Область техники, к которой относится изобретение
Представленное изобретение относится к соединениям, обладающим активностью в качестве ингибиторов гепарансульфат-связывающих белков и в качестве ингибиторов ферментной гепараназы. Более точно, изобретение относится к сульфатированным производным олигосахаридов, хотя область настоящего изобретения не ограничивается только ими. В частности, изобретение относится к полисульфатированным производным олигосахаридов, причем получение производных предпочтительно проводится по С-1 восстанавливающему концу и/или С-6 невосстанавливающему концу моносахаридного звена. Изобретение относится также к способам получения соединений, композициям, включающим указанные соединения, и применению соединений и их композиций для противоангиогенного, противометастазного, противовоспалительного, противомикробного, противокоагулянтного и/или противомикробного лечения млекопитающего субъекта. Соединения и их композиции могут также применяться для снижения уровня содержания триглицеридов в крови и ингибирования сердечно-сосудистого заболевания у млекопитающего субъекта. Кроме того, соединения могут также применяться для профилактики вышеуказанных расстройств при введении их млекопитающему субъекту.
Уровень техники
Было показано, что сульфатированное олигосахаридное лекарственное средство, известное как PI-88 [1, 2] (см. соединение 1 ниже), является перспективным ингибитором роста опухоли и метастаза [1, 3] и проходит II фазу клинических испытаний на пациентах, больных раком [4]. PI-88 проявляет противоангиогенные эффекты посредством ингибирования взаимодействий факторов роста кровеносных сосудов (главным образом, FGF-1, FGF-2 и VEGF) и их рецепторов с гепарансульфатом [1, 5]. Кроме того, PI-88 является сильным ингибитором ферментной гепараназы, гликозидазы, расщепляющей боковые гепарансульфатные цепи протеогликанов, которые являются основными компонентами внеклеточного матрикса (ЕСМ) и базальных мембран, окружающих опухолевые клетки [1, 2]. Гепараназа в значительной степени вовлечена в развитие кровеносных сосудов: она обладает способностью высвобождать активные гепарансульфат-связанные факторы роста кровеносных сосудов из ЕСМ и вовлечена в разложение ЕСМ и последующее трансформирование тканей, связанное с развитием новых кровеносных сосудов [6]. Разложение ЕМС гепараназой также играет важную роль в распространении опухолевых клеток (метастаз), обеспечивая возможность их поступления в кровяной поток и размещая в удаленных сайтах, где они могут образовывать вторичные опухоли [6, 7].
В дополнение к противоангиогенным эффектам PI-88 ингибирует каскад коагулирования крови через (i) ингибирование протеаз в гемокоагуляции, обусловленной эндогенными факторами, (ii) стимулирование высвобождения ингибитора пути метаболизма тканевого фактора (TFPI) и (iii) активацию ингибирования тромбина, проводимого кофактором гепарина. Однако PI-88 не взаимодействует с АТ III и, следовательно, не проявляет противодействующей активности в отношении Ха или противодействующей АТ III-проводимой активности в отношении IIa [8, 9]. Исследования, проведенные in vivo на обезьянах, показали, что применение PI-88 в низких дозах стимулирует высвобождение всех гепарансульфат-связанных TFPI из клеточной стенки сосуда [9]. Ранее было показано, что помимо влияния на коагулирование TFPI является лекарственным средством для лечения развития кровеносных сосудов [10] и ингибитором метастаза [11]. Было также показано, что PI-88 блокирует пролиферацию гладких миоцитов сосудов и начальное утолщение [12], ингибирует инфицирование клеток вирусом простого герпеса (herpes simplex virus - HSV) и распространение HSV-1 и HSV-2 от клетки к клетке [13], а также ингибирует протеинурию в пассивном нефрите Гейманна [14].
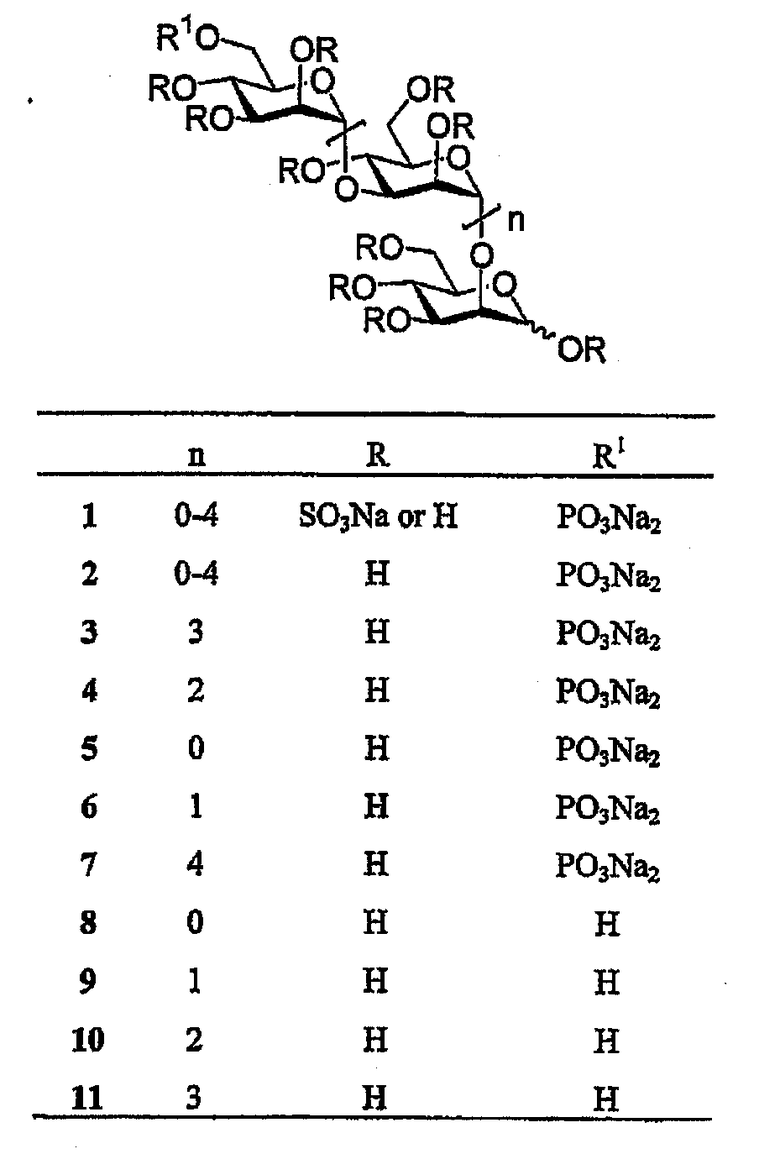
PI-88 представляет собой смесь высокосульфатированных монофосфорилированных маннозоолигосахаридов, размер которых изменяется в интервале от ди- до гексасахарида [15, 16]. PI-88 получен исчерпывающим сульфированием [2, 16] олигосахаридфосфатной фракции (2) (см. формулу I ниже), полученной в результате гидролиза фосфоманнана дрожжей Pichia (Hansenula) holstii NRRL Y-2448 в мягких условиях с использованием кислоты в качестве катализатора [17, 18]. Основными его компонентами являются пента- и тетрасахаридфосфаты 3 (~60%) и 4 (~30%) соответственно, в то время как оставшиеся 10% составляют ди-, три- и гексасахаридфосфаты (5-7), а также тетрасахариламин (не показан) [15, 16].
Формула I
Хорошо известно, что другие полисульфатированные олиго- и полисахариды и их производные также проявляют биологические активности, аналогичные активностям PI-88 [19-25]. Эти биологические активности относятся к ингибированию различных гепарансульфат(HS)-cвязывающих белков. Предметом настоящего изобретения является разработка производных PI-88, которые обладают аналогичными биологическими активностями, но характеризуются улучшенными свойствами, например улучшенными фармакокинетическим и/или ADME (абсорбция, распределение, метаболизм, экскрекция) профилями. Еще одним предметом настоящего изобретения является предоставление соединений, содержащих единый углеродный скелет для облегчения их синтеза и идентификации.
Сущность изобретения
В соответствии с первым вариантом осуществления изобретения, предоставлено соединение общей формулы

где Х, Y и Z, каждый представляет собой моносахаридный фрагмент, в котором к каждому атому углерода, не связывающему группы Х, Y и Z, посредством одинарной или кратной связи присоединена группа UR за исключением атома углерода в положении 1 моносахарида Z, к которому посредством одинарной или кратной связи присоединена группа UR1;
n представляет собой целое число и принимает значения от 0 до 6;
каждый U независимо представляет собой атомы С, N, S или О или указанные атомы с более высокой степенью окисления, включая СО, СОО, NO, NO2, S(O), S(O)O;
каждый R независимо представляет собой SO3M или Н, где М представляет собой любой фармацевтически приемлемый катион или любую алкильную, арильную, ацильную, ароильную, алкилсульфонильную, арилсульфонильную группу, группу PEG или ее производную группу, Н или группу

где независимо в каждой АВ группе А представляет собой О или NH, В представляет собой Н или М, где М принимает значения, определенные выше, или алкил, арил или любую другую подходящую группу;
R1 представляет собой SO3M, H, алкил, арил, ацил, ароил, алкилсульфонил, арилсульфонил, PEG или PEG-производное, или R1 вместе с U представляет собой N3 или замещенный триазол или его производное, замещенный тетразол или его производное, замещенный арил или его производное или замещенный гетероарил или его производное;
при условии, что когда все UR1 и UR группы представляют собой OSO3M или OH (за исключением экзоциклической метиленовой группы моносахаридного Х), экзоциклическая метиленовая группа моносахарида Х не может представлять собой OSO3M2 группу.
В соответствии со вторым вариантом осуществления изобретения, предоставлена фармацевтическая или ветеринарная композиция для профилактики или лечения расстройства у млекопитающего субъекта, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенного уровня содержания в крови триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания, указанная композиция включает, по меньшей мере, одно соединение согласно первому варианту осуществления изобретения вместе с фармацевтически или ветеринарно приемлемым носителем или разбавителем для указанного, по меньшей мере, одного соединения.
Третий вариант осуществления настоящего изобретения включает применение соединения согласно первому варианту осуществления изобретения для производства лекарственного средства для профилактики или лечения расстройства у млекопитающего субъекта, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенного уровня содержания в крови триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания.
В соответствии с четвертым вариантом осуществления изобретения, предоставлен способ профилактики или лечения расстройства млекопитающего субъекта, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенных уровней содержания в крови триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания, указанный способ включает введение субъекту эффективного количества, по меньшей мере, одного соединения согласно первому варианту осуществления изобретения или композиции, включающей, по меньшей мере, одно соединение.
Дополнительный вариант осуществления изобретения включает новые промежуточные продукты и путь синтеза, который приводит к получению сульфатированных олигосахаридов согласно первому варианту осуществления изобретения.
Предпочтительные соединения согласно изобретению, в которых моносахаридные молекулы представляет собой исключительно D-маннозу, и гликозидные мостики представляют собой α(1→2) и α(1→3), представлены на следующей структуре:
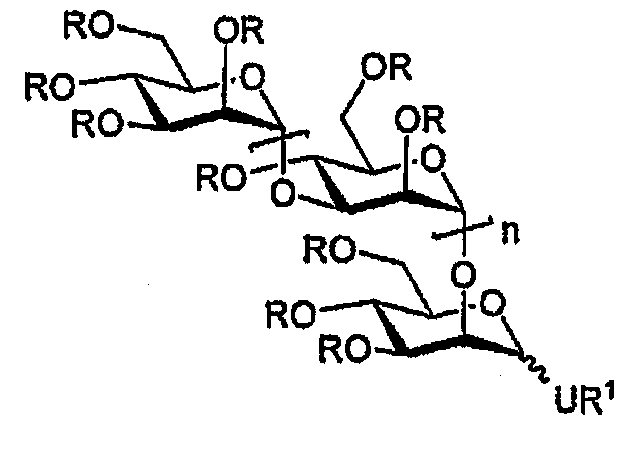
где R, R1, U и n принимают значения, определенные выше.
Для простоты понимания настоящего изобретения и внедрения изобретения в практику далее будут описаны, только в качестве примеров, один или несколько предпочтительных вариантов его осуществления со ссылкой на прилагающуюся фигуру.
Краткое описание чертежей
На чертеже проиллюстрировано влияние PI-88-подобных соединений на способности заражения HSV-1 (А) и распространение HSV-2 от клетки к клетке (В). На графике А результаты представлены в виде количества колониеобразующих вирусных единиц (PFU), сформировавшихся в клетках, зараженных обработанными соединением вирионами, которое выражено в процентах от количества, полученного в контроле, обработанном имитатором. На графике В результаты представлены в виде средней площади 20 вирусных бляшек, которые сформировались при длительном присутствии соединения, выраженной в процентах от площади бляшек в клетках, обработанных в холостом опыте.
Подробное описание предпочтительных вариантов осуществления изобретения
Авторами настоящего изобретения было обнаружено, что большое количество сульфатированных производных олигосахаридов могут быть синтезированы в соответствии с рядом различных стратегий, которые в общих чертах описаны ниже и проиллюстрированы в примерах. Данные соединения могут применяться для профилактики или лечения заболевания млекопитающих субъектов, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенного уровня содержания в крови триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания. Возможность такого применения является результатом способности соединений блокировать связывание гепарансульфат-связывающих белков с их рецепторами или ингибировать активность ферментной гепараназы.
Что касается рассматриваемых соединений формулы II, моносахаридные фрагменты Х, Y и Z могут представлять собой, например, любую гексозу или пентозу и могут являться D- или L изомером. Такие гексозы включают глюкозу, маннозу, алтрозу, аллозу, талозу, галактозу, идозу и галозу. Такие пентозы включают рибозу, арабинозу, ксилозу и ликсозу. Гликозидные мостики, связывающие моносахаридные фрагменты, по конфигурации и типу мостиковой связи могут быть исключительно одного типа или различных типов.
Фармацевтически приемлемый катион М предпочтительно представляет собой натрий.
Что касается целого числа n, его предпочтительное значение равно 3, так как оно обеспечивает соединение, которое представляет собой пентасахарид.
Предпочтительной подходящей группой R1 является н-октил.
Аномерная конфигурация при UR1 соединений формулы II, когда это применимо, может быть α- или β-конфигурацией или представлять собой аномерную α/β смесь.
Что касается заместителей, приведенных выше в определении соединений формулы II, термин «алкил», когда используется сам по себе или в сложных словах, таких как «арилалкил», относится к углеводородной группе с прямой цепью, разветвленной или циклической, предпочтительно C1-20, такой как C1-10. Например, термин «C1-C6 алкил» относится к углеводородной группе с прямой, разветвленной или циклической цепью, содержащей от 1 до 6 атомов углерода. Примеры «C1-6 алкила» включают метил, этил, изопропил, н-пропил, н-бутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2,2-диметилпропил, н-гексил, 2-метилпентил, 2,2-диметилбутил, 3-метилпентил и 2,3-диметилпропил, н-гексил, 2-метилпентил, 2,2-диметилбутил, 3-метилпентил и 2,3-диметилбутил. Примеры циклического C1-6 алкила включают циклопропил, циклобутил, циклопентил и циклогексил. Другие примеры алкила включают гептил, 5-метилгексил, 1-метилгексил, 2,2-диметилпентил, 3,3-диметилпентил, 4,4-диметилпентил, 1,2-диметилпентил, 1,3-диметилпентил, 1,4-диметилпентил, 1,2,3-триметилбутил, 1,1,2-триметилбутил, 1,1,3-триметилбутил, октил, 6-метилгептил, 1-метилгептил, 1,1,3,3-тетраметилбутил, нонил, 1-, 2-, 3-, 4-, 5-, 6- или 7-метилоктил, 1-, 2-, 3-, 4- или 5-этилгептил, 1-, 2- или 3-пропилгексил, децил, 1-, 2-, 3-, 4-, 5-, 6-, 7- и 8- метилнонил, 1-, 2-, 3-, 4-, 5- или 6-этилоктил, 1-, 2-, 3- или 4-пропилгептил, ундецил, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-метилдецил, 1-, 2-, 3-, 4-, 5-, 6- или 7-этилнонил, 1-, 2-, 3-, 4- или 5-пропилоктил, 1-, 2- или 3-бутилгептил, 1-пентилгексил, додецил, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8,- 9- или 10-метилундецил, 1-, 2-, 3-, 4-, 5-, 6-, 7- или 8-этилдецил, 1-, 2-, 3-, 4-, 5- или 6-пропилнонил, 1-, 2-, 3- или 4-бутилоктил, 1-2-пентилгептил и т.п. Алкильная группа может быть необязательно замещенной одним или несколькими необязательными заместителями, которые определены в описании. Необязательно, прямая разветвленная или циклическая углеводородная группа (содержащая, по меньшей мере, 2 атома углерода) может содержать ненасыщенную связь первой, второй или более высокой степени ненасыщения, что приводит к получению алкенильной или алкинильной группы, предпочтительно C2-20 алкенила, более предпочтительно C2-6 алкенила, или C2-20 алкинила, более предпочтительно C2-6 алкинила. Примеры таких групп включают углеводородный остаток, содержащий одну, две или более двойных связей или одну, две или более тройных связей. Таким образом, в данном описании подразумевается, что термин «алкил» включает алкенил и алкинил.
Термин «арил», используемый сам по себе или в сложных словах, таких как «арилалкил», означает моноциклический, полициклический, сопряженный или конденсированный остатки ароматических углеводородных или ароматических гетероциклических (гетероарильных) систем, в которых один или более атомов углерода циклического углеводородного остатка замещен гетероатомом с получением ароматического остатка. Когда замещены два или более атомов углерода, два или более гетероатомов могут быть одинаковыми или разными. Подходящие гетероатомы включают атомы О, N, S и Se.
Примеры «арила» включают фенил, бифенил, трифенил, квартфенил, нафтил, тетрагидронафтил, антраценил, дигидроантраценил, бензантраценил, дибензантраценил, фенантренил, флуоренил, пиренил, иденил, азуленил, хризенил, пиридил, 4-фенилпиридил, 3-фенилпиридил, тиенил, фурил, пирролил, индолил, пиридазинил, пиразолил, пиразинил, тиазолил, пиримидинил, хинолинил, изохинолинил, бензофуранил, бензотиенил, пуринил, хиназолинил, феназинил, акридинил, беноксазолил, бензотиазолил и т.п. Предпочтительные углеводородные арильные группы включают фенил и нафтил. Предпочтительные гетероциклические арильные группы включают пиридил, тиенил, фурил, пирролил. Арильная группа может быть необязательно замещенной одним или несколькими необязательными заместителями, которые определены в описании.
Термин «ацил» относится к группе -C(O)-R, где R представляет собой алкильную или арильную группу. Примеры ацила включают алканоил с прямой или разветвленной цепью, такой как ацетил, пропаноил, бутаноил, 2-метилпропаноил, пентаноил, 2,2-диметилпропаноил, гексаноил, гептаноил, октаноил, нонаноил, деканоил, ундеканоил, додеканоил, тридеканоил, тетрадеканоил, пентадеканоил, гексадеканоил, гептадеканоил, октадеканоил, нонадеканоил и икозаноил; циклоалкилкарбонил, такой как циклопропилкарбонил, циклобутилкарбонил, циклопентилкарбонил и циклогексилкарбонил; ароил, такой как бензоил, толуоил и нафтоил; аралканоил, такой как фенилaлканоил (например, фенилацетил, фенилпропаноил, фенилбутаноил, фенилизобутиноил, фенилпентаноил и фенилгексаноил) и нафтилалканоил (например, нафтилацетил, нафтилпропаноил и нафтилбутаноил). Поскольку R группа может быть необязательно замещенной, как описано выше, подразумевается, что термин «ацил» включает необязательно замещенный ацил.
Необязательные заместители для алкила, арила или ацила включают галоген (бром, фтор, хлор, йод), гидроксильную группу, C1-6алкил (например, метил, этил, пропил (н- и изо-)), C1-6алкокси (например, метокси, этокси, пропокси (н- и изо-), бутокси (н-, втор- и трет-изомеры), нитрогруппу, аминогруппу, C1-6алкиламино (например, метиламино, этиламино, пропил(н- и изо-)амино), C1-6 диалкиламино (например, диметиламино, диэтиламино, диизопропиламино), гелогенметил (например, трифторметил, трибромметил, трихлорметил), гелогенметокси (например, трифторметокси, трибромметокси, трихлорметокси) и ацетил.
5-6-Членная гетероциклильная группа включает ароматические 5-6-членные гетероциклические группы (гетероарил), которые определены выше, и неароматические 5-6-членные гетероциклические группы, содержащие один или несколько гетероатомов (предпочтительно 1 или 2), независимо выбранные из О, N, S и Se. Примеры таких групп включают диоксанил, пиранил, тетрагидрофуранил, пиперидил, морфолино, пиперазинил, тиоморфолино и сахариды.
Степень сульфатации соединений согласно изобретению обычно составляет, по меньшей мере, 50%. То есть, по меньшей мере, 50% R групп производного олигосахарида включает SO3M. Обычно степень сульфатации составляет от 70 до 100%, предпочтительно, по меньшей мере, 90%.
