ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к классу замещенных шестичленных арил- или гетероарилбензамидных соединений, их солям, содержащим их фармацевтическим композициям и к их применению для терапии человеческого организма. В частности, настоящее изобретение относится к классу замещенных гетероарилбензамидных соединений, которые представляют собой ингибиторы протеинкиназ семейства тропомиозин-зависимых рецепторных киназ (Trk), а потому применимы при лечении боли, воспаления, злокачественной опухоли, рестеноза, атеросклероза, псориаза, тромбоза, заболевания, нарушения, повреждения или дисфункции, связанных с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактор роста нервов (NGF).
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Trk-рецепторы представляют собой протеинкиназные рецепторы с высокой аффинностью связывания, которые активируются нейротрофинами (NT), группой растворимых факторов роста, включающей в себя фактор роста нервов (NGF), нейротрофический фактор головного мозга (BDNF) и нейротрофин 3-5 (NT 3-5). Trk-рецепторы представлены тремя представителями семейства (TrkA, TrkB и TrkC), которые связывают нейротрофины и опосредуют полученную в результате сигнальную трансдукцию. NGF активирует TrkA, BDNF и NT-4/5 активируют TrkB, и NT3 активирует TrkC.
Было показано, что ингибиторы Trk/нейротрофинового пути являются высокоэффективными в многочисленных доклинических моделях боли на животных. Было показано, что анти-NGF и анти-TrkA антитела эффективны в моделях воспалительной и нейропатической боли на животных и в клинических испытаниях на человеке (См. Woolf, C.J. et al. (1994) Neuroscience 62, 327-331; Zahn, P.K. et al. (2004) J. Pain 5, 157-163; McMahon, S. B. et al., (1995) Nat. Med. 1, 774-780; Ma, Q.P. and Woolf, C. J. (1997) Neuroreport 8, 807-810; Shelton, D. L. et al. (2005) Pain 116, 8-16; Delafoy, L. et al. (2003) Pain 105, 489-497; Lamb, K. et al. (2003) Neurogastroenterol. Motil. 15, 355-361; и Jaggar, S. I. et al. (199) Br. J. Anaesth. 83, 442-448). Посредством исследований с деструкцией генов у мышей было обнаружено, что взаимодействие TrkA-NGF необходимо для выживания определенных популяций периферических нейронов, участвующих в опосредовании передачи болевого сигнала в случае злокачественной опухоли поджелудочной железы; было показано, что повышение экспрессии TrkA коррелирует с увеличением уровня передачи болевого сигнала (Zhu et al., Journal of Clinical oncology, 17:2419-2428 (1999)). Повышенная экспрессия NGF и TrkA также наблюдалась в хондроцитах человека при остеоартрите (Iannone et al, Rheumatology 41:1413-1418 (2002)). В частности, было продемонстрировано, что анти-TrkA антитела и анти-NGF антитела представляют собой эффективные анальгетики в моделях воспалительной и нейропатической боли in vivo (См. WO2006/131952, WO2005/061540, EP1181318 и WO01/78698, WO2004/058184 и WO2005/019266, соответственно; См. также WO2004/096122 и WO2006/137106, в которых описано применение анти-TrkA антител в сочетании с опиоидным анальгетиком для лечения или профилактики боли).
Ингибиторы Trk, которые могут индуцировать апоптоз пролиферирующих остеобластов, могут быть применимы для лечения заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, таких как остеопороз, ревматоидный артрит и костные метастазы. Наблюдалась экспрессия TrkA и TrkC рецепторов в области формирования кости в моделях перелома кости на мышах и локализация NGF практически во всех формирующих кость клетках (K. Asaumi, et al., Bone (2000) 26(6) 625-633); См. также Expert Opin. Ther. Patents (2009) 19(3)), WO2006/115452 и WO2006/087538, WO6123113, WO10033941, WO10077680, WO2005110994, Investigational New Drugs (2004), 22, 449-458 и R. Tripathy, et al., Bioorg. Med. Chem. Lett., 2008, 18, 3551-3555). Связь между повышенной экспрессией, активацией, амплификацией и/или мутацией Trk-рецепторов и некоторыми злокачественными опухолями, которая видна из исследований, проведенных для нейробластомы (Brodeur, G.M., Nat. Rev. Cancer 2003, 3, 203-216), злокачественной опухоли яичников (Kruettgen et al., Brain Pathology 2006, 16: 304-310), злокачественной опухоли предстательной железы (Dionne et al., Clin. Cancer Res. 1998, 4(8): 1887-1898), злокачественной опухоли поджелудочной железы (Dang et al., J of Gastroenterology and Hepatology 2006, 21(5): 850-858), крупноклеточных нейроэндокринных опухолей (Marchetti et al., Human Mutation 2008, 29(5), 609-616) и колоректальной злокачественной опухоли (Bardelli, A., Science 2003, 300, 949), поддерживает соображение, что терапевтическое значение эффективного ингибитора Trk может простираться далеко за пределы лечения боли (См. также WO2005/030128, WO2012158413, WO07013673, WO07025540, WO08052734, WO2012028579, WO2012159565, WO2012107434, WO2012003387, WO2010111653, WO2008124610, WO2004098518, EP1388341, WO2012028579, WO2008003770, WO2012161879, WO2012100223, WO2009046802, WO2009003999, WO2007042321, US2005143384, WO2009003998, WO2007069773, WO2005/030128 и US2010120862).
Также многообещающей является возможность применения ингибиторов Trk для лечения воспалительных заболеваний легких, таких как бронхиальная астма (Freund-Michel, V; et al., Pharmacology & Therapeutics (2008), 117(1), 52-76), интерстициального цистита (Hu Vivian Y; et. al., J of Urology (2005, 173(3), 1016-21), воспалительного заболевания кишечника, включая язвенный колит и болезнь Крона (Di Mola, F. F., et al., Gut (2000), 46(5), 670-678), и воспалительных заболеваний кожи, таких как атопический дерматит (Dou, Y.C., et. Al., Archives of Dermatological Research (2006), 298(1), 31-37), экзема и псориаз (Raychaudhuri, S. P. et. al., J of Investigative Dermatology (2004), 122(3), 812-819).
Также было показано, что модулирование пути нейротрофин/Trk оказывает эффект на этиологию нейродегенеративных заболеваний, включая рассеянный склероз, болезнь Паркинсона и болезнь Альцгеймера (Sohrabji, et. al., Neuroendocrinology (2006), 27(4), 404-414).
Поэтому считается, что соединения согласно настоящему изобретению, которые представляют собой ингибиторы Trk, применимы для лечения разнообразных типов острой и хронической боли, включая без ограничения воспалительную боль, нейропатическую боль и боль, ассоциированную со злокачественной опухолью, хирургическим вмешательством и переломом кости. Такие соединения также могут быть применимы при лечении злокачественной опухоли, воспаления, нейродегенеративных заболеваний и определенных инфекционных заболеваний.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям общей формулы (I), приведенной ниже, или к их фармацевтически приемлемым солям, которые применимы в качестве Trk-киназного медиатора NGF-зависимых биологических ответов, ингибитора TrkA, а также других Trk-киназ.
