Изобретение относится к области медицины и фармацевтической промышленности и касается способов получения 1-алкил-2-алкилкарбамоилглицеринов, которые могут быть использованы для медикаментозной терапии артериальной гипертензии.
Известно, что средство на основе 1-алкил-2-алкилкарбамоилглицерина (АКГ), являющегося структурным аналогом предшественника фактора активации тромбоцитов (ФАТ) - фосфолипида, имеющего структуру согласно формуле: 1-О-алкил-2-ацетил-sn-глицеро-3-фосфохолин, вызывает долгосрочное снижение артериального давления (АД) у крыс со спонтанной генетической и реноваскулярной гипертензией [Malekin S.I. Long-term normalization of blood pressure in SHR and 1-kidney 1-clip rats by synthetic precursor of stable PAF analogue without systemic effects in normotensive rats / S.I. Malekin, S.V. Kotelevtsev, S.A. Gavrilova et al // Pathophysiology. - 2011. - 18(2). - P. 151-157].
В качестве исходных субстратов для синтеза 1-O-алкил-2-ацетил-sn-глицерофосфолипидов использовались различные производные глицерина. Так, 1-O-алкил-2-O-ацетил-sn-глицеролы были получены из D-маннитола в 8 стадий, которые включали 1,2-O-изопропилидин-sn-глицерин как ключевой промежуточный продукт в схеме синтеза [Chacko G.K. Chemical synthesis of i-O-(D)-and 3-O-(L)-glyceryl monoethers, diethers and derivatives: glycerides, monoester phospholipids and diether phospholipids / Chacko G.K., Hanahan D.J. // Biochim. Biophys. Acta. - 1968. - 164. - Р.252]. 1-О-алкил-2-О-бензилглицерофосфохолин готовили из 1,3-бензилиденглицерола и 1-деокси-1-йодо-3-О-алкилглицерола. Встречающийся в природе О-алкилглицерины, также использовали как исходные материалы для синтеза алкил-фосфолипидов с ограниченным количеством алкильных цепей в положении sn-1 и sn-3.
Известен синтез 1-O-алкил-2-ацетил-sn-глицеро-3-фосфохолинов - аналогов фактора активации тромбоцитов в 4 стадии из эпихлорогидрина. На первой стадии проводят этерификацию эпихлорогидрина с различными спиртами (тетрадецилом, гексадецилом, октадецилом) с получением глицидиловых эфиров (выход 78-80%). На второй стадии проводят раскрытие эпоксидого цикла уксусным ангидридом с получением ацетилированных продуктов (выход 76-78%). Ключевой стадией является методика, использующая 1,3-специфичную липазу для получения 1-O-алкил-2- ацетилглицерина (выход 45-50%). Гидролизированные продукты фосфорилировали с получением 1-O-алкил-2-ацетил-sn-глицеро-3-фосфохолинов (выход 80-85%) [Vijeeta Т. Chemo enzymatic synthesis of rac 1 О alkyl 2 acetyl sn glycero 3 phosphocholine and its analogues / Vijeeta Т., Balakrishna M, Karuna M.S.L., Rao B.W.S.K., Prasad R.B.N. // J Oleo Sci. - 2014. - 63(9). - Р.933-938].
При проведении скрининговых исследований было обнаружено, что ряд структурных гомологов предшественника ФАТ перспективны для разработки на их основе антигипертензивных лекарственных средств. Из полученных кандидатов отобран АКГ [патент RU 2414453 С2, опубликован 10.08.2010] и проведено детальное изучение его влияния на АД крыс. Показано, что АКГ вызывает долгосрочное снижение АД у крыс со спонтанной генетической и реноваскулярной гипертензией, при этом продолжительность гипотензивного действия АКГ при его однократном введении была выше, чем у широко используемых в настоящее время лекарственных препаратов, включая кальциевые антагонисты, диуретики, ингибиторы ангиотензин-превращающего фермента.
Известно несколько способов получения алкилкарбамоилглицеринов.
