Настоящее изобретение относится к способу получения N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида. Объектом настоящего изобретения являются также новые твердые формы этого соединения, их применение при лечении заболеваний, вызванных дерегулируемой активностью протеинкиназы, и содержащие их фармацевтические композиции.
Нарушение функции протеинкиназ (PK) является отличительным признаком многочисленных заболеваний. Большая часть онкогенов и протоонкогенов, принимающих участие в злокачественных новообразованиях у человека, кодирует PK. Повышенная активность PK также имеет место при многих доброкачественных заболеваниях, таких как доброкачественная гиперплазия предстательной железы, наследственный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация васкулярных гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
PK также участвуют в воспалительных состояниях и в размножении вирусов и паразитов. PK также могут играть важную роль в патогенезе и развитии нейродегенеративных расстройств.
В качестве общих ссылок, описывающих нарушения нормальной работы и дерегуляции PK, см., например, Current Opinion in Chemical Biology 1999, 3, 459-465; Nature Rev. Drug Discov. 2002; и Carcinogenesis 2008, 29, 1087-1091.
Подмножество PK представляет собой группу мембранных рецепторов с внутренней активностью протеин-тирозинкиназы (RPTK). После связывания с факторами роста RPTK становятся активированными и фосфорилируют себя и серии субстратов в цитоплазме. Посредством этого механизма они могут преобразовывать внутриклеточную передачу сигналов для пролиферации, дифференцировки или других биологических изменений. Структурные аномалии, сверхэкспрессия и активация RTPK часто наблюдаются в опухолях человека, предполагая, что конститутивная инициация сигнальной трансдукции, приводящая к клеточной пролиферации, может привести к злокачественной трансформации. Киназа анапластической лимфомы (ALK) представляет собой рецептор тирозинкиназы, принадлежащий к подсемейству рецепторов инсулина RTK: ген ALK расположен на хромосоме 2 и экспрессируется в основном в нервных клетках, особенно в период развития. Ген ALK принимает участие в сбалансированной хромосомной транслокации с геном нуклеофосмина (NPM) на хромосоме 5 в большой субпопуляции анапластических крупноклеточных лимфом (ALCL). В ALK+ ALCL в результате транслокации убиквитарный промотор NPM регулирует эктопическую экспрессию слитого белка, в которой часть NPM димеризуется, и домен киназы ALK подвергается аутофосфорилированию и становится конститутивно активным.
Многие литературные данные продемонстрировали, что слитый белок NPM-ALK обладает сильным онкогенным потенциалом, и его эктопическая экспрессия является причиной клеточной трансформации. Кроме того, конститутивная экспрессия NPM-ALK человека в мышиных Т-клеточных лимфоцитах является достаточной для развития лимфоидной неоплазии у трансгенных животных с коротким периодом латентности.
ALCL является установленным заболеванием, характеризующимся поверхностной экспрессией антигена CD30 (Ki-1), и является причиной неходжкинских лимфом у 2% взрослых пациентов и 13% детей, поражая преимущественно молодых пациентов мужского пола. ALK+ ALCL составляет 70% всех ALCL и является агрессивным заболеванием с системными симптомами и частым экстралимфатическим поражением (костного мозга, кожи, кости, мягких тканей).
Обнаружено, что приблизительно 15-20% ALK-экспрессирующих ALCL имеют различные хромосомальные транслокации, включающие цитоплазматическую часть ALK с разными N-концевыми частями, приводящими к конститутивной активации домена ALK-киназы.
Кроме того, установлено, что клеточные линии, происходящие из солидных опухолей эктодермального происхождения, подобных меланоме, раку молочной железы, а также нейробластоме, глиобластоме, саркоме Юинга, ретинобластоме, экспрессируют рецептор ALK.
В заключение, препятствие передачи сигнала ALK, вероятно, представляет собой специфический и эффективный путь блокирования пролиферации опухолевых клеток при ALCL и возможных других показаниях.
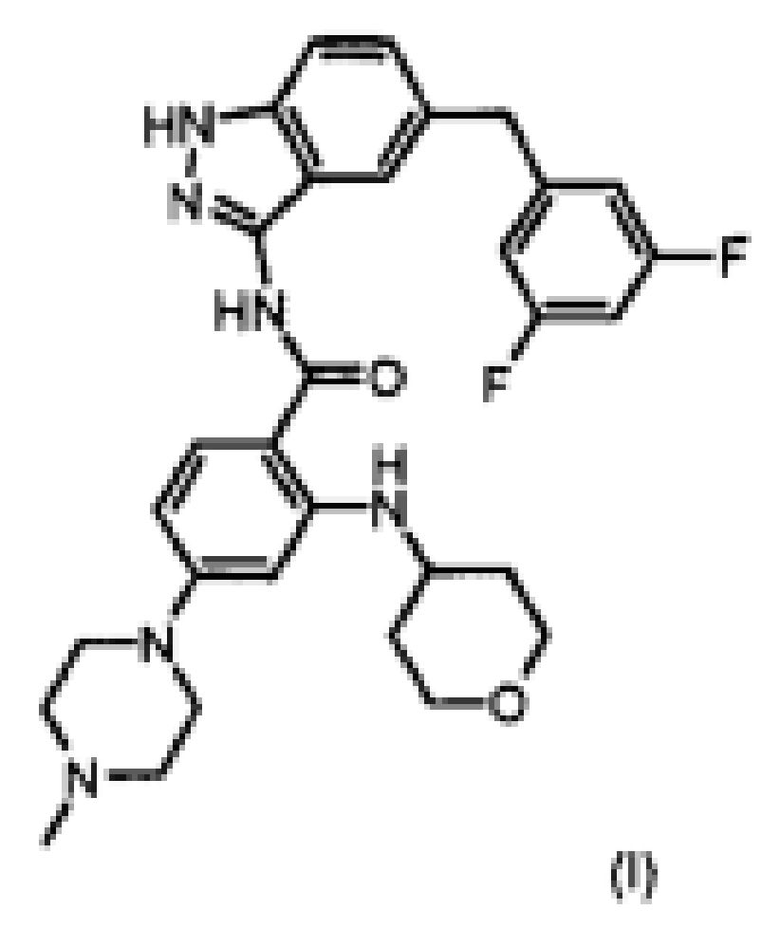
В международной патентной заявке WO 2009/013126 (Nerviano Medical Sciences Sri.) описана и заявлена форма свободного основания N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида, которая имеет формулу (I)
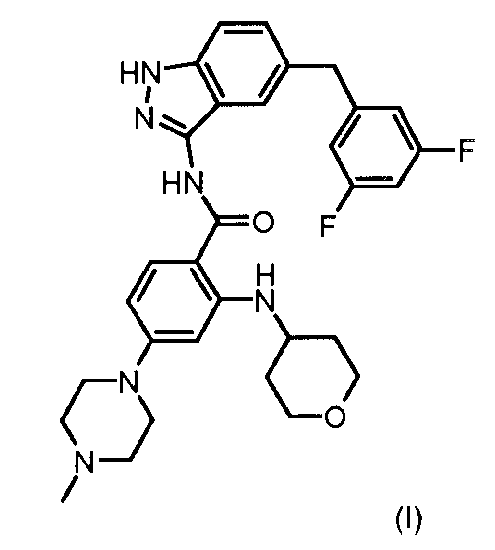
и сообщается, что соединение действует как ингибитор киназы, в частности как ингибитор ALK и, таким образом, может быть использовано при лечении различных видов злокачественного новообразования и клеточных пролиферативных нарушений.
Способы получения этого соединения описаны в примере 2 (стадия i′) и в примере 7 вышеуказанной патентной заявки.
Известное получение соединения формулы (I), как описано в примере 2 (стадия i′) вышеуказанной патентной заявки, включает, по существу, добавление раствора 5-(3,5-дифторбензил)-1Н-индазол-3-иламина к хлорангидриду 4-(4-метилпиперазин-1-ил)-2-[(тетрагидропиран-4-ил)-(2,2,2-трифторацетил)амино]бензойной кислоты и затем удаление из полученного соединения защитной группы с помощью органического основания при высокой температуре, чтобы получить требуемый амид формулы (I) после очистки с помощью колоночной хроматографии и кристаллизации.
Известный способ получения соединения формулы (I), как описано в примере 7 вышеуказанной патентной заявки, включает, по существу, взаимодействие 2-амино-N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)бензамида с тетрагидропиран-4-оном в присутствии трифторуксусной кислоты и триацетоксиборгидрида тетраметиламмония с получением желаемого амида формулы (I) после очистки с помощью колоночной хроматографии.
В связи с этим авторы настоящего изобретения неожиданно обнаружили, что соединение формулы (I) может быть удобным образом получено посредством способа, который позволяет получить желаемый продукт в промышленном масштабе легко воспроизводимым способом, с высокой степенью чистоты, с характеристиками, пригодными для введения людям и с меньшими затратами. Кроме того, новый способ больше подходит для применения в крупносерийном производстве. И, наконец, указанное соединение получают в определенных твердых формах.
