Техническая область
Настоящее изобретение относится к области медицинской химии и, в частности, к ингибитору FAK и комбинациям лекарственных средств с ним.
Уровень техники
Киназа фокальной адгезии (FAK) представляет собой нерецепторную тирозинкиназу, содержащуюся в клетках, которая была впервые обнаружена в трансфицированных фибробластах эмбрионов кур линии V-Src. FAK имеет более высокую степень экспрессии в большинстве тканей, и ее белковая последовательность имеет более высокую гомологию у многих видов (у мышей, жаб, людей и др.). FAK представляет собой место пересечения нескольких путей передачи сигналов в клетке, участвующее в образовании, пролиферации, метастазировании и апоптозе опухолей, сердечно-сосудистых заболеваниях и других биологических процессах. В настоящее время она является одной из противоопухолевых мишеней, которой стали уделять большое внимание.
В ходе недавних исследований было установлено, что FAK может быть активирована целым рядом различных факторов, включая интегрины, рецепторы, связанные с G-белком и др. В то же время FAK регулирует внутриклеточные пути передачи сигналов Р53 и PI3K-AKT-mTOR с помощью зависимых и независимых от киназ путей, а также участвует в биологических процессах жизнеобеспечения, пролиферации и метастазирования опухолевых клеток. Первоначальная попытка заключалась в подавлении роста опухоли путем подавления экспрессии FAK в опухолевых клетках. Трансфекция FAK с инактивированным карбоксильным концом (FAK-CD) вызывает подавление экспрессии FAK, снижение клеточной адгезии и пролиферации, а в экспериментах in vivo достигается подавляющее действие на рост клеток рака молочной железы. Трансфекция плазмиды, содержащей PFIK с подавленной экспрессией FAK (FAK-siRNA), вызывает подавление роста раковой опухоли в условиях in vivo. Одновременное подавление экспрессии FAK и сигнальных молекул, расположенных после FAK по пути сигнала (таких как Src), может усиливать противоопухолевый эффект.
Ввиду важности функций FAK в опухолевых клетках, надежности трансфекции генов и безопасности вирусных векторов начали появляться низкомолекулярные ингибиторы, действующие на основе сигнального пути FAK, и в последние годы были достигнуты хорошие результаты. В настоящее время существует множество ингибиторов FAK, действующих как противоопухолевые лекарственные средства, которые находятся на стадии доклинических исследований или клинических испытаний. Согласно данным из литературы (новая противоопухолевая мишень - киназа фокальной адгезии FAK и результаты исследования ее ингибиторов, Chen Ying и др.), ТАЕ226, также известный как NVP-226, может блокировать сайт соединения FAK и АТФ, а также сайты фосформирования Y397 и Y861 в FAK, и участвовать в подавлении активности FAK. Однако в данной области техники по-прежнему существует потребность в разработке ингибиторов FAK с лучшей ингибирующей активностью или лучшими фармакодинамическими свойствами.
Термин «дейтерированные лекарственные средства» означает, что часть атомов водорода в молекулах этих лекарственных средств замещена дейтерием. Дейтерий (D) является стабильным изотопом водорода. Так как форма и объем дейтерия в лекарственном средстве в основном такие же, как у водорода, некоторые атомы водорода в молекуле лекарственного средства замещены дейтерием, однако активность молекулы данного лекарственного средства в основном остается неизменной. Кроме того, поскольку масса атомов дейтерия вдвое больше, чем у водорода, вибрационная энергия нулевой точки углерод-дейтериевых связей (CD) меньше, чем у углерод-водородных связей (СН), и, таким образом, углерод-дейтериевая связь является более стабильной. Замещение части атомов водорода в молекулах лекарственного средства дейтерием может замедлить процесс разложения лекарственного средства, продлить действие дейтерированного лекарственного средства в организме и достичь цели по изменению скорости метаболизма или метаболического пути лекарственного средства, тем самым улучшая фармакокинетику и снижая метаболическую токсичность лекарственного средства. Ввиду важности применения ингибиторов FAK в области лечения опухолей и связанных с ними ограничений, объединение их с технологией получения дейтерированных лекарственных средств, открытием новых химических соединений, снижением их воздействия на работу печени и почек, а также повышение безопасности и эффективности лекарственного средства является направлением исследований, способствующим дальнейшему развитию данного вида лекарственных средств и имеющим большие прикладное значение.
Совместное применение лекарственных средств является эффективным способом улучшения терапевтических эффектов лекарственных средств. Данные о совместном применении дейтерированных ингибиторов FAK с другими противораковыми препаратами или противораковыми методами отсутствуют.
Описание изобретения
Для решения вышеупомянутых задач в настоящем изобретении предложено дейтерированное соединение и его применение в качестве ингибитора FAK, а также схема применения указанного дейтерированного соединения в комбинации с другими противораковыми лекарственными средствами.
В настоящем изобретении предложено соединение формулы (I) или его оптический изомер, таутомер, фармацевтически приемлемая соль, пролекарство, гидрат или сольват:

в котором кольцо S выбрано из ароматического кольца или пятичленного гетероциклического кольца; каждый А, В, X, Y и Z независимо выбран из С или N; Е отсутствует или представляет собой метилен;
и/или R6 выбран из Н или отсутствует; R7 выбран из Н, N или отсутствует; R8 выбран из галогеналкила или галогена, или R7 и R8 связаны друг с другом с образованием кольца; и/или R9 выбран из -NMeSO2Me, -CONHOMe, -CONHMe, амида, водорода или отсутствует; R10 выбран из Н или отсутствует; R11 выбран из -NHSO2Me, галогена, замещенного пиперазина или водорода, и заместитель в пиперазине представляет собой этанольную группу; R12 выбран из -SO2Me или Н; R13 выбран из -CONHMe, -CONHOMe, N-алкилсульфонамида, Н или отсутствует, или R11 и R13 связаны друг с другом с образованием кольца:
Dx в формуле (I) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-A):
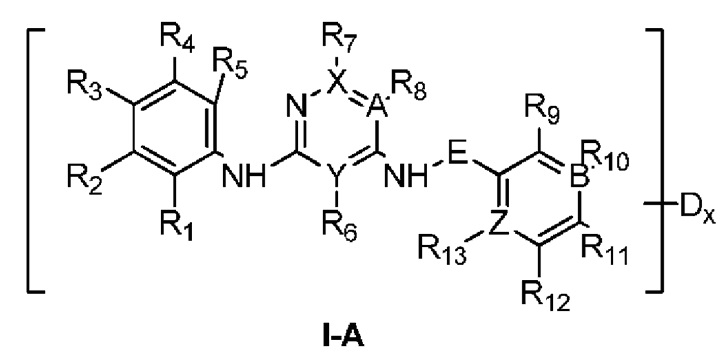
