Данное изобретение относится к области использования защитных групп в синтетической химии углеводов. Синтетические производные олигосахаридов находят в настоящее время широкое применение в биохимии и медицинской практике.
Ключевой стадией при синтезе и направленной модификации полигидроксильных органических соединений является избирательное введение и удаление временных защит гидроксильных групп (ОН-групп), так называемые О-защиты, при удалении которых происходит высвобождение ОН-групп. В настоящее время известен широкий спектр защитных групп, различающихся устойчивостью в различных реакционных условиях (T.W. Greene, P.G.M. Wuts. Protective group in organic synthesis. - 3rd edition, John Wiley & Sons, 1999, pp. 17-245). Однако многие из предложенных ранее защитных групп требуют использования весьма дорогих и малодоступных реагентов для их введения или удаления. Это вносит ограничения для их препаративного использования в практических синтезах органических соединений, включая промышленные синтезы.
К видам наиболее широко используемых О-защит относятся ацильные, прежде всего ацетильные и бензоильные группы. Введение О-ацетильных и О-бензоильных защит осуществляется при ацилировании соответствующим реагентом соединений со свободными ОН-группами. Удаление же О-цетильных и О-бензоильных защит с образованием соответствующих производных со свободными ОН-группами может быть проведено в щелочных (карбонат калия в метаноле, цианид калия в этаноле, аммиак в метаноле, метилат натрия в метаноле, гидроксид натрия в метаноле и др.) или кислых условиях. Помимо методов исчерпывающего удаления О-ацетильных и О-бензоильных групп, особую практическую значимость имеют методы, позволяющие избирательно удалять О-ацетильные группы в присутствии О-бензоильных групп.
N.E. Byramova, M.V. Ovchinnikov, L.V. Backinowsky, N.K. Kochetkov [Carbohydr. Res., 1983, 124, C8] описали удаление О-ацетильных групп в присутствии О-бензоильных при обработке разбавленным раствором хлористого водорода в метаноле. V. Pozsgay [J. Am. Chem. Soc, 1995, 117, 6673] описал аналогичное превращение под действием 0.3 М HBF4 в метаноле. Ограничение данного метода состоит в несовместимости условий снятия ацетильных защит с наличием в молекуле других кислотолабильных групп, таких как силильные, ацетальные, тритильные и др.
Y.-C. Xu, A. Bizuneh и С. Walker [Tetrahedron Lett., 1996, 37, 455] предложили использовать раствор метоксида магния в метаноле для избирательного удаления О-ацетильной защиты в присутствии О-бензоильной или других О-ацильных групп. Недостатком данного метода является его малая эффективность применительно к сложно построенным углеводным системам.
N. Kunesch, С. Meit и J. Poisson [Tetrahedron Lett., 1987, 28, 3569] использовали для селективного дезацетилирования раствор гуанидина в этаноле, Т. Neilson и E.S. Werstiuk [Can. J. Chem. 1971, 49, 493] - раствор аммиака в метаноле, a L.Н.В. Baptistella, J.F. Dos Santos, К.С. Ballabio и A.J. Marsaioli [Synthesis, 1989, 436-438] - раствор 1,8-диазабицикло[5.4.0]ундец-7-ена (ДБУ) в бензоле. В этих трех перечисленных примерах недостатком была также невысокая эффективность процесса.
В патенте US 5629433 «Избирательный процесс для дезацилирования и дезацетилирования таксола и таксанов» описывается одностадийный процесс, при котором под действием пероксида водорода в THF в присутствии различных основных реагентов (NaHCO3, Na2CO3, CaCO3, BaCO3 и др.) ацильные группы, расположенные у атомов углерода 2', 10 и 7 таксола и других таксановых соединений, могут быть избирательно удалены.
