместитель циклоалкила может иметь цис- или транс-конфигурацию.
Любые полученные смеси стереоизомеров могут разделяться на основе физико-химических различий компонентов на чистые или преимущественно чистые геометрические изомеры, энантиомеры, диастереомеры, например, путем хроматографии и/или фракционной кристаллизации.
Любые полученные рацематы конечных продуктов или промежуточных соединений могут разделяться на оптические антиподы методами, известными специалистам в данной области техники, например, путем разделения их диастереомерных солей. Рацемические продукты также могут быть разделяться с путем хиральной хроматографии, например, высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хирального адсорбента. Предпочтительные энантиомеры также могут быть получены путем асимметрического синтеза. См. например, Jacques, и др., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Principles of Asymmetric Synthesis (2nd Ed. Robert E. Gawley, Jeffrey  , Elsevier, Oxford, UK, 2012); Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007).
, Elsevier, Oxford, UK, 2012); Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007).
Соединения, описанные в настоящем описании, необязательно могут быть замещены одним или несколькими заместителями, такими, как в целом проиллюстрированы далее, или примерами конкретных классов, подклассов и видов согласно настоящему изобретению.
Термин "замещенный" обычно относится к замене одного или нескольких водородных радикалов в заданной структуре радикалом указанного заместителя. Если не указано иное, замещенная группа может иметь заместитель в каждом замещаемом положении группы. Когда несколько положений в заданной структуре может быть замещено несколькими заместителями, выбранным из указанной группы, заместитель может являться одинаковым или различаться в каждом положении.
Термин "незамещенный" относится к указанной группе, которая не содержит заместителей.
Термин "необязательно замещенный…" может использоваться взаимозаменяемо с термином "незамещенный или замещенный…", то есть, структура является незамещенной или замещенной одним или несколькими указанными заместителями. Заместители согласно настоящему изобретению, включают без ограничения D, F, Cl, Br, I, N3, CN, NO2, ОН, SH, NH2, алкил, галогеналкил, алкенил, алкинил, алкокси, алкиламино, циклоалкил, гетероциклил, арил, гетероарил и т.п.
Кроме того, следует пояснить, что фраза "каждый… независимо" и "каждый из… и… независимо", если не указано иное, следует интерпретировать в широком смысле. Конкретные варианты, обозначаемые одним и тем же символом, не зависят друг от друга в различных группах; обозначаемые одним и тем же символом, не зависят друг от друга в одних и тех же группах.
В различных местах настоящего описания заместители описанных соединений описаны в группах или в диапазонах. Специально указано, что изобретение включает каждую индивидуальную подкомбинацию членов таких групп и диапазонов. Например, термин "C1-6-алкил" специально предназначен для обозначения в отдельности метила, этила, С3-алкил, С4-алкил, С5-алкила и С6-алкила.
В различных местах настоящего описания описаны связывающие заместители. Там, где структура четко требует связывающей группы, под соединительными группами понимаются переменные Маркуша, перечисленные для этой группы. Например, если структура требует связывающей группы, и в определении группы Маркуша для этой переменной указан "алкил" или "арил", подразумевается, что "алкил" или "арил" представляет собой связывающую алкиленовую группу или ариленовую группу, соответственно.
Термин "алкил" или "алкильная группа" относится к одновалентной углеводородной группе с насыщенной линейной или разветвленной цепью, при этом алкильная группа необязательно замещена одним или несколькими описанными заместителями. Если не указано иное, алкильная группа содержит 1-20 атомов углерода. В некоторых вариантах осуществления алкильная группа содержит 1-12 атомов углерода. В других вариантах осуществления алкильная группа содержит 3-12 атомов углерода. В других вариантах осуществления алкильная группа содержит 1-6 атомов углерода. В других вариантах осуществления, в которых алкильная группа содержит 1-4 атома углерода.
Некоторые не ограничивающие примеры алкильных групп включают метил (Me, -СН3), этил (Et, -СН2СН3), 1-пропил (n-Pr, n-пропил, -СН2СН2СН3), 2-пропил (i-Pr, изопропил, -СН(СН3)2), 1-бутил (n-Bu, n-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (i-Bu, изобутил, -СН2СН(СН3)2), 2-бутил (s-Bu, s-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (t-Bu, третбутил, -С(СН3)3), 1-пентил (n-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3) СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1-бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3), 2-гексил (-СН(СН3) СН2СН2 СН2СН3), 3-гексил (-СН(СН2СН3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2 СН2СН2 СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН (СН3)СН2 СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН (СН2СН3)СН (СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С (СН3)3, 1-гептил, 1-октил, и т.п.
Термин "алкилен" относится к насыщенной двухвалентной углеводородной группе, полученной из насыщенного углеводорода с прямой или разветвленной цепью путем удаления двух атомов водорода. Если не указано иное, алкиленовая группа содержит 1-12 атомов углерода. В некоторых вариантах осуществления алкиленовая группа содержит 1-6 атомов углерода. В других вариантах осуществления алкиленовая группа содержит 1-4 атома углерода. В других вариантах осуществления алкиленовая группа содержит 1-3 атома углерода. В других вариантах осуществления алкиленовая группа содержит 1-2 атома углерода. Примером алкиленовой группы является метилен (-СН2-), этилен (-СН2СН2-), изопропилен (-СН(СН3)СН2-) и т.п.
Термин "алкенил" относится к одновалентному углеводородному радикалу с линейной или разветвленной цепью, содержащему, по меньшей мере, один участок ненасыщенной углерод-углеродную двойной связи (sp2), при этом алкенильный радикал может быть необязательно независимо замещен одним или несколькими описанными заместителями и включает радикалы, имеющие "цис" и "транс" ориентации, или, в качестве альтернативы, "Е" и "Z" ориентации. В некоторых вариантах осуществления алкенил содержит от 2 до 12 атомов углерода. В других вариантах осуществления алкенил содержит от 3 до 12 атомов углерода. В других вариантах осуществления алкенил содержит от 2 до 6 атомов углерода. В других вариантах осуществления алкенил содержит от 2 до 4 атомов углерода. Примеры алкенильных групп включают без ограничения этиленил или винил (-СН=СН2), аллил (-СН2СН=СН2) и т.п.
Термин "алкинил" относится к одновалентному углеводородному радикалу с линейной или разветвленной цепью, содержащему от 2 до 12 атомов углерода, по меньшей мере, с одним участком ненасыщенности, т.е. углерод-углеродной тройной sp-связью, при этом алкинильный радикал может быть необязательно независимо замещен одним или несколькими описанными заместителями. В некоторых вариантах осуществления алкинил содержит от 3 до 12 атомов углерода. В других вариантах осуществления алкинил содержит от 2 до 6 атомов углерода. В других вариантах осуществления алкинил содержит от 2 до 4 атомов углерода. Примеры таких групп включают без ограничения этинил (-С≡СН), пропаргил (-СН2С≡СН), 1-пропинил (-С≡С-СН3) и т.п.
Термин "алкокси" относится к алкильной группе согласно данному выше определению, присоединенной к исходному молекулярному фрагменту посредством атом кислорода. Если не указано иное, алкоксильная группа содержит 1-12 атомов углерода. В одном из вариантов осуществления алкоксильная группа содержит 1-6 атомов углерода. В другом варианте осуществления алкоксильная группа содержит 1-4 атома углерода. В еще другом варианте осуществления алкоксильная группа содержит 1-3 атома углерода. Алкоксильная группа может быть необязательно замещена одним или несколькими описанными заместителями.
Некоторые не ограничивающие примеры алкоксильной группы включают без ограничения метокси (МеО, -ОСН3), этокси (EtO, -ОСН2СН3), 1-пропокси (n-PrO, n-пропокси, -ОСН2СН2СН3), 2-пропокси (i-PrO, изопропокси, -ОСН(СН3)2), 1-бутокси (n-BuO, n-бутокси, -ОСН2СН2СН2СН3), 2-метил-1-пропокси (i-BuO, изобутокси, -OCH2CH(СН3)2), 2-бутокси (s-BuO, втор-бутокси, -ОСН(СН3)СН2СН3), 2-метил-2-пропокси (трет-BuO, трет-бутокси, -ОССН3)3), 1-пентокси (n-пентокси, -ОСН2СН2СН2СН2СН3), 2-пентокси (-ОСН(СН3)СН2СН2СН3), 3-пентокси (-ОСН(СН2СН3)2), 2-метил-2-бутокси (-ОС(СН3)2СН2СН3), 3-метил-2-бутокси (-ОСН(СН3)СН(СН3)2), 3-метил-1-бутокси (-ОСН2СН2СН(СН3)2), 2-метил-1-бутокси (-ОСН2СН(СН3)СН2СН3) и т.п.
Термин "алкиламино" охватывает "N-алкиламино" и "N,N-диалкиламино", при этом аминогруппы независимо друг от друга замещены одним алкильным радикалом или двумя алкильными радикалами, соответственно, а алкильная группа имеет значение согласно данному ей определению. В некоторых вариантах осуществления алкиламиногруппой является низшая алкиламиногруппа, имеющая одну или две алкильные группы из 1-6 атомов углерода, присоединенных к атому азота. В других вариантах осуществления алкиламиногруппой является алкиламиногруппа, имеющая одну или две низшие алкильные группы из 1-4 атомов углерода, присоединенных к атому азота. Некоторые неограничивающие примеры применимого алкиламинорадикала включают моно- или диалкиламины. Некоторые примеры включают без ограничения N-метиламино, N-этиламино, N,N-диметиламино, N,N-диэтиламино и т.п.
Термин "галоалкил", "галоалкокси" или "галогенированный алкиламино", соответственно, относится к алкильной, алкоксильной или алкиламиногруппе, в зависимости от обстоятельств, замещенной одним или несколькими атомами галогена, при этом каждая из алкильной, алкоксильной или алкиламиногруппы имеет значение согласно данному ей определению. Примеры таких групп включают без ограничения трифторметил, 2,2,3,3-тетрафторпропил, дифторметокси, трифторметокси, трифторметиламино и т.п.
Термин "циклоалкил" относится к одновалентному или поливалентному насыщенному кольцу, содержащему от 3 до 12 атомов углерода, в качестве моноциклической, бициклической или трициклической системы углеводородов. В некоторых вариантах осуществления циклоалкильная группа содержит от 7 до 12 атомов углерода. В других вариантах осуществления циклоалкильная группа содержит от 3 до 8 атомов углерода. В других вариантах осуществления циклоалкильная группа содержит от 3 до 6 атомов углерода. Циклоалкильная группа может быть необязательно замещена одним или несколькими описанными заместителями.
Термин "карбоциклил" относится к одновалентному или поливалентному, неароматическому, насыщенному или частично ненасыщенному кольцу, содержащему от 3 до 12 атомов углерода, в качестве моноциклической, бициклической или трициклической системой углеводородов. Карбобициклильная группа включает спиральную карбобициклильную группу или слитую карбобициклильную группу. Применимые карбоциклильные группы включают без ограничения циклоалкил, циклоалкенил и циклоалкинил. В некоторых вариантах осуществления карбоциклильная группа содержит от 3 до 8 атомов углерода. В других вариантах осуществления карбоциклильная группа содержит от 3 до 6 атомов углерода. Дополнительные примеры карбоциклильных групп включают циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-цикогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и т.п. Карбоциклильная группа может быть необязательно замещена одним или несколькими описанными заместителями.
Термин "карбоцикл" или "карбоциклическое кольцо" относится к неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему от 3 до 12 атомов углерода, в качестве моноциклической, бициклической или трициклической системы углеводородов. Применимый карбоцикл или карбоциклическое кольцо включает без ограничения циклопарафин, циклоолефин и циклоалкин. В некоторых вариантах осуществления карбоцикл или карбоциклическое кольцо содержит от 3 до 8 атомов углерода. В других вариантах осуществления карбоцикл или карбоциклическое кольцо содержит от 3 до 6 атомов углерода. Дополнительные примеры карбоцикла или карбоциклического кольца включают циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексен, циклогексадиен, циклогептан, циклооктан, циклононан, циклодекан, циклоундекан, циклододекан и т.п. Карбоцикл или карбоциклическое кольцо может быть необязательно замещено одним или несколькими описанными заместителями.
Термин "гетероциклил" относится к одновалентному или поливалентному, насыщенному или частично ненасыщенному моноциклическому, бициклическому или трициклическому кольцу, содержащему от 3 до 12 кольцевых атомов, по меньшей мере, один из которых выбран из азота, серы или кислорода. Если не указано иное, гетероциклильной группой может являться углерод или азот, а группой -СН2- может быть необязательно заменена группой -С(=O)-, в которой, сера может быть необязательно окислена до S-оксида, а азот может быть необязательно окислен до N-оксида. Некоторые неограничивающие примеры гетероциклической группы включают оксиранил, азетидинил, оксетанил, тиетанил, пирролидинил, 2-пирролинил, 3-пирролинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, дигидротиенили, 1,3-диоксоланил, дитиоланил, тетрагидропиранил, дигидропиранил, 2Н-пиранил, 4Н-пиранил, тетрагидротиопиранил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, диоксанил, дитианил, тиоксанил, гомопиперазинил, гомопиперидинил, диазепанил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил и 2-окс-5-азабицикло[2.2.1]гепт-5-ил. Некоторые неограничивающие примеры гетероциклила, у которого группа -СН2-заменена фрагментом -С(=O)-, включают 2-оксопирролидинил, оксо-1,3-тиазолидинил, 2-пиперидинонил, 3,5-диоксопиперидинил и пиримидиндионил. Некоторые неограничивающие примеры гетероциклила, у которого кольцевой атом серы окислен, включают сульфоланил, 1,1-диоксо-тиоморфолинил. Гетероциклильная группа может быть необязательно замещена одним или несколькими описанными заместителями.
Термин "гетероцикл" или "гетероциклическое кольцо" относится к насыщенному или частично ненасыщенному моноциклическому, бициклическому или трициклическому кольцу, содержащее от 3 до 12 кольцевых атомов, по меньшей мере, один из которых выбран из азота, серы или кислорода. Если не указано иное, группа -СН2- гетероцикла или гетероциклического кольца может быть необязательно заменена фрагментом -С(=O)-, в которой, сера может быть необязательно окислена до S-оксида, а азот может быть необязательно окислен до N-оксида. Некоторые неограничивающие примеры гетероцикла или гетероциклического кольцо включают оксиран, азетидин, оксетан, тиациклобутан, пирролидин, пирролин, пиразолин, пиразолидин, имидазолин, имидазолидин, тетрагидрофуран, дигидрофуран, тиофан, дигидротиофен, 1,3-диоксолан, дитиолан, тетрагидропиран, дигидропиран, 2Н-пиран, 4Н-пиран, тетрагидротиапиран, пиперидин, морфолин, тиоморфолин, пиперазин, диоксан, дитиан, тиоксан, гомопиперазин, гомопиперидин, диазепан, оксепан, тиациклогептан, оксазепин, диазепин, тиазепин и 2-окс-5-азабицикло[2.2.1]гептан. Некоторые неограничивающие примеры гетероцикла или гетероциклического кольца, у которого группа -СН2- заменена фрагментом -С(=O)-, включают пирролидон, тиазолидон, пиперидон, 3,5-диоксопиперидин и пиримидиндион. Некоторые неограничивающие примеры гетероцикла или гетероциклического кольца, у которого кольцевой атом серы окислен, включают сульфолан, 1,1-диоксо-тиоморфолин. Гетероцикл или гетероциклическое кольцо может быть необязательно замещено одним или несколькими описанными заместителями.
Термин "r-членный", где r представляет собой целое число, обычно описывает число образующих кольца атомов во фрагменте, при этом r является число образующих кольца атомов. Например, пиперидинил является одним из примеров 6-членного гетероциклоалкила, а декалинил является одним из примеров 10-членной циклоалкильной группы.
Термин "ненасыщенный" относится к фрагменту, имеющему один или несколько звеньев ненасыщенности.
Термин "гетероатом" относится к одному или нескольким из следующего: кислороду, сере, азоту, фосфору и кремнию, включая любую окисленную форму азота, серы или фосфора; кватернизированной форме любого основного азота; или замещаемому азоту гетероциклического кольца, например, N (как в 3,4-дигидро-2H-пирролиле), NH (как в пирролидиниле) или NR (как в N-замещенном пирролидиниле).
Термин "галоген" относится к фтору (F), хлору (Cl), брому (Br) или йоду (I).
Термин "циано" или "CN" относится к цианоструктуре. Такая группа может быть связана с другими группами.
Термин "нитро" или "NO2" относится к нитроструктуре. Такая группа может быть связана с другими группами.
Термин "арил" относится к одновалентным или поливалентным моноциклическим, бициклическим и трициклическим карбоциклическим кольцевым системам, имеющим в общей сложности от шести до четырнадцати кольцевых членов, или от шести до двенадцати кольцевых членов или от шести до десяти кольцевых членов, при этом, по меньшей мере, одно кольцо в системе является ароматическим и имеет одну точку или множество точек присоединения к остальной молекуле. В одном из вариантов осуществления арильной группой является одновалентная или поливалентная карбоциклическая кольцевая система, имеющая от шести до десяти кольцевых членов, при этом, по меньшей мере, одно кольцо в системе является ароматическим. Примеры арильного кольца могут включать фенил, нафтил и антрил. Арильная группа может быть необязательно и независимо замещена одним или несколькими описанными заместителями.
Термин "ароматическое кольцо" относится к моноциклическим, бициклическим и трициклическим карбоциклическим кольцевым системам, имеющим в общей сложности от шести до четырнадцати кольцевых членов или от шести до двенадцати кольцевых членов или шести до десяти кольцевых членов, при этом, по меньшей мере, одно кольцо в системе является ароматическим. В одном из вариантов осуществления арильной группой является карбоциклическая кольцевая система, имеющая от шести до десяти кольцевых элементов, при этом, по меньшей мере, одно кольцо в системе является ароматическим. Примеры ароматического кольца могут включать фен, нафталин и антрацен. Ароматическое кольцо может быть необязательно и независимо замещено одним или несколькими описанными заместителями.
Термин "гетероарил" относится к одновалентным или многовалентным моноциклическим, бициклическим и трициклическим карбоциклическим кольцевым системам, имеющим в общей сложности пяти до двенадцати кольцевых членов или от пяти до десяти кольцевых членов или от пяти до шести кольцевых членов, при этом, по меньшей мере, одно кольцо в системе является ароматическим, и, по меньшей мере, один кольцевой атом выбран из гетероатома и имеет одну точку или множество точек присоединения к остальной молекуле. Гетероарильная группа необязательно замещена одним или несколькими описанными заместителями. В одном из вариантов осуществления гетероарильной группой является 5- до 12-членный гетероарил, содержащий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N. В другом варианте осуществления гетероарильной группой является 5- или 6-членный гетероарил, содержащий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N.
Некоторые неограничивающие примеры гетероарила включают 2-фуранил, 3-фуранил, N-имидазолил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, оксадиазолил (например, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил), оксатриазолил (например, 1,2,3,4-оксатриазолил), 2-тиазолил, 4-тиазолил, 5-тиазолил, изотиазолил, 2-тиадиазолил (например, 1,3,4-тиадиазолил, 1,2,3-тиадиазолил, 1,2,5-тиадиазолил), тиатриазолил (например, 1,2,3,4-тиазолтриазолил), тетразолил (например, 2Н-1,2,3,4-тетразолил, 1Н-1,2,3,4-тетразолил), триазолил (например, 2Н-1,2,3-триазолил, 1Н-1,2,4-триазолил, 4Н-1,2,4-триазолил), 2-тиенил, 3-тиенил, 1Н-пиразолил (например, 1Н-пиразол-3-ил, 1Н-пиразол-4-ил, 1Н-пиразол-5-ил), N-пирролил, 2-пирролил, 3-пирролил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пиридазинил (например, 3-пиридазинил, 4-пиридазинил), 2-пиразинил, триазинил (например, 1,3,5-триазин), тетразинил (например, 1,2,4,5-тетразинил, 1,2,3,5-тетразинил); а также следующие бициклы: бензимидазолил, бензофурил, бензотиофенил, индолил (например, 2-индолил), пуринил, хинолил (например, 2-хинолил, 3-хинолил, 4-хинолил), изохинолил (например, 1-изохинолил, 3-изохинолил или 4-изохинолил), имидазо [1,2-а] пиридил, пиразол [1,5-а] пиридил, пиразол [1,5-а] пиримидил, имидазо[1,2-b] пиридазинил, [1,2,4]триазоло[4,3-b]пиридазинил, [1,2,4]триазоло[1,5-а]пиримидинил или [1,2,4]триазоло[1,5-a]пиридил и т.п.
Термин "гетероароматическое кольцо" или "гетероароматическое соединение" относится к моноциклическим, бициклическим и трициклическим карбоциклическим кольцевым системам, имеющим в общей сложности от пяти до двенадцати членов кольца или от пяти до десяти членов кольца или от пяти до шести членов кольца, при этом, по меньшей мере, одно кольцо в системе является ароматическим, и, по меньшей мере, один кольцевой атом выбран из гетероатома. Гетероароматическое кольцо или гетероароматическое соединение необязательно замещено одним или несколькими описанными заместителями. В одном из вариантов осуществления гетероароматическим кольцом или гетероароматическим соединение является от 5- до 12-членный гетероарил, содержащий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N. В другом варианте осуществления гетероароматическим кольцом или гетероароматическим соединение является от 5- до 6-членный гетероарил, содержащий 1, 2, 3 или 4 гетероатома, независимо выбранных из О, S и N.
Некоторые неограничивающие примеры гетероароматического кольца или гетероароматических соединений включают фуран, имидазол, изоксазол, оксазол, оксадиазол, оксатризол, тиазол, изотиазол, тиадиазол, тиатриазол, тетразол, триазол, тиофен, 1Н-пиразол, пиррол, пиридин, пиримидин, пиридазин, пиразин, триазин, тетразин, а также следующие бициклы: бензимидазол, бензофуран, бензотиофен, индол, пурин, хинолин, изохинолин, имидазо[1,2-a]пиридин, пиразол [1,5-a]пиридин, пиразол[1,5-а]пиримидин, имидазо[1,2-b]пиридазин, [1,2,4]триазоло[4,3-6]пиридазин, [1,2,4]триазоло[1,5-а]пиримидин или [1,2,4]триазоло[1,5-а]пиридин и т.п.
Термины "карбокси" или "карбоксил" отдельно или с другими терминами, такими как "карбокси", представляет собой -СО2Н. Термин "карбонил" отдельно или с другими терминами, такими как "аминокарбонил" или "ацилокси", представляет собой -(С=O)-.
Как описано в изобретении, связь заместителя с центром одного кольца в кольцевой системе (как показано в Формуле b) отображает замещение заместителя в любом замещаемом или обоснованном положении в кольце. Например, Формула b отображает однократные или многократные замещения заместителя R в любом замещаемом или обоснованном положении в кольце С, как показано в Формулах с1-с19.
Как описано в изобретении, связь с центром одного кольца в кольцевой системе (как показано в Формуле d) отображает возможность связи в любом обоснованном и соединяемом положении кольца с остальной молекулой. Формула d отображает возможность связи в любом обоснованном и соединяемом положении кольца D с остальной молекулой.
Как описано в изобретении, связь заместителя R с центром одного кольца в пределах кольцевой системы отображает замещение заместителя R в любом замещаемом положении в кольце. Например, Формула е отображает кольцо В, которое может быть замещено в любом замещаемом положении заместителем R, как показано в Формулах f, g, h и i.
Термин "защитная группа" или "PG" относится к заместителю, который обычно используется для блокирования или защиты конкретной функциональной группы во время реакции с другими функциональными группами соединения. Например, "аминозащитная группа" представляет собой заместитель, присоединенный к аминогруппе, который блокирует или защищает функциональную аминогруппу в соединении. Применимые аминозащитные группы включают ацетил, трифторацетил, трет-бутокси-карбонил (ВОС, Boc), бензилоксикарбонил (CBZ, Cbz) и 9-флуоренилметиленокси-карбонил (Fmoc). Аналогичным образом, "гидроксизащитная группа" относится к заместителю гидроксигруппы, который блокирует или защищает гидроксильную функциональную группу. Применимые защитные группы включают ацетил и силил. "Карбоксизащитная группа" относится к заместителю карбоксильной группы, который блокирует или защищает функциональную карбоксильную группу. Обычные карбоксизащитные группы включают -CH2CH2SO2Ph, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этокси-метил, 2-(р-толуолсульфонил)-этил, 2-(р-нитрофенилсульфенил)-этил, 2-(дифенилфосфино)этил, нитроэтил и т.п. Общее описание защитных групп и их использования приведено у T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991; и P.J. Kocienski, Protecting Groups, Thieme, Stuttgart, 2005.
Термин "пролекарство" относится к соединению, которое преобразуется in vivo в соединение Формулы (I). Такое преобразование может осуществляться, например, путем гидролиза пролекарственной формы в крови или ферментативного превращения в исходную форму в крови или ткани. Пролекарствами описанных соединений могут являться, например, сложные эфиры. Некоторыми обычными сложными эфирами, которые используются в качестве пролекарств, являются фенильные сложные эфиры, алифатические (С1-С24) сложные эфиры, ацилоксиметиловые сложные эфиры, карбонаты, карбаматы и аминокислотные сложные эфиры. Например, описанное соединение, которое содержит гидроксильную группу, может быть ацилировано в этом положении в форму его пролекарства. Другие пролекарственные формы включают фосфаты, такие как фосфатные соединения, полученные путем фосфонирования гидроксильной группы в исходном соединении. Подробное описание пролекарств приведено у Т. Higuchi и V. Stella, Pro-drugs as Novel Delivery Systems, том 14, A.C.S. Symposium Series, Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, J. Rautio и др. Prodrugs: Design and Clinical Applications, Nature Review Drug Discovery, 2008, 7, 255-270, и S.J. Hecker и др., Prodrugs of Phosphates and Phosphonates, Journal of Medicinal Chemistry, 2008, 51, 2328-2345.
"Метаболит" представляет собой продукт, образующийся в результате метаболизма указанного соединения или его соли в организме. Метаболиты соединения могут идентифицироваться обычными способами, известными в данной области техники, а их активность может определяться путем испытаний, таких как описаны в настоящем изобретении. Такие продукты могут быть получены, например, в результате окисления, восстановления, гидролиза, амидирования, дезаминирования, этерификации, деэтерификации, ферментативного расщепления и т.п. введенного соединения. Соответственно, настоящее изобретение включает метаболиты описанных соединений, в том числе метаболиты, образующиеся в результате контакте описанного соединения с млекопитающим в течение достаточного периода времени.
Термин "фармацевтические приемлемые соли" относится к органическим или неорганическим солям описанного соединения. Фармацевтически приемлемые соли хорошо известны из техники. Например, фармацевтически приемлемые соли подробно описаны в статье S.М. Berge и др. в J. Pharmaceutical Sciences, 1977, 66: 1-19, которая в порядке ссылки включена в настоящую заявку. Некоторые неограничивающие примеры фармацевтически приемлемых и нетоксичных солей включают соли аминогруппы с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота и малоновая кислота, или другими способами, используемыми в данной области техники, такими как ионный обмен. Другие фармацевтически приемлемые соли включают адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, стеарат, тиоцианат, р-толуолсульфонат, ундеканоат, валерат и т.п. Соли, полученные из соответствующих оснований, включают соли щелочных металлов, щелочноземельных металлов, аммония и N+(С1-4-алкила)4. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп описанных соединений. Путем такой кватернизации могут быть получены растворимые в воде или масле или диспергируемые продукты. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Дополнительные фармацевтически приемлемые соли включают, когда это уместно, нетоксичный аммоний, четвертичный аммоний и катионы аминов, сформированные с использованием противоионов, таких как галид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, C1-8 сульфонат или арилсульфонат.
Термин "сольват" относится к ассоциации или комплексу одной или нескольких молекул растворителя и описанного соединения. Примеры растворителей, которые образуют сольваты, включают без ограничения воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, где молекулой растворителя является молекула воды.
Термин "гидрат" может использоваться, когда растворителем является вода. В одном из вариантов осуществления одна молекула растворителя ассоциирована с одной молекулой описанных соединений с образованием гидрата. В другом варианте осуществления несколько молекул растворителя может быть ассоциировано с одной молекулой описанных соединений с образованием дигидрата. В еще одном из вариантов, менее одной молекулы растворителя может быть ассоциировано с одной молекулой описанных соединений с образованием полугидрата. Кроме того, все сольваты согласно изобретению сохраняют биологическую эффективность негидратной формы описанных соединений.
Используемые термины "лечить" или "лечение" любого заболевания или нарушения относятся ко всем процессам, в которых возможно замедление, прерывание, купирование, регулирование или прекращение прогрессирования заболевания или нарушения, но не обязательно с полным устранением всех симптомов нарушения, а также профилактическую терапию упомянутых состояний, в частности, у пациента, который предрасположен к такому заболеванию или нарушению. В некоторых вариантах осуществления термины "лечить" или "лечение" относятся к облегчению заболевания или нарушения (т.е. замедлению или прекращению или ослаблению развития заболевания или, по меньшей мере, одного из его клинических симптомов). В других вариантах осуществления термины "лечить" или "лечение" относятся к облегчению или улучшению, по меньшей мере, одного физического параметра, включая параметры, неразличимые пациентом. В других вариантах осуществления термины "лечить" или "лечение" относятся к модулированию заболевания или нарушения физически (например, путем стабилизация различимого симптома), физиологически (например, путем стабилизации физического параметра) или обоими способами. В другом варианте осуществления термины "лечить" или "лечение" относятся к предотвращению или задержке начала развития или прогрессирования заболевания или нарушения.
Используемый термин "терапевтически эффективное количество" или "терапевтически эффективная доза" относится к количеству соединения согласно изобретению, которое способно вызывать биологическую или медицинскую реакцию (такую как снижение или ингибирование активности фермента или белка или ослабление симптомов, облегчение симптомов, замедление или задержка развития заболевания или предотвращение заболевания и т.д.) у индивидуума. В одном неограничивающем варианте осуществления изобретения субъекту вводят "терапевтически эффективное количество", когда соединение согласно настоящему изобретению: (1) по меньшей мере, частично облегчает, ингибирует, предотвращает и/или облегчает заболевание или нарушение, (i) опосредованное ксантиноксидазой или переносчиком 1 уратов (URAT1), или (ii) связанное с активностью ксантиноксидазы или переносчика 1 уратов, или (iii) характеризующееся аномальной активностью ксантиноксидазы или переносчика 1 уратов; или (2) снижает или ингибирует активность ксантиноксидазы или переносчика 1 уратов; или (3) уменьшает или ингибирует экспрессию ксантиноксидазы или переносчика 1 уратов. В другом варианте осуществления изобретения термин "терапевтически эффективное количество" относится к эффективному количеству соединений согласно настоящему изобретению при их введении в клетку или орган или неклеточный биологический материал или среду, которое может, по меньшей мере, частично снижать или ингибировать активность ксантиноксидазы или переносчика 1 уратов, или, по меньшей мере, частично уменьшать или ингибировать экспрессию ксантиноксидазы и переносчика 1 уратов.
Используемый термин "введение" соединения следует понимать как введение нуждающемуся в этом индивидууму соединения согласно изобретению или пролекарства соединения согласно изобретению. Очевидно, что специалист в данной области может осуществлять лечение пациента с высоким содержанием мочевой кислоты или профилактическое лечение пациента, страдающего нарушениями, эффективным количеством соединения согласно настоящему изобретению.
Используемый термин "композиция" предназначен для обозначения продукта, содержащего указанные ингредиенты в указанных количествах, а также любого продукта, который прямо или косвенно, получен в результате комбинирования определенных ингредиентов в определенных количествах. Применительно к фармацевтической композиции такой термин предназначен для обозначения продукта, содержащего активный(-е) ингредиент(-ы) и инертный(-е) ингредиент(-ы), которые образуют носитель, а также любого продукта, который прямо или косвенно, получен в результате комбинирования, комплексообразования или агрегации любых двух или более ингредиентов или в результате диссоциации одного или нескольких ингредиентов или в результате реакций или взаимодействий других типов одного или нескольких ингредиентов. Соответственно, фармацевтические композиции согласно настоящему изобретению включают любую композицию, полученную путем смешивания соединения согласно настоящему изобретению и фармацевтически приемлемого носителя.
ОПИСАНИЕ СОЕДИНЕНИЙ СОГЛАСНО ИЗОБРЕТЕНИЮ
В настоящем изобретении предложены производные карбоксизамещенных (гетеро) ароматических колец, фармацевтически приемлемые соли, фармацевтические составы и композиции, которые могут использоваться качестве ингибиторов ксантиноксидазы и переносчика 1 уратов, и их потенциальное применение при лечении симптомов или заболеваний, связанных с высоким содержанием мочевой кислоты в крови человека, например, гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией и мочекаменной болезни.
Согласно одной из особенностей изобретения, предложено соединение Формулы (I) или его стереоизомер, N-оксид, сольват, метаболит, фармацевтически приемлемая соль или пролекарство,
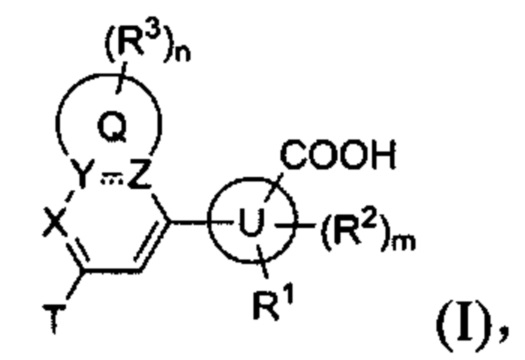
где Q, U, Т, X, Y, Z, R1, каждый R2, каждый R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления U представляет собой фенил или от 5- до 6-членный гетероарил.
В некоторых вариантах осуществления изобретения каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, C1-6-алкил, С2-6-алкенил, С2-6-алкинил, C1-6-галогеналкил, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членный циклоалкил или от 3- до 8-членный гетероциклил, при этом каждый из C1-6-алкила, С2-6-алкенила, С2-6-алкинила, C1-6-галогеналкила, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членного циклоалкила и от 3- до 8-членного гетероциклила независимо друг от друга и необязательно замещены 1, 2, 3, 4 или 5 заместителями, выбранными из ОН, оксо (=O), NH2, NO2 или CN.
В некоторых вариантах осуществления, Т представляет собой Н, D, F, Cl, Br, NO2, CN или CF3.
В некоторых вариантах осуществления, X представляет собой CR4 или N; и
при этом R4 имеет значение согласно данному ему определению.
В некоторых вариантах осуществления R4 представляет собой Н, D, галоген, C1-6-алкил, C1-6-галогеналкил, C1-6-алкокси, C1-6-алкиламино или С1-6-галогеналкокси.
В некоторых вариантах осуществления каждый из Y и Z независимо представляет собой С, СН или N.
В некоторых вариантах осуществления  представляет собой одинарную связь или двойную связь.
представляет собой одинарную связь или двойную связь.
В некоторых вариантах осуществления Q представляет собой фен, С4-7 карбоцикл, от 4- до 7-членный гетероцикл или 5-или 6-членное гетероароматическое кольцо.
В некоторых вариантах осуществления изобретения каждый R3 независимо представляет собой Н, D, галоген, оксо (=O), ОН, NH2, NO2, CN, C1-6-алкил, С2-6-алкенил, С2-6-алкинил, C1-6-галогеналкил, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членный циклоалкил, от 3- до 8-членный гетероциклил, от 5- до 10-членный гетероарил, фенил, нафтил или G, при этом каждый из C1-6-алкила, С2-6-алкенила, С2-6-алкинила, C1-6-галогеналкила, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членного циклоалкила, от 3- до 8-членного гетероциклила, от 5- до 10-членного гетероарила независимо и необязательно замещен 1, 2, 3, 4 или 5 заместителями, выбранными из ОН, оксо (=O), NH2, NO2, CN или G; и
где G имеет значение согласно данному ему определению.
В некоторых вариантах осуществления G замещен C1-6 алифатическим углеводородом, при этом каждая из метиленовых групп алифатического углеводорода С1-6 необязательно и независимо замещена J; и
где J имеет значение согласно данному ему определению.
В некоторых вариантах осуществления, J представляет собой -NH-, -S-, -О-, -С(=O)-, -C(=O)NH-, -SO-, -SO2-, -NHC(=O)-, -С(=O)O-, -SO2NH- или -NHC(=O)NH-.
В некоторых вариантах осуществления, m равно 0, 1, 2 или 3.
В некоторых вариантах осуществления, n равно 0, 1, 2, 3 или 4.
В некоторых вариантах осуществления предложено соединение при условии, что:
когда Т представляет собой F, Cl, Br или CF3, R1 представляет собой ОН.
В некоторых вариантах осуществления предложено соединение при условии, что:
когда Т представляет собой Н, 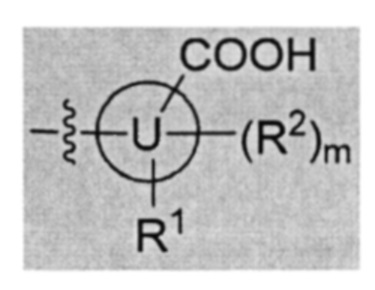 представляет собой
представляет собой 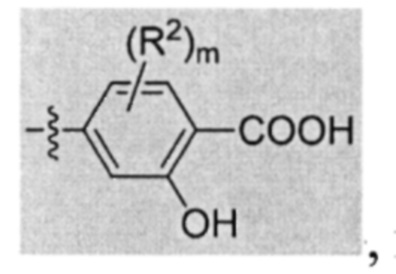 и Q не представляет собой фен, иными словами, Q представляет собой С4-7 карбоцикл, от 4- до 7-членный гетероцикл или 5- или 6-членное гетероароматическое кольцо.
и Q не представляет собой фен, иными словами, Q представляет собой С4-7 карбоцикл, от 4- до 7-членный гетероцикл или 5- или 6-членное гетероароматическое кольцо.
В некоторых вариантах осуществления предложено соединение при условии, что:
когда Т представляет собой NO2, R1 не означает Н, иными словами, R1 представляет собой D, галоген, ОН, NH2, NO2, CN, C1-6-алкил, С2-6-алкенил, С2-6-алкинил, C1-6-галогеналкил, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членный циклоалкил или от 3- до 8-членный гетероциклил, при этом каждый из C1-6-алкила, С2-6-алкенила, С2-6-алкинила, C1-6-галогеналкила, C1-6-алкокси, C1-6-галогеналкокси, C1-6-алкиламино, C1-6-галоалкиламино, от 3- до 8-членного циклоалкила и от 3- до 8-членного гетероциклил а независимо и необязательно замещен 1, 2, 3, 4 или 5 заместителями, выбранными из ОН, оксо (=O), NH2, NO2 или CN.
В некоторых вариантах осуществления предложенным соединением Формулы (I) является соединение Формулы (II) или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль и пролекарство,
где Q, U, X, Y, Z, R1, каждый из R2, каждый из R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления U представляет собой фенил, пиридинил, пиримидинил, пиразинил, пиридазинил, 1,3,5-триазинил, пирролил, фуранил, тиазолил, тиенил, оксазолил или изоксазолил.