где каждый А, В, X, Y и Z независимо выбран из С или N; Е отсутствует или представляет собой метилен:
R1 и R5 выбраны из Н или метокси-группы; R2 и R4 выбраны из Н или метокси-группы; R3 выбран из Н, -CONHMe, алкоксиамида, морфолина, метокси-группы, этиламина или сульфонамида, или R2 и R3 связаны друг с другом с образованием кольца, или R3 и R4 связаны друг с другом с образованием кольца;
и/или R6 выбран из Н или отсутствует; R7 выбран из Н, N или отсутствует; R8 выбран из галогеналкила или галогена, или R7 и R8 связаны друг с другом с образованием кольца; и/или R9 выбран из NMeSO2Me, -CONHOMe, -CONHMe, амида, водорода или отсутствует; R10 выбран из Н или отсутствует; R11 выбран из -NHSO2Me, галогена, замещенного пиперазина или водорода, и заместитель в пиперазине представляет собой этанольную группу; R12 выбран из -SO2Me или Н; R13 выбран из -CONHMe, N-алкилсульфонамида, Н или отсутствует, или R11 и R13 связаны друг с другом с образованием кольца, или R13, R3 и R4 связаны друг с другом с образованием кольца;
Dx в формуле (I-A) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-B):

где А, X и Y выбраны из С или N; Е отсутствует;
каждый R14, R15 и R16 независимо выбран из Н, алкила C1-6, циклоалкила С3-6, метила, этила или изопропила;
и/или R6 представляет собой Н; R7 представляет собой Н; R8 представляет собой галоген; R9 и R13 выбраны из -CONHOMe, -CONHMe или Н; R10 и R12 представляют собой Н;
R11 выбран из галогена, Н или замещенного пиперазина, и заместитель в пиперазине представляет собой этанольную группу;
Dx в формуле (I-B) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥ 1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-C):
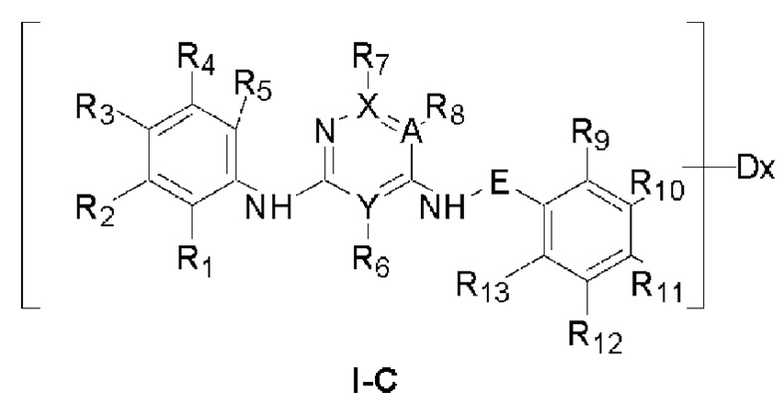
где А, X и Y выбраны из С или N; Е представляет собой метилен или отсутствует;
R9 и R13 выбраны из Н, -NMeSO2Me, -CONHOMe или -CONHMe; R10 и R12 представляют собой Н;
R11 выбран из Н, замещенного пиперазина или галогена, и заместитель в пиперазине представляет собой этанольную группу;
и/или R6 выбран из Н; R7 выбран из Н; R8 выбран из галогеналкила или галогена; или R7 и R8 связаны друг с другом с образованием кольца;
и/или R1, R4 и R5 выбраны из Н или метокси-группы; R2 выбран из Н или метокси-группы; R3 выбран из Н, -CONHMe, алкоксиамида, морфолина, метокси-группы, этиламина или сульфонамида; или R2 и R3 связаны друг с другом с образованием кольца;
Dx в формуле (I-C) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥ 1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-D):
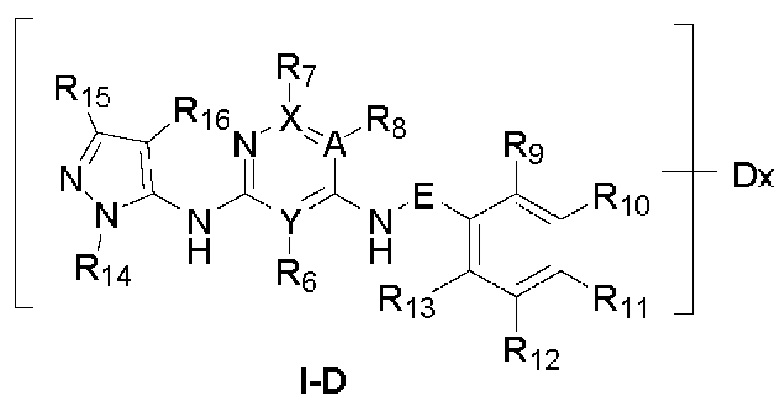
где R9 и R13 выбраны из -CONHOMe, -CONHMe или Н; R10 и R12 представляют собой Н; R11 выбран из галогена, Н или замещенного пиперазина, и заместитель в пиперазине представляет собой этанольную группу;
А, X и Y выбраны из С или N; Е отсутствует;
и/или, R6 выбран из Н; R7 выбран из Н; R8 представляет собой галоген;
и/или, R14 выбран из метила, этила или изопропила; R15 выбран из метила или Н; R16 представляет собой Н;
Dx в формуле (I-D) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-Е):
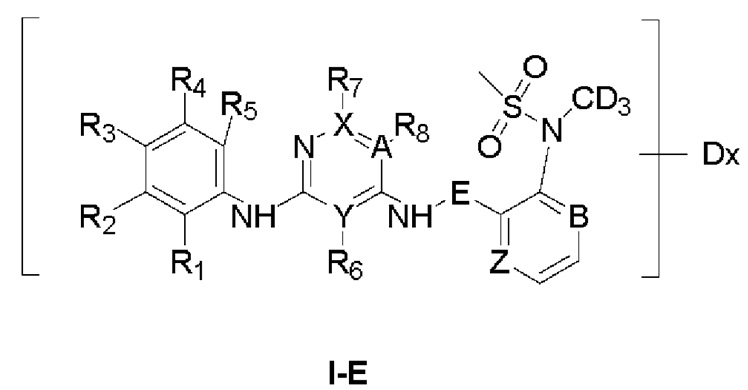
где В и Z выбраны из С или N; Е представляет собой метилен; Y представляет собой N; X и А представляют собой С:
и/или, R6 отсутствует; R7 представляет собой Н; R8 представляет собой галогеналкил:
и/или, R1 и R2 представляют собой Н; R3 представляет собой -CONHMe; R4 представляет собой Н; или R3 и R4 связаны друг с другом с образованием кольца;
Dx в формуле (1-Е) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-F):
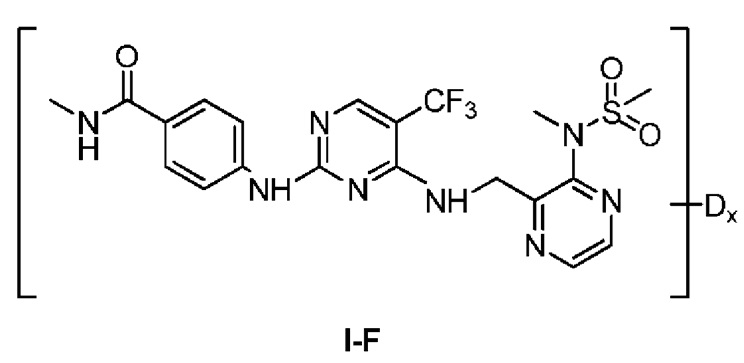
Где Dx в формуле (I-F) указывает на то, что водород по меньшей мере на одном атоме углерода соединения, заключенного в скобки, замещен дейтерием, а х представляет собой целое число ≥1.
В предпочтительном варианте реализации указанное соединение имеет структуру формулы (I-G):

где один или более из R1, R5, R17, R18, R19, R20, R21, R22, R23 и R24 замещены дейтерием. В предпочтительном варианте реализации указанное соединение выбрано из одного из следующих соединений, замещенных дейтерием, но не ограничивается ими:

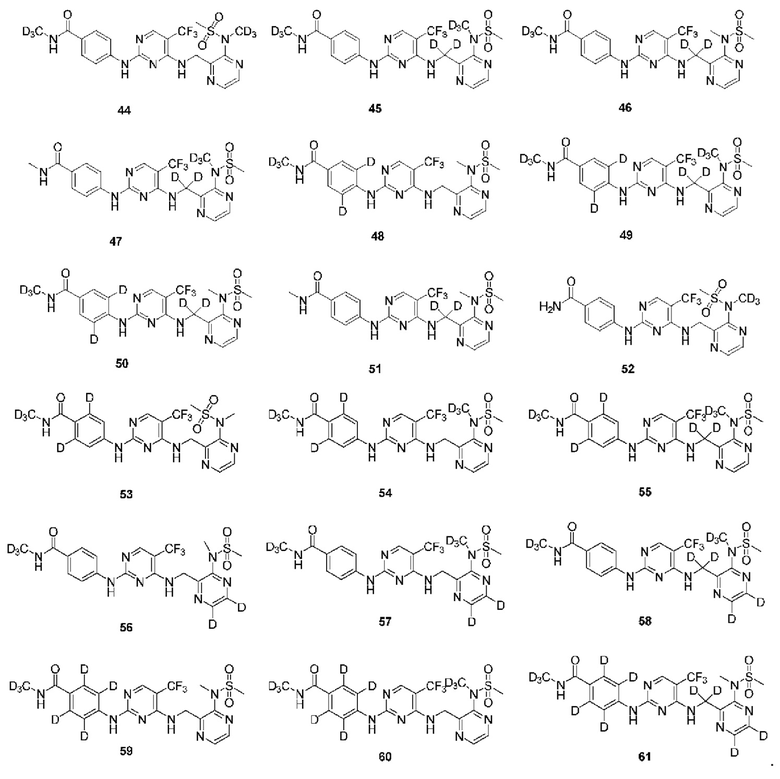
В предпочтительном варианте реализации указанное соединение выбрано из одного из следующих соединений или из одного из следующих соединений, замещенных дейтерием, но не ограничивается ими:
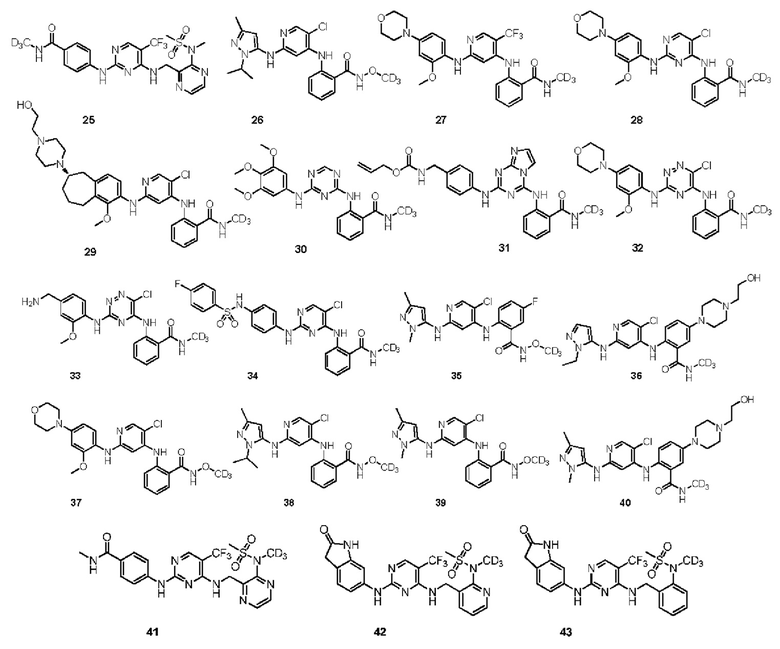
Настоящее изобретение также относится к применению вышеуказанного соединения или его оптического изомера, таутомера, фармацевтически приемлемой соли, пролекарства, гидрата или сольвата для получения ингибитора FAK; предпочтительно ингибитор FAK представляет собой лекарственное средство для лечения рака.
При этом указанный рак представляет собой солидные опухоли;
Солидные опухоли включают мезотелиому, рак поджелудочной железы, опухоли мягких тканей, метастазы, не являющиеся солидными раковые заболевания, саркомы, аденокарциному, рак легких, рак молочной железы, лимфому, рак желудочно-кишечного тракта, рак мочеполовой системы, рак предстательной железы и рак яичников; рак желудочно-кишечного тракта включает рак толстой кишки; рак мочеполовой системы включает опухоли почек, уротелия или яичек; и рак яичников включает распространенный рак яичников:
Мезотелиома включает нейрофибромы, рак почек, рак легких, мелкоклеточный рак легких, немелкоклеточный рак легких, KRAS-мутантный немелкоклеточный рак легких, рак печени, рак щитовидной железы, рак молочной железы, опухоли нервной системы, шванному, менингиому, неврому, аденоидную кистозную карциному, эпендимому, опухоли эпендимы, злокачественный плеврит, злокачественную мезотелиому плевры, тройной негативный рак молочной железы, негематологические злокачественные новообразования, меланому, колоректальную карциному, лейкоз, аденокарциному, солидные опухоли;
Меланома включает местно-распространенную меланому, меланому, вызванную мутантным NRAS, метастатическую злокачественную меланому кожи; колоректальный рак включает метастатический колоректальный рак; и лейкоз включает острый миелогенный лейкоз; аденокарцинома включает аденокарциному; и солидные опухоли включают местно-распространенные солидные опухоли, метастатические солидные опухоли и гепатоцеллюлярную карциному.
