ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По настоящей заявке заявляется приоритет, согласно Кодексу законов США, раздел 35, §119(е), предварительной заявки на патент США №61/453,011, поданной 15 марта 2011 года, которая включена в настоящий документ путем ссылки.
ЗАЯВЛЕНИЕ ОТНОСИТЕЛЬНО ФЕДЕРАЛЬНО СПОНСИРОВАННЫХ НАУЧНЫХ ИССЛЕДОВАНИЙ И РАЗРАБОТОК
[0002] Настоящее изобретение сделано при государственной поддержке по контракту №HHSN272200800042C, присужденному Национальным институтом по изучению аллергических и инфекционных заболеваний. Государственная власть имеет определенные права на данное изобретение.
СТОРОНЫ СОВМЕСТНОГО ИССЛЕДОВАТЕЛЬСКОГО СОГЛАШЕНИЯ
[0003] Настоящее изобретение сделано в соответствии с совместным исследовательским соглашением между компаниями Trius Therapeutics, Inc. и Lawrence Livermore National Security, LLC по контракту Министерства энергетики США №TC02128.0.
УРОВЕНЬ ТЕХНИКИ
Область
[0004] Настоящее описание относится к области медицинской химии и, в частности, к соединениям и их фармацевтическим композициями, применимым в качестве антибиотиков. В частности, трициклические соединения гиразы ингибируют ферменты ДНК-гиразу В (GyrB) и топоизомеразу IV (ParE). Рассмотрены также родственные способы лечения бактериальных инфекций и способы получения указанных соединений с использованием новых промежуточных веществ.
Описание области техники
[0005] Бактериальные инфекции представляют собой постоянную медицинскую проблему, поскольку антибактериальные лекарства со временем вызывают устойчивость в бактериях, в отношении которых они используются. Следовательно, существует необходимость в новых лекарствах, эффективных против патогенных бактерий для применения при терапии и профилактике бактериальных инфекций.
[0006] Одной мишенью для разработки антибактериальных лекарств являются ферменты ДНК-гираза В (GyrB) и топоизомераза IV (ParE), необходимые для репликации ДНК. Ингибиторы гиразы описаны в публикации RE40245, которая включена в настоящий документ путем ссылки в полном объеме.
[0007] Ферментативный карман GyrB подробно описан в публикации Wigley, D.B. et al., Nature, 351(6328), 624-629, 1991. Смотри также Tsai FT, et al., The high-resolution crystal structure of a 24-kDa gyrase В fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin, Proteins. 1997 May; 28(1):41-52.
[0008] Ферментативный карман ParE подробно описан в публикации Bellon, S., et al. Crystal structures of Escherichia coli topoisomerase IV ParE subunit (24 and 43 kilodaltons): а single residue dictates differencesin novobiocin potency against topoisomerase IV and DNA gyrase, Antimicrob. Agents Chemother. 48: 1856-1864 (2004). Эти ссылки включены в настоящий документ путем ссылки в полном объеме.
[0009] Напротив, патентные публикации авторов изобретений Hurley et al. относятся к ингибиторам протеинкиназы, применимым для опосредованных протеинкиназой заболеваний и состояний, таких как рак. Смотри, например, US 2008/0051414, US 2009/0143399 и US 2009/0099165.
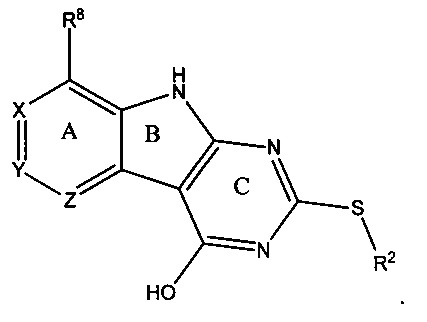
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
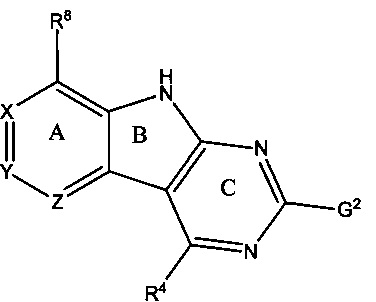
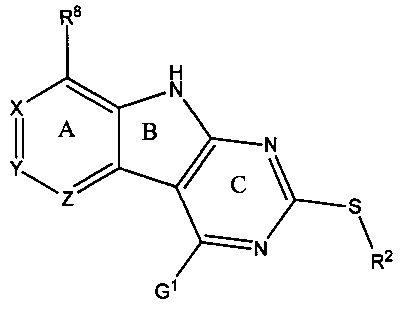
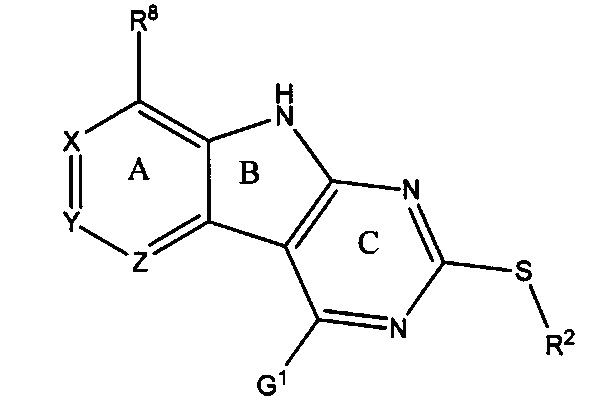
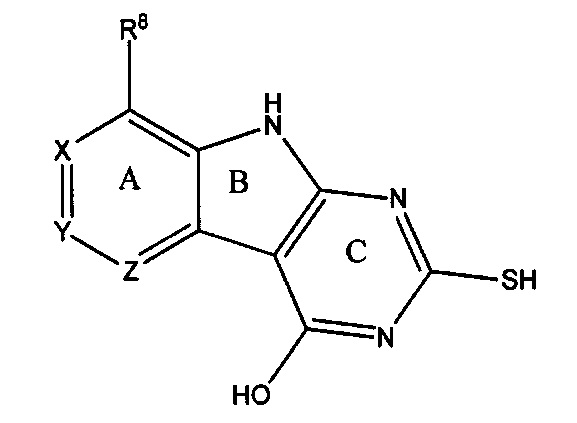
[0010] Трициклические соединения гиразы Формулы (I) ингибируют ферменты ДНК-гиразу В (GyrB) и топоизомеразу IV (ParE). Соединение Формулы I имеет структуру
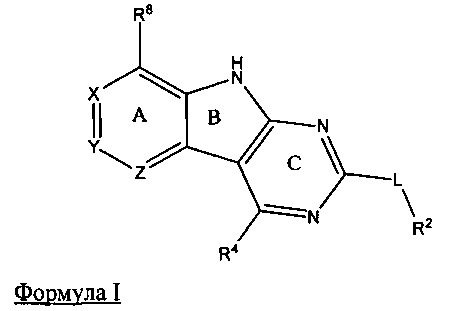
[0011] Подразумеваются также его фармацевтически применимые соли, сложные эфиры и пролекарства. Ниже представлены переменные в Формуле I.
[0012] L может быть О или S.
[0013] R8 может быть H или взаимодействующим заместителем, имеющим длину от около 1 Å до около 5 Á от точки присоединения к углероду в Кольце А до концевого атома в R8, и ширину около 3,3 Å или менее.
[0014] Χ, Y и Ζ могут быть независимо выбраны из группы, состоящей из Ν, CRX, CRY и CRZ, при условии что не более двух из Χ, Y и Ζ представляют собой N. RX может быть H или взаимодействующим заместителем, имеющим длину от около 1 Å до около 2 Å от атома углерода в CRX до концевого атома в RX. RY может быть H или взаимодействующим заместителем, имеющим длину от около 1 Å до около 3 Å от атома углерода в CRY до 10 концевого атома в RY. RZ может быть Η или взаимодействующим заместителем, имеющим длину от около 1 Å до около 2 Å от атома углерода в CRZ до концевого атома в RZ.
[0015] R2 может быть 6-членным ариловым или гетероариловым кольцом, содержащим 0-3 гетероатома О, S или N, необязательно замещенным 0-3 не мешающими заместителями, причем 2 соседних не мешающих заместителя у R2 могут образовывать одно 15 или несколько конденсированных колец с 6-членным ариловым или гетероариловым кольцом. В некоторых аспектах 6-членное ариловое или гетероариловое кольцо R2 имеет СН в положениях, непосредственно смежных с положением присоединения R2 к L.
[0016] R4 может быть:
Н;
необязательно замещенным ORa;
необязательно замещенным вторичным или третичным амином, присоединенным к Кольцу С через N вторичного или третичного амина; или
необязательно замещенным 5-10-членным ненасыщенным циклическим или гетероциклическим остатком, содержащим 0-3 гетероатома N, О или S.
[0017] Необязательный заместитель может быть 0-3 не мешающими заместителями. Ra может быть 5-6-членным арилом или гетероарилом, содержащим 0-3 гетероатома О, S или N, необязательно замещенным 0-3 не мешающими заместителями. В некоторых аспектах заместитель R4 выступает не более чем примерно на 3 Å ниже плоскости Колец А, В и С в сторону дна связывающего кармана GyrB/ParE в связанной конформации. Кроме того, в некоторых аспектах R4 стерически не сталкивается с R2 или Z, если соединение находится в связанной конформации. Рассмотрены также способы применения указанных соединений для лечения антибактериальных инфекций и способы получения указанных соединений с использованием новых промежуточных веществ.
[0018] Эти и другие родственные аспекты более подробно изложены ниже.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0019] На Фигуре 1 представлено схематическое изображение рецепторных ограничений соединения, в частности, способы связывания трициклических ингибиторов с карманом активного сайта GyrB/ParE (по кристаллографическим данным). Размеры, представленные для длин, были измерены от центра атома члена Кольца А до центра атома ближайшего неводородного атома в кармане активного сайта. На этой фигуре указана длина от около 6 Å до около 8 Å от атома С, присоединенного к R8, до атома в кармане активного сайта; от около 4 Å до около 5 Å от атома Кольца А в X до атома в кармане активного сайта; от около 4 Å до около 6 Å от атома Кольца А в Y до атома в кармане активного сайта; и от около 4 Å до около 6 Å от атома Кольца А в Ζ до атома в кармане активного сайта. Показаны относительные положения заместителей R8, R4 и циклического R2. Показана приблизительная форма поперечного сечения иллюстративного кармана активного сайта GyrB/ParE в плоскости трициклической структуры и над ней (то есть Кольца А, В и С). Заштрихованная область из сплошных линий изображает участки ингибитора, закрытые на обеих поверхностях карманом активного сайта. Кроме того, показана приблизительная форма поперечного сечения иллюстративного кармана активного сайта GyrB/ParE под плоскостью трициклической структуры. Заштрихованная область из пунктирных линий отображает участки ингибитора, контактирующие с поверхностью дна кармана активного сайта, тогда как на поверхность над трициклической кольцевой системой воздействует растворитель. На Фигуре 1 показано приблизительное положение консервативной побочной цепи Asp, связывающей субстрат, и структурная молекула воды, а также совокупность потенциальных водородных связей (изображенных пунктирными линиями), наблюдаемых между трициклической структурой и Asp, и водой. Выделены поверхности кармана активного сайта, на которые воздействует растворитель и которые закрыты от растворителя. Растворитель относится к in vivo окружению активного сайта GyrB/ParE как части белка, которое обычно включает водное окружение, в котором белок находится внутри клетки. Также в некоторых аспектах атомы фрагмента R4 выступают не более, чем примерно на 3 Å под плоскостью трициклической кольцевой системы в сторону дна связывающего кармана GyrB/ParE в связанном состоянии.
[0020] На Фигуре 2 представлено схематическое изображение внутриклеточных ограничений соединения, если R2 представляет собой 6-членное кольцо. В частности, молекулярная геометрия и конформации R-групп, необходимые для обеспечения возможности связывания трициклических ингибиторов с карманами активных сайтов GyrB/ParE, ограничивают размер и состав заместителей в некоторых положения структуры ингибитора. На этой фигуре показаны области потенциального стерического столкновения между заместителем R4 и заместителем R2 или RZ в связанной конформации.
[0021] На Фигуре 3 показан пример относительных расположений первичного амина, охваченного определением R4, при его связывании с GyrB/ParE. Эта иллюстрация относится также к вторичному амину, который не показан на Фигуре 3. Объем, занимаемый амином R4 в отношении трициклической структуры, в целом по аминам, определили с использованием приема четырехточечной трилатерации на основании расстояний между амином R4 и четырьмя различными атомами трициклической структуры из 17 различных кристаллических структур комплексов Е. faecalis GyrB с трициклическими ингибиторами, содержащими разные наборы аминов R4, включающих вторичный или третичный амин, присоединенный к Кольцу С через N вторичного или третичного амина, и первичный или вторичный амин, не присоединенный к Кольцу С.Относительное положение первичного (или вторичного, не показано) амина должно быть над плоскостью трициклической структуры, во избежание столкновения с дном активного сайта.
ПОДРОБНОЕ ОПИСАНИЕ
[0022] Ниже подробно разработаны некоторые аспекты соединений Формулы I. В Формуле I выше L представляет собой линкер, который связывает R2 с кольцом С. L может быть О или S. В некоторых аспектах L представляет собой О. В некоторых аспектах L представляет собой S.
[0023] При использовании в настоящем документе термин «арил» относится к необязательно замещенному моноциклическому и конденсированному бициклическому углеводородному фрагменту. В это определение входят любые моноциклические или конденсированные кольцевые бициклические системы, которые имеют свойства ароматичности с точки зрения распределения электронов в кольцевой системе. Как правило, кольцевые системы содержат 5-12 атомов в кольце. «Гетероарил» относится к необязательно замещенным ароматическим моноциклическим и конденсированным бициклическим гетероциклам, содержащим один или несколько гетероатомов, выбранных из N, О и S. Включение гетероатома допускает включение 5-членных колец, а также 6-членных колец.
[0024] При использовании в настоящем документе, термин «алкил» включает прямые и разветвленные, а также циклические одновалентные заместители. Примеры включают метил, этил, пропил, изопропил и циклопропил. Если указано, то алкиловые заместители могут содержать 1-10С (от 1 до 10 атомов углерода), как, например, 1-3С, 1-6С или 1-8С.
[0025] При использовании в настоящем документе, «углеводородный остаток» относится к остатку, который содержит только атомы углерода и водорода. Углеводородный остаток может быть насыщенным или ненасыщенным, алифатическим или ароматическим, прямым, разветвленным или циклическим, включая одно кольцо, конденсированную кольцевую систему, мостиковую кольцевую систему или спирокольцевую систему, или комбинацию углеводородных групп. Углеводородный остаток, определенный таким образом, тем не менее может содержать гетероатомы помимо и сверх углеродных и водородных членов остатка указанного заместителя. Так, если специально указано содержание таких гетероатомов, то указанный углеводородный остаток может содержать гетероатомы, такие как О, S или N в «скелете» углеводородного остатка. Углеводородная группа может включать комбинацию углеводород-содержащих фрагментов, таких как гетероциклическая группа, связанная с гетероалкилом, содержащим комбинацию прямой алкиловой и циклоалкиловой группы.
[0026] При использовании в настоящем документе, «циклический остаток» относится к циклическому углеводородному остатку, который содержит только атомы углерода и водорода. Циклический остаток, определенный таким образом, тем не менее может содержать гетероатомы помимо и сверх углеродных и водородных членов остатка указанного заместителя. Так, если специально указано содержание таких гетероатомов, то указанный гетероциклический остаток может содержать гетероатомы, такие как О, S или N в «скелете» циклического остатка. В некоторых аспектах, если это указано, циклический остаток является циклоалифатическим или циклогетероалифатическим остатком. Насыщенный циклоалифатический или насыщенный циклогетероалифатический остаток относится к кольцу, содержащему насыщенные связи между всеми членами кольца.
[0027] При использовании в настоящем документе, «ненасыщенный циклический остаток» относится по меньшей мере к частично ненасыщенному или ароматическому циклическому углеводородному остатку, который содержит только атомы углерода и водорода. Ненасыщенный циклический остаток, определенный таким образом, тем не менее может содержать гетероатомы помимо и сверх углеродных и водородных членов остатка указанного заместителя. Так, если специально указано содержание таких гетероатомов, то указанный ненасыщенный гетероциклический остаток может содержать гетероатомы, такие как О, S или N в «скелете» ненасыщенного циклического остатка.
[0028] Термин «члены» или «-членный» в контексте гетероциклических и гетероариловых групп относится к общему количеству атомов, углерода и гетероатомов N, О и/или S, которые образуют кольцо. Так, примером 6-членного насыщенного циклогетероалифатического кольца является пиперидин, а примером 6-членного гетероарилового кольца является пиридин.
[0029] Связанная конформация относится к такой конформации (то есть пространственному расположению атомов) трициклического соединения гиразы, которую можно предположить, если бы оно было связано с карманом активного сайта GyrB/ParE во внутренней части указанного фермента. На практике, указанное соединение может взаимодействовать с карманом активного сайта и ингибировать активность АТФазы. Если соединение связано с карманом активного сайта GyrB/ParE, то некоторые заместители взаимодействуют с определенными аминокислотами, и поэтому способность заместителей свободно вращаться вокруг связи ограничена. Следовательно, могут быть сделаны более применимые измерения для определения расстояний, имеющих отношение к определению размеров соответствующих заместителей. Если указано, то измерения основаны на относительных положениях заместителей в соединении, когда оно гипотетически связано с карманом активного сайта GyrB/ParE. Упоминание связанной конформации в отношении соединения не следует толковать как буквально охватывающее карман активного сайта GyrB/ParE в комбинации с соединением. Связанная конформация характеризуется измерениями, полученными из трехмерной структуры по данным рентгеновской кристаллографии ингибитора, связанного в комплекс с белковой структурой, которая обычно охватывает АТФ-связывающий домен размером от 24 до 46 кДа одного или нескольких иллюстративных бактериальных ортологов GyrB или ParE. С учетом высокой степени идентичности последовательностей между ферментами GyrB и ParE в большинстве рассматриваемых патогенных организмов, клинически релевантная структурная информация, полученная из белкового ортолога из любого патогена, является достаточной для описания связанной конформации. Вкратце, кристаллографические структуры созданы с использованием следующих способов: рассматриваемые белки (например, Е. faecalis GyrB, Ε. coli GyrB, F. tularensis ParE или Ε. coli ParE) генерируют в стандартной системе экспрессии Ε. coli. Открытые рамки считывания клонируют в экспрессионную плазмиду (например, рЕТ28а) и экспрессируют в соответствующем экспрессионном штамме Е. coli (например, BL21 (DE3)). Для кристаллографии клонируют АТФ-связывающие домены размером 24 кДа и 46 кДа с меткой C(His)6, чтобы облегчить очистку металл-аффинной хроматографией. Эта надежная стадия хроматографии обычно дает белок чистотой более 80%. При необходимости выполняют стадии доочистки, включая ионный обмен и эксклюзионную хроматографию, до достижения удовлетворительной чистоты (>95%). Когда получен очищенный белок, создают комплексы GyrB или ParE и заданной молекулы ингибитора путем смешивания стехиометрического избытка рассматриваемого ингибитора с мишенью рекомбинантного белка в растворе и кристаллизации комплекса с использованием устоявшихся методик кристаллизации (как правило, диффузия из паровой фазы, как описано в публикации Drenth J. (1999) In Principles of protein x-ray crystallography. 2nd ed. Springer, New York). После кристаллизации собирают данные рентгеновской дифракции на монокристаллах комплексов белок-ингибитор, используя монохроматические рентгеновские лучи, создаваемые вращающимся анодом или источником синхротронного излучения. Обработку, анализ рентгеновских данных, а также последующую расшифровку и уточнение структуры выполняют с использованием хорошо устоявшихся вычислительных способов (рассмотрены в публикации Drenth J. (1999) In Principles of protein x-ray crystallography. 2nd ed. Springer, New York).
[0030] Взаимодействующие заместители соединения, которые взаимодействуют с карманом активного сайта GyrB/ParE, включают те заместители, которые расположены во внутренней части белка, когда соединение находится в связанной конформации. Взаимодействия взаимодействующих заместителей обычно включают гидрофобные взаимодействия (которые способствуют аппозиции липофильных поверхностей ингибитора и кармана активного сайта) и электростатические взаимодействия, такие как взаимодействия Ван-дер-Ваальса, диполь-дипольные, кулоновские взаимодействия или водородное связывание между атомами соединения и атомами кармана активного сайта GyrB/ParE. Например, R8, RX, RY и RZ взаимодействуют с различными участками внутренней части белка. Если R8, RX, RY или RZ представляет собой NH2 или NHR (причем R, например, представляет собой небольшую алкильную группу), то атом(ы) H у азота могут взаимодействовать с электроотрицательными атомами, такими как азот или кислород, 5 расположенными вблизи кармана активного сайта GyrB/ParE, с которым может связываться указанное соединение. Если R8, RX, RY и RZ являются неполярными (например, метальными группами), то взаимодействующий заместитель может также электростатически взаимодействовать с атомом во внутренней части белка за счет взаимодействий Ван-дер-Ваальса и дополнительно десольватировать липофильные поверхности в кармане активного сайта с образованием благоприятных гидрофобных взаимодействий. Кроме того, в некоторых аспектах форма и размер активного сайта могут накладывать ограничения на размеры заместителей соединения, которые должны быть стерически совместимы с карманом активного сайта.
[0031] Если указано, то могут быть представлены размеры заместителя, и они связаны с размерами кармана, в котором должно располагаться соединение, будучи в связанной конформации. Например, может быть дана длина заместителя на основании его расстояния от атома в трициклической структуре до атома заместителя, расположенного дальше всего от трициклической структуры, то есть концевого атома. Это расстояние измеряют на основании расстояния от центра первого атома, такого как С, в трициклической структуре до центра концевого атома. Это расстояние измеряют от точки до точки по прямой линии, независимо от того факта, что связи в заместителе расположены не линейно, как в этильном или ОН-заместителе.
[0032] Ширину заместителя R8 можно понимать в отношении размера кармана активного сайта, в котором находится R8 (карман R8), а также в отношении заместителя R8, когда он принимает конформацию кармана R8, если соединение находится в связанной конформации. Заместитель R8, как правило, выступает в карман R8 вдоль оси, которая проходит через атом С Кольца А, присоединенного к R8, и атом С того же кольца в мета-положении, которое делит общий атом С с кольцом В, когда соединение находится в связанной конформации. Ширина заместителя R8 относится к ширине в его самой широкой точке, измеренной от центра атома до центра атома, находящихся на самом большом удалении друг от друга, примерно перпендикулярно относительно такой оси, когда соединение находится в связанной конформации. Таким образом, заместитель R8 может быть способен принимать такую конформацию, если соединение находится в связанной конформации, которая имеет ширину, не превышающую около 3,3 Å. Например, фрагмент NHMe у R8 имеет ширину около 2,8 Å. Эта ширина получена суммированием расстояния от центра атома метильного протона, транс-ориентированного относительно протона N-H, перпендикулярно оси, описанной выше, с расстоянием от центра протона N-H перпендикулярно той же оси. Далее, ширина циклопропильного заместителя может составлять около 3,1 Å, которую измеряют как расстояние между центрами протонов у соседних углеродных атомов на противоположных поверхностях циклопропильного кольца.
[0033] R8 может быть H или взаимодействующим заместителем, имеющим длину от около 1 Å до около 5 Å от точки присоединения к углероду в Кольце А до концевого атома в R8, и ширину около 3,3 Å или менее. Длина R8 соответствует длине от углерода трициклической структуры до кармана активного сайта, на основании кристаллографических данных, которая составляет от около 6 Å до около 8 Å, как показано на Фигуре 1. В некоторых аспектах R8 представляет собой H, Cl, F, Br, NH2, ОН, 1-3С алкил, амино-1-3С алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH, причем D представляет собой дейтерий. В некоторых аспектах R8 представляет собой H, Cl, F, Br, NH2, 1-3С алкил, амино-1-3С алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH. Например, R8 может быть H, СН3, СН2СН3, Cl, ОСН3, NHCD3, NHCH3, NHCH2CH3 или NH2, таким как NHCH3.
[0034] X, Y и Ζ могут быть независимо выбраны из группы, состоящей из Ν, CRX, CRY и CRZ, при условии что не более двух из Χ, Y и Ζ представляют собой N. RX может быть H или представляет собой взаимодействующий заместитель, имеющий длину от около 1 Å до около 2 Å от атома углерода в CRX до концевого атома в RX. RY может быть H или взаимодействующим заместителем, имеющим длину от около 1 Å до около 3 Å от атома углерода в CRY до концевого атома в RY. Например, RY не может быть метокси-заместителем, поскольку метокси-заместитель длиннее 3 Å. RZ может быть H или представляет собой взаимодействующий заместитель, имеющий длину от около 1 Å до около 2 Å от атома углерода в CRZ до концевого атома в RZ. Эти длины CRX, CRY и CRZ являются подходящими по сравнению с длинами от углерода трициклической структуры до кармана активного сайта, на основании кристаллографических данных, представленных на Фигуре 1. В некоторых аспектах Χ, Y и Ζ представляют собой CRX, CRY и CRZ, соответственно. RX может быть Н, СН3, Cl, Br или F, таким как H или F. RY может быть H, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F, таким как H, F, Cl или CF3. RZ может быть H, СН3, Cl, Br или F, таким как H, СН3 или F.
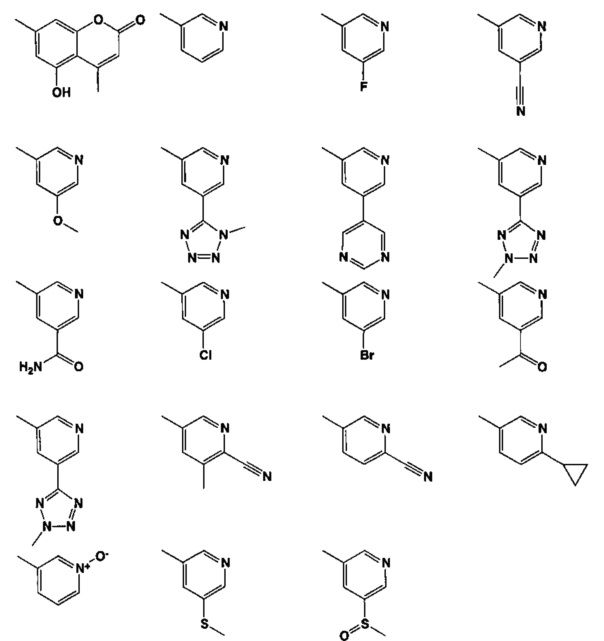
[0035] Не ограничиваясь теорией, R2 может быть применим для обеспечения селективности и эффективности против эукариотических АТФ-связывающих белков, таких как киназы и HSP90. Так, одно из преимуществ соединений настоящего изобретения включает избегание токсичности за счет нецелевого связывания, такого как связывание с киназой, частично благодаря селективности R2 как части указанного соединения. Как правило, в некоторых аспектах соединения не являются сильнодействующими ингибиторами эукариотических киназ. В некоторых аспектах R2 представляет собой 6-членное ариловое или гетероариловое кольцо, содержащее 0-3 гетероатома О, S или N, необязательно замещенное 0-3 не мешающими заместителями, причем 2 соседних не мешающих заместителя у R2 могут образовывать одно или несколько конденсированных колец с 6-членным ариловым или гетероариловым кольцом. Например, R2 может быть необязательно замещенным 6-членным ариловым или гетероариловым кольцом, содержащим 0-3 гетероатома О, S или N, таким как необязательно замещенный пиримидинил, фенил или пиридил. В некоторых аспектах R2 является гетероариловым кольцом, таким как 6-членный гетероарил. В некоторых аспектах R2 может быть присоединен к L через атом углерода в 6-членном ариловом или гетероариловом кольце. Не ограничиваясь теорией, растворитель, закрывающий поверхности карманов активного сайта GyrB/ParE, ограничивает размер заместителей соединения, расположенных вблизи этих поверхностей, закрытых растворителем. Таким образом, в отношении R2, 6-членное ариловое или гетероариловое кольцо может содержать СН в кольцевых положениях, непосредственно смежных с положением кольца, в котором R2 присоединяется к L. В некоторых аспектах не существует N в 6-членном ариловом или гетероариловом кольце R2 в кольцевом положении, непосредственно смежном с положением кольца, в котором R2 присоединяется к L.
[0036] На Фигуре 2 R2 показан как необязательно замещенное 6-членное гетероариловое кольцо, хотя положение заместителей относится также к 6-членному ариловому кольцу. На этом изображении А и Ε представляют собой С. Rb и Rc граничат с растворителем в связанной конформации, и поэтому эти заместители в этом положении могут варьироваться и могут включать пролекарства. Может быть допустима циклизация между Rb и Rc. На Rd растворитель действует частично, и может быть допустима циклизация между Rc и Rd (например, с акцептором водородной связи в положении Rd). Крупные заместители, такие как крупные разветвленные группы, у Rd могут сталкиваться с наружным краем кармана.
[0037] В некоторых аспектах необязательно замещенное 6-членное ариловое или гетероариловое кольцо из R2 в комбинации с одним или несколькими конденсированными кольцами, образованными из необязательных заместителей, может быть выбрано из группы, состоящей из необязательно замещенного индолила, азаиндолила, пиримидопиридила, хиназолила, хиноксалинила, нафтиридинила, пуринила, имидизопиридинила, фуропиридинила, изоиндолилинила, бензодиоксинила, дигидробензодиоксинила, бензотиазолила, пирролопиридинила, дигидропирролопиридинила, бензоимидазолила, имидазопиридинила, дигидроимидазопиридинила, тетрагидроизоиндолила, хроменила, бензтиофена, бензтриазолила, бензфуранила, бензоксадиазолила, индазолила, хинолинила, изохинолинила, индолина, азаиндолинила или  .
.
[0038] Поверхности карманов активного сайта GyrB/ParE, на которые воздействует растворитель, обеспечивают возможность воздействия окружающего растворителя на части указанного соединения, при таком его использовании, как показано на Фигуре 1. В некоторых аспектах не мешающие заместители могут быть водорастворимыми для обеспечения совместимости со средой водного растворителя. Относительные размеры заместителей в направлении потенциальной среды растворителя не являются критичными, но специалистам в данной области понятно, что пригодны стерически не затрудненные заместители. Таким образом, относительные размеры заместителей, на которые воздействует растворитель, могут быть разными.
[0039] В отличие от «взаимодействующих заместителей», некоторые положения молекулы могут быть описаны как допускающие «не мешающие заместители». Эта терминология используется, поскольку заместители в этих положениях, вообще говоря, имеют меньшее отношение к активности молекулы в целом. В этих положениях могут быть использованы самые различные заместители, и специалисты в данной области могут легко определить, является ли какой-либо конкретный заместитель взаимодействующим или «не мешающим».
[0040] При использовании в настоящем документе, «не мешающий заместитель» представляет собой заместитель, который оставляет качественно не поврежденной способность соединения Формулы I ингибировать бактериальный рост по меньшей мере одного типа бактерий Например, не мешающий заместитель сохраняет способность соединения обеспечивать антибактериальную эффективность на основании минимальной ингибирующей концентрации (MIC) менее 32 мкг/мл, или на основании ингибирования активности АТФазы ДНК гиразы В (GyrB) или топоизомеразы IV (ParE) менее 10 нм. Следовательно, этот заместитель может изменять степень ингибирования на основании MIC или активности АТФазы. Однако поскольку соединение Формулы I сохраняет способность ингибировать бактериальную активность/активность АТФазы, этот заместитель классифицируют как «не мешающий». В данной области техники существует множество анализов для определения MIC или способности любого соединения ингибировать активность АТФазы ДНК-гиразы В (GyrB) или топоизомеразы IV (ParE), и некоторые представлены ниже в Примерах. Например, связанный спектрофотометрический анализ, в котором измеряют фермент-зависимое высвобождение неорганического фосфата при гидролизе АТФ, определяет ингибирующую активность произвольно выбранного соединения при инкубировании с GyrB или ParE при добавлении АТФ. Свойства, связанные с активностью молекулы, строго определены. Положения, которые занимают «не мешающие заместители» могут быть замещены стандартными фрагментами, известными в данной области. Испытывать внешние пределы таких замещений неуместно. Релевантными свойствами соединений являются те, которые подробно изложены далее в настоящем документе.
[0041] R2 может иметь 0-3 не мешающих заместителя в 6-членном ариловом или гетероариловом кольце. Например, R2 может иметь заместитель, выбранный из группы, состоящей из ОН, CO2H, CN, NH2, Br, Cl, F, SO3H, SO2NH2, SO2CH3, SOCH3, NHOH, NHOCH3 и NO2. R2 также может иметь заместитель, который является необязательно замещенным С1-С15 углеводородным остатком, содержащим 0-5 гетероатомов О, S или N, необязательно замещенным ОН, CN, =O, NH2, NHOH, =NOH, =NNH2, =NOCH3, Br, F, Cl, SO3H или NO2. Замещения могут быть у атома углерода или гетероатома, что допускает такие группы как S=O. В случаях если гетероарил содержит пиридиновое кольцо, атом азота может быть окислен до пиридина-оксида; следовательно, заместитель ОН может быть в форме оксида, что допускает, например, пиридил, имеющий N-оксид, причем N является кольцевым гетероатомом.
[0042] Углеводородный остаток С1-С15, содержащий 0-5 гетероатомов О, S или N может включать комбинацию углеводородных групп, такую как комбинация алифатических колец или цепей и ароматический колец, связанных вместе.
[0043] В некоторых аспектах два соседних не мешающих заместителя у R2 образуют одно или несколько конденсированных колец. Например, комбинация одного или нескольких конденсированных колец с 6-членным ариловым или гетероариловым кольцом R2 содержит 5-15 членов и 0-5 гетероатомов О, S или N, необязательно замещенным заместителями, такими как ОН, =O, CN, NH2, Br, F или Cl.
[0044] Необязательные заместители могут занимать все положения кольцевой структуры R2, которые не являются соседними к линкеру, такие как положение один, положения 1-2 или положения 1-3. В некоторых аспектах одно положение является необязательно замещенным. Эти заместители могут быть необязательно замещены заместителями, аналогичными перечисленным заместителям. Конечно, некоторые заместители, такие как галоген, не являются дополнительно замещенными, как известно специалистам в данной области.
[0045] В некоторых аспектах R2 может быть пиримидинилом или пиридинилом, необязательно замещенным СН(ОН)СН3, С(ОН)(СН3)2, ОСН3, CN, СН3, СН2СН3, О-циклопропилом, SCH3, Br, Cl, F или NH2.
[0046] Не мешающие заместители у 6-членного арилового или гетероарилового кольца R2, которые могут подвергаться воздействию растворителя в связанной конформации, могут включать крупные заместители, такие как пролекарства.
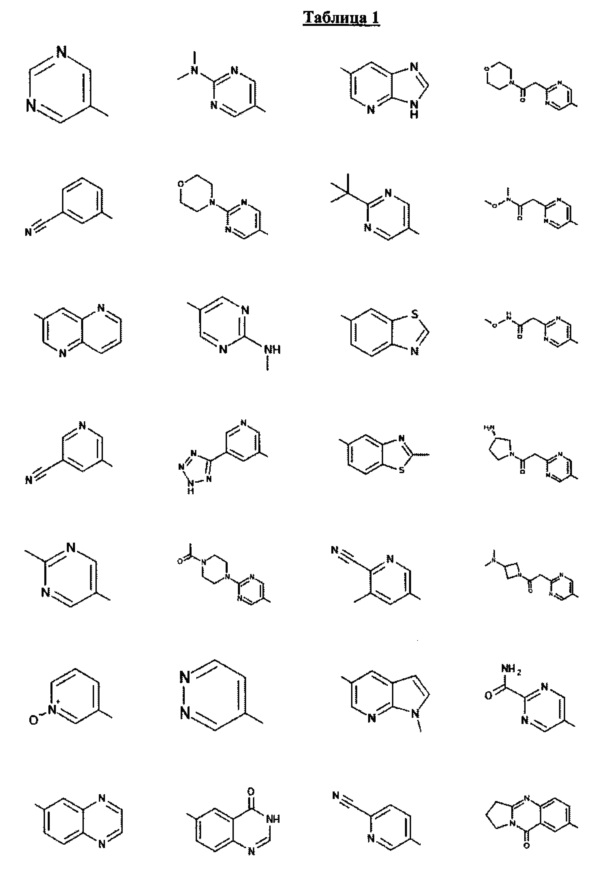
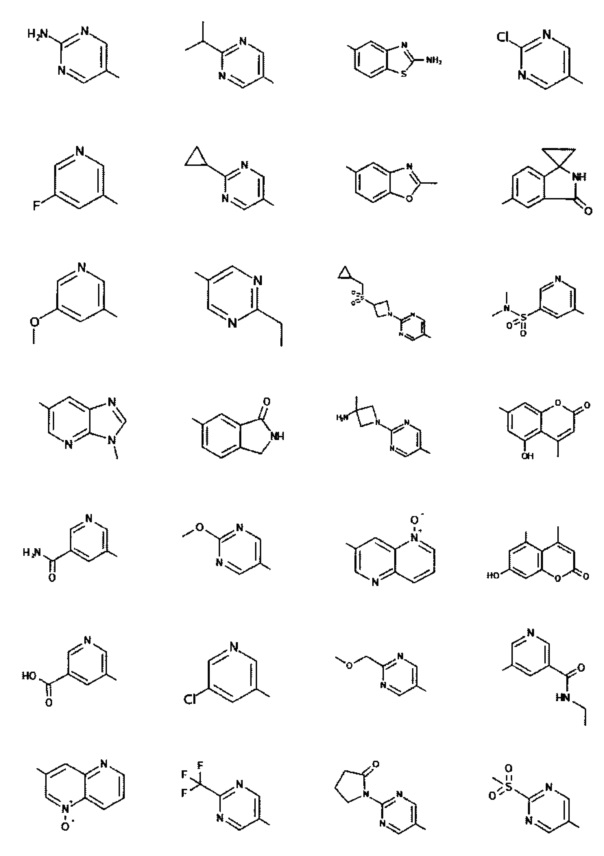
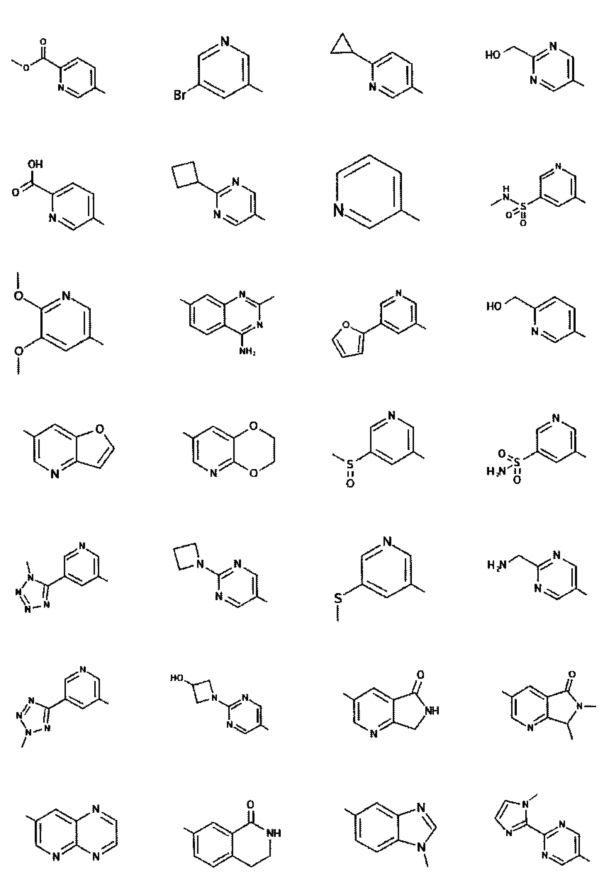
[0047] В некоторых аспектах R2 может быть выбран из заместителей в следующей Таблице 1.
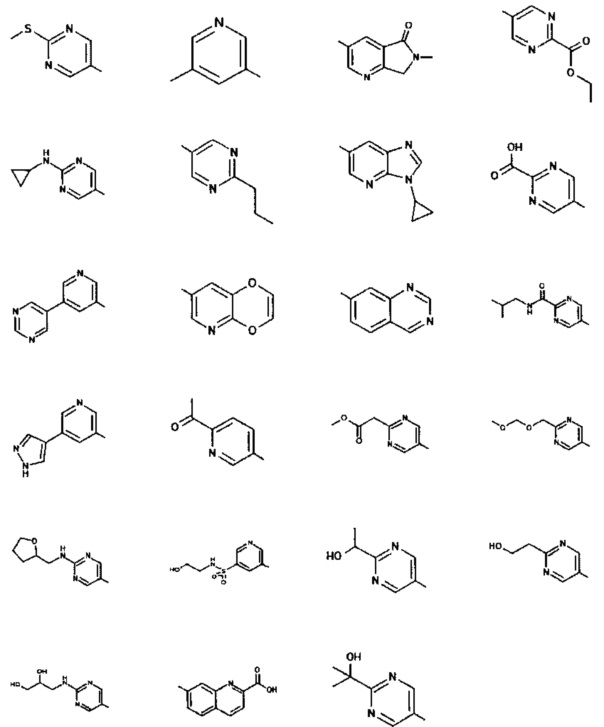
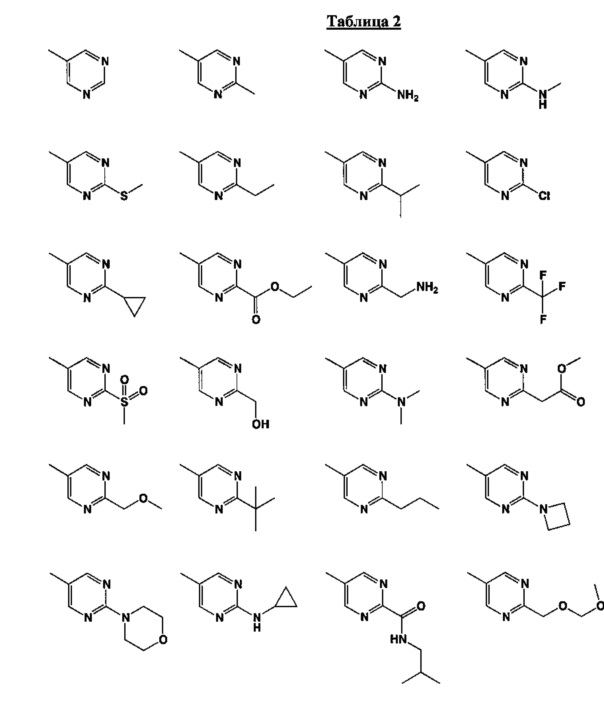
[0048] В некоторых аспектах R2 может быть выбран из заместителей в следующей Таблице 2.
[0049] На Фигурах 1 и 2 показано, что соединение подвергается воздействию растворителя в связанной конформации вдоль оси связи R4 и с поворотом против часовой стрелки на 0-90° от оси связи R4. Следовательно, выбор пролекарств и заместителей у R4 может варьироваться. При выборе заместителя R4, в некоторых аспектах, группы R4 стерически не сталкиваются с группами R2 или Z в связанной конформации, как показано на Фигуре 2. Опытным специалистам понятно, что во избежание стерического столкновения атомы у R4 не должны приближаться к атомам R2 или Rz (в связанной конформации) так, чтобы межатомные расстояния ближайших атомов составляли менее суммы их Ван-дер-Ваальсовских радиусов.
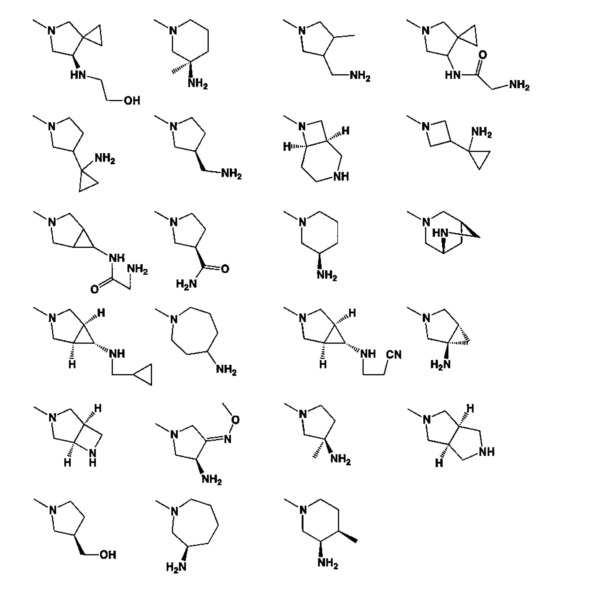
[0050] Кроме того, в некоторых аспектах заместитель R4 выступает не более чем примерно на 3 Å ниже плоскости Колец А, В и С в сторону связывающего кармана GyrB/ParE в связанной конформации. «В сторону дна связывающего кармана GyrE/ParE» относится к отсутствию выступания более, чем на 3 Å ниже плоскости в пределах около 5-6 связей от точки присоединения R4 к указанной структуре. Следовательно, те части R4, которые распространяются более, чем примерно на 5-6 связей от точки присоединения R4 к Кольцу С, могут выступать больше, чем примерно на 3 Å под плоскостью Колец А, В и С, поскольку эти части не ограничены дном связывающего кармана GyrE/ParE.
[0051] Это расстояние определяют как перпендикулярное расстояние от плоскости, построенной между центрами атомов трициклической структуры, до центра наиболее удаленного атома (от этой плоскости) у заместителя R4 в связанной конформации.
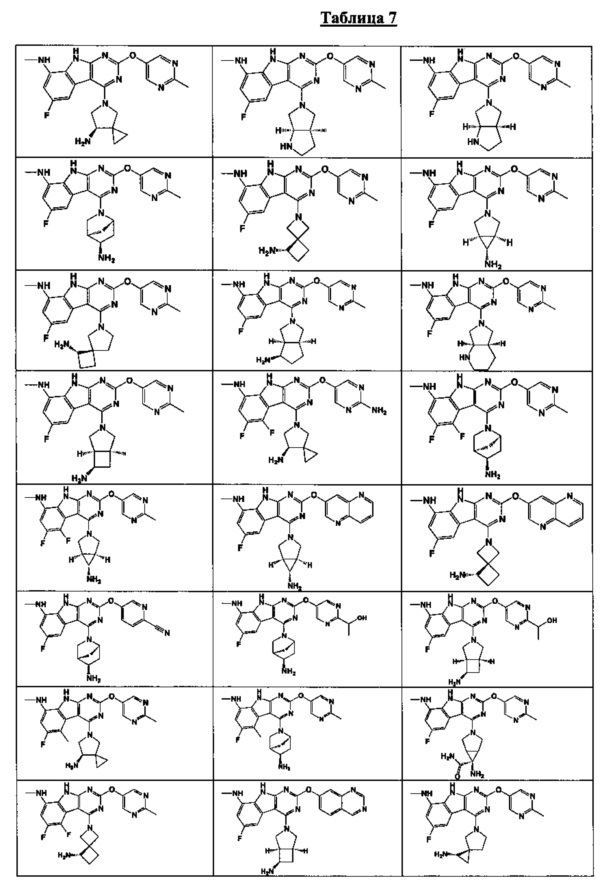
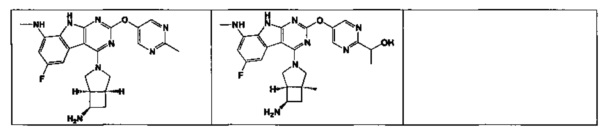
[0052] В некоторых аспектах R4 может быть Н.
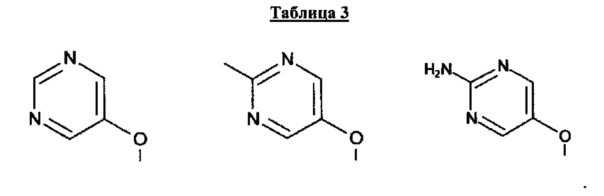
[0053] В некоторых аспектах R4 также может быть необязательно замещенным ORa; причем Ra представляет собой 5-6-членный арил или гетероарил, содержащий 0-3 гетероатома О, S или N, необязательно замещенный 0-3 не мешающими заместителями. В некоторых аспектах кольцевые положения, соседние с положением, в котором О присоединяется к Ra, могут быть замещены небольшими заместителями, такими как те, которые имеют 2 атома в скелете, такими как OCH3, CH3, CH2CH3, OH, NH2, F, Cl, Br, I или NO. В остальных положениях заместители могут быть крупнее и разнообразнее, поскольку заместители в этих положениях подвергаются воздействию растворителя в связанной конформации. В некоторых аспектах Ra представляет собой необязательно замещенный пиримидинил или пиридинил, такой как незамещенный пиримидинил или пиримидинил, замещенный CH3 или NH2. В некоторых аспектах ORa представляет собой один из следующих заместителей в Таблице 3.
[0054] В некоторых аспектах R4 может быть необязательно замещенным вторичным или третичным амином, присоединенным к Кольцу С через N вторичного или третичного амина. «Вторичный амин» относится к N-содержащему заместителю, который содержит один Н, присоединенный к N вторичного амина, когда этот заместитель присоединен к остальной части молекулы. «Третичный амин» относится к N-содержащему заместителю, который не содержит Н, присоединенного к N третичного амина, когда этот заместитель присоединен к остальной части молекулы.
[0055] Если R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к Кольцу С через N вторичного или третичного амина, то R4 может дополнительно включать первичный или вторичный амин, причем указанный первичный или вторичный амин не присоединен напрямую к Кольцу С. «Первичный амин» относится к аминной группе, которая содержит два атома Н, присоединенных к N первичного амина, при присоединении к остальной части заместителя. В отношении «вторичного амина», который не присоединен напрямую к Кольцу С, в этом случае вторичный амин относится к аминной группе, которая содержит один атом Н, присоединенный к N вторичного амина, при присоединении к остальной части заместителя. Первичный или вторичный амин, который напрямую не присоединен к Кольцу С, может располагаться в указанном соединении в связанной конформации, если:
a) расстояние между атомом С или N в Y и атомом N первичного или вторичного амина составляет от около 7 Å до около 10,5 Å;
b) расстояние между атомом С, к которому присоединен R8, и атомом N первичного или вторичного амина составляет от около 6 Å до около 9 Å;
c) расстояние между атомом С, к которому присоединен R4, и атомом N первичного или вторичного амина составляет от около 3,5 Å до около 6 Å; и
d) расстояние между атомом С, к которому присоединен R2, и атомом N первичного или вторичного амина составляет от около 5 Å до около 7,5 Å.
«Напрямую не присоединен к Кольцу С» в отношении первичного или вторичного амина относится к отсутствию связи, объединяющей первичный или вторичный амин с Кольцом С.
[0056] В некоторых аспектах R4 может быть необязательно замещенным третичным амином, который представляет собой необязательно замещенную 4-14-членную насыщенную циклогетероалифатическую кольцевую систему третичного амина, содержащую 1-3 атома N, 0-3 атома О и 0-1 атом S; и при этом указанная 4-14-членная насыщенная циклогетероалифатическая кольцевая система представляет собой одно кольцо, конденсированную кольцевую систему, мостиковую кольцевую систему или спирокольцевую систему.
[0057] В некоторых аспектах R4 может быть необязательно замещенным третичным амином, присоединенным к Кольцу С через N третичного амина, причем необязательно замещенный третичный амин содержит по меньшей мере один дополнительный N, отделенный от N третичного амина 2-3 атомами. Атомы, отделяющие N, не обязательно должны быть расположены в том же кольце. Например, один атом, отделяющий N, может быть в кольце, а второй атом может находиться в заместителе, или оба атома, отделяющие N, могут быть в скелете или у заместителя одного или различных колец.
