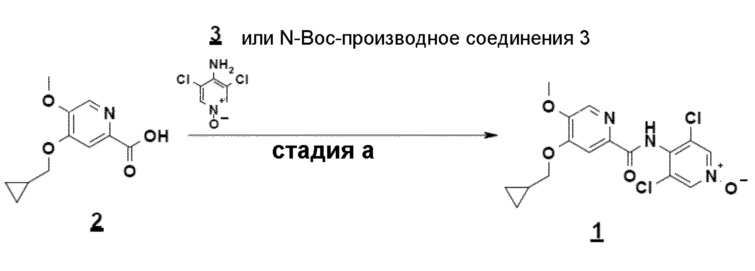
Настоящее изобретение относится к новому способу получения соединения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида, а также некоторых новых синтетических промежуточных соединений, полезных в этом способе получения.
Соединение 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамид и способы получения описаны в документе WO 95/04045. Объект настоящего изобретения составляет новый надежный способ синтеза, который может быть адаптирован к промышленному масштабу, для производства заданного соединения в больших количествах и для получения этого соединения в достаточно чистой форме, т.е. без образования нежелательных побочных продуктов.
Настоящее изобретение относится к новому способу получения соединения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида (соединения 1), отвечающего следующей формуле:
 ,
,
в основной или гидратной форме, или в форме фармацевтически приемлемых солей.
Документ WO 95/04045 описывает несколько общих путей подхода к получению серии соединений, соответствующих общей формуле (I), которая, согласно конкретной комбинации заместителей, может определять соединение формулы 1.
Согласно первому подходу, описанному в WO 95/04045, по приведенной ниже схеме, соединение общей формулы (I) может быть получено взаимодействием соединения формулы (II) с соединением формулы (III).
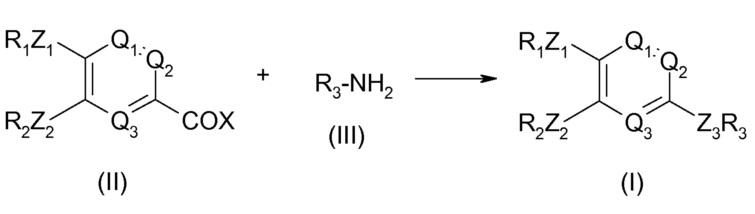
В этой схеме R1, R2, R3, Q1, Q2, Q3, Z1 и Z2 являются такими, как описано в WO 95/04045. X означает атом галогена. Взаимодействие происходит в присутствии основания, такого как гидрид щелочного металла, такой как гидрид натрия, или амин, предпочтительно третичный амин, такой как триэтиламин или пиридин, необязательно в инертном растворителе, таком как дихлорметан или диметилформамид, или простой эфир, такой как простой диэтиловый эфир или тетрагидрофуран.
Альтернативно, соединение общей формулы (I) может быть получено взаимодействием соединения формулы (II) с соединением следующей формулы (IV) R4CONHR3, в которой R4 означает алкильную или циклоалкильную группу, содержащую до 5 атомов углерода.
Соединение общей формулы (II) может быть получено из соединения формулы (XIX), приведенной ниже:
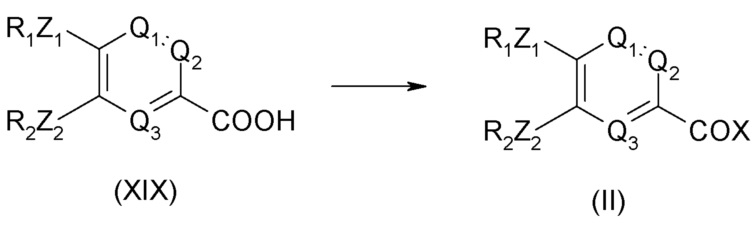
Соединение общей формулы (XIX) может быть получено из соединения формулы (XVIII), приведенной ниже:
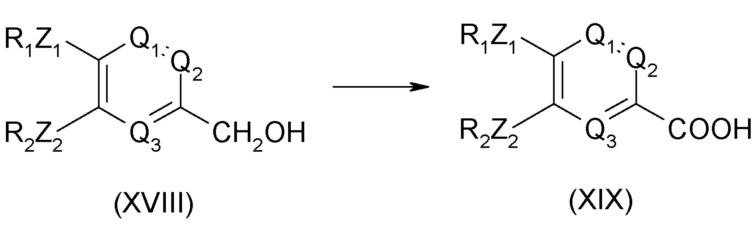
Условия технологического процесса синтеза соединений формулы (I), как они описаны в документе WO 95/04045, являются ограничительными и не могут быть перенесены на промышленные масштабы из соображений безопасности, и, кроме того, они сопровождаются образованием треххлористого примесного компонента соединения 1, который трудно удалить.
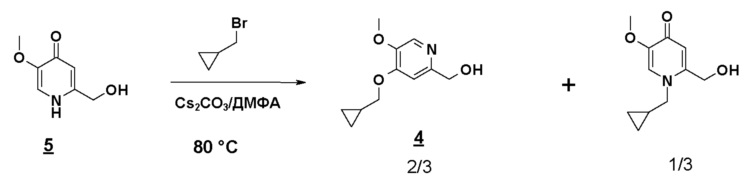
Соединение формулы 1 может также быть получено по 11-стадийному способу, иллюстрируемому схемой 1. Согласно этому способу конечное соединение получают на стадии алкилирования сложного дигидропиридинонового эфира (5) с помощью бромметилциклопропана, с последующим, после омыления, амидированием моноацетилированным N-оксидом 3,5-дихлораминопиридина.
Схема 1
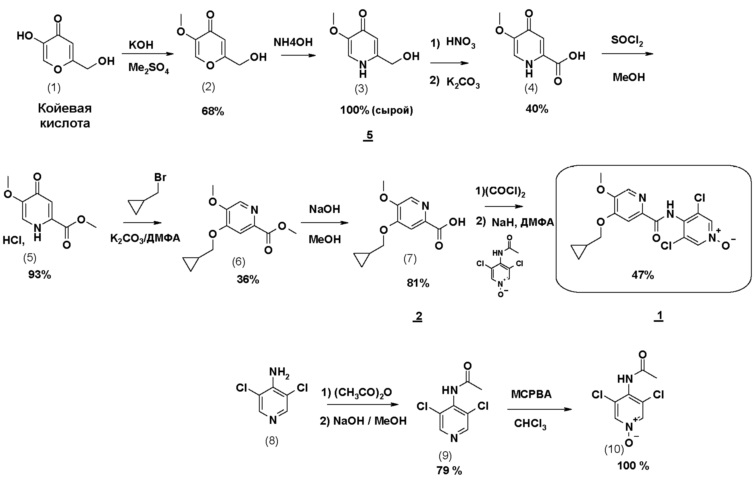
Различные особенности настоящего изобретения дают возможность оптимизировать способ изготовления соединения формулы 1, сокращая число стадий, делая способ промышленно применимым в крупных масштабах и снижая количество примесей.