Настоящее изобретение также относится к способам лечения у пациента (предпочтительно человека) заболеваний или нарушений, в которые вовлечены связывающие NGF рецепторные Trk-киназы, в частности TrkA. Настоящее изобретение дополнительно относится к использованию соединений в качестве ингибитора и/или антагонистов связывающей NGF рецепторной TrkA для приготовления лекарственного средства для лечения и/или профилактики заболеваний, ассоциированных с ингибированием TrkA, которые включают в себя боль, злокачественную опухоль, рестеноз, атеросклероз, псориаз, тромбоз или заболевание, нарушение или повреждение, связанное с дисмиелинизацией или демиелинизацией. Настоящее изобретение также относится к фармацевтическим композициям, которые включают в себя эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, и к применению соединений и фармацевтических композиций согласно настоящему изобретению для лечения таких заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Согласно одному варианту осуществления, настоящее изобретение относится к соединениям общей формулы (I)
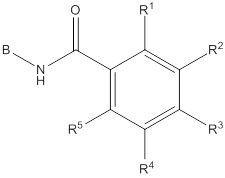 I
I
или к их фармацевтически приемлемым солям, где:
B представляет собой фенил, тетрагидронафтиридинил, дигидропирролопиридинил или шестичленный гетероарил, содержащий, по меньшей мере, один гетероатом, который представляет собой азот, причем упомянутый фенил, тетрагидронафтиридинил, дигидропирролопиридинил и гетероарил необязательно замещен 1-3 группами Ra;
R представляет собой водород, OH или C1-6алкил, причем упомянутый алкил необязательно замещен 1-3 группами Rf;
R1 и R5 независимо выбирают из группы, состоящей из водорода, CN, OH, C1-6алкила и галогена;
R2 и R4 независимо выбирают из группы, состоящей из водорода, галогена, C1-6алкила, (CHR)nC6-10арила и (CHR)nC5-10гетероцикла, причем упомянутый алкил, арил и гетероцикл необязательно замещен 1-3 группами Ra,
R3 представляет собой водород, C1-6алкил, C1-4галогеналкил, -OC1-4галогеналкил и галоген;
Ra выбирают из группы, состоящей из водорода, –CN, NO2, -(CH2)nC(O)ORf, -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, C1-6алкенила, C1-6алкинила, -(CH2)nC3-6циклоалкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -(CH2)nC(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, SO2Rd, (CH2)nNHSO2Rd, -(CH2)nSO2N(Rd)2, S(O)(NH)Rg, -C(O)CF3, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNRC(O)NHRd, -(CH2)nNHC(O)ORd, (CHR)nC(O)N(Rd)2OC1-6алкила, -O- и –OH, причем упомянутый алкил, циклоалкил, арил или гетероцикл необязательно замещен 1-3 группами Rb; где если две группы Rd присоединены к атому азота, то они могут формировать вместе с этим атомом азота 4-8-членный гетероцикл, который необязательно замещен 1-3 группами Rf;
Rb выбирают из группы, состоящей из галогена, C1-6алкила, C1-6алкилOR, C1-4галогеналкила, -(CH2)nN(Rd)2, ORc, -O-, -CN, S(O)(NH)Rg, -SO2R, -SO2N(Rd)2, -(CH2)nC(O)N(Rd)2, -(CH2)nNHC(O)Rd, COR, C(O)OR, C3-6циклоалкила, -O-(CH2)nC4-10гетероцикла и C1-6алкилN(Rd)2, причем упомянутый алкил и гетероцикл необязательно замещен 1-3 группами Rf;
Rc выбирают из группы, состоящей из водорода, C1-6алкилORg, C1-4галогеналкила и C1-6алкила;
Rd независимо выбирают из группы, состоящей из водорода, галогена, C1-4галогеналкила, C1-6алкила, COR, -(CH2)nSO2R, -(CH2)nNRfC4-10гетероцикла, (CH2)nC3-6циклоалкила, -(CH2)nC4-10гетероцикла, упомянутый алкил, циклоалкил и гетероцикл необязательно замещен 1-3 группами Rf; где если две группы Rd присоединены к атому азота, то они могут формировать вместе с этим атомом азота 4-8-членный гетероцикл, который необязательно замещен 1-3 группами Rf;
Rf выбирают из группы, состоящей из водорода, C1-6алкила, ORc, CN, -N(Rc)2, C(O)N(Rg)2, C(O)C1-6алкила, -SO2Rg, -O-, -C1-6алкилSO2Rg, -C1-6алкилORg, -C1-6алкилN(Rg)2,
Rg выбирают из группы, состоящей из водорода, C1-6алкила; и
n равно 0-6.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой незамещенный или замещенный фенил. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда B представляет собой незамещенный фенил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда B представляет собой замещенный фенил.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой необязательно замещенный шестичленный гетероарил, выбранный из группы, состоящей из пиридила, пиримидинила, пиридазинила и пиразинила. Вариант осуществления этого аспекта настоящего изобретения формулы I реализуется, когда B представляет собой замещенный пиридил. Дополнительный вариант осуществления этого аспекта настоящего изобретения формулы I реализуется, когда B представляет собой незамещенный пиридил. Еще один вариант осуществления этого аспекта настоящего изобретения формулы I реализуется, когда B представляет собой замещенный пиримидинил. Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой незамещенный пиримидинил. Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой необязательно замещенный пиразинил. Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой незамещенный пиразинил. Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой незамещенный или замещенный пиридазинил.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой необязательно замещенный тетрагидронафтиридинил.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой необязательно замещенный дигидропирролопиридинил.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда Ra выбирают из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда Ra выбирают из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -(CH2)nN(Rd)2, COR, -(CH2)nгалогена и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда Rb выбирают из группы, состоящей из галогена, C1-6алкила, C1-6алкилOR, C1-4галогеналкила, -(CH2)nN(Rd)2, ORc, -(CH2)nC(O)N(Rd)2, -(CH2)nNHC(O)Rd.
Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда R1 и R5 оба представляют собой водород. Другой вариант осуществления настоящего изобретения формулы I реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой галоген. Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда R1 и R5 оба представляют собой галоген. Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой хлор, фтор, CN, OH или C1-6алкил. Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой C1-6алкил. Еще один вариант осуществления настоящего изобретения формулы I реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой хлор или фтор.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда R2 и R4 оба представляют собой водород.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл, причем упомянутый гетероцикл является незамещенным или замещен 1-3 группами Ra. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда значение n в гетероцикле (CHR)nC5-10 равно 0. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда необязательно замещенный гетероцикл представляет собой пяти- или шестичленное кольцо, содержащее один или несколько гетероатомов, по меньшей мере, один из которых представляет собой азот. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда необязательно замещенный гетероцикл представляет собой пятичленное кольцо, содержащее один или несколько гетероатомов, по меньшей мере, один из которых представляет собой азот. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда необязательно замещенный гетероцикл представляет собой шестичленное кольцо, содержащее один или несколько гетероатомов, по меньшей мере, один из которых представляет собой азот. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл выбирают из группы, состоящей из пиразолила, пиридила, тиазолила, триазолила, оксазолила, оксадиазолила и пиримидинила, причем упомянутые группы являются необязательно замещенными. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный пиразолил. Другой подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой замещенный пиразолил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный тиазолил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный пиридил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный оксадиазолил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный оксазолил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный триазолил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл представляет собой необязательно замещенный пиримидинил. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда гетероцикл необязательно замещен 1-3 группами Ra, выбранными из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -C(O)CF3, C(O)R, C(O)N(R)2, -(CH2)nгалогена и –OR.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда R3 выбирают из группы, состоящей из водорода, CF3, OCF3, CH3, брома, хлора и фтора. Подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 представляет собой CF3. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 представляет собой OCF3. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 представляет собой CH3. Еще один подвариант осуществления данного аспекта настоящего изобретения реализуется, когда R3 представляет собой водород.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой галоген, один из R2 и R4 представляет собой водород, а другой выбирают из необязательно замещенного пиразолила, пиридила, тиазолила, триазолила, оксазолила, оксадиазолила и пиримидинила, и R3 выбирают из группы, состоящей из водорода, CF3, OCF3, CH3, брома, хлора и фтора. Аспект данного варианта осуществления реализуется, когда один из R1 и R5 представляет собой водород, а другой представляет собой фтор, один из R2 и R4 представляет собой водород, а другой выбирают из необязательно замещенного пиразолила и пиримидинилп, и R3 представляет собой CF3 или OCF3.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой пиримидинил замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда пиримидинил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на пиримидиниле представляет собой необязательно замещенный фенил.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой пиридил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда пиридил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на пиридиле представляет собой необязательно замещенный фенил.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой фенил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда фенил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на фениле представляет собой необязательно замещенный фенил.
Другой вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой пиразинил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда пиразинил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на пиразиниле представляет собой необязательно замещенный фенил.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой тетрагидронафтиридинил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда тетрагидронафтиридинил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на тетрагидронафтиридиниле представляет собой необязательно замещенный фенил.
Вариант осуществления настоящего изобретения формулы I реализуется, когда B представляет собой дигидропирролопиридинил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb, где n=0-2. Дополнительный подвариант осуществления данного аспекта настоящего изобретения реализуется, когда дигидропирролопиридинил замещен 1-3 группами, выбранными из галогена, CH2OH, CH3, хлора и необязательно замещенного фенила или пиразолила. Другой вариант осуществления данного аспекта настоящего изобретения реализуется, когда один из заместителей на дигидропирролопиридиниле представляет собой необязательно замещенный фенил.
Еще один вариант осуществления настоящего изобретения формулы I представлен структурной формулой Ia:
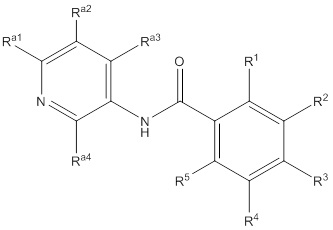 Ia
Ia
или ее фармацевтически приемлемой солью, где значения R1, R2, R3, R4 и R5 описаны ранее, значения Ra1, Ra2, Ra3 и Ra4 все соответствуют Ra, и значение Ra описано ранее. Подвариант осуществления настоящего изобретения формулы Ia реализуется, когда Ra1, Ra2, Ra3 и Ra4 независимо выбирают из водорода, C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb. Другой подвариант осуществления настоящего изобретения формулы Ia реализуется, когда, по меньшей мере, два из Ra1, Ra2, Ra3 и Ra4 на пиридиле представляют собой водород. Другой подвариант осуществления настоящего изобретения формулы Ia реализуется, когда два из Ra1, Ra2, Ra3 и Ra4 на пиридиле представляют собой водород, а два не представляют собой водород. Другой подвариант осуществления настоящего изобретения формулы Ia реализуется, когда один из Ra1, Ra2, Ra3 и Ra4 всегда представляет собой фенил. Другой подвариант осуществления настоящего изобретения формулы Ia реализуется, когда Ra4 представляет собой фенил. Еще один подвариант осуществления настоящего изобретения формулы Ia реализуется, когда R1 и R5 независимо выбирают из водорода и галогена, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл. Другой подвариант осуществления настоящего изобретения формулы Ia реализуется, когда в R2 и R4 гетероцикл представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил.
