ОБЛАСТЬ И ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение, в некоторых его вариантах, относится к новым окисленным липидам и способам, в которых используются окисленные липиды, для лечения или профилактики воспаления, ассоциированного с эндогенными окисленными липидами. Способы по настоящим вариантам могут быть использованы для лечения или профилактики заболеваний и нарушений, связанных с воспалением, таких как, например, атеросклероз и родственные нарушения, аутоиммунные заболевания или нарушения, а также пролиферативные заболевания или нарушения.
Ранее, окисленные фосфолипиды были описаны как эффективные для лечения состояний, таких как, например, сердечно-сосудистые заболевания, цереброваскулярные заболевания и воспалительные заболевания и нарушения.
Международная заявка на патент PCT/IL2004/000453 (Публикация 04/106486), настоящего патентообладателя, описывает окисленные липиды, которые могут быть использованы для профилактики и лечения воспалений, ассоциированных с эндогенными окисленными липидами. Пример такого соединения описан и известен как CI-201 (также называемый в данной области как VB-201).
Международная заявка на патент PCT/IL01/01080 (Публикация WO 02/41827), настоящего патентообладателя, описывает окисленные липиды, которые могут быть использованы для профилактики и лечения атеросклероза и родственных заболеваний.
Международная заявка на патент PCT/IL05/000735 (Публикация WO 06/006161), настоящего патентообладателя, описывает синтетические подходы, которые могут быть использованы для промышленного приготовления терапевтически эффективных окисленных фосфолипидов без применения колоночной хроматографии.
Дополнительный уровень техники включает Международную заявку на патент PCT/IL02/00005 (Публикация WO 02/053092) и PCT/IL08/000013 (Публикация WO 08/084472), принадлежащие настоящему патентообладателю.
Все указанные выше публикации включены в виде ссылки, как если бы они были изложены здесь полностью.
Все указанные выше публикации описывают этерифицированные окисленные липиды, которые содержат углеродный скелет, к которому присоединена алкильная цепь, при этом алкильная цепь является замещенной окисленной группой, и фосфат-содержащая группа. Алкильная цепь, которая является замещенной окисленной группой, предпочтительно присоединена к углеродному скелету эфирной связью (поэтому соединения называются «этерифицированными окисленными липидами»), поскольку такая связь наделяет соединения требуемыми фармакологическими свойствами, которые подробно описаны в упомянутых выше публикациях.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения разработали, успешно приготовили и протестировали новые окисленные фосфолипиды.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено соединение, имеющее формулу:
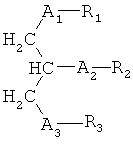
или его фармацевтически приемлемые соли, где:
(i) каждый A1, А2 и А3 независимо является выбранным из группы, состоящей из О и S.
(ii) R1 является выбранным из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и
 ,
,
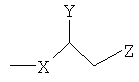
где Х представляет собой С1-25 цепь, Y является выбранным из группы, состоящей из 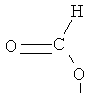 , -ОН,-Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп;и
, -ОН,-Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп;и
Z является выбранным из группы, состоящей из:
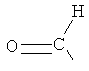 ,
, 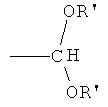 , ,
, , 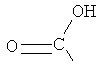 ,
, 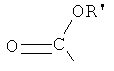 и -OH,
и -OH,
где R' представляет собой С1-4 алкил; и
(iii) R2 является выбранным из группы, состоящей из (4-метилкарбокси)бутила, (3 -карбокси)пропила, (6-карбокси)гексанила, (2-карбокси)этила, 5,6-дигидроксигексанила, 5,5-диэтоксипентила и диметоксипентила; и
(iv) R3 является выбранным из группы, состоящей из Н, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты, фосфосерина и фосфоинозитола.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено соединение, имеющее формулу:
или его фармацевтически приемлемые соли, где;
(i) каждый A1, A2 и А3 независимо является выбранным из группы, состоящей из О и S;
(ii) каждый R1 и R2 независимо является выбранным из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и Y
при условии, что по меньшей мере один из R1 и R2 представляет собой Y
, где Х является С1-25 цепью, Y является выбранным из группы, состоящей из , -ОН, -Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iii) Кз является выбранным из группы, состоящей из Н, фосфата, фосфоэтаноламина, фосфоэтаноламин-М-глутаровой кислоты и фосфосерина.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено соединение, имеющее формулу:
или его фармацевтически приемлемые соли, где:
(i) каждый A1, А2 и А3 независимо является выбранным из группы, состоящей из О и S;
(ii) R1 является выбранным из группы, состоящей из додецила, октадецила, октила, эйкозанила, цис-9-гексадеценила, (2-октил)додецила и (15-карбокси)пентадецила;
(iii) R2 является выбранным из группы, состоящей алкильной цепи длиной 2-28 атомов углерода, и
,
при условии, если R1 является отличным от (15-карбокси)пентадецила, то R2 представляет собой
где X представляет собой С1-25 цепь, Y является выбранным из группы, состоящей из
, -OH, -H, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iv) R3 является выбранным из группы, состоящей из Н, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-Н-глутаровой кислоты, фосфосерина и фосфоинозитола.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено соединение, имеющее формулу:
или его фармацевтически приемлемые соли, где:
(i) A1 представляет собой S, и каждый А2 и А3 представляет собой О;
(ii) каждый R1 и R2 независимо является выбранным из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и Y
при условии, что, по меньшей мере, один из R1 и R2 является , где Х представляет собой С1-25 цепь, Y является выбранным из группы, состоящей из:
, -ОН, -Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iii) R3 является выбранным из группы, состоящей из Н, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты, фосфосерина и фосфоинозитола.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено соединение, выбранное из группы, состоящей из следующих соединений:
1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфат (CI-201-РА);
1-гексадепил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламин;
1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолин (CI-208);
1 -гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-202);
1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламин (CI-206);
1 -гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолин (CI-205);
1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолин(С1-203);
1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-209);
1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-М-глутаровая
кислота (CI-210);
1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3 -фосфохолин (CI-213);
1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3 -фосфоэтаноламин (CI-214);
1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-215);
1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-216);
1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолин (CI-217);
1-8-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (1 -S-CI-201);
1-8-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин(1-8-С1-202);
1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолин (di-OH);
1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолин;
1-гексадецил-2-(4-карбокси)бутил-глицерин;
1-гексадецил-2-(5',5'-диэтоксипентил)глицеро-3-фосфохолин (diEtAc);
1-гексадецил-2-(5',5'-диметоксипентил)глицеро-3 -фосфохолин (diMeAc);
1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-207);
1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин;
1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-219);
1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-220);
1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (VB-221);
1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (VB-221); и
1 -гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфосерин (VB-223). Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечена фармацевтическая композиция, включающая, в качестве активного ингредиента, указанное соединение и фармацевтически приемлемый носитель.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечен способ лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, при этом способ включает введение нуждающемуся субъекту терапевтически эффективного количества описанного здесь соединения, осуществляя тем самым лечение или профилактику воспаления, ассоциированного с эндогенным окисленным липидом у субъекта.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечен способ снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта, при этом способ включает введение субъекту эффективного количества описанного здесь соединения.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечен способ лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным, при этом способ включает введение нуждающемуся субъекту эффективного количества описанного здесь соединения.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено использование описанного здесь соединения в производстве лекарственных средств, предназначенных для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено использование описанного здесь соединения в производстве лекарственных средств, предназначенных для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта.
Согласно одному аспекту некоторых вариантов по настоящему изобретению, обеспечено использование описанного здесь соединения в производстве лекарственных средств, предназначенных для лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Согласно некоторым вариантам изобретения, r) представляет собой алкильную цепь длиной 2-28 атомов углерода.
Согласно некоторым вариантам изобретения, описанное здесь соединение определено для использования в способе лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом.
Согласно некоторым вариантам изобретения, описанное здесь соединение определено для использования в способе снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23.
Согласно некоторым вариантам изобретения, описанное здесь соединение определено для использования в способе лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Согласно некоторым вариантам изобретения, фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском в упаковочном материале или надписью, нанесенной на указанный упаковочный материал, как препарат, предназначенный для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом.
Согласно некоторым вариантам изобретения, фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском в упаковочном материале или надписью, нанесенной на указанный упаковочный материал, как препарат, предназначенный для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23.
Согласно некоторым вариантам изобретения, фармацевтическая композиция упакована в упаковочный материал и идентифицирована оттиском в упаковочном материале или надписью, нанесенной на указанный упаковочный материал, как препарат, предназначенный для лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Согласно некоторым вариантам изобретения, воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечнососудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
Если иное не оговорено, все технические и/или научные термины, использованные здесь, имеют то же самое значение, которое обычно понимается средним специалистом в данной области, к которой относится изобретение. Несмотря на то, что способы и материалы, аналогичные или эквивалентные приведенным в данном описании, могут использоваться при осуществлении на практике или проверке вариантов изобретения, ниже представлены примеры способов и/или материалов, описанных здесь. В случае возникновения противоречия необходимо обратиться к описанию изобретения, включающему определения терминов. Кроме того, материалы, способы и примеры носят только иллюстративный характер, и их не следует рассматривать как ограничивающие объем изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Некоторые варианты изобретения описаны здесь только в виде примера со ссылкой на прилагаемые чертежи. При подробном рассмотрении чертежей необходимо подчеркнуть, что отдельные детали приведены только в качестве примера и для иллюстрации вариантов изобретения. В данной связи, описание изобретения в сочетании с чертежами наглядно показывает опытным в данной области специалистам, каким образом можно осуществить на практике варианты данного изобретения.
На чертежах:
На Фиг.1 изображена диаграмма, показывающая продукцию IL 12/23 р40 клетками, обработанными разными дозами CI-202 (каждый столбик представляет 3 образца); Р<0.004 для каждой дозы по сравнению с контролем (0 мкг/мл);
На Фиг.2 изображена диаграмма, показывающая экспрессию мРНК IL 12/23 р40 в клетках, обработанных CI-201, CI-202 и фосфатидилхолином (PC), через 2, 3 и 4 часа после обработки;
На Фиг.3 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (18.5 или 37 мкМ) CI-202, 10 или 20 мкг/мл (17 или 34 мкМ) CI-201, фосфатидилхолином (PC) и смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.4А и 4В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-202;
На Фиг.5 изображена диаграмма, показывающая развитие МОГ-индуцированного экспериментального аутоиммунного энцефаломиелита у мышей, обработанных PBS или 4 мг/кг CI-202;
На Фиг.6 изображена диаграмма, показывающая развитие коллаген-индуцированного артрита у мышей, обработанных CI-202 (этаноламиновый аналог CI-201), и у контрольных мышей;
На Фиг.7 изображена диаграмма, показывающая продукцию IL 12/23 р40 клетками, обработанными разными дозами CI-203 (каждый столбик представляет 6 образцов), и Р величины по сравнению с контролем (0 мкг/мл);
На Фиг.8 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 1 или 20 мкг/мл (1.6 или 33 мкМ) (R)-CI-203 (R-CI-203) и рацемическим CI-203 (рац-CI-203), 1 или 20 мкг/мл (1.7 или 34 мкМ) (R)-CI-201 (R-CI-201) и (S)-CI-20l (S-CI-201), и 1 или 20 мкг/мл (1.3 или 26 мкМ) фосфатидилхолина (Ph.Ch.); ERK1/2 показана как контроль загрузки белка;
На Фиг.9А и 9В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-203;
На Фиг.10 изображена диаграмма, показывающая продукцию IL 12/23 р40 клетками, обработанными разными дозами CI-209 (каждый столбик представляет 5 образцов), и Р величины (Р<0.008 для доз 10 и 20 мкг/мл, и Р<0.016 для доз 1, 2.5 и 5 мкг/мл) по сравнению с контролем (0 мкг/мл);
На Фиг.11 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 20 мкг/мл (38 мкМ) CI-209, CI-201 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.12А и 12В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-209;
На Фиг.13 изображена диаграмма, показывающая продукцию IL 12/23 р40 клетками, обработанными разными дозами CI-210 (каждый столбик представляет 4 образца), и Р величины (Р<0.029 для доз 10 и 20 мкг/мл и Р 0.057 для доз 2.5 и 5 мкг/мл) по сравнении с контролем (0 мкг/мл);
На Фиг.14 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 20 мкг/мл (31 мкМ) CI-210, CI-201 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.15А и 15В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-210;
На Фиг.16 изображена диаграмма, показывающая продукцию IL 12/23 р40 клетками, обработанными разными дозами CI-216 (каждый столбик представляет 4 образца), и Р величины по сравнению с контролем (0 мкг/мл);
На Фиг.17 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (17 или 34 мкМ) CI-215 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.18 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (15 или 30 мкМ) CI-216, 10 или 20 мкг/мл (17 или 34 мкМ) CI-201, фосфатидилхолином (PC) или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.19 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 20 мкг/мл (38 мкМ) CI-206 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.20 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 20 мкг/мл (35 мкМ) CI-205, CI-201 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.21А и 21В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-206;
На Фиг.22А и 22В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-205;
На Фиг.23 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 20 мкг/мл (34 мкМ) CI-208, CI-201 или фосфатидилхолином (PC); α-тубулин (α-Туб) показан как контроль загрузки белка;
На Фиг.24А и 24В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-208;
На Фиг.25А и 25В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-213;
На Фиг.26А и 26В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-214;
На Фиг.27 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (18 или 36 мкМ) CI-217, 10 или 20 мкг/мл (17 или 34 мкМ) CI-201, фосфатидилхолином (PC) или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.28 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (31 мкМ) CI-219 и 20 мкг/мл (34 мкМ) CI-201 или фосфатидилхолином (PC) или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.29 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (34 мкМ) CI-220 и 20 мкг/мл (34 мкМ) CI-201 или фосфатидилхолином (PC), или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.30А и 30В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз CI-201-РА;
На Фиг.31 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (17 или 34 мкМ) l-S-CI-201 (CI-S-201), 10 или 20 мкг/мл (17 или 34 мкМ) CI-201, фосфатидилхолином (PC) или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.32 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 10 или 20 мкг/мл (18 или 36 мкМ) l-S-CI-202 (CI-S-202), 10 или 20 мкг/мл (18.5 или 37 мкМ) CI-202, 10 или 20 мкг/мл (17 или 34 мкМ) CI-201, фосфатидилхолином (PC) или смесью PBS/1% этанол (раствор);
ERK1/2 показана как контроль загрузки белка;
На Фиг.33А и 33В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз di-OH;
На Фиг.34 изображена диаграмма, показывающая область атеросклеротических изменений у мышей, обработанных di-OH, и у контрольных мышей;
На Фиг.35 изображена диаграмма, показывающая область атеросклеротических изменений у мышей, обработанных diMeAc, и у контрольных мышей;
На Фиг.36А и 36В изображены диаграммы, показывающие результаты отдельного эксперимента по тестированию токсичности разных доз diEtAc;
На Фиг.37 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 1,5, 10 или 20 мкг/мл (1.7, 8.3, 16.7 или 33.3 мкМ) VB-223 или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.38 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 1, 5, 10 или 20 мкг/мл (1.6, 8, 16 или 32 мкМ) VB-221 или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка;
На Фиг.39 представлена фотография Вестерн-блот анализа, показывающая фосфотирозин (р-Тирозин) в образцах, обработанных 1, 5, 10 или 20 мкг/мл (1.7, 8.4,
16.8 или 33.6 мкМ) VB-222 или смесью PBS/1% этанол (раствор); ERK1/2 показана как контроль загрузки белка.
ОПИСАНИЕ КОНКРЕТНЫХ ВАРИАНТОВ ИЗОБРЕТЕНИЯ
Настоящее изобретение, в некоторых его вариантах, относится к новым окисленным липидам и способам, в которых используются окисленные липиды, для лечения или профилактики воспаления, ассоциированного с эндогенными окисленными липидами. Окисленные липиды, описанные здесь, могут применяться для лечения или профилактики заболеваний и нарушений, ассоциированных с воспалением, таких как, например, атеросклероз и родственные нарушения, аутоиммунные заболевания или нарушения, а также пролиферативные заболевания или нарушения.
Принципы и способы осуществления настоящего изобретения могут быть лучше поняты при обращении к фигурам и прилагаемым описаниям.
Прежде чем перейти к подробному объяснению, по меньшей мере, одного варианта изобретения, необходимо отметить, что изобретение не ограничивается подробной информацией, изложенной в последующем описании или проиллюстрированной Примерами. Возможны другие варианты изобретения, которые можно осуществить на практике или выполнить разными способами. Также, должно быть понятно, что фразеология и терминология, используемая здесь, служит для описания и не ограничивает объем изобретения.
Экспериментальные и клинические данные свидетельствуют о причинной роли окисленного LDL (ox LDL) и компонентов LDL в этиологии повышенной воспалительной реакции в случае атеросклероза. Была продемонстрирована как клеточная, так и гуморальная иммунная реактивность в отношении окисленного LDL, связанного с бляшкам, что позволяет предположить возникновение важного аутоиммунного компонента против окисленного LDL в процессе атерогенеза. Таким образом, против LDL, окисленного LDL и его компонентов были направлены многочисленные методы профилактики и лечения болезней сердца, сердечнососудистых заболеваний и заболеваний периферических сосудов.
Роль окисленных фосфолипидов в лечении воспаления является раскрытой, например, в Международной патентной заявке PCT/IL2004/000453 (Публикация WO 04/106486) и Американской заявке 11/528,657 (Публикация 2007-0099868) настоящего патентообладателя, обе из которых включены здесь в виде ссылки так, как если бы они были изложены здесь полностью.
CI-201 (также называемый здесь и в данной области как VB-201) является многообещающим окисленным фосфолипидом, который в настоящее время проходит расширенные клинические испытания по применению для лечения воспалительных состояний, таких как атеросклероз.
В попытке улучшить лечение воспаления, а также заболеваний и нарушений, ассоциированных с окисленными липидами, авторы настоящего изобретения приготовили новые окисленные фосфолипиды и структурно родственные соединения, которые демонстрируют улучшенное противовоспалительное действие и/или улучшенные фармакологические характеристики.
Улучшенное противовоспалительное действие может быть легко определено с помощью известных in vitro и in vivo моделей воспалительных процессов, а также может проявляться улучшенным терапевтическим действием на подлежащее лечению заболевание, как подробно описано далее. Улучшенные фармакологические характеристики включают улучшенную биостабильность, биодоступность, пониженную токсичность и, более того, улучшенную стабильность в процессе производства, составления и/или хранения. Данные характеристики могут быть также определены с помощью экспериментов, которые легко распознают опытные в данной области специалисты, и как подробно описано далее.
Как показано в последующем разделе «Примеры», при осуществлении настоящего изобретения на практике было действительно подтверждено, что новые разработанные окисленные липиды, описанные здесь, модулируют продукцию цитокина, ассоциированную с иммунной и/или воспалительной реакцией на эндогенный окисленный LDL, тем самым проявляя способность к снижению воспалительной реакции в воспалительных заболеваниях, таких как, но не ограничиваясь ими, атеросклероз и ревматоидный артрит.
Кроме того, как показано в последующем разделе «Примеры», новые разработанные окисленные липиды, описанные здесь, модулируют фосфорилирование тирозина аналогично CI-201, что свидетельствует о том, что данные новые разработанные окисленные липиды разделяют биологические эффекты (например, противовоспалительные действия), которые, как было ранее показано, проявлял CI-201.
Кроме того, как показано в последующем разделе «Примеры», описанные здесь соединения проявляют минимальную токсичность и проявляют биологические эффекты при дозах, при которых соединения являются в значительной степени не токсичными.
На Фиг.1 и 2 показано ингибирование иллюстративным соединением CI-202 продукции субъединицы р40 провоспалительных цитокинов, интерлейкина-12 и интерлейкина-23. На Фиг.3 показана модуляция фосфорилирования тирозина с помощью CI-202, аналогичная модуляции, проявляемой с помощью CI-201. На Фиг.4А и 4В показаны профили токсичности CI-202. На Фиг.5 показано, что CI-202 является терапевтически эффективным в мышиной модели аутоиммунного энцефаломиелита (экспериментальная модель человеческого множественного склероза и острого рассеянного энцефаломиелита). На Фиг.6 показано, что CI-202 является терапевтически эффективным в мышиной модели артрита.
На Фиг.7 показано ингибирование иллюстративным соединением CI-203 продукции субъединицы р40 провоспалительных цитокинов, интерлейкина-12 и интерлейкина-23. На Фиг.8 показана модуляция фосфорилирования тирозина с помощью CI-203, аналогичная модуляции, проявляемой с помощью CI-201. На Фиг.9А и 9В показаны профили токсичности CI-203.
На Фиг.10 показано ингибирование иллюстративным соединением CI-209 продукции субъединицы р40 провоспалительных цитокинов, интерлейкина-12 и интерлейкина-23. На Фиг.11 показана модуляция фосфорилирования тирозина с помощью CI-209. На Фиг.12А и 12В показаны профили токсичности CI-209.
На Фиг.13 показано ингибирование иллюстративным соединением CI-210 продукции субъединицы р40 провоспалительных цитокинов, интерлейкина-12 и интерлейкина-23. На Фиг.14 показана модуляция фосфорилирования тирозина с помощью CI-210. На Фиг.15А и 15В показаны профили токсичности CI-210.
На Фиг.16 показано ингибирование иллюстративным соединением CI-216 продукции субъединицы р40 провоспалительных цитокинов, интерлейкина-12 и интерлейкина-23. На Фиг.18 показана модуляция фосфорилирования тирозина с помощью CI-216, аналогичная модуляции, проявляемой с помощью CI-201.
На Фиг.17 показана модуляция фосфорилирования тирозина с помощью CI-215. На Фиг.19 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-206. На Фиг.21А и 21В показаны профили токсичности CI-206.
На Фиг.20 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-205, аналогичная модуляции, проявляемой с помощью CI-201. На Фиг.22А и 22В показаны профили токсичности CI-205.
На Фиг.23 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-208, аналогичная модуляции, проявляемой с помощью CI-201. На Фиг.24А и 24В показаны профили токсичности CI-208.
На Фиг.25А и 25В, а также 26А и 26В показаны профили токсичности иллюстративных соединений CI-213 и CI-214, соответственно.
На Фиг.27 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-217, аналогичная модуляции, проявляемой с помощью CI-201.
На Фиг.28 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-219, аналогичная модуляции, проявляемой с помощью CI-201.
На Фиг.29 показана модуляция фосфорилирования тирозина иллюстративным соединением CI-220, аналогичная модуляции, проявляемой с помощью CI-201.
На Фиг.30А и 30В показаны профили токсичности иллюстративного соединения CI-201-РА.
На Фиг.31 показана модуляция фосфорилирования тирозина иллюстративным соединением 1-S-CI-201, аналогичная модуляции, проявляемой с помощью CI-201.
На Фиг.32 показана модуляция фосфорилирования тирозина иллюстративным соединением 1-S-CI-202, аналогичная модуляции, проявляемой с использованием CI-201.
На Фиг.34 показано, что иллюстративное соединение di-OH является терапевтически эффективным в мышиной модели атеросклероза. На Фиг.33А и 33В показаны профили токсичности di-OH.
На Фиг.35 показано, что иллюстративное соединение diMeAc является терапевтически эффективным в мышиной модели атеросклероза. На Фиг.36А и 36В показаны профили токсичности diEtAc, соединение тесно связано с diMeAc.
На Фиг.37-39 показана модуляция фосфорилирования тирозина иллюстративными соединениями VB-223, VB-221 и VB-222, соответственно.
Таким образом, было показано, что описанные здесь иллюстративные соединения являются биологически активными по результатам тестов in vitro, и было подтверждено, что некоторые соединения являются терапевтически эффективными in vivo. Свойства окисленного липида, которые не были тестированы in vivo, могут быть далее проверены на пригодных животных моделях, например таких, которые описаны здесь далее в разделе «Примеры», в Международной патентной заявке PCT/IL2004/000453 (Публикация WO 04/106486) и Американской патентной заявке 11/528,657 (Публикация 2007-0099868), и моделях, разработанных, как описано, например, в Singh et al.. Clinical Chemistry 51:12, 2252-2256 (2005), которая включена здесь полностью в виде ссылки, как если бы она была изложена здесь полностью.
Биостабильность окисленных липидов, описанных здесь, является улучшенной благодаря присутствию эфирных и/или сульфидных связей вместо сложноэфирных связей, присутствующих в большинстве липидов. Биостабильность обычно улучшает терапевтическое действие соединения. Биостабильность окисленных липидов можно определить, например, анализом их ферментативного расщепления фосфолипазой-С с использованием ELISA или измерением абсорбции.
Таким образом, описанные здесь окисленные липиды могут быть успешно признаны проявляющими улучшенное действие при лечении или профилактике воспаления, ассоциированного с эндогенными окисленными липидами, в отношении улучшенных терапевтических и/или фармакокинетических параметров.
Таким образом, согласно одному аспекту вариантов по настоящему изобретению, обеспечены новые окисленные липиды (например, окисленные фосфолипиды), как описано здесь.
Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфат (также называемый здесь как «CI-201-PA»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламин. Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «CI-208»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (также называемый здесь как «CI-202»), Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламин (также называемый здесь как «CI-206»).
Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолин (также называемый здесь как «CI-205»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолин (также называемый здесь как «CI-203»), Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «CI-209»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-1-глутаровую кислоту (также называемую здесь как «CI-210»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «CI-213»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -(15' -карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3 -фосфоэтаноламин (также называемый здесь как «CI-214»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «CI-215»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (также называемый здесь как «CI-216»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолин (также называемый здесь как «CI-217»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-8-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «1-S-CI-201»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-8-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (также называемый здесь как «1-S-CI-202»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолин (также называемый здесь как «di-ОН»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолин. Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(4-карбокси)бутил-глицерин. Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолин (также называемый здесь как «diEtAc»).
Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -гексадецил-2-(5',5' -диметоксипентил)-глицеро-3 -фосфохолин (также называемый здесь как «diMeAc»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-октил-2-(4-карбокси)бутил-глицеро-3 -фосфохолин (также называемый здесь как «CI-207»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин. Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3 -фосфохолин (также называемый здесь как «CI-219»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (также называемый здесь как «CI-220»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (также называемый здесь как «VB-221»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1 -(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (также называемый здесь как «VB-222»). Согласно одному иллюстративному варианту, окисленный липид представляет собой 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфосерин (также называемый здесь как «VB-223»),
Использующийся здесь префикс «1-S-» относится к соединению, в котором атом кислорода в положении 1 глицеринового остова (.sn-1) заменен на атом серы таким образом, что соединение является производным 1-тиоглицерина вместо производного глицерина.
Префиксы «CI-» и «VB-» используются здесь взаимозаменяемо.
В зависимости от заместителей, некоторые атомы углерода в каждом соединении, описанном здесь, могут быть хиральными или нехиральными. Таким образом, в иллюстративных соединениях, описанных здесь выше, атом углерода в положении 2 глицеринового остова является хиральным. Любой хиральный атом углерода, который присутствует в соединениях, описанных здесь, может быть либо в R-конфигурации, S-конфигурации, или в виде рацемата. Таким образом, настоящие варианты охватывают любую комбинацию хиральных и рацемических атомов углерода, включая все возможные стереоизомеры, оптические изомеры и энантиомеры.
Как показано в последующем разделе «Примеры», соединения вариантов по настоящему изобретению могут быть синтезированы при сохранении конфигурации исходного материала. Соединения настоящих вариантов могут быть, кроме того, избирательно синтезированы на основании стереохимии окисленной группы. Таким образом, с помощью выбора соответствующих исходных материалов и соответствующих условий синтеза могут быть определены оптическая чистота (например, включение хиральных и/или рацемических атомов углерода) и полученные стереоизомеры конечных соединений. В случаях, когда получены рацемические смеси, могут быть использованы известные методики разделения оптических или стерео-изомеров. Такие методики описаны, например, в "Organic chemistry, fourth Edition by Paula Yurkanis Bruice, page 180-185 and page 214, Prentice Hall, Upper Sadde R1ver, NJ 07458".
Вышеуказанные соединения могут быть охарактеризованы согласно их определенным новым структурным элементам.
Таким образом, некоторые из вышеуказанных окисленных липидов содержат глицериновый остов, к которому присоединена окисленная боковая цепь в положении 2, при этом окисленная боковая цепь является выбранной из группы, состоящей из (4-метилкарбокси)бутила, (3-карбокси)пропила, (6-карбокси)гексанила, (2-карбокси)этила, 5,6-дигидроксигексанила, 5,5-диэтоксипентила и 5,5-диметоксипентила.
Таким образом, согласно некоторым вариантам настоящего изобретения, обеспечены соединения, в совокупности представленные формулой:
или их фармацевтически приемлемые соли, где:
(i) каждый A1, А2 и А3 независимо является выбранным из группы, состоящей из О и S.
(ii) R1 является выбранным из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и
где X представляет собой С1-25 цепь, Y является выбранным из группы, состоящей из
, -OH,-H, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(in) R2 является выбранньш из группы, состоящей из (4-метилкарбокси)бутила, (3-карбокси)пропила, (б-карбокси)гексанила, (2-карбокси)этила, 5,6-дигидроксигексанила, 5,5-диэтоксипентила и 5,5-диметоксипентила; и
(iv) R3 является выбранным из группы, состоящей из И, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты, фосфосерина и фосфоиноситола.
Согласно некоторым вариантам, R1 представляет собой алкильную цепь длиной 2-28 атомов углерода.
Некоторые из окисленных липидов, описанных здесь выше, могут отличаться содержанием фосфорильной группы в 3-м положении соединения, выбранного из группы, состоящей из фосфата, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты и фосфосерина, или являются нефосфорилированными и незамещенными в положении 3 (т.е. атом водорода присутствует в положении 3).
Таким образом, согласно некоторым вариантам настоящего изобретения обеспечены соединения, в совокупности представленные формулой:
или их фармацевтически приемлемые соли, где:
(i) каждый A1, A2 и А3 независимо является выбранным из группы, состоящей из О и S;
(ii) каждый R1 и R2 независимо является выбранным из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и Y
при условии, что по меньшей мере один из R1 и R2 представляет собой Y
, где Х является С 1-25 цепью, Y является выбранным из группы, состоящей из , -ОН, -Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iii) R3 является выбранным из группы, состоящей из Н, фосфата, фосфоэтаноламина, фосфоэтаноламин-Н-глутаровой кислоты и фосфосерина.
Согласно некоторым вариантам, R1 представляет собой алкильную цепь длиной 2-28 атомов углерода. Предпочтительно, чтобы в таких вариантах R2 представлял собой .
Некоторые из окисленных липидов, описанных здесь выше, могут отличаться содержанием в боковой цепи в положении 1 соединения, выбранного из группы, состоящей из додецила, октадецила, октила, эйкозанила, цис-9-гексадеценила, (2-октил)додецила и (15-карбокси)пентадецила.
Таким образом, согласно некоторым вариантам настоящего изобретения обеспечены соединения, в совокупности представленные формулой:
или их фармацевтически приемлемые соли, где:
(i) каждый A1, A2 и А3 независимо является выбранным из группы, состоящей из О и S;
(ii) R1 является выбранным из группы, состоящей из додецила, октадецила, октила, эйкозанила, цис-9-гексадеценила, (2-октил)додецила и (15-карбокси)пентадецила;
(iii) R2 является выбранным из группы, состоящей алкильной цепи длиной 2-28 атомов углерода, и
при условии, если R1 является отличным от (15-карбокси)пентадецила, то R2 представляет собой
где Х представляет собой C1-25 цепь, Y является выбранным из группы, состоящей из
, -ОН, -Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iv) R3 является выбранным из группы, состоящей из Н, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты, фосфосерина и фосфоинозитола.
Предпочтительно, чтобы (15-карбокси) пентадецильная группа, описанная здесь, Y соответствовала группе, описанной здесь, где Х представляет собой алкильную цепь, состоящую из 13 атомов углерода. Y представляет собой водород и Z является -С(=O)ОН.
Аналогично, (4-метилкарбокси)бутил, (3-карбокси)пропил, (6-карбокси)гексанил, (2-карбокси)этил, 5,6-дигидроксигексанил, 5,5-диэтоксипентил и 5,5-диметоксипентил, соответствующий группе, в которой Z является -С(=0)ОН (как для (3-карбокси)пропил, (6-карбокси)гексанил и (2-карбокси)этил), где Z является -CH(OR')2 (как для 5,5-диэтоксипентил и 5,5-диметоксипентил), или где Z является -ОН (как для 5,6-дигидроксигексанил).
Некоторые из окисленных липидов, описанных здесь выше, могут характеризоваться тем, что они содержат атом серы в положении 1 и атомы кислорода в положениях 2- и 3-.
Следовательно, согласно некоторым вариантам настоящего изобретения обеспечены соединения, представленные формулой:
или их фармацевтически приемлемые соли, где:
(i) A1 представляет собой S, и каждый A2 и А3 представляет собой О;
(ii) каждый R1 и R1 независимо являются выбранными из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и
при условии, что по меньшей мере один из R1 и R2 является , где Х представляет собой С 1.25 цепь, Y является выбранным из группы, состоящей из:
, -ОН, -Н, алкила, алкокси, галогена, ацетокси и ароматических функциональных групп; и
Z является выбранным из группы, состоящей из:
, , , , и -OH,
где R' представляет собой С1-4 алкил; и
(iii) R3 является выбранным из группы, состоящей из Н, ацила, алкила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты, фосфосерина и фосфоинозитола.
Согласно некоторым вариантам R1 представляет собой алкильную цепь длиной 2-28 атомов углерода.
Согласно некоторым вариантам настоящего изобретения переменная Z, описанная здесь выше, является выбранной из группы, состоящей из:
,, и -OH.
Согласно факультативным вариантам настоящего изобретения, переменная Y является выбранной из группы, состоящей из Н и -ОН. В некоторых вариантах Y представляет собой -ОН, когда Z является -ОН и/или -О-С(=О)Н. В некоторых вариантах Y представляет собой Н, когда Z является -С(=О)Н, -CH(OR')2, -C(=О)OH и/или-C(=О)OR'.
Согласно иллюстративным вариантам, R' представляет собой насыщенный, ненасыщенный С 1-4 алкил. Факультативно, R' является выбранным из группы, состоящей из этила и метила.
Согласно факультативным вариантам, алкильная цепь длиной 2-28 атомов углерода, описанная здесь, является насыщенной, если специально не указано иное. Факультативно, алкильная цепь является ненасыщенной, если специально не указано иное.
Согласно факультативным вариантам переменная X, описанная здесь, является насыщенной алкильной цепью, состоящей из 1-25 атомов углерода, если специально не указано иное. Факультативно, алкильная цепь является ненасыщенной, если специально не указано иное.
Использующийся здесь термин «алкил» относится к насыщенному или ненасыщенному алифатическому углеводороду, включающему группы с прямой и разветвленной цепью. Предпочтительно, чтобы алкильная группа содержала от 1 до 20 атомов углерода. Во всех случаях указанный числовой интервал, например, «1-20», означает, что группа, в данном случае алкильная группа, может содержать 1 атом углерода, 2 атома углерода, 3 атома углерода, и т.д. до 20 атомов углерода включительно. Более предпочтительно, чтобы алкил имел цепь средней длины, включающую от 1 до 10 атомов углерода. Наиболее предпочтительно, за исключением особо оговоренных случаев, чтобы алкил являлся низшим алкилом, включающим от 1 до 4 атомов углерода. Алкильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, циклоалкил, арил, гетероарил, гетероалициклик, гало, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, O-карбокси и амино, в указанных здесь значениях. В некоторых вариантах алкил является незамещенным.
«Циклоалкильная» группа означает группу с моноциклическим или конденсированным кольцом, состоящим из атомов углерода (т.е. с кольцами, которые имеют общую смежную пару атомов углерода), в которой одно или более колец не имеют полностью конъюгированную систему пи-электронов. Неограничивающие примеры циклоалкильных групп включают циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексадиен, циклогептан, циклогептатриен и адамантан. Циклоалкильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может являться, например, алкил, арил, гетероарил, гетероалициклик, гало, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, O-карбокси и амино, в указанных здесь значениях.
«Арильная» группа, также называемая здесь как «ароматическая функциональная группа», означает группу с моноциклическим или полициклическим кольцом, состоящим из атомов углерода (т.е. с кольцами, которые имеют общую смежную пару атомов углерода), которая имеет полностью конъюгированную систему пи-электронов. Неограничивающие примеры арильных групп включают фенил, нафталенил и антраценил. Арильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, алкил, циклоалкил, арил, гетероарил, гетероалициклик, гало, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, O-карбокси и амино, в указанных здесь значениях.
«Гетероарильная» группа означает группу с моноциклическим или конденсированным кольцом (т.е. с кольцами, которые имеют общую смежную пару атомов), которая содержит в кольце(ах) один или более атомов, таких как, например, азот, кислород и сера, и дополнительно включает полностью конъюгированную систему пи-электронов. Неограничивающие примеры гетероарильных групп включают пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиридин, пиримидин, хинолин, изохинолин и пурин. Гетероарильная группа может быть замещенной или незамещенной. В случае замещения замещающей группой может быть, например, алкил, циклоалкил, арил, гетероарил, гетероалициклик, гало, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, O-карбокси и амино, в указанных здесь значениях.
«Гетероалициклическая» группа означает группу с моноциклическим или конденсированным кольцом, которая содержит в кольце(ах) один или более атомов, таких как азот, кислород и сера. Кольца могут также содержать одну или более двойных связей. Однако кольца не имеют полностью конъюгированную систему пи-электронов. Гетероалициклическая группа может быть замещенной или незамещенной. В случае замещения замещающей группой могут быть, например, электроны неподеленной пары, алкил, циклоалкил, арил, гетероарил, Гетероалициклическая группа, гало, гидрокси, алкокси, арилокси, тиогидрокси, тиоалкокси, тиоарилокси, циано, нитро, азид, сульфонил, сульфинил, сульфонамид, фосфонил, фосфинил, оксо, карбонил, тиокарбонил, мочевина, тиомочевина, O-карбамил, N-карбамил, O-тиокарбамил, N-тиокарбамил, С-амидо, N-амидо, С-карбокси, O-карбокси и амино в указанных здесь значениях. Типичными примерами являются пиперидин, пиперазин, тетрагидрофуран, тетрагидропиран, морфолин и тому подобные.
«Гидроксильная» группа означает -ОН группу.
«Азид» означает -N=N группу.
«Алкоксильная» группа означает -O-алкильную и -O-циклоалкильную группу в указанных здесь значениях.
«Арилокси» группа означает -O-арильную и -O-гетероарильную группу в указанных здесь значениях.
«Тиогидроксильная» группа означает -SH группу.
«Тиоалкоксильная» группа означает -S-алкильную и -S-циклоалкильную группу в указанных здесь значениях.
«Тиоарилокси» группа означает -S-арильную и -S-гетероарильную группу в указанных здесь значениях.
«Карбонильная» или «ацильная» группа означает -C(=0)-R группу, где R представляет собой водород, алкил, алкенил, циклоалкил, арил, гетероарил (связанный атомом углерода в кольце) или гетероалициклическую группу (связанную атомом углерода в кольце) в указанных здесь значениях.
«Альдегидная» группа означает карбонильную группу, где R представляет собой водород.
«Тиокарбонильная» группа означает -C(=S)-R группе, где R имеет указанные здесь значения.
«С-карбокси» группа означает -C(=O)-O-R группы, где R имеет указанные здесь значения.
«O-карбокси» группа означает RC(=0)-O- группу, где R имеет указанные здесь значения.
«Ацетокси» группа означает СН3С(=O)-O-. «Оксо» группа означает=O группу.
Группа «карбоновой кислоты» означает С-карбоксильную группу, в которой R представляет собой водород.
«Гало» или «галоген» группа означает фтор, хлор, бром или йод.
«Сульфинильная» группа означает -S(=O)-R группу, где R имеет указанные здесь значения.
«Сульфонильная» группа означает -S(=O)2-R группу, где R имеет указанные здесь значения.
«Сульфонамидная» группа означает -S(=O)2-NR2 группу или RS(=O)2-NR-группу, при этом каждый из R имеет указанные здесь значения.
«O-карбамильная» группа означает -OC(=O)-NR2 группу, где каждый из R имеет указанные здесь значения.
«N-карбамильная» группа означает ROC(=O)-NR- группу, где каждый из R имеет указанные здесь значения.
«O-тиокарбамильная» группа означает -OC(=S)-NR2 группу, где каждый из R имеет указанные здесь значения.
«N-тиокарбамильная» группа означает ROC(=S)NR- группу, где каждый из R имеет указанные здесь значения.
«Амино» группа означает -NR2 группу, где каждый из R имеет указанные здесь значения.
«С-амидо» группа означает -C(=O)-NR2 группу, где каждый из R имеет указанные здесь значения.
«N-амидо» группа означает RC(=O)-NR- группу, где каждый из R имеет указанные здесь значения.
Группа «мочевина» означает -NRC(=O)-NR2 группу, где каждый из R имеет указанные здесь значения.
«Нитро» группа означает -NO2 группу.
«Циано» группа означает -C≡N группу.
Термин «фосфонил» или «фосфонат» означает -P(=O)(OR)2 группу, где каждый из R имеет указанные выше значения.
Термин «фосфинил» описывает -PR2 группу, где каждый из R имеет указанные здесь значения.
Термин «тиомочевина» описывает -NR-C(=S)-NR- группу, где каждый из R имеет указанные выше значения.
Настоящие варианты, кроме того, охватывают любые фармацевтически приемлемые соли, пролекарства, гидраты и сольваты соединений, описанных здесь выше.
Термин «пролекарство» относится к агенту, который превращается в активное соединение (активное родительское лекарство) in vivo. Пролекарства являются обычно эффективными для облегчения введения родительского лекарственного препарата. Пролекарства могут, например, быть биодоступными при оральном введении, в то время как родительский лекарственный препарат не может быть пригоден для такого введения. Пролекарство может также иметь улучшенную растворимость по сравнению с родительским лекарственным препаратом в фармацевтических композициях. Пролекарства также часто используются для достижения замедленного высвобождения активного соединения in vivo. Неограничивающим примером пролекарства может являться описанное здесь соединение, содержащее одну или более групп карбоновой кислоты, которое вводится в виде сложного эфира («пролекарство»). Такое пролекарство гидролизуется in vivo, обеспечивая тем самым свободное соединение (родительское пролекарство). Выбранный сложный эфир может воздействовать как на характеристики растворимости, так и на скорость гидролиза пролекарства.
Выражение «фармацевтически приемлемая соль» означает заряженную разновидность родительского соединения и его противоион, который обычно используют для изменения характеристик растворимости родительского соединения и/или для уменьшения любого значительного раздражения организма родительским соединением без изменения биологической активности и свойств вводимого соединения. Неограничивающим примером фармацевтически приемлемой соли может являться карбоксилатный ион и катион, который включает, не ограничиваясь ими, аммоний, натрий, калий и тому подобные.
Термин «сольват» относится к комплексу с переменной стехиометрией (например, ди-, три-, тетра-, пента-, гекса- и т.д.), который образуется растворенным веществом (соединение по настоящим вариантам) и растворителем, при этом растворитель не влияет на биологическую активность растворенного вещества. Пригодные растворители включают, например, этанол, уксусную кислоту и тому подобные.
Термин «гидрат» относится к сольвату, как определено здесь выше, в котором растворителем является вода.
Как подробно описано здесь далее, новые разработанные соединения по настоящим вариантам обладают высокой иммуномодулирующей активностью и, следовательно, могут быть использованы в разных терапевтических применениях. Использование этих соединений в терапевтическом применении включает их введение per se или в составе фармацевтической композиции, в которой соединения смешаны с приемлемыми носителями или вспомогательными веществами.
Таким образом, согласно другому аспекту вариантов настоящего изобретения обеспечена фармацевтическая композиция, которая содержит в качестве активного ингредиента любое из описанных здесь соединений и фармацевтически приемлемый носитель.
Использующийся здесь термин «фармацевтическая композиция» означает препарат, содержащий один или более описанных здесь активных ингредиентов с другими химическими компонентами, такими как физиологически приемлемые носители и вспомогательные вещества. Целью создания фармацевтической композиции является облегчение введения соединения в организм субъекта.
Использующийся здесь термин «активный ингредиент» относится к соединениям (окисленным липидам), описанным здесь выше, ответственным за биологическое действие.
Использующиеся здесь далее выражения «физиологически приемлемый носитель» и «фармацевтически приемлемый носитель», которые могут использоваться взаимозаменяемо, относятся к носителю или разбавителю, который не вызывает значительного раздражения в организме и не ухудшает биологическую активность и свойства вводимого соединения. Адъювант входит в определение указанного выражения.
Использующийся здесь термин «вспомогательное вещество» относится к инертному веществу, добавляемому в фармацевтическую композицию для дальнейшего облегчения введения активного ингредиента. Неограничивающие примеры вспомогательных веществ включают карбонат кальция, фосфат кальция, разные сахара и виды крахмала, производные целлюлозы, желатин, растительные масла и пропиленгликоли.
Методики составления и введения лекарственных препаратов можно найти в "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, latest edition, которая включена здесь в виде ссылки.
Приемлемые режимы введения могут включать, например, оральное, ректальное, введение через слизистую оболочку, особенно в нос, или парентеральное введение, включающее внутримышечные, подкожные и интрамедуллярные инъекции, а также интратекальные, прямые интравентрикулярные, внутривенные, интраперитонеальные, интраназальные или внутриглазные инъекции.
Или же, фармацевтическую композицию можно вводить локально вместо системного введения, например, в виде инъекции фармацевтической композиции непосредственно в участок ткани субъекта.
В дополнительном варианте настоящего изобретения фармацевтическая композиция предназначена для модуляции иммунной и/или воспалительной реакции при введении через слизистую оболочку.
В другом дополнительном варианте настоящего изобретения фармацевтическая композиция предназначена для модуляции иммунной и/или воспалительной реакции при оральном введении.
Факультативно, фармацевтические композиции по вариантам настоящего изобретения предназначены для назального или интраперитонеального введения, как подробно описано здесь далее.
Фармацевтические композиции по вариантам настоящего изобретения могут быть приготовлены хорошо известными в данной области способами, например, с использованием обычных процессов смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, заключения в оболочку или лиофилизации.
Фармацевтические композиции, предназначенные для применения по настоящим вариантам, таким образом, могут быть составлены обычными способами с использованием одного или более физиологически приемлемых носителей, включающих наполнители и вспомогательные вещества, которые облегчают обработку активных ингредиентов в препаратах и могут быть использованы фармацевтически. Требуемый состав зависит от выбранного режима введения.
Для получения инъекционных препаратов активные ингредиенты фармацевтической композиции могут быть составлены в водных растворах, например, в физиологически совместимых буферах, таких как раствор Ханка, раствор Рингера или физиологический раствор. Для введения через слизистую оболочку в составе используют проникающие вещества, соответствующие преодолеваемому барьеру. Такие проникающие вещества в целом известны в данной области.
Для орального введения фармацевтическая композиция может быть легко составлена путем объединения активных соединений с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители позволяют получить фармацевтическую композицию в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, суспензий, взвесей и тому подобных, которые предназначены для приема внутрь субъектом. Фармакологические составы для орального введения можно приготовить с использованием твердых вспомогательных веществ, факультативного измельчения полученной смеси и получения гранул из указанной смеси после добавления, при желании, приемлемых вспомогательных веществ для изготовления таблеток или сердцевины драже. Приемлемыми вспомогательными веществами, в частности, являются наполнители, такие как сахара, включающие лактозу, сахарозу, маннит или сорбит; составы целлюлозы, такие как, например, маисовый крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбоксиметилцеллюлоза; и/или физиологически приемлемые полимеры, такие как поливинилпирролидон (PVP). При желании могут быть добавлены вещества, улучшающие распадаемость, такие как поперечно-сшитый поливинилпирролидон, агар, альгиновая кислота или ее соль, такая как альгинат натрия.
На сердцевины драже наносят приемлемые покрытия. Для этого можно использовать концентрированные растворы сахара, которые могут факультативно содержать аравийскую камедь, тальк, поливинилпирролидон, гелеобразный карбополь, полиэтиленгликоль, диоксид титана, растворы лака и приемлемые органические растворители или смеси растворителей. В покрытия таблеток или драже могут быть добавлены красители или пигменты для идентификации или характеристики разных комбинаций доз активного соединения.
Фармацевтические композиции, которые используются орально, включают жесткие капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Жесткие капсулы могут содержать активные ингредиенты с добавками наполнителя, такого как лактоза, связующего, такого как крахмалы, смазывающего вещества, такого как тальк или стеарат магния, и факультативно со стабилизаторами. В мягких капсулах активные ингредиенты могут быть растворены или суспендированы в приемлемых жидкостях, таких как жирные масла, жидкий парафин или жидкий полиэтиленгликоль. Кроме того, могут быть добавлены стабилизаторы. Все составы для орального введения должны быть в дозированной форме, пригодной для выбранного режима введения.
Композиции для буккального введения могут быть в форме таблеток или таблеток для рассасывания, составленных обычным способом.
Для введения путем назальной ингаляции активные ингредиенты для использования согласно вариантам настоящего изобретения обычно вводят в виде аэрозоля из баллона, находящегося под давлением, или ингалятора с использованием приемлемого пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана или диоксида углерода. В случае находящегося под давлением аэрозоля устройство может быть снабжено клапаном для подачи отмеренного количества. Капсулы и кассеты, изготовленные, например, из желатина, которые предназначены для использования в дозирующем устройстве, могут содержать порошковую смесь соединения и приемлемую порошковую основу, такую как лактоза или крахмал.
Описанная здесь фармацевтическая композиция может быть составлена для парентерального введения, например, болюсного введения или непрерывного вливания. Составы для инъекций могут быть представлены в дозированной лекарственной форме, например, в ампулах или в многодозовых контейнерах, в некоторых случаях с добавленным консервантом. Композиции могут представлять собой суспензии, растворы или эмульсии в масляных или водных носителях, а также могут содержать образующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Фармацевтические композиции для парентерального введения включают водные растворы активных составов в водорастворимой форме. Кроме того, суспензии активных ингредиентов могут быть приготовлены в виде соответствующих суспензий для инъекций на масляной или водной основе. Приемлемые липофильные растворители или носители включают жирные кислоты, такие как кунжутное масло, или эфиры синтетических жирных кислот, такие как этилолеат, триглицериды или липосомы. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Факультативно, суспензия может также содержать приемлемые стабилизаторы или агенты, которые повышают растворимость активных ингредиентов для приготовления высококонцентрированных растворов.
