ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к усовершенствованному способу получения мелфлуфена или его соли и мелфалана или его соли. Изобретение также относится к новым промежуточным соединениям, полученным в способе по изобретению.
УРОВЕНЬ ТЕХНИКИ
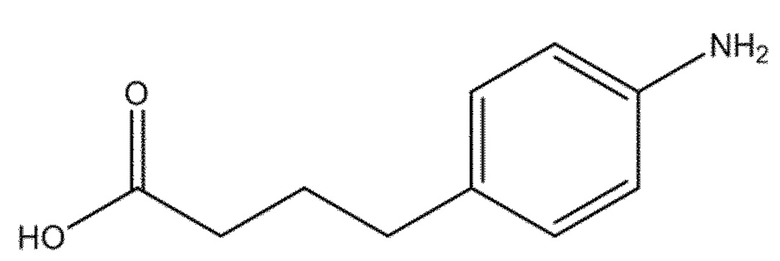
Алкилирующие агенты, такие как препараты, полученные из азотистого иприта, то есть производные бис(2-хлорэтил)амина, применяются в качестве химиотерапевтических средств для лечения широкого ряда злокачественных опухолей. Мелфалан или p-бис-(2-хлорэтил)-амино-L-фенилаланин (соединение (Id), CAS № 148-82-3) является алкилирующим агентом, который представляет собой конъюгат азотистого иприта и аминокислоты фенилаланина (US 3032584). Мелфалан используется клинически при лечении метастатических меланом, при этом имеет ограниченную эффективность, может проявлять ограничивающую дозу токсичность и резистентность.
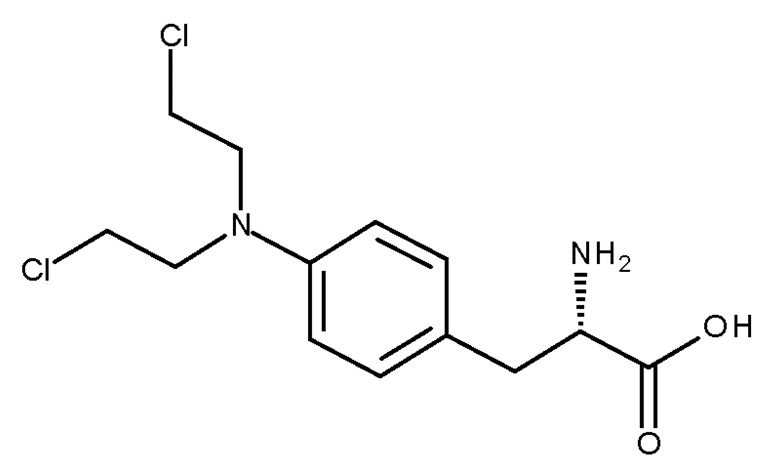
(Id)
Сложный этиловый эфир мелфалан-флуфенамида (сложный этиловый эфир L-мелфаланил-L-п-фторфенилаланина, мелфлуфен, соединение (Ib)) является производным мелфалана, конъюгированного с аминокислотой фенилаланином, с образованием дипептида (WO 01/96367):
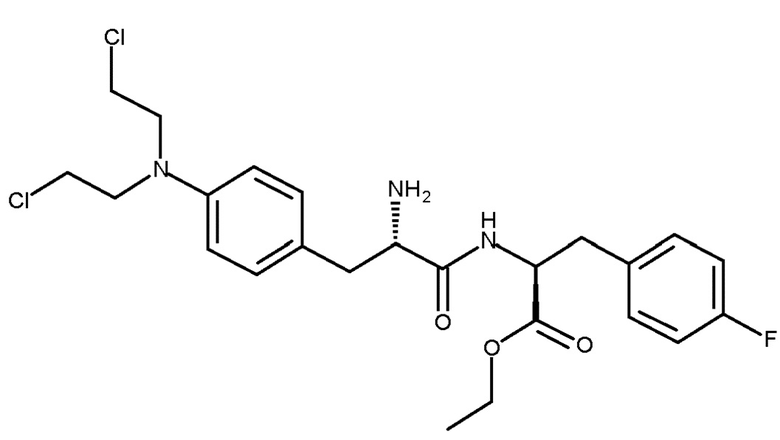
(Ib)
Монохлористоводородная соль мелфлуфена (моногидрохлорид этилового эфира L-мельфаланил-L-p-фторфенилаланина, хлористоводородная соль (Ib); CAS № 380449-54-7) известна под названием мелфлуфена гидрохлорид.
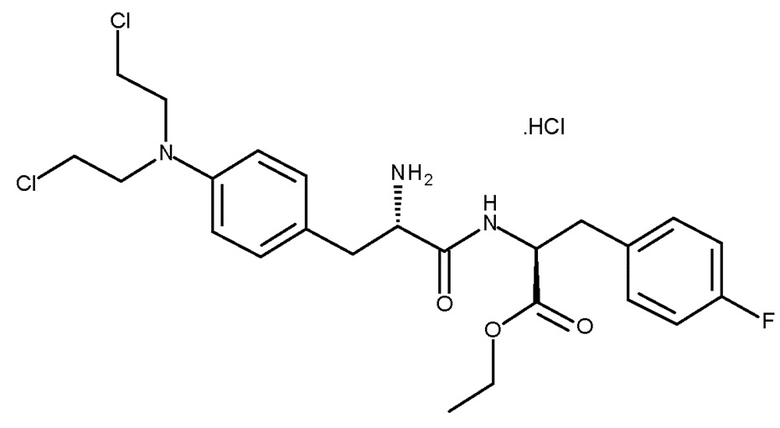
хлористоводородная соль (Ib)
При исследовании культур опухолевых клеток человека, представляющих приблизительно 20 различных диагнозов злокачественных опухолей человека, включая миелому, мелфлуфен показал в 50-100 раз более высокую эффективность по сравнению с эффективностью мелфалана (http://www.oncopeptides.se/products/melflufen, по состоянию на 26 марта 2015 года). Данные, предоставленные в Arghya, et al, abstract 2086 ʺA Novel Alkylating Agent Melphalan Flufenamide Ethyl Ester Induces an Irreversible DNA Damage in Multiple Myeloma Cellsʺ (2014) 5th ASH Annual Meeting and Exposition, свидетельствуют о том, что мелфлуфен вызывает быстрое, надежное и необратимое повреждение ДНК, что может обусловливать его способность преодолевать резистентность к мелфалану в клетках множественной миеломы. В настоящее время мелфлуфен проходит фазу I/IIa клинических испытаний при множественной миеломе.
Способ получения мелфлуфена в форме хлористоводородной соли описан в WO 01/96367 и проиллюстрирован на схеме 1 ниже. В этом способе N-трет-бутоксикарбонил-L-мелфалан подвергают взаимодействию со сложным этиловым эфиром p-фторфенилаланина с получением сложного этилового эфира N-трет-бутоксикарбонил-L-мелфаланил-L-p-фторфенилаланина. После очистки с помощью градиентной колоночной хроматографии выход на этой стадии составляет 43%.
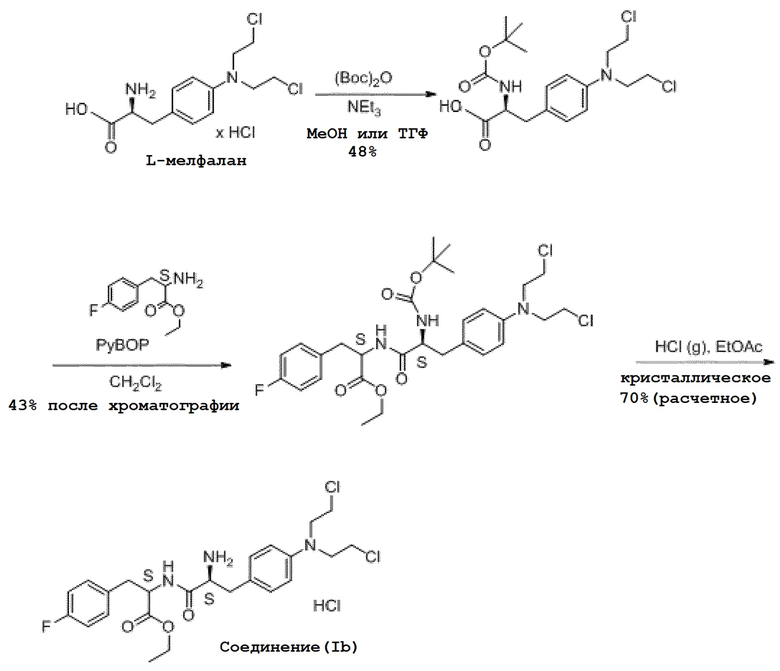
Схема 1. Существующий в настоящее время способ получения мелфлуфена (в форме хлористоводородной соли)
Как показано на схеме 1, в известном способе получения мелфлуфена (в форме хлористоводородной соли), в качестве исходного вещества, используется цитотоксический агент мелфалан, а мелфлуфен получают методом последовательного многоэтапного синтеза. Мелфалан обладает высокой токсичностью и соответственно исходные вещества и все промежуточные продукты, а также образующиеся отходы, являются чрезвычайно токсичными. Это является серьезным недостатком с точки зрения безопасности, воздействия на окружающую среду и затрат при использовании этого процесса в больших масштабах. Поэтому весьма желателен усовершенствованный и более безопасный способ, в частности для производства мелфлуфена в больших масштабах. Кроме того, чистота коммерчески доступного мелфалана является низкой вследствие его плохой стабильности, выход продукта на каждой стадии процесса является низким, а чистота конечного продукта, полученного известным способом, не является высокой.
Способ получения мелфалана описан в WO 2014/141294. В WO 2014/141294 стадия введения группы бис(2-хлорэтил) в молекулу включает превращение первичного фениламина в третичный фениламиндиол путем взаимодействия с газом этиленоксидом. Это обеспечивает выход, составляющий 52,6%. Затем аминдиол превращают в бис(2-хлорэтил)фениламин путем взаимодействия с фосфорилхлоридом. Использование этиленоксида, или хлорэтанола, для превращения ароматического амина в соответствующий бис-(2-гидроксиэтил)амин с последующим хлорированием этого промежуточного соединения является общим способом получения ароматических бис-(2-хлорэтил)аминов. Известно также, что он начинается с хлорарена и обеспечивает осуществление SNAr-взаимодействия с диэтаноламином. Авторы настоящего изобретения использовали эти способы получения мелфлуфена (в форме соли), показанные на схеме 2 ниже.
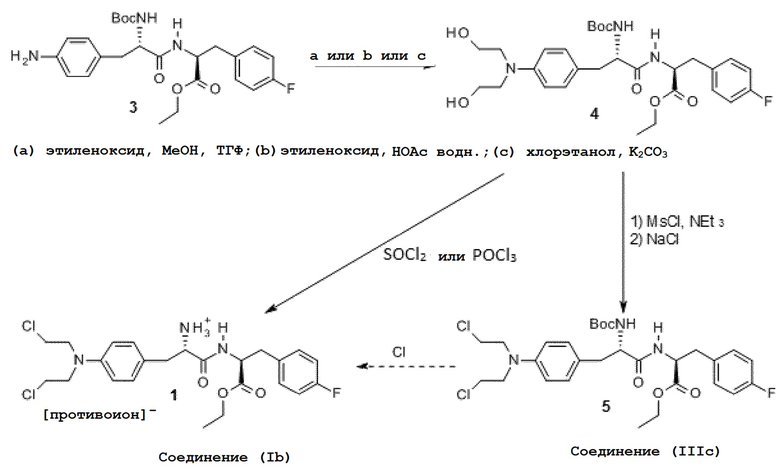
Схема 2. Альтернативные способы получения мелфлуфена
Авторы настоящего изобретения обнаружили, что при использовании этиленоксида в ТГФ (способ (а) схемы 2) алкилирование не происходит при 55°С; повышение температуры до 60°С приводит к образованию диалкилированного промежуточного соединения, однако взаимодействие осуществлялось очень медленно. Для повышения выхода продукта и скорости взаимодействия для взаимодействия потребовались бы высокие температуры, но это могло бы вызвать повышение давления, вследствие чего взаимодействие должно выполняться в реакторе под давлением. Такие условия, вероятно, приводят к образованию побочных продуктов. Аналогичные условия взаимодействия, но с использованием смеси этиленоксида и уксусной кислоты 50:50 (способ (b) схемы 2), обеспечивают более короткое время взаимодействия, однако приводит к образованию побочных продуктов. Использование карбоната калия и хлороэтанола (способ (с) схемы 2) также приводит к образованию побочного продукта, вероятно, из-за того, что хлороэтанол подвергается частичной трансэтерификации с этиловым эфиром.
Авторы настоящего изобретения также предприняли попытку хлорирования диалкилированного соединения. Хлорирование бис-(2-гидроксиэтил) соединения (4) схемы 2 с использованием тионилхлорида в дихлорметане привело к значительному образованию лишенного защиты побочного продукта. Хлорирование бис-(2-гидроксиэтил) соединения (4) схемы 2 с использованием POCl3 требовало высокой температуры и продолжительности взаимодействия. Кроме того, как тионилхлорид, так и POCl3 сложнее обрабатывать в больших масштабах из-за проблем с безопасностью. Авторы настоящего изобретения также превратили бис-(2-гидроксиэтил) соединение (4) схемы 2 в соответствующий димезилат путем обработки метансульфонилхлоридом и триэтиламином. Далее димезилат обрабатывали хлоридом натрия в ДМФ при 120°С. Однако необработанный продукт этого взаимодействия содержал значительное количество побочных продуктов, делающих этот способ непригодным с экономической точки зрения для использования в необходимом масштабе.
Таким образом, ни один из этих способов не был признан подходящим для масштабного производства мелфлуфена с высокой степенью чистоты. Они не обеспечивают удовлетворительный синтез мелфлуфена, что приводит к плохому выходу продукта, и являются неэффективными. Кроме того, способы, показанные на схеме 2, требуют нескольких стадий для образования N,N-бис-хлорэтиламина и использования токсичных реагентов.
Авторы настоящего изобретения обнаружили усовершенствованный способ получения мелфлуфена (в частности мелфлуфена в форме его хлористоводородной соли), который обеспечивает соединение с превосходным выходом и очень высокой степенью чистоты.
