Область изобретения
Данное изобретение относится к новому классу 17β -амино и гидроксиламиностероидов, которые, как полагают, связываются с рецептором прогестина и которые проявляют высокую антигестагенную активность, промежуточным стероидным соединениям, которые пригодны для получения вышеупомянутых стероидов и способам получения промежуточных стероидных соединений. Такие соединения пригодны для лечения фибром, эндометриоза и некоторых опухолей как факторы, вызывающие созревание шейки матки перед родоразрешением, в гормонозаместительной терапии и для регулирования фертильности и размножения.
Обсуждение предпосылок
Прогестерон играет основную роль в обеспечении репродуктивного здоровья и функционирования репродуктивной системы. Его воздействия, например, на матку, молочную железу, шейку матки и гипоталамо-гипофизарную единицу хорошо установлены. Он также обладает не связанными с репродукцией активностями, которые изучены хуже, такими как воздействия на мозг, иммунную систему, эндотелиальную сосудистую систему и на метаболизм липидов. При таком широком множестве эффектов, очевидно, что соединения, которые имитируют некоторые эффекты прогестерона (агонисты), противодействуют этим эффектам (антагонисты) или проявляют смешанные эффекты (неполные агонисты или смешанные агонисты/антагонисты) могут применяться для лечения ряда заболеваний и патологических состояний.
Стероидные гормоны проявляют свои эффекты частично благодаря связыванию с внутриклеточными рецепторами. Соединения, которые связываются с соответствующими рецепторами и являются антагонистами или неполными агонистами эстрогенных и андрогенных гормонов давно известны, но приблизительно до 1982 г. не было заявлено об открытии соединений, которые связываются с рецептором прогестерона и оказывают действие, противоположное действию прогестерона. После этого, в научной и патентной литературе было сообщено о ряде таких соединений, и были изучены их действия in vitro, на животных и человеке. Хотя такие соединения как эстрогены и некоторые ингибиторы ферментов могут препятствовать физиологическим действиям эндогенного прогестерона, в данном обсуждении термин "антипрогестин" ограничен такими соединениями, которые связываются с рецептором прогестина.
В настоящее время имеется информация, свидетельствующая о том, что антипрогестины могут быть эффективны при многих патологических состояниях. Эта информация суммирована в сообщении Медицинского Института (Donaldson, Molly S.; Dorflinger, L.; Brown, Sarah S.; Benet, Leslie Z., Editors, Clinical Applications of Mifepristone (RU 486) and Other Antiprogestins, Committee on Antiprogestins: Assessing the Science, Institute of Medicine, National Academy Press, 1993). Принимая во внимание центральную роль, которую прогестерон играет в репродукции, не удивительно, что антипрогестины могли бы играть роль в регулировании фертильности, включая контрацепцию (долговременную и в непредвиденных случаях или после полового акта), индукцию менструации и медицинское прерывание беременности, кроме этого существуют многие другие возможные применения, которые подтверждены немногочисленными клиническими или доклиническими исследованиями. Среди таких применений - следующие случаи:
1. Роды и родоразрешение - антипрогестины могут применяться для созревания шейки матки перед стимуляцией родов, как, например, при наступлении срока или когда роды должны быть стимулированы из-за гибели плода. Они могут также применяться как вспомогательные средства при стимуляции родов в срок или при переношенной беременности.
2. Лечение лейомиом матки (фибром) - эти не злокачественные опухоли могут поражать до 20% женщин в возрасте свыше 30 лет и являются одной из наиболее распространенных причин операций у женщин в репродуктивном возрасте. Гистерэктомия, общепринятое лечение при стойких симптомах, конечно приводит к стерильности.
3. Лечение эндометриоза - это распространенное (охватывает от 5 до 15% женщин, гораздо больше среди бесплодных женщин) и часто болезненное состояние в настоящее время лечится лекарственными средствами, такими как даназол или аналоги гонадотропин-высвобождающего гормона, которые обладают существенными побочными действиями, или надо обращаться к хирургической операции.
4. Гормонозаместительная терапия, при которой антипрогестины могут предлагаться, чтобы прервать или сократить активность прогестинов.
5. Раковые болезни, особенно рак молочной железы, - присутствие рецепторов прогестина во многих случаях заболевания раком молочной железы, наводит на мысль о применении антипрогестинов для лечения рака с метастазами или для профилактики рецидива или начального развития рака.
6. Другие опухоли, такие как менингиомы - эти опухоли оболочки мозга, хотя и не злокачественны, но приводят к смерти пациентов, и нехирургические способы лечения отсутствуют.
7. Мужская контрацепция - антипрогестины могут отрицательно влиять на жизнеспособность спермы, хотя существует такой эффект антипрогестинов или нет, является спорным, так как это может быть связано с антиглюкокортикоидной активностью таких соединений.
8. Антиэстрогенные эффекты - по меньшей мере, некоторые антипрогестины препятствуют действию эстрогенов в некоторых тестах, но очевидно посредством механизма, в который не вовлечены классические рецепторы гормонов. Это открывает множество возможностей для их применения в медицине.9. Антиглюкокортикоидные эффекты - это распространенное побочное действие антипрогестинов, которое может быть использовано в некоторых случаях, таких как лечение синдрома Кушинга, и может играть роль, например, при иммунных заболеваниях. В других случаях желательно минимизировать такие эффекты.
Эффекты и применения агонистов прогестерона хорошо известны в данной области. Кроме того, недавно было показано, что некоторые соединения, структурно близкие к известным антипрогестинам, обладают сильной агонистической активностью в определенных биологических системах (например, классические эффекты прогестина в примированной эстрогеном недоразвитой матке кролика; сравни, С.Е.Cook et al., Life Sciences, 52, 155-162 (1993)). Такие соединения являются неполными агонистами в рецепторных системах, полученных на основе клеток человека, в которых они связываются с сайтом, отличным от сайта связывания прогестина и от сайта связывания антипрогестина (Wagner et al., Proc. Natl. Acad. Sci., 93, 8739-8744 (1996)). Таким образом, общий класс антипрогестинов может иметь подклассы, которые могут отличаться по совокупности своих клинических параметров.
В общем антигестагенная активность связана с наличием 11β -арильного заместителя в ядре стероида, наряду с Δ 4,9-3-кетонным или Δ 4-3-кетонным фрагментом. Однако показано, что заместители в D-кольце стероида могут оказывать заметное влияние на биологические параметры этих соединений (смотри выше). Ранее предложенные антипрогестины были замещены 17β -гидроксильной группой и различными 17α -заместителями. (Смотри, например, Teutsch, Jean G.; Costerousse, Germain; Philibert, Daniel, and Deraedt, Roger. Novel steroids. US 4386085. 1983; Philibert, Daniel; Teutsch, Jean G.; Costerousse, Germain, and Deraedt, Roger. 3-Keto-19-nor-Δ -4,9-steroids. US 4477445. 1983; Teutsch, Jean G.; Pantin, Germain; Costerousse, Saint-Maurice; Daniel Philibert; La Varenne Saint Hilaire; Roger Deraedt, inventors. Steroid derivatives. Roussel Uclaf, assignee. US 4447424. 1984; Cook, С. Edgar; Tallent, C. Ray; Reel, Jerry R., and Wani, Mansukh C. 17α -(Substituted-methyl)-17β -hydroxy/esterified hydroxy steroids and pharmaceutical compositions containing them. US 4774236 (1988) и 4861763 (1989)). Затем было обнаружено, что 17β -ацетил, 17α -ацилоксигруппа также могут оказывать антигес-тагенные действия (Cook, С, Edgar; Lee, Y.-W.; Reel, Jerry R.; Wani, Mansukh C., Rector, Douglas. lip-Substituted Progesterone Analogs. U.S. Patent Nos. 4954490 (1990) и 5073548 (1991)), и также были выполнены различные перестановки этих найденных вариантов. Однако, введение 16α -этильной группы или заместителя водорода в Цех-положение в серии соединений, содержащих 17β -ацил, приводит к агонистической или неполной агонистической активности (С.Е.Cook et al., Life Science, 52, 155-162 (1993)). Таким образом, изменения в D-кольце стероида приводит к широкому ряду воздействий на биологическую активность. Соответственно остается необходимость в соединениях антипрогестинов, которые проявляют более высокую специфичность.
Можно видеть, что 17β -положение данных антипрогестинов характеризуется замещением атомом углерода или атомом кислорода. Не сообщалось о влиянии на гормональную и антигормональную активность азотных заместителей, таких как амины, амиды аминов и гидроксиламины в 17β -положении 11β -арилстероидов. До настоящего изобретения не существовало способов их синтеза. В общей химической литературе и в патентах сообщалось об очень немногих 17β -амино и гидроксиламиностероидах и совсем не сообщалось о 11β -замещении. Действительно, в одном из немногочисленных сообщений о таком типе 17β -замещения (Р.Kaspar and Н.Witzel, J.Steroid. Biochem., 23:259(1985)) показано, что этот тип замещения в области эстрогена приводит к появлению соединений, которые на порядок и на несколько порядков величин менее эффективны (как измерено по связыванию с рецептором или стандартными тестами in vivo на эстрогенную активность), чем соответствующие 17β -гидроксисоединения. Одной из новых особенностей данного изобретения является обнаружение того, что 17β -азотные заместители в 11β -арилстероидах приводят к появлению соединений с хорошей связывающей способностью с рецептором прогестина и с неожиданно высокоэффективной антигестагенной активностью, или с высокой антигестагенной активностью и такой же сопутствующей гестагенной активностью. Другой новой особенностью данного изобретения является обнаружение того, что 17β -азотные заместители в 11β -арилстероидах приводят к появлению соединений, обладающих необычной антиэстрогенной активностью.
Кроме того, данное изобретение предоставляет группу новых 11,17-спироциклотетрагидропирролстероидов. Хотя известны очень немногие 17,17-спироциклотетрагидропирролстероиды (сравни, Keana, John F.W.; Tamura, Toshinari; McMillen, Debra A., and Jost, Patricia C. Synthesis and characterization of novel cholesterol nitroxide spin label. Application to the molecular organization of human high-density lipoprotein. J. Am. Chem. Soc. 1981; 103 (16):4904-4912), они были использованы для создания спиновых меток, а не для применения их биологических свойств. Не сообщалось о таких соединениях с 11β -арильными заместителями. Кроме того, новой особенностью данного изобретения является обнаружение того, что эти соединения неожиданно хорошо связываются с рецептором прогестина и проявляют антигестагенную активность.
Поэтому целью данного изобретения являются новые и эффективные антагонисты прогестина (антипрогестинов) и смешанные или неполные агонисты прогестина, способы их применения в медицинских целях на млекопитающих, включая человека, и способы их синтеза.
Несмотря на перспективное клиническое применение антипрогестинов, по состоянию на 1 мая 1998 г. не было зарегистрировано антипрогестинов в качестве лекарственных средств ни в Соединенных Штатах, ни в многих других странах. Только одно антипрогестинное лекарственное средство одобрено и доступно для клинического применения повсеместно в мире и это лекарственное средство, мифепристон, главным образом используется для медицинского прерывания беременности. Причиной такой ситуации является ряд факторов, но несомненно существует необходимость в новых антигестагенных лекарственных средствах, которые могут применяться при состояниях, описанных выше.
Поэтому целью данного изобретения являются новые и эффективные антагонисты прогестина (антипрогестинов) и смешанные или неполные агонисты прогестина, а также способы их применения в медицинских целях на млекопитающих, включая человека.
Краткое изложение сути изобретения
Данное изобретение относится к группе новых 17β -амино и гидроксиаминостероидов, которые характеризуются 11β -замещением, в частности замещением 11β -арилом.
Согласно данному изобретению одним из вариантов является гормональное или антигормональное стероидное соединение структуры I,
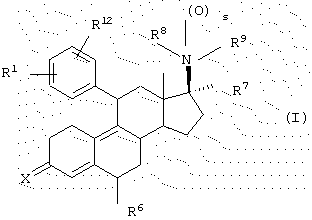
где R1 означает (R2R3N(О)r)-, где r равно 0 или 1 и каждый из R2 и R3 независимо представляет Н, C1-6алкил, С3-8циклоалкил, С2-6алкенил или С2-6алкинил, любой из которых может быть необязательно замещен; или
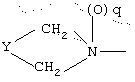
R1 означает
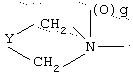
где q равно 0 или 1, Y означает -(СН2)m-, где m является целым числом от 0 до 5, или Y означает (СН2)n-Z-(СН2)р-, где n является целым числом от 0 до 2, p является целым числом от 0 до 2, и Z означает гетероатом (необязательно замещенный) и где СН2 группы могут быть необязательно замещены; или
R1 означает N-имидазолил, N-пирролил-, Н, атом галогена, НО-, СF3SО2О-, C1-6алкил-О-, C1-6алкил-S-, C1-6алкил-S(О)-, C1-6алкил-S(O)2-, C1-6алкил-СО-, C1-6алкил-СН(ОН)-, NC-, НСС-, С6Н5СС-, 2'-фурил, 3'-фурил, 2'-тиофенил, 3'-тиофенил, 2'-пиридил, 3'-пиридил, 4'-пиридил, 2'-тиазолил, 2'-N-метилимида-золил, 5'-пиримидинил, С6Н5-, Н2С=СН-, C1-6алкил или МеС(=СН2)-;
R12 является Н или атомом галогена; или
R1 и R12 объединяются с образованием кольца
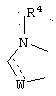
где W означает CH2, CH, NH, N, О или S, и R4 означает Н или C1-6алкил;
Х означает О или NOR5, где R5 представляет собой Н или C1-6алкил, С3-8циклоалкил, С2-6алкенил, С2-6алкинил, С6-12арил или гетероарил, любой из которых может быть необязательно замещен; или
Х означает (Н,Н), (Н,ОН), (Н, OSi (C1-6алкил)3) или (H, OCOR5), где R5 представляет собой C1-6алкил, С3-8циклоалкил, С2-6алкенил, С2-6алкинил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен; или
Х означает
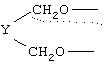
где Y представляет -(CH2)m-, где m является целым числом 0 до 3, или Y представляет собой (CH2)n-Z-(CH2)р-, где n является целым числом от 0 до 2, p является целым числом от 0 до 2 и Z является гетероатомом (необязательно замещенным) или Z является атомом углерода, замещенным одной или двумя C1-6алкильными группами;
R6 означает Н, C1-6алкил или галоген;
R7 означает Н, C1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен, CN, COOR10 или CONHR10, где R10 представляет собой Н, С1-18алкил, С2-18алкенил, С2-18алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен;
s равно 0 или 1;
каждый из R8 и R9 независимо означает Н, C1-6алкил, С2-6алкенил или С2-6алкинил, R10'CO, OR11, любой из которых может быть необязательно замещен,
где R10' означает Н, С1-18алкил, С2-18алкенил, С2-18алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен, и
где R11 означает Н, C1-6алкил, Si(C1-6алкил)3, 2'-тетрагидропиранил или R10CO, где R10 имеет значение, определенное выше;
где, когда s равно 0, R8 может также быть О- и R9 представляет собой =СН2 или =C(H, C1-6), =С(Н, арил) или =C(C1-6)2 и азот, присоединенный в положении 17, является положительно заряженным;
и его фармацевтически приемлемые соли.
Согласно данному изобретению другим вариантом является гормональное или антигормональное стероидное соединение структуры II,
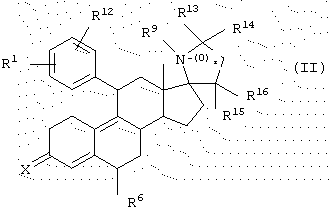
где R1 означает (R2R3N(О)r)-, где r равно 0 или 1 и каждый из R2 и R3 независимо представляет Н, C1-6алкил, С3-8циклоалкил, С2-6алкенил или С2-6алкинил, любой из которых может быть необязательно замещен; или
R1 означает
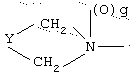
где q равно 0 или 1, Y означает -(CH2)m-, где m является целым числом от 0 до 5, или Y означает -(CH2)n-Z-(СН2)р-, где n является целым числом от 0 до 2, р является целым числом от 0 до 2, и Z означает гетероатом (необязательно замещенный), и где СН2 группы могут быть необязательно замещены; или
R1 означает N-имидазолил-, N-пирролил-, атом галогена, НО-, СF3SO2О-, C1-6алкил О-, C1-6алкил S-, C1-6алкил S(O)-, C1-6алкил-S(О)2-, C1-6алкил СО-, C1-6алкил СН(ОН)-, NC-, НСС-, C6H5CC-, 2'-фурил, 3'-фурил, 2'-тиофенил, 3'-тиофенил, 2'-пиридил, 3'-пиридил, 4'-пиридил, 2'-тиазолил, 2'-N-метил- имидазолил, 5'-пиримидинил, C6H5-, H2C=CH-, C1-6алкил или МеС(=СН2)-;
R12 является Н или атомом галогена; или
R1 и R12 объединяются с образованием кольца
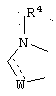
где W означает СН2, СН, NH, N, О или S, и R4 означает Н или С1-6алкил;
Х означает О или NOR5, где R5 представляет Н или C1-6алкил, С3-8циклоалкил, С2-6алкенил, С2-6алкинил, С6-12арил или гетероарил, любой из которых может быть необязательно замещен; или
Х означает (Н,Н), (Н,ОН), (Н, OSi(C1-6алкил)3) или (H,OCOR5), где R5 представляет C1-6алкил, С3-8циклоалкил, С2-6алкенил, С2-6алкинил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен; или
Х означает

где Y представляет собой -(СН2)m-, где m является целым числом 0 до 3, или Y представляет собой -(СН2)n-Z-(CH2)р-, где n является целым числом от 0 до 2, р является целым числом от О до 2 и Z является гетероатомом (необязательно замещенным) или Z является атомом углерода, замещенным одним или двумя C1-6алкилами;
R6 означает Н, C1-6алкил или галоген;
s равно 0 или 1;
R9 означает Н, C1-6алкил, С2-6алкенил или С2-6алкинил, R10CO, OR11, любой из которых может быть необязательно замещен,
где R10 означает Н, С1-18алкил, С2-18алкенил, С2-18алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен, и
где R11 означает Н, C1-6алкил, Si(C1-6алкил)3, 2'-тeтpaгидропиранил или R10CO, где R10 имеет значение, определенное выше;
каждый из R13 и R14 независимо представляет собой Н, С1-18алкил, С2-18алкенил, С2-18алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил или аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен; или
R13R14 является О; и
каждый из R15 и R16 является Н, или они объединяются с образованием группы =СН2, необязательно замещенной,
и его фармацевтически приемлемые соли.
Краткое описание чертежей
Более полное понимание изобретения и многих присущих ему преимуществ будет легко достигнуто, поскольку вышеупомянутое становится более понятным благодаря обращению к следующему детальному описанию, которое обсуждается в связи с сопровождающими чертежами, где:
на фиг. 1 - схема реакции получения соединений аминов и гидроксиламинов согласно данному изобретению;
на фиг. 2 - схема реакции получения соединений циклических аминов согласно данному изобретению;
на фиг. 3 - схема реакции получения соединений циклических гидроксиламинов согласно данному изобретению. Детальное описание предпочтительных вариантов
Идентифицированные выше соединения формулы I и II в частности включают соединения, которые замещены в 3-положении А-кольца двумя атомами водорода.
Полагают, что эти соединения подвергаются окислению in vivo до соответствующего карбонильного соединения.
В рамках данного изобретения термин гетероатом означает кислород, азот, серу, кремний или бор. Галоген означает фтор, хлор, бром или иод, и Halo означает фторо, хлоро, бромо или иодо. Аралкил, аралкенил или аралкинил означают C1-C4aлкильную, С2-С4алкенильную или С2-С4алкинильную группу, несущую арильный заместитель. Низший алкил означает C1-С6алкильную группу. Гетероарил означает единицу из 5-12 не водородных атомов, состоящую из одной или большего количества циклических структур, которые могут быть конденсированы или связаны друг с другом, которые содержат от 1 до 5 гетероатомов и которые специалистами в данной области общепринято считаются единицами, обладающими электронной системой ароматического характера.