PI-88 производные формулы II могут быть получены постадийным методом синтеза или исходя из готового скелета PI-88 (с использованием легко доступных соединений 8-11; см. формулу I выше) с получением их желаемых модификаций. Заявители настоящего изобретения, изучая структуры PI-88 (1) и его предшественника (2), установили, что существуют две предпочтительные реакционноспособные точки получения его производных: на восстанавливающемся конце (A) и в концевом положении 6 на невосстанавливающемся конце (B), как показано на приведенной далее структурной формуле.

SO3Na или H, R1=PO3Na2, n=0-4
Следует отметить, что ди-, три-, тетра- и пентасахаридные (или более высокие) производные могут быть получены одинаковым химическим способом. Однако пентасахаридные производные предпочтительны, поскольку они являются наиболее биологически активными [1,2,5,8,13]. Далее из полученных производных удаляются защитные группы (обычно деацетилированием с NaOMe), и полученный полиол подвергается сульфатированию с использованием сульфатирующего реагента, такого как комплекс триоксида серы и пиридина или комплекс триоксида серы и триметиламина.
Как показано выше, соединения согласно изобретению применимы в профилактике или лечении расстройства млекопитающих субъектов, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенных уровней содержания триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания. Соединения особенно применимы для лечения перечисленных выше расстройств у людей. Соединения обычно вводятся в виде компонента фармацевтической композиции, как описано далее. Как показано ниже, соединения проявляют активность, аналогичную или превосходящую активность PI-88.
Фармацевтические композиции для перорального введения могут быть представлены в форме таблеток, капсул, порошка или жидкости. Таблетка может включать твердый носитель, такой как желатин, или адъювант или инертный разбавитель. Жидкие фармацевтические композиции обычно включают жидкий носитель, такой как вода, нефтяные, животные или растительные масла, минеральное масло или синтетическое масло. В композиции может включаться физиологический раствор или гликоли, такие как этиленгликоль, пропиленгликоль или полиэтиленгликоль. Такие композиции и препараты обычно будут содержать, по меньшей мере, 0,1% мас. соединения.
Парентеральное введение включает введение следующими путями: внутривенно, кожно или подкожно, назально, внутримышечно, внутриглазным путем, трансэпителиально, интраперитонеально и местным введением. Местное введение включает чрескожное, глазное, ректальное, назальное, а также введение ингаляцией или с помощью аэрозольных устройств. Для внутривенной, кожной или подкожной инъекции или инъекции в место, где желательно лечение, активный ингредиент будет находиться в форме парентерально приемлемого водного раствора, который не содержит пирогена и характеризуется подходящими рН, изотоничностью и стабильностью. Квалифицированный специалист в данной области техники будет способен приготовить подходящие растворы, используя, например, растворы рассматриваемых соединений или их производные.
Помимо, по меньшей мере, одного соединения и носителя или разбавителя композиции согласно изобретению могут дополнительно включать фармацевтически или ветеринарно приемлемый наполнитель, буферный раствор, стабилизатор, добавку, регулирующую изотоничность, консервант или антиоксидант или любой другой материал, известный специалисту в данной области техники. Квалифицированному специалисту в данной области техники будет понятно, что эти материалы должны быть нетоксичными и не должны оказывать вредного воздействия на эффективность соединения(ий). Точная природа любой добавки будет зависеть от способа введения композиции, то есть от того, перорально или парентерально вводится композиция. Что касается буферных растворов, водные композиции обычно включают такие вещества для сохранения значения рН композиции на уровне, близком к физиологическому, или, по меньшей мере, в интервале примерно от 5,0 до 8,0.
Композиции согласно изобретению помимо, по меньшей мере, одного соединения могут также включать дополнительные активные ингредиенты. Такие ингредиенты будут выбраны, главным образом, в зависимости от их эффективности в качестве лекарственных средств, обладающих противоангиогенной, противометастазной, противовоспалительной, антикоагулянтной, противомикробной и противотромботической активностью, и лекарственных средств, применяемых при повышенном уровне содержания триглицеридов в крови, а также их эффективности в качестве лекарственных средств для лечения сердечно-сосудистого заболевания, но могут быть выбраны и в зависимости от их эффективности в отношении любого состояния, связанного с указанными заболеваниями.
Фармацевтическая или ветеринарная композиция согласно изобретению будет вводиться субъекту в профилактически эффективном или терапевтически эффективном количестве, как это необходимо в конкретной рассматриваемой ситуации. Фактическое количество, по меньшей мере, одного соединения, вводимого посредством композиции, а также дозировка и схема введения будут зависеть от природы и тяжести состояния, подлежащего лечению или требующего профилактики. Назначение лечения, такое как назначение дозировки и т.д., будет определяться лечащим врачом или ветеринаром, ответственным за медицинский уход за субъектом. Однако обычно композиции, вводимые человеку, будут включать в интервале примерно от 0,01 до 100 мг соединения на кг массы тела, более предпочтительно в интервале от примерно 0,1 до 10 мг/кг массы тела.
Соединения могут быть включены в композиции в виде их фармацевтически или ветеринарно приемлемых производных. Термин «производные» соединения, используемый в описании, включает соли, координационные комплексы с ионами металлов, такими как Mn2+ и Zn2+, сложные эфиры, такие как гидролизуемые in vivo сложные эфиры, свободные кислоты или основания, гидраты или пролекарства. Соединения, содержащие кислотные группы, такие как фосфаты или сульфаты, могут образовывать соли с щелочными или щелочно-земельными металлами, такими как Na, K, Mg и Са, с органическими аминами, такими как триэтиламин и трис(2-гидроксиэтил)амин. Соли могут также образовываться между соединениями, содержащими основные группы, такие как аминогруппы, и неорганическими кислотами, такими как соляная кислота, фосфорная кислота или серная кислота, или органическими кислотами, такими как уксусная кислота, лимонная кислота, бензойная кислота, фумаровая кислота или винная кислота. Соединения, содержащие одновременно кислотные и основные группы, могут образовывать внутренние соли.
Сложные эфиры могут быть образованы между гидроксильной группой или группой карбоновой кислоты, присутствующими в соединении, и подходящей карбоновой кислотой или спиртом в качестве второго реагента с использованием методик, которые хорошо известны квалифицированному специалисту в данной области техники.
Пролекарственные производные соединений настоящего изобретения могут подвергаться превращению in vivo или in vitro в исходные соединения. Обычно, по меньшей мере, одна из биологических активностей исходного соединения может быть подавлена в форме пролекарства соединения и может активироваться превращением пролекарства в исходное соединение или его метаболит. Примерами пролекарств являются гликолипидные производные, в которых один или несколько липидных фрагментов присутствуют в виде заместителей на фрагментах, которые высвобождают свободную форму соединения после расщепления под действием фермента, обладающего фосфолипазной активностью. Пролекарства соединений настоящего изобретения включают применение защитных групп, которые могут удаляться in vivo для высвобождения активного соединения или служат для ингибирования расщепления лекарственного средства. Подходящие защитные группы хорошо известны квалифицированному специалисту данной области техники и включают ацетатную группу.
Как показано выше, соединения согласно настоящему изобретению применимы для производства лекарственного средства для профилактики или лечения у млекопитающего субъекта расстройства, которое является результатом развития кровеносных сосудов, метастаза, воспаления, коагуляции/тромбоза, повышенного уровня содержания в крови триглицеридов, заражения организма болезнетворными микроорганизмами, и/или сердечно-сосудистого заболевания. Способы производства таких лекарственных средств известны квалифицированному специалисту данной области техники и включают способы, применяемые для производства фармацевтических композиций, описанных выше.
Далее будет приведено общее описание путей синтеза соединений согласно изобретению. Для простоты во всех схемах, чертеже и таблицах, приведенных далее, за исключением особо оговоренных случаев, R1 будет представлять (1→3)-связанную Man4 тетрасахаридную часть (с концевой 6-О-фосфогруппой или без нее).
Общие методики
Гликозид-производные PI-88 (O-, S- и С-гликозиды)
Гликозид-производные могут быть легко получены активацией олигосахарида (с концевой 6-О-фосфогруппой или без нее) для гликозилирования и конденсацией его с подходящим спиртом. Подходящим способом является реакция перацетилированного сахара, например, 12, катализируемая или ускоряемая кислотой Льюиса, со спиртовым акцептором с получением, например, 13 или 14.
Схема 1
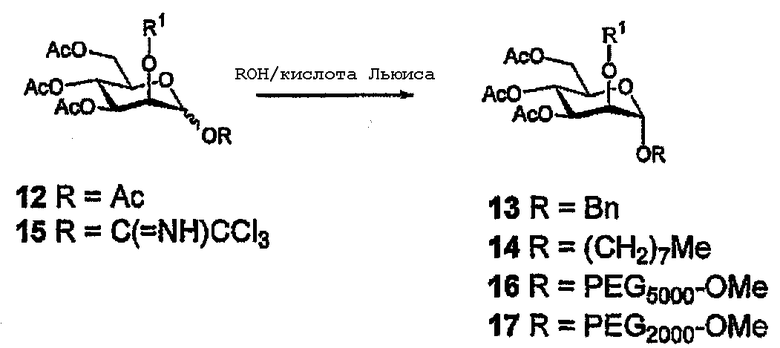
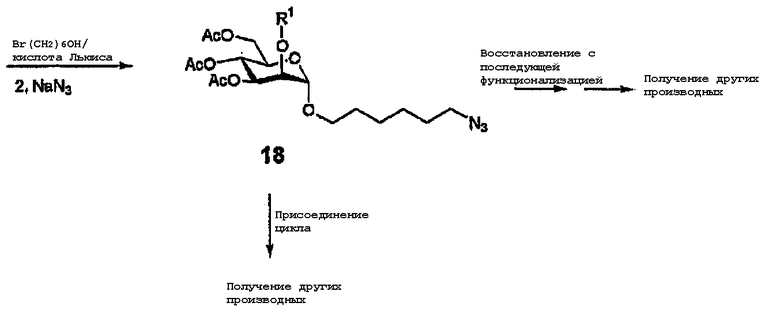
В данной области известны различные типы доноров, и подходящими в качестве доноров являются, например, тиогликозиды, галогениды, н-пентенилгликозиды, селенгликозиды и т.д. Квалифицированному специалисту данной области техники ясно, что S- и С-гликозиды могут быть получены аналогичными или сходными способами, описанными в литературе, например, с использованием подходящего тиола (или тиол-производного) или известного углеродного нуклеофила (например, аллилтриметилсилана или подходящего фенола) с приемлемо активированным донором. Продукт затем может легко деацетилироваться или сульфатироваться. Продукт гликозилирования может представлять собой единственный аномер (α или β) или смесь, содержащую оба аномера. И чистые α и β аномеры и аномерная смесь подходят для последующих превращений. Это также применимо и для других производных, получаемых в результате взаимодействия по аномерному центру, описанного в последующих разделах. Следовательно, когда обозначен единственный аномер, подразумевается, что заявляется также его антипод или смесь двух аномеров. Квалифицированному специалисту также понятно, что полученный исходный гликозид может подвергаться последующему превращению в зависимости от природы агликона. Например, если в качестве спирта используется 2-бромгексанол, продукт может подвергаться превращению в азид (18). То есть он является весьма реакционно-способным соединением (схема 2) и может далее функционализироваться, например, циклоприсоединением с соединением, содержащим подходящий диполярофил. Альтернативно, азид может подвергаться восстановлению с получением амина и затем подвергаться последующей функционализации, например, алкилированием, ацилированием, 4-компонентной конденсацией по Уги и т.д.
Схема 2
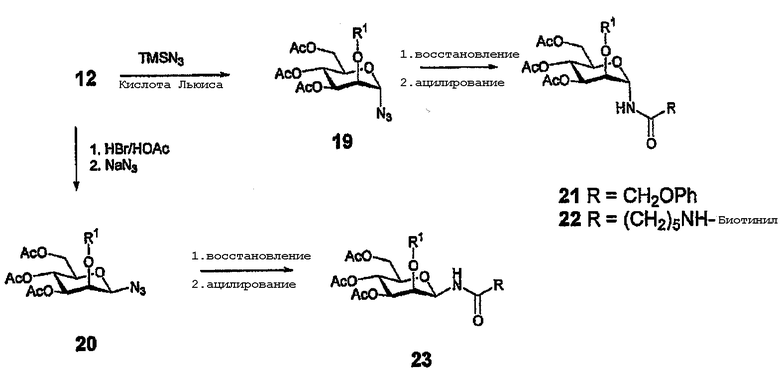
N-присоединенные производные
Катализируемое кислотой Льюиса взаимодействие соединения 12, используемого в качестве исходного соединения, с TMSN3 приводит к получению азида 19 (главным образом, α). Альтернативно, β-азид 20 может быть получен исключительно с образованием сначала α-бромида с последующим замещением NaN3 (схема 3). Бромид также может использоваться в качестве промежуточного продукта для получения, например, тиогликозидов или изотиоцианатов. Азиды могут быть подвергнуты реакции удаления защитной группы и сульфироваться как таковые или могут подвергаться восстановлению и ацилированию хлорангидами различных кислот для получения ряда гликозиламидов (схема 3).
Схема 3
Производные, полученные взаимодействием по невосстанавливающемуся концу
Получение производных может осуществляться взаимодействием по невосстанавливающему концу, например, с применением фосфорилированных олигосахаридов (отдельных или в виде смеси) и с получением производных через фосфатную группу, например, с использованием сложных фосфат-эфиров или фосфорамидов. В самом деле, подходящие соединения могут быть получены осуществлением реакции по восстанавливающему концу либо с аналогичной, либо с другой функциональной группой.
Приведенное выше описание является общим, и далее будут приведены примеры конкретных соединений, их синтеза и их биологических активностей, но без ограничения области настоящего изобретения только данными примерами.
Примеры
Нейтральные манно-олигосахариды
(а) Манно-олигосахариды: (8) α-D-Ман-(1→2)-D-Ман, (9) α-D-Ман-(1→3)-α-D-Ман-(1→2)-D-Ман, (10) α-D-Ман-(1→3)-α-D-Ман-(1→3)-α-D-Ман-(1→2)-D-ман и (11) α-D-Ман-(1→3)-α-D-Ман-(1→3)α-D-Ман-(1→3)-α-D-Ман-(1→2)-D-ман - выделяют распределительной хроматографией из нейтральной фракции, полученной в результате гидролиза внеклеточного фосфоманнана из P. holstii NRRL Y-2448, проводимого в мягких условиях с использованием в качестве катализатора кислоты в соответствии с методикой, описанной в литературе [17]. Альтернативно, олигосахариды 8-11 синтезируют постадийным способом из составляющих моносахаридных блоков, как описано в примере 1 (см. ниже).
(b) Альтернативно, нейтральную фракцию подвергают прямому ацилированию (избыток смеси Ас2О/пиридин), индивидуальные перацетилированные олигосахариды выделяют флэш-хроматографией (силикагель) и сразу используют в следующей стадии без дополнительной очистки.
(с) В соответствии с другим подходом, перацетилированную смесь со стадии (b) сразу используют в следующей стадии, а затем индивидуальные продукты затем выделяют флэш-хроматографией.
Общая методика деацетилирования
Раствор перацетата в безводном метаноле (0,1 М) обрабатывают раствором метоксида натрия в метаноле (1,35 М, 0,2-0,6 экв.). Смесь перемешивают при комнатной температуре в течение 1-3 часов (контролируя ход реакции ТСХ). Для доведения рН до 6-7 добавляют кислотную смолу AG®-50W-X8 (H+ форма), смесь фильтруют и смолу подвергают разложению метанолом. Фильтрат и промывной раствор объединяют, смесь концентрируют в вакууме и тщательно сушат, получая полиол.
Общая методика сульфирования
Смесь полиола и SO3·триметиламинового или SO3·пиридинового комплекса (2 экв. на спирт) в ДМФА нагревают (60°С, в течение ночи). Охлажденную (до комнатной температуры) смесь обрабатывают МеОН и затем подщелачивают (до рН>10) добавлением Na2CO3 (10% мас./мас.). Смесь фильтруют, и фильтрат упаривают и затем подвергают совместному выпариванию (Н2О). Сырой полисульфатированный продукт подвергают вытеснительной хроматографии. Если необходимо, после лиофилизации продукт пропускают через колонку с ионно-обменной смолой (AG®-50W-X8, Na+ форма, 1х4 см, деионизированная Н2О, 15 мл) для полного превращения продукта в форму натриевой соли. Собранный раствор упаривают и лиофилизуют с получением конечного продукта в виде бесцветного стеклообразного вещества или белого порошка.
Распределительная хроматография
Распределительную хроматографию проводят на Bio-Gel P-2 в колонке 5х100 см с расходом элюента 2,8 мл/мин 0,1М NH4 +·HCO3 -, собирая фракции 2,8 мин (7,8 мл). Фракции анализируют на содержание углевода нанесением пятна на пластины силикагеля и визуализацией с помощью обработки углем и/или анализируют на полизаряженные виды пробой диметилметиленовым синим. И, наконец, фракции проверяют на чистоту с помощью СЕ15, и фракции, которые свободные от соли, объединяют и лиофилизуют. В случаях, когда присутствуют несульфатированные побочные продукты или другие органические соли (обычно лишь в небольших количествах, но, тем не менее, в действительности часто обнаруживаемые), для полного их удаления используют LH20 колоночную хроматографию (2х95 см,1,2 мл/мин, 3,5 мин на пробирку).
Пример 1: Общий синтез нейтральных манноолигосахаридов (8-11) из Pichia
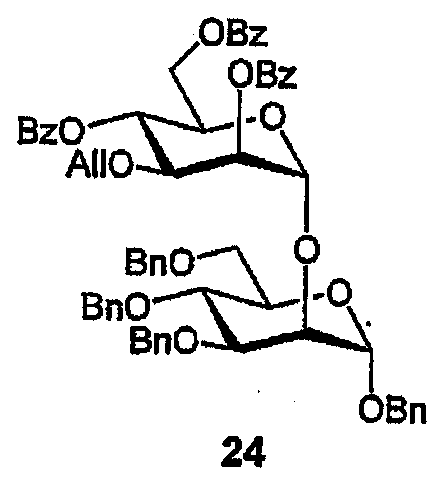
Бензил-2-О-(3-О-Аллил-2,4,6-три-О-бензоил-α-D-маннопиранозил)-3,4,6-три-О-бензил-α-D-маннопиранозид (24)
Смесь 3-O-аллил-2,4,6-три-O-бензоил-α-D-маннопиранозилтрихлорацетимидата [26] (902 мл, 1,21 ммоль) и бензил-3,4,6-три-O-бензил-α-D-маннопиранозида [27] (723 мл, 1,34 ммоль) в 1,2-ДХЭ (10 мл) перемешивают в присутствии молекулярных сит (1,0 г 3Е порошка) в атмосфере аргона (30 мин). Смесь охлаждают (0°C) при непрерывном перемешивании (10 мин) перед добавлением TMSOTf (219 мкл, 1,21 ммоль). По истечении некоторого времени (10 мин) добавляют Et3N (100 мкл) и полученную смесь фильтруют. Растворитель выпаривают и остаток очищают ФХ (10-50% EtOAc/гексан), получая трибензоат (24) в виде бесцветного масла (1,14 г, 84%). 1H ЯМР (CDCl3) δ 3,67-3,81, 3,88-3,95, 4,06-4,15, 4,30-4,35 (4м, 12H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,OCH2), 4,94-4,70 (м, 7H; CH2Ph), 4,84 (д, 1H, JA,B 10,8 Hz; A AB квартета), 4,93-4,96, 5,04-5,09 (2м, 2H; =CH2), 5,02 (д, 1H, J1,2 1,9 Hz; H-1I), 5,24 (д, 1H; J1,2 1,9 Ha; H-1II), 5,59-5,69 (м, 1H; =CH), 5,72 (дд, 1H, J2,3 3,1 Hz; H-2II), 5,75 (дд, 1H, J3,4 9,8, J4,5 9,9 Hz; H-4II), 7,09-7,58, 7,97-8,06 (2м, 35 H; Ar). 13C ЯМР (CDCl3) δ 61,50, 63,49 (2C; C-6I,-6II), 68,63, 69,17, 69,31, 69,46, 69,64, 71,08, 72,04, 72,64, 73,60, 74,73, 75,30, 75,38, (13 C; C-3I,-4I,-5I,-2II,-3II,-4II,-5II,OCH2, CH2Ph), 79,97 (C-2I), 98,52, 99,60 (C-1I,-1II), 117,67 (=CH2), 127,70-138,43 (43 C; =CH, Ar), 165,61, 165,69, 166,42 (3 C; C=O).