Еще один вариант осуществления настоящего изобретения формулы Ia реализуется, когда один из Ra1, Ra2, Ra3 и Ra4 всегда представляет собой фенил, один из R1 и R5 представляет собой водород, а другой представляет собой галоген, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил. Дополнительный аспект данного подварианта осуществления реализуется, когда Ra4 представляет собой фенил, R1 представляет собой водород, R3 представляет собой CF3, R5 представляет собой фтор, R2 представляет собой необязательно замещенный пиразолил, и R4 представляет собой водород
Еще один вариант осуществления настоящего изобретения формулы I представлен структурной формулой II:
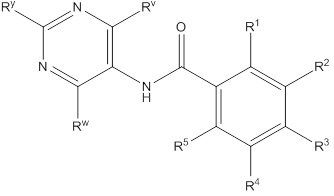 II
II
или ее фармацевтически приемлемой солью, где значения R1, R2, R3, R4 и R5 описаны ранее, значения Ry, Rv и Rw все соответствуют Ra, и значение Ra описано ранее Подвариант осуществления настоящего изобретения формулы II реализуется, когда Ry, Rv и Rw независимо выбирают из водорода, C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда, по меньшей мере, два из Ry, Rv и Rw на пиримидиниле представляют собой водород. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда два из Ry, Rv и Rw на пиримидиниле представляют собой водород. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда один из Ry, Rv и Rw всегда представляет собой фенил. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда Rw представляет собой фенил. Еще один подвариант осуществления настоящего изобретения формулы II реализуется, когда R1 и R5 независимо выбирают из водорода и галогена, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл. Другой подвариант осуществления настоящего изобретения формулы II реализуется, когда в R2 и R4 гетероцикл представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил.
Еще один вариант осуществления настоящего изобретения формулы II реализуется, когда один из Ry, Rv и Rw всегда представляет собой фенил, один из R1 и R5 представляет собой водород, а другой представляет собой галоген, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил. Дополнительный аспект данного подварианта осуществления реализуется, когда Rw представляет собой фенил, R1 представляет собой водород, R3 представляет собой CF3, R5 представляет собой фтор, R2 представляет собой необязательно замещенный пиразолил, и R4 представляет собой водород
Еще один вариант осуществления настоящего изобретения формулы I представлен структурной формулой III:
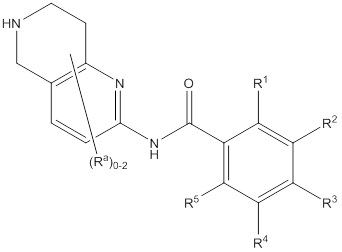 III
III
или ее фармацевтически приемлемой солью, где значения R1, R2, R3, R4, Ra и R5 описаны ранее. Подвариант осуществления настоящего изобретения формулы III реализуется, когда Ra независимо выбирают из водорода, C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутый алкил, арил или гетероцикл необязательно замещен 1-3 группами Rb. Другой подвариант осуществления настоящего изобретения формулы III реализуется, когда присутствуют два Ra, и они не представляют собой водород. Другой подвариант осуществления настоящего изобретения формулы III реализуется, когда один из присутствующих Ra всегда представляет собой фенил. Другой подвариант осуществления настоящего изобретения формулы III реализуется, когда Ra, который представляет собой фенил, присутствует на пиридинильной части кольца. Еще один подвариант осуществления настоящего изобретения формулы II реализуется, когда R1 и R5 независимо выбирают из водорода и галогена, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл. Другой подвариант осуществления настоящего изобретения формулы III реализуется, когда в R2 и R4 гетероцикл представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил.
Еще один вариант осуществления настоящего изобретения формулы III реализуется, когда присутствуют два Ra, один из которых всегда представляет собой фенил, причем упомянутый фенил присоединен к пиридинильной части кольца, один из R1 и R5 представляет собой водород, а другой представляет собой галоген, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой необязательно замещенный оксодиазолил, пиразолил, пиридил, тиазолил, оксазолил и пиримидинил. Дополнительный аспект данного подварианта осуществления реализуется, когда присутствуют два Ra, один из которых всегда представляет собой фенил, причем упомянутый фенил присоединен к пиридинильной части кольца, R1 представляет собой водород, R3 представляет собой CF3, R5 представляет собой фтор, R2 представляет собой необязательно замещенный пиразолил, и R4 представляет собой водород.
Настоящее изобретение также относится к способам лечения у пациента (предпочтительно человека) заболеваний или нарушений, в которые вовлечен TrkA-рецептор, таких как боль, воспаление, злокачественная опухоль, рестеноз, атеросклероз, псориаз, тромбоз, заболевание, нарушение, повреждение или дисфункция, связанная с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактора роста нервов (NGF), посредством введения пациенту терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к применению соединения согласно настоящему изобретению для лечения заболевания или нарушения, в которое вовлечен TrkA-рецептор, такого как боль, воспаление, злокачественная опухоль, рестеноз, атеросклероз, псориаз, тромбоз, заболевание, нарушение, повреждение или дисфункция, связанная с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактора роста нервов (NGF), посредством введения пациенту соединения согласно настоящему изобретению или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к лекарственным средствам или фармацевтическим композициям для лечения у пациента (предпочтительно человека) заболеваний или нарушений, в которые вовлечен TrkA-рецептор, таких как боль, воспаление, злокачественная опухоль, рестеноз, атеросклероз, псориаз, тромбоз, заболевание, нарушение, повреждение или дисфункция, связанная с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактора роста нервов (NGF), которые содержат соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способу производства лекарственного средства или фармацевтической композиции для лечения заболеваний, в которые вовлечен TrkA-рецептор, таких как боль, воспаление, злокачественная опухоль, рестеноз, атеросклероз, псориаз, тромбоз, заболевание, нарушение, повреждение или дисфункция, связанная с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактора роста нервов (NGF), включающему в себя объединение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя.
Если в любой формуле согласно настоящему изобретению или в любом ее заместителе переменное значение встречается более одного раза, то отдельные случаи появления этого переменного значения независимы друг от друга, если не указано иное. Кроме того, комбинации заместителей или переменных значений являются допустимыми, только если такие комбинации приводят к стабильным соединениям.
Используемый в настоящем документе термин «алкил», сам по себе или как часть другого заместителя, означает насыщенный неразветвленный или разветвленный углеводородный радикал, характеризующийся указанным числом атомов углерода (например, C1-10алкил означает алкильную группу, содержащую от одного до десяти атомов углерода). Предпочтительные алкильные группы для использования согласно настоящему изобретению представляют собой C1-6алкильные группы, содержащие от одного до шести атомов углерода. Типовые алкильные группы включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, и т. п. C0алкил означает химическую связь.
Используемый в настоящем документе термин «алкенил», сам по себе или как часть другого заместителя, означает неразветвленный или разветвленный углеводородный радикал, характеризующийся единственной двойной углерод-углеродной связью и указанным числом атомов углерода (например, C2-10алкенил означает алкенильную группу, содержащую от двух до десяти атомов углерода). Предпочтительные алкенильные группы для использования согласно настоящему изобретению представляют собой C2-6алкенильные группы, содержащие от двух до шести атомов углерода. Типовые алкенильные группы включают в себя этенил и пропенил.
Используемый в настоящем документе термин «циклоалкил», сам по себе или как часть другого заместителя, означает насыщенный циклический углеводородный радикал, характеризующийся указанным числом атомов углерода (например, C3-12циклоалкил означает циклоалкильную группу, содержащую от трех до двенадцати атомов углерода). Используемый в настоящем документе термин «циклоалкил» включает в себя моно-, би- и трициклические насыщенные карбоциклы, спироциклы и связанные мостиковой связью и конденсированные карбоциклы, а также оксозамещенные циклоалкильные группы.
Предпочтительные циклоалкильные группы для использования согласно настоящему изобретению представляют собой моноциклические C3-8циклоалкильные группы, содержащие от трех до восьми атомов углерода. Типовые моноциклические циклоалкильные группы включают в себя циклопропил, циклобутил, циклопентил, циклогексил и т. п. Типовые связанные мостиковой связью циклоалкильные группы включают в себя адамантил и норборнил. Типовые конденсированные циклоалкильные группы включают в себя декагидронафталин.
Термин «гетероатом» означаает O, S или N, выбранные самостоятельно.
Используемый в настоящем документе термин «арил», сам по себе или как часть другого заместителя, означает ароматический циклический углеводородный радикал. Предпочтительные арильные группы содержат от шести до десяти атомов углерода. Термин «арил» включает в себя системы с нексколькими кольцами, а также системы с одним кольцом. Предпочтительные арильные группы для использования согласно настоящему изобретению включают в себя фенил и нафтил.
Термин «арил» также включает в себя конденсированные циклические углеводородные кольца, которые являются частично ароматическими (т.е., одно из конденсированных колец является ароматическим, а другое является неароматическим). Типовая арильная группа, которая является частично ароматическими, представляет собой инданил.
Используемые в настоящем документе термины «гетероциклил», «гетероцикл» или «гетероциклический», представляют собой стабильное 5-7-членное моноциклическое или стабильное 8-11-членное бициклическое гетероциклическое кольцо, которое является насыщенным или ненасыщенным и которое состоит из атомов углерода и 1-4 гетероатомов, выбранных из группы, состоящей из N, O и S, включая любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, если это приводит к формированию стабильной структуры. Термины «гетероциклил», «гетероцикл» или «гетероциклический» включают в себя гетероарильные фрагменты. Примеры таких гетероциклических фрагментов включают в себя без ограничения азепинил, бензодиоксолил, бензимидазолил, бензизоксазолил, бензофуразонил, бензопиранил, бензотиопиранил, бензофурил, бензотиазолил, бензотиенил, бензотриазолил, бензоксазолил, хроманил, циннолинил, дигидробензофурил, дигидробензотиенил, дигидробензотиопиранил, дигидробензотиопиранилсульфон, 1,3-диоксоланил, фурил, имидазолидинил, имидазолинил, имидазолил, индолинил, индолил, изохроманил, изоиндолинил, изохинолинил, изотиазолидинил, изотиазолил, изотиазолидинил, морфолинил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, пиперидил, пиперазинил, пиридил, пиразинил, пиразолидинил, пиразолил, пиразолопиридинил, пиридазинил, пиримидинил, пирролидинил, пирроилил, хиназолинил, хинолинил, хиноксалинил, тетрагидрофурил, тетрагидроизохинолинил, тетрагидрохинолинил, тиаморфолинил, тиаморфолинилсульфоксид, тиазолил, тиазолинил, тиенофурил, тиенотиенил, тиенил, триазолил, их N-оксиды и –C=O производные.