Гетероаралкил, гетероаралкенил или гетероаралкинил означают С1-С4алкильную, С2-С4алкенильную или С2-С4алкинильную группу, несущую гетероарильный заместитель.
"Необязательно замещенный" означает незамещенный или замещенный одним или большим количеством гетероатомов и/или галогенами, и/или алкильными группами, в которые входят от 1 до 4 атомов углерода, и/или алкенильными, и/или алкинильными группами, в которые входят от 2 до 4 атомов углерода, и/или циклоалкильными группами, в которые входят от 3 до 7 атомов углерода, и/или арильными группами, в которые входят от 6 до 12 атомов углерода, и/или гетероарильными группами, и в которых алкильная, алкенильная, алкинильная, циклоалкильная, арильная или гетероарильная группа может быть, кроме того, замещена одним или большим количеством гетероатомов и/или галогенов. Замещение может осуществляться непосредственно в СН2 группах гетероциклов циклических аминов. Там, где позволяет валентность, замещение гетероатомами может быть либо в пределах углеродной цепи, либо путем присоединения к ней с помощью одинарной или двойной связей. Например, - CH2CH2C(=O)Н, -СН2(С=O)СН3, -СН2СН2ОСН3, -СН2СН2СН2OН, СН3СН2СН2O-, CH2CH2C(=O)NH2, СН3СН2С(=O)NН-, -СН2СН2СООСН3, СН3СН2СОО-, и СF3СС- все подпадают под это определение.
Во всех случаях, где позволяет валентность и стерические соображения, алкильные, алкенильные, алкинильные и циклоалкильные группы могут содержать дополнительные двойные или тройные связи и/или разветвленные цепи.
В структурах I и II группа R6, как она показана, при С6 может быть либо в α , либо в β -положении. В предпочтительном варианте группа R6 находится в α -положении.
В другом варианте С11β-арильная группа может быть заменена группой пиридина, замещенной группами R1 и R12, как описано ранее.
В предпочтительном варианте стероид, имеющий структуру I, замещен следующим образом:
где R1-Ph означает 4-аминофенил, 4-(N-метиламино)фенил, 4-(N,N-диметиламино)фенил, 4-(N-пиперидино)фенил, 4-(N-пирролидино)фенил, 4-(N-морфолино)фенил, 1-метилиндол-5-ил или 1-метил-2,3-дигидроиндол-5-ил или R1-Ph означает N-оксид 4-(N,N-диметил)фенила, 4-(N-пиперидино)фенила, 4-(N-пирролидино)фенила, 4-(N-морфолино)фенила;
Х означает О, NOH или NОСН3;
R6 означает Н, СН3, F или Сl;
R7 означает Н, метил, этинил, 1-пропинил, 3-пропинил, 3-гидроксипропил, 3-гидрокси-1-пропенил (Е- или Z-), 3,3,3-трифторпропин-1-ил, 3-гидроксипропин-1-ил, (СН2)2СООСН3, (СН2)2СООС2Н5, (СН2)2СОСН3, СС-С6Н5, СH2С6Н5, CN или СООСН3;
R8 означает Н, СН3 или CH2C6H5; и
R9 означает Н, ОН, ОСН3, СНО, СН3СО, C6H5CO или С6Н5СН2СО.
В предпочтительном варианте соединение структуры II является замещенным, где
R1Ph означает 4-аминофенил, 4-(N-метиламино)фенил, 4-(N,N-димeтилaминo)фенил, 4-(N-пиперидино)фенил, 4-(N-пирролидино)фенил, 4-(N-морфолино)фенил, 1-метилиндол-5-ил или 1-метил-2,3-дигидроиндол-5-ил;
Х означает О, NOH или NOCH3;
R6 означает Н, СН3, F или С1;
R9 означает Н, ОН, СНО, СН3СО, С6Н5СО или C6H5CH2CO.
R13 и R14 являются О, (Н, Н), (Н, СН3) или (СН3, СН3); и
R15 и R16 являются (Н, Н) или объединяются с образованием (=СН2).
В предпочтительном варианте спироциклической аминогруппой у C17 атома соединения структуры II является такая группа, в которой азот амина расположен с передней β стороны относительно плоскости соединения, в син-положении относительно С11 арильной группы.
Соединение структуры I также может нести функциональную группу нитрона, когда s равно О, R8 представляет собой О- и R9 является алкеном. В этом случае азот в 17 положении несет положительный заряд.
Конкретные, не ограничивающие изобретение примеры включают следующие соединения:
11β -(4-(N,N-диметиламино)фенил)-17β -(N-гидроксиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он; 11β -(4-(N-пиперидино)фенил)-17β -(N-гидроксиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-гидрокси-N-метиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он; 11β -(4-(N-пиперидино)фенил)-17β -(N-гидрокси-N-метиламино)-17α -(1-пропинил)-эстра-4,9-диен-3-он; 17β -амино-11β -(4-(N,N-диметиламино)фенил)-17α -(1-пропинил)эстра-4,9-диен-3-он; 17β -амино-11β -(4-(N-пиперидино)фенил)-17α -(1-пропинил)эстра-4,9-диен-3-он; 17β -(N-ацетамидо)-11β -(4-(N,N-диметиламино)фенил)-17α -(1-пропинил)эстра-4,9-диен-3-он; 17β -(N-ацетиламидо)-11β -(4-(N-пиперидино)фенил)-17α -(1-пропинил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-формамидо)-17α -(1-пропинил)-эстра-4,9-диен-3-он и его N-оксид; 17β -(N-формамидо)-11β -(4- (N-пиперидино)фенил)-11α -(1-пропинил)эстра-4,9-диен-3-он и его N-оксид; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-гидроксиламино)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 11β -(4-(N-пиперидино)фенил)-17β -(N-гидроксиламино)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-гидрокси-N-метиламино)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 11β -(4-(N-пиперидино)фенил)-17β -(N-гидрокси-N-метиламино)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 17β -амино-11β -(4-(N,N-диметиламино)фенил)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 17β -амино-17α -(3-гидроксипропил)-11β -(4-(N-пиперидино)фенил)эстра-4,9-диен-3-он; 17β -(N-ацетамидо)-11β -(4-(N,N-диметиламино)фенил)-17α -(3-гидроксипропил)эстра-4, 9-диен-3-он; 17β -(N-ацетамидо)-17α -(3-гидроксипропил)- 11β -(4-(N-пиперидино)фенил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-формамидо)-17α -(3-гидроксипропил)эстра-4,9-диен-3-он; 17β -(N-формамидо)-17α -(3-гидроксипропил)-11β -(4-(N-пиперидино)фенил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-17β -(N-формамидо)-17α -(3-формилокси-1-пропил)эстра-4,9-диен-3-он; 17β -(N-формамидо)-17α -(3-формилокси-1-пропил)-11β -(4-(N-пиперидино)фенил)эстра-4,9-диен-3-он; 11β -(4-(N,N-диметиламино)фенил)-1'-гидрокси-5'-метилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N-пиперидино)фенил)-1'-гидрокси-5'-метилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N,N-диметиламино)фенил)-1'-гидроксиспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N-пиперидино)фенил)-1'-гидроксиспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N,N-диметиламино)фенил)-5'-метил-спиро [эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N-пиперидино)фенил)-5'-метилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N,N-диметиламино)фенил)спиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N-пиперидино)фенил)-спиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N,N-диметиламино)фенил)-5'-оксоспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N-пиперидино)фенил)-5'-оксоспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он; 11β -(4-(N,N-диметиламино)-фенил)-1'-формилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он и 11β -(4-(N-пиперидино)фенил)-1'-формилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он.
К соединениям данного изобретения, которые несут аминогруппу, также может относиться соль, образованная с амином. Подходящие фармацевтически приемлемые соли известны специалистам в данной области и включают карбоксилаты, сульфаты, фосфаты и галогениды.
Амино- и гидроксиламиносоединения данного изобретения могут быть получены из промежуточного гидроксинитросоедиения структуры (III)
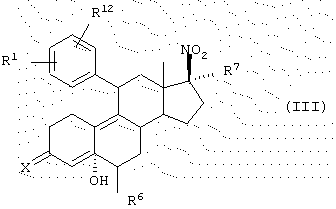
где R1 означает (R2R3N(О)r)-, где r равно 0 или 1 и каждый из R2 и R3 независимо представляет Н, C1-6алкил, С3-8циклоалкил, С2-6алкенил или С2-6алкинил, любой из которых может быть необязательно замещен; или
R1 является
где q равно 0 или 1, Y означает - (CH2)m-, где m является целым числом от 0 до 5, или Y означает -(CH2)n-Z-(СН2)р-, где n является целым числом от 0 до 2, р является целым числом от О до 2, и Z означает гетероатом (необязательно замещенный) и где CH2 группы могут быть необязательно замещены; или
R1 означает N-имидазолил-, N-пирролил-, Н, атом галогена, НО-, СF3SO2O-, C1-6алкил-О-, C1-6алкил-S-, C1-6алкил-S(О)-, C1-6алкил-S(O)2-, C1-6алкил-СО-, C1-6алкил-СН(ОН)-, NC-, НСС-, С6Н5СС-, 2'-фурил, 3'-фурил, 2'-тиофенил, 3'-тиофенил, 2'-пиридил, 3'-пиридил, 4'-пиридил, 2,-тиaзoлил, 2'-N-метилимидазолил, 5'-пиримидинил, С6Н5-, Н2C=СН-, C1-6алкил или МеС(=СН2)-;
R12 является Н или атомом галогена; или
R1 и R12 объединяются с образованием кольца
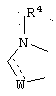
где W означает СН2, СН, NH, N, О или S, и R4 означает Н или C1-6алкил;
X означает
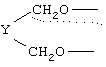
где Y представляет собой -(СН2)m-, где m является целым числом от 0 до 3, или Y представляет собой -(CH2)n-Z-(СН2)p-, где n является целым числом от 0 до 2, р является целым числом от 0 до 2 и Z является гетероатомом (необязательно замещенным) или Z является атомом углерода, замещенным одной или двумя C1-6алкильными группами;
R6 означает Н, C1-6алкил или галоген;
R7 означает Н, C1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен, CN, COOR10 или CONHR10, где R10 представляет собой Н, С1-18алкил, С2-18алкенил, С2-18алкинил, С3-8циклоалкил, С6-12арил, аралкил, аралкенил, аралкинил, гетероарил, гетероаралкил, гетероаралкенил или гетероаралкинил, любой из которых может быть необязательно замещен, путем восстановления нитрогруппы с последующим гидролизом кеталя и элиминированием гидроксильной группы. Такой способ может быть выполнен с помощью традиционных методов, известных специалистам в данной области. Например, нитрогруппа может быть восстановлена до амина или гидроксиламина путем обработки цинком и хлоридом аммония. Можно преимущественно получать в качестве продукта амин или гидроксиламин, подбирая соотношение между цинком и нитросоединением, при более низкой молярной доле цинка (2-18) преимущественно идет образование гидроксиламина и при более высокой молярной доле (20-80) преимущественно идет образование амина. Соединения структуры III могут быть получены способом, описанным в заявке на патент США, озаглавленной "17β -Nitro-11β -aryl Steroids and Their Derivatives having Agonist or Antagonist Hormonal Properties" C.E.Cook, J.A.Kepler, R.S.Shetty, G.S.Bartley and D.Lee, зарегистрированной одновременно с этой заявкой, (Attorney Docket №2025-0133-77), части которой, относящиеся к данному вопросу, включены таким образом в качестве ссылки.
Стероиды, обладающие гестагенной, антигестагенной и/или антиглюкокортикоидной активностью, имеют применение для регулирования фертильности у людей и других млекопитающих, таких как приматы, домашние животные и сельскохозяйственные животные, и для лечения патологических состояний у животных или людей, на которых эти активности могут оказывать целебное действие. Таким образом, дополнительно к их применению для регулирования фертильности и репродукции, они могут быть пригодны для лечения таких состояний, как фибромы, синдром Кушинга, глаукома, эндометриоз, созревание шейки матки перед родоразрешением, гормонозаместительная терапия, предменструальный синдром и рак.
Соединения данного изобретения могут вводиться различными способами. Поэтому, те продукты изобретения, которые активны при пероральном пути введения, могут вводиться в виде растворов, суспензий, эмульсий, таблеток, включая подъязычные и внутриротовые таблетки, мягких желатиновых капсул, включая растворы, используемые в мягких желатиновых капсулах, водных или масляных суспензий, эмульсий, пилюлей, лепешек, пастилок, таблеток, сиропов или эликсиров и им подобных. Продукты изобретения, активные при парентеральном введении, могут вводиться путем инъекции веществ замедленного всасывания, имплантатов, включая имплантаты Silastic™ и биодеградируемые имплантаты, внутримышечных и внутривенных инъекций.
Композиции могут быть приготовлены в соответствии с любым способом производства фармацевтических композиций, известным в этой области, и такие композиции могут содержать один или большее количество агентов, выбранных из группы, состоящей из подсластителей, вкусовых агентов, красителей и консервантов. Приемлемы таблетки, содержащие активный ингредиент с добавлением нетоксичных фармацевтически приемлемых эксципиентов, которые пригодны для производства таблеток. Такими эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия, агенты гранулирования и дизинтегрирующие агенты, такие как кукурузный крахмал или альгиновая кислота; связывающие агенты, такие как крахмал, желатин или акация; и смазывающие агенты, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрыты или могут быть покрыты с помощью известных способов, чтобы замедлить разрушение и адсорбцию в желудочно-кишечном тракте и таким образом обеспечить замедленное действие в течение более длительного периода. Например, могут быть использованы такие замедляющие высвобождение материалы, как глицерилмоностеарат или глицерилдистеарат, причем отдельно или вместе с воском.
Композиции для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например с карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, такой как арахисовое масло, жидкий парафин или оливковое масло.
Водные суспензии изобретения содержат активные материалы с добавлением эксципиентов, пригодных для производства водных суспензий. Такие эксципиенты включают в себя суспендирующее средство, такое как натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксипропилэтилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие или увлажняющие агенты, такие как фосфатиды природного происхождения (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с алифатическим спиртом с длинной цепью (например, гептадекаэтиленоксицетанол), продукт конденсации этиленоксида с неполным сложным эфиром, полученным из жирной кислоты и гексита (например, сорбитполиоксиэтиленмоноолеат), или продукт конденсации этиленоксида с неполным сложным эфиром, полученным из жирной кислоты и гекситангидрида (например, сорбитанполиокси-этиленмоноолеат). Водная суспензия также может содержать один или большее количество консервантов, таких как этил- или н-пропил-пара-гидроксибензоат, один или большее количество красителей, один или большее количество вкусовых агентов и один или большее количество подсластителей, таких как сахароза, аспартам или сахарин. В композициях для офтальмологии, как известно в этой области, следует регулировать осмотическое давление.
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Могут быть добавлены подсластители для того, чтобы снабдить пероральный препарат приятным вкусом. Эти композиции могут сохраняться за счет добавления антиоксиданта, такого как аскорбиновая кислота.
Дисперсные порошки и гранулы изобретения, пригодные для приготовления водной суспензии путем добавления воды, могут быть приготовлены из активных ингредиентов с добавлением диспергирующего, суспендирующего и/или увлажняющего агента, и одного или большего количества консервантов. Примерами приемлемых диспергирующих или увлажняющих агентов и суспендирующих агентов являются агенты, указанные выше. Также могут присутствовать дополнительные эксципиенты, например подсластители, вкусовые агенты и красители.
Фармацевтическая композиция изобретения также может быть в форме эмульсий масла в воде. Масляной фазой может быть растительное масло, такое как оливковое масло или арахисовое масло, минеральное масло, такое как жидкий парафин, или их смесь. Приемлемые эмульгирующие агенты включают камеди природного происхождения, такие как аравийская камедь и трагакантовая камедь, фосфатиды природного происхождения, такие как лецитин сои, сложные эфиры и неполные сложные эфиры, полученные из жирных кислот и гекситангидридов, такие как сорбитанмоноолеат, и продукты конденсации этих неполных сложных эфиров с этиленоксидом, такие как сорбитанполиоксиэтиленмоноолеат. Эмульсия также может содержать подсластители и вкусовые агенты.
Сиропы и эликсиры могут быть приготовлены с добавлением подсластителей, таких как глицерин, сорбит или сахароза. Такие композиции могут также содержать агент, уменьшающий раздражение, консервант, вкусовой агент или краситель.
Фармацевтические композиции изобретения могут быть в форме стерильных инъекционных препаратов, таких как стерильная инъекционная водная или масляная суспензия. Эта суспензия может быть приготовлена в соответствии с известными в данной области способами при использовании тех приемлемых диспергирующих или увлажняющих агентов и суспендирующих агентов, которые указаны выше. Стерильный, инъекционный препарат может также быть в виде стерильного инъекционного раствора или суспензии в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, таком как раствор 1,3-бутандиола. В число приемлемых наполнителей и растворителей, которые могут применяться, входят вода и раствор Рингера, изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно могут применяться стерильные нелетучие масла. Для этой цели может применяться легкое нелетучее масло, включая синтетические моно- и диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, также могут использоваться в препаратах для инъекций. Стерилизация может проводиться традиционными способами, известными специалистам в данной области, такими как стерильная фильтрация, облучение или термическая стерилизация (например, автоклавирование).
Водные композиции (т.е. эмульсии масла в воде, сиропы, эликсиры и инъекционные препараты) могут быть приготовлены при доведении рН до рН оптимальной стабильности. Определение оптимума рН может быть осуществлено традиционными способами, известными специалистам в данной области. Для поддержания рН композиции также могут быть использованы приемлемые буферы.
Соединения данного изобретения также могут вводиться в форме суппозиториев для ректального введения лекарственного средства. Такие композиции могут быть приготовлены смешиванием лекарственного средства с приемлемым нераздражающим эксципиентом, который при обычных температурах является твердым, но становится жидким при ректальных температурах и, поэтому, будет плавиться в прямой кишке, высвобождая лекарственное средство. Неограничивающими примерами таких материалов являются масло какао и полиэтиленгликоли.
Они также могут быть введены интраназальным, внутриглазным, интравагинальным и интраректальным путями, включая суппозитории, инсуффляцию, порошки и аэрозольные композиции.
Продукты изобретения, которые предпочтительно вводятся локально, в виде аппликаторных полосок, растворов, суспензий, эмульсий, гелей, кремов, мазей, паст, желе, в виде наносимых кисточкой лекарственных веществ в вязком носителе, порошков и аэрозолей.
Продукты, обладающие антиглюкокортикоидной активностью, имеют особое значение для патологических состояний, для которых характерен избыток эндогенного глюкокортикоида, таких как синдром Кушинга, гирсутизм и в особенности для тех случаев, когда состояние связано с андрогенитальным синдромом, для глазных болезней, связанных с избытком глюкокортикоида, таких как глаукома, симптомов стресса, связанных с избытком секреции глюкокортикоидов и им подобных состояний.
Продукты, обладающие гестагенной активностью имеют особое значение как гестагенные средства, ингибиторы овуляции, регуляторы менструации, контрацептивные средства, агенты синхронизации периодов фертильности у крупного рогатого скота, при эндометриозе и подобных состояниях. При использовании в целях контрацепции приемлемым может являться добавление к ним эстрогенных агентов, таких, например, как этинилэстрадиол или сложные эфиры эстрадиола.
Продукты, обладающие антигестагенной активностью, характеризуются действием, противоположным эффекту прогестерона. Как таковые, они могут иметь значение для регулирования гормональных отклонений от нормального менструального цикла и синхронизации периодов фертильности у крупного рогатого скота.
Соединения изобретения могут быть использованы для регулирования фертильности в течение всего репродуктивного цикла. Они имеют особое значение как посткоитальные контрацептивы, для придания матке свойств, неблагоприятных для имплантации, и как контрацептивы на "один раз в месяц". Они могут использоваться вместе с простагландинами, средствами, стимулирующими родовую деятельность, эстрогенами и им подобными средствами.
