ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ Данная заявка является не предварительной заявкой, которая подана в соответствии с 37 CFR §1.53 (b), и в соответствии с 35 USC §119(e) испрашивает приоритет предварительной заявки США номер 61/711900, поданной 10 октября 2012 года, которая включена в настоящий документ посредством ссылки в полном объеме.
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к способам получения соединения GDC-0980, являющегося ингибитором PI3K.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Фосфоинозитид 3-киназы (PI3K) представляют собой киназы липидов, которые фосфорилируют липиды по 3-гидроксильному остатку кольца инозита (Whitman et al (1988) Nature, 332: 664). 3-фосфорилированные фосфолипиды (PIP3), образуемые PI3-киназами, действуют как вторичные посредники, привлекая киназы со связывающими липиды доменами (включая гомологичные плекстрину (РН) районы), такие как Akt и фосфоинозитид-зависимая киназа-1 (PDK1). Связывание Akt с мембранными PIP3 вызывает перемещение Akt на плазматическую мембрану, в результате чего Akt контактирует с PDK1, который отвечает за активацию Akt. Опухоль-супрессорная фосфатаза PTEN дефосфорилирует PIP3 и, следовательно, действует как негативный регулятор активации Akt. PI3-киназы Akt и PDK1 играют важную роль в регуляции многих клеточных процессов, включая регуляцию клеточного цикла, пролиферацию, выживание, апоптоз и подвижность клеток, и являются значимыми компонентами молекулярных механизмов таких заболеваний как рак, диабет и иммунное воспаление (Vivanco et al (2002) Nature Rev. Cancer 2: 489; Phillips et al (1998) Cancer 83: 41).
Наиболее важной изоформой PI3-киназ при раке является PI3-киназа класса I, р110α (альфа) (US 5824492; US 5846824; US 6274327). Другие изоформы вовлечены в сердечно-сосудистые и иммунно-воспалительные заболевания (Workman Р (2004) Biochem Soc Trans 32: 393-396; Patel et al (2004) Proceedings of the American Association of Cancer Research (Abstract LB-247) 95th Annual Meeting, March 27-31, Orlando, Florida, USA; Ahmadi K and Waterfield MD (2004) Encyclopedia of Biological Chemistry (Lennarz W J, Lane M D eds) Elsevier/Academic Press). Путь PI3 киназа/Akt/PTEN является привлекательной мишенью для разработки лекарств против рака, так как, как ожидается, такие модулирующие или ингибирующие агенты будут способны ингибировать пролиферацию, устранять устойчивость к апоптозу и преодолевать устойчивость к цитотоксическим агентам раковых клеток (Folkes et al (2008) J. Med. Chem. 51: 5522-5532; Yaguchi et al (2006) Jour, of the Nat. Cancer Inst. 98(8): 545-556). Регуляция сигнального пути PI3K-PTEN-AKT нарушена при самых разнообразных видах рака (Samuels Y, Wang Z, Bardellil A et al. High frequency of mutations of the PIK3CA gene in human cancers. (2004) Science; 304 (5670): 554; Carpten J, Faber AL, Horn C. "A transforming mutation in the pleckstrin homology domain of AKT1 in cancer" (2007) Nature; 448: 439-444).
GDC-0980 (Genentech, Inc., Roche, RG-7422) демонстрирует активность в широком спектре доклинических моделей ксенотрансплантатов рака, включая рак молочной железы, яичников, легких и предстательной железы, и в настоящее время ведутся исследования данного соединения в качестве потенциального перорального лекарственного средства для лечения рака, включая солидные опухоли и неходжкинскую лимфому (Wagner AJ; Burris III НА; de Bono JS et al AACR-NCI-EORTC International Congress (2009), 21st: November 17 (Abs B137) "Pharmacokinetics and Pharmacodynamic biomarkers for the dual PI3K/mTOR inhibitor GDC-0980: initial phase I evaluation"; US 7888352; US 2009/0098135; US 2010/0233164). В марте 2009 года было начато исследование I фазы на пациентах с солидными опухолями или NHL; в апреле 2009 года было начато второе исследование I фазы; эти исследования продолжались в апреле 2010 г. В декабре 2010 г. было начато исследование фазы 1b комбинированного лечения метастатического рака молочной железы. В июле 2010 года исследование II фазы лечения метастатического рака молочной железы было запланировано провести в первой половине 2011 года; пациенты должны были получать GDC-0980 в комбинации с гормональной терапией. Клинические результаты на сегодняшний день показывают, что GDC-0980 может принести пользу пациентам с солидными опухолями или гематологическими злокачественными новообразованиями (Sutherlin DP, Belvin М, Bao L et al, American Association for Cancer Research Annual Meeting, (2011) 102nd: April 04 (Abs 2787)).
GDC-0980 является мощным селективным пероральным ингибитором PI3K класса I и киназы mTOR, который характеризуется следующими значениями IC50 против изоформ PI3K класса I, измеренными биохимическими методами in vivo: р110а (альфа) - 4,8 нМ; р110β (бета) - 26,8 нМ; р110 (гамма) - 13,8 нм; p110d (дельта) - 6,7 нМ; Ki для mTOR - 17,3 нМ. GDC-0980 был селективным для PI3K по сравнению с большим набором киназ (>145), в том числе по сравнению с другими членами семейства фосфатидилинозит киназ. В клеточных линиях РСЗ и MCF7-neo/HER2 соединение продемонстрировало значения IC50, равные 307 и 320 нМ соответственно. GDC-0980 был стабилен в человеческих микросомах и гепатоцитах, проявлял низкую активность против hERG, IC50>100 μМ (мкМ) и не давал значительный ответ в анализе скрининга рецепторов (n=68; GDC-0980=10 мкМ). У грызунов и собак значения клиренса были от умеренных до высоких, 60 мл/мин/кг и 12 мл/мин/кг соответственно. Терминальный период полувыведения соединения составлял от 6 до 18 ч, значения AUC и Cmax пропорционально увеличивались с увеличением единичной пероральной дозы. GDC-0980 (от 25 до 150 мг/кг в день, перорально) был эффективен в нескольких моделях ксенотрансплантатов, в том числе в моделях ксенотрансплантатов РС3 PTEN-рака простаты и MCF7.1 E545K рака молочной железы на мышах. В модели ксенотрансплантата рака молочной железы MDA-IVIB-361.1 GDC-0980 давал значительное ингибирование роста при минимальной дозе 1,0 мг/кг в день.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
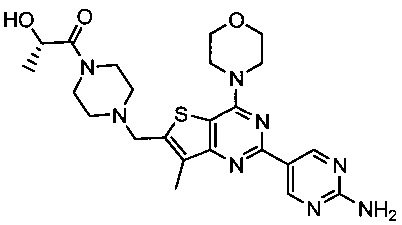
Изобретение относится к способам получения двойного ингибитора mTOR/PI3K GDC-0980, имеющего химическое название (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он, и имеющего структуру:
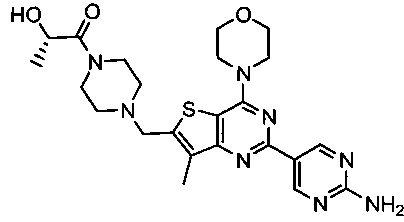
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей.
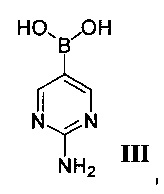
Другой аспект настоящего изобретения относится к способам получения интермедиата, 2-аминопиримидин-5-илбороновой кислоты III, который полезен для получения GDC-0980, имеющего структуру:

Другой аспект настоящего изобретения относится к новому интермедиату, оксалату (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V, который полезен для получения GDC-0980, имеющему структуру:
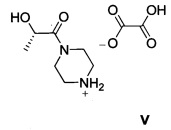
ОПРЕДЕЛЕНИЯ
Термин "хиральный" относится к молекулам, которые обладают свойством неналожимости на свое зеркальное отображение, тогда как термин "ахиральный" относится к молекулам, которые являются наложимыми на их зеркальные отображения.
Термин "стереоизомеры" относится к соединениям, которые имеют одинаковый химический состав, но отличаются по расположению атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, чьи молекулы не являются зеркальным отображениями друг друга. Диастереомеры имеют различные физические свойства, например, точки плавления, точки кипения, спектральные свойства и реакционную способность. Смеси диастереомеров можно разделить с помощью аналитических процедур с высоким разрешением, таких как электрофорез и хроматография.
"Энантиомеры" относятся к двум стереоизомерам соединения, которые являются не наложимыми зеркальными отражениями друг друга.
Стереохимические определения и условные термины, используемые в настоящем документе, как правило, соответствуют S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; and Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметричные или хиральные центры и поэтому могут существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, включены в настоящее изобретение. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоско-поляризованного света. При описании оптически активного соединения префиксы D и L или R и S используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра (центров). Префиксы d и I или (+) и (-) используются для обозначения знака вращения плоскости поляризованного света соединением, причем (-) или 1 означает, что соединение является левовращающим. Соединение с префиксом (+) или D является правовращающим. Для данной химической структуры эти стереоизомеры идентичны за исключением того, что они являются зеркальными отражениями друг друга. Конкретный стереоизомер может также называться энантиомером, и смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров в соотношении 50:50 называют рацемической смесью или рацематом, такая смесь может быть получена в том случае, когда в ходе химической реакции или процесса не было стереоизбирательности или стереоспецифичности. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомеров, лишенной оптической активности.
Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с различной энергией, которые являются взаимопревращаемыми за счет низкого энергетического барьера. Например, протонные таутомеры (также известные как прототропные таутомеры) осуществляют взаимопревращения за счет миграции протона, такой как процессы кето-енольной и имин-енаминной изомеризации. Валентные таутомеры включают взаимопревращения за счет реорганизации некоторых связывающих электронов.
Фраза "фармацевтически приемлемая соль" в настоящем документе относится к фармацевтически приемлемым органическим или неорганическим солям соединений настоящего изобретения. Примеры солей включают, но ими не ограничиваются ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентисинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат "мезилат", этансульфонат, бензолсульфонат, пара-толуолсульфонат, памоат (то есть 1,1'-метилен-бис(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может содержать включения других молекул, таких как ионы ацетата, ионы сукцината или другие противоионы. Указанный противоион может быть любым органическим или неорганическим фрагментом, который стабилизирует заряд на исходном соединении. Кроме того, фармацевтически приемлемая соль может содержать более одного заряженного атома в своей структуре. В случаях, когда фармацевтически приемлемая соль включает несколько заряженных атомов, такая соль может иметь несколько противоионов. Следовательно, фармацевтически приемлемая соль может содержать один или более из заряженных атомов и/или один или более из противоионов.
Если соединение настоящего изобретения представляет собой основание, нужную фармацевтически приемлемую соль можно получить любым подходящим способом, известным специалистам в данной области, например обработкой свободного основания неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, гликолевая кислота, малоновая кислота, щавелевая кислота, пировиноградная кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как пара-толуолсульфоновая кислота или этансульфоновая кислота или т.п.
Если соединение настоящего изобретения представляет собой кислоту, нужную фармацевтически приемлемую соль можно получить любым подходящим способом, например, обрабатывая свободную кислоту неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксидом щелочного металла или гидроксидом щелочноземельного металла или подобными веществами. Иллюстративные примеры подходящих солей включают, но не ограничиваются ими, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов, циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные с натрием, кальцием, калием, магнием, марганцем, железом, медью, цинком, алюминием и литием.
Термин "сольват" относится к ассоциации или комплексу молекул одного или более растворителей и соединения настоящего изобретения. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются ими, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, в котором молекулой растворителя является вода.
ПОЛУЧЕНИЕ GDC-0980
Настоящее изобретение включает процессы, способы, реагенты и интермедиаты для синтеза GDC-0980, низкомолекулярного ингибитора PI3K и mTOR (CAS Reg. No. 1032754-93-0), который имеет структуру:
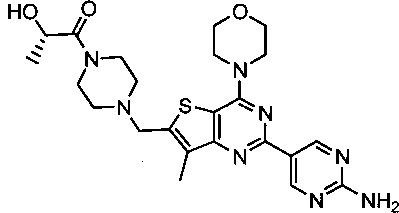
и может быть назван: (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он (US 7888352, US 2009/0098135, US 2010/0233164). В настоящем документе GDC-0980 включает все стереоизомеры, геометрические изомеры, таутомеры и их фармацевтически приемлемые соли.
Соединения по изобретению могут включать асимметричные или хиральные центры и поэтому могут существовать в различных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, включены в настоящее изобретение. Кроме того, настоящее изобретение охватывает все геометрические изомеры и изомеры положения. Если в структурах, показанных в данном описании, не указана стереохимия любого конкретного хирального атома, то все их стереоизомеры рассматриваются и включены в качестве соединений по изобретению. Если стереохимия показана с помощью закрашенного клина или пунктирной линии, что указывает на конкретную конфигурацию, то определен и указан этот конкретный стереоизомер.