Используемый в настоящем документе термин «гетероарил», за исключением отмеченных случаев, представляет собой стабильную 5-7-членную моноциклическую или стабильную 9-10-членную конденсированную бициклическую гетероциклическую кольцевую систему, которая содержит ароматическое кольцо, любое из колец которой может быть насыщенным, такую как пиперидинил, частично насыщенным или ненасыщенным, такую как пиридинил, и которая состоит из атомов углерода и 1-4 гетероатомов, выбранных из группы, состоящей из N, O и S, и где гетероатомы азота и серы могут быть необязательно окислены, и гетероатом азота может быть необязательно кватернизован, включая любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому гетероатому или атому углерода, если это приводит к формированию стабильной структуры. Примеры таких гетероарильных групп включают в себя без ограничения бензимидазол, бензизотиазол, бензизоксазол, бензофуран, бензотиазол, бензотиофен, бензотриазол, бензоксазол, карболин, циннолин, фуран, фуразан, имидазол, индазол, индол, индолизин, изохинолин, изотиазол, изоксазол, нафтиридин, оксадиазол, оксазол, фталазин, птеридин, путин, пиран, пиразин, пиразол, пиридазин, пиридин, пиримидин, пиррол, хиназолин, хинолин, хиноксалин, тетразол, тиадиазол, тиазол, тиофен, триазин, триазол, их N-оксиды и –C=O производные. Подходящими гетероарильными группами являются имидазопиридинил, индазолил, имидазотиазолил, имидазопиримидинил, имидазопиридазинил, имидазотиадиазолил, хиноксалинил и имидазопирроилил.
Если определенная в настоящем документе гетероциклильная группа является замещенной, то заместитель может быть связан с атомом углерода кольца гетероарильной группы или с гетероатомом кольца (т.е., азот, кислород или сера), который имеет валентность, позволяющую замещение. Предпочтительно, заместитель связан с атомом углерода кольца. По аналогии, если гетероарильная группа определена в настоящем документе в качестве заместителя, то место присоединения может находиться на атоме углерода кольца гетероарильной группы или на гетероатоме кольца (т.е., азот, кислород или сера), который имеет валентность, позволяющую замещение. Предпочтительно, место присоединения находится на атоме углерода кольца.
Используемый в настоящем документе термин «галоид» или «галоген» включает в себя фтор, хлор, бром или йод.
Используемый в настоящем документе символ –O- включает в себя оксо (например, в кольце -CH-, замещенный оксо, представляет собой -C(O) или карбонил).
Соединения согласно настоящему изобретению могут содержать один или несколько центров асимметрии. Соединения с центрами асимметрии приводят к образованию энантиомеров (оптических изомеров), диастереоизомеров (конфигурационных изомеров), или и тех и других, и предполагается, что все возможные энантиомеры и диастереоизомеры в смесях и в виде чистых или частично очищенных соединений включены в объем настоящего изобретения. Предусмотрено, что настоящее изобретение охватывает все такие изомерные формы соединений согласно настоящему изобретению. Настоящее изобретение включает в себя все стереоизомеры формулы (I) и их фармацевтически приемлемые соли.
Независимый синтез энантиомерно или диастереоизомерно обогащенных соединений или их хроматографическое разделение, может осуществляться, как известно в данной области техники, путем соответствующей модификации методологии, раскрытой в настоящем документе. Их абсолютная стереохимия может быть определена методом рентгеновской кристаллографии кристаллических продуктов или кристаллических промежуточных продуктов, которые при необходимости дериватизуют реагентом, содержащим центр асимметрии с известной абсолютной конфигурацией.
При желании, рацемические смеси соединений могут быть разделены, так что могут быть выделены отдельные энантиомеры или диастереоизомеры. Разделение может проводиться способами, хорошо известными в данной области техники, такими как сочетание рацемической смеси соединений с энантиомерно чистым соединением с получением диастереоизомерной смеси, с последующим разделением отдельных диастереоизомеров стандартными способами, такими как фракционная кристаллизация или хроматографии. Реакция сочетания часто представляет собой формирование солей с использованием энантиомерно чистой кислоты или основания. Диастереоизомерные производные могут быть затем преобразованы до чистых энантиомеров путем отщепления добавляемого хирального остатка. Рацемические смеси соединений могут быть также разделены непосредственно хроматографическими способами с использованием хиральных неподвижных фаз, и такие способы хорошо известны в данной области техники.
В качестве альтернативы, любой энантиомер или диастереомер соединения может быть получен путем стереоселективного синтеза с использованием оптически чистых исходных веществ или реагентов с известной конфигурацией способами, хорошо известными в данной области техники.
В соединениях согласно настоящему изобретению атомы могут проявлять свой природный изотопный состав, или один или несколько из атомов могут быть искусственно обогащены конкретным изотопом, имеющим тот же атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, преимущественно обнаруживаемых в природе. Предусмотрено, что настоящее изобретение включает в себя все подходящие изотопные варианты соединений общей формулы (I). Например, различные изотопные формы водорода (H) включают в себя протий (1H) и дейтерий (2H). Протий является преимущественным изотопом водорода, обнаруживаемым в природе. Обогащение дейтерием может наделять определенными терапевтическими преимуществами, такими как увеличение полураспада in vivo или снижение требований к дозировке, или может предоставить соединение, применимое в качестве стандарта для охарактеризования биологических образцов. Обогащенные изотопами соединения общей формулы (I) могут быть получены без проведения излишней экспериментальной работы традиционными методиками, хорошо известными специалистам в данной области техники, или в ходе процессов, аналогичных описанным на схемах и в примерах, представленных в настоящем документе, с использованием соответствующих изотопно обогащенных реагентов и/или промежуточных продуктов.
Термин «по существу чистый» означает, что выделенное вещество характеризуется чистотой, по меньшей мере, 90%, предпочтительно 95%, и еще более предпочтительно 99%, согласно анализу с использованием аналитических методик, известных в данной области техники.
Используемый в настоящем документе термин «TrkA» относится к одному из протеинкиназных Trk-рецепторов с высокой аффинностью связывания, который активируется нейротрофинами (NT), группой растворимых факторов роста, включающей в себя фактор роста нервов (NGF), нейротрофический фактор головного мозга (BDNF) и нейротрофин 3-5 (NT 3-5). Trk-рецепторы представлены тремя представителями семейства (TrkA, TrkB и TrkC), которые связывают нейротрофины и опосредуют полученную в результате сигнальную трансдукцию. Было показано, что ингибиторы Trk/нейротрофинового пути являются высокоэффективными в многочисленных доклинических моделях боли на животных. Соединения согласно настоящему изобретению представляют собой модуляторы Trk-рецепторов, в частности TrkA.
Термин «фармацевтически приемлемые соли» относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соединения согласно настоящему изобретению могут представлять собой моно-, ди- или тризамещенные соли, в зависимости от числа функциональных групп кислоты, присутствующих в свободной основной форме соединения. Свободные основания и соли, полученные из неорганических оснований, включают в себя соли алюминия, аммония, кальция, меди(I), железа(II), железа(III), лития, магния, марганца(III), марганца(II), калия, натрия, цинка и т. п.
Соли в твердой форме могут существовать более чем в одной кристаллической форме, а также могут существовать в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают в себя соли первичных, вторичных и третичных аминов, замещенных аминов, включая замещенные амины, встречающиеся в природе, циклические амины и основные ионообменные смолы, такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, тебромин, триэтиламин, триметиламин, трипропиламин, трометамин и т. п.
Если соединение согласно настоящему изобретению представляет собой основание, то соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают в себя уксусную, трифторуксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, соляную, изэтиновую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, пара-толуолсульфоновую кислоту и т. п.
Настоящее изобретение относится к применению соединений формулы (I), раскрытых в настоящем документе, в качестве ингибиторов TrkA у нуждающегося в таком лечении пациента или субъекта, такого как млекопитающее, включающему в себя введение эффективного количества соединения. В дополнение к людям, лечению в соответствии со способом согласно настоящему изобретению может подвергаться целый ряд других млекопитающих.
Соединения согласно настоящему изобретению применимы при лечении или уменьшении интенсивности болевых нарушений (включая боль, ассоциированную со злокачественной опухолью, хирургическим вмешательством и переломом кости, острую боль, воспалительную боль и нейропатическую боль). Соединения формулы I также применимы при лечении злокачественных опухолей, включая нейробластому, злокачественную опухоль яичников, поджелудочной железы и колоректальную злокачественную опухоль. Другие состояния, которые можно лечить соединениями согласно настоящему изобретению, включают в себя воспаление и определенные инфекционные заболевания, интерстициальный цистит, синдром болезненного мочевого пузыря, недержание мочи, бронхиальную астму, анорексию, атопический дерматит и псориаз. С использованием соединений согласно настоящему изобретению также возможно лечение демиелинизации и дисмиелинизации путем стимулирования миелинизации, выживания нейронов и дифференцировки олигодендроцитов посредством блокирования Sp35-TrkA взаимодействия.