Сущность изобретения
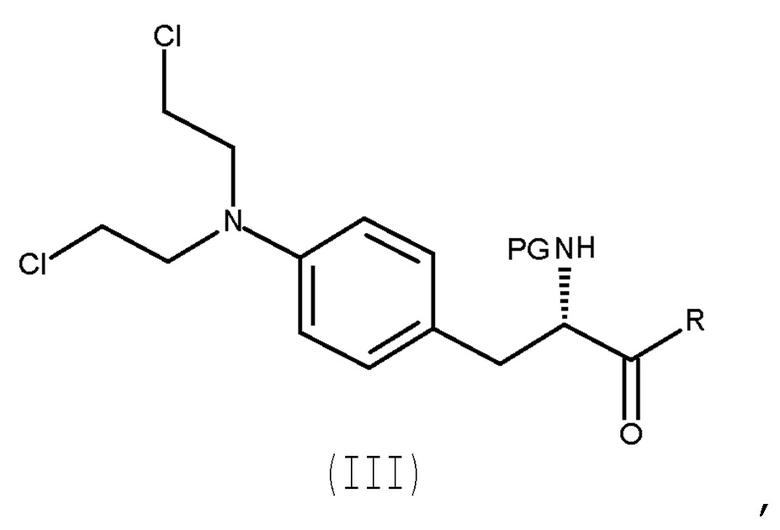
Настоящее изобретение относится к способу получения соединения (III) или его лишенного защиты продукта:
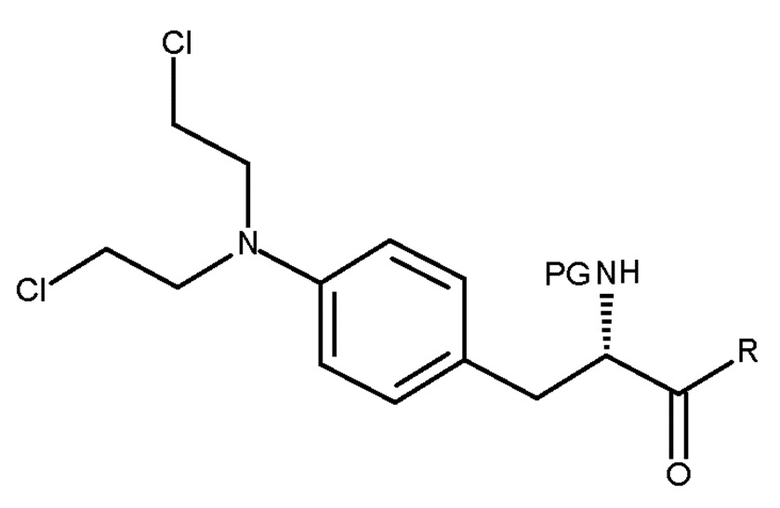
(III)
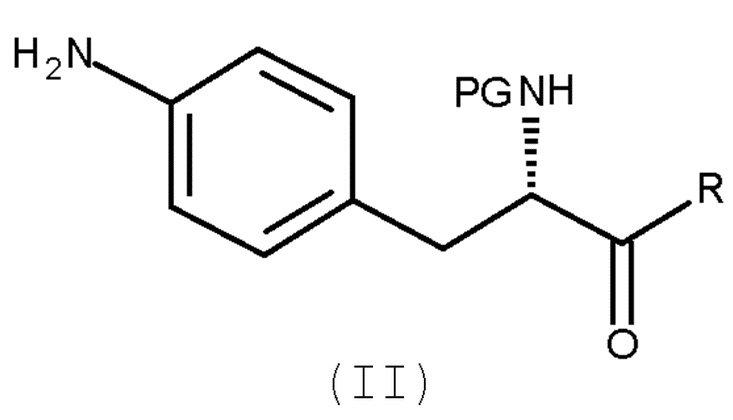
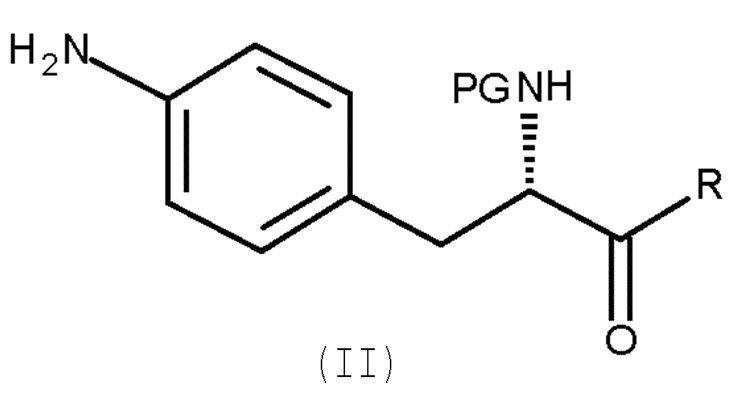
включающему взаимодействие соединения (II)
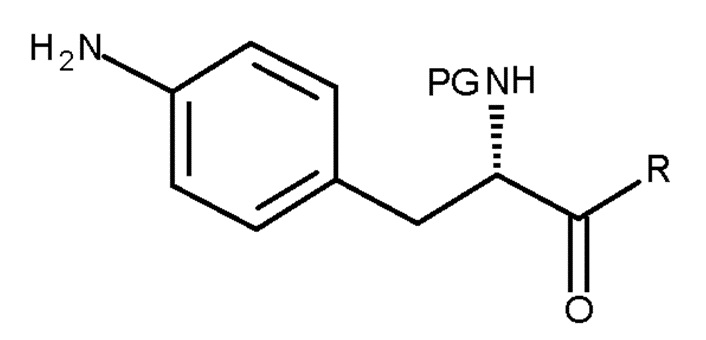
(II)
с хлоруксусной кислотой, в присутствии восстанавливающего агента;
где PG представляет собой защитную группу, и R представляет собой ОН в соответствующим образом защищенной форме или 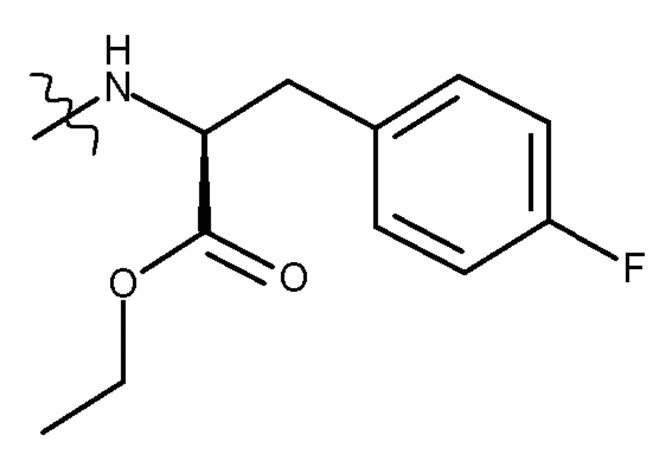 .
.
Авторы настоящего изобретения обнаружили, что превращение соединения ароматического амина (II) в азотистый иприт может быть достигнуто в течение одной стадии с высоким выходом и с высокой степенью чистоты.
Настоящее изобретение также относится к способу получения соединения (I) или его соли:
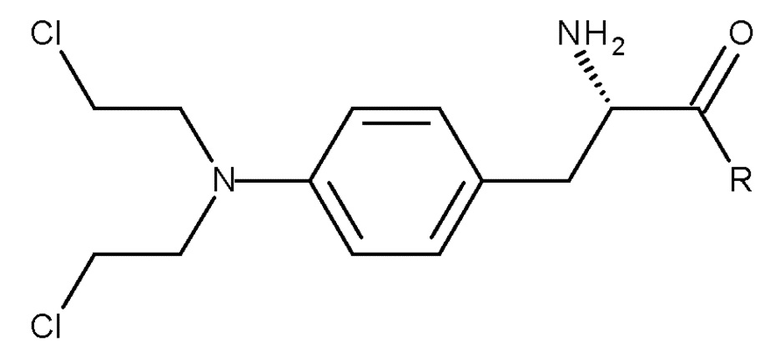
(I)
который включает осуществление способа получения соединения (III), описанного выше, и последующее удаление защитной группы соединения (III) с получением соединения (I) или его соли,
где R представляет собой OH необязательно в соответствующим образом защищенной форме или .
Кроме того, настоящее изобретение относится к способу получения соединения (VIb):
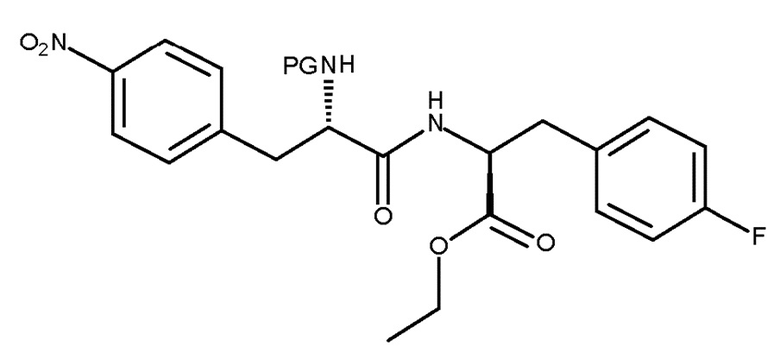
(VIb)
включающему взаимодействие соединения (IV):
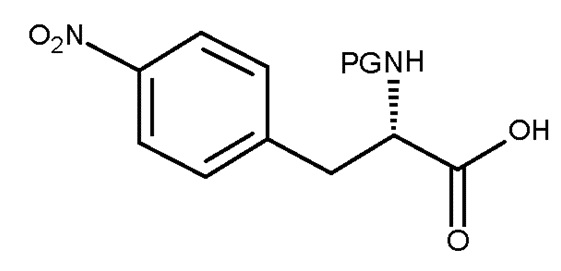
(IV)
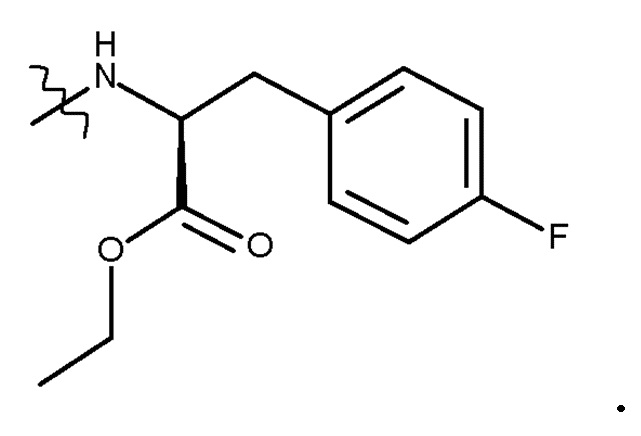
с соединением (V):
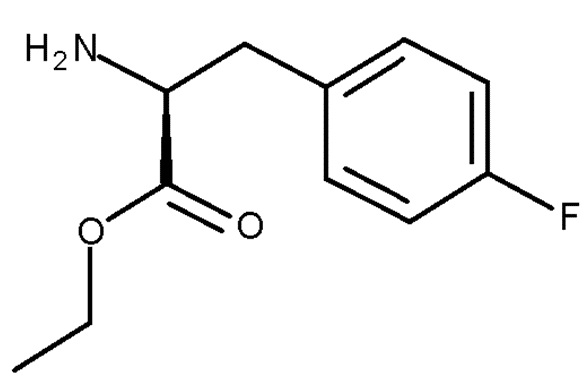
(V)
где PG представляет собой защитную группу.
Кроме того, настоящее изобретение относится к способу получения соединения (II):
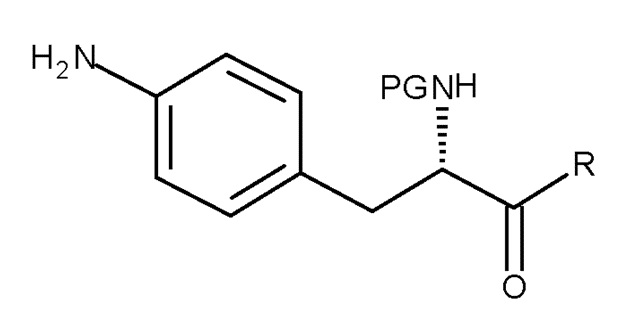
(II)
включающему взаимодействие соединения (VI):
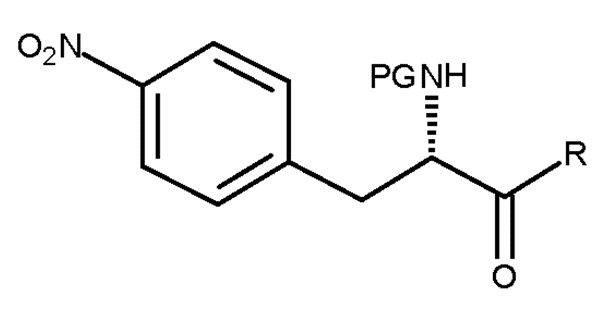
(VI)
с восстанавливающим агентом,
где PG представляет собой защитную группу, и R представляет собой OH необязательно в соответствующим образом защищенной форме или .
Кроме того, настоящее изобретение относится к соединению, имеющему следующую структуру:
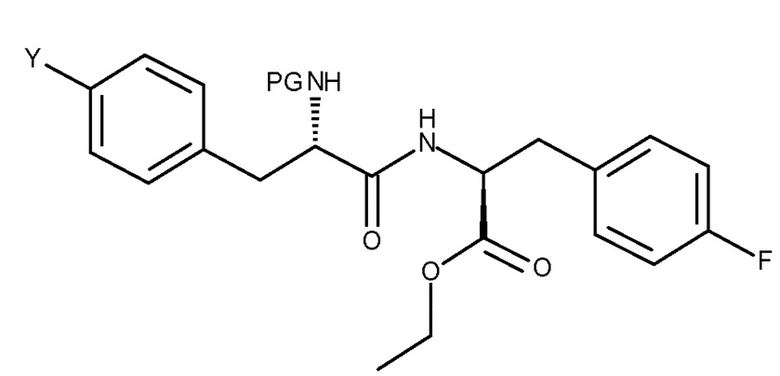
где Y представляет собой NH2 или NO2, и PG представляет собой защитную группу.
Подробное описание изобретения
Настоящее изобретение обеспечивает усовершенствованный способ синтеза мелфлуфена или его соли, или мелфалана или его соли, включающий превращение первичного ароматического амина (соединения (II)) в ароматический N,N-бис-хлорэтил амин за одну стадию с использованием хлоруксусной кислоты и восстанавливающего агента. Этот способ работает очень хорошо и дает хороший выход продукта с высокой степенью чистоты. Этот способ особенно эффективен, поскольку две отдельные стадии, бис-гидроксиалкилирование и хлорирование, заменяются одной операцией в одном сосуде.
Во избежание неоднозначности толкования, в случае, когда в настоящем документе упоминается ʺмелфлуфенʺ, если явно не указано иное, этот термин может относиться к мелфлуфену или его соли (например, гидрохлориду мелфлуфена).
Во избежание неоднозначности толкования, вариант осуществления или предпочтительный аспект любого одного признака способа по изобретению, или соединение, описанное в настоящем документе, могут быть объединены с любым вариантом или предпочтительным аспектом другого признака способа по изобретению или соединения, описанного в настоящем документе, для образования дополнительного варианта осуществления.
Мелфалан имеет стереохимию ʺLʺ; мелфлуфен имеет стереохимию ʺLLʺ, и это является стереохимией ʺLʺ и ʺLLʺ, которая находится в структурах, изображенных в настоящей заявке. Способы по настоящему изобретению и описанные здесь соединения в равной степени применимы к изомерам ʺDʺ или ʺDLʺ, ʺLDʺ и ʺDDʺ или смесям (включая рацемические смеси) изомеров.
Настоящее изобретение относится к способу получения соединения (III) или его лишенного защиты продукта:
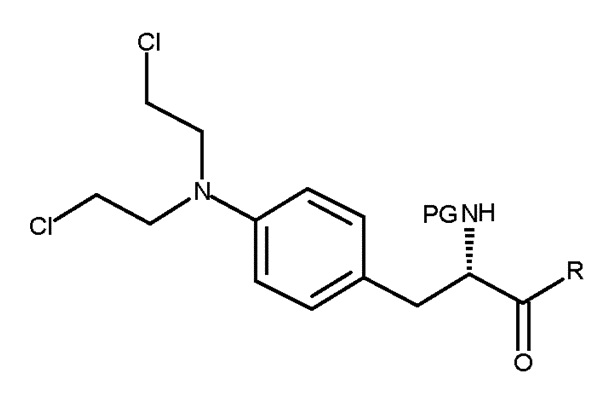
(III)
включающему следующую стадию:
(c) взаимодействие соединения (II)
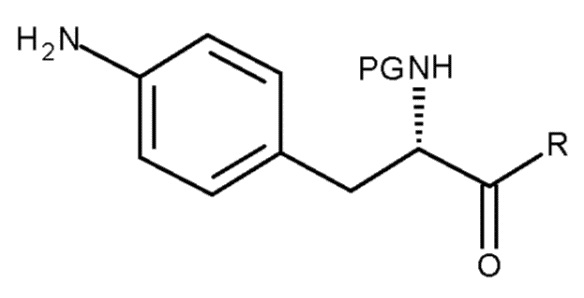
(II)
с хлоруксусной кислотой в присутствии восстановителя;
где PG представляет собой защитную группу; и R представляет собой OH в соответствующим образом защищенной форме или .
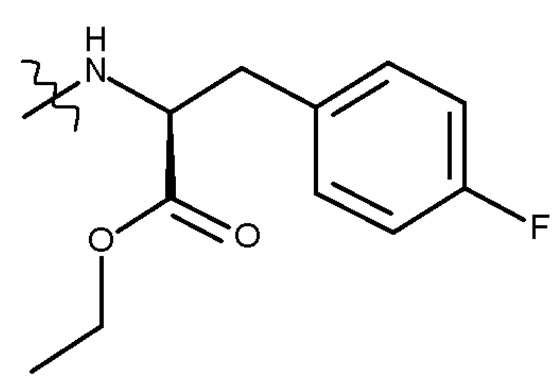
В предпочтительных вариантах осуществления изобретения, R представляет собой . Таким образом, изобретение относится к способу получения соединения (IIIb) или его лишенного защиты продукта:
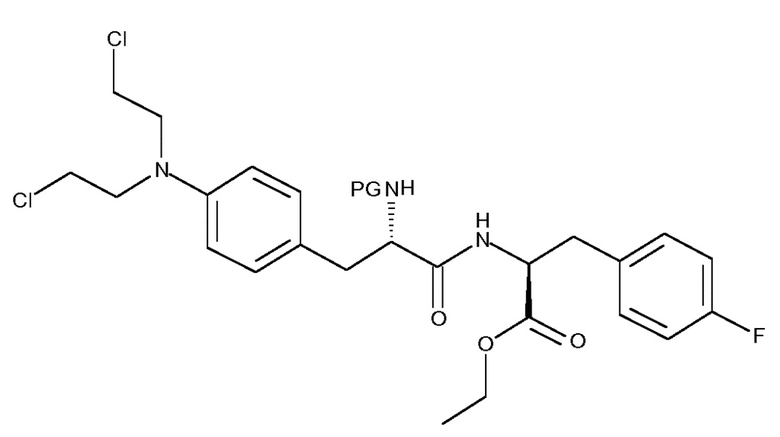
(IIIb)
включающему следующую стадию:
(c) взаимодействие соединения (IIb)
(IIb)
с хлоруксусной кислотой в присутствии восстановителя;
где PG представляет собой защитную группу.