Важное дальнейшее применение продуктов изобретения основано на их способности замедлять рост гормонозависимых раковых заболеваний. Такие виды рака включают в себя рак почки, молочной железы, эндометрия, яичника и простаты, которые характеризуются наличием рецепторов прогестерона и для которых можно ожидать ответной реакции на продукты данного изобретения. Другие применения антигестагенных средств включают в себя лечение кистозно-фиброзной мастопатии. Некоторые раковые заболевания и, в частности, меланомы, могут положительно реагировать на кортикоидную/антикортикоидную терапию.
Соединения согласно данному изобретению могут вводиться любому теплокровному млекопитающему, такому как человек, домашние животные и сельскохозяйственные животные. К домашним животным относятся собаки, кошки и т.д. К сельскохозяйственным животным относятся коровы, лошади, свиньи, овцы, козы и т.д.
Количество активного ингредиента, которое может быть объединено с материалом носителя, чтобы получить разовую лекарственную форму будет варьироваться в зависимости от заболевания, которое лечится, вида млекопитающего и конкретного способа введения. Терапевтически эффективное количество может быть определено с помощью стандартных экспериментов и по аналогии с количествами, используемыми при лечении тех же патологических состояний аналогичными стероидными соединениями. Например, стандартная доза стероида предпочтительно может содержать от 0,1 миллиграмма до 1 грамма активного ингредиента. Более предпочтительна стандартная доза от 0,001 до 0,5 грамм. В конкретных случаях лечения эндометриоза или фиброзных опухолей может вводиться количество от 0,01 до 10 мг/кг веса тела, предпочтительно от 0,1 до 3 мг/кг. Сходные дозы этих соединений могут применяться для других терапевтических целей. Обычно соединения могут вводиться ежедневно от 1 до 4 раз в сутки, предпочтительно от 1 до 2 раз в сутки, но при таком применении, как, например, в гормонозаместительной терапии, они могут вводиться по схеме с циклическими стадиями. В некоторых случаях частота и выбор определенного времени дозирования будет зависеть от таких факторов, как полупериод существования в организме конкретного соединения, состава дозы и пути введения. Однако следует понимать, что конкретный уровень дозы для каждого конкретного пациента будет зависеть от различных факторов, включая активность конкретных применяемых соединений; возраст, вес тела, общее состояние здоровья, пол и диету человека во время лечения и путь введения; скорость выведения; другие лекарственные средства, которые были предварительно введены; и тяжести конкретного заболевания, подвергающегося терапии, что хорошо понятно специалистам в данной области.
Такие соединения пригодны для лечения эндометриоза, лейомиом матки (фибром) и некоторых раковых заболеваний и опухолей, в гормонозаместительной терапии, а также в регуляции различных этапов репродукции и фертильности, такой как контрацепция. Более детальное описание потенциальных применений таких соединений дано в Donaldson, Molly S.; Dorflinger, L.; Brown, Sarah S.; Benet, Leslie Z., Editors, Clinical Application of Mifepristone (RU 486) and Other Antiprogestins, Committee on Antiprogestins: Assessing the Science, Institute of Medicine, National Academy Press, 1993. Они также применяются как промежуточные продукты для синтеза других стероидов.
Имея общее описание данного изобретения, дальнейшее понимание может быть получено при обращении к некоторым общим примерам, которые предоставляются здесь только в целях иллюстрации и не предназначены как ограничивающие примеры, если не оговорено особо.
Процедуры синтеза
Соединения изобретения могут быть приготовлены согласно процедурам, таким как процедуры, схематично показанные на фиг. с 1 по 3, исходя из 11β -арил-3,3-[1,2-этандиилбис-(окси)]-5α -гидрокси-17(3-нитроэстр-9-енов (например, соединение А-1 на фиг. 1 или аналогичные соединения) или из 3',4'-дигидро-11β -арил-3,3-[1,2-этандиилбис(окси)]-1'-оксо-спиро-[эстр-9-ен-17β ,2'(2'Н)-пиррол]-5α -олов (например, соединение С-1 на фиг. 3 или аналогичные соединения). Соединения типа А-1 или С-1 могут быть получены согласно процедурам, приведенным в заявке на патент США, озаглавленной "17β -Nitro-11β -aryl Steroids and Their Derivatives having Agonist or Antagonist Hormonal Properties" C.E.Cook, J.A.Kepler, R.S.Shetty, G.S.Bartley and D.Lee, зарегистрированной одновременно с этой заявкой, (Attorney Docket №2025-0133-77).
Таким образом, например, обработка А-1 избытком цинковой пыли (предпочтительно около 9 атом-эквивалентов) и хлоридом аммония в растворе спирта/воды при температуре окружающей комнатной среды приводит к хорошему выходу 17β -N-гидроксил-аминосоединения А-2, которое при обработке водным раствором кислоты (предпочтительно трифторуксусная кислота в воде и CH2Cl2) претерпевает гидролиз кеталя и подвергается дегидратации до 4,9-диен-3-она А-3. Если промежуточный продукт гидроксиламин А-2 обрабатывается формалином и NaBH3CN, то азот метилируется с образованием промежуточного 17β -N-метил-N-гидроксисоединения А-5, которое может быть гидролизовано и дегидратировано тем же способом, что и А-2, чтобы получить диенон А-7.
Если соединение А-1 обрабатывается избытком цинковой пыли (предпочтительно около 60 атом-эквивалентов) и хлоридом аммония в растворе этанола/воды/тетрагидрофурана (ТГФ) при повышенных температурах (предпочтительно около 70° С), восстановление нитрогруппы приводит к 17β -аминам А-4. Эти соединения могут быть гидролизованы и дегидратированы, как описано выше до получения диенонов А-6. Далее воздействием на аминогруппу А-6 могут быть получены производные, например путем превращения в амиды способами, хорошо известными специалистам в данной области. Например, обработка А-6 муравьиной кислотой и N,N-дициклогексилкарбодиимидом (ДЦК) приводит к формамидосоединениям, тогда как результатом обработки ацетангидридом в пиридине является образование ацетамидосоединений.
Если R7 в соединении А-1 оканчивается карбоксилом или сложноэфирной карбоксильной функцией и если позволяет длина цепи, то восстановление нитрогруппы может приводить к циклизации до амида. Таким образом, если R7 является CH2CH2COOEt (соединение В-1) и для восстановления используется избыток цинка при повышенной температуре, образуется циклический амид В-2. Затем кислотный гидролиз и дегидратация приводит к образованию диенона В-3. Если R7 соединения А-1 содержит карбонильную группу (кетон или альдегид) в соответствующем положении в цепи, то восстановление при более мягких условиях приводит к спиронитронам, таким как соединение С-1, получение которого описано в заявке на патент США, озаглавленной "17β -Nitro-11β -aryl Steroids and Their Derivatives having Agonist or Antagonist Hormonal Properties" C.E.Cook, J.A.Kepler, R.S.Shetty, G.S.Bartley and D.Lee, зарегистрированной одновременно с этой заявкой (Attorney Docket №2025-0133-77). Результатом восстановления нитрона, например, борогидридом натрия, является N-гидроксиспиропирролидины С-2, которые также могут быть гидролизованы и дегидратированы до диенонов, как описано выше.
Такие соединения как В-2 и С-2 являются многоцелевыми промежуточными продуктами, которые могут быть превращены во множество новых соединений способами, известными специалистам в данной области, включая N- или O-алкилирование и/или восстановление гидридными реагентами, чтобы получить, например, циклические амины, которые могут быть далее модифицированы, например, путем N-алкилирования или N-ацилирования. Таким образом, могут быть получены различные аналоги спиропирролидинов В-2, В-3, С-2 и С-3.
Имея общее описание данного изобретения, дальнейшее понимание можно получить, обратившись к некоторым конкретным примерам, которые предоставлены здесь только в целях иллюстрации, и не предназначены как ограничивающие, если не оговорено особо.
Общие процедуры. Если не оговорено особо, химически чистые реактивы были получены из коммерческих источников и использовались без дальнейшей очистки. Простой эфир и тетрагидрофуран (ТГФ) перегоняли непосредственно перед использованием из натрийбензофенонкетильной пары в атмосфере азота. Переносы всех чувствительных к влаге и воздуху реакций и реактивов выполняли в атмосфере сухого азота или аргона. Тонкослойную хромотографию выполняли на предварительно покрытых силикагелем 60 F-254 пластинках ЕМ Science. Обычно соединения выявляли в Уф-свете (254 нм) или при опрыскивании анисовым пара-альдегидом. Для препаративной колоночной хроматографии использовали силикагель ЕМ Science, 60  (230-400 меш). Концентрирование растворов проводили с использованием роторного испарителя при давлении, создаваемом водоструйным насосом при температуре окружающей среды. Точки плавления определяли на Mel-Temp II без исправления. Если не оговорено особо, спектры 1Н ЯМР получали при 250 МГц на спектрометре Bruker AC 250 при использовании в качестве растворителя СОСl3 и тетраметилсилана (ТМС) в качестве внутреннего стандарта. Химические сдвиги выражали в единицах м.д. сдвига по полю от ТМС. Масс спектры обычно получали при ионизации электронным ударом при 70 эВ на приборе Hewlett Packard 5989A. Элементные анализы были выполнены Atlantic Microlab. Inc., Atlanta, GA.
(230-400 меш). Концентрирование растворов проводили с использованием роторного испарителя при давлении, создаваемом водоструйным насосом при температуре окружающей среды. Точки плавления определяли на Mel-Temp II без исправления. Если не оговорено особо, спектры 1Н ЯМР получали при 250 МГц на спектрометре Bruker AC 250 при использовании в качестве растворителя СОСl3 и тетраметилсилана (ТМС) в качестве внутреннего стандарта. Химические сдвиги выражали в единицах м.д. сдвига по полю от ТМС. Масс спектры обычно получали при ионизации электронным ударом при 70 эВ на приборе Hewlett Packard 5989A. Элементные анализы были выполнены Atlantic Microlab. Inc., Atlanta, GA.
Пример 1
Синтез 11β -[4-(N,N-диметиламино)фенил]-17β -(N-гидроксил-амино)-17α -(1-пропинил)эстра-4,9-диен-3-он
[А-3 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H)].
11β -[4-(N,N-диметиламино)фенил]-3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17β -(N-гидроксиламино)-17α -(1-пропинил)эстр-9-ен
[A-2 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H)]. К гомогенному раствору 6,34 г (12,2 ммоль) нитропропина А-1 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) и 1,37 г (25,6 ммоль) NH4Cl в 160 мл ТГФ, 80 мл EtOH и 80 мл воды при комнатной температуре добавляли 7,17 г (110 ммоль) цинковой пыли (-325 меш). После перемешивания в течение 1,5 часа, смесь фильтровали через подушку целита с помощью EtOAc. Фильтрат три раза промывали соляным раствором, сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении, получая белое аморфное твердое вещество (6,42 г). Хроматография на силикагеле (70% EtOAc в гексанах) давала гидроксиламин A-2 (R1= 4-Me2N-, R7=СН3СС-, R6=R12=Н) (3,60 г, выход 60%). 1H ЯМР (250 МГц, СDСl3) δ 7,05 (2Н, д, J=8,6 Гц), 6,64 (2Н, д, J=8,8 Гц), 5,02 (1Н, уш.с), 4,43 (1Н, с), 4,21 (1Н, д, J=6,2 Гц), 4,02-3,92 (4Н, м), 2,90 (6Н, с), 1,91 (1Н, с), 0,48 (3Н, с).
11β -[4-(N,N-диметиламино)фенил]-17β -(N-гидроксиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он [А-3 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н)]. Смесь 3,60 г (7,11 ммоль) гидроксикеталя A-2 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) в 316 мл СН2Сl2 и 6,3 мл воды энергично перемешивали при охлаждении в бане с ледяной водой в течение 1,5 часа. К быстро перемешиваемой дисперсии по каплям добавляли 8,80 мл (114 ммоль) трифторуксусной кислоты. После энергичного перемешивания в течение 3 часов, медленно добавляли избыток насыщенного водного раствора NaHCO3 и позволяли смеси перемешиваться при комнатной температуре в течение 20 минут. Водный слой отделяли и три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали соляным раствором, сушили над Na2SO4, фильтровали и удаляли растворитель при пониженном давлении, чтобы получить желтое твердое вещество (3,11 г). Материал хроматографировали на силикагеле (70% EtoAc в гексанах). Объединение полученных в результате фракций с чистотой >97% (как определено с помощью ВЭЖХ анализа) давало диенон гидроксиламина А-3 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) (2,37 г, выход 75%) [т.пл. 100-117° С (аморфный)]. Образец может быть высушен в вакууме при 92° С в течение 14 часов, чтобы получить свободный от растворителя продукт без потери чистоты. 1H ЯМР (250 МГц, СDСl3) δ 7,01 (2Н, д, J=8,5 Гц), 6,65 (2Н, д, J=8,9 Гц), 5,75 (1Н, с), 5,16 (1Н, уш. с), 4,65 (1Н, с), 4,31 (1Н, уш., с), 2,91 (6Н, с), 1,94 (3Н, с), 0,55 (3Н, с). Аналитически вычислено для C29H36N2O2·0,5H2O: С, 76,79; Н, 8,22; N, 6,18. Найдено: С, 76,82; Н, 8,29; N, 6,12. Масс-спектр m/z (относительная интенсивность) 444 (М+, 12), 428 (12), 411 (23), 134 (51), 121 (100).
Пример 2
Синтез 11β -[4-(N,N-диметиламино)фенил]-17β -(N-гидрокси-N-метиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он
[А-7 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н)].
11β -[4-(N,N-диметиламино)фенил]-3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17β -(N-гидрокси-N-метиламино)-17α -(1-пропинил)-эстр-9-ен [А-5 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) ]. К раствору 232 мг (0,458 ммоль) гидроксиламина А-2 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) в 4,6 мл CH3CN при комнатной температуре добавляли 0,19 мл (2,3 ммоль) формалина, затем 49 мг (0,733 ммоль) NaBH3CN. Через 45 мин основную реакционную смесь, содержащую белый осадок [рН 8-9 (как определено с помощью предварительно смоченной водой индикаторной бумаги для определения рН)] доводили до рН 7 добавлением трех небольших порций (в общем, приблизительно 1 капля) ледяной уксусной кислоты, достигая, таким образом, гомогенности. Через 1,5 часа, измеряли значение рН раствора, которое было равно 8, затем еще добавляли небольшое количество (приблизительно 0,25 капли) ледяной уксусной кислоты, затем, спустя 40 мин, были добавлены насыщенный водный NаНСО3 и EtOAc. Водный слой отделяли и три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали раствором соли, сушили над Na2SO4, фильтровали, и растворитель удаляли при пониженном давлении. Полученную в результате белую пену очищали хроматографией на силикагеле (60% EtOAc в гексанах), получая N-метилгидроксиламин А-5 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) (74 мг, выход 31%). 1H ЯМР (250 МГц, СDСl3) δ 7,04 (2Н, д, J=8,7 Гц), 6,63 (2Н, д, J=8,7 Гц), 4,42 (1Н, с), 4,16 (1Н, уш. с), 4,02-3,92 (4Н, м), 2,89 (6Н, с), 2,54 (3Н, с), 1,96 (3Н, с), 0,51 (3Н, с).
11β -[4-(N,N-диметиламино)фенил]-17β -(N-гидрокси-N-метиламино)-17α -(1-пропинил)эстра-4,9-диен-3-он [А-7 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H)]. К энергично перемешиваемой смеси 122 мг (234 ммоль) кеталя N-метилгидроксиламина А-5 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) в 10,2 мл CH2Cl2 и 0,5 мл CDCl3 и 0,21 мл воды при 0° C по каплям добавляли 0,29 мл (3,76 ммоль) трифторуксусной кислоты. После энергичного перемешивания в течение 5,5 час добавляли насыщенный водный NaHCO3, и смесь перемешивали в течение 30 мин, затем разбавляли EtOAc. Водный слой отделяли и три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали соляным раствором, сушили над Na2SO4, фильтровали и удаляли растворитель при пониженном давлении, получая желтую пену (118 мг). Продукт очищали с помощью флэш-хроматографии на силикагеле (80% EtOAc в гексанах), затем флэш-хроматографией на силикагеле (1,1:30:68,9, МеОН-ТГФ-гексаны), затем хроматографией среднего давления на силикагеле (гексаны, затем 30% ТГФ в гексанах), и затем обращенно-фазной препаративной ВЭЖХ (20% Н2О в МеОН), чтобы получить диенон метилгидроксиламина А-7 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) (29,0 мг, выход 27%) с чистотой >97%, как было определено с помощью ВЭЖХ анализа. 1H. ЯМР (250 МГц, CDCl3) δ 7,00 (2Н, д, J=8, 5 Гц), 6,65 (2Н, д, J=8,8 Гц), 5,75 (1Н, с), 4,28 (1H, д, J=6,4 Гц), 2,91 (6Н, с), 2,57 (3Н, с), 1,98 (3Н, с), 0,58 (3Н, с). Аналитически вычислено для C30H38N2O2·0,75 H2O: С, 76,32; H, 8,43; N, 5,93. Найдено: С, 76,62; H, 8,17; N, 5,87. Масс-спектр m/z (относительная интенсивность) 458 (М+, 13), 441 (32), 411 (21), 320 (24), 278 (23), 225 (23), 121 (100).
Пример 3
Синтез 17β -амино-11β -[4-(N,N-диметиламино)фенил]-17α -(1-пропинил)эстра-4,9-диен-3-он
[А-6 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н)].
17β -амино-11β -[4-(N,N-диметиламино)фенил]-3,3-[1,2-этандиилбис (окси)]-5α -гидрокси-17α -(1-пропинил)эстр-9-ен [А-4 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н)]. К гомогенному раствору 5,00 г (9,60 ммоль) нитропропина А-1 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) и 10,3 г (192 ммоль) NH4Cl в 100 мл ТГФ, 50 мл EtOH и 50 мл воды при 70° С добавляли 37,7 г (576 ммоль) цинковой пыли (-325 меш). После эффективного перемешивания в течение 7,5 час, смеси позволяли остыть до комнатной температуры и фильтровали всасыванием через подушку целита с помощью EtOAc. Фильтрат три раза промывали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая желтую пену (4,95 г). Хроматография на силикагеле (20% МеОН в EtOAc) давала аминопропин А-4 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) (3,48 г, выход 74%). 1H ЯМР (250 МГц, СDСl3) δ 7,06 (2Н, д, J=8,5 Гц), 6,64 (2Н, д, J=8,8 Гц), 4,42 (1Н, уш. с), 4,25 (1Н, д, J=6,9 Гц), 4,03-3,90 (4Н, м), 2,91 (6Н, с), 1,84 (3Н, с), 0,40 (3Н, с).
17β -амино-11β -[4-(N,N-диметиламино)фенил]-17α -(1-пропинил)-эстра-4,9-диен-3-он [А-6 (R1=4-Me2N-, R7=СН3СС-, R6= R12=Н)]. Смесь 265 мг (0,540 ммоль) кеталя аминопропина А-4 (R1= 4-Me2N-, R7=СН3СС-, R6=R12=Н) в 24 мл СН2Сl2 и 0,48 мл воды энергично перемешивали в бане с ледяной водой в течение 1,5 час. К быстро перемешиваемой дисперсии по каплям добавляли 0,67 мл (8,69 ммоль) трифторуксусной кислоты. После энергичного перемешивания в течение 4,5 час, медленно добавляли избыток насыщенного водного раствора NaHCO3, затем смесь перемешивали при комнатной температуре в течение 30 минут. Водный слой отделяли и три раза экстрагировали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, чтобы получить желтое масло (258 мг). Полученный таким образом материал был высокой чистоты, судя по анализу 1H ЯМР, и в аналогичных экспериментах мог бы быть использован для последующих превращений без дальнейшей очистки. Образец с чистотой >97% (как было определено с помощью ВЭЖХ анализа) получали благодаря хроматографии на силикагеле (12% МеОН в EtoAc), затем хроматографией на силикагеле (10% МеОН в EtoAc), затем хроматографией на дезактивированном EtsN силикагеле (50% EtoAc в гексанах) и затем обращенно-фазной препаративной ВЭЖХ [30% Н2O в МеОН, содержащем Et3N (50 мМ)], что давало диенон А-6 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) (44,6 мг, выход 19%). 1H ЯМР (250 МГц, CDCl3) δ 7,03 (2Н, д, J=8,7 Гц), 6,66 (2Н, д, J=8,9 Гц), 5,76 (1Н, с), 4,36 (1Н, д, J-7,3 Гц), 2,92 (6Н, с), 1,84 (3Н, с), 0,48 (1Н, с). Аналитически вычислено для С29H36N2О: С, 81,27; H, 8,47; N, 6,54. Найдено: С, 81,21; H, 8,50; N, 6,49. Масс-спектр m/z (относительная интенсивность) 428 (М+ 61), 411 (97), 278 (33), 134 (100).