В 1988 г. Surles J.R. с соавторами использовали для получения 1-О-гексадецил-2-0-(метилкарбамоил)глицерина метод, предложенный Gupta С.М. и Bali А., позволяющий присоединить метилкарбаматный фрагмент. Колбу перед началом реакции продували азотом. К раствору 1-О-гексадецил-3-О-тритилглицерина (9.0 г, 16 ммоль) и 4-(диметиламино)пиридина (2.07 г, 17 ммоль) в CH2Cl2 (50 мл) добавляли метилизоционат (4.85 г, 85 ммоль). Емкость, в которой проходила реакция, продували N2 и герметизировали. Содержимое выдерживали в атмосфере азота при постоянном перемешивании в темноте при комнатной температуре в течение 72 ч, после чего, удаляли летучие компоненты реакции. Получали 15.2 г сырого продукта, который хроматографировали на силикагеле. Далее из полученного продукта (массой 10.1 г) удаляли тритильную защиту и повторно очищали методом хроматографии. Выход продукта (1-O-гексадецил-2-O-(метилкарбамоил)глицерина) составил 62% [Surles J.R. Synthesis and Evidence for the Stability of a Glycerophosp hochloridate: rac -1-O-Hexadecyl-2-O-(methylcarbamyl)-sn -glycero-3-phosphoroc hloridocholine / J.R. Surles, M.H. Marx, C. Piantadosi. // J. Org. Chem. 1988, Vol. 53. P. 901-904 // J. Org. Chem. 1988, Vol. 53. P. 901-904].
Патенты фирмы Takeda Chemical Industries, Ltd., (Япония) - посвящены синтезу производных глицерина. В патенте US 4576933 (А) [опубликован 18.03.1986] описано получение производных глицерина, обладающих гипотензивной активностью. В числе прочих описано получение 2-алкилкарбомоилглицеринов -2-(N,N-диметилкарбамоил)-3-октадецилглицерола (1) и 2-(N,N-дибутирилкарбамоил)-3-октадецилглицерола (2) по следующим методикам:
1) получение 2-(N,N-диметилкарбамоил)-3-октадецилглицерола: (2-(N,N-диметилкарбамоил)-3-октадецил-1-тритилглицерин (2,92 мг, 4,43 ммоль) добавляли к 29 мл 80%-ной уксусной кислоты и полученную смесь перемешивали при 100°С в течение часа, а затем концентрировали досуха при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (13 г) с использованием н-гексан-этилацетат (4:1) в качестве элюента. Получали 1,6 г продукта (выход реакции 86,9%).
2) 2-(N,N-дибутилкарбамоил)-3-октадецилглицерола: 2-(N,N-дибутилкарбамоил)-1-Октадецил-3-триглицерол (2.7 г) растворяли в 30 мл 80% -ной уксусной кислоты, и раствор перемешивали при температуре 103°С в течение 1,5 ч и концентрировали досуха при пониженном давлении, остаток очищали на колонке с силикагелем с использованием н-гексан-этилацетат (4:1) в качестве элюента. Выход продукта составил 1,70 г (92,5%).
В патенте ЕР 0685457 В1 [опубликован 15.12.1999] японской фирмы Nippon Shinyaku Company, Limited описаны способы получения производных глицерина (1,3-О-диолеоил-2-O-(2-бромоэтил)карбомолиглицерол) из 1,3-О-диолеилглицерола, растворенного в пиридине, к которому добавляли 120 мг (0.740 ммоль) N,N'-карбонилдимидазола и смесь выдерживали при температуре окружающей среды в течение 3 ч. После удаления растворителя остаток растворяли в метилен хлориде, отмывали 5%-м водным раствором дигидрофосфата натрия, высушивали и концентрировали. Остаток растворяли в N,N-диметилформамида и добавляли 2-аминоэтанол, смесь выдерживали в течение ночи. После завершения реакции растворитель удаляли и остаток растворяли в метилен хлориде, отмывали 5%-м раствором дигидрофосфата натрия, высушивали, концентрировали. Остаток пропускали через хроматографическую колонку. В зависимости от получаемого производного, выход вещества составил от 80 до 100%.
Наиболее близким к предлагаемому является способ получения 1-алкил-карбомоилглицеринов, описанный в патенте RU 2414453 С2 [опубликован 10.08.2010], согласно которому, исходные 1-алкил-глицерины подвергают взаимодействию с триметилхлорсиланом в присутствии триэтиламина в среде толуола при -20÷0°С, затем с алкилизоцианатом и с последующей обработкой реакционной массы бифторидом аммония в среде метанола при комнатной температуре.
В качестве исходных 1-алкилглицеринов использованы соединения общей формулы
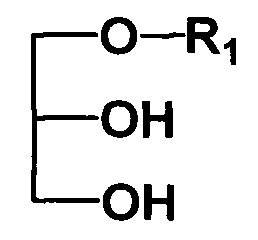
где R1 - углеводородный радикал -(СН2)nCH3 (n=10-18).
Получение 1-гексадецил-2-метилкарбамоил-глицерина.