Соединения по настоящему изобретению могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и предполагается, что настоящее изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения по настоящему изобретению могут также существовать в различных таутомерных формах, и все такие формы охватываются объемом настоящего изобретения. Термины "таутомер" или "таутомерная форма" относятся к структурным изомерам с различной энергией, которые являются взаимопревращаемыми за счет низкого энергетического барьера. Например, протонные таутомеры (также известные под названием прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как кето-енольная и имин-енаминная изомеризация. Валентные таутомеры включают взаимопревращения путем реорганизации некоторых из связывающих электронов.
Соединения по изобретению также включают меченные изотопами соединения, которые идентичны соединениям, приведенным в настоящем документе, за исключением того, что один или более атомов замещены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, встречающихся в природе. Все изотопы какого-либо конкретного атома или элемента, как указано в настоящем документе, включены в объем соединений данного изобретения и их применения. Примеры изотопов, которые могут быть включены в соединения изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17О, 18О, 32Р, 33P, 35S, 18F, 36Cl, 123I и 125I. Некоторые меченные изотопами соединения по настоящему изобретению (например, меченные 3Н или 14С), применимы в анализах распределения лекарственного вещества и/или субстрата в ткани. Меченные тритием (3Н) и углеродом-14 (14С) изотопы являются в особенности предпочтительными вследствие простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2Н), может дать определенные терапевтические преимущества вследствие большей метаболической стабильности (например, за счет увеличения периода полувыведения in vivo или уменьшения требуемых дозировок), и, следовательно, может быть предпочтительным при некоторых обстоятельствах. Излучающие позитроны изотопы, такие как 15О, 13N, 11С и 18F, полезны для исследований методом позитронно-эмиссионной томографии (ПЭТ) для изучения связывания рецептора субстратом. Меченые изотопами соединения данного изобретения могут быть обычно получены согласно процедурам, аналогичным тем, которые приведены в представленных ниже примерах, путем замещения не меченного изотопом реагента на реагент, меченный изотопом.
Исходные материалы и реагенты для получения GDC-0980, как правило, доступны из коммерческих источников, таких как Sigma-Aldrich Chemical (Милуоки, Висконсин), или могут быть легко получены с использованием способов, которые хорошо известны специалистам в данной области техники (например, получены способами, которые в общем описаны Louis F. Fieser and Mary Fieser, Reagenfs for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.), or Beilsteins handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения к этим изданиям (также доступны через онлайн-базу данных Beilstein).
Следующие Схемы 1-8 иллюстрируют химические реакции, процессы, методики синтеза GDC-0980 формулы I, а также некоторые интермедиаты и реагенты. Понятно, что для достижения тех же превращений могут быть использованы другие реагенты, растворители и условия реакции, чем проиллюстрировано на Схемах 1-8.
Схема 1:
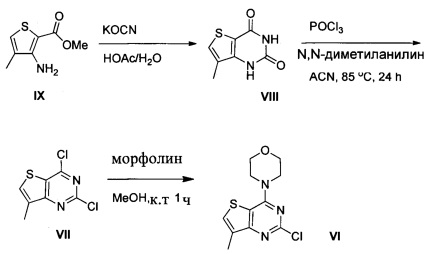
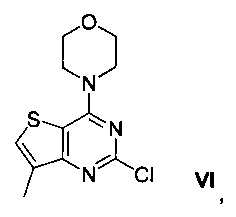
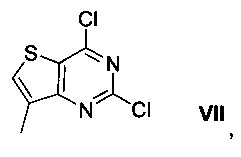
Схема 1 показывает синтез интермедиата 4-(2-хлор-7-метилтиено[3,2-0]пиримидин-4-ил)морфолина VI из метил-3-амино-4-метилтиофен-2-карбоновой кислоты IX. Циклизация IX с цианатом калия в уксусной кислоте и воде давала 7-метилтиено[3,2-d]пиримидин-2,4(1Н,3Н)-дион VIII (Пример 1). Хлорирование VIII оксихлоридом фосфора в присутствии N,N-диметиланилина в ацетонитриле (ACN) давало 2,4-дихлор-7-метилтиено[3,2-d]пиримидин VII (Пример 2). Замещение 4-хлор группы VII морфолином в метаноле давало соединение VI (Пример 3).
Схема 2:
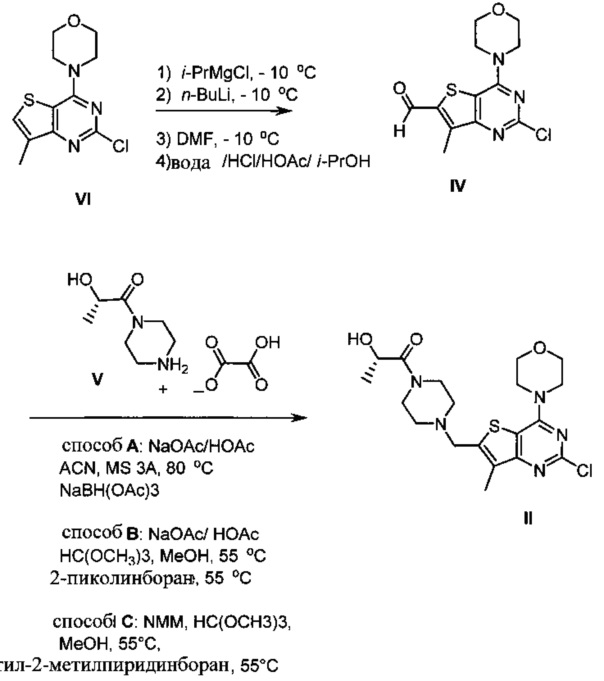
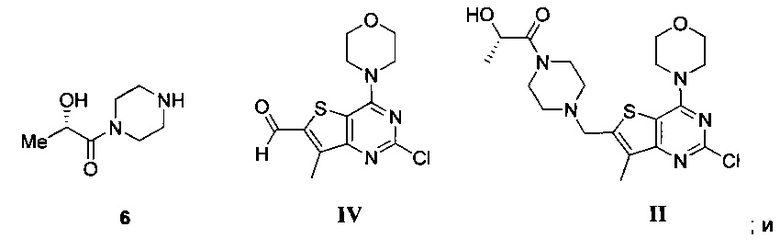
Схема 2 показывает синтез интермедиата (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II из 4-(2-хлор-7-метилтиено[3,2-D]пиримидин-4-ил)морфолина VI. Обработка VI реактивом Гриньяра изопропилмагнийхлоридом, а затем н-бутиллитием при -10°C с последующим добавлением диметилформамида и быстрым разбавлением водным раствором кислоты давала формилированный интермедиат 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV (Пример 4). Восстановительное аминирование IV проводили путем смешивания IV с оксалатом (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V с последующей обработкой восстановительным агентом, таким как триацетоксиборгидрид натрия (способ А), 2-пиколин боран (способ В) или 5-этил-2-метилпиридин боран (Способ С) с получением II, который кристаллизуют в смеси толуол/гептан (Пример 5) или Ме-ТГФ/гептан.
Схема 3:
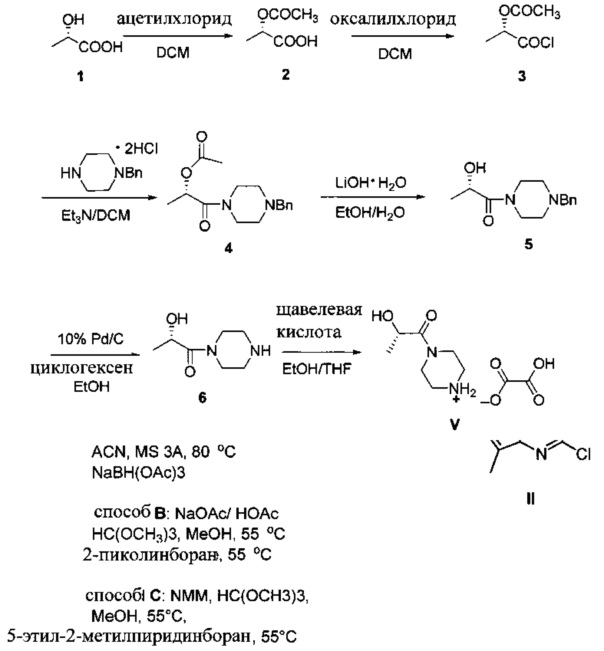
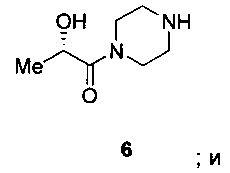
Схема 3 показывает синтез интермедиата, оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V, из (S)-2-гидроксипропановой кислоты (L-молочной кислоты) 1. ацетилирование 1 с получениеми (S)-2-ацетоксипропановой кислоты 2, с последующей обработкой хлорирующим реагентом, таким как оксалилхлорид, с получением хлорангидрида кислоты, (S)-1-хлор-1-оксопропан-2-ил ацетата 3 (Пример 6). Реакция 3 с дигидрохлоридом 1-бензилпиперазина в дихлорметане в присутствии триэтиламина давала (S)-1-(4-бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетат 4 (Пример 7). С помощью гидролиза ацетата 4 гидроксидом лития получали (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1 5 (Пример 8), который затем гидрировали для удаления N-бензильной группы, чтобы получить (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-он 6 (Пример 9). Оксалат получали из 6 с помощью обработки щавелевой кислотой в этаноле и тетрагидрофуране с получением V (Пример 9).
Схема 4:
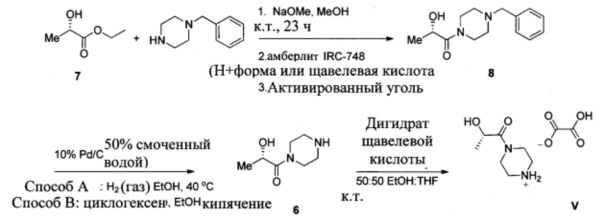
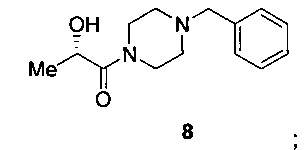
Схема 4 показывает синтез интермедиата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V из (S)-этил 2-гидроксипропаноата 7. 1-бензилпиперазин и 7 подвергали взаимодействию в смеси метоксида натрия и метанола с получением (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1-она 8, который выделяли из смолы Amberlite® IRC-748 или щавелевой кислоты с последующей обработкой активированным углем (Пример 10). Восстановительное удаление бензильной группы из 8 осуществляли с помощью катализа палладием либо обработкой газообразным водородом (способ А), либо циклогексеном (способ В), что давало интермедиат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1 6 (Пример 11). Оксалат получали из 6 с помощью обработки щавелевой кислотой в этаноле и тетрагидрофуране с получением V (Пример 11).
Схема 5:
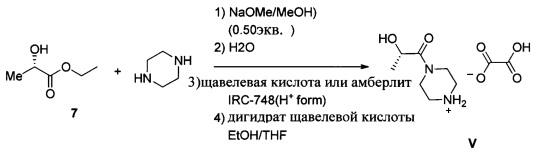
Схема 5 показывает альтернативный одностадийный синтез интермедиата оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V из (S)-этил 2-гидроксипропаноата 7. Незащищенный 7 и пиперазин реагируют с метилатом натрия в метаноле с образованием амида V, после чего проводят обработку щавелевой кислотой или смолой Amberlite® IRC-748 для удаления примесей, и получают соль щавелевой кислоты (Пример 12).
Схема 6:
Способ А
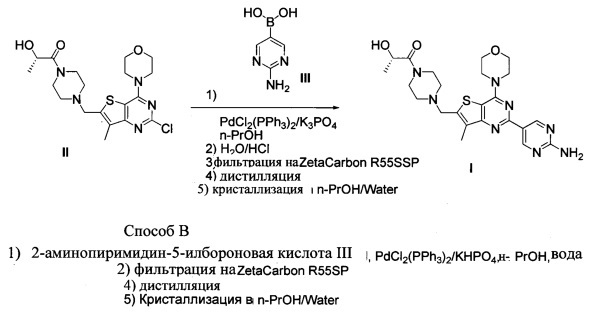
Схема 6 показывает синтез (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она, GDC-0980, Формула I, из интермедиата (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II. Сочетание II и 2-аминопиримидин-5-илбороновой кислоты III с палладиевым катализом по Сузуки-Мияура дает неочищенное соединение I (Пример 13). Для гашения реакционной смеси добавляли воду, после чего проводят рециркуляционную фильтрацию через активированный уголь, чтобы удалить палладий. Летучие вещества удаляли при пониженном давлении и I кристаллизовали из н-пропанола и воды, чтобы получить свободное основание, GDC-0980, формула I. В способе В реакцию проводят с использованием KHPO4 в качестве основания в смеси н-пропанол/вода в качестве растворителя.