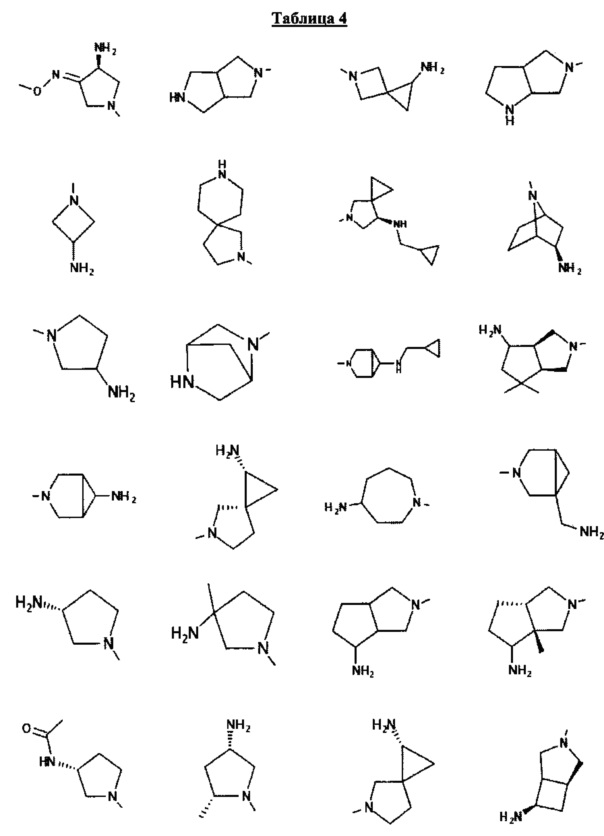
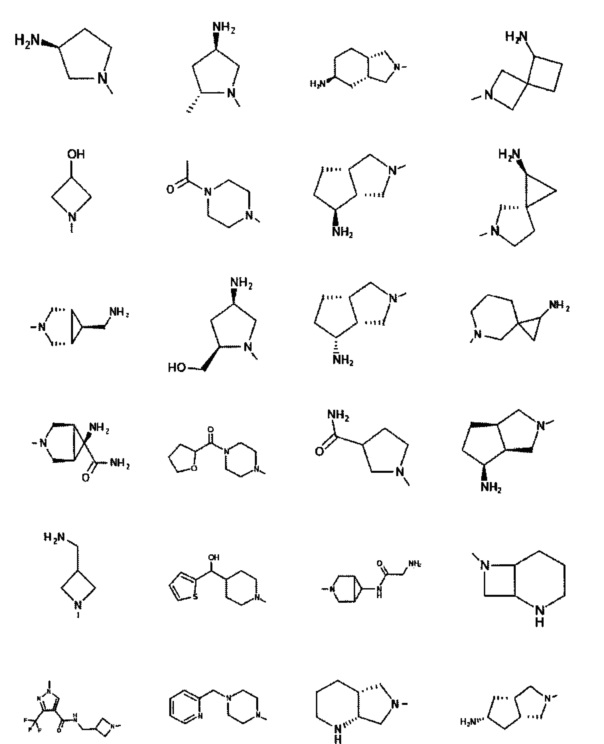
[0058] В некоторых аспектах необязательно замещенный вторичный или третичный амин R4 представляет собой один из следующих заместителей в Таблице 4.
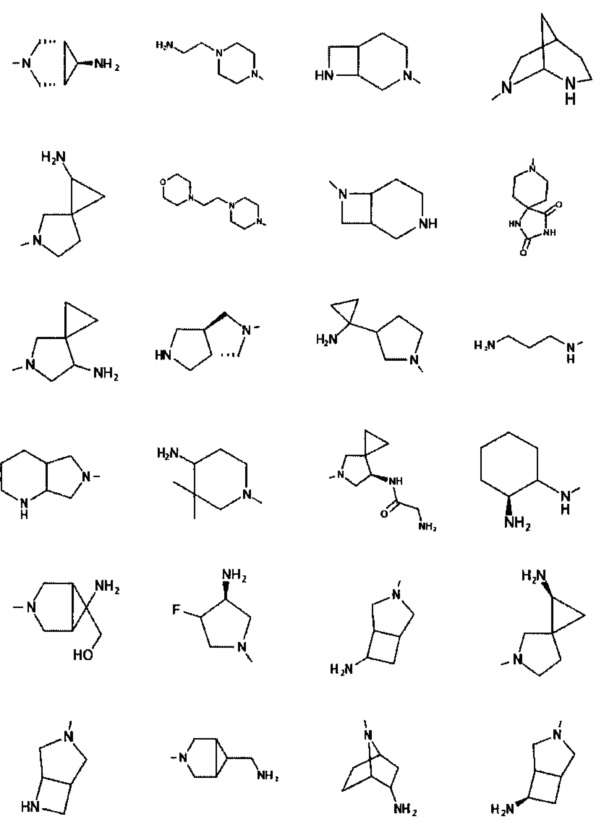
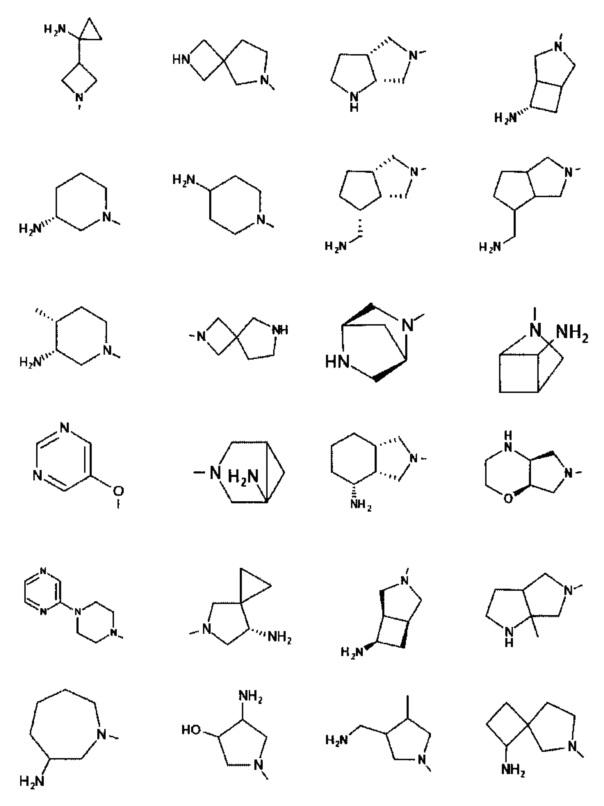
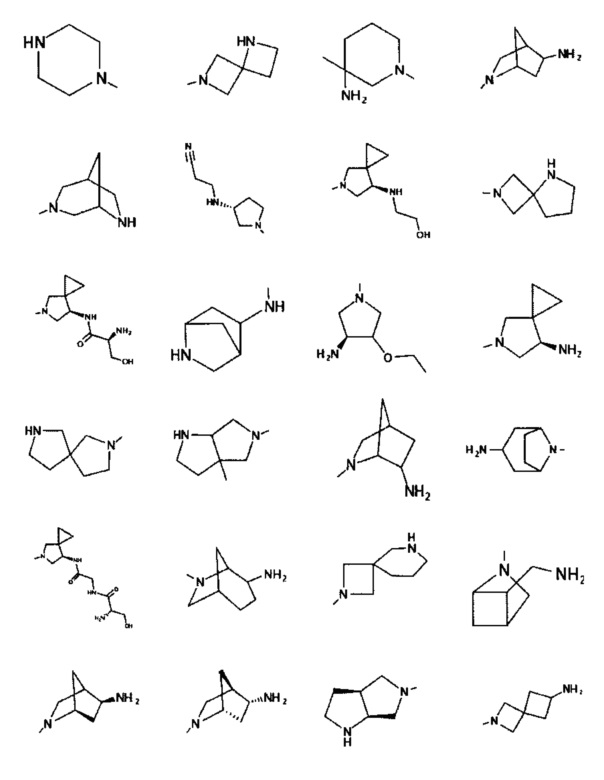
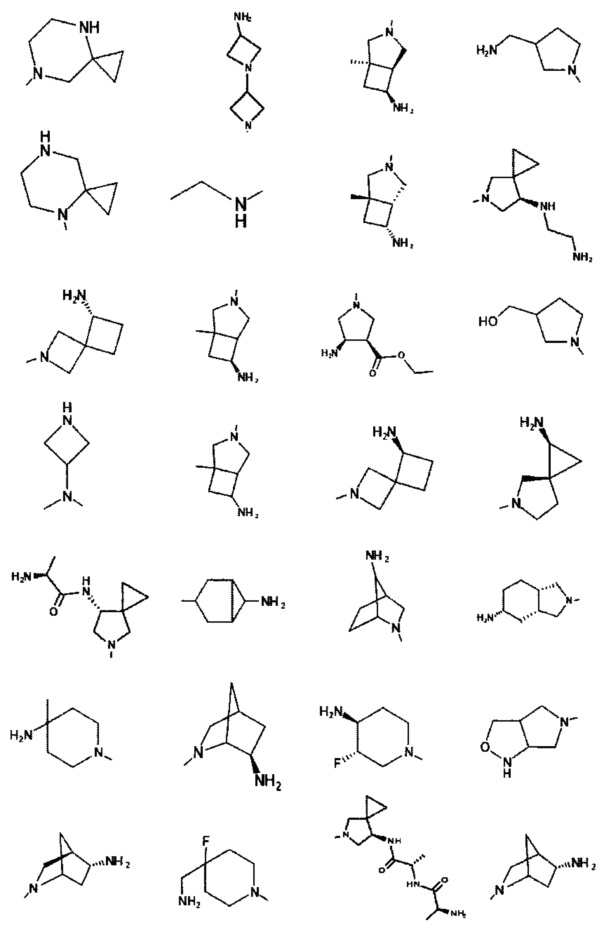
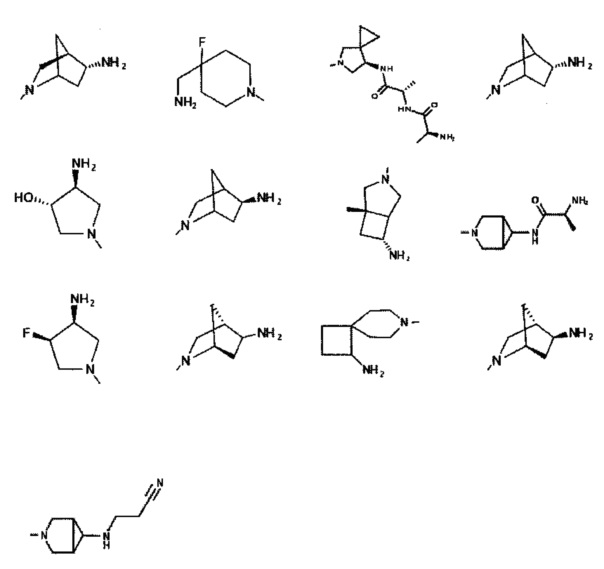
[0059] В некоторых аспектах R4 может быть нециклическим вторичным или третичным амино, замещенным 1-2 не мешающими заместителями.
[0060] В некоторых аспектах R4 может быть выбран из группы, состоящей из необязательно замещенного пиразолила, фенила, пиперазинила, пиридинила и тетрагидропиридинила.
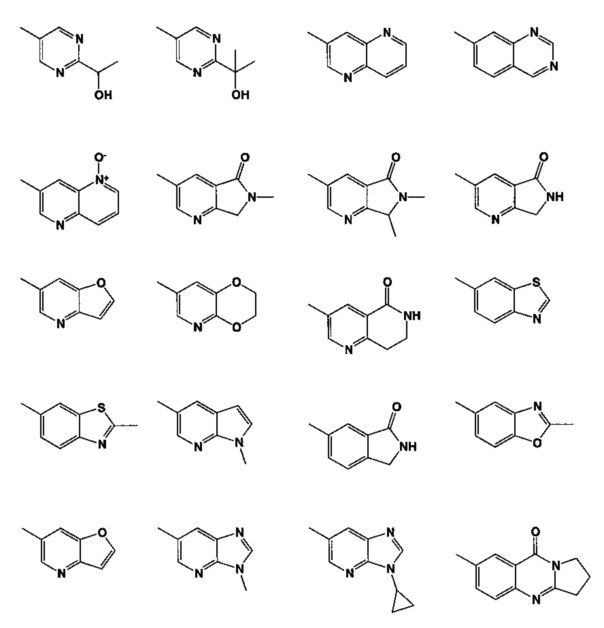
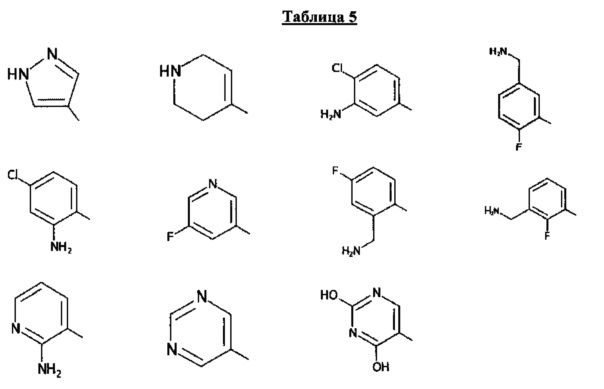
[0061] В некоторых аспектах R4 может быть необязательно замещенным 5-10-членным ненасыщенным циклическим или гетероциклическим остатком, содержащим 0-3 гетероатома N, О или S. Необязательные заместители могут включать 0-2 необязательных заместителя, выбранных из группы, состоящей из CH3, NH2, F, Cl, и CH2NH2. В некоторых аспектах необязательно замещенный 5-10-членный ненасыщенный циклический или гетероциклический остаток, содержащий 0-3 гетероатома N, О или S, у R4 представляет собой один из следующих заместителей в Таблице 5.
[0062] Необязательный заместитель у R4 может включать 0-3 не мешающих заместителя. Не мешающий заместитель у R4 может быть заместителем, выбранным из группы, состоящей из ОН, NO, CO2H, CN, NH2, Br, Cl, F, SO3H и NO2, или представляет собой С1-15 углеводородный остаток, содержащий 0-5 гетероатомов О, S или N, необязательно замещенный ОН, СН=O, NH2, =NOH, =NNH2, =NOCH3, Br, F, Cl, SO3H или NO2. Замещения могут быть у атома С или у гетероатома, что допускает такие группы как S=O. Кроме того, заместитель ОН может быть в форме оксида, допуская таким образом, например, пиридил, имеющий N-оксид, причем N является кольцевым гетероатомом. Углеводородный остаток C1-С15, содержащий 0-5 гетероатомов О, S или N может включать комбинацию углеводородных групп, такую как комбинация алифатических колец или цепей и ароматический колец, связанных вместе.
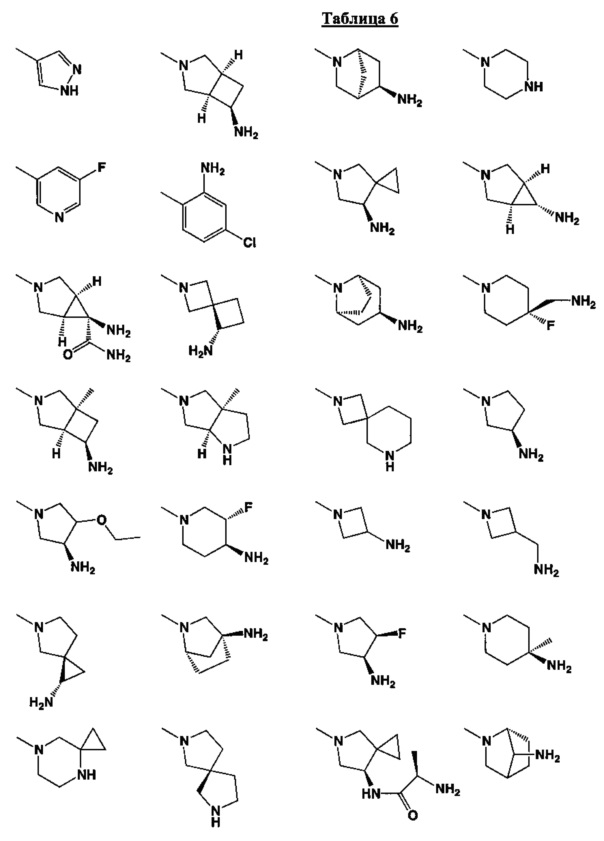
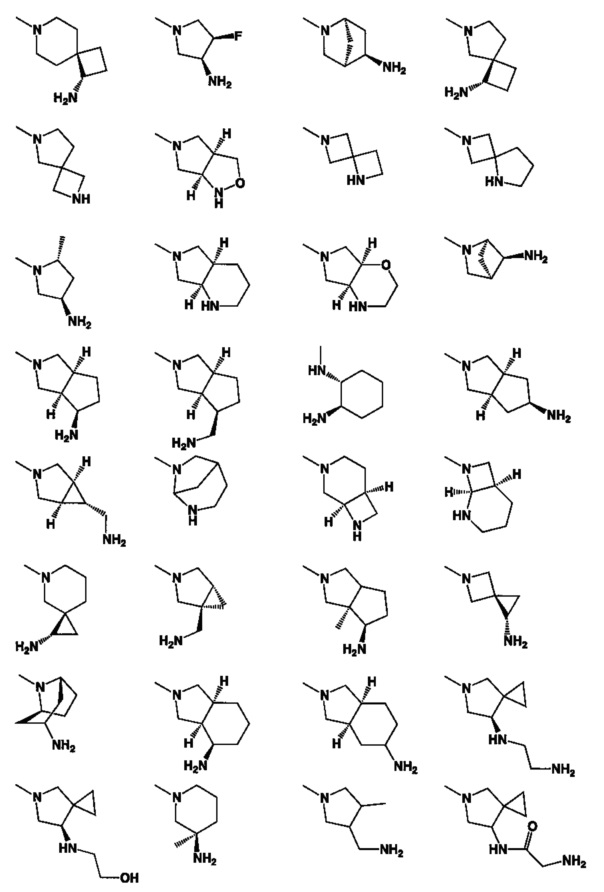
[0063] В некоторых аспектах R4 может быть выбран из заместителей в следующей Таблице 6.
[0064] Соединение может быть одним из соединений, представленных в Примерах.
[0065] В некоторых аспектах соединение может быть соединением в Таблице 7.
[0066] Если соединения Формулы I содержат один или несколько хиральных центров, то предполагаются оптически чистые формы, а также смеси стереоизомеров или энантиомеров.
[0067] Рассмотрены также различные способы получения указанных соединений. Заместители, если не указано, являются такими же заместителями, как в Формуле I. В некоторых аспектах, если R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к Кольцу С через N вторичного или третичного амина, способ включает обработку
соединением HR4 с образованием соединения Формулы I; и необязательно дополнительно включает, перед стадией обработки, защиту R8 защитной группой или защиту амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и необязательно снятие защитных групп после указанной стадии обработки.
[0068] Защитные группы применимы для хемоселективности, и они известны в данной области. Типичные защитные группы включают трет-бутилоксикарбонил (BOC) и карбобензилокси (Cbz). Если защитной группой является BOC, то для снятия защиты может быть использована кислота, если защитной группой является Cbz, то для снятия защиты может быть использовано каталитическое гидрирование.
[0069] Перед стадией обработки, описанной непосредственно выше, способ может дополнительно включать взаимодействие соединения Формулы II
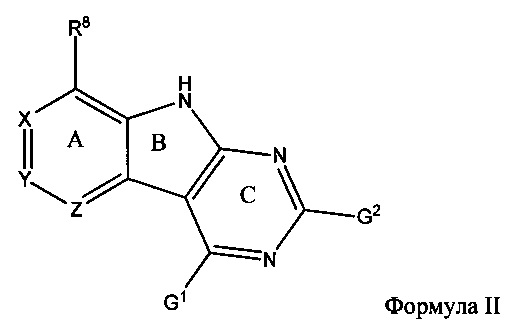
с R2LH в щелочных условиях, причем G1 и G2 являются уходящими группами, независимо выбранными из группы, состоящей из Cl, Br, F, I, SR, SOR, SO2R, OSO2R и O-бензотриазола (OBt); при этом R может быть С1-8 алкилом, арилом или гетероарилом, содержащим 0-5 атомов О, S или N, необязательно замещенным С1-4 алкилом, С1-4 алкокси, Cl, Br, F, I или NO2, таким как метил, бензил и п-метоксибензил, с образованием соединения, имеющего структуру
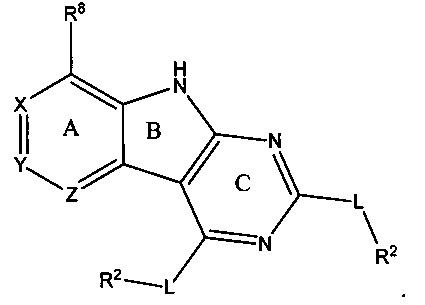
[0070] В некоторых аспектах соединения, в которых R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к Кольцу С через N вторичного или третичного амина, также могут быть получены с использованием способа, включающего обработку
соединением R2LH в щелочных условиях, таких как с анионом фенола, тиофенола, гетероарилгидрокси или гетероарилтиола, причем G2 представляет собой уходящую группу, выбранную из группы, состоящей из Cl, Br, F и I; и необязательно дополнительно включающего, перед стадией обработки, описанной непосредственно выше, защиту R8 защитной группой или защитой амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и снятие защиты с R8 и R4 после стадии обработки.
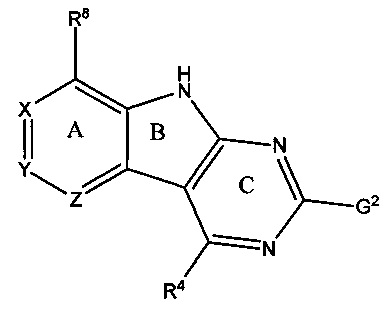
[0071] Перед стадией обработки, описанной непосредственно выше, способ может дополнительно включать взаимодействие соединения Формулы II
с HR4 с образованием
причем G1 представляет собой уходящую группу, выбранную из группы, состоящей из Cl, Br, F и I.
[0072] В некоторых аспектах, если L представляет собой S, то способ получения соединения, в котором R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к Кольцу С через N вторичного или третичного амина, может включать обработку
причем G1 представляет собой уходящую группу, полученную из SO2галогенида, бис(2-оксо-3-оксазолидинил)фосфина (BOP) или бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата (pyBOP), соединением HR4 с образованием описанных в настоящем документе соединений. Этот способ может также необязательно дополнительно включать, перед стадией обработки, описанной непосредственно выше, защиту R8 защитной группой или защиту амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и снятие защиты с R8 и R4 после стадии обработки.
[0073] Перед стадией обработки, описанной непосредственно выше, способ может дополнительно включать взаимодействие
с G1X1 с образованием
причем G1X1 представляет собой SO2галогенид, бис(2-оксо-3-оксазолидинил)фосфин (BOP) или бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфат (pyBOP).
[0074] Перед стадией обработки, описанной непосредственно выше, способ может дополнительно включать связывание
с R2X2, причем X2 представляет собой Br или I, с образованием
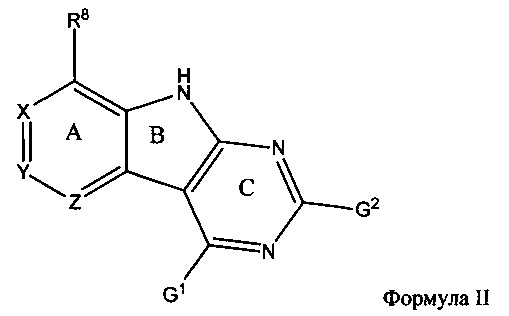
[0075] В другом аспекте промежуточное соединение имеет структуру Формулы II:
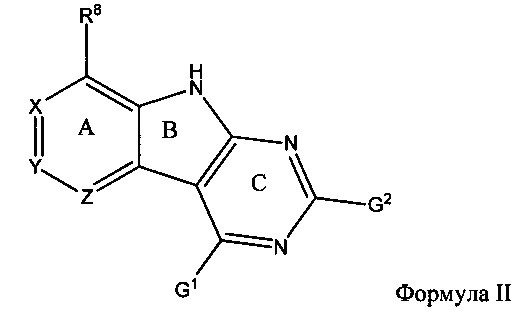
или его амино-защищенное промежуточное соединение; причем: G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из SH, OH, Cl, Br, F, I, SR, SOR, SO2R, OSO2R, OAr и OBt; R представляет собой С1-8 алкил, арил или гетероарил; Ar представляет собой арил или гетероарил, содержащий 0-5 атомов O, S или N, необязательно замещенный С1-4 алкилом, С1-4 алкокси, галогеном или NO2; Bt представляет собой бензотриазол; R8 представляет собой взаимодействующий заместитель, имеющий длину от около 1 Å до около 5 Å от точки присоединения углерода в Кольце А до концевого атома в R8 и ширину около 3,3 Å или менее; и X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, соответственно, при условии, что не более двух из X, Y и Z представляют собой N, причем RX представляет собой Н или взаимодействующий заместитель, имеющий длину от около 1 Å до около 2 Å от углерода в CRX до концевого атома в RX; причем RY представляет собой Н или взаимодействующий заместитель, имеющий длину от около 1 Å до около 3 Å от углерода в CRY до концевого атома в RY; причем RZ представляет собой H или взаимодействующий заместитель, имеющий длину от около 1 Å до около 2 Å от углерода в CRZ до концевого атома в RZ; при условии, что R8 не является CH3, и при условии, что если R8 представляет собой OCH3, то RX и RY не являются OH.
[0076] Если промежуточное соединение представляет собой амино-защищенное промежуточное соединение, то один или несколько атомов азота в этом соединении могут быть защищены карбобензилокси-группой (Cbz) или ВОС. G1 и G2 могут быть уходящими группами, независимо выбранными из группы, состоящей из тозилата, мезилата, трифлата, О-пиримидина, О-фенила и О-пиридина.
[0077] На следующих схемах изображены аспекты реакционных стадий для получения исходных материалов, промежуточных веществ и соединений, описанных в настоящем документе, которые подробно описаны в Примерах. Исходные материалы для заместителей R2 и R4 имеются в продаже или могут быть получены опытным специалистом с использованием способов, описанных в литературе.
1. Общие способы получения трициклического пиримидо[4,5-b]индольного ядра
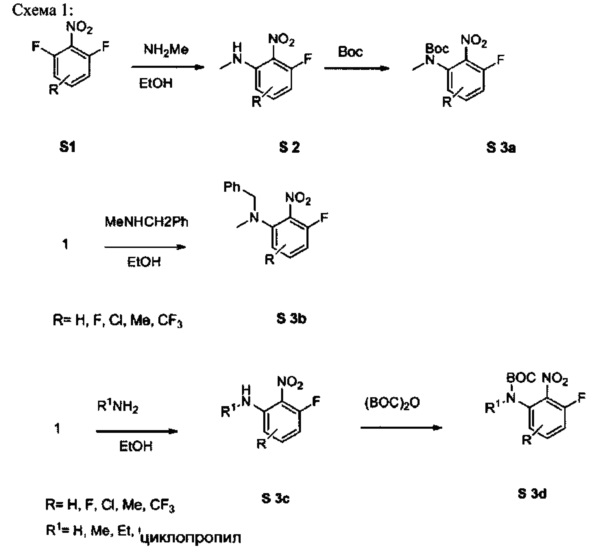
[0078] В Кольцо А пиримидоиндольной системы может быть внедрено большое количество аминов и замещенных аминов, как показано на Схеме 1. Орто-фтор-нитробензолы S1 могут быть легко замещены аминами с образованием ортоамино-аналогов S2. Защитная группа может быть введена путем внедрения в исходный материал (как в S 3b) или путем внедрения после реакции замещения фторарила (как в S 3c). При наличии алкильной или алкокси-группы R8, быть использовано нитрование для внедрения нитрогруппы в орто-положение к группе R8 S3d. Если реакция нитрования обеспечивает смеси региоизомеров, то может быть использована хроматография для выделения заданного изомера.
Схема 1: общий способ получения замещенных фениловых исходных материалов
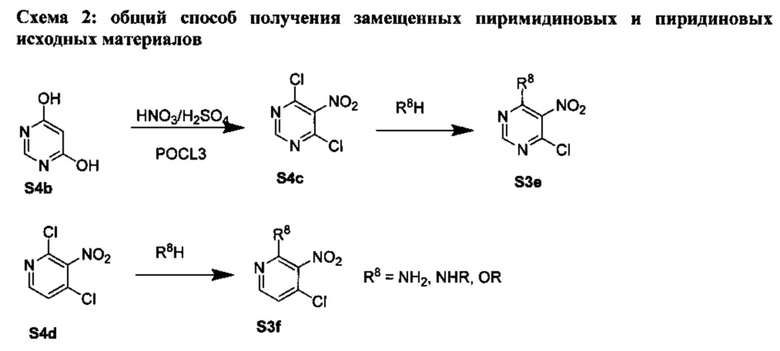
[0079] На Схеме 2 представлены общие способы получения широкого ряда пиридиновых и пиримидиновых исходных материалов. Нитрование 4,6-дигидроксипиримидина с последующим превращением гидроксильных групп в хлор-группу при помощи POCl3 дает промежуточное соединение S4c. Хлор легко замещается аминами и спиртами с образованием заданного промежуточного соединения S3e. Таким же образом имеющийся в продаже пиридин S4d легко замещают аминами и спиртами с образованием промежуточного соединения S3f.
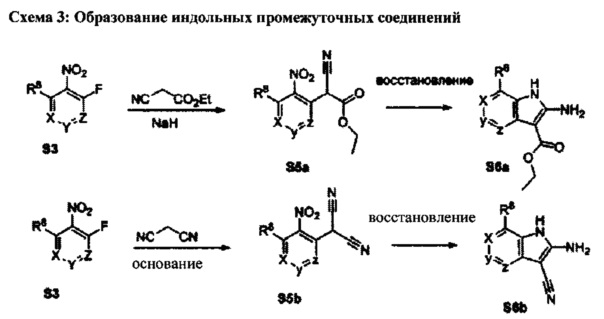
[0080] Ортофтор-нитроароматические вещества S3 превращают (Схема 2) в индолы и азот-замещенные индолы S6a и S6b (пирролопиримидины и пирролопиридины) обработкой цианоэтилацетатом или цианомалонатом с последующим восстановлением цинком в уксусной кислоте, альтернативно, нитро-группа может быть восстановлена многими альтернативными восстановителями, такими как бисульфит натрия.
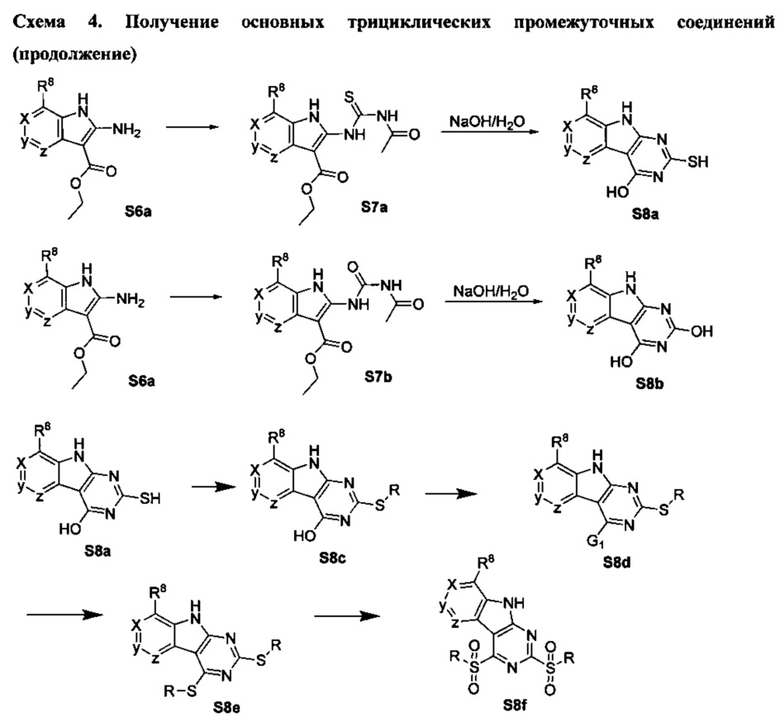
[0081] Индольные промежуточные соединения превращают в трициклические промежуточные соединения, как показано на Схеме 4. Реакция амино-эфирного индола S6a с ацилизотиоцианатом с последующей обработкой основанием дает трицикл S8a с SH в положении 2 и OH в положении 4. Альтернативно, обработка ацилизоцианатом с последующей обработкой основанием дает S8b с заместителем OH в обоих положениях 2 и 4 указанного трицикла. Эти разнообразные промежуточные соединения, такие как S8a, могут быть превращены в бис-сульфон сначала алкилированием серы в положении 2 с последующей активацией положения 4 таким реагентом как ВОР или мезилхлорид, с последующим замещением сульфида, затем окислением до бис-сульфона S8f таким реагентом как сульфон.
Схема 4. Получение основных трициклических промежуточных соединений
[0082] Альтернативно, дигидрокси-ядро S8b может быть превращено в дихлор-трицикл S8g. Амино-нитрил-индольные промежуточные соединения S6b могут быть превращены в бис-сульфон обработкой дисульфидом углерода и алкоксидом с образованием аниона 2,4-дитиол-трицикла. Это промежуточное соединение может быть алкилировано in situ, а затем окислено с образованием бис-сульфона S8f.
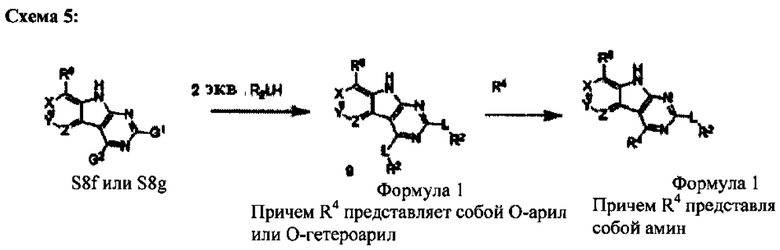
2. Общие способы превращения трициклических ядер в соединения Формулы I
[0083] Существует множество способов превращения основных трициклических промежуточных веществ в соединения Формулы I.
[0084] На Схеме 5 любое из промежуточных соединений S8f или S8g может быть превращено в бис-арилокси-соединение 9. Арилокси-группа в положении 4 может быть замещена аминами или спиртами с образованием заданного соединения Формулы I, причем R4 представляет собой один из аминов алкоксида. В некоторых случаях необходимо использовать защитные группы для промежуточных соединений S8 и/или для группы R4. В этих случаях может потребоваться дополнительная стадия для снятия защитной группы.
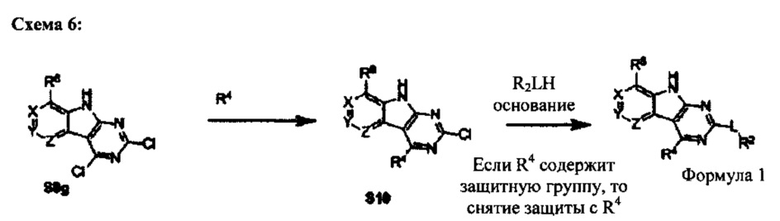
[0085] В качестве альтернативного способа, дихлор-трициклическое промежуточное соединение S8g может быть обработано сначала группой R4 с последующим замещением положения 2 алкоксидом R2OH (Схема 6). Как правило, для этого способа необходимы защитные группы, особенно если в качестве группы R4 используют диамин. В этих случаях снятие защитных групп дает соединение Формулы I. Этот способ особенно применим, если используют ценную группу R2OH, или группа R2 богата электронами.
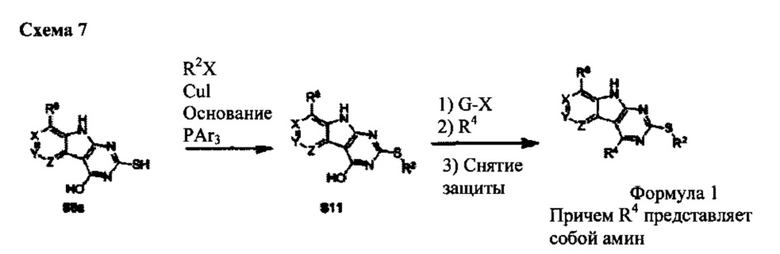
[0086] В случаях, если L представляет собой S, то соединения Формулы I могут быть получены напрямую из S8a по способу на Схеме 7. В этом способе сульфид связывают с арилгалогенидом (предпочтительно йод- или бром-ароматическим соединением). Активация гидроксильной группы в положении 4 такими реагентами как сульфонилгалогенид, или связывающим агентом, таким как ВОР, с последующим замещением амина обеспечивает заданное соединение Формулы I.
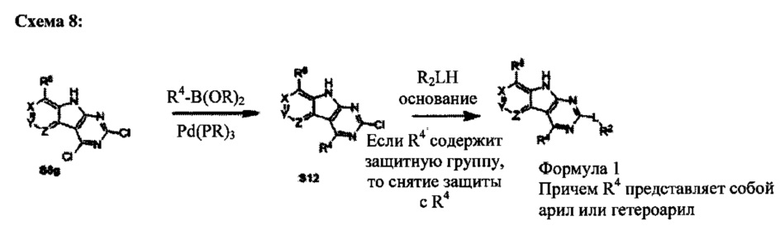
[0087] Соединения Формулы I, в которых R4 представляет собой арил или гетероарил, могут быть получены так, как показано на Схеме 8. В этом случае промежуточное дихлор-соединение S8g связывают с бороновой кислотой, используя условия сочетания Сузуки. Полученный продукт затем обрабатывают алкоксидом с образованием соединения Формулы I.
[0088] Пролекарства также могут быть получены из соединений Формулы I или II. Термин «пролекарство», при использовании в настоящем документе, представляет соединения, которые могут быть преобразованы in vivo в активные исходные соединения, описанные в настоящем документе.
[0089] Примеры пролекарств, например, у R4, включают NHNHCH3,
[0090] Подразумеваются также фармацевтически приемлемые соли, сложные эфиры или пролекарства соединений, описанных в настоящем документе. Специалистам в данной области понятно, что многочисленные пролекарства, соли, гидраты, сольваты и полиморфы могут быть получены из соединений, описанных в настоящем документе, и что также легко могут быть получены различные изотопно-замещенные варианты (например, путем замены водорода дейтерием, углерода на 13C, азота на 15N или фосфора на 32P), известные как «изотопомеры». Все такие производные подразумеваются входящими в рамки настоящего описания.
[0091] Многие из соединений настоящего документа описаны как гидрохлоридные или другие соли, однако специалистам в области медицинской химии понятно, что выбор соли не является критическим, и что могут быть получены другие фармацевтически приемлемые соли по хорошо известным способам. В публикации Handbook of Pharmaceutical Salts: Properties, Selection and Use. (P. Heinrich Stahl and Camille G. Wermuth, eds.) International Union of Pure and Applied Chemistry, Wiley-VCH 2002 and L.D. Bighley, S.M. Berge, D.C. Monkhouse, в книге "Encyclopedia of Pharmaceutical Technology'. Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc., New York, Basel, Hong Kong 1995, pp. 453-499 подробно рассмотрены такие соли.
[0092] Соединения, описанные в настоящем документе, включают такие структуры, которые представлены в примерах, а также их фармацевтически приемлемые соли, сложные эфиры и пролекарства. В некоторых вариантах реализации соединение находится в фармацевтической композиции или лекарственной форме, причем указанная фармацевтическая композиция или лекарственная форма обеспечивает эффективное антибиотическое количество соединения для лечения или предупреждения инфекции.
[0093] В другом аспекте настоящее описание относится к фармацевтической композиции, включающей один или несколько физиологически приемлемых поверхностно-активных агентов, дополнительных носителей, разбавителей, формообразующих средств, сглаживающих агентов, суспендирующих средств, пленкообразующих веществ и вспомогательных добавок для покрытий, или их комбинацию; и композицию, описанную в настоящем документе. Приемлемые дополнительные носители или разбавители для терапевтического применения хорошо известны в фармацевтической области, и они описаны, например, в публикации Remington's Pharmaceutical Sciences, 18е издание, Mack Publishing Co., Easton, PA (1990), которая включена в настоящий документ путем ссылки в полном объеме. В фармацевтической композиции могут быть представлены консерванты, стабилизаторы, красители, подсластители, ароматизаторы, вкусовые агенты и тому подобные. Например, в качестве консервантов могут быть добавлены бензоат натрия, аскорбиновая кислота и сложные эфиры п-гидроксибензойной кислоты. Кроме того, могут быть использованы антиоксиданты и суспендирующие агенты. В различных вариантах реализации в качестве поверхностно-активных агентов могут быть использованы спирты, сложные эфиры, сульфированные алифатические спирты и тому подобные; в качестве формообразующих средств могут быть использованы сахароза, глюкоза, лактоза, крахмал, микрокристаллическая целлюлоза, кристаллическая целлюлоза, маннит, легкий безводный силикат, алюминат магния, магния метасиликат-алюминат, синтетический силикат алюминия, карбонат кальция, гидрокарбонат натрия, гидрофосфат кальция, кальций-карбоксиметилцеллюлоза и тому подобные; в качестве сглаживающих агентов могут быть использованы стеарат магния, тальк, отвержденный жир и тому подобные; в качестве суспендирующих средств или смазывающих агентов могут быть использованы кокосовое масло, оливковое масло, кунжутное масло, арахисовое масло, соя; в качестве суспендирующих средств могут быть использованы фталат ацетата целлюлозы как производное углевода, такого как целлюлоза или сахар, или метилацетат-метакрилатный сополимер как производное поливинила; и в качестве суспендирующих средств могут быть использованы пластификаторы, такие сложноэфирные фталаты и тому подобные.
[0094] Термин «фармацевтическая композиция» относится к смеси соединения, описанного в настоящем документе, с другими химическими компонентами, такими как разбавители или дополнительные носители. Фармацевтическая композиция облегчает введение соединения в организм. В данной области существует множество методик введения фармацевтических композиций, включая, но не ограничиваясь этим, пероральное, инъекционное, аэрозольное, парентеральное и локальное введение. В некоторых вариантах реализации представлены фармацевтически приемлемые соли соединений, описанных в настоящем документе.
[0095] Термин «носитель» относится к химическому соединению, которое облегчает введение соединения в клетки или ткани.
[0096] Термин «разбавитель» относится к химическим соединениям, разбавленным в воде, которые растворяют рассматриваемую композицию, а также стабилизируют биологически активную форму соединения. В качестве разбавителей в данной области техники используют соли, растворенные в буферных растворах. Одним широко применяемым буферным раствором является фосфатно-солевой буферный раствор, поскольку он имитирует солевые условия крови человека. Поскольку буферные соли способны контролировать pH раствора в низких концентрациях, буферные разбавители редко изменяют биологическую активность соединения. При использовании в настоящем документе, «формообразующее средство» относится к инертной субстанции, которую добавляют к композиции для обеспечения композиции, без исключения, объема, консистенции, устойчивости, связывающей способности, смазывания, способности к распадаемости и так далее. «Разбавитель» является типом формообразующего средства.
[0097] Термин «физиологически приемлемый» относится к носителю или разбавителю, который не аннулирует биологическую активность и свойства соединения.
[0098] Фармацевтические соединения, описанные в настоящем документе, могут быть введены пациенту, являющемуся человеком, per se или в фармацевтических композициях, где они смешаны с другим активным ингредиентом(ами), как в комплексной терапии, или с соответствующими носителями или формообразующим средством(ами). В некоторых вариантах реализации лекарственная форма включает такие формы, в которых соединение вводят per se. Кроме того, лекарственная форма может включать фармацевтическую композицию. В любом случае, лекарственная форма может включать достаточное количество димерного соединения для лечения бактериальной инфекции как часть определенного протокола введения, что является понятным для специалистов в данной области. Методики составления композиций и введения соединений настоящего изобретения можно найти в публикации "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18e издание, 1990.
[0099] Пригодные способы введения могут включать, например, пероральное, ректальное, трансмукозальное, локальное или кишечное введение; парентеральную доставку, включая внутримышечные, подкожные, внутривенные, интрамедуллярные инъекции, а также интратекальные, прямые интравентрикулярные, интрапериотнеальные, интраназальные или внутриглазные инъекции. Соединение настоящего изобретение также может быть введено в лекарственных формах устойчивого или контролируемого высвобождения, включая инъекции депо, осмотические насосы, пилюли, трансдермальные (включая электротранспортные) пластыри и тому подобные, для пролонгированного и/или заданного по времени, импульсного введения с определенной скоростью.
[0100] Фармацевтические композиции, описанные в настоящем документе, могут быть выпущены любым известным способом, например, путем традиционного смешивания, растворения, гранулирования, приготовления драже, растирания в порошок, эмульгирования, инкапсулирования, включения в гель или таблетирования.
[0101] Фармацевтические композиции могут быть составлены любым стандартным образом с использованием одного или нескольких физиологически приемлемых носителей, включая формообразующие средства и вспомогательные вещества, которые облегчают переработку активных соединений в препараты, которые могут быть использованы фармацевтически. Правильная композиция зависит от выбранного способа введения. Любой из хорошо известных способов, разбавителей, носителей и формообразующих средств может быть использован так, как это применимо и понимается в данной области; например, в публикации Remington's Pharmaceutical Sciences, выше.
[0102] Композиции для инъекций могут быть получены в стандартных формах, таких как жидкие растворы или суспензии, или в твердых формах, пригодных для получения растворов или суспензий перед инъекцией, а также в виде эмульсий. Пригодные формообразующие средства включают, например, воду, солевой раствор, декстрозу, маннит, лактозу, лецитин, альбумин, глутамат натрия, гидрохлорид цистеина и тому подобные. Кроме того, при необходимости фармацевтические композиции для инъекций могут также содержать небольшие количества нетоксичных вспомогательных веществ, таких как увлажняющие средства, pH буферные агенты и тому подобные. Физиологически совместимые буферы включают, но не ограничиваясь этим, раствор Хэнкса, раствор Рингера или физиологический солевой буферный раствор. При необходимости могут быть использованы препараты, усиливающие абсорбцию.
[0103] Для трансмукозального введения в рецептуре могут быть использованы пенетранты, соответствующие препятствию, через которое должна пройти композиция.
[0104] Фармацевтические композиции для парентерального введения, например, болюсной инъекции или непрерывной инфузии, включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений могут быть приготовлены в виде соответствующих масляных инъекционных суспензий. Водные суспензии для инъекций могут содержать вещества, увеличивающие вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия может содержать также подходящие стабилизаторы или агенты, увеличивающие растворимость соединения настоящего изобретения для обеспечения возможности приготовления высококонцентрированных растворов. Композиции для инъекций могут быть представлены в форме разовой дозы, например, в ампулах, или в упаковках для многократного приема с добавлением консерванта. Композиции могут принимать такие формы как суспензии, растворы или эмульсии в масляных или водных средах, и могут содержать агенты образования этих форм, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в порошковой форме для разбавления соответствующим жидким носителем, например стерильной апирогенной водой, перед применением.
[0105] Для перорального введения композиция может быть легко составлена путем смешивания рассматриваемых композиций с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители, которые могут быть использованы, помимо катионного полимерного носителя, обеспечивают возможность составления композиций в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и тому подобного, для перорального употребления пациентом, подлежащим лечению. Фармацевтические препараты для перорального применения могут быть получены путем смешивания активного соединения с твердым формообразующим средством, необязательного измельчения полученной смеси и переработки этой смеси в гранулы после добавления соответствующих вспомогательных веществ, при необходимости, для получения таблеток или ядер драже. Подходящими формообразующими средствами, в частности, являются наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметил-целлюлоза, натрий-карбоксиметилцеллюлоза и/или поливинилпирролидон (ПВП), например, повидон. При необходимости могут быть добавлены агенты для усиления распадаемости таблеток, такие как поперечно сшитый поливинилпирролидон (например, кросповидон), агар или альгиновая кислота или ее соль, такая как альгинат натрия. Ядра драже покрывают соответствующими покрытиями. Для этой цели могут быть использованы концентрированные растворы сахара, которые могут необязательно содержать гуммиарабик, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или диоксид титана, растворы глазури и соответствующие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к покрытиям таблеток или драже для идентификации или характеристики различных композиций доз активного соединения.
[0106] Фармацевтические препараты, которые могут быть использованы перорально, включают плотно набитые капсулы из желатина, а также мягкие, герметичные капсулы из желатина и пластификатора, такого как глицерин или сорбит. Плотно набитые капсулы могут содержать активные компоненты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или смазочными веществами, такими как тальк или стеарат магния и необязательно со стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все композиции для перорального введения должны быть приготовлены в виде доз, пригодных для такого введения.
[0107] Для буккального введения композиции могут принимать форму таблеток или пастилок, составленных стандартным способом. Подразумевается введение в слизистую оболочку и сублингвально.
[0108] Для введения ингаляцией, композиция может быть соответствующим образом быть доставлена в форме аэрозольного спрея из упаковок под давлением или распылителя, с использованием соответствующего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого соответствующего газа. В случае аэрозоля под давлением разовая доза может определяться за счет обеспечения клапана для доставки дозированного количества. Капсулы и картриджи, например, желатиновые, для использования в ингаляторе или инсуфляторе, могут содержать порошковую смесь соединения и соответствующей порошковой основы, например, лактозы или крахмала.
[0109] Далее в настоящем документе описаны различные фармацевтические композиции, хорошо известные в фармацевтической области для применений, включающих внутриглазную, интраназальную и ушную доставку. Для этих применений в данной области общеизвестны соответствующие пенетранты. Такие пригодные фармацевтические композиции наиболее часто и предпочтительно готовят в стерильной, изотоничной и буферной форме для устойчивости и комфорта. Фармацевтические композиции для интраназальной доставки могут также включать капли и спреи, которые часто готовят для имитации, во многих отношениях, носовых секреций для обеспечения сохранения нормального цилиарного действия. Как описано в публикации Remington's Pharmaceutical Sciences, 18е издание, Mack Publishing Co., Easton, PA (1990), которая включена в настоящий документ путем ссылки в полном объеме, и как хорошо известно специалистам в данной области, применимые композиции наиболее часто и предпочтительно являются изотоничными, немного буферными для сохранения pH от до 5,5 до 6,5, и наиболее часто и предпочтительно включают антимикробные консерванты и соответствующие стабилизаторы лекарств. Фармацевтические композиции для ушной доставки включают суспензии и мази для локального нанесения в ухо. Обычные растворители для таких ушных композиций включают глицерин и воду.
[0110] Композиции могут быть также составлены в ректальные композиции, такие как суппозитории или удерживающие клизмы, например, содержащие стандартную основу суппозиториев, такую как какао-масло или другие глицериды.
[0111] Кроме вышеописанных композиций, композиции могут входить в состав композиций в виде препарата депо. Такие долгодействующие композиции могут быть введены путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Так, например, соединения настоящего изобретения могут быть смешаны с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде слабо растворимых производных, например, слабо растворимых солей.
[0112] Для гидрофобных соединений соответствующий фармацевтический носитель может быть системой совместных растворителей, включающей бензиловый спирт, неполярное поверхностно-активное вещество, смешивающийся с водой органический полимер и водную фазу. Стандартной используемой системой совместных растворителей является система совместных растворителей VPD, которая представляет собой раствор 3% вес/об. бензилового спирта, 8% вес/об. неполярного поверхностно-активного вещества Polysorbate 80™, и 65% вес/об. полиэтиленгликоля 300, доведенная до заданного объема абсолютным этанолом. Естественно, пропорции системы совместных растворителей могут существенно варьироваться без ухудшения ее растворимости и характеристик токсичности. Более того, может варьироваться природа компонентов совместного растворителя: например, могут быть использованы другие низкотоксичные неполярные поверхностно-активные вещества вместо POLYSORBATE 80™; может варьироваться размер фракции полиэтиленгликоля; полиэтиленгликоль может быть заменен другими биосовместимыми полимерами, например, поливинилпирролидоном; и вместо декстрозы могут быть использованы другие сахара или полисахариды.
[0113] Способы лечения бактериальных инфекций могут включать введение терапевтически эффективного количества терапевтических соединений, описанных в настоящем документе. Лечение бактериальной инфекции также может включать профилактическое введение терапевтических соединений для предупреждения инфекции или распространения инфекции в организме субъекта с неизбежным риском инфекции, такого как субъект, перенесший или подлежащий хирургическому вмешательству, субъект с ослабленным иммунитетом или субъект, иным образом имеющий риск инфицирования, если не было введено соединение настоящего изобретения. Соединения настоящего изобретения демонстрируют ингибирующую активность в широком диапазоне бактерий, включая Н. influenzae, Ε. coli, S. aureus, Ε. faecalis, Ε. facium, Κ. pneumonia, A. baumannii, S. pneumoniae и P. aeruginosa. Соединения демонстрируют активность против большинства устойчивых штаммов, например, метициллин-устойчивого Staphylococcus aureus (MRSA). Кроме того, соединения демонстрируют широкий спектр действия против всех бактериальных биозащитных патогенов категорий А, В и С, включая В. anthracis, В. pseudomallei, В. mallei, F. tularensis и Y. psetis. Смотри Примеры. Соединения обладают превосходной относительной антибиотической активностью в относительно низких концентрациях. Кроме того, соединения могут оказывать потенциальное антибактериальное действие против различных патогенов людей и животных, включая грамположительные и грамотрицательные бактерии. В одном варианте реализации бактериальной инфекцией, которую можно лечить или улучшать, является метициллин-резистентный золотистый стафилококк (MRSA).