В настоящем изобретении дополнительно предложена комбинация лекарственных средств для лечения опухолей, которая содержит вышеуказанное соединение и противораковое лекарственное средство в тех же или других специфицированных препаративных формах для одновременного или раздельного введения, и фармацевтически приемлемый носитель. Противораковое лекарственное средство представляет собой лекарственное средство для иммунотерапии, лекарственное средство для химиотерапии или лекарственное средство для лучевой терапии.
Иммунотерапевтические лекарственные средства выбраны из ингибиторов контрольных точек иммунного ответа, ингибиторов PD-1, ингибиторов PD-L1, антител, ингибирующих CTLA-4, антител, ингибирующих TIM3, антител, ингибирующих LAG3, антител, ингибирующих TIGIT, антител, блокирующих мишени контрольных точек, костимулирующих антител или клеток для CAR-T-клеточной терапии. Указанный ингибитор PD-1 или ингибитор PD-L1 включает, но не ограничивается ими: ниволумаб, СТ-011; АМР-224, пембролизумаб, пидилизумаб, MK-3475, BMS936559, MEDI4736, MSB001071 8С, MPDL-3280A, SHR-1210, ГВ1308, BGB-A317, JS001, GLS-010, GB226 гептанолимаб, HLX10, AK103, AK104, AK105, AK112, SSI-361, JY034, KN035, SHR1316, TQB2450, KL-A167, CS1001, STI-A1014, JS003, AK106, HLX-09, антитело mPD-1;
Антитела, блокирующие мишени контрольных точек, включают IMP321 и MGA271; Костимулирующее антитело включает антитело к 4-1ВВ, антитело к ОХ40, антитело к GITR, антитело к CD27 и антитело к CD40.
Указанные химиотерапевтические лекарственные средства представляют собой токсические препараты, алкилирующие препараты, антиметаболические препараты, антибиотики, препараты гормональной терапии, противораковые препараты из натурального продукта, препараты, ингибирующие топоизомеразу, иммунные препараты, комплексные препараты платины, ингибиторы киназ, антипролиферативные препараты, антитела, интерфероны, или препараты, регулирующие сигнальные пути андрогенов. Указанные токсические препараты включают, но не ограничиваются ими: гемцитабин, паклитаксел и доцетаксел;
Указанные ингибиторы киназ включают, но не ограничиваются ими: ингибиторы MEK (митоген-активируемых протеинкиназ), ингибиторы c-Met, ингибиторы VEGFR2 и ингибиторы EGFR;
Указанные лекарственные средства, регулирующие сигнальные пути андрогенов, включают, но не ограничиваются ими: ингибиторы синтеза андрогенов, ингибиторы CYP17A, ингибиторы рецепторов андрогенов, ингибиторы BET, ингибиторы BRD4, ингибиторы RORγ, ингибиторы СВР/Р300, ингибиторы ВМХ, ингибиторы PARP; в предпочтительном варианте реализации, ингибиторы рецепторов андрогенов включают, но не ограничиваются ими: энзалутамид, апалутамид, бикалутамид, абиратерон, ODM-201, EPI-001, ONC1-13B, ЕМ-5854, JNJ-63576, TAS-3681, НС-1119, прокрутамид, SHR3680. В настоящем изобретении дополнительно предложено применение вышеуказанной комбинации лекарственных средств для получения лекарственных средств для лечения онкологических заболеваний.
При этом указанные онкологические заболевания представляет собой солидные опухоли; Солидные опухоли включают мезотелиому, рак поджелудочной железы, опухоли мягких тканей, метастазы, не являющиеся солидными раковые заболевания, саркомы, аденокарциному, рак легких, рак молочной железы, лимфому, рак желудочно-кишечного тракта, рак мочеполовой системы, рак предстательной железы и рак яичников; рак желудочно-кишечного тракта, включая рак толстой кишки; рак мочеполовой системы, включая опухоли почек, уротелия или яичек; а также рак яичников, включая распространенный рак яичников;
Мезотелиома включает нейрофибромы, рак почек, рак легких, мелкоклеточный рак легких, немелкоклеточный рак легких, KRAS-мутантный немелкоклеточный рак легких, рак печени, рак щитовидной железы, рак молочной железы, опухоли нервной системы, шванному, менингиому, неврому, аденоидную кистозную карциному, эпендимому, опухоли эпендимы, злокачественный плеврит, злокачественную мезотелиому плевры, тройной негативный рак молочной железы, негематологические злокачественные новообразования, меланому, колоректальную карциному, лейкоз, аденокарциному, солидные опухоли;
Меланома включает местно-распространенную меланому, меланому, вызванную мутантным NRAS, метастатическую злокачественную меланому кожи; колоректальный рак включает метастатический колоректальный рак; и лейкоз включает острый миелогенный лейкоз; аденокарцинома включает аденокарциному; и солидные опухоли включают местно-распространенные солидные опухоли, метастатические солидные опухоли и гепатоцеллюлярную карциному.
В настоящем изобретении понятие «алкил» включает в себя линейный или разветвленный алкил.
В настоящем изобретении термин «соединение по настоящему изобретению» означает соединение формулы (I). Данный термин также включает различные кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты, оптические изомеры, таутомеры и пролекарства соединения формулы (I).
В настоящем изобретении термин «фармацевтически приемлемая соль» означает соль, пригодную для применения в качестве лекарственного средства, которая образована соединением по настоящему изобретению и кислотой или основанием. Фармацевтически приемлемые соли включают неорганические соли и органические соли. Предпочтительным классом солей является соль соединения по настоящему изобретению с щелочным металлом. Щелочные металлы, пригодные для солеобразования, включают, но не ограничиваются ими: литий, натрий, калий, кальций, магний и т.п.
Способ введения соединения или фармацевтической композиции по настоящему изобретению не ограничивается каким-либо определенным образом, и стандартные способы введения включают (но не ограничиваются ими): пероральное, парентеральное (внутривенное, внутримышечное или подкожное) и местное введение. В настоящем изобретении предложено дейтерированное соединение, которое в сравнении с данным соединением до дейтерирования, демонстрирует лучшую фармакокинетику, более высокую максимальную концентрацию в плазме крови, более высокую степень экспозиции и более длительный период полувыведения, а также имеет еще более высокие метаболические характеристики. Более того, дейтерированное соединение по настоящему изобретению может эффективно ингибировать активность FAK и имеет очень хорошие перспективы применения для получения ингибиторов FAK и/или лекарственных средств для лечения рака. В то же время применение дейтерированного соединения по настоящему изобретению в комбинации с противораковыми лекарственными средствами (такими как ингибиторы PD-1) может вызывать синергетический эффект, значительно улучшить подавляющее действие на опухоли и обеспечить лучший выбор для клинического лечения рака.
Естественно, на основании вышеприведенного содержания настоящего изобретения, в соответствии с общеизвестными техническими знаниями и общепринятыми способами в данной области, могут быть дополнительно произведены иные всевозможные модификации, замены или изменения без отступления от технической сущности настоящего изобретения. Содержание настоящего изобретения дополнительно проиллюстрировано конкретными примерами указанных вариантов реализации. Однако это не означает, что объем вышеуказанного объекта настоящего изобретения ограничивается нижеследующими примерами. Все способы, осуществленные на основе вышеизложенного содержания настоящего изобретения, входят в объем настоящего изобретения.
Описание чертежей
Фиг. 1. Фармакодинамический эксперимент по изучению действия дейтерированного соединения по настоящему изобретению на животной модели с опухолью МС38.
Фиг. 2. Фармакодинамический эксперимент по изучению действия дейтерированного соединения по настоящему изобретению на животной модели с опухолью PAN02.
Примеры
Исходные материалы и оборудование, используемые в настоящем изобретении, могут быть приобретены посредством покупки имеющихся в продаже продуктов.
Пример 1. Синтез N-тридейтерометил-4-((4-(((3-(N-метансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 25)
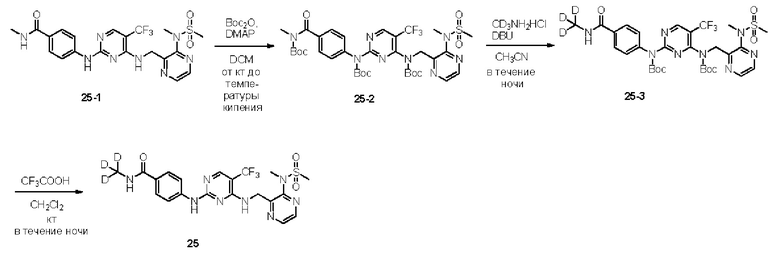
Этап 1: Синтез соединения 25-2
25-1 (200 мг, 0,39 ммоль) и ДМАП (1,29 г, 10,57 ммоль) добавляли к 10 мл дихлорметана, к которому затем по каплям добавляли (Вос)2О (1,71 г, 7,83 ммоль). Систему нагревали с обратным холодильником на масляной бане в течение 24 часов. На следующий день реакционную смесь охладили до комнатной температуры и добавили в нее дихлорметан и 0,1Н раствор HCl. Смесь экстрагировали, а затем отстаивали для разделения слоев. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали отсасыванием и упаривали для удаления растворителя. Первичный продукт отделяли методом колоночной хроматографии с получением твердого вещества белесого цвета 25-2 (136 мг, выход: 42,8%). МС (М+1): 811.2.
Этап 2: Синтез соединения 25-3
25-2 (136 мг, 0,17 ммоль) и дейтерированный гидрохлорид метиламина (189 мг, 2,68 ммоль) добавили к 5 мл ацетонитрила и данную смесь перемешивали при комнатной температуре. Затем добавили DBU (диазабициклоундецен) (613 мг, 4,03 ммоль) и постепенно растворяли до осветления реакционного раствора. После этого систему на ночь поместили на масляную баню для нагревания с обратным холодильником и протекания реакции. На следующий день реакционную смесь охладили до комнатной температуры и упаривали на ротационном испарителе для удаления растворителя. В систему добавили дихлорметан и 0,1Н раствор HCl, реакционную смесь активно перемешивали и отстаивали для разделения слоев.
