ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По данной заявке испрашивается приоритет в соответствии с предварительными заявками на выдачу патента США №№ 61/438685, поданной 2 февраля 2011 года, 61/440987, поданной 9 февраля 2011 года, и 61/495538, поданной 10 июня 2011 года, полное содержание которых включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, применимым в качестве ингибиторов ионных каналов. Настоящее изобретение также относится к фармацевтически приемлемым композициям, содержащим соединения согласно настоящему изобретению, и к способам применения композиций для лечения различных нарушений.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Боль представляет собой защитный механизм, который позволяет здоровым животным избежать повреждения тканей и предотвратить дальнейшее повреждение пораженных тканей. Тем не менее, существует множество состояний, при которых боль продолжается, несмотря на отсутствие ее практической ценности, или при которых ингибирование боли могло бы оказать благоприятное воздействие на пациента. Предполагается, что потенциалзависимые натриевые каналы играют ключевую роль в передаче болевого сигнала. Это предположение основывается на известной роли таких каналов в нормальной физиологии, в патологических состояниях, возникающих вследствие мутаций в генах натриевого канала, из доклинического исследования заболевания в модели на животных и клинической применимости известных модуляторов натриевых каналов (Cummins, T. R., Sheets, P. L., and Waxman, S. G., The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 131 (3), 243 (2007); England, S., Voltage-gated sodium channels: the search for subtype-selective analgesics. Expert Opin Investig Drugs 17 (12), 1849 (2008); Krafte, D. S. and Bannon, A. W., Sodium channels and nociception: recent concepts and therapeutic opportunities. Curr Opin Pharmacol 8 (1), 50 (2008)).
Потенциалзависимые натриевые каналы (NaV) представляют собой ключевые биологические медиаторы передачи электрического сигнала. NaV являются основными медиаторами быстрого возрастания потенциала действия во многих типах возбудимых клеток (например, нейроны, скелетные миоциты, сердечные миоциты), а потому являются ключевыми для инициации передачи сигнала этими клетками (Hille, Bertil, Ion Channels of Excitable Membranes, Third ed. (Sinauer Associates, Inc., Sunderland, MA, 2001)). В связи с участием NaV в процессе инициации и передачи сигналов нейронами, антагонисты, которые уменьшают токи через NaV, могут предотвращать или сжать передачу сигнала нейронами. Таким образом, NaV каналы считаются подходящими мишенями при патологических состояниях, при которых снижение возбудимости предсказуемо облегчает клинические симптомы, такие как боль, эпилепсия и некоторые сердечные аритмии (Chahine, M., Chatelier, A., Babich, O., and Krupp, J. J., Voltage-gated sodium channels in neurological disorders. CNS Neurol Disord Drug Targets 7 (2), 144 (2008)).
NaV образуют подсемейство в суперсемействе потенциалзависимых ионных каналов и включают 9 изоформ, обозначаемых NaV1.1-NaV1.9. Тканевая локализация девяти изоформ очень сильно различается. NaV1.4 представляет собой основной натриевый канал скелетной мускулатуры, а NaV1.5 представляет собой основной натриевый канал сердечных миоцитов. NaV1.7, NaV1.8 и NaV1.9 локализованы, главным образом, в периферической нервной системе, тогда как NaV1.1, NaV1.2, NaV1.3 и NaV1.6 представляют собой нейронные каналы, обнаруживаемые как в центральной, так и в периферической нервной системе. Функциональное поведение этих девяти изоформ сходно, но отличается особенностями их потенциалзависимого и кинетического поведения (Catterall, W. A., Goldin, A. L., and Waxman, S. G., International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57 (4), 397 (2005)).
NaV каналы были определены в качестве основной мишени для некоторых клинически применимых фармацевтических средств, которые уменьшают боль (Cummins, T. R., Sheets, P. L., and Waxman, S. G., The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 131 (3), 243 (2007)). Местные анестезирующие лекарства, такие как лидокаин, блокируют боль путем ингибирования NaV каналов. Такие соединения обеспечивают превосходное местное обезболивание, но обладают таким недостатком, как блокирование нормальной острой боли и афферентной сигнализации. Системное введение таких соединений приводит к ограничивающим дозу побочным эффектам, которые обычно объясняются блокировкой нейронных каналов в ЦНС (тошнота, седативный эффект, спутанность сознания, атаксия). Также могут возникать сердечные побочные эффекты, и более того такие соединения также применяют в качестве антиаритмических средств 1 класса, очевидно вследствие блокировки NaV1.5 каналов в сердце. Считается, что другие соединения, которые доказали свою эффективность при снижении боли, включая карбамазепин, ламотрагин и трициклические антидепрессанты, также действуют посредством блокады натриевых каналов (Soderpalm, B., Anticonvulsants: aspects of their mechanisms of action. Eur J Pain 6 Suppl A, 3 (2002); Wang, G. K., Mitchell, J., and Wang, S. Y., Block of persistent late Na+ currents by antidepressant sertraline and paroxetine. J Membr Biol 222 (2), 79 (2008)). По аналогии, такие соединения лимитируются по дозе побочными эффектами, сходными с наблюдаемыми при использовании местных анестетиков. Предполагается, что антагонисты, которые специфически блокируют только ключевую(ые) для ноцицепции изоформу(ы), обладают повышенной эффективностью, поскольку уменьшение побочных эффектов, обусловленных блокированием не являющихся мишенями каналов, делает возможным более высокое дозирование, а потому более полное блокирование изоформ каналов, являющихся мишенями.
Четыре изоформы NaV (NaV1.3, NaV1.7, NaV1.8 и NaV1.9) были особо отмечены как предпочтительные мишени для снижения боли. Обычно, NaV1.3 обнаруживаются в болевых сенсорных нейронах в дорсальных корешковых ганглиях (DRG) только на раннем этапе развития и быстро исчезают после рождения, как у людей, так и у грызунов. Тем не менее, было обнаружено, что сопровождающиеся повреждением нервов травмы приводят к повторному появлению NaV1.3 каналов в DRG нейронах, и это может приводить к аномальной передаче болевого сигнала при различных хронических болевых состояниях, возникающих вследствие повреждения нервов (нейропатическая боль). Эти данные явились основанием для предположения, что фармацевтическое блокирование NaV1.3 может представлять собой эффективное лечение для нейропатической боли. В противовес этой идее, полное генетическое нокаутирование мышей по NaV1.3 не предотвращает развитие аллодинии в моделях на мышах нейропатической боли (Nassar, M. A. et al., Nerve injury induces robust allodynia and ectopic discharges in NaV1.3 null mutant mice. Mol Pain 2, 33 (2006)). Остается неизвестным, допускают ли компенсаторные изменения в других каналах нормальную нейропатическую боль у нокаутных по NaV1.3 мышей, хотя сообщалось, что нокаутирование по NaV1.1 приводит к резкому увеличению экспрессии NaV1.3. Обратный эффект у нокаутов по NaV1.3 мог бы объяснить такие результаты.
NaV1.7, NaV1.8 и NaV1.9 высоко экспрессируются в DRG нейронах, включая нейроны, аксоны которых формируют С-волокна и Aδ нервные волокна, которые считаются передающими большинство болевых сигналов от ноцицептивных окончаний к центральным нервам. Подобно NaV1.3, экспрессия NaV1.7 увеличивается после повреждения нервов и может приводить к нейропатическим болевым состояниям. Локализация NaV1.7, NaV1.8 и NaV1.9 в ноцицепторах привела к гипотезе, что снижение токов натрия через указанные каналы могло бы облегчить боль. И действительно было доказано, что специфические воздействия, которые уменьшают активность таких каналов, являются эффективными в моделях боли на животных.
Специфическое снижение NaV1.7 у грызунов посредством множества различных методик приводило к уменьшению наблюдаемого болевого поведения у модельных животных. Инъекция вирусного антисмыслового NaV1.7 кДНК-конструкта значительно уменьшает нормальные болевые импульсы, вызванные воспалением или механическим повреждением (Yeomans, D. C. et al., Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of NaV1.7 sodium channels in primary afferents. Hum Gene Ther 16 (2), 271 (2005)). По аналогии, генетическое нокаутирование по NaV1.7 в субпопуляции ноцицепторных нейронов снижает острую и воспалительную боль в моделях на мышах (Nassar, M. A. et al., Nociceptor-specific gene deletion reveals a major role for NaV1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A 101 (34), 12706 (2004)). Полное нокаутирование мышей по NaV1.7 приводило животных к смерти в первые сутки после рождения. Такие мыши неспособны питаться, и это является предполагаемой причиной смерти.
Воздействия, которые специфически снижают активность NaV1.8 каналов в моделях на грызунах, эффективно уменьшают болевую чувствительность. Нокдаун крыс по NaV1.8 путем интратекальной инъекции антисмысловых олигодезоксинуклеотидов уменьшает нейропатическое болевое поведение, но оставляет ощущение острой боли интактным (Lai, J. et al., Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 95 (1-2), 143 (2002); Porreca, F. et al., A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A 96 (14), 7640 (1999)). Полное генетическое нокаутирование мышей по NaV1.8 или специфическое разрушение экспрессирующих NaV1.8 нейронов значительно снижает перцепцию острой механической, воспалительной и висцеральной боли (Akopian, A. N. et al., The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2 (6), 541 (1999); Abrahamsen, B. et al., The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321 (5889), 702 (2008); Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F., Deficits in visceral pain and referred hyperalgesia in NaV1.8 (SNS/PN3)-null mice. J Neurosci 22 (19), 8352 (2002)). В противоположность экспериментам на крысах с антисмысловыми последовательностями, после повреждения нервов у генетически нокаутных мышей нейропатическое болевое поведение развивается, по-видимому, нормально (Lai, J. et al., Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 95 (1-2), 143 (2002); Akopian, A. N. et al., The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2 (6), 541 (1999); Abrahamsen, B. et al., The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321 (5889), 702 (2008); Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F., Deficits in visceral pain and referred hyperalgesia in NaV1.8 (SNS/PN3)-null mice. J Neurosci 22 (19), 8352 (2002)).
Полностью нокаутные по NaV1.9 мыши обладали сниженной чувствительностью к индуцированной воспалением боли, несмотря на нормальное острое и нейропатическое болевое поведение (Amaya, F. et al., The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci 26 (50), 12852 (2006); Priest, B. T. et al., Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci U S A 102 (26), 9382 (2005)). Спинальный нокдаун по NaV1.9 не оказывал очевидного эффекта на болевое поведение у крыс (Porreca, F. et al., A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci U S A 96 (14), 7640 (1999)).
Понимание роли NaV каналов в физиологии и патологии человека было в значительное мере развито посредством открытия и анализа естественных мутаций у человека. Мутации NaV1.1 и NaV1.2 приводят к различным формам эпилепсии (Fujiwara, T., Clinical spectrum of mutations in SCN1A gene: severe myoclonic epilepsy in infancy and related epilepsies. Epilepsy Res 70 Suppl 1, S223 (2006); George, A. L., Jr., Inherited disorders of voltage-gated sodium channels. J Clin Invest 115 (8), 1990 (2005); Misra, S. N., Kahlig, K. M., and George, A. L., Jr., Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49 (9), 1535 (2008)). Мутации NaV1.4 обуславливают мышечные нарушения, такие как врожденная парамиотония (Vicart, S., Sternberg, D., Fontaine, B., and Meola, G., Human skeletal muscle sodium channelopathies. Neurol Sci 26 (4), 194 (2005)). Мутации NaV1.5 приводят к сердечным аномалиям, таким как синдром Бругада и синдром удлиненного интервала QT (Bennett, P. B., Yazawa, K., Makita, N., and George, A. L., Jr., Molecular mechanism for an inherited cardiac arrhythmia. Nature 376 (6542), 683 (1995); Darbar, D. et al., Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117 (15), 1927 (2008); Wang, Q. et al., SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80 (5), 805 (1995)).
В недавних открытиях было продемонстрировано, что мутации в гене, который кодирует NaV1.7 канал (SCN9A), могут вызывать как усиление, так и ослабление болевых синдромов. В работах группы Ваксмана и сотр. были идентифицированы, по меньшей мере, 15 мутаций, которые приводят к усилению тока через NaV1.7 и которые связаны с доминантными врожденными болевыми синдромами. Мутации, которые снижают порог активации NaV1.7, обуславливают наследственную эритромелалгию (IEM). Пациенты с IEM испытывают аномальную жгучую боль в своих конечностях. Мутации, которые препятствуют нормальной инактивации NaV1.7, приводят к пролонгированию токов натрия и вызывают пароксизмальное нарушение, характеризующееся чрезмерной болью (PEPD). Пациенты с PEPD испытывают симптомы периокулярной, перимандибулярной и ректальной боли, которые прогрессируют на протяжении жизни (Drenth, J. P. et al., SCN9A mutations define primary erythermalgia as a neuropathic disorder of voltage gated sodium channels. J Invest Dermatol 124 (6), 1333 (2005); Estacion, M. et al., NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci 28 (43), 11079 (2008)).
Недавно несколькими группами были описаны нуль-мутации NaV1.7 у пациентов-людей (Ahmad, S. et al., A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet 16 (17), 2114 (2007); Cox, J. J. et al., An SCN9A channelopathy causes congenital inability to experience pain. Nature 444 (7121), 894 (2006); Goldberg, Y. P. et al., Loss-of-function mutations in the NaV1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71 (4), 311 (2007)). Во всех случаях, пациенты испытывали врожденную нечувствительность к боли. Такие пациенты не испытывают никакой боли ни при каких обстоятельствах. Многие из этих пациентов страдают от тяжелых травм с раннего детства, поскольку они не имеют защитной, нормальной боли, которая помогает предотвратить повреждения тканей и развивает соответствующее защитное поведение. Если не считать четко выраженной потери болевой чувствительности и сниженного или отсутствующего обоняния (Goldberg, Y. P. et al., Loss-of-function mutations in the NaV1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71 (4), 311 (2007)), такие пациенты являются абсолютно нормальными. Несмотря на нормально высокую экспрессию NaV1.7 в симпатических нейронах (Toledo-Aral, J. J. et al., Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 94 (4), 1527 (1997)) и надпочечниковых феохромоцитах (Klugbauer, N., Lacinova, L., Flockerzi, V., and Hofmann, F., Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J 14 (6), 1084 (1995)), такие пациенты с нуль-мутацией NaV1.7 не проявляют ни одного признака дисфункции нейроэндокринной или симпатической нервной системы.
Приобретение функциональных мутаций NaV1.7, которые вызывают боль, в сочетании с утратой функциональных мутаций NaV1.7, которые отменяют боль, обеспечивают убедительное доказательство того, что NaV1.7 играет важную роль в передаче болевого сигнала у людей. Относительно хорошее здоровье пациентов с нуль-мутацией NaV1.7 указывает на то, что разрушение NaV1.7 хорошо переносится такими пациентами.
К сожалению, эффективность блокаторов натриевых каналов, используемых в настоящий момент для описанных выше болезненных состояний, была в значительной степени ограничена целым рядом побочных эффектов. Такие побочные эффекты включают различные нарушения ЦНС, такие как расфокусированное зрение, головокружение, тошнота и седативный эффект, а также потенциально более угрожающие жизни сердечные аритмии и сердечная недостаточность. Соответственно, остается необходимость в разработке дополнительных антагонистов натриевых каналов, предпочтительно антагонистов, обладающих повышенной эффективностью и меньшим количеством побочных эффектов.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Теперь, было обнаружено, что соединения согласно настоящему изобретению и их фармацевтически приемлемые композиции применимы в качестве ингибиторов потенциалзависимых натриевых каналов. Такие соединения или их фармацевтически приемлемые соли характеризуются общей формулой I:

Указанные соединения и фармацевтически приемлемые композиции применимы для лечения или ослабления тяжести ряда заболеваний, нарушений или состояний, включая без ограничения острую, хроническую, нейропатическую или воспалительную боль, артрит, мигрень, кластерную головную боль, тригеминальную невралгию, герпетическую невралгию, обычные невралгии, эпилепсию или эпилептические состояния, нейродегенеративные нарушения, психиатрические нарушения, такие как тревожность и депрессия, миотонию, аритмию, двигательные нарушения, нейроэндокринные нарушения, атаксию, рассеянный склероз, синдром раздраженного кишечника, недержание, висцеральную боль, остеоартритную боль, постгерпетическую невралгию, диабетическую нейропатию, корешковый болевой синдром, ишиас, боль в спине, боль в голове и шее, тяжелую или некупируемую боль, ноцицептивную боль, прорыв боли, послеоперационную боль или раковую боль.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно одному варианту осуществления, настоящее изобретение относится к соединениям формулы I:

или к их фармацевтически приемлемой соли,
где, независимо в каждом случае:
R1 представляет собой H, C1-С8алкил, C3-С8циклоалкил, галоген, CN, NR8SO2R8, SO2R8, SR8, SOR8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, гетероциклоалкил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8, или два R1 образуют вместе оксогруппу, или 3-7-членное конденсированное циклоалкильное кольцо, или 3-7-членное спироциклическое кольцо;
R2 представляет собой H, C1-С8алкил, галоген, C1-С8галогеналкил, CN, OH, SO2R8, SR8, SOR8, CO2R8, CON(R8)2, COR8, SO2N(R8)2, CF3, CHF2 или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2, CF2 или NR8;
R3 представляет собой H, C1-С8алкил, C3-С8циклоалкил, CO2R8, COR8, СОН, CON(R8)2, CF3, CH2CF3, CH2CHF2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2, CF2 или NR8;
R4 представляет собой H, C1-С8алкил, галоген, C3-С8циклоалкил, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8, или два R4 образуют вместе конденсированное 3-7-членное циклоалкильное кольцо;
R8 представляет собой H, C1-С8алкил, CF3, C3-С8циклоалкил, фторалкил, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR, или два R8 образуют вместе с атомами, к которым они присоединены, кольцо;
R9 представляет собой H, CF3, CO2R, OH, арил, гетероарил, C3-С8циклоалкил, гетероциклоалкил, N(R)2, NRCOR, CON(R)2, CN, галоген или SO2R;
R представляет собой H, C1-С8алкил, арил, гетероарил, C3-С8циклоалкил или гетероциклоалкил;
A представляет собой необязательно замещенный арил, гетероарил или гетероциклическое соединение;
m равно целому числу от 0 до 4 включительно;
n равно целому числу от 0 до 3 включительно; и
о равно целому числу от 0 до 4 включительно.
Согласно другому аспекту, настоящее изобретение относится к соединениям формулы I или к их фармацевтически приемлемой соли, где, независимо в каждом случае:
R1 представляет собой H, C1-С6алкил, C3-С8циклоалкил, галоген, CN, NR8SO2R8, SO2R8, SR8, SOR8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, гетероциклоалкил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8, или два R1 образуют вместе оксогруппу, или 3-7-членное конденсированное циклоалкильное кольцо, или 3-7-членное спироциклическое кольцо;
R2 представляет собой H, C1-С6алкил, C1-С6галогеналкил, CN, OH, SO2R8, SR8, SOR8, CO2R8, CON(R8)2, SO2N(R8)2, CF3, CHF2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2, CF2 или NR8;
R3 представляет собой H, C1-С6алкил, C3-С8циклоалкил, CO2R8, COR8, СОН, CON(R8)2, CF3, CH2CF3, CH2CHF2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8;
R4 представляет собой H, C1-С6алкил, галоген, C3-С8циклоалкил, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8, или два R4 образуют вместе конденсированное 3-7-членное циклоалкильное кольцо;
R8 представляет собой H, C1-С6алкил, CF3, C3-С8циклоалкил или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR, или два R8 образуют вместе с атомами, к которым они присоединены, кольцо;
R9 представляет собой H, CF3, CO2R, OH, арил, гетероарил, C3-С8циклоалкил, гетероциклоалкил, N(R)2, NRCOR, CON(R)2, CN или SO2R;
R представляет собой H, C1-С6алкил, арил, гетероарил, C3-С8циклоалкил или гетероциклоалкил;
A представляет собой необязательно замещенный арил, гетероарил или гетероциклическое соединение;
m равно целому числу от 0 до 4 включительно;
n равно целому числу от 0 до 3 включительно; и
о равно целому числу от 0 до 4 включительно.
Для целей настоящего изобретения, химические элементы определены в соответствии с Периодической Таблицей элементов, версия CAS, Handbook of Chemistry and Physics, 75th Ed. Кроме того, основные принципы органической химии описаны в «Organic Chemistry», Thomas Sorrell, University Science Books, Sausalito: 1999, и «March's Advanced Organic Chemistry», 5th Ed., Ed.: Smith, M.B. and March, J., John Wiley & Sons, New York: 2001, полное содержание которых включено в настоящий документ посредством ссылки.
Описанные в настоящем документе соединения согласно настоящему изобретению необязательно могут быть замещены одним или несколькими заместителями, такими как в общем смысле представленные выше, или представленные на примере отдельных классов, подклассов и видов согласно настоящему изобретению. Выражение «необязательно замещенный» используют взаимозаменяемо с выражением «замещенный или незамещенный». Используемые в настоящем документе переменные R1-R9 в формуле I охватывают конкретные группы, такие как, например, алкил и арил. Если не указано иное, то каждая из конкретных групп для переменных R1-R8 может быть необязательно замещена одним или несколькими заместителями из галогена, циано, оксоалкокси, гидрокси, амино, нитро, арила, галогеналкила и алкила. Например, алкильная группа может быть необязательно замещена одним или несколькими из галогена, циано, оксоалкокси, гидрокси, амино, нитро, арила, галогеналкила и алкила. В качестве дополнительного примера, арильная группа может быть необязательно замещена одним или несколькими из галогена, циано, алкокси, гидрокси, нитро, галогеналкила и алкила. Средний специалист в данной области техники должен осознавать, что комбинации заместителей, предусмотренные настоящим изобретением, представляют собой такие комбинации, которые приводят к образованию стабильных или химически возможных соединений. Используемый в настоящем документе термин «стабильный» относится к соединениям, которые по существу не изменяются при воздействии условий, предусматривающих их получение, определение и предпочтительно их восстановление, очистку и применение для одной или нескольких из раскрытых в настоящем документе целей. Согласно некоторым вариантам осуществления, стабильное соединение или химически возможное соединение представляет собой соединение, которое по существу не изменяется при хранении при температуре 40°C или менее в отсутствие влаги или в других химически реакционноспособных условиях, по меньшей мере, в течение недели. Если две алкоксигруппы связаны с одним и тем же атомом или смежными атомами, то две алкоксигруппы могут образовывать кольцо вместе с атомом(ами), с которым(и) они связаны.
В общем, термин «замещенный», с предшествующим термином «необязательно» или без него, относится к замещению водородных радикалов в представленной структуре радикалом конкретного заместителя. Конкретные заместители описаны выше в определениях и ниже в описании соединений и их примеров. Если не указано иное, то необязательно замещенная группа может содержать заместитель в каждом способном к замещению положении группы, и если более чем одно положение в представленной структуре может быть замещено более чем одним заместителем, выбранным из конкретной группы, то в каждом положении заместитель может быть тем же самым или другим. Циклический заместитель, такой как гетероциклоалкил, может быть связан с другим кольцом, таким как циклоалкил, с образованием спиробициклической кольцевой системы, например, оба кольца имеют один общий атом. Средний специалист в данной области техники должен осознавать, что комбинации предусмотренных в настоящем документе заместителей представляют собой такие комбинации, которые приводят к образованию стабильных или химически возможных соединений.
Используемое в настоящем документе выражение «до» относится к нулю или любому целому числу, которое равно или меньше числа в последующей фразе. Например, «до 3» означает любое из 0, 1, 2 и 3.
Используемый в настоящем документе термин «алифатический», «алифатическая группа» или «алкил» означает неразветвленную (т.е., линейную) или разветвленную, замещенную или незамещенную углеводородную цепь, которая является полностью насыщенной, или которая содержит одну или несколько единиц ненасыщенности. Если не указано иное, то алифатическая группа содержит 1-20 алифатических атомов углерода. Согласно некоторым вариантам осуществления, алифатические группы содержат 1-10 алифатических атомов углерода. Согласно другим вариантам осуществления, алифатические группы содержат 1-8 алифатических атомов углерода. Согласно другим вариантам осуществления, алифатические группы содержат 1-6 алифатических атомов углерода, и согласно другим вариантам осуществления, алифатические группы содержат 1-4 алифатических атомов углерода. Подходящие алифатические группы включают без ограничения неразветвленные или разветвленные, замещенные или незамещенные алкильные, алкенильные, алкинильные группы. Термин «циклоалифатический» или «циклоалкил» означает моноциклический, бициклический или трициклический углеводород, который является полностью насыщенным или который содержит одну или несколько единиц ненасыщенности, но который не является ароматическим и содержит одну точку присоединения к остатку молекулы. Согласно некоторым вариантам осуществления, термин «циклоалифатический» относится к моноциклическому C3-С8углеводороду или бициклическому C8-С12углеводороду, который является полностью насыщенным или который содержит одну или несколько единиц ненасыщенности, но который не является ароматическим и который содержит одну точку присоединения к остатку молекулы, причем каждое отдельное кольцо в указанной бициклической кольцевой системе содержит 3-7 членов.
Используемый в настоящем документе термин «электроноакцепторная группа» означает атом или группу, которая является электроотрицательной относительно водорода (см., например, «Advanced Organic Chemistry: Reactions, Mechanisms, and Structure», Jerry March, 4th Ed., John Wiley & Sons (1992), e.g., pp. 14-16, 18-19, и т.д.). Примеры таких заместителей включают галоген, такой как Cl, Br или F, CN, COOH, CF3, и т.д.
Если не указано иное, то используемый в настоящем документе термин «гетероцикл», «гетероциклил», «гетероциклоалифатический», «гетероциклоалкил» или «гетероциклическое соединение» означает неароматические, моноциклические, бициклические или трициклические кольцевые системы, в которых один или несколько кольцевых атомов в одном или нескольких кольцах представляют собой независимо выбранный гетероатом. Гетероциклическое кольцо может быть насыщенным или может содержать одну или несколько ненасыщенных связей. Согласно некоторым вариантам осуществления, «гетероцикл», «гетероциклильная», «гетероциклоалифатическая», «гетероциклоалкильная» или «гетероциклическая» группа содержит от трех до четырнадцати кольцевых атомов, где один или несколько кольцевых атомов представляет собой гетероатом, независимо выбранный из кислорода, серы, азота или фосфора, и каждое кольцо в кольцевой системе содержит от 3 до 7 кольцевых атомов.
Термин «гетероатом» означает кислород, серу, азот, фосфор или кремний (включая любую окисленную форму азота, серы, фосфора или кремния; четвертичную форму любого основного азота или способный к замещению азот в гетероциклическом кольце, например N (как в 3,4-дигидро-2Н-пирролиле), NH (как в пирролидиниле) или NR+ (как в N-замещенном пирролидиниле)).
Используемый в настоящем документе термин «ненасыщенный» означает, что фрагмент содержит одну или несколько единиц ненасыщенности, но не является ароматическим.
Используемый в настоящем документе термин «алкокси» или «тиоалкил» относится к определенной выше алифатической группе, присоединенной к основной углеродной цепи через атом кислорода («алкокси») или серы («тиоалкил»). Используемая в настоящем документе алкоксигруппа включает алкенилоксигруппы и алкинилоксигруппы.
Используемый в настоящем документе термин «арил», отдельно или как часть большего фрагмента, как в «аралкил», «аралкокси» или «арилоксиалкил», относится к моноциклическим, бициклическим и трициклическим кольцевым системам с общим числом кольцевых атомов углерода от пяти до четырнадцати, где, по меньшей мере, одно кольцо в системе является ароматическим, и где, каждое кольцо в системе содержит от 3 до 7 кольцевых атомов углерода. Термин «арил» может использоваться взаимозаменяемо с термином «арильное кольцо».
Используемый в настоящем документе термин «гетероарил», отдельно или как часть большего фрагмента, как в «гетероаралкил» или «гетероарилалкокси», относится к моноциклическим, бициклическим и трициклическим кольцевым системам с общим числом кольцевых атомов от пяти до четырнадцати, где, по меньшей мере, одно кольцо в системе является ароматическим, по меньшей мере, одно кольцо в системе содержит один или несколько гетероатомов, и где, каждое кольцо в системе содержит от 3 до 7 кольцевых атомов. Термин «гетероарил» может использоваться взаимозаменяемо с термином «гетероарильное кольцо» или термином «гетероароматический».
Термин «алкилиденовая цепь» относится к неразветвленной или разветвленной углеродной цепи, которая может быть полностью насыщенной или содержать одну или несколько единиц ненасыщенности и иметь две точки присоединения к остатку молекулы.
Если не указано иное, то подразумевается, что изображенные в настоящем документе структуры также включают все изомерные (например, энантиомерные, диастереоизомерные и геометрические (или конформационные)) формы структуры; например, R и S конфигурации для каждого центра асимметрии, изомеры (Z) и (E) по двойной связи и конформационные (Z) и (E) изомеры. Таким образом, под объем настоящего изобретения подпадают простые стереохимические изомеры, а также энантиомерные, диастереоизомерные и геометрические (или конформационные) смеси соединений согласно настоящему изобретению.
Если не указано иное, то все таутомерные формы соединений согласно настоящему изобретению подпадают под объем настоящего изобретения. Таким образом, таутомеры соединений формулы I подпадают под объем настоящего изобретения.
Кроме того, если не указано иное, то подразумевается, что изображенные в настоящем документе структуры также включают соединения, которые отличаются только наличием одного или нескольких изотопно обогащенных атомов. Например, под объем настоящего изобретения подпадают соединения формулы I, в которых один или несколько атомов водорода заменены на дейтерий или тритий, или один или несколько атомов углерода заменены 13C- или 14C-обогащенным углеродом. Такие соединения применимы, например, в качестве аналитических средств, зондов в биологических методах анализа, или блокаторов натриевых каналов с улучшенным терапевтическим профилем.
В формулах и рисунках линия, пересекающая кольцо и связанная с R группой, например, как в
означает, что R группа может быть связана с любым атомом углерода или, в соответствующих случаях, гетероатомом, таким как N, этого кольца, где это позволяет валентность.
Подразумевается, что в пределах определения термина, такого как, например, R1, R2, R3, R4, R5, R6 или R7, если CH2 группа или, взаимозаменяемо, метиленовая группа, могут быть заменены O, CO, S, SO, SO2 или NR8, то она включает любую CH2 группу, включая CH2 в составе концевой метильной группы. Например, -CH2CH2CH2SH находится в пределах определения C1-С8алкила, в котором до двух CH2 групп могут быть заменены S, поскольку CH2 группа концевой метильной группы была заменена S.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R1 представляет собой C1-С8алкил, или два R1 образуют вместе с атомами, к которым они присоединены, 3-7-членное конденсированное циклоалкильное или спироциклическое кольцо. Согласно другому варианту осуществления, R1 представляет собой CH3, или два R1 образуют вместе конденсированное циклогексильное кольцо.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R2 представляет собой H, C1-С8алкил, галоген, CF3, CN, CON(R8)2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2, CF2 или NR8. Согласно другому варианту осуществления, R2 представляет собой COCF3, COtBu, Cl, COCH3, CF2CF3, CH2CF3, CF3, CN, Br, COCH(CH3)2, COCH2CH3, CH(OH)CF3, SO2CH3,  COPh,
COPh, 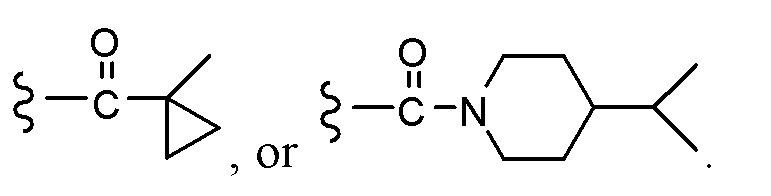 или .
или .
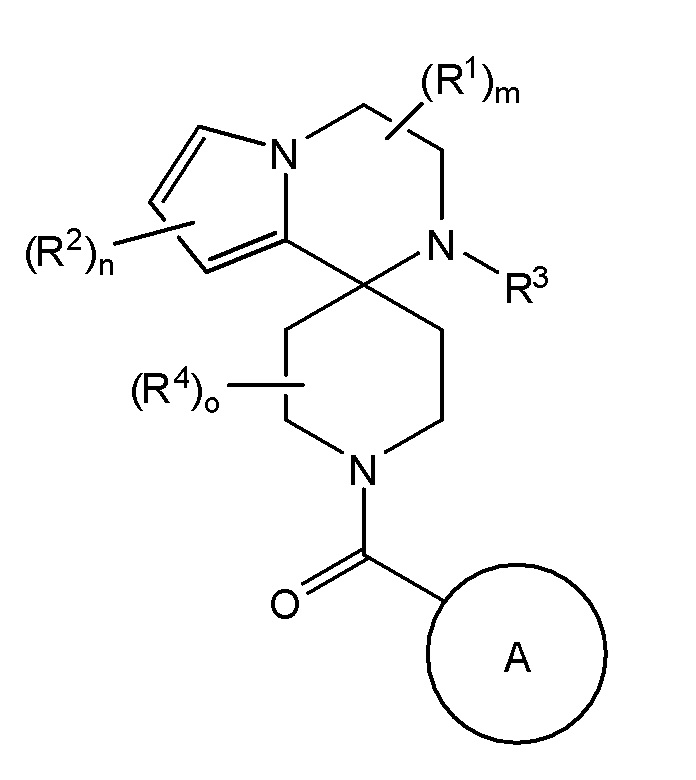
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R3 представляет собой H, C1-С8алкил, CO2R8, COR8, СОН, CON(R8)2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, CF2, S, SO, SO2 или NR8. Согласно другому варианту осуществления, R3 представляет собой H, CH3, CH2CH3, CH2CH2OCH3, бензил, CH2CH(CH2)2, CH(CH2)2, циклобутил, COCH3, CO2CH3, CO2CH2CH3, CH2CF3, CH2CHF2, СОН, CON(CH3)2 или CONHCH3.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R4 представляет собой H, галоген или C1-С8алкил. Согласно другому варианту осуществления, R4 представляет собой H, F или CH3.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где m равно 0, 1 или 2. Согласно другому варианту осуществления, n равно 0, 1 или 2. Согласно другому варианту осуществления, o равно 0 или 1.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где A представляет собой
 ,
,
где:
R5 представляет собой H, C1-С8алкил, C3-С8циклоалкил, C1-С8алкокси, галоген, CN, OH, OR8, N(R8)2, NR8SO2R8, SO2R8, SOR8, SR8, CO2R8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CHF2, CF3, OCF3, OCHF2, R9, гетероциклоалкил, гетероциклоалкокси, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8;
R6 представляет собой H, C1-С8алкил, C3-С8циклоалкил, C1-С8алкокси, C3-С8циклоалкокси, галоген, CN, OH, OR8, N(R8)2, NR8SO2R8, SO2R8, SOR8, SR8, CO2R8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, OCF3, OCHF2, R9, гетероциклоалкил, гетероциклоалкокси, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8;
R7 представляет собой H, C1-С8алкил, C3-С8циклоалкил, C1-С8алкокси, галоген, CN, OH, OR8, N(R8)2, NR8SO2R8, SO2R8, OSO2R8, SOR8, SR8, CO2R8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, OCF3, OCHF2, R9, гетероциклоалкил, гетероциклоалкокси, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-(R9)P, где p равно 1 или 2, и где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8; или
R5 и R6, или R6 и R7, оба представляют собой C1-С8алкил, и образуют вместе с атомами углерода, к которым они присоединены, необязательно замещенное кольцо, содержащее до 2 гетероатомов.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R5 представляет собой H, C1-С8алкил, C1-С8алкокси, галоген, OCF3, OCHF2, R9, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR7. Согласно другому варианту осуществления, R5 представляет собой H, CH3, OCH3, OCF3, OPh, Ph, OCHF2 или F.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R6 представляет собой H, C1-С8алкил, C1-С8алкокси, галоген, R9, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8. Согласно другому варианту осуществления, R6 представляет собой H, CH3, OCH3, OCH2CH3, OCH2CH2CH3, OCH(CH3)2, CF3, CN, Ph, SO2CH3, OH, CH(CH3)2, OCH2CH2CH2CH3, F, Cl или CH2OH.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где R7 представляет собой H, C1-С8алкил, C1-С8алкокси, SO2R8, OSO2R8, SO2N(R8)2, R9, или неразветвленный, разветвленный или циклический (C1-C8)-(R9)P, где p равно 1 или 2, и где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8. Согласно другому варианту осуществления, R7 представляет собой H, CH2CH3, tBu, Cl, F, OH, C(=CH2)CH3, OC(=CH2)CH3, OCH3, OCH2CH2CH2CH3, CH2OH, OCH2CH2OH, OCH2CH2CH2OH, OtBu, OCH(CH3)(CH2CH3), OCH2C(CH3)2OH, C(CH3)2OH, CH2C(CH3)2OH, CH(OH)CH(CH3)2, C(CH3)2CH2OH, OCH2CH2CH(CH3)2, OCH2CH2CH3, OCH(CH3)2, OCH2CH2OCH3,  SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2,
SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2,  SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,
SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2, 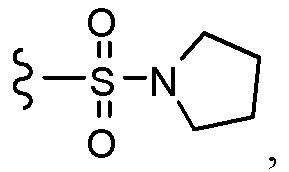 Ph,
Ph, 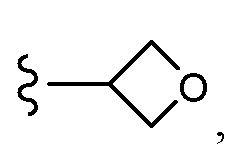 OCH2CH2OCH3, CH(CH3)2, SO2N(CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OPh, OCH2CH2CH2CH3, CH2OPh,
OCH2CH2OCH3, CH(CH3)2, SO2N(CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OPh, OCH2CH2CH2CH3, CH2OPh, 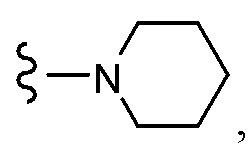 OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2CH3, CH2Ph,
OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2CH3, CH2Ph, 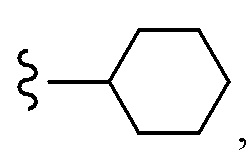 CCCH2OCH3, SO2CHF2, OCF3,
CCCH2OCH3, SO2CHF2, OCF3, 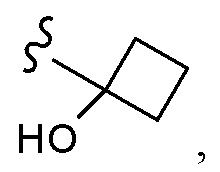 OCHF2,
OCHF2, 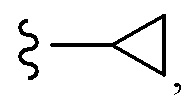 CH2CH(CH3)2, OCH2tBu,
CH2CH(CH3)2, OCH2tBu,  OCH2CF3,
OCH2CF3, 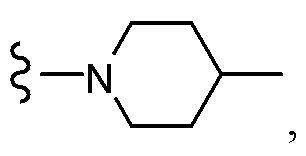
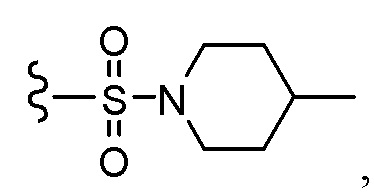 CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, C(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2,
CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, C(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2, 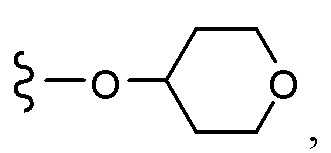 CF3, OCH2C(CH3)2F,
CF3, OCH2C(CH3)2F, 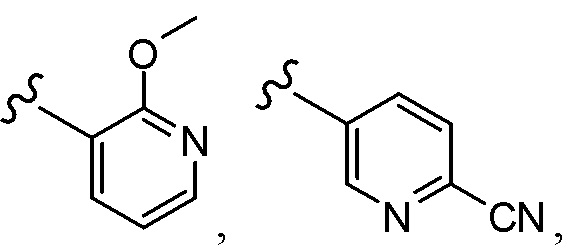
 CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,
CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,  или OCH2CH2OCF3.
или OCH2CH2OCF3.
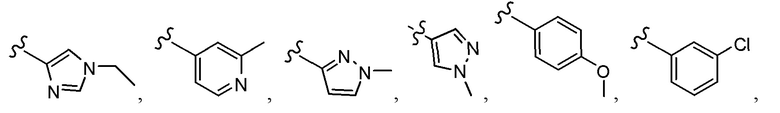
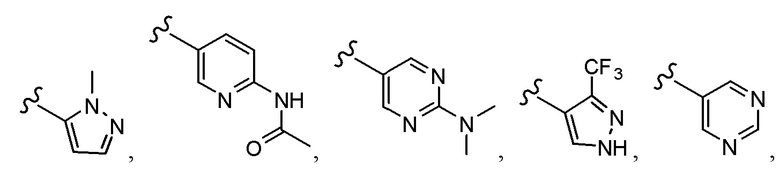
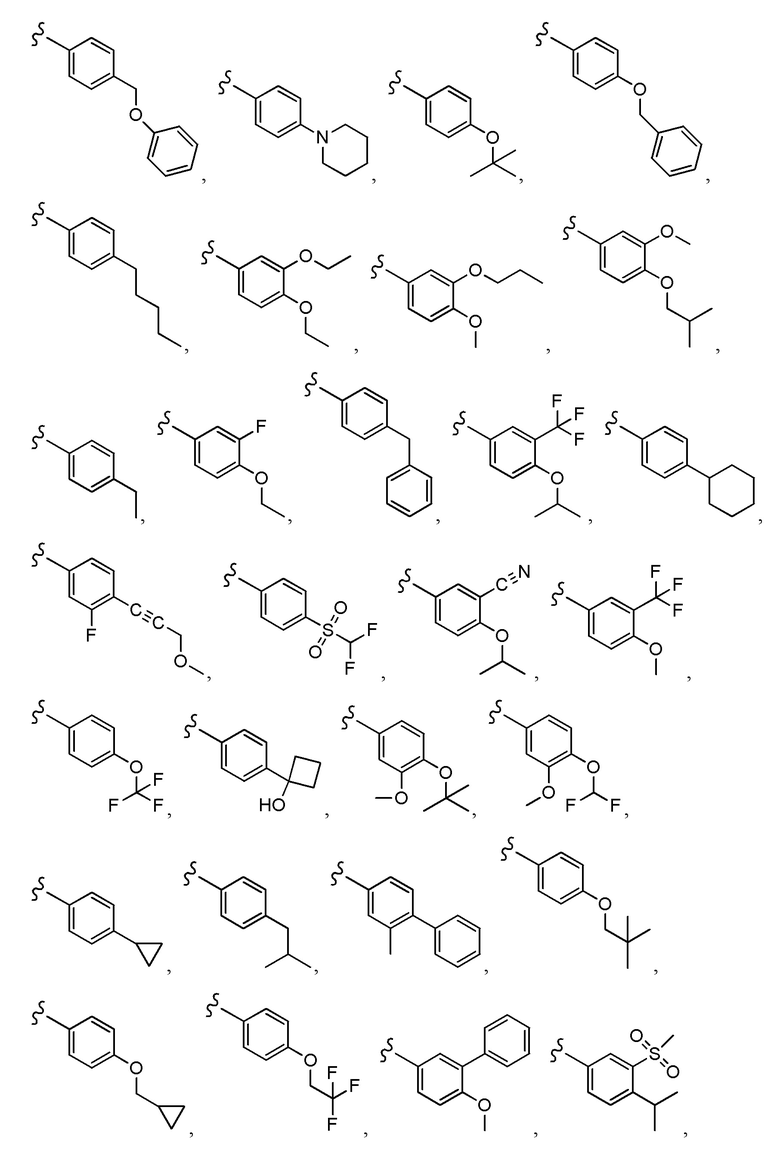
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где A представляет собой 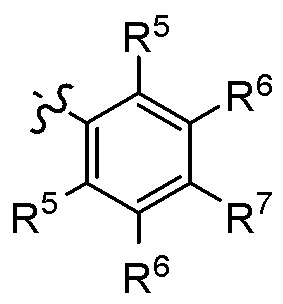 и выбрано из:
и выбрано из:



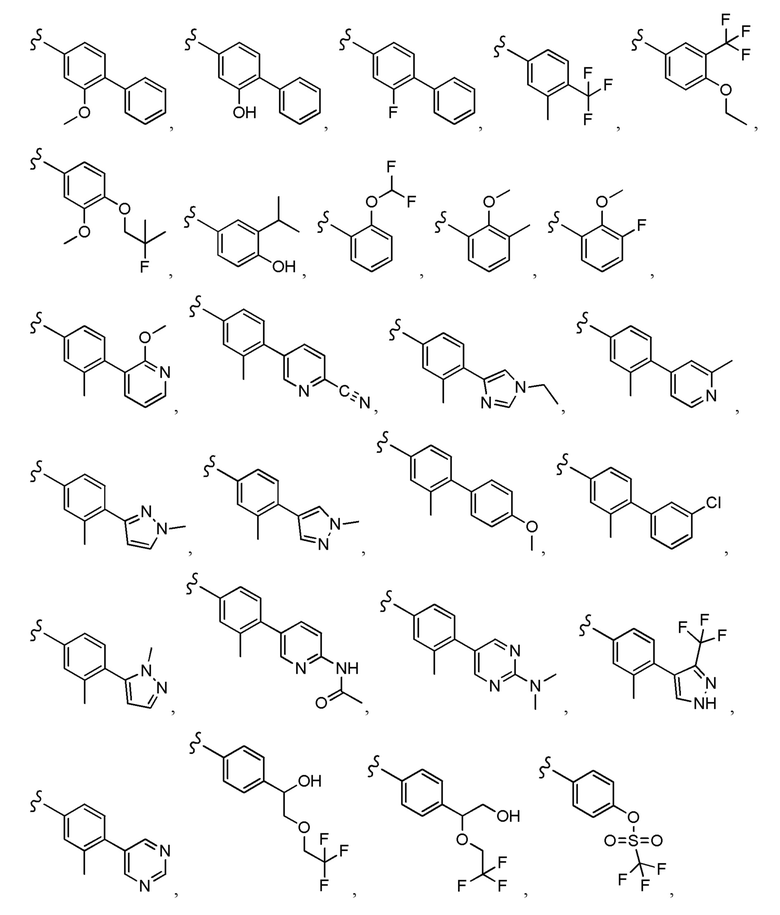
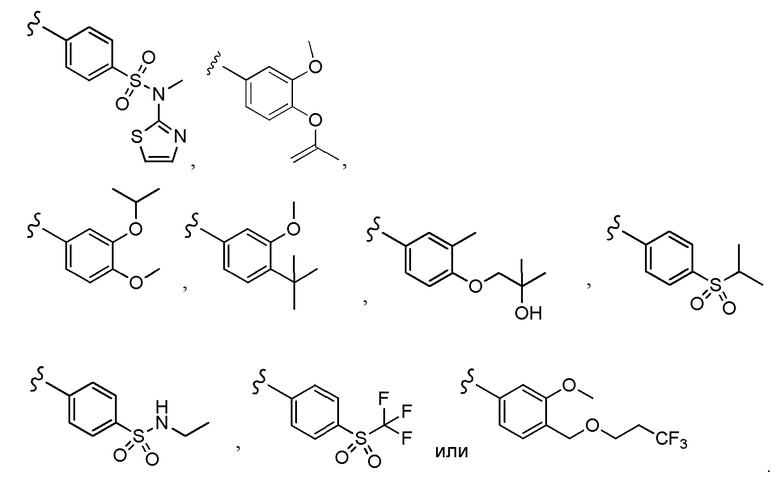
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где A представляет собой гетероарил или гетероциклическое соединение. Согласно другому варианту осуществления, A представляет собой моноциклический гетероарил, содержащий от 1 до 3 гетероатомов, независимо выбранных из N, O или S. Согласно другому варианту осуществления, A выбирают из бициклического гетероарила, содержащего от 1 до 3 гетероатомов, выбранных из N, O или S.
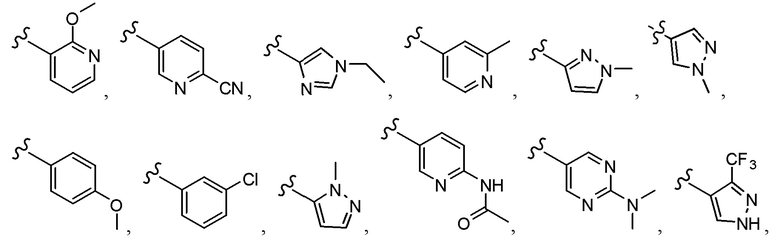
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I и сопутствующим определениям, где A выбирают из следующего:
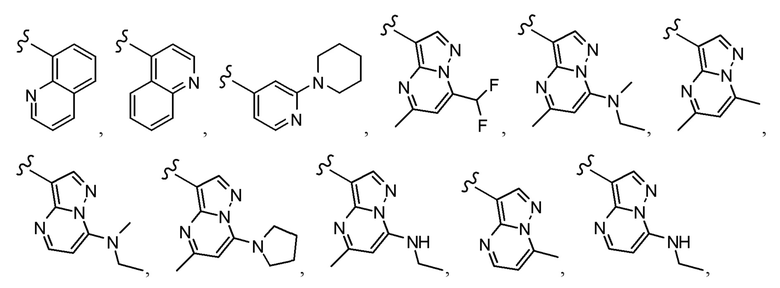
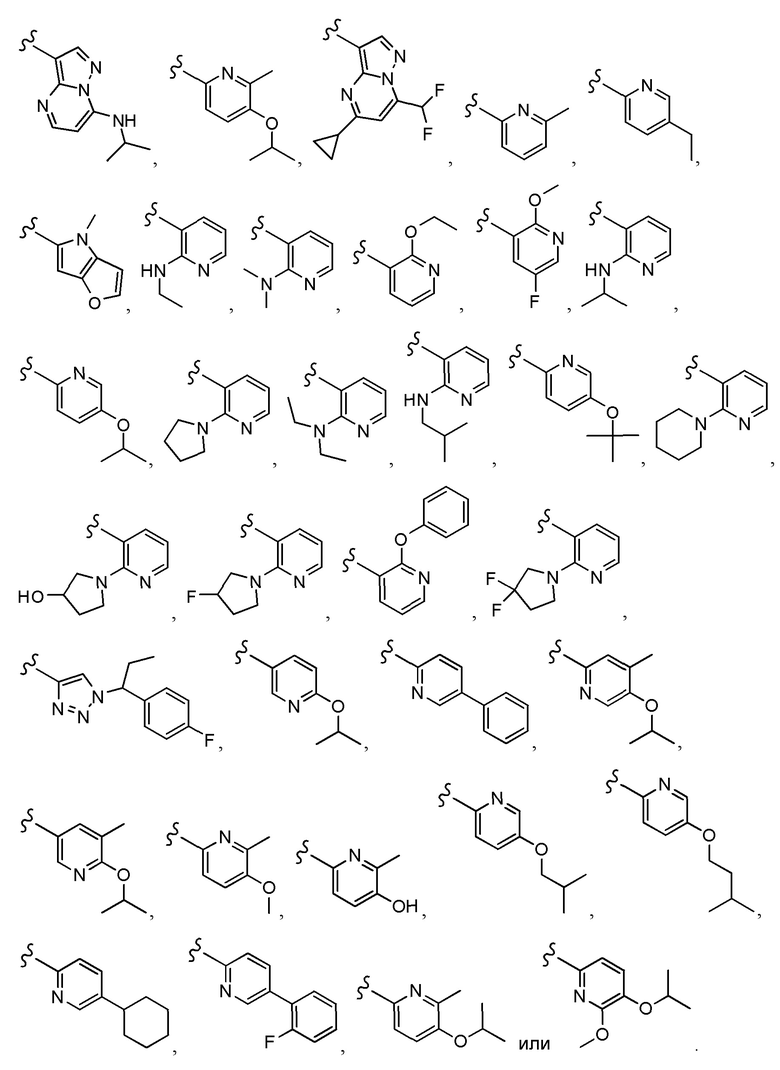
Согласно другому варианту осуществления, соединения согласно настоящему изобретению характеризуются формулой IA:
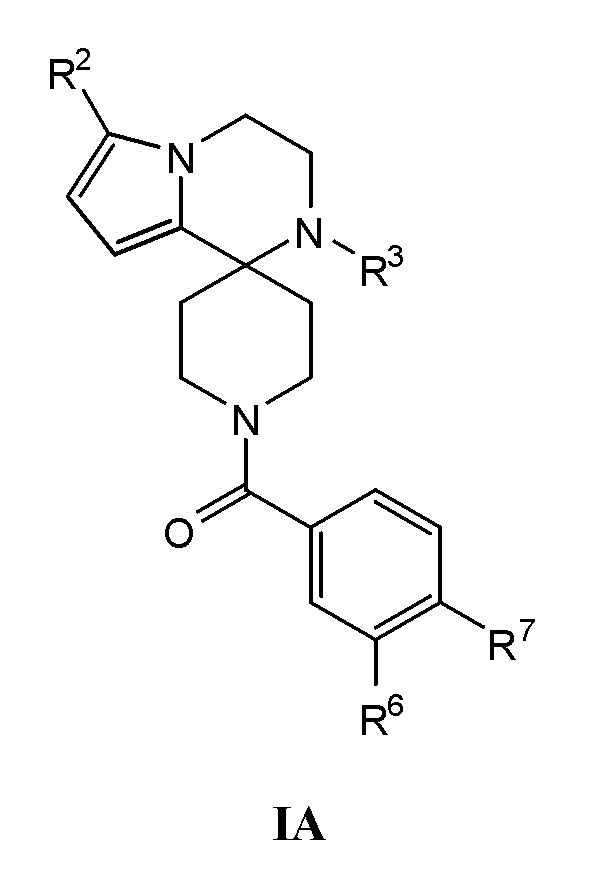
где:
R2 представляет собой H, C1-С8алкил, галоген, C1-С8галогеналкил, CN, OH, SO2R8, SR8, SOR8, COR8, CO2R8, CON(R8)2, SO2N(R8)2, CF3, CHF2, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, S, SO, SO2 CF2 или NR8;
R3 представляет собой H, C1-С8алкил, CO2R8, COR8, СОН, CON(R8)2, CF3, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до двух CH2 групп могут быть заменены O, CO, CF2, S, SO, SO2 или NR8;
R6 представляет собой H, C1-С8алкил, C3-С8циклоалкил, C1-С8алкокси, C3-С8циклоалкокси, галоген, CN, OH, OR8, N(R8)2, NR8SO2R8, SO2R8, SOR8, SR8, CO2R8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, OCF3, OCHF2, R9, гетероциклоалкил, гетероциклоалкокси, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8;
R7 представляет собой H, C1-С8алкил, C3-С8циклоалкил, C1-С8алкокси, галоген, CN, OH, OR8, N(R8)2, NR8SO2R8, SO2R8, SOR8, SR8, CO2R8, NR8COR8, NR8CO2R8, CON(R8)2, SO2N(R8)2, CF3, OCF3, OCHF2, R9, гетероциклоалкил, гетероциклоалкокси, арил, гетероарил, или неразветвленный, разветвленный или циклический (C1-C8)-R9, где до трех CH2 групп могут быть заменены O, CO, S, SO, SO2 или NR8.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где R2 представляет собой H, COCF3, COtBu, Cl, COCH3, CF2CF3, CH2CF3, CF3, CN, Br, COCH(CH3)2, COCH2CH3, CH(OH)CF3, SO2CH3,  COPh,
COPh,  или
или 
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где R3 представляет собой H, CH3, CH2CH3, CH2CH2OCH3, CH2CH2OH CH2CO2CH2CH3, CH2CON(CH3)2, CH2CONH2, CH2CN, бензил, циклобутил, CH2CH(CH2)2, CH(CH2)2, CH2CF3, CH2CHF2, COCH3, COCH2CH3, CO2CH3, CO2CH2CH3, СОН, CONH(CH3)2 или CONHCH3.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где R6 представляет собой H, CH3, OCH3, OCH2CH3, OCH2CH2CH3, OCH(CH3)2, CF3, CN, Ph, SO2CH3, ОН, CH(CH3)2, OCH2CH2CH2CH3, F, Cl или CH2OH.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где R7 представляет собой H, CH3, CH2CH3, tBu, Cl, F, OH, C(=CH2)CH3, OC(=CH2)CH3, OCH3, OCH2CH2CH2CH3, CH2OH, OCH2CH2OH, OCH2CH2CH2OH, OtBu, OCH(CH3)(CH2CH3), OCH2C(CH3)2OH, C(CH3)2OH, CH2C(CH3)2OH, CH(OH)CH(CH3)2, C(CH3)2CH2OH, OCH2CH2CH(CH3)2, OCH2CH2CH3, OCH(CH3)2, OCH2CH2OCH3, 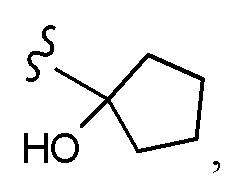 SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2,
SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2, 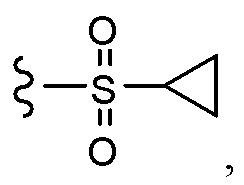 SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,
SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2, 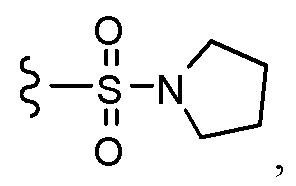 OPh, Ph,
OPh, Ph, 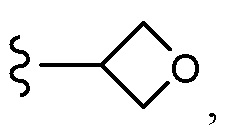 OCH2CH2OCH3, CH(CH3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OCH2CH2CH2CH3, CH2OPh,
OCH2CH2OCH3, CH(CH3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OCH2CH2CH2CH3, CH2OPh,  OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2Ph,
OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2Ph, 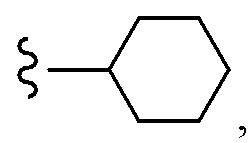 CCCH2OCH3, SO2CHF2, OCF3,
CCCH2OCH3, SO2CHF2, OCF3, 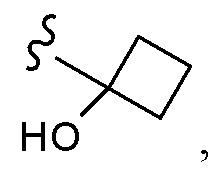 OCHF2,
OCHF2, 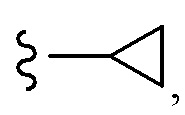 CH2CH(CH3)2, OCH2tBu,
CH2CH(CH3)2, OCH2tBu,  OCH2CF3,
OCH2CF3,  CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, C(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2,
CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, C(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2,  CF3, OCH2C(CH3)2F,
CF3, OCH2C(CH3)2F,
 CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,
CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,  или OCH2CH2OCF3.
или OCH2CH2OCF3.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где R2 представляет собой H, CF3, COCF3, COtBu, Cl, COCH3, CF2CF3, CH2CF3 или CN; R3 представляет собой H, CH2CH2OCH3, бензил, CH3, CH2CH3, CH2CH(CH2)2, циклобутил, COCH3, CO2CH3, СОН, CH(CH2)2, CH2CF3, CH2CHF2, CO2CH2CH3, CON(CH3)2 или CONHCH3; R6 представляет собой СН3, ОСН3, ОСН2СН3, OCH2CH2CH2CH3, CH2OH, F или Cl; и R7 представляет собой F, CH2CH3, tBu, OH, OCH3, OCH2CH2CH2CH3, OtBu, OCH(CH3)(CH2CH3), OCH2CH2OH, OCH2CH2CH2OH, OCH2C(CH3)2OH, C(CH3)2OH, C(=CH2)CH3, OC(=CH2)CH3, CH2OH, C(CH3)2CH2OH,  OCH2CH2CH(CH3)2, OCH2CH2CH3, OCH(CH3)2, OCH2CH2OCH3, SO2CH3, SO2CH2CH3, SO2CH(CH3)2,
OCH2CH2CH(CH3)2, OCH2CH2CH3, OCH(CH3)2, OCH2CH2OCH3, SO2CH3, SO2CH2CH3, SO2CH(CH3)2,  SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2 или OCH2CH2OCF3.
SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2 или OCH2CH2OCF3.
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы IA и сопутствующим определениям, где фрагмент 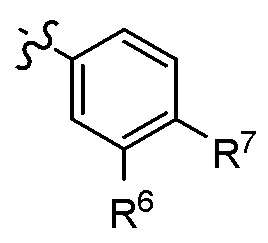 выбирают из:
выбирают из:


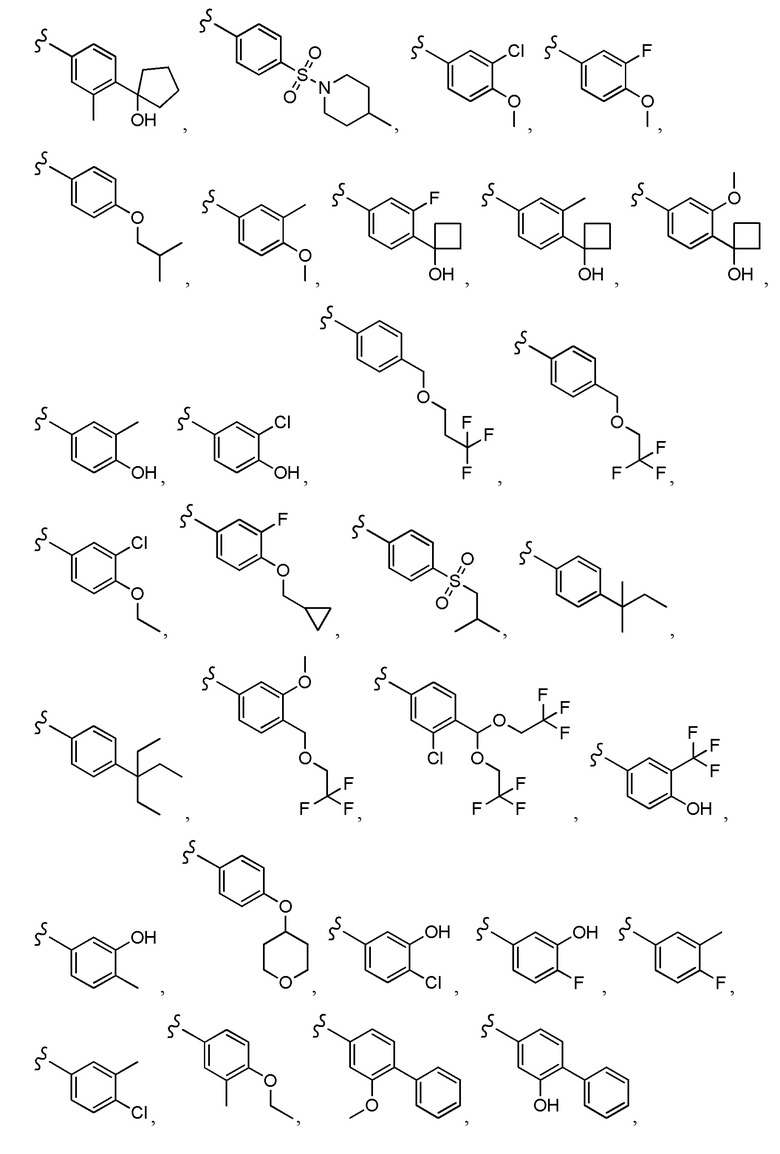
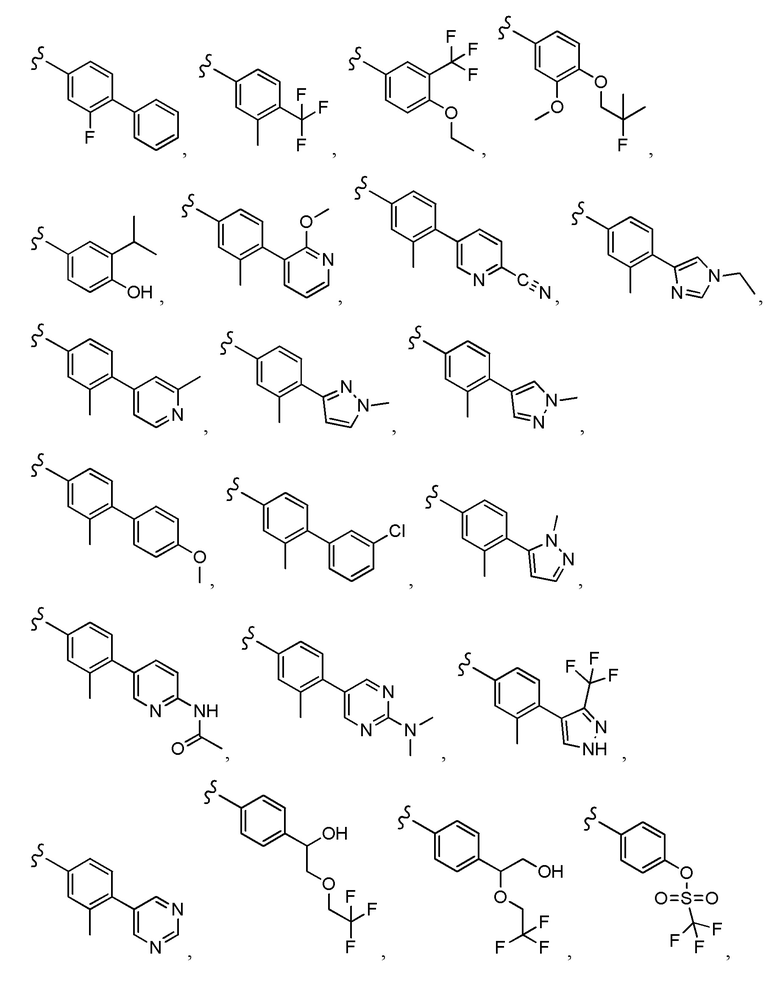
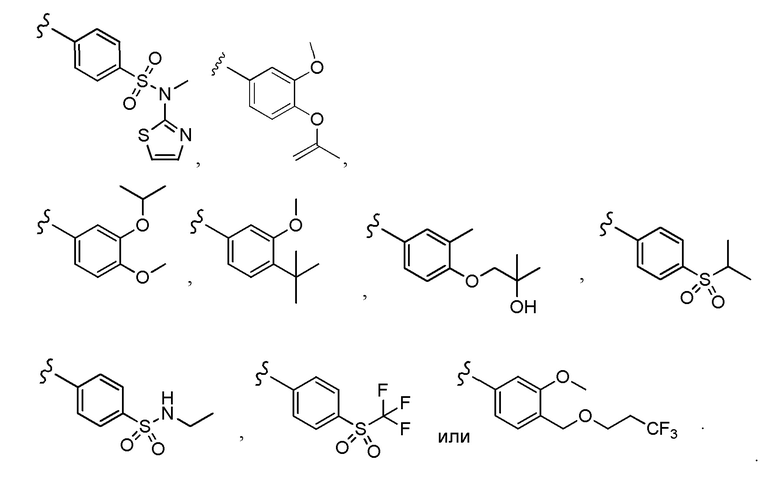
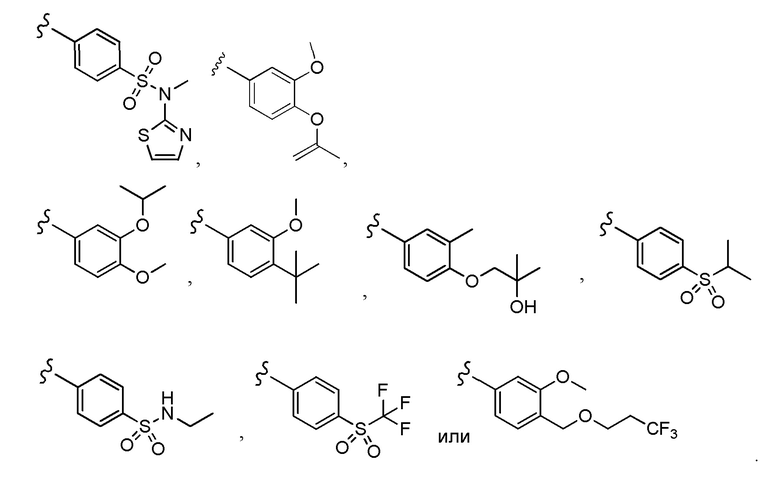
Согласно другому варианту осуществления, настоящее изобретение относится к соединению формулы I, где соединение выбирают из таблицы 1:
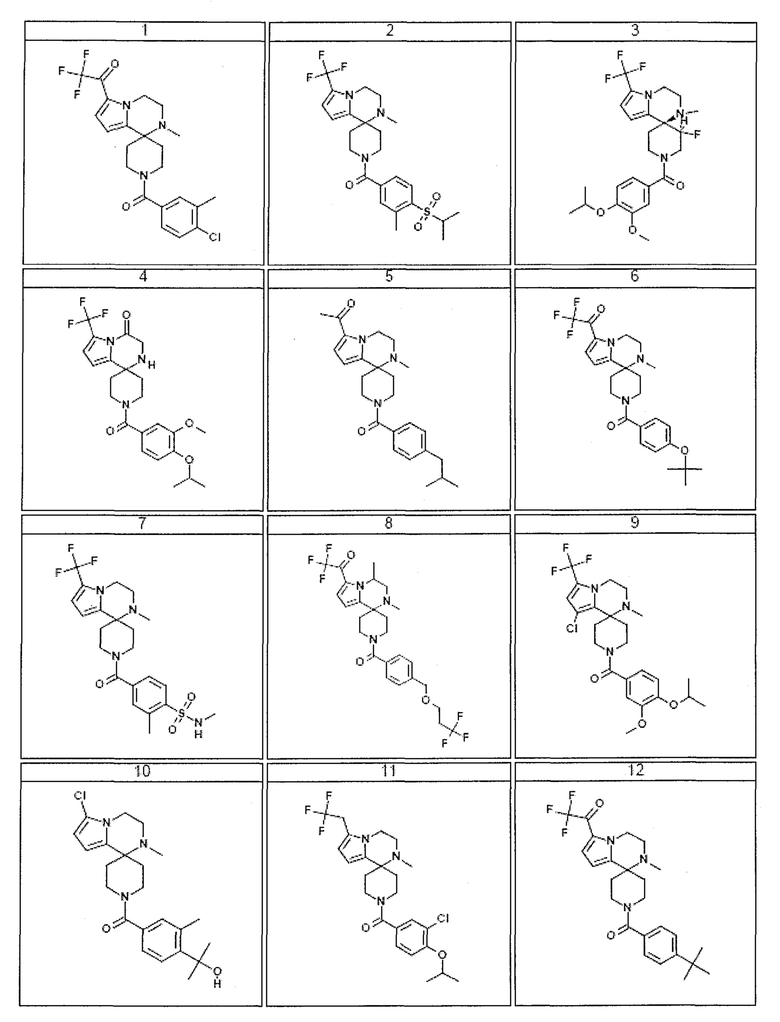

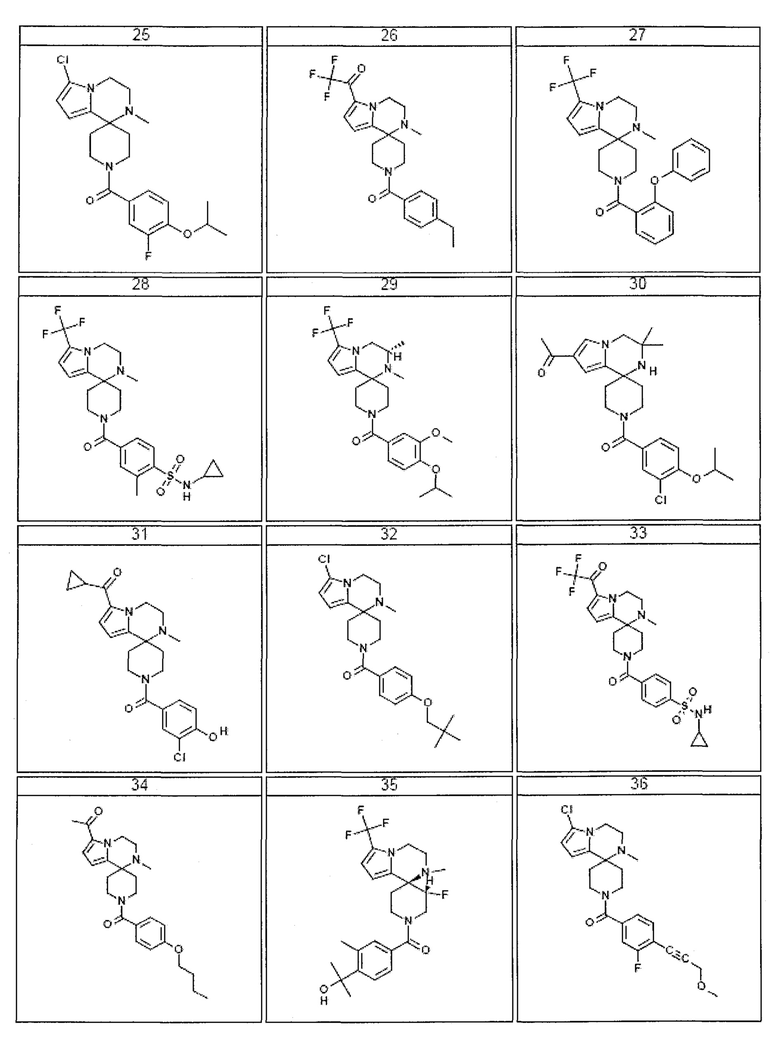

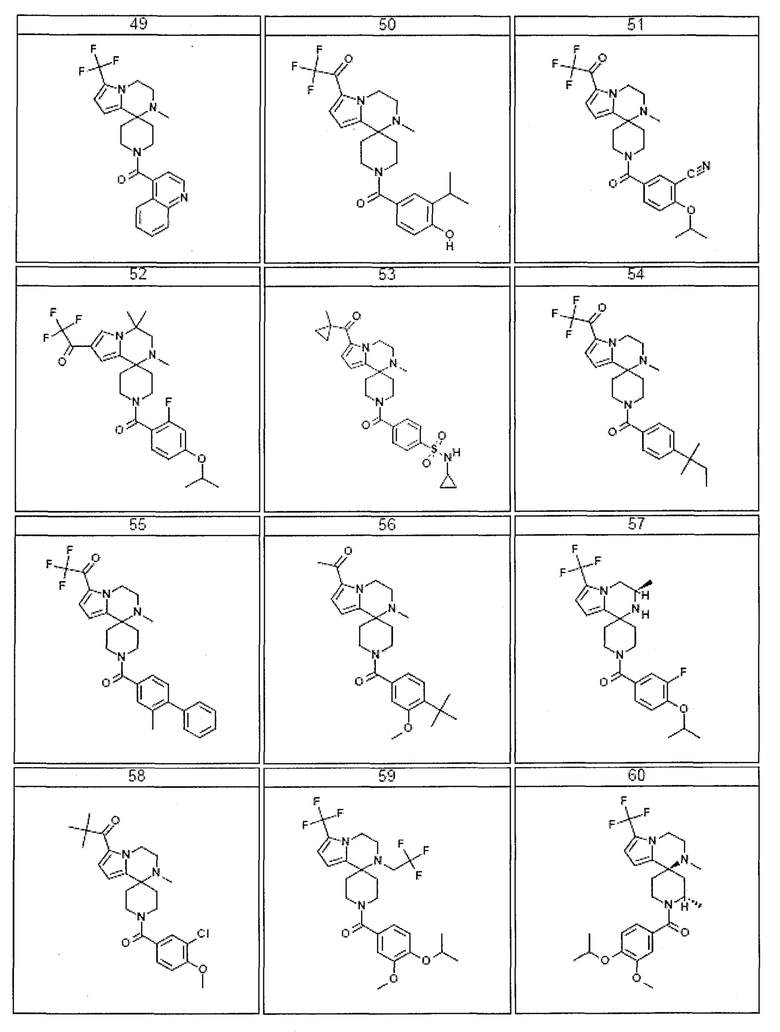
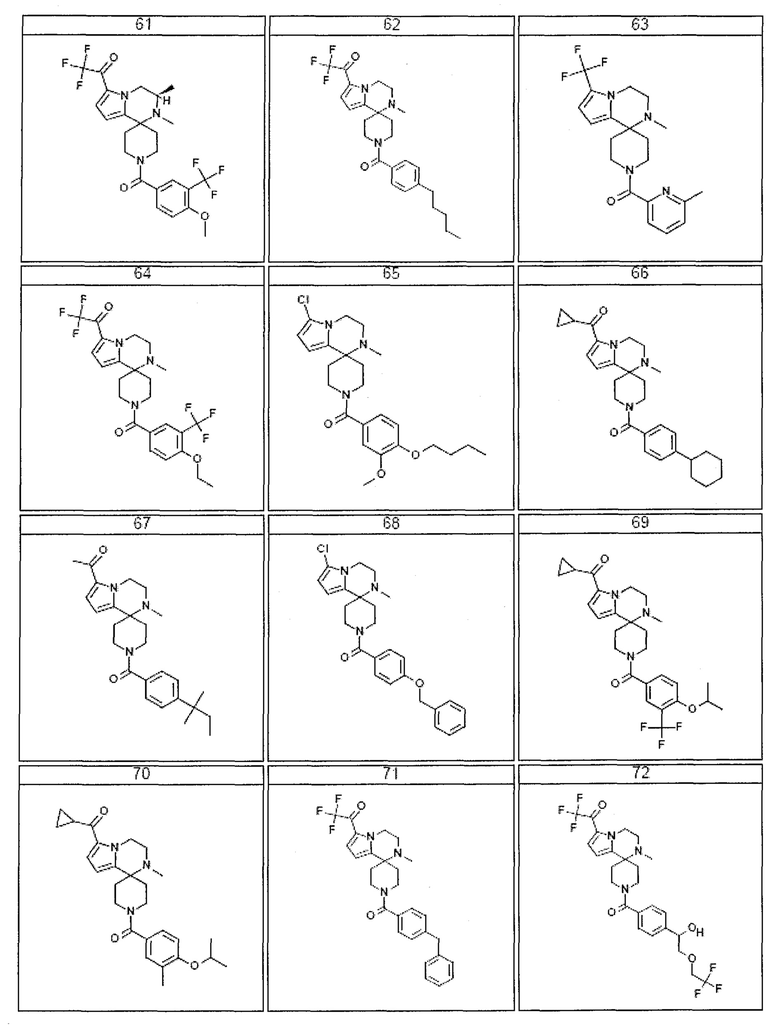
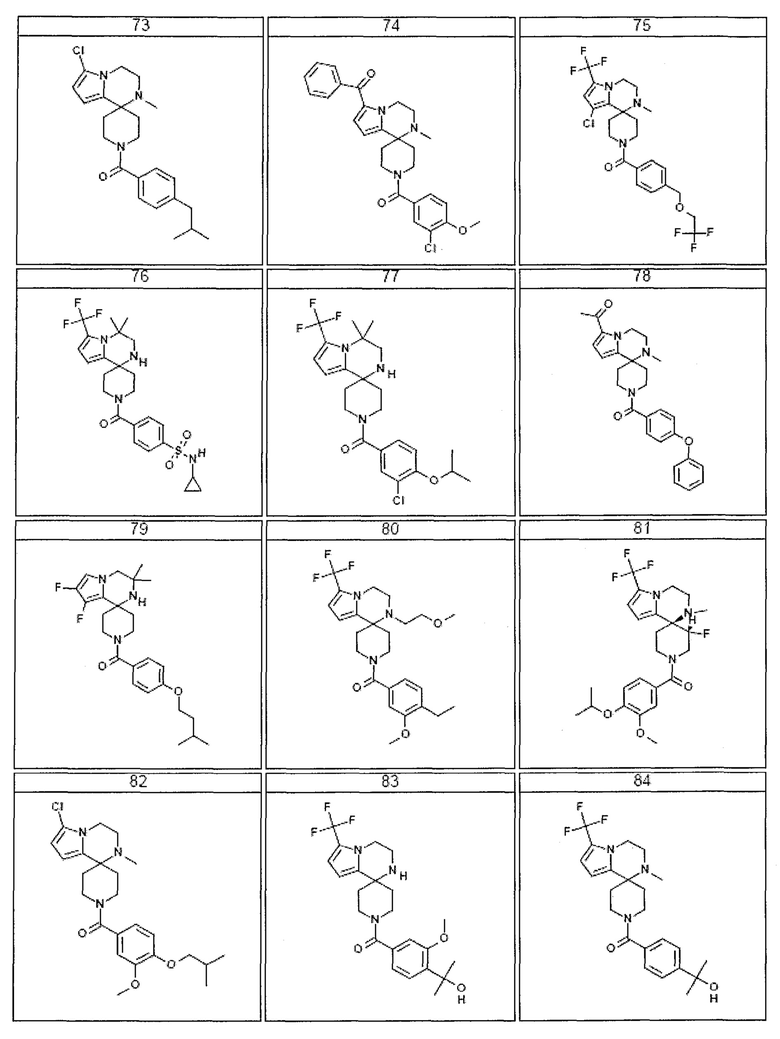
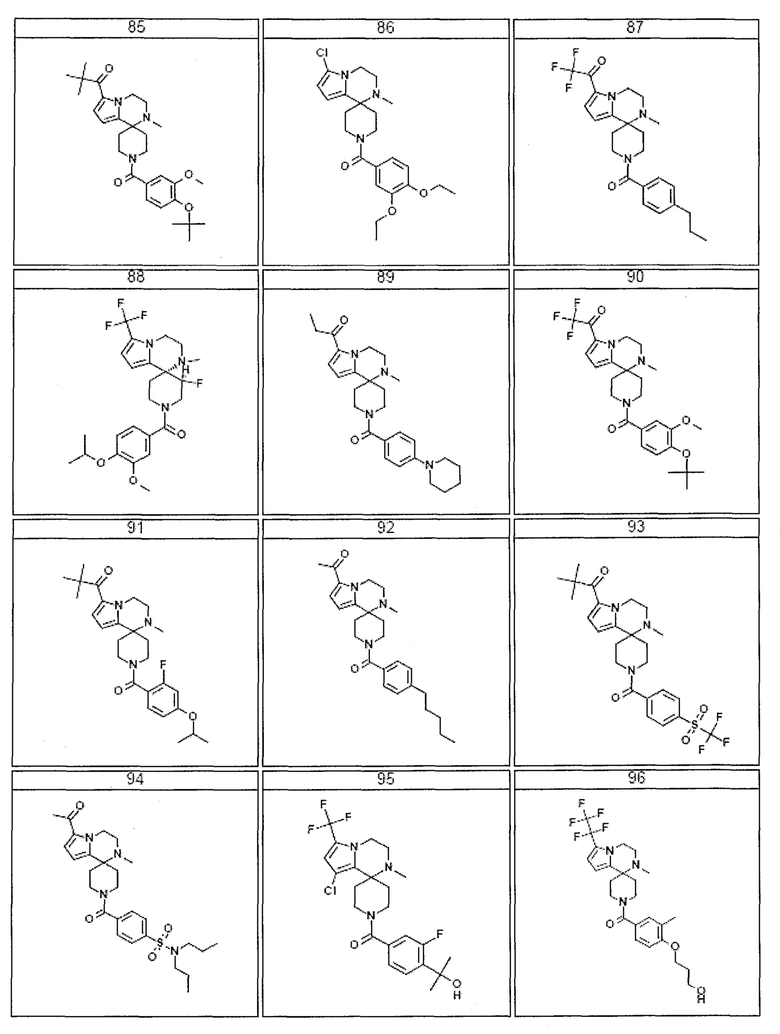
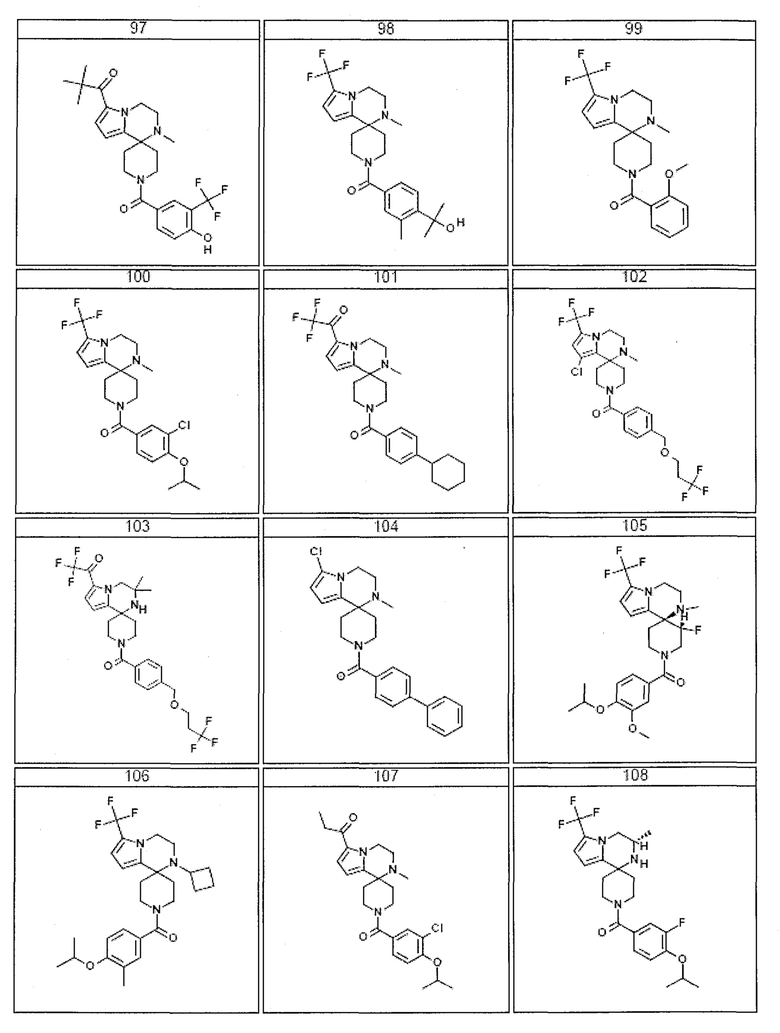
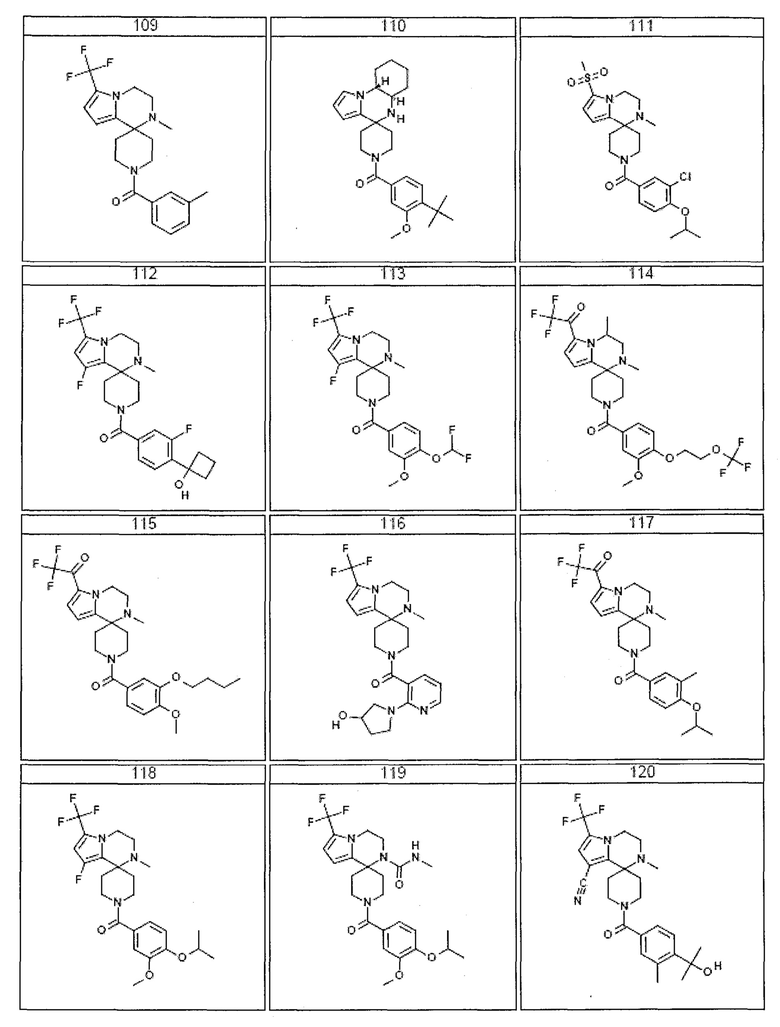
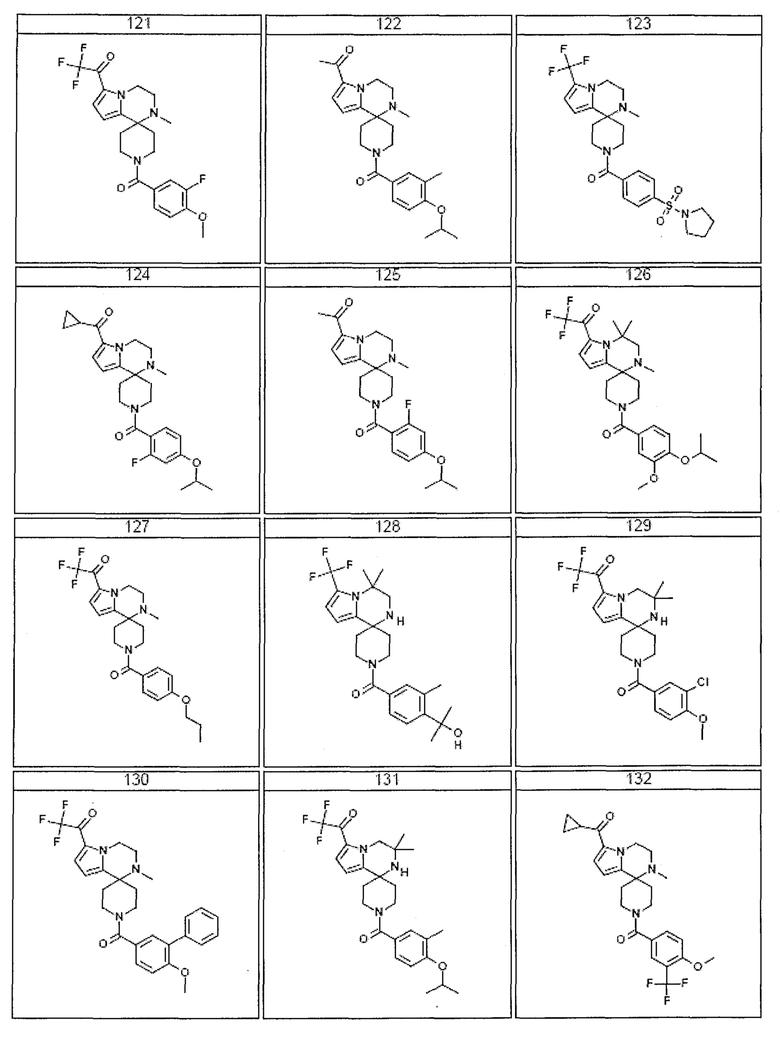
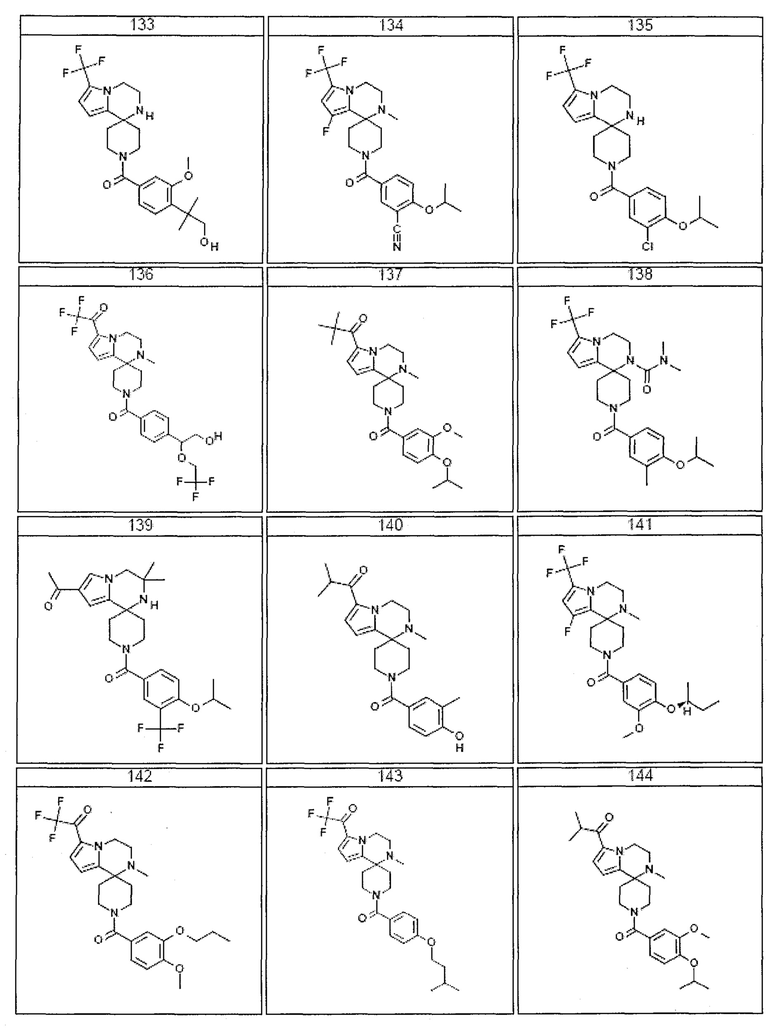
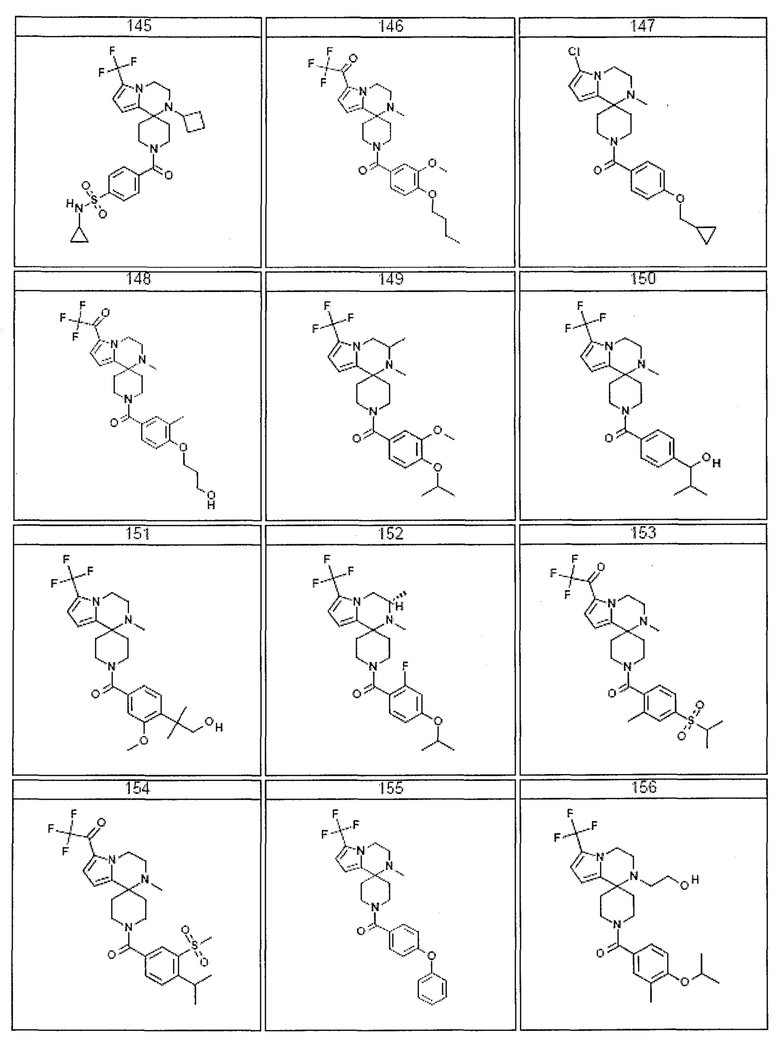
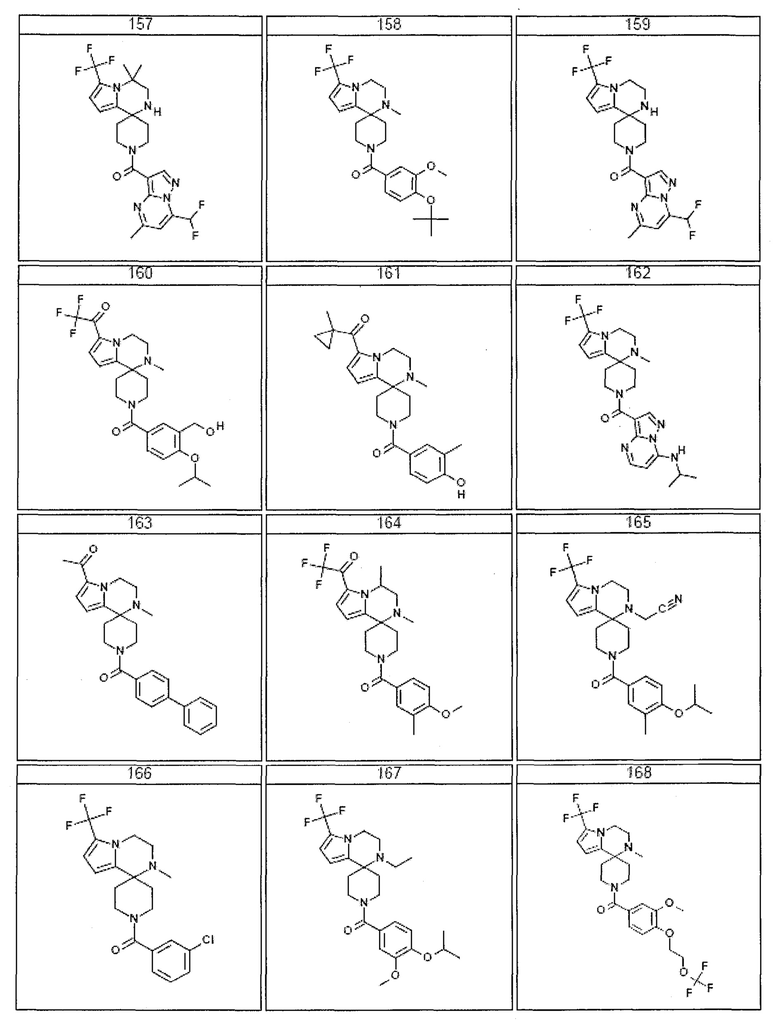
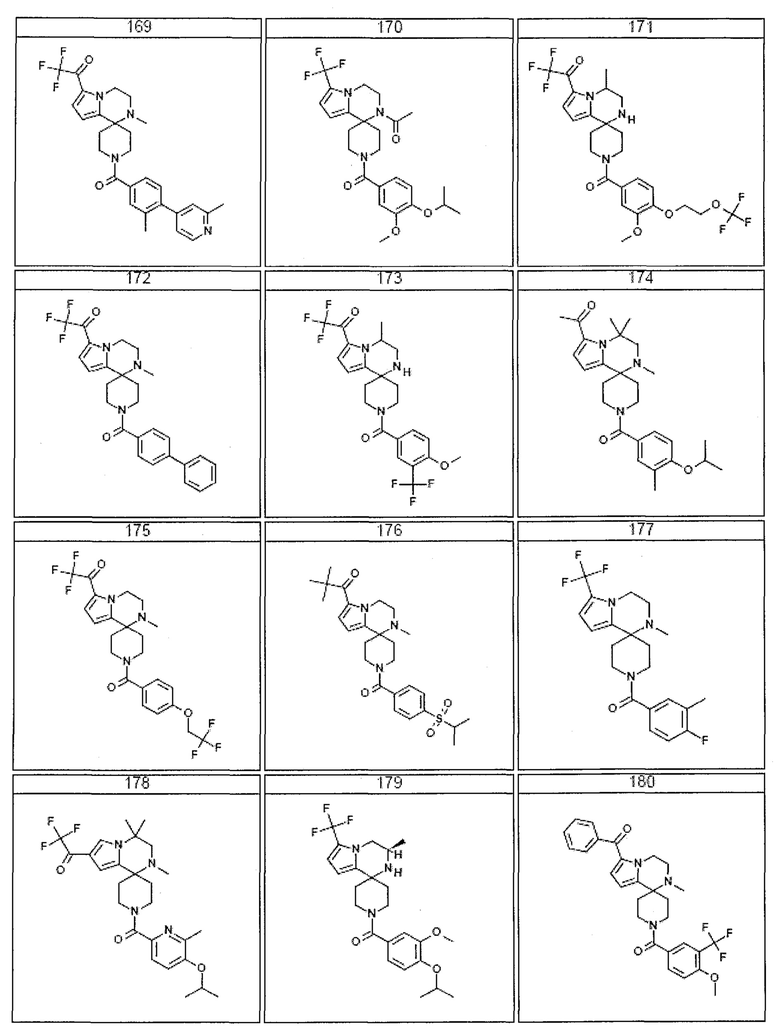
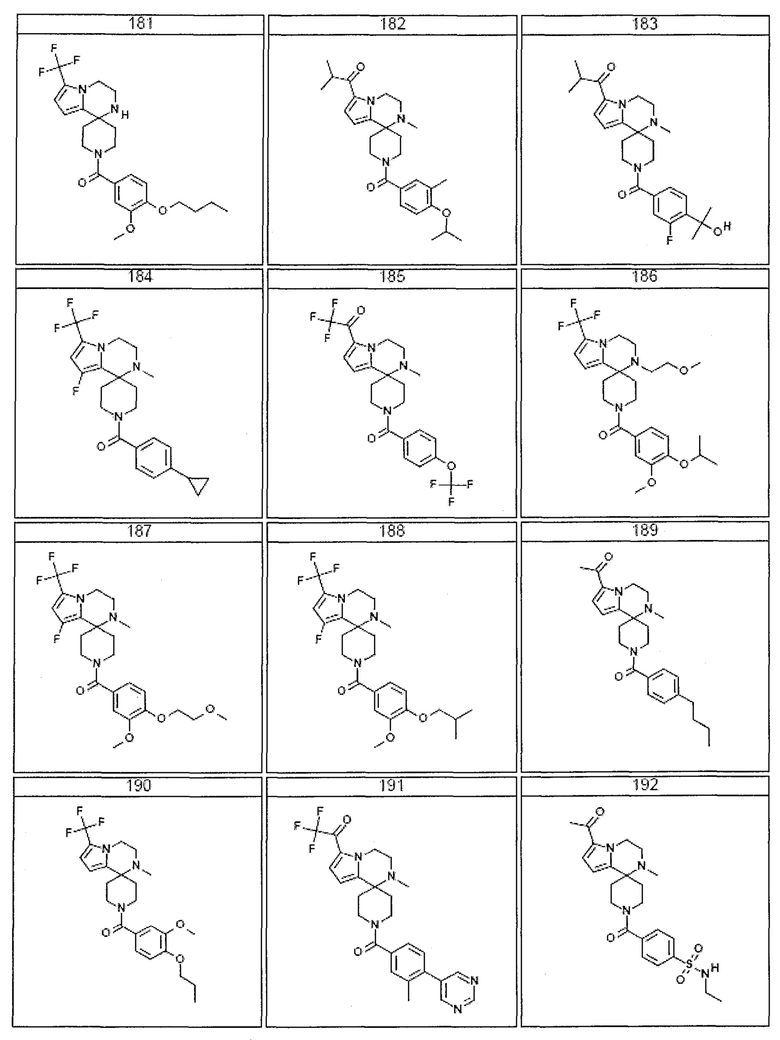
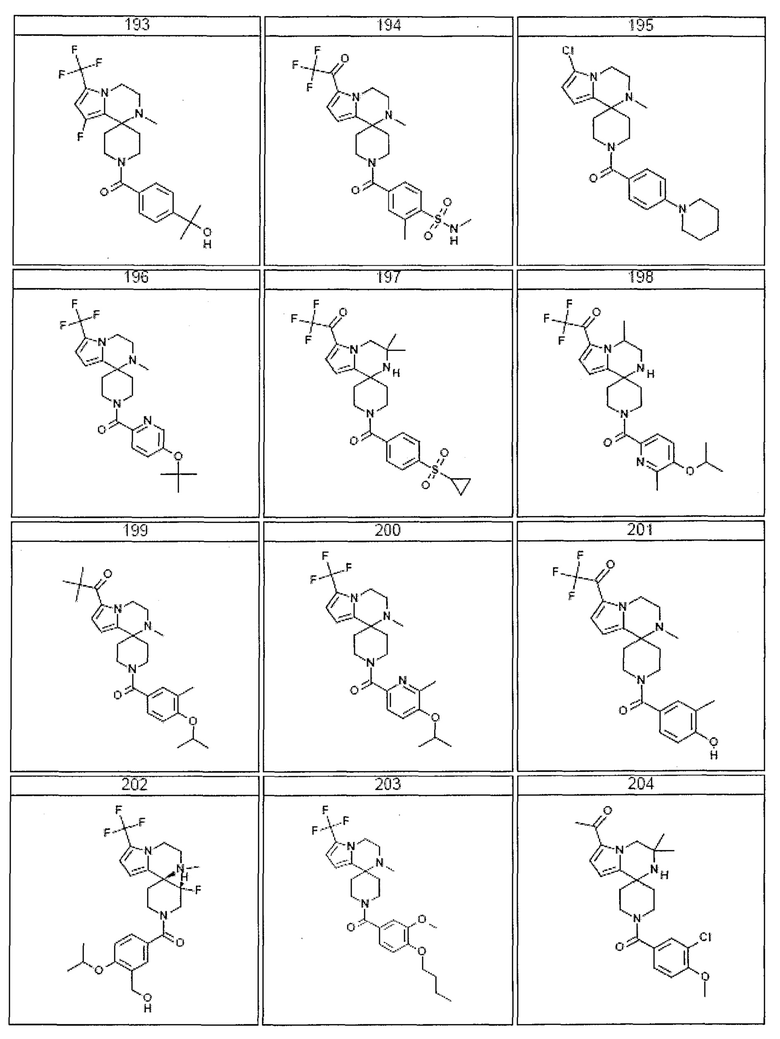
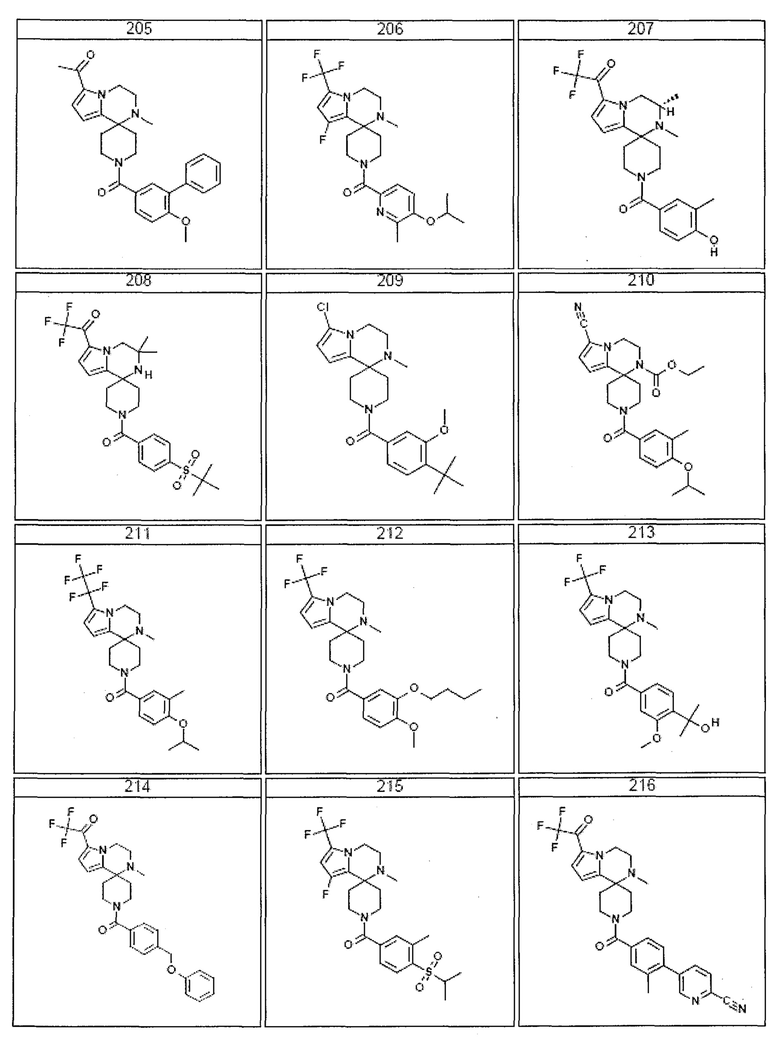
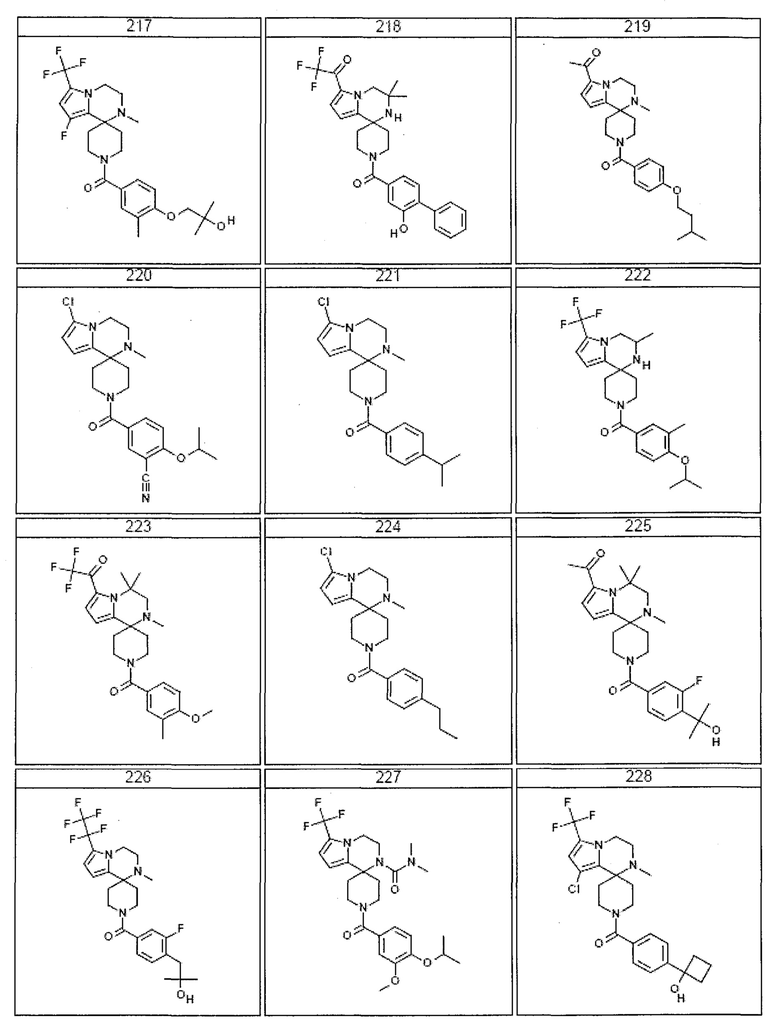
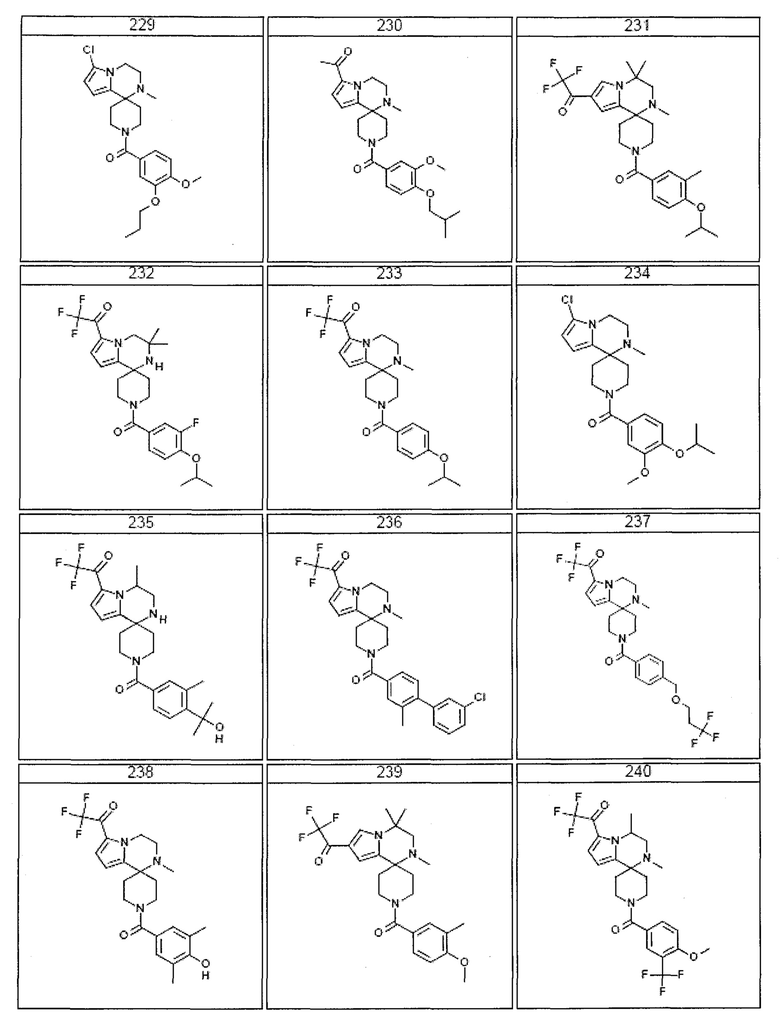
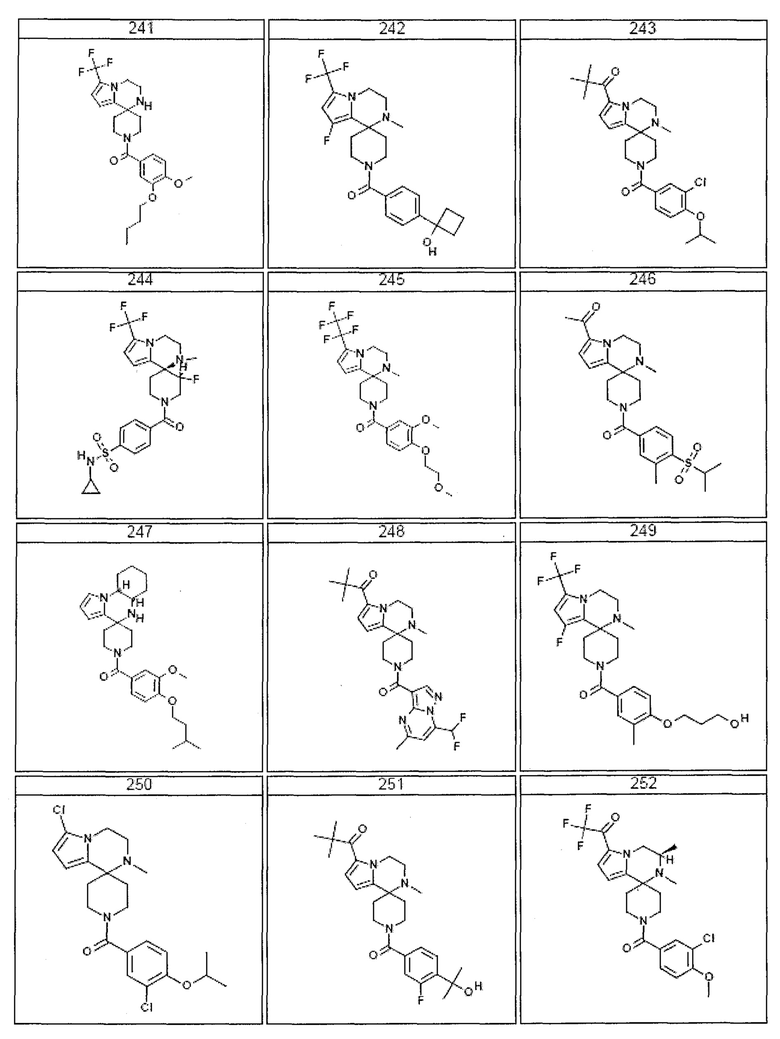
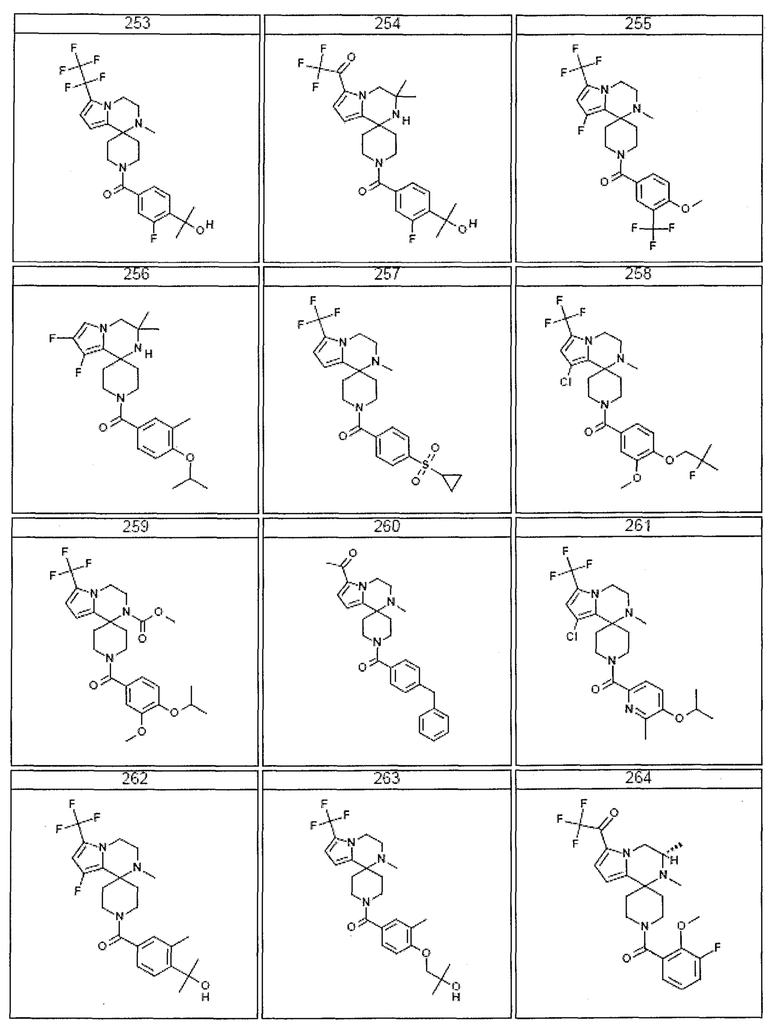
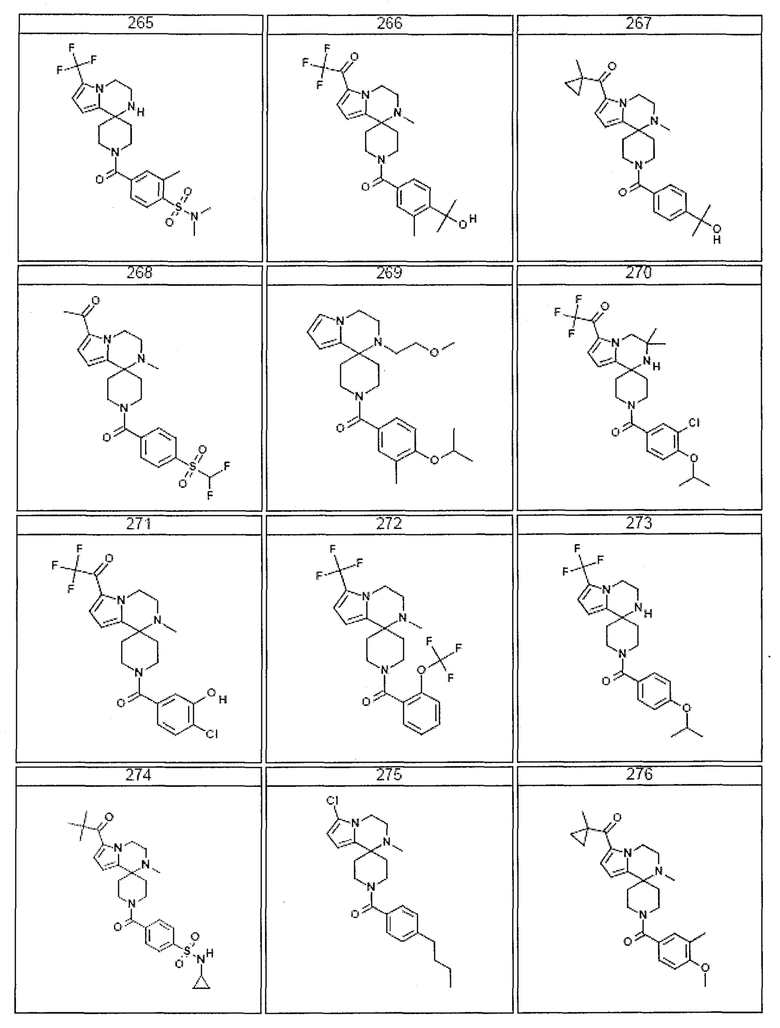
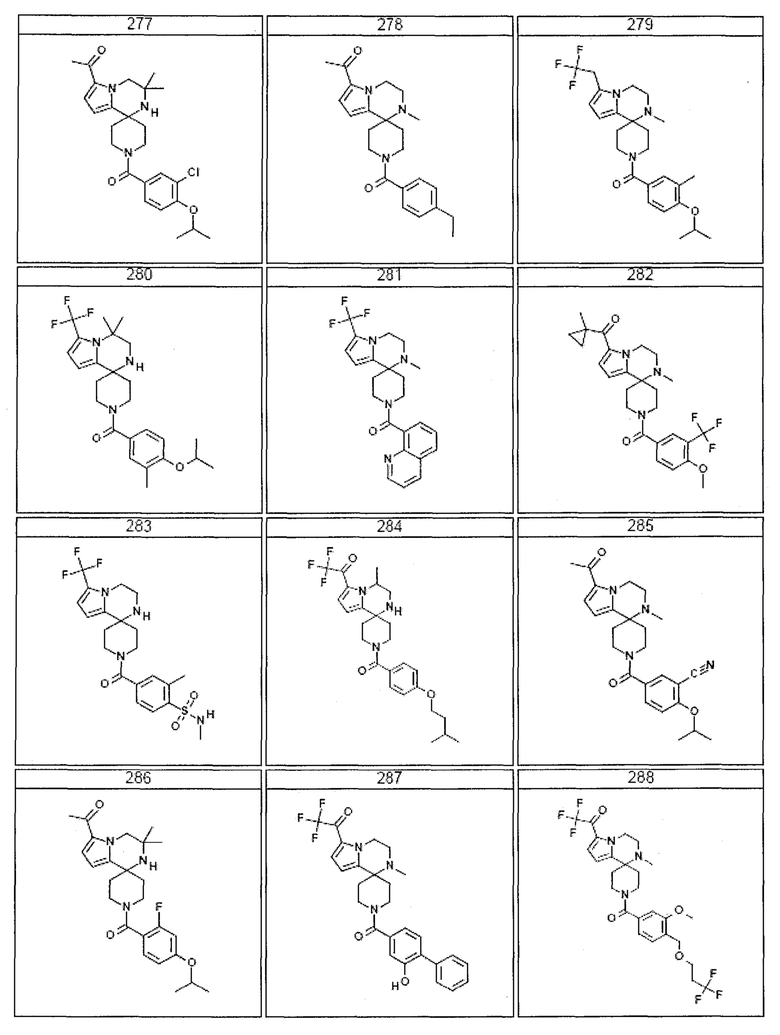
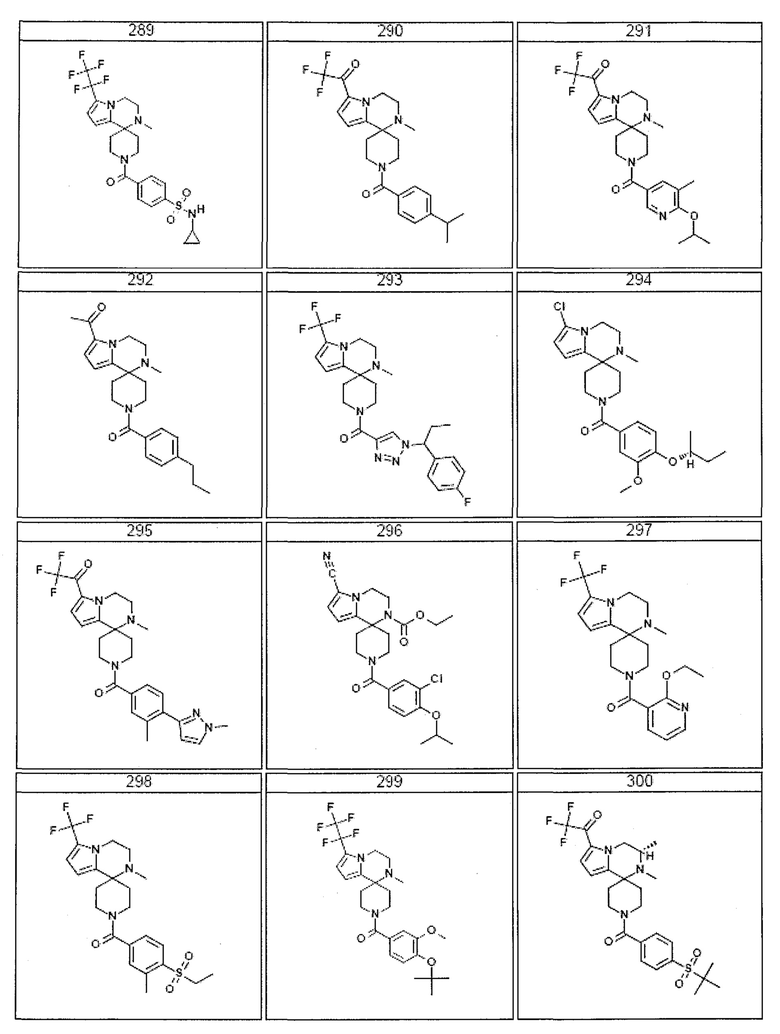
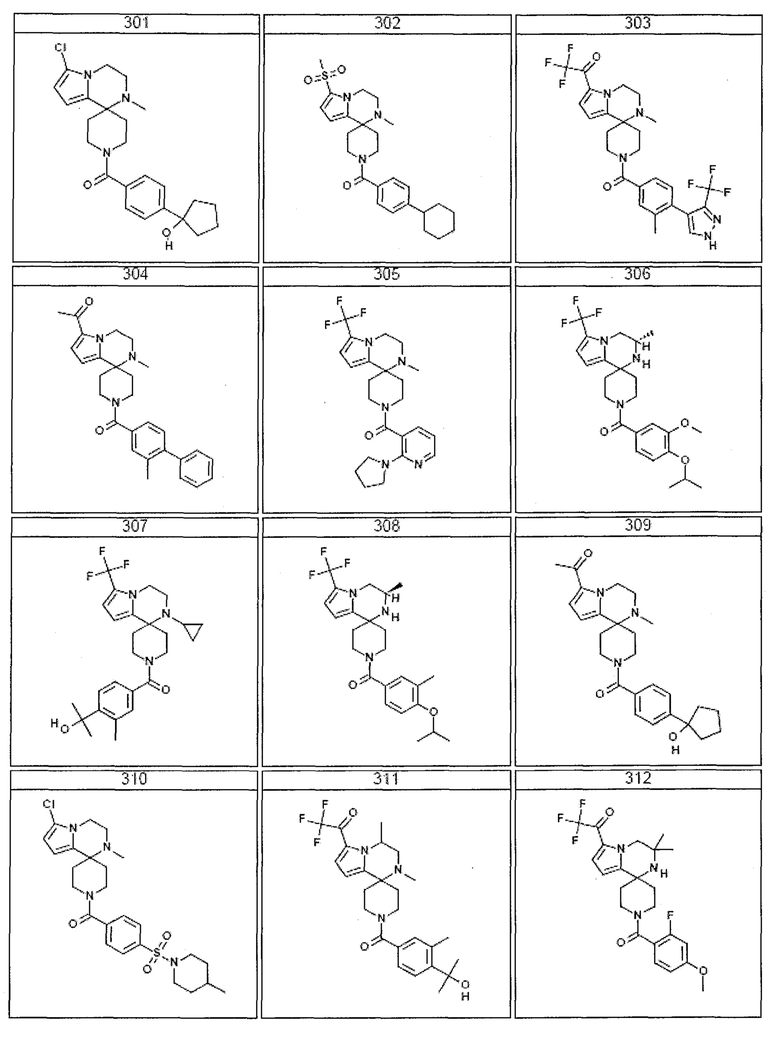