К раствору 3.67 г (0.012 моля) 1-гексадецилглицерина и 1.17 г (0.012 моля) триэтиламина в 150 мл толуола в атмосфере аргона при -20÷0°С прибавляли раствор 1.26 г (0.012 моля) триметилхлорсилана в 20 мл толуола. Реакционную смесь выдерживали при 0°С, прибавляли 2 мл метилизоцианата и оставляли на ночь. Затем фильтруют, удаляют в вакууме растворитель, остаток растворяют в 40 мл гексана, прибавляли 0.66 г (0.012 моля) бифторида аммония в 40 мл метанола и выдерживают в течение 4 часов при комнатной температуре. Смесь фильтруют через слой окиси алюминия, удаляют в вакууме растворитель, добавляют 150 мл гексана, фильтруют, удаляют в вакууме гексан и получали 3.33 г (77%) 1-гексадецил-2-метилкарбамоил-глицерина, т.пл. 59°С, Rf=0.3 (силикагель, эфир-гексан = 4:1).
Недостатками способа является низкий выход 1-алкил-2-алкилкарбамоилглицеринов - 65-85%, использование токсичного триэтиламина, низкие загрузки компонентов, что создает сложности при получении большого количества соединения для доклинических и клинических испытаний.
Новый технический результат изобретения заключается в увеличении выхода целевого продукта, разработке метода получения целевого продукта в больших количествах, и расширении арсенала способов получения лекарственных средств, которые могут быть использованы для медикаментозной терапии артериальной гипертензии.
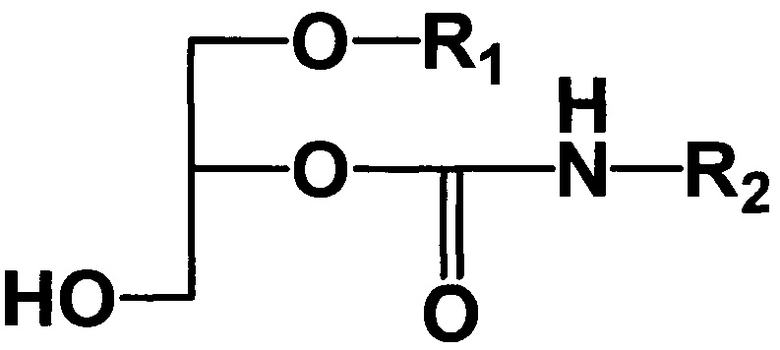
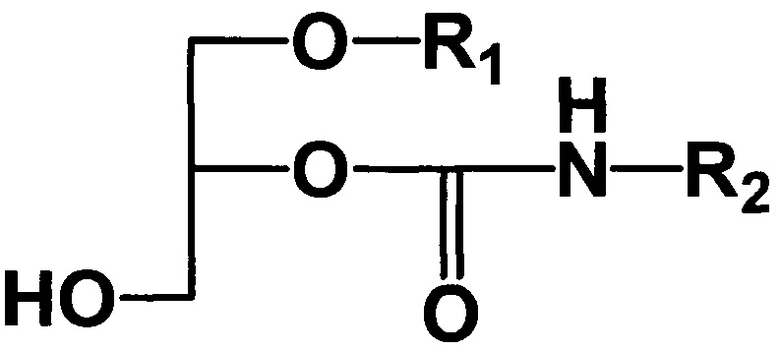
Для достижения технического результата в способе получения 1-алкил-2-алкилкарбамоилглицеринов общей формулы I
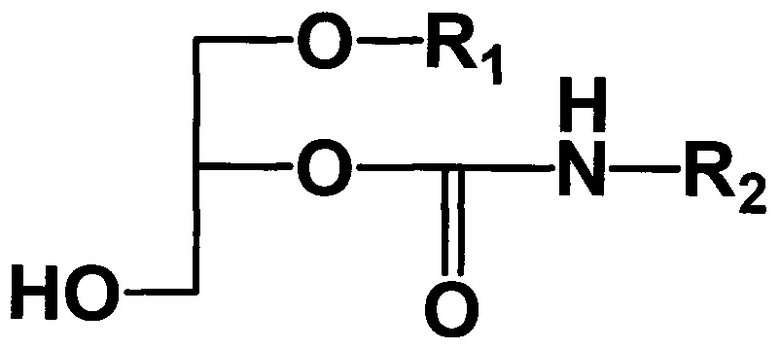
где R1 - углеводородный остаток С16-С18; R2 - метил, этил, заключающемся в том, что подвергают взаимодействию исходные 1-алкилглицерины, общей формулы II
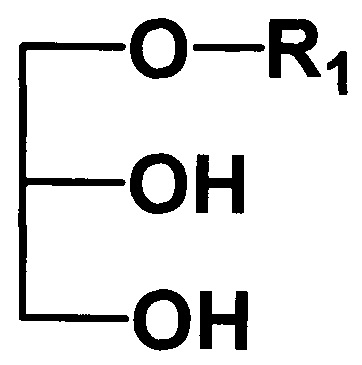
с триметилхлорсиланом в присутствии основания в среде растворителя, отличающийся тем, что, исходные 1-алкилглицерины подвергают взаимодействию с триметилхлорсиланом в присутствии пиридина, а в качестве растворителя используют метилен хлорид, при этом, реакцию проводят в атмосфере воздуха при 0° с дальнейшим присоединением соответствующего алкилизоцианата и последующей обработкой смесью метанол-хлороформ.