Органическую фазу промывали сначала очищенной водой, затем насыщенным солевым раствором, сушили над безводным сульфатом натрия и упаривали на ротационном испарителе для удаления растворителя. Остаток разделяли и очищали методом предварительной ТСХ (РЕ/ЕА (петролейный эфир/этилацетат)=2:1) с получением белого твердого вещества 25-3 (42 мг, выход 35,3%). МС (М-Вос+1): 614.2.
Этап 3: Синтез соединения 25
25-3 (42 мг, 0,06 ммоль) добавили к 2 мл дихлорметана и перемешивали при комнатной температуре (до полного растворения и осветления раствора), затем добавили 0,1 мл трифторметансульфоновой кислоты. Реакционный раствор постепенно становился прозрачным и чистым, и смесь оставили на ночь при комнатной температуре для перемешивания и протекания реакции. На следующий день растворитель удалили ротационным упариванием, затем в систему добавили этилацетат и насыщенный раствор NaHCO3. Полученную смесь активно перемешивали и затем отстаивали для разделения слоев. Было зафиксировано, что значение рН водной фазы составляет примерно от 7 до 8. Органический слой дважды промывали сначала водой, затем насыщенным солевым раствором и сушили над безводным сульфатом натрия.
Растворитель удаляли ротационным упариванием до получения белого твердого соединения 25 (24 мг, выход: 80,0%).
1Н ЯМР (400 Гц, ДМСО-d6(диметилсульфоксид-D6): δ 9,863 (1H, s), 8,688 (1H, d, J=2,4 Гц), 8,581 (1H, d, J=2,8 Гц), 8,316 (1Н, s), 8 8,180 (1H, s), 7,665-7,594 (4Н, dd, J1=19,6 Гц, J2=8,8 Гц), 7,476-7,450 (1Н, t, J=5,2 Гц), 5,001 (2Н, d, J=4,8 Гц), 3,221 (3Н, s), 3,199 (3Н, s). ЖХ-МС(М+Н+): 514.2.
Соединения 26-40 получали, используя исходные материалы, соответствующие соединению, и способ получения, аналогичный синтезу соединения 25.
Пример 2. Синтез N-метил-4-((4-(((3-(N-тридейтерметилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 41)
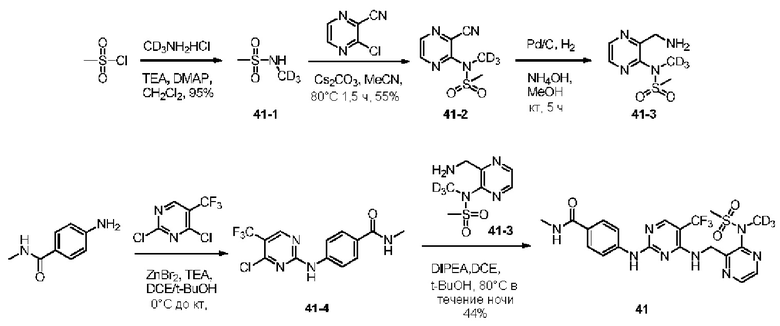
Этап 1: Синтез N-тридейтерометилметансульфонамида (соединение 41-1) Дейтерированный гидрохлорид метиламина (7,75 г, 109,99 ммоль) поместили в одногорлую круглодонную колбу объемом 250 мл с одновременным добавлением дихлорметана (120 мл). Данную смесь перемешивали при комнатной температуре. Затем систему поместили на водяную баню со льдом для охлаждения и перемешивали. Через 15 мин добавляли последовательно триэтиламин (21,73 г, 214,75 ммоль) и ДМАП (128 мг, 1,05 ммоль). Затем смесь дополнительно перемешивали в течение 10 мин на водяной бане со льдом. После этого в систему добавили метансульфонилхлорид (12,0 г, 104,76 ммоль) и затем сняли с ледяной бани. Систему оставили на ночь при комнатной температуре для перемешивания и протекания реакции. На следующий день реакцию завершили детектированием методом ТСХ и систему отфильтровали. Осадок с фильтра несколько раз промыли этилацетатом, фильтрат объединили и растворитель удалили ротационным упариванием. Затем к системе добавили этилацетат (90 мл) и энергично перемешивали в течение 10 мин. После этого систему снова отфильтровали отсасыванием и осадок с фильтра несколько раз промыли этилацетатом в небольших количествах. Фильтрат объединили и концентрировали в вакууме до получения N-тридейтерометилметансульфонамида (11,16 г) в виде бесцветной прозрачной маслянистой жидкости, который сразу же использовали на следующем этапе без дополнительной очистки. Выход: 94,9%. ЖХ/МС (ИЭР+): масса/заряд 113,3 [М+Н]+ (рассчитано для C2H4D3NO2S, 113,1).
Этап 2: Синтез N-(3-цианопиразин-2-ил)-N-тридейтерометилметансульфонамида (соединение 41-2)
N-тридейтерометилметансульфонамид (6,0 г, 53,54 ммоль) и 2-хлор-3-цианопиразин (6,23 г, 44,62 ммоль) поместили в одногорлую круглодонную колбу объемом 500 мл с одновременным добавлением ацетонитрила (300 мл). Полученную смесь перемешивали при комнатной температуре. Далее в систему добавили карбонат цезия (24,71 г, 75,85 ммоль). Затем систему поместили на масляную баню при 80°С и далее реакционную смесь нагревали и перемешивали. Через 1,5 часа взяли образец и подвергли его ТСХ. ТСХ показала полное отсутствие исходных веществ. Нагревание прекратили и оставили систему охлаждаться естественным путем до комнатной температуры с последующей фильтрацией. Осадок с фильтра несколько раз промыли ацетонитрилом в небольших количествах и затем фильтрат объединили. Растворитель удалили ротационным упариванием. После этого в систему добавили этилацетат (150 мл) и воду (150 мл). Полученную смесь энергично перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (50 мл⋅3). Органические фазы объединили, промывали последовательно очищенной водой (30 мл × 3) и насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме для получения первичного продукта, который разделяли и очищали методом колоночной хроматографии с получением N-(3-цианопиразин-2-ил)-N-дейтерометилметансульфонамида (5,29 г) в виде бледно-коричнево-красной маслянистой жидкости. Выход: 55,1%. ЖХ/МС (ИЭР+): м/з 233,1 [М+H2O] (рассчитано для C7H5D3N4O2S [М+Н]+, 216,1).
1Н ЯМР (400 МГц, ДМСО-D6) δ 8,65-8,63 (dd, J=6,0, 2,4 Гц, 2Н), 3,26 (s, 3Н).
Этап 3: Синтез N-(3-(аминометил)пиразин-2-ил)-N-тридейтерометилметансульфонамида (соединение 41-3)
N-(3-цианопиразин-2-ил)-N-дейтерометилметансульфонамид (2,0 г, 9,30 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 500 мл, в которую добавили метанол (270 мл), и затем данную смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Далее в систему добавили влажный палладированный уголь (1 г) и аммиачную воду (20 мл). После этого из системы откачали воздух и произвели 5-кратную продувку газообразным аргоном для создания в системе среды из инертного газа. В системе снова произвели замену на водород, и после этого систему продолжали перемешивать и выдерживать при комнатной температуре для протекания реакции. Через 5 часов взяли образец и подвергли его ТСХ. ТСХ показала, что исходные вещества израсходованы. Реакцию остановили и отсоединили устройство гидрогенизации. Систему отфильтровали отсасыванием и осадок с фильтра несколько раз элюировали метанолом. Фильтрат объединили и ротационным упариванием удалили растворитель. Оставшуюся в системе воду удалили ротационным упариванием в несколько приемов с получением N-(3-(аминометил)пиразин-2-ил)-N-дейтерометилметансульфонамида в виде светло-желто-коричневой прозрачной маслянистой жидкости, который сразу же использовали на следующем этапе без дополнительной очистки. ЖХ/МС (ИЭР+): м/з 220,1 [М+Н]+ (рассчитано для C7H9D3N4O2S, 220,1).
Этап 4: Синтез сложного трет-бутилового эфира (4-(метилкарбамил)фенил)карбаминовой кислоты
4-((трет-бутоксикарбонил)амино)бензойную кислоту (3,0 г, 12,65 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 250 мл, в которую добавили 50 мл ДМФА, и перемешивали данную смесь при комнатной температуре. Далее в систему добавили последовательно EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) (4,8 г, 25,29 ммоль), TEA (триэтаноламин) (4,5 г, 44,28 ммоль), гидрохлорид метиламина (1,3 г, 18,98 ммоль) и ДМАП (16,0 мг, 0,13 ммоль). После этого систему перемешивали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день, определив, что исходные вещества израсходованы, в систему добавили этилацетат (70 мл) и воду (50 мл), и реакционную смесь энергично перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (20 мл ⋅ 3). Органические слои объединили, промыли сначала водой (20 мл ⋅ 3), затем насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. Растворитель удалили ротационным упариванием для получения первичного продукта, который затем разделили методом колоночной хроматографии с получением трет-бутилового эфира (4-(метилкарбамил)фенил)карбаминовой кислоты в виде твердого вещества белесого цвета (2,1 г, выход: 66,5%). МС (ИЭР): м/з 251,2 [М+Н]+.
Этап 5: Синтез соединения трифторацетат 4-амино-N-метилбензамида Отвесили 500,0 мг (2,00 ммоль) трет-бутилового эфира (4-(метилкарбамоил)фенил)карбаминовой кислоты и поместили в одногорлую круглодонную колбу объемом 50 мл, в которую добавили 10 мл дихлорметана и перемешивали данную смесь при комнатной температуре. После этого в систему добавили трифторуксусную кислоту (1 мл), и затем систему перемешивали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день ТСХ показала, что реакция завершена. Реакционную смесь концентрировали для удаления растворителя и излишков трифторуксусной кислоты, а остатки трифторуксусной кислоты в системе удаляли ротационным упариванием с дихлорметаном в несколько приемов до полного превращения системы в твердое вещество с получением трифторацетата 4-амино-№метилбензамида в виде твердого вещества белесого цвета (510,0 мг), который сразу же использовали на следующем этапе без дополнительной очистки.