[0114] Композиции или фармацевтические композиции, описанные в настоящем документе, могут быть введены субъекту любым пригодным способом. Не ограничивающие примеры способов введения включают, среди прочих, (a) введение через ротовые пути, и это введение включает введение в виде капсул, таблеток, гранул, спреев, сиропов или в других таких формах; (b) введение не через ротовые пути, такое как ректальное, вагинальное, интрауретральное, внутриглазное, интраназальное или ушное, и это введение включает введение в виде водной суспензии, масляного препарата или тому подобного, или в виде капель, спреев, суппозиториев, мазей, притирок и тому подобного; (с) введение путем инъекции, подкожно, интраперитонеально, внутривенно, внутримышечно, интрадермально, интраорбитально, интракапсулярно, интраспинально, интрастернально или тому подобно, включая доставку инфузионным насосом; а также (d) введение локально; на усмотрение специалиста в данной области для надлежащего приведения активного соединения в контакт с живой тканью.
[0115] Фармацевтические композиции, применимые для введения, включают композиции, в которых активные ингредиенты содержатся в количестве, эффективном для достижения заданной цели. В некоторых вариантах реализации терапевтически эффективное количество соединения представляет собой количество, эффективное для лечения бактериальной инфекции, например, у млекопитающего субъекта (например, человека). Терапевтически эффективное количество соединений, описанных в настоящем документе, необходимое в качестве дозы, зависит от способа введения, типа животного, включая человека, подлежащего лечению, и физических характеристик конкретного рассматриваемого животного. Доза может быть подобрана для достижения заданного эффекта, но она зависит от таких факторов как вес, рацион питания, сопутствующее лечение и другие факторы, которые известны специалистам в медицинской области. Более конкретно, терапевтически эффективное количество обозначает количество соединения, эффективное для предупреждения, облегчения или улучшения симптомов заболевания или увеличения продолжительности существования субъекта, подлежащего лечению. Определение терапевтически эффективного количества входит в возможности специалиста в данной области, особенно в свете подробного описания, представленного в настоящем документе.
[0116] Как легко понятно специалистам в данной области, применимая доза in vivo для введения и конкретного способа введения варьируется в зависимости от возраста, веса и вида млекопитающего, подлежащего лечению, конкретных используемых соединений и конкретного применения, для которого используют эти соединения. Определение уровней эффективной дозы, то есть уровней дозы, необходимых для достижения заданного результата, может быть выполнено специалистом в данной области при помощи стандартных фармакологических способов. Как правило, медицинские клинические применения продуктов начинают с более низких уровней дозы, с повышением уровня дозы до достижения заданного эффекта. Альтернативно, могут быть использованы приемлемые исследования in vitro для определения пригодных доз и способов введения композиций, определенных представленными способами, с использованием существующих фармакологических способов.
[0117] В исследованиях не на людях, а на животных применение потенциальных продуктов начинают с более высоких уровней доз, с понижением дозы до исчезновения заданного эффекта и исчезновения нежелательных побочных эффектов. Доза может варьироваться в широком диапазоне, в зависимости от заданного эффекта и терапевтического показания. Как правило, дозы могут составлять от около 10 микрограмм/кг до около 100 мг/кг веса тела, предпочтительно, от около 100 микрограмм/кг до около 10 мг/кг веса тела. Альтернативно, дозы могут быть основаны и рассчитаны по площади поверхности пациента, что является понятным специалистам в данной области.
[0118] Точная композиция, способ введения и доза фармацевтических композиций могут быть выбраны индивидуальным врачом с учетом состояния пациента. (Смотри, например, публикацию Fingl et al. 1975, в "The Pharmacological Basis of Therapeutics", которая включена в настоящий документ путем ссылки в полном объеме, с особой ссылкой на Главу 1, с. 1). В некоторых вариантах реализации диапазон доз композиции, введенной пациенту, может составлять от около 0,5 до около 1000 мг/кг веса тела пациента. Доза может быть однократной или серией из двух или более доз, введенных курсом от одного до нескольких дней, в зависимости от потребностей пациента. В случаях, если дозы соединений для людей были определены по меньшей мере для некоторых состояний, то могут быть использованы эти же дозы или дозы, которые составляют от около 0,1% до около 500%, предпочтительно, от около 25% до около 250% определенной дозы для человека. Если доза для человека не была определена, как в случае новых открытых фармацевтических композиций, соответствующая доза для человека может быть предположена из значений ED50 или ID50, или других соответствующих значений, полученных из исследований in vitro или in vivo, определенных по испытаниям токсичности и испытаниям эффективности на животных.
[0119] Следует отметить, что лечащий врач должен знать как и когда остановить, прервать или скорректировать введение из-за токсичности или дисфункции органов. И наоборот, лечащий врач должен также знать, как скорректировать лечение до более высоких уровней, если клинический ответ не достаточен (не допуская токсичности). Диапазон вводимых доз при лечении рассматриваемого расстройства варьируется в зависимости от тяжести состояния, подлежащего лечению, и способа введения. Тяжесть состояния может быть оценена, например, отчасти, стандартными способами прогностической оценки. Кроме того, доза и, возможно, частота введения доз варьируются также в соответствии с возрастом, весом тела и реакцией индивидуального пациента. Программа, сравнимая с программой, рассмотренной выше, может быть использована в ветеринарной медицине.
[0120] Хотя точную дозу определяют на основании постепенного введения лекарства, в большинстве случаев могут быть сделаны некоторые обобщения относительно указанной дозы. Ежедневная схема дозирования для взрослого человека может быть, например, пероральной дозой от около 0,1 мг до 2000 мг активного ингредиента, предпочтительно от около 1 мг до около 500 мг, например, от 5 до 200 мг. В других вариантах реализации используют внутривенную, подкожную или внутримышечную дозу активного ингредиента от около 0,01 мг до около 100 мг, предпочтительно от около 0,1 мг до около 60 мг, например, от около 1 до около 40 мг. В случаях введения фармацевтически приемлемой соли, дозы могут быть рассчитаны как для свободной кислоты. В некоторых вариантах композицию вводят от 1 до 4 раз в день. Альтернативно, композиции могут быть введены непрерывной внутривенной инфузией, предпочтительно в дозе до 1000 мг в день. Специалистам в данной области понятно, что в некоторых ситуациях может быть необходимо вводить соединения, описанные в настоящем документе, в количествах, которые превышают и даже сильно превышают указанный выше предпочтительный диапазон доз для эффективного и активного лечения особенно агрессивных заболеваний или инфекций. В некоторых вариантах соединения вводят в течение периода непрерывной терапии, например, в течение недели или более, или месяцев или лет.
[0121] Количество дозы и интервал могут быть подобраны индивидуально для обеспечения уровней активного фрагмента в плазме, достаточных для поддержания антибиотического эффекта, или минимальной эффективной концентрации (MEC). МЕС варьируется для каждого соединения, но может быть оценена по данным in vitro. Дозы, необходимые для достижения МЕС, зависят от индивидуальных характеристик и способа введения. Однако для определения концентраций в плазме может быть использован анализ ВЭЖХ или биоанализ.
[0122] Интервалы доз также могут быть определены с использованием значения МЕС. Композиции следует вводить с использованием схемы, которая поддерживает уровни в плазме выше МЕС в течение 10-90% времени, предпочтительно, между 30-90%, и наиболее предпочтительно между 50-90%.
[0123] В случаях локального введения или селективного поглощения эффективная локальная концентрация лекарства может быть не связана с концентрацией в плазме.
[0124] Количество вводимой композиции может зависеть от субъекта, подлежащего лечению, веса субъекта, тяжести инфекции, способа введения и решения врача, прописывающего лекарство.
[0125] Композиции, описанные в настоящем документе, могут быть оценены на эффективность и токсичность с использованием известных способов. Например, токсикология соединения может быть установлена определением in vitro токсичности в отношении клеточной линии, такой как клеточная линия млекопитающего и, предпочтительно, человека. Результаты таких испытаний зачастую являются прогнозными для токсичности у животных, таких как млекопитающие или, более конкретно, люди. Альтернативно, с использованием известных способов может быть определена токсичность определенных соединений в животной модели, такой как мыши, крысы, кролики или обезьяны. Эффективность конкретного соединения может быть определена с использованием нескольких общепризнанных способов, таких как способы in vitro, животные модели или клинические испытания на людях. Практически для каждого класса состояний существуют общепризнанные in vitro модели. Точно так же, приемлемые животные модели могут быть использованы для определения эффективности химических веществ для лечения таких состояний. При выборе модели для определения эффективности, опытный специалист может руководствоваться существующим уровнем техники для выбора соответствующей модели, дозы и способа введения, а также схемы введения. Конечно, для определения эффективности соединения на людях могут быть также использованы клинические испытания на людях.
[0126] Композиции, при необходимости, могут быть представлены в упаковке или распылительном устройстве, которое может содержать одну или несколько разовых лекарственных форм, содержащих активный ингредиент. Упаковка может включать, например, металлическую фольгу или пластиковую пленку, как в блистерной упаковке. Упаковка или распылительное устройство могут сопровождаться инструкциями по применению. Упаковка или распылительное устройство могут также сопровождаться уведомлением, находящемся в контейнере, в форме, предписанной государственным органом, регулирующим производство, применение или продажу фармацевтических средств, и это уведомление отражает одобрение указанным органом данной формы лекарства для введения человеку или для ветеринарного применения. Такое уведомление может быть, например, маркировкой, одобренной Управлением США по надзору за качеством пищевых продуктов и лекарственных средств, для предписания лекарств или вкладышем одобренного продукта. Композиции, включающие соединение, составленное в композицию с совместимым фармацевтическим носителем, также может быть получено, помещено в соответствующий контейнер и этикетировано для лечения указанного состояния.
[0127] В некоторых вариантах реализации, в фармацевтической промышленности стандартной практикой является обеспечение в значительной степени чистого материала при составлении фармацевтических композиций. Следовательно, в некоторых вариантах реализации, «в значительной степени» относится к степени чистоты, необходимой для составления фармацевтических средств, которые могут включать, например, небольшое количество другого материала, который не ухудшает пригодности для фармацевтического применения. В некоторых вариантах реализации в значительной степени чистое соединение содержит по меньшей мере около 96% соединения по весу, как, например, по меньшей мере около 97%, 98%, 99% или 100% соединения.
[0128] Термины «примерно», «около» и «в значительной степени», используемые в настоящем документе, представляют количество, близкое к указанному количеству, которое все еще выполняет заданную функцию или дает заданный результат. Например, термины «примерно», «около» и «в значительной степени» могут относиться к количеству, которое находится в пределах менее 10%, в пределах менее 5%, в пределах менее 1%, в пределах менее 0,1% и в пределах менее 0,01% от указанного количества.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
РАЗДЕЛ А:
Синтез уникальных частиц R2
[0129] Все 2-замещенные пиримидинолы, отсутствующие в продаже, были получены в соответствии со способами, описанными в публикации US 5162529 или опубликованном документе Tetrahedron, 65(4), 757-764; 2009.
Примеры:
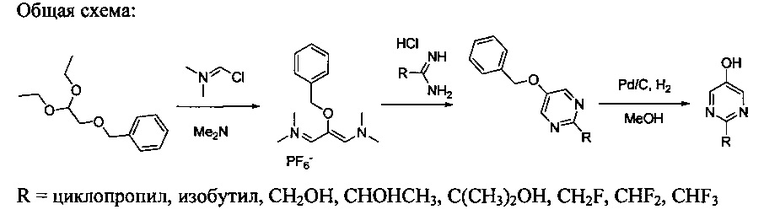
Экспериментальная часть:
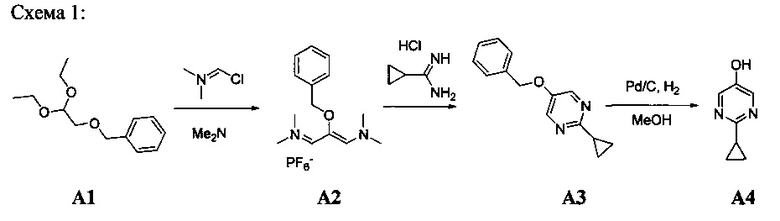
[0130] Получение соединения А2: Оксихлорид фосфора (96 г, 0,62 моль) добавили к безводному ДМФ (46 г, 0,62 моль) при 0°C и перемешивали смесь при комнатной температуре в течение 1 часа. Затем добавили CHCl3 (500 мл) и по каплям добавили бензилоксиацетальдегида диэтилацетат (40 г, 0,18 моль). После завершения реакции реакционную смесь нагревали с дефлегматором в течение 2,5 часов, а затем оставили остывать до комнатной температуры. Оранжевый раствор медленно вылили в холодную воду (500 мл) при 0°C и перемешивали двухфазную смесь в течение 15 минут. Органическую фазу промыли водой (500 мл). Объединенные водные слои по каплям добавили к раствору диметиламина гидрохлорида (59 г, 0,72 моль) в воде (200 мл). pH довели до 8,5 добавлением 5 н. водного раствора гидроксида натрия, поддерживая температуру около 15°C. Раствор перемешивали в течение 1 часа и добавили гексафторфосфат натрия (40 г, 0,23 моль) в воде (100 мл). Образовавшийся осадок собрали фильтрацией, промыли водой и высушили под высоким вакуумом для получения соединения 2 (22 г, выход: 30%) в виде бледно-бежевого твердого вещества, которое использовали на следующей стадии без какой-либо дополнительной очистки.
1H ЯМР (400 МГц, ДМСО-d6): δ: 7,42-7,39 (m, 5Н), 4,74 (s, 2Н), 3,32 (s, 3Н), 3,21 (s, 3Н).
[0131] Получение соединения A3: К перемешанной суспензии соединения А2 (14 г, 39 ммоль) и циклопропанкарбоксимидамида гидрохлорида (5,65 г, 47 ммоль) в CH3CN (100 мл) добавили карбонат калия (16,2 г, 117 ммоль). Реакционную смесь нагревали при 90°C в течение 12 часов, затем охладили до комнатной температуры, вылили в ледяную воду, экстрагировали этилацетатом (2×50 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения A3 (2,5 г, выход: 26%) в виде твердого желтого вещества.
1H ЯМР (400 МГц, CDCl3): δ: 8,44 (s, 2Н), 7,46-7,28 (m, 5Н), 5,24 (s, 2Н), 2,18-2,12 (m, 1H), 0,99-0,90 (m, 2H), 0,89-0,86 (m, 2H).
[0132] Получение соединения A4: К раствору соединения A3 (3,50 г, 15,8 ммоль) в МеОН (30 мл) добавили 10% палладий на углероде (350 мг) и перемешивали смесь под атмосферой водорода в течение 4 часов. Твердое вещество отфильтровали, а фильтрат концентрировали для получения соединения A4 (2,0 г, выход: 98%).
1H ЯМР (300 МГц, ДМСО-d6): δ: 10,05 (s, 1H), 8,17 (s, 2H), 2,12-2,05 (m, 1H), 0,93-0,91 (m, 2H), 0,86-0,83 (m, 2H). ЖХМС [подвижная фаза: 2-60% ацетонитрил-0,05% ТФК за 6 минут, в конце при этих условиях в течение 0,5 минуты] чистота >95%, Rt=2,564 минуты; МС, рассчитано: 136,1; МС, найдено: 137,1 ([М+1]+)
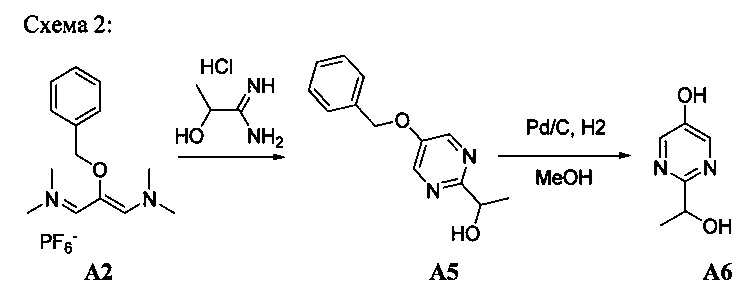
[0133] Получение соединения А5: К перемешанной суспензии соединения А2 (14 г, 39 ммоль) и 2-гидроксипропанимидамида гидрохлорида (5,65 г, 47 ммоль) в CH3CN (100 мл) добавили карбонат калия (16,2 г, 117 ммоль). Реакционную смесь нагревали при 90°C в течение 12 часов, затем охладили до комнатной температуры, вылили в ледяную воду, экстрагировали этилацетатом (2×50 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А5 (2,5 г, выход: 26%) в виде твердого желтого вещества.
1Н ЯМР (300 МГц, CDCl3): δ: 8,84 (s, 2Н), 7,48 (m, 3Н), 7,37 (m, 2Н), 5,20 (s, 2Н), 4,68 (m, 1H), 3,25 (m, 1Н), 1,48 (d, 3Н).
[0134] Получение соединения А6: К раствору соединения А5 (3,50 г, 15,8 ммоль) в МеОН (30 мл) добавили 10% палладий на углероде (350 мг) и перемешивали смесь под атмосферой водорода в течение 4 часов. Твердое вещество отфильтровали, а фильтрат концентрировали для получения соединения А6 (2,0 г, выход: 98%).
1Н ЯМР (400 МГц, CDCl3): δ: 8,84 (s, 2Н), 5,40 (широкий, 1H), 4,66 (m, 1H), 3,25 (m, 1Н), 1,46 (d, 3Н). ЖХМС, найдено: 141,1 ([М+1]+)
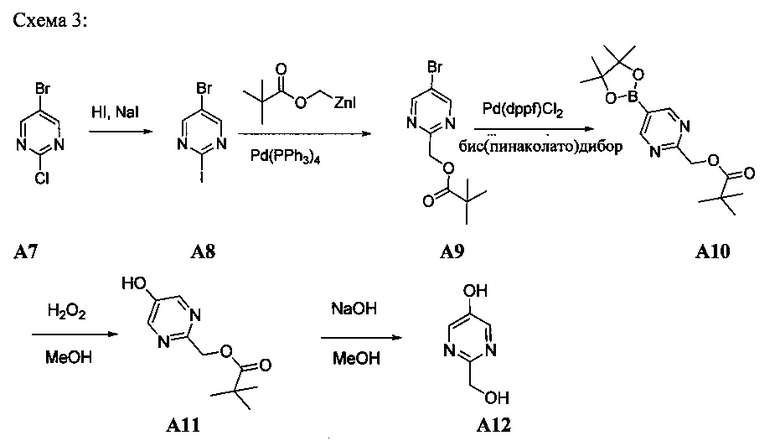
[0135] Получение соединения А8: К раствору соединения А7 (50 г, 0,26 моль) в ДХМ (300 мл) добавили NaI (80 г, 0,52 моль) при комнатной температуре, затем добавили HI (75 г, 0,52 моль). После перемешивания при 50°C в течение 5 часов, смесь вылили в ледяную воду и осторожно нейтрализовали добавлением твердого бикарбоната натрия до обесцвечивания смеси. Затем смесь экстрагировали ДХМ (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А8 (60 г, выход: 81%) в виде твердого белого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 8,54 (s, 2Н).
[0136] Получение соединения А9: К раствору соединения А8 (50 г, 0,18 моль) в ТГФ (300 мл) добавили Pd(PPh3)4 (11,5 г, 0,01 моль), затем добавили раствор цинкового реагента 3 (только что приготовленного из йодметил-2,2-диметилпропаноата) в ТГФ (500 мл, 0,36 моль) и перемешивали при комнатной температуре в течение 12 часов. Затем добавили ледяную воду и экстрагировали смесь этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта. Остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 10:1) для получения соединения А9 (41 г, выход: 85%) в виде твердого желтого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 8,75 (s, 2Н), 5,26 (s, 2Н), 5,06 (s, 1Н), 1,28 (s, 9Н).
[0137] Получение соединения А10: К перемешанному раствору соединения А9 (15,0 г, 54,9 ммоль) в диоксане (100 мл) добавили бис(пинаколато)дибор (17,0 г, 65,4 ммоль) под азотом, затем Pd(dppf)Cl2 (2,20 г, 2,72 ммоль) и KOAc (16 г, 163 ммоль). Реакционную смесь нагревали при 85°C в течение 3 часов. Черную суспензию охладили до комнатной температуры, отфильтровали, концентрировали для получения неочищенного продукта. Остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 15:1) для получения соединения А10 (15,4 г) в виде твердого белого вещества, с примесью производных пинакола.
1Н ЯМР (400 МГц, CDCl3): δ: 8,97 (s, 2Н), 5,30 (s, 2Н), 1,35 (s, 9Н), 1,28 (s, 9Н).
[0138] Получение соединения A11: К раствору соединения А10 (15,6 г, 48,7 ммоль) в МеОН (100 мл) добавили Н2О2 (16,0 г, 140 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. Добавили 2 н. раствор тиосульфата натрия (200 мл) и экстрагировали смесь этилацетатом (200 мл). Водную фазу довели до pH 4-5 при помощи 2 н. HCl; затем смесь экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения A11 (9,4 г, выход: 82% за две стадии).
1H ЯМР (400 МГц, ДМСО-d6): δ: 10,48 (s, 1Н), 8,31 (s, 2H), 5,11 (s, 2H), 1,21 (s, 9H).
[0139] Получение соединения А12: К раствору соединения A11 (10 г, 30 ммоль) в МеОН (200 мл) добавили MeONa (50 мл, 1 M в МеОН). После перемешивания при комнатной температуре в течение 12 часов, смесь вылили в воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А12 (7,3 г, выход: 98%) в виде твердого белого вещества.
1H ЯМР (300 МГц, CDCl3): δ: 8,43 (s, 2Н), 7,35 (d, J=8,8 Гц, 2H), 6,93 (d, J=8,8 Гц, 2H), 5,09 (s, 2Н), 4,78 (s, 2Н).
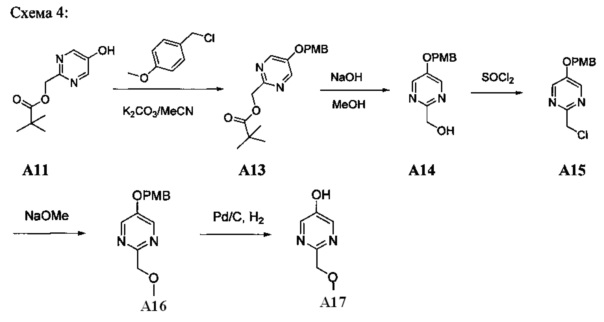
[0140] Получение соединения А13: К раствору соединения A11 (12,3 г, 58,5 ммоль) в CH3CN (100 мл) добавили K2CO3 (10,5 г, 76 ммоль) и PMBCl (12 г, 76 ммоль), и перемешивали смесь при комнатной температуре в течение 12 часов, и нагревали до 50°C в течение 3 часов. Затем смесь вылили в воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали, остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 10:1) для получения соединения А13 (10,0 г, выход: 52%) в виде твердого белого вещества.
1H ЯМР (300 МГц, CDCl3): δ: 8,41 (s, 2Н), 7,34 (d, J=8,8 Гц, 2H), 6,93 (d, J=8,8 Гц, 2H), 5,24 (s, 2Н), 5,07 (s, 2Н), 3,82 (s, 3Н), 1,26 (s, 9Н).
[0141] Получение соединения А14: К раствору соединения А13 (10 г, 30 ммоль) в МеОН (200 мл) добавили MeONa (50 мл, 1M в МеОН). После перемешивания при комнатной температуре в течение 12 часов, смесь вылили в воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А14 (7,3 г, выход: 98%) в виде твердого белого вещества.
1Н ЯМР (300 МГц, CDCl3): δ: 8,43 (s, 2Н), 7,35 (d, J=8,8 Гц, 2H), 6,93 (d, J=8,8 Гц, 2H), 4,78 (s, 2Н).
[0142] Получение соединения А16: К раствору соединения А14 (15 г, 61 ммоль) в 25 ДХМ (200 мл) добавили тионилхлорид (10,8 г, 91 ммоль). После перемешивания при комнатной температуре в течение 2 часов, смесь затем вылили в воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А15 (16 г) в виде твердого белого вещества. К раствору соединения А15 (15 г) в МеОН (200 мл) добавили MeONa (50 мл, 50% в MeOH). Смесь перемешивали при 50°C в течение 5 часов, затем охладили до комнатной температуры, концентрировали для получения неочищенного продукта. Остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения соединения А16 (12,5 г, выход: 80%) в виде твердого желтого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 8,45(8, 2Н), 7,34 (d, J=8,8 Гц, 2Н), 6,92 (d, J=8,8 Гц, 2Н), 5,08 (s, 2Н), 4,64 (s, 2Н), 3,82 (s, 3Н), 3,52 (s, 3Н).
[0143] Получение соединения А17: К раствору соединения А16 (3,0 г) в МеОН (30 мл) добавили 10% палладий на углероде (350 мг) и перемешивали смесь под атмосферой водорода в течение 4 часов. Твердое вещество отфильтровали, а фильтрат концентрировали; остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 1:1) для получения соединения А17 (1,2 г, выход: 74%) в виде твердого белого вещества.
1Н ЯМР (400 МГц, ДМСО-d6): δ: 10,45 (s, 1Н), 8,33 (s, 2Н), 4,44 (s, 2Н), 3,31 (s, 3Н). ЖХМС [подвижная фаза: 95-5% ацетонитрил-0,02% NH4Ac за 6 минут, в конце при этих условиях в течение 0,5 минуты] чистота >95%, Rt=3,3 минуты; МС, рассчитано: 140,1,1; МС, найдено: 141,1 ([М+1]+).
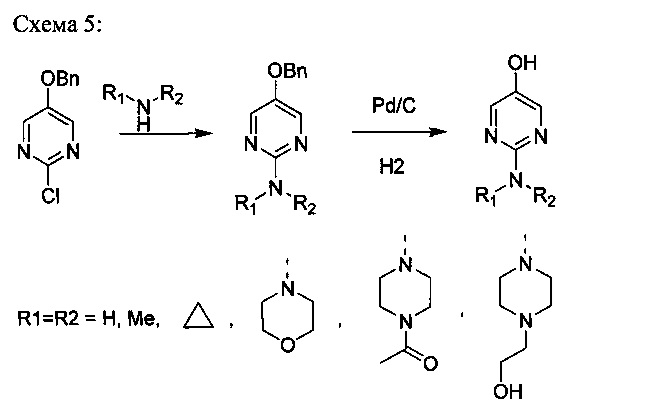
Примеры:
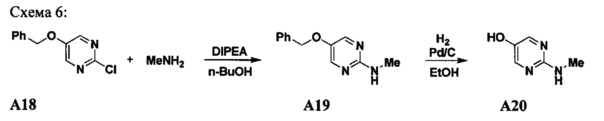
[0144] Синтез 2-(метиламино)пиримидин-5-ола: Смесь 5-(бензилокси)-2-хлорпиримидина А18 (0,500 г, 2,27 ммоль), метиламина (1,25 мл, 2,50 ммоль, 2,0 М раствор в МеОН) и DIPEA (0,594 мл, 3,41 ммоль) в n-BuOH (5,0 мл) перемешивали в течение 48 часов при 100°C. После перемешивания в течение 48 часов, реакцию проверили по ЖХ/МС. Полученную смесь охладили до 23°C и концентрировали под пониженным давлением. Неочищенный материал очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:1 (об./об.)) для получения 5-(бензилокси)-N-метилпиримидин-2-амина А19 (0,355 г, 1,65 ммоль, 73%) в виде бесцветных кристаллов. ЖХ/МС (М+Н+)=216. Смесь палладия на углероде (0,176 г, 0,165 ммоль, 10,0 мол.%) и 5-(бензилокси)-N-метилпиримидин-2-амина А19 (0,355 г, 1,65 ммоль) в этаноле (7,0 мл) перемешивали в течение 20 часов под атмосферой водорода при 23°C. Полученную смесь отфильтровали через Целит и промыли слой фильтра метанолом (25 мл). Фильтрат концентрировали под пониженным давлением для получения указанного в заголовке соединения, 2-(метиламино)пиримидин-5-ола А20 (0,196 г, 1,57 ммоль, 95%) в виде светло-желтого твердого вещества. ЖХ/МС (М+Н+)=126.
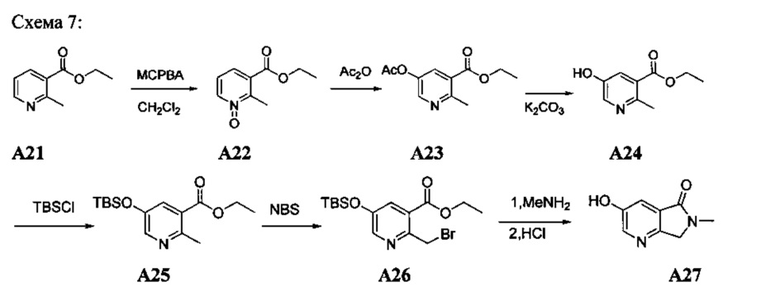
[0145] Получение соединения А22: К раствору А21 (50,0 г, 0,303 моль) в ДХМ (200 мл) добавили m-CPBA (80,0 г, 0,465 моль) при 0°C. После перемешивания при 0°C в течение 1 часа, при комнатной температуре в течение ночи, смесь вылили в ледяную воду. Добавили 2 н. NaOH, чтобы довести pH до 8-9, и полученную смесь экстрагировали ДХМ (3×200 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А22 (50,0 г, выход: 91%) в виде твердого желтого вещества.
[0146] Получение соединения А23: Раствор А22 (50,0 г, 0,276 ммоль) в уксусном ангидриде (300 мл) нагревали до 90°C в течение 1,5 часов. Затем смесь концентрировали, а остаток вылили в ледяную воду; добавили 2 н. NaOH, чтобы довести pH до 8-9, и полученную смесь экстрагировали этилацетатом (3×100 мл). Органический слой высушили над Na2SO4 и концентрировали для получения неочищенного вещества, которое очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения соединения А23 (10,0 г, выход: 16%) в виде маслянистого желтого вещества.
1H ЯМР (400 МГц, CDCl3): δ: 8,43 (d, J=2,4 Гц, 1H), 7,99 (d, J=1,6 Гц, 1H), 4,41-4,35 (q, J=3,2 Гц, 3H), 2,83 (s, 3Н), 2,34 (s, 3Н), 1,42-4,39 (t, J=3,2 Гц, 3Н).
[0147] Получение соединения А24: К раствору А23 (10,0 г, 44,8 ммоль) в МеОН (300 мл) добавили карбонат калия (12,4 г, 89,8 ммоль). После перемешивания при комнатной температуре в течение 12 часов, смесь вылили в ледяную воду. Добавили 2 н. HCl, чтобы довести pH до 8-9, и экстрагировали смесь этилацетатом (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А24 (8,00 г, выход 99%) в виде твердого желтого вещества.
1H ЯМР (400 МГц, ДМСО-d6): δ: 10,0 (s, 1H), 8,18 (d, J=2,4 Гц, 1H), 7,54 (d, J=2,8 Гц, 1Н), 4,32-4,26 (q, J=3,2 Гц, 3Н), 2,57 (s, 3Н), 1,33-1,29 (t, J=3,2 Гц, 3Н).
[0148] Получение соединения А25: К раствору соединения А24 (2,50 г, 13,8 ммоль) в ДХМ (50 мл) добавили имидазол (3,00 г, 44,1 ммоль) и трет-бутилдиметилсилил хлорид (2,50 г, 16,7 ммоль), и перемешивали смесь при комнатной температуре в течение 3 часов. Затем выпарили растворитель, остаток очистили хроматографией (петролейный эфир/этилацетат = 5:1) для получения соединения А25 (2,80 г, выход 69%) в виде маслянистого желтого вещества.
1H ЯМР (400 МГц, CDCl3): δ: 8,12 (d, J=2,8 Гц, 1Н), 7,54 (d, J=2,8 Гц, Ш), 4,30-4,26 (q, J=3,2 Гц, 3Н), 2,64 (s, 3Н), 1,32-1,28 (t, J=3,2 Гц, 3Н), 0,92 (s, 9H), 0,12 (s, 6H).
[0149] Получение соединения A26: К раствору соединения А25 (2,80 г, 9,48 ммоль) в CCl4 (100 мл) добавили азодиизобутиронитрил (280 мг) и NBS (1,80 г, 10,1 ммоль) и перемешивали смесь при 70°C в течение 15 часов, затем растворитель выпарили, остаток очистили хроматографией (петролейный эфир/этилацетат = 5:1) для получения соединения А26 (1,60 г, выход 45%) в виде маслянистого желтого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 8,28 (d, J=3,2 Гц, 1H), 7,68 (d, J=3,2 Гц, 1H), 4,98 (s, 3Н), 4,45-4,40 (q, J=3,2 Гц, 3Н), 1,45-1,42 (t, J=2,8 Гц, 3Н), 1,00 (s, 9Н), 0,26 (s, 6Н).
[0150] Получение соединения А27: К раствору соединения А26 (1,60 г, 4,27 ммоль) в EtOH (100 мл) добавили раствор метиламина в EtOH (1,24 г, 12,0 ммоль, 30% вес/вес.) и перемешивали смесь при комнатной температуре в течение 3 часов. Затем растворитель выпарили, а остаток очистили хроматографией (петролейный эфир/этилацетат = 5:1) для получения соединения А27а (300 мг, выход: 25%) в виде твердого желтого вещества.
1Н ЯМР (400 МГц, ДМСО-d6): δ: 8,34 (d, J=2,8 Гц, 1H), 7,43 (d, J=2,8 Гц, 1H), 4,42 (s, 2Н), 3,06 (s, 3Н), 0,95 (s, 9Н), 0,20 (s, 6Н).
[0151] К раствору соединения А27а (300 мг, 1,14 ммоль) в ТГФ (5 мл) добавили 6 н. HCl (0,5 мл). После перемешивания при комнатной температуре в течение 1 часа, смесь концентрировали для получения соединения А27 (150 мг, выход 80%) в виде твердого желтого вещества.
1Н ЯМР (400 МГц, ДМСО-d6): δ: 10,27 (s, 1Н), 8,27 (d, J=2,4 Гц, 1H), 7,32 (d, J=2,8 Гц, 1Н), 4,37 (s, 2Н), 3,0 (s, 3Н). ЖХМС, подвижная фаза: мт 40% воды (0,05% ТФК) и 60% CH3CN до 10% воды (0,05% ТФК) и 90% CH3CN за 6 минут, в конце при этих условиях в течение 0,5 минуты] Чистота >95%, Rt=3,7 минуты; МС, рассчитано: 164,1; МС, найдено: 165,1 ([М+1]+).
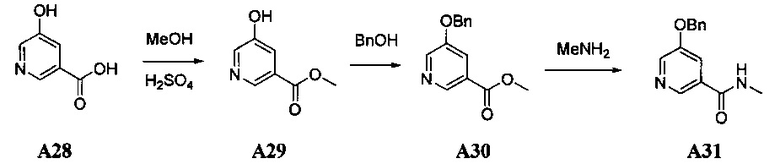
[0152] Получение соединения А29: Смесь соединения А28 (25,0 г, 180 ммоль) и концентрированной H2SO4 (10 мл) в СН3ОН (100 мл) нагревали с дефлегматором в течение ночи. Смесь концентрировали, остаток промыли водным раствором NaHCO3 (50 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А29 (18,7 г, выход: 68%).
1Н ЯМР (300 МГц, ДМСО-d6): δ: 10,42 (s, 1H), 8,60 (d, J=1,6 Гц, 1Н), 8,36 (d, J=2,8 Гц, 1H), 7,60-7,61 (m, 1H), 3,87 (s, 3Н).
[0153] Получение соединения А30: BnOH (3,90 г, 36,1 ммоль, 1,1 экв.) и PPh3 (17,1 г, 65,4 ммоль, 2,0 экв.) добавили к раствору соединения А29 (5,00 г, 32,7 ммоль) в ТГФ (100 мл), затем добавили DEAD (6,80 г, 39,2 ммоль, 1,2 экв.) при 0°C. Смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпарили, остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 10:1) для получения соединения А30 (5,70 г, выход: 71%) в виде твердого белого вещества.
1Н ЯМР (300 МГц, CDCl3): δ: 8,83 (d, J=1,6 Гц, 1Н), 8,54 (d, J=2,8 Гц, 1H), 7,85-7,86 (m, 1Н), 7,27-7,46 (m, 5Н), 5,15 (s, 2Н), 3,95 (s, 3Н).
[0154] Получение соединения А31: Раствор соединения А30 (12,8 г, 52,9 ммоль) в спиртовом растворе метиламина в закрытой пробирке перемешивали при 70°C в течение ночи. Затем смесь охладили до комнатной температуры, а растворитель выпарили для получения соединения А31 (12,0 г, выход: 100%).
1Н ЯМР (300 МГц, CDCl3): δ: 8,50 (d, J=1,6 Гц, 1Н), 8,48 (d, J=2,8 Гц, 1H), 7,73-7,74 (m, 1H), 7,73-7,74 (m, 5Н), 6,16 (s, 1Н), 3,15 (s, 2Н), 3,04 (d, J=4,4 Гц, 3Н).
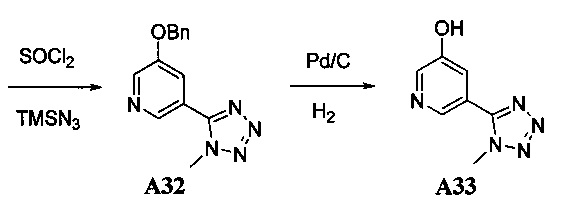
[0155] Получение соединения А32: Раствор соединения А31 (11,0 г, 45,5 ммоль) в SOCl2 (100 мл) нагревали с дефлегматором в течение 4 часов. Затем SOCl2 удалили под вакуумом, а остаток растворили в MeCN (200 мл). Медленно добавили TMSN3 (12,5 г, 90,0 ммоль, 2,0 экв.) и перемешивали смесь при 90°C в течение 3 часов. Затем растворитель выпарили, остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 2:3) для получения соединения А32 (9,50 г, выход: 78%).
1Н ЯМР (300 МГц, CDCl3): δ: 8,59 (d, J=2,8 Гц, 1Н), 8,56 (d, J=1,6 Гц, 1H), 7,68-7,69 (m, 1H), 7,3-7,46 (m, 5Н), 5,21 (s, 2Н), 4,17 (s, 3Н).
[0156] Получение соединения А33: К раствору соединения А32 (5,00 г, 18,7 ммоль) в СН3ОН (100 мл) добавили Pd(OH)2 (0,50 г). Смесь перемешивали при комнатной температуре под атмосферой Н2 в течение 3 часов. Твердое вещество отфильтровали, а фильтрат концентрировали для получения соединения А33 (1,60 г, выход: 48%).
1Н ЯМР (300 МГц, ДМСО-d6): δ: 10,56 (s, 1H), 8,49 (d, J=1,6 Гц, 1H), 8,36 (d, J=2,8 Гц, 1Н), 7,61-7,62 (m, 1H), 4,19 (s, 3Н).
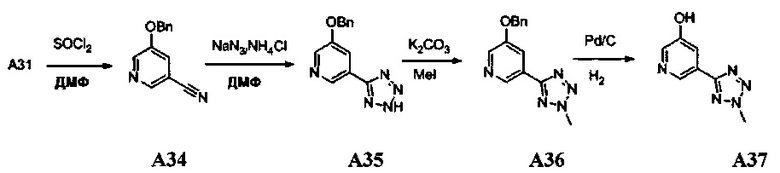
[0157] Получение соединения А34: Тионилхлорид (15,0 г, 107 ммоль) добавили к ДМФ (200 мл) при 0°C и перемешивали смесь при 0°C в течение 30 минут, затем к смеси добавили А31 (12,2 г, 53,5 ммоль) и перемешивали при 0°C в течение 1 часа. Затем реакционную смесь вылили в ледяную воду и экстрагировали этилацетатом (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А34 (11,5 г, выход: 100%).
1Н ЯМР (300 МГц, CDCl3): δ: 8,57 (d, J=2,8 Гц, 1Н), 8,48 (d, J=1,6 Гц, 1Н), 7,45-7,39 (m, 6Н), 5,15 (s, 2Н).
[0158] Получение соединения А35: К раствору А34 (12,0 г, 57,1 ммоль) в ДМФ (200 мл) добавили NH4Cl (5,20 г, 97,1 ммоль) и NaN3 (6,31 г, 97,1 ммоль). Полученную смесь нагревали до 100°C в течение 14 часов, охладили до комнатной температуры, вылили в ледяную воду, добавили 2 н. HCl, чтобы довести pH до 3-4, и экстрагировали этилацетатом (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения соединения А35 (13,0 г, выход: 90%).
1Н ЯМР (300 МГц, ДМСО-d6): δ: 8,82 (d, J=1,6 Гц, 1Н), 8,57 (d, J=2,8 Гц, 1Н), 8,04-8,02 (m, 1Н), 7,52-7,35 (m, 5Н), 5,30 (s, 2Н).
[0159] Получение соединения А36: Соединение А35 (7,00 г, 27,7 ммоль) растворили в ацетоне (150 мл), к смеси добавили карбонат калия (5,70 г, 41,2 ммоль) и перемешивали при комнатной температуре в течение 20 минут, затем к смеси добавили йодметан (5,89 г, 41,2 ммоль) и нагревали до 45°C в течение 1 часа, охладили до комнатной температуры, вылили в ледяную воду, экстрагировали этилацетатом (2×100 мл). Органический слой высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта, остаток очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 3:1) для получения соединения А36 (4,5 г, выход: 61%) в виде твердого белого вещества.
1Н ЯМР (300 МГц, CDCl3): δ: 8,97 (d, J=1,6 Гц, 1Н), 8,48 (d, J=2,4 Гц, 1Н), 8,00-7,99 (m, 1Н), 7,47-7,26 (m, 5Н), 5,19 (s, 2Н), 4,43 (s, 3Н).
[0160] Получение соединения А37: К раствору соединения А36 (7,5 г, 28,0 ммоль) в CH3OH (100 мл) добавили Pd(OH)2 (500 мг). Смесь перемешивали при комнатной температуре под атмосферой Н2 в течение 3 часов. Твердое вещество отфильтровали, а фильтрат концентрировали для получения соединения А37 (4,3 г, выход: 87%). ЖХ-МС: М+1: 178,16.
1Н ЯМР (300 МГц, ДМСО-d6): δ: 10,42 (s, 1Н), 8,68 (d, J=1,6, 1Н), 8,28 (d, J=2,8, 1Н), 7,74-7,73 (m, 1Н), 4,45 (s, 3Н).
РАЗДЕЛ В:
Синтез уникальных частиц R4
Асимметричный синтез (1R,4R,5R)трет-бутил 5-амино-2-азабицикло[2.2.1]гептан-2-карбоксилата
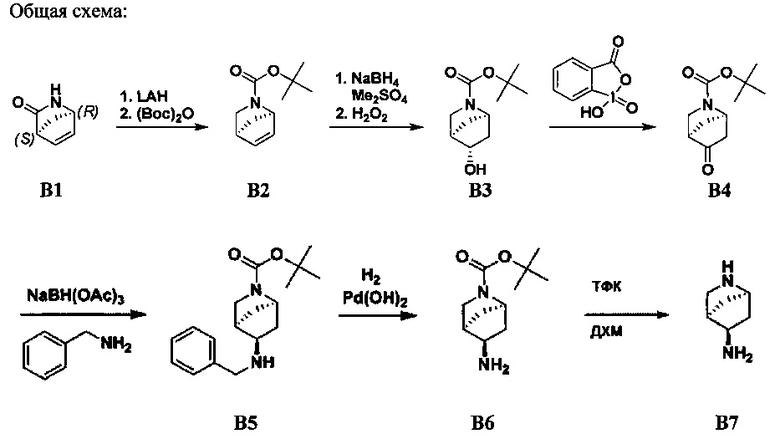
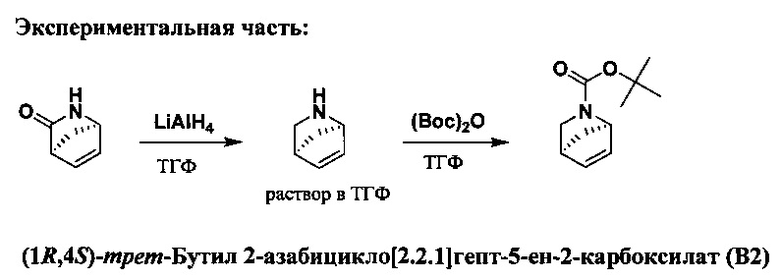
[0161] (1R)-(-)-2-Азабицикло[2.2.1]гепт-5-ен-3-он (5,00 г, 45,8 ммоль, э.и. = 99%), растворенный в безводном ТГФ (15,0 мл), медленно добавили к раствору алюмогидрида лития (57,3 мл, 57,3 ммоль, 1 М раствор в ТГФ) в безводном ТГФ (35,0 мл) под атмосферой азота при 0°C. После успешного завершения добавления смесь перемешивали в течение 3 часов при 23°C, а затем нагревали при 60°C в течение 12 часов. Полученную гетерогенную смесь охладили до 0°C, и к этой смеси осторожно добавили H2O (5,00 мл) через шприц. Суспензию белого цвета отфильтровали через фильтрующий материал Целит, а слой фильтра промыли безводным диэтиловым эфиром (50,0 мл). Затем фильтрат обработали (Boc)2O (15,0 г, 68,7 ммоль) и перемешивали в течение 24 часов при 23°C. Смесь концентрировали in vacuo, а неочищенный материал очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:7 (об./об.)) для получения указанного в заголовке соединения B2 в виде бесцветных кристаллов. (После выпаривания растворителя на ротационном испарителе, полученное бесцветное маслянистое вещество быстро кристаллизовалось при 23°C)
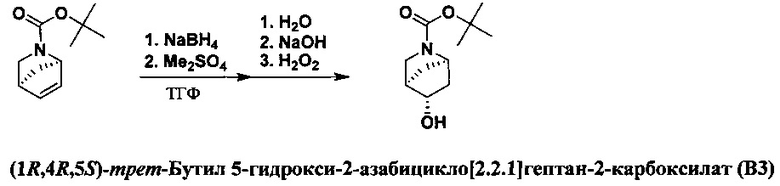
[0162] Смесь (1R,4S)-трет-бутил 2-азабицикло[2.2.1]гепт-5-ен-2-карбоксилата (1,50 г, 7,68 ммоль) и боргидрида натрия (0,24 г, 6,30 ммоль) в ТГФ (9,5 мл) перемешивали в течение 0,5 часа под атмосферой азота при 23°C. После перемешивания в течение 0,5 часа смесь нагрели до 35°C, а затем по каплям добавили диметилсульфат (0,57 мл, 6,30 ммоль), растворенный в ТГФ (2,0 мл), через шприц. Полученную смесь перемешивали в течение 4 часов при 35°C, затем охладили до 0°C и погасили добавлением по каплям Н2О (5,0 мл). Добавили раствор гидроксида натрия (15,0 мл, 15,0 ммоль, 1 М раствор NaOH) при 0°C, затем добавили перекись водорода (0,96 мл, 30 вес.% в H2O). Смесь нагрели до 23°C и перемешивали еще 1 час. Полученный бесцветный раствор разбавили диэтиловым эфиром (75,0 мл) и отделили органический слой, промыли насыщенным солевым раствором (50,0 мл) и высушили над сульфатом магния. Смесь концентрировали на ротационном испарителе, а полученное бесцветное маслянистое вещество в виде неочищенного продукта очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:1 (об./об.)) для получения указанного в заголовке соединения В3 (1,00 г, 4,69 ммоль, 61%) в виде бесцветного маслянистого вещества.
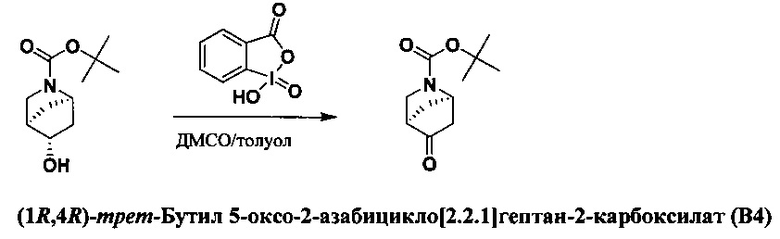
[0163] 2-Йодоксибензойную кислоту (3,43 г, 5,52 ммоль, 45 вес.% (SIBX)) добавили к раствору (1R,4R,5S)-трет-бутил 5-гидрокси-2-азабицикло[2.2.1]гептан-2-карбоксилата (0,87 г, 4,09 ммоль), растворенного в диметилсульфоксиде (5,0 мл) и толуоле (10,0 мл), под атмосферой азота при 23°C. Смесь перемешивали в течение 3 часов при 60°C и охладили до 23°C. Полученную смесь обработали насыщенным раствором карбоната натрия (водным) (50,0 мл) и отфильтровали под пониженным давлением для удаления твердого белого вещества. Фильтрат экстрагировали этилацетатом (75,0 мл × 3), а органические экстракты промыли насыщенным солевым раствором, высушили над сульфатом магния и концентрировали in vacuo. Неочищенный материал в виде бесцветного маслянистого вещества очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:2 (об./об.)) для получения указанного в заголовке соединения В4 (0,62 г, 2,91 ммоль, 71%) в виде белого твердого вещества.
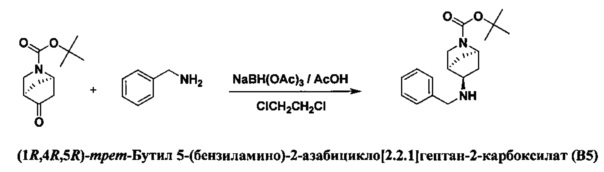
[0164] Триацетоксиборгидрид натрия (23,4 г, 105 ммоль) и ледяную уксусную кислоту (4,66 г, 77,6 ммоль) добавили к раствору (1R,4R)-трет-бутил 5-оксо-2-азабицикло[2.2.1]гептан-2-карбоксилата (16,4 г, 77,6 ммоль) и бензиламина (8,32 г, 77,6 ммоль) в 1,2-дихлорэтане (250 мл) под атмосферой азота при 23°C. Полученную смесь перемешивали в течение 5 часов при 23°C, а затем погасили насыщенным раствором бикарбоната натрия (водным) (300 мл). Смесь экстрагировали этилацетатом (350 мл × 3), а органические экстракты промыли насыщенным солевым раствором, высушили над сульфатом магния и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 9:1 (об./об.)) для получения указанного в заголовке соединения В5 (20,0 г, 66,1 ммоль, 85%) в виде бесцветного маслянистого вещества.
1Н ЯМР (300 МГц, CDCl3): δ 7,35-7,27 (m, 5Н), 4,21 (s, 0,5Н), 4,08 (s, 0,5Н), 3,80-3,68 (m, 2Н), 3,58 (d, J=10,0 Гц, 1Н), 3,28-3,22 (m, 1Н), 3,20-3,11 (m, 1Н), 2,62 (m, 1Н), 2,05-1,97 (m, 1H), 1,76-1,69 (m, 1Н), 1,55-1,51 (m, 1Н), 1,48 (s, 9Н), 1,30-1,14 (m, 1Н).
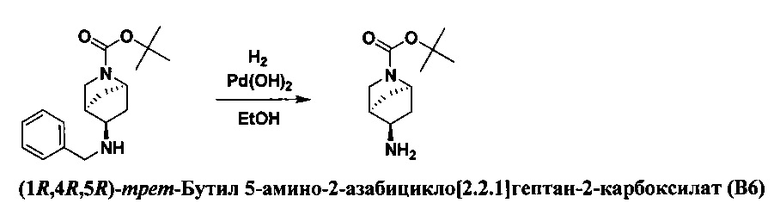
[0165] К смеси гидроксида палладия (4,30 г, 6,12 ммоль, 10,0 мол. %, 20 вес. % на углероде, влажность 50%) и (1R,4R,5R)-трет-бутил 5-(бензиламино)-2-азабицикло[2.2.1]гептан-2-карбоксилата (18,5 г, 61,2 ммоль) в этаноле (100 мл) перемешивали в течение 36 часов под атмосферой водорода при 23°C. Полученную смесь отфильтровали через Целит, а слой фильтра промыли этилацетатом (500 мл). Фильтрат концентрировали под пониженным давлением для получения указанного в заголовке соединения В6 (12,8 г, 60,3 ммоль, 99%) в виде бесцветных кристаллов.
1Н ЯМР (300 МГц, MeOD): δ 4,11 (s, 1Н), 3,56-3,51 (m, 1H), 3,43-3,39 (m, 1Н), 3,18-3,15 (m, 1Н), 2,49 (bs, 1Н), 2,14-2,05 (m, 1Н), 1,74-1,68 (m, 1Н), 1,61 (d, J=10,0 Гц, 1H), 1,48 (s, 9Н), 1,18-1,10 (m, 1H).
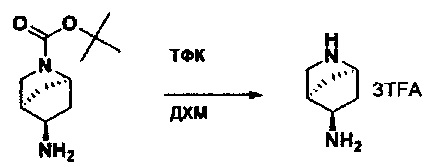
[0166] Получение (1R,4R,5R)-2-азабицикло[2.2.1]гептан-5-амина (В7): К Вос-защищенному амину (200 мг, 0,94 ммоль) в CH2Cl2 (10 мл) по каплям добавили ТФК (5 мл) и перемешивали смесь при комнатной температуре в течение 10 минут. Растворитель удалили под вакуумом, а амин (100 мг, 99%) использовали для следующих реакций без дополнительной очистки.