В стадии сочетания по Сузуки-Мияура с образованием соединения I может быть использовано множество палладиевых катализаторов. Сочетание по Сузуки-Мияура представляет собой опосредованную палладием реакцию перекрестного сочетания арилгалогенида, такого как II, с бороновой кислотой, такой как III. Для получения I могут быть использованы низковалентные Pd (II) и Pd (0) катализаторы, в том числе PdCl2(PPh3)2, Pd(t-Bu)3, PdCl2 dppf CH2Cl2, Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Rd[(Pet3)]2, Pd(DIPHOS)2, Cl2Pd(Bipy), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[P(o-tol)3]2, Pd2(dba)3/P(o-tol)3, Pd2(dba)/P(фурил)3, Cl2Pd[P(фурил)3]2, Cl2Pd(PMePh2)2, Cl2Pd[P(4-F-Ph)3]2, Cl2Rd[P(C6F6)3]2, Cl2Pd[P(2-COOH-Ph)(Ph)2]2, Cl2Pd[P(4-COOH-Ph)(Ph)2]2, а также инкапсулированные катализаторы Pd EnCat™ 30, Pd EnCat™ TPP30 и Pd(II)EnCat™ BINAP30 (US 2004/0254066).
Для удаления палладия после стадии сочетания по Сузуки-Мияура с образованием соединения I может быть использовано множество твердых адсорбентов поглотителей палладия. Примеры вариантов осуществления включают поглотители палладия FLORISIL®, SILIABOND® Thiol и SILIABOND® Thiourea. Другие поглотители палладия включают силикагель, стекло с контролируемым размером пор (TosoHaas) и дериватизированный слабосшитый полистирол QUADRAPURE™ АЕА, QUADRAPURE™ IMDAZ, QUADRAPURE™ МРА, QUADRAPURE™ TU (Reaxa Ltd., Sigma-Aldrich Chemical Co.).
Реакцию арилгалогенида, такого как II, и бороновой кислоты, такой как III, с образованием соединения I также можно проводить в условиях катализа палладием по Бухвальду с использованием предкатализатора палладацикла Бухвальда и реагентов-лигандов из таблицы 1, и как описано в ссылках: Biscoe et al (2008) J. Am. Chem. Soc. 130: 6686-6687; Kinzel et al (2010) J. Am. Chem. Soc. 132:14073-14075; Molander et al (2012) J. Am. Chem. Soc. 134: 11667-11673; Walker et al (2004) Angew. Chem. Int. Ed. 43: 1871; Billingsley et al (2007) Angew. Chem. Int. Ed. 46: 5359-5363; US 6946560; US 7026498; US 7247731; US 7560582; US 6307087; US 6395916; US 7223879; US 7858784, которые включены в настоящий документ посредством ссылки. Такие реагенты коммерчески доступны (Johnson Matthey Inc., Wayne, PA; Sigma Aldrich Fine Chemical, St. Louis, MO; Strem Chemicals, Inc., Newburyport, MA).
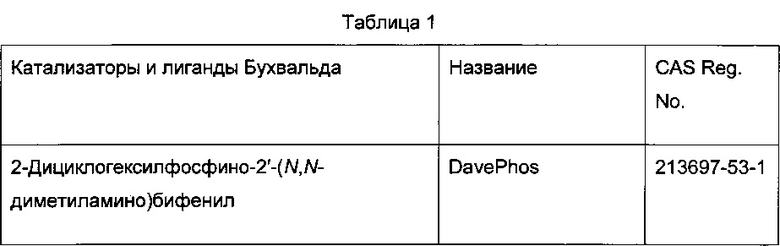
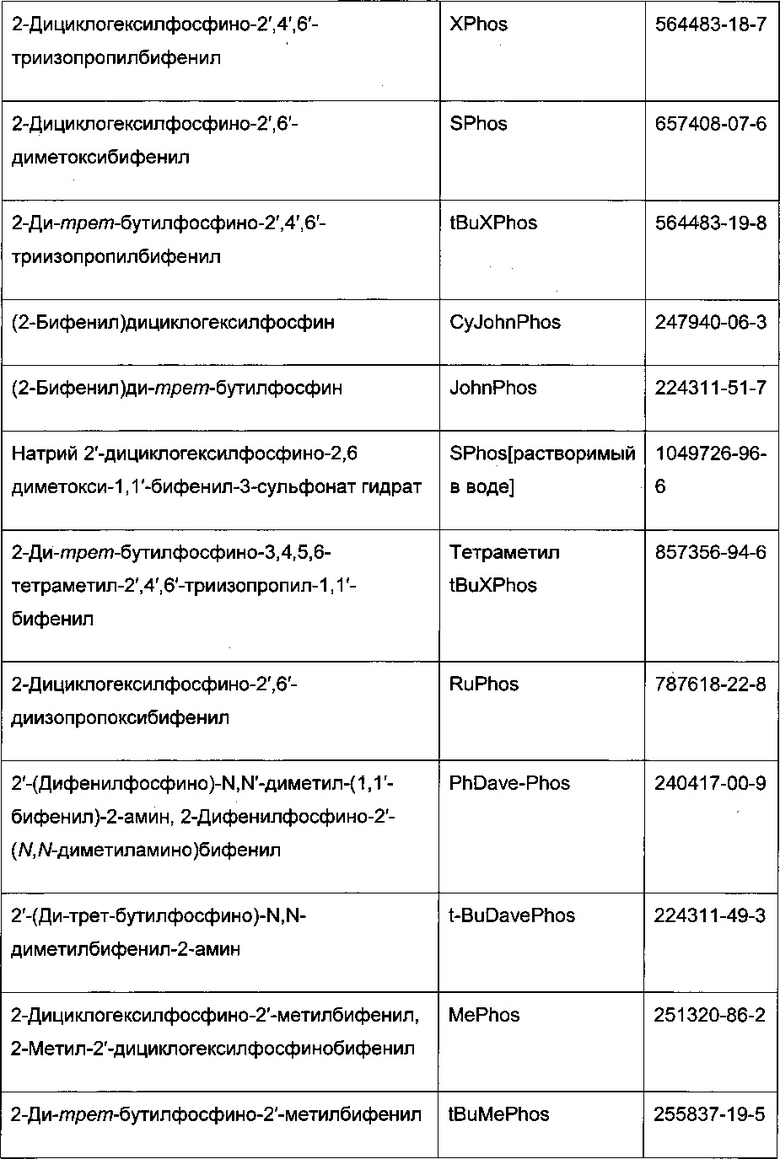
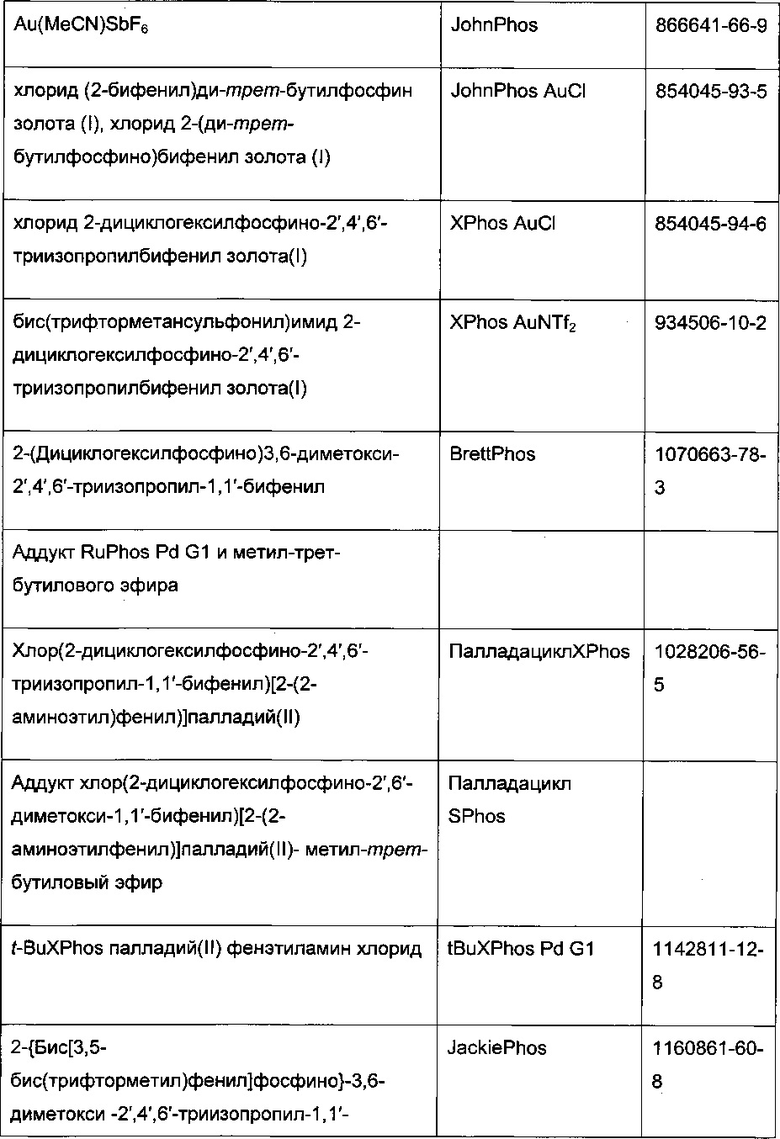
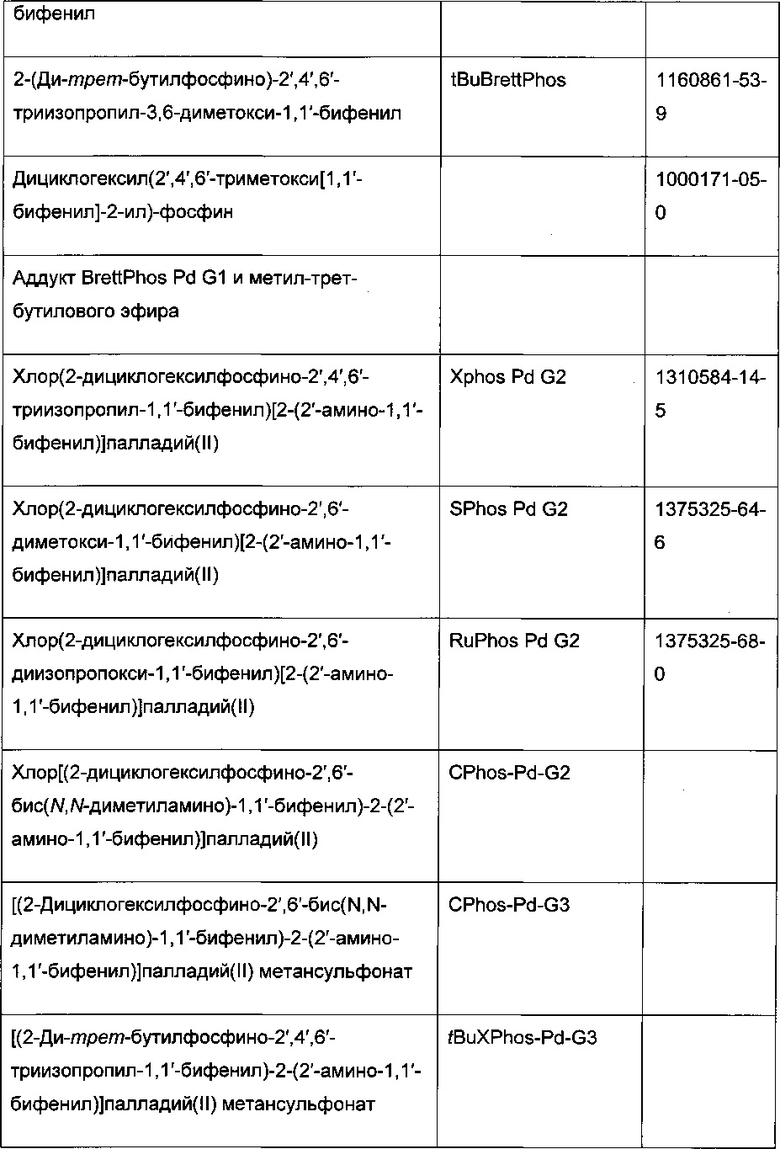
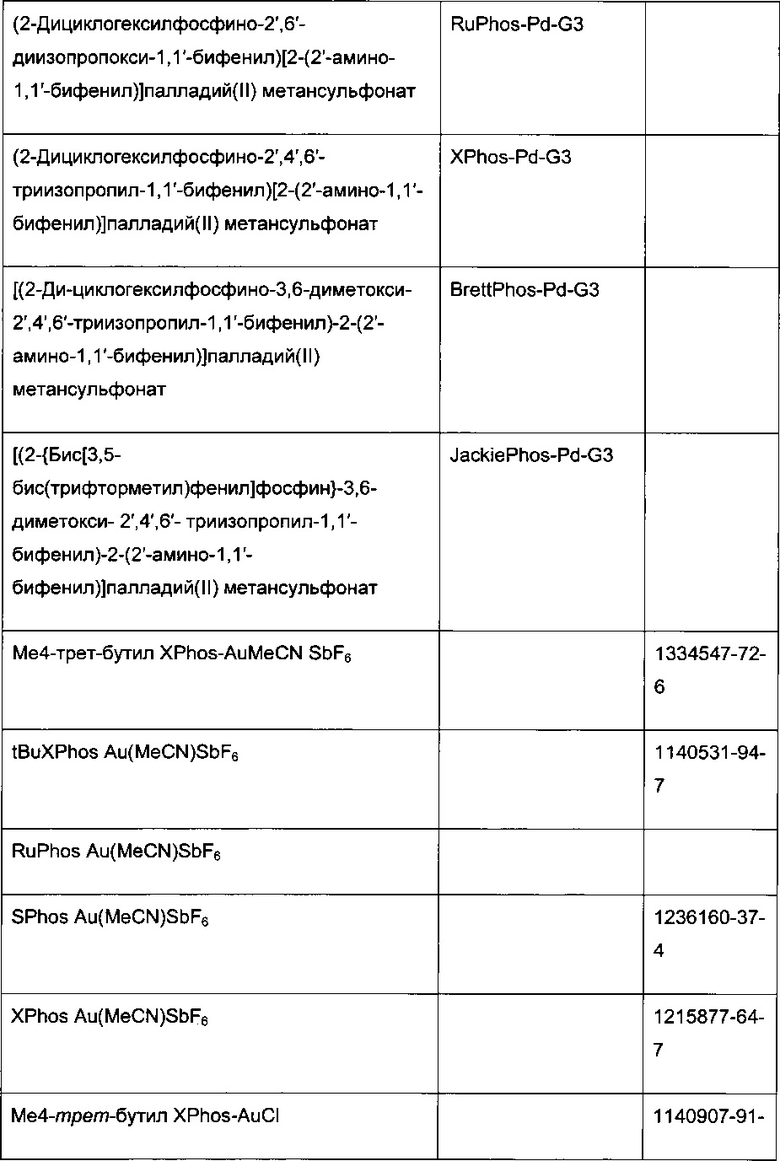
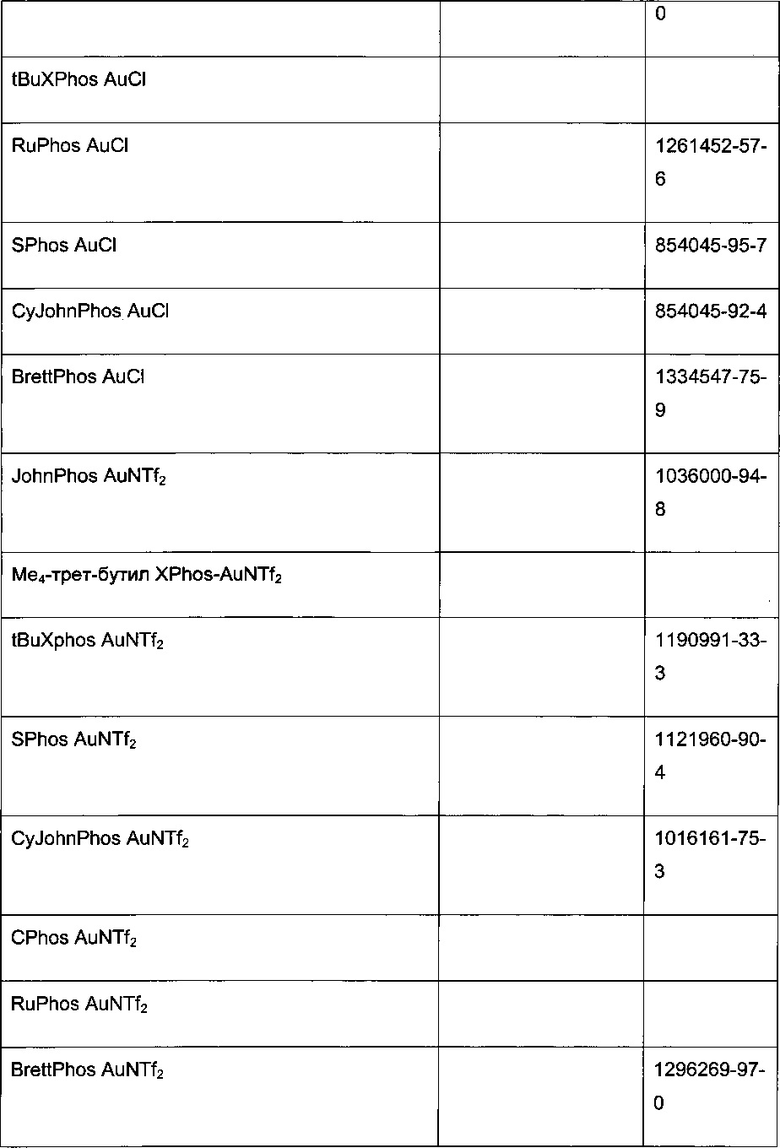
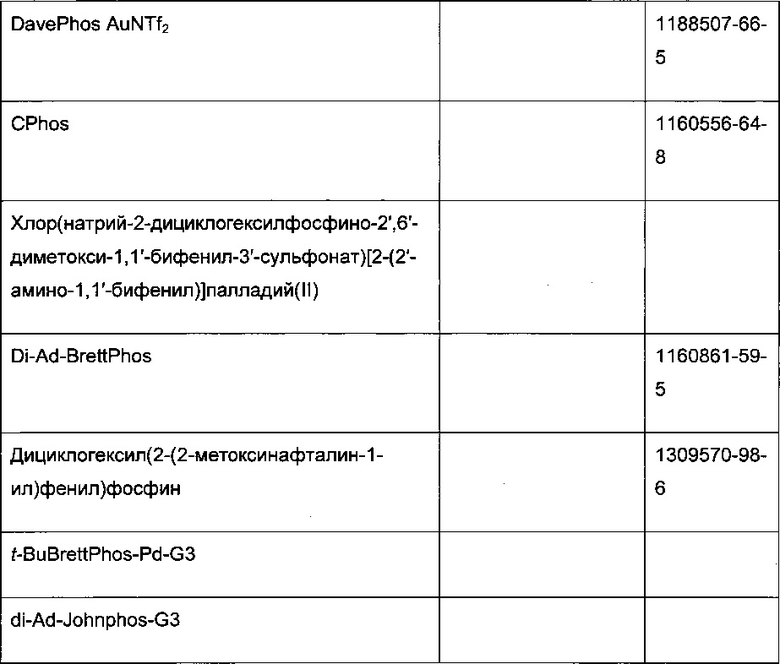
Схема 7:
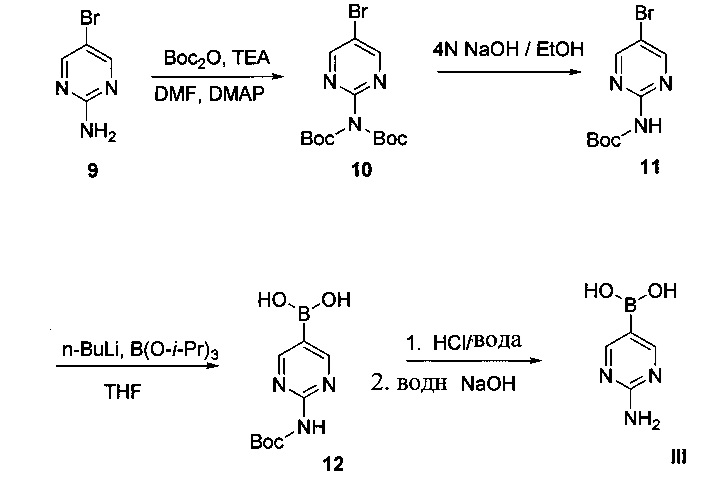
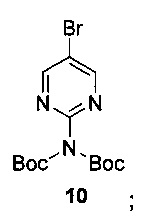
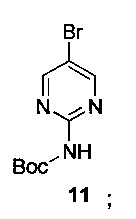
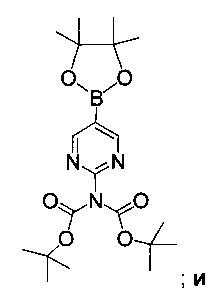
Схема 7 показывает синтез 2-аминопиримидин-5-илбороновой кислоты III из 5-бромпиримидин-2-амина 9. Защиту 2-аминогруппы с помощью Вос-защитного реагента, такого как ди-трет-бутилдикарбонат (Boc2O), проводили через бис-Вос-защищенный интермедиат, бис-трет-бутил-5-бромпиридин-2-ил-дикарбамат 10 (Пример 14) с последующим основным гидролизом одной группы Воc с получением моно-Вос-защищенного трет-бутил 5-бромпиримидин-2-илкарбамата 11 (Пример 15). Основный гидролиз может быть проведен с использованием гидроксида щелочноземельного металла, такого как гидроксид калия, гидроксид натрия или гидроксид лития. Металлирование 11 алкиллитиевым реагентом, таким как н-бутиллитий, и борилирование триалкилборатным реагентом, таким как триизопропилборат, дает 2-(трет-бутоксикарбониламино)пиримидин-5-илбороновую кислоту 12 (Пример 16). Удаление защитной группы с помощью водного кислотного гидролиза и подщелачивания или нейтрализации дает III (Пример 17).
Схема 8:

Схема 8 иллюстрирует альтернативный синтез 2-аминопиримидин-5-илбороновой кислоты III из 5-бромпиримидин-2-амина 9. Металлирование брома в незащищенном 9 с использованием н-бутиллития и борилирование триизопропилборатом дает III (Пример 17).