В других вариантах осуществления U представляет собой фенил, 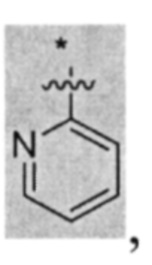
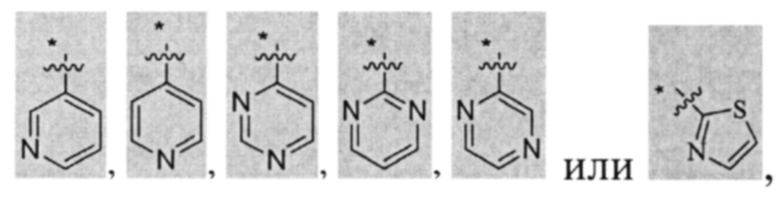 при этом "*" представляет собой положение U-кольца, присоединенного к
при этом "*" представляет собой положение U-кольца, присоединенного к 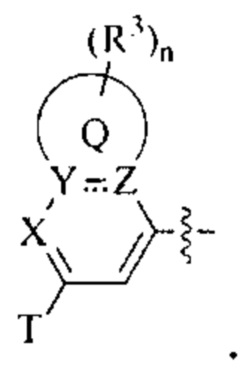
В некоторых вариантах осуществления предложенным соединением Формулы (I) является соединение Формулы (III) или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль и пролекарство,
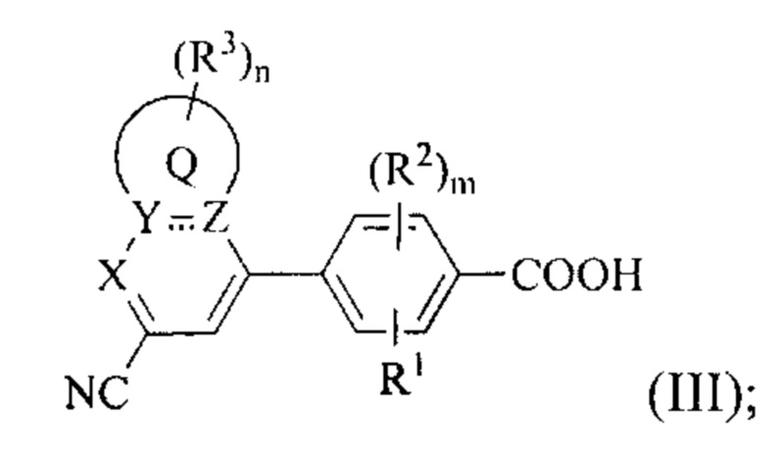
где Q, X, Y, Z, R1, каждый из R2, каждый из R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления предложенным соединением Формулы (I) является соединение Формулы (IV) или его стереоизомер, геометрический изомер, таутомер, N-оксид, гидрат, сольват, метаболит, сложный эфир, фармацевтически приемлемая соль и пролекарство,
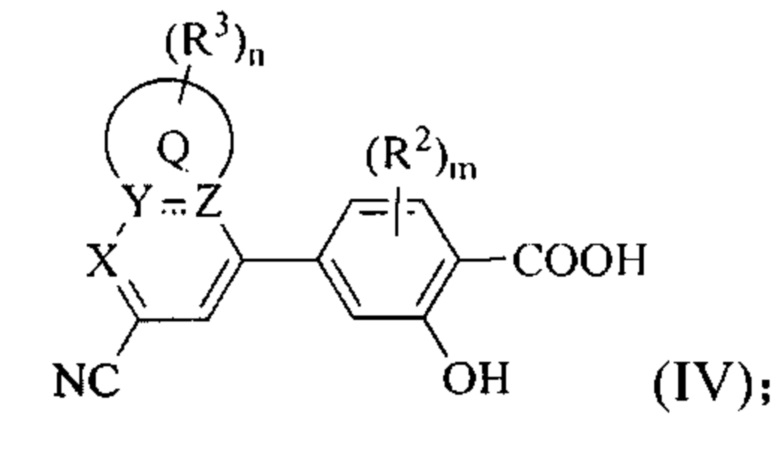
где Q, X, Y, Z, каждый из R2, каждый из R3, m и n имеют значения согласно данным им определениям.
В некоторых вариантах осуществления изобретения каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, С1-4-алкил, С2-4-алкенил, С2-4-алкинил, C1-4-галогеналкил, С1-4-алкокси, С1-4-галогеналкокси, С1-4-алкиламино, C1-4-галоалкиламино, от 3- до 6-членный циклоалкил или от 3- до 6-членный гетероциклил, при этом каждый из С1-4-алкила, С2-4-алкенила, С2-4-алкинила, C1-4-галогеналкила, С1-4-алкокси, С1-4-галогеналкокси, C1-4-алкиламино, С1-4-галоалкиламино, от 3- до 6-членного циклоалкила или от 3- до 6-членного гетероциклила независимо и необязательно замещен 1, 2 или 3 заместителями, выбранными из ОН, оксо (=O), NH2, NO2 или CN.
В других вариантах осуществления изобретения каждый R1 и R2 независимо представляет собой Н, D, галоген, ОН, NH2, NO2, CN, метил, этил, изопропил, бутил, гидроксиметил, гидроксиэтил, аминометил, дифторметил, трифторметил, метокси, этокси, изо-пропокси, трет-бутокси, n-бутокси, метиламино, этиламино, дифторметокси, трифторметокси, ацетил, ацетокси, ацетиламино, циклопропил, циклобутил, циклопентил, циклогексил, оксиранил, пирролидинил или тетрагидрофуранил.
В некоторых вариантах осуществления изобретения каждый R3 независимо представляет собой Н, D, галоген, оксо (=O), ОН, NH2, NO2, CN, метил, этил, изопропил, дифторметил, трифторметил, метокси, этокси, изо-пропокси, дифторметокси, трифторметокси, формил, карбокси, формамидо, ацетил, карбамоил, пропилсульфонамидо, циклопропил, циклобутил, имидазолил, пиразолил, тиазолил, оксазолил, пиридил, пиримидинил, хинолил, индолил, фенил или нафтил.
В некоторых вариантах осуществления изобретения каждый R4 представляет собой Н, D, галоген, метил, этил, изопропил, трет-бутил, n-бутил, дифторметил, трифторметил, метокси, этокси, трет-бутокси, метиламино, дифторметокси или трифторметокси.
В некоторых вариантах осуществления  представляет собой
представляет собой 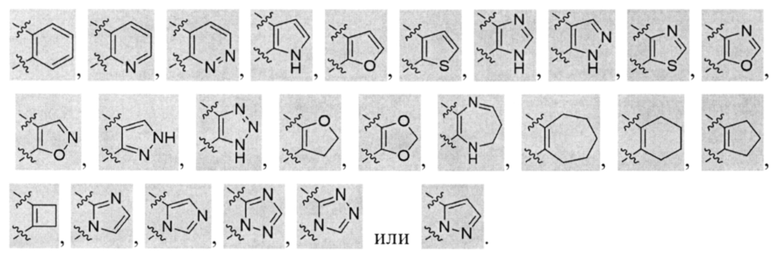
В некоторых вариантах осуществления 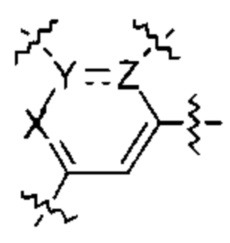 представляет собой
представляет собой 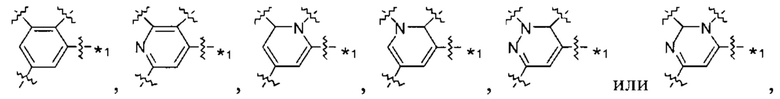
при этом *1
представляет собой положение, прикрепленное к U-кольцу.
В других вариантах осуществления изобретения, предложено соединение, имеющее одну из следующих формул, или его стереоизомер, N-оксид, сольват, метаболит, фармацевтически приемлемая соль или пролекарство, включая без ограничения: 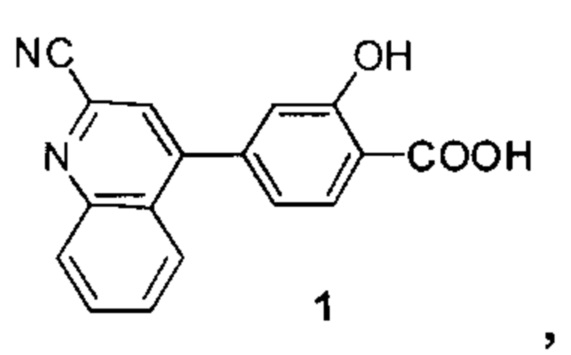
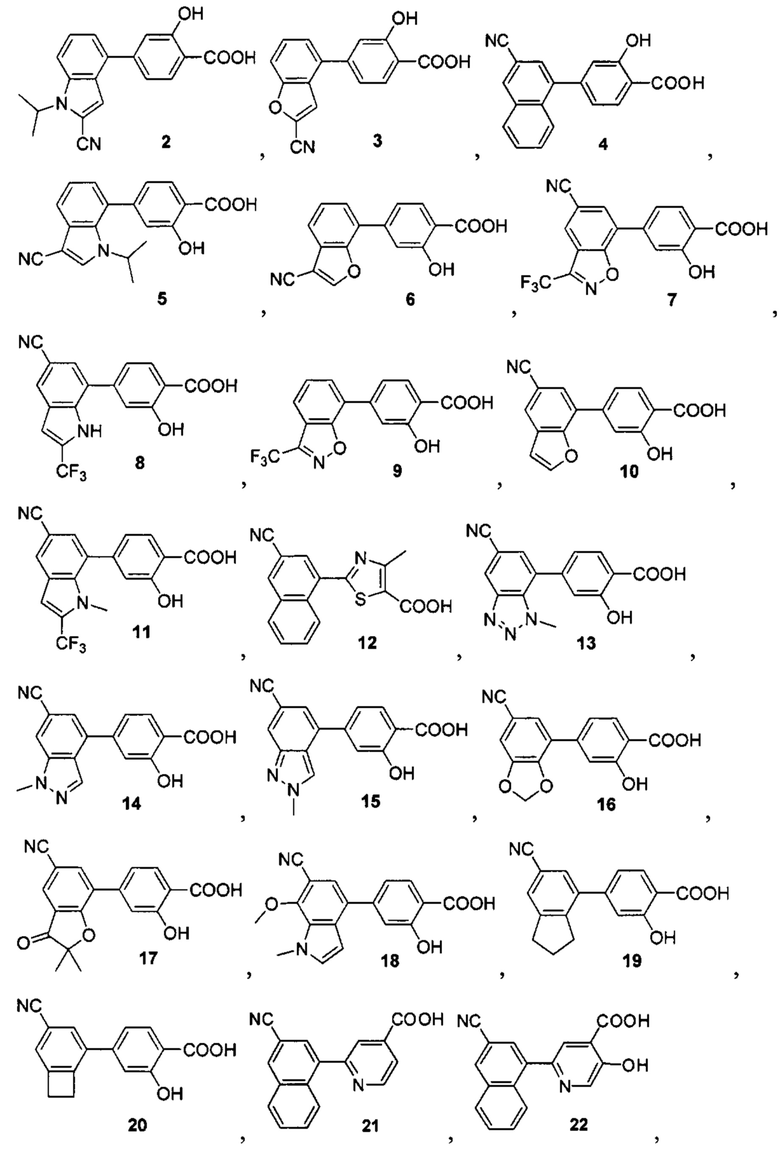
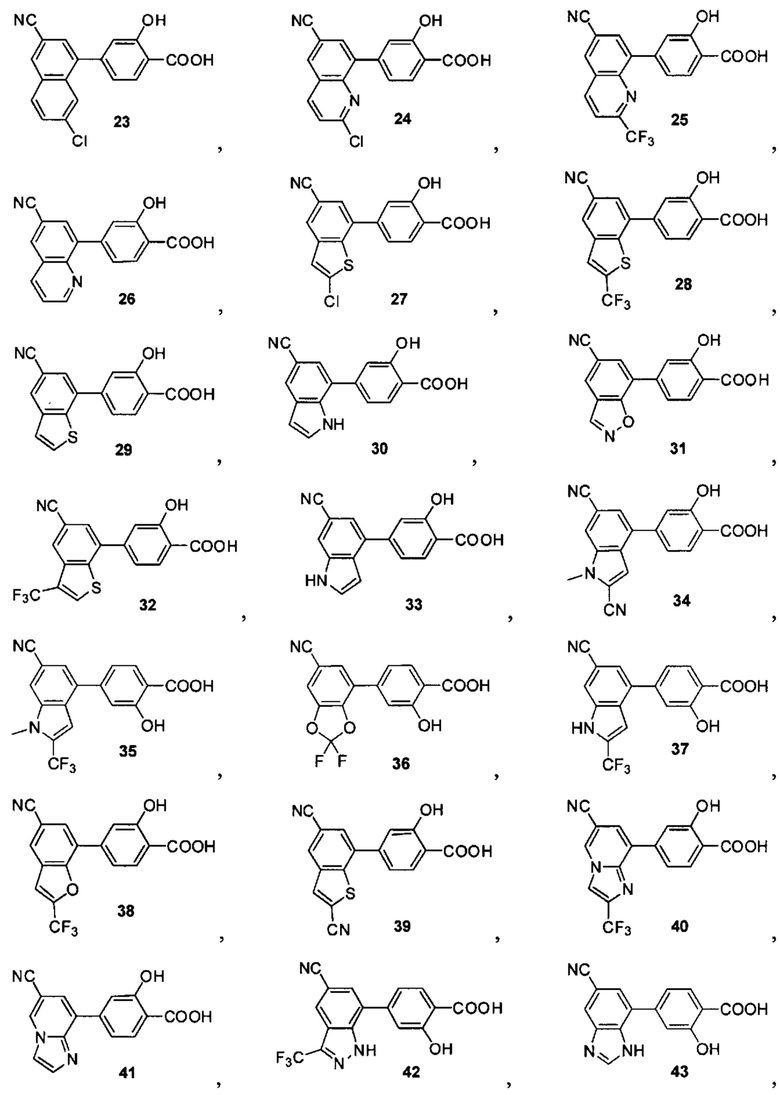
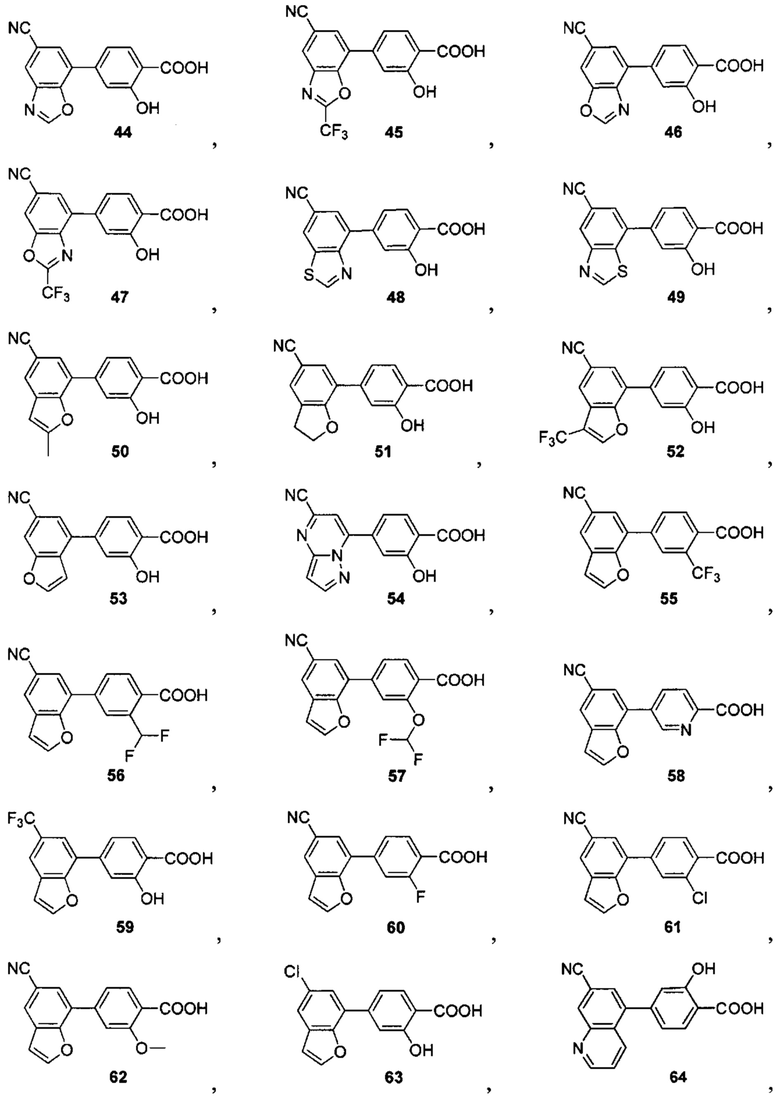
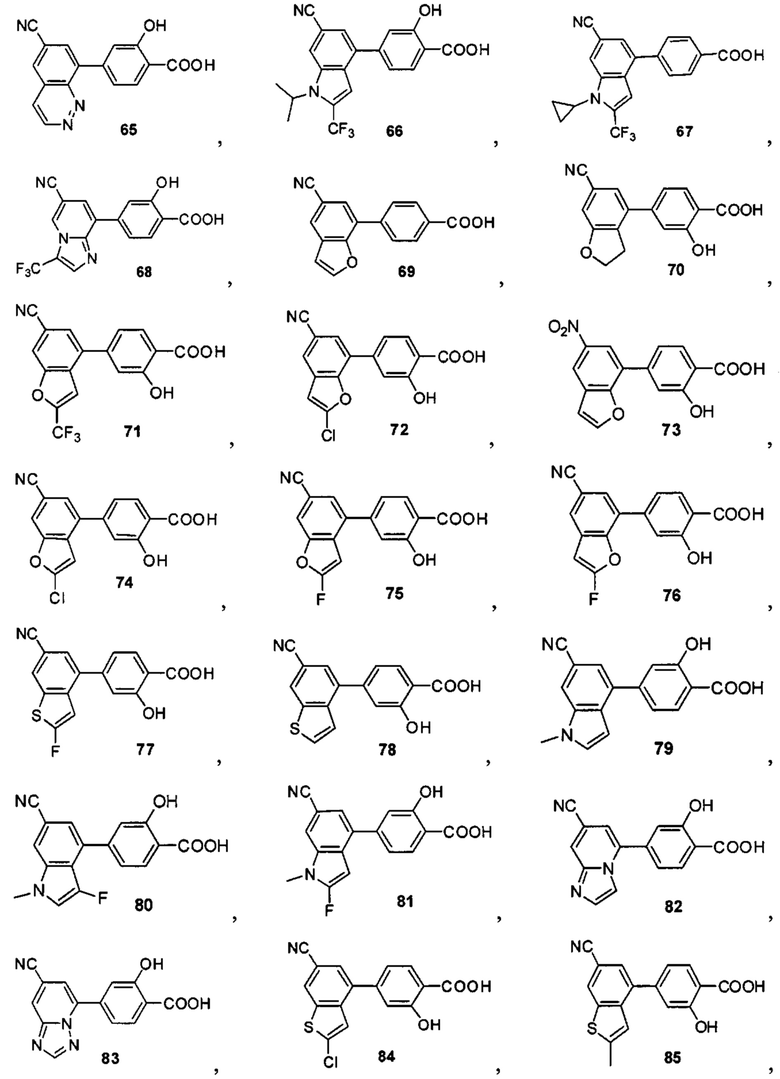
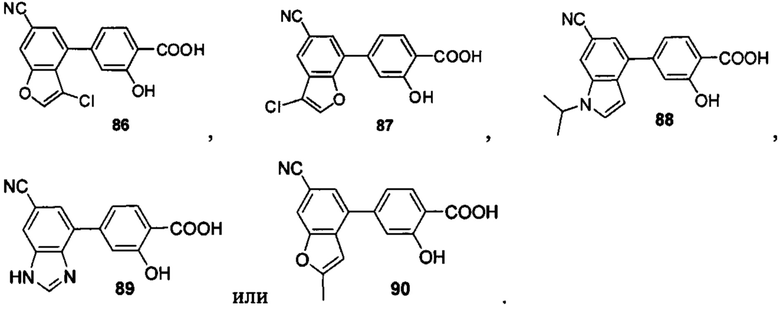
Если не указано иное, в объем настоящего изобретения входят все стереоизомеры, сольваты, метаболиты, фармацевтически приемлемые соли или пролекарства соединения Формул (I), (II), (III) или (IV).
Описанные соединения могут содержать асимметричный или хиральный центр и, следовательно, могут существовать в качестве различных стереоизомеров. Предполагается, что в объем настоящего изобретения входят все стереоизомерные формы описанных соединений Формул (I), (II), (III) или (IV), включая без ограничения диастереомеры, энантиомеры, атропизомеры и геометрические (или конформационные) изомеры, а также их смеси, такие как рацемические смеси.
Когда стереохимия любого конкретного хирального атома не указана, настоящим изобретением предусмотрены все стереоизомеры описанной структуры, которые, как и описанные соединения, входят в объем в объем настоящего изобретения. Когда для обозначения специфической конфигурации стереохимии используется сплошная или пунктирная линия, обозначенной, структура стереоизомеров является ясной и определенной.
Соединение Формул (I), (II), (III) или (IV) могут существовать в различных таутомерных формах, и все эти таутомеры входят в объем настоящего изобретения.
Соединение Формул (I), (II), (III) или (IV) могут существовать в форме соли. В одном из вариантов осуществления соль является фармацевтически приемлемой солью. Термин "фармацевтический приемлемый" представляет собой, что соединение или композиция должна быть химически и/или токсикологически совместимой с другими ингредиентами, входящими в композицию и/или используемыми для лечения млекопитающего. В другом варианте осуществления соль необязательно является фармацевтически приемлемой солью и может представлять собой соединение для получения и/или очистки Формул (I), (II), (III) или (IV) и/или для отделения энантиомеров Формул (I), (II), (III) или (IV).
В результате взаимодействия описанного соединения с неорганическими кислотами или органическими кислотами могут образовываться фармацевтически приемлемые соли присоединения кислот, например, ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллинат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидроиодид/иодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат, субсалицилат, тартрат, тозилат и трифторацетат.
Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородная кислота, серную кислоту, азотную кислоту, фосфорную кислоту и т.п.
Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т.п.
В результате взаимодействия с неорганическими или органическими основаниями могут образовываться фармацевтически приемлемые соли присоединения оснований.
Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлы из столбцов I-XII периодической таблицы. В некоторых вариантах осуществления соли получают из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; особо применимые соли включают соли аммония, калия, натрия, кальция и магния.
Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т.п. Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин.
Фармацевтически приемлемые соли согласно настоящему изобретению могут быть синтезированы из основного или кислотного фрагмента обычными химическими методами. Обычно такие соли могут быть получены введения этих соединений в форме свободных кислот в реакцию со стехиометрическим количеством соответствующего основания (такого как Na, Са, Mg или K гидроксид, карбонат, бикарбонат или т.п.), или путем введения этих соединений в форме свободных оснований в реакцию со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде или в органическом растворителе или в смеси того и другого. Обычно желательно использование неводных сред, таких как эфир, этилацетат, этанол, изопропанол или ацетонитрил, если это возможно практически. еречни дополнительных применимых солей можно найти, например, в "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Кроме того, описанные соединения, включая их соли, также могут применяться в форме их гидратов или могут включать другие растворители, такие как этанол, ДМСО и т.п., используемые для их кристаллизации. Соединения согласно настоящему изобретению в силу своей природы или структуры могут образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду), соответственно, предполагается, что в изобретение входят как сольватированные, так и несольватированные формы описанных соединений.
Также предполагается, что любая приведенная формула отображает изотопически необогащенные формы, а также изотопически обогащенные формы соединений. Примеры изотопов, которые могут быть включены в соединения согласно изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н (дейтерий, D) 3Н, 11С, 13С, 14С, 15N, 17O, 18O, 18F, 31Р, 32Р, 35S, 36Cl, 125I, соответственно.
Согласно другой особенности соединения согласно настоящему изобретению включают изотопически обогащенные соединения согласно данному им определению, например, соединения, в которые включены радиоактивные изотопы, такие как 3Н, 14С и 18F, или соединения, в которых присутствуют нерадиоактивные изотопы, такие, как 2Н и 13С. Такие изотопически обогащенные соединения применимы в исследованиях метаболизма (с 14С), кинетических реакций (например, с 2Н или 3Н), методах обнаружения или визуализации, таких как позитронно-эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография (ОФЭКТ), включая анализ распределения лекарств или субстратов в тканях, или в радиотерапии пациентов. В частности, особо желательным для исследований ПЭТ или ОФЭКТ может являться 18F-обогащенное соединение. Изотопически обогащенные соединения Формулы (I) обычно могут быть получены традиционными способами, известными специалистам в данной области техники, или способами, аналогичными тем, которые описаны в прилагаемых разделах "Примеры" и "Препараты", с использованием соответствующего изотопного реактива вместо ранее использовавшегося немеченого реактива.
Кроме того, замещение более тяжелыми изотопами, в частности дейтерием (т.е. 2Н или D) может обеспечивать определенные терапевтические преимущества за счет более высокой метаболической стабильности, например, увеличенного периода полураспада in vivo или меньшей требуемой дозировки или повышения терапевтического индекса. Ясно, что дейтерий в этом контексте рассматривается в качестве заместителя соединения Формулы (I). Концентрация такого тяжелого изотопа, в частности, дейтерия, может определяться на основании коэффициента изотопного обогащения. Используемый термин "коэффициент изотопного обогащения" представляет собой соотношение между распространенностью изотопа и распространенностью в природе элемента определенного изотопа. Если заместителем в соединении согласно изобретению является дейтерий, такое соединение имеет коэффициент изотопного обогащения для каждого указанного атома дейтерия, по меньшей мере, 3500 (52,5% включения дейтерия в каждом указанном атоме дейтерия), по меньшей мере, 4000 (60% включение дейтерия), по меньшей мере, 4500 (67,5% включения дейтерия), по меньшей мере, 5000 (75% включения дейтерия), по меньшей мере, 5500 (82,5% включения дейтерия), по меньшей мере, 6000 (90% включения дейтерия), по меньшей мере, 6333,3 (95% включения дейтерия), по меньшей мере, 6466,7 (97% включения дейтерия), по меньшей мере, 6600 (99% включения дейтерия), или, по меньшей мере, 6633,3 (99,5% включения дейтерия). Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, в которых растворитель кристаллизации может быть изотопически замещен, например, D2O, d6-ацетон, ДМСО-d6.
Согласно другой особенности предложено получение промежуточного соединения Формул (I), (II), (III) или (IV).
Согласно другой особенности изобретения, предложен способ получения, отделения или очистки соединения Формул (I), (II), (III) или (IV).
Согласно другой особенности изобретения, предложена фармацевтическая композиция, содержащая описанное соединение. В некоторых вариантах осуществления предложена фармацевтическая композиция, дополнительно содержащая фармацевтически приемлемый носитель, эксципиент, адъювант, растворитель или их сочетание. В других вариантах осуществления фармацевтическая композиция может являться жидкой, твердой, полутвердой, желеобразной или гель или распыляемой.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ СОЕДИНЕНИЯ СОГЛАСНО ИЗОБРЕТЕНИЮ, ПОЛУЧЕНИЕ И ВВЕДЕНИЕ
В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению, например, соединение по примерам, и фармацевтически приемлемый эксципиент, носитель, адъювант, растворитель или их сочетание.
В настоящем изобретении предложен способ лечения, предупреждения или облегчения заболевания или нарушения, включающий введение безопасного и эффективного количества комбинации лекарственных средств, содержащей соединения согласно изобретению и одно или несколько терапевтических действующих веществ. Комбинация лекарственных средств включает одно или несколько дополнительных лекарственных средства для лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, при этом действующий ингредиент дополнительных лекарственных средств отличается от соединения согласно настоящему изобретению.
Другие лекарственные средства для лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, включают без ограничения колхицин, нестероидные противовоспалительные препараты, глюкокортикоиды, средства против мочевой кислоты, средства выведения мочевой кислоты, средства подщелачивания мочи или любое их сочетание.
Другие лекарственные средства для профилактики или лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией и мочекаменной болезнью, включают колхицин, индометацин, эторикоксиб, диклофенак, ибупрофен, рофекоксиб, целекоксиб, мелоксикам, преднизон, сукцината гидрокортизон, аллопуринол, пробенецид, сульфинпиразон, бензбромарон, аллопуринол, фебуксостат, рекомбинантную уратоксидазу Aspergillus Flavus, пегилированную рекомбинантную уратоксидазу, бикарбонат натрия в таблетках, смесь цитрата калия и натрия или любое их сочетание.
Количество соединения в описанной фармацевтической композиции относится к количеству, которое может эффективно определяться как ингибирующее ксантиноксидазу и переносчик 1 уратов в биологическом образце и организме пациента. Доза действующего ингредиента в композициях согласно настоящему изобретению может варьировать; однако, необходимо, чтобы количеством действующего ингредиента являлось количество, из которого может быть получена применимая лекарственная форма. Действующий ингредиент может вводиться нуждающимся в таком лечении пациентам (животным или человеку) в дозировке, которая будет обеспечивать оптимальную фармацевтическую эффективность. Выбранная доза зависит от желаемого терапевтического эффекта, пути введения и продолжительности лечения. Дозировка будет варьировать для различных пациентов в зависимости от природы и тяжести заболевания, веса пациента, особого рациона пациента, сопутствующей лекарственной терапии, а также других факторов, известных специалистам в данной области техники. Диапазон доз обычно составляет от около 0,5 мг до 1,0 г в сутки на пациента с возможностью введения в форме одной или нескольких доз. В одном из вариантов осуществления диапазон доз составляет от около 0,5 мг до 500 мг в сутки на пациента, в другом варианте осуществления от около 0,5 мг до 200 мг в сутки на пациента, а в еще одном из вариантов осуществления от около 5 мг до 50 мг в сутки на пациента.
Следует также иметь в виду, что некоторые соединения согласно настоящему изобретению могут существовать в свободной форме или, когда это допустимо, в форме фармацевтически приемлемых производных. Фармацевтически приемлемое производное включают фармацевтически приемлемые пролекарства, соли, сложные эфиры, соли таких сложных эфиров или любые другие продукты присоединения или производные, которые после введения нуждающемуся в этом пациенту способны прямо или косвенно обеспечивать описанное соединение или его метаболит или остаток.
Фармацевтические композиции согласно настоящему изобретению могут приготавливаться и упаковываться в бестарную форму с возможностью извлечения безопасного и эффективного количества описанного соединения Формулы (I) и затем его введения пациенту, например, в форме порошков или сиропов. Чтобы получить эффективный антагонистический эффект в отношении ксантиноксидазы и переносчика 1 уратов, пациенту обычно вводят дозы на уровне от 0,0001 до 10 мг/кг массы тела. В качестве альтернативы, фармацевтические композиции согласно изобретению могут приготавливаться и упаковываться в стандартную лекарственную форму, в которой каждая физически дискретная единица содержит безопасное и эффективное количество описанного соединения Формулы (I). При приготовлении в виде стандартной лекарственной формы фармацевтические композиции согласно изобретению обычно содержат от около 0,5 мг до 1 г, или от 1 мг до 700 мг, или от 5 мг до 100 мг соединения согласно настоящему изобретению.
Когда фармацевтические композиции согласно настоящему изобретению также содержат один или несколько других действующих ингредиентов помимо соединения согласно настоящему изобретению, весовое соотношение соединения согласно настоящему изобретению и второго действующего ингредиента может варьировать и зависит от эффективной дозы каждого ингредиента. Обычно используется эффективная доза каждого из них. Так, например, когда соединение согласно настоящему изобретению сочетается с другим средством, весовое соотношение соединения согласно настоящему изобретению и другого средства обычно находится интервале от около 1000:1 до около 1:1000, например, от около 200:1 до 1:200. Сочетания соединения согласно настоящему изобретению и других действующих ингредиентов обычно также находиться в пределах указанного выше диапазона, но в каждом случае следует использовать эффективную дозу каждого действующего ингредиента.
Используемый термин "фармацевтически приемлемый эксципиент" представляет собой фармацевтически приемлемый материал, композицию или носитель, участвующий в придании формы или консистенции фармацевтической композиции. Каждый эксципиент должен являться совместимым с другими ингредиентами фармацевтической композиции при смешивании во избежание взаимодействий, которые могло бы существенно снизить эффективность соединения согласно настоящему изобретению при введении пациенту и сделать композиции фармацевтически неприемлемыми. Кроме того, каждый эксципиент, разумеется, должен иметь достаточно высокую чистоту, чтобы сделать его фармацевтически приемлемым.
Применимые фармацевтически приемлемые эксципиенты варьируют в зависимости от конкретной выбранной лекарственной формы. Кроме того, могут выбираться фармацевтически приемлемые эксципиенты, применимые для конкретной функции, которую они могут выполнять в композиции. Например, некоторые фармацевтически приемлемые эксципиенты могут выбираться с учетом их способности способствовать изготовлению однородных лекарственных форм. Некоторые фармацевтически приемлемые эксципиенты могут выбираться с учетом их способности способствовать изготовлению стабильных лекарственных форм. Некоторые фармацевтически приемлемые эксципиенты могут выбираться с учетом их способности способствовать переносу или транспортировке соединения согласно настоящему изобретению после его введения пациенту от одного органа или части тела до другого органа или части тела. Некоторые фармацевтически приемлемые эксципиенты могут выбираться с учетом их способности улучшать соблюдение больным режима и схемы лечения.
Применимые фармацевтически приемлемые эксципиенты включают эксципиенты следующих типов: разбавители, наполнители, связующие вещества, разрыхлители, смазывающие вещества, вещества, способствующие скольжению, гранулирующие вещества, вещества для нанесения покрытий, смачивающие вещества, растворители, сорастворители, суспендирующие вещества, эмульгаторы, подсластители, ароматизаторы, маскирующие запах вещества, красители, вещества, предотвращение спекания, увлажнители, хелатирующие вещества, пластификаторы, вещества, повышающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные вещества. Специалисту в данной области ясно, что некоторые фармацевтически приемлемые эксципиенты могут выполнять несколько функций и могут выполнять альтернативные функции в зависимости от количества эксципиента, присутствующего в композиции, и от того, какие другие ингредиенты присутствуют в композиции.
Специалисты в данной области техники обладают знаниями и навыками, которые позволяют им выбрать применимые фармацевтически приемлемые эксципиенты в соответствующих количествах для использования в настоящем изобретении. Кроме того, существует целый ряд доступных специалистам в данной области ресурсов, которые подробно описывают фармацевтически приемлемые эксципиенты и могут быть полезны при выборе применимых фармацевтически приемлемых эксципиентов. Их примеры включают "Фармацевтику" Ремингтона (Remington, Pharmaceutical Sciences, издательство Mack Publishing Company), "Справочник фармацевтических добавок" (издательство Gower Publishing Limited) и "Справочник фармацевтических эксципиентов" (издательство Американской фармацевтической ассоциации и Pharmaceutical Press).
У Ремингтона, The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, и в Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J.C. Boylan, 1988-1999, Marcel Dekker, New York, содержание каждого из которых в порядке ссылки включено в настоящую заявку, описаны различные носители, используемые при составлении фармацевтически приемлемых композиций, и известные способы их получения. За исключением тех случаев, когда обычная среда носителя несовместима с соединениями согласно настоящему изобретению, например, вследствие создания какого-либо нежелательного биологического эффекта или иного взаимодействия вредным образом с любым другим компонентом(-ами) фармацевтически приемлемой композиции, предполагается, что его использование входит в объем настоящего изобретения.
Фармацевтические композиции согласно настоящему изобретению получают способами и методами, известными специалистам в данной области техники. Некоторые из способом, обычно используемых в данной области техники, описаны в "Фармацевтике" Ремингтона (издательство Mack Publishing Company).
Таким образом, согласно еще одной особенности настоящего изобретения, предложен способ получения фармацевтической композиции. Фармацевтическая композиция содержит описанное соединение и фармацевтический приемлемый эксципиент, носитель, адъювант, носитель или их сочетание, при этом способ включает смешивание различных ингредиентов. Фармацевтическая композиция, содержащая описанное соединение, может быть получена, например, при температуре окружающей среды и атмосферном давлении.
Соединение согласно настоящему изобретению обычно содержится в лекарственной форме, приспособленной для введения пациенту желаемым способом. Например, лекарственные формы включают формы, которые приспособлены для (1) перорального введения, такие как таблетки, капсулы, таблетки с покрытием, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, саше и облатки; (2) парентерального введения, такие как стерильные растворы, суспензии и порошки для восстановления; (3) чрескожного введения, такие как трансдермальные пластыри; (4) ректального введения, такие как суппозитории; (5) ингаляции, такие как аэрозоли, растворы и сухие порошки; и (6) местного применения, такие кремы, мази, лосьоны, растворы, пасты, спреи, пены и гели.
В одном из вариантов осуществления описанные соединения могут быть приспособлены для перорального введения. В другом варианте осуществления описанные соединения могут быть приспособлены для ингаляции. В еще одном из вариантов осуществления описанные соединения могут быть приспособлены для интраназального введения. В еще одном из вариантов осуществления описанные соединения могут быть приспособлены для чрескожного введения. В других вариантах осуществления изобретения описанные соединения могут быть приспособлены для местного введения.
Предложенные фармацевтические композиции могут применяться в виде прессованных таблеток, растертых в порошок таблеток, жевательных пастилок, быстрорастворимых таблеток, многократно спрессованных таблеток и таблеток с энтеросолюбильным, сахарным или пленочным покрытием. Таблетки с энтеросолюбильным покрытием представляют собой прессованные таблетки, покрытые веществами, которые устойчивы к воздействию желудочной кислоты, но растворяются или распадаются в кишечнике, защищая тем самым действующие ингредиенты от кислой среды желудка. Энтеросолюбильные покрытия включают без ограничения жирные кислоты, жиры, фенилсалицилат, воски, шеллак, аммонизированный шеллак и фталаты ацетилцеллюлозы. Таблетки с сахарным покрытием представляют собой прессованные таблетки, окруженные сахарным покрытием, которое может быть выгодным для маскирования неприятного вкуса или запаха и для защиты таблетки от окисления. Таблетки с пленочным покрытием представляют собой прессованные таблетки, которые покрыты тонким слоем или пленкой из растворимого в воде материала. Пленочные покрытия включают без ограничения гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу натрия, полиэтиленгликоль 4000 и фталат ацетилцеллюлозы. Пленочное покрытие придает такие же общие характеристики, как и сахарное покрытие. Многократно прессованные таблетки представляют собой таблетки, изготовленные с использованием нескольких циклов прессования, включая многослойные таблетки и таблетки с прессованным покрытием или сухим покрытием.
Таблетированные формы могут изготавливаться из действующего ингредиента в порошковой, кристаллической или гранулированной форме по отдельности или в сочетании с одним или несколькими описанными носителями или эксципиентами, включая связующие вещества, разрыхлители, полимеры с контролируемым высвобождением, смазывающие вещества, разбавители и/или красители. При изготовлении жевательных таблеток и пастилок особенно применимыми являются ароматизаторы и подсластители.