В работах Н.М. Verheij, P.F. Smith, P.P.М. Bonsen и L.L.М. Van Deenen [Biochim. Biophys. Acta, 1970, 218, 97], а также P. Greimel, M. Lapeyre, Y. Nagatsuka, Y. Hirabayashi и Y. Ito [Bioorg. Med. Chem. 2008, 16, 7210] для селективного снятия ацетильных защит в присутствии сложных эфиров жирных кислот были использованы моногидроацетат гидразина или смесь гидразингидрат - уксусная кислота (молярное соотношение 4:1) соответственно. Данные методы имеют явное ограничение, поскольку подразумевали первоначальное де-О-ацетилирование первичного гидроксила с последующим деблокированием вторичных гидроксильных групп за счет внутримолекулярного соучастия освободившегося первичного, а затем и вторичных гидроксильных групп.
Наиболее близкой к данному изобретению является работа W.R. Roush и X.-F. Lid [J. Am. Chem. Soc. 1995, 117, 2236], в которой описан пример селективного снятия аномерного ацетата в присутствии аксиального ацетата с использованием 1.5 эквивалента гидразингидрата в метаноле. Однако в данном случае происходит селективное снятие ацетильной группы с полуацетального гидроксила, не затрагивая при этом ацетильной группы на вторичном гидроксиле. Подобные селективные превращения широко известны в литературе и легко реализуются под действием широкого круга реагентов.
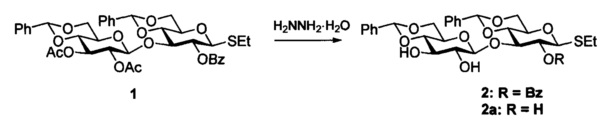
Перечисленные методы не имеют общего характера и поэтому имеют ограниченное практическое применение. Так, например, проведенные нами попытки получения диола 2 путем обработки диацетата-монобензоата 1 указанными выше реагентами (разбавленный раствор метилата натрия в метаноле, метоксид магния [Y.-C. Xu, A. Bizuneh, С. Walker, Tetrahedron Lett., 1996, 37, 455], триэтиламин в метаноле, ДБУ в бензоле, моногидроацетат гидразина [Н.М. Verheij, P.F. Smith, P.P.М. Bonsen, L.L. M. Van Deenen, Biochim. Biophys. Acta 1970, 218, 97] или смесь гидразингидрата - уксусная кислота (молярное соотношение 4:1) [P. Greimel, М. Lapeyre, Y. Nagatsuka, Y. Hirabayashi, Y. Ito, Bioorg. Med. Chem. 2008, 16, 7210], гидразингидрат 2-5 эквивалентов), для которых в научной литературе сообщалось о применении для селективного снятия ацетильной группы в присутствии бензоильной, приводили к неселективному превращению либо, как в случае моногидроацетата гидразина, - к малой (~10-20%) конверсии исходного соединения 1 (см. Примеры 1а-д).
Использование небольшого избытка (2-3 эквивалента) гидразингидрата в смеси метанол - хлористый метилен при комнатной температуре в течение нескольких часов приводили к образованию смеси продуктов неизбирательного снятия как ацетильных, так и бензоильных групп со значительным количеством непрореагировавшего исходного 1.
Предметом настоящего изобретения является метод избирательного удаления О-ацетильных групп в присутствии О-бензоильных при обработке большими избытками гидразингидрата - от 30 до 100 мольных эквивалентов (преимущественно 50) В интервале 1-15 эквивалентов гидразингидрата наблюдается неполная конверсия (<50%), при этом через несколько часов наблюдается образование побочного продукта снятия бензоильной группы - триола 2а. Оптимальным интервалом соотношения субстрат - реагент является значение 40-50. Использование большего (проверено вплоть до 1:100) количества гидразингидрата незначительно сокращает время реакции, но не рационально с точки зрения экономии реагентов.
Так, в результате обработки 1 50 экв. гидразингидрата в смеси хлористого метилена и метанола (1:2 по объему) при комнатной температуре в течение 25 минут получили 8.5% непрореагировавшего 1 и 91.5% продукта дезацетилирования 2. При повторной обработке выделенного из реакционной смеси непрореагировавшего исходного 1 в аналогичных условиях образуется дополнительное количество диола 2, суммарный выход которого практически количественный (см. Пример 2Г).