Другой альтернативный синтез 2-аминопиримидин-5-илбороновой кислоты III может быть проведен с помощью реакции бис-трет-бутил-5-бромпиримидин-2-ил-дикарбамата 10 и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), также известного как бис(пинаколато)дибор, B2Pin2, пинакол диборан, в соответствии с условиями катализа палладием по Бухвальду с использованием предкатализатора палладацикла Бухвальда и реагентов-лигандов из таблицы 1 (Пример 18) с получением бис-трет-бутил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) пиримидин-2-илдикарбамата 13.
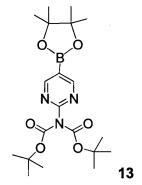
Кислотный гидролиз обеих групп Воc и пинаколиновой группы дает 2-аминопиримидин-5-илбороновую кислоту III.
Лекарственные формы
GDC-0980 может быть получен в виде лекарственных форм в соответствии со стандартной фармацевтической практикой для использования в терапевтических комбинациях для терапевтического лечения (включая профилактическое лечение) гиперпролиферативных заболеваний у млекопитающих, включая человека. Настоящее изобретение относится к фармацевтической композиции, содержащей GDC-0980 в сочетании с одним или более фармацевтически приемлемыми носителями, веществами, способствующими скольжению, разбавителями или эксципиентами.
Подходящие носители, разбавители, вещества, способствующие скольжению, и эксципиенты хорошо известны специалистам в данной области и включают такие материалы как углеводы, воски, растворимые в воде и/или набухающие в воде полимеры, гидрофильные или гидрофобные материалы, желатин, масла, растворители, воду и т.п.
Лекарственные формы могут быть получены с использованием обычных процедур растворения и смешивания. Соединениям по настоящему изобретению, как правило, придают вид фармацевтических дозированных форм, чтобы обеспечить легко контролируемое дозирование лекарства и обеспечить соблюдение пациентом предписанного режима.
Фармацевтическая композиция (или лекарственная форма) может быть упакована в различных формах в зависимости от способа, используемого для введения лекарственного средства. Как правило, изделие для продажи включает контейнер, содержащий фармацевтический препарат в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники и включают бутылки (из пластика и стекла), саше, ампулы, пластиковые мешки, металлические цилиндры и тому подобное. Контейнер может также включать приспособление для контроля первого вскрытия, чтобы предотвратить нежелательный доступ к содержимому упаковки. Кроме того, контейнер несет на себе этикетку, которая описывает содержимое контейнера. Этикетка может также включать соответствующие предупреждения.
Фармацевтические лекарственные формы соединений по настоящему изобретению могут быть получены для различных путей и типов введения с фармацевтически приемлемыми разбавителями, носителями, эксципиентами, глидантами или стабилизаторами (Remington's Pharmaceutical Sciences (1995) 18th edition, Mack Publ. Co., Easton, PA) в форме лиофилизированной лекарственной формы, размолотого порошка или водного раствора. Получение лекарственной формы может быть проведено при перемешивании при комнатной температуре при соответствующем рН и при желаемой степени чистоты с физиологически приемлемыми носителями, то есть с носителями, которые являются нетоксичными для реципиентов в используемых дозах и концентрациях. рН лекарственной формы зависит в основном от конкретного применения и концентрации соединения и может быть в диапазоне от около 3 до около 8.
Фармацевтическая лекарственная форма предпочтительно является стерильной. В частности, лекарственные формы, которые будут использоваться для введения in vivo, должны быть стерильными. Такую стерилизацию легко осуществить путем фильтрации через стерильные фильтрационные мембраны.
Фармацевтическую лекарственную форму обычно можно хранить в виде твердой композиции, таблетки, пилюли, капсулы, лиофилизированной лекарственной формы или в виде водного раствора.