Таким образом, первым объектом настоящего изобретения является способ получения соединения формулы (I), как описано выше, где способ включает:
a) добавление в стехиометрическом соотношении ацилхлорида формулы (II):
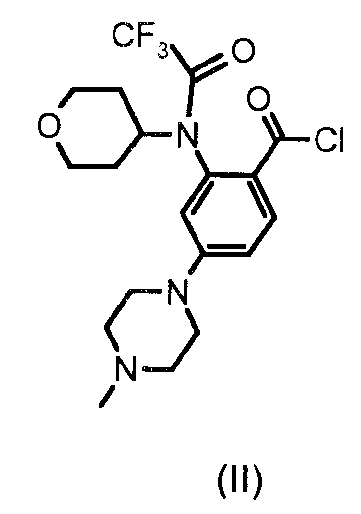
к индазол-3-иламину формулы (III):
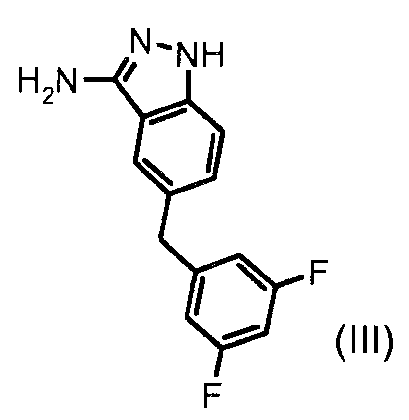
прекращение добавления, когда индазол-3-иламин формулы (III) полностью прореагировал;
b) удаление защитной группы в мягких основных условиях из соединения формулы (IV):
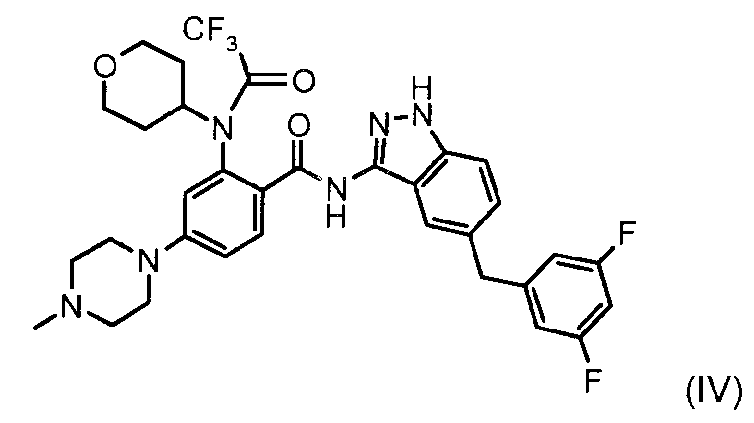
с получением желаемого соединения формулы (I), которое выделяют в аморфной форме;
желаемую кристаллическую форму затем получают либо
c1) обработкой полученного аморфного соединения формулы (I) смесью этанола и воды в присутствии затравочных кристаллов с получением желаемого соединения формулы (I) в кристаллической форме 1, либо
c2) обработкой полученного аморфного соединения формулы (I) смесью этанола и воды с получением желаемого соединения формулы (I) в кристаллической форме 2, и,
необязательно,
d) преобразование соединения, полученного на стадии b), на стадии с1) или на стадии c2) в фармацевтически приемлемую соль.
Новый способ позволяет получить соединения формулы (I) с высокой степенью чистоты без хроматографической очистки и контролируя вид твердой формы.
Благодаря порядку добавления и стехиометрическому количеству ацилпроизводного формулы (II) к индазолиламиновому производному формулы (III) с последующим выделением обработкой соответствующей смесью растворителей, защищенное промежуточное соединение формулы (IV), полученное на стадии а), является более чистым, чем в известном способе. Действительно, этот новый способ значительно снижает образование примесей, таких как, например, образование нежелательных региоизомеров и продуктов двойного присоединения, таким образом избегая необходимости очищать полученный таким образом продукт формулы (IV) с использованием хроматографических колонок, что является не подходящим для масштабного получения из-за времени и затрат, связанных с этим способом.
Далее на стадии b) преобразование продукта формулы (IV) в конечный продукт формулы (I) в мягких условиях удаления защитной группы, предполагающих низкотемпературный водный гидролиз с неорганическими основаниями, предотвращает образование побочных продуктов преобразования, наблюдаемых в известном способе из-за высокой температуры обработки органическими основаниями в метаноле.
Наконец, на стадии c1) или стадии c2) соединение формулы (I), получаемое сначала в аморфной форме, затем преобразуют соответственно в кристаллическую форму 1 путем внесения затравки или в кристаллическую форму 2 путем обработки соответствующими растворителями. В известном способе в этот момент чистота соединения и методы выделения путем инсолюбилизации и/или путем хроматографической очистки были такими, чтобы предотвратить преобразование либо в кристаллическую форму 1 или кристаллическую форму 2.
В соответствии со стадией a) соединение формулы (II) суспендируют в растворителях, таких как ТГФ или DCM, предпочтительно, суспендируют в сухом DCM и затем суспензию медленно и постепенно добавляют к раствору соединения формулы (III) в пиридине.
Предпочтительно, реакцию проводят при температуре в интервале между -20°C и -40°C, предпочтительно при температуре в интервале между -30°C и -40°C.
В конце реакции растворители выпаривают и остаток обрабатывают растворителями, такими как DCM, MTBE, MeOH, в предварительно определенном соотношении в интервале между 1/1/1 и 30/30/1, предпочтительно обработку проводят при соотношении DCM/MTBE/MeOH в интервале между 8/8/1 и 30/30/1 с получением осадка чистого соединения формулы (IV).
Согласно стадии b) удаление защитной группы соединения формулы (IV) может быть проведено в мягких основных условиях, таких как водные или водно/метанольные растворы щелочных карбонатов или гидроксидов, предпочтительно, используется раствор К2СО3 в воде/метаноле.
Предпочтительно, реакцию проводят при температуре в интервале между 20°C и 5°C, предпочтительно при температуре около 10°C. Желаемое соединение формулы (I) затем выделяют в аморфной форме путем прибавления по каплям в воду при температуре в интервале между 5°C и 25°C, предпочтительно при температуре в интервале между 5°C и 10°C.
Согласно стадии c1) аморфное соединение формулы (I) обрабатывают сначала этанолом, нагревая при кипячении с обратным холодильником и отгоняя часть растворителя, затем водой и затравкой кристаллической формы 1 при температуре в интервале между 10°C и 30°C, предпочтительно при температуре в интервале между 20°C и 25°C. Полученное соединение формулы (I) является кристаллической формой 1.
Согласно стадии c2) продукт, полученный согласно стадии b), обрабатывают последовательно этанолом при температуре в интервале между 10°C и 30°C, предпочтительно при температуре в интервале между 20°C и 25°C, а затем водой при температуре в интервале между 10°C и 30°C, предпочтительно при температуре в интервале между 20°C и 25°C. Полученное соединение формулы (I) является кристаллической формой 2.
Исходные соединения и реагенты, используемые в способе по настоящему изобретению, являются известными соединениями или могут быть получены из известных соединений с помощью хорошо известных методов. В частности, получение соединений формул (II) и (III), определенных выше, описано в вышеупомянутой патентной заявке.
В примере 2 (стадия i′) и в примере 7 вышеуказанной патентной заявки нет указаний на твердую форму, аморфную или кристаллическую. Авторы настоящего изобретения провели исследования и обнаружили, что соединение формулы (I), полученное как описано в примере 2 (стадия i′), представляет собой кристаллический сольват, который далее в настоящем документе указывается для удобства как кристаллическая форма 3; соединение формулы (I), полученное как описано в примере 7, является аморфным и далее указывается как аморфная форма.
Кроме того, авторы настоящего изобретения обнаружили, что соединение формулы (I), полученное как описано в примере 1, стадия b) настоящей заявки, является аморфным; соединение формулы (I), полученное как описано в примере 1, стадия с1) настоящей заявки, является кристаллическим, которое далее называется кристаллической формой 1; наконец, соединение формулы (I), полученное как описано в примере 1, стадия c2) настоящей заявки, является кристаллическим, которое далее называется кристаллической формой 2.
Затем, в дальнейшем аспекте, настоящее изобретение относится к новым и стабильным кристаллическим формам соединения формулы (I), т.е. к кристаллической форме 1 и кристаллической форме 2, полученным способом, описанным выше.
Кристаллическая форма 3 представляет собой сольват с EtOAc и н-гексаном и не подходит для введения человеку в связи с наличием недопустимых количеств растворителей; аморфная форма представляет собой гигроскопичное твердое вещество, которое является менее пригодным для разработки перорального препарата.
Поглощение влаги является значительной проблемой для фармацевтических порошков. Было показано, что влажность имеет существенное влияние, например, на физические, химические и производственные свойства лекарственных средств, вспомогательных веществ и препаратов. Она также является ключевым фактором в принятии решения, касающегося упаковки, хранения, транспортировки и срока годности, и для успешной разработки необходимо глубокое понимание гигроскопических свойств.
Например, преобразование из безводной в гидратированную форму может наблюдаться, когда относительная влажность превышает критический уровень и содержание влаги быстро увеличивается в твердом веществе. Это имеет воздействие не только на физические и фармацевтические свойства лекарственных средств per se, но и на их биофармацевтическую перспективу. Кроме того, хорошо известно, что гидратные формы, как правило, имеют тенденцию быть менее растворимыми относительно гомологичной безводной формы, с потенциальным губительным влиянием также на свойства скорости растворения активного соединения per se и на его профиль абсорбции в желудочно-кишечном тракте. Таким же образом, преобразование из аморфной формы в кристаллическую форму может наблюдаться в присутствии влаги, с потенциальными недостатками в плане физической стабильности. Аморфное активное лекарственное средство, если оно гигроскопично, например, может абсорбировать относительно большое количество воды из атмосферы вплоть до его растворения, при этом также может быть затронута его химическая стабильность, поскольку аморфная структура, будучи термодинамически активированной, более подвержена химической деградации и химическому взаимодействию с другими химическими веществами. Таким образом, производительность и эффективность как препарата, так и активного ингредиента могут быть существенно изменены.
Соответственно, существует необходимость в терапии твердыми формами соединения формулы (I), пригодными для введения человеку, которые не содержат неприемлемые количества остаточных растворителей и обладают низкой гигроскопичностью, а также хорошими и воспроизводимыми биофармацевтическими свойствами, позволяющими более безопасное и эффективное пероральное введение.
Авторы настоящего изобретения решили описанную вышеуказанную техническую задачу путем предложения новых кристаллических форм соединения формулы (I), подходящих для введения человеку и обладающих улучшенными физико-химическими свойствами. Действительно, в новых кристаллических формах не содержатся остатки растворителей, и они менее гигроскопичны, чем аморфная форма, помимо всех других имеющихся преимуществ, в частности, терапевтических преимуществ, проявляемых известными формами.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Изобретение также иллюстрируется со ссылкой на прилагаемые чертежи, описываемые далее.
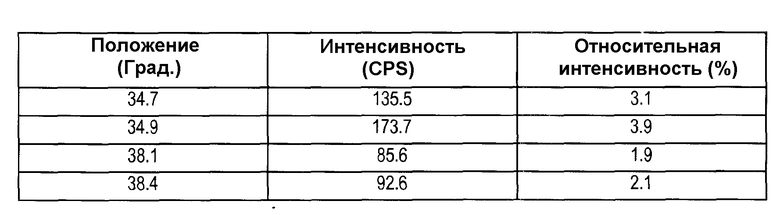
На фиг. 1 представлены рентгеновские дифрактограммы кристаллической формы 3, где 2-тета углы (град) показаны на оси x, а интенсивность (CPS) представлена на оси y.