Синтез октагидроциклопента[с]пиррол-4-амина:
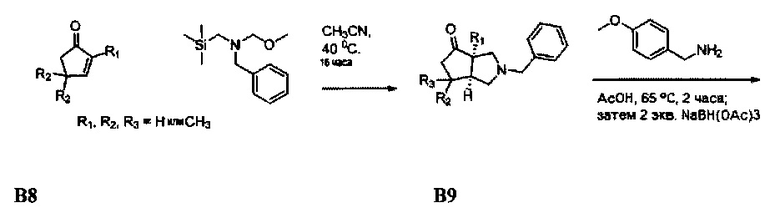
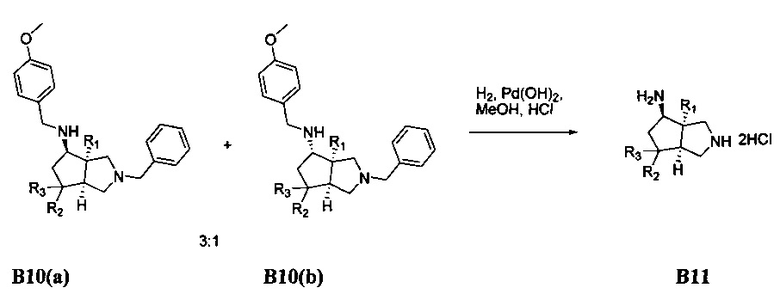
[0167] (3aR,6aS)-2-бензилгексагидроциклопента[c]пирро-4-(5H)-он (В9): К раствору N-метоксиметил)-N-(триметилсилилметил)бензиламина (50 г, 0,21 моль) в ацетонитриле (134 мл) добавили 2-циклопентен-1-он. Смесь перемешивали под аргоном при 45°C в течение ночи. После удаления растворителя на ротационном испарителе, остаток очистили хроматографией не колонке С18 для получения указанного в заголовке соединения в виде прозрачного маслянистого вещества (30 г, 66,4%). Хиральность определили по хиральной ВЭЖХ для получения заданного энантиомера (В9) с э.и. >99%.
[0168] (3aR,4R,6aS)-2-бензил-N-(4-метоксибензил)октагидроциклопента[с]пиррол-4-амин В10(a) и В10(b): К раствору соединения (В9) (2,9 г, 13,43 ммоль) в уксусной кислоте (25 мл) добавили молекулярные сита 4 Å (5,7 г) и 4-метоксибензиламин (2,76 г, 20,15 ммоль). После перемешивания смеси при 75°C в течение одного часа, к ней частями добавили триацетоксиборгидрид натрия, в общем, 1,2 эквивалента (285 мг, 1,35 ммоль с интервалами каждые 20 минут). Реакцию продолжали при температуре от 75°C до комнатной температуры в течение ночи. Молекулярные сита отфильтровали и промыли MeOH. Раствор концентрировали на ротационном испарителе, а полученный остаток очистили хроматографией через колонку С18. pH объединенных собранных элюентов довели до слабощелочного значения добавлением карбоната натрия, и экстрагировали ДХМ (150 мл × 3). Объединенные органические слои высушили над безводным сульфатом натрия и концентрировали на ротационном испарителе для получения указанного в заголовке продукта В10(а) в виде желтого маслянистого вещества (2,56 г, 56,7%).
[0169] (3aR,4R,6aS)-октагидроциклопента[с]пиррол-4-амина HCl соль (В11): К раствору соединения В10(а) (2,56 г, 7,61 ммоль) в MeOH (100 мл) добавили Pd(OH)2 на 20% углерода-50% воды (2 г), затем медленно добавили концентрированную HCl, 37% (3 г). Водород из аппарата двойного слоя пропускали через реакционную смесь в течение 16 часов. Палладий на углероде отфильтровали и промыли MeOH (10 мл). Фильтрат концентрировали на ротационном испарителе, а избыток HCl удалили азеотропной перегонкой с МеОН-толуолом для получения указанного в заголовке соединения (В11) в виде светло-желтой HCl соли (1,51 г, выход 100%).
[0170] Асимметричный синтез трет-бутил (1R,4R,5R)-2-азабицикло[2.2.1]гептан-5-илкарбамата
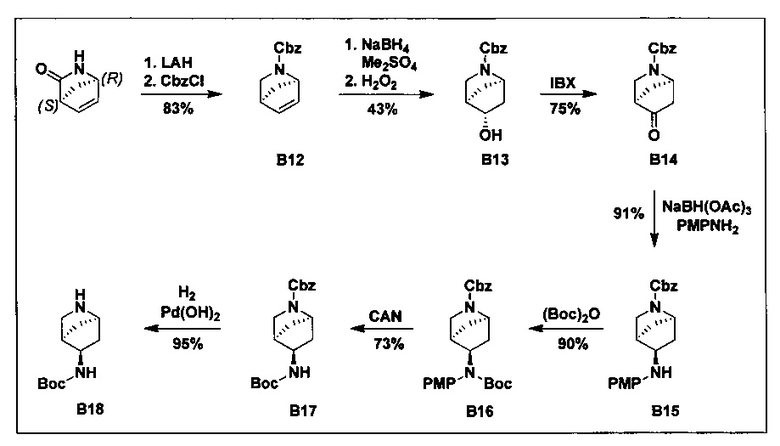
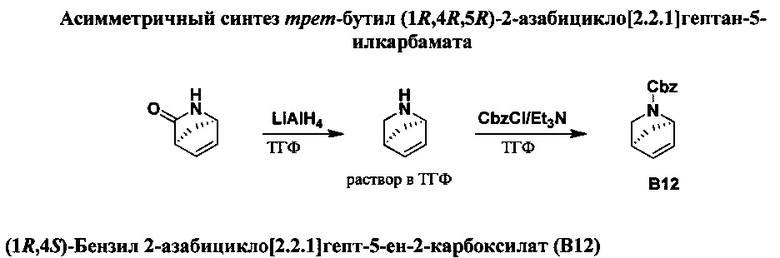
[0171] (1R)-(-)-2-Азабицикло[2.2.1]гепт-5-ен-3-он (5,00 г, 45,8 ммоль, э.и. = 99%), растворенный в безводном ТГФ (45,0 мл), медленно добавили к раствору алюмогидрида лития (28,7 мл, 57,3 ммоль, 2 М раствор в ТГФ) в безводном ТГФ (50,0 мл) под атмосферой азота при 0°C. После успешного завершения добавления смесь перемешивали в течение 3 часов при 23°C, а затем нагревали при 60°C в течение 24 часов. Полученную гетерогенную смесь охладили до 0°C и осторожно добавили к смеси H2O (5,00 мл) через шприц. Белую суспензию отфильтровали через фильтрующий материал Целит, а слой фильтра промыли безводным ТГФ (250,0 мл). Фильтрат в виде прозрачного раствора охладили до 0°C, а затем обработали триэтиламином (12,8 мл, 91,6 ммоль) и CbzCl (10,3 мл, 68,7 ммоль), в этом порядке. Полученную гетерогенную смесь, включающую белый осадок, медленно нагрели до 23°C и оставили перемешиваться в течение 48 часов. Белый осадок отфильтровали под пониженным давлением, а полученный прозрачный раствор концентрировали in vacuo. Неочищенный материал в виде светло-желтого маслянистого вещества очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:4 (об./об.)) для получения указанного в заголовке соединения В12 (8,68 г, 37,9 ммоль, 83%) в виде бесцветного маслянистого вещества.
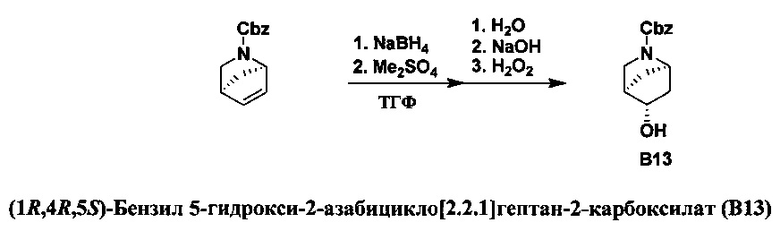
[0172] Смесь (1R,4S)-бензил 2-азабицикло[2.2.1]гепт-5-ен-2-карбоксилата (8,679 г, 37,86 ммоль) и боргидрида натрия (1,17 г, 31,0 ммоль) в ТГФ (60,0 мл) перемешивали в течение 0,5 часа под атмосферой азота при 23°C. После перемешивания в течение 0,5 часа смесь нагрели до 35°C, а затем по каплям добавили диметилсульфат (2,93 мл, 31,0 ммоль), растворенный в ТГФ (2,0 мл) через шприц. (Примечание: диметилсульфат добавляли медленно из-за выделения газа). Полученную гетерогенную смесь перемешивали в течение 4 часов при 35°C, затем охладили до 0°C и погасили добавлением по каплям H2O (5,0 мл). Добавили раствор гидроксида натрия (80,0 мл, 80,0 ммоль, 1 М раствор NaOH) при 0°C, затем добавили перекись водорода (5,0 мл, 30 вес. % в H2O). Смесь нагрели до 23°C и перемешивали еще 1 час. Полученный бесцветный раствор разбавили этилацетатом (250 мл) и отделили органический слой, промыли насыщенным солевым раствором (150 мл) и высушили над сульфатом магния. Смесь концентрировали на ротационном испарителе, а полученное бесцветное маслянистое вещество в виде неочищенного продукта очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:1 (об./об.)) для получения указанного в заголовке соединения В13 (4,02 г, 16,3 ммоль, 43%) в виде бесцветного маслянистого вещества.
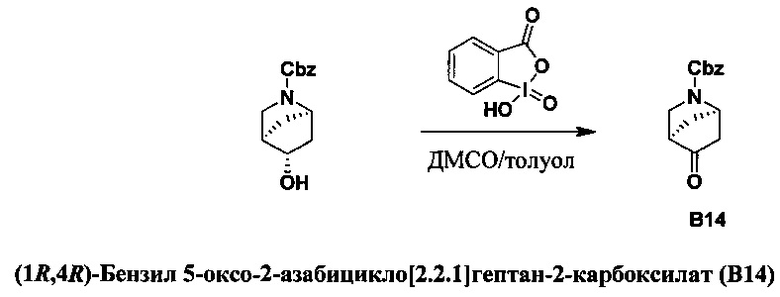
[0173] 2-Йодоксибензойную кислоту (13,7 г, 22,0 ммоль, 45 вес. % (SIBX)) добавили к раствору (1R,4R,5S)-трет-бутил 5-гидрокси-2-азабицикло[2.2.1]гептан-2-карбоксилата (4,02 г, 16,3 ммоль), растворенного в диметилсульфоксиде (20,0 мл) и толуоле (40,0 мл), под атмосферой азота при 23°C. Смесь перемешивали в течение 3 часов 30 минут при 60°C, а затем охладили до 23°C. Полученную гетерогенную смесь обработали насыщенным раствором карбоната натрия (водным) (250 мл) и отфильтровали под пониженным давлением для удаления твердого белого вещества. Фильтрат экстрагировали этилацетатом (250 мл × 3), а органические экстракты промыли насыщенным солевым раствором, высушили над сульфатом магния и концентрировали in vacuo. Неочищенный материал в виде бесцветного маслянистого вещества очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:2 (об./об.)) для получения указанного в заголовке соединения В14 (2,99 г, 12,2 ммоль, 75%) в виде бесцветного маслянистого вещества.
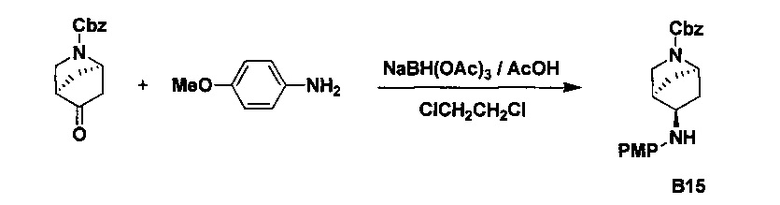
(1R,4R,5R)-Бензил 5-(4-метоксифениламино)-2-азабицикло[2.2.1]гептан-2-карбоксилат (В15)
[0174] Триацетоксиборгидрид натрия (0,904 г, 4,05 ммоль) и ледяную уксусную кислоту (0,180 г, 3,00 ммоль) добавили к раствору (1R,4R)-бензил 5-оксо-2-азабицикло[2.2.1]гептан-2-карбоксилата (0,736 г, 3,00 ммоль) и п-анизидина (0,370 г, 3,00 ммоль) в 1,2-дихлорэтане (10,0 мл) под атмосферой азота при 23°C. Полученную смесь перемешивали в течение 3 часов при 23°C. Гетерогенную смесь охладили до 0°C и погасили насыщенным раствором бикарбоната натрия (водным) (150 мл). Смесь экстрагировали этилацетатом (200 мл × 3), а органические экстракты промыли насыщенным солевым раствором, высушили над сульфатом магния и концентрировали in vacuo. Неочищенный материал в виде прозрачного желтого маслянистого вещества очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:2 (об./об.)) для получения указанного в заголовке соединения В15 (0,964 г, 2,73 ммоль, 91%) в виде белого твердого вещества.
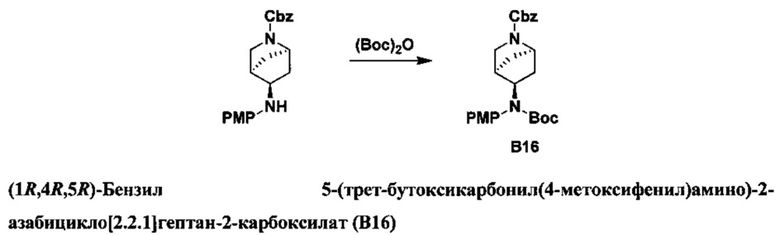
[0175] Смесь (1R,4R,5R)-бензил 5-(4-метоксифениламино)-2-азабицикло[2.2.1]гептан-2-карбоксилата (0,352 г, 1,00 ммоль) и KHMDS (1,30 мл, 1,30 ммоль, 1,0 М раствор в ТГФ) в безводном ТГФ (15,0 мл) перемешивали в течение 15 минут под атмосферой азота при 23°C. Полученную зеленоватую смесь обработали (Boc)2O (0,470 г, 2,15 ммоль), а затем перемешивали в течение 16 часов при 23°C. Смесь концентрировали под пониженным давлением для получения желтого маслянистого вещества. Неочищенный материал очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:2 (об./об.)) для получения указанного в заголовке соединения В16 (0,408 г, 0,901 ммоль, 90%) в виде бесцветного маслянистого вещества.
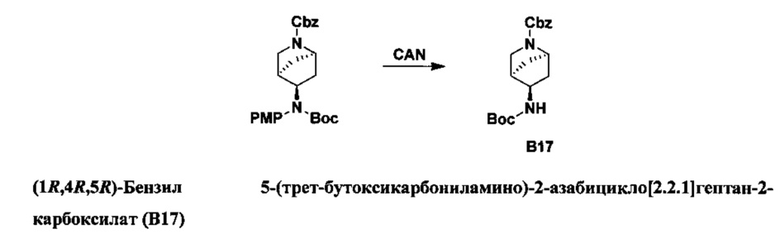
[0176] Церия-аммония нитрат (1,73 г, 3,15 ммоль), растворенный в Н2О (5,0 мл), добавили к раствору (1R,4R,5R)-бензил 5-(трет-бутоксикарбонил(4-метоксифенил)амино)-2-азабицикло[2.2.1]гептан-2-карбоксилата (0,408 г, 0,901 ммоль) в ацетонитриле (25 мл) под атмосферой азота при 0°C. Полученную смесь перемешивали в течение 1 часа при 0°C, а затем разбавили Н2О (100 мл), экстрагировали этилацетатом (150 мл × 3). Объединенную органическую фазу промыли 1 н. раствором Ns2SO3 (75 мл), высушили над MgSO4 и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией (SiO2, EtOAc:n-Hex 1:2 (об./об.)) для получения указанного в заголовке соединения В17 (0,229 г, 0,661 ммоль, 73%) в виде бесцветного маслянистого вещества.
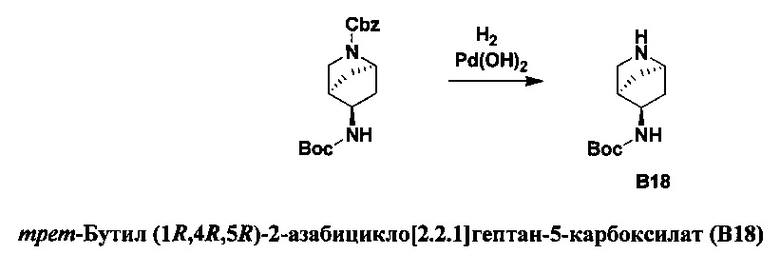
[0177] Смесь гидроксида палладия (0,015 г, 0,022 ммоль, 10,0 мол. %, 20 вес. % на углероде, влажность 50%) и (1R,4R,5R)-бензил 5-(трет-бутоксикарбониламино)-2-азабицикло[2.2.1]гептан-2-карбоксилата (0,077 г, 0,222 ммоль) в этаноле (5,0 мл) перемешивали в течение 3 часов 30 минут под атмосферой водорода при 23°C. Полученную смесь отфильтровали через Целит, а слой фильтра промыли этилацетатом (100 мл). Фильтрат концентрировали под пониженным давлением для получения указанного в заголовке соединения В18 (0,045 г, 0,212 ммоль, 95%) в виде бесцветного маслянистого вещества.
1Н ЯМР (300 МГц, MeOD): δ 3,89 (d, J=11,2 Гц, 1Н), 3,42 (s, 1Н), 3,01 (d, J=10,4 Гц, 1Н), 2,74-2,69 (m, 1Н), 2,58 (bs, 1Н), 2,12-2,02 (m, 1Н), 1,64 (s, 2Н), 1,46 (s, 9Н), 1,19-1,13 (m, 1Н).
РАЗДЕЛ С:
Раздел для соединений, в которых L=S
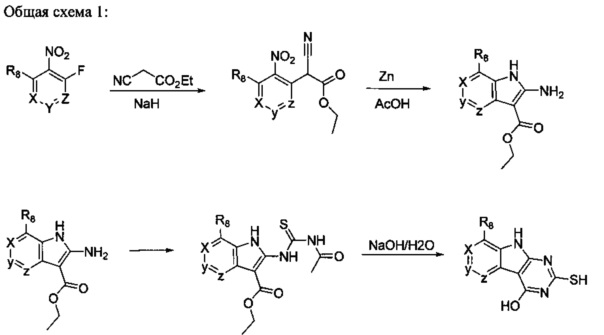
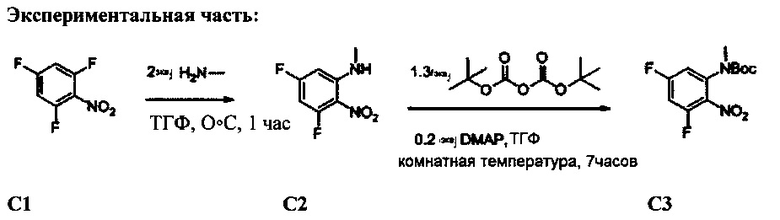
[0178] 3,5-дифтор-N-метил-2-нитроанилин (С2): 1,3,5-Трифтор-2-нитробензол (35,16 г, 0,2 моль) растворили в 100 мл ТГФ и охладили на бане изо льда и воды. К этому раствору по каплям добавили 40% водный раствор метиламина (23,25 г, 0,3 моль) за ~20 минут через капельную воронку. Реакционную смесь перемешивали в течение 1 часа. Затем ее разбавили гексаном (50 мл), а растворители разделили на два слоя. Водный раствор удалили, а органический слой промыли водой (20 мл). Раствор концентрировали осторожным выпариванием на ротационном испарителе при комнатной температуре, и дополнительно высушили под высоким вакуумом для получения неочищенного продукта (С2) в виде оранжевого твердого вещества (36 г, 96%).
1H ЯМР (CDCl3, 300 МГц): δ=6,97-6,88 (m, 2Н), 3,27 (s, 3Н).
[0179] Трет-бутил 3,5-дифтор-2-нитрофенил(метил)карбамат (С3): К раствору неочищенного 3,5-дифтор-N-метил-2-нитроанилина (С2) (36 г, 0,191 моль) в 100 мл ТГФ добавили ди-трет-бутил-дикарбонат (54,3 г, 0,249 моль), затем 4-диметиламинопиридин (4,68 г, 0,038 моль). Реакционную смесь перемешивали при комнатной температуре в течение 7 часов. Затем добавили воду (50 мл), и полученный раствор перемешивали в течение 1,5 часов. После разбавления гексаном (100 мл) раствор разделили на два слоя, а водную фазу удалили через экстракционную воронку и снова экстрагировали этилацетатом (50 мл). Затем объединенный органический слой промыли сначала 5% раствором NH4Cl (100 мл), а затем 5% раствором K2CO3 (100 мл). После концентрирования объединенного органического растворителя на ротационном испарителе при комнатной температуре, полученный остаток повторно растворили в MeOH ((~50 мл), а затем по каплям добавили в 600 мл ~0,01% раствора K2CO3. Твердый оранжевый продукт (С3) отфильтровали, промыли водой и высушили под высоким вакуумом (46,78 г, 85%).
1Н ЯМР (CDCl3, 300 МГц): δ=6,93-6,85 (m, 2Н), 3,20 (s, 3Н), 1,32 (s, 9Н).
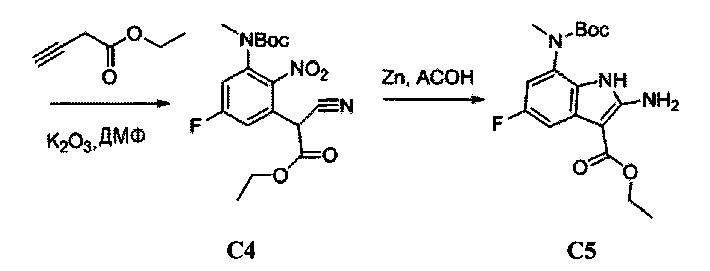
[0180] Синтез соединения С4: К раствору С3 (40 г, 0,14 моль) в ДМФ (200 мл) добавили карбонат калия (19 г, 0,14 моль), затем порцию этилцианоацетата (15 г, 0,14 моль). Смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавили дополнительную порцию карбоната калия (19 г, 0,14 моль) и порцию этилцианоацетата (15 г, 0,14 моль). После перемешивания смеси при комнатной температуре в течение 4 часов, добавили карбонат калия (19 г, 0,14 моль) и перемешивали смесь при комнатной температуре еще 12 часов. Затем смесь вылили в ледяную воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали, концентрировали и очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения соединения С4 (33 г, выход: 63%) в виде твердого желтого вещества.
1Н ЯМР (CDCl3, 300 МГц): δ=6,93-6,85 (m, 2Н), 4,88 (m, 1Н), 4,33 (m, 2Н), 3,20 (s, 3Н), 1,32 (s, 9Н), 1,28 (t, 3Н).
[0181] Синтез соединения С5: К раствору С4 (20 г, 52 ммоль) в толуоле (100 мл) и уксусной кислоте (100 мл) добавили цинковый порошок (30 г, 0,46 моль) и перемешивали смесь при 75°C в течение 2 часов. Затем добавили дополнительное количество порошка Zn (10 г, 0,15 моль). После перемешивания при 75°C еще 0,5 часа, смесь охладили до комнатной температуры, отфильтровали и вылили в ледяную воду. Добавили 2 н. NaOH, чтобы довести pH до 8-9, и экстрагировали полученную смесь этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали, концентрировали и очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения соединения С5 в виде твердого коричневого вещества (8,3 г, выход: 45%).
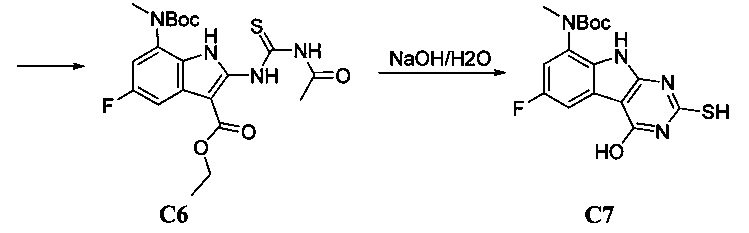
[0182] Синтез соединения С7: К перемешанной суспензии соединения С5 (7,4 г, 20 ммоль) в ацетоне (140 мл) по каплям добавили раствор ацетилтиоизоцианата (12 мл, 140 ммоль) в ацетоне (50 мл) при комнатной температуре. Реакционную смесь нагревали с дефлегматором в течение 16 часов. Данные ЖХМС показали, что реакция завершена. Реакционную смесь концентрировали для следующей стадии без очистки. ЖХ-МС: М+1: 453,21.
[0183] Полученный выше остаток растворили в 50 мл метанола и 50 мл H2O, затем добавили 10 мл 10% раствора KOH, раствор смеси нагревали с дефлегматором в течение 30 минут. Когда данные ЖХМС показали, что реакция завершена, реакционную смесь охладили до комнатной температуры, подкислили до pH 5 при помощи 1 М водного раствора HCl, осадок собрали фильтрацией для получения соединения С7 в виде твердого вещества (5 г, 65,4% за две стадии). ЖХ-МС: М+1: 365,13.
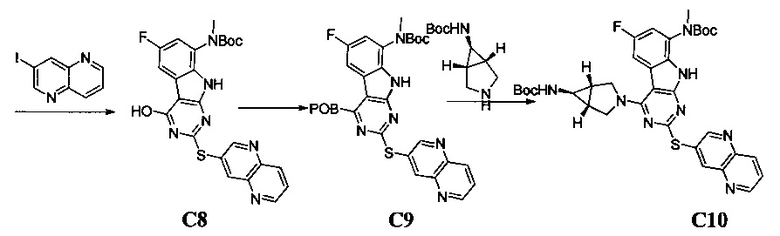
[0184] Синтез соединения С10: Раствор CuI (67 мг, 0,35 ммоль), диметилциклогексан-1,2-диамина (100 мг, 0,70 ммоль) в 9 мл NMP добавили к перемешанной суспензии трет-бутил (4-гидрокси-2-меркапто-9Н-пиримидо[4,5-b]индол-8-ил)(метил)карбамата (5, 350 мг, 1,0 ммоль) и соответствующий I-Ar (1,17 ммоль), K2CO3 (324 мг, 2,35 ммоль) и PPh3 (400 мг, 1,53 ммоль) в NMP (9 мл). Смесь нагревали до 130°C в течение от 2 до 12 часов, контролируя завершение реакции по ЖХ-МС. Когда реакция была завершена, смесь охладили до 0°C, добавили BOP (621 мг, 1,40 ммоль) и Et3N (0,41 мл, 2,93 ммоль), перемешивали в течение 30 минут при 0°C, затем нагрели до комнатной температуры и добавили соответствующий Вос-защищенный диамин (2,34 ммоль). Реакционную смесь нагревали до 50°C в течение 30 минут. Данные ЖХ-МС показали, что реакция завершена. После завершения реакции смесь разделили между этилацетатом и водой, водный слой дважды экстрагировали этилацетатом, объединенный органический слой высушили и очистили флэш-хроматографией для получения продуктов соединения С10 в виде твердого вещества (420 мг, 63% за две стадии). ЖХ-МС: М+1: 673,25.
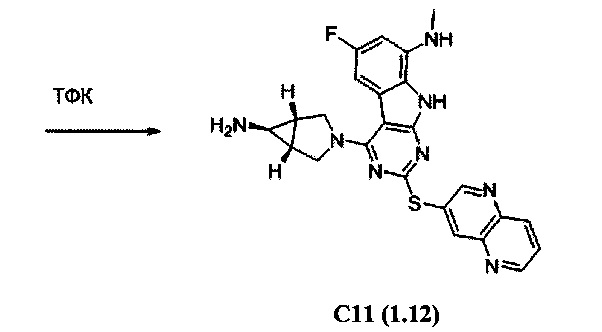
[0185] Синтез соединения С11: Полученное выше соединение (420 мг, 0,63 ммоль) растворили в 10 мл ТФК и перемешивали в течение 30 минут при комнатной температуре. После удаления растворителей остаток повторно растворили в 10 мл метанола и 10 мл H2O, затем добавили 1 н. NaOH для нейтрализации раствора до pH 14, затем щелочной раствор разбавили дополнительно 100 мл H2O, и этот раствор энергично перемешивали еще 1 час, собрали осадок и высушили для получения конечных соединений в виде твердого белого вещества (200 мг, 70%). ЖХ-МС: М+1: 473,13.
1H ЯМР (300 МГц, ДМСО) δ (ppm): 11,75 (s, 1Н), 8,09 (d, 1H), 8,95 (s, 1H), 8,52 (m, 1H), 8,35 (s, 1H), 7,75 (m, 1Н), 7,01 (d, J=11,2, 1Н), 5,96 (d, 1Н), 4,10 (s, 1H), 2,98 (s, 3Н), 2,85 (m, 2Н), 2,67 (m, 2Н), 1,38 (m, 1H), 0,75 (br m, 2Н).
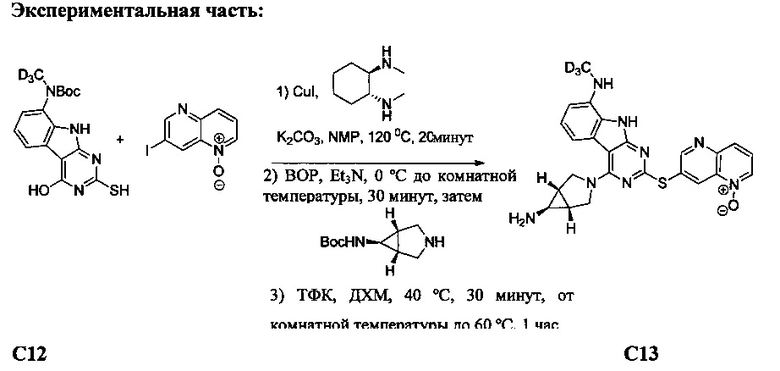
[0186] 7-(4-(6-амино-3-азабицикло[3.1.0]гексан-3-ил)-8-(дейтерированный метиламино)-9H-пиримидо[4,5-b]индол-2-илтио)-1,5-нафтиридин 1-оксид С13 (CD3 аналог соединения 1.13): К смеси CuI (76 мг, 0,4 ммоль) и K2CO3 (112 мг, 0,8 ммоль) в NMP (1 мл) добавили транс-N,N'-диметилциклогексан-1,2-диамин (113,6 мг, 0,8 ммоль). Смесь перемешивали при 120°C в течение 10 минут. Затем к ней добавили соединение (1) (70 мг, 0,2 ммоль) и 7-йод-1,5-нафтиридин 1-оксид (59,8 г, 0,22 ммоль). Реакцию продолжали при 120°C в течение 20 минут. Затем смесь охладили до ~4°C, а затем добавили Et3N (0,3 мл), затем [бензотриазол-1-ил-окси-трис-(диметиламино)фосфония гексафторфосфат] (реагент ВОР) (97,3 мг, 0,22 ммоль). После перемешивания при температуре от ~4°C до комнатной температуры в течение 30 минут, к реакционной смеси добавили амин (3) (79,3 мг, 0,4 ммоль), а затем нагревали при 60°C в течение одного часа. Затем ее очистили при помощи ВЭЖХ. Воду в собранных элюентах Вос-аддукта удалили экстракцией с ДХМ (20 мл × 2). Объединенные органические слои концентрировали на ротационном испарителе. Остаток повторно растворили в ДХМ (2 мл) и трифторуксусной кислоте (~0,2 мл). Смесь перемешивали при 40°C в течение 30 минут для снятия ВОС-защиты. Реакционную смесь очистили флэш-хроматографией ВЭЖХ для получения указанного в заголовке соединения (С13) в виде твердого белого вещества (52,1 мг, 55%).
1H ЯМР (300 МГц, ДМСО) δ (ppm): 11,75 (s, 1Н), 8,09 (d, 1H), 8,95 (s, 1H), 8,52 (m, 1H), 8,35 (s, 1H), 7,75 (m, 1H), 7,01 (d, J=11,2, 1H), 5,96 (d, 1H), 4,10 (s, 1H), 2,85 (m, 2H), 2,67 (m, 2H), 1,38 (m, 1H), 0,75 (br m, 2H).
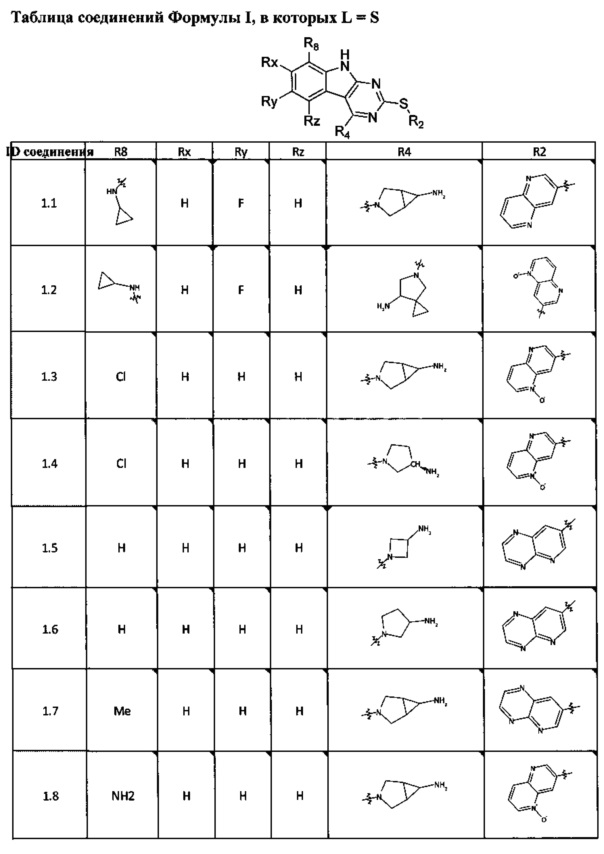
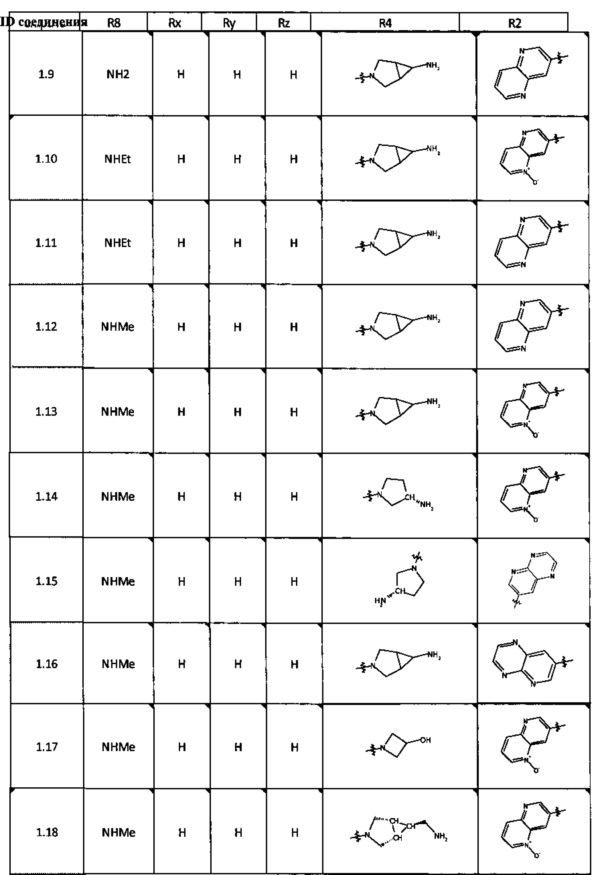
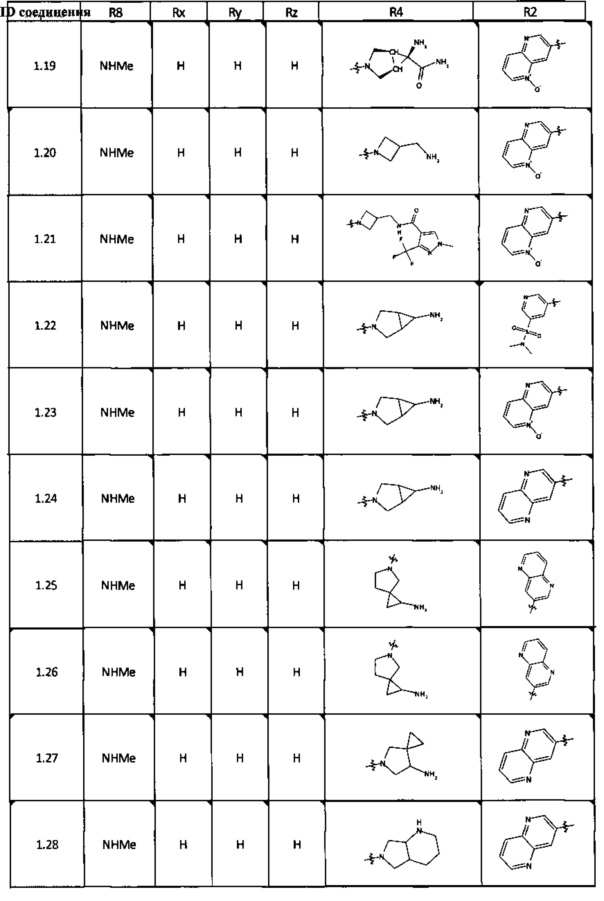
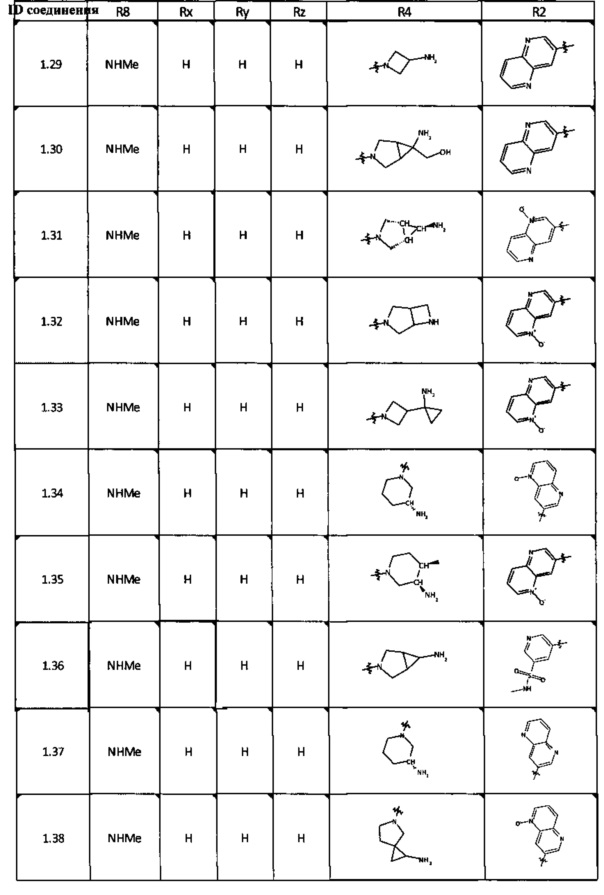
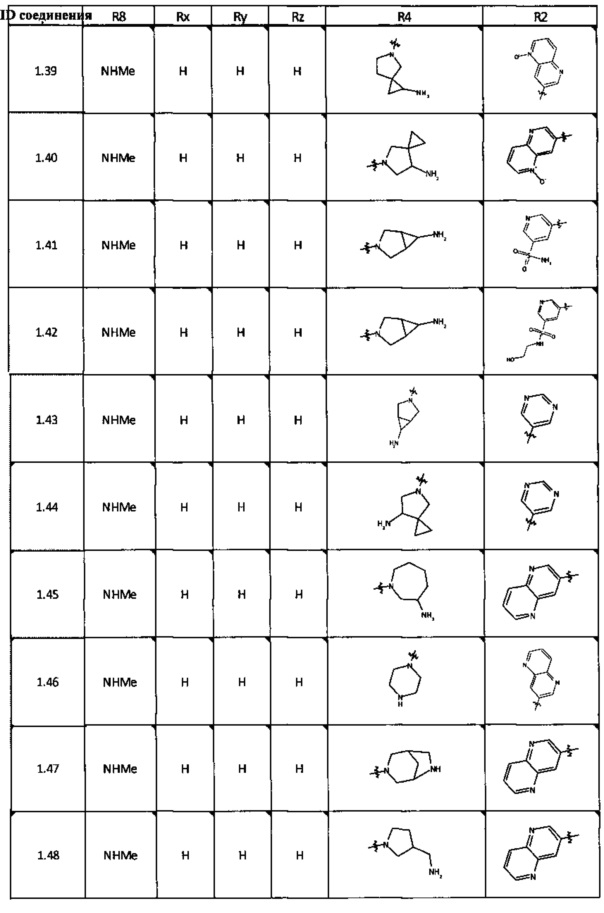
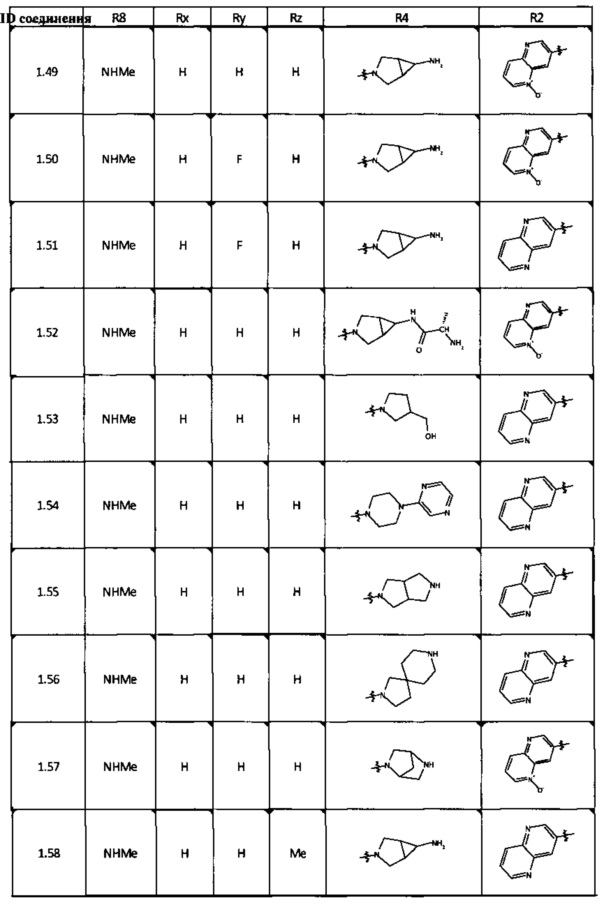
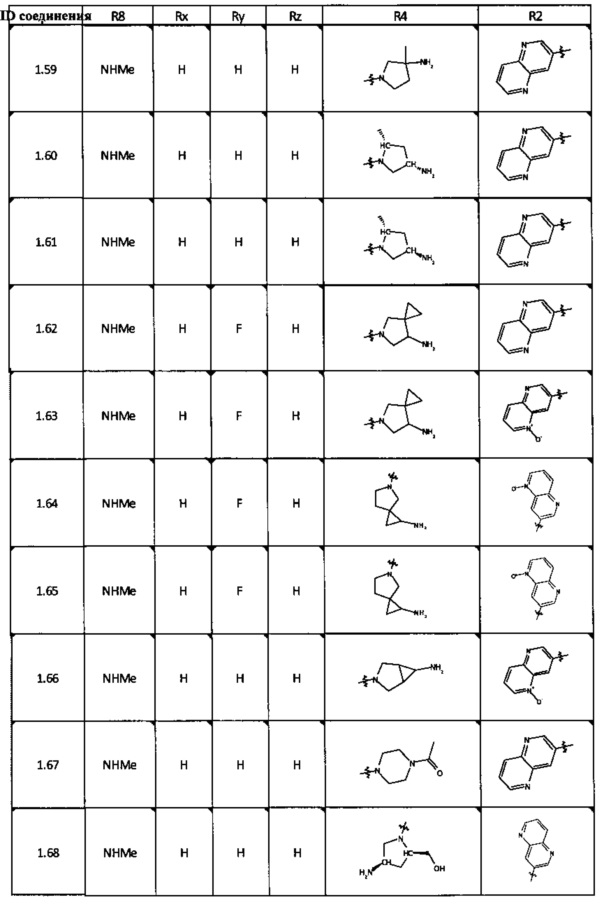
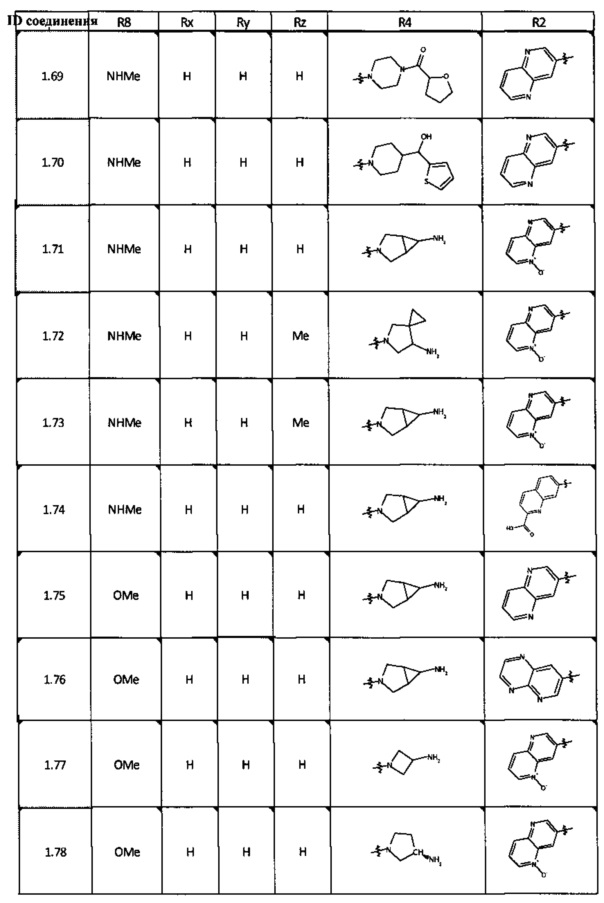
РАЗДЕЛ D: СИНТЕЗ СОЕДИНЕНИЙ ФОРМУЛЫ I, В КОТОРЫХ L=O
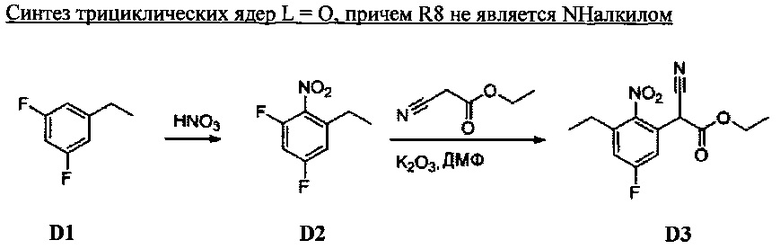
[0187] Синтез соединения D2: К раствору D1 (40 г, 0,28 моль) в H2SO4 (200 мл) добавили HNO3 (26 г, 0,42 моль) при 0°C. После перемешивания при 0°C в течение 1 часа, смесь вылили в ледяную воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали, концентрировали и очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 15:1) для получения соединения D2 (37 г, выход: 70%) в виде маслянистого желтого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 6,93 (s, 1Н), 6,91 (s, 1Н), 4,33-4,27 (m, 2Н), 2,73-2,68 (m, 2Н), 1,29-1,25 (t, J=7,6 Гц, 2Н).
[0188] Синтез соединения D3: К раствору 2 (37 г, 0,20 моль) в ДМФ (200 мл) добавили карбонат калия (54,8 г, 0,40 моль), затем порцию этилцианоацетата (22,3 г, 0,20 моль). Смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавили дополнительную порцию карбоната калия (54,8 г, 0,40 моль) и порцию этилцианоацетата (22,3 г, 0,20 моль). После перемешивания смеси при комнатной температуре в течение 4 часов, добавили карбонат калия (27,4 г, 0,2 моль) и перемешивали смесь при комнатной температуре еще 12 часов. Затем смесь вылили в ледяную воду и экстрагировали этилацетатом (2×200 мл). Органический слой высушили над Ыаг804, отфильтровали, концентрировали и очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения соединения D3 (25 г, выход: 67%) в виде твердого желтого вещества.
1Н ЯМР (400 МГц, CDCl3): δ: 7,33-7,04 (dd, J=4,4, 2,4 Гц, 1H), 7,16-7,13 (dd, J=4,4, 2,4 Гц, 1Н), 5,06 (s, 1Н), 4,32-4,27 (m, 2Н), 2,74-2,68 (m, 2Н), 1,35-1,26 (m, 6Н).
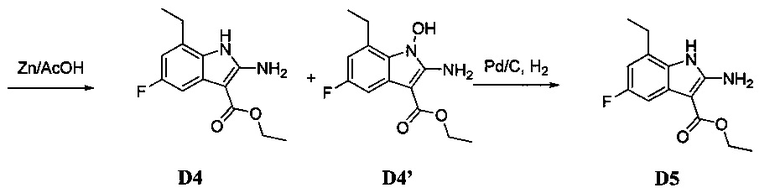
[0189] Синтез соединения D4 и D4': К раствору D3 (22 г, 79 ммоль) в толуоле (100 мл) и уксусной кислоте (100 мл) добавили цинковый порошок (30 г, 0,46 моль) и перемешивали смесь при 75°C в течение 2 часов. Затем добавили дополнительное количество порошка Zn (10 г, 0,15 моль). После перемешивания при 75°C еще 0,5 часа, смесь охладили до комнатной температуры, отфильтровали и вылили в ледяную воду. Добавили 2 н. NaOH, чтобы довести pH до 8-9, и экстрагировали полученную смесь этилацетатом (2×200 мл). Органический слой высушили над Na2SO4, отфильтровали, концентрировали и очистили хроматографией на силикагеле (петролейный эфир/этилацетат = 5:1) для получения коричневого твердого вещества, которое перекристаллизовали из петролейного эфира/EtOAc (10:1) для получения смеси соединения D4 и D4' (7,2 г, выход: 35%) в виде твердого коричневого вещества.
[0190] Синтез соединения D5: Раствор смеси соединения D4 и D4' (5,8 г) в EtOH (100 мл)/HOAc (5 мл) гидрогенировали с катализатором 10% Pd/C (580 мг) в течение ночи под давлением 50 Psi (3,4 атмосферы). Катализатор отфильтровали, а фильтрат концентрировали для получения соединения D5 (5,3 г, выход: 93%).
1Н ЯМР (400 МГц, ДМСО-d6): δ: 10,75 (s, 1H), 7,08 (dd, J=9,6, 2,4 Гц, 1Н), 6,55 (dd, J=10,8, 2,4 Гц, 1H), 6,44 (s, 2Н), 4,21 (q, J=7,2 Гц, 2Н), 2,71 (q, J=7,6 Гц, 2Н), 1,31 (t, J=6,8 Гц, 3Н), 1,20 (t, J=7,6 Гц, 3Н). ЖХМС [подвижная фаза: 30%-95% ацетонитрил-0,02% NH4Ac за 6 минут, в конце при этих условиях в течение 0,5 минуты] чистота >95%, Rt=2,953 минуты; МС, рассчитано: 250; МС, найдено: 251 ([М+1]+).
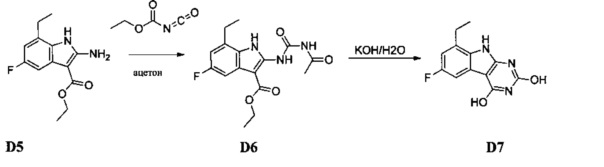
[0191] К перемешанной суспензии соединения D5 (7,4 г, 20 ммоль) в ацетоне (140 мл) по каплям добавили раствор ацетилтиоизоцианата (12 мл, 140 ммоль) в ацетоне (50 мл) при комнатной температуре. Реакционную смесь нагревали с дефлегматором в течение 16 часов. Данные ЖХМС показали, что реакция завершена. Реакционную смесь концентрировали для следующей стадии без очистки. ЖХ-МС: М+1: 453,21.