Соединения формулы I также могут быть применимы при лечении заболеваний костей (например, заболеваний, вовлеченных в резорбцию кости). Примеры заболеваний костей включают в себя метастатическое заболевание костей, индуцированную лечением потерю костной массы, остеопороз, ревматоидный артрит, анкилозирующий спондилит, болезнь Педжета и пародонтоз. Другое нарушение или заболевание костей, которое можно лечить соединениями согласно настоящему изобретению, представляет собой индуцированный опухолью метастатический остеолиз. Известные злокачественные опухоли, вызывающие индуцированный опухолью остеолиз, представляют собой гематологические злокачественные новообразования, такие как миелома и лимфома, и солидные опухоли, такие как злокачественные опухоли молочной железы, предстательной железы, легкого, почек и щитовидной железы.
Болевые нарушения, для которых могут быть применимы соединения согласно настоящему изобретению, включают в себя нейропатическую боль (такую как постгерпетическая невралгия, повреждение нервов, «динии», например, вульводиния, фантомная боль конечности, корешковые авульсии, болезненная диабетическая нейропатия, болезненная травматическая мононейропатия, болезненная полинейропатия); центральные болевые синдромы (потенциально обусловленные по существу любым повреждением на любом уровне нервной системы); послеоперационные болевые синдромы (например, синдром постмастэктомии, синдром постторакотомии, боль культи); боль в костях и суставах (остеоартрит), боль при повторяющихся движениях, зубную боль, боль при злокачественной опухоли, миофасциальную боль (мышечное повреждение, фибромиалгия); периоперационную боль (общая хирургия, гинекологическая), хроническую боль, дисменорею, а также боль, ассоциированную со стенокардией, и воспалительную боль различного происхождения (например, остеоартрит, ревматоидный артрит, ревматизм, теносиновит и подагра), головную боль, мигрень и кластерную головную боль, головную боль, первичную гиперальгезию, вторичную гиперальгезию, первичную аллодинию, вторичную аллодинию и другую боль, обусловленную повышением центральной чувствительности.
Соединения согласно настоящему изобретению также можно применять для лечения или профилактики дискинезий. Кроме того, соединения согласно настоящему изобретению можно применять для снижения толерантности и/или зависимости от опиоидного лечения боли, и для лечения абстинентного синдрома, вызванного, например, алкоголем, опиоидами и кокаином.
Субъект или пациент, которому вводят соединения согласно настоящему изобретению, как правило, представляет собой млекопитающее, такое как человек, мужского или женского пола, для которых желательно модулирование TrkA и/или TrkB. Таким образом, аспект настоящего изобретения представляет собой способ лечения заболеваний ингибитором TrkA и/или TrkB, включающий в себя введение упомянутому млекопитающему одного или нескольких соединений формулы I или его фармацевтически приемлемой соли в количестве, эффективном для лечения или профилактики упомянутого нарушения. Конкретный аспект настоящего изобретения относится к способу лечения боли, злокачественной опухоли, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi путем введения упомянутому млекопитающему терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Еще один аспект настоящего изобретения относится к способу лечения у млекопитающего остеолитического заболевания путем введения терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В рамках настоящего изобретения млекопитающие включают в себя собак, кошек, мышей, крыс, крупный рогатый скот, лошадей, овец, кроликов, обезьян, шимпанзе и других человекообразных обезьян и приматов, для которых желательно лечение от вышеуказанных нарушений.
Соединения согласно настоящему изобретению можно применять в сочетании с одним или несколькими другими лекарствами для лечения заболеваний или состояний, для которых применимы соединения согласно настоящему изобретению, причем сочетанное применение лекарств вместе является более безопасным или более эффективным, чем каждого из лекарств по отдельности. Кроме того, соединения согласно настоящему изобретению можно применять в сочетании с одним или несколькими лекарствами, которые лечат, предотвращают, контролируют, уменьшают интенсивность или снижают риск побочных эффектов или токсичность соединений согласно настоящему изобретению. Такие другие лекарства можно вводить путями и в количестве, обычно используемыми для этого, одновременно или последовательно с соединениями согласно настоящему изобретению. Соответственно, фармацевтические композиции согласно настоящему изобретению включают в себя композиции, которые в дополнение к соединениям согласно настоящему изобретению содержат одно или несколько других активных ингредиентов. Сочетания можно вводить как часть комбинированного продукта в виде стандартной лекарственной формы или в виде набора или лечебного протокола, когда одно или несколько дополнительных лекарств вводят в отдельных лекарственных формах, как часть схемы лечения.
Примеры сочетаний соединений включают в себя сочетания со средствами для лечения боли, например, стероидами, такими как дексаметазон, кортизон и флутиказон, нестероидными противовоспалительными средствами, такими как аспирин, диклофенак, дифлунизал, фенопрофен, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, напроксен, оксапрозин, пироксикам, сулиндак и толметин; ингибиторами COX-2, такими как целококсиб, рофекоксиб и вальдекоксиб; агонистами CB-2; антагонистами VR-1; антагонистами рецептора брадикинина B1; блокаторами и антагонистами натриевых каналов; ингибиторами синтазы оксида азота (NOS) (включая ингибиторы iNOS и nNOS); антагонистами глициновых рецепторов, включая лакозамид; нейрональными никотиновыми агонистами; антагонистами NMDA; открывателями калиевых каналов; антагонистами AMPA/каинатного рецептора; блокаторами кальциевых каналов, такими как зиконотид; модуляторами IO рецептора GABA-A (например, агонист рецептора GABA-A); ингибиторами матриксной металлопротеазы (MMP); тромболитическими средствами; химиотерапевтическими средствами, опиоидными анальгетиками, такими как кодеин, фентанил, гидроморфон, леворфанол, меперидин, метадон, морфин, оксикдон, оксиморфон, пентазоцин, пропоксифен; фактором ингибирующим нейтрофилы (NIF); прамипексолом, ропиниролом; антихолинергическими средствами; амантадином; ингибиторами моноаминоксидазы B15 («MAO-B»); агонистами или антагонистами рецептора 5HT; антагонистами mGlu5; альфа агонистами; нейрональными никотиновыми агонистами; агонистами или антагонистами рецептора NMDA; антагонистами NK1; ингибиторами селективного обратного захвата серотонина («SSRI») и/или ингибиторами селективного обратного захвата серотонина и норэпинефрина («SSNRI»), такими как дулоксетин; трициклическими антидепрессантами, модуляторами норэпинефрина; литием; вальпроатом; габапентином; прегабалином; ризатриптаном; золмитриптаном; наратриптаном и суматриптаном.
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель. Еще один аспект настоящего изобретения относится к соединению формулы I или его фармацевтически приемлемой соли для применения при лечении состояния, поддающегося лечению ингибитором TrkA и/или TrkB, такого как нарушения, состояния и/или заболевания, описанные в настоящем документе. Еще один аспект относится к применению соединения формулы I или его фармацевтически приемлемой соли для лечения боли, злокачественной опухоли, воспаления, нейродегенеративного заболевания и инфекции Trypanosoma cruzi.
Используемый в настоящем документе термин «композиция» предназначен для охвата продукта, содержащего конкретные ингредиенты в определенных количествах или соотношениях, а также любого продукта, который, прямо или опосредованно, является следствием сочетания конкретных ингредиентов в определенных количествах. Этот термин в отношении фармацевтических композиций предназначен для охвата продукта, содержащего один или несколько активных ингредиентов и необязательно носитель, содержащий инертные ингредиенты, а также любого продукта, который, прямо или опосредованно, является следствием сочетания, комплексообразования или агрегации любых двух или нескольких из ингредиентов, или следствием диссоциации одного или нескольких из ингредиентов, или следствием других типов реакций или взаимодействий одного или нескольких из ингредиентов.
Обычно, фармацевтические композиции приготавливают путем равномерного и тщательного приведения активного ингредиента в контакт с жидким носителем или мелкодисперсным твердым носителем, или с обоими, с последующим, при необходимости, формованием продукта в желаемый состав. В фармацевтической композиции активное соединение, которое представляет собой соединение формулы (I), включают в количестве, достаточном для получения желаемого эффекта на процесс или состояние заболевания. Соответственно, фармацевтические композиции согласно настоящему изобретению охватывают любую композицию, приготовленную путем смешивания соединения согласно настоящему изобретению и фармацевтически приемлемого носителя.
Носитель может принимать большое разнообразие форм в зависимости от формы препарата, желательной для введения, например, пероральной или парентеральной (включая внутривенную). Таким образом, фармацевтические композиции согласно настоящему изобретению могут быть представлены в виде дискретных элементов, подходящих для перорального введения, таких как капсулы, облатки или таблетки, каждый из которых содержит определенное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в виде неводной жидкости, в виде эмульсии типа «масло-в-воде» или в виде эмульсии типа «вода-в-масле». В дополнение к обычным лекарственным формам, приведенным выше, соединения согласно настоящему изобретению или их фармацевтически приемлемые соли также можно вводить посредством контролируемого высвобождения и/или устройствами доставки.