PG представляет собой защитную группу, подходящую для защиты первичного амина. Такие защитные группы хорошо известны специалисту в данной области техники, см. например, Wuts, P.G. M., Greene's Protective Groups in Organic Synthesis, 5th Edition (2014) John Wiley & Sons, Inc. Выбор защитной группы является общепринятым и находится в рамках нормальной практики специалиста в данной области техники. Например, PG может быть выбрана из группы, состоящей из метилоксикарбонила, этилоксикарбонила, 9-флуоренилметил-оксикарбонила (Fmoc), трет-бутилоксикарбонила (Boc), бензилоксикарбонила (Cbz), p-метоксибензилоксикарбонила (Moz), 1-адамантилоксикарбонила (Adoc), p-бромбензилоксикарбонила, трифторацетила, хлорацетила, фенилацетила, бензацетила, p-толуолсульфонила (тозил, Ts), 2-нитробензолсульфонила (Nps), трет-бутилсульфонила (Bus), 2- или 4-нитробензолсульфонила (нозил), 2,4-динитронзенсульфонила (DNs) и 2-нафталинсульфонила.
Например, соединение формулы (IIIb) может представлять собой: метилоксикарбонил-L-мелфалан, этилоксикарбонил-L-мелфалан, 9-флуоренилметилоксикарбонил-L-мелфалан, трет-бутилоксикарбонил-L-мелфалан, бензилоксикарбонил-L-мелфалан, p-метоксибензилоксикарбонил-L-мелфалан, 1-адамантилоксикарбонил-L-мелфалан, p-бромбензилоксикарбонил-L-мелфалан, трифторацетил-L-мелфалан, хлорацетил-L-мелфалан, фенилацетил-L-мелфалан, бензацетил-L-мелфалан, p-толуолсульфонил-L-мелфалан, 2-нитробензолсульфонил-L-мелфалан, трет-бутилсульфонил-L-мелфалан, 2- или 4-нитробензолсульфонил-L-мелфалан, 2,4-динитробензолсульфонил-L-мелфалан, 2-нафталинсульфонил-L-мелфалан; сложный этиловый эфир метилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир этилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир 9-флуоренилметилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир трет-бутилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир бензилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир p-метоксибензилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир 1-адамантилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир p-бромбензилоксикарбонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир трифторацетил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир хлорацетил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир фенилацетил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир бензацетил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир p-толуолсульфонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир 2-нитробензолсульфонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир трет-бутилсульфонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир 2- или 4-нитробензолсульфонил-L-мелфаланил-L-p-фторфенилаланина, сложный этиловый эфир 2,4-динитробензолсульфонил-L-мелфаланил-L-p-фторфенилаланина или сложный этиловый эфир 2-нафталинсульфонил-L-мелфаланил-L-p-фторфенилаланина.
Например, соединение формулы (II) может представлять собой этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(метилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(этилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(9-флуоренилметилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(трет-бутоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(бензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(p-метоксибензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(1-адамантилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(p-бромбензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(трифторацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(хлорацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(фенилацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(бензацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(p-толуолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(2-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(t-бутилсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(4-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(2-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(2,4-динитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат или этил (2S)-2-[[(2S)-3-(4-аминофенил)-2-(2-нафталинсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат.
Предпочтительно PG выбран из группы, состоящей из Fmoc, Boc, Cbz, Moz, Adoc, бромбензилкарбамата и трифторацетамида. Более предпочтительно PG представляет собой Boc.
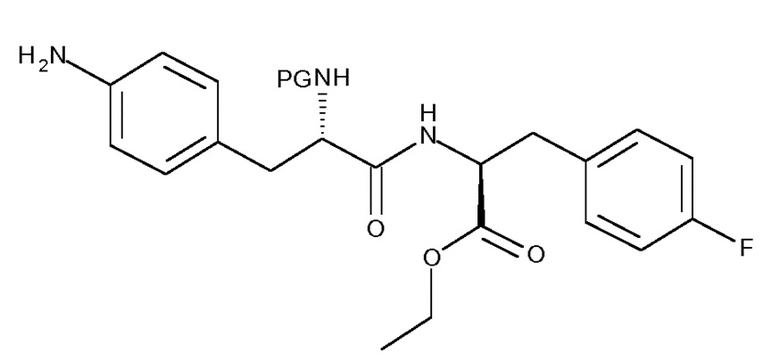
Таким образом, изобретение относится к способу получения соединения (IIIa) или его лишенного защиты продукта:
(IIIa)
включающему следующую стадию:
(c) взаимодействие соединения (IIa)
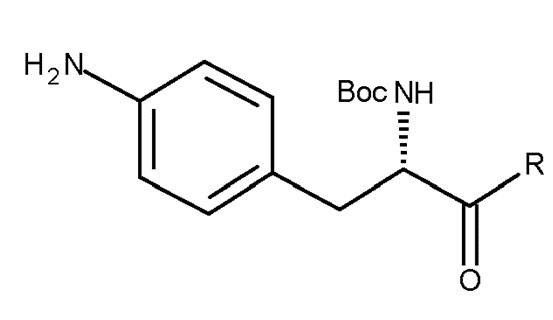
(IIa)
с хлоруксусной кислотой в присутствии восстановителя;
где R представляет собой OH в соответствующим образом защищенной форме или .
В вариантах осуществления, где R представляет собой ОН в подходящей защищенной форме, группа ОН (и необязательно смежная карбонильная группа) может быть защищена любой группой, подходящей для защиты карбоновой кислоты. Такие защитные группы хорошо известны специалисту в данной области техники, см., например, Wuts, P.G. M., Greene's Protective Groups in Organic Synthesis, 5th Edition (2014) John Wiley & Sons, Inc. Выбор защитной группы для карбоновой кислоты является общепринятым и находится в рамках нормальной практики специалиста в данной области техники. Например, защитная группа может быть выбрана из группы, состоящей из метилового сложного эфира, метоксиметилового сложного эфира, 9-флуоренилметилового сложного эфира, трет-бутилового сложного эфира, бензилового сложного эфира, дифенилметилового сложного эфира, трифенилметилового сложного эфира, 2,6-диметилфенилового сложного эфира, триметилсилилового сложного эфира, триэтилсилилового сложного эфира, 2-(триметилсилил)этоксиметилового сложного эфира, 2-(триметилсилил)этилового сложного эфира, S-трет-бутилового сложного эфира и 2-алкил-1,3-оксазолина.
В особенно предпочтительном варианте осуществления, PG представляет собой Boc, и R представляет собой .
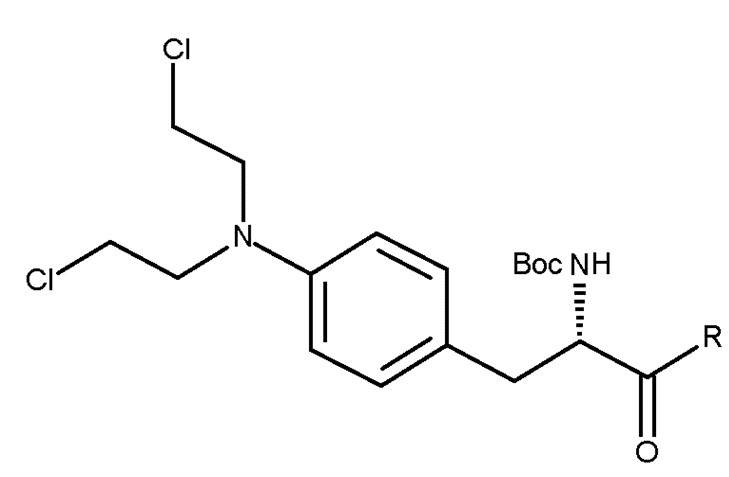
Таким образом, изобретение относится к способу получения соединения (IIIc) или его лишенного защиты продукта:
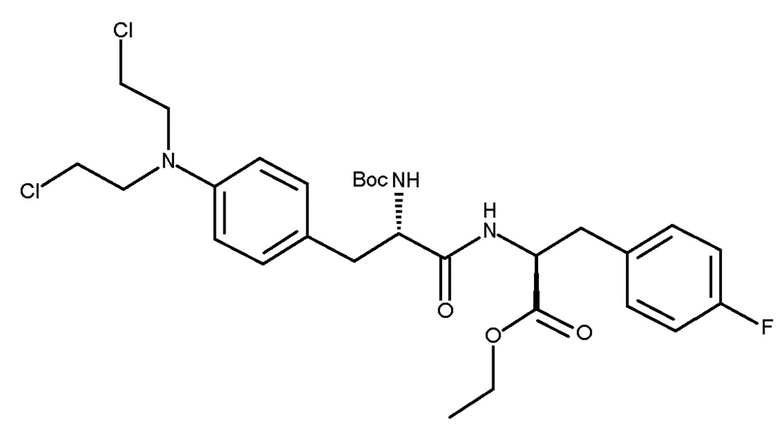
(IIIc)
включающему следующую стадию:
(c) взаимодействие соединения (IIc)
(IIc)
с хлоруксусной кислотой в присутствии восстановителя.
Восстановителем для использования на стадии (с) настоящего изобретения может быть, например, восстановитель, подходящий для использования в реакции восстановительного аминирования или восстановительного алкилирования. Предпочтительно восстанавливающим агентом является гидридный донор, например, восстановитель, выбранный из группы, состоящей из борана, комплекса боран-основание по Льюису, боргидрида, гидрида металла и Н2 в присутствии металлического катализатора. В некоторых вариантах осуществления изобретения, восстанавливающий агент выбран из группы, состоящей из B2H6, B10H14, BH3SMe2 (боран-диметилсульфид, BMS), BH3ТГФ, NaBH4, LiBH4, NaBH3CN, алюмогидрида (алан), бис(2-метоксиэтокси)алюмогидрида натрия и Н2 в присутствии металлического катализатора (например, катализатора, выбранного из группы, состоящей из палладия, платины, никеля, рутения, родия и его соединения (например, его оксида), необязательно на носителе, например угле). Если восстанавливающим агентом является H2, то катализатором является палладий (H2/Pd). Более предпочтительно восстанавливающий агент выбран из группы, состоящей из борана и комплекса боран-основание по Льюису, например B2H6, B10H14, BH3SMe2, BMS или BH3ТГФ. Наиболее предпочтительно восстанавливающий агент выбран из группы, состоящей из BMS и BH3ТГФ. Еще более предпочтительно восстанавливающим агентом является BMS.
Взаимодействие предпочтительно проводят в присутствии растворителя. Выбор подходящего растворителя является общепринятым и находится в рамках нормальной практики специалиста в данной области техники. Предпочтительно растворитель представляет собой полярный апротонный растворитель. Например, растворитель может быть растворителем, выбранным из группы, состоящей из тетрагидрофурана (ТГФ), 2-метилтетрагидрофурана (2-MeТГФ), метилциклопентилового эфира и дибутилового эфира и их смесей. Более предпочтительно растворитель выбран из группы, состоящей из ТГФ и 2-MeТГФ. В одном предпочтительном варианте осуществления растворитель представляет собой ТГФ. В другом предпочтительном варианте растворитель представляет собой 2-MeТГФ.
Предпочтительно температура реакции находится в диапазоне от 1 до 80°C. В некоторых предпочтительных вариантах осуществления температура реакции находится в диапазоне от 1 до 50, предпочтительно от 4 до 45, более предпочтительно от 5 до 40°C, например, в диапазоне от 5 до 30°C. В одном предпочтительном варианте осуществления реакцию проводят в диапазоне от 5 до 20°C, например реакцию можно проводить, начиная с 5-7°C, а затем, в процессе реакции, температуру повышают приблизительно до 20-30°C, например, приблизительно до 20°C.
В другом предпочтительном варианте осуществления реакцию проводят в диапазоне от 1 до 50°C (более предпочтительно от 4 до 45°C), например реакцию можно проводить, начиная приблизительно с 1-10°C (например, 3-7°C, предпочтительно 4-6°C), а затем, в процессе реакции, температуру повышают приблизительно до 20-30°C, например, приблизительно до 20-25°C. Более предпочтительно реакцию можно проводить, начиная приблизительно с 1-10°C (например, 3-7°C, предпочтительно 4-6°C), а затем, в процессе реакции, температуру повышают примерно до 5-15°C, например, до 5-13°C, затем приблизительно до 20-30°C, например, приблизительно до 20-25°C.
В одном из вариантов осуществления температуру реакции первоначально повышают до приблизительно 40-50°C. Например, температуру реакции повышают до 45°C, затем снижают приблизительно до 1°C (например, 3-7°C, например, 3, 4, 5, 6 или 7°C, предпочтительно около 4-6°C), а затем, в процессе реакции, температуру повышают приблизительно до 20-30°C, например, приблизительно 20-25°C. Более предпочтительно температуру реакции повышают до 45°C, потом охлаждают приблизительно до 1-10°C (например, до 3-7°C, например, 3, 4, 5, 6 или 7°C, предпочтительно приблизительно до 4-6°C), а затем температуру, в процессе реакции, повышают приблизительно до 5-15°C, например до 5-13°C, а далее приблизительно до 20-30°C, например, приблизительно до 20-25°C.
Предпочтительно молярное соотношение соединение (II):хлоруксусная кислота равно или меньше 1:2, предпочтительно равно или меньше 1:5, более предпочтительно равно или меньше 1:10, наиболее предпочтительно равно или меньше 1:20. В некоторых предпочтительных вариантах осуществления молярное соотношение соединение (II):хлоруксусная кислота составляет от 1:2 до 1:100; предпочтительно от 1:5 до 1:40; более предпочтительно 1:10 до 1:35; еще более предпочтительно 1:15 до 1:30; и наиболее предпочтительно от 1:20 до 1:28, например 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 1:27 или 1:28, предпочтительно 1:26).
Предпочтительно молярное соотношение соединение (II):восстанавливающий агент равно или меньше 1:1, предпочтительно равно или меньше 1:3, и более предпочтительно равно или меньше 1:7. В некоторых предпочтительных вариантах осуществления молярное соотношение соединение (II):восстанавливающий агент составляет от 1:1 до 1:50; предпочтительно от 1: 3 до 1:30; более предпочтительно 1:5 до 1:20; еще более предпочтительно 1:8 до 1:18; и наиболее предпочтительно от 1:10 до 1:15, например 1:10, 1:11, 1:12, 1:13, 1:14 или 1:25, предпочтительно 1:13.
Авторы настоящего изобретения также неожиданно обнаружили, что взаимодействие соединения (II) с получением соединения (III), как описано выше, дополнительно усовершенствуется, при условии ее осуществления в присутствии буферного агента, например буферного агента, обеспечиваемого солью хлоруксусной кислоты, который действует как буфер в сочетании с хлоруксусной кислотой. Авторы настоящего изобретения обнаружили, что использование соли хлоруксусной кислоты в качестве буферного агента дает в результате соединение (III) с еще более высоким выходом, с более высокой чистотой, по сравнению с процессом, в котором используют только хлоруксусную кислоту. Было отмечено, что в случае, когда взаимодействие осуществляют в присутствии соли хлоруксусной кислоты, возникает меньше побочных продуктов, в частности, меньше побочных продуктов, возникающих в результате удаления защитной группы PG. Например, когда соединение (II) представляет собой соединение (IIc), отмечается, что количество побочных продуктов, возникающих в результате удаления группы Boc, меньше.
В некоторых предпочтительных вариантах осуществления взаимодействие проводят в присутствии буферного агента. Буферным агентом является агент, который может оказывать действие для поддержания рН раствора около выбранного значения после добавления другой кислоты или основания. В неводном растворителе, обычно используемом для восстановления по изобретению, любое соединение, которое может удалять кислоту (то есть протоны) из раствора, можно считать буферным агентом. Например, буферный агент может быть слабой кислотой или основанием в сочетании с солью слабой кислоты или основания, например фосфорной кислотой и фосфатной солью, такой как фосфат натрия и/или гидрофосфат натрия и/или дигидрофосфат натрия. В случае, когда в реакции присутствует хлоруксусная кислота, буферным агентом может быть соль хлоруксусной кислоты.