Этап 6: Синтез соединения 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-метилбензамид
Отвесили 499,0 мг (2,30 ммоль) 2,4-дихлор-5-(трифторметил)пиримидина и поместили в одногорлую круглодонную колбу объемом 50 мл, в которую добавили 1,2-дихлорэтан (5 мл) и трет-бутанол (5 мл), и затем данную смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. После этого систему поместили на водяную баню со льдом для продолжения охлаждения и перемешивания. Через 15 минут в систему добавили бромистый цинк (1,4 г, 6,00 ммоль). Затем систему дополнительно перемешивали в течение 30 минут на водяной бане со льдом. Затем в систему добавили трифторацетат 4-амино-N-метилбензамида, синтезированный на предыдущем этапе, и триэтиламин (648,0 мг, 6,40 ммоль). После их добавления систему сняли с ледяной бани, перемешали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день после завершения реакции детектированием растворитель удалили ротационным упариванием. В систему добавили этилацетат (30 мл) и воду (20 мл), и затем реакционную смесь энергично перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (10 мл ⋅ 3). Органические слои объединили, промывали последовательно водой (15 мл ⋅ 3) и насыщенным солевым раствором (15 мл) и сушили над безводным сульфатом натрия. Реакционную смесь концентрировали при пониженном давлении для получения первичного продукта, который затем разделяли методом колоночной хроматографии с получением 4-((4-хлор-5-(трифторметил)гшримидин-2-ил)амино)-N-метилбензамида в виде твердого вещества белесого цвета (280,0 мг, выход: 42,3%). МС (ИЭР): м/з 331,0 [М+Н]+.
Этап 7: Синтез соединения N-метил-4-((4-(((3-(N-дейтерометансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамид
В одногорлую круглодонную колбу объемом 25 мл, содержащую N-(3-(аминометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (соединение 41-3, 65,8 мг, 0,30 ммоль), добавили 5 мл 1,2-дихлорэтана и 5 мл трет-бутанола и затем смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Далее в систему добавили 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-метилбензамид (100,0 мг, 0,30 ммоль) и диизопропилэтиламин (116,3 мг, 0,90 ммоль). После их добавления систему поместили на масляную баню при 80°С и нагревали с обратным холодильником для протекания реакции. Через 8 часов с помощью ТСХ зафиксировали полный расход исходных веществ. Нагревание прекратили и затем систему охладили до комнатной температуры, растворитель удалили ротационным упариванием для получения первичного продукта, который затем отделяли и очищали методом предварительной ТСХ с получением N-метил-4-((4-(((3-(N-тридейтерометилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде твердого вещества белесого цвета (соединение 41, 12 мг). Выход: 7,8%. МС (ИЭР) m/z 514,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1Н), 8,69 (s, 1H), 8,59 (s, 1H), 8,32 (s, 1H), 8,20 (d, J=4,0 Гц, 1H), 7,68-7,61 (dd, J=14,4, 8,4 Гц, 4ГГ), 7,41-7,39 (t, J=4,4 Гц, 1H), 5,01 (d, J=3,6 Гц, 2H), 3,20 (s, 3H), 2,76 (d, J=4,0 Гц, 3Н).
Соединения 42 и 43 получали, используя исходные вещества, соответствующие соединению, и способ получения, аналогичный синтезу соединения 41.
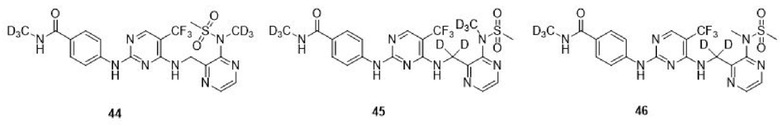
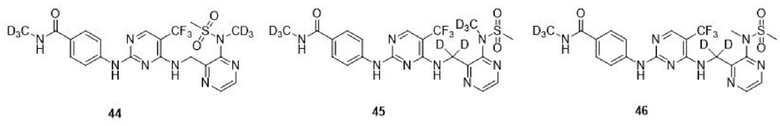
Пример 3. Синтез N-тридейтерметил-4-((4-(((3-(N-тридейтерметилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 44) и его гидрохлорида

Этап 1: Синтез трет-бутилового эфира (4-(тридейтерометилкарбамоил)фенил)карбаминовой кислоты (соединение 44-1)
N-Boc-4-аминобензойную кислоту (6,0 г, 25,29 ммоль) и EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) (7,27 г, 37,93 ммоль) соответственно отвесили и поместили в одногорлую круглодонную колбу объемом 250 мл с одновременным добавлением ДМФА (50 мл). Данную смесь перемешивали при комнатной температуре. Затем в систему добавили триэтиламин (6,40 г, 63,22 ммоль) и дейтерированный гидрохлорид метиламина (1,96 г, 27,82 ммоль). После их добавления систему перемешивали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день взяли образец и подвергли его ТСХ, и когда ТСХ показала, что реакция завершена, в систему добавили этилацетат (50 мл) и воду (50 мл). Реакционную смесь энергично перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (50 мл⋅3). Органические слои объединили, промыли сначала водой (30 мл⋅3), затем насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. Реакционную смесь концентрировали в вакууме для получения первичного продукта, который затем разделяли и очищали методом колоночной хроматографии с получением трет-бутилового эфира (4-(дейтерометилкарбамоил)фенил)карбаминовой кислоты в виде твердого вещества белесого цвета (4,92 г, выход: 76,8%). ЖХ/МС (ИЭР+): м/з 254,2 [М+Н]+ (рассчитано для C13H15D3N2O3, 254,2).
Этап 2: Синтез трифторацетата 4-амино-N-тридейтерометилбензамида (соединение 44-2) Трет-бутиловый эфир (4-(дейтерометижарбамоил)фенил)карбаминовой кислоты (3,0 г, 11,84 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 100 мл с одновременным добавлением дихлорметана (15 мл). Данную смесь перемешивали при комнатной температуре. После этого в систему добавили трифторуксусную кислоту (7 мл) и затем систему перемешивали и выдерживали при комнатной температуре для протекания реакции. Через 5 часов взяли образец и подвергли его ТСХ. ТСХ показала, что реакция завершена. Реакционную смесь упаривали на ротационном испарителе для удаления растворителя и излишков трифторуксусной кислоты, и остатки трифторуксусной кислоты в системе удаляли ротационным упариванием с дихлорметаном в несколько приемов с получением трифторацетата 4-амино-N-дейтерометилбензамида в виде твердого вещества белесого цвета, который сразу же использовали на следующем этапе без дополнительной очистки.
Этап 3: Синтез 4-((4-хлор-5-(трифторметил)гшримидин-2-ил)амино)-N-тридейтерометилбензамида (соединение 44-3)
2,4-дихлор-5-трифторметилпиримидин (2,83 г, 13,02 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 100 мл, в которую добавили 1,2-дихлорэтан (30 мл) и трет-бутанол (30 мл) и затем смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. После этого систему поместили на водяную баню со льдом для продолжения охлаждения и перемешивания. Когда температура внутри системы снизилась примерно до 0°С, в систему добавили бромид цинка (8,0 г, 35,52 ммоль). Затем систему дополнительно выдерживали на водяной бане со льдом в течение 30 минут для протекания реакции. Затем в систему добавили трифторацетат 4-амино-N-дейтерометилбензамида, синтезированный на предыдущем этапе, и триэтиламин (3,83 г, 37,89 ммоль). После их добавления систему сняли с ледяной бани, перемешивали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день взяли образец и подвергали его ТСХ. ТСХ показала, что исходные вещества полностью израсходованы, и реакция была прекращена. Растворитель удалили ротационным упариванием. В систему добавили этилацетат (50 мл) и воду (30 мл), и реакционную смесь интенсивно перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (30 мл⋅3). Органические слои объединили, промывали последовательно водой (30 мл*3) и насыщенным солевым раствором (30 мл) и сушили над безводным сульфатом натрия. Реакционную смесь концентрировали в вакууме для получения первичного продукта, который разделяли и очищали методом колоночной хроматографии с получением 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамида в виде твердого вещества белесого цвета (3,23 г). Выход двухэтапной реакции со 2-го на 3-й этап: 81,8%. ЖХ/МС (ИЭР+): м/з 334,0 [М+Н]+ (рассчитано для C13H7D3ClF3N4O, 334,0).
1Н ЯМР (400 МГц, ДМСО-d6) δ 10,89 (s, 1Н), 8,87 (s, 1H), 8,31 (s, 1H), 7,84-7,77 (m, 4H).
Этап 4: Синтез N-тридейтерометил-4-((4-(((3-(N-тридейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 44)
4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамид (3,1 г, 9,29 ммоль) и N-(3-цианопиразин-2-ил)-N-дейтерометилметансульфонамид (2,0 г, 9,29 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 250 мл, в которую затем добавили 1,2-дихлорэтан (80 мл) и трет-бутанол (80 мл) и затем смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Затем в систему добавили диизопропилэтиламин (3,6 г, 27,87 ммоль). После этого систему поставили на масляную баню при 80°С, нагревали с обратным холодильником, перемешивали и оставили на ночь для протекания реакции. На следующий день взяли образец и подвергли его ТСХ. Как только ТСХ показала, что реакция завершена, растворитель удалили ротационным упариванием. В систему добавили этилацетат (100 мл) и воду (50 мл), и реакционную смесь интенсивно перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (50 мл⋅3). Органические слои объединили, промывали последовательно водой (30 мл⋅3) и насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. Реакционную смесь концентрировали в вакууме для получения первичного продукта, который разделяли и очищали методом колоночной хроматографии с получением целевого соединения в виде твердого вещества белесого цвета (2,36 г). Затем указанное твердое вещество поместили в одногорлую круглодонную колбу объемом 250 мл, в которую добавили этилацетат (75 мл) и данную смесь перемешивали при комнатной температуре до образования суспензии. Через 3 часа ее отфильтровали отсасыванием. Осадок с фильтра несколько раз промыли этилацетатом (45 мл) в небольшом количестве и затем поместили в вакуумную сушильную печь для сушки при низкой температуре до получения N-[-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (2,11 г) в виде белого твердого вещества. Выход: 44,0%. ЖХ/МС (ИЭР+): м/з 517,2 [М+Н]+ (рассчитано для C20H15D6F3N8O3S, 517,2).