Предложенные фармацевтические композиции могут применяться в виде мягких или твердых капсул, которые могут быть изготовлены из желатина, метилцеллюлозы, крахмала или альгината кальция. Твердые желатиновые капсулы, также известные как капсулы с сухим наполнителем (DFC), состоят из двух частей, одна из которых способна плавно входить в другую, полностью охватывая тем действующий ингредиент. Мягкая эластичная капсула (SEC) является мягкой, шаровидной оболочкой, такой как желатиновая оболочка, которая пластифицирована путем добавления глицерина, сорбита или подобного полиола. Мягкие желатиновые оболочки могут содержать консервант для предотвращения роста микроорганизмов. Применимыми консервантами являются описанные консерванты, включая метил- и пропил-парабены и сорбиновую кислоту. Предложенные жидкие, полутвердые и твердые лекарственные формы могут быть заключены в капсулу. Применимые жидкие и полутвердые лекарственные формы включают растворы и суспензию в пропиленкарбонате, растительных маслах или триглицеридах. Капсулы, содержащие такие растворы, могут быть получены, как описано в патентах US 4,328,245; 4,409,239; и 4,410,545. Капсулы также могут быть покрыты, как это известно специалистам в данной области техники, с целью изменения или поддержания растворения действующего ингредиента.
Предложенные фармацевтические композиции могут применяться в жидких и полутвердых лекарственных формах, включая эмульсии, растворы, суспензии, эликсиры и сиропы. Эмульсией является двухфазная система, в которой одна жидкость диспергирована в виде сферических частиц в другой жидкости, как в случае масло-в-воде или вода-в-масле. Эмульсии могут включать фармацевтически приемлемые неводные жидкости или растворитель, эмульгатор и консервант. Суспензии могут включать фармацевтически приемлемое суспендирующее средство и консервант. Водные спиртовые растворы могут включать фармацевтически приемлемый ацеталь, такой как ди(низший алкил)ацеталь низшего алкил-альдегида, например диэтилацеталь ацетальдегида, и смешиваемый с водой растворитель, имеющий одну или несколько гидроксильных групп, такой как пропиленгликоль и этанол. Эликсиры являются прозрачными, подслащенными и водно-спиртовыми растворами. Сиропы являются концентрированными водные растворы сахара, например, сахарозы, и также могут содержать консервант. Для получения жидкой лекарственной форме, например, раствор в полиэтиленгликоле может быть разбавлен достаточным количеством фармацевтически приемлемого жидкого носителя, например, воды, удобно дозированной для введения.
Другие применимые жидкие и полутвердые лекарственные формы включают без ограничения формы, которые содержат предложенный(-е) действующий(-е) ингредиент(-ы) и диалкилированный моно- или полиалкиленгликоль, включая 1,2-диметоксиметан, диглим, триглим, тетраглим, диметиловый эфир полиэтиленгликоля-350, диметиловый эфир полиэтиленгликоля-550, диметиловый эфир полиэтиленгликоля-750, при этом 350, 550 и 750 означают приблизительную среднюю молекулярную массу полиэтиленгликоля. Эти композиции могут дополнительно содержать один или несколько антиоксидантов, таких как бутилированный гидрокситолуол (ВНТ), бутилированный гидроксианизол (ВНА), пропилгаллат, витамин Е, гидрохинон, гидроксикумарины, этаноламин, лецитин, цефалин, аскорбиновая кислота, яблочная кислота, сорбит, фосфорная кислота, бисульфит, метабисульфит натрия, тиодипропионовая кислота и их сложные эфиры и дитиокарбаматы.
Там, где это приемлемо, стандартные лекарственные составы для перорального введения могут быть микроинкапсулированы. Также может быть получен состав, продлевающий или поддерживающий высвобождение, например, путем нанесения покрытия или внедрения материала в виде частиц в полимеры, воск или т.п.
Предложенные фармацевтические композиции для перорального введения также могут применяться в форме липосом, мицелл, микросфер или наносистем. Мицеллярные лекарственные формы могут быть получены, как описано в патенте US 6,350,458.
Предложенные фармацевтические композиции могут применяться в форме нешипучих или шипучих гранул и порошков для восстановления в жидкую лекарственной форму. Фармацевтически приемлемые носители и наполнители, используемые в нешипучих гранулах или порошках, могут включать разбавители, подсластители и смачивающие средства. Фармацевтически приемлемые носители и наполнители, используемые в шипучих гранулах или порошках, могут включать органические кислоты и источник диоксида углерода.
Во всех перечисленных выше лекарственных формах могут использоваться окрашивающие и ароматизирующие средства.
Описанные соединения также могут быть связаны с растворимыми полимерами в качестве нацеленных носителей лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксидполилизин, замещенный пальмитоильными радикалами. Кроме того, соединения могут быть связаны биоразлагаемыми полимерами, применимыми для обеспечения контролируемого высвобождения лекарственного средства, например полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидроксипиранами, полицианоакрилатами и сшитыми или амфипатическими блок-сополимерами гидрогелей.
Предложенные фармацевтические композиции могут применяться как лекарственные формы с немедленным или модифицированным высвобождением, включая формы с замедленным, устойчивым, импульсным, контролируемым, нацеленным и запрограммированным высвобождением.
Предложенные фармацевтические композиции могут содержать другие действующие ингредиенты, которые не снижают желательное терапевтическое действие, или вещества, которые дополняют желательное действие.
Предложенные фармацевтические композиции могут вводиться парентерально путем инъекции, инфузии или имплантации с целью местного или системного введения. Парентеральное введение включают внутривенное, внутриартериальное, внутрибрюшинное, внутриоболочечное, внутрижелудочковое, внутриуретральное, интрастернальное, внутричерепное, внутримышечное, внутрисуставное и подкожное введение.
Из предложенных фармацевтических композиций могут быть созданы любые лекарственные формы, применимые для парентерального введения, включая растворы, суспензии, эмульсии, мицеллы, липосомы, микросферы, наносистемы и твердые формы, применимые растворения или суспендирования в жидкости перед инъекцией. Такие лекарственные формы могут быть получены традиционными способами, известными специалистам в фармацевтике (смотри выше, Remington: The Science and Practice of Pharmacy).
Фармацевтические композиции, предназначенные для парентерального введения, могут содержать один или несколько фармацевтически приемлемых носителей и эксципиентов, включая без ограничения водные носители, смешиваемые с водой носители, безводные носители, противомикробные средства или консерванты против роста микроорганизмов, стабилизаторы, усилители растворимости, изотонические средства, буферные средства, антиоксиданты, местные анестетики, суспендирующие и диспергирующие средства, смачивающие или эмульгирующие средства, комплексообразователи, связывающие или хелатирующие средства, криопротекторные средства, лиопротекторы, загустители, регуляторы рН и инертные газы.
Применимые водные носители включают без ограничения воду, солевой раствор, физиологический раствор или забуференный фосфатом физиологический раствор (PBS), хлорид натрия для инъекций, раствор Рингера для инъекций, изотоническую декстрозу для инъекций, стерильную воду для инъекций, декстрозу и лактированный раствор Рингера для инъекций. Безводные носители включают без ограничения нелетучие масла растительного происхождения, касторовое масло, кукурузное масло, хлопковое масло, оливковое масло, арахисовое масло, мятное масло, сафлоровое масло, кунжутное масло, соевое масло, гидрогенизированные растительные масла, гидрогенизированное соевое масло и среднецепочечные триглицериды кокосового масла и масла из семян пальмы. Смешиваемые с водой носители включают без ограничения этанол 1,3-бутандиол, жидкий полиэтиленгликоль (например, полиэтиленгликоль 300 и полиэтиленгликоль 400), пропиленгликоль, глицерин, N-метил-2-пирролидон, N,N-диметилацетамид и диметилсульфоксид.
Применимые противомикробные средства или консерванты включают без ограничения фенолы, крезолы, ртутные препараты, бензиловый спирт, хлорбутанол, метил- и пропил-р-гидроксибензоаты, тимерозал, хлорид бензалкония (например, хлорид бензетония), метил- и пропилпарабены и сорбиновую кислоту. Применимые изотонические средства включают без ограничения хлорид натрия, глицерин и декстрозу. Применимые буферные средства включают без ограничения фосфат и цитрат. Применимыми антиоксидантами являются описанные антиоксиданты, включая бисульфит и метабисульфит натрия. Применимые местные анестетики включают без ограничения гидрохлорид прокаина. Применимыми суспендирующими и диспергирующими средствами являются описанные средства, включая натрий-карбоксиметилцеллюлозу, гидроксипропилметилцеллюлоза и поливинилпирролидон. Применимыми эмульгаторами являются описанные эмульгаторы, включая полиоксиэтиленсорбитан монолаурат, полиоксиэтиленсорбитан моноолеат 80 и триэтаноламин олеат. Применимые связывающие или хелатирующие средства включают без ограничения ЭДТА. Применимые средства, регулирующие рН, включают без ограничения гидроксид натрия, соляную кислоту, лимонную кислоту и молочную кислоту. Применимые комплексообразующие средства включают без ограничения циклодекстрины, включая α-циклодекстрин, β-циклодекстрин, гидроксипропил-β-циклодекстрин, β-циклодекстрин сульфобутилового эфира и 7-β-циклодекстрин сульфобутилового эфира (CAPTISOL®, CyDex, Ленекса, штат Канзас, США).
Предложенные фармацевтические композиции могут быть предназначены для введения однократной или многократной дозы. Однократные дозы могут содержаться в ампуле, пробирке или шприце. Многократные дозы для парентерального введения должны содержать противомикробное средство в бактериостатических или фунгистатических концентрациях. Все составы для парентерального введения с должны являться стерильными, как это известно и принято в данной области техники.
В одном из вариантов осуществления фармацевтические композиции применяются в форме готовых к употреблению стерильных растворов. В другом варианте осуществления фармацевтические композиции применяются в форме стерильных сухих растворимых продуктов, включая лиофилизированные порошки и таблетки для подкожных инъекций, которые должны восстанавливаться носителем перед применением. В еще одном из вариантов осуществления фармацевтические композиции применяются в форме готовых к употреблению стерильных суспензий. В еще одном из вариантов осуществления фармацевтические композиции применяются в форме стерильных сухих нерастворимых продуктов, которые должны восстанавливаться носителем перед применением. В еще одном из вариантов осуществления фармацевтические композиции применяются в форме готовых к употреблению стерильных эмульсий.
Фармацевтические композиции могут представлять собой суспензию, твердую, полутвердую или тиксотропную жидкость для введения в качестве имплантируемого депо. В одном из вариантов осуществления предложенные фармацевтические композиции диспергированы в твердой внутренней матрицы, которая окружена наружной полимерной мембраной, нерастворимой в жидкостях организма, но позволяющей диффундировать через нее действующему ингредиенту фармацевтических композиций.
Применимые внутренние матрицы включают полиметилметакрилат, полибутилметакрилат, пластифицированный или непластифицированный поливинилхлорид, пластифицированный нейлон, пластифицированный полиэтилентерефталат, натуральный каучук, полиизопрен, полиизобутилен, полибутадиен, полиэтилен, сополимер этилена и винилацетата, кремнийорганические каучуки, полидиметилсилоксаны, сополимеры силиконов и карбонатов, гидрофильные полимеры, такие как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, поперечно-сшитый поливиниловый спирт и поперечно-сшитый частично гидролизованный поливинилацетат.
Применимые наружные полимерные мембраны включают полиэтилен, полипропилен, сополимеры этилена и пропилена, сополимеры этилена и этилакрилата, сополимеры этилена и винилацетата, кремнийорганические каучуки, полидиметилсилоксаны, неопреновый каучук, хлорированный полиэтилен, поливинилхлорид, сополимеры винилхлорида с винилацетатом, винилиденхлоридом, этиленом и пропиленом, иономерный полиэтилентерефталат, бутилкаучук, эпихлоргидриновые каучуки, сополимер этилена и винилового спирта, тройной сополимер этилена, винилацетата и винилового спирта и сополимер этилена и винилоксиэтанола.
Согласно другой особенности из фармацевтической композиции согласно изобретению получают лекарственную форму для введения пациенту путем ингаляции, например, в виде сухого порошка, аэрозоля, суспензии или состава для растворения. В одном из вариантов осуществления настоящее изобретение относится к лекарственной форме для введения пациенту путем ингаляции в виде сухого порошка. Сухие порошковые композиции для доставки в легкие путем ингаляции обычно содержат описанное соединение или его фармацевтически приемлемую соль в виде тонкоизмельченного порошка вместе с одним или несколькими фармацевтически приемлемыми эксципиентами в виде тонко измельченных порошков. Фармацевтически приемлемые эксципиенты, особо применимые для использования в сухих порошках, известны специалистам в данной области техники и включают лактозу, крахмал, маннит, и моно-, ди- и полисахариды. Тонко измельченный порошок может быть получен, например, путем микронизации и измельчения. Обычно соединение с частицами уменьшенного размера (например, микронизированными) может быть охарактеризовано значением D50 от около 1 до около 10 мкм (например, измеренным методом лазерной дифракции).
Путем суспендирования или растворения описанного соединения или его фармацевтически приемлемой соли в сжиженном пропелленте могут быть получены аэрозоли. Применимые пропелленты включают галогенуглеводороды, углеводороды и другие сжиженные газы. Типичные пропелленты включают трихлорфторметан (пропеллент 11), дихлорфторметан (пропеллент 12), дихлортетрафторэтан (пропеллент 114), тетрафторэтан (HFA-134а), 1,1-дифторэтан (HFA-152a), дифторметан (HFA-32), пентафторэтан (HFA-12), гептафторпропан (HFA-227a), перфторпропан, перфторбутан, перфторпентан, бутан, изобутан и пентан. Аэрозоли, содержащие соединение Формулы (I) или его фармацевтически приемлемую соль, обычно вводят пациенту с помощью дозирующего ингалятора (MDI). Такие устройства известны специалистам в данной области техники.
Аэрозоль может содержать дополнительные фармацевтически приемлемые эксципиенты, обычно используемые с МДИ, такие как поверхностно-активные вещества, смазывающие вещества, растворители и другие эксципиенты для повышения физической стабильности композиции, повышения эффективности клапана, улучшения растворимости или улучшения вкуса.
Фармацевтические композиции для чрескожного введения могут применяться в форме отдельных пластырей, рассчитанных на то, чтобы оставаться в тесном контакте с эпидермисом пациента в течение длительного периода времени. Например, действующий ингредиент может доставляться из пластыря путем ионофореза, как в целом описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические композиции для местного применения представлять собой мази, кремы, суспензии, лосьоны, порошки, растворы, пасты, гели, спреи, аэрозоли, или масла. Мази, кремы и гели могут иметь, например, водную или масляную основу с добавлением применимых загустителей и/или желирующих средств и/или растворителей. Такие основы могут включать, например, воду и/или масло, такое как жидкий парафин или растительное масло, такое как арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загустители и желирующие средства, которые могут использоваться в зависимости от характера основы, включают мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, ланолин, пчелиный воск, карбоксиполиметилен и производные целлюлозы и/или глицерил моностеарат и/или неионогенные эмульгирующие средства.
Лосьоны содержать водную или масляную основу и обычно также содержат одно или несколько эмульгирующих средств, стабилизирующих средств, диспергирующих средств, суспендирующих средств или загустителей.
Порошки для наружного применения могут быть содержать любую применимую порошковую основу, например, тальк, лактозу или крахмал. Капли могут содержать водную или безводную основу, также содержащую одно или несколько диспергирующих средств, солюбилизирующих средств, суспендирующих средств или консервантов.
Препараты для местного применения могут накладываться один или нескольких раз в день на пораженный участок; поверх участков кожи преимущественно могут использоваться окклюзионные повязки. За счет адгезивной резервуарной системы может достигаться непрерывная или длительная доставка.
ПРИМЕНЕНИЕ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ СОГЛАСНО ИЗОБРЕТЕНИЮ
Описанные соединения или фармацевтические композиции согласно изобретению могут применяться при изготовлении лекарственного средства для лечения, профилактики, облегчения, контроля или ослабления гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, у млекопитающих, включая человека, а также других лекарственных средств для ингибирования как ксантиноксидазы, так и переносчика 1 уратов.
В частности, композиции согласно настоящему изобретению могут эффективно и с возможностью обнаружения ингибировать как ксантиноксидазу, так и переносчик 1 уратов, а описанные соединения могут применяться в качестве лекарственных средств для профилактики или лечения гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, у людей.
Описанные соединения или композиции применимы без ограничения для профилактики или лечения или ослабления гиперурикемии, отложения солей, подагрического артрита, заболеваний почек, связанных с гиперурикемией или мочекаменной болезнью, у млекопитающих, включая человека путем введения субъекту описанного соединения или композиции в эффективном количестве.
Помимо того применимости для лечения человека эти соединения и фармацевтические композиции также могут применимы в ветеринарии для лечения животных, таких как домашние животные, экзотические животные и сельскохозяйственные млекопитающие. В других вариантах осуществления животные включают лошадей, собак и кошек. Описанные соединения включают их фармацевтически приемлемые производные.
ТЕРАПИИ
В одном из вариантов осуществления описанные терапии включают введение безопасного и эффективного количества соединения согласно настоящему изобретению или фармацевтической композиции, содержащей соединение согласно настоящему изобретению, нуждающимся в этом пациентам. Каждый описанный пример представляет собой способ лечения описанных выше заболеваний, включающий введение безопасного и эффективного количества соединения согласно изобретению или фармацевтической композиции, содержащей соединение согласно настоящему изобретению, нуждающимся в этом пациентам.
В одном из вариантов осуществления соединение согласно настоящему изобретению или его фармацевтическая композиция может вводиться любым применимым способом, включая как системное введение, так и местное введение. Системное введение включает пероральное введение, парентеральное введение, чрескожное введение и ректальное введение. Парентеральное введение относится к путям введения помимо энтерального или чрескожного и обычно осуществляется путем инъекции или инфузии. Парентеральное введение включает внутривенную, внутримышечную и подкожную инъекцию или инфузию. Местное введение включает нанесение на кожу, а также внутриглазное, ушное, интравагинальное, ингаляционное и интраназальное введение. В одном из вариантов осуществления соединение согласно изобретению или его фармацевтическая композиция может вводиться перорально. В другом варианте осуществления соединение согласно настоящему изобретению или его фармацевтическая композиция может вводиться путем ингаляции. В одном из дополнительных вариантов осуществления соединение согласно изобретению или его фармацевтическая композиция может вводиться интраназально.
В одном из вариантов осуществления соединение согласно изобретению или его фармацевтическая композиция может вводиться один раз или в соответствии со схемой приема, когда определенное число доз вводится с различными интервалами в течение определенного периода времени. Например, дозы могут вводиться один, два, три или четыре раза в день. В одном из вариантов осуществления доза вводится один раз в день. В еще одном из вариантов осуществления доза вводится дважды в день. Дозы могут вводиться, пока не будет достигнут желаемый терапевтический эффект, или неопределенно долго для поддержания желаемого терапевтического эффекта. Применимые схемы приема соединения согласно настоящему изобретению или его фармацевтической композиции зависят от фармакокинетических свойств этого соединения, таких как абсобрция, распределение и период полураспада, которые могут быть определены специалистом в данной области. Кроме того, применимые схемы приема соединения согласно настоящему изобретению или его фармацевтической композиции, включая продолжительность их применения, зависят от заболевания, подлежащего лечению, тяжести заболевания, подлежащего лечению, возраста и физического состояния пациента, подлежащего лечению, истории болезни пациента, подлежащего лечению, характера сопутствующей терапии, желаемого терапевтического эффекта и подобных факторов в пределах знаний и опыта специалиста в данной области техники. Специалистам в данной области техники также ясно, что такие применимые схемы приема могут требовать корректировки с учетом индивидуальной реакции пациента на схему приема или с течением времени по мере изменения индивидуальных потребностей пациента.
Соединения согласно настоящему изобретению могут вводиться одновременно с с одним или несколькими другими терапевтическими средствами или до или после их введения. Соединения согласно настоящему изобретению и другие средства могут вводиться по отдельности одним и тем же путем или вместе в одной и той же фармацевтической композиции.
Фармацевтическая композиция или комбинация согласно настоящему изобретению может содержать унифицированную дозу около 1-1000 мг действующих ингредиентов для субъекта весом около 50-70 кг, предпочтительно около 1-500 мг или около 1-250 мг или около 1-150 мг или около 0,5-100 мг или около 1-50 мг действующих ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, массы тела, возраста и индивидуального состояния, подлежащего лечению нарушения или заболевания, или его тяжести. Врач, клиницист или ветеринар обычной квалификации может легко определить эффективное количество каждого из действующих ингредиентов, необходимых для профилактики, лечения или ингибирования прогресса нарушения или заболевания.
Свойства приведенных выше доз могут быть продемонстрированы путем испытаний in vitro и in vivo с использованием преимущественно млекопитающих, например, мышей, крыс, собак, обезьян или выделенных органов, тканей и их образцов. Соединения согласно настоящему изобретению могут применяться in vitro в форме растворов, например, предпочтительно водных растворов и in vivo энтерально или парентерально, преимущественно внутривенно, например, в виде суспензии или в водном растворе.
В одном из вариантов осуществления изобретения терапевтически эффективная доза описанного соединения составляет от около 0,1 мг до около 2000 мг в день. Фармацевтическая композиция должна обеспечить дозировку соединения от около 0,1 мг до около 2000 мг. В одном из особых вариантов осуществления предусмотрены унифицированные лекарственные формы фармацевтической композиции, обеспечивающие от около 1 мг до около 2000 мг, от около от 10 мг до около 1000 мг, от около от 20 мг до около 500 мг или от около от 25 мг до около 250 мг действующего ингредиента или сочетания основных ингредиентов в унифицированной лекарственной форме. В одном из особых вариантов осуществления предусмотрены унифицированные лекарственные формы фармацевтической композиции, обеспечивающие около 10 мг, 20 мг, 25 мг, 50 мг, 100 мг, 250 мг, 500 мг, 1000 мг или 2000 мг действующего ингредиента.
Кроме того, соединения согласно настоящему изобретению могут вводиться в качестве пролекарств. Используемый термин "пролекарство" соединения согласно изобретению представляет собой функциональное производное соединения, которое при введении пациенту, в конечном счете, высвобождает соединение согласно изобретению in vivo. Введение соединения согласно настоящему изобретению в качестве пролекарства может позволять специалисту в данной области одно или несколько из следующего: (а) изменять начало действия соединения in vivo; (б) изменять продолжительность действия соединения in vivo; (в) модифицировать перенос или распределение соединения in vivo, (г) модифицировать растворимость соединения in vivo, и (д) преодолевать побочный эффект или другие сложности, возникающие с соединением. Типичные функциональные производные, используемые для получения пролекарств, включают модификации соединения, которые химически или ферментативно расщепляется in vivo. Такие модификации, которые включают препараты фосфатов, амидов, сложных эфиров, тиоэфиров, карбонатов и карбаматов, хорошо известны специалистам в данной области техники.
ОБЩИЕ ПРОЦЕДУРЫ СИНТЕЗА
Следующие примеры осуществления приведены в целях обеспечения возможности более полного понимания изобретения. Тем не менее, подразумевается эти варианты осуществления просто обеспечивают один из способов практической реализации настоящего изобретения, и настоящее изобретение не ограничено ими.
Обычно описанные соединения могут быть получены описанными способами, в которых заместители имеют значения согласно определениям, данным выше для Формул (I), (II), (III) или (IV), за исключением указанных далее случаев. Следующие неограничивающие схемы и примеры представлены в целях дополнительной иллюстрации изобретения.
Специалистам в данной области техники ясно, что описанные химические реакции могут быть легко адаптированы для получения ряда других описанных соединений, при этом альтернативные способы получения описанных соединений считаются входящими в объем в объем изобретения. Так, например, не проиллюстрированные примерами соединения согласно изобретению могут успешно синтезироваться путем очевидных для специалистов в данной области техники модификаций, например, путем соответствующей защиты интерферирующих групп, использования других известных в данной области техники применимых реагентов, отличающихся от описанных, и/или путем обычных модификаций условий реакции. В качестве альтернативы, известные условия реакций, описанные в настоящем изобретении, считаются применимыми для получения других описанных соединений.
В описанных далее примерах, если не указано иное, все значения температуры приведены в градусах Цельсия. У компаний-поставщиков, таких как Aldrich Chemical Company, Arco Chemical Company и Alfa Chemical Company, были реагенты приобретены и использованы без дополнительной очистки, если не указано иное. У компаний-поставщиков, таких как Shantou XiLong Chemical Factory, Guangdong Guanghua Chemical Reagent Factory, Guangzhou Reagent Chemical Factory, Tianjin YuYu Fine Chemical Ltd., Tianjin Fuchen Chemical Reagent Factory, Wuhan XinHuaYuanm Technology Development Co. Ltd., Qingdao Tenglong Reagent Chemical Ltd. и Qingdao Ocean Chemical Factory, были приобретены обычные растворители.
Путем дефлегмации растворителя с натрием были получены безводные ТГФ, диоксан, толуол, и эфир. Путем дефлегмации растворителя СаН2 были получены безводные CH2Cl2 и CHCl3. Перед использованием EtOAc, ПЭ, гексан, DMAc и DMF были обработаны безводным Na2SO4.
Приведенные далее реакции были осуществлены в основном при положительном давлении азота или аргона или с осушительной трубкой (если не указано иное) в безводных растворителях, а реактивные склянки были обычно снабжены каучуковыми мембранами для введения субстратов и реагентов с помощью шприца. Стеклянную посуду сушили в печи и/или подвергали термической сушке.
Для осуществления колоночной хроматографии использовали колонку с силикагелем. Силикагель (300-400 меш) был приобретен у компании Qingdao Ocean Chemical Factory.
Были определены 1Н ЯМР-спектры с помощью 400-МГц или 600-МГц спектрометра Bruker с использованием CDCl3, ДМСО-d6, CD3OD или d6-ацетона в качестве растворителей (указаны в миллионных долях), а также с использованием TMS (0 миллионных долей) или хлороформа (7,26 миллионных долей) в качестве эталона. Для обозначения пиковых кратностей были использованы следующие сокращения: s (синглет), d (дублет), t (триплет), m (мультиплет), br (расширенный), dd (дублет дублетов) и dt (дублет триплетов). Константы связывания, когда они приведены, указаны в герцах (Гц).
Условия масс-спектрометрии (МС) низкого разрешения: Agilent 6120 Quadrupole HPLC-M (тип колонки: Zorbax SB-C18; 2,1×30 мм; 3,5 мкм; 6 мин, скорость потока 0,6 мл/мин. Подвижные фазы состояли из сочетания А (0,1% муравьиной кислоты в CH3CN) и В (0,1% муравьиной кислоты в Н2О) в градиентном режиме (от 5% до 95%), при этом был использован источник ESI. Для получения ВЭЖХ использовали детектор длин волн UV-Vis на волне 210/254 нм.
Определяли чистоту соединений путем высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием Agilent 1260 HPLC (модель колонки: Agilent Zorbax Eclipse Plus С18) и детектора DAD. Рассчитывали чистоту соединений методом нормализации площади.
В описании используются следующие сокращения:
АсОН уксусная кислота
CDCl3 дейтерохлороформ
CD3OD метанол-D4
ДБУ 1,8-диазабицикло[5.4.0]ундец-7-ен
ДХМ дихлорметан
ДМАП 4-диметиламинопиридин
ДМФ N,N-диметилформамид
ДМСО диметилсульфоксид
ДМСО-d6 диметилсульфоксид-d6
г грамм
ч час
мин минута
ммоль миллимоль
М моль на литр
°С по Цельсию
H2SO4 серная кислота
HATU 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат
НБС N-бромсукцинимид
MeCN, CH3CN ацетонитрил
МеОН метанол
мл миллилитр
НМП N-метил-2-пирролидон
к.т. комнатная температура
об/мин оборотов в минуту
в.у. время удержания
ТФК трифторуксусная кислота
ТГФ тетрагидрофуран
Типичные процедуры получения соединений согласно настоящему изобретению, показаны на примере следующей схемы синтеза, где L представляет собой уходящую группу, включая без ограничения атом галогена и трифторметансульфонилокси группу; Ra представляет собой Н или С1-4-алкил, или два Ra вместе с атомами, к которым они присоединены, образуют кольцо d; Rb представляет собой С1-4-алкил; Rc представляет собой С1-4-алкил; Re, Rd вместе с атомами, к которым они присоединены, могут образовывать Q-кольцо посредством соответствующих химических реакций, при этом некоторые примеры Re и Rd включают без ограничения случаи, когда Re представляет собой С2-3 алкенил, Rd представляет собой ОН, или когда Re представляет собой  , Rd представляет собой 2,2,2-трифторацетамидо; Rg, Rh вместе с атомами, к которым они присоединены, могут образовывать Q-кольцо посредством взаимодействия с триэтилортоформиатом, при этом некоторые примеры Rg и Rh включают без ограничения случаи, когда Rg представляет собой ОН или NH2, Re представляет собой NH2. Если не указано иное, Q, U, Т, X, Y, Z, R1, каждый из R2, каждый из R3, m и n имеет значение согласно данному ему определению.
, Rd представляет собой 2,2,2-трифторацетамидо; Rg, Rh вместе с атомами, к которым они присоединены, могут образовывать Q-кольцо посредством взаимодействия с триэтилортоформиатом, при этом некоторые примеры Rg и Rh включают без ограничения случаи, когда Rg представляет собой ОН или NH2, Re представляет собой NH2. Если не указано иное, Q, U, Т, X, Y, Z, R1, каждый из R2, каждый из R3, m и n имеет значение согласно данному ему определению.
Схема 1;
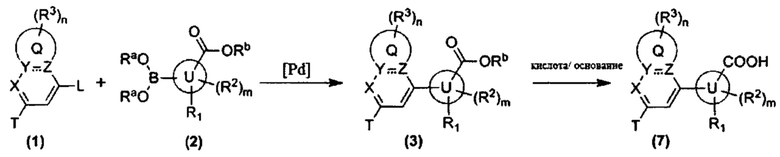
Соединение (7) может быть получено следующим путем.
Замещенные гетероциклическое или карбоциклическое соединение (1) может вступать в реакцию с соединением, содержащим бороновый сложный эфир (2), в присутствии катализатора [Pd] с образованием соединения (3) согласно реакции сочетания Сузуки; соединение (3) может быть преобразовано в соединение (7) в присутствии кислоты или основания.
Схема 1 получения промежуточного соединения
Соединение (1а) может быть получено следующим путем.
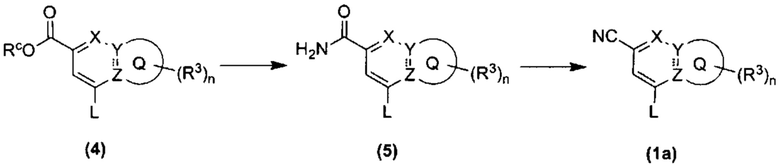
Замещенное соединение (4) может вступать в реакцию с раствором аммиака в метаноле с образованием соединения (5); соединение (5) может быть преобразовано в соединение (1а) в присутствии оксихлорида фосфора.
Схема 2 получения промежуточного соединения
Соединение (1а) также может быть получено следующим путем.
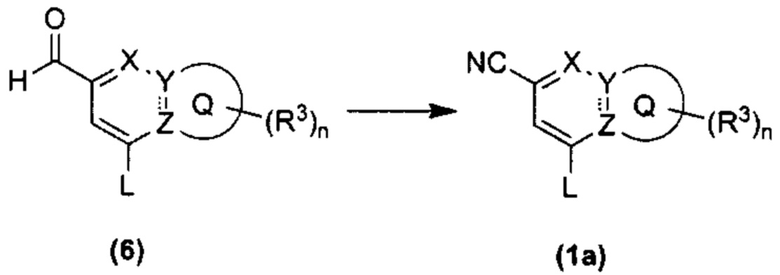
Замещенное соединение (4) может вступать в реакцию с гидроксидом аммония и йода с образованием соединения (1а).
Схема 3 получения промежуточного соединения
Соединение (1а) также может быть получено следующим путем.
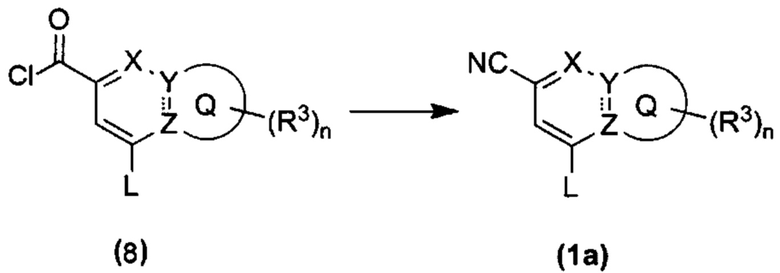
Замещенное соединение (8) может вступать в реакцию с гидроксидом аммония с образованием амидного продукта, который может быть преобразован в соединение (1а) в присутствии оксихлорида фосфора путем реакции дегидратации.
Схема 4 получения промежуточного соединения
Соединение (1а) также может быть получено следующим путем.
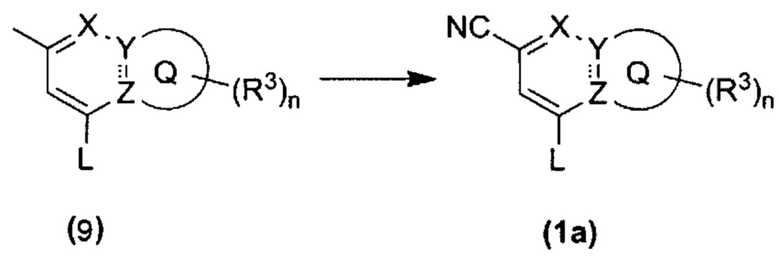
Замещенное соединение (9) может взаимодействовать с азидом натрия или трет-бутилнитритом с образованием соединения (1а).
Схема 5 получения промежуточного соединения
Замещенные соединение (10) может вступать в реакцию в применимых условиях с образованием соединения (1b).
Схема 2
Замещенное соединение (11) может взаимодействовать с триэтилортоформиатом с образованием соединения (3). Затем соединение (3) может быть преобразовано в соединение (7) в присутствии кислоты или основания.
Предложенные соединения и фармацевтические композиции, а также их применение дополнительно проиллюстрировано следующими примерами.
ПРИМЕРЫ
Пример 1
4-(5-цианобензофуран-7-ил)-2-гидроксибензойная кислота
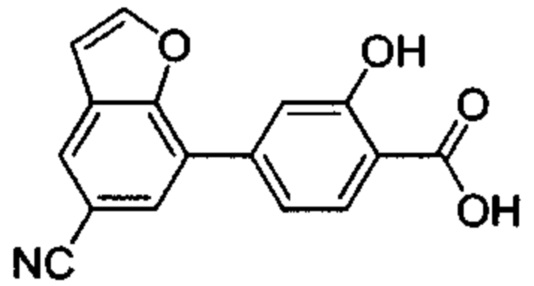
Стадия 1) Синтез метил-3-бром-4-гидроксибензоата
Добавили в 500-мл одногорлую колбу 3-бром-4-гидроксибензойную кислоту (10,0 г, 46,1 ммоль) и метанол (120 мл), затем по каплям добавили тионилхлорид (7,35 мл, 101 ммоль) при 0°С. Нагрели смесь до 90°С и в течение 12 часов перемешивали в среде азота. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный водный раствор бикарбоната натрия (200 мл), и экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (10,0 г, 94%).
МС m/z (ES-API, с отрицательными ионами): 228,0 [М-2]-.
Стадия 2) Синтез метил-3-бром-4-(2,2-диэтоксиэтокси)бензоата
Последовательно добавили в одногорлую 250-мл колбу метил-3-бром-4-гидроксибензоат (10,0 г, 43,3 ммоль), 2-бром-1,1-диэтоксиэтан (8,06 мл, 52,0 ммоль), карбонат цезия (28,2 г, 86,6 ммоль) и безводный N,N-диметилформамид (60 мл), затем нагрели реакционную смесь до 160°С и в течение 6 часов перемешивали в среде азота. Охладили полученную смесь до комнатной температуры, разбавили насыщенным водным раствором хлорида аммония (200 мл) и экстрагировали этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/50), и получили указанное соединение в виде бледно-желтого твердого вещества (5,5 г, 37%).
МС m/z (ES-API, с положительными ионами): 348,1 [М+2]+.
Стадия 3) Синтез метил-7-бромбензофуран-5-карбоксилата
Добавили в 100-мл одногорлую колбу полифосфорную кислоту (5,00 г) и хлорбензол (40 мл), и нагрели реакционную смесь с целью дефлегмации в среде азота, затем по каплям добавили раствор метил-3-бром-4-(2,2-диэтоксиэтокси)бензоата (3,68 г, 10,6 ммоль) в хлорбензоле (40 мл). В течение 2 часов перемешивали реакционную смесь при 145°С. Охладили полученную смесь до комнатной температуры. Слили верхнюю органическую фазу, а затем концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/100), и получили указанное соединение в виде бледно-желтого твердого вещества (0,40 г, 15%).
1Н ЯМР (400 МГц, CDCl3) δ: 8,31 (d, J=1,4 Гц, 1H), 8,22 (d, J=1,3 Гц, 1H), 7,78 (d, J=2,1 Гц, 1H), 6,95 (d, J=2,2 Гц, 1H), 3,97 (s, 3H).
Стадия 4) Синтез 7-бромбензофуран-5-карбоксамида
Последовательно добавили в 50-мл герметично закрытую пробирку метил-7-бромбензофуран-5-карбоксилат (400 мг, 1,56 ммоль) и раствор аммиака (20 мл, 7 М) в метаноле. Нагрели смесь до 150°С и перемешивали в течение 24 часов. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении)=1/50), и получили указанное соединение в виде желтого твердого вещества (0,26 г, 70%).
МС m/z (ES-API, с положительными ионами): 241,0 [М+2]+.
Стадия 5) Синтез 7-бромбензофуран-5-карбонитрила
По каплям добавили в 50-мл одногорлую колбу 7-бромбензофуран-5-карбоксамид (0,39 г, 1,62 ммоль) и толуол (20 мл), а затем оксихлорид фосфора (0,74 мл, 8,1 ммоль). В течение 24 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры, быстро охладили насыщенным рассолом (80 мл) и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/50), и получили указанное соединение в виде желтого твердого вещества (0,32 г, 90%).
МС m/z (ES-API, с положительными ионами): 222,9 [М+2]+.
Стадия 6) Синтез метил-4-(5-цианобензофуран-7-ил)-2-гидроксибензоата
Последовательно добавили в двугорлую 50-мл колбу 7-бромбензофуран-5-карбонитрил (0,32 г, 1,44 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,36 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл), а затем добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде. Нагрели реакционную смесь до 90°С и перемешивали в течение 0,5 часа. Охладили полученную смесь до комнатной температуры, разбавили насыщенным рассолом (80 мл) и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (0,29 г, 76%).
МС m/z (ES-API, с положительными ионами): 294,1 [М+1]+.
Стадия 7) Синтез 4-(5-цианобензофуран-7-ил)-2-гидроксибензойной кислоты
Последовательно добавили в 100-мл одногорлую колбу метил-4-(5-цианобензофуран-7-ил)-2-гидроксибензоат (0,29 г, 0,99 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), затем добавили гидроксид натрия (0,40 г, 9,9 ммоль), и в течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли водную фазу диэтиловым эфиром (50 мл), затем подкислили до рН 1 с помощью 2 N разбавленной соляной кислоты. Экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении)=1/50), и получили указанное соединение в виде белого твердого вещества (0,18 г, 65%).
МС m/z (ES-API, с положительными ионами): 280,1 [М+1]+;
ВЭЖХ: чистота = 97%; и
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8,28 (s, 2Н), 8,05 (s, 1Н), 7,94 (d, J=8,1 Гц, 1Н), 7,52 (s, 1Н), 7,47 (d, J=7,9 Гц, 1Н), 7,19 (s, 1Н).