В некоторых особенно предпочтительных вариантах осуществления буферный агент представляет собой соль хлоруксусной кислоты. Соль хлоруксусной кислоты может быть выбрана из группы, состоящей из хлорацетата натрия, хлорацетата калия, хлорацетата магния, хлорацетата кальция и их смесей. Предпочтительно соль хлоруксусной кислоты представляет собой хлорацетат натрия, то есть взаимодействие осуществляют в присутствии хлорацетата натрия.
Предпочтительно, когда взаимодействие осуществляют в присутствии соли хлоруксусной кислоты, количество соли хлоруксусной кислоты и хлоруксусной кислоты является таким, что получается буферный раствор. Буферный раствор представляет собой раствор, который препятствует изменению рН или, в неводных растворителях, количеству кислоты (то есть протонов) в растворе, при добавлении к нему небольшого количества кислоты или щелочи.
В некоторых вариантах осуществления, где взаимодействие осуществляют в присутствии соли хлоруксусной кислоты, авторы настоящего изобретения обнаружили, что образование побочного продукта минимизируется при использовании определенных молярных соотношений соединение (II):соль хлоруксусной кислоты. Предпочтительно, когда взаимодействие осуществляют в присутствии соли хлоруксусной кислоты, молярное соотношение соединение (II):соль хлоруксусной кислоты равно или меньше 1:3; более предпочтительно равно или меньше 1:4; и еще более предпочтительно равно или меньше 1:7, например равно или меньше 1:9, равно или меньше 1:12, равно или меньше 1:15. В некоторых предпочтительных вариантах осуществления молярное соотношение соединение (II):соль хлоруксусной кислоты составляет от 1:4 до 1:50; предпочтительно от 1:5 до 1:30; более предпочтительно от 1:7 до 1:20; и еще более предпочтительно от 1:8 до 1:15, например 1:10.
В некоторых предпочтительных вариантах осуществления молярное соотношение хлоруксусная кислота:соль хлоруксусной кислоты составляет по меньшей мере 1:1, предпочтительно по меньшей мере 2:1. В некоторых предпочтительных вариантах осуществления молярное соотношение хлоруксусная кислота:соль хлоруксусной кислоты составляет от 1:1 до 10:1, предпочтительно от 1:1 до 6:1; более предпочтительно от 2:1 до 5:1; и еще более предпочтительно от 2:1 до 4:1, например, 2,6:1).
Предпочтительно, когда взаимодействие осуществляется в присутствии соли хлоруксусной кислоты, для каждого молярного эквивалента соединения (I), имеется, по меньшей мере, 2 молярных эквивалента хлоруксусной кислоты, по меньшей мере, 1 молярный эквивалент соли хлоруксусной кислоты; и, по меньшей мере, 1 молярный эквивалент восстановителя. Более предпочтительно, для каждого молярного эквивалента соединения (I) имеется, по меньшей мере, 8 молярных эквивалентов хлоруксусной кислоты, по меньшей мере, 4 молярных эквивалента соли хлоруксусной кислоты; и, по меньшей мере, 4 молярных эквивалента восстановителя. Еще более предпочтительно, для каждого молярного эквивалента соединения (I) имеется, по меньшей мере, 14 молярных эквивалентов хлоруксусной кислоты, по меньшей мере, 7 молярных эквивалентов соли хлоруксусной кислоты; и, по меньшей мере, 7 молярных эквивалентов восстановителя. Еще более предпочтительно, для каждого молярного эквивалента соединения (I) имеется, по меньшей мере, 20 молярных эквивалентов хлоруксусной кислоты, по меньшей мере, 7 молярных эквивалентов соли хлоруксусной кислоты; и, по меньшей мере, 10 молярных эквивалентов восстановителя. Наиболее предпочтительно, для каждого молярного эквивалента соединения (I) имеется, по меньшей мере, 24 молярных эквивалента хлоруксусной кислоты (например, 24, 26, 28 или 30 молярных эквивалентов), по меньшей мере 9 молярных эквивалентов соли хлоруксусной кислоты (например, 9, 10, 12 или 15 молярных эквивалентов); и, по меньшей мере, 12 молярных эквивалентов восстановителя (например, 12, 13 или 15 молярных эквивалентов).
В некоторых предпочтительных вариантах осуществления, для каждого одного молярного эквивалента соединения (I), существует от 2 до 60 молярных эквивалентов хлоруксусной кислоты, от 1 до 50 молярных эквивалентов соли хлоруксусной кислоты; и от 1 до 30 молярных эквивалентов восстановителя. Более предпочтительно, от 8 до 60 молярных эквивалентов хлоруксусной кислоты, от 4 до 50 молярных эквивалентов соли хлоруксусной кислоты; и от 4 до 30 молярных эквивалентов восстановителя. Еще более предпочтительно, для каждого молярного эквивалента соединения (I), существует от 14 до 40 молярных эквивалентов хлоруксусной кислоты, от 7 до 25 молярных эквивалентов соли хлоруксусной кислоты; и от 7 до 20 молярных эквивалентов восстановителя; и наиболее предпочтительно, для каждого молярного эквивалента соединения (I), существует от 24 до 30 (например, 26) молярных эквивалентов хлоруксусной кислоты, от 9 до 15 (например, 10) молярных эквивалентов соли хлоруксусной кислоты; и от 12 до 15 (например, 13) молярных эквивалентов восстановителя.
Предпочтительно, как только стадия (с) реакции завершается, реакцию гасят полярным протонным растворителем, например спиртом или водой. В одном предпочтительном варианте осуществления, реакцию гасят этанолом. В другом предпочтительном варианте осуществления, реакцию гасят водой.
В некоторых предпочтительных вариантах осуществления, соединение (III), образованное в соответствии с настоящим изобретением, перекристаллизовывается в одном или нескольких растворителях (например, растворитель, выбранный из этанола, этилацетата, ацетона, 2-MeТГФ и их смесей) и одном или нескольких антирастворителях (например, гептан) для удаления примесей. В вариантах осуществления, где соединение (III) представляет собой соединение (IIIc), авторы настоящего изобретения обнаружили, что растворение соединения (IIIc) в этаноле, этилацетате, ацетоне или 2-MeТГФ при повышенных температурах, понижение температуры и добавление, например, гептана, может повысить степень чистоты. Например, повторная кристаллизация при 50°C приблизительно в семи объемах ацетона с последующим добавлением гептана и охлаждением может повысить чистоту продукта с 96,8 до 98,6% по площади ВЭЖХ. В некоторых вариантах осуществления, соединение (III) (например, соединение (IIIc)) подвергается повторной кристаллизации в смеси ацетон/гептан или смеси 2-MeТГФ/гептан.
В некоторых вариантах осуществления, в случае, если реакцию гасят водой, после добавления воды соединение (III) может быть осаждено из реакционного раствора (например, путем охлаждения реакционного раствора приблизительно до 4-8°C), а затем повторно растворено в реакционном растворе при повышенных температурах (например, около 30-40°C). Потом органическая и водная фазы могут быть отделены и органическая фаза промыта (например, промыта водным солевым раствором (например, водным NaCl, предпочтительно 20% водным раствором NaCl)). Далее органическая фаза может быть концентрирована с осаждением соединения (III). Предпочтительно соединение (III) затем может быть промыто растворителем, например апротонным растворителем или смесью апротонного растворителя и, более предпочтительно, смесью 2-MeТГФ/гептан. В некоторых вариантах осуществления, в случае, если соединение (III) представляет собой соединение (IIIc), авторы настоящего изобретения обнаружили, что эти стадии выделения обеспечивают удивительно высокую чистоту соединения (IIIc) без необходимости проведения дополнительных стадий очистки (например, повторная кристаллизация).
Лишенный защиты продукт соединения (III) или (IIIa) в соответствии с настоящим изобретением представляет собой соединение формулы (III) или (IIIc), в котором удалена защитная группа PG или, когда R представляет собой ОН в соответствующим образом защищенной форме, удалена защитная группа ОН. Предпочтительно удалена защитная группа PG (например, соединение формулы (I)). Более предпочтительно удалена защитная группа PG и, когда R представляет собой ОН в соответствующим образом защищенной форме, удалена защитная группа ОН.
Лишенный защиты продукта соединения (IIIb) или (IIIc) в соответствии с настоящим изобретением представляет собой соединение формулы (IIIb) или (IIIc), в котором удалена защитная группа PG (например, соединение формулы (Ib)).
В некоторых вариантах осуществления изобретения образуется соль соединения (III) или соль лишенного защиты продукта соединения (III). Таким образом, изобретение также предоставляет способ получения соли соединения (III) (например, соли соединения (IIIa), (IIIb) или (IIIc)) или соли лишенного защитного продукта соединения (III) (например, соль лишенного защитного продукта соединения (IIIa), (IIIb) или (IIIc)), включая способ получения соединения (III) (например, (IIIa), (IIIb) или (IIIc)), как описано выше, и стадию образования соли, например стадию образования хлористоводородной соли.
Стадия образования соли соединения (III) или соли лишенного защитного продукта соединения (III) может быть отдельной стадией на стадии образования соединения (III) или его лишенного защиты продукта; или стадия образования соли может быть осуществлена как часть стадии образования соединения (III) или его лишенного защиты продукта.
Способ может необязательно включать дополнительную стадию (d) удаления защитной группы PG соединения (III) с получением соединения (I) или его соли:
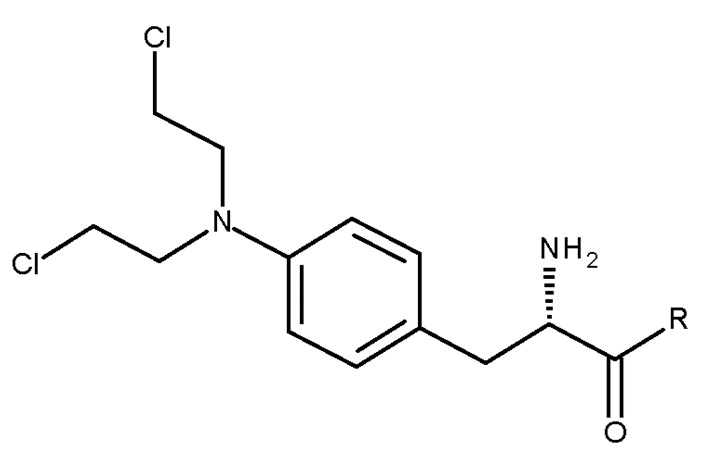
(I);
где PG и R являются такими, как определено выше для стадии (с).
Предпочтительно, соль соединения (I) представляет собой фармацевтически приемлемую соль (то есть соли соединения (I), которые являются фармацевтически приемлемыми, пригодны для использования в медицине, являются такими солями, где противоион является фармацевтически приемлемым). Предпочтительно соль соединения (I) представляет собой фармацевтически приемлемую кислую соль и, более конкретно, хлористоводородную соль.
В частности, подходящие соли, образованные с помощью кислот по изобретению, включают соли, образованные минеральными кислотами, сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, содержащие 1-4 атома углерода, которые являются незамещенными или замещенными, например, галогеном, такими как насыщенные или ненасыщенные дикарбоновые кислоты, такие как гидроксикарбоновые кислоты, такие как аминокислоты, или органическими сульфоновыми кислотами, такими как (C1-C4)-алкил- или арилсульфоновые кислоты, которые являются незамещенными или замещенными, например галогеном. К фармацевтически приемлемым кислотно-аддитивным солям относятся соли, образованные соляной, бромистоводородной, серной, азотной, лимонной, винной, уксусной, фосфорной, молочной, пировиноградной, уксусной, трифторуксусной, янтарной, перхлорной, фумаровой, малеиновой, гликолевой, молочной, салициловой, оксалоуксусной, метансульфоновой, этансульфоновой, п-толуолсульфоновой, муравьиной, бензойной, малоновой, нафталин-2-сульфоновой, бензолсульфоновой, изэтионовой, аскорбиновой, яблочной, фталевой, аспарагиновой и глутаминовой кислотами, лизином и аргинином.
В предпочтительных вариантах изобретения получают соль соединения (I). Более предпочтительно получают фармацевтически приемлемую соль и, наиболее предпочтительно, фармацевтически приемлемую кислую соль и, более конкретно, хлористоводородную соль.
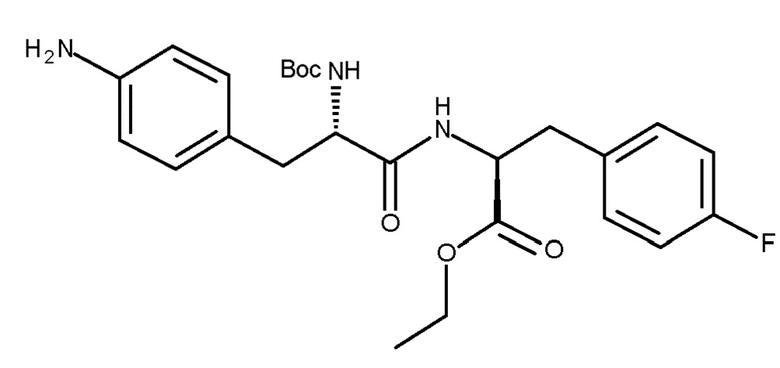
В предпочтительном варианте осуществления R представляет собой , и стадия (d) включает удаление защитной группы PG соединения (IIIb) с получением соединения (Ib) (мелфлуфен) или его соли; где PG является такой, как определено для стадии (c) выше.
Предпочтительно получают соль соединения (Ib). Более предпочтительно получают фармацевтически приемлемую соль, наиболее предпочтительно, фармацевтически приемлемую кислую соль и, более конкретно, хлористоводородную соль.
Для осуществления удаления защитной группы PG на стадии (d) может быть использован очень широкий диапазон условий реакции. Необходимые условия реакции на стадии (d) зависят от природы защитной группы PG. Выбор условий для удаления защитной группы, в соответствии с приведенном выше определением PG на стадии (с), является обычным и находится в рамках обычной практики специалиста в данной области техники. Например, если защитная группа представляет собой карбаматную группу, для удаления защитной группы может быть использована кислота, такая как хлористоводородная кислота (HCl) или трифторуксусная кислота (TFA) (предпочтительно HCl). Подходящие условия для удаления защитных групп рассмотрены, например, в Wuts, P.G. M., Greene's Protective Groups in Organic Synthesis, 5th Edition (2014) John Wiley & Sons, Inc.
Например, если PG представляет собой Boc (т.е. соединение (III) представляет собой соединение (IIIa)), стадия (d) включает взаимодействие соединения (IIIa) в кислых условиях реакции с получением соединения (I) (мелфлуфен или мелфалан) или его соли (предпочтительно его хлористоводородной соли); где PG и R являются такими, как определено выше для стадии (с).
В другом предпочтительном варианте осуществления PG представляет собой Boc, и R представляет собой , и стадия (d) включает удаление защитной группы PG соединения (IIIc) с получением соединения (Ib) (мелфлуфена) или его соли. Предпочтительно получают соль соединения (Ib). Более предпочтительно получают фармацевтически приемлемую соль соединения (Ib), наиболее предпочтительно фармацевтически приемлемую кислую соль соединения (Ib) и, более конкретно, хлористоводородную соль соединения (Ib):
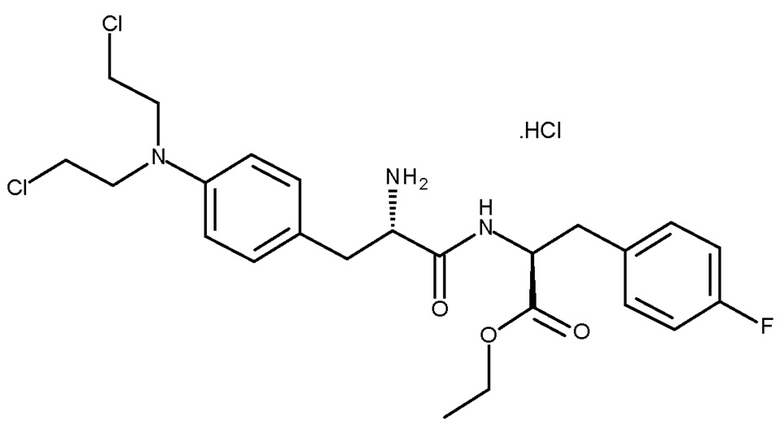 .
.