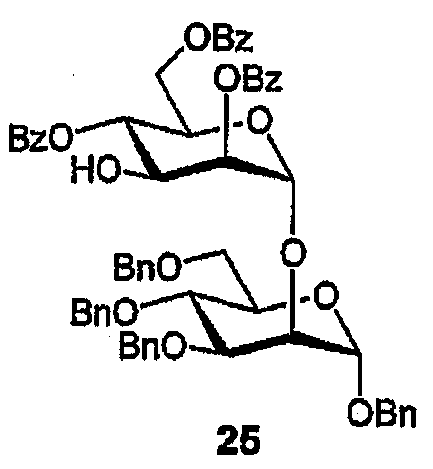
Бензил-2-О-(2,4,6-три-О-бензоил-α-D-маннопиранозил)-3,4,6-три-О-бензил-α-D-маннопиранозид (25)
PdCl2 (40 мл) добавляют к раствору аллилового эфира (24) (1,09 г, 0,97 ммоль) в MeOH (10 мл) и 1,2-ДХЭ (10 мл) и полученную смесь нагревают (70°С, 40 мин). По истечении указанного времени растворители выпаривают, и остаток очищают ФХ (20-30% EtOAc/гексаны) с получением спирта (25) в виде бесцветного масла (0,96 г, 91%). 1H ЯМР (CDCl3) δ 3,68-3,81, 3,97-4,06, 4,32-4,71, (3м, 18H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,CH2Ph), 4,84 (д, 1H, JA,B 12 Hz; A AB квартета), 5,05 (д, 1H, J1,2 1,9 Hz; H-1I), 5,26 (д, 1H; J1,2 1,9 Ha; H-1II), 5,61 (дд, 1H, J2,3 3,3 Hz; H-2II), 5,67 (дд, 1H, J3,4 9,8, J4,5 9,9 Hz; H-4II), 7,13-7,40, 7,48-7,59, 7,98-8,06 (3м, 35 H; Ar). 13C ЯМР (CDCl3) δ 60,61, 63,32 (2C; C-6I,-6II), 69,06, 69,12, 69,25, 69,44, 70,45, 72,14, 72,65, 72,77, 73,48, 74,79, 75,48, 75,47, 76,23 (13 C; C-3I,-4I,-5I,-2II,-3II,-4II,-5II,OCH2, CH2Ph), 79,66 (C-2I), 98,34, 99,40 (C-1I,-1II), 127,70-138,47 (42 C; Ar), 165,97, 166,36, 166,97 (3 C; C=O).

Бензил-2-O-[(3-О-аллил-2,4,6-три-O-бензоил-α-D-маннопиранозил)-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-3,4,6-три-O-бензил-α-D-маннопиранозид (26)
Смесь 3-О-аллил-2,4,6-три-О-бензоил-α-D-маннопиранозилтрихлорацетимидата (742 мл, 1,01 ммоль) и спирта (25) (908 мл, 0,84 ммоль) в 1,2-ДХЭ (10 мл) перемешивают в присутствии молекулярных сит (1,0 г 3Е порошка) в атмосфере аргона (30 мин). Смесь охлаждают (0°C) при непрерывном перемешивании (10 мин) перед добавлением TMSOTf (181 мкл, 1,01 ммоль). По истечении некоторого времени (10 мин) вводят Et3N (100 мкл) и полученную смесь фильтруют. Растворитель выпаривают, и остаток очищают ФХ (10-50% EtOAc/гексан), получая гексабензоат (26) в виде бесцветного масла (1,26 г, 90%). 1H ЯМР (CDCl3) δ 3,51-3,56, 3,66-4,06, 4,23-4,27, 4,30-4,42, 4,47-4,72, 4,78-4,86 (6м, 26H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,-3III,-5III,-6aIII,-6bIII, OCH2, =CH2, CH2Ph), 5,04 (д, 1H, J1,2 1,7 Hz; H-1I), 5,15 (дд, 1H, J1,2 1,8, J2,3 2,7 Hz; H-2II), 5,26 (д, 1H; H-1 II), 5,28 (д, 1H, J1,2 1,7 Hz; H-1III), 5,33-5,43 (м, 1H; =CH), 5,77-5,82 (м, 2H; H-4II,-2III), 5,92 (дд, 1H, J3,4 9,5, J4,5 9,8 Hz; H-4III), 7,00-7,61, 7,80-8,19 (2м, 50H; Ar).

Бензил-2-О-[(2,4,6-три-O-бензоил-α-D-маннопиранозил)-(1→3)-(2,4,6-три-O-бензоил-α-D-маннопиранозил)]-3,4,6-три-O-бензил-α-D-маннопиранозид (27)
PdCl2 (40 мл) добавляют к раствору простого аллилового эфира (26) (394 мл, 241 мкмоль) в MeOH (10 мл) и 1,2-ДХЭ (10 мл), и полученную смесь нагревают (70°С, 60 мин). По истечении указанного времени растворители выпаривают, и остаток очищают ФХ (20-30% EtOAc/гексаны), получая спирт (27) в виде бесцветного масла (317 мл, 84%). 1H ЯМР (CDCl3) δ 3,67-3,82, 3,91-3,99, 4,01-4,21, 4,29-4,71 (4м, 21H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,-3III,-5III,-6aIII,-6bIII, CH2Ph), 4,83 (д, 1H, JA,B 10,9 Hz; A AB квартета), 5,03-5,05 (м, 2H; H-1I,-2II), 5,25-5,28 (м, 2H; H-1II,-1III), 5,63 (дд, 1H, J3,4=J4,5 9,9 Hz; H-4II), 5,77 (дд, 1H, J1,2 2,0, J2,3 3,1 Hz; H-2III), 5,92 (дд, 1H, J3,4 9,7, J4,5 9,9 Hz; H-4III), 6,99-7,62, 7,80-8,16 (2м, 50H; Ar).
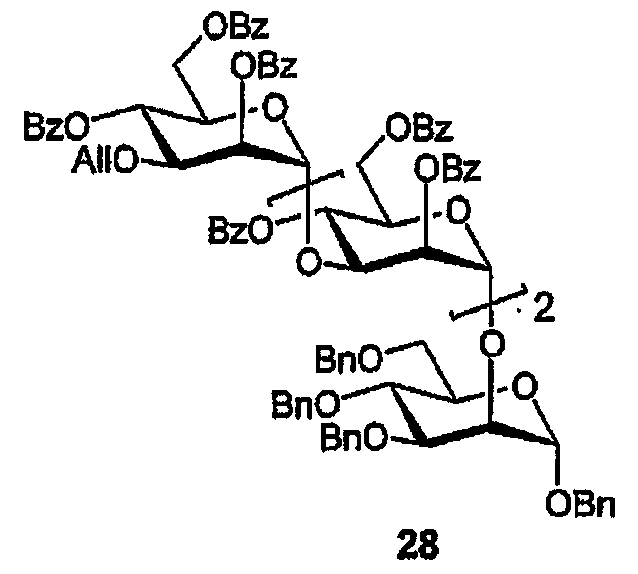
Бензил-2-О-[(3-О-аллил-2,4,6-три-О-бензоил-α-D-маннопиранозил)-(1→3)-(2,4,6-три-О- бензоил-α-D-маннопиранозил)]-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-3,4,6-три-О-бензил-α-D-маннопиранозид (28)
Смесь 3-O-аллил-2,4,6-три-O-бензоил-α-D-маннопиранозилтрихлорацетимидата (102 мл, 138 мкмоль) и спирта (27) (135 мл, 86,5 мкмоль) в 1,2-ДХЭ (6 мл) перемешивают в присутствии молекулярных сит (100 мг 3Е порошка) в атмосфере аргона (30 мин). Смесь охлаждают (0°) при непрерывном перемешивании (10 мин) перед добавлением TMSOTf (25 мкл, 138 мкмоль). По истечении некоторого времени (10 мин) вводят Et3N (100 мкл), и смесь фильтруют. Растворитель выпаривают, и остаток очищают ФХ (10-50% EtOAc/гексан), получая нонабензоат (28) в виде бесцветного масла (173 мл, 94%). 1H ЯМР (CDCl3) δ 3,44-3,49, 3,60-3,99, 4,05-4,16, 4,42-4,44, 4,48-4,68, 4,73-4,77 (6м, 30H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,-3III,-5III,-6aIII,-6bIII,-3IV,-5IV,-6aIV,-6bIV, OCH2, =CH2 CH2Ph), 4,83 (д, 1H, JA,B 10,9 Hz; A AB квартета), 5,01-5,04 (м, 2H; H-1I,-2III), 5,19-5,23 (м, 1H; H-2II), 5,27-5,40 (м, 4H; H-1I,-1II,-1III, =CH2), 5,61 (дд, 1H, J3,4=4,5 9,9 Hz; H-4IV), 5,77 (дд, 1H, J1,2 2,0, J2,3 3,1 Hz; H-2IV), 5,90-5,96 (м, 2H; H-4II,-4III), 7,01-7,56, 7,70-8,16 (2м, 65H; Ar).
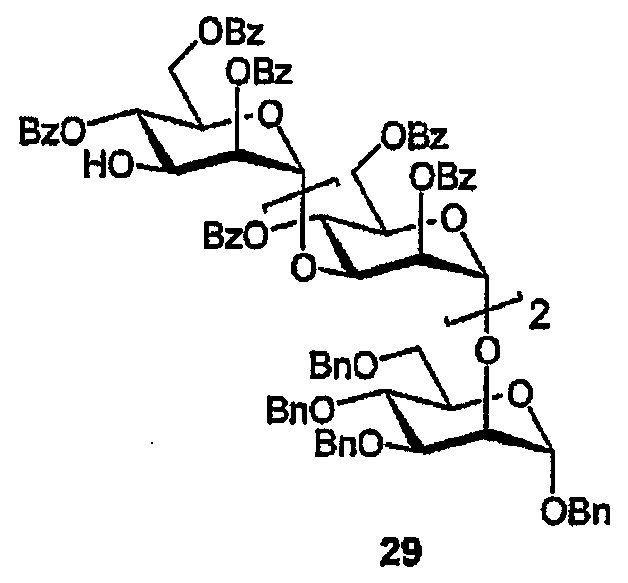
Бензил-2-О-[(2,4,6-три-O-бензоил-α-D-маннопиранозил)-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-3,4,6-три-О-бензил-α-D-маннопиранозид (29)
PdCl2 (30 мл) добавляют к раствору простого аллилового эфира (28) (155 мл, 70,4 мкмоль) в MeOH (5 мл) и 1,2-ДХЭ (5 мл), и полученную смесь нагревают (70°С, 40 мин). По истечении указанного времени растворители выпаривают, и остаток очищают ФХ (20-40% EtOAc/гексаны), получая спирт (29) в виде бесцветного масла (97 мл, 64%). 1H ЯМР (CDCl3) δ 3,67-3,82, 3,90-4,10, 4,24-4,68 (3м, 26H; H-2I,-3I,-4I,-5I,-6aI,-6bI,-3II,-5II,-6aII,-6bII,-3III,-5III,-6aIII,-6bIII,-3IV,-5IV,-6aIV,-6bIV, CH2Ph), 4,84 (д, 1H, JA,B 11,2 Hz; A of AB квартет), 4,86 (д, J1,2 1,8 Hz; H-1I), 4,90 (дд, 1H, J1,2 1,8, J2,3 3,1 Hz; H-2III), 5,03 (д, 1H, J1,2 1,5 Hz; H-1IV), 5,22 (дд, 1H, J1,2 2,1, J2,3 2,6 Hz; H-2II), 5,27-5,29 (м, 2H; H-1III,-1IV), 5,46 (дд, 1H, J3,4 9,7, J4,5 9,9 Hz; H-4IV), 5,79 (дд, 1H, J2,3 2,9, Hz; H-2IV), 5,90-5,96 (м, 2H; H-4II,-4III), 7,01-7,56, 7,68-8,16 (2м, 65H; Ar).
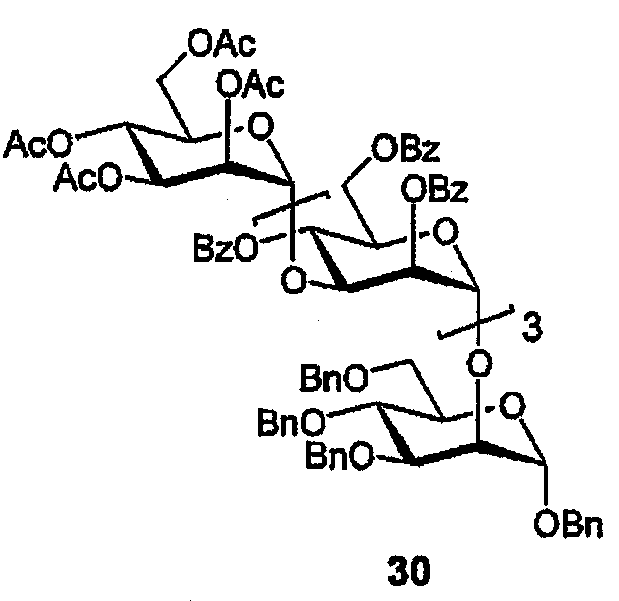
Бензил-2-О-[(2,3,4,6-тетра-О-ацетил-α-D-маннопиранозил)-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-(1→3)-(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-(1→3)(2,4,6-три-О-бензоил-α-D-маннопиранозил)]-3,4,6-три-О-бензил-α-D-маннопиранозид (30)
Смесь 2,3,4,6-тетра-O-ацетил-α-D-маннопиранозилтрихлорацетимидата [28] (39 мл, 78 мкмоль) и спирта (29) (85 мл, 39 мкмоль) в 1,2-ДХЭ (3 мл) перемешивают в присутствии молекулярных сит (100 мг 3Е порошка) в атмосфере аргона (30 мин). Смесь охлаждают (0°С) при непрерывном перемешивании (10 мин) перед добавлением TMSOTf (14,2 мкл, 78 мкмоль). По истечении некоторого времени (30 мин) вводят Et3N (100 мкл) и полученную смесь фильтруют. Растворитель выпаривают и остаток очищают ФХ (30-60% EtOAc/гексан), получая тетраацетат (30) в виде бесцветного масла (85 мл, 87%). 1H ЯМР (CDCl3) δ 1,82-2,04 (4s, 3H each; CH3CO), 3,67-3,95, 4,05-4,72, 4,82-5,03, 5,21-5,28, 5,69-5,50 (м, 43H; H-1I-IV,-2I-IV,-3I-IV,-4I-IV,-5I-IV,-6abI-IV, CH2Ph), 7,01-7,56, 7,68-8,16 (2м, 65H; Ar).
Общая методика удаления защитных групп из манноолигосахаридов
(А) Небольшой кусочек натрия добавляют к раствору простого тетрабензилового эфира (25, 27, 29, 30) в МеОН и ТГФ и полученную смесь перемешивают (при комнатной температуре в течение ночи). По истечении указанного времени смесь нейтрализуют добавлением смолы Dowex 50X8 в форме (Н+) и фильтруют. Растворитель выпаривают, остаток подвергают совместному выпариванию (МеОН) и используют в следующей реакции без дополнительной очистки.
(В) Pd(OH)2 (10% на C) добавляют к раствору сырого продукта со стадии (A) в ТГФ и Н2О, содержащему небольшое количество АсОН (50 мкл), и объединенную смесь энергично перемешивают в атмосфере водорода (100 фунтов на кв. дюйм, 3 часа). По истечении указанного времени смесь фильтруют и растворитель выпаривают. Остаток очищают вытеснительной хроматографией (Biogel P2; H2O; 60 мл/час), получая после лиофилирования манноолигосахарид (8-11) в виде бесцветного порошка. Соединения 8-11 идентичны во всех аспектах веществам, полученным в результате гидролиза Pichia, как описано выше.
Пример 2: Полисульфат бензилгликозида(PG500)
Перацетат 12
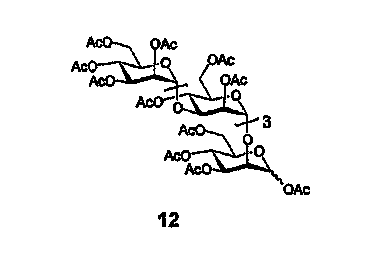
Пентасахарид 11 (1,03 г, 95% М5), ацетат натрия (1,2 г) и уксусный ангидрид (50 мл) нагревают при перемешивании до 140°С и перемешивают при указанной температуре в течение ночи в сушильном шкафу. Смесь охлаждают до комнатной температуры, упаривают досуха, остаток переносят в EtOAc, промывают насыщенным раствором соли (х3), очищают флэш-хроматографией (40 г силикагеля, 80:20 EtOAc:гексан), получая 810 мг перацетата 12 в виде стекловидного вещества с примесями менее чистого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6,14 (д, 0,84H, J=2,0, αHII), 5,71 (д, 0,16H, J=0,9, βHII), 5,30-5,10 (м, 8H), 5,00-4,85 (м, 7H), 4,25-3,70 (м, 19H), 2,20-1,90 (м, 51H). HRMS (масс-спектр высокого разрешения):
вычислено для C64H87O43 [M+H]+ 1543,4623, найдено 1543,4599.
Общая методика прямого гликозилирования перацетилированных олигосахаридов:
К раствору перацетата (например, 12) (1 экв.) в ДХМ (0,03 М), высушенном с помощью 3Е молекулярных сит, добавляют спирт (6 экв.). В некоторых случаях добавляют небольшое количество порошкообразных 3Е МС. Добавляют этерат трифторида бора (4 экв.) и смесь перемешивают в атмосфере аргона при 60°С или 75°С в течение периода времени в интервале от 2 до 26 часов. Смесь охлаждают и к смеси добавляют триэтиламин. Смесь разбавляют дихлорметаном, промывают насыщенным водным раствором карбоната натрия и сушат (безв. MgSO4). Высушенный раствор фильтруют и осадок на фильтре промывают дихлорметаном. Фильтрат и промывной раствор объединяют, концентрируют, загружают на силикагель и очищают флэш-хроматографией (силикагель, элюирование с градиентом, гексан-EtOAc от 6:1 до 1:4), получая после упаривания и сушки в высоком вакууме целевой гликозид.
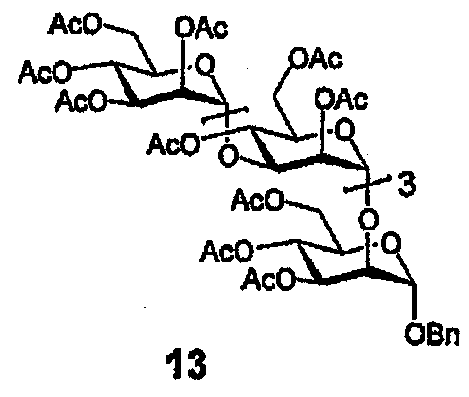
Бензилгликозид 13
Гликозилирование проводят с использованием 12 и бензилового спирта с получением продукта (13) в виде бесцветной смолы: 108 мг, 46% (Rf=0,32, гексан-EtOAc=1:3). 1H ЯМР (CDCl3, 400 МГц) δ 7,35-7,27 (м, 5H, C6H5), 5,30-5,12 (м, 8H), 5,00-4,85 (м, 8H), 4,68 (AB квартет, 1H, J=11,8) и 4,50 (AB квартет, 1H, J=11,8, PhCH2O), 4,27-3,74 (м, 19H), 2,14(4), 2,13(5), 2,13, 2,10, 2,08(4), 2,07(9), 2,07(6), 2,06(9), 2,06(6), 2,06(2x), 2,02, 2,00, 1,99, 1,97, 1,94 (15с, 48H, 16 x Ac); 13C ЯМР (CDCl3, 100 МГц) δ 171,0, 170,5(3), 170,5(1), 170,5(0), 170,4, 170,3, 170,2, 170,0(4), 170,0(2), 169,8(9), 169,8(8), 169,7, 169,6, 169,5(6), 169,4(6) и 169,3 (общ. 16 x CO), 136,1 (ipso-C6H5), 128,5, 128,2 и 127,9 (o, m, p-C6H5), 99,2 (2C), 98,9, 98,8, 97,3 (5 х сахар-Cl), 76,7, 75,1, 74,9(9), 74,9(7), 71,1, 70,9, 70,8, 70,2, 69,7, 69,5(9), 69,5(6), 69,4(2), 69,3(7), 69,2, 68,6, 68,3, 67,1, 66,7(3), 66,6(7), 66,1, 65,5, 62,4, 62,1, 61,9, 61,6, и 60,2 (26С, 25 атомов С сахара минус 5 атомов С-Cl сахара и 6 атомов С СН2-групп бензила), 20,9, 20,8(2), 20,8(0), 20,7(8), 20,7, 20,6, 20,5(4), 20,5(1), 20,4(9) и 20,4(6) (10C, 16 x Ac).
