Область техники, к которой относится изобретение
В настоящем изобретении описаны соединения, фармацевтические композиции, содержащие одно или больше из описанных соединений или их фармацевтически приемлемые соли, которые эффективно подавляют связывание различных хемокинов, таких как MIP-1α, лейкотактин, MPIF-1 и RANTES, с CCR1 рецептором. В качестве антагонистов или модуляторов CCR1 рецептора, описанные соединения и композиции могут применяться в лечении воспалительных и иммунных заболеваний и нарушений.
Уровень техники
Здоровье человека зависит от способности тела обнаруживать и уничтожать чуждые патогены, которые могут отбирать ценные ресурсы у человека и/или вызывать заболевания. Иммунная система, включающая лейкоциты (белые кровяные тельца (БКТ): Т- и В-лимфоциты, моноциты, макрофаги, гранулоциты, нормальные клетки-киллеры, лаброциты, дендритные клетки и клетки, являющиеся производными клеток иммунной системы (например, остеокласты)), лимфоидные ткани и сосуды, является защитной системой организма. В целях борьбы с инфекцией белые кровяные тельца циркулируют по организму, обнаруживая патогены. При обнаружении патогена, врожденные иммунные клетки, и в особенности цитотоксичные Т-клетки, поступают к инфицированному сайту для уничтожения патогена. Хемокины играют роль молекулярных маяков для привлечения и активации иммунных клеток, таких как лимфоциты, моноциты и гранулоциты, идентифицируя места наличия патогенов.
Несмотря на контроль за патогенами со стороны иммунной системы, может развиваться неправильная подача сигналов хемокинами, что приписывают возникновению и развитию воспалительных заболеваний, таких как ревматоидный артрит, рассеянный склероз и другие. Например, при ревматоидном артрите неконтролируемое накопление хемокинов в суставах привлекает и активирует инфильтрующиеся макрофаги и Т-клетки. Активность этих клеток индуцирует разрастание синовиальных клеток, которое приводит, по меньшей мере частично, к воспалению и, в конечном итоге, к разрушению костей и суставов (см. DeVries, М.Е., et al., Semin Immunol 11(2):95-104 (1999)). Отличительной чертой некоторых демиелинизирующих заболеваний, таких как рассеянный склероз, является осуществляемое с участием хемокинов привлечение моноцитов/макрофагов и Т-клеток в центральную нервную систему (см. Kennedy, et al., J. Clin. Immunol. 19(5):273-279 (1999)). Привлечение хемокинами разрушительных белых кровяных телец в трансплантаты приводит к их последующему отторжению. Смотри статью DeVries, М.Е., et al., ibid. Поскольку хемокины играют жизненно-важную роль в процессах воспаления и развития лимфоцитов, возможность точно управлять их активностью оказывает огромное влияние на смягчение и лечение заболеваний, для которых в настоящее время не существует удовлетворительных методов лечения. Кроме того, можно минимизировать отторжение трансплантата, избегая общих и тяжелых побочных эффектов дорогих иммуносупрессантов.
Хемокины, группа из более чем 40 небольших пептидов (7-10 кДа), связываются с рецепторами, экспрессированными главным образом на белых кровных тельцах или клетках, являющихся производными иммунной системы, и генерируют сигналы посредством GPSR-каскадов, для осуществления своих хемоаттрактантных и хемостимулирующих функций. Рецепторы могут связываться с более чем одним лигандом; например, рецептор CCR1 связывается с RANTES (regulated on activation normal T cell expressed, регуляция активации, экспрессии и секреции нормальных Т-клеток), MIP-1α (macrophage inflammatory protein, воспалительный белок макрофагов), MPIF-1/CKβ8 и лейкотактином (среди других, обладающих меньшим сродством). В настоящий момент известно 24 хемокиновых рецептора. Огромное количество хемокинов, рецепторов, связывающихся с несколькими лигандами, и разные профили рецепторов на иммунных клетках делают возможным тонко управляемые и специфичные иммунные ответы. См. Rossi, et al., Ann. Rev. Immunol. 18(1):217-242 (2000). Активностью хемокинов можно управлять путем модулирования их соответствующих рецепторов, вылечивая зависимые от них воспалительные и иммунологические заболевания и обеспечивая возможность пересадки органов и тканей.
Рецептор CCR1 и его хемокиновые лиганды, включая, например, MIP-1α, MPIF-1/СКβ8, лейкотактин и RANTES, представляют собой важные терапевтические мишени (см. Saeki, et al., Current Pharmaceutical Design 9:1201-1208 (2003)), поскольку они участвуют в развитии ревматоидного артрита, отторжении трансплантата (см. DeVries, М.Е., et al., ibid.) и рассеянного склероза (см. Fischer, et al., J Neuroimmunol. 110(1-2):195-208 (2000); Izikson, et al., J. Exp. Med. 192(7):1075-1080 (2000); и Rottman, et al., Eur. J. Immunol. 30(8):2372-2377 (2000). На самом деле, были открыты антитела, блокирующие работу хемокинов, модифицированные лиганды и низкомолекулярные органические молекулы для хемокиновых рецепторов, некоторые из которых показали успешное лечение или профилактику некоторых хемокин-опосредованных заболеваний (см. обзор Rossi, et al., ibid.). Следует отметить, что в экспериментальной модели ревматоидного артрита развитие заболевания замедляется при введении модифицированного RANTES-лиганда, блокирующего проведение сигнала (см. Plater-Zyberk, et al., Immunol Lett. 57(1-3): 117-120 (1997)). Хотя терапия блокирующими работу антителами и низкомолекулярными пептидами является многообещающей, их недостатками является риск разрушения, очень короткие времена полужизни после введения и фактически запретительные затраты на разработку и производство, что характерно для большинства белков. Низкомолекулярные органические соединения предпочтительны, поскольку они часто имеют большие времена полужизни in vivo, требуют более низких дозировок для достижения эффективности, часто могут вводиться перорально и менее дорогие. Некоторые органические антагонисты CCR1 были описаны раньше (см. Hesselgesser, et al., J. Biol. Chem. 273(25): 15687-15692 (1998); Ng, et al., J. Med. Chem. 42(22):4680-4694 (1999); Liang, et al., J. Biol. Chem. 275(25): 19000-19008 (2000); и Liang, et al., Eur. J. Pharmacol. 389(1):41-49 (2000)). В свете эффективности, продемонстрированной в лечении заболевания на животных моделях (см. Liang, et al., J. Biol. Chem. 275(25):19000-19008 (2000)), продолжается поиск, направленный на выявление дополнительных соединений, которые могут применяться для лечения заболеваний, опосредованных CCR1-сигнальной системой.
Раскрытие изобретения
В настоящей заявке описаны соединения, имеющие формулу:
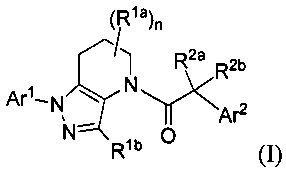
и их соли, ротамеры и оптические изомеры, где подстрочный символ n, и заместители R1a, R1b, R2a, R2b, Ar1 и Ar2 имеют значения, указанные в описании и формуле изобретения.
Некоторыми группами соединений являются соединения формул Ia, Ia1, Ia2, II, III и IV:
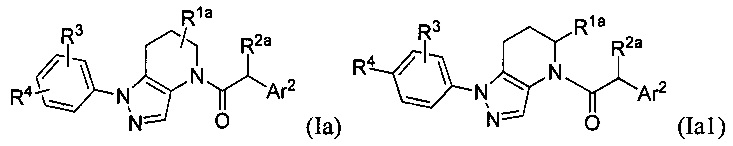

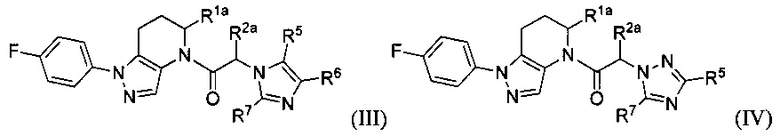
Помимо указанных соединений, в настоящем изобретении описаны также фармацевтические композиции, содержащие одно или больше из указанных соединений, а также способы применения указанных соединений в терапевтических методах, главным образом в лечении заболеваний, связанных с CCR1 сигнальной активностью.
Осуществление изобретения
I. Сокращения и определения
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин "циклоалкил" относится к углеводородным циклам, имеющим указанное число атомов в цикле (например, С3-6циклоалкил) и являющимся полностью насыщенными или имеющими не более одной двойной связи между вершинами цикла. "Циклоалкил" относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Термин "гетероциклоалкил" относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, О, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкил может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничивающие примеры гетероциклоалкильных групп включают пирролидин, пиперидинил, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п. Гетероциклоалкильная группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле.
Термин "алкилен" сам по себе и как часть другого заместителя означает двухвалентный радикал, являющийся производным алкана, примером которого может служить -СН2СН2СН2СН2-. В типичном случае, алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, при этом группы с 10 атомами углерода или меньше являются предпочтительными по настоящему изобретению. "Низший алкил" или "низший алкилен" представляет собой алкильную или алкиленовую группу с более короткой цепочкой, обычно содержащую четыре или меньше атомов углерода. Аналогично, «алкенилен» или «алкинилен» относится к ненасыщенным формам «алкилена», содержащим двойные или тройные связи, соответственно.
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламино-групп, алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил," включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Термин «гетероарил» относится к арильным группам (или циклам), содержащим от одного до пяти гетероатомов, выбранных из N, О, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, в то время как неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждом из перечисленных выше арильных или гетероарильных циклических систем выбраны из группы приемлемых заместителей, описанных выше.
Для краткости, термин "арил" в случае применения в комбинации с другими терминами (например, арилокси, арилтиокси, арилалкил) включает как арильные, так и гетероарильные циклы, как описано выше. Так, термин "арилалкил" означает радикалы, в которых арильная группа соединена с алкильной группой (например, бензил, фенетил, пиридилметил и т.п.).
Указанные выше термины (например, "алкил", "арил" и "гетероарил") в некоторых вариантах осуществления включают как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикала перечислены ниже. Для краткости, термины «арил» и «гетероарил» относятся к замещенным или незамещенным версиям, как описано ниже, в то время как термин "алкил" и родственные алифатические радикалы относятся к незамещенным версиям, если не указано, что они имеют заместители.
Заместителями в алкильных радикалах (включая группы, которые часто именуются алкилен, алкенил, алкинил и циклоалкил) могут быть различные группы, выбранные из: -галоген, -OR', -NR'R'', -SR', -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'-C(O)NR''R''', -NR''C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -CN и -NO2, в количестве от нуля до (2m'+1), где m' это общее число атомов углерода в таком радикале. R', R'' и R''' каждый независимо означают атом водорода, незамещенный C1-8 алкил, незамещенный гетероалкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный С1-8 алкил, C1-8 алкокси или C1-8 тиоалкокси группу, или незамещенные арил-С1-4 алкильные группы. Когда R' и R'' присоединены к одному и тому же атому азота, они могут объединяться с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного цикла. Например, -NR'R'' включает 1-пирролидинил и 4-морфолинил.
Аналогично, заместители в арильных и гетероарильных группах варьируются и обычно выбраны из: -галоген, -OR', -OC(O)R', -NR'R'', -SR', -R', -CN, -NO2, -CO2R', -CONR'R'', -C(O)R', -OC(O)NR'R'', -NR''C(O)R', -NR''C(O)2R', -NR'-C(O)NR''R''', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NR'S(O)2R'', -N3, перфтор(С1-С4)алкокси, и перфтор(С1-С4)алкил, в количестве от нуля до общего числа незанятых валентностей в ароматической циклической системе; и где R', R'' и R''' независимо выбраны из атома водорода, C1-8 алкила, С3-6 циклоалкила, С2-8 алкенила, С2-8 алкинила, незамещенного арила и гетероарила, (незамещенный арил)-С1-4 алкила и незамещенный арилокси-С1-4 алкила. Другие подходящие заместители включают каждый из перечисленных выше заместителей для арила, присоединенных к атому в цикле алкиленовым мостиком из 1-4 атомов углерода.
Два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -T-C(O)-(CH2)q-U-, где Т и U независимо представляют собой -NH-, -О-, -СН2- или одинарную связь, и q представляет собой целое число от 0 до 2. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -А-(СН2)r-В-, где А и В независимо представляют собой -СН2-, -О-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'- или одинарную связь, и r представляет собой целое число от 1 до 3. Одна из простых связей в новом цикле, образующемся таким образом, может опционально быть заменена на двойную связь. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -(CH2)s-X-(CH2)t-, где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -О-, -NR'-, -S-, -S(O)-, -S(O)2- или -S(O)2NR'-. Заместитель R' в -NR'- и -S(O)2NR'- выбран из атома водорода или незамещенного С1-6 алкила.
При использовании в настоящем тексте, термин "гетероатом" включает в себя кислород (О), азот (N), серу (S) и кремний (Si).
Термин "фармацевтически приемлемые соли" включает соли действующих веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, тиэтиламин, триметиламин, трипропиламин, трометамин и т.п. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная, азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, йодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическим кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, "Pharmaceutical Salts", Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм, в настоящем изобретении описаны соединения, представляющие собой пролекарственные формы. Пролекарства описанных в настоящем тексте соединений представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства можно превратить в соединения по настоящему изобретению химическими или биохимическими методами в ex vivo условиях. Например, пролекарства можно медленно превратить в соединения по настоящему изобретению при помещении их в резервуар пластыря для чрезкожного введения с подходящим ферментативным или химическим реагентом.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3Н), иод-125 (125I) или углерод-14 (14С). Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения.
II. Общие положения
Настоящее изобретение является производным открытия того, что соединения формулы I (а также подформулы Ia, Ia1, Ia2, II, III и IV) работают как потенциальные антагонисты рецептора CCR1. Данные соединения обладают in vivo противовоспалительной активностью. Соответственно, описанные в настоящем тексте соединения могут применяться в фармацевтических композициях, способах лечения CCR1-опосредованных заболеваний и в качестве контроля в исследованиях по идентификации конкурентоспособных CCR1 антагонистов.
III. Соединения
В одном аспекте, в настоящем изобретении описаны соединения, имеющие формулу:
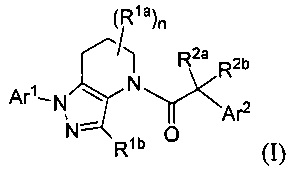
и их соли, ротамеры и оптические изомеры.
В формуле I, подстрочный индекс n представляет собой целое число от 0 до 3; каждый Rla и Rlb является представителем, независимо выбранным из группы, состоящей из Н, C1-8 алкила, C1-8 галогеналкила, С3-6 циклоалкила, -CORa, -CO2Ra, -CONRaRb, -NRaRb, -NRaCORb, -ORa, -X1CORa, -X1CO2Ra, -X1CONRaRb, -X1NRaCORb, -X1NRaRb и -X1ORa, где X1 выбран из группы, состоящей из С1-4 алкилена, и каждый Ra и Rb независимо выбран из группы, состоящей из атома водорода, С1-8 алкила, C1-8 галогеналкила и С3-6 циклоалкила, и опционально две группы Rla у соседних атомов углерода объединены с образованием 5-, 6- или 7-членного карбоциклического или гетероциклического кольца; каждый из R2a и R2b является представителем, независимо выбранным из группы, состоящей из Н, гидроксила, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси, С1-4 алкокси-С1-4 алкила, C1-8 гидроксиалкила, С1-4 алкокси-С1-4 алкокси, С3-6 циклоалкила, С3-6 циклоалкил-С1-4 алкила, 3-7-членного гетероциклоалкила, 3-7-членный гетероциклоалкил-С1-4 алкила, -X'CO2Ra, -X1CONRaRb, -X1NRaCORb, -X1NRaRb, где X1, Ra и Rb соответствуют данным выше определениям.
Символ Ar1 представляет собой шести- или десятичленное моноциклическое или конденсированное бициклическое арильное кольцо, или 5-10-членное моноциклическое или конденсированное бициклическое гетероарильное кольцо; каждое из которых имеет от одного до пяти заместителей R3, R3a, R3b, R4 и R4a, которые независимо выбраны из группы, состоящей из Н, галогена, -ORc, -OC(O)Rc, -NRcRd, -SRc, -Re, -CN, -NO2, -CO2Rc, -CONRcRd, -C(O)Rc, -OC(O)NRcRd, -NRdC(O)Rc, -NRdC(O)2Re, -NRc-C(O)NRcRd, -NH-C(NH2)=NH, -NReC(NH2)=NH, -NH-C(NH2)=NRe, -NH-C(NHRe)=NH, -S(O)Re, -S(O)2Re, -NRcS(O)2Re, -S(O)2NRcRd, -N3, -X2ORc, -O-X2ORc, -X2OC(O)Rc, -X2NRcRd, -O-X2NRcRd, -X2SRc, -X2CN, -X2NO2, -X2CO2Rc, -O-X2CO2Rc, -X2CONRcRd, -O-X2CONRcRd, -X2C(O)Rc, -X2OC(O)NRcRd, -X2NRdC(O)Rc, -X2NRdC(O)2Re, -X2NRcC(O)NRcRd, -X2NH-C(NH2)=NH, -X2NReC(NH2)=NH, -X2NH-C(NH2)=NRe, -X2NH-C(NHRe)=NH, -X2S(O)Re, -X2S(O)2Re, -X2NRcS(O)2Re, -X2S(O)2NRcRd, -X2N3, -NRd-X2ORc, -NRd-X2NRcRd, -NRd-X2CO2Rc и -NRd-X2CONRcRd, где каждый X2 является представителем, независимо выбранным из группы, состоящей из С1-4 алкилена, и каждый Rc и Rd независимо выбран из атома водорода, C1-8 алкила, С1-8 гидроксиалкила, C1-8 галогеналкила и С3-6 циклоалкила, или опционально Rc и Rd, когда они присоединены к одному и тому же атому азота, могут объединяться с атомом азота с образованием пяти- или шестичленного цикла, содержащего от 0 до 2 дополнительных гетероатомов в качестве членов цикла; и каждый Re независимо выбран из группы, состоящей из C1-8 алкила, C1-8 гидроксиалкила, C1-8 галогеналкила и С3-6 циклоалкила.
Символ Ar2 представляет собой шести- или десятичленное моноциклическое или конденсированное бициклическое арильное кольцо, или 5-10-членное моноциклическое или конденсированное бициклическое гетероарильное кольцо; каждое из которых имеет от одного до пяти заместителей R5, R6, R7, R8 и R9, независимо выбранных из группы, состоящей из Н, галогена, -ORf, -OC(O)Rf, -NRfRg, -SRf, -Rh, -CN, -NO2, -CO2Rf, -CONRfRg, -C(O)Rf, -OC(O)NRfRg, -NRgC(O)Rf, -NRgC(O)2Rh, -NRf-C(O)NRfRg, -NH-C(NH2)=NH, -NRhC(NH2)=NH, -NH-C(NH2)=NRh, -NH-C(NHRh)=NH, -S(O)Rh, -S(O)2Rh, -NRfS(O)2Rh, -S(O)2NRfRg, -NRfS(O)2NRfRg, -N3, -X3ORf, -X3OC(O)Rf, -X3NRfRg, -X3SRf, -X3CN, -X3NO2, -X3CO2Rf, -X3CONRfRg, -X3C(O)Rf, -X3OC(O)NRfRg, -X3NRgC(O)Rf, -X3NRgC(O)2Rh, -X3NRf-C(O)NRfRg, -X3NH-C(NH2)=NH, -X3NRhC(NH2)=NH, -X3NH-C(NH2)=NRh, -X3NH-C(NHRh)=NH, -X3S(O)Rh, -X3S(O)2Rh, -X3NRfS(O)2Rh, -X3S(O)2NRfRg, -Y, -X3Y, -S(O)2Y, -C(O)Y, -X3N3, -O-X3ORf, -O-X3NRfRg, -O-X3CO2Rf, -O-X3CONRfRg, -NRg-X3ORf, -NRg-X3NRfRg, -NRg-X3CO2Rf и -NRg-X3CONRfRg, где Y представляет собой пяти- или шестичленное арильное, гетероарильное или гетероциклическое кольцо, опционально имеющее 1-3 заместителя, выбранных из группы, состоящей из галогена, -ORf, -OC(O)Rf, -NRfRg, -Rh, -SRf, -CN, -NO2, -CO2Rf, -CONRfRg, -C(O)Rf, -NRgC(O)Rf, -NRgC(O)2Rh, -S(O)Rh, -S(O)2Rh, -NRfS(O)2Rh, -S(O)2NRfRg, -X3ORf, X3SRf, -X3CN, -X3NO2, -X3CO2Rf, -X3CONRfRg, -X3C(O)Rf, -X3OC(O)NRfRg, -X3NRgC(O)Rf, -X3NRgC(O)2Rh, -X3NRf-C(O)NRfRg, -X3OC(O)Rf, -X3S(O)Rh, -X3S(O)2Rh, -X3NRfRg, -X3NRfS(O)2Rh, -X3S(O)2NRfRg, -O-X3ORf, -O-X3NRfRg, -O-X3CO2Rf, -O-X3CONRfRg, -NRg-X3ORf, -NRg-X3NRfRg, -NRg-X3CO2Rf и -NRg-X3CONRfRg, и где каждый X3 независимо выбран из группы, состоящей из С1-4 алкилена, и каждый Rf и Rg независимо выбран из атома водорода, C1-8 алкила, C1-8 гидроксиалкила, C1-8 галогеналкила и С3-6 циклоалкила, или, когда они присоединены к одному и тому же атому азота, они могут объединяться с атомом азота с образованием пяти- или шестичленного цикла, содержащего от 0 до 2 дополнительных гетероатомов в качестве членов цикла, и каждый Rh независимо выбран из группы, состоящей из C1-8 алкила, C2-8 алкенила, C2-8 алкинила, C1-8 гидроксиалкила, С1-8 галогеналкила и С3-6 циклоалкила; или когда два из R5, R6, R7, R8 и R9 присоединены к соседним атомам цикла Ar2, они опционально объединяются с образованием пяти- или шестичленного цикла, содержащего ноль, один или два гетероатома, выбранных из О и N, в качестве членов цикла.
В некоторых вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила, пиридила, пиразинила, пиридазинила, пиримидинила, триазинила, хинолинила, хиноксалинила и пуринила, каждый из которых необязательно имеет заместители R3, R3a, R3b, R4 и R4a.
В других вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила и пиридила, каждый из которых необязательно имеет заместители R3, R3a, R3b, R4 и R4a.
В других вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar2 выбран из группы, состоящей из фенила, пиразолила, имидазолила, триазолила, тетразолила, оксазолила, изоксазолила, оксадиазолила, оксатиадиазолила, пирролила, тиазолила, изотиазолил, бензимидазолила, бензоксазолила, бензопиразолила, бензотриазолила, пиразоло[3,4-b]пиридина, пиразоло[3,4-d]пиримидина, имидазо[4,5-b]пиридина, имидазо[1,5-а]пиридина и пирроло[2,3-b]пиридина, каждый из которых необязательно имеет заместители R5, R6 и R7.
В других вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила, каждый из которых имеет заместители R5, R6 и R7.
В некоторых вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar1 выбран из группы, состоящей из фенила, нафтила и пиридила, каждый из которых имеет от одного до пяти заместителей R3, R3a, R3b, R4 и R4a; и Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила, каждый из которых имеет заместители R5, R6 и R7.
В частных вариантах осуществления, соединения формулы I представляют собой соединения, в которых Ar1 представляет собой фенил, имеющий от одного до пяти заместителей R3, R3a, R3b, R4 и R4a, и Ar2 выбран из группы, состоящей из пиразолила, имидазолила, бензимидазолила, бензопиразолила, пиразоло[3,4-b]пиридина, пиразоло[3,4-d] пиримидина, имидазо[4,5-b]пиридина, имидазо[1,5-а]пиридина и пирроло[2,3-b]пиридина, каждый из которых необязательно имеет заместители R5, R6 и R7.
Другими вариантами осуществления настоящего изобретения являются соединения формул Ia, Ia1, Ia2, II, III и IV.
Соответственно, в некоторых вариантах осуществления, указанные соединения представляют собой соединения формулы Ia:
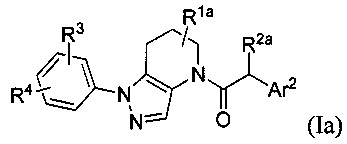
или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где R3 и R4 независимо выбраны из группы, состоящей из Н, галогена, -Re, -CN и -SO2Re; и группы R1a, R2a и Ar2 имеют значения, указанные выше для формулы I, или другие описанные варианты осуществления.
В других вариантах формулы I или Ia, Ar2 представляет собой гетероарильную группу; в других вариантах осуществления, Ar2 представляет собой гетероарильную группу, необязательно замещенную и присоединенную к остальной части молекулы через атом азота в цикле; и. в других вариантах осуществления, Ar2 имеет формулу:
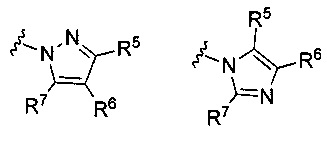 или
или  ,
,
где R5, R6 и R7 независимо выбраны из группы, состоящей из Н, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y, где -Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В одной группе частных вариантов осуществления, соединения имеют формулу:
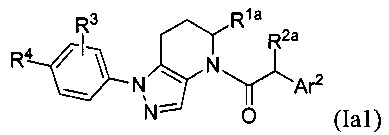
или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где R4 выбран из группы, состоящей из F и Cl; и группы R1a, R2a, R3 и Ar2 имеют значения, указанные выше для формулы I или Ia, или другие описанные варианты осуществления.
В другой группе частных вариантов осуществления, соединения имеют формулу:
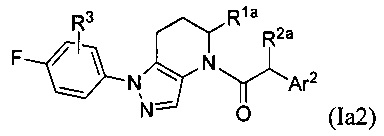
или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где R3 выбран из группы, состоящей из Н, галогена, C1-8 алкила, C1-8 галогеналкила и C1-8 алкокси; R1a и R2a независимо выбраны из группы, состоящей из Н, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и Ar2 имеет значения, указанные выше для формулы I или Ia, или другие описанные варианты осуществления.
В другой группе частных вариантов осуществления, соединения имеют формулу:

или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где Rla и R2a независимо выбраны из группы, состоящей из Н, C1-8 алкила, С1-8 галогеналкила, С1-8 алкокси и С1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из Н, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где -Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В другой группе частных вариантов осуществления, соединения имеют формулу:
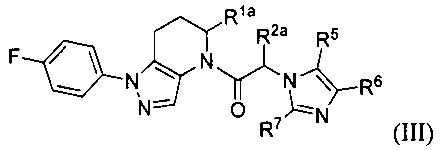
или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где R1a и R2a независимо выбраны из группы, состоящей из Н, C1-8 алкила, С1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из Н, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где -Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В другой группе частных вариантов осуществления, соединения имеют формулу:
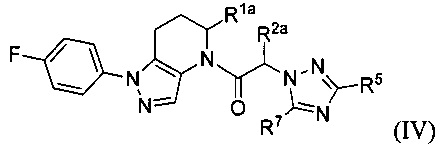
или их фармацевтически приемлемые соли, ротамеры или оптические изомеры, где Rla и R2a каждый независимо выбраны из группы, состоящей из Н, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и R5 и R7 каждый независимо выбраны из группы, состоящей из Н, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y; где -Rh, Rf, Rg и Y имеют значения, указанные выше для формулы I.
В любом из описанных выше вариантов осуществления, в случае наличия Y, частные варианты осуществления представляют собой варианты, в которых Y выбран из группы, состоящей из пиридила, пиримидинила, имидазолила, оксазолила, оксадиазолила, триазолила, тиазолила, имидазолинила и пиразолила.
Некоторые особенно интересные соединения представляют собой соединения, представленные в таблице 1, вместе с их фармацевтически приемлемыми солями, гидратами или N-оксидами, ротамерами и стереоизомерами.
Получение соединений
Как описано ниже в примерах, соединения и интермедиаты по настоящему изобретению могут быть получены квалифицированным специалистом в данной области по принципу сборки из компонентов.
IV. Фармацевтические композиции
Помимо описанных выше соединений, композиции для модулирования активности CCR1 у человека и животных в типичном случае содержат фармацевтический носитель или разбавитель.
Термин "композиция" при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, получающийся напрямую или косвенно при комбинации указанных ингредиентов в указанных количествах. Термин «фармацевтически приемлемый» означает, что носитель, разбавитель или вспомогательное вещество должны быть совместимы с другими ингредиентами в препарате и не наносить вреда пациенту, принимающему препарат.
Фармацевтические композиции для введения соединений по настоящему изобретению удобно выпускать в единичной лекарственной форме, и их можно приготовить любым из методов, хорошо известных в области фармацевтики и введения лекарственных средств. Все методы включают стадию соединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции готовят путем однородного и равномерного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, формования продукта в желаемый препарат. В фармацевтическую композицию действующее вещество включают в количестве, достаточном для достижения желаемого эффекта при болезненном процессе или состоянии.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например, форму таблеток, пастилок, ромбовидных таблеток, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся составов, как описано в заявке на патент США 2002-0012680, твердых или мягких капсул, сиропов, эликсиров, растворов, буккальных пластырей, гелей для перорального применения, жевательной резинки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции для перорального применения можно приготовить согласно любым методам, известным в области производства фармацевтических композиций, и такие композиции могут содержать одно или больше средств, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, для создания фармацевтически удачных и приятных на вид препаратов. Таблетки содержат действующее вещество в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые подходят для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид кремния, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия, гранулирующие средства и разрыхлители, например, кукурузный крахмал или альгиновая кислота; связующие средства, например, поливинилпирролидон, целлюлоза, ПЭГ, крахмал, желатин или камедь акации, и лубриканты, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут иметь нанесенное покрытие, которое растворяется в кишечнике или другим известным образом замедляет распад и всасывание в желудочно-кишечном тракте, тем самым обеспечивая продолжительное действие в течение длительного периода времени. Например, можно применять замедляющее вещество, такое как глицерил моностеарат или глицерил дистеарат. Также таблетки могут иметь покрытие, нанесенное по методике, описанной в патентах США 4256108; 4166452 и 4265874, с формированием осмотических терапевтических таблеток с замедленным высвобождением.
Препараты для перорального применения могут также иметь вид твердых желатиновых капсул, в которых действующее вещество смешано с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или вид мягких желатиновых капсул, в которых действующее вещество смешано с водной или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, эмульсии могут быть приготовлены с несмешивающимся с водой ингредиентом, таким как масло, и стабилизированы поверхностно-активными веществами, такими как моно- и диглицериды, ПЭГ-эфиры и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательными веществами, подходящими для производства водных суспензий. Такими вспомогательными веществами являются суспендирующие средства, например, натрия карбоксиметилцеллюлоза, метилцеллюлоза, гидрокси-пропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и смола акации; диспергирующие и смачивающие средства, которые могут представлять собой природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтилен стеарат, продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитол моноолеат, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситол-ангидридами, например, полиэтилен сорбитан моноолеат. Водные суспензии могут также содержать один или больше консервантов, например, этил или н-пропил пара-гидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно приготовить суспендированием действующего вещества в растительном масле, например, в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например, пчелиный воск, твердый парафин или цетиловый спирт. Можно добавлять подсластители, такие как описанные выше, и ароматизаторы для получения приятного препарат для перорального приема. Такие композиции можно консервировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водных суспензий путем добавления воды, содержат действующее вещество в смеси с диспергирующим или смачивающим средством, суспендирующим средством и одним или больше консервантами. Примерами подходящих диспергирующих и смачивающих средств могут являться вещества, уже упомянутые выше. Также могут присутствовать дополнительные вспомогательные вещества, например, подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, жидкий парафин, или их смесь. Подходящими эмульгаторами могут быть природные смолы, например, смола акации или трагакантовая камедь, природные фосфатиды, например, соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и гекситол-ангидридов, например, сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например, полиоксиэтилен сорбитан моноолеат. Эмульсии могут также содержать подсластители и ароматизаторы.
В сиропы и эликсиры можно добавлять подсластители, например, глицерин, пропиленгликоль, сорбит и сахарозу. Такие препараты могут также содержать мягчитель, консервант, ароматизатор и краситель. Композиции для перорального приема можно готовить в комбинации с циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить согласно методам из существующего уровня техники, применяя перечисленные выше подходящие диспергирующие или смачивающие средства, а также суспендирующие средства. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парэнтерально приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые могут применяться, можно упомянуть воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные жирные масла широко применяются в качестве растворителя или суспендирующей среды. Для этой цели можно применять любую марку жирного масла, включая синтетические моно- и диглицериды. Кроме того, в препаратах для инъекций нашли применение жирные кислоты, такие как олеиновая кислота.
Соединения по настоящему изобретению можно также вводить в форме суппозиториев для ректального введения лекарственных препаратов. Такие композиции можно готовить смешиванием лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому плавится в заднем проходе, высвобождая лекарственное средство. Такие вещества включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить через глаза посредством растворов или мазей. Кроме того, можно осуществлять чрезкожное введение рассматриваемых соединений посредством ионофоретических пластырей и т.п. Для местного нанесения применяют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединения по настоящем изобретению. При использовании в настоящем тексте, местное нанесение включает применение жидкостей для промывания и полоскания полости рта.
Соединения по настоящему изобретению можно также присоединять к носителю, который представляет собой подходящий полимер, в качестве нацеливаемого переносчика лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополимеры пирана, полигидрокси-пропил-метакриламид-фенол, полигидроксиэтил-аспартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоиловыми остатками. Кроме того, соединения по настоящему изобретению можно присоединять к носителю из класса биоразлагаемых полимеров, которые могут применяться для достижения замедленного высвобождения лекарственного средства, например, полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, поли-эпсилон-капролактон, полигидроксибутановая кислота, полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатные блок-сополимеры гидрогелей. Полимеры и полупроницаемые полимерные матриксы можно формовать в изделия, такие как клапаны, стенты, трубочки, протезы и т.п. В одном варианте осуществления настоящего изобретения, соединение по настоящему изобретению присоединяют к полимеру или полупроницаемому полимерному матриксу, который формуют в стент или стент-графт.
V. Способы лечения заболеваний, модулируемых CCR1
В другом аспекте, в настоящем изобретении описаны способы лечения CCR1-опосредуемых состояний или заболеваний путем введения субъекту, страдающему таким заболеванием или состоянием, терапевтически эффективного количества соединения, соответствующего приведенной выше формуле I. Термин "субъект" в настоящем тексте включает животных, таких как млекопитающие, включая (но не ограничиваясь только ими) приматов (например, человека), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и т.п.
CCR1 представляет собой мишень для вмешательства в работу или промотирования частных аспектов функций иммунных клеток, или, в более общем смысле, для вмешательства в работу функций, связанных с экспрессированием CCR1 на широком наборе типов клеток млекопитающих, таких как человек. Соединения, ингибирующие CCR1, особенно подходят для модулирования работы моноцитов, макрофагов, лимфоцитов, гранулоцитов, NK клеток, тучных клеток, дендритных клеток, нейтрофилов и определенных клеток, являющихся производным иммунных (например, остеокласты), в терапевтических целях. Соответственно, настоящее изобретение касается соединений, которые могут применяться для профилактики и/или лечения широкого ряда воспалительных и иммунорегуляторных заболеваний и нарушений (смотри Saeki, et al., Current Pharmaceutical Design 9:1201-1208 (2003)).
Например, быстродействующее соединение, которое ингибирует одну или больше функций CCR1, можно вводить для ингибирования (т.е. уменьшения или профилактики) воспаления или инфильтрации клеток, связанной с иммунным нарушением. В результате можно подавлять один или больше воспалительных процессов, таких как эмиграция или инфильтрация лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождение медиаторов воспаления. Например, по настоящему способу можно подавлять инфильтрацию моноцитов в место воспаления (например, в поврежденный сустав при артрите, или в ЦНС при рассеянном склерозе).
Аналогично, быстродействующее соединение, промотирующее одну или больше функций CCR1, вводят для стимулирования (индуцирования или усиления) воспалительного ответа, такого как миграция лейкоцитов, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождения медиаторов воспаления, что приводит к благоприятному стимулированию воспалительных процессов. Например, моноциты могут привлекаться для борьбы с бактериальными инфекциями.
Заболевания и состояния, вызванные воспалением, иммунными нарушениями и инфекцией, можно лечить способом по настоящему изобретению. В предпочтительном варианте осуществления, заболевание или состояние является таким, при котором действие иммунных клеток, таких как моноциты, макрофаги, лимфоциты, гранулоциты, NK клетки, тучные клетки, дендритные клетки или некоторые клетки, являющиеся производными иммунных клеток (например, остеокласты), необходимо подавлять или промотировать, для модулирования воспалительного или аутоиммунного ответа.
В одной группе вариантов осуществления, заболевания или состояния, включая хронические заболевания, человека или других видов, можно лечить модуляторами работы CCR1. Такие заболевания или состояния включают: (1) аллергические заболевания, такие как системные анафилактические или гиперсензитивные ответы, аллергия на лекарственные препараты, аллергия на укусы насекомых и пищевые аллергии, (2) воспалительное заболевание кишечника, такое как болезнь Крона, язвенный колит, илеит и энтерит, (3) вагинит, (4) псориаз и воспалительный дерматоз, такой как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница и зуд, (5) васкулит, (6) спондилоортропатия, (7) склеродермия, (8) астма и аллергические респираторные заболевания, такие как аллергическая астма, аллергический ринит, гиперчувствительность легких и т.п., (9) аутоиммунные заболевания, такие как фибромиалгия, склеродермия, анкилозирующий спондилоартрит, юношеский ревматоидный артрит, синдром Стилла, многосуставный юношеский ревматоидный артрит, олигосуставный юношеский ревматоидный артрит, ревматическая полимиалгия, ревматоидный артрит, псориазный артрит, остеоартрит, многосуставный артрит, множественный склероз, системная красная волчанка, диабет I типа, диабет II типа, гломерулонефрит и т.п., (10) отторжение трансплантата (включая отторжение аллотрансплантата и реакция «трансплантат против хозяина»), и (11) другие заболевания, при которых необходимо подавить нежелательные воспалительные ответы или иммунные нарушения, такие как сердечно-сосудистые заболевания, включая атеросклероз и рестеноз, миозит, нейродегенеративные заболевания (например, болезнь Альцгеймера), энцефалит, менингит, гепатит, нефрит, сепсис, саркоидоз, аллергический конъюнктивит, отит, хроническое обструктивное заболевание легких, синусит, синдром Бехчета и подагра, (12) остеопороз и другие нарушения костной ткани, (13) иммунно-опосредованные пищевые аллергии, такие как глютеновая болезнь, и (14) радиационное заболевание легких (RIPD). Смотри, например, Yang et al, Am J Respir Cell Mol Biol. 45(1):127-35 (2011).
В другой группе вариантов осуществления, заболевания или состояния можно лечить добавлением модуляторов работы CCR1. Примеры заболеваний, которые лечат модуляторами работы CCR1, включают раковые заболевания, такие как множественная миелома и родственное остеолитическое заболевание костей, сердечно-сосудистые заболевания, заболевания, при которых играют определенную роль ангиогенез и образование новых сосудов (неопластические заболевания, ретинопатия и мышечная дегенерация), инфекционные заболевания (вирусные инфекции, например, инфекции ВИЧ и PC-вирус, бактериальные инфекции) и иммунодепрессивные заболевания, такие как состояние после пересадки органа и состояние после пересадки кожи. Термин «состояние после пересадки органа» включает состояния после пересадки костного мозга и состояния после пересадки солидного органа (например, почки, печени, легкого, сердца, поджелудочной железы или их комбинации).
Соединения по настоящему изобретению, соответственно, могут применяться в профилактике и лечении широкого ряда воспалительных и иммуннорегуляторных состояний и заболеваний.
В зависимости от заболевания, которое необходимо вылечить, и от состояния пациента, соединения по настоящему изобретению можно вводить перорально, парэнтерально (например, внутримышечно, интраперитонеально, внутривенно, интрацеребрально, интрацистерниальными инъекциями или инфузиями, подкожной инъекцией или в виде импланта), имплантацией (например, когда соединение вводят в связке со стентом), в виде спрея для ингаляций, назально, вагинально, ректально, сублингвально, или местно, и их можно вводить в состав препаратов индивидуально или совместно, в составе подходящих дозированных лекарственных форм, содержащих общеупотребимые нетоксичные фармацевтически приемлемые носители, адъюванты и растворители, подходящие для каждого способа введения.
При лечении или профилактике состояний, требующих модулирования хемокинового рецептора, подходящий уровень дозировок в целом составляет примерно от 0,001 до 100 мг на килограмм веса тела пациента в день, которые можно вводить в виде одной или нескольких доз. Предпочтительно, уровень дозировки составляет от примерно 0,01 до примерно 25 мг/кг в день; более предпочтительно от примерно 0,05 до примерно 10 мг/кг в день. Подходящий уровень дозировки может составлять примерно от 0,01 до 25 мг/кг в день, примерно от 0,05 до 10 мг/кг в день, или примерно от 0,1 до 5 мг/кг в день. В указанном диапазоне, дозировка может составлять от 0,005 до 0,05, от 0,05 до 0,5 или от 0,5 до 5,0 мг/кг в день. При пероральном введении, композиции предпочтительно выпускаются в форме таблеток, содержащих от 1,0 до 1000 миллиграммов действующего вещества, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 миллиграммов действующего вещества, для симптоматического регулирования дозировки для пациента, проходящего лечение. Соединения можно вводить в режиме от 1 до 4 раз в день, предпочтительно один или два раза в день.
Однако, следует понимать, что конкретная дозировка и частота введения для каждого конкретного пациента могут варьироваться и зависят от различных факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и длительность действия соединения, возраст, вес тела, наследственность, общее состояние здоровья, пол и диету пациента, а также путь и время введения, скорость выведения, комбинацию с другими лекарственными средствами и тяжесть конкретного заболевания у пациента, проходящего терапию.
Описанными соединениями, композициями и способами можно лечить заболевания и состояния, связанные с воспалением, иммунным нарушением, инфекцией и раковым заболеванием.
Соединения и композиции по настоящему изобретению можно комбинировать с другими соединениями и композициями, имеющими соответствующее применение в целях профилактики и лечения интересующих состояний или заболеваний, таких как воспалительные или аутоиммунные нарушения, состояния и заболевания, включая воспалительное заболевание кишечника, ревматоидный артрит, остеоартрит, псориазный артрит, многосуставный артрит, множественный склероз, аллергические заболевания, псориаз, атопический дерматит и астма, а также перечисленные выше патологии.
Например, при лечении или профилактике воспаления или аутоиммунных заболеваний, или, например, артрита в комбинации с дегенерацией костной ткани, описанные соединения и композиции можно применять в комбинации с противовоспалительным средством или анальгетиком, такими как опиатный агонист, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, NMDA антагонист, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или цитокин-супрессирующее противовоспалительное средство, например, с таким соединением как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п. Аналогично, описанные соединения и композиции можно вводить совместно с перечисленными выше анальгетиками: потенциатором, таким как кофеин, Н2 антагонистом (например, ранитидином), симетиконом, гидроксидом алюминия или магния, противоотечным средством, таким как фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или лево-дезоксиэфедрин; противокашлевыми средствами, такими как кодеин, гидрокодон, карамифен, карбетапентан или декстрометорфан; диуретиками; и седативными или неседативными антигистаминными средствами.
Сходным образом, соединения и композиции по настоящему изобретению можно применять в комбинации с другими лекарственными средствами, которые используются для лечения, профилактики, приостановки или облегчения тяжести заболеваний или состояний, при которых применяются соединения и композиции по настоящему изобретению. Эти другие лекарственные соединения можно вводить обычно применяющимися способами и в обычно применяющихся дозировках, одновременно или последовательно с соединением или композицией по настоящему изобретению. Когда соединение или композиция по настоящему изобретению применяется одновременно с одним или больше другими лекарственными средствами, предпочтительна фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению или композиции по настоящему изобретению. Соответственно, фармацевтические композиции по настоящему изобретению включают композиции, которые содержат также одно или больше других действующих веществ или терапевтических средств, помимо соединения или композиции по настоящему изобретению. Примеры других терапевтических средств, которые можно комбинировать с соединением или композицией по настоящему изобретению, при введении по отдельности или в составе одной и той же фармацевтической композиции, включают (но не ограничиваются только ими): (a) VLA-4 антагонисты, (b) кортикостероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, преднизолон, дексаметазон, флутиказон, гидрокортизон, бутезонид, триамцинолон, сальметерол, сальбутамол, форметерол; (c) иммуносупрессанты, такие как циклоспорин (циклоспорин А, Sandimmune®, Neoral®), такролимус (FK-506, Prograf®), рапамицин (сиролимус, Rapamune®) и другие иммуносупрессанты типа FK-506, и микофенолят, например, микофенолят мофетил (CellCept®); (d) антигистаминные средства (антагонисты Н1-гистамина), такие как бромфенирамин, хлорфенирамин, дексхлорфенирамин, триплоидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатидин, ципрогептадин, антазолин, фенирамин пириламин, астемизол, терфенадин, лоратадин, цетиризин, фексофенадин, дезкарбоэтоксилоратидин и т.п.; (e) нестероидные антиастматические средства (например, тербуталин, метапротеренол, фенотерол, изоэтарин, альбутерол, битолтерол и пирбутерол), теофиллин, кромолин натрия, атропин, ипратория бромид, лейкотриеновые антагонисты (например, зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст и SKB-106,203), ингибиторы синтеза лейкотриена (зилеутон, BAY-1005); (f) нестероидные противовоспалительные средства (НСПВ), такие как производные пропионовой кислоты (например, алминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, рниропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например, индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозовая кислота, фетиазак, фурофенак, ибуфенак, озоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенамовой кислоты (например, флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота и толфенамовая кислота), производные бифенилкарбоновой кислоты (например, дифлунизал и флуфенизал), оксикамы (например, изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (например, ацетилсалициловая кислота и сульфазалазин) и пиразолоны (например, апазон, бензпиперилон, фепразон, мофебутазон, оксифенбутазон и фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб (Celebrex®) и рофекоксиб (Vioxx®); (h) ингибиторы фосфодиэстеразы IV типа (PDE IV); (i) соединения золота, такие как ауранофин и ауротиоглюкоза, (j) пептидные модуляторы и антитела-модуляторы иммунных регуляторных молекул, такие как этанерцепт (Enbrel®), инфликсимаб (Remicade®), адалимумаб (Humira®), цертолизумаб пегол (Cimzia®), абатацепт (Orencia®) и голимумаб (Simponi®), (k) средства терапии антителами, такие как ортоклон (OKT3), даклизумаб (Zenapax®), базиликсимаб (Simulect®) и инфликсимаб (Remicade®), (l) другие антагонисты хемокиновых рецепторов, в особенности CCR5, CXCR2, CXCR3, CCR2, CCR3, CCR4, CCR7, CX3CR1 и CXCR6; (m) лубриканты или мягчители, такие как петролатум и ланолин, (n) кератолитические средства (например, тазаротен), (o) производные витамина D3, например, кальципотриен или кальципотриол (Dovonex®), (p) PUVA, (q) антралин (Drithrocreme®), (r) этретинат (Tegison®) и изотретиноин, и (s) средства терапии множественного склероза, такие как интерферон β-1β (Betaseron®), интерферон (β-1α (Avonex®), азатиоприн (Imurek®, Imuran®), глатирамер ацетат (Capoxone®), глюкокортикоид (например, преднизолон) и циклорфосфамид (t) DMAPvDS, такие как метотрексат, (u) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства; гидроксихлорохин; D-пеницилламин; антиметаболиты, такие как азатиоприн, 6-меркаптопурин и метотрексат; ингибиторы синтеза ДНК, такие как гидроксимочевина, и вещества, нарушающие работу микротрубочек, такие как колхицин. Весовое соотношение соединения по настоящему изобретению и второго действующего вещества может варьироваться и зависит от эффективной дозировки каждого ингредиента. Обычно применяют эффективную дозировку каждого соединения. Так, например, когда соединение по настоящему изобретению комбинируют с НСПВ, весовое соотношение соединения по настоящему изобретению и НСПВ обычно находится в диапазоне от примерно 1000:1 до примерно 1:1000, предпочтительно от примерно 200:1 до примерно 1:200. Комбинации соединения по настоящему изобретению и других действующих веществ также обычно находятся в указанном выше диапазоне, но в каждом случае должна применяться эффективная дозировка каждого действующего вещества.
VI. Примеры
Приведенные далее примеры предназначены для иллюстрации, а не для ограничения заявленного изобретения.
Реагенты и растворители, использовавшиеся в описанных ниже примерах, можно получить из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1Н-ЯМР спектры записывали на ЯМР-спектрометре Varian Mercury 400 МГц. Химические сдвиги приведены относительно ТМС и табулированы в таком порядке: мультиплетность (с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет) и число протонов. Результаты масс-спектрометрии выражены как соотношение массы к заряду, с последующим указанием относительной интенсивности каждого иона (в скобках). В примерах приведено одно значение т/е для иона М+Н (или если указано, М-Н), содержащего наиболее распространенные изотопы атомов. Во всех случаях картина распределения изотопных пиков соответствует ожидаемой формуле. Масс-спектральный анализ методом электроспрея (ESI) проводили на электроспреевом масс-спектрометре Hewlett-Packard MSD с применением ВЭЖХ HP1100 для ввода образца. Обычно аналит растворяли в метаноле в концентрации 0,1 мг/мл, и 1 мкл вводили с растворителем в масс-спектрометр, сканирующий в интервале от 100 до 1500 дальтон. Все соединения можно исследовать методом ESI с детектированием положительных ионов, используя смесь ацетонитрил/вода с 1% муравьиной кислоты в качестве раствора для ввода. Описанные ниже соединения можно также анализировать методом ESI с детектированием отрицательных ионов, используя 2 мМ раствор NH4OAC в смеси ацетонитрил/вода в качестве раствора для ввода.
Соединения, входящие в объем настоящего изобретения, можно синтезировать как описано ниже, используя различные реакции, известные квалифицированным специалистам в данной области. Примеры подходящих путей синтеза азаиндольных производных и некоторых соединений по настоящему изобретению описаны ниже или в тексте настоящей заявки. В приведенных далее описаниях синтезов, некоторые предшественники арилпиперазиновых и гетероароматических субъединиц были получены из коммерческих источников. Коммерческие источники включают Aldrich Chemical Co., Acros Organics, Ryan Scientific Incorporated, Oakwood Products Incorporated, Lancaster Chemicals, Sigma Chemical Co., Lancaster Chemical Co., TCI-America, Alfa Aesar, Davos Chemicals и GFS Chemicals. Некоторые арилпиперазиновые соединения можно приобрести в коммерческих источниках. Другие можно получить как описано в заявке на патент США №11/008,774, содержание которой полностью включено в настоящий текст в рамках целей настоящего изобретения. Стандартные химические реакции применяли для соединения арилпиперазиновых и гетероароматических субъединиц (полученных коммерчески или описанными ниже способами), применяя подходящий оптимизированный линкер, такой как ацетильный фрагмент, описанный в тексте настоящего изобретения.
Квалифицированному специалисту в данной области будет также понятно, что для синтеза целевых соединений по настоящему изобретению могут применяться альтернативные методы, и что подходы, описанные в тексте настоящего документа, не являются исчерпывающими, но обеспечивают широко применимые практичные способы синтеза заявленных соединений.
Некоторые молекулы, заявленные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, и все такие варианты указанных соединений входят в объем настоящего изобретения.
Региоизомерия является распространенным явлением в органической химии, и она особенно распространена среди некоторых описанных в настоящем тексте структурных типов. Квалифицированным специалистам в данной области техники будет понятно, в отношении описанных в настоящем тексте соединений, что реакции сочетания с гетероароматическими циклическими системами могут давать либо один, либо смесь детектируемых региоизомеров.
Подробное описание экспериментальных методик, использованных для синтеза ключевых соединений в настоящем тексте, приводит к молекулам, которые описаны характеризующими их физическими данными, а также относящимися к ним структурными изображениями.
Квалифицированным специалистам в данной области также будет понятно, что в стандартных методиках обработки реакционных смесей часто используются кислоты и основания. В ходе экспериментальных методик, описанных в настоящем патенте, иногда образуются соли материнских соединений, если эти соединения обладают необходимой кислотностью или основностью.
Примеры
Общий метод А. Сочетание с применением HATU. Соответствующий амин (1,2 экв) и карбоновую кислоту объединяли в ТГФ (0,2 М) с Et3N (1-2 экв), затем добавляли HATU (1,3 экв) при комнатной температуре. По окончании реакции смесь разбавляли этилацетатом, промывали насыщенным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении.
Общий метод В. Образование ацилхлорида и сочетание. Соответствующую карбоновую кислоту разбавляли хлороформом (0,25 М) с 2 каплями ДМФА. При комнатной температуре добавляли оксалилхлорид (1,2 экв), наблюдая выделение газа из реакционной смеси. Через 20-30 минут добавляли соответствующий амин (1,2 экв) и затем Et3N (2,5 экв) или равный объем (в сравнении с CHCl3) насыщенного раствора NaHCO3. По окончании реакции смесь разбавляли этилацетатом, промывали раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении.
Пример 1. Синтез 2-[4-амино-3-(1H-имидазол-2-ил)пиразоло[3,4-d]пиримидин-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

a) В круглодонную колбу объемом 500 мл помещали 4-фторфенилгидразина гидрохлорид (18,9 г, 116 ммоль), 3-фтор-2-формилпиридин (14,2 г, 113 ммоль), K2CO3 (47,0 г, 340 ммоль), и ДМФА (150 мл). Полученную суспензию нагревали до 120°С на масляной бане 41 час, после чего реакционную смесь охлаждали и выливали в 1,2 л Н2О. Коричневый осадок отделяли посредством фильтрования под вакуумом и трижды промывали водой. Полученное твердое вещество растворяли в CH2Cl2, и полученный раствор промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая коричневое твердое вещество. Полученное твердое вещество растирали в 200 мл смеси 9:1 гексан : EtOAc и сушили, получая 1-(4-фторфенил)-1H-пиразоло[4,3-b]пиридин (16,3 г, 68%) в виде коричневого порошка. MS: (ES) m/z вычислено для C12H9FN3 [М+Н]+ 214,1, найдено 214,1.
b) Полученный выше гетероцикл (16,3 г, 77 ммоль) растворяли в 380 мл метанола и добавляли 20% Pd(OH)2/C (10,7 г) в аппарате Парра объемом 2 л. Систему продували и заполняли водородом (3х) и затем встряхивали под давлением водорода 56 фунт/кв. дюйм. Еще 8,0 г катализатора добавляли через 1 день. После исчезновения исходного вещества колбу продували азотом, и реакционную смесь фильтровали через слой целита, промывая несколькими порциями метанола. Фильтрат упаривали при пониженном давлении, получая 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридин (13,7 г, 82%) в виде светло-коричневого твердого вещества. MS: (ES) m/z вычислено для C12H13FN3 [М+Н]+ 218,1, найдено 218,1.
c) Данное соединение получали из 2-(4-амино-3-(1H-имидазол-2-ил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)уксусной кислоты и описанного выше 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу А. Полученный продукт очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (6,4 мг). 1Н-ЯМР (400 МГц, МеОН-d4) δ 8,35 (с, 1 Н), 8,15 (с, 1 Н), 7,54 (м, 2 Н), 7,27 (м, 4 Н), 5,70 (с, 2 Н), 4,00 (м, 2 Н), 2,91 (т, J=6,2 Гц, 2 Н), 2,18 (м, 2 Н); MS: (ES) m/z вычислено для C22H20FN10O [М+Н]+ 459,18, найдено 459,1.
Синтез 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусной кислоты
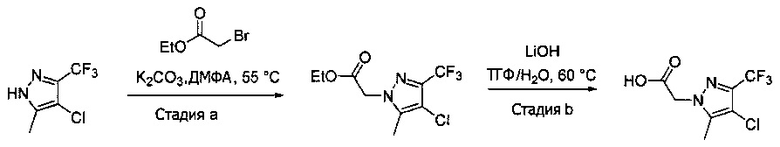
a) К раствору 4-хлор-5-метил-3-(трифторметил)пиразола (15 г, 81,3 ммоль) в ДМФА (82 мл) добавляли этил 2-бромацетат (13,6 г, 81,3 ммоль) и карбонат калия (12,4 г, 89,4 ммоль). Полученную смесь нагревали при 55°С в течение 10 часов при перемешивании. После охлаждения до комнатной температуры, реакционную смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 0-30% этилацетата в гексане), получая целевой продукт (18,5 г, 68,4 ммоль, 84%).
b) К раствору этил 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)ацетата (7,5 г, 27,7 ммоль) в ТГФ (100 мл) и воде (50 мл) добавляли LiOH⋅H2O (2,33 г, 55,4 ммоль). Полученную смесь перемешивали при 60°С 5 часов. После охлаждения до комнатной температуры, реакционную смесь подкисляли 1н. раствором HCl (80 мл) и экстрагировали смесью 1:2 изопропанол/CHCl3 (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали и упаривали в вакууме, получая целевое соединение (6 г, 24,7 ммоль, 89%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 4,98 (с, 2 Н), 2,21 (с, 3 Н); MS: (ES) m/z вычислено для C7H6ClF3N2O2 [М+Н]+ 243,0, найдено 243,0.
Пример 2. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