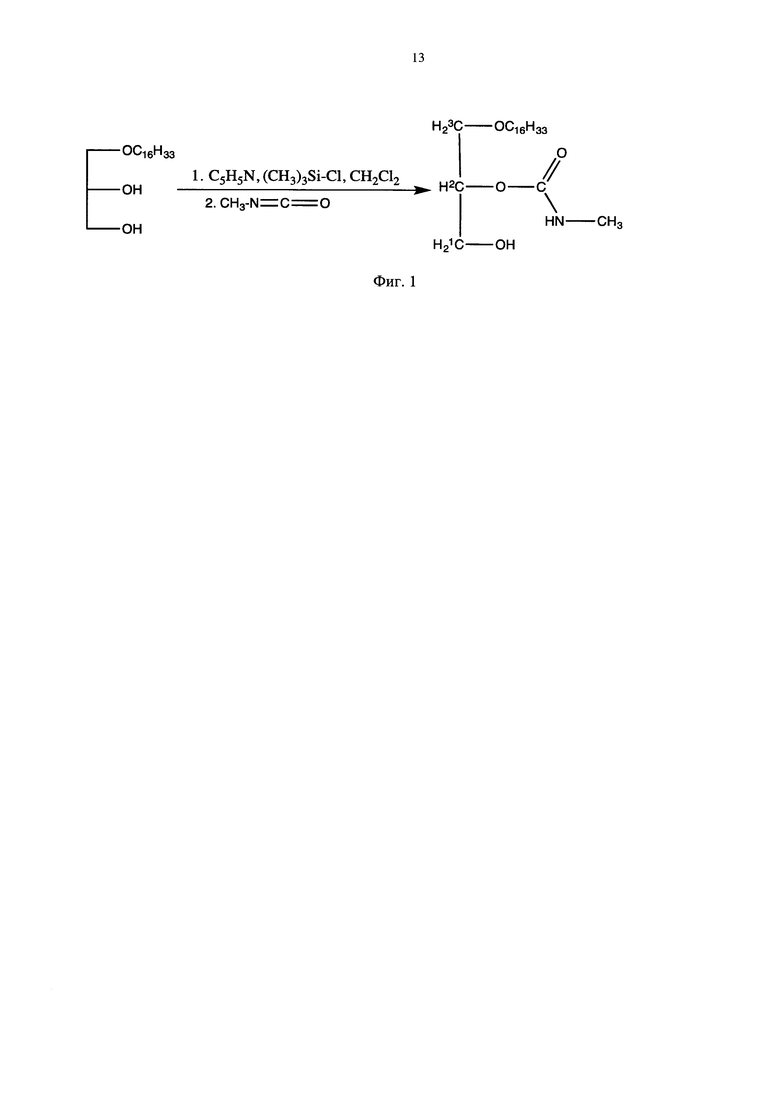
Схема реакции приведена на Фиг. 1.
Предлагаемый способ получения 1-алкил-2-алкилкарбамоил-глицеринов позволил увеличить выход целевого продукта до 98% и чистоту целевого продукта до 98,9%.
Новым в предлагаемом способе является то, что в качестве основания вместо триэтиламина используют менее токсичный пиридин, в качестве растворителя используют метилен хлорид. Реакцию проводят при комнатной температуре в атмосфере воздуха. В предлагаемом способе не требуется стадия гидролиза метанольным раствором бифторида аммония.
В результате синтеза получают 1-алкил-2-алкилкарбамоилглицерины с выходом до 98% и чистотой от 98,9%.
Режим нового способа получения 1-алкил-2-алкилкарбамоил-глицеринов был основан на результатах экспериментальных исследований.