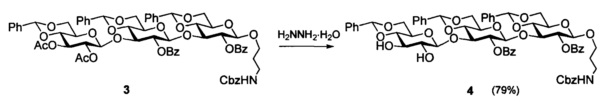
Аналогично эффективно могут быть превращены и другие соединения, одновременно содержащие О-ацетильные и О-бензоильные защиты, например, трисахарид 3, в котором содержится по две О-ацетильные и О-бензоильные защиты. Обработка этого соединения гидразингидратом (~44 экв.) в метаноле при комнатной температуре в течение 25 минут (Пример 3) дает продукт избирательного де-О-ацетилирования 4 с высоким выходом 79%.
Пример 1. Де-О-ацетилирование дисахарида 1 под действием известных реагентов.
А) Де-О-ацетилирование дисахарида 1 под действием разбавленного раствора метилата натрия в метаноле:
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 5 мкл (0.005 ммоль, 0.07 экв.) 1 М раствора метилата натрия в метаноле при комнатной температуре в течение 25 минут. Смесь нейтрализовали ионообменной смолой Amberlite IR-120 (Н+), фильтровали, упаривали досуха и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1, диола 2 и триола 2а примерно в равных количествах.
Б) Де-О-ацетилирование дисахарида 1 под действием метоксида магния [Y.-С. Xu, A. Bizuneh, С. Walker, Tetrahedron Lett, 37, 455 (1996)]:
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 5 мкл (0.005 ммоль, 0.07 экв.) свежеприготовленного 1 М раствора метоксида магния в метаноле при комнатной температуре в течение 12 часов. Смесь нейтрализовали ионообменной смолой Amberlite IR-120 (Н+), фильтровали, упаривали досуха и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1, диола 2 и триола 2а примерно в равных количествах.
В) Де-О-ацетилирование дисахарида 1 под действием триэтиламина в метаноле
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 50 мкл (0.68 ммоль, 10 экв.) триэтиламина при комнатной температуре в течение 2 часов. Смесь упаривали досуха и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1, диола 2 и триола 2а примерно в равных количествах.
Г) Де-О-ацетилирование дисахарида 1 под действием ДБУ в бензоле [Synthesis}
Раствор 0.05 г (0.067 ммоль) 1 в 2.0 мл бензола обрабатывали 15 мкл (0.097 ммоль, 1.44 экв.) ДБУ при комнатной температуре в течение 2.5 часов. Смесь разбавили хлористым метиленом, промыли 1 М водным раствором соляной кислоты, сушили фильтрованием через вату, упаривали досуха и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1, диола 2 и триола 2а примерно в равных количествах.
Д) Де-О-ацетилирование дисахарида 1 под действием моногидроацетата гидразина [Н.М. Verheij, P.F. Smith, P.P.М. Bonsen, L.L. М. Van Deenen, Biochim. Biophys. Acta, 1970, 218, 97] или смеси гидразингидрат - уксусная кислота (молярное соотношение 4:1) [P. Greimel, М. Lapeyre, Y. Nagatsuka, Y. Hirabayashi, Y. Ito, Bioorg. Med. Chem. 2008, 16 7210].
Раствор 0.05 г (0.067 ммоль) 1 в 0.5 мл диметилформамида обрабатывали 18 мг (0.2 ммоль, 1.5 экв.) моногидроацетата гидразина (или смесью 25 мг гидразингидрата и 9 мкл уксусной кислоты) при комнатной температуре в течение 2 часов. Смесь экстрагировали этилацетатом, сушили, упаривали досуха и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1 и диола 2, с явным преобладанием исходного дисахарида 1 (>90%).
Пример 2. Де-О-ацетилирование дисахарида 1 под действием гидразингидрата в метаноле.
А) Де-О-ацетилирование дисахарида 1 под действием 1.5 эквивалентов гидразингидрата в метаноле.