Фармацевтические лекарственные формы по изобретению можно дозировать и вводить в соответствии с хорошей медицинской практикой, то есть их количества, концентрации, график введения, курс введения, носители и способ введения соответствуют хорошей медицинской практике. Факторы, подлежащие рассмотрению в данном контексте, включают конкретное расстройство, которое подвергают лечению, клиническое состояние конкретного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные практикующим врачам.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гексаметония; хлорид бензалкония, хлорид бензетония; фенол, бутиловый спирт, этанол или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол, а также м-крезол); низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, Zn-протеиновые комплексы); и/или неионные поверхностно-активные вещества, такие как TWEEN™, в том числе Tween 80, PLURONIC™ или полиэтиленгликоль (PEG), в том числе PEG400. Активные фармацевтические ингредиенты могут быть также заключены в микрокапсулы, которые получены, например, способами коацервации или путем межфазной полимеризации, например, микрокапсулы из гидроксиметилцеллюлозы или желатина и микрокапсулы из поли(метилметакрилата) соответственно в коллоидные системах доставки лекарственных средств (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие способы описаны в Remington's Pharmaceutical Sciences 18th edition, (1995) Mack Publ. Co., Easton, PA. Другие примеры лекарственных форм можно найти в Liberman, Н. A. and Lachman, L, Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol 3, 2nd Ed., New York, NY.
Фармацевтически приемлемые глиданты могут быть выбраны из диоксида кремния, порошкообразной целлюлозы, микрокристаллической целлюлозы, стеаратов металлов, алюмосиликата натрия, бензоата натрия, карбоната кальция, силиката кальция, кукурузного крахмала, карбоната магния, свободного от асбеста талька, Stearowet С, крахмала, крахмала 1500, лаурилсульфата магния, оксида магния и их комбинаций.
Фармацевтические лекарственные формы включают лекарственные формы, пригодные для путей введения, которые описаны в данном документе. Лекарственные формы могут быть удобно представлены в единичной дозированной форме и могут быть получены любым из способов, хорошо известных в области фармации. Соответствующие способы и лекарственные формы в целом описаны в Remington's Pharmaceutical Sciences 18th Ed. (1995) Mack Publishing Co., Easton, PA. Такие способы включают стадию ассоциации активного ингредиента с носителем, который состоит из одного или более вспомогательных ингредиентов. В общем, лекарственные формы получают путем однородного и тщательного смешивания активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями, или с ними обоими с последующим формованием продукта, если это необходимо.
Фармацевтические композиции могут быть в форме стерильного инъецируемого препарата, например в виде стерильной инъекционной водной или масляной суспензии. Эти суспензии могут быть получены в соответствии с известным уровнем техники с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые были указаны выше. Стерильный инъецируемый препарат может представлять собой раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле, или может быть получен из лиофилизированного порошка. Приемлемые носители и растворители, которые могут быть использованы, включают воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно могут быть использованы стерильные нелетучие масла. С этой целью могут быть использованы любые мягкие нелетучие масла, в том числе синтетические моно- или диглицериды. Кроме того, в инъецируемых препаратах аналогичным образом могут быть использованы жирные кислоты, такие как олеиновая кислота.
ПРИМЕРЫ
Пример 1. 7-метилтиено[3,2-d]пиримидин-2,4 (1Н,3Н)-дион VIII
Метиловый эфир 3-амино-4-метилтиофен-2-карбоновой кислоты IX (100 г, 0,584 моль) и уксусную кислоту (750 мл, 13,1 моль) перемешивали в течение 5 мин с получением прозрачного раствора. К раствору в течение 20 мин медленно добавляли раствор цианата калия (56,8 г, 0,70 моль) в воде (120 мл) и смесь перемешивали в течение 1,5 ч. Затем в течение 20 мин дополнительно медленно добавляли раствор цианата калия (56,8 г, 0,70 моль) в воде (120 мл) и смесь перемешивали в течение 2 ч. Добавляли воду (600 мл) и смесь охлаждали до 10°С и перемешивали в течение 2 ч. Твердое вещество собирали фильтрацией и промывали холодной водой (250 мл). Твердое вещество затем перемешивали в течение 12 ч в растворе гидроксида натрия (79,4 г, 1,99 моль) в воде (1,4 л). рН доводили до 6-7 путем медленного добавления водного концентрированного раствора хлористоводородной кислоты (35% по весу, 110 мл), а затем перемешивали в течение 5 мин. Полученное твердое вещество собирали фильтрацией, промывали водой (2% 250 мл) и сушили при пониженном давлении при 50°С в течение 24 ч с получением 7-метилтиено[3,2-d]пиримидин-2,4(1Н,3Н)-диона VIII в виде грязно-белого твердого вещества (89,6 г, выход 84%).
1H ЯМР (500 МГц, ДМСО-d6) δ 7,68 (с, 1Н), 2,20 (с, 3Н); жидкостная хроматография - масс-спектрометрия LCMS (ионизация электрораспылением, режим положительных ионов, ESI pos) m/z [М+Н] 183.
Пример 2. 2,4-дихлор-7-метилтиено[3,2-d]пиримидин VII
К смеси 7-метилтиено[3,2-d]пиримидин-2,4(1Н,3Н)-диона VIII (89,4 г, 0,491 моль) и N,N-диметиланилина (44,6 г, 0,368 моль) в ацетонитриле (450 мл) добавляли оксихлорид фосфора (312 г, 2,04 моль) в течение 10 мин. Реакционную смесь нагревали до 85°С и перемешивали в течение 24 ч. После охлаждения до комнатной температуры смесь медленно переносили в смесь льда (900 г) и воды (300 мл), поддерживая температуру ниже 10°С. Смесь перемешивали при этой температуре в течение 30 мин. Твердое вещество собирали фильтрацией, промывали водой (450 мл) и сушили при пониженном давлении при 50°С в течение 24 ч с получением 2,4-дихлор-7-метилтиено[3,2-d]пиримидина VII в виде грязно-белого твердого вещества (97,0 г, выход 90%). 1Н ЯМР (400 МГц, CDCl3) δ 7,75 (s, 1Н), 2,50 (s, 3Н); LCMS (ESI pos) m/z [M+H] 220.
Пример 3. 4-(2-хлор-7-метилтиено[3,2-d]пиримидин-4-ил) морфолин VI
Смесь 2,4-дихлор-7-метилтиено[3,2-d]пиримидина VII (90 г, 0,411 моль) и метанола (900 мл) охлаждали до 10°C. Добавляли морфолин (89,5 г, 1,03 моль), поддерживая температуру ниже 15°C. Реакционную смесь перемешивали в течение 2 ч и затем охлаждали до 5°C, и дополнительно перемешивали в течение 1 ч. Твердое вещество собирали фильтрацией, промывали водой (450 мл) и сушили при пониженном давлении при 50°C в течение 24 ч с получением 4-(2-хлор-7-метилтиено[3,2-d]пиримидин-4-ил)морфолина VI виде белого твердого вещества (105 г, выход 95%). 1Н-ЯМР (400 МГц, CDCl3) δ 7,94 (s, 1Н), 3,95-3,86 (m, 4Н), 3,80-3,71 (m, 4Н), 2,9 (s, 3Н); LCMS (ESI pos) m/z [M+H] 270.
Пример 4. 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV
4-(2-хлор-7-метилтиено[3,2-d]пиримидин-4-ил)морфолин VI (27,0 г, 100 ммоль) загружали в подходящего размера реактор и добавляли тетрагидрофуран (безводный, 270 мл). Реакционную смесь охлаждали до температуры ниже -10°С и медленно добавляли 20% (по весу) раствор i-PrMgCl в тетрагидрофуране (25,7 г, 50,0 ммоль) с последующим медленным добавлением 25%-ного раствора н-BuLi в гептане (30,0 г, 117 ммоль), при этом внутреннюю температуру поддерживали ниже -10°C. Смесь оставляли перемешиваться при температуре ниже -10°С в течение 2 ч. При поддержании внутренней температуры ниже -10°С медленно добавляли безводный N,N-диметилформамид (14,6 г, 200 ммоль). Реакционную смесь перемешивали в течение 1-2 ч и переносили в смесь 80% уксусной кислоты, 37%-ного водного раствора соляной кислоты, изопропанола и воды. Полученную суспензию нагревали до 50-55°С и перемешивали в течение 1-3 ч. Суспензию концентрировали при пониженном давлении, чтобы удалить тетрагидрофуран. Затем суспензию охлаждали до комнатной температуры, фильтровали и промывали водой. Осадок сушили при пониженном давлении при 40-60°С с получением 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегида IV в виде желтого твердого вещества (29,2 г, выход 98%), 1Н-ЯМР (400 МГц, CDCl3) δ 10,38 (s, 1Н), 4,03-4,05 (m, 4Н), 3,85-3,87 (m, 4Н), 2,76 (s, 3Н)
Пример 5. (S)-1-(4-((2-Хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1 -ил)-2-гидроксипропан-1-он II
Способ А. В реактор подходящего размера загружали 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV (68,9 г, 231 ммоль), а затем ацетонитрил (870 мл), оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она (V) (86,2 г, 347 ммоль), ацетат натрия (57,0 г, 695 ммоль) и ледяную уксусную кислоту (6,90 г, 115 ммоль). В реактор добавляли порошок молекулярных сит  (75 г) и суспензию нагревали до 80°C и перемешивали в течение как минимум 2 ч. Смесь охлаждали до 40°C и добавляли триацетоксиборгидрид натрия (59,0 г, 278 ммоль). После перемешивания в течение 2 ч медленно добавляли воду (690 мл) и CELITE® (35 г) и смесь нагревали до 50°C и перемешивали в течение 1 ч, фильтровали и промывали ацетонитрилом (210 мл). Фильтрат концентрировали при пониженном давлении для удаления ацетонитрила. Добавляли толуол (689 мл) и рН доводили до 7,5-8,0 с помощью 10% водного раствора карбоната натрия. Органическую фазу отделяли и экстрагировали смесью воды и серной кислоты. Водную фазу отделяли и добавляли толуол (483 мл). рН доводили до 7,5-8,0 с помощью 10% водного раствора карбоната натрия. Смесь нагревали до 20°С, органическую фазу отделяли, концентрировали при пониженном давлении для удаления ацетонитрила, промывали толуолом, и полученную смесь охлаждали до 0-5°C. Медленно добавляли н-гептан (344 мл) и полученную суспензию фильтровали и промывали смесью толуола и н-гептана. Осадок сушили при пониженном давлении с получением (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II (76,3 г, выход 75%). 1H ЯМР (300 МГц, ДМСО-d6) δ 4,84 (d, J=6,80 Гц, 1Н), 4,37-4,47 (m, 1Н), 3,79-3,97 (m, 4Н), 3,65-3,78 (m, 4Н) 3,35-3,64 (m, 4Н), 3,30 (s, 2Н), 2,33-2,64 (m, 4Н), 2,24 (s, 3Н), 1,17 (d, J=6,80 Гц, 3Н).
(75 г) и суспензию нагревали до 80°C и перемешивали в течение как минимум 2 ч. Смесь охлаждали до 40°C и добавляли триацетоксиборгидрид натрия (59,0 г, 278 ммоль). После перемешивания в течение 2 ч медленно добавляли воду (690 мл) и CELITE® (35 г) и смесь нагревали до 50°C и перемешивали в течение 1 ч, фильтровали и промывали ацетонитрилом (210 мл). Фильтрат концентрировали при пониженном давлении для удаления ацетонитрила. Добавляли толуол (689 мл) и рН доводили до 7,5-8,0 с помощью 10% водного раствора карбоната натрия. Органическую фазу отделяли и экстрагировали смесью воды и серной кислоты. Водную фазу отделяли и добавляли толуол (483 мл). рН доводили до 7,5-8,0 с помощью 10% водного раствора карбоната натрия. Смесь нагревали до 20°С, органическую фазу отделяли, концентрировали при пониженном давлении для удаления ацетонитрила, промывали толуолом, и полученную смесь охлаждали до 0-5°C. Медленно добавляли н-гептан (344 мл) и полученную суспензию фильтровали и промывали смесью толуола и н-гептана. Осадок сушили при пониженном давлении с получением (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II (76,3 г, выход 75%). 1H ЯМР (300 МГц, ДМСО-d6) δ 4,84 (d, J=6,80 Гц, 1Н), 4,37-4,47 (m, 1Н), 3,79-3,97 (m, 4Н), 3,65-3,78 (m, 4Н) 3,35-3,64 (m, 4Н), 3,30 (s, 2Н), 2,33-2,64 (m, 4Н), 2,24 (s, 3Н), 1,17 (d, J=6,80 Гц, 3Н).