Согласно настоящему изобретению способ синтеза соединения формулы 1 включает стадии a)-c), представленные приведенной ниже схемой 2.
Схема 2

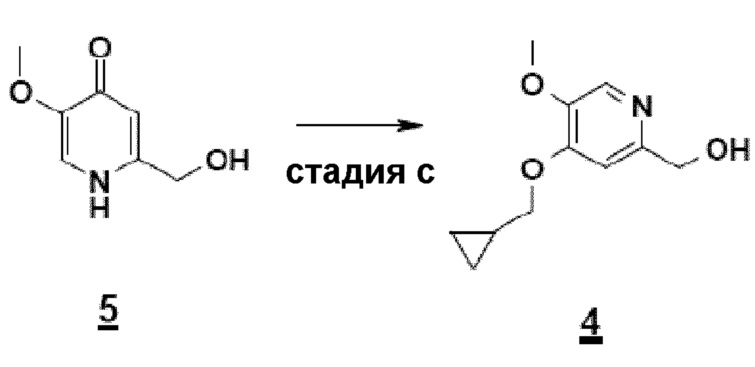
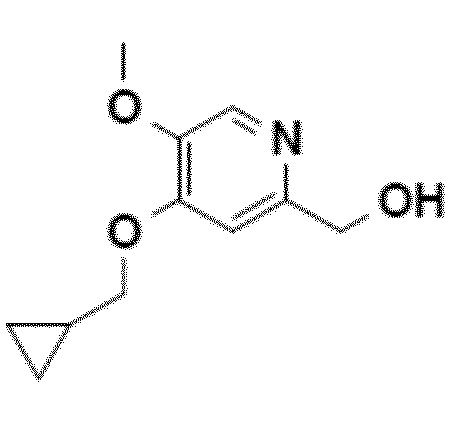
Согласно первому аспекту настоящего изобретения синтез соединения формулы 1 упрощен осуществлением прямого O-алкилирования соединения 5 (2-гидроксиметил-5-метокси-4-пирид-(1H)-она) метилциклопропановым производным, в частности бромидом метилциклопропана (схема 3). Это дает возможность избежать последовательности из метилирования и деметилирования карбоксилата (схема 1) и, таким образом, уменьшить число стадий, приводящих к соединению 2 (4-циклопропилметокси-5-метоксипиридин-2-карбоновой кислоте).
Схема 3
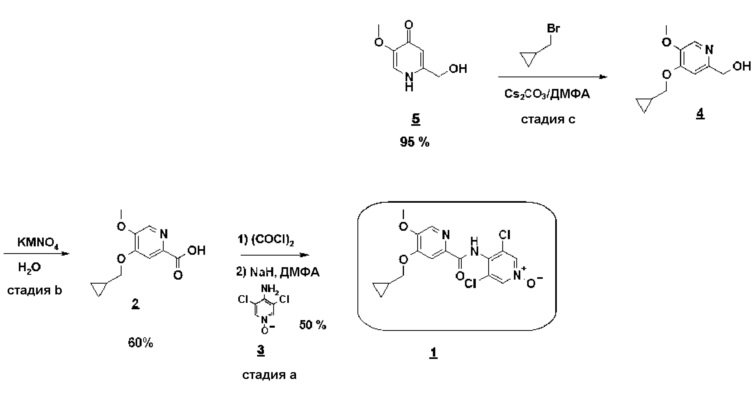
Синтез по схеме 3 также ставит задачу селективности между N-алкилированием и O-алкилированием этого типа дигидропиримидинонового аналога 5. Заявители настоящего изобретения показали, что, как функция условий технологического процесса, возможна ориентация взаимодействия на значительно преобладающее образование продукта O-алкилирования 4. В частности, заявители продемонстрировали, что высокотемпературное нагревание, например при 80°C, смеси, как указано для стадии c) схемы 3, позволяет получить больше 60% продукта O-алкилирования. Заявители также показали, что этот выход возрастает, когда бромид метилциклопропана вводят горячим в реакционную смесь, предварительно нагретую до высокой температуры, например до 80°C. В последнем способе равновесие "енолят/амид" явно сдвинуто в сторону образования алкоксида, приводя почти исключительно к продукту O-алкилирования 4.
Этот новый путь синтеза (схема 3), таким образом, позволяет получать соединение формулы 1 при сокращении числа стадий от 11 до 7 и с общим выходом 15% вместо 3,5% (схема 1).
Другое преимущество способа по настоящему изобретению состоит в том, что повышение селективности стадии O-алкилирования дигидропиридинонового промежуточного соединения (соединения 5) ведет к сравнительно чистому продукту, который не требует очистки хроматографией. Эти результаты делают синтез совместимым с возможными совокупностями функциональных единиц (OU).
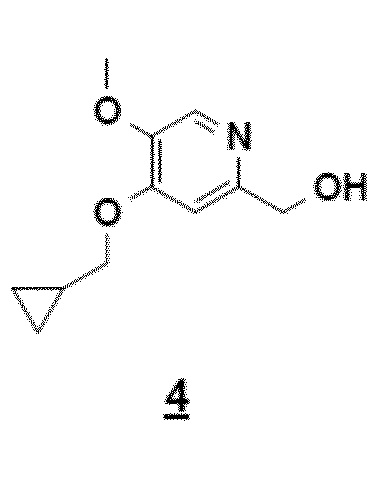
Согласно этому первому аспекту настоящее изобретение также относится к соединению 4, CH2OH-промежуточному соединению, 4-циклопропилметокси-5-метокси-2-гидроксиметилпиридину, полезному в качестве промежуточного продукта.