Пример 2
4-(3-цианонафталин-1-ил)-2-гидроксибензойная кислота
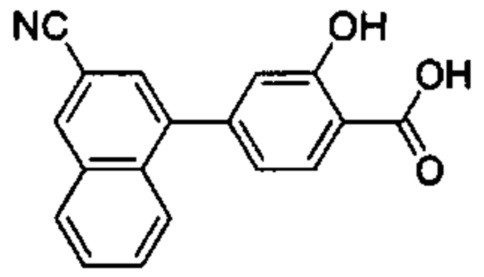
Стадия 1) Синтез 3-амино-4-бром-2-нафтойной кислоты
Последовательно добавили в 250-мл двугорлую колбу 3-амино-2-нафтойную кислоту (7,48 г, 40 ммоль) и безводный N,N-диметилформамид (100 мл), охладили смесь до 0°С, а затем порциями добавили N-бромсукцинимид (7,46 г, 42 ммоль). В течение 16 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь воду (200 мл) и этилацетат (160 мл), затем разделили смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении)=1/4), и получили указанное соединение в виде желтого твердого вещества (10,4 г, 98%).
МС m/z (ES-API, с положительными ионами): 267,0 [М+2]+.
Стадия 2) Синтез 4-бром-2-нафтойной кислоты
Последовательно добавили в 100-мл одногорлую колбу 3-амино-4-бром-2-нафтойную кислоту (5,32 г, 20 ммоль), толуол (100 мл) и этанол (35 мл). Охладили смесь до 0°С, и медленно по каплям добавили серную кислоту (3,5 мл, 98%), затем порциями добавили нитрит натрия (3,0 г, 43 ммоль). В течение 0,5 часа перемешивали реакционную смесь при 0°С, затем нагрели с целью дефлегмации и проводили реакцию в течение 1,5 часа. Охладили смесь до комнатной температуры, и добавили воду (200 мл) и этилацетат (200 мл).
Разделили полученную смесь, промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде желтого твердого вещества (3,57 г, 71%).
МС m/z (ES-API, с отрицательными ионами): 247,9 [М-2]-.
Стадия 3) Синтез метил-4-бром-2-нафтоата
Добавили в 100-мл одногорлую колбу 4-бром-2-нафтойную кислоту (3,57 г, 14,2 ммоль) и безводный метанол (50 мл). Охладили смесь до 0°С, а затем медленно по каплям добавили тионилхлорид (2,53 г, 1,53 мл, 21,3 ммоль). Постепенно нагрели реакционную смесь до 80°С и перемешивали в течение 12 часов. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (150 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтого твердого вещества (1,1 г, 34%).
1H ЯМР (400 МГц, CDCl3) δ: 8,57 (s, 1H), 8,38 (d, J=1,3 Гц, 1Н), 8,28 (d, J=8,5 Гц, 1H), 7,96 (d, J=8,2 Гц, 1H), 7,72 (t, J=7,7 Гц, 1H), 7,61 (t, J=7,5 Гц, 1H), 3,99 (s, 3H).
Стадия 4) Синтез 4-бром-2-нафтамида
Добавили в 100-мл герметично закрытую пробирку метил-4-бром-2-нафтоат (3,5 г, 13,2 ммоль) и аммиак (50 мл, 7 М в метаноле). В течение 12 часов перемешивали реакционную смесь при 130°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (2,8 г, 85%).
МС m/z (ES-API, с положительными ионами): 250,9 [М+2]+.
Стадия 5) Синтез 4-бром-2-нафтонитрила
Добавили оксихлорид фосфора (8,0 г, 52 ммоль) в 100-мл одногорлую колбу с раствором 4-бром-2-нафталин карбоксамида (2,6 г, 10,4 ммоль) в толуоле (25 мл). В течение 12 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (100 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (2,2 г, 91%).
1H ЯМР (400 МГц, CDCl3) δ: 8,29 (d, J=8,5 Гц, 1H), 8,20 (s, 1H), 7,92-7,90 (m, 2H), 7,77 (t, J=7,7 Гц, 1H), 7,67 (t, J=7,5 Гц, 1H).
Стадия 6) Синтез метил-4-(3-цианонафталин-1-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 4-бром-2-нафталин-карбонитрил (0,417 г, 1,8 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,42 г, 1,5 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино) ферроцена и дихлорида палладия(II) (122 мг, 0,15 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,5 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, и добавили насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/3), и получили указанное соединение в виде белого твердого вещества (0,18 г, 40%).
МС m/z (ES-API, с положительными ионами): 304,2 [М+1]+.
Стадия 7) Синтез 4-(3-цианонафталин-1-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(3-цианонафталин-1-ил)-2-гидроксибензоат (0,18 г, 0,59 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем добавили гидроксид натрия (0,12 г, 3,0 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли полученную смесь эфиром (50 мл), подкисляли до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде белого твердого вещества (0,06 г, 35%).
МС m/z: (ES-API, с отрицательными ионами): 288,1 [М-1]-;
ВЭЖХ: чистота = 97%; и
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8,65 (s, 1H), 8,18-8,15 (m, 1H), 7,98-7,80 (m, 2H), 7,74-7,72 (m, 3Н), 7,08-6,86 (m, 2H).
Пример 3
4-(6-цианобензо[d][1,3]диоксол-4-ил)-2-гидроксибензойная кислота
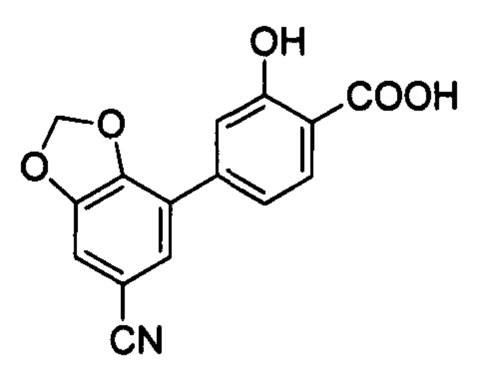
Стадия 1) Синтез 3-бром-4-гидрокси-5-метоксибензонитрила
Добавили в 100-мл одногорлую колбу 3-бром-4-гидрокси-5-метоксибензальдегид (6,93 г, 30,0 ммоль), гидроксид аммония (100 мл, 28%) и тетрагидрофуран (100 мл), а затем порциями добавили йод (7,46 г, 42 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Быстро охладили смесь насыщенным тиосульфатом натрия (100 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (2,9 г, 43%).
МС m/z (ES-API, с отрицательными ионами): 225,0 [М-2]-.
Стадия 2) Синтез 3-бром-4,5-дигидроксибензонитрила
Добавили в 100-мл одногорлую колбу 3-бром-4-гидрокси-5-метоксибензонитрил (2,28 г, 10,0 ммоль) и дихлорметан (30 мл), затем по каплям добавили трибромид бора (3,1 мл, 33 ммоль) при -70°С в среде азота. В течение 3 часов перемешивали реакционную смесь при -70°С, и затем в течение 6 часов перемешивали при -50°С. Быстро охладили полученную смесь водой (100 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали под вакуумом. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (1,6 г, 75%).
MC m/z (ES-API, с отрицательными ионами): 210,9 [М-2]-.
Стадия 3) Синтез 7-бромбензол [d][1,3]диоксол-5-карбонитрила
Постепенно добавили в 100-мл одногорлую колбу 3-бром-4,5-дигидроксибензонитрил (0,26 г, 1,2 ммоль), дийодметан (0,39 г, 1,46 ммоль), карбонат цезия (1,37 г, 4,2 ммоль) и безводный N,N-диметилформамид (8 мл). В течение 12 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили смесь до комнатной температуры, и добавили насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (60 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,23 г, 85%).
MC m/z (ES-API, с положительными ионами): 227,0 [М+2]+.
Стадия 4) Синтез метил-4-(6-цианобензо[d][1,3]диоксол-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бромбензол[d|[1,3]диоксол-5-карбонитрил (0,30 г, 1,32 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,31 г, 1,1 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (90 мг, 0,11 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,1 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, и добавили насыщенный рассол (80 мл). Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (0,16 г, 49%).
МС m/z (ES-API, с отрицательными ионами): 296,0 [М-1]-.
Стадия 5) Синтез 4-(6-цианобензо[d][1,3]диоксо-4-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(6-цианобензо[d][1,3]диоксол-4-ил)-2-гидроксибензоат (0,16 г, 0,54 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем добавили гидроксид натрия (0,11 г, 2,7 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли полученную смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты, а затем экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,09 г, 59%).
МС m/z: (ES-API, с отрицательными ионами): 282,1 [М-1]-;
ВЭЖХ: чистота = 98%; и
1H ЯМР (400 МГц, ДМСО-d6) δ: 7,86 (d, J=8,2 Гц, 1Н), 7,75 (s, 1H), 7,45 (s, 1H), 7,40-7,26 (m, 2H), 6,27 (s, 2H).
Пример 4:
4-(5-циано-2,3-дигидробензофуран-7-ил)-2-гидроксибензойная кислота
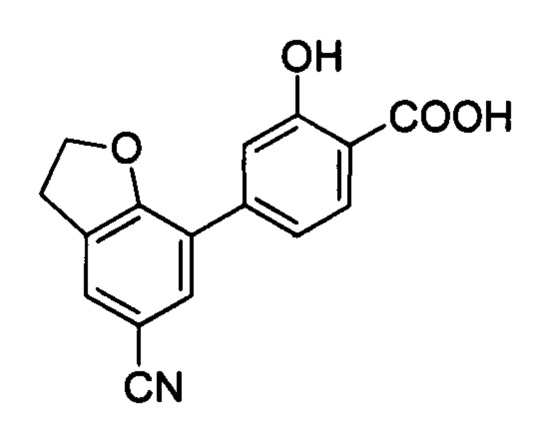
Стадия 1) Синтез 7-бром-2,3-дигидробензофуран-5-карбальдегида
Добавили в 100-мл одногорлую колбу 2,3-дигидробензофуран-5-карбальдегид (2,96 г, 2,0 ммоль), ацетат натрия (1,97 г, 24 ммоль) и уксусную кислоту (40 мл), а затем добавили бром (6,39 г, 40 ммоль) при 10°С. В течение 1 часа перемешивали реакционную смесь при комнатной температуре. Добавили в смесь ледяную воду (100 мл) и насыщенный водный раствор тиосульфата натрия (10 мл). Экстрагировали полученную смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (4,36 г, 96%).
1H ЯМР (400 МГц, CDCl3) δ: 9,78 (s, 1H), 7,82 (s, 1H), 7,66 (d, J=0,8 Гц, 1H), 4,79 (t, J=8,8 Гц, 2H), 3,38 (t, J=8,8 Гц, 2Н).
Стадия 2) Синтез 7-бром-2,3-дигидробензофуран-5-карбонитрила
Добавили в 100-мл одногорлую колбу 7-бром-2,3-дигидробензофуран-5-карбальдегид (1,82 г, 8,0 ммоль), гидроксид аммония (20 мл, 28%) и тетрагидрофуран (20 мл), а затем порциями добавили йод (2,23 г, 8,8 ммоль). В течение 4 часов перемешивали реакционную смесь при комнатной температуре. Быстро охладили смесь насыщенным водным раствором тиосульфата натрия (100 мл). Экстрагировали полученную смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде белого твердого вещества (1,52 г, 85%).
МС m/z (ES-API, с положительными ионами): 225,0 [М+2]+.
Стадия 3) Синтез трет-бутил-4-(5-циано-2,3-дигидробензофуран-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-2,3-дигидробензофуран-5-карбонитрил (0,29 г, 1,0 ммоль), трет-бутил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,32 г, 1,0 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (82 мг, 0,10 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,0 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (0,18 г, 53%).
МС m/z (ES-API, с положительными ионами): 338,0 [М+1]+.
Стадия 4) Синтез 4-(5-циано-2,3-дигидробензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-2,3-дигидробензофуран-7-ил)-2-гидроксибензоат (0,18 г, 0,53 ммоль) и дихлорметан (12 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,11 г, 73%).
МС m/z: (ES-API, с отрицательными ионами): 280,1 [М-1]-;
ВЭЖХ: чистота = 95%; и
1Н ЯМР (400 МГц, ДМСО-d6) δ: 7,89 (s, 1H), 7,84 (d, J=8,3 Гц, 1Н), 7,73 (s, 1H), 7,34 (d, J=1,4 Гц, 1H), 7,30 (m, 1H), 4,82-4,65 (m, 2H), 3,32-3,28 (m, 2H).
Пример 5:
4-(6-цианоимидазо[1,2-а]пиридин-8-ил)-2-гидроксибензойная кислота
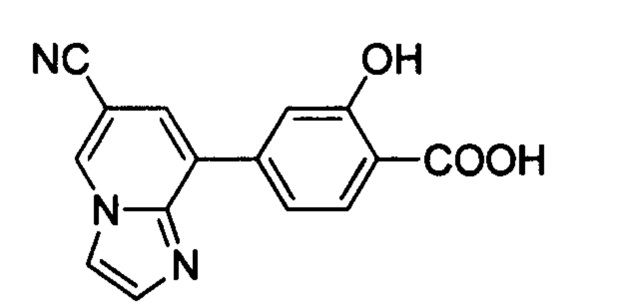
Стадия 1) Синтез метил-8-бромимидазо[1,2-а]пиридин-6-карбоксилата
Добавили в 100-мл одногорлую колбу метил-6-амино-5-бромникотинат (2,50 г, 10,8 ммоль), бикарбонат натрия (1,55 г, 18,5 ммоль) и этанол (25 мл), а затем 40% хлор формальдегид (8,6 мл, 54 ммоль). Нагрели реакционную смесь с целью дефлегмации, и проводили реакцию в течение 12 часов. Охладили полученную смесь до комнатной температуры и разбавили насыщенным водным раствором бикарбоната натрия (100 мл). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (100% дихлорметан), и получили указанное соединение в виде белого твердого вещества (2,19 г, 79%).
МС m/z (ES-API, с положительными ионами): 256,0 [М+2]+.
Стадия 2) Синтез 8-бромимидазо[1,2-а]пиридин-6-карбоксамида
Последовательно добавили в 100-мл герметично закрытую пробирку метил-8-бромимидазо[1,2-a]пиридин-6-карбоксилат (2,5 г, 9,8 ммоль) и аммиак (20 мл, 7 М в метаноле). Нагрели смесь до 130°С и перемешивали в течение 48 часов. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан = 1/50), и получили указанное соединение в виде белого твердого вещества (1,64 г, 70%).
МС m/z (ES-API, с положительными ионами): 241,0 [М+2]+.
Стадия 3) Синтез 8-бромимидазо[1,2-a]пиридин-6-карбонитрила
Добавили в 100-мл одногорлую колбу 8-бромимидазо[1,2-a]пиридин-6-карбоксамид (0,55 г, 2,3 ммоль) и толуол (25 мл), и по каплям добавили оксихлорид фосфора (1,5 мл, 16 ммоль). В течение 12 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (100 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (0,42 г, 83%).
МС m/z (ES-API, с положительными ионами): 222,9 [М+2]+.
Стадия 4) Синтез метил-4-(6-цианоимидазо[1,2-а]пиридин-8-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 8 бромимидазо[1,2-a]пиридин-6-карбонитрил (0,40 г, 1,9 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,46 г, 1,7 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (120 мг, 0,16 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,7 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, разбавили насыщенным рассолом (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (100% дихлорметан), и получили указанное соединение в виде белого твердого вещества (0,21 г, 43%).
МС m/z (ES-API, с положительными ионами): 294,0 [М+1]+.
Стадия 5) Синтез 4-(6-цианоимидазо[1,2-a]пиридин-8-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(6-цианоимидазо[1,2-а]пиридин-8-ил)-2-гидроксибензоат (0,21 г, 0,72 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воды (8 мл), а затем гидроксид натрия (0,14 г, 3,6 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли полученную смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты, а затем экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,053 г, 27%).
МС m/z: (ES-API, с положительными ионами): 280,1 [М+I]+;
ВЭЖХ: чистота = 98%; и
1Н ЯМР (400 МГц, ДМСО-d6) δ: 9,40 (s, 1Н), 8,15 (s, 1H), 7,89-7,83 (m, 4H), 7,65 (d, J=8,1 Гц, 1Н).
Пример 6
4-(6-циано-1-метил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензойная кислота
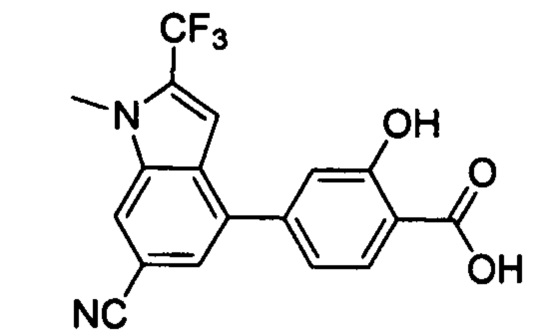
Стадия 1) Синтез 3-бром-4-метил-5-нитробензойной кислоты
Добавили в 250-мл двугорлую колбу 4-метил-3-нитробензойную кислоту (17,7 г, 94,8 ммоль) и концентрированную серную кислоту (80 мл, 98%), а затем порциями добавили 1,3-дибром-5,5-диметилгидантоин (13,8 г, 47,3 ммоль). В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Вылили полученную смесь в ледяную воду. Профильтровали выпавшее в осадок желтое твердое вещество, высушили осадок на фильтре, и получили указанное соединение в виде желтого твердого вещества (22,3 г, 86%).
МС m/z (ES-API, с отрицательными ионами): 257,0 [M-2]-.
Стадия 2) Синтез метил-3-бром-4-метил-5-нитробензоата
Добавили в 500-мл одногорлую колбу 3-бром-4-метил-5-нитробензойную кислоту (22,3 г, 85,8 ммоль) и безводный метанол (200 мл), а затем тионилхлорид (13,7 мл, 189 ммоль) при 0°С. Нагрели реакционную смесь до 80°С, и проводили реакцию в течение 6 часов. Охладили смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (200 мл) и этилацетат (300 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтого твердого вещества (21,3 г, 77%).
1Н ЯМР (400 МГц, CDCl3) δ: 8,45 (d, J=1,4 Гц, 1Н), 8,36 (d, J=1,3 Гц, 1Н), 3,98 (s, 3H), 2,64 (s, 3H).
Стадия 3) Синтез метил-3-амино-5-бром-4-метилбензоата
Добавили в 250-мл одногорлую колбу метил-3-бром-4-метил-5-нитробензоат (7,65 г, 27,9 ммоль), уксусную кислоту (20 мл), метанол (80 мл) и тетрагидрофуран (20 мл), а затем порциями добавили цинк (11,0 г, 168 ммоль). Перемешивали реакционную смесь в течение 1 ч при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (200 мл) и этилацетат (200 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде желтого твердого вещества (4,3 г, 63%).
МС m/z (ES-API, с положительными ионами): 245,0 [М+2]+.
Стадия 4) Синтез метил-3-бром-4-метил-5-(2,2,2-трифторацетамидо)бензоата
Добавили в 100-мл двугорлую колбу метил-3-амино-5-бром-4-метилбензоат (1,15 г, 4,62 ммоль), триэтиламин (1,28 мл, 9,21 ммоль) и дихлорметан (20 мл), а затем по каплям добавили трифторуксусный ангидрид (0,79 мл, 5,6 ммоль) при 0°С. В течение 1 часа перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме и получили указанное соединение в виде желтого твердого вещества (1,5 г, 95%).
МС m/z (ES-API, с положительными ионами): 341,0 [М+2]+.
Стадия 5) Синтез метил-3-бром-4-(хлорметил)-5-(2,2,2-трифторацетамидо) бензоата
Добавили в 100-мл двугорлую колбу метил-3-бром-4-метил-5-(2,2,2-трифторацетамидо)бензоат (1,5 г, 4,4 ммоль), сульфурилхлорид (1,46 мл, 17,6 ммоль), перекись бензоила (0,21 г, 0,88 ммоль) и четыреххлористый углерод (20 мл). В течение 3 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде твердого вещества желтого цвета (1,45 г, 88%).
МС m/z (ES-API, с отрицательными ионами): 370,9 [М-2]-.
Стадия 6) Синтез (2-бром-4-(метоксикарбонил)-6(2,2,2-трифторацетамидо)бензил)трифенилфосфонийхлорида
Добавили в 500-мл одногорлую колбу метил-3-бром-4-(хлорметил)-5-(2,2,2-трифторацетамидо)бензоат (12,1 г, 32,4 ммоль), трифенилфосфин (9,36 г, 35,7 ммоль) и толуол (200 мл). В течение 6 часов перемешивали реакционную смесь при 100°С в среде азота. Охладили смесь до комнатной температуры и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде желтого твердого вещества (13,0 г, 63%).
МС m/z (ES-API, с отрицательными ионами): 598,0 [М-2]-.
Стадия 7) Синтез метил-4-бром-2-(трифторметил)-1H-индол-6-карбоксилата
Добавили в 100-мл одногорлую колбу (2-бром-4-(метоксикарбонил)-6(2,2,2-трифторацетамидо)бензил)трифенилфосфонийхлорид (13,0 г, 20,4 ммоль) и N,N-диметилформамид (40 мл). В течение 26 часов перемешивали реакционную смесь при 140°С. Охладили полученную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (60 мл). Экстрагировали смесь этилацетатом (60 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого твердого вещества (2,5 г, 38%).
МС m/z (ES-API, с отрицательными ионами): 319,1 [М-2]-.
Стадия 8) Синтез 4-бром-2-(трифторметил)-1H-индол-6-карбоксамида
Последовательно добавили в 100-мл герметично закрытую пробирку метил-4-бром-2-(трифторметил)-1H-индол-6-карбоксилат (2,7 г, 8,4 ммоль) и аммиак (20 мл, 7 М в метаноле). В течение 72 часов перемешивали реакционную смесь при 130°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде желтого твердого вещества (1,5 г, 58%).
МС m/z (ES-API, с положительными ионами): 308,0 [М+2]+.
Стадия 9) Синтез 4-бром-2-(трифторметил)-1H-индол-6-карбонитрила
Добавили в 100-мл одногорлую колбу 4-бром-2-(трифторметил)-1H-индол-6-карбоксамид (1,5 г, 4,9 ммоль) и толуол (25 мл), затем по каплям добавили оксихлорид фосфора в (2,3 мл, 24,5 ммоль), и в течение 12 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (100 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде желтого твердого вещества (0,78 г, 55%).
МС m/z (ES-API, с отрицательными ионами): 286,0 [М-2]-.
Стадия 10) Синтез 4-бром-1-метил-2-(трифторметил)-1H-индол-6-карбонитрила
Добавили в 50-мл двугорлую колбу 4-бром-2-(трифторметил)-1H-индол-6-карбонитрил (0,50 г, 1,73 ммоль) и N,N-диметилформамид (10 мл), а затем гидрид натрия (0,50 г, 1,95 ммоль, 60%). В течение 0,5 часа перемешивали смесь при 0°С, а затем по каплям добавили йодистый метил (0,20 мл, 3,2 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде желтого твердого вещества (0,36 г, 69%).
МС m/z (ES-API, с положительными ионами): 304,0 [М+2]+.
Стадия 11) Синтез метил-4-(6-циано-1-метил-2-(трифторметил)-1Н-индол-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 4-бром-1-метил-2-(трифторметил)-1H-индол-6-карбонитрил (0,36 г, 1,19 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,30 г, 1,08 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (48 мг, 0,059 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,1 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, разбавили насыщенным рассолом (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (70 мг, 17%).
МС m/z (ES-API, с положительными ионами): 375,2 [М+1]+.
Стадия 12) Синтез 4-(6-циано-1-метил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(6-циано-1-метил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензоат (70 мг, 0,19 ммоль), метанол (6 мл), тетрагидрофуран (6 мл) и воду (6 мл), а затем гидроксид натрия (75 мг, 1,88 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли полученную смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (45 мг, 67%).
МС m/z (ES-API, с положительными ионами): 361,2 [М+1]+;
ВЭЖХ: чистота = 98%; и
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8,41 (s, 1Н), 7,93 (s, 1H), 7,65 (s, 1H), 7,23-7,19 (m, 3H), 3,98 (s, 3H).
Пример 7
4-(5-циано-1H-индол-7-ил)-2-гидроксибензойная кислота
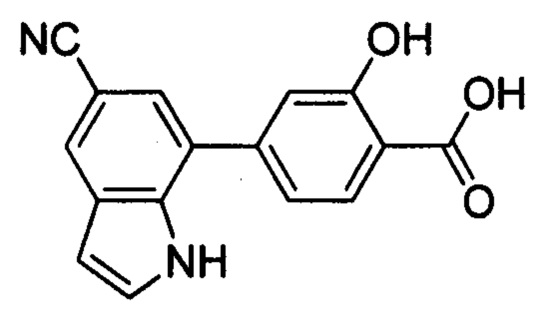
Стадия 1) Синтез 7-бром-1H-индол-5-карбоновой кислоты
Добавили в 100-мл двугорлую колбу 3-бром-4-нитробензойную кислоту (0,30 г, 1,2 ммоль) и безводный тетрагидрофуран (10 мл), а затем по каплям добавили винилмагнийбромид (4,3 мл, 4,3 ммоль, 1 моль/л в тетрагидрофуране) при -75°С в среде азота. В течение 2 часов перемешивали реакционную смесь при -75°С. Добавили к полученной смеси насыщенный хлорид аммония (60 мл) и этилацетат (80 мл), и разделили смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, затем очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтого твердого вещества (0,12 г, 41%).
МС m/z (ES-API, с положительными ионами): 241,0 [М+2]+.
Стадия 2) Синтез 7-бром-1H-индол-5-карбоксамида
Постепенно добавили в 50-мл двугорлую колбу 7-бром-1H-индол-5-карбоновую кислоту (0,50 г, 2,1 ммоль), хлорид аммония (0,22 г, 4,1 ммоль) и безводный N,N-диметилформамид (12 мл), а затем 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (0,95 г, 2,5 ммоль) и N,N-диизопропилэтиламин (1,0 мл, 6,1 ммоль) при 0°С. В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Добавили к полученной смеси насыщенный хлорид аммония (100 мл) и этилацетат (100 мл). Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,42 г, 84%).
МС m/z (ES-API, с отрицательными ионами): 236,0 [М-2]-.
Стадия 3) Синтез 7-бром-1H-индол-5-карбонитрила
По каплям добавили в 100-мл одногорлую колбу 7-бром-177-индол-5-карбоновую кислоту (0,91 г, 3,8 ммоль) и толуол (20 мл), а затем оксихлорид фосфора (2,4 мл, 26 ммоль), после чего в течение 12 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (100 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде белого твердого вещества, 0,49 г, 58%).
МС m/z (ES-API, с отрицательными ионами): 218,0 [М-2]-.
Стадия 4) Синтез трет-бутил-7-бром-5-циано-1H-индол-1-карбоксилата
Добавили в 100-мл одногорлую колбу 7-бром-1H-индол-5-карбонитрил (0,49 г, 2,2 ммоль), ди-трет-бутилдикарбонат (0,59 мл, 2,6 ммоль) и дихлорметан (20 мл), а затем 4-диметиламинопиридин (0,027 г, 0,22 ммоль). В течение 3 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,67 г, 94%).
Стадия 5) Синтез трет-бутил-7-(4-трет-бутоксикарбонил)-3-гидроксифенил)-5-циано-1H-индол-1-карбоксилата
Постепенно добавили в 50-мл двугорлую колбу трет-бутил-7-бром-5-циано-1H-индол-1-карбоксилат (0,67 г, 2,1 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,53 г, 1,7 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (140 мг, 0,19 ммоль) и N,N-диметилформамид (12 мл). Добавили в среде азота раствор карбоната калия (1,1 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры, разбавили насыщенным рассолом (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,61 г, 85%).
МС m/z (ES-API, с отрицательными ионами): 433,1 [М-1]-.
Стадия 6) Синтез 4-(5-циано-1H-индол-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-7-(4-трет-бутоксикарбонил)-3-гидроксифенил)-5-циано-1H-индол-1-карбоксилат (0,61 г, 1,4 ммоль) и дихлорметан (12 мл), а затем трифторуксусную кислоту (5 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (78 мг, 20%),
МС m/z (ES-API, с положительными ионами): 279,1 [М+1]+;
ВЭЖХ: чистота 99%; и
1H ЯМР (400 МГц, ДМСО) δ: 11,61 (s, 1H), 8,13 (s, 1H), 7,91 (d, J=7,8 Гц, 1H), 7,53 (s, 1H), 7,49 (s, 1H), 7,14-7,12 (m, 2H), 6,70 (s, 1H).
Пример 8
4-(5-циано-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензойная кислота
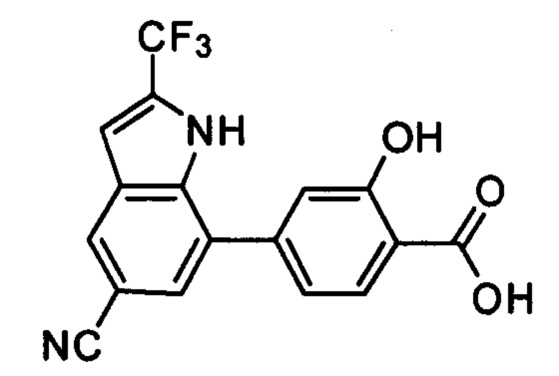
Стадия 1) Синтез N-(4-циано-2-метилфенил)-2,2,2-трифторацетамида
Добавили в 100-мл двугорлую колбу 4-амино-3-метилбензонитрил (0,611 г, 4,62 ммоль), триэтиламин (1,28 мл, 9,21 ммоль) и дихлорметан (20 мл), а затем трифторуксусный ангидрид (0,79 мл, 5,6 ммоль) при 0°С. В течение 1 часа перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде желтого твердого вещества (1,0 г, 95%).
MC m/z (ES-API, с положительными ионами): 229,0 [М+1]+.
Стадия 2) Синтез N-(2-(хлорметил)-4-цианофенил)-2,2,2-трифторацетамида
Добавили в 100-мл одногорлую колбу N-(4-циано-2-метилфенил)-2,2,2-трифторацетамид (1,0 г, 4,4 ммоль), сульфурилхлорид (1,46 мл, 17,6 ммоль), перекись бензоила (0,21 г, 0,88 ммоль) и четыреххлористый углерод (20 мл). Нагрели реакционную смесь до 80°С и в течение 3 часов перемешивали в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде желтого масла (1,14 г, 99%).
MC m/z (ES-API, с положительными ионами): 263,0 [М+1]+.
Стадия 3) Синтез (5-циано-2-(2,2,2-трифторацетамидо)бензил)трифенилфосфонийхлорида
Добавили в 500-мл одногорлую колбу N-(2-(хлорметил)-4-цианофенил)-2,2,2-трифторацетамид (8,51 г, 32,4 ммоль), трифенилфосфин (9,36 г, 35,7 ммоль) и толуол (200 мл). Нагрели реакционную смесь до 100°С и в течение 6 часов перемешивали в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде желтого твердого вещества (7,99 г, 47%).
MC m/z (ES-API, с положительными ионами): 525,1 [М+1]+.
Стадия 4) Синтез (3-бром-5-циано-2-(2,2,2-трифторацетамидо)бензил)трифенилфосфонийбромида
Добавили в 100-мл одногорлую колбу (5-циано-2-(2,2,2-трифторацетамидо) бензил)трифенилфосфонийхлорид (1,00 г, 1,91 ммоль), активированный уголь (1,00 г) и N,N-диметилформамид (20 мл). В течение 6 часов перемешивали реакционную смесь при комнатной температуре. Профильтровали полученную смесь, и промыли осадок на фильтре N,N-диметилформамидом (10 мл), добавили к объединенным фильтратам N-бромсукцинимид (1,04 г, 5,73 ммоль), и в течение 24 часов перемешивали реакционную смесь при комнатной температуре. Затем добавили в смесь N-бромсукцинимид (1,04 г, 5,73 ммоль), и в течение 36 часов перемешивали реакционную смесь при комнатной температуре. Добавили к полученной смеси насыщенный водный раствор тиосульфата натрия (80 мл) и этилацетат (80 мл), затем разделили смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/100), и получили указанное соединение в виде желтого твердого вещества (0,384 г, 31%).
МС m/z (ES-API, с положительными ионами): 648,9 [М+3]+.
Стадия 5) Синтез 7-бром-2-(трифторметил)-1H-индол-5-карбонитрила
Добавили в 100-мл одногорлую колбу (3-бром-5-циано-2-(2,2,2-трифторацетамидо)бензил)трифенилфосфонийбромид (1,3 г, 2,0 ммоль) и N,N-диметилформамид (20 мл), и в течение 3 часов перемешивали реакционную смесь при 130°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (100 мл). Экстрагировали смесь этилацетатом (60 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (0,40 г, 70%).
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 13,15 (s, 1H), 8,32 (s, 1H), 8,01 (s, 1H), 7,33 (s, 1H).
Стадия 6) Синтез метил-4-(5-циано-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-2-(трифторметил)-1H-индол-5-карбонитрил (340 мг, 1,19 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (300 мг, 1,08 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (48 мг, 0,059 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,1 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С.Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (240 мг, 57%).
МС m/z (ES-API, с положительными ионами): 361,0 [М+1]+.
Стадия 7) Синтез 4-(5-циано-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(5-циано-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензоат (240 мг, 0,67 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (80 мг, 2,0 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (144 мг, 62%).
МС m/z (ES-API, с положительными ионами): 347,0 [М+1]+; и
1H ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 12,69 (s, 1H), 8,31 (s, 1H), 7,95 (d, J=7,9 Гц, 1H), 7,67 (s, 1H), 7,30 (s, 1H), 7,25-7,05 (m, 2H).
Пример 9
4-(5-циано-1-метил-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензойная кислота
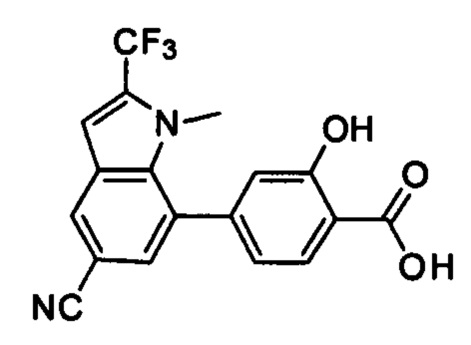
Стадия 1) Синтез 7-бром-1-метил-2-(трифторметил)-1H-индол-5-карбонитрила
Добавили в 50-мл двугорлую колбу 7-бром-2-(трифторметил)-1H-индол-5-карбонитрил (500 мг, 1,73 ммоль) и N,N-диметилформамид (10 мл), а затем 60% гидрид натрия (500 мг, 1,95 ммоль). В течение 0,5 часа перемешивали смесь при 0°С, и по каплям добавили йодметан (0,20 мл, 3,2 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный рассол (80 мл) и этилацетат (80 мл), и разделили смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде желтого твердого вещества (500 мг, 96%).
МС m/z (ES-API, с положительными ионами): 304,0 [М+2]+.
Стадия 2) Синтез метил-4-(5-циано-1-метил-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-1-метил-2-(трифторметил)-1H-индол-5-карбонитрил (360 мг, 1,19 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (300 мг, 1,08 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (48 мг, 0,059 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,1 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (140 мг, 32%).
МС m/z (ES-API, с положительными ионами): 375,1 [М+1]+.
Стадия 3) Синтез 4-(5-циано-1-метил-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(5-циано-1-метил-2-(трифторметил)-1H-индол-7-ил)-2-гидроксибензоат (140 мг, 0,37 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (45 мг, 1,12 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь эфиром (50 мл), а затем подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (85 мг, 64%).
МС m/z (ES-API, с положительными ионами): 361,0 [М+1]+; и
1H ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,34 (s, 1H), 7,89 (s, 1H), 7,51 (s, 1H), 7,40 (s, 1H), 7,12-7,05 (m, 2H), 3,40 (s, 3H).
Пример 10
4-(6-циано-1-метил-1H-индазол-4-ил)-2-гидроксибензойная кислота
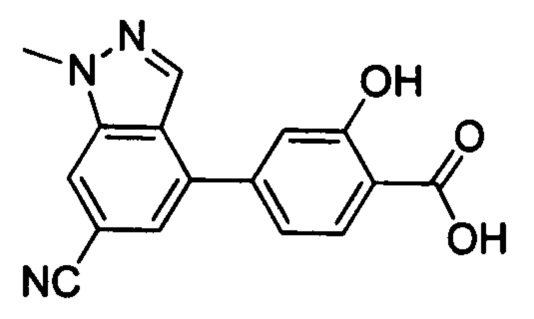
Стадия 1) Синтез 3-бром-4-метил-5-нитробензойной кислоты
Добавили в 250-мл двугорлую колбу 4-метил-3-нитробензойную кислоту (5,43 г, 30 ммоль) и концентрированную серную кислоту (45 мл, 98%), а затем порциями добавили 1,3-дибром-5,5-диметилгидантоин (4,29 г, 15 ммоль). В течение 16 часов перемешивали реакционную смесь при комнатной температуре. Вылили полученную смесь в воду со льдом (1000 мл). Экстрагировали смесь этилацетатом (150 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде белого твердого вещества (7,1 г, 91%).
МС m/z (ES-API, с положительными ионами): 260,9 [М+2]+.
Стадия 2) Синтез метил-3-бром-4-метил-5-нитробензоата
Добавили в 250-мл одногорлую колбу 3-бром-4-метил-5-нитробензойную кислоту (7,10 г, 27,3 ммоль) и метанол (100 мл), а затем по каплям добавили дихлорсульфоксид (2,96 мл, 41,0 ммоль) при 0°С. В течение 12 часов перемешивали реакционную смесь при 90°С. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный бикарбонат натрия (200 мл), и экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтого твердого вещества (6,36 г, 85%).
Стадия 3) Синтез метил-3-амино-5-бром-4-метилбензоата
Добавили в 500-мл одногорлую колбу метил-3-бром-4-метил-5-нитробензоат (8,50 г, 31,0 ммоль), уксусную кислоту (20 мл) и тетрагидрофуран (170 мл), а затем порциями добавили железо (8,68 г, 155 ммоль). В течение 12 часов перемешивали реакционную смесь при 75°С. Охладили полученную смесь до комнатной температуры, профильтровали с целью удаления нерастворимого твердого вещества, и концентрировали фильтрат в вакууме. Добавили к остатку воду (500 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого твердого вещества (5,52 г, 73%).
МС m/z (ES-API, с положительными ионами): 244,9 [М+2]+.
Стадия 4) Синтез метил-4-бром-1H-индазол-6-карбоксилата
Добавили в 250-мл одногорлую колбу 3-амино-5-бром-4-метилбензоат (6,20 г, 25,4 ммоль) и уксусную кислоту (110 мл), и по каплям добавили раствор нитрита натрия (1,90 г, 27,5 ммоль) в воде (11 мл). В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный бикарбонат натрия (500 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде оранжевого твердого вещества (4,08 г, 63%).
МС m/z (ES-API, с положительными ионами): 255,9 [М+2]+.
Стадия 5) Синтез метил-4-бром-1-метил-1H-индазол-6-карбоксилата
Добавили в 100-мл двугорлую колбу метил-4-бром-1H-индазол-6-карбоксилат (1,53 г, 6,0 ммоль), карбонат цезия (3,95 г, 12,1 ммоль) и N,N-диметилформамид (20 мл), а затем йодометан (1,1 г, 7,7 ммоль). В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Профильтровали полученную смесь, чтобы удалить нерастворимые вещества. Добавили к фильтрату насыщенный хлорид аммония (150 мл). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (1,1 г, 68%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,10 (s, 1H), 8,02 (s, 1H), 7,95 (d, J=0,7 Гц, 1H), 4,13 (s, 3H), 3,97 (s, 3H).