Или же, активный ингредиент может находиться в порошковой форме, предназначенной для растворения непосредственно перед применением в приемлемом растворителе, например, стерильном апирогенном водном растворе.
Фармацевтическая композиция по вариантам настоящего изобретения может также быть составлена в виде композиции для ректального введения, такого как суппозитории или удерживающие клизмы, с использованием, например, обычных основ для суппозиториев, таких как масло какао или другие глицериды.
Фармацевтические композиции, пригодные для применения в контексте настоящих вариантов, включают композиции, в которых активные ингредиенты присутствуют в эффективном количестве, достаточном для достижения поставленной цели. В частности, терапевтически эффективное количество означает количество активных ингредиентов, необходимых для предупреждения, смягчения или ослабления симптомов заболевания (например, атеросклероза) или продления жизни субъекта, подвергаемого лечению.
Специалисты в данной области могут легко определить терапевтически эффективное количество, особенно с учетом обеспеченного здесь подробного раскрытия.
Для любого препарата, используемого в способах по вариантам изобретения, терапевтически эффективное количество или дозу можно сначала определить с помощью анализов in vitro и исследования культуры клеток. Например, дозу можно определить при помощи животных моделей, позволяющих установить требуемую концентрацию или титр. Такую информацию можно использовать для более точного определения эффективных доз для человека.
Токсичность и терапевтическую эффективность активных ингредиентов, описанных здесь, можно определить стандартными фармацевтическими методами in vitro с использованием культур клеток или экспериментальных животных (например, как проиллюстрировано здесь далее в разделе «Примеры»). Данные, полученные в результате указанных анализов in vitro, исследования культур клеток и животных, можно использовать для определения диапазона доз для лечения человека. Дозы могут изменяться в зависимости от используемой лекарственной формы и применяемого режима введения. Точный состав, режим введения и доза могут быть выбраны индивидуально лечащим врачом с учетом состояния субъекта. (См., например, Fingi, et al; 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.1).
Величина дозы и интервал между приемами могут регулироваться индивидуально для обеспечения в плазме или головном мозге уровня активного ингредиента (минимальная эффективная концентрация, МЕС), достаточного для индукции или подавления воспаления (например, ангиогенезис). МЕС будет различаться для каждого состава, но может быть определена на основании данных, полученных in vitro. Дозы, необходимые для достижения МЕС, будут зависеть от индивидуальных характеристик и режима введения. Для определения концентраций в плазме можно использовать известные методы обнаружения.
В зависимости от тяжести заболевания и реакции на проводимое лечение можно производить однократное или многократное введение дозы, при этом курс лечения может продолжаться от нескольких дней до нескольких недель, до полного выздоровления или ослабления симптомов заболевания.
Количество вводимой композиции, несомненно, зависит от состояния субъекта, нуждающегося в лечении, тяжести поражения, способа введения, заключения лечащего врача, и др.
Композиции по вариантам настоящего изобретения при желании могут быть представлены в упаковке или дозирующем устройстве, таком как набор, утвержденный Управлением по контролю за продуктами и лекарствами (FDA), который может содержать одну или более дозированных лекарственных форм, содержащих активный ингредиент. Упаковка может, например, включать металлическую или пластиковую фольгу, такую как блистерная упаковка. К упаковке или дозирующему устройству может прилагаться инструкция по введению. К упаковке или дозирующему устройству может также прилагаться предупреждение, соответствующее форме, утвержденной правительственным органом, регулирующим производство, применение или продажу фармацевтических препаратов, которое должно содержать информацию о санкционировании данным органом формы композиций или пригодности для человека или животных. Такое предупреждение, например, может быть в виде маркировки, утвержденной Управлением по контролю за продуктами и лекарствами (США) для рецептурных лекарственных средств или в виде утвержденного листка-вкладыша. Могут быть получены композиции, содержащие препарат по настоящему изобретению вместе с совместимым фармацевтическим носителем, которые помещают в соответствующий контейнер и маркируют с указанием заболевания, для лечения которого они предназначены, как подробно описано далее.
Таким образом, в факультативном варианте настоящего изобретения фармацевтическую композицию упаковывают в упаковочный материал и указывают в печатном вкладыше или на упаковочном материале, что данная композиция предназначена для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом. Перечень типичных примеров заболеваний и нарушений, ассоциированных с таким воспалением, обеспечен здесь ниже.
Альтернативно или дополнительно, фармацевтическую композицию упаковывают в упаковочный материал и указывают в печатном вкладыше или на упаковочном материале, что данная композиция предназначена для уменьшения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, и/или для лечения заболевания или нарушения, в котором уменьшение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Как подробно описано далее, фармацевтическая композиция по настоящим вариантам может кроме того включать дополнительное соединение, которое является эффективным для лечения или профилактики описанного здесь воспаления.
Как подробно описано в нижеследующем разделе «Примеры», типичные примеры новых разработанных соединений по настоящему изобретению позволяют эффективно модулировать уровень цитокинов, ассоциированных с иммунной реакцией и с воспалением. Полученные результаты указывают на то, что данные соединения являются эффективными для ингибирования иммунной реакции и воспаления, ассоциированного с эндогенным окисленным липидом.
Таким образом, другим объектом вариантов по настоящему изобретению является способ лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом. Способ по данному аспекту настоящих вариантов осуществляется путем введения нуждающемуся субъекту терапевтически эффективного количества одного или более окисленных липидов, как описано здесь.
Использующееся здесь выражение «эндогенный окисленный липид» относится к одному или более окисленным липидам, которые присутствуют или образованы in vivo в результате воспалительных или других процессов, опосредуемых клетками или гуморальной системой.
Термин «способ» означает способы, средства, методы и процедуры, выполняемые для достижения поставленной цели, которые включают, не ограничиваясь ими, способы, средства, методы и процедуры, которые известны в данной области или могут быть легко разработаны на основе известных способов, средств, методов и процедур специалистами в химической, фармакологической, биологической, биохимической и медицинской областей.
Использующееся здесь выражение «лечение или профилактика» означает устранение, значительное ингибирование, замедление или реверсию прогрессирования заболевания, значительное ослабление клинических симптомов заболевания или значительное предупреждение появления клинических симптомов заболевания.
Примеры субъектов, которым может быть назначено такое лечение, включают субъекты, страдающие заболеваниями или нарушениями, ассоциированными с воспалением, как определено здесь выше. Подходящие субъекты согласно настоящим вариантам включают млекопитающих, таких как представители семейства псовых, кошачьих, овцы, свиньи, лошади и жвачные животные. Факультативно, отдельными субъектами согласно настоящим вариантам является человек.
Использующееся здесь выражение «воспаление, ассоциированное с эндогенным окисленным липидом» означает воспаление, которое ассоциировано с in vivo образованием или присутствием одного или более окисленных липидов (например, окисленный LDL, окисленные мембранные липиды, и т.д.).
Воспаление представляет собой защитную реакцию организма на повреждение. Некоторые цитокины играют ключевую роль в опосредовании воспалительных реакций, к которым относятся интерлейкины 12 и 23 (IL-12 и IL-23). Избыточное воспаление часто является разрушительным, включающим или приводящим к большому числу заболеваний и нарушений. Как подробно объясняется здесь выше, повышенная воспалительная реакция обычно ассоциирована с эпитопами окисленных липидов.
Таким образом, согласно факультативным вариантам настоящего изобретения обеспечен способ уменьшения цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23 у субъекта.
Согласно дополнительным факультативным вариантам настоящего изобретения обеспечен способ лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Вышеуказанные способы осуществляются путем введения нуждающемуся субъекту терапевтически эффективного количества одного или более окисленных липидов, как описано здесь.
Согласно другому аспекту вариантов настоящего изобретения обеспечено применение, по меньшей мере, одного окисленного липида, описанного здесь, в производстве лекарственных препаратов. Факультативные составы для лекарственных препаратов описаны здесь.
В некоторых вариантах лекарственный препарат предназначен для лечения или предупреждения воспаления, ассоциированного с эндогенным окисленным липидом, как описано здесь далее более подробно.
В некоторых вариантах лекарственный препарат предназначен для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23 у субъекта.
В некоторых вариантах лекарственных препарат предназначен для лечения заболевания или нарушения, в котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
Противовоспалительное действие окисленных липидов, описанных здесь, можно использовать для лечения или профилактики заболевания или нарушения, обусловленного воспалением, в которое вовлечены эндогенные окисленные LDL или любые другие эндогенные окисленные липиды. Такие заболевания и нарушения включают, например, заболевания или нарушения, обусловленные образованием бляшек, которые включают, не ограничиваясь ими, атеросклероз, атеросклеротическое сердечно-сосудистое заболевание, цереброваскулярное заболевание, болезнь периферических сосудов, стеноз, рестеноз и стеноз в стенте, а также аутоиммунные заболевания или нарушения, нейродегенеративные заболевания или нарушения, пролиферативные заболевания или нарушения, и процессы, связанные со старением.
Таким образом, типичные примеры заболеваний или нарушений, обусловленных воспалением, которое в свою очередь ассоциировано с эндогенными окисленными липидами, и поэтому пригодны для лечения способом по вариантам настоящего изобретения, включают, например, идиопатические воспалительные заболевания или нарушения, хронические воспалительные заболевания или нарушения, острые воспалительные заболевания или нарушения, аутоиммунные заболевания или нарушения, инфекционные заболевания или нарушения, воспалительные злокачественные заболевания или нарушения, воспалительные заболевания или нарушения, связанные с трансплантацией, воспалительные дегенеративные заболевания или нарушения, заболевания или нарушения, обусловленные гиперчувствительностью, воспалительные сердечно-сосудистые заболевания или нарушения, воспалительные цереброваскулярные заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительные заболевания или нарушения желез, воспалительные желудочно-кишечные заболевания или нарушения, воспалительные кожные заболевания или нарушения, воспалительные заболевания или нарушения печени, воспалительные неврологические заболевания или нарушения, воспалительные заболевания или нарушения костно-мышечной системы, воспалительные заболевания или нарушения почек, воспалительные заболевания или нарушения репродуктивной системы, воспалительные системные заболевания или нарушения, воспалительные заболевания или нарушения соединительной ткани, воспалительные опухоли, некрозы, воспалительные заболевания или нарушения, связанные с имплантатом, воспалительные процессы, связанные во старением, заболевания или нарушения, обусловленные иммунодефицитом, пролиферативные заболевания и нарушения, а также воспалительные легочные заболевания или нарушения легких, как определено здесь ниже.
Неограничивающие примеры гиперчувствительности включают гиперчувствительность Типа I, гиперчувствительность Типа II, гиперчувствительность Типа III, гиперчувствительность Типа IV, гиперчувствительность немедленного типа, антителоопосредованную гиперчувствительность, гиперчувствительность, опосредованную иммунным комплексом, гиперчувствительность, опосредованную Т-лимфоцитами, гиперчувствительность замедленного типа, гиперчувствительность, опосредованную Т-лимфоцитами-хелперами, гиперчувствительность, опосредованную цитотоксическими Т-лимфоцитами, гиперчувствительность, опосредованную ТН1-лимфоцитами и гиперчувствительность, опосредованную ТН2-лимфоцитами.
Неограничивающие примеры воспалительных сердечно-сосудистых заболеваний или нарушений включают обтурирующие заболевания или нарушения, атеросклероз, порок клапан сердца, стеноз, рестеноз, стеноз в стенте, коронарную болезнь сердца, острые коронарные синдромы, застойную сердечную недостаточность, болезнь Руньон-Гебердена, ишемическую болезнь миокарда, тромбоз, гранулематоз Вегенера, синдром Такаясу, синдром Кавасаки, аутоиммунное заболевание или нарушение, направленное против фактора VII, некротический васкулит маленьких сосудов, микроскопическую ангиопатию, синдром Чарга-Стросса, олигоиммунный очаговый некротический гломерулонефрит, серповидный гломерулонефрит, антифосфолипидный синдром, антителоиндуцированную сердечную недостаточность, тромбоцитопеническую пурпуру, аутоиммунную гемолитическую анемию, аутоиммунное заболевание сердца, болезнь или нарушение Чагаса и аутоиммунитет против Т-лимфоцитов-хелперов.
Стеноз представляет собой окклюзивное заболевание сосудистой системы, обычно вызванное атеросклеротической бляшкой и повышенной активностью тромбоцитов, особенно тяжело поражающее коронарную сосудистую систему.
Рестеноз представляет собой прогрессирующую реокклюзию, часто возникающую после удаления окклюзии в стенозированной сосудистой системе. В случаях, когда раскрытое состояние сосуда требует механической поддержки стентом, может возникнуть стеноз в стенте, вызывая повторную обтурацию восстановленного сосуда.
Неограничивающие примеры цереброваскулярных заболеваний или нарушений включают инсульт, воспаление сосудов головного мозга, кровоизлияние в мозг и позвоночно-артериальную недостаточность.
Неограничивающие примеры заболеваний или нарушений периферических сосудов включают гангрену, диабетический васкулит, ишемическую болезнь кишечника, тромбоз, диабетическую ретинопатию и диабетическую нефропатию.
Неограничивающие примеры аутоиммунных заболеваний или нарушений включают все заболевания, вызванные иммунной реакцией, такой как аутоантитело -или клеточно-опосредуемая иммунная реакция на аутоантиген и тому подобные. Типичными примерами являются хронический ревматоидный артрит, болезнь Стилла, системная красная волчанка, склеродермия, смешанное поражение соединительной ткани, узелковый периартериит, полимиозит/дерматомиозит, синдром Шегрена, болезнь Бехчета, рассеянный склероз, аутоиммунный диабет, зоб Хасимото, псориаз, первичная мексидема, пернициозная анемия, миастения гравис, хронический активный гепатит, аутоиммунная гемолитическая анемия, идиопатическая тромбоцитопеническая пурпура, увеит, васкулит и гепарин-индуцированная тромбоцитопения.
Неограничивающие примеры воспалительных заболеваний или нарушений желез включают заболевания или нарушения поджелудочной железы, диабет Типа I, заболевания или нарушения щитовидной железы, болезнь Грейвса, тиреоидит, спонтанный аутоиммунный тиреоидит, тиреоидит Хашимото, идиопатическую микседему, аутоиммунное заболевание яичника, аутоиммунное мужское бесплодие, аутоиммунный простатит и аутоиммунный полигландулярный синдром Типа I.
Неограничивающие примеры воспалительных желудочно-кишечных заболеваний или нарушений включают колит, илеит, болезнь Крона, хроническое воспалительное заболевание тонкой кишки, синдром раздраженной толстой кишки, глютеиновую болезнь, неспецефический язвенный колит, язву, кожную язву, пролежни, язву желудка, пептическуто язву, щечную язву, носоглоточную язву, язву пищевода, язву двенадцатиперстной кишки и язву желудочно-кишечного тракта.
Неограничивающие примеры воспалительных кожных заболеваний или нарушений включают угревую сырь, аутоиммунное буллезное заболевание кожи, обыкновенную пузырчатку, буллезный пемфигоид, листовидную пузырчатку, контактный дерматит и лекарственную сыпь.
Неограничивающие примеры воспалительных заболеваний или нарушений печени включают аутоиммунный гепатит, цирроз печени и билиарный цирроз печени.
Неограничивающие примеры воспалительных неврологических заболеваний или нарушений включают рассеянный склероз, болезнь Альцгеймера, болезнь Паркинсона, миастению гравис, двигательную невропатию, синдром Гийена-Барре, аутоиммунную нейропатию, синдром Итона-Ламберта, паранеопластическое неврологическое заболевание или нарушение, паранеопластическую атрофию мозжечка, непаранеопластический синдром мышечной скованности, прогрессирующая атрофия мозжечка, энцефалит Расмусена, амиотрофический боковой склероз, хорею Сиденгама, синдром Жилль де ла Туретта, аутоиммунную полиэндокринопатию, дисиммунную нейропатию, приобретенную нейромиотонию, врожденный артрогриппоз, болезнь Хантингтона, деменцию, обусловленную СПИДом, амиотрофический латеральный склероз (AML), рассеянный склероз, инсульт, воспалительное заболевание или нарушение сетчатки, воспалительное заболевание или нарушение глаз, неврит зрительного нерва, губкообразную энцефалопатию, мигрень, головную боль, гистаминовую головную боль и синдром скованного человека.
Неограничивающие примеры воспалительных заболеваний или нарушений соединительной ткани включают аутоиммунный миозит, первичный синдром Шегрена, аутоиммунное заболевание или нарушение гладких мышц, миозит, тендинит, лигаментное воспаление, хондрит, воспаление суставов, синовиальное воспаление, запястный синдром, артрит, ревматоидный артрит, остеоартрит, анкилозирующий спондилоартрит, костное воспаление, аутоиммунное заболевание или нарушение уха и аутоиммунное заболевание или нарушение внутреннего уха.
Неограничивающие примеры воспалительных заболеваний или нарушений почек включают аутоиммунный интерстециальный нефрит и/или рак почки.
Неограничивающие примеры воспалительных заболеваний или нарушений репродуктивной системы включают повторную потерю плода, кисту яичника или заболевание или нарушение, связанное с менструальным циклом.
Неограничивающие примеры воспалительных системных заболеваний или нарушений включают системную красную волчанку, системный склероз, септический шок, синдром токсического шока и кахексию.
Неограничивающие примеры инфекционного заболевания или нарушения включают хронические инфекционные заболевания или нарушения, подострое инфекционное заболевание или нарушение, острое инфекционное заболевание или нарушение, вирусное заболевание или нарушение, бактериальное заболевание или нарушение, заболевание или нарушение, вызванное простейшими, паразитарное заболевание или нарушение, грибковое заболевание или нарушение, микоплазматическое заболевание или нарушение, гангрену, сепсис, заболевание или нарушение, вызванное насекомыми, грипп, туберкулез, малярию, синдром приобретенного иммунодефицита и атипичную пневмонию.
Неограничивающие примеры воспалительных заболеваний или нарушений, связанных с трансплантацией, включают отторжение трансплантата, хроническое отторжение трансплантата, подострое отторжение трансплантата, острое отторжение трансплантата, гиперострое отторжение трансплантата и заболевание или нарушение «трансплантат против хозяина». Типичные имплантаты включают протезный имплантат, грудной имплантат, силиконовый имплантат, зубной имплантат, имплантат полового члена, сердечный имплантат, искусственный сустав, восстанавливающее устройство при переломе кости, имплантат, заменяющий кость, доставляющий лекарственный препарат имплантат, катетер, кардиостимулятор, искусственное сердце, искусственный сердечный клапан, высвобождающий лекарственный препарат имплантат, электрод и дыхательную трубку.
Неограничивающие примеры воспалительных опухолей включают злокачественную опухоль, доброкачественную опухоль, солидную опухоль, метастатическую опухоль и несолидную опухоль.
Неограничивающие примеры воспалительных легочных заболеваний или нарушений включают астму, аллергическую астму, эмфизему, хроническое обструктивное заболевание или нарушение легких, саркоидоз и бронхит.
Примером пролиферативного заболевания или нарушения является рак.
В научной литературе было описано применение фосфолипидов и метаболитов фосфолипидов для лечения или профилактики заболеваний или синдромов, таких как, например, окислительный стресс в процессе старения (Onorato JM, et al, Annal N Y Acad Sci 1998 Nov 20;854:277-90), ревматоидный артрит (RA)(Paimela L, et al. Ann Rheum Dis 1996 Aug;55(8):558-9), болезнь Стилла (Savolainen A, et al, 1995;24(4):209-11), воспалительное заболевание кишечника (IBD) (Sawai T, et al, Pediatr Surg Int 2001 May;17(4):269-74) и рак почек (Noguchi S, et al, Biochem Biophys Res Commun 1992 Jan 31;182(2):544-50), что еще более подтверждает пригодность аналогов окисленных липидов окисленных фосфолипидов для лечения или профилактики указанных заболеваний или нарушений.
В соответствии со способом по вариантам настоящего изобретения, окисленные липиды можно вводить субъекту разными способами, которые включают, например, оральное, ректальное введение, введение через слизистую оболочку, особенно в нос, кишечное или парентеральное введение, включающее внутримышечные, подкожные и интрамедуллярные инъекции, а также интратекальный, прямые интравентикулярные, внутривенные, интраперитонеальные, интраназальные или внутриглазные инъекции. Однако, как подробно описано здесь и показано в последующем разделе «Примеры», предпочтительными способами введения являются оральный, через слизистую оболочку, назальный, интрадермальный (подкожный) и интраперитонеальный способы введения.
Следовательно, в одном варианте интраперитонеально вводят 0.1-100 мг/кг окисленного липида в приемлемом носителе, как описано здесь, который включает, не ограничиваясь ими, PBS или глицерин, от одного до трех раз в неделю в соответствии со схемой постоянного или чередующегося введения.
В другом варианте в нос вводят 0.1-100 мг/кг окисленного липида, как описано здесь, в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, от одного до трех раз в неделю, в соответствии со схемой постоянного или чередующегося введения.
В еще другом варианте подкожно вводят 0.1-100 мг/кг окисленного липида, как описано здесь, в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, от одного до трех раз в неделю, в соответствии со схемой постоянного или чередующегося введения,
В еще другом варианте орально вводят 0.1-100 мг/кг окисленного липида, как описано здесь, в приемлемом носителе, который включает, не ограничиваясь ими, PBS или глицерин, от одного до трех раз в неделю, в соответствии со схемой постоянного или чередующегося введения.
Фармацевтические композиции и способы, описанные здесь, могут кроме того включать введение одного или более дополнительных соединений, предназначенных для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, как описано здесь выше.
Способы согласно вариантам настоящего изобретения могут, следовательно, включать совместное введение, осуществляемое до, одновременно или после введения окисленных липидов, терапевтически эффективного количества одного или более таких дополнительных соединений, при этом фармацевтическая композиция согласно настоящим вариантам может содержать помимо соединений, описанных здесь, такие дополнительные соединения.
Типичные примеры дополнительных соединений, описанные здесь выше, которые предназначены для лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, и поэтому пригодны для использования в контексте данного варианта настоящего изобретения, включают, не ограничиваясь ими, ингибиторы HMGCoA-редуктазы (статины), адъюванты слизистой оболочки, кортикостероиды, стероидные противовоспалительные средства, нестероидные противовоспалительные средства, аналгетики, факторы роста, токсины, ингибиторы транспортного белка холестериновых эфиров (СЕТР), агонисты пролиферирующего активированного рецептора пероксисомы (PPAR), противоатеросклеротические лекарственные средства, антипролиферативные средства, эзетимид, никотиновую кислоту, ингибиторы сквалена, АроЕ Milano, HSPs, бета-2-гликопротеин-I и любое их производное или аналог.
Ингибиторы HMGCoA-редуктазы (статины) являются хорошо известными лекарственными средствами, которые эффективно снижают уровни LDL-холестерина в результате ингибирования фермента, который регулирует скорость продуцирования холестерина, и усиления выведения печенью LDL-холестерина, присутствующего в крови. Неограничивающие примеры обычно прописываемых статинов включают аторвастатин, флувастатин, ловастатин, правастатин и симвастатин.
Эзетимиб является первым средством в новом классе ингибиторов абсорбции холестерина, которые эффективно и избирательно ингибирует поглощение пищевого и билиарного холестерина у щеточной каемки кишечного эпителия, не влияя на поглощение триглицерида или жирорастворимых витаминов. Таким образом, эзетимиб уменьшает общее поступление холестерина в печень, и индуцирует повышенную экспрессию рецепторов LDL, что способствует большему удалению LDL-C из плазмы.
Пероксисома представляет собой органеллу с одиночной мембраной, присутствующую практически во всех эукариотических клетках. Одним из самых важных метаболических процессов, опосредуемых пероксисомой, является (3-окисление жирных кислот с длинными и очень длинными цепями. Пероксисома также участвует в синтезе желчной кислоты, холестерина, плазмалогена, метаболизме аминокислот и пурина.
Никотиновая кислота является известным агентом, который снижает уровни общего холестерина, LDL-холестерина и триглицерида, и повышает уровни HDL-холестерина. Существует три типа лекарственных средств на основе никотиновой кислоты: средства с немедленным высвобождением, с выдержкой по времени и с пролонгированным высвобождением. Никотиновая кислота или ниацин, водорастворимый витамин В, улучшает все липопротеины при введении в дозах, значительно превышающих потребность в данном витамине.
Сквален, изопреноидное соединение, аналогичное по структуре бета-каротину, представляет собой промежуточный метаболит в процессе синтеза холестерина. Организм человека поглощает примерно 60 процентов сквалина, поступающего с пищей. Сквалин переносится в сыворотке, будучи связанным с липопротеинами, имеющими очень низкую плотность, и распределяется убиквитарно по тканям человека с достижением наибольшей концентрации в коже, где он является одним из основных компонентов поверхностных липидов кожи. Ингибиторы сквалина (например, монооксигеназа и синтаза) служат в качестве ингибиторов биосинтеза холестерина.
Агонисты пролифератор-активирующего рецептора (PPAR), например, фибраты, представляют собой активированные жирной кислотой члены надсемейства нуклеарных рецепторов, которые играют важные роли в метаболизме липидов и глюкозы, и применяются для лечения заболеваний обмена веществ, связанных с ожирением, таких как гиперлипемия, устойчивость к инсулину и ишемическая болезнь сердца. Фибраты в целом эффективно снижают повышенные уровни триглицеридов и холестерина в плазме и действуют в качестве агонистов PPAR. Наиболее выраженным действием фибратов является снижение концентрации липопротеинов с высоким содержанием триглицеридов (TRLs) в плазме. Уровни LDL-холестерина (LDL-C) обычно снижаются у субъектов с повышенными исходными концентрациями в плазме, и уровни HDL-холестерина (HDL-C) обычно повышаются при низких исходных концентрациях в плазме. Неограничивающие примеры обычно прописываемых фибратов включают безафибрат, гемфиброзил и фенофибрат.
Ингибиторы транспортного белка холестериновых эфиров (СЕТР) играют важную роль в атерогенезе, уменьшая накопление сложного холестерилового эфира в макрофагах и стенках артерий, в результате чего сокращается образование ксантомных клеток и поглощение холестерина. Самым перспективным в настоящее время ингибитором СЕТР является авизимиб.
ApoA-I Milano обычно используется в виде рекомбинантного комплекса с фосфолипидом (ЕТС-216) и вызывает значительную регрессию атеросклероза коронарной артерии.
Совместное введение адъювантов слизистой оболочки имеет важное значение для предупреждения инвазии инфекционных агентов через поверхность слизистой оболочки. На ранних стадиях индукции иммунной реакции слизистой оболочки поглощение орально или назально вводимых антигенов достигается благодаря уникальной совокупности антиген-отбирающих клеток, М клеток, расположенных в организованных лимфоидных фолликулах (FAE) индуктивных сайтов. После успешного поглощения антигены немедленно процессируются и представляются подстилающими дендритными клетками (DC).
Неограничивающие примеры нестероидных противовоспалительных средств включают оксикамы, такие как пироксикам, изоксикам, теноксикам, судоксикам и СР-14,304; салицилаты, такие как аспирин, дизальцид, бенорилат, трилизат, сафаприн, солприн, дифлунизал и фендозал; производные уксусной кислоты, такие как диклофенак, фенклофенак, индометацин, сулиндак, толметин, изоксепак, фурофенак, тиопинак, зидометацин, ацематацин, фентиазак, зомепирак, клинданак, оксепинак, фелбинак и кеторолак; фенаматы, такие как мефенаминовая, мелкофенаминовая, флуфенаминовая, нифлуминовая и толфенаминовая кислоты; производные пропионовой кислоты, такие как ибупрофен, напроксен, беноксапрофен, флурбипрофен, кетопрофен, фенопрофен, фенбуфен, индопрофен, пирпрофен, карпрофен, оксапрозин, пранопрофен, миропрофен, тиоксапрофен, супрофен, алминопрофен и тиапрофеновая кислота; пиразолы, такие как фенилбутазон, оксифенбутазон, фепразон, азапропазон и триметазон.
Неограничивающие примеры стероидных противовоспалительных средств включают, не ограничиваясь ими, кортикостероиды, такие как гидрокортизон, гидроксилтриамцинолон, альфа-метил-дексаметазон, фосфат дексаметазона, дипропионаты беклометазона, валерат клобетазола, дезонид, дезоксиметазон, ацетат дезоксикортикостерона, дексаметазон, дихлоризон, диацетат дифторазона, валерат дифлукортолона, флуадренолон, ацетонид флуклоролона, флудрокортизон, пивалат флуметазона, ацетонид флузинолона, флуцинонид, сложные бутиловые эфиры флукортина, флуокортолон, ацетат флупреднидена (флупреднилиден), флурандренолон, гальцинонид, ацетат гидрокортизона, бутират гидрокортизона, метилпреднизолон, ацетонид триамцинолона, кортизон, кортодоксон, флуцетонид, флудрокортизон, диацетат дифлорозона, флуадренолон, флудрокортизон, диацетат дифлурозона, ацетонид флурадренолона, медризон, амцинафел, амценафид, бетаметазон и баланс его сложных эфиров, хлорпреднизон, ацетат хлорпреднизона, клокортелон, клесцинолон, дихлоризон, дифлурпреднат, флуклоронид, флунизолид, фторметалон, флуперолон, флупреднизолон, валерат гидрокортизона, циклопентилпропионат гидрокортизона, гидрокортамат, мепреднизон, параметазон, преднизолон, преднизон, дипропионат беклометазона, триамцинолон и их смеси.
Неограничивающие примеры аналгетиков (болеутоляющих средств) включают аспирин и другие салицилаты (такие как салицилат холина или магния), ибупрофен, кетопрофен, напроксен-натрия и ацетаминофен.
Факторы роста являются гормонами, выполняющими целый ряд функций, включающих регулирование образования молекул межклеточной адгезии, изменение клеточной пролиферации, увеличение васкуляризации, повышение синтеза коллагена, регулирование костного метаболизма и изменение миграции клеток на данном участке. Неограничивающие примеры факторов роста включают инсулиноподобный фактор роста (IGF-1), трансформирующий β-фактор роста (TGF-β), костный морфогенетический белок (BMP) и тому подобные.
Неограничивающие примеры токсинов включают холероген, который служит также в качестве адъюванта.
Неограничивающие примеры антипролиферативных средств включают алкилирующий агент, такой как азотистый иприт, этиленимин и метилмеламин, алкилсульфонат, нитрозомочевину и триазен; антиметаболит, такой как аналог фолиевой кислоты, аналог пиримидина и аналог пурина; природный продукт, такой как алкалоид барвинка, эпидофиллотоксин, антибиотик, фермент, таксан и модификатор биологической реакции; разные агенты, такие как платиновый координационный комплекс, антрацендион, антрациклин, замещенная мочевина, производное метилгидразина или адренокортикальный супрессор; гормон или антагонист, такой как адренокортикостероид, прогестин, эстроген, антиэстроген, андроген, антиандроген или аналог гонадолиберина. Конкретные примеры хемиотерапевтических средств включают, например, азотистый иприт, эпиподофиллотоксин, антибиотик, платиновый комплексообразующий комплекс, блеомицин, доксорубицин, паклитаксел, 4-ОН циклофосфамид и цисплатин.
Семейство HSP состоит примерно из 25 белков, отличающихся молекулярной массой с высококонсервативными структурами. Почти у всех людей возникают клеточные и гуморальные иммунные реакции против микробного хитшокового белка 60 (HSP60). Из-за высокой степени антигенной гомологии, существующей между микробным (бактериальным и паразитарным) и человеческим HSP60, иммунитет в отношении микробов чреват опасностью перекрестной реактивности с HSP60 человека, экспрессируемым эндотелиальными клетками артерий в условиях стресса. Действительный аутоиммунитет против измененного аутологичного HSP60 может также запустить данный процесс (Wick et al. Atherosclerosis as an autoimmune disease: an update. TRENDS in Immunology. 2001;22(12):665-669). HSP участвует в качестве аутоантигена-мишени в некоторых экспериментальных аутоиммунных заболеваниях (артрит, диабет типа I). Антитела против HSP65 и против HSP60 были наглядно ассоциированы с атероматозными поражениями у человека. Исследования, выполненные на кроликах и мышах, показали, что возникновение HSP65-индуцируемой иммунной реакцией в результате иммунизации рекомбинантным белком или препаратом Mycobacterium tuberculosis с высоким содержанием HSP65 усиливала атерогенез. Используя аутоиммунные процессы в отношении HSP65 в качестве возможного антигена-кандидата, который вызывает состояние иммунологической толерантности в результате «толеризации» слизистой оболочки, для блокировки указанных реакций, авторы настоящего изобретения обнаружили ослабление начальной стадии атеросклероза у мышей, которым перорально вводили HSP65, по сравнению с мышами, которым перорально вводили BSA или PBS (Harats et al. Oral tolerance with heat shock protein 65 attenuates mycobacterium tuberculosis induced and high fat diet driven atherosclerosis lesions. J Am Coil Cardiol. 2002;40:1333-1338), полученный результат был подтвержден Мароном, который показал, что назальная вакцинация HSP уменьшает воспалительный процесс, ассоциированный с атеросклерозом (Maron et al. Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low density lipoprotein receptor-deficient mice. Circulation. 2002; 106:1708-1715).
Оказалось, что бета-2-гликопротеин I (бета2GРI) является фосфолипид-связывающим белком, который служит мишенью для протромботических антител против фосфолипида. Недавно было установлено, что данный белок вызывает иммуноопосредуемую реакцию и усиливает атеросклероз у мышей. β-Антитела к бета-2-GPI обладают способностью активировать моноциты и эндотелиальные клетки и могут индуцировать иммунную реакцию на бета2GРI у мышей с повышенным риском возникновения атеросклероза, ускоряя развитие данного заболевания. Когда бета2GРI-реактивный лимфатический узел и клетки селезенки переносили в организм мышей с отсутствием LDL-рецептора, они стимулировали образование жировых полосок, подтверждая непосредственную проатерогенную роль бета2GРI-специфических лимфоцитов. Индукция иммунологической толерантности к бета2GРI в результате предварительного орального введения антигена позволяла значительно уменьшить степень атеросклеротических поражений. Таким образом, бета2GРI является предполагаемым модулятором атеросклеротических бляшек и поэтому, вероятно, может быть использован в качестве иммуномодулятора развития бляшек. Пероральное введение бета20Р1 ингибирует реактивность клеток лимфатического узла в отношении бета2GРI у мышей, иммунизированных против белка человека. В клетках лимфатического узла бета2GРI-толерантных мышей, иммунизированных против бета2GРI, увеличилось продуцирование IL-4 и IL-10 при инициировании соответствующим белком. Таким образом, пероральное введение бета2GРI является эффективным средством подавления атеросклероза у мышей (George et al. Suppression of early atherosclerosis in LDL-receptor deficient mice by oral tolerance with beta2-glycoprotein I. Cardiovasc Res. 2004;62(3):603-9).
Описанные здесь окисленные липиды можно приготовить в соответствии с любыми пригодными способами, известными в области химии. Например, описанные здесь фосфолипиды можно приготовить в соответствии с методиками, описанными в Международной патентной заявке PCT/IL05/000735 (Публикация WO 06/006161) или Американская патентная заявка 11/650,973 (Публикация 2007-0112211).
Должно быть понятно, что некоторые признаки изобретения, которые, для ясности, описаны в отношении отдельных вариантов, также могут быть обеспечены вместе в одном варианте. Напротив, разные признаки изобретения, которые для краткости описаны в отношении одного варианта, могут также быть обеспечены по-отдельности или в любой приемлемой подкомбинации, или как удобно в любом другом описанном варианте изобретения. Некоторые признаки, описанные в отношении разных вариантов, не должны рассматриваться как существенные признаки таких вариантов, за исключением случаев, когда вариант является наработоспособным без этих элементов.
Разные варианты и аспекты настоящего изобретения, как описано здесь выше и как заявлено в формуле ниже, находят экспериментальное подтверждение в следующих примерах.
ПРИМЕРЫ
Далее приведены примеры, которые вместе с вышеуказанными описаниями иллюстрируют некоторые варианты изобретения, не ограничивая его объем.
МАТЕРИАЛЫ И СПОСОБЫ
Материалы:
Уксусная кислота (ледяная) была получена от фирмы Bio-Lab;
Кристаллический фиолетовый был получен от фирмы Sigma;
Дитиотреитол (DTT) был получен от фирмы Bio-Lab;
Фетальная бычья сыворотка (термоинактивированная) была получена от фирмы Biological Industries (Израиль);
Метанол (чистый) был получен от фирмы Bio-Lab;
MOG пептид 35-55 был получен от фирмы Sigma-Aldrich;
Мышиный GM-CSF (гранулоцитарно-монопитарный колониестимулирующий фактор) был получен от фирмы Peprotech (Израиль);
Раствор пенициллин/стрептомицин был получен от фирмы Biological Industries (Израиль);
Лизирующий буфер для эритроцитов был получен от фирмы Biological Industries (Израиль); и
Среда RPMI-1640 с L-глутамином была получена от фирмы Biological Industries (Израиль).
24-луночные обработанные культурой тканей планшеты COSTAR® Sterile, были получены от фирмы Corning.
Фосфатно-солевой буферный раствор (PBS) был приготовлен путем разбавления 10х концентрата фосфатного буфера Дюльбекко без кальция или магния (Biological Industries, Израиль) бидистиллированной водой.
Клетки были инкубированы при 37°С в атмосфере с 5% CO2.
Спектрометрические измерения выполняли с использованием планшет-ридера Tecan SUNRISE и программного обеспечения для сбора данных Magellan Version 6.3. Абсорбцию при 595 нм определяли с использованием полосного фильтра Tecan SpectraFluor 595 nm.
Для исследований in vitro тестируемые соединения растворяли в этаноле до концентрации 100 мг/мл и затем разбавляли в PBS до концентрации 1 мг/мл.
Анализ фосфорилирования тирозина:
Анализировали фосфорилирование тирозина в макрофагах или клетках костного мозга (BMDCs) мышей.
Первичные мышиные макрофаги изолировали из брюшной полости 7-8 недельных самок мышей линии C57BL/6 с последующим активированием тиогликолятом. Клетки предварительно обедняли 0.5%-ной фетальной бычьей сывороткой (FBS) в среде RPMI-1640 в течение ночи. Мышиные первичные клетки костного мозга изолировали вымыванием костного мозга из бедренной кости и голени самок мышей линии SJL охлажденной средой RPMI-1640. Готовили клеточную суспензию и удаляли эритроциты лизирующим буфером, и инкубировали при 4°С в течение 15 минут в буфере, содержащем фосфатно-солевой буферный раствор (PBS) и 0.5% бычий сывороточный альбумин (BSA) с микросферами В220 и CD90 (Miltenyi Biotech). Клетки затем отмывали, ресуспендировали в том же буфере и отделяли от В и Т клеток на сепараторе Midi-Macs через LD или LS колонки (Miltenyi Biotech). Обедненные клетки костного мозга подсчитывали, отмывали и засевали в концентрации 106 клеток/мл в среде RPMI-1640 с добавлением L-глутамина, β-меркаптоэтанола, 10%-ной фетальной бычьей сыворотки, антибиотиков (пенициллин/стрептомицин) и 2 нг/мл гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) мыши. Среду меняли через день. После 5-6 дней культивирования клетки собирали, подсчитывали и засевали (106 клеток/мл) в среду на 24 часа с последующим культивированием 0.5%-ной фетальной бычьей сыворотки (FBS) в среде RPMI-1640 в течение ночи.
Макрофаги или BMDCs затем обрабатывали в течение 10 минут 1, 10 или 20 мкг/мл тестируемого соединения в фосфатно-солевом буферном растворе (PBS) с 1% этанола. В качестве негативного контроля использовали обработку 1 или 20 мкг/мл фосфатидилхолина или растворителем (PBS с 1% этанола). В качестве негативного контроля использовали обработку с помощью CI-201 (1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин) в концентрации 1,10 или 20 мкг/мл.
Белки с фосфорилированным тирозином затем наблюдали с помощью Вестерн-блот анализа с использованием моноклональных антител к фосфотирозиновым белкам. Вестерн-блот анализ для ERK1/2 или α-тубулина выполняли как контроль загрузки белка.
Оценка токсичности in vitro:
Активированные тиогликолятом мышиные перитонеальные макрофаги отмывали, подсчитывали и засевали (от 2х105 до 3х105 клеток на ячейку в 24-луночные планшеты) в ячейки в 3-х повторностях в среде, содержащей RPMI-1640, L-глутамин, 10% FBS и антибиотики (пенициллин/стрептомицин). После восстановления в течение 24 часов тестируемые соединения (или контрольные соединения) добавляли в клеточную среду в дозах 2, 10, 20, 50, 100 или 150 мкг/мл, сохраняя добавленный объем одинаковым при всех обработках путем доведения объема растворителем.
После добавления соединений клетки инкубировали еще в течение 24 часов, после чего клетки отмывали, фиксировали раствором 10% метанол/10% уксусная кислота и окрашивали кристаллическим фиолетовым (0.4% в 20% этаноле). Количество клеток измеряли определением оптической плотности при 595 нм. Клетки, инкубированные с носителем (PBS с 1% этанола), использовали в качестве контроля, по которым нормализовали количество клеток в обработанных образцах.
Данные представлены как среднее±стандартное отклонение. Статистическую значимость относительно клеток, обработанных носителем, рассчитывали с использованием критерия Стъюдента, при этом р-значения менее 0.05 указывали статистическую значимость.
Анализ in vitro продукции IL12/23p40:
Клетки костного мозга (BMDCs) получали из бедренной кости и голени самок мышей линии C57BL, культивировали и засевали при концентрации 106 клеток/мл через 5-6 дней после культивирования, как описано здесь выше, для анализа фосфорилирования тирозина.
Тестируемое соединение затем добавляли в клетки в течение 1 часа при концентрации 1, 2.5, 5, 10 или 20 мкг/мл. Клетки активировали для продукции IL12/23p40 путем инкубирования в течение 24 часов с 10 мкг/мл пептидокликана (PGN). Продукцию цитокина из супернатанта измеряли с использованием ELISA. В качестве контроля использовали активированные клетки без тестируемого соединения.
Анализ in vitro экспрессии MPHK IL12/23 р40:
Культуры клеток костного мозга были приготовлены, как описано здесь выше. На 5-6 день после культивирования культуры клеток обогащали дендритными клетками CD11c+(>90%) с помощью магнитных шариков Mouse CD 11 с Microbeads через MS или LS колонки (Miltenyi Biotech). Дендритные клетки CD11c+стимулировали в течение 1, 2 и 3 часов только 10 мкг\мл пептидогликана или в присутствии 20 мкг/мл тестируемого соединения, добавленного за 1 час до активирования. РНК экстрагировали из клеток с использованием набора RNeasy mini kit (Qiagen, Valencia, CA). Для приготовления кДНК, 1 мкг РНК связывали с Oligo dT в течение 10 минут при 70°С, 1-м стандартным буфером. Дитиотреитол, dNTP и обратную транскриптазу Superscript (SS-II) (Invitrogen, Carlsbad, CA) добавляли в течение 50 минут при 42°С и реакцию завершали инкубированием еще в течение 15 минут при 70°С. Все реакции PCR проводили в режиме реального времени с использованием LightCycler Taqman master (Roche Diagnostics, Mannheim, Германия) и выполняли на LightCycler machine (Roche). Готовые наборы пробы с праймерами использовали для анализов IL 12/23 р40 и GAPDH (Applied Biosystems, анализы #Mm01288992_ml и Mm99999915_gl, соответственно), с последним, предназначенным для нормализации уровней РНК. 20 мкг/мл CI-201 и фосфатидилхолин (PC) использовали в качестве положительного и негативного контроля соответственно.
Анализ in vivo миелин-олигодендроцитарный гликопротеин (MOG)-индуцированного экспериментального аутоиммунного энцефаломиелита (ЕАЕ):
Мышам линии C57BL/6 орально вводили указанное количество тестируемого соединения в конечном объеме 200 мкл в течение 5 последовательных дней за 5 дней до иммунизации.
Для индуцирования ЕАЕ мышей иммунизировали подкожно эмульсией, содержащей 1.5 мг/мл MOG пептида 35-55 (MEVGWYRSPFSRVVHLYRNGK) и 2.5 мг/мл CFA (полный адъювант Фрейнда), при этом 100 мкл эмульсии вводили в каждый бок. Сразу же после этого и снова через 48 часов мышам вводили внутрибрюшинную инъекцию коклюшного токсина (500 нг в 500 мкл PBS).
Начало ЕАЕ оценивали по среднему клиническому показателю. Анализ in vivo коллаген-индуцированного артрита (CIA):
Мышиных самцов линии DBA/I иммунизировали для индуцирования артрита инъекцией коллагена, содержащей адъювант Фрейнда (CFA), в основание хвоста (0 день) и повторно в бок (21 день). Наблюдение за развитием артрита у мышей проводили в течение 36 дней. Введение через зонд тестируемого соединения и контрольных веществ начинали на 22 день и проводили на ежедневной основе (6 раз в неделю).
Начало артрита оценивали по клиническому показателю артрита.
Анализ in vivo атеросклеротического поражения:
12-16-недельным мышиным самцам линии LDL-RD вводили орально 0.2 мл PBS или PBS с указанным количеством тестируемого соединения, один раз в сутки, через день, в течение 5 обработок. Мышей содержали на высокожировой диете в течение 5 недель и затем умерщвляли.
Или же, 14-16-недельным мышам АроЕ КО орально вводили 0.2 мл PBS или PBS с указанным количеством тестируемого соединения, один раз в сутки, через день, в течение 5 обработок и умерщвляли по окончании 8 недель.
Количественную оценку атеросклеротических повреждений производили расчетом размера повреждения в синусе аорты, как было описано ранее [Paigen et al., Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 1987; 68:231-240], с некоторыми изменениями. Сердце и аорту удаляли из животных и тщательно очищали от периферического жира. Сердца заливали гелем, обеспечивающим оптимальные условия разрезания при данной температуре (Optimal Cutting Temperature, ОСТ) и замораживали. Повреждение синуса аорты определяли по окрашенным Oil red О 3-8 серийным срезам (10 мкм толщина), по всему синусу аорты (400 мкм). Область повреждения подсчитывали с использованием компьютерного способа анализа (Image Pro Plus software [version 4.5.1.29], Medical Cybernetics Corporation).
ПРИМЕР 1
1-Гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (Cl-202) и 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламин
(R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин и (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламин синтезировали, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1 -гексадецил-2-(4-карбокси)бутил- глицеро-3-фосфоэтаноламин и (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламин синтезировали с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-1-гексадецил-глицерина: 11 г (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 20 г порошкового гидроксида калия и 27.96 г гексадецилбромида перемешивали в 150 мл толуола и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 40 мл. Реакционную смесь охлаждали до комнатной температуры, добавляли 100 мл воды и экстрагировали трижды 75 мл дихлорметана. Объединенную органическую фазу промывали 50 мл воды, и растворитель удаляли при пониженном давлении. Остаток растворяли в 200 мл смеси 90:10:5 (объем/объем) метанол:вода:концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры добавляли 100 мл воды. Продукт экстрагировали трижды 100 мл дихлорметана, промывали последовательно 100 мл воды, 100 мл насыщенного водного раствора карбоната натрия и снова 100 мл воды. Растворитель удаляли при пониженном давлении, и продукт кристаллизовали из гексана (200 мл) для получения 21.69 г чистого (S)-1-гексадецил-глицерина, который высушивали в десикаторе при пониженном давлении. Выход составил 82%.
Синтез (R)-1-гексадецил-3-тритил-глицерина: 20 г (S)-1-гексадецил-глицерина и 21.29 г трифенилхлорметана растворяли в 369 мл сухого тетрагидрофурана (THF) и 93 мл сухого ацетонитрила. Добавляли 17.75 мл триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (100 грамм), переносили в делительную воронку и экстрагировали метил-трет-бутиловым эфиром. Органическую фазу последовательно промывали 200 мл воды, дважды 200 мл разбавленной (1.5%) серной кислоты, 200 мл воды, 200 мл насыщенного водного раствора бикарбоната натрия и снова 200 мл воды. Органическую фазу высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 36.86 г сырого продукта в виде остатка. Данный остаток растворяли в горячем гексане (200 мл) и полученный раствор охлаждали при 4°С в течение ночи. Осажденный продукт фильтровали с получением 30.71 г (R)-1-гексадецил-3-тритил-глицерина.
Синтез (R)-1-гексадецил-2-(5'-гексенил)-3-тритил-глицерина: 19.94 г 1-гексадецил-3-тритил-глицерина, 6.98 г 6-бром-1-гексена и 15 г порошкового гидроксида калия перемешивали в 350 мл гексана и кипятили в течение 8 часов с азеотропным удалением воды. Реакционную смесь охлаждали до комнатной температуры, переносили в делительную воронку, промывали дважды 200 мл воды, и растворитель затем удаляли при пониженном давлении. Остаток растворяли в 150 мл гексана и снова промывали дважды 200 мл воды. Органический раствор выдерживали при 4°С в течение ночи, при этом возникало осаждение побочных продуктов. Фильтрация и удаление растворителя при пониженном давлении позволили получить 19,86 г (R)-1-гексадецил-2-(5'-гексенил)-3-тритил-глицерина. Выход составил 86.6%.