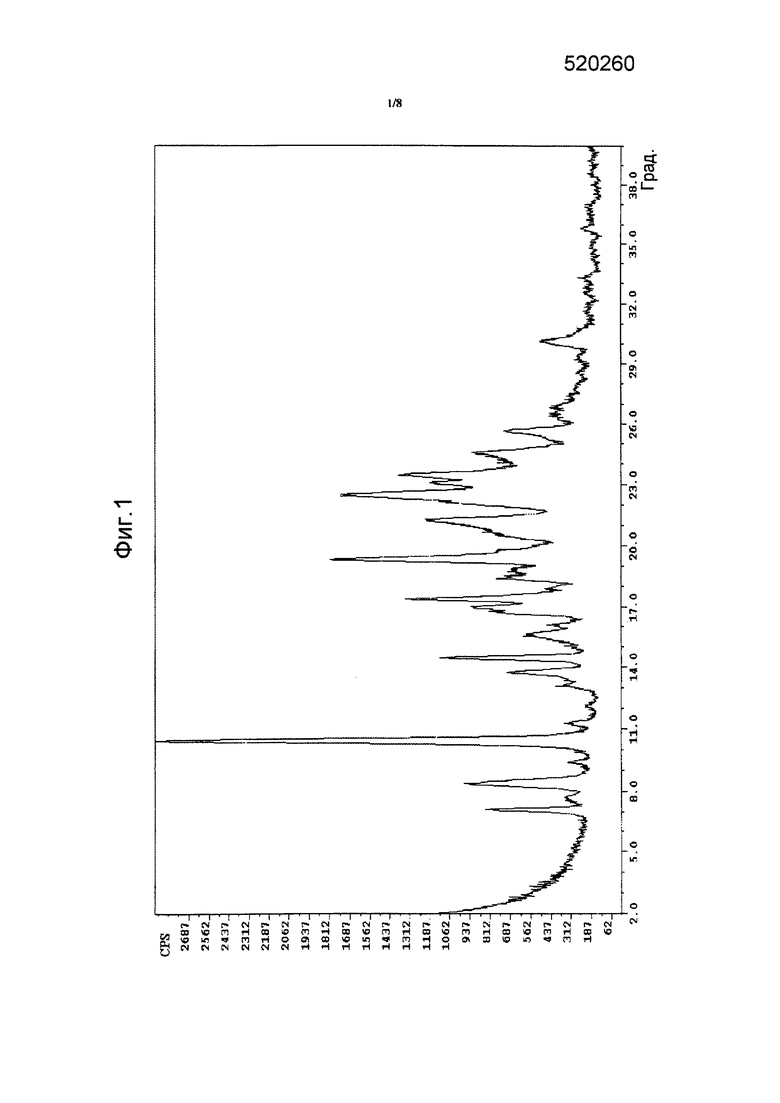
На фиг. 2 приведены рентгеновские дифрактограммы аморфной формы, где 2-тета углы (град) показаны на оси x, а интенсивность (CPS) представлена на оси y.
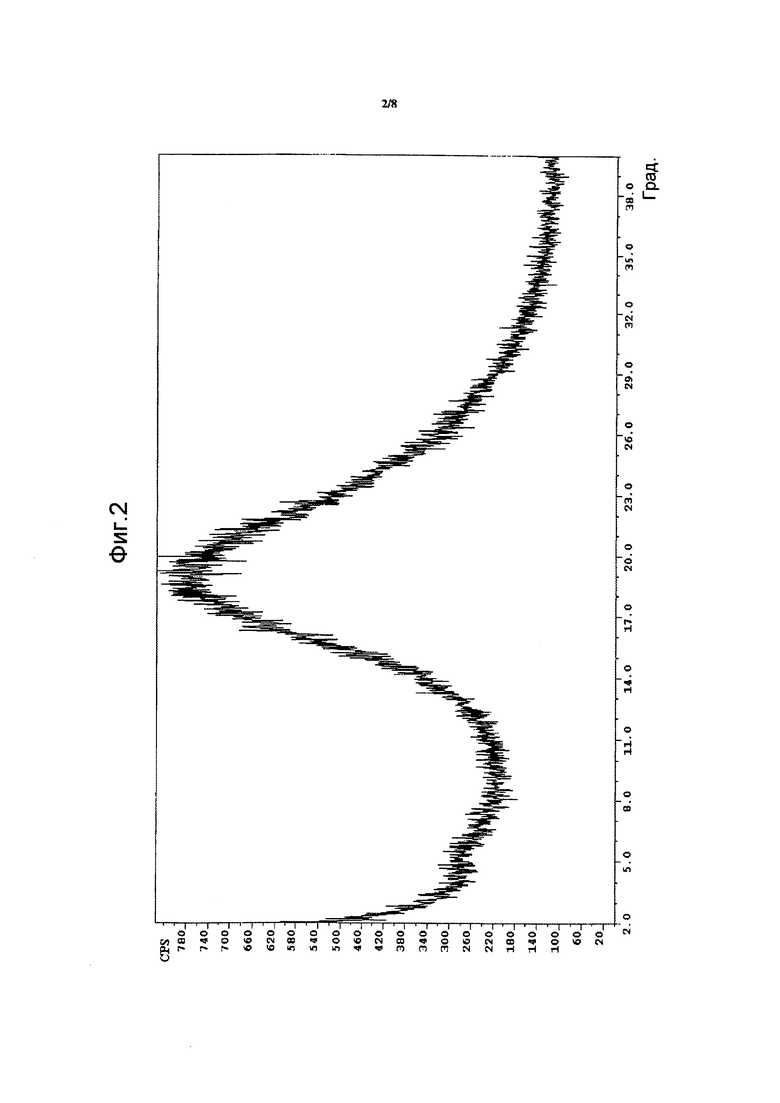
На фиг. 3 приведены рентгеновские дифрактограммы кристаллической формы 1, где 2-тета углы (град) показаны на оси x, а интенсивность (CPS) представлена на оси y.
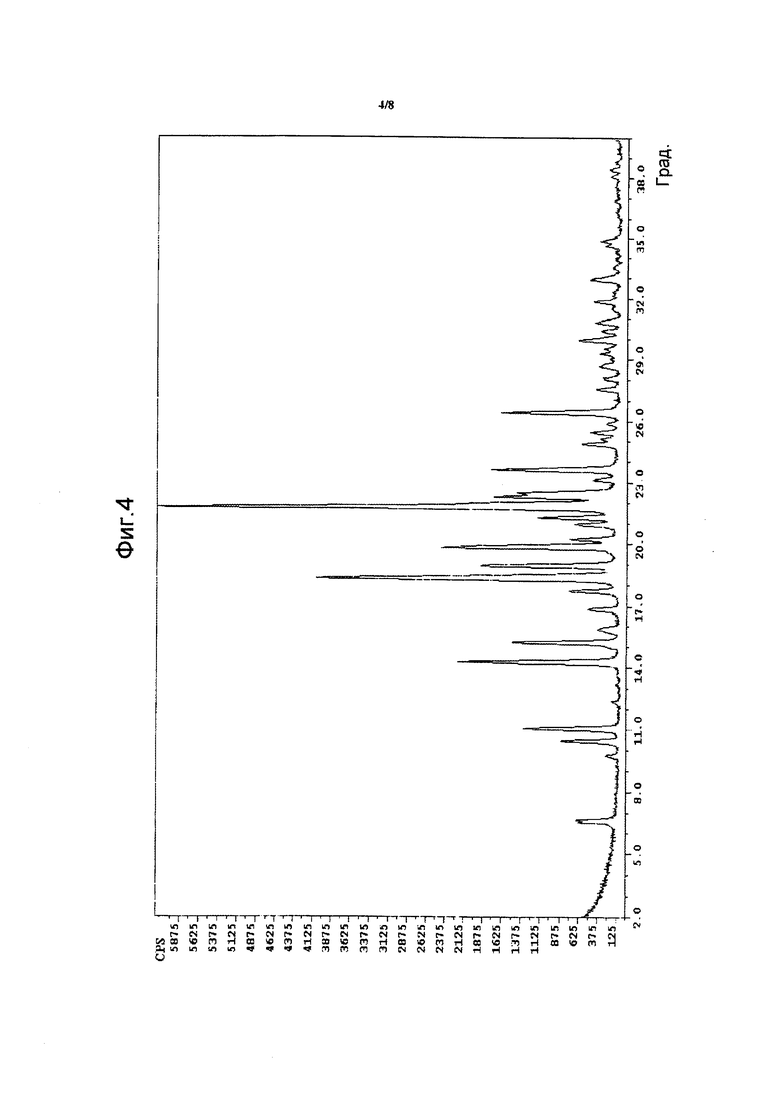
На фиг. 4 приведены рентгеновские дифрактограммы кристаллической формы 2, где 2-тета углы (град) показаны на оси x, а интенсивность (CPS) представлена на оси y.
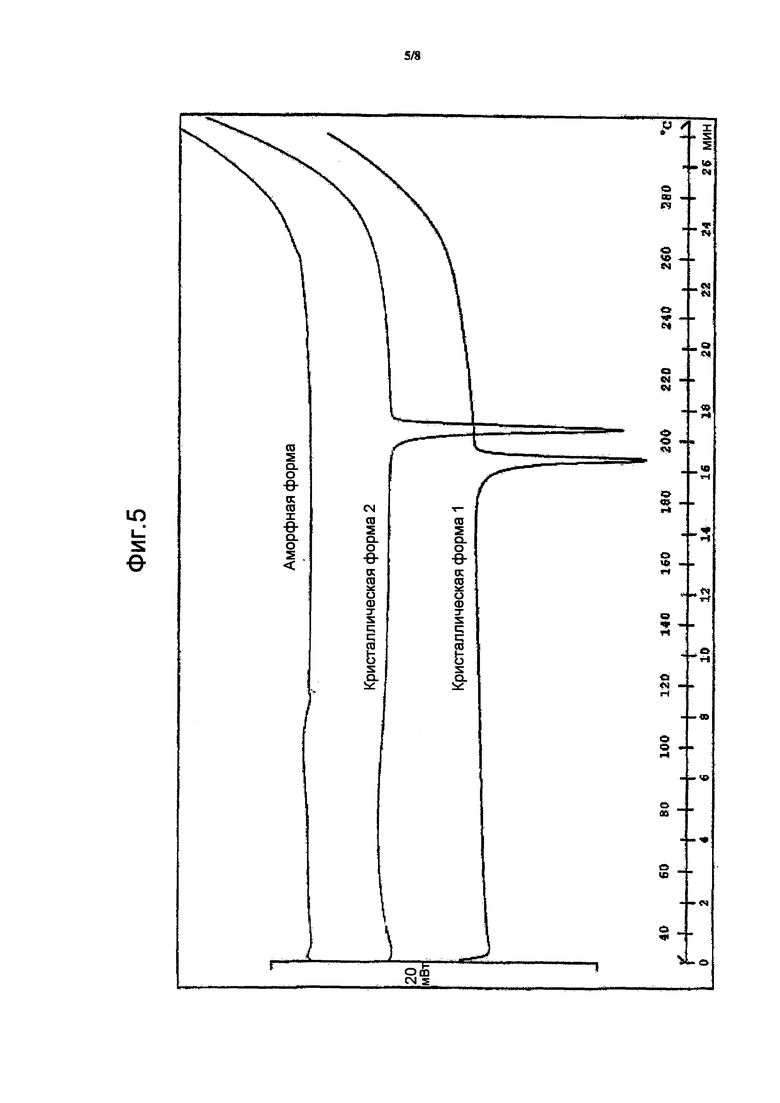
На фиг. 5 показаны термограммы DSC аморфной формы, кристаллической формы 1 и кристаллической формы 2.
