Перекрестная ссылка на родственные заявки
Данная заявка, не являющаяся предварительной, поданная согласно 37 Свода федеральных правил (CFR) §1.53(b), претендует на приоритет согласно 35 Свода законов США (USC) §119(e) в соответствии с предварительной заявкой на патент США №61/779619, поданной 13 марта 2013 года, содержание которой включено в настоящее описание полностью посредством ссылки на нее.
Область изобретения
Изобретение относится к способам получения ингибитора PI3K соединения GDC-0032.
Предшествующий уровень техники
Фосфоинозитид-3-киназы (PI3K от phosphoinositide 3-kinases) представляют собой липидкиназы, которые фосфорилируют липиды по 3-гидроксильному остатку инозитольного кольца (Whitman et al (1988) Nature, 332: 664). 3-Фосфорилированные фосфолипиды (PIP3s от 3-phosphorylated phospholipids), синтезированные PI3-киназами, действуют в качестве вторичных мессенджеров, вовлекающих киназы с липид-связывающими доменами (включая плекстрин-гомологичные (РН от plekstrin homology) области), такие как Akt и фосфоинозитид-зависимая киназа-1 (PDK1 от phosphoinositide-dependent kinase-1). Связывание Akt с мембранными PIP3s вызывает транслокацию Akt к клеточной мембране, приводя в контакт Akt с PDK1, который ответственен за активацию Akt. Опухоль-супрессорная фосфатаза, PTEN, дефосфорилирует PIP3 и поэтому действует в качестве отрицательного регулятора активации Akt. PI3-киназы Akt и PDK1 имеют большое значение в регуляции многих клеточных процессов, включая регуляцию клеточного цикла, пролиферацию, продолжительность существования, апоптоз и подвижность, и являются важными компонентами молекулярных механизмов заболеваний, таких как рак, диабет и иммунное воспаление (Vivanco et al (2002) Nature Rev. Cancer 2: 489; Phillips et al (1998) Cancer 83: 41).
Основной изоформой PI3-киназы при раке является PI3-киназа класса I, p110 α (альфа) (US 5824492; US 5846824; US 6274327). Другие изоформы участвуют в сердечно-сосудистых и иммуновоспалительных заболеваниях (Workman Р (2004) Biochem Soc Trans 32: 393-396; Patel et al (2004) Proceedings of the American Association of Cancer Research (Abstract LB-247) 95th Annual Meeting, March 27-31, Orlando, Florida, USA; Ahmadi K and Waterfield MD (2004) Encyclopedia of Biological Chemistry (Lennarz W J, Lane M D eds) Elsevier/Academic Press). Путь PI3 киназа/Akt/PTEN является привлекательной мишенью для разработки противораковых лекарственных средств, поскольку ожидается, что такие модулирующие или ингибирующие агенты будут ингибировать пролиферацию, изменять подавление апоптоза и преодолевать сопротивляемость к цитотоксическим средствам в раковых клетках (Folkes et al (2008) J. Med. Chem. 51: 5522-5532; Yaguchi et al (2006) Jour, of the Nat. Cancer Inst. 98(8): 545-556). Сигнальный путь PI3K-PTEN-AKT дерегулируется при большом числе раковых заболеваний (Samuels Y, Wang Z, Bardellil A et al. High frequency of mutations of the PIK3CA gene in human cancers. (2004) Science; 304 (5670): 554; Carpten J, Faber AL, Horn C. "A transforming mutation in the pleckstrin homology domain of AKT1 in cancer" (2007) Nature; 448: 439-444).
GDC-0032, также известный как 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид, оказывает сильное действие на PI3K (WO 2011/036280; US 8242104) и исследуется на пациентах с местно-распространенными или метастатическими солидными опухолями.
Сущность изобретения
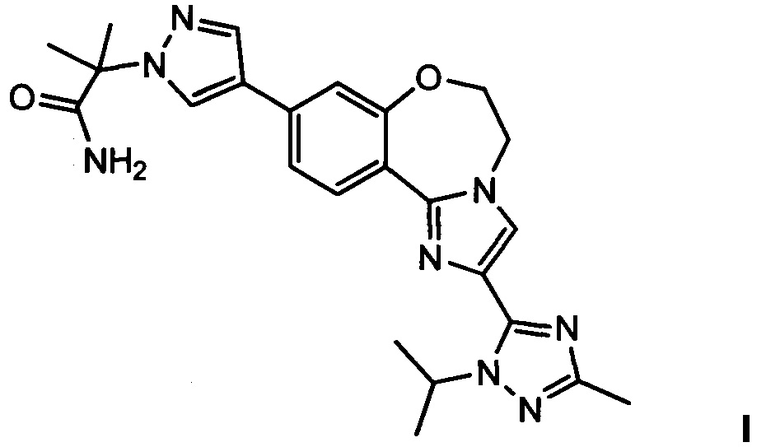
Изобретение относится к способам получения PI3K ингибитора I (GDC-0032), называемого 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид, имеющего структуру:
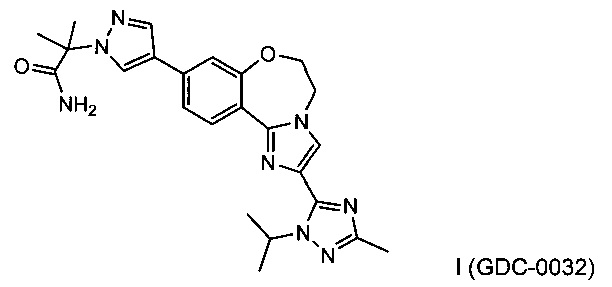
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей.
Другой аспект изобретения включает новые промежуточные соединения, полезные для получения GDC-0032 и имеющие структуры:
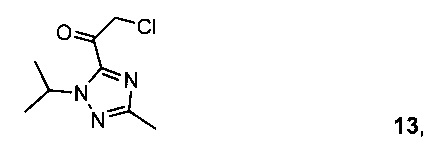
 и
и
Определения
Термин "хиральный" относится к молекулам, которые обладают свойством неналожения зеркального изображения партнера, тогда как термин "ахиральный" относится к молекулам, которые являются налагающимися на зеркальное изображение их партнера.
Термин "стереоизомер" относится к соединениям, которые имеют одинаковый химический состав, но отличаются расположением атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и чьи молекулы не являются зеркальными изображениями друг друга. Диастереомеры имеют разные физические свойства, например, температуры плавления, температуры кипения, спектральные свойства и реакционные способности. Смеси диастереомеров можно разделить в ходе аналитических методик с высоким разрешением, таких как электрофорез и хроматография.
"Энантиомеры" относятся к двум стереоизомерам соединения, которые являются неналагающимися зеркальными изображениями друг друга.
Стереохимические определения и обозначения, используемые в данном документе, как правило применяют согласно S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать центры асимметрии или хиральные центры, и, следовательно, существуют в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, являются частью настоящего изобретения. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения приставки D и L, или R и S, используют для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов). Приставки d и I или (+) и (-) используют для обозначения знака направления вращения плоскополяризованного света соединением, так (-) или I означает, что соединение является левовращающим. Соединение с приставкой (+) или d является правовращающим. Для приведенной химической структуры эти стереоизомеры являются одинаковыми за исключением того, что они представляют собой зеркальные изображения друг друга. Также конкретный стереоизомер может называться энантиомером, и смесь таких изомеров часто называется энантиомерной смесью. Смесь 50:50 энантиомеров называется рацемической смесью или рацемат, которая может встречаться, когда отсутствует стереоселективность или стереоспецифичность в химической реакции или способе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных соединений без оптической активности.
Термин "таутомер" или "таутомерная форма" относится к структурным изомерам разных энергий, которые являются взаимопревращаемыми при переходе через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения в ходе перемещения протона, такие как кето-енольные и имин-енаминовые изомеризации. Валентные таутомеры включают взаимопревращения в ходе перегруппировки некоторых из связывающих электронов.
Фраза "фармацевтически приемлемая соль", как используется в данном документе, относится к фармацевтически приемлемым органическим или неорганическим солям соединения по изобретению. Примерные соли включают, но не ограничиваются этим, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат "мезилат", этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может содержать включение другой молекулы, такой как ион ацетата, ион сукцината или другой противоион. Противоион может представлять собой органическую или неорганическую группировку, которая стабилизирует заряд в исходном соединении. Кроме того, фармацевтически приемлемая соль может иметь более чем один заряженный атом в своей структуре. В тех случаях, когда многочисленные заряженные атомы являются частью фармацевтически приемлемой соли, они могут содержать многочисленные противоионы. Следовательно, фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более чем один противоион.
Если соединение по изобретению представляет собой основание, то требуемую фармацевтически приемлемую соль можно получить любым подходящим способом, применимым в данной области техники, например, обработкой свободного основания неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и подобные, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильная кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота, или подобные.
Если соединение по изобретению представляет собой кислоту, то требуемую фармацевтически приемлемую соль можно получить любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла, или подобные. Иллюстративные примеры подходящих солей включают, но не ограничиваются этим, органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
"Сольват" относится к ассоциации или комплексу одной или более молекул растворителя и соединения по изобретению. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются этим, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, где молекулой растворителя является вода.
Фраза "фармацевтически приемлемая соль", как используется в данном документе, относится к фармацевтически приемлемым органическим или неорганическим солям соединения по изобретению. Примерные соли включают, но не ограничиваются этим, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат "мезилат", этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может содержать включение другой молекулы, такой как ион ацетата, ион сукцината или другой противоион. Противоион может представлять собой органическую или неорганическую группировку, которая стабилизирует заряд в исходном соединении. Кроме того, фармацевтически приемлемая соль может иметь более чем один заряженный атом в своей структуре. В тех случаях, когда многочисленные заряженные атомы являются частью фармацевтически приемлемой соли, они могут содержать многочисленные противоионы. Следовательно, фармацевтически приемлемая соль может иметь один или более заряженных атомов и/или один или более чем один противоион.
Получение GDC-0032
Настоящее изобретение включает процессы, способы, реактивы и промежуточные соединения для синтеза GDC-0032, формулы I, низкомолекулярного ингибитора PI3K и mTOR, (Roche RG7604, CAS рег. №1282512-48-4), который имеет структуру:
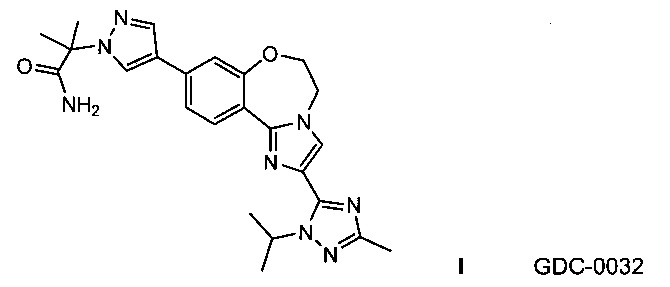
и может быть назван: 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид (US 8242104; WO 2011/036280, которые явным образом включены посредством ссылки). Как используется в данном документе, GDC-0032 включает все стереоизомеры, геометрические изомеры, таутомеры и фармацевтически приемлемые соли.
Соединения по изобретению могут содержать центры асимметрии или хиральные центры, и, следовательно, существуют в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, являются частью настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. В структурах, показанных в данном документе, где стереохимия конкретного хирального атома не определена, все стереоизомеры предусмотрены и включены в качестве соединений по изобретению. Когда стереохимия обозначена цельным клином или пунктирной линией, изображающей конкретную конфигурацию, тогда этот стереоизомер таким образом установлен и определен.
Соединения по изобретению могут существовать в несольватированных, а также сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные, и предполагается, что изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения по изобретению могут также существовать в разных таутомерных формах, и все такие формы находятся в объеме данного изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам разных энергий, которые являются взаимопревращаемыми при переходе через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения в ходе перемещения протона, такие как кето-енольные и имин-енаминовые изомеризации. Валентные таутомеры включают взаимопревращения в ходе перегруппировки некоторых из связывающих электронов.
Соединения по изобретению также включают изотопно-меченые соединения, которые являются такими же как те, что перечислены в данном документе, за исключением того факта, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличающееся от атомной массы или массового числа, обычно находящегося в природе. Все изотопы конкретного атома или элемента, как указано, находятся в объеме соединений по изобретению, и их использования. Примерные изотопы, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14С, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I и 125I. Конкретные изотопно-меченые соединения по настоящему изобретению (например, меченные 3Н и 14С) полезны в анализах распределения в тканях соединения и/или субстрата. Меченные тритием (3Н) и углеродом-14 (14С) изотопы полезны из-за легкости их получения и обнаружения. Кроме того, замещение тяжелыми изотопами, такими как дейтерий (т.е. 2Н), может давать определенные терапевтические преимущества, возникающие вследствие большей метаболической устойчивости (например, увеличенный in vivo период полураспада или уменьшенные требования к дозировке) и, следовательно, может быть предпочтительно при некоторых обстоятельствах. Позитронно-активные изотопы, такие как 15O, 13N, 11С и 18F, полезны для изучений позитронно-эмиссионной томографии (ПЭТ), чтобы проверить степень занятости рецептора субстратом. Изотопно-меченые соединения по настоящему изобретению как правило можно получить, соблюдая способы, аналогичные тем, что раскрыты в разделе примеры, приведенном ниже в данном документе, замещая изотопно-меченым реагентом неизотопно-меченый реагент.
Исходные вещества и реактивы для получения GDC-0032 как правило есть в наличии у поставщиков, таких как Sigma-Aldrich Chemical (Milwaukee, WI), или их легко получают, используя способы, хорошо известные квалифицированным специалистам в данной области техники (например получают способами, в общем описанными у Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступные через электронную базу данных Бейльштейн).
Следующие схемы 1-15 иллюстрируют химические реакции, способы, методику синтеза GDC-0032, формулы I, и конкретные промежуточные соединения и реактивы.