Данное соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1Н-пиразол-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М NaHSO4 и дважды 1,5М KOH и вымывали органические соединения через слой силикагеля. Полученную суспензию упаривали до получения белого твердого вещества, которое растирали в смеси 1:1 гексан : EtOAc, получая указанное в заголовке соединение в виде белого кристаллического вещества (75,9 мг). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,34 (с, 0,7 Н), 7,57 (с, 0,3 Н), 7,44 (м, 2 Н), 7,15 (м, 2 Н), 5,24 (с, 0,4 Н), 5,13 (с, 1,6 Н), 3,92 (м, 0,5 Н), 3,84 (м, 1,5 Н), 2,87 (м, 2 Н), 2,33 (с, 2,3 Н), 2,32 (с, 0,7 Н), 2,12 (м, 1,5 Н), 2,05 (м, 0,5 Н); MS: (ES) m/z вычислено для C19H17ClF4N5O [М+Н]+ 442,1, найдено 442,0.
Пример 3. Синтез 2-(4-хлорфенил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
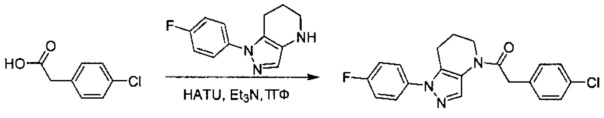
Данное соединение получали из 4-хлорфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина по Общему методу А. Полученную смесь продуктов кристаллизовали из смеси 4:1 CH3CN:Н2О, получая указанное в заголовке соединение в виде не совсем белого кристаллического соединения (37,6 мг). 1Н-ЯМР (400 МГц, MeOD-d4, смесь ротамеров) δ 8,18 (с, 0,9 Н), 7,84 (с, 0,1 Н), 7,56 (м, 2 Н), 7,25-7,37 (м, 6 Н), 4,02 (с, 0,2 Н), 3,92 (с, 1,8 Н), 2,81 (т, J=6,2 Гц, 2 Н), 2,49 (м, 2 Н), 1,90 (м, 2 Н); MS: (ES) m/z вычислено для C20H18ClFN3O [М+Н]+ 370,1, найдено 370,1.
Пример 4. Синтез 1-[1-(4-хлор-3-метокси-фенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]этанона и 1-(2-(4-хлор-3-метоксифенил)-6,7-дигидро-2H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)этанона
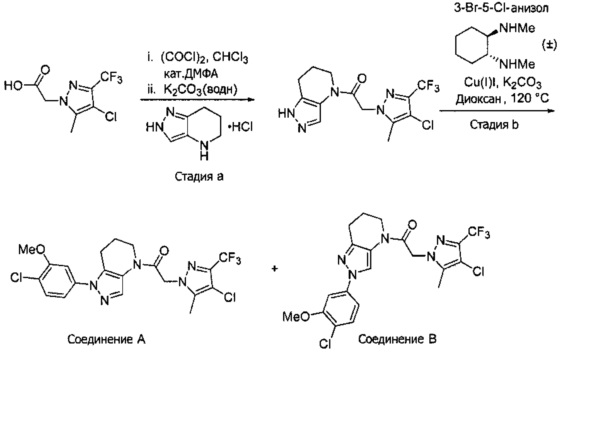
a) 2-(4-Хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусную кислоту (255 мг, 1,05 ммоль) разбавляли в 5 мл CHCl3 и 2 каплях ДМФА. Медленно добавляли оксалилхлорид (105 мкл, 1,2 ммоль), наблюдая выделение газа. Через 30 минут добавляли 160 мг (1 ммоль) 4,5,6,7-тетрагидро-2Н-пиразоло[4,3-b]пиридин⋅HCl, затем 0,75 мл 3М раствора K2CO3. Через 30 минут добавляли в реакционную суспензию 1 мл 3М раствора KOH и 1 мл МеОН, и полученную смесь перемешивали 90 минут. Реакционную суспензию промывали насыщенным водным раствором хлорида натрия, и органический слой сушили над MgSO4, фильтровали и упаривали, получая амид в виде коричневого твердого вещества (184 мг, 53%), которое использовали далее без дополнительной очистки. MS: (ES) m/z вычислено для C13H14ClF3N5O [М+Н]+ 348,1, найдено 348,0.
b) Полученный выше амид (104 мг, 0,3 ммоль) объединяли с 5-бром-2-хлоранизолом (199 мг, 0,9 ммоль), 0,3 мл диоксана, Cu(I)I (11,4 мг, 0,06 ммоль, 20%), (±)-транс-N,N'-диметилциклогексан-1,2-диамином (21,3 мг, 0,15 ммоль, 50%) и K2CO3 (82,9 мг, 0,6 ммоль). Полученную суспензию нагревали при 120°С до окончания реакции. Полученный продукт очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая два региоизомера. Элюирующееся первым, Соединение А: 1-[1-(4-хлор-3-метокси-фенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]этанон выделяли в виде белой пены (8,1 мг). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,37 (с, 0,7 Н), 7,60 (с, 0,3 Н), 7,41 (д, J=8,4 Гц, 1 Н), 7,19 (д, J=2,6 Гц, 1 Н), 6,92 (дд, J=2,2, 8,4 Гц, 1 Н), 5,24 (с, 0,5 Н), 5,14 (с, 1,5 Н), 3,97 (с, 0,6 Н), 3,95 (с, 2,4 Н), 3,85 (м, 2 Н), 2,92 (м, 2 Н), 2,34 (с, 2,6 Н), 2,33 (с, 0,4 Н), 2,13 (м, 1,4 Н), 2,05 (м, 0,6 Н); MS: (ES) m/z вычислено для C20H19F3N5O2 [М+Н]+ 488,1, найдено 488,1. Элюирующееся вторым, Соединение В: 1-(2-(4-хлор-3-метоксифенил)-6,7-дигидро-2H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)этанон выделяли в виде белого твердого вещества (26,8 мг). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,57 (с, 1 Н), 7,35 (д, J=8,4 Гц, 1 Н), 7,26 (д, J=2,2 Гц, 1 Н), 7,11 (дд, J=2,2, 8,4 Гц, 1 Н), 5,22 (с, 0,2 Н), 5,16 (с, 1,8 Н), 3,95 (с, 2,5 Н), 3,86 (с, 0,5 Н) 3,84 (м, 2 Н), 2,93 (т, J=6,6 Гц, 2 Н), 2,32 (с, 3 Н), 2,19 (м, 2 Н); MS: (ES) m/z вычислено для C20H19F3N5O2 [М+Н]+ 488,1, найдено 488,0.
Пример 5а. Синтез 1-[1-(4-хлорфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]этанона и 1-(2-(4-хлорфенил)-6,7-дигидро-2H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)этанона
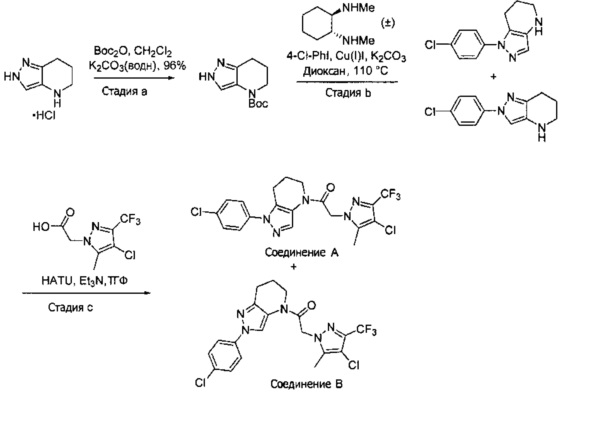
a) 4,5,6,7-Тетрагидро-2Н-пиразоло[4,3-b]пиридин⋅HCl (319 мг, 2 ммоль) объединяли с Boc2O (480 мг, 2,2 ммоль) и K2CO3 (1,3 мл, 3М водный раствор, 2 экв.) в CH2Cl2 (10 мл). Через 15 часов исходный амин исчез (LCMS). Реакционную суспензию промывали насыщенным водным раствором хлорида натрия, и органический слой сушили над MgSO4, фильтровали и упаривали, получая вязкое желтое масло (446 мг, 96%), которое использовали далее без дополнительной очистки. MS: (ES) m/z вычислено для C11H18N3O2 [М+Н]+ 224,1, найдено 224,2.
b) Полученный выше Вос-амин (112 мг, 0,5 ммоль) объединяли с 4-хлориодбензолом (239 мг, 1 ммоль), 6 мл диоксана, Cu(I)I (12 мг, 0,06 ммоль, 10%), (±)-транс-N,N'-диметилциклогексан-1,2-диамином (18,5 мг, 0,13 ммоль, 25%) и K2CO3 (138 мг, 1 ммоль). Полученную суспензию нагревали до 110°С в течение ночи. Два региоизомера получали примерно в равных количествах. Реакционную суспензию разбавляли 20 мл EtOAc и промывали насыщенным водным раствором хлорида натрия и дважды 1М раствором NaHSO4. Органический слой сушили над MgSO4, фильтровали и упаривали, получая оранжевое масло, которое использовали далее без дополнительной очистки.
c) Полученную выше смесь региоизомеров (66 мг, в сумме примерно 0,25 ммоль) объединяли с 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусной кислотой (67 мг, 0,275 ммоль), HATU (114 мг, 0,3 ммоль) и Et3N (52 мкл, 0,38 ммоль) в ТГФ (1 мл). Через 90 минут реакционную суспензию разбавляли EtOAc, промывали водой и упаривали. Полученный остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая два региоизомера. Элюирующееся первым, Соединение А: 1-[1-(4-хлорфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]этанон выделяли в виде белого твердого вещества (1,7 мг). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,36 (с, 0,8 Н), 7,59 (с, 0,2 Н), 7,43 (с, 4 Н), 5,24 (с, 0,4 Н), 5,13 (с, 1,6 Н), 3,92 (м, 0,4 Н), 3,85 (м, 1,6 Н), 2,90 (м, 1,8 Н), 2,77 (м, 0,2 Н), 2,34 (с, 2,4 Н), 2,32 (с, 0,6 Н), 2,13 (м, 1,5 Н), 2,05 (м, 0,5 Н); MS: (ES) m/z вычислено для C19H17Cl2F3N5O [М+Н]+ 458,1, найдено 458,0. Элюирующееся вторым, Соединение В: 1-(2-(4-хлорфенил)-6,7-дигидро-2H-пиразоло[4,3-b]пиридин-4(5Н)-ил)-2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)этанон выделяли в виде белого твердого вещества (31,0 мг). 1H-ЯМР (400 МГц, CDCl3) δ 8,56 (с, 1 Н), 7,56 (м, 2 Н), 7,36 (м, 2 Н), 5,16 (с, 2 Н), 3,84 (с, 2 Н), 2,93 (т, J=6,6 Гц, 2 Н), 2,32 (с, 3 Н), 2,19 (м, 2 Н); MS: (ES) m/z вычислено для C19H17Cl2F3N5O [М+Н]+ 458,1, найдено 458,0.
Пример 5b. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-пиразоло[3,4-b]пиридин-1-ил-этанона
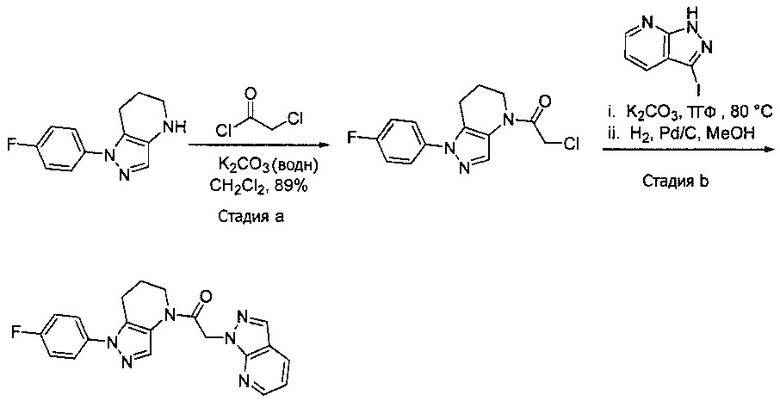
a) В колбу, содержащую 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридин (0,869 г, 4 ммоль) и 3М раствор K2CO3 (водн.) (2,6 мл, 8 ммоль) в 10 мл CH2Cl2, по каплям добавляли хлорацетилхлорид (0,35 мл, 4,4 ммоль). Через 20 минут суспензию разбавляли 5 мл H2O и разделяли слои. Органический слой промывали насыщенным водным раствором хлорида натрия и дважды 1М раствором NaHSO4, сушили над MgSO4, фильтровали и упаривали с получением коричневой твердой пены (1,05 г, 89%). MS: (ES) m/z вычислено для C14H14ClFN3O [М+Н]+ 294,1, найдено 294,1.
b) Полученный выше α-хлорацетамид (147 мг, 0,5 ммоль) разбавляли в 1,5 мл ТГФ и добавляли K2CO3 (138 мг, 1 ммоль) и 3-иод-1H-пиразоло[3,4-b]пиридин (130 мг, 0,53 ммоль). Полученную суспензию нагревали до 75°С. По окончании реакции добавляли в колбу 4 мл метанола и 50 мг 10% Pd/C. Реакционную смесь затем встряхивали в аппарате Парра под давлением водорода 50 фунт/кв. дюйм в течение ночи. Реакционную смесь фильтровали через целит, промывая метанолом, и упаривали. Полученный остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 30,9 мг продукта в виде белого кристаллического вещества. 1H-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,54 (дд, J=1,5, 4,4 Гц, 1 Н), 8,35 (с, 0,8 Н), 8,14 (с, 1 Н), 8,11 (м, 1H), 7,75 (с, 0,2 Н), 7,43-7,51 (м, 2 Н), 7,12-7,21 (м, 3 Н), 5,70 (с, 0,4 Н), 5,59 (с, 1,6 Н), 3,91 (м, 2 Н), 2,87 (м, 2 Н), 2,15 (м, 1,6 Н), 2,05 (м, 0,4 Н); MS: (ES) m/z вычислено для C20H18FN6O [М+Н]+ 377,1, найдено 377,1.
Пример 6. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-пиразоло[3,4-b]пиридин-1-ил-этанона
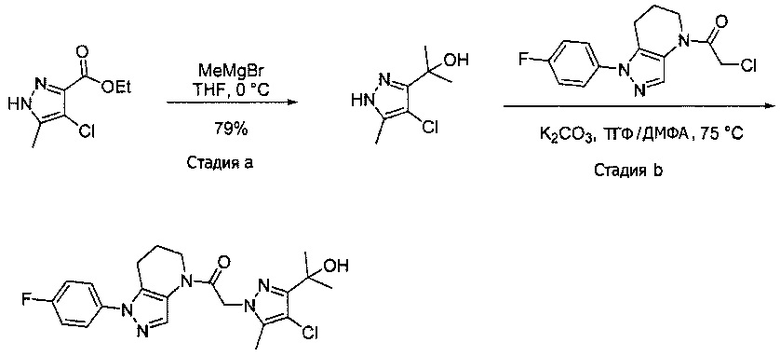
a) Закрытую септой виалу с магнитной мешалкой промывали безводным ТГФ, и помещали в нее этил 4-хлор-5-метил-1H-пиразол-3-карбоксилат (94 мг, 0,5 ммоль) и 1 мл сухого ТГФ. В атмосфере азота реакционный сосуд охлаждали на ледяной бане, после чего прикапывали 520 мкл MeMgBr (1,55 ммоль, 3М раствор в Et2O). Через 40 минут добавляли еще 150 мкл MeMgBr (0,45 ммоль). По окончании реакции, в смесь добавляли 3 мл 1М раствора NaHSO4 и трижды экстрагировали EtOAc. Органические слои объединяли, сушили над MgSO4, фильтровали и упаривали, получая желто-коричневое твердое вещество (69 мг, 79%), которое использовали далее без дополнительной очистки. MS: (ES) m/z вычислено для C7H12ClN2O [М+Н]+ 175,1, найдено 175,1.
b) Полученный выше карбинол (69 мг, 0,4 ммоль) разбавляли 1,2 мл ТГФ и 500 мкл ДМФА, и добавляли K2CO3 (111 мг, 0,8 ммоль) и 2-хлор-1-(1-(4-фторфенил)-6,7-дигидро-1Н-пиразоло[4,3-b]пиридин-4(5Н)-ил)этанон (118 мг, 0,4 ммоль). Полученную суспензию нагревали до 75°С. Реакционную суспензию разбавляли EtOAc и промывали насыщенным водным раствором хлорида натрия, сушили над MgSO4, фильтровали и упаривали. Полученный остаток очищали методом обращенно-фазной ВЭЖХ (С 18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 69 мг целевого продукта (43%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,37 (с, 0,8 Н), 7,63 (с, 0,2 Н), 7,42-7,46 (м, 2 Н), 7,13-7,19 (м, 2 Н), 5,14 (с, 0,4 Н), 5,03 (с, 1,6 Н), 3,90 (м, 0,4 Н), 3,87 (м, 1,6 Н), 2,86 (м, 2 Н), 2,28 (с, 2,4 Н), 2,25 (с, 0,6 Н), 2,05-2,13 (м, 2 Н), 1,62 (с, 6 Н); MS: (ES) m/z вычислено для C21H24ClFN5O2 [М+Н]+ 432,2, найдено 432,1.
Пример 7. Синтез соли 2-(4-хлор-5-метил-3-пиримидин-2-ил-пиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой и соли 2-(4-хлор-3-метил-5-пиримидин-2-ил-пиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой
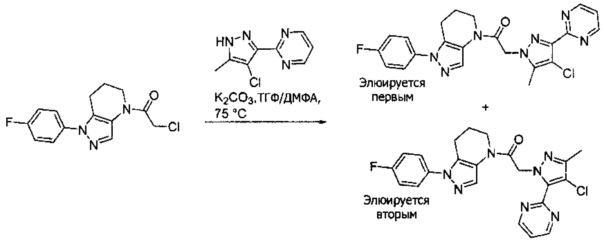
2-Хлор-1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанон (118 мг, 0,4 ммоль) разбавляли 1,2 мл смеси 2:1 ТГФ : ДМФА и добавляли K2CO3 (111 мг, 0,8 ммоль) и 2-(4-хлор-5-метил-1H-пиразол-3-ил)пиримидин (78 мг, 0,4 ммоль). Полученную суспензию нагревали до 75°С и перемешивали в течение ночи. Присутствовали два изомера с одинаковым молекулярным весом, по данным LCMS. Реакционную суспензию разбавляли EtOAc и промывали насыщенным водным раствором хлорида натрия, сушили над MgSO4, фильтровали и упаривали. Полученный остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая два региоизомера. Элюирующийся первым изомер: Соль 2-(4-Хлор-5-метил-3-пиримидин-2-ил-пиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой: получено 25 мг белых игольчатых кристаллов. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,86 (д, J=5,1 Гц, 2 Н), 8,13 (с, 1 Н), 7,59 (м, 2 Н), 7,44 (т, J=4,8 Гц, 1 Н), 7,34 (м, 2 Н), 5,50 (с, 2 Н), 3,84 (м, 2 Н), 2,87 (т, J=6,2 Гц, 2 Н), 2,24 (с, 3 Н), 2,02 (м, 2 Н); MS: (ES) m/z вычислено для C22H20ClFN7O [М+Н]+ 452,1, найдено 452,1. Элюирующийся вторым изомер: Соль 2-(4-Хлор-3-метил-5-пиримидин-2-ил-пиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой: получено 8,8 мг светло-оранжевого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6, смесь ротамеров) δ 8,87 (д, J=5,1, 1,6 Н), 8,81 (д, J=5,1 Гц, 0,4 Н), 7,96 (с, 1 Н), 7,66 (м, 0,4 Н), 7,56 (м, 1,6 Н), 7,42 (т, J=4,8, 1 Н), 7,33 (м, 2 Н), 5,72 (с, 2 Н), 3,84 (м, 2 Н), 2,85 (т, J=6,2 Гц, 2 Н), 2,23 (с, 3 Н), 1,99 (м, 2 Н); MS: (ES) m/z вычислено для C22H20ClFN7O [М+Н]+ 452,1, найдено 452,1.
Пример 8. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]этанона
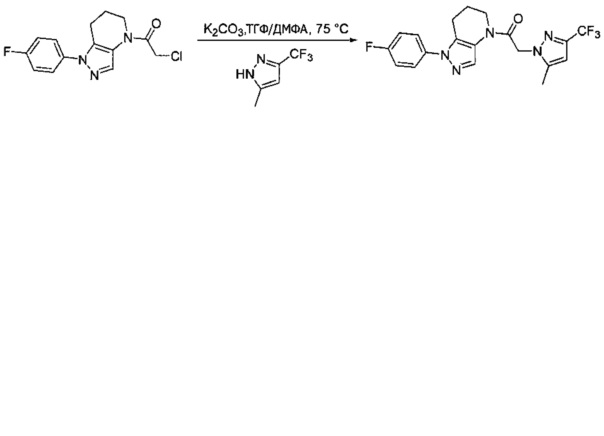
2-Хлор-1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанон (118 мг, 0,4 ммоль) разбавляли 1,2 мл смеси 2:1 ТГФ : ДМФА и добавляли K2CO3 (111 мг, 0,8 ммоль) и 5-метил-3-(трифторметил)-1H-пиразол (60 мг, 0,4 ммоль). Полученную суспензию нагревали до 75°С 2 часа. Реакционную суспензию очищали методом флэш-хроматографии на нормальной фазе (24 г колонка, элюирование 10-80% EtOAc в гексане), получая 109 мг (67%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,36 (с, 0,8 Н), 7,59 (с, 0,2 Н), 7,42-7,47 (м, 2 Н), 7,12-7,25 (м, 2 Н), 5,25 (с, 0,4 Н), 5,14 (с, 1,6 Н), 3,92 (м, 0,4 Н), 3,86 (м, 1,6 Н), 2,86 (т, J=6,2 Гц, 2 Н), 2,37 (с, 2,4 Н), 2,36 (м, 0,6 Н), 2,11 (м, 1,6 Н), 2,04 (м, 0,4 Н); MS: (ES) m/z вычислено для C19H18F4N5O [М+Н]+ 408,1, найдено 408,1.
Пример 9. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]пропан-1-она
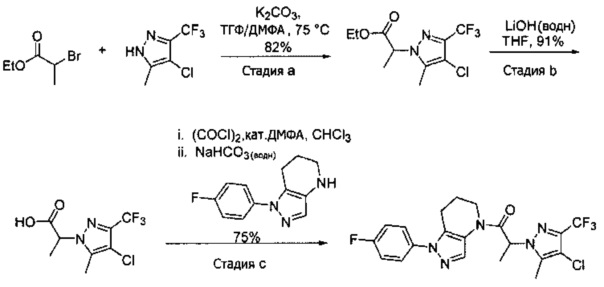
a) Этил бромпропионат (1,448 г, 8 ммоль), 4-хлор-5-метил-3-(трифторметил)-1H-пиразол (1,476 г, 8 ммоль) и K2CO3 (2,211 г, 16 ммоль) объединяли в 24 мл смеси 2:1 ТГФ : ДМФА и нагревали при 75°С в течение 3 часов. Летучие вещества упаривали, остаток суспендировали в 20 мл смеси 4:1 гексан : EtOAc и трижды промывали водой и дважды насыщенным водным раствором хлорида натрия. Органический слой сушили и очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 5-80% EtOAc в гексане), получая 1,859 г (82%) указанного в заголовке соединения в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4,95 (кв, J=7,3 Гц, 1 Н), 4,21 (кв, J=7,3 Гц, 2 Н), 2,27 (с, 3 Н), 1,83 (д, J=7,3 Гц, 3 Н), 1,81 (т, J=7,3 Гц, 1 Н); MS: (ES) m/z вычислено для C10H13ClF3N2O2 [М+Н]+ 285,1, найдено 285,0.
b) Полученный выше сложный эфир суспендировали в 13 мл ТГФ и 6,5 мл 2М раствора LiOH. По окончании реакции, летучие вещества упаривали и добавляли 9 мл 2М раствора НСl для высаживания продукта. Осадок промывали водой (3×5 мл) и сушили в вакууме до постоянного объема. Указанное в заголовке соединение получали в виде белого твердого вещества (1,53 г, 82%). MS: (ES) m/z вычислено для C8H9ClF3N2O2 [М+Н]+ 257,0, найдено 257,0.
c) Указанное в заголовке соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)пропановой кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 137 мг (75%) указанного в заголовке соединения в виде белой пены. 1Н-ЯМР (400 МГц, CDCl3) δ 8,41 (с, 1 Н), 7,43 (м, 2 Н), 7,15 (м, 2 Н), 5,52 (кв, J=7,0 Гц, 1 Н), 3,75 (м, 1 Н), 3,41 (м, 1 Н), 2,81 (м, 2 Н), 2,28 (с, 3 Н), 1,85-1,99 (м, 2 Н), 1,81 (д, J=7 Гц, 1 Н); MS: (ES) m/z вычислено для C20H19ClF4N5O [М+Н]+ 456,1, найдено 456,1.
Пример 10. Синтез 2-(4-хлорфенил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
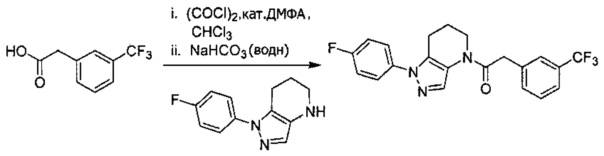
Данное соединение получали из 3-трифторметилфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 83 мг (69%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,45 (с, 1 Н), 7,42-7,55 (м, 6 Н), 7,14 (м, 2 Н), 4,08 (с, 0,3 Н), 3,95 (с, 1,7 Н), 3,77 (м, 2 Н), 2,83 (т, J=6,6 Гц, 2 Н), 2,01 (м, 2 Н); MS: (ES) m/z вычислено для C21H18F4N3O [М+Н]+ 404,1, найдено 404,1.
Пример 11. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-3-метил-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

a) В круглодонную колбу объемом 100 мл добавляли 4-фторфенилгидразин гидрохлорид (3,33 г, 20,5 ммоль), K2CO3 (8,29 г, 60 ммоль) и ДМФА (27 мл). В полученную суспензию добавляли 2,78 г (20 ммоль) 1-(3-фторпиридин-2-ил)этанона, получая желтую суспензию. Полученную смесь нагревали до 120°С 24 часа, после чего все исходные вещества исчезали. Полученную суспензию охлаждали до комнатной температуры и добавляли 70 мл H2O для высаживания продукта. Полученное твердое вещество собирали на стеклянном фильтре средней пористости и трижды промывали водой. Полученное твердое вещество сушили при 60°С, получая продукт в виде коричневого твердого вещества (3,82 г, 84%). MS: (ES) m/z вычислено для C13H11FN3 [М+Н]+ 228,1, найдено 228,1.
b) Метанол (80 мл), 20% Pd(OH)2/C (472 мг, около 0,33 ммоль) и 1-(4-фторфенил)-3-метил-1H-пиразоло[4,3-b]пиридин (3,82 г, 16,8 ммоль) объединяли в аппарате Парра и продували водородом. Колбу заполняли водородом под давлением 55 фунт/кв. дюйм H2 и затем встряхивали полученную суспензию в течение ночи. Полученную смесь фильтровали через целит, промывая несколькими порциями метанола. Фильтрат затем упаривали, получая 3,73 г (96% выход неочищенного продукта) оранжевого масла, затвердевающего при стоянии. MS: (ES) m/z вычислено для C13H15FN3 [М+Н]+ 232,1, найдено 232,1.
c) Указанное в заголовке соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусной кислоты и 1-(4-фторфенил)-3-метил-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 75 мг (41%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 7,47 (м, 0,8 Н), 7,40 (м, 1,2 Н), 7,14 (м, 2 Н), 5,11 (с, 1,2 Н), 5,01 (с, 0,8 Н), 3,81 (м, 2 Н), 2,85 (м, 2 Н), 2,29-2,40 (м, 6 Н), 2,07 (м, 2 Н); MS: (ES) m/z вычислено для C20H19ClF4N5O [М+Н]+ 456,1, найдено 456,1.
Пример 12. Синтез 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(2-(трифторметил)фенил)этанона
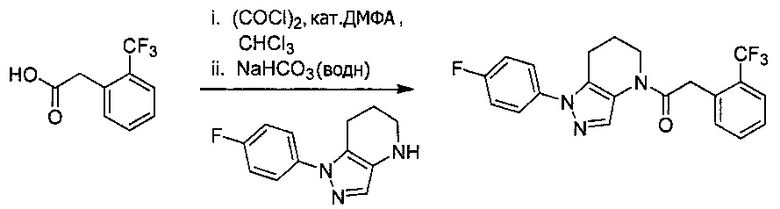
Данное соединение получали из 2-трифторметилфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 72 мг (59%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,46 (с, 0,8 Н), 7,70 (м, 1 Н), 7,55 (м, 1,2 Н), 7,41-7,50 (м, 4 Н), 7,15 (м, 2 Н), 4,19 (с, 0,3 Н), 4,06 (с, 1,7 Н), 3,95 (м, 0,3 Н), 3,76 (м, 1,7 Н), 2,82-2,88 (м, 2 Н), 2,03 (м, 2 Н); MS: (ES) m/z вычислено для C21H18F4N3O [М+Н]+ 404,1, найдено 404,1.
Пример 13. Синтез 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(4-(трифторметил)фенил)этанона

Данное соединение получали из 4-трифторметилфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 77 мг (64%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,45 (с, 0,8 Н), 7,62 (д, J=7,8 Гц, 2 Н), 7,55 (с, 0,2 Н), 7,40-7,49 (м, 4 Н), 7,15 (м, 2 Н), 4,08 (с, 0,3 Н), 3,96 (м, 2 Н), 3,75 (м, 1,7 Н), 2,80-2,86 (м, 2 Н), 2,00 (м, 2 Н); MS: (ES) m/z вычислено для C21H18F4N3O [М+Н]+ 404,1, найдено 404,1.
Пример 14. Синтез 2-(4-хлор-3-(трифторметил)фенил)-1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанона
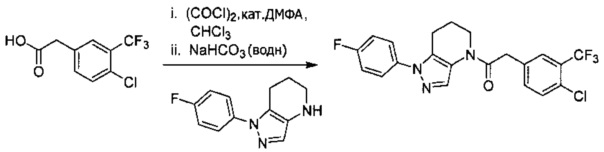
Данное соединение получали из 4-хлор-3-трифторметилфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов промывали насыщенным водным раствором хлорида натрия и дважды 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 54 мг (41%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,44 (с, 0,8 Н), 7,59 (м, 1 Н), 7,57 (с, 0,2 Н), 7,41-7,51 (м, 4 Н), 7,17 (м, 2 Н), 4,03 (с, 0,3 Н), 3,95 (м, 0,3 Н), 3,91 (с, 1,7 Н), 3,78 (м, 1,7 Н), 2,82-2,85 (м, 2 Н), 2,05 (м, 2 Н); MS: (ES) m/z вычислено для C21H17ClF4N3O [М+Н]+ 438,1, найдено 438,1.
Пример 15. Синтез 2-(3-хлор-4-(трифторметил)фенил)-1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанона
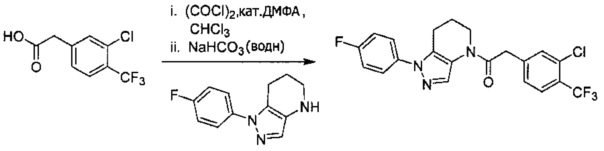
Данное соединение получали из 3-хлор-4-трифторметилфенилуксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов промывали насыщенным водным раствором хлорида натрия и дважды 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-60% EtOAc в гексане), получая 68 мг (52%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,45 (с, 0,8 Н), 7,67 (м, 1 Н), 7,55 (с, 0,2 Н), 7,44-7,49 (м, 3 Н), 7,30 (м, 1 Н), 7,14-7,21 (м, 2 Н), 4,04 (с, 0,4 Н), 3,94 (м, 0,4 Н), 3,92 (с, 1,6 Н), 3,77 (м, 1,6 Н), 2,82-2,87 (м, 2 Н), 2,04 (м, 2 Н); MS: (ES) m/z вычислено для C21H17ClF4N3O [М+Н]+ 43 8,1, найдено 438,1.
Пример 16. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-3-метил-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
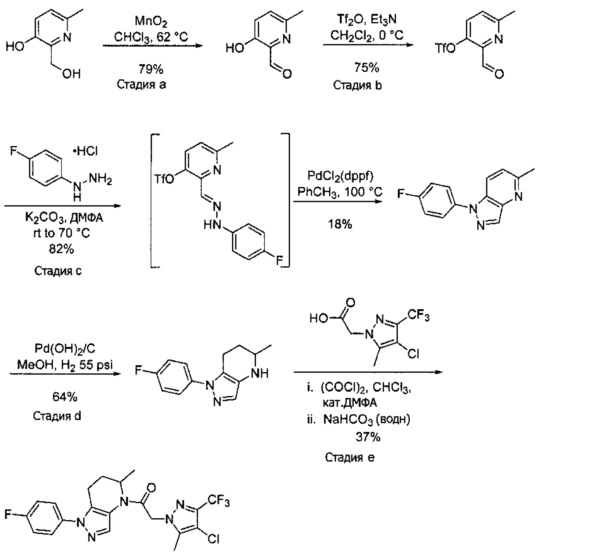
а) 2,6-Лутидин-α-2,3-диол (4,17 г, 30 ммоль) растворяли в 60 мл CHCl3 и добавляли MnO2 (15,65 г, 180 ммоль). Полученную суспензию нагревали до 62°С на 90 минут, после чего реакция была завершена. Реакционную смесь фильтровали через тонкий слой целита и промывали несколькими порциями CHCl3. Фильтрат упаривали, получая продукт в виде коричневого твердого вещества (3,24 г, 79%). MS: (ES) m/z вычислено для C7H8NO2 [М+Н]+ 138,0, найдено 138,0.
b) Полученный выше альдегид растворяли в CH2Cl2 (20 мл) и Et3N (2,3 мл, 16,5 ммоль). Прикапывали ангидрид трифторметансульфокислоты (Tf2O, 2,78 мл, 16,5 ммоль) в течение 5 минут. Через 30 минут добавляли еще Et3N (500 мкл) и Tf2O (200 мкл). Через 75 минут, реакционную смесь дважды промывали водой и дважды насыщенным раствором NaHCO3. Органический слой сушили над MgSO4, фильтровали и упаривали, получая 3,02 г (75%) оранжевого масла, которое напрямую использовали в следующей стадии.
c) Описанный выше трифлат (1,08 г, 4 ммоль) растворяли в 5 мл ДМФА в закрытой септой виале. Добавляли карбонат калия (1,66 г, 12 ммоль) и 4-фторфенилгидразин гидрохлорид (678 мг, 4,17 ммоль) и полученную суспензию перемешивали при комнатной температуре, в ходе перемешивания она приобретала глубокий оранжево-красный цвет. Через 3 часа LCMS показал полное исчезновение исходного вещества с формированием гидразинового интермедиата (с трифлатом и без трифлата). В полученную реакционную смесь добавляли 400 мг (0,55 ммоль) PdCl2(dppf) и 5 мл PhCH3. Колбу помещали в термошкаф с температурой 100°С на ночь. Реакционную смесь выливали в 100 мл Н2О и 80 мл EtOAc. Полученную смесь фильтровали через слой целита и разделяли слои. Органический слой упаривали, и остаток очищали методом флэш-хроматографии на нормальной фазе (40 г колонка, элюирование 15-70% EtOAc в гексане), получая 160 мг (18%) указанного в заголовке соединения в виде маслянистого оранжевого твердого вещества. MS: (ES) m/z вычислено для C13H11FN3 [М+Н]+ 228,1, найдено 228,1.
d) Метанол (3,5 мл), 20% Pd(OH)2/C (200 мг, около 0,14 ммоль) и 1-(4-фторфенил)-5-метил-1H-пиразоло[4,3-b]пиридин (160 мг, 0,7 ммоль) объединяли в аппарате Парра и дважды продували водородом. Колбу заполняли водородом под давлением 55 фунт/кв. дюйм и встряхивали полученную суспензию 2 дня. Полученную смесь фильтровали через целит, промывая несколькими порциями метанола. Фильтрат упаривали до 103 мг (64% выход неочищенного продукта) маслянистого оранжевого твердого вещества. MS: (ES) m/z вычислено для C13H15FN3 [М+Н]+ 232,1, найдено 232,1.
e) Данное соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусной кислоты и 1-(4-фторфенил)-5-метил-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 37 мг (37%) указанного в заголовке соединения в виде белой пены. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,37 (с, 0,8 Н), 7,58 (с, 0,2 Н), 7,49 (м, 2 Н), 7,17 (м, 2 Н), 5,08-5,31 (м, 2 Н), 4,51 (м, 1 Н), 2,78-2,99 (м, 2 Н), 2,34 (с, 2,3 Н), 2,31 (с, 0,7 Н), 1,97-2,09 (м, 2 Н), 1,36 (д, J=6,7 Гц, 2,4 Н), 1,15 (д, J=6,6 Гц, 0,6 Н); MS: (ES) m/z вычислено для C20H19ClF4N5O [М+Н]+ 456,1, найдено 456,1.
Общая схема для примеров 17-20:
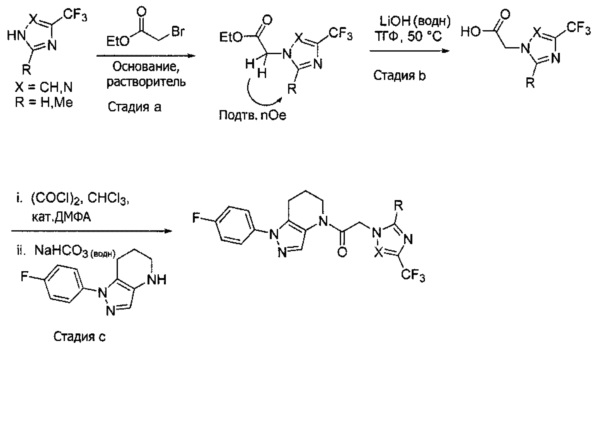
Пример 17. Синтез соли 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(4-(трифторметил)-1H-имидазол-1-ил)этанона с трифторуксусной кислотой.
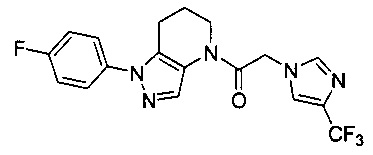
а) В виалу добавляли 4-трифторметил-1H-имидазол (953 мг, 7 ммоль), 7 мл этанола и этоксид натрия (524 мг, 7,7 ммоль). В полученную смесь медленно добавляли этилбромацетат (1,29 г, 7,7 ммоль). Через 18 часов добавляли еще 100 мкл этилбромацетата и примерно 100-200 мг NaOEt. Через 90 минут реакцию гасили добавлением NaHCO3 и трижды экстрагировали EtOAc. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 15-90% EtOAc в гексане), получая 746 мг (48%) указанного в заголовке соединения в виде твердого кристаллического вещества. MS: (ES) m/z вычислено для C8H10F3N2O2 [М+Н]+ 223,1, найдено 223,1.
b) Полученный выше сложный эфир (746 мг, 3,35 ммоль) растворяли в 10 мл ТГФ и добавляли 3,5 мл 2М раствора LiOH (водн.). Полученную смесь нагревали на бане с температурой 50°С 5 часов. Летучие вещества упаривали и добавляли 1 мл 6н. раствора HCl (водн.) и 1 мл 1М раствора NaHSO4. Выпавший осадок собирали и промывали водой, растворяли в EtOAc, и упаривали до получения белого твердого вещества (200 мг). Водные промывные растворы упаривали до половины объема и получали вторую порцию продукта (141 мг). 1Н-ЯМР (400 МГц, CD3OD) δ 7,80 (с, 1 Н), 7,64 (с, 1 Н), 4,94 (с, 2 Н); MS: (ES) m/z вычислено для C6H6F3N2O2 [М+Н]+ 195,0, найдено 195,1. Региохимию системы подтверждали экспериментом nOe между α-метиленовым протоном и обоими протонами имидазольного кольца.
c) Указанное в заголовке соединение получали из 2-(4-(трифторметил)-1H-имидазол-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали насыщенным водным раствором хлорида натрия. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 29 мг указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 8,25 (с, 0,9 Н), 7,85 (с, 0,1 Н), 7,82 (с, 1 Н), 7,65 (с, 1 Н), 7,55 (м, 2 Н), 7,27 (м, 2 Н), 5,45 (с, 0,2 Н), 5,33 (с, 1,8 Н), 3,90 (м, 0,2 Н), 3,87 (м, 1,8 Н), 2,88 (т, J=6,6 Гц, 2 Н), 2,13 (м, 1,8 Н), 2,03 (м, 0,2 Н); MS: (ES) m/z вычислено для C18H16F4N5O [М+Н]+ 394,1, найдено 394,1.
Пример 18. Синтез соли 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(2-метил-4-(трифторметил)-1H-имидазол-1-ил)этанона с трифторуксусной кислотой.