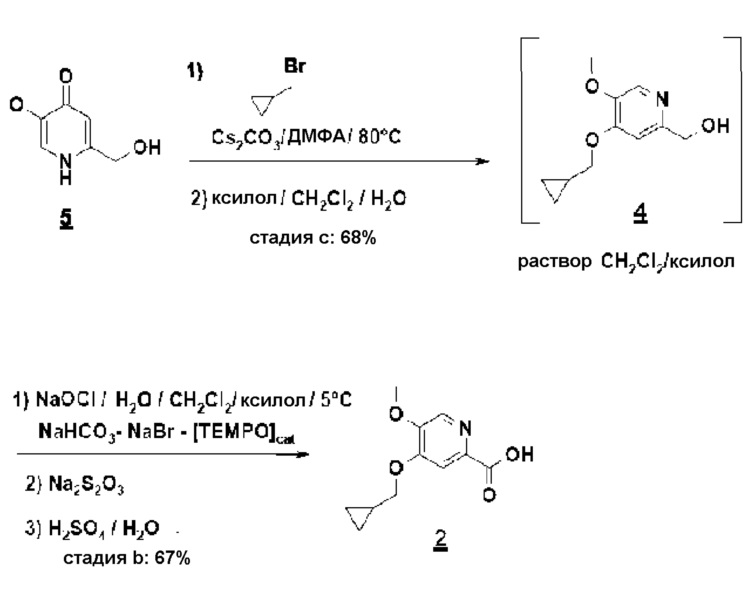
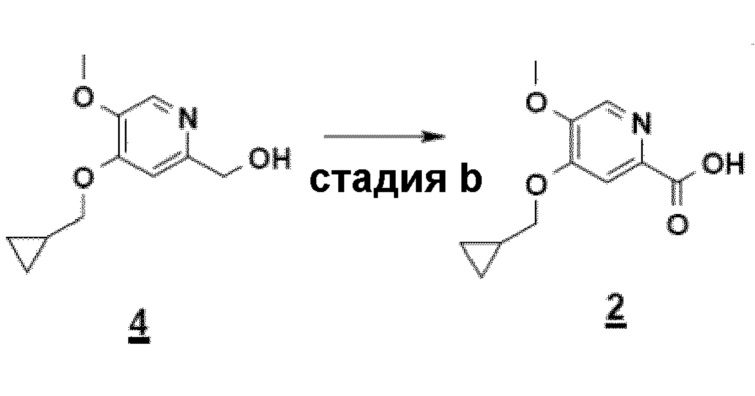
Согласно второму аспекту настоящего изобретения, заявители также оптимизировали стадию b) способа (схема 4), ведущую от соединения 4 к соединению 2 посредством "однореакторного" способа в соответствии с приведенной ниже реакцией.
Схема 4
Согласно третьему аспекту настоящего изобретения, для стадии a) были разработаны новые альтернативные способы, предназначенные для промышленного производства.
Первый способ включает реакцию амидирования с циклическим тримером н-пропилфосфонового ангидрида (T3P) между кислотой и N-оксидом (схема 5):
Схема 5

T3P представляет собой промышленно выпускаемый реагент (Clariant), который коммерчески доступен в чистой форме или в виде раствора (например, в диметилформамиде (ДМФА), этилацетате или любом другом совместимом растворителе).
При этом в присутствии 4-диметиламинопиридина (DMAP/по меньшей мере 0,2 эквивалента), приблизительно при 75°C весь диацил превращается в конечный продукт, который выделяют простым осаждением в водной среде и перекристаллизацией из изопропилового спирта с выходом около 80% и микроаналитической степенью чистоты (схема 6).
Использование реагента T3P и катализа с помощью DMAP обеспечивает качество, достаточное для изготовления партий, предназначенных для применения в медицинских целях.
Схема 6

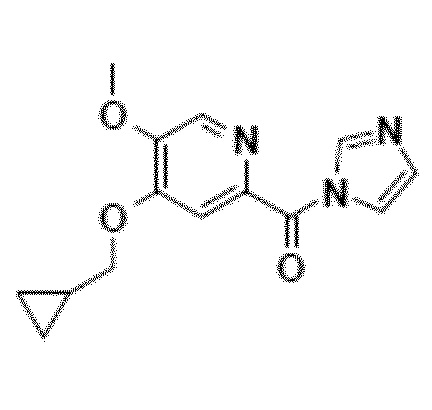
Альтернативно, второй способ включает реакцию сочетания между кислотным имидазолидом и N-оксидом (схема 7).
Схема 7
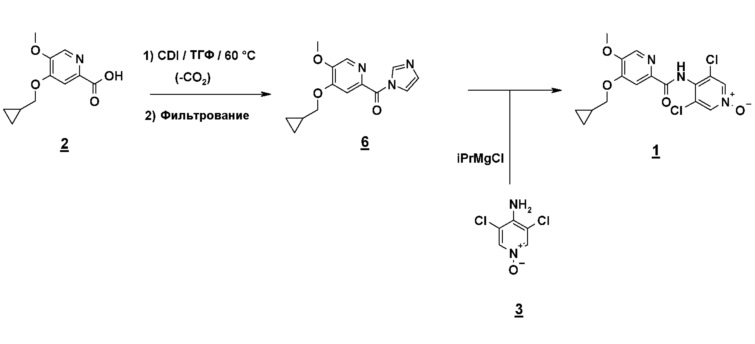
Этот очень удобный метод активации позволяет избежать использования коррозионных реагентов, таких как тионилхлорид, и работать в менее жестких условиях исключения воды, поскольку имидазолид менее чувствителен к воде, чем соответствующий хлорангидрид кислоты.
Имидазолид получают нагреванием кислоты в присутствии карбонилдиимидазола (CDI) в тетрагидрофуране (ТГФ) или метил-ТГФ и выделяют холодным фильтрованием. Образовавшийся имидазол остается растворимым и таким образом удаляется. Анион N-оксида получают депротонированием, используя магнийорганический реагент. Подходящим для применения является хлорид циклогексилмагния (CyMgCl) или хлорид изопропилмагния (iPrMgCl), и эти реагенты поставляются дешево в промышленном масштабе. Из соображений безопасности CyMgCl предпочтительней, чем iPrMgCl, который во время депротонирования N-оксида приводит к образованию пропана, воспламеняющегося газа.
Сочетание с имидазолидом осуществляют нагреванием до температуры кипения с обратным холодильником в ТГФ (4-5 часов). Ожидаемый продукт экстрагируют после водного гидролиза (водный NH4Cl), который предпочтителен для фосфатов вследствие риска соосаждения фосфатов магния) и очищают перекристаллизацией (iPrOH или н-PrOH).
Другой объект настоящего изобретения относится к имидазолидному промежуточному соединению формулы 6, имеющему следующую формулу:
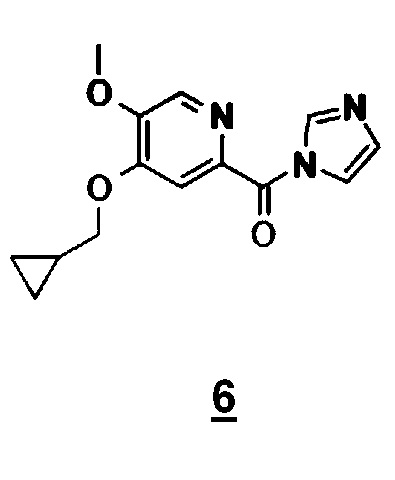
Альтернативно третий способ включает амидирование карбодиимидом соединения формулы 2 и N-Boc-производного соединения 3 (N-Boc-дихлорпиридоксида). Соединение формулы 1 получают гидролизом Boc-группы в кислотной среде (схема 8).