[0192] К перемешанной суспензии D6 (9,13 г, 20,0 ммоль) в воде/EtOH (75 мл/25 мл) добавили раствор KOH в 20 мл воды при комнатной температуре. После добавления полученную смесь нагревали с дефлегматором в течение 4 часов. Данные ТСХ показали, что реакция завершена, затем реакционную смесь охладили до комнатной температуры, подкислили 1 М раствором HCl до pH=5, осадок собрали фильтрацией, промыли водой (200 мл × 1), затем этилацетатом (200 мл × 1) для получения продукта D7 в виде бледно-желтого твердого вещества (5,90 г, выход 87,1%). ТСХ: Rf=0,05 (силикагель, метанол : ДХМ = 1:10, об./об.). ЖХ-МС: М-1: 248,10
1Н ЯМР (400 МГц, ДМСО-d6): δ: 11,44 (s, 1Н), 10,75 (s, 1Н), 7,22 (s, 1H), 7,08 (dd, J=9,6, 2,4 Гц, 1Н), 6,55 (dd, J=10,8, 2,4 Гц, 1Н), 2,70 (q, J=7,6 Гц, 2Н), 1,22 (t, J=7,6 Гц, 3Н).
[0193] Соединение D7 (2 г, 8,06 ммоль) поместили с раствором POCl3 (50 мл) в пробирку для работы под давлением и добавили несколько капель N-этилдиизопропиламина. Реакционную смесь нагревали при 185°C в закрытом состоянии в течение 10 часов. Смесь охладили и вылили в ледяную воду, а твердое желтое вещество собрали фильтрацией, высушили под пониженным давлением для получения D8 (2,1 г, выход 95%) в виде твердого желтого вещества. ЖХ-МС: М+1: 285,01
[0194] К перемешанному раствору соединения D8 (250 мг, 0,88 ммоль) в 2 мл NMP при 110°C добавили (R)-трет-бутил 5-азаспиро[2.4]гептан-7-илкарбамат (98 мг, 0,88 ммоль) и K2CO3 (7 мг, 0,05 ммоль). После завершения реакции через 10 минут, к реакционной смеси добавили 2-метилпиримидин-5-ол (28 мг, 0,25 ммоль) в пробирке для микроволновых реакций. Реакционную смесь закрыли и поместили в микроволновый реактор при 180°C на 10 минут. Заданный продукт получили очисткой ВЭЖХ с образованием D9 (115 мг, 30%) в виде твердого белого вещества. ЖХ-МС: М+1: 434,25.
1Н ЯМР (300 МГц, ДМСО-d6): δ: 11,44 (s, 1Н), 10,75 (s, 1Н), 7,22 (s, 1Н), 7,08 (dd, J=9,6, 2,4 Гц, 1Н), 6,55 (dd, J=10,8, 2,4 Гц, 1Н), 2,70 (q, J=7,6 Гц, 2Н), 2,64 (m, 2Н), 2,62 (m, 2Н), 2,01-2,41 (m, 4Н), 1,22 (t, J=7,6 Гц, 3Н).
[0195] Синтез соединения D11 (2.06): Указанное в подзаголовке соединение синтезировали по тому же способу, который описан для соединения 1629, исходя из 2,4-дихлор-6-фтор-8-метил-9Н-пиримидо[4,5-b]индола и (R)-трет-бутил 5-азаспиро[2.4]гептан-7-илкарбамата. ЖХ-МС: М+1: 434,25.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 11,75 (s, 1Н), 8,72 (s, 2Н), 8,09 (br s, 3Н), 7,01 (d, J=11,2, 1Н), 6,31 (d, J=9,1, 1H), 4,40 (d, J=9,9, 1Н), 4,32 (dd, J=7,6, 4,5, 1H), 4,03 (d, J=12,3, 1Н), 3,50 (d, J=9,8, 2Н), 2,67 (s, 3Н), 2,05 (s, 3Н), 1,09 (m, 1Н), 0,81 (br m, 3Н).
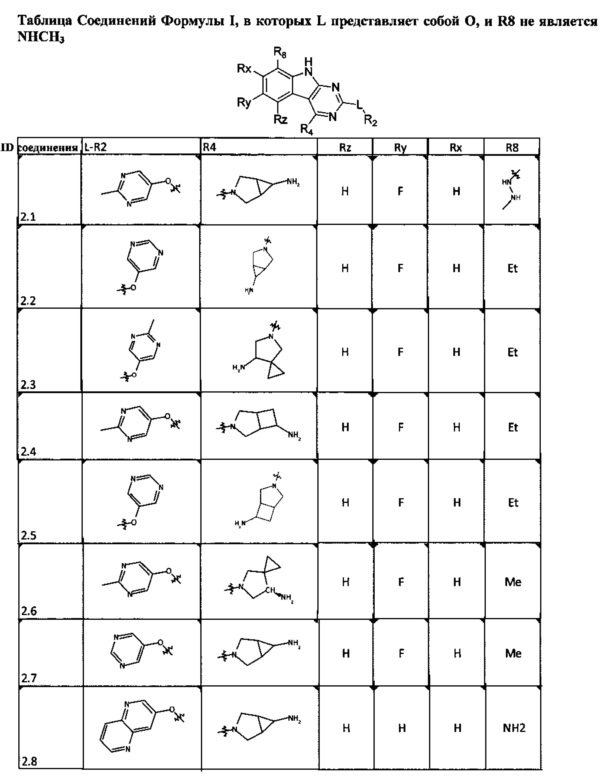
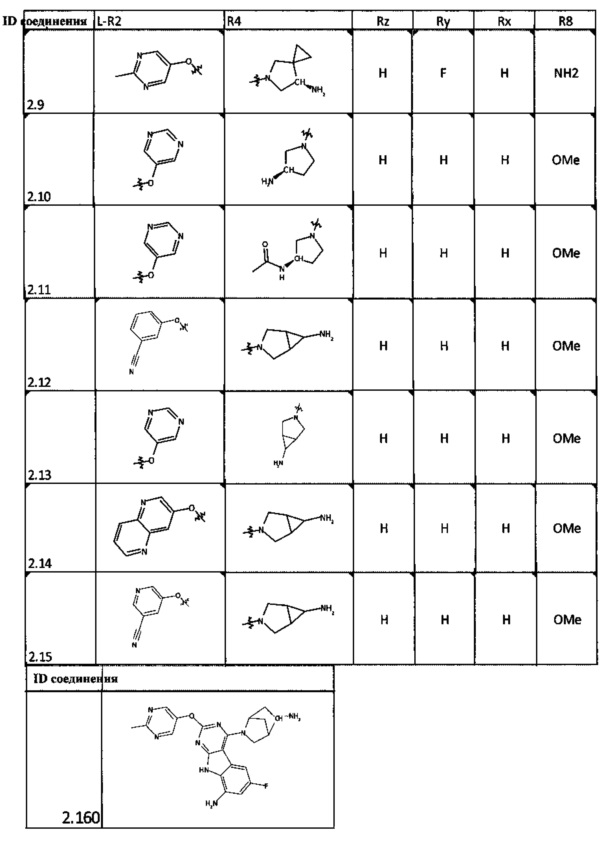
Синтез соединений Формулы I, в которых L=O, и R8 является NH алкилом
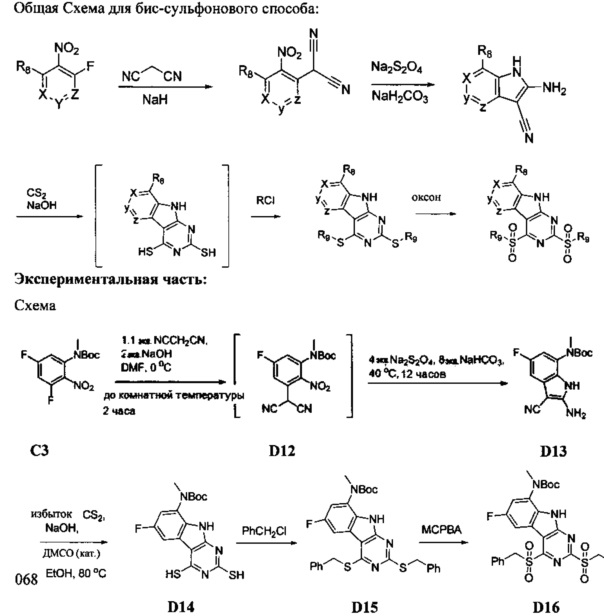
[0196] Трет-бутил 2-амино-3-циано-5-фтор-1H-индол-7-ил(метил)карбамат (D13): Неочищенный трет-бутил 3,5-дифтор-2-нитрофенил(метил)карбамат (С3) (46,12 г, 0,162 моль) растворили в ДМФ (80 мл) и охладили на бане изо льда и воды. К нему добавили малононитрил (11,8 г, 179 ммоль), затем добавили раствор NaOH (12,98 г, 325 ммоль) в воде (20 мл). После перемешивания экзотермической реакционной смеси в течение одного часа, убрали баню изо льда и воды, а реакционную смесь перемешивали еще один час. Затем ее разбавили ДМФ (80 мл) и водой (80 мл), и заменили атмосферу аргоном. Добавили бикарбонат натрия (109 г, 1,3 моль), затем гидросульфит натрия (123 г, 649 ммоль). Смесь хорошо перемешивали под аргоном при 40°C в течение 12 часов (если для завершения реакции требуется больше времени, можно добавить дополнительное количество гидросульфита натрия). После охлаждения реакционной смеси до комнатной температуры, ее разбавили EtOAc (100 мл), затем отфильтровали через стеклянную фильтровальную воронку. Твердые вещества промыли EtOAc/гексаном (1:1, 400 мл). Водный слой отделили, а органический слой экстрагировали 10% буферным раствором 7 (3×100 мл). Объединенные водные слои снова экстрагировали EtOAc/гексаном (1:1, 200 мл). Объединенные органические фазы промыли 5% раствором K2CO3 (300 мл). Экстракты затем высушили над сульфатом натрия и концентрировали на ротационном испарителе для получения неочищенного соединения (D13) в виде твердого вещества коричневого цвета (32,6 г, 66%). ЖХ-МС: М+1: 305,16.
1H ЯМР (ДМСО, 300 МГц): δ=10,77 (s, 1H), 6,84-6,80 (m, 1Н), 6,69 (s, 2H), 6,69-6,66 (m, 1H), 3,14 (s, 3H), 1,33 (s, 9H).
[0197] Трет-бутил 2,4-бис(бензилтио)-6-фтор-9H-пиримидо[4,5-b]индол-8-ил(метил)карбамат (D15): Неочищенный трет-бутил 2-амино-3-циано-5-фтор-1H-индол-7-ил(метил)кабамат (D13) (4 г, 13,14 ммоль), гидроксид натрия (756 мг, 18,9 ммоль) и EtOH (40 мл) поместили в 350 мл закрытую пробирку. Смесь перемешивали при 50°C в течение 15 минут для растворения всего NaOH, а затем охладили до комнатной температуры. После замены атмосферы аргоном, к раствору добавили дисульфид углерода (10 мл) и диметилсульфоксид (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем нагревали с дефлегматором при 80°C в течение 42 часов. Затем ее охладили до комнатной температуры и поместили на баню из льда и воды. Добавили воду (20 мл), затем добавили бензилхлорид (3,33 г, 26,27 ммоль). Баню изо льда и воды убрали, а реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Добавили дополнительное количество бензилхлорида (1,66 г, 13,13 ммоль), и полученный раствор перемешивали при комнатной температуре в течение ночи. Смесь разбавили EtOAc (60 мл) и водой (100 мл). Полученный раствор разделили на два слоя, а водную фазу удалили через экстракционную воронку и снова экстрагировали 50 мл этилацетата. Объединенные органические слои концентрировали на ротационном испарителе, а остаток очистили силикагелевой колоночной хроматографией (15% EtOAc в гексане) для получения указанного в заголовке соединения (D15) в виде желтого пенистого вещества (2,65 г, 36%). ЖХ-МС: М+1: 561,05.
1Н ЯМР (CDCl3, 300 МГц): δ=8,72 (s, 1Н), 7,66-7,62 (dd, J=8,37, 2,28 Гц, 1Н), 7,48-7,27 (m, 10Н), 7,05-7,01 (dd, J=10,14, 2,28 Гц, 1Н), 4,69 (s, 2Н), 4,55 (s, 2Н), 3,37 (s, 3Н), 1,48 (s, 9Н).
[0198] Трет-бутил 2,4-бис(бензилсульфонил)-6-фтор-9H-пиримидо[4,5-b]индол-8-ил(метил)карбамат (D16): Раствор трет-бутил 2,4-бис(бензилтио)-6-фтор-9H-пиримидо[4,5-b]индол-8-ил(метил)карбамата (D15) (2,28 г, 4,07 ммоль) в ДХМ (50 мл) охладили на бане изо льда и воды и добавили 3-хлорпероксибензойную кислоту, 77% (2,01 г, 8,95 ммоль). После перемешивания реакционной смеси в течение 1 часа, баню изо льда и воды убрали и добавили дополнительное количество mCPBA (2,01 г). Полученный раствор перемешивали при комнатной температуре в течение 7 часов. Затем его экстрагировали 5% раствором K2CO3 (100 мл), а водный слой снова экстрагировали ДХМ (100 мл). Объединенные органические слои промыли сначала 5% раствором K2CO3 (100 мл), затем 5% раствором NaCl (50 мл). Смесь высушили над сульфатом натрия и концентрировали на ротационном испарителе для получения неочищенного указанного в заголовке соединения (D16) в виде ярко-желтого твердого вещества (2,54 г, количественный выход). ЖХ-МС: М+1: 625,05.
1Н ЯМР (CDCl3, 300 МГц): δ=10,07 (s, 1Н), 8,49-8,46 (dd, J=8,64, 2,22 Гц, 1H), 7,54-7,51 (m, 1Н), 7,38-7,27 (m, 10Н), 4,95 (s, 2Н), 4,84 (s, 2Н), 3,40 (s, 3Н), 1,52 (s, 9Н).
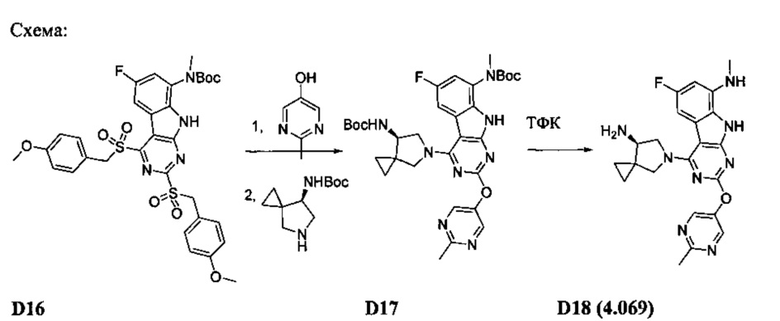
[0199] Получение D17: Бис-сульфон 2 (11,80 г, 17,23 ммоль) растворили в NMP (60 мл), затем добавили 2-метилпиримидин-5-ол 1 (7,59 г, 68,93 ммоль). Получили гомогенный раствор. Добавили K2CO3 (9,53 г, 68,93 ммоль), и полученную суспензию нагревали до 100°C в течение 1 часа, затем добавили Вос-защищенный амин (7,32 г, 34,46 ммоль), и полученную смесь нагревали до 100°C еще один час, охладили до комнатной температуры и вылили в эту смесь воду (450 мл) при перемешивании. Смесь охладили до 0°C, отфильтровали и промыли осадок водой (2×25 мл), высушили для получения около 12 г неочищенного продукта в виде твердого белого вещества. Неочищенное твердое вещество растворили в дихлорметане и добавили силикагель. Растворители удалили. В результате флэш-хроматографии остатка на силикагеле (EtOAc/гексан: от 20% до 50% до 90%) получили чистый D17 в виде твердого белого вещества (7,76 г, 75%). ЖХ-МС: М+1: 635,30.
[0200] Получение D18 (4.069): Соединение D17 растворили в 50 мл ТФК и перемешивали в течение 1 минуты при комнатной температуре. После удаление растворителя добавили воду (50 мл) и EtOH (25 мл). Гомогенный раствор нейтрализовали 1 н. NaOH (около 150 мл, pH>10). Образовалось вязкое твердое вещество, его отделили. Вязкое твердое вещество суспендировали в воде (50 мл) и разбили вязкое твердое вещество на мелкие кусочки при помощи шпателя. Осадок отфильтровали, дважды промыли водой и высушили на воздухе для получения 4,40 грамм чистого D18 (4.069) в виде слегка белого твердого вещества (85%, общий выход 63% из D6). ЖХ-МС: М+1: 435,24.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 11,75 (s, 1Н), 8,72 (s, 2Н), 8,09 (br s, 3Н), 7,01 (d, J=11,2, 1Н), 6,31 (d, J=9,7, 1Н), 4,40 (d, J=9,9, 1Н), 4,32 (dd, J=7,6, 4,5, 1Н), 4,03 (d, J=12,3, 1H), 3,50 (d, J=9,8, 2Н), 2,85 (s, 3Н), 2,67 (s, 3Н), 1,09 (m, 1Н), 0,81 (br m, 3Н).
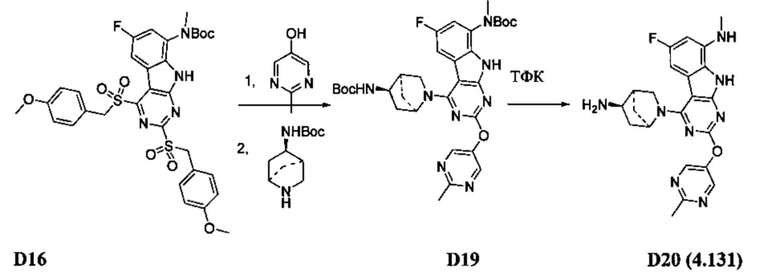
[0201] Получение D20 (4.131): Указанное в подзаголовке соединение синтезировали по способу, описанному выше, исходя из трет-бутил (1R,4R,5R)-2-азабицикло[2.2.1]гептан-5-илкарбамата.
ЖХ-МС: М+1: 435,24.
1Н ЯМР (500 МГц, ДМСО) δ (ppm): 11,75 (brm, 1Н), 8,92 (brm, 1Н), 8,66 (brs, 1Н), 7,44 (d, J=9,7, 1Н), 7,04 (d, J=5,2), 6,31 (d, J=12,2, 1H), 5,56 (s, 1H), 4,38 (m, 1H), 4,04 (s, 1Н), 3,37 (m, 1Н), 3,01 (m, 1H), 2,87 (m, 1Н), 2,85 (m, 3Н), 2,66 (s, 3Н), 2,16 (m, 1Н), 1,86 (m, 1Н), 1,79 (m, 1Н), 1,75 (m, 1Н).

[0202] Получение D22 (4.408): Указанное в подзаголовке соединение синтезировали по способу, описанному выше, исходя из 2-(1-гидроксиэтил)пиримидин-5-ола. ЖХ-МС: М+1: 465,22.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 11,75 (s, 1Н), 8,72 (s, 2Н), 7,01 (d, J=11,2, 1Н), 6,31 (d, J=9,7, 1Н), 4,82 (brm, 1Н), 4,02 (m, 1Н), 3,81 (m, 1Н), 3,49 (m, 1Н), 2,85 (s, 3Н), 2,63 (brs, 1Н), 2,14 (m, 1Н), 1,65-182 (m, 2Н), 1,47 (d, 3Н), 1,38 (m, 1Н).
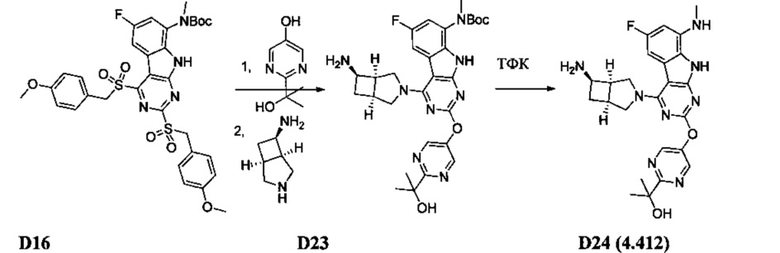
[0203] Получение D24 (4.412): Указанное в подзаголовке соединение синтезировали по способу, описанному выше, исходя из 2-(2-гидроксипропан-2-ил)пиримидин-5-ола и (6R)-3-азабицикло[3.2.0]гептан-6-амина.
ЖХ-МС: М+1: 479,25.
1Н ЯМР (500 МГц, ДМСО) δ (ppm): 11,35 (brm, 1Н), 8,82 (s, 2Н), 7,07 (d, J=9,7, 1Н), 6,31 (d, J=12,2, 1Н), 5,63 (m, 2Н), 5,11 (brs, 1Н), 4,67 (m, 1H), 3,96 (m, 1H), 3,33-3,53 (m, 6Н), 3,01 (m, 1Н), 2,85 (s, 3Н), 2,70 (m, 1Н), 2,51 (m, 1Н), 1,55 (s, 6Н).
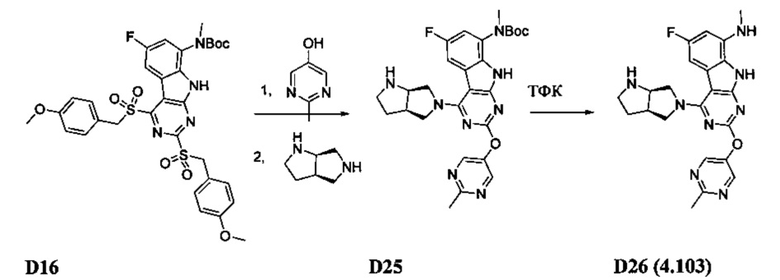
[0204] Получение D26 (4.103): Указанное в подзаголовке соединение синтезировали по способу, описанному выше, исходя из (3aR,6aR)-октагидропирроло[3,4-b]пиррола.
ЖХ-МС: М+1: 435,21.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 8,71 (s, 2Н), 6,96 (d, J=11,2, 1Н), 6,28 (d, J=11,9, 1Н), 5,56 (m, 1Н), 3,85 (m, 1Н), 3,73 (m, 1Н), 3,68 (d, J=11,2, 1Н), 3,60 (d, J=11,3, 1Н), 2,92 (m, 1Н), 2,83 (m, 4Н), 2,77 (m, 1H), 2,67 (s, 3Н), 1,85 (m, 1Н), 1,62 (m, Ш).
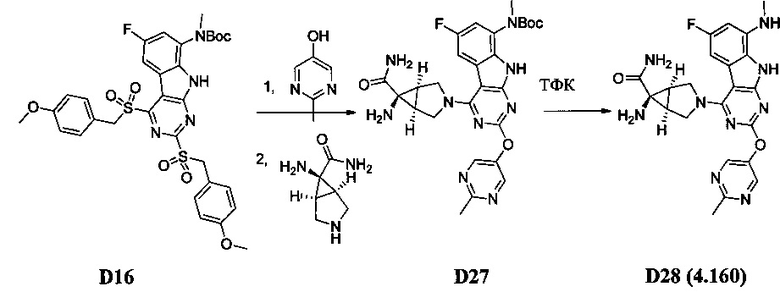
[0205] Получение D28 (4.160): Указанное в подзаголовке соединение синтезировали по способу, описанному выше, исходя из (1R,5S,6r)-6-амино-3-азабицикло[3.1.0]гексан-6-карбоксамида. ЖХ-МС: М+1: 435,24.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 11,05 (s, 1H), 8,72 (s, 2Н), 7,21 (s, 2Н), 7,01 (d, J=11,2, 1H), 6,11 (d, J=9,7, 1Н), 5,01 (s, 2Н), 4,03 (d, J=12,3, 1Н), 2,95 (s, 3Н), 2,81 (m, 2Н), 2,75 (m, 2Н), 2,67 (s, 3Н), 0,85 (br m, 2Н).
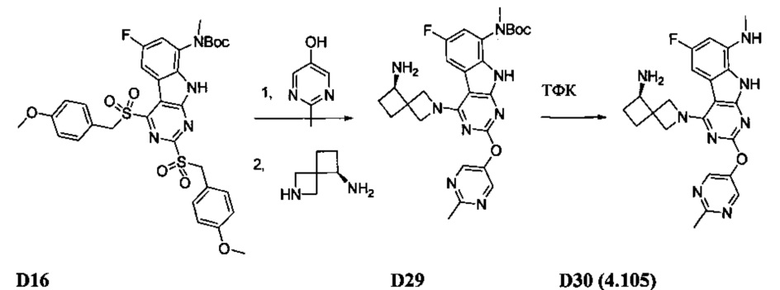
[0206] Указанное в подзаголовке соединение D30 синтезировали по способу, описанному для представленного выше соединения, исходя из бис-сульфона и (R)-2-азаспиро[3.3]гептан-5-амина (этот диамин получили хиральным колоночным разделением из имеющегося в продаже рацемата). ЖХ-МС: М+1: 435,21.
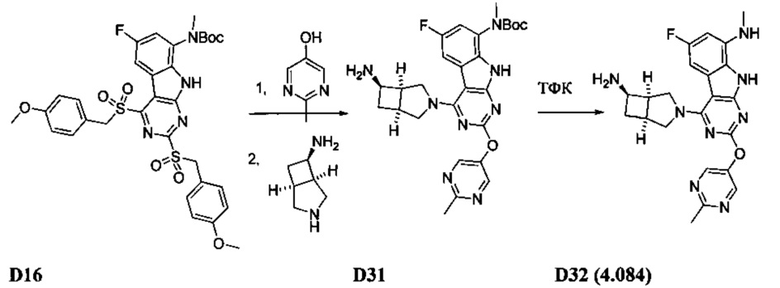
[0207] Указанное в подзаголовке соединение D32 синтезировали по способу, описанному для представленного выше соединения, исходя из бис-сульфона и (1S,5R,6R)-3-азабицикло[3.2.0]гептан-6-амина (этот диамин получили в соответствии с патентным способом Международной заявки PCT (1994), WO 9415933 A1 19940721 и выделили на хиральной колонке). ЖХ-МС: М+1: 435,21.
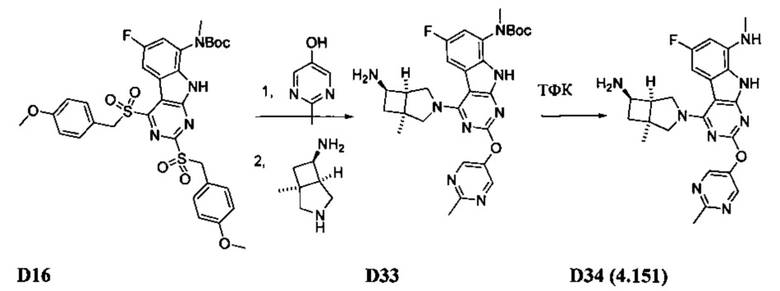
[0208] Указанное в подзаголовке соединение D34 синтезировали по способу, описанному для представленного выше соединения, исходя из бис-сульфона и (1S,5R,6R)-1-метил-3-азабицикло[3.2.0]гептан-6-амина (этот диамин получили в соответствии с патентным способом WO 2001053273 A1 и выделили на хиральной колонке). ЖХ-МС: М+1: 449,25.
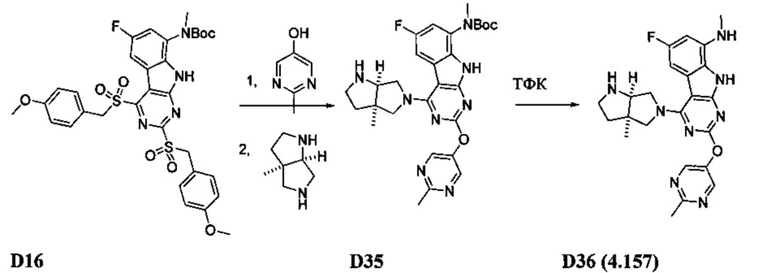
[0209] Указанное в подзаголовке соединение D36 синтезировали по способу, описанному для представленного выше соединения, исходя из бис-сульфона и (3aR,6aR)-3a-метилоктагидропирроло[3,4-b]пиррола (этот диамин получили в соответствии с патентным способом US 5202337 (A) и выделили на хиральной колонке). ЖХ-МС: М+1: 449,23.
Дихлор-способ
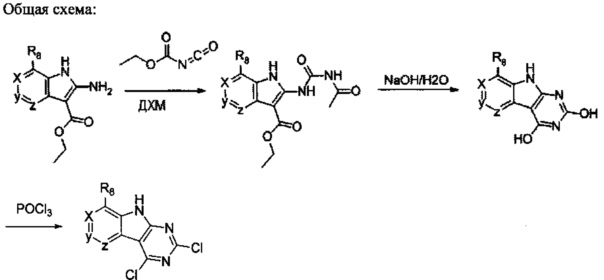
Экспериментальная часть:
Примеры соединений, полученных первоначальным присоединением R4, а затем R2
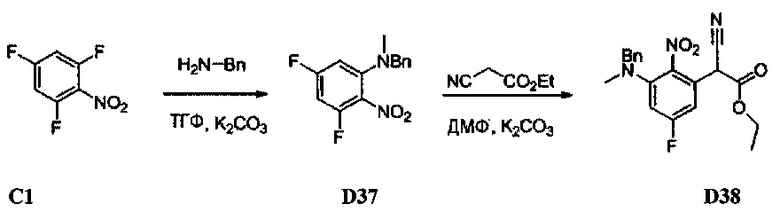
[0210] К перемешанной суспензии BnNHMe (34,2 г, 0,282 моль) и K2CO3 (50,6 г, 0,367 моль) в 400 мл ТГФ по каплям добавили раствор соединения 1 (50,0 г, 0,282 моль) в 100 мл ТГФ при температуре ниже 10°C. После добавления реакционную смесь медленно нагрели до комнатной температуры и перемешивали в течение ночи. Данные ТСХ показали, что реакция завершена; реакционную смесь концентрировали под вакуумом. Остаток разделили между этилацетатом (300 мл) и водой (500 мл), органический слой промыли насыщенным солевым раствором (300 мл × 3), высушили над Na2SO4, отфильтровали и концентрировали под вакуумом. Неочищенный продукт очистили флэш-хроматографией (петролейный эфир/EtOAc, от 100/1 до 50/1, об./об.) для получения продукта D37 в виде бледно-желтого твердого вещества (69,0 г, выход 87,9%). ЖХ-МС: М+1: 279
1Н-ЯМР (400 МГц, CDCl3) δ (ppm): = 7,37 (5Н, m), 6,43 (2Н, m), 4,40 (2Н, s), 2,84 (3Н, s).
[0211] К перемешанной суспензии K2CO3 (57,6 г, 0,417 моль) и этилцианоацетата (35,4 г, 0,313 моль) в 200 мл ДМФ добавили раствор соединения D37 (58,0 г, 0,208 моль) в 100 мл ДМФ под защитой N2. После добавления реакционную смесь перемешивали при комнатной температуре в течение двух дней. Данные ТСХ показали, что исходный материал израсходован, затем реакционную смесь разбавили этилацетатом (400 мл) и водой (1500 мл), органический слой отделили, водный слой экстрагировали этилацетатом (200 мл). Объединенный органический слой промыли насыщенным солевым раствором (300 мл × 3), высушили над Na2SO4, отфильтровали и концентрировали в вакууме. Неочищенный продукт очистили хроматографией (петролейный эфир/EtOAc, от 100/1 до 20/1, об./об.) для получения продукта Б38 в виде бледно-желтого твердого вещества (61,0 г, выход 79,2%). ЖХ-МС: М+1: 371
1Н-ЯМР (400 МГц, CDCl3) δ (ppm): 7,33 (5Н, m), 6,92 (1H, d, J=8 Гц), 6,84 (1Н, d, J=8 Гц), 5,13 (1Н, s), 4,37 (2Н, s), 4,30 (2Н, dd, J=14,4 Гц), 2,78 (3Н, s), 1,35 (3Н, t, J=7,2 Гц).
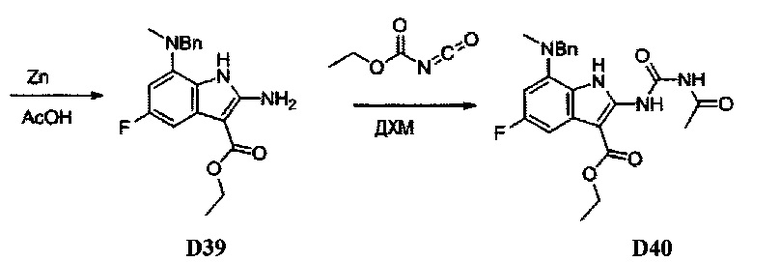
[0212] К перемешанному раствору соединения D38 (61,0 г, 0,164 моль) в 400 мл AcOH, охлажденному на ледяной бане, частями добавили цинковый порошок. После добавления реакционную смесь нагрели до 60°C и перемешивали при этой температуре в течение 5 часов. Данные ТСХ показали, что реакция завершена. Реакционную смесь охладили до комнатной температуры, отфильтровали, фильтрат концентрировали под вакуумом, остаток растворили в этилацетате (400 мл), подщелочили насыщенным водным раствором NaHCO3 (400 мл), затем отделили органический слой, промыли насыщенным солевым раствором (200 мл × 3), высушили над Na2SO4, отфильтровали и концентрировали in vacuo для получения темного маслянистого вещества, которое очистили хроматографией (петролейный эфир/ДХМ, от 5/1 до ДХМ, об./об.) для получения продукта D39 в виде бледно-желтого твердого вещества (26,0 г, выход 46,4%). ЖХ-МС: М+1: 342
1Н-ЯМР (400 МГц, CDCl3) δ (ppm): 8,02 (1Н, S), 7,33 (5Н, m), 6,52 (1H, d, J=2,4 Гц), 6,49 (1H, d, J=2,4 Гц), 5,73 (2Н, s), 4,35 (2Н, dd, J=15,2 Гц). 4,19 (2Н, s), 2,73 (3Н, s), 1,44 (3Н, t, J=7,2 Гц).
[0213] К перемешанной суспензии D39 (16,0 г, 46,9 ммоль) в 200 мл ДХМ по каплям добавили этил изоцианофаормиат (растворенный в 50 мл ДХМ) с охлаждением на ледяной бане. После добавления полученную смесь перемешивали при комнатной температуре. Исходный материал постепенно растворился, затем из реакционной смеси образовался осадок. Через 4 часа данные ТСХ показали, что реакция завершена. Реакционную смесь отфильтровали. Фильтрат концентрировали in vacuo. Остаток суспендировали в 50 мл ДХМ, перемешали, затем отфильтровали. Две партии осадков на фильтре объединили, высушили in vacuo для получения продукта D40 в виде бледно-желтого твердого вещества (14,4 г, выход 67,3%). ЖХ-МС: М+1: 457
1Н-ЯМР (400 МГц, ДМСО-d6) δ (ppm): 12,01 (1Н, S), 11,12 (1Н, S), 11,06 (1Н, S), 10,41 (1Н, S), 7,33 (5Н, m), 6,63 (1H, d, J=2,0 Гц), 6,60 (1H, d, J=2,4 Гц), 4,34 (2Н, dd, J=7,2 Гц), 4,28 (2Н, s), 4,24 (2Н, dd, J=7,2 Гц), 4,14 (2Н, dd, J=7,2 Гц), 2,75 (3Н, s), 1,37 (3Н, t, J=7,2 Гц) 1,27 (3Н, t, J=7,2 Гц), 1,22 (3Н, t, J=6,8 Гц).
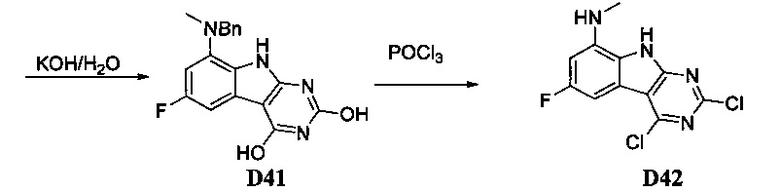
[0214] К перемешанной суспензии D40 (9,13 г, 20,0 ммоль) в воде/EtOH (75 мл/25 мл) добавили раствор KOH в 20 мл воды при комнатной температуре. После добавления полученную смесь нагревали с дефлегматором в течение 4 часов. Данные ТСХ показали, что реакция завершена, затем реакционную смесь охладили до комнатной температуры, подкислили 1 М раствором HCl до pH=5, осадок собрали фильтрацией, промыли водой (200 мл × 1), затем этилацетатом (200 мл × 1) для получения продукта D41 в виде бледно-желтого твердого вещества (5,90 г, выход 87,1%). ЖХ-МС: М-1: 337.
1Н-ЯМР (400 МГц, ДМСО-d6) δ (ppm): 7,25 (5Н, m), 7,01 (1Н, dd, J=8,8 Гц), 6,35 (1H, d, J=12,0 Гц), 4,45 (2Н, s), 2,76 (3Н, s).
[0215] Соединение D41 (2 г, 5,75 ммоль) поместили с раствором POCl3 (100 мл) в пробирку для работы под давлением и добавили несколько капель N-этилдиизопропиламина. Реакционную смесь нагревали при 185°C в закрытом состоянии в течение 10 часов. Смесь охладили и вылили в ледяную воду, а твердое желтое вещество собрали фильтрацией, высушили под пониженным давлением для получения D42 (1,6 г, выход 98%) в виде твердого желтого вещества. ЖХ-МС: М+1: 286,02
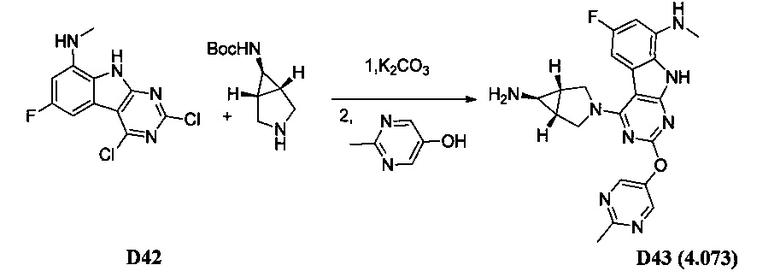
[0216] К перемешанному раствору соединения D42 (250 мг, 0,87 ммоль) в 5 мл NMP при 110°C добавили (R)-трет-бутил 5-азаспиро[2.4]гептан-7-илкарбамат (175 мг, 0,88 ммоль) и K2CO3 (7 мг, 0,05 ммоль). После завершения реакции через 10 минут, реакционную смесь добавили к раствору 2-метилпиримидин-5-ола (90 мг, 0,90 ммоль) в пробирке для микроволновых реакций. Реакционную смесь закрыли и поместили в микроволновый реактор при 220°C на 10 минут. Заданный продукт получили очисткой ВЭЖХ с образованием D43 (90 мг, 25%) в виде твердого белого вещества. ЖХ-МС: М+1: 421,18.
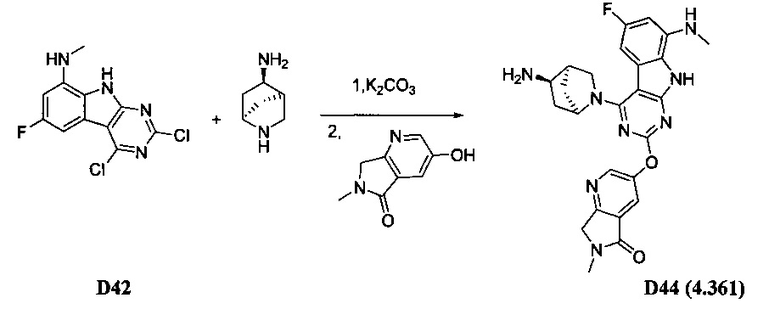
[0217] Указанное в подзаголовке соединение D44 синтезировали по способу, описанному выше, исходя из (1R,4R,5R)-2-азабицикло[2.2.1]гептан-5-амина и 3-гидрокси-6-метил-6,7-дигидро-5Н-пирроло[3,4-b]пиридин-5-она. ЖХ-МС: М+1: 489,22.
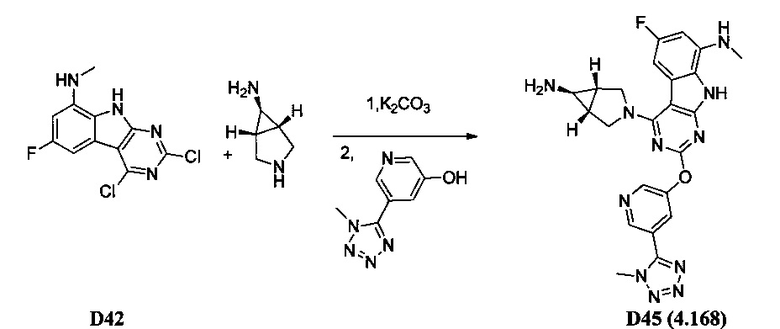
[0218] Указанное в подзаголовке соединение D45 синтезировали по способу, описанному выше, исходя из трет-бутил 3-азабицикло[3.1.0]гексан-6-илкарбамата и 5-(1-метил-1Н-тетразол-5-ил)пиридин-3-ола. ЖХ-МС: М+1: 488,20.
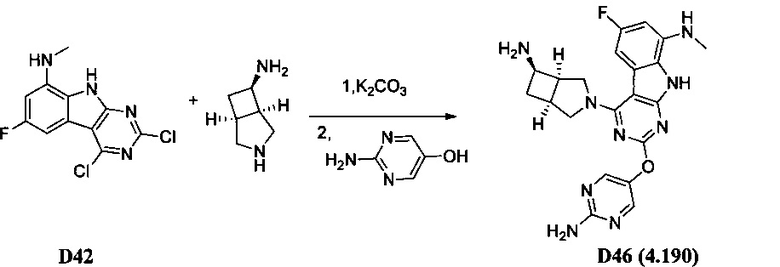
[0219] Указанное в подзаголовке соединение D46 синтезировали по способу, описанному выше, исходя из (6R)-3-азабицикло[3.2.0]гептан-6-амина и 2-аминопиримидин-5-ола. ЖХ-МС: М+1: 436,20.
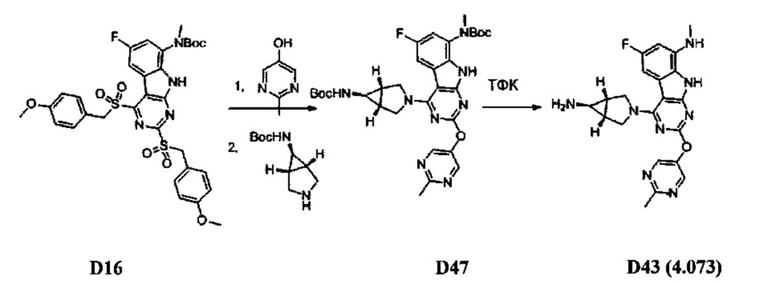
[0220] Указанное в подзаголовке соединение D43 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона и трет-бутил 3-азабицикло[3.1.0]гексан-6-илкарбамата. ЖХ-МС: М+1: 421,18.
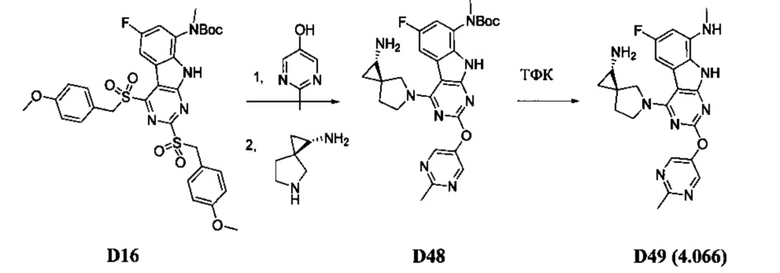
[0221] Указанное в подзаголовке соединение D49 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона и (1R)-5-азаспиро[2.4]гептан-1-амина. ЖХ-МС: М+1: 435,23.
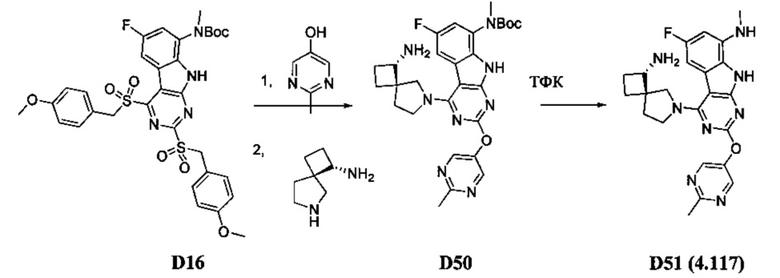
[0222] Указанное в подзаголовке соединение D51 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона и (1S,4R)-6-азаспиро[3.4]октан-1-амина. ЖХ-МС: М+1: 449,25.
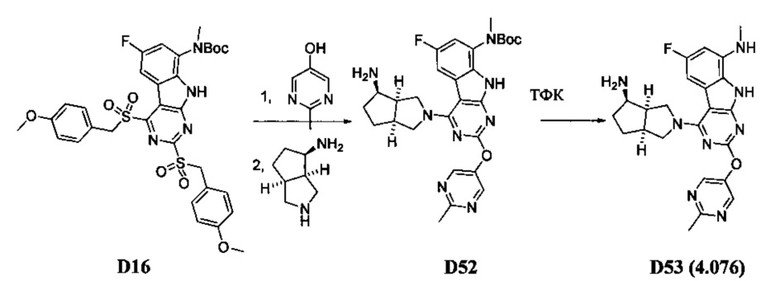
[0223] Указанное в подзаголовке соединение D53 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона и (3aR,4R,6aS)-октагидроциклопента[с]пиррол-4-амина. ЖХ-МС: М+1: 449,21.
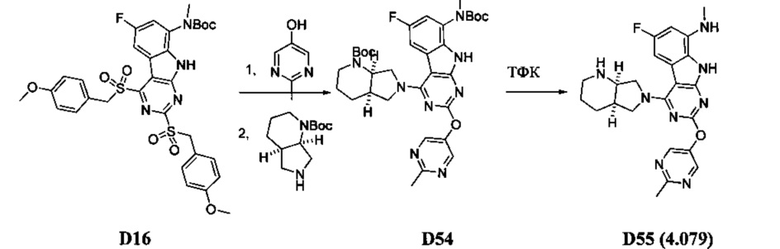
[0224] Указанное в подзаголовке соединение D55 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона и (4aR,7aR)-трет-бутил октагидро-1Н-пирроло[3,4-b]пиридин-1-карбоксилата. ЖХ-МС: М+1: 449,23.
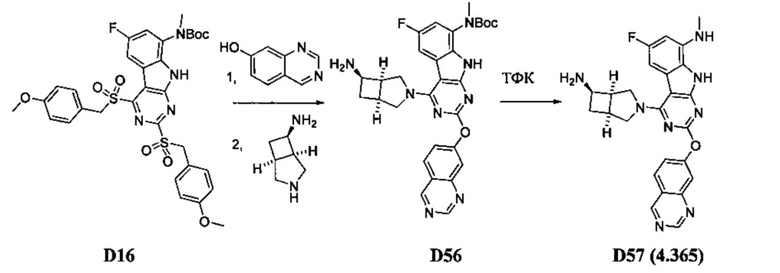
[0225] Указанное в подзаголовке соединение D57 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона, хиназолин-7-ола и (1S,5R,6R)-3-азабицикло[3.2.0]гептан-6-амина. ЖХ-МС: М+1: 471,26.
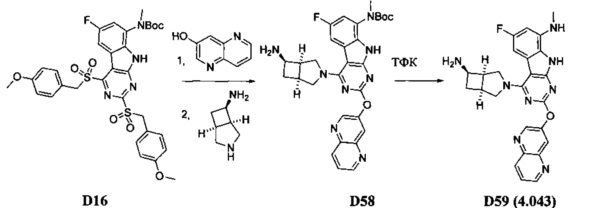
[0226] Указанное в подзаголовке соединение D59 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона, 1,5-нафтиридин-3-ола и (1S,5R,6R)-3-азабицикло[3.2.0]гептан-6-амина. ЖХ-МС: М+1: 471,20.
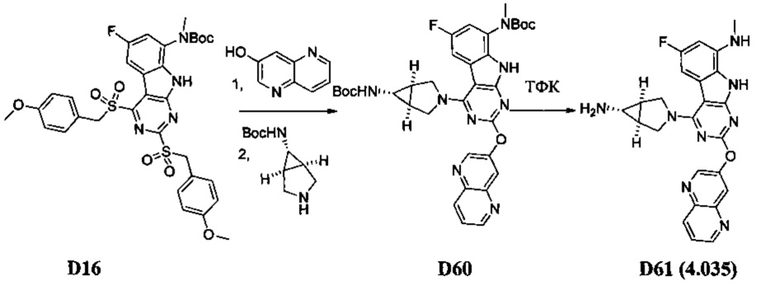
[0227] Указанное в подзаголовке соединение D61 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона, 1,5-нафтиридин-3-ола и трет-бутил 3-азабицикло[3.1.0]гексан-6-илкарбамата. ЖХ-МС: М+1: 457,20.
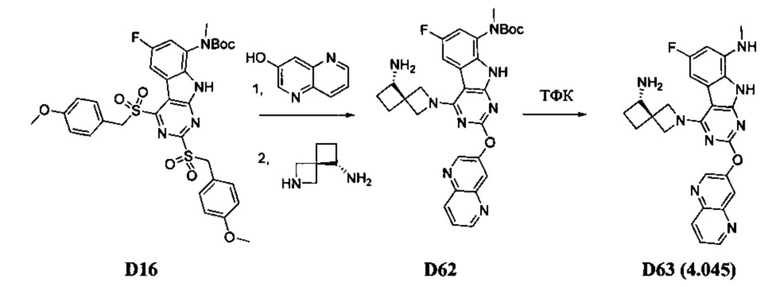
[0228] Указанное в подзаголовке соединение D63 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона, 1,5-нафтиридин-3-ола и (S)-2-азаспиро[3.3]гептан-5-амина. ЖХ-МС: М+1: 471,22.
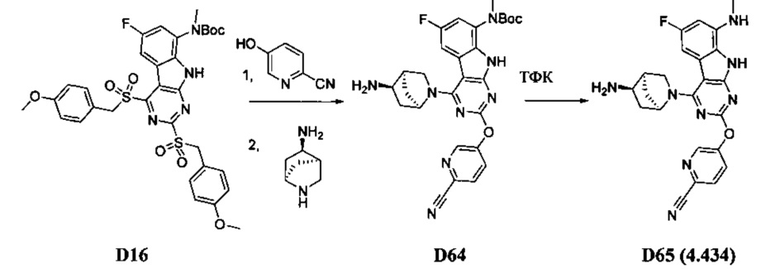
[0229] Указанное в подзаголовке соединение D65 синтезировали по такому же способу, как описан для представленного выше соединения, исходя из бис-сульфона, 5-гидроксипиколинонитрила и (1R,4R,5R)-2-азабицикло[2.2.1]гептан-5-амина. ЖХ-МС: М+1: 445,18.
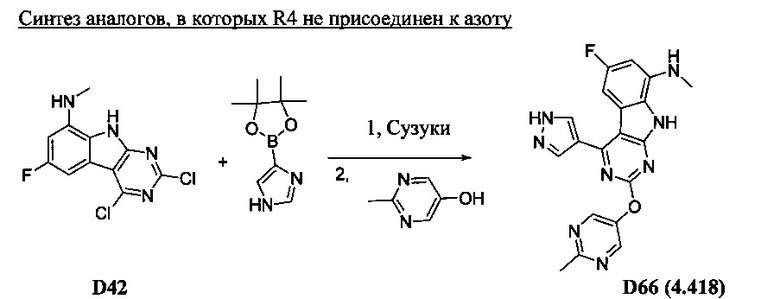
[0230] 2-хлор-6-фтор-4-(1Н-имидазол-4-ил)-N-метил-9H-пиримидо[4,5-b]индол-8-амин: Смесь соединения (1) (150 мг, 0,52 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-имидазола (2) (100 мг, 0,52 ммоль), K2CO3 (100 мг, 0,5 ммоль) и каталитического количества Pd[(PPh3)]Cl2 растворили в ДМФ (3 мл) и воде (0,3 мл). Смесь нагревали при 150°C под микроволновым излучением в течение 10 минут. Затем смесь очистили при помощи ВЭЖХ для получения указанного в заголовке соединения в виде твердого желтого вещества (91 мг; выход 55%). ЖХ-МС: М+1: 317,08.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 14,01 (S, 1H), 11,71 (s, 1H), 7,98 (s, 2Н), 7,51 (d, J=11,2, 1Н), 6,30 (d, J=9,7, 1H), 4,12 (s, 1H), 3,15 (s, 3Н).
[0231] 6-фтор-4-(1Н-имидазол-4-ил)-N-метил-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-амин D66: К раствору соединения (3) (80 мг, 2,52 ммоль) в NMP (5 мл) добавили 2-метилпиримидин-5-ол (33 мг, 3,0 ммоль) и карбонат калия (43,6 мг, 0,31 ммоль). Затем смесь нагревали при 160°C в микроволновых условиях в течение 15 минут. Затем смесь очистили при помощи ВЭЖХ для получения указанного в заголовке соединения в виде твердого желтого вещества (59 мг, 60%). ЖХ-МС: М+1: 391,15.