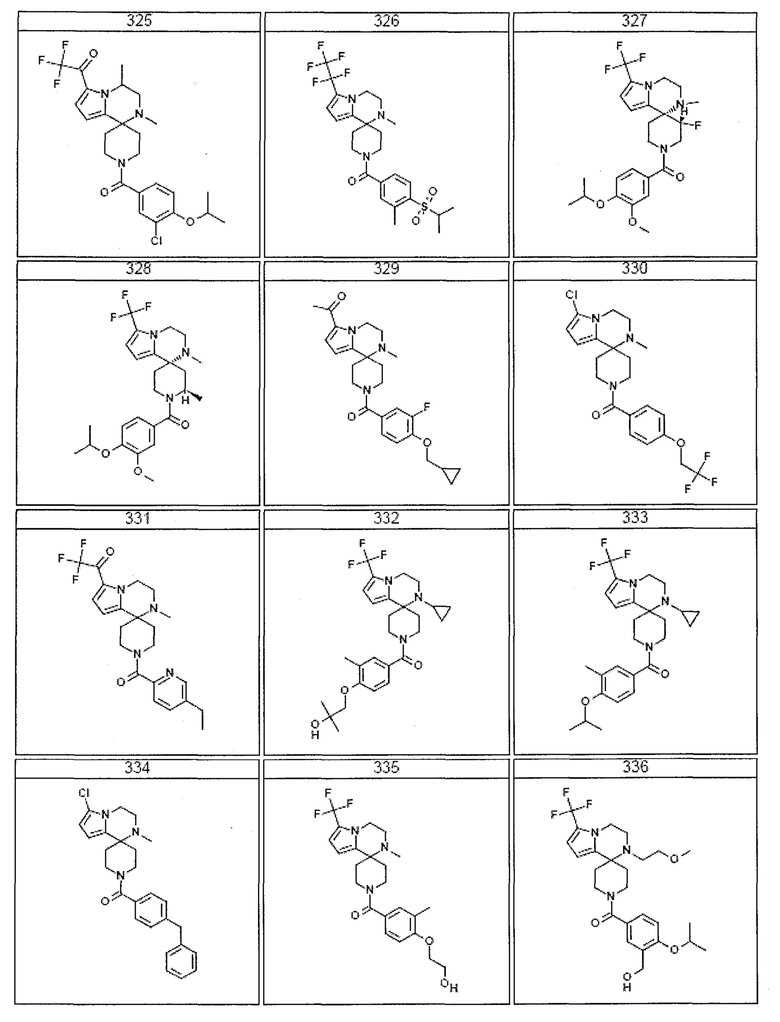
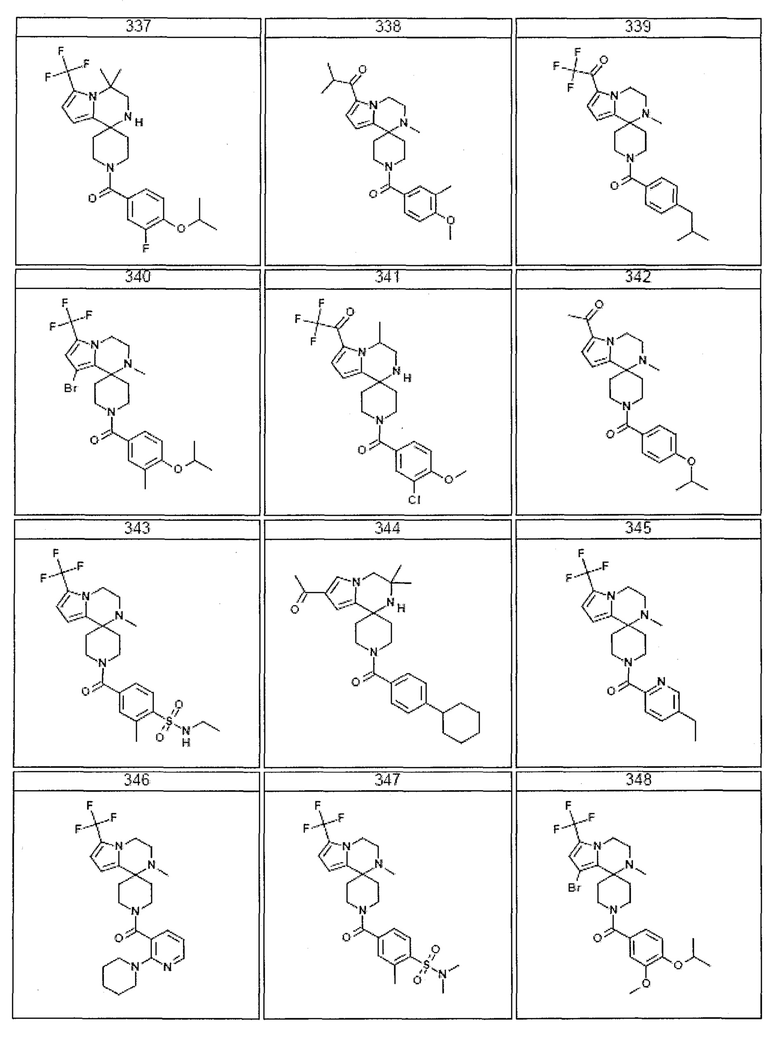
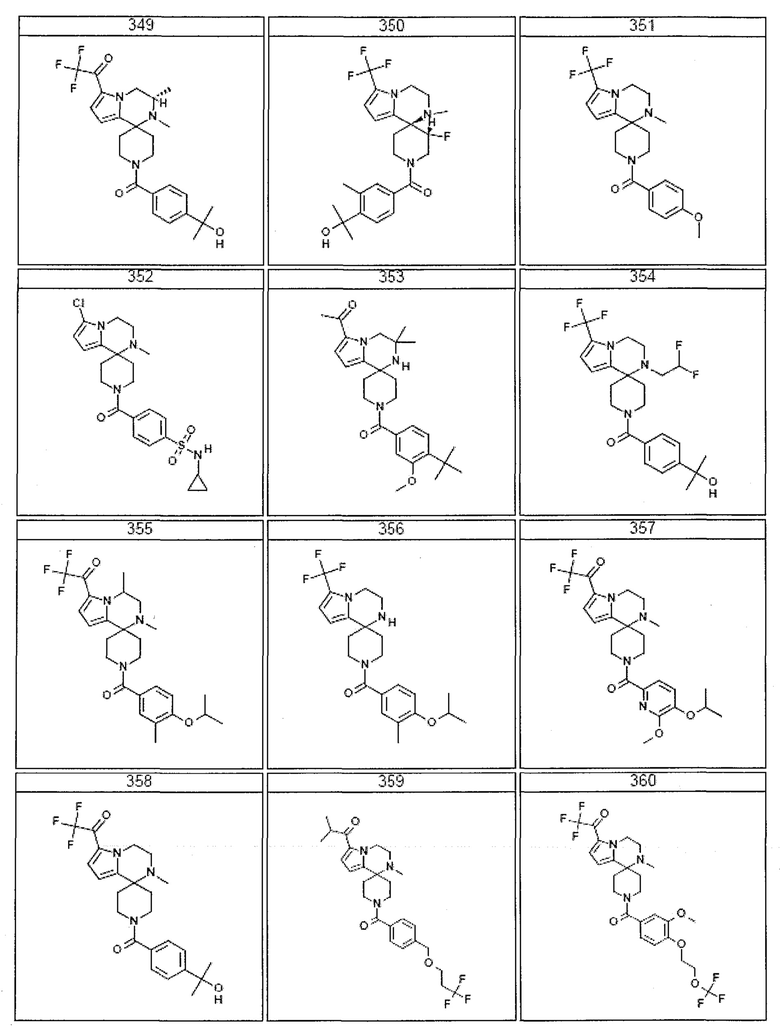
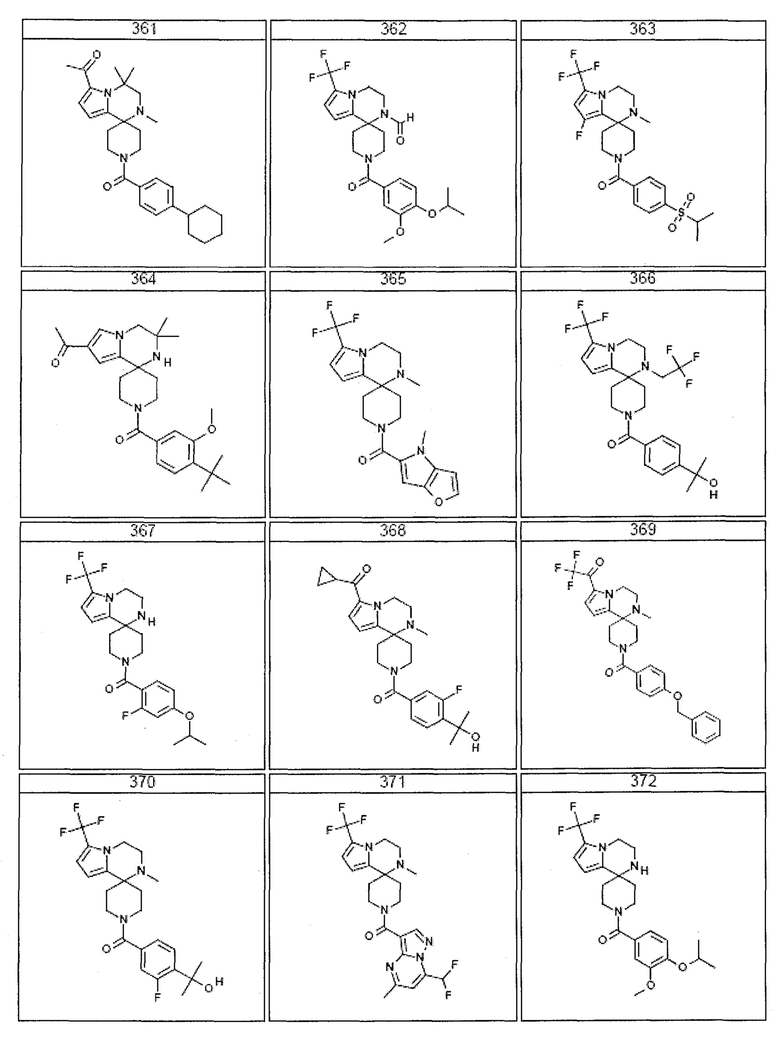
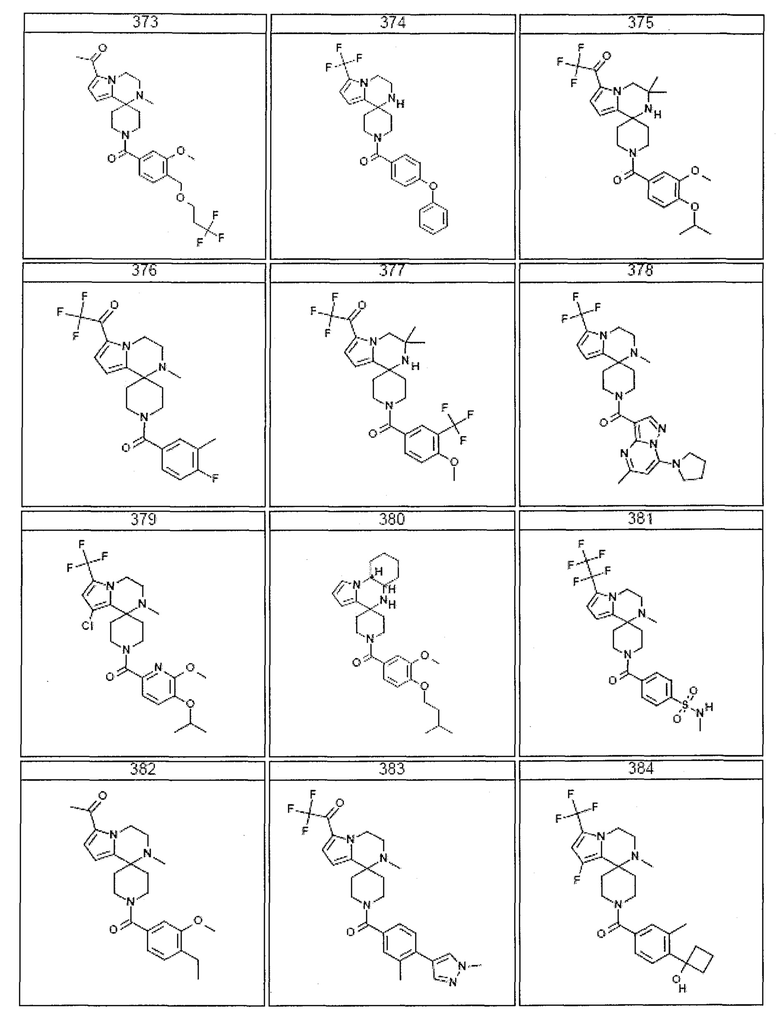

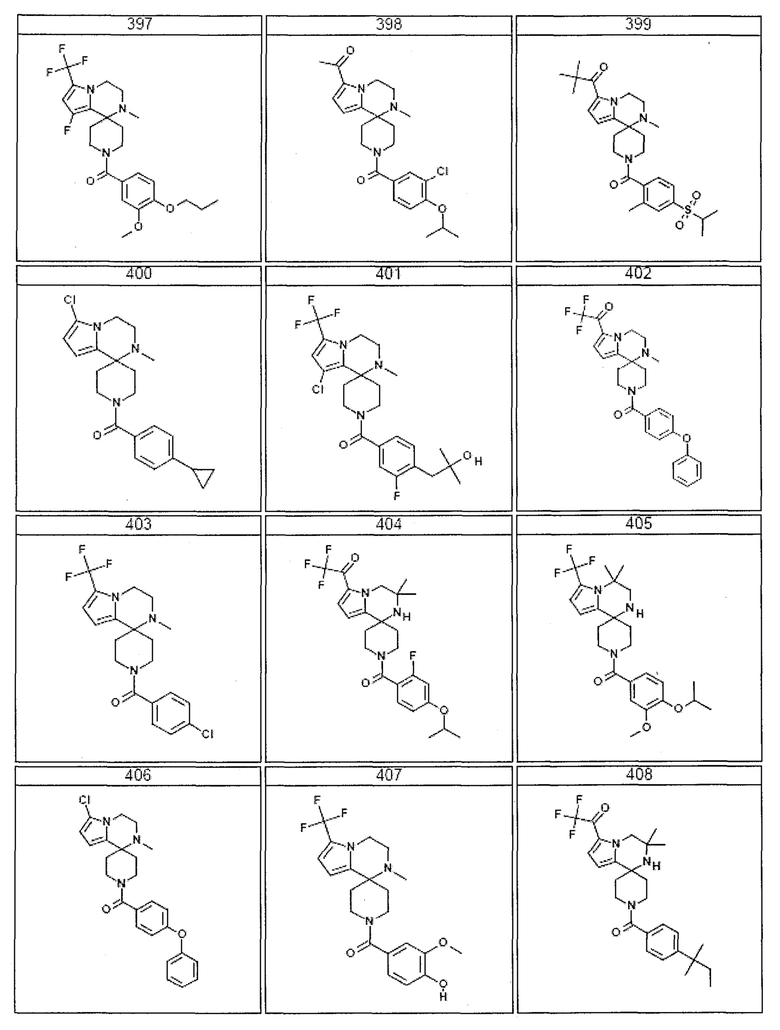
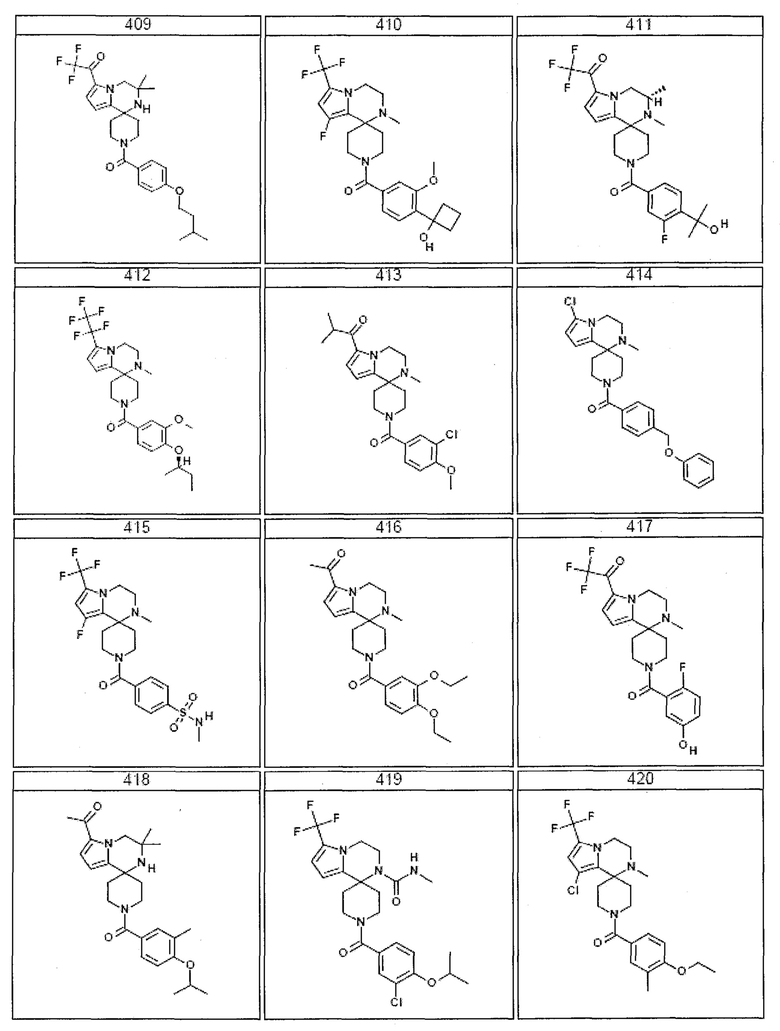
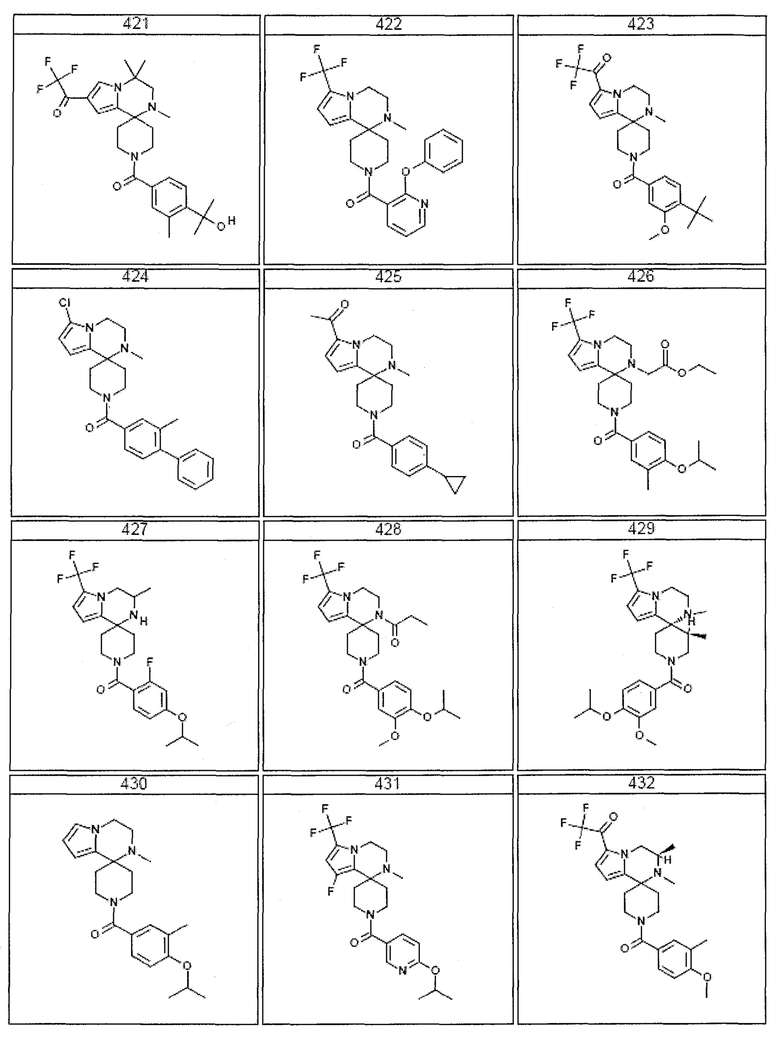
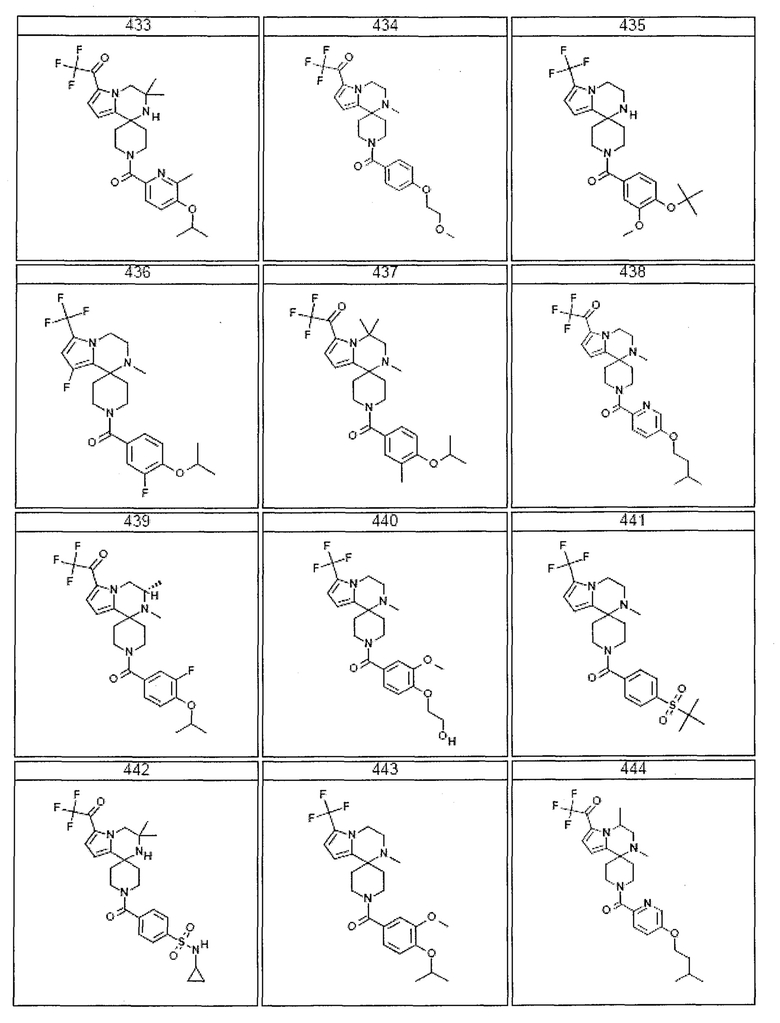
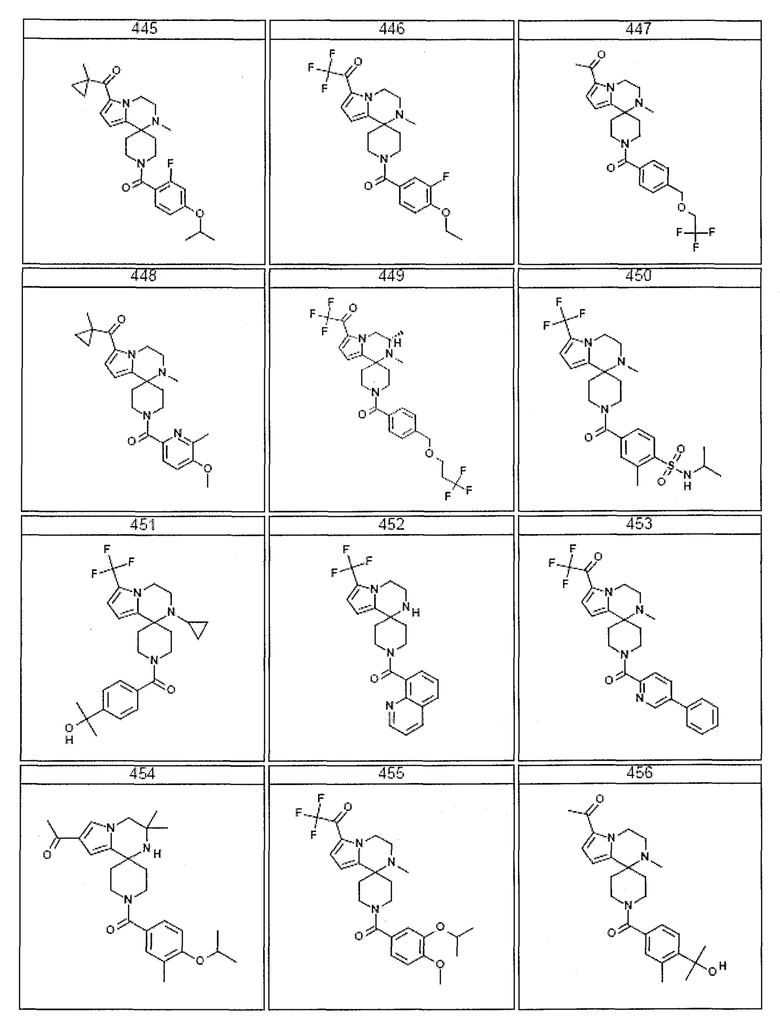

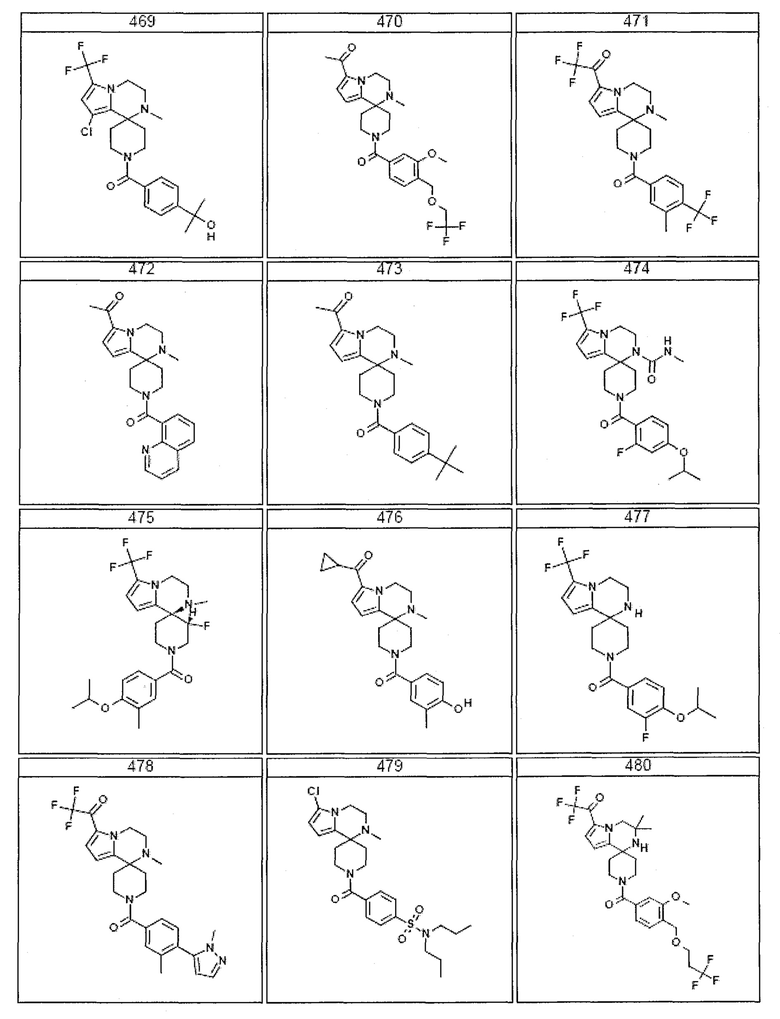
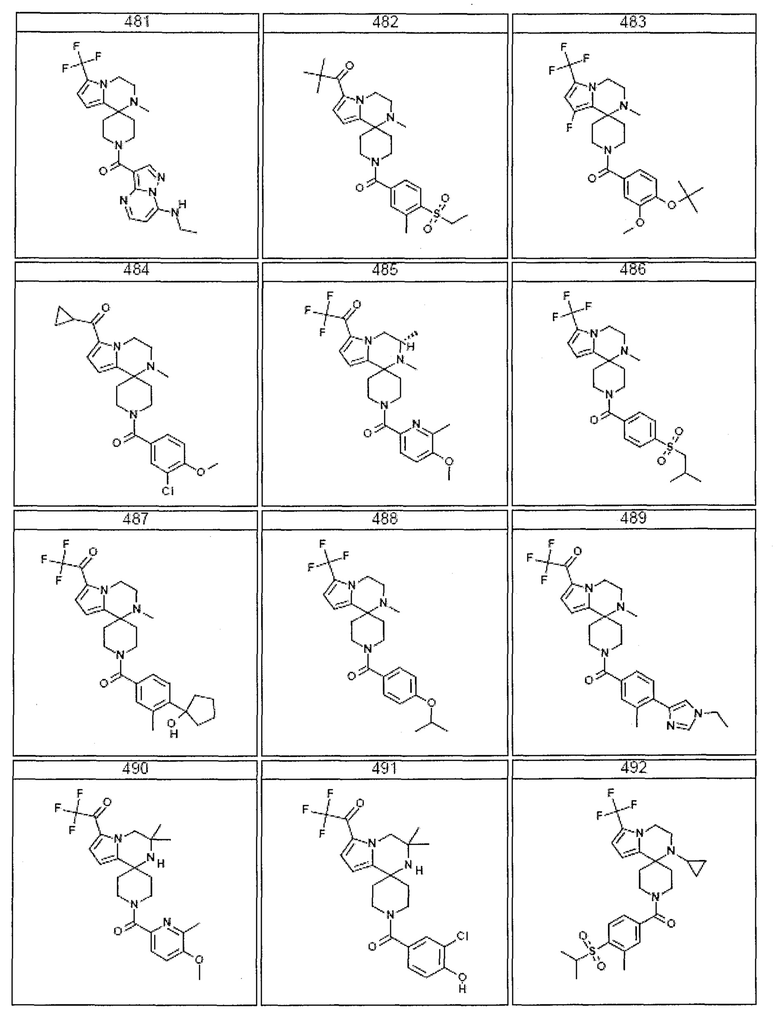
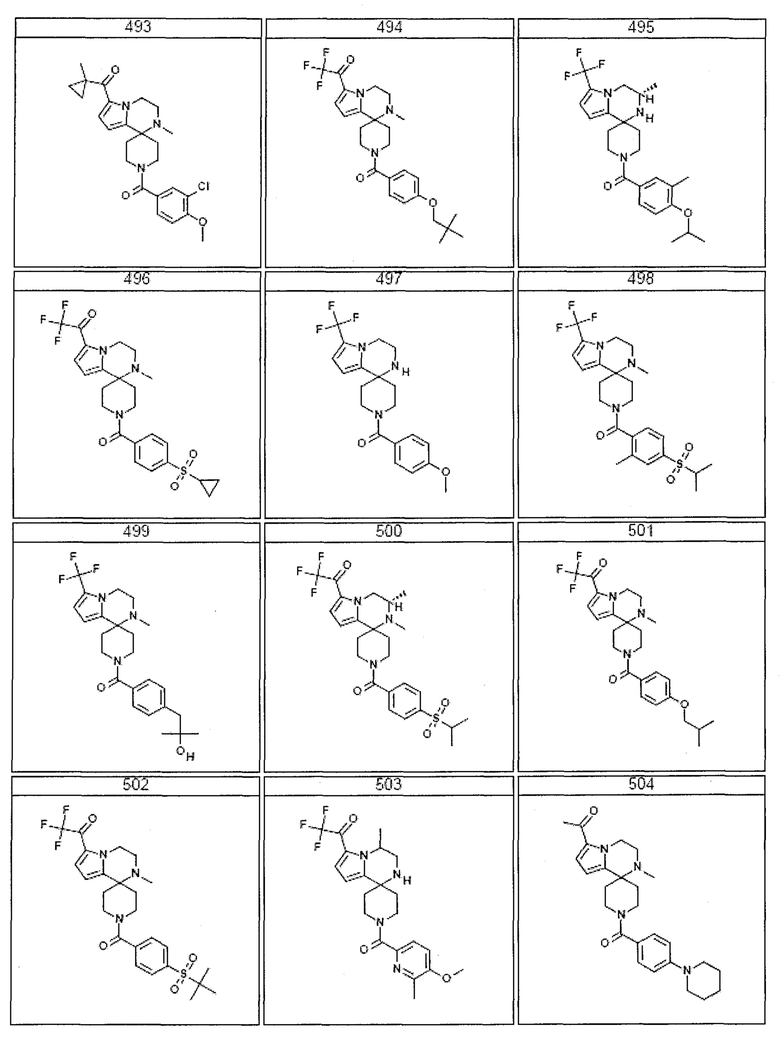
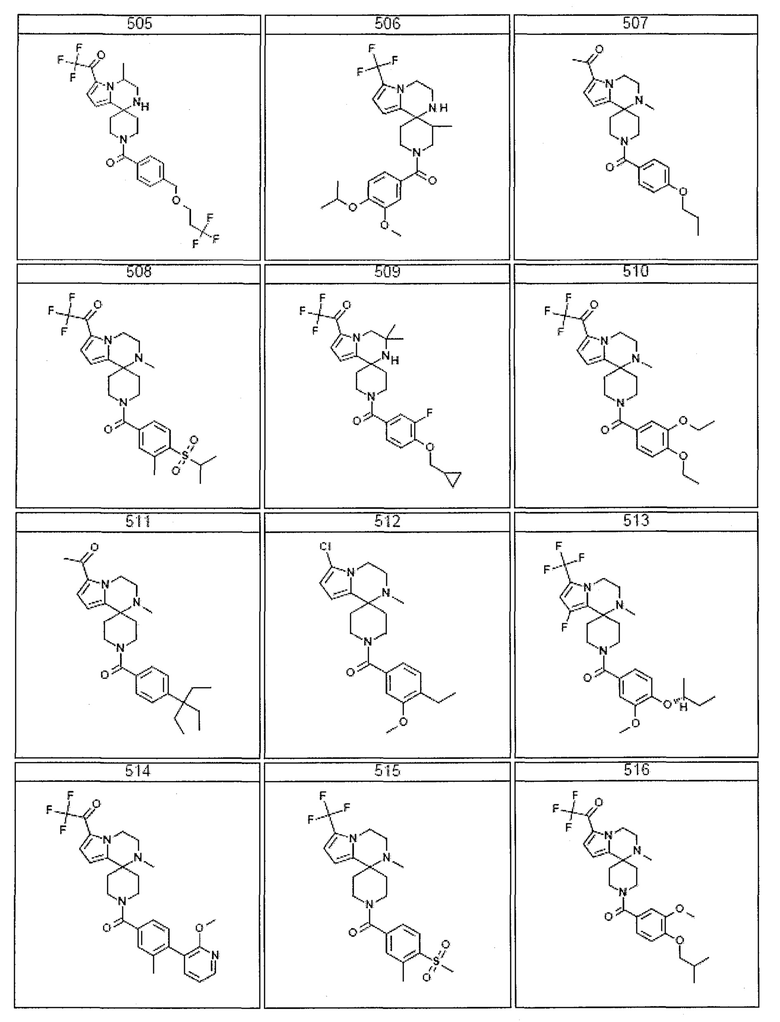
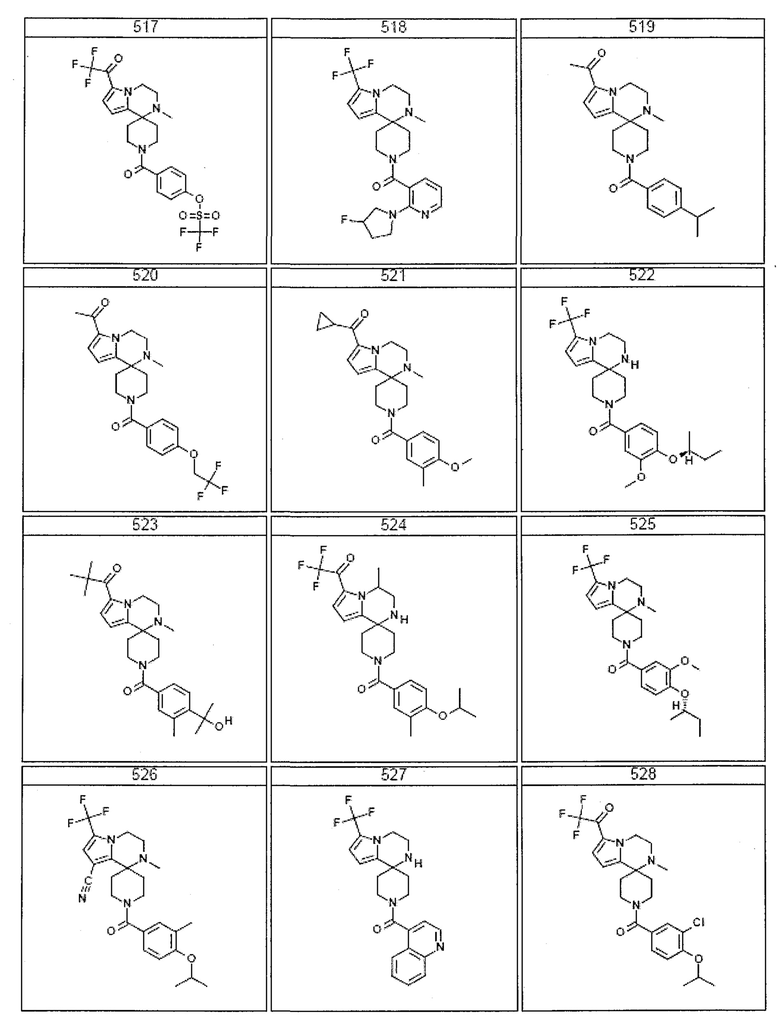
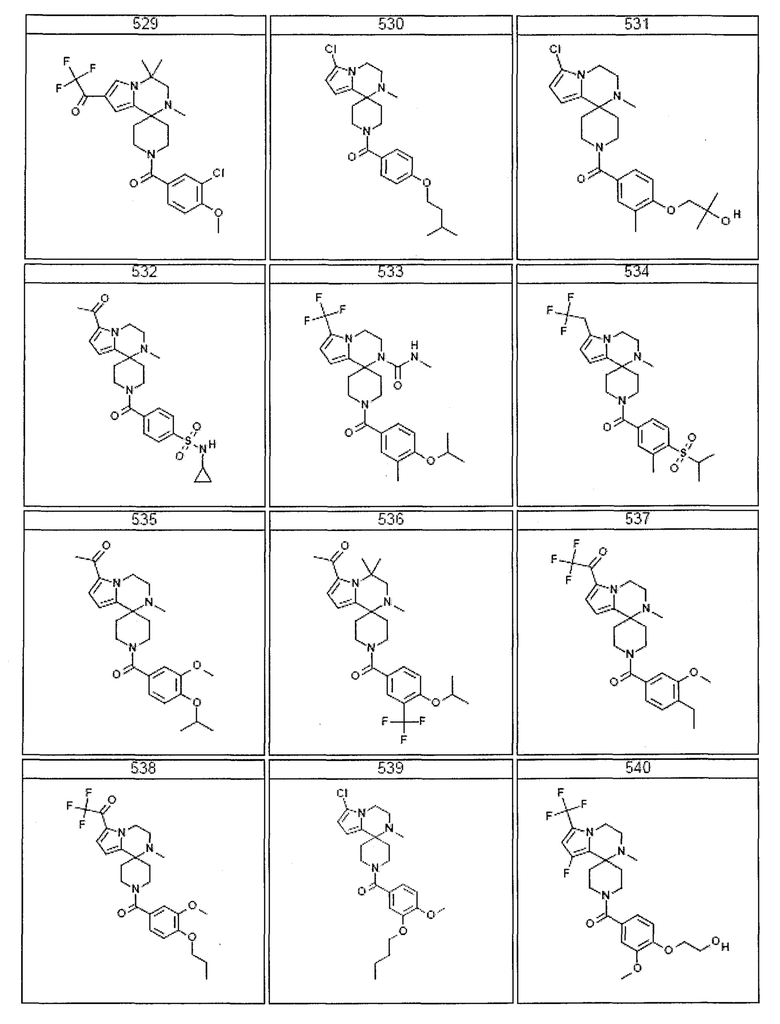
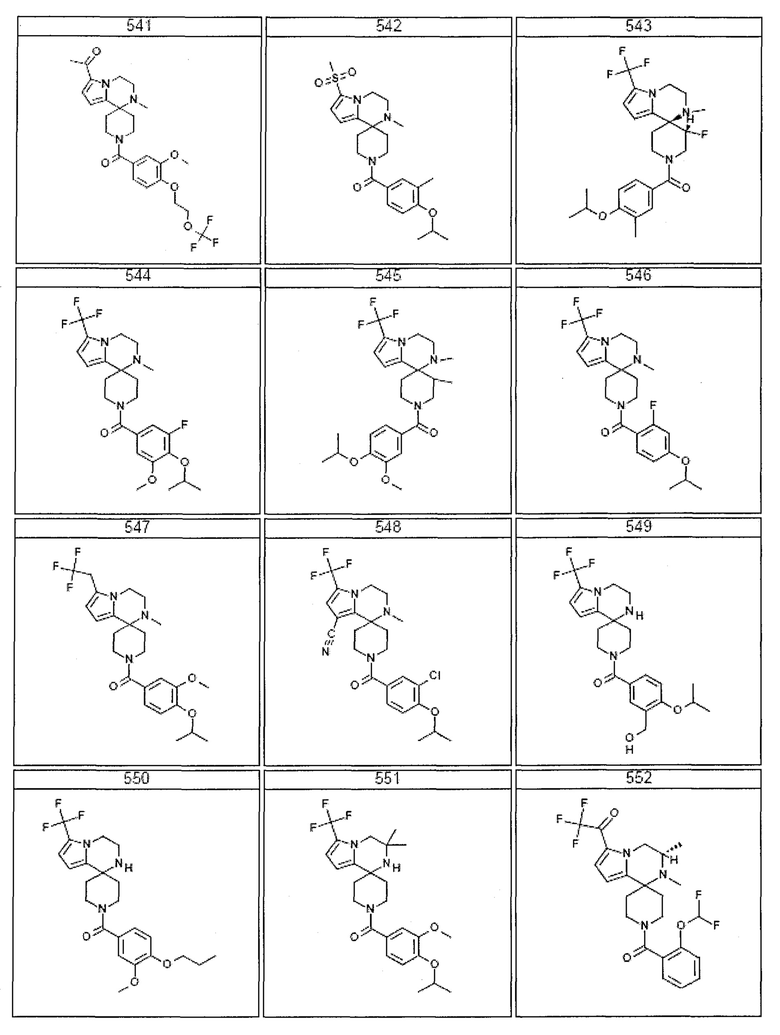
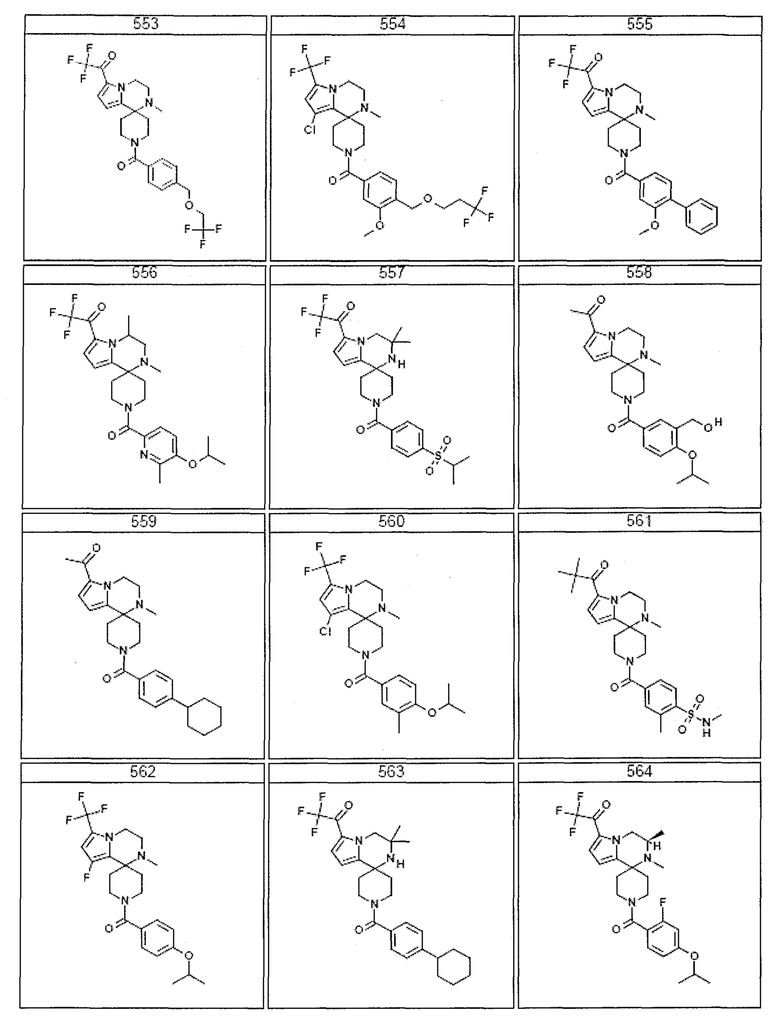
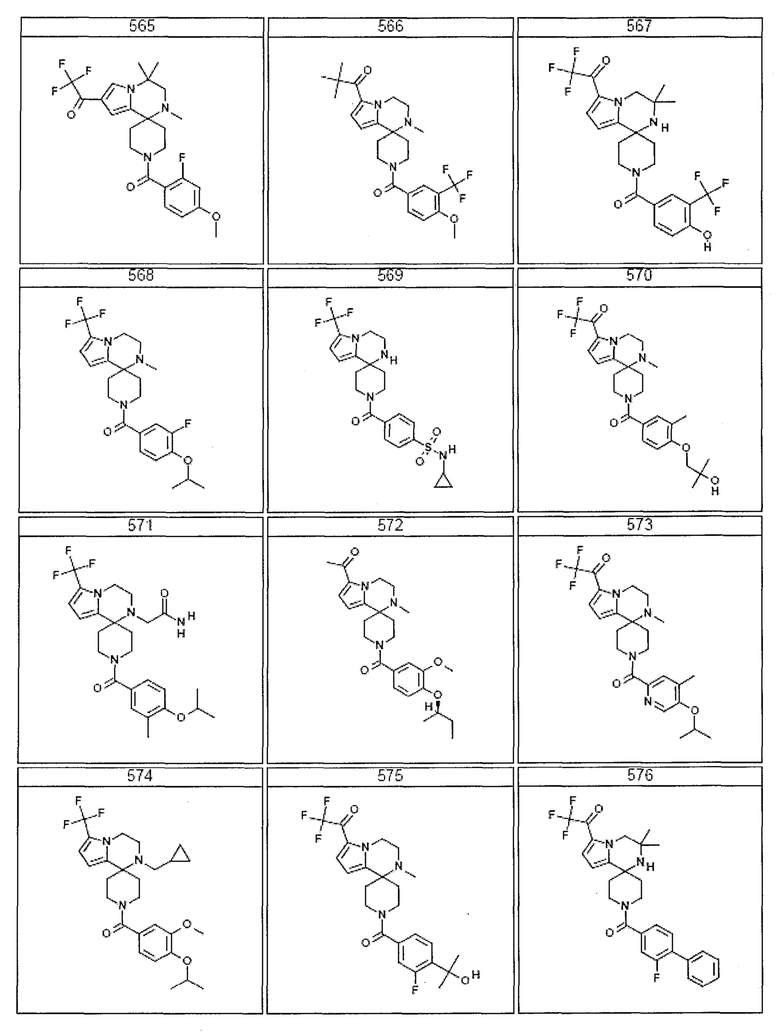
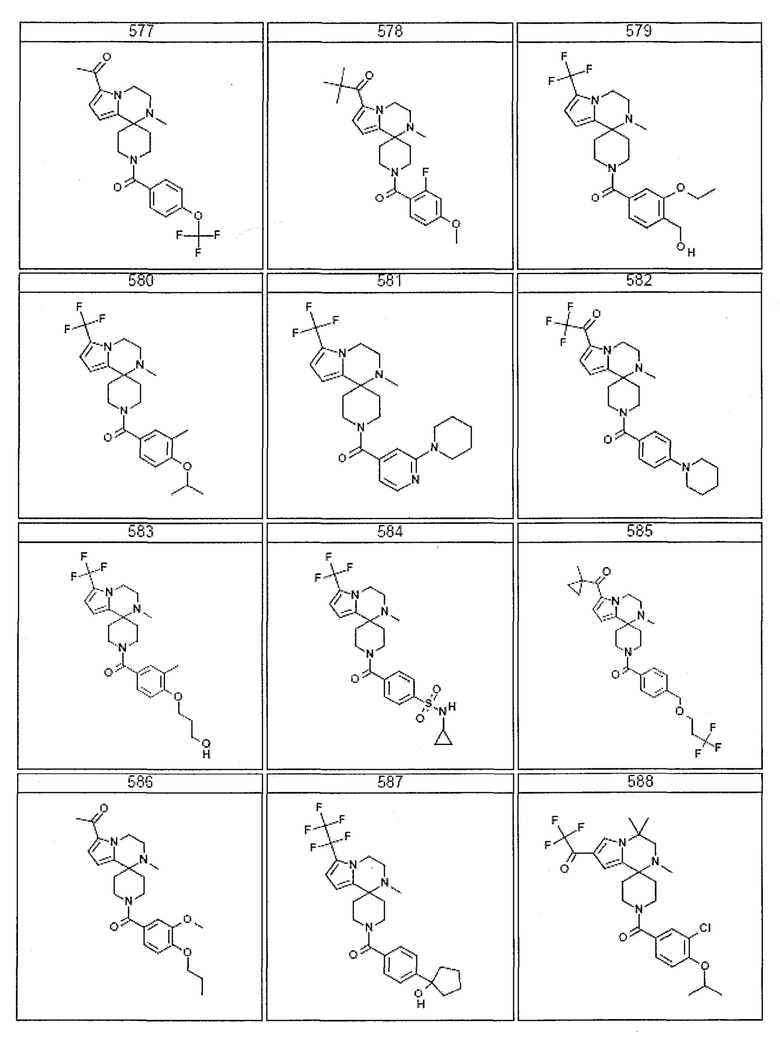
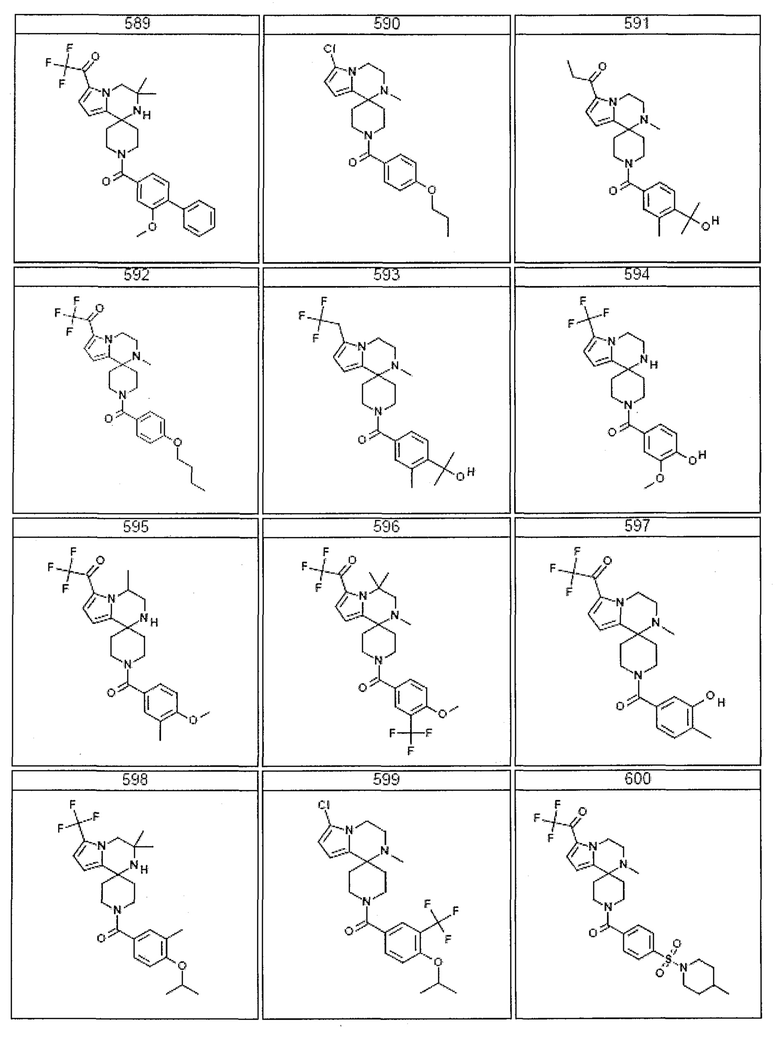
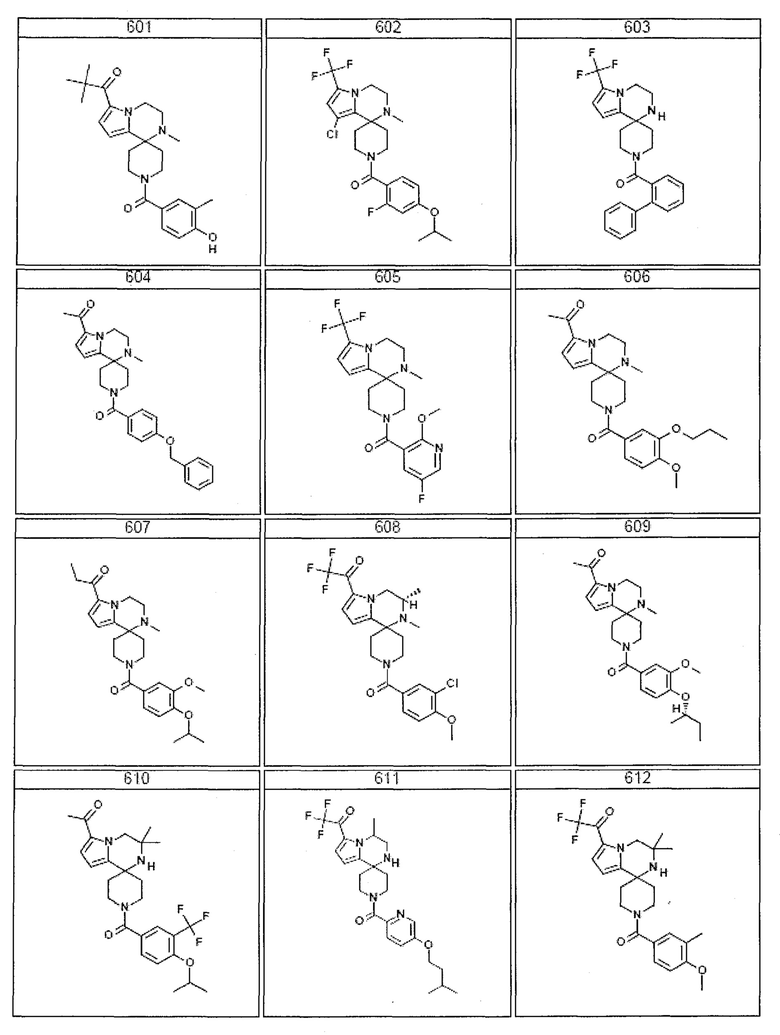
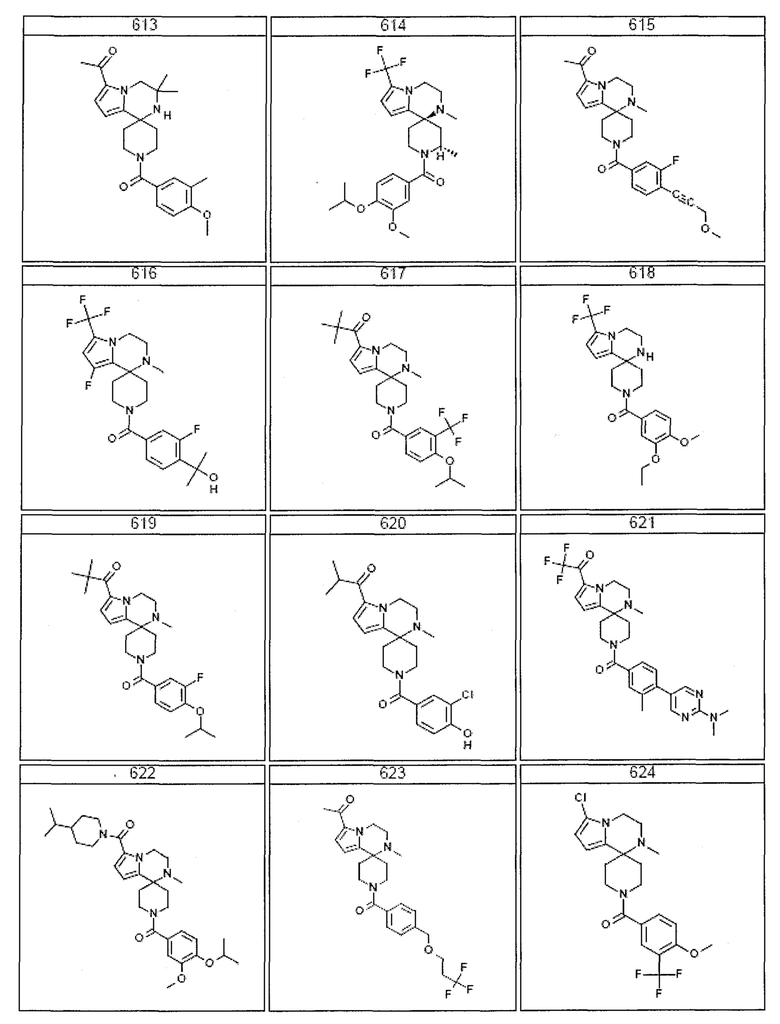
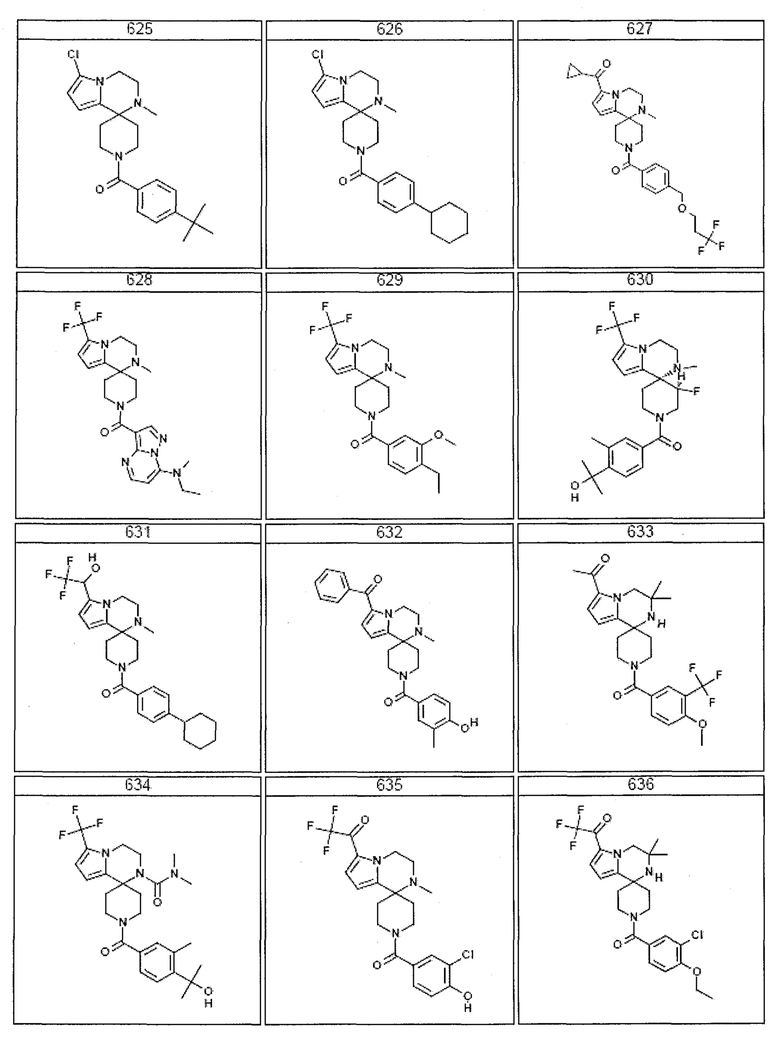
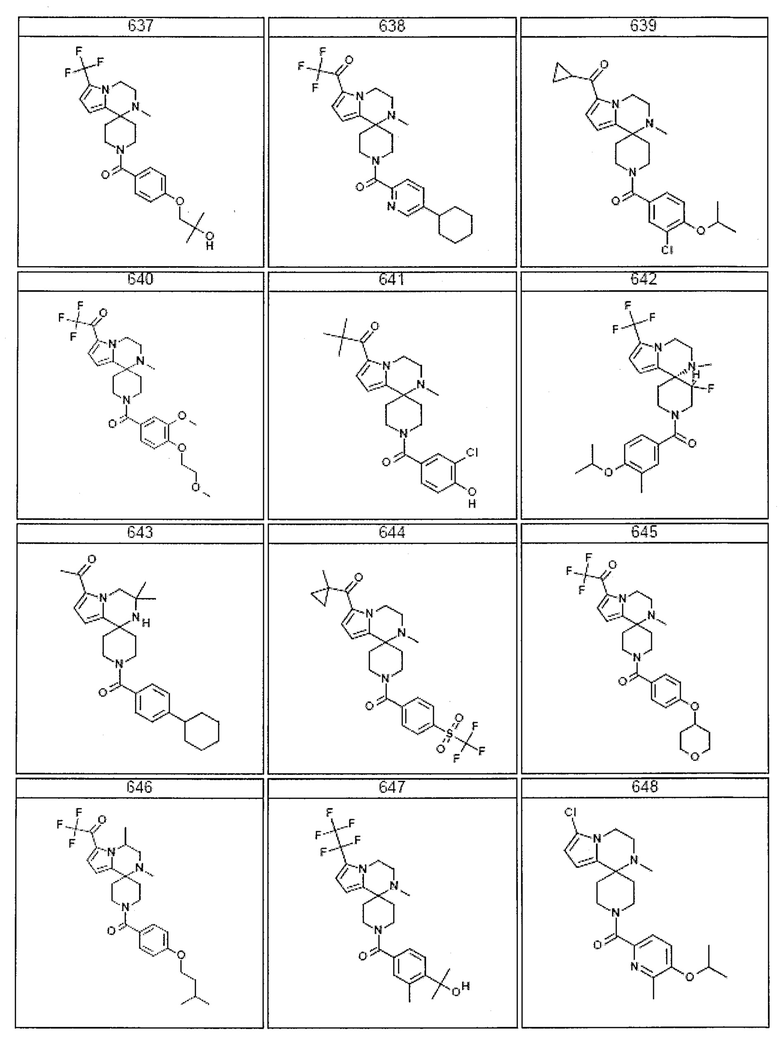