Фармацевтические композиции, предназначенные для перорального использования, можно приготовить в соответствии с любым способом, известным в области техники производства фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подсластителей, вкусоароматизаторов, красителей и консервантов, с целью обеспечения фармацевтически привлекательных и приятных на вкус препаратов. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые подходят для производства таблеток. Указанные наполнители могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция и фосфат натрия; гранулирующие средства и разрыхлители, например, кукурузный крахмал или альгиновую кислоту; связующие вещества, например, крахмал, желатин или акация и смазки, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть не покрытыми, или на них может быть нанесено покрытие посредством известных методик для отсрочки разрыхления и всасывания в желудочно-кишечном тракте, обеспечивая тем самым пролонгированное действие в течение более длительного периода времени.
Таблетка, содержащая композицию согласно настоящему изобретению, может быть приготовлена путем прессования или формования, необязательно с одним или несколькими вспомогательными ингредиентами или адъювантами. Прессованные таблетки могут быть получены путем прессования в подходящей машине активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом, смазкой, инертным разбавителем, поверхностно-активным веществом или средством, способствующим диспергированию. Формованные таблетки могут быть получены путем формования в подходящей машине смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит приблизительно от 0,1 мг приблизительно до 500 мг активного ингредиента, и каждый каше или капсула предпочтительно содержит приблизительно от 0,1 мг приблизительно до 500 мг активного ингредиента.
Композиции для перорального применения также могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешивают с инертным твердым разбавителем, например, с карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешивают с водной или масляной средой, например, с арахисовым маслом, жидким парафином или оливковым маслом.
Другие фармацевтические композиции включают в себя водные суспензии, которые содержат активные вещества в смеси с наполнителями, подходящими для производства водных суспензий. Кроме того, масляные суспензии могут быть составлены путем суспендирования активного ингредиента в растительном масле, например, в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии также могут содержать различные наполнители. Фармацевтические композиции согласно настоящему изобретению также могут быть в форме эмульсий типа «масло-в-воде», которые также могут содержать наполнители, такие как подсластители и вкусоароматизаторы.
Фармацевтические композиции могут быть форме стерильных инъекционных водных или масляных суспензий, или в форме стерильных порошков для немедленного приготовления таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная инъекционная форма должна быть стерильной и должна быть действительно жидкой для легкого введения через шприц. Фармацевтические композиции должны быть стабильными в условиях производства и хранения; поэтому, их следует предпочтительно предохранять от контаминационного действия микроорганизмов, таких как бактерии и грибы.
Фармацевтические композиции согласно настоящему изобретению могут быть в форме, подходящей для местного применения, например, такой как аэрозоль, крем, мазь, лосьон, присыпка и т. п. Кроме того, композиции могут быть в форме подходящей для использования в чрескожных устройствах. Указанные составы могут быть приготовлены посредством общепринятых технологических способов. В качестве примера, крем или мазь приготавливают путем смешивания гидрофильного вещества и воды вместе с приблизительно от 5 масс.% приблизительно до 10 масс.% соединения с получением крема или мази, характеризующейся желаемой консистенцией.
Фармацевтические композиции согласно настоящему изобретению могут быть также в форме, подходящей для ректального введения, в которой носитель представляет собой твердое вещество. Предпочтительно, когда смесь образует суппозитории с однократной дозой. Подходящие носители включают в себя какао-масло и другие вещества, обычно используемые в данной области техники.
Под термином «фармацевтически приемлемый» подразумевается что носитель, разбавитель или наполнитель должен быть совместим с другими ингредиентами состава и безвреден для его реципиента.
Термины «введение» или «вводить» применительно к соединению следует понимать как предоставление соединения согласно настоящему изобретению нуждающемуся в лечении индивидууму в форме, которая может быть введена в организм индивидуума в терапевтически применимой форме и терапевтически применимом количестве, включая без ограничения: пероральные лекарственные формы, такие как таблетки, капсулы, сиропы, суспензии и т. п.; инъекционные лекарственные формы, такие как внутривенные, внутримышечные или интраперитонеальные инъекции и т. п.; чрескожные лекарственные формы, включая кремы, желе, порошки или пластыри; буккальные лекарственные формы; ингаляционные порошки, спреи, суспензии и т. п.; и ректальные суппозитории.
Термины «эффективное количество» или «терапевтически эффективное количество» означают количество рассматриваемого соединения, которое будет вызывать биологический или медицинский ответ в ткани, системе, животном или человеке, которые находятся под наблюдением исследователя, ветеринара, врача или другого клинициста.
Используемый в настоящем документе термин «лечение» или «проведение лечения» означает любое введение соединения согласно настоящему изобретению и включает в себя (1) ингибирование заболевания у животного, которое испытывает или демонстрирует патологию или симптоматику заболевания (т.е., остановку дальнейшего развития патологии и/или симптоматики) или (2) уменьшение интенсивности заболевания у животного, которое испытывает или демонстрирует патологию или симптоматику заболевания (т.е., реверсию патологии и/или симптоматики).
Композиции, содержащие соединения согласно настоящему изобретению, могут быть представлены подходящим образом в виде стандартной лекарственной формы и могут быть приготовлены любыми способами хорошо известными в области техники фармацевтики. Термин «стандартная лекарственная форма» используют для обозначения однократной дозы, в которой все активные и неактивные ингредиенты объединены в подходящую систему, так что пациент или субъект, который вводит лекарство пациенту, может открыть отдельный контейнер или упаковку с цельной дозой, содержащейся в ней, и ему не нужно смешивать вместе любые компоненты из двух или нескольких контейнеров или упаковок. Типичные примеры стандартных лекарственных форм представляют собой таблетки или капсулы для перорального введения, флаконы для инъекций с однократной дозой или суппозитории для ректального введения. Этот перечень стандартных лекарственных форм не предназначен для какого-либо ограничения, а лишь представляет собой типичные примеры стандартных лекарственных форм.
Композиции, содержащие соединения согласно настоящему изобретению, могут быть соответствующим образом представлены в виде набора, в котором два или несколько компонентов, которые могут представлять собой активные или неактивные ингредиенты, носители, разбавители и т. п., снабженного инструкциями для приготовления действующей лекарственной формы пациентом или субъектом, который вводит лекарство пациенту. Такие наборы могут быть снабжены всеми необходимыми веществами и ингредиентами, содержащимися в нем, или могут содержать инструкции по применению или приготовлению веществ или компонентов, которые должны быть получены независимо пациентом или субъектом, который вводит лекарство пациенту.
При лечении или уменьшении интенсивности нарушения или заболевания, для которого предписаны соединения согласно настоящему изобретению, удовлетворительные результаты обычно получают, если соединения согласно настоящему изобретению вводят в суточной дозе приблизительно от 0,1 мг приблизительно до 100 мг на килограмм массы тела животного, предпочтительно в виде однократной суточной дозы или отдельными дозами от двух до шести раз в сутки, или в форме с замедленным высвобождением. Общая суточная дозировка составляет приблизительно от 1 мг приблизительно до 2000 мг, предпочтительно приблизительно от 0,1 мг приблизительно до 20 мг на килограмм массы тела. В случае взрослого человека массой 70 кг общая суточная доза будет обычно приблизительно от 7 мг приблизительно до 1400 мг. Такая схема дозирования может быть адаптирована для обеспечения оптимального терапевтического ответа. Соединения можно вводить по схеме от 1 до 4 раз в сутки, предпочтительно однократно или дважды в сутки.
Количество активного ингредиента, которое можно сочетать с веществами-носителями, для получения однократной лекарственной формы будет варьировать в зависимости от подвергаемого лечению реципиента и конкретного пути введения. Например, состав, предназначенный для перорального введения людям, может соответствующим образом содержать приблизительно от 0,005 мг приблизительно до 2,5 г активного средства, смешанного с подходящим и удобным количеством вещества-носителя. Стандартные лекарственные формы обычно будут содержать приблизительно от 0,005 мг приблизительно до 1000 мг активного ингредиента, обычно 0,005, 0,01 мг, 0,05 мг, 0,25 мг, 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг, вводимого один, два или три раза в сутки.
Однако следует понимать, что для каждого конкретного пациента конкретный уровень дозы и частоту дозирования можно варьировать, и они будут зависеть от ряда факторов, включая активность конкретного используемого соединения, метаболическую стабильность и длительность действия такого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион питания, путь и время введения, скорость выведения, сочетание лекарств, тяжесть конкретного состояния и реципиента, подвергающегося лечению.
Несколько способов получения соединений согласно настоящему изобретению проиллюстрированы на последующих схемах и примерах. Исходные вещества и требуемые промежуточные продукты являются в некоторых случаях коммерчески доступными или могут быть получены в соответствии с описанными в литературе методиками или как проиллюстрировано в настоящем документе.
Соединения согласно настоящему изобретению могут быть получены с использованием реакций, представленных на последующих схемах, в дополнение к другим стандартным манипуляциям, которые хорошо известны из литературы или описаны на примерах в экспериментальной части. Нумерация заместителей, представленная на схемах, необязательно коррелирует с таковой, приведенной в формуле изобретения, и часто, для наглядности, присоединенным к соединению показан единственный заместитель, тогда как согласно представленным выше определениям допускается наличие нескольких заместителей. Реагенты, использованные для получения соединений согласно настоящему изобретению, получают с использованием реакций, приведенных на схемах и в примерах, представленных в настоящем документе, в дополнение к другим стандартным манипуляциям, таким как гидролиз сложных эфиров, отщепление защитных групп, и т. д., и могут быть известны из литературы или описаны на примерах в экспериментальной части.