Стадия 6) Синтез 4-бром-1-метил-1H-индазол-6-карбоксамида
Добавили в 50-мл герметично закрытую пробирку метил-4-бром-1-метил-1H-индазол-6-карбоксилат (1,0 г, 3,7 ммоль) и аммиак (20 мл, 7 М в метаноле). В течение 24 часов осуществляли реакцию смеси при 110°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (500 мг, 53%).
МС m/z (ES-API, с положительными ионами): 254,9 [М+2]+.
Стадия 7) Синтез 4-бром-1-метил-1H-индазол-6-карбонитрила
Добавили в 100-мл одногорлую колбу 4-бром-1-метил-1H-индазол-6-карбоксамид (500 мг, 1,97 ммоль) и толуол (20 мл), а затем по каплям добавили оксихлорид фосфора (3,0 г, 19,7 ммоль). В течение 12 часов перемешивали реакционную смесь при 120°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде белого твердого вещества (440 мг, 95%).
МС m/z (ES-API, с положительными ионами): 236,9 [М+2]+.
Стадия 8) Синтез трет-бутил-4-(6-циано-1-метил-1H-индазол-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 4-бром-1-метил-1Н-индазол-6-карбонитрил (340 мг, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис (дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 2/1), и получили указанное соединение в виде белого твердого вещества (394 мг, 86%).
МС m/z (ES-API, с положительными ионами): 350,1 [М+1]+.
Стадия 9) Синтез 4-(6-циано-1-метил-1H-индазол-4-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-1-метил-1H-индазол-4-ил)-2-гидроксибензоат (394 мг, 1,13 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (222 мг, 67%).
МС m/z (ES-API, с положительными ионами): 294,1 [М+1]+; и
1H ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,39 (s, 1H), 8,27 (s, 1H), 7,88 (d, J=8,5 Гц, 1H), 7,58 (s, 1H), 7,11 (d, J=7,1 Гц, 2Н), 4,16 (s, 3H).
Пример 11
4-(6-циано-2,3-дигидро-1H-инден-4-ил)-2-гидроксибензойная кислота
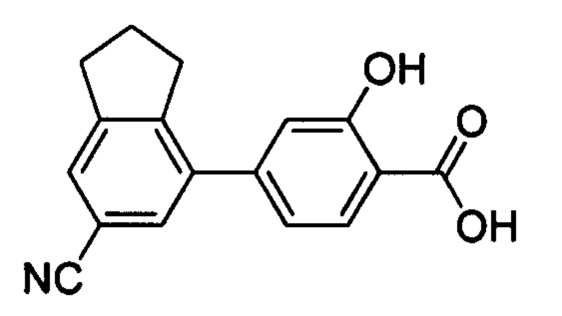
Стадия 1) Синтез 1-(7-бром-2,3-дигидро-1H-инден-5-ил)этанона
Добавили в 250-мл одногорлую колбу 1-(2,3-дигидро-1H-инден-5-ил)этанон (4,90 г, 30,6 ммоль) и дихлорметан (100 мл), и порциями добавили трихлорид алюминия (10,2 г, 76,5 ммоль) при 0°С. В течение 0,2 часа перемешивали смесь при комнатной температуре в среде азота. Затем по каплям добавили бром (2,35 мл, 45,9 ммоль), и в течение 2 часов осуществляли реакцию смеси при комнатной температуре. Медленно вылили полученную смесь в воду со льдом (500 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/200), и получили указанное соединение в виде желтой жидкости (2,56 г, 35%).
МС m/z (ES-API, с положительными ионами): 240,0 [М+2]+.
Стадия 2) Синтез 7-бром-2,3-дигидро-1H-инден-5-карбоновой кислоты
Добавили в 250-мл одногорлую колбу 1-(7-бром-2,3-дигидро-1H-инден-5-ил)этанон (2,53 г, 10,6 ммоль), 10% водный раствор гидроксида натрия (25,4 мл), 10% водный раствор гипохлорита натрия (63,5 мл). В течение 27 часов осуществляли реакцию смеси при 50°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный водный раствор тиосульфата натрия (50 мл), и экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде желтого твердого вещества (1,71 г, 67%).
МС m/z (ES-API, с отрицательными ионами): 237,9 [М-2]-.
Стадия 3) Синтез 7-бром-2,3-дигидро-1H-инден-5-карбонилхлорида
Добавили в 100-мл одногорлую колбу 7-бром-2,3-дигидро-1H-инден-5-карбоновую кислоту (1,70 г, 7,05 ммоль) и тионилхлорид (20 мл). В течение 12 часов осуществляли реакцию смеси при 90°С. Концентрировали полученную смесь в вакууме с целью удаления тионилхлорида, и получили указанное соединение в виде желтого твердого вещества (1,83 г, 100%).
Стадия 4) Синтез 7-бром-2,3-дигидро-1H-инден-5-карбоксамида
По каплям добавили раствор 7-бром-2,3-дигидро-1H-инден-5-карбонилхлорида (1,83 г, 7,05 ммоль) в дихлорметане (20 мл) к гидроксиду аммония (20 мл, 28%) в 100-мл одногорлой колбе. В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь воду (100 мл). Экстрагировали полученную смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтого твердого вещества (0,85 г, 50%).
МС m/z (ES-API, с положительными ионами): 241,0 [М+2]+.
Стадия 5) Синтез 7-бром-2,3-дигидро-1H-инден-5-карбонитрила
Добавили в 50-мл одногорлую колбу 7-бром-2,3-дигидро-1H-инден-5-карбоксамид (0,850 г, 3,54 ммоль) и толуола (20 мл), а затем оксихлорид фосфора (2,71, 17,7 ммоль). Нагрели реакционную смесь до 120°С и перемешивали в течение 12 ч. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде желтого твердого вещества (566 мг, 72%).
МС m/z (ES-API, с положительными ионами): 222,9 [М+2]+.
Стадия 6) Синтез метил-4-(6-циано-2,3-дигидро-1H-инден-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-2,3-дигидро-1H-инден-5-карбонитрил (320 мг, 1,44 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (360 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (238 мг, 62%).
МС m/z (ES-API, с положительными ионами): 294,1 [М+1]+.
Стадия 7) Синтез 4-(6-циано-2,3-дигидро-1H-инден-4-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(6-циано-2,3-дигидро-1H-инден-4-ил)-2-гидроксибензоат (238 мг, 0,81 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (162 мг, 4,05 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (195 мг, 86%).
МС m/z (ES-API, с положительными ионами): 280,1 [М+1]+; и
1H ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 7,84 (s, 1H), 7,68-7,63 (m, 2H), 7,02 (s, 2H), 2,95 (s, 4H), 2,00 (s, 2H).
Пример 12
4-(5-циано-2-(трифторметил)бензо[b]тиофен-7-ил)-2-гидроксибензойная кислота
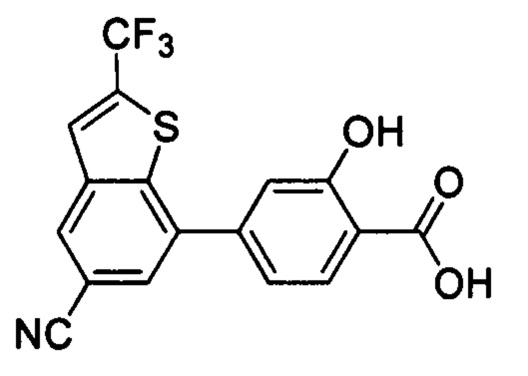
Стадия 1) Синтез 2-бром-3-метокси-5-метилбензальдегида
Добавили в 250-мл двугорлую колбу N,N,N'-триметилэтилендиамин (3,80 г, 37,2 ммоль) и тетрагидрофуран (10 мл) и, по каплям добавили n-бутиллитий (16 мл, 38,4 ммоль, 2,4 М в ТГФ) при -65°С. В течение 0,5 часа перемешивали смесь при -65°С. По каплям добавили раствор 3-метокси-5-метилбензальдегида (5,33 г, 35,3 ммоль) в тетрагидрофуране (35 мл). В течение 0,8 часа перемешивали реакционную смесь при -65°С. Затем по каплям добавили n-бутиллитий (16 мл, 38,4 ммоль, 2,4 М в ТГФ). В течение 14 часов перемешивали смесь при -20°С. Затем по каплям добавили 1,2-дибром-1,1,2,2-тетрафторэтанэтан (41,3 г, 159 ммоль), и в течение 1,5 часа перемешивали смесь при комнатной температуре. Медленно вылили полученную смесь в воду со льдом (300 мл). Подкислили смесь до pH 1-2 разбавленной соляной кислотой и экстрагировали этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтого твердого вещества (6,39 г, 79%).
1H ЯМР (400 МГц, CDCl3) δ (миллионные доли): 10,39 (s, 1H), 7,33 (s, 1H), 6,94 (s, 1H), 3,93 (s, 3Н), 2,37 (s, 3H),
Стадия 2) Синтез 1-(2-бром-3-метокси-5-метилфенил)-2,2-дихлор-3,3,3-трифторпропан-1-ола
Добавили в 100-мл двугорлую колбу 2-бром-3-метокси-5-метилбензальдегид (5,91 г, 25,8 ммоль), 1,1,1-трихлор-2,2,2-трифторэтан (9,71 г, 51,8 ммоль) и диметилсульфоксид (30 мл), и порции добавили безводный хлорид олова (7,80 г, 41,1 ммоль). В течение 4 часов перемешивали реакционную смесь при комнатной температуре. Быстро охладили полученную смесь насыщенным водным раствором хлорида аммония (200 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого масла (4,93 г, 50%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,28 (s, 1H), 6,78 (s, 1H), 6,06 (d, J=6,0 Гц, 1H), 3,90 (s, 3H), 2,39 (s, 3H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -75,35 (s, 3F).
Стадия 3) Синтез 1-(2-бром-3-метокси-5-метилфенил)-2,2-дихлор-3,3,3-трифторпропилметансульфоната
Добавили в 250-мл двугорлую колбу 1-(2-бром-3-метокси-5-метилфенил)-2,2-дихлор-3,3,3-трифторпропан-1-ол (6,11 г, 16 ммоль), триэтиламин (3,24 г, 32 ммоль) и дихлорметан (50 мл), а затем по каплям добавили метансульфонилхлорид (2,75 г, 24 ммоль) при 0°С. В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Добавили к полученной смеси насыщенный рассол (200 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (6,18 г, 84%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,32 (s, 1H), 6,83 (s, 1H), 6,81 (s, 1H), 3,91 (s, 3H), 2,81 (s, 3H), 2,41 (s, 3H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -75,41 (s, 3F).
Стадия 4) Синтез (Z)-2-бром-1-(2-хлор-3,3,3-трифторпроп-1-ен-1-ил)-3-метокси-5-метилбензола
Добавили в 100-мл одногорлую колбу 1-(2-бром-3-метокси-5-метилфенил)-2,2-дихлор-3,3,3-трифторпропилметансульфонат (4,78 г, 10,4 ммоль) и N,N-диметилформамид (25 мл), порциями добавили цинк (0,820 г, 12,5 ммоль) в среде азота, поддерживая при этом температуру на уровне не выше 50°С. В течение 0,5 часа перемешивали смесь при комнатной температуре, затем нагрели до 50°С и перемешивали в течение 0,5 часа. Охладили полученную смесь до комнатной температуры. Добавили в смесь насыщенный водный раствор хлорида аммония (200 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтого твердого вещества, 3,22 г, 94%).
1H ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,50 (s, 1H), 7,13 (s, 1H), 6,75 (s, 1H), 3,91 (s, 3Н), 2,37 (s, 3H);
и 19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -68,96.
Стадия 5) Синтез 7-метокси-5-метил-2-(трифторметил)бензо[b]тиофена
Постепенно добавили в 100-мл одногорлую колбу (Z)-2-бром-1-(2-хлор-3,3,3-трифторпроп-1-ен-1-ил)-3-метокси-5-метилбензол (4,09 г, 12,4 ммоль), нонагидросульфид натрия (5,98 г, 24,9 ммоль), йодид меди (236 мг, 1,24 ммоль) и N,N-диметилформамид (20 мл). В течение 24 часов перемешивали смесь при 80°С. Охладили полученную смесь до комнатной температуры. Добавили к смеси насыщенный рассол (200 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде белого твердого вещества (2,14 г, 70%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,58 (d, J=0,9 Гц, 1Н), 7,26 (s, 1Н), 6,70 (s, 1Н), 3,99 (s, 3Н), 2,48 (s, 3H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -56,32 (s, 3F).
Стадия 6) Синтез 7-метокси-2-(трифторметил)бензо[b]тиофен-5-карбонитрила
Постепенно добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения в среде азота 7-метокси-5-метил-2-(трифторметил)бензо[b]тиофен (1,01 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (0,669 г, 4,1 ммоль), ацетат палладия (0,045 г, 0,20 ммоль) и ацетонитрил (20 мл). В течение 48 часов перемешивали реакционную смесь при 80°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (610 мг, 58%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,81 (s, 1Н), 7,72 (d, J=0,8 Гц, 1Н), 7,02 (s, 1Н), 4,05 (s, 3Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -56,67 (s, 3F).
Стадия 7) Синтез 7-гидрокси-2-(трифторметил)бензо[b]тиофен-карбонитрила
Добавили в 100-мл одногорлую колбу 7-метокси-2-(трифторметил)бензо[b]тиофен-5-карбонитрил (2,57 г, 10 ммоль) и дихлорметан (20 мл), по каплям добавили трибромид бора (7,5 г, 30 ммоль) при -70°С. В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь ледяную воду (200 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде бледно-желтого твердого вещества (0,827 г, 34%).
МС m/z (ES-API, с отрицательными ионами): 242,0 [М-1]-.
Стадия 8) Синтез 5-циано-2-(трифторметил)бензо[b]тиофен-7-ил трифторметансульфоната
Добавили в 250-мл одногорлую колбу 7-гидрокси-2-(трифторметил)бензо[b]тиофен-карбонитрил (1,0 г, 4,1 ммоль), пиридин (0,97 г, 12,3 ммоль) и дихлорметан (80 мл) и по каплям добавили ангидрид трифторметансульфокислоты (1,35 г, 4,8 ммоль) при 0°С. В течение 1 часа перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/3), и получили указанное соединение в виде белого твердого вещества (1,31 г, 85%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,24 (s, 1H), 7,87 (s, 1H), 7,70 (d, J=0,7 Гц,1Н);и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -56,84 (s, 3F), -72,67 (s, 3F).
Стадия 9) Синтез метил-4-(5-циано-2-(трифторметил)бензо[b]тиофен-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 5-циано-2-(трифторметил)бензо [b]тиофен-7-ил трифторметансульфонат (540 мг, 1,44 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан -2-ил)бензоат (360 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль), карбонат калия (362 мг, 2,62 ммоль) и безводный 1,4-диоксан (15 мл). В течение 5 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,336 г, 68%).
МС m/z (ES-API, с положительными ионами): 378,0 [М+1]+.
Стадия 10) Синтез 4-(5-циано-2-(трифторметил)бензо[b]тиофен-7-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-(5-циано-2-(трифторметил)бензо[b]тиофен-7-ил)-2-гидроксибензоат(0,306 г, 0,81 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (0,162 г, 4,05 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь эфиром (50 мл), и подкислили водную фазу до pH 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде белого твердого вещества (0,118 г, 40%).
МС m/z (ES-API, с положительными ионами): 364,0 [М+1];
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,61 (s, 1H), 8,35 (s, 1H), 8,07 (s, 1H), 7,97 (d, J=7,9 Гц, 1H), 7,32 (d, J=9,1 Гц, 2Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -55,49 (s, 3F).
Пример 13
4-(5-циано-бензо[b]тиофен-7-ил)-2-гидроксибензойная кислота
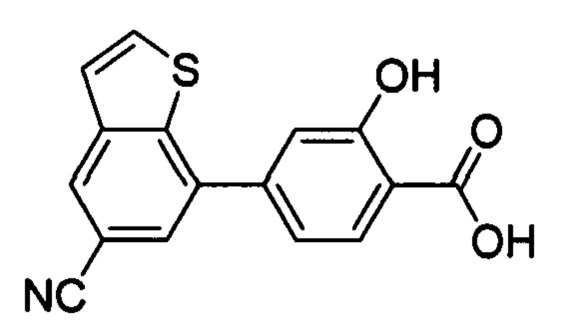
Стадия 1) Синтез метил-7-бром-5-метилбензо[b]тиофен-2-карбоксилата
Постепенно добавили в 100-мл одногорлую колбу 3-бром-2-фтор-5-метилбензальдегид (4,34 г, 20 ммоль), карбонат калия (5,53 г, 40 ммоль), этил тиогликолят (2,88 г, 24 ммоль) и N,N-диметилформамид (40 мл). В течение 12 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили полученную смесь до комнатной температуры и быстро охладили насыщенным водным раствором хлорида аммония (150 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде бледно-желтого твердого вещества (5,3 г, 93%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,04 (s, 1H), 7,60 (s, 1H), 7,45 (s, 1H), 4,41 (d, J=7,1 Гц, 2Н), 2,46 (s, 3H), 1,42 (t, J=7,1 Гц, 3Н).
Стадия 2) Синтез 7-бром-5-метилбензо[b]тиофен-2-карбоновой кислоты
Постепенно добавили в 250-мл одногорлую колбу метил-7-бром-5-метилбензо[b]тиофен-2-карбоксилат (2,0 г, 7,0 ммоль), метанол (25 мл), тетрагидрофуран (25 мл) и воду (25 мл), а затем гидроксид натрия (0,84 г, 21 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (120 мл). Промыли смесь эфиром (80 мл), подкислили водную фазу рН 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде твердого вещества белого цвета (1,8 г, 95%).
МС m/z (ES-API, с отрицательными ионами): 271,9 [М+2]+.
Стадия 3) Синтез 7-бром-5-метилбензо[b]тиофена
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-метилбензо[b]тиофен-2-карбоновую кислоту (1,79 г, 6,6 ммоль), медь (0,83 г, 13,1 ммоль) и хинолин (15 мл), нагрели смесь до 200°С и перемешивали в течение 0,5 часа. Охладили полученную смесь до комнатной температуры. Добавили в смесь концентрированную соляную кислоту (100 мл, 12 М). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (100% петролейный эфир), и получили указанное соединение в виде бесцветной жидкости (1,41 г, 94%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,56 (s, 1Н), 7,47 (d, J=5,4 Гц, 1Н), 7,35 (d, J=5,4 Гц, 2Н), 2,46 (s, 3Н).
Стадия 4) Синтез 7-бромбензо[b]тиофен-5-карбонитрила
Постепенно добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-метилбензо[b]тиофен (930 мг, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (669 мг, 4,1 ммоль), ацетат палладия (45 мг, 0,20 ммоль) и ацетонитрил (20 мл). Нагрели реакционную смесь до 80°С и перемешивали в течение 48 часов в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (185 мг, 19%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,10 (d, J=1,1 Гц, 1Н), 7,7-7,67 (m, 3,2 Гц, 2Н), 7,52 (d, J=5,5 Гц, 1Н).
Стадия 5) Синтез метил-4-(5-цианобензо[b]тиофен-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бромбензо[b]тиофен-5-карбонитрил (340 мг, 1,44 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (360 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде бледно-желтого твердого вещества (134 мг, 33%).
МС m/z (ES-API, с положительными ионами): 310,0 [М+1]+.
Стадия 6) Синтез 4-(5-цианобензо[b]тиофен-7-ил)-2-гидроксибензойной кислоты
Постепенно добавили в 100-мл одногорлую колбу метил 4-(5-цианобензо[b]тиофен-7-ил)-2-гидроксибензоат (134 мг, 0,433 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (87 мг, 2,16 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь эфиром (50 мл), и подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/40), и получили указанное соединение в виде белого твердого вещества (93 мг, 73%).
МС m/z (ES-API, с положительными ионами): 296,0 [М+1]+; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,48 (s, 1Н), 8,04 (d, J=5,3 Гц, 1Н), 7,95 (d, J=7,9 Гц, 1Н), 7,84 (s, 1Н), 7,68 (d, J=5,4 Гц, 1Н), 7,29-7,26 (m, 2Н).
Пример 14
4-(6-циано-1-изопропил-1H-индол-4-ил)-2-гидроксибензойная кислота
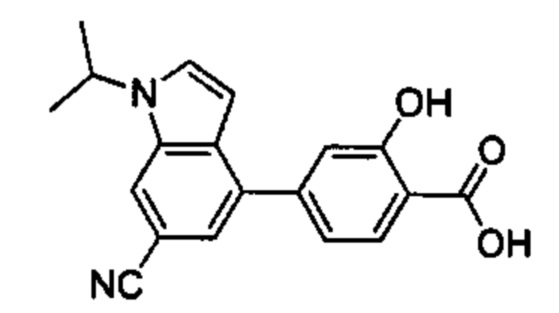
Стадия 1) Синтез 2-(2,6-дибром-4-метилфенил)ацетальдегида
Добавили в 500-мл одногорлую колбу 2,6-дибром-4-метиланилин (21,2 г, 80,0 ммоль), разбавленную соляную кислоту (100 мл, 2 М) и ацетон (150 мл), и по каплям добавили раствор нитрита натрия (6,07 г, 88 ммоль) в воде (30 мл) при 0°С. В течение 1 часа перемешивали смесь при 0°С. Затем добавили раствор этилового эфира (57,6, 800 ммоль) и ферроцена (2,98 г, 16 ммоль) в ацетоне (30 мл). После того, как в течение 1 часа перемешивали смесь при 0°С, в течение 12 часов перемешивали смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления ацетона. Добавили к остатку насыщенный водный раствор хлорида аммония (300 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого твердого вещества (10 г, 43%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 9,72 (s, 1Н), 7,40 (s, 2Н), 4,16 (s, 2Н), 2,31 (s, 3Н).
Стадия 2) Синтез 4-бром-6-метил-1H-индола
Постепенно добавили в 100-мл двухгорлую колбу 2-(2,6-дибром-4-метилфенил)ацетальдегид (10,0 г, 34 ммоль), гидроксид аммония (40 мл, 28%), оксид меди (0,49 г, 3,4 ммоль) и 1-метил-2-пирролидон (60 мл). В течение 3 часов перемешивали реакционную смесь при 60°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили в смесь насыщенный рассол (150 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде бледно-желтого твердого вещества (5,57 г, 78%).
МС m/z (ES-API, с положительными ионами): 210,9 [М+2]+.
Стадия 3) Синтез 1-(4-бром-6-метил-1Н-индол-1-ил)этанона
Постепенно добавили в 250-мл двугорлую колбу 4-бром-6-метил-1H-индол (3,6 г, 17,2 ммоль), пиридин (8, 16, 103 ммоль), 4-диметиламинопиридин (0,40 г, 3,3 ммоль) и безводный дихлорметан (80 мл), а затем добавили в среде азота уксусный оксид (7,09 г, 68,8 ммоля) при 0°С. В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде светло-желтого масла (3,9 г, 90%).
МС m/z (ES-API, с положительными ионами): 252,9 [М+2]+.
Стадия 4) Синтез 1-ацетил-4-бром-1H-индол-6-карбонитрила
Постепенно добавили в 250-мл пробирку для реакций под воздействием СВЧ-излучения 1-(4-бром-6-метил-1H-индол-1-ил)этанон (1,03 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (669 мг, 4,1 ммоль), диацетат палладия (45 мг, 0,20 ммоль) и ацетонитрил (20 мл). В течение 48 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтого твердого вещества (378 мг, 35%).
МС m/z (ES-API, с положительными ионами): 263,9 [М+2]+.
Стадия 5) Синтез 4-бром-1H-индол-6-карбонитрила
Постепенно добавили в 100-мл одногорлую колбу 1-ацетил-4-бром-1H-индол-6-карбонитрил (526 мг, 2,0 ммоль), 1,8-диазабицикло[5.4.0]-ундец-7-ен (1,22 г, 8,0 ммоль) и ацетонитрил (20 мл). В течение 24 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя, затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде бледно-желтого твердое вещество (186 мг, 42%).
МС m/z (ES-API, с положительными ионами): 221,9 [М+2]+.
Стадия 6) Синтез 4-бром-1-изопропил-1H-индол-6-карбонитрила
Добавили в 100-мл одногорлую колбу 4-бром-1H-индол-6-карбонитрил (180 мг, 0,81 ммоль) и безводный N,N-диметилформамид (10 мл), а затем добавили гидрид натрия (65 мг, 1,62 ммоль, 60%) при 0°С. В течение 0,5 часа перемешивали смесь при 0°С, а затем добавили изопропилбромид (200 мг, 1,62 ммоль). В течение 12 часов перемешивали смесь при комнатной температуре. Добавили в смесь насыщенный хлорид аммония (80 мл) и этилацетат (80 мл), и разделили полученную смесь. Промыли органическую фазу насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (81 мг, 38%).
МС m/z (ES-API, с положительными ионами): 264,0 [М+2]+.
Стадия 7) Синтез трет-бутил-4-(6-циано-1-изопропил-1H-индол-4-ил)-2-гидроксибензоата
постепенно добавили в 50-мл двугорлую колбу 4-бром-1-изопропил-1Н-индол-6-карбонитрил (380 мг, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (300 мг, 62%).
МС m/z (ES-API, с положительными ионами): 377,1 [М+1]+.
Стадия 8) Синтез 4-(6-циано-1-изопропил-1H-индол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-1-изопропил-1H-индол-4-ил)-2-гидроксибензоат (230 мг, 0,62 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя, и очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (85 мг, 43%).
МС m/z (ES-API, с отрицательными ионами): 319,1 [М-1]-; и
1Н ЯМР (600 МГц, ДМСО-d6) δ (миллионные доли): 8,18 (s, 1Н), 7,89 (d, J=3,1 Гц, 1Н), 7,85 (d, J=7,7 Гц, 1Н), 7,40 (s, 1Н), 7,0-6,99 (m, 2Н), 6,70 (d, J=3,0 Гц, 1Н), 4,96-4,91 (m, 1Н), 1,50 (d, J=6,6 Гц, 6Н).
Пример 15
4-(6-циано-1H-индол-4-ил)-2-гидроксибензойная кислота
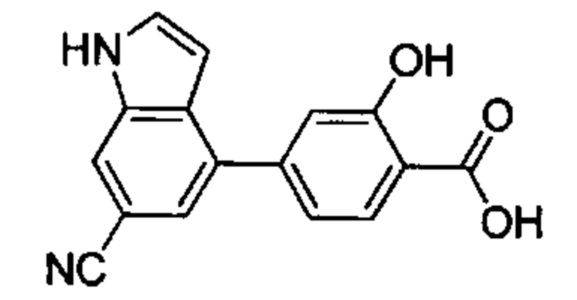
Стадия 1) Синтез трет-бутил-4-(6-циано-1H-индол-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 4-бром-1H-индол-6-карбонитрил (318 мг, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис (дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде белого твердого вещества (145 мг, 33%).
МС m/z (ES-API, с положительными ионами): 335,1 [М+1]+.
Стадия 2) Синтез 4-(6-циано-1H-индол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-1H-индол-4-ил)-2-гидроксибензоат (207 мг, 0,62 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме, чтобы удалить растворитель, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (135 мг, 78%),
MC m/z (ES-API, с отрицательными ионами): 277,1 [M-1]-; и
1Н ЯМР (600 МГц, ДМСО-d6) δ (миллионные доли): 11,91 (s, 1Н), 7,94 (s, 1Н), 7,90 (d, J=7,9 Гц, 1Н), 7,74-7,71 (m, 1Н), 7,42 (s, 1Н), 7,14-7,03 (m, 2Н), 6,68 (s, 1Н).
Пример 16
4-(6-циано-1H-бензо[d]имидазол-4-ил)-2-гидроксибензойной кислоты
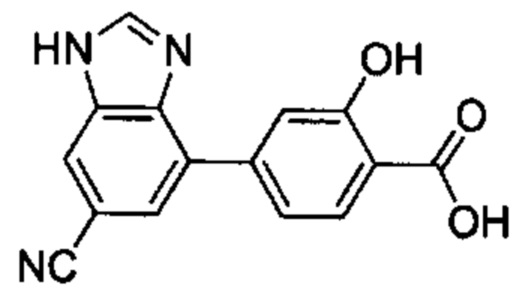
Стадия 1) Синтез трет-бутил-2'-амино-5'-циано-3-гидрокси-3'-нитро-[1,1'-бифенил]-4-карбоксилата
Постепенно добавили в 50-мл двугорлую колбу 4-амино-3-бром-5-нитробензонитрил (0,317 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде желтого твердого вещества (93 мг, 20%).
МС m/z (ES-API, с положительными ионами): 356,1 [М+1]+.
Стадия 2) Синтез трет-бутил-2',3'-диамино-5'-циано-3-гидрокси-[1,1'-бифенил]-4-карбоксилата
Добавили в 250-мл одногорлую колбу трет-бутил-2'-амино-5'-циано-3-гидрокси-3'-нитро-[1,1'-бифенил]-4-карбоксилат (0,782 г, 2,20 ммоль), дигидрохлорид олова (1,60 г, 7,10 ммоль) и этанол (80 мл). Нагрели реакционную смесь до 90°С и перемешивали в течение 12 часов. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси воду (120 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде желтого твердого вещества (0,680 г, 95%).
МС m/z (ES-API, с положительными ионами): 326,1 [М+1]+.
Стадия 3) Синтез трет-бутил-4-(6-циано-1H-бензо[d]имидазол-4-ил)-2-гидроксибензоата
Добавили в 100-мл одногорлую колбу трет-бутил-2',3'-диамино-5'-циано-3-гидрокси-[1,1'-бифенил]-4-карбоксилат (0,740 г, 2,27 ммоль) и триэтилортоформиат (20 мл). Нагрели смесь до 160°С и перемешивали в течение 3 часов. Охладили смесь до комнатной температуры и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,412 г, 54%).
МС m/z (ES-API, с положительными ионами): 336,1 [М+1]+.
Стадия 4) Синтез 4-(6-циано-1H-бензо[d]имидазол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-1H-бензо[d]имидазол-4-ил)-2-гидроксибензоат (0,369 г, 1,10 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (261 мг, 85%).
МС m/z (ES-API, с отрицательными ионами): 278,1 [М-1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 13,18 (s, 1Н), 8,56 (s, 1Н), 8,16 (s, 1Н), 7,89-7,83 (m, 3Н), 7,64 (s, 1Н).
Пример 17
4-(6-циано-бензо[d]оксазол-4-ил)-2-гидроксибензойная кислота
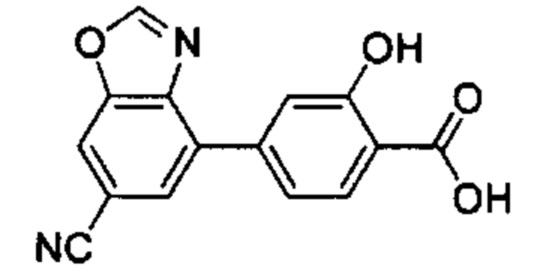
Стадия 1) Синтез 4-амино-3-бром-5-метоксибензонитрила
Добавили в 250-мл одногорлую колбу 4-амино-3-метоксибензонитрил (5,93 г, 40 ммоль), уксусную кислоту (40 мл) и метанол (100 мл), а затем по каплям добавили бром (7,03 г, 44 ммоль) при 0°С. В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь насыщенный водный раствор карбоната натрия (200 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого твердого вещества (7,63 г, 84%).
МС m/z (ES-API, с положительными ионами): 227,9 [М+2]+.
Стадия 2) Синтез 4-амино-3-бром-5-гидроксибензонитрила
Добавили в 100-мл одногорлую колбу 4-амино-3-бром-5-метоксибензонитрил (2,27 г, 10 ммоль) и дихлорметан (20 мл), а затем по каплям добавили трибромид бора (7,5 г, 30 ммоль) при -25°С. В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь насыщенный водный раствор бикарбоната натрия (200 мл) и разделили полученную смесь. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, а затем очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде бледно-желтого твердого вещества (2,02 г, 95%).
МС m/z (ES-API, с положительными ионами): 213,9 [М+2]+.
Стадия 3) Синтез трет-бутил-2'-амино-5'-циано-3,3'-дигидрокси[1,1'-бифенил]-4-карбоксилата
Постепенно добавили в 50-двугорлую колбу 4-амино-3-бром-5-гидроксибензонитрил (0,279 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде желтого твердого вещества (0.205 г, 48%).
МС m/z (ES-API, с положительными ионами): 327,1 [М+1]+.
Стадия 4) Синтез трет-бутил-4-(6-цианобензо[d]оксазол-4-ил)-2-гидроксибензоата
Добавили в 100-мл одногорлую колбу трет-бутил-2'-амино-5'-циано-3,3'-дигидрокси[1,1'-бифенил]-4-карбоксилат (0,741 г, 2,27 ммоль) и триэтилортоформиат (20 мл). Нагрели смесь до 130°С и перемешивали в течение 3 часов. Охладили смесь до комнатной температуры и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде белого твердого вещества (92 мг, 12%).
МС m/z (ES-API, с положительными ионами): 337,1 [М+1]+.
Стадия 5) Синтез 4-(6-цианобензо[d]оксазол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-цианобензо[d]оксазол-4-ил)-2-гидроксибензоат (0,370 г, 1,10 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме, чтобы удалить растворитель, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,185 г, 60%),
МС m/z (ES-API, с отрицательными ионами): 279,1 [М-1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 9,10 (s, 1Н), 8,50 (s, 1Н), 8,22 (s, 1Н), 7,93 (d, J=8,5 Гц, 1Н), 7,72 (s, 1Н), 7,63 (d, J=8,2 Гц, 1Н).
Пример 18
4-(6-циано-2-метилбензофуран-4-ил)-2-гидроксибензойная кислота
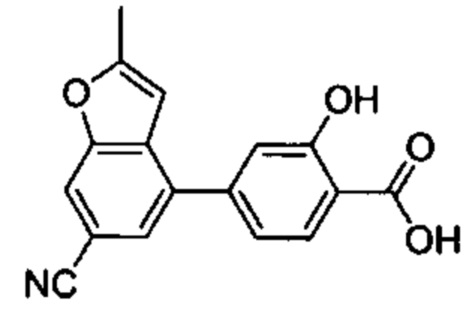
Стадия 1) Синтез метил-4-гидрокси-2-метилбензофуран-6-карбоксилата
Постепенно добавили в 500-мл двугорлую колбу метил-3,5-дигидрокси-4-иодбензоат (4,12 г, 14 ммоль), йодид меди (130 мг, 0,68 ммоль), бис(трифенилфосфин)палладий(II)хлорид (477 мг, 0,68 ммоль), пиперидин (3,58 г, 42 ммоль), 3% пропин (2,2 г, 55 ммоль) в гептане (100 мл), N,N-диметилацетамид (60 мл) и тетрагидрофуран (50 мл). В течение 24 часов перемешивали реакционную смесь при 38°С. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку насыщенный водный раствор хлорида аммония (300 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/3), и получили указанное соединение в виде коричневого твердого вещества (2,22 г, 77%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,72 (s, 1Н), 7,39 (s, 1Н), 6,51 (s, 1Н), 3,93 (s, 3Н), 2,48 (s, 3Н),
Стадия 2) Синтез 4-гидрокси-2-метилбензофуран-6-карбоновой кислоты
Постепенно добавили в 250-мл одногорлую колбу метил-4-гидрокси-2-метилбензофуран-6-карбоксилат (2,06 г, 10 ммоль), метанол (20 мл), тетрагидрофуран (20 мл) и воду (20 мл), а затем натрия гидроксида (2,0 г, 50 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (120 мл). Промыли смесь эфиром (80 мл), и подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты и экстрагировали этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде белого твердого вещества (1,84 г, 96%).
МС m/z (ES-API, с отрицательными ионами): 191,0 [М-1]+.
Стадия 3) Синтез 4-гидрокси-2-метилбензофуран-6-карбонилхлорида
Добавили в 100-мл одногорлую колбу 4-гидрокси-2-метилбензофуран-6-карбоновую кислот (1,35 г, 7,05 ммоль) и тионилхлорид (20 мл). В течение 12 часов перемешивали реакционную смесь при 90°С. Концентрировали смесь в вакууме с целью удаления тионилхлорида, и получили указанное соединение в виде коричневой вязкой жидкости (1,48 г, 100%).
Стадия 4) Синтез 4-гидрокси-2-метилбензофуран-6-карбоксамида
По каплям добавили раствор 4-гидрокси-2-метилбензофуран-6-карбонилхлорида (1,48 г, 7,05 ммоль) в дихлорметане (20 мл) к гидроксиду аммония (20 мл, 28%) в 100-мл одногорлой колбе. В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь воду (100 мл), и разделили полученную смесь. Экстрагировали водную фазу этилацетатом (80 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтого твердого вещества (1,31 г, 97%).
МС m/z (ES-API, с положительными ионами): 192,0 [М+1]+.
Стадия 5) Синтез 4-гидрокси-2-метилбензофуран-6-карбонитрила
Добавили в 50-мл одногорлую колбу 4-гидрокси-2-метилбензофуран-6-карбоновой кислоты (0,677 г, 3,54 ммоль) и толуол (20 мл), а затем по каплям добавили оксихлорид фосфора (2,71, 17,7 ммоль). В течение 12 часов перемешивали смесь при 120°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/3), и получили указанное соединение в виде бледно-желтого твердого вещества (0,490 г, 80%).
МС m/z (ES-API, с положительными ионами): 174,0 [М+1]+.
Стадия 6) Синтез 6-циано-2-метилбензофуран-4-ил трифторметансульфоната
Добавили в 250-мл одногорлую колбу 4-гидрокси-2-метилбензофуран-6-карбонитрил (0,710 г, 4,1 ммоль), пиридин (0,970 г, 12,3 ммоль) и дихлорметан (80 мл), и по каплям добавили ангидрид трифторметансульфоновой кислоты (1,35 г, 4,8 ммоль) при 0°С. В течение 1 часа перемешивали смесь при комнатной температуре. Концентрировали смесь в вакууме, и очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества, 0,726 г, 58%).
МС m/z (ES-API, с положительными ионами): 306,0 [М+1]+.
Стадия 7) Синтез трет-бутил-4-(6-циано-2-метилбензофуран-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 6-циано-2-метилбензофуран-4-ил трифторметансульфонат (0,440 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия(II) (53 мг, 0,065 ммоль), карбонат калия (0,362 г, 2,62 ммоль) и безводный 1,4 диоксан (15 мл). В течение 5 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества, 0,307 г, 67%).
МС m/z (ES-API, с положительными ионами): 350,1 [М+1]+.
Стадия 8) Синтез 4-(6-циано-2-метилбензофуран-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-2-метилбензофуран-4-ил)-2-гидроксибензоат (0,217 г, 0,62 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,167 г, 92%),
MC m/z (ES-API, с отрицательными ионами): 292,0 [M-1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 11,60 (s, 1Н), 8,21 (s, 1Н), 8,01-7,92 (m, 1Н), 7,83 (s, 1Н), 7,27 (d, J=5,1 Гц, 2Н), 6,93 (s, 1Н), 2,57 (s, 3Н).