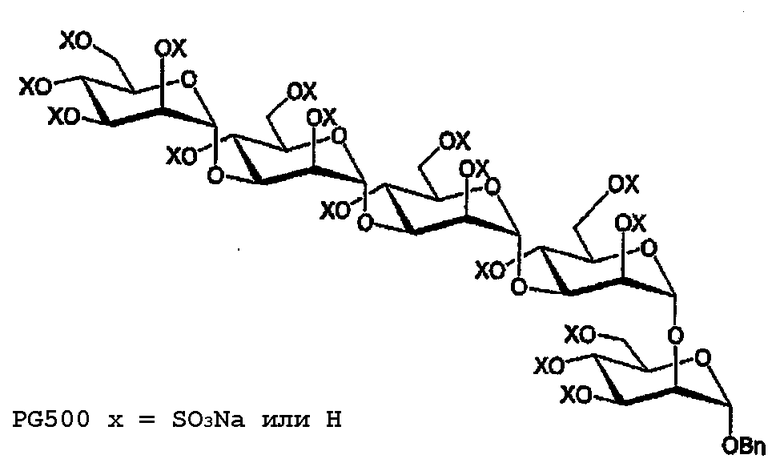
Полисульфат бензилгликозида (PG500)
Cоединение 13 деацетилируют (HRMS: вычислено для полиола C37H59O26 [M+H]+ 919,3296, найдено 919,3279) и сульфируют в соответствии с общими методиками с получением продукта (PG500) в виде белого порошка, 76,1 мг, 44%. 1H ЯМР (D2O, 400 МГц) δ 7,35-7,26 (м, 5H, C6H5), 5,32 (с, 1H), 5,30 (д, 1H, J=1,2), 5,26 (д, 1H, J=2,0), 5,24 (д, 1H, J=1,6), 5,05 (дд, 1H, J=2,8, 2,0), 5,00 (д, 1H, J=2,0), 4,87-4,85 (м, 2H), 4,68-4,34 (м, 12H), 4,32-3,86 (м, 17H); 13C ЯМР (D2O, 100 МГц) δ 137,0, 129,5, 129.4, 129,1, 100,5(9), 100,5(6), 100,2, 97,9, 93,8, 76,9, 76,8, 75,6, 75,5(3), 75,4(8), 74,4, 73,8, 73,1, 73,0, 72,8, 72,7, 71,8, 71,3, 70,7, 70,6, 70,4, 69,9, 69,8, 69,7, 68,0, 67,8, 67,5, 66,6, 66,3(7), 66,3(5).
Пример 3: Полисульфат октилгликозида (PG501)
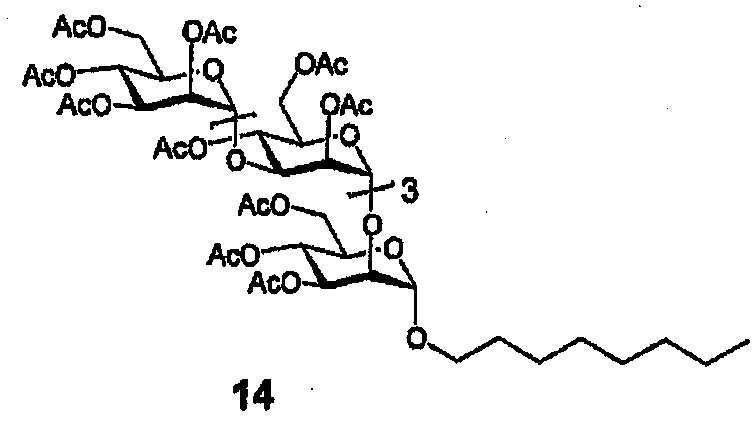
Октилгликозид 14
Гликозилирование проводят с использованием 12 и октанола с получением продукта (14) в виде бесцветной смолы, 207 мг, 66% (Rf=0,41, гексан-EtOAc=1:3). 1H ЯМР (CDCl3, 400 МГц) δ 5,23-5,09 (м, 8H), 4,96-4,82 (м, 8H), 4,23-3,71 (м, 19H), 3,59 (дт, 1H, J=9,4, 6,8, OCH2R), 3,35 (дт, 1H, J=9,4, 6,8, OCH2R), 2,11, 2,10(2), 2,09(8), 2,06, 2,05, 2,04(4), 2,04(1), 2,03(8), 2,03, 2,02, 2,01, 1,99(3), 1,98(8), 1,96, 1,94 и 1,90 (16с, 48H, 16 x Ac), 1,52 (квинтет, 2H, J=7,2, CH2), 1,27-1,18 (м, 10H, (CH2)5), 0,80 (т, 3H, J=7,2, CH3); 13C ЯМР (CDCl3, 100 МГц) δ 170,4(0) (2C), 170,3(8) (2C), 170,3, 170,2, 170,1, 169,9 (2C), 169,8(2), 169,7(5), 169,6, 169,5, 169,4(4), 169,3(5), 169,3 (16 x CO, 3 наложенные), 99,1 (2C), 98,8, 98,7, 98,0 (5 x сахар-Cl), 77,0, 75,0, 74,8(3), 74,7(5), 71,0, 70,8, 70,7, 70,1, 69,4(9), 69,4(7), 69,3(0), 69,2(7), 69,2, 68,3, 68,2(0), 68,1(6), 67,2, 66,6(4), 66,6(0),66,1, 65,4, 62,4, 62,3, 61,8 и 61,5 (25 атомов С сахара минус 5 атомов С-Cl сахара и атомы С октил-СН2О), 31,5, 29,1, 29,0, 28,9, 25,9, 22,4 (6 x октил-CH2), 20,7(3), 20,7(0), 20,6(7), 20,6, 20,5, 20,4(3), 20,4(0), 20,3(9), 20,3(7) (9C, 16 x Ac), 13,85 (октил-CH3).
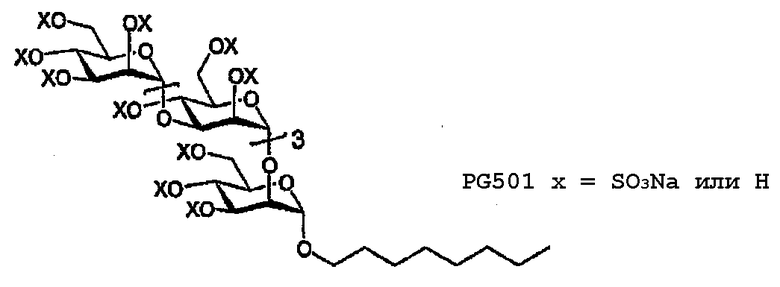
Соединение 14 деацетилируют (HRMS: вычислено для полиола C38H69O26 [M+H]+ 941,40784, найдено 941,4060) и сульфируют в соответствии с общей методикой с получением продукта (PG501) в виде белого порошка, 195 мг, 72%. 1H ЯМР (D2O, 400 МГц) δ 5,33 (с, 1H), 5,29 (д, 1H, J=1,6), 5,24 (д, 1H, J=1,6), 5,21 (д, 1H, J=1,6), 5,03 (дд, 1H, J=2,8, 2,0), 4,87 (д, 1H, J=1,6), 4,86-4,83 (м, 2H), 4,70-3,92 (м, 27H), 3,59 (дт, 1H, J=9,6, 7,0), 3,44 (дт, 1H, J=9,6, 7,0), 1,48-1,40 (м, 2H), 1,21-1,08 (м, 10H), 0,678 (т, 3H, J=7,2); 13C ЯМР (D2O, 100 МГц) δ 100,5, 100,4, 100,1, 100,0, 99,0, 98,4(1), 98,3(8), 98,3(6), 98,3(5), 76,8(5), 76,7(9), 76,7, 76,6, 76,5(2), 76,4(7), 76,0, 75,4(0), 75,3(5), 75,3, 75,2, 74,3, 73,0(5), 72,9(9), 72,7, 72,6, 71,7, 70,4, 70,2, 69,8(4), 69,7(5), 69,6, 69,1, 67,8(5), 67,7(7), 66,5, 66,2, 31,5, 30,0, 28,8, 25,8, 22,5, 14,0.
Пример 4: PEG 5000 -полисульфат (PG504)
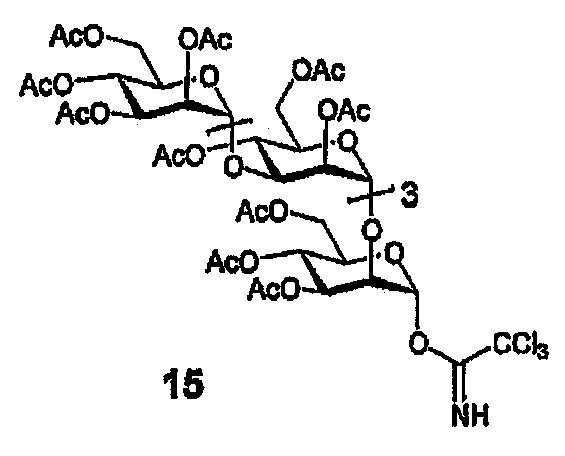
Имидат 15
(А) Смесь ацетата (12) (68 мг, 51 мкмоль) и BnNH2 (17 мкл, 152 мкмоль) в ТГФ (2 мл) перемешивают (при комнатной температуре) в течение некоторого времени (2 дня). Смесь разбавляют СНCl3 (20 мкл) и подвергают дальнейшей обработке. Органическую фазу выпаривают, затем подвергают совместному испарению (2х10 мл MeCN) и используют в следующей реакции без дополнитеьной очистки.
(В) К раствору сырого продукта (со стадии А) и трихлорацетонитрила (1,0 мл, 10 ммоль) в 1,2-ДХЭ (4 мл) добавляют DBU (10 мкл, 6,7 мкмоль), и полученную смесь перемешивают (0°С→12°С, в течение ночи). Смесь концентрируют, и остаток очищают ФХ (50-90% EtOAc/гексаны), получая 15 в виде масла бледно-желтого цвета (35 мг, 48%, 2 стадии). 1H ЯМР (400 МГц, CDCl3) δ 8,70 (с, 1H, NH), 6,32 (д, 1H, J=2,0, HII), 5,36-5,13 (м, 8H), 5,00-4,90 (м, 6H), 4,26-3,75 (м, 20H), 2,15-1,94 (м, 48H).
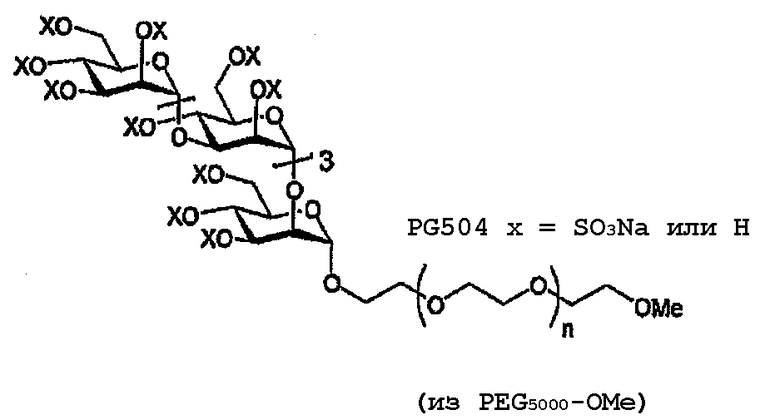
PEG 5000 -полисульфат (PG504)
(А) Смесь имидата 15 (33 мг, 20,2 мкмоль) и PЕG5000-монометилового эфира (151 мг, 30,3 мкмоль) в 1,2-ДХЭ (3 мл) перемешивают в присутствии молекулярных сит (50 мг 3Е порошка) в атмосфере аргона (10 мин). Смесь охлаждают (-20°С) при непрерывном перемешивании (10 мин) перед добавлением TMSOTf (5 мкл, 2,8 мкмоль). По истечении некоторого времени (20 мин) вводят Et3N (10 мкл) и смесь фильтруют. Растворитель выпаривают и остаток очищают ФХ (0-7,5% MeOH/CHCl3), получая 16 в виде бесцветного стеклообразного вещества (104 мг, 80% из расчета на среднюю Mr 6483). 1H ЯМР (400 МГц, CDCl3) δ 5,28-4,87 (м, 14Н), 4,43-3,42 (м, 829 Н), 3,34 (с, 3Н, ОМе), 2,15-1,94 (м, 48Н).
(В) Соединение 16 (104 мг, 16 мкмоль) деацетилируют в соответствии с общей методикой, получая Man5-PЕG5000-ОМе в виде бесцветного воскообразного вещества (82 мг, 89% из расчета на среднюю Mr 5769).
(С) Man5-PЕG5000-ОМе (82мг, 14 мкмоль) сульфируют в соответствии с общей методикой, получая PG504 в виде бесцветной пены (45 мг, 42% из расчета на среднюю Mr 7401). 1H ЯМР (400 МГц, D2O) δ 5,34-4,87 (м, 7H), 4,71-3,97 (м, 20H), 3,76-3,35 (м, 432H), 3,23 (с, 3H, OMe).
Пример 5: PЕG 5000 -полисульфат (PG506)
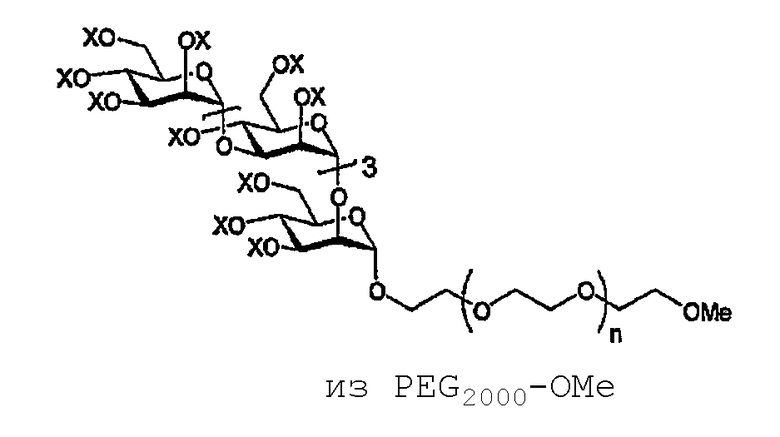
PG506 X = SO 3 Na или Н
(А) Смесь имидата (15) (60 мг, 36,5 мкмоль) и PЕG2000-ОМе (110 мг, 55,0 мкмоль) обрабатывают TMSOTf, как описано для PЕG5000-ОМе, получая соединение 17 в виде бесцветного стеклообразного вещества (96 мг, 74%). 1H ЯМР (400 МГц, CDCl3) δ 5,28-5,13, 5,00- 4,87, 4,27-3,40 (3м, множество H, Н1I-V, 2I-V, 3I-V, 4I-V, 5I-V, 6aI-V, 6bI-V, OCH2CH2O), 3,34 (с, 3H, OMe), 2,15-1,94 (16c, 3H каждый, COMe).
(D) Соединение 17 деацетилируют в соответствии с общей методикой получения полиола PЕG2000-ОМе в виде бесцветного воскообразного вещества (63 мг, 81%). Полученный остаток используют в следующей реакции без дополнительной очистки или идентификации.
(С) Продукт со стадии (В), описанной выше, сульфируют в соответствии с общей методикой с получением указанного в заголовке соединения (PG506) в виде бесцветного порошка (47 мг, 68%). 1H ЯМР (400 МГц, D2O) δ 5,34-3,97 (м, 498Н), 3,80-3,35 (м, 81Н), 3,23 (с, 3Н, ОМе).
Пример 6: PG502
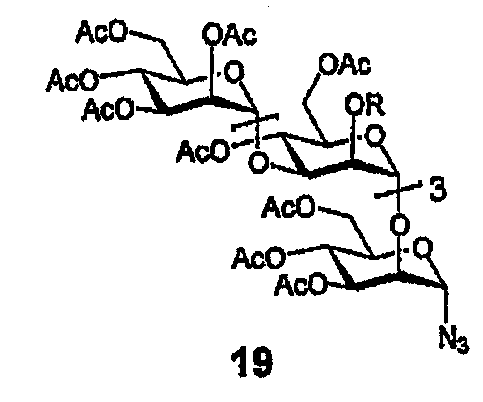
Азид 19
Раствор перацетата 12 (270 мг, 175 мкмоль), TMSN3 (60 мг, 525 мкмоль) и SnCl4 (20 мкл 1М в ДХМ) в безв. ДХМ (20 мл) перемешивают в течение ночи в темноте. Добавляют дополнительные количества TMSN3 (3 экв.) и SnCl4 и перемешивание продолжают в темноте в течение ночи. Добавляют лед и NaHCO3 (нас. водн.), и смесь экстрагируют EtOAc, дважды промывают, упаривают, и остаток очищают флэш-хроматографией (10 г силикагеля, элюирование с градиентом, EtOAc/гексан от 50:50 до 75:25), получая 218 мг (82%) азида. 1H ЯМР (400 МГц, CDCl3) δ 5,52 (д, 1H, J=2,0, H1I), 5,29-5,12 (м, 8H), 5,02-4,87 (м, 7H), 4,29-3,76 (м, 19H), 2,18-1,95 (м, 48H); 13C ЯМР (100 МГц, CDCl3) δ 170,5(9), 170,5(7), 170,5(6), 170,4, 170,3, 170,2, 170,1, 169,9(9), 169,9(8), 169,9(5), 169,7(3), 169,6(9), 169,6(6), 169,6, 169,5, 169,3, 99,3(0), 99,2(7), 99,1, 99,0, 88,1, 75,2, 75,1, 74,8, 71,1, 70,9, 70,8, 70,6, 69,7, 69,5, 69,4, 69,2, 68,3, 67,3, 66,8, 66,7, 65,5(9), 65,5(8), 62,6, 62,2, 62,0, 61,7, 20,8(8), 20,8(6), 20,8, 20,7, 20,6(2), 20,5(8), 20,5(7), 20,5.
HRMS: вычислено для C62H84N3O41 [M+H]+ 1526,4583, найдено 1526,4557.
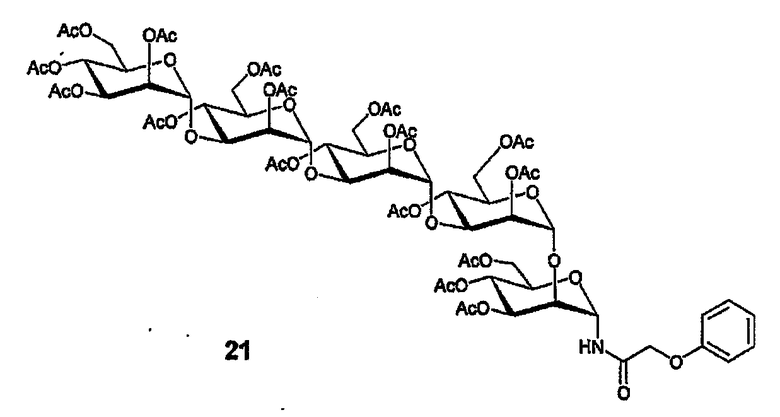
1-Деокси-1-α-феноксиацетамидоперацетат 21
Раствор 19 (32 мг, 21 мкмоль), PPh3 (11 мг, 42,6 мкмоль) и феноксиацетилхлорида (7,3 мг, 43 мкмоль) в безв. ацетонитриле (5 мл) перемешивают при 0°С в течение 4 часов и затем при комнатной температуре в течение ночи. К смеси добавляют EtOAc и NaHCO3 (нас.водн) и органический слой промывают насыщенным раствором соли, затем сушат (MgSO4), очищают флэш-хроматографией (элюирование с градиентом EtOAc/гексан от 60:40 до 90:10), получая 11,4 мг (33%) амида 21 с некоторым остаточным количеством PPh3/PPh3O. 1H ЯМР (400 МГц, CDCl3) δ 7,36-7,32 (м, 2H), 7,18 (уш. д, 1H, J=8,1, NH), 7,00-6,90 (м, 3H), 5,79 (дд, 1H, J=3,8, 8,2, H1I), 5,32-4,97 (м, 15H), 4,60-3,76 (м, 21H), 2,20-1,95 (м, 48H, AcO). HRMS: вычисл. для C70H92NO43 [M+H]+ 1634,5045, найдено 1634,5002.
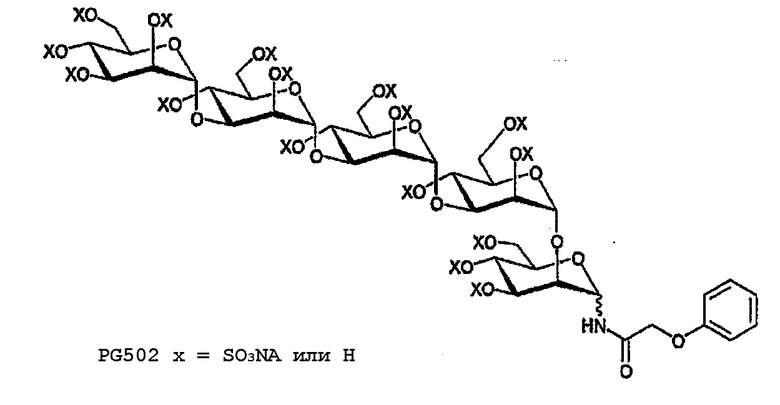
PG502
Перацетат 21 (11 мг, 6,7 мкмоль) деацетилируют и сульфируют в соответствии с общими методиками с получением 6 мг (34% для 2 стадий) PG502 после лиофилизации. 1H ЯМР (400 МГц, D2O, растворитель подавлен) δ: 7,30-7,21 (м, 2H, ArHm), 6,96-6,84 (м, 3H, ArH0,p), 5,56-3,59 (м, 30H, подавлен).