На термограмме представлены температура (°C) и время (мин) на оси x, при этом тепловой поток (мВт) показан на оси y.
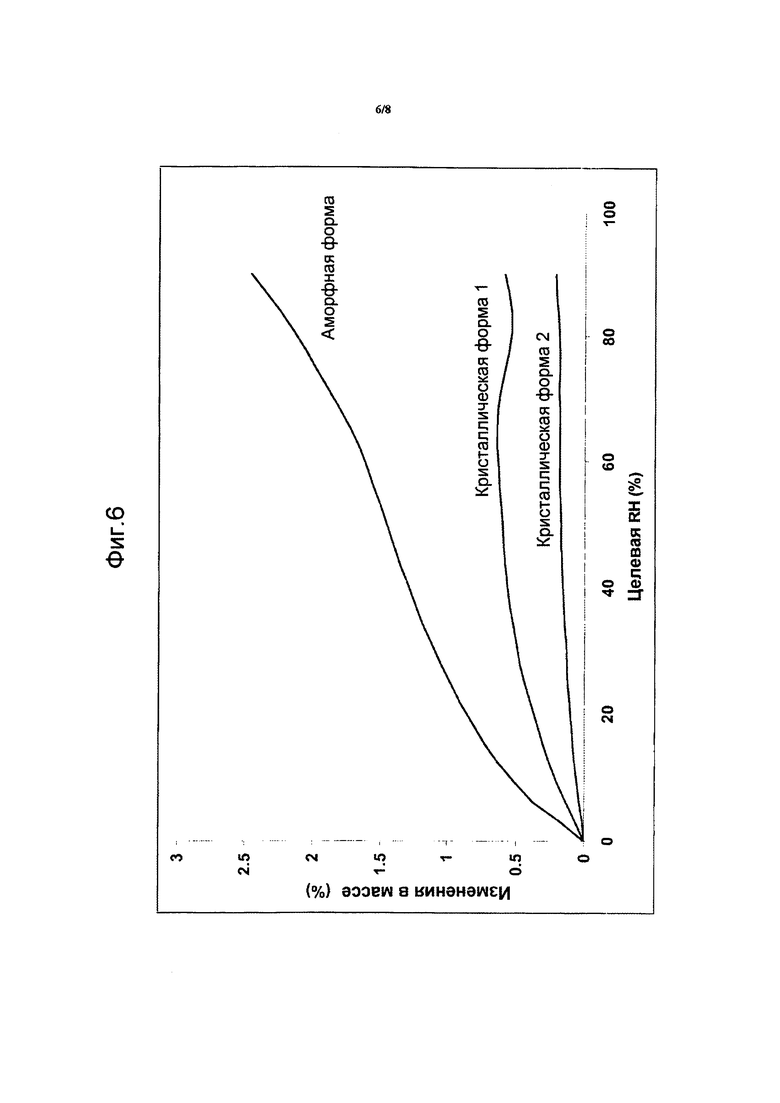
На фиг. 6 показана изотерма DVS участка аморфной формы, кристаллической формы 1 и кристаллической формы 2.
Значения относительной влажности воздуха (RH, %) приведены на оси x, а изменение массы (%) представлено на оси y. Кривые относятся к стадии сорбции в интервале между 0% RH и 90% RH при 25°C.
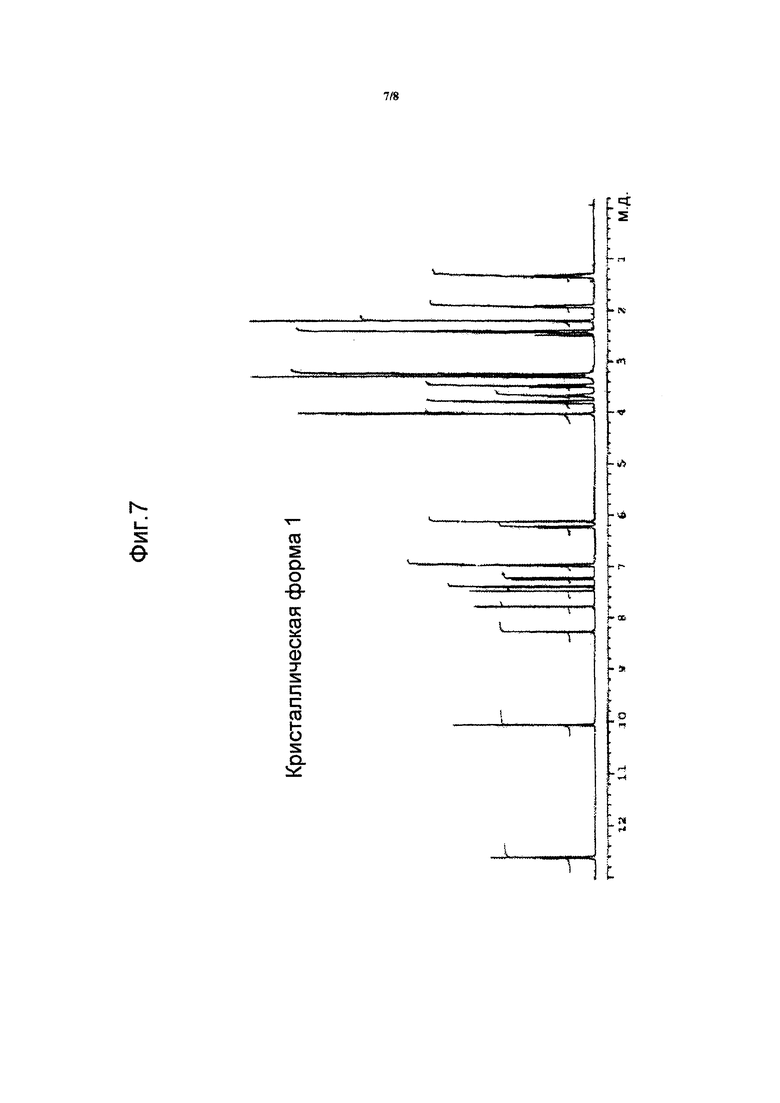
На фиг. 7 представлен 1H ЯМР спектр кристаллической формы 1, где химические сдвиги (м.д.) указаны на оси x.
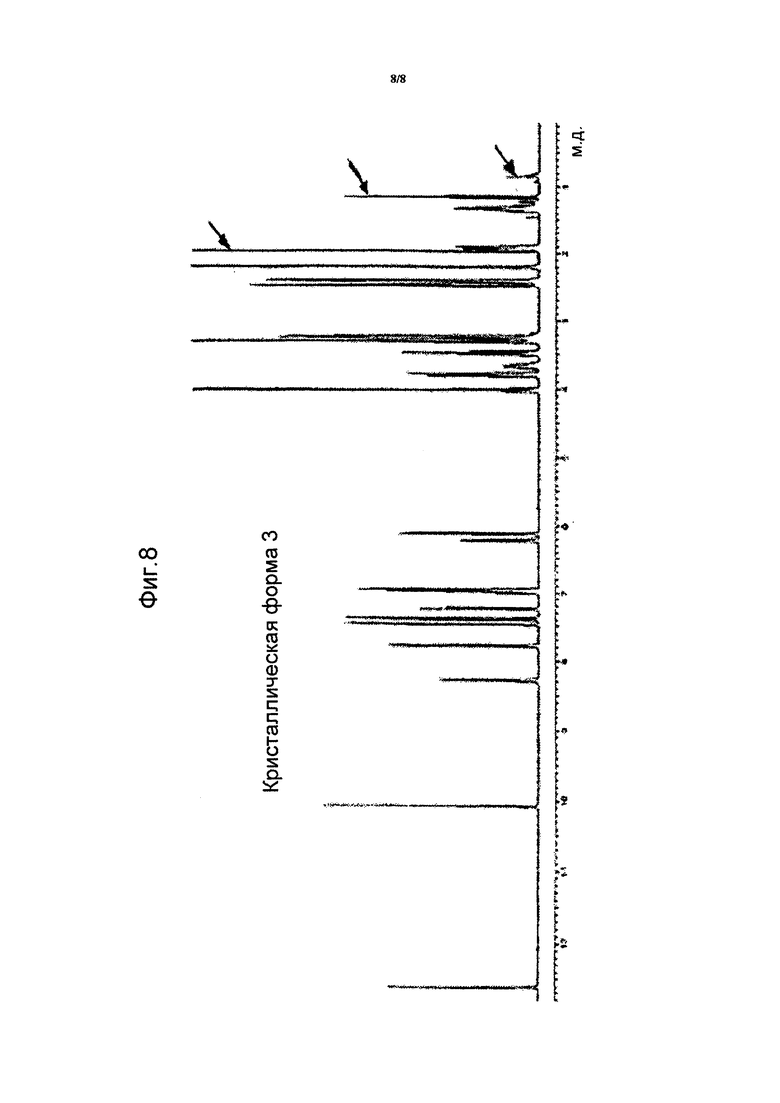
На фиг. 8 представлен 1H ЯМР спектр кристаллической формы 3, где химические сдвиги (м.д.) указаны на оси x.