Схема 1:
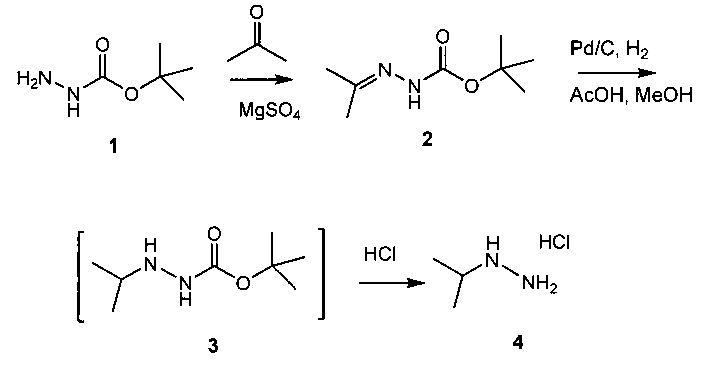
Схема 1 демонстрирует синтез промежуточного изопропилгидразина гидрохлорида 4 из Boc-гидразина 1. Конденсация 1 с ацетоном и сульфатом магния давала Boc-гидразон, трет-бутил 2-(пропан-2-илиден)гидразинкарбоксилат 2 (пример 1). Катализируемое палладием гидрирование 2 в уксусной кислоте и метаноле давало Boc-изопропил-гидразин 3 (пример 2), который обрабатывали в реакционной смеси газообразным хлороводородом, получая 4 (пример 3).
Альтернативно, двойную связь в 2 можно восстановить гидридным реагентом, таким как цианоборгидрид натрия (пример 2).
Схема 2:
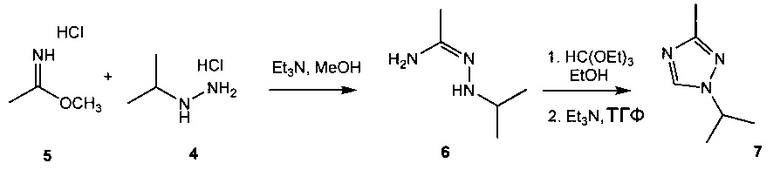
Схема 2 демонстрирует синтез 1-изопропил-3-метил-1Н-1,2,4-триазола 7 из метилацетимидата гидрохлорида 5 и изопропилгидразина гидрохлорида 4. Реакция 5 и 4 в триэтиламине и метаноле с последующей циклизацией продукта конденсации, N'-изопропилацетогидразонамида 6 (пример 4), с триэтилортоформиатом (триэтоксиметаном) давала 7 (пример 5). Альтернативно, 4 и ацетамидин можно подвергнуть взаимодействию с получением 6.
Или 4 можно подвергнуть взаимодействию с ацетонитрилом и кислотой с образованием соответствующей соли 6.
Схема 3:
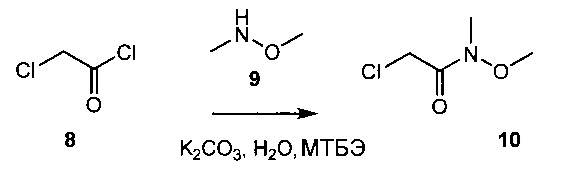
Схема 3 демонстрирует синтез промежуточного соединения, 2-хлор-N-метокси-N-метилацетамида 10. Реакция 2-хлорацетилхлорида 8 и N,O-диметилгидроксиламина гидрохлорида 9 в водном карбонате калия и метил-трет-бутиловом эфире (МТБЭ) давала 10 (пример 6).
Схема 4:
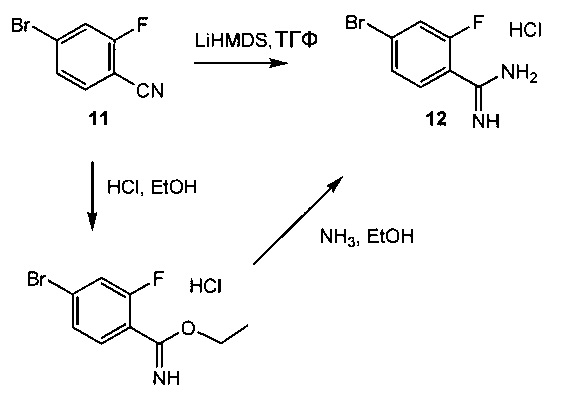
Схема 4 демонстрирует синтез промежуточного 4-бром-2-фторбензимидамида гидрохлорида 12, образующегося в ходе реакции 4-бром-2-фторбензонитрила 11 с гексаметилдисилазидом лития (LiHMDS) в тетрагидрофуране (пример 7). Альтернативно, 11 обрабатывают хлороводородом в спирте, таком как этанол, с образованием имидата, этил 4-бром-2-фторбензимидата гидрохлорида, затем аммиаком в спирте, таком как этанол, с образованием 12 (пример 7).
Схема 5:
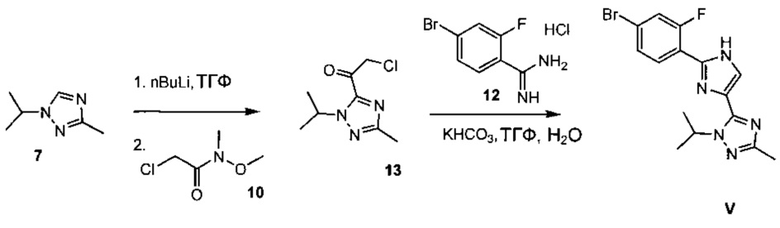
Схема 5 демонстрирует синтез 5-(2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил)-1-изопропил-3-метил-1Н-1,2,4-триазола V из 1-изопропил-3-метил-1Н-1,2,4-триазола 7. Депротонирование 7 с н-бутиллитием и ацилирование с 2-хлор-N-метокси-N-метилацетамидом 10 давало промежуточный 2-хлор-1-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)этанон 13 (пример 8). В ходе циклизации 13 с 4-бром-2-фторбензимидамида гидрохлоридом 12 и гидрокарбонатом калия в воде и ТГФ (тетрагидрофуране) образовывался имидазол V (пример 9).
Схема 6:
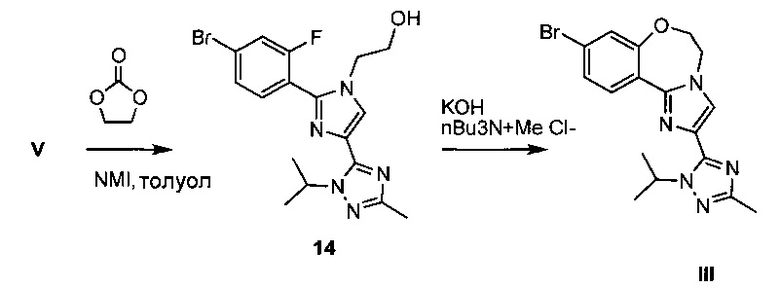
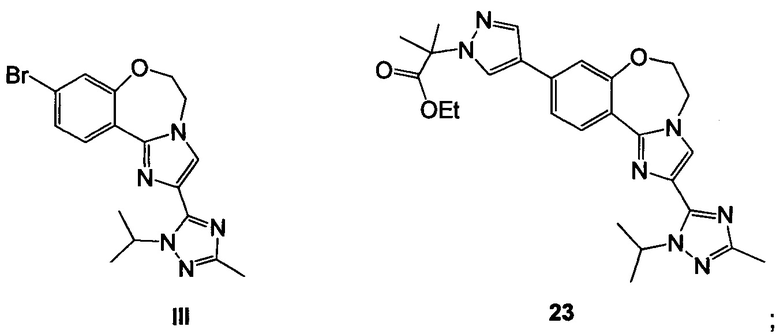
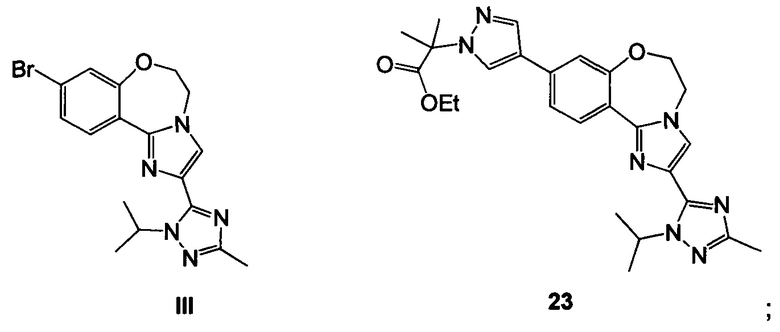
Схема 6 демонстрирует синтез 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина III из V. Алкилирование имидазольного азота V 2-гидроксиэтилирующим реагентом, таким как 1,3-диоксолан-2-он, давало 2-(2-(4-бром-2-фторфенил)-4-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-1Н-имидазол-1-ил)этанол 14 (пример 10). Циклизация 14 с водным основным реагентом, таким как метилтрибутиламмония хлорид, в водном гидроксиде калия, давало III, который может кристаллизоваться из этанола и воды (пример 11).
Схема 7:
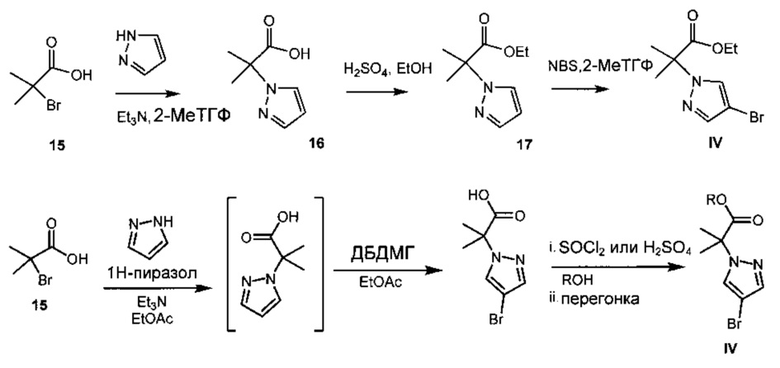
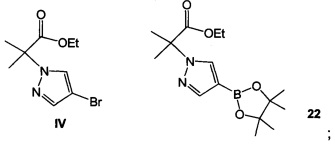
Схема 7 демонстрирует синтез этил 2-(4-бром-1H-пиразол-1-ил)-2-метилпропионата IV, исходя из 2-бром-2-метилпропионовой кислоты 15. Алкилирование пиразола с 15 давало 2-метил-2-(1H-пиразол-1-ил)пропионовую кислоту 16 (пример 12). Этерификация 16 с серной кислотой в этаноле давала этил 2-метил-2-(1H-пиразол-1-ил)пропионат 17 (пример 13). Региоспецифическое бромирование 17 с N-бромсукцинимидом (NBS) давало IV (пример 14). Альтернативно, 16 обрабатывали в реакционной смеси бромирующим реагентом, таким как 1,3-дибром-5,5-диметилгидантоин (ДБДМГ), получая 2-(4-бром-1Н-пиразол-1-ил)-2-метилпропионовую кислоту, которую этерифицировали, получая IV, где R представляет собой этил. Также можно получить другие эфиры, такие как метиловый, изопропиловый, или любой алкиловый, бензиловый или ариловый эфир.
Схема 8:
 г
г
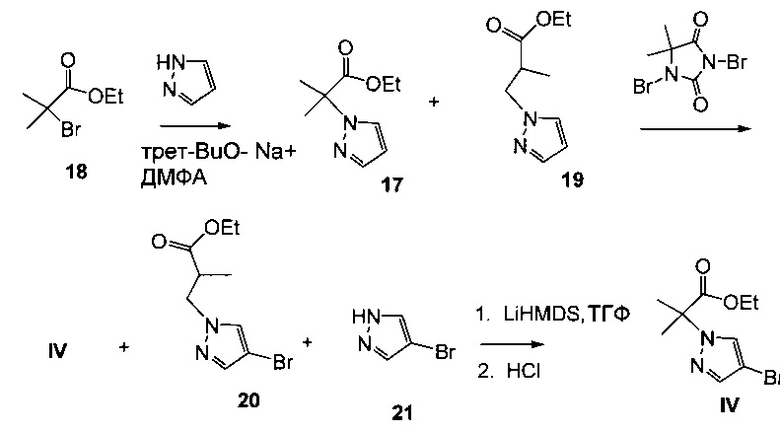
Схема 8 демонстрирует альтернативный синтез этил 2-(4-бром-1Н-пиразол-1-ил)-2-метилпропионата IV, исходя из этил 2-бром-2-метилпропионата 18. Алкилирование пиразола с 18 в присутствии основания, такого как трет-бутилат натрия или карбонат цезия, давало смесь этил 2-метил-2-(1H-пиразол-1-ил)пропионата 17 и этил 2-метил-3-(1H-пиразол-1-ил)пропионата 19. Бромирование смеси 1,3-дибром-5,5-диметилимидазолидин-2,4-дионом (ДБДМГ) давало смесь, содержащую IV, этил 3-(4-бром-1H-пиразол-1-ил)-2-метилпропионат 20 и 4-бром-1Н-пиразол 21, которую обрабатывали сильным основанием в безводных условиях, таких как гексаметилдисилазид лития в тетрагидрофуране. Подкисление хлористоводородной кислотой давало IV.