В вариантах осуществления, где PG представляет собой кислото-неустойчивую защитную группу, например Boc, предпочтительно соединение (III) (например, соединение (IIIa) или (IIIc)) подвергают взаимодействию в условиях кислой реакции, предпочтительно с HCl, для удаления защитной группы, с образованием соединения (I) (например, (Ib)) или его соли. Предпочтительно образуется хлористоводородная соль соединения (I) (например, (Ib)).
Примеры подходящего источника HCl для стадии (d) включают 1,3 М HCl/EtOH, 2,5 М HCl/EtOH, 1 М HCl/EtOAc, 3 М HCl/EtOH и 5-6 М HCl/iPrOH. Предпочтительно молярное соотношение соединение (III):HCl составляет от 1:1: до 1:50; более предпочтительно от 1:3 до 1:30; еще более предпочтительно 1:5 до 1:20; наиболее предпочтительно от 1:7 до 1:17, например 1:10.
Предпочтительно растворитель для удаления защитной группы выбран из группы, состоящей из этанола (EtOH), изопропилового спирта, этилацетата, ТГФ, ацетона и их смесей. Например, реакцию можно проводить в смеси HCl/EtOH, этилацетат/HCl/EtOH, ацетон/HCl/EtOH или ТГФ/HCl/EtOH. Например, HCl/EtOH, ацетон/HCl/EtOH или ТГФ/HCl/EtOH. Предпочтительно реакцию проводят в смеси HCl/EtOH или этилацетат/HCl/EtOH.
Продукт стадии (d) может быть очищен, например, очищен путем промывки растворителем (например, одной или несколькими промывками этанолом, например 3 промывками этанолом) и/или кристаллизацией (например, перекристаллизацией в этаноле и/или перекристаллизацией в метил-трет-бутиловый эфир, например, перекристаллизацию в метил-трет-бутиловом эфире или, например, перекристаллизацией в метил-трет-бутиловом эфире, с последующей перекристаллизацией в этаноле).
Предпочтительно удаление защитной группы представляет собой отдельную стадию после стадии (с), на которой образуется N,N-бис-хлорэтил амин. Однако в некоторых вариантах осуществления удаление защитной группы может быть осуществлено как часть стадии (с) в синтезе, осуществляемом в одном реакционном сосуде. В этом случае, способ по изобретению непосредственно обеспечивает лишенный защиты продукт, соединение (I).
В некоторых вариантах осуществления изобретения образуется соль соединения (I) (например, соль соединения (Ib)). Таким образом, изобретение также обеспечивает способ получения соли соединения (I) (например, соли соединения (Ib)), включающий способ получения соединения (I) (например, (Ib)), как описано выше, и стадию образования соли соединения (I) (например, соли соединения (Ib)), например стадию образования хлористоводородной соли.
Стадия образования соли соединения (I) (например, соли соединения (Ib)) может представлять собой отдельную стадию после стадии удаления защитной группы, или стадия образования соли может быть проведена как часть стадии удаления защитной группы (d). В одном предпочтительном варианте осуществления стадию образования соли соединения (I) (например, соли соединения (Ib), предпочтительно хлористоводородной соли соединения (Ib)), выполняют как часть стадии удаление защитной группы (d). В таком варианте осуществления предпочтительно PG представляет собой кислото-неустойчивую защитную группу, например Boc; и предпочтительно соединение (III) (например, соединение (IIIa) или (IIIc)) подвергается взаимодействию в условиях кислой реакции и более предпочтительно с HCl, для удаления защитной группы с образованием соединения (I) (например, (Ib)) или его соли. Более предпочтительно, образуется хлористоводородная соль соединения (I) (например, (Ib)).
В другом варианте осуществления, стадия образования соли соединения (I) (например, соли соединения (Ib)) и стадия удаления защитной группы может быть проведена как часть стадии (с) в синтезе, осуществляемом в одном реакционном сосуде. В этом случае способ согласно изобретению непосредственно обеспечивает соль лишенного защиты продукта, соединение (I).
Авторы настоящего изобретения дополнительно обнаружили новый способ, включающий взаимодействие двух замещенных фенилаланинов: PG-p-нитро-L-фенилаланин (соединение (IV)) и сложный этиловый эфир p-фтор-L-фенилаланина (соединение (V)), где PG является таким, как определено для стадии (с) выше, с получением соединения (VIb):
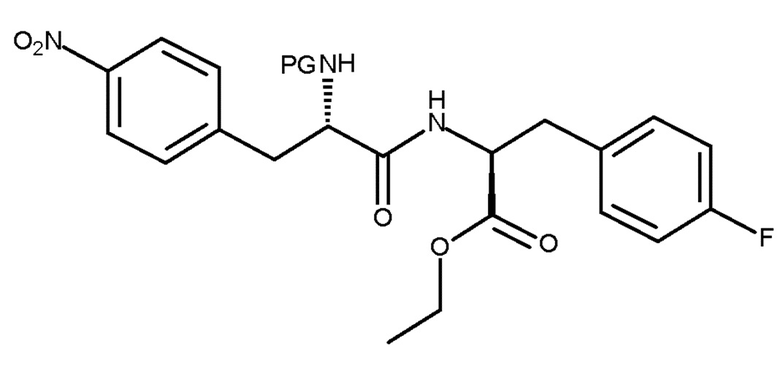
(VIb)
Аминокислоты связываются с образованием соединения (VIb). Затем соединение (VIb) может быть восстановлено с образованием соединения ароматического амина (IIb). В способе получения соединения (IIb) используются нетоксичные исходные вещества и, следовательно, при использовании в способе для синтеза мелфлуфена, не допускается образование токсичных продуктов до проведения заключительных стадий. Как таковой этот способ является намного безопаснее, чем применение исходного вещества, содержащего бис-(2-хлорэтил), используемого в известном способе синтеза мелфлуфена.
Таким образом, настоящее изобретение также обеспечивает способ получения соединения (VIb), включающий следующую стадию:
(a) взаимодействие соединения (IV):
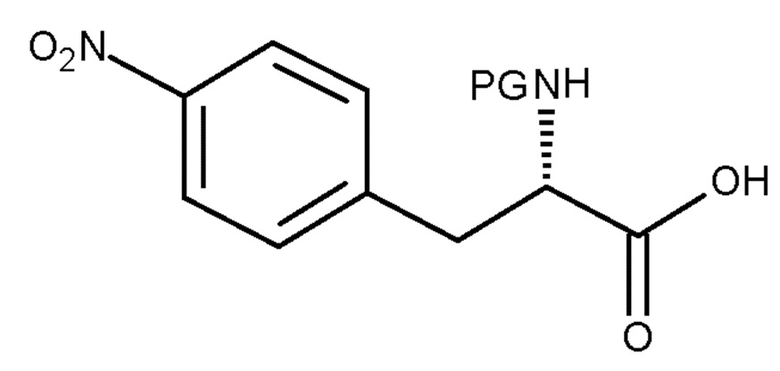
(IV)
с соединением (V):
(V)
где PG является таким, как определено выше для стадии (c).
В некоторых предпочтительных вариантах осуществления, PG представляет собой Boc, и способ включает следующую стадию:
(a) взаимодействие соединения (IVc):
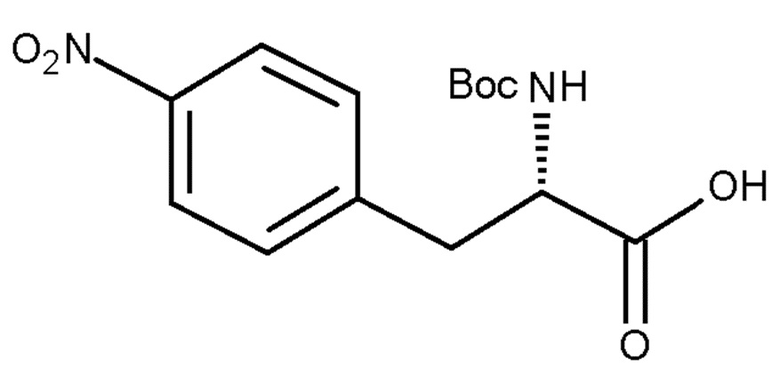
(IVc)
с соединением (V) с получением соединения (VIc):
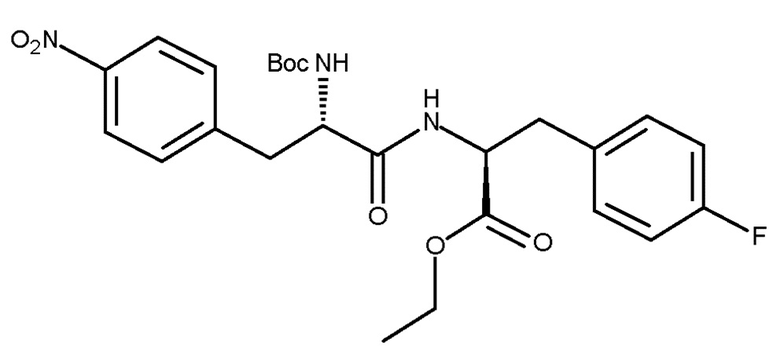
(VIc).
Стадию (а) способа по изобретению можно проводить в любых условиях, подходящих для реакции амидного связывания. Выбор условий реакции амидного связывания является общепринятым и находится в рамках обычной практики специалиста в данной области техники. Необходимые условия реакции могут зависеть от природы защитной группы PG.
Реагенты для реакции амидного связывания, подходящие для использования в настоящем изобретении, включают карбодиимиды, например дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC), этил-(N',N'-диметиламино)пропилкарбодиимид гидрохлорид (EDC); реагенты на основе фосфония, например гексафторфосфат (бензотриазол-1-илокси)трис(диметиламино)фосфония (BOP), гексафторфосфат бензотриазол-1-ил-окситрипирролидинофосфония (PyBOP); на основе аминов, например, гексафторфосфат N,N,N',N'-тетраметил-O-(1H-бензотриазол-1-ил)урония (HBTU), гексафторфосфат 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида (HATU), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(6-хлорбензотриазол-1-ил)-N,N,N',N'-тетраметилфурония (HCTU), тетрафторборат O-(3,4-дигидро-4-оксо-1,2,3-бензотриазин-3-ил)-N,N,N',N'-тетраметилурония (TDBTU); реагенты на основе на основе иммония аминия, например гексахлорантимонат (1H-бензотриазол-1-илокси)-N,N-диметилметаниминия (BOMI), гексахлорантимонат 5-(1Н-бензотриазол-1-илокси)-3,4-дигидро-1-метил 2Н-пирролия (BDMP) и гексахлорантимонат 5-(7-азабензотриазол-1-илокси)-3,4-дигидро-1-метил-2-пирролия (AOMP); агенты, образующие хлорангидриды, например тионилхлорид, пентахлорид фосфора, трифосген, триазины (например, цианурхлориды, цианурфториды и их производные), гексафторфосфат тетрафторформамидиния (TFFH), бис(тетраметилен)фторформамидиний (BTFFH) и гексафторфосфат 1,3-диметил-2-фтор-4,5-дигидро-1H-имидазолия (DFIH); или другие реагенты для реакции связывания, например 3-(диэтилфосфорилокси)-1,2,3-бензотриазин-4(3H)-он (DEPBT), карбонилдиимидазол (CDI), пропилфосфоновый ангидрид (T3P) и N-этоксикарбонил-2-этокси-1,2-дигидрохинолин (EEDQ).
В условия амидного связывания, для подавления рацемизации, может быть включен дополнительный агент, например гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt), этил-2-циано-2-(гидроксиимино)ацетат (оксима). Предпочтительно, дополнительный агент для подавления рацемизации представляет собой HOBt.
В некоторых предпочтительных вариантах осуществления,
амидное связывание выполняется с использованием HATU или HBTU с N,N-диизопропилэтиламином (DIPEA) или N-метилморфолином (NMM); с использованием EDC и гидроксибензотриазола (HOBt) с DIPEA или NMM; с использованием пропилфосфонового ангидрида (T3P) с DIPEA; или с использованием PyBOP с DIPEA.
Предпочтительно стадия (а) осуществляется в присутствии EDC. Более предпочтительно стадия (а) осуществляется в присутствии EDC, HOBt и NMM. Используя эти условия реакции, соединение (VIb) можно получить с очень высокой исходной чистотой, например, с чистотой 96%. Наиболее предпочтительно стадия (а) осуществляется в присутствии 1,1 эквивалента EDC, 0,1 эквивалента HOBt, 3,5 эквивалента NMM и с использованием приблизительно 1 эквивалента каждого из соединения (IV) и соединения (V).
Выбор растворителя для конкретных условий реакции амидного связывания является общепринятым и находится в рамках обычной практики специалиста в данной области техники. Предпочтительно растворителем для реакции является апротонный растворитель. Например, растворитель может быть выбран из группы, состоящей из этилацетата, ацетона, ТГФ, 2-MeТГФ, дихлорметана, диметилформамида; циклопентил метиловый сложный эфир и их смесей. Предпочтительно растворитель выбран из группы, состоящей из ДМФ, этилацетата, ацетона, 2-MeТГФ и их смесей. Более предпочтительно растворителем является этилацетат или ацетон. Авторы настоящего изобретения обнаружили, что ацетон является неожиданно эффективным растворителем для реакции амидного связывания по изобретению, сохраняя реакционную смесь в виде раствора в процессе реакции. Таким образом, наиболее предпочтительным растворителем является ацетон.
Продукт, полученный на стадии (а), может быть также очищен до следующей стадии реакции, например, очищен кристаллизацией. Альтернативно, продукт, полученный на стадии (а), может быть использован непосредственно на стадии (b) (описанной ниже) без дополнительного выделения или очистки соединения (VIb).
Было обнаружено, что чистота продукта, на каждой стадии реакции от (а) до (d), имеет важное значение, поскольку примеси на каждой стадии могут проходить через этот процесс, например, чистота продукта на стадии (а) оказывает влияние на чистоту последующих стадий реакции (b), (с) и (d). Поэтому важно, чтобы каждая стадия способа обеспечивала продукт с максимально высокой степенью чистоты, чтобы получить наиболее высокую чистоту соединения (III) и, следовательно, соединения (I). Каждая из стадий (а)-(d) по настоящему изобретению обеспечивает продукты с высокой степенью чистоты, что делает общий способ реакции особенно эффективным для получения соединения (III) и, следовательно, соединения (I), имеющего высокую чистоту.
Настоящее изобретение также относится к способу по изобретению для получения соединения (II), включающему следующую стадию:
(b) взаимодействие соединения (VI)
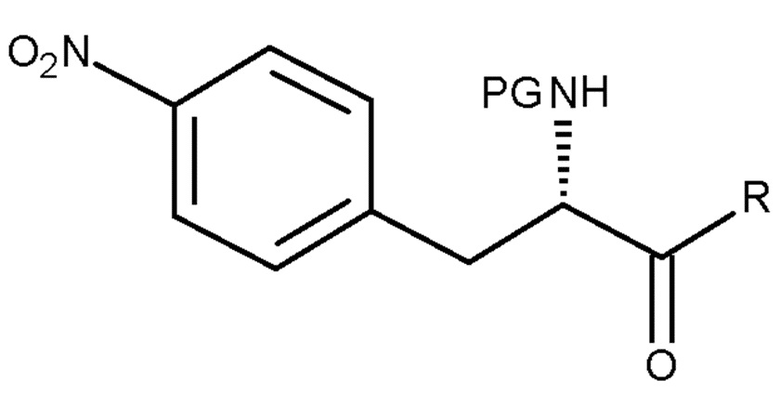
(VI)
с восстанавливающим агентом,
где PG является таким, как определено выше для стадии (c), и R представляет собой OH необязательно в соответствующим образом защищенной форме или .
Например, соединение формулы (VI) может представлять собой: этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(метилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(этилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(9-флуоренилметилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(трет-бутоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(бензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(p-метоксибензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(1-адамантилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(p-бромбензилоксикарбониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(трифторацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(хлорацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(фенилацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(бензацетиламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(p-толуолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(2-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(t-бутилсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(4-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(2-нитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат, этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(2,4-динитробензолсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат или этил (2S)-2-[[(2S)-3-(4-нитрофенил)-2-(2-нафталинсульфониламино)пропаноил]амино]-3-(4-фторфенил)пропаноат.