Схема 8
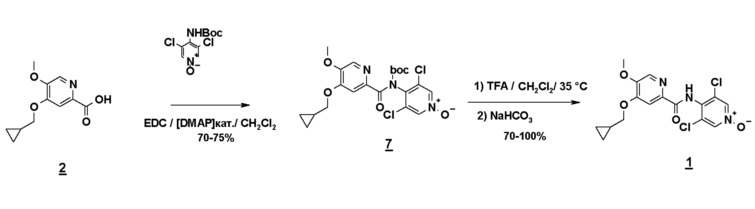
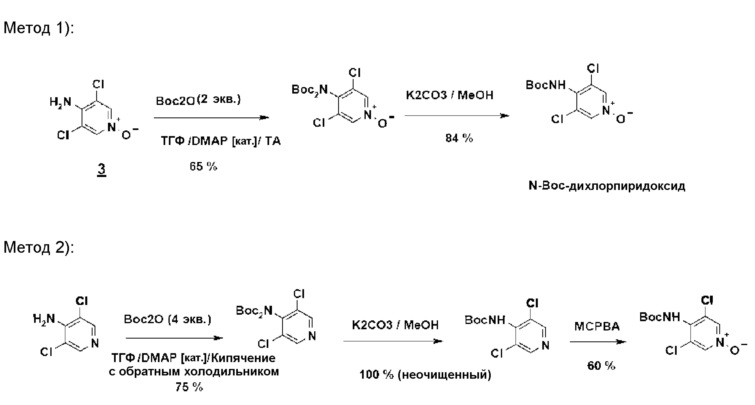
Предшественник N-Boc-дихлорпиридоксида может быть синтезирован следующими двумя методами (схема 9):
1) из дихлорпиридоксида посредством обработки Boc 20 (2 эквивалента/комнатная температура/3 часа) с последующим снятием монозащиты путем метанолиза: этот метод предпочтительней метода 2), поскольку дихлорпиридоксид значительно более реакционноспособен, чем аминодихлорпиридин по отношению к Boc 20.
2) из 4-амино-3,5-дихлорпиридина путем обработки Boc 20 (4 эквивалента/нагревание до температуры кипения ТГФ/30 часов), снятием монозащиты путем метанолиза и затем N-окислением мета-хлорбензойной кислотой.
Схема 9
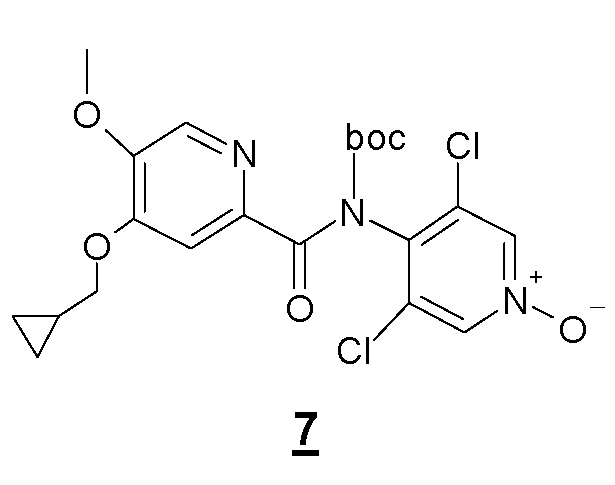
Другой объект настоящего изобретения относится к промежуточному соединению формулы 7, N-Boc-производному соединения 1, имеющему следующую формулу:
Согласно изобретению способ синтеза соединения формулы 1 включает стадии a)-c), представленные приведенной ниже схемой 2.
Схема 2
Согласно изобретению способ синтеза соединения формулы 1 отличается тем, что включает следующие стадии:
- стадию a): реакцию сочетания, необязательно в присутствии катализа с помощью DMPA,
с циклическим тримером н-пропилфосфонового ангидрида T3P или
с карбонилдиимидазолом (CDI), с соединением 3 или его N-Boc-производным;
- стадию b): "однореакторную" реакцию окисления;
- стадию c) O-алкилирования: взаимодействия соединения 2-гидроксиметил-5-метокси-4-пирид-(1H)-она формулы 5 с метилциклопропановым производным, таким как бромид метилциклопропана, и основанием, таким как карбонат цезия, в полярном апротонном растворителе, таком как ДМФА (диметилформамид).
Альтернативно стадии b) и c) могут быть осуществлены без выделения соединения 4.
Определения
Термин "основание" означает магнийорганические реагенты, такие как хлорид циклогексилмагния, хлорид изопропилмагния или хлорид бензилмагния; гексиллитий; гидриды или карбонат цезия.
Термин "полярный апротонный растворитель" означает простые эфиры, такие как ТГФ (тетрагидрофуран), Me-ТГФ (метилтетрагидрофуран), DME (диметоксиэтан), MTBE (простой метил-трет-бутиловый эфир) или диоксан, хлорированные растворители, такие как дихлорметан, 1,2-дихлорэтан, нитрилы, такие как ацетонитрил, кетоны, такие как ацетон, метилэтилкетон или метилизобутилкетон, амиды, такие как ДМФА (диметилформамид), DMAC (диметилацетамид) или NMP (N-метилпирролидон).
Термин "аполярный апротонный растворитель" означает ароматические растворители, такие как толуол, ксилол или хлорбензол, и сложные эфиры, такие как этилацетат или бутилацетат.
Термин "полярный протонный растворитель" означает спирты, такие как метанол, этанол, изопропанол или бутанол.
Термин "сильная кислота" означает хлористоводородную кислоту, серную кислоту, сульфоновую кислоту, метансульфоновую кислоту, пара-толуолсульфоновую кислоту, фосфорную кислоту или уксусную кислоту.
Другой объект настоящего изобретения относится к способу получения соединения формулы 1, отличающемуся тем, что включает по меньшей мере одну из стадий a), b) или c), как описано выше и ниже, или отличающемуся тем, что включает все стадии a)-c).
Другой объект настоящего изобретения относится к новому способу получения соединения формулы 1, отличающемуся тем, что стадия c) представляет собой прямое O-алкилирование.