1Н ЯМР (400 МГц, ДМСО) δ 9,83 (s, 1H), 8,69 (d, J=2,8 Гц 1H), 8,58 (d, J=2,4 Гц, 1H), 8,31 (s, 1H), 8,17 (s, 1H), 7,67-7,61 (dd, J=15,4, 8,6 Гц, 4H), 7,41 (t, J=5,0 Гц 1H), 5,00 (d, J=4,8 Гц 2H), 3,20 (s, 3H).
Этап 5: Синтез гидрохлорида N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида
Соединение 44 (500 мг, 0,97 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 100 мл, в которую добавили метанол (25 мл) и данную смесь перемешивали при комнатной температуре. Затем в систему медленно по каплям добавляли раствор HCl в этаноле (2,25 мл, 2,0 М). После этого систему продолжали перемешивать и выдерживать при комнатной температуре для протекания реакции. Через 1,5 часа систему отфильтровали отсасыванием и осадок с фильтра несколько раз промыли метанолом (15 мл) в небольших количествах, а затем поместили в вакуумную сушильную печь для сушки при низкой температуре до получения гидрохлорида N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензоила (517 мг) в виде твердого вещества белесого цвета. Выход: 96,6%. ЖХ/МС (ИЭР+): м/з 517,2 [М+Н]+ (рассчитано для C20H16D6ClF3N8O3S, 517,2).
1Н ЯМР (400 МГц ДМСО) δ 10,02 (s, 1H), 8,68 (d, J=2,8 Гц, 1H), 8,58 (d, J=2,4 Гц, 1H), 8,35 (s, 1H), 8,21 (s, 1H), 7,67-7,57 (m, 5Н), 5,26 (br, 6Н), 5,00 (d, J=4,8 Гц, 2Н), 3,19 (s, 3Н).
Пример 4. Синтез N-дейтерометил-4-((4-(((3-(N-дейтерометансульфонамид)пиразин-2-ил)дейтерометил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 45)
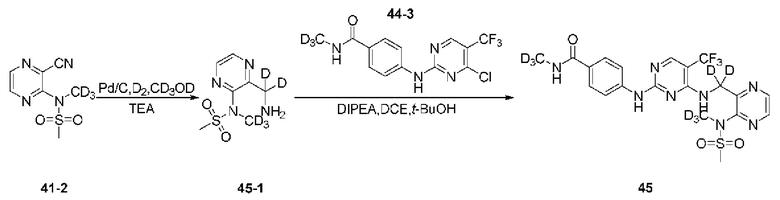
Этап 1: Синтез соединения N-(3-(аминодидейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (45-1)
Соединение 41-2 (100,0 мг, 0,46 ммоль) отвесили и поместили в одногорлую круглодонную колбу объемом 25 мл, в которую затем добавили 5 мл дейтерированного метанола (188,2 мг, 1,86 ммоль) и затем данную смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Затем в систему добавили последовательно 20,0 мг влажного палладированного угля (обработанного тяжелой водой) и триэтиламин (188,2 мг, 1,86 ммоль). Над системой произвели действия по замене дейтерия и повторяли эти действия десять раз. После этого систему перемешивали и выдерживали при комнатной температуре для протекания реакции. Через 72 часа реакцию завершали детектированием. Затем систему отфильтровали отсасыванием. Осадок с фильтра несколько раз промыли дейтерированным метанолом (10 мл) в небольших количествах. Фильтрат объединили и растворитель удалили ротационным упариванием с получением N-(3-(аминодейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамида в виде светло-желто-коричневой маслянистой жидкости, который сразу же использовали на следующем этапе без дополнительной очистки. МС (ИЭР): м/з 222,2 [М+Н]+.
Этап 2: Синтез соединения N-дейтерометил-4-((4-(((3-(N-дейтерометансульфонамид)пиразин-2-ил)дейтерометил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамид (45)
В одногорлую круглодонную колбу объемом 25 мл, содержащую N-(3-(аминодейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (22,1 мг, 0,10 ммоль), добавили 2 мл 1,2-дихлорэтана и 2 мл трет-бутанола и затем смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Затем в систему добавили соединение 44-3 (33,4 мг, 0,10 ммоль) и диизопропилэтиламин (30,6 мг, 0,30 ммоль). После их добавления систему поместили на масляную баню при 80°С и нагревали с обратным холодильником для протекания реакции. Через 8 часов с помощью ТСХ зафиксировали полный расход исходных веществ. Нагревание прекратили и после охлаждения системы до комнатной температуры растворитель удалили ротационным упариванием для получения первичного продукта, который затем разделяли и очищали методом предварительной ТСХ с получением N-дейтерометил-4-((4-(((3-(N-дейтерометансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде твердого вещества белесого цвета (8,1 мг) с выходом 15,6%. МС (ИЭР): м/з 519,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1H), 8,69 (d, J=2,0 Гц, 1H), 8,58 (d, J=2,4 Гц, 1H), 8,31 (s, 1H), 8,17 (s, 1Н), 7,67-7,61 (dd, J=15,2, 8,8 Гц, 4Н), 7,39 (s, 1Н), 3,20 (s, 3Н).
Пример 5. Синтез 4-((4-(((3-N-тридейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 52)

Этап 1: Синтез соединения 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)бензамид (52-2)
2,4-дихлор-5-(трифторметил)пиримидин (434 мг, 2,00 ммоль) отвесили и растворили в одногорлой круглодонной колбе объемом 25 мл, в которую добавили 1,2-дихлорэтан (5 мл) и трет-бутанол (5 мл) и затем данную смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. После этого систему поместили на водяную баню со льдом для продолжения охлаждения и перемешивания. Через 15 минут в систему добавили бромид цинка (1,2 г, 5,22 ммоль). Затем систему дополнительно перемешивали в течение 30 минут на водяной бане со льдом. Затем в систему добавили 4-аминобензамид (237 мг, 1,74 ммоль), синтезированный на предыдущем этапе, и триэтиламин (564 мг, 5,57 ммоль). После их добавления систему сняли с ледяной бани, перемешивали и оставили на ночь при комнатной температуре для протекания реакции. На следующий день после завершения реакции детектированием растворитель удалили ротационным упариванием. В систему добавили этилацетат (30 мл) и воду (20 мл), и реакционную смесь энергично перемешивали и отстаивали для разделения слоев. Водную фазу обратно экстрагировали этилацетатом (10 мл⋅3). Органическую фазу объединили, промывали последовательно водой (15 мл⋅3) и насыщенным солевым раствором (15 мл) и сушили над безводным сульфатом натрия. Реакционную смесь концентрировали при пониженном давлении для получения первичного продукта, который затем разделяли методом колоночной хроматографии с получением 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде твердого вещества белесого цвета (294 мг, выход: 53,4%). МС (ИЭР): м/з 317,0 [М+Н]+.
Этап 2: Синтез соединения 4-((4-(((3-(N-тридейтерометилметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамид (52)
В одногорлую круглодонную колбу объемом 25 мл, содержащую соединение 52-2, полученное на предыдущем этапе, добавили 5 мл 1,2-дихлорэтана и 5 мл трет-бутанола и затем данную смесь перемешивали при комнатной температуре до растворения реагентов и осветления раствора. Затем в систему добавили соединение 44-3 (63,0 мг, 0,20 ммоль) и диизопропилэтиламин (78 мг, 0,60 ммоль). После их добавления систему поместили на масляную баню при 80°С и нагревали с обратным холодильником для протекания реакции.
На следующий с помощью ТСХ зафиксировали полный расход исходных веществ. Нагревание прекратили и после охлаждения системы до комнатной температуры растворитель удалили ротационным выпариванием для получения первичного продукта, который затем разделяли и очищали методом предварительной ТСХ с получением 4-((4-(((3-(N-дейтеромешлметансульфонамид)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (22 мг) в виде твердого вещества белесого цвета с выходом 22,0%. МС (ИЭР) м/з 500,1 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1Н), 8,69 (s, 2Н), 8,60 (s, 1H), 8,31 (s, 1Н), 8,20 (d, J=4,0 Гц, 1H), 7,68-7,60 (dd, J=15,4, 8,4 Гц, 4Н), 7,41-7,39 (t, J=4,4 Гц, 1H), 5,03 (d, J=3,6 Гц, 2Н), 3,20 (s, 3Н).
В качестве исходных веществ использовали известные соединения 4-амино-3,5-дидейтеробензойной кислоты и 4-амино-2,6-дидейтеробензойной кислоты (Journal of Tabel Compounds and Radiopharmaceuticals, 53(11-12), 668-673; 2010), а соединения 48-50 и 53-55 получали способами, описанными в вышеприведенных примерах.
Соединения 59-61 получали способом, описанным в вышеприведенных примерах, используя известное соединение 4-амино-2,3,5,6-тетрадейтеробензойнаякислота (Journal of Natural Products, 79(6), 1532-1537; 2016) в качестве исходного вещества. Положительный эффект настоящего изобретения продемонстрирован в нижеследующих экспериментальных примерах.
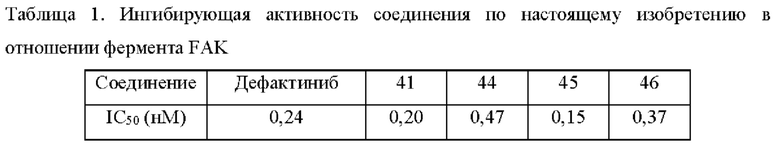
Экспериментальный пример 1. Ингибирующая активность дейтерированного соединения по настоящему изобретению в отношении FAK
(1) Методика проведения эксперимента
Согласно методике, описанной в литературе (Cancer Res. 2008, 68, 1935), был проведен эксперимент по изучению ингибирующей активности в отношении фермента FAK. Подробнее: изучаемое соединение разбавляли до 1000 нМ а затем подвергали серийному разведению 1:3 ДМСО (диметилсульфоксидом). 0,1 л раствора поместили на 384-луночный планшет и для каждой концентрации отвели по 2 лунки с одинаковыми вариантами. Добавили 5 л раствора фермента FAK 2, затем центрифугировали при 1000 об/мин в течение 1 минуты и инкубировали при 25°С в течение 15 минут. Добавили 5 л раствора субстрата 2х и инкубировали при 25°С в течение 60 минут. Затем добавили 5 л раствора Sa-XL665 и 5 мкл ТК (тимидинкиназного) антитела Eu3+ и центрифугировали при 1000 об/мин в течение 1 минуты, затем инкубировали при 25°С в течение 60 минут. В завершение, использовали планшетный ридер Envision 2104 для считывания сигнала флуоресценции и вычисления концентрации полумаксимального ингибирования IC50 каждого соединения в отношении фермента FAK. В качестве контроля использовали известный ингибитор FAK дефактиниб.