1Н ЯМР (300 МГц, ДМСО) δ (ppm): 14,01 (S, 1Н), 11,71 (s, 1Н), 7,98 (s, 2Н), 7,69 (s, 2Н), 7,51 (d, J=11,2, 1Н), 5,98 (d, J=9,7, 1H), 4,02 (s, 1Н), 3,10 (s, 3Н), 2,65 (s, 3Н).
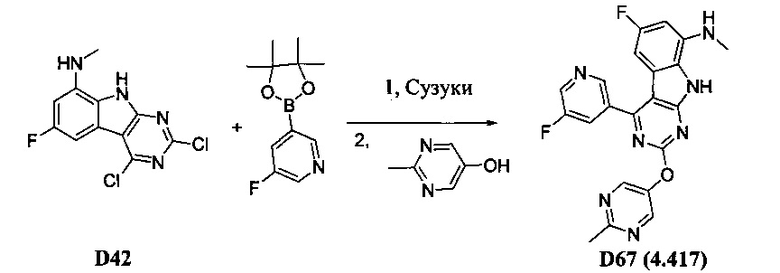
[0232] Указанное в подзаголовке соединение D67 синтезировали по способу, описанному выше, исходя из 3-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина. ЖХ-МС: М+1: 420,16.
1H ЯМР (300 МГц, ДМСО) δ (ppm): 11,71 (s, 1Н), 9,10 (s, 1Н), 8,52 (d, 1Н), 7,63-7,80 (m, 3Н), 7,31 (brs, 1Н), 5,98 (d, J=9,7, 1Н), 4,10 (s, 1H), 2,98 (s, 3Н), 2,66 (s, 3Н).
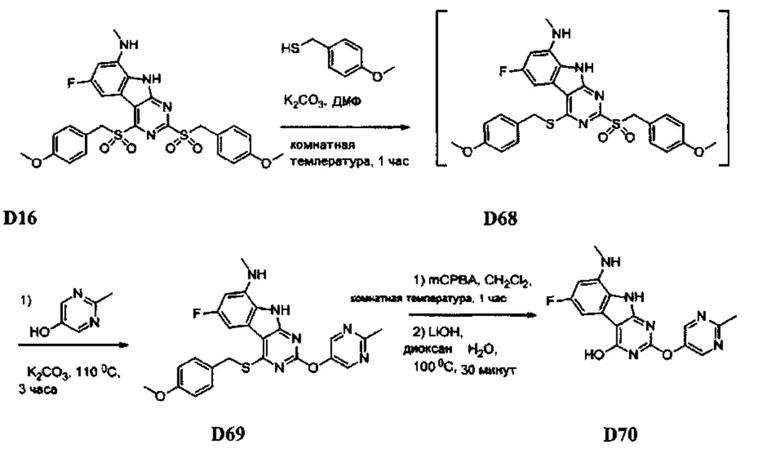
[0233] 6-Фтор-4-(4-метоксибензилтио)-N-метил-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-амин (D69): К раствору соединения (1) (2,923 г, 5 ммоль) в NMP (12 мл) добавили карбонат калия (2,073 г, 15 ммоль), затем 4-метоксифенил)метантиол (0,771 г, 5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа. Затем добавили 2-метилпиримидин-5-ол (1,101 г, 10 ммоль). Полученную смесь нагревали при 100°C в течение 3 часов. Смесь очистили С18 колоночной хроматографией для получения указанного в заголовке соединения в виде светло-желтого твердого вещества (2,4 г, 83%).
[0234] 6-Фтор-8-(метиламино)-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-4-ол (D70): К раствору соединения (3) (2,48 г, 4,3 ммоль) в диоксане (12 мл) частями добавили 3-хлорпероксибензойную кислоту (1,484 г, 8,6 ммоль) за 10 минут. После перемешивания реакционной смеси при комнатной температуре в течение 30 минут, добавили гидроксид лития (1,8 г, 75 ммоль) и воду (5 мл). Полученный раствор перемешивали при температуре от комнатной до 100°C в течение одного часа. Затем смесь очистили С18 колоночной хроматографией для получения указанного в заголовке соединения в виде белого твердого вещества (1,39 г, 95%).
[0235] 4-Хлор-6-фтор-N-метил-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-амин (D71): Соединение (D70) (1,06 г, 2,407 ммоль) растворили в POCl3 (20 мл) и N-этил-изопропилпропан-2-амине (0,43 г, 3,33 ммоль). Смесь нагревали при 50°C в течение 4 часов. После охлаждения реакционной смеси до комнатной температуры, ее вылили в 1 л колбу, содержащую лед (~500 г) и NaOH (20 г), и полученную смесь выдерживали в течение одного часа. Затем смесь экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои высушили над Na2SO4 и концентрировали на ротационном испарителе для получения указанного в заголовке соединения в виде белого твердого вещества (492 мг, 57%).
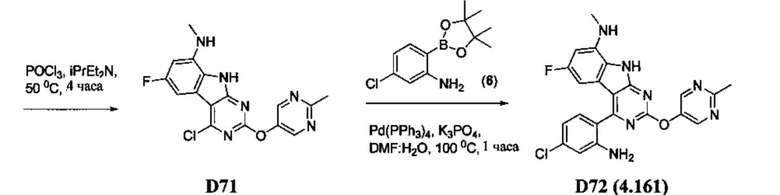
[0236] 4-(2-амино-4-хлорфенил)-6-фтор-N-метил-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-амин (D72): Смесь соединения (D71) (36 мг, 0,1 ммоль), пинаколового эфира бороновой кислоты (6) (38 мг, 0,15 ммоль), фосфата калия (64 мг, 0,3 ммоль) и каталитического количества Pd(PPh3)4 растворили в ДМФ (1 мл) и воде (0,3 мл). Реакционную смесь нагревали с дефлегматором при 100°C в течение одного часа. Затем смесь очистили при помощи ВЭЖХ для получения указанного в заголовке соединения в виде твердого желтого вещества (17 мг, 37,8%).
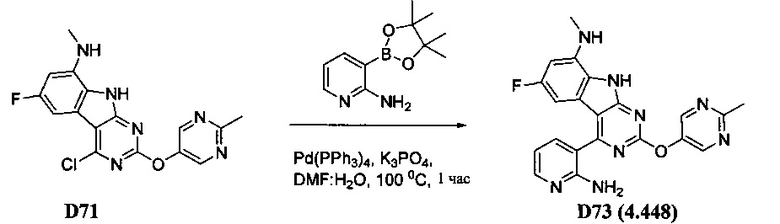
[0237] Указанное в подзаголовке соединение D73 синтезировали по способу, описанному выше, исходя из 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина.
Синтез пролекарств у R4
(S)-2-Амино-N-((R)-5-(6-фтор-8-(метиламино)-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-4-ил)-5-азаспиро[2.4]гептан-7-ил)пропанамид D76 (4.424)
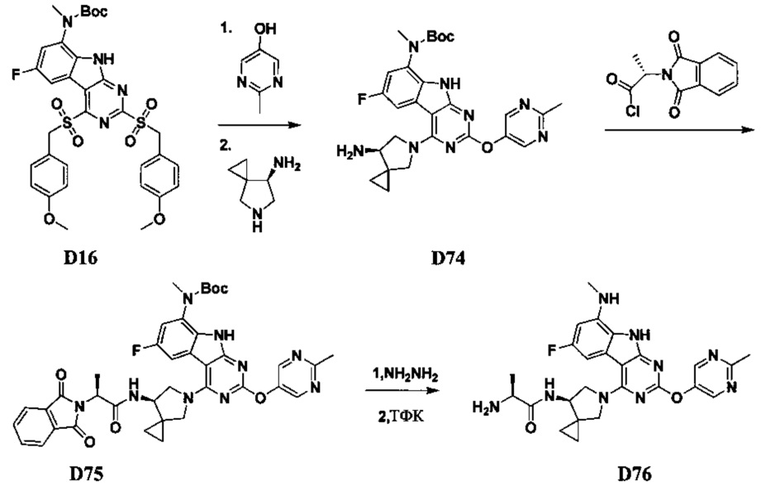
[0238] Смесь D16 (0,342 г, 0,500 ммоль), 2-метилпиримидин-5-ола (0,165 г, 1,50 ммоль) и K2CO3 (0,276 г, 2,00 ммоль) в NMP (5,0 мл) перемешивали в течение 1 часа 30 минут при 100°C. После перемешивания в течение 1 часа 30 минут, реакцию проверили по ЖХ/МС. Сразу добавили (R)-5-азаспиро[2.4]гептен-7-амин (0,168 г, 1,50 ммоль), смесь оставили перемешиваться в течение 1 часа 30 минут при 100°C. Полученную гетерогенную смесь охладили до 23°C и очистили ВЭЖХ для получения D74 (0,100 г, 0,187 ммоль) в виде светло-желтого твердого вещества. ЖХ/МС (ESI, М+H+)=535. К раствору D74 (0,100 г, 0,187 ммоль) и K2CO3 (0,052 г, 0,374 ммоль) в CH2Cl2 (8,0 мл) добавили (S)-2-(1,3-диоксоизоиндолин-2-ил)пропаноил хлорид (0,089 г, 0,374 ммоль), растворенный в CH2Cl2 (2,0 мл) при 23°C. Смесь оставили перемешиваться в течение 1 часа 30 минут при 60°C, затем охладили до 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения D75 в виде твердого желтого вещества. ЖХ/МС (ESI, М+Н+)=736. К раствору D75 в этаноле (7,0 мл) через шприц добавили гидразин (1,5 мл, 30 вес. % раствор в воде) при 23°C. Смесь перемешивали в течение 1 часа при 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения D76 в виде твердого светло-желтого вещества. ЖХ/МС (Е81, М+Н+)=606. Смесь D76 в трифторуксусной кислоте (1,00 мл) перемешивали в течение 1 часа при 23°C. Неочищенный материал очистили ВЭЖХ для получения указанного в заголовке соединения D76 (0,026 г, 0,051 ммоль) в виде белого твердого вещества. ЖХ/МС (ESI, М+Н+)=506.
Синтез пролекарств у R8
(S)-2-Амино-N-(4-((R)-7-амино-5-азаспиро[2.4]гептан-5-ил)-6-фтор-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-ил)-N-метилпропанамид
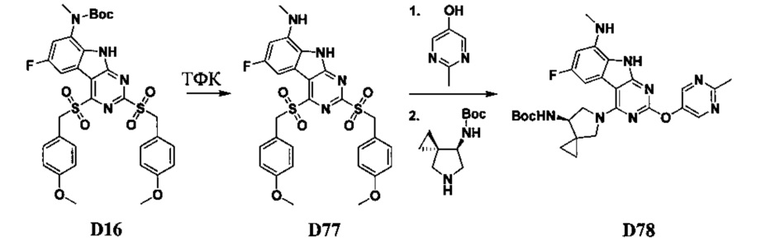
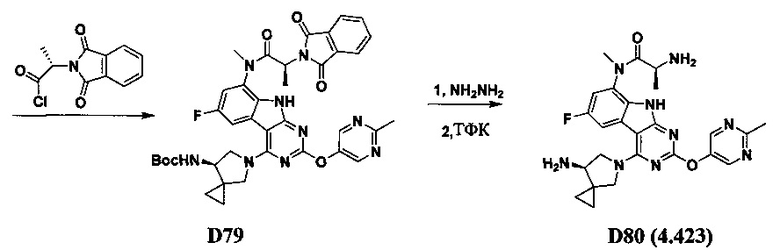
[0239] Смесь D16 (1,00 г, 1,46 ммоль) в трифторуксусной кислоте (3,0 мл) перемешивали в течение 30 минут при 23°C. Трифторуксусную кислоту выпарили под пониженным давлением для получения D77 (количественный выход) в виде темно-оранжевого твердого вещества. Этот неочищенный материал использовали для следующей реакции без дополнительной очистки. ЖХ/МС (ESI, М+Н+)=585. Смесь D77 (0,292 г, 0,50 ммоль), 2-метилпиримидин-5-ола (0,165 г, 1,50 ммоль) и K2CO3 (0,276 г, 2,00 ммоль) в NMP (5,0 мл) перемешивали в течение 2 часов при 100°C. После перемешивания в течение 2 часов, реакцию проверили по ЖХ/МС. Сразу добавили (R)-трет-бутил 5-азаспиро[2.4]гептен-7-илкарбамат (0,318 г, 1,50 ммоль), смесь оставили перемешиваться в течение 1 часа 30 минут при 100°C. Полученную гетерогенную смесь охладили до 23°C и очистили ВЭЖХ для получения D78 (0,182 г, 0,34 ммоль) в виде желтого твердого вещества. ЖХ/МС (ESI, М+Н+)=535. К раствору D78 (0,182 г, 0,34 ммоль) и K2CO3 (0,094 г, 0,68 ммоль) в CH2Cl2 (10,0 мл) добавили (S)-2-(1,3-диоксоизоиндолин-2-ил)пропаноил хлорид (0,161 г, 0,68 ммоль), растворенный в CH2Cl2 (2,0 мл) при 23°C. Смесь оставили перемешиваться в течение 2 часов при 60°C, а затем охладили до 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения D79 в виде твердого желтого вещества. ЖХ/МС (ESI, М+Н+)=736. К раствору D79 в этаноле (7,0 мл) через шприц добавили гидразин (1,5 мл, 30 вес. % раствор в воде) при 23°C. Смесь перемешивали в течение 1 часа при 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения 5 в виде твердого светло-желтого вещества. ЖХ/МС (ESI, М+Н+)=606. Смесь 5 в трифторуксусной кислоте (1,50 мл) перемешивали в течение 30 минут при 23°C. Неочищенный материал очистили ВЭЖХ для получения указанного в заголовке соединения D80 (0,031 г, 0,061 ммоль) в виде белого твердого вещества. ЖХ/МС (ESI, М+Н+)=506.
Пролекарство у R4 и R8:
(R)-2-Амино-N-(4-(7-(2-аминоацетамидо)-5-азаспиро[2.4]гептан-5-ил)-6-фтор-2-(2-метилпиримидин-5-илокси)-9H-пиримидо[4,5-b]индол-8-ил)-N-метилацетамид
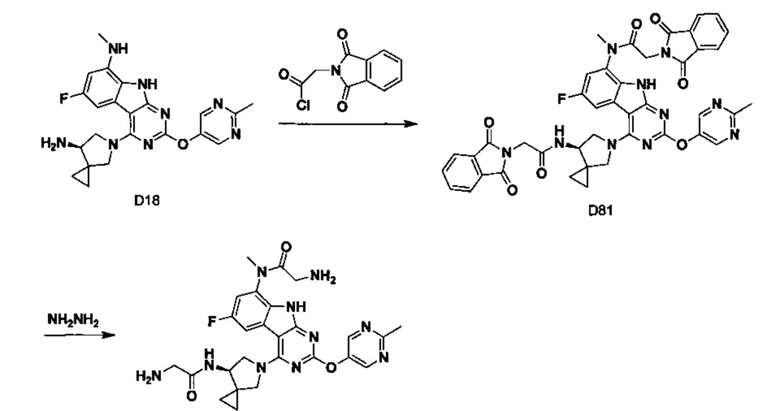
[0240] К раствору D16 (0,075 г, 0,173 ммоль) и K2CO3 (0,084 г, 0,606 ммоль) в CH2Cl2 (8,0 мл) добавили 2-(1,3-диоксоизоиндолин-2-ил)ацетилхлорид (0,136 г, 0,606 ммоль), растворенный в CH2Cl2 (2,0 мл) при 23°C. Смесь оставили перемешиваться в течение 3 часа 30 минут при 60°C, затем охладили до 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения D81 в виде твердого светло-желтого вещества. ЖХ/МС (ESI, М+Н+)=809. К раствору D81 в этаноле (5,0 мл) через шприц добавили гидразин (1,0 мл, 30 вес. % раствор в воде) при 23°C. Смесь перемешивали в течение 1 часа при 23°C. Реакционную смесь концентрировали на ротационном испарителе, а неочищенный материал очистили ВЭЖХ для получения указанного в заголовке соединения D82 (0,084 г, 0,153 ммоль) виде твердого белого вещества. ЖХ/МС (ESI, М+Н+)=549.
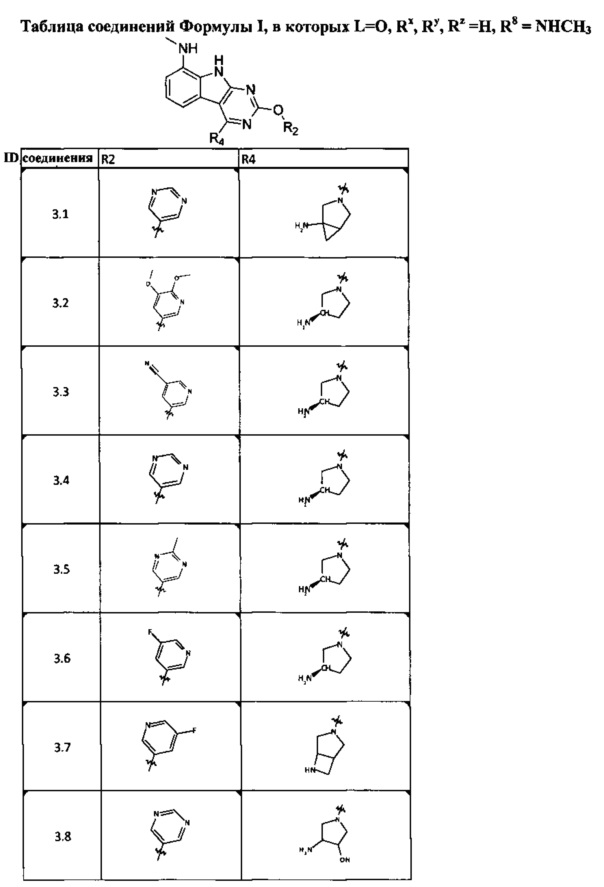
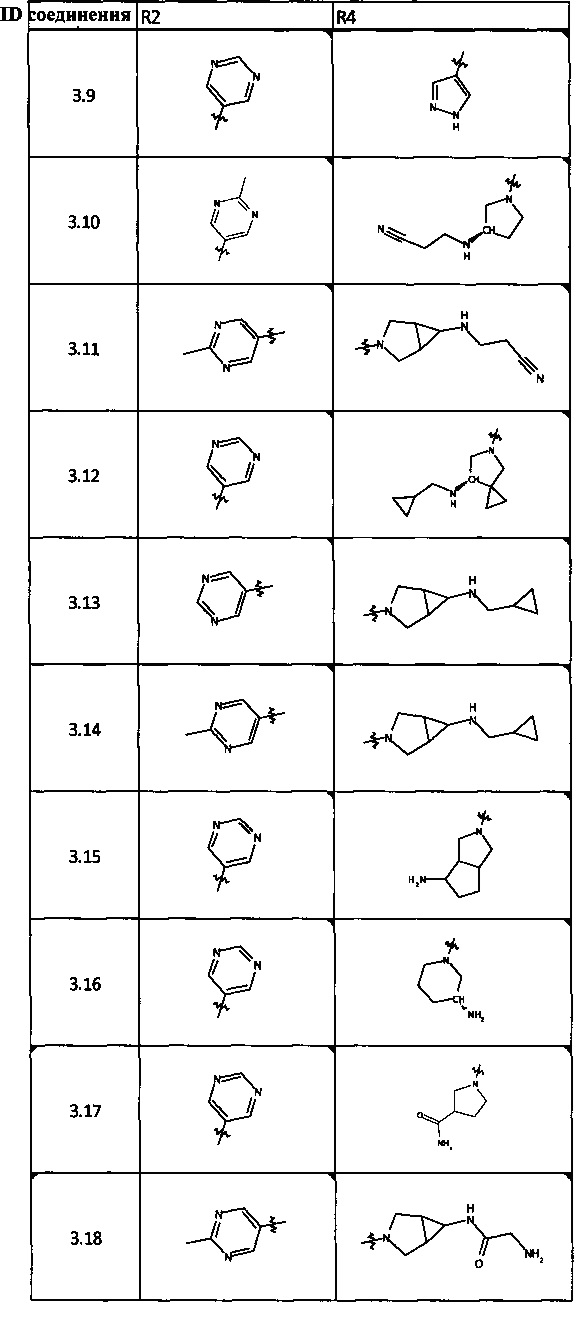
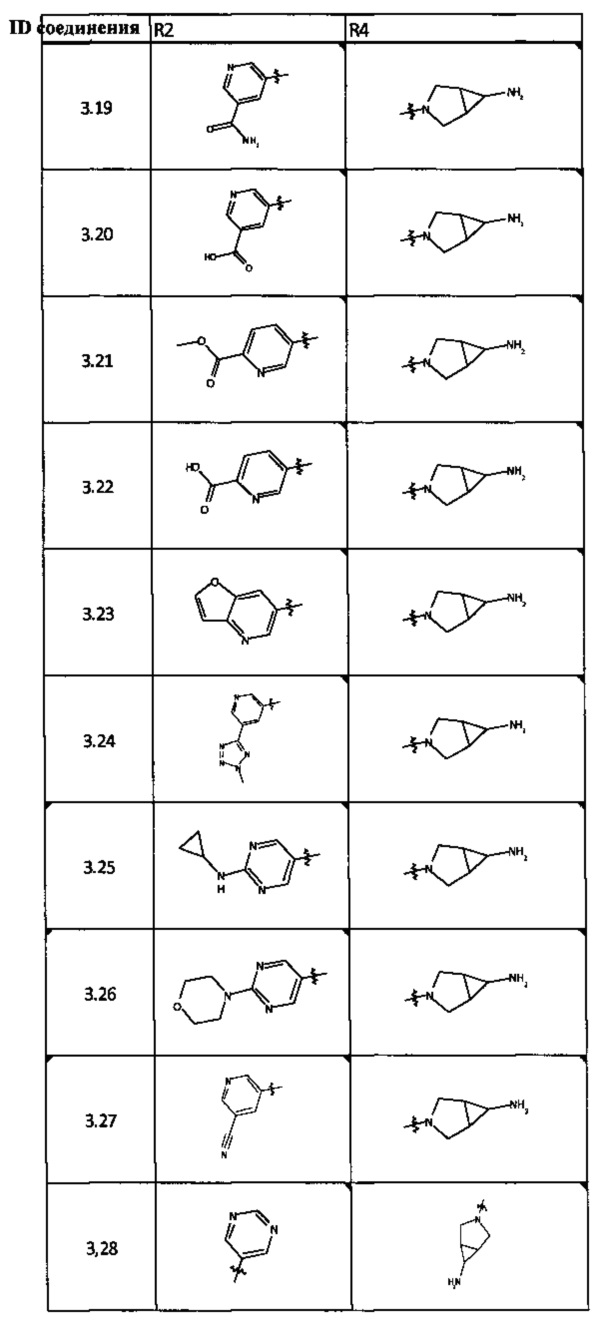
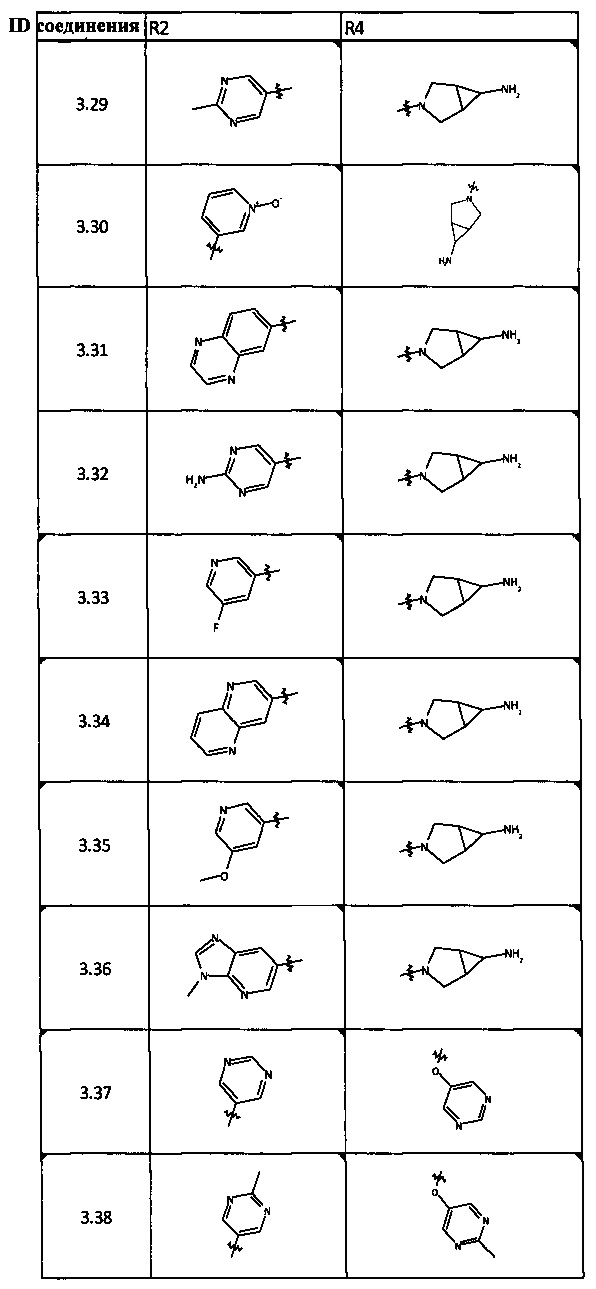
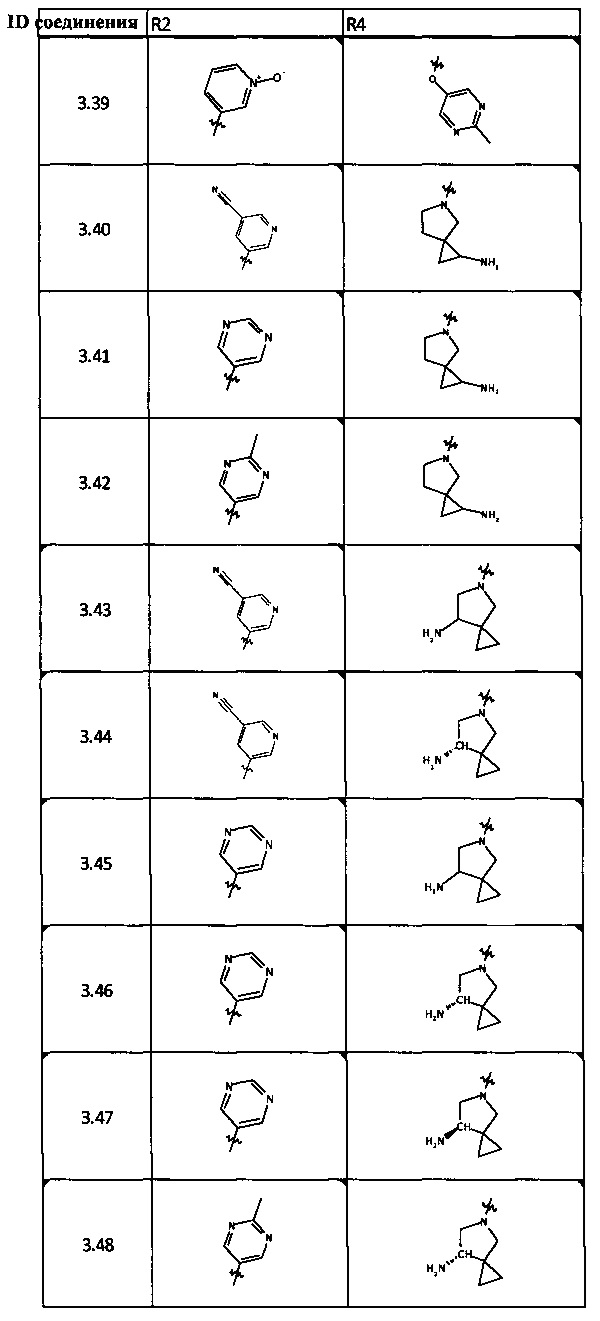
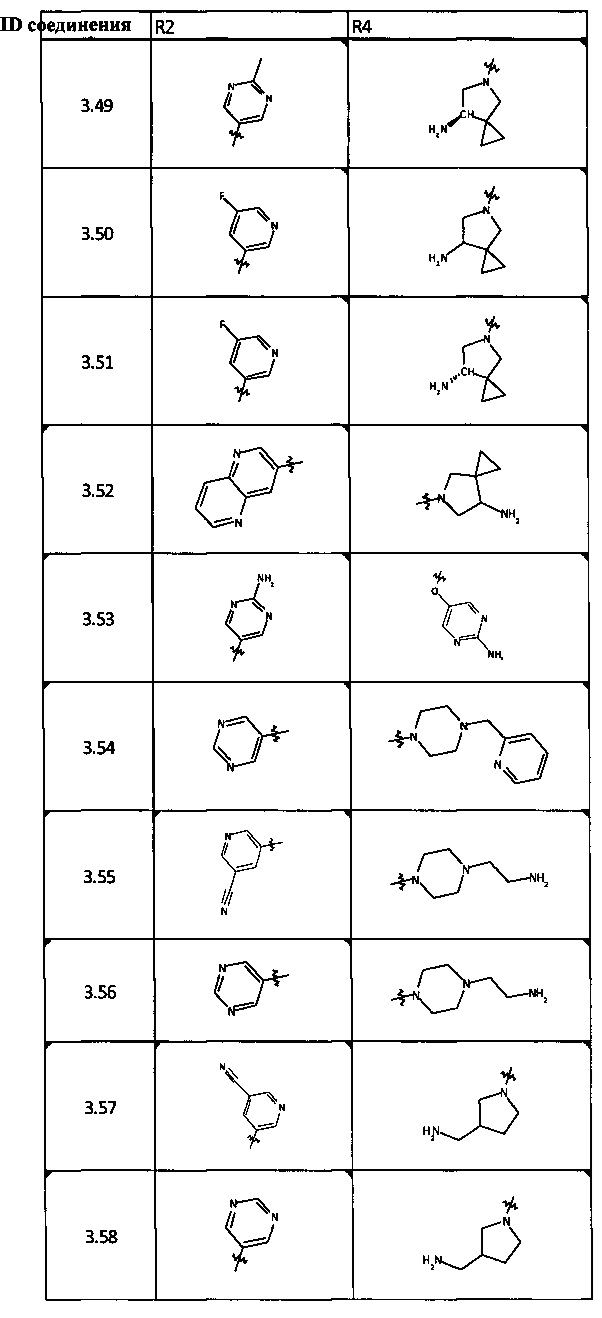
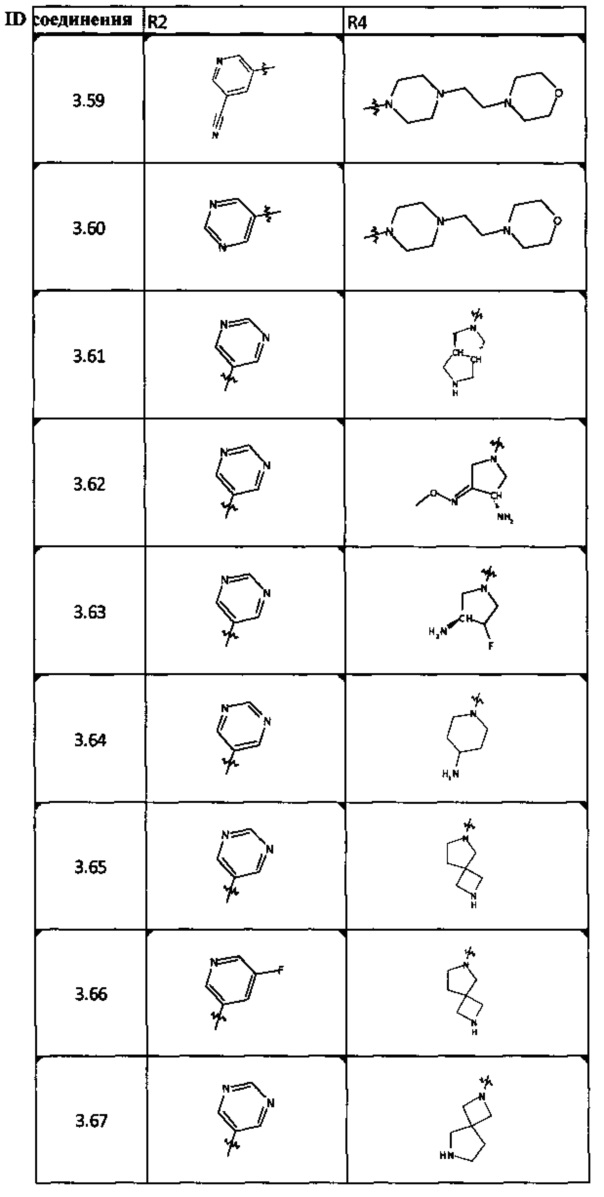
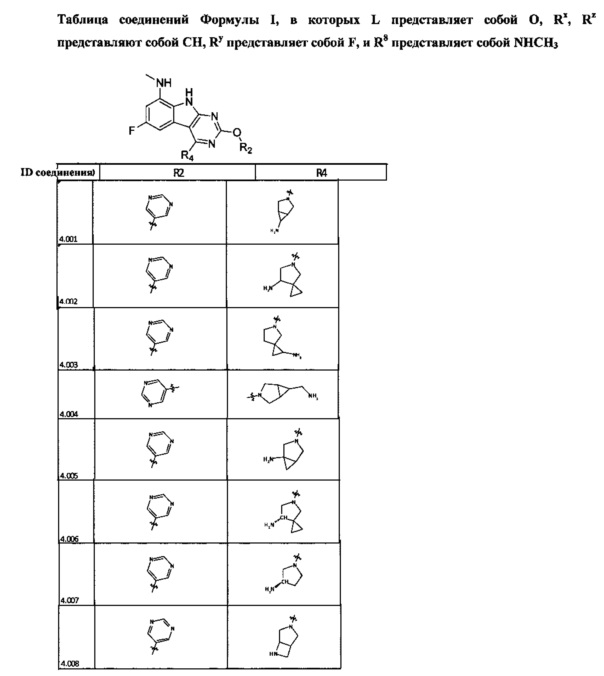
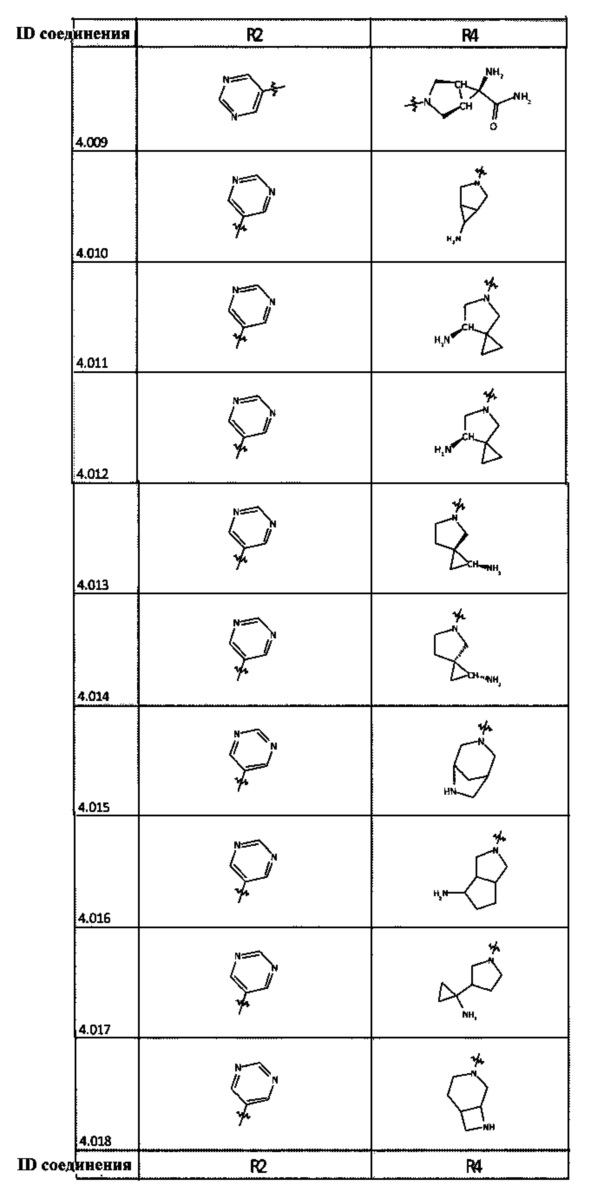
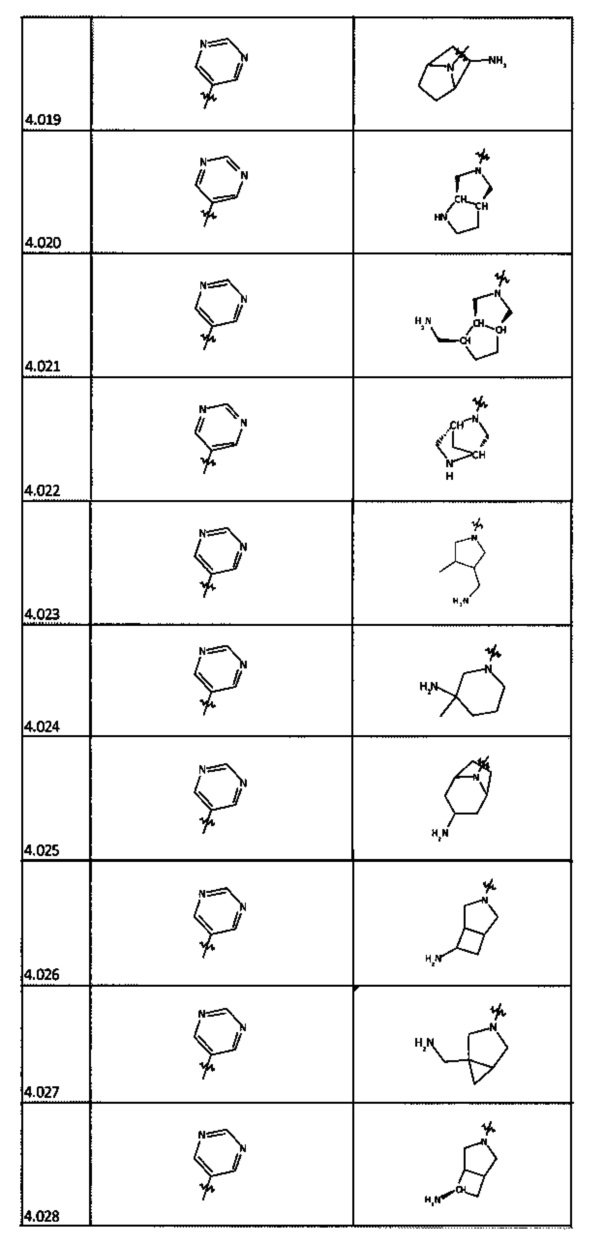
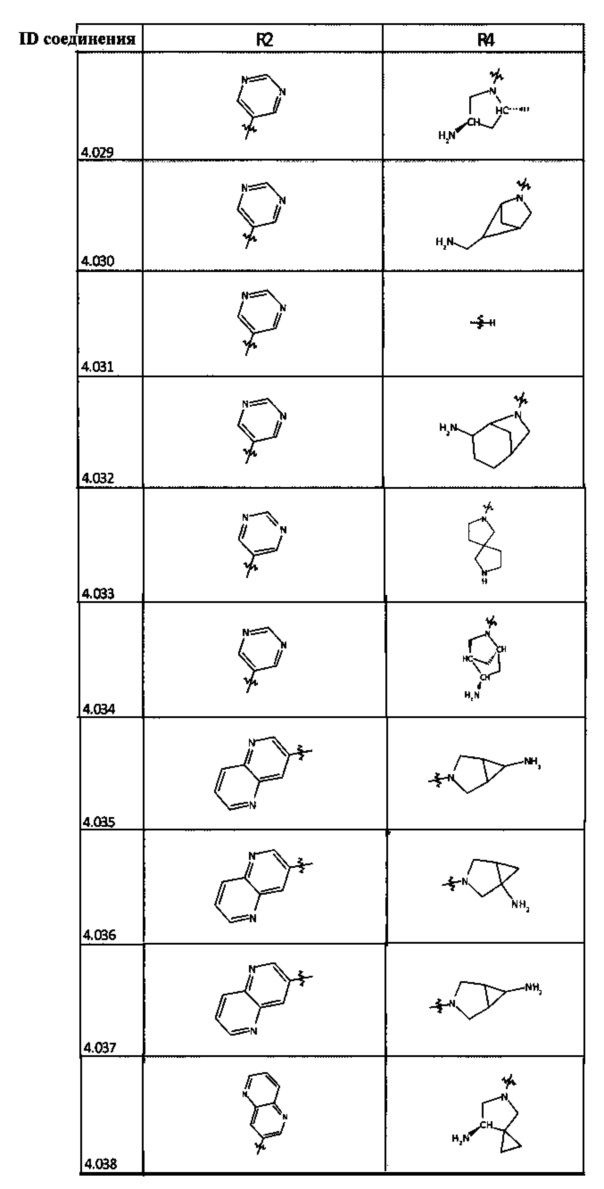
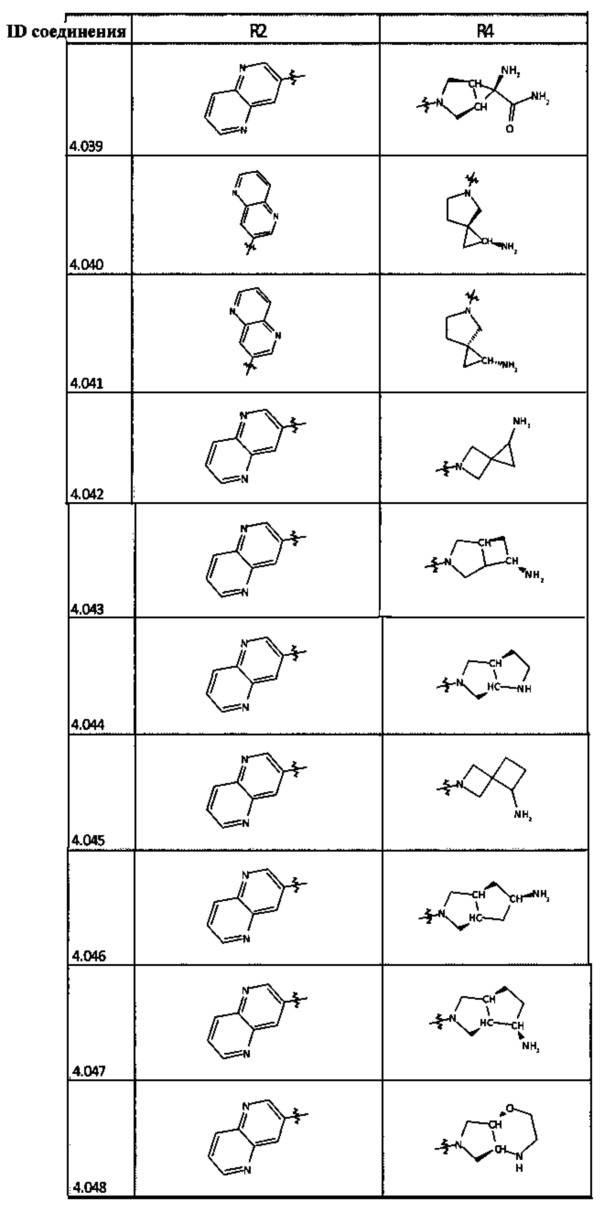
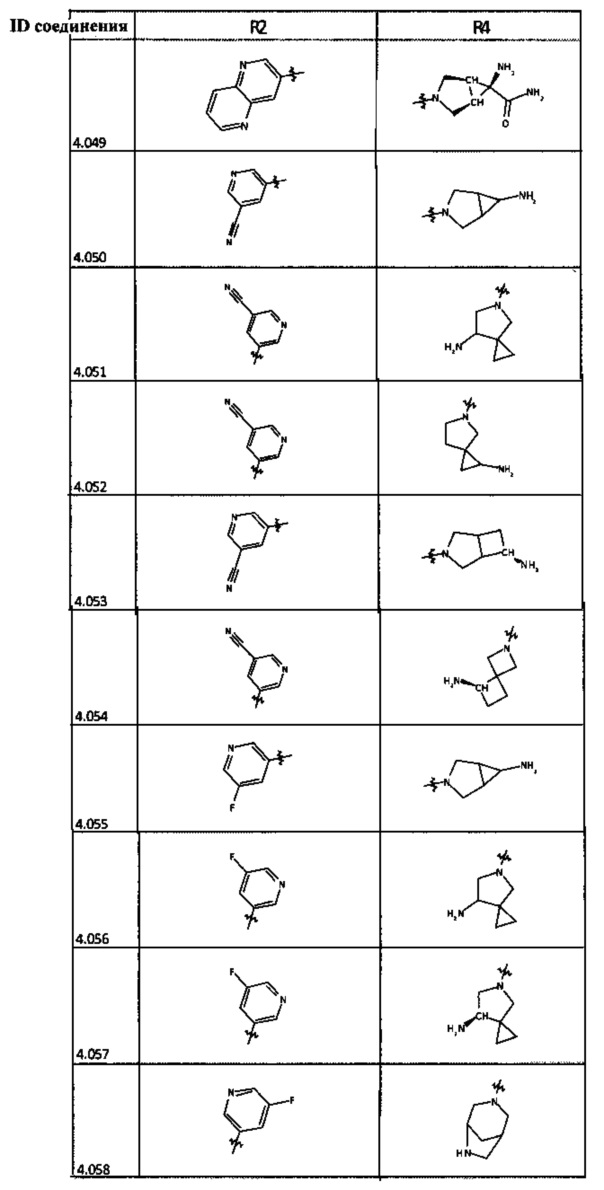
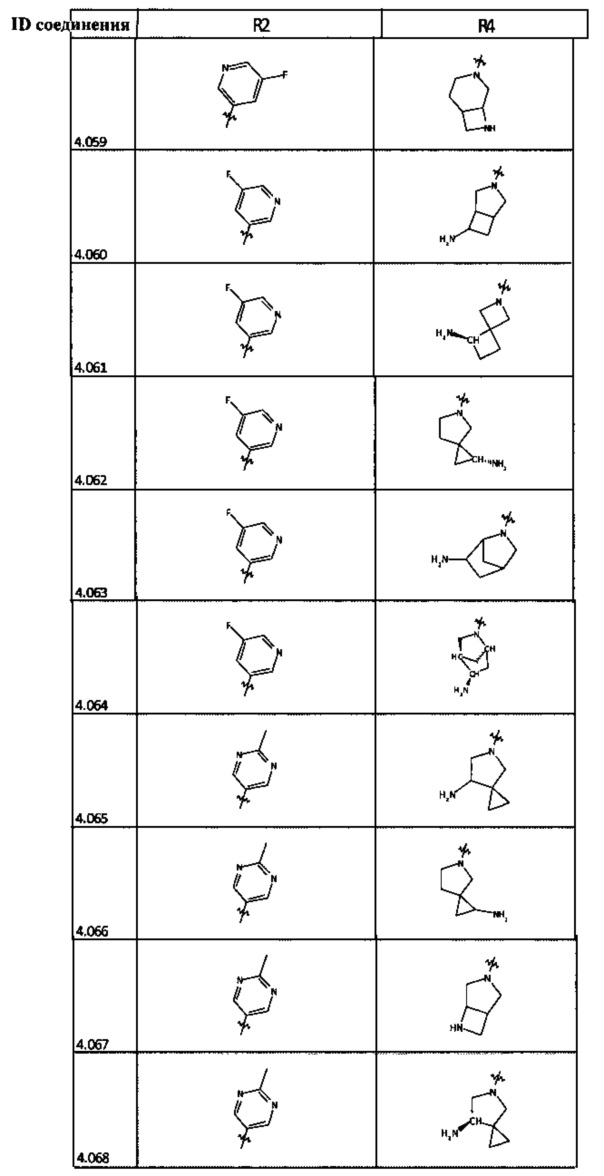
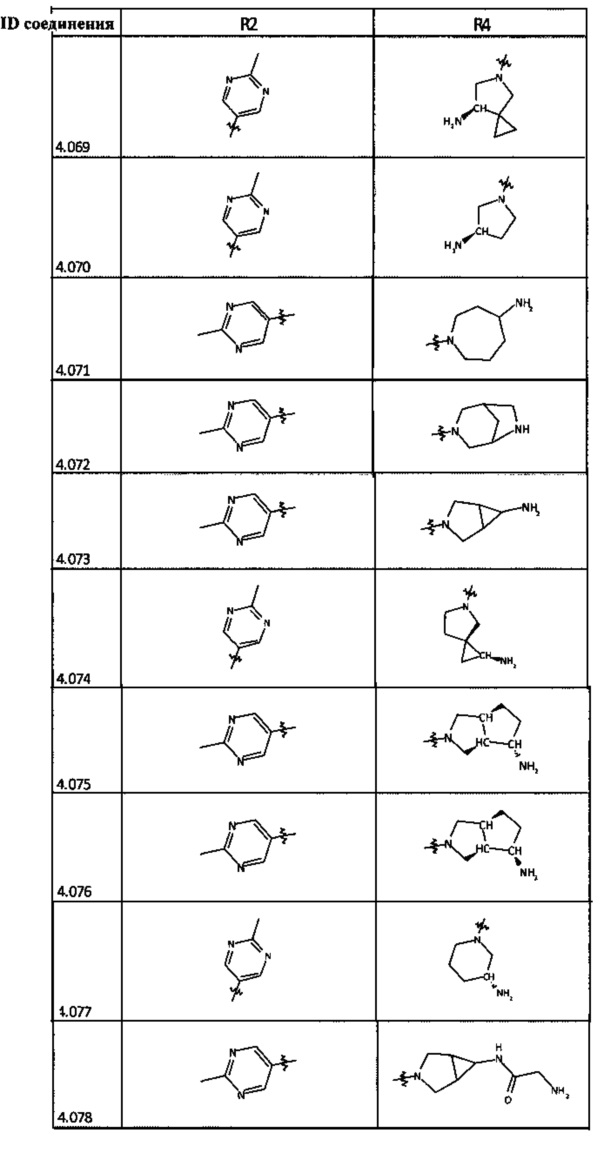
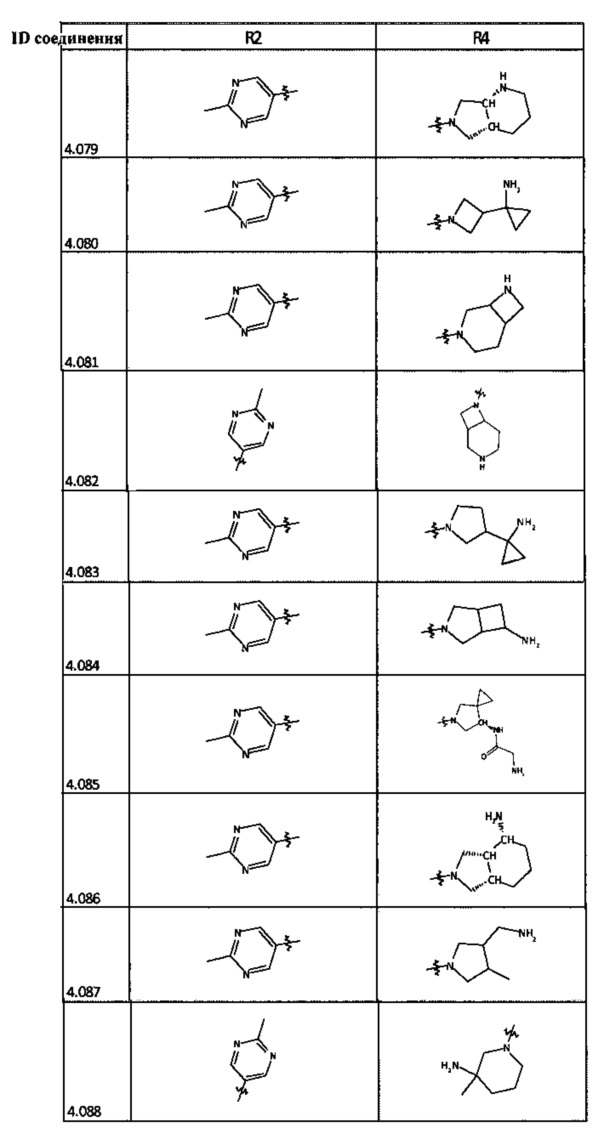
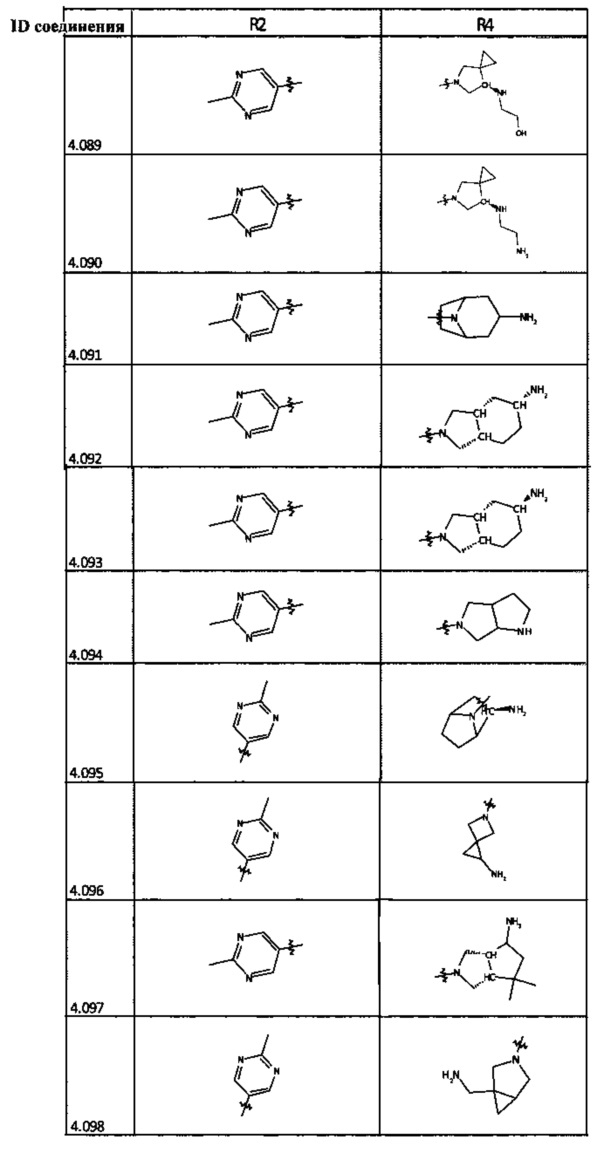
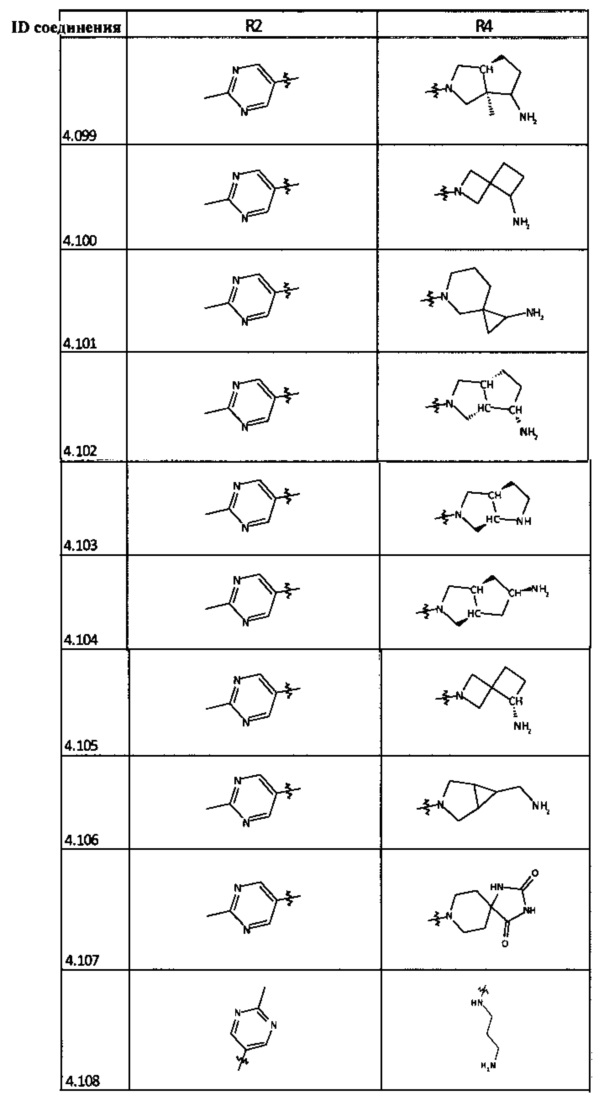
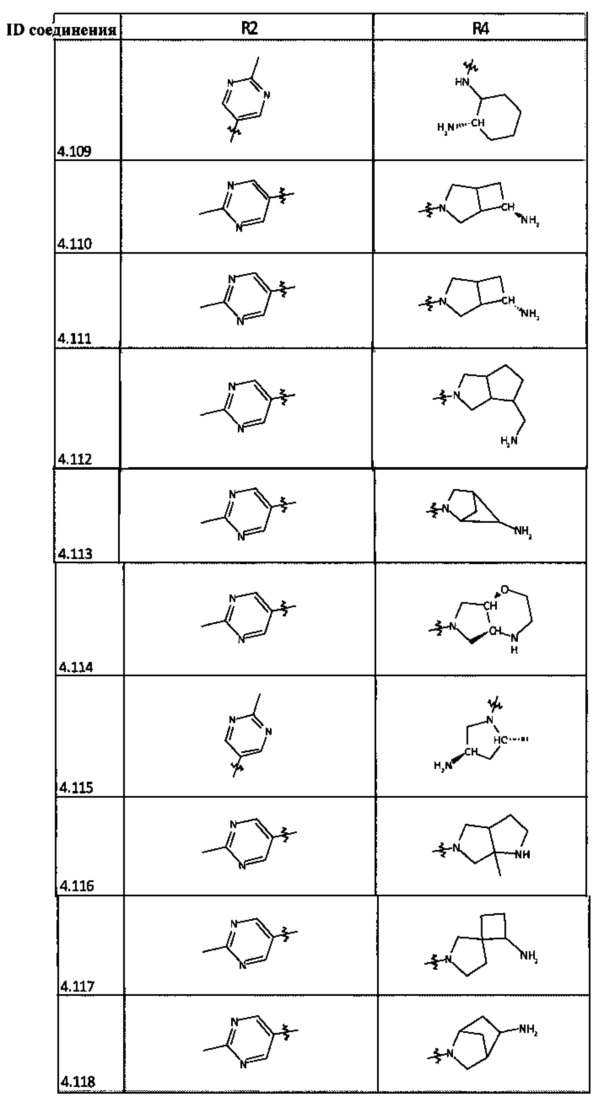
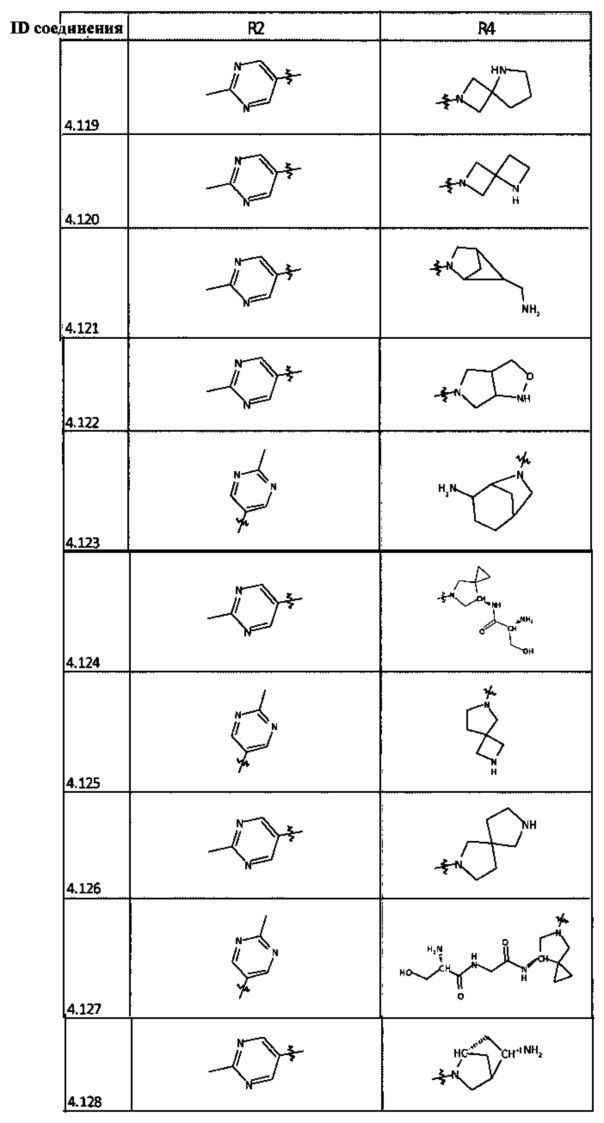
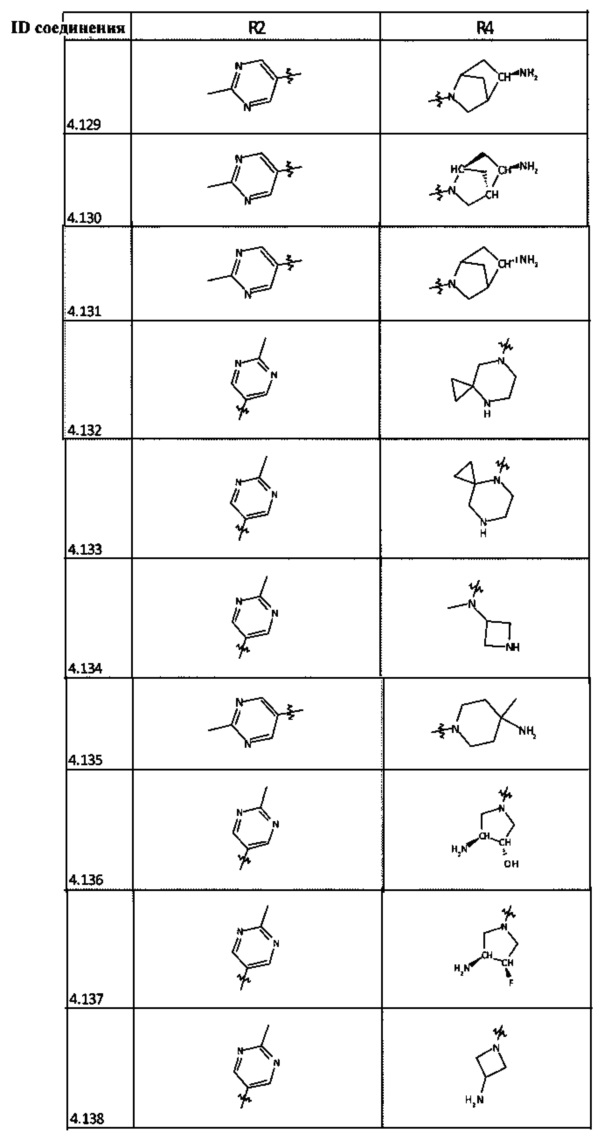
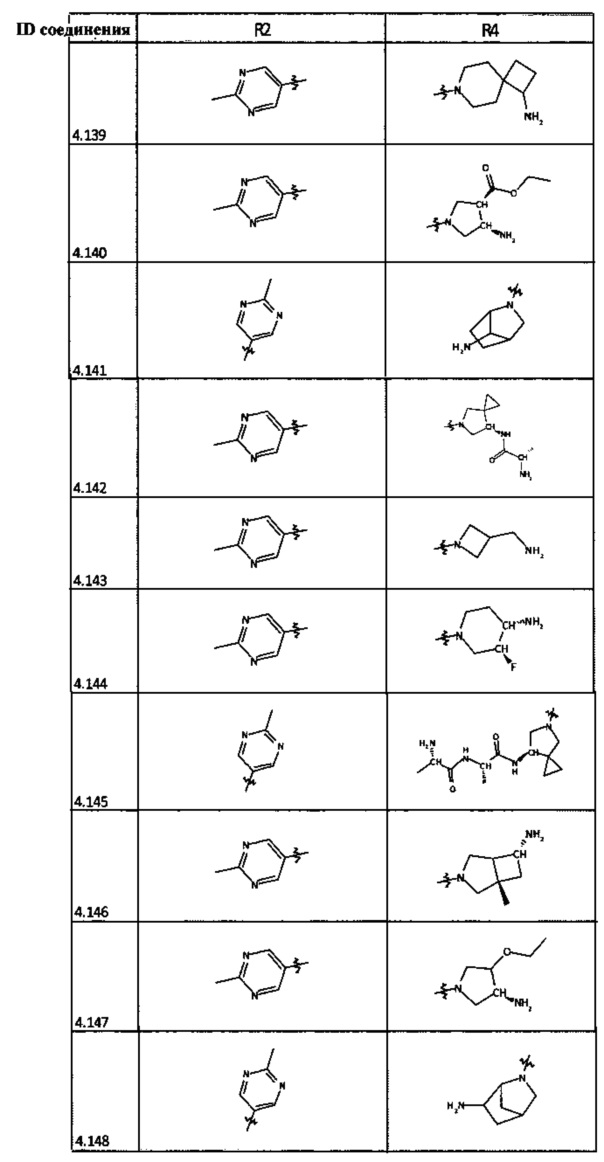
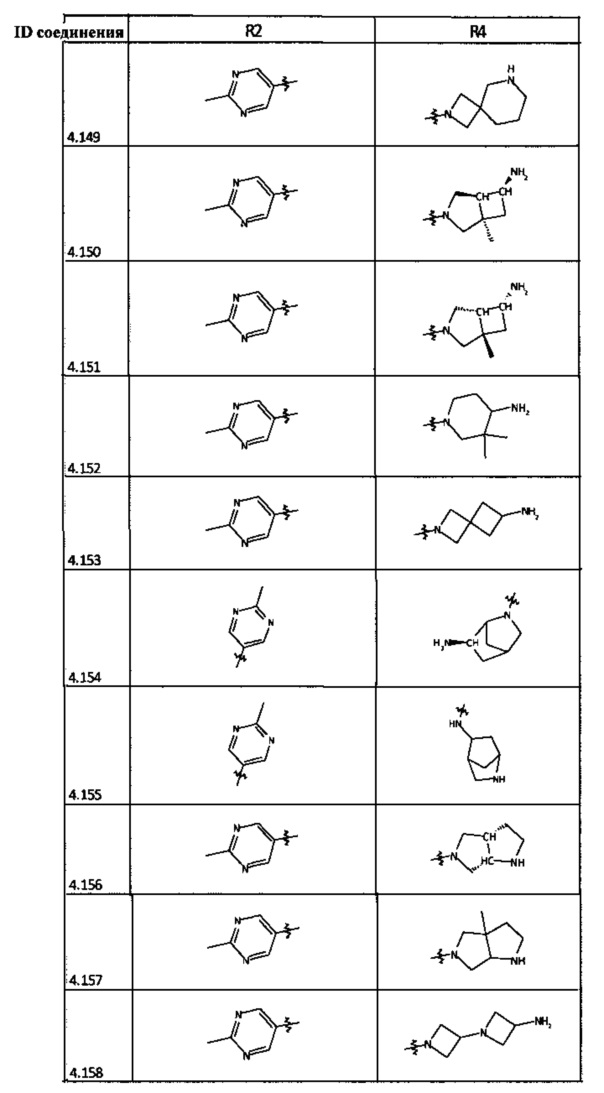
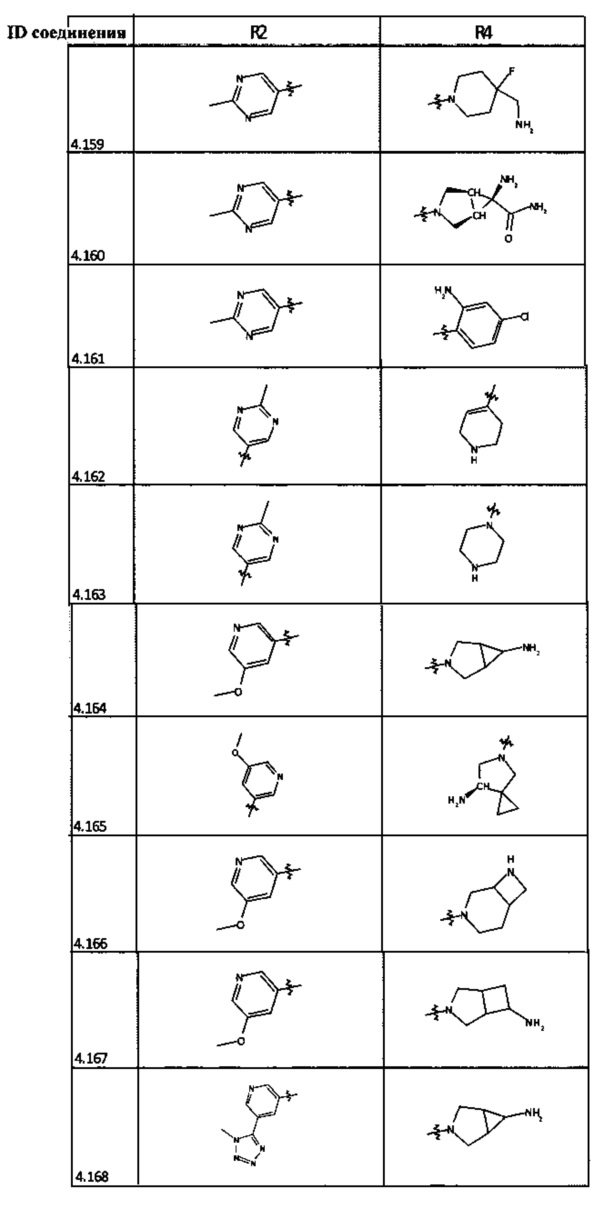
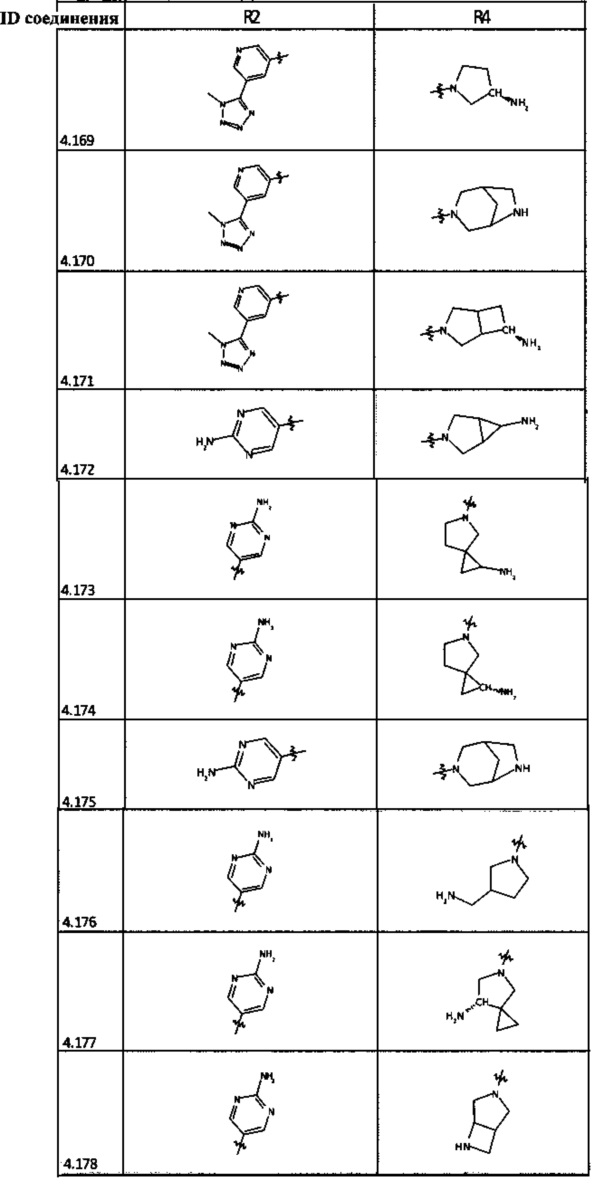
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2683325C2 |
| ИНГИБИТОР FAK И КОМБИНАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ С НИМ | 2019 |
|
RU2820555C2 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ 2Н-ПИРАЗОЛА | 2016 |
|
RU2722363C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
| СОЕДИНЕНИЯ ТИЕНИЛ[3, 2-d]ПИРИМИДИН-4-ОН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2624021C2 |
| Бензотиофены и родственные соединения в качестве агонистов STING | 2019 |
|
RU2806274C2 |
| Спироконденсированные пирролидиновые производные в качестве ингибиторов деубиквитилирующих ферментов (DUB) | 2017 |
|
RU2730552C2 |
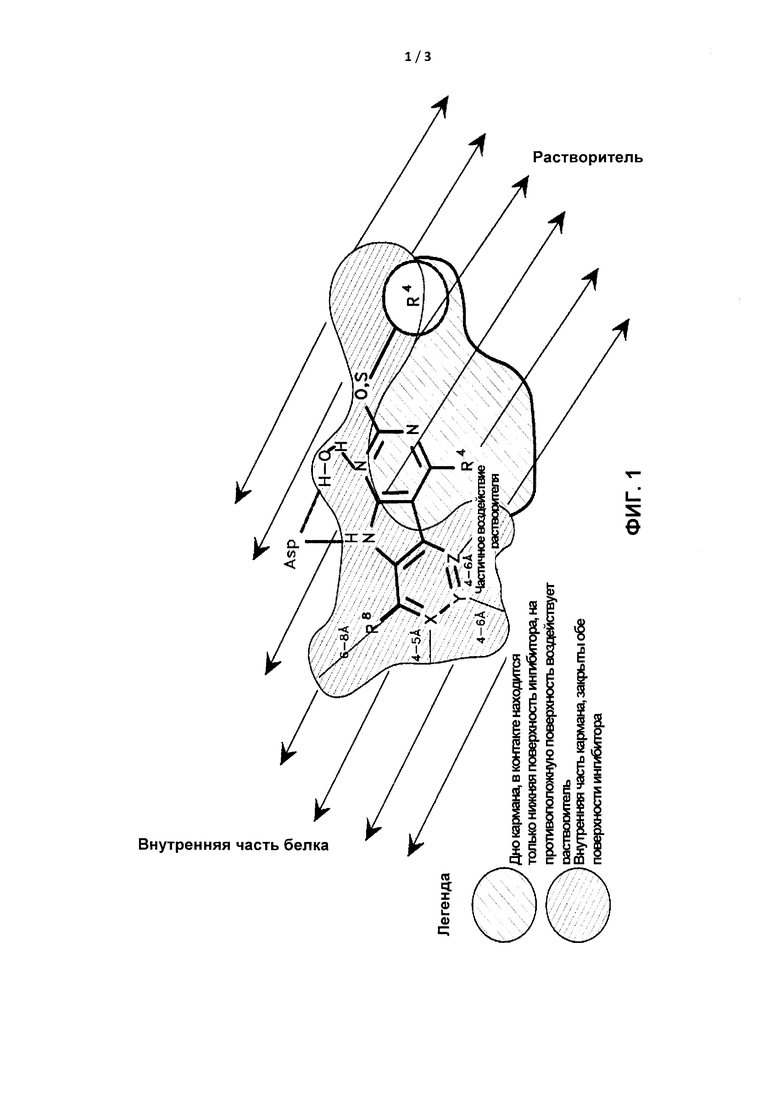
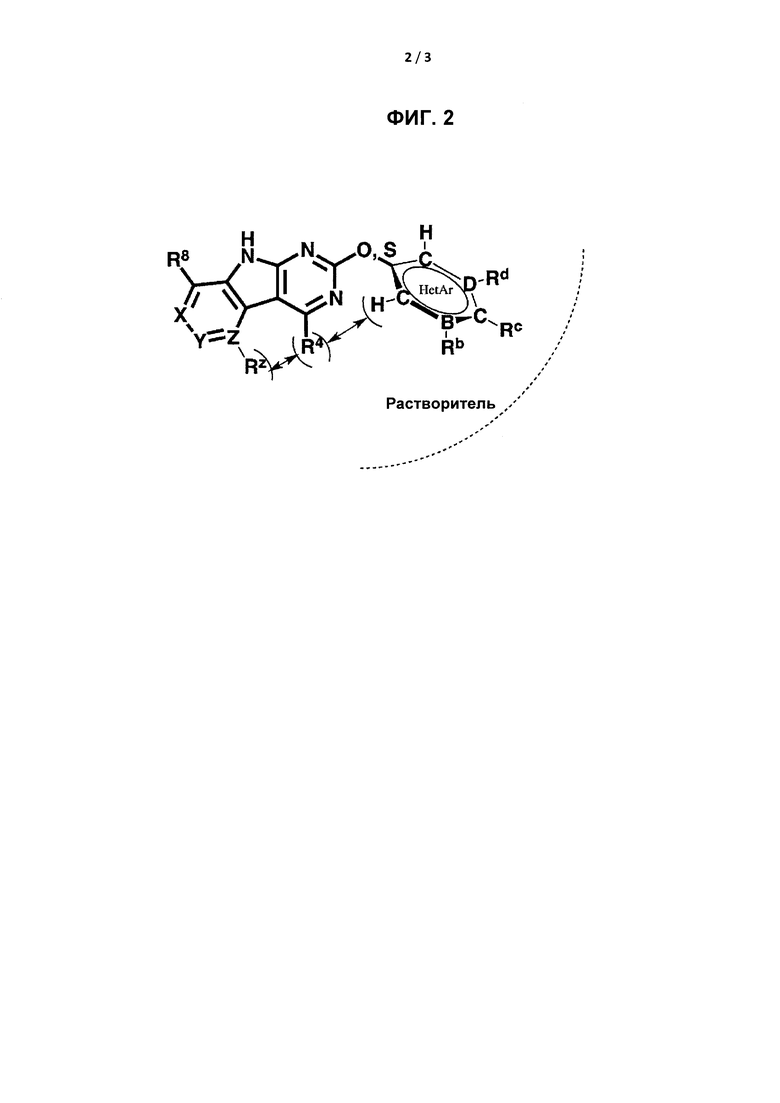
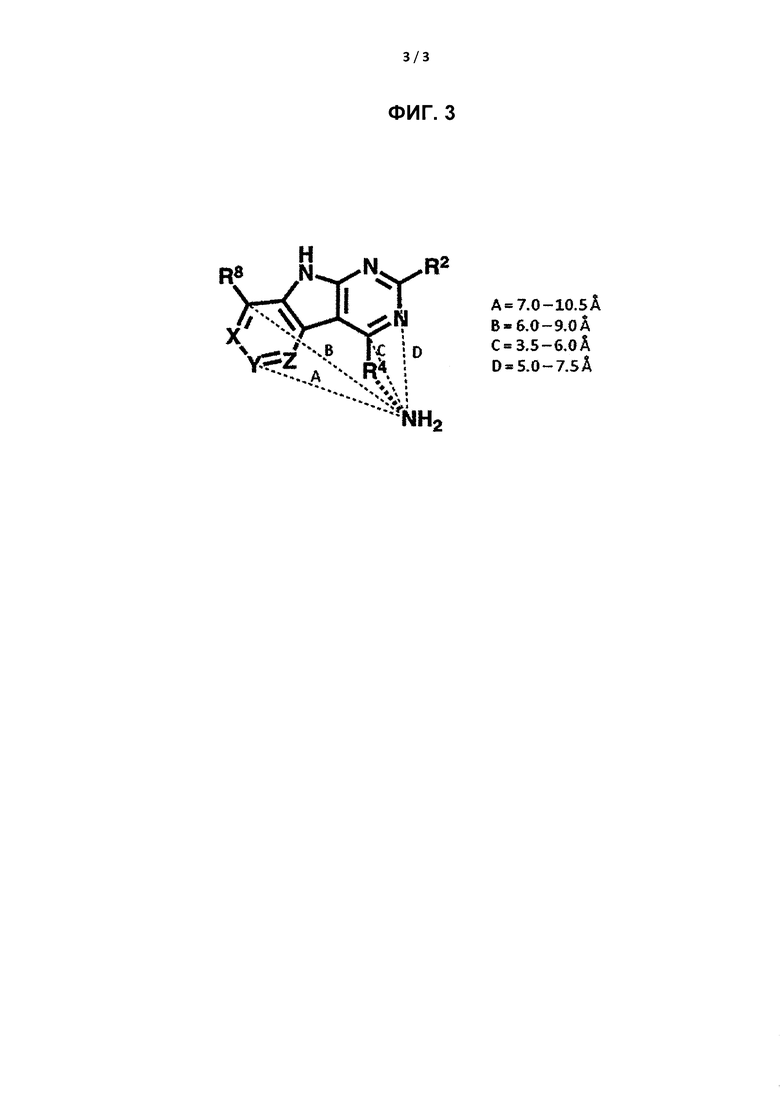
Изобретение относится к новым трициклическим соединениям формулы I и их фармацевтически применимым солям, обладающим активностью ингибиторов фермента гиразы В (GyrB) и топоизомеразы IV (ParE). Соединения могут найти применение для лечения бактериальных инфекций. Изобретение также относится к способу получения соединений формулы I и промежуточным продуктам формулы (1) для их получения. В формуле I:
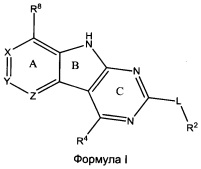
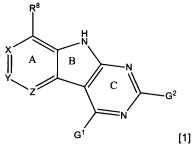
L представляет собой О или S; R8 представляет собой Н, Cl, F, Br, ОН, NH2, C1-3алкил, амино-C1-3алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH; X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N, причем RX представляет собой Н, СН3, Cl, Br или F; причем RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F; причем RZ представляет собой H, СН3, Cl, Br или F;
R2 представляет собой 6-членное моноциклическое ариловое или 6-членное моно- или 9-10-членное бициклическое гетероариловое кольцо, где гетероариловое кольцо содержит 1-3 гетероатома, выбранных из О, S и N, причем указанные 6-членное моноциклическое ариловое кольцо, 6-членное моноциклическое гетероариловое кольцо и 9-10-членное бициклическое гетероариловое кольцо могут быть необязательно замещенными 0-3 заместителями, выбранными из ОН, CO2H, СО2СН3, C(O)NH2, С(O)СН3, =O, CN, NH2, Br, Cl, F, SO3H, SO2NH2, SO2CH3, SOCH3, NHOH, C(O)NHOCH3, СН(ОН)СН3, С(ОН)(СН3)2, SCH3, или C1-С6алкила или С3-С6циклоалкила, содержащего 0-2 гетероатома, выбранных из О, S или N, необязательно замещенного С1-С4алкилом, ОН, CN, =O, NH2, Br, F, Cl; при этом бициклическое гетероариловое кольцо может быть спироконденсировано с циклопропилом; или
R2 представляет собой  или
или  ; R4 представляет собой: Н; необязательно замещенный ORa, где Ra представляет собой пиридил, пиримидинил, необязательно замещенный СН3 и NH2; необязательно замещенный насыщенный вторичный или третичный амин, выбранный из 4-6-членного алифатического амина, 4-6-членного моноциклического амина, 5-10-членного бициклического амина с 1-3 гетероатомами, выбранными из N, О или S, причем указанный необязательно замещенный насыщенный 5-10-членный бициклический амин может представлять собой спиро- или ортоконденсированный бициклический амин, или необязательно замещенного 5-6-членного ненасыщенного циклического остатка, содержащего 0-2 гетероатома N; причем указанный необязательно замещенный насыщенный вторичный или третичный амин присоединен к кольцу С через N вторичного или третичного амина, и необязательный заместитель выбран из OH, галогена, NH2, C(O)CH2NH2, C(O)NH2, CH2NHC(O), С5-6гетероциклила, возможно замещенного СН3 и CF3, СН2ОН, -С3-6циклоалкила, возможно замещенного NH2, -С3-6гетероциклила, содержащего от 0-2 атомов N, возможно замещенного NH2, -СН3, -С(O)СН3, -С(O)С3-6гетероциклила, содержащего от 1 до 3 атомов О, -СН(ОН)С3-6гетероциклила, содержащего от 1 до 3 атомов S, -NHCOCH3, -NHCH2CH2CN, -NHCH2C3-6циклоалкила, -С1-4алкилС3-6гетероциклила, содержащего от 0-2 гетероатомов, выбранных из О и N, -С1-4алкилNH2, =NOCH3, -NHC(O)CH2NH2, -NHCH2CH2OH, -NHCH2CH2NH2, -NHC(O)CH(NH2)CH2OH, -NHC(O)CH2NHC(O)CH(NH2)CH2OH, -C(O)OCH2CH3, -ОС1-4алкила. В промежуточных соединениях формулы (1) G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из SH, ОН, Cl, Br, F, I, SR, SOR, SO2R, OSO2R и OBt; R представляет собой C1-8алкил, Ar, где Ar представляет собой фенил, необязательно замещенный С1-4алкилом, С1-4алкокси, галогеном или NO2; Bt представляет собой бензотриазол; R8 представляет собой Н, Cl, F, Br, OH, NH2, C1-3алкил, амино-С1-3алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH; и X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ соответственно, при условии, что не более двух из X, Y и Z представляют собой N. 15 н. и 20 з.п. ф-лы, 3 ил., 11 табл., 7 пр.