Пример 19
4-(5-циано-2-метилбензофуран-7-ил)-2-гидроксибензойная кислота
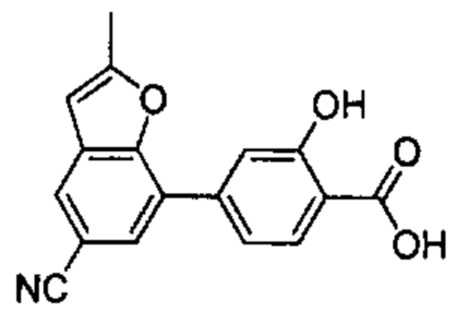
Стадия 1) Синтез 4-(аллилокси)-3-бромбензонитрила
Постепенно добавили в 250-мл двугорлую колбу 3-бром-4-гидроксибензонитрил (1,98 г, 10 ммоль), аллилбромид (2,78 г, 23 ммоль), карбонат калия (4,15 г, 30 ммоль) и ацетон (100 мл), В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Профильтровали смесь, и промыли осадок на фильтре ацетоном (50 мл). Концентрировали объединенные фильтраты в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде бесцветного масла (2,29 г, 96%).
МС m/z (ES-API, с положительными ионами): 238,9 [М+2]+.
Стадия 2) Синтез 3-аллил-5-бром-4-гидроксибензонитрила
Добавили в 50-мл одногорлую колбу 4-(аллилокси)-3-бромбензонитрил (1,50 г, 6,3 ммоль) и 1-метил-2-пирролидинон (15 мл). В течение 18 часов перемешивали реакционную смесь при 190°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде бесцветной жидкости (1,30 г, 87%).
МС m/z (ES-API, с положительными ионами): 238,9 [М+2]+.
Стадия 3) Синтез 7-бром-2-(бромметил)-2,3-дигидробензофуран-5-карбонитрила
Добавили 50-мл одногорлую колбу 3-аллил-5-бром-4-гидроксибензонитрил (0,476 г, 2,0 ммоль) и безводный тетрагидрофуран (10 мл), и добавили N-бромсукцинимид (0,399 г, 2,24 ммоль) при 0°С. В течение 18 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде белого твердого вещества (0,609 г, 96%).
МС m/z (ES-API, с положительными ионами): 317,9 [М+3]+.
Стадия 4) Синтез 7-бром-2-метилбензофуран-5-карбонитрила
Добавили в 50-мл одногорлую колбу 7-бром-2-(бромметил)-2,3-дигидробензофуран-5-карбонитрил (0,600 г, 1,89 ммоль), 1,8-диазабицикло[5.4.0] ундец-7-ен (0,320 г, 2,10 ммоль) и безводный N,N-диметилформамид (10 мл). В течение 8 часов перемешивали реакционную смесь при 55°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде белого твердого вещества (0,299 г, 67%).
1Н ЯМР (600 МГц, CDCl3) δ (миллионные доли): 7,77 (d, J=1,2 Гц, 1Н), 7,67 (d, J=1,2 Гц, 1Н), 6,55 (d, J=1,0 Гц, 1Н), 2,57 (s, 3Н).
Стадия 5) Синтез трет-бутил-4-(5-циано-2-метилбензофуран-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-2-метилбензофуран-5-карбонитрил (0,309 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М), а затем в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде бледно-желтого твердого вещества (0,416 г, 91%).
МС m/z (ES-API, с положительными ионами): 350,1 [М+1]+.
Стадия 6) Синтез 4-(5-циано-2-метилбензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-2-метилбензофуран-7-ил)-2-гидроксибензоат (0,416 г, 1,19 ммоль), дихлорметан (15 мл) и трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,286 г, 82%).
МС m/z (ES-API, с отрицательными ионами): 292,0 [М-1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 11,40 (s, 1H), 8,12 (s, 1Н), 7,95 (s, 1Н), 7,93 (d, J=8,3 Гц, 1Н), 7,50 (s, 1Н), 7,46 (d, J=8,3 Гц, 1Н), 6,80 (s, 1Н), 2,53 (s, 3Н).
Пример 20
4-(5-циано-3-(трифторметил)бензофуран-7-ил)-2-гидроксибензойная кислота
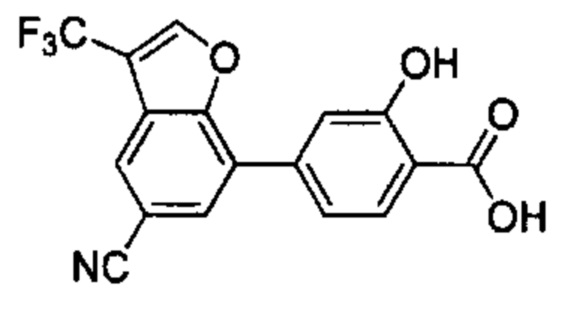
Стадия 1) Синтез 7-бром-5-метил-3-(трифторметил)-2,3-дигидробензофуран-2-ола
Добавили в 250-мл двугорлую колбу 2,2,2-трифторэтиламин (8,13 г, 60 ммоль) и дихлорметан (90 мл), и по каплям добавили раствор нитрита натрия (4,97 г, 72 ммоль) в воде (3 мл) при 0°С. В течение 1 часа перемешивали смесь при 0°С. Затем постепенно добавили 3-бром-5-метил-2-гидроксибензальдегид (2,15 г, 10,0 ммоль) и трифторэтерат бора (5,68 г, 40 ммоль) при -78°С. В течение 12 часов перемешивали реакционную смесь при -78°С, а затем в течение 12 часов перемешивали при комнатной температуре. Добавили к полученной смеси метанол (40 мл), насыщенный раствор бикарбоната натрия (200 мл) и этилацетат (200 мл), и разделили смесь. Промыли органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде желтого твердого вещества (1,93 г, 65%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,28 (s, 1Н), 7,09 (s, 1Н), 6,16-6,14 (m, 1Н), 4,04-4,02 (m, 1Н), 2,31 (s, 3Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -70,45 (s, 3F).
Стадия 2) Синтез 7-бром-5-метил-3-(трифторметил)бензофурана
Добавили в 250-мл одногорлую колбу 7-бром-5-метил-3-(трифторметил)-2,3-дигидробензофуран-2-ол (2,97 г, 10,0 ммоль) и концентрированную серную кислоту (20 мл, 98%). В течение 0,5 часа перемешивали смесь при комнатной температуре. Медленно вылили полученную смесь в воду со льдом (200 мл). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде белого твердого вещества (2,48 г, 89%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,98 (d, J=1,3 Гц, 1Н), 7,41 (s, 2Н), 2,46 (s, 3Н);и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -59,68 (s, 3F).
Стадия 3) Синтез 7-бром-3-(трифторметил)бензофуран-5-карбонитрила
Постепенно добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-метил-3-(трифторметил)бензофуран (1,14 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (669 мг, 4,1 ммоль), диацетат палладия (45 мг, 0,20 ммоль) и ацетонитрил (20 мл). В течение 48 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/30), и получили указанное соединение в виде белого твердого вещества (0,309 г, 26%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли) 8,18 (d, J=1,3 Гц, 1Н), 8,00 (s, 1Н), 7,88 (d, J=1,2 Гц, 1Н).
Стадия 4) Синтез трет-бутил-4-(5-циано-3-((трифторметил)бензофуран-7-ил)-2-гидроксибензоата
Постепенно добавили на 50-мл двугорлую колбу 7-бром-3-(трифторметил)бензофуран-5-карбонитрил (0,380 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М), а затем в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,328 г, 62%).
МС m/z (ES-API, с положительными ионами): 404,1 [М+1]+.
Стадия 5) Синтез 4-(5-циано-3-(трифторметил)бензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-3-((трифторметил)бензофуран-7-ил)-2-гидроксибензоат (0,480 г, 1,19 ммоль), дихлорметан (15 мл) и трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,347 г, 84%).
МС m/z (ES-API, с отрицательными ионами): 346,0 [М-1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 9,09 (d, J=1,4 Гц, 1Н), 8,36 (s, 1Н), 8,25 (d, J=1,3 Гц, 1Н), 7,95 (d, J=8,2 Гц, 1Н), 7,52 (d, J=1,5 Гц, 1Н), 7,48-7,45 (m, 1Н); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -58,00 (s, 3F).
Пример 21
4-(6-цианобензофуран-4-ил)-2-гидроксибензойная кислота
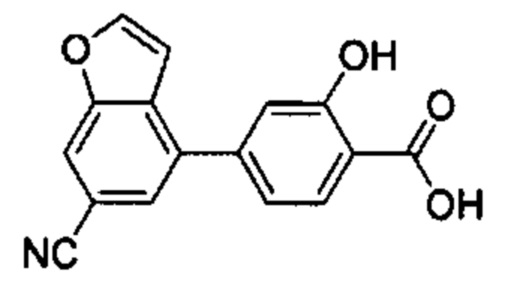
Стадия 1) Синтез 3-(этоксикарбонил)-4-(фуран-2-ил)-3-бутеновой кислоты
Добавили в 1000-мл двугорлую колбу трет-бутоксид калия (126,0 г, 1123 ммоль) и трет-бутанол (350 мл), а затем по каплям добавили смесь диэтилового эфира янтарной кислоты (294 г, 1688 ммоль) и фуран-2-карбальдегида (36,0 г, 375 ммоль). В течение 3 часов перемешивали реакционную смесь при 110°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления трет-бутанола. Добавили к остатку разбавленную соляную кислоту (1000 мл, 6 М). Экстрагировали смесь диэтиловым эфиром (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде бледно-желтого твердого вещества (83,2 г, 99%).
МС m/z (ES-API, с отрицательными ионами): 223,1 [М-1]-.
Стадия 2) Синтез этил-4-ацетоксибензофуран-6-карбоксилата
Добавили в 1000-мл одногорлую колбу 3-(этоксикарбонил)-4-(фуран-2-ил)-3-бутеновую кислоту (84,1 г, 375 ммоль), ацетат натрия (123 г, 1499 ммоль) и уксусный ангидрид (350 мл). В течение 5 часов перемешивали реакционную смесь при 180°С. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме с целью удаления уксусного ангидрида. Добавили к остатку 15% водный раствор карбоната натрия с целью доведения рН до слабощелочного уровня. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде светло-желтого масла (55,6 г, 60%).
МС m/z (ES-API, с положительными ионами): 249,1 [М+1]+.
Стадия 3) Синтез 4-гидроксибензофуран-6-карбоновой кислоты
Добавили в 1000-мл одногорлую колбу этил-4-ацетоксибензофуран-6-карбоксилат (24,8 г, 100 ммоль), метанол (100 мл), тетрагидрофуран (100 мл), воду (100 мл) и гидроксид натрия (12 г, 300 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (600 мл). Промыли смесь диэтиловым эфиром (200 мл), подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты и профильтровали. Промыли осадок на фильтре водой (300 мл × 2), высушили, и получили указанное соединение в виде белого твердого вещества (16,9 г, 95%).
МС m/z (ES-API, с отрицательными ионами): 177,0 [М-1]-.
Стадия 4) Синтез метил-4-метоксибензофуран-6-карбоксилата
Добавили в 100-мл одногорлую колбу 4-гидроксибензофуран-6-карбоновую кислоту (1,78 г, 10 ммоль), карбонат цезия (8,15 г, 25 ммоль) и N,N-диметилформамид (20 мл), и по каплям добавили йодометан (3,12 г, 22 ммоль). В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Быстро охладили смесь насыщенным раствором хлорида аммония (200 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (1,92 г, 93%).
МС m/z (ES-API, с положительными ионами): 207,1 [М+1]+.
Стадия 5) Синтез 4-метоксибензофуран-6-карбоновой кислоты
Постепенно добавили в 100-мл одногорлую колбу метил-4-метоксибензофуран-6-карбоновую кислоту (2,06 г, 10 ммоль), метанол (10 мл), тетрагидрофуран (10 мл) и воду (10 мл), а затем гидроксид натрия (1,2 г, 30 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (80 мл). Промыли смесь эфиром (80 мл), подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты и профильтровали. Промыли осадок на фильтре водой (60 мл × 2), высушили, и получили указанное соединение в виде белого твердого вещества (1,75 г, 91%).
МС m/z (ES-API, с отрицательными ионами): 191,0 [М-1]-.
Стадия 6) Синтез 4-метоксибензофуран-6-карбонилхлорида
Добавили в 100-мл одногорлую колбу 4-метоксибензофуран-6-карбоновую кислоту (1,35 г, 7,05 ммоль) и тионилхлорид (20 мл). В течение 12 часов перемешивали реакционную смесь при 90°С. Концентрировали смесь в вакууме с целью удаления тионилхлорида, и получили указанное соединение в виде коричневой вязкой жидкости (1,48 г, 100%).
Стадия 7) Синтез 4-метоксибензофуран-6-карбоксамида
По каплям добавили раствор 4-метоксибензофуран-6-карбонилхлорида (1,48 г, 7,05 ммоль) в дихлорметане (20 мл) к гидроксиду аммония (20 мл, 28%) в 100-мл одногорлой колбе. В течение 2 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь воду (100 мл). Экстрагировали водную фазу этилацетатом (80 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/дихлорметан (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтого твердого вещества (1,31 г, 97%).
МС m/z (ES-API, с положительными ионами): 192,0 [М+1]+.
Стадия 8) Синтез 4-метоксибензофуран-6-карбонитрила
Добавили в 50-мл одногорлую колбу 4-метоксибензофуран-6-карбоксамид (0,677 г, 3,54 ммоль) и толуол (20 мл), а затем по каплям добавили оксихлорид фосфора (2,71, 17,7 ммоль). В течение 12 часов перемешивали смесь при 120°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл), и разделили смесь. Экстрагировали водную фазу этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/3), и получили указанное соединение в виде бледно-желтого твердого вещества (0,490 г, 80%).
МС m/z (ES-API, с положительными ионами): 174,0 [М+1]+.
Стадия 9) Синтез 4-гидроксибензофуран-6-карбонитрила
Добавили в 100-мл одногорлую колбу 4-метоксибензофуран-6-карбонитрил (1,73 г, 10 ммоль) и дихлорметан (20 мл), а затем по каплям добавили трибромид бора (7,5 г, 30 ммоль) при -70°С. В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь ледяную воду (200 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде бледно-желтого твердого вещества (1,42 г, 89%).
МС m/z (ES-API, с отрицательными ионами): 158,0 [М-1]-.
Стадия 10) Синтез 6-цианобензофуран-4-ил трифторметансульфонат
Добавили в 250-мл одногорлую колбу 4-гидроксибензофуран-6-карбонитрил (0,652 г, 4,1 ммоль), пиридин (0,970 г, 12,3 ммоль) и дихлорметан (80 мл), а затем по каплям добавили ангидрид трифторметансульфоновой кислоты (1,35 г, 4,8 ммоль) при 0°С. В течение 1 часа перемешивали смесь при комнатной температуре. Концентрировали смесь в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде белого твердого вещества (0,991 г, 83%).
МС m/z (ES-API, с положительными ионами): 291,9 [М+1]+.
Стадия 11) Синтез трет-бутил-4-(6-цианобензофуран-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 6-цианобензофуран-4-ил трифторметансульфонат (0,419 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль), карбонат калия (0,362 г, 2,62 ммоль) и безводный 1,4-диоксан (15 мл). В течение 7 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества, 0,185 г, 42%).
МС m/z (ES-API, с положительными ионами): 336,1 [М+1]+.
Стадия 12) Синтез 4-(6-цианобензофуран-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-цианобензофуран-4-ил)-2-гидроксибензоат (0,185 г, 0,552 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,128 г, 83%),
МС m/z (ES-API, с отрицательными ионами): 278,0 [М-1]-; и
1Н ЯМР (600 МГц, ДМСО-d6) δ (миллионные доли): 11,44 (s, 1Н), 8,39 (d, J=2,2 Гц, 1Н), 8,33 (s, 1Н), 7,95 (d, J=8,5 Гц, 1Н), 7,88 (d, J=1,0 Гц, 1H), 7,27-7,26 (m, 2Н), 7,22 (d, J=1,3 Гц, 1Н).
Пример 22
4-(5-цианобензофуран-7-ил)-2-(трифторметил)бензойная кислота
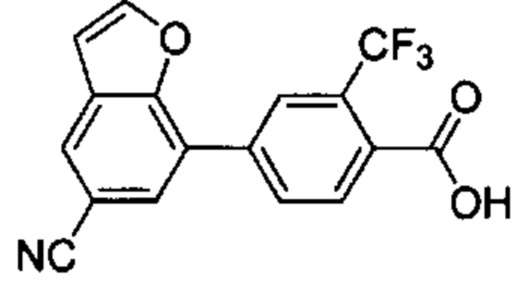
Стадия 1) Синтез 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензофуран-5-карбонитрила
Постепенно добавили в 50-мл двугорлую колбу 7-бром-5-карбонитрил (2,2 г, 10 ммоль), бис(пинаколато)дибор (3,30 г, 13 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (816 мг, 1,0 ммоль), безводный ацетат калия (3,43 г, 35 ммоль) и N,N-диметилформамид (15 мл). В течение 6 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (100 мл). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (2,29 г, 85%).
МС m/z (ES-API, с положительными ионами): 270,1 [М+1]+.
Стадия 2) Синтез метил-4-(5-цианобензофуран-7-ил)-2-(трифторметил)бензоата
Постепенно добавили в 50-мл двугорлую колбу метил-4-бром-2-(трифторметил)бензоат (0,408 г, 1,44 ммоль), 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензофуран-5-карбонитрил (0,353 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде белого твердого вещества (0,348 г, 77%).
МС m/z (ES-API, с положительными ионами): 346,1 [М+1]+.
Стадия 3) Синтез 4-(5-цианобензофуран-7-ил)-2-(трифторметил)бензойной кислоты
Добавили в 100-мл одногорлую колбу метил-4-(5-цианобензофуран-7-ил)-2-(трифторметил)бензоат (0,345 г, 0,99 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем добавили гидроксид натрия (0,4 г, 9,9 ммоля). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь диэтиловым эфиром (50 мл), и подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты. Экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,243 г, 74%).
МС m/z (ES-API, с положительными ионами): 332,0 [М+1]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,34-8,30 (m, 4Н), 8,18 (s, 1Н), 7,99 (d, J=7,1 Гц, 1Н), 7,20 (s, 1Н); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -58,05.
Пример 23
4-(5-цианобензофуран-7-ил)бензойная кислота
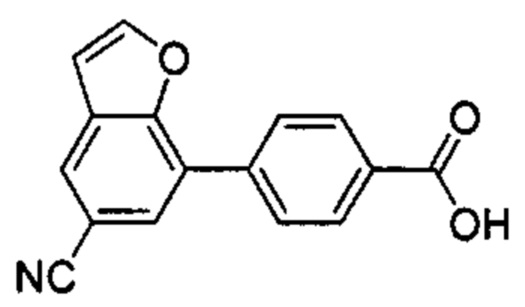
Стадия 1) Синтез метил-4-(5-цианобензофуран-7-ил)бензоата
Постепенно добавили в 50-мл двугорлую колбу метил-4-бромбензоат (0,310 г, 1,44 ммоль), 7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензофуран-5-карбонитрил (0,353 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М), а затем в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,272 г, 75%).
МС m/z (ES-API, с положительными ионами): 278,1 [М+1]+.
Стадия 2) Синтез 4-(5-цианобензофуран-7-ил)бензойной кислоты
Добавили в 100-мл одногорлую колбу метил-4-(5-цианобензофуран-7-ил)бензоат (0,274 г, 0,99 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем гидроксид натрия (0,4 г, 9,9 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (60 мл). Промыли смесь диэтиловым эфиром (50 мл), и подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты. Экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,227 г, 87%).
МС m/z (ES-API, с положительными ионами): 264,1 [М+1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 13,13 (s, 1Н), 8,31-8,29 (m, 2Н), 8,12-8,10 (m, 2Н), 8,07-8,05 (m, 3Н), 7,21 (d, J=2,0, 1Н).
Пример 24
2-гидрокси-4-(5-(трифторметил)бензофуран-7-ил)бензойная кислота
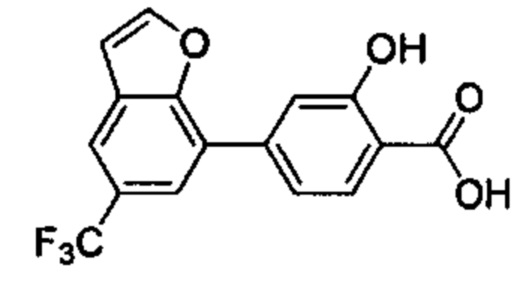
Стадия 1) Синтез 2-бром-1-(2,2-диэтоксиэтокси)-4-(трифторметил)бензола
Постепенно добавили в 100-мл одногорлую колбу 2-бром-4-(трифторметил)фенол (10,4 г, 43,3 ммоль), 2-бром-1,1-диэтоксиэтан (8,06 мл, 52,0 ммоль), карбонат цезия (28,2 г, 86,6 ммоль) и безводный N,N-диметилформамид (60 мл). В течение 18 часов перемешивали реакционную смесь при 150°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный водный раствор хлорида аммония (200 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/30), и получили указанное соединение в виде бледно-желтой жидкости (10,4 г, 67%).
Стадия 2) Синтез 7-бром-5-(трифторметил)бензофурана
Добавили в 250-мл одногорлую колбу полифосфорную кислоту (5,00 г) и хлорбензол (40 мл), и нагрели смесь с целью дефлегмации в среде азота. Затем по каплям добавили раствор 2-бром-1-(2,2-диэтоксиэтокси)-4-(трифторметил)бензола (3,79 г, 10,6 ммоль) в хлорбензоле (30 мл). В течение 2 часов перемешивали реакционную смесь при 145°С. Охладили полученную смесь до комнатной температуры. Вылили верхний органический слой и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/30), и получили указанное соединение в виде бесцветной жидкости (0,478 г, 17%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,87 (s, 1Н), 7,81 (s, 1Н), 7,76 (s, 1Н), 6,95 (d, J=2,0 Гц, 1Н).
Стадия 3) Синтез трет-бутил-2-гидрокси-4-(5-(трифторметил)бензофуран-7-ил)бензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-5-(трифторметил)бензофуран (0,347 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,287 г, 58%).
МС m/z (ES-API, с положительными ионами): 379,1 [М+1]+.
Стадия 4) Синтез 2-гидрокси-4-(5-(трифторметил)бензофуран-7-ил)бензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-2-гидрокси-4-(5-(трифторметил)бензофуран-7-ил)бензоат (0,287 г, 0,76 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,171 г, 70%),
МС m/z (ES-API, с отрицательными ионами): 321,0 [М-1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,27 (s, 1Н), 8,15 (s, 1Н), 7,94 (s, 1Н), 7,85 (s, 1Н), 7,51-7,47 (m, 2Н), 7,19 (s, 1Н); и
19F ЯМР (376 МГц, ДМСО-(d6) δ (миллионные доли): -59,32 (s, 3F).
Пример 25
4-(6-циано-2-(трифторметил)бензофуран-4-ил)-2-гидроксибензойная кислота
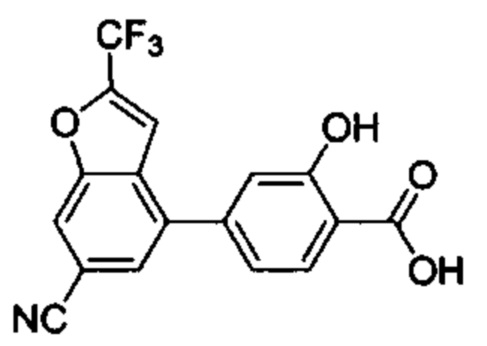
Стадия 1) Синтез этил(Z)-3-бром-2-(2-хлор-3,3,3-трифтор-1-пропен-1-ил)-5-метил-фенилметилацетата
Постепенно добавили в 100-мл одногорлую колбу 2-бром-6-гидрокси-4-метилбензальдегид (5,60 г, 26 ммоль), цинк (8,50 г, 130 ммоль), уксусный ангидрид (7,96 г, 78 ммоль) и N,N-диметилформамид (30 мл), а затем по каплям добавили 1,1,1-трихлортрифторэтан (7,80 г, 41,1 ммоль) в среде азота при поддержании температуры реакции не выше 50°С. В течение 4 часов перемешивали реакционную смесь при комнатной температуре. Добавили к полученной смеси насыщенный водный раствор хлорида аммония (200 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде бледно-желтой жидкости (1,49 г, 16%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,36 (s, 1Н), 7,13 (s, 1Н), 6,98 (s, 1Н), 2,37 (s, 3Н), 2,23 (s, 3Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -69,00 (s, 3F).
Стадия 2) Синтез 4-бром-6-метил-2-(трифторметил)бензофурана
Добавили в 100-мл одногорлую колбу (Z)-3-бром-2-(2-хлор-3,3,3-трифтор-1-пропен-1-ил)-5-метилфенилацетат (4,00 г, 11,2 ммоль) и N,N-диметилформамид (25 мл), а затем порциями добавили трет-бутоксид калия (3,77 г, 33,6 ммоль) при 0°С. В течение 6 часов перемешивали реакционную смесь при комнатной температуре. Добавили к полученной смеси насыщенный хлорид аммония (200 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде белого твердого вещества (1,66 г, 53%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,34 (s, 1Н), 7,31 (s, 1Н), 7,16 (s, 1Н), 2,48 (s, 3Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -64,90 (s, 3F).
Стадия 3) Синтез 4-бром-2-(трифторметил)бензофуран-6-карбонитрила
Постепенно добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 4-бром-6-метил-2-(трифторметил)бензофуран (1,14 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (669 мг, 4,1 ммоль), диацетат палладия (45 мг, 0,20 ммоль) и ацетонитрил (20 мл). В течение 48 часов перемешивали реакционную смесь при 80°С в среде азота. Охладили полученную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде белого твердого вещества (0,333 г, 28%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,88 (s, 1Н), 7,78 (s, 1Н), 7,31 (s, 1Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -65,20 (s, 3F).
Стадия 4) Синтез трет-бутил-4-(6-циано-2-(трифторметил)бензофуран-4-ил)-2-гидроксибензоата
Постепенно добавили на 50-мл двугорлую колбу 4-бром-2-(трифторметил) бензофуран-6-карбонитрил (0,380 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде белого твердого вещества (0,380 г, 72%).
МС m/z (ES-API, с положительными ионами): 404,1 [М+1]+.
Стадия 5) Синтез 4-(6-циано-2-(трифторметил)бензофуран-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-2-(трифторметил)бензофуран-4-ил)-2-гидроксибензоат (0,307 г, 0,76 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества, 0,185 г, 70%).
МС m/z (ES-API, с отрицательными ионами): 346,0 [М-1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,54 (s, 1Н), 7,98 (m 8,03-7,94, 3Н), 7,28 (m, 7,30-7,27, 2Н); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -63,70 (s, 3F).
Пример 26
2-гидрокси-4-(5-нитробензофуран-7-ил)бензойная кислота
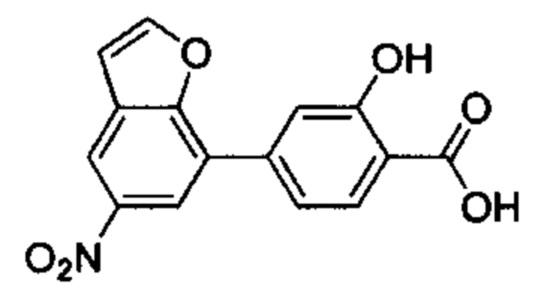
Стадия 1) Синтез 3-бром-2-гидрокси-5-нитробензальдегида
Добавили в 100-мл двугорлую колбу 2-гидрокси-5-нитро-бензальдегид (2,0 г, 12 ммоль) и дихлорметан (30 мл), а затем по каплям добавили бром (2,24 г, 14 ммоль). В течение 1 часа перемешивали смесь при комнатной температуре. Быстро охладили смесь насыщенным водным раствором тиосульфата натрия (100 мл), и разделили полученную смесь. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде твердого вещества желтого цвета (2,89 г, 98%).
МС m/z (ES-API, с положительными ионами): 246,9 [М+2]+.
Стадия 2) Синтез этил-7-бром-5-нитробензофуран-2-карбоксилата
Постепенно добавили в 250-мл одногорлую колбу 3-бром-2-гидрокси-5-нитробензальдегид (4,92 г, 20 ммоль), карбонат калия (5,53 г, 40 ммоль), диэтил броммалонат (5,74 г, 24 ммоль) и бутанон (40 мл). В течение 2 часов перемешивали реакционную смесь при 85°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный хлорид аммония (150 мл). Экстрагировали смесь этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде бледно-желтого твердого вещества (3,77 г, 60%).
МС m/z (ES-API, с положительными ионами): 314,9 [М+2]+.
Стадия 3)
Синтез 7-бром-5-нитробензофуран-2-карбоновой кислоты
Добавили в 250-мл одногорлую колбу этил-7-бром-5-нитробензофуран-2-карбоксилат (2,20 г, 7,0 ммоль), метанол (25 мл), тетрагидрофуран (25 мл) и воду (25 мл), а затем гидроксид натрия (0,840 г, 21 ммоль). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя. Добавили к остатку воду (120 мл). Промыли смесь диэтиловым эфиром (80 мл), и подкислили водную фазу до рН 1 с помощью 2 N разбавленной соляной кислоты. Экстрагировали полученную смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, и получили указанное соединение в виде желтого твердого вещества (1,90 г, 95%).
МС m/z (ES-API, с отрицательными ионами): 282,9 [М-2]-.
Стадия 4) Синтез 7-бром-5-нитробензофурана
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-нитробензофуран-2-карбоновую кислоту (1,89 г, 6,6 ммоль), медь (0,83 г, 13,1 ммоль) и хинолин (15 мл), нагрели смесь до 200°С и перемешивали в течение 0,5 часа. Охладили полученную смесь до комнатной температуры. Добавили в смесь в концентрированную соляную кислоту (100 мл, 12 М). Экстрагировали смесь этилацетатом (80 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде оранжевого твердого вещества (0,671 г, 42%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,50 (d, J=2,0 Гц, 1Н), 8,43 (d, J=2,0 Гц, 1Н), 7,86 (d, J=2,1 Гц, 1Н), 7,02 (d, J=2,2 Гц, 1Н).
Стадия 5) Синтез трет-бутил-2-гидрокси-4-(5-нитробензофуран-7-ил)бензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-5-нитробензофуран (0,317 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия(II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,275 г, 59%).
МС m/z (ES-API, с положительными ионами): 356,1 [М+1]+.
Стадия 6) Синтез 2-гидрокси-4-(5-нитробензофуран-7-ил)бензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-2-гидрокси-4-(5-нитробензофуран-7-ил)бензоат (0,275 г, 0,77 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,177 г, 77%).
МС m/z (ES-API, с отрицательными ионами): 298,0 [М-1]-.
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,69 (d, J=2,1 Гц, 1Н), 8,38 (d, J=2,1 Гц, 1Н), 8,33 (d, J=1,9 Гц, 1Н), 7,97 (d, J=8,2 Гц, 1Н), 7,53 (d, J=1,1 Гц, 1Н), 7,49 (d, J=8,3 Гц, 1H), 7,30 (d, J=2,0 Гц, 1Н).
Пример 27
4-(2-хлор-6-цианобензофуран-4-ил)-2-гидроксибензойная кислота
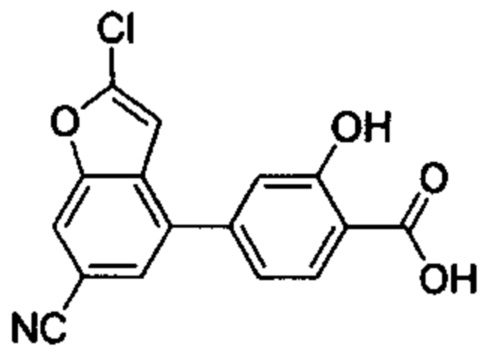
Стадия 1) Синтез 2-хлор-4-метоксибензофуран-6-карбонитрила
Добавили в 100-мл двугорлую колбу 4-метоксибензофуран-6-карбонитрил (1,73 г, 10 ммоль) и тетрагидрофуран (30 мл), а затем по каплям n-бутиллитий (5,0 мл, 12 ммоль, 2,4 М в ТГФ) добавили при -70°С. В течение 1,5 часа перемешивали смесь при -70°С в среде азота. По каплям добавили раствор гексахлорэтана (4,73 г, 20 ммоль) в тетрагидрофуране (30 мл). В течение 12 часов перемешивали смесь при комнатной температуре. Добавили в смесь насыщенный водный раствор хлорида аммония (800 мл). Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (1,35 г, 65%).
МС m/z (ES-API, с положительными ионами): 208,0 [М+1]+.
Стадия 2) Синтез 2-хлор-4-гидроксибензофуран-6-карбонитрила
Добавили в 100-мл одногорлую колбу 2-хлор-4-метоксибензофуран-6-карбонитрил (2,08 г, 10 ммоль) и дихлорметан (20 мл), и по каплям добавили трибромид бора (7,5 г, 30 ммоль) при -70°С С. В течение 24 часов
перемешивали реакционную смесь при комнатной температуре. Добавили в смесь ледяную воду (200 мл), и разделили полученную смесь. Экстрагировали водную фазу этилацетатом (100 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде бледно-желтого твердого вещества (1,78 г, 92%).
МС m/z (ES-API, с отрицательными ионами): 191,9 [М-1]-.
Стадия 3) Синтез 2-хлор-6-цианобензофуран-4-ил трифторметансульфоната
Добавили в 250-мл одногорлую колбу 2-хлор-4-гидроксибензофуран-6-карбонитрил (0,794 г, 4,1 ммоль), пиридин (0,970 г, 12,3 ммоль) и дихлорметан (80 мл), а затем по каплям добавили ангидрид трифторметансульфоновой кислоты (1,35 г, 4,8 ммоль) при 0°С. В течение 1 часа перемешивали смесь при комнатной температуре. Концентрировали смесь в вакууме, и очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде белого твердого вещества (1,13 г, 85%).
МС m/z (ES-API, с положительными ионами): 325,9 [М+1]+.
Стадия 4) Синтез трет-бутил-4-(2-хлор-6-цианобензофуран-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 2-хлор-6-цианобензофуран-4-ил-трифторметансульфонат (0,469 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль), карбонат калия (0,362 г, 2,62 ммоль) и безводный 1,4-диоксан (15 мл).
В течение 7 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,194 г, 40%).
МС m/z (ES-API, с положительными ионами): 370,1 [М + 1]+.
Стадия 5) Синтез 4-(2-хлор-6-цианобензофуран-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(2-хлор-6-цианобензофуран-4-ил)-2-гидроксибензоат (0,194 г, 0,524 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,141 г, 86%).
МС m/z (ES-API, с отрицательными ионами): 312,0 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,31 (s, 1H), 7,91 (s, 2H), 7,32 (s, 1H), 7,24 (s, 2H).
Пример 28
4-(6-циано-2-фторбензофуран-4-ил)-2-гидроксибензойная кислота
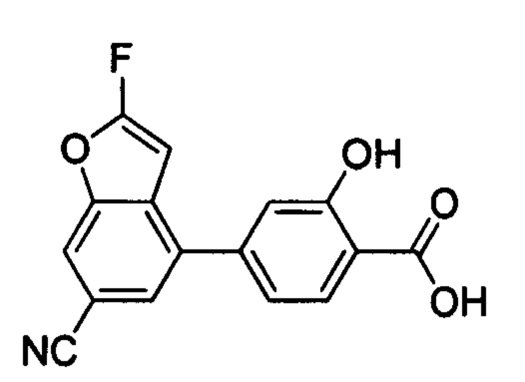
Стадия 1) Синтез 2-фтор-4-метоксибензофуран-6-карбонитрила
Добавили в 1000-мл двугорлую колбу 4-метоксибензофуран-6-карбонитрил (17,3 г, 100 ммоль) и тетрагидрофуран (300 мл), а затем по каплям добавили n-бутиллитий (50 мл, 120 ммоль, 2,4 М в ТГФ) при -70°С. В течение 1,5 часа перемешивали смесь при -70°С в среде азота. Раствор N-фторбензолсульфонимида (63,1 г, 200 ммоль) в тетрагидрофуране (200 мл) по каплям добавили. В течение 12 часов перемешивали смесь при комнатной температуре. Добавили в смесь насыщенный водный раствор хлорида аммония (800 мл). Экстрагировали смесь этилацетатом (300 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (300 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили продукт. Дополнительно очистили неочищенный продукт путем препаративной хроматографии с обращенной фазой (H2O/CH3CN, 0,1% ТФК), и получили указанное соединение в виде белого твердого вещества (0,956 г, 5%).
МС m/z (ES-API, с положительными ионами): 192,0 [М + 1]+.
Стадия 2) Синтез 2-фтор-4-гидроксибензофуран-6-карбонитрила
Добавили в 100-мл одногорлую колбу 2-фтор-4-метоксибензофуран-6-карбонитрил (0,208 г, 1,0 ммоль) и дихлорметан (10 мл), а затем по каплям добавили трибромид бора (0,75 г, 3,0 ммоль) при -70°С. В течение 24 часов перемешивали реакционную смесь при комнатной температуре. Добавили в смесь ледяную воду (80 мл), и разделили полученную смесь. Экстрагировали водную фазу этилацетатом (60 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме, очистили остаток путем хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/6), и получили указанное соединение в виде белого твердого вещества (62 мг, 35%).
МС m/z (ES-API, с отрицательными ионами): 176,0 [М - 1]-.
Стадия 3) Синтез 6-циано-2-фторбензофуран-4-ил трифторметансульфоната
Добавили в 250-мл одногорлую колбу 2-фтор-4-гидроксибензофуран-6-карбонитрил (0,726 г, 4,1 ммоль), пиридин (0,970 г, 12,3 ммоль) и дихлорметан (80 мл), а затем по каплям добавили ангидрид трифторметансульфоновой кислоты (1,35 г, 4,8 ммоль) при 0°С. В течение 1 часа перемешивали смесь при комнатной температуре. Концентрировали смесь в вакууме, очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде твердого вещества белого цвета (1,09 г, 86%).
МС m/z (ES-API, с положительными ионами): 309,9 [М + 1]+.
Стадия 4) Синтез трет-бутил-4-(6-циано-2-фторбензофуран-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 6-циано-2-фторбензофуран-4-ил трифторметансульфонат (0,405 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль), карбонат калия (0,362 г, 2,62 ммоль) и безводный 1,4-диоксан (15 мл). В течение 7 часов перемешивали реакционную смесь при 90°С в среде азота. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,222 г, 48%).
МС m/z (ES-API, с положительными ионами): 354,1 [М + 1]+.
Стадия 5) Синтез 4-(6-циано-2-фторбензофуран-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-2-фторбензофуран-4-ил)-2-гидроксибензоат (0,222 г, 0,629 ммоль) и дихлорметан (15 мл), а затем трифторацетат (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,141 г, 86%).
МС m/z (ES-API, с отрицательными ионами): 296,0 [М - 1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,31 (s, 1H), 7,93 (s, 2H), 7,23-7,21 (m, 2H), 6,70 (d, J=5,9 Гц, 1H); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): - 106,40 (s, 1F).