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 5 мкл (0.1 ммоль, 1.5 экв.) гидразингидрата при комнатной температуре в течение 25 минут. Смесь разбавили 5 мл воды, экстрагировали хлористым метиленом (3×3 мл), объединенный экстракт промыли насыщенным раствором хлористого натрия, сушили фильтрованием через вату, концентрировали в вакууме и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1 и диола 2, с явным преобладанием исходного дисахарида 1 (>80%).
Б) Де-О-ацетилирование дисахарида 1 под действием 5 эквивалентов гидразингидрата в метаноле.
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 17 мкл (0.33 ммоль, 5 экв.) гидразингидрата при комнатной температуре в течение 25 минут. Смесь разбавили 5 мл воды, экстрагировали хлористым метиленом (3×3 мл), объединенный экстракт промыли насыщенным раствором хлористого натрия, сушили фильтрованием через вату, концентрировали в вакууме и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1 и диола 2, с явным преобладанием исходного дисахарида 1 (>80%). Кроме того наблюдалось образование триола 2а в видимых количествах.
В) Де-О-ацетилирование дисахарида 1 под действием 15 эквивалентов гидразингидрата в метаноле.
Раствор 0.05 г (0.067 ммоль) 1 в смеси 0.5 мл хлористого метилена и 1.0 мл метанола обрабатывали 50 мкл (1 ммоль, 15 экв.) гидразингидрата при комнатной температуре в течение 25 минут. Смесь разбавили 5 мл воды, экстрагировали хлористым метиленом (3×3 мл), объединенный экстракт промыли насыщенным раствором хлористого натрия, сушили фильтрованием через вату, концентрировали в вакууме и остаток анализировали методом ТСХ на пластинке с силикагелем в системе толуол-этилацетат (2:1). Продуктом данного превращения явилась смесь исходного дисахарида 1 и диола 2, с явным преобладанием исходного дисахарида 1 (>70%). Кроме того, наблюдалось образование триола 2а в видимых количествах.
Г) Де-О-ацетилирование дисахарида 1 под действием 50 и более (проверено вплоть до значений 100) эквивалентов гидразингидрата в метаноле.
Раствор 1.42 г (1.89 ммоль) 1 в смеси 14 мл хлористого метилена и 28 мл метанола обрабатывали 4.7 мл (97 ммоль, ~50 экв.) гидразингидрата при комнатной температуре в течение 25 минут. Смесь разбавили 150 мл воды, экстрагировали хлористым метиленом (3×50 мл), объединенный экстракт промыли насыщенным раствором хлористого натрия, сушили фильтрованием через вату, концентрировали в вакууме и остаток очищали на силикагеле в градиенте ацетон - хлористый метилен (0→15%), получая 120 мг (8.5%) непрореагировавшего 1 и 1.15 (91.5%) 2. Повторная обработка регенерированного 1 в условиях, аналогичных указанным выше, привела к получению дополнительно 110 мг 2. Суммарный выход 2 - 1.26 г (колич.), твердое аморфное вещество, [α]D - 54 (с 1, CHCl3). Спектр 1Н ЯМР (CDCl3, 600 МГц): δ 1.26 (т, 3H, J=7.4 Гц, CH3CH2S), 2.53 (с, 1Н, ОН-32), 2.76 (м, 2Н, CH3CH2S), 2.90 (д, 1H, JOH,2=2.4 Гц, ОН-22), 3.30 (м, 1Н, Н-52), 3.46 (м, 3H, Н-22, 42, 62а), 3.62 (м, 2Н, Н-32, 51), 3.81 (т, 1Н, J=9.6 Гц, Н-41), 3.85 (м, 2Н, H-61а, 62b), 4.21 (т, 1Н, J=9.0 Гц, Н-31), 4.43 (дд, 1H, J6b,5=5.1 Гц, J6b,6а=10.5 Гц,, Н-61b), 4.47 (д, 1Н, J1,2=7.8 Гц, Н-12), 4.73 (д, 1Н, J1,2=10.2 Гц, Н-11), 5.37 (т, 1Н, J=9.3 Гц, Н-21), 5.43, 5.63 (2 с, каждый 1Н, 2 PhCH), 7.32-8.08 (м, 15Н, 3 Ph). Спектр 13С ЯМР (CDCl3, 150 МГц): δ 14.8 (CH3CH2S), 24.1 (CH3CH2S), 66.6 (С-52), 68.4 (С-62), 68.5 (С-61), 70.9 (С-51), 71.9 (С-21), 72.8 (С-32), 73.4 (С-22), 79.1 (С-41), 79.3 (С-31), 80.2 (С-42), 84.3 (С-11), 101.6, 101.8 (2 PhCH), 103.0 (С-12), 126.1-136.9 (3 Ph), 156.8 (PhCO). Данные масс-спектра: рассчитано для C35H38O11S [М+К]+ 705.1766; найдено - 705.1778.