Схема 9:
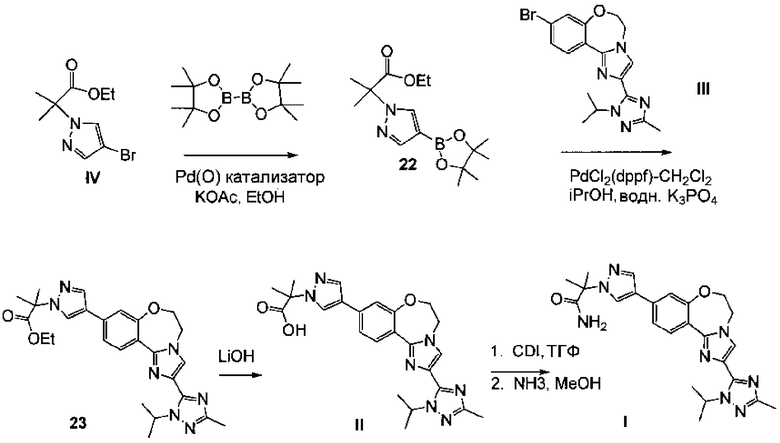
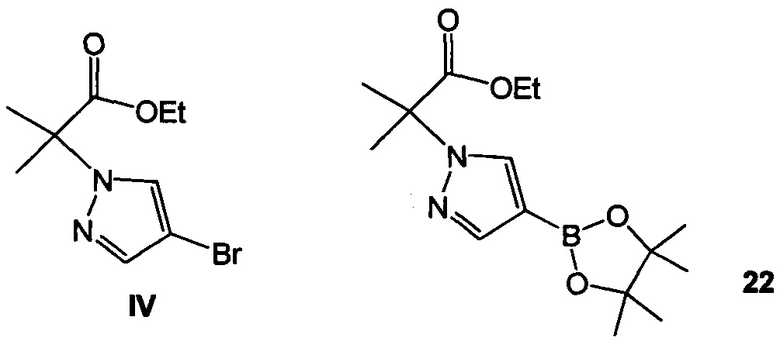
Схема 9 демонстрирует синтез 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамида, GDC-0032, I из этил 2-(4-бром-1H-пиразол-1-ил)-2-метилпропионата IV (регистрационный номер CAS: 1040377-17-0, WO 2008/088881) и 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина III (регистрационный номер CAS: 1282514-63-9, US 2012/0245144, US 8242104). Также кроме этилового можно использовать другие эфиры, которые могут гидролизоваться с водным основанием, такие как метиловый, изопропиловый, или любой алкиловый, бензиловый или ариловый эфир. При однореакторном борилировании Мияура / Сузуки, система Бухвальда, этил 2-(4-бром-1H-пиразол-1-ил)-2-метилпропионат IV подвергают взаимодействию с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан), CAS рег. №73183-34-3, также называемым B2PiN2, и палладиевым катализатором, таким как XPhos (2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, CAS рег. №564483-18-7), с солью, такой как ацетат калия, в растворителе, таком как этанол, приблизительно при 75°C с образованием промежуточного этил 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропионата 22 (пример 15, регистрационный номер CAS: 1201657-32-0, US 8242104, US 8263633, WO 2009/150240).
Промежуточное соединение 22 может быть выделено или подвергнуто взаимодействию в реакционной смеси (один реактор) с III с образованием 23.
Целый ряд низковалентных Pd(II) и Pd(0) палладиевых катализаторов можно использовать на стадии сочетания Сузуки с образованием 23 (пример 16) из 22 и III, включая PdCl2(PPh3)2, Pd(t-Bu)3, PdCl2 dppf CH2Cl2, Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Pd[(Pet3)]2, Pd(DIPHOS)2, Cl2Pd(Bipy), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[P(o-толил)3]2, Pd2(dba)3/P(o-толил)3, Pd2(dba)/P(фурил)3, Cl2Pd(фурил)3]2, Cl2Pd(PMePh2)2, Cl2Pd[P(4-F-Ph)3]2, Cl2Pd[P(C6F6)3]2, Cl2Pd[P(2-COOH-Ph)(Ph)2]2, Cl2Pd[P(4-COOH-Ph)(Ph)2]2, и инкапсулированные катализаторы Pd EnCat™ 30, Pd EnCat™ TPP30 и Pd(II)EnCat™ BINAP30 (US 2004/0254066).
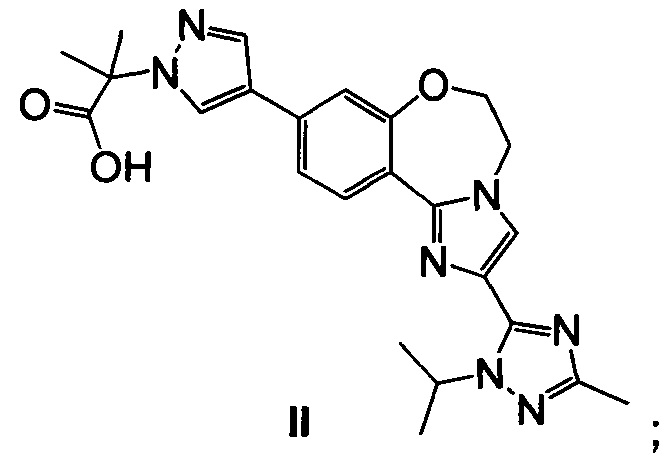
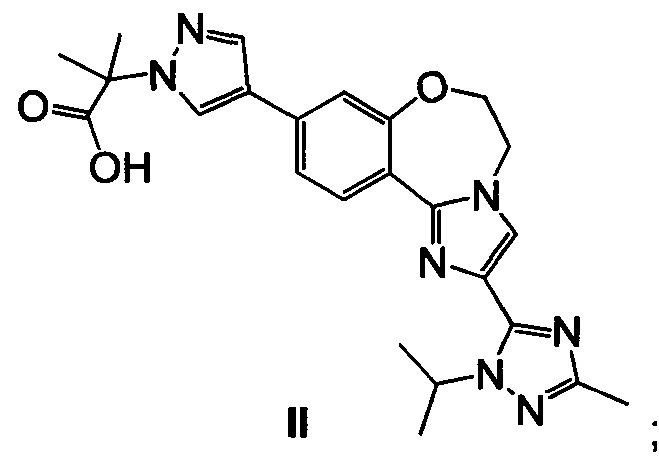
Эфирную группу 23 омыляют водным основным реагентом, таким как гидроксид лития, получая 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропионовую кислоту II (пример 17). Промежуточное соединение 23 можно выделить или дополнительно подвергнуть взаимодействию в реакционной смеси с водным основным реагентом с образованием II. Карбоксильную группу II активируют с ацил-активирующим реагентом, таким как ди(1Н-имидазол-1-ил)метанон (карбонилдиимидазол, CDI) или N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония гексафторфосфат (HATU), и затем подвергают взаимодействию со спиртовым раствором аммиака, таким как аммиак, растворенный в метаноле, этаноле или изопропаноле, водным гидроксидом аммония, водным хлоридом аммония или аммиаком, растворенным в ТГФ, получая I (пример 18).
Целый ряд твердых адсорбирующих акцепторов палладия можно использовать, чтобы удалить палладий после стадии сочетания Сузуки с образованием соединения I. Примерные воплощения акцепторов палладия включают FLORISIL®, SILIABOND®Thiol и SILIABOND® Thiourea. Другие акцепторы палладия включают силикагель, стекло с контролируемым размером пор (TosoHaas) и дериватизированный слабосшитый полистирол QUADRAPURE™ АЕА, QUADRAPURE™ IMDAZ, QUADRAPURE™ МРА, QUADRAPURE™ TU (Reaxa Ltd., Sigma-Aldrich Chemical Co.).
Схема 10:
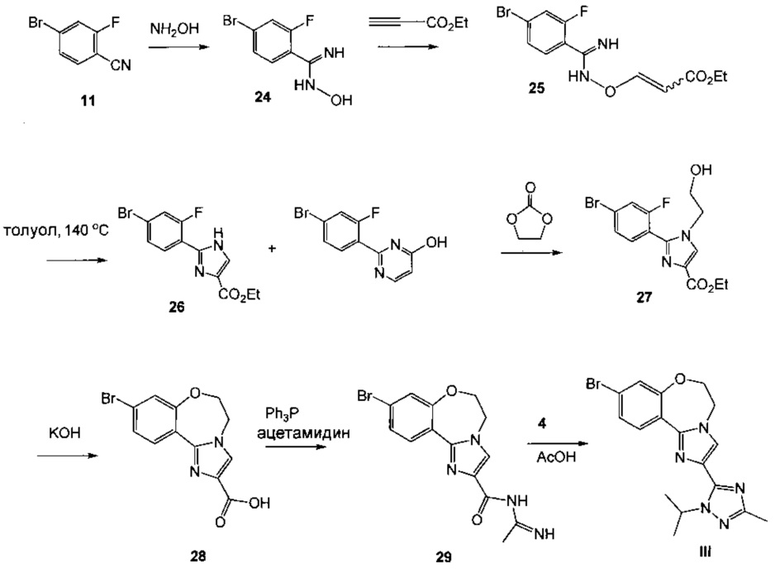
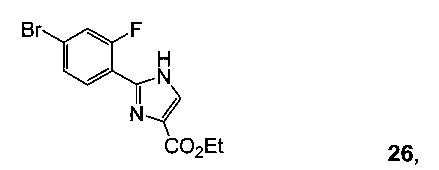
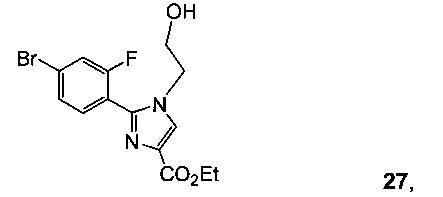
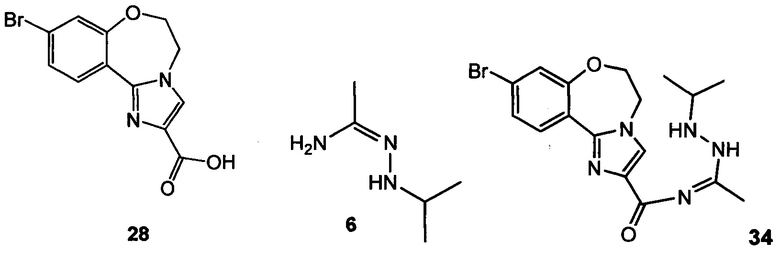
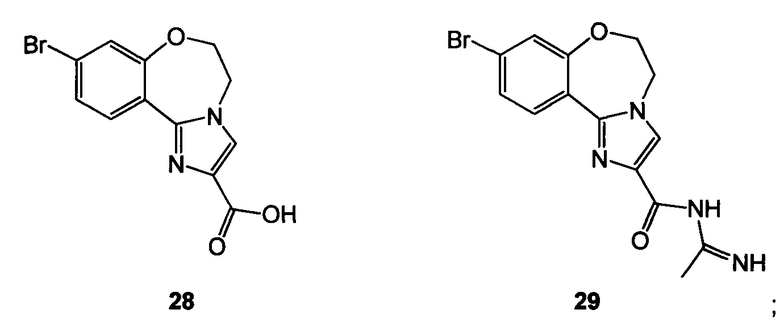
Схема 10 демонстрирует синтез 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина III из 4-бром-2-фторбензонитрила 11. Присоединение гидроксиламина к нитрилу 11 давало 4-бром-2-фтор-N-гидроксибензимидамид 24. Присоединение по Михаэлю 24 к этилпропиолату давало этил 3-(4-бром-2-фторбензимидамидоокси)акрилат 25. Нагревание 25 в высококипящем растворителе, таком как толуол, ксилол, этилбензол или дифенилоксид, давало циклизованный имидазол, этил 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоксилат 26, наряду с побочным продуктом пиримидина, 2-(4-бром-2-фторфенил)пиримидин-4-олом. Альтернативно, 25 может циклизоваться до 26 с каталитическими кислотами Льюиса, такими как соли Cu(I) или Cu(II). Алкилирование 26 с 2-гидроксиэтилирующим реагентом, таким как 1,3-диоксолан-2-он, в основании, таком как N-метилимидазол или карбонат цезия, давало этил 2-(4-бром-2-фторфенил)-1-(2-гидроксиэтил)-1H-имидазол-4-карбоксилат 27. Кольцевая циклизация 27 с водным основным реагентом, таким как гидроксид калия, гидроксид лития и метилтрибутиламмония гидрохлорид, давала 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновую кислоту 28. Присоединение ацетамидина к 28 с трифенилфосфином давало 9-бром-N-(1-иминоэтил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 29. Кольцевая циклизация 29 с изопропилгидразина гидрохлоридом 4 в уксусной кислоте давала 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III.
Альтернативно, 28 можно подвергнуть взаимодействию с N'-изопропилацетогидразонамидом 6, получая III (Схема 12).
Схема 11:
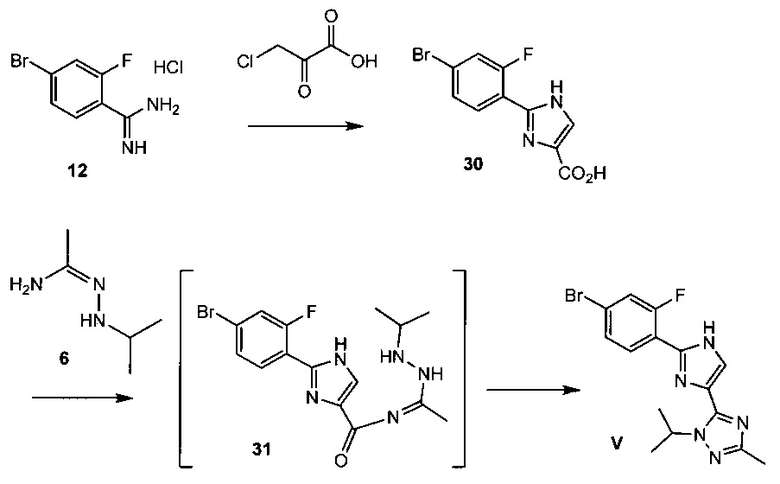
Схема 11 демонстрирует синтез 5-(2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил)-1-изопропил-3-метил-1Н-1,2,4-триазола V из 4-бром-2-фторбензимидамида гидрохлорида 12. 3-Хлор-2-оксопропионовую кислоту и 12 подвергают взаимодействию с основанием, получая 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоновую кислоту 30. Альтернативно, 3-бром-2-оксопропионовую кислоту можно подвергнуть взаимодействию с 12, получая 30. Реакция 30 с N'-изопропилацетогидразонамидом 6 и связующим реагентом HBTU (N,N,N',N'-тетраметил-O-(1Н-бензотриазол-1-ил)урония гексафторфосфат, О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, CAS рег. №94790-37-1) в ДМФА дает промежуточное соединение, 2-(4-бром-2-фторфенил)-N-(1-(2-изопропилгидразинил)этилиден)-1Н-имидазол-4-карбоксамид 31, который не обязательно выделять, и он циклизуется при нагревании, что дает V.