Синтез (S)-1-гексадецил-2-(4-карбокси)бутил-sn-глицерина: 150.16 г (702 ммоль) периодата натрия суспендировали в 500 мл воды в трехгорлой колбе с круглым дном, снабженной термометром и капельной воронкой. Добавляли 7.21 г (85.8 ммоль) бикарбоната натрия и 2.47 г (15.6 ммоль) перманганата калия, после чего суспензию нагревали до температуры 40°С.50 г (78 ммоль) (R)-1-гексадецил-2-(5'-гексенил)-3-тритил-глицерина растворяли в 500 мл трет-бутанола, и полученный раствор добавляли в смесь периодата натрия и перманганата калия в течение 1 часа. После добавления (R)-1-гексадецил-2-(5'-гексенил)-3-тритил-глицерина в периодат натрия и перманганат калия реакционную смесь нагревали при температуре 40°С в течение 3 часов. Через 1.5 часа добавляли еще 0.62 г (3.9 ммоль) перманганата калия для сохранения розового цвета реакционной смеси. По окончании 3 часового периода реакционную смесь охлаждали до комнатной температуры, переносили в делительную воронку и экстрагировали 200 мл гексана. Органическую фазу промывали раствором, содержащим 15 г Na2S2O5 в 100 мл воды. Разбавленную соляную кислоту (0.65 мл концентрированной HCl в 13 мл воды) добавляли в органическую фазу и 200 мл растворителя перегоняли при пониженном давлении. Оставшийся прозрачный раствор нагревали до температуры 80°С в течение 6 часов и отгоняли еще 250 мл растворителя. Остаток обрабатывали 100 мл воды и 10 мл 30% раствора NaOH, устанавливая рН 12. Осажденный трифенилметанол отфильтровывали и промывали 4 раза 10 мл воды. Фильтрат экстрагировали смесью 50 мл гексана и 50 мл этилацетата для удаления оставшегося трифенилметанола и других примесей. В водной фазе натриевую соль 1-гексадецил-2-(4-карбокси)бутил-sn-глицерина протонировали 8.45 мл (101.4 ммоль) концентрированной соляной кислоты. Полученную свободную карбоновую кислоту экстрагировали 100 мл гексана. Выпаривание до сухого состояния и совместное выпаривание с 100 мл гексана позволило получить 27.00 г сырого (S)-1-гексадецил-2-(4-карбокси)бутил-sn-глицерина. Сырой продукт кристаллизовали растворением в смеси, содержащей 7 мл ацетона и 68 мл гексана, и охлаждали до 0°С. Осадок фильтровали, промывали дважды 7 мл охлажденного гексана и высушивали. 20.90 г (S)-1-гексадецил-2-(4-карбокси)бутил-5п-глицерина было получено в виде грязно-белого твердого вещества. Выход составил 64.3%.
Синтез (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина:. 15.0 г (36.0 ммоль) (S)-1-гексадецил-2-(4-карбокси)бутил-sn-глицерина растворяли в 100 мл метанола и добавляли 3 мл концентрированной соляной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли триэтиламин до установления рН реакционной смеси, равным 7. Раствор переносили в делительную воронку и дважды экстрагировали 200 мл гексана. Органическую фазу отмывали водой. Выпаривание до сухого состояния и совместное выпаривание с 100 мл гексана позволило получить 14.92 г (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина. Выход составил 96.2%.
Синтез (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 2.88 г (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина и 3 мл триэтиламина растворяли в 30 мл THF. Данный раствор по каплям добавляли в ледяной раствор, содержащий 2 мл POCl3 в 20 мл тетрагидрофурана, в течение 15 минут при перемешивании. Перемешивание продолжали еще в течение 10 минут при охлаждении и при комнатной температуре еще в течение 45 минут. Реакционную смесь охлаждали льдом и в течение 20 минут по каплям добавляли раствор, содержащий 1.21 мл этаноламина и 5.6 мл триэтиламина в 50 мл THF. Перемешивание продолжали в течение 10 минут при охлаждении и при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси 24 мл уксусной кислоты и 10 мл воды, и нагревали до 70°С в течение 1 часа. Смесь трижды экстрагировали 50 мл хлороформа, органическую фазу дважды промывали 50 мл воды, и растворитель удаляли при пониженном давлении для получения 4.0 г (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина в виде желтого воска.
ЯМР характеристика 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец 1 -гексадецил-2-(4-метилкарбокси)бутил-глицеро-3 -фосфоэтаноламина растворяли в дейтерированном хлороформе (CDCl3). Спектры затем измеряли при 300 МГц. Образцы исследовали с помощью 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1 -гексадецил-2-(4-метилкарбокси)бутил-глицеро-3 -фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.28 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
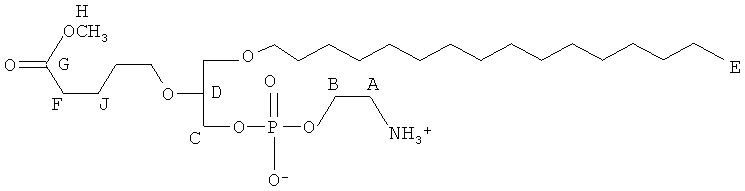
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.0 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(4-метилкарбокси) бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина (C27H56NO8P) составила 553.7092.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=554, соответствующего протонированному молекулярному иону [М+Н]+. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина.
Синтез (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-202): 3.5 г (R)-1-гексадецил-2-(4-метилкарбокси) бутил-sn-глицеро-3-фосфоэтаноламина растворяли в 100 мл смеси 8:2 (об/об) метанол: 10% раствор гидроксида натрия. Смесь перемешивали при комнатной температуре в течение 5 часов. РН реакционной смеси доводили до 4 добавлением однозамещенного фосфорнокислого натрия и муравьиной кислоты. Добавляли воду (100 мл) и хлороформ (100 мл). После экстрагирования фазы разделяли, и растворитель удаляли из органической фазы при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении для получения 3,0 г сырого (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина. Сырой продукт очищали хроматографией на силикагеле (120 г). Продукт элюировали смесью хлороформ: метанол и водой при объемном соотношении 60:35:5. Растворитель из фракций, содержащих требуемый продукт, удаляли при пониженном давлении, остаток растворяли в хлороформе и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 2.11 г чистого (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина в виде воска. Воск высушивали при пониженном давлении над пентоксидом фосфора.
ЯМР характеристика CI-202:
Образец CI-202 растворяли в дейтерированном хлороформе (CDCl3). Спектры затем измеряли при 300 МГц. Образцы измеряли с помощью 1Н и 13С ЯМР спектроскопии. Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (CI-202) и, таким образом, полностью подтвердили данную структуру. Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом: 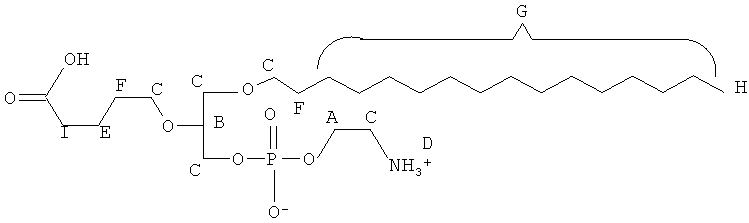
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.26 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
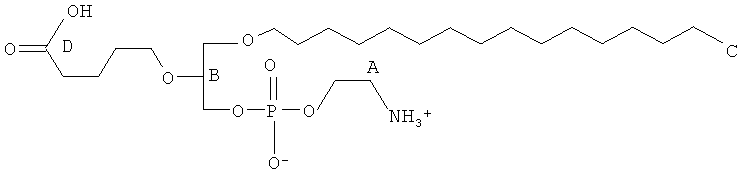
13C ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.004 м.д.)
Масс-спектрометрическая характеристика CI-202:
Вычисленная масса для 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C26H54NO8P) составила 539.36.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=538, соответствующего депротонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (CI-202).
Продукция in vitro IL12/23 р40:Действие CI-202 на продукцию in vitro IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.1, CI-202 ингибировало продукцию IL12/23 р40 клетками костного мозга дозозависимым образом.
Экспрессия мРНК IL12/23p40:
Действие CI-202 на экспрессию in vitro мРНК IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.2, CI-202 ингибировало экспрессию мРНК IL12/23 р40 во время всего периода тестирования (2-4 часа после введения CI-202), и ингибирование с использованием CI-202 было сравнимо с ингибированием с использованием CI-201.
Фосфорилирование тирозина:
Действие CI-202 на фосфорилирование тирозина in vitro в первичных макрофагах определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.3, обработка с помощью 10 мкг/мл (18.5 мкМ) CI-202 приводит к индуцированию фосфорилирования тирозина, в то время как воздействие 20 мкг/мл (37 мкМ) CI-202 вызывает снижение уровней фосфотирозина. Данные изменения схожи с действием, вызванным соответственно 10 мкг/мл (17 мкМ) и 20 мкг/мл (34 мкМ) положительного контроля CI-201.
Токсичность CI-202:
Токсичность CI-202 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.4А и 4В, значительная токсичность CI-202 была обнаружена только при дозах выше 50 мкг/мл, и LD50 CI-202 находилась в диапазоне от 50 до 100 мкг/мл.
Развитие экспериментального аутоиммунного энцефаломиелита (ЕАЕ), индуцированного миелин-олигодендроцитарным гликопротеином (MOG):
Действие CI-202 на развитие in vivo экспериментального аутоиммунного энцефаломиелита (ЕАЕ) у мышей, индуцированного миелин-олигодендроцитарным гликопротеином (MOG) определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.5, введение 4 мг/кг CI-202 замедляло начало заболевания и снижало клиническое проявление ЕАЕ.
Развитие коллаген-индуцированного артрита (CIA):
Действие CI-202 на развитие in vivo коллаген-индуцированного артрита у мышей определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.6, введение 0.4 мг/кг CI-202 значительно снижало тяжесть артрита во время периода исследования. Максимальный клинический показатель артрита понизился на 42% по сравнению с контрольными мышами.
ПРИМЕР 2
1-Гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолин (CI-203)
(R)-1-гексадецил-2-(6-карбокси)гексанил-sn-глицеро-3-фосфохолин синтезировали, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S-1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолин синтезировали с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (R)-1-гексадецил-3-тритил-глицерина: (R)-1 -гексадецил-3-тритил-глицерин был приготовлен, как описано в Примере 1, сначала приготовлением (S)-1-гексадецил-глицерина с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола.
Синтез (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-3-тритил-глицерина: 5 г (R)-1-гексадецил-3-тритил-глицерина и 2 мл этил 7-бром-гептаноата растворяли в 70 мл бензола. Добавляли 23 г порошкового КОН, реакционную смесь перемешивали и кипятили в течение 14 часов с азеотропным удалением воды. Реакционную смесь охлаждали до комнатной температуры, трижды промывали 70 мл воды и высушивали над сульфатом натрия. Растворитель удаляли при пониженном давлении, полученный остаток растворяли в 25 мл горячего гексана и раствор охлаждали до 4°С в течение ночи. Осажденный побочный продукт фильтровали и растворитель удаляли при пониженном давлении для получения 5 г (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-3-тритил-глицерина в виде белого твердого вещества.
Синтез (S)-1-гексадецил-2-(6-этилкарбокси)гексанил-глицерина: 5 г (R}-1-гексадецил-2-(6-этилкарбокси)гексанил-3-тритил-глицерина растворяли в 90 мл этанола. Медленно добавляли 20 мл концентрированной соляной кислоты, смесь перемешивали и кипятили с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед и экстрагировали трижды 100 мл диэтилового эфира. Органическую фазу промывали 100 мл воды, 100 мл насыщенного раствора бикарбоната натрия и снова 100 мл воды, и высушивали над сульфатом натрия. После фильтрации сульфата натрия растворитель удаляли при пониженном давлении. Полученный остаток растворяли в горячем n-гексане, и смесь затем выдерживали при 4°С в течение ночи. После фильтрации осадка растворитель удаляли при пониженном давлении для получения 3.1 г желтого масла. Остаток очищали хроматографией на колонке с силикагелем (140 г). Продукт элюировали 300 мл CHCl3:этилацетат (6:4 об/об). Удаление растворителя при пониженном давлении позволило получить 1.34 г бесцветного масла. Сушка при пониженном давлении над пентоксидом фосфора позволила получить (S)-1-гексадецил-2-(6-этилкарбокси)гексанил-глицерин в виде бесцветного твердого вещества.
Синтез (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфоэтаноламина: 1.34 г (S)-1-гексадецил-2-(6-этилкарбокси)гексанил-глицерина и 1.2 мл триэтиламина растворяли в 15 мл THF. Данный раствор добавляли по каплям в течение 15 минут в ледяной раствор, содержащий 0.8 мл POCl3 в 10 мл THF. Раствор перемешивали еще 10 минут при охлаждении и еще 45 минут при комнатной температуре. Реакционную смесь охлаждали льдом и в течение 15 минут по каплям добавляли раствор, содержащий 0.52 мл этаноламина и 2.4 мл триэтиламина в 25 мл THF. Перемешивание реакционной смеси продолжали в течение 10 минут при охлаждении в ледяной ванне и затем в течение ночи при комнатной температуре. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси, состоящей из 24 мл уксусной кислоты и 10 мл воды, и затем нагревали до температуры 70°С в течение 1 часа. Смесь затем экстрагировали трижды 50 мл хлороформа и промывали дважды 50 мл воды. Удаление растворителя при пониженном давлении позволило получить 1.87 г (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфоэтаноламина в виде желтого масла.
Синтез (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфохолина: 1.87 г (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси, состоящей из 50 мл изопропанола и 18 мл дихлорметана. Раствор, содержащий 2.17 г карбоната калия в 10 мл воды, добавляли по каплям в течение 5 минут при поддержании температуры при 35-40°С.Раствор 1.52 мл диметилсульфата в 10 мл изопропанола добавляли по каплям при температуре 40°С в течение 10 минут.Реакционную смесь выдерживали при температуре 40°С в течение 90 минут, затем добавляли воду и смесь экстрагировали дважды 50 мл хлороформа.Органическую фазу промывали 50 мл воды и растворитель удаляли при пониженном давлении для получения 1.8 г (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфохолина в виде воска.
Синтез (R)-1-гексадецил-2-(6-карбокси)гексанил-sn-глицеро-3-фосфохолина (CI-203): 1.8 г (R)-1-гексадецил-2-(6-этилкарбокси)гексанил-sn-глицеро-3-фосфохолина растворяли в 50 мл метанола. Добавляли 10% раствор гидроксида натрия и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакционной смеси доводили до 4-5 добавлением однозамещенного фосфорнокислого натрия. Добавляли 70 мл воды и 70 мл хлороформа. Водную и органическую фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении для получения 1.29 г белого воска. Остаток очищали хроматографией на силикагеле (62 г). Продукт элюировали смесью CHCl3: метанол: Н2О при объемном соотношении 60:35:5. После удаления растворителя при пониженном давлении остаток растворяли в хлороформе, высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении для получения 1.0 г (R)-1-гексадецил-2-(6-карбокси)гексанил-sn-глицеро-3-фосфохолина в виде белого воска.
ЯМР характеристика 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3). Спектры 1Н ЯМР и 13С ЯМР измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина, были обозначены следующим образом: 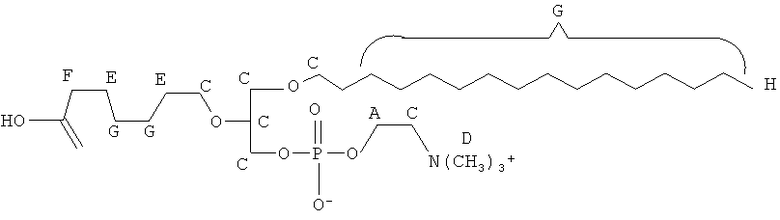
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.260 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина, были обозначены следующим образом: 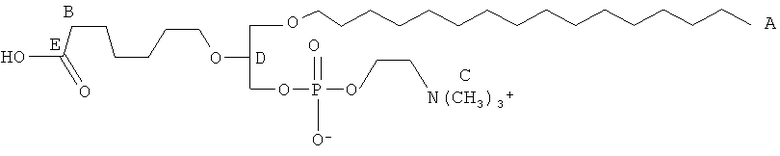
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.002 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина:Вычисленная масса для 1-гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина (C31H64NO8P) составила 609.82.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=609, соответствующего депротонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1 -гексадецил-2-(6-карбокси)гексанил-глицеро-3-фосфохолина. Продукция in vitro IL12/23 р40:
Действие CI-203 на продукцию in vitro IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.7, CI-203 ингибировало продукцию IL12/23 р40 клетками костного мозга дозозависимым образом.
Фосфорилирование тирозина:
Действие CI-203 на in vitro фосфорилирование тирозина в клетках костного мозга определяли, как описано здесь выше в разделе «Материалы и Способы». Тестировали R энантиомер CI-203 и рацемический CI-203. Оба, R энантиомер и S энантиомер, использовали в качестве контроля. Как показано на Фиг.8, обработка с использованием 20 мкг/мл (33 мкМ) (R)-CI-203 и рацемическим CI-203 вызывала снижение уровней фосфотирозина, в то время как обработка с использованием 1 мкг/мл (1.7 мкМ) CI-203 не имела выраженного эффекта. Действие (R)-CI-203 и рацемического CI-203 было аналогичным действию, индуцированному R и S энантиомерами CI-201. Токсичность CI-203:
Токсичность CI-203 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.9А и 9В, значительную токсичность CI-203 наблюдали только при дозе 100 мкг/мл, и LD50 CI-203 находилась в диапазоне от 50 до 100 мкг/мл.
ПРИМЕР 3
1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-209)(R)-1-додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин был синтезирован, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин синтезировали с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-додецил-глицерина: 11 г (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 20.6 г порошкового гидроксида калия и 24.08 г додецилбромида перемешивали в 300 мл бензола и кипятили в течение 14 часов с азеотропным удалением воды. Реакционную смесь затем охлаждали до комнатной температуры и добавляли 150 мл воды. Реакционную смесь затем экстрагировали трижды 150 мл метил-трет-бутилового эфира (МТВЕ), объединенную органическую фазу промывали 100 мл воды, и растворитель затем удаляли при пониженном давлении, получая 29.71 г светло-коричневого масла. Данный остаток растворяли в 100 мл метанола. Добавляли 6 мл концентрированной соляной кислоты и полученный раствор кипятили с обратным холодильником до получения прозрачного раствора, с последующим охлаждением до комнатной температуры и добавлением 100 мл воды. Продукт экстрагировали 150 мл хлороформа, последовательно промывали 150 мл воды, 150 мл насыщенного водного раствора бикарбоната натрия и снова 150 мл воды. Растворитель высушивали над безводным Na2SO4, фильтровали и удаляли при пониженном давлении, получая 23.77 г сырого продукта. Сырой продукт рекристаллизовали из 200 мл гексана при 4°С для получения 19.83 г чистого (5)-1-додецил-глицерина в виде белых кристаллов. Синтез (R)-1-додецил-3-тритил-глицерина: 19.83 г (S)-1-додецил-глицерина и 21.0 г трифенилхлорметана добавляли в смесь, содержащую 250 мл сухого тетрагидрофурана (THF) и 60 мл сухого ацетонитрила. Добавляли 22 мл сухого триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов в атмосфере азота. Реакционную смесь затем охлаждали до комнатной температуры и добавляли 5 мл триэтиламина и 10 г льда. Смесь переносили в делительную воронку и экстрагировали 100 мл МТВЕ. Органическую фазу последовательно промывали 150 мл воды, 150 мл разбавленной (1.5%) H2S04, 150 мл воды, 150 мл концентрированного водного раствора бикарбоната натрия и снова 150 мл воды. Раствор затем высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Остаток, 40.63 г коричневого масла, растворяли в 200 мл этилацетата и охлаждали до -20°С в течение двух дней. Смесь центрифугировали при температуре -10°С и сливали маточную жидкость. Твердый продукт расплавляли при комнатной температуре и очищали хроматографией на колонке с силикагелем (195 г). 18.57 г чистого (R)-1-додецил-3-тритил-глицерина элюировали смесями хлороформа и гексана с последующей смесью 9:1 (об/об) хлороформ:этилацетат. Выход составил 48.5%.
Синтез (S)-1-додецил-2-(5'-гексенил)-глицерина: 18.57 г (R)-1-додецил-3-тритил-глицерина, 4 мл 6-бром-1-гексена и 22.57 г порошкового гидроксида калия перемешивали в 100 мл бензола и кипятили в течение 9 часов с азеотропным удалением воды. Реакционную смесь охлаждали до комнатной температуры, добавляли 100 мл воды и раствор переносили в делительную воронку. Раствор экстрагировали четыре раза 50 мл диэтилового эфира и растворитель из объединенной органической фазы удаляли при пониженном давлении, получая 19.31 г остатка. Остаток растворяли в 100 мл метанола и добавляли 6 мл концентрированной HCl (37%). Реакционную смесь кипятили с обратным холодильником в течение 4 часов, охлаждали до комнатной температуры и перемешивали при этой температуре в течение 96 часов. Реакционную смесь концентрировали примерно до 50 мл с помощью удаления растворителя при пониженном давлении и добавляли 50 мл воды. Раствор переносили в делительную воронку и экстрагировали дважды 100 мл МТВЕ. Растворитель затем удаляли при пониженном давлении. Остаток (18.85 г) растворяли в гексане, и полученный раствор охлаждали в ледяной ванне в течение 30 минут. Осадок отфильтровывали, отмывали охлажденным гексаном, и растворитель из фильтрата удаляли при пониженном давлении. Остаток (15.23 г) растворяли снова в горячем гексане и охлаждали до 4°С в течение ночи. После фильтрации и удаления растворителя из фильтрата, фильтрат очищали хроматографией на силикагеле (77.34 г). Элюирование выполняли смесью 1:1 хлороформа и этилацетата с последующим чистым хлороформом и затем хлороформом с 3% ацетона. Получали 10.03 г чистого (S)-1-додецил-2-(5'-гексенил)-глицерина. Выход составил 78.1%.
Синтез (R)-1-додецил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина: 5.75 г
(S)-1-додецил-2-(5'-гексенил)-sn-глицерина (который высушивали в десикаторе над P2O5) и 3.11 мл триэтиламина растворяли в 50 мл THF. Данный раствор добавляли по каплям в течение 30 минут в ледяной раствор, содержащий 1.7 мл POCl3 в 20 мл THF. Перемешивание продолжали еще в течение 30 минут при охлаждении и затем еще в течение 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и затем добавляли по каплям раствор 1.3 мл этаноламина и 3.3 мл триэтиламина в THF в течение 15 минут. Перемешивание продолжали в течение 30 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель из фильтрата удаляли при пониженном давлении. Остаток растворяли в смеси 36 мл уксусной кислоты и 15 мл воды, нагревали до 70°С в течение 1 часа и охлаждали до комнатной температуры. Раствор экстрагировали дважды 50 мл смеси 2:1 (об/об) хлороформ:метанол, отмывали разбавленным раствором бикарбоната натрия и растворитель удаляли при пониженном давлении, получая 8.54 г сырого продукта. Этот сырой продукт очищали хроматографией на силикагеле (55 г). 4.99 г чистого (R)-1-додецил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина элюировали хлороформом с последующими смесями хлороформа с 2.5%-40% метанола. Выход составил 63.85%.
Синтез (R)-1-додецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина: 4.99 г (R)-1-додецил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина растворяли в смеси, состоящей из 35 мл метанола и 100 мл дихлорметана. Добавляли раствор, содержащий 10 г карбоната калия в 20 мл воды. Затем добавляли 2.5 мл диметилсульфата по каплям в течение 1 часа и реакционную смесь перемешивали при комнатной температуре в течение ночи. Как было определено тонкослойной хроматографией, в реакционной смеси все еще присутствовало некоторое количество исходного материала. Добавляли еще 1 мл диметилсульфата и реакционную смесь нагревали до 40°С в течение 5 часов. Реакционную смесь затем охлаждали до комнатной температуры, добавляли 100 мл воды с последующей экстракцией смеси трижды 100 мл смеси 2:1 (об/об) хлороформ:метанол. Растворитель из органической фазы удаляли при пониженном давлении, получая 5.8 г сырого (R)-1-додецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина.
Синтез (R)-1-додецил-2-(4-карбокси)бутил-8sn-глицеро-3-фосфохолина (CI-209): В раствор, содержащий 5.18 г (R)-1-додецил-2-(5'-гексенил)-5и-глицеро-3-фосфохолина в 100 мл воды, добавляли 2.76 г бикарбоната натрия. Затем добавляли раствор 20.4 г периодата натрия в 100 мл воды. В капельную воронку помещали раствор, содержащий 270 мг перманганата калия в 82 мл воды, и добавляли по каплям для сохранения розового цвета реакционной смеси. Всего во время реакции было добавлено 58 мл раствора перманганата. Реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили примерно до 4 добавлением 20 г однозамещенного фосфорнокислого натрия и затем 3 мл 80% фосфорной кислоты. Реакционную смесь экстрагировали трижды 50 мл смеси 2:1 хлороформ:метанол и растворитель удаляли из органической фазы при пониженном давлении. Остаток растворяли в хлороформе и отмывали водой. Органический раствор высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении, получая 4.89 г сырого продукта. Сырой продукт очищали хроматографией на силикагеле (58.2 г). 1.81 г чистого (R)-1-додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина элюировали хлороформом с последующими смесями хлороформа с 10%-60% метанола. Выход составил 33.76%.
Дополнительные синтетические способы синтеза CI-209 выполняли следующим образом:
Синтез (R)-1-додецил-2-(5'-гексенил)-3-ацетил-глицерина: (R)-1-додецил-3-тритил-глицерин (67 г), приготовленный как описано здесь выше, 6-бром-1-гексен (26.14 г) и порошковый КОН (35 г) перемешивали в бензоле (200 мл) и кипятили в течение 9 часов с азеотропным удалением воды. Объем растворителя постепенно был уменьшен примерно до 100 мл. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (150 мл). Раствор переносили в делительную воронку и экстрагировали диэтиловым эфиром (3×150 мл). Объединенную органическую фазу промывали водой (3×150 мл) и затем растворитель удаляли при пониженном давлении. Остаток (78 г) растворяли в уксусной кислоте (200 мл) и раствор охлаждали в ледяной ванне. В этот охлажденный раствор добавляли смесь, содержащую 40 мл уксусного ангидрида (40 мл) и 1 мл 70% перхлорной кислоты. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем перемешивали при комнатной температуре в течение ночи. Добавляли лед, затем диэтиловый эфир (400 мл) и воду (400 мл). Органическую фазу разделяли, и растворитель удаляли при пониженном давлении. Остаток растворяли в горячем гексане, и раствор выдерживали в течение ночи при температуре 4°С. Осажденные побочные продукты отфильтровывали, и растворитель удаляли из фильтрата при пониженном давлении для получения 55 г масляного коричневого продукта. Сырой продукт очищали хроматографией на колонке с силикагелем (350 г). Чистый (R)-1-додецил-2-(5'-гексенил)-3-ацетил-глицерин элюировали смесью 1:1 (об/об) хлороформ тексан (1500 мл) с последующим хлороформом (1500 мл). Удаление растворителя из фракций, содержащих продукт, позволило получить 52 г продукта. Синтез (8)-1-додецил-2-(4-карбокси)бутил-глицерина: Периодат натрия (150 г), перманганат калия (2.5 г) и бикарбонат натрия (10 г) суспендировали в воде (500 мл). В водную суспензию в течение 1 часа добавляли раствор (R)-1-до децил-2-(5'-гексенил)-3-ацетил-глицерина (52 г) в трет-бутаноле (500 мл) и реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Смесь фильтровали через слой Целита, который далее отмывали трет-бутанолом. Раствор экстрагировали гексаном (3×100 мл). Объединенную органическую фазу промывали дважды водным раствором бисульфита натрия (15 г в 100 мл воды) и затем водой (100 мл). Растворитель концентрировали при пониженном давлении и обрабатывали 100 мл воды и 10 мл 30% NaOH для достижения рН 12. Смесь перемешивали при комнатной температуре в течение ночи. Смесь экстрагировали смесью 8:2 (об/об) гексан: МТВЕ (200 мл) для удаления оставшихся примесей. Водный щелочной раствор подкисляли HCl (6 мл) до рН 1 и затем экстрагировали смесью 7:3 (об/об) гексан:этилацетат (3×100 мл). Объединенную органическую фазу высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении. Остаток (30 г желтого масла) очищали хроматографией на колонке с силикагелем (500 г). Чистый (S)-1-додецил-2-(4-карбокси)бутил-глицерин элюировали смесью 1:1 (об/об) хлороформ: гексан (1000 мл) с последующим хлороформом (1000 мл) и затем смесями хлороформ:этилацетат (от 9:1 до 1:1, об/об). Удаление растворителя из фракций, содержащих продукт, позволило получить 13.4 г продукта.
Синтез (S)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицерина: (S)-1-додецил-2-(4-карбокси)бутил-sn-глицерин (13.38 г) растворяли в 100 мл метанола и добавляли 2 мл концентрированной соляной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Добавляли воду (50 мл), раствор переносили в делительную воронку и экстрагировали хлороформом (3×100 мл). Объединенную органическую фазу промывали водой (100 мл), концентрированным бикарбонатом натрия (100 мл) и снова водой (100 мл). Сушка над безводным Na2SO4 и выпаривание растворителя при пониженном давлении позволило получить 13.9 г сырого продукта в виде остатка. Остаток очищали хроматографией на колонке с силикагелем (200 г). Чистый (S)-1-додецил-2-(4-метилкарбокси)бутил-глицерин элюировали хлороформом с последующими смесями хлороформ:этилацетат (от 9:1 до 7:3, об/об). Удаление растворителя из фракций, содержащих продукт, позволило получить 10.7 г чистого продукта.
Синтез (R)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина:10.7 г (S)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицерина (который был высушен азеотропной перегонкой с бензолом) и 5.2 мл сухого триэтиламина растворяли в THF (50 мл). Полученный раствор добавляли по каплям в течение 90 минут в ледяной раствор POCl3 (3.2 мл) в 50 мл THF при перемешивании. Перемешивание продолжали еще 15 минут при охлаждении и затем еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и при перемешивании добавляли по каплям в течение 60 минут раствор, содержащий этаноламин (22 мл) и триэтиламин (7.2 мл) в 50 мл THF. Перемешивание продолжали в течение 30 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли из фильтрата при пониженном давлении. Остаток (14 г желтого масла) растворяли в смеси уксусной кислоты (120 мл) и воды (50 мл), нагревали до 70°С в течение 1 часа и охлаждали до комнатной температуры. Раствор экстрагировали смесью 2:1 (об/об) хлороформ:метанол (3×100 мл), объединенную органическую фазу промывали водой (2×100 мл) и растворитель удаляли при пониженном давлении, получая 15 г (R)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина в виде оранжевого масла. (R)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламин (14 г) растворяли в смеси изопропанола (100 мл) и дихлорметана (55 мл). Раствор карбоната калия (22 г) в воде (100 мл) добавляли по каплям при поддержании температуры реакции 35-40°С. Раствор диметилсульфата (14 мл) в изопропаноле (50 мл) добавляли по каплям при 40°С. Смесь перемешивали при 40°С в течение 2 часов, охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли воду (80 мл) и смесь экстрагировали хлороформом (3×100 мл). Объединенную органическую фазу промывали водой (100 мл) и растворитель удаляли при пониженном давлении, получая сырой (R)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолин (14.6 г) в виде оранжевого воска.
Синтез (R)-1-додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-209): (R)-1-додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолин (14.6 г) растворяли в смеси 8:2 (об/об) метанол: 10% раствор NaOH (100 мл) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакционной смеси устанавливали до 5 путем добавления однозамещенного фосфорнокислого натрия и муравьиной кислоты. Добавляли воду (150 мл) и метанол (50 мл). Водную и органическую фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении, получая 15 г в виде воска. Воск очищали хроматографией на силикагеле (224 г). Продукт элюировали хлороформом (600 мл) с последующей смесью 8:2 (об/об) хлороформ:метанол (600 мл) и затем смесью хлороформ: метанол: Н2О (60:35:5, об/об). После удаления растворителя при пониженном давлении из фракций, содержащих требуемый продукт, остаток растворяли в хлороформе, высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении, получая 10.5 г (R)-1-додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина в виде белого воска.
ЯМР характеристика 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры 1Н ЯМР и 13С ЯМР измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
1H ЯМР
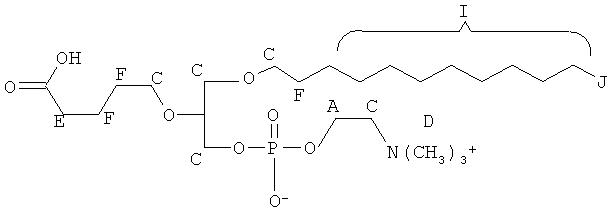
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.282 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
13С ЯМР
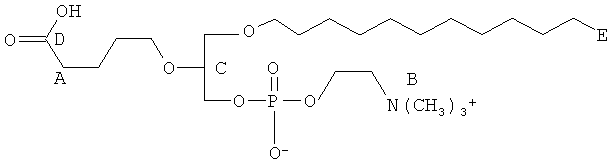
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=78.020 м.д.)
Масс-спектрометрическая характеристика 1-додецил-2-(4-карбокси) бутил-глицеро-3-фосфохолина:
Вычисленная масса для 1-додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C25H52NO8P) составила 525.3431.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=524, соответствующего депротонированному молекулярному иону [М-Н]". Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1 -додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
Продукции in vitro IL12/23 p40:
Действие CI-209 на продукцию in vitro IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.10, CI-209 ингибировало продукцию IL12/23 р40 клетками костного мозга дозозависимым образом.
Фосфорилирование тирозина:
Действие CI-209 на фосфорилирование тирозина in vitro в клетках первичных макрофагов определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.11, обработка с использованием 20 мкг/мл CI-209 индуцировала повышение уровней фосфотирозина, в то время как обработка с использованием 20 мкг/мл CI-201 вызывала снижение уровней фосфотирозина.
Токсичность CI-209:
Токсичность CI-209 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.9А и 9В, в двух экспериментах, которые были выполнены, токсичность CI-209 не наблюдалась ни при каких тестируемых дозах. Таким образом, LD50 CI-209 была выше 150 мкг/мл (286 мкМ).
ПРИМЕР 4
1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровая кислота (CI-210)
(R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин-N-глутаровую кислоту синтезировали, как описано здесь ниже, с использованием (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина в качестве исходного материала. (S)-1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровую кислоту синтезировали с помощью аналогичных методик, но с использованием (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина в качестве исходного материала.
Синтез (R)- и (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфоэтаноламина описан здесь выше в Примере 1.
1.85 г (3.33 ммоль) (R)-1-гексадецил-2-(4-метилкарбокси)бутил-уи-глицеро-3-фосфоэтаноламина растворяли в 175 мл дихлорметана и добавляли 1.39 мл триэтиламина. Этот раствор добавляли по каплям в течение 15 минут в раствор 0.42 г глутарового ангидрида в 175 мл дихлорметана. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли раствор, содержащий 20 г динатрийфосфата в 100 мл воды, и реакционную смесь энергично перемешивали в течение 20 минут. Реакционную смесь переносили в делительную воронку, фазы разделяли и водную фазу экстрагировали дважды 100 мл хлороформа. Объединенную органическую фазу высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Получали 1.16 г сырого продукта, который очищали на силикагеле (60 г). Продукт элюировали 200 мл хлороформа с последующими смесями хлороформ:метанол в соотношении 9:1, 8:2 и 7:3 (об/об), и затем 200 мл хлороформ:метанол (1:1 объемное соотношение). Растворитель из фракций, содержащих продукт, удаляли при пониженном давлении, остаток растворяли в хлороформе и высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении для получения 131.4 мг чистой (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты (CI-210) в виде грязно-белого воска.
ЯМР характеристика 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры 1Н ЯМР и 13С ЯМР измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3- фосфоэтаноламин-N-глутаровой кислоты, были обозначены следующим образом:
1H ЯМР
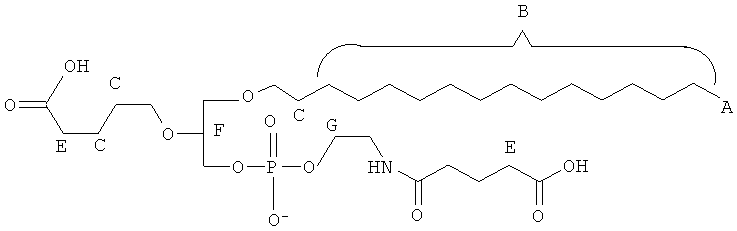
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.28 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты, были обозначены следующим образом:
13С ЯМР
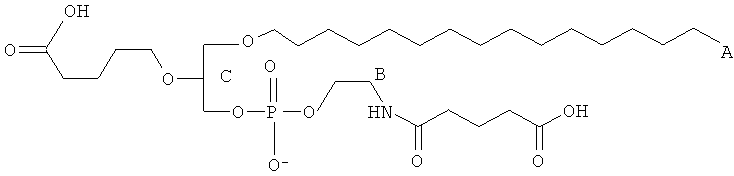
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.062 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты:
Вычисленная масса для 1-гексадецил-2(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты (C31H59NO11P) составила 652.7746.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=652, соответствующего депротонированному молекулярному иону [М-Н]'. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1 -гексадецил-2(4-карбокси)бутил-глицеро-3-фосфоэтаноламин-N-глутаровой кислоты (CI-210).
Продукция in vitro IL12/23 р40:
Действие CI-210 на продукцию in vitro IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.13, CI-210 ингибировало продукцию IL12/23 р40 клетками костного мозга дозозависимым образом.
Фосфорилирование тирозина:
Действие CI-210 на in vitro фосфорилирование тирозина в клетках первичных макрофагов определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.14, обработка с использованием 20 мкг/мл CI-210 индуцировала повышение уровней фосфотирозина, в то время как обработка с с использованием 20 мкг/мл CI-201 вызывала снижение уровней фосфотирозина.
Токсичность CI-210:Токсичность CI-210 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.15А и 15В, значительная токсичность CI-210 наблюдалась при дозе 100 мкг/мл (156.6 мкМ) или выше, и LD50 CI-210 составила приблизительно 100 мкг/мл (235 мкМ).
ПРИМЕР 5
1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-216) и 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-215)
(R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин и (R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин синтезировали, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин и (S)-1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин синтезировали с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-1-октадецил-глицерина: 20 мл (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 27 г порошкового гидроксида калия и 59 г 1-бромоктадекана перемешивали в 250 мл бензола и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшился примерно до 200 мл. Реакционную смесь затем охлаждали до комнатной температуры и перемешивали при этой температуре в течение ночи. Добавляли 200 мл воды, реакционную смесь дважды экстрагировали 200 мл диэтилового эфира, объединенную органическую фазу промывали 200 мл воды, и растворитель затем удаляли при пониженном давлении. Полученный остаток растворяли в 100 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником в течение 1 часа с последующим охлаждением до комнатной температуры и добавлением 200 мл воды. Продукт экстрагировали дважды 200 мл хлороформа, промывали последовательно 200 мл воды, 200 мл насыщенного водного раствора карбоната натрия и снова 200 мл воды. Растворитель затем удаляли при пониженном давлении, и сырой продукт кристаллизовали из 500 мл гексана для получения 39.5 г чистого (S)-1-октадецил-глицерина, который сушили в десикаторе при пониженном давлении с оксидом фосфора.
Синтез (R)-1-октадецил-3-тритил-глицерина: 39 г (113 ммоль) (S)-1-октадецил-глицерина и 40 г (137 ммоль) трифенилхлорметана добавляли в смесь 500 мл сухого THF и 130 мл сухого ацетонитрила. Добавляли 32 мл сухого триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь затем охлаждали до комнатной температуры, заливали на лед (1 кг), переносили в делительную воронку и дважды экстрагировали 200 мл диэтилового эфира. Органическую фазу промывали последовательно 200 мл воды, дважды 100 мл разбавленной (1.5%) H2SO4, 200 мл воды, 200 мл концентрированного водного раствора бикарбоната натрия и снова 200 мл воды. Раствор затем высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Остаток, коричневое масло, растворяли в 250 мл этилацетата и охлаждали до -20°С в течение ночи. Смесь центрифугировали (3500 оборотов в минуту) при температуре -10°С (3500 об/мин) и маточную жидкость сливали. Оставшееся твердое вещество растворяли в гексане и охлаждали (5±3°С) в течение ночи. Фильтрация осадка позволила получить 50 г чистого (К)-1-октадецил-3-тритил-глицерина.
Синтез (R)-1-октадецил-2(5'-гексенил)-3-тритил-глицерина: 50 г (89.2 ммоль) (R)-1-октадецил-3-тритил-глицерина и 18 г (102 ммоль) 5-гексенил-1-метан сульфоната растворяли в 150 мл бензола. Добавляли 20 г порошкового КОН и реакционную смесь перемешивали и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 50 мл. Реакционную смесь охлаждали до комнатной температуры и добавляли 200 мл воды. Смесь трижды экстрагировали 200 мл диэтилового эфира, объединенную органическую фазу трижды промывали 200 мл воды и растворитель удаляли при пониженном давлении, получая 50 г (R)-1-октадецил-2-(5'-гексенил)-3-тритил-глицерина в виде оранжевого масла.
Синтез (S)-1-октадецил-2-(4-карбокси)бутил-глицерина: 145 г NaIO4 растворяли в 500 мл воды. В этот раствор добавляли 14 г K2CO3 и 2.4 г KMnO4, и суспензию нагревали до температуры 40°С. Раствор 50 г (R)-1-октадецил-2-(5'-гексенил)-3-тритил-глицерина в 500 мл трет-бутанола добавляли по каплям в течение 1.5 часов, и смесь нагревали еще в течение 4 часов. Добавляли дополнительное количество раствора KMnO4 для сохранения розового цвета. Реакционную смесь охлаждали до комнатной температуры и перемешивали при этой температуре в течение ночи. Бисульфит натрия добавляли порциями до исчезновения розового и затем коричневого цвета, и появления желтого цвета реакционной смеси. После перемешивания раствора в течение 30 минут при комнатной температуре по каплям добавляли 100 мл 10% серной кислоты, раствор переносили в делительную воронку и дважды экстрагировали 200 мл гексана. Органическую фазу дважды промывали раствором, содержащим 15 г Na2S2O5 в 100 мл воды, и затем 200 мл воды. Органическую фазу концентрировали удалением примерно 500 мл растворителя при пониженном давлении. В оставшийся раствор добавляли 15 мл воды и 1.5 мл концентрированной соляной кислоты, и полученную смесь кипятили с обратным холодильником в течение 6 часов, затем охлаждали до комнатной температуры и снова концентрировали путем удаления растворителя при пониженном давлении. РН остатка доводили до 12 добавлением 100 мл воды и 10 мл 30% раствора NaOH. Осадок отфильтровывали и промывали четыре раза 10 мл воды. Фильтрат экстрагировали 100 мл смеси 1:1 (об/об) гексан:этилацетат. Водную фазу подкисляли до рН 1 добавлением 8 мл концентрированной соляной кислоты и затем экстрагировали 100 мл гексана. Сушка над безводным NaSO4, удаление растворителя при пониженном давлении и рекристаллизация в течение ночи сырого продукта из смеси 1:9 (об/об) ацетон тексан при 5±3°С позволило получить 19 г чистого N-октадецил-2-(4-карбокси)бутил-sn-глицерина в виде грязно-белого твердого вещества.
Синтез (S)-1-октадецил-2-(4-метилкарбокси) бутил-глицерина: 17 г (S)-1-октадецил-2-(4-карбокси)бутил-sn-глицерина растворяли в 100 мл метанола. Добавляли 2 мл концентрированной HCl (37%) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и в полученный остаток добавляли 100 мл воды. Смесь экстрагировали трижды 70 мл хлороформа. Объединенную органическую фазу промывали 70 мл воды, 70 мл концентрированного раствора бикарбоната натрия и снова 70 мл воды. Раствор затем высушивали над сульфатом натрия, фильтровали и выпаривали при пониженном давлении для получения 14 г (S)-1-октадецил-2-(4-метилкарбокси)бутил-глицерина в виде белого воска.
Синтез (R)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 1 г (S)-1-октадецил-2-(4-метилкарбокси)бутил-глицерина (который был высушен азеотропной перегонкой с бензолом) и 7 мл триэтиламина растворяли в 60 мл THF. Раствор добавляли по каплям в течение 30 минут в ледяной раствор, содержащий 4.3 мл POCl3 в 40 мл THF. Перемешивание продолжали еще в течение 15 минут при охлаждении, и еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и затем добавляли по каплям в течение 30 минут раствор 3 мл этаноламина и 13 мл триэтиламина в 60 мл THF. Перемешивание продолжали в течение 15 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси 72 мл уксусной кислоты и 30 мл воды, и нагревали до температуры 70°С в течение 1 часа. Смесь трижды экстрагировали 80 мл хлороформа и дважды промывали 100 мл воды. Удаление растворителя при пониженном давлении позволило получить 10 г (R)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламинав виде желтого масла.
Синтез (R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-216): 3 г (S)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в 100 мл смеси 8:2 (об/об) метанол:10% водный раствор гидроксида натрия и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакционной смеси доводили приблизительно до 4 добавлением муравьиной кислоты. Добавляли 100 мл воды и 100 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель затем удаляли при пониженном давлении. Полученный остаток (3 г) очищали хроматографией на силикагеле (55 г). Смесь хлороформа и гексана с последующими смесями хлороформа и метанола, и в конце смеси хлороформа, метанола и воды использовали для элюирования 760 мг (R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина из колонки.
ЯМР характеристика 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры 1Н ЯМР и 13С ЯМР измеряли при 600 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1H соответствующие структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
1Н ЯМР
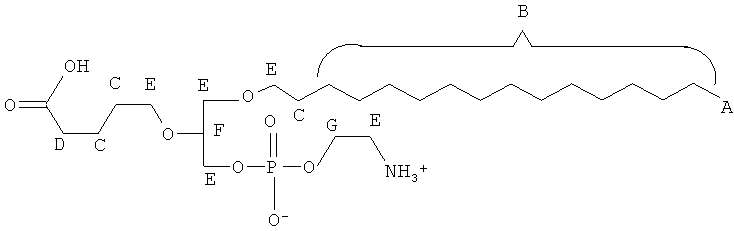
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.341 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
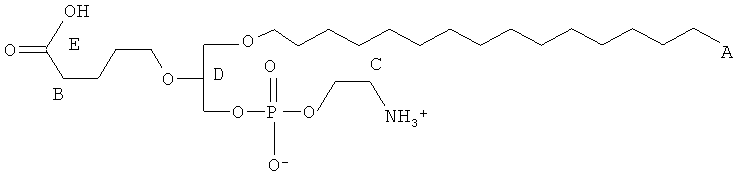
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.281 м.д.)
Масс-спектрометрическая характеристика 1-октадецил-2-(4-карбокси) бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-октадецил-2(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C28H58NO8P) составила 567.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=566, соответствующего депротонированному молекулярному иону [М-Н]-. Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=590, соответствующего катионированному молекулярному иону [M+Na]+. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (CI-216).
Синтез (R)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина: 6 г (R)-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси 50 мл изопропанола и 18 мл дихлорметана, и смесь нагревали до температуры в диапазоне 35-40°С. Добавляли по каплям раствор, содержащий 7.5 г карбоната калия в 10 мл воды, при поддержании температуры 35-40°С. Затем при 40°С по каплям добавляли раствор, содержащий 5 мл диметилсульфата в 10 мл изопропанола. Реакцию поддерживали при 40°С в течение 2 часов и затем при комнатной температуре в течение ночи. Добавляли 100 мл воды с последующей экстракцией смеси трижды 100 мл дихлорметана. Органическую фазу промывали 100 мл воды и растворитель удаляли при пониженном давлении для получения 6 г (R)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина в виде желтого масла.
Синтез (R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-215): 6 г (R)-1-октадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина растворяли в 100 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия, и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакционной смеси доводили приблизительно до 4 добавлением муравьиной кислоты. Затем добавляли 100 мл воды и 100 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель затем удаляли при пониженном давлении. Полученный остаток (5.3 г) очищали хроматографией на силикагеле (112 г). Смесь хлороформа и гексана с последующими смесями хлороформа и метанола, и в конце смесями хлороформа, метанола и воды использовали для элюирования продукта. Удаление растворителя при пониженном давлении из фракций, содержащих продукт, позволило получить 2.8 г (R)-1-октадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин (CI-215) в виде белого воска.
ЯМР характеристика 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры 1Н ЯМР и 13С ЯМР измеряли при 600 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
1H ЯМР
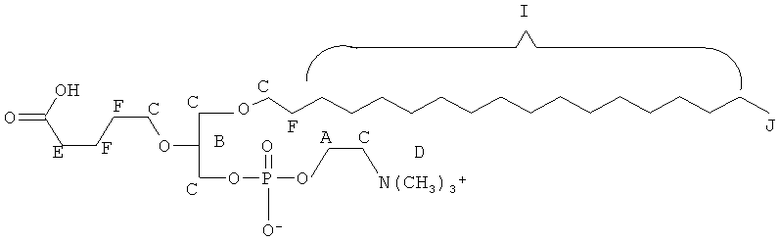
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.343 м.д.)
Наблюдаемые пики С, соответствующие структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
13С ЯМР
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.285 м.д.)
Масс-спектрометрическая характеристика 1-октадецил-2-(4-карбокси) бутил-глицеро-3-фосфохолина:
Вычисленная масса для 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C31H64NO8P) составила 609.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=610, соответствующего протонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-октадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (CI-215).
Продукция in vitro IL12/23 р40:Действие CI-216 на продукцию т vitro IL12/23 р40 определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.16, 20 мкг/мл CI-216 ингибировало продукцию IL12/23 р40 клетками костного мозга.
Фосфорилирование тирозина:
Действие CI-215 и CI-216 на in vitro фосфорилирование тирозина в клетках первичных макрофагов определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.17, обработка с использованием 10 мкг/мл (17 мкМ) CI-215 индуцировала повышение уровней фосфотирозина, в то время как обработка с использованием 20 мкг/мл (34 мкМ) CI-215 вызывала снижение уровней фосфотирозина.
Аналогично, как показано на Фиг.18, обработка с использованием 10 мкг/мл (15 мкМ) CI-216 привела к индуцированию фосфорилирования тирозина, в то время как воздействие 20 мкг/мл (30 мкМ) CI-216 вызвало снижение уровней фосфотирозина. Эти изменения были аналогичны эффекту, вызванному 10 мкг/мл (17 мкМ) и 20 мкг/мл (34 мкМ) положительного контроля CI-201.
ПРИМЕР 6
1-гексадецил-2-(3-карбонси)пропил-глицеро-3-фосфоэтаноламин (CI-206) и 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолин (CI-205)
(R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфоэтаноламин и (R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфохолин были синтезированы, как описано здесь далее, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламин и (S)-1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолин были синтезированы с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (R)-1-гексадецил-3-тритил-глицерина: (R)-1-гексадецил-3-тритил-глицерин был приготовлен, как описано в Примере 1, сначала приготовлением (S)-1-гексадецил-глицерина с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола.