Например, если PG представляет собой Boc, способ может включать стадию:
(b) взаимодействие соединения (VIa):
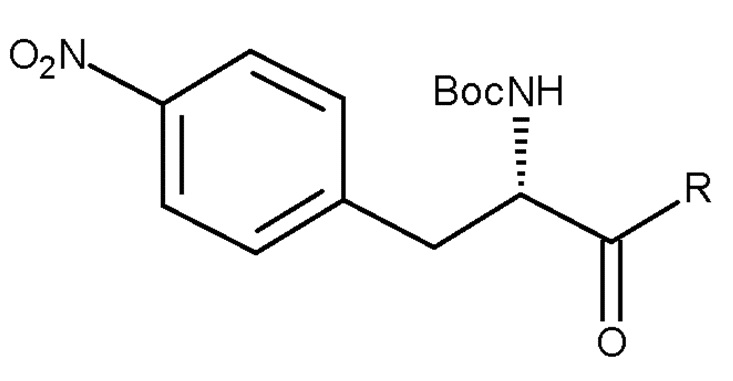
(VIa)
с восстанавливающим агентом, например H2/Pd/C,
с получением соединения (IIa), где R представляет собой OH необязательно в соответствующим образом защищенной форме или .
В некоторых предпочтительных вариантах осуществления, в случае, если R представляет собой ,
способ может включать стадию:
(b) взаимодействие соединения (VIb) с восстанавливающим агентом, например H2/Pd/C,
с получением соединения (IIb)
где PG является таким, как определено на стадии (с) выше.
В некоторых предпочтительных вариантах осуществления, если PG представляет собой Boc, и R представляет собой , способ может включать стадию:
(b) взаимодействие соединения (VIc) с восстанавливающим агентом, например H2/Pd/C,
с получением соединения (IIc).
Стадию (b) способа по изобретению можно проводить в условиях, подходящих для восстановления нитрогруппы до аминогруппы. Необходимые условия реакции могут зависеть от природы защитной группы PG. Выбор условий реакции восстановления является общепринятым и находится в рамках нормальной практики специалиста в данной области техники.
В некоторых предпочтительных вариантах осуществления, реакция восстановления на стадии (b) может представлять собой реакцию гидрирования, более предпочтительно реакцию каталитического гидрирования, то есть реакцию гидрирования проводят в присутствии водорода и катализатора. Следовательно, восстановителем на стадии (b) может быть водород (Н2) и катализатор. Предпочтительно катализатор представляет собой металлический катализатор, например катализатор, выбранный из группы, состоящей из платины, палладия, родия, рутения, никеля (например, никеля Ренея или никеля Урушибары), железа и его соединения (например, его оксида). Катализатор может быть гомогенным или гетерогенным; предпочтительно он является гетерогененным. Предпочтительно катализатор находится на носителе катализатора, например носителе катализатора, выбранного из группы, состоящей из углерода, оксида алюминия, диоксида кремния и карбоната кальция. Наиболее предпочтительно носителем катализатора является углерод.
В предпочтительном варианте осуществления изобретения катализатором является палладий (Pd) и более предпочтительно палладий на угле (Pd/C), то есть в реакции восстановления (b) используется газ H2 с катализатором Pd/C (H2/Pd/C) (т.е. восстановитель на стадии (b) представляет собой H2/Pd/C). Более предпочтительно, катализатор Pd/C представляет собой Pd, с содержанием 1-15%, на активированном угле, предпочтительно 1-10% Pd на активированном угле, более предпочтительно 3-6% Pd на активированном угле, наиболее предпочтительно 3-5% Pd на активированном угле. Предпочтительно катализатором Pd/C является катализатор с влажностью приблизительно 50%.
Предпочтительно, давление Н2 составляет от 1 до 8 бар, более предпочтительно от 1 до 3 бар. Предпочтительно, катализатор присутствует в количестве от 1 до 30 масс%, более предпочтительно от 3 до 20 масс%; еще более предпочтительно от 3 до 10 масс% и наиболее предпочтительно от 3 до 6 масс%, например 3, 4, 4,5, 5 или 6 масс%.
Выбор растворителя для реакции гидрирования является общепринятым и находится в рамках обычной практики специалиста в данной области техники. Примеры подходящих растворителей включают 2-MeТГФ, этилацетат, этанол и их смеси. Предпочтительно растворителем является 2-MeТГФ.
После завершения, катализатор может быть удален фильтрованием, например, фильтрованием через углеродный фильтр.
Для удаления примеси перед использованием на стадии (с) способа, продукт стадии (b) (соединение (II)) может быть кристаллизован в одном или нескольких растворителях (например, этаноле, этилацетате, 2-MeТГФ и их смеси) и одном или нескольких антирастворителях (например, гептан). Авторы настоящего изобретения обнаружили, что растворение соединения (II) в, например, этаноле, этилацетате или 2-MeТГФ, или их смесях при повышенных температурах, понижении температуры и добавлении, например, гептана, повышает чистоту. Предпочтительно, соединение (II) кристаллизуется из смеси 2-MeТГФ/гептан.
Стадия (b), как описано выше, может использоваться в сочетании со стадиями (с) и/или (d), описанными выше. Таким образом, настоящее изобретение обеспечивает способ, включающий одну или несколько следующих стадий:
(b) Способ получения соединения (II), включающий взаимодействие соединения (VI) с восстанавливающим агентом;
(c) Способ получения соединения (III) или его лишенного защиты продукта или соли его лишенного защиты продукта, включающий взаимодействие соединения (II) с хлоруксусной кислотой в присутствии восстановителя; и/или
(d) Способ получения соединения (I) или его соли, включающий удаление защитной группы соединения (III) с получением соединения (I) или его соли;
где PG представляет собой защитную группу, и R представляет собой OH необязательно в соответствующим образом защищенной форме или . Предпочтительно PG представляет собой Boc.
В одном предпочтительном варианте осуществления, стадия (с) представляет собой способ получения лишенного защиты продукта соединения (III), и способ дополнительно включает стадию образования соли лишенного защиты продукта соединения (III). В другом варианте осуществления, стадия (d) представляет собой способ получения соли соединения (I), и способ дополнительно включает стадию образования соли соединения (I).
В одном предпочтительном варианте осуществления, где R представляет собой , стадии (a) и/или (b), как описано выше, можно использовать в сочетании со стадиями (c) и/или (d), описанными выше. Таким образом, настоящее изобретение обеспечивает способ, включающий одну или несколько из следующих стадий:
(a) Способ получения соединения (VIb), включающий взаимодействие соединения (IV) с соединением (V); и/или
(b) Способ получения соединения (IIb), включающий взаимодействие соединения (VIb) с восстанавливающим агентом; и/или
(c) Способ получения соединения (IIIb) или его лишенного защиты продукта, включающий взаимодействие соединения (IIb) с хлоруксусной кислотой в присутствии восстановителя; и/или
(d) Способ получения соединения (Ib) или его соли, включающий удаление защитной группы соединения (IIIb) с образованием соединения (Ib) или его соли,
где PG является таким, как определено выше для стадии (c).
В таком варианте осуществления предпочтительно PG представляет собой Boc.
В одном предпочтительном варианте осуществления стадия (с) представляет собой способ получения лишенного защиты продукта соединения (IIIb), и этот способ дополнительно включает стадию образования соли лишенного защиты продукта соединения (IIIb). В другом предпочтительном варианте осуществления, стадия (d) представляет собой способ получения соли соединения (Ib), и этот способ дополнительно включает стадию образования соли соединения (Ib).
Изобретение также относится к соединению (III), (IIIa), (IIIb), (IIIc) или его лишенному защиты продукту или соли его лишенного защиты продукта, полученному описанным здесь способом; соединению формулы (I), (Ib) или его соли, полученному описанным здесь способом; соединению формулы (VIb), полученному описанным здесь способом; или соединению формулы (II) или (IIc), полученное описанным здесь способом.
Также в настоящем документе описан способ получения соединения (X), или его соли, необязательно в надлежащим образом защищенной форме:
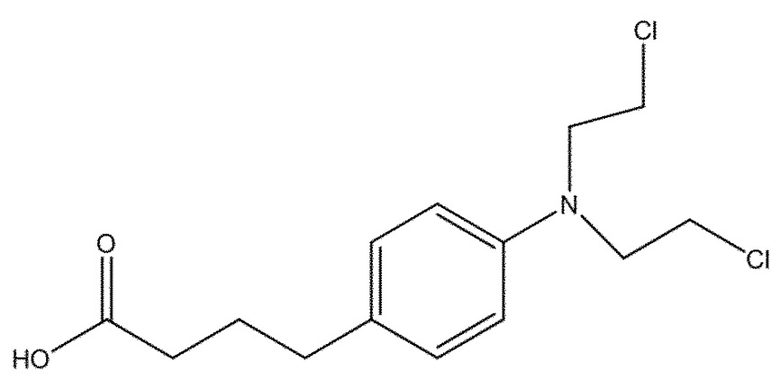
(X)
включающий следующую стадию:
(c) взаимодействие соединения (XI)
(XI)
с хлоруксусной кислотой, в присутствии восстановителя и необязательно в подходящем растворителе.
Предпочтительными условиями реакции являются условия, предпочтительные для стадии (с), описанные выше.
Примеры
Общие экспериментальные данные
Если не указано иное, все реагенты/растворители были приобретены из коммерческих источников и использованы без дополнительной очистки.
Соединения очищали на системе ВЭЖХ, оборудованной C18 колонкой с обращенной фазой, и использовали УФ-детектирование при длине волны 262 нм. Все соединения разделяли с градиентом ацетонитрила в воде.
Пример 1 - Синтез соединения (VIc)
(VIc)
В реактор, оборудованный устройством с верхним перемешиванием, впускным отверстием для азота и обратным холодильником, загружали Boc-нитрофениланенанин (соединение (IVc)) (35,0 г, 112,8 ммоль, 1 экв.), затем ацетон (420 мл), N-метилморфолин (43,4 мл, 394,8 ммоль, 3,5 экв.), гидрохлорид сложного этилового эфира фтор-L-фенилаланина (соединение (V)) (28,5 г, 115 ммоль, 1,02 экв.), EDC (23,8 г, 124,1 ммоль, 1,1 экв.) и HOBt⋅H2O (1,7 г, 11,3 ммоль, 0,1 экв.). Взвесь перемешивали при комнатной температуре в течение 18,5 ч, что обеспечивало, по данным ВЭЖХ, полный расход соединения (IVc). Добавляли воду (180 мл) и 2-MeТГФ (965 мл). Затем из прозрачной двухфазной смеси оранжевого цвета упариванием удаляли приблизительно 640 г растворителя (TJ:35°C). Потом добавляли 360 мл 2-MeТГФ и дважды упаривали. Водную фазу подкисляли до значения рН, составляющего 3, добавлением 58 мл 2М серной кислоты. Органический слой нагревали до 35-40°С и затем последовательно промывали водой (90 мл), дважды насыщенным водным раствором NaHCO3 (90 мл) и далее насыщенным солевым раствором (90 мл) и, наконец, водой (90 мл). Перед тем как температуру смеси доводили до значения комнатной температуры в течение ночи при перемешивании, к растворенному 2-MeТГФ продукту добавляли по каплям гептан (270 мл) при 35-40°С. Перед тем как взвесь бежевого цвета охлаждали до 10°С, добавляли по каплям еще 135 мл гептана. Продукт выделяли и промывали 100 мл холодной смеси 2-MeТГФ/гептана 6/4. Продукт (VIc) хранили влажным (82,5 г). Небольшой образец продукта анализировали по пределу обнаружения (LOD), который показал, что твердое вещество содержит 43,8% остатков растворителя. Исходя из этого, очищенный продукт получали с выходом 82%. Чистоту определяли посредством ВЭЖХ: 99,4% по площади.
1H-ЯМР (300 МГц, ДМСО-D6) δ 8,48 (ушир. д, 1H, J=7,5 Гц), 8,16 (2H, д, J=8,7 Гц), 7,55 (2H, д, J=9 Гц), 7,28 (2H, дд, J=8,7, 8,1 Гц), 7,12-7,02 (3H, м), 4,49 (1H, дд, J=14,4, 7,2 Гц), 4,32-4,24 (1 H, м), 4,04 (2H, дд, J=14,4, 7,2 Гц), 3,08-2,95 (3H, м), 2,84 (1H, дд, J=13,2, 10,8 Гц), 1,27 (с, 9H), 1,11 (3H, т, J=7,2Гц)
13C-ЯМР (75 МГц, ДМСО-D6) δ 171,4 (C=O), 171,2 (C=O), 161,2 (C-F, д, J=242,3 Гц), 155,2 (C=O), 146,6 (C), 146,2 (C), 133,1 (C), 131,1 (2 атома углерода, CH, д, J=8,3 Гц), 130,6 (2 атома углерода, CH), 123,1 (C), 114,9 (2 атома углерода, CH, J=20,4 Гц), 78,1 (C), 60,6 (CH2), 55,1 (CH), 53,6 (CH), 37,3 (CH2), 35,9 (CH2), 28,0 (3 атома углерода, CH3), 14,0 (CH3)
Пример 2 - Синтез соединения (IIc)
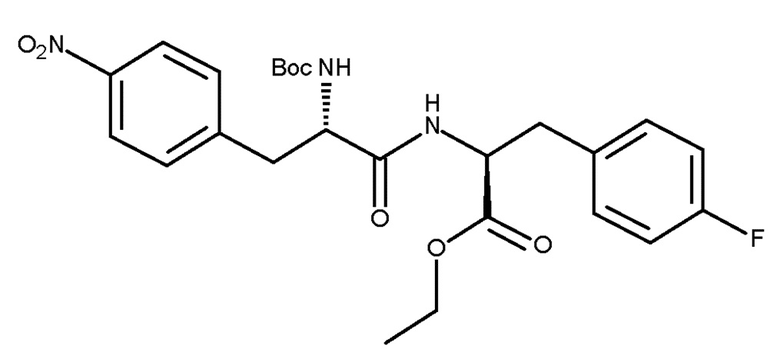
(IIc)
В автоклав для гидрирования добавляли увлажненное твердое соединение (VIc) (приблизительно 4,9 г сухой массы, 9,7 ммоль, 1 экв.), 2-MeТГФ (75 мл) и 3 масc% 5% Pd/C-катализатора (147 мг, влажность 50%). Реакционную смесь дегазировали азотом и затем заполняли газообразным водородом при давлении 1 бар (0,1 мПа). Перемешивание устанавливали на 600 об/мин и TJ равной 36°C. Взаимодействие осуществляли в течение четырех часов. Автоклав для гидрирования промывали 10 мл 2-MeТГФ и промывочную часть добавляли к реакционному раствору в колбе Эрленмейера. Затем добавляли древесный уголь (250 мг, 5 масc%) и полученную смесь, перед ее фильтрацией, перемешивали в течение 15 минут при комнатной температуре. Фильтр промывали 10 мл 2-MeТГФ и промывочную часть добавляли к фильтру. Фильтрат светло-желтого/розового цвета содержал осажденный продукт белого цвета. Перед тем, как по каплям в течение одного часа добавляли гептан (42 мл), для растворения твердого вещества взвесь нагревали до примерно 40°С. Нагревание прекращали, и смеси обеспечивали нагрев до комнатной температуры с перемешиванием в течение ночи. Перед тем, как смесь охлаждали до приблизительно 7°C (лед/водяная баня), дополнительно добавляли 21 мл гептана. Твердое вещество выделяли и промывали 10 мл холодной смеси 2-MeТГФ/гептан 6/4. Увлажненное твердое вещество (5,7 г) сушили в вакууме при 35°С в течение ночи с получением сухой массы соединения (IIc), составляющей 4,2 г, что соответствует выходу 91%. Чистоту, составляющую 99,1% по площади, определяли по ВЭЖХ.
1H-ЯМР (300 МГц, ДМСО-D6) δ 8,26 (1H, д, J=7,5 Гц), 7,26 (дд, 2H, J=8,1, 5,7 Гц), 7,09 (2H, т, J=8,7 Гц), 6,86 (2H, д, J=8,1 Гц), 6,71 (1H, д, J=8,7 Гц), 6,45 (1H, д, J=8,1 Гц), 4,87 (2H, с), 4,45 (1H, дд, J=14,4, 7,5 Гц), 4,07-4,00 (3H, м), 3,06-2,91 (2H, м), 2,71 (1H, дд, J=13,8, 3,9 Гц), 2,54-2,46 (1H, м), 1,31 (с, 9H), 1,11 (3H, т, J=6,9 Гц).