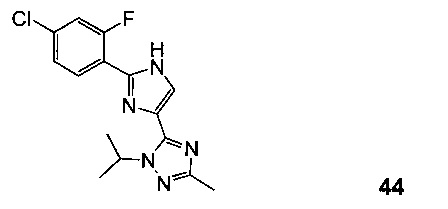
Альтернативно, 5-(2-(4-хлор-2-фторфенил)-1H-имидазол-4-ил)-1-изопропил-3-метил-1Н-1,2,4-триазол 44, вариант V с хлором, можно получить из 4-хлор-2-фторбензонитрила 38 (Схема 15).
Схема 12:
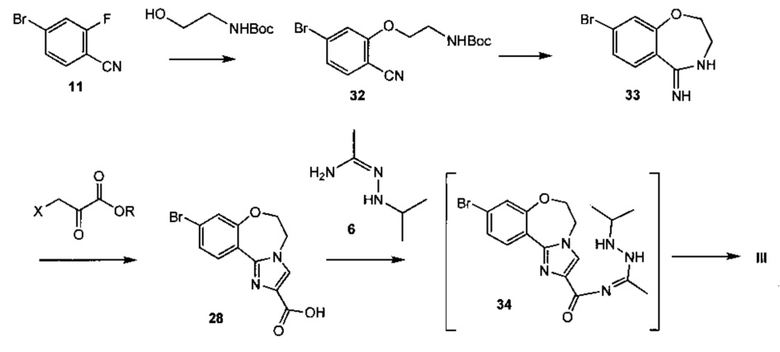
Схема 12 демонстрирует альтернативный синтез 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина III из 4-бром-2-фторбензонитрила 11. Алкилирование 11 с трет-бутил 2-гидроксиэтилкарбаматом дает трет-бутил 2-(5-бром-2-цианофенокси)этилкарбамат 32. Циклизация 32 при кислотных условиях, таких как хлористоводородная кислота в этаноле, дает 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имин 33. Следует отметить, что 33 имеет альтернативную таутомерную форму, когда двойная связь находится внутри кольца оксазепина. Образование имидазольного кольца происходит в ходе реакции 3-бром-2-оксопропионовой кислоты (X = Br, R = ОН) или другой 3-гало-2-оксопропионовой кислоты, или эфира (R = алкил), и 33, что дает 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновую кислоту 28. Связывание 28 с N'-изопропилацетогидразонамидом 6 и связующим реагентом, таким как HBTU, HATU или CDI, в ДМФА дает промежуточное соединение, 9-бром-N-(1-(2-изопропилгидразинил)этилиден)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 34, который не обязательно выделять, и из него образуется 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III при нагревании.
Альтернативно, N'-изопропилацетогидразонамид 6 используют в виде моногидрохлорида, который следует выделить при реакционных условиях с подходящим основанием, таким как K2CO3.
Схема 13:
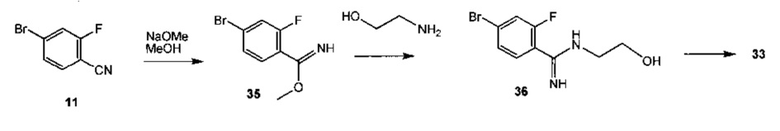
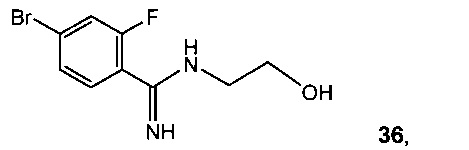
Схема 13 демонстрирует альтернативный синтез 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имина 33 из 4-бром-2-фторбензонитрила 11. Реакция 11 с метилатом натрия в метаноле дает метил 4-бром-2-фторбензимидат 35. Алкилирование 35 с 2-аминоэтанолом дает 4-бром-2-фтор-N-(2-гидроксиэтил)бензимидамид 36 с последующей циклизацией до 33.
Схема 14:
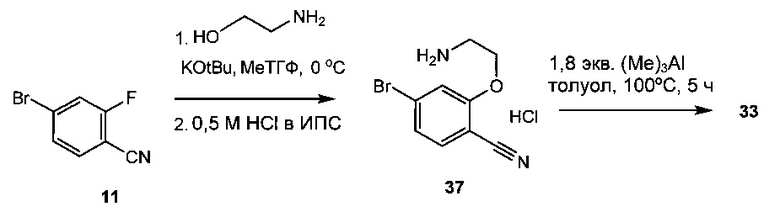
Схема 14 демонстрирует другой альтернативный синтез 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имина 33 из 4-бром-2-фторбензонитрила 11. В ходе реакции 11 с 2-аминоэтанолом и трет-бутилатом калия замещается фтор, что дает 2-(2-аминоэтокси)-4-бромбензонитрила гидрохлорид 37. Замыкание кольца 37 с триметилалюминием давало 33. Альтернативно, можно использовать другие триалкилалюминиевые реагенты или алкоголяты магния, такие как этилат магния (диэтоксид магния, CAS рег. №2414-98-4), чтобы циклизовать 37 до 33.
Схема 15:
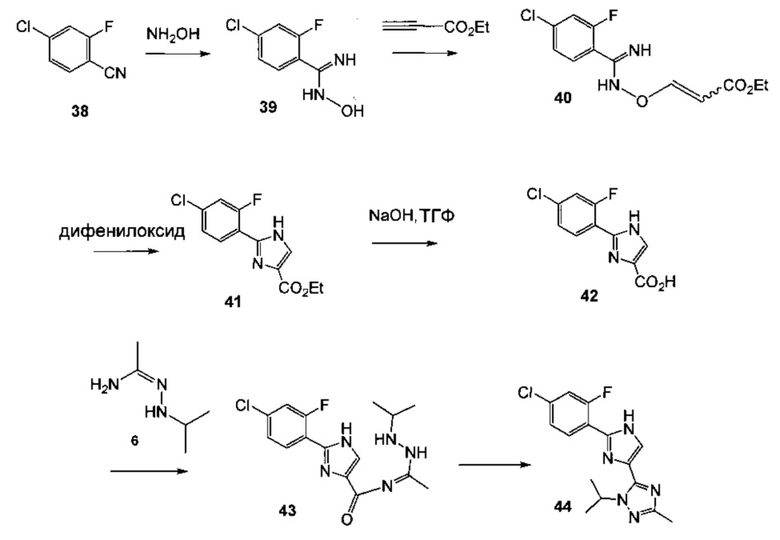
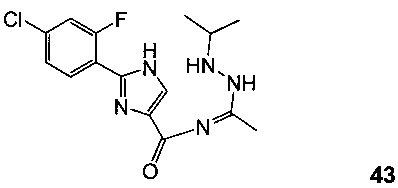
Схема 15 демонстрирует синтез 5-(2-(4-хлор-2-фторфенил)-1Н-имидазол-4-ил)-1-изопропил-3-метил-1Н-1,2,4-триазола 44 из 4-хлор-2-фторбензонитрила 38. Присоединение гидроксиламина к нитрилу 38 давало 4-хлор-2-фтор-N-гидроксибензимидамид 39. Присоединение по Михаэлю 39 к этилпропиолату давало этил 3-(4-хлор-2-фторбензимидамидоокси)акрилат 40. Нагревание 40 в дифенилоксиде давало циклизованный имидазол, этил 2-(4-хлор-2-фторфенил)-1H-имидазол-4-карбоксилат 41. Омыление эфира 41 с водным гидроксидом натрия в тетрагидрофуране давало 2-(4-хлор-2-фторфенил)-1Н-имидазол-4-карбоновую кислоту 42. Реакция 42 с N'-изопропилацетогидразонамидом 6 и связующим реагентом HBTU в ДМФА дает промежуточное соединение, 2-(4-хлор-2-фторфенил)-N-(1-(2-изопропилгидразинил)этилиден)-1Н-имидазол-4-карбоксамид 43, который циклизуется при нагревании с образованием 44.
Лекарственные препараты
GDC-0032, формулы I, можно изготовить в соответствии с общепринятой фармацевтической практикой для применения в терапевтическом сочетании для терапевтического лечения (включая профилактическое лечение) гиперпролиферативных заболеваний у млекопитающих, включая людей. Согласно изобретению предложена фармацевтическая композиция, включающая GDC-0032 в сочетании с одним или более чем одним фармацевтически приемлемым носителем, скользящем веществом, разбавителем или эксципиентом.
Подходящие носители, разбавители, скользящие вещества и эксципиенты хорошо известны квалифицированным специалистам в данной области техники и включают вещества, такие как углеводы, воски, водорастворимые и/или разбухающие полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, вода и подобные.
Препараты можно получить, используя традиционные процедуры растворения и перемешивания. Соединение по настоящему изобретению обычно входит в состав фармацевтических лекарственных форм, обеспечивая тем самым легко регулируемую дозировку лекарства и соблюдение пациентом предписанного режима.
Фармацевтическая композиция (или препарат) для применения может быть упакован разными способами в зависимости от способа, используемого для введения лекарства. Как правило, изделие для распространения включает контейнер с вложенным в него фармацевтическим препаратом в подходящей форме. Подходящие контейнеры хорошо известны квалифицированным специалистам в данной области техники и включают материалы, такие как флаконы (пластиковые и стеклянные), пакетики, ампулы, пластиковые пакеты, металлические цилиндры и подобные. Также контейнер может включать устройство контроля первого вскрытия, чтобы предотвратить неосторожный доступ к содержимому упаковки. Кроме того, на контейнер наносится этикета, которая описывает содержимое контейнера. Также этикета может включать соответствующие предупреждения.
Фармацевтические препараты соединений по настоящему изобретению можно получить для разных способов и типов введения с фармацевтически приемлемыми разбавителями, носителями, экципиентами, скользящими веществами или стабилизаторами (Remington's Pharmaceutical Sciences (1995) 18th edition, Mack Publ. Co., Easton, PA) в форме лиофилизированного препарата, полученного размолом порошка или водного раствора. Препарат можно производить, смешивая при температуре окружающей среды при соответствующем pH и при требуемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые нетоксичны к реципиентам при используемых дозах и концентрациях. pH препарата в основном зависит от конкретного применения и концентрации соединения, но может изменяться от приблизительно 3 до приблизительно 8.
Фармацевтический препарат предпочтительно является стерильным. В частности, препараты, используемые для введения in vivo, должны быть стерильными. Такую стерилизацию легко выполнить в ходе фильтрации через стерилизующие фильтрующие мембраны.
Фармацевтический препарат как правило можно хранить в виде твердой композиции, таблетки, пилюли, капсулы, лиофилизированного препарата или в виде водного раствора.
Фармацевтические препараты по изобретению дозируют и вводят до известной степени, т.е. количества, концентрации, режимы, курс, наполнители и способ введения согласуются с надлежащей медицинской практикой. Рассматриваемые в этой связи факторы включают конкретное заболевание, подвергаемое лечению, клиническое состояние отдельного пациента, причину заболевания, место доставки лекарства, способ введения, планирование введения и другие факторы, известные врачам.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы не токсичны для реципиентов при используемых дозах и концентрациях, и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметония хлорид; бензалкония хлорид, бензетония хлорид; фенол, бутил, этанол или бензиловый спирт; алкилпарабены, такие как метил или пропилпарабен; катехол; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как ЭДТА; сахара, такие как лактоза, сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплексы Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, включая Твин 80, PLURONICS™ или полиэтиленгликоль (ПЭГ), включая ПЭГ400. Активные фармацевтические ингредиенты также могут быть включены в микрокапсулы, полученные, например, способами коацервации или межфазной полимеризацией, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и микрокапсулы из поли-(метилметацилат), соответственно, в коллоидных системах доставки лекарственного средства (например, липосомах, альбуминовых микросферах, микроэмульсиях, наночастицах и нанокапсулах) или в макроэмульсиях. Такие методики раскрыты в Remington's Pharmaceutical Sciences 18th edition, (1995) Mack Publ. Co., Easton, PA. Другие примеры лекарственных препаратов можно найти у Liberman, Н.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol 3, 2nd Ed., New York, NY.
Фармацевтически приемлемые скользящие вещества могут быть выбраны из диоксида кремния, порошкообразной целлюлозы, микрокристаллической целлюлозы, стеаратов металлов, алюмосиликата натрия, бензоата натрия, карбоната кальция, силиката кальция, кукурузного крахмала, карбоната магния, не содержащего асбеста талька, стеаровета C, крахмала, крахмала 1500, лаурилсульфата магния, оксида магния и их комбинаций.
Фармацевтические препараты включают те, что подходят для способов введения, подробно описанных в данном документе. Удобно, когда препараты находятся в стандартной лекарственной форме, и их можно получить любым из способов, хорошо известных в области фармации. Как правило методики и препараты находятся в Remington's Pharmaceutical Sciences 18th Ed. (1995) Mack Publishing Co., Easton, PA. Такие способы включают стадию, согласно которой объединяют активный ингредиент с носителем, который составляет один или более вспомогательных ингредиентов. Как правило препараты получают, равномерно и тщательно объединяя активный ингредиент с жидкими носителями или тонкоизмельченными твердыми носителями, или обоими, и затем, если требуется, формуя продукт.
Фармацевтические композиции могут находится в форме стерильного инъекционного препарата, такого как стерильная инъекционная водная или маслянистая суспензия. Эту суспензию можно изготовить согласно известной области техники, используя те подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты, которые были упомянуты выше. Стерильный инъекционный препарат может представлять собой раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле или полученный из лиофилизированного порошка. Приемлемыми наполнителями и растворителями, которые можно использовать, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стандартно можно использовать стерильные жирные масла в качестве растворителя или суспендирующей среды. Для этой цели можно использовать любое легкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, подобным образом можно использовать жирные кислоты, такие как олеиновая кислота, при получении инъекционных препаратов.