Пример 4
Синтез 17β -(N-ацетамидо)-11β -[4-(N,N-диметиламино)фенил]-17α -(1-пропинил)эстра-4,9-диен-3-он
[А-8 (R1=4-Me2N-, R7=СН3СС-, R10=СН3, R6=R12=H)]
К раствору 72 мг (0,168 ммоль) аминодиенона А-6 (R1=4-Me2N-, R7=СН3СС-, R6=R12=Н) в 1,6 мл пиридина при 0° С добавляли 18,0 мл (185 ммоль) Ас2О. Через 2,5 час добавляли одну каплю Ас20, и через 30 мин раствор немного концентрировали при медленном пропускании азота. Полученное в результате желто-коричневое масло объединяли с 19 мг предварительно полученного продукта, затем хроматографировали на силикагеле (90% EtOAc в гексанах), получая ацетамид диенона А-8 (Rl=4-Me2N-, R7=СН3СС-, R10=СН3, R6=R12=Н) (64,8 мг) в виде твердого вещества. Три последовательных очистки с помощью обращенно-фазной СЭЖХ (80:20, МеОН-Н2O, 75:25, МеОН-Н2O, и 77,5:22,5, МеОН-Н2O) давали диенон А-8 (R1=4-Me2N-, R10=СН3, R7=СН3СС-, R6=R12=Н) (37,7 мг, выход 38% [скорректировано]) с чистотой >97%, как было определено с помощью ВЭЖХ анализа. 1H ЯМР (250 МГц, СDСl3) δ 7,03 (2Н, д, J=8,6 Гц), 6,70 (2Н, д, J=8,8 Гц), 5,67 (1Н, с), 5,55 (1Н, уш. с), 4,36 (1Н, д, J=6,2 Гц), 2,92 (6Н, с), 1,93 (3Н, с), 1,87 (3Н, с), 0,49 (3Н, с). Аналитически вычислено для С31Н38N2O2·0,5 Н2O: С, 77,63; Н, 8,20; N, 5,84. Найдено: С, 77,30; Н, 8,20; N, 5,77. Масс-спектр m/z (относительная интенсивность) 470 (М+, 100), 411 (9), 280 (44), 121 (41).
Пример 5
Синтез 11β -[4-(N,N-диметиламино)фенил]-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-он
[А-8 (R1=4-Me2N-, R7=СН3СС-, R10=R6=R12=Н)].
К раствору 2,11 г (10,2 ммоль) дициклогексилкарбодиимида в 11 мл СНСl3 при комнатной температуре добавляли 20,4 мл (20,4 ммоль) 1,00 М муравьиной кислоты в СНСl3, что вызывало появление белого осадка. Через 45 мин смесь добавляли к раствору 2,19 г (5,11 ммоль) аминопропина А-6 (R1=4-Me2N-, R7=СН3СС-, R6=R12=H) и 2,47 мл (30,7 ммоль) пиридина в 22 мл СНСl3. Через 20 мин добавляли 66 мл эфира и полученную в результате смесь фильтровали через подушку целита, промывая шесть раз 10 мл эфира. Фильтрат концентрировали при пониженном давлении, затем разбавляли 22 мл EtOAc и перемешивали в течение 10 мин. Полученную в результате смесь фильтровали через подушку целита, восемь раз промывая 3 мл EtOAc. Фильтрат концентрировали при пониженном давлении. Три раза повторяли разбавление остатка 11 мл толуола и концентрировали при пониженном давлении в роторном испарителе. После дополнительного удаления растворителя в вакууме, светло-зеленый остаток не содержал пиридина, судя по 1H ЯМР анализу, и его хроматографировали на силикагеле (75% EtOAc в гексанах), получая формамид А-8 (R1=4-Me2N-, R7=СН3СС-, R10=R6=R12=H) (1,56 г, выход 67%) [т.пл. 128-142° С (аморфный)]. Чистота продукта, определенная ВЭЖХ анализом была >97%, и его можно нагревать в вакууме при 90° С в течение 16 часов для получения продукта, не содержащего растворителя, с сохранением чистоты >97% по данным ВЭЖХ анализа. Это соединение существовало в виде смеси равновесных форм (по наблюдениям с помощью 1H ЯМР). 1H ЯМР (250 МГц, СDСl3, интегрирование регулировали для определения соотношения главной и минорной форм) δ 8,52 (2Н, д, J=11,7 Гц), 6,98 (2Н, д, J=8,6 Гц), 6,64 (2Н, д, J=8,8 Гц), 6,21 (1Н, д, J=11,8 Гц), 5,77 (1Н, с), 4,37 (1Н, д, J=6,7 Гц), 2,91 (6Н, с), 1,90 (3Н, с), 0,47 (3Н, с); минорная форма: 8,08 (1Н, с), 7,04 (плечо), 5,68 (1Н, с) 1,88 (3Н, с), 0,50 (плечо). Аналитически вычислено для C30H36N2O2·l, 25 Н2O: С, 75,20; Н, 8,10; N, 5,85. Найдено: С, 75,17; Н, 7,56; N, 5,80. Масс-спектр m/z (относительная интенсивность) 456 (М+, 94), 280 (44), 134 (51), 121 (100).
Пример 6
Синтез 17β -амино-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]-эстра-4,9-диен-3-он
[А-6 (R1=4-(N-пиперидино)-, R7=- (СН2)3ОН, R6=R12=Н)].
3,3-[1,2-этандиилбис(окси)]-5α ,10α -оксидоэстр-9(11)-ен-17-он. К раствору 32,0 г (102 ммоль) 3,3-[1,2-этандиилбис(окси)]эстра-5(10),9(11)-диен-17-она в 192 мл СН2Сl2 при 0° С добавляли 7,04 мл (50,9 ммоль) тригидрата гексафторацетона (Lancaster Synthesis, Inc.), после чего 2,46 г (17,3 ммоль) Na2HPO4 и затем по каплям добавляли 8,64 мл (153 ммоль) 50% Н2O2 к эффективно перемешиваемой смеси (механическое перемешивание сверху). Эффективное перемешивание продолжали в течение 18 час, за это время позволяли температуре постепенно подняться до комнатной температуры, затем добавляли 192 мл насыщенного водного раствора Nа2S2O3. После 20 мин перемешивания смесь объединяли с другой партией (32,0 г), которую получали идентично до этого момента в параллели. Водный слой (нижний) отделяли и три раза экстрагировали 80 мл EtOAc. Объединенные органические растворы разбавляли 240 мл EtOAc и дважды промывали 80 мл насыщенного водного раствора NaHCO3, дважды 80 мл раствора соли, сушили над МgSO4, фильтровали и растворитель удаляли при пониженном давлении. Желтое твердое вещество (76,1 г) растирали с 320 мл диэтилового эфира с помощью магнитной мешалки в течение 12 час в закрытом сосуде. Полученную в результате белую суспензию объединяли с тремя другими партиями (3× 32,0 г), которые были приготовлены в параллели идентично (и пропорционально) до этого момента, затем фильтровали отсасыванием через крупнопористую полученную спеканием стеклянную воронку, три раза промывая 40 мл диэтилового эфира, затем отсасывали досуха в течение 1,5 час. Полученный в результате белый отжатый на фильтре осадок осторожно соскребали в виде тонко дисперсного порошка и сушили в вакууме, получая желаемый эпоксид высокой чистоты (89,5 г, выход 53%). 1H ЯМР (250 МГц, СDСl3) δ 6,06 (1Н, уш. с), 3,98-3,88 (4Н, м), 2,52-2,44 (2Н, м), 1,32-1,12 (1Н, м), 0,88 (3Н, с).
1-(4-Бромфенил)пиперидин. К раствору 320 г (1,86 ммоль, 1,00 экв.) 4-броманилина в 1,20 л толуола при комнатной температуре в 5 л круглодонном сосуде, снабженном верхней механической мешалкой, добавляли 648 мл (3,72 ммоль, 2,00 экв.) диизопропилэтиламина, затем 253 мл (1,86 ммоль, 1,00 экв) 1,5-дибромпентана, после чего промывали 200 мл толуола. Эффективно перемешиваемый раствор, накрытый согревающим кожухом, нагревали до 100-115° С, как было определено по термометру, погруженному в реакционный раствор. Через 10 час наблюдалась высокая степень превращения в целевой продукт по данным ТСХ анализа. Полученной в результате объемной коричневой смеси, содержащей осадок, давали остыть до комнатной температуры. Смесь разминали шпателем до суспензии, которую можно было перенести, с помощью 390 мл толуола. Твердые частицы гидрохлорида диизопропилэтиламина удаляли вакуумным фильтрованием через крупнопористую фриттованную воронку с последующим промыванием твердых частиц толуолом (3 х 320 мл). (Дальнейшие промывки толуолом полученных в результате коричневых твердых частиц давали только незначительное количество материала). Роторное испарение коричневого фильтрата при пониженном давлении с последующим дополнительным удалением растворителя в вакууме в течение 12 час при комнатной температуре давало мягкое коричневое сухое вещество (398 г, грубый выход 89%), которое дробили на маленькие кусочки. 20,0 г этого неочищенного материала отбирали для экспериментального подбора оптимальных условий, результатом которого является следующий протокол очистки.
К остатку неочищенного продукта (378 г) пятью порциями по 300 мл добавляли 1,50 л диэтилового эфира при эффективном перемешивании магнитной мешалкой. Через 30 мин при комнатной температуре перемешивание прекращали и вынимали стержень мешалки. Темному нерастворимому тонкодисперсному твердому веществу давали возможность осесть и коричневый раствор осторожно декантировали, промывая коричневые твердые частицы диэтиловым эфиром (3× 100 мл). К объединенным коричневым эфирным растворам при комнатной температуре добавляли 38,5 мл (406 ммоль) уксусного ангидрида. После перемешивания при комнатной температуре в течение 3 час добавляли 300 мл 10%-ного водного раствора соляной кислоты (т.е., 3,7% НСl(водн)) при комнатной температуре и смесь энергично перемешивали в течение 5 мин, за это время образовывалось небольшое количество желтого осадка. Эфирный слой отделяли и экстрагировали водной 10%-ной соляной кислотой (5× 300 мл). Объединенные кислые водные растворы декантировали, чтобы удалить небольшое количество желтого твердого вещества и затем один раз повторно экстрагировали 150 мл диэтилового эфира. Водный раствор подщелачивали до рН 10 при энергичном перемешивании при комнатной температуре путем медленного добавления 235 мл концентрированной гидроокиси аммония в течение 20 мин. К полученной в результате смеси, содержащей желтый/белый осадок, при быстром перемешивании добавляли 600 мл диэтилового эфира, таким образом, полностью растворяя твердые частицы примерно через 10 мин. Водный слой отделяли и экстрагировали диэтиловым эфиром (2× 150 мл). Объединенные эфирные растворы промывали раствором соли (2× 150 мл), сушили над Nа2SO4, фильтровали и растворитель удаляли роторным испарением при пониженном давлении. Дальнейшее удаление растворителя в вакууме в течение 12 часов давало высоко очищенный продукт в виде не совсем белого твердого вещества [338 г, выход 80% (с поправкой на изъятие 20,0 г)]. 1H ЯМР (250 МГц, СDСl3) δ 7,29 (2Н, д, J=9,1 Гц), 6,76 (2Н, д, J=9,1 Гц), 3,11-3,07 (4Н, м), 1,73-1,66 (4Н, м), 1,59-1,51 (2Н, м).
3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пипери-дино)фенил]эстр-9-ен-17-он. Круглодонную колбу объемом 3 л, оборудованную верхней механической мешалкой и наполненную 9,67 г (398 ммоль) магниевой стружкой, сушили пламенем в потоке сухого азота. После охлаждения до комнатной температуры, добавляли 333 мл ТГФ, и затем несколько кристаллов йода, придавая, таким образом, светло-коричневую окраску. К эффективно перемешиваемой смеси добавляли 40 мл раствора 91,9 г (383 ммоль) 1-(4-бромфенил)пиперидина в 333 мл ТГФ. После нагревания смеси до температуры кипения с обратным холодильником в течение примерно 5 мин йодная окраска быстро исчезала до обесцвечивания, в это время смеси давали остыть примерно до комнатной температуры. В течение 1,5 час по каплям добавляли остаток раствора бромида. Затем смесь охлаждали в бане с ледяной водой в течение 1,8 час, затем одной порцией добавляли 15,1 г (153 ммоль) тонкодисперсного порошкообразного CuCl. После эффективного перемешивания смеси в течение 60 сек добавляли (вливали) за 30 сек раствор 50,6 г (153 ммоль) 3,3-[1,2-этандиилбис(окси)]-5α ,10α -оксидоэстр-9(11)-ен-17-она в 380 мл ТГФ, что вызывало образование объемного светло-желтого осадка. Спустя 10 мин медленно добавляли 250 мл насыщенного водного раствора NH4Cl, затем 630 мл EtOAc. После перемешивания в течение 30 мин смесь разбавляли 300 мл воды и перемешивали. Водный слой отделяли и три раза экстрагировали 250 мл EtOAc. Объединенные органические растворы три раза промывали 250 мл раствора соли, сушили над MgSO4, фильтровали и растворитель удаляли при пониженном давлении. Полученный в результате материал (116 г) объединяли с аналогично приготовленной партией неочищенного продукта (11,3 г) в 55 мл CH2Cl2, затем хроматографировали на силикагеле (элюирование побочного продукта пиперидинофенильного реагента с помощью CH2Cl2, затем элюирование продукта 60% EtOAc в гексанах), получая целевой продукт, в котором некоторые фракции содержали незначительное количество бис-аддукта, в котором произошло присоединение реагента Гриньяра к 17-карбонильной группе. Таким образом, еще одна хроматографическая процедура концентрированных, содержащих примеси фракций на силикагеле (60% EtOAc в гексанах) и объединение полученных в результате чистых фракций с чистыми фракциями, полученными при первом хроматографическом разделении, давали продукт, свободный от примесей (65,2 г, скорректированный выход 79%). 1H ЯМР (250 МГц, СDСl3) δ 7,07 (2Н, д, J=8,6 Гц), 6,83 (2Н, д, J=8,7 Гц), 4,53 (1Н, с), 4,30 (1Н, д, J=6,8 Гц), 4,02-3,92 (4Н, м), 3,13-3,08 (4Н, м), 0,50 (3Н, с).
3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пипери-дино)фенил]эстр-9-ен-17-оксим.
К раствору 65,1 г (132 ммоль) 3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пиперидино)фенил]эстр-9-ен-17-она в 450 мл водного пиридина при комнатной температуре в атмосфере азота добавляли 15,2 г (218 ммоль) гидрохлорида гидроксиламина. После перемешивания в течение 19,5 час добавляли 1,5 л воды и 475 мл EtOAc. После 10 мин перемешивания водный слой отделяли и три раза экстрагировали 275 мл EtOAc. Объединенные органические растворы дважды промывали 275 мл раствора соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Три раза повторяли упаривание полученной в результате пены в роторном испарителе при пониженном давлении с 275 мл толуола при 40° С, за это время присоединяли 4,93 г аналогично полученного материала. Затем растворитель удаляли в вакууме, получая, таким образом, целевой оксим в виде желтой пены (81,7 г), свободный от пиридина, судя по 1H ЯМР анализу. Материал использовали далее без дальнейшей очистки. 1H ЯМР (250 МГц, СDСl3) δ 8,37 (1Н, уш. с), 7,08 (2Н, д, J=9,0 Гц), 6,81 (2Н, д, J=8,6 Гц), 4,37 (1Н, с), 4,22 (1Н, д, J=6,6 Гц), 4,08-3,89 (4Н, м), 3,12-3,08 (4Н, м), 0,54 (3Н, с).
3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пиперидино-N-оксид)фенил]эстр-9-ен-17-оксим. К раствору 81,7 г (предположительно 148 ммоль) 3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пиперидино)фенил]эстр-9-ен-17-оксима в 290 мл СН2Сl2 при 0° С добавляли 10,3 мл (73,7 ммоль) тригидрата гексафторацетона. При энергичном перемешивании по каплям добавляли 17,8 мл (310 ммоль) 50% Н2O2. Смесь энергично перемешивали в течение 14,5 час, за это время смесь постепенно нагревалась до комнатной температуры. Добавляли воду (414 мл) и EtOAc (1,65 л) и полученную в результате смесь хорошо перемешивали в течение 20 мин. Органический слой отделяли и пять раз экстрагировали 125 мл воды. Объединенные водные растворы использовали на следующем этапе без дополнительной обработки. Небольшое количество может быть сконцентрировано в вакууме для определения характеристик: 1H ЯМР (250 МГц, CDCl3) δ 10,7 (1H, уш. с), 7,96 (2Н, д, J=8,6 Гц), 7,36 (2Н, д, J=8,6 Гц), 4,39-4,32 (1H, м), 4,05-3,97 (4Н, м), 3,68-3,56 (2Н, м), 3,44-3,39 (2Н, м), 0,49 (3Н, с).
17-Бром-3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17-нитро-11β -[4-(N-пиперидино)-фенил]эстр-9-ен [А-1 (R1=4-(N-пиперидино)-, R7=Вr, R6=R12=Н)]. К раствору 65,9 г (369 ммоль) N-бромсукцинимида (N-БСИ) в 375 мл 1,4-диоксана при комнатной температуре добавляли раствор 37,0 г (369 ммоль) КНСО3 в 375 мл воды. Через 5 мин указанный выше водный раствор 3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-11β -[4-(N-пиперидино-N-оксид)-фенил]эстр-9-ен-17-оксима при комнатной температуре разбавляли 720 мл 1,4-диоксана, затем медленно добавляли к раствору N-БСИ-КНСО3, с такой скоростью, которая позволяла избежать чрезмерное выделение газа и пенообразование. После 16 час перемешивания раствора при комнатной температуре добавляли 262 г (944 ммоль) FeSO4·7H2O, что вызывало образование объемного коричневого осадка, затем добавляли 375 мл EtOAc и 650 мл воды. После 30 мин эффективного перемешивания водный слой отделяли и четыре раза экстрагировали 300 мл EtOAc. Объединенные органические растворы дважды промывали 300 мл раствора соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая А-1 (R1=4-(N-пиперидино)-, R7=Br, R6=R12=H) в виде коричневой пены (69,3 г), которая была непосредственно использована на следующем этапе без очистки. 1H ЯМР (250 МГц, СDСl3) δ 7,07 (2Н, д, J=7,5 Гц), 6,87 (2Н, м), 4,42 (1Н, с), 4,32 (1Н, д, J=6,0 Гц), 4,03-3,93 (4Н, м), 3,43-3,28 (1Н, м), 3,12 (4Н, уш. с), 0,48 (3Н, с).