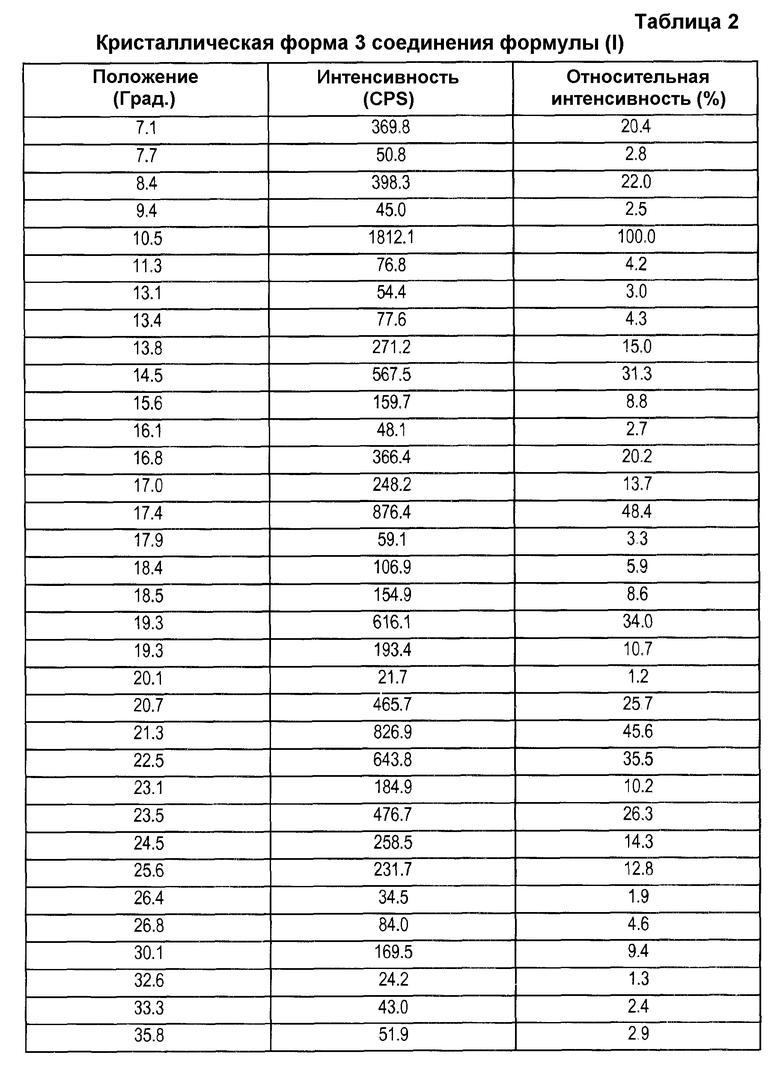
Кристаллическая форма 3 характеризуется диаграммой дифракции рентгеновских лучей, которая является по существу такой же, как и диаграмма, представленная на фиг. 1, со значительными интенсивностями пиков при значениях 2-тета (градусы), описанных в таблице 1. В образцах, не содержащих каких-либо дополнительных веществ (другие кристаллические формы, инертные вспомогательные вещества), можно наблюдать дифракционные пики при значениях 2-тета (градусы), описанные в таблице 2.
Аморфная форма характеризуется диаграммой дифракции рентгеновских лучей, которая является по существу такой же, как и диаграмма, представленная на фиг. 2.
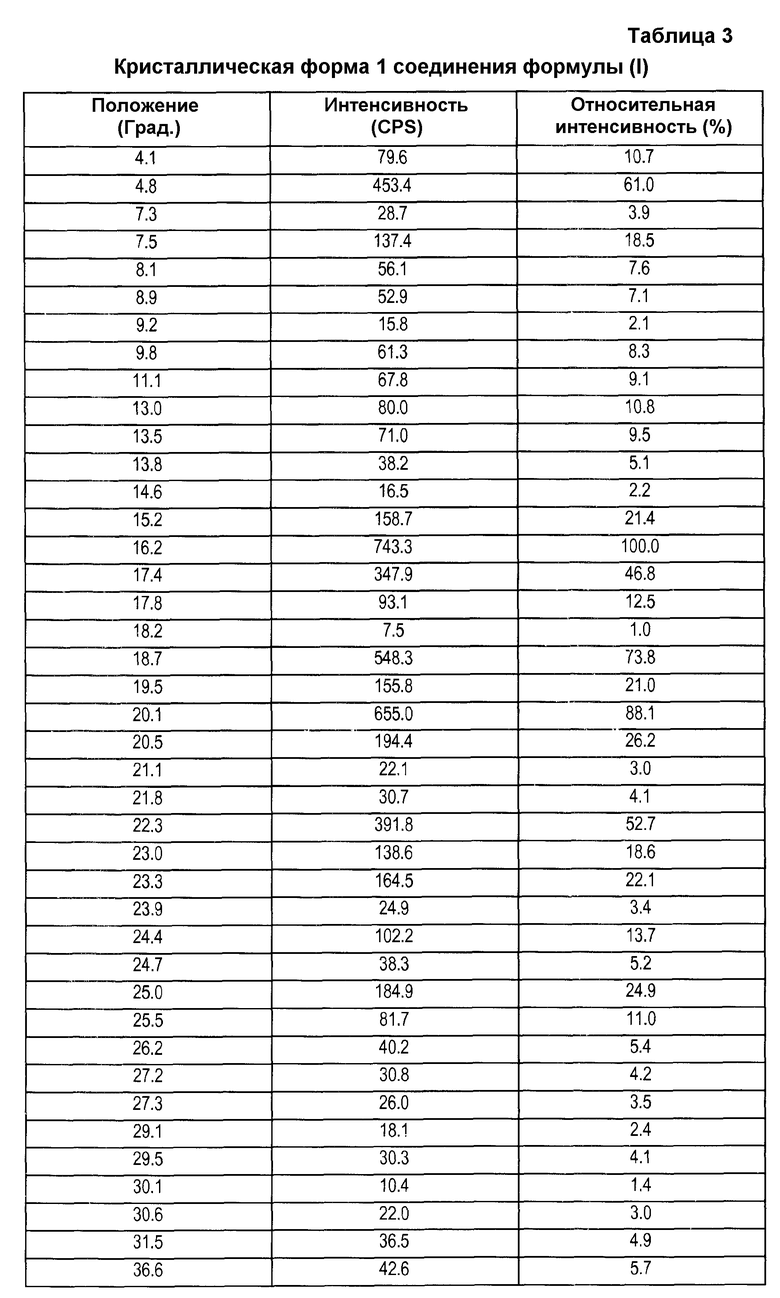
Кристаллическая форма 1 характеризуется диаграммой дифракции рентгеновских лучей, которая является по существу такой же, как и диаграмма, представленная на фиг. 3, со значительными интенсивностями пиков при значениях 2-тета (градусы), описанными в таблице 1. В образцах, не содержащих каких-либо дополнительных веществ (другие кристаллические формы, инертные вспомогательные вещества), можно наблюдать дифракционные пики при значениях 2-тета (градусы), описанные в таблице 3.
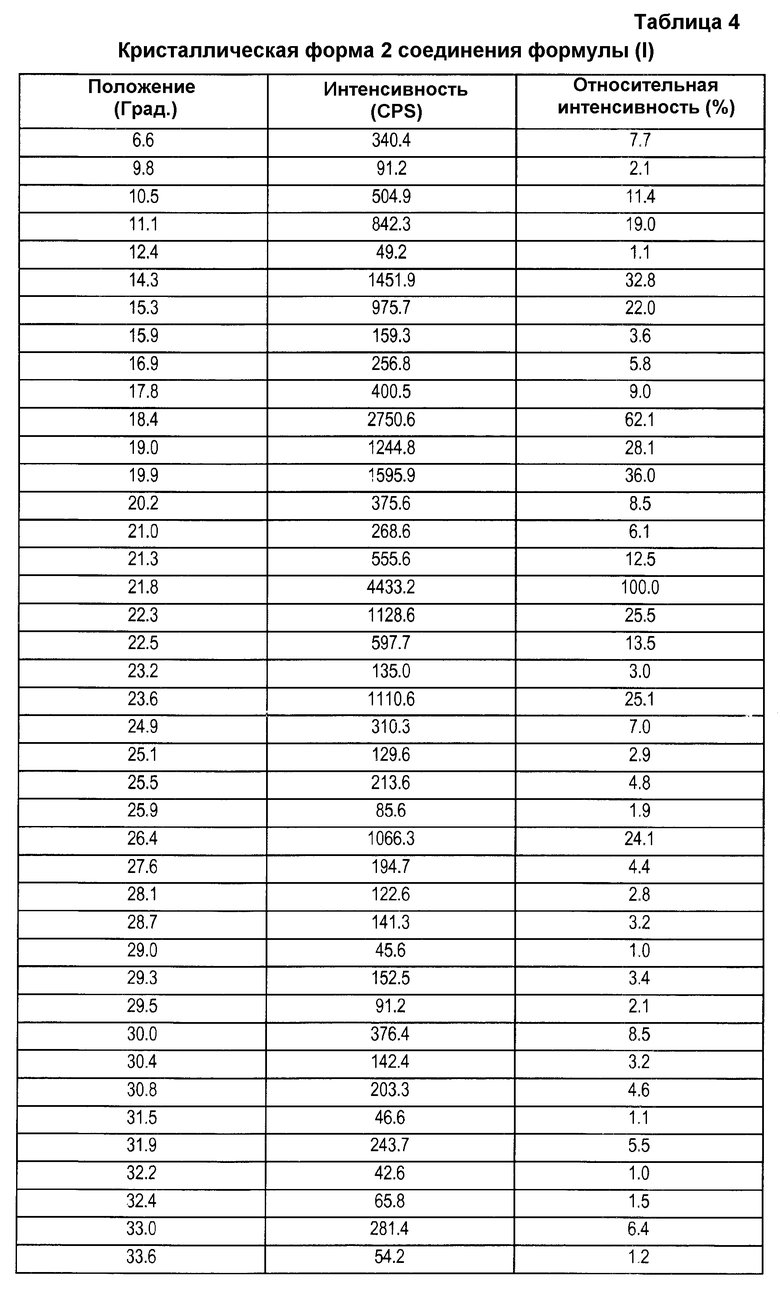
Кристаллическая форма 2 характеризуется диаграммой дифракции рентгеновских лучей, которая является по существу такой же, как и диаграмма, представленная на фиг. 4, со значительными интенсивностями пиков при значениях 2-тета (градусы), описанными в таблице 1. В образцах, не содержащих каких-либо дополнительных веществ (другие кристаллические формы, инертные вспомогательные вещества), можно наблюдать дифракционные пики при значениях 2-тета (градусы), описанные в таблице 4.
В еще одном аспекте было обнаружено, что кристаллическая форма 3 представляет собой кристаллическую форму соединения формулы (I) с высокой температурой плавления, проявляющей свойство сольватации с этилацетатом и н-гексаном (профиль PXRD: фиг. 1; другие данные, касающиеся PXRD, описаны в таблице 1).