Способ В. В реактор подходящего размера загружали 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV (14,9 г, 50,0 ммоль), а затем метанол (298 мл), оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V (18,6 г, 74,9 ммоль), ацетат натрия (12,3 г, 150 ммоль), ледяную уксусную кислоту (3,0 г, 50,0 ммоль) и триметилортоформиат (53,1 г, 500 ммоль). Суспензию нагревали до 55-60°C и перемешивали в течение 4 ч. Медленно добавляли 30%-ный раствор 2-пиколин-борана в ТГФ (21,4 г, 60,0 ммоль) и суспензию перемешивали в течение 1-2 ч. Реакционную смесь частично концентрировали при пониженном давлении. Добавляли толуол (230 мл) и реакционную смесь концентрировали при пониженном давлении. Добавляли толуол (114 мл) и реакционную смесь снова частично концентрировали при пониженном давлении. К остатку добавляли толуол (218 мл) и смесь охлаждали до 20-30°C. Добавляли воду (431 мл) и рН доводили до 7,5-8,5 с помощью 10%-ного водного раствора карбоната натрия (162 мл). Органическую фазу отделяли, охлаждали до 0-5°C и экстрагировали смесью воды (180 мл) и 96%-ной серной кислоты (6,1 г). Водную фазу отделяли и добавляли толуол (118 мл). рН доводили до 7,5-8,5 10%-ным водным раствором карбоната натрия (110 мл) при 0-5°C. Смесь нагревали до 20°C и отделяли органическую фазу. Органическую фазу разбавляли толуолом (100 мл) и концентрировали при пониженном давлении до ее первоначального объема (приблизительно 150 мл). Раствор нагревали до 53-57°C и добавляли н-гептан (26 мл). В раствор вносили затравку (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II и суспензию перемешивали при 53-57°С в течение 30 мин. Медленно добавляли гептан (82 мл) и полученную суспензию охлаждали до 0-5°С, фильтровали и промывали смесью толуола и н-гептана, а затем н-гептаном. Осадок сушили при 30-45°С при пониженном давлении с получением (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II (18,6 г, выход 84%)
Способ С. В реактор подходящего размера загружали 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV (15,0 г, 50 ммоль), а затем метанол (306 мл), оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V (18,8 г, 75 ммоль), N-метилморфолин (10,2 г, 100 ммоль) и триметилортоформиат (53,1 г, 500 ммоль). Суспензию нагревали до 55-60°С и перемешивали в течение 4 часов. Медленно добавляли 5-этил-2-метилпиридин боран (8,7 г, 60,0 ммоль) и раствор перемешивали в течение 2 ч. Затем реакционную смесь частично концентрировали при пониженном давлении. Добавляли Ме-ТГФ (350 мл) и реакционную смесь концентрировали при пониженном давлении до конечного объема 300 мл. Смесь охлаждали до 5°C. Для достижения рН 1,6 добавляли 3,3%-ный раствор серной кислоты в воде (401 г). Органическую фазу удаляли. Значение рН водной фазы доводили до 7,9 с помощью 10%-ного водного раствора карбоната натрия (300 г) при 0-5°C. Смесь нагревали до 25°C и отделяли органическую фазу. Органическую фазу концентрировали при пониженном давлении до 75 мл. Раствор нагревали до 35°С и вносили затравку 41 мг (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II. Суспензию охлаждали до 0°С и добавляли н-гептан (200 г). Полученную суспензию выдерживали при температуре -5°С, фильтровали и промывали н-гептаном. Осадок сушили при 70°С при пониженном давлении с получением (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II (17,8 г, выход 81%)
Пример 6. (S)-1-хлор-1-оксопропан-2-ил ацетат 3
Раствор (S)-2-гидроксипропановой кислоты (L-молочной кислоты) 1 (35,0 кг, 388 моль) в дихлорметане (50,0 кг) охлаждали до 5-10°С, и к нему добавляли ацетилхлорид (75,0 кг, 955 моль) при поддержании температуры реакции 10-20°С. Реакционную смесь перемешивали при 10-20°С в течение 4 ч. Добавляли дихлорметан (240 кг), а затем оксалилхлорид (139 кг, 1095 моль), причем добавление реагентов проводили при скорости, которая позволяла поддерживать температуру реакционной смеси 0-15°С. Реакционную смесь выдерживали при 10-20°С в течение 10 ч и смесь концентрировали при пониженном давлении с получением остатка, содержащего (S)-1-хлор-1-оксопропан-2-ил ацетат 3.
Пример 7. (S)-1-(4-Бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетат 4
В сосуд загружали дихлорметан (260 кг), а затем добавляли триэтиламин (65,0 кг, 642 моль). Смесь охлаждали до 0-10°С и добавляли 4-бензилпиперазин дигидрохлорид (31,8 кг, 128 моль). К смесь добавляли (S)-1-хлор-1-оксопропан-2-ил ацетат 3 из примера 6 при поддержании температуры реакции 5-15°С. Реакционную смесь перемешивали при 10-20°С в течение 10 ч. Добавляли воду со льдом (50 кг) и разделяли слои. Водный слой экстрагировали дихлорметаном (2×50 кг). Органические слои объединяли и охлаждали до 5-10°С. Для доведения рН до 6-7 медленно добавляли водный раствор HCl (4 н). Слои разделяли, и водный слой экстрагировали дихлорметаном (2×50 кг). Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка, содержащего (S)-1-(4-бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетат 4.
Пример 8. (S)-1-(4-Бензилпиперазин-1-ил)-2-гидроксипропан-1 5
К остатку из примера 7, содержащему (S)-1-(4-бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетат 4, добавляли метанол (300 кг) и смесь охлаждали до 0-10°С. Добавляли раствор моногидрата гидроксида лития (13,6 кг, 324 моль) в воде (100 кг), причем скорость добавления была такой, чтобы поддерживать температуру реакционной смеси 0-15°С. Смесь выдерживали в течение 2 ч, затем рН доводили до 7 при 5-15°C с помощью уксусной кислоты (4,5 кг, 75 моль). Смесь концентрировали при пониженном давлении. К остатку добавляли дихлорметан (150 кг) и разделяли слои. Водный слой экстрагировали дихлорметаном (2×150 кг). Органические слои объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К остатку добавляли этилацетат (31 кг), после чего медленно добавляли циклогексан (183 кг). Смесь нагревали до 40-50°С и перемешивали в течение 1 ч. Смесь охлаждали до 0-10°С и выдерживали в течение 8 ч. Твердое вещество собирали фильтрованием, промывали холодным циклогексаном и сушили при пониженном давлении при 50°С в течение 12 ч с получением (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропана-1 5 (46 кг, выход 71%, чистота 98% по данным ВЭЖХ). 1Н-ЯМР (300 МГц, CDCl3) δ 7,42-7,20 (m, 5Н), 4,43 (d, J=6,4 Гц, 1Н), 3,84 (широкий s, 1Н), 3,76-3,55 (т, 2Н), 3,53 (s, 2Н), 3,48-3,32 (m, 2Н), 2,46 (s, 4Н), 1,32 (dd, J=6,6, 3,9 Гц, 3Н).
Пример 9. Оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V
Этанол (350 кг), палладий (10% на активированном угле) (8,40 кг, 7,89 моль) и (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1 5 (70,0 кг, 282 моль) загружали в реактор, и смесь продували азотом и нагревали с обратным холодильником. Медленно добавляли циклогексен (70,0 кг, 852 моль). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 24 ч. После охлаждения до комнатной температуры смесь фильтровали через слой CELITE®, и осадок на фильтре промывали этанолом (20 кг). Фильтрат охлаждали до 10-15°С и медленно добавляли раствор дигидрата щавелевой кислоты (36,0 кг, 286 моль) в тетрагидрофуране (156 кг) со скоростью, позволяющей поддерживать температуру реакционной смеси 10-20°С. Смесь выдерживали в течение 2 ч, затем твердое вещество собирали путем фильтрации, промывали этанолом (100 кг) и сушили при пониженном давлении при 50-55°C с получением оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V (58,2 кг, 83%). 1Н-ЯМР (300 МГц, D2O) δ 4,73-4,51 (m, 1Н), 3,93-3,59 (m, 4Н), 3,26 (dd, J=8,8, 4,0 Гц, 4Н), 1,27 (d, J=6,7 Гц, 3Н).
Пример 10. (S)-1-(4-Бензилпиперазин-1-ил)-2-гидроксипропан-1 8
В колбу загружали 1-бензилпиперазин (5,0 г, 28,40 ммоль 1,00 экв.), и колбу охлаждали до 10°С. Добавляли этил (28)-2-гидроксипропаноат 7 (10,1 г, 85,1 ммоль, 3,00 экв.), причем скорость добавления была такой, чтобы поддерживать температуру ниже 20°С. При поддержании температуры ниже 20°С добавляли метоксид натрия (25% по массе в метаноле) (4,9 мл, 21,3 ммоль, 0,75 экв.). Баню с холодной водой убирали, и реакционную смесь оставляли нагреваться до комнатной температуры и выдерживали в течение 16 ч. Смесь разбавляли этанолом (25 мл) и обрабатывали смолой Amberlite® IRC-748 (Dow Chemical Co., Na+ форма, 31,6 г, 1,8 мэкв/г, 2 экв., предварительно превращена в Н* форму с использованием 5%-ного водного раствора HCl). Суспензию перемешивали при комнатной температуре в течение 2 ч. Смолу удаляли фильтрацией через слой CELITE® (3,5 г) и слой промывали этанолом (2×33,8 мл). Фильтрат и промывные воды объединяли и концентрировали при пониженном давлении до 50 мл. К раствору добавляли активированный уголь (DARCO® KB-WJ, Norit Inc., 50% по массе в пересчете на 100% теоретического выхода продукта, 3,52 г). Суспензию перемешивали при комнатной температуре в течение 18 ч. Суспензию фильтровали через слой CELITE® (7 г) и слой промывали EtOH (2×33,6 мл). Фильтрат и промывные растворы соединяли и концентрировали в вакууме с получением остатка, содержащего (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1 8.
Пример 11. Оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V
Способ А. Гидрогенолиз газообразным водородом. К остатку, содержащему (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1 8 из примера 10, добавляли этанол (22,5 мл) и палладий (10% на активированном угле, смочен 56,14% воды) (2,07 г, 0,85 ммоль, 0,03 экв.). Смесь продували аргоном, и в сосуд нагнетали водород до давления 50 фунтов на квадратный дюйм. Смесь перемешивали в атмосфере водорода при 40°С в течение 21 ч. После охлаждения до комнатной температуры смесь фильтровали через слой CELITE® (7 г) и промывали этанолом (33 мл). Фильтрат и промывные воды объединяли и концентрировали при пониженном давлении с получением остатка, содержащего неочищенный (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-он 6 в виде масла. Неочищенный (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-он 6 растворяли в смеси этанол/тетрагидрофуран 50:50 (объем/объем) (17,5 мл каждого) и охлаждали до 10°С. Медленно добавляли раствор дигидрата щавелевой кислоты (7,17 г, 56,8 ммоль, 2 экв.) в смеси этанол: тетрагидрофуран 50:50 (объем/объем) (14 мл каждого). Суспензию оставляли нагреваться до комнатной температуры и выдерживали в течение 18 ч. Суспензию охлаждали до 10°С, и твердое вещество собирали фильтрованием, промывали холодным этанолом (2×27,5 мл), сушили при пониженном давлении при 40°С в течение 24 ч с получением оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V в виде белого твердого вещества (5,24 г, 74%).
Способ В. Гидрогенолиз путем переноса водорода. Остаток, содержащий (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1-он 8 из примера 10, разбавляли этанолом (22,5 мл) и раствор дегазировали, повторяя цикл азот/вакуум три раза. Добавляли палладий (10% на активированном угле, смочен 56,14% воды) (2,07 г, 0,85 ммоль, 0,03 экв.) и смесь дегазировали, повторяя цикл азот/вакуум пять раз. Смесь нагревали до 55°С и медленно добавляли циклогексен (11,6 г, 142 ммоль, 5,00 экв.). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 6 ч. После охлаждения до комнатной температуры смесь фильтровали через слой CELITE® (14 г), промывали этанолом (33 мл). Фильтрат и промывные воды объединяли и концентрировали при пониженном давлении с получением остатка, содержащего (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-он 6 в виде масла, которое растворяли в смеси этанол: тетрагидрофуран 50:50 (объем/объем) (35 мл каждого) и охлаждали до 10°С. Медленно добавляли раствор дигидрата щавелевой кислоты (7,17 г, 56,8 ммоль, 2 экв.) в смеси этанол: тетрагидрофуран 50:50 (объем/объем) (14 мл каждого). Суспензию оставляли нагреваться до комнатной температуры и выдерживали в течение 18 ч. Суспензию охлаждали до 10°С и твердое вещество собирали фильтрованием. Твердое вещество промывали холодным этанолом (2×27,5 мл) и сушили в вакууме при 40°С в течение 24 ч с получением оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V в виде белого твердого вещества (4,16 г, 59%). 1Н-ЯМР (300 МГц, D2O) δ 4,73-4,51 (m, 1Н), 3,93-3,59 (m, 4Н), 3,26 (dd, J=8,8, 4,0 Гц, 4Н), 1,27 (d, J=6,7 Гц, 3Н).