3,3-[1,2-этандиилбис (окси)]-5α -гидрокси-17β -нитро-11β -[4-(N-пиперидино)фенил]эстр-9-ен [А-1 (R1=4-(N-пиперидино)-, R6=R7=R12=H)]. К хорошо перемешиваемому раствору 69,3 г указанного выше неочищенного бромида [А-1 (R1=4-(N-пиперидино)-, R7=Br, R6=R12=H) ] в 1,17 л ТГФ и 230 мл воды при комнатной температуре небольшими порциями добавляли 14,4 г (380 ммоль) NaBH4 в течение 1 час периода. Еще через 1 час осторожно добавляли 84,3 г (1,21 моль) гидрохлорида гидроксиламина в 585 мл воды. Через 15 мин водный слой отделяли и три раза экстрагировали 116 мл EtOAc. Объединенные органические растворы три раза промывали 116 мл раствора соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая желтую пену (56,2 г). Материал брали в минимальном количестве СН3Сl2 и хроматографировали на силикагеле (55% EtOAc в гексанах), получая промежуточное нитросоединение А-1 (R1=4-(N-пиперидино)-, R6=R7=R12=H) (33,3 г, выход после 5 этапов 45%) в виде желтого твердого вещества. 1H ЯМР (250 МГц, CDCl3) δ 7,04 (2Н, д, J=8,6 Гц), 6,81 (2Н, д, J=8,8 Гц), 4,38 (1Н, с), 4,33 (1Н, т, J=11 Гц), 4,23 (1Н, д, J=6, 6 Гц), 3,12-3,07 (4Н, м), 2,71 (1Н, д, J=13 Гц), 0,36 (3Н, с).
17α -(2-карбометоксиэтил)-3,3-[1,2-этандиилбис(окси)]-5α - гидрокси-17β -нитро-11β -[4-(N-пиперидино)фенил]эстр-9-ен [А-1 (R1=4-(N-пиперидино)-, R7=-(СН3)2СООСН3, R6=R12=Н)]. К смеси 12,0 г (23,0 ммоль) промежуточного нитросоединения А-1 (R1=4-(N-пиперидино)-, R6=R7=R12=Н)] в 65 мл трет-ВUОН при комнатной температуре добавляли 41,2 мл (460 ммоль) метилакрилата, после чего по каплям добавляли 13,2 мл (30,0 ммоль) 40% вес/вес Тритона В в МеОН. Через 1 час при комнатной температуре, добавляли 132 мл насыщенного водного раствора NH4Cl и 132 мл EtOAc. Водный слой отделяли и три раза экстрагировали 30 мл EtOAc. Объединенные органические растворы дважды промывали 132 мл раствора соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Хроматография на силикагеле (60% EtOAc в гексанах) давала сложный нитроэфир А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)2СООСН3, R6=R12=Н) 11,8 г, (выход 85%). 1H ЯМР (250 МГц, CDCl3) δ 7,04 (2Н, д, J=8,6 Гц), 6,81 (2Н, д, J=8,7 Гц), 4,35 (1Н, с), 4,30 (1Н, д, J=6,1 Гц), 4,03-3,93 (4Н, м), 3,68 (3Н, с), 3,11-3,07 (4Н, м), 0,38 (3Н, с).
3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17α -(3-гидрокси-пропил)-17β -нитро-11β -[4-(N-пиперидино)фенил]эстр-9-ен [А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)]. К раствору 8,13 г (13,3 ммоль) сложного эфира А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)2СООСН3, R6=R12=Н)] в 145 мл ТГФ при 0° С добавляли 67,0 мл (67,0 ммоль) 1,0 М DIBAL-H в гексанах. Через 15 мин добавляли 55 мл насыщенного водного раствора смешанного виннокислого калия-натрия, вызывая, таким образом, формирование геля. После перемешивания смеси при комнатной температуре в течение 2 час гель рассеивался, давая прозрачную смесь. Водный слой отделяли и три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая нитропропанол А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н) в виде желтой пены (7,83 г, выход 100%). 1H ЯМР (250 МГц, CDCl3) δ 7,05 (2Н, д, J=8,6 Гц), 6,81 (2Н, д, J=8,7 Гц), 4,38 (1Н, с), 4,29 (1Н, д, J=6,4 Гц), 4,02-3,93 (4Н, м), 3,68-3,50 (2Н, м), 3,11-3,07 (4Н, м), 2,90-2,75 (1Н, м), 0,37 (3Н, с).
17β -Амино-3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстр-9-ен [А-4 (R1 - 4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)]. К раствору 7,25 г (12,5 ммоль) нитропропанола А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)] и 13,4 г (250 ммоль) NH4Cl в 70 мл EtOH, 70 мл воды и 140 мл ТГФ при 70° С осторожно добавляли 49,0 г (749 ммоль) цинковой пыли (-325 меш) примерно в течение 3 мин. После этого полученную в результате смесь эффективно перемешивали в течение 18 час, позволяли ей остыть до комнатной температуры и затем фильтровали через подушку целита с помощью EtOAc. Фильтрат дважды промывали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая аминопропанол А-4 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н) в виде белого аморфного твердого вещества (9,16 г), которое использовали для следующего этапа без очистки. 1H ЯМР (250 МГц, CDCl3) δ 7,15 (2Н, д, J=8,2 Гц), 6,85 (2Н, д, J=8,3 Гц), 4,40-4,20 (2Н, м), 4,05-3,82 (4Н, м), 3,46 (2Н, уш. с), 3,10 (4Н, уш. с), 0,54 (3Н, с).
17β -Амино-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстра-4,9-диен-3-он [А-6 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)]. К энергично перемешиваемой молочно-белой смеси 9,45 г (предположительно 13 ммоль) кеталя А-4; (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)], 575 мл СН2Сl2 и 12 мл воды при 0° С по каплям добавляли 16,6 мл (215 ммоль) трифторуксусной кислоты, которая постепенно вызывала бледно-голубое окрашивание, которое постепенно переходило в бледно-желтое в течение примерно 15 мин. После энергичного перемешивания в течение 2 час осторожно добавляли насыщенный водный раствор NaHCO3 и смесь осторожно перемешивали при комнатной температуре в течение 14 час. Смесь три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали раствором соли, сушили над Nа2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая диенон А-6 (R1=4-(N-пиперидино)-, R7=-(CH2)3ОН, R6=R12=Н) в виде желтой пены (6,18 г, скорректированный выход после 3 этапов 74%), в высокоочищенном состоянии по данным 1H ЯМР анализа. 1H ЯМР (250 МГц, СDСl3) δ 7,04 (2Н, д, J=8,6 Гц), 6,85 (2Н, д, J=8,7 Гц), 5,76 (1Н, с), 4,36 (1Н, д, J=6, 1 Гц), 3,62-3,42 (2Н, м), 3,13-3,09 (4Н, м), 0,54 (3Н, с). Масс спектр m/z (относительная интенсивность) 488 (М+, 21), 470 (37), 387 (100), 320 (25), 162 (52), 96 (35).
Пример 7
Синтез 17β -гидроксиламино-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстра-4,9-диен-3-он [А-3 (R1=4-(N-пиперидино)-, R7=-(CH2)3зОН, R6=R12=H)].
3,3-[1,2-этандиилбис(окси)]-5α -гидрокси-17β -гидроксиламино-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстр-9-ен [А-2 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=H)]. К раствору 4,34 г (7,47 ммоль) нитропропанола А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=H)] и 840 мг (15,7 ммоль) NH4C1 в 49 мл EtOH, 49 мл воды и 99 мл ТГФ при комнатной температуре добавляли 4,40 г (67,2 ммоль) цинковой пыли (-325 меш). После эффективного перемешивания в течение 2,5 час смесь фильтровали через подушку целита с помощью EtOAc. Фильтрат дважды промывали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении. Хроматография на силикагеле (6% МеОН в EtOAc) давала исходный нитропанол А-1 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=H) (420 мг, 10% получено обратно) и требуемый гидроксила-минопропанол А-2 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=H) (2,95 г, выход 70%). 1H ЯМР (250 МГц, СDСl3) δ 7,07 (2Н, д, J=8,5 Гц), 6,82 (2Н, д, J=8,7 Гц), 5,29 (1Н, уш. с), 4,35 (1Н, с), 4,18-4,16 (1Н, уш. с), 4,01-3,92 (4Н, м), 3,75-3,65 (1Н, м), 3,60-3,51 (1H, м), 3,10-3,05 (4Н, м), 1,04-0,93 (1H, м), 0,57 (3Н, с).
17β -Гидроксиламино-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстра-4,9-диен-3-он [А-3 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)]. К энергично перемешиваемой смеси 2,95 г (5,20 ммоль) гидроксиламина А-2 (R1= 4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н)], 4,80 мл воды и 230 мл CH2Cl2 при 0° С по каплям добавляли 6,50 мл (84,2 ммоль) трифторуксусной кислоты. После энергичного перемешивания при 0° С в течение 3 час осторожно добавляли избыток насыщенного раствора NaHCO3. После перемешивания при комнатной температуре в течение 30 мин смесь три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали раствором соли, сушили над Na2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая желтую пену (2,77 г). Материал объединяли с аналогично полученным материалом (205 мг) в минимальном количестве СН2Сl2 и хроматографировали на силикагеле (5,5% МеОН в EtOAc), получая желтую пену (1,99 г). Ее объединяли с подобным дополнительным материалом (393 мг) в минимальном количестве CH2Cl2 и хроматографировали на силикагеле (5,0% МеОН в EtOAc). Объединение фракций, имеющих чистоту >97%, на основании определения с помощью ВЭЖХ анализа, давало диенон А-3 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н) (1,69 г, скорректированный выход 52%). Большая часть этого материала (1,68 г) была высушена при температуре от 92 до 94° С в вакууме в течение 38 час, давая продукт, который по данным 1H ЯМР анализа не содержал растворителя, и имел чистоту >97% по данным ВЭЖХ анализа. 1H ЯМР (250 МГц, СDСl3) δ 7,03 (2Н, д, J=8,6 Гц), 6,84 (2Н, д, J-8,7 Гц), 5,75 (1H, с), 5,21 (1Н, уш. с, обмениваемый с D2O), 4,28 (1Н, д, J=6,0 Гц), 3,78-3,74 (1Н, м), 3,64-3,61 (1H, м), 3,10-3,02 (4Н, м), 1,08-0,93 (1H, м), 0,64 (3Н, с). Масс спектр m/z: LC-MS 505 (M+, 1); MS-MS 505 (М+ 1). Аналитически вычислено для C32H44N2O3: С, 76,15; Н, 8,79; N, 5,55. Найдено: С, 75,90; Н, 8,77; N, 5,50.
Пример 8
17β -(N-Формамидо)-17α -[3-(формилокси)пропил]-11β -[4-(N-пиперидино)фенил]эстра-4,9-диен-3-он
[А-8 (Rl=4-(N-пипepидинo)-, R7=-(CH2)3OCHO, R10=R6=R12=Н)].
К раствору 10,4 г (50,6 ммоль) дициклогексилкарбодиимида в 28 мл СНСl3 при комнатной температуре добавляли 101 мл (101 ммоль) 1,00 М муравьиной кислоты в СНСl3, что вызывало появление белого осадка. Через 5 мин смесь добавляли к раствору 6,18 г (12,7 ммоль) аминоспирта А-6 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R6=R12=Н) и 12,4 мл (152 ммоль) пиридина в 55 мл СНСl3. Через 15 мин добавляли 4-диметиламинопиридин на кончике шпателя. Через 2,5 час еще раз добавляли 4-диметиламинопиридин на кончике шпателя. Дополнительное количество смеси 2,60 г (12,7 ммоль) дициклогексилкарбодиимида и 25,3 мл (25,3 ммоль) 1,00 М муравьиной кислоты в СНСl3 в 10 мл СНСl3 перемешивали в течение 5 мин, затем добавляли к реакционной смеси. Спустя 1 час реакционную смесь разбавляли 600 мл диэтилового эфира и эффективно перемешивали в течение 12 час, затем фильтровали через подушку целита с помощью промывок диэтиловым эфиром. Фильтрат концентрировали в роторном испарителе при пониженном давлении, затем в вакууме. Остаток перемешивали со 100 мл EtOAc в течение 45 мин, затем полученное в результате твердое вещество отфильтровывали, используя подушку целита с помощью промывок EtOAc. Фильтрат объединяли с меньшей партией требуемого грубого продукта (0,688 ммоль, теоретически), которая была получена сходным образом вплоть до этого момента. Растворитель удаляли при пониженном давлении. Три раза повторяли разбавление остатка 30 мл толуола и концентрировали при пониженном давлении, с помощью роторного испарителя (чтобы удалить пиридин), получая 8,42 г оранжевой/желтой пены. Материал дважды хроматографировали на силикагеле (85% EtOAc в гексанах), получая 4,66 г желтого аморфного твердого вещества. Образец этого материала массой 1,63 г сушили в вакууме в течение 21 час при 95° С, получая желтое аморфное твердое вещество (1,51 г, скорректированный выход 51%). Обнаружено, что группа сложного эфира муравьиной кислоты подвергалась медленному отщеплению в растворе МеОН. Анализ с помощью аналитической обращенно-фазной ВЭЖХ (колонка С-18, YMC, inc.) показал, что чистота требуемого продукта, существующего в двух равновесных формах, составляла >97%. Подтверждение такого взаимопревращения было получено путем разделения индивидуальных форм аналитической ВЭЖХ с последующим повторным введением этих форм, чтобы получить фактически идентичные хроматограммы. Такой же характер превращения наблюдался при экспериментах с применением двумерной ТСХ, а также на спектрах 1H ЯМР (соотношение примерно 2:1). 1H ЯМР (250 МГц, CDCl3 интегрирование регулировали для определения соотношения главной и минорной форм) δ 8,03 (1Н, с), 7,03 (2Н, д, J=8,5 Гц), 6,83 (2Н, д, J=8,6 Гц), 5,75 (1Н, с), 5,32 (1Н, - с), 4,44-4,30 (1Н, м), 4,25-4,15 (2Н, м), 3,10 (4Н, уш. с), 0,50(3 Н, с); минорная форма: 8,14 (1Н, д, J=12,4 Гц), 6,99 (2Н, перекрывающийся дублет), 5,95 (1Н, д, J=12,4 Гц). Аналитически вычислено для C34H44N2O4: С, 74,97; Н, 8,14; N, 5,14. Найдено: С, 74,72; Н, 8,26; N, 5,07. Масс-спектр m/z (относительная интенсивность) 544 (М+, 25), 320 (23), 161 (100).
Пример 9
17β -(N-Формамидо)-17α -(3-гидроксипропил)-11β -[4-(N-пиперидино)фенил]эстра-4,9-диен-3-он
[А-8 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R10=R6=R12=H)].
К смеси 1,43 г (2,62 ммоль) сложного эфира муравьиной кислоты А-8 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОСНО, R10=R6=R12=Н)] и 24 мл МеОН при комнатной температуре и хорошем перемешивании по каплям добавляли 0,48 мл концентрированной гидроокиси аммония, что вызывало постепенное образование гомогенного раствора. Через 1,2 час добавляли 24 мл насыщенного водного раствора хлорида аммония, 24 мл воды и 24 мл EtOAc. Водный слой отделяли и три раза экстрагировали EtOAc. Объединенные органические растворы дважды промывали раствором соли, сушили над Nа2SO4, фильтровали и растворитель удаляли при пониженном давлении, получая 1,35 г (выход 100%) продукта. Материал объединяли с 474 мг аналогично полученного неочищенного продукта в минимальном количестве CH2Cl2 и хроматографировали на силикагеле (8% МеОН в EtOAc), получая желтую пену (1,59 г). Три раза повторяли разведение материала 15 мл CH2Cl2 и концентрировали при пониженном давлении в роторном испарителе, получая материал свободный от всех других растворителей, судя по данным 1Н ЯМР анализа. Материал сушили при 95° С в течение 23,5 час в вакууме, получая 1,36 г в высокой степени очищенного от растворителей целевого формамидопропанола А-8 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОН, R10=R6=R12=Н) [1,45 г, выход 79% (с поправкой на добавку)] с чистотой >97% по данным ВЭЖХ анализа. Равновесные формы, аналогичные указанному выше формамиду А-8 (R1=4-(N-пиперидино)-, R7=-(СН2)3ОСНО, R10=R6=R12=Н) сходным образом наблюдали с помощью аналитической ВЭЖХ и lH ЯМР (соотношение примерно 1:1). 1H ЯМР (250 МГц, СDСl3, интегрирование регулировали для определения соотношения форм), δ 8,12 (1Н, д, J=12,3 Гц), 7,03 (2Н, д, J=8,5 Гц), 6,83 (2Н, д, J=8,6 Гц), 6,39 (1Н, д, J=12,9 Гц), 5,75 (1Н, с), 4,40-4,31 (1Н, м), 3,65-3,62 (2Н, м), 3,10-3,08 (4Н, м), 0,50 (3Н, с); другая форма, не полная: 8,01 (1Н, с), 7,00 (2Н, д, J=8,6 Гц), 5,39 (1Н, с), 0,49 (3Н, с). Аналитически вычислено для C33H44N2O3·0,25Н2O: С, 76,04; Н, 8,61; N, 5,37. Найдено: С, 75,95; Н, 8,62; N, 5,38. Масс спектр m/z (относительная интенсивность): 516 (М+, 44), 387 (16), 320 (38), 161 (100).
Пример 10
Синтез 5'-оксо-11β -[4-(N-пиперидино)фенил]-спиро[эстра-4,9-диeн-17β ,2'-пиppoлидинo]-3-он [B-3 (R1=4-(N-пиперидино)-, R6=R12=Н)].
3,3-[1,2-Этандиилбис(окси)]-5α -гидрокси-5'-оксо-11β -[4-(N-пиперидино)фенил]-спиро[эстр-9-ен-17(5,2'-пирролидин] [В-2 (R1=4-(N-пиперидино)-, R6=R12=Н)]. Получали раствор сложного нитроэфира В-1 (R1=4-(N-пиперидино)-, R6=R12=Н, R=СН3') (3,13 г, 5,10 ммоль) в 63 мл 50% водного раствора этанола и 32 мл ТГФ. К этому раствору добавляли 1,93 г (73,4 ммоль) хлорида аммония и 20 г (306 ммоль) цинковой пыли. Реакционную смесь нагревали до 70° С в течение 20 час. Реакционную смесь фильтровали сквозь целит и фильтрат концентрировали. Грубый продукт хроматографировали на силикагеле, элюируя этилацетатом-гексаном-метанолом 6:3:1, получая 1,97 г (выход 70%) чистого В-2 (R1H-(N-пиперидино)-, R6=R12=H): ИК-спектр-(раствор, CDCl3) 3495, 2995, 2855, 1685, 1506, 1438, 1384, 1226 см-1; 1H ЯМР (250 МГц, CDCl3), δ 7,03 (д, 2, J=8,5 Гц, ArH), 6,83 (д, 2, J=8,8 Гц, ArH), 5,57 (с, 1, NH), 4,39 (с, 1, С5 ОН), 4,25 (д, 1, J=5,9 Гц, С11α H), 3,98-4,02 (м, 4, (ОСН2)2), 3,09 (м, 4, N(CH2)2), 0,42 (с, 3, C18H).
5'-оксо-11β -[4-(N-пиперидино)фенил]-спиро[эстра-4,9-диен-17β ,2' -пирролидин]-3-он [B-3 (R1=4-(N-пиперидино)-, R6=R12=H)]. К раствору 120 мг (0,23 ммоль) В-2 (R1 =4-(N-пиперидино)-, R6=R12=Н)] в 2,5 мл CH2Cl2 добавляли 0,1 мл воды и смесь охлаждали до 0° С. К раствору по каплям добавляли 0,5 мл трифторуксусной кислоты (ТФУ). Реакционную смесь перемешивали при 0° С в течение 1 час. Реакцию гасили насыщенным раствором бикарбоната натрия и экстрагировали СН2Сl2. Слой CH2Cl2 промывали водой, после чего раствором соли и сушили над безводным MgSO4. Высушенный раствор затем фильтровали и концентрировали в вакууме. Грубый продукт хроматографировали на силикагеле, используя в качестве элюента хлористый метилен-гексан-метанол 5:5:1, получая 79 мг (выход 76%) чистого В-3 (R1=4-(N-пиперидино)-, R6=R12=Н). 1H ЯМР (250 МГц, СDСl3) δ 6,99 (д, 2, J=8,7 Гц, ArH), 6,64 (д, 2, J=8,8 Гц, ArH), 5,79 (с, 1, NH), 5,77 (с, 1, С4Н), 4,36 (д, 1, J=6,4 Гц, С11α Н), 3,12 (м, 4, N(CH2)2), 0,49 (с, 3, C18H). Масс спектр m/z: (относительная интенсивность) 484 (75), 374 (5), 320 (16), 213 (6), 174 (17), 161 (100); Аналитически вычислено для C32H40N2O2: С, 79,30; Н, 8,32; N, 5,78. Найдено: С, 79,12; Н, 8,26; N, 5,72.