Пример 7: PG503
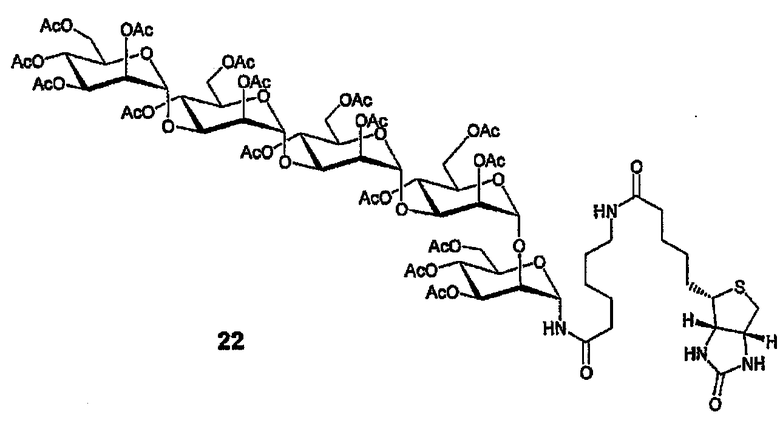
1-Деокси-1-α-биотинамидокапроамидоперацетат 22
Смесь 19 (70 мг, 46 мкмоль) и катализатора Адама (2 мг) в смеси EtOAc:EtOH (2:1, 3 мл) перемешивают в атмосфере Н2 (100 фунтов на кв. дюйм) в течение ночи, затем фильтруют, упаривают и остаток подвергают совместному выпариванию с безв. пиридином. Сложный эфир биотинамидокапроат-N-гидроксисукцинимид (31 мг, 68 мкмоль) и 1 мл безводного пиридина добавляют к смеси и полученную смесь выдерживают при 60°С в течение 3 дней при перемешивании. Раствор упаривают и остаток очищают флэш-хроматографией (9,4 г промытого Et3N, элюирование с градиентом от EtOAc:гексан 75:25 до МеОН:EtOAc 30:70) c получением 30,8 мг (36% для двух стадий) амида 22. 1H ЯМР (400 МГц, CDCl3) δ 7,41 (уш. д, 1Н, J=9,4, NH), 6,47, 6,17 (2x уш. с, 2х1Н, имидные NH), 5,40 (уш. д, 1Н, J=9,4, H1I), 5,40-4,90 (м, 16Н), 4,52 (дд, 1Н, J=4,9, 7,5, биотин-Н4), 4,36-3,72 (м, 20H), 3,25- 3,12 (м, 3H), 2,91 (дд, 1H, J=5,0, 13,0, биотин-H5A), 2,75 (д, 1H, J=12,9, биотин-H5B), 2,27-1,96 (м, 52H), 1,82-1,29 (м, 12H, алкильные цепи).
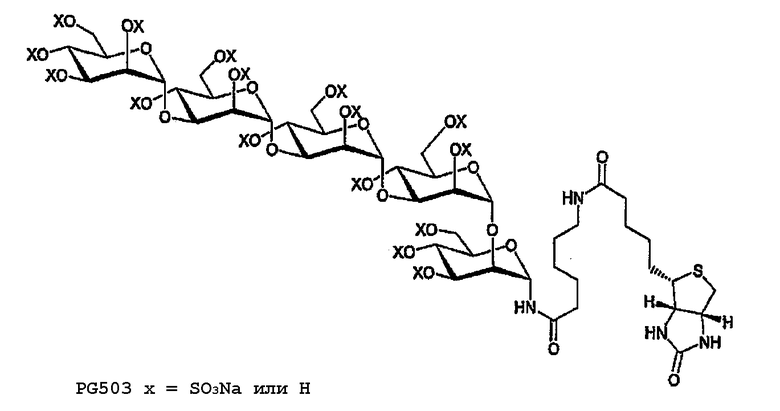
PG503
Перацетат 22 (30 мг, 16,3 мкмоль) деацетилируют и сульфируют в соответствии с общими методиками для получения после лиофилизации 28 мг (61% для 2 стадий) PG503. 1H ЯМР (400 МГц, D2O, подавление растворителя воздействием амидных ротамеров) δ 5,60-4,75 (м, 7H, атомы Н сахаров), 4,68 (дд, 1H, J=4,7, 7,2, биотин-H4), 4,60-3,60 (м, 26H, атомы Н сахаров), 421 (дд, 1H, J=4,4, 7,2, биотин-H3), 3,33-3,16 (м, 1H, биотин-H2), 3,07-2,97 (м, 3H, биотин-H5A+CH2N), 2,92 (дд, 1H, J=4,9, 13,5, биотин-H5B), 2,33-2,14 (м, 2H, COCH2B), 2,09 (т, 2H, J=7A, COCH2A), 1,63-1,15 (м, 12H, алкильные цепи).
Пример 8: PG505
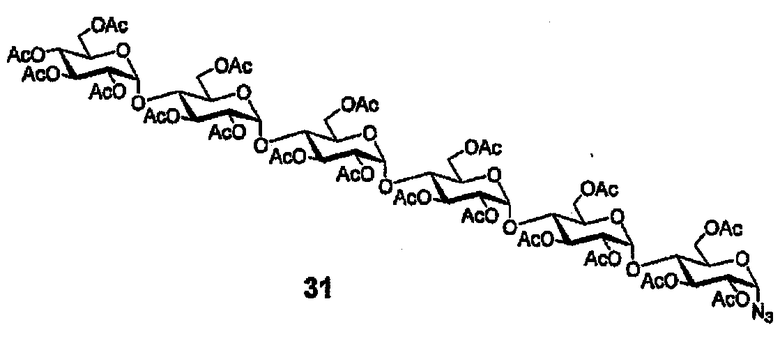
Азид 31
Раствор малтогексаозперацетата (500 мг, 273 мкмоль), TMSN3 (83 мг, 726 мкмоль) и SnCl4 (145 мкл 1М в ДХМ) в безв. ДХМ (20 мл) перемешивают в течение ночи в темноте. После этого добавляют дополнительные количества TMSN3 (50 мкл) и SnCl4 (100 мкл 1М в ДХМ) и перемешивание снова продолжают в темноте в течение ночи. Добавляют лед и NaHCO3 (насыщ. водн.), полученную смесь экстрагируют EtOAc, промывают насыщенным раствором соли, упаривают и остаток очищают флэш-хроматорафией (10 г силикагеля, элюирование с градиентом, EtOAc:гексан от 75:20 до 80:20), получая 488 мг (98%) азида 31. 1H ЯМР (400 МГц, CDCl3) δ 5,30-5,11 (м, 11H), 4,93 (т, 1H, J=9,9), 4,72 (дд, 1H, J=4,0, 10,5), 4,68-4,57 (м, 6H), 4,44-3,67 (м, 23H), 2,09-1,85 (м, 57H). 13C ЯМР (100 МГц, CDCl3) δ: 170,3(4), 170,3(1), 170,2(7), 170,2, 170,1(4), 170,1(0), 170,0(7), 170,0, 169,6, 169,4, 169,3, 169,2(3), 169,2(2), 169,1(7), 169,1(4), 169,1(1), 95,5(0), 95,4(5), 95,4, 95,3, 87,1, 74,7, 73,9, 73,3, 73,2, 72,2, 71,4, 71,3, 71,2(4), 71,2(1), 70,2, 70,1, 69,8, 69,0, 68,8, 68,7, 68,2, 67,7, 62,4, 62,3, 62,1(8), 62,1(6), 62,0, 61,1, 30,0, 20,5(5), 20,5(3), 20,5(0), 20,4(6), 20,3(3), 20,2(8), 20,2(4), 20,2(2).

PG505
Азид 31 (97 мг, 54 мкмоль) деацетилируют и сульфируют в соответствии с общими методиками с получением после лиофилизации 66 мг (41% для 2 стадий) PG505. 1H ЯМР (400 МГц, D2O, растворитель подавленный) δ: 3,69-5,78 (м, 42Н подвержен действию подавления растворителя).
Пример 9: PG515
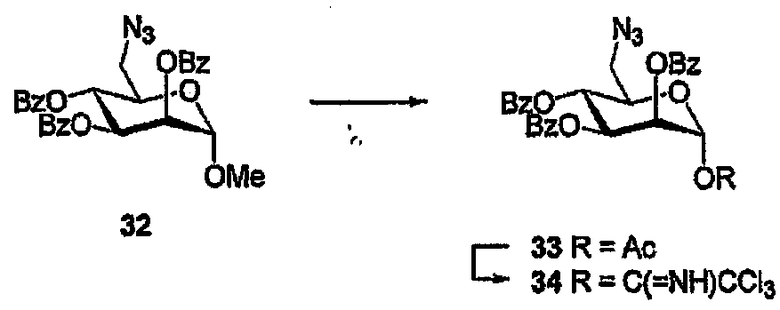
6-Азидо-6-деокси-2,3,4-три-О-бензоил-α-D-маннопиранозилтрихлорацетимидат(34)
(А) H2SO4 (0,5 мл) добавляют к охлажденному (0°С) раствору метилгликозида (32) [29] (1,52 г, 2,9 ммоль) и Ас2О (10 мл) в АсОН (5 мл), и полученную смесь перемешивают (0°С→комнатная температура, в течение ночи). Небольшими порциями добавляют NaOAc (1,0 г) до достижения рН>5,0 и затем смесь обрабатывают МеОН (2 мл). Смесь фильтруют и растворитель выпаривают, остаток подвергают совместному выпариванию (толуол) перед обработкой (EtOAc) и RSF (10-20% EtOAc/гексан) с получением предположительно ацетата (33) в виде бесцветной пены (1,12 г, 70%).
(В) Гидразинацетат (196 мг, 2,13 ммоль) при перемешивании добавляют к раствору ацетата (33) (1,08 г, 1,94 ммоль) в ДМФА (10 мл) и полученную смесь нагревают (55°С, 15 мин). Смесь выливают в насыщенный раствор NaCl и экстрагируют (EtOAc). Органический слой выпаривают и подвергают RSF (10-30% EtOAc/гексан) c получением бесцветного масла (888 мг). Остаток подвергают совместному испарению (2 х 100 мл CH3CN) и применяют в следующей реакции без дополнительной очистки или идентификации.
(С) К раствору сырого продукта, полученного на стадии (В), описано выше, (888 мг) и Cl3CN (2,0 мл, 20 ммол) в 1,2-ДСЭ (8 мл) добавляют DBU (3 капли) и полученную смесь перемешивают (0°С→комнатная температура, 1 час). Смесь фильтруют, растворитель выпаривают и остаток очищают ФХ (10-30% EtOAc/гексан) с получением имидата (34) в виде бесцветного масла (777 мг, 61%, 2 стадии). 1H ЯМР (400 МГц, CDCl3) δ 8,88 (уш. с, 1H, NH), 8,10-7,22 (м, 15H, ArH), 6,56 (д, 1H, J1,2 2,0 Гц, H1), 5,99 (дд, 1H, J3,4-4,5 9,6 Гц, H4), 5,94-5,88 (м, 2H, H2,3), 4,44 (ддд, 1H, J5,6 2,8, 5,6 Гц, H5), 3,54 (дд, 1H, J6,6 13,6 Гц, H6), 3,47 (дд, 1H, H6). 13C ЯМР (100 МГц, CDCl3) δ 165,61, 165,37, 159,95, 134,00, 133,92, 133,58, 130,25, 130,05, 129,12, 129,04, 128,97, 128,91, 128,76, 128,74, 128,57, 94,62, 73,03, 69,69, 68,90, 67,05, 51,06.
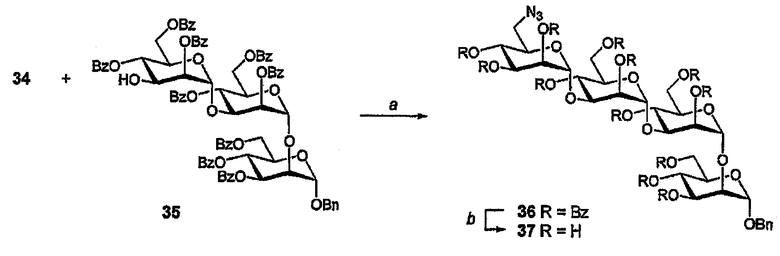
Бензил-(6-азидо-6-деокси-α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозил)-(1→2)-(α-D-маннопиранозид) (37)
(А) Смесь имидата (34) (93 мг, 141 мкмоль), спирта (35) (90 мг, 94,1 мкмоль) и молекулярный сит (50 мг 4А порошка) в 1,2-ДХЭ (2 мл) обрабатывают TMSOTf (10 мкл, 55,1 мкмоль) и полученную смесь перемешивают (0°С→комнатная температура, 20 мин). Вводят Et3N (100 мкл), смесь фильтруют и растворитель выпаривают. Остаток очищают ФХ (10-40% EtOAc/гексан) с получением азида (36) в виде бесцветного масла (68 мг, 57%). 1H ЯМР (400 МГц, CDCl3) δ 8,80-7,12 (м, 65H, ArH), 6,01 (дд, 1H, J 3,4-4,5 9,9 Гц, H4III), 5,96 (дд, 1H, J3,4-4,5 9,9 Гц, H4I), 5,92 (дд, 1H, J3,4-4,5 9,6 Гц, H4II), 5,83 (дд, 1H, J2,3 3,3 Гц, H3I), 5,79 (дд, 1H, J1,2 2,0, J2,3 3,3 Гц, H2II), 5,70 (дд, 1H, J3,4-4,5 9,9 Гц, H4IV), 5,50 (дд, 1H, J2,3 3,3 Гц, H3IV), 5,36 (д, 1H, J1,2 1,7 Гц, H3III), 5,29 (дд, 1H, J2,3 3,0 Гц, H2III), 5,23 (д, 1H, H1II), 5,18 (д, 1H, J1,2 1,9 Гц, H2IV), 5,16 (д, 1H, J1,2 1,6 Гц, H1I), 4,87 (д, 1H, H1IV), 4,72-4,24 (м, 14H, H2I, H3II,III, H5I-III, H6I-III), 3,99 (ддд, 1H, J5,6 2,9, 3,4 Гц, H5IV), 3,02 (дд, 1H, J6,6 13,5 Гц, H6IV), 2,83 (дд, 1H, H6IV).
(В) Бензоат (36) (63 мг, 31 мкмоль) обрабатывают в соответствии с общей методикой и полученный продукт очищают хроматографией (С18, 0-10% МеОН/Н2О) с получением тетрасахарида (37) в виде бесцветного стеклообразного вещества 915 мг, 62%). 1H ЯМР (400 МГц, МеОD) δ 7,34-7,22 (м, 5H, ArH), 5,12 (д, 1H, J1,2 1,5 Гц, H1a), 5,09 (д, 1H, J1,2 1,7 Гц, H1b), 5,07 (д, 1H, J1,2 1,6 Гц, H1c), 4,92 (д, 1H, J1,2 1,9 Гц, H1d), 4,71, 4,48 (AB AB квартета, J 11,7 Гц, CH2Ph), 4,14 (дд, 1H, J2,3 3,0 Гц, H2a), 4,19 (дд, 1H, J2,3 3,2 Гц, H2b), 3,96 (дд, 1H, J2,3 3,4 Гц, H2c), 3,94 (дд, 1H, J3,4 9,4 Гц, H3b), 3,88-3,52 (м, 19H, H2d, H3a,c,d, H4a-d, H5a-d, H6a-d), 3,44 (дд, 1H, J5,6 6,3 J6,6 10,1 Гц, H6IV).
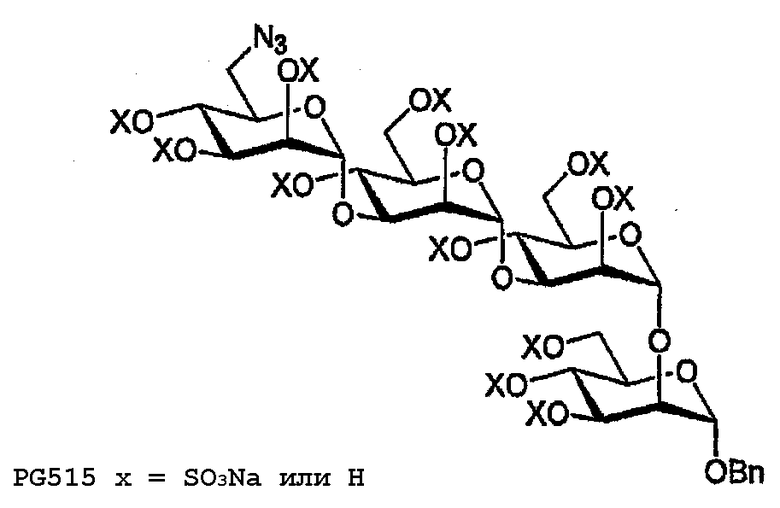
PG515
Тетрасахарид 37 (12 мг, 15,3 мкмоль) сульфируют в соответствии с общими методиками с получением после лиофилизации 14 мг (38% для 2 стадий) PG515. 1H ЯМР (500 МГц, D2O) δ 7,47-7,37 (м, 1H, ArH), 5,45-4,02 (м, 29H, C1I-IV, 2I-IV, 3I-IV, 4I-IV 5I- V, 6аI-IV, 6bI-III, CH2Ph), 3,69-3,67 (м, 1H, H6bIV).
Пример 10: PG509
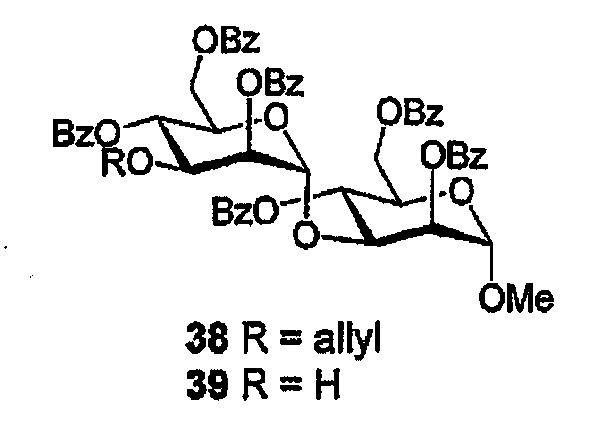
Метил-3-О-(2,4,6-три-О-бнзоил-α-D-маннопиранозил)-2,4,6-три-О-
бензоил-α-D-манопиранозид (39)
(А) Смесь 3-О-аллил-2,4,6-три-О-бензоил-α-D-маннопиранозилтрихлорацетимидат [26] (410 мг, 0,57 ммоль) и метил-2,4,6-три-О-бензоил-α-D-маннопиранозида [26] (200 мг, 0,51 ммоль) в 1,2-ДХЭ(6 мл) в присутствии молекулярный сит (700 мг 3Е порошка) обрабатывают TMSOTf (30 мкл, 0,17 ммоль) и полученную смесь перемешивают (0°С→комнатная температура, 30 мин). Вводят Et3N (100 мкл), смесь фильтруют и растворитель выпаривают. Остаток очищают ФХ (10-50% EtOAc/гексан) с получением предположительно дисахарида 38 в виде бесцветного масла.
(В) PdCl2 (40 мг) добавляют к раствору продукта, полученного на стадии (А), в МеОН (10 мл) и 1,2-ДХЭ (10 мл), и полученную смесь нагревают (70°С, 40 мин). Растворители выпаривают, и остаток очищают ФХ (10-50% EtOAc/гексан) с получением спирта (39) в виде бесцветного масла (316 мг, 68%, 2 стадии). 1Н и 13С ЯМР (CDCl3) спектры аналогичны спектрам, приведенным в литературе [26].
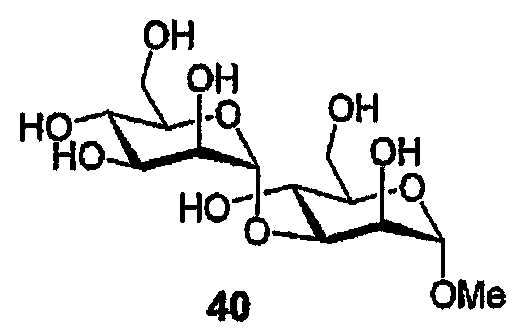
Метил-(α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозид) (40)
Спирт (39) (10 мг, 0,10 ммоль) подвергают реакции трансэтерификации в соответствии с общей методикой с получением дисахарида (40) в виде бесцветного масла (3 мг, 85%), который идентифицируют спектрами ЯМР, аналогичными приведенным в литературе [30, 31].
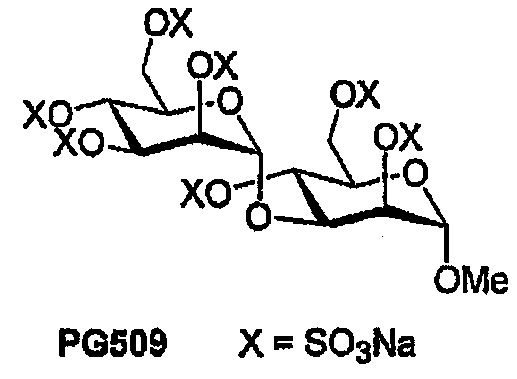
PG509
Дисахарид 40 (25 мг, 70 мкмоль) сульфируют в соответствии с общими методиками с получением после лиофилизации 27 мг (36%) PG509. 1H ЯМР (400 МГц, D2O) δ 5,26 (д, 1H, J1,2 1,8 Гц; H1II), 4,98 (дд, 1H, J2,3 2,4 Гц; H2II), 4,87 (д, 1H, J1,2 1,9 Гц; H1I), 4,60-4,55 (м, 1H, H3II), 4,53 (дд, 1H, J2,3 2,3 Гц; H2I), 4,41-4,19 (м, 5H; H4I,4II,6aI,6aII,6bII), 4,15 (дд, 1H, J3,4 9,3 Гц; H3I), 4,06-3,91 (м, 3H; H5I,5II,6bI), 3,29 (с, 3H; OCH3).