Пример 3. Де-О-ацетилирование трисахарида 3 под действием гидразингидрата в метаноле.
Раствор 1.55 г (1.24 ммоль) 3 в смеси 8 мл хлористого метилена и 16 мл метанола обрабатывали 2.6 мл (54 ммоль, ~44 экв.) гидразингидрата при комнатной температуре в течение 25 минут. Смесь разбавили 100 мл воды, экстрагировали хлористым метиленом (3×30 мл), объединенный экстракт промыли насыщенным раствором хлористого натрия, сушили фильтрованием через вату, концентрировали в вакууме и остаток очищали на силикагеле в градиенте хлористый метилен - ацетон (0→15%), получая 200 мг (13%) непрореагировавшего 3 и 1.03 (71%) 4. Повторная обработка регенерированного 3 в условиях, аналогичных указанным выше, привела к получению дополнительно 110 мг 4. Суммарный выход 4 - 1.14 г (79%), твердое аморфное вещество, [α]D - 21 (с 1, CHCl3). Спектр 1Н ЯМР (CDCl3, 600 МГц, характеристичные сигналы): δ 1.64 (м, 2Н, NHCH2CH2CH2O), 3.08, 3.12 (2 м, 2Н, NHCH2CH2CH2O), 3.26 (м, 1H, Н-53), 3.43 (м, 4Н, Н-23, 43, 63а, NHCH2CH2CHaHbO), 3.48 (м, 1Н, Н-51), 3.62 (м, 2Н, Н-33, Н-52), 3.68 (т, 1Н, J=10.2 Гц, Н-61а), 3.83 (м, 3H, Н-62а, 63b, NHCH2CH2CHaHbO), 3.86 (т, 1Н, J=9.1 Гц, Н-41), 4.91 (т, 1H, J=9.2 Гц, Н-42), 4.04 (т, 1Н, J=8.1 Гц, Н-31), 4.20 (м, 2Н, Н-32, 61b), 4.39 (дд, 1Н, J6b,5=4.7 Гц, J6b,6a=10.5 Гц, Н-62b), 4.44 (д, 1Н, J1,2=7.8 Гц, Н-13), 4.62 (д, 1Н, J1,2=7.0 Гц, Н-11), 4.97 (уширенный т, 1Н, NH), 5.03 (д, 1Н, J1,2=6.6 Гц, Н-12), 5.07 (с, 2Н, PhCH2), 5.25 (м, 2Н, Н-21, 22), 5.35, 5.41, 5.58 (3 с, каждый 1Н, PhCH), 7.30-8.89 (м, 30Н, 6 Ph). Спектр 13С ЯМР (CDCl3, 150 МГц): δ 29.4 (NHCH2CH2CH2O), 38.0 (NHCH2CH2CH2O), 65.9 (С-51), 66.2 (С-52), 66.4 (С-53), 66.6 (PhCH2), 67.2 (NHCH2CH2CH2O), 68.3 (С-63), 68.6 (С-61), 68.8 (С-62), 72.7 (С-33), 73.5 (2С), 73.6 (С-21, 22, 23), 77.9 (С-32), 78.2 (С-31), 78.5 (С-41), 79.3 (С-42), 80.2 (С-43), 100.0 (С-12), 100.9 (С-11), 101.3, 101.7, 101.8 (3 PhCH), 102.5 (С-13), 126.0-137.0 (6 Ph), 156.4 (OCONH), 164.9, 165.3 (2 PhCO). Данные масс-спектра: рассчитано для C64H65NO20 [М+Na]+ 1190.3992; найдено - 1190.4002.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения (2R,3R,4R,5S)-1-бутил-2-(гидроксиметил)пиперидин-3,4,5-триола | 2020 |
|
RU2762605C1 |
| Способ получения пентасахарида | 1983 |
|
SU1694065A3 |
| (22ζ)-6b-МЕТОКСИ-3a,5-ЦИКЛО-5a-ХОЛЕСТАН-24-ОН-22-ОЛ В КАЧЕСТВЕ ПОЛУПРОДУКТА В СИНТЕЗЕ (22R,23R)-3b-АЦЕТОКСИ-22,23-ИЗОПРОПИЛИДЕНДИОКСИ-24-МЕТИЛХОЛЕСТ-5-ЕНА | 1991 |