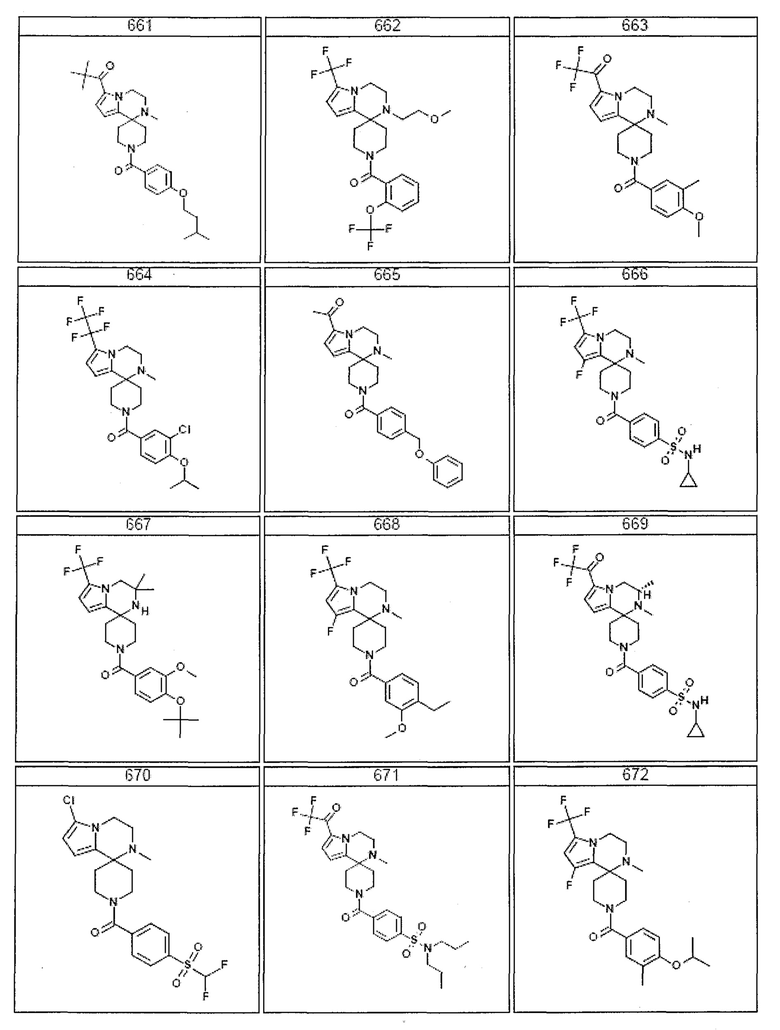
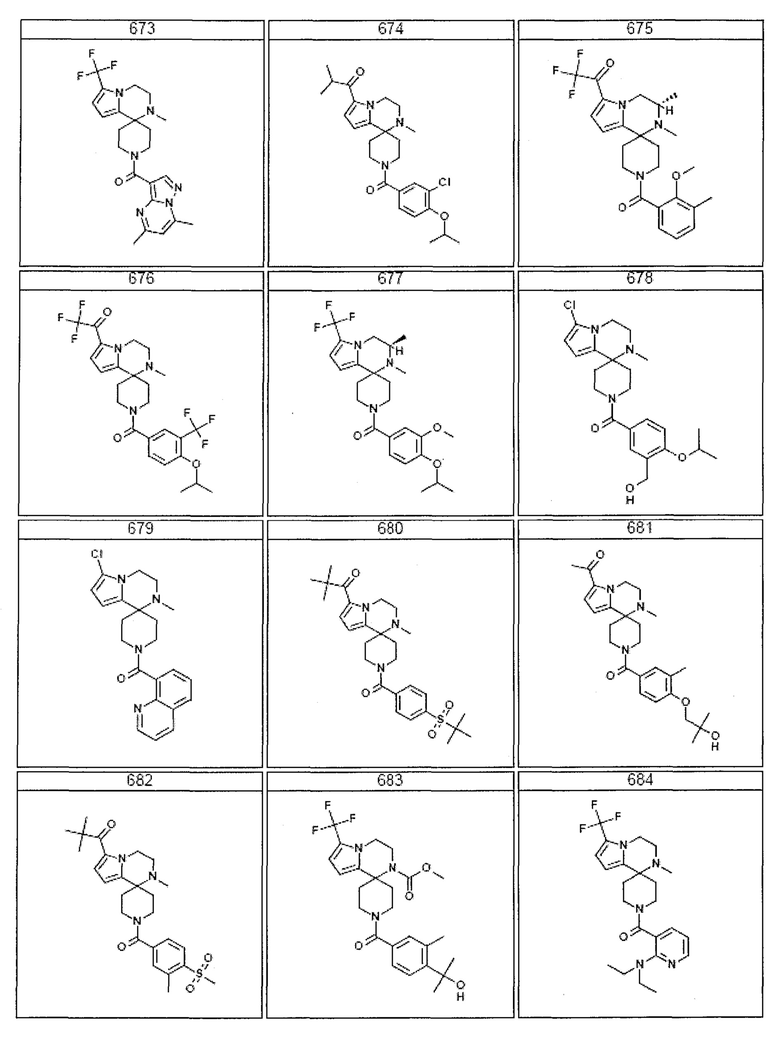
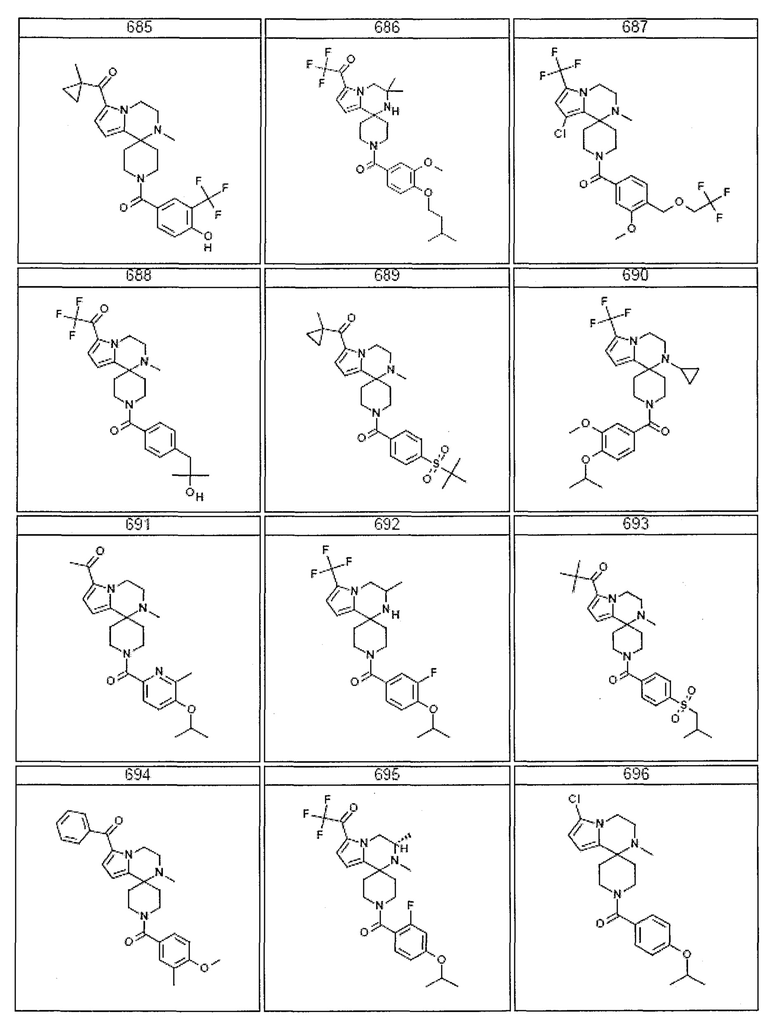
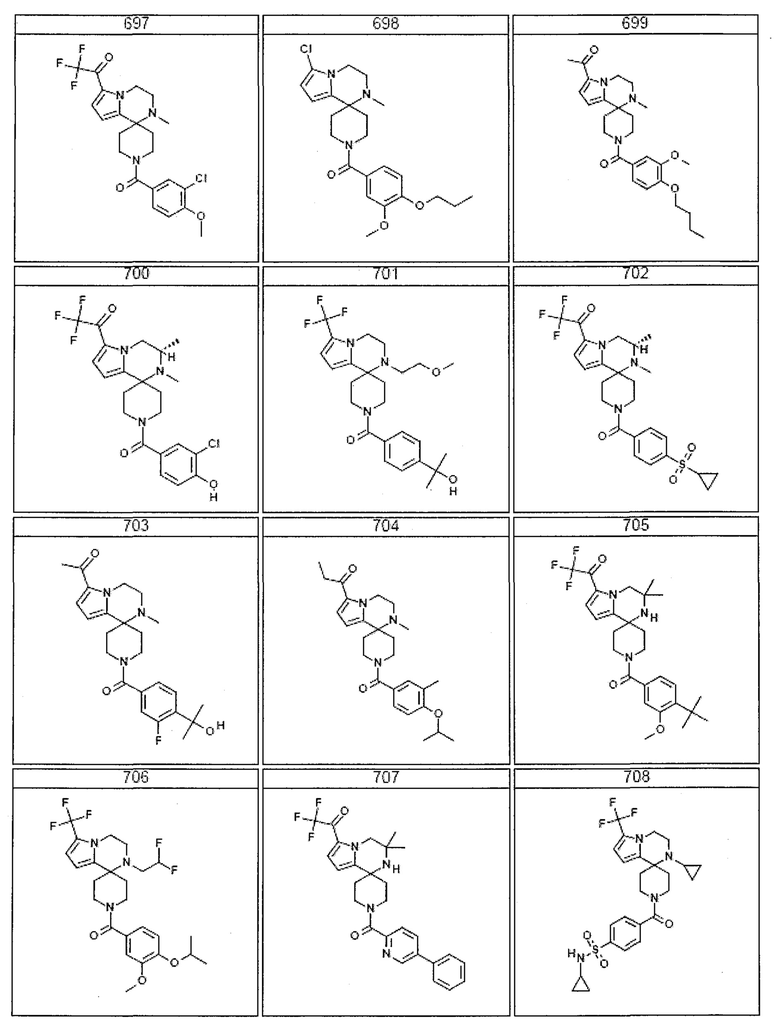

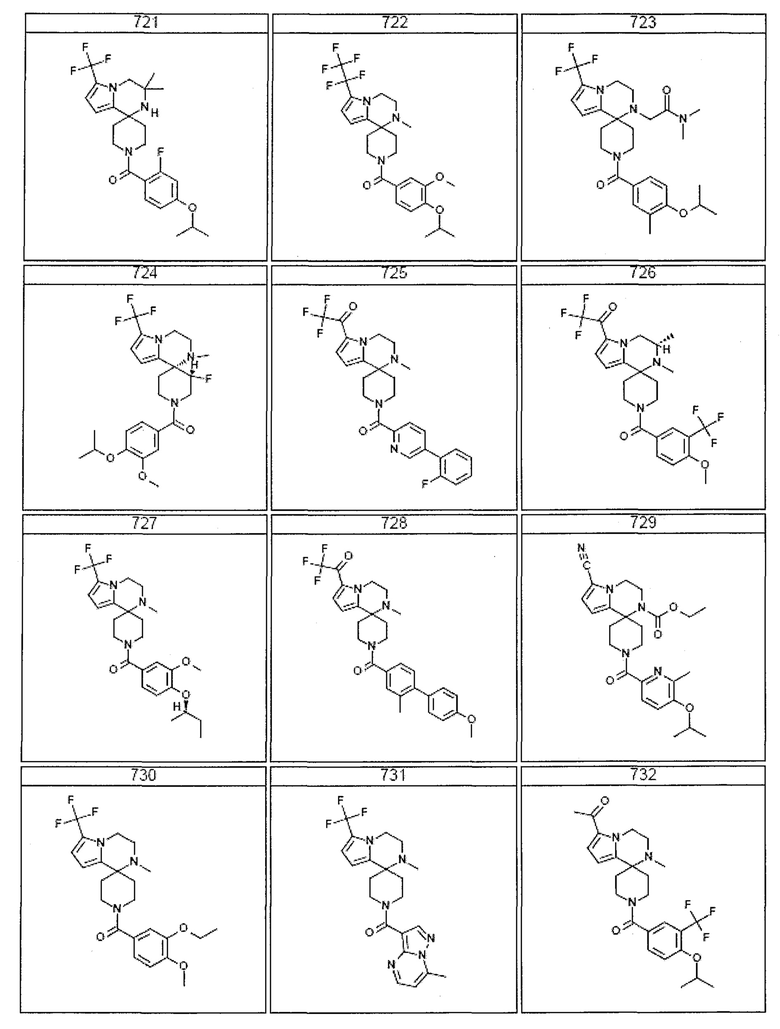
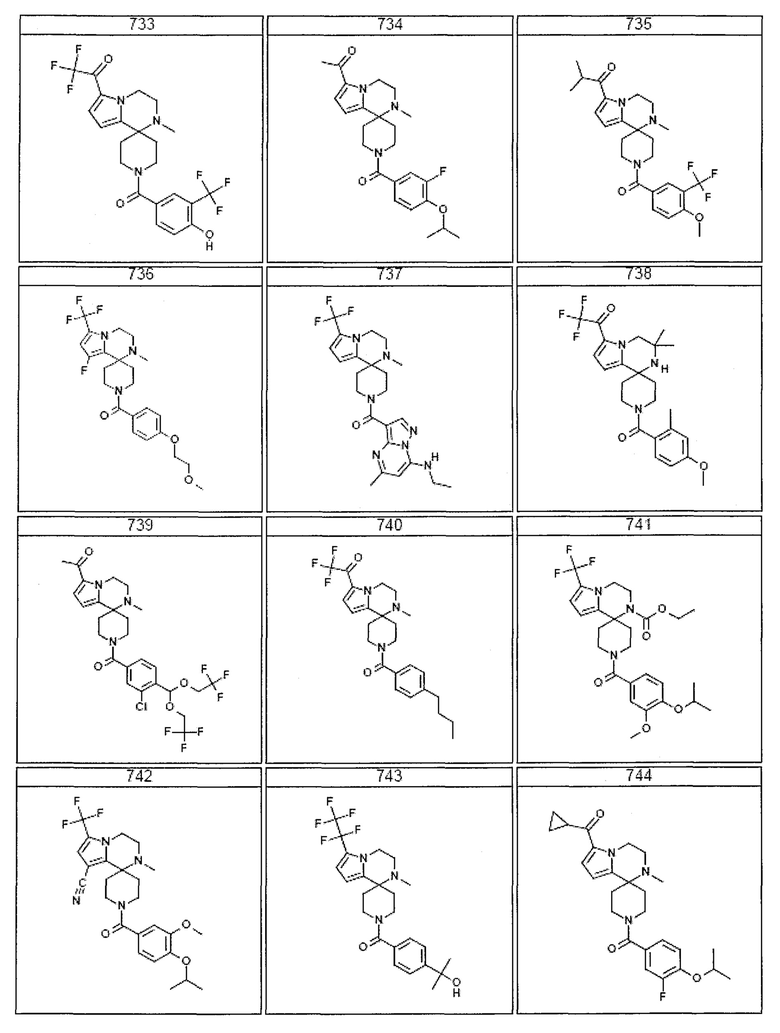
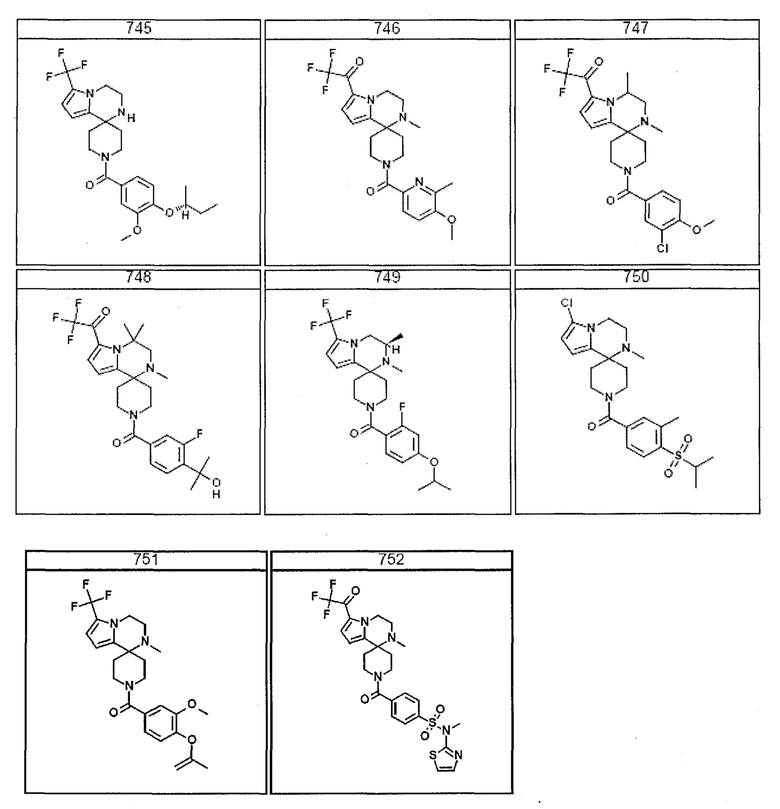
Согласно другому аспекту, настоящее изобретение относится к фармацевтической композиции, содержащей соединение согласно настоящему изобретению и фармацевтически приемлемый носитель.
Согласно другому аспекту, настоящее изобретение относится к способу ингибирования потенциалзависимого натриевого ионного канала:
у пациента; или
в биологическом образце;
включающему введение пациенту соединения или композиции согласно настоящему изобретению или приведение биологического образца в контакт с ним. Согласно другому варианту осуществления, потенциалзависимый натриевый ионный канал представляет собой NaV1.7.
Согласно другому аспекту, настоящее изобретение относится к способу лечения или ослабления тяжести острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерных головных болей, тригеминальной невралгии, герпетической невралгии, обычных невралгий, эпилепсии или эпилептических состояний, нейродегенеративных нарушений, психиатрических нарушений, тревоги, депрессии, биполярного нарушения, миотонии, аритмии, двигательных нарушений, нейроэндокринных нарушений, атаксии, рассеянного склероза, синдрома раздраженного кишечника, недержания, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, корешкового болевого синдрома, ишиаса, боли в спине, боли в голове и шее, тяжелой или некупируемой боли, ноцицептивной боли, прорыва боли, послеоперационной боли, раковой боли, инсульта, церебральной ишемии, травматического повреждения головного мозга, бокового амиотрофического склероза, индуцированной стрессом или физической нагрузкой стенокардии, учащенного сердцебиения, артериальной гипертензии, мигрени или аномальной моторики желудочно-кишечного тракта у субъекта, включающему введение эффективного количества соединения или композиции согласно настоящему изобретению.
Согласно другому варианту осуществления, способ используют для лечения или ослабления тяжести боли при злокачественной опухоли бедренной кости; незлокачественной хронической костной боли; ревматоидного артрита; остеоартрита; спинального стеноза; нейропатической поясничной боли; нейропатической поясничной боли; миофасциального болевого синдрома; фибромиалгии; боли в височно-нижнечелюстном суставе; хронической висцеральной боли, абдоминальной боли; боли в поджелудочной железе; боли при IBS; хронической и острой головной боли; мигрени; головной боли напряжения, включая кластерные головные боли; хронической и острой нейропатической боли, постгерпетической невралгии; диабетической нейропатии; HIV-ассоциированной нейропатии; тригеминальной невралгии; нейропатии Шарко-Мари-Тута; наследственных сенсорных нейропатий; повреждения периферических нервов; болезненных невром; эктопических разрядов в проксимальных и дистальных отделах спинного мозга; радикулопатии; нейропатической боли, индуцированной химиотерапией; нейропатической боли, индуцированной лучевой терапией; постмастэктомической боли; центральной боли; боли при травме спинного мозга; постинсультной боли; таламической боли; комплексного регионального болевого синдрома; фантомной боли; некупируемой боли; острой боли; острой послеоперационной боли; острой скелетно-мышечной боли; суставной боли; механической поясничной боли; боли в шее; тендинита; боли, вызванной повреждением/физической нагрузкой; острой висцеральной боли, абдоминальной боли; пиелонефрита; аппендицита; холецистита; кишечной непроходимости; грыж; боли в груди, сердечной боли; тазовой боли, боли при почечной колике, острой акушерской боли, родовых схваток; боли при кесаревом сечении; острой воспалительной, ожоговой и травматической боли; острой периодической боли, эндометриоза; острой боли при опоясывающем герпесе; серповидно-клеточной анемии; острого панкреатита; прорыва боли; орофациальной боли, включая боль при синусите, зубную боль; боли при рассеянном склерозе (MS); боли при депрессии; лепрозной боли; боли при болезни Бехчета; адипозалгии; флебитической боли; боли при синдроме Гийена-Барре; болезненности в ногах и при движении пальцами ног; синдрома Хаглунда; эритромелалгической боли; боли при болезни Фабри; заболеваний мочевого пузыря и урогенитального тракта, включая недержание мочи; гиперактивного мочевого пузыря; синдрома болезненного мочевого пузыря; интерстициального цистита (IC); простатита; комплексного регионального болевого синдрома (CRPS) I типа и II типа; распространенной боли, пароксизмальной чрезмерной боли, прурита, шума в ушах или боли, индуцированной стенокардией.
Соединения согласно настоящему изобретению могут быть легко получены с использованием следующих способов. На представленных ниже схемах 1-4 проиллюстрированы способы получения соединений согласно настоящему изобретению.
Схема 1
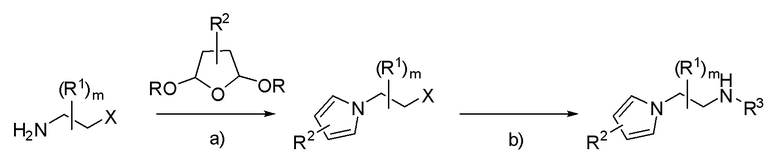
X = уходящая группа или NH2; R3 = алкил.
a) H+: протонсодержащая кислота, такая как уксусная кислота или пара-толуолсульфоновая кислота, NaOAc; b) H2NR3, растворитель (например: EtOH или CH3CN).
Схема 2
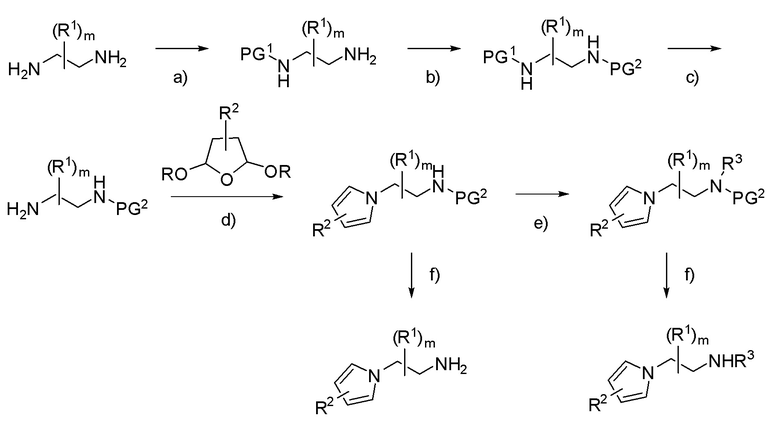
PG1 = неустойчивая в кислой среде защитная группа (например: Boc); PG2 = устойчивая в кислой среде защитная группа (например: карбобензокси, бензил); R3 = алкил.
a) PG1 = Boc; Boc2O, основание (например: Et3N), растворитель (например: THF); b) PG2 = карбобензокси; бензил-2,5-диоксопирролидин-1-илкарбонат, основание (например: Et3N), растворитель (например: THF); c) PG1 = Boc; H+ (например: HCl или TFA), растворитель (например: iPrOH, EtOH, CH3CN или CH2Cl2); d) H+: протонсодержащая кислота, такая как уксусная кислота или пара-толуолсульфоновая кислота, NaOAc; e) R3-X, основание (например: NaH или K2CO3), растворитель (например: DMF, THF или CH3CN); f) PG2 = карбобензокси; Pd/C, H2, растворитель (например: iPrOH, EtOH или CH3CN).
Схема 3
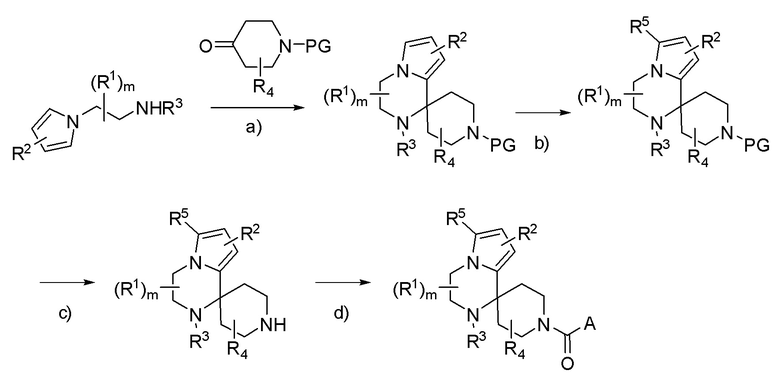
PG = защитная группа, такая как Boc, бензил, карбобензокси; R3 = H или алкил.
a) кат. H+: протонсодержащая кислота, такая как трифторуксусная кислота, пара-толуолсульфоновая кислота или дихлоруксусная кислота, растворитель (например: EtOH); b) R5 = CF3, 5-(трифторметил)-5H-дибензо[b,d]тиофентрифторметансульфонат, основание (например: K2CO3), растворитель (например: CH3CN) или R5= галогеналкил; галогеналкилйодид (например: CF3I, CF3CH2I или CF3CF2I), FeSO4·6H2O, H2O2, растворитель (например: DMSO); R5 = CN, хлорсульфонилизоцианат, растворитель (например: THF или DMF); R5 = Cl, CF3SO2Cl, растворитель (например: CH2Cl2); R5 = R6C(О), ацилирующий агент (например: R6C(О)2О, R6C(О)Cl), основание (например: пиридин, Et3N или DBN), растворитель (например: CH2Cl2; DCE или THF) или i) NBS, CH2Cl2; ii); CH3(CH2)nOCH=CHR7, катализатор (например: Pd2dba3·CHCl3), растворитель (например: диоксан); c) PG = Boc, H+ (например: HCl или TFA), растворитель (например: iPrOH, EtOH, CH3CN или CH2Cl2); PG = карбобензокси; Pd/C, H2, растворитель (например: iPrOH, EtOH или CH3CN); d) A-CO2H; агент сочетания (например: HATU или EDCI), основание (например: Et3N или iPr2NEt), растворитель (например: DMF, CH3CN или CH2Cl2); или A-C(О)-Cl, NaOH, растворитель (например: вода и MTBE).
Схема 4
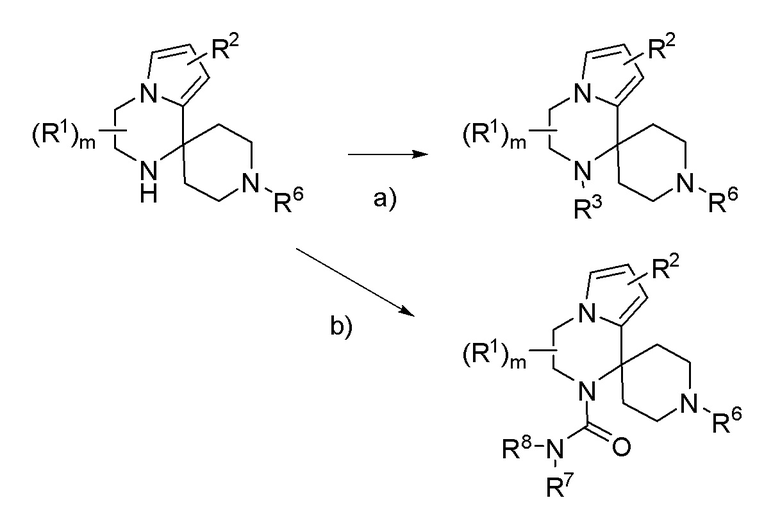
R3= ацил; R6 = PG или C(О)A; R7 = алкил.
a) R3-X (X = уходящая группа например: галоген, OTs), основание (например: K2CO3, Et3N или пиридин), растворитель (например: DMF, THF, ACN, CH2Cl2 или пиридин); b) R8 = H; R7-NCO, основание (например: Et3N), растворитель (например: THF) или ClC(О)NR7R8, основание (например: пиридин).
Применения, приготовление рецептур и введение
Фармацевтически приемлемые композиции
Как обсуждалось выше, настоящее изобретение относится к соединениям, которые представляют собой ингибиторы потенциалзависимых натриевых ионных каналов, а потому соединения согласно настоящему изобретению применимы для лечения заболеваний, нарушений и состояний, включая без ограничения острую, хроническую, нейропатическую или воспалительную боль, артрит, мигрень, кластерные головные боли, тригеминальную невралгию, герпетическую невралгию, обычные невралгии, эпилепсию или эпилептические состояния, нейродегенеративные нарушения, психиатрические нарушения, такие как тревожность и депрессия, миотонию, аритмию, двигательные нарушения, нейроэндокринные нарушения, атаксию, рассеянный склероз, синдром раздраженного кишечника и недержание. Соответственно, согласно другому аспекту настоящего изобретения, предусмотрены фармацевтически приемлемые композиции, причем такие композиции содержат любое из соединений, описанных в настоящем документе, и необязательно содержат фармацевтически приемлемый носитель, адъювант или основу. Согласно некоторым вариантам осуществления, такие композиции необязательно также содержат одно или несколько дополнительных терапевтических средств.
Также следует принимать во внимание, что некоторые из соединений согласно настоящему изобретению могут существовать для лечения в свободной форме, или, в соответствующих случаях, в качестве их фармацевтически приемлемого производного. Согласно настоящему изобретению, фармацевтически приемлемые производные включают без ограничения фармацевтически приемлемые соли, сложные эфиры, соли таких сложных эфиров, или любой другой аддукт или производное, которое после введения нуждающемуся в этом субъекту способно обеспечивать, прямо или опосредованно, описанное в настоящем документе соединение или его метаболит или остаток.
Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к таким солям, которые подходят в рамках здравого медицинского суждения для использования в контакте с тканями людей и низших животных без проявления чрезмерной токсичности, раздражения, аллергической реакции и т.п., и которые соразмерны с приемлемым соотношением польза/риск. Термин «фармацевтически приемлемая соль» означает любую нетоксичную соль или соль сложного эфира соединения согласно настоящему изобретению, которая после введения реципиенту способна обеспечивать, прямо или опосредованно, соединение согласно настоящему изобретению или его активный в плане ингибирования метаболит или остаток. Используемый в настоящем документе термин «его активный в плане ингибирования метаболит или остаток» означает, что его метаболит или остаток также представляет собой ингибитор потенциалзависимого натриевого ионного канала.
Фармацевтически приемлемые соли хорошо известны в данной области техники. Например, S. M. Berge и соавт. подробно описаны фармацевтически приемлемые соли в работе J. Pharmaceutical Sciences, 1977, 66, 1-19, включенной в настоящий документ посредством ссылки. Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают соли, полученные из подходящих неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых, нетоксичных кислотно-аддитивных солей являются соли аминогруппы, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или путем использования других способов, применяемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают адипаты, альгинаты, аскорбаты, аспартаты, бензолсульфонаты, бензоаты, бисульфаты, бораты, бутираты, камфораты, камфорсульфонаты, цитраты, циклопентанпропионаты, диглюконаты, додецилсульфаты, этансульфонаты, формиаты, фумараты, глюкогептонаты, глицерофосфаты, глюконаты, гемисульфаты, гептаноаты, гексаноаты, гидроиодиды, 2-гидроксиэтансульфонаты, лактобионаты, лактаты, лаураты, лаурилсульфаты, малаты, малеаты, малонаты, метансульфонаты, 2-нафталинсульфонаты, никотинаты, нитраты, олеаты, оксалаты, пальмитаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, фосфаты, пикраты, пивалаты, пропионаты, стеараты, сукцинаты, сульфаты, тартраты, тиоцианаты, паратолуолсульфонаты, ундеканоаты, валераты и т.п. Соли, полученные из подходящих оснований, включают соли щелочных металлов, щелочноземельных металлов, соли аммония и N+(C1-4алкил)4 соли. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп соединения, раскрытого в настоящем документе. Путем такой кватернизации могут быть получены продукты, растворимые в воде или в масле, или диспергируемые продукты. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Кроме того, фармацевтически приемлемые соли включают, в соответствующих случаях, нетоксичные соли аммония, четвертичного аммония и аминокатионов, образованных с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, сульфонат низшего алкила и арилсульфонат.
Как описано выше, фармацевтически приемлемые композиции согласно настоящему изобретению дополнительно содержат фармацевтически приемлемый носитель, адъювант или основу, которые в данном контексте включают всевозможные растворители, разбавители или другую жидкую основу, способствующие диспергированию и суспендированию вещества, поверхностно-активные вещества, изотонические средства, загустители или эмульгаторы, консерванты, твердые связующие вещества, смазки и т.п., подбираемые в зависимости от конкретной желаемой лекарственной формы. В работе Remington’s Pharmaceutical Sciences, Sixteenth Edition, E. W. Martin (Mack Publishing Co., Easton, Pa., 1980) раскрыты различные носители, используемые при составлении фармацевтически приемлемых композиций, и известные методики для их приготовления. За исключением случаев, когда любая общепринятая среда-носитель является несовместимой с соединениями согласно настоящему изобретению, как например при индукции любого нежелательного биологического эффекта или иного другого вредного взаимодействия с любым(и) другим(и) компонентом(ами) фармацевтически приемлемой композиции, ее применение предусмотрено как подпадающее под объем настоящего изобретения. Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей включают без ограничения ионообменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческий сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота или сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, двузамещенный фосфорнокислый натрий, вторичный кислый фосфат калия, хлорид натрия, соли цинка, коллоидную окись кремния, трисиликат магния, поливинилпирролидон, полиакрилаты, воски, блок-сополимеры полиэтилена/полиоксипропилена, ланолин, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетатцеллюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как какао-масло и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль или полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический солевой раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазки, такие как лаурилсульфат натрия и стеарат магния, а также красители, рилизинг-агенты, покровные средства, подсластители, вкусоароматизаторы и ароматизирующие добавки, консерванты и антиоксиданты также могут присутствовать в композиции, в соответствии с решением разработчика рецептур.
Использование соединений и фармацевтически приемлемых композиций
Согласно еще одному аспекту, настоящим изобретением предусмотрен способ лечения или облегчения тяжести острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, герпетической невралгии, обычных невралгий, эпилепсии или эпилептических состояний, нейродегенеративных нарушений, психиатрических нарушений, таких как тревожность и депрессия, биполярного нарушения, миотонии, аритмии, двигательных нарушений, нейроэндокринных нарушений, атаксии, рассеянного склероза, синдрома раздраженного кишечника, недержания, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, корешкового болевого синдрома, ишиаса, боли в спине, боли в голове и шее, тяжелой или некупируемой боли, ноцицептивной боли, прорыва боли, послеоперационной боли или раковой боли, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту.
Согласно некоторым вариантам осуществления, настоящим изобретением предусмотрен способ лечения или облегчения тяжести инсульта, церебральной ишемии, травматического повреждения головного мозга, амиотрофического бокового склероза, индуцированной стрессом или физической нагрузкой стенокардии, учащенного сердцебиения, артериальной гипертензии, мигрени или аномальной моторики желудочно-кишечного тракта, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту.
Согласно некоторым вариантам осуществления, настоящим изобретением предусмотрен способ лечения или облегчения тяжести острой, хронической, нейропатической или воспалительной боли, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту. Согласно некоторым другим вариантам осуществления, настоящим изобретением предусмотрен способ лечения или облегчения тяжести корешкового болевого синдрома, ишиаса, боли в спине, головной боли или боли в шее, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту. Согласно другим вариантам осуществления, настоящим изобретением предусмотрен способ лечения или облегчения тяжести тяжелой и некупируемой боли, острой боли, послеоперационной боли, боли в спине, шума в ушах или раковой боли, включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту.
Согласно некоторым вариантам осуществления, настоящим изобретением предусмотрен способ лечения или облегчения тяжести боли при злокачественной опухоли бедренной кости; незлокачественной хронической костной боли; ревматоидного артрита; остеоартрита; спинального стеноза; нейропатической поясничной боли; нейропатической поясничной боли; миофасциального болевого синдрома; фибромиалгии; боли в височно-нижнечелюстном суставе; хронической висцеральной боли, включая абдоминальную боль; боли в поджелудочной железе; боли при IBS; хронической и острой головной боли; мигрени; головной боли напряжения, включая кластерные головные боли; хронической и острой нейропатической боли, включая постгерпетическую невралгию; диабетической нейропатии; HIV-ассоциированной нейропатии; тригеминальной невралгии; нейропатии Шарко-Мари-Тута; наследственных сенсорных нейропатий; повреждения периферических нервов; болезненных невром; эктопических разрядов в проксимальных и дистальных отделах спинного мозга; радикулопатии; нейропатической боли, индуцированной химиотерапией; нейропатической боли, индуцированной лучевой терапией; постмастэктомической боли; центральной боли; боли при травме спинного мозга; постинсультной боли; таламической боли; комплексного регионального болевого синдрома; фантомной боли; некупируемой боли; острой боли; острой послеоперационной боли; острой скелетно-мышечной боли; суставной боли; механической поясничной боли; боли в шее; тендинита; боли, вызванной повреждением/физической нагрузкой; острой висцеральной боли, включая абдоминальную боль; пиелонефрита; аппендицита; холецистита; кишечной непроходимости; грыж и т.п.; боли в груди, включая сердечную боль; тазовой боли, боли при почечной колике, острой акушерской боли, включая родовые схватки; боли при кесаревом сечении; острой воспалительной, ожоговой и травматической боли; острой периодической боли, включая эндометриоз; острой боли при опоясывающем герпесе; серповидно-клеточной анемии; острого панкреатита; прорыва боли; орофациальной боли, включая боль при синусите, зубную боль; боли при рассеянном склерозе (MS); боли при депрессии; лепрозной боли; боли при болезни Бехчета; адипозалгии; флебитической боли; боли при синдроме Гийена-Барре; болезненности в ногах и при движении пальцами ног; синдрома Хаглунда; эритромелалгической боли; боли при болезни Фабри; заболеваний мочевого пузыря и урогенитального тракта, включая недержание мочи; гиперактивного мочевого пузыря; синдрома болезненного мочевого пузыря; интерстициального цистита (IC); или простатита; комплексного регионального болевого синдрома (CRPS) I типа и II типа; боли, индуцированной стенокардией; включающий введение эффективного количества соединения или фармацевтически приемлемой композиции, содержащей соединение, нуждающемуся в этом субъекту.
Согласно некоторым вариантам осуществления настоящего изобретения термин «эффективное количество» соединения или фармацевтически приемлемой композиции означает, что такое количество эффективно для лечения или облегчения тяжести одного или нескольких нарушений из острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, герпетической невралгии, обычных невралгий, эпилепсии или эпилептических состояний, нейродегенеративных нарушений, психиатрических нарушений, таких как тревожность и депрессия, миотонии, аритмии, двигательных нарушений, нейроэндокринных нарушений, атаксии, рассеянного склероза, синдрома раздраженного кишечника, недержания, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, корешкового болевого синдрома, ишиаса, боли в спине, боли в голове и шее, тяжелой или некупируемой боли, ноцицептивной боли, прорыва боли, послеоперационной боли, шума в ушах или раковой боли.
В соответствии со способом согласно настоящему изобретению, соединения и композиции могут быть введены с использованием любого количества и любого пути введения, эффективного для лечения или облегчения тяжести одного или нескольких нарушений из острой, хронической, нейропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, герпетической невралгии, обычных невралгий, эпилепсии или эпилептических состояний, нейродегенеративных нарушений, психиатрических нарушений, таких как тревожность и депрессия, миотонии, аритмии, двигательных нарушений, нейроэндокринных нарушений, атаксии, рассеянного склероза, синдрома раздраженного кишечника, недержания, висцеральной боли, остеоартритной боли, постгерпетической невралгии, диабетической нейропатии, корешкового болевого синдрома, ишиаса, боли в спине, боли в голове и шее, тяжелой или некупируемой боли, ноцицептивной боли, прорыва боли, послеоперационной боли, шума в ушах или раковой боли. Точное необходимое количество будет варьировать для разных субъектов в зависимости от биологического вида, возраста и общего состояния здоровья субъекта, тяжести инфекции, конкретного средства, способа его введения и т.п. Для простоты введения и однородности дозирования соединения согласно настоящему изобретению предпочтительно включают в состав стандартной лекарственной формы. Используемое в настоящем документе выражение «стандартная лекарственная форма» относится к физически дискретной единице средства, подходящего для субъекта, подлежащего лечению. Однако следует понимать, что общая суточная дозировка соединений и композиций согласно настоящему изобретению будет определяться лечащим врачом в рамках здравого медицинского суждения. Конкретная величина эффективной дозы для каждого конкретного субъекта или организма будет зависеть от ряда факторов, включая подлежащее лечению нарушение и тяжесть этого нарушения; активность конкретно используемых соединения; конкретно используемые композиции; возраст, массу тела, общее состояние здоровья, пол и рацион питания субъекта; время введения, путь введения и скорость выведения конкретного используемого соединения; длительность лечения; лекарства, используемые в сочетании или одновременно с конкретно используемым соединением, и подобные факторы, хорошо известные в области медицинской техники. Используемые в настоящем документе термины «субъект» или «пациент» означают животное, предпочтительно млекопитающее, и наиболее предпочтительно человека.
В зависимости от тяжести подлежащей лечению инфекции фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (как, например, посредством порошков, мазей или капель), буккально, в виде перорального или назального спрея или т.п. Согласно некоторым вариантам осуществления, соединения согласно настоящему изобретению могут быть введены перорально или парентерально в дозах приблизительно от 0,01 мг/кг приблизительно до 50 мг/кг и предпочтительно приблизительно от 1 мг/кг приблизительно до 25 мг/кг массы тела субъекта в сутки, однократно или несколько раз в сутки, для получения желаемого терапевтического эффекта.
Жидкие лекарственные формы для перорального введения включают без ограничения фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло ростков пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана и их смеси. Помимо инертных разбавителей пероральные композиции также могут содержать адъюванты, такие как увлажнители, эмульгаторы и способствующие суспендированию средства, подсластители, вкусоароматизаторы и ароматизирующие средства.
Инъекционные препараты, например, стерильные инъекционные водные или масляные суспензии могут быть составлены в соответствии с известным уровнем техники с использованием подходящих способствующих диспергированию средств или увлажнителей и способствующих суспендированию средств. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. В число приемлемых основ и растворителей, которые могут быть использованы, включены вода, раствор Рингера, соответствующий U.S.P. и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или среды для суспендирования традиционно используются стерильные нелетучие масла. Для этой цели может быть использовано любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, в приготовлении инъецируемых препаратов используют жирные кислоты, такие как олеиновая кислота.
Инъецируемые лекарственные формы могут быть подвергнуты стерилизации, например, путем фильтрования через задерживающий бактерии фильтр или путем включения в состав стерильных твердых композиций стерилизующих средств, которые перед применением могут быть растворены или диспергированы в стерильной воде или другой стерильной инъецируемой среде.
Для пролонгирования эффекта соединения согласно настоящему изобретению часто является желательным замедлить всасывание соединения при подкожной или внутримышечной инъекции. Это может быть осуществлено посредством применения жидкой суспензии кристаллического или аморфного вещества с низкой растворимостью в воде. В таком случае, скорость всасывания соединения зависит от скорости растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. В качестве альтернативы, замедленного всасывания парентерально введенной формы соединения достигают путем растворения или суспендирования соединения в масляной основе. Инъецируемые депо-формы приготавливают путем формирования микроинкапсулированных матриц соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения соединения и конкретного используемого полимера и его природы, скорость высвобождения соединения можно регулировать. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые лекарственные депо-формы также приготавливают путем включения соединения в липосомы или микроэмульсии, которые являются совместимыми с тканями тела.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть приготовлены путем смешивания соединений согласно настоящему изобретению с подходящими не вызывающими раздражения наполнителями или носителями, такими как какао-масло, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при комнатной температуре, но жидкими при температуре тела, а потому разжижаются в прямой кишке или влагалище и высвобождают активное соединение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано, по меньшей мере, с одним инертным фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или дикальция фосфат и/или а) начинками или разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и акация, с) увлажнителями, такими как глицерин, d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, е) замедлителями растворения, такими как парафин, f) усилителями всасывания, такими как соединения четвертичного аммония, g) увлажнителями, такими как, например, цетиловый спирт и глицеролмоностеарат, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазками, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смеси. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные средства.
Твердые композиции сходного типа также могут быть использованы в качестве содержимого мягких и твердых желатиновых капсул с использованием таких наполнителей, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть снабжены покрытиями и оболочками, такими как кишечнорастворимые покрытия и другие покрытия, хорошо известные в области разработки фармацевтических рецептур. Они могут необязательно содержать замутняющие средства, а также могут представлять собой композицию, которая высвобождает активный(ые) ингредиент(ы) исключительно, или предпочтительно, в конкретном отделе желудочно-кишечного тракта, необязательно в процессе замедленного высвобождения. Примеры имплантируемых композиций, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции сходного типа также могут быть использованы в качестве содержимого мягких и твердых желатиновых капсул с использованием таких наполнителей, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п.
Активные соединения также могут находиться в микроинкапсулированной форме с одним или несколькими указанными выше наполнителями. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы могут быть снабжены покрытиями и оболочками, такими как кишечнорастворимые покрытия, покрытия, регулирующие высвобождение, и другие покрытия, хорошо известные в области разработки фармацевтической рецептур. В таких твердых лекарственных формах активное соединение может быть смешано, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы также могут содержать (что является обычной практикой) дополнительные вещества, отличные от инертных разбавителей, например, смазки для таблетирования и другие средства для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, лекарственные формы также могут содержать буферные средства. Они могут необязательно содержать замутняющие средства, а также могут представлять собой композицию, которая высвобождает активный(ые) ингредиент(ы) исключительно, или предпочтительно, в конкретном отделе желудочно-кишечного тракта, необязательно в процессе замедленного высвобождения. Примеры имплантируемых композиций, которые могут быть использованы, включают полимерные вещества и воски.
Лекарственные формы для местного или чрескожного введения соединения согласно настоящему изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Активные компоненты смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми нужными консервантами или буферами, которые могут быть необходимы. Офтальмическая лекарственная форма, ушные капли и глазные капли также рассматриваются как подпадающие под объем настоящего изобретения. Кроме того, настоящим изобретением предусмотрены чрескожные пластыри, которые обладают дополнительным преимуществом, обеспечивая контролируемую доставку соединения в организм. Такие лекарственные формы приготавливают путем растворения или диспергирования соединения в подходящей среде. Для увеличения поступления соединения через кожу также могут быть использованы усилители всасывания. Скорость можно регулировать как посредством снабжения регулирующей скорость мембраной, так и посредством диспергирования соединения в полимерном матриксе или геле.
Как в целом описано выше, соединения согласно настоящему изобретению применимы в качестве ингибиторов потенциалзависимых натриевых ионных каналов. Согласно одному варианту осуществления, соединения и композиции согласно настоящему изобретению представляют собой ингибиторы одного или нескольких каналов из NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9, а потому, не желая быть связанным какой-либо конкретной теорией, соединения и композиции являются особенно применимыми для лечения или облегчения тяжести заболевания, состояния или нарушения, при котором активация или гиперактивность одного или нескольких каналов из NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9 вовлечена в заболевание, состояние или нарушение. Если активация или гиперактивность NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9 вовлечена в конкретное заболевание, состояние или нарушение, то заболевание, состояние или нарушение также может быть отнесено к «заболеванию, состоянию или нарушению, опосредованному NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9». Соответственно, согласно другому аспекту, настоящее изобретение относится к способу лечения или облегчения тяжести заболевания, состояния или нарушения, при котором в болезненное состояние вовлечена активация или гиперактивность одного или нескольких каналов из NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9.
Активность соединения, используемого в настоящем изобретении в качестве ингибитора NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9, может быть оценена в соответствии со способами, описанными в общих чертах в настоящем документе в разделе «Примеры», или в соответствии со способами, доступными специалисту в данной области техники.
Согласно некоторым типичным вариантам осуществления, соединения согласно настоящему изобретению применимы в качестве ингибиторов NaV1.7 и/или NaV1.8.
Также следует принимать во внимание, что соединения и фармацевтически приемлемые композиции согласно настоящему изобретению могут быть использованы при сочетанной терапии, а именно, соединения и фармацевтически приемлемые композиции могут быть введены одновременно, до, или после одного или нескольких других желаемых терапевтических средств или медицинских процедур. Конкретное сочетание терапий (терапевтических средств или процедур), применяемых при сочетанной схеме лечения, будет принимать во внимание совместимость желаемых терапевтических средств и/или процедур и поставленный целью желаемый терапевтический эффект. Также следует принимать во внимание, что применяемые терапии могут достигать желаемого эффекта для того же самого нарушения (например, соединение настоящего изобретения может быть введено одновременно с другим средством, используемым для лечения того же самого нарушения), или они могут достигать различных эффектов (например, контроль неблагоприятного эффекта). Как используется в данной заявке, дополнительные терапевтические средства, которые обычно вводятся для лечения или предупреждения конкретного заболевания или состояния, известны как “подходящие для заболевания или состояния, которое лечат.” Например, типичные дополнительные терапевтические средства включают без ограничения: неопиоидные анальгетики (индолы, такие как этодолак, индометацин, сулиндак, толметин; нафтилалканоны, такие как набуметон; оксикамы, такие как пироксикам; производные парааминофенола, такие как ацетаминофен; пропионовые кислоты, такие как фенопрофен, флурбипрофен, ибупрофен, кетопрофен, напроксен, напроксен натрий, оксапрозин; салицилаты, такие как аспирин, холин-магний трисалицилат, дифлунизал; фенаматы, такие как меклофенамовая кислота, мефенамовая кислота; и пиразолы, такие как фенилбутазон); или опиоидные (наркотические) агонисты (такие как кодеин, фентанил, гидроморфон, леворфанол, меперидин, метадон, морфин, оксикодон, оксиморфон, пропоксифен, бупренорфин, буторфанол, дезоцин, налбуфин и пентазоцин). Дополнительно, могут быть использованы нелекарственные анальгетические подходы в сочетании с введением одного или нескольких соединений согласно настоящему изобретению. Например, могут быть использованы анестезиологический (интраспинальная инфузия, блокада нервов), нейрохирургический (невролиз путей ЦНС), нейростимуляторный (чрескожная электрическая стимуляция нервов, стимуляция заднего столба спинного мозга), физиотерапевтический (физиотерапия, ортопедические устройства, диатермия) или психологический (когнитивные способы - гипноз, способы биологической обратной связи или поведенческие способы) подходы. Дополнительные подходящие терапевтические средства или подходы описаны в общих чертах в The Merck Manual, Seventeenth Edition, Ed. Mark H. Beers and Robert Berkow, Merck Research Laboratories, 1999, и на веб-сайте Food and Drug Administration www.fda.gov, полное содержание которых включено в настоящий документ посредством ссылки.
Согласно другому варианту осуществления, дополнительные подходящие терапевтические средства выбирают из следующего:
(1) опиоидного анальгетика, например, морфин, героин, гидроморфон, оксиморфон, леворфанол, леваллорфан, метадон, меперидин, фентанил, кокаин, кодеин, дигидрокодеин, оксикодон, гидрокодон, пропоксифен, налмефен, налорфин, налоксон, налтрексон, бупренорфин, буторфанол, налбуфин или пентазоцин;
(2) нестероидного противовоспалительного средства (NSAID), например, аспирин, диклофенак, дифлузинал, этодолак, фенбуфен, фенопрофен, флуфенизал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, меклофенамовая кислота, мефенамовая кислота, мелоксикам, набуметон, напроксен, нимесулид, нитрофлурбипрофен, олсалазин, оксапрозин, фенилбутазон, пироксикам, сульфасалазин, сулиндак, толметин или зомепирак;
(3) барбитуратного седативного средства, например, амобарбитал, апробарбитал, бутабарбитал, бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобарбитал, секобарбитал, талбутал, тиамилал или тиопентал;
(4) обладающего седативным действием бензодиазепина, например, хлордиазепоксид, клоразепат, диазепам, флуразепам, лоразепам, оксазепам, темазепам или триазолам;
(5) обладающего седативным действием антагониста H1, например, дифенгидрамин, пириламин, прометазин, хлорфенирамин или хлорциклизин;
(6) седативного средства, такого как глутетимид, мепробамат, метаквалон или дихлоралфеназон;
(7) релаксанта скелетной мускулатуры, например, балофен, карисопродол, хлорзоксазон, циклобензаприн, метокарбамол или орфренадин;
(8) антагониста NMDA рецептора, например, декстрометорфан ((+)-3-метокси-N-метилморфинан) или его метаболит декстрорфан ((+)-3-гидрокси-N-метилморфинан), кетамин, мемантин, пирролохинолин хинин, цис-4-(фосфонометил)-2-пиперидинкарбоновая кислота, будипин, EN-3231 (MorphiDex®, сочетанная лекарственная форма морфина и декстрометорфана), топирамат, нерамексан или перзинфотел, включая антагонист NR2B, например, ифенпродил, траксопродил или (-)-(R)-6-{2-[4-(3-фторфенил)-4-гидрокси-1-пиперидинил]-1-гидроксиэтил-3,4-дигидро-2(1H)-хинолинон;
(9) α-адреноблокатора, например, доксазозин, тамсулозин, клонидин, гуанфацин, дексметатомидин, модафинил или 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин;
(10) трициклического антидепрессанта, например, дезипрамин, имипрамин, амитриптилин или нортриптилин;
(11) антиконвульсанта, например, карбамазепин, ламотригин, топирамат или вальпроат;
(12) антагониста тахикининов (NK), в частности антагонист NK-3, NK-2 или NK-1, например, (αR,9R)-7-[3,5-бис(трифторметил)бензил]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7H-[1,4]диазоцино[2,1-g][1,7]-нафтиридин-6-13-дион (TAK-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-бис(трифторметил)фенил]этокси-3-(4-фторфенил)-4-морфолинил]метил]-1,2-дигидро-3H-1,2,4-триазол-3-он (MK-869), апрепитант, ланепитант, дапитант или 3-[[2-метокси-5-(трифторметокси)фенил]метиламино]-2-фенилпиперидин (2S,3S);
(13) антагониста мускаринового рецептора, например, оксибутинин, толтеродин, пропиверин, тропсия хлорид, дарифенацин, солифенацин, темиверин и ипратропиум;
(14) селективного ингибитора COX-2, например, целекоксиб, рофекоксиб, парекоксиб, вальдекоксиб, деракоксиб, эторикоксиб или лумиракоксиб;
(15) анальгетика-производного анилина, в частности парацетамол;
(16) нейролептика, такого как дроперидол, хлорпромазин, галоперидол, перфеназин, тиоридазин, мезоридазин, трифлуоперазин, флуфеназин, клозапин, оланзапин, рисперидон, зипрасидон, кветиапин, сертиндол, арипипразол, сонепипразол, блонансерин, илоперидон, пероспирон, раклоприд, зотепин, бифепрунокс, азенапин, лурасидон, амисульприд, балаперидон, палиндор, эпливансерин, осанетант, римонабант, меклинертант, Miraxion® или саризотан;
(17) агониста ваниллоидного рецептора (например, резинфератоксин) или его антагониста (например, капсазепин);
(18) β-адреноблокатора, такого как пропранолол;
(19) местного анестетика, такого как мексилетин;
(20) кортикостероида, такого как дексаметазон;
(21) агониста или антагониста 5-HT рецептора, в частности агониста 5-HT1B/1D, такого как элетриптан, суматриптан, наратриптан, золмитриптан или ризатриптан;
(22) антагониста 5-HT2A рецептора, такого как R(+)-α-(2,3-диметоксифенил)-1-[2-(4-фторфенилэтил)]-4-пиперидинметанол (MDL-100907);
(23) холинэргического (никотинового) анальгетика, такого как изпрониклин (TC-1734), (E)-N-метил-4-(3-пиридинил)-3-бутен-1-амин (RJR-2403), (R)-5-(2-азетидинилметокси)-2-хлорпиридин (ABT-594) или никотин;
(24) Tramadol®;
(25) ингибитора PDEV, такого как 5-[2-этокси-5-(4-метил-1-пиперазинилсульфонил)фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (силденафил), (6R,12aR)-2,3,6,7,12,12a-гексагидро-2-метил-6-(3,4-метилендиоксифенил)-пиразино[2',1':6,1]-пиридо[3,4-b]индол-1,4-дион (IC-351 или тадалафил), 2-[2-этокси-5-(4-этилпиперазин-1-ил-1-сульфонил)-фенил]-5-метил-7-пропил-3H-имидазо[5,1-f][1,2,4]триазин-4-он (варденафил), 5-(5-ацетил-2-бутокси-3-пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-(5-ацетил-2-пропокси-3-пиридинил)-3-этил-2-(1-изопропил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 4-[(3-хлор-4-метоксибензил)амино]-2-[(2S)-2-(гидроксиметил)пирролидин-1-ил]-N-(пиримидин-2-илметил)пиримидин-5-карбоксамид, 3-(1-метил-7-оксо-3-пропил-6,7-дигидро-1H-пиразоло[4,3-d]пиримидин-5-ил)-N-[2-(1-метилпирролидин-2-ил)этил]-4-пропоксибензолсульфонамид;
(26) α-2-δ лиганда, такого как габапентин, прегабалин, 3-метилгабапентин, (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусная кислота, (3S,5R)-3-аминометил-5-метилгептановая кислота, (3S,5R)-3-амино-5-метилгептановая кислота, (3S,5R)-3-амино-5-метилкаприловая кислота, (2S,4S)-4-(3-хлорфенокси)пролин, (2S,4S)-4-(3-фторбензил)пролин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусная кислота, 3-(1-аминометилциклогексилметил)-4H-[1,2,4]оксадиазол-5-он, C-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусная кислота, (3S,5R)-3-аминометил-5-метилкаприловая кислота, (3S,5R)-3-амино-5-метилпеларгоновая кислота, (3S,5R)-3-амино-5-метилкаприловая кислота, (3R,4R,5R)-3-амино-4,5-диметилгептановая кислота и (3R,4R,5R)-3-амино-4,5-диметилкаприловая кислота;
(27) каннабиноида;
(28) антагониста метаботропного глутаматного рецептора подтипа 1 (mGluR1);
(29) ингибитора обратного захвата серотонина, такого как сертралин, метаболит сертралина десметилсертралин, флуоксетин, норфлуоксетин (метаболит флуоксетиндесметила), флувоксамин, пароксетин, циталопрам, метаболит циталопрама десметилциталопрам, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон, церикламин и тразодон;
(30) ингибитора обратного захвата норадреналина (норэпинефрина), такого как мапротилин, лофепрамин, миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупроприон, метаболит бупроприона гидроксибупроприон, номифензин и вилоксазин (Vivalan®), в частности селективного ингибитора обратного захвата норадреналина, такого как ребоксетин, в частности (S,S)-ребоксетин;
(31) ингибитора обратного захвата серотонина и норадреналина, такого как венлафаксин, метаболит венлафаксина O-десметилвенлафаксин, кломипрамин, метаболит кломипрамина десметилкломепрамил, дулоксетин, милнаципран и имипрамин;
(32) ингибитора индуцибельной синтазы оксида азота (iNOS), такого как S-[2-[(1-иминоэтил)амино]этил]-L-гомоцистеин, S-[2-[(1-иминоэтил)амино]этил]-4,4-диоксо-L-цистеин, S-[2-[(1-иминоэтил)амино]этил]-2-метил-L-цистеин, (2S,5Z)-2-амино-2-метил-7-[(1-иминоэтил)амино]-5-гептеновая кислота, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-S-хлор-S-пиридинкарбонитрил; 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-4-хлорбензонитрил, (2S,4R)-2-амино-4-[[2-хлор-5-(трифторметил)фенил]тио]-5-тиазолбутанол, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-6-(трифторметил)-3-пиридинкарбонитрил, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-5-хлорбензонитрил, N-[4-[2-(3-хлорбензиламино)этил]фенил]тиофен-2-карбоксамидин или гуанидинэтилдисульфид;
(33) ингибитора ацетилхолинэстеразы, такого как донепезил;
(34) антагониста простагландина E2 подтипа 4 (EP4), такого как N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)карбонил]-4-метилбензолсульфонамид или 4-[(15)-1-({[5-хлор-2-(3-фторфенокси)пиридин-3-ил]карбонил}амино)этил]бензойная кислота;
(35) антагониста лейкотриена B4; такого как 1-(3-бифенил-4-илметил-4-гидроксихроман-7-ил)-циклопентанкарбоновая кислота (CP-105696), 5-[2-(2-карбоксиэтил)-3-[6-(4-метоксифенил)-5E-гексенил]оксифенокси]валериановая кислота (ONO-4057) или DPC-11870;
(36) ингибитора 5-липоксигеназы, такого как зилеутон, 6-[(3-фтор-5-[4-метокси-3,4,5,6-тетрагидро-2H-пиран-4-ил])феноксиметил]-1-метил-2-хинолон (ZD-2138) или 2,3,5-триметил-6-(3-пиридилметил)-1,4-бензохинон (CV-6504);
(37) блокатора натриевых каналов, такого как лидокаин;
(38) антагониста 5-HT3, такого как ондансетрон;
и их фармацевтически приемлемых солей и сольватов.
Количество дополнительного терапевтического средства, содержащегося в композициях согласно настоящему изобретению, не должно превышать количество, которое обычно вводилось бы в композиции, содержащей такое терапевтическое средство в качестве единственного активного средства. Предпочтительно, количество дополнительного терапевтического средства в композициях, раскрытых в настоящем документе, будет варьировать приблизительно от 50% до 100% от количества, обычно содержащегося в композиции, содержащей такое средство в качестве единственного терапевтически активного средства.
Соединения согласно настоящему изобретению или их фармацевтически приемлемые композиции также могут быть включены в состав композиций для покрытия имплантируемых медицинских изделий, таких как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Соответственно, согласно другому аспекту, настоящее изобретение включает композицию для покрытия имплантируемых изделий, содержащую соединение согласно настоящему изобретению, описанное в общих чертах выше и в классах и подклассах настоящего документа, и носитель, подходящий для покрытия упомянутого имплантируемого изделия. Согласно другому аспекту, настоящее изобретение включает имплантируемое изделие покрытое композицией, содержащей соединение согласно настоящему изобретению, описанное в общих чертах выше и в классах и подклассах настоящего документа, и носитель, подходящий для покрытия упомянутого имплантируемого изделия. Подходящие покрытия и общий способ изготовления снабженных покрытием имплантируемых изделий описаны в патентах США №№ 6099562; 5886026 и 5304121. Покрытия обычно представляют собой биосовместимые полимерные вещества, такие как полимерные гидрогели, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. В дальнейшем, покрытия могут быть необязательно снабжены подходящим верхним покрытием из фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их сочетаний для наделения композиции характеристиками регулируемого высвобождения.
Другой аспект настоящего изобретения относится к ингибированию активности одного или нескольких каналов из NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9 в биологическом образце или субъекте, причем способ включает введение субъекту соединения формулы I или содержащей упомянутое соединение композиции или приведение упомянутого биологического образца в контакт с ним(ней). Используемый в настоящем документе термин «биологический образец» включает без ограничений клеточные культуры или их экстракты; биопсионный материал, полученный из млекопитающих или его экстракты; и кровь, слюну, мочу, кал, семенную жидкость, слезы и другие физиологические жидкости или их экстракты.
Ингибирование активности одного или нескольких каналов из NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8 или NaV1.9 в биологическом образце применимо для ряда целей, известных специалисту в данной области техники. Примеры таких целей включают без ограничения исследование ионных натриевых каналов в биологических и патологических процессах; и сравнительную оценку новых ингибиторов натриевых ионных каналов.
ПРИМЕРЫ
Общие методы. 1H-ЯМР (400 МГц) и 13C-ЯМР (100 МГц) спектры получали в растворах в дейтероацетонитриле (CD3CN), хлороформе-d (CDCl3) или диметилсульфоксиде-D6 (DMSO). Масс-спектры (MS) получали с использованием ЖХ/МС системы Applied Biosystems API EX, оснащенной колонкой Phenomenex 50×4,60 мм luna-5μ C18. Элюентами для ЖХ/МС являлись 1-99% или 10-99% ацетонитрил в H2O с добавлением 0,035% об./об. трифторуксусной кислоты, 0,035% об./об. муравьиной кислоты, 5 мМ HCl или 5 мМ формиата аммония; использовали 3- или 15-минутный линейный градиент и скорость потока 12 мл/мин. Хроматографию на силикагеле проводили с использованием силикагеля 60 с размером частиц 230-400 меш. Пиридин, дихлорметан (CH2Cl2), тетрагидрофуран (THF), диметилформамид (DMF), ацетонитрил (ACN), метанол (MeOH) и 1,4-диоксан производства Aldrich находились в бутылях Sure-Seal в атмосфере осушенного азота. Все реакционные смеси перемешивались на магнитной мешалке, если иное не указано особо.
транс-5a',6',7',8',9',9a'-гексагидро-5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]дигидрохлорид