а) В виалу добавляли 2-метил-4-(трифторметил)-1H-имидазол (750 мг, 5 ммоль), 5 мл ТГФ, 2,5 мл ДМФА и K2CO3 (2,07 г, 15 ммоль). В полученную смесь медленно добавляли этилбромацетат (1,00 г, 6 ммоль). Через 2 часа полученную суспензию разбавляли EtOAc, фильтровали через целит и упаривали. Полученный остаток очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 15-100% EtOAc в гексане), получая 476 мг (40%) указанного в заголовке соединения в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 7,20 (с, 1 Н), 4,61 (с, 2 Н), 4,28 (кв, J=7,0 Гц, 2 Н), 1,63 (с, 3 Н), 1,31 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C9H12F3N2O2 [М+Н]+ 237,1, найдено 237,1. Региохимию системы подтверждали экспериментом nOe между α-метиленовым протоном и протонами имидазольного кольца и метила.
b) Полученный выше сложный эфир (476 мг, 2 ммоль) растворяли в 6 мл ТГФ и добавляли 2 мл 2М раствора LiOH, полученную смесь перемешивали при комнатной температуре в течение ночи. Затем упаривали летучие компоненты и добавляли 670 мкл 6н. раствора HCl (водн.). Выпавший осадок собирали, промывали водой и сушили в вакууме до постоянного объема, получая указанное в заголовке соединение в виде белого порошка (284 мг, 68%). MS: (ES) m/z вычислено для C6H6F3N2O2 [М+Н]+ 209,0, найдено 209,0.
c) Указанное в заголовке соединение получали из 2-(2-метил-4-(трифторметил)-1H-имидазол-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов разбавляли 1 мл CHCl3 и промывали водой. Органический слой упаривали, и остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 31 мг указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 8,24 (с, 0,9 Н), 7,88 (с, 0,1 Н), 7,52-7,60 (м, 3 Н), 7,24-7,32 (м, 2 Н), 5,36 (с, 0,2 Н), 5,26 (с, 1,8 Н), 3,89 (м, 2 Н), 2,89 (т, J=6,2 Гц, 2 Н), 2,38 (с, 2,7 Н), 2,36 (м, 0,3 Н), 2,14 (м, 2 Н); MS: (ES) m/z вычислено для C19H18F4N5O [М+Н]+ 408,1, найдено 408,1.
Пример 19. Синтез 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(3-(трифторметил)-1H-1,2,4-триазол-1-ил)этанона
а) В виалу добавляли 3-(трифторметил)-1H-1,2,4-триазол (358 мг, 2,6 ммоль), 2,6 мл этанола и этоксид натрия (354 мг, 5,2 ммоль). В полученную смесь медленно добавляли этилбромацетат (320 мкл, 2,9 ммоль). Через 18 часов реакционную смесь упаривали и подкисляли добавлением 1 мл 6н. раствора HCl (водн.). Полученную суспензию разбавляли добавлением EtOAc и Н2О и разделяли слои. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 40 г колонка, элюирование 2-80% EtOAc в гексане), получая 423 мг (73%) указанного в заголовке соединения в виде твердого кристаллического вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,30 (с, 1 Н), 5,05 (с, 2 Н), 4,29 (кв, J=7,3 Гц, 2 Н), 1,32 (т, J=7,1 Гц, 3 Н); MS: (ES) m/z вычислено для C8H10F3N2O2 [М+Н]+ 224,1, найдено 224,1. Региохимию системы подтверждали экспериментом nOe между α-метиленовым протоном и протоном имидазольного кольца.
b) Полученный выше сложный эфир (142 мг, 0,64 ммоль) растворяли в 2 мл ТГФ и добавляли 650 мкл 2М раствора LiOH (водн.). Полученную смесь перемешивали при 50°С 75 минут. Летучие вещества упаривали и добавляли 500 мкл 6н. раствора HCl (водн.). Полученный раствор дважды экстрагировали EtOAc, сушили над MgSO4, фильтровали и упаривали, получая указанное в заголовке соединение в виде бесцветного масла (108 мг, 76%). MS: (ES) m/z вычислено для C5H3F3N3O2 [М-Н]- 194,1, найдено 194,1.
c) Указанное в заголовке соединение получали из 2-(3-(трифторметил)-1H-1,2,4-триазол-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали насыщенным водным раствором хлорида натрия. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 33 мг указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 8,41 (с, 1 Н), 8,35 (с, 0,75 Н), 7,60 (с, 0,25 Н), 7,43-7,50 (м, 2 Н), 7,15-7,23 (м, 2 Н), 5,41 (с, 0,5 Н), 5,30 (с, 1,5 Н), 3,94 (м, 0,5 Н), 3,84 (м, 1,5 Н), 2,89 (м, 2 Н), 2,15 (м, 1,5 Н), 2,07 (м, 0,5 Н); MS: (ES) m/z вычислено для C17H15F4N6O [М+Н]+ 395,1, найдено 395,1.
Пример 20. Синтез 1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)-2-(5-метил-3-(трифторметил)-1H-1,2,4-триазол-1-ил)этанона

а) В колбу помещали 5-метил-3-(трифторметил)-1H-1,2,4-триазол (2,27 г, 15 ммоль), 15 мл EtOH и этоксид натрия (2,04 г, 30 ммоль). В полученную смесь медленно добавляли этилбромацетат (2,76 г, 16,5 ммоль). Через 5 часов реакцию гасили добавлением 3 мл 6н. раствора HCl (водн.) и доводили значение pH до 7 насыщенным раствором NaHCO3. Упаривали до половины объема и разбавляли полученную суспензию добавлением 30 мл EtOAc. Разделяли слои и промывали органический слой водой. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 5-60% EtOAc в гексане), получая 2,00 г (56%) указанного в заголовке соединения в виде светло-желтого масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4,91 (с, 2 Н), 4,28 (кв, J=7,0 Гц, 2 Н), 2,50 (с, 3 Н), 1,31 (т, J=7,3 Гц, 3 Н); MS: (ES) m/z вычислено для C8H11F3N3O2 [М+Н]+ 238,1, найдено 238,1. Региохимию системы подтверждали экспериментом nOe между α-метиленовым протоном и протонами триазольного метила.
b) Полученный выше сложный эфир (2,00 г, 8,4 ммоль) растворяли в 10 мл ТГФ и добавляли 8 мл 2М раствора LiOH (водн.). Полученную смесь перемешивали при 50°С 2 часа. Летучие вещества упаривали и добавляли 2,5 мл 6н. раствора HCl (водн.). Полученную суспензию экстрагировали 20 мл EtOAc, сушили над MgSO4, фильтровали и упаривали, получая указанное в заголовке соединение (1,59 г, 90%) в виде желтого масла, затвердевающего при стоянии. MS: (ES) m/z вычислено для C6H5F3N3O2 [М-Н]- 208,0, найдено 208,1.
c) Указанное в заголовке соединение получали из 2-(5-метил-3-(трифторметил)-1H-1,2,4-триазол-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов разбавляли 1 мл CHCl3 и промывали водой. Органический слой упаривали, и остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 37 мг указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,34 (с, 0,8 Н), 7,59 (с, 0,2 Н), 7,44-7,50 (м, 2 Н), 7,14-7,27 (м, 2 Н), 5,29 (с, 0,4 Н), 5,18 (с, 1,6 Н), 3,93 (м, 0,4 Н), 3,86 (м, 1,6 Н), 2,88 (м, 2 Н), 2,56 (с, 2,4 Н), 2,55 (м, 0,6 Н), 2,14 (м, 1,6 Н), 2,06 (м, 0,4 Н); MS: (ES) m/z вычислено для C18H17F4N6O [М+Н]+ 409,1, найдено 409,1.
Пример 21. Синтез 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-1-(1-(5-фторпиридин-2-ил)-6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанона
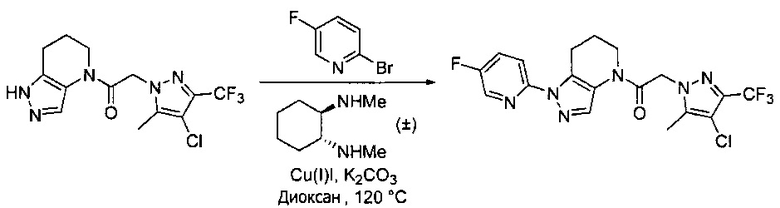
2-(4-Хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-1-(6,7-дигидро-1H-пиразоло[4,3-b]пиридин-4(5H)-ил)этанон (104 мг, 0,3 ммоль) объединяли с 5-фтор-2-бромпиридином (106 мг, 0,6 ммоль), 0,3 мл диоксана, Cu(I)I (11,4 мг, 0,06 ммоль, 20%), (±)-транс-N,N'-диметилциклогексан-1,2-диамином (21,3 мг, 0,15 ммоль, 50%) и K2CO3 (83 мг, 0,6 ммоль). Полученную суспензию нагревали до 120°С до завершения реакции (примерно 3 часа). Реакционную смесь разбавляли добавлением 4 мл EtOAc и дважды промывали водой. Органический слой упаривали, и остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая указанное в заголовке соединение в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,91 (с, 0,8 Н), 8,33 (д, J=2,7 Гц, 0,2 Н), 8,28 (д, J=2,9 Гц, 0,8 Н), 7,87-7,94 (м, 1 Н), 7,68-7,77 (м, 1 Н), 5,42 (с, 1,6 Н), 5,41 (с, 0,4 Н), 3,93 (с, 1,6 Н), 3,88 (с, 0,4 Н), 2,88 (т, J=6,4 Гц, 2 Н), 2,29 (с, 0,6 Н), 2,28 (с, 2,4 Н), 2,18 (м, 2 Н); MS: (ES) m/z вычислено для C18H16ClF4N6O [М+Н]+ 443,1, найдено 443,0.
Пример 22. Синтез этил 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропаноата и этил 3-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропаноата
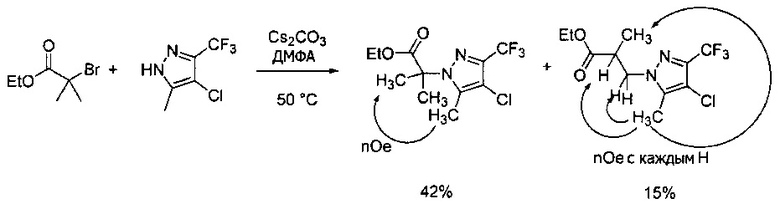
К 4-хлор-5-метил-3-(трифторметил)-1H-пиразолу (1,846 г, 2,22 мл, 10 ммоль) и карбонату цезия (6,516 г, 20 ммоль), суспендированным в ДМФА (20 мл), в один прием добавляли этил α-бромизобутират (2,926 г, 15 ммоль). Колбу помещали на масляную баню и нагревали систему до 50°С. Через 2 часа добавляли еще этил α-бромизобутират (1 мл) и продолжали нагревание. По окончании реакции, которое определяли по исчезновению пиразола, полученную суспензию разбавляли этилацетатом и фильтровали через целит, промывая еще 150 мл этил ацетата. Фильтрат трижды промывали насыщенным водным раствором хлорида натрия (полунасыщенный), сушили над MgSO4, фильтровали и упаривали с получением желтого масла. Полученный продукт очищали методом флэш-хроматографии (SiO2, 150 г колонка, элюирование 5-60% EtOAc в гексане), получая 1,24 г (42%) геминального диметильного соединения и 434 мг (15%) α-метильного соединения в виде бесцветных масел. Этил 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропаноат. 1Н-ЯМР (400 МГц, CDCl3) δ 4,25 (кв, J=7,1 Гц, 2 Н), 2,19 (с, 3 Н), 1,81 (с, 6 Н), 1,27 (т, J=7,1 Гц, 3 Н); MS: (ES) m/z вычислено для C11H15ClF3N2O2 [М+Н]+ 299,1, найдено 299,0. Региохимию системы подтверждали экспериментом nOe между α-метиленовым протоном и протонами пиразольного метила. Этил 3-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропаноат. 1Н-ЯМР (400 МГц, CDCl3) δ 4,35 (дд, J=8,2, 13,8 Гц, 1 Н), 4,11 (кв, J=7,1 Гц, 2 Н), 4,02 (дд, J=6,5, 13,8 Гц, 1 Н), 3,18 (м, 1 Н), 2,30 (с, 3 Н), 1,21 (т, J=7,0 Гц, 3 Н), 1,21 (д, J=7,1 Гц, 3 Н); MS: (ES) m/z вычислено для C11H15ClF3N2O2 [М+Н]+ 299,1, найдено 299,0. Региохимию системы подтверждали экспериментом nOe между протонами пиразольного метила и соседними протонами.
Пример 23. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-метил-пропан-1-она
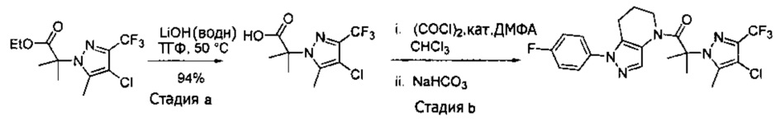
a) К этил 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропаноату (1,24 г, 4,2 ммоль) добавляли 12 мл ТГФ и 4,2 мл 2М раствора LiOH (водн.). Полученную суспензию нагревали при 50°С 80 минут и затем уменьшали объем до половины исходного. Полученный раствор подкисляли добавлением HCl, получая белый осадок, который промывали 1М раствором NaHSO4 и дважды водой. После сушки получали 1,06 г (94%) указанного в заголовке соединения. MS: (ES) m/z вычислено для C9H11ClF3N2O2 [М+Н]+ 271,0, найдено 271,0.
b) Указанное в заголовке соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-2-метилпропановой кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 80 мг (68%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (м, 2 Н), 3,08 (м, 2 Н), 2,74 (т, J=6,6 Гц, 2 Н), 2,21 (с, 3 Н), 1,91 (с, 6 Н), 1,76 (ддд, J=5,8, 6,2, 10,5 Гц, 2 Н); MS: (ES) m/z вычислено для C21H21ClF4N5O [М+Н]+ 470,1, найдено 470,1.
Общая схема для примеров 24 и 25

Пример 24. Синтез 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-1-(1-(4-фторфенил)-6,7-дигидро-1H-пиразоло[4,3-b]пирвдин-4(5H)-ил)бутан-1-она
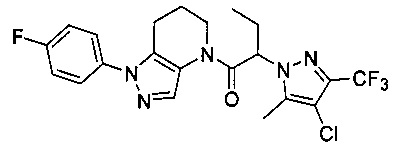
a) Метил бромбутират (996 мг, 5,5 ммоль), 4-хлор-5-метил-3-(трифторметил)-1H-пиразол (923 мг, 5 ммоль) и K2CO3 (1,382 г, 10 ммоль) объединяли в 15 мл смеси 2:1 ТГФ : ДМФА при 50°С и выдерживали в течение ночи. Твердый осадок отфильтровывали, промывали этилацетатом и упаривали летучие вещества. Полученный остаток разбавляли этилацетатом, трижды промывали водой и дважды насыщенным водным раствором хлорида натрия. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 40 г колонка, элюирование 2-50% EtOAc в гексане), получая 1,06 г (74%) продукта в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4,27 (дд, J=6,7, 9,0 Гц, 1 Н), 3,74 (с, 3 Н), 2,34 (м, 2 Н), 2,27 (с, 3 Н), 0,89 (т, J=6,5 Гц, 3 Н); MS: (ES) m/z вычислено для C10H13ClF3N2O2 [М+Н]+ 285,1, найдено 285,0. Региохимию системы подтверждали экспериментом nOe между α-метиновым протоном и протонами пиразольного метила (и протонами этильной группы).
b) Полученный выше сложный эфир (1,06 г, 3,72 ммоль) суспендировали в 10 мл ТГФ и 3,7 мл 2М раствора LiOH (водн.) при 50°С. Через 70 минут летучие вещества упаривали и добавляли 1,25 мл 6М HCl (водн.). Полученную суспензию дважды экстрагировали CH2Cl2 и сушили органический слой над MgSO4, фильтровали и упаривали с получением светло-желтого твердого вещества (0,844 мг, 84%). MS: (ES) m/z вычислено для C9H11ClF3N2O2 [М+Н]+ 270,0, найдено 270,0.
c) Указанное в заголовке соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)бутановой кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 81 мг (69%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,43 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (м, 2 Н), 5,30 (дд, J=6,7, 8,6 Гц, 1 Н), 3,81 (ддд, J=3,2, 8,6, 11,5 Гц, 1 Н), 3,50 (ддд, J=3,1, 7,4, 10,2 Гц, 1 Н), 2,79 (дт, J=2,8, 6,3 Гц, 2 Н), 2,33 (м, 1 Н), 2,30 (с, 3 Н), 2,22 (м, 1 Н), 1,96 (м, 1 Н), 1,86 (м, 1 Н), 0,98 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C20H19ClF4N5O [М+Н]+ 470,1, найдено 470,1.
Пример 25. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-метил-бутан-1-она

a) Этил 2-бром-3-метилбутаноат (836 мг, 4 ммоль), 4-хлор-5-метил-3-(трифторметил)-1H-пиразол (738 мг, 4 ммоль) и K2CO3 (1,106 г, 8 ммоль) объединяли в 12 мл смеси 2:1 ТГФ : ДМФА при 50°С на ночь. Добавляли еще 400 мкл алкилирующего агента и продолжали перемешивание в течение ночи. Твердый осадок отфильтровывали, промывая этилацетатом, и летучие вещества упаривали. Полученный остаток разбавляли этилацетатом и дважды промывали насыщенным водным раствором хлорида натрия (полунасыщенный). Органический слой упаривали, и остаток очищали методом флэш-хроматографии на нормальной фазе (40 г колонка, элюирование 5% EtOAc в гексане), получая 870 мг (70%) указанного в заголовке соединения в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4,42 (д, J=10,2 Гц, 1 Н), 4,20 (кв, J=7,4 Гц, 2 Н), 2,83 (м, 1 Н), 2,31 (с, 3 Н), 1,24 (т, J=7,4 Гц, 3 Н), 1,09 (д, J=6,7 Гц, 3 Н), 0,82 (д, J=6,6 Гц, 3 Н); MS: (ES) m/z вычислено для C12H17ClF3N2O2 [М+Н]+ 313,1, найдено 313,1. Региохимию системы подтверждали экспериментом nOe между α-метиновым протоном и протонами пиразольного метила (а также протонами изопропильной группы).
b) Полученный выше сложный эфир (870 мг, 2,8 ммоль) суспендировали в 8,3 мл ТГФ и 2,8 мл 2М раствора LiOH (водн.) при 50°С. Через 80 минут летучие вещества упаривали и добавляли 1 мл 6М HCl (водн.). Выпавший осадок собирали, трижды промывали водой и сушили, получая указанное в заголовке соединение в виде белого твердого вещества (844 мг, 94%). MS: (ES) m/z вычислено для C10H13ClF3N2O2 [М+Н]+ 285,1, найдено 285,0.
c) Указанное в заголовке соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)-3-метилбутановой кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-50% EtOAc в гексане), получая 89 мг (74%) указанного в заголовке соединения в виде стекловидного белого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (м, 2 Н), 5,03 (д, J=10,9 Гц, 1 Н), 3,89 (ддд, J=2,7, 8,2, 10,9 Гц, 1 Н), 3,78 (ддд, J=2,7, 7,8, 10,9 Гц, 1 Н), 2,96 (м, 1 Н), 2,79 (м, 2 Н), 2,37 (с, 3 Н), 1,99 (м, 1 Н), 1,82 (м, 1 Н), 1,12 (д, J=6,3 Гц, 3 Н), 0,79 (д, J=7,1 Гц, 3 Н); MS: (ES) m/z вычислено для C22H23ClF4N5O [М+Н]+ 484,1, найдено 484,1.
Пример 26. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)пиразоло[3,4-b]пиридин-1-ил]этанона
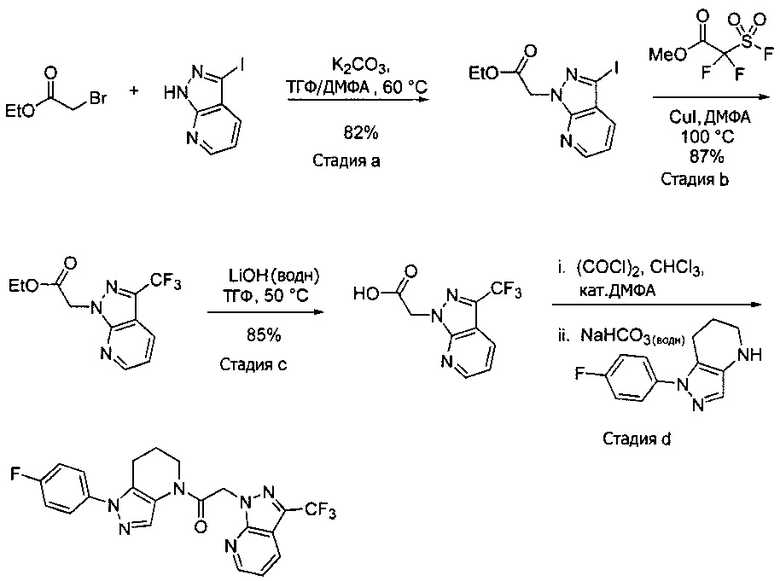
a) Этилбромацетат (1,336 г, 8 ммоль), 3-иод-1H-пиразоло[3,4-b]пиридин (1,953 мг, 8 ммоль) и K2CO3 (2,211 г, 16 ммоль) объединяли в 32 мл смеси 2:1 ТГФ : ДМФА при 60°С. Через 4 часа удаляли летучие компоненты, остаток разбавляли добавлением 50 мл EtOAc, и полученную суспензию трижды промывали насыщенным водным раствором хлорида натрия (полунасыщенный). Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 5-20% EtOAc в гексане), получая 2,172 г (82%) продукта в виде белых твердых гранул. MS: (ES) m/z вычислено для C10H11IN3O2 [М+Н]+ 332,0, найдено 332,0.
b) Метил фторсульфонилдифторацетат (MFSDA, 1,25 мл, 10,5 ммоль) прикапывали к суспензии этил 2-(3-иод-1Н-пиразоло[3,4-b]пиридин-1-ил)ацетата (994 мг, 3 ммоль), CuI (571 мг, 3 ммоль) и ДМФА (30 мл). Полученную смесь нагревали до 100°С на 1 час и разбавляли добавлением 50 мл EtOAc. Реакционную смесь фильтровали через слой целита, промывая дополнительным количеством этилацетата. Фильтрат упаривали до половины объема и добавляли равный объем насыщенного водного раствора хлорида натрия, что приводило к формированию осадка. Полученное твердое вещество собирали, промывали водой и сушили при 60°С, получая указанное в заголовке соединение в виде объемного кристаллического вещества (1,366 г, 87%).
c) Полученный выше сложный эфир (546 мг, 2 ммоль) суспендировали в 6 мл ТГФ и 1,5 мл 2М раствора LiOH (водн.) при 50°С. После завершения реакции, летучие вещества упаривали и добавляли 3 мл 1М раствор NaHSO4 (водн.). Выпавший осадок собирали, трижды промывали водой и сушили, получая указанное в заголовке соединение в виде белого порошка (415 мг, 85%). MS: (ES) m/z вычислено для C9H7F3N3O2 [М+Н]+ 246,0, найдено 246,0.
d) Указанное в заголовке соединение получали из 2-(3-(трифторметил)-1H-пиразоло[3,4-b]пиридин-1-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Реакционную суспензию очищали методом флэш-хроматографии (SiO2, 24 г колонка, элюирование 5-70% EtOAc в гексане), получая 58 мг указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,65 (дд, J=2,6, 4,7 Гц, 1 Н), 8,32 (с, 0,8 Н), 8,24 (д, J=8,2 Гц, 1 Н), 7,72 (с, 0,2 Н), 7,44-7,52 (м, 2 Н), 7,33 (дд, J=4,3, 8,2 Гц, 1 Н), 7,13-7,23 (м, 2 Н), 5,76 (с, 0,4 Н), 5,65 (с, 1,6 Н), 3,93 (м, 2 Н), 2,89 (т, J=6,3 Гц, 2 Н), 2,17 (м, 1,6 Н), 2,06 (м, 0,4 Н); MS: (ES) m/z вычислено для C21H17F4N6O [М+Н]+ 445,1, найдено 445,1.
Пример 27. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)-4,5,6,7-тетрагидропиразоло[3,4-b]пиридин-1-ил]этанона
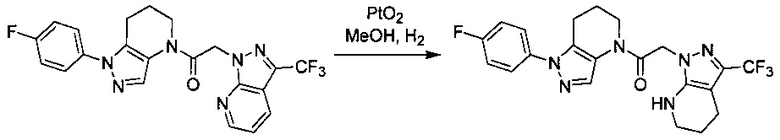
1-[1-(4-Фторфенил)-6,7-дигидро-5Н-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)пиразоло[3,4-b]пиридин-1-ил]этанон (22,2 мг, 0,05 ммоль), получение которого описано выше, растворяли в 1 мл метанола и добавляли ~20 мг PtO2 в закрытой септой виале. Виалу продували и заполняли водородом дважды, затем помещали в атмосферу водорода (шарик) и перемешивали в течение ночи. Полученную суспензию фильтровали и фильтрат очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая указанное в заголовке соединение (раствор нейтрализовывали и экстрагировали, получая свободное основание) в виде белого твердого вещества (13 мг, 60%). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,36 (с, 0,85 Н), 7,71 (с, 0,15 Н), 7,43-7,49 (м, 2 Н), 7,14-7,19 (м, 2 Н), 5,15 (с, 0,3 Н), 5,00 (с, 1,7 Н), 4,29 (ушир.с, 1 Н), 3,88-3,93 (м, 2 Н), 3,29 (м, 2 Н), 2,85 (т, J=6,3 Гц, 2 Н), 2,63 (т, J=6,2 Гц, 2 Н), 2,09 (м, 2 Н), 1,85 (м, 2 Н); MS: (ES) m/z вычислено для C21H21F4N6O [М+Н]+ 449,1, найдено 449,1.
Пример 28. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)-5,6-дигидро-4H-пирано[2,3-с]пиразол-1-ил]этанона
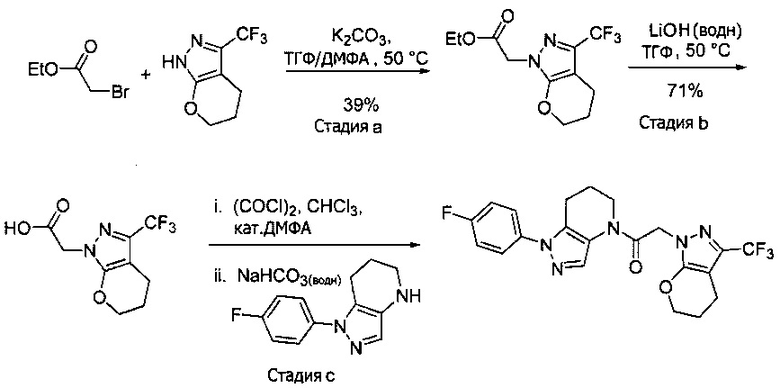
a) 3-(Трифторметил)-1,4,5,6-тетрагидропирано[2,3-с]пиразол (96 мг, 0,5 ммоль), K2CO3 (885 мг, 6,4 ммоль) и 8 мл смеси 3:1 ТГФ : ДМФА объединяли и добавляли 71 мкл (0,64 ммоль) этилбромацетата. Полученную суспензию перемешивали при 50°С в течение ночи и упаривали летучие компоненты. Полученный остаток разбавляли в EtOAc и Н2О и разделяли слои. Органический слой промывали насыщенным водным раствором хлорида натрия и упаривали. Полученный остаток очищали методом флэш-хроматографии (SiO2, 40 г колонка, элюирование 5-20% EtOAc в гексане), получая 70 мг (39%) указанного соединения в виде прозрачного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 4,73 (с, 2 Н), 4,31 (дд, J=5,1, 6,1 Гц, 2 Н), 4,23 (кв, J=7,0 Гц, 2 Н), 2,65 (т, J=6,7 Гц, 2 Н), 2,00 (м, 2 Н), 1,28 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C11H14F3N2O3 [М+Н]+ 279,1, найдено 279,1.
b) Полученный выше сложный эфир (70 мг, 0,25 ммоль) суспендировали в 1 мл ТГФ и 0,5 мл 2М раствора LiOH (водн.) при 50°С. Через 90 минут летучие вещества упаривали и добавляли 2 мл 1М раствором NaHSO4 (водн.). Полученную смесь экстрагировали дважды этилацетатом. Органический слой сушили над Na2SO4, фильтровали и упаривали, получая целевое вещество в виде твердой пены (45 мг, 71%). MS: (ES) m/z вычислено для C9H10F3N2O3 [М+Н]+ 251,1, найдено 251,1,
c) Указанное в заголовке соединение получали из 2-(3-(трифторметил)-5,6-дигидропирано[2,3-с]пиразол-1(4H)-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 32 мг (70%) указанного в заголовке соединения в виде белой пены. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,40 (с, 0,8 Н), 7,56 (с, 0,2 Н), 7,43-7,48 (м, 2 Н), 7,13-7,21 (м, 2 Н), 5,12 (с, 0,4 Н), 5,00 (с, 1,6 Н), 4,34 (дд, J=5,1, 5,1 Гц, 2 Н), 3,91 (м, 0,4 Н), 3,80 (м, 1,6 Н), 2,86 (т, J=6,2 Гц, 1,6 Н), 2,83 (т, J=6,2 Гц, 0,4 Н), 2,67 (т, J=6,2 Гц, 2 Н), 2,09 (м, 1,6 Н), 2,00-2,05 (м, 2,4 Н); MS: (ES) m/z вычислено для C21H20F4N5O2 [М+Н]+ 450,1, найдено 450,1.
Пример 29. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(2,4-дифторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
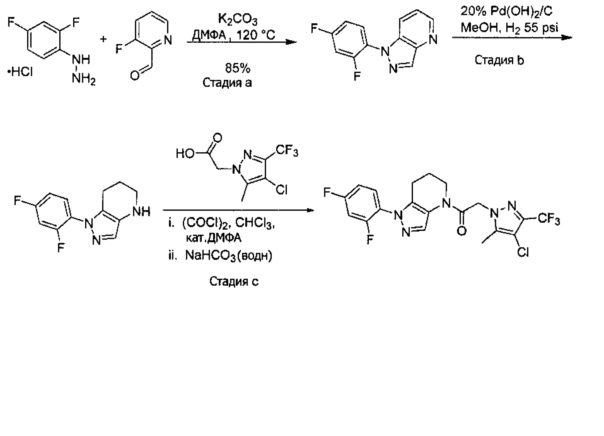
a) В круглодонную колбу объемом 100 мл добавляли 2,4-дифторфенилгидразина гидрохлорид (1,48 г, 8,2 ммоль), 3-фтор-2-формилпиридин (1,00 g, 8 ммоль), K2CO3 (3,32 г, 24 ммоль) и ДМФА (10 мл). Полученную суспензию нагревали до 120°С на масляной бане 16 часов, после чего реакционную смесь охлаждали и выливали в 25 мл Н2О. Коричневый осадок отфильтровывали и трижды промывали водой. Полученное твердое вещество растворяли в CH2Cl2, промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали с получением коричневого твердого вещества. Его растворяли в EtOAc, пропускали через слой силикагеля и упаривали с получением коричневого твердого вещества (1,58 г, 85%). MS: (ES) m/z вычислено для C12H8F2N3 [М+Н]+ 232,1, найдено 232,1.
b) Полученный выше гетероцикл (1,58 г, 6,8 ммоль) растворяли в 34 мл метанола и добавляли 20% Pd(OH)2/C (1,00 г) в аппарате Парра объемом 500 мл. Систему продували и заполняли водородом (3х) и затем встряхивали под давлением водорода 55 фунт/кв. дюйм до исчезновения исходного соединения. Колбу продували азотом, и реакционную смесь фильтровали через слой целита, промывая несколькими порциями метанола. Фильтрат упаривали, получая продукт (1,10 г) в виде светлого оранжево-коричневого масла. MS: (ES) m/z вычислено для C12H12F2N3 [М+Н]+ 236,1, найдено 236,1.
c) Указанное соединение получали из 2-(4-хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)уксусной кислоты и 1-(2,4-дифторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу В. Полученную смесь продуктов дважды промывали 1М раствором NaHSO4. Органический слой упаривали, и остаток очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая указанное в заголовке соединение в виде белой твердой пены (47 мг, 70%). 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,39 (с, 0,8 Н), 7,64 (с, 0,2 Н), 7,48 (м, 2 Н), 7,00 (м, 2 Н), 5,25 (с, 0,4 Н), 5,14 (с, 1,6 Н), 3,92 (м, 0,4 Н), 3,85 (м, 1,6 Н), 2,70 (м, 2 Н), 2,33 (с, 2,4 Н), 2,32 (с, 0,6 Н), 2,11 (м, 1,6 Н), 2,03 (м, 0,4 Н); MS: (ES) m/z вычислено для C19H16ClF5N5O [М+Н]+ 460,1, найдено 460,1.
Примеры 30 и 31. Синтез (2R)- и (2S)-2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]бутан-1-она