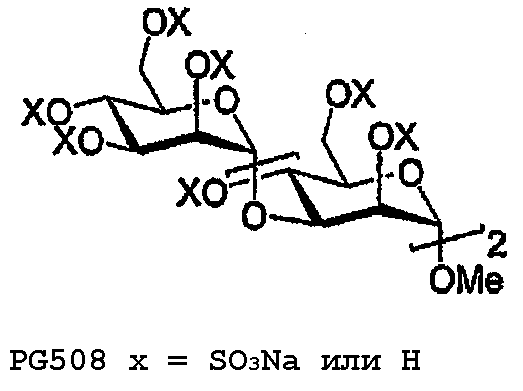
Пример 11: PG508
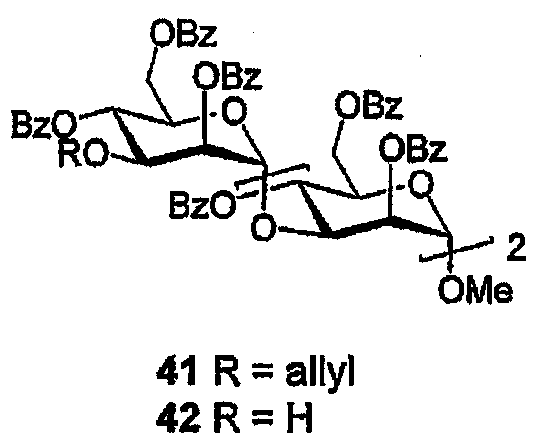
Метил-3-О-[3-O-(2,4,6-три-О-бензоил-α-D-маннопиранозил)-2,4,6-три-О-бензоил-α-D-маннопиранозил]-2,4,6-три-О-бензоил-α-D-маннопиранозид (42)
(А) Смесь 3-О-аллил-2,4,6-три-О-бензоил-α-D-маннопиранозилтрихлорацетимидата (269 мг, 0,37 ммоль) и спирта (39) (306 мг, 0,31 ммоль) в 1,2-ДХЭ (5 мл) в присутствии молекулярный сит (100 мг 3Е порошка) обрабатывают TMSOTf (20 мкл, 0,11 ммоль) и полученную смесь перемешивают (0°С→комнатная температура, 30 мин). Вводят Et3N (100 мкл), смесь фильтруют, и растворитель выпаривают. Остаток очищают ФХ (10-50% EtOAc/гексан) с получением предположительно трисахарида 41 в виде бесцветного масла.
(В) PdCl2 (40 мг) добавляют к раствору продукта, полученного на стадии (А), в МеОН (10 мл) и 1,2-ДХЭ (10 мл), и объединенную смесь нагревают (70°С, 40 мин). Растворители выпаривают и остаток очищают ФХ (10-50% EtOAc/гексан) с получением спирта (42) в виде бесцветного масла (316 г, 70%, 2 стадии). 1H ЯМР (400 МГц, CDCl3) δ 8,14-7,22 (м, 45H, ArH), 6,63 (дд, 1H, J1III,2III 1,8, J2III,3III 3,3 Гц, H2III), 5,94 (дд, 1H, J3III,4III 10,0, J4III,5III 10,0 Гц, H4III), 5,84 (дд, 1H, J3II,4II 9,9, J4II,5II 9,9 Гц, H4II), 5,48 (дд, 1H, J3I,4I 9,8, J4I,5I 9,8 Гц, H4I), 5,26 (д, 1H, J1I,2I 1,9 Гц, H1I), 5,22 (дд, 1H, J1II,2II 2,1, J2II,3II 3,0 Гц, H2II), 4,91 (д, 1H, H1III), 4,90 (дд, 1H, J2I,3I 3,2 Гц, H2I), 4,86 (дд, 1H, J1II,2II 1,7 Гц, H1II), 4,67-4,63 (12H, H3I,3II,3III,5I,5II,5III,6I,6II,6III). 13C ЯМР (100 МГц, CDCl3) δ 166,49, 166,38, 166,25, 166,07, 165,94, 165,77, 165,63, 165,19, 165,15, 133,80, 133,60, 133,61, 133,58, 133,52, 133,06, 130,22, 130,16, 130,09, 130,05, 130,16, 129,97, 129,9, 129,88, 129,84, 129,51, 129,17, 129,01, 128,85, 128,63, 128,53, 128,5, 128,46, 99,35, 99,24, 98,73, 76,48, 76,12, 72,45, 71,77, 71,64, 69,93, 69,7, 69,01, 68,86, 68,6, 68,53, 67,82, 63,17, 62,79, 62,41, 55,66; ESMS: m/z 1373,4 [M-Bz+H+Na]+, 1269,4 [M-2Bz+2H+Na]+.
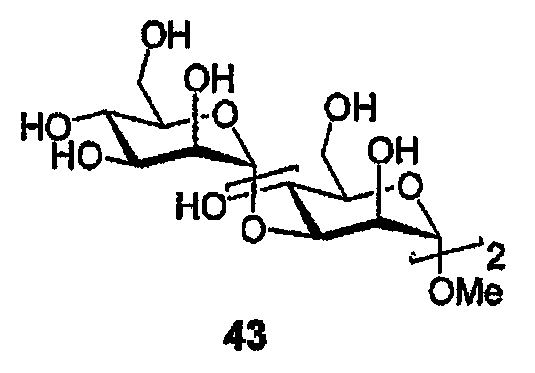
Метил-(α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозид) (43)
Спирт (42) (115 мг, 0,79 ммоль) подвергают реакции трансэтерификации в соответствии с общей методикой с получением трисахарида (43) в виде бесцветного масла (35 мг, 86%), ЯМР спектр полученного продукта аналогичен описанному в литературе [32]. HRMS: m/z 519,1862 [M+H]+, 541,1646 [M+Na]+.
PG508
Трисахарид 43 (25 мг, 49 мкмоль) сульфируют в соответствии с общими методиками с получением после лиофилизации 36 мг (48%) PG508. 1H ЯМР (400 МГц, D2O) δ 5,26 (д, 1H, J1,2 1,9 Гц; H1III), 5,22 (д, 1H, J1,2 1,8 Гц; H1II), 5,04 (дд, 1H, J2,3 2,4 Гц; H2III), 4,89 (д, 1H, J1,2 1,6 Гц; H1I), 4,76-4,75 (м, 1H; H2II), 4,60-4,55 (м, 1H; H3III), 4,55 (дд, 1H, J2,3 3,1 Гц; H2I), 4,50 (дд, 1H, J3,4 9,6, J4,5 9,7 Гц; H4III), 4,41-4,12, 4,04-3,91 (м, 12H; H3II,4I,4II,5I-III,6aI-III,6bI-III), 4,10 (дд, 1H, J3,4 9,5 Гц; H3I), 3,29 (с, 3H; OCH3).
Пример 12: PG512
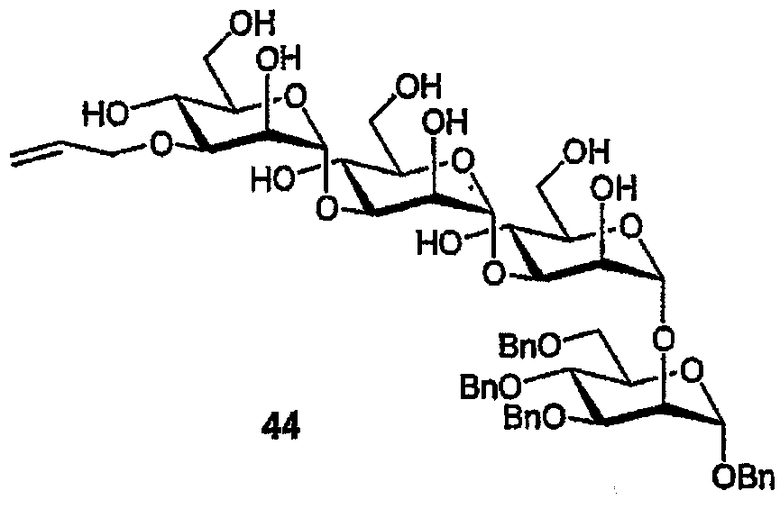
Бензил-(3-О-аллил-α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозил)-(1→3)-(α-D-маннопиранозил)-(1→2)-(3,4,6-три-О-бензил-α-D-маннопиранозид) (44)
Натрий (небольшой кусочек) добавляют к нанобензоату (28) (115 мг, 0,79 ммоль) в МеОН (6 мл) и объединенную смесь перемешивают (комнатная температура, в течение ночи). Смесь нейтрализуют (Dowex 50X8, H+), фильтруют, фильтрат концентрируют и остаток очищают ФХ (0-10% МеОН/CH2Cl2) c получением тетрабензилового эфира (44) в виде бесцветного масла (89 мг, 64%). 1H ЯМР (400 МГц, СD3ОD) δ 7,33-7,13 (м, 20H, ArH), 6,02-5,92 (м, 1H, CH=CH2), 5,32-5,27, 5,11-5,09 (2м, 2H, CH=CH2), 5,10 (д, 1H, J1,2 1,4 Гц, H1a), 5,09 (д, 1H, J1,2 1,5 Гц, H1b), 5,03 (д, 1H, J1,2 1,2 Гц, H1c), 4,97 (д, 1H, J1,2 1,4 Гц, H1d), 4,74, 4,49 (2d, AB of ABq, JH,H 10,9 Гц, PhCH2-a), 4,67, 4,48 (2d, AB of ABq, JH,H 11,8 Гц, PhCH2-b), 4,65, 4,58 (2d, AB of ABq, JH,H 11,6 Гц, PhCH2-c), 4,57, 4,51 (2d, AB of ABq, JH,H 12,4 Гц, PhCH2-d), 4,21-3,62 (м, 26H, H2I-IV,3I-IV,4I-IV,5I-IV,6aI-IV,6bI-IV, OCH2CH=).
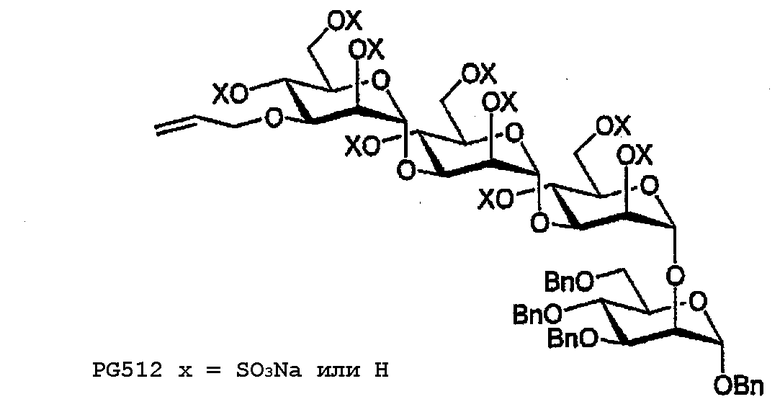
PG512
Тетрасахарид 44 (23 мг, 21,5 мкмоль) сульфируют в соответствии с общими методиками с получением PG512 в виде бесцветного порошка (26 мг, 61%). 1H ЯМР (400 МГц, CDCl3) δ 7,32-7,18, 7,00-6,98 (2м, 20H, ArH), 5,88-5,78 (м, 1H, CН=CH2), 5,30-5,23, 5,08-5,04, 4,91-4,90, 4,83- 4,82, 4,71-4,08, 4,00-3,89, 3,73-3,70, 3,62- 3,45 (8м, 40H, CH=CH2, OCН2CH, H1-6I-IV, PhCH2 I-IV).
Пример 13: PG513
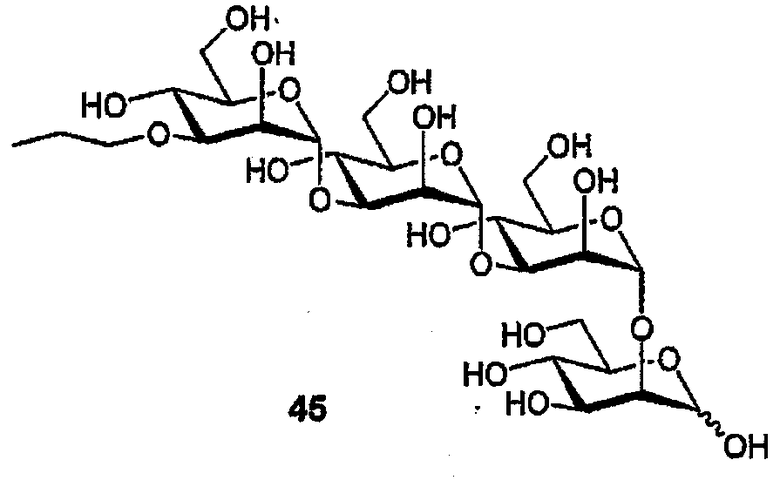
Смесь тетрабензилового эфира (44) (62 мг, 50 мкмоль) и Pd(OH)2 (10 мг 10% на С) в ТГФ (1 мл) и Н2О (1 мл) перемешивают в атмосфере Н2 (10 фунтов на кв. дюйм) (комнатная температура, в течение ночи). Смесь фильтруют, концентрируют, и остаток очищают ФХ (SiO2; H2O) с получением пропилового эфира (45) в виде бесцветного стеклообразного вещества (32 мг, 73%). 1H ЯМР (D2O) δ 5,22 (уш. с, 1H, H1a), 5,00 (д, 1H, J1,2 1,7 Гц, H1b), 4,97 (д, 1H, J1,2 1,6 Гц, H1c), 4,87 (д, 1H, J1,2 1,8 Гц, H1d), 4,11-4,07, 3,91-3,35 (2м, 26H, H2I-IV,3I-IV,4I-IV,5I-IV,6aI-IV,6bI-IV, OCH2), 1,50-1,42 (м, 2H, CH2CH3), 0,76 (т, 3H, JH,H 7,2 Гц, CH2CH3).
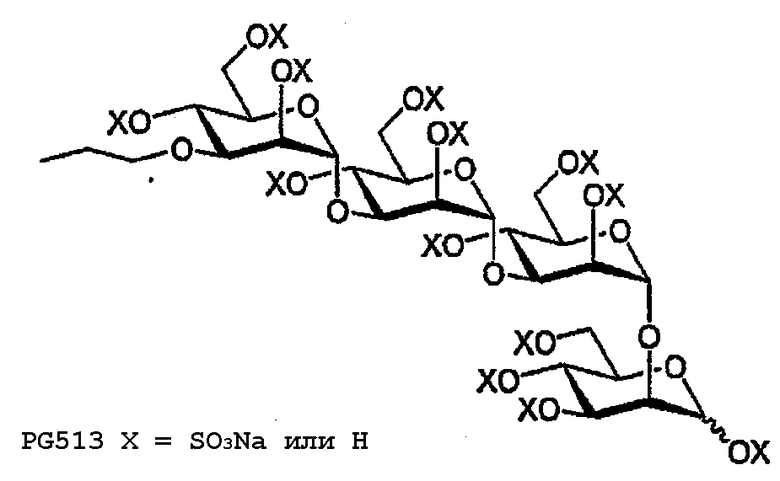
PG513
Тетрасахарид 45 (21 мг, 29,6 мкмоль) сульфируют в соответствии с общими методиками с получением PG513 в виде бесцветного порошка (29 мг, 34%). 1H ЯМР (D2O) δ 5,61 (д, 1H, J1,2 2,3 Гц; H1a), 5,61 (уш. с, 1H, H1b), 5,32 (д, 1H, J1,2 1,8 Гц; H1c), 5,26 (д, 1H, J1,2 2,0 Гц; H1d), 4,90-4,88, 4,77-4,31, 4,23-4,04, 3,98-3,81, 3,57-3,51, 3,41-3,36 (6м, 26H, OCH2CH2, H2-6I-IV), 1,48-1,39 (м, 1H, CH2CH3), 0,76 (дд, 1H, JH,H 7,4 Гц; CH2CH3).
Пример 14: PG510
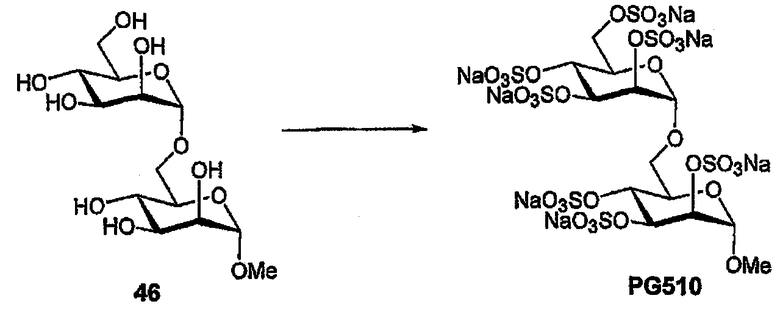
Полиол 46 [31] (22 мг, 61,7 мкмоль) сульфируют в соответствии с общей методикой с получением PG510 в виде бесцветного порошка (46 мг, 70%). 1H ЯМР (D2O) δ 5,10 (д, 1H, J1,2 2,0 Гц; H1II), 4,90 (д, 1H, J1,2 2,0 Гц; H1I), 4,78 (дд, 1H, J2,3 3,0 Гц; H2II), 4,73 (дд, 1H, J2,3 3,1 Гц; H2I), 4,64-4,40 (м, 1H; H3II), 4,52 (дд, 1H, J3,4 9,5 Гц; H3I), 4,33-4,30 (м, 2H; H4II,6aII), 4,22 (дд, 1H, J4,5 9,7 Гц; H4I), 4,12-4,04 (м, 2H; H5II,6bII), 3,96-3,90 (м, 2H; H5I,6aI), 3,76 (дд, 1H, J5,6b 8,6, J6a,6b 11,3 Гц; H6bI), 3,31 (с, 3H, OCH3).
Пример 15: PG511
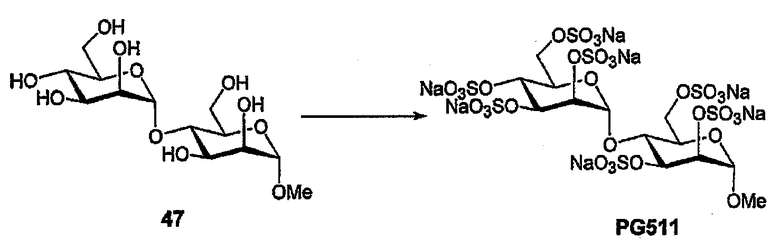
Полиол 47 ([31] (20 мг, 56 мкмоль) сульфируют в соответствии с общей методикой с получением PG511 в виде бесцветного порошка (29 мг, 48%). 1H ЯМР (D2O) δ 5,36 (д, 1H, J1,2 2,2 Гц; H1II), 4,90 (уш. с, 1H; H2II), 4,87 (д, 1H, J1,2 2,1 Гц; HII), 4,74 (дд, 1H, J2,3 3,0 Гц; H2II), 4,58-4,40, 4,29-4,10, 3,88-3,85 (3м, 10H, H3-6I,II), 3,30 (с, 3H; OCH3).