Стадия 1:
трет-Бутил-N-[(1R,2R)-2-аминоциклогексил]карбамат (1,06 г, 4,93 ммоль), ацетат натрия (1,70 г, 20,7 ммоль) и 2,5-диметокситетрагидрофуран (764 мкл, 5,91 ммоль) объединяли в уксусной кислоте (10,6 мл). Реакционную смесь нагревали при 80°C в течение 16 часов. Затем, реакционную смесь упаривали досуха, и распределяли остаток между этилацетатом и насыщенным водным раствором бикарбоната натрия. Слои разделяли, органический слой дважды промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и упаривали досуха с получением коричневого твердого вещества. Затем, это твердое вещество растворяли в HCl в диоксане (10,3 мл 4,0M раствора, 41,1 ммоль) и оставляли отстаиваться в течение 3 часов. Затем, удаляли растворитель с получением транс-2-(1H-пиррол-1-ил)циклогексанамина гидрохлорида (989 мг, 99%) в виде коричневого твердого вещества. ESI-MS m/z рассчитано 164,1, обнаружено 165,2 (M+1)+; время удерживания: 0,27 минуты (цикл 4 мин).
Стадия 2:
транс-2-(1H-пиррол-1-ил)циклогексанамина гидрохлорид (989 мг, 4,93 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (982 мг, 4,93 ммоль) и малеиновую кислоту (56,2 мг, 0,493 ммоль) объединяли в этаноле (12 мл). Реакционную смесь нагревали при 80°C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и выпаривали растворитель. Остаток растворяли в дихлорметане, а затем очищали на 80 г силикагеля с использованием градиента 0-10% метанола в дихлорметане, с получением транс-трет-бутил-5a',6',7',8',9',9a'-гексагидро-5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-карбоксилата. ESI-MS m/z рассчитано 345,2, обнаружено 346,2 (M+1)+; время удерживания: 1,63 минуты (цикл 4 мин).
Стадия 3:
транс-трет-Бутил-5a',6',7',8',9',9a'-гексагидро-5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалин]-1-карбоксилат (0,311 г, 0,901 ммоль) суспендировали в хлороводороде в диоксане (2,0 мл 4,0M раствора, 8,0 ммоль). Реакционную смесь оставляли отстаиваться в течение 2 часов. Затем, реакционную смесь упаривали досуха с получением транс-5a',6',7',8',9',9a'-гексагидро-5'H-спиро[пиперидин-4,4'-пирроло[1,2-a]хиноксалина]. ESI-MS m/z рассчитано 245,2, обнаружено 246,3 (M+1)+; время удерживания: 0,32 минуты (цикл 3 мин).
2'-Метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид
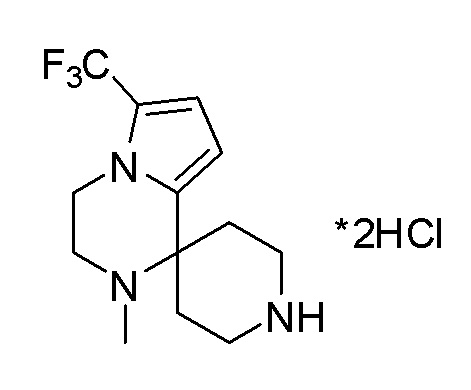
Стадия 1:
Смесь 2,5-диметокситетрагидрофурана (15 г, 113,5 ммоль), 2-хлорэтанамина гидрохлорида (44,76 г, 385,9 ммоль), и ацетата натрия (46,55 г, 567,5 ммоль) в уксусной кислоте (55 мл) нагревали при 110°C. Спустя 2 часа, реакционную смесь вливали в солевой раствор и экстрагировали продукт дихлорметаном. Органические фазы промывали солевым раствором, насыщенным Na2CO3, и снова солевым раствором. Органические фазы сушили над сульфатом натрия и упаривали. Неочищенное вещество фильтровали через слой Florisil (80 г) с использованием гексана в качестве элюента с получением 1-(2-хлорэтил)пиррола (10,1 г, 69%). 1H ЯМР (400 МГц, CDCl3) δ 6,70 (т, J=1,9 Гц, 2H), 6,18 (т, J=1,9 Гц, 2H), 4,20 (т, J=6,5 Гц, 2H), 3,73 (т, J=6,5 Гц, 2H).
Стадия 2:
1-(2-Хлорэтил)пиррол (2,0 г, 15,43 ммоль) объединяли с раствором 33% метиламина в этаноле (7,3 мл 33% масс./об. раствора, 77,15 ммоль). Смесь нагревали при 90°C в течение 16 часов, после чего ее концентрировали в условиях пониженного давления с получением N-метил-2-пиррол-1-илэтанамина (2,19 г, 88%) который использовали непосредственно в последующей реакции. ESI-MS m/z рассчитано 124,1, обнаружено 125,3 (M+1)+; время удерживания: 0,22 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 6,73-6,68 (м, 2H), 6,22-6,14 (м, 2H), 4,05 (т, J=5,9 Гц, 2H), 2,94 (т, J=5,9 Гц, 2H), 2,45 (с, 3H).
Стадия 3:
N-Метил-2-пиррол-1-илэтанамин (2,19 г, 17,64 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (3,51 г, 17,64 ммоль) и pTsOH∙H2O (0,334 г, 1,76 ммоль) объединяли в этаноле (87,60 мл) и нагревали при 70°C в течение 4 часов. Реакционную смесь концентрировали, и растворяли остаток в дихлорметане. Органические фазы промывали насыщенным NaHCO3 раствором и солевым раствором. Органические фазы сушили над сульфатом натрия и упаривали. Неочищенное вещество очищали методом хроматографии на силикагеле, элюируя 0-10% метанолом в дихлорметане с 2% триэтиламина, с получением трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (4,2 г, 78%). ESI-MS m/z рассчитано 305,4, обнаружено 306,3 (M+1)+; время удерживания: 0,97 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 6,55-6,52 (м, 1H), 6,15-6,11 (м, 1H), 5,92-5,89 (м, 1H), 3,92 (т, J=6,0 Гц, 2H), 3,91-3,75 (м, 2H), 3,29 (т, J=6,0 Гц, 2H), 3,26-3,12 (м, 2H), 2,36 (с, 3H), 2,10-1,99 (м, 2H), 1,83-1,69 (м, 2H), 1,47 (с, 9H).
Стадия 4:
Способ A: трет-Бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилат (1,0 г, 3,27 ммоль), карбонат калия (497,7 мг, 3,60 ммоль) и трифторметансульфонат 5-(трифторметил)дибензотиофен-5-ия (1,32 г, 3,27 ммоль) объединяли в ацетонитриле (10 мл). Реакционную смесь нагревали при 60°C в течение 16 часов. Реакционную смесь упаривали досуха, и растворяли остаток в дихлорметане. Органические фазы промывали водой и солевым раствором, сушили над сульфатом натрия и упаривали. Неочищенное вещество очищали методом хроматографии на силикагеле, элюируя 0-50% этилацетата в гексанах, с получением трет-бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (812 мг, 66%). ESI-MS m/z рассчитано 373,2, обнаружено 374,5 (M+1)+; время удерживания: 1,21 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 6,52 (д, J=3,8 Гц, 1H), 5,91 (д, J=3,8 Гц, 1H), 3,98 (т, J=6,0 Гц, 2H), 3,93-3,76 (м, 2H), 3,32 (т, J=6,0 Гц, 2H), 3,26-3,08 (м, 2H), 2,36 (с, 3H), 2,11-1,99 (м, 2H), 1,81-1,65 (м, 2H), 1,47 (с, 9H).
Способ B: К трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (10,0 г, 32,7 ммоль) в DMSO (164 мл) добавляли гептагидрат сульфата железа(II) (9,8 мл 1,0M раствора, 9,8 ммоль), а затем CF3I (6,41 г, 32,7 ммоль) путем медленного барботирования через раствор и измерения изменения массы баллона. Смесь охлаждали на бане со льдом, после чего по каплям в течение 15 мин добавляли H2O2 (3,71 мл 30% масс./об., 32,7 ммоль), поддерживая внутреннюю температуру <20°C. Смесь вливали в 300 мл воды со льдом и экстрагировали EtOAc (2×400 мл). Объединенные органические фазы промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали в условиях вакуума. Неочищенное вещество очищали методом колоночной хроматографии, элюируя 0-10% метанолом в дихлорметане с 2% iPr2Net, с получением трет-бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (7,8 г, 64%).
Стадия 5:
трет-Бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилат (7,8 г, 20,89 ммоль) перемешивали в 4M HCl в диоксане (26,10 мл 4M раствора, 104,4 ммоль) и метаноле (22 мл) при комнатной температуре в течение 1 часа. Реакционную смесь упаривали досуха, и совместно упаривали остаток с 100 мл MTBE с получением 2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорида в виде желтой пены/твердого вещества (7,23 г, количественный выход). ESI-MS m/z рассчитано 273,2, обнаружено 274,5 (M+1)+; время удерживания: 0,44 минуты (цикл 3 мин).
2'-Метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-карбонитрила дигидрохлорид
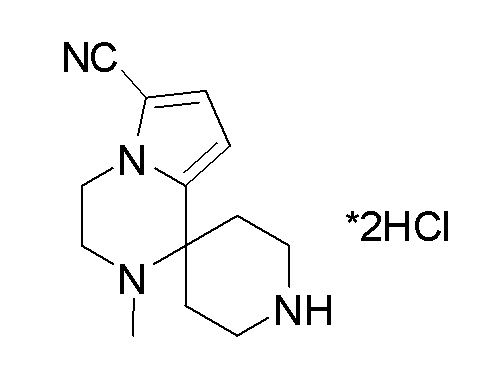
Стадия 1:
К раствору трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1020 мг, 3,340 ммоль) в тетрагидрофуране (9 мл), поддерживаемом при -78°C (температура бани) в атмосфере аргона медленно добавляли раствор хлорсульфонилизоцианата (590,9 мг, 363,4 мкл, 4,175 ммоль) в тетрагидрофуране (2 мл). Реакционную смесь оставляли перемешиваться в течение 1 часа при -78°C. Затем, к холодной реакционной смеси медленно добавляли N,N-диметилформамид (732,4 мг, 775,8 мкл, 10,02 ммоль). Затем, реакционную смесь оставляли медленно нагреваться до комнатной температуры. После перемешивания в течение 3 часов при комнатной температуре, неочищенное вещество разбавляли 25 мл тетрагидрофурана, промывали 1M раствором гидроксида натрия, а затем трижды промывали насыщенным водным раствором хлорида натрия. Органический слой сушили над сульфатом натрия, фильтровали и упаривали досуха с получением неочищенного продукта. Неочищенное вещество очищали на 80 г силикагеля с использованием градиента 0-70% этилацетата в гексанах с получением трет-бутил-6-циано-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (280 мг, 25%) в виде белого твердого вещества. ESI-MS m/z рассчитано 330,2, обнаружено 331,1 (M+1)+; время удерживания: 0,94 минуты. 1H ЯМР (400 МГц, CDCl3) δ 6,76 (д, J=4,0 Гц, 1H), 5,97 (д, J=4,0 Гц, 1H), 4,01 (т, J=6,0 Гц, 2H), 3,98-3,76 (м, 2H), 3,36 (т, J=6,0 Гц, 2H), 3,30-3,08 (м, 2H), 2,36 (с, 3H), 2,09-1,98 (м, 2H), 1,84-1,66 (м, 2H), 1,47 (с, 9H).
Стадия 2:
трет-Бутил-6-циано-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилат (280 мг, 0,8474 ммоль) растворяли в смеси соляной кислоты в диоксане (8 мл 4M раствора, 32,00 ммоль) и диоксана (8 мл). Реакционную смесь оставляли перемешиваться в течение 30 минут, а затем упаривали досуха с получением 2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-карбонитрила дигидрохлорида (258 мг, 99%) в виде белого твердого вещества. ESI-MS m/z рассчитано 230,2, обнаружено 231,5 (M+1)+; время удерживания: 0,50 минуты (цикл 3 мин). 1H ЯМР (400 МГц, D2O) δ 7,10 (д, J=4,2 Гц, 1H), 6,59 (д, J=4,3 Гц, 1H), 4,51 (т, J=6,4 Гц, 2H), 4,02 (т, J=6,3 Гц, 2H), 3,66-3,56 (м, 2H), 3,49-3,36 (м, 2H), 2,95 (с, 3H), 2,69-2,59 (м, 2H), 2,54-2,40 (м, 2H).
N-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксамида гидрохлорид
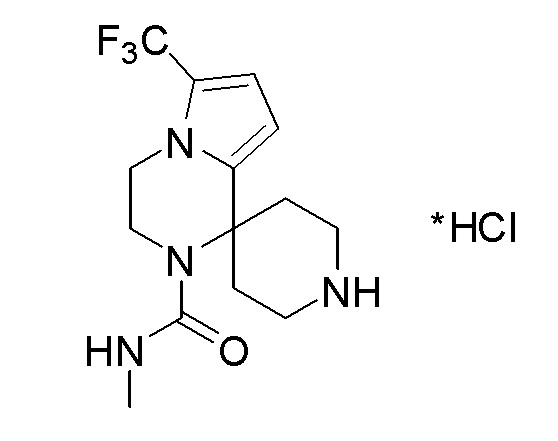
Стадия 1:
К трет-бутил-6-(трифторметил)спиро[3,4-дигидро-2H-пирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (600 мг, 1,67 ммоль), THF (3 мл) и Et3N (698 мкл, 5,01 ммоль) добавляли метилизоцианат (199 мкл, 3,34 ммоль). Смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. К смеси дополнительно добавляли Et3N (698 мкл, 5,01 ммоль) и метилизоцианат (199 мкл, 3,34 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 3 суток. Растворитель выпаривали в условиях пониженного давления. Остаток растворяли в этилацетате (40 мл) и промывали водой (3×10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях вакуума с получением трет-бутил-2-(метилкарбамоил)-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (680 мг, 97%). ESI-MS m/z рассчитано 416,2, обнаружено 417,4 (M+1)+; время удерживания: 1,73 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 7,14 (кв., J=4,3 Гц, 1H), 6,56 (д, J=3,4 Гц, 1H), 6,03 (д, J=3,9 Гц, 1H), 3,88 (т, J=5,4 Гц, 2H), 3,73 (т, J=5,4 Гц, 2H), 3,69-3,55 (м, 2H), 3,23-3,03 (м, 2H), 2,78-2,62 (м, 2H), 2,56 (д, J=4,4 Гц, 3H), 1,76-1,60 (м, 2H), 1,40 (с, 9H).
Стадия 2:
К трет-бутил-2-(метилкарбамоил)-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (0,66 г, 1,6 ммоль) и ацетонитрилу (5 мл) добавляли раствор HCl в диоксане (5,2 мл 4,0M раствора, 21 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 60 минут. Растворитель выпаривали в условиях пониженного давления с получением N-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-2-карбоксамида гидрохлорида в виде коричневого твердого вещества (99%). ESI-MS m/z рассчитано 316,2, обнаружено 317,2 (M+1)+; время удерживания: 0,77 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методик, описанных выше:
2'-(2-метоксиэтил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-бензил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-(2-метоксиэтил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-этил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
3',3'-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
3'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
4',4'-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2',3'-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
3-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-циклопропил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-(циклопропилметил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2',3-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-метил-6'-(2,2,2-трифторэтил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-метил-6'-(перфторэтил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
N,N-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксамид,
2'-(2,2,2-трифторэтил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]гидрохлорид,
3-фтор-2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
2,2'-диметил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
метил-2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-4'-карбоксилат,
2'-(2,2-дифторэтил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]гидрохлорид,
2'-циклобутил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид,
этил-2-(6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-ил)ацетат, и
метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксилат.
2,2,2-Трифтор-1-(2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил)этанона дигидрохлорид
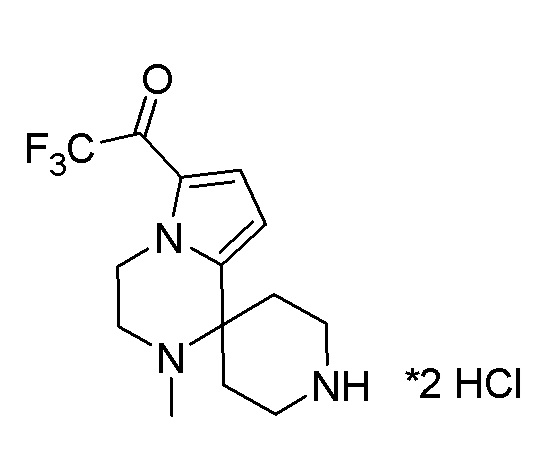
Стадия 1:
К раствору трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,0 г, 3,27 ммоль), пиридина (1,06 мл, 13,10 ммоль) и CH2Cl2 (6,5 мл) при комнатной температуре по каплям добавляли (2,2,2-трифторацетил)-2,2,2-трифторацетат (910 мкл, 6,55 ммоль). Смесь нагревали при 35°C в течение 2 часов. Реакционную смесь распределяли между 1н HCl и CH2Cl2. Слои разделяли, и экстрагировали водный слой CH2Cl2 (2×). Объединенные органические фазы сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали с получением трет-бутил-2-метил-6-(2,2,2-трифторацетил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,38 г, 94%) в виде желтоватого твердого вещества. ESI-MS m/z рассчитано 401,2, обнаружено 402,5 (M+1)+; время удерживания: 1,36 минуты. 1H ЯМР (400 МГц, CDCl3) δ 7,21 (дд, J=4,4, 2,1 Гц, 1H), 6,14 (д, J=4,5 Гц, 1H), 4,34 (т, J=6,0 Гц, 2H), 3,93 (с, 2H), 3,33 (т, J=6,0 Гц, 2H), 3,19 (с, 2H), 2,39 (с, 3H), 2,13-2,05 (м, 2H), 1,79 (т, J=11,6 Гц, 2H), 1,48 (с, 9H).
Стадия 2:
К раствору трет-бутил-2-метил-6-(2,2,2-трифторацетил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,38 г, 3,44 ммоль) в CH2Cl2 (9,7 мл) при комнатной температуре добавляли хлористый водород (6,01 мл 4M раствора, 24,07 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали в условиях пониженного давления с получением 2,2,2-трифтор-1-(2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил)этанона дигидрохлорида (1,34 г, 99%) в виде каштанового твердого вещества. ESI-MS m/z рассчитано 301,1, обнаружено 302,5 (M+1)+ ; время удерживания: 1,02 минуты.
2,2-Диметил-1-(2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил)пропан-1-она дигидрохлорид
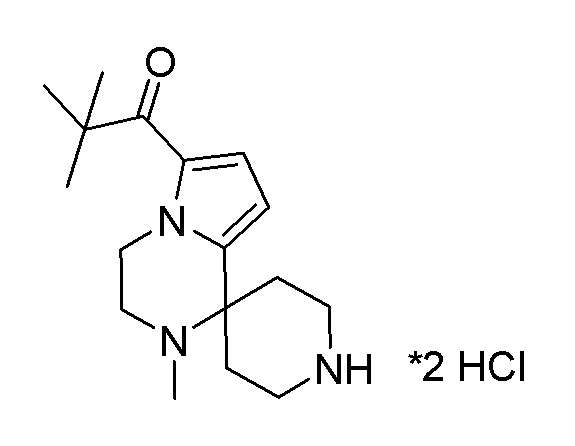
Стадия 1:
К смеси трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (2,75 г, 9,0 ммоль), DBN (1,22 мл, 9,90 ммоль) и дихлорэтана (6,9 мл) при комнатной температуре добавляли 2,2-диметилпропаноилхлорид (1,22 мл, 9,90 ммоль). Смесь оставляли перемешиваться в течение 18 часов при 115°C. Смесь охлаждали до комнатной температуры, после чего ее распределяли между CH2Cl2 и 1н HCl. Слои разделяли, и промывали органический слой 1н NaOH. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали методом колоночной хроматографии (0-100% этилацетат в гексанах) с получением трет-бутил-6-(2,2-диметилпропаноил)-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,8 г, 41%) в виде не совсем белого твердого вещества. ESI-MS m/z рассчитано 389,3, обнаружено 390,5 (M+1)+; время удерживания: 1,46 минуты.
Стадия 2:
К раствору трет-бутил-6-(2,2-диметилпропаноил)-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,75 г, 4,49 ммоль) в CH2Cl2 (12,3 мл) при комнатной температуре добавляли хлороводород (5,1 мл 4M раствора, 20,22 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали в условиях пониженного давления с получением 2,2-диметил-1-(2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил)пропан-1-она дигидрохлорида (1,8 г, 99%) в виде каштанового твердого вещества. ESI-MS m/z рассчитано 289,2, обнаружено 290,5 (M+1)+; время удерживания: 0,97 минуты.
1-(2'-Метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид
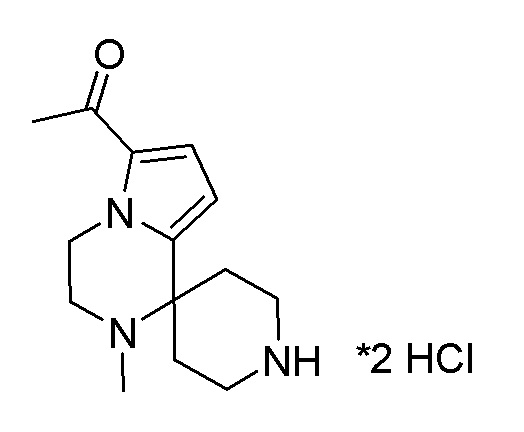
Стадия 1:
К трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (10 г, 32,74 ммоль) в хлористом метилене (73,5 мл) при 0°C порциями добавляли N-бромсукцинимид (5,53 г, 31,10 ммоль). Реакционную смесь перемешивали при 0°C. Спустя 30 минут, дополнительно добавляли N-бромсукцинимид (291,4 мг, 1,64 ммоль), и перемешивали реакционную смесь в течение 1 часа. Реакционную смесь разбавляли 0,5M Na2S2O3 (135 мл), и удаляли водную фазу. Органический слой промывали солевым раствором (135 мл). Органический слой сушили над сульфатом натрия, фильтровали, и выпаривали растворитель в условиях пониженного давления с получением трет-бутил-6-бром-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата в виде красной вязкой жидкости, которую использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, DMSO) δ 6,09 (д, J=3,7 Гц, 1H), 5,99 (д, J=3,7 Гц, 1H), 3,78-3,65 (м, 4H), 3,27 (т, J=6,0 Гц, 2H), 3,17-2,92 (м, 2H), 2,21 (с, 3H), 2,03-1,93 (м, 2H), 1,65-1,53 (м, 2H), 1,40 (с, 9H). ESI-MS m/z рассчитано 383,1, обнаружено 386,0 (M+1)+; время удерживания: 1,13 минуты (цикл 3 минуты).
Стадия 2:
Раствор трет-бутил-6-бром-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (2 г, 5,2 ммоль) и N-циклогексил-N-метилциклогексанамина (1,67 мл, 7,81 ммоль) в 1,4-диоксане (8,0 мл) продували N2 в течение 5 минут. Добавляли 1-винилоксибутан (7,04 мл, 52,04 ммоль), Pd(dba)3 (1,078 г, 1,04 ммоль) и три-трет-бутилфосфан (642,0 мкл, 2,60 ммоль), и нагревали реакционную смесь при 80°C в течение 5 часов в толстостенном сосуде. Реакционную смесь фильтровали через слой целита с использованием этилацетата. Растворитель выпаривали в условиях пониженного давления. Неочищенный продукт очищали методом хроматографии на силикагеле с использованием градиента 1-100% этилацетата в гексане с получением трет-бутил-6-ацетил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (672,1 мг, 1,93 ммоль, 37%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ 7,04 (д, J=4,1 Гц, 1H), 6,12 (д, J=4,2 Гц, 1H), 4,17 (т, J=6,0 Гц, 2H), 3,83-3,68 (м, 2H), 3,21 (т, J=6,0 Гц, 2H), 3,17-2,91 (м, 2H), 2,31 (с, 3H), 2,24 (с, 3H), 2,07-1,97 (м, 2H), 1,72-1,59 (м, 2H), 1,41 (с, 9H). ESI-MS m/z рассчитано 347,2, обнаружено 348,5 (M+1)+; время удерживания: 0,95 минуты (цикл 3 минуты).
Стадия 3:
К трет-бутил-6-ацетил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (300 мг, 0,86 ммоль) и хлористому метилену (1,7 мл) добавляли хлороводород в диоксане (1,60 мл 4M раствора, 6,40 ммоль) Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часа. Растворитель выпаривали в условиях пониженного давления с получением 1-(2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил)этанона дигидрохлорида с количественным выходом в виде светло-зеленого твердого вещества. ESI-MS m/z рассчитано 247,2, обнаружено 248,2 (M+1)+; время удерживания: 0,17 минуты (цикл 3 минуты).
Следующие соединения синтезировали с использованием методик, описанных выше:
1-(2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)пропан-1-она дигидрохлорид,
2-метил-1-(2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)пропан-1-она дигидрохлорид,
циклопропил(2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)метанона дигидрохлорид,
(2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)(фенил)метанона дигидрохлорид,
1-(3',3'-диметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)-2,2,2-трифторэтанона дигидрохлорид,
1-(3',3'-диметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид,
1-(3',3'-диметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-7'-ил)этанона дигидрохлорид,
2,2,2-трифтор-1-(2',4',4'-триметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид,
2,2,2-трифтор-1-(2',4',4'-триметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-7'-ил)этанона дигидрохлорид,
1-(2',4',4'-триметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид,
1-(2',3'-диметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)-2,2,2-трифторэтанона дигидрохлорид,
2,2,2-трифтор-1-(3'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид,
2,2,2-трифтор-1-(4'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанона дигидрохлорид,
1-(2',4'-диметил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)-2,2,2-трифторэтанона дигидрохлорид, и
(2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)(1-метилциклопропил)метанон.
2'-Метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-8'-карбонитрила дигидрохлорид

Стадия 1:
К раствору трет-бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,86 г, 5,0 ммоль) в ацетонитриле (50 мл) добавляли N-бромсукцинимид (930,4 мг, 5,25 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали в условиях пониженного давления. Остаток распределяли между этилацетатом и водой. Слои разделяли, и экстрагировали водный слой этилацетатом (2×). Объединенные органические слои промывали солевым раствором, сушили над MgSO4 и концентрировали досуха. Неочищенное вещество очищали методом колоночной хроматографии (10-20% этилацетат в гексанах) с получением продукта в виде светло-желтого твердого вещества (1,7 г, 75%). ESI-MS m/z рассчитано 451,1, обнаружено 452,1 (M+1)+; время удерживания: 1,59 минуты (цикл 3 минуты).
Стадия 2:
Смесь трет-бутил-8-бром-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,67 г, 3,7 ммоль) и дицианоцинка (234,9 мкл, 3,7 ммоль) в DMF (10 мл) продували N2 в течение 5 мин. Добавляли Pd(PPh3)4 (427,6 мг, 0,37 ммоль). Смесь нагревали в течение ночи при 150°C в герметично закрытом сосуде для микроволновой обработки. Смесь распределяли между этилацетатом и водой. Слои разделяли. Водный слой экстрагировали этилацетатом (3×). Все водные слои объединяли, промывали водой (3×), солевым раствором, сушили над MgSO4, фильтровали и концентрировали досуха. Неочищенное вещество очищали методом колоночной хроматографии (10-20% EtOAc в гексанах) с получением трет-бутил-8-циано-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (510 мг, 35%) в виде белого твердого вещества. ESI-MS m/z рассчитано 398,2, обнаружено 399,3 (M+1)+; время удерживания: 1,56 минуты (цикл 3 минуты).
Стадия 3:
К раствору трет-бутил-8-циано-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (278,9 мг, 0,7 ммоль) в DCM (4 мл) добавляли HCl в диоксане (2 мл 4M раствора, 8,0 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали, и использовали неочищенное вещество непосредственно на следующей стадии без дополнительной очистки. ESI-MS m/z рассчитано 298,1, обнаружено 299,5 (M+1)+; время удерживания: 0,88 минуты (цикл 3 минуты).
6'-Хлор-2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид
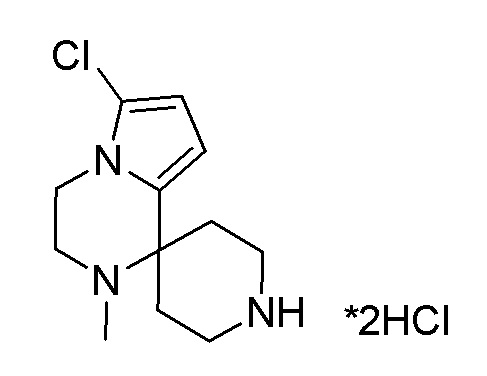
Стадия 1:
К трет-бутил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (5 г, 16,37 ммоль) в дихлорметане (50,00 мл) при 0°C добавляли трифторметансульфонилхлорид (3,64 мл, 34,38 ммоль), и перемешивали реакционную смесь в течение ночи при температуре от 0°C до комнатной температуры. Реакционную смесь разбавляли дихлорметаном и промывали водой. Слои разделяли, органические фазы сушили над сульфатом натрия, фильтровали и концентрировали. Путем очистки остатка методом хроматографии на силикагеле, элюируя 10-100% этилацетатом в гексанах, получали трет-бутил-6-хлор-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилат (4,3 г, 77%), в виде желтого твердого вещества. ESI-MS m/z рассчитано 339,2, обнаружено 340,3 (M+1)+; время удерживания: 1,13 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 6,01 (д, J=3,8 Гц, 1H), 5,91 (д, J=3,7 Гц, 1H), 3,77 (т, J=6,1 Гц, 2H), 3,34 (с, 2H), 3,24 (с, 2H), 2,34 (с, 3H), 2,03 (д, J=13,1 Гц, 2H), 1,74 (т, J=11,1 Гц, 2H), 1,47 (с, 9H).
Стадия 2:
К раствору трет-бутил-6-хлор-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (624 мг, 1,84 ммоль) в дихлорметане (2 мл) добавляли HCl (1,84 мл 4M в диоксане, 7,34 ммоль), и перемешивали при 40°C в течение 1 часа. Реакционную смесь упаривали досуха с получением 6'-хлор-2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорида (количественный выход), который использовали без дополнительной очистки. ESI-MS m/z рассчитано 239,1, обнаружено 240,3 (M+1)+; время удерживания: 0,22 минуты (цикл 3 мин).
Этил-6'-циано-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксилата гидрохлорид
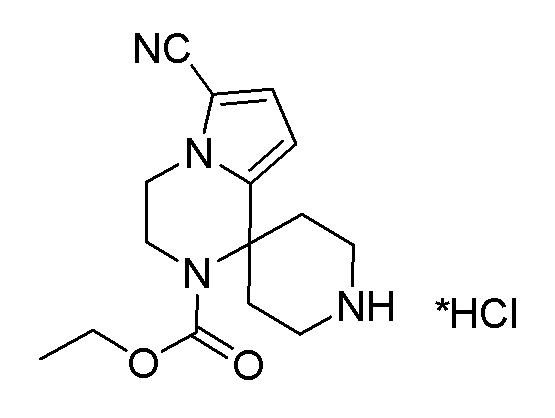
Стадия 1:
К раствору трет-бутилспиро[3,4-дигидро-2H-пирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (500 мг, 1,72 ммоль) и K2CO3 (474,3 мг, 3,43 ммоль) в ацетонитриле (5,0 мл) добавляли этилхлороформиат (328,2 мкл, 3,43 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь фильтровали с использованием ацетонитрила, и выпаривали растворитель в условиях пониженного давления. Соединение растворяли в этилацетате и промывали 1н соляной кислотой и солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением 1-трет-бутил-2'-этил 3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1,2'-дикарбоксилата (395 мг, 63%) в виде янтарного масла, которое использовали на следующей стадии без дополнительной очистки. ESI-MS m/z рассчитано 363,2, обнаружено 364,3 (M+1)+; время удерживания: 1,78 минуты (цикл 3 минуты).
Стадия 2:
К раствору 1-трет-бутил-2'-этил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1,2'-дикарбоксилата (100 мг, 0,27 ммоль) в THF (1,0 мл) при -78°C в атмосфере азота медленно добавляли раствор N-(оксометилен)сульфамоилхлорида (23,9 мкл, 0,27 ммоль) в THF (200,0 мкл). Реакционную смесь перемешивали в течение 1 часа при -78°C. Затем, к холодному раствору медленно добавляли N,N-диметилформамид (39,9 мкл, 0,51 ммоль). Затем, реакционную смесь оставляли медленно нагреваться до комнатной температуры. Реакционную смесь фильтровали и очищали методом ЖХ с обращенной фазой/масс-спектрометрии (10-99% CH3CN/H2O) с использованием HCl в качестве модификатора с получением 1-трет-бутил-2'-этил-6'-циано-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1,2'-дикарбоксилата. ESI-MS m/z рассчитано 388,2, обнаружено 389,3 (M+1)+; время удерживания: 1,82 минуты (цикл 3 минуты).
Стадия 3:
К раствору 1-трет-бутил-2'-этил-6'-циано-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1,2'-дикарбоксилата (0,27 ммоль) в дихлорметане (5 мл) добавляли 4н HCl в диоксане (8,7 мл, 34,7 ммоль), и перемешивали смесь при 40°C в течение 1 часа. Реакционную смесь упаривали досуха с получением этил-6'-циано-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксилата гидрохлорида. ESI-MS m/z рассчитано 288,2, обнаружено 289,3 (M+1)+; время удерживания: 0,75 минуты (цикл 3 минуты).
8'-Фтор-2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид
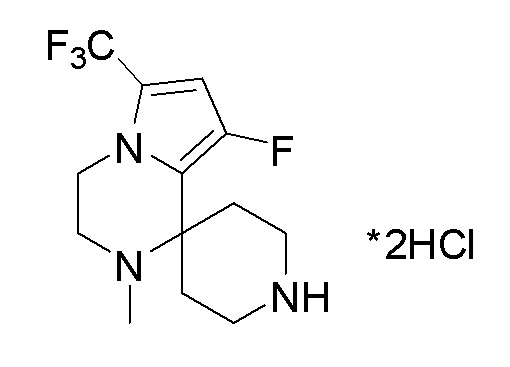
Стадия 1:
К раствору трет-бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (5,60 г, 15,0 ммоль) в ацетонитриле (50 мл) добавляли NBS (2,80 г, 15,8 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и распределяли остаток между EtOAc и водой. Водный слой экстрагировали EtOAc (2×). Объединенные органические слои промывали солевым раствором, сушили над MgSO4 и концентрировали досуха. Неочищенное вещество очищали методом колоночной хроматографии (10-20% EtOAc-Hex) с получением трет-бутил-8-бром-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (5,40 г, 73%) в виде светло-желтого твердого вещества. ESI-MS m/z рассчитано 452,3 обнаружено 454,5 (M+1)+; время удерживания: 1,60 минуты (цикл 3 минуты).
Стадия 2:
Раствор трет-бутил-8-бром-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (15,0 г, 33,2 ммоль) в THF (200 мл) продували аргоном в течение 5 мин. Смесь охлаждали до -78°C, после чего по каплям добавляли n-BuLi (42,5 мл 1,6M раствора, 68 ммоль). Смесь перемешивали при -78°C в течение 30 мин, после чего по каплям добавляли раствор N-(бензолсульфонил)-N-фторбензолсульфонамида (20,9 г, 66,3 ммоль) в THF (100 мл). Смесь оставляли нагреваться до комнатной температуры в течение ночи. Реакционную смесь гасили добавлением насыщенного водного NH4Cl. Слои разделяли, и экстрагировали водный слой EtOAc (2×). Органические слои объединяли, промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали досуха. Добавляли CH2Cl2, и удаляли твердое вещество путем фильтрования. Фильтрат концентрировали досуха, и очищали остаток методом колоночной хроматографии (10-20% EtOAc-Hex) с получением трет-бутил-8-фтор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (5,52 г, 43%) в виде светло-коричневого масла, которое отверждалось при отстаивании. ESI-MS m/z рассчитано 391,4, обнаружено 392,5 (M+1)+; время удерживания: 1,35 минуты (цикл 3 минуты).
Стадия 3:
К трет-бутил-8-фтор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (310 мг, 0,80 ммоль) в CH2Cl2 (2 мл) добавляли раствор HCl в 1,4-диоксане (2,0 мл 4M раствора, 8,0 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Летучие вещества удаляли в условиях пониженного давления с получением 8-фтор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]дигидрохлорида (290 мг, 99%) в виде розового твердого вещества. ESI-MS m/z рассчитано 291,1, обнаружено 292,3 (M+1)+; время удерживания: 0,75 минуты (цикл 3 минуты).
8'-Хлор-2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорид
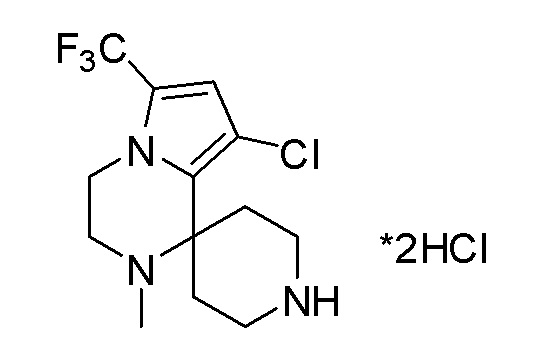
Стадия 1:
К раствору трет-бутил-8-бром-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (5,00 г, 11,1 ммоль) в безводном THF (125 мл) при -78°C медленно добавляли nBuLi (8,84 мл 2,5M раствора, 22,1 ммоль). Реакционную смесь оставляли перемешиваться при -78°C в течение 20 минут, после чего по каплям добавляли 1,1,1,2,2,2-гексахлорэтан (5,36 г, 22,7 ммоль) в виде раствора в THF (12 мл). Реакционную смесь оставляли медленно нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили добавлением насыщенного водного хлорида аммония (100 мл). Летучие вещества удаляли в условиях пониженного давления до половины объема. Оставшуюся водную суспензию экстрагировали EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением густого коричневого масла. Неочищенный продукт очищали методом колоночной хроматографии на силикагеле (градиент 10-20% EtOAc в гексане) с получением трет-бутил-8-хлор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (2,40 г, 53%) в виде желтого масла, которое кристаллизовалось при отстаивании. ESI-MS m/z рассчитано 407,2, обнаружено 407,9 (M+1)+; время удерживания: 1,70 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 6,47 (с, 1H), 3,97 (т, J=5,5 Гц, 4H), 3,28 (с, 2H), 3,13 (с, 2H), 2,51-2,28 (м, 5H), 1,93 (д, J=13,7 Гц, 2H), 1,47 (д, J=9,5 Гц, 9H).
Стадия 2:
К раствору трет-бутил-8-хлор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (750 мг, 1,84 ммоль) в CH2Cl2 (2 мл) добавляли полученный 1/1 раствор трифторуксусной кислоты (2,0 мл, 26 ммоль) в CH2Cl2 (2 мл). После перемешивания при комнатной температуре в течение 2 часов, медленно добавляли насыщенный водный раствор бикарбоната натрия (75 мл). Смесь экстрагировали EtOAc (2×75 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением 8-хлор-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидина] (555 мг, 98%) в виде светло-коричневого твердого вещества. ESI-MS m/z рассчитано 307,1, обнаружено 307,9 (M+1)+; время удерживания: 1,12 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 9,72 (с, 1H), 9,38 (с, 1H), 6,49 (с, 1H), 3,98 (т, J=5,6 Гц, 2H), 3,36 (д, J=11,6 Гц, 2H), 3,26 (д, J=6,1 Гц, 4H), 2,78 (с, 2H), 2,40 (с, 3H), 2,11 (д, J=14,4 Гц, 2H), 1,69 (с, 2H).
2'-Метил-6',7'-бис(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]
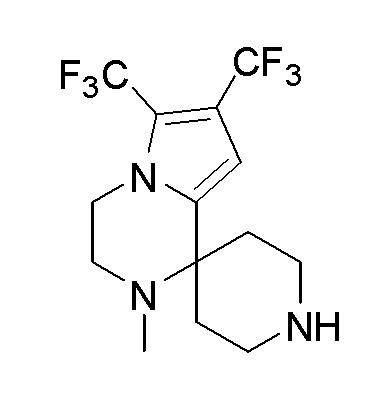
Стадия 1:
К смеси трет-бутил-2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,00 г, 2,68 ммоль) и DMSO (15 мл) при комнатной температуре добавляли гептагидрат сульфата железа(II) (803 мкл 1,00M раствора, 0,803 ммоль). В сосуд загружали CF3I (газ), после чего по каплям добавляли H2O2 (304 мкл 30% масс./об., 2,68 ммоль). Смесь оставляли перемешиваться в течение ночи при комнатной температуре, после чего ее распределяли между этилацетатом и водой. Слои разделяли, и экстрагировали водный слой этилацетатом (3×). Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии (0-100% этилацетат в гексанах) с получением трет-бутил-2-метил-6,7-бис(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата. ESI-MS m/z рассчитано 441,2, обнаружено 442,5 (M+1)+; время удерживания: 1,36 минуты (цикл 3 мин).
Стадия 2:
трет-Бутил-2-метил-6,7-бис(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилат (полученный на стадии 1) переносили в CH2Cl2 (2 мл), и добавляли HCl (1,7 мл 4M раствора, 6,8 ммоль). Смесь оставляли перемешиваться в течение 30 мин, после чего ее концентрировали в условиях пониженного давления. Остаток переносили в этилацетат и промывали насыщенным водным NaHCO3, а затем солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением 2-метил-6,7-бис(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидина] (22 мг, 2% в 2 этапа). ESI-MS m/z рассчитано 341,1, обнаружено 342,3 (M+1)+; время удерживания: 1,29 минут.
2'-Метил-6'-(метилсульфонил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]
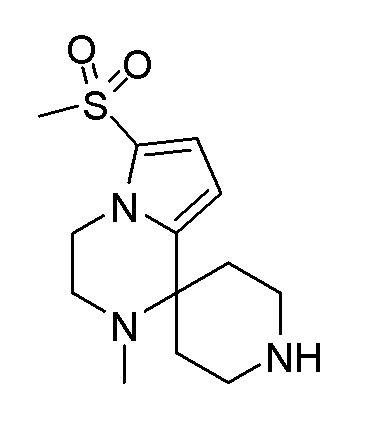
Стадия 1:
К бензил-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилату (3,00 г, 8,84 ммоль) в CH2Cl2 (30 мл) при 0°C порциями добавляли NBS (1,57 г, 8,84 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов. Дополнительно добавляли NBS (157 мг), и перемешивали реакционную смесь при 0°C в течение 15 минут (повторяли еще 6 раз до расходования исходного вещества). Реакционную смесь разбавляли 0,5M Na2S2O3 (30 мл), и удаляли водную фазу. Органическую фазу промывали солевым раствором (30 мл). Органический слой сушили над сульфатом натрия и фильтровали, и выпаривали растворитель в условиях пониженного давления. Неочищенный продукт очищали методом колоночной хроматографии с использованием градиента 0-30% этилацетата в CH2Cl2 с получением бензил-6-бром-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (2,34 г, 63%) в виде кремового твердого вещества. ESI-MS m/z рассчитано 417,1, обнаружено 418,1 (M+1)+; время удерживания: 1,49 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 7,43-7,26 (м, 5H), 6,09 (д, J=3,7 Гц, 1H), 5,99 (д, J=3,7 Гц, 1H), 5,08 (с, 2H), 3,89-3,74 (м, 2H), 3,68 (т, J=5,8 Гц, 2H), 3,27 (т, J=5,8 Гц, 2H), 3,22-2,99 (м, 2H), 2,21 (с, 3H), 2,09-1,94 (м, 2H), 1,73-1,52 (м, 2H).
Стадия 2:
Смесь метансульфината натрия (293 мг, 2,87 ммоль), бензил-6-бром-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (1,00 г, 2,39 ммоль), CuI (296 мг, 1,55 ммоль) и DMSO (5 мл) нагревали при 90°C в толстостенном сосуде в течение 20 часов. Смесь охлаждали и распределяли между эфиром (10 мл) и водой (10 мл). Органический слой разделяли, и экстрагировали водный слой эфиром (3×5 мл). Объединенные органические слои промывали солевым раствором (2×10 мл), сушили над сульфатом натрия, фильтровали и концентрировали в условиях вакуума. Неочищенный продукт очищали методом колоночной хроматографии с использованием градиента 0-50% этилацетата в CH2Cl2 с получением бензил-2-метил-6-метилсульфонилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (520 мг, 52%) в виде белого твердого вещества. ESI-MS m/z рассчитано 417,2, обнаружено 418,3 (M+1)+; время удерживания: 1,27 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,45-7,30 (м, 5H), 6,88 (д, J=4,1 Гц, 1H), 6,01 (д, J=3,9 Гц, 1H), 5,15 (с, 2H), 4,28-4,15 (м, 2H), 4,09-3,89 (м, 2H), 3,40-3,16 (м, 4H), 3,10 (с, 3H), 2,37 (с, 3H), 2,20-2,02 (м, 2H), 1,89-1,66 (м, 2H).
Стадия 3:
К раствору бензил-2-метил-6-метилсульфонилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбоксилата (520 мг, 1,25 ммоль) в EtOH (13 мл) в атмосфере азота добавляли 10% палладированный уголь (66 мг, 0,062 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере водорода. Смесь фильтровали через слой целита, и выпаривали растворитель в условиях пониженного давления с получением 2-метил-6-метилсульфонилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидина] (342 мг, 97%) в виде не совсем белого твердого вещества. ESI-MS m/z рассчитано 283,1, обнаружено 284,3 (M+1)+; время удерживания: 0,30 минуты (цикл 3 мин).
3-Метил-4-(метилсульфамоил)бензойная кислота
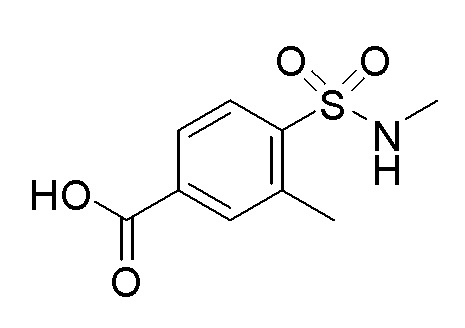
Стадия 1:
Раствор 4-амино-3-метилбензойной кислоты (4,00 г, 26,5 ммоль) в конц. HCl (15 мл) охлаждали до 0°C на бане со льдом, после чего по каплям добавляли раствор нитрита натрия (1,97 г, 910 мкл, 28,6 ммоль) в воде (5 мл), поддерживая температуру ниже 5°C. Раствор уксусной кислоты (60 мл) и CuCl2 (889 мг, 6,62 ммоль) барботировали SO2 в течение 20 мин, после чего добавляли холодный раствор диазония. Реакционную смесь перемешивали в течение 1 часа, после чего ее выливали на лед. Твердое вещество собирали путем фильтрования и промывали водой. Твердое вещество дополнительно сушили с получением 4-хлорсульфонил-3-метилбензойной кислоты (2,30 г, 37%).1H ЯМР (400 МГц, DMSO) δ 7,81 (д, J=7,8 Гц, 1H), 7,74-7,66 (м, 2H), 2,57 (с, 3H).
Стадия 2:
4-Хлорсульфонил-3-метилбензойную кислоту (0,99 г, 4,2 ммоль), метанамин (2,0 мл, 33% масс./об., 21 ммоль) и триэтиламин (1,8 мл, 13 ммоль) перемешивали в течение 45 мин при комнатной температуре Реакционную смесь упаривали, и распределяли полученное масло между этилацетатом и 1н HCl. Органические фазы разделяли и промывали дополнительной порцией 1н HCl, а затем солевым раствором. Органические фазы сушили над сульфатом натрия и упаривали с получением 3-метил-4-(метилсульфамоил)бензойной кислоты (841 мг, 87%). ESI-MS m/z рассчитано 229,0, обнаружено 230,5 (M+1)+; время удерживания: 0,64 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методик, описанных выше:
4-(N-этилсульфамоил)-3-метилбензойная кислота,
4-(N-циклопропилсульфамоил)-3-метилбензойная кислота,
4-(N-изопропилсульфамоил)-3-метилбензойная кислота, и
4-(N,N-диметилсульфамоил)-3-метилбензойная кислота.
7-(Этил(метил)амино)пиразоло[1,5-a]пиримидин-3-карбоновая кислота
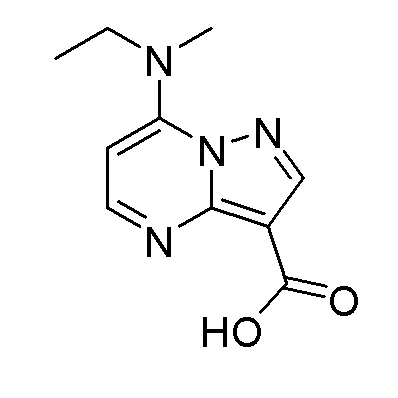
Стадия 1:
К перемешанной суспензии этил-7-гидроксипиразоло[1,5-a]пиримидин-3-карбоксилата (0,95 г, 4,6 ммоль) в POCl3 (8,0 мл, 86 ммоль) добавляли диметиланилин (0,8 мкл, 0,006 ммоль), и нагревали смесь при 80°C в течение 2 часов. Смесь медленно выливали на лед, и осторожно корректирования значение pH до ~7 добавлением 1н NaOH, а затем до pH 10 добавлением твердого Na2CO3. Затем, смесь экстрагировали дихлорметаном (3×). Органические фазы объединяли, промывали солевым раствором, сушили (MgSO4) и упаривали досуха. Твердые вещества растирали с гексанами с получением этил-7-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (260 мг, 25%) в виде каштанового твердого вещества. ESI-MS m/z рассчитано 225,0, обнаружено 226,5 (M+1)+; время удерживания: 0,87 минуты (цикл 3 мин).
Стадия 2:
К раствору этил-7-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (68 мг, 0,30 ммоль) в CH3CN (1 мл) добавляли N-метилэтанамин (18 мг, 0,30 ммоль), и перемешивали смесь при температуре окружающей среды в течение 16 часов. Смесь упаривали, и очищали остаток методом колоночной хроматографии (0 - 10% MeOH в дихлорметане) с получением твердого вещества. Твердое вещество переносили в EtOH (0,5 мл) и воду (0,1 мл), после чего добавляли NaOH (12 мг, 0,30 ммоль). Смесь перемешивали при 50°C в течение 4 часов. Значение pH смеси корректировали до 4 добавлением конц. HCl, и удаляли растворители. Остаток упаривали совместно с MeOH (3×) с получением 7-(этил(метил)амино)пиразоло[1,5-a]пиримидин-3-карбоновой кислоты. ESI-MS m/z рассчитано 220,1, обнаружено 221,5 (M+1)+; время удерживания: 0,29 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методик, описанных выше:
7-(этиламино)пиразоло[1,5-a]пиримидин-3-карбоновая кислота,
7-(изопропиламино)пиразоло[1,5-a]пиримидин-3-карбоновая кислота,
7-(этил(метил)амино)-5-метилпиразоло[1,5-a]пиримидин-3-карбоновая кислота,
5-метил-7-(пирролидин-1-ил)пиразоло[1,5-a]пиримидин-3-карбоновая кислота, и
7-(этиламино)-5-метилпиразоло[1,5-a]пиримидин-3-карбоновая кислота.
4-(1-Гидрокси-1-метилэтил)-3-метилбензойная кислота
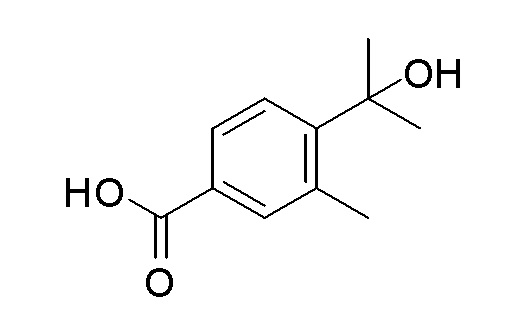
4-Бром-3-метилбензойную кислоту (3,96 г, 18,4 ммоль) растворяли в тетрагидрофуране (100 мл), и охлаждали раствор до -78°C. По каплям в течение 20 минут добавляли н-бутиллитий в гексанах (16,2 мл 2,5M раствора, 41 ммоль). Реакционную смесь оставляли перемешиваться в течение 30 минут при -78°C, а затем по каплям добавляли ацетон (1,35 мл, 18,4 ммоль). Реакционную смесь оставляли перемешиваться в течение 30 минут при -78°C, а затем оставляли ее нагреваться до комнатной температуры. Затем, реакционную смесь разбавляли 100 мл 1M водного гидроксида натрия. Органический слой отбрасывали, а затем подкисляли водный слой добавлением 4M водной соляной кислоты. Затем, водный слой трижды экстрагировали этилацетатом. Объединенные экстракты сушили над сульфатом натрия, а затем упаривали досуха. Неочищенное вещество дополнительно очищали на силикагеле с использованием градиента 0-10% метанола в дихлорметане с получением 4-(1-гидрокси-1-метилэтил)-3-метилбензойной кислоты (1,51 г, 42%). 1H ЯМР (400 МГц, DMSO) δ 12,74 (с, 1H), 7,68 (дд, J=3,9, 2,5 Гц, 2H), 7,55 (д, J=8,7 Гц, 1H), 5,06 (с, 1H), 2,56 (с, 3H), 1,51 (с, 6H).
Следующие соединения синтезировали с использованием методик, описанных выше:
4-(1-гидроксициклопентил)-3-метилбензойная кислота,
4-(1-гидроксициклопентил)бензойная кислота,
4-(1-гидроксициклогексил)-3-метилбензойная кислота,
4-(1-гидроксициклогексил)бензойная кислота,
3-фтор-4-(1-гидроксициклогексил)бензойная кислота, и
4-(1-гидроксициклогексил)-3-метоксибензойная кислота.
5-Изопропокси-6-метилпиколиновая кислота
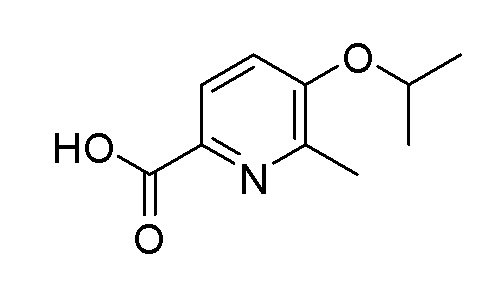
Стадия 1:
2-Метил-3-пиридинол (8,3 г, 76,1 ммоль) суспендировали в ацетонитриле (125 мл). К суспензии по каплям в течение 1 часа добавляли раствор NBS (27,7 г, 155,6 ммоль, 2,05 эквив.) в ацетонитриле (275 мл). Смесь нагревали при температуре кипения с обратным холодильником в течение 1,5 часов. Смесь концентрировали, и очищали остаток методом колоночной хроматографии (DCM) с получением 4,6-дибром-2-метилпиридин-3-ола (15,8 г, 78%) в виде желтого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6) 2,41 (с, 3H), 7,70 (с, 1H), 9,98 (с, 1H).
Стадия 2:
4,6-Дибром-2-метилпиридин-3-ол (15,8 г, 59,4 ммоль) растворяли в THF (200 мл). Раствор охлаждали до -78°C, и по каплям добавляли n-BuLi (50 мл, 125 ммоль, 2,5M в гексане), поддерживая температуру ниже -78°C. Смесь оставляли перемешиваться при этой температуре в течение 2 часов. Смесь гасили добавлением воды (50 мл) и нейтрализовали добавлением 2н HCl. Водную смесь экстрагировали дихлорметаном (2×). Объединенные органические слои сушили (Na2SO4) и концентрировали с получением 6-бром-2-метилпиридин-3-ола (10,5 г, 95%) в виде желтого масла. 1H-ЯМР (300 МГц, DMSO-d6) 2,29 (с, 3H), 7,08 (д, 1H), 7,26 (д, 1H), 10,08 (с, 1H).
Стадия 3:
6-Бром-2-метилпиридин-3-ол (10,5 г, 55,9 ммоль) растворяли в DMF (100 мл). К раствору добавляли K2CO3 (19,3 г, 139,6 ммоль) и 2-бромпропан (13,1 мл, 139,6 ммоль), и нагревали смесь при 100°C в течение ночи. Смесь вливали в смесь воды и EtOAc (200 мл). Слои разделяли, и экстрагировали водный слой EtOAc (2×). Объединенные органические слои сушили (Na2SO4) и концентрировали. Неочищенное масло очищали методом колоночной хроматографии (0-20% этилацетат в гептанах) с получением 6-бром-3-изопропокси-2-метилпиридина (10,9 г, 85) в виде желтого масла. 1H-ЯМР (300 МГц, CDCl3) 1,42 (д, 6H), 2,48 (с, 3H), 4,65 (м, 1H), 7,20 (д, 1H), 8,04 (д, 1H).
Стадия 4:
К MeOH (5,2 мл) и ацетонитрилу (20 мл) в реакторе Berghoff добавляли 6-бром-3-изопропокси-2-метилпиридин (2,00 г, 8,70 ммоль), PdCl2(PPh3)2 (0,18 г, 0,26 ммоль) и Et3N (1,8 мл, 13,04 ммоль). Реактор заполняли CO (газ., 10 бар) и нагревали при 60°C в течение ночи. Смесь концентрировали, и распределяли остаток между DCM и водой. Слои разделяли, органический слой промывали солевым раствором и сушили (Na2SO4). Смесь концентрировали и очищали методом колоночной хроматографии с получением метил 5-изопропокси-6-метилпиколината (1,3 г, 71%) в виде желтого масла. 1H-ЯМР (300 МГц, CDCl3) 1,40 (д, 6H), 2,53 (с, 3H), 3,98 (с, 3H), 4,62 (м, 1H), 7,12 (д, 1H), 7,98 (д, 1H).
Стадия 5:
5-Изопропокси-6-метилпиколинат (1,3 г, 6,22 ммоль) растворяли в THF/воде (2/1, 9 мл). Добавляли LiOH·H2O (0,26 г, 6,22 ммоль), и перемешивали смесь при комнатной температуре в течение ночи. Смесь вливали в смесь воды и EtOAc, и разделяли слои. Водный слой подкисляли до pH 4 добавлением 2н HCl и экстрагировали EtOAc (2×). Объединенные органические фазы сушили (Na2SO4) и концентрировали с получением 5-изопропокси-6-метилпиколиновой кислоты (860 мг, 74%) в виде бежевого твердого вещества. 1H-ЯМР (300 МГц, DMSO-d6) 1,31 (д, 6H), 4,73 (м, 1H), 7,44 (д, 1H), 7,86 (д, 1H). 1H ЯМР (400 МГц, DMSO) δ 12,61 (с, 1H), 7,88 (д, J=8,5 Гц, 1H), 7,44 (д, J=8,7 Гц, 1H), 4,74 (дт, J=12,0, 6,0 Гц, 1H), 2,37 (с, 3H), 1,32 (д, J=6,0 Гц, 6H).
Следующее соединение синтезировали с использованием методик, описанных выше:
5-метокси-6-метилпиколиновая кислота.
4-Изопропокси-3-метоксибензойная кислота
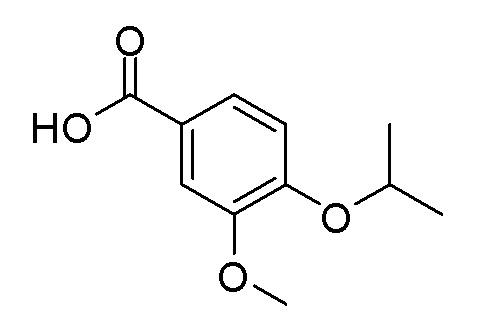
Стадия 1:
В атмосфере подаваемого из баллона N2 к раствору 4-бром-1-изопропокси-2-метоксибензола (400 мг, 1,63 ммоль) в THF (6,0 мл) при -78°C по каплям добавляли трет-бутиллитий (2,14 мл 1,6M раствора, 3,43 ммоль). Смесь оставляли перемешиваться в течение 1 часа при -78°C, после чего ее по каплям добавляли в колбу с твердым CO2 (сухой лед) (1,80 г, 40,8 ммоль) в THF (2,0 мл). Смесь оставляли перемешиваться в течение 30 мин, в процессе чего она нагревалась до комнатной температуры (осторожно: выделение газообразного CO2). Добавляли воду (20 мл), и удаляли летучие вещества в условиях пониженного давления. Полученный водный слой подкисляли до pH~1-2 добавлением 1н HCl, и экстрагировали смесь этилацетатом (3×15 мл). Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением 4-изопропокси-3-метоксибензойной кислоты (чистота ≥94%, 310 мг, 85%) в виде белого твердого вещества. ESI-MS m/z рассчитано 210,1, обнаружено 210,9 (M+1)+; время удерживания: 1,23 мин. 1H ЯМР (400 МГц, DMSO) δ 12,63 (с, 1H), 7,53 (дд, J=8,4, 2,0 Гц, 1H), 7,44 (д, J=2,0 Гц, 1H), 7,04 (д, J=8,7 Гц, 1H), 4,67 (дт, J=12,1, 6,0 Гц, 1H), 3,78 (с, 3H), 1,28 (д, J=6,0 Гц, 6H).
3-Метокси-4-(2-(трифторметокси)этокси)бензойная кислота