Синтез (R)-1-гексадецил-2-(4'-пентенил)-3-тритил-глицерина: 7.35 г (R)-1-гексадецил-3-тритил-глицерина и 1.87 мл 5-бром-1-пентена растворяли в 150 мл бензола. Добавляли 3 г порошкового КОН и реакционную смесь перемешивали и кипятили в течение 10 часов с азеотропным удалением воды. Бензол перегоняли до почти сухого состояния. Реакционную смесь охлаждали до комнатной температуры. Добавляли 100 мл диэтилового эфира и смесь промывали водой (3×50 мл), и сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении. Полученный остаток (7.8 г) растворяли в 20 мл гексана и охлаждали до 4°С в течение ночи. Осажденный побочный продукт отфильтровывали и растворитель удаляли при пониженном давлении для получения 7.75 г продукта в виде желтого масла.
Синтез (К)-1-гексадецил-2-(3-карбокси)пропил-3-тритил-глицерина: 7.75 г (R)-гексадецил-2-(4'-пентенил)-3-тритил-глицерина растворяли в 280 мл t-бутанола. Добавляли раствор, содержащий 3.2 г карбоната калия в 90 мл воды. Затем добавляли 50 мл раствора, содержащего 34 г периодата натрия в 250 мл воды, и 2 мл раствора, содержащего 470 мг перманганата калия в 10 мл воды. Смесь перемешивали, и оставшиеся части раствора периодата добавляли в течение 10 минут. Добавляли дополнительное количество раствора перманганата, требуемое для сохранения розового цвета. Смесь нагревали до 40°С в течение 4,5 часов, охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли бисульфит натрия порциями, и цвет смеси становился коричневым, затем твердые вещества исчезали и цвет становился желтым. Смесь затем перемешивали в течение 30 минут и по каплям добавляли 25 мл 10% раствора серной кислоты. Раствор экстрагировали диэтиловым эфиром (3 х 100 мл). Объединенные органические фазы промывали 50 мл воды, дважды 20 мл раствора бисульфита натрия (приготовленного из 5 грамм бисульфита натрия в 20 мл воды) и дважды 50 мл воды, затем высушивали над сульфатом натрия, фильтровали и выпаривали при пониженном давлении для получения 11 г продукта в виде желтого воска.
Синтез (S)-1-гексадецил-2-(3-карбокси)пропил-глицерина: 11 г 1-гексадецил-2-(3-карбокси)пропил-3-тритил-глицерина растворяли в 100 мл муравьиной кислоты и смесь перемешивали при комнатной температуре в течение 2 часов. Муравьиную кислоту затем удаляли при пониженном давлении. Добавляли 100 мл раствора 1:1 толуол:гексан и смесь перемешивали при комнатной температуре. Раствор экстрагировали дважды 100 мл смеси 8:2 (об/об) метанол: 10% водный раствор NaOH. Щелочной раствор подкисляли однозамещенным фосфорнокислым натрием до установления рН в диапазоне 4-5. Добавляли 100 мл диэтилового эфира и 100 мл воды, фазы разделяли и водную фазу дважды промывали 100 мл диэтилового эфира. Объединенную органическую фазу затем промывали 100 мл воды и 100 мл солевого раствора, высушивали над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении для получения 8 г продукта. Продукт кристаллизовали из смеси 1:9 ацетонтексан для получения 2.4 г (S)-1-гексадецил-2-(3-карбокси)пропил-глицерина.
Синтез (5)-1-гексадецил-2-(3-метилкарбокси)пропил-глицерина: 2.4 г (S)-1-гексадецил-2-(3-карбокси)пропил-глицерина растворяли в 50 мл метанола. Добавляли 1 мл концентрированной HCl и смесь перемешивали при комнатной температуре в течение 6 часов, и затем выдерживали при 4°С в течение ночи. Растворитель удаляли при пониженном давлении, добавляли 50 мл воды и смесь экстрагировали трижды 50 мл хлороформа. Органическую фазу промывали 50 мл воды, 50 мл концентрированного бикарбоната натрия и снова 50 мл воды. Смесь высушивали над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении для получения 2.9 г продукта, который высушивали при пониженном давлении пентоксидом фосфора.
Синтез (R)-1-гексадецил-2-(3-метилкарбокси)пропил-5п-глицеро-3-фосфоэтаноламина: 2.9 г (S)-1-гексадецил-2-(3-метилкарбокси)пропил-глицерина и 3 мл триэтиламина растворяли в 30 мл THF. Этот раствор добавляли по каплям в течение 15 минут в ледяной раствор, содержащий 2 мл POCl3 в 20 мл THF. Перемешивание продолжали еще в течение 10 минут при охлаждении и еще в течение 45 минут при комнатной температуре. Раствор, содержащий 1.3 мл этаноламина и 6 мл триэтиламина в 50 мл THF, добавляли по каплям в течение 15 минут в ледяную реакционную смесь. Перемешивание продолжали в течение 10 минут при 0°С и затем в течение ночи при комнатной температуре. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Остаток растворяли в смеси 24 мл уксусной кислоты и 10 мл воды, и нагревали до 70°С в течение 1 часа. Смесь концентрировали трижды 50 мл хлороформа и промывали дважды 50 мл воды. Удаление растворителя при пониженном давлении позволило получить 3.8 г (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфоэтаноламина в виде коричневого масла.
Синтез (R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфоэтаноламина (CI-206) 0.8 г (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфоэтаноламина растворяли в 20 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия. Добавляли 10% раствор гидроксида натрия и смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили до 4-5 добавлением однозамещенного фосфорнокислого натрия. Добавляли 50 мл воды и 50 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении для получения 684 мг сырого продукта в виде остатка. Этот остаток очищали хроматографией на силикагеле (30 г). Продукт элюировали смесью хлороформ:метанол:вода при объемном соотношении 60:35:5. Раствор удаляли при пониженном давлении, остаток растворяли в хлороформе и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 314 мг (R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфоэтаноламина в виде белого воска, который высушивали при пониженном давлении пентоксидом фосфора.
ЯМР характеристика 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3). Спектры 1Н ЯМР и 13С ЯМР измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1 -гексадецил-2-(3 -карбокси)пропил-глицеро-3 -фосфоэтаноламина (CI-206) и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре CI-206, были обозначены следующим образом:
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.338 м.д.)
13/Наблюдаемые пики С, соответствующие структуре CI-206, были обозначены следующим образом:
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.256 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(3-карбокси) пропил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-гексадецил-2(3-карбокси)пропил-глицеро-3-фосфоэтаноламина (C25H52NO8P) составила 525.6560.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=524, соответствующего депротонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2(3-карбокси)пропил-глицеро-3-фосфоэтаноламина (CI-206).
Синтез (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфохолина: 2.8 г (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси 50 мл изопропанола и 18 мл дихлорметана. Добавляли по каплям раствор, содержащий 3.7 г карбоната калия в 10 мл воды, при поддержании температуры реакционной смеси в диапазоне 35-40°С. По каплям добавляли раствор, содержащий 2.52 мл диметилсульфата в 10 мл изопропанола, при 40°С в течение 5 минут. Реакционную смесь затем выдерживали при 40°С в течение 90 минут, охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли воду и смесь экстрагировали трижды 50 мл хлороформа. Органическую фазу промывали 50 мл воды и растворитель удаляли при пониженном давлении для получения 3 г (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфохолина в виде коричневого масла.
Синтез (R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфохолина (CI-205) 3 г (R)-1-гексадецил-2-(3-метилкарбокси)пропил-sn-глицеро-3-фосфохолина растворяли в 50 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия и смесь перемешивали при комнатной температуре в течение ночи. РН реакции доводили до 4-5 добавлением однозамещенного фосфорнокислого натрия. Добавляли 50 мл воды и 50 мл хлороформа, и полученный раствор переносили в делительную воронку. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении для получения 2.3 г сырого продукта в виде остатка. Полученный остаток очищали хроматографией на силикагеле (110 г). Продукт элюировали смесью хлороформ:метанол:вода при объемном соотношении 60:35:5. После удаления растворителя при пониженном давлении остаток растворяли в хлороформе и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 677 мг (R)-1-гексадецил-2-(3-карбокси)пропил-sn-глицеро-3-фосфохолина (CI-205) в виде белого воска, который высушивали при пониженном давлении пентоксидом фосфора.
ЯМР характеристика 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3). Спектры 1Н ЯМР и 13С ЯМР измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфохолина, были обозначены следующим образом:
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.338 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
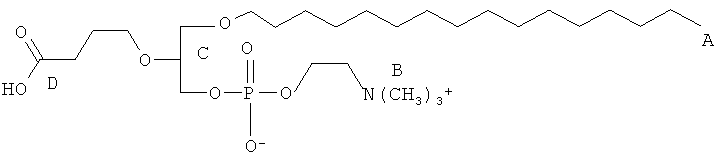
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.231 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(3-карбокси)пропил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-гексадецил-2(3-карбокси)пропил-глицеро-3-фосфоэтаноламина (C28H58NO8P) составила 567.7358.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESF-MS), показал наличие молекулярного иона с m/z=566, соответствующего депротонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2(3-карбокси)пропил-глицеро-3-фосфохолина.
Фосфорилирование тирозина:
Действие CI-205 и CI-206 на т vitro фосфорилирование тирозина в клетках первичных макрофагов определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.19, обработка 20 мкг/мл CI-206 вызвала снижение уровней фосфотирозина.
Аналогично, как показано на Фиг.20, обработка 20 мкг/мл CI-205 вызывала снижение уровней фосфотирозина, так же как при обработке 20 мкг/мл положительного контроля, CI-201.
Токсичность CI-205 и CI-206:
Токсичности С1-205 и CI-206 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.21А и 21В, значительная токсичность CI-206 была обнаружена при дозах 50 мкг/мл или выше, с LD50 CI-206 между 50 и 100 мкг/мл.
Как показано на Фиг.22А и 22В, значительная токсичность CI-205 была обнаружена при дозе 100 мкг/мл в двух экспериментах и при дозе 20-50 мкг/мл только в одном эксперименте с LD50 С1-205, находящейся между 50 и 100 мкг/мл.
ПРИМЕР 7
1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-207) и 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин
(R)-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин и (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин были синтезированы, как описано здесь ниже, с использованием (S)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолин и (S)-1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин были синтезированы с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-1-октил-глицерина: 21 мл (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 29 г порошкового гидроксида калия и 32 мл 1-бромоктана перемешивали в 150 мл бензола и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 100 мл. Реакционную смесь затем охлаждали до комнатной температуры и добавляли 200 мл воды. Реакционную смесь затем экстрагировали трижды 150 мл диэтилового эфира, объединенную органическую фазу промывали 100 мл воды, и растворитель затем удаляли при пониженном давлении. Полученный остаток растворяли в 100 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником в течение 2 часов с последующим охлаждением до комнатной температуры и добавлением 100 мл воды. Продукт экстрагировали трижды 150 мл хлороформа, промывали последовательно 150 мл воды, 150 мл насыщенного водного раствора бикарбоната натрия и снова 100 мл воды. Растворитель высушивали над безводным Na2SO4, фильтровали и удаляли при пониженном давлении с получением 34 г (S)-1-октил-глицерина.
Синтез (R)-1-октил-3-тритил-глицерина: 34 г (S)-1-октил-глицерина и 61 г трифенилхлорметана добавляли в смесь, состоящую из 500 мл сухого THF и 130 мл сухого ацетонитрила. Добавляли 46 мл сухого триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь затем охлаждали до комнатной температуры и заливали на лед (1 кг). Смесь переносили в делительную воронку и экстрагировали трижды 200 мл диэтилового эфира. Органическую фазу промывали последовательно 150 мл воды, дважды 100 мл разбавленной (1.5%) H2SO4, 200 мл воды, 200 мл концентрированного водного раствора бикарбоната натрия и снова 200 мл воды. Раствор затем высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Полученный остаток, 80 г коричневого масла, растворяли в 500 мл горячего гексана и выдерживали при температуре 4°С в течение ночи. Осадок отфильтровывали, и растворитель из фильтрата удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле. Полученный чистый (R)-1-октил-3-тритил-глицерин элюировали смесью хлороформа с 10% гексана, хлороформа с 5% гексана с последующей смесью хлороформа с этилацетатом (5% и 10%). Выход составил 73%.
Синтез (R)-1-октил-2-(5'-гексенил)-3-тритилглицерин: 18.7 г (R)-1-октил-3-тритил-глицерина, 5.5 г 6-бром-1-гексена и 22 г порошкового гидроксида калия перемешивали в 100 мл бензола и кипятили в течение 9 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 30 мл. Реакционную смесь охлаждали до комнатной температуры и добавляли 100 мл воды. Полученную смесь переносили в делительную воронку и экстрагировали диэтиловым эфиром. Объединенную органическую фазу дважды промывали 200 мл воды и растворитель удаляли при пониженном давлении, что позволило получить 20.2 г (R)-1-октил-2-(5'-гексенил)-3-тритилглицерина.
Синтез (S)-1-октил-2-(5'-гексенил)-sn-глицерина: 20.2 г (R)-1-октил-2-(5'-гексенил)-3-тритилглицерина растворяли в 100 мл метанола, добавляли 10 мл концентрированной соляной кислоты (32%) и полученную реакционную смесь кипятили с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли 100 мл воды и раствор экстрагировали дважды 100 мл диэтилового эфира. Объединенную органическую фазу промывали 100 мл воды, 100 мл насыщенного водного раствора бикарбоната натрия и снова 100 мл воды. Растворитель удаляли при пониженном давлении. Полученный остаток (20.1 г) растворяли в 250 мл гексана и полученный раствор выдерживали при температуре 4°С в течение 96 часов, во время которого происходило осаждение почти всего трифенилкарбинола. После фильтрации и удаления растворителя из фильтрата оставшийся продукт (12 г) очищали хроматографией на силикагеле (91.4 г). Чистый (S)-1-октил-2-(5'-гексенил)-sn-глицерин (5.7 г) элюировали хлороформом с последующей смесью хлороформа с 5% ацетона. Выход составил 52%.
Синтез (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина: 4.9 г (S)-1-октил-2-(5'-гексенил)-sn-глицерина (который был высушен в десикаторе над P2O5) и 2.65 мл триэтиламина растворяли в 40 мл THF. Этот раствор добавляли по каплям в течение 30 минут в ледяной раствор, содержащий 1.4 мл POCl3 в 20 мл THF, при перемешивании. Перемешивание продолжали еще 30 минут при охлаждении и еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и добавляли по каплям раствор 1.1 мл этаноламина и 2.8 мл триэтиламина в 30 мл THF в течение 15 минут при перемешивании. Перемешивание продолжали еще 35 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали и твердую фазу промывали дважды 15 мл THF, и растворитель из фильтрата удаляли при пониженном давлении. Полученный остаток (5.6 г) растворяли в смеси, состоящей из 36 мл уксусной кислоты и 15 мл воды, и нагревали до температуры 70°С в течение 1 часа. После охлаждения до комнатной температуры раствор переносили в делительную воронку и экстрагировали дважды смесью 2:1 (об/об) хлороформ:метанол и отмывали разбавленным раствором бикарбоната натрия, и растворитель затем удаляли при пониженном давлении, с получением 4.6 г сырого (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина. Сырой продукт очищали хроматографией на силикагеле (59 г). 2.6 г чистого продукта элюировали хлороформом с последующими смесями хлороформа с 10%-40% метанола. Выход составил 75.9%.
Синтез (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина: 2.1 г (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина растворяли в растворе 100 мл этанола с 6 г карбоната калия. Добавляли 8 мл диметилсульфата и реакционную смесь нагревали до температуры 40°С в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли 100 мл воды. Смесь затем экстрагировали дважды 100 мл хлороформа. Растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе и высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Сырой продукт очищали хроматографией на силикагеле (59 г). 2.0 г чистого (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина элюировали хлороформом с последующими смесями хлороформа с 20%-60% метанола. Выход составил 86.4%.
Синтез (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-207): Раствор, содержащий 172 мг бикарбоната натрия в 17 мл воды, добавляли в раствор, содержащий 700 мг (R)-1-октил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина в 28 мл воды. Затем добавляли раствор, содержащий 3.0 г периодата натрия в 28 мл воды. Раствор, содержащий 40 мг перманганата калия в 12 мл воды, помещали в капельную воронку и добавляли по каплям в реакционную смесь в количестве, требуемом для сохранения розового цвета реакционной смеси. Во время реакции было добавлено около половины раствора перманганата. После перемешивания при комнатной температуре в течение 3 часов добавляли 6 г однозамещенного фосфорнокислого натрия, и реакционную смесь экстрагировали трижды 50 мл смеси 2:1 (об/об) хлороформ:метанол. Объединенную органическую фазу высушивали над безводным Na2SO4 и растворитель удаляли при пониженном давлении с получением 360 мг сырого продукта. Сырой продукт очищали хроматографией на силикагеле (12.23 г). 119 мг чистого (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина элюировали хлороформом с последующей смесью хлороформа с 10%-60% метанола.
Аналогично, синтез с использованием (S)-1-октил-2-(5'-гексенил)-3-тритил-глицерина, приготовленного как описано здесь выше, выполняли следующим образом:
Синтез (S)-1-октил-2-(4-карбокси)бутил-sn-глицерина: 11 г NaIO4 растворяли в 300 мл воды. В этот раствор добавляли 9 г NaHCO3 и 1.26 г KMnO4, и суспензию нагревали до 40°С. Раствор, содержащий 21 г (S)-1-октил-2-(5'-гексенил)-3-тритил-глицерина в 300 мл трет-бутанола, добавляли по каплям в реакционную смесь в течение 1 часа, и смесь нагревали еще в течение 3 часов. Дополнительное количество раствора КМп04 добавляли для сохранения розового цвета. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит, и целит промывали wpew-бутанолом. Добавляли по каплям 100 мл 10% раствора серной кислоты и раствор переносили в делительную воронку и экстрагировали трижды 200 мл гексана. Органическую фазу промывали раствором, содержащим 20 г Na2S2O5 в 100 мл воды, и затем 200 мл воды. Органическую фазу концентрировали путем удаления растворителя при пониженном давлении до уменьшения объема примерно до 150 мл. В оставшийся раствор добавляли 15 мл воды и 2 мл концентрированной НС1, и полученную смесь кипятили с обратным холодильником в течение 6 часов, затем охлаждали до комнатной температуры и снова концентрировали путем удаления растворителя при пониженном давлении. РН остатка доводили до 12 добавлением 100 воды и 10 мл 30% раствора NaOH. Осадок отфильтровывали и промывали четыре раза 20 мл воды. Фильтрат экстрагировали 100 мл смеси 1:1 (об/об) гексан:этилацетат. Водную фазу подкисляли до рН 1 добавлением 10 мл концентрированной HCl и экстрагировали трижды 100 мл гексана. Сушка над безводным NaSO4 и удаление растворителя при пониженном давлении позволило получить 7.4 г сырого продукта в виде желтого масла. Сырой продукт очищали хроматографией на силикагеле (100 г). 4.8 г чистого(S)-1-октил-2-(4-карбокси)бутил-sn-глицерина элюировали хлороформом с последующей смесью хлороформа с 5%-50% этилацетата. Выход составил 39.7%.
Синтез (S)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицерин: 1.14 г (S)-1-октил-2-(4-карбокси)бутил-sn-глицерина растворяли в 20 мл дихлорметана. Добавляли 748 мг дифенилдиазометана, приготовленного как описано в J. Organic Chem. (1959) 24: 560-561, и темно-красную реакционную смесь перемешивали при комнатной температуре в течение примерно 3 часов до тех пор, пока не происходило изменение цвета раствора. Растворитель удаляли при пониженном давлении. Остаток (1.9 г) очищали хроматографией на силикагеле (43 г). 1.08 г чистого (S)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицерина элюировали хлороформом с последующей смесью хлороформа с 5%-20% этилацетата. Выход составил 61.4%.
Синтез (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 1 г (S)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицерина (который сушили азеотропной перегонкой с бензолом) и 0.885 мл триэтиламина растворяли в 30 мл THF. Этот раствор по каплям добавляли в течение 15 минут в ледяной раствор, содержащий 0.235 мл POCl3 в 20 мл THF, при перемешивании. Перемешивание продолжали еще 15 минут при охлаждении и еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и затем по каплям добавляли раствор 0.154 мл этаноламина и 0.885 мл триэтиламина в 50 мл THF в течение 15 минут при перемешивании. Перемешивание продолжали в течение 15 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси 24 мл уксусной кислоты и 10 мл воды, и нагревали до 70°С в течение 1 часа. Смесь экстрагировали трижды 80 мл хлороформа и дважды промывали 50 мл воды. Удаление растворителя при пониженном давлении позволило получить 1.12 г (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-уи-глицеро-3-фосфоэтаноламина в виде желтого масла.
Синтез (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфохолина: 1.12 г (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси 65 мл метанола и 18 мл дихлорметана, и смесь нагревали до температуры в диапазоне 35-40°С. По каплям добавляли раствор, содержащий 1.3 г карбоната калия в 10 мл воды, при поддержании температуры смеси 35-40°С. По каплям добавляли раствор, содержащий 7.2 мл диметилсульфата в 10 мл метанола, при 40°С. Реакционную смесь выдерживали при температуре 40°С в течение 2 часов и затем при комнатной температуре в течение ночи. Добавляли 50 мл воды, и смесь затем экстрагировали трижды 50 мл хлороформа. Органическую фазу промывали 50 мл воды и растворитель удаляли при пониженном давлении для получения 1 г (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфохолинав виде желтого масла.
Синтез (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-207): Газообразную HCl пропускали в течение 90 минут через ледяной раствор 1 г (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфохолина в 40 мл хлороформа. Полученный раствор перемешивали в ледяной ванне еще в течение 2 часов. Затем РН реакционной смеси доводили примерно до 6 добавлением однозамещенного фосфорнокислого натрия. Добавляли 50 мл воды и смесь экстрагировали трижды 60 мл хлороформа. Объединенную органическую фазу промывали 60 мл воды и растворитель удаляли при пониженном давлении, получая 0.370 г (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина. Выход составил 50.2%.
Синтез (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина:
Газообразную HCl пропускали через ледяной раствор, содержащий 5 г (R)-1-октил-2-(4-бензгидрилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина, приготовленного как описано здесь выше, в 40 мл хлороформа, в течение 90 минут. После завершения добавления HCl реакционную смесь перемешивали в ледяной ванне еще в течение 2 часов. РН реакционной смеси доводили примерно до 6 добавлением водного раствора двузамещенного фосфорнокислого натрия. Смесь экстрагировали хлороформом (3×50 мл) и объединенную органическую фазу промывали водой (100 мл). Растворитель удаляли при пониженном давлении с получением 3.5 г коричневого масла. Данное масло очищали хроматографией на силикагеле (68.5 г). Продукт элюировали хлороформом с последующей смесью 8:2 (об/об) хлороформ:метанол, и затем смесью 700:26:45 (об/об) хлороформ:метанол:Н20. После удаления растворителя при пониженном давлении из фракции, содержащей требуемый продукт, полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении с получением 150 мг (R)-1-октил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина в виде желтого воска.
ЯМР характеристика 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (CI-207):
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OO). Спектры измеряли при 300 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
1Н ЯМР
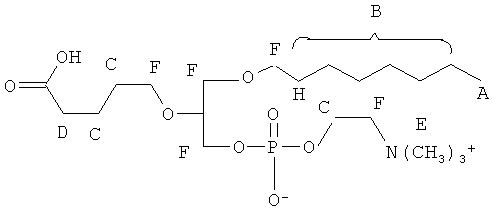
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.33 м.д.)
13C ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
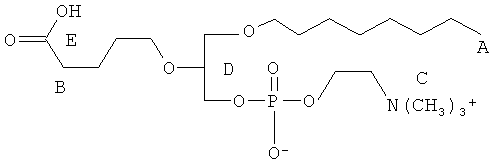
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=76.99 м.д.)
Масс-спектрометрическая характеристика 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Вычисленная масса для 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C21H44NO8P) составила 469.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI--MS), показал наличие молекулярного иона с m/z=468, соответствующего депротонированному молекулярному иону [М-Н]-. Таким образом, спектр, полученный масс-спектрометрии, соответствует химической структуре 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
ЯМР характеристика 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
1H ЯМР
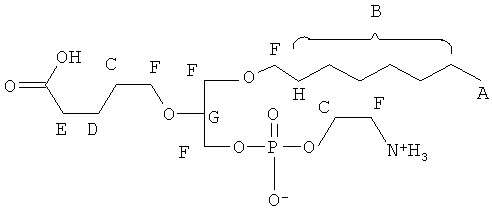
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.361 м.д.)
13С ЯМР
Наблюдаемые пики С, соответствующие структуре 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
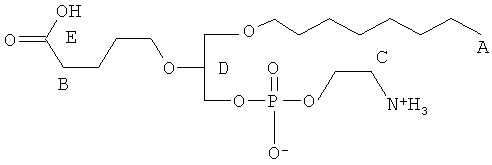
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=79.344 м.д.)
Масс-спектрометрическая характеристика 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-октил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C18H38NO8P) составила 427.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI-MS), показал наличие молекулярного иона с m/z=426, соответствующего депротонированному молекулярному иону [М-Н]-. Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=428, соответствующего протонированному молекулярному иону [М+Н]+ и иону с m/z=450, соответствующему катионированному молекулярному иону [M+Na]+. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1 -октил-2-(4-карбокси)бутил-глицеро-3 -фосфоэтаноламина.
ПРИМЕР 8
1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолин (CI-208)
(R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолин был синтезирован, как описано здесь ниже, из (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина. (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолин был синтезирован из (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицерина с использованием таких же методик.
Синтез (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина и (R)-1-гексадецил-2-(4-метилкарбокси)бутил-глицерина описан в Примере 1.
Синтез (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина (CI-208): Раствор, содержащий 8.60 г (19.97 ммоль) (S)-1-гексадецил-2-(4-метилкарбокси)бутил-глицерина (приготовленного как описано в Примере 1) и 2.63 г (26 ммоль) триэтиламина в 500 мл THF, добавляли по каплям в течение 25 минут в ледяной раствор, содержащий 3.90 г (26 ммоль) POCl3 в 100 мл THF. Перемешивание продолжали еще в течение 10 минут в ледяной ванне и еще в течение 45 минут при комнатной температуре. Раствор 1.6 мл этаноламина и 6.4 мл триэтиламина в 500 мл THF добавляли по каплям при энергичном перемешивании в ледяную реакционную смесь. Перемешивание продолжали еще в течение 10 минут в ледяной ванне и продолжали в течение ночи при комнатной температуре. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Остаток растворяли в смеси 24 мл уксусной кислоты и 100 мл воды, и нагревали до 70°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и экстрагировали дважды 250 мл дихлорметана. Растворитель затем удаляли при пониженном давлении. Остаток растворяли в смеси 500 мл изопропанола и 180 мл дихлорметана. Раствор, содержащий 50 г карбоната калия в 100 мл воды, добавляли таким образом, чтобы получить рН выше 11. Раствор выдерживали при температуре в диапазоне 35-40°С при добавлении по каплям 11.15 г метилтозилата в 100 мл изопропанола в течение 45 минут. Еще через 90 минут смесь подкисляли соляной кислотой. Добавляли 100 мл воды и 550 мл дихлорметана, и фазы разделяли. Органическую фазу промывали 100 мл воды, и растворитель удаляли при пониженном давлении. Сырой продукт очищали хроматографией на колонке с силикагелем. 11.0 г (18.46 ммоль) (R)-1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина элюировали хлороформом с последующей смесью хлороформа, метанола и воды. Выход составил 92.45%.
ЯМР характеристика 1-гексадецил-2-(4-метилкарбокси)бутип-глицеро-3-фосфохолина (CI-208):
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 300 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
1Н ЯМР
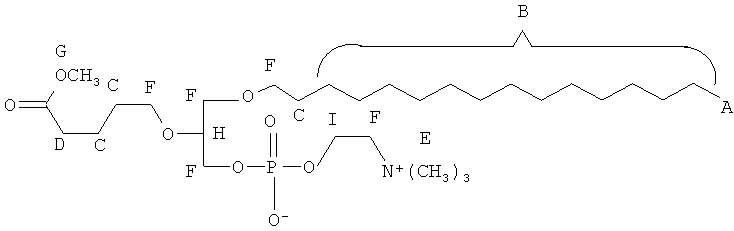
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.27 м.д.)
13С ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
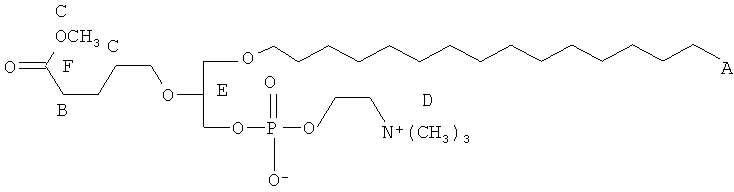
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.03 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина (CI-208).
Вычисленная масса для 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина (C30H62NO8P) составила 595.79.
Масс-спектр, полученный методом бомбардировки ускоренными атомами (FAB+), показал наличие молекулярного иона с m/z=596.324, соответствующего протонированному молекулярному иону [М+Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(4-метилкарбокси)бутил-глицеро-3-фосфохолина.
Фосфорилирование тирозина:
Действие CI-208 на in vitro фосфорилирование тирозина в клетках первичных макрофагов определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.23, обработка 20 мкг/мл CI-208 вызывала уменьшение уровней фосфотирозина, которое было более интенсивным, чем снижение, вызванное обработкой 20 мкг/мл контрольного CI-201.
Токсичность CI-208:
Токсичность CI-208 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.24А и 24В, значительная токсичность CI-208 была обнаружена при дозах 50 мкг/мл или выше, с токсичностью при дозе 20 мкг/мл, которую обнаружили только в одном из двух экспериментов. LD50 CI-208 оказалась в диапазоне от 50 до 100 мкг/мл.
ПРИМЕР 9
1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-213) и 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-214)
(R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин и (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин были синтезированы, как описано здесь ниже, с использованием (R)-(+)-3-бензилокси-1,2-пропандиола в качестве исходного материала. (S)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин и (S)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин были синтезированы с помощью таких же методик, но с использованием (S)-(-)-3-бензилокси-1,2-пропандиола в качестве исходного материала.
Синтез 1-тритил-3-бензил-sn-глицерина: 5 г (27.44 ммоль) (R)-(+)-3-бензилокси-1,2-пропандиола и 10 г (35.87 ммоль) трифенилхлорметана добавляли в 100 мл сухого THF и 25 мл сухого ацетонитрила. Добавляли 8 мл сухого триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (100 г), переносили в делительную воронку и экстрагировали трижды 100 мл диэтилового эфира. Органическую фазу последовательно промывали 100 мл воды, дважды 100 мл разбавленной (1.5%) серной кислоты, 100 мл воды, 100 мл концентрированного раствора бикарбоната натрия и снова 100 мл воды. Органическую фазу высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, получая 11 г 1-тритил-3-бензил-уп-глицерина в виде желтого масла. Выход составил 94%.
Синтез 1-тритил-2-(5'-гексенил)-3-бензил-5п-глицерина: 11 г 1-тритил-3-бензил-.уи-глицерина и 5.7 г 5-гексенил-1-метан сульфоната растворяли в 110 мл бензола. Добавляли 6 г порошкового КОН и реакционную смесь перемешивали и кипятили в течение 12 часов с азеотропным удалением воды. Реакционную смесь охлаждали до комнатной температуры и трижды промывали 100 мл воды. Органическую фазу высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Остаток растворяли в 150 мл горячего гексана, охлаждали и выдерживали при 4°С в течение ночи, при этом происходило осаждение побочных продуктов. Фильтрация и удаление растворителя из фильтрата при пониженном давлении позволила получить 13 г 1-тритил-2-(5'-гексенил)-3-бензил-sn-глицерина в виде коричневого масла.
Синтез 2-(5'-гексенил)-3-бензил-5п-глицерина: 13 г 1-тритил-2-(5'-гексенил)-3-бензил-sn-глицерина растворяли в 100 мл метанола. Добавляли 4 мл концентрированной соляной кислоты (37%) и раствор кипятили с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (100 г), переносили в делительную воронку и экстрагировали трижды 100 мл диэтилового эфира. Органическую фазу последовательно промывали 100 мл воды, 100 мл насыщенного раствора бикарбоната натрия и снова 100 мл воды. Органическую фазу высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 14.5 г сырого продукта. Сырой продукт очищали хроматографией на колонке с силикагелем (150 г). 3.17 г 2-(5'-гексенил)-3-бензил-sn-глицерина элюировали дихлорметаном с последующей смесью дихлорметана и этилацетата. Выход составил 46.8%.
Синтез 1-(15 '-карбокси)пентадецил-2-(5'-гексенил)-3-бензил-sn-глицерина: 3 г 2-(5'-гексенил)-3-бензил-sn-глицерина растворяли в 100 мл бензола. Добавляли 3 г КОН и реакционную смесь высушивали азеотропной перегонкой в течение 2 часов. В эту смесь добавляли раствор, содержащий 5.35 г трет-бутил-16-бромгексадеканоата в 100 мл бензола, по каплям в течение 3 часов. После завершения добавления смесь кипятили с обратным холодильником еще в течение 12 часов. Объем растворителя постепенно уменьшался примерно до 20 мл. Реакционную смесь охлаждали до комнатной температуры, добавляли 100 мл воды и 100 мл трет-бутанола. РН реакционной смеси доводили примерно до 1 добавлением концентрированной HCl. Смесь перемешивали при комнатной температуре в течение 2 часов и экстрагировали трижды 100 мл диэтилового эфира. Объединенную органическую фазу промывали 100 мл воды порциями до тех пор, пока рН не стало нейтральным. Сушка над безводным Na2SO4 и удаление растворителя при пониженном давлении позволила получить 6 г коричневого масла. Данное масло растворяли в 100 мл смеси 1:1 (об/об) гексан:этилацетат. Раствор экстрагировали дважды 100 мл смеси 8:2 (об/об) метанол: 10% водный раствор NaOH. Основной раствор подкисляли до рН 4-5 добавлением NaH2PO4. Добавляли 100 мл диэтилового эфира и 100 мл воды, и фазы затем разделяли. Водную фазу экстрагировали дважды 100 мл диэтилового эфира и объединяли с органической фазой. Органическую фазу промывали 100 мл воды и 100 мл солевого раствора. Сушка над безводным Na2SO4 и удаление растворителя при пониженном давлении позволила получить 5.3 г 1-(15'-карбокси)пентадецил-2-(5'-гексенил)-3-бензил-5'и-глицерина в виде желтого масла. Выход составил 90%.
Синтез 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-3-бензил-sn-глицерина: 5.3 г 1-(15'-карбокси)пентадецил-2-(5'-гексенил)-3-бензил-sn-глицерина растворяли в 130 мл трет-бутанола и добавляли раствор 1.2 г бикарбоната натрия в 40 мл воды. Затем добавляли 54 мг KMnO4 в 1 мл воды. Растворяли 20 г NaIO4 в 115 мл воды и этот раствор затем добавляли в реакционную смесь в течение 10 минут. Добавляли дополнительное количество раствора 216 мг KMnO4 в 4 мл воды, требуемое для сохранения розового цвета реакции. Перемешивание продолжали в течение 6 часов и затем реакционную смесь выдерживали при 4°С в течение ночи. Добавляли бисульфит натрия до тех пор, пока цвет реакционной смеси не становился светло-желтым. Смесь перемешивали в течение 30 минут, добавляли по каплям 25 мл 10% раствора серной кислоты и смесь экстрагировали трижды 100 мл диэтилового эфира. Объединенную органическую фазу последовательно промывали 100 воды, дважды 100 мл раствора, содержащего 25 грамм бисульфита натрия в 100 мл воды, и дважды 100 мл воды. Сушка над безводным Na2SO4 и удаление растворителя при пониженном давлении позволила получить 5.3 г сырого 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-3-бензил-sn-глицерина в виде желтого воска.
Синтез (S)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицерина:5.0 г 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-3-бензил-sn-глицерина растворяли в 50 мл метанола и добавляли 10 мл 85% муравьиной кислоты. Добавляли 5 г палладиевой черни и реакционную смесь нагревали до 60°С в атмосфере азота в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через Целит. Целит промывали метанолом и затем водой. Промывочные растворы объединяли с фильтратом, и растворитель из фильтрата удаляли при пониженном давлении. Остаток (5 г) растворяли в смеси 10 мл 10% водного раствора гидроксида натрия и 40 мл метанола, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь промывали 50 мл смеси 1:1 (об/об) гексан:толуол. РН реакционной смеси доводили до 5 добавлением раствора однозамещенного фосфорнокислого натрия, и смесь затем экстрагировали трижды 50 мл хлороформа. Органическую фазу промывали 50 мл солевого раствора, высушивали над безводным Na2SO4 и растворитель удаляли при пониженном давлении. Сырой продукт (4 г) рекристаллизовали из смеси 9:1 (об/об) гексан: ацетон с получением 3 г (S)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицерина в виде бесцветного твердого вещества. Выход составил 72%.
Синтез (S)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицерина: 3 г (S)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицерина растворяли в 50 мл метанола. Добавляли 1 мл концентрированной соляной кислоты (37%), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали примерно до 10 мл при пониженном давлении, добавляли 50 мл воды, и смесь затем экстрагировали трижды 50 мл хлороформа. Объединенную органическую фазу последовательно промывали 50 мл воды, 50 мл концентрированного раствора бикарбоната натрия и 50 мл воды. Сушка над безводным Na2SO4 и удаление растворителя при пониженном давлении позволило получить 3 г (S)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицерин в виде желтого масла.
Синтез (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 2.8 г (S)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицерина и 2.5 мл сухого триэтиламина растворяли в 30 мл THF. Этот раствор добавляли по каплям в течение 20 минут в ледяной раствор, содержащий 1.65 мл POCl3 в 20 мл THF, при перемешивании. Перемешивание продолжали еще в течение 10 минут в ледяной ванне и еще в течение 45 минут при комнатной температуре. Раствор 1.1 мл этаноламина и 5 мл триэтиламина в 50 мл THF добавляли по каплям в течение 60 минут в ледяную реакционную смесь при перемешивании. Перемешивание продолжали еще в течение 10 минут в ледяной ванне и продолжали при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси 24 мл уксусной кислоты и 100 мл воды, и нагревали до 70°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, экстрагировали трижды 50 мл хлороформа, и органическую фазу дважды промывали 50 мл воды. Удаление растворителя при пониженном давлении позволило получить 4 г сырого (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина в виде коричневого масла.
Синтез (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-214): 1.5 г (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в 50 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия, и полученную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакции доводили до 4 добавлением однозамещенного фосфорнокислого натрия и муравьиной кислоты. Добавляли 100 мл воды и 100 мл хлороформа. После экстракции фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе и высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении для получения 500 мг сырого (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-фосфоэтаноламина. Сырой продукт очищали хроматографией на силикагеле (15 г). Элюировали 100 мл хлороформа, с последующими смесями хлороформ:метанол в объеме 100 мл (объемное соотношение 9:1 и 8:2) и затем смесями хлороформ:метанол в объеме 200 мл (объемное соотношение 70:26:4 и 60:35:5). Растворитель из фракций, содержащих требуемый продукт, удаляли при пониженном давлении, остаток растворяли в хлороформе и высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении для получения 88 мг сырого (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-214) в виде желтого воска.
ЯМР характеристика 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
1H ЯМР
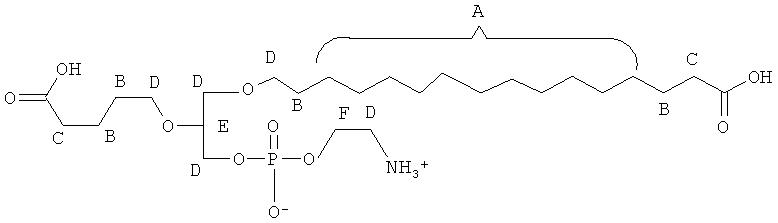
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.44 м.д.)
1C ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-(15'-карбокси) пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
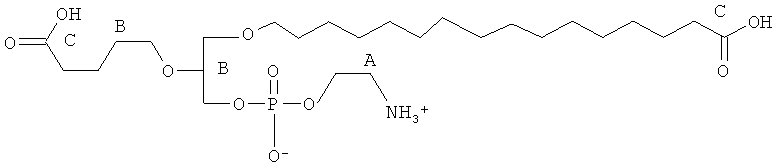
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.614 м.д.)
Масс-спектрометрическая характеристика 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C26H52NO10P) составила 569.6655.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES-MS), показал наличие молекулярного иона с m/z=568, соответствующего депротонированному молекулярному иону [М-Н]-.
К тому же, масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES+-MS), показал наличие молекулярного катиона с m/z=570, соответствующего протонированному молекулярному иону [М+Н]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина.
Синтез (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина: 1.2 г (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-уп-глицеро-3-фосфоэтаноламина растворяли в смеси 60 мл метанола и 20 мл дихлорметана. Добавляли раствор, содержащий 2 г карбоната калия в 10 мл воды, после чего раствор нагревали и выдерживали при температуре в диапазоне 35-40°С. По каплям добавляли раствор, содержащий 1.25 мл диметилсульфата в 10 мл метанола. После завершения добавления реакционную смесь перемешивали при 40°С еще в течение 90 минут, затем охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли 100 мл воды и экстрагировали 100 мл хлороформа три раза. Объединенную органическую фазу промывали 100 мл воды и растворитель удаляли при пониженном давлении, получая 900 мг (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-уп-глицеро-3-фосфохолина в виде коричневого масла.
Синтез (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-213): 1.88 г (R)-1-(15'-метилкарбокси)пентадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина растворяли в 50 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия, и полученную смесь перемешивали при комнатной температуре в течение 5 часов. РН реакции доводили до 4 добавлением однозамещенного фосфорнокислого натрия и муравьиной кислоты. Добавляли 100 мл воды и 100 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе и высушивали над безводным Na2SO4, после чего растворитель удаляли при пониженном давлении для получения 860 мг сырого (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-фосфохолина. Сырой продукт очищали хроматографией на силикагеле (20 г). Элюирование выполняли 100 мл хлороформа с последующими 100 мл смеси хлороформ:метанол (объемное соотношение 9:1 и 8:2), и затем 200 мл смеси хлороформ:метанол:вода (объемное соотношение (70:26:4 и 60:35:5). Растворитель из фракций, содержащих требуемый продукт, удаляли при пониженном давлении, остаток растворяли в хлороформе и высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении для получения 105 мг чистого (R)-1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-5 м-глицеро-3-фосфохолина (CI-213) в виде желтого воска.
ЯМР характеристика 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемого сигнала для спектральных элементов 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
1Н ЯМР
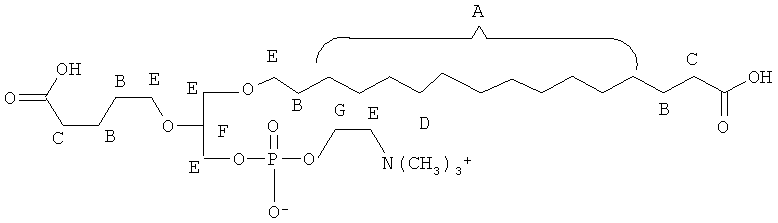
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.38 м.д.)
13С ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-(15'-карбокси) пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
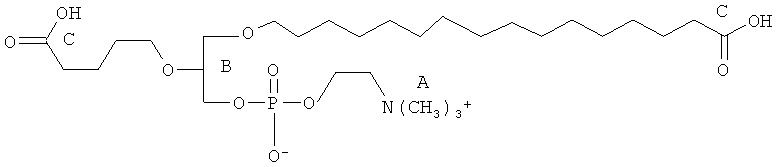
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.35 м.д.)
Масс-спектрометрическая характеристика 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C29H58NO10P) составила 611.7453.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES-MS), показал наличие молекулярного иона с m/z=610, соответствующего иону [М-Н]-.
Дополнительно, масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES+-MI) показал наличие молекулярного иона с m/z=612, соответствующего протонированному молекулярному иону [М+Н]+, сопровождаемому молекулярным катионом с m/z=634, соответствующим катионированному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-(15'-карбокси)пентадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
Токсичность CI-213 и CI-214:
Токсичности CI-213 и CI-214 оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.25А, CI-213 не четко проявлял значительную токсичность в пределах тестируемых доз (т.е. до 150 мкг/мл, 245.2 мкМ). Статистически значимое понижение в количестве клеток было обнаружено только в одном эксперименте при дозе 100 мкг/мл (163.5 мкМ).
Аналогично, как показано на Фиг.26А и 26В, CI-214 не показал четко значительной токсичности в пределах диапазона тестируемых доз (т.е. до 150 мкг/мл, 263.3 мкМ). Статистически значимое понижение в количестве клеток было обнаружено только в одном эксперименте при дозе 100 мкг/мл (175.5 мкМ).
Данные результаты показали, что LD50 CI-213 и CI-214 выше, чем 150 мкг/мл.
ПРИМЕР 10
1-Гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолин (CI-217)
(R)-1-Гексадецил-2-(2-карбокси)этил-sn-глицеро-3-фосфохолин был синтезирован, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S-1-Гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолин синтезировали с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-1-гексадецил-5п-глицерина: 40 г (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 80 г порошкового гидроксида калия и 100 г гексадецилбромида перемешивали в 350 мл бензола и кипятили в течение 7 часов а азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 50 мл. Реакционную смесь охлаждали до комнатной температуры, добавляли 300 мл ледяной воды, и смесь экстрагировали 4 раза 150 мл дихлорметана. Объединенную органическую фазу отмывали водой, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в 500 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры добавляли 200 мл воды. Продукт экстрагировали три раза 200 мл хлороформа и последовательно промывали 200 мл воды, 200 мл насыщенного водного раствора карбоната натрия и снова 200 мл воды. Растворитель удаляли при пониженном давлении, и продукт кристаллизовали из 450 мл петролейного эфира при 4°С, получая 69.93 г чистого (S)-1-гексадецил-sn-глицерина. Выход составил 73%.
Синтез 1-гексадецил-3-тритил-глицерина: 18.47 г (S)-1-гексадецил-sn-глицерина и 19 г трифенилхлорметана растворяли в смеси, состоящей из 250 мл сухого THF и 58 мл сухого ацетонитрила. Добавляли 17 мл триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (20 г) и триэтиламин (5 мл), переносили в делительную воронку и экстрагировали диэтиловым эфиром. Органическую фазу последовательно промывали 200 мл воды, дважды 200 мл разбавленной (1.5%) серной кислоты, 200 мл насыщенного водного раствора бикарбоната натрия и снова 200 мл воды. Органическую фазу высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 41.79 г сырого продукта в виде остатка. Данный остаток растворяли в 300 мл этилацетата и охлаждали при -20°С в течение ночи. Смесь центрифугировали (3500 оборотов в минуту) при -10°С и маточную жидкость сливали. Полученное твердое вещество расплавляли и растворяли в гексане, и охлаждали (5±3°С) в течение ночи. Фильтрация осадка позволила получить 18.97 г чистого (R)-1-гексадецил-3-тритил-sn-глицерина в виде грязно-белого твердого вещества.
Синтез 3-гексенил-1-метан сульфоната: Смесь 20 мл цис-3-гексен-1-ола и 40 мл сухого триэтиламина в 100 мл сухого дихлорметана охлаждали в ледяной ванне. Раствор 20 мл метансульфонил хлорида в 50 мл дихлорметана добавляли по каплям в течение 75 минут, и смесь затем выдерживали при 4°С в течение 2 часов. Добавляли лед (50 г) и смесь перемешивали при комнатной температуре в течение 30 минут, и затем дважды экстрагировали 100 мл хлороформа. Органическую фазу промывали 50 мл воды, 50 мл 10% водной серной кислоты, 50 мл 10% водного бикарбоната натрия и затем дважды 50 мл воды. Растворитель высушивали над безводным Na2SO4 и удаляли при пониженном давлении. Полученный остаток (34.90 г) очищали хроматографией на силикагеле (85.37 г). 18.83 г 3-гексенил-1-метан сульфоната получали путем элюирования смесью 1:1 (об/об) хлороформа и гексана.
Синтез (S)-1-гексадецил-2-(3'-гексенил)-глицерина: 10.60 г (R)-1-гексадецил-3-тритил-sn-глицерина растворяли в смеси, состоящей из 50 мл бензола и 50 мл петролейного эфира. Добавляли 16.65 г порошкового КОН и реакционную смесь нагревали до кипения в атмосфере азота. Раствор, содержащий 10 мл 3-гексенил-1-метан сульфоната в 50 мл бензола, и 200 мл петролейного эфира добавляли по каплям в кипящую реакционную смесь в течение 10 часов с азеотропным удалением воды. После завершения добавления перемешивание продолжали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли 200 мл воды. Смесь экстрагировали трижды 200 мл диэтилового эфира, объединенную органическую фазу трижды промывали 200 мл воды, и растворитель удаляли при пониженном давлении, получая 11.88 г сырого (R)-1-гексадецил-2-(3-гексенил)-3-тритил-sn-глицерина. 1-Гексадецил-2-(3-гексенил)-3-тритил-глицерин растворяли в 100 мл метанола, добавляли 4 мл концентрированной HCl и полученный раствор кипятили с обратным холодильником в течение 4 часов. Добавляли 100 мл воды и смесь экстрагировали четыре раза 100 мл диэтилового эфира. Объединенную органическую фазу промывали 100 мл воды и высушивали над безводным Na2SO4, после чего растворитель удаляли при пониженном давлении. Объединенный остаток растворяли в гексане и хранили при 4°С в течение ночи. Фильтрация осадка и удаление растворителя при пониженном давлении позволила получить 10.08 г сырого продукта. Этот продукт очищали хроматографией на силикагеле (95.91 г). 3.71 г продукта, (S)-1-гексадецил-2-(3'-гексенил)-sn-глицерина, элюировали смесью 1:1 (об/об) гексана и хлороформа с последующим хлороформом и затем хлороформом с 2% ацетона.