а) 2-(4-Хлор-5-метил-3-(трифторметил)-1H-пиразол-1-ил)бутановую кислоту (677 мг, 2,5 ммоль) растворяли в 10 мл CHCl3 и 3 каплях ДМФА. Прикапывали оксалилхлорид (262 мкл, 3 ммоль), при этом выделялся газ. Через 30 минут добавляли (R)-фенилглицинол (377 мг, 2,75 ммоль) затем 8 мл насыщенного раствора NaHCO3. Через 45 минут реакционную смесь промывали насыщенным водным раствором хлорида натрия и очищали методом флэш-хроматографии (SiO2, 40 г колонка, элюирование 10-70% EtOAc в гексане). Элюирующийся первым диастереомер: 1Н-ЯМР (400 МГц, CDCl3) δ 7,62 (д, J=7,8 Гц, 1 Н), 7,23-7,39 (м, 3 Н), 7,08-7,11 (м, 2 Н), 5,00-5,04 (м, 1 Н), 4,66 (т, J=7,4 Гц, 1 Н), 3,84 (м, 2 Н), 2,31 (т, J=7,4 Гц, 3 Н), 2,26 (с, 3 Н), 0,91 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C17H20ClF3N3O2 [М+Н]+ 390,1, найдено 390,1. Элюирующийся вторым диастереомер: 1Н-ЯМР (400 МГц, CDCl3) δ 7,58 (д, J=7,0 Гц, 1 Н), 7,38 (м, 2 Н), 7,32 (м, 1 Н), 7,23-7,25 (м, 2 Н), 5,00 (дт, J=5,1, 7,0 Гц, 1 Н), 4,69 (т, J=7,8 Гц, 1 Н), 3,75 (т, J=5,5 Гц, 2 Н), 2,32 (с, 3 Н), 2,10-2,27 (м, 3 Н), 0,84 (т, J=7,4 Гц, 3 Н); MS: (ES) m/z вычислено для C17H20ClF3N3O2 [М+Н]+ 390,1, найдено 390,1.
b) Полученные как описано выше диастереомеры независимо нагревали до 80°С в 0,25 М диоксане с 15 экв. 6М H2SO4. Суспензии реакционных смесей упаривали примерно до половины объема и трижды экстрагировали EtOAc. Органический слой сушили над Na2SO4, фильтровали и упаривали, получая целевые кислоты с выходом 82-89%.
c) (2R)-2-[4-Хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]бутан-1-он и (2S)-2-[4-Хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]бутан-1-он: Кислоту, полученную из элюирующегося первым диастереомера на стадии а, вводили в реакцию сочетания с 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридином по Общему методу А. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 73 мг (62%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,43 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (м, 2 Н), 5,29 (дд, J=6,7, 8,6 Гц, 1 Н), 3,81 (ддд, J=2,7, 7,4, 10,6 Гц, 1 Н), 3,50 (ддд, J=2,8, 8,6, 11,8 Гц, 1 Н), 2,79 (дт, J=2,7, 6,3 Гц, 2 Н), 2,33 (м, 1 Н), 2,30 (с, 3 Н), 2,22 (м, 1 Н), 1,96 (м, 1 Н), 1,86 (м, 1 Н), 0,98 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C21H21ClF4N5O [М+Н]+ 470,1, найдено 470,1. Кислоту, полученную из элюирующегося вторым диастереомера на стадии а, вводили в реакцию сочетания с 1-(4-фторфенил)-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридином по Общему методу А. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 71 мг (60%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,43 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (м, 2 Н), 5,29 (дд, J=6,7, 8,6 Гц, 1 Н), 3,81 (ддд, J=2,7, 7,4, 10,6 Гц, 1 Н), 3,50 (ддд, J=2,8, 8,6, 11,8 Гц, 1 Н), 2,79 (дт, J=2,7, 6,3 Гц, 2 Н), 2,33 (м, 1 Н), 2,30 (с, 3 Н), 2,22 (м, 1 Н), 1,96 (м, 1 Н), 1,86 (м, 1 Н), 0,98 (т, J=7,0 Гц, 3 Н); MS: (ES) m/z вычислено для C21H21ClF4N5O [М+Н]+ 470,1, найдено 470,1.
Пример 32. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[1-(трифторметил)имидазо[1,5-а]пиридин-3-ил]этанона
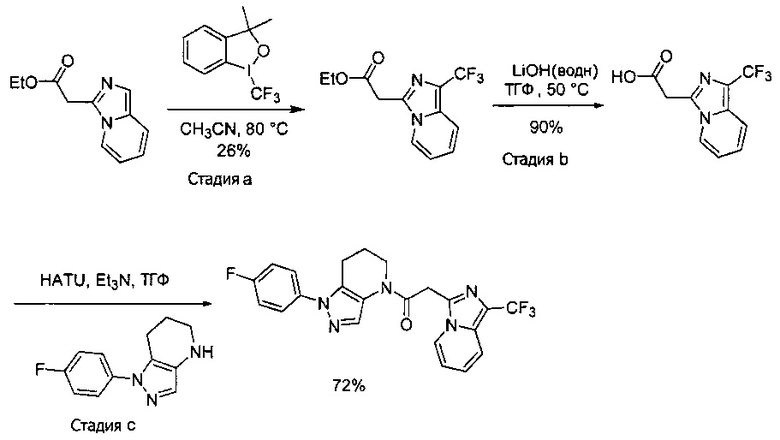
a) К этил 2-(имидазо[1,5-а]пиридин-3-ил)ацетату (131 мг, 0,64 ммоль) в CH3CN добавляли 3,3-диметил-1-(трифторметил)-1,2-бензиодоксол (212 мг, 0,64 ммоль). Полученную суспензию нагревали до 80°С 95 минут, затем охлаждали до комнатной температуры. Реакционную смесь очищали методом флэш-хроматографии (SiO2, 40 г колонка, элюирование 15-75% EtOAc в гексане), получая продукт (46 мг, 26%) в виде оранжевого масла затвердевающего при стоянии. MS: (ES) m/z вычислено для C12H12F3N2O2 [М+Н]+ 273,1, найдено 273,0.
b) Полученный выше сложный эфир (36 мг, 0,13 ммоль) суспендировали в 1 мл ТГФ и 0,125 мл 2М раствора LiOH (водн.) при 50°С. По окончании реакции, летучие вещества упаривали и добавляли 500 мкл 1М раствора NaHSO4 (водн.) и 4 мл EtOAc. Разделяли слои и промывали органическую фазу насыщенным водным раствором хлорида натрия. Органический слой сушили над Na2SO4, фильтровали и упаривали с получением коричневого твердого вещества (29 мг, 90%). MS: (ES) m/z вычислено для C10H8F3N2O2 [М+Н]+ 245,1, найдено 245,0.
c) Указанное в заголовке соединение получали из 2-(1-(трифторметил)имидазо[1,5-а]пиридин-3-ил)уксусной кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу А. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 32 мг (72%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3, смесь ротамеров) δ 8,36 (д, J=6,6 Гц, 1 Н), 8,35 (с, 0,9 Н), 8,06 (с, 0,1 Н), 7,63 (д, J=9,0 Гц, 1 Н), 7,50 (м, 0,3 Н), 7,42 (м, 1,7 Н), 7,12-7,22 (м, 2 Н), 7,01 (м, 1 Н), 6,79 (т, 6,6 Гц, 1 Н), 4,51 (с, 0,2 Н), 4,38 (с, 1,8 Н), 4,08 (м, 1,7 Н), 3,90 (м, 0,3 Н), 2,86 (т, J=6,6 Гц, 0,3 Н), 2,80 (т, J=6,6 Гц, 1,7 Н), 2,02 (м, 2 Н); MS: (ES) m/z вычислено для C22H18F4N5O [М+Н]+ 444,1, найдено 444,1.
Пример 33. Синтез 1-[1-(4-фторфенил)-5-метил-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]бутан-1-она

Указанное в заголовке соединение получали из 2-(5-метил-3-(трифторметил)-1H-пиразол-1-ил)бутановой кислоты и 1-(4-фторфенил)-5-метил-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу А. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н2О с 0,1% ТФУК в качестве элюента), получая 13 мг (46%) указанного в заголовке соединения в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1 Н), 7,47 (м, 2 Н), 7,16 (м, 2 Н), 6,35 (с, 1 Н), 5,37 (дд, J=6,7, 8,6 Гц, 1 Н), 4,63 (м, 1 Н), 2,86 (м, 1 Н), 2,73 (ддд, J=1,9, 5,1, 11,3 Гц, 1 Н), 2,37 (с, 3 Н), 2,27 (м, 2 Н), 1,92-1,97 (м, 2 Н), 0,93 (т, J=6,5 Гц, 3 Н), 0,57 (т, J=6,7 Гц, 3 Н); MS: (ES) m/z вычислено для C22H24F4N5O [М+Н]+ 450,2, найдено 450,2.
Пример 34. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(1-гидрокси-1-метил-этил)-5-метил-пиразол-1-ил]бутан-1-она
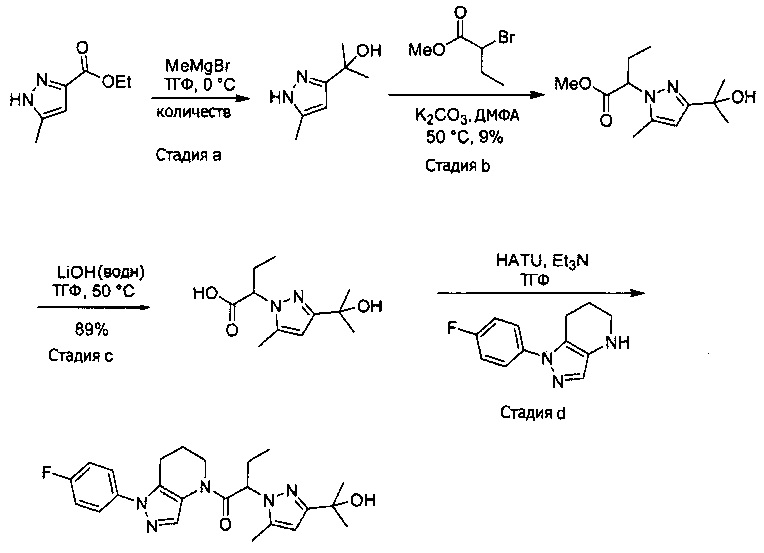
a) В промытую тетрагидрофураном круглодонную колбу объемом 250 мл помещали 80 мл ТГФ и 6,17 г (40 ммоль) этил 5-метил-1H-пиразол-3-карбоксилата. На колбу устанавливали капельную воронку и охлаждали в ледяной бане в атмосфере N2. Прикапывали метилмагния бромид (3М раствор в Et2O, 41 мл, 124 ммоль). Через 70 минут добавляли еще 4 мл реагента Гриньяра. Через 2 часа реакцию гасили медленным добавлением 2 мл 1М раствора NaHSO4. Реакционную суспензию подкисляли медленным добавлением 6М HCl (водн.). Летучие вещества упаривали и трижды экстрагировали смесь этилацетатом. Органический слой сушили над MgSO4, фильтровали и упаривали, получая вязкое желто-оранжевое масло (6,0 г, колич.), затвердевающего при стоянии. MS: (ES) m/z вычислено для C7H13N2O [М+Н]+ 141,1, найдено 141,1.
b) Метил бромбутират (1,068 г, 5,9 ммоль), 2-(5-метил-1H-пиразол-3-ил)пропан-2-ол (752 мг, 5,36 ммоль) и K2CO3 (1,520 г, 11 ммоль) объединяли в 16 мл смеси 2:1 ТГФ : ДМФА при 70°С в течение ночи. Реакционную смесь охлаждали и осадок отфильтровывали, промывая этилацетатом. Фильтрат дважды промывали водой и насыщенным водным раствором хлорида натрия. Органический слой упаривали, и остаток очищали методом флэш-хроматографии (SiO2, 80 г колонка, элюирование 10-65% EtOAc в гексане), получая 114 мг (9%) указанного в заголовке соединения в виде бесцветного масла. 1Н-ЯМР (400 МГц, CDCl3) δ 5,93 (с, 1 Н), 4,62 (дд, J=5,8, 9,8 Гц, 1 Н), 3,72 (с, 3 Н), 2,95 (ушир.с, 1 Н), 2,29 (м, 2 Н), 2,24 (с, 3 Н), 1,52 (с, 6 Н), 0,85 (т, J=7,4, 3 Н); MS: (ES) m/z вычислено для C12H21N2O3 [М+Н]+ 241,2, найдено 241,2.
c) Полученный выше сложный эфир (113 мг, 0,47 ммоль) суспендировали в 1 мл ТГФ и 310 мкл 2М раствора LiOH при 50°С. Через 30 минут летучие вещества упаривали и добавляли 1 мл 1M раствора NaHSO4. Полученную суспензию экстрагировали 3×EtOAc, и органический слой сушили над Na2SO4, фильтровали и упаривали, получая прозрачное масло (95 мг, 89%).
d) Указанное в заголовке соединение получали из 2-(3-(2-гидроксипропан-2-ил)-5-метил-1H-пиразол-1-ил)бутановой кислоты и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина по Общему методу А. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 3,7 мг указанного в заголовке соединения в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,46 (с, 1 Н), 7,44 (м, 2 Н), 7,15 (т, J=8,6 Гц, 2 Н), 5,92 (с, 1 Н), 5,17 (дд, J=7,4, 7,4 Гц, 1 Н), 3,73 (м, 1 Н), 3,48 (м, 1 Н), 2,75 (м, 2 Н), 2,33 (м, 1 Н), 2,23 (с, 3 Н), 2,17 (с, 1 Н), 2,14 (м, 1 Н), 1,87 (м, 1 Н), 1,73 (м, 1 Н), 1,52 (с, 6 Н), 0,96 (т, J=7,4 Гц, 3 Н); MS: (ES) m/z вычислено для C23H29FN5O2 [М+Н]+ 426,2, найдено 426,2.
Пример 35. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-(3-изопропенил-5-метил-пиразол-1-ил)бутан-1-она
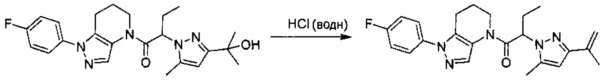
К части 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(1-гидрокси-1-метил-этил)-5-метил-пиразол-1-ил]бутан-1-она добавляли 6М HCl до окончания реакции. Реакционную суспензию очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 12 мг указанного в заголовке соединения в виде не совсем белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1 Н), 7,44 (м, 2 Н), 7,14 (т, J=8,6 Гц, 2 Н), 6,14 (с, 1 Н), 5,42 (ушир.с, 1 Н), 5,22 (дд, J=7,1, 8,2 Гц, 1 Н), 5,02 (ушир.с, 1 Н), 3,80 (ддд, J=3,1, 7,8, 10,1 Гц, 1 Н), 3,47 (ддд, J=2,8, 9,0, 12,1 Гц, 1 Н), 2,74 (м, 2 Н), 2,33 (м, 1 Н), 2,23 (с, 3 Н), 2,15 (м, 1 Н), 2,10 (с, 3 Н), 1,87 (м, 1 Н), 1,78 (м, 1 Н), 0,98 (т, J=7,4 Гц, 3 Н); MS: (ES) m/z вычислено для C23H27FN5O [М+Н]+ 408,2, найдено 408,2.
Пример 36. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]пропан-1-она
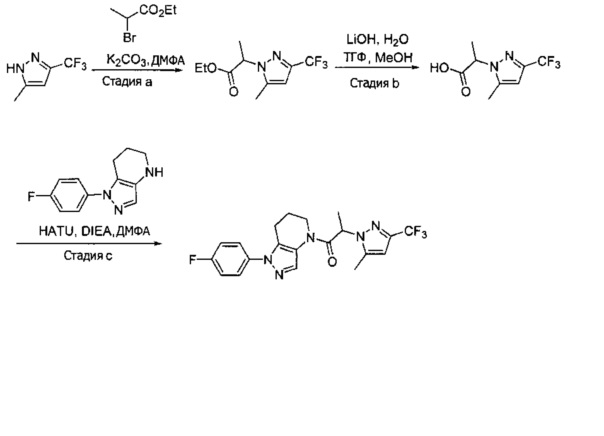
а) Смесь карбоната калия (922 мг, 6,67 ммоль), 2-метил-5-(трифторметил)-1H-пиразола (500 мг, 3,33 ммоль) и этил 2-бромпропаноата (0,48 мл, 3,7 ммоль) в смеси диметилформамид: тетрагидрофуран (3 мл: 6 мл) нагревали при 60°С 5 часов при перемешивании. После охлаждения до комнатной температуры, большую часть тетрагидрофурана удаляли пропусканием тока азота над реакционной смесью. Добавляли этилацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 10-20% этилацетата в гексане), получая целевой продукт (735 мг, 2,94 ммоль, 88%).
b) Этил 2-[5-метил-3-(трифторметил)пиразол-1-ил]пропаноат (734 мг, 2,94 ммоль) растворяли в тетрагидрофуране (4 мл) при комнатной температуре. Добавляли водный раствор гидроксида лития (1,5н., 2 мл, 3,0 ммоль), затем метанол (~2 мл) с получением однородного раствора. Через 2 часа большую часть тетрагидрофурана и метанол удаляли осторожным пропусканием азота над реакционной смесью. Водный раствор соляной кислоты (5н., 0,6 мл, 3,0 ммоль) добавляли в реакционную смесь, и продукт экстрагировали дихлорметаном три раза. Объединенные органические слои сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт (625 мг, 2,82 ммоль, 96%) в виде белого твердого вещества, которое использовали далее без дополнительной очистки.
c) Диметилформамид (1 мл) добавляли в смесь, содержащую 2-[5-метил-3-(трифторметил)пиразол-1-ил]пропановую кислоту (49,6 мг, 0,223 ммоль) и HATU (89,1 мг, 0,234 ммоль). Диизопропилэтиламин (98 мкл, 0,56 ммоль) добавляли при комнатной температуре и перемешивали реакционную смесь 1 минуту. 1-(4-Фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (49,5 мг, 0,228 ммоль) добавляли в один прием, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли этил ацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 20-38% этилацетата в гексане), получая целевой продукт в виде белого твердого вещества (83,5 мг, 0,198 ммоль, 89%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,45 (с, 1 Н), 7,44 (ддд, J=9,2, 5,2, 2,0 Гц, 2Н), 7,15 (дт, J=8,8, 2,0 Гц, 2 Н), 6,31 (с, 1 Н), 5,55 (кв, J=7,2 Гц, 1 Н), 3,73 (ддд, J=13, 7,6, 3,2 Гц, 1 Н), 3,37 (ддд, J=12, 8,8, 3,2 Гц, 1 Н), 2,72-2,79 (м, 2 Н), 2,29 (с, 3 Н), 1,86 - 1,97 (м, 1 Н), 1,76-1,85 (м, 1 Н), 1,80 (д, J=7,2 Гц, 3Н); MS: (ES) m/z вычислено для C20H19N5OF4 [М+Н]+ 422, найдено 422.
Пример 37. Синтез 1-[1-(4-фторфеннл)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]бутан-1-она

a) Смесь карбоната калия (924 мг, 6,69 ммоль), 2-метил-5-(трифторметил)-1H-пиразола (501 мг, 3,34 ммоль) и метил 2-бромбутирата (0,42 мл, 3,7 ммоль) в смеси диметилформамид: тетрагидрофуран (3 мл: 6 мл) нагревали при 60°С 5 часов при перемешивании. После охлаждения до комнатной температуры, большую часть тетрагидрофурана удаляли пропусканием тока азота над реакционной смесью. Добавляли этилацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 12-17% этилацетата в гексане), получая целевой продукт (704 мг, 2,81 ммоль, 84%).
b) Метил 2-[5-метил-3-(трифторметил)пиразол-1-ил]бутират (703 мг, 2,81 ммоль) растворяли в тетрагидрофуране (4 мл) при комнатной температуре. Добавляли водный раствор гидроксида лития (1,5н., 2 мл, 3,0 ммоль), затем метанол (~2 мл) с получением однородного раствора. Через 2 часа большую часть тетрагидрофурана и метанола удаляли осторожным пропусканием азота над реакционной смесью. Водный раствор соляной кислоты (5н., 0,6 мл, 3,0 ммоль) добавляли в реакционную смесь, и полученную смесь экстрагировали дихлорметаном три раза. Объединенные органические слои сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт (661 мг, 2,80 ммоль, 100%) в виде белого твердого вещества, которое использовали далее без дополнительной очистки.
c) Диметилформамид (0,7 мл) добавляли в смесь, содержащую 2-[5-метил-3-(трифторметил)пиразол-1-ил]бутановую кислоту (50,5 мг, 0,214 ммоль) и HATU (84,1 мг, 0,221 ммоль). Диизопропилэтиламин (92 мкл, 0,53 ммоль) добавляли при комнатной температуре, и реакционную смесь перемешивали 1 минуту. 1-(4-Фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (44,1 мг, 0,203 ммоль) затем добавляли в один прием, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 17-33% этилацетата в гексане), получая целевой продукт в виде белого твердого вещества (80,9 мг, 0,186 ммоль, 92%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,44 (ддд, J=8,8, 5,2, 2,0 Гц, 2Н), 7,15 (ддд, J=8,4, 8,4, 2,0 Гц, 2 Н), 6,30 (с, 1 Н), 5,32 (дд, J=8,4, 6,8 Гц, 1 Н), 3,78 (ддд, J=12, 7,2, 2,8 Гц, 1 Н), 3,47 (ддд, J=12, 8,8, 3,2 Гц, 1 Н), 2,69-2,81 (м, 2 Н), 2,31 (с, 3 Н), 2,18-2,38 (м, 2 Н), 1,88-1,97 (м, 1 Н), 1,74-1,83 (м, 1Н), 0,98 (т, J=7,2 Гц, 3Н); MS: (ES) m/z вычислено для C21H21N5OF4 [М+Н]+ 436, найдено 436.
Пример 38. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[2-метил-4-(трифторметил)имидазол-1-ил]пропан-1-она

a) Смесь карбоната калия (832 мг, 6,02 ммоль), 2-метил-5-(трифторметил)-1Н-имидазола (450 мг, 3,00 ммоль) и этил 2-бромпропаноата (0,43 мл, 3,3 ммоль) в смеси диметилформамид: тетрагидрофуран (2 мл: 4 мл) нагревали при 50°С 7 часов при перемешивании и затем при комнатной температуре в течение ночи. Большую часть тетрагидрофурана удаляли пропусканием тока азота над реакционной смесью. Добавляли этилацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 17-50% этилацетата в гексане), получая целевой продукт (697 мг, 2,79 ммоль, 93%).
b) Этил 2-[2-метил-4-(трифторметил)имидазол-1-ил]пропаноат (697 мг, 2,79 ммоль) растворяли в тетрагидрофуране (5 мл) при комнатной температуре. Добавляли водный раствор гидроксида лития (1,5н., 2,5 мл, 3,8 ммоль), затем метанол (~2 мл) с получением однородного раствора. Через 2 часа большую часть тетрагидрофурана и метанола удаляли осторожным пропусканием тока азота над реакционной смесью. Водный раствор соляной кислоты (5н., 0,75 мл, 3,8 ммоль) добавляли в реакционную смесь, и продукт экстрагировали дихлорметаном три раза. Объединенные органические слои сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт (562 мг, 2,52 ммоль, 91%) в виде белой пены, которую использовали далее без дополнительной очистки.
c) 2-[2-Метил-4-(трифторметил)имидазол-1-ил]пропановую кислоту (151 мг, 0,679 ммоль) растворяли в дихлорметане (2 мл) при комнатной температуре. Добавляли оксалилхлорид (120 мкл, 1,38 ммоль) и каталитическое количество диметилформамида (1 мкл), и реакционную смесь перемешивали при комнатной температуре 2 часа. Растворитель удаляли при пониженном давлении, и полученный ацилхлорид сушили в вакууме. Неочищенный ацилхлорид растворяли в дихлорметане (2,1 мл). К части полученного раствора (0,7 мл, 0,22 ммоль) добавляли 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (41,5 мг, 0,191 ммоль) и пиридин (55 мкл, 0,68 ммоль) при комнатной температуре, и реакционную смесь перемешивали 2 часа. Добавляли воду и разделяли слои. Водный слой еще два раза экстрагировали дихлорметаном. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 40-100% этилацетата в гексане с 0,1% водного раствора гидроксида аммония), получая целевой продукт в виде белого твердого вещества (70,1 мг, 0,166 ммоль, 87%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,45 (ддд, J=9,2, 4,8, 2,0 Гц, 2Н), 7,17 (дт, J=9,2, 2,0 Гц, 2 Н), 5,21 (кв, J=6,8 Гц, 1 Н), 3,75 (ддд, J=12, 7,2, 3,2 Гц, 1 Н), 3,51 (ддд, J=12, 8,0, 3,2 Гц, 1 Н), 2,84 (т, J=6,6 Гц, 2 Н), 2,47 (с, 3 Н), 1,96-2,09 (м, 2 Н), 1,72 (д, J=6,4 Гц, 3Н); MS: (ES) m/z вычислено для C20H19N5OF4 [М+Н]+ 422, найдено 422.
Пример 39. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[2-метил-4-(трифторметил)имидазол-1-ил]бутан-1-она

a) Смесь карбоната калия (834 мг, 6,04 ммоль), 2-метил-5-(трифторметил)-1H-имидазола (449 мг, 2,99 ммоль) и метил 2-бромбутирата (0,38 мл, 3,3 ммоль) в смеси диметилформамид: тетрагидрофуран (2 мл: 4 мл) нагревали при 50°С 7 часов при перемешивании и затем при комнатной температуре в течение ночи. Большую часть тетрагидрофурана удаляли осторожным пропусканием тока азота над реакционной смесью. Добавляли этилацетат и воду и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 20-50% этилацетата в гексане), получая целевой продукт (641 мг, 2,56 ммоль, 86%).
b) Метил 2-[2-метил-4-(трифторметил)имидазол-1-ил]бутират (641 мг, 2,56 ммоль) растворяли в тетрагидрофуране (5 мл) при комнатной температуре. Добавляли водный раствор гидроксида лития (1,5н., 2,2 мл, 3,3 ммоль), затем метанол (~2 мл) с получением однородного раствора. Через 2 часа большую часть тетрагидрофурана и метанола удаляли пропусканием тока азота над реакционной смесью. Водный раствор соляной кислоты (5н., 0,66 мл, 3,3 ммоль) добавляли в реакционную смесь, и продукт три раза экстрагировали дихлорметаном. Объединенные органические слои сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт (497 мг, 2,11 ммоль, 82%) в виде белой пены, которую использовали далее без дополнительной очистки.
c) 2-[2-Метил-4-(трифторметил)имидазол-1-ил]бутановую кислоту (149 мг, 0,632 ммоль) растворяли в дихлорметане (2 мл) при комнатной температуре. Добавляли оксалилхлорид (110 мкл, 1,26 ммоль) и каталитическое количество диметилформамида (1 мкл), и реакционную смесь перемешивали при комнатной температуре 2 часа. Растворитель удаляли при пониженном давлении, и полученный ацилхлорид сушили в вакууме. Неочищенный ацилхлорид растворяли в ацетонитриле (1,5 мл). К части полученного раствора (0,5 мл, 0,21 ммоль) добавляли 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (38,7 мг, 0,178 ммоль) и пиридин (55 мкл, 0,68 ммоль) при комнатной температуре, и реакционную смесь перемешивали 2 часа. Затем добавляли воду и этилацетат и разделяли слои. Водный слой еще два раза экстрагировали этилацетатом. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 50~80% этилацетата в гексане с 0,1% водного раствора гидроксида аммония), получая целевой продукт, загрязненный небольшим количеством 2-[2-метил-4-(трифторметил)имидазол-1-ил]бутановой кислоты. Полученный продукт растворяли в этилацетате и один раз промывали 1н. водным раствором гидроксида натрия. После сушки над безводным сульфатом натрия, фильтрования и удаления растворителя при пониженном давлении, и сушки в вакууме получали целевой продукт в виде белого твердого вещества (60,4 мг, 0,139 ммоль, 78%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,20 (с, 1 Н), 7,84 (д, J=1,2 Гц, 1H), 7,58 (ддд, J=9,2, 5,2, 2,0 Гц, 2Н), 7,35 (ддд, J=9,2, 8,4, 0,8 Гц, 2 Н), 5,47 (дд, J=9,2, 5,6 Гц, 1 Н), 3,81-3,98 (м, 2 Н), 2,84 (т, J=6,2 Гц, 2 Н), 2,34 (с, 3 Н), 2,03-2,11 (м, 2 Н), 1,96-2,01 (м, 2 Н), 0,85 (т, J=7,2 Гц, 3Н); MS: (ES) m/z вычислено для C21H21N5OF4 [М+Н]+ 436, найдено 436.
Пример 40. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-(трифторметокси)фенил]этанона
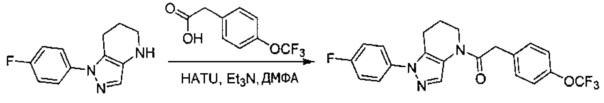
К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,023 г, 0,10 ммоль), 4-(трифторметокси)фенилуксусной кислоты (0,025 г, 0,11 ммоль) и Et3N (0,060 мл, 0,43 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,040 г, 95%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,51 (с, 1 Н), 7,43 (м, 2 Н), 7,30 (м, 2 Н), 7,18 (м, 4 Н), 3,91 (с, 2 Н), 3,76 (м, 2 Н), 2,79 (т, J=6,4 Гц, 2 Н), 2,00 (м, 2 Н); MS: (ES) m/z вычислено для C21H17F4N3O2 [М+Н]+ 420,1, найдено 420,1.
Пример 41. Синтез 2-[4-хлор-3-(трифторметокси)фенил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
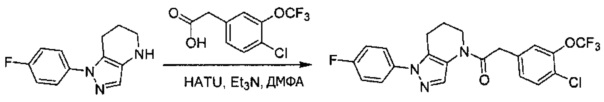
К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,023 г, 0,10 ммоль), 4-хлор-3-(трифторметокси)фенилуксусной кислоты (0,025 г, 0,10 ммоль) и Et3N (0,060 мл, 0,43 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой и разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,039 г, 85%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1 Н), 7,44 (м, 3 Н), 7,12~7,22 (м, 4 Н), 3,90 (с, 2 Н), 3,76 (м, 2 Н), 2,80 (т, J=6,2 Гц, 2 Н), 2,02 (м, 2 Н); MS: (ES) m/z вычислено для C21H16ClF4N3O2 [М+Н]+ 454,1, найдено 454,1.
Пример 42. Синтез 2-[4-хлор-3-(трифторметил)фенил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-метил-пропан-1-она
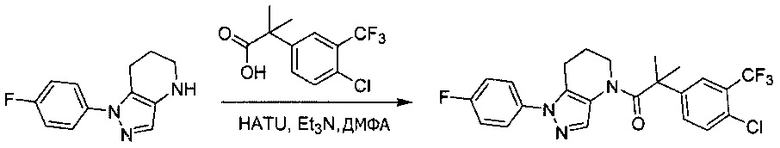
К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,030 г, 0,11 ммоль), 2-[4-хлор-3-(трифторметил)фенил]-2-метил-пропановой кислоты (0,030 г, 0,11 ммоль) и NEt3 (0,070 мл, 0,50 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой и разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,005 г, 10%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,50 (с, 1 Н), 7,60 (д, J=2,0 Гц, 1 Н), 7,30~7,51 (м, 4 Н), 7,14 (дд, J=8,8, 8,4 Гц, 2 Н), 3,22 (м, 2 Н), 2,69 (т, J=6,4 Гц, 2 Н), 1,64 (с, 6 Н); 1,62 (м, 2 Н); MS: (ES) m/z вычислено для C23H20ClF4N3O [М+Н]+ 466,1, найдено 466,1.
Пример 43. Синтез 2-(4-хлорпиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
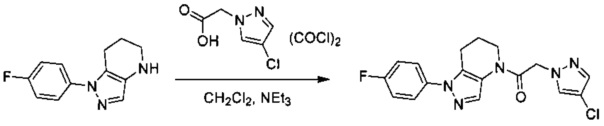
Смесь 2-[(4-хлор-пиразол-1-ил)]уксусной кислоты (0,050 г, 0,31 ммоль), (COCl)2 (0,060 мл, 0,70 ммоль) и ДМФА (1 капля) в CH2Cl2 (1 мл) перемешивали 30 минут при комнатной температуре. Затем упаривали досуха на высоковакуумном насосе. Полученное масло добавляли в смесь, содержащую 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридин (0,020 г, 0,092 ммоль) и NEt3 (0,070 мл, 0,50 ммоль) в CH2Cl2 (1 мл). Полученную смесь затем перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой и разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,012 г, 36%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,41 (с, 1 Н), 7,60 (с, 1 Н), 7,51 (с, 1 Н), 7,44 (дд, J=8,8, 4,6 Гц, 2 Н), 7,17 (дд, J=9,2, 8,8 Гц, 2 Н), 5,16 (с, 2 Н), 3,82 (м, 2 Н), 2,85 (т, J=6,2 Гц, 2 Н), 2,09 (м, 2 Н); MS: (ES) m/z вычислено для C17H15ClFN5O [М+Н]+ 360,1, найдено 360,1.
Пример 44. Синтез 2-[4-хлор-3-(дифторметил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,023 г, 0,10 ммоль), 2-(4-хлор-3-дифторметил-5-метил-пиразол-1-ил)уксусной кислоты (0,023 г, 0,10 ммоль) и NEt3 (0,065 мл, 0,46 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой и разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,032 г, 75%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1 Н), 7,44 (м, 2 Н), 7,16 (дд, J=8,8, 8,4 Гц, 2 Н), 6,67 (д, J=54 Гц, 1 Н), 5,11 (с, 2 Н), 3,83 (м, 2 Н), 2,85 (т, J=6,2 Гц, 2 Н), 2,30 (с, 3 Н), 2,10 (м, 2 Н); MS: (ES) m/z вычислено для C19H17ClF3N5O [М+Н]+ 424,1, найдено 424,1.
Пример 45. Синтез 2-[4-хлор-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
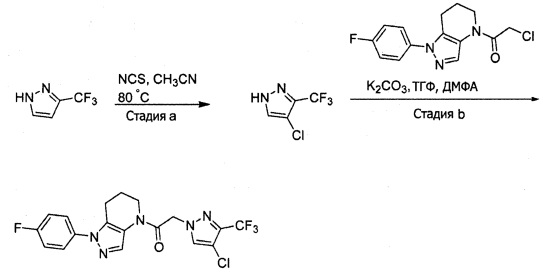
a) Смесь 3-(трифторметил)пиразола (1,58 г, 11,6 ммоль) и NCS (1,55 г, 11,6 ммоль) в CH3CN (20 мл) нагревали при 80°С в течение 3 часов. После этого охлаждали до комнатной температуры, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2) элюируя градиентом от 0 до 15% EtOAc/CH2Cl2, получая 4-хлор-3-(трифторметил)пиразол (1,52 г, 77%).
b) Смесь 4-хлор-3-(трифторметил)пиразола (0,026 г, 0,15 ммоль), 2-хлор-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,045 г, 0,15 ммоль) и K2CO3 (0,043 г, 0,31 ммоль) в ТГФ (0,6 мл) и ДМФА (0,3 мл) нагревали при 60°С в течение 1 часа. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом флэш-хроматографии на SiO2, элюируя градиентом 0-60% EtOAc/гексан, получая целевой продукт (0,056 г, 87%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1 Н), 7,70 (с, 1 Н), 7,45 (м, 2 Н), 7,16 (дд, J=8,4, 8,4 Гц, 2 Н), 5,17 (с, 2 Н), 3,81 (м, 2 Н), 2,87 (т, J=6,2 Гц, 2 Н), 2,12 (м, 2 Н); MS: (ES) m/z вычислено для C18H14ClF4N5O [М+Н]+ 428,1, найдено 428,1.
Пример 46. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)пиразол-1-ил]этанона
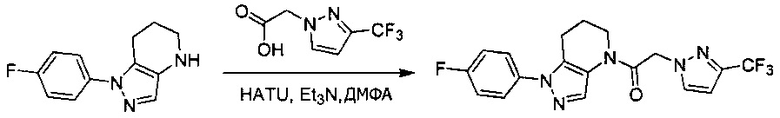
К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,026 г, 0,12 ммоль), 2-(3-(трифторметил)-пиразол-1-ил)уксусной кислоты (0,024 г, 0,12 ммоль) и NEt3 (0,060 мл, 0,43 ммоль) в ДМФА (0,5 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,035 г, 74%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1 Н), 7,64 (м, 1 Н), 7,43 (м, 2 Н), 7,15 (дд, J=8,8, 8,4 Гц, 2 Н), 6,62 (д, J=2,4 Гц, 1 Н), 5,23 (с, 2 Н), 3,82 (м, 2 Н), 2,84 (т, J=6,4 Гц, 2 Н), 2,08 (м, 2 Н); MS: (ES) m/z вычислено для C18H15F4N5O [М+Н]+ 394,1, найдено 394,1.
Пример 47. Синтез 2-(4-хлор-5-метил-пиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