Стадия 1:
К гидриду натрия (200 мг, 5,0 ммоль) в DMF (6 мл) в атмосфере N2 добавляли метил-4-гидрокси-3-метоксибензоат (920 мг, 5,0 ммоль), и перемешивали смесь в течение 10 мин. Затем, по каплям добавляли 2-(трифторметокси)этилтрифторметансульфонат (1,2 г, 4,6 ммоль), и перемешивали раствор при комнатной температуре в течение 2 часов, а затем при 50°C в течение 2 часов. Смесь концентрировали до состояния твердого вещества, остаток переносили в 50 мл CH2Cl2, после чего его промывали солевым раствором (20 мл), сушили над MgSO4 и очищали методом колоночной хроматографии (0-25% EtOAc в гексане) с получением метил-3-метокси-4-(2-(трифторметокси)этокси)бензоата в виде белого твердого вещества. ESI-MS m/z рассчитано 294,1, обнаружено 295,3 (M+1)+; время удерживания: 1,63 минуты (цикл 3 мин).
Стадия 2:
Метил-3-метокси-4-(2-(трифторметокси)этокси)бензоат (полученный на стадии 1) растворяли в THF (5 мл), и добавляли суспензию LiOH (550 мг, 23 ммоль) в воде (5 мл). Смесь энергично перемешивали и нагревали при 60°C в течение 6 часов, после чего ее концентрировали до половины объема. Добавляли воду (5 мл), и экстрагировали смесь диэтиловым эфиром (1×10 мл). Водный слой подкисляли до pH 2 добавлением 4н HCl. Полученную смесь экстрагировали этилацетатом (3×10 мл), объединенные органические фазы промывали (1×10 мл H2O, 1×10 мл солевого раствора), сушили над MgSO4 и упаривали с получением 3-метокси-4-(2-(трифторметокси)этокси)бензойной кислоты (1,0 г, 82%) в виде белого твердого вещества. ESI-MS m/z рассчитано 280,1, обнаружено 281,3 (M+1)+; время удерживания: 1,34 минуты (цикл 3 мин).
4-трет-Бутокси-3-метоксибензойная кислота
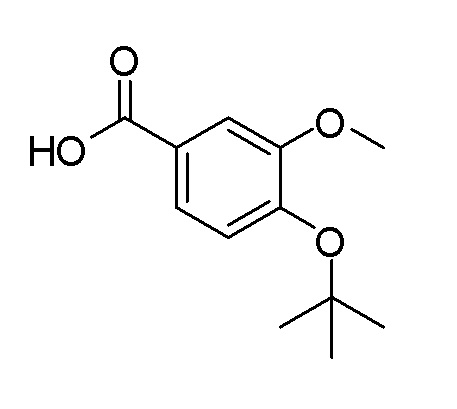
Стадия 1:
4-Гидрокси-3-метоксибензальдегид (500 мг, 3,29 ммоль), Boc2O (1,74 г, 7,97 ммоль) и Sc(OTf)3 (0,080 г, 0,16 ммоль) объединяли в дихлорметане (5 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 24 часов. Добавляли воду (5 мл) и дихлорметан (5 мл), и разделяли две фазы. Водный слой экстрагировали дихлорметаном (3×5 мл), объединенные органические фазы перемешивали с 10% водным гидроксидом калия до исчезновения в органической фазе всего исходного вещества (согласно методу TLC, 40% этилацетат в гексанах). Две фазы разделяли, затем дихлорметановый слой дважды промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и упаривали досуха с получением 4-трет-бутокси-3-метоксибензальдегида (130 мг, 19%) в виде желтого масла. Rf = 0,66 (SiO2, 40% этилацетат в гексанах); ESI-MS m/z рассчитано 208,1, обнаружено 209,2 (M+1)+; время удерживания: 0,96 минут (цикл 6 мин).
Стадия 2:
4-трет-Бутокси-3-метоксибензальдегид (130 мг, 0,62 ммоль) суспендировали в смеси диоксана (520 мкл) и гидроксида калия (6,5 мл 0,20M раствора, 1,3 ммоль). Добавляли KMnO4 (150 мг, 0,93 ммоль), и энергично перемешивали реакционную смесь в течение 16 часов. Реакционную смесь фильтровали, а затем концентрировали до 3 мл. Добавляли соляную кислоту (1M, 4 мл), полученный осадок фильтровали (после отстаивания в течение 15 минут) и промывали 1M HCl и небольшим количеством воды с получением 4-трет-бутокси-3-метоксибензойной кислоты (68 мг, 49%) в виде белого твердого вещества. Rf = 0,23 (SiO2, 40% этилацетат в гексанах); ESI-MS m/z рассчитано 224,1, обнаружено 225,2 (M+1)+; время удерживания: 1,66 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 12,80 (с, 1H), 7,66-7,41 (м, 2H), 7,09 (д, J=8,8 Гц, 1H), 3,78 (с, 3H), 1,32 (с, 9H).
Следующие соединения синтезировали с использованием методик, описанных выше:
4-трет-бутокси-3-метилбензойная кислота из 4-гидрокси-3-метилбензальдегида,
2-фтор-4,5-диметоксибензойная кислота из 2-фтор-4,5-диметоксибензальдегида,
4-трет-бутокси-2-метоксибензойная кислота из 4-гидрокси-2-метоксибензальдегида,
4-трет-бутокси-2-фторбензойная кислота из 2-фтор-4-гидроксибензальдегида, и
4-трет-бутоксибензойная кислота из 4-гидроксибензальдегида.
4-Этил-3-метоксибензойная кислота
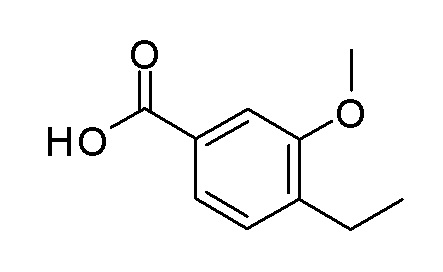
Смесь 4-бром-3-метоксибензойной кислоты (2,49 г, 10,9 ммоль) и Pd(dppf)Cl2 (158 мг, 0,216 ммоль) перемешивали в диоксане (25 мл), и добавляли Et2Zn (22 мл, 1M в гексанах, 22 ммоль). Реакционную смесь нагревали при 70°C в течение 1 часа. Смесь охлаждали до комнатной температуры и гасили добавлением MeOH (1,1 мл). Раствор разбавляли этилацетатом (20 мл) и промывали 1н HCl (10 мл). Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия и упаривали досуха с получением 4-этил-3-метоксибензойной кислоты. ESI-MS m/z рассчитано 180,1, обнаружено 179,1 (M-1)-; время удерживания: 1,77 минуты (цикл 3 мин).
4-(Изопропилсульфонил)-3-метилбензойная кислота
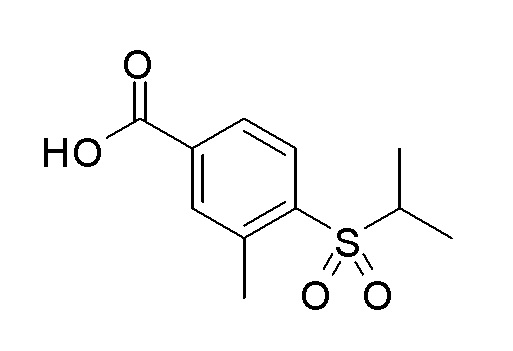
Стадия 1:
К смеси 4-бром-3-метилбензойной кислоты (2,5 г, 12 ммоль) и THF (63 мл) при -78°C по каплям добавляли бутиллитий (16 мл 1,6M раствора, 26 ммоль). Смесь оставляли перемешиваться при -78°C в течение 30 минут, после чего по каплям добавляли раствор 2-изопропилдисульфанилпропан (1,7 г, 12 ммоль) в THF (2 мл). Смесь оставляли перемешиваться при -78°C в течение 30 мин, затем 30 мин при комнатной температуре. Затем, реакционную смесь разбавляли 100 мл 1M водного гидроксида натрия. Органический слой отбрасывали, и подкисляли водный слой добавлением 4M водной соляной кислоты. Затем, водный слой трижды экстрагировали этилацетатом. Объединенные экстракты сушили над сульфатом натрия, а затем упаривали досуха. Неочищенное вещество очищали методом колоночной хроматографии с использованием градиента 0-5% MeOH в дихлорметане с получением 4-(изопропилтио)-3-метилбензойной кислоты (870 мг, 18%). MS m/z рассчитано 210,3, обнаружено 211,2 (M+1)+; время удерживания: 2,32 минуты (цикл 3 мин).
Стадия 2:
К смеси 4-(изопропилтио)-3-метилбензойной кислоты (250 мг, 1,2 ммоль) и дихлорметана (5,0 мл) при 25°C добавляли 3-хлорбензолкарбопероксовую кислоту (930 мг, 4,2 ммоль). Смесь оставляли перемешиваться при 25°C в течение 2 часов, после чего ее концентрировали в условиях вакуума. Белое твердое вещество переносили в дихлорметане и подвергали колоночной хроматографии (0-2% MeOH в дихлорметане) с получением 4-изопропилсульфонил-3-метилбензойной кислоты (90 мг, 31%) в виде белого твердого вещества. ESI-MS m/z рассчитано 242,3, обнаружено 243,2 (M+1)+; время удерживания: 1,57 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 13,50 (с, 1H), 8,50-7,66 (м, 3H), 3,50-3,47 (м, 1H), 2,67 (с, 3H), 1,19 (д, J=1,16 Гц, 6H).
Следующие соединения синтезировали с использованием методик, описанных выше:
4-(изопропилсульфонил)-2-метилбензойная кислота и
4-(этилсульфонил)-3-метилбензойная кислота,
4-(2-Гидроксипропан-2-ил)-3-метоксибензойная кислота

4-Бром-3-метоксибензойную кислоту (2,00 г, 8,67 ммоль) растворяли в THF (50 мл), и охлаждали раствор до -78°C. В течение 15 минут по каплям добавляли n-BuLi в гексанах (7,6 мл 2,5M раствора, 19 ммоль). Реакционную смесь оставляли перемешиваться в течение 30 минут при -78°C, а затем по каплям добавляли ацетон (640 мкл, 8,9 ммоль). Реакционную смесь оставляли перемешиваться в течение 30 минут при -78°C, а затем оставляли ее нагреваться до комнатной температуры. Затем, реакционную смесь разбавляли 100 мл 1M водного гидроксида натрия. Органический слой отбрасывали, и подкисляли водный слой добавлением 4M водной соляной кислоты. Затем, водный слой трижды экстрагировали этилацетатом. Объединенные экстракты сушили над сульфатом натрия, а затем упаривали досуха. Неочищенное вещество очищали методом колоночной хроматографии с использованием градиента 0-5% метанола в дихлорметане с получением 4-(2-гидроксипропан-2-ил)-3-метоксибензойной кислоты (618 мг, 34%). ESI-MS m/z рассчитано 210,1, обнаружено 209,1 (M-1)-; время удерживания: 0,68 минуты (цикл 3 мин).
3-Фтор-4-изопропоксибензойная кислота
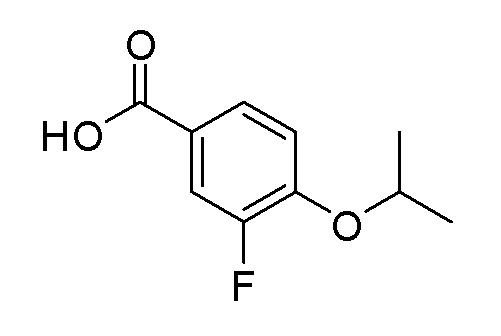
Стадия 1:
К метил-3-фтор-4-гидроксибензоату (2,00 г, 11,8 ммоль) в DMF (12,5 мл) добавляли K2CO3 (6,50 г, 47,0 ммоль), а затем 2-йодпропан (2,35 мл, 23,5 ммоль). Смесь нагревали при 60°C в течение 1,5 часов. Смесь фильтровали с использованием EtOAc, и упаривали фильтрат в условиях пониженного давления. Остаток растворяли в EtOAc и промывали водой и солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением метил-3-фтор-4-изопропоксибензоата. ESI-MS m/z рассчитано 212,1, обнаружено 213,3 (M+1)+; время удерживания: 1,70 минуты (цикл 3 мин).
Стадия 2:
Метил-3-фтор-4-изопропоксибензоат (полученный на стадии 1), 1,4-диоксан (31 мл) и NaOH (31 мл 1,0M раствора, 31 ммоль) объединяли и нагревали смесь при 80°C в течение 20 мин. Растворитель выпаривали в условиях пониженного давления. Неочищенную смесь растворяли в воде и промывали EtOAc (3×). Объединенные органические фазы отбрасывали. Водный слой подкисляли и экстрагировали EtOAc (3×). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением 3-фтор-4-изопропоксибензойной кислоты (1,25 г, 72%) в виде белого твердого вещества. ESI-MS m/z рассчитано 198,1, обнаружено 199,3 (M+1)+; время удерживания: 1,34 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методик, описанных выше:
2-фтор-4-изопропоксибензойная кислота,
4-изопропокси-3-метилбензойная кислота,
3-циано-4-изопропоксибензойная кислота и
4-изопропокси-3-(трифторметил)бензойная кислота.
4-(трет-Бутилсульфонил)бензойная кислота
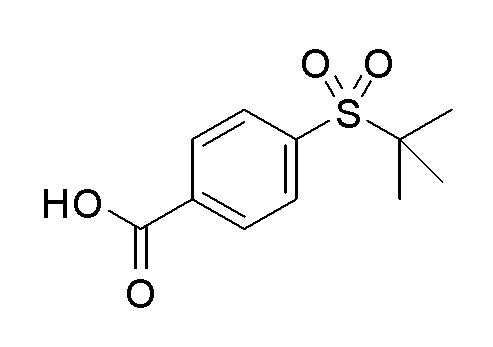
Стадия 1:
Этил-4-фторбензоат (1,5 г, 8,9 ммоль) и трет-бутилсульфанилнатрий (2,00 г, 17,8 ммоль) объединяли в DMF (10 мл). Реакционную смесь нагревали при 80°C в течение 2 часов. Образовывалось небольшое количество осадка, дополнительно добавляли 15 мл DMF, и перемешивали реакционную смесь еще в течение 20 часов при 80°C. Реакционную смесь распределяли между этилацетатом (100 мл) и водой (100 мл). Органический слой отбрасывали, и подкисляли водный слой добавлением 4M соляной кислотой. Водный слой дважды экстрагировали этилацетатом. Объединенные экстракты сушили над сульфатом натрия, фильтровали и упаривали досуха с получением 4-(трет-бутилтио)бензойной кислоты в виде бесцветного масла. ESI-MS m/z рассчитано 210,3, обнаружено 211,1 (M+1)+; время удерживания: 1,74 минуты (цикл 3 мин).
Стадия 2:
4-(трет-Бутилтио)бензойную кислоту (полученную на стадии 1) растворяли в AcOH (10 мл), и добавляли к реакционной смеси пероксид водорода (5,0 мл 30% масс./масс., 52 ммоль). Полученную смесь нагревали при 80°C в течение 2 часов. Затем, реакционную смесь оставляли охлаждаться до комнатной температуры, и разбавляли 50 мл воды и 100 мл этилацетата. Слои разделяли, и экстрагировали водный слой этилацетатом. Объединенные этилацетатные экстракты сушили над сульфатом натрия, фильтровали и упаривали досуха с получением белого твердого вещества. Затем, белое твердое вещество растворяли в дихлорметане и упаривали досуха. Затем, твердое вещество сушили в условиях вакуума в течение 16 часов с получением 4-трет-бутилсульфонилбензойной кислоты (2,2 г, 92%) в виде белого твердого вещества. ESI-MS m/z рассчитано 242,1, обнаружено 243,1 (M+1)+; время удерживания: 1,15 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 8,18 (д, J=8,0 Гц, 2H), 7,94 (д, J=7,6 Гц, 2H), 1,25 (с, 9H).
Следующее соединение синтезировали с использованием методик, описанных выше:
4-(этилсульфонил)бензойная кислота.
4-(Изобутилсульфонил)бензойная кислота
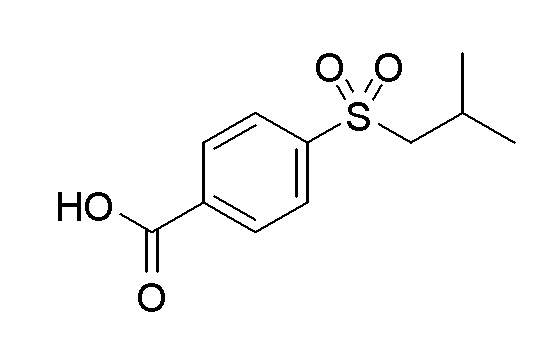
Стадия 1:
К смеси метил-4-сульфанилбензоата (1,00 г, 5,95 ммоль), 1-бром-2-метилпропана (970 мкл, 8,92 ммоль) и DMF (10 мл) при комнатной температуре добавляли K2CO3 (1,23 г, 8,92 ммоль). Смесь оставляли перемешиваться в течение 4 часов при комнатной температуре, после чего отфильтровывали твердые вещества. Твердые вещества промывали этилацетатом, а затем отбрасывали. Объединенные фильтраты распределяли между этилацетатом (100 мл) и водой (100 мл). Слои разделяли, органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением метил-4-(изобутилтио)бензоата (82%) в виде прозрачного масла. ESI-MS m/z рассчитано 224,1, обнаружено 225,2 (M+1)+; время удерживания: 1,59 минуты (цикл 3 мин).
Стадия 2:
К раствору метил-4-(изобутилсульфанил)бензоата (1,00 г, 4,46 ммоль) в CH2Cl2 (20 мл) при комнатной температуре добавляли m-CPBA (3,59 г, 15,6 ммоль). Смесь оставляли перемешиваться в течение 2 часов, после чего ее концентрировали в условиях вакуума. Путем колоночной хроматографии остатка (0-100% этилацетат в гексанах) получали метил-4-(изобутилсульфонил)бензоат. ESI-MS m/z рассчитано 256,1, обнаружено 257,2 (M+1)+; время удерживания: 1,96 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 8,23 (д, J=8,4 Гц, 2H), 8,00 (д, J=8,3 Гц, 2H), 3,98 (с, 3H), 3,02 (д, J=6,5 Гц, 2H), 2,25 (дп, J=13,3, 6,6 Гц, 1H), 1,07 (д, J=6,7 Гц, 6H).
Стадия 3:
Смесь метил-4-изобутилсульфонилбензоата (1,00 г, 3,90 ммоль), NaOH (10 мл 1,0M раствора, 10 ммоль) и 1,4-диоксана (10 мл) нагревали при 80°C в течение 1,5 часов. Смесь охлаждали до к.т., после чего ее концентрировали в условиях вакуума. Твердый остаток переносили в воду и промывали этилацетатом, который отбрасывали. Водный слой подкисляли добавлением 1н HCl и экстрагировали этилацетатом (2×). Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия и концентрировали в условиях вакуума. Путем колоночной хроматографии остатка (0-100% этилацетат в гексанах) получали 4-(изобутилсульфонил)бензойную кислоту (98%). ESI-MS m/z рассчитано 242,1, обнаружено 243,2 (M+1)+; время удерживания: 1,73 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 8,30 (д, J=8,3 Гц, 2H), 8,05 (д, J=8,3 Гц, 2H), 3,03 (д, J=6,5 Гц, 2H), 2,27 (дт, J=13,3, 6,6 Гц, 1H), 1,08 (д, J=6,7 Гц, 6H).
3-(Гидроксиметил)-4-изопропоксибензойная кислота
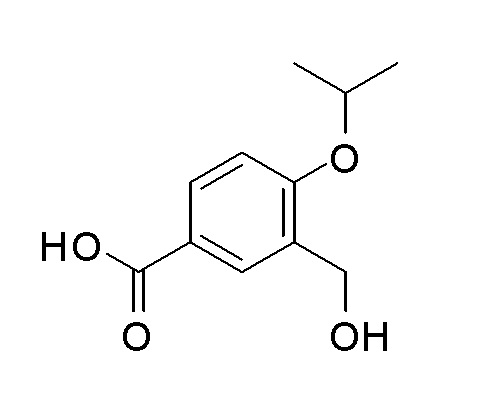
Стадия 1:
К метил-3-формил-4-гидроксибензоату (10,0 г, 55,5 ммоль), карбонату калия (30,7 г, 222 ммоль) и DMF (63 мл) добавляли 2-йодпропан (11,1 мл, 111 ммоль). Смесь нагревали при 60°C в течение 18 часов. Смесь фильтровали с использованием этилацетата (200 мл), и выпаривали растворитель в условиях пониженного давления. Остаток растворяли в этилацетате (150 мл) и промывали водой (3×75 мл) и насыщенным водным раствором хлорида натрия (1×75 мл). Органический слой сушили над сульфатом натрия, фильтровали, и выпаривали растворитель в условиях пониженного давления с получением метил-3-формил-4-изопропоксибензоата (98%) в виде вязкой желтой жидкости. ESI-MS m/z рассчитано 222,2, обнаружено 223,3 (M+1)+; время удерживания: 1,51 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 10,35 (с, 1H), 8,23 (д, J=2,3 Гц, 1H), 8,17 (дд, J=8,8, 2,3 Гц, 1H), 7,39 (д, J=8,9 Гц, 1H), 4,98-4,83 (м, 1H), 3,85 (с, 3H), 1,38 (д, J=6,0 Гц, 6H).
Стадия 2:
Метил-3-формил-4-изопропоксибензоат (180 мг, 0,81 ммоль) растворяли в тетрагидрофуране (4,8 мл), и добавляли LiBH4 (35 мг, 1,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, после чего ее гасили добавлением метанола (3 мл). Реакционную смесь нейтрализовали добавлением насыщенного водного раствора бикарбоната натрия (3 мл), а затем экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (1×10 мл), сушили над сульфатом натрия, фильтровали, и выпаривали растворитель в условиях пониженного давления с получением метил-3-(гидроксиметил)-4-изопропоксибензоата (99%) в виде вязкой жидкости. ESI-MS m/z рассчитано 224,3, обнаружено 225,3 (M+1)+; время удерживания: 1,26 минут (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 8,09 (с, 1H), 7,89 (д, J=8,6 Гц, 1H), 7,13 (д, J=8,6 Гц, 1H), 5,25 (т, J=5,6 Гц, 1H), 4,86-4,68 (м, 1H), 4,54 (д, J=5,6 Гц, 2H), 3,87 (с, 3H), 1,35 (д, J=6,0 Гц, 6H).
Стадия 3:
К метил-3-(гидроксиметил)-4-изопропоксибензоату (180 мг, 0,80 ммоль) и 1,4-диоксану (1,895 мл) добавляли гидроксид натрия (2,1 мл 1,0M раствора, 2,1 ммоль), и нагревали смесь при 80°C в течение 50 минут. Растворитель выпаривали в условиях пониженного давления. Неочищенную смесь растворяли в воде (10 мл) и промывали этилацетатом (3×10 мл), который отбрасывали. Водный слой подкисляли добавлением соляной кислоты. Водный слой экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы сушили над сульфатом натрия, фильтровали, и выпаривали растворитель в условиях пониженного давления с получением 3-(гидроксиметил)-4-изопропоксибензойной кислоты (89%) в виде белого твердого вещества. ESI-MS m/z рассчитано 210,2, обнаружено 211,3 (M+1)+; время удерживания: 1,01 минут (цикл 3 мин).
Следующие соединения синтезировали с использованием методик, описанных выше:
4-этокси-3-(гидроксиметил)бензойная кислота,
4-(2-гидрокси-2-метилпропокси)-3-метилбензойная кислота,
4-изопропокси-3-метокси-5-метилбензойная кислота,
5-изобутоксипиколиновая кислота,
5-(изопентилокси)пиколиновая кислота,
5-изопропокси-4-метилпиколиновая кислота.
3-Метил-4-(оксетан-3-ил)бензойная кислота
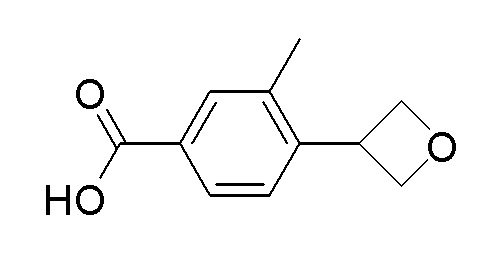
Стадия 1:
(4-Циано-2-метилфенил)бороновая кислота (1,75 г, 10,87 ммоль), йодид никеля(II) (102 мг, 0,326 ммоль), (1S,2S)-2-аминоциклогексан-1-ола гидрохлорид (50 мг, 0,33 ммоль) и NaHMDS (2,01 г, 11,0 ммоль) объединяли в изопропаноле (10 мл) в атмосфере N2 в толстостенном сосуде. Добавляли раствор 3-йодоксетана (1,00 г, 5,44 ммоль) в изопропаноле (1 мл). Сосуд погружали в предварительно нагретую до 90°C масляную баню и перемешивали в течение 2 часов, а затем охлаждали, разбавляли этанолом (20 мл), фильтровали через целит, концентрировали, а затем абсорбировали на целите и очищали методом колоночной хроматографии на силикагеле (0-60% EtOAc в гексане) с получением 3-метил-4-(оксетан-3-ил)бензонитрила (616 мг, 3,556 ммоль, 65,42%) в виде белого твердого вещества. ESI-MS m/z рассчитано 173,1, обнаружено 174,3 (M+1)+; время удерживания: 1,09 минуты (цикл 3 мин).
Стадия 2:
К 3-метил-4-(оксетан-3-ил)бензонитрилу (500 мг, 2,89 ммоль) в этаноле (7,5 мл) добавляли NaOH (3,0 мл 5M раствора, 15 ммоль), и погружали смесь в нагретую до 85°C масляную баню. Смесь нагревали и перемешивали в течение 1 часа, концентрировали, а затем разбавляли этилацетатом (20 мл). Добавляли 6н HCl (~3 мл) для корректировки pH до 6. Водный слой экстрагировали этилацетатом (2×20 мл), затем объединенные органические фазы промывали солевым раствором (10 мл), сушили над MgSO4 и концентрировали с получением белого твердого вещества, которое растирали с эфиром с получением смеси 3-метил-4-(оксетан-3-ил)бензойной кислоты (500 мг, 14%) и ее амида (2/3 согласно ЯМР-анализу). ESI-MS m/z рассчитано 192,2, обнаружено 193,3 (M+1)+; время удерживания: 0,87 минуты (цикл 3 мин).
5-Изопропокси-6-метоксипиколиновая кислота
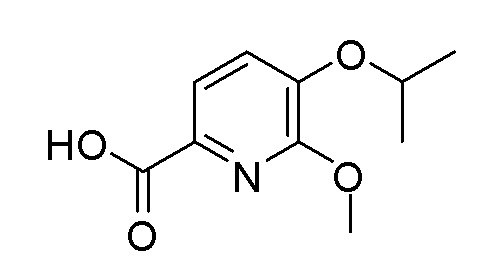
Стадия 1:
К раствору 2-хлор-6-йодпиридин-3-ола (5,00 г, 19,57 ммоль) в DMF добавляли хорошо измельченный карбонат калия (5,409 г, 39,14 ммоль), а затем 2-бромпропан (4,814 г, 3,675 мл, 39,14 ммоль). Реакционную смесь оставляли перемешиваться при 70°C в течение ночи. Реакционную смесь концентрировали в условиях пониженного давления. Остаток растворяли/суспендировали в EtOAc (75 мл) и промывали водой (1×75 мл). Водный слой дополнительно экстрагировали EtOAc (1×75 мл). Оба органических слоя объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением желтого масла, которое очищали методом колоночной хроматографии на силикагеле (градиент 0-30% EtOAc в гексане) с получением 2-хлор-6-йод-3-изопропоксипиридина (5,68 г, 97.%) в виде прозрачного бесцветного маловязкого масла. ESI-MS m/z рассчитано 296,9, обнаружено 298,4 (M+1)+; время удерживания: 1,74 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,55 (д, J=8,3 Гц, 1H), 6,90 (д, J=8,3 Гц, 1H), 4,53 (дт, J=12,1, 6,1 Гц, 1H), 1,39 (д, J=6,1 Гц, 6H).
Стадия 2:
2-Хлор-6-йод-3-изопропоксипиридин (2,00 г, 6,722 ммоль) растворяли в DMF (15 мл). Добавляли Zn(CN)2 (592 мг, 5,04 ммоль), барботировали смесь азотом, после чего добавляли Pd(PPh3)4 (600 мг, 0,519 ммоль). Реакционную систему герметично закрывали и нагревали в условиях обработки микроволнами при 100°C в течение 30 минут. Реакционную смесь разбавляли EtOAc (75 мл) и промывали насыщенным водным раствором бикарбоната натрия (75 мл), а затем солевым раствором (75 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением прозрачного масла, которое очищали методом колоночной хроматографии на силикагеле (градиент 0-30% EtOAc в гексане) с получением 6-хлор-5-изопропоксипиридин-2-карбонитрила (1,17 г, 88%) в виде прозрачного бесцветного масла, которое кристаллизовалось при отстаивании. ESI-MS m/z рассчитано 196,0, обнаружено 197,3 (M+1)+; время удерживания: 1,46 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,61 (д, J=8,4 Гц, 1H), 7,21 (д, J=8,4 Гц, 1H), 4,67 (дт, J=12,1, 6,1 Гц, 1H), 1,45 (д, J=6,1 Гц, 6H).
Стадия 3:
6-Хлор-5-изопропоксипиридин-2-карбонитрил (1,10 г, 5,59 ммоль) растворяли в метаноле (11 мл). К раствору добавляли раствор HCl в 1,4-диоксане (11 мл 4M раствора, 44,00 ммоль). Реакционную смесь оставляли перемешиваться при 70°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и концентрировали в условиях пониженного давления. Оставшиеся твердые вещества суспендировали в EtOAc (75 мл) и промывали насыщенным водным раствором бикарбоната натрия (1×75 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Концентрат очищали методом колоночной хроматографии на силикагеле (0-50% EtOAc в гексане) с получением метил-6-хлор-5-изопропоксипиридин-2-карбоксилата (894 мг, 69%) в виде прозрачного бесцветного масла, которое кристаллизовалось при отстаивании. ESI-MS m/z рассчитано 229,1, обнаружено 230,3 (M+1)+; время удерживания: 1,23 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 8,06 (д, J=8,4 Гц, 1H), 7,25 (д, J=8,5 Гц, 1H), 4,68 (дт, J=12,1, 6,1 Гц, 1H), 3,97 (с, 3H), 1,44 (д, J=6,1 Гц, 6H).
Стадия 4:
Метил-6-хлор-5-изопропоксипиридин-2-карбоксилат (330 мг, 1,44 ммоль) растворяли в диоксане (12 мл), и добавляли раствор метоксида натрия в метаноле (5,75 мл 0,5M раствора, 2,87 ммоль). Реакционную смесь нагревали в условиях обработки микроволнами при 140°C в течение 1,5 часов. Добавляли воду (52 мкл, 2,9 ммоль), и нагревали реакционную смесь в условиях обработки микроволнами при 100°C в течение 30 минут. Реакционную смесь разбавляли 1н раствором HCl (50 мл) и экстрагировали EtOAc (2×50 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления с получением 5-изопропокси-6-метоксипиридин-2-карбоновой кислоты (300 мг, 98%) в виде бежевого твердого вещества. ESI-MS m/z рассчитано 211,1, обнаружено 211,9 (M+1)+; время удерживания: 0,97 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,83 (д, J=8,1 Гц, 1H), 7,17 (д, J=8,1 Гц, 1H), 4,72-4,61 (м, 1H), 4,06 (с, 3H), 1,45 (т, J=7,4 Гц, 7H).
5-трет-Бутоксипиколиновая кислота
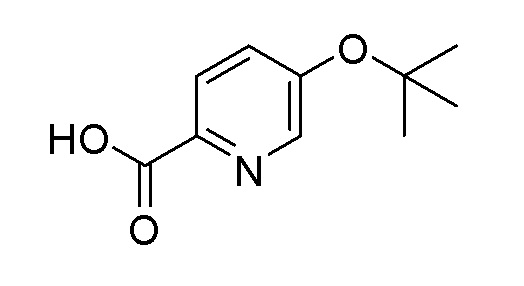
Стадия 1:
К NaOtBu (1,57 г, 16,4 ммоль) в HMPA (6 мл) добавляли DMF (6 мл) (для облегчения перемешивания). Добавляли 5-фторпиридин-2-карбонитрил (1,00 г, 8,19 ммоль), и перемешивали темную смесь в течение ночи. Смесь разбавляли водой (100 мл), экстрагировали DCM (3×50 мл), органические фазы промывали водой (50 мл) и насыщенным водным NaHCO3 (50 мл), сушили над MgSO4, упаривали и очищали методом колоночной хроматографии (0-50% EtOAc в гексане) с получением 5-трет-бутоксипиридин-2-карбонитрила (0,90 г, 62%) в виде желтого твердого вещества. ESI-MS m/z рассчитано 211,1, обнаружено 211,9 (M+1)+; время удерживания: 0,97 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 8,38 (дд, J=2,7, 0,5 Гц, 1H), 7,67-7,56 (м, 1H), 7,41-7,31 (м, 1H), 1,52-1,38 (м, 10H).
Стадия 2:
К 5-трет-бутоксипиридин-2-карбонитрилу (751 мг, 4,26 ммоль) в этаноле (10 мл) добавляли NaOH (4,262 мл 5M раствора, 21,31 ммоль) и погружали смесь в нагретую до 85°C баню. Смесь нагревали и перемешивали в течение 1 часа, концентрировали, а затем разбавляли этилацетатом (50 мл). Добавляли 10 мл солевого раствора и ~3 мл 6н HCl (для корректировки pH до 6). Органический слой разделяли, сушили над MgSO4 и концентрировали с получением 5-трет-бутоксипиридин-2-карбоновой кислоты (820 мг, 98%) в виде желтого твердого вещества. ESI-MS m/z рассчитано 195,1, обнаружено 196,1 (M+1)+; время удерживания: 0,62 минуты (цикл 3 мин). 1H ЯМР (400 МГц, DMSO) δ 12,74 (с, 1H), 8,29 (д, J=32,6 Гц, 1H), 7,99 (с, 1H), 7,60 (д, J=6,5 Гц, 1H), 3,37 (с, 1H), 1,39 (с, 11H).
4-(N-Метил-N-(тиазол-2-ил)сульфамоил)бензойная кислота
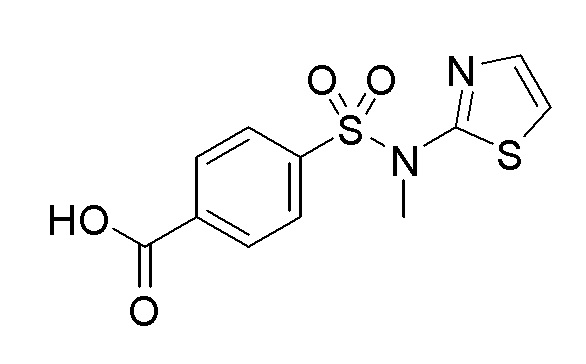
Стадия 1:
К метил-4-хлорсульфонилбензоату (4 г, 17,05 ммоль) добавляли N-метилтиазол-2-амин (1,95 г, 17,05 ммоль), 1,2-дихлорэтан (20 мл) и триэтиламин (2,38 мл, 17,05 ммоль), и нагревали реакционную смесь при 100°C в толстостенном сосуде в течение 20,5 часов на термоблоке. Растворитель выпаривали в условиях пониженного давления. Неочищенное соединение растворяли в дихлорметане и фильтровали. Фильтрат очищали методом хроматографии на силикагеле с использованием градиента 0-30% этилацетата в дихлорметане с получением метил-4-[метил(тиазол-2-ил)сульфамоил]бензоата (4,22 г, 79,2%). 1H ЯМР (400 МГц, DMSO) δ 8,16 (д, J=8,5 Гц, 2H), 7,95 (д, J=8,5 Гц, 2H), 7,46 (д, J=3,6 Гц, 1H), 7,44 (д, J=3,6 Гц, 1H), 3,89 (с, 3H), 3,39 (с, 3H). ESI-MS m/z рассчитано 312,0, обнаружено 313,3 (M+1)+; время удерживания: 1,52 минуты (цикл 3 мин).
Стадия 2:
К метил-4-[метил(тиазол-2-ил)сульфамоил]бензоату (4,22 г, 13,5 ммоль) и 1,4-диоксану (32 мл) добавляли водный NaOH (62 мл 2,5M раствора, 155 ммоль) и перемешивали смесь при 50°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры. Добавляли этилацетат (135 мл), после чего смесь подкисляли до pH 1 добавлением HCl (37%). Органический и водный слои разделяли. Водный слой экстрагировали этилацетатом (1×50 мл). Органические слои сушили над сульфатом натрия, фильтровали, и выпаривали растворитель в условиях пониженного давления с получением 4-[метил(тиазол-2-ил)сульфамоил]бензойной кислоты (3,63 г, 87%) в виде белого твердого вещества. ESI-MS m/z рассчитано 298,0, обнаружено 299,1 (M+1)+; время удерживания: 1,31 минуты (цикл 3 минуты). 1H ЯМР (400 МГц, DMSO) δ 13,56 (с, 1H), 8,14 (д, J=8,6 Гц, 2H), 7,92 (д, J=8,5 Гц, 2H), 7,46 (д, J=3,6 Гц, 1H), 7,44 (д, J=3,6 Гц, 1H), 3,39 (с, 3H).
4-(2-Фтор-2-метилпропокси)-3-метоксибензойная кислота
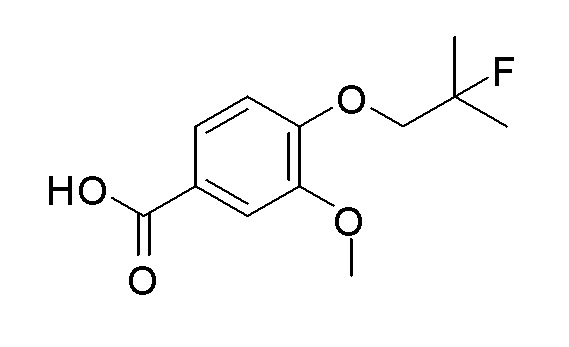
Стадия 1:
К раствору метил-4-(2-гидрокси-2-метилпропокси)-3-метоксибензоата (509 мг, 2,00 ммоль) в DCM (5 мл) при 0°C по каплям добавляли 2-метокси-N-(2-метоксиэтил)-N-(трифтор-сульфанил)этанамин (406 мкл, 2,20 ммоль). Смесь перемешивали при 0°C в течение 1 часа, после чего удаляли охлаждающую баню, и перемешивали смесь в течение 1 часа при комнатной температуре. Смесь вливали в воду и экстрагировали EtOAc (3×). Органические фазы объединяли, промывали водой, солевым раствором, сушили (Na2SO4) и упаривали досуха. Путем очистки остатка методом колоночной хроматографии (0-20% EtOAc в гексане) получали метил-4-(2-фтор-2-метилпропокси)-3-метоксибензоат (450 мг, 70%). ESI-MS m/z рассчитано 256,1, обнаружено 257,1 (M+1)+; время удерживания: 1,57 минуты (цикл 3 мин).
Стадия 2:
К раствору метил-4-(2-фтор-2-метилпропокси)-3-метоксибензоата (450 мг, 1,76 ммоль) в MeOH (3,6 мл) и воде (900 мкл) добавляли NaOH (210 мг, 5,27 ммоль), и перемешивали смесь при 40°C в течение 1 часа. Выпаривали MeOH, и корректировали значение pH раствора до 3 добавлением 1н HCl. Осадок фильтровали, промывали водой и осушали с получением 4-(2-фтор-2-метилпропокси)-3-метоксибензойной кислоты. ESI-MS m/z рассчитано 242,2, обнаружено 243,7 (M+1)+; время удерживания: 1,25 минуты (цикл 3 мин).
3-Метокси-4-((2,2,2-трифторэтокси)метил)бензойная кислота
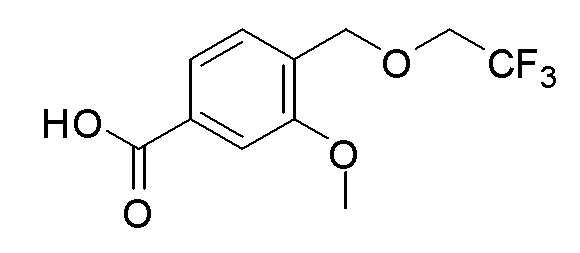
К раствору 2,2,2-трифторэтанола (874 мкл, 12,0 ммоль) при 0°C добавляли NaH (60%, 520 мг, 13,0 ммоль), и перемешивали смесь при этой температуре в течение 10 мин, затем при комнатной температуре в течение 10 мин. Смесь охлаждали до 0°C, после чего добавляли метил-4-(бромметил)-3-метоксибензоат (2,59 г, 10,0 ммоль). Охлаждающую баню удаляли, и перемешивали смесь при комнатной температуре в течение 3 часов. Смесь вливали в воду и экстрагировали EtOAc (3×). Органические фазы объединяли, промывали водой, солевым раствором, сушили (MgSO4) и упаривали досуха. К остатку добавляли измельченный NaOH, а затем воду (4 мл) и MeOH (20 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. Выпаривали MeOH, переносили остаток в 1н NaOH (30 мл), и корректировали pH до 2 добавлением конц. HCl. Осадок фильтровали, промывали водой (2×), а затем гексанами (2×), и осушали с получением 3-метокси-4-((2,2,2-трифторэтокси)метил)бензойной кислоты. 1H ЯМР (400 МГц, CDCl3) δ 7,79 (дд, J=7,8, 1,4 Гц, 1H), 7,61 (д, J=1,4 Гц, 1H), 7,53 (д, J=7,8 Гц, 1H), 4,80 (с, 2H), 4,00-3,91 (м, 5H).
Следующее соединение синтезировали с использованием методик, описанных выше:
метил-3-метокси-4-((3,3,3-трифторпропокси)метил)бензоат.
4-(1-Гидрокси-2-метилпропан-2-ил)-3-метоксибензойная кислота
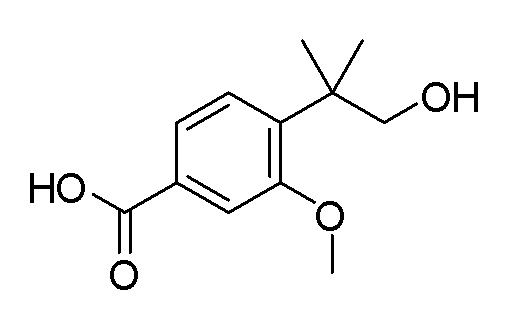
Стадия 1:
4-Бром-3-метоксибензойную кислоту (1,50 г, 6,49 ммоль), K2CO3 (2,69 г, 19,5 ммоль) и DMF (10 мл) объединяли, и оставляли смесь перемешиваться в течение 10 минут. По каплям добавляли бромметилбензол (849 мкл, 7,14 ммоль), и оставляли смесь перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь гасили добавлением солевого раствора и экстрагировали EtOAc (3×). Органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом хроматографии на силикагеле (5%-70% EtOAc в гексанах) с получением бензил-4-бром-3-метоксибензоата (91%). ESI-MS m/z рассчитано 320,0, обнаружено 321,0/323,0 (M+I)+; время удерживания: 3,24 минуты (цикл 4 мин).
Стадия 2:
В продутую азотом колбу добавляли Pd(tBu3P)2 (26 мг, 0,050 ммоль), ZnF2 (52 мг, 0,50 ммоль) и DMF (4 мл). Реакционную смесь оставляли перемешиваться в течение 10 минут, после чего добавляли бензил-4-бром-3-метоксибензоат (323 мг, 1,01 ммоль), а затем триметил(2-метилпроп-1-енокси)силан (277 мкл, 1,51 ммоль). Реакционную смесь нагревали при 80°C в течение ночи. Неочищенную смесь гасили добавлением солевого раствора и трижды экстрагировали EtOAc. Органический слой сушили над сульфатом натрия и выпаривали растворитель с получением бензил-3-метокси-4-(2-метил-1-оксопропан-2-ил)бензоата. ESI-MS m/z рассчитано 312,4, обнаружено 313,4 (M+1)+; время удерживания: 3,27 минуты (цикл 4 мин).
Стадия 3:
Затем, 3-метокси-4-(2-метил-1-оксопропан-2-ил)бензоат (неочищенный, полученный на стадии 2) растирали с MeOH (2 мл), а затем добавляли NaBH4 (190 мг, 5,03 ммоль). Реакционную смесь перемешивали в течение 1 часа, после чего ее гасили добавлением солевого раствора и экстрагировали EtOAc. Органические слои объединяли, сушили над сульфатом натрия и упаривали с получением бензил-4-(1-гидрокси-2-метилпропан-2-ил)-3-метоксибензоата.
Стадия 4:
К неочищенному бензил-4-(1-гидрокси-2-метилпропан-2-ил)-3-метоксибензоату (полученному на стадии 3) добавляли THF (2 мл), а затем водный NaOH (1,7 мл 3,0M раствора, 5,0 ммоль). Реакционную смесь перемешивали в течение 3 часов. Реакционную смесь подкисляли до pH 3 и трижды экстрагировали EtOAc. Органические слои сушили над сульфатом натрия, и выпаривали растворитель с получением 4-(1-гидрокси-2-метилпропан-2-ил)-3-метоксибензойной кислоты. ESI-MS m/z рассчитано 224,1, обнаружено 224,2 (M+1)+; время удерживания: 2,46 минуты (цикл 4 мин).
3-Фтор-4-(2-гидрокси-2-метилпропил)бензойная кислота
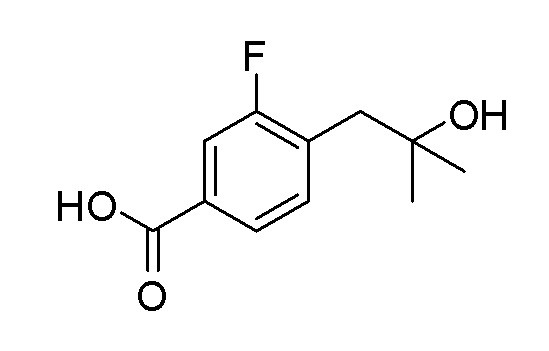
Стадия 1:
К раствору 2-(4-бром-2-фторфенил)уксусной кислоты (4,50 г, 19,3 ммоль) в толуоле (7,7 мл) и MeOH (7,7 мл) в атомсфере азота при комнатной температуре по каплям добавляли диазометилтриметилсилан (11,6 мл 2,0M раствора, 23,2 ммоль). После завершения добавления диазометана сохранялось постоянное желтое окрашивание. Затем, реакционную смесь гасили добавлением нескольких капель уксусной кислоты, и удаляли растворители в условиях пониженного давления. Остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-10% EtOAc в гексанах с получением метил-2-(4-бром-2-фторфенил)ацетата (4,32 г, 91%). 1H ЯМР (400 МГц, CDCl3) δ 7,28-7,22 (м, 2H), 7,15 (т, J=8,0 Гц, 1H), 3,71 (с, 3H), 3,63 (д, J=1,0 Гц, 2H).
Стадия 2:
Метил-2-(4-бром-2-фторфенил)ацетат (4,00 г, 16,2 ммоль) в THF (56 мл) охлаждали на бане со льдом в атомсфере азота, после чего по каплям в течение 30 минут добавляли бромметилмагний (16,2 мл 3M раствора, 48,6 ммоль). Реакционную смесь продолжали перемешивать в течение 2 часов при охлаждении на бане со льдом. Затем, реакционную смесь гасили добавлением насыщенного водного хлорида аммония и разбавляли EtOAc. Слои разделяли, и экстрагировали водный слой EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-15% EtOAc в гексанах с получением 1-(4-бром-2-фторфенил)-2-метилпропан-2-ола (3,0 г, 75%) в виде прозрачного бесцветного масла. ESI-MS m/z рассчитано 246,0, обнаружено 231,1 (M+1)+; время удерживания: 1,53 минуты (цикл 3 мин). При анализе методом ЖХ/МС наблюдали не характерное для массы исходного соединения значение m/z, а фрагмент M-17. 1H ЯМР (400 МГц, CDCl3) δ 7,26-7,21 (м, 2H), 7,14 (т, J=8,1 Гц, 1H), 2,78 (д, J=1,4 Гц, 2H), 1,24 (д, J=0,8 Гц, 6H).
Стадия 3:
1-(4-Бром-2-фторфенил)-2-метилпропан-2-ол (2,35 г, 9,51 ммоль), диацетоксипалладий (214 мг, 0,951 ммоль), 3-дифенилфосфанилпропилдифенилфосфан (404 мг, 0,951 ммоль) и Et3N (4,24 мл, 30,4 ммоль) в DMF (26 мл) обрабатывали MeOH (20,0 мл, 495 ммоль). Сосуд заполняли CO до давления 50 ф/дюйм2 (266 мг, 9,51 ммоль), а затем выпускали газ. Эту операцию повторяли дважды. В сосуд подавали давлением 50 ф/дюйм2 и нагревали при 80°C в течение 15 часов. Реакционную смесь оставляли охлаждаться, выпускали из нее газ и распределяли между EtOAc и солевым раствором. Слои разделяли, и экстрагировали водный слой EtOAc. Объединенные органические фазы дважды промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали до состояния оранжевого масла. Остаток очищали методом колоночной флэш-хроматографии на силикагеле с использованием 0-30% EtOAc в гексанах с получением метил-3-фтор-4-(2-гидрокси-2-метилпропил)бензоата (1,83 г, 85%). ESI-MS m/z рассчитано 226,1, обнаружено 227,5 (M+1)+; время удерживания: 1,29 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,78 (дд, J=7,9, 1,7 Гц, 1H), 7,71 (дд, J=10,3, 1,6 Гц, 1H), 7,34 (т, J=7,6 Гц, 1H), 3,92 (с, 3H), 2,88 (д, J=1,3 Гц, 2H), 1,26 (с, 6H).
Стадия 4:
Метил-3-фтор-4-(2-гидрокси-2-метилпропил)бензоат (1,59 г, 7,03 ммоль) растворяли в THF (40 мл), воде (200 мл) и MeOH (21 мл), после чего добавляли LiOH (1,01 г, 42,2 ммоль). Реакционную смесь нагревали при 55°C в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры, и удаляли растворители в условиях пониженного давления. Остаток растворяли в воде, охлаждали на бане со льдом и обрабатывали 1M HCl до pH 3. Полученный осадок собирали путем вакуумного фильтрования, промывали водой и сушили в условиях высокого вакуума с получением 3-фтор-4-(2-гидрокси-2-метилпропил)бензойной кислоты (999 мг, 67%) в виде белого твердого вещества. ESI-MS m/z рассчитано 212,1, обнаружено 211,1 (M+1)+; время удерживания: 0,98 минуты (цикл 3 мин, в режиме ионизации и сканирования положительно заряженных ионов). 1H ЯМР (400 МГц, CDCl3) δ 7,84 (дд, J=7,9, 1,6 Гц, 1H), 7,77 (дд, J=10,1, 1,6 Гц, 1H), 7,39 (д, J=15,1 Гц, 1H), 2,91 (с, 2H), 1,28 (с, 6H).
3-Фтор-4-(3-метоксипроп-1-инил)бензойная кислота