Более предпочтительно другой объект настоящего изобретения относится к способу синтеза соединения формулы 1, включающему стадию c) или включающему стадии a)-c), отличающемуся тем, что алкилирующий агент представляет собой бромид метилциклопропана. Более предпочтительно другой объект настоящего изобретения относится к способу синтеза соединения формулы 1, включающему стадию c) или включающему стадии a)-c), отличающемуся тем, что основание представляет собой карбонат цезия. Более предпочтительно другой объект настоящего изобретения относится к способу синтеза соединения формулы 1, включающему стадию c) или включающему стадии a)-c), отличающемуся тем, что полярный апротонный растворитель представляет собой диметилформамид.
Более предпочтительно другой объект настоящего изобретения относится к способу синтеза соединения формулы 1, включающему стадию c) или включающему стадии a)-c), отличающемуся тем, что реакция O-алкилирования происходит при температуре не ниже 80°C.
Более предпочтительно другой объект настоящего изобретения относится к способу синтеза соединения формулы 1, включающему стадию c) или включающему стадии a)-c), отличающемуся тем, что диметилформамид и карбонат цезия предварительно нагревают до температуры не ниже 80°C и бромид метилциклопропана вводят горячим.
Другой объект настоящего изобретения относится к новому способу получения соединения формулы 1, отличающемуся тем, что стадия b) является "однореакторной" стадией.
Другой объект настоящего изобретения относится к новому способу получения соединения формулы 1, отличающемуся тем, что стадия a) представляет собой стадию амидирования циклическим тримером н-пропилфосфонового ангидрида (T3P). Более предпочтительно эту стадию осуществляют в присутствии 4-диметиламинопиридина.
Другой объект настоящего изобретения относится к новому способу получения соединения формулы 1, отличающемуся тем, что стадия a) представляет собой стадию сочетания между имидазолидом соединения 2 (имидазолидом 4-циклопропилметокси-5-метокси-2-пиридинкарбоновой кислоты) и моноацетилированным N-оксидом 3,5-дихлораминопиридина. Более предпочтительно эту стадию осуществляют в присутствии хлорида циклопропилмагния.
Другой объект настоящего изобретения относится к новому способу получения соединения формулы 1, отличающемуся тем, что стадия a) представляет собой стадию опосредованного карбодиимидом амидирования между соединением 2 (4-циклопропилметокси-5-метокси-2-пиридинкарбоновой кислотой) и N-Boc-производным соединения 3 (N-оксида 3,5-дихлораминопиридина). Более предпочтительно эту стадию осуществляют в присутствии 4-диметиламинопиридина.
Другой объект настоящего изобретения относится к соединению формулы 4.
Другой объект настоящего изобретения относится к соединению формулы 6.
Другой объект настоящего изобретения относится к соединению формулы 7.
Другой объект настоящего изобретения относится к применению одного или более соединений формул 4, 6 и 7 в качестве промежуточного соединения в способе получения соединения формулы 1.
Согласно одному из аспектов настоящего изобретения, в частности, возможно идентифицировать следующие примеси:
- Соединение I.1: 4-(циклобутилокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамид
Молекулярная формула: C16H15Cl2N3O4, относительная молекулярная масса: 384,22
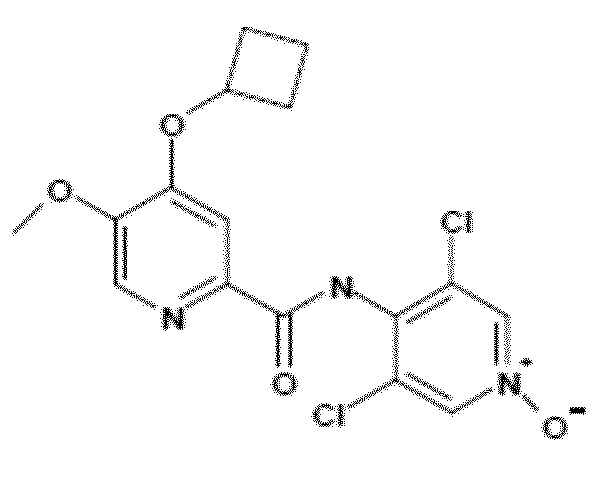
- Соединение I.2: 4-циклопропилметокси-5-метоксипиридин-2-карбоновая кислота (4-циклопропилметокси-5-метоксипиридин-2-карбонил)-(3,5-дихлор-1-окси-4-пиридил)амид
Молекулярная формула: C27H26Cl2N4O7, относительная молекулярная масса: 589,44

- Соединение I.3: 4-(циклопропилметокси)-N-(3,5-дихлор-4-пиридил)-5-метоксипиридин-2-карбоксамид
Молекулярная формула: C16H15Cl2N3O3, относительная молекулярная масса: 368,22
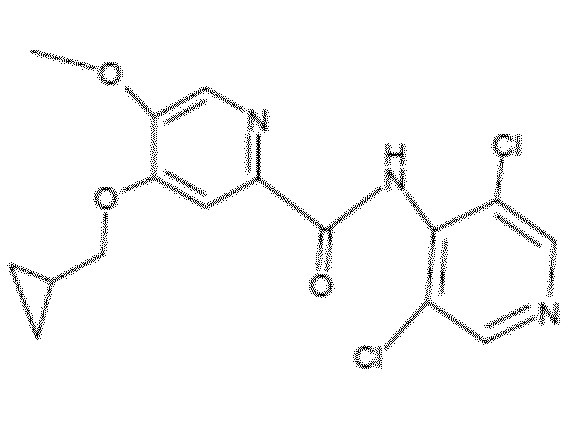
Идентификация одного или более из этих соединений может быть полезна в качестве признака способа получения соединения формулы 1 по настоящему изобретению.
Подробные примеры получения согласно заявленным способам описаны ниже. Эти примеры не являются ограничивающими и только иллюстрируют настоящее изобретение.