a) Смесь 2-(5-метилпиразол-1-ил)уксусной кислоты (0,700 г, 5 ммоль) и NCS (0,668 г, 5 ммоль) в CH3CN (10 мл) нагревали при 80°С в течение 1,5 часов. После этого охлаждали до комнатной температуры, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2, элюируя градиентом 0~20% МеОН/CH2Cl2) с получением 2-(4-хлор-5-метилпиразол-1-ил)уксусной кислоты (0,870 г, 100%).
b) К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,026 г, 0,12 ммоль), 3-(трифторметил)-пиразол-1-ил)уксусной кислоты (0,024 г, 0,12 ммоль) и NEt3 (0,060 мл, 0,43 ммоль) в ДМФА (0,5 мл) добавляли HATU (0,060 г, 0,15 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,025 г, 55%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1 Н), 7,48 (с, 1 Н), 7,44 (дд, J=8,8, 4,6 Гц, 2 Н), 7,16 (дд, J=8,4, 8,4 Гц, 2 Н), 5,16 (с, 2 Н), 3,84 (м, 2 Н), 2,84 (т, J=6,4 Гц, 2 Н), 2,29 (с, 3 Н), 2,10 (м, 2 Н); MS: (ES) m/z вычислено для C18H17ClFN5O [М+Н]+ 374,1, найдено 374,1.
Пример 48. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-(трифторметил)пиразол-1-ил]этанона
Смесь 4-(трифторметил)-1H-пиразола (0,030 г, 0,22 ммоль), 2-хлор-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,025 г, 0,085 ммоль) и K2CO3 (0,060 г, 0,43 ммоль) в ТГФ (0,8 мл) и ДМФА (0,4 мл) нагревали при 65°С в течение 1 часа. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,038 г, 100%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,40 (с, 1 Н), 7,89 (с, 1 Н), 7,77 (с, 1 Н), 7,44 (дд, У=8,8, 4,6 Гц, 2 Н), 7,16 (дд, J=8,8, 8,4 Гц, 2 Н), 5,20 (с, 2 Н), 3,83 (м, 2 Н), 2,85 (т, J=6,2 Гц, 2 Н), 2,10 (м, 2 Н); MS: (ES) m/z вычислено для C18H15F4N5O [М+Н]+ 394,1, найдено 394,1.
Пример 49. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)-6,7-дигидро-4H-пирано[4,3-с]пиразол-1-ил]этанона
Смесь 3-(трифторметил)-1,4,6,7-тетрагидропирано[4,3-с]пиразола (0,050 г, 0,26 ммоль), 2-хлор-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,050 г, 0,17 ммоль) и K2CO3 (0,130 г, 0,94 ммоль) в ТГФ (0,8 мл) и ДМФА (0,4 мл) нагревали при 55°С в течение 1 часа. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между EtOAc (50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,035 г, 55%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1 Н), 7,44 (дд, J=8,8, 4,6 Гц, 2 Н), 7,17 (дд, J=8,8, 8,0 Гц, 2 Н), 5,13 (с, 2 Н), 4,75 (с, 2 Н), 3,98 (т, J=5,4 Гц, 2 Н), 3,87 (м, 2 Н), 2,85 (т, J=6,2 Гц, 2 Н), 2,79 (т, J=5,6 Гц, 2 Н), 2,10 (м, 2 Н); MS: (ES) m/z вычислено для C21H19F4N5O2 [М+Н]+ 450,1, найдено 450,1.
Пример 50. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[4-(1,2,4-оксадиазол-3-ил)пиразол-1-ил]этанона
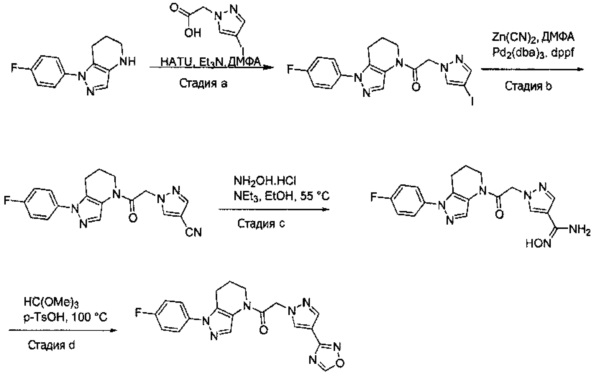
a) К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,400 г, 1,83 ммоль), 2-(4-иод-пиразол-1-ил)уксусной кислоты (0,554 г, 2,2 ммоль) и NEt3 (0,642 мл, 4,59 ммоль) в ДМФА (5 мл) добавляли HATU (0,836 г, 2,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 40 минут. Затем реакцию гасили водой. Смесь разделяли между EtOAc (100 мл) и насыщенным раствором NaHCO3 (50 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2, элюируя градиентом 0~30% EtOAc/CH2Cl2), получая 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-(4-иодпиразол-1-ил)этанон (0,82 г, 100%).
b) Смесь 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-(4-иодпиразол-1-ил)этанона (0,388 г, 0,86 ммоль), Zn(CN)2 (0,151 г, 1,29 ммоль), Pd2(dba)3 (0,079 г, 0,086 ммоль) и dppf (0,072 г, 0,13 ммоль) в ДМФА (10 мл) нагревали при 90°С в течение 1 часа. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между EtOAc (10 мл) и насыщенным раствором NaHCO3 (50 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом флэш-хроматографии, элюируя градиентом 0-100% EtOAc/CH2Cl2 с получением 2-(4-цианопиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,205 г, 68%).
c) Смесь 2-(4-цианопиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5Н-пиразоло[4,3-b]пиридин-4-ил]этанона (0,175 г, 0,50 ммоль), NH2OH-HCl (0,500 г, 7,2 ммоль) и NEt3 (1,00 мл, 7,1 ммоль) в ЕtOН (3 мл) нагревали при 90°С 5 часов. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между смесью изопропанол/CHCl3 (1:2, 50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4 и упаривали в вакууме, получая 1-[2-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-оксо-этил]-N'-гидрокси-пиразол-4-карбоксамидин (0,115 г, 60%).
d) Смесь 1-[2-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-оксо-этил]-N'-гидрокси-пиразол-4-карбоксамидина (0,115 г, 0,30 ммоль) и p-TsOH.H2O (0,030 г, 0,15 ммоль) в HC(OMe)3 (3 мл) нагревали при 100°С 1,5 часа. После этого охлаждали до комнатной температуры. Полученную смесь разделяли между смесью изопропанол/CHCl3 (1:2, 50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,055 г, 21%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,71 (с, 1 Н), 7,65-8,50 (м, 3 Н), 7,44 (дд, J=8,4, 4,6 Гц, 2 Н), 7,17 (дд, J=9,2, 7,6 Гц, 2 Н), 5,27 (м, 2 Н), 3,85 (м, 2 Н), 2,85 (м, 2 Н), 2,14 (м, 2 Н); MS: (ES) m/z вычислено для C19H16FN7O2 [М+Н]+ 394,1, найдено 394,1.
Пример 51. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]этанона
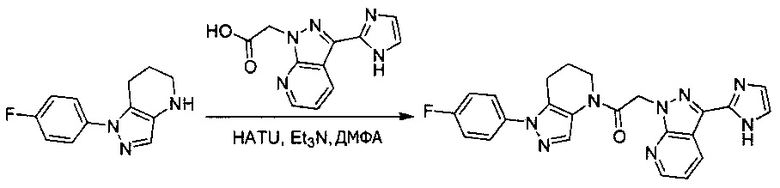
К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,050 г, 0,23 ммоль), 2-[3-(1H-имидазол-2-ил)пиразоло[3,4-b]пиридин-1-ил]уксусной кислоты (0,050 г, 0,21 ммоль) и Et3N (0,15 мл, 1,07 ммоль) в ДМФА (0,7 мл) добавляли HATU (0,130 г, 0,33 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Полученную смесь разделяли между смесью изопропанол/CHCl3 (1:2, 100 мл) и насыщенным раствором NaHCO3 (40 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,090 г, 96%). 1Н-ЯМР (соль с ТФУК) (400 МГц, CD3OD) δ 8,73 (дд, J=4,8, 1,6 Гц, 1 Н), 8,69 (дд, J=8,0, 1,6 Гц, 1 Н), 8,14 (с, 1 Н), 7,72 (с, 2 Н), 7,55 (м, 3 Н), 7,28 (дд, J=9,2, 8,4 Гц, 2 Н), 5,89 (с, 2 Н), 4,06 (м, 2 Н), 2,92 (т,J=6,2 Гц, 2 Н), 2,20 (м, 2 Н); MS: (ES) m/z вычислено для C23H19FN8O [М+Н]+ 443,1, найдено 443,1.
Пример 52. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)-4,5,6,7-тетрагидроиндазол-1-ил]этанона

К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,023 г, 0,11 ммоль), 2-[3-(трифторметил)-4,5,6,7-тетрагидроиндазол-1-ил]уксусной кислоты (0,027 г, 0,11 ммоль) и NEt3 (0,050 мл, 0,36 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,050 г, 0,13 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между смесью изопропанол/CHCl3 (1:2, 50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,027 г, 55%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,43 (с, 1 Н), 7,42 (дд, J=8,8, 4,4 Гц, 2 Н), 7,17 (дд, J=9,2, 8,0 Гц, 2 Н), 5,15 (с, 2 Н), 3,87 (м, 2 Н), 2,82 (т, J=6,2 Гц, 2 Н), 2,61 (м, 4 Н), 2,11 (м, 2 Н), 1,87 (м, 2 Н), 1,79 (м, 2 Н); MS: (ES) m/z вычислено для C22H21F4N5O [М+Н]+ 448,1, найдено 448,1.
Пример 53. Синтез 2-(1,3-бензоксазол-2-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,023 г, 0,11 ммоль), бензооксазол-2-ил-уксусной кислоты (0,023 г, 0,13 ммоль) и NEt3 (0,060 мл, 0,43 ммоль) в ДМФА (0,6 мл) добавляли HATU (0,050 г, 0,13 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Полученную смесь разделяли между смесью изопропанол/CHCl3 (1:2, 50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,022 г, 53%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,46 (с, 1 Н), 7,71 (м, 1 Н), 7,53 (м, 1 Н), 7,45 (дд, J=8,8, 4,8 Гц, 2 Н), 7,34 (м, 2 Н), 7,15 (дд, J=8,8, 8,4 Гц, 2 Н), 4,25 (с, 2 Н), 3,88 (м, 2 Н), 2,83 (т, J=6,4 Гц, 2 Н), 2,08 (м, 2 Н); MS: (ES) m/z вычислено для C21H17FN4O2 [М+Н]+ 377,1, найдено 377,1.
Пример 54. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-метокси-пропан-1-она

К смеси 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,045 г, 0,10 ммоль) и NaH (0,030 г, 0,75 ммоль, 60% в минеральном масле) в ДМФА (0,8 мл) добавляли сухой параформальдегид (0,015 г, 0,50 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,006 г, 12%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,44 (дд, J=8,8, 4,6 Гц, 2 Н), 7,16 (дд, J=8,4, 8,4 Гц, 2 Н), 5,51 (дд, J=8,0, 7,2 Гц, 1 Н), 4,18 (м, 1 Н), 4,08 (м, 1 Н), 3,75 (м, 1 Н), 3,37 (с, 3 Н), 3,35 (м, 1 Н), 2,78 (м, 2 Н), 2,33 (с, 3 Н), 2,09 (м, 2 Н); MS: (ES) m/z вычислено для C21H20ClF4N5O2 [М+Н]+ 486,1, найдено 486,1.
Пример 55. Синтез 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-гидрокси-пропан-1-она
К смеси 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (0,100 г, 0,23 ммоль) в ТГФ (3 мл) при -78°С добавляли LDA (0,15 мл, 0,3 ммоль, 2М раствора в ТГФ). После перемешивания в течение 10 минут при -78°С, добавляли сухой параформальдегид (0,020 г, 0,66 ммоль, суспензия в ТГФ). Полученную смесь затем нагревали до комнатной температуры на 8 минут, гасили насыщенным раствором NH4Cl. Полученную смесь разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,060 г, 56%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,45 (с, 1 Н), 7,43 (дд, J=8,4, 4,6 Гц, 2 Н), 7,17 (дд, J=8,8, 8,4 Гц, 2 Н), 5,40 (т, J=5,4 Гц, 1 Н), 4,90 (с, 1 Н), 4,30 (м, 1 Н), 4,20 (м, 1 Н), 3,71 (м, 1 Н), 3,29 (м, 1 Н), 2,79 (м, 2 Н), 2,28 (с, 3 Н), 1,95 (м, 2 Н); MS: (ES) m/z вычислено для C20H18ClF4N5O2 [М+Н]+ 472,1, найдено 472,1.
Пример 56. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-гидрокси-2-[5-метил-3-(трифторметил)пиразол-1-ил]пропан-1-она
К смеси 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]этанона (0,100 г, 0,25 ммоль) в ТГФ (3 мл) при -78°С добавляли LDA (0,15 мл, 0,3 ммоль, 2М раствора в ТГФ). После перемешивания в течение 10 минут при -78°С, добавляли сухой параформальдегид (0,025 г, 0,82 ммоль, суспензия в ТГФ). Полученную смесь затем нагревали до комнатной температуры на 8 минут, после чего добавляли насыщенный раствор NH4Cl. Смесь разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,056 г, 55%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,50 (с, 1 Н), 7,42 (м, 2 Н), 7,18 (дд, J=8,4, 8,4 Гц, 2 Н), 6,36 (с, 1 Н), 5,41 (т, J=5,6 Гц, 1 Н), 4,36 (м, 1 Н), 4,20 (м, 1 Н), 3,69 (м, 1 Н), 3,26 (м, 1 Н), 2,76 (м, 2 Н), 2,31 (с, 3 Н), 1,92 (м, 2 Н); MS: (ES) m/z вычислено для C20H19F4N5O2 [М+Н]+ 438,1, найдено 438,1.
Пример 57. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-4-гидрокси-4-метил-2-[5-метил-3-(трифторметил)пиразол-1-ил]пентан-1-она

а) К смеси 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]этанона (0,150 г, 0,37 ммоль) в ТГФ (4 мл) при -78°С добавляли LDA (0,247 мл, 0,50 ммоль, 2М раствора в ТГФ). После перемешивания в течение 10 минут при -78°С, добавляли этилбромацетат (0,061 мл, 0,55 ммоль). Полученную смесь затем нагревали до комнатной температуры на 10 минут и гасили насыщенным раствором NH4Cl. Полученную смесь разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2, элюируя градиентом 0-60% EtOAc/гексан), получая этил 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутаноат (0,120 г, 83%).
b) Смесь этил 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутаноата (0,028 г, 0,10 ммоль) и MeMgCl (0,100 мл, 0,30 ммоль, 3М раствор в ТГФ) в ТГФ (1 мл) перемешивали при комнатной температуре в течение 10 минут. Затем реакцию гасили насыщенным раствором NH4Cl и разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,005 г, 17%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,45 (с, 1 Н), 7,43 (м, 2 Н), 7,16 (дд, J=8,8, 8,4 Гц, 2 Н), 6,31 (с, 1 Н), 5,72 (т, J=6,4 Гц, 1 Н), 3,88 (м, 1 Н), 3,52 (м, 1 Н), 2,75 (м, 3 Н), 2,32 (с, 3 Н), 2,25 (м, 2 Н), 1,96 (м, 1 Н), 1,85 (м, 1 Н), 1,34 (с, 3 Н), 1,18 (с, 3 Н); MS: (ES) m/z вычислено для C23H25F4N5O2 [М+Н]+ 480,1, найдено 480,1.
Пример 58. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]-3-тетрагидропиран-4-ил-пропан-1-она
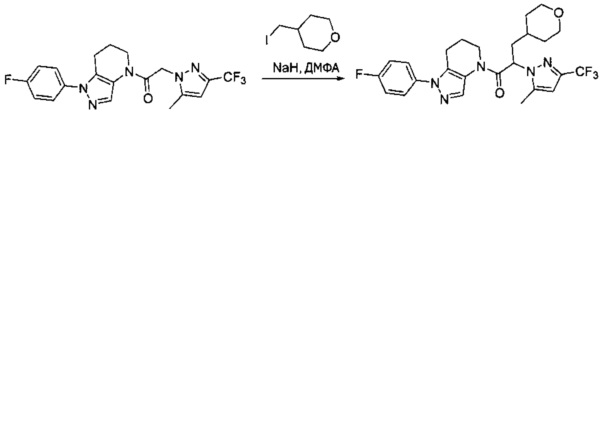
Смесь 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)пиразол-1-ил]эталона (0,100 г, 0,25 ммоль), 4-(иодметил)тетрагидро-2H-пирана (0,165 г, 0,75 ммоль) и NaH (0,030 г, 0,75 ммоль, 60% в минеральном масле) в ДМФА (0,8 мл) перемешивали при комнатной температуре в течение 1,5 часов. Затем реакцию гасили насыщенным раствором NH4Cl. Смесь разделяли между EtOAc (50 мл) и насыщенным водным раствором хлорида натрия (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,007 г, 6%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,43 (дд, J=8,8,4,8 Гц, 2 Н), 7,16 (дд, J=8,8, 8,4 Гц, 2 Н), 6,31 (с, 1 Н), 5,54 (м, 1 Н), 3,95 (м, 2 Н), 3,78 (м, 1 Н), 3,47 (м, 1 Н), 3,34 (м, 2 Н), 2,75 (м, 2 Н), 2,30 (с, 3 Н), 2,20 (м, 3 Н), 1,88 (м, 3 Н), 1,40 (м, 3 Н); MS: (ES) m/z вычислено для C25H27F4N5O2 [М+Н]+ 506,2, найдено 506,2.
Пример 59. Синтез 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутанамида

a) Смесь этил 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутаноата (0,030 г, 0,060 ммоль), LiOH⋅H2O (0,020 г, 0,47 ммоль), ТГФ (0,4 мл), МеОН (0,4 мл) и H2O (0,2 мл) перемешивали 30 минут при комнатной температуре. Затем подкисляли добавлением 1М водного раствора HCl (2 мл) и экстрагировали смесью изопропанол/CHCl3 (1:2, 50 мл). Органический слой отделяли, сушили над Na2SO4, и упаривали в вакууме, получая 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутановую кислоту (0,029 г, 100%).
b) К смеси 4-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-3-[5-метил-3-(трифторметил)пиразол-1-ил]-4-оксо-бутановой кислоты (0,029 г, 0,08 ммоль) и NH3 (0,3 мл, насыщенный раствор в CH2Cl2) в ДМФА (0,6 мл) добавляли HATU (0,040 г, 0,10 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакцию гасили водой. Смесь разделяли между смесью изопропанол/CHCl3 (1:2, 50 мл) и насыщенным раствором NaHCO3 (30 мл). Органический слой отделяли, сушили над Na2SO4, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ, получая целевой продукт (0,014 г, 37%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,42 (дд, J=9,2, 4,6 Гц, 2 Н), 7,16 (дд, J=8,4, 8,0 Гц, 2 Н), 6,53 (с, 1 Н), 6,34 (с, 1 Н), 6,32 (с, 1 Н), 5,92 (м, 1 Н), 3,80 (м, 1 Н), 3,41 (м, 1 Н), 3,33 (м, 1 Н), 3,00 (м, 1 Н), 2,76 (м, 2 Н), 2,37 (с, 3 Н), 1,98 (м, 1 Н), 1,88 (м, 1 Н); MS: (ES) m/z вычислено для C21H20F4N6O2 [М+Н]+ 465,1, найдено 465,1.
Пример 60. Синтез метил 4-[2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]ацетил]-1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-5-карбоксилата
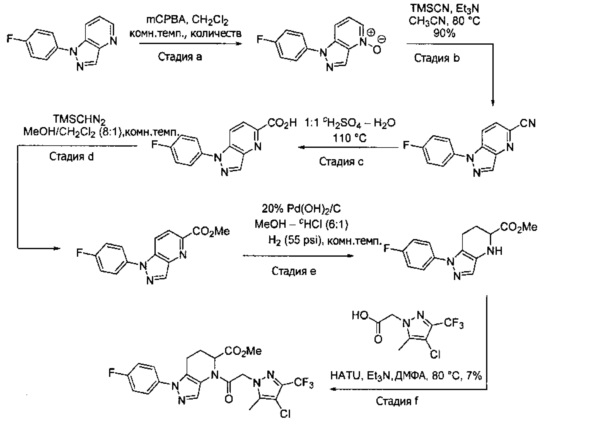
a) К охлажденному раствору 1-(4-фторфенил)пиразоло[4,3-b]пиридина (1 г, 4,67 ммоль) в CH2Cl2 (50 мл) в атмосфере азота добавляли mCPBA (75%, 1,2 г, 5,14 ммоль) в один прием. Полученный раствор нагревали до комнатной температуры и перемешивали 12 часов. Реакционную смесь затем разбавляли добавлением CH2Cl2 (100 мл) и промывали насыщенным раствором NaHCO3 (100 мл). Водный слой экстрагировали CH2Cl2 (50 мл). Объединенные органические слои сушили (MgSO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 20% МеОН в CH2Cl2), получая целевой продукт (1,1 г, 4,7 ммоль, количественный выход).
b) К раствору 1-(4-фторфенил)-4-оксидо-пиразоло[4,3-b]пиридин-4-ия (916 мг, 4 ммоль) в CH3CN (10 мл) добавляли TMSCN (800 мкл, 6 ммоль) и Et3N (556 мкл, 4 ммоль) и перемешивали при 80°С в течение ночи. Реакционную смесь затем охлаждали до комнатной температуры и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 80% EtOAc в гексане), получая целевой продукт (857 мг, 3,6 ммоль, 90%).
c) Воду (10 мл) добавляли к 1-(4-фторфенил)пиразоло[4,3-b]пиридин-5-карбонитрилу (600 мг, 2,52 ммоль), и полученную смесь охлаждали до 0°С, после чего медленно прикапывали концентрированную H2SO4 (10 мл). Полученный прозрачный желтоватый раствор затем перемешивали при 110°С 12 часов. Реакционную смесь охлаждали до 0°С, после чего медленно добавляли 10н. раствор NaOH по каплям при перемешивании до значения pH 5-6. Выпавший белый твердый осадок отфильтровывали и промывали водой (30 мл) и гептаном (50 мл), после чего сушили в высоком вакууме, получая целевой неочищенный продукт (700 мг), который использовали в следующей стадии без дополнительной очистки.
d) К 1-(4-фторфенил)пиразоло[4,3-b]пиридин-5-карбоновой кислоте (650 мг, 2,5 ммоль) по каплям добавляли CH2Cl2 (40 мл) и МеОН (5 мл), затем TMSCHN2 (2 М раствор в Et2O, 10 мл, избыток). Результирующую желтую суспензию перемешивали при комнатной температуре 1 час. Реакционную смесь разбавляли добавлением CH2Cl2 (40 мл), промывали насыщенным раствором NaHCO3 (50 мл), сушили (MgSO4) и упаривали в вакууме, получая целевой неочищенный продукт (750 мг) который напрямую использовали в следующей стадии без дополнительной очистки.
e) МеОН (6 мл) и 12н. раствор HCl (1 мл) добавляли к метил 1-(4-фторфенил)пиразоло[4,3-b]пиридин-5-карбоксилату (100 мг, 0,37 ммоль), затем добавляли 20% Pd(OH)2 на угле (100 мг, избыток), и полученную смесь перемешивали в атмосфере водорода (55 фунт/кв. дюйм) в аппарате Парра при комнатной температуре 36 часов. Реакционную смесь затем фильтровали через небольшой слой целита, промывали метанолом (20 мл) и упаривали в вакууме. Полученный остаток растворяли в EtOAc (50 мл) и промывали насыщенным раствором NaHCO3 (20 мл), сушили (MgSO4) и упаривали в вакууме, получая целевой неочищенный продукт (30 мг), который напрямую использовали в следующей стадии без дополнительной очистки.
f) К раствору 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]уксусной кислоты (20 мг, 0,073 ммоль) в ДМФА (2 мл) добавляли Et3N (50 мкл, 0,219 ммоль), HATU (55 мг, 0,145 ммоль), затем добавляли метил 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин-5-карбоксилат (20 мг, 0,073 ммоль) при комнатной температуре. Полученный раствор затем перемешивали при 80°С 5 часов. Реакционную смесь затем охлаждали до комнатной температуры, добавляли Н2О (10 мл) и полученную смесь экстрагировали EtOAc (2×15 мл). Объединенные этилацетатные слои сушили (MgSO4), фильтровали, упаривали в вакууме и полученный неочищенный продукт очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого порошка (0,004 г, 0,008 ммоль, 7%). 1Н-ЯМР (400 МГц, CD3OD) δ 8,28 (с, 1H), 7,52 (2Н, дд, J=5, 9 Гц), 7,26 (2Н, т, 8,6, J=17,2 Гц), 5,3 (2Н, дд, J=17,2, 69,2 Гц), 3,83 (с, 3Н), 2,80-2,82 (м, 2Н), 2,29 (с, 3Н), 2,15-2,17 (м, 2Н); MS: (ES) m/z вычислено для C21H18ClF4N5O3 [М+Н]+ 500,1, найдено 500,1.
Пример 61. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5Н-пиразоло[4,3-b]пиридин-4-ил]-2-[3-(трифторметил)-5,6-дигидро-4Н-циклопента[с]пиразол-1-ил]этанона