Пример 11
Синтез 11β -[4-(N,N-диметиламино)фенил]-1'-гидрокси-5'-метилспиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он [С-3 (R1=4-(Me2N-, R6=R12=Н, R13=СН3)].
1',5α -Дигидрокси-11β -[4-(N,N-диметиламино)фенил]-3,3-[1,2-этандиилбис(окси)]-5'-метил-спиро[эстр-9-ен-17β ,2'-пирролидин] [С-2 R1=4-Me2N-)-, R6=R12=Н, R13 =СН3]. К перемешиваемому раствору С-1 (R1=4-Me2N-, R6=R12=Н, R13=СН3) (200 мг, 0,38 ммоль) и(3,58 ммоль) NaBH3CN в 4 мл метанола добавляли 0,5 мл АсОН. Реакционную смесь перемешивали при комнатной температуре в течение 2 час. Реакцию гасили насыщенным раствором NH4Cl и экстрагировали СН2Сl2. Органический слой промывали водой, затем раствором соли и сушили над безводным Na2SO4. Органический слой фильтровали и концентрировали, получая неочищенный С-2 (R1=4-Me2N-, R6=R12=Н, R13=СН3), который использовали для следующего этапа без дополнительной очистки. 1H ЯМР (250 МГц, СDС13), δ 7,06 (д, 2, J=8,7 Гц, ArH), 6,64 (д, 2, J=8,7 Гц, ArH), 4,35 (с, 1, С5 ОН), 4,19 (д, 1, J=6,2 Гц, С11α H), 3,98-4,02 (м, 4, (ОСН2)2), 2,88 (с, 6, N(CH3)2), 1.19 (д, 3, J=6,5 Гц, НОNСНСН3), 0,62 (с, 3, C18H).
11β -[4-(N,N-Диметиламино)фенил]-1'-гидрокси-5'-метилспиро[эстра-4,9-диeн-17β ,2'-пиppoлидин]-3-он [С-3 (R1 =4-Me2N-, R6=R12=Н, R13=СН3)]. К раствору 200 мг (0,38 ммоль) С-2 (R1=4-Me2N-, R6=R12=Н, R13=СН3) в 2,0 мл СН2Сl2 добавляли 0,1 мл воды и смесь охлаждали до 0° С. К охлажденному раствору по каплям добавляли примерно 0,5 мл ТФУ. Реакционную смесь перемешивали при 0° С в течение 1 час. Реакцию гасили насыщенным раствором бикарбоната натрия и экстрагировали СН2Сl2. Слой СН2Сl2 промывали водой, после чего раствором соли и сушили над безводным Na2SO4. Высушенный раствор фильтровали и концентрировали в вакууме. Грубый продукт хроматографировали на силикагеле, используя в качестве элюента 3:1 этилацетат-гексан, получая 114 мг (выход 65%) чистого С-3 (R1=4-Me2N-, R6=R12=Н, R13=СН3). 1H ЯМР (250 МГц, СDСl3) δ 7,02 (д, 2, J=8,6 Гц, ArH), 6,65 (д, 2, J=8,7 Гц, ArH), 5,74 (с, 1, С4Н), 4,30 (д, 1, J=6,8 Гц, C11α H), 2,89 (с, 6, N(СН3)2), 1,19 (д, 3, J=6,4 Гц, НОNСНСН3), 0,69 (с, 3, С18СН3); масс спектр m/z (относительная интенсивность) 459 (8), 458 (23), 442 (31), 280 (12), 134 (100), 121 (33), 96 (7); Аналитически вычислено для C30H40N2O2·0,25H2O: С, 77,45; Н, 8,83; N, 5,61. Найдено: С, 77,46; Н, 8,78; N, 6,02.
Пример 12
Синтез 11β -[4-(N,N-диметиламино)фенил]-1'-гидрокси-спиро-[эстра-4,9-диен-17β ,2'-пирролидин]-3-он [С-3 (R1=4-(Me2N)-, R6=R12=Н, R13=Н)]
1',5α -Дигидрокси-11β -[4-(N,N-диметиламино)фенил]-3,3-[1,2-этандиилбис(окси)]-спиро[эстр-9-ен-17β ,2'-пирролидин] [С-2 (R1=4-Me2N-)-, R6=R12=Н, R13=Н]. К перемешиваемому раствору С-1 (R1=4-Me2N-, R6=R12=H, R13=H) (200 мг, 0,39 ммоль) и 234 мг (3,77 ммоль) NaBH3CN в 5 мл метанола добавляли 0,5 мл АсОН. После перемешивания при комнатной температуре в течение 2 час реакцию гасили насыщенным раствором NH4Cl и экстрагировали СН2Сl2. Органический слой промывали водой, затем раствором соли и сушили над безводным Na2SO4. Органический слой фильтровали и концентрировали, получая неочищенный продукт, который был использован для следующего этапа без дополнительной очистки. 1H ЯМР (250 МГц, СDСl3), δ 7,04 (д, 2, J=8,7 Гц, ArH), 6,64 (д, 2, J=8,8 Гц, ArH), 4,37 (с, 1, С5 ОН), 4,16 (д, 1, J=7,4 Гц, С11α Н), 3,92-4,02 (м, 4, (ОСН3)2), 2,90 (с, 6, N(CH3)2), 0,64 (с, 3, C18H).
11β -[4-(N,N-Диметиламино)фенил]-1'-гидрокси-спиро[эстра-4,9-диен-17β ,2'-пирролидин]-3-он [С-3 (R1=4-Me2N-, R6=R12=H, R13= H) ]. К раствору 200 мг (0,39 ммоль) С-2 (R1=4-Me2N-, R6=R12=Н, R13=Н) в 5,0 мл CH2Cl2 добавляли 0,1 мл воды и смесь охлаждали до 0° С. К охлажденному раствору по каплям добавляли примерно 0,5 мл ТФУ. После перемешивания при 0° С в течение 1 час реакцию гасили насыщенным раствором бикарбоната натрия и экстрагировали CH2Cl2. Слой СН2Сl2 промывали водой, после чего раствором соли и сушили над безводным Na2SO4. Высушенный раствор фильтровали и концентрировали в вакууме. Грубый продукт хроматографировали на силикагеле, используя в качестве элюента этилацетат-гексан 3:1, получая 115 мг (выход 65%) чистого С-3 (R1=4-Ме2N-, R6=R12=H, R13=H). 1H ЯМР (250 МГц, CDCl3) δ 7,00 (д, 2, J=8,7 Гц, ArH), 6,65 (д, 2, J=8,8 Гц, ArH), 5,75 (с, 1, С4Н), 4,29 (д, 1, J=6,7 Гц, С11α Н), 2,91 (с, 6, М(СН3)2), 0,66 (с, 3, C18H); масс спектр m/z (относительная интенсивность) 446 (11), 428 (66), 347 (82), 280 (27), 226 (31), 134 (80), 121 (87), 96 (100), 83 (42); Аналитически вычислено для C29H38N2O2·0,25H2O: С, 77,44; Н, 8,84; N, 5,84. Найдено: С, 77,20; Н, 8,60; N, 6,21.
Биологическая активность соединений данного изобретения была проверена методами тестирования in vitro и in vivo.
Связывание с рецептором:
Сродство соединений с рецептором гормона прогестерона человека определяли стандартными методами, сходными с методами, которые описаны Horwitz, et al.. Cell, 28:633-42 (1982) и Mockus, et al., Endocrinology, 110:1564-71 (1982). Рецептор получали во фракции цитозоля из клеток молочной железы человека T-47D, и в качестве радиолиганда использовали [3H]-R5020. Клетки T47D (1 биллион, мл) гомогенизировали в буфере ТЭДГ (10 мМ трис, 1,5 мМ ЭДТА, 1 мМ дитиотреитол, 1 мМ молибдат натрия и 10% глицерин), используя гомогенизатор с пестиком Даунса А, и гомогенат центрифугировали при 34000 × g в течение 1 час. Надосадочную жидкость хранили при - 80° С. Аликвоту препарата рецептора смешивали с тестируемым соединением, 0,4 нМ [3H]-R5020 и буфером ТЭДГ до конечного объема 150 мкл, и инкубировали в течение 4 час при 4° С в планшетах для микротитрования. В конце инкубации к инкубируемой смеси добавляли 40 мкл 40% полиэтиленгликоля и 15 мкл 1% гамма глобулина человека, и содержимое каждой лунки собирали на фильтровальные вкладыши В двойной толщины (Wallac LKB) с помощью устройства для сбора клеток ТоmТес. На высушенные фильтровальные вкладыши накладывали пленку сцинтилляционного воска Meltilux и вкладыши просчитывали в сцинтилляционном счетчике, определяя ингибирование связывания [3H]-R5020. Данные выражали в виде значений ИК50, т.е. в виде концентрации соединения, которая ингибирует связывание радиолиганда на 50%.
В таблице 1 показано, что соединения данного изобретения сильно связываются с рецептором прогестина, но с различной степенью сродства.
Чтобы далее охарактеризовать биологическую активность соединений изобретения также были выполнены тесты на животных.
Определение гестагенной и антигестагенной активности in vivo:
Гестагенную активность и антигестагенную активность определяли на кроликах с помощью теста МакГинти (только тестируемое соединение само по себе, метод McGinty et al. Endocrinology, 24:829-832 (1939)) или с помощью теста анти-МакГинти (тестируемое соединение плюс прогестерон, метод Tamara et al., Jpn. J. Fertil. Steril. 24:48-81 (1979)). Результаты количественно оценивали согласно методу МакФейла (McPhail, J. Physiol., 83:146 (1934)). Эти способы являются стандартными, хорошо известными специалистам в данной области. Результаты этих исследований показаны в таблицах 2 (агонистическая активность) и 3 (антагонистическая активность). Большинство соединений показали выраженную антигестагенную активность. Некоторые соединения очень эффективны в этом отношении. Например, соединения I (R1=4-Me2N-C6H4, Х=О, R6=R8=R12.= Н, R7=СН3СС, R9=СНО) и I (R1=4-Ме2N-С6Н4, Х=О, R6=R8=R12=Н, R7 =С Н3СС, R9=ОН) были эффективны, полностью блокируй действие прогестерона при дозе всего в 0,3 мкг при исследовании в тесте анти-МакГинти (таблица 3). Однако последнее соединение в высоких дозах также проявляло некоторую гестагенную (агонистическую) активность при исследовании в тесте МакГинти (таблица 2) и в высоких дозах при исследовании в тесте анти-МакГинти. Таким образом, среди соединений данного изобретения может быть обнаружено множество соединений, проявляющих свойства агонистов и антагонистов.
Антиэстрогенная активность:
Некоторые соединения проявляли не конкурентную антиэстрогенную активность такого типа, которая сообщалась для мифепристона, например Wolf et al. (Fertil. Steril. 52:1055-1060 (1989). Неожиданным было то, что они проявляли эту активность, несмотря на тот факт, что они не имеют 17β -гидроксильного заместителя, характерного как для мифепристона, так и для эстрогенов, таких как эстрадиол, но вместо этого имеют 17β -азотные заместители. Таким образом, если недоразвитым самкам кроликов перорально вводили 11β -(4-(N,N-диметиламино)фенил)-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-он в дозе 10 мг/сутки одновременно с 5 мкг эстрадиола в сутки, извлекали и взвешивали матку, вес матки, который возрастал с 216,7± 37,2 (стандартное отклонение) мг без эстрадиола до 1337± 105 мг в присутствии одного эстрадиола, при этом снижался до 716± 96,6 мг.
Пример 13
17β -(N-формамидо)-11β -[4-(N-пиперидино)фенил]-17β -(1-пропинил)эстра-4,9-диен-3-он (R1 = пиперидиновая группа) (соединение 6617-031, или 069528-301-А).
lH-ЯMP-cпeктp этого соединения показал, что в растворе оно существует в двух взаимопревращающихся формах.
1H ЯМР (300 МГц, CDCl3): основная форма: δ 8,52 (д, 1, J=11,7 Гц, NHCHO), 6,98 (д, 1, J=8,7 Гц, АrН), 6,83 (д, 1, J=8,7 Гц, АrН), 5,99 (д, 1, J=11,7 Гц, NHCHO), 5,77 (с, 1, 4-Н), 4,37 (д, 1, J=6,7 Гц, 11-Н), 3,12 (м, 4), 1,90 (с, 3, С+С-Ме), 0,45 (с, 3, 18-Ме); минорная форма: 8,08 (с, 1), 7,04 (плечо, АrН), 5,60(с, 1), 0,47 (плечо, 18-Ме); Масс спектр: m/z (относительная интенсивность)496 (М'1, 43), 320 (25), 174 (27), 161 (100).
Аналитически вычислено для С33Н40N2O2·0,25Н2О: С, 79,08; Н, 8,14; N, 5,59. Найдено: С, 78,97; Н, 8,12; N, 5,56.
Сравнение активности производных с 11-диметиламинофенильной и 11-пиперидинфенильной группой дано а таблице 4.
Условия определения агонистов: Стандартное соединение (прогестерон, подкожно) или испытываемое соединение (перорально) вводили сенсибилированным эстрогеном кроликам. Матки окрашивали и подсчитывали индекс МакФейла (0-4; где 4 соответствует наибольшему гестагенному ответу).
Условия определения антагонистов: Оба соединения стандартное соединение (прогестерон, подкожно) и испытываемое соединение (перорально) вводили сенсибилированным эстрогеном кроликам. Матки окрашивали и подсчитывали индекс МакФейла (0-4; где 4 соответствует наибольшему гестагенному ответу).
Как можно видеть из представленных данных, тестируемые соединения (6617-026 с 11β -диметиламинофенильной группой и 6617-031 с 11β -пиперидинофенильной группой) не проявляют агонистической активности, но оба соединения были способны полностью блокировать агонистическую активность прогестерона.
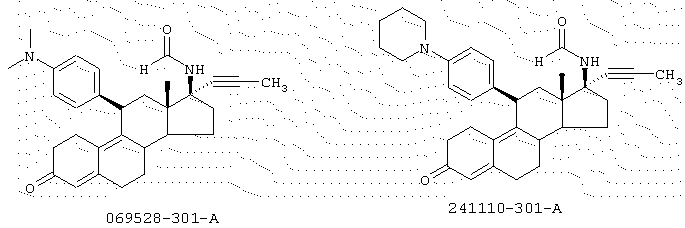
Пример 14
Синтез 11β -[4-(ацетил)фенил]-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-она (7) осуществляли согласно следующей схеме:
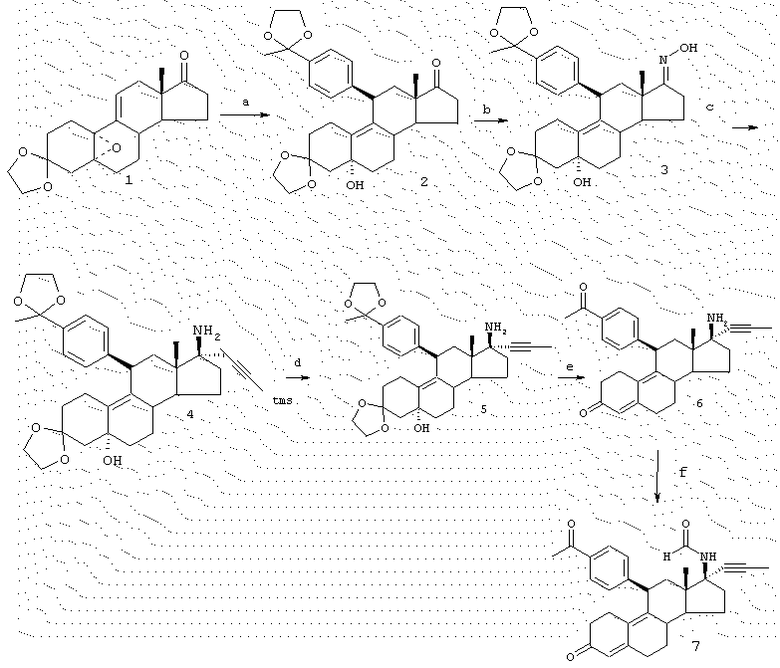
a) этиленгликолькеталь 4-бромацетофенона, Мg, CuI;
b) H2NOH-HCl, пиридин;
c) 1) (PhS)2, Вu3Р, т-BuMgCl; 2) Mg, 3-бром-1-(триметилсилил)-1-пропин;
d) KOt-Bu, DMSO;
e) TFA, H2O, CH2Cl2, 0° C;
f) HCOOH, Aс2O.
3,3-[1,2-Этандиилбис(окси)]-11β -{4-{1,1-[1,2-этандиилбис(окси)] этил}фенил}эстр-9-ен-5α -ол-17-он (2)
Магниевые стружки (2,5 г; 103 ммоль) и небольшой кристалл йода помещали в двугорлую колбу объемом один литр, оборудованную холодильником и капельной воронкой. К перемешиваемому магнию добавляли 10 мл раствора кеталя 4-бромацетофенонэтиленгликоля (24,0 г; 98,8 ммоль) в ТГФ (100 мл). Реакцию инициировали добавлением этиленбромида (100 мкл) и нагреванием смеси феном. Остаток кеталя добавляли в течение 1 час, поддерживая температуру реакции 50° С с помощью масляной бани. Добавляли дополнительное количество ТГФ (30 мл), чтобы растворить не совсем белое твердое вещество, которое образовалось. Реакционную смесь нагревали при 55-60° С еще в течение 2 час и затем охлаждали до -10° С. Йодид меди (I) (7,80 г; 41,0 ммоль) добавляли одной порцией. Реакционную смесь перемешивали в течение 30 сек и затем добавляли эпоксид 1 (12,9 г; 39,0 ммоль), растворенный в ТГФ (120 мл), в течение 1 мин. После перемешивания в течение 30 мин при 0° С реакционную смесь вливали в лед (200 мл), насыщенный раствор NH4Cl (200 мл) и NH4OH (25 мл). Добавляли этилацетат (300 мл) и смесь энергично перемешивали на открытом воздухе в течение 1 час. Слои разделяли и водный слой экстрагировали EtOAc (3Х). Все EtOAc-экстракты объединяли и промывали насыщенным NH4Cl (1X), H2O (1X), насыщенным раствором соли (1X), сушили (Na2SO4) и выпаривали, получая масло цвета густого меда. Масло обрабатывали ультразвуком с изопропанолом (70 мл) в течение 15 мин и оставляли на 1 час. Полученные в результате кристаллы собирали фильтрованием и промывали холодным изопропанолом (3Х).После сушки в условиях глубокого вакуума в течение 18 час получали 9,97 г соединения 2 в виде белых кристаллов. Маточный раствор хранили в холодильнике в течение 10 дней, дополнительно получая 3,5 г соединения 2, общий выход 70%.
1H ЯМР (300 МГц-СDСl3) δ 7,35 (д, 2, J=8,2 Гц, Аr-Н), 7,19 (д, 2, J=8,2 Гц, АrН), 4,38 (с, 1,5-ОН), 4,32 (д, 1, J=7,1, 11-Н), 3,97 (м, 6, кетали), 3,78 (м, 2, кеталь), 1,64 (с, 3, СН3 кеталя ацетофенона), 0,47 (с, 3, 18-СН3).