; R4 представляет собой: Н; необязательно замещенный ORa, где Ra представляет собой пиридил, пиримидинил, необязательно замещенный СН3 и NH2; необязательно замещенный насыщенный вторичный или третичный амин, выбранный из 4-6-членного алифатического амина, 4-6-членного моноциклического амина, 5-10-членного бициклического амина с 1-3 гетероатомами, выбранными из N, О или S, причем указанный необязательно замещенный насыщенный 5-10-членный бициклический амин может представлять собой спиро- или ортоконденсированный бициклический амин, или необязательно замещенного 5-6-членного ненасыщенного циклического остатка, содержащего 0-2 гетероатома N; причем указанный необязательно замещенный насыщенный вторичный или третичный амин присоединен к кольцу С через N вторичного или третичного амина, и необязательный заместитель выбран из OH, галогена, NH2, C(O)CH2NH2, C(O)NH2, CH2NHC(O), С5-6гетероциклила, возможно замещенного СН3 и CF3, СН2ОН, -С3-6циклоалкила, возможно замещенного NH2, -С3-6гетероциклила, содержащего от 0-2 атомов N, возможно замещенного NH2, -СН3, -С(O)СН3, -С(O)С3-6гетероциклила, содержащего от 1 до 3 атомов О, -СН(ОН)С3-6гетероциклила, содержащего от 1 до 3 атомов S, -NHCOCH3, -NHCH2CH2CN, -NHCH2C3-6циклоалкила, -С1-4алкилС3-6гетероциклила, содержащего от 0-2 гетероатомов, выбранных из О и N, -С1-4алкилNH2, =NOCH3, -NHC(O)CH2NH2, -NHCH2CH2OH, -NHCH2CH2NH2, -NHC(O)CH(NH2)CH2OH, -NHC(O)CH2NHC(O)CH(NH2)CH2OH, -C(O)OCH2CH3, -ОС1-4алкила. В промежуточных соединениях формулы (1) G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из SH, ОН, Cl, Br, F, I, SR, SOR, SO2R, OSO2R и OBt; R представляет собой C1-8алкил, Ar, где Ar представляет собой фенил, необязательно замещенный С1-4алкилом, С1-4алкокси, галогеном или NO2; Bt представляет собой бензотриазол; R8 представляет собой Н, Cl, F, Br, OH, NH2, C1-3алкил, амино-С1-3алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH; и X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ соответственно, при условии, что не более двух из X, Y и Z представляют собой N. 15 н. и 20 з.п. ф-лы, 3 ил., 11 табл., 7 пр.
1. Соединение, имеющее структуру Формулы I:
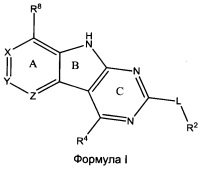
или его фармацевтически применимые соли,
причем
L представляет собой О или S;
R8 представляет собой Н, Cl, F, Br, ОН, NH2, C1-3алкил, амино-C1-3алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
причем RX представляет собой Н, СН3, Cl, Br или F;
причем RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F;
причем RZ представляет собой H, СН3, Cl, Br или F;
R2 представляет собой 6-членное моноциклическое ариловое или 6-членное моно- или 9-10-членное бициклическое гетероариловое кольцо, где гетероариловое кольцо содержит 1-3 гетероатома, выбранных из О, S и N,
причем указанные 6-членное моноциклическое ариловое кольцо, 6-членное моноциклическое гетероариловое кольцо и 9-10-членное бициклическое гетероариловое кольцо могут быть необязательно замещенными 0-3 заместителями, выбранными из
ОН, CO2H, СО2СН3, C(O)NH2, С(O)СН3, =O, CN, NH2, Br, Cl, F, SO3H, SO2NH2, SO2CH3, SOCH3, NHOH, C(O)NHOCH3, СН(ОН)СН3, С(ОН)(СН3)2, SCH3, или
C1-С6алкила или С3-С6циклоалкила, содержащего 0-2 гетероатома, выбранных из О, S или N, необязательно замещенного С1-С4алкилом, ОН, CN, =O, NH2, Br, F, Cl;
при этом бициклическое гетероариловое кольцо может быть спироконденсировано с циклопропилом;
или
R2 представляет собой  или
или  ;
;
R4 представляет собой:
Н;
необязательно замещенный ORa, где Ra представляет собой пиридил, пиримидинил, необязательно замещенный СН3 и NH2;
необязательно замещенный насыщенный вторичный или третичный амин, выбранный из
4-6-членного алифатического амина,
4-6-членного моноциклического амина,
5-10-членного бициклического амина с 1-3 гетероатомами, выбранными из N, О или S, причем указанный необязательно замещенный насыщенный 5-10-членный бициклический амин может представлять собой спиро- или ортоконденсированный бициклический амин, или
необязательно замещенного 5-6-членного ненасыщенного циклического остатка, содержащего 0-2 гетероатома N;
причем указанный необязательно замещенный насыщенный вторичный или третичный амин присоединен к кольцу С через N вторичного или третичного амина, и
необязательный заместитель выбран из
OH,
галогена,
NH2,
C(O)CH2NH2,
C(O)NH2,
CH2NHC(O),
С5-6гетероциклила, возможно замещенного СН3 и CF3,
СН2ОН,
-С3-6циклоалкила, возможно замещенного NH2,
-С3-6гетероциклила, содержащего от 0-2 атомов N, возможно замещенного NH2,
-СН3,
-С(O)СН3,
-С(O)С3-6гетероциклила, содержащего от 1 до 3 атомов О,
-СН(ОН)С3-6гетероциклила, содержащего от 1 до 3 атомов S,
-NHCOCH3,
-NHCH2CH2CN,
-NHCH2C3-6циклоалкила,
-С1-4алкилС3-6гетероциклила, содержащего от 0-2 гетероатомов, выбранных из О и N,
-С1-4алкилNH2,
=NOCH3,
-NHC(O)CH2NH2,
-NHCH2CH2OH,
-NHCH2CH2NH2,
-NHC(O)CH(NH2)CH2OH,
-NHC(O)CH2NHC(O)CH(NH2)CH2OH,
-C(O)OCH2CH3,
-ОС1-4алкила.
2. Соединение по п. 1, отличающееся тем, что L представляет собой О.
3. Соединение по п. 1, отличающееся тем, что L представляет собой S.
4. Соединение по п. 3, отличающееся тем, что R8 представляет собой Н, СН3, СН2СН3, Cl, ОСН3, NHCD3, NHCH3, NHCH2CH3 или NH2.
5. Соединение по п. 4, отличающееся тем, что R8 представляет собой NHCH3.
6. Соединение по любому из пп. 1-3, отличающееся тем, что X, Y и Z представляют собой CRX, CRY и CRZ соответственно.
7. Соединение по любому из пп. 1-3, отличающееся тем, что R2 представляет собой необязательно замещенное гетероариловое кольцо.
8. Соединение по любому из пп. 1-3, отличающееся тем, что R2 выбран из группы, состоящей из необязательно замещенного пиримидинила, фенила, пиридила, индолила, азаиндолила, пиримидопиридила, хиназолинила, хиноксалинила, нафтиридинила, пуринила, фуропиридинила, изоиндолинила, бензодиоксинила, дигидробензодиоксинила, бензотиазолила, пирролопиридинила, дигидропирролопиридинила, бензоимидазолила, имидазопиридинила, дигидроимидазопиридинила, тетрагидроизоиндолила, хроменила, бензтиофена, бензтриазолила, бензфуранила, бензоксадиазолила, индазолила, хинолинила, изохинолинила, индолина, азаиндолинила и
 .
.
9. Соединение по любому из пп. 1-3, отличающееся тем, что R2 представляет собой пиримидинил или пиридинил, необязательно замещенный СН(ОН)СН3, С(ОН)(СН3)2, ОСН3, CN, СН3, СН2СН3, О-циклопропилом, SCH3, Br, Cl, F или NH2.
10. Соединение по любому из пп. 1-3, отличающееся тем, что R2 выбран из группы, состоящей из

11. Соединение по любому из пп. 1-3, отличающееся тем, что R4 необязательно замещен ORa, a Ra представляет собой необязательно замещенный пиримидинил или пиридинил.
12. Соединение по п. 11, отличающееся тем, что Ra представляет собой незамещенный пиримидинил или пиридинил, замещенный СН3 или NH2.
13. Соединение, имеющее структуру Формулы I:
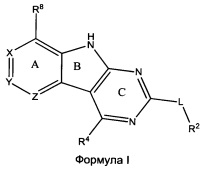
или его фармацевтически применимые соли,
в котором
L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX, RY, RZ, R2 имеют значения, указанные в п. 1;
R4 представляет собой необязательно замещенный вторичный или третичный С4-С6циклический амин, присоединенный к кольцу С через N вторичного или третичного амина, дополнительно включающий первичный или вторичный амин, причем указанный первичный или вторичный амин не присоединен напрямую к кольцу С.
14. Соединение, имеющее структуру Формулы I:
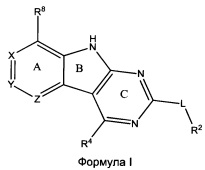
или его фармацевтически применимые соли,
в котором L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX представляет собой Н, СН3, Cl, Br или F;
RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F;
RZ представляет собой Н, СН3, Cl, Br или F;
R2 представляет собой 6-членное моноциклическое арильное или 6-членное моно- или 9-10-членное бициклическое гетероарильное кольцо, где гетероарильное кольцо содержит 1-3 гетероатома, выбранных из О, S, или N,
где указанное 6-членное моноциклическое арильное или 6-членное моно- или 9-10-членное бициклическое гетероарильное кольцо необязательно замещено 0-3 заместителями, выбранными из
ОН, CO2H, СО2СН3, C(O)NH2, С(O)СН3, =O, CN, NH2, Br, Cl, F, SO3H, SO2NH2, SO2CH3, SOCH3, NHOH, C(O)NHOCH3, CH(OH)CH3, C(OH)(CH3)2, SCH3; или
C1-С6алкила или С3-С6циклоалкила, содержащего 0-2 гетероатома, выбранных из О, S или N, необязательно замещенных С1-С4алкилом, ОН, CN, =O, NH2, Br, F, Cl;
где бициклическое гетероарильное кольцо может быть спироконденсировано с циклопропилом; или
R2 представляет собой  или
или  ;
;
R4 представляет собой необязательно замещенный бициклический спиро- или орто-конденсированный азотсодержащий 6-10-членный насыщенный гетероциклический остаток, содержащий 1-3 гетероатома, выбранных из N, О или S, при этом остаток присоединен к кольцу С через N.
15. Соединение, имеющее структуру Формулы I:
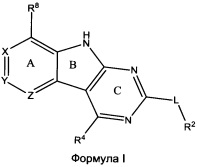
или его фармацевтически применимые соли,
в котором L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX представляет собой Н, СН3, Cl, Br или F;
RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F;
RZ представляет собой H, СН3, Cl, Br или F;
R2 представляет собой 6-членное моноциклическое арильное или 6-членное моно- или 9-10-членное бициклическое гетероарильное кольцо, где гетероарильное кольцо содержит 1-3 гетероатома, выбранных из О, S, или N,
R4 представляет собой необязательно замещенный третичный амин, присоединенный к кольцу С через N третичного амина, причем необязательно замещенный третичный амин содержит по меньшей мере один дополнительный N, отделенный от N третичного амина 2-3 атомами углерода.
16. Соединение, имеющее структуру Формулы I:
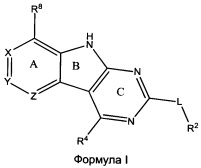
или его фармацевтически применимые соли,
в котором L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX, RY, RZ, R2 имеют значения, указанные в п. 1;
R4 представляет собой нециклический вторичный или третичный амин, замещенный 1-2 заместителями, выбранными из =O, NH2, CH2NH2, C(O)CH2NH2.
17. Соединение по любому из пп. 1-3, отличающееся тем, что R4 выбран из группы, состоящей из необязательно замещенного пиразолила, фенила, пиперазинила, пиридинила и тетрагидропиридинила.
18. Соединение, имеющее структуру Формулы I:
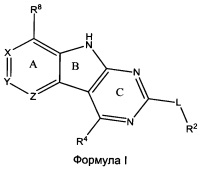
или его фармацевтически применимые соли,
в котором L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX, RY, RZ, R2 имеют значения, указанные в п. 1;
R4 представляет собой необязательно замещенный 5-10-членный ненасыщенный циклический или гетероциклический остаток, содержащий 0-3 гетероатома N, О или S, включает 0-2 необязательных заместителя, выбранных из группы, состоящей из СН3, NH2, F, Cl и CH2NH2.
19. Соединение, имеющее структуру Формулы I:
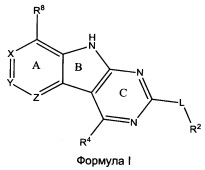
или его фармацевтически применимые соли,
в котором L, R8 имеют значения, указанные в п. 1;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, при условии, что не более двух из X, Y и Z представляют собой N,
RX, RY, RZ, R2 имеют значения, указанные в п. 1;
R4 представляет собой Н или заместитель, выбранный из группы, состоящей из

 .
.
20. Соединение, имеющее структуру Формулы I:
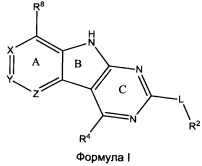
или его фармацевтически применимые соли,
в котором L представляет собой О;
R8 представляет собой NHCH3;
X, Y и Z представляют собой CRX, CRY или CRZ соответственно, причем RX представляет собой Н или F; RY представляет собой Н, F, Cl или CF3; и RZ представляет собой Н, СН3 или F;
R2 выбран из группы, состоящей из
 ;
;
и
R4 выбран из группы, состоящей из
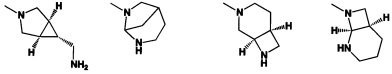
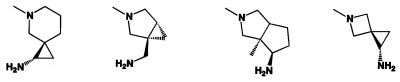
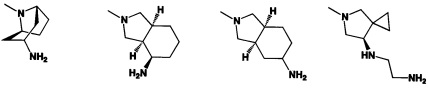
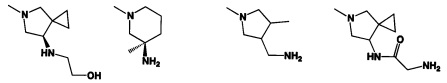
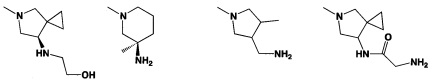
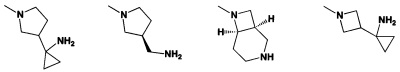
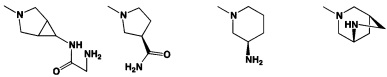
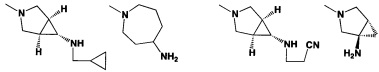
 .
.