Новый способ получения 1-алкил-2-алкилкарбамоил-глицеринов для специалиста явным образом не вытекает из уровня техники и описание способа получения 1-алкил-2-алкилкарбамоил-глицеринов, обладающего идентичной совокупностью существенных признаков не обнаружено авторами в патентной и научно-медицинской литературе. Предлагаемый способ апробирован в лабораторных условиях. Таким, образом, предлагаемое техническое решение соответствует критериям изобретения, а именно - «новизна», «изобретательский уровень» и «промышленная применимость».
Способ осуществляют следующим образом:
Пример 1. Получение 1-гексадецил-2-метилкарбамоилглицерина. К раствору 21,6 г (0.068 моль) 1-гексадецилглицерина и 9,9 мл (0.123 моль) пиридина в 150 мл хлористого метилена прибавляют раствор 7,4 г (0.068 моль) триметилхлорсилана в 20 мл хлористого метилена. Реакционную смесь выдерживают при 0°С, прибавляют 11,4 мл (0.1845 моль) метилизоцианата и оставляют на ночь. Затем фильтруют, удаляют в вакууме растворитель, остаток растворяют в 200 мл 5%-ого раствора метанола в хлороформе, добавляют 50 мл воды и интенсивно встряхивают в течение часа. После чего на делительной воронке отделяют органический слой. После к отделенному органическому слою добавляют смесь хлороформ: бензол и удаляют растворитель под вакуумом. Остаток растворяют в достаточном для полного растворения объеме хлороформа и фильтруют через слой оксида кремния, который затем промывают смесью этиловый эфир: хлороформ: ацетонитрил. Фильтрат сушат на вакууме. Получают 24,96 г (98%) 1-гексадецил-2-метилкарбамоилглицерина, т.пл. 59°С, Rf=0.3 (силикагель, эфир-гексан = 4:1). 1Н ЯМР (500 МГц, CDCl3, δ, м.д.): 0.9 (3Н, т, - СН3), 1.2 (28 Н, м, -CH2-), 1-6 (2Н, м, -OCH2); 2.8 (3Н, с, -CONHCH3), 3.0 (1H, м, -ОН), 3.4-3.6 (4Н, м, - 1CH2СН-3CH3); 3.8 (1H, с, -CONH-); 4.9 (1H, м, -2СН-ОСО-). ИК-спектр (νmax, см-1): 1700 (СО); 3300 (NH, ОН). Найдено, %: С 67.12; Н 11.45; N 2.94. C21H43NO4. Вычислено, % 67.52; Н, 11.60; N, 3.75; О, 17.13 Количественное содержание: 99,7%.
Пример 2. Получение 1-гексадецил-2-этилкарбамоилглицерина. К раствору 21,6 г (0.068 моль) 1-гексадецилглицерина и 9,9 мл (0.123 моль) пиридина в 150 мл хлористого метилена прибавляют раствор 7,4 г (0.068 моль) триметилхлорсилана в 20 мл хлористого метилена. Реакционную смесь выдерживают при 0°С, прибавляют 14,6 мл (0.1845 моль) этилизоцианата и оставляют на ночь. Затем фильтруют, удаляют в вакууме растворитель, остаток растворяют в 200 мл 5%-ого раствора метанола в хлороформе, добавляют 50 мл воды и интенсивно встряхивают в течение часа. После чего на делительной воронке отделяют органический слой. После к отделенному органическому слою добавляют смесь хлороформ: бензол, и удаляют растворитель под вакуумом. Остаток растворяют в достаточном для полного растворения объеме хлороформа и фильтруют через слой оксида кремния, который затем промывают смесью этиловый эфир: хлороформ: ацетонитрил. Фильтрат сушат на вакууме. Получают 25,65 г (выход 97%) 1-гексадецил-2-этилкарбамоилглицерина. Т.пл. 50°С, Rf=0.34 (силикагель, эфир-гексан=4:1). 1Н ЯМР (500 МГц, CDCl3, δ, м.д.): 0.9 (3Н, т, - СН3); 1.2 (28 Н, м, -СН2); 1.6 (2Н, м, -ОСH2); 2.4 (3Н, с, -CONHCH3); 3.4-3.6 (4Н, м, 1СH2СН-3СH2); 5.2 (1H, м, -2СН-ОСО-). Найдено, %: С 67.91; Н 10.82; N 3.24. C22H45NO4. Вычислено, %: С 68.21; Н 11.62; N 3.62. Количественное содержание: 98,9%.