3,3-[1r2-Этандиилбис(окси)]-11β -{4-{1,1-[1,2-этандиилбис(окси)]этил)фенил}-17-оксиминоэстр-9-ен-5α -ол (3)
Кетон 2 (18,65 г, 37/68 ммоль) растворяли в пиридине (145 мл) при комнатной температуре и добавляли гидрохлорид гидроксиламина (4,2 г). Анализ ТСХ (SiO2:75% EtOAc-гексан) реакционной смеси через 3,5 час показал, что еще присутствует некоторое количество 2. Добавляли еще гидрохлорид гидроксиламина (2,5 г)и реакционную смесь перемешивали в течение 2 час. Смесь вливали в H2O и экстрагировали EtOAc (3Х). Объединенный экстракт промывали Н2O (3Х), насыщенным NaHCO3 (IX) и насыщенным раствором соли (2Х). Раствор сушили (Na2SO4) и выпаривали при пониженном давлении. Остаток растворяли в толуоле (50 мл) и выпаривали (Н2O-баня при 35° С) при пониженном давлении. Обработку толуолом повторяли еще два раза, получая 21,4 г белой пены. Пену растворяли в теплом EtOAc (125 мл) и раствору давали возможность медленно остыть до комнатной температуры. Кристаллы собирали фильтрованием, промывали холодным EtOAc (3Х) и сушили, получая 14,98 г (выход 78%) соединения 3. Маточный раствор концентрировали и остаток перекристаллизовывали из EtOH, дополнительно получая 2 (1,25 г). Получали общий выход соединения 2: 85%.
1H ЯМР (300 МГц-СDСl3) δ 7,72 (с, 1, N-OH), 7,34 (д, 2, J=8,2 Гц, Ar-H), 7,19 (д, 2, J 8,2 Гц, Ar-H), 4,38 (С, 1, 5-ОН), 4,30 (д, 1, J=7,1, 11-Н), 1,64 (с, 3, СН3 кеталя ацетофенона), 0,51 (с, 3,18-СН3).
17β -Амино-3,3-[1,2-этандиилбис(окси)]-11β -{4-{1,1-[1,2-этандиилбис(окси)]этил)фенил}-17α -[1-(триметилсилил)пропин-3-ил]эстра-9-ен-5α -ол (4)
Магниевые стружки (1,14 г; 47 ммоль) помещали в 3-горлую колбу объемом 250 мл и энергично перемешивали в течение 10-15 мин. Добавляли небольшой кристалл йода и 5 мл эфира. Реакционный сосуд охлаждали до 0° С на бане со льдом. 3-Бром-1-(триметилсилил)-1-пропин (2,29 г; 12 ммоль) растворяли в 20 мл эфира и 1 мл добавляли к перемешиваемой Мg-смеси. Инициация не происходила, поэтому добавляли 50 мкл дибромэтана. Спустя десять минут происходила инициация, что было видно по исчезновению йодного окрашивания. Реакционную смесь охлаждали до -20° С, используя баню, содержащую сухой лед-ацетонитрил. Остальной раствор брома добавляли медленно в течение 45 мин. Реакционную смесь перемешивали при -20° С в течение 1 час.
Оксим 3 (512 мг, 1,0 ммоль) растворяли в 7 мл ТГФ перед добавлением дифенилдисульфида (349 мг; 1,6 ммоль) и трибутил-фосфина (0,88 мл; 3,5 ммоль). Полученную смесь перемешивали в течение 2 час при комнатной температуре, охлаждали до 0° С и добавляли трет-бутилмагнийхлорид (2,5 мл; 5,0 ммоль). Смесь перемешивали еще в течение 30 мин и с помощью шприца добавляли описанный выше раствор Гриньяра. После перемешивания в течение ночи при комнатной температуре реакционную смесь вливали в насыщенный раствор хлорида аммония (100 мл) и лед. Добавляли этилацетат (100 мл) и смесь энергично перемешивали в течение 15 мин. Полученную смесь экстрагировали этилацетатом (3х), объединенные органические слои промывали насыщенным раствором соли (2х), сушили сульфатом магния и выпаривали, получая неочищенный продукт. Продукт подвергали хроматографии на SiO2 (50 г), используя ступенчатый градиент 3:1 гексан-этилацетат (400 мл), 2:1 (300 мл), 1:1 (400 мл), 1:2 (300 мл). Фракции, содержащие чистый 4 объединяли и выпаривали, получая 377 мг (выход 62%) продукта.
1H ЯМР (300 МГц-СDСl3) δ 7,34 (д, 2, J=9 Гц, Ar-H), 7,17 (д, 2, J=9 Гц, Ar-H), 4,37 (с, 1,5-ОН), 4,31 (д, 1,9 Гц, 11-Н), 3,98 (м, 6, кетали), 3,78 (с, 2, кеталь), 1,64 (с, 3, СН3 кеталя ацетофенона), 0,43 (с, 3, 18-СН3), 0,17 (с, 9, Si СН3).
17β -Амино-3,3-[1,2-этандиилбис(окси)]-11β -{4-{1,1-[1,2-этандиилбис(окси)]этил}фенил}-17α -(1-пропинил)эстра-9-ен-5α -ол (5)
Соединение 4 (266 мг; 0,439 ммоль) помещали в круглодонную колбу объемом 5 мл и подсоединяли вакуумный насос на 2,5 часа. Колбу убирали, помещали в атмосферу Аr и добавляли трет-бутоксид калия (173 мг; 1,54 ммоль). Смесь растворяли в 2 мл безводного ДМСО, получая в результате оранжево-коричневый раствор. Раствор перемешивали в течение ночи при комнатной температуре, вливали в воду и экстрагировали этилацетатом (3х). Объединенные органические слои промывали водой (1х), насыщенным раствором соли (2х), сушили сульфатом магния и выпаривали, получая 224 мг соединения 5 (выход 96%). Соединение использовали без дальнейшей очистки.
1H ЯМР (300 МГц-СDСl3) δ 7,34 (д, 2, J=9 Гц, Ar-H), 7,19 (д, 2, J=9 Гц, Ar-H), 4,50 (С, 1,5-ОН), 4,34 (д, 1,9 Гц, 11-Н), 3,98 (м, 6, кетали), 3,78 (с, 2, кеталь), 1,84 (с, 3, СН3 пропинила), 1,64 (с, 3, СН3 кеталя ацетофенона), 0,35 (с, 3, 18-СН3). 11β -[4-(Ацетил)фенил]-17β -амино-17α -(1-пропинил)эстра-4,9-диен-3-он (6)
Раствор неочищенного соединения 5 (224 мг; 0,420 ммоль) в метиленхлориде (15 мл) и воде (1,5 мл) охлаждали до 0° С. Добавляли трифторуксусную кислоту (0,425 мл) и смесь перемешивали при 0° С в течение 2 час. Реакционную смесь вливали в делительную воронку и медленно добавляли насыщенный раствор бикарбоната натрия. Смесь экстрагировали этилацетатом (3х). Объединенные экстракты промывали насыщенным бикарбонатом натрия (1х), насыщенным раствором соли (2х), сушили сульфатом магния и выпаривали, получая 162 мг соединения 6 (выход 92%). Соединение использовали без дальнейшей очистки.
1H ЯМР (300 МГц-СDСl3) δ 7,88 (д, 2, J=9 Гц, Ar-H), 7,30 (д, 2, J 9 Гц, Ar-H), 5,80 (с, 1,4-Н), 4,48 (д, 1, 9 Гц, 11-Н), 2,58 (с, 3, СН3 ацетофенона), 1,86 (с, 3, СН3 пропинила), 0,41 (с, 3, 18-СН3).
11β -[4-(Ацетил)фенил]-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-он (7)
Неочищенное соединение 6 (80 мг; 0,187 ммоль) растворяли в муравьиной кислоте (0,8 мл; 21 ммоль) и добавляли уксусный ангидрид (2,0 мл; 21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 час, вливали в делительную воронку, разбавляли этилацетатом и медленно добавляли насыщенный раствор бикарбоната натрия. Полученную смесь экстрагировали этилацетатом (3х). Объединенные экстракты промывали насыщенным бикарбонатом натрия (1х), насыщенным раствором соли (2х), сушили сульфатом магния и выпаривали, получая 81 мг неочищенного соединения 7. Полученный продукт очищали препаративной ВЭЖХ (YMC ODS-A, 20× 250 мм, 70% МеОН-30% Н2О), получая чистое соединение 7 (43 мг; выход 51%). ВЭЖХ-анализ (YMC-ODS, 4,6× 150 мм, 70% МеОН-30% Н2O) показал, что чистота составляла 99,1% AUC.
1H ЯМР (300 МГц-СDСl3). 1H-ЯМР соединения 7 показал, что в растворе оно существует в двух формах. Основная форма: δ 8,54 (д, 1, J=12 Гц, СНО), 7,88 (д, 2, J=9 Гц, Ar-H), 7,26 (д, 2, J=9 Гц, Ar-H), 5,81 (с, 1,4-Н), 5,67 (д, 1, J=12 Гц, N-H), 4,49 (д, 1,9 Гц, 11-Н), 2,59 (с, 3, СН3 ацетофенона), 1,92 (с, 3, СН3 пропинила), 0,41 (с, 3, 18-СН3). Минорная форма: δ 8,08 (с, 1, СНО), 5,48 (с, 1, N-H), 0,42 (с, 3, 18-СН3), Масс-спектр, m/z 455 (М+), 410,346, 235. Анализ: Вычислено для С30Н33NО3·0,75Н2O: С, 76,81; Н, 7,41; N, 2,99, Найдено: С, 76,93; Н, 7,30; N, 3,07.
Биологические данные для 11β -[4-(ацетил)фенил]-17β -(N-формамидо) -17α -(1-пропинил)эстра-4,9-диен-3-она (7)
Указанное соединение демонстрировало связывание с рецептором прогестина с относительной аффинностью связывания (IС50) 51 нМ. Оно антагонизировало гестагенное действие прогестерона в клетках T47D с IC50, равной 1 нМ.
Пример 15
Синтез 11β -(4-Аминофенил)-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-он осуществляли согласно следующей схеме:
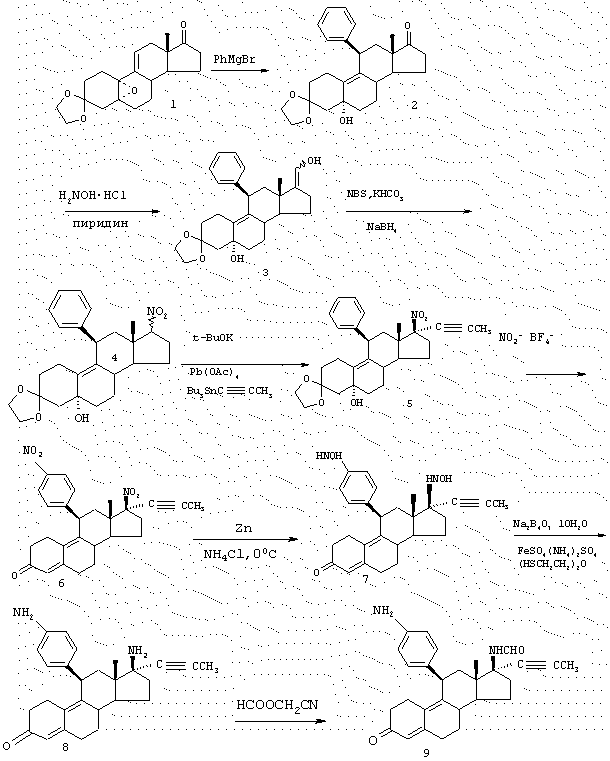
3,3-[1,2-Этандиилбис(окси)]-5α -гидрокси-11β -фенилэстр-9-ен-17-он (2)
В атмосфере сухого аргона фенилмагнийбромид в ТГФ (120 мл; 1 М раствор) разбавляли ТГФ (80 мл) и охлаждали до -10° С и перемешивали. Одной порцией добавляли CuI (11,43 г), продолжали перемешивание в течение 1 мин, в течение 3 мин добавляли эпоксид 1 19,8 г (0,06 моль) в ТГФ (120 мл). Реакционную смесь перемешивали в течение 45 мин при -5° С, вливали в смесь насыщенного NH4Cl (400 мл) и колотого льда (750 мл), содержащую концентрированный NH4OH (19 мл), и перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь экстрагировали EtOAc (3× 200 мл). Объединенные органические слои промывали насыщенным NаНСО3 (1X), H2O (1X) и насыщенным раствором соли (1X). Раствор сушили (Na2SO4) и растворитель удаляли при пониженном давлении. Полученный в результате продукт подвергали хроматографии на колонке с силикагелем (500 г), используя смесь EtOAc-гексан-CH2Cl2-Et3N (1:1:0,1:0,01), получая 179 г (выход 73,3% соединения 2).
1H ЯМР (300 МГц-СDСl3) δ 7,11-7,28 (м, 5, АrН), 4,38 (С, 1, ОН), 4,30 (д, 1, J=6 Гц, 11-Н), 3,92-4,02 (м, 4, 3-кеталь), 0,49 (с, 3, 18-СН3).
3,3-[1,2-Этандиилбис(окси)]-5α -гидрокси-17-гидроксиимино-11β -фенилэстр-9-ен (3)
К раствору 17,67 г (43,3 ммоль) кетона 2 в пиридине (100 мл) добавляли 3,16 г (45,5 ммоль) гидрохлорида гидроксиламина. После перемешивания в течение ночи при комнатной температуре добавляли воду (400 мл) и EtOAc (500 мл). После 10 мин перемешивания водный слой отделяли и экстрагировали EtOAc (3× 300 мл). Объединенный органический раствор дважды промывали 300 мл насыщенного раствора соли, сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении. Полученную в результате пену выпаривали в роторном испарителе при пониженном давлении со 100 мл толуола. Оставшийся растворитель удаляли в вакууме, получая 19,6 г (грубый выход 107%) оксима 3 в виде пены. Полученный продукт использовали без дальнейшей очистки.
1H ЯМР (300 МГц-СDСl3) δ 7,96 (с, 1, =N-OH), 7,11-7,31 (м, 5, ArH), 4,38 (с, 1, ОН), 4,30 (д, 1, J=6 Гц, 11-Н), 3,80-3,99 (м, 4, кеталь), 0,53 (с, 3, 18-СН3).
3,3-[1,2-Этандиилбис(окси)]-17-нитро-5α -гидрокси-11β -фенилэстр-9-ен (4)
NBS (25 г; 0,14 моль) растворяли в диоксане (230 мл). Добавляли раствор КНСО3 (13,9 г; 0,14 моль) в воде (230 мл). Добавляли оксим 3 (19,6 г; 46,3 ммоль), растворенный в диоксане (230 мл), и смесь перемешивали в течение ночи. Порциями добавляли NaBH4 (12,3 г; 0,33 моль). Через 2 час добавляли раствор гидрохлорида гидроксиламина (150 г в 300 мл Н2O) и перемешивание продолжали в течение 0,5 час. Затем экстрагировали EtOAc (3× 300 мл). Объединенный органический раствор промывали насыщенным NaHCO3 (3× 100 мл), насыщенным раствором соли (2× 100 мл), сушили (Na2SO4), фильтровали и растворитель удаляли при пониженном давлении. Полученный в результате продукт подвергали хроматографии на колонке с силикагелем (700 г), используя в качестве элюента 15-25% EtOAc-гексан, получая нитросоединение 4 (17,9 г; выход 73,5%).
1H ЯМР (300 МГц-СDСl3) δ 7,11-7,28 (м, 5, ArH), 4,38 (с, 1, ОН), 4,30 (д, 1, J=6 Гц, 11-Н), 3,91-4,02 (м, 4, 3-кеталь), 0,49 (С, 3, 18-СН3).
3,3-[1,2-Этандиилбис(окси)]-5α -гидрокси-17β -нитро-11β -фенил-17α -(1-пропинил)эстр-9-ен (5)
Нитросоединение 4 (13,2 г; 30,0 ммоль) растворяли в безводном ДМСО (100 мл). Добавляли KOBut (3,7 г; 33,0 ммоль) и смесь перемешивали при комнатной температуре в течение 1 час. В другой колбе растворяли Рb(ОАс)4 (30 г; 67,72 ммоль) в безводном ДМСО (135 мл). Вu3SnС+С-СН3 (25 г; 76,0 ммоль), растворенные в ДМСО (50 мл), добавляли к раствору тетраацетата свинца и тщательно перемешивали в течение 3 мин 15 сек, получая в результате осадок. К осадку добавляли раствор 4 в ДМСО и смесь перемешивали в течение 0,5 час. Реакционную смесь вливали в смесь насыщенный раствор NH4Cl-H2O (240 мл каждого), затем добавляли KF (52,4 г) и EtOAc (300 мл). Полученную смесь энергично перемешивали в течение 45 мин и фильтровали через подушку целита. Остаток промывали EtOAc. Водный слой отделяли и экстрагировали EtOAc (3× 250 мл). Объединенный органический слой промывали насыщенным раствором соли (2× 100 мл), сушили (Na2SO4), фильтровали и растворитель удаляли при пониженном давлении. Грубый продукт очищали хроматографией на колонке с силикагелем (400 г), используя в качестве элюента 15-20% EtOAc-гексан, получая 8,3 г (выход 58%) соединения 5.
1H ЯМР (300 МГц-СDСl3) δ 7,09-7,26 (м, 5, ArH), 4,49 (с, 1, ОН), 4,38 (д, 1, J=6 Гц, 11-Н), 3,92-4,02 (м, 4, 3-кеталь), 1,94 (с, 3, С+С-СН3), 0,34 (с, 3, 18-СН3).
11β -(4-Нитрофенил)-17β -нитро-17α -(1-пропинил)эстра-4,9-диен-3-он (6)
Соединение 5 (2,7 г; 6,5 ммоль) растворяли в CH3CN (60 мл). Двумя порциями добавляли тетрафторборат нитрония (1,5 г; 11,1 ммоль). После перемешивания в течение 1,5 час растворитель удаляли при пониженном давлении. Неочищенный продукт растворяли в EtOAc (250 мл), промывали насыщенным раствором соли (3× 100 мл), сушили (Na2SO4), фильтровали и выпаривали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (100 г), используя в качестве элюента 15-30% EtOAc-гексан, получая 2,2 г (выход 73%) соединения 6.
1H ЯМР (300 МГц-СDСl3) δ 8,15 (д, 2, J=9 Гц, АrН), 7,36 (д, 2, J=9 Гц, АrН), 5,84 (с, 1, 4-Н), 1,95 (с, 3, С+С-СН3), 0,38 (с, 3, 18-СН3).
11β -[4-(N-гидроксиамино)фенил)]-17β -(N-гидроксиамино)-17α -(1-пропинил)эстра-4,9-диен-3-он (7)
Промежуточное динитросоединение 6 (500 мг; 1,1 ммоль) растворяли в смеси Tro-EtOH-H2O (10:5:5 мл). Охлаждали до 0° С и в течение 2 мин добавляли Zn (5,0 г; 77,0 ммоль). Реакционную смесь энергично перемешивали в течение 10 мин и затем быстро фильтровали через подушку целита, промывали Н2О, EtOH и EtOAc (100 мл). EtOAc-слой промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и выпаривали при пониженном давлении, получая 447 мг неочищенного соединения 7.
1H ЯМР (300 МГц-СDСl3) δ 6,86 (д, 2, J=9 Гц, АrН), 6,53 (д, 2, J=9 Гц, АrН), 5,69 (с, 1, 4-Н), 4,20 (д, 1, 11-Н), 1,85 (с, 3, С+С-СН3), 0,48 (с, 3, 18-СН3).
17β -Амино-11β -(4-аминофенил)-17α -(1-пропинил)эстра-4,9-диен-3-он (8)
Na2B4O7·10H2O (3,17 г; 8,31 ммоль) растворяли в H2O (40 мл). Добавляли 0,1 N НСl (2,34 мл), затем (HSCH2CH2)2O (394 мкл) и сульфат железа (18 мг в 5 мл Н2О). Добавляли промежуточное дигидроксиаминосоединение 7 (500 мг; 1,16 ммоль), растворенное в смеси EtOH (10 мл)ТГФ (5 мл) и реакционную смесь нагревали при 90-100° С в течение 6 час. Смесь охлаждали до комнатной температуры и экстрагировали EtOAc (3× 150 мл). Объединенный органический слой промывали насыщенным раствором соли (3× 60 мл), сушили (Na2SO4), фильтровали и растворитель удаляли при пониженном давлении. Грубый продукт очищали хроматографией на колонке с силикагелем (50 г), используя в качестве элюента 15-80% EtOAc-гексан, получая 190 мг (выход 40%) соединения 8.