Стадия 1:
К метил-4-бром-3-фторбензоату (2,50 г, 10,7 ммоль), йодиду меди(I) (204 мг, 1,07 ммоль) и Pd(PPh3)2Cl2 (753 мг, 1,07 ммоль) в продутой аргоном колбе добавляли DMF (дегазированный в течение 30 мин), и охлаждали реакционную смесь до 0°C. Добавляли Et3N (1,95 мл, 14,0 ммоль), а затем 3-метоксипроп-1-ин (997 мкл, 11,8 ммоль), и оставляли смесь перемешиваться при 60°C в течение 70 мин. Смесь охлаждали, разбавляли этилацетатом и фильтровали. Фильтрат последовательно промывали 1M HCl, 10% NH4OH и солевым раствором. Органический слой разделяли, сушили и очищали на силикагеле с использованием этилацетата в гексанах с получением метил-3-фтор-4-(3-метоксипроп-1-инил)бензоата. ESI-MS m/z рассчитано 222,2, обнаружено 223,2 (M+1)+; время удерживания: 1,53 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,75 (ддд, J=11,2, 8,8, 1,5 Гц, 2H), 7,50 (дд, J=7,9, 7,0 Гц, 1H), 4,37 (с, 2H), 3,92 (с, 3H), 3,47 (с, 3H).
Стадия 2:
К метил-3-фтор-4-(3-метоксипроп-1-инил)бензоату (1,40 г, 6,30 ммоль) в 15 мл THF/MeOH (2/1) при комнатной температуре добавляли NaOH (1,89 мл 4M раствора, 7,56 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, после чего удаляли летучие вещества. Остаток экстрагировали эфиром. Водный слой подкисляли добавлением 1M HCl и экстрагировали эфиром (3×). Объединенные эфирные экстракты сушили и концентрировали с получением 3-фтор-4-(3-метоксипроп-1-инил)бензойной кислоты в виде белого твердого вещества. ESI-MS m/z рассчитано 208,2, обнаружено 209,2 (M+1)+; время удерживания: 1,22 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,85 (дд, J=8,0, 1,4 Гц, 1H), 7,80 (дд, J=9,5, 1,3 Гц, 1H), 7,59-7,49 (м, 1H), 4,39 (с, 2H), 3,48 (с, 3H).
4-(бис(2,2,2-трифторэтокси)метил)-3-хлорбензойная кислота
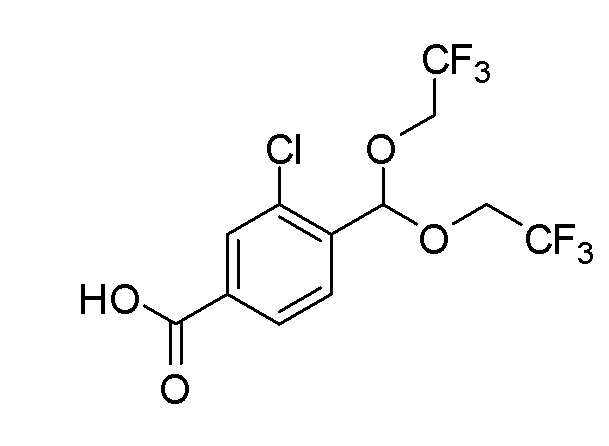
Стадия 1:
К раствору метил-3-хлор-4-метилбензоата (2,00 г, 10,8 ммоль) и NBS (2,05 г, 11,5 ммоль) в CCl4 (9 мл) при нагревании с обратным холодильником добавляли AIBN (178 мг, 1,08 ммоль). Смесь нагревали при температуре кипения с обратным холодильником в течение 16 часов, после чего дополнительно добавляли AIBN (178 мг, 1,08 ммоль). Смесь нагревали при температуре кипения с обратным холодильником в течение 72 часов, после чего охлаждали. Смесь концентрировали досуха, остаток распределяли между Et2O и водой, разделяли слои, и экстрагировали водный слой Et2O (2×). Органические фазы объединяли, промывали водой, солевым раствором, сушили (MgSO4) и концентрировали досуха. Путем очистки остатка методом колоночной хроматографии (0-30% EtOAc в гексане) получали метил-3-хлор-4-(дибромметил)бензоат. ESI-MS m/z рассчитано 342,5, обнаружено 342,9 (M+1)+; время удерживания: 1,96 минуты (цикл 3 мин).
Стадия 2:
К раствору 2,2,2-трифторэтанола (109 мкл, 1,50 ммоль) в DMF (2,63 мл) добавляли NaH (64 мг, 1,6 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, после чего добавляли метил-3-хлор-4-(дибромметил)бензоат (342 мг, 1,00 моль). Смесь перемешивали при комнатной температуре в течение 2 часов, после чего ее вливали в воду и экстрагировали EtOAc (3×). Органические фазы объединяли, промывали водой, солевым раствором, сушили (Na2SO4) и упаривали досуха. Вещество переносили в MeOH, добавляли хорошо измельченный NaOH (160 мг, 4,00 моль), и перемешивали смесь при комнатной температуре в течение 1 часа. Смесь упаривали и растирали с 1н HCl (до сильного закисления раствора). Выпавший в осадок продукт промывали водой и сушили с получением 4-(бис(2,2,2-трифторэтокси)метил)-3-хлорбензойной кислоты. ESI-MS m/z рассчитано 394,4, обнаружено 394,5 (M+1)+; время удерживания: 1,74 минуты (цикл 3 мин).
4-(1-Гидрокси-2-(2,2,2-трифторэтокси)этил)бензойная кислота и 4-(2-гидрокси-1-(2,2,2-трифторэтокси)этил)бензойная кислота
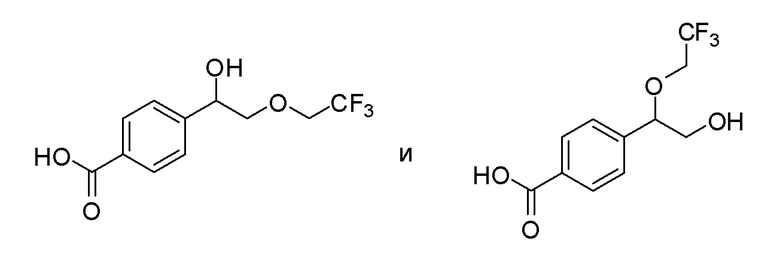
Стадия 1:
К раствору метил-4-ацетилбензоата (8,91 г, 50,0 ммоль) в AcOH (80 мл) по каплям добавляли Br2 (2,71 мл, 52,5 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Смесь охлаждали до 0°C, и фильтровали твердое вещество. Осадок промывали MeOH в воде (1/1) и осушали с получением метил-4-(2-бромацетил)бензоата (10,6 г, 82%) в виде каштанового твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,19-8,12 (м, 2H), 8,04 (д, J=8,5 Гц, 2H), 4,47 (с, 2H), 3,96 (с, 3H).
Стадия 2:
К перемешанному раствору метил-4-(2-бромацетил)бензоата (0,59 г, 2,3 ммоль) в MeOH (6 мл) при 0°C порциями добавляли NaBH4 (92 мг, 2,4 ммоль). Охлаждающую баню удаляли, и перемешивали смесь при комнатной температуре в течение 1 часа. Добавляли K2CO3 (317 мг, 2,30 ммоль), и перемешивали смесь при комнатной температуре в течение 72 часов. Смесь вливали в воду и экстрагировали Et2O (3×). Органические фазы объединяли, промывали водой, солевым раствором, сушили (MgSO4) и упаривали досуха с получением метил-4-(оксиран-2-ил)бензоата (370 мг, 90%) в виде белого твердого вещества. ESI-MS m/z рассчитано 178,2, обнаружено 179,1 (M+1)+; время удерживания: 1,14 минуты (цикл 3 мин).
Стадия 3:
К 2,2,2-трифторэтанолу (0,50 мл, 6,9 ммоль) добавляли NaH (10 мг, 0,24 ммоль). Смесь перемешивали в течение 5 мин, после чего добавляли метил-4-(оксиран-2-ил)бензоат (36 мг, 0,20 ммоль) и нагревали при 70°C в течение 2 часов. Добавляли измельченный NaOH (40 мг, 1,0 ммоль), а затем воду (0,1 мл), и перемешивали смесь при 40°C в течение 3 часов. Смесь упаривали, и распределяли остаток между 1н HCl и EtOAc. Слои разделяли, и экстрагировали водный слой EtOAc (2×). Органические фазы объединяли и упаривали досуха с получением смеси 4-[1-гидрокси-2-(2,2,2-трифторэтокси)этил]бензойной кислоты и 4-(2-гидрокси-1-(2,2,2-трифторэтокси)этил)бензойной кислоты (30 мг, 57%) в виде масла. ESI-MS m/z рассчитано 264,1, обнаружено 265,1 (M+1)+; время удерживания: 1,07 минуты (цикл 3 мин).
(4-Изопропокси-3-метоксифенил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанон

Смесь 2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]дигидрохлорида (69 мг, 0,20 ммоль), 4-изопропокси-3-метоксибензойной кислоты (42 мг, 0,20 ммоль), HATU (76 мг, 0,20 ммоль), Et3N (112 мкл, 0,80 ммоль) и DMF (2 мл) оставляли перемешиваться при комнатной температуре в течение 3 часов. Смесь фильтровали и очищали методом препаративной HPLC с обращенной фазой (10-99% ACN в воде). Целевые фракции концентрировали с получением (4-изопропокси-3-метоксифенил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанона в виде аморфного белого твердого вещества. ESI-MS m/z рассчитано 465,2, обнаружено 466,3 (M+1)+; время удерживания: 1,23 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,01 (д, J=1,7 Гц, 1H), 6,97 (дд, J=8,2, 1,7 Гц, 1H), 6,88 (д, J=8,3 Гц, 1H), 6,53 (д, J=3,0 Гц, 1H), 5,94 (д, J=3,5 Гц, 1H), 4,57 (дт, J=12,2, 6,1 Гц, 1H), 4,52 (с, 1H), 3,99 (с, 2H), 3,87 (с, 3H), 3,69 (с, 1H), 3,40 (с, 1H), 3,34 (т, J=5,4 Гц, 3H), 2,39 (с, 3H), 2,11 (с, 2H), 1,82 (с, 2H), 1,38 (д, J=6,1 Гц, 6H).
(4-Изопропокси-3-метоксифенил)(6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанон
К перемешанному раствору 6-(трифторметил)спиро[3,4-дигидро-2H-пирроло[1,2-a]пиразин-1,4'-пиперидин]дигидрохлорида (475 мг, 1,43 ммоль), 4-изопропокси-3-метоксибензойной кислоты (361 мг, 1,72 ммоль) и 1-гидроксибензотриазола (232 мг, 1,72 ммоль) в CH2Cl2 (5,5 мл) при температуре окружающей среды добавляли 3-(этилиминометиленамино)-N,N-диметилпропан-1-амин (266 мг, 1,72 ммоль), а затем 4-метилморфолин (786 мкл, 7,15 ммоль). Смесь перемешивали в течение 16 часов при комнатной температуре. Смесь вливали в воду и экстрагировали EtOAc (3×). Органические фазы объединяли, промывали 0,1н HCl, 1н NaOH, водой, солевым раствором, сушили (Na2SO4) и упаривали досуха. Остаток очищали методом колоночной хроматографии (0-100% этилацетат в гексанах) с получением (4-изопропокси-3-метоксифенил)-[6-(трифторметил)спиро[3,4-дигидро-2H-пирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанона (623 мг, 96%). ESI-MS m/z рассчитано 451,2, обнаружено 452,3 (M+1)+; время удерживания: 1,30 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,03 (д, J=1,9 Гц, 1H), 6,98 (дд, J=8,2, 1,9 Гц, 1H), 6,88 (д, J=8,3 Гц, 1H), 6,54 (д, J=3,7 Гц, 1H), 5,93 (д, J=3,8 Гц, 1H), 4,56 (дд, J=12,2, 6,1 Гц, 1H), 4,52 (с, 1H), 3,94 (с, 2H), 3,88 (с, 3H), 3,69 (с, 1H), 3,47 (с, 2H), 3,27 (с, 2H), 1,87 (с, 4H), 1,38 (д, J=6,1 Гц, 6H).
Следующие соединения получали с использованием методик, описанных выше:
1-(1-(4-Изопропокси-3-метоксибензоил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-ил)этанон
К смеси (4-изопропокси-3-метоксифенил)-[6-(трифторметил)спиро[3,4-дигидро-2H-пирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанона (100 мг, 0,222 ммоль) и пиридина (1 мл) при комнатной температуре по каплям добавляли ацетилхлорид (158 мкл, 2,22 ммоль). Смесь оставляли перемешиваться в течение 16 часов при комнатной температуре, после чего ее распределяли между этилацетатом и 1н HCl. Слои разделяли, и промывали органический слой 1н HCl, водой, а затем солевым раствором. Органический слой сушили над сульфатом натрия и концентрировали в условиях пониженного давления. Остаток переносили в DMF и очищали методом препаративной HPLC (10-99% ACN в воде с формиатом аммония в качестве модификатора) с получением 1-[1'-(4-изопропокси-3-метоксибензоил)-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-2-ил]этанона (60 мг, 53%) в виде белого твердого вещества. ESI-MS m/z рассчитано 493,2, обнаружено 494,7 (M+1)+; время удерживания: 1,70 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CDCl3) δ 7,06 (д, J=1,8 Гц, 1H), 7,05-6,98 (м, 1H), 6,88 (д, J=8,2 Гц, 1H), 6,55 (д, J=3,3 Гц, 1H), 6,15 (д, J=3,9 Гц, 1H), 4,57 (д, J=6,1 Гц, 1H), 4,19-4,10 (м, 2H), 3,88 (м, s, 5H), 3,79 (с, 2H), 3,70-3,52 (м, J=31,5 Гц, 2H), 3,11 (с, 2H), 2,24 (с, 3H), 1,92-1,75 (м, 2H), 1,38 (д, J=6,1 Гц, 6H).
Следующие соединения синтезировали с использованием методики, описанной выше:
метил-1-(4-изопропокси-3-метоксибензоил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксилат и
этил-1-(4-изопропокси-3-метоксибензоил)-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-2'-карбоксилат.
2,2,2-Трифтор-1-[1'-[4-(2-метокси-3-пиридил)-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон

В микропробирку с Pd(dppf)Cl2 (5,5 мг, 0,075 ммоль) добавляли 2-метоксипиридин-3-илбороновую кислоту (0,10 ммоль) в NMP (0,2 мл), а затем раствор 1-(1-(4-бром-3-метилбензоил)-2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)-2,2,2-трифторэтанона (0,75 ммоль) в DMF (0,3 мл) и водном Na2CO3 (2M, 4 ммоль). Реакционную смесь перемешивали встряхиванием при 80°C в течение 16 часов. Путем фильтрования с последующей очисткой методом препаративной HPLC (1-99% ACN в воде (модификатор - HCl)) получали 2,2,2-трифтор-1-[1'-[4-(2-метокси-3-пиридил)-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон. ESI-MS m/z рассчитано 526,2, обнаружено 527,3 (M+1)+; время удерживания: 1,38 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методики, описанной выше:
5-[2-метил-4-[2-метил-6-(2,2,2-трифторацетил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбонил]фенил]пиридин-2-карбонитрил,
1-[1'-[4-(1-этилимидазол-4-ил)-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]-2,2,2-трифторэтанон,
2,2,2-трифтор-1-[2-метил-1'-[3-метил-4-(2-метил-4-пиридил)бензоил]спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон,
2,2,2-трифтор-1-[2-метил-1'-[3-метил-4-(1-метилпиразол-3-ил)бензоил]спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон,
2,2,2-трифтор-1-[2-метил-1'-[3-метил-4-(1-метилпиразол-4-ил)бензоил]спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон,
2,2,2-трифтор-1-[1'-[4-(4-метоксифенил)-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон,
1-[1'-[4-(3-хлорфенил)-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]-2,2,2-трифторэтанон,
2,2,2-трифтор-1-[2-метил-1'-[3-метил-4-(2-метилпиразол-3-ил)бензоил]спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон,
N-[5-[2-метил-4-[2-метил-6-(2,2,2-трифторацетил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-карбонил]фенил]-2-пиридил]ацетамид,
1-[1'-[4-[2-(диметиламино)пиримидин-5-ил]-3-метилбензоил]-2-метилспиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]-2,2,2-трифторэтанон,
2,2,2-трифтор-1-[2-метил-1'-[3-метил-4-[3-(трифторметил)-1H-пиразол-4-ил]бензоил]спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон, и
2,2,2-трифтор-1-[2-метил-1'-(3-метил-4-пиримидин-5-илбензоил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-6-ил]этанон.
(2-(3-Гидроксипирролидин-1-ил)пиридин-3-ил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанон
Смесь (2-хлор-3-пиридил)-[2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанона (0,1 моль) и пирролидин-3-ола (0,3 ммоль) в DMF (0,5 мл) перемешивали при 80°C в течение 16 часов. Дополнительно добавляли пирролидин-3-ол (0,5 ммоль), и перемешивали смесь при 150°C в течение 16 часов. Смесь фильтровали и подвергали препаративной HPLC (10-90% ACN в воде) с получением (2-(3-гидроксипирролидин-1-ил)пиридин-3-ил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанона. ESI-MS m/z рассчитано 463,5, обнаружено 464,3 (M+1)+; время удерживания: 0,75 минуты (цикл 3 мин).
Следующие соединения синтезировали с использованием методики, описанной выше:
(2-(3-фторпирролидин-1-ил)пиридин-3-ил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанон и
(2-(3,3-дифторпирролидин-1-ил)пиридин-3-ил)(2'-метил-6'-(трифторметил)-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-1-ил)метанон.
2,2,2-Трифтор-1-(1-(3-метокси-4-(проп-1-ен-2-илокси)бензоил)-2'-метил-3',4'-дигидро-2'H-спиро[пиперидин-4,1'-пирроло[1,2-a]пиразин]-6'-ил)этанон
Стадия 1:
К смеси 2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]дигидрохлорида (1,00 г, 2,89 ммоль), 4-гидрокси-3-метоксибензойной кислоты (486 мг, 2,89 ммоль), EDCI (831 мг, 4,33 ммоль), HOBt (585 мг, 4,33 ммоль) и DMF (10 мл) при комнатной температуре добавляли 4-метилморфолин (1,59 мл, 14,4 ммоль). Смесь нагревали при 60°C в течение ночи, после чего ее охлаждали до комнатной температуры и распределяли между этилацетатом и 1н HCl. Слои разделяли, и экстрагировали водный слой этилацетатом (3×). Объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток подвергали колоночной хроматографии (0-100% этилацетат в гексанах) с получением (4-гидрокси-3-метоксифенил)-[2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанона (690 мг, 58%). ESI-MS m/z рассчитано 423,2, обнаружено 424,1 (M+1)+; время удерживания: 1,16 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CD3CN) δ 7,04 (д, J=1,8 Гц, 1H), 6,94 (дд, J=8,1, 1,9 Гц, 1H), 6,87 (д, J=8,1 Гц, 1H), 6,82 (с, 1H), 6,62-6,56 (м, 1H), 6,05 (д, J=3,9 Гц, 1H), 4,35 (с, 1H), 4,00 (т, J=6,0 Гц, 2H), 3,89 (с, 3H), 3,64 (с, 2H), 3,36 (т, J=6,0 Гц, 2H), 3,29 (с, 1H), 2,36 (с, 3H), 2,16-2,04 (м, 2H), 1,81 (дд, J=17,1, 7,3 Гц, 2H),26-7,21 (м, 2H), 7,14 (т, J=8,1 Гц, 1H), 2,78 (д, J=1,4 Гц, 2H), 1,24 (д, J=0,8 Гц, 6H).
Стадия 2:
Смесь Cs2CO3 (115 мг, 0,354 ммоль), ацетилацетона (12 мкл, 0,12 ммоль), CuCl (5,8 мг, 0,059 ммоль) и THF (2,5 мл) перемешивали во флаконе при комнатной температуре в течение 5 мин, после чего добавляли (4-гидрокси-3-метоксифенил)-[2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанон (100 мг, 0,236 ммоль), а затем 2-бромпроп-1-ен (27 мкл, 0,31 ммоль). Флакон закрывали крышкой и нагревали при 70°C в течение ночи. Смесь охлаждали до комнатной температуры, фильтровали и концентрировали в условиях пониженного давления. Остаток подвергали колоночной хроматографии (0-100% этилацетат в гексанах) с получением (4-изопропенилокси-3-метоксифенил)-[2-метил-6-(трифторметил)спиро[3,4-дигидропирроло[1,2-a]пиразин-1,4'-пиперидин]-1'-ил]метанон (8,9 мг, 8%). ESI-MS m/z рассчитано 463,2, обнаружено 464,3 (M+1)+; время удерживания: 1,47 минуты (цикл 3 мин). 1H ЯМР (400 МГц, CD3CN) δ 7,12 (д, J=1,8 Гц, 1H), 7,07 (д, J=8,0 Гц, 1H), 7,01 (дд, J=8,0, 1,8 Гц, 1H), 6,60 (д, J=3,4 Гц, 1H), 6,07 (д, J=3,7 Гц, 1H), 4,42 (с, 1H), 4,12 (дд, J=1,6, 0,9 Гц, 1H), 4,01 (т, J=6,0 Гц, 2H), 3,85 (с, 3H), 3,79 (д, J=1,7 Гц, 1H), 3,67-3,40 (м, 2H), 3,36 (т, J=5,6 Гц, 2H), 3,20 (с, 1H), 2,36 (с, 3H), 2,18 (с, 2H), 2,00 (д, J=0,7 Гц, 3H), 1,83 (с, 2H).
В представленной ниже таблице 2 приведены аналитические данные для соединений, представленных в таблице 1.
МЕТОДЫ АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ И ИЗМЕРЕНИЯ ИНГИБИРУЮЩИХ NaV СВОЙСТВ СОЕДИНЕНИЯ
E-VIPR оптический метод анализа мембранного потенциала с электрической стимуляцией
Натриевые каналы представляют собой потенциалзависимые белки, которые могут быть активированы путем индукции изменений мембранного потенциала посредством применения электрических полей. Прибор для электрической стимуляции и способы его применения описаны в разделе «Методы исследования ионных каналов» в документе PCT/US01/21652, включенном в настоящий документ посредством ссылки и называемым E-VIPR. Прибор включает устройство для манипулирования титрационным микропланшетом, оптическую систему для возбуждения кумаринового красителя при одновременной регистрации испусканий кумарина и оксонола, синхрогенератор, управляемый током или напряжением усилитель и устройство для введения электродов в лунку. При интегрированном управлении компьютером этот прибор сообщает клеткам, находящимся в лунках титрационного микропланшета, электрические стимулы согласно запрограммированному пользователем протоколу.
За 24 часа до анализа на E-VIPR, клетки HEK, экспрессирующие подтип NaV человека, такой как NaV1.7, высеивают в покрытые полилизином 384-луночные микропланшеты в количестве 15000-20000 клеток на лунку. С другими подтипами действуют аналогичным образом на клеточных линиях, экспрессирующих интересующий NaV. Клетки HEK выращивают в среде (точный состав специфичен для каждого типа клеток и подтипа NaV) с добавлением 10% FBS (пригодная к использованию фетальная бычья сыворотка; GibcoBRL #16140-071) и 1% Pen-Strep (пенициллин/стрептомицин; GibcoBRL #15140-122). Клетки выращивают во флаконах с вентилируемыми крышками при 90% влажности и 10% CO2 до 100% конфлуентности. Обычно, их отделяют путем обработки трипсином 1/10 или 1/20 (в зависимости от требований запланированного анализа) и выращивают в течение 2-3 суток до следующего разделения.
Реагенты и растворы:
100 мг/мл Pluronic F-127 (Sigma #P2443) в безводном DMSO
Планшеты для соединения: 384-луночные круглодонные планшеты, например 384-луночные полипропиленовые круглодонные планшеты Corning #3656
Культивационные планшеты: 384-луночные обработанные планшеты для тканевых культур, например, Greiner #781091-1B
10 мМ DiSBAC6(3) (Aurora #00-100-010) в безводном DMSO
10 мМ CC2-DMPE (Aurora #00-100-008) в безводном DMSO
200 мМ ABSC1 в H2O
Буфер Bath1: глюкоза 10 мМ (1,8 г/л), хлорид магния (безводный) 1 мМ (0,095 г/л), хлорид кальция 2 мМ (0,222 г/л), HEPES 10 мМ (2,38 г/л), хлорид калия 4,5 мМ (0,335 г/л), хлорид натрия 160 мМ (9,35 г/л).
Раствор гексилового красителя: Буфер Bath1 + 0,5% β-циклодекстрин (смешивают перед применением, Sigma #C4767), 8 мкМ CC2-DMPE + 2,5 мкМ DiSBAC6(3). Для приготовления раствора добавляют 10% маточный Pluronic F127 в объеме, равном объему CC2-DMPE + DiSBAC6(3). Порядок приготовления: сначала смешивают Pluronic и CC2-DMPE, затем при перемешивании на вортексе добавляют DiSBAC6(3), а затем добавляют Bath1 + β-циклодекстрин.
Протокол анализа:
1) В планшеты для соединений предварительно вносят соединения (в чистом DMSO). В каждую лунку в 160-кратном относительно желаемой конечной концентрации количестве добавляют контроль с растворителем (чистый DMSO), положительный контроль (20 мМ маточный раствор тетракаина в DMSO, конечная концентрация при анализе 125 мкМ) и тестируемые соединения в чистом DMSO. Конечный объем в планшете для соединений будет составлять 80 мкл (80-кратное промежуточное разбавление 1 мкл внесенного соединения в DMSO; 160-кратное конечное разбавление после перемещения в клеточный планшет). Конечная концентрация DMSO для всех лунок при анализе составляет 0,625%.
2) Приготавливают раствор гексилового красителя
3) Приготавливают клеточные планшеты. В день анализа среду отбирают путем аспирации, и трижды промывают клетки 100 мкл буфера Bath1, оставляя в каждой лунке остаточный объем 25 мкл.
4) В каждую лунку культивационного планшета раскапывают по 25 мкл раствора гексилового красителя. Инкубируют в течение 20-35 минут при комнатной температуре/в условиях окружающей среды.
5) В каждую лунку планшета для соединений раскапывают по 80 мкл Bath1. Добавляют краситель Кислотный Желтый 17 (1 мМ), и в зависимости от подтипа NaV и чувствительности анализа содержание хлорида калия может быть изменено до 4,5-20 мМ.
6) Каждую лунку культивационного планшета трижды промывают 100 мкл Bath1, оставляя в каждой лунке остаточный объем 25 мкл. Затем, из каждой лунки планшетов для соединений переносят 25 мкл в культивационные планшеты. Инкубируют в течение 20-35 минут при комнатной температуре/в условиях окружающей среды.
7) Проводят считывание с планшета на E-VIPR. Для сообщения стимулирующих волновых импульсов (обычно продолжительностью 9 секунд и при скорости сканирования в 400 Гц) используют управляемый током усилитель. Для получения базовой линии интенсивностей без стимуляции перед стимуляцией проводят регистрацию в течение 0,5 секунд. В течение 9 секунд подают стимулирующий импульс с последующей 0,5 секундной пост-стимуляционной регистрацией для проверки процесса релаксации до состояния покоя. Электрический стимулирующий импульс является специфичным для каждого типа клеток, и для обеспечения оптимального для анализа импульса может варьироваться амплитуда, длительность и частота подаваемого тока.
Анализ данных
Данные анализируют и приводят в виде нормированных соотношений значений интенсивности испускания, измеренных в каналах регистрации при 460 нм и 580 нм, за вычетом фонового значения. Из полученного для каждого канала значения вычитают фоновые значения интенсивности. Фоновые значения интенсивности получают путем измерения значений интенсивности испускания в течение тех же периодов времени в идентично обработанных лунках, в которых отсутствуют клетки. Затем, кривую ответа во времени приводят в виде соотношений, полученных с использованием следующей формулы:
Данные дополнительно преобразовывают путем вычисления начального (Ri) и конечного (Rf) соотношений. Они представляют собой средние значения соотношений в течение части или всего престимуляционного периода и в каждой точке измерения в течении периода стимуляции. Затем рассчитывают ответ на стимул R = Rf/Ri и приводят его в виде функции во времени.
Ответы в контрольных группах получают путем проведения анализа в присутствии соединения с желаемыми свойствами (положительный контроль), такого как тетракаин, и в отсутствие фармакологических средств (отрицательный контроль). Ответы в отрицательном (N) и положительном (Р) контролях рассчитывают, как представлено выше. Активность А соединения-антагониста определяют как:
 ,
,
где R представляет собой ответ на стимул для тестируемого соединения.
ЭЛЕКТРОФИЗИОЛОГИЧЕСКИЕ МЕТОДЫ АНАЛИЗА АКТИВНОСТИ NaV И ИНГИБИРОВАНИЯ ТЕСТИРУЕМЫМИ СОЕДИНЕНИЯМИ
Для оценки эффективности и селективности блокаторов натриевых каналов в нейронах спинномозговых ганглиев использовали электрофизиологический метод пэтч-кламп. Нейроны крыс выделяли из спинномозговых ганглиев и поддерживали в культуре в течение 2-10 суток в присутствии NGF (50 нг/мл) (культуральная среда представляет собой среду Neurobasal A с добавлением B27, глутамина и антибиотиков). Нейроны с малым диаметром (ноцицепторы, 8-12 мкм в диаметре) визуально идентифицировали и зондировали стеклянными электродами с тонким концом, присоединенными к усилителю (Axon Instruments). Для оценки IC50 соединения использовали метод «фиксации потенциала» клетки на уровне – 60 мВ. Кроме того, для тестирования эффективности блокирования соединениями генерации потенциала действия в ответ на подаваемый ток использовали способ «фиксации тока». Результаты указанных экспериментов способствовали определению профиля эффективности соединений.
Метод анализа IonWorks
Натриевые токи регистрировали с использованием автоматизированной пэтч-кламп системы IonWorks (Molecular Devices Corporation, Inc.). Клетки, экспрессирующие подтипы NaV, собирали из тканевой культуры и помещали в суспензию в количестве 0,5-4 миллиона клеток на мл буфера Bath1. Аппарат IonWorks измеряет изменения натриевых токов в ответ на «фиксацию напряжения» по аналогии с традиционным методом пэтч-кламп, но в 384-луночном формате. С использованием IonWorks были определены дозовые зависимости ответа при «фиксации напряжения» путем деполяризации клетки от определенного в эксперименте исходного потенциала до тестируемого потенциала, равного приблизительно 0 мВ, до и после добавления тестируемого соединения. Влияние соединения на токи измеряли при тестируемом потенциале.
Анализ связывания 1-бензазепин-2-она
Способности соединений согласно настоящему изобретению ингибировать натриевые каналы также могут быть определены путем методов анализа, описанных в работе Williams, B. S. et al., “Characterization of a New Class of Potent Inhibitors of the Voltage-Gated Sodium Channel NaV1.7,” Biochemistry, 2007, 46, 14693-14703, полное содержание которой включено в настоящий документ посредством ссылки.
Согласно измерениям с использованием методов анализа, описанных выше в настоящем документе, типичные соединения, представленные в настоящем документе в таблице 1, являются активными в отношении одного или нескольких натриевых каналов, как представлено в таблице 3.
Как очевидно специалистам в данной области техники, без выхода за рамки объема настоящего изобретения могут быть произведено множество модификаций и изменений вариантов осуществления, описанных в настоящем документе. Конкретные варианты осуществления, описанные в настоящем документе, приводятся лишь в иллюстративных целях.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2692766C1 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2811402C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ХИНОКСАЛИНЫ И АЗАХИНОКСАЛИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА CRTH | 2011 |
|
RU2589709C2 |
| МОДУЛЯТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2797323C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| СОЕДИНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТА АДЕНОЗИНОВОГО РЕЦЕПТОРА A2a И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2840068C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| СПИРОЦИКЛИЧЕСКИЕ АМИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ S1P | 2011 |
|
RU2602800C2 |
Изобретение относится к соединению формулы I или его фармацевтически приемлемым солям
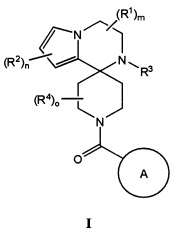
В формуле I R1 представляет собой C1-С8алкил, С1-С8гидроксиалкил, CO2R8, или два R1 образуют вместе оксогруппу, или 3-7-членное конденсированное циклоалкильное кольцо; R2 представляет собой галоген, C1-С8галогеналкил, C1-С8гидроксигалогеналкил, CN, SO2CH3, CO2R8, CON(R8)2 или COR8; R3 представляет собой Н, C1-С8алкил, С3-С8циклоалкил, CO2R8, COR8, CON(R8)2, CF3, CH2CF3, CH2CHF2, СН2-фенил, CH2-CN, CH2-CO2R8, CH2-CON(R8)2, СН2-С3-С8циклоалкил, С1-С8алкокси-С1-С8алкил или C1-С8гидроксиалкил; R4 представляет собой C1-С8алкил или галоген; А представляет собой фенил, замещенный 1-2 заместителями; или 5-членный гетероарил, где три кольцевых атома представляют собой N, замещенный фторфенил-С1-С8алкилом; или 6-членный гетероарил, где один кольцевой атом представляет собой N, замещенный 1-2 заместителями; или 9-10-членный бициклический гетероарил, где 2-3 кольцевых атома независимо представляют собой гетероатомы, выбранные из N, О, необязательно замещенный 1-2 заместителями; m равно целому числу от 1 до 2; n равно целому числу от 1 до 2; и о равно целому числу от 0 до 1; значения остальных заместителей указаны в формуле изобретения. Изобретение также относится к фармацевтическим композициям, к способам ингибирования потенциалзависимого натриевого ионного канала, к способам лечения, к индивидуальным соединениям. Технический результат: получены новые соединения, обладающие ингибирующей активностью в отношении потенциалзависимого натриевого ионного канала. 10 н. и 32 з.п. ф-лы, 3 табл., 752 пр.
1. Соединение формулы I:
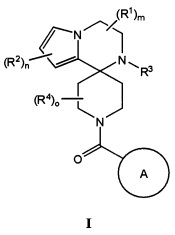
или его фармацевтически приемлемая соль,
где, независимо в каждом случае:
R1 представляет собой C1-С8алкил, С1-С8гидроксиалкил, CO2R8, или два R1 образуют вместе оксогруппу, или 3-7-членное конденсированное циклоалкильное кольцо;
R2 представляет собой галоген, C1-С8галогеналкил, C1-С8гидроксигалогеналкил, CN, SO2CH3, CO2R8, CON(R8)2 или COR8;
R3 представляет собой Н, C1-С8алкил, С3-С8циклоалкил, CO2R8, COR8, CON(R8)2, CF3, CH2CF3, CH2CHF2, СН2-фенил, CH2-CN, CH2-CO2R8, CH2-CON(R8)2, СН2-С3-С8циклоалкил, С1-С8алкокси-С1-С8алкил или C1-С8гидроксиалкил;
R4 представляет собой C1-С8алкил или галоген;
R8 представляет собой Н, C1-С8алкил, С3-С8циклоалкил, С3-С8циклоалкил-С1-С3алкил, C1-С8фторалкил, фенил, 5-членный гетероарил, где 2 кольцевых гетероатома независимо представляют собой, N, S, или два R8 образуют вместе с атомами, к которым они присоединены, 5-6-членное кольцо, необязательно замещенное C1-С8алкилом, гидрокси или галогеном;
А представляет собой
фенил, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С2-С8алкенила, С3-С8циклоалкила, С3-С8гидроксициклоалкила, C1-С8галогеналкила, галогена, гидроксила, C1-С8алкокси, О-С2-С8алкенила, С1-С8алкокси-С3-С8циклоалкила, C1-С8галогеналкокси, С1-С8алкокси-С1-С8алкокси, CN, SO2R8, SO2NH(R8)2, OSO2R8, С1-С8галогеналкокси-С1-С8алкил, С2-С8алкинил-О-С1-С8алкила, С1-С8гидроксиалкил-О-С1-С8галогеналкила, C1-С8гидроксиалкила, C1-С8гидроксиалкокси, С1-С8галогеналкокси-С1-С8алкокси, фенила, необязательно замещенного галогеном, C1-С8алкокси, фенокси, бензила, бензилокси, С1-С8алкокси-фенила, тетрагидропиранилокси, N(R8)2, 4-членного гетероциклила, где 1 кольцевой гетероатом представляет собой О, 5-6-членного гетероарила, где 1-2 кольцевых гетероатома представляют собой N, необязательно замещенного C1-С8алкилом, C1-С8алкокси, CN, CF3, NHC(O)СН3, N(СН3)2; или
5-членный гетероарил, где три кольцевых атома представляют собой N, замещенный фторфенил-С1-С8алкилом; или
6-членный гетероарил, где один кольцевой атом представляет собой N, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С3-С8циклоалкила, C1-С8алкокси, N(R8)2, C1-С8галогеналкила, гидроксила, фенила, фенокси; или
9-10-членный бициклический гетероарил, где 2-3 кольцевых атома независимо представляют собой гетероатомы, выбранные из N, О, необязательно замещенный 1-2 заместителями, выбранными из C1-С8алкила, C1-С8галогеналкила, С3-С8циклоалкила, N(R8)2;
m равно целому числу от 1 до 2;
n равно целому числу от 1 до 2; и
о равно целому числу от 0 до 1.
2. Соединение по п. 1, где, независимо в каждом случае:
R1 представляет собой C1-С6алкил, или два R1 образуют вместе оксогруппу, или 3-7-членное конденсированное циклоалкильное кольцо;
R2 представляет собой C1-С6галогеналкил, CN, SO2CH3, CO2R8 или CON(R8)2;
R3 представляет собой Н, C1-С6алкил, С3-С8циклоалкил, CO2R8, COR8, CON(R8)2, CF3, CH2CF3 или CH2CHF2;
R4 представляет собой C1-С6алкил или галоген;
R8 представляет собой Н, C1-С6алкил, С3-С8циклоалкил, или два R8 образуют вместе с атомами, к которым они присоединены, 5-6-членное кольцо;
А представляет собой
А представляет собой фенил, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С2-С8алкенила, С3-С8циклоалкила, С3-С8гидроксициклоалкил, C1-С8галогеналкила, галогена, гидроксила, C1-С8алкокси, О-С2-С8алкенила, С1-С8алкокси-С3-С8циклоалкила, C1-С8галогеналкокси, С1-С8алкокси-С1-С8алкокси, CN, SO2R8, SO2N(R8)2, OSO2R8, С1-С8галогеналкокси-С1-С8алкил, С2-С8алкинил-O-С1-С8алкила, С1-С8гидроксиалкил-O-С1-С8галогеналкила, C1-С8гидроксиалкила, C1-С8гидроксиалкокси, С1-С8галогеналкокси-С1-С8алкокси, фенила, необязательно замещенного галогеном, С1-С8алкокси, фенокси, бензила, бензилокси, C1-С8алкокси-фенила, тетрагидропиранилокси, N(R8)2, 4-членного гетероциклила, где 1 кольцевой гетероатом представляет собой О, 5-6-членного гетероарила, где 1-2 кольцевых гетероатома представляют собой N, необязательно замещенного C1-С8алкилом, C1-С8алкокси, CN, CF3, NHC(O)СН3, N(СН3)2; или
5-членный гетероарил, где три кольцевых атома представляют собой N, замещенный фторфенил-С1-С8алкилом; или
6-членный гетероарил, где один кольцевой атом представляет собой N, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С3-С8циклоалкила, С1-С8алкокси, N(R8)2, С1-С8галогеналкила, гидроксила, фенила, фенокси; или
9-10-членный бициклический гетероарил, где 2-3 кольцевых атома независимо представляют собой гетероатомы, выбранные из N, О, необязательно замещенный 1-2 заместителями, выбранными из C1-С8алкила, C1-С8галогеналкила, С3-С8циклоалкила, N(R8)2.
3. Соединение по п. 1, где R1 представляет собой C1-С8алкил, или два R1 образуют вместе с атомами, к которым они присоединены, 3-7-членное конденсированное циклоалкильное кольцо.
4. Соединение по п. 1, где R1 представляет собой СН3, или два R1 образуют вместе конденсированное циклогексильное кольцо.
5. Соединение по п. 1, где R2 представляет собой галоген, CF3, CN, COR8 или CON(R8)2.
6. Соединение по п. 1, где R2 представляет собой COCF3, COtBu, Cl, СОСН3, CF2CF3, CH2CF3, CF3, CN, Br, COCH(CH3)2, COCH2CH3, CH(OH)CF3, SO2CH3, 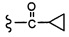 , COPh,
, COPh, 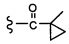 или
или 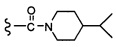 .
.
7. Соединение по п. 1, где R3 представляет собой Н, C1-С8алкил, CO2R8, COR8 или CON(R8)2.
8. Соединение по п. 1, где R3 представляет собой Н, СН3, СН2СН3, СН2СН2ОСН3, СН2СН2ОН, CH2CO2CH2CH3, CH2CON(СН3)2, CH2CONH2, CH2CN, бензил, циклобутил, СН2СН(СН2)2, СН(СН2)2, CH2CF3, CH2CHF2, СОСН3, СОСН2СН3, CO2CH3, CO2CH2CH3, СОН, CON(CH3)2 или CONHCH3.
9. Соединение по п. 1, где R4 представляет собой галоген или C1-С6алкил.
10. Соединение по п. 1, где R4 представляет собой F или СН3.
11. Соединение по п. 1, где m равно 1.
12. Соединение по п. 1, где n равно 2.
13. Соединение по п. 1, где о равно 0.
14. Соединение по п. 1, где А представляет собой фенил, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С2-С8алкенила, С3-С8циклоалкила, С3-С8гидроксициклоалкила, C1-С8галогеналкила, галогена, гидроксила, C1-С8алкокси, O-С2-С8алкенила, С1-С8алкокси-С3-С8циклоалкила, C1-С8галогеналкокси, C1-С8алкокси-С1-С8алкокси, CN, SO2R8, SO2N(R8)2, OSO2R8, C1-С8галогеналкокси-С1-С8алкил, С2-С8алкинил-O-С1-С8алкила, C1-С8гидроксиалкил-O-С1-С8галогеналкила, C1-С8гидроксиалкила, C1-С8гидроксиалкокси, С1-С8галогеналкокси-С1-С8алкокси, фенила, необязательно замещенного галогеном, C1-С8алкокси, фенокси, бензила, бензилокси, C1-С8алкокси-фенила, тетрагидропиранилокси, N(R8)2, 4-членного гетероциклила, где 1 кольцевой гетероатом представляет собой О, 5-6-членного гетероарила, где 1-2 кольцевых гетероатома представляют собой N, необязательно замещенного С1-С8алкилом, C1-С8алкокси, CN, CF3, NHC(O)CH3 или N(CH3)2.
15. Соединение по п. 14, где заместитель фенила представляет собой C1-С8алкил, C1-С8алкокси, галоген, OCF3, OCHF2 или С3-С8гидроксициклоалкил.
16. Соединение по п. 14, где заместитель фенила представляет собой СН3, ОСН3, OCF3, фенокси, фенил, OCHF2 или F.
17. Соединение по п. 14, где заместитель фенила представляет собой C1-С8алкил, C1-С8алкокси или галоген.
18. Соединение по п. 14, где заместитель фенила представляет собой СН3, ОСН3, ОСН2СН3, ОСН2СН2СН3, ОСН(СН3)2, CF3, CN, фенила, SO2CH3, ОН, СН(СН3)2, ОСН2СН2СН2СН3, F, Cl или СН2ОН.
19. Соединение по п. 14, где заместитель фенила представляет собой C1-С8алкил, C1-С8алкокси, SO2R8, OSO2R8, SO2N(R8)2, OCHF2 или OCF3.
20. Соединение по п. 14, где заместитель фенила представляет собой СН3, СН2СН3, tBu, Cl, F, ОН, С(=СН2)СН3, ОС(=СН2)СН3, ОСН3, ОСН2СН2СН2СН3, СН2ОН, ОСН2СН2ОН, ОСН2СН2СН2ОН, OtBu, ОСН(СН3)(СН2СН3), ОСН2С(СН3)2ОН, С(СН3)2OH, СН2С(СН3)2ОН, СН(ОН)СН(СН3)2, С(СН3)2СН2ОН, OCH2CH2CH(CH3)2, OCH2CH2CH3, ОСН(СН3)2, ОСН2СН2ОСН3, 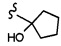 , SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2,
, SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2, 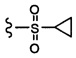 , SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,
, SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,  , фенокси, фенил,
, фенокси, фенил, 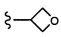 , СН(СН3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, ОСН2СН2СН2СН3, CH2OPh,
, СН(СН3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, ОСН2СН2СН2СН3, CH2OPh, 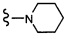 , OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, ОСН2СН(СН3)2, CH2Ph,
, OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, ОСН2СН(СН3)2, CH2Ph, 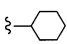 , СССН2ОСН3, SO2CHF2, OCF3,
, СССН2ОСН3, SO2CHF2, OCF3, 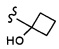 , OCHF2,
, OCHF2,  , CH2CH(CH3)2, OCH2tBu,
, CH2CH(CH3)2, OCH2tBu, 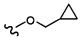 , OCH2CF3,
, OCH2CF3,  ,
, 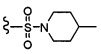 , CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, С(CH3)2CH2CH3, С(СН2СН3)3, CH(OCH2CF3)2,
, CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, С(CH3)2CH2CH3, С(СН2СН3)3, CH(OCH2CF3)2, 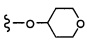 , CF3, OCH2C(CH3)2F,
, CF3, OCH2C(CH3)2F, 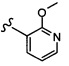 ,
, 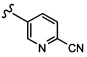 ,
,
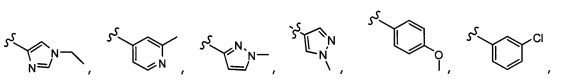

CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3, 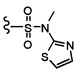 или OCH2CH2OCF3.
или OCH2CH2OCF3.
21. Соединение по п. 14, где А представляет собой:
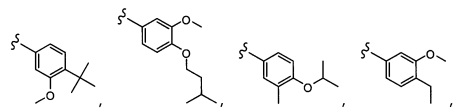
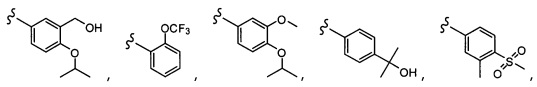
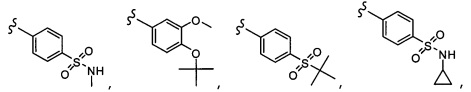
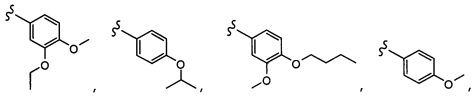
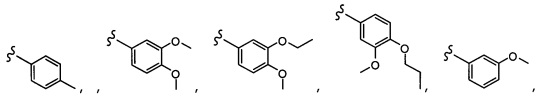
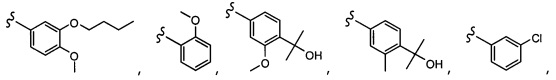
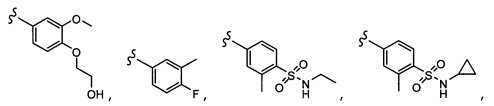

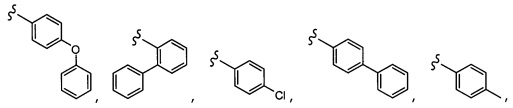
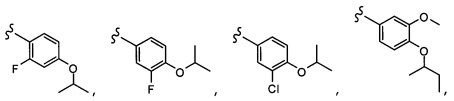
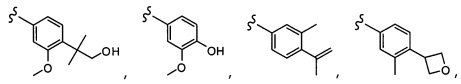
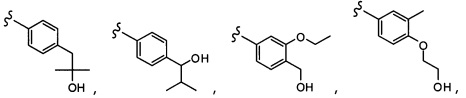

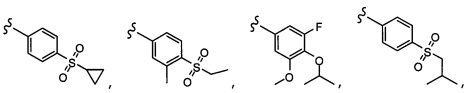
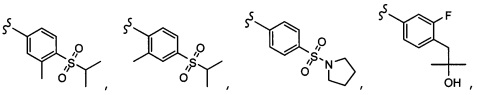
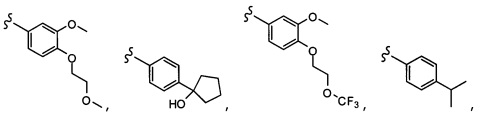


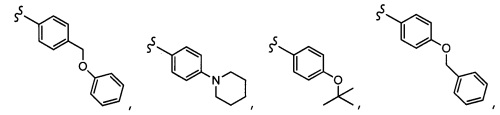


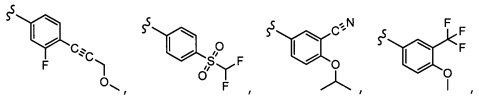
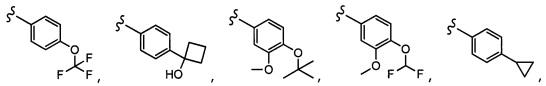

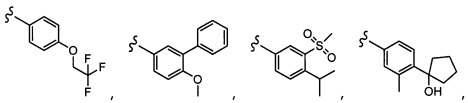
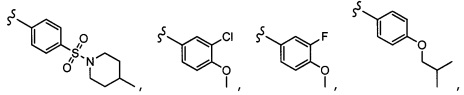
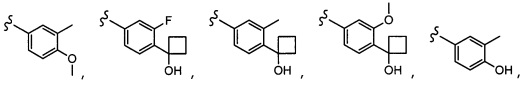
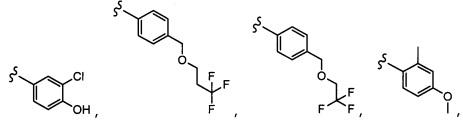

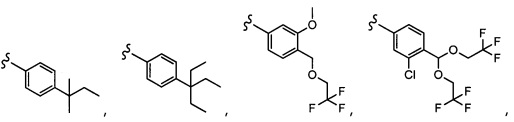
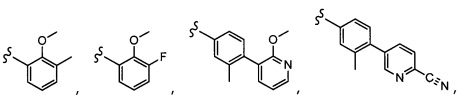
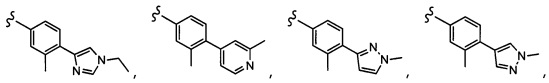

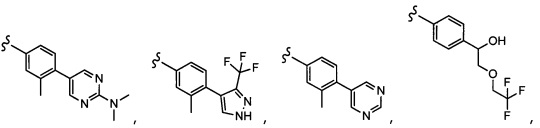
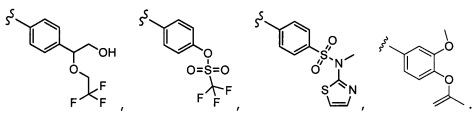
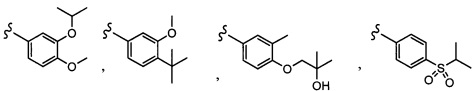
 .
.
22. Соединение по п. 1, где А представляет собой 5-членный гетероарил, где три кольцевых атома представляют собой N, замещенный фторфенил-С1-С8алкилом; 6-членный гетероарил, где один кольцевой атом представляет собой N, замещенный 1-2 заместителями, выбранными из С1-С8алкила, С3-С8циклоалкила, C1-С8алкокси, N(R8)2, C1-С8галогеналкила, гидроксила, фенила, фенокси; 9-10-членный бициклический гетероарил, где 2-3 кольцевых атома независимо представляют собой гетероатомы, выбранные из N, О, необязательно замещенный 1-2 заместителями, выбранными из C1-С8алкила, C1-С8галогеналкила, С3-С8циклоалкила или N(R8)2.
23. Соединение по п. 22, где А представляет собой 6-членный гетероарил, где один кольцевой атом представляет собой N, замещенный 1-2 заместителями, выбранными из C1-С8алкила, С3-С8циклоалкила, C1-С8алкокси, N(R8)2, C1-С8галогеналкила, гидроксила, фенила или фенокси.
24. Соединение по п. 22, где А представляет собой 9-10-членный бициклический гетероарил, где 2-3 кольцевых атома независимо представляют собой гетероатомы, выбранные из N, О, необязательно замещенный 1-2 заместителями, выбранными из C1-С8алкила, C1-С8галогеналкила, С3-С8циклоалкила или N(R8)2.
25. Соединение по п. 22, где А представляет собой:
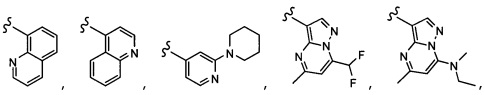

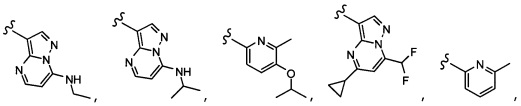
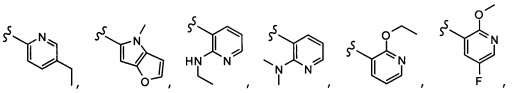

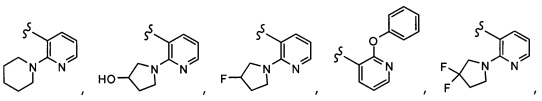
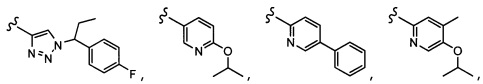

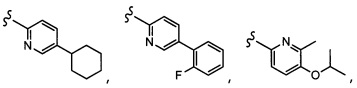 или
или 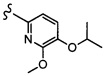
26. Соединение по п. 1, где соединение характеризуется формулой IA:
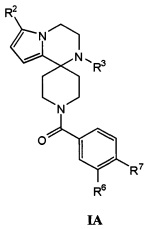
где:
R2 представляет собой галоген, C1-С8галогеналкил, CN, SO2CH3, COR8, CO2R8, CON(R8)2 или C1-С8гидроксигалогеналкил;
R3 представляет собой Н, C1-С8алкил, CO2R8, COR8, С3-С8циклоалкил, CON(R8)2, CF3, СН2-фенил, CH2-CN, CH2-CO2R8, СН2-CON(R8)2, СН2-С3-С8циклоалкил, С1-С8лкокси-С1-С8алкил или C1-С8гидроксиалкил;
R6 представляет собой Н, C1-С8алкил, С3-С8циклоалкил, C1-С8алкокси, С1-С8алкокси-С3-С8циклоалкил, С1-С8гидроксиалкил, галоген, CN, ОН, SO2R8, SO2N(R8)2, CF3, OCF3, OCHF2, 4-членный гетероциклил, где 1 кольцевой гетероатом представляет собой О, фенил, 5-6-членный гетероарил, где 1-2 кольцевых гетероатома представляют собой N, необязательно замещенный C1-С8алкилом, C1-С8алкокси, CN, CF3, NHC(O)CH3 или N(CH3)2;
R7 представляет собой Н, С1-С8алкил, С2-С8алкенил, С3-С8циклоалкил, C1-С8алкокси, O-С2-С8алкенил, галоген, CN, ОН, N(R8)2, SO2R8, OSO2R8, C1-С8гидроксиалкил, C1-С8галогеналкил, C1-С8гидроксиалкокси, С3-С8гидроксициклоалкил, С2-С8алкинил-O-С1-С8алкил, C1-С8галогеналкокси, С1-С8галогеналкокси-С1-С8алкил, C1-С8галогеналкокси-С1-С8алкокси, С1-С8алкокси-С3-С8циклоалкил, C1-С8гидроксиалкил-О-С1-С8галогеналкил, SO2N(R8)2, 4-членный гетероциклил, где 1 кольцевой гетероатом представляет собой О, фенил, необязательно замещенный галогеном, С1-С8алкокси, фенокси, бензил, бензилокси, 5-6-членный гетероарил, где 1-2 кольцевых гетероатома представляют собой N, необязательно замещенный C1-С8алкилом, С1-С8алкокси, CN, CF3, NHC(O)CH3 или N(CH3)2.
27. Соединение по п. 26, где R2 представляет собой COCF3, COtBu, Cl, СОСН3, CF2CF3, CH2CF3, CF3, CN, Br, COCH(CH3)2, COCH2CH3, CH(OH)CF3, SO2CH3,  , COPh,
, COPh,  или
или  .
.
28. Соединение по п. 26, где R3 представляет собой Н, СН3, СН2СН3, СН2СН2ОСН3, СН2СН2ОН, CH2CO2CH2CH3, CH2CON(CH3)2, CH2CONH2, CH2CN, бензил, циклобутил, CH2CH(СН2)2, СН(СН2)2, CH2CF3, CH2CHF2, СОСН3, СОСН2СН3, CO2CH3, СО2СН2СН3, СОН, CONH(CH3)2 или CONHCH3.
29. Соединение по п. 26, где R6 представляет собой Н, СН3, ОСН3, ОСН2СН3, ОСН2СН2СН3, ОСН(СН3)2, CF3, CN, Ph, SO2CH3, ОН, СН(СН3)2, ОСН2СН2СН2СН3, F, Cl или СН2ОН.
30. Соединение по п. 26, где R7 представляет собой Н, СН3, СН2СН3, tBu, Cl, F, ОН, С(=СН2)СН3, ОС(=СН2)СН3, ОСН3, ОСН2СН2СН2СН3, СН2ОН, ОСН2СН2ОН, ОСН2СН2СН2ОН, OtBu, ОСН(СН3)(СН2СН3), ОСН2С(СН3)2ОН, С(СН3)2ОН, СН2С(СН3)2ОН, СН(ОН)СН(СН3)2, С(СН3)2СН2ОН, ОСН2СН2СН(СН3)2, ОСН2СН2СН3, ОСН(СН3)2, ОСН2СН2ОСН3,  , SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2,
, SO2CH3, SO2tBu, SO2CH2CH3, SO2CH2CH(CH3)2, SO2CH(CH3)2, 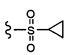 , SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,
, SO2NH(CH3), SO2NH(CH(CH2)2), SO2NH(CH2CH3), SO2NH(CH(CH3)2), SO2N(CH3)2,  , OPh, Ph,
, OPh, Ph, 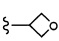 , CH(CH3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OCH2CH2CH2CH3, CH2OPh,
, CH(CH3)2, SO2N(CH2CH2CH3)2, CH2CH2CH2CH3, CH2CH2CH3, OCH2CH2CH2CH3, CH2OPh, 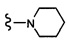 , OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2Ph,
, OCH2Ph, CH2CH2CH2CH2CH3, OCH2CH3, OCH2CH(CH3)2, CH2Ph,  , CCCH2OCH3, SO2CHF2, OCF3,
, CCCH2OCH3, SO2CHF2, OCF3,  , OCHF2,
, OCHF2, 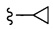 , CH2CH(CH3)2, OCH2tBu,
, CH2CH(CH3)2, OCH2tBu, 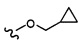 , OCH2CF3,
, OCH2CF3, 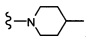 ,
, 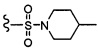 , CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, С(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2,
, CH2OCH2CH2CF3, CH2OCH2CF3, SO2CF3, С(CH3)2CH2CH3, C(CH2CH3)3, CH(OCH2CF3)2, 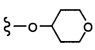 , CF3, OCH2C(CH3)2F,
, CF3, OCH2C(CH3)2F,  ,
,  ,
,

 , CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,
, CH(OH)CH2OCH2CF3, CH(OCH2CF3)CH2OH, OSO2CF3,  или OCH2CH2OCF3.
или OCH2CH2OCF3.
31. Соединение по п. 26, где  фрагмент представляет собой:
фрагмент представляет собой:

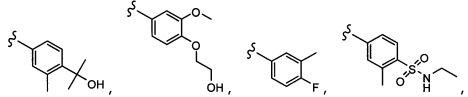
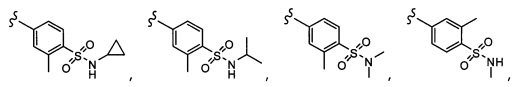
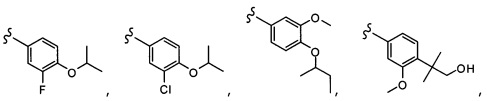
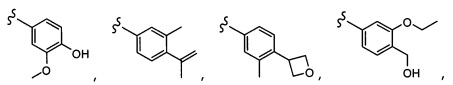
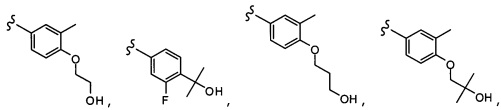
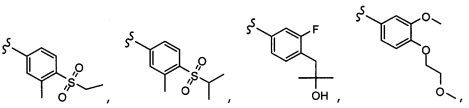
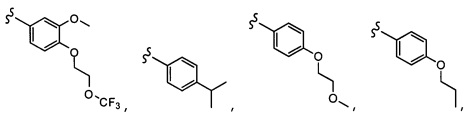
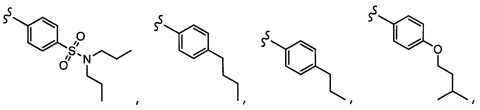

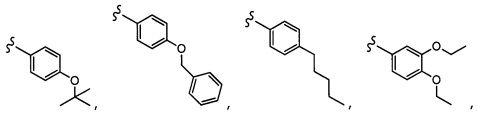
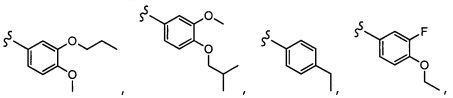

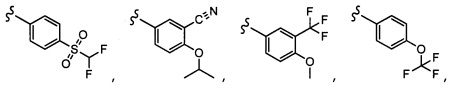
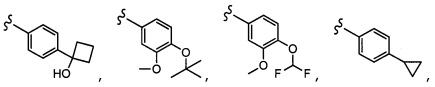
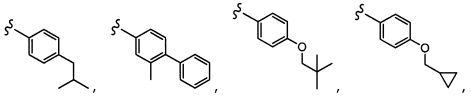
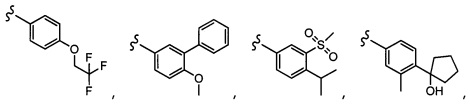

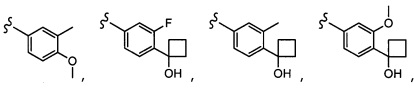
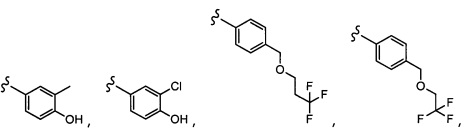

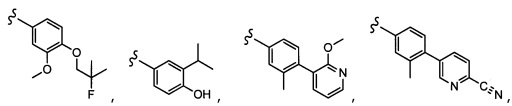
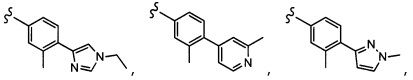
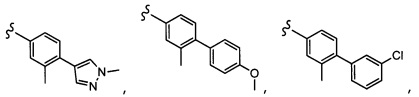