Пример 16: PG514
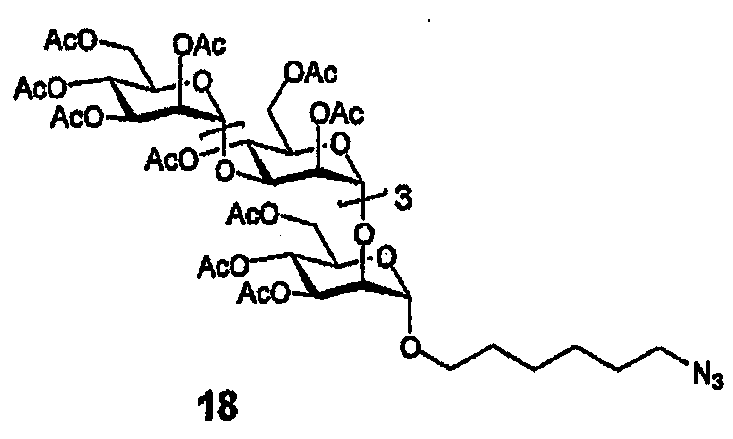
Азид 18
(А) Диэтилэтерат трифторида бора (257 мг, 1,81 ммоль) медленно добавляют к раствору перацетата 12 (700 мг, 0,453 ммоль) и 6-бром-1-гексанола (492,7 мг, 2,721 ммоль) в ДХЭ (20 мл, 3Е молекулярные сита), и смесь перемешивают в атмосфере аргона при 60°С в течение 72 часов. Раствор охлаждают, нейтрализуют Et3N, разбавляют ДХМ (30 мл), промывают насыщенным раствором NaHCO3, сушат (MgSO4) и очищают флэш-хроматографией (диоксид кремния, элюирование с градиентом, EtOAc/гексан от 40:60 до 100:0) с получением 340 мг (0,204 ммоль, 45,0%) 6-бромгексилгликозида. 1H ЯМР (400 МГц, CDCl3) δ 5,25-5,08 (м, 8H), 4,98-4,81 (м, 8H), 4,25-3,70 (м, 19H), 3,607 (дт, 1H, J=9,553, J=6,635, OCH2A), 3,354 (дт, 1H, J=9,641, J=6,637, OCH2B), 3,33 (т, 2H, J=6,700, CH2Br), 2,104, 2,096, 2,09, 2,06, 2,043, 2,038, 2,036, 2,033, 2,029, 2,02, 2,01, 1,97, 1,95, 1,94 и 1,90 (16x S, 48H, OAc), 1,85-1,74 (м, 2H, CH2), 1,59-1,46 (м, 2H, CH2), 1,44-1,35 (м, 2H, CH2), 1,35-1,25 (м, 2H, CH2); 13C ЯМР (CDCl3, 100 МГц): 170,42, 170,41, 170,39, 170,28, 170,16, 170,07, 169,96, 169,94, 169,83, 169,77, 169,58, 169,52, 169,45, 169,36, 169,25 (19xCO), 99,10, 98,83, 98,75, 98,01 (C-Cl сахара), 76,96, 75,00, 74,83, 74,75, 70,96, 70,82, 70,70, 70,08, 69,49, 69,28, 69,16, 68,24, 68,17, 68,04, 67,20, 66,65, 66,60, 66,09, 65,44, 62,41, 62,31, 61,86, и 61,54 (атомы С сахара минус атомы С-Cl сахара и атомы С группы бромгексил-СН2-О), 33,49, 32,32, 29,43, 28,92, 27,59, 25,12, (6x бромгексил-CH2), 20,73, 20,71, 20,68, 20,62, 20,56, 20,47, 20,44, 20,41, (Ac-CH3), 13,85 (CH2Br).
(В) Раствор 6-бромгексигликозида, полученного на стадии (А), (340 мг, 0,204 ммоль) и азида натрия (66 мг, 1,02 ммоль) в ДМФА (4 мл) нагревают до 100°С и выдерживают при данной температуре в течение 48 часов. После этого анализ сырой смеси ТСХ показывает, что смесь остается неизменной. К смеси добавляют йодид тетрабутиламмония (20 мг) и смеси дают возможность взаимодействовать в течение дополнительных 48 часов. Сырую смесь охлаждают и очищают флэш-хроматографией (от 0:100 до 5:95 ДХМ:МеОН) с получением 21,1 мг (0,013 ммоль, 6,4%) азида 18.
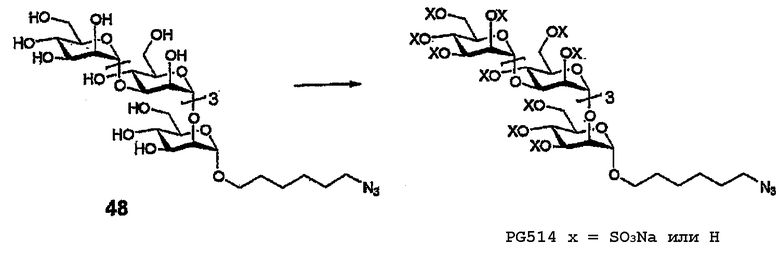
PG514
(А) Азид 18 (21,1 мг, 0,013 ммоль) деацетилируют в стандартных условиях Цемплена (2 мл МеОН) с получением 12,6 мг (0,013 ммоль, 102%) полиола 48.
(В) Полиол 48 (12,6 мг,13,2 мкмоль) обрабатывают SO3-триметиламином в соответствии с общей методикой сульфатирования с получением PG514 в виде бесцветного порошка (18,4 мг, 54%). 1H ЯМР (400 МГц, D2O) δ 5,4-4,69 (м, 8 H), 4,68-3,41 (м, 27H), 3,22 (т, 2H, J=6,5), 1,51 (уш. с, 5H), 1,29 (уш. с, 5H).
Биологические испытания соединений
Анализ связывания фактора роста
Сродство лигандов к связыванию факторов роста FGF-1, FGF-2 и VEGF определяют с использованием анализа сродства к раствору, основанного на поверхностном резонансе плазмона (SPR). Основным принципом этого испытания является тот факт, что гепарин, иммобилизированный на поверхность чувствительного элемента, различает свободный и связанный фактор роста в равновесном растворе фактора роста и лиганда. После инъекции раствора свободный фактор роста связывается с иммобилизированным гепарином, что фиксируется повышением SPR ответа, и таким образом определяют его концентрацию. Снижение концентрации свободного фактора роста в зависимости от концентрации лиганда позволяет вычислить константу диссоциации Kd. Важно заметить, что связывание лиганда с факторами роста можно обнаружить только в том случае, когда во взаимодействие вовлечен HS-связывающий сайт, что исключает таким образом возможность учета неспецифического связывания с другими сайтами на белке. Для всех взаимодействий белок:лиганд стехиометрическое соотношение принимают равным 1:1.
Для количественного испытания активности связывания фактора роста используют чувствительные элементы, покрытые гепарином. Их получение посредством иммобилизации биотинилированного BSA-гепарина на стрептавидин-покрытый чувствительный элемент описано в литературе [5]. Гепарин также иммобилизируют посредством альдегидного связывания с использованием либо дигидразида адипиновой кислоты, либо 1,4-диаминобутана. Для каждого измерения Кd приготавливают растворы, содержащие фиксированную концентрацию белка и изменяющиеся концентрации лиганда в буфере. Содержание лигандов, связывающихся с FGF-1 и VEGF, определяют в HBS-EP буфере (10 mM HEPES, pH 7,4, 150 мM NaCl, 3,0 мM EDTA и 0,005% (об./об.) полисорбата 20), в то время как количественное определение FGF-2 проводят в HBS-EP буфере, содержащем 0,3М NaCl [5]. Перед инъекцией образцы выдерживают при 4°С для получения максимальной стабильности белка. В каждую экспериментальную смесь вводят 50-200 мкл раствора с объемной скоростью 5-40 мкл/мин и определяют относительный ответ связывания. Все эксперименты поверхностного связывания проводят при 25°С. Поверхность регенерируют инъекцией 40 мкл 4М NaCl со скоростью 40 мкл/мин с последующей инъекцией 40 мкл буфера с объемной скоростью 40 мкл/мин.
Данные сенсограммы анализируют с использованием программного обеспечения BIAevaluation (BIAcore). Фоновые сенсограммы вычитают из экспериментальных сенсограмм для получения кривых специфического связывания и исходный уровень затем доводят до нуля для всех кривых. Стандартные кривые, относящиеся к относительному значению ответа на вводимую концентрацию белка, являются линейными, что говорит о прямой зависимости реакции на связывание от концентрации белка и таким образом подтверждает, что эксперименты связывания проводятся в условиях массопереноса [34]. Поэтому на основании значения относительной реакции связывания для каждой инъекции концентрация свободного белка может быть вычислена в соответствии со следующим уравнением
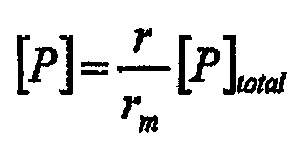
где r представляет собой относительную реакцию связывания, и rm представляет собой максимальную реакцию связывания.
Стехиометрическое соотношение равновесного связывания, установленное в растворе перед инъекцией, принимается равным 1:1. Следовательно, для состояния равновесия

где Р соответствует белку ростового фактора, L представляет собой лиганд, P-L представляет собой комплекс белок:лиганд, тогда уравнение равновесия принимает вид
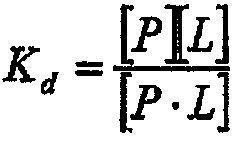
и уравнение связывания [5] может быть представлено следующим образом
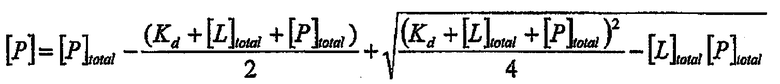
Значения Кd являются значениями, которые подставляют с использованием уравнения связывания в график зависимости [P] от [L]общ. Когда значения Кd определяют в повторных опытах, значения представляют в виде средней величины повторных измерений. Было показано, что GAG имитаторы, которые прочно связываются этими ростовыми факторами, например, PI-88, проявляют биологический ответ в условиях in vivo [5].
Исследования ингибирования гепараназы
Исследование ингибирования гепараназы проводят с использованием ультрафильтрационного анализа Микрокона (Microcon). Исследования основаны на принципе физического отделения гепарансульфата (HS), который потребляется гепараназой, от неизмененного HS, для определения активности гепараназы. В эксперименте используют ультрафильтрационные устройства (Microcon YM010) для отделения от чистого НS меньших по размеру фрагментов HS, расщепленного гепараназой.
Реакцию проводят в объеме 90 мкл,
40мМ ацетатного буфера (рН 5,0)
0,1 мг/мл BSA
90 нг гепараназы
2,5 мкМ 3Н меченого HS
ингибиторы в различных концентрациях.
Подготавливают реакционные смеси, включающие все компоненты за исключением 3Н меченого HS, и уравновешивают их в течение 10 мин при 22°С. После этого опыты инициируют добавлением HS и сразу же отбирают 20 мкл смеси, смешивают с 80 мкл 10мМ фосфата (рН 7,0), и 100 мкл полученной смеси переносят в аппарат для концентрирования Microcon YM-10, которую затем центрифугируют в течение 5 мин при приблизительно 14000g. Раствор, пропущенный через мембрану (фильтрат), сохраняют. Этот образец принимают за образец, полученный в момент, когда время =0. Экспериментальным растворам (теперь объемом 70 мкл) дают возможность взаимодействовать при 22°С в течение 2,5 часа, и затем стадию фильтрации повторяют для трех аликвот объемом 20 мкл из каждого эксперимента.
Фильтрат, полученный, когда время =0, и три образца фильтрата, полученные, когда время =2,5 часа, обсчитывают на содержание 3Н. Разность значений для образца времени =0 и средним значением образцов времени =2,5 часа дает значение гепараназной активности. Все эксперименты по ингибированию проводят также со стандартным образцом, который идентичен анализируемой композиции, описанной выше, но не содержит ингибитора, и значение ингибирования гепараназы в других анализируемых образцах определяют сравнением с этим стандартом. Значение IC50 для PI-88 в данном опыте равно 0,98 мкМ.
Количественное определение противовирусной активности
В опыте используют монослойные культуры клеток почки африканской зеленой мартышки [35] и штамма вируса простого герпеса (HSV-1) KOS321. Количественное определение противовирусной активности на соединениях проводят, как описано в публикации Nyberg et al. [13]. В целом, влияния соединений на заражение клеток экзогенно введенным вирусом исследуют смешением последовательным пятикратным разбавлением соединения (при 0,032-20 мкМ) с приблизительно 200 единицами вируса, образующими блашки. После инкубирования вируса и соединения в течение 120 минут при комнатной температуре смесь добавляют к клеткам и оставляют на клеточном монослое на 2 часа при 37°С. Затем инокулят аспирируют и замещают верхнюю среду 1% раствором метилцеллюлозы в минимальной поддерживающей среде Игла (Eagle's minimum essential medium - EMEM). Вирусные бляшки после инкубирования клеток в течение 3 дней при 37°С подкрашивают 1% раствором кристаллического фиолетового красителя и считывают. Влияние соединений на распространение HSV-1 от клетки к клетке проверяют добавлением последовательных пятикратных разбавлений соединения (при 0,032-20 мкМ) в не содержащей сыворотки верхней среде к клеткам после их инфицирования HSV-1. После инкубирования соединения с клетками в течение 3 дней при 37°С снимают изображения 20 бляшек и подвергают плоскостной детерминации с использованием программного обеспечения IM500 (Leica). Результаты вирусного заражения клеток и распространения вируса от клетки к клетке представлены на чертеже (А и В), соответственно, а полученные значения IC50 представлены в таблице 1.
Результаты
Результаты испытаний, описанных в предыдущем разделе, представлены в таблице 1.
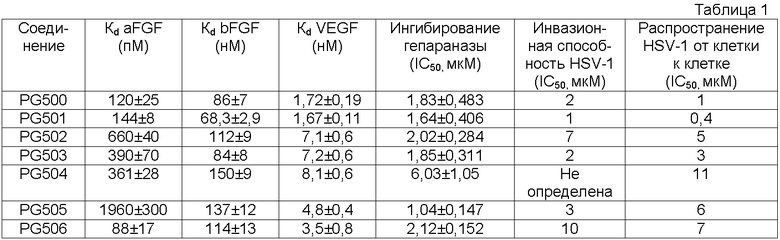
Фармакокинетическая оценка
Получение [35S]-меченых соединений
Предшественники PG500, 501, 503, 504, 506 и PI-66, которые представляют собой многоатомные спирты (2 мг каждого), сушат в вакуумном эксикаторе над Р2О5 в течение 3 дней. В каждую емкость впрыскивают основной раствор 1,77 мг (2,0 mCi) комплекса 35SO3·пиридин и 2 мг SO3·Me3N в 300 мкл безводного ДМФА (Aldrich, повторно высушен над свежепрокаленными молекулярными ситами 3Е). Дополнительные 600 мкл безводного ДМФА добавляют в SO3 емкость и распределяют в каждую емкость с образцом. Образцы нагревают до 60°С и выдерживают при этой температуре в течение 66 часов. В каждую емкость добавляют SO3·Me3N (14 мг в 300 мкл безводного ДМФА), и полученные растворы выдерживают при 60°С в течение ночи. Емкости охлаждают до комнатной температуры и хранят при -80°С перед очисткой.
Каждый образец гасят добавлением Na2CO3 (нас. водн. раствор для достижения рН 8-9), упаривают досуха и подвергают SEC (Biogel P2, 2,6x90 м, скорость истечения 30 мк/час, 5 мин/фракция). Фракции, содержащие целевой материал, обнаруживают с использованием G-M счетчика и DMB теста с последующим СЕ. Результаты представлены в таблице 2.
Краткие результаты экспериментов введения радиоактивной метки
(мг)
(мкКи/мг)
Фармакокинетические исследования
В исследовании используют самцов крыс Sprague Dawley. Животным предоставляют свободный доступ к пище и воде до и в процессе проведения экспериментов, во время которых они содержатся в свободном состоянии в метаболических клетках. Крыс наркотизируют изофлураном (Forthane®). Катетер вставляют в наружную яремную вену через надрез на шее и пропускают под кожу во второй надрез на коже спины (вблизи средней линии между лопаток). Затем его выводят наружу с защитой из легкой металлической пружины. Надрез закрывают и пружину фиксируют на коже нитями Михеля таким образом, чтобы движения крыс имели полную амплитуду. Животных внимательно наблюдают в процессе пробуждения после наркоза (1-4 часа).
Растворы для дозированного введения приготавливают смешением подходящих количеств немеченого и меченого лекарственного средства (растворенного в фосфатно-буферном растворе) для получения общей концентрации лекарственного средства, равной 1,25 мг/мл. Все дозы вводят в форме болюсной внутривенной инъекции 2,5 мг/кг в объеме 2 мл/кг. Общее количество радиоактивности, вводимой каждой крысе, составляет 0,5-10 мкКи. Уровень дозировки, используемый в данном исследовании, в 10 раз ниже, чем не вызывающая эффекта доза, предварительно установленная для острой токсичности PI-88. Образцы крови (~250 мкл) отбирают перед введением дозы и через 5, 15, 30, 45 минут, а также через 1, 1,5, 2, 4, 8, 12, 24, 36 и 48 часов после ее введения. Образцы крови сразу центрифугируют и плазму собирают. По завершении экспериментов животных умерщвляют введением смертельной сверхдозы IV пентобарбитонового анестетика (Nembutal®). От каждого животного собирают урину в интервалах 0-12 часов, 12-24 часа и 24-48 часов после введения дозы. Промывные воды из клеток (~15 мл деионизированной воды) также собирают. В конце эксперимента содержимое мочевого пузыря каждого животного аспирируют и добавляют к опорожнениям, собранным в интервале 24-48 часов. Экскременты собирают с такими же временными интервалами, что и урину.
Аликвоты плазмы (100 мкл), урины и промывных вод, полученных после промывки клеток (500 мкл), переносят непосредственно в 6 мл полипропиленовые сцинтилляционные емкости для определения радиоактивности. Экскременты, собранные в течение каждого временного периода (от одного животного с введенной дозой каждого соединения), взвешивают и гомогенизируют в 4 объемах деионизированной воды с использованием механического гомогенизатора. Приблизительно 1 г (точная навеска) такой взвеси переносят в 20 мл стеклянную сцинтилляционную емкость, добавляют 2 мл тканевого солюбилизатора и емкости закрывают и инкубируют при 60°С в течение, по меньшей мере, 24 часов. Радиоактивность измеряют после смешения образцов с жидким сцинтилляционным коктейлем Packard Ultima Gold (2 мл для плазмы и дозы, 5,0 мл для урины и промывных вод, 10 мл для экскрементов). Подсчет проводят на жидком сцинтилляционном счетчике Packard Tr-Carb. Любой результат, который меньше чем утроенный фон, рассматривают как значение меньше нижнего предела вычисления и не используют в вычислениях. Плазму, урину и промывные воды считывают трижды в течение 5 дней после сбора и не корректируют для устранения радиохимического разложения. Экскременты подвергают технологической переработке один раз по завершении исследования и результаты обсчета этих образцов корректируют с учетом радиохимического разложения. Фармакокинетические параметры плазмы, вычисленные с использованием программного обеспечения PK Solution 2.0 (Summit Research Services, Ohio), представлены в таблице 3.
Фармакокинетические параметры, определенные для 35S-меченных соединений, после внутривенного введения их самцам крыс Sprague Dawley
(мкг-экв./мл)
(мкг-экв./мл)
*Наблюдаемые значения
аВычислены только для интервала 0-8 часов после введения дозы
bВычислены только для интервала 0-4 часа после введения дозы
сВычислены только для интервала 0,75-4,0 часа после введения дозы для PI-88, PG500, PG501, PG503 и PG506; вычислены только для интервала 4,0-12 часов после введения дозы для PG504.
Результаты, представленные в таблице 1, показывают, что широкий спектр соединений согласно изобретению обладают ингибиторной активностью в отношении гепараназы и обладают сильным сродством к GAG-связывающим ростовым факторам, следовательно могут служить в качестве модуляторов активности таких факторов аналогично PI-88. Кроме того, соединения обладают антивирусной активностью, аналогичной PI-88. Результаты, представленные в таблице 3, показывают, что соединения обладают фармакокинетическими свойствами, отличающимися от фармакокинетических свойств PI-88.
Представленные выше варианты иллюстрируют только основные положения изобретения, и различные модификации и изменения легко реализуются квалифицированным специалистом данной области техники. Изобретение может быть внедрено в практику и осуществлено различными способами и в других вариантах его осуществления. Следует представлять, что терминология, применяемая в изобретении, предназначена для описания и не должна рассматриваться как ограничивающая.
Термин «включать» и варианты этого термина, такие как «включает» или «включающий», используются для обозначения включения заявленного целого числа или заявленных целых чисел, но не исключает любое другое целое или любые другие целые значения, за исключением случаев, когда в контексте или применении требуется ограничивающая интерпретация термина.
Любая ссылка на публикации, цитированные в данном описании, не означает, что предметы изобретения общеизвестны в Австралии.
Список использованных источников
[1] Parish, C.R.; Freeman, C.; Brown, K J.; Francis, D. J.; Cowden, W.B. Cancer Res. 1999, 59,3433.
[2] Parish, C.R.; Cowden, W.B. 6,143,730, 2000.
[3] Iversen, P.O.; Sorenson, D.R.; Benestad, H.B. Leukemia 2002, 16,376.
[4] Ferro, V.; Don, R. Australas. Biotechnol. 2003, 13,38.
[5] Cochran, S.; Li, C.; Fairweather, J.K.; Kett, W.C.; Coombe, D.R.; Ferro, V. J. Med. Chem. 2003, 40,4601.
[6] Vlodavsky, L.; Friedmann, Y. J. Clin. Invest. 2001, 108,341.
[7] Parish, C.R.; Freeman, C.; Hulett, M.D. Biochim. Biophys. Acta 2001, 1471, M99.
[8] Wall, D.; Douglas, S.; Ferro, V.; Cowden, W.; Parish, C. Thromb. Res. 2001, 103,325.
[9] Demir, M.; Iqbal, O.; Hoppensteadt, D.A.; Piccolo, P.; Ahmad, S.; Schultz, C.L.; Linhardt, R.J.; Fareed, J. Clin. Appl. Thromb. Hemost. 2001, 7,131.
[10] Hembrough, T.A.; Ruiz, J. F.; Papathanassiu, A.E.; Green, S. J.; Strickland, D.K. J. Biol. Chem. 2001, 276, 12241.