Пример 29
4-(5-циано-2-фторбензофуран-7-ил)-2-гидроксибензойная кислота
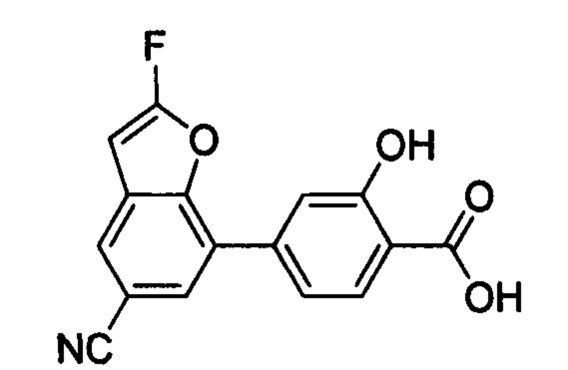
Стадия 1) Синтез 7-бром-2-фторбензофуран-5-карбонитрила
Добавили в 1000-мл двугорлую колбу 7-бромбензофуран-5-карбонитрил (22,2 г, 100 ммоль) и тетрагидрофуран (300 мл), а затем по каплям добавили диизопропиламид лития (60 мл, 120 ммоль, 2,0 М в ТГФ) при -70°С. В течение 1,5 часа перемешивали смесь при -70°С в среде азота. Затем добавили раствор N-фторбензолсульфонимида (63,1 г, 200 ммоль) в тетрагидрофуране (200 мл) по каплям добавили. В течение 12 часов перемешивали смесь при комнатной температуре. Добавили в смесь насыщенный хлорид аммония (800 мл). Экстрагировали смесь этилацетатом (300 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (300 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), получили неочищенный продукт. Дополнительно очистили неочищенный продукт путем препаративной хроматографии с обращенной фазой (H2O/CH3CN, 0,1% ТФК), и получили указанное соединение в виде белого твердого вещества (1,44 г, 6%).
1H ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,77 (s, 1Н), 7,71 (s, 1H), 6,07 (d, J = 6,6 Гц, 1Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -106,46 (s, 1F).
Стадия 2) Синтез трет-бутил-4-(5-циано-2-фторбензофуран-7-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 7-бром-2-фторбензофуран-5-карбонитрил (0,338 г, 1,41 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,245 г, 53%).
МС m/z (ES-API, с положительными ионами): 354,1 [М + 1]+.
Стадия 3) Синтез 4-(5-циано-2-фторбензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-2-фторбензофуран-7-ил)-2-гидроксибензоат (0,245 г, 0,69 ммоль) и дихлорметан (15 мл), а затем трифторуксусную кислоту (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,177 г, 77%),
МС m/z (ES-API, с отрицательными ионами): 296,0 [М - 1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,18 (s, 1H), 8,04 (s, 1H), 7,93 (d, J = 8,2 Гц, 1H), 7,44 (s, 1H), 7,41 (d, J = 8,2 Гц, 1H), 6,58 (d, J = 6,2 Гц, 1H); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -109,18 (s, 1F).
Пример 30
4-(6-циано-2-фторбензо[b]тиофен-4-ил)-2-гидроксибензойная кислота
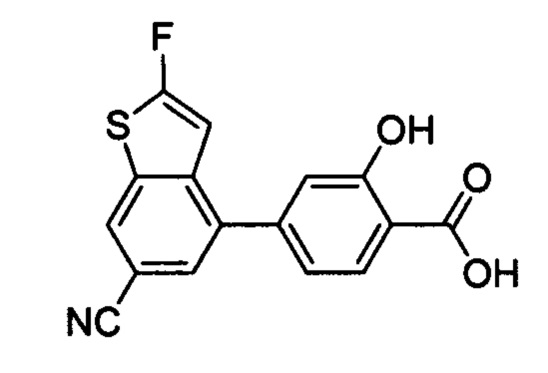
Стадия 1) Синтез 4-бром-2-фторбензо [в] тиофен-6-карбонитрила
Добавили в 1000-мл двугорлую колбу 4-бромбензо[b]тиофен-6-карбонитрил (23,8 г, 100 ммоль) и тетрагидрофуран (300 мл), а затем по каплям добавили диизопропиламид лития (60 мл, 120 ммоль, 2,0 М в ТГФ) -70°С. В течение 1,5 часа перемешивали смесь при -70°С в среде азота. Затем по каплям добавили раствор N-фторбензолсульфонимида (63,1 г, 200 ммоль) в тетрагидрофуране (200 мл). В течение 12 часов перемешивали смесь при комнатной температуре. Добавили в смесь насыщенный водный раствор хлорида аммония (800 мл). Экстрагировали водную фазу этилацетатом (300 мл × 2). Промыли объединенные органические фазы насыщенным рассолом (300 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили неочищенный продукт. Дополнительно очистили неочищенный продукт путем препаративной хроматографии с обращенной фазой (H2O/CH3CN, 0,1% ТФК), и получили указанное соединение в виде твердого вещества белого цвета (2,05 г, 8%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,95 (s, 1H), 7,78 (s, 1H), 6,99 (d, J = 2,2 Гц, 1Н); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -114,38 (s, 1F).
Стадия 2) Синтез трет-бутил-4-(6-циано-2-фторбензо[b]тиофен-4-ил)-2-гидроксибензоата
Постепенно добавили в 50-мл двугорлую колбу 4-бром-2-фторбензо[b]тиофен-6-карбонитрил (0,361 г, 1,41 ммоль), трет-бутил-2-гидрокси-4-(4,4,5,5-тетраметил-1,2,3-диоксаборолан-2-ил)бензоат (420 мг, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили в среде азота раствор карбоната калия (1,3 мл, 2 М) в воде, после чего в течение 0,5 часа перемешивали реакционную смесь при 90°С. Охладили полученную смесь до комнатной температуры. Добавили к полученной смеси насыщенный рассол (80 мл). Экстрагировали смесь этилацетатом (40 мл × 2). Промыли объединенную органическую фазу насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия, профильтровали и концентрировали в вакууме. Очистили остаток путем хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,290 г, 60%).
МС m/z (ES-API, с положительными ионами): 354,1 [М + 1]+.
Стадия 3) Синтез 4-(6-циано-2-фторбензо[b]тиофен-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-2-фторбензо[b]тиофен-4-ил)-2-гидроксибензоат (0,290 г, 0,786 ммоль) и дихлорметан (15 мл), а затем трифторуксусную кислоту (2 мл). В течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали полученную смесь в вакууме с целью удаления растворителя, очистили остаток путем хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,177 г, 77%),
МС m/z (ES-API, с отрицательными ионами): 312,0 [М - 1]-;
1H ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли) 8,60 (s, 1H), 7,92 (d, J = 8,0 Гц, 1H), 7,83 (s, 1H), 7,15 (m, 7,17-7,12, 3Н); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -117,54 (s, 1F).
Пример 31
4-(6-цианобензо [b]тиофен-4-ил)-2-гидроксибензойная кислота
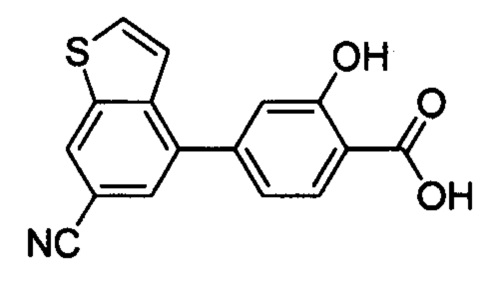
Стадия 1) Синтез 2-бром-6-фтор-4-метиланилина
Добавили в 250-мл одногорлую колбу 2-фтор-4-метиланилин (12,5 г, 100 ммоль), ледяную уксусную кислоту (80 мл) и метанол (80 мл), а затем медленно по каплям добавили к смеси в колбе бром (17,6 г, 110 ммоль) при 0°С. После этого в течение 0,5 часа перемешивали реакционную смесь при 0°С, а затем в течение 4 часов перемешивали при комнатной температуре. Добавили к реакционной смеси насыщенный водный сульфит натрия (300 мл), и концентрировали полученную смесь под вакуумом с целью удаления органического растворителя. Экстрагировали остаток этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде оранжевого твердого вещества (17,5 г, 86%).
МС m/z (ES-API, с положительными ионами): 204,9 [М + 2]+.
Стадия 2) Синтез 1-бром-3-фтор-5-метилбензола
Добавили в 500-мл одногорлую колбу 2 бром-6-фтор-4-метиланилин (11,2 г, 55 ммоль), разбавленную соляную кислоту (28,5 мл, 6 М) и ацетонитрил (300 мл), а затем медленно по каплям добавили к смеси в колбе раствор нитрита натрия (5,69 г, 82,4 ммоль) в воде (40 мл) при 0°С. После этого в течение 1 часа перемешивали реакционную смесь при 0°С, затем добавили в колбу раствор йодида калия (18,3 г, 110 ммоль) в воде (30 мл). После этого в течение 0,5 часа перемешивали смесь при 0°С, а в течение ночи перемешивали при комнатной температуре. Добавили к реакционной смеси насыщенный водный сульфит натрия (300 мл), и концентрировали полученную смесь в вакууме с целью удаления ацетонитрила. Экстрагировали остаток этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде белого твердого вещества (13,2 г, 76%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,29 (s, 1H), 6,81 (d, J = 8,4 Гц, 1Н), 2,31 (s, 3Н).
Стадия 3) Синтез 2-бром-6-фтор-4-метилбензальдегида
Добавили в 500-мл одногорлую колбу 1-бром-3-фтор-5-метилбензол (18,5 г, 58,7 ммоля) и безводный тетрагидрофуран (250-мл), а затем медленно по каплям добавили к смеси в колбе раствора хлористого изопропилмагния (35,2 мл, 2 М в ТГФ) при -45°С. После этого в течение 1,0 часа перемешивали реакционную смесь при -45°С, а затем добавили в колбу безводный N,N-диметилформамид (17,2 г, 235 ммоль). После этого медленно нагрели реакционную смесь до комнатной температуры и перемешивали в течение ночи. Добавили к реакционной смеси разбавленную соляную кислоту (200 мл, 4 М), и в течение 2 часов перемешивали полученную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления тетрагидрофурана. Экстрагировали остаток этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/200), и получили указанное соединение в виде светло-желтого твердого вещества (12,3 г, 96%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 10,31 (s, 1H), 7,31 (s, 1H), 6,95 (d, J = 11,2 Гц, 1H), 2,39 (s, 3H).
Стадия 4) Синтез этил-4-бром-6-метилбензо[b]тиофен-2-карбоксилата
Добавили в 100-мл одногорлую колбу 2-бром-6-фтор-4-метилбензальдегид (4,34 г, 20 ммоль), карбонат калия (5,53 г, 40 ммоль), этил-2-меркаптоацетат (2,88 г, 24 ммоль) и N,N-диметилформамид (40 мл). В течение 12 часов перемешивали полученную смесь при 80°С в среде азота. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный водный раствор хлорида аммония (150 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде белого твердого вещества (5,09 г, 85%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,10 (s, 1H), 7,57 (s, 1H), 7,42 (s, 1H), 4,41 (d, J = 7,1 Гц, 2H), 2,46 (s, 3H), 1,42 (t, J=7,1 Гц, 3Н).
Стадия 5) Синтез 4-бром-6-метилбензо[b]тиофен-2-карбоновой кислоты
Добавили в 250-мл одногорлую колбу этил-4-бром-6-метилбензо[b]тиофен-2-карбоксилат (2,0 г, 7,0 ммоль), метанол (25 мл), тетрагидрофуран (25 мл) и воду (25 мл), а затем добавили к полученной смеси в колбе гидроксид натрия (0,840 г, 21 ммоль). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали реакционную смесь в вакууме с целью удаления растворителя, и добавили к остатку воду (120 мл). Промыли полученную смесь этиловым эфиром (80 мл). Затем подкислили водный слой разбавленной соляной кислотой (2 N) до рН 1, и экстрагировали полученную смесь этилацетатом (80 мл × 2). Промыли объединенные органические слои насыщенным рассолом (80 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме, и получили указанное соединение в виде белого твердого вещества (1,80 г, 95%).
МС m/z (ES-API, с отрицательными ионами): 271,9 [М + 2]+.
Стадия 6) Синтез 4-бром-6-метилбензо[b]тиофена
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 4-бром-6-метилбензо[b]тиофен-2-карбоновую кислоту (1,79 г, 6,6 ммоль), медный порошок (0,83 г, 13,1 ммоль) и хинолин (15 мл), и в течение 0,5 часа перемешивали полученную смесь при 200°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь концентрированную соляную кислоту (100 мл, 12 М). Экстрагировали полученную смесь этилацетатом (80 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (100% петролейный эфир), и получили указанное соединение в виде бесцветной жидкости (1,33 г, 89%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,60 (s, 1H), 7,41 (s, 2H), 7,39 (s, 1H), 2,45 (s, 3Н).
Стадия 7) Синтез 4-бромбензо[b]тиофен-6-карбонитрила
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 4-бром-6-метилбензо[b]тиофен (0,931 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (0,669 г, 4,1 ммоль), ацетат палладия (0,045 г, 0,20 ммоль) и ацетонитрил (20 мл), и в течение 48 часов перемешивали полученную смесь при 80°С. Охладили реакционную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,420 г, 43%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,15 (s, 1H), 7,82-7,75 (m, 2Н), 7,57 ((d, J = 5,5 Гц, 1H).
Стадия 8) Синтез трет-бутил-4-(6-цианобензо[b]тиофен-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бромбензо[b]тиофен-6-карбонитрил (0,340 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,420 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 M) в среде азота, и перемешивали полученную смесь при 90°С в течение 0,5 часа. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,313 г, 68%).
МС m/z (ES-API, с положительными ионами): 352,1 [M + 1]+.
Стадия 9) Синтез 4-(6-цианобензо[b]тиофен-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-цианобензо[b]тиофен-4-ил)-2-гидроксибензоат (0,313 г, 0,891 ммоль) и дихлорметан (15 мл), а затем добавили к смеси в колбе трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали с целью удаления растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,145 г, 55%).
МС m/z (ES-API, с отрицательными ионами): 294,0 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,72 (s, 1H), 8,19 (d, J = 5,5 Гц, 1H), 7,94 (d, J = 7,9 Гц, 1H), 7,81 (s, 1H), 7,54 (d, J = 5,5 Гц, 1H), 7,19-7,7 (m, 2H).
Пример 32
4-(6-циано-1-метил-1H-индол-4-ил)-2-гидроксибензойная кислота
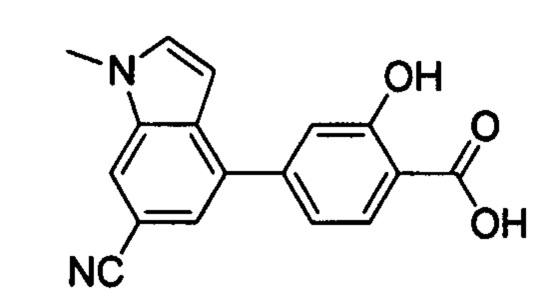
Стадия 1) Синтез трет-бутил-4-(6-циано-1-метил-1H-индол-4-ил)-2-метоксибензоата
Добавили в 50-мл двугорлую колбу трет-бутил-4-(6-циано-1H-индол-4-ил)-2-гидроксибензоат (0,578 г, 1,73 ммоль) и безводный тетрагидрофуран (10 мл), затем медленно по каплям добавили к смеси в колбе гидрид натрия (0,500 г, 1,95 ммоль, 60%) при 0°С. После этого в течение 0,5 часа перемешивали реакционную смесь при 0°С, затем добавили в колбу йодид калия (18,3 г, 110 ммоль). После этого в течение 12 часов перемешивали смесь при комнатной температуре. Добавили к реакционной смеси насыщенный водный хлорид аммония (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,464 г, 74%).
МС m/z (ES-API, с положительными ионами): 363,1 [М + 1]+.
Стадия 2) Синтез 4-(6-циано-1-метил-1H-индол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-1-метил-1Н-индол-4-ил)-2-метоксибензоат (0,362 г, 1,0 ммоль) и дихлорметан (10 мл), затем по каплям добавили к смеси в колбе трибромид бора (0,750 г, 3,0 ммоль) при -20°С. После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси ледяную воду (80 мл), и экстрагировали полученную смесь этилацетатом (60 мл × 2). Промыли объединенные органические слои насыщенным рассолом (80 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме с целью удаления растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,091 г, 31%).
МС m/z (ES-API, с отрицательными ионами): 291,1 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 11,43 (s, 1H), 8,20 (s, 1H), 7,96 (d, J = 8,1 Гц, 1H), 7,79 (d, J = 3,1 Гц, 1H), 7,55 (s, 1H), 7,35-7,23 (m, 2H), 6,71 (d, J = 3,0 Гц, 1H), 3,96 (s, 3H).
Пример 33
4-(2-хлор-6-цианобензо[b]тиофен-4-ил)-2-гидроксибензойной кислоты
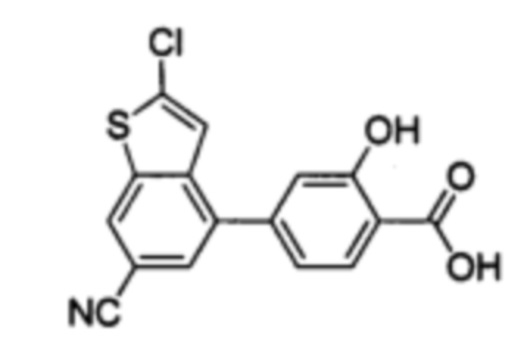
Стадия 1) Синтез 4-бром-2-хлорбензо[b]тиофен-6-карбонитрила
Добавили в 100-мл двугорлую колбу 4-бромбензо[b]тиофен-6-карбонитрил (2,38 г, 10 ммоль) и безводный тетрагидрофуран (30 мл), а затем медленно по каплям добавили к смеси в колбе раствор диизопропиламида лития (6,0 мл, 12 ммоль, 2,0 М в ТГФ) при -70°С. После этого в течение 1,5 часа перемешивали реакционную смесь при -70°С, затем добавили в колбу раствор гексахлорэтана (4,73 г, 20 ммоль) в тетрагидрофуране (20 мл). После этого медленно нагрели реакционную смесь до комнатной температуры и перемешивали в течение ночи. Добавили к реакционной смеси насыщенный водный хлорид аммония (100 мл). Экстрагировали полученную смесь этилацетатом (80 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (2,34 г, 86%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли) 7,97 (s, 1H), 7,76 (s, 1H), 7,42 (s, 1H).
Стадия 2) Синтез трет-бутил-4-(2-хлор-6-цианобензо[b]тиофен-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бром-2-хлорбензо[b]тиофен-6-карбонитрил (0,384 г, 1,41 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,420 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис (дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) в среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме.
Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/1), и получили указанное соединение в виде белого твердого вещества (0,212 г, 42%).
МС m/z (ES-API, с положительными ионами): 386,0 [М + 1]+.
Стадия 3) Синтез 4-(2-хлор-6-цианобензо[b]тиофен-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(2-хлор-6-цианобензо[b]тиофен-4-ил)-2-гидроксибензоат (0,212 г, 0,55 ммоль) и дихлорметан (15 мл), а затем добавили к смеси в колбе трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления органического растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,177 г, 77%).
МС m/z (ES-API, с отрицательными ионами): 327,9 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,62 (s, 1H), 7,93 (d, J = 8,0 Гц, 1H), 7,84 (d, J = 1,2 Гц, 1H), 7,53 (s, 1H), 7,21-7,07 (m, 2H).
Пример 34
4-(2-цианохинолин-4-ил)-2-гидроксибензойная кислота
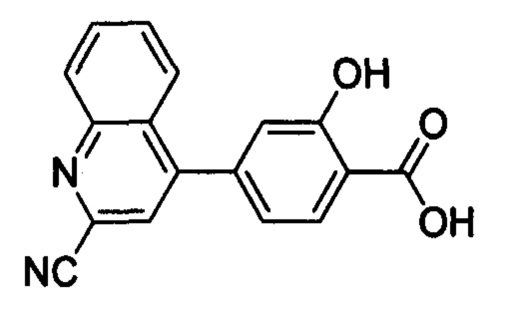
Стадия 1) Синтез 2-метилхинолин-4-ола
Добавили в 250-мл одногорлую колбу анилин (3,73 г, 40,0 ммоль) и полифосфорную кислоту (50 мл), а затем по каплям добавили к смеси в колбе этиловый эфир ацетоуксусной кислоты (6,25 г, 48 ммоль) при 80°С. После этого в течение 16 часов перемешивали реакционную смесь при 120°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь ледяную воду (300 мл) и гидроксид аммония (100 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме и концентрировали с целью удаления органического растворителя, а затем очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (4,46 г, 70%).
МС m/z (ES-API, с положительными ионами): 160,1 [М + 1]+.
Стадия 2) Синтез 4-бром-2-метилхинолина
Добавили в 250-мл одногорлую колбу2-метилхинолин-4-ол (4,46 г, 28,0 ммоль) и N,N-диметилформамид (80 мл), а затем по каплям добавили к смеси в колбе трибромид фосфора (11,37 г, 42 ммоль) при 0°С. После этого в течение 3 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси ледяную воду (100 мл) и гидроксид аммония (100 мл, 25%), а затем экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде светло-желтой жидкости (0,995 г, 16%).
МС m/z (ES-API, с положительными ионами): 222,9 [М + 2]+.
Стадия 3) Синтез 4-бромхинолин-2-карбальдегида
Добавили в 100-мл одногорлую колбу 4-бром-2-метилхинолин (0,995 г, 4,48 ммоль), диоксид селен (0,99 г, 8,9 ммоль) и 1,4-диоксан (25 мл). После этого в течение 2 часов перемешивали реакционную смесь при 80°С. Охладили реакционную смесь до комнатной температуры и профильтровали с целью удаления нерастворимого вещества, затем Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде бледно-желтого твердого вещества (0,825 г, 78%).
МС m/z (ES-API, с положительными ионами): 236,9 [М+2]+.
Стадия 4) Синтез 4-бромхинолин-2-карбонитрила
Добавили в 100-мл одногорлую колбу4-бромхинолин-2-карбальдегид (1,89 г, 8,0 ммоль), гидроксид аммония (20 мл, 28%) и тетрагидрофуран (20 мл), а затем порциями добавили к смеси в колбе йод (2,23 г, 8,8 ммоль). После этого в течение 4 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси насыщенный водный раствор тиосульфата натрия (80 мл), и экстрагировали полученную смесь этилацетатом (60 мл × 2). Промыли объединенные органические слои насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (1,51 г, 81%).
МС m/z (ES-API, с положительными ионами): 233,9 [М + 2]+.
Стадия 5) Синтез трет-бутил-4-(2-цианохинолин-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бромхинолин-2-карбонитрил (0,305 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,42 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) в среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (0,263 г, 58%).
МС m/z (ES-API, с положительными ионами): 347,1 [М + 1]+.
Стадия 6) Синтез 4-(2-цианохинолин-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(2-цианохинолин-4-ил)-2-гидроксибензоат (0,263 г, 0,760 ммоль) и дихлорметан (15 мл), а затем добавили к смеси в колбе трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления органического растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,174 г, 79%).
МС m/z (ES-API, с отрицательными ионами): 289,0 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,20 (d, J=8,4 Гц, 1Н), 8,00-7,93 (m, 4H), 7,79 (t, J = 7,6 Гц, 1Н), 7,05-6,87 (m, 2H).
Пример 35
4-(5-циано-3-(трифторметил)бензо[d]изоксазол-7-ил)-2-гидроксибензойная кислота
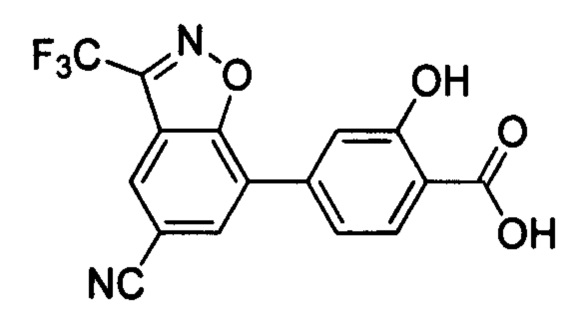
Стадия 1) Синтез 3-бром-2-фтор-5-метилбензальдегида
Добавили в 500-мл одногорлую колбу 2-бром-1-фтор-4-метилбензол (18,9 г, 100 ммоль) и безводный тетрагидрофуран (150 мл), а затем медленно по каплям добавили к смеси в колбе раствор диизопропиламида лития (60 мл, 2 М в ТГФ) при -70°С. После этого в течение 3 часов перемешивали реакционную смесь при -70°С, затем добавили в колбу безводный N,N-диметилформамида (17,2 г, 235 ммоль). После этого медленно нагрели реакционную смесь до комнатной температуры и перемешивали в течение ночи. Добавили к реакционной смеси разбавленную соляную кислоту (200 мл, 4 М), и в течение 2 часов перемешивали полученную смесь при комнатной температуре. Концентрировали смесь в вакууме с целью удаления тетрагидрофурана. Экстрагировали остатокэтилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), и получили указанное соединение в виде оранжевого твердого вещества (10,9 г, 50%).
МС m/z (ES-API, с положительными ионами): 217,9 [М + 2]+.
Стадия 2) Синтез 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтанола
Добавили в 500-мл одногорлую колбу 3-бром-2-фтор-5-метилбензальдегид (13,0 г, 59,9 ммоль), (трифторметил)триметилсилан (10,2 г, 71,7 ммоль) и тетрагидрофуран (150 мл), затем по каплям добавили к смеси в колбе раствор фторида тетрабутиламмония (1,2 мл, 1,0 М в ТГФ) при 0°С. После этого в течение 2 часов перемешивали реакционную смесь при комнатной температуре, затем по каплям добавили в колбу раствор фторида тетрабутиламмония (1,2 мл, 1,0 М в ТГФ) при 0°С. После этого в течение 3 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси разбавленную соляную кислоту (300 мл), и экстрагировали полученную смесь этилацетатом (100 мл × 2). Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде светло-желтой жидкости (13,4 г, 78%).
МС m/z (ES-API, с положительными ионами): 287,9 [М + 2]+.
Стадия 3) Синтез 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтанона
Добавили в 250-мл одногорлую колбу 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтанол (9,85 г, 34,3 ммоль), 2-йодоксибензойную кислоту (30,3 г, 103 ммоль) и этилацетат (120 мл). В течение 24 часов перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры и профильтровали, затем концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде светло-желтого масла (7,53 г, 77%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,72 (дд, J = 6,0, 1,7 Гц, 1Н), 7,60 (d, J = 5,7 Гц, 1Н), 2,42 (s, 3Н).
Стадия 4) Синтез 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтаноноксима
Добавили в 100-мл одногорлую колбу 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтанон (2,50 г, 8,77 ммоль), ацетат натрия (10,2 г, 106 ммоль), гидрохлорид гидроксиламина (6,35 г, 87,7 ммоль) и метанол (30 мл). В течение 30 часов перемешивали полученную смесь при 80°С в защитной среде азота. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный водный раствор хлорида аммония (150 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме, и получили указанное соединение в виде светло-желтого масла (2,60 г, 99%).
Стадия 5) Синтез 7-бром-5-метил-3-(трифторметил)бензо[d]изоксазола
Добавили в 20-мл пробирку для реакций под воздействием СВЧ-излучения 1-(3-бром-2-фтор-5-метилфенил)-2,2,2-трифторэтаноноксим (2,60 г, 8,7 ммоль), 1,8-диазабицикло[5.4.0] ундец-7-ен (1,55 мл, 10,4 ммоль) и тетрагидрофуран (15 мл). В течение 0,5 часа перемешивали полученную смесь при 150°С в условиях СВЧ-излучения. Охладили реакционную смесь до комнатной температуры, затем добавили в смесь разбавленную соляную кислоту (80 мл, 1 М). Экстрагировали полученную смесь этилацетатом (60 мл × 2). Промыли объединенные органические слои насыщенным рассолом (60 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме и получили указанное соединение в виде белого твердого вещества (2,10 г, 87%).
1H ЯМР (400 МГц, CDCl3) δ (миллионные доли) 7,68 (s, 1H), 7,53 (s, 1H), 2,53 (s, 3Н).
Стадия 6) Синтез 7-бром-3-(трифторметил)бензо[d]изоксазол-5-карбонитрила
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-метил-3-(трифторметил)бензо[d]изоксазол (1,15 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (0,669 г, 4,1 ммоль), ацетат палладия (0,045 г, 0,20 ммоль) и ацетонитрил (20 мл), и в течение 48 часов перемешивали полученную смесь при 80°С. Охладили реакционную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,538 г, 42%).
МС m/z (ES-API, с положительными ионами): 291,9 [М + 2]+.
Стадия 7) Синтез трет-бутил-4-(5-циано-3-(трифторметил)бензо[d]изоксазол-7-ил)-2-гидрокси-бензоата
Добавили в 50-мл двугорлую колбу 7-бром-3-(трифторметил)бензо[d]изоксазол-5-карбонитрил (0,419 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,420 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (П) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) в среде азота, и течение 0,5 часа перемешивали полученную смесь при 90°С в. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили указанное соединение в виде белого твердого вещества (0,212 г, 40%).
МС m/z (ES-API, с положительными ионами): 405,1 [М + 1]+.
Стадия 8) Синтез 4-(5-циано-3-(трифторметил)бензо[d]изоксазол-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-3-(трифторметил)бензо[d]изоксазол-7-ил)-2-гидроксибензоат (0,212 г, 0,524 ммоль) и дихлорметан (15 мл), а затем добавили к смеси в колбе трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления органического растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,119 г, 65%).
МС m/z (ES-API, с отрицательными ионами): 347,0 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,66 (s, 1H), 8,48 (s, 1H), 7,90 (s, 1H), 7,30-7,26 (m, 2H).
Пример 36
4-(6-циано-2-метил-2H-индазол-4-ил)-2-гидроксибензойная кислота
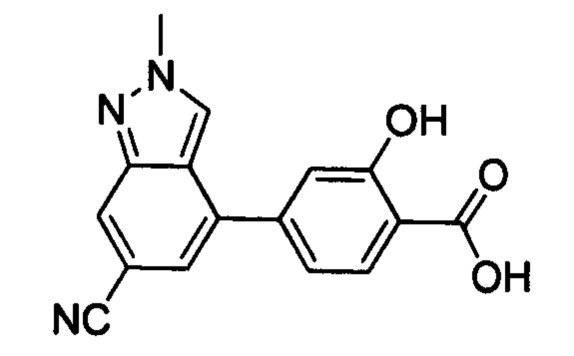
Стадия 1) Синтез метил-4-бром-2-метил-2H-индазол-6-карбоксилата
Добавили в 100-мл двугорлую колбу метил-4-бром-1H-индазол-6-карбоксилат (1,53 г, 6,0 ммоль), карбонат цезия (3,95 г, 12,1 ммоль) и N,N-диметилформамид (20 мл), затем добавили к смеси в колбе йодометан (1,1 г, 7,7 ммоль). После этого в течение 24 часов перемешивали реакционную смесь при комнатной температуре. Профильтровали реакционную смесь, чтобы удалить нерастворимые вещества, и добавили к фильтрату насыщенный водный хлорид аммония (150 мл). Экстрагировали полученную смесь этилацетатом (80 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде светло-желтого твердого вещества (0,517 г, 32%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,42 (s, 1H), 7,96 (s, 1H), 7,88 (s, 1H), 4,26 (s, 3Н), 3,95 (s, 3H),
Стадия 2) Синтез 4-бром-2-метил-2H-индазол-6-карбоксамида
Добавили в 50-мл герметично закрывающуюся пробирку метил-4-бром-2-метил-2H-индазол-6-карбоксилат (1,0 г, 3,7 ммоль) и раствор аммиака в метаноле (20 мл, 7 М). Герметично закрыли пробирку, и в течение 24 часов перемешивали реакционную смесь в закрытой пробирке при 110°С. Охладили реакционную смесь до комнатной температуры и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/50), и получили указанное соединение в виде белого твердого вещества (0,50 г, 53%).
МС m/z (ES-API, с положительными ионами): 254,9 [М + 2]+.
Стадия 3) Синтез 4-бром-2-метил-2H-индазол-6-карбонитрила
Добавили в 100-мл одногорлую колбу 4-бром-2-метил-2H-индазол-6-карбоксамид (0,50 г, 1,97 ммоль) и толуол (20 мл), а затем добавили к смеси в колбе оксихлорид фосфора (3,0 г, 19,7 ммоль). После этого в течение 12 часов перемешивали реакционную смесь при 120°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/5), и получили указанное соединение в виде белого твердого вещества (0,44 г, 95%).
МС m/z (ES-API, с положительными ионами): 236,9 [М + 2]+.
Стадия 4) Синтез трет-бутил-4-(6-циано-2-метил-2H-индазол-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бром-2-метил-2H-индазол-6-карбонитрил (0,340 г, 1,44 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,420 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис (дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) под в защитной среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 2/1), и получили указанное соединение в виде белого твердого вещества (0,384 г, 84%).
МС m/z (ES-API, с положительными ионами): 350,1 [М + 1]+.
Стадия 5) Синтез 4-(6-циано-2-метил-2H-индазол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(6-циано-2-метил-2H-индазол-4-ил)-2-гидроксибензат (0,384 г, 1,10 ммоль) и дихлорметан (15 мл), затем добавили к смеси трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,210 г, 65%).
МС m/z (ES-API, с положительными ионами): 294,1 [М + 1]+; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,74 (s, 1H), 8,35 (s, 1H), 8,03-7,85 (m, 1H), 7,50 (s, 1H), 7,31 (d, J = 4,4 Гц, 2H), 4,27 (s, 3H).
Пример 37
4-(6-циано-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензойная кислота
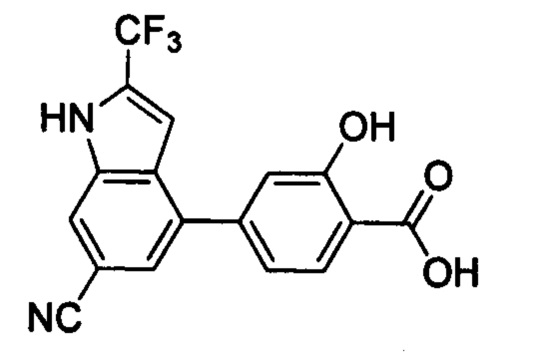
Стадия 1) Синтез метил-4-(6-циано-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бром-2-(трифторметил)-1H-индол-6-карбонитрил (0,344 г, 1,19 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,300 г, 1,08 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (48 мг, 0,059 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,1 мл, 2 М) в среде азота, и перемешивали полученную смесь при 90°С в течение 0,5 ч. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), сушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/10), и получили указанное соединение в виде белого твердого вещества (0,17 г, 39%).
МС m/z (ES-API, с положительными ионами): 361,0 [М + 1)+.
Стадия 2) Синтез 4-(6-циано-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу метил-4-(6-циано-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензоат (0,133 г, 0,37 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем добавили в смесь гидроксид натрия (45 мг, 1,12 ммоль). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали реакционную смесь в вакууме с целью удаления органического растворителя, и добавили к остатку воду (60 мл). Промыли полученную смесь этиловым эфиром (50 мл). Затем подкислили водный слой разбавленной соляной кислотой (2 N) до рН 1, и экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические слои насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме, очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (89 мг, 67%).
МС m/z (ES-API, с отрицательными ионами): 345,1 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 13,21 (s, 1H), 8,06 (s, 1H), 7,93 (d, J = 7,4 Гц, 1H), 7,61 (s, 1H), 7,25-7,16 (m, 3H).
Пример 38
4-(5-циано-2-(трифторметил)бензофуран-7-ил)-2-гидроксибензойная кислота
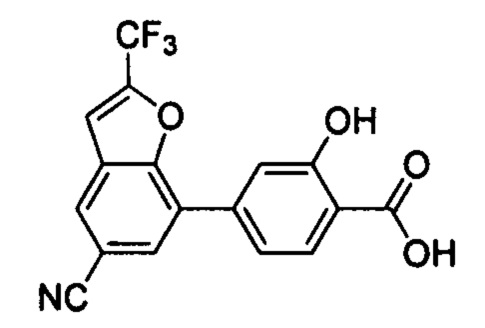
Стадия 1) Синтез (Z)-2-бром-4-(2-хлор-3,3,3-трифторпроп-1-ен-1-ил)-6-метилфенилацетата
Добавили в 100-мл одногорлую колбу 3-бром-2-гидрокси-5-метилбензальдегид (5,60 г, 26 ммоль), порошок цинка (8,50 г, 130 ммоль), уксусный ангидрид (7,96 г, 78 ммоль) и N,N-диметилформамид (30 мл), а затем медленно по каплям добавили в смесь 1,1,1-трихлортрифторэтан (7,80 г, 41,1 ммоль) в среде азота, поддерживая температуру смеси не выше 50°С. После этого в течение 4 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси насыщенный водный хлорид аммония (200 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/50), и получили указанное соединение в виде светло-желтой жидкости (4,83 г, 52%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,56 (s, 1H), 7,49 (s, 1H), 7,20 (s, 1H), 2,38 (s, 3Н), 2,35 (s, 3H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -69,06 (s, 3F).
Стадия 2) Синтез 7-бром-5-метил-2-(трифторметил)бензофурана
Добавили в 100-мл одногорлую колбу (Z)-2-бром-4-(2-хлор-3,3,3-трифторпроп-1-ен-1-ил)-6-метилфенилацетат (4,00 г, 11,2 ммоль) и N,N-диметилформамид (25 мл), а затем порциями добавили к смеси в колбе трет-бутоксид калия (3,77 г, 33,6 ммоль) при 0°С. После этого в течение 6 часов перемешивали реакционную смесь при комнатной температуре. Добавили к реакционной смеси насыщенный водный хлорид аммония (200 мл). Экстрагировали полученную смесь этилацетатом (100 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (100 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/100), получая указанное соединение в виде бесцветной жидкости (1,78 г, 57%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,43 (s, 1H), 7,37 (s, 1H), 7,14 (s, 1H), 2,44 (s, 3H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -64,77 (s, 3F).
Стадия 3) Синтез 7-бром-2-(трифторметил)бензофуран-5-карбонитрила
Добавили в 25-мл пробирку для реакций под воздействием СВЧ-излучения 7-бром-5-метил-2-(трифторметил)бензофуран (1,14 г, 4,1 ммоль), трет-бутилнитрит (1,26 г, 12,2 ммоль), N-гидроксифталимид (0,669 г, 4,1 ммоль), ацетат палладия (0,045 г, 0,20 ммоль) и ацетонитрил (20 мл), и в течение 48 часов перемешивали полученную смесь при 80°С. Охладили реакционную смесь до комнатной температуры и концентрировали в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/8), и получили указанное соединение в виде белого твердого вещества (0,166 г, 14%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 8,00 (d, J=1,1 Гц, 1H), 7,90 (d, J = 1,1 Гц, 1H), 7,33 (s, 1H); и
19F ЯМР (376 МГц, CDCl3) δ (миллионные доли): -65,01 (s, 3F).
Стадия 4) Синтез трет-бутил-4-(5-циано-2-(трифторметил)бензофуран-7-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 7-бром-2-(трифторметил)бензофуран-5-карбонитрил (0,380 г, 1,31 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2ил)бензоат (0,42 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) в защитной среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/40), и получили указанное соединение в виде белого твердого вещества (0,391 г, 74%).