В еще одном аспекте было обнаружено, что аморфная форма показывает поглощение воды в 2,5% при 25°C/90% RH, что является обратимым путем снижения RH при постоянной температуре 25°C (профиль PXRD: фиг. 2; профиль DSC: фиг. 5; профиль DVS: фиг. 6; другие данные, касающиеся профилей PXRD, DSC, DVS, приведены в таблице 1).
В еще одном аспекте было обнаружено, что кристаллическая форма 1 представляет собой кристаллическую форму соединения формулы (I) с высокой температурой плавления, что показывает поглощение воды в 0,6% при 25°C/90%RH, что ниже, чем для аморфной формы, и является обратимым путем снижения RH при постоянной температуре 25°C (профиль PXRD: фиг. 3; профиль DSC: фиг. 5; профиль DVS: фиг. 6; другие данные, касающиеся профилей PXRD, DSC, DVS, приведены в таблице 1).
В еще одном аспекте было обнаружено, что кристаллическая форма 2 имеет высокую температуру плавления кристаллической формы соединения формулы (I), что показывает поглощение воды в 0,2% при 25°C/90% RH, что ниже, чем для аморфной формы, и является обратимым путем снижения RH при постоянной температуре 25°C (профиль PXRD: фиг. 4; профиль DSC: фиг. 5; профиль DVS: фиг. 6; другие данные, касающиеся профилей PXRD, DSC, DVS, приведены в таблице 1).
Описание свойств твердого состояния и ссылки на фигуры/таблицы кристаллической формы 3, аморфной формы, кристаллической формы 1 и кристаллической формы 2 соединения формулы (I)
Дальнейшим объектом изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество кристаллической формы 1 или кристаллической формы 2, описанной выше, или ее фармацевтически приемлемой соли в качестве активного ингредиента, и фармацевтически приемлемый эксципиент, носитель или разбавитель.
Кристаллическая форма 1 или кристаллическая форма 2, описанная выше, или ее фармацевтически приемлемая соль легко всасывается перорально, поэтому, предпочтительно, ее вводят перорально. Излишне говорить, что соединения по настоящему изобретению могут быть введены любым путем введения, например парентерально, местно, ректально и назальным путем.
Композиции по изобретению могут быть в форме, подходящей для перорального применения. Примерами этих форм являются: таблетки, твердые или мягкие капсулы, водные или масляные суспензии, эмульсии, диспергируемые порошки или гранулы. Композиции по изобретению могут также быть в виде, подходящем для местного применения. Примерами таких форм являются: кремы, мази, гели или водные или масляные растворы или суспензии. Композиции по изобретению могут также быть в виде, подходящем для введения путем ингаляции, таком как, например, тонкоизмельченный порошок или жидкий аэрозоль. Композиции по изобретению могут также быть в виде, подходящем для введения путем инсуффляции, таком как, например, мелкодисперсный порошок. Композиции по изобретению могут также быть в виде, подходящем для парентерального введения (таком как, например, стерильный водный или масляный раствор для внутривенного, подкожного, внутримышечного введения), или в виде суппозиториев для ректального введения. Композиции по изобретению могут быть получены обычными способами с использованием обычных фармацевтических эксципиентов, хорошо известных в данной области техники.
Таким образом, композиции, предназначенные для перорального введения, могут содержать одну или несколько добавок, таких как, например, окрашивающие, подслащивающие, ароматизирующие и консервирующие агенты.
Например, твердые пероральные формы могут содержать вместе с активным соединением разбавители, например лактозу, декстрозу, сахарозу, сахаробиозу, маннит, целлюлозу, кукурузный крахмал или картофельный крахмал; лубриканты, например диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; глиданты, например коллоидный диоксид кремния; связующие вещества, например крахмалы, гуммиарабик, желатин метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезинтегранты, например крахмал, альгиновую кислоту, альгинаты или натрий крахмалгликолят; шипучие смеси; красители; подсластители; смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, обычно, нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических препаратах. Эти фармацевтические препараты могут быть изготовлены известным способом, например путем смешивания, гранулирования, таблетирования, покрытия сахаром или покрытия пленкой.
Жидкими дисперсиями для перорального применения могут быть, например, сиропы, эмульсии и суспензии.
В качестве примера, сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннитом и сорбитом.
Суспензии и эмульсии могут содержать в качестве примеров носителей, камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт.
Суспензии или растворы для внутримышечных инъекций могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль, и, если необходимо, соответствующее количество гидрохлорида лидокаина.
Растворы для внутривенных инъекций или вливаний могут содержать в качестве носителя стерильную воду, или, предпочтительно, они могут быть в виде стерильных водных изотонических солевых растворов, или они могут содержать пропиленгликоль в качестве носителя.
Суппозитории могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например, масло какао, полиэтиленгликоль, поверхностно-активный сложный эфир жирной кислоты и полиоксиэтиленсорбитана или лецитин.
Следующим объектом изобретения является кристаллическая форма 1 или кристаллическая форма 2, описанная выше, или ее фармацевтически приемлемая соль для применения в качестве лекарственного средства.
Следующим объектом изобретения является кристаллическая форма 1 или кристаллическая форма 2, описанная выше, или ее фармацевтически приемлемая соль, либо отдельно, либо в сочетании с другими терапевтическими агентами или с лучевой терапией, для применения в лечении болезненного состояния, поддающегося лечению путем ингибирования ALK, такого как злокачественное новообразование и клеточные пролиферативные нарушения.
Следующим объектом изобретения является способ лечения млекопитающего, включая человека, при необходимости ингибирования ALK, включающего введение указанному млекопитающему терапевтически эффективного количества кристаллической формы 1 или кристаллической формы 2, описанной выше, или ее фармацевтически приемлемой соли.
Наконец, еще одним объектом изобретения является применение кристаллической формы 1 или кристаллической формы 2, описанной выше, или ее фармацевтически приемлемой соли, либо отдельно, либо в сочетании с другими терапевтическими агентами или с лучевой терапией, при получении лекарственного средства для лечения болезненного состояния, поддающегося лечению путем ингибирования ALK, такого как злокачественное новообразование и клеточные пролиферативные нарушения.
Термин «болезненное состояние, поддающееся лечению» означает, что лечение согласно изобретению обеспечивает ремиссию болезненного состояния, или, по крайней мере, состояние здоровья и качество жизни у млекопитающих, находящихся на лечении, улучшаются.
Примерами таких болезненных состояний, в частности, являются различные виды злокачественных новообразований, которые могут включать конкретные виды злокачественных новообразований, включая карциному, плоскоклеточный рак, гематопоэтические опухоли миелоидной или лимфоидной линии, опухоли мезенхимального происхождения, опухоли центральной и периферической нервной системы, меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератокарциному, фолликулярный рак щитовидной железы и саркому Капоши.
Другими предпочтительными болезненными состояниями являются конкретные виды злокачественного новообразования, такие как, но не ограничиваясь ими, рак молочной железы, рак легкого, колоректальный рак, рак предстательной железы, рак яичников, рак эндометрия, рак желудка, светлоклеточный тип почечно-клеточного рака почек, увеальная меланома, множественная миелома, рабдомиосаркома, саркома Юинга, саркома Капоши и медуллобластома.
Другими предпочтительными болезненными состояниями являются ALK+ анапластическая крупноклеточная лимфома (ALCL), и, возможно, другие показания, при которых может играть роль активность ALK, такие как нейробластома, рабдомиосаркома, глиобластома, воспалительная миофибробластическая опухоль и некоторые виды меланомы, карцинома молочной железы, саркома Юинга и немелкоклеточный рак (NSCLC).
Следующими предпочтительными болезненными состояниями являются клеточные пролиферативные нарушения, такие как, но этим не ограничиваясь, доброкачественная гиперплазия предстательной железы, семейный аденоматоз полипоз, нейрофиброматоз, псориаз, сосудистая пролиферация клеток гладких мышц, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
Термин «другие терапевтические агенты» может включать, но этим не ограничивается, антигормональные агенты, такие как антиэстрогены, антиандрогены и ингибиторы ароматазы, ингибиторы топоизомеразы I, ингибиторы топоизомеразы II, агенты, нацеленные на микротрубочки, агенты на основе платины, алкилирующие агенты, повреждающие ДНК или интеркалирующие агенты, противоопухолевые антиметаболиты, другие ингибиторы киназ, другие антиангиогенные агенты, ингибиторы кинезинов, терапевтические моноклональные антитела, ингибиторы mTOR, ингибиторы гистондеацетилазы, ингибиторы фарнесилтрансферазы и ингибиторы гипоксической реакции.
Эффективная доза соединения формулы (I), кристаллической формы 1 или кристаллический формы 2, описанной выше, или фармацевтически приемлемой соли может различаться в зависимости от заболевания, тяжести заболевания и состояния пациента, проходящего лечение. Поэтому врач, как всегда, должен установить оптимальную дозу для каждого пациента. В любом случае диапазон эффективной дозировки может быть от примерно 10 мг до примерно 1 г на дозу (в расчете на свободное основание), от 1 до 3 раз в день.
ПРИМЕРЫ
Следующие примеры иллюстрируют изобретение.
Температура измеряется в градусах Цельсия (°C).
Если не указано иное, реакции или эксперименты осуществляют при комнатной температуре.
Сокращения:
RT: комнатная температура
RH: относительная влажность
PXRD: порошковая рентгеновская дифракция
DSC: дифференциальная сканирующая калориметрия
DVS: динамическая сорбция паров
TGA: термогравиметрический анализ
ACN (ацетонитрил)
EtOAc (этилацетат)
DCM (дихлорметан)
DMA (N,N-диметилацетамид)
DMF (N,N-диметилформамид)
ДМSО (диметилсульфоксид)
МТBE (метил-трет-бутиловый эфир)
THF (тетрагидрофуран)
TFA (трифторуксусная кислота)
Пример 1: Получение кристаллической формы 1 и кристаллической формы 2 соединения формулы (I).