К раствору 2-[3-(трифторметил)-5,6-дигидро-4H-циклопента[с]пиразол-1-ил]уксусной кислоты (83 мг, 0,35 ммоль) в ДМФА (2 мл) добавляли Et3N (75 мкл, 0,525 ммоль), HATU (160 мг, 0,42 ммоль), затем 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (77 мг, 0,35 ммоль) при комнатной температуре. Полученный раствор затем перемешивали при 50°С 30 минут. Реакционную смесь затем охлаждали до комнатной температуры, добавляли Н2О (10 мл), и полученную смесь экстрагировали EtOAc (2×20 мл). Объединенные этилацетатные слои сушили (MgSO4), фильтровали и упаривали в вакууме. Очистка методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-Н32О с 0,1% ТФУК в качестве элюента) давала целевой продукт в виде белого порошка (0,011 г, 0,025 ммоль, 7%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,15 (с, 1Н), 7,6 (м, 2Н), 7,35 (т, 2Н, J=8,6, 17,6 Гц), 5,38 (с, 2Н), 3,77 (м, 2Н), 2,85 (т, 2Н, J=6,3, 12,5 Гц), 2,66 (м, 4Н), 2,53 (м, 2Н), 1,97 (м, 2Н); MS: (ES) m/z вычислено для C21H19F4N5O [М+Н]+ 434,2, найдено 434,1.
Пример 62. Синтез 2-[4-хлор-3-(трифторметил)фенил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]пропан-1-она
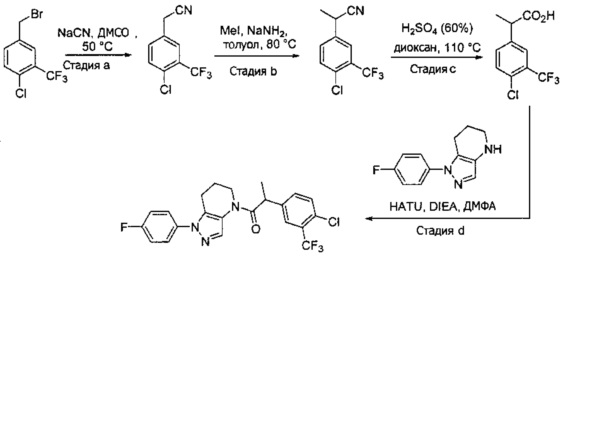
а) К раствору 4-(бромметил)-1-хлор-2-(трифторметил)бензола (3,94 г, 14,4 ммоль) в ДМСО (29 мл) добавляли NaCN (1,06 г, 21,6 ммоль). Полученную смесь нагревали при 50°С 1 час при перемешивании. После охлаждения до комнатной температуры, реакционную смесь выливали в 100-мл стакан с ледяной водой (25 мл). Водный слой экстрагировали дихлорметаном (4×10 мл), объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили (Na2SO4), фильтровали и упаривали в вакууме, получая целевой продукт (2,5 г, 79%).
b) К смеси 2-[4-хлор-3-(трифторметил)фенил]ацетонитрила (2,0 г, 9,1 ммоль) и метилиодида (1,29 г, 9,1 ммоль) в толуоле (20 мл) при 80°С медленно добавляли NaNH2 (428 мг, 11 ммоль). Полученную смесь перемешивали при 80°С 1 час. После охлаждения до комнатной температуры добавляли воду (10 мл). Полученную смесь экстрагировали этилацетатом (2×15 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили (Na2S04), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 0-15% этилацетата в гексане), получая целевой продукт (500 мг, 24%).
c) К раствору 2-[4-хлор-3-(трифторметил)фенил]пропаннитрила (450 мг, 1,93 ммоль) в диоксане (6 мл) добавляли серную кислоту (60%, 6 мл). Полученную смесь нагревали при 110°С 14 часов. После охлаждения до комнатной температуры, реакционную смесь разделяли между водой и дихлорметаном. Водный слой экстрагировали дихлорметаном (2×10 мл), и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали и упаривали в вакууме, получая целевой продукт (350 мг, 72%).
d) К смеси 2-[4-хлор-3-(трифторметил)фенил]пропановой кислоты (47 мг, 0,184 ммоль) и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (40 мг, 0,184 ммоль) в ДМФА (1 мл) добавляли основание Хунига (59 мг, 0,46 ммоль) и 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия (HATU) (77 мг, 0,2 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час и затем разделяли между водой (4 мл) и этилацетатом (6 мл). Органический слой промывали насыщенным водным раствором хлорида натрия и сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С 18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (24 мг, 29%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,51 (с, 1 Н), 7,62 (д, J=2,0 Гц, 1 Н), 7,48-7,42 (м, 4 Н), 7,16-7,11 (м, 2 Н), 4,11 (м, 1 Н), 3,76 (м, 1 Н), 3,52 (м, 1 Н), 2,77 (м, 2 Н), 1,96 (м, 1 Н), 1,80 (м, 1 Н), 1,55 (д, J=7,2 Гц, 3 Н); MS: (ES) m/z вычислено для C22H18ClF4N3O [М+Н]+ 452,1, найдено 452,1.
Пример 63. Синтез 2-[2-хлор-4-(трифторметил)фенил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
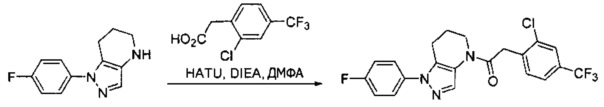
К смеси 2-[2-хлор-4-(трифторметил)фенил]уксусной кислоты (59 мг, 0,23 ммоль) и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (50 мг, 0,23 ммоль) в ДМФА (1 мл) добавляли основание Хунига (59 мг, 0,46 ммоль) и 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия (HATU) (96 мг, 0,2 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час, и затем разделяли между водой (4 мл) и этилацетатом (6 мл). Органический слой промывали насыщенным водным раствором хлорида натрия и сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С 18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (25 мг, 25%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 0,8 Н), 7,70 (с, 1 Н), 7,56-7,44 (м, 4 Н), 7,38 (с, 0,2 Н), 7,18-7-14 (м, 2 Н), 4,14 (с, 2 Н), 3,82 (м, 2 Н), 2,85 (м, 2 Н), 2,08 (м, 2 Н); MS: (ES) m/z вычислено для C21H16ClF4N3O [М+Н]+ 438,1, найдено 438,1.
Пример 64. Синтез 2-[1H-бензимидазо-2-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
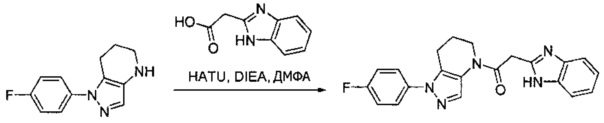
К смеси (1H-бензоимидазол-2-ил)уксусной кислоты (41 мг, 0,23 ммоль) и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (50 мг, 0,23 ммоль) в ДМФА (1 мл) добавляли основание Хунига (59 мг, 0,46 ммоль) и 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия (HATU, 96 мг, 0,2 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час и затем разделяли между водой (4 мл) и этилацетатом (6 мл). Органический слой промывали насыщенным водным раствором хлорида натрия и сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (0,020 г, 23%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,38 (с, 0,8 Н), 8,00 (с, 1 Н), 7,85 (м, 1 Н), 7,61 (с, 0,2 Н), 7,52-7,43 (м, 2 Н), 7,36-7-29 (м, 3 Н), 7,23-7,14 (м, 2 Н), 5,18 (с, 2 Н), 3,84 (м, 2 Н), 2,88 (м, 2 Н), 2,13 (м, 2 Н); MS: (ES) m/z вычислено для C21H16ClF4N3O [М+Н]+ 376,1, найдено 376,1.
Пример 65. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-пирроло[2,3-b]пиридин-1-ил-этанона

a) Смесь 1H-пирроло[2,3-b]пиридина (471,2 мг, 4,0 ммоль), этил 2-бромацетата (530 мкл, 4,8 ммоль) и карбоната калия (667,9 мг, 4,8 ммоль) в ДМФА (5 мл) нагревали при 60°С 2 часа при перемешивании. После охлаждения до комнатной температуры, реакционную смесь разбавляли 20 мл воды и экстрагировали EtOAc (2×30 мл). Объединенные органические слои затем промывали водой (2×20 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 40% этилацетата в гексане), получая целевой продукт (371,5 мг, 1,8 ммоль, 45%).
b) Смесь этил 2-пирроло[2,3-b]пиридин-1-илацетата (371,5 мг, 1,8 ммоль) и гидроксида лития (87,9 мг, 3,6 ммоль) в смеси ТГФ (4 мл) и H2O (1 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали в вакууме, и подкисляли до pH 5 3н. водным раствором соляной кислоты. Выпавший осадок отфильтровывали и промывали водой (2×10 мл), и сушили в вакууме, получая целевой продукт (200,2 мг, 1,1 ммоль, 62%).
c) Смесь 2-пирроло[2,3-b]пиридин-1-илуксусной кислоты (51,2 мг, 0,24 ммоль), 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридина (50,3 мг, 0,29 ммоль), триэтиламина (200 мкл, 1,4 ммоль) и HATU (110,7 мг, 0,29 ммоль) в ДМФА (1 мл) перемешивали при комнатной температуре 2 часа. Реакционную смесь разбавляли добавлением 10 мл насыщенного водного раствора бикарбоната натрия, и экстрагировали EtOAc (2×15 мл). Объединенные органические слои затем промывали водой (2×20 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 70% этилацетата в гексане), получая целевой продукт (25,9 мг, 0,07 ммоль, 28%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,40 (с, 0,8 Н), 8,30 (дд, J=4,8, 1,6 Гц, 1 Н), 7,94 (дд, J=7,2, 1,6 Гц, 1 Н), 7,81 (с, 0,2 Н), 7,45 (м, 2 Н), 7,36 (д, J=3,6 Гц, 0,8 Н), 7,31 (д, J=3,6 Гц, 0,2 Н), 7,18 (м, 2 Н), 7,09 (дд, J=7,6, 4,8 Гц, 1 Н), 6,58 (д, J=3,6 Гц, 1 Н), 5,44 (с, 0,2 Н), 5,32 (с, 0,8 Н), 3,91 (м, 2Н), 2,84 (м, 2Н), 2,07 (м, 2Н); MS: (ES) m/z вычислено для C21H18FN5O [М+Н]+ 376,1, найдено 376,1.
Пример 66. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-имидазо[4,5-b]пиридин-3-ил-этанона
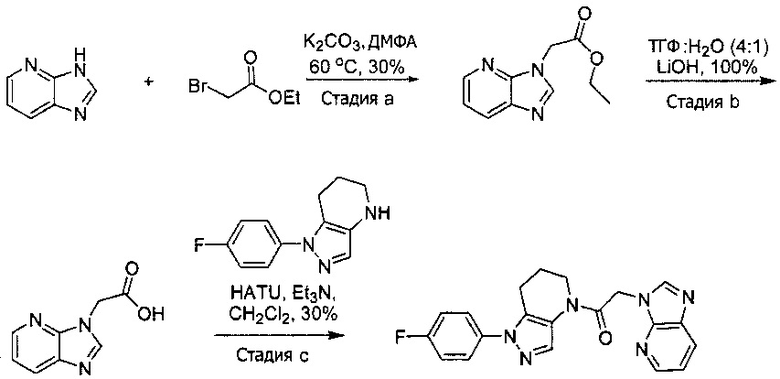
a) Смесь 3H-имидазо[4,5-b]пиридина (952,7 мг, 8,0 ммоль), этил 2-бромацетата (1,0 мл, 9,6 ммоль) и карбоната калия (1,37 г, 9,6 ммоль) в ДМФА (10 мл) нагревали при 60°С 2 часа при перемешивании. После охлаждения до комнатной температуры, реакционную смесь разбавляли 20 мл воды и экстрагировали EtOAc (2×30 мл). Объединенные органические слои промывали водой (2×20 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, от 100% этилацетат до 10% МеОН в EtOAc), получая целевой продукт (489,3 мг, 2,4 ммоль, 30%).
b) Смесь этил 2-имидазо[4,5-b]пиридин-3-илацетата (400,3 мг, 1,9 ммоль) и гидроксида лития (96,7 мг, 4,0 ммоль) в смеси ТГФ (4 мл) и Н2О (1 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали в вакууме и подкисляли до pH 5 3н. водным раствором соляной кислоты. Полученный водный раствор затем сушили в вакууме, получая сырой продукт, который напрямую использовали в следующей стадии.
c) Смесь 2-имидазо[4,5-b]пиридин-3-илуксусной кислоты (251,7 мг, избыток), 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридина (42,7 мг, 0,23 ммоль), триэтиламина (200 мкл, 1,4 ммоль) и HATU (87,9 мг, 0,23 ммоль) в ДМФА (1 мл) перемешивали при комнатной температуре 2 часа. Реакционную смесь разбавляли 10 мл насыщенного водного раствора бикарбоната натрия и экстрагировали EtOAc (2×15 мл). Объединенные органические слои промывали водой (2×20 мл), сушили (Na2SO4), фильтровали и упаривали в вакууме. Неочищенный остаток очищали методом флэш-хроматографии (SiO2, 10% МеОН в EtOAc), получая целевой продукт (26,1 мг, 0,07 ммоль, 30%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,38 (д, J=7,2 Гц, 1 Н), 8,50 (с, 0,8 Н), 8,25 (с, 0,2 Н), 8,12 (д, J=5,6 Гц, 1 Н), 7,45 (м, 2 Н), 7,28 (м, 1Н), 7,15 (м, 2Н), 5,46 (с, 0,2 Н), 5,33 (с, 0,8 Н), 3,95 (м, 2Н), 2,88 (м, 2Н), 2,15 (м, 2Н); MS: (ES) m/z вычислено для C20H17FN6O [М+Н]+ 377,1, найдено 377,1.
Пример 67. Синтез 2-[4-хлор-5-метил-3-(метилсульфонил)пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
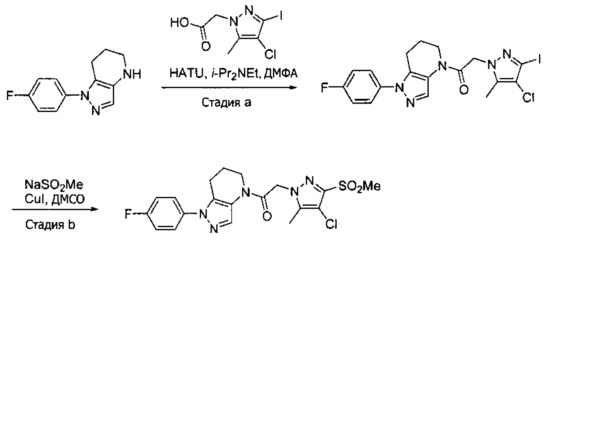
a) К смеси 1-(4-фторфенил)-4,5,6,7-тетрагидро-1Н-пиразоло[4,3-b]пиридина (303 мг, 1,40 ммоль), 2-[4-хлор-5-метил-3-(трифторметил)пиразол-1-ил]уксусной кислоты (420 мг, 1,40 ммоль) и изо-Pr2NEt (1 мл, 7,0 ммоль) в ДМФА (5 мл) добавляли HATU (583 мг, 1,54 ммоль). Полученный раствор затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением EtOAc (50 мл) и промывали водой (3×10 мл). Этилацетатный слой сушили (Na2SO4), фильтровали и упаривали в вакууме. Сырой продукт (480 мг, 70%) напрямую использовали в следующей стадии.
b) Смесь 2-(4-хлор-5-метил-3-иодпиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5Н-пиразоло[4,3-b]пиридин-4-ил]этанона (85 мг, 0,17 ммоль), NaSO2Me (52 мг, 0,51 ммоль), CuI (98 мг, 0,51 ммоль) в ДМСО (3 мл) нагревали при 110°С в течение ночи. После охлаждения до комнатной температуры, реакционную смесь разбавляли EtOAc (50 мл) и промывали водой (3×10 мл). Этилацетатный слой сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С 18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого порошка (42 мг, 55%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,15 (с, 1 Н), 7,60 (м, 2Н), 7,38 (дд, J=8,9, 8,1 Гц, 2 Н), 5,58 (с, 2 Н), 3,80 (м, 2 Н), 3,28 (с, ЗН), 2,83 (т, J=5,7 Гц, 2 Н), 2,22 (с, 3 Н), 2,00 (м, 2 Н); MS: (ES) m/z вычислено для C19H20ClFN5O3S [М+Н]+ 452,1, найдено 452,0.
Пример 68. Синтез 2-(4-хлор-5-метил-3-цианопиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона

Смесь 2-(4-хлор-5-метил-3-иодпиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (430 мг, 0,86 ммоль), Zn(CN)2 (151 мг, 1,3 ммоль), dppf (72 мг, 0,14 ммоль) и Pd2(dba)3 (79 мг, 0,09 ммоль) в ДМФА (10 мл) и воде (0,5 мл) нагревали при 90°С в течение ночи. После охлаждения до комнатной температуры, реакционную смесь разбавляли EtOAc (30 мл) и промывали водой (3×5 мл). Этилацетатный слой сушили (Na2SO4), фильтровали и упаривали в вакууме. Полученный остаток растирали в смеси 1:1 CH2Cl2/МеОН (3×5 мл), получая целевой продукт в виде белого порошка (250 мг, 72%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,15 (с, 1 Н), 7,58 (м, 2Н), 7,35 (дд, J=8,7, 8,1 Гц, 2 Н), 5,58 (с, 2 Н), 3,80 (м, 2 Н), 2,84 (т, J=5,4 Гц, 2 Н), 2,25 (с, 3 Н), 2,00 (м, 2 Н); MS: (ES) m/z вычислено для C19H17ClF4N5O [М+Н]+ 399,1, найдено 399,1.
Пример 69. Синтез соли 2-[4-хлор-3-(4,5-дигидро-1H-имидазол-2-ил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой
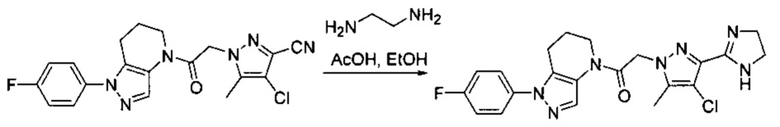
Смесь 2-(4-хлор-5-метил-3-цианопиразол-1-ил)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (150 мг, 0,38 ммоль), этилендиамина (2 мл) и уксусной кислоты (0,3 мл) в этаноле (5 мл) нагревали при 100°С 2 часа. После охлаждения до комнатной температуры, реакционную смесь разбавляли CH2Cl2 (50 мл) и промывали водой (3×10 мл). Органический слой сушили (Na2S04), фильтровали и упаривали в вакууме. Очистка методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента) дала соль 2-[4-хлор-3-(4,5-дигидро-1H-имидазол-2-ил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона с трифторуксусной кислотой в виде белого порошка (84 мг, 51%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 10,21 (с, 2Н), 8,15 (с, 1 Н), 7,58 (м, 2 Н), 7,37 (дд, J=8,7, 8,1 Гц, 2 Н), 5,60 (с, 2 Н), 3,92 (с, 4Н), 3,82 (м, 2 Н), 2,89 (т, J=4,9 Гц, 2 Н), 2,29 (с, 3 Н), 2,01(м, 2 Н); MS: (ES) m/z вычислено для C21H22ClFN7O [М+Н]+ 442,2, найдено 442,1.
Пример 70. Синтез 2-[4-хлор-3-(1H-имидазол-2-ил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона
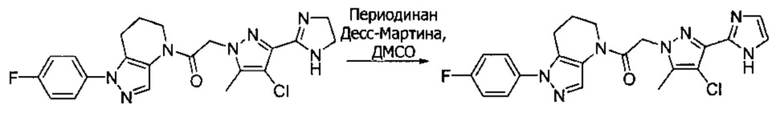
Смесь 2-[4-хлор-3-(4,5-дигидро-1H-имидазол-2-ил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанона (45 мг, 0,1 ммоль), периодинана Десс-Мартина (80 мг, 0,2 ммоль) в ДМСО (1 мл) нагревали при 80°С 1 час. После охлаждения до комнатной температуры, реакционную смесь разбавляли CH2Cl2 (20 мл) и промывали водой (3×5 мл). Органический слой сушили (Na2SO4), фильтровали, упаривали е вакууме и очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 2-[4-хлор-3-(1H-имидазол-2-ил)-5-метил-пиразол-1-ил]-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]этанон в виде белого порошка (32 мг, 73%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,09 (с, 1 Н), 7,60 (с, 2Н), 7,52 (м, 2 Н), 7,30 (дд, J=8,8, 8,3 Гц, 2 Н), 5,49 (с, 2 Н), 3,90 (м, 2 Н), 2,82 (т, J=5,1 Гц, 2 Н), 2,22 (с, 3 Н), 1,95 (м, 2 Н); MS: (ES) m/z вычислено для C21H20ClFN7O [М+Н]+ 440,1, найдено 440,1.
Пример 71. Синтез (2S)-1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[2-метил-4-(трифторметил)имидазол-1-ил]пропан-1-она
К раствору (2S)-2-[2-метил-4-(трифторметил)имидазол-1-ил]пропановой кислоты (0,020 г, 0,09 ммоль) в CH2Cl2. (3 мл) добавляли оксалилхлорид (0,040 мл, 0,46 ммоль) и ДМФА (1 капля). Через 20 минут перемешивания при комнатной температуре, смесь упаривали в вакууме, и остаток добавляли в другую колбу, содержащую 1-(4-фторфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (0,028 г, 0,13 ммоль) и NEt3 (0,060 мл, 0,43 ммоль) в CH2Cl2 (3 мл). Полученную смесь перемешивали при комнатной температуре 20 минут, гасили насыщенным водным раствором NaHCO3 (30 мл) и экстрагировали этилацетатом (50 мл). Органический слой сушили над безводным сульфатом натрия, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2, 0-8% MeOH/EtOAc), получая указанное в заголовке соединение (0,030 г, 44%, соль с ТФУК) в виде белого твердого вещества. 1Н-ЯМР (соль с ТФУК) (400 МГц, CDCl3) δ 9.70 (с, 1 Н), 8,43 (с, 1 Н), 7,78 (д, J=0,8 Гц, 1 Н), 7,42 (дд, J=9,2, 8,8 Гц, 2 Н), 7,20 (дд, J=8,8, 8,4 Гц, 2 Н), 5,61 (кв, J=7,2 Гц, 1 Н), 3,89 (м, 2 Н), 2,84 (дд, J=6,2, 6,2 Гц, 2 Н), 2,65 (с, 3 Н), 2,15 (м, 2 Н), 1,84 (д, J=7,2 Гц, 3 Н). MS: (ES) m/z вычислено для C20H19F4N5O [М+Н]+ (свободная форма) 422,1, найдено 422,1; Указанные в заголовке соединения анализировали методом хиральной хроматографии на нормальной фазе (RegisPack, 25 см × 4,6 мм, 5 микрон, кат. №793104, 0,1% DEA/IPA, 0,7 мл/мин). (S)-энантиомер (преобладающий) имел время удерживания 7,2 мин, а (R)-энантиомер (минорный) имел время удерживания 6,2 мин (выделены в энантиомерном соотношении 19:1).
Пример 72. Синтез (2S)-1-[1-(4-хлорфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[2-метил-4-(трифторметил)имидазол-1-ил]пропан-1-она
К раствору (2S)-2-[2-метил-4-(трифторметил)имидазол-1-ил]пропановой кислоты (0,020 г, 0,09 ммоль) в CH2Cl2 (1,5 мл) добавляли оксалилхлорид (0,050 мл, 0,58 ммоль) и ДМФА (1 капля). Через 20 минут при комнатной температуре, смесь упаривали в вакууме. И остаток добавляли в другую колбу, содержащую 1-(4-хлорфенил)-4,5,6,7-тетрагидропиразоло[4,3-b]пиридин (0,028 г, 0,13 ммоль) и NEt3 (0,050 мл, 0,35 ммоль) в CH2Cl2 (1 мл). Полученную смесь перемешивали при комнатной температуре 20 минут, гасили насыщенным водным раствором NaHCO3 (30 мл) и экстрагировали этилацетатом (50 мл). Органический слой сушили над безводным сульфатом натрия, упаривали в вакууме и очищали методом флэш-хроматографии (SiO2, 0-6% MeOH/EtOAc), получая указанное в заголовке соединение (0,022 г, 32%, соль с ТФУК) в виде белого твердого вещества. 1Н-ЯМР (свободная форма) (400 МГц, CDCl3) δ 8,46 (с, 1 Н), 7,44 (м, 4 Н), 7,42 (дд, J=9,2, 8,8 Гц, 2 Н), 7,37 (д, J=1,2 Гц, 1 Н), 5,21 (кв, J=7,2 Гц, 1 Н), 3,75 (м, 1 Н), 3,53 (м, 1 Н), 2,86 (дд, J=6,4, 6,4 Гц, 2 Н), 2,46 (с, 3 Н), 1,72 (д, J=6,8 Гц, 3 Н); MS: (ES) m/z вычислено для C20H19ClF3N5O [М+Н]+ (свободная форма) 438,1, найдено 438,1; Указанные в заголовке соединения анализировали методом хиральной хроматографии на нормальной фазе (RegisPack, 25 см × 4,6 мм, 5 микрон, кат. №793104, 0,1% DEA/IPA, 0,7 мл/мин). (S)-энантиомер (преобладающий) имел время удерживания 8,4 мин, a (R)-энантиомер (минорный) имел время удерживания 6,4 мин (выделены в энантиомерном соотношении 18:1).
Пример 73. Синтез 1-[1-(4-фторфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)-1,2,4-триазол-1-ил]пропан-1-она
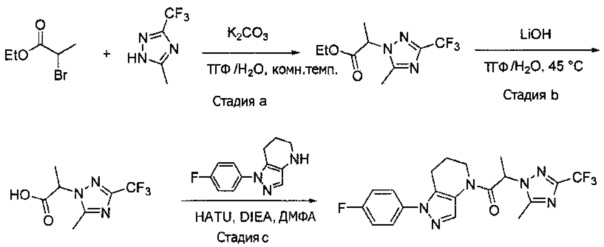
a) Раствор этил 2-бромпропионата (3,16 г, 17,5 ммоль), 5-метил-3-(трифторметил)-1Н-1,2,4-триазола (2,20 г, 14,6 ммоль) и K2CO3 (4,00 г, 29,1 ммоль) в ТГФ/H2O (2:1, 30 мл) перемешивали при комнатной температуре 5 часов. Полученную смесь разбавляли добавлением EtOAc (50 мл). Органический слой промывали водой и насыщенным водным раствором хлорида натрия, сушили (MgSO4), фильтровали и упаривали в вакууме, получая целевой продукт (4,0 г) в виде бесцветного масла, которое использовали в следующей стадии без дополнительной очистки.
b) В неочищенный продукт стадии а в ТГФ (40 мл) добавляли 2н. раствор LiOH (15 мл, 30 ммоль) при 45°С, выдерживали 1 час и упаривали. Полученный остаток разбавляли водой (20 мл), доводили значение pH до 2 добавлением 1М раствора H2SO4 и экстрагировали EtOAc (50 мл). Органический слой сушили (MgSO4), фильтровали и упаривали в вакууме, получая целевую кислоту (2,56 г, 10,2 ммоль, 70% за две стадии) в виде бесцветного твердого вещества.
c) К смеси 2-[5-метил-3-(трифторметил)-1,2,4-триазол-1-ил]пропановой кислоты (0,051 г, 0,23 ммоль) и 1-(4-фторфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,050 г, 0,23 ммоль) в ДМФА (1 мл) добавляли основание Хунига (0,059 г, 0,46 ммоль) и 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия (HATU) (0,096 г, 0,25 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час и затем разделяли между водой (4 мл) и этилацетатом (6 мл). Органический слой промывали насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (0,029 г, 0,069 ммоль, 30%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,42 (с, 1 Н), 7,44 (ддд, J=10,4, 5,2, 2,8 Гц, 2 Н), 7,21-7,10 (м, 2 Н), 5,61 (кв, J=7,1 Гц, 1 Н); 3,77 (ддд, J=12,6, 7,5, 3,0 Гц, 1 Н), 3,52 (ддд, J=12,5, 8,4, 3,0 Гц, 1 Н), 2,82 (тд, J=6,4, 2,4 Гц, 2 Н), 2,52 (с, 3Н), 2,10-1,80 (м, 5 Н); MS: (ES) m/z вычислено для C19H18ClF3N6O [М+Н]+ 423,1, найдено 422,9.
Пример 74. Синтез 1-[1-(4-хлорфенил)-6,7-дигидро-5H-пиразоло[4,3-b]пиридин-4-ил]-2-[5-метил-3-(трифторметил)-1,2,4-триазол-1-ил]пропан-1-она
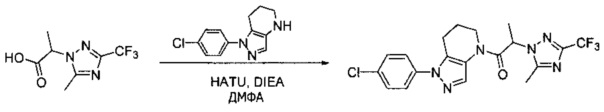
К смеси 2-[5-метил-3-(трифторметил)-1,2,4-триазол-1-ил]пропановой кислоты (0,048 г, 0,21 ммоль) и 1-(4-хлорфенил)-4,5,6,7-тетрагидро-1H-пиразоло[4,3-b]пиридина (0,050 г, 0,21 ммоль) в ДМФА (1 мл) добавляли основание Хунига (0,055 г, 0,42 ммоль) и 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминия (HATU) (0,090 г, 0,23 ммоль). Полученную смесь перемешивали при комнатной температуре 1 час и затем разделяли между водой (4 мл) и этилацетатом (6 мл). Органический слой промывали насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали, упаривали в вакууме и очищали методом обращенно-фазной ВЭЖХ (С18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая целевой продукт в виде белого твердого вещества (0,028 г, 0,063 ммоль, 30%). 1Н-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1 Н), 7,43 (с, 4 Н), 5,60 (кв, J=7,1 Гц, 1 Н), 3,77 (ддд, J=12,6, 7,5, 3,0 Гц, 1 Н), 3,52 (ддд, J=12,3, 8,3, 3,0 Гц, 1 Н), 2,84 (тт, J=6,9, 3,3 Гц, 2 Н), 2,52 (с, 3Н), 2,09-1,81 (м, 5 Н); MS: (ES) m/z вычислено для C19H18ClF3N6O [М+Н]+ 439,1, найдено 438,9.
Пример 75
В данном примере проиллюстрирована оценка биологической активности соединений по настоящему изобретению (соединений-кандидатов).
Материалы и методы
А. Клетки
1. CCR1 экспрессирующие клетки
а) ТНР-1 клетки
ТНР-1 клетки получали от АТСС (TIB-202) и выращивали в виде суспензии в среде RPMI-1640 с добавлением 2 мМ L-глютамина, 1,5 г/л бикарбоната натрия, 4,5 г/л глюкозы, 10 мМ HEPES, 1 мМ пирувата натрия, 0,05% 2-меркаптоэтанола и 10% FBS. Клетки выращивали в атмосфере 5% CO2/95% воздух, 100% влажности при 37°С и пересевали дважды в неделю при соотношении 1:5 (клетки выращивали при плотности от 2×105 до 2×106 клеток/мл) и отбирали на анализ с плотностью 1×106 клеток/мл. ТНР-1 клетки экспрессируют CCR1 и могут применяться в анализе связывания с CCR1 в функциональном тесте.
b) Выделенные моноциты человека
Моноциты выделяли из лейкотромбоцитарного слоя человека с применением системы выделения частиц Miltenyi (Miltenyi, Auburn, СА). Вкратце, после фиколл-градиентного разделения с выделением моноядерных клеток из периферической крови, клетки промывали PBS, и красные кровяные тельца лизировали по стандартной методике. Оставшиеся клетки метили анти-CD14 антителами, соединенными с магнитными частицами (Miltenyi Biotech, Auburn, СА). Меченые клетки пропускали через AutoMACS (Miltenyi, Auburn, СА) и собирали положительно заряженную фракцию. Моноциты экспрессируют CCR1 и могут применяться в анализе связывания с CCR1 и функциональном тесте.
В. Тесты
1. Подавление связывания CCR1 с лигандом
CCR1-экспрессирующие клетки центрифугировали и заново суспендировали в буфере для анализа (20 мМ HEPES pH 7,1, 140 мМ NaCl, 1 мМ CaCl2, 5 мМ MgCl2, с 0,2% альбумина бычьей сыворотки) до концентрации 5×106 клеток/мл для клеток ТНР-1 и 5×105 для моноцитов. Анализ связывания проводили следующим образом: 0,1 мл клеток (5×105 ТНР-1 клеток/лунку или 5×104 моноцитов) помещали в планшеты для анализа, содержащие испытуемые соединения, до конечной концентрации ~2-10 мкМ каждого соединения, для скрининга (или для определения зависимости ответа от дозировки, с целью определения значения IC50 для испытуемых соединений). Затем добавляли 0,1 мл 125I меченого MIP-1α (получен от Perkin Elmer Life Sciences, Boston, MA) или 0,1 мл 125I меченого CCL15/лейкотактин (получен как помеченный по заказу от Perkin Elmer Life Sciences, Boston, MA), разбавленного в буфере для анализа до конечной концентрации ~50 пкМ, что дает ~30000 имп/мин на лунку (применяя 125I меченый MIP-1α с ТНР-1 клетками и 125I меченый CCL15/лейкотактин с моноцитами), планшеты герметично закрывали и инкубировали примерно 3 часа при 4°С на качающейся платформе. Реакционные смеси фильтровали при отсасывании через стеклянные фильтры GF/B, предварительно смоченные 0,3%-ным раствором полиэтиленимина (PEI), на вакуумном коллекторе клеток (Packard Instruments; Meriden, CT). В каждую лунку добавляли сцинтилляционную жидкость (40 мкл; Microscint 20, Packard Instruments), планшеты герметично закрывали и замеряли радиоактивность на сцинтилляционном счетчике Topcount (Packard Instruments). Контрольные лунки, содержащие только разбавитель (для общего подсчета) или избыток MIP-1α или MIP-1β (1 мкг/мл, для неспецифического связывания), использовали для подсчета процента общего подавления для соединения. Компьютерную программу Prism от GraphPad, Inc. (San Diego, Ca) использовали для подсчета значений IC50. Значения IC50 представляют собой концентрации, необходимые для уменьшения связывания меченого MIP-1α с рецептором на 50%. (Дальнейшее описание анализа связывания лигандов и других функциональных тестов смотри в Dairaghi, et al., J. Biol. Chem. 274:21569-21574 (1999), Penfold, et al., Proc. Natl. Acad. Sci. USA. 96:9839-9844 (1999), и Dairaghi, et al,. J. Biol. Chem. 272:28206-28209(1997)).
2. Мобилизация кальция
Для детектирования высвобождения кальция из внутриклеточных хранилищ, клетки (ТНР-1 или моноциты) инкубировали с 3 мкМ красителя INDO-1AM (Molecular Probes; Eugene, OR) в клеточной среде в течение 45 минут при комнатной температуре и промывали фосфатно-солевым буферным раствором (PBS). После загрузки INDO-1AM, клетки суспендировали в буферном растворе (сбалансированный солевой раствор Хэнкса (HBSS) и 1% FBS). Мобилизацию кальция замеряли с помощью спектрофотометра Photon Technology International (Photon Technology International; New Jersey) с длиной волны возбуждения 350 нм и двойной одновременной регистрацией флуоресценции испускания при 400 нм и 490 нм. Относительные концентрации внутриклеточного кальция выражали в виде соотношения эмиссии 400 нм/490 нм. Эксперименты проводили при 37°С и постоянном перемешивании в кюветах, каждая из которых содержала 106 клеток в 2 мл буферного раствора. Хемокиновые лиганды можно применять в диапазоне от 1 до 100 нМ. Эмиссионное соотношение откладывали на графике относительно времени (в типичном случае 2-3 минуты). Соединения-кандидаты, блокирующие лиганды (до 10 мкМ) добавляли через 10 секунд, затем добавляли хемокины через 60 секунд (т.е., MIP-1α; R&D Systems; Minneapolis, MN) и контрольный хемокин (т.е., SDF-loc; R&D Systems; Minneapolis, MN) через 150 секунд.
3. Исследование хемотаксиса
Исследования хемотаксиса проводили с использованием поликарбонатных покрытых поливинилпирролидоном фильтров с диаметром пор 5 мкм, в 96-луночных камерах для исследования хемотаксиса (Neuroprobe; Gaithersburg, MD), применяя хемотаксис-буфер (сбалансированный солевой раствор Хэнкса (HBSS) и 1% FBS). Использовали CCR1 хемокиновые лиганды (т.е., MIP-1α, CCL15/Лейкотактин; R&D Systems; Minneapolis, MN) для оценки подавления исследуемым соединением миграции, вызываемой CCR1. Другие хемокины (т.е., SDF-1α; R&D Systems; Minneapolis, MN) использовали в качестве контроля специфичности. В нижнюю камеру помещали 29 мкл хемокина (т.е., 0,1 нМ CCL15/Лейкотактин) и различные количества соединения, верхняя камера содержала 100000 ТНР-1 или моноцитов в 20 мкл. Камеры инкубировали 1-2 часа при 37°С, и число клеток в нижней камере подсчитывали либо путем прямого подсчета клеток в пяти полях высокой мощности на лунку, либо посредством CyQuant анализа (Molecular Probes), метода флуоресцентного окрашивания, в котором замеряют содержание нуклеиновой кислоты, и замерами с помощью Spectrafluor Plus (Tecan). Компьютерную программу Prism для GraphPad, Inc. (San Diego, Ca) применяли для вычисления значений IC50. Значения IC50 представляют собой концентрации соединений, необходимые для 50%-ного уменьшения числа клеток, отвечающих на CCR1 агонист.
4. Эффективность in vivo
a) Животная модель «кролик» деструктивного воспаления сустава
LPS исследование на кроликах проводили по существу как описано в статье Podolin, et al. J. Immunol. 169(11):6435-6444 (2002). Самкам новозеландского кролика (примерно 2 килограмма) вводили внутрисуставно в оба колена LPS (10 нг). Исследуемое соединение, например, 1,016 (в 1%-ном растворе метилцеллюлозы) или растворитель (1%-ный раствор метилцеллюлозы) дозировали перорально по 5 мл/кг в два приема (за 2 часа до внутрисуставной инъекции LPS и через 4 часа после внутрисуставной инъекции LPS). Через 16 часов после инъекции LPS, проводили лаваж колена и подсчитывали число клеток. Положительный эффект лечения определяли по уменьшению числа воспалительных клеток, привлеченных в синовиальную жидкость воспаленного коленного сустава. Обработка исследуемым соединением приводила к значительному уменьшению числа привлеченных воспалительных клеток.
b) Оценка исследуемого соединения в крысиной модели коллаген-индуцируемого артрита
Проводили 17-дневное исследование развивающегося артрита от коллагена II типа для оценки действия исследуемого соединения на вызываемое артритом клиническое распухание голеностопного сустава. Коллагеновый артрит у крыс является экспериментальной моделью полиартрита, которая широко используется для доклинических испытаний многих противоревматических средств (см. Trentham, et al., J. Exp. Med. 146(3):857-868 (1977), Bendele, et al., Toxicologic Pathol. 27:134-142 (1999), Bendele, et al., Arthritis Rheum. 42:498-506 (1999)). Отличительными чертами данной модели являются надежное наступление симптомов и прогресс устойчивого, легко измеряемого многосуставного воспаления, выраженное разрушение хряща в комбинации, с формированием паннуса, и от умеренной до средней резорбция костной ткани и разрастание околохрящевой кости.
Самок крыс Льюиса (примерно 0,2 килограмма) усыпляли изофлураном и инъецировали им неполный адъювант Фрейнда, содержащий 2 мг/мл телячьего коллагена II типа, в основание хвоста и в две точки на спине в день 0 и день 6 в ходе 17-дневного исследования. Исследуемое соединение дозировали подкожно ежедневно со дня 0 до дня 17 в эффективной дозировке. Проводили измерение диаметра голеностопного сустава, и уменьшение опухоли сустава считали мерилом эффективности.
Мышиная модель дерматологического заболевания
Соединения по настоящему изобретению можно исследовать в мышиной модели кожной гиперчувствительности замедленного типа, индуцируемой оксазолоном. Вкратце, мышей BALB/c возраста 8-10 недель сенсибилизировали местным нанесением 1%-ного раствора оксазолона в этаноле, в день 0. На 6-й день мышам, прошедшим сенсибилизацию, перорально вводили растворитель или увеличивающиеся дозировки соединения по настоящему изобретению, непосредственно перед и через 4 часа после местного нанесения 0,5%-ного раствора оксазолона в этаноле на правое ухо. На следующий день (день 7) замеряли толщину уха с помощью измерения каверномером. Животные, которым вводили исследуемое соединение, имели значительно меньшее опухание уха, в сравнении с контрольной группой, получавшей только растворитель, демонстрируя обуславливаемое соединением уменьшение оксазолон-индуцируемой кожной гиперчувствительности.
Мышиная модель астмы
Соединения по настоящему изобретению можно протестировать в мышиной модели аллергической астмы. Астму вызывают у мышей BALB/c возраста 8-10 недель посредством сенсибилизирования мышей овальбумином в алюминиевом адъюванте в дни 0 и 10. На 20-й день мышам вводили овальбумин в PBS назально, вызывая воспаление дыхательных путей. Группам мышей вводили либо носитель, либо возрастающие дозировки соединения по настоящему изобретению, начиная с 20-го дня и продолжая до 23-го дня. Животных исследовали на 23-й день после назального введения овальбумина на предмет клеточных инфильтратов в бронхоальвеолярных выделениях (BAL). Значительное уменьшение числа лейкоцитов в BAL относительно мышей, которым вводили носитель, указывает на эффективность соединения в данной модели.
Мышиная модель рака
В данном примере описана методика оценки эффективности CCR1 антагонистов в лечении злокачественных опухолей. Нормальным линиям мышей можно пересадить различные хорошо описанные линии рака мышей, включая мышиную тимому EL4, трансфицированную овальбумином, что позволяет легко оценивать опухоль-специфичные антигенные ответы после вакцинации овальбумином. Три серии групп мышей из любой из моделей опухолей тестировали на эффективность CCR1 антагонистов следующим способом: одной серии мышей дополнительно вводили PBS и Tween 0,5% интраперитонеально, вскоре после трансплантации опухоли и далее с различными режимами дозирования. Вторая серия состояла из групп мышей, которым вводили различные дозировки CCR1 антагониста интраперитонеально, внутривенно, подкожно, внутримышечно, перорально или каким-либо другим образом, вскоре после пересадки опухоли и далее с различными режимами дозирования. Третья серия мышей, выступающая в роли положительного контроля, состоит из групп, которым вводили анти-IL4 антитела, анти-IFNg антитела, IL4 или TNF, интраперитонеально, вскоре после пересадки опухоли и далее с различными режимами дозирования. Эффективность отслеживали по росту опухоли относительно регрессии. В случае OVA-трансфицированной модели EL4 тимомы, цитолитические OVA-специфические ответы можно измерить путем стимуляции клеток дренирующих лимфатических узлов овальбумином in vitro и измерения антиген-специфической цитотоксичности через 72 часа.
Мышиная модель воспалительного заболевания кишечника
У MDR1a-нокаут мышей, не имеющих гена Р-гликопротеина, спонтанно развивался колит в условиях отсутствия специфического патогена. Патология у данных животных характеризовалась как опосредованное Т-клетками воспаление Th1-типа, сходное с язвенным колитом у человека. Заболевание обычно начинает развиваться примерно через 8-10 недель после рождения. Однако возраст, в котором данное заболевание проявляется, и максимальный уровень его распространения часто сильно различается между различными вивариями. В исследовании с использованием MDR1a-нокаут мышей, CCR1 антагонист можно оценивать с профилактической или терапевтической точки зрения, в зависимости от времени введения. Самкам мыши (n=34) ежедневно подкожно вводили тестируемое соединение в эффективной дозировке. В данном исследовании оценивали связанные с воспалительным заболеванием кишечника параметры: замедление роста, выраженность анальных выделений и раздражения. Соединение, которое уменьшает анальные выделения и раздражение или ингибирует связанное с воспалительным заболеванием кишечника замедление роста, указывает на эффективность соединения в данном показании.
Мышиная модель солидных опухолей
Мышиная модель опухоли RENCA точно имитирует развитие карциномы клеток почек у человека, в особенности в части спонтанных метастазов в легкие, и служит моделью солидных опухолей. Самкам мышей Balb/c 6-8-недельного возраста инокулировали примерно 5е5 клеток RENCA (аденокарцинома почек у мышей; АТСС cat# CRL-2947) под почечную капсулу и наблюдали рост почечной опухоли в течение 22 дней, при этом метастазы в легкие наблюдали уже на 15-й дней. Животным вводили носитель или соединение по настоящему изобретению, например, каждый день подкожно, начиная со дня пересадки опухоли, для мониторинга влияния на первичный рост, или в более позднее время (например, день 7) для мониторинга действия соединения на метастазы. Площади первичной опухоли замеряют дважды в неделю с использованием механического кавернометра. Объемы опухолей вычисляют по формуле v=pab2/6, где а это самый длинный диаметр, и b представляет собой второй по длине диаметр, перпендикулярный а. Уменьшение объема опухоли или частоты появления метастазов указывает на эффективность соединения по настоящему изобретению.
Мышиная модель легочных заболеваний, вызванных облучением
Для оценки эффективности соединения по настоящему изобретению в восстановлении после RIPD можно использовать различные модели. Например, Tokuda et al описывает модель фиброза легких, вызванного блеомицином (Tokuda et al, J Immunol. 164(5):2745-51 (2000)). Альтернативно, Yang et al описывает модель заболевания, вызванного прямым облучением (Yang et al, Am J Respir Cell Mol Biol. 45(1): 127-35 (2011)). Вкратце, обездвиживали мышей возраста 10-12 недель, обработанных или необработанных анестетическим соединением, закрывали все, за исключением грудной полости, и животных облучали единовременной дозировкой в 14,5 Гр от цезиевого источника с мощностью излучения 1,65 Гр/мин. В этой дозировке выживаемость достаточна для обеспечения достаточного числа животных для долговременного анализа. Эффективность соединения можно оценить многими методами, известными в данной области, включая механический осмотр легких, уровень гидроксипролинового геля и др.
В приведенной ниже таблице представлены структуры и показатели активности для описанных в настоящем тексте репрезентативных соединений. Показатели активности для описанных выше тестов на хемотаксис и на связывание приведены в следующем формате:
+, IC50>12,5 мкМ;
++, 2500 нМ<IC50<12,5 мкМ;
+++, 1000 нМ<IC50<2500 нМ; и
++++, IC50<1000 нМ.
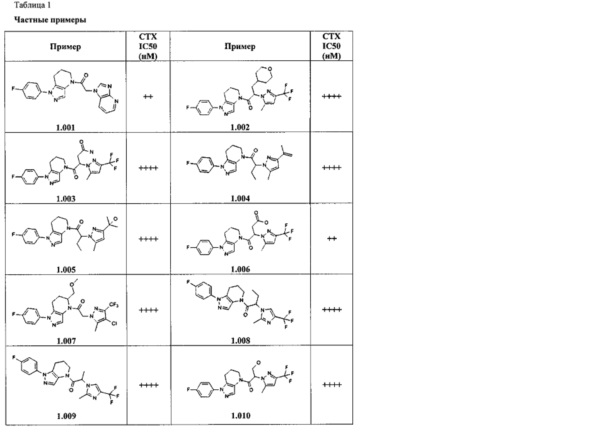
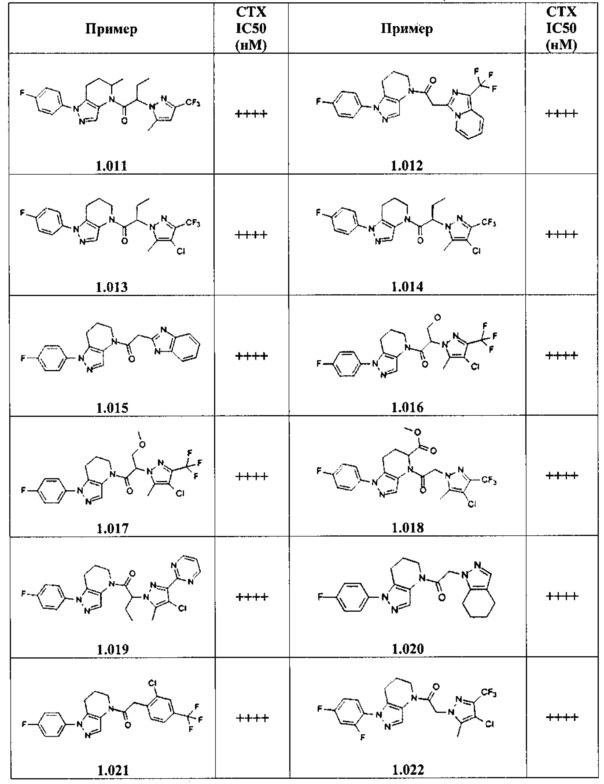
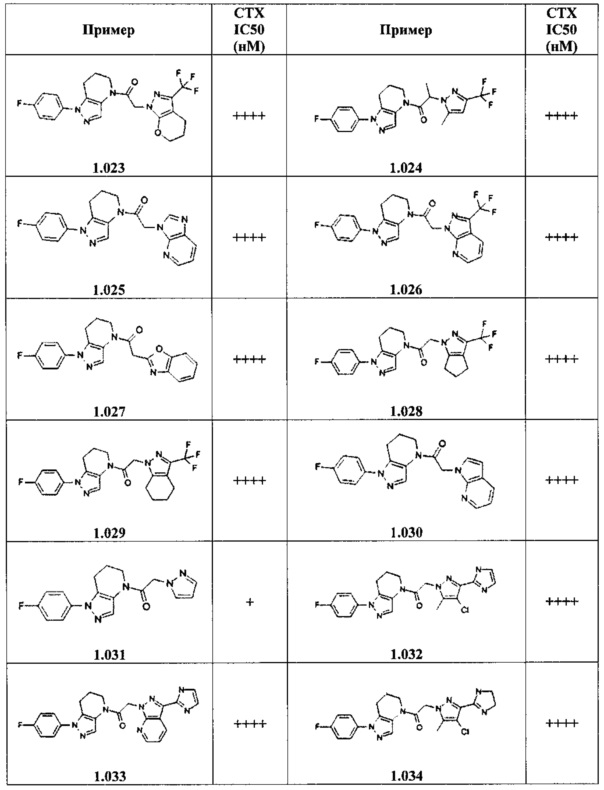
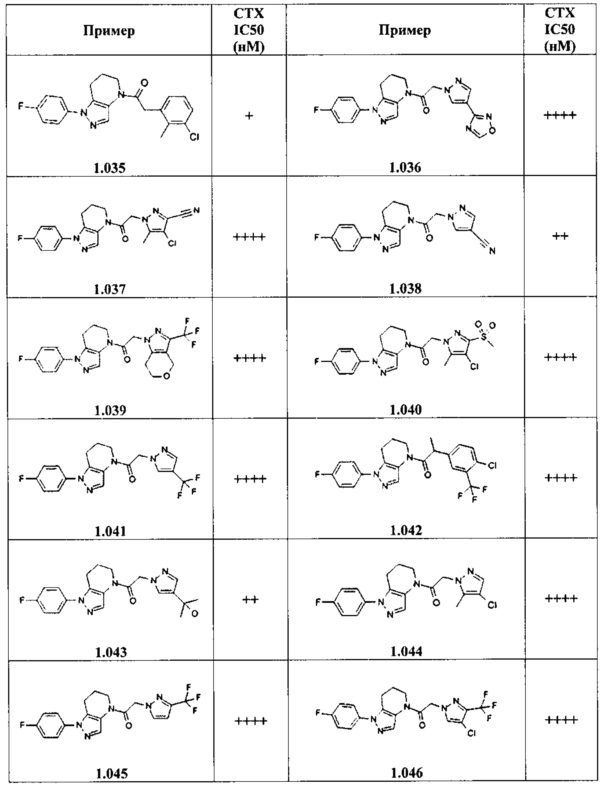
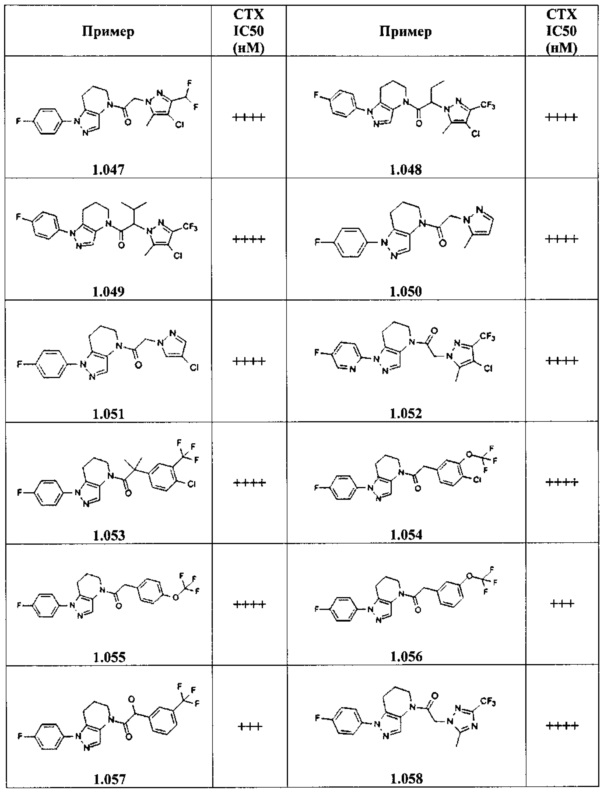
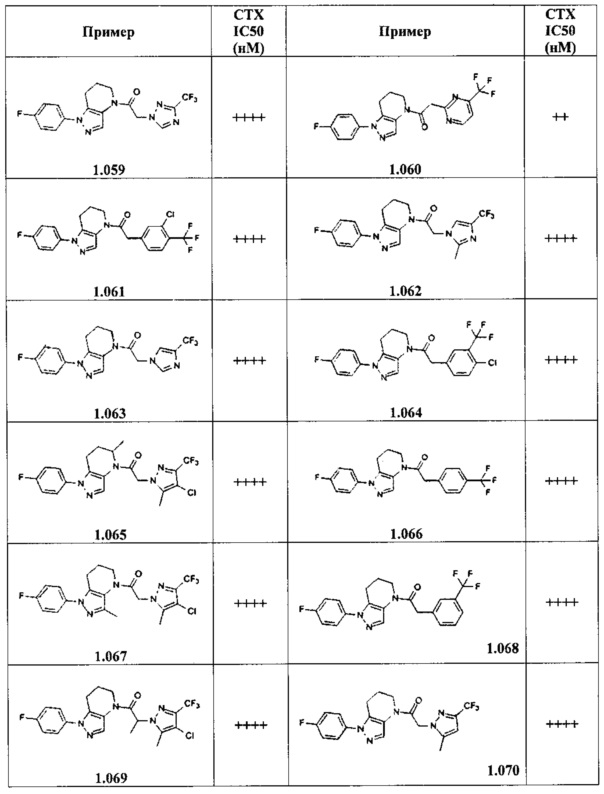
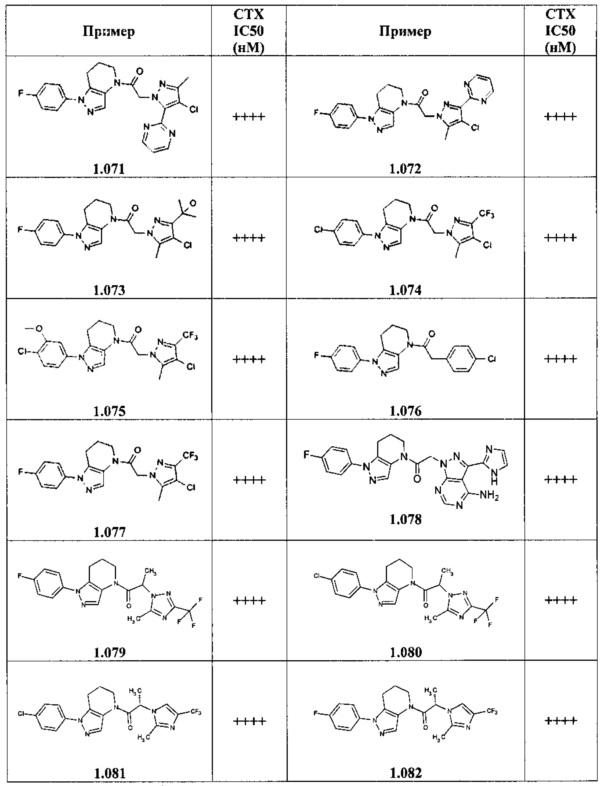
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-a]ПИРАЗОЛ-1-ОНЫ (ВАРИАНТЫ), ИНГИБИРУЮЩИЕ ВЫСВОБОЖДЕНИЕ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ | 2002 |
|
RU2299885C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ДИАЗОЛЬНЫЕ ЛАКТАМЫ | 2013 |
|
RU2666730C2 |
| 6,7-ДИГИДРО-5H-ПИРАЗОЛО[1,2-А]ПИРАЗОЛ-1-ОНЫ, РЕГУЛИРУЮЩИЕ ВОСПАЛИТЕЛЬНЫЕ ЦИТОКИНЫ (ВАРИАНТЫ), И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2003 |
|
RU2289584C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПИРРОЛОПИРАЗИН(ПИПЕРИДИН)АМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2012 |
|
RU2634900C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| ДИАРИЛ-ЗАМЕЩЕННЫЕ 6,5-СОПРЯЖЕННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ C5aR | 2018 |
|
RU2796983C2 |
Изобретение относится к соединениям формулы (I) или к их фармацевтически приемлемым солям и оптическим изомерам. В формуле (I) Ar1 выбран из группы, состоящей из фенила, пиридила, пиразинила, пиридазинила, пиримидинила и триазинила; каждый из которых необязательно имеет от одного до двух заместителей, Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила; каждый из которых имеет от одного до трех заместителей, и остальные группы и заместители имеют определения, приведенные в формуле изобретения. Данные соединения являются антагонистами CCR1 рецептора и проявляют in vivo противовоспалительную активность. Они могут применяться в фармацевтических композициях, способах лечения CCR1-опосредованных заболеваний и в качестве контроля в тестах на идентификацию конкурентоспособных антагонистов CCR1. 3 н. и 21 з.п. ф-лы, 1 табл., 75 пр.
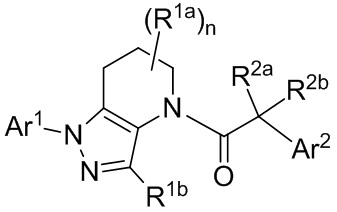 (I)
(I)
1. Соединение, имеющее формулу:
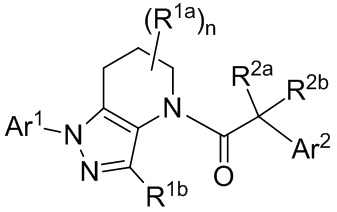 (I),
(I),
где подстрочный индекс n представляет собой целое число от 0 до 1;
каждый R1a и R1b является представителем, независимо выбранным из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, -CORa, -CO2Ra, -CONRaRb, -NRaRb, -NRaCORb, -ORa, и -X1ORa, где X1 выбран из группы, состоящей из C1-4 алкилена, и каждый Ra и Rb независимо выбран из группы, состоящей из атома водорода, C1-8 алкила и C1-8 галогеналкила;
каждый из R2a и R2b является представителем, независимо выбранным из группы, состоящей из H, гидроксила, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси, C1-4 алкокси-
C1-4 алкила, C1-8 гидроксиалкила, C1-4 алкокси-C1-4 алкокси, C3-6 циклоалкила, C3-6 циклоалкил-C1-4 алкила, 3-7-членного гетероциклоалкила, 3-7-членного гетероциклоалкил-C1-4 алкила, -X1CO2Ra, -X1CONRaRb, -X1NRaCORb, -X1NRaRb, где X1, Ra и Rb соответствуют данным выше определениям;
Ar1 выбран из группы, состоящей из фенила, пиридила, пиразинила, пиридазинила, пиримидинила и триазинила; каждый из которых необязательно имеет от одного до двух заместителей R3, R3a, R3b, R4 и R4a, которые независимо выбраны из группы, состоящей из H, галогена, -ORc, -Re, -CN, каждый Rc независимо выбран из атома водорода, C1-8 алкила, C1-8 гидроксиалкила, и C1-8 галогеналкила; и каждый Re независимо выбран из группы, состоящей из C1-8 алкила, C1-8 гидроксиалкила и C1-8 галогеналкила;
Ar2 выбран из группы, состоящей из пиразолила, имидазолила и триазолила; каждый из которых имеет от одного до трех заместителей R5, R6 и R7, независимо выбранных из группы, состоящей из H, галогена, -ORf, -Rh, -CN, -NO2, -CO2Rf, -CONRfRg, -S(O)Rh, -S(O)2Rh, -Y, где Y представляет собой пяти- или шестичленное арильное, гетероарильное или гетероциклическое кольцо, опционально имеющее 1-3 заместителя, выбранных из группы, состоящей из галогена, и -Rh, и каждый Rh независимо выбран из группы, состоящей из C1-8 алкила, C2-8 алкенила, C2-8 алкинила, C1-8 гидроксиалкила, C1-8 галогеналкила и C3-6 циклоалкила;
или его фармацевтически приемлемая соль или оптические изомеры.
2. Соединение по п. 1, где Ar1 представляет собой фенил, имеющий от одного до двух заместителей R3, R3a, R3b, R4 и R4a, и Ar2 выбран из группы, состоящей из пиразолила и имидазолила, каждый из которых необязательно имеет заместители R5, R6 и R7.
3. Соединение по п. 1, где указанное соединение имеет формулу:
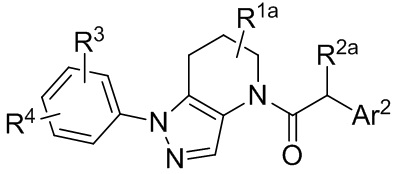 (Ia)
(Ia)
или его фармацевтически приемлемые соли или оптические изомеры, где R3 и R4 независимо выбраны из группы, состоящей из H, галогена, -Re, -CN.
4. Соединение по п. 1, где указанный Ar2 имеет формулу:
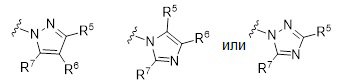
или его фармацевтически приемлемые соли или оптические изомеры, где R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y.
5. Соединение по п. 1, имеющее формулу:
 (Ia1)
(Ia1)
или его фармацевтически приемлемые соли или оптические изомеры, где R4 выбран из группы, состоящей из F и Cl.
6. Соединение по п. 1, где Y выбран из группы, состоящей из пиридила, пиримидинила, имидазолила, оксазолила, оксадиазолила, триазолила, тиазолила, имидазолинила и пиразолила.
7. Соединение по п. 1, имеющее формулу:
 (Ia2)
(Ia2)
или его фармацевтически приемлемые соли или оптические изомеры, где R3 выбран из группы, состоящей из H, галогена, C1-8 алкила, C1-8 галогеналкила и C1-8 алкокси; R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила.
8. Соединение по п. 1, имеющее формулу:
 (II)
(II)
или его фармацевтически приемлемые соли или оптические изомеры, где R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y.
9. Соединение по п. 1, имеющее формулу:
 (III)
(III)
или его фармацевтически приемлемые соли или оптические изомеры, где R1a и R2a независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и R5, R6 и R7 независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y.
10. Соединение по п. 1, имеющее формулу:
 (IV)
(IV)
или его фармацевтически приемлемые соли или оптические изомеры, где R1a и R2a каждый независимо выбраны из группы, состоящей из H, C1-8 алкила, C1-8 галогеналкила, C1-8 алкокси и C1-8 гидроксиалкила; и R5 и R7 каждый независимо выбраны из группы, состоящей из H, галогена, -Rh, -CN, -SO2Rh, -CO2Rf, -CONRfRg и Y.
11. Соединение по п. 1, выбранное из группы, состоящей из:
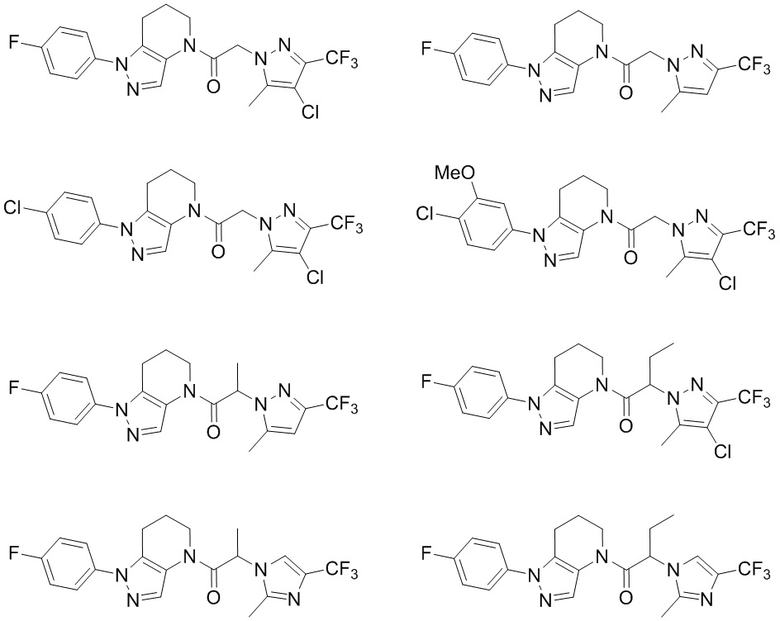 .
.
12. Соединение по п. 1, где указанное соединение выбрано из группы, состоящей из:

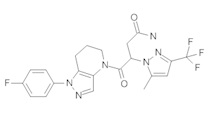
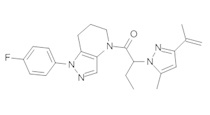

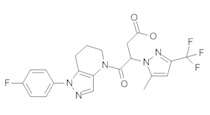

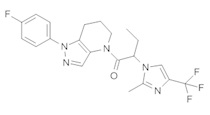
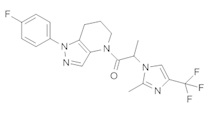
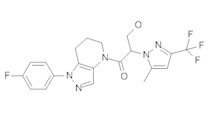
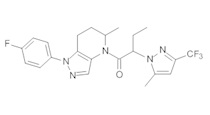
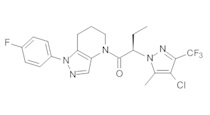
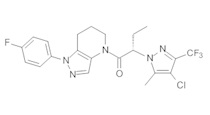
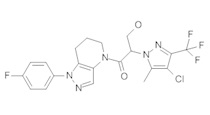


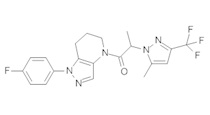
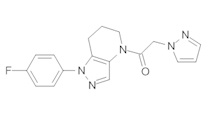

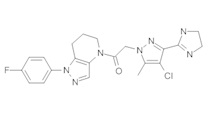

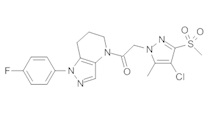





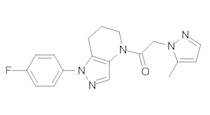
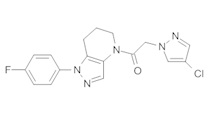
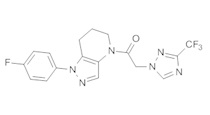
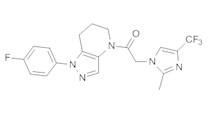
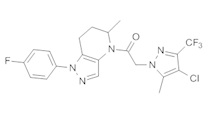

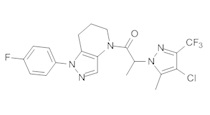

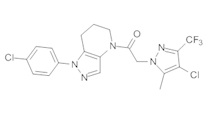



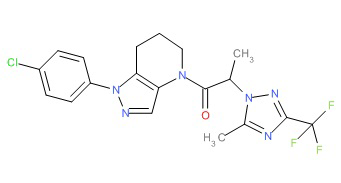
 и
и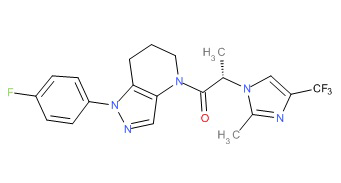
или его фармацевтически приемлемая соль.
13. Соединение по п. 1, имеющее структуру:
или его фармацевтически приемлемая соль.
14. Соединение по п. 1, имеющее структуру:
или его фармацевтически приемлемая соль.
15. Соединение по п. 1, имеющее структуру:
или его фармацевтически приемлемая соль.
16. Фармацевтическая композиция для лечения CCR1-опосредованных заболеваний, содержащая фармацевтически приемлемый наполнитель или носитель и соединение по п. 1.
17. Фармацевтическая композиция по п. 16, где указанную композицию формируют в виде стент-устройства или стент-графт-устройства.
18. Способ лечения CCR1-опосредованных заболеваний или состояний, включающий введение нуждающемуся пациенту терапевтически эффективного количества соединения или композиции по любому из пп. 1-17.
19. Способ по п. 18, где указанное CRR1-опосредуемое заболевание или состояние представляет собой воспалительное состояние.
20. Способ по п. 18, где указанное CRR1-опосредуемое заболевание или состояние представляет собой иммунорегуляторное нарушение.
21. Способ по п. 18, где указанное CRR1-опосредуемое заболевание или состояние выбрано из группы, состоящей из ревматоидного артрита, рассеянного склероза, отторжения трансплантата, дерматита, экземы, уртикарной сыпи, васкулита, воспалительной болезни кишечника, пищевой аллергии, астмы, болезни Альцгеймера, болезни Паркинсона, псориаза, красной волчанки, остеоартрирта, удара, рестеноза, вызванного облучением легочного заболевания, энцефаломиелита, множественной миеломы и остеопороза.
22. Способ по п. 18, где указанное введение представляет собой пероральное, парэнтеральное или ректальное.
23. Способ по п. 18, где указанное введение представляет собой чрескожное, подъязычное или назальное.
24. Способ по п. 18, где указанное соединение вводят в комбинации с противовоспалительным средством, болеутоляющим средством, антипролиферативным средством, ингибитором метаболизма, ингибитором миграции лейкоцитов или иммуномодулятором.
| WO 2012087782 A1, 28.06.2012 | |||
| Винтовой клапан | 1929 |
|
SU15529A1 |