В процессе любой из последовательностей синтеза может быть необходимо или желательно защищать чувствительные или реакционноспособные группы на любой из рассматриваемых молекул. Это может достигаться посредством использования традиционных защитных групп, таки как описанные в Protective Groups in Organic Chemistry, ed. J.F.W.McOmie, Plenum Press, 1973, и в T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 1999. Защитные группы могут быть удалены на подходящей последующей стадии с использованием способов, известных из уровня техники.
В некоторых случаях, конечный продукт может быть дополнительно модифицирован, например, путем манипуляций с заместителями. Указанные манипуляции могут включать в себя без ограничений реакции восстановления, окисления, алкилирования, ацилирования и гидролиза, которые обычно известны специалистам в данной области техники. В некоторых случаях, порядок проведения реакций на представленных выше схемах реакций может варьировать для облегчения реакции или недопущения образования нежелательных продуктов реакций. Последующие примеры представлены таким образом, что изобретение может стать более понятным. Указанные примеры представлены исключительно с иллюстративной целью и не должны истолковываться как ограничивающие каким-либо образом настоящее изобретение.
Следующие сокращения используются по всему тексту:
Me: метил
Et: этил
Bu: бутил
t-Bu: трет-бутил
Ar: арил
Ph: фенил
Bn: бензил
Ac: ацетил
DMF⋅DMA: N,N-диметилформамида диметилацеталь
DMSO: диметилсульфоксид
DMF: N,N-диметилформамид
THF: тетрагидрофуран
TEA: триэтиламин
водн.: водный
HPLC: высокоэффективная жидкостная хроматография
MS: масс-спектрометрия
CDI: 1,1'-карбонилдиимидазол
DCE: 1,2-дихлорэтан
HCl: соляная кислота
°C: градусы Цельсия
BINAP: 2,2'-бис(дифенилфосфино)-1,1'-бинафталин
ATP: аденозинтрифосфат
i-Pr: изопропил
Py: пиридил
OAc: ацетат
TFA: трифторуксусная кислота
TFAA: трифторуксусный ангидрид
Boc: трет-бутоксикарбонил
Boc2O: ди-трет-бутилдикарбонат
BOP: (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат
DEA: диэтиламин
DIEA: N,N-диизопропилэтиламин
DIPEA: N,N-диизопропилэтиламин
HOBT: 1-гидроксибензотриазол
EDC: N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид
EDCI: N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид
PyCLU: хлордипирролидинокарбений
n-BuLi: н-бутиллитий
HATU: O-(7-азабензотриазол-1-ил)-N,N,N'N'-тетраметиурония гексафторфосфат
EDTA: этилендиаминтетрауксусная кислота
HMDS: гексаметилдисилазан
мин: минуты
ч: часы
LCMS: жидкостная хроматография/масс-спектрометрия
SFC: сверхкритическая жидкостная хроматография
TLC: тонкослойная хроматография
NMP: 1-метил-2-пирролидинон
MTBE: метил-трет-бутиловый эфир
DMA: N,N-диметилацетамид
NBS: N-бромсукцинимид
CAN: аммоний-церия(IV) нитрат
dppf: 1,1'-бис(дифенилфосфино)ферроцен
dtbpf: 1,1′-бис(ди-трет-бутилфосфино)ферроцен
dba: дибензилиденацетон
DMAP: 4-(диметиламино)пиридин
PMBCl: 4-метоксибензилхлорид
DIBAL: диизобутилалюминия гидрид
DAST: (диэтиламино)серы трифторид
DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен
AIBN: 2-2'-азобисизобутиронитрил
m-CPBA: 3-хлорпероксибензойная кислота
DABCO: диазабицикло[2.2.2]октан
LDA: лития диизопропиламид
HOAt: 1-гидрокси-7-азабензотриазол
LAH: лития алюмогидрид
AOP: 7-(азабензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат
PyAOP: 7-(азабензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат
DCM: дихлорметан
PE: петролейный эфир
TMS: триметилсилил
конц.: концентрированный
TIPS: триизопропилсилил
OTf: трифторметансульфонат
bis-pin: 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан)
NCS: N-хлорсукцинимид
DPPA: дифенилфосфорилазид
PCC: пиридиния хлорхромат
DME: 1,2-диметоксиэтан
PMB: 4-метоксибензил
NMO: 4-метилморфолин N-оксид
PyBop: бензотриазол-1-илокситрипирролидинофосфония гексафторфосфат
PS: полистирол
Схемы реакций
Соединения согласно настоящему изобретению могут быть легко получены в соответствии с последующими схемами и конкретными примерами или их модификациями, с использованием легкодоступных исходных веществ, реагентов и традиционных методик синтеза. В этих реакциях, также возможно использование вариантов, которые сами по себе известны средним специалистам в данной области техники, но не представлены в деталях. Общие методики для получения соединений, приведенных в формуле настоящего изобретения, станут хорошо понятны и будут высоко оценены специалистом в данной области техники в результате изучения последующих схем.
Схема 1 иллюстрирует общую стратегию получения соединений согласно настоящему изобретению, при которой промежуточная карбоновая кислота (1.1) может быть активирована (например, посредством обработки POCl3, (COCl)2 или SOCl2 с получением хлорангидрида) с последующим сочетанием с амином (1.2) с получением целевого продукта-амида 1.3. Различные промежуточные карбоновые кислоты, такие как кислоты, описанные в настоящем документе (см. ниже), могут быть подвергнуты сочетанию с самыми разнообразными аминами с получением соединений согласно настоящему изобретению. Существует много известных стратегий для осуществления таких химических реакций сочетания, включая использование агентов сочетания, таких как EDC с HOBT, PyBOP, HATU, AOP, PyAOP, CDI и т. п.
Схема 1
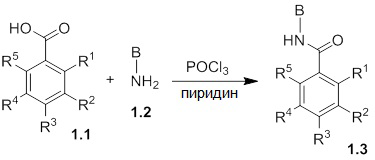
В некоторых случаях, для получения конкретного соединения согласно настоящему изобретению могут применяться различные стратегии использования защитных групп, известные специалистам в области органического синтеза. Этот общий подход может быть успешен при получении целого ряда амидных фрагментов, используя самые разнообразные промежуточные кислоты и амины.
Схема 2
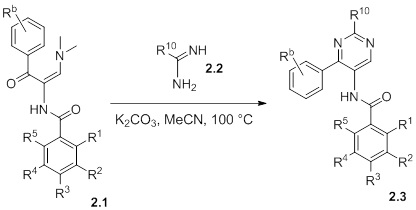
Схема реакций 2 иллюстрирует способ получения соединений типа 2.3, в котором кето-енамин 2.1 нагревают с амидином 2.2 в присутствии основания с получением амида 2.3.
Схема 3
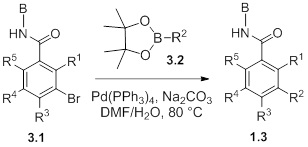
Схема реакций 3 иллюстрирует альтернативный способ получения соединений типа 1.3. Путем кросс-сочетания бромида 3.1 с арил- или гетероарилбороновым эфиром 3.2 (или с другим подходящим промежуточным продуктом) в водной смеси растворителей в присутствии подходящего катализатора и системы оснований (например, Pd(PPh3)4 и Na2CO3) получают амид 1.3.
Схемы реакций 4-6 иллюстрируют получение промежуточных аминов типа 1.2, которые используют для получения соединений согласно настоящему изобретению, как описано выше.
Схема 4

Схема реакций 4 иллюстрирует получение промежуточных аминов типа 4.6. Пиридин 4.1 депротонируют t-BuLi при низкой температуре, а затем подвергают воздействию йода с получением йодида 4.2. Путем селективного кросс-сочетания йодида 4.2 с фенилбороновой кислотой (или с другим подходящим промежуточным продуктом) в условиях по Судзуки в этом случае получают пиридин 4.3. В последующей реакции кросс-сочетания 4.3 с алкил- или арилбороновым эфиром 4.4 (или с другим подходящим промежуточным продуктом) образуется пиридин 4.5. Удаление Boc-группы проводят путем воздействия HCl в EtOAc с получением амина 4.6.
Схема 5
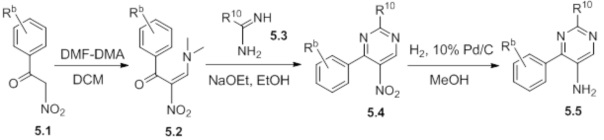
Схема реакций 5 иллюстрирует получение промежуточных аминов типа 5.5. Осуществляют взаимодействие бензоилнитрометана 5.1 с диметилацеталем N,N-диметилформамида с получением соединения 5.2. Преобразование до пиримидина 5.4 проводят путем воздействия на енамин 5.2 амидином 5.3 в этаноле в щелочных условиях. Нитрогруппу 5.4 восстанавливают H2 в присутствии 10% Pd/C в MeOH с получением амина 5.5.