(2) Результаты эксперимента
Ингибирующая активность каждого соединения в отношении FAK представлена в Таблице 1. Видно, что соединение, полученное согласно настоящему изобретению, может эффективно ингибировать активность фермента FAK, и по сравнению с недейтерированным соединением «дефактиниб», дейтерированные соединения 41 и 45 по настоящему изобретению обладают более высокой ингибирующей активностью в отношении фермента FAK.
Экспериментальный пример 2. Фармакокинетический тест дейтерированного соединения по настоящему изобретению на крысах
(1) Методика проведения эксперимента
С точностью отвесили соответствующее количество исследуемого препарата (10 мг) и сначала добавили 0,25 мл N,N-диметилацетамида (ДМА) для его растворения, затем медленно добавляли 0,5% карбоксиметилцеллюлозы натрия (CMC-Na) к 5 мл. Данную смесь обрабатывали ультразвуком, перемешивали на вортексе и дополнительно хорошо перемешивали. 0,2 мл полученного раствора извлекли и хранили при -20°С для определения концентрации. После голодания в течение ночи (питьевая вода была доступна без ограничений) трем здоровым взрослым самцам крыс линии SD (180-250 г, приобретены у Chengdu Dossy Experimental Animal Co., Ltd.) вводили исследуемые препараты в количестве 5 мл/кг веса через желудочный зонд; перед введением, а также через 0,5, 1, 2, 4, 6, 8, 12 и 24 часа после введения производили забор 0,1 мл крови из ретроорбитального венозного сплетения, центрифугировали при 4°С в течение 5 минут для отделения плазмы и хранили при -20°С для тестирования. Затем методом ЖХ/МС/МС определяли концентрацию тестируемого соединения в плазме. В качестве контроля использовали известный ингибитор FAK дефактиниб.
(2) Результаты эксперимента
Таблица 2. Фармакокинетические параметры соединений по настоящему изобретению
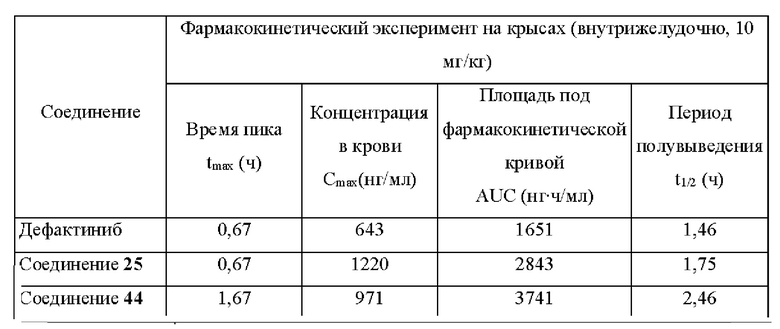
Как показано в Таблице 2, дейтерированные соединения 25 и 44, полученные согласно настоящему изобретению, демонстрировали лучшую фармакокинетику по сравнению с дефактинибом. Соединение по настоящему изобретению имеет более высокую максимальную концентрацию Cmax в плазме крови, большую площадь под фармакокинетической кривой экспозиции и более длительный период полу выведения. Следовательно, дейтерированное соединение, полученное согласно настоящему изобретению, будет иметь лучшие перспективы применения в качестве ингибиторов FAK или лекарственных средств для лечения рака.
Экспериментальный пример 3. Фармакодинамический эксперимент по изучению действия дейтерированного соединения по настоящему изобретению в комбинации с ингибитором PD-1 на модели опухоли животного
1. Модель опухоли МС38:
(1) Методика проведения эксперимента
Клеточная культура: Клетки МС-38 культивировали на среде DMEM, содержащей 10% фетальной бычьей сыворотки (FBS). Производили забор клеток МС-38 в логарифмической фазе роста и ресуспендировали их в HBSS до концентрации, пригодной для подкожной инокуляции опухоли у мышей линии C57BL/6.
Подопытные животные: 96 самок мышей линии C57BL/6 в возрасте 6-8 недель, весом около 18-20 г, приобретенные у Beijing Vital River Experimental Animal Technology Co., Ltd. Инокуляция опухолевых клеток: Производили забор опухолевых клеток в логарифмической фазе роста и при помощи HBSS доводили концентрацию клеток до 5×106/мл, затем 0,1 мл инокулировали подкожно в правый бок каждой мыши возле спины, то есть 5×105/мышь. Затем наблюдали за ростом и измеряли объем опухоли. Когда средний объем опухоли у мышей вырос до 50-100 мм3, мышей с опухолью случайным образом разбили на группы в соответствии с объемом опухоли и вводили им препарат. Подробная информация представлена в Таблице 3, а день разбиения на группы и введения препарата обозначен как день 0.
Вычисление объема опухоли: На 18-е суши мышей умертвили, извлекли опухоли измерили объем опухолей и вычислили степень подавления опухоли у каждой группы.
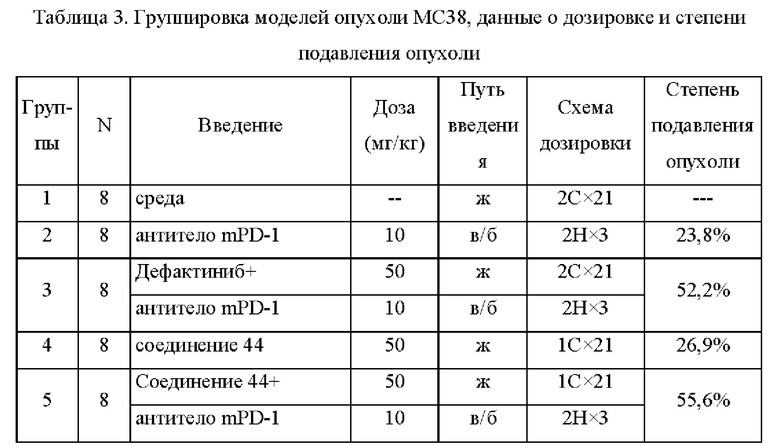
Примечание: N: количество использованных животных; в/б: внутрибрюшинная инъекция; ж: внутрижелудочное введение; 2С: два раза в сутки; 1С: один раз в сутки; 2Н: два раза в неделю.
(2) Результаты эксперимента
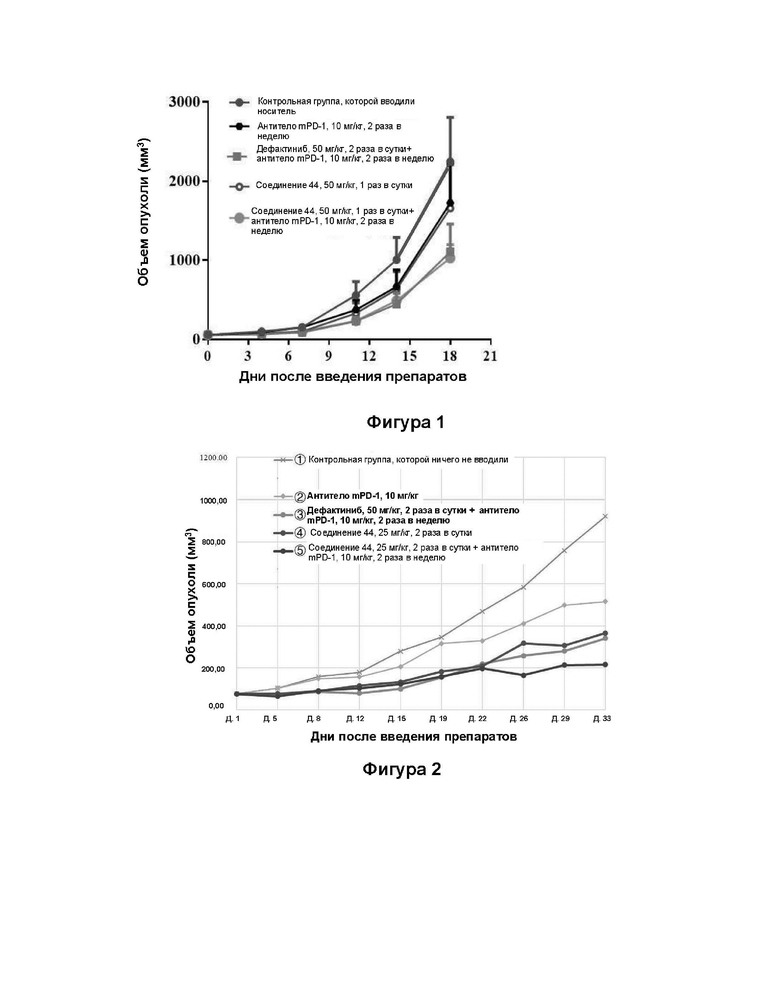
Фармакодинамика животных после 18 дней введения показана на Фиг. 1, а вычисленная степень подавления опухоли показана в Таблице 3. Видно, что эффективность соединения 44, вводимого в отдельности, была выше, чем эффективность антитела mPD-1 в отдельности, что указывает на то, что соединение по настоящему изобретению оказывало терапевтический эффект на модель опухоли МС38 у мышей. Кроме того, по сравнению с введением соединения 44 в отдельности (группа 4) или антитела mPD-1 в отдельности (группа 2), введением соединения 44 в комбинации с антителом mPD-1 (группа 5) достигалось значительно лучшее подавляющее действие на опухоли, и проявлялся синергетический эффект.