Пример 3. Получение 1-октадецил-2-метилкарбамоилглицерина. К раствору 23,5 г (0.068 моль) 1-октадецилглицерина и 9,9 мл (0.123 моль) пиридина в 150 мл хлористого метилена прибавляют раствор 7,4 г (0.068 моль) триметилхлорсилана в 20 мл хлористого метилена. Реакционную смесь выдерживают при 0°С, прибавляют 11,4 мл (0.1845 моль) метилизоцианата и оставляют на ночь. Затем фильтруют, удаляют в вакууме растворитель, остаток растворяют в 200 мл 5%-ого раствора метанола в хлороформе, добавляют 50 мл воды и интенсивно встряхивают в течение часа. После чего на делительной воронке отделяют органический слой. После к отделенному органическому слою добавляют смесь хлороформ: бензол, и удаляют растворитель под вакуумом. Остаток растворяют в достаточном для полного растворения объеме хлороформа и фильтруют через слой оксида кремния, который затем промывают смесью этиловый эфир: хлороформ: ацетонитрил. Фильтрат сушат на вакууме. Получают 26,31 г (выход 96%) 1-октадецил-2-метилкарбамоилглицерина. Количественное содержание: 99,3%. Т.пл. 46°С, Rf=0.42 (силикагель, эфир-петролейный эфир=3:1). 1Н ЯМР (500 МГц, CDCl3, δ, м.д.): 0.9 (3Н, т, -СН3); 1-2 (30 Н, м, -СН2); 1-6 (2Н, м, -ОСH2); 2.8 (3Н, с, -CONHCH3); 3.0 (1H, м, -ОН); 3.4-3.6 (4Н, м, -1СH2СН-3CH2-); 3.8 (1H, с, -CONH-); 4.9 (1H, м, -2СН-ОСО-). Найдено, %: С 67.81; Н 11.21; N 3.10. C22H45NO4. Вычислено, %: С 68.21, Н 11.62, N 3.62.
Пример 4. Получение 1-октадецил-2-этилкарбамоилглицерина. К раствору 23,5 г (0.068 моль) 1-октадецилглицерина и 9,9 мл (0.123 моль) пиридина в 150 мл хлористого метилена прибавляют раствор 7,4 г (0.068 моль) триметилхлорсилана в 20 мл хлористого метилена. Реакционную смесь выдерживают при 0°С, прибавляют 14,6 мл (0.1845 моль) этилизоцианата и оставляют на ночь. Затем фильтруют, удаляют в вакууме растворитель, остаток растворяют в 200 мл 5%-ого раствора метанола в хлороформе, добавляют 50 мл воды и интенсивно встряхивают в течение часа. После чего на делительной воронке отделяют органический слой. После к отделенному органическому слою добавляют смесь хлороформ: бензол, и удаляют растворитель под вакуумом. Остаток растворяют в достаточном для полного растворения объеме хлороформа и фильтруют через слой оксида кремния, который затем промывают смесью этиловый эфир: хлороформ: ацетонитрил. Фильтрат сушат на вакууме. Получают 27,80 г (выход 98%) 1-октадецил-2-этилкарбамоилглицерина. Т.пл. 42°С, Rf=0.44 (силикагель, эфир-петролейный эфир=3:1). 1Н ЯМР (500 МГц, СDCl3, δ, м.д.): 0.9 (3Н, т, - СН3); 1.2 (30 Н, м, - СH2-); 1.6 (2Н, м, - OCH2); 2.4 (3H, с, - CONHCH2CH3); 3.4-3.6 (4Н, м, -1CH2CH-3CH2-); 5.2 (1H, м, -2СН-ОСО- ). Найдено, %: С 68,11; Н 11.11; N 3.30. C23H47NO4. Вычислено, %: С 68.82, Н 11.72, N 3.49. Количественное содержание: 99,1%.
Количественное определение и определение примесей
Хроматографическая система Dionex Ultimate 3000 («Тпептю», Германия), включающая следующие составляющие: насос LPG-3400SD (или аналогичный); автосемплер WPS-3000SL(rthi аналогичный); термостат TCC-3000SD (или аналогичный); детектор ОАЕ)-3000(или аналогичный).
Хроматографическая колонка для обращенно-фазной хроматографии Hypersyl ODS С18, 5 мкм 120 4,6×250 mm (или аналогичная).
4,6×250 mm (или аналогичная).
Скорость потока элюента 1 мл/мин; объем вводимой пробы 20 мкл; температура термостата колонки 30°С; длина волны детектирования 200 нм.
Колонку уравновешивают подвижной фазой до достижения стабильной базовой линии в течение 30 минут.Последовательно анализируют стандартный раствор и испытуемый раствор.