21. Соединение, имеющее структуру Формулы I:
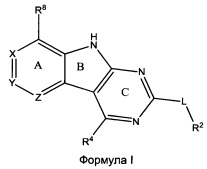
или его фармацевтически применимые соли,
причем
L представляет собой S;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ, и
R8, Rx, Ry, Rz, R4 и R2 представляют собой следующие группы:
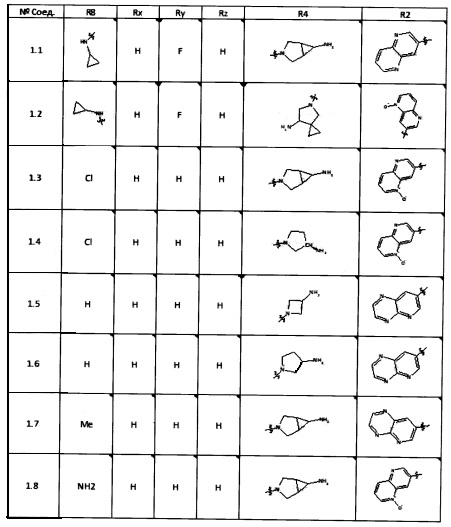
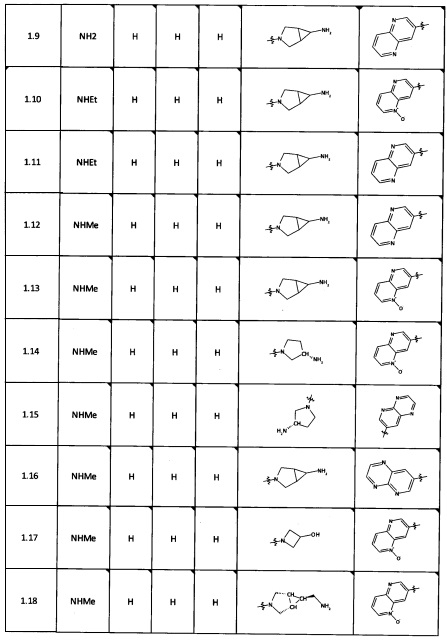
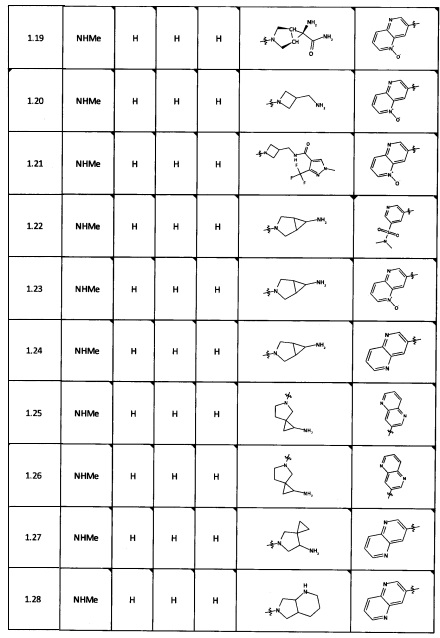
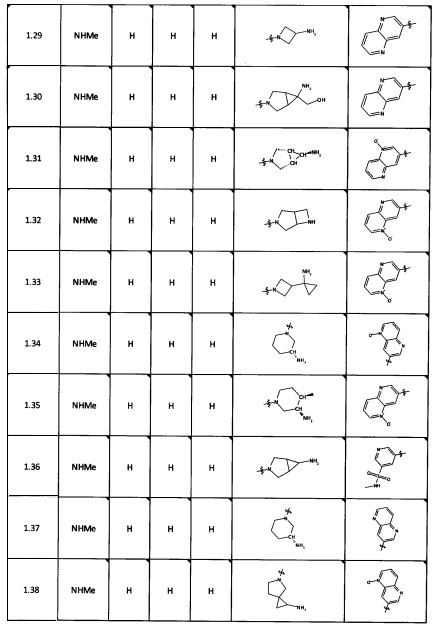
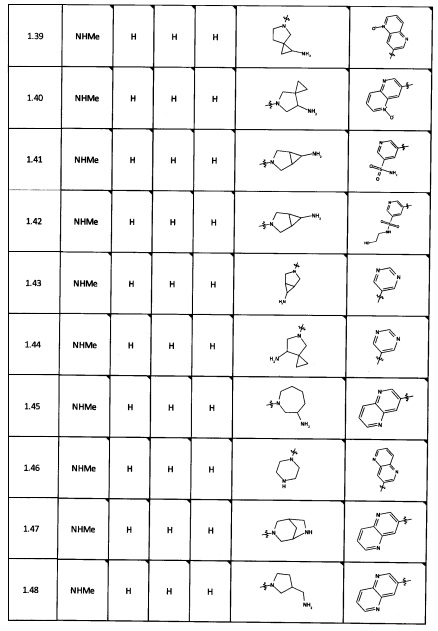
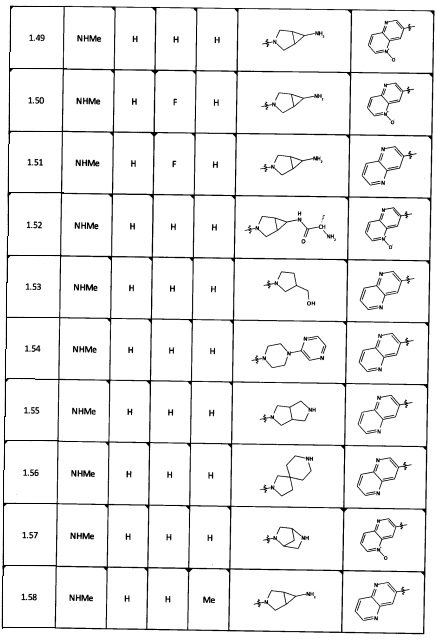
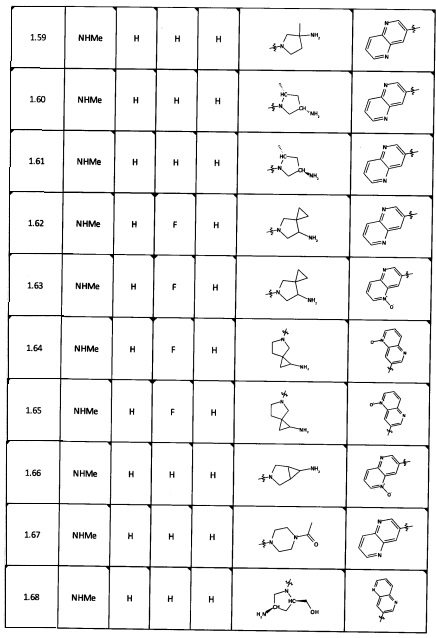
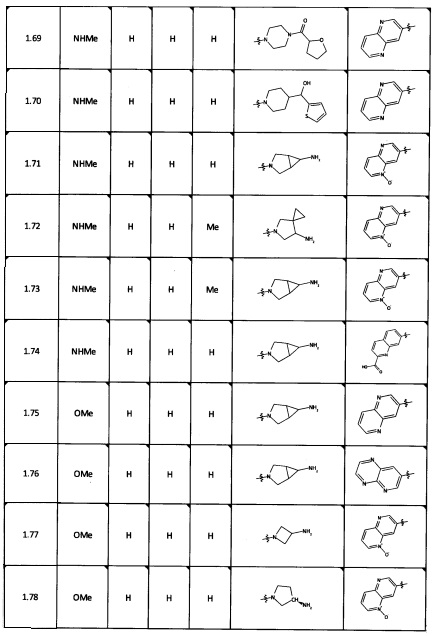 ;
;
или L представляет собой О и L-R2, R4, Rx, Ry, Rz и R8 представляют собой следующие группы:
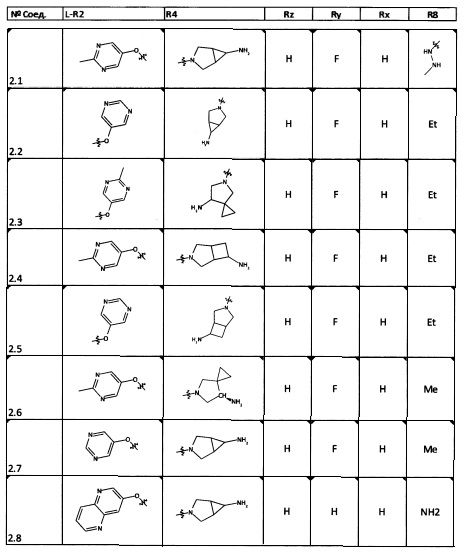
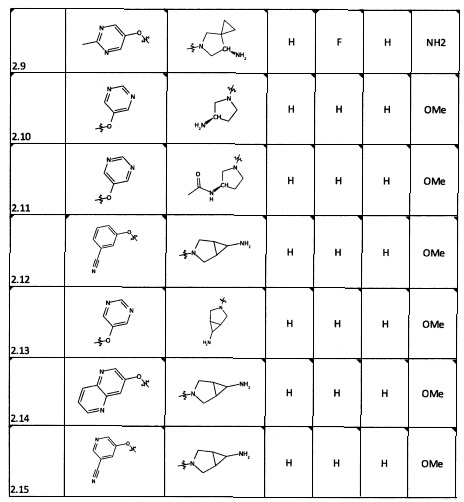 ;
;
или
соединение Формулы I, представляющее собой
 ;
;
или L представляет собой О, Rx, Ry, Rz представляют собой Н, R8 представляет собой NHCH3 и R2 и R4 представляют собой следующие группы:
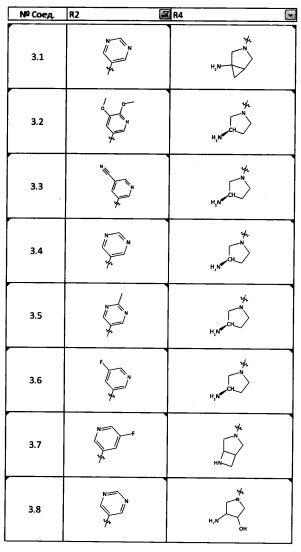
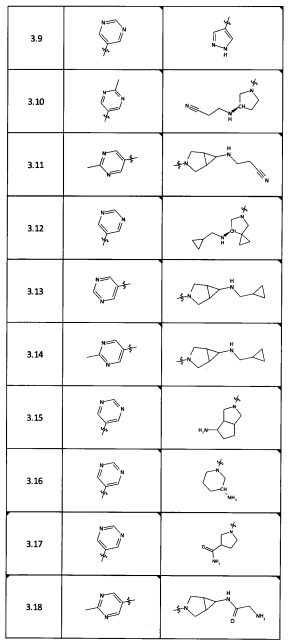
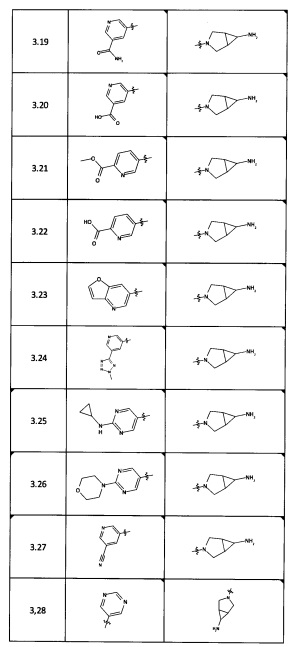
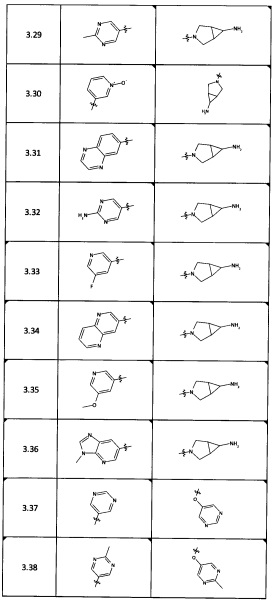
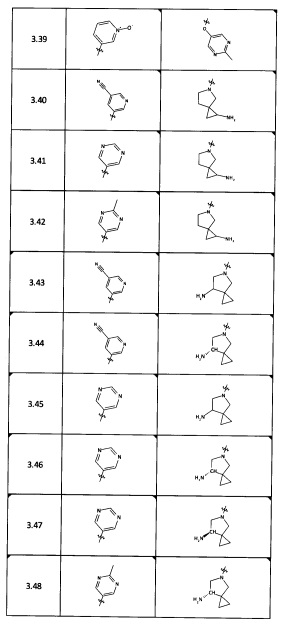
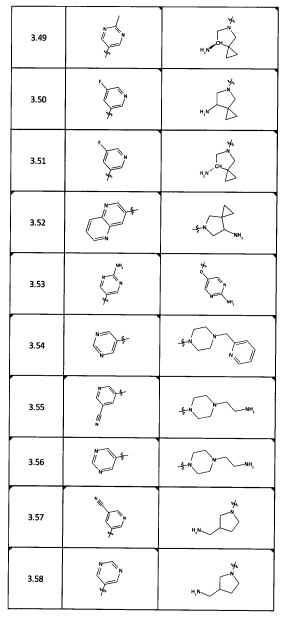
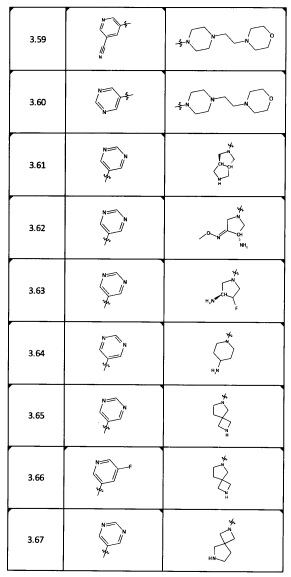 ;
;
или L представляет собой О, Rx, Rz представляют собой СН, Ry представляет собой F, R8 представляет собой NHCH3 и R2 и R4 представляют собой следующие группы:

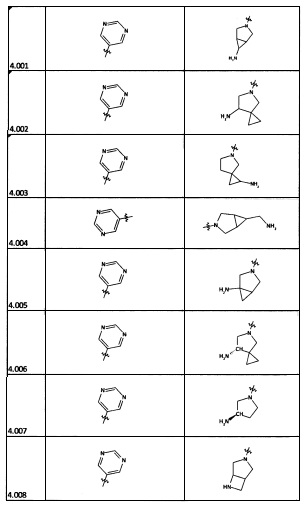
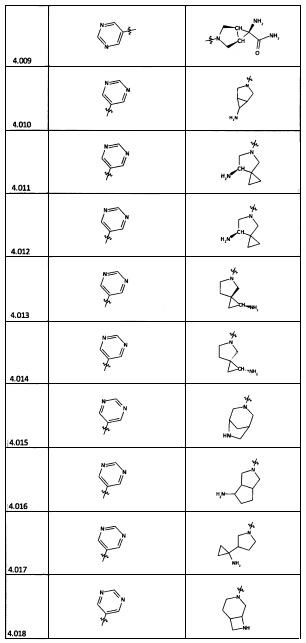
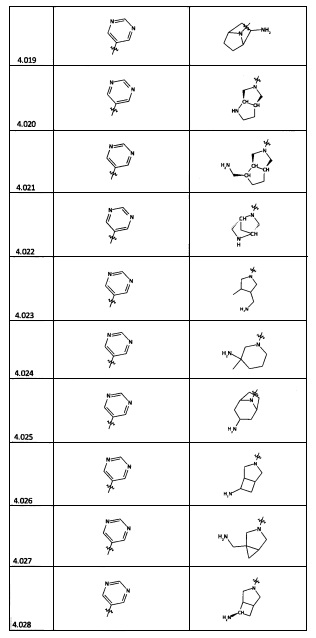
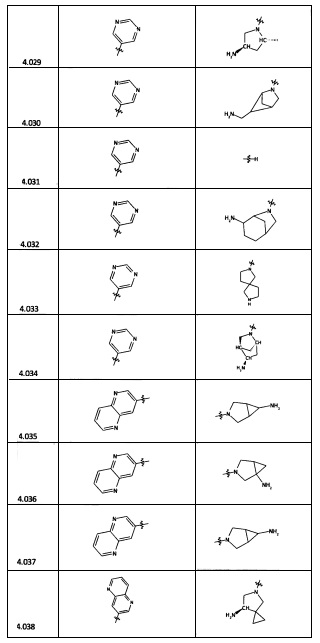
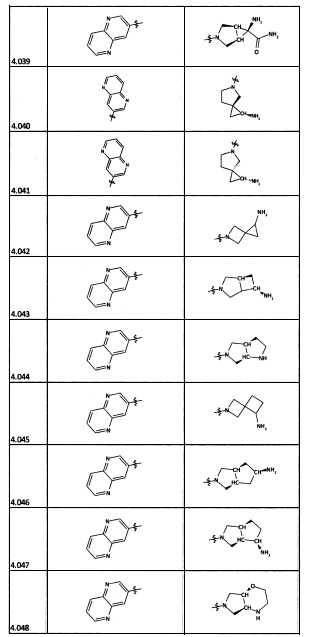
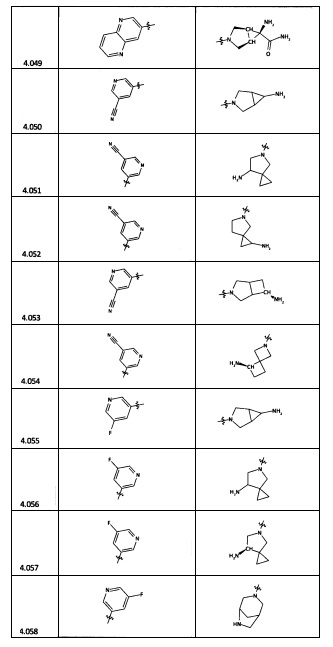
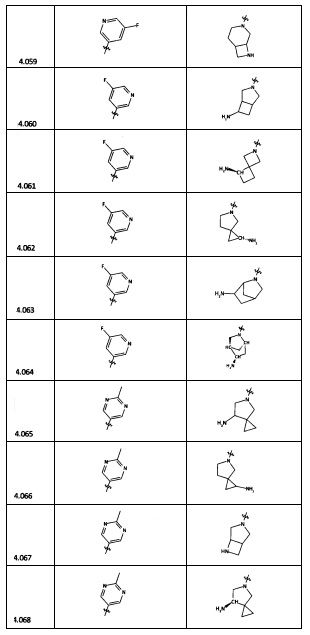
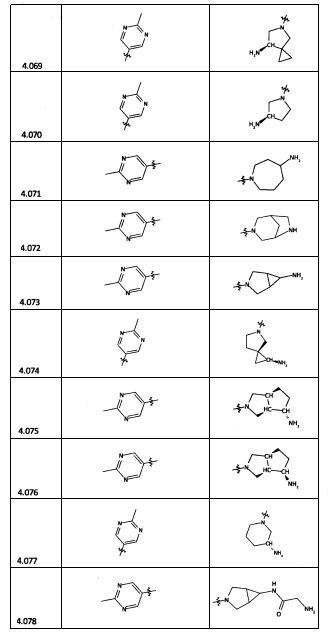
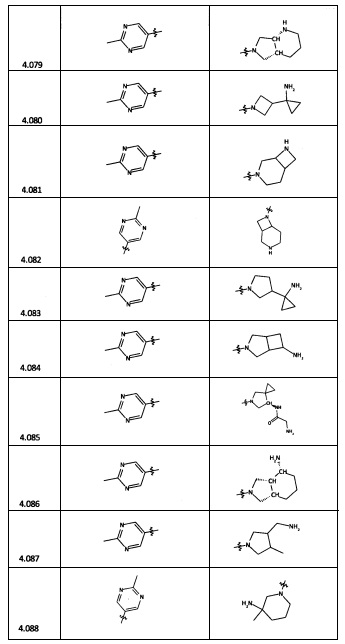
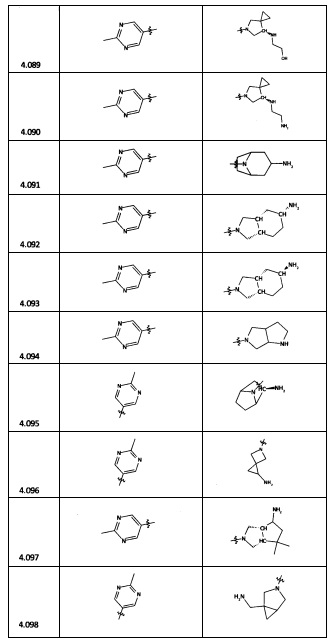
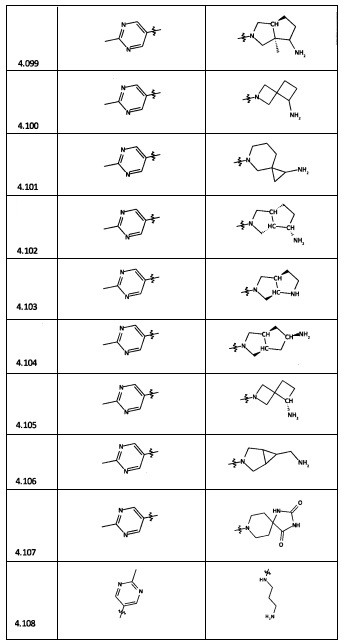
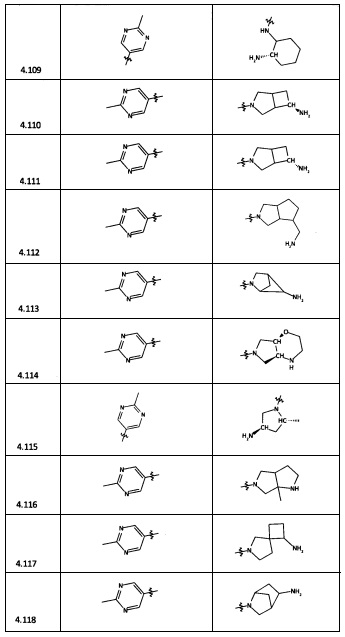
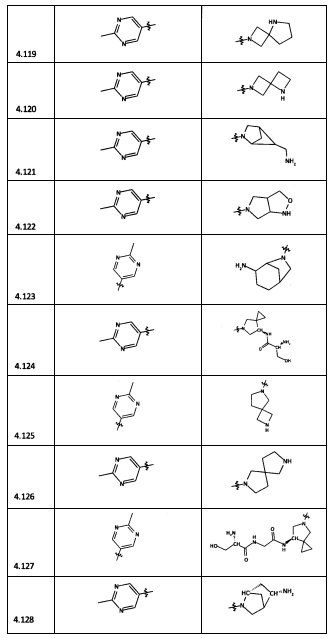
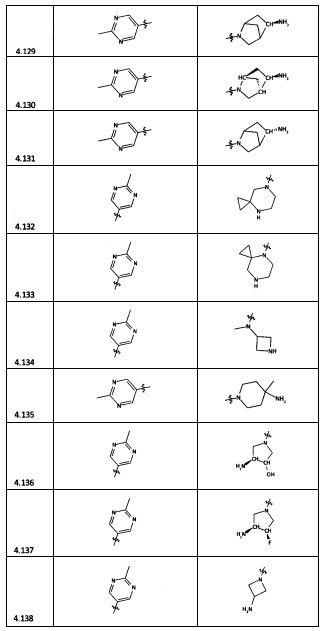
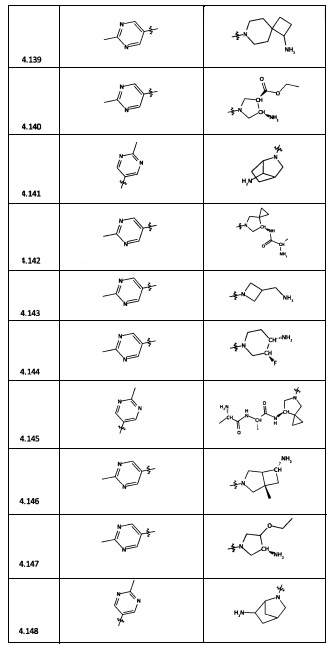
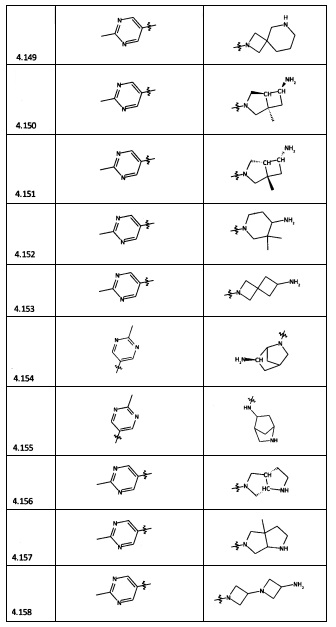
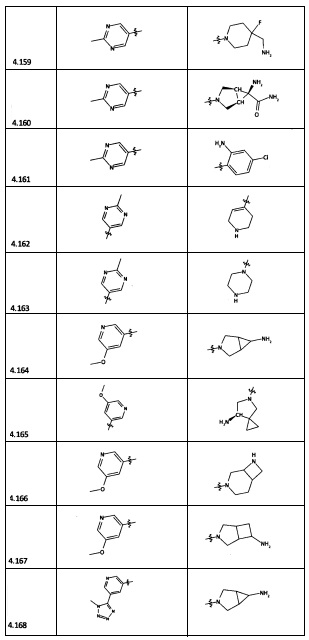
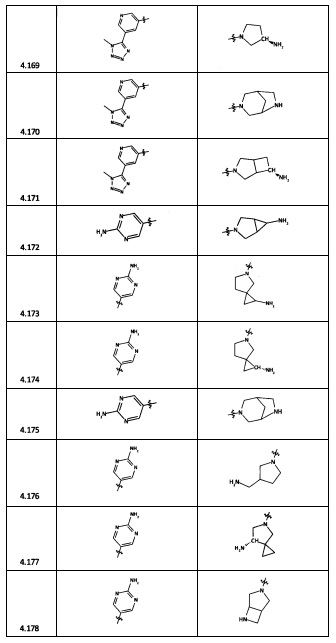
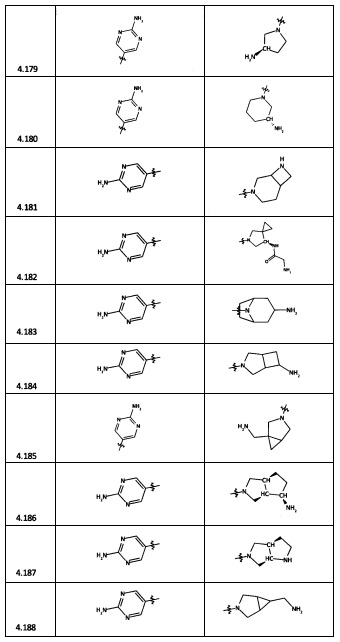
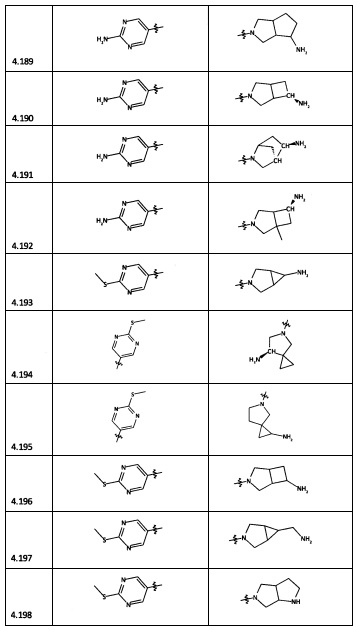
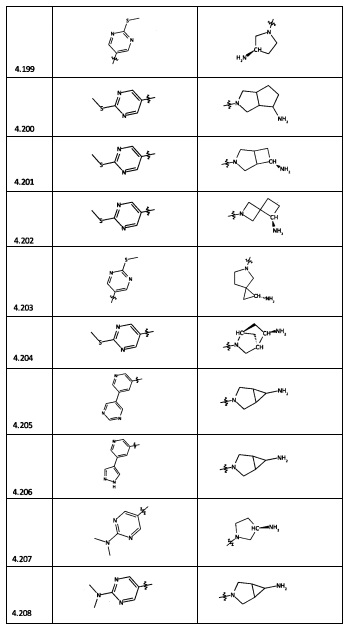
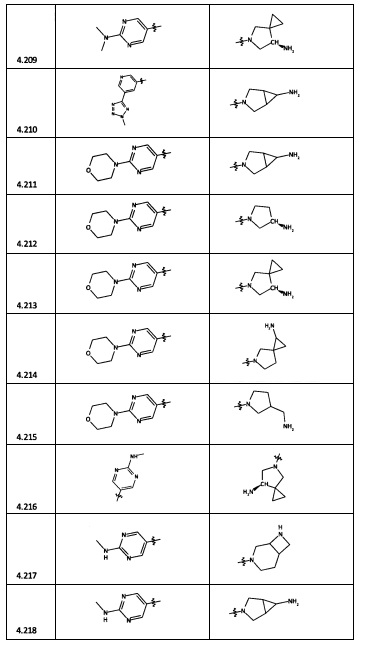
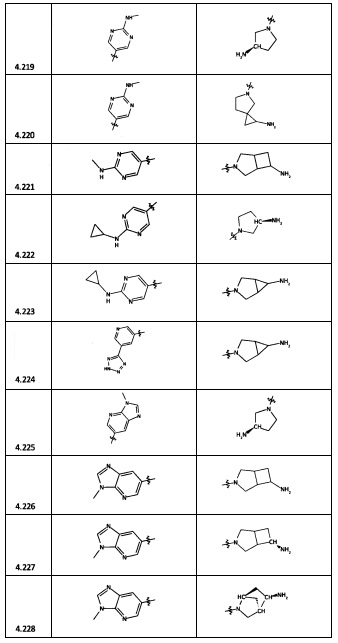
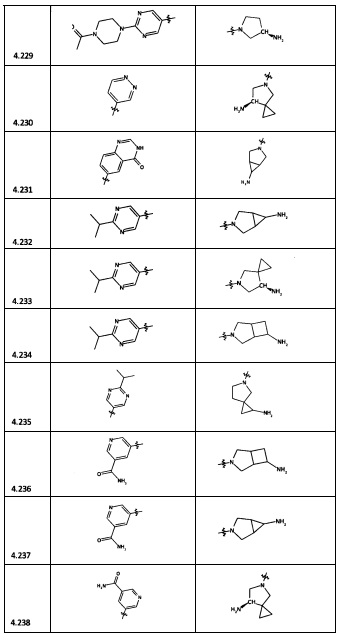
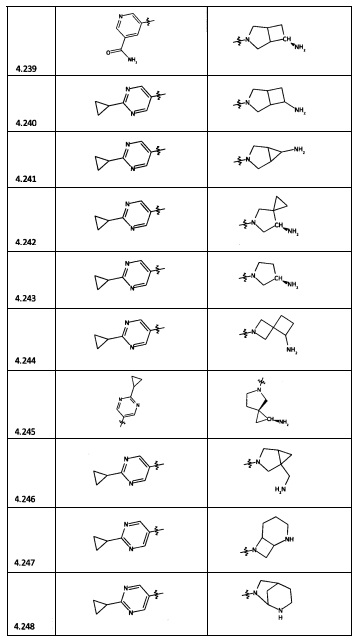
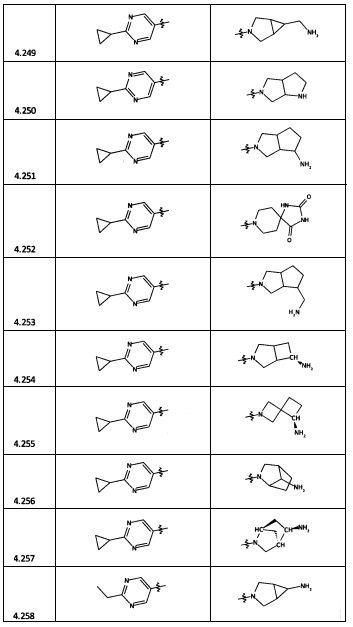
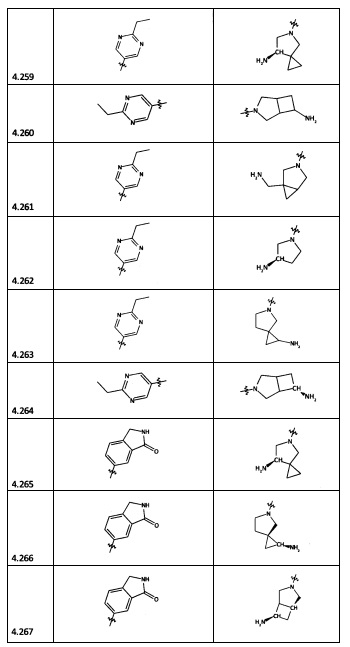
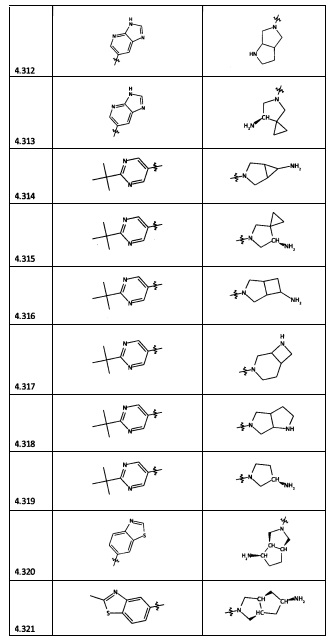
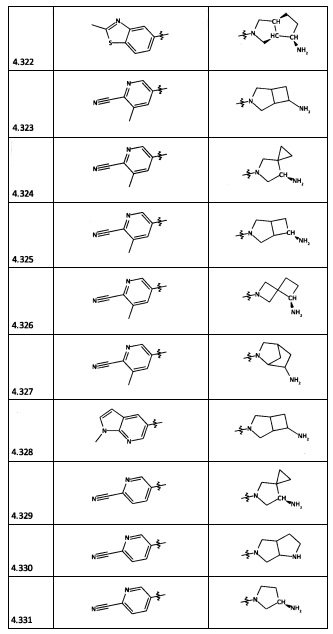
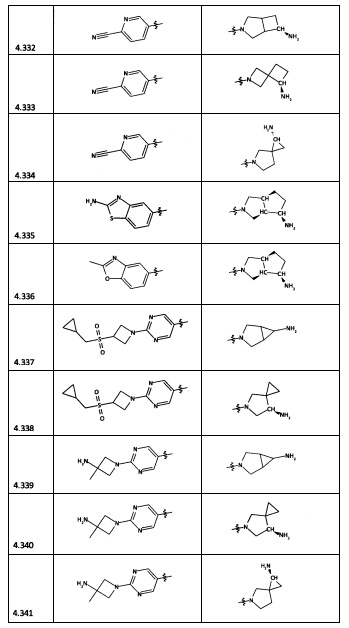
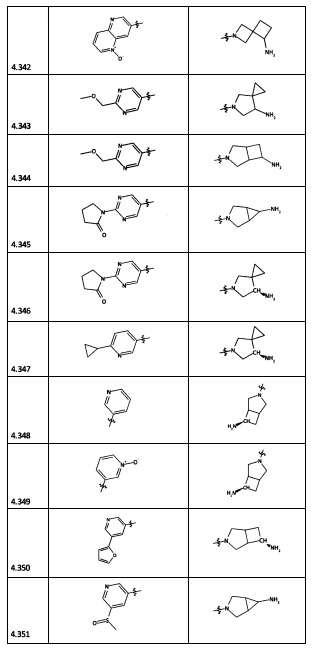
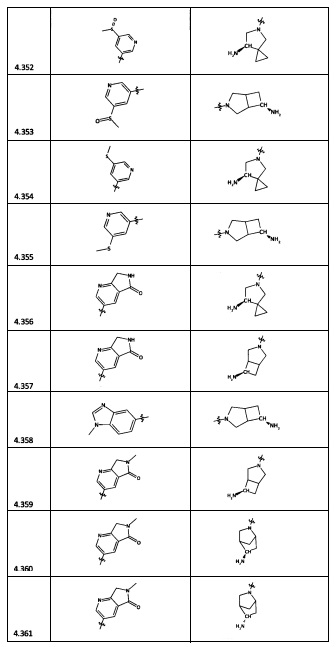
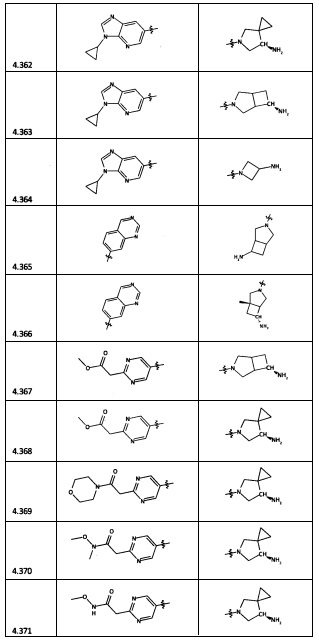
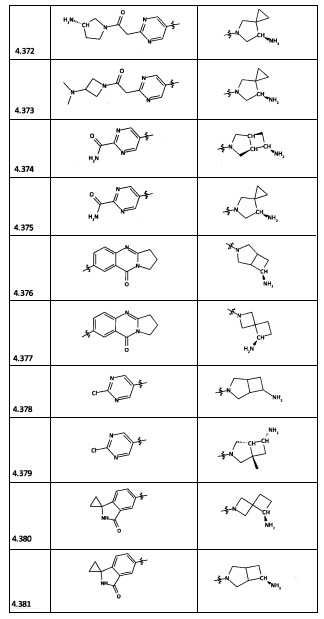
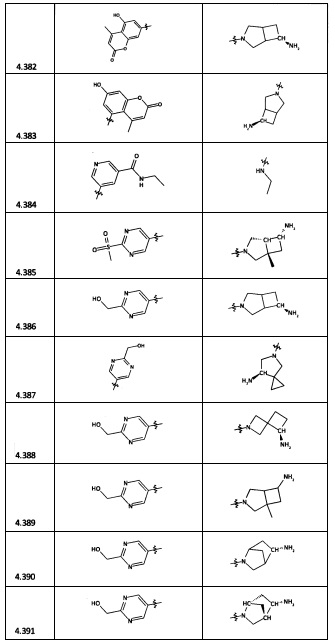
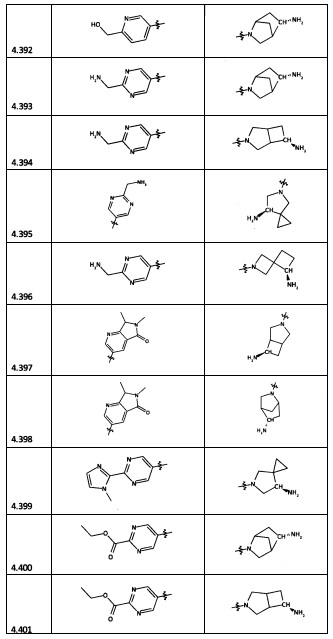
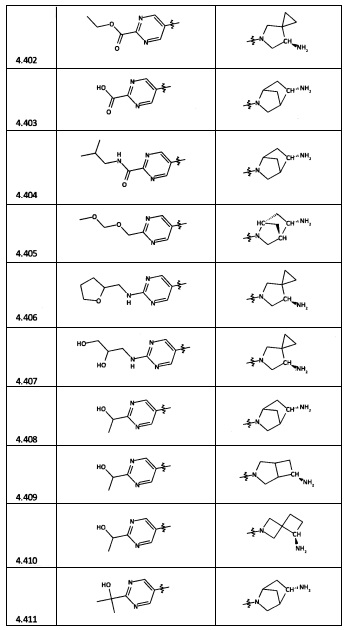
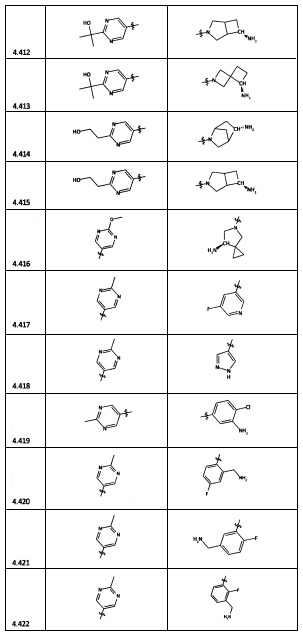 ;
;
или соединение формулы I выбрано из:
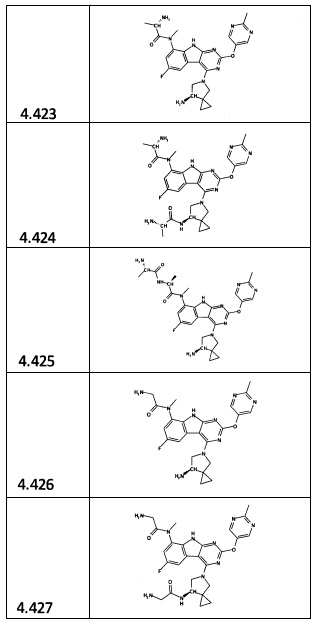 ;
;
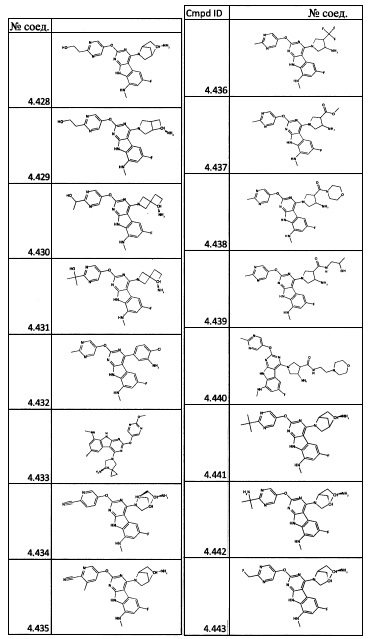
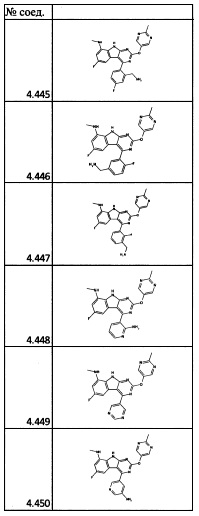
или L представляет собой О, Rx представляет собой СН, Ry, Rz представляют собой F, R8 представляет NHCH3 и где R2 и R4 представляют собой следующие группы:

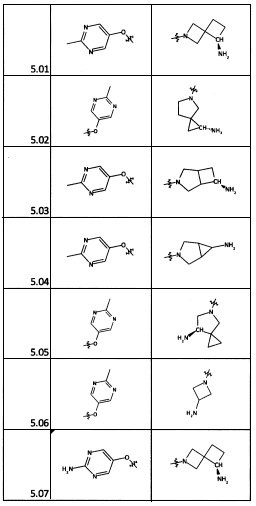
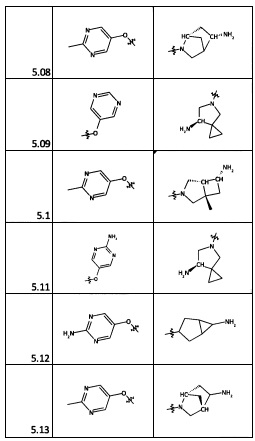 ;
;
или соединение формулы I выбрано из группы:
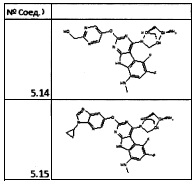 ;
;
или L представляет собой О, и R2, R4, Rx, Ry, Rz и R8 представляют собой следующие группы:
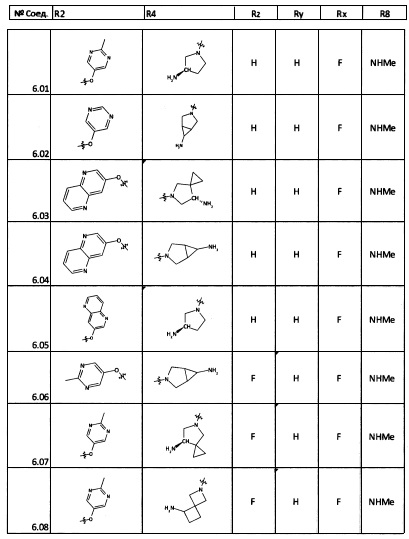
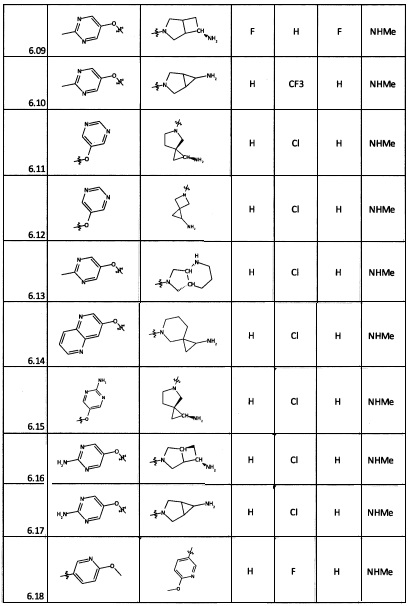
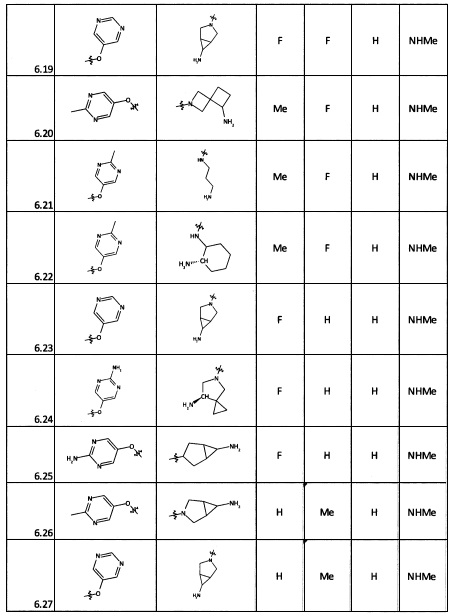
или соединение формулы I выбрано из группы:
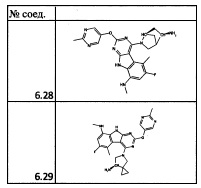
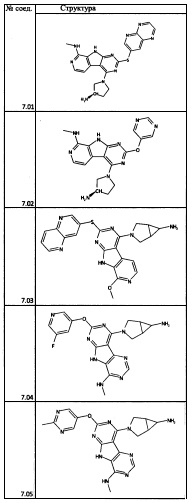
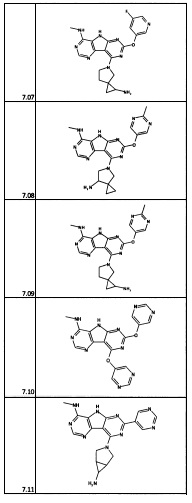
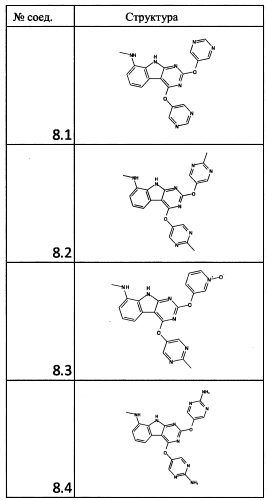
22. Соединение, выбранное из группы, состоящей из
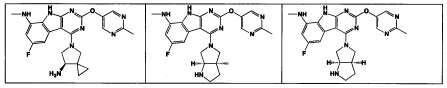
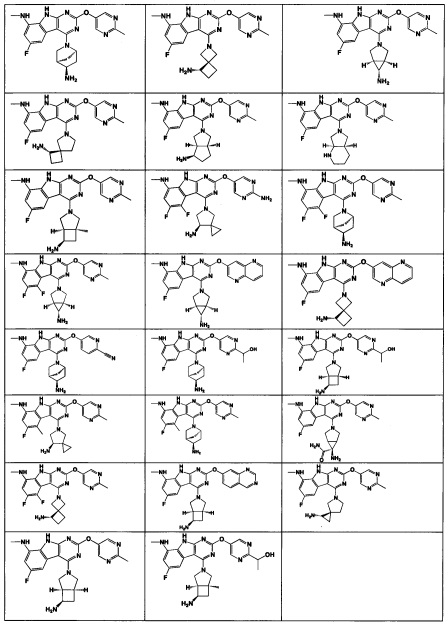
23. Способ получения соединения по п. 1, включающий:
обработку
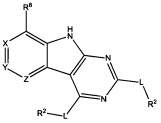 ,
,
где L и R2 такие же, как в п. 1,
соединением HR4 с образованием соединения Формулы I;
необязательно дополнительно включающий, перед стадией обработки, защиту R8 защитной группой или защиту амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и
необязательно снятие защитной группы после стадии обработки.
24. Способ по п. 23, отличающийся тем, что перед стадией обработки указанный способ дополнительно включает:
взаимодействие
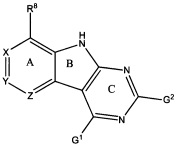
с R2LH в щелочных условиях,
причем
G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из Cl, Br, F, I, SR, SOR, SO2R, OSO2R и OBt,
R представляет собой С1-8 алкил, арил или гетероарил, содержащий 0-5 атомов О, S или N, необязательно замещенный С1-4 алкилом, С1-4 алкокси, Cl, Br, F, I или NO2; и
Bt представляет собой бензотриазол;
с образованием соединения, имеющего структуру
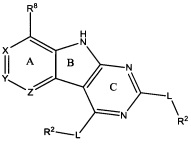 .
.
25. Способ получения соединения по п. 1, в котором R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к кольцу С через N вторичного или третичного амина, включающий:
обработку
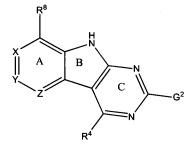
соединением R2LH в щелочных условиях, причем G2 представляет собой уходящую группу, выбранную из группы, состоящей из Cl, Br, F и I;
L, R2 и R8 имеют значения, указанные в п. 1; и
необязательно дополнительно включающий, перед стадией обработки, защиту R8 защитной группой или защиту амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и снятие защиты с R8 и R4 после стадии обработки.
26. Способ по п. 25, отличающийся тем, что перед стадией обработки указанный способ дополнительно включает:
взаимодействие
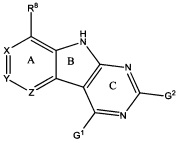
с HR4 с образованием
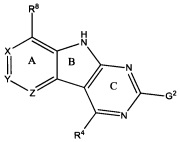
причем G1 представляет собой уходящую группу, выбранную из группы, состоящей из Cl, Br, F и I.
27. Способ получения соединения по п. 1, в котором R4 представляет собой необязательно замещенный вторичный или третичный амин, присоединенный к кольцу С через N вторичного или третичного амина, включающий:
обработку
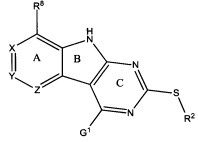
причем G1 представляет собой уходящую группу, выбранную из SО2галогенида, бис(2-оксо-3-оксазолидинил)фосфина (ВОР) или бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата (руВОР),
R2 и R8 имеют значения, указанные в п. 1;
соединением HR4 с образованием соединения Формулы I;
необязательно дополнительно включающий, перед стадией обработки, защиту R8 защитной группой или защиту амина в R4, который не является N вторичного или третичного амина, при его наличии, защитной группой; и снятие защиты с R8 и R4 после стадии обработки.
28. Способ по п. 27, отличающийся тем, что перед стадией обработки указанный способ дополнительно включает:
осуществление взаимодействия
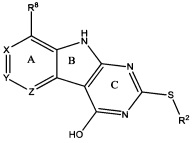
с G1X1 с образованием
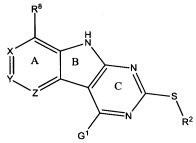
причем G1X1 представляет собой SO2галогенид, бис(2-оксо-3-оксазолидинил)фосфин (ВОР) или бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфат (руВОР).
29. Способ по п. 28, отличающийся тем, что перед стадией обработки указанный способ дополнительно включает:
перед указанной стадией взаимодействия связывание
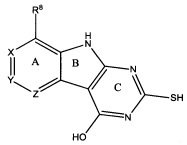
с R2X2, причем X2 представляет собой Br или I, с образованием
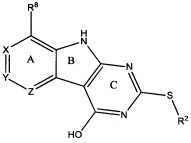 .
.
30. Промежуточное соединение для получения любого из соединений по пп. 1-22, имеющее структуру:
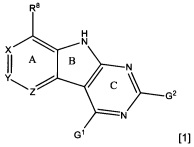
где
G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из SH, ОН, Cl, Br, F, I, SR, SOR, SO2R, OSO2R и OBt;
R представляет собой C1-8алкил, Ar, где Ar представляет собой фенил, необязательно замещенный С1-4алкилом, С1-4алкокси, галогеном или NO2;
Bt представляет собой бензотриазол;
R8 представляет собой Н, Cl, F, Br, OH, NH2, C1-3алкил, амино-С1-3алкил, аминоциклопропил, ОСН3, ОСН2СН3, циклопропил, СН2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH; и
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY и CRZ соответственно, при условии, что не более двух из X, Y и Z представляют собой N,
причем RX представляет собой Н, СН3, Cl, Br или F;
причем RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F;
причем RZ представляет собой H, СН3, Cl, Br или F;
при условии, что R8 не является СН3, и
при условии, что если R8 представляет собой ОСН3, то RX и RY не являются ОН;
причем один или более атомов азота в указанном соединении защищены карбобензилоксигруппой (Cbz) или ВОС.
31. Промежуточное соединение по п. 30, отличающееся тем, что G1 и G2 представляют собой уходящие группы, независимо выбранные из группы, состоящей из тозилата и мезилата.
32. Соединение, имеющее структуру Формулы I:
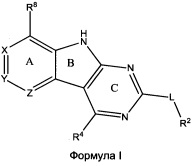
или его фармацевтически приемлемая соль,
где L представляет собой О или S;
R8 представляет собой Н, Cl, F, Br, ОН, NH2, C1-3aлкил, амино-C1-3aлкил, аминоциклопропил, ОСН3, OCH2CH3, циклопропил, CH2циклопропил, CH2Cl, CHCl2, CCl3, CH2CH2Cl, CH2Br, CHBr2, CH2F, CHF2, CF3, CH2CH2F, CH2CHF2, CH2CF3, NHNH2, NHOH, NHNHCH3, NHOCH3, NHCD3, SCH3 или NHCOH;
X, Y и Z независимо выбраны из группы, состоящей из N, CRX, CRY, и CRZ, при условии, что не более чем два из X, Y и Z представляют собой N,
причем RX представляет собой Н, СН3, Cl, Br или F;
причем RY представляет собой Н, СН3, CHF2, CF3, CN, СН2СН3, Cl, Br или F;
причем RZ представляет собой Н, СН3, Cl, Br или F;
R2 представляет собой  ,
, 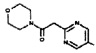 ,
, 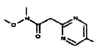 ,
,
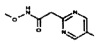 ,
, 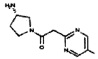 ,
, 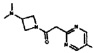 ,
, 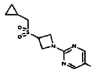 ,
,
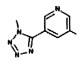 , или
, или 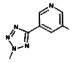 ;
;
R4 представляет собой:
Н;
необязательно замещенный ORa, где Rа представляет собой пиридил, пиримидил, необязательно замещенный СН3 и NH2;
необязательно замещенный насыщенный вторичный или третичный амин, выбранный из
4-6-членного алифатического амина,
4-6-членного моноциклического амина или
5-10-членного бициклического амина с 1-3 гетероатомами, выбранными из N, О или S, где указанный необязательно замещенный насыщенный 5-10-членный бициклический амин представляет собой необязательно спиро- или ортоконденсированный бициклическй амин; или
необязательно замещенный 5-6-членный ненасыщенный циклический остаток, содержащий 0-2 N гетероатома;
причем указанный необязательно замещенный насыщенный вторичный или третичный амин присоединен к С кольцу через N вторичного или третичного амина; и
необязательный заместитель выбран из
ОН,
галогена,
NH2,
C(O)CH2NH2,
C(O)NH2,
CH2NHC(O),
C5-6гетероциклил, возможно замещенный CH3 или CF3,
CH2OH,
-С3-6циклоалкил, возможно замещенный NH2,
-C3-6гетероциклил, содержащий 0-2 N атома, возможно замещенный NH2,
-СН3,
-C(O)CH3,
-C(O)C3-6гетероциклил, содержащий 1-3 О атома,
-CH(OH)C3-6гетероциклил, содержащий 1-3 S атома,
-NHCOCH3,
-NHCH2CH2CN,
-NHCH2C3-6циклоалкил,
-C1-4aлкилC3-6гетероциклил, содержащий 0-2 гетероатома, выбранных из О и N,
-C1-4aлкилNH2,
=NOCH3,
-NHC(O)CH2NH2,
-NHCH2CH2OH,
-NHCH2CH2NH2,
-NHC(O)CH(NH2)CH2OH,
-NHC(O)CH2NHC(O)CH(NH2)CH2OH,
-C(O)OCH2CH3,
-ОС1-4алкил.
33. Соединение по п. 1, представляющее собой
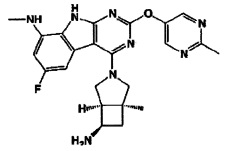 .
.
34. Соединение по п. 1, представляющее собой
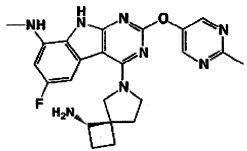 .
.
35. Соединение по п. 1, представляющее собой
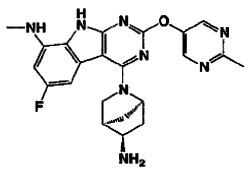 .
.
| US 7326712 B2, 2008-02-05 (колонка 81, соединение 32-25; колонка 64, схема 6, соединение 7; колонка 156, схема 14, соединение 22) | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| US 7326712 B2 (MONTIGEN PHARMACEUTICALS INC ), 2008-02-05 (колонка 81, соединение 32-25; колонка 64, схема 3, соединение 7, схема 6, соединение 7, 17; колонка 156, схема 14, соединение 22, 23) | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| WO 03062443 A2 (ANTEX PHARMA INC), 2003-07-31 (схема 2, формула изобретения) | |||
| US 2009143399 A1 (UNIV ARIZONA), 2009-06-04 (табл | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| СПОСОБЫ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА, АЗОТСОДЕРЖАЩИЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРЕДНАЗНАЧЕННАЯ ДЛЯ ВВЕДЕНИЯ ИНГИБИТОРА ТИРОЗИНКИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА, НАПРИМЕР, ERB-B2, ERB-B3 ИЛИ ERB-B4, И ПРОТИВОЗАЧАТОЧНАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2158127C2 |
| Способ образования азотной кислоты | 1921 |
|
SU1428A1 |
| Способ контроля прочностных характеристик ферромагнитных материалов и устройство для его осуществления | 1990 |
|
SU1749822A1 |