Схема 1, приведенная ниже, показывает получение кристаллической формы 1 и кристаллической формы 2 соединения формулы (I)
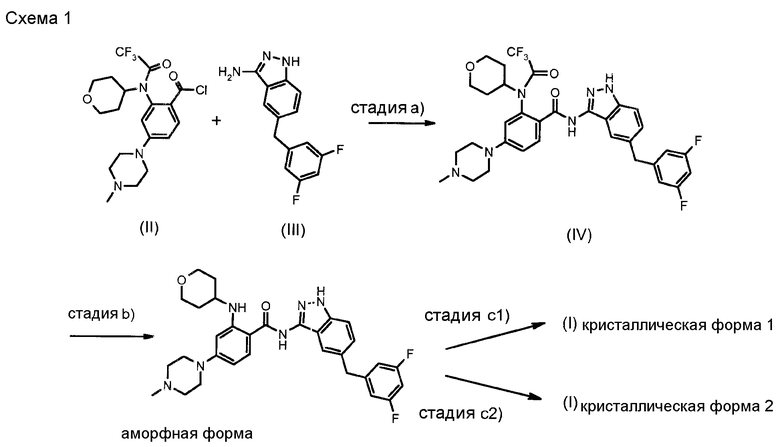
К суспензии трифторацетата 4-(4-метилпиперазин-1-ил)-2-[тетрагидро-2H-пиран-4-ил(трифторацетил)амино]бензойной кислоты (3,7 кг, 7 моль) в сухом DCM (36 л) и N,N-диметилформамиде (14 мл) добавляли оксалилхлорид (1,78 л, 21 моль). Смесь перемешивали в течение около 1,5 часов и выпаривали до маслянистого остатка; затем добавляли сухой DCM и дважды упаривали.
Ацилхлорид формулы (II) суспендировали в сухом DCM и суспензию медленно и постепенно добавляли к раствору 5-(3,5-дифторбензил)-1Н-индазол-3-иламина (1,6 кг, 6,1 моль) в сухом пиридине (16 л) при -40/-30°C. Добавление прекращали, когда 5-(3,5-дифторбензил)-1Н-индазол-3-иламин полностью реагировал. Примерно через 1 час растворитель упаривали и последовательно добавляли DCM (55 л), метанол (6,5 л) И MTBE (55 л). Очищенное защищенное соединение формулы (IV) фильтровали, промывали смесью 10/10/1 DCM/MTBE/MeOH и сушили в вакууме (3,8 кг).
Полученный таким образом сырой N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-[(тетрагидропиран-4-ил)-2,2,2-трифторацетил)амино]бензамид с ВЭЖХ чистотой >95% растворяли в метаноле и добавляли раствор К2СО3 в смеси вода/метанол при температуре 10°С. Раствор фильтровали и добавляли по каплям в воду; осадок аморфного N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида фильтровали, промывали водой и сушили в вакууме (2,88 кг).
5,5 г сухого аморфного N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида суспендировали в 130 мл этанола и нагревали при кипячении с обратным холодильником в течение 10 минут; отгоняли около 70 мл этанола и охлаждали до комнатной температуры. Добавляли 110 мл воды и в суспензию вносили затравку 55 мг кристаллической формы 1. Суспензию перемешивали в течение примерно 72 часов, отбирая пробы для контроля преобразования в кристаллическую форму 1 с помощью DSC. Суспензию затем фильтровали и сушили с получением 4,3 г желаемой кристаллической формы 1.
Сухой аморфный N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамид (2,88 кг) переводили во взвесь в примерно 10 объемах этанола, чтобы дать возможность преобразования в желаемую кристаллическую форму 2; затем добавляли 20 объемов воды и суспензию фильтровали. Продукт окончательно сушили в вакууме, получая таким образом около 2,6 кг N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида (4,6 моль) в желаемой кристаллической форме 2.
Пример 2: Результаты анализа с помощью порошковой рентгеновской дифракции (PXRD)
Кристаллическая форма 3, аморфная форма, кристаллическая форма 1 и кристаллическая формы 2 соединения (I) были охарактеризованы с помощью порошковой рентгеновской дифракции (PXRD), выполненной на дифрактометре Thermo/ARL XTRA, путем облучения образцов порошка источником CuKα (45 кВ, 40 мА, 1,8 кВт - Kα1 излучение, длина волны λ=1,54060 ангстрем) в диапазоне между 2° и 40° 2-тета при комнатной температуре.
Скорость сканирования была 1,20°/мин (шаг 0,020° с отсчетом времени в 1 сек на шаг).
На диаграммах дифракции рентгеновских лучей углы дифракции 2-тета нанесены на горизонтальной оси (ось x), и интенсивность - на вертикальной (ось y).
В пунктах, определяющих пики порошковой рентгеновской дифракции кристаллических форм соединения формулы (I), термин «примерно» используется в выражении «…при примерно углах 2-тета, представленных в таблице…», чтобы указать, что точные положения пиков (т.е. указанные значения угла 2-тета) не следует рассматривать в качестве абсолютных значений, поскольку, как это понятно специалисту в данной области, точное положение пиков может незначительно отличаться между данными одного прибора и другого, от одного образца к другому или в результате незначительных отклонений в используемых условиях измерения.
В предыдущих абзацах указано также, что аморфная форма и кристаллические формы соединения формулы (I) показывают порошковые рентгеновские дифрактограммы, по существу такие же, как порошковые рентгеновские дифрактограммы, показанные на фиг. 1, 2, 3 и 4, и имеют существенно наиболее значительные пики при значениях угла 2-тета, указанных в таблицах 1, 2, 3 и 4. Должно быть понятно, что использование термина «существенно» в данном контексте также предназначено, чтобы указать, что значения угла 2-тета в порошковых рентгеновских дифрактограммах могут незначительно отличаться между данными одного прибора и другого, от одного образца к другому или в результате незначительных отклонений в используемых условиях измерения, поэтому положения пиков, показанные на чертежах или приведенные в таблицах, опять-таки, не следует рассматривать как абсолютные.
В связи с этим, в данной области техники известно, что порошковая рентгеновская дифракция может быть получена с одной или несколькими ошибками измерения, зависящими от условий измерения (таких, как, например, оборудование и/или подготовка образца). В частности, общеизвестно, что интенсивности в порошковой рентгеновской дифрактограмме могут отличаться в зависимости от условий измерений и подготовки образца.
Например, специалистам в области техники рентгеновской порошковой дифракции будет понятно, что относительные интенсивности пиков могут зависеть, например, от зерна более 30 мкм по размеру и неунитарности пропорций, что может повлиять на анализ образцов.
Специалистам в данной области техники будет также понятно, что положение отражений может зависеть от точной высоты, на которой закреплен образец в дифрактометре и от нулевой калибровки дифрактометра.
Плоскостность поверхности образца также может повлиять на результат.
Следовательно, специалисту в данной области техники будет понятно, что данные дифракционной картины, представленные в настоящем документе, не следует рассматривать как абсолютные (для дальнейшей информации см. «Fundamentals of Powder Diffraction and Structural Characterization», Pecharsky and Zavalij, Kluwer Academic Publishers, 2003). Поэтому, должно быть понятно, что аморфная форма и кристаллические формы соединения формулы (I), описанные в настоящем изобретении, не ограничиваются аморфностью и кристалличностью, которые показывает порошковая рентгеновская дифрактограмма, идентичная порошковым рентгеновским дифрактограммам, показанным на фиг. 1, 2, 3 и 4, и любой образец или партия аморфной формы или кристаллических форм соединения формулы (I), показывающих порошковые рентгеновские дифрактограммы, по существу такие же, как показано на фиг. 1, 2, 3 и 4, входят в объем настоящего изобретения. Специалист в области порошковой дифракции может судить о существенной идентичности рентгеновских порошковых дифрактограмм.
Обычно погрешность измерения дифракционного угла на рентгеновской порошковой дифрактограмме составляет примерно 2-тета = 0,5 град. или меньше (или, что более подходяще, около 2-тета = 0,2 град. или меньше), и такая степень погрешности измерений должна быть учтена при рассмотрении порошковой рентгеновской дифрактограммы на фиг. 1, 2, 3 и 4 и при сравнении образцов или интерпретации положений пиков, упомянутых в тексте и в таблицах 1, 2, 3 и 4.
Поэтому, когда указано, например, что кристаллические формы соединения формулы (I) имеют порошковую рентгеновскую дифракцию с по меньшей мере одним специфическим пиком при 2-тета = 20,1 град. (или любом из других упомянутых углов), то это может быть истолковано как 2-тета = 20,1 град. плюс или минус 0,5 град., или 2-тета = 20,1 град. плюс или минус 0,2 град.
Диаграммы рентгеновской дифракции кристаллической формы 3, аморфной формы, кристаллической формы 1 и кристаллической формы 2 представлены на фиг. 1, 2, 3 и 4 соответственно. Положения рентгеновских дифракционных пиков кристаллической формы 3, кристаллической формы 1 и кристаллической формы 2 представлены в таблицах 2, 3 и 4 соответственно.
Пример 3: Данные анализа, полученные с помощью дифференциальной сканирующей калориметрии (DSC)
DSC анализы осуществляли на приборе Mettler Toledo Star system. В алюминиевые тигли DSC помещали 2-4 мг образца. Диапазон температур был между 25°C и максимальным значением 300°С. Образцы анализировали в статическом состоянии в атмосфере азота при скорости нагрева 10°C/мин.
На фиг. 5. представлены термограммы DSC аморфной формы, кристаллической формы 1 и кристаллической формы 2.
Наблюдаемая эндотерма плавления для кристаллической формы 1 находится в диапазоне примерно 188°C-196°C (пиковая температура) при дельта H в диапазоне 54-64 Дж/г. Наблюдаемая эндотерма плавления для кристаллической формы 2 находится в диапазоне примерно 197°C-198,5°С (пиковая температура) при дельта H в диапазоне 72-78,5 Дж/г. Будет понятно, что нарастание и/или пиковые значения температуры DSC могут незначительно отличаться между данными одного прибора и другого, от одного метода к другому или от одного образца к другому, и поэтому указанные значения не следует рассматривать как абсолютные. Действительно, наблюдаемые температуры будут зависеть от скорости изменения температуры, а также от метода подготовки образца и конкретного используемого прибора. Будет учтено и принято во внимание, что значения температуры, полученные с применением таких различных условий, могут меняться в диапазоне примерно плюс или минус 4°C.
Пример 4: Результаты анализа методом динамической сорбции паров (DVS)
Наблюдаемое поглощение воды исследовали, подвергая образец таких веществ исследованию на гигроскопичность с помощью DVS 1000 (SMS). Прибор представляет собой «контролируемое атмосферное микроравновесие», где взвешенный образец подвергается запрограммированному изменению относительной влажности (RH) при постоянной и контролируемой температуре. Измеренные параметры (масса, время и RH), представленные в таблицах Excel, позволяют получить кривые гигроскопичности относительно исследуемого диапазона RH. Например, циклы сорбции/десорбции между 0% и 90% RH могут быть выполнены при контролируемой температуре 25°C. Прогрессивные изменения RH могут быть, например, 10% и 3% и управляются с помощью программного обеспечения при уравновешивании массы образца. Это условие может быть определено при постоянной скорости изменения массы в процентах, такой как, например, 0,005%/мин.