Проверка пригодности хроматографической системы
Хроматографическая система считается пригодной, если на хроматограмме стандартного раствора 1-O-алкил-2-O-алкилкарбомоилглицерина:
- коэффициент удерживания 1-O-алкил-2-O-алкилкарбомоилглицерина не менее 1,00;
- эффективность колонки рассчитанная по пику 1-O-алкил-2-O-алкилкарбомоилглицерина - не менее 5000 теоретических тарелок;
- относительное стандартное отклонение значений времен удерживания и площадей пика 1-O-алкил-2-O-алкилкарбомоилглицерина, рассчитанное по 5 последовательным хроматограммам, не превышает 2%;
- коэффициент асимметрии пика 1-O-алкил-2-O-алкилкарбомоилглицерина 0,80-1,20.
Содержание 1-O-алкил-2-O-алкилкарбомоилглицерина рассчитывается по формуле:
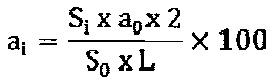
где ai- содержание 1-O-алкил-2-O-алкилкарбомоилглицерина в субстанции;
Si - площадь пика 1-O-алкил-2-O-алкилкарбомоилглицерина на хроматограмме испытуемого раствора;
S0 - площадь пика 1-O-алкил-2-O-алкилкарбомоилглицерина на хроматограмме стандартного раствора;
2- коэффициент разбавления;
а0 - навеска 1-O-алкил-2-O-алкилкарбомоилглицерина, взятая для приготовления стандартного раствора в граммах;
L - навеска субстанции, взятая для приготовления испытуемого раствора в граммах;
100 - коэффициент для перевода в проценты.
Результатом определения считается среднее арифметическое не менее трех последовательных определений. Содержание 1-O-алкил-2-O-алкилкарбомоилглицерина в 1 г должно быть от 98,5% до 101,5%.
По хроматограмме испытуемого раствора вычисляют площадь пика каждой индивидуальной примеси - Si, сумму площадей всех пиков ΣSi, площадь пика 1-О-алкил-2-О-алкилкарбомоилглицерина - S. Системные пики (коэффициент удерживания менее 1) и пики площадью менее 0,02% не учитываются.
Содержание примесей рассчитывают по формуле:
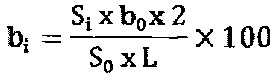
где bi- содержание примеси (в %) от содержания 1-O-алкил-2-O-алкилкарбомоилглицерина;
Si - площадь пика примеси на хроматограмме испытуемого раствора;
S0 - площадь пика 1-O-алкил-2-O-алкилкарбомоилглицерина на хроматограмме стандартного раствора;
2- коэффициент разбавления;
b0 - навеска 1-O-алкил-2-O-алкилкарбомоилглицерина, взятая для приготовления стандартного раствора в граммах;
L - навеска субстанции, взятая для приготовления испытуемого раствора в граммах;
100 - коэффициент для перевода в проценты.
Приготовление растворов
Подвижная фаза: в мерную колбу вместимостью 1000 мл помещают 500 мл воды для хроматографии и 300 мл ацетонитрила (HLPC grade, Panreac, Cat.No. 221881.1612) и 200 мл изопропилового спирта (HLPC grade, Panreac, Cat.No. 221090.1612), перемешивают и фильтруют через мембранный фильтр с диаметром пор 0,45 мкм. Допускается менять состав подвижной фазы для достижения пригодности хроматографической системы.
Стандартный раствор 1-O-алкил-2-O-алкилкарбомоилглицерина готовят путем растворения точной навески (12,5 мг) РСО в подвижной фазе в мерной колбе на 50 мл.
Испытуемый раствор: точную навеску образца (25 мг) переносят в колбу на 100 мл, растворяют в подвижной фазе и доводят ей до метки.
Обоснование режима способа
Использование более слабого основания (пиридина рКа 5.25) позволяет увеличить выход конечного продукта т.к. это способствует селективному присоединению защитной группировки по 3 атому углерода молекулы 1-О-алкилглицерина, что в свою очередь приводит к более селективному введению алкилкарбомоильной группы по второму атому углерода. Кроме того, токсичность пиридина (LD50 1580 мг/кг по данным National Institute for Occupational Safety and Health) в 3 раза ниже, чем у триэтиламина (LD50 460 мг/кг по данным National Institute for Occupational Safety and Health), что повышает безопасность производства.
Замена растворителя для проведения химической реакции на более полярный (метиленхлорид), за счет улучшения сольватации интермедиатов реакции, протекающей по Sn2-механизму, также способствовует увеличению скорости протекания процесса замещения и повышению выхода конечного продукта за счет улучшения сольватации интермедиатов реакции, протекающей по Sn2-механизму.