Синтез (R)-1-гексадецил-2-(3'-гексенил)-5п-глицеро-3-фосфоэтаноламина: 2.08 г (S)-1-гексадецил-2-(3'-гексенил)-$т7-глицерина (который был высушен в десикаторе над Р2О5) и 1.2 мл триэтиламина растворяли в 60 мл THF. Этот раствор добавляли по каплям в течение 55 минут в ледяной раствор, состоящий из 0.84 мл РОС1з и 0.7 мл триэтиламина в 40 мл THF, при перемешивании. Перемешивание продолжали еще в течение 10 минут при охлаждении и еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и раствор 0.55 мл этаноламина и 2.0 мл триэтиламина в 15 мл THF затем добавляли по каплям в течение 25 минут. Перемешивание продолжали в течение 30 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Добавляли еще 0.18 нл этаноламина и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали, и растворитель из фильтрата удаляли при пониженном давлении. Полученный остаток растворяли в смеси, состоящей из 48 мл уксусной кислоты и 20 мл воды, нагревали до 77°С в течение 1 часа, и охлаждали до комнатной температуры. Раствор экстрагировали трижды 100 мл смеси 2:1 (об/об) хлороформ:метанол, последовательно отмывали разбавленным раствором бикарбоната натрия и 100 мл воды, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над безводным Na2SO4 и растворитель удаляли при пониженном давлении, получая 2.57 г (R)-1-гексадецил-2-(3'-гексенил)-sn-глицеро-3-фосфоэтаноламина.
Синтез (R)-1-гексадецил-2-(3'-гексенил)-sn-глицеро-3-фосфохолина: 6.8 г карбоната калия добавляли в раствор, содержащий 2.54 г (R)-1-гексадецил-2-(3'-гексенил)-sn-глицеро-3-фосфоэтаноламина в 100 мл метанола и 100 мл дихлорметана. Затем по каплям добавляли 3 мл диметилсульфата и реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Добавляли еще 1 мл диметилсульфата и реакционную смесь перемешивали при комнатной температуре в течение ночи. В реакционную смесь добавляли 26.4 г однозамещенного фосфорнокислого натрия, затем 100 мл воды и после этого смесь экстрагировали трижды 100 мл смеси 2:1 (об/об) хлороформ:метанол. Объединенную органическую фазу промывали 100 мл воды, и растворитель удаляли при пониженном давлении, получая 3.44 г сырого (R)-1-гексадецил-2-(3'-гексенил)-sn-глицеро-3-фосфохолина.
Синтез (R)-1-гексадецил-2-(2-карбокси)этил-sn-глицеро-3-фосфохолина (CI-217): Раствор, содержащий 3.4 г (R)-1-гексадецил-2-(3'-гексенил)-sn-глицеро-3-фосфохолина в 200 мл воды, нагревали до 35°С и добавляли 4.33 г бикарбоната натрия. Раствор, содержащий 13.5 г периодата натрия в 90 мл воды, затем помещали в капельную воронку и добавляли по каплям. Раствор, содержащий 180 мг перманганата калия в 10 мл воды, помещали во вторую капельную воронку и добавляли по каплям для сохранения розового цвета реакционной смеси. Всего было добавлено примерно 4 мл раствора перманганата. Реакционную смесь перемешивали в течение 5 часов при 35-40°С и затем при комнатной температуре в течение ночи. РН реакционной смеси доводили примерно до 3 добавлением однозамещенного фосфорнокислого натрия и затем фосфорной кислоты (80%). Реакционную смесь экстрагировали трижды 100 мл хлороформа и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе и промывали дважды 50 мл раствора бисульфита натрия, и затем 50 мл воды. Органический раствор сушили над безводным Na2SO4, и растворитель удаляли при пониженном давлении, получая 2.78 г сырого продукта. Сырой продукт очищали хроматографией на силикагеле (30.89 г). 1.98 г чистого (R)-1-гексадецил-2-(2-карбокси)этил-sn-глицеро-3-фосфохолина элюировали смесями гексана с 20%-50% хлороформа с последующим хлороформом и затем смесями хлороформа с 10%-80% метанола.
ЯМР характеристика 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола. Спектры измеряли при 600 МГц.
Результаты показали наличие предполагаемого сигналя для спектральных элементов 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
1H ЯМР
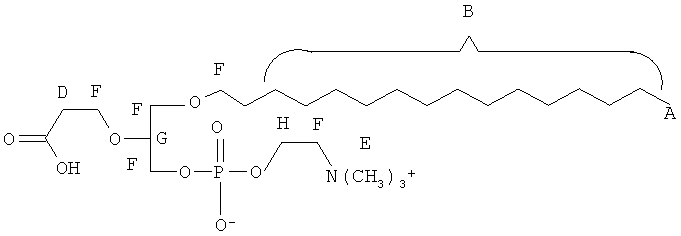
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.27 м.д.)
13C ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолина, были обозначены следующим образом:
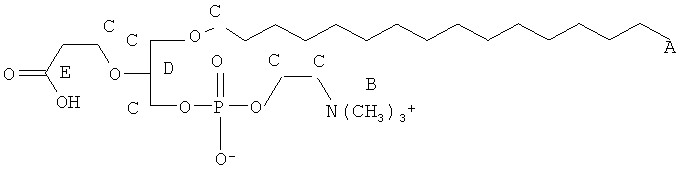
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.817 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(2-карбокси) этил-глицеро-3-фосфохолина:
Вычисленная масса для 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолина (C27H56NO8P) составила 554.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES-MS), показал наличие молекулярного иона с m/z=553, соответствующего депротонированному иону [М-Н]-.
Дополнительно, масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES+-MI), показал наличие молекулярного иона с m/z=555, соответствующего протонированному молекулярному иону [M+H]+, сопровождаемому молекулярным ионом с m/z=577, соответствующим катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(2-карбокси)этил-глицеро-3-фосфохолина.
Фосфорилирование тирозина:
Действие CI-217 на in vitro фосфорилирование тирозина в первичных макрофагах определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.27, обработка 20 мкг/мл (36 мкМ) CI-217 вызывала снижение уровней фосфотирозина, в то время как обработка 10 мкг/мл (18 мкМ) CI-217 не имела действия. Эти результаты были очень похожи на результаты соответствующей обработки 20 мкг/мл (34 мкМ) и 10 мкг/мл (17 мкМ) положительного контроля, CI-201.
ПРИМЕР 11
1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (CI-219) и 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (CI-220)
(R)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин и (R)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин были синтезированы, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолин и (S)-1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин были синтезированы с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез (S)-1-зйкозанил-т-глицерина: 8.6 мл (76.08 ммоль) (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 15 г порошкового гидроксида калия и 27.5 г (76.08 ммоль) 1-бромэйкозана перемешивали в 150 мл бензола и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 70 мл. Реакционную смесь затем охлаждали до комнатной температуры и добавляли 150 мл воды. Реакционную смесь затем экстрагировали трижды 150 мл диэтилового эфира, объединенную органическую фазу промывали 100 мл воды, и растворитель затем удаляли при пониженном давлении. Полученный остаток растворяли в 105 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником до тех пор, пока раствор не становился прозрачным. Раствор затем охлаждали до комнатной температуры и добавляли 100 мл воды. Продукт экстрагировали 150 мл хлороформа, последовательно промывали 150 мл воды, 150 мл насыщенного раствора карбоната натрия и снова 150 мл воды. Растворитель затем удаляли при пониженном давлении, и продукт кристаллизовался из 200 мл гексана с получением 21.0 г чистого 1-эйкозанил-sn-глицерина, который высушивали в десикаторе при пониженном давлении с оксидом фосфора. Выход составил 81.5%.
Синтез (R)-1-эйкозанил-3-тритил-sn-глицерина: 20 г 1-эйкозанил-sn-глицерина и 18 г трифенилхлорметана добавляли в смесь 400 мл сухого тетрагидрофурана (THF) и 100 мл сухого ацетонитрила. Добавляли 15 мл сухого триэтиламина и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь затем охлаждали до комнатной температуры, заливали на лед (500 г), переносили в делительную воронку и экстрагировали трижды 200 мл диэтилового эфира. Органическую фазу последовательно промывали 150 мл воды, дважды 150 мл разбавленной (1.5%) H2SO4, 150 мл воды, 150 мл концентрированного водного бикарбоната натрия и снова 150 мл воды. Раствор затем высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Полученный остаток, коричневое масло, растворяли в 300 мл этилацетата и охлаждали при -20°С в течение ночи. Смесь центрифугировали (3500 оборотов в минуту) при -10°С, и маточную жидкость сливали. Полученное твердое вещество растворяли в гексане и охлаждали (5±3°С) в течение ночи. Фильтрация осадка позволила получить 26.0 г чистого (R)-1-эйкозанил-3-тритил-sn-глицерина в виде грязно-белого твердого вещества. Выход составил 79%.
Синтез (R)-1-эйкозанил-2-(5'-гексенил)-3-тритил-sn-глицерина: 26 г (R)-1-эйкозанил-3-тритил-sn-глицерина и 10 г 5-гексенил-1-метан сульфоната растворяли в 150 мл бензола. Добавляли 12 г порошкового КОН и реакционную смесь перемешивали, и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 75 мл. Реакционную смесь охлаждали до комнатной температуры и добавляли 200 мл воды. Смесь экстрагировали трижды 150 мл диэтилового эфира, объединенную органическую фазу трижды промывали 150 мл воды и растворитель удаляли при пониженном давлении. Полученный остаток, 28 г коричневого масла, очищали на колонке с силикагелем (200 г). 28 г продукта элюировали хлороформом в виде светло-желтого масла. Выход составил 87.5%.
Синтез (S)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицерина: 70 г NaIO4 растворяли в 250 мл воды. В этот раствор добавляли 6 г NaHCO3 и 1.2 г KMnO4, и суспензию нагревали до 40°С. Раствор, содержащий 25 г (R)-1-эйкозанил-2-(5'-гексенил)-3-тритил-sn-глицерина в 250 мл трет-бутанола, добавляли по каплям в течение 90 минут, и смесь нагревали еще в течение 6 часов. Добавляли дополнительное(количество раствора KMnO4, требующееся для сохранения розового цвета. Реакционную смесь охлаждали до комнатной температуры, фильтровали через целит, и целит промывали затем 50 мл трет-буганола. Добавляли по каплям 100 мл 10% раствора серной кислоты и раствор переносили в делительную воронку, и трижды экстрагировали 200 мл гексана. Органическую фазу промывали раствором, содержащим 20 г Na2S2O5 в 100 мл воды, и затем 100 мл воды. Органическую фазу концентрировали путем удаления примерно 400 мл растворителя при пониженном давлении. В оставшийся раствор добавляли 15 мл воды и 2 мл концентрированной HCl, и полученную смесь кипятили с обратным холодильником в течение 6 часов. Смесь затем охлаждали до комнатной температуры и концентрировали снова удалением растворителя при пониженном давлении. РН остатка доводили до 12 добавлением 100 мл воды и 10 мл 30% раствора NaOH. Осадок отфильтровывали и промывали четыре раза 20 мл воды. Фильтрат экстрагировали 100 мл смеси 1:1 (об/об) гексан:этилацетат. Водную фазу подкисляли до рН 1 добавлением 10 мл концентрированной HCl и затем три раза экстрагировали 100 мл гексана. Сушка над безводной NaSO4 и удаление растворителя при пониженном давлении, с последующей рекристаллизацией в течение ночи сырого продукта из смеси 1:9 (об/об) ацетонгексан при 5±3°С, позволила получить 9.0 г чистого (S)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицерина. Выход составил 53.1%.
Синтез (S)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицерина: 8.9 г 1-эйкозанил-2-(4-карбокси)бутил-sn-глицерина растворяли в 50 мл метанола. Добавляли 1 мл концентрированной HCl (32%) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и в полученный остаток добавляли 50 мл воды. Смесь экстрагировали трижды 50 мл хлороформа. Объединенную органическую фазу промывали 50 мл воды, 50 мл насыщенного раствора бикарбоната натрия и снова 50 мл воды. Раствор затем высушивали над сульфатом натрия, фильтровали и выпаривали при пониженном давлении для получения 8.9 г (S)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицерина в виде белого воска.
Синтез (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 8.9 г 1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицерина (который был высушен азеотропной перегонкой с бензолом) и 8 мл триэтиламина растворяли в 70 мл THF. Раствор добавляли по каплям в течение 30 минут в ледяной раствор, содержащий 5.36 мл POCl3 в 40 мл THF, при перемешивании. Перемешивание продолжали еще в течение 30 минут при охлаждении и еще в течение 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне, и затем добавляли по каплям раствор 3.5 мл этаноламина и 16 мл триэтиламина в 50 мл THF в течение 30 минут при перемешивании. Перемешивание продолжали в течение 30 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток (14 г желтого масла) растворяли в смеси 72 мл уксусной кислоты и 30 мл воды, и нагревали до 70°С в течение 1 часа. Полученную смесь экстрагировали трижды 150 мл хлороформа и промывали дважды 150 мл воды. Удаление растворителя при пониженном давлении позволило получить 13 г (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина в виде желтого масла.
Синтез (R)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-220): 4 г (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-5п-глицеро-3-фосфоэтаноламина растворяли в 100 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем рН реакционной смеси доводили примерно до 4 добавлением муравьиной кислоты. Добавляли 150 мл воды и 150 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (70 г). Смесь хлороформа и гексана с последующими смесями хлороформа и метанола, и в конце смесями хлорофома, метанола и воды использовали для элюирования 835 мг (R)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (CI-220) из колонки. Выход составил 21%.
ЯМР характеристика 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для спектральных элементов 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом
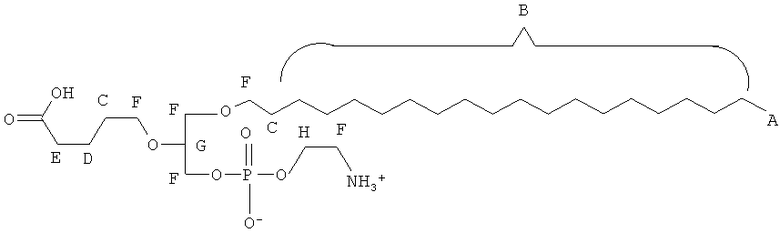
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.357 м.д.)
Наблюдаемые пики ' С, соответствующие структуре 1 -эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
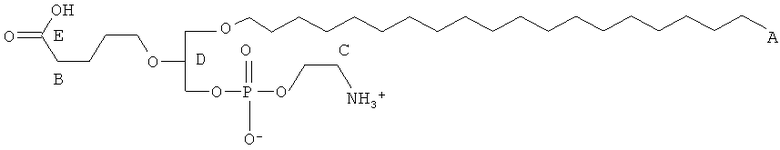
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.318 м.д.)
Масс-спектрометрическая характеристика 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-эйкозанил-2-(4-карбокси)этил-глицеро-3-фосфоэтаноламина (C30H62NO8P) составила 595.42.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES-MS), показал наличие молекулярного иона с m/z=594, соответствующего депротонированному иону [М-Н]-.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES+-MI) показал наличие молекулярного иона с m/z=596, соответствующего протонированному молекулярному иону [М+Н]+, сопровождаемому молекулярным с m/z=618, соответствующим катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина.
Синтез (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина: 9 г (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси, содержащей 40 мл изопропанола и 18 мл дихлорметана, и смесь нагревали до 35-40°С. Раствор, содержащий 10.3 г карбоната калия в 20 мл воды, добавляли по каплям при поддержании температуры смеси 35-40°С. Затем по каплям добавляли раствор, содержащий 7.2 мл диметилсульфата в 10 мл изопропанола (10 мл), при 40°С. Реакцию проводили при 40°С в течение 2 часов и затем при комнатной температуре в течение ночи. Добавляли 150 мл воды и смесь экстрагировали трижды 150 мл хлороформа. Органическую фазу промывали 150 мл воды и растворитель удаляли при пониженном давлении для получения 8 г (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина в виде воска.
Синтез (R)-1-эйкозанил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина (CI-219): 8 г (R)-1-эйкозанил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина растворяли в 100 мл смеси 8:2 (об/об) метанол: 10% водный раствор гидроксида натрия, и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Затем рН реакционной смеси доводили примерно до _ добавлением однозамещенного фосфорнокислого натрия и муравьиной кислоты. Добавляли 100 мл воды и 150 мл хлороформа. Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель затем удаляли при пониженном давлении. Полученный остаток (7.5 г) очищали хроматографией на силикагеле (150 г). Хлороформ, с последующими смесями хлороформа и метанола, и в конце смесями хлороформа, метанола и воды использовали для элюирования продукта. Удаление растворителя при пониженном давлении из фракций, содержащих продукт, позволило получить 4 г (R)-1-эйкозанил-2-(4-карбокси)бутил-ди-глицеро-3-фосфохолина в виде белого твердого вещества. Выход составил 51.1%.
ЯМР характеристика 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
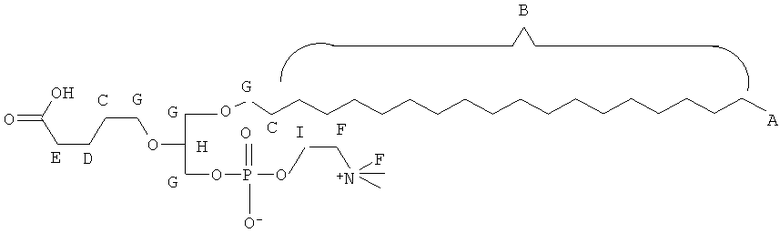
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.246 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
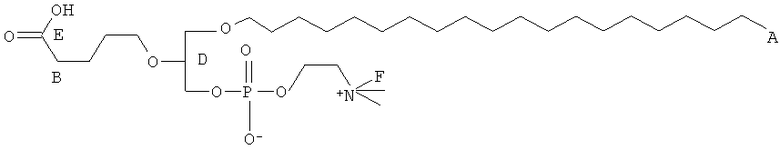
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.386 м.д.)
Масс-спектрометрическая характеристика 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Вычисленная масса для 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C33H68NO8P) составила 637.47.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES-MS), показал наличие молекулярного иона с m/z=637, соответствующего депротонированному молекулярному иону [М-Н]+.
Дополнительно, масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES+-MI) показал наличие молекулярного иона с m/z=638, соответствующего протонированному молекулярному иону [М+Н]+, сопровождаемому молекулярным ионом с m/z=660, соответствующим катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-эйкозанил-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
Фосфорилирование тирозина:
Действие CI-219 и CI-220 на in vitro фосфорилирование тирозина в первичных макрофагах определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.29, обработка 20 мкг/мл (36 мкМ) CI-219 вызывала снижение уровней фосфотирозина, в то время как обработка 10 мкг/мл CI-219 дала незначительный эффект, в случае его наличия. Действие 20 мкг/мл CI-219 было аналогичным действию 20 мкг/мл CI-201.
Аналогично, как показано на Фиг.29, обработка 20 мкг/мл CI-220 вызвала снижение уровней фосфотирозина, в то время как обработка 10 мкг/мл CI-220 дала незначительный эффект, в случае его присутствия. Действие 20 мкг/мл CI-220 было аналогично действию 20 мкг/мл CI-20I.
ПРИМЕР 12
1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфат (CI-201-PA)
(R) -1 -гексадецил-2-(4-карбокси)бутил-sn-глицеро-3 -фосфат синтезировали, как описано здесь ниже, из (R)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина. (S)-1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфат синтезировали с помощью таких же методик, но из (R)-1-гексадецил-2-(4-метилкарбокси)бутил-глицерина.
Синтез (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина и (R)-1-гексадецил-2-(4-метилкарбокси)бутил-глицерина описан в Примере 1.
Синтез (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфата:
Раствор (S)-1-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина (1.44 г), приготовленный согласно методикам, описанным в Примере 1, и триэтиламина (1.5 мл) в THF (15 мл) добавляли по каплям в течение 20 минут в ледяной раствор POCl3 (1 мл) в THF (15 мл) при перемешивании. Перемешивание продолжали еще в течение 30 минут при охлаждении и при комнатной температуре еще в течение 1 часа. Растворитель выпаривали при пониженном давлении. Полученный остаток растворяли в ледяном растворе насыщенного бикарбоната натрия (100 мл) и реакционную смесь перемешивали в ледяной ванне в течение 45 минут. РН раствора доводили до 4-5 добавлением HCl (32%). Смесь экстрагировали хлороформом (3×50 мл), органическую фазу промывали водой (50 мл) и растворитель удаляли при пониженном давлении. Смесь растворяли в хлороформе и очищали на силикагеле (30 г). Хлороформ с последующими смесями хлороформа с 10%-50% метанола использовали для элюирования 470 мг чистого 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата.
ЯМР характеристика 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата (CI-201-PA):
Образец растворяли в дейтерированном хлороформе (CDCl3). 1H и 13С ЯМР спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата, были обозначены следующим образом:
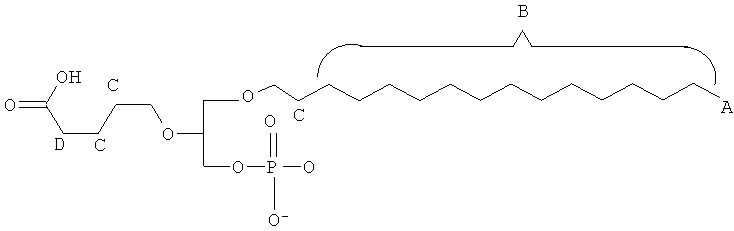
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.29 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата, были обозначены следующим образом:
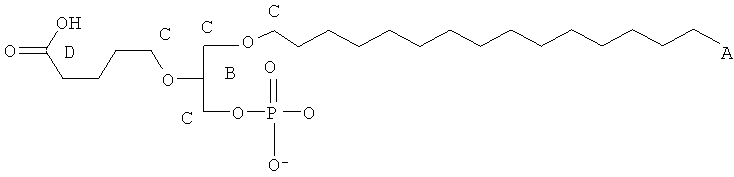
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.113 м.д.)
Масс-спектрометрическая характеристика 1-гекадецил-2-(4-карбокси)бутил-глицеро-3-фосфата:
Вычисленная масса для 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата (C24H49O8P) составила 496.3165.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES'MS), показал наличие молекулярного иона с m/z=495, соответствующего депротонированному иону [М-Н]-.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфата(CI-201-РА).
Токсичность CI-201-PA:
Токсичность CI-201-PA оценивали, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.30А и 30В, токсичность определяли для CI-201-PA при дозах 20 мкг/мл или выше, с LD50 примерно 50 мкг/мл. При концентрации 10 мкг/мл токсичность наблюдали только в одно из двух экспериментов.
ПРИМЕР 13
1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (1-S-CI-201) и 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (1-S-CI-202)
1-S-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин и 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин синтезировали, как описано здесь ниже, с использованием (R)-2,2-диметил-1,3-диоксолан-4-метанол р-толуолсульфоната в качестве исходного материала. 3-S-гексадецил-2-(4-карбокси)бутил-глицеро-1-фосфохолин и 3-S-гексадецил-2-(4-карбокси)бутил-глицерол-1-фосфоэтаноламин синтезировали с помощью таких же методик с использованием (5)-2,2-диметил-1,3-диоксолан-4-метанол p-толуолсульфоната в качестве исходного материала.
Синтез 1-8-гексадецил-sn-глицерина: 48 мл 1-гексадекантиола перемешивали в 150 мл бензола и кипятили с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 125 мл. Добавляли 58 мл раствора этилата натрия в этаноле, и смесь перемешивали в течение 30 минут в атмосфере азота. Добавляли 30 г (R)-2,2-диметил-1,3-диоксолан-4-метанол p-толуолсульфоната в 100 мл сухого бензола, и смесь перемешивали в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Реакционную смесь заливали на лед и экстрагировали трижды 150 мл диэтилового эфира. Органическую фазу промывали дважды 150 мл воды, высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в 100 мл смеси 9:1:0.5 (об/об) метанол: во да: концентрированная HCl, и полученный раствор кипятили с обратным холодильником в течение 2 часов, затем охлаждали до комнатной температуры. Реакционную смесь заливали на лед и трижды экстрагировали 200 мл хлороформа, затем промывали 200 мл воды, 200 мл насыщенного раствора карбоната натрия и снова 200 мл воды. Растворитель удаляли при пониженном давлении, получая 70 г оранжевого твердого вещества. Остаток рекристаллизовали дважды из 400 мл гексана для получения 30 г 1-S-гексадецил-sn-глицерина (30 г) в виде белого твердого вещества.
Синтез 1-S-гексадецил-3-тритил-sn-глицерина: 28 г 1-S-гексадецил-sn-глицерина и 31 г хлортрифенилметана помещали в смесь, содержащую 370 мл сухого THF и 100 мл сухого ацетонитрила. Добавляли 25 мл сухого триэтиламина, и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (1 килограмм), переносили в делительную воронку и экстрагировали трижды 200 мл диэтилового эфира. Органическую фазу промывали 200 мл воды, дважды 200 мл разбавленной серной кислоты (1.5%), 200 мл воды, 200 мл концентрированного раствора бикарбоната натрия и 200 мл воды. Раствор затем высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 60 г коричневого масла. Данное масло растворяли в 150 мл этилацетата, и полученный раствор выдерживали при -20°С в течение ночи. Смесь затем центрифугировали (при -10°С), и маточную жидкость сливали. Полученное твердое вещество рекристаллизовали из гексана при 4°С.
После фильтрации получали 36 г 1-S-гексадецил-3-тритил-sn-глицерина в виде белого твердого вещества.
Синтез 1-S-гексадецил-2-(4-1-бутил-карбокси)бутил-sn-глицерина: 10 г 1-S-гексадецил-3-тритил-sn-глицерина растворяли в смеси, состоящей из 150 мл бензола и 50 мл петролейного эфира, и затем добавляли 20.48 г порошкового КОН. Реакционную смесь затем перемешивали и кипятили с обратным холодильником. Раствор, содержащий 10 мл t-бутил валерата в 200 мл петролейного эфира, по каплям добавляли в кипящий раствор в течение 10 часов с азеотропным удалением воды. После завершения добавления реакционную смесь кипятили с обратным холодильником еще в течение 1 часа для уменьшения объема растворителей. Реакционную смесь затем охлаждали до комнатной температуры и добавляли смесь, содержащую 100 мл ледяной воды и 20 мл муравьиной кислоты. Реакционную смесь затем экстрагировали дважды в 100 мл хлороформа, высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 15.65 г светло-коричневого масла. Полученное масло растворяли в смеси, содержащей 160 мл трет-бутиловото эфира (МТВЕ) и 20 мл метанола. Добавляли 3 мл концентрированной HCl, и полученный раствор кипятили с обратным холодильником в течение 4 часов, затем охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали 100 мл насыщенного водного раствора карбоната натрия и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении для получения 14.10 г светло-коричневого масла. Масло очищали хроматографией на колонке с силикагелем (91 г). Гексан с последующими смесями гексана и хлороформа, и затем хлороформа и ацетона использовали для элюирования продукта из колонки. Растворитель удаляли при пониженном давлении для получения 6.84 г 1-S-гексадецил-2-(4-1-бутил-карбокси)бутил-sn-глицерина.
Синтез 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицерина: 6.84 г 1-S-гексадецил-2-(4-t-бутил-карбокси)бутил-sn-глицерина растворяли в 80 мл этанола. Добавляли раствор, содержащий 3.7 г КОН в 5 мл воды, и смесь перемешивали, и кипятили с обратным холодильником в течение 6 часов. После охлаждения смеси до комнатной температуры добавляли 16 мл воды и 100 мл смеси 8:2 (об/об) гексан:этилацетат. Фазы разделяли и в органическую фазу добавляли 50 мл воды и 5 мл муравьиной кислоты. Экстракция хлороформом, сушка над сульфатом натрия и удаление растворителей при пониженном давлении позволили получить 3.89 г 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицерина в виде светло-коричневого масла.
Синтез 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина: 3.89 г 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицерина растворяли в 100 мл метанола и добавляли 1 мл концентрированной соляной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи и дважды экстрагировали 100 мл хлороформа. Органическую фазу дважды промывали 50 мл воды и высушивали над сульфатом натрия, и растворитель затем удаляли при пониженном давлении для получения 3.75 г остатка. Остаток очищали хроматографией на колонке с силикагелем (49 г). Хлороформ, с последующими смесями хлороформа и ацетона использовали для элюирования продукта из колонки. Растворитель удаляли при пониженном давлении для получения 2.94 г чистого 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина.
Синтез 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 2.83 г 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицерина и 1.7 мл триэтиламина растворяли в смеси, состоящей из 20 мл бензола и 120 мл THF. Этот раствор добавляли по каплям в течение 60 минут в ледяной раствор, содержащий 1.14 мл POCl3 и 0.98 мл триэтиламина в 20 мл THF, при перемешивании. Перемешивание продолжали еще в течение 10 минут при охлаждении и еще в течение 45 минут при комнатной температуре. Затем добавляли по каплям раствор 1.02 мл этаноламина и 2.8 мл триэтиламина в 50 мл THF в течение 40 минут в ледяную реакционную смесь. Перемешивание продолжали в течение 10 минут при 0°С и затем при комнатной температуре в течение ночи. Реакционную смесь затем фильтровали, и растворитель удаляли при пониженном давлении. Остаток (4.23 г) растворяли в смеси, содержащей 48 мл уксусной кислоты и 20 мл воды, и нагревали до 70°С в течение 1 часа. Раствор затем экстрагировали 50 мл смеси 2:1 (об/об) хлороформ: метанол и органический растворитель удаляли при пониженном давлении для получения 4.05 г сырого 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина.
Синтез 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина: 0.97 г 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в 50 мл метанола. Добавляли 7 мл 10% раствора гидроксида натрия, и полученный раствор перемешивали при комнатной температуре в течение 8 часов. Добавляли 2 мл муравьиной кислоты, и смесь затем экстрагировали трижды 50 мл хлороформа. Объединенный органический растворитель удаляли при пониженном давлении для получения 0.70 г воскообразного остатка. Данный остаток очищали хроматографией на силикагеле (32 г). Хлороформ с последующими смесями хлороформа и метанола использовали элюирования продукта из колонки. Растворитель удаляли при пониженном давлении для получения 0.625 г сырого 1-S-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламина (1-S-CI-202).
ЯМР характеристика 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (1-S-CI-202):
Образец измеряли методом 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (1-S-CI-202) и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 13С, соответствующие структуре 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (1-S-CI-202), были обозначены следующим образом:
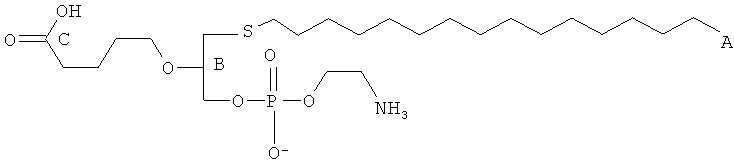
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.724 м.д.)
Масс-спектрометрическая характеристика 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201):Вычисленная масса для 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C26H54NO7PS) составила 555.75.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES'MS), показал наличие молекулярного иона с m/z=554, соответствующего депротонированному иону [М-Н]-.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=556, соответствующего протонированному молекулярному иону [М+Н]+ и иона с m/z=578, соответствующего катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (1-S-CI-202).
Синтез 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина: 3.48 г 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина растворяли в смеси, состоящей из 35 мл метанола и 100 мл дихлорметана. Добавляли раствор, содержащий 10 г карбоната калия в 20 мл воды. Затем добавляли 3.5 мл диметилсульфата, и реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили до 4 добавлением 1 мл муравьиной кислоты. Смесь трижды экстрагировали 75 мл смеси 2:1 (об/об) хлороформ:метанол с последующим удалением растворителя, что позволило получить 3.72 г сырого 1-5-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина.
Синтез (R)-1-S-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина:3.72 г 1-S-гексадецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина растворяли в 100 мл метанола. Добавляли 10 мл 10% раствора гидроксида натрия, и полученный раствор перемешивали при комнатной температуре в течение ночи. Добавляли 1.2 мл муравьиной кислоты, и смесь экстрагировали трижды 100 мл смеси 2:1 (об/об) хлороформ:метанол. Объединенный органический растворитель удаляли при пониженном давлении для получения 2.92 г остатка. 2.46 г этого остатка очищали хроматографией на силикагеле (54 г). Смесь хлороформа и гексана с последующим хлороформом, и затем смеси хлороформа и метанола использовали для элюирования продукта из раствора. Растворитель удаляли при пониженном давлении для получения 1.21 г чистого 1-S-гексадецил-2-(4-карбокси)бутил-sm-глицеро-3-фосфохолина (1-S-CI-201).
ЯМР характеристика 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201):
Образец измеряли методом 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201) и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 13С, соответствующие структуре 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201), были обозначены следующим образом:
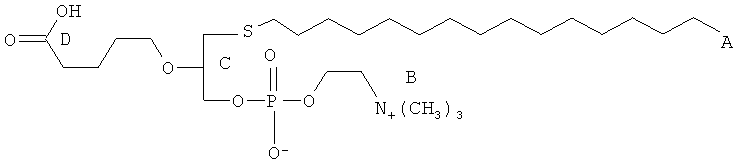
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=75.948 м.д.)
Масс-спектрометрическая характеристика 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201):
Вычисленная масса для 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C29H60NO7PS) составила 597.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ES'MS), показал наличие молекулярного иона с m/z=596, соответствующего депротонированному иону [М-Н]-.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=598, соответствующего протонированному молекулярному иону [М+Н]+, и иона с m/z=620, соответствующего катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-S-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина (1-S-CI-201).
Фосфорилирование тирозина.
Действие 1-S-CI-201 и 1-S-CI-202 на in vitro фосфорилирование тирозина в первичных макрофагах определяли, как описано здесь выше.
Как показано на Фиг.31, обработка 20 мкг/мл (34 мкМ) 1-S-CI-201 вызывала снижение уровней фосфотирозина, также как обработка 20 мкг/мл (34 мкМ) положительного контроля, CI-201. Однако обработка 10 мкг/мл (17 мкМ) 1-S-CI-201 дала незначительный эффект, в случае его наличия, в то время как 10 мкг/мл (34 мкМ) CI-201 вызывало повышение уровней фосфотирозина.
Как показано на Фиг.32, обработка 20 мкг/мл (36 мкМ) 1-S-CI-202 вызывала снижение уровней фосфотирозина, в то время как обработка 10 мкг/мл (18 мкМ) 1-S-CI-202 вызывала повышение уровней фосфотирозина. Эти результаты аналогичны результатам, полученным для 10 мкг/мл и 20 мкг/мл каждого из CI-201 и CI-202 соответственно.
ПРИМЕР 14
1-Гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолин (di-OH)
(R)-1-Гексадецил-2-(5,6-дигидрокси)гексанил-sn-глицеро-3-фосфохолин был синтезирован, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. S-1-Гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолин синтезировали с помощью таких же методик с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез 1-гексадецил-sn-глицерина: 19.4 г (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 49 г порошкового КОН и 4.8 г гексадецилбромида перемешивали в 500 мл толуола (500 мл) и кипятили в течение 6 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 100 мл, и реакционную смесь охлаждали до комнатной температуры. Охлажденную реакционную смесь растворяли в 500 мл дихлорметана, промывали дважды 200 мл воды, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в 500 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная H2SO4, и полученный раствор кипятили с обратным холодильником в течение 30 минут, охлаждали до комнатной температуры и экстрагировали 500 мл дихлорметана. Экстракт промывали дважды 100 мл воды, 100 мл водного 5% карбоната натрия и снова 100 мл воды до тех пор, пока экстракт не становился нейтральным. Растворитель удаляли при пониженном давлении, и сырой продукт рекристаллизовали из гексана при 4°C с получением 35.3 г сырого 1-гексадецил-sn-глицерина. Выход составил 76%.
Синтез 1-гексадецил-3-тритил-sn-глицерина: Раствор, содержащий 35.3 г 1-гексадецил-sn-глицерина, 37.3 г трифенилхлорметана и 22.44 г сухого триэтиламина в смеси 470 мл сухого тетрагидрофурана и 120 мл сухого ацетонитрила кипятили с обратным холодильником в течение 15 часов в атмосфере азота. После охлаждения до комнатной температуры реакционную смесь фильтровали. Фильтрат заливали на лед (500 г) и затем экстрагировали трижды 200 мл хлороформа. Органическую фазу последовательно промывали 500 мл воды, 500 мл 0.15 N HCl, 500 мл насыщенного водного NaHCO3, и снова водой. Экстракт высушивали над Na2SO4, и растворитель удаляли при пониженном давлении. Полученный желтый остаток растворяли в 500 мл теплого гексана, и полученный прозрачный раствор охлаждали (5±3°С) в течение ночи. За это время происходило осаждение. После фильтрации осадка растворитель из фильтрата удаляли при пониженном давлении, получая 58.3 г 1-гексадецил-3-тритил-sn-глицерина. Выход составил 95%.
Синтез (S)-1-гексадецил-2-(5'-гексенил)-sn-глицерина: 36.3 г 1-гексадецил-3-тритил-sn-глицерина и 11.5 г 5-гексенил-метансульфоната (приготовленного из S-гексен-1-ола и метансульфонилхлорида в сухом пиридине) растворяли в 500 мл толуола. Добавляли 20 г порошкового КОН, и реакционную смесь перемешивали и кипятили в течение 8 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 100 мл, и реакционную смесь охлаждали до комнатной температуры. Охлажденную реакционную смесь растворяли в 500 мл дихлорметана, промывали дважды 200 мл воды, и растворитель удаляли при пониженном давлении. Полученный 1-гексадецил-2-(5'-гексенил)-3-тритилглицерол растворяли в 500 мл смеси 90:10:5 (об/об) метанол:вода:концентрированная (32%) HCl и полученный раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли 500 мл воды и затем смесь экстрагировали трижды 250 мл дихлорметана. Объединенную органическую фазу промывали дважды 100 мл воды, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в 250 мл гексана, и полученный раствор хранили при -20°С в течение 48 часов, вызывая осаждение почти всего трифенил карбинола. После фильтрации и удаления растворителя из фильтрата оставшийся продукт очищали хроматографией на силикагеле. 11.65 г чистого (S)-1-гексадецил-2-(5'-гексенил)-sn-глицерина элюировали смесью 1:1 (об/об) хлороформ:петролейный эфир. Выход составил 45%.
Синтез (R)-1-гексадеццл-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина:11.65 г 1-гексадецил-2-(5'-гексенил)-sn-глицерина и 3.23 г триэтиламина растворяли в 650 мл сухого THF. Этот раствор добавляли по каплям в ледяной раствор, содержащий 5.34 г оксихлорида фосфора в 130 мл THF, при перемешивании. Добавление выполняли с такой скоростью, чтобы температура не превышала 15°С. Перемешивание продолжали еще в течение 10 минут при охлаждении и еще 45 минут при комнатной температуре. Реакционную смесь затем охлаждали в ледяной ванне и по каплям добавляли раствор 2.10 мл этаноламина и 9.73 мл триэтиламина в THF в течение 30 минут при перемешивании. Перемешивание продолжали в течение 20 минут в ледяной ванне и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток (15.93 г) растворяли в смеси 240 мл уксусной кислоты и 100 мл воды. Полученный раствор нагревали при 70°С в течение 1 часа, охлаждали до комнатной температуры и экстрагировали дважды 250 мл смеси 2:1 (об/об) хлороформ:метанол. Растворитель из органической фазы удаляли при пониженном давлении, получая 12.50 г R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфоэтаноламина.
Синтез (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина: (R)-1-гексадецил-2-(5'-гексенил)-3-фосфоэтаноламин растворяли в 650 мл изопропанола и 220 мл дихлорметана. Добавляли раствор, содержащий 66.5 г K2CO3 в 130 мл воды, и реакционную смесь нагревали до 40°С. Раствор, содержащий 13.3 мл диметилсульфата в 130 мл изопропанола, добавляли по каплям (в течение 45 минут) с такой скоростью, при которой температура реакции не превышала 35-40°С. После завершения добавления перемешивание продолжали при 40°С в течение 90 минут. Реакционную смесь затем охлаждали до комнатной температуры, экстрагировали трижды 500 мл смеси 2:1 (об/об) хлороформ:метанол, и растворитель из органической фазы удаляли при пониженном давлении, получая 12.50 г (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина.
Синтез (R)-1-гексадецил-2-(5,6-дигидрокси-гексил)-sn-глицеро-3-фосфохолина: 8.57 г (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфохолина растворяли в 80 мл муравьиной кислоты. Добавляли 18.7 мл 30% пероксида водорода, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли 250 мл воды, и раствор переносили в делительную воронку и экстрагировали 5 раз 100 мл смеси 2:1 (об/об) хлороформ:метанол. Растворитель из органической фазы удаляли при пониженном давлении и полученный остаток растворяли в 150 мл метанола. Добавляли 55 мл водного гидроксида натрия (10%) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли 3 мл холодной смеси концентрированной HCl (32%) в 150 мл воды и полученный раствор переносили в делительную воронку, и продукт экстрагировали 5 раз 100 мл смеси 2:1 (об/об) хлороформ:метанол. Растворитель из органической фазы удаляли при пониженном давлении, и полученный сырой продукт очищали хроматографией на силикагеле. 4.5 г сырого (R)-1-гексадецил-2-(5,6-дигидрокси-гексил)-sn-глицеро-3-фосфохолина (di-OH) элюировали смесями хлороформа и 4%-60% метанола. Выход составил 50%.
ЯМР характеристика 1-гексадецил-2-(5,6-дигидрокси-гексил)-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3). 1Н ЯМР и 13С ЯМР спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина, были обозначены следующим образом:
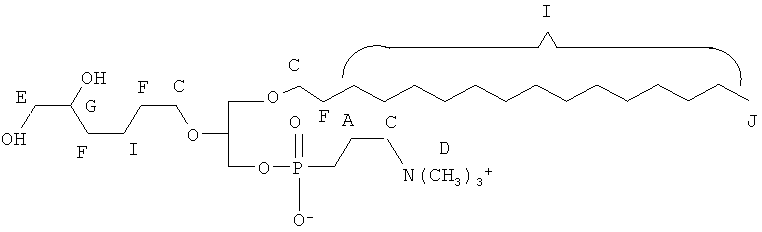
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.299 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина, были обозначены следующим образом:
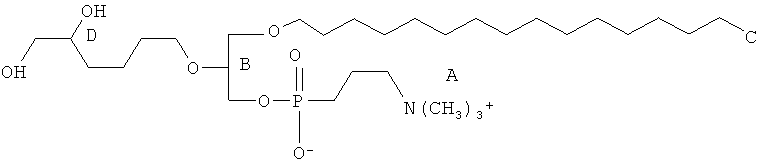
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.700 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина:
Вычисленная масса для 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина (C30H64NO8P) составила 597.4370
Масс-спектр, полученный методом бомбардировки ускоренными атомами (FAB+), показал наличие молекулярного иона с m/z=598.400, соответствующего протонированному молекулярному иону [М+Н]-.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(5,6-дигидрокси)гексанил-глицеро-3-фосфохолина (di-OH).
Токсичность di-OH:
Токсичность di-OH оценивали, как описано здесь выше.
Как показано на Фиг.33А и 33В, токсичность di-OH обнаруживали при дозах 20 мг/мл или выше, с LD50 di-OH в диапазоне между 20 и 50 мкг/мл.
Анализ атеросклеротического поражения:
Эффективность in vivo di-OH против атеросклеротических поражений тестировали на мышиных самцах линии LDL-RD, как описано здесь выше в разделе «Материалы и Способы». Di-OH вводили в дозе 1 мг на мышь, эквивалентную дозе 60 мг/кг.
Как показано на Фиг.34, 1 мг/мышь di-OH уменьшал область атеросклеротического повреждения на 25% по сравнению с контролем (PBS).Эти результаты указывают на то, что di-OH является эффективным против атеросклероза.
ПРИМЕР 15
1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолин (R)-1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин синтезировали, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолин синтезировали с помощью таких же методик с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Синтез 1-(цис-9-гексадеценил)-sn-глицерина: 5.32 г (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола, 12.26 г порошкового гидроксида калия и 10 г цис-9-гексадеценил-метансульфоната перемешивали в 250 мл бензола и кипятили в течение 10.5 часов с азеотропным удалением вод. Объем растворителя постепенно уменьшался примерно до 50 мл. Реакционную смесь охлаждали до комнатной температуры, добавляли 50 мл воды и смесь экстрагировали трижды 100 мл диэтилового эфира. Объединенную органическую фазу промывали 100 мл воды, и растворитель удаляли при пониженном давлении. Остаток (12.01 г) растворяли в 200 мл смеси 90:10:3 (об/об) метанол: вода: концентрированная соляная кислота, и полученный раствор перемешивали при комнатной температуре в течение ночи и затем кипятили с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры добавляли 100 мл воды. Продукт трижды экстрагировали 75 мл диэтилового эфира, последовательно промывали 100 мл воды, 100 мл насыщенного водного раствора бикарбоната натрия и снова 100 мл воды. После сушки над Na2SO4 растворитель удаляли при пониженном давлении, получая 9.104 г сырого 1-(цис-9-гексадеценил)-sn-глицерина. Сырой продукт очищали хроматографией на колонке с силикагелем (30 г). 9.07 г чистого 1-(цис-9-гексадеценил)-глицерина элюировали хлороформом с последующей смесью хлороформа и 5% метанола. Выход составил 91.8%.
Синтез (S)-1-(цис-9-гексадеценил)-3-тритил-sn-глицерина: 9.07 г 1-(цис-9-гексадеценил)-глицерина растворяли в смеси сухого THF (160 мл) и сухого ацетонитрила (40 мл). Добавляли 10.52 г трифенилхлорметана и 10 мл триэтиламина, и реакционную смесь кипятили с обратным холодильником в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (100 г), переносили в делительную воронку и экстрагировали трижды 100 мл хлороформа. Органическую фазу последовательно промывали 100 мл воды, 100 мл разбавленной (1.0%) серной кислоты, 100 мл воды, 100 мл насыщенного водного бикарбоната натрия и снова 100 мл воды. Органическую фазу сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в горячем гексане (100 мл) и полученный раствор охлаждали при 4°С в течение 36 часов. Фильтрация осажденных побочных продуктов и удаление растворителя при пониженном давлении позволило получить 14.57 г сырого продукта. Сырой продукт очищали хроматографией на силикагеле (200 г). 9.81 г сырого 1-(цис-9-гексадеценил)-3-тритил-sn-глицерина элюировали хлороформом. Выход составил 61.1%.
Синтез (S)-1-(цис-9-гексадеценил)-2-(4-трет-бутил-карбокси)бутил-sn-глицерина: 8.83 г 1-(цис-9-гексадеценил)-3-тритил-5и-глицерина растворяли в смеси, состоящей из бензола (170 мл) и петролейного эфира (100 мл). Добавляли порошковый КОН (23.1 г) и реакционную смесь нагревали до мягкого кипения. Раствор трет-бутил-валерата (20 мл) в петролейном эфире (420 мл) добавляли по каплям в течение 25 часов с азеотропным удалением воды. После охлаждения до комнатной температуры рН реакционной смеси доводили примерно до 6 добавлением муравьиной кислоты (10 мл). Смесь экстрагировали диэтиловым эфиром (3×100 мл) и органическую фазу промывали водой (100 мл). Удаление растворителя при пониженном давлении позволило получить 17.72 г масляного продукта. Этот остаток растворяли в метаноле (50 мл), добавляли 4 мл концентрированной HCl (32%), и реакционную смесь кипятили с обратным холодильником в течение 5.5 часов. После охлаждения до комнатной температуры добавляли 50 мл воды, и смесь экстрагировали диэтиловым эфиром (3×50 мл). Объединенную органическую фазу промывали водой (50 мл) и растворитель удаляли при пониженном давлении, получая 14.26 г сырого продукта. 2.93 г чистого 1-(цис-9-гексадеценил)-2-(4-трет-бутил-карбокси)бутил-sn-глицерина очищали хроматографией на силикагеле (110 г). Продукт элюировали смесью хлороформ гексан (1:1 объемное соотношение) с последующими смесями хлороформа с этилацетатом до 3%. Растворитель из фракций, содержащих требуемый продукт, удаляли при пониженном давлении. Выход составил 39.2%.
Синтез (R)-1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина: 1.59 г (S)-1-(цис-9-гексадеценил)-2-(4-трет-бутил-карбокси)бутил-sn-глицерина (высушенного азеотропной перегонкой с бензолом) и 0.7 мл триэтиламина растворяли в THF (45 мл). Этот раствор добавляли по каплям в течение 18 минут в ледяной раствор РОС1з (0.5 мл) и триэтиламина (0.05 мл) в THF (20 мл) при перемешивании. Перемешивание продолжали еще в течение 10 минут при охлаждении и еще в течение 45 минут при комнатной температуре. Реакционную смесь охлаждали в ледяной ванне, и по каплям добавляли раствор этаноламина (0.38 мл) и триэтиламина (3.25 мл) в THF (54 мл) в течение 65 минут при перемешивании. Перемешивание продолжали еще в течение 10 минут при охлаждении и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, промывали THF (2×10 мл), и растворитель удаляли при пониженном давлении. Остаток (2.1 г) растворяли в смеси уксусной кислоты (48 мл) и воды (20 мл), и нагревали до 70°С в течение 1 часа. Экстрагирование диэтиловым эфиром (2×50 мл), промывка водой (2×50 мл) и удаление растворителя при пониженном давлении позволило получить 2.15 г сырого продукта в виде светло-коричневого масла. Это масло растворяли в смеси метанола (35 мл) и дихлорметана (100 мл). Добавляли раствор карбоната калия (10 г) в воде (20 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли диметилсульфат (2.5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли еще 1 мл диметилсульфата и реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Смесь экстрагировали хлороформом (3×50 мл) и растворитель из объединенной органической фазы удаляли при пониженном давлении для получения 2.48 г воскообразного продукта. Этот остаток растворяли в метаноле (100 мл), добавляли раствор (рН ≈11) гидроксида лития (0.19 г) в воде (6 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили до 4-5 добавлением муравьиной кислоты, и смесь затем экстрагировали хлороформом (3×100 мл). Растворитель из объединенной органической фазы удаляли при пониженном давлении. Анализ тонкослойной хроматографией показал примерно 60% превращения. Полученный остаток растворяли в этаноле (100 мл), добавляли раствор (рН ≈11) гидроксида лития (0.2 г) в воде (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили до 4-5 добавлением муравьиной кислоты (0.22 мл) и смесь затем экстрагировали смесью 2:1 (об/об) хлороформ:метанол (3×100 мл). Объединенную органическую фазу сушили над безводным Na2SO4 и растворитель удаляли при пониженном давлении, получая 2.18 г сырого продукта. 1.14 г чистого (R)-1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-.уи-глицеро-3-фосфохолина элюировали хлороформом с последующими смесями хлороформа с 10%-80% метанола.