Пример 1: синтез CH2OH-промежуточного соединения (4-циклопропилметокси-5-метокси-2-гидроксиметилпиридина, соединения 4) путем прямого O-алкилирования
Нагревая до 80°C (15 минут) смесь 2-гидроксиметил-5-метокси-4-пирид-(1H)-она (соединения 5), карбоната цезия (1,5 эквивалента) и бромметилциклопропана (1 эквивалент) в 10 объемах ДМФА получают смесь продукта N-алкилирования (30%-40%) и продукта O-алкилирования (60%-70%):
С другой стороны, если соединение 5 предварительно нагрето до 80°C в ДМФА, в присутствии Cs2CO3, бромид метилциклопропана вводится горячим, почти исключительно образуется ожидаемый продукт O-алкилирования, равновесие енолят/амид явно сдвинуто в этих условиях в сторону образования алкоксида:
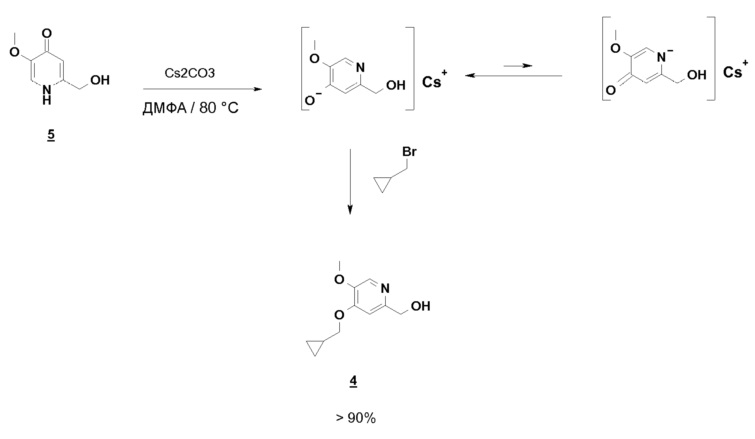
Возможные следы продукта N-алкилирования, который может появиться, удаляют во время обработки путем промывки водой, что делает этот способ совместимым с OU-совокупностями. Выход соединения 4 больше или равен 75%.
Пример 2: синтез 4-циклопропилметокси-5-метоксипиридин-2-карбоновой кислоты
Суспензию, содержащую моногидрат метоксипиридона (OU) в 7 объемах ДМФА. концентрируют и сушат до 2 объемов дистиллированием в вакууме с ДМФА. После добавления карбоната цезия (1,1 эквивалент/сухой) эту суспензию предварительно нагревают до 85°C. При этой температуре добавляют бромметилциклопропан приблизительно за 30 минут (1,05 эквивалента/сухой - экзотермическая реакция). После охлаждения до комнатной температуры минеральные соли удаляют фильтрованием и ДМФА дистиллируют азеотропным захватом ксилолом. Концентрат поглощают метиленхлоридом и промывают водой. Хлорметиленовую экстракционную фазу, содержащую ожидаемый продукт 4 (8 объемов приблизительно при 0,5 М/выход около 80%), используют в полученном виде на следующей стадии.
К двухфазной системе метиленхлорид (около 8 объемов приблизительно при 15 масс./масс.% ксилола)/вода (20 объемов), содержащей циклопропилпиридинол (OU), бикарбонат натрия (1,0 эквивалента), бромид натрия (0,5 эквивалента) и 4-ацетамидо-TEMPO (0,04 эквивалента), добавляют приблизительно за 1 час при 0-5°C, при энергичном перемешивании, водный раствор гипохлорита натрия приблизительно при 2M (2,4 эквивалента/5,9 объемов). После перемешивания в течение 3 часов при 0-5°C, избыток окисляющего агента нейтрализуют 2M водным раствором тиосульфата натрия (достаточным количеством, т.е. 0,6 объема в описанном случае). После охлаждения до комнатной температуры и разделения фаз отстаиванием водную фазу концентрируют (12 объемов) при стандартном давлении, осадок карбоксилата натрия отфильтровывают и затем повторно растворяют в воде (около 8 объемов) при 40°C. После подкисления до pH около 4-5 с помощью серной кислоты, соединение 2 выделяют, после кристаллизации из воды при 40°C, с выходом около 69%.
1H-ЯМР-спектр (400 МГц, δ в м.д., ДМСО-d6): 0,36 (м, 2H), 0,59 (м, 2H), 1,24 (м, 1H), 3,95 (с, 3H), 3,97 (д, J=6,8 Гц, 2H), 7,57 (с, 1H), 8,25 (с, 1H).
Пример 3: синтез 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида
К суспензии кислоты соединения 2 (4-циклопропилметокси-5-метоксипиридин-2-карбоновой кислоты) и N-оксида соединения 3 (3,5-дихлор-4-аминопиридин1-оксида, 1,25 эквивалента) в этилацетате (EtOAc) (около 10 объемов) добавляют раствор T3P при 50% в EtOAc (1,2 эквивалента), триэтиламин (2,5 эквивалента) и DMAP (0,2 эквивалента). Смесь нагревают до температуры кипения с обратным холодильником около десяти часов, дают охладиться, гидролизуют водой, фильтруют и затем промывают водой и EtOAc. Сырое соединение 1 выделяют с выходом около 80% и микроаналитической чистотой свыше 99%.
1H-ЯМР-спектр (500 МГц, δ в м.д., ДМСО-d6): 0,37 (м, 2H), 0,60 (м, 2H), 1,25 (м, 1H), 3,99 (с, 3H), 4,01 (д, J=7,1 Гц, 2H), 7,62 (с, 1H), 8,29 (с, 1H), 8,71 (с, 2H), 10,51 (ушир.с, 1H).
Пример 4: синтез 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида
Раствор карбонилдиимидазола (CDI, 1,1 эквивалента) в метил-ТГФ (7 объемах) выливают в суспензию циклопропилпиридиновой кислоты (OU=50 г) в метил-ТГФ (7 объемов) и нагревают до 60°C. Выделение CO2 регулируют скоростью введения CDI. После нагревания в течение 1-2 часов при 60°C реакционную среду охлаждают приблизительно до 5°C. Имидазолидный осадок отфильтровывают в атмосфере азота, промывают минимальным количеством метил-ТГФ (2 объема) и сушат в вакуумном сушильном шкафу. Имидазолид выделяют с выходом около 80% и чистотой свыше 95%.
1H-ЯМР-спектр (400 МГц, δ в м.д., ДМСО-d6): 0,39 (м, 2H), 0,62 (м, 2H), 1,28 (м, 1H), 4,01 (с, 3H), 4,04 (д, J=7,0 Гц, 2H), 7,12 (ушир.с, 1H), 7,73 (с, 1H), 7,95 (ушир.с, 1H), 8,40 (с, 1H), 8,76 (ушир.с, 1H).
Суспензию дихлорпиридоксида (1,5 эквивалента) в 20 объемах ТГФ обрабатывают при 20-30°C хлоридом циклогексилмагния в виде 1,3 М раствора в смеси ТГФ/толуол (1,2 эквивалента). После перемешивания в течение 3 часов при комнатной температуре имидазолид (OU=20 г), суспендированный в 5 объемах ТГФ, выливают в полученный таким образом амид. После 5 часов при 60°C и затем охлаждения до комнатной температуры смесь гидролизуют молярным водным раствором хлорида аммония (30 объемов), экстрагируют этилацетатом (30 объемов), промывают водой (10 объемов) и кристаллизуют из изопропанола (15 объемов), после замены растворителя; соединение 1 выделяют фильтрованием и при сушке получают выход около 70% и микроаналитическую чистоту.