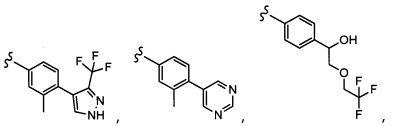


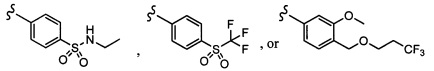 .
.
32. Соединение по п. 1, где соединение выбирают из следующей таблицы:
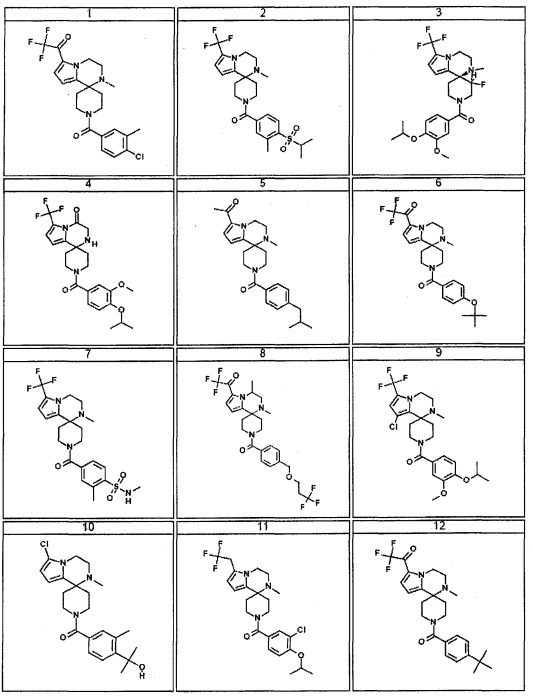
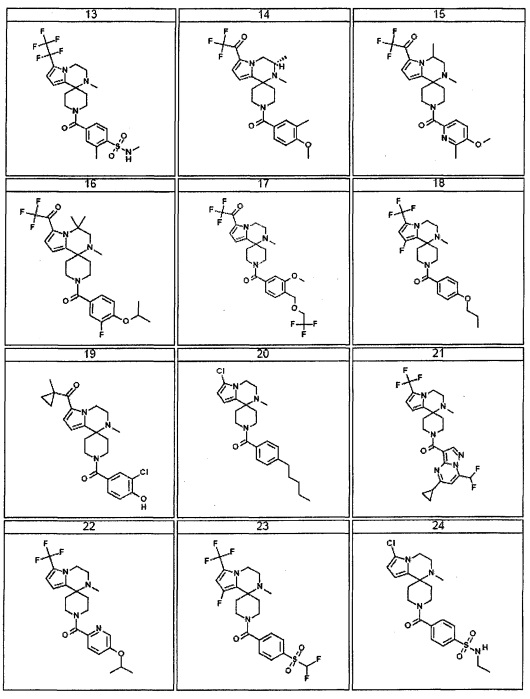
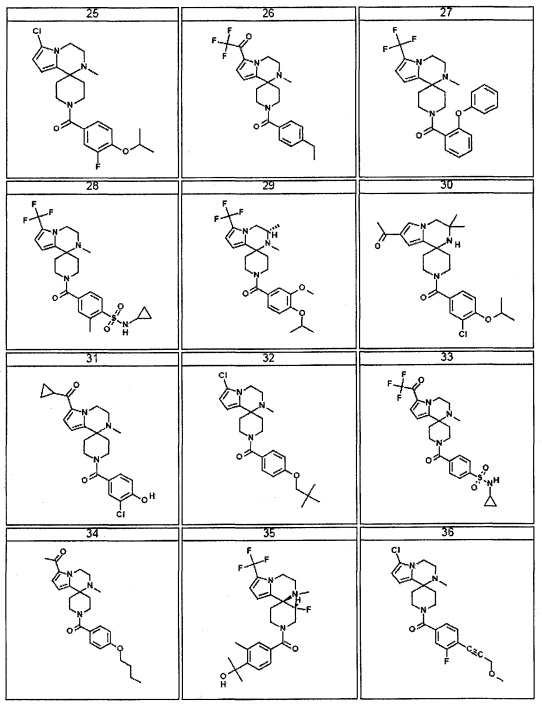
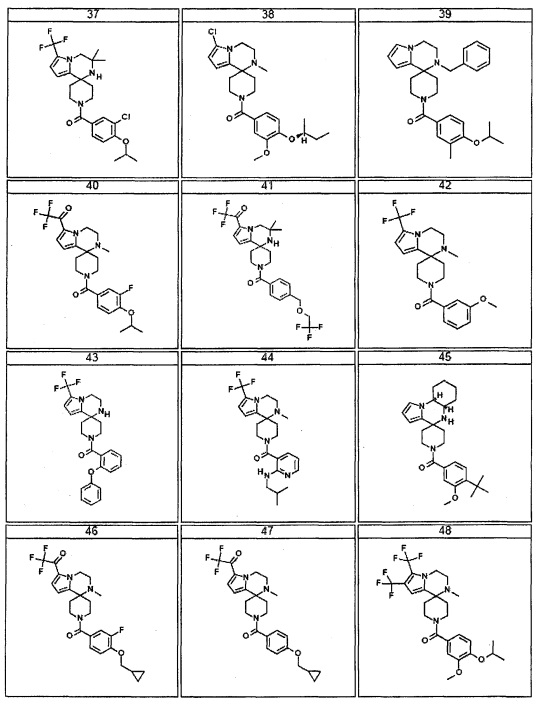
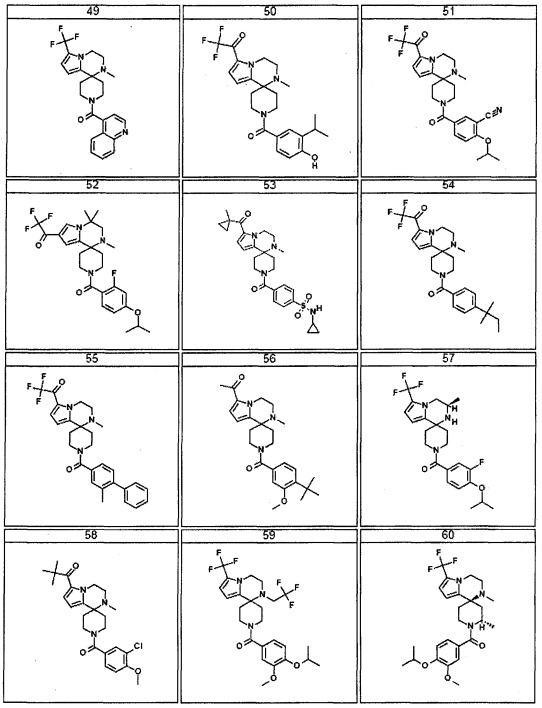
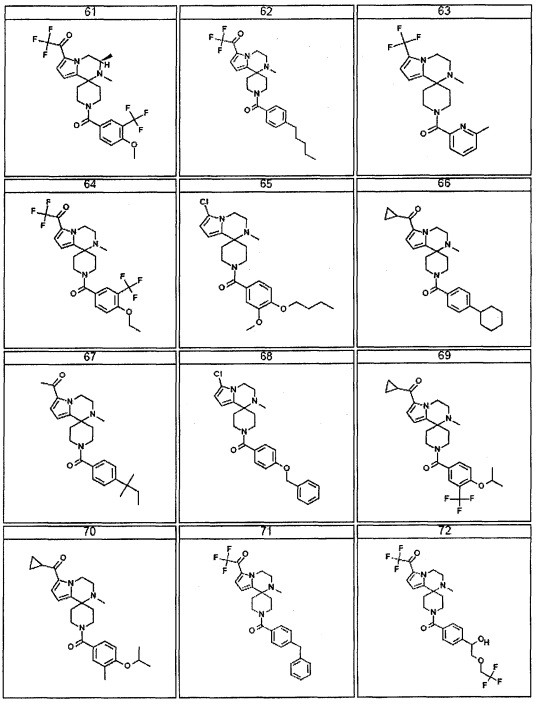
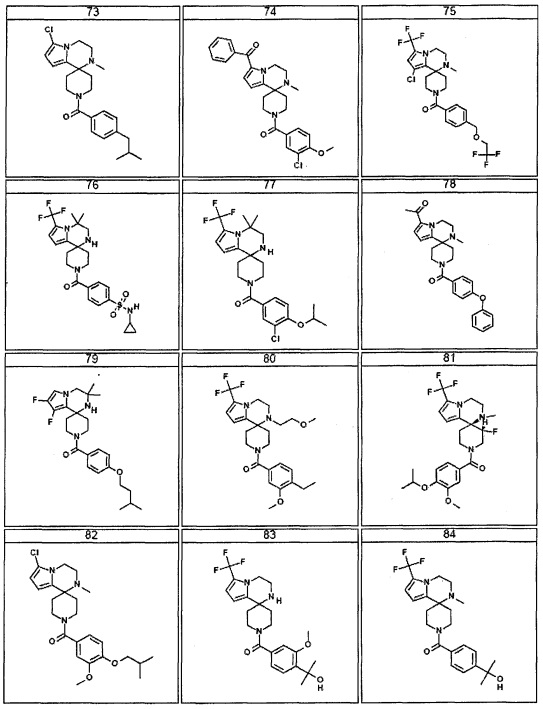
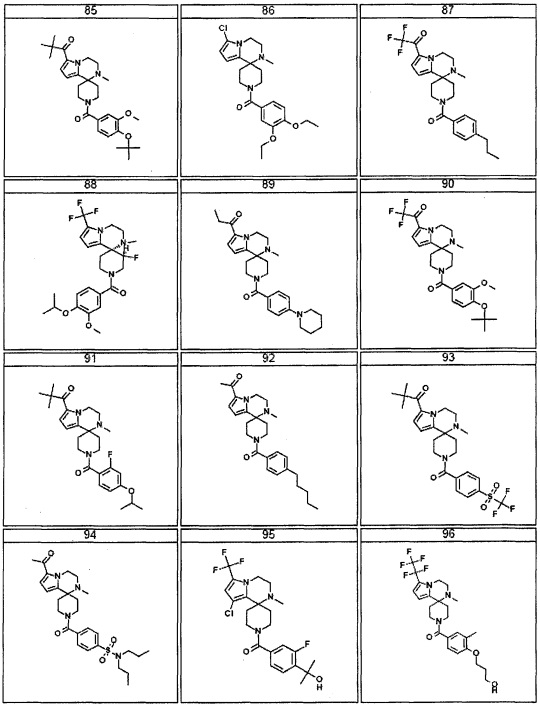
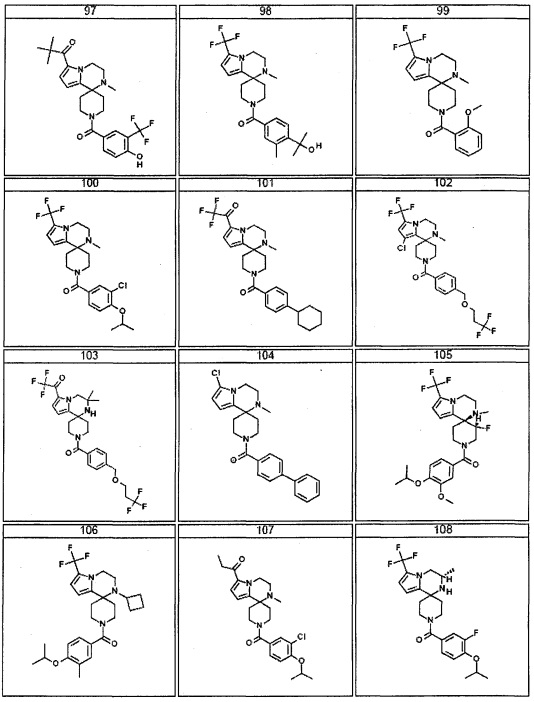
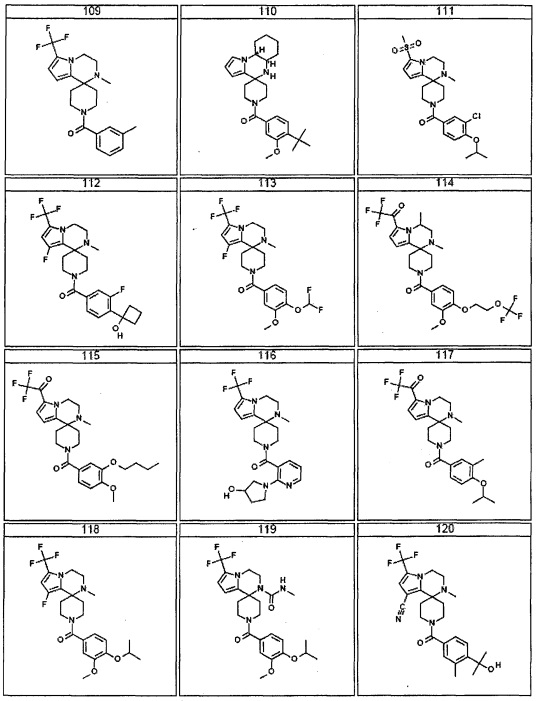
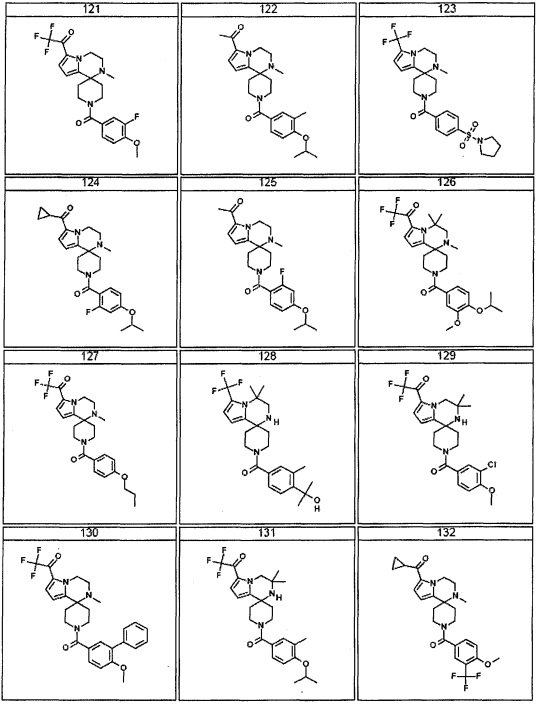

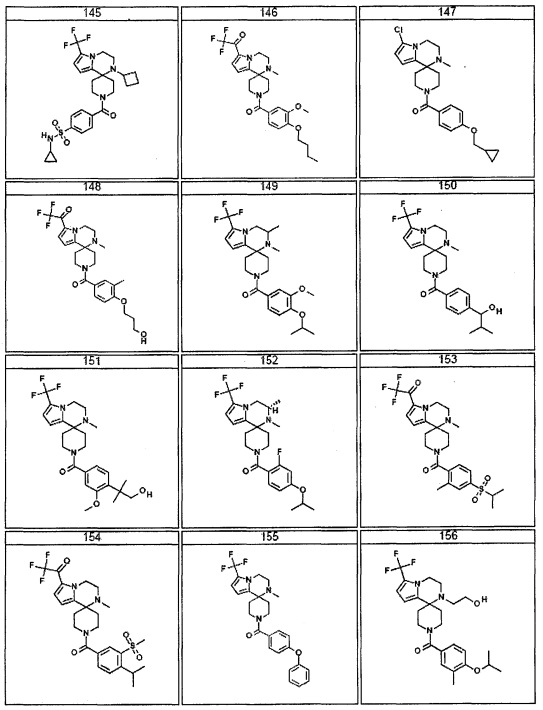
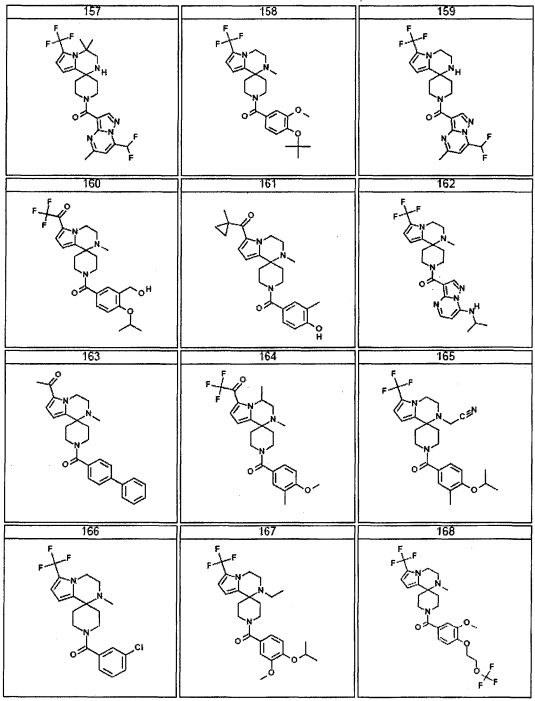

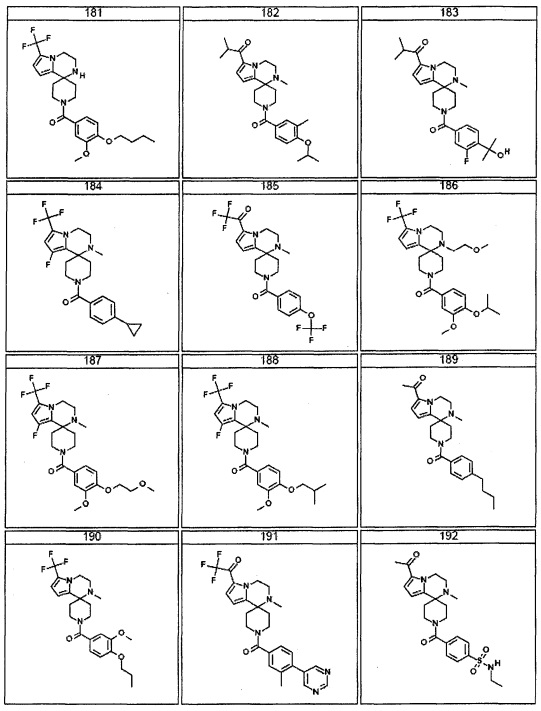
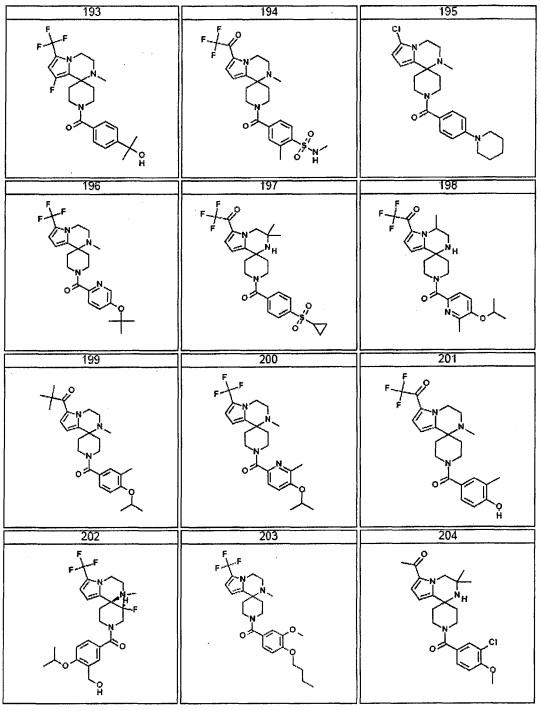
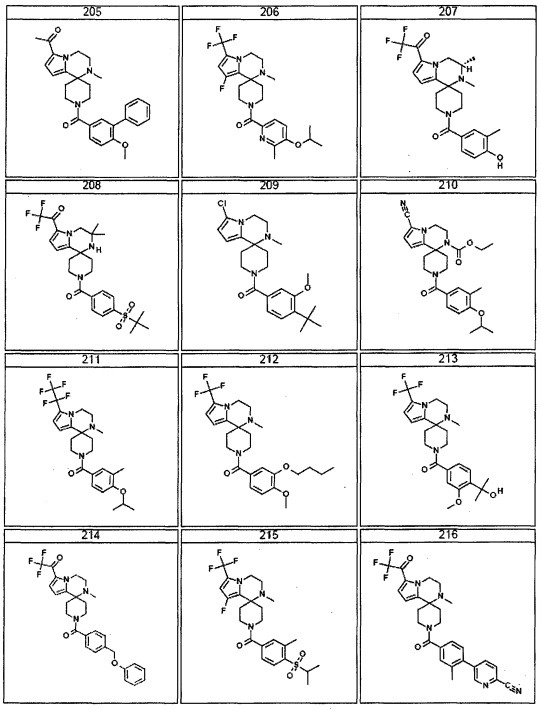
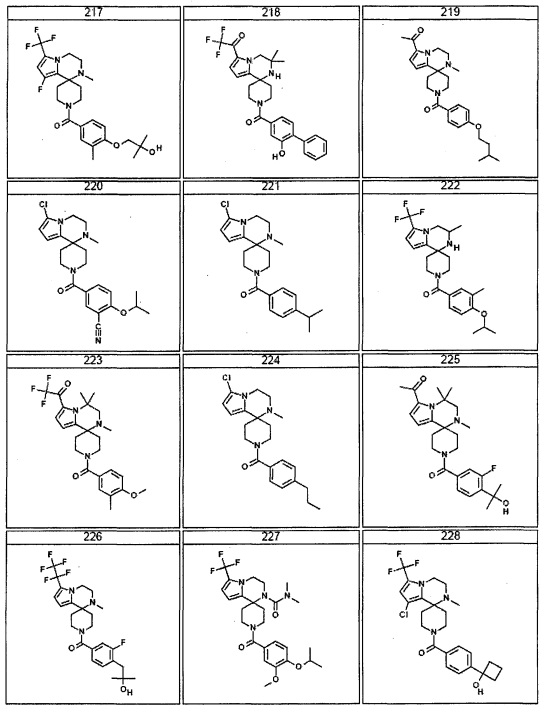
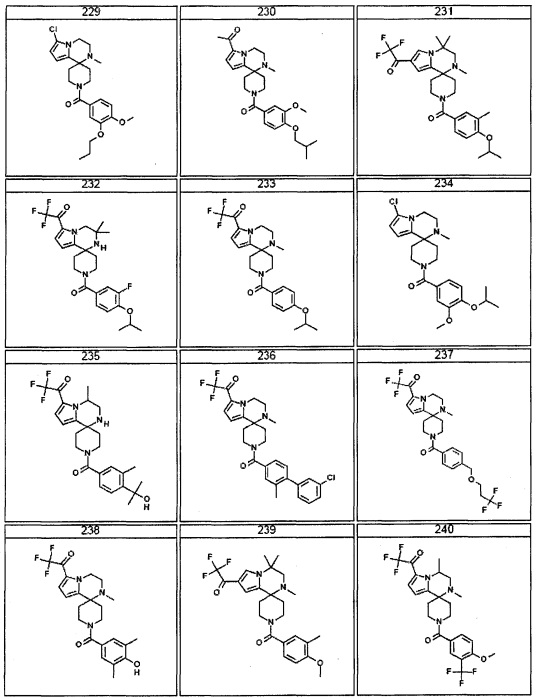
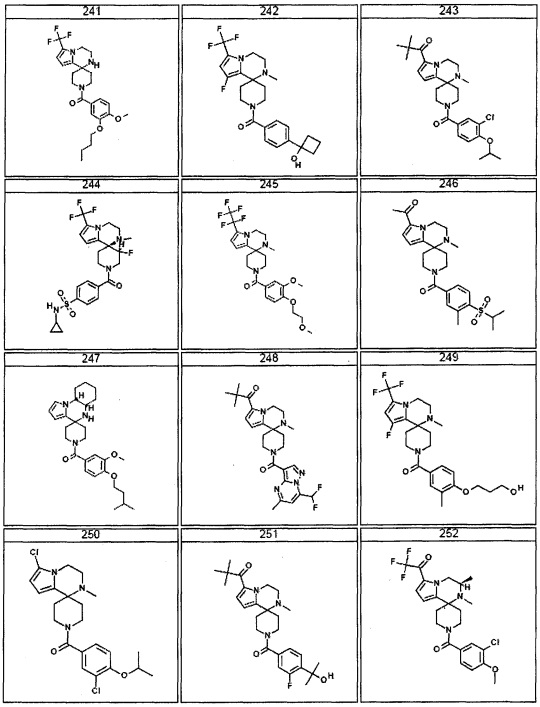



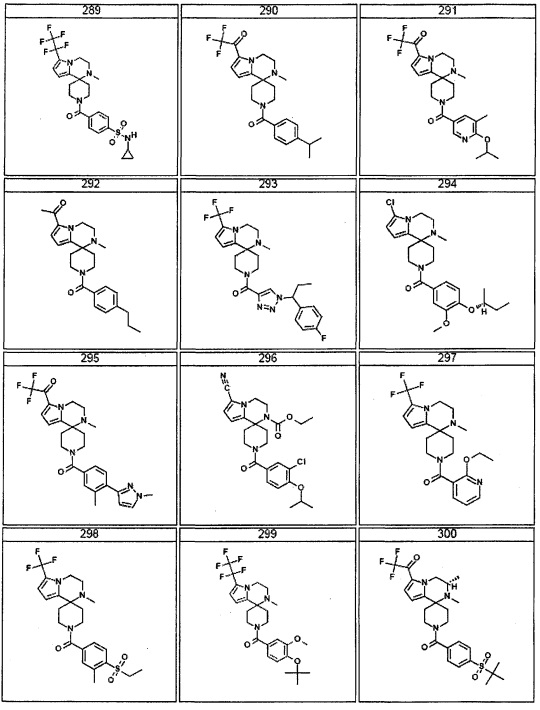
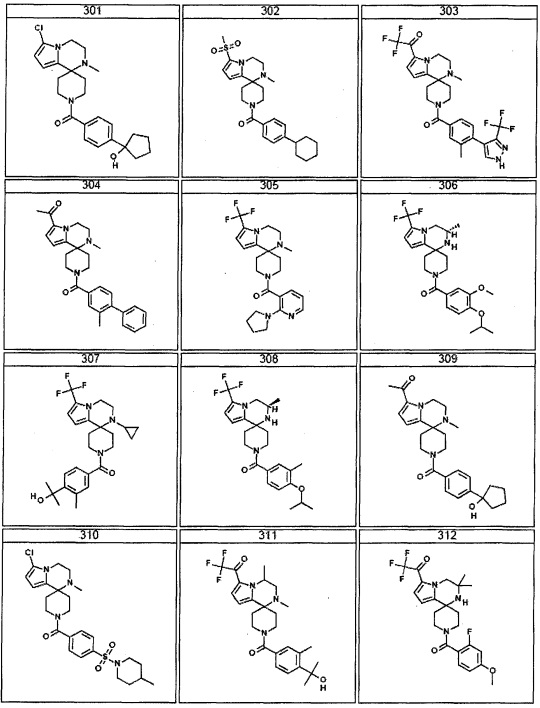
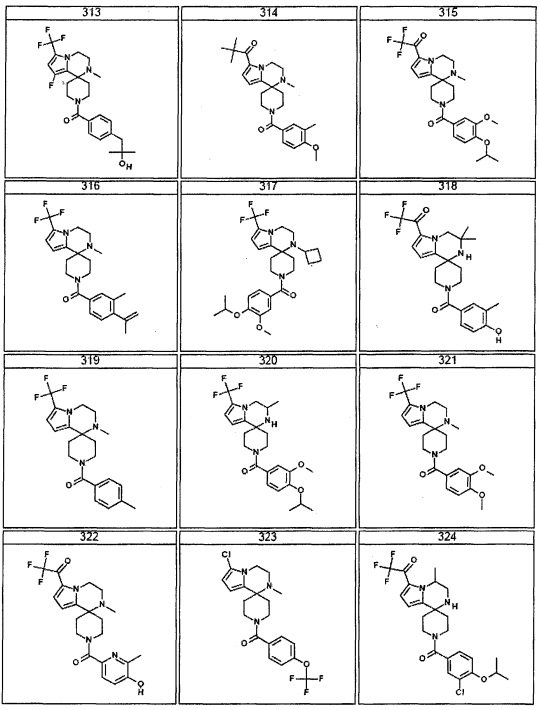
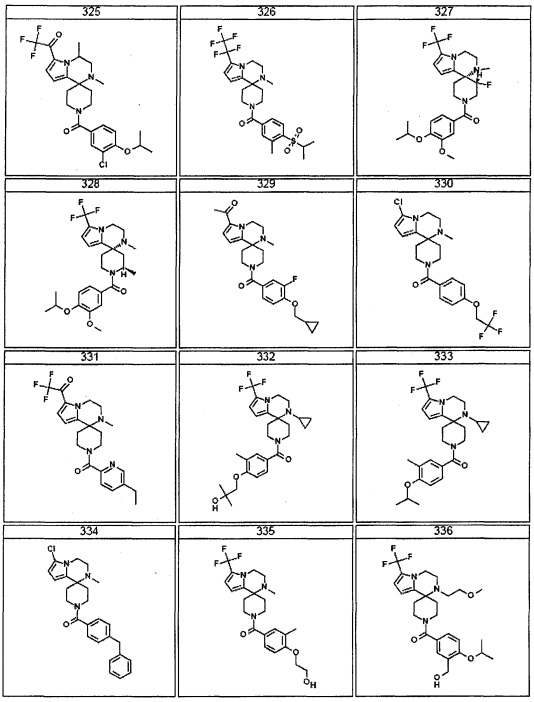
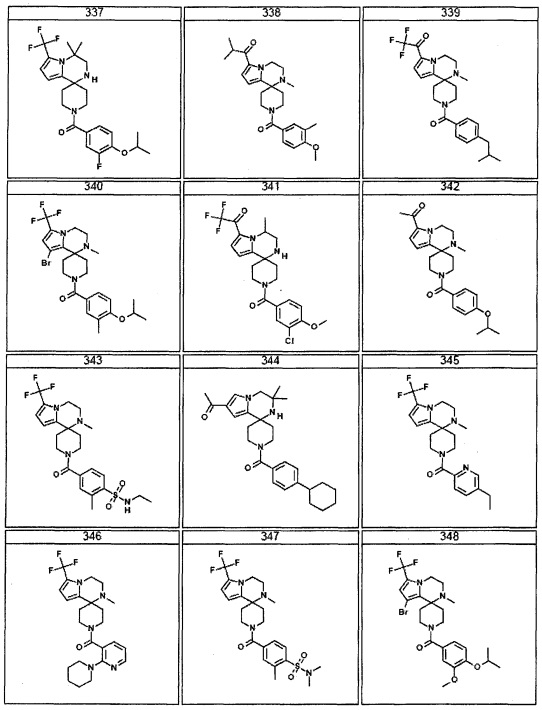
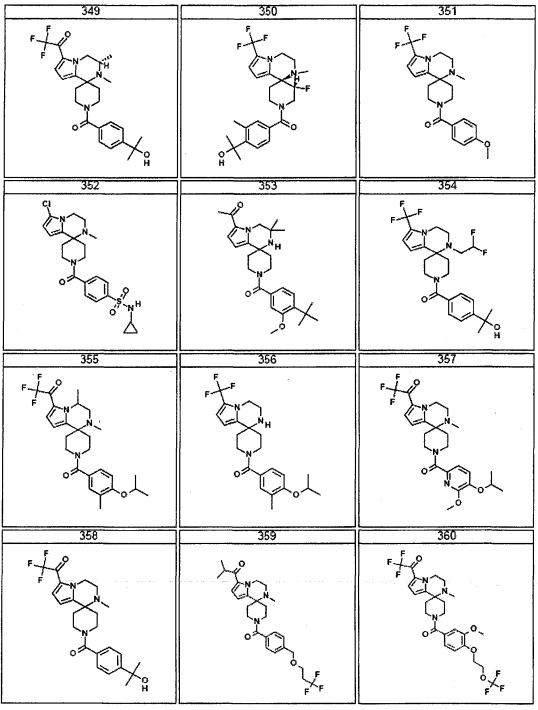
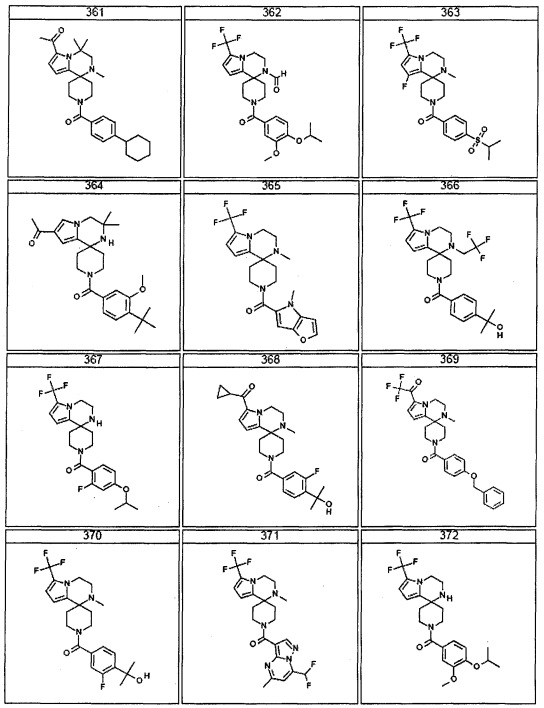


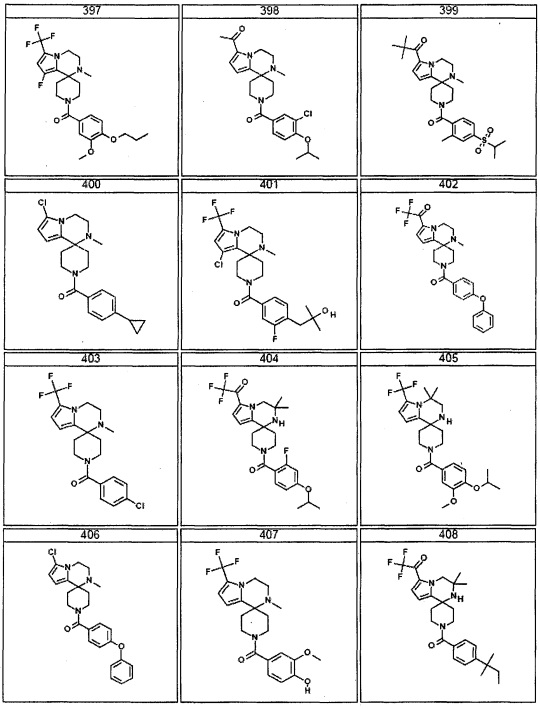
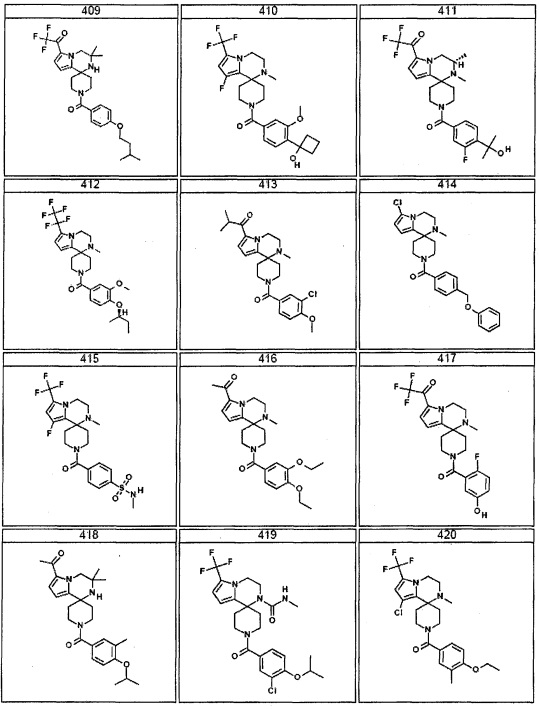
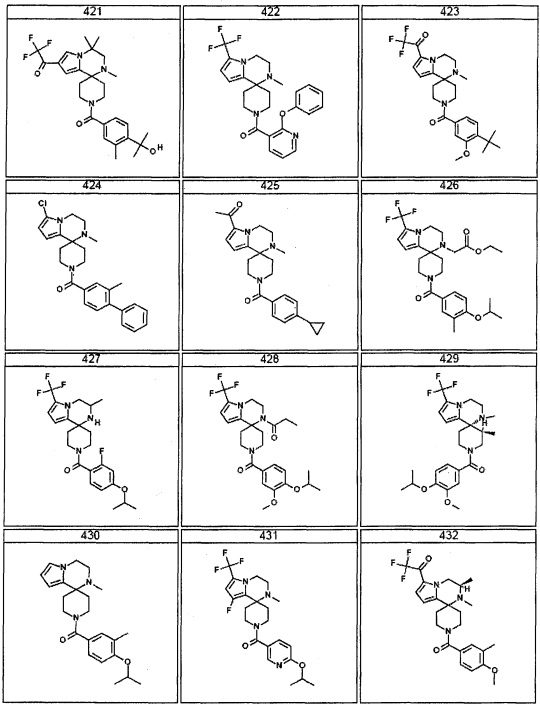


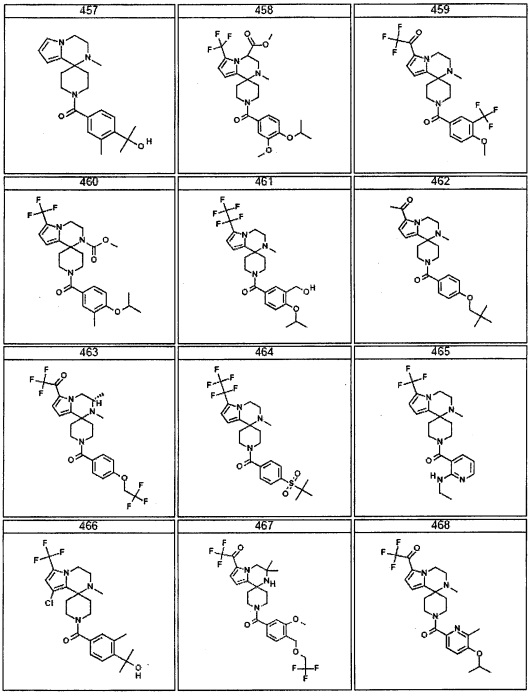
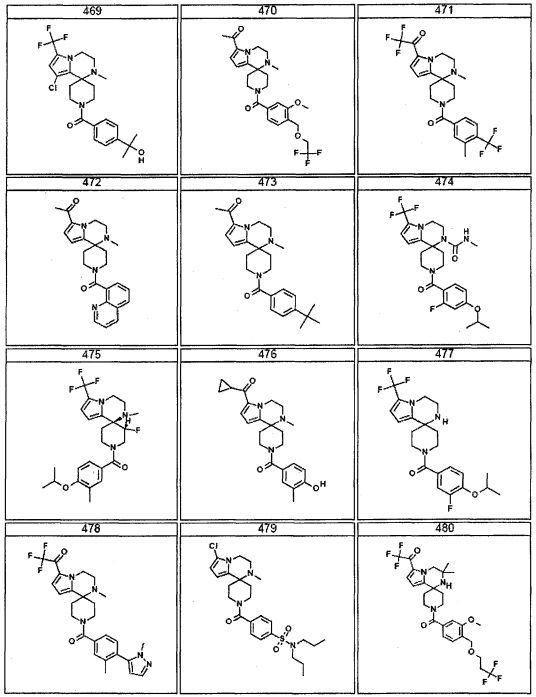
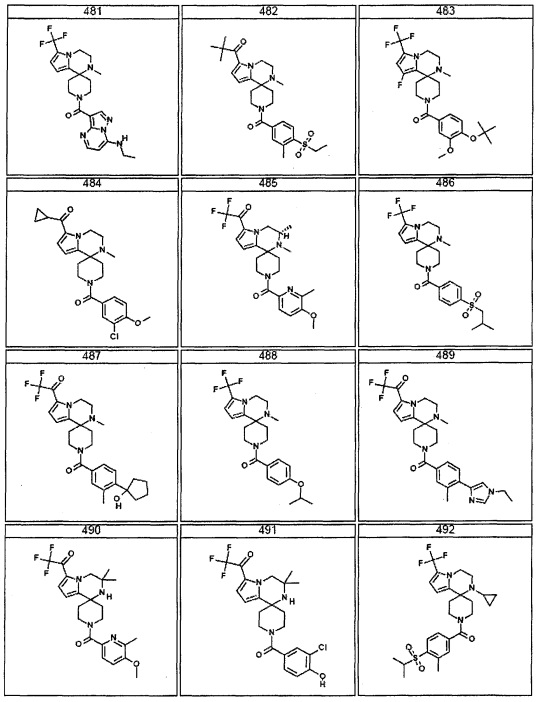
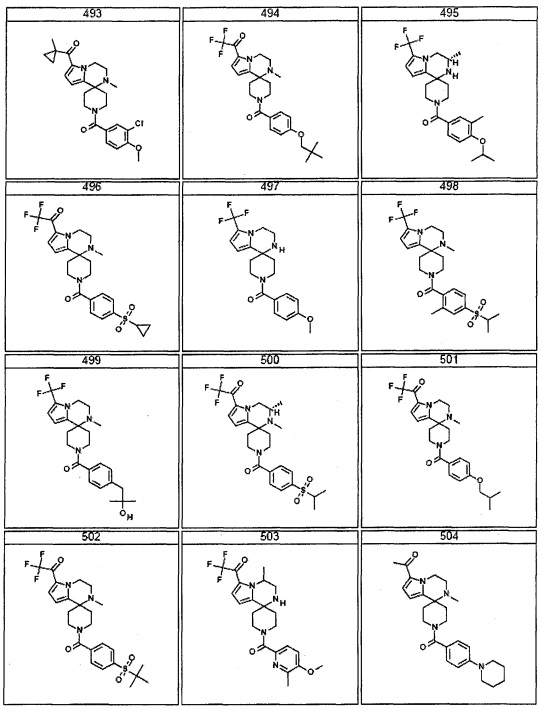
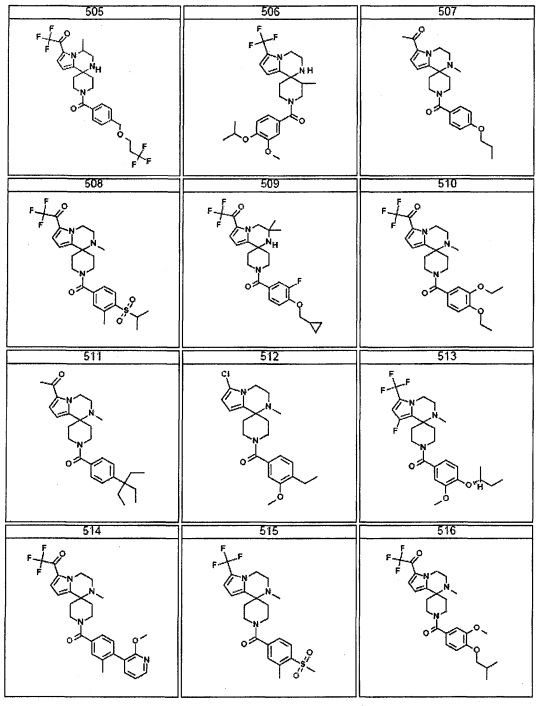
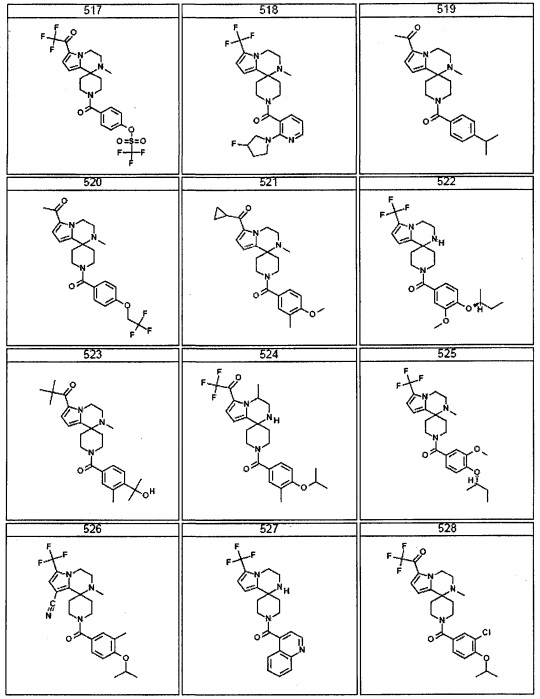
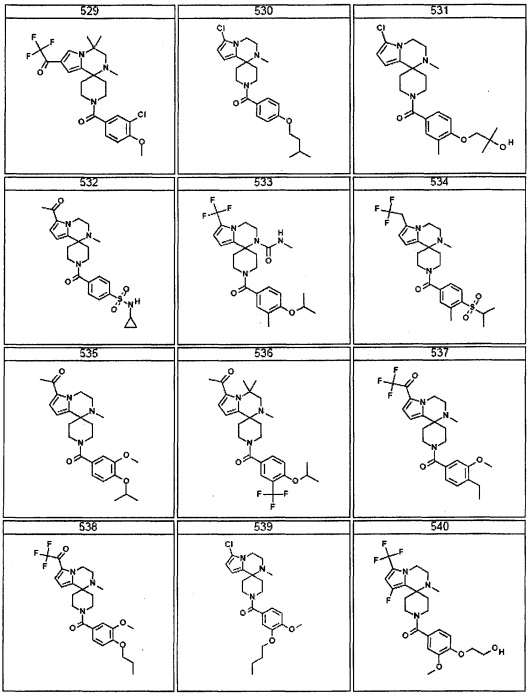
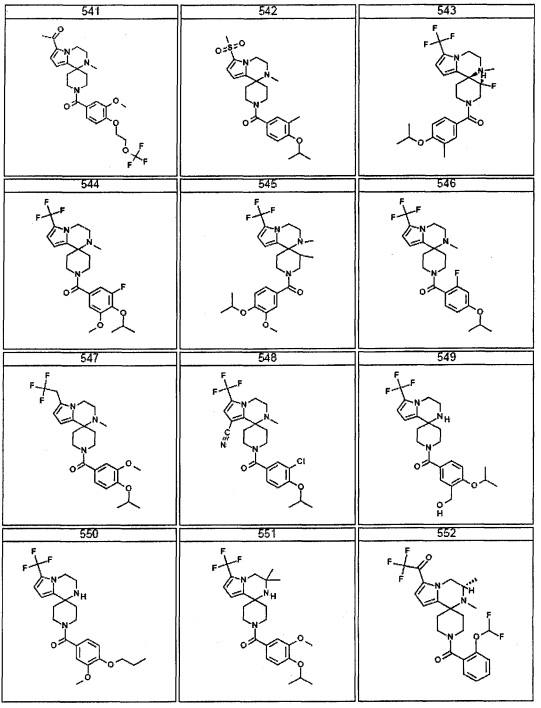
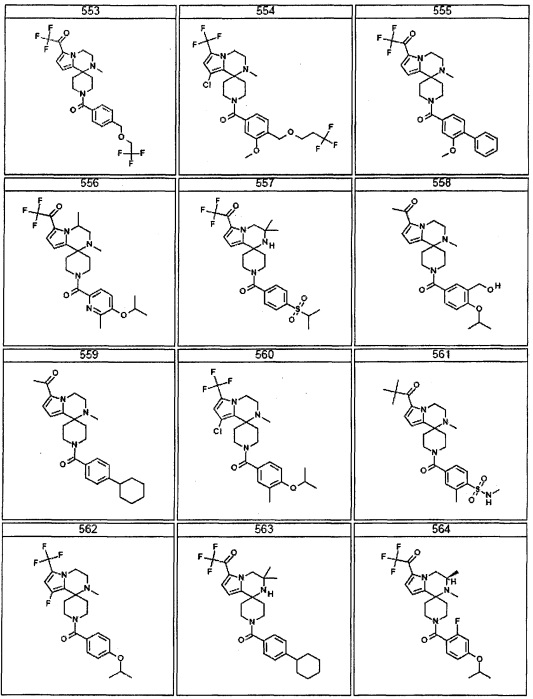
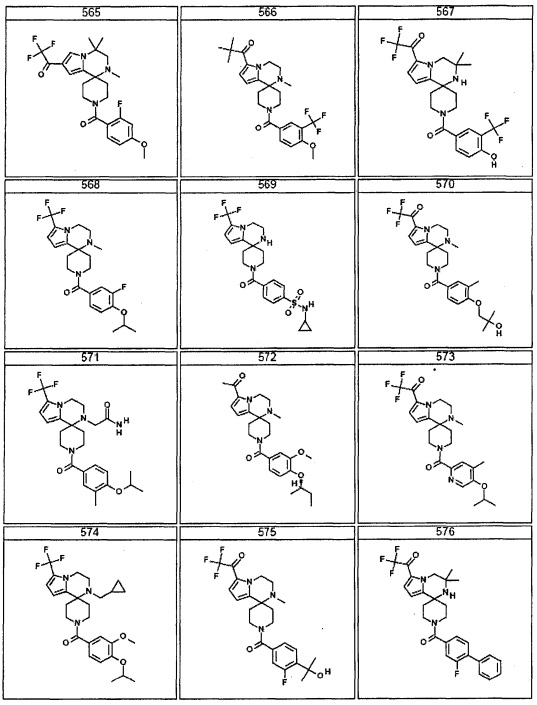
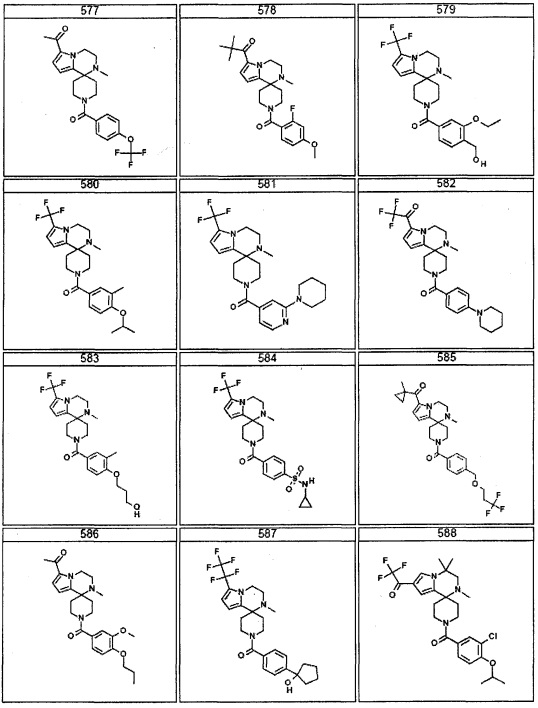
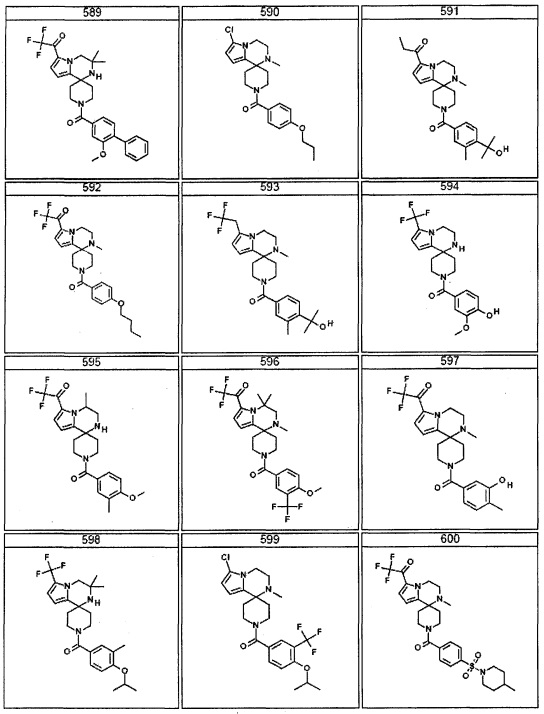

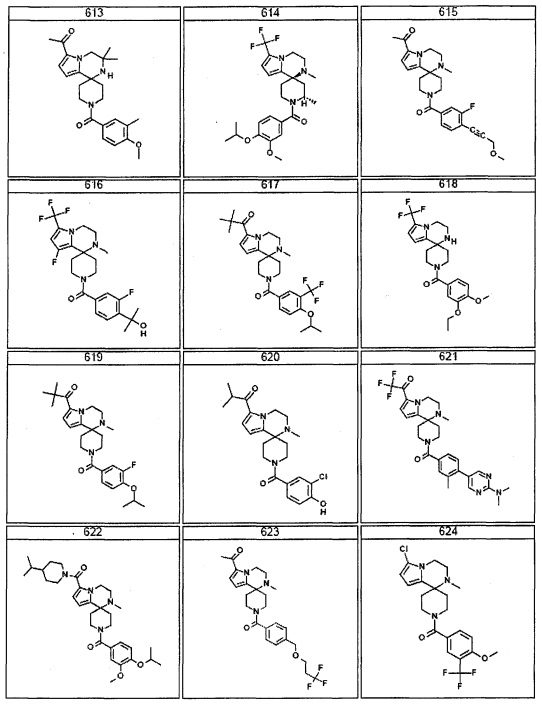
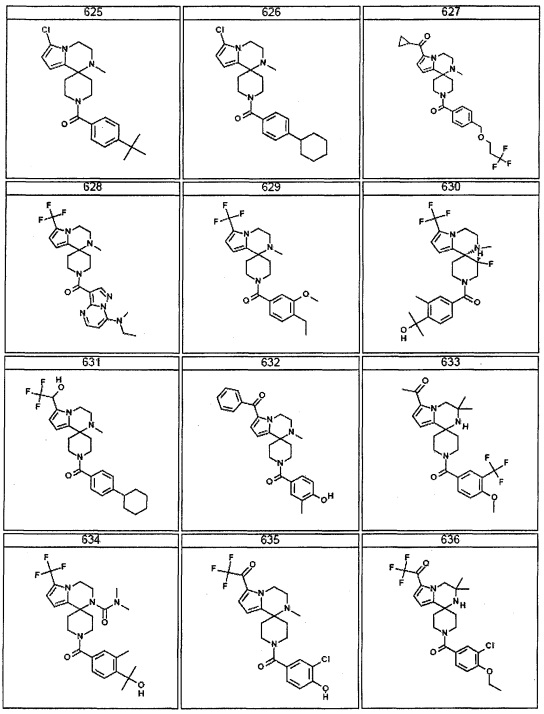
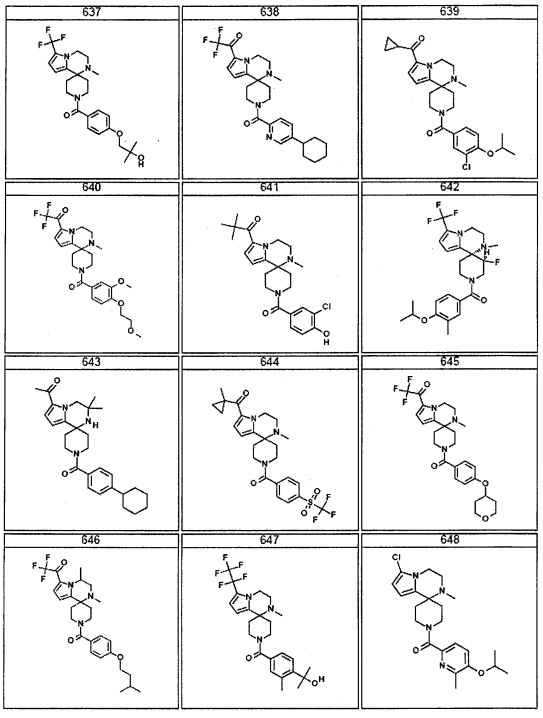
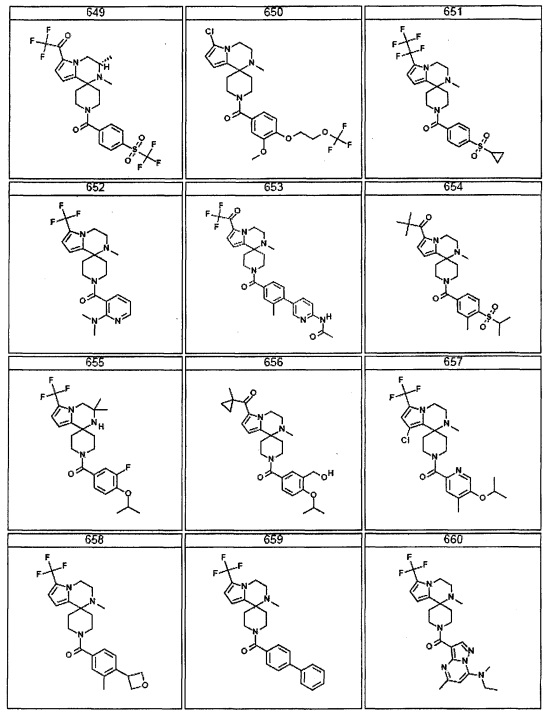
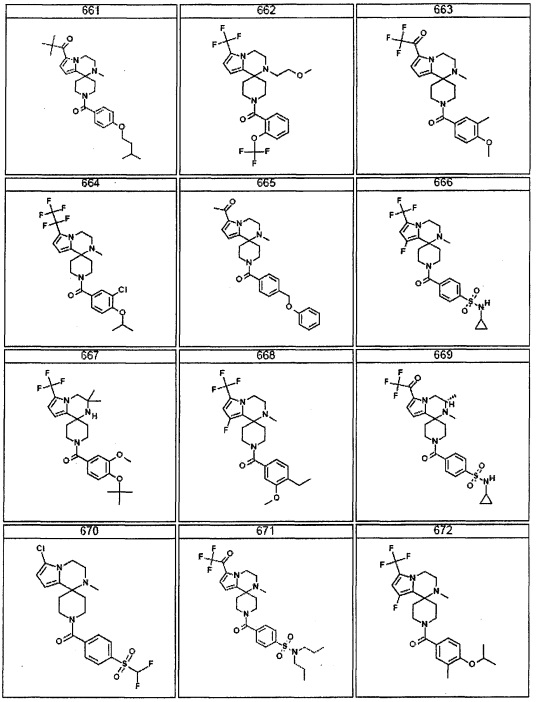


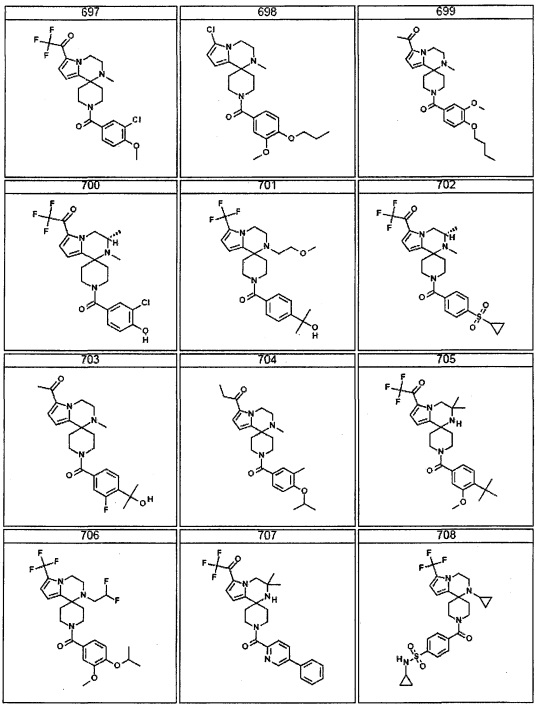
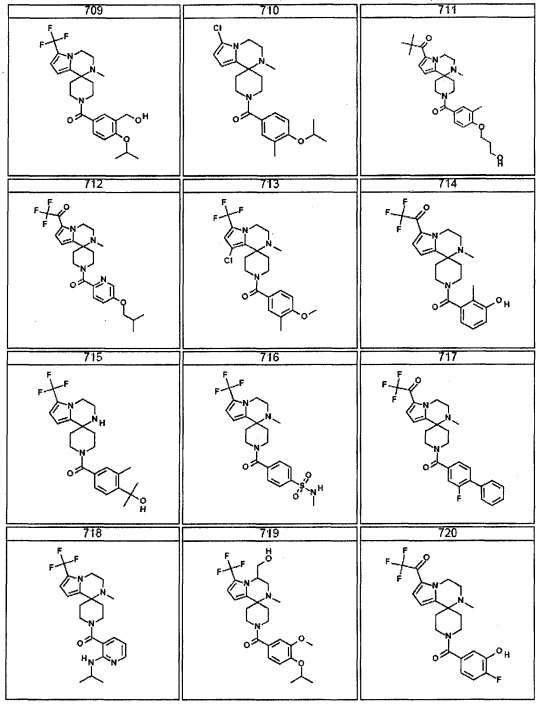
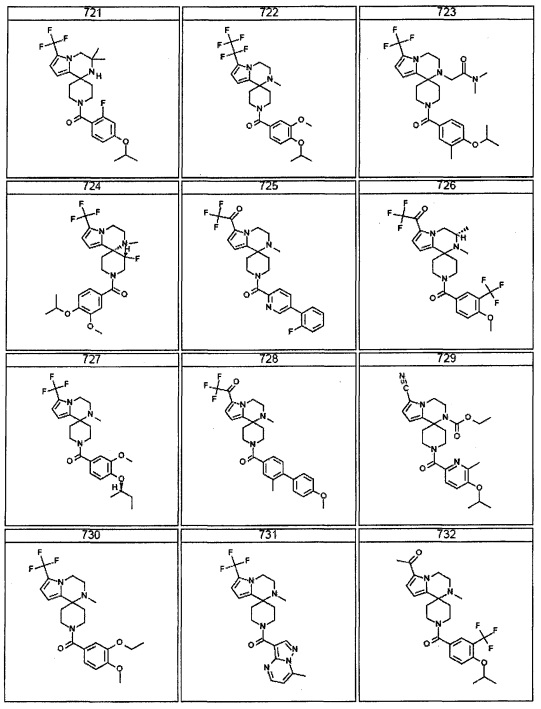
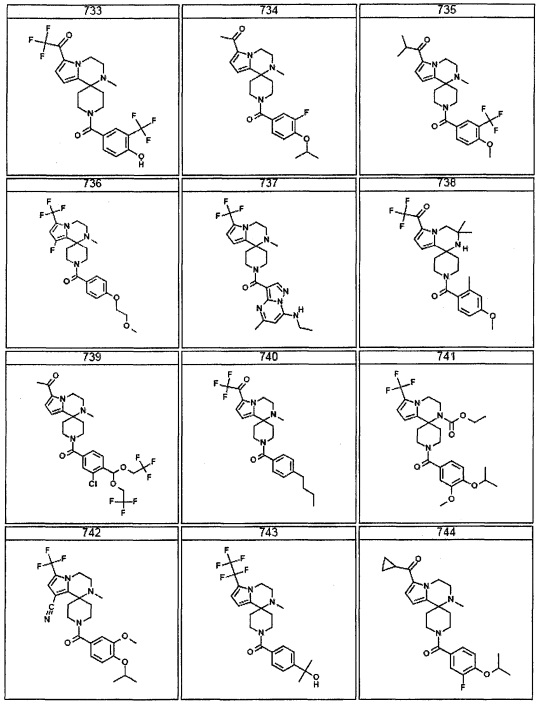

33. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении потенциалзависимого натриевого ионного канала, содержащая соединение по п. 1 и фармацевтически приемлемый носитель.
34. Способ ингибирования потенциалзависимого натриевого ионного канала:
у пациента; или
в биологическом образце;
включающий введение пациенту соединения по п. 1 или приведение биологического образца в контакт с ним.
35. Способ лечения или ослабления тяжести воспалительной боли, эпилепсии или эпилептических состояний, аритмии, остеоартритной боли, постгерпетической невралгии, раковой боли, нейропатической боли, острой воспалительной, ожоговой боли; эритромелалгической боли у субъекта, включающий введение эффективного количества соединения по п. 1.
36. Способ по п. 35, где упомянутый способ используют для лечения или ослабления тяжести боли при злокачественной опухоли бедренной кости; остеоартрита; спинального стеноза; нейропатической поясничной боли; хронической и острой нейропатической боли, постгерпетической невралгии; диабетической нейропатии; HIV-ассоциированной нейропатии; тригеминальной невралгии; нейропатии Шарко-Мари-Тута; наследственных сенсорных нейропатий; нейропатической боли, индуцированной химиотерапией; нейропатической боли, индуцированной лучевой терапией; острой воспалительной и ожоговой боли; эритромелалгической боли.
37. Соединение формулы

или его фармацевтически приемлемая соль.
38. Соединение формулы
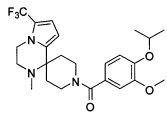
39. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении потенциалзависимого натриевого ионного канала, содержащая соединение по п. 37 или 38 и фармацевтически приемлемый носитель.
40. Способ ингибирования потенциалзависимого натриевого ионного канала:
у пациента; или
в биологическом образце;
включающий введение пациенту соединения по п. 37 или 38 или приведение биологического образца в контакт с ним.
41. Способ лечения воспалительной боли, эпилепсии или эпилептических состояний, аритмии, остеоартритной боли, постгерпетической невралгии, раковой боли, нейропатической боли, острой воспалительной, ожоговой боли; эритромелалгической боли у субъекта, включающий введение эффективного количества соединения по п. 37 или 38.
42. Способ лечения или ослабления тяжести боли при злокачественной опухоли бедренной кости; остеоартрита; спинального стеноза; нейропатической поясничной боли; хронической и острой нейропатической боли, постгерпетической невралгии; диабетической нейропатии; HIV-ассоциированной нейропатии; тригеминальной невралгии; нейропатии Шарко-Мари-Тута; наследственных сенсорных нейропатий; нейропатической боли, индуцированной химиотерапией; нейропатической боли, индуцированной лучевой терапией; острой воспалительной и ожоговой боли; эритромелалгической боли у субъекта, включающий введение эффективного количества соединения и фармацевтической композиции соединения по п. 37 или 38.
| WO 2010151595 A1, 29.12.2010 | |||
| Способ изготовления из листового асбестоцемента тонкостенных строительных плит и тому подобных изделий с термоизоляционным заполнением | 1948 |
|
SU75116A1 |
| RU 99112573 A, 27.04.2001 | |||
| RU 2009123134 A, 27.01.2011. | |||