ЯМР характеристика 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина:
Образцы растворяли в дейтерированном хлороформе (CDCl3). 1Н ЯМР и 13С ЯМР спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-(цис-9-гексадеценил-2-(4-карбокси)бутил-глицеро-3-фосфохолина и, таким образом, полностью подтвердили структуру 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
Наблюдаемые пики 1Н, соответствующие структуре 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
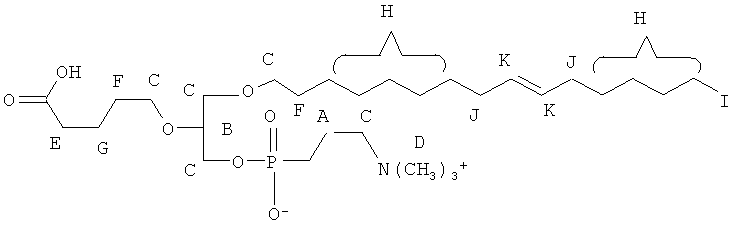
ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.26 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина, были обозначены следующим образом:
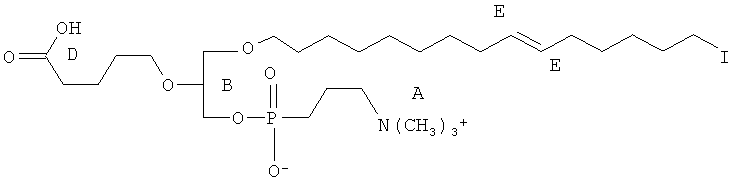
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.257 м.д.)
Масс-спектрометрическая характеристика 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина:Вычисленная масса для 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина (C29H58NO8P) составила 579.3900
Масс-спектр, полученный методом бомбардировки ускоренными атомами (FAB+), показал наличие молекулярного иона с m/z=580.3995, соответствующего протонированному молекулярному иону [М+Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-(цис-9-гексадеценил)-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
ПРИМЕР 16
(S)-1-гексадецил-2-(4-карбокси)бутил-глицерин
(S)-1-гексадецил-2-(4-карбокси)бутил-sn-глицерин синтезировали, как описано в Примере 1, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. Синтез (R)-1-гексадецил-2-(4-карбокси)бутил-глицерина, с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала также описан в Примере 1.
ЯМР характеристика (S)-1-гексадецил-2-(4-карбокси)бутил-глицерина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(4-карбокси)бутил-глицерина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицерина, были обозначены следующим образом:
1H ЯМРВ.
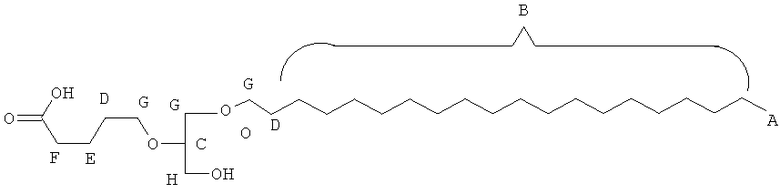
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.26 м.д.)
13С ЯМР
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(4-карбокси)бутил-глицерина, были обозначены следующим образом:
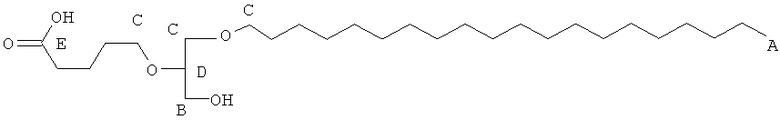
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.014 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(4-карбокси)бутил-глицерина:
Вычисленная масса для 1 -гексадецил-2-(4-карбокси)бутил-глицерина (C24H48O5) составила 416.635.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=417.0, соответствующего протонированному молекулярному иону [М+Н]-. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(4-карбокси)бутил-глицерина.
ПРИМЕР 17
1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолин (diMeAc) и 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолин (diEtAc)
(R)-1-гексадецил-2-(5',5'-диэтоксипентил)-sn-глицеро-3-фосфохолин и (R)-1-гексадецил-2-(5',5'-диметоксипентил)-sn-глицеро-3-фосфохолин были приготовлены из (R)-1-гексадецил-2-(5,6-дигидроксигексанил)-sn-глицеро-3-фосфохолина, как описано ниже. С помощью таких же методик, (R)-1-гексадецил-2-(5',5'-диэтоксипентил)-глиперо-3-фосфохолин и (S)-1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолин были приготовлены из (S)-1-гексадецил-2-(5,6-дигидроксигексанил)-глицеро-3-фосфохолина.
Синтез (R)-1-гексадецил-2-(5,6-дигидроксигексанил)-sn-глицеро-3-фосфохолина и (S)-1-гексадецил-2-(5,6-дигидроксигексанил)-глицеро-3-фосфохолина описан в Примере 14.
Синтез (R)-1-гексадецил-2-(5'-оксо-пентил)-sn-глицеро-3-фосфохолина: 2 г периодата натрия добавляли в ледяной раствор (R)-1-гексадецил-2-(5',6'-дигидроксигексанил)-sn-глицеро-3-фосфохолина (2 г) в воде (200 мл), и реакционную смесь перемешивали в течение 30 минут при охлаждении и при комнатной температуре в течение ночи. Реакционную смесь переносили в делительную воронку и экстрагировали смесью 2:1 (об/об) хлороформ:метанол (3×100 мл), и растворитель из объединенной органической фазы удаляли при пониженном давлении. Объединенный остаток растворяли в хлороформе, высушивали над безводным Na2SO4 и фильтровали, после чего растворитель удаляли при пониженном давлении. Объединенный сырой продукт очищали хроматографией на силикагеле (21 г). 1.2 г чистого (R)-1-гексадецил-2-(5'-оксо-пентил)-sn-глицеро-3-фосфохолина элюировали с последующими смесями хлороформа с 40%-60% метанола. Выход составил 63%.
Синтез (R)-1-гексадецил-2-(5',5'-диэтоксипентил)-sn-глицеро-3-фосфохолина (diEtAc): Реакцию проводили в атмосфере азота. (R)-1-гексадецил-2-(5'-оксо-пентил)-уп-глицеро-3-фосфохолин (50 мг, 0.088 ммоль) растворяли в этаноле (10 мл). Добавляли триэтил-орто-формиат (0.053 мл, 0.0476 г, 0.32 ммоль) и 3 капли концентрированной серной кислоты (95-97%), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь переносили с добавлением дихлорметана (75 мл) в делительную воронку. Раствор промывали подои (75 мл), водным 2.5% раствором бикарбоната натрия (75 мл) и водой (75 мл), и высушивали над безводным сульфатом натрия. Фильтрация и удаление растворителя при пониженном давлении позволила получить 50 мг (R)-1-гексадецил-2-(5',5'-диэтоксипентил)-sn-глицеро-3-фосфохолина. Выход составил 88.4%.
Синтез (R)-1-гексадецил-2-(5',5'-диметоксипентил)-sn-глицеро-3-фосфохолина (diMeAc): Реакцию проводили в атмосфере азота. (R)-1-гексадецил-2-(5'-оксо-пентил)-sn-глицеро-3-фосфохолин (35 мг, 0.097 ммоль) растворяли в метаноле (10 мл). Добавляли триметил-орто-формиат (0.043 мл, 0.414 г, 0.39 ммоль) и 3 капли концентрированной серной кислоты (95-97%), и и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дихлорметан (75 мл), и реакционную смесь переносили в делительную воронку. Раствор последовательно промывали водой (75 мл), водным 2.5% раствором бикарбоната натрия (75 мл) и снова водой (75 мл). Сушка над безводным сульфатом натрия и удаление растворителя при пониженном давлении позволила получить 36.8 мг (R)-1-гексадецил-2-(5',5'-диметоксипентил)-sn-глицеро-3-фосфохолина. Выход составил 62%.
ЯМР характеристика 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина (DiEtAc):
Образец растворяли в дейтерированном хлороформе (CDCl3). 1Н ЯМР и 13С ЯМР спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина, были обозначены следующим образом:
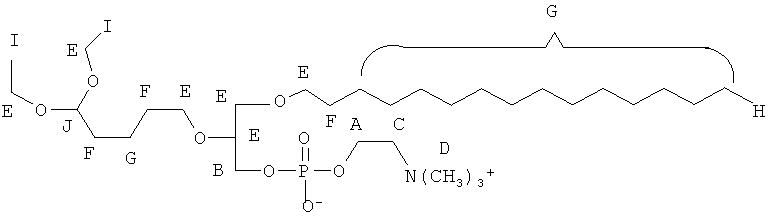
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.270 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(5',5' диэтоксипентил)-глицеро-3-фосфохолина, были обозначены следующим образом:
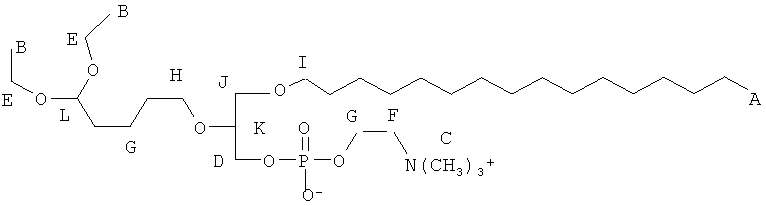
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.0046 м.д.)
Масс-спектрометрическая характеристика 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина (DiEtAc):
Вычисленная масса для 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина (C33H70NO8P) составила 639.88.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=640, сопровождаемого ионом с m/z=662, соответствующим катионированному молекулярному иону [M+Na]+, и иона с m/z=594, соответствующего деэтоксилированному молекулярному иону [M-OEt]+. Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-гексадецил-2-(5',5'-диэтоксипентил)-глицеро-3-фосфохолина.
ЯМР характеристика 1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолина (DiMeAc):
Образец растворяли в дейтерированном хлороформе (CDCl2). 1Н ЯМР и 13С ЯМР спектры измеряли при 300 МГц.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолина (DiMeAc) и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолина, были обозначены следующим образом:
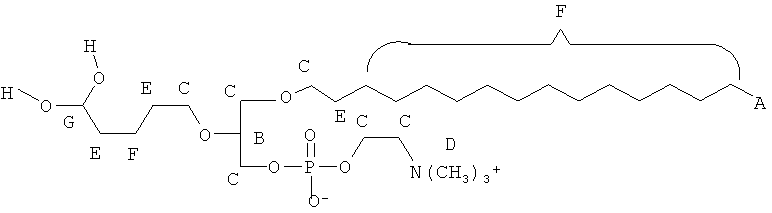
1H ЯМР (300 МГц, эталонный растворитель (CDCl3)=7.28 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-гексадецил-2-(5',5'-диметоксипентил)-глицеро-3-фосфохолина, были обозначены следующим образом:
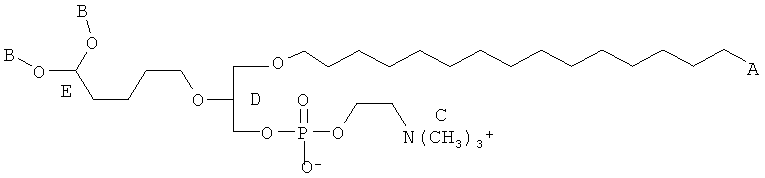
13С ЯМР (300 МГц, эталонный растворитель (CDCl3)=77.000 м.д.)
Анализ атеросклеротического поражения:
Эффективность in vivo diMeAc против атеросклеротических поражений тестировали на мышах линии АроЕ KO, как описано здесь выше в разделе «Материалы и Способы». diMeAc вводили в дозе 1 мг на мышь, эквивалентной дозе 40 мг/кг.
Как показано на Фиг.35, 1 мг/мышь diMeAc уменьшало область атеросклеротического поражения на 23% по сравнению с контролем (PBS).
Эти результаты указывают на то, что diMeAc является эффективным против атеросклероза.
Токсичность diMeAc
Токсичность diMeAc оценивали, как описано здесь выше.
Как показано на Фиг.36А и 36В, diMeAc проявлял токсичность при дозах 20 мкг/мл или выше с LD50 diEtAc в диапазоне между 20 и 50 мкг/мл. При концентрации 10 мкг/мл токсичность наблюдали только в одном из двух экспериментов.
ПРИМЕР 18
1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфосерин (VB-223)
(R)-1-гексадецил-2-(4-карбокси)бутил-глицеро-3-фосфосерин был приготовлен из (S)-1-гексадецил-2-(5'-гексенил)-sn-глицерина, как описано здесь ниже. С помощью таких же методик (S)-1-гексадецил-2-(5'гексенил)-глицеро-3-фосфосерин был приготовлен из (R)-1-гексадецил-2-(5'-гексенил)-глицерина.
Синтез (S)-1-гексадецил-2-(5'-гексенил)-sn-глицерина и (R)-1-гексадецил-2-(5'-гексенил)-глицерина описан в Примере 14.
Синтез (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфата: 1.0 г (S)-1-гексадецил-2-(5'-гексенил)-глицерина (высушенного азеотропной перегонкой с бензолом) и сухой пиридин (1 мл) растворяли в THF (60 мл). Этот раствор добавляли по каплям в ледяной раствор POCl3 (0.3 мл) в THF (12 мл) при перемешивании. Перемешивание продолжали еще в течение 3 часов при охлаждении. В охлажденную реакционную смесь добавляли раствор бикарбоната натрия (2.44 г) в воде (24 мл), и смесь перемешивали в ледяной ванне еще в течение 30 минут. РН реакционной смеси доводили примерно до 1 медленным добавлением 10% соляной кислоты. Экстрагирование диэтиловым эфиром (3×150 мл), промывка объединенной органической фазы (2×150 мл), сушка над безводным Na2SO4 и удаление растворителя при пониженном давлении позволило получить 1.43 г (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфата.
Синтез (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфо-N-Вос-L-серин-бензгидрилового эфира: 1.30 г (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфата и 0.95 г N-Boc-серин-бензгидрилового эфира (который высушивали в десикаторе с P2O5) растворяли в пиридине (30 мл). Добавляли 2,4,6-триизопропил-бепзол сульфонил хлорид (2.99 г), и реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 20 часов. Добавляли воду (50 мл), и смесь переносили в делительную воронку. Экстрагирование проводили смесью 8:2 (об/об) гексан:этилацетат (3×50 мл), объединенную органическую фазу высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси 8:2 (об/об) гексан:этилацетат (50 мл) и отмывали охлажденной разбавленной уксусной кислотой (5%). Растворитель затем удаляли при пониженном давлении, получая 1.33 г сырого (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфо-N-Вос-L-серин-бензгидрилового эфира.
Синтез (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфо-N-Вос-L-серин-бензгидрилового эфира: Периодат натрия (3.0 г), перманганат калия (0.05 г), карбонат натрия (0.15 г) и карбонат калия (0.03 г) растворяли в воде (100 мл). В этот раствор добавляли раствор (R)-1-гексадецил-2-(5'-гексенил)-sn-глицеро-3-фосфо-N-Boc-L-серин-бензгидрилового эфира (0.90 г) в трет-бутаноле (100 мл) по каплям при комнатной температуре в течение 30 минут. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли дополнительное количество перманганата калия (0.02 г), и реакционную смесь перемешивали в течение 90 минут. Добавляли раствор однозамещенного фосфорнокислого натрия (10 г) в воде (100 мл), и реакционную смесь экстрагировали хлороформом (3×100 мл). Объединенную органическую фазу промывали солевым раствором (100 мл), и растворитель удаляли при пониженном давлении, получая 1.02 г сырого (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфо-N-Вос-L-серин-бензгидрилового эфира.
Синтез (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфо-L-серина (VB-223): 1.02 г (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфо-N-Вос-L-серин-бензгидрилового эфира растворяли в дихлорметане (100 мл). Раствор охлаждали в ледяной ванне и насыщали газом HCl в течение 30 минут. Реакционную смесь перемешивали еще в течение 1 часа. Реакционную смесь затем нейтрализовали путем добавления водного раствора однозамещенного фосфорнокислого натрия и затем экстрагировали смесью 2:1 (об/об) хлороформ:метанол (3×100 мл). Органическую фазу высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении. Объединенный сырой продукт (0.72 г) очищали хроматографией на силикагеле (12.60 г). 0.60 г чистого (R)-1-гексадецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфо-L-серина элюировали смесью 1:1 (об/об) гексан: хлороформ с последующим хлороформом и затем смесью хлороформа с 10% метанола.
Фосфорилирование тирозина:
Действие VB-223 на in vitro фосфорилирование тирозина в первичных макрофагах определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано на Фиг.37, обработка 5, 10 и 20 мкг/мл (8.3, 16.7 и 33.3 мкМ) VB-223 и возможно также 1 мкг/мл (1.7 мкМ) VB-223 индуцировала фосфорилирование тирозина.
ПРИМЕР 18
1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин (VB-221) и 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин (VB-222)
(R)-1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин и (R)-1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин были синтезированы, как описано здесь ниже, с использованием (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала. (S)-1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламин и (S)-1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолин были синтезированы с помощью таких же методик, но с использованием (S)-(+)-2,2-диметил-1,3-диоксолан-4-метанола в качестве исходного материала.
Метансульфоновой кислоты 2-октил-додециловый эфир: 2-0ктил-1-додеканол (20 мл, 56.14 ммоль) и безводный триэтиламин (16 мл, 112.28 ммоль) растворяли в сухом дихлорметане (60 мл). Раствор охлаждали до 0°С и по каплям добавляли метансульфонил хлорид (5.2 мл, 67.36 ммоль) в сухом дихлорметане (40 мл), После завершения добавления смесь перемешивали при 0°С в течение 3 часов и затем охлаждали (2-8°С) в течение ночи. Реакционную смесь заливали на лед (500 г), оставляли нагреваться до комнатной температуры и экстрагировали эфиром (3×150 мл). Органическую фазу последовательно промывали водой (150 мл), 2% H2SO4 (150 мл), водой (150 мл), насыщенным бикарбонатом натрия (150 мл) и снова водой (150 мл). Органическую фазу высушивали над безводным Na2SO4, и растворитель удаляли при пониженном давлении, получая 22.8 г метансульфоновой кислоты 2-октил-додециловый эфир в виде желтого масла.
1-(2-октил)додецил-глицерин: (R)-(-)-2,2-диметил-1,3-диоксолан-4-метанол (8.3 мл, 66.59 ммоль), порошковый гидроксид калия (12 г) и метансульфоновой кислоты 2-октил-додециловый эфир (22.77 г, 60.50 ммоль) перемешивали в бензоле (250 мл) и кипятили в течение 5 часов а азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 150 мл. Реакционную смесь охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли 200 мл воды и смесь экстрагировали диэтиловым эфиром (3×200 мл). Объединенную органическую фазу промывали водой (200 мл), и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в 105 мл смеси 90:10:5 (об/об) метанол: вода: концентрированная соляная кислота, и полученный раствор кипятили с обратным холодильником в течение 30 минут. После охлаждения до комнатной температуры добавляли воду (100 мл). Продукт экстрагировали хлороформом (3×100 мл) и последовательно промывали водой (100 мл), насыщенным водным раствором карбоната натрия (100 мл) и снова водой (100 мл). Растворитель удаляли при пониженном давлении, и сырой продукт очищали хроматографией на колонке с силикагелем (400 г). 17 г чистого 1-(2-октил)додецил-глицерина элюировали хлороформом с последующими смесями хлороформа и 10%-30% этилацетата с получением бесцветного масла. Выход составил 75.5%.