13C-ЯМР (75 МГц, ДМСО-D6) δ 171,4 (C=O), 171,2 (C=O), 161,2 (C-F, д, J=242,3 Гц), 155,1 (C=O), 146,9 (C), 133,2 (C, д, J=3,0 Гц), 131,1 (2 атома углерода, CH, д, J=8,3 Гц), 129,5 (2 атома углерода, CH), 124,8 (C), 114,8 (2 атома углерода, CH, J=21,1 Гц), 113,6 (2 атома углерода, CH), 77,9 (C), 60,5 (CH2), 56,0 (CH), 53,5 (CH), 36,7 (CH2), 35,9 (CH2), 28,1 (3 атома углерода, CH3), 13,9 (CH3)
Авторы настоящего изобретения неоднократно повторяли пример 2 с использованием необработанного соединения (VIc) или перекристаллизованного соединения (VIc) (чистота: 99,1% по площади) в качестве исходного вещества и изменяли различные условия реакции, например давление H2, масс/масс%, Pd/C, растворитель и температуру. Исходная чистота (97,2% по площади) была немного выше, если в качестве исходного вещества использовали перекристаллизованное соединение (VIc), чем при использовании необработанного соединения (VIc), и в этом случае исходная чистота обычно составляет 95-96% по площади. Конечный выход и чистота также немного выше, чем при использовании необработанного соединения (VIc) (98-98,5% по площади).
Авторы настоящего изобретения также повторяли пример 2 несколько раз, изменяя Pd/C масс/масс%, температуру, давление H2 и концентрацию, используя 2-MeТГФ в качестве растворителя. Высокая конверсия соединения (VIc) (>99,5% по площади) достигается при Pd/C масс/масс% от 3 до 6 бар; температура изменяется от 30 до 40°С, давление H2 от 1 до 6 бар и при изменении концентраций реакции. Полученная исходная чистота была одинаковой во всех попытках (95,3-96,2% по площади), как и чистота выделенного продукта после кристаллизации из смеси 2-MeТГФ/гептан (98,0-98,5% по площади).
Пример 3 - Получение соединения (IIIc)
(i) осуществляли с использованием BH3SMe2 в присутствии соли хлоруксусной кислоты
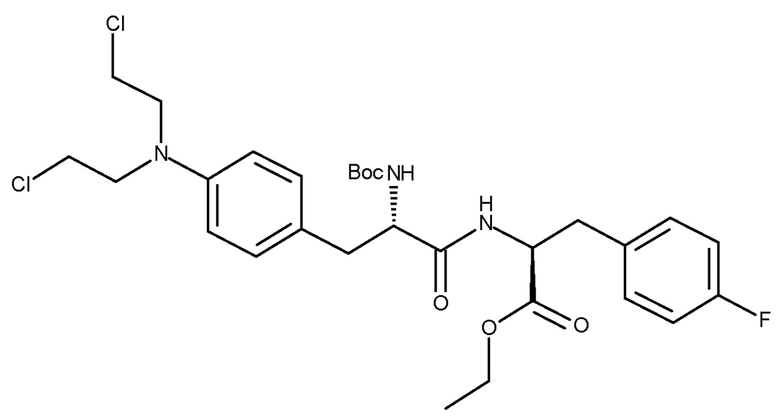
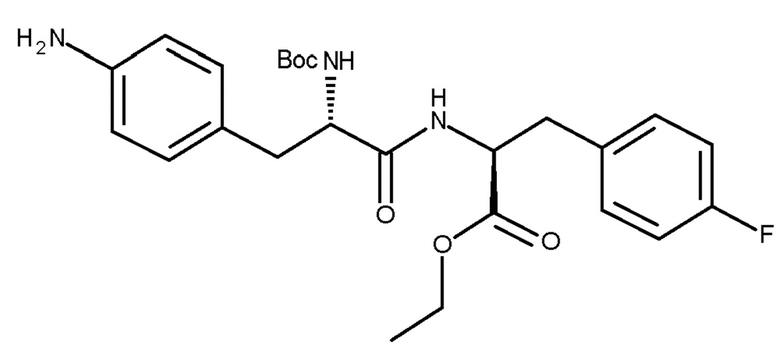
(IIIc)
В 0,5-литровый высушенный реактор, оборудованный верхней мешалкой, добавляли соединение (IIc) (6,99 г, 14,76 ммоль), затем безводный тетрагидрофуран (46 мл), хлоруксусную кислоту (36,3 г, 383,8 ммоль), натриевую соль хлоруксусной кислоты (17,2 г, 147,6 ммоль) при TI=5-13°С. Потом в течение 45 минут добавляли раствор BH3SMe2 (14,6 г, 191,9 ммоль, 18,2 мл). После добавления, температуру реакции доводили до TI=25-30°С и, после достижения этой температуры, ее поддерживали в течение 2 часов. Реакцию медленно гасили этанолом (17,7 г, 383,8 ммоль, 22,4 мл) и перемешивали в течение ночи при TJ=5°С и затем, для осаждения продукта, соединения (IIIc), медленно разбавляли дистиллированной водой (138 мл). Температуру доводили до TI=15°С и повышали скорость перемешивания перед добавлением водного раствора K2CO3 (8,0 М, 27 мл) для доведения рН=7,0-7,5. Реакционную взвесь собирали на фильтре, а реакционный сосуд и фильтровальную лепешку промывали водой (2×40 мл). Фильтровальную лепешку повторно промывали водой (200 мл) в течение 1 часа при TJ=20°С и затем снова фильтровали. После промывки водой (50 мл) с последующим высушиванием при TJ=35°С в условиях высокого вакуума получали неочищенный продукт белого цвета, соединение (IIIc), с нескорректированным выходом 7,85 г (88,8%). ВЭЖХ чистота составляла 97,5% по площади.
Необработанное соединение (IIIc) (7,5 г), полученное в соответствии с описанным способом, загружали в реактор и промывали 2-MeТГФ (80 мл). Нагревание при TJ=50°С обеспечивало растворение вещества. Добавляли гептан (80 мл) с перемешиванием при TI=45-50°С и затем перемешивали перед доведением температуры до TJ=10°С. Осажденное твердое вещество собирали фильтрованием и сушили при TJ=35°С в условиях высокого вакуума, в результате чего получали продукт белого цвета, соединение (IIIc), с выходом 6,86 г (91,5%). ВЭЖХ чистота составляла 99,1% по площади.
1H-ЯМР (300 МГц, ДМСО-D6) δ 8,30 (1H, д, J=7,8 Гц), 7,26 (2H, дд, J=8,1, 6 Гц), 7,09-7,05 (3H, м), 6,79 (1H, д, J=8,9 Гц), 6,63 (2H, д, J=8,4 Гц), 4,49-4,42 (1H, дд, J=14,7, 7,5 Гц), 4,07-3,99 (3H, м), 3,68 (8H, с), 3,06-2,91 (2H, м), 2,76 (1H, дд, J=13,8, 4,2 Гц), 2,56 (1H, м), 1,29 (9H, с), 1,1 (3H, т, J=6,6 Гц)
13C-ЯМР (75 МГц, ДМСО-D6) δ 172,1 (C=O), 171,3 (C=O), 161,2 (C-F, д, J=242,3 Гц), 155,2 (C=O), 144,7 (C), 133,2 (C, д, J=3,0 Гц), 131,1 (2 атома углерода, CH, д, J=7,5 Гц), 130,2 (2 атома углерода, CH), 126,1 (C), 114,9 (2 атома углерода, CH, J=21,1 Гц), 111,6 (2 атома углерода, CH), 78,0 (C), 60,6 (CH2), 55,9 (CH), 53,5 (CH), 52,2 (CH2), 41,2 (CH2), 36,4 (CH2), 35,9 (CH2), 28,1 (3 атома углерода, CH3), 14,0 (CH3)
(ii) осуществляли с использованием BH3SMe2 в присутствии соли хлоруксусной кислоты
В 0,5-литровый высушенный реактор, оборудованный верхней мешалкой, добавляли соединение (IIc) (7,5 г, 15,84 ммоль) с последующим добавлением 2-MeТГФ (150 мл). Смесь нагревали до 45°С с образованием прозрачного раствора. Раствор охлаждали до 4°С и добавляли хлоруксусную кислоту (38,9 г, 411,8 ммоль), затем натриевую соль хлоруксусной кислоты (18,4 г, 158,4 ммоль) при TI=5-13°С. Потом добавляли раствор BH3SMe2 (15,6 г, 205,9 ммоль, 19,5 мл) в течение 90 минут. После добавления, температуру реакции доводили до TI=20-25°С и, после достижения этой температуры, ее поддерживали в течение 5 часов. Реакцию медленно гасили водой при TI=15-25°С (150 г, 8333 ммоль, 150 мл), рН=3,5 в водной фазе, и оставляли на ночь без перемешивания при TI=6°С. Продукт, соединение (IIIc), осаждали в органической фазе и температуру доводили до TI=35°С при перемешивании и образовывались две прозрачные фазы. Обеспечивали разделение фаз и водную фазу удаляли. Органическую фазу трижды промывали 20% NaCl (водн.). Значение рН в трех водных фазах соответствовало: 1,7, 1,1 и 1,1. После удаления третьей водной фазы, органическую фазу переносили в круглодонную колбу и концентрировали до половины ее объема на испарителе. Продукт, соединение (IIIc), начинал осаждаться и взвесь продукта оставляли созревать при 6°С в течение 19 часов. Взвесь собирали на фильтре, а круглодонную колбу и фильтровальную лепешку промывали смесью 2-MeТГФ:н-гептан (2×40 мл) с последующей сушкой при TJ=35°С в высоком вакууме с получением неочищенного продукта белого цвета, соединение (IIIc), с нескорректированным выходом 8,3 г (87,6%). ВЭЖХ чистота составляла 99,4% по площади.
(iii) осуществляли с использованием боран-тетрагидрофурана в присутствии соли хлоруксусной кислоты
В 100 мл-овую высушенную круглодонную колбу, оборудованную магнитной мешалкой, добавляли соединение (IIc) (0,75 г, 1,58 ммоль) в медленном токе азота, затем безводный тетрагидрофуран (6 мл), хлоруксусную кислоту (3,89 г, 41,2 ммоль) и натриевую соль хлоруксусной кислоты (1,84 г, 15,8 ммоль). При TI=5-13°С в течение 30 минут добавляли 1 М раствор BH3ТГФ (20,6 ммоль, 20,6 мл). После добавления, температуру реакции устанавливали в диапазоне TI=23-28°С и поддерживали в течение 2 часов после достижения указанной температуры. В процессе выполнения контрольный образец (ВЭЖХ) показал не полностью завершенную реакцию, и температуру рубашки устанавливали равной TJ=40°С, и когда внутренняя температура достигала TI=40°С, температуру реакции поддерживали при этом значении в течение 2 часов, когда образец в процессе выполнения (ВЭЖХ) показал 6,7% по площади исходного вещества, 7,1% аддукта реакции ацилирования (примесь) и 84,1% соединения (IIIc). Реакцию осуществляли при TI=23°С и реакционную смесь оставляли в течение 4 дней, затем медленно гасили этанолом (2,4 г, 3 мл). Добавляли воду (100 мл) и рН смеси, с помощью 1 М водного раствора K2CO3, доводили до значения, равного 7. Реакционную взвесь собирали на фильтре, а реакционный сосуд и осадок на фильтре промывали водой (2×20 мл) и затем сушили при TJ=35°С в условиях высокого вакуума с получением неочищенного бесцветного продукта с нескорректированным выходом, составляющим 0,85 г (89,6%). ВЭЖХ чистота составляла 94,3% по площади, при этом одна основная примесь относилась к аддукту хлорацилирования исходного вещества в количестве 3,8% по площади.
(iv) осуществляли с использованием BH3SMe2 без добавления соли хлоруксусной кислоты
В 100 мл-овую высушенную круглодонную колбу с магнитной мешалкой добавляли соединение (IIc) (0,75 г, 1,58 ммоль) в медленном токе азота, затем безводный тетрагидрофуран (6 мл) и хлоруксусную кислоту (3,89 г, 41,2 ммоль). При TI=5-16°C добавляли раствор BH3SMe2 (1,56 г, 20,6 ммоль, 2,0 мл). После добавления, температуру реакции устанавливали в диапазоне TI=25°C и поддерживали в течение 2,5 ч после достижения этой температуры. В процессе выполнения контрольный образец (ВЭЖХ) показал мелфлуфен (соединение (Ib)), лишенную защитной группы Boc форму соединения (IIIc), в количестве 66% по площади. Реакцию медленно гасили этанолом (2,9 г, 3,7 мл). С помощью 1 М водного раствора K2CO3, рН смеси доводили до значения 8 с последующим добавлением EtOAc (40 мл). Слои разделяли, и водный слой повторно экстрагировали EtOAc (50 мл). Органические слои объединяли и восстанавливали при <30 мбар/35°C до масла. Масло повторно отгоняли из EtOAc (30 мл) дважды и остаток сушили при TJ=23°C/5 мбар с получением 1,6 г масла коричневатого цвета. ВЭЖХ чистота соединения (Ib) составляла 66,1% по площади.
Пример 4 - Получение соединения (Ib) в виде хлористоводородной соли
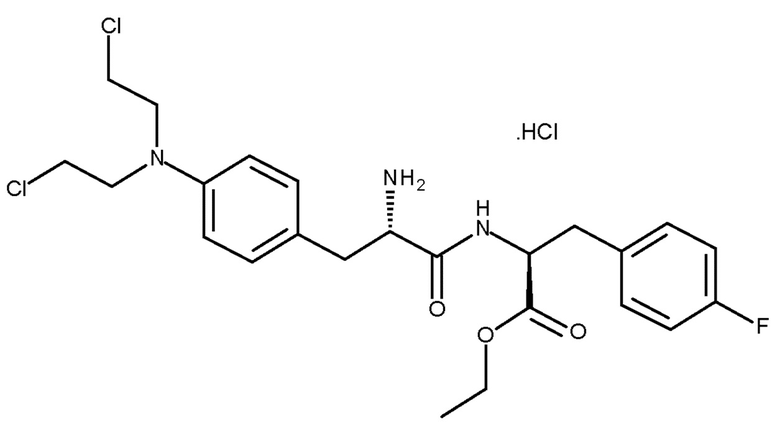 .
.
хлористоводородная соль (Ib)
Boc-мелфлуфен (соединение (IIIc)) (5,0 г, 8,3 ммоль) загружали в круглодонную колбу, снабженную магнитной мешалкой и впускным отверстием для азота. Добавляли 1,3 М HCl (безводная) в этаноле (64 мл, 83,5 ммоль, 10 экв.). Через 19 ч конверсия составляла 99,4%. Растворители частично отгоняли при TJ=33°C на роторном испарителе с последующим добавлением этанола (18 мл). Эту процедуру повторяли дважды. Добавляли затравочные кристаллы и через 30 минут продукт осаждался. Взвесь перемешивали в течение 21 ч и затем концентрировали. Добавляли метил трет-бутиловый эфир (MTBE) (108 мл) при комнатной температуре с равномерной скоростью в течение 30 минут. Через 100 минут перемешивания при комнатной температуре осадок собирали фильтрованием в вакууме и промывали 2×25 мл этанола:MTBE (1:6). Сушку осуществляли в течение ночи при TJ=35°C/5 мбар в вакуумной печи. Выход соединения (Ib) в виде его хлористоводородной соли составлял 4,0 г (90%). ВЭЖХ чистота составляла 98,7% по площади.
1H-ЯМР (300 МГц, MeOH-D4) δ 7,26 (2H, дд, J=8,4, 8,1 Гц), 7,17 (2H, д, J=8,4 Гц), 7,02 (2H, дд, J=9, 8,4 Гц), 6,74 (2H, д, J=8,4 Гц), 4,69 (1H, дд, J=7,8, 6,3 Гц), 4,15 (2H, дд, J=14,1, 7,2 Гц), 4,04 (1H, дд, J=8,4, 5,4 Гц), 3,76 (4H, дд, J=6,3, 6 Гц), 3,67 (4H, дд, 6,6, 5,7 Гц), 3,17 (2H, дд, J=14,4, 6 Гц), 3,06-2,88 (2H, м), 1,22 (3H, т, J=7,2 Гц)
13C-ЯМР (75 МГц, MeOH-D4) δ 172,2 (C=O), 169,8 (C=O), 163,4 (C-F, д, J=244,5 Гц), 147,4 (C), 133,9 (C, д, J=3 Гц), 132,1 (2 атома углерода, CH, д, J=7,5 Гц), 131,8 (2 атома углерода, CH), 123,4 (C), 116,2 (2 атома углерода, CH, д, J=21,9 Гц), 113,7 (2 атома углерода, CH), 62,6 (CH2), 55,6 (CH), 55,5 (CH), 54,3 (CH2), 41,6 (CH2), 37,6 (CH2), 37,6 (CH2), 14,5 (CH3)
Пример 4 успешно повторяли в присутствии этилацетата и с различными концентрациями HCl, от 1,3 до 2,5 М, и при различных температурах, от 6°C до комнатной температуры.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ТИОНУКЛЕОЗИДА ИЛИ ЕГО СОЛЬ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2015 |
|
RU2669806C1 |
| ПИПЕРАЗИНОВОЕ ПРОИЗВОДНОЕ | 2016 |
|
RU2731913C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 4-АЛКОКСИ-3-(АЦИЛ ИЛИ АЛКИЛ)ОКСИПИКОЛИНАМИДОВ | 2017 |
|
RU2742005C2 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| 2-АРИЛПРОПИОНОВЫЕ КИСЛОТЫ И ПРОИЗВОДНЫЕ, И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2520212C2 |
| ПРОЛЕКАРСТВА ЛЕВОДОПА, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2365580C2 |
| СПОСОБ ПОЛУЧЕНИЯ NO-ДОНОРНЫХ СОЕДИНЕНИЙ, ТАКИХ КАК NO-ДОНОРНЫЙ ДИКЛОФЕНАК | 2003 |
|
RU2322434C2 |
| ЗАМЕЩЕННОЕ ПРОПАНАМИДНОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2394560C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
Изобретение относится к способу получения соединения (III) или его лишенного защиты продукта, который может найти применение при синтезе мелфалана и мелфлуфена. Способ включает взаимодействие соединения (II) с хлоруксусной кислотой в присутствии восстановителя. При этом PG представляет собой защитную группу и R представляет собой OH в соответствующим образом защищенной форме или
 .