Примеры
Пример 1 трет-бутил 2-(пропан-2-илиден)гидразинкарбоксилат 2
К раствору трет-бутил гидразинкарбоксилата 1 (CAS рег. №870-46-2) (25,1 г, 0,190 моль) в ацетоне (185 мл) добавляли сульфат магния (6 г) и 12 капель уксусной кислоты (Wu et al (2012) Jour. Med. Chem. 55(6): 2724-2736; WO 2007/056170; Zawadzki et al (2003) Polish Jour. Chem. 77(3): 315-319). Смесь нагревали с обратным холодильником в течение 2,5 часов и охлаждали до к.т., и фильтровали. Фильтрат концентрировали, получая трет-бутил 2-(пропан-2-илиден)гидразинкарбоксилат 2 (CAS рег. №16689-34-2) в виде твердого вещества белого оттенка (32 г, 98%) (использовали на следующей стадии без дополнительной очистки). ЖХ-МС [М+Н]+ = 172,9, RT = 2,11 минут. 1Н ЯМР 300 МГц (CDCl3) d 7,35 (br s, 1Н, NH), 2,04 (s, 3H), 1,82 (s, 3H), 1,54 (s, 9H); 13C ЯМР 300 МГц (CDCl3) d 152,9, 149,7, 80,7, 28,1, 25,3, 15,9.
Пример 2 трет-бутил 2-изопропилгидразинкарбоксилат 3 трет-Бутил 2-(пропан-2-илиден)гидразинкарбоксилат 2 восстанавливали с палладиевым катализатором на углероде газообразным водородом в уксусной кислоте и метаноле, получая трет-бутил 2-изопропилгидразинкарбоксилат 3 (CAS рег. №16689-35-3).
Альтернативно, трет-бутил 2-(пропан-2-илиден)гидразинкарбоксилат 2 (0,51 г, 3,0 ммоль) растворяли в 20 мл ТГФ, обрабатывали NaBH3CN (0,19 г, 3,0 ммоль) и несколько мг бромкрезолового зеленого, затем раствором п-толуолсульфоновой кислоты (0,57 г, 3,0 ммоль) в 1,5 мл ТГФ, который добавляли по каплям в течение приблизительно 1 часа, чтобы поддерживать pH реакции между 3,5-5,0. После перемешивания при комнатной температуре в течение дополнительного часа растворитель удаляли вращательным испарением и остаток разделяли между EtOAc (30 мл) и солевым раствором. Органическую фазу экстрагировали насыщ. NaHCO3, 20 мл, и солевым раствором, выпаривали до остатка и растворяли в 10 мл этанола. Этанольный раствор обрабатывали 3,6 мл 1 М NaOH раствора (3,6 ммоль) и оставляли перемешиваться при к.т. в течение 30 минут. Растворитель удаляли вращательным испарением, и остаток переносили в этилацетат и экстрагировали водой. Органический слой выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, используя 5% MeOH в ДХМ в качестве элюента, чтобы собрать трет-бутил 2-изопропилгидразинкарбоксилат 3 (0,4 г, выход 77%): т.пл. = 47-49°C; Rf = 0,44 (5% MeOH в ДХМ); 1Н ЯМР 300 МГц (CDCl3) d 6,03 (s, N-H, 1Н), 3,92 (s, N-H, 1H), 3,14 (m, 1H), 1,46 (s, 9H), 1,02 (d, 6H, J=6 Гц); 13C ЯМР 300 МГц (CDCl3) d 157,2, 80,8, 51,2, 28,7, 21,0.
Пример 3 изопропилгидразина гидрохлорид 4
трет-Бутил 2-изопропилгидразинкарбоксилат 3 обрабатывали хлористоводородной кислотой, чтобы удалить защитную группу Boc, и получали 4 (CAS рег. №16726-41-3).
Пример 4 N'-изопропилацетогидразонамид 6
Метилацетимидата гидрохлорид 5 (CAS рег. №14777-27-6), изопропилгидразина гидрохлорид 4 и триэтиламин подвергали взаимодействию в метаноле, получая 6 (CAS рег. №73479-06-8).
Пример 5 1-изопропил-3-метил-1H-1,2,4-триазол 7
N'-Изопропилацетогидразонамид 6 обрабатывали триэтилортоформиатом в этаноле, затем триэтиламином и тетрагидрофураном, получая 7 (CAS рег. №1401305-30-3).
Пример 6 2-хлор-N-метокси-N-метилацетамид 10
К раствору 21,2 кг карбоната калия K2CO3 (153,7 моль, 3,0 экв.) в 30 л H2O добавляли N,O-диметилгидроксиламин 9 (CAS рег. №1117-97-1) (5,0 кг, 51,3 моль, 1.0 экв.) при 15~20°C. Реакционную смесь перемешивали при к.т. в течение 30 минут и добавляли 30 л метил-трет-бутилового эфира (ТБМЭ). После перемешивания в течение 30 минут смесь охлаждали до 5°C и медленно добавляли 11,6 кг 2-хлорацетилхлорида 8 (CAS рег. №79-04-9 (102,7 моль, 2,0 экв.). Реакционную смесь перемешивали при к.т. в течение ночи. Органические вещества отделяли от водной среды и водную среду экстрагировали ТБМЭ (30 л). Объединенные органические вещества промывали H2O (50 л), солевым раствором (50 л) и сушили над Na2SO4. Фильтровали и концентрировали в вакууме, получая 5.1 кг 2-хлор-N-метокси-N-метилацетамида 10 (CAS рег. №67442-07-3) в виде белого твердого вещества.
Пример 7 4-бром-2-фторбензимидамида гидрохлорид 12
К 35,0 л гексаметилдисилазида лития LiHMDS (35,0 моль, 1,4 экв., 1,0 М в ТГФ) в атмосфере N2 добавляли ТГФ раствор 4-бром-2-фторбензонитрила 11 (CAS рег. №105942-08-3) (5,0 кг в 10 л ТГФ) при 10°C, смесь перемешивали при к.т. в течение 3 часов. Охлаждали до -20°C и добавляли 8,3 л HCl-EtOH (6,6 М). Смесь перемешивали при -10°C в течение дополнительного 1 часа, фильтровали. Влажный осадок промывали ЭА (10 л) и H2O (6 л). Сушка в вакууме давала 5,8 кг 4-бром-2-фторбензимидамида гидрохлорида 12 (CAS рег. №1187927-25-8) в виде твердого вещества белого оттенка.
Альтернативно, в 200-л сосуд загружали 4-бром-2-фторбензонитрил 11 (10 кг, 50,00 моль, 1,00 эквив.) и этанол (100 л), затем продували 40 кг хлороводорода (газ) при -10°C, перемешивая (Схема 4). Полученный в результате раствор взаимодействовал в течение дополнительных 36 часов при 10°C. Ход реакции контролировали с помощью ТСХ до расхода всего 11. Полученную в результате смесь концентрировали в вакууме, поддерживая температуру ниже 60°C. Объем концентрировали до 10~15 л, затем добавляли 60 л МТБЭ, чтобы осадить продукт. Выпавшие в осадок вещества собирали в ходе фильтрации, получая 12 кг этил 4-бром-2-фторбензимидата гидрохлорида 12 в виде белого твердого вещества, (выход: 85%). 1Н ЯМР δ 7,88-7,67 (m), 4,89 (br s), 4,68 (q), 3,33 (m), 1,61 (t). MC M+1: 245,9, 248,0.
В 200 л сосуд загружали этил 4-бром-2-фторбензимидата гидрохлорид (12,5 кг, 44 моль, 1,00 эквив., 99%) и этанол (125 л), затем продували NH3 (газ) при -5°C в течение 12 часов. Полученный в результате раствор перемешивали при 30°C в течение дополнительных 24 часов. Ход реакции контролировали с помощью ТСХ до расхода всего SM. Выпавшие в осадок вещества фильтровали и фильтрат концентрировали в вакууме. Продукт осаждали и собирали в ходе фильтрации, получая 6,1 кг (54,5%) 4-бром-2-фторбензамидина гидрохлорида 12 в виде белого твердого вещества. 1Н ЯМР δ 9,60 (br), 7,91-7,64 (m), 3,40 (s), 2,50 (m). МС М+1: 216,9, 219,9.
Пример 8 2-хлор-1-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)этанон 13
В 10 л четырехгорлую колбу загружали 1-изопропил-3-метил-1Н-1,2,4-триазол 7 (400 г) в ТГФ (2,5 л). Полученный в результате раствор охлаждали до -40°C и добавляли 2,5 М н-бутиллитий BuLi в н-гексанах (1,41 л), сохраняя при этом внутреннюю темп, ниже -20°C. Полученную в результате желтую суспензию перемешивали при -40°C в течение 1 часа, затем переносили. В 20 л колбу загружали 2-хлор-N-метокси-N-метилацетамид 10 (485 г) в ТГФ (4 л). Полученный в результате раствор охлаждали до -40°C, в результате чего получали белую суспензию, и к ней добавляли раствор литированного триазола 7, поддерживая внутреннюю темп, ниже -20°C. На данном этапе получали желто-оранжевый раствор, который перемешивали при -30°C в течение 1 часа. Добавляли пропионовую кислоту (520 мл), поддерживая внутреннюю темп, ниже -20°C. Полученную в результате бело-желтоватую суспензию нагревали до -5°C в течение 30 минут. Добавляли лимонную кислоту (200 г) в воде (0,8 л) и после перемешивания в течение 5 минут получали прозрачную двухфазную смесь. На данном этапе останавливали перемешивание и нижний водный слой удаляли. Органическую фазу промывали 20 масс. % раствором K3PO4 (1 л), 20 масс. % раствором K2HPO4 (2 л) и 20 масс. % раствором NaCl (1 л). Органические вещества уменьшали до приблизительно 4 л в ходе вакуумной перегонки, получая 2-хлор-1-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)этанон 13 в виде темно-янтарной жидкости, которую использовали "как есть" на следующей стадии.
Пример 9 5-(2-(4-бром-2-фторфенил)-1H-имидазол-4-ил)-1-изопропил-3-метил-1H-1,2,4-триазол V
В 10 л четырехгорлую колбу загружали ТГФ (5,6 л), 4-бром-2-фторбензимидамида гидрохлорид 12 (567 г), KHCO3 (567 г) и воду (1,15 л). Полученную в результате белую суспензию нагревали до 60°C в течение 2 часов. На данном этапе получали мутный раствор, к которому добавляли раствор 2-хлор-1-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)этанона 13 в ТГФ (2 л). Этот раствор перемешивали при 60-65°C в течение 24 часов. Затем удаляли водный нижний слой. Органический слой концентрировали в вакууме. Остаток суспендировали в смеси МИБК (1,25 л) и толуола (0,7 л), и осажденный продукт фильтровали, получая 552 г 5-(2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил)-1-изопропил-3-метил-1H-1,2,4-триазола V (чистота 98,0%, 254 нм) в виде коричневого твердого вещества.
Пример 10 2-(2-(4-бром-2-фторфенил)-4-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-1Н-имидазол-1-ил)этанол 14
5-(2-(4-Бром-2-фторфенил)-1Н-имидазол-4-ил)-1-изопропил-3-метил-1Н-1,2,4-триазол V (2,75 кг, 7,55 моль) добавляли к раствору 3-диоксолан-2-она (этиленкарбонат, 3,99 кг, 45,3 моль) в N-метилимидазоле (12 л) при 50°C. Суспензию нагревали при 80°C в течение 7 часов до завершения реакции согласно ВЭЖХ. Раствор 14 охлаждали до 35°C и сразу использовали в последующей циклизации.
Пример 11 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III
К раствору 2-(2-(4-бром-2-фторфенил)-4-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-1Н-имидазол-1-ил)этанола (7,55 ммоль) 14 в N-метилимидазоле (12 л) при 35°C добавляли метилтрибутиламмония хлорид (115 г, 0,453 моль), толуол (27,5 л) и 35% раствор гидроксида калия (10,6 кг, 25 моль в 22 л воды). Двухфазный раствор энергично перемешивали при 65°C в течение 18 часов до завершения реакции согласно ВЭЖХ. Останавливали перемешивание, но продолжали нагревание и удаляли нижний водный слой. Добавляли изопропилацетат (13,8 л) и органическую фазу дважды промывали водой (13,8 л и 27,5 л). Растворитель удаляли в ходе вакуумной перегонки и затем удаляли 30 л, добавляли изопропанол (67,6 л). Вакуумную перегонку возобновляли до удаления дополнительных 30 л растворителя. Добавляли дополнительный изопропанол (28,8 л) и продолжали вакуумную перегонку до уменьшения объема на 42 л. Добавляли изопропанол (4 л) и температуру увеличивали до >50°C. Добавляли воду (28 л), так чтобы внутренняя температура поддерживалась выше 50°C, затем нагревали до 75°C, получая прозрачный раствор. Смесь медленно охлаждали и продукт выкристаллизовывался из раствора. Полученную в результате суспензию охлаждали до 0°C, выдерживали в течение 1 часа, затем фильтровали и осадок на фильтре промывали водой (5,5 л). Осадок на фильтре сушили при 45°C при обдувке азотом, получая III в виде желтовато-коричневого твердого вещества (3,30 кг, 71,6 масс. %, выход 80,6%).
Пример 12 2-метил-2-(1H-пиразол-1-ил)пропионовая кислота 16
2-Бром-2-метилпропионовую кислоту 15 и пиразол подвергали взаимодействию в триэтиламине и 2-метилтетрагидрофуране, получая 16.
Пример 13 этил 2-метил-2-(1H-пиразол-1-ил)пропионат 17 2-Метил-2-(1H-пиразол-1-ил)пропионовую кислоту 16 обрабатывали серной кислотой в этаноле, получая 17.
Альтернативно, пиразол (10 г, 147 ммоль, 1,0 экв.) растворяли в ДМФА (500 мл) при комнатной температуре (Схема 8). 2-Бромизобутират 18 (22 мл, 147 ммоль, 1,0 экв.), карбонат цезия Cs2CO3 (53 г, 162 ммоль, 1,1 экв.) и каталитический иодид натрия NaI (2,2 г, 15 ммоль, 0,1 экв.) добавляли к смеси, которую затем нагревали до 60°C в течение 24 часов. Реакцию контролировали с помощью 1Н ЯМР и через 24 часа пиразол не обнаружили. Реакционную смесь гасили насыщенным раствором NaHCO3 (200 мл) и добавляли этилацетат EtOAc (150 мл), и органические вещества отделяли от водной среды. Органические вещества сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая масло, которое очищали с помощью флэш-хроматографии, получая 17.