(S)-1-(2-октил)додецил-3-тритил-глицерин: 17 г 1-(2-октил)додецил-глицерина (высушенного азеотропной перегонкой с бензолом) растворяли в смеси сухого THF (400 мл) и сухого ацетонитрила (160 мл). Добавляли трифенилхлорметан (15.8 г) и сухой триэтиламин (14 мл), и реакционную смесь кипятили с обратным холодильником в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, заливали на лед (1 кг), переносили в делительную воронку и экстрагировали диэтиловым эфиром (3×200 мл). Объединенную органическую фазу последовательно промывали водой (200 мл), разбавленной (1.5%) серной кислотой (2×100 мл), водой (200 мл), концентрированным водным бикарбонатом натрия (200 мл) и снова водой (200 мл). Органическую фазу высушивали над безводным сульфатом натрия, растворитель удаляли при пониженном давлении, и полученный сырой продукт очищали хроматографией на колонке с силикагелем (350 г). 26 г чистого (S)-1-(2-октил)додецил-3-тритил-глицерина элюировали гексаном с последующими смесями гексана и хлороформа (50-100%) с получением желтого масла. Выход составил 92.7%.
1-(2-октил)додецил-2-(5'-гексенил)-3-тритил-глицерин: 1-(2-октил)додецил-3-тритил-глицерин (26 г, 42.28 ммоль) и 5-гексенил-1-метан сульфонат (9.4 г, 50.73 ммоль) растворяли в бензоле (150 мл). Добавляли порошковый КОН (17 г), и реакционную смесь кипятили в течение 5.5 часов с азеотропным удалением воды. Объем растворителя постепенно уменьшался примерно до 50 мл. После охлаждения реакционной смеси до комнатной температуры добавляли 200 мл воды, и смесь экстрагировали диэтиловым эфиром (3×100 мл). Объединенную органическую фазу промывали солевым раствором (3×100 мл), и растворитель удаляли при пониженном давлении, получая 22.8 г сырого продукта. 21 г чистого 1-(2-октил)додецил-2-(5'-гексенил)-3-тритил-глицерина получали очисткой сырого продукта хроматографией на силикагеле (300 г). Продукт элюировали хлороформом в виде желтого масла. Выход составил 71.2%.
(S)-1-(2-октил)додецил-2-(4-карбокси)бутил-глицерин: Периодат натрия (58 г), перманганат калия (960 мг) и карбонат калия (7 г) суспендировали в воде (250 мл). По каплям добавляли раствор 1-(2-октил)додецил-2-(5'-гексенил)-3-тритил-глицерина (21 г) в трет-бутаноле (250 мл) в течение 2.5 часов. Реакционную смесь затем перемешивали в течение ночи (раствор перманганата добавляли в количестве, требуемом для сохранения розового цвета). Смесь фильтровали через слой целита, который затем отмывали wpew-бутанолом. Добавляли по каплям 10 мл разбавленной серной кислоты (10%), и полученный раствор затем экстрагировали гексаном (3×200 мл). Объединенную органическую фазу дважды промывали раствором бисульфита натрия (20 г) в воде (100 мл) и затем водой (200 мл). Растворитель концентрировали при пониженном давлении до объема 150 мл. Добавляли 20 мл воды и 5 мл концентрированной HCl, и полученную смесь кипятили с обратным холодильником в течение 6 часов. После охлаждения до комнатной температуры растворитель концентрировали при пониженном давлении, и полученный остаток обрабатывали смесью 30% гидроксида натрия (10 мл) и воды (100 мл), и рН реакционной смеси достигала 12. Осажденный трифенил метанол фильтровали и промывали водой (4×10 мл). Фильтрат экстрагировали смесью 1:1 (об/об) гексан:этилацетат (100 мл). Щелочной раствор подкисляли концентрированной HCl (10 мл) до рН 1 и экстрагировали гексаном (100 мл). Органический раствор высушивали над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, получая 8.5 г (S)-1-(2-октил)додецил-2-(4-карбокси)бутил-глицерина в виде желтого масла. Выход составил 60%.
(S)-1-(2-октил)додецил-2-(4'-карбоксиметил)бутил-глицерин: (S)-1 -(2-октил)додецил-2-(4-карбокси)бутил-глицерина (8.39 г) растворяли в метаноле (100 мл). Добавляли 2 мл концентрированной НС1 (32%), и раствор перемешивали при комнатной температуре в течение ночи. Добавляли воду (100 мл), и смесь экстрагировали хлороформом (3×100 мл). Объединенную органическую фазу последовательно промывали водой (100 мл), концентрированным раствором бикарбоната натрия (100 мл) и водой (100 мл), и затем высушивали над безводным сульфатом натрия. Удаление растворителя при пониженном давлении позволило получить 8.48 г (S)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-глицерина в виде желтого масла. Выход составил 98%.
(R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламин: (S)-1-(2-октил)додецил-2-(4'-карбоксиметил)бутил-глицерин (8.48 г) и триэтиламин (7.3 мл) растворяли в сухом THF (50 мл). Данный раствор добавляли по каплям в течение 60 минут в ледяной раствор POCl3 (4.85 мл) в THF (50 мл) при перемешивании. Перемешивание продолжали еще в течение 15 минут при охлаждении и еще в течение 45 минут при комнатной температуре. Эту реакционную смесь охлаждали на льду, и по каплям добавляли раствор этаноламина (3.2 мл) и триэтиламина (15 мл) в сухом THF (50 мл) в течение 60 минут при перемешивании. Перемешивание продолжали в течение 10 минут на льду и затем при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в смеси уксусной кислоты (24 мл) и воды (10 мл), и нагревали до 70°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и экстрагировали хлороформом (3×80 мл). Объединенную органическую фазу промывали водой (2×50 мл), и растворитель удаляли при пониженном давлении. Остаток (11 г) очищали хроматографией на силикагеле (220 г). 4.25 г чистого (R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламина элюировали хлороформом с последующими смесями хлороформа с 5%-20% метанола и в конце смесью 70:26:4 хлороформ:метанол:вода. Выход составил 40%.
(R)-1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфоэтаноламин (VB-222): (R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламин (1.2 г) растворяли в 100 мл смеси 8:2 (об/об) метанол: 10% раствор гидроксида натрия. Реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакционной смеси доводили до 5 добавлением однозамещенного фосфорнокислого натрия. Добавляли воду (100 мл) и хлороформ (100 мл). Фазы разделяли, и растворитель из органической фазы удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, и растворитель удаляли при пониженном давлении. Сырой продукт (1.2 г) очищали хроматографией на силикагеле (23 г). Продукт элюировали смесями 8:2 (об/об) хлороформ:метанол с последующими смесями хлороформ: вода в объемном соотношении 70:26:4 и затем 60:35:5. Растворитель из фракций, содержащих требуемый продукт, удаляли при пониженном давлении, полученный остаток растворяли в хлороформе и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении, получая 500 мг чистого (7?М-(2-октил)додецил-2-(4-карбокси)бутил-.уи-глицеро-3-фосфоэтаноламина в виде воска. Выход составил 42.65%.
ЯМР характеристика 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
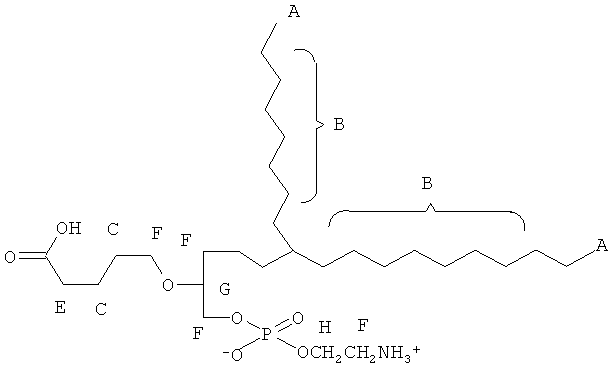
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.313 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина, были обозначены следующим образом:
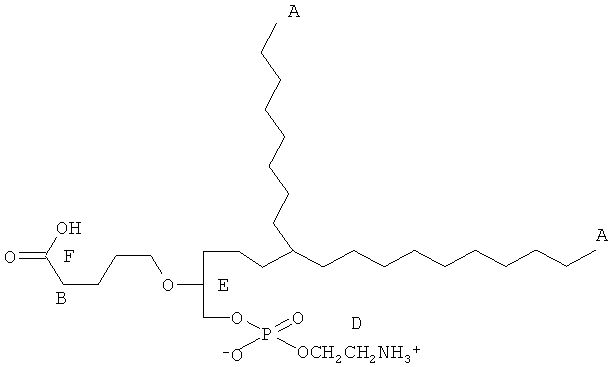
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.189 м.д.)
Масс-спектрометрическая характеристика 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина:
Вычисленная масса для 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина (C30H62NO8P) составила 595.42.
Масс-спектр, полученный методом масс-спектрометрии с электрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=596, соответствующего протонированному молекулярному иону [М+Н]+, сопровождаемому молекулярным ионом с m/z=618, соответствующим катионированному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфоэтаноламина.
(R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолин: (R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфоэтаноламин (2.62 г) растворяли в смеси изопропанола (18 мл) и дихлорметана (40 мл). Раствор карбоната калия (3 г) в воде (10 мл) добавляли по каплям при поддержании температуры реакционной смеси 35-40°С. Раствор диметилсульфата (2.1 мл) в изопропаноле (10 мл) добавляли по каплям при 40°С. Реакционную смесь выдерживали при 40°С в течение 2 часов, затем охлаждали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Добавляли воду (100 мл), и смесь экстрагировали хлороформом (3×100 мл). Объединенную органическую фазу промывали водой (100 мл), и растворитель удаляли при пониженном давлении, получая 2.78 (R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолина в виде белого воска.
(R)-1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолин (VB-221): (R)-1-(2-октил)додецил-2-(4-метилкарбокси)бутил-sn-глицеро-3-фосфохолин (2.78 г) растворяли в 100 мл смеси 8:2 (об/об) метанола и 10% водного раствора гидроксида натрия, и реакционную смесь перемешивали при комнатной температуре в течение ночи. РН реакции доводили до 5 добавлением однозамещенного фосфорнокислого натрия. Добавляли воду (100 мл) и хлороформ (100 мл). Фазы разделяли, и растворитель удаляли при пониженном давлении. Полученный остаток растворяли в хлороформе, высушивали над сульфатом натрия и фильтровали, после чего растворитель удаляли при пониженном давлении. Остаток (2.7 г) очищали хроматографией на силикагеле (50 г). Неполярные примеси элюировали смесью 8:2 (об/об) хлороформ:метанол. Продукт затем элюировали смесями хлороформ:метанол:вода при объемном соотношении 70:26:4, с последующим 60:35:5. После удаления растворителя при пониженном давлении полученный остаток растворяли в хлороформе и высушивали над сульфатом натрия, и растворитель удаляли при пониженном давлении, получая 800 мг чистого (R)-1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина в виде белого воска. Выход составил 29.4;.
ЯМР характеристика 1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина:
Образец растворяли в дейтерированном хлороформе (CDCl3) с несколькими каплями дейтерированного метанола (CD3OD). Спектры измеряли при 600 МГц. Образцы измеряли методом 1Н и 13С ЯМР-спектроскопии.
Результаты показали наличие предполагаемых сигналов для структурных элементов 1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина и, таким образом, полностью подтвердили данную структуру.
Наблюдаемые пики 1Н, соответствующие структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина, были обозначены следующим образом:
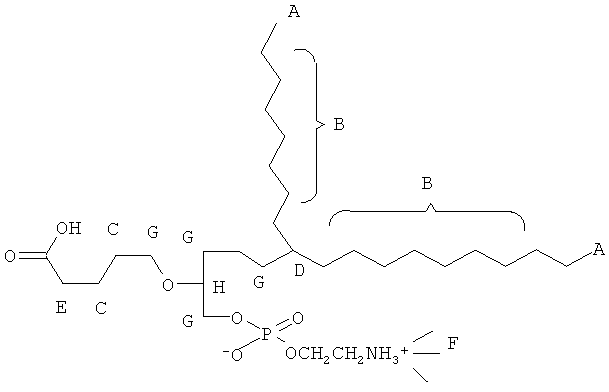
1H ЯМР (600 МГц, эталонный растворитель (CDCl3)=7.352 м.д.)
Наблюдаемые пики 13С, соответствующие структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-sn-глицеро-3-фосфохолина, были обозначены следующим образом:
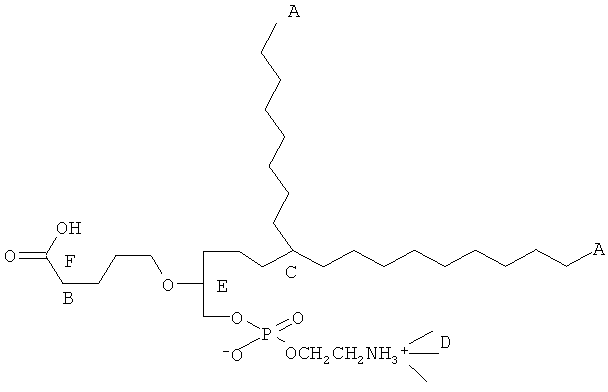
13С ЯМР (600 МГц, эталонный растворитель (CDCl3)=77.308 м.д.)
Масс-спектрометрическая характеристика:
Вычисленная масса для 1-(2-октил)додецил-2-(4-карбокси)бутил-зп-глицеро-3-фосфохолина (C33H68NO8P) составила 637.87.
Масс-спектр, полученный методом масс-спектрометрии с элетрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=636, соответствующего депротонированному молекулярному иону [М-Н]+.
Масс-спектр, полученный методом масс-спектрометрии с элетрораспылительной ионизацией (ESI+-MS), показал наличие молекулярного иона с m/z=638, соответствующего протонированному молекулярному иону [М+Н]+ сопровождаемому ионом с m/z=660, соответствующим катионированному молекулярному иону [M+Na]+.
Таким образом, спектр, полученный методом масс-спектрометрии, соответствует химической структуре 1-(2-октил)додецил-2-(4-карбокси)бутил-глицеро-3-фосфохолина.
Фосфорилирование тирозина:
Действие VB-221 и VB-222 на in vitro фосфорилирование тирозина в первичных макрофагах определяли, как описано здесь выше в разделе «Материалы и Способы».
Как показано в Фиг.38, обработка 5, 10 и 20 мкг/мл (8, 16 и 32 мкМ) VB-221 вызывала фосфорилирование тирозина.
Аналогично, как показано на Фиг.39, обработка 10 и 20 мкг/мл (16.8 и 33.6 мкМ) VB-222 приводит к индуцированию фосфорилированию тирозина.
Несмотря на то, что изобретение было описано в сочетании с их конкретными вариантами, очевидно, что много альтернативных вариантов, модификаций и вариаций будут понятны опытным в данной области специалистам. Соответственно, предполагается включение всех таких альтернативных вариантов, модификаций и вариаций, которые соответствуют духу и объему прилагаемой формулы.
Все публикации, патенты и патентные заявки, упомянутые в данном описании, включены полностью в виде ссылки в описание, в таком же объеме, как если отдельная публикация, патент или патентная заявка были специально и индивидуально были включены здесь в виде ссылки. К тому же, размещение или идентификация любой ссылки в данной заявке не должно рассматриваться как допущение того, что такая ссылка существует как уровень техники для настоящего изобретения. В той части, в какой такие заголовки разделов используются, они не должны рассматриваться как обязательное ограничение.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКИСЛЕННЫЕ ЛИПИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2004 |
|
RU2362567C2 |
| ОКИСЛЕННЫЕ ЛИПИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2009 |
|
RU2482854C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ОКИСЛЕННЫХ ФОСФОЛИПИДОВ | 2005 |
|
RU2399626C2 |
| Способ получения трифенилметильных производных S @ -глицеро-3-фосфохолина и S @ -глицеро-3-фосфоэтаноламина | 1985 |
|
SU1422999A3 |
| Способ получения производных глицерофосфолипидов | 1985 |
|
SU1400511A3 |
| СПОСОБ ПОЛУЧЕНИЯ 1-АЛКИЛ-2-АЛКИЛКАРБАМОИЛГЛИЦЕРИНОВ | 2016 |
|
RU2621347C1 |
| Способ получения модифицированных кристаллов магнетита | 2018 |
|
RU2689392C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУПРАМОЛЕКУЛЯРНЫХ ПЛАТИНОВЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2795256C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЛАТИНЫ И ЛИПИДОВ И НАНОЧАСТИЦЫ | 2014 |
|
RU2737735C2 |
| ЛИПИДНОЕ СОЕДИНЕНИЕ И КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2021 |
|
RU2825571C1 |
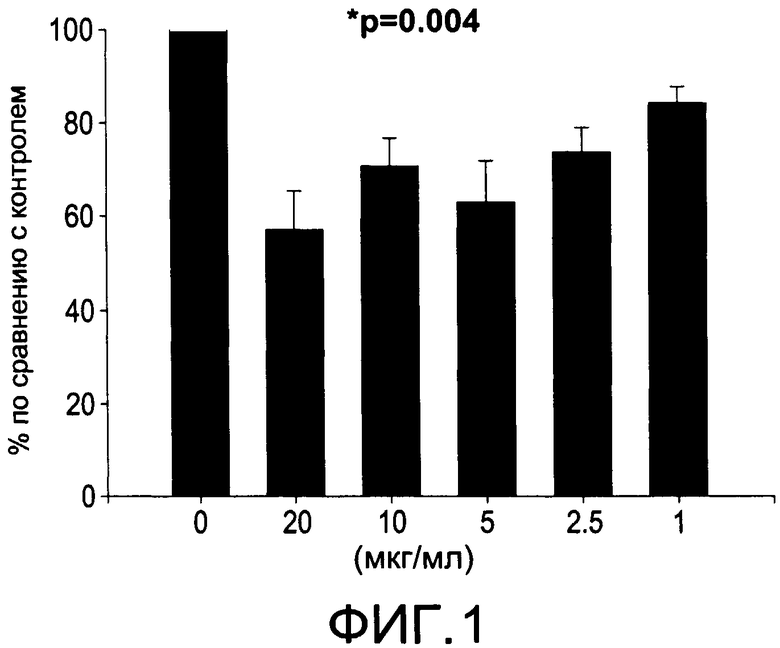
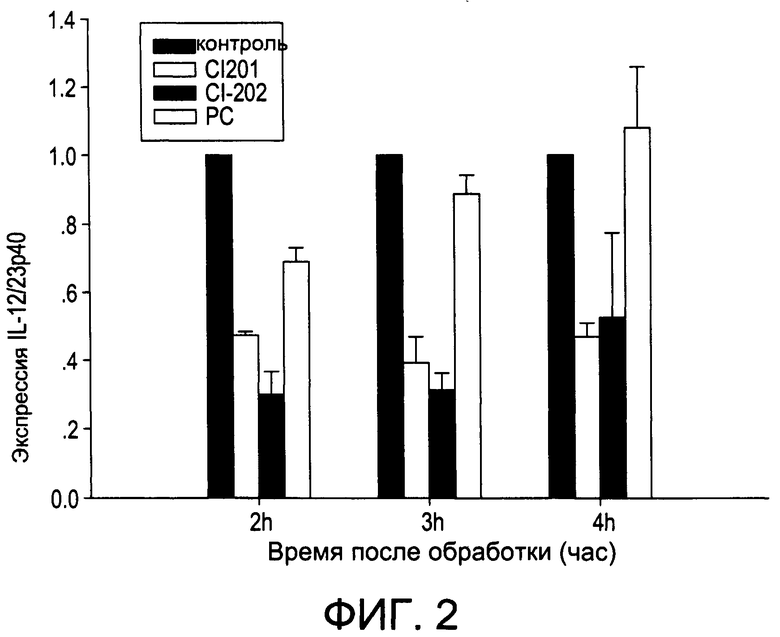
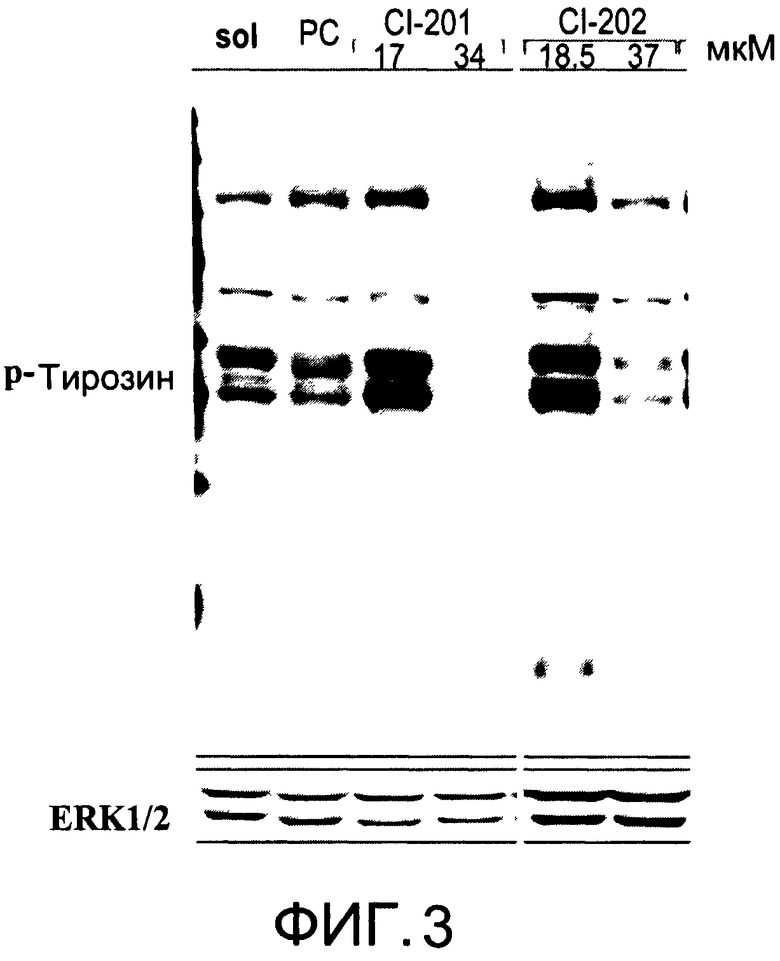
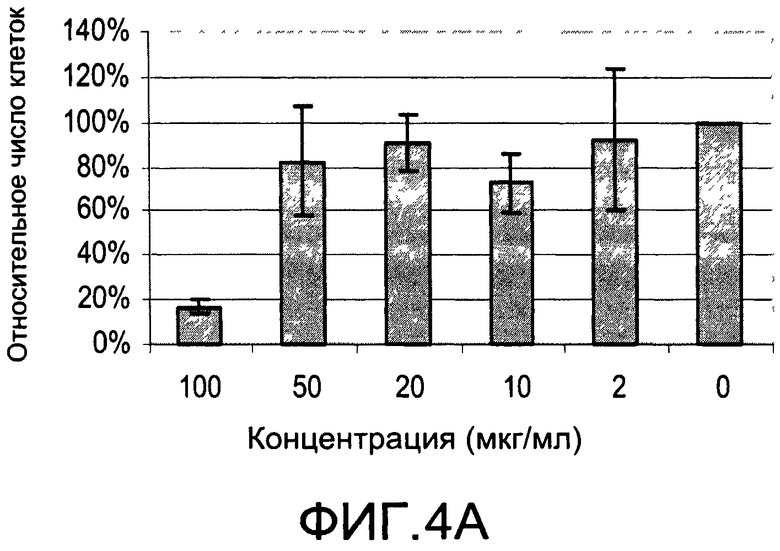
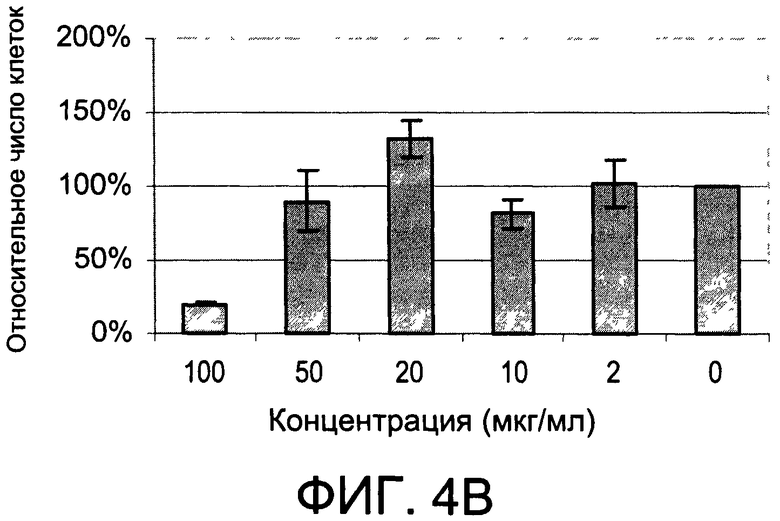
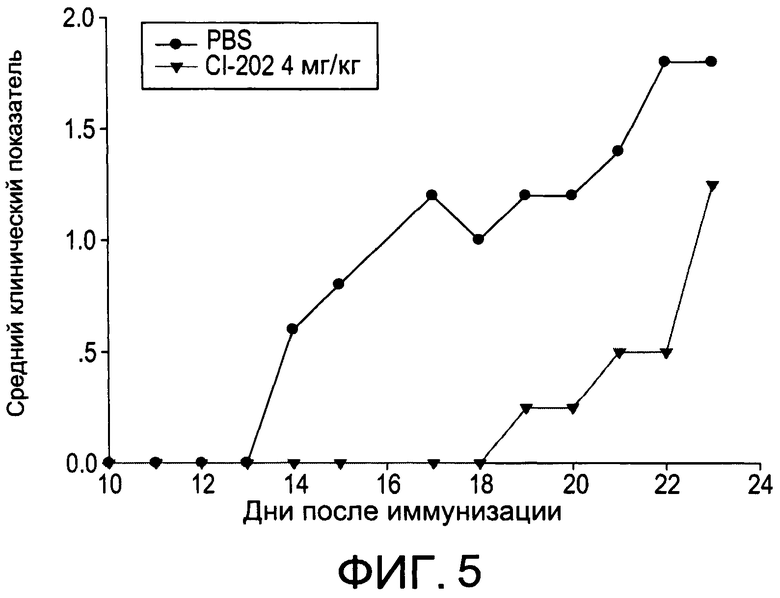
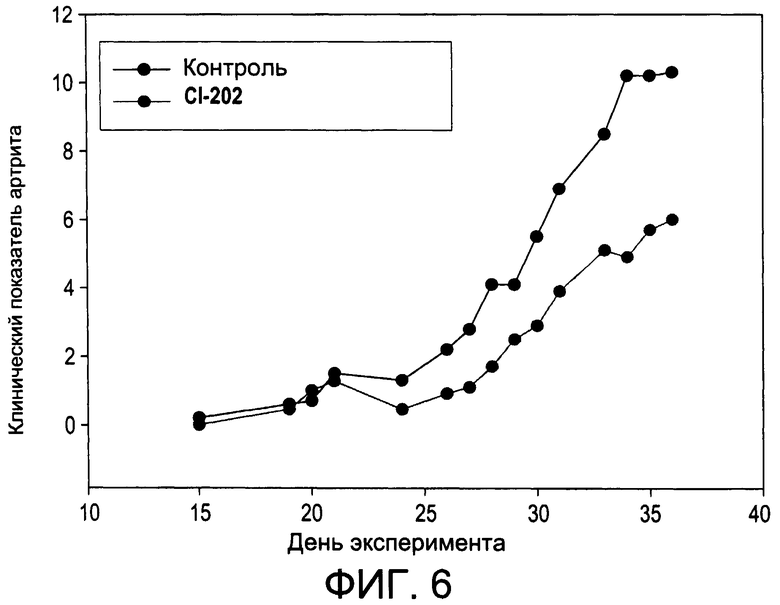
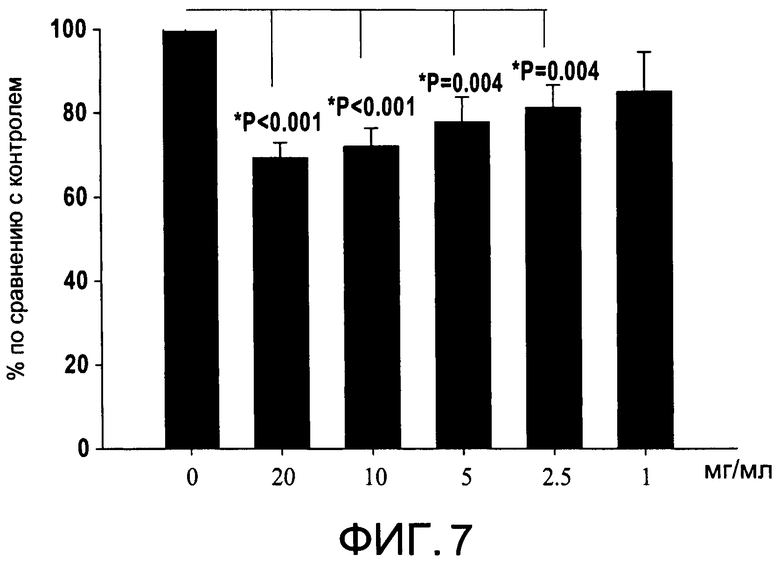
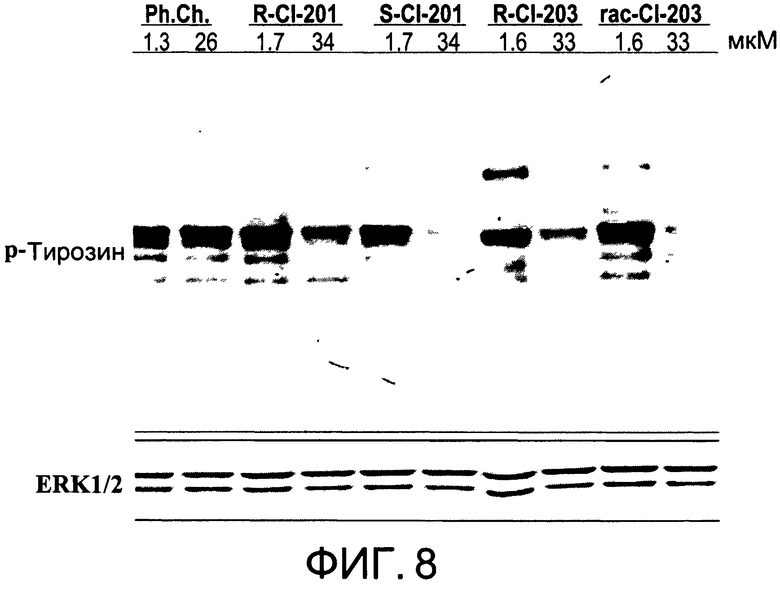
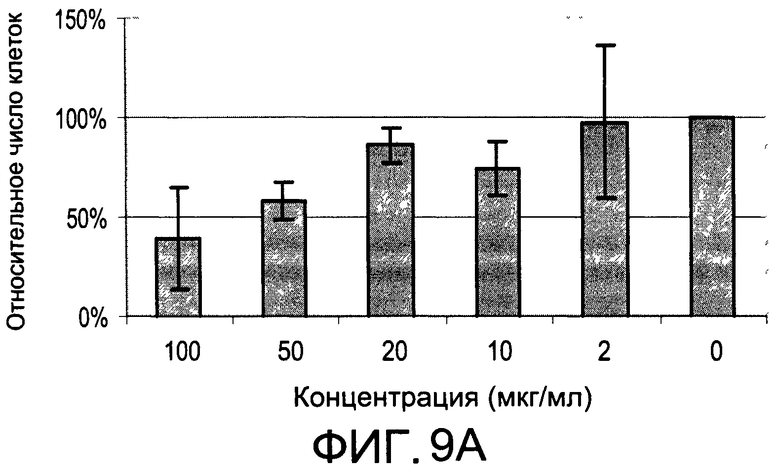
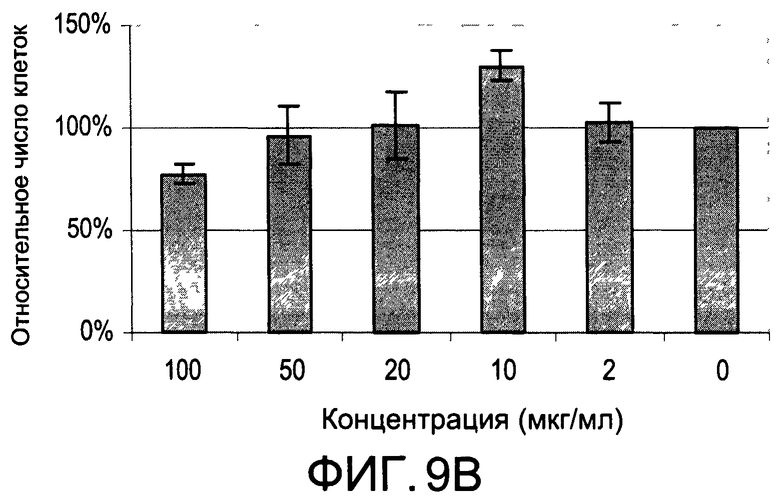
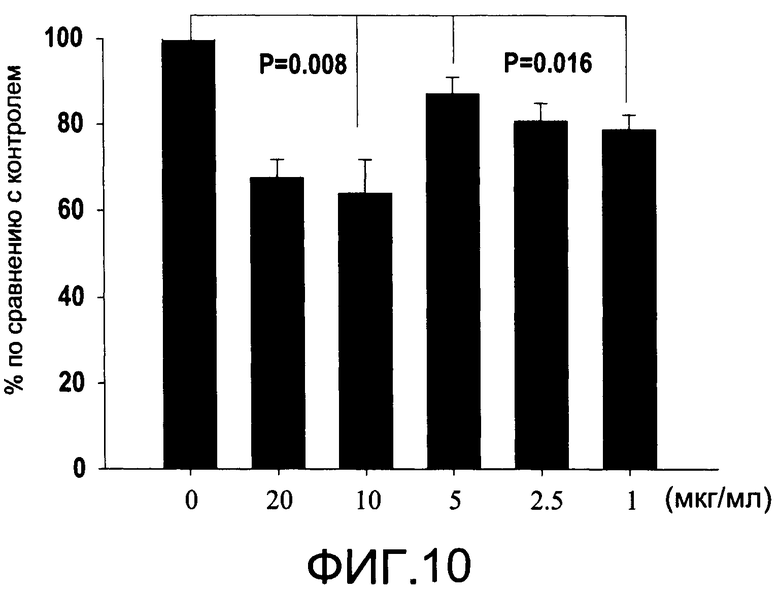
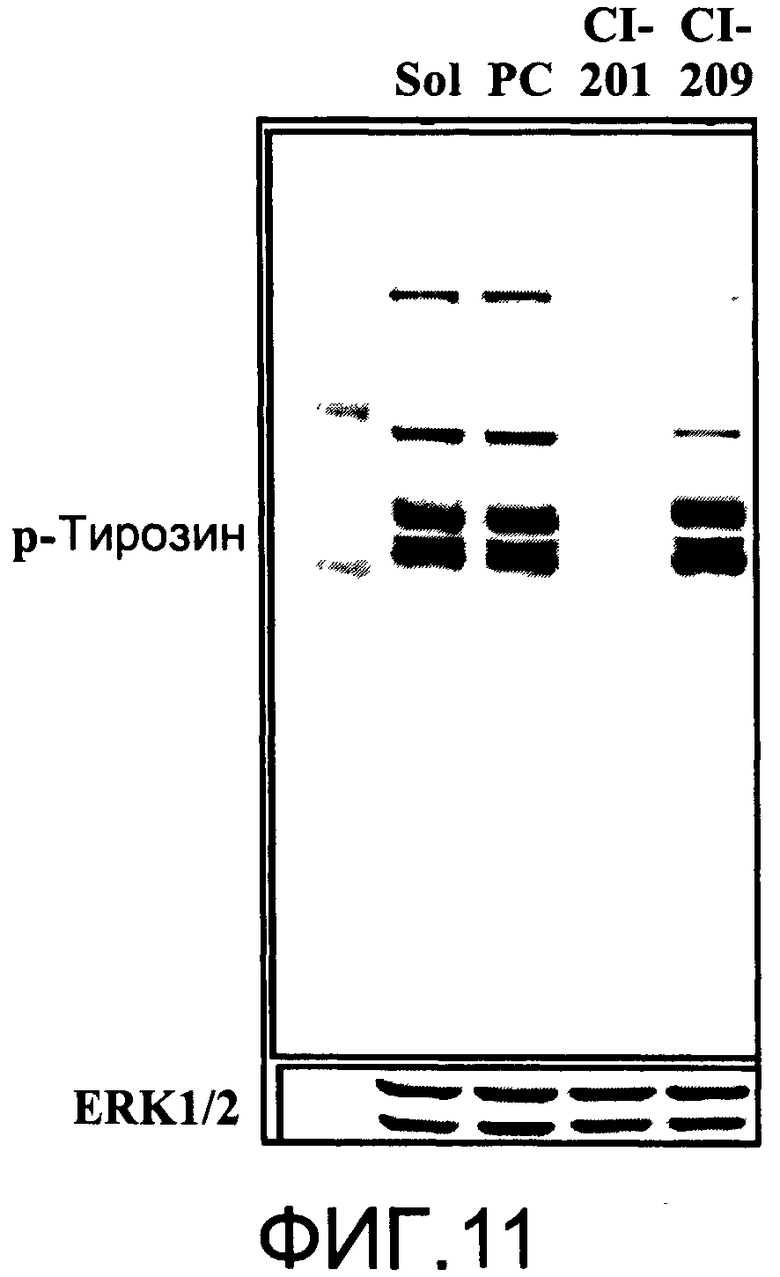
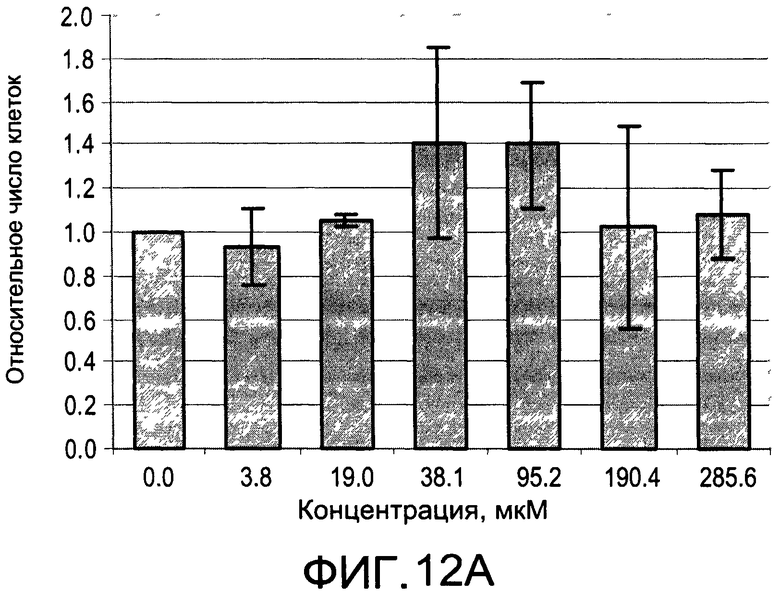
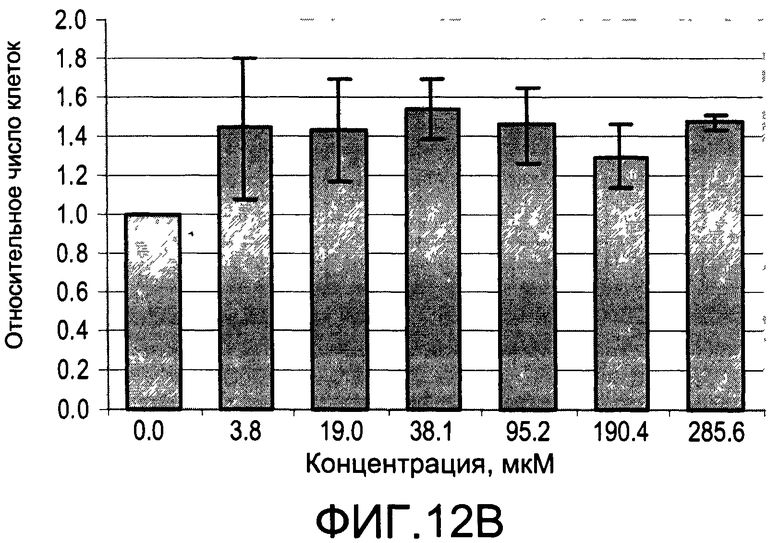
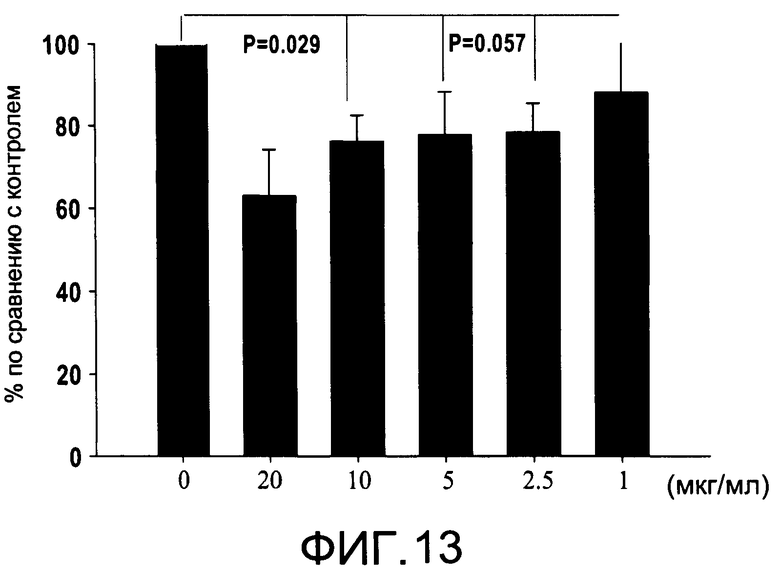
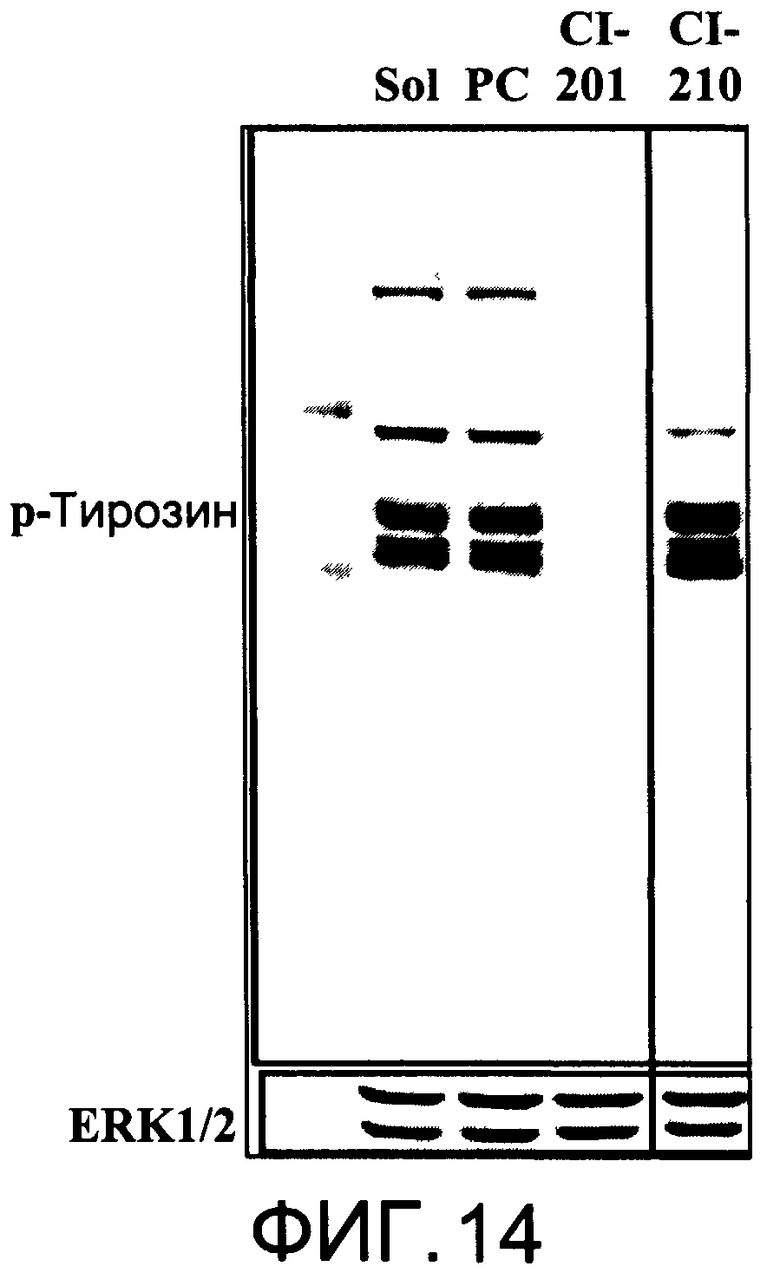
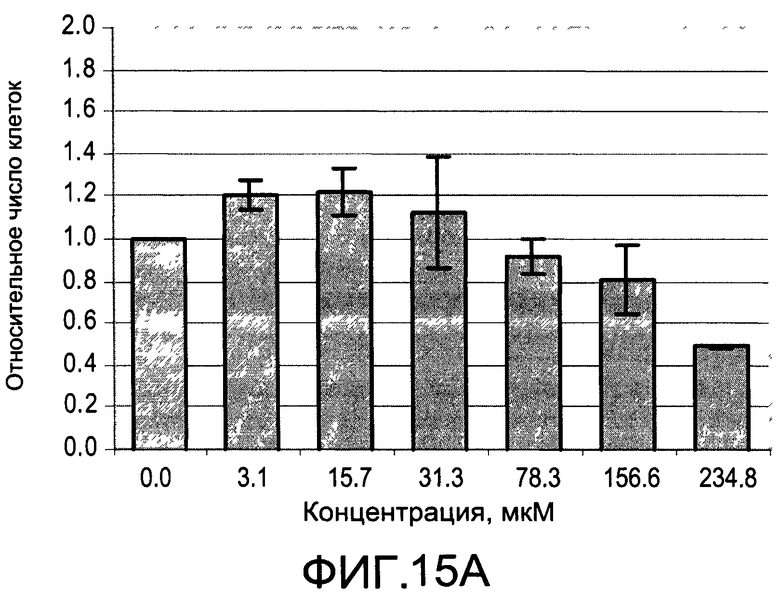
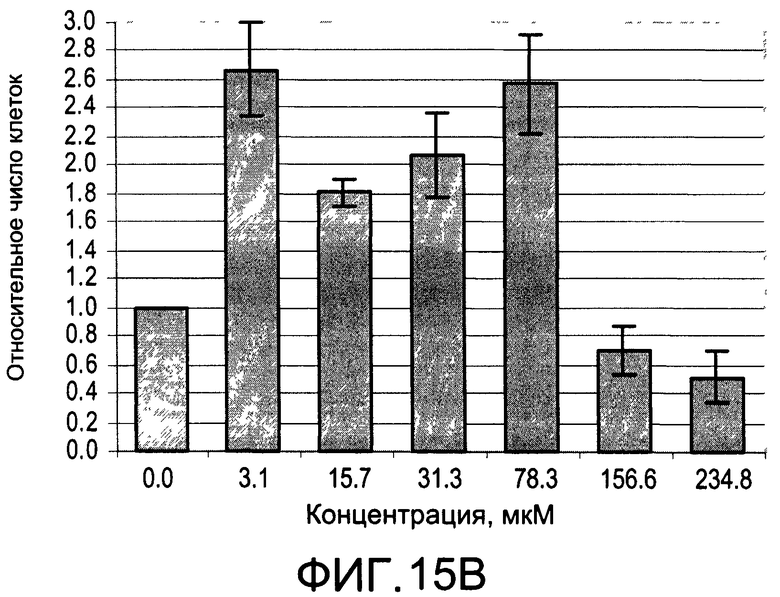
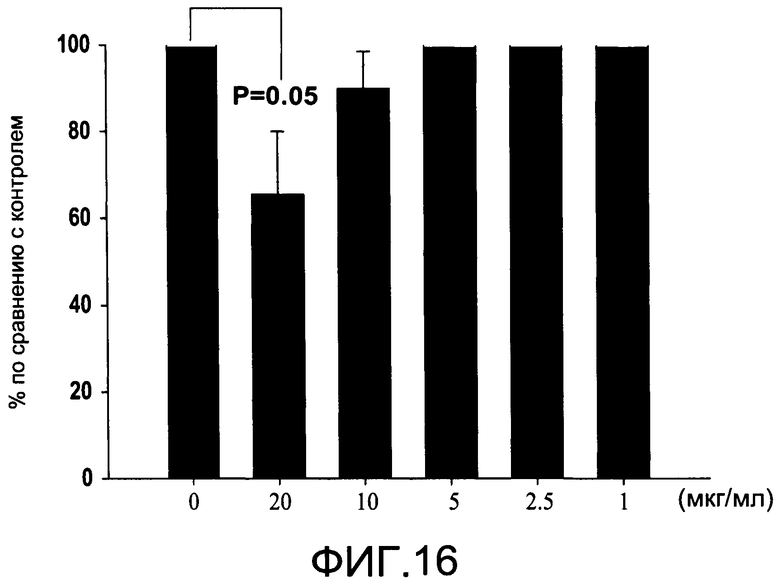
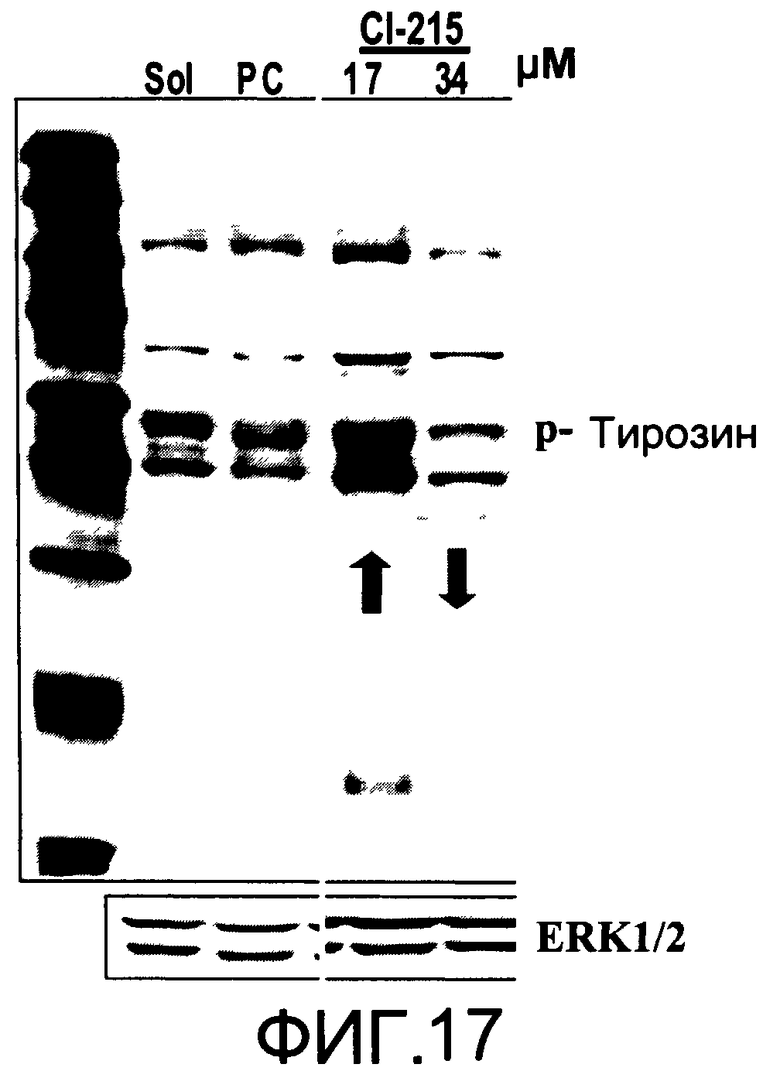
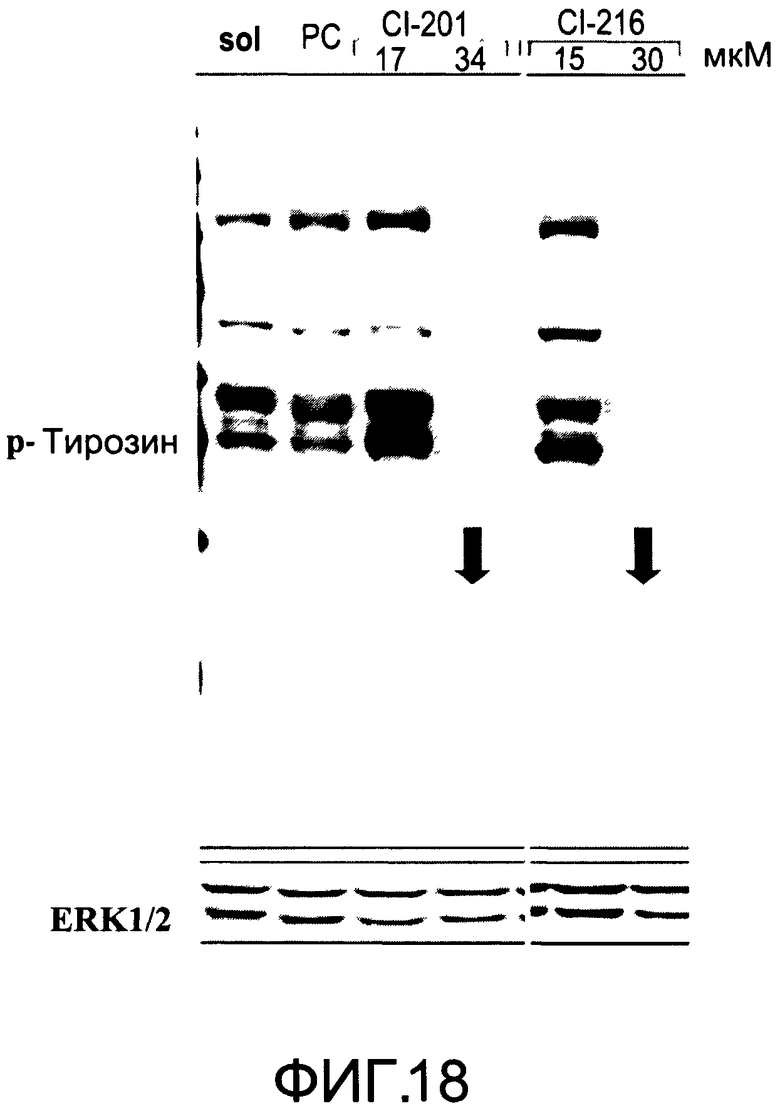
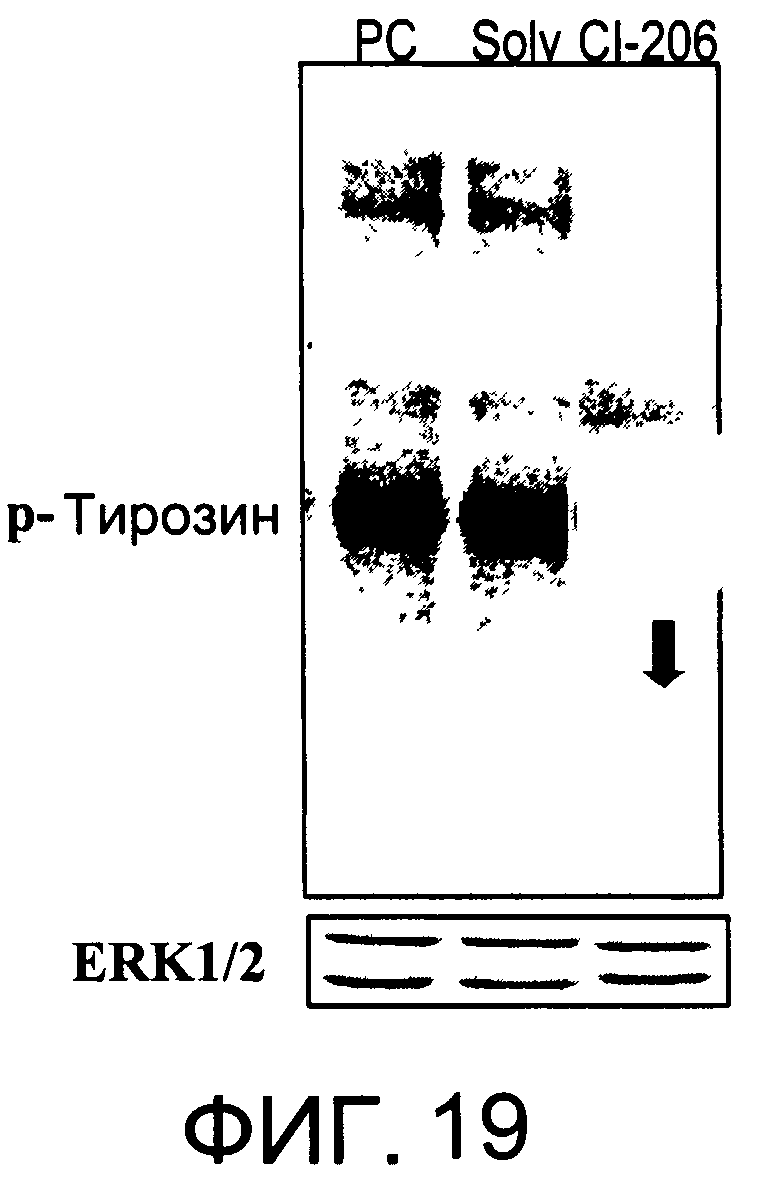
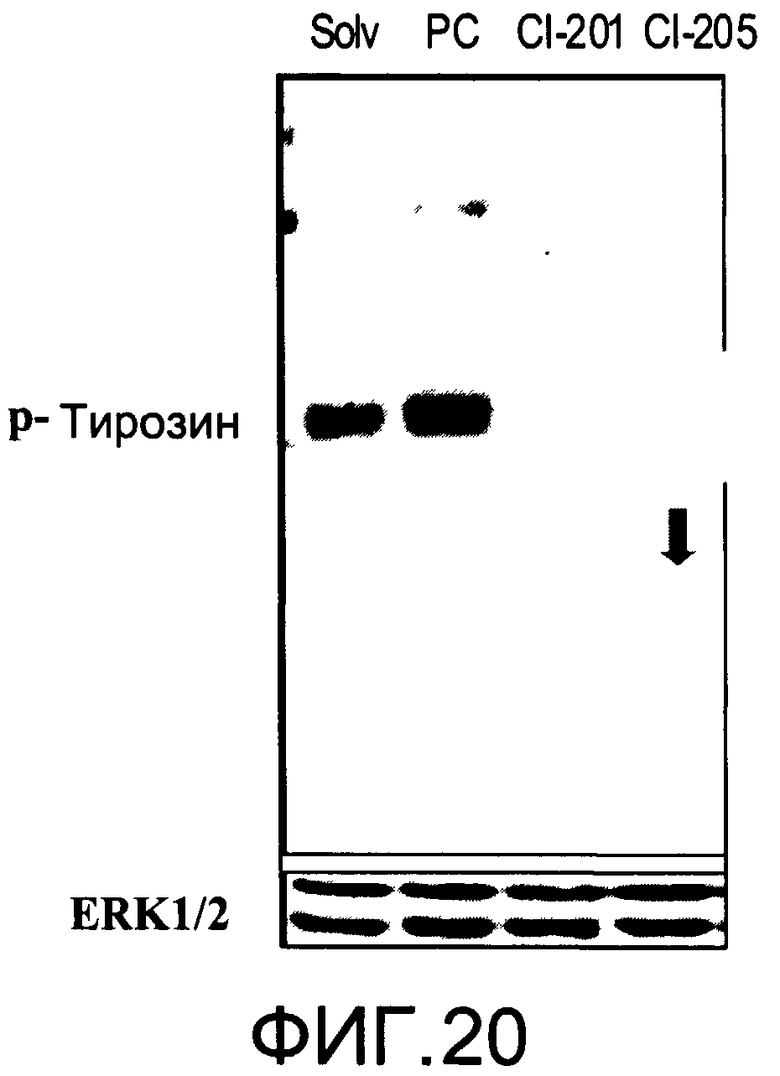
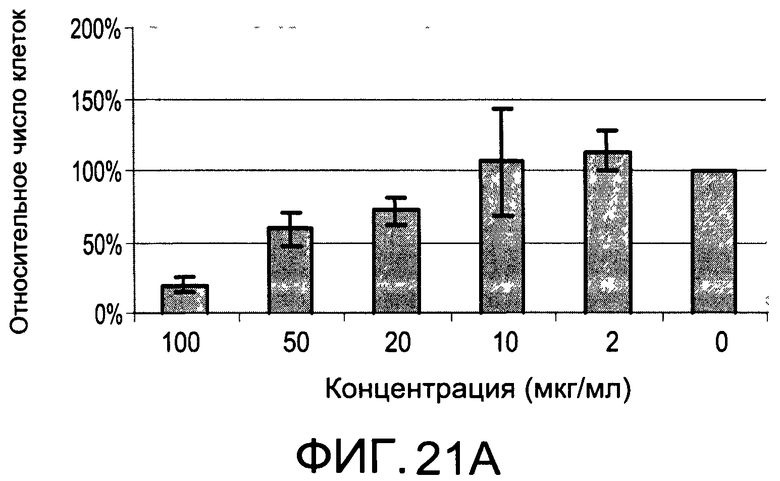
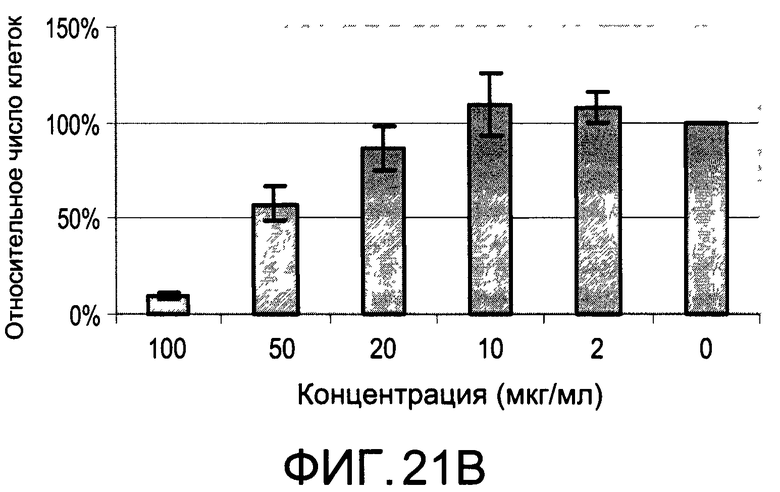