.
Кроме того, изобретение относится к способам получения других промежуточных соединений, которые включают указанный выше способ, а также к промежуточным соединениям. 6 н. и 19 з.п. ф-лы, 4 пр.
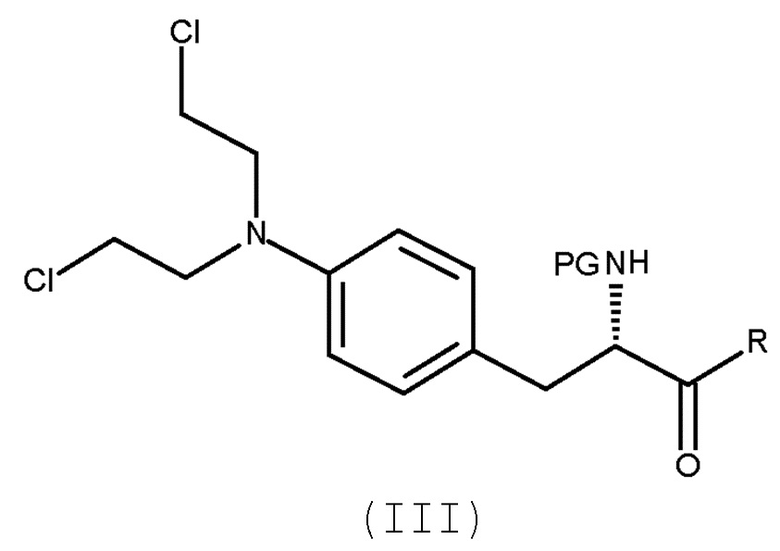
1. Способ получения соединения (III) или его лишенного защиты продукта:
включающий взаимодействие соединения (II)
с хлоруксусной кислотой в присутствии восстановителя,
где PG представляет собой защитную группу и R представляет собой OH в соответствующим образом защищенной форме или
2. Способ по п.1, который осуществляют в присутствии восстановителя, выбранного из группы, состоящей из борана, комплекса боран-основание по Льюису, боргидрида, гидрида металла и Н2, в присутствии металлического катализатора.
3. Способ по п.2, где восстановителем является BH3 или боран-диметилсульфид.
4. Способ по любому из пп.1-3, где PG выбран из группы, состоящей из метилоксикарбонила, этилоксикарбонила, 9-флуоренилметилоксикарбонила, трет-бутилоксикарбонила, бензилоксикарбонила, p-метоксибензилоксикарбонила, 1-адамантилоксикарбонила, p-бромбензилоксикарбонила, трифторацетила, хлорацетила, фенилацетила, бензацетила, p-толуолсульфонила, 2-нитробензолсульфонила, трет-бутилсульфонила, 2- или 4-нитробензолсульфонила, 2,4-динитробензолсульфонила и 2-нафталинсульфонила.
5. Способ по п.4, где PG представляет собой трет-бутилоксикарбонил.
6. Способ по любому из пп.1-5, который осуществляется при температуре в диапазоне от 3 до 50°C, например от 4 до 45°C.
7. Способ по любому из пп.1-6, который осуществляется при температуре в диапазоне от 5 до 40°C.
8. Способ по любому из пп.1-7, который осуществляется в присутствии буферного агента.
9. Способ по п.8, в котором буферным агентом является соль хлоруксусной кислоты, например хлорацетат натрия.
10. Способ по п.9, где молярное соотношение хлоруксусная кислота:соль хлоруксусной кислоты составляет от 2:1 до 5:1.
11. Способ по п.9 или 10, где молярное соотношение соединение (II) : соль хлоруксусной кислоты составляет от 1:7 до 1:20.
12. Способ по любому из пп.1-11, где R представляет собой
.
13. Способ получения соли соединения (III) или соли лишенного защиты продукта соединения (III), включающий способы по любому из пп.1-12 и стадию образования соли, например стадию образования хлористоводородной соли.
14. Способ по любому из пп.1-13, где соединение (II) было получено путем взаимодействия соединения (VI)
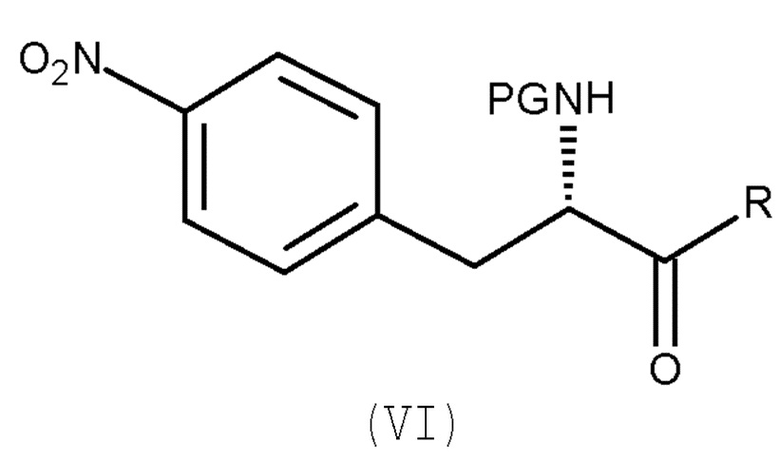
с восстанавливающим агентом,
где PG является таким, как определено для соединения (II), и R представляет собой OH необязательно в соответствующим образом защищенной форме или
.
15. Способ по п.14, где восстановителем является водород и катализатор, например H2/Pd/C.
16. Способ по п.14 или 15, где R представляет собой
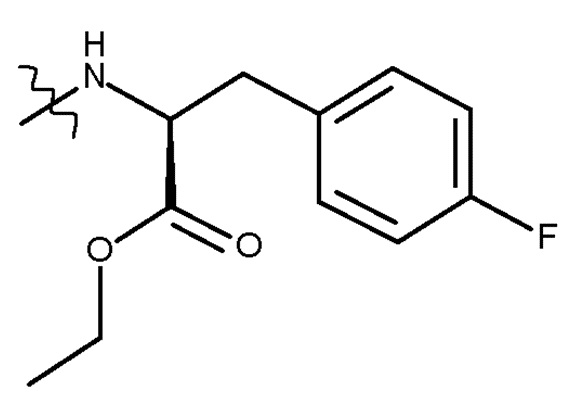
и соединение (VI) было получено путем взаимодействия соединения (IV)
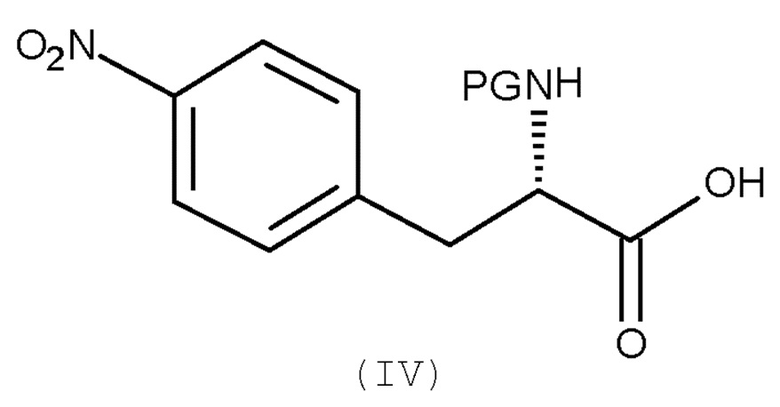
с соединением (V)
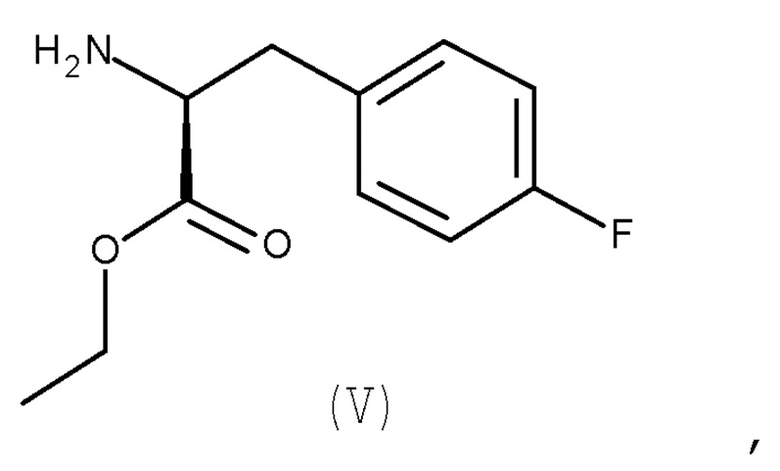
где PG является таким, как определено для соединения (II).
17. Способ получения соединения (I) или его соли:

который включает осуществление способа по любому из пп.1-16 и последующее удаление защитной группы соединения (III) с образованием соединения (I) или его соли,
где R представляет собой OH необязательно в соответствующим образом защищенной форме или
18. Способ по п.17, где соединение (I) представляет собой соединение (Ib) или его соль:
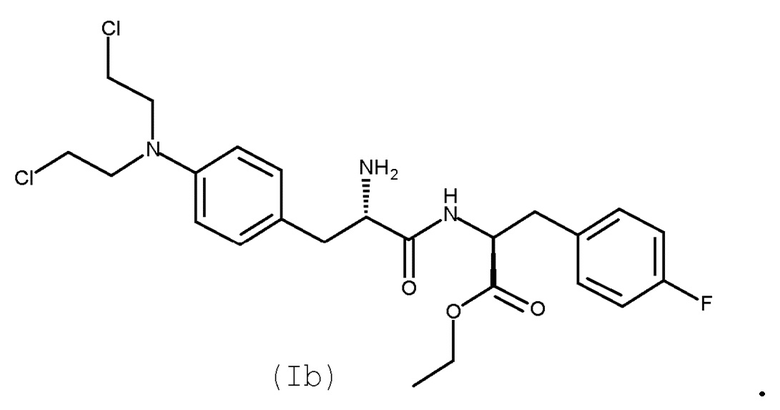
19. Способ получения соли соединения (I), включающий способ по п.17 или 18 и стадию образования соли, например стадию образования хлористоводородной соли.
20. Способ по п.19, где способ предназначен для производства хлористоводородной соли соединения (Ib)
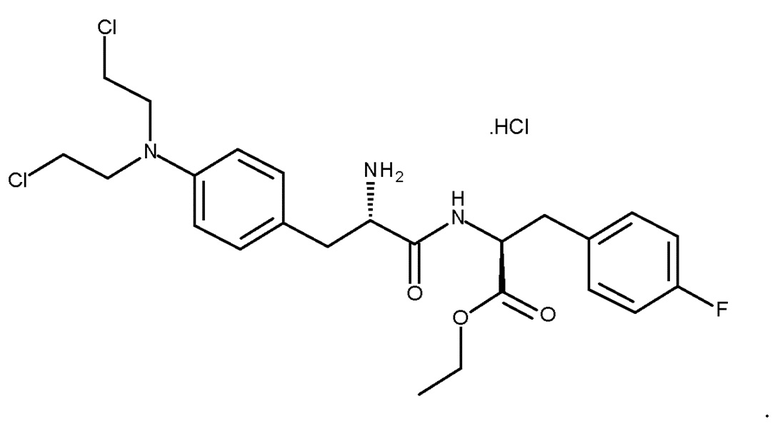
21. Способ получения соединения (VIb)

включающий взаимодействие соединения (IV)
с соединением (V)
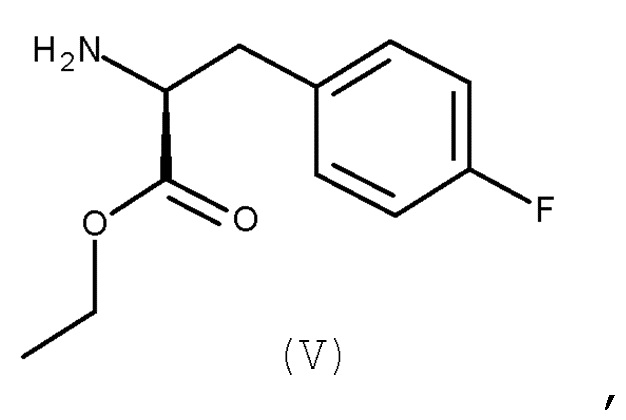
где PG представляет собой защитную группу.
22. Соединение, имеющее следующую структуру:
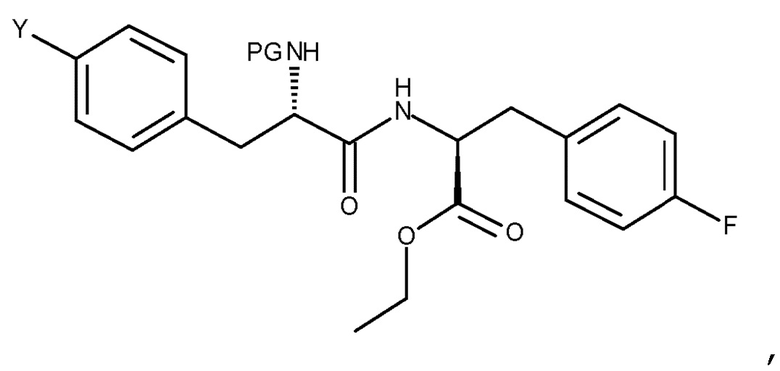
где Y представляет собой NH2 или NO2 и PG представляет собой защитную группу.
23. Соединение по п.22, где PG представляет собой трет-бутилоксикарбонил.
24. Соединение по п.22, где PG выбран из группы, состоящей из метилоксикарбонила, этилоксикарбонила, 9-флуоренилметил-оксикарбонила, трет-бутилоксикарбонила, бензилоксикарбонила, p-метоксибензилоксикарбонила, 1-адамантилоксикарбонила, p-бромбензилоксикарбонила, трифторацетила, хлорацетила, фенилацетила, бензацетила, p-толуолсульфонила, 2-нитробензолсульфонила, трет-бутилсульфонила, 2- или 4-нитробензолсульфонила, 2,4-динитронзенсульфонила и 2-нафталинсульфонила.
25. Способ по пп.1-11, 13-15 или 17, где R представляет собой ОН в подходящей защищенной форме и где защитная группа для ОН выбрана из группы, состоящей из метилового сложного эфира, метоксиметилового сложного эфира, 9-флуоренилметилового сложного эфира, трет-бутилового сложного эфира, бензилового сложного эфира, дифенилметилового сложного эфира, трифенилметилового сложного эфира, 2,6-диметилфенилового сложного эфира, триметилсилилового сложного эфира, триэтилсилилового сложного эфира, 2-(триметилсилил)этоксиметилового сложного эфира, 2-(триметилсилил)этилового сложного эфира, S-трет-бутилового сложного эфира и 2-алкил-1,3-оксазолина.
| L | |||
| KUPCZYK-SUBOTKOWSKA et al., Modulation of Melphalan Resistance in Glioma Cells with a Peripheral Benzodiazepine Receptor Ligand-Melphalan Conjugate, J | |||
| MED | |||
| CHEM., 1997, Vol | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Телефонная трансляция с катодным реле | 1920 |
|
SU1726A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| M | |||
| WICKSTROM et al., The novel melphalan prodrug J1 inhibits neuroblastoma | |||