Пример 14 Этил 2-(4-бром-1H-пиразол-1-ил)-2-метилпропионат IV Способ А: Этил 2-метил-2-(1H-пиразол-1-ил)пропионат 17 подвергали взаимодействию с N-бромсукцинимидом (NBS) в 2-метилтетрагидрофуране, получая IV(CAS рег. №1040377-17-0).
Способ В: Этил 2-бром-2-метилпропионат 18 и пиразол подвергали взаимодействию с трет-бутилатом натрия в диметилформамиде (ДМФА), получая смесь этил 2-метил-2-(1H-пиразол-1-ил)пропионата 17 и этил 2-метил-3-(1Н-пиразол-1-ил)пропионата 19, которую обрабатывали 1,3-дибром-5,5-диметилимидазолидин-2,4-дионом, получая смесь IV, этил 3-(4-бром-1H-пиразол-1-ил)-2-метилпропионата 20 и 4-бром-1H-пиразола 21. Смесь обрабатывали каталитическим количеством гексаметилдисилазида лития в тетрагидрофуране, затем подкисляли хлористоводородной кислотой, получая IV.
Пример 15 этил 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропионат 22
В 50 л стеклянный реакционный сосуд загружали этил 2-(4-бром-1Н-пиразол-1-ил)-2-метилпропионат IV (1,00 кг, 3,85 моль, 1,00 эквив.), ацетат калия KOAc (0,47 кг, 4,79 моль, 1,25 эквив.), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан), бис(пинаколато)диборон B2Pin2 (1,22 кг, 4,79 моль, 1,25 эквив.) и этанол (10 л, 10 об.) и смесь перемешивали до получения прозрачного раствора. Раствор дегазировали в вакууме 3х с азотом. К этой смеси добавляли лиганд XPhos (0,023 кг, 0,048 моль, 1,0 мол. %) и Pd предварительный катализатор (0,018 кг, 0,022 моль, 0,5 мол. %), получая в результате гомогенный оранжевый раствор. Раствор дегазировали в вакууме один раз с азотом. Внутреннюю температуру реакционной смеси доводили до 75°C и отбирали пробу реакционной смеси каждые 30 минут, как только установленная температура была достигнута, и контролировали с помощью ЖХ (IPC способ: XTerra MS Boronic). Через 5 часов превращение до 22 (CAS рег. №1201657-32-0) почти завершалось с 1,3% оставшегося IV.
Пример 16 этил 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропионат 23
Этил 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пропионат 22 и 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III подвергали взаимодействию в условиях реакции Сузуки с палладиевым катализатором в изопропаноле и водном фосфатном буфере, получая 23.
1 М раствор K3PO4 (1,60 кг в 7,6 л воды, 7,54 моль, 2,00 эквив.) загружали в вышеприведенную реакционную смесь из примера 15, затем добавляли раствор III в ТГФ (1,33 кг в 5,0 л, 3,43 моль, 0,90 эквив.) в течение 2 минут. Реакционную смесь нагревали до 75°C (внутренняя температура) в течение 45 минут и перемешивали в течение 13 часов при 75°C, затем анализировали с помощью ВЭЖХ (III не обнаружен), что показывало образование 23.
Пример 17 2-(4-(2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропионовая кислота II
Этил 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропионат 23 обрабатывали водным гидроксидом лития, получая II.
Реакцию омыления эфира вызывали, добавляя 3,5 М водный LiOH (0,74 кг в 5,0 л, 17,64 моль, 5 эквив.) к реакционной смеси из примера 16 и нагревая до 75°C. Отбирали образец смеси каждые 30 минут (IPC способ: XTerra MS Boronic) и омыление завершалось через 4,5 часа (с менее чем 0,3% оставшегося 23). Реакционную смесь концентрировали в ходе перегонки до приблизительно половины объема (начальный объем = 37 л; конечный объем = 19 л), чтобы удалить EtOH и ТГФ, получая в результате желтовато-коричневую взвесь. Воду (5 л, 5 об.) загружали в смесь и затем перегоняли (начальный объем = 25 л; конечный объем = 21 л). Температуру доводили до 60°C (контроль кожуха) и затем загружали изопропилацетат, ИПАц (4 л, 4 об.). Двухфазную смесь перемешивали минимум 5 минут и затем слои разделяли в течение минимум 5 минут. Нижний водный слой удаляли в чистую бутыль и органические вещества собирали во вторую бутыль. Процесс экстракции повторяли всего четыре раза до получения заметно прозрачного органического слоя. Водную смесь переносили обратно в реакционный сосуд и затем охлаждали до 15°C. 6 М раствор HCl (6,4 л, 38,40 моль, 10 эквив.) медленно загружали до получения конечного рН=1. Затем гетерогенную смесь фильтровали. Полученные в результате твердые вещества дважды промывали 5 л (2×5 об.) воды. Затем фильтр нагревали до 80°C и устанавливали вакуум -10 Psi (с отводом азота), и твердые вещества сушили в течение 24 часов (КФ = 2,0% H2O), получая 1,54 кг (95% скорректированный выход) II в виде белого твердого вещества; 98 масс. %, 97,3% чистоты.
Пример 18 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид I (GDC-0032)
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропионовую кислоту II обрабатывали ди(1Н-имидазол-1-ил)метаноном (карбонилдиимидазолом, CDI) в тетрагидрофуране, затем метанольным аммиаком, получая неочищенное соединение I.
Твердое вещество II (1,44 кг, 3,12 моль, 1,00 эквив.) переносили в 20 л сосуд и затем загружали ТГФ (10 л, 7 об.). Взвесь переносили при пониженном давлении во второй 50 л реакционный сосуд и добавляли дополнительный ТГФ (5 л, 3 об.) для промывки. Внутреннюю температуру взвеси доводили до 22°C и 1'1-карбонилдиимидазол, CDI (0,76 кг, 5,12 моль, 1,50 эквив.) загружали в смесь, и через 5 минут наблюдали прозрачный раствор. Брали образец реакционной смеси каждые 30 минут и анализировали с помощью ВЭЖХ (IPC: XTerra MS Boronic способ), что показывало почти полное превращение в ацил-имидазольное промежуточное соединение и 1,2% оставшегося II после 30 минут. Добавляли дополнительную порцию CDI (0,07 кг, 0,15 моль, 0,14 эквив.), и реакционную смесь перемешивали в течение 1 часа и затем анализировали с помощью ВЭЖХ (IPC: XTerra MS Boronic способ), что показывало 0,8% оставшегося II.
Во второй 50-л реакционный сосуд добавляли NH3/MeOH (1,5 л, 10,5 моль, 3,37 эквив.) и ТГФ (5 л, 3 об.). Ацил-имидазольное промежуточное соединение переносили во второй реакционный сосуд при пониженном давлении (время переноса ~10 минут). Затем внутреннюю температуру доводили до 45°C и объем растворителя перегоняли с 35 л до 12 л. Затем добавляли воду (6 л, 4 об.) к смеси, которую дополнительно перегоняли с 18 л до 11 л. В конце добавляли другую порцию воды (6 л, 4 об.) и растворители перегоняли последний раз с 17 л до 14 л, пока больше не выделялся ТГФ. Реакционную смесь затем охлаждали до 10°C (внутренняя температура). Белую взвесь фильтровали и осадок на фильтре промывали водой (2×6 л, 2×4 об.). Затем твердые вещества сушили при 80°C (темп. кожуха) на фильтре Aurora в течение 24 часов (КФ = 1,5% H2O) в вакууме, получая 1,25 кг неочищенного соединения I, GDC-0032 (84% скорректированный выход, 96 масс. %, 97,3% чистоты согласно ВЭЖХ) в виде белого твердого вещества.
Получали взвесь неочищенного соединения I (1,15 кг, 2,50 моль) в MeOH (6 л, 5 об.) и затем ее загружали в 50 л стеклянный реакционный сосуд. Дополнительный MeOH (24 л, 21 об.) добавляли к смеси, которую затем нагревали до 65°C. Получали однородную смесь. Si-тиол (Silicycle, Inc., 0,23 кг, 20 масс. %) добавляли к раствору через дополнительное отверстие и смесь перемешивали в течение 3 часов. Затем ее фильтровали теплой через фильтр Aurora (температура кожуха = 60°C), осветляли фильтрованием и переносили сразу во второй 50 л реакционный сосуд с пониженным давлением. Раствор затем снова нагревали до 65°C внутренней температуры (BT). Однородный раствор охлаждали до 54°C и затравочные кристаллы I (12 г, 1 масс. %) в MeOH (50 мл) добавляли при пониженном давлении применительно к реакционному сосуду. Смесь затем охлаждали до 20°C в течение 16 часов. Потом твердые вещества фильтровали через фильтр Aurora и сушили при 80°C в течение 72 часов, получая 921 г, выход 80%, I в виде сольвата метаноата (форма A согласно XRPD), и переносили в предварительно взвешенную емкость точечного заряда.
В разделителе твердые вещества суспендировали в ИПАц (8 л, 7 об.) и переносили в прозрачный 10 л реакционный сосуд. Смесь перемешивали в течение 1 часа при 60°C (ВТ). Затем твердые вещества фильтровали через систему Aurora и сушили при 80°C (кожух) в течение 96 часов. Образец I отбирали и анализировали с помощью ГХ (ИПАц = 1%). Пытаясь осуществить более эффективную сушку, АФИ переносили в две стеклянные ванночки в разделителе и закрывали с сушильной емкостью перед сушкой в вакуумной печи при 100°C в течение 16 часов. ГХ (IPC: Q12690V2) показала, что 1% растворителя еще присутствует. Способ давал 760 г (68% скорректированный выход, 68 масс. %, 99,9% чистоты согласно ЖХ) белого твердого вещества (форма B согласно XRPD).
Неочищенное соединение I (340,7 г) загружали в 2-л ПЭНД колбу и суспендировали с 0,8 л изоамилового спирта (ИАС). Взвесь переносили в 20 л реакционный сосуд и разбавляли в 6,7 л круглодонной колбе (общий объем 22). Белую взвесь нагревали до получения раствора (внутренняя температура поднималась до 118°C, и затем охлаждали до 109°C). Раствор осветляли фильтрованием (0,2 мкМ фильтр). Колбу оборудовали верхнеприводной мешалкой и фильтрат суспендировали в изоамиловом спирте (344 мл, 21 об.). Смесь нагревали до 95°C (внутренняя) до растворения твердых веществ. Загружали взвесь угля (10 масс. %, 0,16 г) и silicycle тиола (10 масс. %, 0,16 г) в изоамиловом спирте (1 об., 16 мл), и смесь перемешивали при 90-95°C в течение 1 часа и затем фильтровали (через прокладку Celite®). Прозрачный янтарно-желтый раствор охлаждали до 73°C (темп. диапазон затравливания = 70±5°C) и добавляли затравочный кристалл GDC-0032 I (10 масс. %, 0,16 г). Колбонагреватель выключали и смесь охлаждали до комнатной температуры в течение ночи, перемешивая (200 об/мин). Через 17 часов белые твердые вещества фильтровали, начиная с медленной гравитационной фильтрации, и затем применяли вакуум. Твердые вещества сушили отсасыванием в течение 20 минут, перемешивая, до получения свободнотекучего порошка. Вес неочищенного вещества до сушки в печи = 16 г. Твердые вещества сушили в печи при 100°C в течение 24 часов и затем брали образец для исследования. Сушка продолжалась при 100°C в течение еще 24 часов. 1Н ЯМР (ДМСО d6) δ 8,38 (t), 8,01 (s), 7,87 (s), 7,44, 7,46 (d), 7,36 (s), 7,18 (br s), 6,81 (br s), 5,82 (m), 3,99 (s), 2,50 (s), 2,26 (s), 1,75 (s), 1,48, 1,46 (d).
Очищенный 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид I (GDC-0032) сухим гранулированием изготавливали в форме таблетки способом вальцевания (Не et al (2007) Jour, of Pharm. Sci., 96(5): 1342-1355) с эксципиентами, включая лактозу, микрокристаллическую целлюлозу (AVICEL® PH 01, FMC BioPolymer, 50 мкМ частица), кроскармеллозу натрия (Ac-Di-Sol®, FMC BioPolymer) и стеарат магния.
Пример 19 4-бром-2-фтор-N-гидроксибензимидамид 24
К раствору 4-бром-2-фторбензонитрила 11 (800 г, 4 моль, 1 экв.), гидроксиламина гидрохлорида (695 г, 10 моль, 2,5 экв.) в MeOH (2 л, 2,5 об.) добавляли Et3N (485 г, 4,8 моль, 1,2 экв.), затем смесь перемешивали при 60°C в течение 40 минут и проверяли с помощью ВЭЖХ (не осталось нитрила). Потом реакционную смесь гасили H2O (30 л) и отделяли большой объем твердого вещества белого оттенка, и затем фильтровали, осадок на фильтре промывали водой (10 л × 2) и получали 1350 г влажного 4-бром-2-фтор-N-гидроксибензимидамида 24 с чистотой 96%.
Пример 20 этил 3-(4-бром-2-фторбензимидамидоокси)акрилат 25
К раствору 4-бром-2-фтор-N-гидроксибензимидамида 24 (800 г, 3,43 моль, 1 экв.) и Amberlyst® А21 (20 масс. %, 160 г) в PhMe (12 л, 15 об.) добавляли этилпропиолат (471 г, 4,8 моль, 1,4 экв.) при 10°C. Реакционную смесь перемешивали при 50°C в течение ночи и проверяли с помощью ЖХ-МС (оставалось около 14А% исходного вещества 24). Затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме, и получали 1015 г этил 3-(4-бром-2-фторбензимидамидоокси)акрилата 25 в виде желтого масла с чистотой 84,9% согласно ЖХ (выход: 89%).