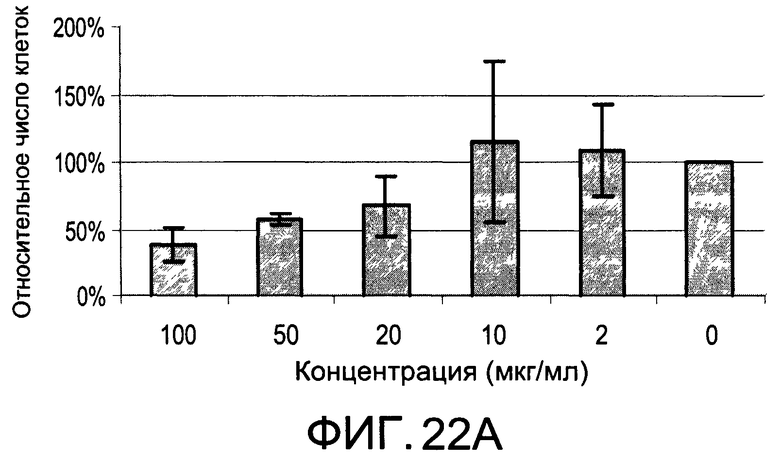
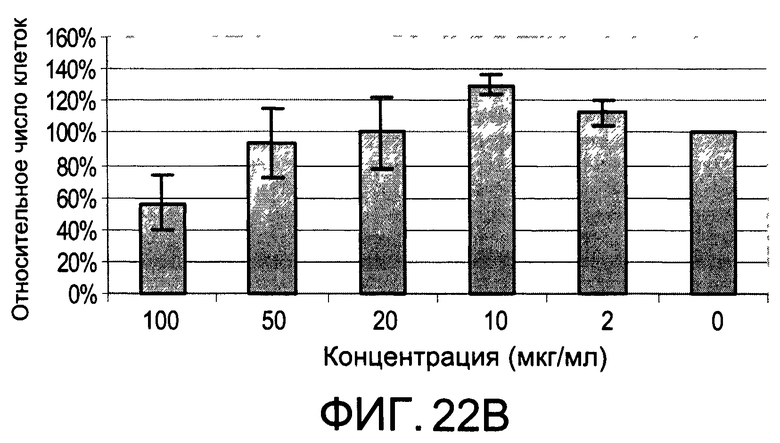
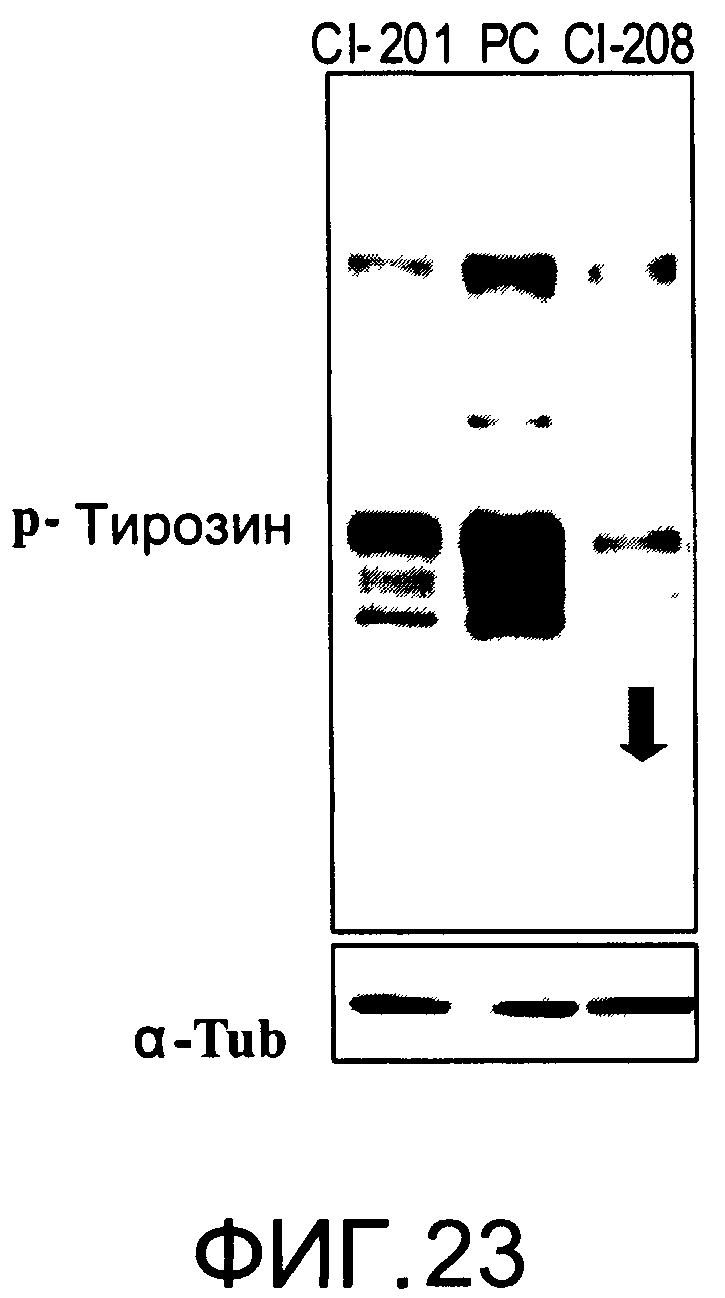
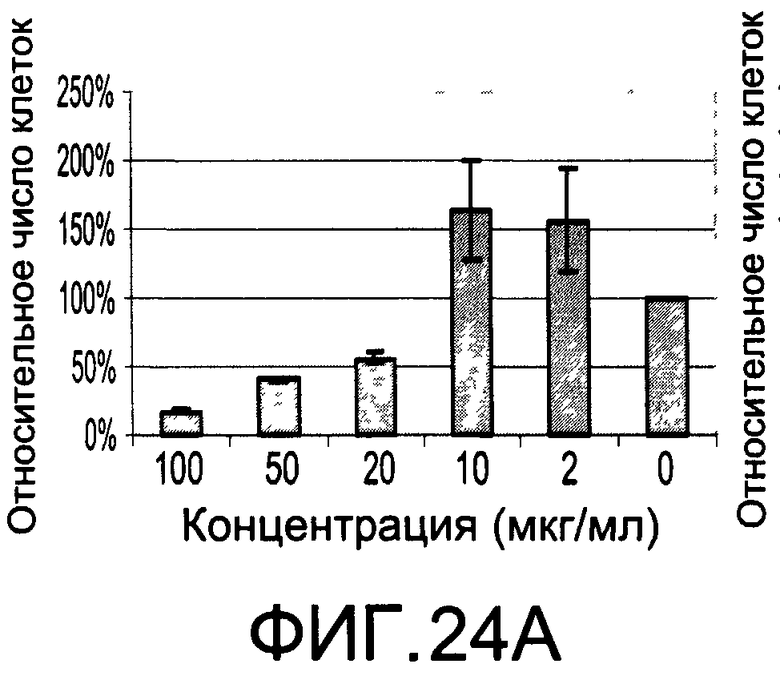
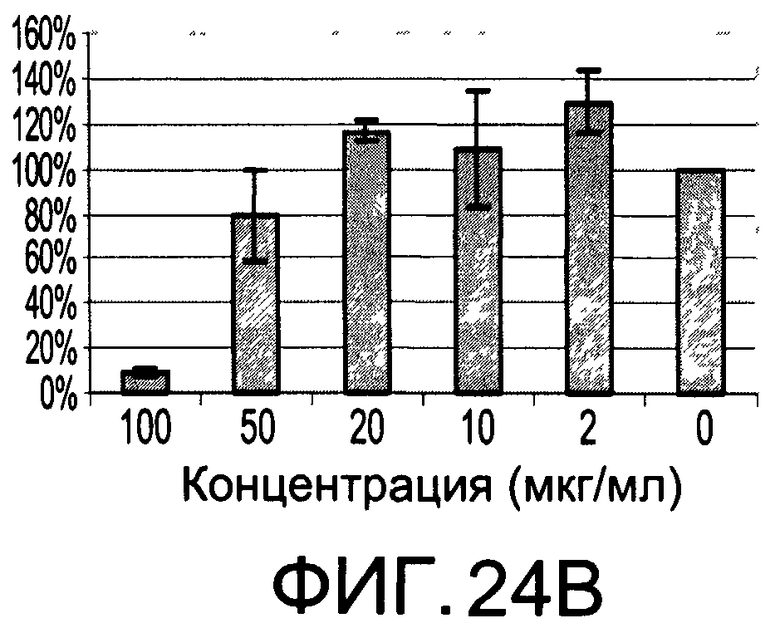
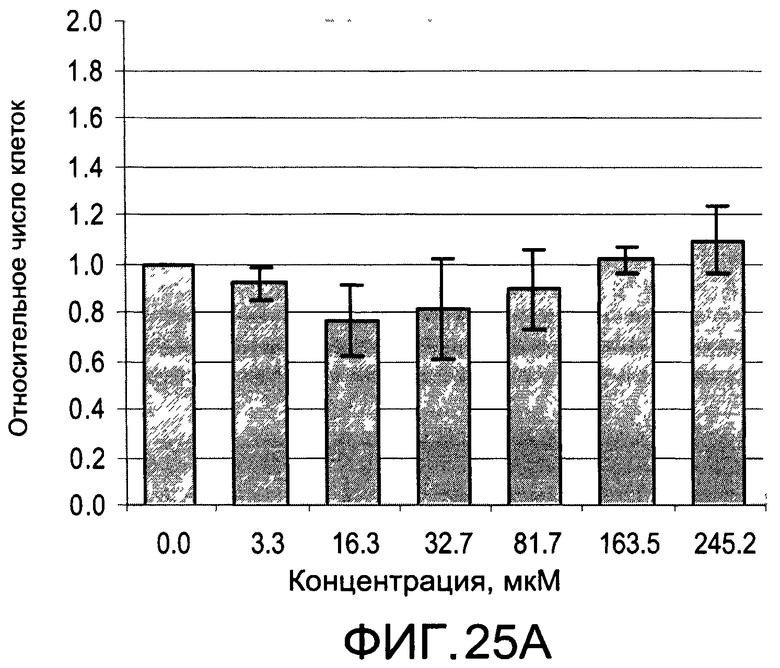
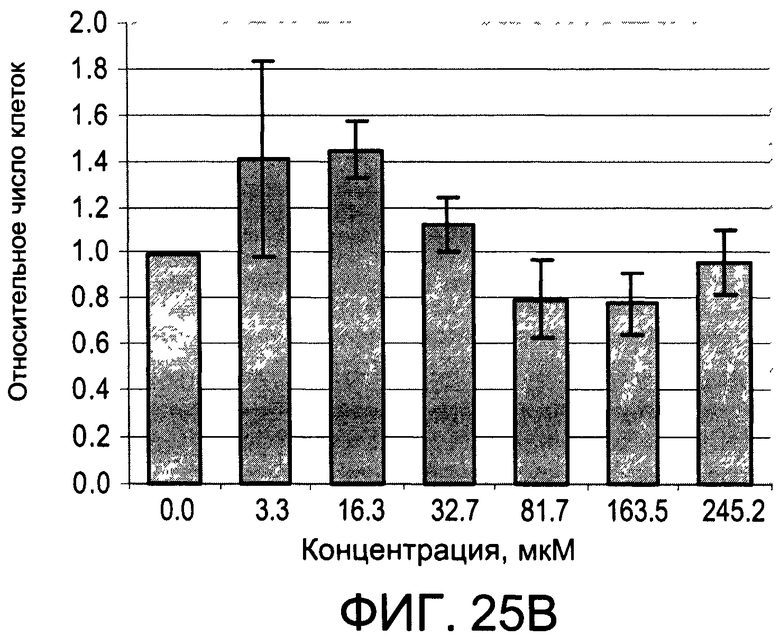
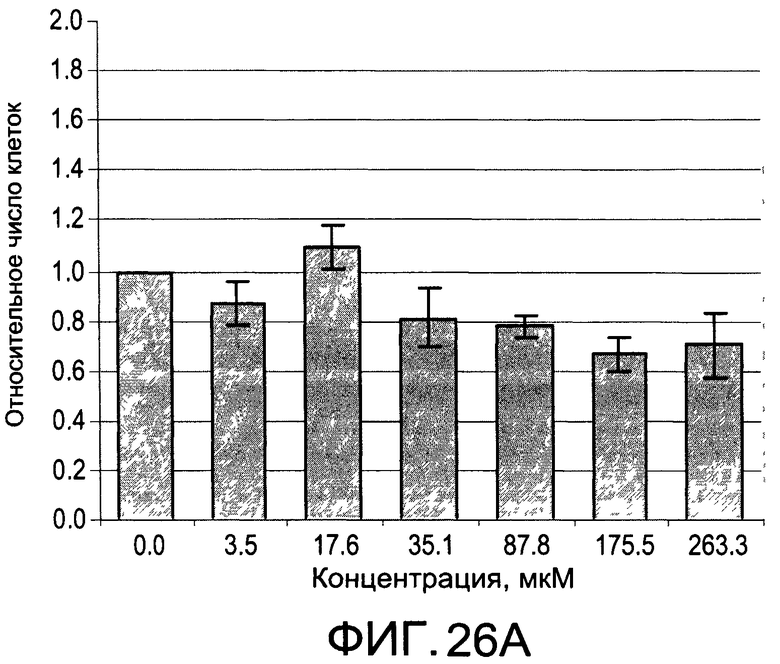
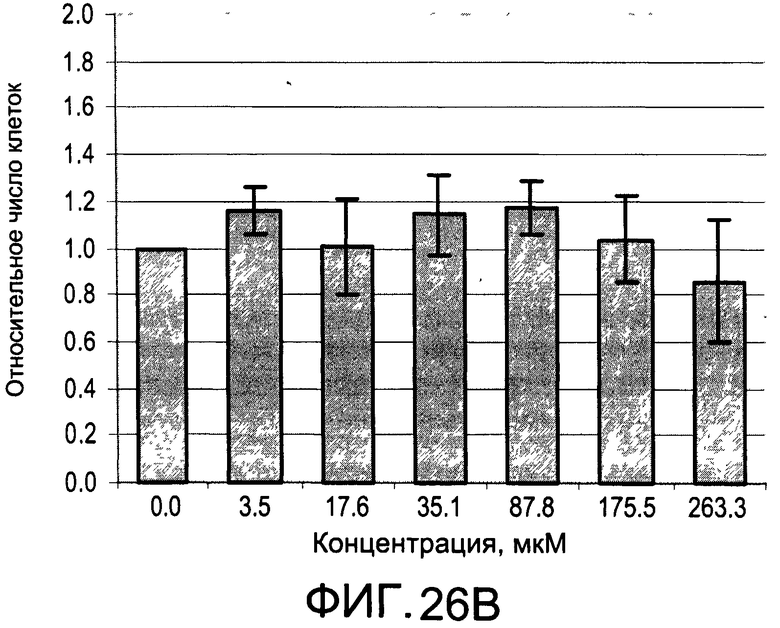
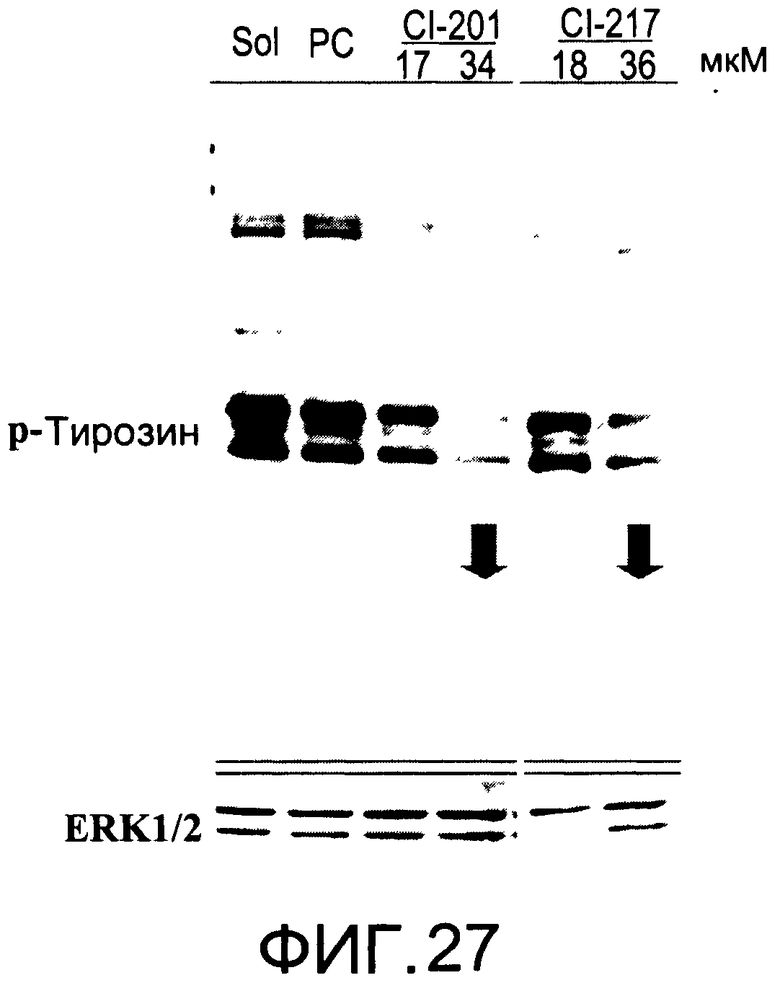
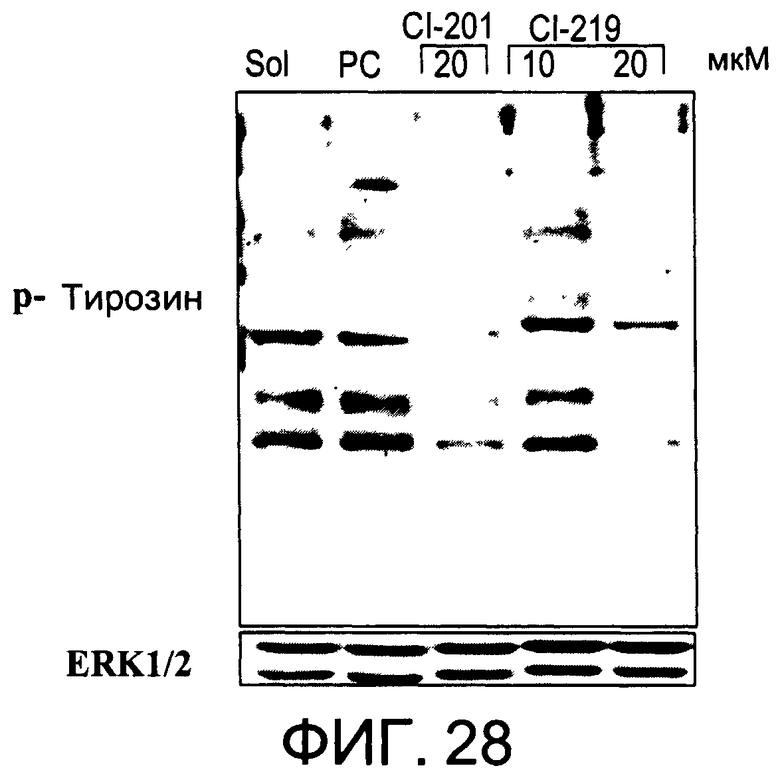
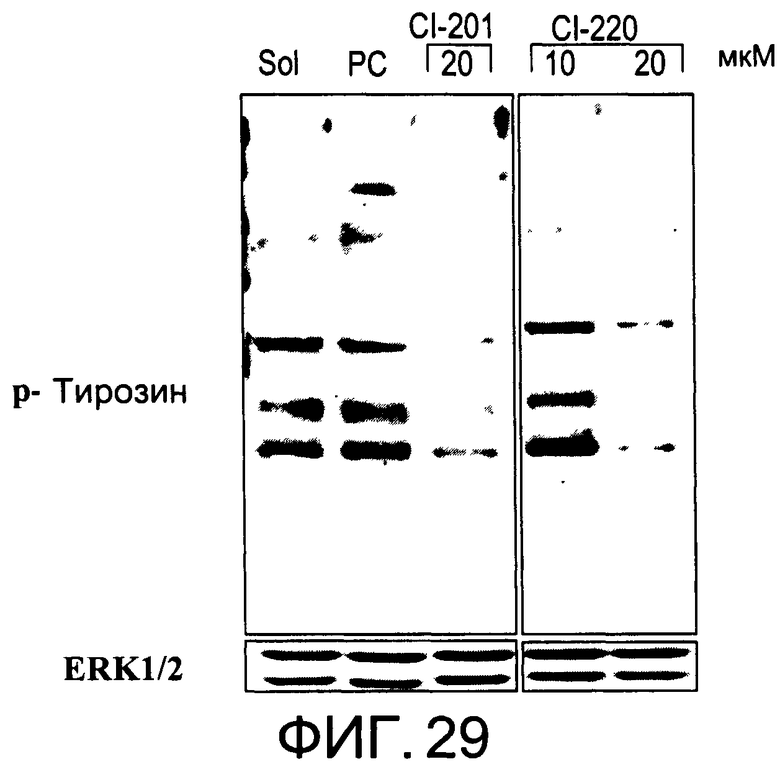
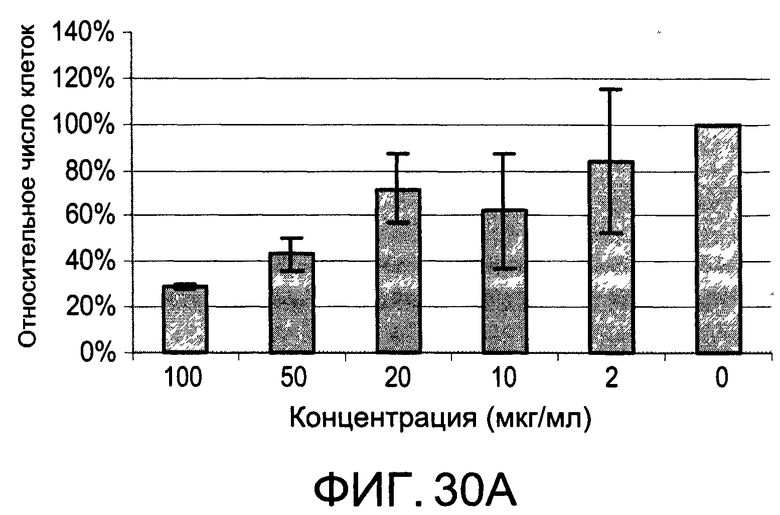
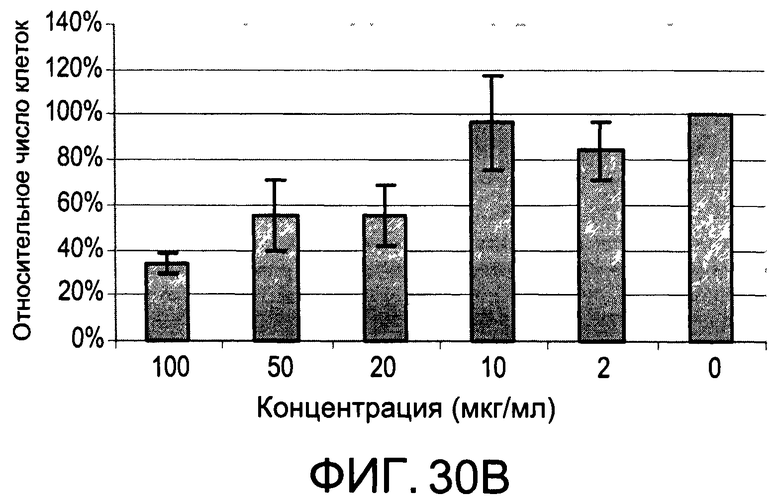
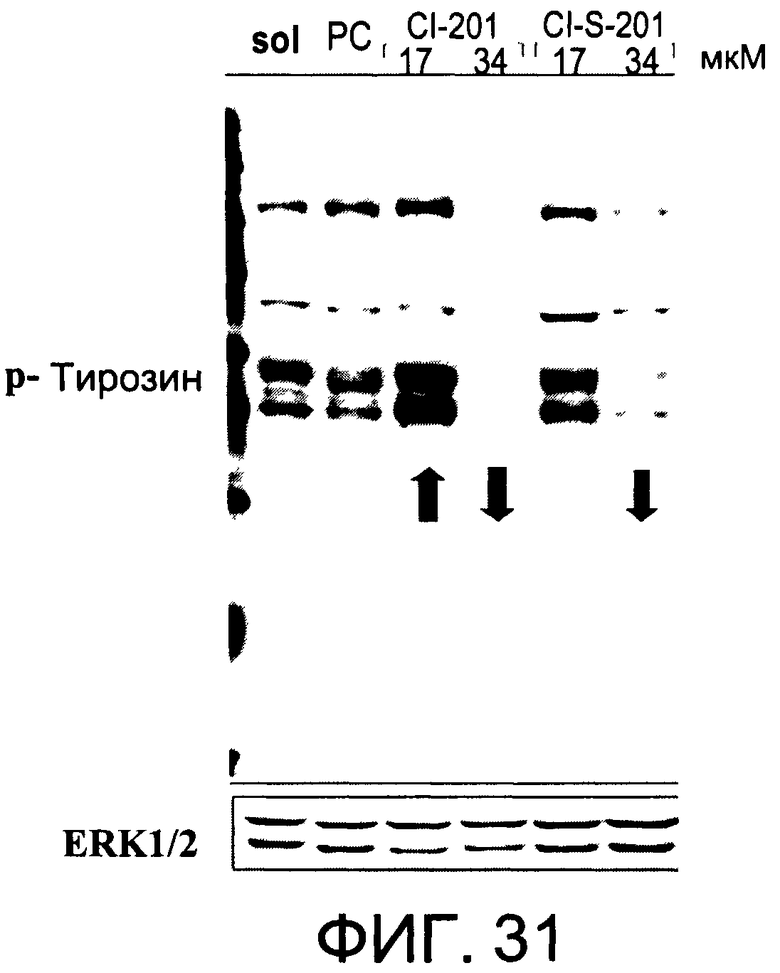
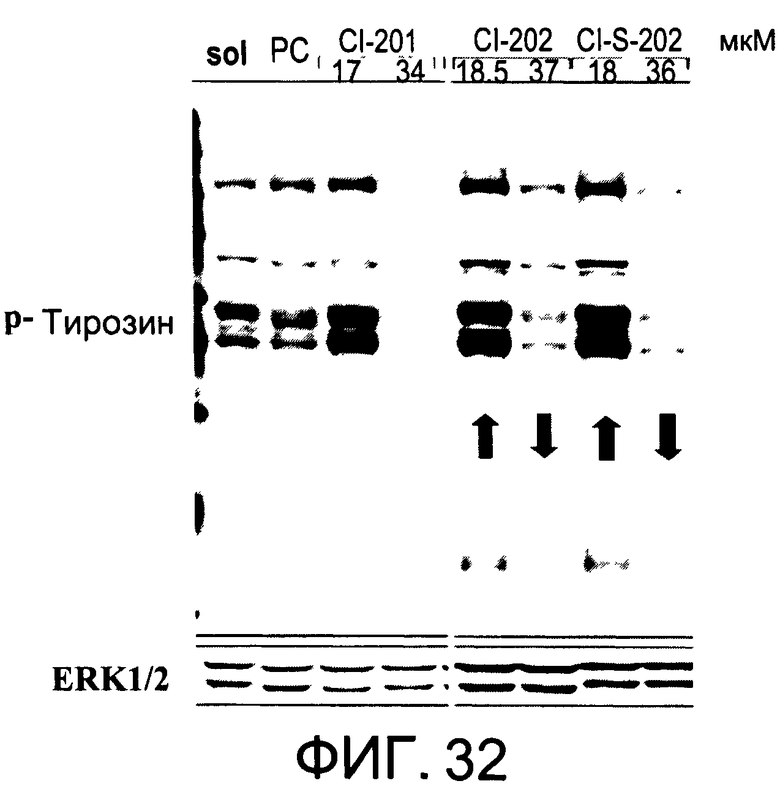
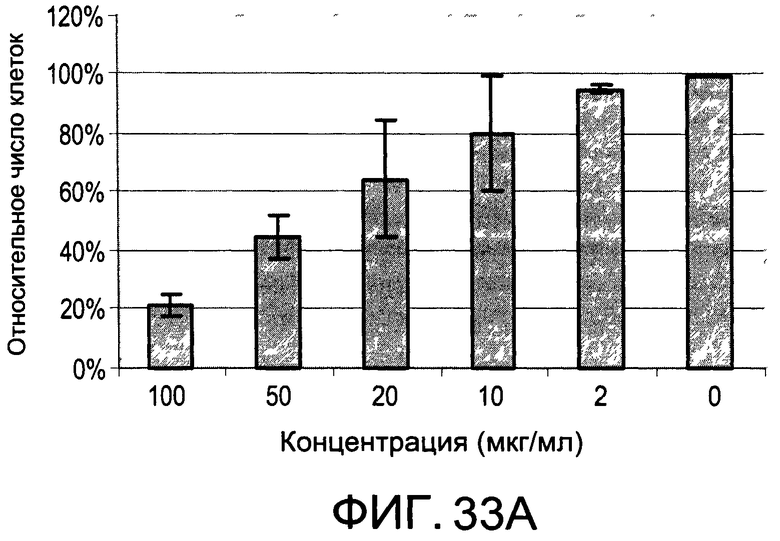
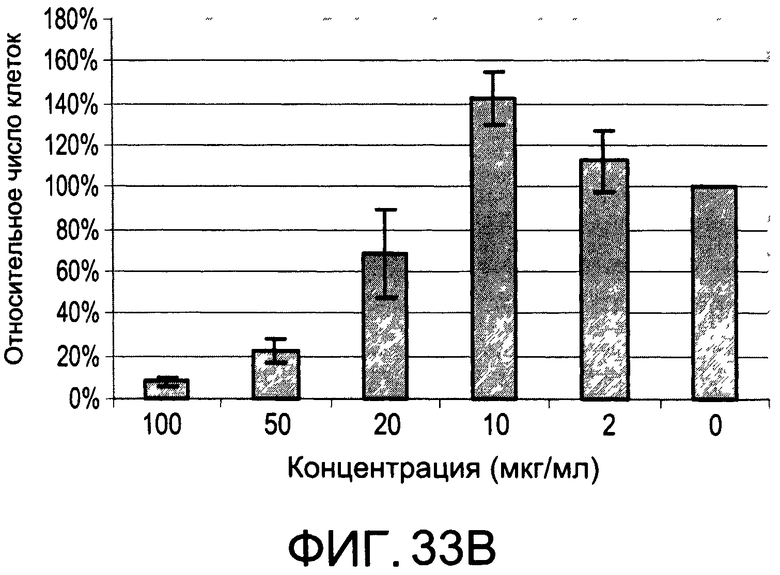
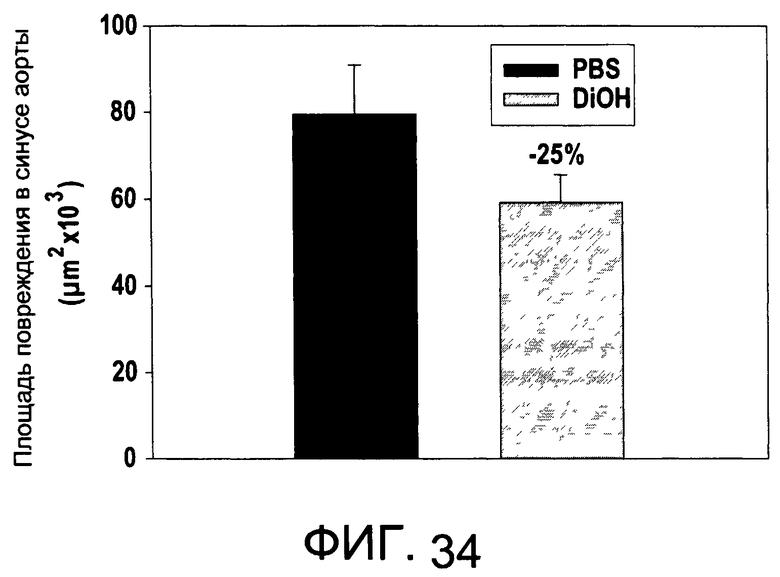
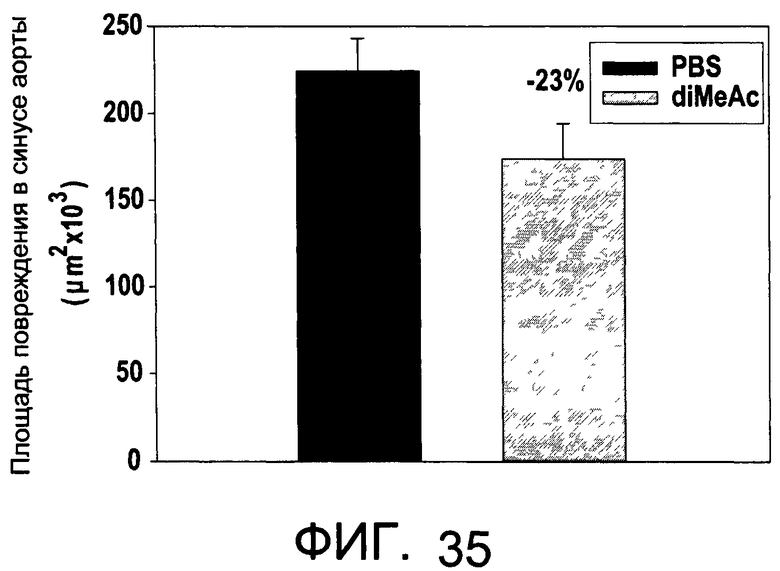
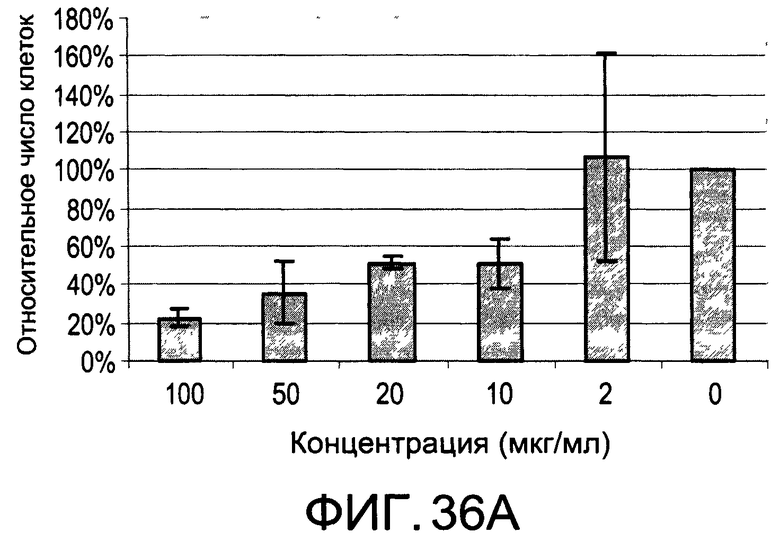
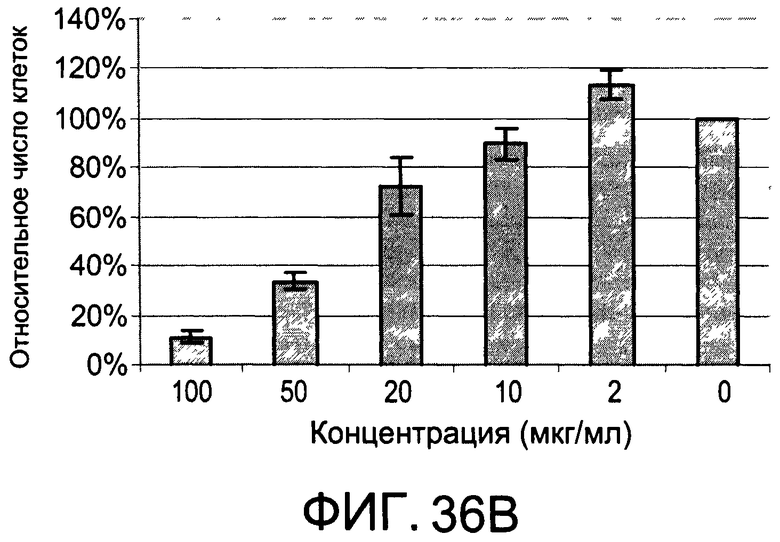
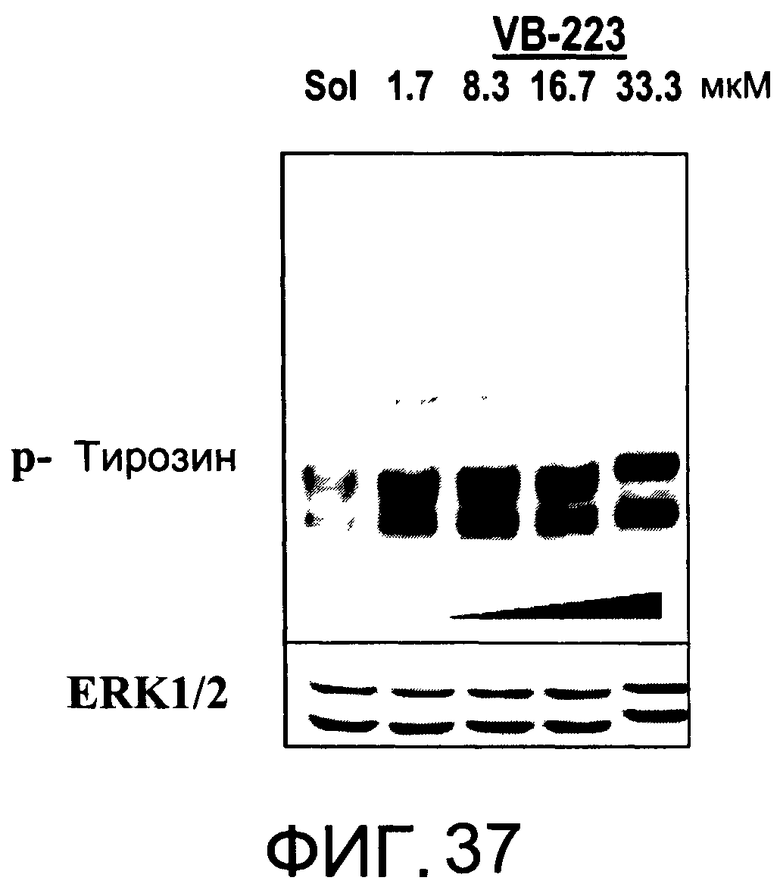
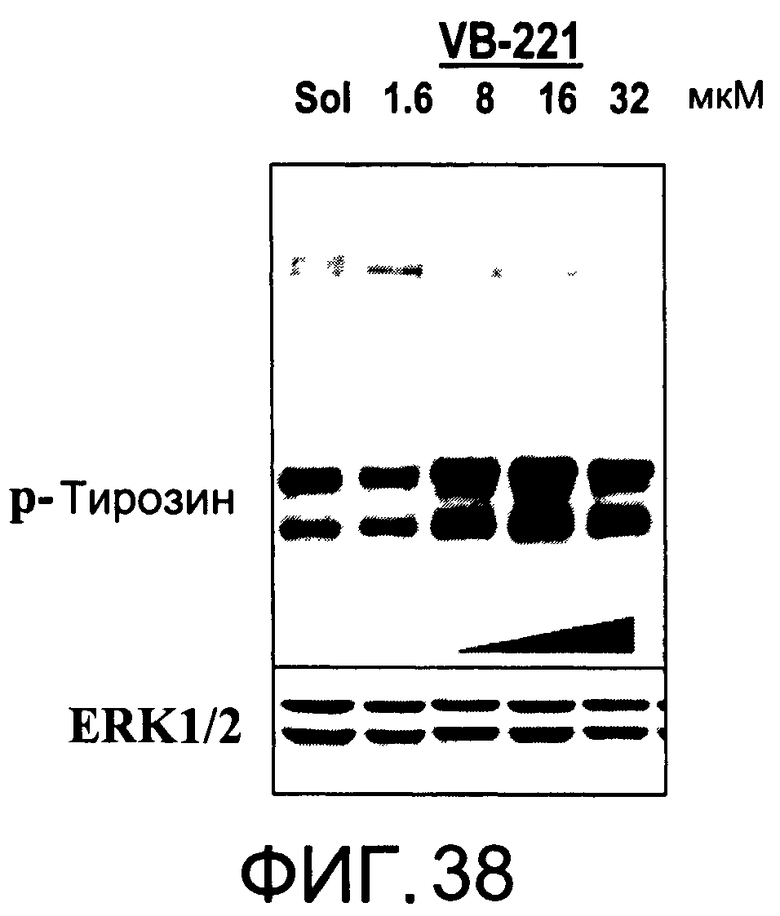
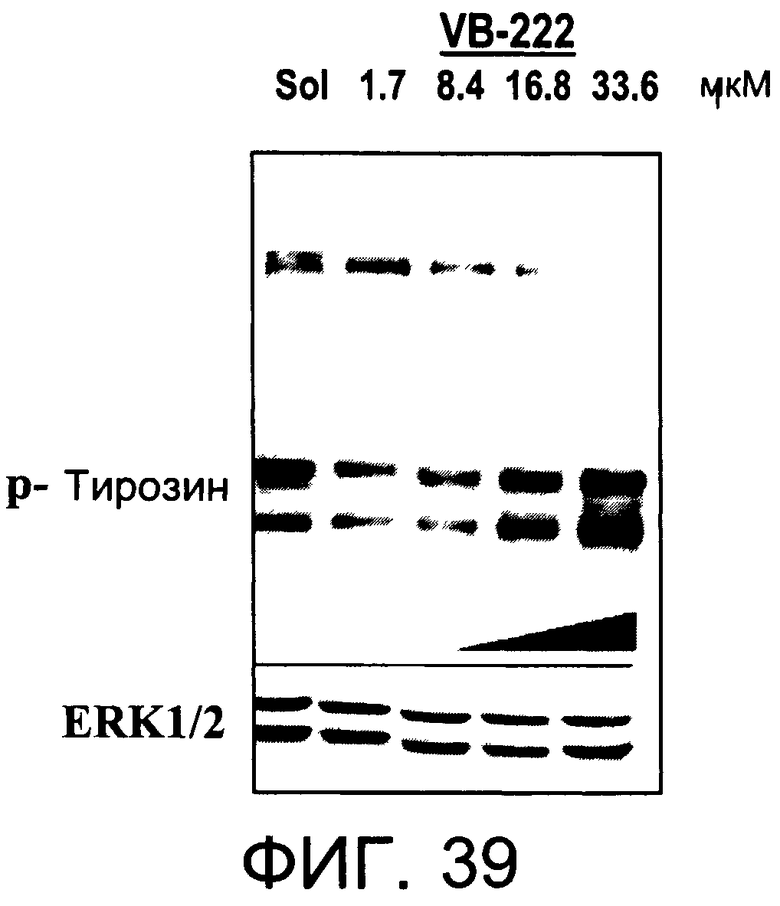
Изобретение относится к новым соединениям структурной формулы (I) , которые могут быть использованы для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением. В формуле (I) каждый из A1, А2 и А3 независимо выбран из группы, состоящей из О и S, R1 представляет собой алкильную цепь длиной 2-28 атомов углерода, R2 выбран из группы, состоящей из (3-карбокси)пропила и (2-карбокси)этила, и R3 выбран из группы, состоящей из Н, С1-20ацила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты и фосфосерина. Изобретение также относится к фармацевтической композиции, содержащей указанные соединения, и к применению соединений для изготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, или для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23. 15 н. и 45 з.п. ф-лы, 39 ил., 18 пр.
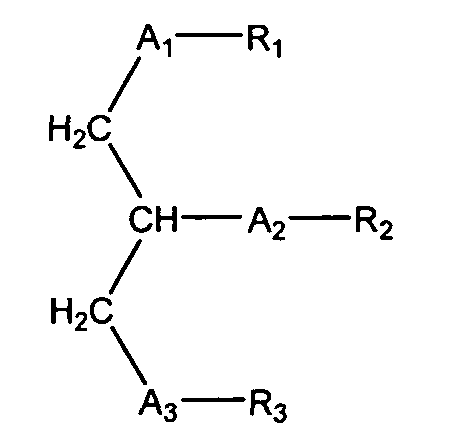 (I)
(I)
1. Соединение, имеющее формулу:
или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь или фармацевтически приемлемая соль, где:
(i) каждый из A1, А2 и А3 независимо выбран из группы, состоящей из О и S;
(ii) R1 представляет собой алкильную цепь длиной 2-28 атомов углерода; и
(iii) R2 выбран из группы, состоящей из (3-карбокси)пропила и (2-карбокси)этила; и
(iv) R3 выбран из группы, состоящей из Н, С1-20ацила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты и фосфосерина.
2. Соединение по п.1 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, причем соединение выбрано из группы, состоящей из:
1-гексадецил-2-(3-карбокси)пропилглицеро-3-фосфоэтаноламина (CI-206);
1-гексадецил-2-(3-карбокси)пропилглицеро-3-фосфохолина (CI-205); и
1-гексадецил-2-(2-карбокси)этилглицеро-3-фосфохолина (CI-217).
3. Соединение по п.1, выбранное из группы, состоящей из:

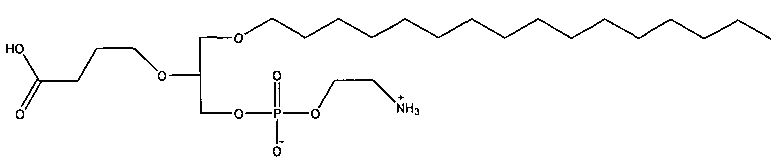 и
и
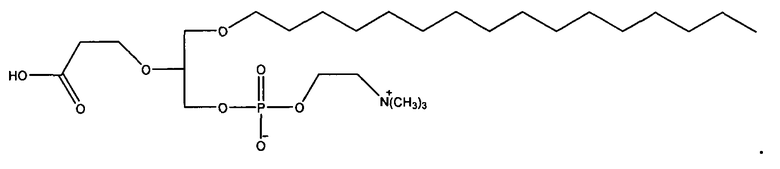
4. Соединение по любому из пп.1-3 для применения в способе лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
5. Соединение по любому из пп.1-3 для применения в способе снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
6. Соединение по любому из пп.1-3 для применения в способе лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
7. Соединение по любому из пп.4-6, где субъектом является человек.
8. Соединение по п.4, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
9. Фармацевтическая композиция для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, включающая в качестве активного ингредиента эффективное количество соединения по любому из пп.1-3 и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п.9, предназначенная для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
11. Фармацевтическая композиция по п.9, предназначенная для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23.
12. Фармацевтическая композиция по п.9, предназначенная для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
13. Фармацевтическая композиция по п.9, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
14. Применение соединения по любому из пп.1-3 для изготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
15. Применение соединения по любому из пп.1-3 для изготовления лекарственного средства, предназначенного для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта.
16. Применение по п.15, где субъектом является человек.
17. Применение соединения по любому из пп.1-3 для изготовления лекарственного средства, предназначенного для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
18. Применение по п.14, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
19. Соединение, характеризующееся формулой:
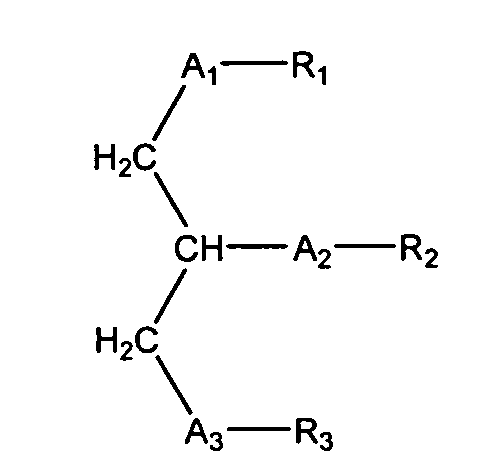
или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь или фармацевтически приемлемая соль, где:
(i) каждый из A1, A2 и А3 независимо выбран из группы, состоящей из О и S;
(ii) R1 представляет собой алкильную цепь длиной 2-28 атомов углерода;
(iii) R2 представляет собой
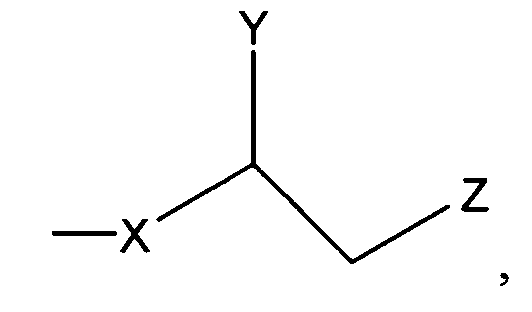
где Х представляет собой C1-25алкил, Y представляет собой -Н, C1-20алкил или галоген; и
Z выбран из группы, состоящей из:
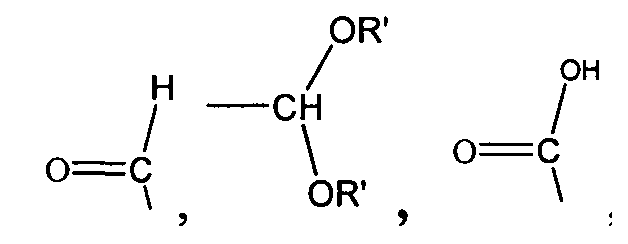 и
и 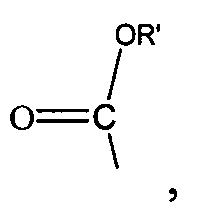
тогда как R′ представляет собой С1-4алкил; и
(iv) R3 выбран из группы, состоящей из фосфоэтаноламин-N-глутаровой кислоты и фосфосерина.
20. Соединение по п.19 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, причем соединение выбрано из группы, состоящей из:
1-гексадецил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламин-N-глутаровой кислоты (CI-210) и 1-гексадецил-2-(4-карбокси)бутилглицеро-3-фосфосерина (VB-223).
21. Соединение по п.19, выбранное из группы, состоящей из:
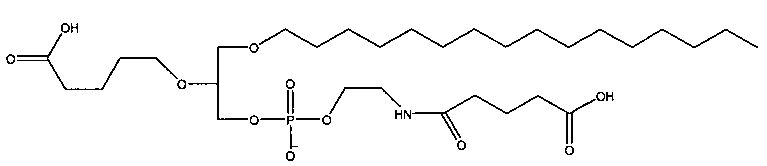 и
и
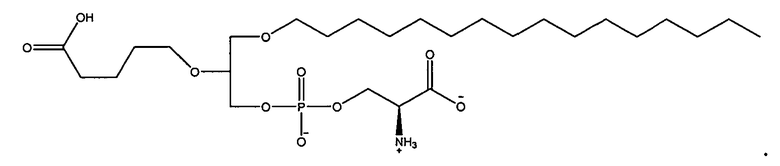
22. Соединение по любому из пп.19-21 для применения в способе лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
23. Соединение по любому из пп.19-21 для применения в способе снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
24. Соединение по любому из пп.19-21 для применения в способе лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
25. Соединение по любому из пп.22-24, где субъектом является человек.
26. Соединение по п.22, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
27. Фармацевтическая композиция для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, включающая в качестве активного ингредиента эффективное количество соединения по любому из пп.19-21 и фармацевтически приемлемый носитель.
28. Фармацевтическая композиция по п.27, предназначенная для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
29. Фармацевтическая композиция по п.27, предназначенная для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23.
30. Фармацевтическая композиция по п.27, предназначенная для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
31. Фармацевтическая композиция по п.27, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
32. Применение соединения по любому из пп.19-21 для изготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
33. Применение соединения по любому из пп.19-21 для изготовления лекарственного средства, предназначенного для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта.
34. Применение по п.33, где субъектом является человек.
35. Применение соединения по любому из пп.19-21 для изготовления лекарственного средства, предназначенного для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
36. Применение по п.32, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
37. Соединение, характеризующееся формулой:
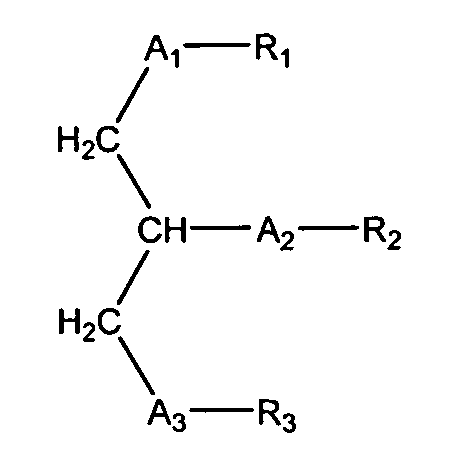
или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь или фармацевтически приемлемая соль, где:
(i) каждый из A1, A2 и А3 независимо выбран из группы, состоящей из О и S;
(ii) R1 выбран из группы, состоящей из додецила, октадецила, октила, эйкозанила, цис-9-гексадеценила, (2-октил)додецила и (15-карбокси)пентадецила;
(iii) R2 выбран из группы, состоящей из алкильной цепи длиной 2-28 атомов углерода, и
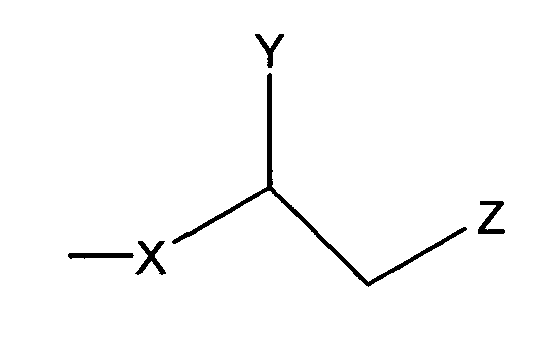
где Х представляет собой С1-25алкил, Y представляет собой -Н, С1-20алкил или галоген; при условии, что если R1 отличен от (15-карбокси)пентадецила, то R2 представляет собой
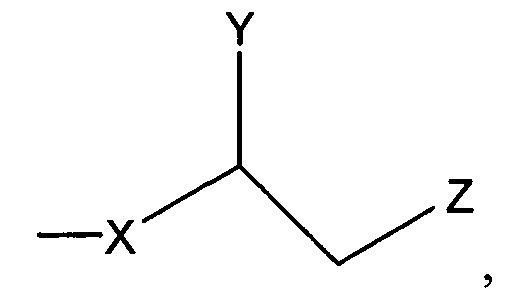 определенный выше; и
определенный выше; и
Z выбран из группы, состоящей из:
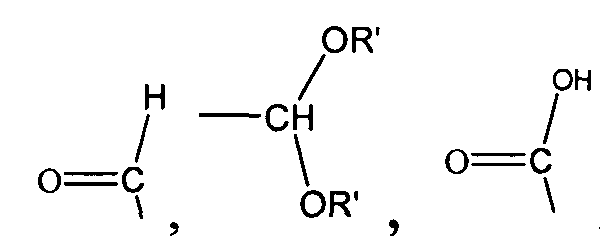 и
и 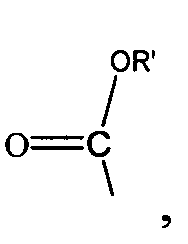
где R′ представляет собой С1-4алкил; и
(iv) R3 выбран из группы, состоящей из Н, С1-20ацила, фосфата, фосфохолина, фосфоэтаноламина, фосфоэтаноламин-N-глутаровой кислоты и фосфосерина.
38. Соединение по п.37 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, где A1 представляет собой S, а каждый из А2 и А3 представляет собой О.
39. Соединение по п.37, где R2 выбран из группы, состоящей из (4-метилкарбокси)бутила, (3-карбокси)пропила, (6-карбокси)гексанила, (2-карбокси)этила, 5,6-дигидроксигексанила, 5,5-диэтоксипентила и 5,5-диметоксипентила.
40. Соединение по п.37 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, причем соединение выбрано из группы, состоящей из:
1-додецил-2-(4-карбокси)бутилглицеро-3-фосфохолина (CI-209);
1-(15′-карбокси)пентадецил-2-(4-карбокси)бутилглицеро-3-фосфохолина (CI-213);
1-(15′-карбокси)пентадецил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламина (CI-214);
1-октадецил-2-(4-карбокси)бутилглицеро-3-фосфохолина (CI-215);
1-октадецил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламина (CI-216);
1-(цис-9-гексадеценил)-2-(4-карбокси)бутилглицеро-3-фосфохолина;
1-октил-2-(4-карбокси)бутилглицеро-3-фосфохолина (CI-207);
1-октил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламина;
1-эйкозанил-2-(4-карбокси)бутилглицеро-3-фосфохолина (CI-219);
1-эйкозанил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламина (CI-220);
1-(2-октил)додецил-2-(4-карбокси)бутилглицеро-3-фосфохолина (VB-221); и
1-(2-октил)додецил-2-(4-карбокси)бутилглицеро-3-фосфоэтаноламина (VB-222).
41. Соединение по п.40 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, причем соединение представляет собой
1-(2-октил)додецил-2-(4-карбокси)бутилглицеро-3-фосфохолин (VB-221).
42. Соединение по п.40 или его стереоизомер, оптический изомер, энантиомер, рацемическая смесь или стереоизомерная смесь, причем соединение представляет собой 1-эйкозанил-2-(4-карбокси)бутилглицеро-3-фосфохолин (CI-219).
43. Соединение по п.37, выбранное из группы, включающей:
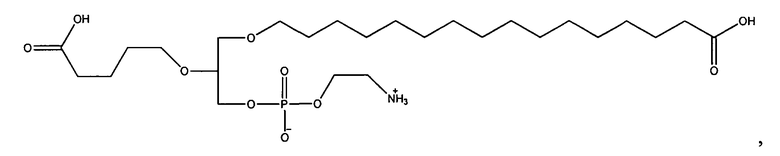
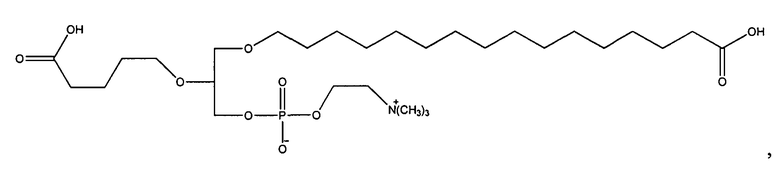
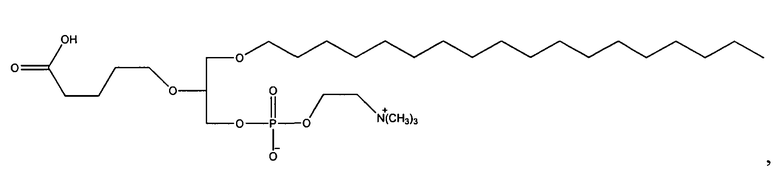
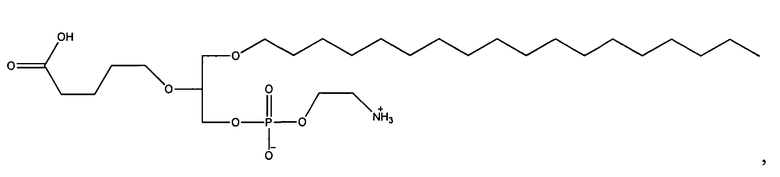
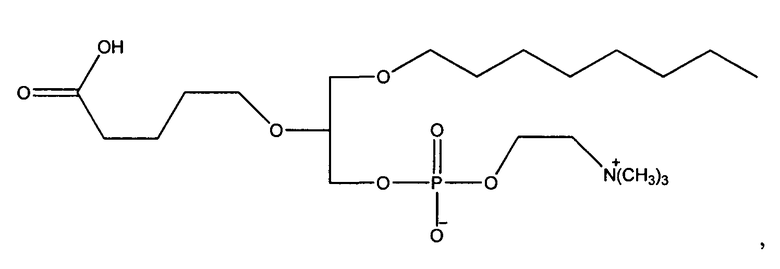
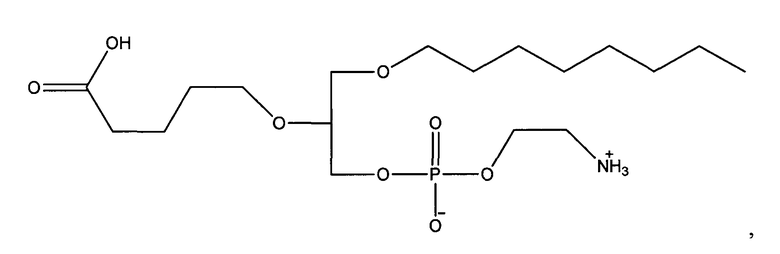
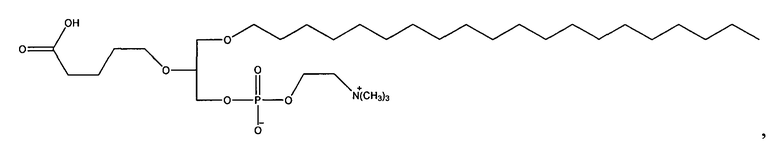
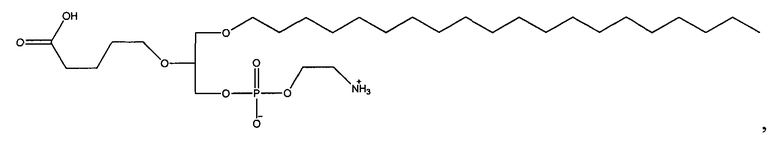
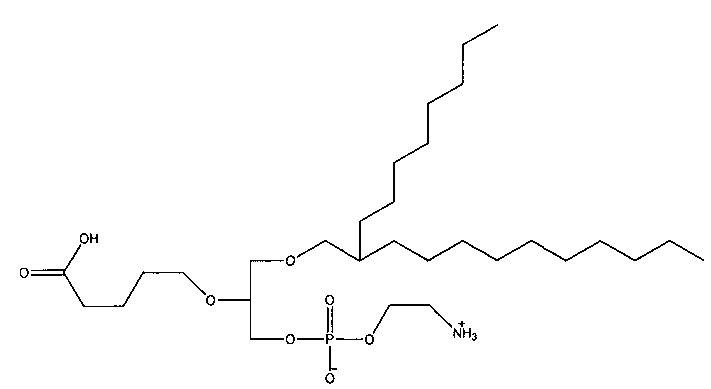 и
и 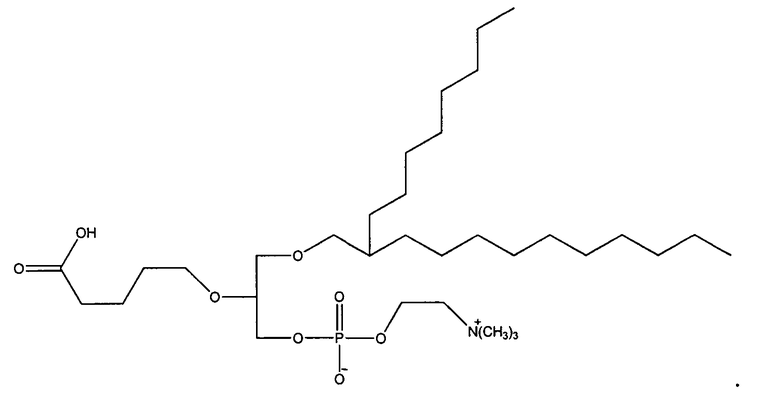
44. Соединение по п.43, имеющее формулу:
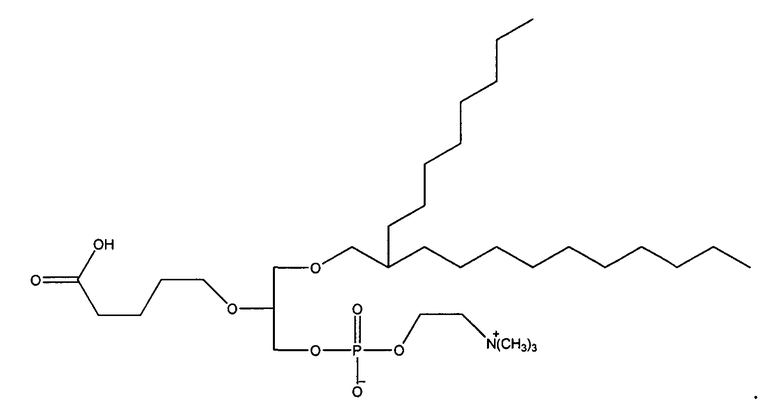
45. Соединение по п.43, имеющее формулу:
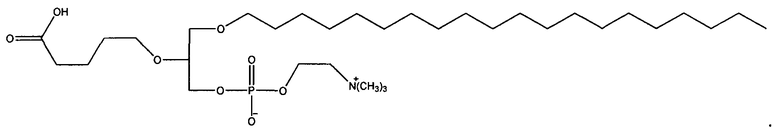
46. Соединение по любому из пп.37-45 для применения в способе лечения или профилактики воспаления, ассоциированного с эндогенным окисленным липидом, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
47. Соединение по любому из пп.37-45 для применения в способе снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
48. Соединение по любому из пп.37-45 для применения в способе лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным, причем способ включает введение нуждающемуся в этом субъекту эффективного количества соединения.
49. Соединение по любому из пп.46-48, где субъектом является человек.
50. Соединение по п.46, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
51. Фармацевтическая композиция для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением, включающая в качестве активного ингредиента эффективное количество соединения по любому из пп.37-45 и фармацевтически приемлемый носитель.
52. Фармацевтическая композиция по п.51, предназначенная для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
53. Фармацевтическая композиция по п.51, предназначенная для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23.
54. Фармацевтическая композиция по п.51, предназначенная для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
55. Фармацевтическая композиция по п.51, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
56. Применение соединения по любому из пп.37-45 для изготовления лекарственного средства, предназначенного для лечения или профилактики заболеваний или нарушений, ассоциированных с воспалением.
57. Применение соединения по любому из пп.37-45 для изготовления лекарственного средства, предназначенного для снижения уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, у субъекта.
58. Применение по п.57, где субъектом является человек.
59. Применение соединения по любому из пп.37-45 для изготовления лекарственного средства, предназначенного для лечения заболевания или нарушения, при котором снижение уровня цитокина, выбранного из группы, состоящей из интерлейкина-12 и интерлейкина-23, является предпочтительным.
60. Применение по п.56, причем воспаление ассоциировано с заболеванием или нарушением, выбранным из группы, состоящей из идиопатического воспалительного заболевания или нарушения, хронического воспалительного заболевания или нарушения, острого воспалительного заболевания или нарушения, аутоиммунного заболевания или нарушения, инфекционного заболевания или нарушения, воспалительного злокачественного заболевания или нарушения, воспалительного заболевания или нарушения, связанного с трансплантацией, воспалительного дегенеративного заболевания или нарушения, заболевания или нарушения, обусловленного гиперчувствительностью, воспалительного сердечно-сосудистого заболевания или нарушения, воспалительного цереброваскулярного заболевания или нарушения, заболевания или нарушения периферических сосудов, воспалительного заболевания или нарушения желез, воспалительного желудочно-кишечного заболевания или нарушения, воспалительного кожного заболевания или нарушения, воспалительного заболевания или нарушения печени, воспалительного неврологического заболевания или нарушения, воспалительного заболевания или нарушения костно-мышечной системы, воспалительного заболевания или нарушения почек, воспалительного заболевания или нарушения репродуктивной системы, воспалительного системного заболевания или нарушения, воспалительного заболевания или нарушения соединительной ткани, воспалительной опухоли, некроза, воспалительного заболевания или нарушения, связанного с имплантатом, воспалительного процесса, связанного со старением, заболевания или нарушения, обусловленного иммунодефицитом, а также воспалительного легочного заболевания или нарушения.
| US6838452 B2, 04.01.2005 | |||
| US20070020691 A1, 25.01.2007 | |||
| ФОСФОНОЛИПИДЫ КАРБОНОВЫХ КИСЛОТ И ИХ СОЛИ И АНТИВИРУСНОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1996 |
|
RU2166508C2 |
| КОВАЛЕНТНЫЕ КОНЪЮГАТЫ ЛИПИДОВ С ФОСФОНОКАРБОНОВОЙ КИСЛОТОЙ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИВИРУСНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 1996 |
|
RU2178418C2 |
| V.N | |||
| BOCHKOV, “Inflammatory profile of oxidized phospholipids”, Journal of Thrombosis and Haemostasis, 2007, vol.97, pp.348-354 | |||
| S.S | |||
| DAVIES ET AL., “Oxidized alkyl phospholipids are specific, high affinity peroxisome | |||