Пример 12. Оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V
В колбу загружали пиперазин (10,0 г, 116 ммоль) и (S)-этил 2-гидроксипропаноат 7 (17,84 г, 151 ммоль, 1,30 экв.), и колбу охлаждали до 10°С.Поддерживая температуру ниже 20°С, медленно добавляли метоксид натрия (25% по массе в метаноле) (12,55 г, 58,1 ммоль, 0,50 экв.). Баню с холодной водой убирали, и реакционную смесь оставляли нагреваться до комнатной температуры и выдерживали ее в течение 19 ч. Добавляли воду (6,23 г, 346 ммоль, 3,0 экв.) и смесь выдерживали в течение 16 ч. Смесь разбавляли этанолом (40 мл) и концентрировали при пониженном давлении. Остаток разбавляли этанолом (40 мл) и обрабатывали раствором дигидрата щавелевой кислоты (6,58 г, 52,2 ммоль) в этаноле (30 мл), чтобы довести рН до 7,5. Суспензию охлаждали до <10°С, фильтровали через слой CELITE® и промывали этанолом (2% 60 мл). Фильтрат и промывные воды объединяли и концентрировали до 50 мл. Раствор охлаждали до 10°С и к нему медленно добавляли раствор дигидрата щавелевой кислоты (16,1 г, 128 ммоль) в этаноле (60 мл). Суспензию оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Суспензию охлаждали до 10°С и твердое вещество собирали фильтрованием, промывали холодным этанолом (2×18 мл) и сушили при пониженном давлении при 50°С в течение 24 ч с получением оксалата (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V в виде белого твердого вещества (15,2 г, 53%). 1Н-ЯМР (300 МГц, D2O) δ 4,73-4,51 (m, 1Н), 3,93-3,59 (m, 4Н), 3,26 (dd, J=8,8, 4,0 Гц, 4Н), 1,27 (d, J=6,7 Гц, 3Н).
Пример 13. (S)-1-(4-((2-(2-Аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он, GDC-0980, Формула I
Способ А
(S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он II (22,0 г, 50,0 ммоль) загружали в реактор подходящего размера, а затем добавляли н-пропанол (198 мл), 2-аминопиримидин-5-илбороновую кислоту III (8,30 г, 59,7 ммоль) и фосфат калия (21,3 г, 100 ммоль). Полученную смесь дегазировали, повторяя цикл вакуум/продувание аргоном три раза. Добавляли хлорид бис(трифенилфосфин)палладия (II) (0,053 г, 0,076 ммоль) и суспензию вновь дегазировали, повторяя цикл вакуум/продувание аргоном три раза. Смесь нагревали в течение 2 ч до 85°С и перемешивали в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры, добавляли воду (200 мл) и рН смеси доводили до 6,0-8,0 с помощью 37%-ного (по массе) водного раствора хлористоводородной кислоты (6,92 мл). Двухфазную смесь нагревали до 80°С и перемешивали в течение 1 ч. Органическую фазу отделяли и медленно фильтровали под давлением через предварительно нагретый фильтр, в который был загружен слой ZETACARBON® R55SP (Cuno Inc., 3М Company, Meriden, СТ). Блок фильтра промывали теплой (80°С) смесью н-пропанола (45 мл) и воды (24 мл). Фильтрат концентрировали при пониженном давлении при сохранении постоянного объема путем добавления воды (150 мл). Полученную суспензию охлаждали до 26-36°С, фильтровали и промывали смесью н-пропанола (15 мл) и воды (108 мл). Осадок сушили при пониженном давлении при 45°C с получением неочищенного продукта в виде желтовато-белого твердого вещества (20,7 г). Неочищенный продукт загружали в реактор подходящего размера с последующим добавлением н-пропанола (116 мл) и воды (62 мл). Суспензию нагревали до 85°С и перемешивали с получением прозрачного раствора. Раствор фильтровали через предварительно нагретый блок фильтра тонкой очистки и промывали смесью н-пропанола (23 мл) и воды (12 мл). Фильтрат охлаждали до -10°С, выдерживали в течение 1 ч и фильтровали. Осадок на фильтре промывали н-пропанолом (77 мл) и сушили при пониженном давлении при 60-70°C с получением (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она, GDC-0980, формула I, в виде твердого вещества от желтовато-белого до белого цвета (18,9 г, 76%). ЯМР (400 МГц, ДМСО-d6) δ 9,15 (s, 2Н), 7,05 (s, 2Н), 4,84 (s, J=6,98 Гц, 1Н), 4,35-4,48 (m, 1Н), 3,89-4,00 (m, 4Н), 3,84 (s, 2Н), 3,67-3,78 (m, 4Н), 3,36-3,64 (m, 4Н), 2,38-2,60 (m, 4Н), 2,34 (s, 3Н), 1,18 (d, J=6,53 Гц, 3Н).
Способ В
(S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-он II (33,0 г, 75 ммоль) загружали в реактор подходящего размера с последующим добавлением н-пропанола (337 г), воды (450 г), 2-аминопиримидин-5-илбороновой кислоты III (12,5 г, 90 ммоль) и гидрофосфата калия (39,2 г, 225 ммоль). Полученную смесь дегазировали, повторяя цикл вакуум/продувание аргоном три раза. Добавляли хлорид бис(трифенилфосфин)палладия (II) (0,079 г, 0,112 ммоль) и суспензию вновь дегазировали, повторяя цикл вакуум/продувание аргоном три раза. Смесь нагревали в течение 2 ч до 65°С и перемешивали в течение 10 часов. Органическую фазу отделяли и медленно фильтровали под давлением через предварительно нагретый фильтр, в который был загружен слой ZETACARBON® R55SP (Cuno Inc., 3М Company, Meriden, СТ). Блок фильтра промывали теплым (80°С) н-пропанолом (45 мл). К фильтрату добавляли воду (750 мл) и полученную суспензию охлаждали до 10°С, выдерживали в течение 1 ч и фильтровали. Осадок на фильтре промывали водой (150 г) и сушили при пониженном давлении при 45°С с получением (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она, GDC-0980, формулы I в виде белого твердого вещества (30,1 г, 79%).
Пример 14. Бис-трет-бутил-5-бромпиримидин-2-ил-дикарбамат 10
К смеси 5-бромпиримидина-2-амина 9 (80,0 кг, 460 моль), ди-трет-бутилдикарбоната (Boc2O) (250 кг, 1140 моль) и триэтиламина (139 кг, 1370 моль) в диметилформамиде (ДМФ) (319 л) медленно добавляли 4-диметиламинопиридин (DMAP) (5,70 кг, 46,6 моль). Реакционную смесь нагревали до 70-90°С и перемешивали в течение 3 ч. После охлаждения до 15-40°С смесь медленно разбавляли водой со льдом (6000 кг) и суспензию перемешивали в течение 1 ч. Твердое вещество собирали фильтрованием и перемешивали с водой (200 кг) в течение 1 ч. Полученное твердое вещество собирали фильтрованием и сушили в вакууме при 50°С в течение 10 ч с получением бис-трет-бутил 5-бромпиримидин-2-ил-дикарбамата 10 (216 кг, >97А% по данным ВЭЖХ, количественный выход). 1Н-ЯМР (500 МГц, CDCl3) δ 8,71 (s, 2Н), 1,40 (s, 18Н); LCMS (ESI): m/z [М-Н] 373
Пример 15. Трет-бутил 5-бромпиримидин-2-илкарбамат 11
К раствору 2-[бис(трет-бутоксикарбонил)амино]-5-бромпиримидина 10 (216 кг неочищенного вещества, 460 моль, при условии количественного выхода в предыдущей стадии) в безводном этаноле (1692 л) медленно добавляли раствор гидроксида натрия (55,2 кг, 1380 моль) в воде (344 л), поддерживая температуру 0-20°С.Смесь перемешивали при этой температуре до содержания 2-[бис(трет-бутоксикарбонил)амино]-5-бромпиримидина (10) ≤0,5% по данным ВЭЖХ. Реакционную смесь охлаждали до 0-5°С и рН доводили до 7 добавлением щавелевой кислоты (86,0 кг, 955 моль), поддерживая температуру ниже 5°C. Затем смесь перегоняли в вакууме до объема 500-600 л, поддерживая температуру ниже 50°C. Добавляли воду (800 кг) и смесь перемешивали в течение 1 ч. Твердое вещество собирали фильтрацией и перемешивали с водой (2×500 л). Полученное твердое вещество собирали фильтрованием и сушили при пониженном давлении при 50°C с получением трет-бутил-5-бромпиримидин-2-илкарбамата (11107 кг, 85%-ный выход для двух стадий). 1Н-ЯМР (500 МГц, CDCl3) δ 8,63 (s, 2Н), 8,12 (s, 1Н), 1,55 (s, 9Н). LCMS (ESI): m/z [М+Н-Вос] 176
Пример 16. 2-(Трет-бутоксикарбониламино)пиримидин-5-илбороновая кислота 12
К смеси трет-бутилового эфира (5-бромпиримидин-2-ил)карбамата 11 (45,0 кг, 164 моль) в тетрагидрофуране (910 л) медленно добавляли триизопропилборат (Sigma-Aldrich, CAS 5419-55-6, 77,4 кг, 412 моль) и смесь охлаждали до -70°С (минус семьдесят градусов по Цельсию). При поддержании температуры ниже -65°С добавляли н-бутиллитий (2,5 М раствор в гексане, 264 л, 660 моль) и реакционную смесь перемешивали до содержания трет-бутил(5-бромпиримидин-2-ил)карбамата 11 ≤0,5% по данным ВЭЖХ. При поддержании температуры ниже 40°С добавляли очищенную воду (5 кг). Смесь охлаждали до 5°С и рН доводили до 7 добавлением 25%-ного водного раствора сульфата натрия (270 кг). Смесь нагревали до 50°С и органические растворители удаляли при пониженном давлении. Добавляли воду (600 кг) и смесь охлаждали до <5°С, после чего рН доводили до 3,5 добавлением 25%-ного водного раствора сульфата натрия (60 кг). Твердое вещество собирали фильтрацией и перемешивали с водой (240 кг) в течение 30 мин. Полученное твердое вещество собирали путем фильтрации и повторно суспендировали в воде (550 кг), и смесь охлаждали до 0-5°С. При поддержании температуры ниже 10°С добавляли 10% водный раствор гидроксида натрия и смесь перемешивали в течение 2 ч. Водную фазу экстрагировали петролейным эфиром (2% 40 кг). Значение рН водной фазы доводили до 3,5 добавлением 25%-ного раствора гидросульфата натри при поддержании температуры 0-10°С. Суспензию фильтровали и твердое вещество повторно суспендировали в воде (400 кг) в течение 1 ч. Твердое вещество собирали фильтрацией и высушивали на фильтре с получением Вос-защищенной 2-(трет-бутоксикарбониламино)пиримидин-5-илбороновой кислоты 12 (40 кг неочищенного вещества, 49% по массе по данным ВЭЖХ, выход 50%). 1H ЯМР (500 МГц, ДМСО-d6) δ 10,08 (s, 1Н), 8,82 (s, 2Н), 8,42 (s, 2Н), 1,46 (s, 9Н).
Пример 17. 2-Аминопиримидин-5-илбороновая кислота III
К смеси [2-[(трет-бутоксикарбонил)амино]пиримидин-5-ил]бороновой кислоты 12 (40,0 кг, 49% по массе по данным ВЭЖХ, 82,0 моль) в воде (245 кг) добавляли концентрированную соляную кислоту (39,6 л), поддерживая температуру ниже 30°С. Реакционную смесь перемешивали в течение 12 ч и затем охлаждали до 10°С. рН смеси доводили до 6,5 добавлением 50% водного раствора гидроксида натрия, поддерживая температуру ниже 15°С, и смесь затем перемешивали в течение 1 ч. Добавляли воду (69,0 кг) и смесь выдерживали в течение 30 мин. Полученную суспензию фильтровали и осадок на фильтре сушили в вакууме при 50°C с получением 2-аминопиримидин-5-илбороновой кислоты III (10,2 кг, выход 90%). ЯМР (300 МГц, ДМСО-d6) δ 8,50 (s, 2Н), 7,97 (s, 2Н), 6,74 (s, 2Н).