[11] Amirkhosravi, A.; Meyer, T.; Chang, J.Y.; Amaya, M.; Siddiqui, F.; Desai, H.; Francis, J.L. Thromb. Haemost. 2002, 87,930.
[12] Francis, D.J.; Parish, C.R.; McGarry, M.; Santiago, F.S.; Lowe, H.C.; Brown, K.J.; Bingley, J.A.; Hayward, I. P.; Cowden, W.B.; Campbell, J.H.; Campbell, G.R.; Chesterman, C.N.; Khachigian, L. M. Circ. Res. 2003, 92, e70.
[13] Nyberg, K.; Ekblad, M.; Bergstrom, T.; Freeman, C.; Parish, C.R.; Ferro, V.; Trybala, E. Antiviral Res. 2004, 63,15.
[14] Levidiotis, V.; Freeman, C.; Punler, M.; Martinello, P.; Creese, B.; Ferro, V.; van der Vlag, J.; Berden, J.H.M.; Parish, C.R.; Power, D.A. J. Am. Soc. Nephrol. 2004, 15, 2882.
[15] Ferro, V.; Li, C.; Fewings, K.; Palermo, M.C.; Linhardt, R.J.; Toida, T. Carbohydr. Res. 2002, 337,139.
[16] Yu, G.; Gunay, N.S.; Linhardt, R.J.; Toida, T.; Fareed, J.; Hoppensteadt, D.A.; Shadid, H.; Ferro, V.; Li, C.; Fewings, K.; Palermo, M.C.; Podger, D. Eur. J. Med. Chem. 2002, 37,783.
[17] Ferro, V.; Fewings, K.; Palermo, M.C.; Li, C. Carbohydr. Res. 2001, 332,183.
[18] Parolis, L.A.S.; Parolis, H.; Kennie, L.; Meidal, M.; Bock, K. Carbohydr. Res. 1998, 309,77.
[19] Gunay, N.S.; Linhardt, R. J. Planta Med. 1999, 65,301.
[20] Ferro, V.; Hammond, E.; Fairweather, J.K. Mini-Rev. Med Chem. 2004, 4,159.
[21] Alban, S.; Franz, G. Biomacromolecules 2001, 2,354.
[22] Foxall, C.; Wei, Z.; Schaefer, M.E.; Casabonne, M.; Fugedi, P.; Peto, C.; Castellot, J.J., Jr; Brandley, B.K. J. Cell. Physiol. 1996, 168,657.
[23] Fugedi, P.; Tyrrell, D.J.; Tressler, R.J.; Stack, R.J.; Ishihara, M. 5,739,115, 1998.
[24] Katsuraya, K.; Nakashima, H.; Yamamoto, N.; Uryu, T. Carbohydr. Res. 1999, 315,234.
[25] Wessel, H.P. Topics Curr. Chem. 1997, 187,215.
[26] Chen, L.; Kong, F. J. Carbohydr. Chem. 2002, 21,341.
[27] Mori, M.; Ito, Y.; Ogawa, T. Carbohydr. Res. 1989, 192,131.
[28] Kerekgyarto, J.; Kamerling, J.P.; Bouwstra, J.B.; Vliegenthart, J.F.; Liptak, A. Carbohydr. Res. 1989, 186,51.
[29] Jacobsen, S. Acta Chem. Scand. Ser. B, Org. Chem. Biochem. 1984, B38,157.
[30] Ogawa, T.; Sasajima. Carbohydr. Res. 1981, 93, 53.
[31] Ogawa, T.; Sasajima. Carbohydr. Res. 1981, 97,205.
[32] Garegg, P. J.; Olsson, L.; Oscarson, S. Bioorg. Med. Chem. 1996, 4,1867.
[33] Fairweather, J.K.; Karoli, T.; Ferro, V. Bioorg. Med Chem. 2004, 12,6063.
[34] Karlsson, R.; Roos, H.; Fagerstam, L.; Persson, B. Methods 1994, 6,99.
[35] Gunalp, A. Proc. Soc. Exp. Biol. Med 1965, 118,185.
[36] Holland, T.C.; Homa, F.L.; Marlin, S.D.; Levine, M.; Glorioso, J. J. Virol. 1984, 52, 566.
| название | год | авторы | номер документа |
|---|---|---|---|
| 5,6-БИС-(1',2':3',4'-ДИ-О-ИЗОПРОПИЛИДЕН-α-D-ГАЛАКТОПИРАНОЗО-6'-ИЛ)-1,3-ДИИМИНОИЗОИНДОЛИН | 2009 |
|
RU2409586C1 |
| 4,5-БИС-(1',2':3',4'-ДИ-О-ИЗОПРОПИЛИДЕН-α-D-ГАЛАКТОПИРАНОЗО-6'-ИЛ)ФТАЛОНИТРИЛ | 2009 |
|
RU2409585C1 |
| СИНТЕЗ ЭПОТИЛОНОВ, ИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ, АНАЛОГОВ И ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2462463C2 |
| СИНТЕЗ ЭПОТИЛОНОВ, ИХ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ, АНАЛОГОВ И ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2311415C2 |
| ФТОРОТАКСОЛЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2131874C1 |
| ПРЕДШЕСТВЕННИКИ ВИТАМИНА D, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2000 |
|
RU2247710C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФУКОЗИЛИРОВАННОГО УГЛЕВОДА, СПОСОБ ПОЛУЧЕНИЯ ФУКОЗИЛИРОВАННОЙ СИАЛИЛИРОВАННОЙ УГЛЕВОДНОЙ МОЛЕКУЛЫ, РЕАКЦИОННАЯ СИСТЕМА IN VITRO | 1992 |
|
RU2125092C1 |
| СПОСОБЫ СИНТЕЗА АПЛИДИНА И НОВЫХ ПРОТИВООПУХОЛЕВЫХ ПРОИЗВОДНЫХ, СПОСОБЫ ИХ ПРОМЫШЛЕННОГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2299887C2 |
| ЧУВСТВИТЕЛЬНЫЕ К ГЛЮКОЗЕ КОНЪЮГАТЫ ИНСУЛИНА | 2014 |
|
RU2676307C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ БЕТА 1,6 ГЛЮКОЗАМИН-ДИСАХАРИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2481352C2 |
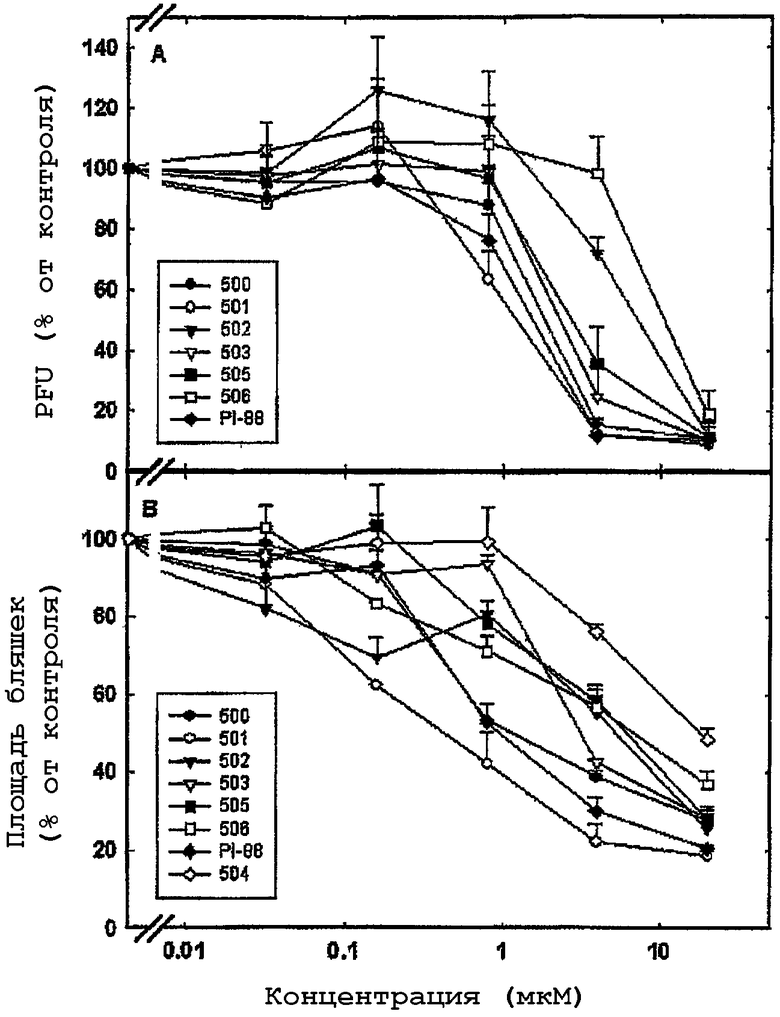
Настоящее изобретение относится к сульфатированным олигосахаридам общей формулы X-[Y]n-Z-UR1, где X, Y и Z, каждый, представляет собой одинаковый гексозный моносахаридный фрагмент, выбранный из группы, включающей глюкозу, маннозу, алтрозу, аллозу, талозу, галактозу, идозу и галозу, при этом соседние моносахаридные фрагменты связаны 1→2, 1→3, 1→4, и/или 1→6 гликозидными связями, а к каждому атому углерода, не связывающему группы X, Y и Z, посредством одинарной связи присоединена группа UR за исключением атома углерода в положении 1 моносахарида Z, к которому посредством одинарной связи присоединена группа UR1; где n целое число и принимает значения от 0 до 6; U представляет собой атом О, или NH; каждый R независимо представляет собой С2-С6-алкенил, бензил, SO3M или Н, где М представляет собой любой фармацевтически приемлемый катион щелочного металла или органического амина, или R вместе с U представляет собой N3; R1 представляет собой С1-С12алкил, бензил, монометиловый эфир PEG или его производное, С1-С12алкилазид,
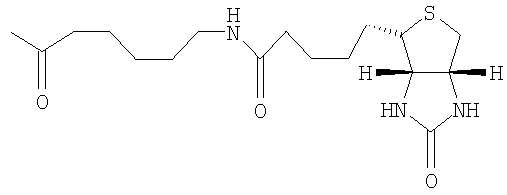 в форме сложного эфира, свободной кислоты или свободного основания, или гидрата. Изобретение относится также к фармацевтической или ветеринарной композиции на основе указанных соединений для профилактики или лечения расстройства млекопитающего субъекта, которое является пролиферативной ретинопатией, твердой опухолью и/или результатом метастаза, коагуляции/тромбоза, и/или заражения организма вирусными инфекциями. Кроме того, изобретение относится к применению указанных соединений для производства лекарственного средства для профилактики или лечения у млекопитающего субъекта расстройства, которое является результатом метастаза, коагуляции/тромбоза и/или заражения организма вирусными инфекциями. 3 н. и 8 з. п. ф-лы, 3 табл., 1 ил.
в форме сложного эфира, свободной кислоты или свободного основания, или гидрата. Изобретение относится также к фармацевтической или ветеринарной композиции на основе указанных соединений для профилактики или лечения расстройства млекопитающего субъекта, которое является пролиферативной ретинопатией, твердой опухолью и/или результатом метастаза, коагуляции/тромбоза, и/или заражения организма вирусными инфекциями. Кроме того, изобретение относится к применению указанных соединений для производства лекарственного средства для профилактики или лечения у млекопитающего субъекта расстройства, которое является результатом метастаза, коагуляции/тромбоза и/или заражения организма вирусными инфекциями. 3 н. и 8 з. п. ф-лы, 3 табл., 1 ил.
1. Сульфатированный олигосахарид общей формулы:
X-[Y]n-Z-UR1,
где X, Y и Z, каждый, представляет собой одинаковый гексозный моносахаридный фрагмент, выбранный из группы, включающей глюкозу, маннозу, алтрозу, аллозу, талозу, галактозу, идозу и галозу, при этом соседние моносахаридные фрагменты связаны 1→2, 1→3, 1→4, и/или 1→6 гликозидными связями, а к каждому атому углерода, не связывающему группы X, Y и Z, посредством одинарной связи присоединена группа UR за исключением атома углерода в положении 1 моносахарида Z, к которому посредством одинарной связи присоединена группа UR1; где n целое число и принимает значения от 0 до 6;
U представляет собой атом О, или NH;
каждый R независимо представляет собой С2-С6-алкенил, бензил, SO3M или Н, где М представляет собой любой фармацевтически приемлемый катион щелочного металла или органического амина, или R вместе с U представляет собой N3;
R1 представляет собой С1-С12алкил, бензил, монометиловый эфир PEG или его производное, С1-С12алкилазид,
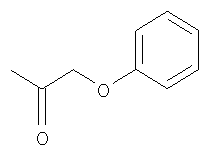 , или
, или
или R1 вместе с U представляет собой N3;
при условии, что:
когда U представляет собой атом О или N, R1 не является группой SO3М или Н,
и, по меньшей мере, 50% R групп представляют собой SO3М;
в форме сложного эфира, свободной кислоты или свободного основания, или гидрата.
2. Соединение по п.1, где М представляет собой натрий.
3. Соединение по п.1, где n равно 3.
4. Соединение по п.1, где R1 представляет собой н-октил.
5. Соединение по п.1, где от 70 до 100% групп R включают SO3М.
6. Соединение по п.1, где указанное соединение представляет собой
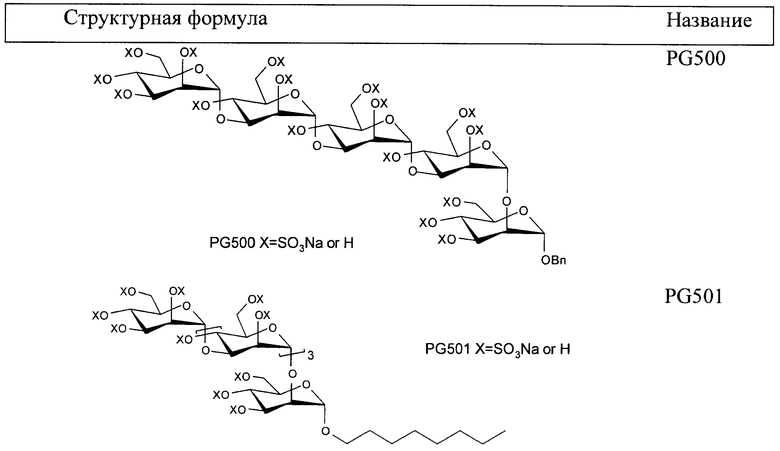
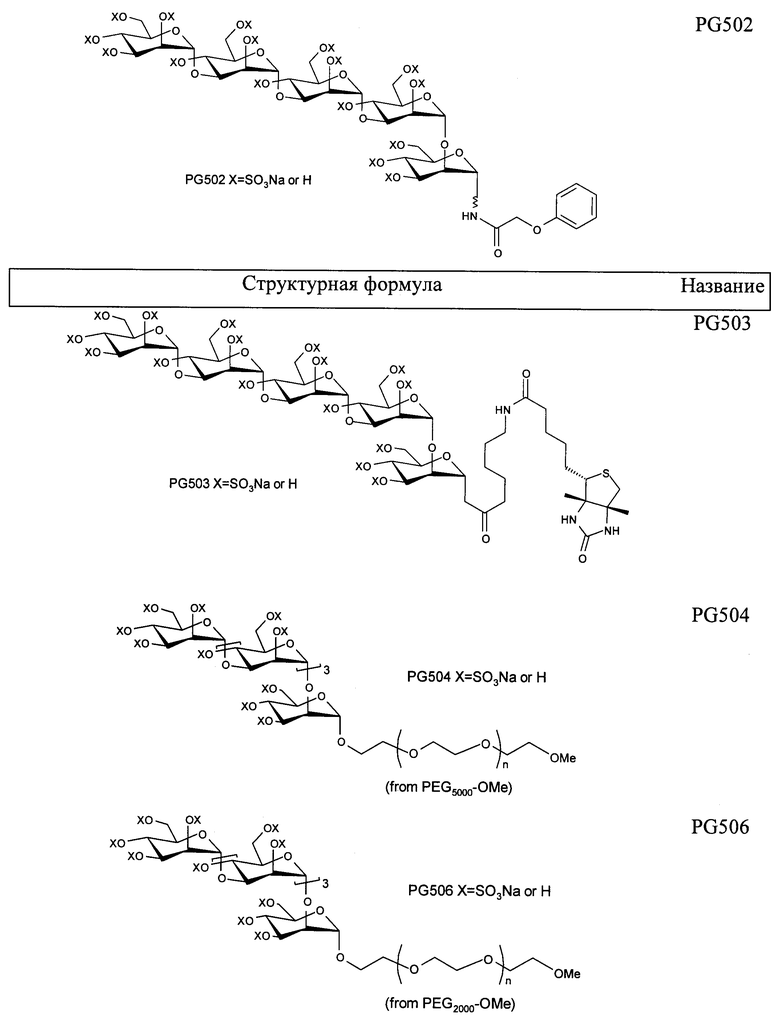
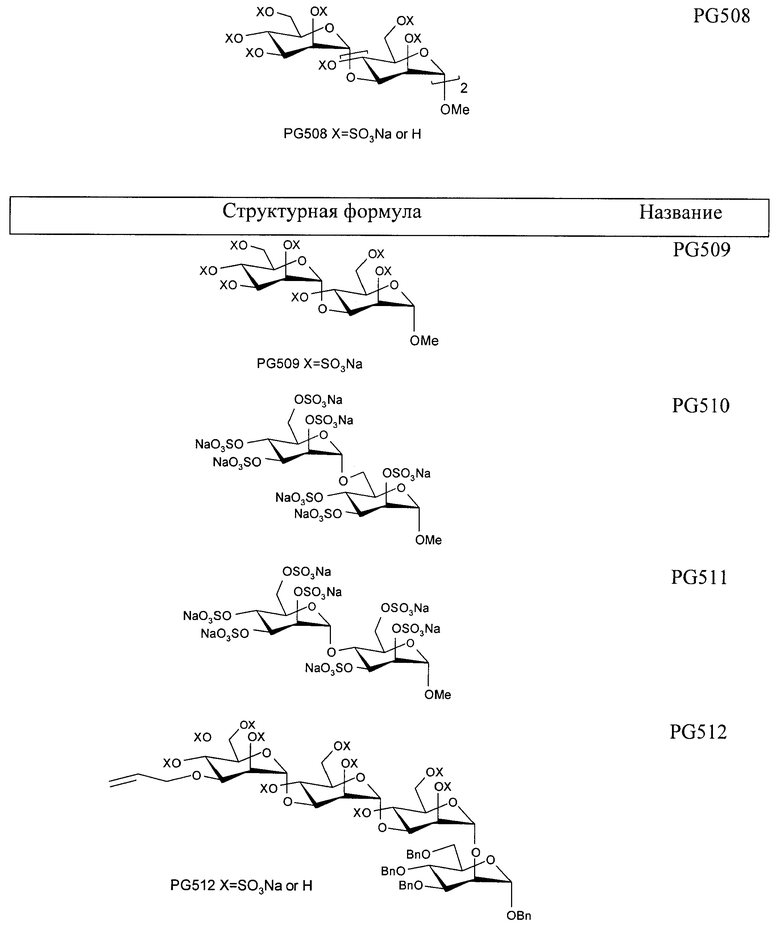

7. Фармацевтическая или ветеринарная композиция для профилактики или лечения расстройства млекопитающего субъекта, которое является пролиферативной ретинопатией, твердой опухолью и/или результатом метастаза, коагуляции/тромбоза, и/или заражения организма вирусными инфекциями, причем указанная композиция включает эффективное количество, по меньшей мере, одного соединения по п.1 вместе с фармацевтически или ветеринарно приемлемым носителем или разбавителем для указанного, по меньшей мере, одного соединения.
8. Композиция по п.7, которая дополнительно включает фармацевтически или ветеринарно приемлемый наполнитель, буфер, стабилизатор, добавку, регулирующую изотоничность, консервант или антиоксидант.
9. Композиция по п.7, в которой указанное соединение присутствует в форме сложного эфира, свободной кислоты или свободного основания, или гидрата.
10. Применение соединения по п.1 для производства лекарственного средства для профилактики или лечения у млекопитающего субъекта расстройства, которое является результатом метастаза, коагуляции/тромбоза и/или заражения организма вирусными инфекциями.
11. Применение по п.10, где указанный млекопитающий субъект является человеком.
| GAREGG PER J., OLSSON L., OSCARSON S | |||
| // BIOORGANIC & MEDICINAL CHEMISTRY, Vol:4, Nr.11, P.1867-1871 | |||
| KHACHIGIAN L.M, PARISH C.R | |||
| CARDIOVASCULAR DRUG REVIEWS, Vol:22, P.1-6 | |||
| RU 99121187 A, 20.07.2001 | |||
| УГЛЕВОДНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2183638C2 |
| УГЛЕВОДНЫЕ ПРОИЗВОДНЫЕ | 1998 |
|
RU2210573C2 |
| СОЕДИНЕНИЕ САХАРА | 1996 |
|
RU2193039C2 |
| ПРОИЗВОДНЫЕ 3-ДЕЗОКСИОЛИГОСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2133752C1 |
| НОВЫЕ ПЕНТАСАХАРИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2193040C2 |