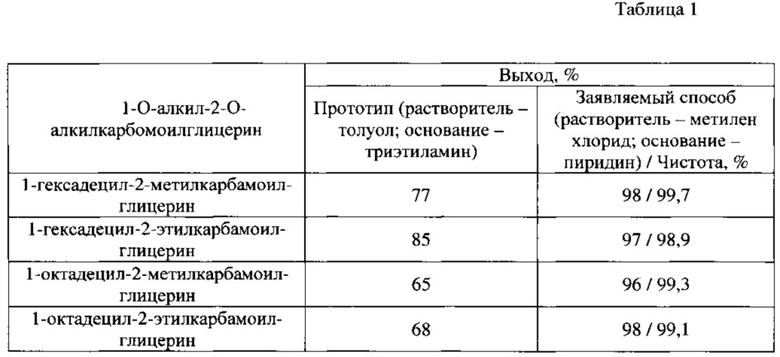
Экспериментальный синтез 1-O-алкил-2-O-алкилкарбомоилглицеринов с использованием указанных изменений, подтвердил увеличение выхода целевого продукта до 98% и чистоты целевого продукта от 98,9% (Таблица 1).
Приложение 1
Способ получения 1-алкил-2-алкилкарбамоилглицеринов
Фигура 1 - Схема получения 1-O-алкил-2-O-алкилкарбомоилглицеринов
Таблица 1 - Изменение выхода целевого продукта при использовании заявленного способа
Изобретение относится к способу получения 1-алкил-2-алкилкарбамоилглицеринов общей формулы I, где R1 - углеводородный остаток С16-С18, R2 - метил, этил, которые могут быть использованы для медикаментозной терапии артериальной гипертензии. Способ заключается в том, что подвергают взаимодействию исходные 1-алкилглицерины общей формулы II с триметилхлорсиланом в присутствии основания в среде растворителя. При этом взаимодействие исходных 1-алкилглицеринов с триметилхлорсиланом осуществляют в присутствии пиридина, а в качестве растворителя используют метиленхлорид. Реакцию проводят в атмосфере воздуха при 0°C с дальнейшим присоединением соответствующего алкилизоцианата и последующей обработкой смесью метанол-хлороформ. Предлагаемый способ позволяет повысить выход и чистоту целевого продукта. 1 ил., 1 табл., 4 пр.
 (I)
(I) 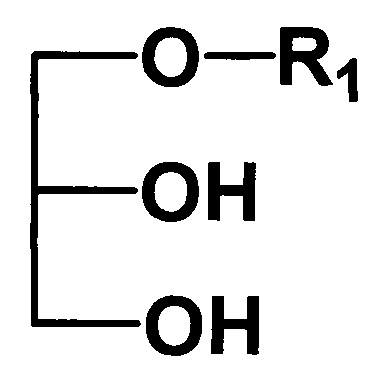 (II)
(II)
Способ получения 1-алкил-2-алкилкарбамоилглицеринов общей формулы I
 ,
,
где R1 - углеводородный остаток С16-С18; R2 - метил, этил, заключающийся в том, что подвергают взаимодействию исходные 1-алкилглицерины общей формулы II

с триметилхлорсиланом в присутствии основания в среде растворителя, отличающийся тем, что исходные 1-алкилглицерины подвергают взаимодействию с триметилхлорсиланом в присутствии пиридина, а в качестве растворителя используют метиленхлорид, при этом реакцию проводят в атмосфере воздуха при 0°C с дальнейшим присоединением соответствующего алкилизоцианата и последующей обработкой смесью метанол-хлороформ.
| 1-АЛКИЛ-2-АЛКИЛКАРБАМОИЛ-ГЛИЦЕРИНЫ, ОБЛАДАЮЩИЕ АНТИГИПЕРТЕНЗИВНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2008 |
|
RU2414453C2 |
| J | |||
| R | |||
| SURLES et al, Synthesis and evidence for the stability of a glycerophosphochloridate: rac-1-O-hexadecyl-2-O-(methylcarbamyl)-sn-glycero-3-phosphorochloridocholine, J | |||
| ORG | |||
| CHEM., 1988, 53, pp | |||
| Ухват | 1923 |
|
SU899A1 |
| US 4731373, 15.03.1988 | |||
| Установка для имитации дождя при климатических испытаниях аппаратуры | 1950 |
|
SU94186A1 |
| Способ тонкого размола | 1949 |
|
SU89635A1 |