В альтернативном способе синтеза, представленном на схеме 8, в 3-литровую колбу в атмосфере азота загружали тетрагидрофуран (1055 мл), а затем 5-бромпиримидин-2-амин 9 (70,0 г, 0,40 моль). Смесь охлаждали до температуры между -60°С и -70°С и при поддержании температуры между -60°С и -70°С в течение 30 мин в колбу загружали бис(триметилсилил)амид (LiHMDS) (11 М раствор в тетрагидрофуране, 483 мл, 0,483 моль). Смесь перемешивали при температуре от -60°С до -70°С в течение 1 ч. В течение 1 ч загружали н-бутиллитий (2,5 М раствор в гексане, 515 мл, 1,29 моль), поддерживая температуру между -60°С и -70°С, и реакционную смесь выдерживали в течение 2 ч. Дополнительно при поддержании температуры между -60°С и -70°С в течение 15 мин в колбу загружали н-бутиллитий (2,5 М в гексане, 48 мл, 0,12 моль), и реакционную смесь перемешивали в течение 1 ч. Поддерживая температуру между -60°С и -70°С, к смеси в течение 1 ч добавляли триизопропилборат (91,0 г, 0,48 моль), и реакционную смесь перемешивали в течение 1 ч. Затем смесь оставляли нагреваться до 0-5°С и к смеси добавляли воду (700 мл) в течение 1 ч. После выдерживания при 0-5°С в течение 30 мин полученные слои разделяли. К водному слою в течение 30 мин добавляли воду (420 мл) с последующим добавлением трет-бутилметилового эфира (822 мл). Смесь оставляли нагреваться до 20-25°С и перемешивали в течение 30 мин. Слои разделяли и водный слой промывали трет-бутилметиловым эфиром (5% 700 мл). Водный слой охлаждали до 0-5°С и в течение 1 ч добавляли 35%-ный водный раствор хлористоводородной кислоты (137 мл), поддерживая температуру между 0-5°С. Смесь перемешивали при 0-5°С в течение 1,5 ч, фильтровали, промывали водой (14 мл) и осадок сушили в вакууме при 45-50°C с получением неочищенного продукта (26,7 г). Неочищенный продукт загружали в 5-литровую колбу, затем добавляли метанол (908 мл) и смесь перемешивали при комнатной температуре (RT) в течение 20 мин. Смесь нагревали до 65°С и перемешивали в течение 1,5 ч. К смеси в течение 2 ч добавляли воду (2136 мл) и суспензию перемешивали в течение 1,5 ч. Смесь охлаждали до 20°С и перемешивали в течение 14 ч. Твердое вещество собирали фильтрацией и осадок на фильтре промывали водой (13 мл) и сушили в вакууме при 45-50°С в течение 12 ч с получением 2-аминопиримидин-5-илбороновой кислоты III (23,6 г, выход 42%)
Пример 18. 2-Аминопиримидин-5-илбороновая кислота III
К раствору 2-[бис(трет-бутоксикарбонил)амино]-5-бромпиримидина 10 (10 г, 27 ммоль), хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладия (II) (101 мг, 0,128 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана), также известного как бис(пинаколато)дибор, BzPing, пинакол диборан (13,6 г, 53,4 ммоль), и ацетата натрия (7,9 г, 80 ммоль) добавляли 50 мл толуола. Смесь нагревали до 85°С в течение 7 часов. После охлаждения до 20°С добавляли 1 н. NaOH в воде (90 мл). Двухфазную смесь фильтровали через целит и органический слой отбрасывали. Органический слой нагревали до 80°С и добавляли 37%-ный раствор HCl в воде (21,2 г). Раствор перемешивали в течение 2 часов и охлаждали до 0°С. К раствору добавляли 28%-ный раствор NaOH в воде (23,7 г) до рН 7. Полученную суспензию фильтровали и промывали водой. Грязно-белое твердое вещество сушили в вакууме при 50°С в течение 16 часов.
Полученные 4,5 г неочищенной 2-аминопиримидин-5-илбороновой кислоты III суспендировали в 144 г метанола и нагревали до 65°С. При этой температуре добавляли воду (73 г). Суспензию охлаждали до 20°С и фильтровали. Белое твердое вещество сушили в вакууме при 50°С в течение 16 часов с получением 2-аминопиримидин-5-илбороновой кислоты III (1,9 г, 97% (вес/вес), выход 49%).
Хотя изобретение было описано выше с указанием некоторых деталей с целью иллюстрации и приведения примеров в целях ясности понимания, описание и примеры не должны быть истолкованы как ограничивающие объем изобретения. Соответственно, все подходящие модификации и эквиваленты подпадают под объем настоящего изобретения, как он определен в приведенной ниже формуле изобретения. Раскрытие всей патентной и научной литературы, приведенной в данном описании, специально включено в настоящий документ во всей своей полноте посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2468027C2 |
| СОЕДИНЕНИЯ-ИНГИБИТОРЫ ФОСФОИНОЗИТИД 3-КИНАЗЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2470936C2 |
| ПУРИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ РI3К, И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2509081C2 |
| КОМБИНАЦИИ ИНГИБИТОРОВ ФОСФОИНОЗИТИД 3-КИНАЗЫ И ХИМИОТЕРАПЕВТИЧЕСКИХ АГЕНТОВ И СПОСОБЫ ПРИМЕНЕНИЯ | 2008 |
|
RU2523890C2 |
| ИНГИБИТОРЫ PI3-КИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2595718C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОКСАЗЕПИНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2649976C2 |
| СОЕДИНЕНИЯ ДИОКСИН- И ОКСАЗИН[2,3-D]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2612251C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2007 |
|
RU2443706C2 |
| СОЕДИНЕНИЯ 8-ФТОРФТАЛАЗИН-1(2Н)-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА | 2012 |
|
RU2622391C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИН-2-ОНА ИЛИ ПИРРОЛОПИРИДИН/ПИРИМИДИН-2-ОНА | 2014 |
|
RU2666532C2 |
Изобретение относится к способу получения (S)-1-(4-((2-(2-аминoпиpимидин-5-ил)-7-мeтил-4-мopфoлинoтиeнo[3,2-d]пиpимидин-6-ил)мeтил)пипepaзин-1-ил)-2-гидpoкcипpoпaн-1-она, имеющего структуру:
 ,
,
а также к новому промежуточному соединению оксалату (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она, имеющему следующую структуру:
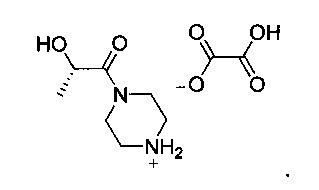
Технический результат: разработан новый способ получения (S)-1-(4-((2-(2-аминoпиpимидин-5-ил)-7-мeтил-4-мopфoлинoтиeнo[3,2-d]пиpимидин-6-ил)мeтил)пипepaзин-1-ил)-2-гидpoкcипpoпaн-1-она, являющегося двойным ингибитором mTOR/PI3K GDC-0980. 2 н. и 14 з.п. ф-лы, 1 табл., 18 пр.
1. Способ получения (S)-1-(4-((2-(2-аминопиримидин-5-ил)-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она I, имеющего структуру:
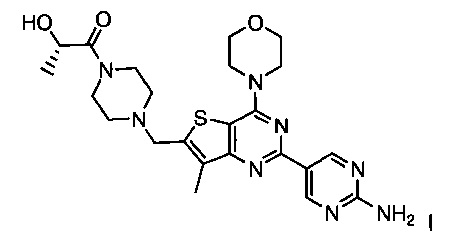
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей, включающий:
(a) взаимодействие соли (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она 6 и 2-хлор-7-метил-4-морфолинотиено[3,2-D]пиримидин-6-карбальдегида IV с восстанавливающим агентом с получением (S)-1-(4-((2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-ил)метил)пиперазин-1-ил)-2-гидроксипропан-1-она II
(b) взаимодействие II, палладиевого катализатора и 2-аминопиримидин-5-илбороновой кислоты III, имеющей структуру:
с получением I.
2. Способ по п. 1, где соль (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она 6 представляет собой оксалат.
3. Способ по п. 2, где оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она получают способом, включающим взаимодействие (S)-этил 2-гидроксипропаноата с пиперазином, а затем с щавелевой кислотой.
4. Способ по п. 2, где оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V, имеющий структуру:
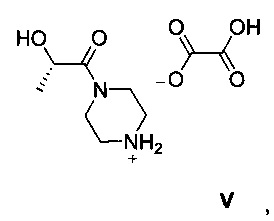
получают способом, включающим:
(a) взаимодействие (S)-этил 2-гидроксипропаноата 7 с 1-бензилпиперазином с получением (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1-она 8, имеющего структуру:
(b) восстановление 8 с помощью палладиевого катализатора с получением (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она 6, имеющего структуру:
(c) взаимодействие (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она с щавелевой кислотой с получением V.
5. Способ по п. 2, где оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она V получают способом, включающим:
(a) ацетилирование (S)-2-гидроксипропановой кислоты (L-молочной кислоты) 1 с получением (S)-2-ацетоксипропановой кислоты;
(b) взаимодействие (S)-2-ацетоксипропановой кислоты с хлорирующим реагентом с получением (S)-1-хлор-1-оксопропан-2-ил ацетата;
(c) взаимодействие (S)-1-хлор-1-оксопропан-2-ил ацетата с 1-бензилпиперазином с получением (S)-1-(4-бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетата;
(d) гидролиз ацетата (S)-1-(4-бензилпиперазин-1-ил)-1-оксопропан-2-ил ацетата с получением (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1-она;
(e) восстановительное удаление бензильной группы (S)-1-(4-бензилпиперазин-1-ил)-2-гидроксипропан-1-она с помощью палладиевого катализатора с получением (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она; и
(f) взаимодействие (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она с щавелевой кислотой с получением V.
6. Способ по п. 1, в котором 2-аминопиримидин-5-илбороновую кислоту III получают способом, включающим:
(a) взаимодействие 5-бромпиримидин-2-амина с защитным реагентом Boc с получением бис-трет-бутил-5-бромпиримидин-2-ил-дикарбамата 10, имеющего структуру:
(b) основной гидролиз одной группы Boc (трет-бутилоксикарбонильная группа) с получением трет-бутил-5-бромпиримидин-2-илкарбамата 11, имеющего структуру:
(c) металлирование 11 алкиллитиевым реагентом и борилирование триалкилборатным реагентом с получением 2-(трет-бутоксикарбониламино)пиримидин-5-илбороновой кислоты 12; и
(d) удаление защитной группы Boc соединения 12 в кислых условиях с получением III.
7. Способ по п. 1, в котором 2-аминопиримидин-5-илбороновую кислоту III получают способом, включающим:
(a) взаимодействие 5-бромпиримидин-2-амина с бис(триметилсилил)амидом лития, затем с н-бутиллитием, а затем триалкилборатом; и
(b) обработку смеси водным раствором кислоты с получением III.
8. Способ по п.1, в котором 2-хлор-7-метил-4-морфолинотиено[3,2-d]пиримидин-6-карбальдегид IV получают способом, включающим взаимодействие 4-(2-хлор-7-метилтиено[3,2-d]пиримидин-4-ил)морфолина VI, имеющего структуру:
с реактивом Гриньяра, алкиллитиевым реагентом и диметилформамидом.
9. Способ по п. 1, в котором 2-аминопиримидин-5-илбороновую кислоту III получают способом, включающим:
(a) взаимодействие 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) и бис-трет-бутил-5-бромпиримидин-2-ил-дикарбамата 10 в условиях катализа палладием по Бухвальду с получением бис-трет-бутил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-илдикарбамата, имеющего структуру:
(b) кислотный гидролиз обеих групп Boc и пинаколиновой группы с получением III.
10. Способ по п. 8, в котором 4-(2-хлор-7-метилтиено[3,2-d]пиримидин-4-ил)морфолин VI получают способом, включающим взаимодействие 2,4-дихлор-7-метилтиено[3,2-d]пиримидина VII, имеющего структуру
с морфолином.
11. Способ по п. 10, в котором 2,4-дихлор-7-метилтиено[3,2-d]пиримидин VII получают способом, включающим взаимодействие 7-метилтиено[3,2-d]пиримидин-2,4 (1Н,3Н)-диона VIII, имеющего структуру:
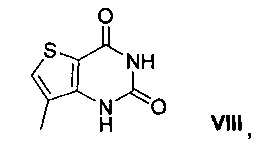
с оксихлоридом фосфора.
12. Способ по п. 11, в котором 7-метилтиено[3,2-d]пиримидин-2,4(1Н,3Н)-дион VIII получают способом, включающим взаимодействие метил-3-амино-4-метилтиофен-2-карбоновой кислоты IX, имеющей структуру:
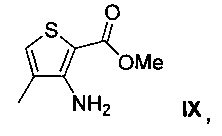
с цианатом калия.
13. Способ по п. 1, в котором палладиевый катализатор выбирают из PdCl2(PPh3)2, Pd(t-Bu)3, PdCl2 dppf CH2Cl2, Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Pd[(Pet3)]2, Pd(DIPHOS)2, Cl2Pd(бипиридин), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[P(o-толуил)3]2, Pd2(dba)3/P(o-толуил)3, Pd2(dba)/P(фурил)3, Cl2Pd[P(фурил)3]2, Cl2Pd(PMePh2)2, Cl2Pd[P(4-F-Ph)3]2, Cl2Pd[P(C6F6)3]2, Cl2Pd[P(2-COOH-Ph)(Ph)2]2 и Cl2Pd[P(4-COOH-Ph)(Ph)2]2.
14. Способ по п. 1, в котором восстанавливающим агентом является триацетоксиборгидрид натрия, 2-пиколинборан или 5-этил-2-метилпиридин боран.
15. Способ по любому из пп. 1-14, дополнительно включающий фильтрацию реакционной смеси через активированный уголь после реакции II, палладиевого катализатора и 2-аминопиримидин-5-илбороновой кислоты III.
16. Оксалат (S)-2-гидрокси-1-(пиперазин-1-ил)пропан-1-она, имеющий следующую структуру:
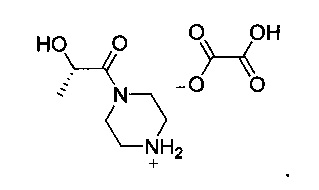
| RU 2009125897 A, 20.01.2011 | |||
| WO 2010105008 A2, 16.09.2010 | |||
| DANIEL P | |||
| SUTHERLIN et al | |||
| "Discovery of a Potent, Selective, and Orally Available Class Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Kinase Inhibitor (GDC-0980) for the Treatment of Cancer", JOURNAL OF MEDICINAL CHEMISTRY, т | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| Локомотив с несколькими двигателями внутреннего горения (тепловоз) | 1926 |
|
SU7579A1 |