Пример 21 этил 2-(4-бром-2-фторфенил)-1H-имидазол-4-карбоксилат 26 Раствор этил 3-(4-бром-2-фторбензимидамидоокси)акрилата 25 (300 г, 0,91 моль, 1 экв.) в дифенилоксиде (900 мл, 3 об.) перемешивали при 190°C в атмосфере N2 в течение 1 часа и проверяли с помощью ЖХ-МС (отсутствовал 25). Охлаждали смесь до к.т. и добавляли ТБМЭ (600 мл, 2 об. относительно 25), и затем по каплям добавляли ПЭ (1,8 л, 6 об. относительно 25), чтобы отделить твердые вещества. Смесь перемешивали при к.т. в течение 20 минут и фильтровали, получая 160 г влажного осадка. Влажный осадок промывали ПЭ (1 л) и сушили, получая 120 г этил 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоксилата 26 с 92% чистотой согласно ЖХ в виде коричневого твердого вещества.
Пример 22 этил 2-(4-бром-2-фторфенил)-1-(2-гидроксиэтил)-1Н-имидазол-4-карбоксилат 27
Этил 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоксилат 26 и 1,3-диоксолан-2-он, и N-метилимидазол подвергали взаимодействию, получая 27.
Пример 23 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновая кислота 28
Этил 2-(4-бром-2-фторфенил)-1-(2-гидроксиэтил)-1H-имидазол-4-карбоксилат 27, гидроксид калия и метилтрибутиламмония гидрохлорид подвергали взаимодействию при 65°C, охлаждали и концентрировали. Смесь растворяли в этаноле и воде, чтобы кристаллизовалось соединение 28.
Пример 24 9-бром-N-(1-иминоэтил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 29
9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновую кислоту 28, трифенилфосфин и ацетамидин подвергали взаимодействию, получая 29.
Пример 25 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III
9-Бром-N-(1-иминоэтил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 29 подвергали взаимодействию с изопропилгидразина гидрохлоридом 4 в уксусной кислоте, получая III.
Пример 26 2-(4-бром-2-фторфенил)-1H-имидазол-4-карбоновая кислота 30.
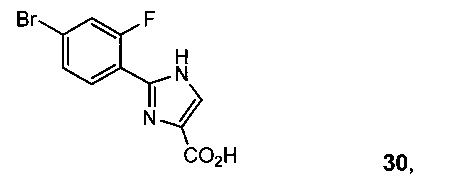
3-Хлор-2-оксопропионовую кислоту и 4-бром-2-фторбензимидамида гидрохлорид 12 подвергали взаимодействию с основанием, получая 2-(4-бром-2-фторфенил)-1H-имидазол-4-карбоновую кислоту 30.
Альтернативно, к раствору этил 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоксилата 26 (1350 г, 4,3 моль) в ТГФ (8,1 л, 6 об.) и H2O (4 л, 3 об.) добавляли NaOH (520 г, 13 моль, 3 экв.) и реакционную смесь перемешивали при 65°C в течение 48 часов до завершения реакции (проверяли с помощью ЖХ-МС). Доводили смесь с 2 М HCl до рН=5, и продукт отделяли в виде желтого твердого вещества, фильтровали, получая 2,2 кг влажного осадка, влажный осадок промывали H2O (1,5 л), ДХМ (1,5 л × 3), ПЭ (1 л) и сушили, получая 970 г чистой 2-(4-бром-2-фторфенил)-1Н-имидазол-4-карбоновой кислоты 30 (Схема 10).
Пример 27 5-(2-(4-бром-2-фторфенил)-1H-имидазол-4-ил)-1-изопропил-3-метил-1H-1,2,4-триазол V
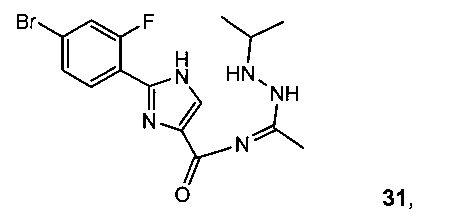
Реакция 30 с N'-изопропилацетогидразонамидом 6 и связующим реагентом HBTU в ДМФА дает промежуточное соединение, 2-(4-бром-2-фторфенил)-N-(1-(2-изопропилгидразинил)этилиден)-1H-имидазол-4-карбоксамид 31, который циклизуется при нагревании с образованием V.
Пример 28 трет-бутил 2-гидроксиэтилкарбамат дает трет-бутил 2-(5-бром-2-цианофенокси)этилкарбамат 32
Алкилирование 4-бром-2-фторбензонитрила 11 с трет-бутил 2-гидроксиэтилкарбаматом дает 32.
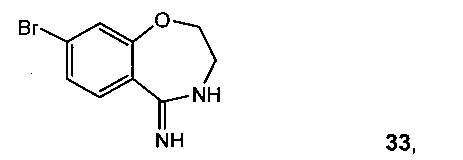
Пример 29 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имин 33
Циклизация трет-бутил 2-гидроксиэтилкарбамата дает трет-бутил 2-(5-бром-2-цианофенокси)этилкарбамат 32 в кислой среде, такой как хлористоводородная кислота в этаноле, с образованием 33.
Пример 30 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновая кислота 28
Реакция 3-бром-2-оксопропионовой кислоты и 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имина 33 дает 28 (CAS рег. №1282516-74-8).
Пример 31 9-бром-2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин III
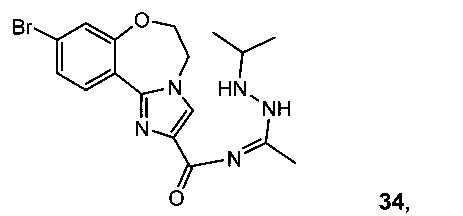
Связывание 28 с N'-изопропилацетогидразонамидом 6 и связующим реагентом HBTU в ДМФА дает промежуточное соединение, 9-бpoм-N-(1-(2-изопропилгидразинил)этилиден)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 34, который образует III при нагревании.
Пример 32 метил 4-бром-2-фторбензимидат 35
Реакция 4-бром-2-фторбензонитрила 11 с метилатом натрия в метаноле дает 35.
Пример 33 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2Н)-имин 33 Алкилирование метил 4-бром-2-фторбензимидата 35 с 2-аминоэтанолом дает 4-бром-2-фтор-N-(2-гидроксиэтил)бензимидамид 36 с последующей циклизацией до 33 (Схема 13).
Альтернативно, реакция 11 с 2-аминоэтанолом и трет-бутилатом калия замещает фтор, что дает 2-(2-аминоэтокси)-4-бромбензонитрила гидрохлорид 37. Замыкание кольца 37 с триметилалюминием давало 33 (Схема 14). Раствор 11 (10 г, 50 ммоль) и 2-аминоэтанола (3,1 мл, 50,8 ммоль) в 2-метилтетрагидрофуране (80 мл) охлаждали до 0°C и медленно добавляли 1 М раствор трет-бутилата калия в тетрагидрофуране (55 мл, 55 ммоль), поддерживая температуру раствора ниже 5°C. Реакционную смесь перемешивали при 0°C в течение 30 минут до завершения реакции согласно ВЭЖХ, после чего ее нагревали до 25°C. Добавляли 0,5 М раствор HCl в изопропаноле (100 мл, 50 ммоль), и требуемая соль HCl 3 сразу кристаллизовалась из раствора. Твердое вещество собирали в ходе фильтрации и сушили в вакууме с отводом азота, что давало 2-(2-аминоэтокси)-4-бромбензонитрила гидрохлорид 37 в виде белого твердого вещества (12,1 г, выход 87%).
В колбу загружали 37 (9,00 г, 32,4 ммоль) и толуол (90,0 мл). Суспензию охлаждали до 0°C и добавляли триметилалюминий (1,8 эквив., 58,4 ммоль, 2Mb толуоле) по каплям в течение 30 минут. Потом суспензию перемешивали при комнатной температуре в течение 1 часа и затем нагревали до 100°C. Через 5 часов раствор охлаждали до 0°C и гасили водным NaOH (2 н, 90,0 мл). Суспензию экстрагировали EtOAc (4×90 мл) и объединенные экстракты сушили, затем фильтровали через Celite®. Раствор концентрировали и остаток растирали с EtOAc, получая 8-бром-3,4-дигидробензо[f][1,4]оксазепин-5(2H)-имин 33 (6,26 г, 26,0 ммоль, выход 80%) в виде белого кристаллического твердого вещества.
Пример 34 4-хлор-2-фтор-N-гидроксибензимидамид 39
К раствору 4-хлор-2-фторбензонитрила 38 (400 г, 2,58 моль, 1,0 экв.), гидроксиламина гидрохлорида (448 г, 6,45 моль, 2,5 экв.) в MeOH (1 л, 2,5 об.) добавляли Et3N (313 г, 3,1 моль, 1,2 экв.), затем смесь перемешивали при 60°C в течение 40 минут и проверяли с помощью ВЭЖХ (нитрила не оставалось). Затем реакцию гасили H2O (10 л), и большое количество твердого вещества белого оттенка отделяли и затем фильтровали, осадок на фильтре промывали водой (10 л × 2) и получали 378 г 4-хлор-2-фтор-N-гидроксибензимидамида 39 с чистотой 93% (Схема 15).
Пример 35 этил 3-(4-хлор-2-фторбензимидамидоокси)акрилат 40
К раствору 4-хлор-2-фтор-N-гидроксибензимидамида 39 (378 г, 2 моль, 1,0 экв.) и Amberlyst® А21 (20 масс. %, 75,6 г) в толуоле PhMe (5,6 л, 15 об.) добавляли этилпропиолат (275 г, 2,8 моль, 1,4 экв.) при 30°C. Реакционную смесь перемешивали при 30°C в течение ночи и проверяли с помощью ЖХ-МС. Затем реакционную смесь фильтровали и фильтрат концентрировали в вакууме, и получали 550 г этил 3-(4-хлор-2-фторбензимидамидоокси)акрилата 40 в виде желтого масла с чистотой 83% согласно ЖХ (Схема 15).
Пример 36 этил 2-(4-хлор-2-фторфенил)-1H-имидазол-4-карбоксилат 41
Раствор этил 3-(4-хлор-2-фторбензимидамидоокси)акрилата 40 (550 г, 1,9 моль, 1,0 экв., 83% чистоты согласно ЖХ) в дифенилоксиде (1,65 л, 3 об.) перемешивали при 190°C в атмосфере N2 в течение 1 часа и проверяли с помощью ЖХ-МС (нет оставшегося 40). Охлаждали смесь до к.т. и добавляли по каплям ПЭ (10 л). Смесь перемешивали при к.т. в течение 20 минут и фильтровали, получая 400 г влажного осадка, затем очищали с помощью хроматографии на силикагеле (ПЭ/ЭА = 1/5), что давало 175 г чистого этил 2-(4-хлор-2-фторфенил)-1Н-имидазол-4-карбоксилата 41 с 98% чистотой согласно ЖХ (Схема 15).
Пример 37 2-(4-хлор-2-фторфенил)-1Н-имидазол-4-карбоновая кислота 42
К раствору этил 2-(4-хлор-2-фторфенил)-1Н-имидазол-4-карбоксилата 41 (175 г, 4,3 моль) в ТГФ (1 л, 6 об.) и H2O (500 мл, 3 об.) добавляли NaOH (78 г, 1,95 моль, 3,0 экв.) и реакционную смесь перемешивали при 65°C в течение 48 часов до завершения реакции (проверяли с помощью ЖХ-МС). Смесь с 2 н HCl доводили до pH=5 и продукт выделяли в виде желтого твердого вещества, фильтровали, получая 210 г влажного осадка, влажный осадок промывали H2O (300 мл), ДХМ (3×300 мл), ПЭ (500 мл) и сушили, получая 110 г чистой 2-(4-хлор-2-фторфенил)-1H-имидазол-4-карбоновой кислоты 42 (CAS рег. №1260649-87-3) (Схема 15). 1Н ЯМР (ДМСО d6) δ: 12,8 (br s), 8,0, 7,9 (br s), 7,46, 7,4 (m).
Несмотря на то что вышеизложенное изобретение описано довольно подробно с помощью иллюстраций и примеров в целях лучшего понимания, описание и примеры не ограничивают объем изобретения. Следовательно, все подходящие изменения и эквиваленты можно считать находящимися в объеме изобретения, как определено формулой изобретения, приведенной далее. Все патенты и научная литература, приведенные в данном документе, явным образом включены в него полностью посредством ссылки на них.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800543C2 |
| БЕНЗОПИРАНОВЫЕ И БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ РI3K И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2506267C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ПРОИЗВОДНЫЕ [(1,5-ДИФЕНИЛ-1H-1,2,4-ТРИАЗОЛ-3-ИЛ)ОКСИ]УКСУСНОЙ КИСЛОТЫ И ИХ СОЛИ, СОДЕРЖАЩИЕ ИХ СРЕДСТВА ДЛЯ ЗАЩИТЫ ПОЛЕЗНЫХ ИЛИ КУЛЬТУРНЫХ РАСТЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЗАЩИТНЫХ СРЕДСТВ | 2020 |
|
RU2829883C1 |
Изобретение относится к способам получения соединения формулы (I):
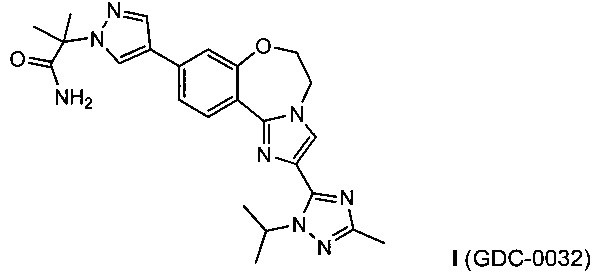 . Технический результат: разработан способ получения соединения GDC-0032, являющегося ингибитором PI3K, который может быть использован для получения соединения формулы I в промышленном масштабе, где осуществлен специальный выбор реакций, комбинация которых привела к снижению массы используемых реактивов при одновременном исключении вредных и опасных реактивов. 3 н. и 5 з.п. ф-лы, 37 пр.