На фиг. 6 представлены профили DVS аморфной формы, кристаллической формы 1 и кристаллической формы 2 соединения формулы (I). Значения относительной влажности (RH, %) приведены на оси x, а изменение массы (%) представлено на оси y. Кривые относятся к шагу сорбции в интервале между 0% RH и 90% RH при 25°C.
Экспериментальные результаты показывают, что кристаллическая форма 1 и кристаллическая форма 2 соединения (I), соответственно, характеризуются поглощением воды в 0,6% и 0,2% при 25°C/90% RH. Такое поглощение воды является обратимым при снижении RH при постоянной температуре 25°С. Кристаллические формы 1 и 2 соединения (I) могут считаться низкогигроскопичными.
Результаты экспериментов также показывают, что аморфная форма соединения (I) характеризуется поглощением воды в 2,5% при 25°C/90% RH, что является обратимым при снижении RH при постоянной температуре 25°C. Аморфная форма соединения (I) характеризуется более высокой гигроскопичностью, чем кристаллические формы 1 и 2. Поглощение воды аморфной формой соединения (I) выше, чем кристаллическими формами 1 и 2. В качестве дополнительного аспекта, поглощение воды аморфной формой соединения (I) больше, чем на 1% от значений RH, что меньше, чем 30% RH, с последующим угловым коэффициентом повышения в области высоких значений RH.
Пример 5: Аналитические данные посредством термогравиметрического анализа (TGA)
TGA анализы проводили на приборе Perkin-Elmer TGA-7. В алюминиевые тигли DSC помещали 5÷10 мг образца. Температурный диапазон анализов был между 30°С и максимальным значением около 250°С. Образцы анализировали в токе азота (для устранения окислительного и пиролитического эффектов) при скорости нагрева 2°C/мин.
Пример 6: ЯМР-анализ
Анализ 1H ЯМР проводили при постоянной температуре 28°C на спектрометре Varian Inova 500 для образца кристаллической формы 3 (см. фиг. 8) и при постоянной температуре 28°C на спектрометре Varian Inova 400 для образца кристаллической формы 1 (см. фиг. 7). Небольшое количество каждого образца растворяли в 0,75 мл ДМСО-d6 и переносили в 5 мм пробирку для ЯМР для последующего анализа.
Так как одинаковый спектр 1H ЯМР получали для различных кристаллических форм, т.е. кристаллические формы 1 и 2 имели одинаковый спектр 1H ЯМР, то представлен только спектр кристаллической формы 1. Спектр кристаллической формы 3 дан лишь для того, чтобы показать наличие остаточных растворителей, сигналы которых четко отличаются от сигналов продукта и обозначены стрелками на фиг. 8.
Пример 7: Процентный состав препарата для перорального применения
Изобретение относится к новым кристаллическим формам N-[5-(3,5-дифторбензил)-1Н-индазол-3-ил]-4-(4-метилпиперазин-1-ил)-2-(тетрагидропиран-4-иламино)бензамида (формула1), а именно формам 1 и 2 , способам их получения, а также к их применению в качестве ингибитора ALK при лечении заболеваний, вызванных дерегулируемой активностью протеинкиназы, и содержащим их фармацевтическим композициям. 4 н. и 12 з.п. ф-лы, 8 ил., 4 табл., 7 пр.
1. Кристаллическая форма 1 соединения формулы (I)
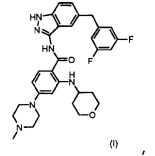
характеризующаяся порошковой рентгенограммой с характерными пиками при значениях угла отражения 2-тета, полученных при использовании Cu-Kα1, равных 4,8, 7,5, 15,2, 16,2, 17,4, 17,8, 18,7, 19,5, 20,1, 20,5, 22,3, 23,0, 23,3, 24,4 и 25,0°±0,5°.
2. Кристаллическая форма 1 по п. 1, характеризующаяся порошковой рентгенограммой с характерными пиками при значениях угла отражения 2-тета, полученных при использовании Cu-Kα1, равных 4,1, 4,8, 7,3, 7,5, 8,1, 8,9, 9,2, 9,8, 11,1, 13,0, 13,5, 13.8, 14,6, 15,2, 16,2, 17,4, 17,8, 18,2, 18,7, 19,5, 20,1, 20,5, 21,1, 21,8, 22,3, 23,0, 23,3, 23,9, 24,4, 24,7, 25,0, 25,5, 26,2, 27,2, 27,3, 29,1, 29,5, 30,1, 30,6, 31,5 и 36,6°±0,5°.
3. Кристаллическая форма 1 по п. 1 или 2, характеризующаяся DSC термограммой, имеющей эндотермический пик плавления в диапазоне 188°С-196°С.
4. Кристаллическая форма 2 соединения формулы (I), представленной в п. 1, характеризующаяся порошковой рентгенограммой с характерными пиками при значениях угла отражения 2-тета, полученных при использовании Cu-Kα1, равных 6,6, 9,8, 10,5, 11,1, 12,4, 14,3, 15,3, 15,9, 16,9, 17,8, 18,4, 19,0, 19,9, 20,2, 21,0, 21,3, 21,8, 22,3, 22,5, 23,2, 23,6, 24,9, 25,1, 25,5, 25,9, 26,4, 27,6, 28,1, 28,7, 29,0, 29,3, 29,5, 30,0, 30,4, 30,8, 31,5, 31,9, 32,2, 32,4, 33,0, 33,6, 34,7, 34,9, 38,1 и 38,4°±0,5°.
5. Кристаллическая форма 2 по п. 4, характеризующаяся DSC термограммой, имеющей эндотермический пик плавления в диапазоне 197°С-198,5°С.
6. Фармацевтическая композиция, обладающая свойствами ALK ингибитора, содержащая терапевтически эффективное количество кристаллической формы 1 по п. 1, или кристаллической формы 2 по п. 4, или ее фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый эксципиент, носитель или разбавитель.
7. Фармацевтическая композиция по п. 6, где указанная композиция представлена в форме таблетки, капсулы, суспензии, эмульсии, диспергируемого порошка или гранул.
8. Фармацевтическая композиция по п. 6 или 7, где указанная композиция содержит от 10 мг до 1 г на дозу кристаллической формы 1 по п. 1 или кристаллической формы 2 по п. 4 или ее фармацевтически приемлемую соль.
9. Кристаллическая форма 1 по п. 1 или кристаллическая форма 2 по п. 4 или ее фармацевтически приемлемая соль для применения в качестве лекарственного средства, обладающего свойствами ALK ингибитора.
10. Кристаллическая форма 1 по п. 1 или кристаллическая форма 2 по п. 4 или ее фармацевтически приемлемая соль, для получения лекарственного средства для лечения болезненного состояния, которое поддается лечению путем ингибирования ALK, где болезненное состояние выбрано из злокачественного новообразования и клеточных пролиферативных нарушений.
11. Кристаллическая форма 1 по п. 1 или кристаллическая форма 2 по п. 4 или ее фармацевтически приемлемая соль для получения лекарственного средства по п. 10, где указанное злокачественное новообразование выбрано из рака молочной железы, рака легкого, колоректального рака, рака предстательной железы, рака яичников, рака эндометрия, рака желудка, светлоклеточного типа почечно-клеточного рака, увеальной меланомы, множественной миеломы, рабдомиосаркомы, саркомы Юинга, саркомы Капоши, медуллобластомы, глиобластомы, анапластической крупноклеточной лимфомы и нейробластомы.
12. Кристаллическая форма 1 по п. 1 или кристаллическая форма 2 по п. 4 или ее фармацевтически приемлемая соль для получения лекарственного препарата по п. 10, где указанным злокачественным новообразованием является немелкоклеточный рак легкого.
13. Кристаллическая форма 1 по п. 1 или кристаллическая форма 2 по п. 4 или ее фармацевтически приемлемая соль для получения лекарственного препарата по п. 10 или 11, где указанным злокачественным новообразованием является колоректальный рак.
14. Способ получения соединения формулы (I)
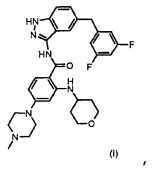
который включает следующие стадии:
a) добавления в стехиометрическом количестве ацилхлорида формулы (II):
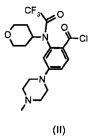
к индазол-3-иламину формулы (III)
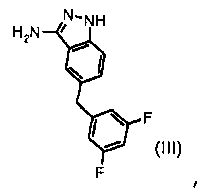
прекращения добавления, когда индазол-3-иламин формулы (III) полностью прореагирует;
b) удаления защитной группы в мягких основных условиях при 10°С из полученного соединения формулы (IV):
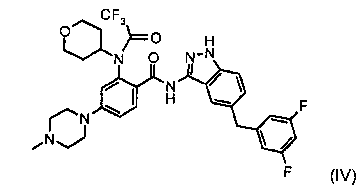
с получением желаемого соединения формулы (I), которое выделяют в аморфной форме;
либо
c1) обработки полученного аморфного соединения формулы (I) сначала этанолом, нагревая при кипении с обратным холодильником и отгоняя часть растворителя, затем водой, и внося затравку кристаллической формы 1 при температуре в интервале между 10°С и 30°С, с получением желаемого соединения формулы (I) в кристаллической форме 1 по п. 1,
либо
с2) обработки полученного аморфного соединения формулы (I) последовательно этанолом при температуре в интервале между 10°С и 30°С и затем водой при температуре в интервале между 10°С и 30°С с получением желаемого соединения формулы (I) в кристаллической форме 2 по п. 4,
и, необязательно,
d) преобразования образовавшегося соединения, полученного на стадии b), на стадии c1) или на стадии с2) в фармацевтически приемлемую соль.
15. Способ по п. 14, где соединение формулы (II) суспендируют в дихлорметане и соединение формулы (III) суспендируют в пиридине.
16. Способ по п. 14, где удаление защитной группы соединения формулы (IV) осуществляют в растворе K2CO3 в смеси вода/метанол.
| WO 2009013126 A1 29.01.2009 | |||
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2194048C2 |
| Способ получения 6,7-диметокси-4амино-2(4,/2-фуроил/-1-пиперазинил)хиназолина | 1977 |
|
SU753360A3 |