МС m/z (ES-API, с положительными ионами): 404,1 [М + 1]+.
Стадия 5) Синтез 4-(5-циано-2-(трифторметил)бензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(5-циано-2-(трифторметил) бензофуран-7-ил)-2-гидроксибензоат (0,391 г, 0,97 ммоль) и дихлорметан (15 мл), а затем добавили к смеси трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления органического растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,257 г, 74%).
МС m/z (ES-API, с отрицательными ионами): 346,0 [М - 1]-;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,44 (d, J = 1,1 Гц, 1Н), 8,27 (d, J = 1,1 Гц, 1H), 8,06-7,89 (m, 2H), 7,56-7,37 (m, 2H); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -63,66 (s, 3F).
Пример 39
4-(6-циано-1-циклопропил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензойная кислота
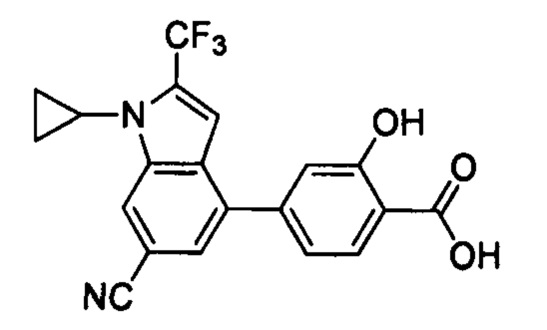
Стадия 1) Синтез 4-бром-1-циклопропил-2-(трифторметил)-1H-индол-6-карбонитрила
Добавили в 50-мл двугорлую колбу 4-бром-2-(трифторметил)-1H-индол-6-карбонитрил (0,500 г, 1,73 ммоль), 2,2'-дипиридил (0,270 г, 1,7 ммоль), циклопропилборную кислоту (0,300 г, 3,5 ммоль), ацетат меди (0,310 г, 1,7 ммоль), карбонат калия (0,480 г, 3,5 ммоль) и 1,2-дихлорметан (12 мл). После этого в течение 16 часов перемешивали реакционную смесь при 70°С в среде кислорода. Охладили реакционную смесь до комнатной температуры, затем добавили в смесь насыщенный рассол (80 мл) и этилацетат (80 мл). Разделили полученную смесь, промыли органический слой насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,400 г, 70%).
МС m/z (ES-API, с положительными ионами): 329,9 [М + 2]+.
Стадия 2) Синтез метил-4-(6-циано-1-циклопропил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 4-бром-1-циклопропил-2-(трифторметил)-1H-индол-6-карбонитрил (0,392 г, 1,19 ммоль), метил-2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,300 г, 1,08 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (48 мг, 0,059 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,1 мл, 2 М) в защитной среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (этилацетат/петролейный эфир (в объемном отношении) = 1/15), и получили указанное соединение в виде желтого твердого вещества (0,377 г, 78%).
МС m/z (ES-API, с положительными ионами): 401,1 [М + 1]+.
Стадия 3) Синтез 4-(6-циано-1-циклопропил-2-(трифторметил)-1H-индол-4-ил)-2-гидрокси-бензойной кислоты
Добавили в 100-мл одногорлую колбу метил-4-(6-циано-1-циклопропил-2-(трифторметил)-1H-индол-4-ил)-2-гидроксибензоат (0,148 г, 0,37 ммоль), метанол (8 мл), тетрагидрофуран (8 мл) и воду (8 мл), а затем добавили к смеси в колбе гидроксид натрия (45 мг, 1,12 ммоль). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре. Концентрировали реакционную смесь в вакууме с целью удаления растворителя, и добавили к остатку воду (60 мл). Промыли полученную смесь этиловым эфиром (50 мл). Затем подкислили водный слой разбавленной соляной кислотой (2 N) до рН 1, и экстрагировали полученную смесь этилацетатом (40 мл × 2). Промыли объединенные органические слои насыщенным рассолом (40 мл × 2), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме, очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде бледно-желтого твердого вещества (0,107 г, 75%).
МС m/z (ES-API, с положительными ионами): 387,1 [М + 1]+;
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,30 (s, 1H), 7,93 (d, J = 8,3 Гц, 1H), 7,67 (s, 1H), 7,21-7,19 (m, 3Н), 3,41-3,35 (m, 1H), 1.31-1.09 (m, 6Н); и
19F ЯМР (376 МГц, ДМСО-d6) δ (миллионные доли): -57,68 (s, 3F).
Пример 40
4-(2-хлор-5-цианобензофуран-7-ил)-2-гидроксибензойная кислота
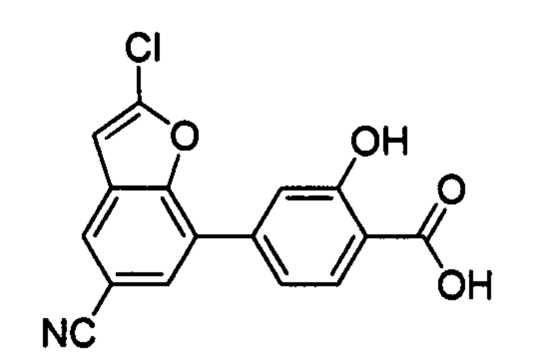
Стадия 1) Синтез 7-бром-2-хлорбензофуран-5-карбонитрила
Добавили в 1000-мл двугорлую колбу 7-бромбензофуран-5-карбонитрил (22,2 г, 100 ммоль) и тетрагидрофуран (300 мл), а затем медленно по каплям добавили к смеси в колбе раствор диизопропиламида лития (60 мл, 120 ммоль, 2,0 М в ТГФ) при -70°С. После этого в течение 1,5 часа перемешивали реакционную смесь при -70°С в среде азота, затем добавили в колбу раствор гексахлорэтана (28,4 г, 120 ммоль) в тетрагидрофуране (200 мл). После этого медленно нагрели реакционную смесь до комнатной температуры и перемешивали в течение ночи. Добавили к реакционной смеси насыщенный водный раствор хлорида аммония (800 мл), и экстрагировали полученную смесь этилацетатом (300 мл × 2). Промыли объединенные органические слои насыщенным рассолом (300 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/4), и получили неочищенный продукт, который затем очистили путем препаративной хроматографии с обращенной фазой (H2O/CH3CN, 0,1% ТФК), и получили указанное соединение в виде белого твердого вещества (16,7 г, 65%).
1Н ЯМР (400 МГц, CDCl3) δ (миллионные доли): 7,79 (d, J = 1,0 Гц, 1Н), 7,73 (d, J = 1,0 Гц, 1Н), 6,75 (s, 1Н).
Стадия 2) Синтез трет-бутил-4-(2-хлор-5-цианобензофуран-7-ил)-2-гидроксибензоата
Добавили в 50-мл двугорлую колбу 7-бром-2-хлорбензофуран-5-карбонитрил (0,362 г, 1,41 ммоль), трет-бутил 2-гидрокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоат (0,420 г, 1,31 ммоль), дихлорметановый комплекс 1,1'-бис(дифенилфосфино)ферроцена и дихлорида палладия (II) (53 мг, 0,065 ммоль) и N,N-диметилформамид (8 мл). Добавили к реакционной смеси водный раствор карбоната калия (1,3 мл, 2 М) в среде азота, и в течение 0,5 часа перемешивали полученную смесь при 90°С. Охладили реакционную смесь до комнатной температуры, и добавили в смесь насыщенный рассол (80 мл). Экстрагировали полученную смесь этилацетатом (40 мл × 2), и объединили органические слои. Промыли объединенные органические слои насыщенным рассолом (60 мл), высушили над безводным сульфатом натрия и профильтровали. Концентрировали фильтрат в вакууме. Очистили остаток путем колоночной хроматографии на силикагеле (дихлорметан/петролейный эфир (в объемном отношении) = 1/2), и получили указанное соединение в виде белого твердого вещества (0,271 г, 56%).
МС m/z (ES-API, с положительными ионами): 370,1 [М + 1]+.
Стадия 3) Синтез 4-(2-хлор-5-цианобензофуран-7-ил)-2-гидроксибензойной кислоты
Добавили в 100-мл одногорлую колбу трет-бутил-4-(2-хлор-5-цианобензофуран-7-ил)-2-гидроксибензоат (0,271 г, 0,734 ммоль) и дихлорметан (15 мл), а затем добавили к смеси трифторуксусную кислоту (2 мл). После этого в течение 12 часов перемешивали реакционную смесь при комнатной температуре и концентрировали в вакууме с целью удаления растворителя. Очистили остаток путем колоночной хроматографии на силикагеле (метанол/дихлорметан (в объемном отношении) = 1/20), и получили указанное соединение в виде белого твердого вещества (0,182 г, 79%).
МС m/z (ES-API, с отрицательными ионами): 312,1 [М - 1]-; и
1Н ЯМР (400 МГц, ДМСО-d6) δ (миллионные доли): 8,21 (s, 1H), 8,07 (s, 1H), 7,95 (d, J = 8,2 Гц, 1H), 7,46 (s, 1H), 7,42 (d, J = 8,2 Гц, 1H), 7,26 (s, 1H).
Количественное определение биологической активности
Пример 1
Анализ ингибирования активности ХО (ксантиноксидазы)
1) Метод испытания
2,5-кратно разбавили соединение 50 мМ буфера KH2PO4, и получили ряд концентраций от 2000 нМ до 0,524 нМ. Затем добавили по 30 мкл/лунку разбавленного раствора на 384-луночный планшет, и после этого добавили в каждую лунку по 30 мкл ксантиноксидазы в концентрации 21 мЕд/мл. В течение 1 мин центрифугировали полученный раствор со скоростью 3000 об/мин, встряхнули и в течение 10 мин инкубировали при комнатной температуре, а затем добавили в каждую лунку по 30 мкл 600 мкМ субстрата (ксантина), при этом использовали лунку, обработанную буфером (без добавления соединения с добавлением фермента и субстрата в таких же концентрациях) и лунку в качестве отрицательного контроля (без добавления соединения и фермента с добавлением субстрата в такой же концентрации) были созданы. После инкубации в течение 5 мин при комнатной температуре измерили оптическую плотность (OD) на волне 290 нм с помощью спектрофотометра PHERAstar FS для прочтения микропланшетов. Для расчета степени ингибирования ксантиноксидазы (ХО) соединением использовали следующую Формулу, а для расчета значений IC50 использовали GraphPad Prism 5. Результаты представлены в Таблице 1:
Степень ингибирования (%) = [1-(ODобработанная лекарством лунка - ODотрицательный контроль)/(ODобработанная буфером лунка - ODотрицательный контроль)] × 100
2) Результаты испытания
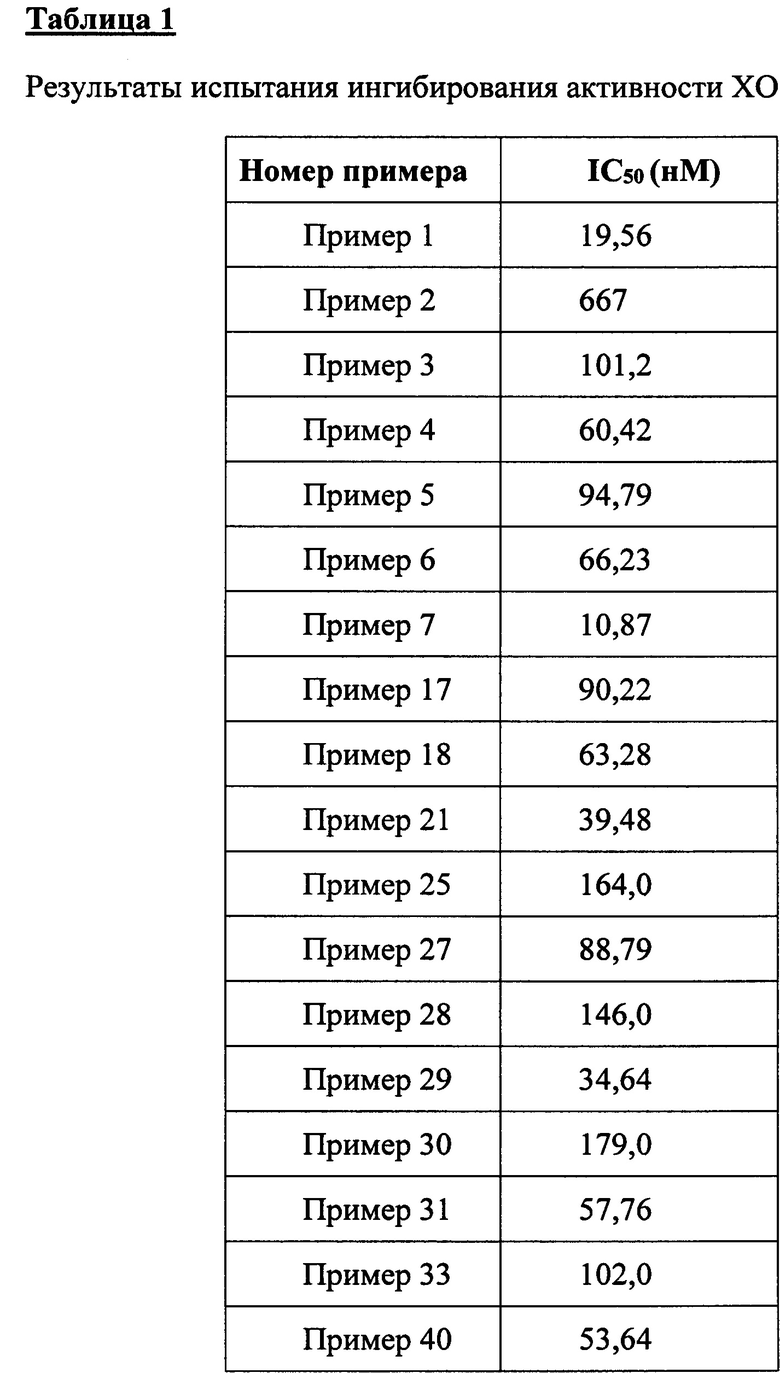
Вывод: Результаты испытания показывают, что соединения согласно изобретению обладают хорошей ингибирующей активностью в отношении ХО.
Пример 2
Анализ ингибирования активности URAT1 (переносчика уратов)
1) Метод испытания
а. Конструирование линии клеток, стабильно экспрессирующих hURAT1
Трансфицировали плазмиду hURAT1 в клетку HEK293T, и с использованием G418 (Генетицина) получили линию клеток, стабильно экспрессирующих hURAT1, для скрининга.
б. Ингибирование поглощения мочевой кислоты
Высеяли полученную на описанных выше стадиях линию клеток, стабильно экспрессирующих hURAT1, на 96-луночный планшет. После инкубации в течение, по меньшей мере, 12 часов удалили среду, а затем промыли клетки не содержащим (Cl-) буфером HBSS. 4-кратно разбавили соединение буфером, и получили растворы соединения в концентрации от 200 мкМ до 0,8 нМ. Равномерно смешали 5 мкл полученного раствора соединения и 45 мкл сливочного масла, содержащего мочевую кислоты [8-14C], а затем добавили полученный смешанный раствор на 96-луночный планшет к линии клеток, стабильно экспрессирующих hURAT1 (т.е. конечная концентрация соединения составляла от 20 мкМ до 0,08 нМ). Одновременно использовали лунку с буфером (линия клеток, стабильно экспрессирующих hURAT1, без лекарственного средства) и отрицательный контроль (клетки HEK293T без лекарственного средства) были созданы. После инкубации в течение 5 мин при 37°С удалили буфер, и промыли клетки буфером. Добавили в каждую лунку пл 50 мкл буфера для лизиса (100 мМ NaOH), чтобы лизировать клетки, и в течение 10 мин встряхивали лунки со скоростью 600 об/мин, в течение 5 мин центрифугировали со скоростью 1000 об/мин, а затем пипеткой накапали 45 мкл супернатанта на микропланшет Isoplate-96. Добавили в каждую лунку по 150 мкл Ultima GoldTM XR и в течение 10 мин встряхивали микропланшет со скоростью 600 об/мин. Использован для подсчета сцинтилляционный/люминесцентный счетчик MicroBeta Trilux (PerkinElmer), с помощью которого определили оставшееся количество мочевой кислоты [8-14C]. Для расчета степени ингибирования абсорбции мочевой кислоты [8-14C] соединением использовали следующую Формулу, а затем с помощью программного обеспечения XLfit вычислили величины IC50, которые показаны в Таблице 2.
Степень ингибирования (%) = [поглощение 1-(14C лункой с лекарством - поглощение 14С отрицательным контролем)/(поглощение 14С лункой с буфером - поглощение 14С отрицательным контролем)] × 100; при этом отрицательным контролем являлась не вакцинированная линия клеток, стабильно экспрессирующих hURAT1.
2) Результаты испытания
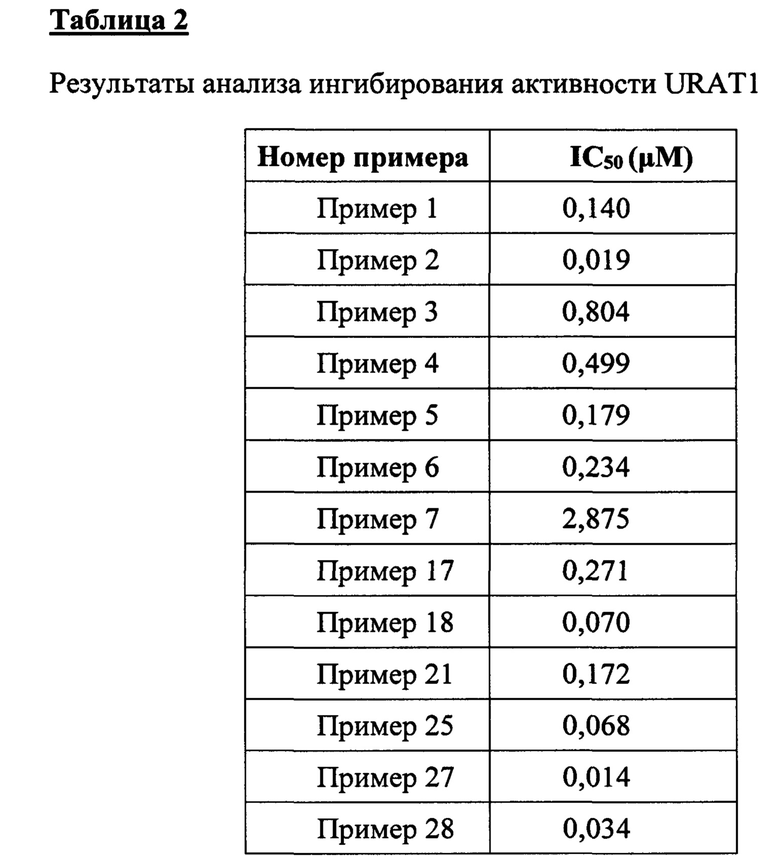
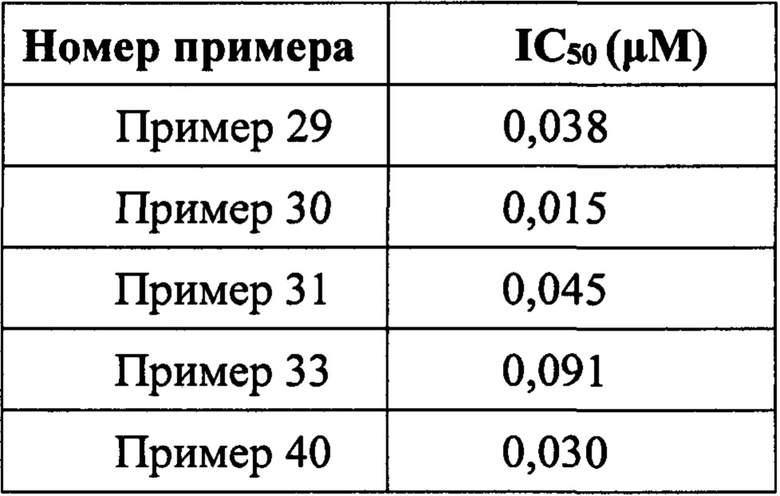
Вывод: Результаты испытания показывают, что соединения согласно изобретению обладает хорошей ингибирующей активностью в отношении URAT1.
Пример 3
Оценка фармакокинетики
1) Метод испытания
Крыс линии SD, которые в течение 15 часов в течение ночи не получали корма, случайным образом распределили по группам в зависимости от массы тела. Растворили испытуемое соединение в растворе из 5% ДМСО + 5% солутола + 90% солевого раствора. Внутривенно ввели животным из опытной группы дозу 1 мг/кг; перорально животным из опытной группы дозу 5 мг/кг. Затем собрали венозную кровь (около 0,2 мл) в момент времени 0, 0,083 (только в группе, получившей внутривенную инъекцию), 0,25, 0,5, 1,0, 2,0, 5,0, 7,0 и 24 ч после введения лекарственного средства. Поместили венозную кровь в пробирку с антикоагулянтом EDTAK2 и в течение 2 мин центрифугировали со скоростью 11000 об/мин. Собрали плазму и хранили ее при -20°С или при -70°С до проведения анализа методом ЖХ/МС/МС. Измерили концентрацию лекарственного средства в плазме в каждый момент времени, и рассчитали фармакокинетические параметры на основании кривой зависимости концентрации лекарственного средства и времени.
Рассчитали указанным выше способом фармакокинетические свойства соединений согласно настоящему изобретению, которые приведены в Таблице 3.
2) Результаты испытания
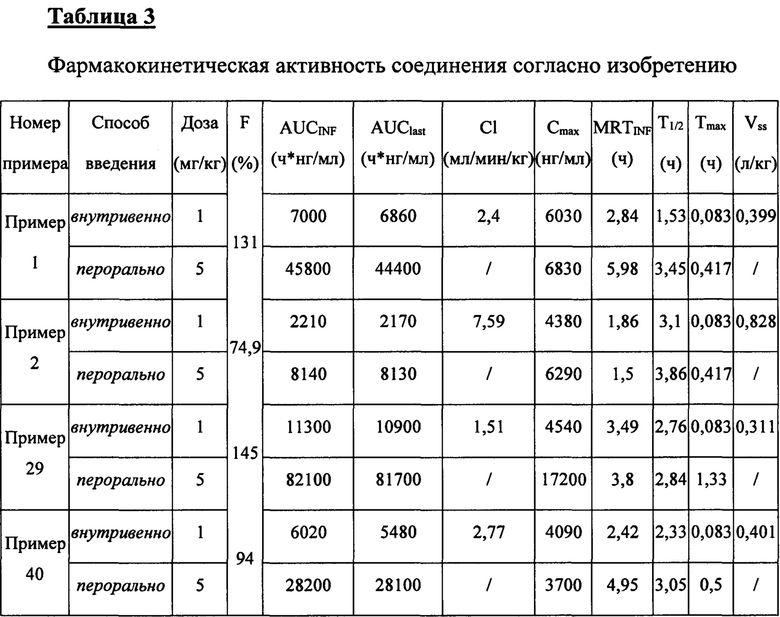
"/" Не испытывалось
Выводы: Результаты испытания, приведенные в Таблице 3, показывают, что соединения согласно настоящему изобретению, имели высокие концентрации в плазме и уровни воздействия на крыс после перорального введения, и имели низкие скорости клиренса и длительный период полураспада. Таким образом, соединения согласно настоящему изобретению, имели хорошие фармакокинетические характеристики.
Наконец, следует отметить, что существуют и другие способы практического осуществления изобретения. Соответственно, хотя были рассмотрены и описаны пояснительные варианты осуществления, специалистам в данной области техники ясно, что эти варианты осуществления не могут считаться ограничивающими настоящее изобретение, и возможны изменения, эквивалентные альтернативы и модификации, не выходящие за пределы объема настоящего изобретения. Все упоминаемые публикации или патенты в порядке ссылки включены в настоящую заявку.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БИС(ГИДРОКСИМЕТИЛ)ЦИКЛОБУТИЛ ПУРИНОВ ИЛИ ПИРИМИДИНОВ | 1989 |
|
RU2041213C1 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ПРОИЗВОДНЫЕ ФЛАВОНА ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ НАРУШЕНИЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2418786C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2724100C2 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
| СОЕДИНЕНИЯ ДИГИДРОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2015 |
|
RU2682672C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| ИМИДАЗОПИРИДАЗИНЫ | 2012 |
|
RU2662443C2 |
Изобретение относится к соединениям общей формулы (I), U представляет собой фенил; каждый из R1 и R2 независимо представляет собой Н, D, ОН или C1-6-галогеналкил; Т представляет собой NO2, CN или CF3; X представляет собой CR4 или N; R4 представляет собой Н; каждый из Y и Z независимо представляет собой С, СН или N;  представляет собой одинарную или двойную связь; Q представляет собой фенил, С4-7 карбоцикл, 5-членный гетероцикл, включающий 1-2 гетероатома, которые независимо выбраны из O, S или N, или 5-6-членное гетероароматическое кольцо, включающее 1-2 гетероатома, которые независимо выбраны из O, S или N; каждый R3 независимо представляет собой Н, D, галоген, C1-6-алкил, C1-6-галогеналкил или 3-8-членный циклоалкил; m равно 0 или 1; и n равно 0, 1, 2 или 3; а также к их сложным эфирам и фармацевтически приемлемым солям. Технический результат – соединения формулы (I) в качестве ингибиторов ксантиноксидазы (XO) и переносчиков 1 уратов (URAT1). 10 з.п. ф-лы, 3 табл., 43 пр.
представляет собой одинарную или двойную связь; Q представляет собой фенил, С4-7 карбоцикл, 5-членный гетероцикл, включающий 1-2 гетероатома, которые независимо выбраны из O, S или N, или 5-6-членное гетероароматическое кольцо, включающее 1-2 гетероатома, которые независимо выбраны из O, S или N; каждый R3 независимо представляет собой Н, D, галоген, C1-6-алкил, C1-6-галогеналкил или 3-8-членный циклоалкил; m равно 0 или 1; и n равно 0, 1, 2 или 3; а также к их сложным эфирам и фармацевтически приемлемым солям. Технический результат – соединения формулы (I) в качестве ингибиторов ксантиноксидазы (XO) и переносчиков 1 уратов (URAT1). 10 з.п. ф-лы, 3 табл., 43 пр.
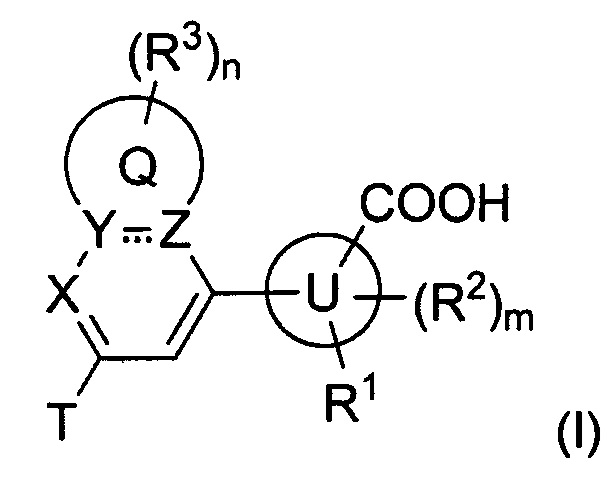
1. Соединение формулы (I) или его сложный эфир, фармацевтически приемлемая соль
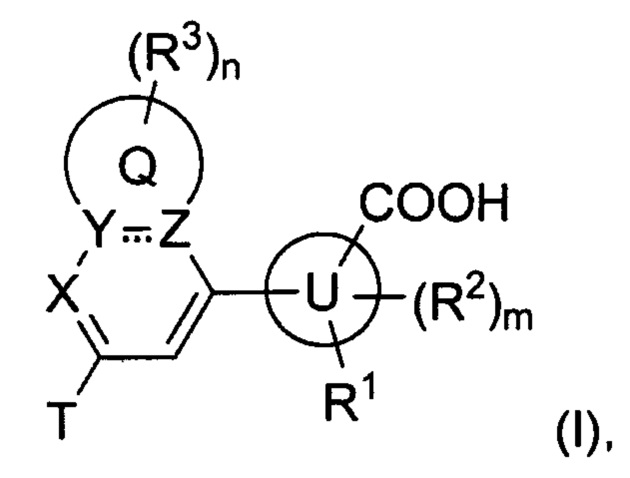
где U представляет собой фенил;
каждый из R1 и R2 независимо представляет собой Н, D, ОН или C1-6-галогеналкил;
Т представляет собой NO2, CN или CF3;
X представляет собой CR4 или N;
R4 представляет собой Н;
каждый из Y и Z независимо представляет собой С, СН или N;
 представляет собой одинарную или двойную связь;
представляет собой одинарную или двойную связь;
Q представляет собой фенил, С4-7 карбоцикл, 5-членный гетероцикл, включающий 1-2 гетероатома, которые независимо выбраны из O, S или N, или 5-6-членное гетероароматическое кольцо, включающее 1-2 гетероатома, которые независимо выбраны из O, S или N;
каждый R3 независимо представляет собой Н, D, галоген, C1-6-алкил, C1-6-галогеналкил, или 3-8-членный циклоалкил;
m равно 0 или 1; и
n равно 0, 1, 2 или 3;
при условии, что:
(1) когда Т представляет собой CF3, R1 представляет собой ОН;
(2) когда Т представляет собой NO2, R1 не означает Н.
2. Соединение по п. 1, имеющее формулу (II), или его фармацевтически приемлемая соль
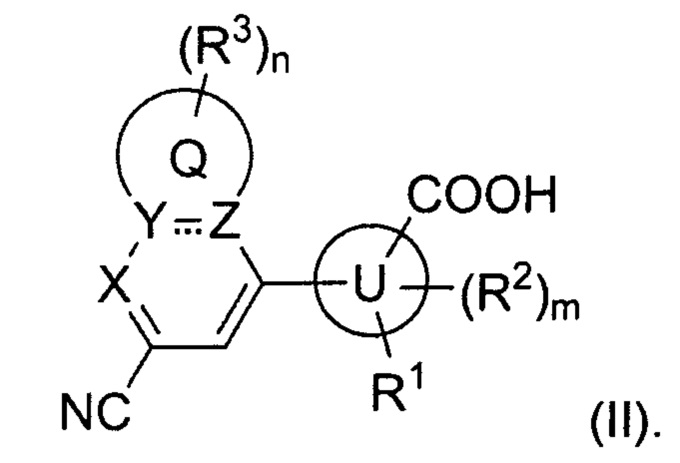
3. Соединение по п. 1, имеющее Формулу (III), или его фармацевтически приемлемая соль
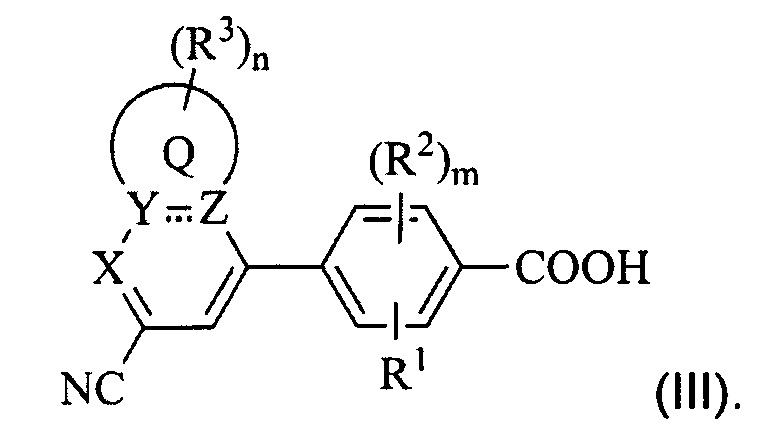
4. Соединение по пп. 1, 2 или 3, где каждый R1 и R2 независимо представляет собой Н, D, ОН или С1-4-галогеналкил.
5. Соединение по п. 4, где каждый R1 и R2 независимо представляет собой Н, ОН, дифторметил или трифторметил.
6. Соединение по пп. 1, 2 или 3, где каждый R3 независимо представляет собой Н, галоген, метил, этил, изопропил, дифторметил, трифторметил, циклопропил или циклобутил.
7. Соединение по пп. 1, 2 или 3, где R4 представляет собой Н и
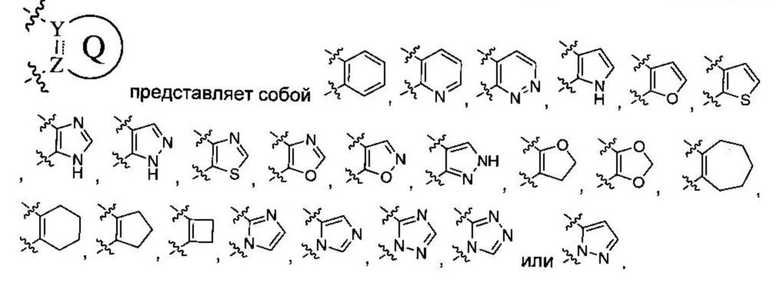
8. Соединение по пп. 1, 2 или 3, где 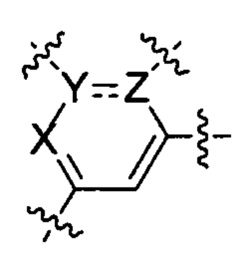 представляет собой
представляет собой 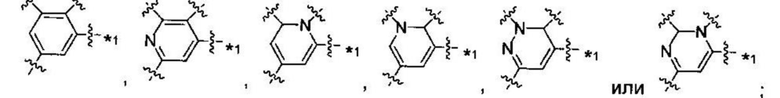
при этом "*1" относится к положению, прикрепленному к U-кольцу.
9. Соединение по п. 1, имеющее одну из следующих формул:
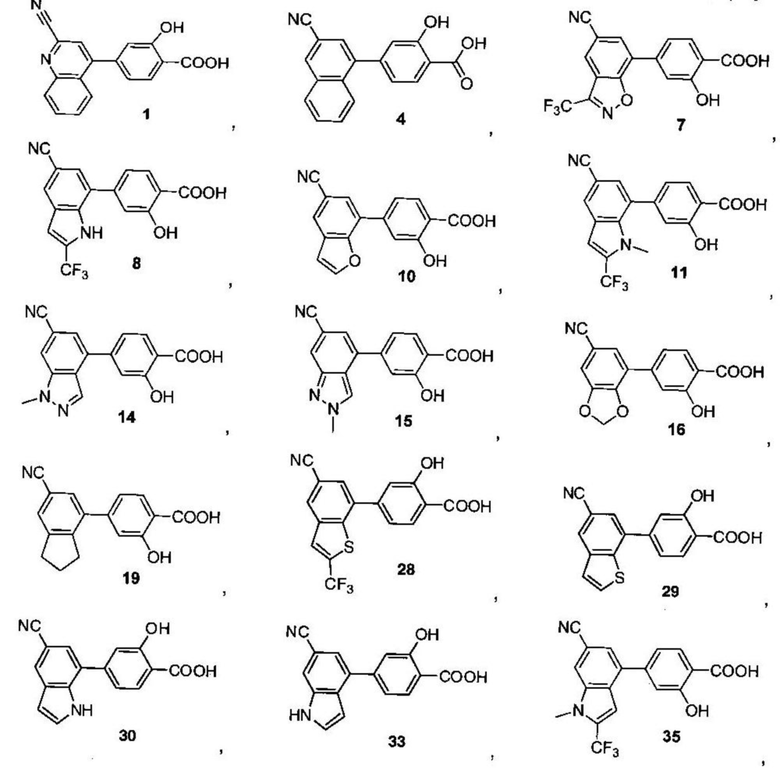
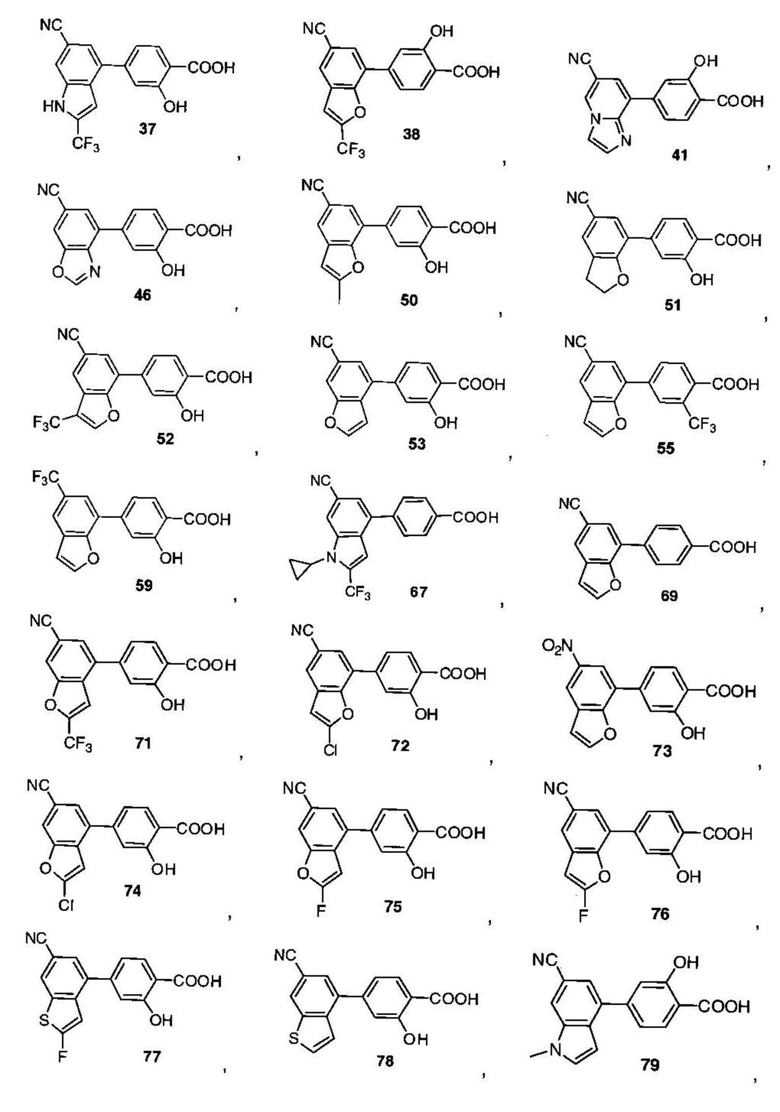
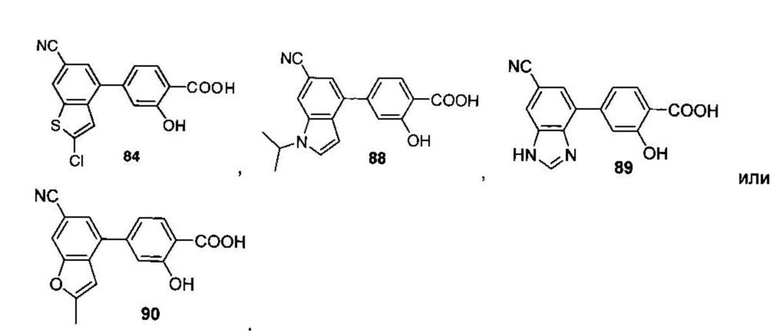
10. Соединение по любому из пп. 1-9 для применения при снижении уровня мочевой кислоты в крови.
11. Соединение по любому из пп. 1-9 для применения при ингибировании ксантиноксидазы и переносчика 1 уратов.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| US 5614520 А1, 25.03.1997 | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| СОДЕРЖАЩЕЕ КОНДЕНСИРОВАННУЮ КОЛЬЦЕВУЮ СТРУКТУРУ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2009 |
|
RU2512547C2 |