1H-ЯМР-спектр (500 МГц, δ в м.д., ДМСО-d6): 0,37 (м, 2H), 0,60 (м, 2H), 1,25 (м, 1H), 3,99 (с, 3H), 4,01 (д, J=7,1 Гц, 2H), 7,62 (с, 1H), 8,29 (с, 1H), 8,71 (с, 2H), 10,51 (ушир.с, 1H).
Пример 5: синтез 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида
N-Boc-производное соединения 1
Хлорметиленовый раствор EDC (гидрохлорид 1-[3-диметиламинопропил]-3-этилкарбодиимида/1,2 эквивалента) добавляют, приблизительно при 10°C, к хлорметиленовому раствору, содержащему циклопропилпиридиновую кислоту (соединение 2/OU), N-Boc-дихлорпиридоксид (1 эквивалент) и DMAP (4-диметиламинопиридин/0,1 эквивалента). После перемешивания в течение 18 часов при 20°C реакционную смесь промывают водой и сушат досуха. N-Boc-продукт соединения 1 выделяют в чистом виде с выходом 70% после перекристаллизации из изопропанола.
1H-ЯМР-спектр (400 МГц, δ в м.д., ДМСО-d6): 0,36 (м, 2H), 0,59 (м, 2H), 1,24 (ушир.с, 10H), 3,96 (с, 3H), 4,00 (д, J=7,0 Гц, 2H), 7,43 (с, 1H), 8,30 (с, 1H), 8,80 (с, 2H).
Гидролиз Boc и получение соединения 1
Хлорметиленовый раствор N-Boc-производного соединения 1 нагревают при 30°C в течение 4 часов в присутствии трифторуксусной кислоты (13 эквивалентов). Соединение 1 выделяют осаждением после обработки реакционной среды водным бикарбонатом натрия с выходом свыше 70%.
1H-ЯМР-спектр (500 МГц, δ в м.д., ДМСО-d6): 0,37 (м, 2H), 0,60 (м, 2H), 1,25 (м, 1H), 3,99 (с, 3H), 4,01 (д, J=7,1 Гц, 2H), 7,62 (с, 1H), 8,29 (с, 1H), 8,71 (с, 2H), 10,51 (ушир.с, 1H).
Получение N-Boc-дихлорпиридоксида
Метод 1
Смесь, содержащую ТГФ (10 объемов), Boc 20 (2,2 эквивалента), дихлорпиридоксид (OU) и DMAP (0,1 эквивалента), перемешивают при комнатной температуре в течение 3 часов. Ожидаемый N-Boc-дихлорпиридоксидный продукт выделяют с выходом 65% после отгонки ТГФ и перекристаллизации из этилацетата.
1H-ЯМР-спектр (400 МГц, δ в м.д., ДМСО-d6): 1,38 (с, 18H), 8,79 (с, 2H).
Метод 2
Смесь, содержащую дихлорпиридоксидное соединение (OU), карбонат калия (3 эквивалента) и метанол (10 объемов), нагревают до температуры кипения с обратным холодильником в течение 3 часов. После фильтрования минеральных солей и дистиллирования MeOH ожидаемый N-Boc-дихлорпиридоксидный продукт выделяют экстракцией этилацетатом и промыванием до нейтральной среды водной HCl. Полученный выход равен 84%.
1H-ЯМР-спектр (400 МГц, δ в м.д., ДМСО-d6): 1,47 (с, 9H), 8,66 (с, 2H), 9,28 (ушир.с, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ НОВЫХ КРИСТАЛЛИЧЕСКИХ ФОРМ-4(ЦИКЛОПРОПИЛМЕТОКСИ)-N-(3, 5-ДИХЛОР-1-ОКСИДОПИРИДИН-4-ИЛ)-5-МЕТОКСИПИРИДИН-2-КАРБОКСАМИДА И ЕГО КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2013 |
|
RU2621894C2 |
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-ПИРИДИНИЛАЛКИЛЬНЫХ СПИРТОВ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ | 2012 |
|
RU2626956C2 |
| ПЕПТИДНЫЕ МАКРОЦИКЛЫ ПРОТИВ ACINETOBACTER BAUMANNII | 2016 |
|
RU2729609C2 |
| НОВЫЕ ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗ | 2008 |
|
RU2495043C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2800292C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| 4-(4-ПИРИДИЛ)БУТИЛОВЫЙ ЭФИР N-СУЛЬФОНИЛТИРОЗИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ПОЛУЧЕНИЯ 4-(4-ПИПЕРИДИЛ)БУТИЛОВОГО ЭФИРА N-СУЛЬФОНИЛТИРОЗИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1993 |
|
RU2114105C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЗОТИСТОГО ИПРИТА | 2016 |
|
RU2715233C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОДИОКСОЛА ИЛИ БЕНЗОДИОКСЕПИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2011 |
|
RU2583787C2 |
| УСИЛИТЕЛЬ ДЕЙСТВИЯ ПРОТИВООПУХОЛЕВОГО СРЕДСТВА | 2010 |
|
RU2548913C2 |
Изобретение относится к способу получения соединения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида формулы (1)
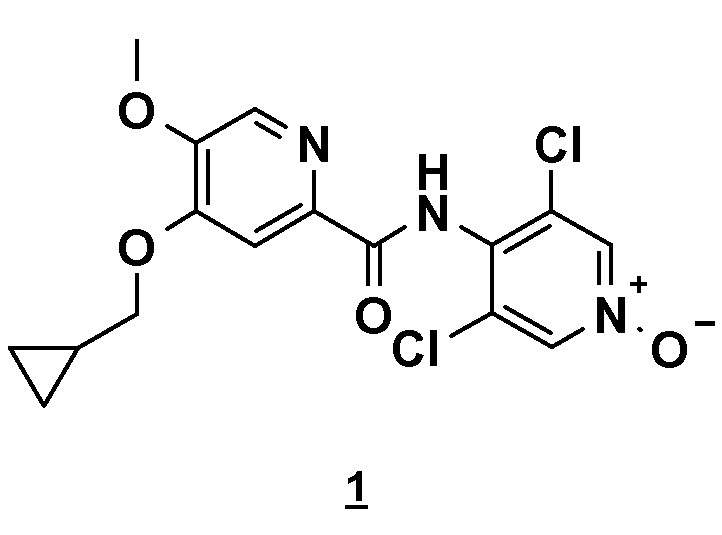 ,
,
в основной или гидратной форме, или в форме фармацевтически приемлемых солей, а также некоторых новых синтетических промежуточных соединений, полезных в этом способе получения. 6 н. и 6 з.п. ф-лы, 5 пр.
1. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида, в основной или гидратной форме, или в форме фармацевтически приемлемых солей, приведенной ниже формулы 1
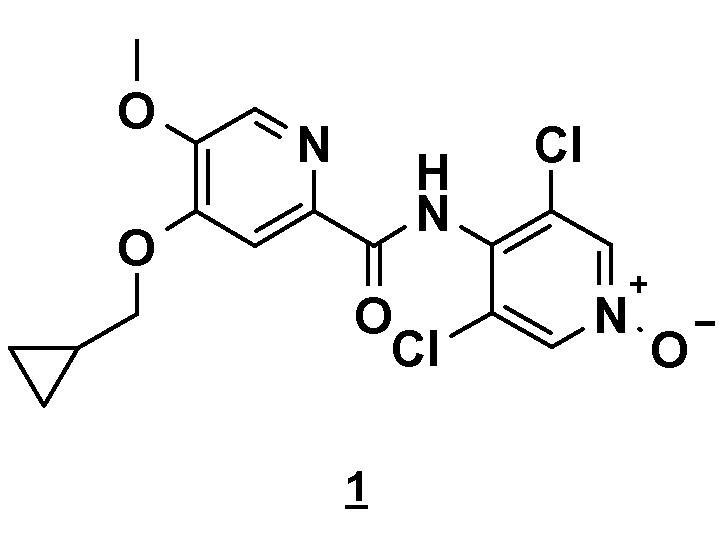
отличающийся тем, что включает:
- стадию c): взаимодействие соединения 2-гидроксиметил-5-метокси-4-пирид-(1H)-она формулы 5 с метилциклопропановым производным и основанием в полярном апротонном растворителе согласно приведенной ниже схеме
- стадию b): реакцию окисления соединения 4, полученного на стадии c), согласно приведенной ниже схеме
- стадию a): реакцию сочетания соединения 2, полученного на стадии b), с N-оксидом 3,5-дихлораминопиридина (соединение 3) или его N-Boc-производным согласно приведенной ниже схеме
 .
.
2. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по п. 1, отличающийся тем, что метилциклопропановое производное представляет собой бромид метилциклопропана, основание представляет собой карбонат цезия и полярный апротонный растворитель представляет собой диметилформамид.
3. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по любому одному из пп. 1 или 2, отличающийся тем, что стадия b) является однореакторной стадией.
4. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по любому одному из пп. 1 или 2, отличающийся тем, что стадия a) представляет собой стадию амидирования циклическим тримером н-пропилфосфонового ангидрида (T3P).
5. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по п. 4, отличающийся тем, что стадию амидирования осуществляют в присутствии 4-диметиламинопиридина.
6. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по любому одному из пп. 1, 2 или 5, отличающийся тем, что стадия a) представляет собой реакцию сочетания между имидазолидом 4-циклопропилметокси-5-метокси-2-пиридинкарбоновой кислоты и N-оксидом 3,5-дихлораминопиридина.
7. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида по любому одному из пп. 1, 2 или 5, отличающийся тем, что стадия a) представляет собой стадию опосредованного карбодиимидом амидирования между 4-циклопропилметокси-5-метокси-2-пиридинкарбоновой кислотой и N-Boc-производным N-оксида 3,5-дихлораминопиридина.
8. Способ получения 4-(циклопропилметокси)-N-(3,5-дихлор-1-оксидо-4-пиридил)-5-метоксипиридин-2-карбоксамида, в основной или гидратной форме, или в форме фармацевтически приемлемых солей, приведенной ниже формулы 1
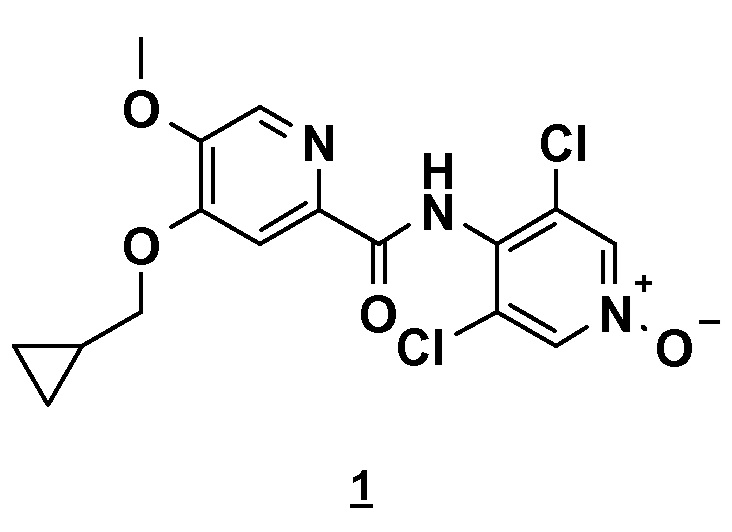
отличающийся тем, что включает реакцию сочетания 4-циклопропилметокси-5-метокси-2-пиридинкарбоновой кислоты (соединения 2) с N-оксидом 3,5-дихлораминопиридина (соединением 3) или его N-Boc-производным согласно приведенной ниже схеме
.
9. Соединение приведенной ниже формулы 4
 .
.
10. Соединение приведенной ниже формулы 6
 .
.
11. Соединение приведенной ниже формулы 7
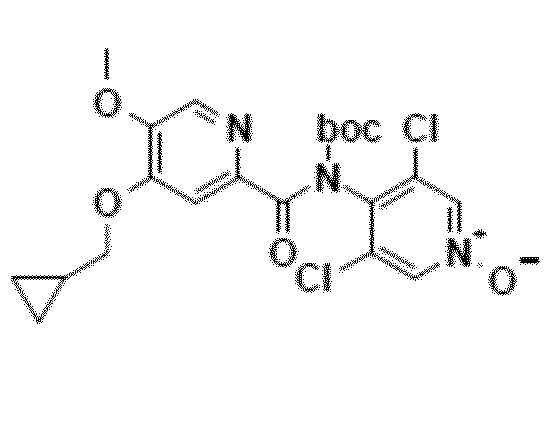 .
.
12. Применение соединений формул 4, 6 и 7 по пп. 9-11 в качестве синтетических промежуточных соединений для получения соединения формулы 1.
| Способ получения эфиров жировых или им подобных кислот | 1928 |
|
SU15355A1 |
| WO2004005258 A1, 15.01.2004 | |||
| WO 1996031476 A1, 10.10.1996 | |||
| WO 2002069905 A2, 12.09.2002. | |||