1H ЯМР (300 МГц-СDСl3) δ 6,95 (д, 2, J=9 Гц, АrН), 6,61 (д, 2, J=9 Гц, АrН), 5,76 (с, 1, 4-Н), 4,33 (д, 1, J=6 Гц, 11-Н), 1,85 (с, 3, С+С-СН3), 0,47 (с, 1, 18-СН3).
11β -(4-Аминофенил)-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-он
Диаминосоединение 8 (190 мг; 0,48 ммоль) растворяли в СН2Сl2 (7 мл). Добавляли цианометилформиат (33,2 мг; 0,328 ммоль) и смесь перемешивали в течение 12 час. После выпаривания при пониженном давлении грубый продукт очищали хроматографией на силикагеле (25 г), используя в качестве элюента 25-50% EtOAc-гексан, получая 41 мг моноформила 9. Дальнейшую очистку проводили препаративной ВЭЖХ (колонка YMC-ODS 250× 20 мм, 60:40 МеОН-Н2O), получая чистое соединение 9 (34 мг). Масс-спектр, m/z 428 (М+). 1H-ЯМР соединения 9 показал, что оно существует в растворе в двух формах.
1H ЯМР (300 МГц-СОСl3) основная форма: δ 8,53 (д, 1, J-12 Гц, NHCHO), 6,90 (д, 2, J=9 Гц, ArH), 6,60 (д, 2, J-9 Гц, ArH), 6,00 (д, 1, J-12 Гц, NHCHO), 5,84 (с, 1, 4-Н), 4,34 (д, 1, J=6 Гц, 11-Н), 1,90 (с, 3, С+С-СН3), 0,47 (с, 3, 18-СН3); минорная форма: 8,09 (с, СНО); 0,49 (1,5, 18-СН3).
Пример 16
Синтез 17β -(N-формамидо)-11β -[4-(N-метиламино)фенил]-17α -(1-пропинил)эстра-4,9-диен-3-он (3) проводили согласно следующей схеме:
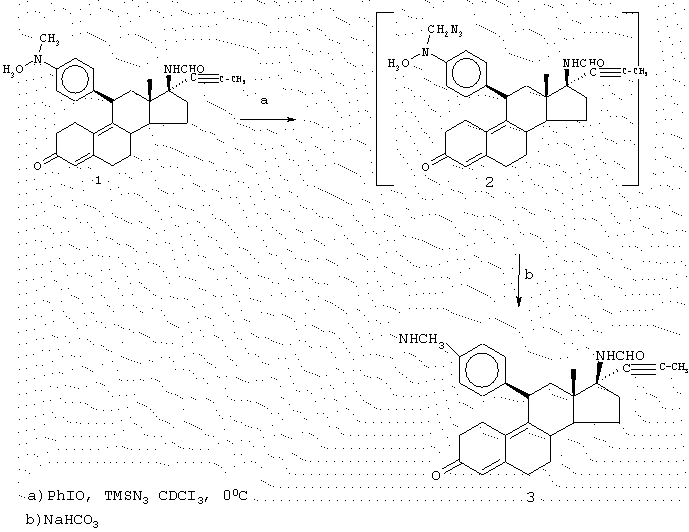
Азидотриметилсилан (799 мг; 6,93 ммоль) добавляли к перемешиваемому раствору 11β -[4-(N,N-диметиламино)фенил]-17β -(N-формамидо)-17α -(1-пропинил)эстра-4,9-диен-3-она (1) (2,1 г; 4,6 ммоль) и йодозобензола (1,21 г; 5,52 ммоль) в CDCl3 (28 мл) при 0° С в атмосфере аргона и перемешивание продолжали еще в течение 15 мин. 1H-ЯМР-спектр смеси показал полное превращение в промежуточный продукт 2. Смесь вливали в энергично перемешиваемую смесь ТГФ и насыщенного раствора NаНСО3 1:1 (220 мл). После перемешивания в течение ночи смесь фильтровали и твердое вещество промывали СНСl3 (50 мл). Фильтрат и промывные воды объединяли и отделяли органический слой. Водный слой экстрагировали СНСl3 (100 мл). Объединенный органический экстракт промывали насыщенным раствором соли (100 мл) и сушили над Na2SO4. Растворитель удаляли, и очистка остатка хроматографией на колонке с SiO2 (75 г, 230-400 меш) (300 мл смеси EtOAc-гексан 6:4, 500 мл смеси EtOAc-гексан 7:3, 1 л смеси EtOAc-гексан 9:1) давала 1,36 г пенообразного твердого вещества. Полученный продукт растворяли в этаноле (4 мл) и раствор по каплям при перемешивании добавляли к теплой (60° С) воде (60 мл). Смесь охлаждали до 5° С и твердое вещество отфильтровывали. Дальнейшая сушка полученного твердого вещества в вакууме давала 1,23 г (выход 60%) соединения 3,
1Н-ЯМР-спектр соединения 3 показал, что оно существует в растворе в двух формах.
1Н ЯМР (300 МГц, CDCl3) основная форма: δ 8,52 (д, 1, J=11,8 Гц, СНО), 6,93 (д, 2, J=8,6 Гц, АrН), 6,53 (д, 2,J=8,6 Гц, АrН), 5,77 (с, 1, 4-Н), 5,72 (д, 1, J=11,8 Гц, NHCHO), 4,36 (д, 1, J=6,8 Гц, 11-Н), 3,66 (шир. с, 1), 2,81 (с, 3, NСН3), 1,91 (с, 3, СССН3), 0,47 (с, 3, 18-СН3); минорная форма: 8,09 (с, СНО), 6,98 (д, АrН), 0,50 (с, 18-СН3); Масс-спектр, m/z 442 (M+), Анализ, Вычислено для С29Н34N2O2·0,25Н2O: С, 77,91; Н, 7,78; N, 6,27. Найдено: С, 77,80; Н, 7,71; N, 6,39.
Очевидно, что в свете приведенных выше данных возможны многочисленные модификации и вариации данного изобретения. Поэтому следует понимать, что в рамках прилагаемой формулы изобретения оно может применяться на практике иначе, чем так, как здесь конкретно описано.
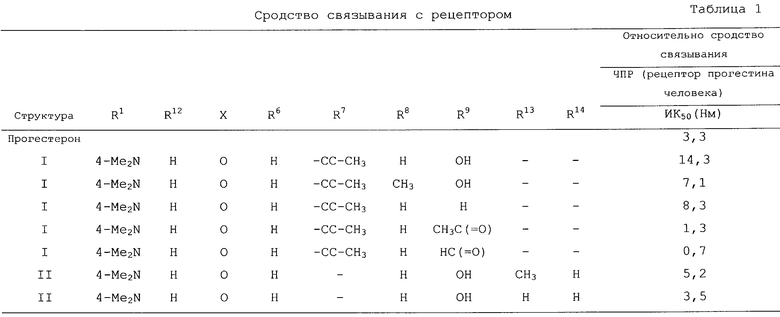
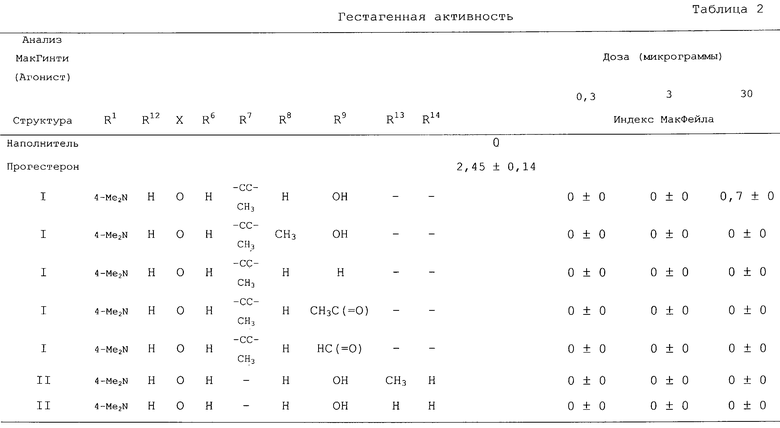

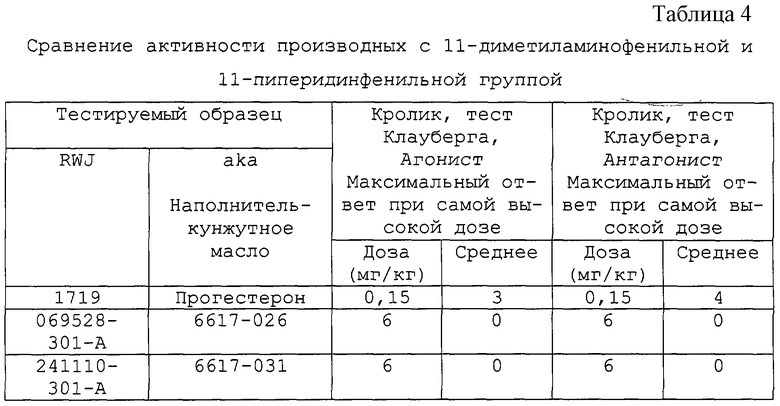
| название | год | авторы | номер документа |
|---|---|---|---|
| ГОРМОНАЛЬНЫЕ ИЛИ НЕГОРМОНАЛЬНЫЕ СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ТЕРАПЕВТИЧЕСКОГО ЛЕЧЕНИЯ | 1999 |
|
RU2230071C2 |
| ПРОИЗВОДНЫЕ 11β-БЕНЗАЛЬДОКСИМ-ЭСТРА-4,9-ДИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2137777C1 |
| АНТАГОНИСТЫ ПРОГЕСТЕРОНА | 2012 |
|
RU2608521C2 |
| ПРОИЗВОДНЫЕ 11,21-БИСФЕНИЛ-19-НОРПРЕГНАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2152952C2 |
| ПРОИЗВОДНЫЕ 16-ГИДРОКСИ-11-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-ЭСТРА-4,9-ДИЕНА, СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2187510C2 |
| ПРОИЗВОДНОЕ 11-(ФЕНИЛЗАМЕЩЕННЫЙ)ЭСТРА-4,9-ДИЕНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2135514C1 |
| МОДУЛЯТОРЫ РЕЦЕПТОРОВ ПРОГЕСТЕРОНА | 2005 |
|
RU2381232C2 |
| ИМИДАЗОЛИЛЬНЫЕ АНТАГОНИСТЫ ПРОГЕСТЕРОНА | 2014 |
|
RU2679445C2 |
| ПРОИЗВОДНЫЕ СУЛЬФАМАТА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 1996 |
|
RU2159774C2 |
| ПРОИЗВОДНЫЕ β БЕНЗАЛЬДОКСИМ-ЭСТРА-4,9-ДИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2130944C1 |

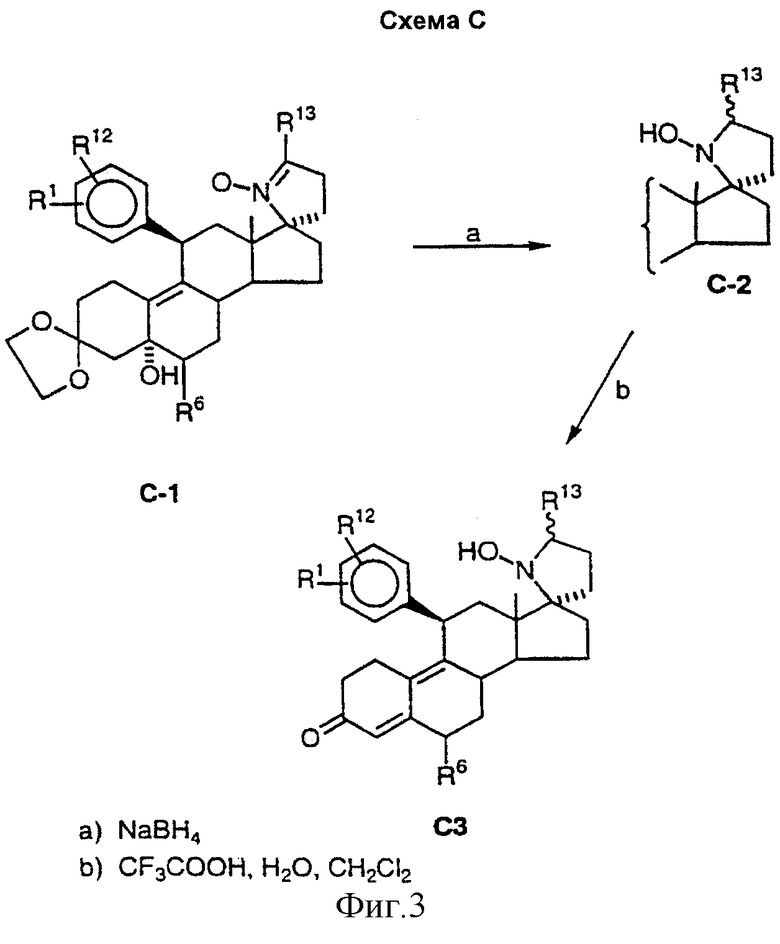
Изобретение относится к новым стероидом общей формулы I, которые проявляют себя, как эффективные антагонисты прогестинов и как смешанные или неполные агонисты прогестина
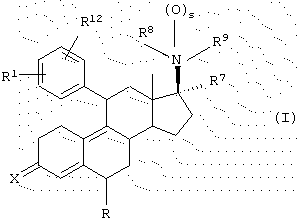
где
R1 - (R2R3N(O)r)-, где r=0, 1, каждый из R2 и R3 независимо - Н, C1-6алкил; или R1 -
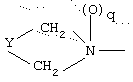
где q=0, - 1, Y-(CH2)m-, где m=1, 2, 3, 4, 5; или
R1 - C1-6алкил-СО-; R12 - Н; Х - О или
 ,
,
где -(СН2)m-, где m=2; R6=Н; R7=Н, C1-6алкил, С2-6алкенил, С2-6алкинил, возможно замещенный ОН или ОСНО; S=0, 1; каждый из R8 и R9 независимо - Н, ОН, C1-6алкил, R10CO, где R10’ - Н, C1-6алкил. Соединения I полезны для лечения фибром, эндометроза, в гормонозаместительной терапии и для регулирования фертильности. 5 с. и 11 з.п. ф-лы, 4 табл., 3 ил.
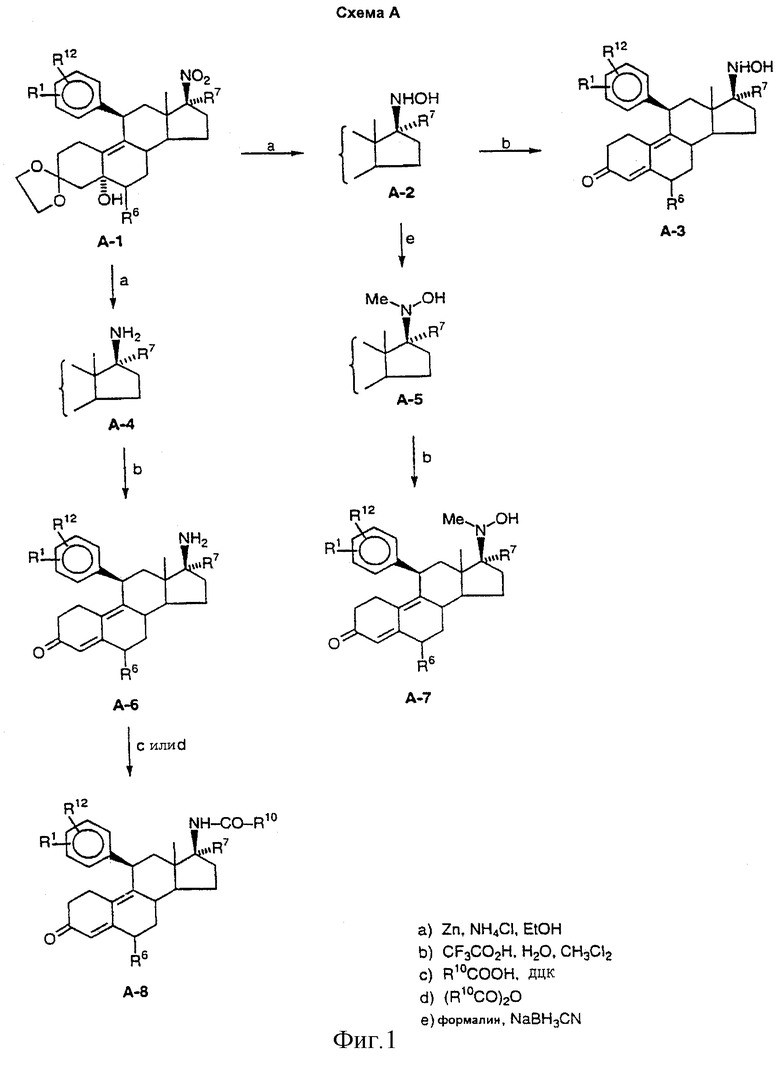
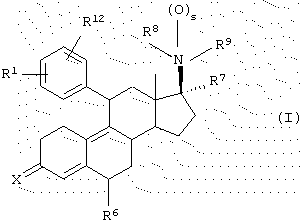
где R1 означает (R2R3N(O)r)-, где r равно 0 или 1 и каждый из R2 и R3 независимо представляет Н, C1-6алкил; или
R1 означает
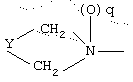
где q равно 0 или 1, Y означает -(CH2)m-, где m является целым числом от 1 до 5, или
R1 означает C1-6алкил-СО-;
R12 является Н;
Х означает О или
Х означает
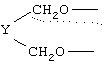
где Y представляет -(СН2)m-, где m равно 2;
R6 означает Н;
R7 означает Н, C1-6алкил, С2-6алкенил, С2-6алкинил, любой из которых может быть необязательно замещен ОН, ОСНО;
s равно 0 или 1;
каждый из R8 и R9 независимо означает Н, ОН, C1-6алкил, R10CO, где R10 означает Н, C1-6алкил,
и его фармацевтически приемлемые соли.
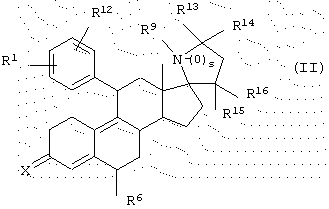
где R1 означает R2R3N-, где каждый из R2 и R3 независимо представляет C1-6алкил, или
R1 означает
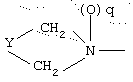
где Y означает -(СН2)m-, где m равно 2, и q равно 0;
s равно 0;
R6 и R12 представляют Н;
R13 и R14 независимо означают Н, C1-6алкил, или R13 и R14 вместе означают =O;
R15 и R16 означают Н;
X означает О;
и его фармацевтически приемлемые соли.
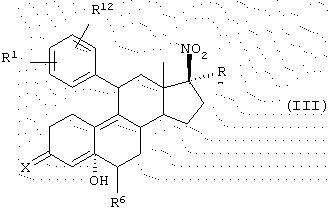
где R1 означает (R2R3N(O)r)-, где r равно 0 или 1 и каждый из R2 и R3 независимо представляет Н, C1-6алкил; или
R1 означает
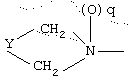
где q равно 0 или 1, Y означает -(CH2)m-, где m является целым числом от 0 до 5, или
R1 означает C1-6алкил-СО-;
R12 является Н;
Х означает О или
Х означает
где Y представляет собой –(СН2)m-, где m равно 2;
R6 означает Н;
R7 означает Н, C1-6алкил, С2-6алкенил, С2-6алкинил, любой из которых может быть необязательно замещен ОН, ОСНО.
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