Вдобавок, по сравнению с введением дефактиниба (50 мг/кг, два раза в сутки) в комбинации с антителом mPD-1 (группа 3), после введения соединения 44 по настоящему изобретению (50 мг/кг, один раз в сутки) в комбинации с антителом mPD-1 (группа 5) подавляющее действие на опухоль было лучше, а также был достигнут еще более высокий уровень подавляющего действия на опухоль. То есть, при использовании в комбинации с ингибиторами PD-1, соединение по настоящему изобретению в дозе, составляющей половину дозы дефактиниба, может достичь лучшего подавляющего действия на опухоль, что указывает на то, что комбинация соединения по настоящему изобретению и ингибиторов PD-1 имеет значительно лучший противоопухолевый эффект на модели опухоли МС38, чем комбинация дефактиниба и ингибиторов PD-1.
2. Модель опухоли PAN02
(1) Методика проведения эксперимента
Произвели забор клеток PAN-02 в логарифмической фазе роста, дважды промыли их PBS (фосфатно-солевым буфером) и затем ресуспендировали в предварительно охлажденном PBS для инокуляции. Подопытными животными были самки мышей линии C57BL/6, приобретенные у Beijing Vital Paver Experimental Animal Technology Co., Ltd. Мыши C57BL/6 адаптировались к лабораторной среде в течение 3 дней. Клетки PAN-02 инокулировали подкожно в ребра правого бока, и количество инокулированных клеток составило 1×106/мышь. Когда опухоль выросла примерно до 100 мм3, мыши были подвергнуты скринингу и случайным образом разбиты на группы. Каждая группа включала в себя 8 мышей. Мышей разбили на группы и вводили им препараты согласно схеме дозировки в соответствии с Таблицей 4. День разбиения на группы и введения препаратов был обозначен как день 1, а период введения составлял 33 дня.
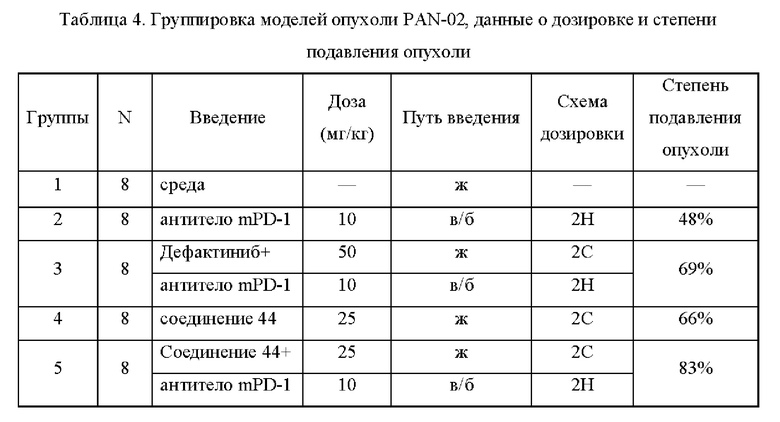
Примечание: N: количество использованных животных; в/б: внутрибрюшинное введение; ж: внутрижелудочное введение; 2С: два раза в сутки; 2Н: два раза в неделю.
(2) Результаты эксперимента
Фармакодинамика животных после 33 дней введения препаратов показана на Фиг. 2, а вычисленная степень подавления опухоли показана в Таблице 4. Видно, что по сравнению с введением соединения 44 в отдельности (группа 4) или антитела mPD-1 в отдельности (группа 2), введением соединения 44 в комбинации с антителом mPD-1 (группа 5) достигалось значительно лучшее подавляющее действие на опухоль.
Кроме того, по сравнению с подавляющим действием на опухоль после введения дефактиниба (50 мг/кг, два раза в сутки) в комбинации с антителом mPD-1 (группа 3), подавляющее действие на опухоль после введения соединения 44 по настоящему изобретению (25 мг/кг, два раза в сутки) в комбинации с антителом mPD-1 (группа 5) было значительно лучше. То есть, при использовании в комбинации с ингибиторами PD-1, соединение по настоящему изобретению в дозе ниже, чем у дефактиниба, может достичь более эффективного подавления опухоли, что указывает на то, что введение соединения по настоящему изобретению в комбинации с ингибиторами PD-1 оказывает значительно лучшее противоопухолевое действие на модель опухоли PAN-02, чем введение дефактиниба в комбинации с ингибиторами PD-1.
Таким образом, в настоящем изобретении предложено дейтерированное соединение, которое продемонстрировало лучшую фармакокинетику, более высокую максимальную концентрацию в плазме крови, более высокую степень экспозиции и более длительный период полувыведения, а также еще более высокие метаболические характеристики по сравнению с данным соединением до дейтерирования. Более того, дейтерированное соединение по настоящему изобретению может эффективно ингибировать активность FAK и имеет очень хорошие перспективы применения для получения ингибиторов FAK и/или лекарственных средств для лечения рака. В то же время применение дейтерированного соединения по настоящему изобретению в комбинации с противораковыми лекарственными средствами (такими как ингибиторы PD-1) может вызвать синергетический эффект, значительно улучшить подавляющее действие на опухоли и обеспечить лучший выбор для клинического лечения рака.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЕЙТЕРИРОВАННОГО ДЕФАКТИНИБА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2761825C1 |
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ, ИХ СПОСОБ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2749437C2 |
| ДЕЙТЕРИРОВАННЫЕ ДИАМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ТАКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2632907C2 |
| ПИРРОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2563644C2 |
| ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРАЗОЛЬНЫЕ ИНГИБИТОРЫ КИНАЗНОЙ АКТИВНОСТИ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2655921C2 |
| ПРОИЗВОДНОЕ СУЛЬФОНИЛБЕНЗАМИДА И ЕГО КОНЬЮГАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2839468C1 |
| ПРОИЗВОДНОЕ ФТАЛАЗИНОНКЕТОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2564527C2 |
| ИНГИБИТОРЫ КИНАЗЫ, ИХ ПРОЛЕКАРСТВЕННЫЕ ФОРМЫ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2010 |
|
RU2568639C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ФЕНИЛАТНОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЯ | 2020 |
|
RU2820475C2 |
Изобретение относится к области органической и медицинской химии, а именно к комбинации лекарственных средств для лечения опухолей, опосредованных FAK. Комбинация характеризуется тем, что содержит соединение, выбранное из соединений формулы 25, 41, 44, 45, 46, или его фармацевтически приемлемую соль, и иммунотерапевтическое лекарственное средство в тех же или разных специфицированных препаративных формах для одновременного или раздельного введения, и фармацевтически приемлемый носитель. Иммунотерапевтическое лекарственное средство представляет собой ингибитор PD-1. Технический результат изобретения заключается в создании новых комбинаций, содержащих соединение, обладающее ингибирующей активностью по отношению к FAK, и иммунотерапевтическое средство, представляющее собой ингибитор PD-1, которые могут найти применение в медицине для лечения FAK-опосредованных опухолей. 2 н. и 2 з.п. ф-лы, 2 ил., 4 табл., 5 пр. 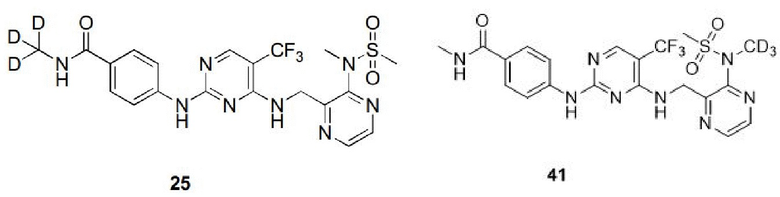
1. Комбинация лекарственных средств для лечения опухолей, опосредованных FAK, характеризующаяся тем, что она содержит соединение, выбранное из

или его фармацевтически приемлемую соль,
и иммунотерапевтическое лекарственное средство в тех же или разных специфицированных препаративных формах для одновременного или раздельного введения, и фармацевтически приемлемый носитель, где иммунотерапевтическое лекарственное средство представляет собой ингибитор PD-1.
2. Комбинация лекарственных средств по п. 1, отличающаяся тем, что ингибитор PD-1 представляет собой антитело mPD-1.
3. Применение комбинации лекарственных средств по п. 1 или 2 для получения лекарственных средств для лечения рака, опосредованного FAK.
4. Применение по п. 3, отличающееся тем, что указанный рак представляет собой солидные опухоли;
при этом солидные опухоли включают мезотелиому, рак поджелудочной железы, опухоли мягких тканей, метастазы, не являющиеся солидными раковые заболевания, саркомы, аденокарциному, рак легких, рак молочной железы, лимфому, рак желудочно-кишечного тракта, рак мочеполовой системы, рак предстательной железы и рак яичников; рак желудочно-кишечного тракта включает рак толстой кишки; рак мочеполовой системы включает опухоли почек, уротелия или яичек; и рак яичников включает распространенный рак яичников;
мезотелиома включает нейрофибромы, рак почек, рак легких, мелкоклеточный рак легких, немелкоклеточный рак легких, KRAS-мутантный немелкоклеточный рак легких, рак печени, рак щитовидной железы, рак молочной железы, опухоли нервной системы, шванному, менингиому, неврому, аденоидную кистозную карциному, эпендимому, опухоли эпендимы, злокачественный плеврит, злокачественную мезотелиому плевры, тройной негативный рак молочной железы, негематологические злокачественные новообразования, меланому, колоректальную карциному, лейкоз, аденокарциному, солидные опухоли;
меланома включает местно-распространенную меланому, меланому, вызванную мутантным NRAS, метастатическую злокачественную меланому кожи; колоректальный рак включает метастатический колоректальный рак; и лейкоз включает острый миелогенный лейкоз; аденокарцинома включает аденокарциному; и солидная опухоль включает местно-распространенную солидную опухоль, метастатическую солидную опухоль и гепатоцеллюлярную карциному.
| WO 2008129380 A1, 30.10.2008 | |||
| WO 2017201043 A1, 23.11.2017 | |||
| WO 2017143842 A1, 31.08.2017 | |||
| US 20110053968 A1, 03.03.2011 | |||
| WO 2010144499 A2, 16.12.2010 | |||
| Карандаш с несколькими цветными иди графитными штифтами | 1929 |
|
SU19966A1 |