. Технический результат: разработан способ получения соединения GDC-0032, являющегося ингибитором PI3K, который может быть использован для получения соединения формулы I в промышленном масштабе, где осуществлен специальный выбор реакций, комбинация которых привела к снижению массы используемых реактивов при одновременном исключении вредных и опасных реактивов. 3 н. и 5 з.п. ф-лы, 37 пр.
1. Способ получения (2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамида I, имеющего структуру:
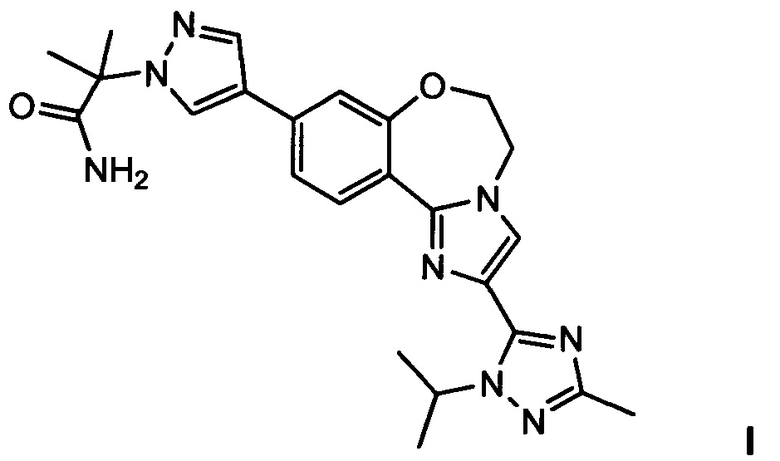
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей, согласно которому:
(а1) подвергают взаимодействию IV и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) с образованием 22
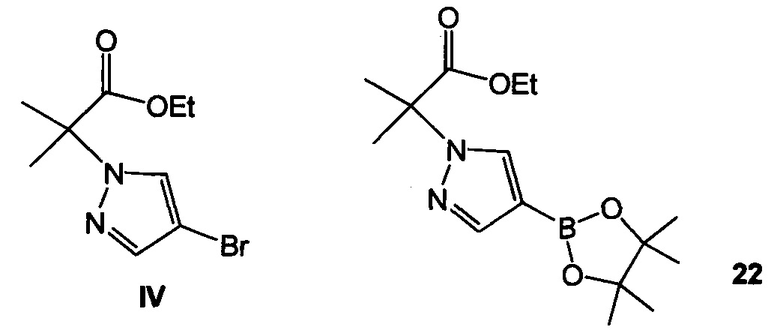 ;
;
(а2) подвергают взаимодействию 28 с N'-изопропилацетогидразонамидом 6 с образованием III через промежуточное соединение 34
(b) подвергают взаимодействию 22, палладиевый катализатор и III с образованием 23
(с) подвергают взаимодействию 23 с водным основным реагентом с образованием II
 и
и
(d) подвергают взаимодействию II с ацил-активирующим реагентом, затем с аммиаком с образованием I.
2. Способ получения (2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамида I, имеющего структуру:
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей, согласно которому:
(а1) подвергают взаимодействию IV и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) с образованием 22
(a2) подвергают взаимодействию V с 2-гидроксиэтилирующим реагентом с образованием 14
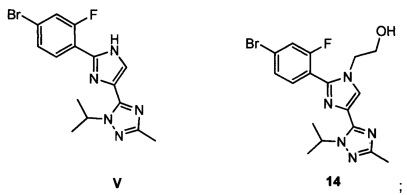 и
и
подвергают взаимодействию 14 с водным основным реагентом с образованием III;
(b) подвергают взаимодействию 22, палладиевый катализатор и III с образованием 23
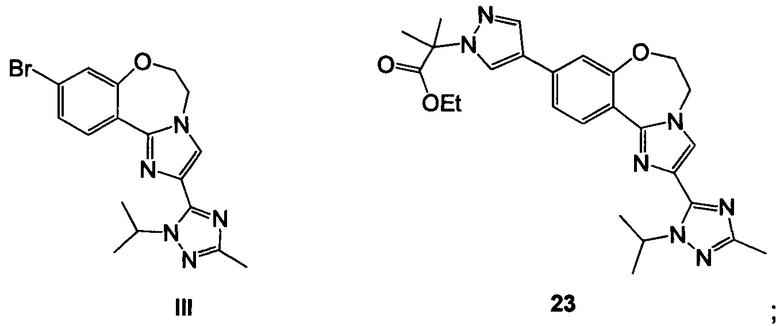
(с) подвергают взаимодействию 23 с водным основным реагентом с образованием II
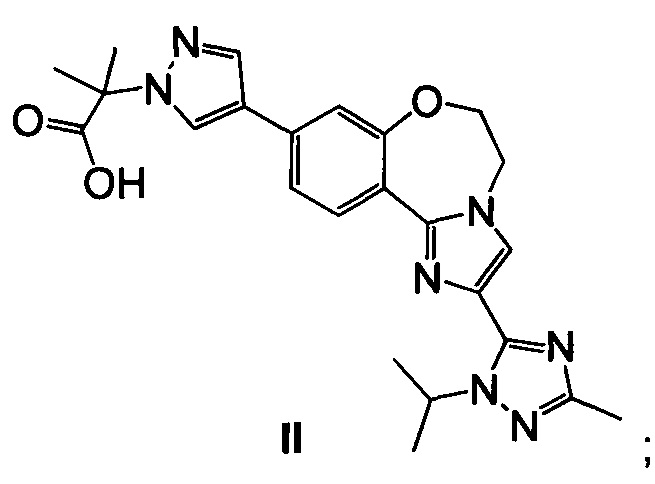 и
и
(d) подвергают взаимодействию II с ацил-активирующим реагентом, затем с аммиаком с образованием I.
3. Способ получения (2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамида I, имеющего структуру:
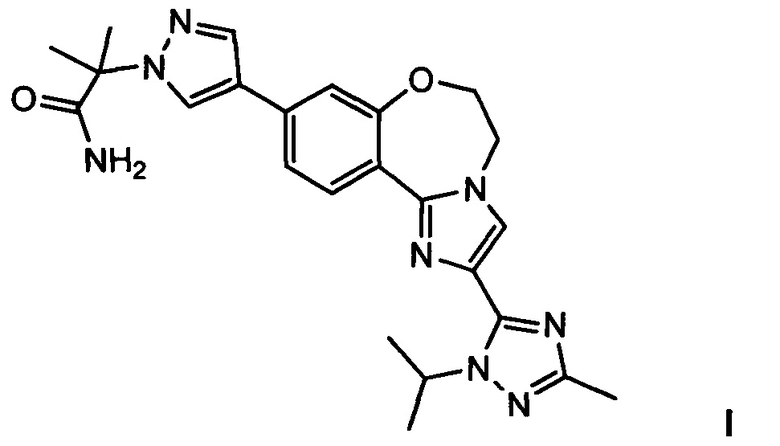
и его стереоизомеров, геометрических изомеров, таутомеров и фармацевтически приемлемых солей, согласно которому:
(а1) подвергают взаимодействию IV и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) с образованием 22
(а2) подвергают взаимодействию 28 с ацетамидином с образованием 29
 и
и
подвергают взаимодействию 29 с изопропилгидразином и кислотным реагентом с образованием III;
(b) подвергают взаимодействию 22, палладиевый катализатор и III с образованием 23
(c) подвергают взаимодействию 23 с водным основным реагентом с образованием II
 и
и
(d) подвергают взаимодействию II с ацил-активирующим реагентом, затем с аммиаком с образованием I.
4. Способ по любому из пп. 1-3, где для получения IV подвергают взаимодействию 17:
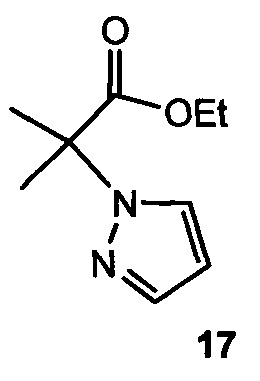
с бромирующим реагентом.
5. Способ по п. 1, где для получения 28
(a) подвергают взаимодействию 4-бромо-2-фторбензонитрил 11 с 2-аминоэтаноломи трет-бутоксидом калия с образованием 37,
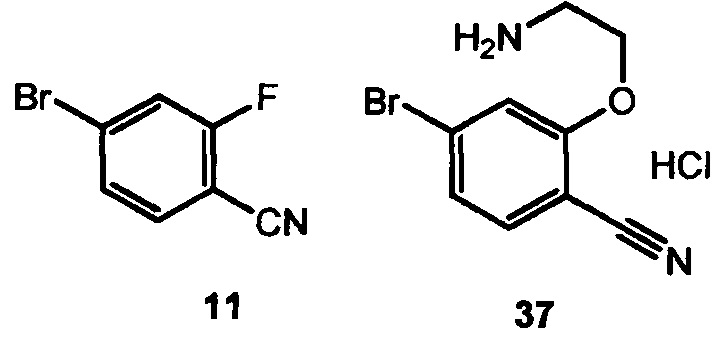
(b) циклизуют 37 с использованием триалкилалюминиевого реагента или магнийалкоксидного реагента с образованием 33,

(c) подвергают взаимодействию 33 с 3-галоген-2-оксопропаноевой кислотой или ее эфиром с образованием 28.
6. Способ по п. 1, где для получения 6 подвергают взаимодействию гидрохлорид изопропилгидразина 4
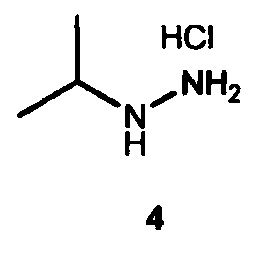
с (i) гидрохлоридом метилацетимидата, (ii) ацетамидином или (iii) ацетонитрилом и кислотой.
7. Способ по п. 1, где для получения IV подвергают взаимодействию пиразол с 2-бром-2-метилпропаноевой кислотой 15 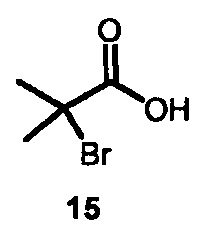
с образованием 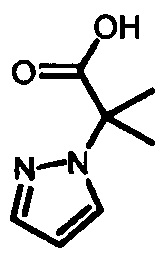 , который затем подвергают взаимодействию с бромирующим агентом с образованием 2-(4-бромо-1Н-пиразол-1-ил)-2-метилпропаноевой кислоты
, который затем подвергают взаимодействию с бромирующим агентом с образованием 2-(4-бромо-1Н-пиразол-1-ил)-2-метилпропаноевой кислоты 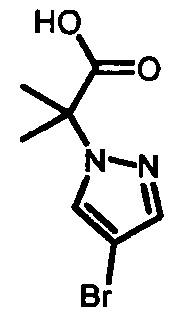 ; и этерифицируют ее с образованием IV.
; и этерифицируют ее с образованием IV.
8. Способ по п. 3, где для получения 28:
(a) подвергают взаимодействию 11 с гидроксиламином с образованием 24
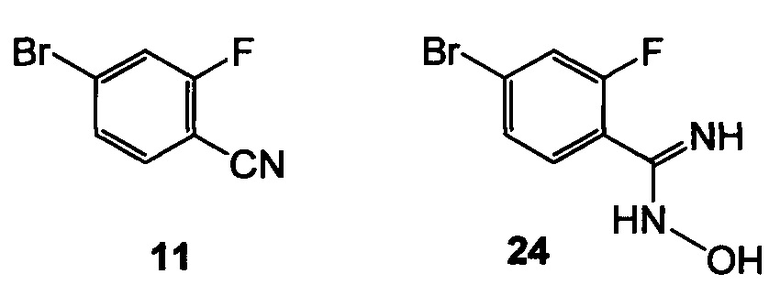 ;
;
(b) подвергают взаимодействию 24 с этилпропиолатом с образованием 25
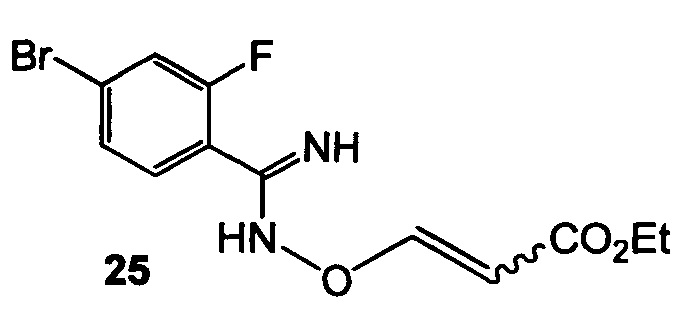 ;
;
(c) нагревают 25 с образованием 26
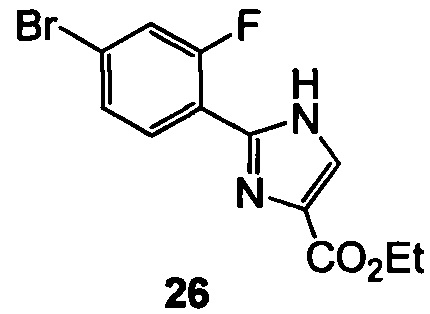 ;
;
(d) подвергают взаимодействию 26 с 2-гидроксиэтилирующим реагентом с образованием 27
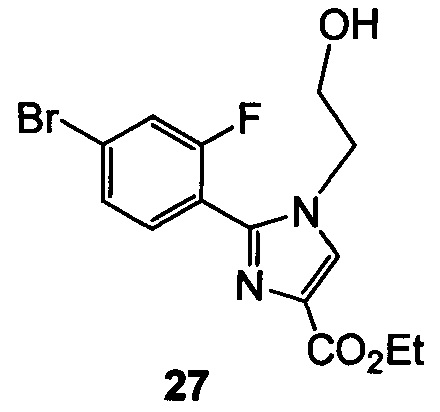 и
и
(е) подвергают взаимодействию 27 с водным основным реагентом с образованием 28.
| US2011076292 A1, 31.03.2011 | |||
| EA200970403 A1, 30.10.2009 | |||
| СПОСОБ ПРЕДПОСЕВНОЙ ОБРАБОТКИ СЕМЯН | 2007 |
|
RU2341066C1 |
| US2012245144 A1, 27.09.2012. | |||