|
RU2024540C1 |
| Способ получения кортикоидных производных | 1984 |
|
SU1561827A3 |
| Способ получения нетилмицина | 1987 |
|
SU1630614A3 |
| СПОСОБЫ ФОРМИРОВАНИЯ ГЛИКОЗИДНЫХ СВЯЗЕЙ, ХИМИЧЕСКАЯ КОМПОЗИЦИЯ, ГЛИКОЗИД И ГЛИКОЗИДНАЯ БИБЛИОТЕКА | 1994 |
|
RU2134693C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛЕНГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | 2017 |
|
RU2664101C1 |
| СТРУКТУРЫ САХАРИДОВ И СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ ТАКИХ СТРУКТУР | 2009 |
|
RU2538973C2 |
| Способ получения 6-деоксиантрациклингликозидов | 1984 |
|
SU1429935A3 |
| СУЛЬФАТИРОВАННЫЕ ПРОИЗВОДНЫЕ ОЛИГОСАХАРИДОВ | 2005 |
|
RU2392281C2 |
Изобретение относится к способу удаления защитных групп в синтетической химии углеводов, который может быть использован в химической промышленности. Предложенный способ состоит в том, что избирательно удаляют О-ацетильные защитные группы в присутствии О-бензоильных в олигосахаридах, обрабатывая производные олигосахаридов гидразингидратом в органическом растворителе, содержащем хлористый метилен и метанол, причем реакцию проводят под действием 30-100 мольных эквивалентов гидразингидрата по отношению к производным олигосахаридов в смеси хлористого метилена и метанола в объемном соотношении 1:2. Предложен новый эффективный способ избирательного удаления О-ацетильных защитных групп в присутствии О-бензоильных, что существенно расширяет возможности эффективного построения олигосахаридных цепей. 1 з.п. ф-лы, 3 пр.
1. Способ избирательного удаления О-ацетильных защитных групп в присутствии О-бензоильных в олигосахаридах, включающий в себя обработку производных олигосахаридов гидразингидратом в органическом растворителе, содержащем хлористый метилен и метанол, отличающийся тем, что реакция проводится под действием 30-100 мольных эквивалентов гидразингидрата по отношению к производным олигосахаридов в смеси хлористого метилена и метанола в объемном соотношении 1:2.
2. Способ по п. 1, отличающийся тем, что реакция проводится под действием 50 мольных эквивалентов гидразингидрата по отношению к производным олигосахаридов.
| W.R | |||
| Roush et al, J | |||
| Am | |||
| Chem | |||
| Soc., 1995, 117, 2236-2250 | |||
| RU 2011144838 A, 20.05.2013 | |||
| WO 2011014793 A2, 03.02.2011. |