Схема 6
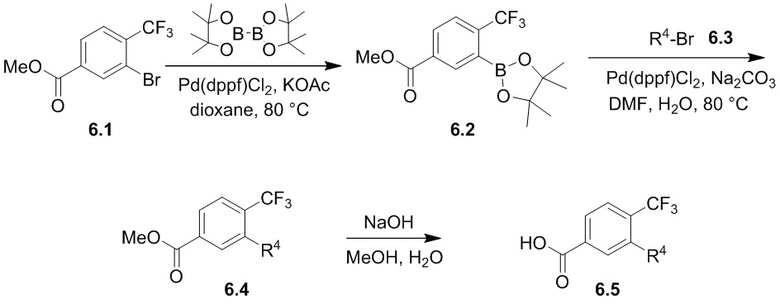
Схема реакций 6 иллюстрирует получение промежуточных кислот типа 6.5, которые используют для получения соединений согласно настоящему изобретению. Бромид 6.1 преобразуют до боронатного эфира 6.2 с использованием 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) в присутствии подходящего катализатора и системы оснований. Путем кросс-сочетания сложного эфира 6.2 с подходящим арил- или гетероарилбромидом (6.3), опосредованного нагреванием в водной системе растворителей в присутствии подходящего катализатора и основания (например, Pd(dppf)Cl2 и Na2CO3 в водном DMF), получают сложный эфир 6.4. Затем, путем гидролиза сложного эфира в щелочных условиях получают кислоту 6.5.
Схема 7
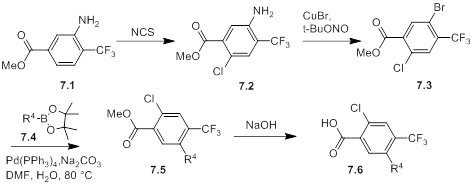
Схема реакций 7 иллюстрирует получение промежуточных кислот типа 7.6, которые используют для получения соединений согласно настоящему изобретению. Осуществляют взаимодействие амина 7.1 с NCS с получением хлорида 7.2, который затем преобразуют до бромида 7.3 путем воздействия трет-бутилнитрита и бромида меди(I). Путем кросс-сочетания бромида 7.3 с арил- или гетероарилбороновым эфиром 7.4 (или с другим подходящим промежуточным продуктом), опосредованного нагреванием в водной системе растворителей в присутствии подходящего катализатора и основания (например, Pd(dppf)Cl2 и Na2CO3 в водном DMF), получают сложный эфир 7.5. Гидролизом сложного эфира в щелочных условиях затем получают кислоту 7.6.
Схема 8
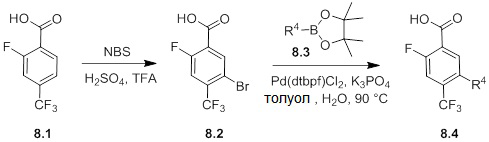
Схема реакций 8 отражает синтез промежуточных кислот типа 8.4. Путем бромирования 8.1 и последующего кросс-сочетания 8.2 с арил- или гетероарилбороновым эфиром 8.3 (или с другим подходящим промежуточным продуктом), опосредованного нагреванием в водной системе растворителей в присутствии подходящего катализатора и основания (например, Pd(dtbpf)Cl2 и K3PO4 в водном толуоле), получают 8.4.
Схема 9
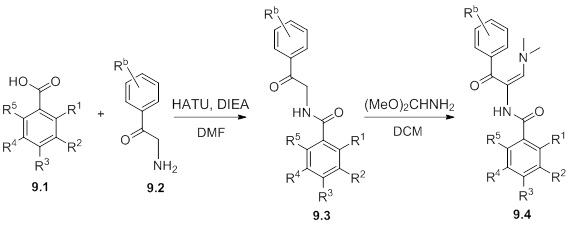
Схема реакций 8 иллюстрирует получение промежуточных енаминов типа 9.4, которые используют для получения соединений согласно настоящему изобретению. Аминоацетофенон 9.2 сочетают с кислотой 9.1 в стандартных условиях формирования амидной связи (например, HATU, DIEA) с получением амида 9.3. Кетоамид 9.3 затем обрабатывают диметилформамида диметилацеталем с получением енамина 9.4.
Схема 10
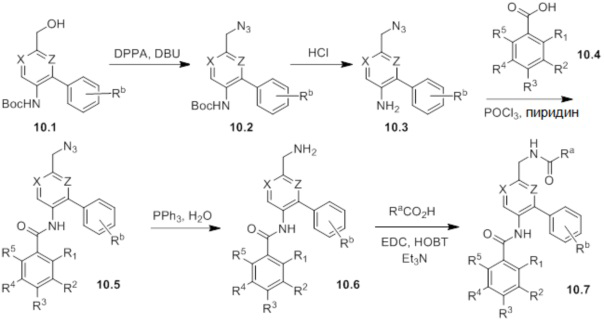
Схема реакций 10 отражает получение бензиловых амидов типа 10.7 согласно настоящему изобретению. Спирт 10.1 преобразуют до азида 10.2 с использованием DPPA и DBU в качестве основания. Затем, соляной кислотой удаляют защитную группу Boc с получением 10.3 в виде гидрохлорида. Путем сочетания 10.3 с бензоилхлоридом, полученным из 10.4, в этом случае путем обработки POCl3, получают бензамид 10.5. Путем восстановления азида в условиях по Штаудингеру получают амин 10.6, который может претерпевать сочетание с карбоновой кислотой в самых разнообразных условиях (например, EDC) с получением 10.7.
Схема 11
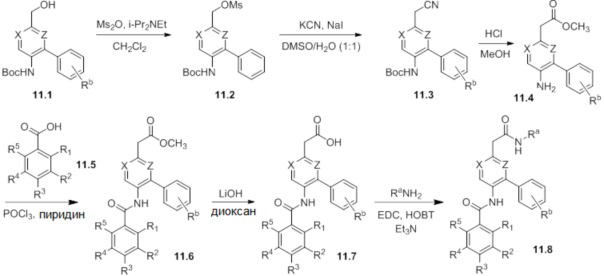
Схема реакций 11 описывает получение ацетамидов типа 11.8 согласно настоящему изобретению. Осуществляют взаимодействие спирта 11.1 с метансульфоновым ангидридом и N,N-диизопропилэтиламином с получением мезилата 11.2, который затем преобразуют до нитрила 11.3 с использованием KCN в присутствии NaI. Гидролиз нитрила с одновременным удалением группы Boc осуществляют путем нагревания 11.3 с соляной кислотой с получением анилина 11.4. Путем сочетания 11.4 с бензоилхлоридом, полученным из 11.5, в этом случае путем обработки POCl3, получают бензамид 11.6. Затем, путем сапонификации получают карбоновую кислоту 11.7, которая может быть сочетана с амином с получением 11.8.
Схема 12
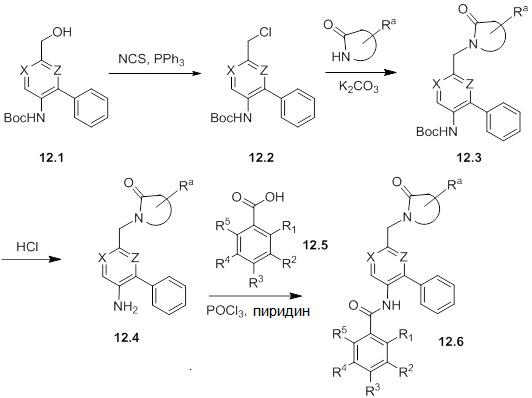
Схема реакций 12 иллюстрирует получение соединений типа 12.6 согласно настоящему изобретению. Спирт 12.1 преобразуют до хлорида 12.2 с использованием NCS и трифенилфосфина. N-алкилирование гетероциклов с использованием 12.2 опосредуется основанием, таким как карбонат калия с получением 12.2. В некоторых случаях, может потребоваться депротониривание гетероцикла более сильным основанием, таким как, например, гидрид натрия. С использованием промежуточных продуктов, содержащих кислотную метиленовую субъединицу, также может проводиться C-алкилирование. Следует отметить, что для оптимальных значений выхода для хлорида 12.2 может потребоваться дополнительная защитная группа Boc на анилиновом фрагменте. Затем, для удаления защитной (защитных) группы (групп) Boc промежуточный продукт 12.3 обрабатывают соляной кислотой с получением анилина 12.4. Путем сочетания 12.4 с бензоилхлоридом, полученным из 12.5, в этом случае путем обработки POCl3, получают бензамид 12.6.
Конкретные варианты осуществления соединений согласно настоящему изобретению и способы их получения описаны в настоящем документе в разделах «Промежуточные продукты» и «Примеры».
Схема реакций для промежуточного продукта A1
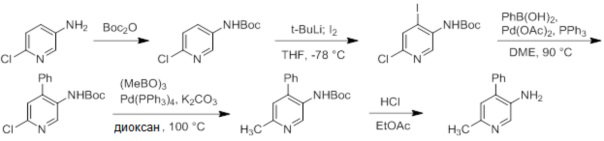
Промежуточный продукт A1
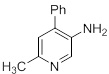
6-Метил-4-фенилпиридин-3-амин
Стадия A: трет-Бутил-(6-хлорпиридин-3-ил)карбамат
Смесь 6-хлорпиридин-3-амина (58,0 г, 451 ммоль) и Boc2O (133 мл, 573 ммоль) в диоксане (600 мл) нагревали при 100°C в течение 20 ч. Дополнительно добавляли Boc2O (17 мл, 72 ммоль), и нагревали смесь при 100°C в течение 7 ч. Смесь охлаждали и концентрировали, и распределяли остаток между EtOAc (500 мл×3) и водой (500 мл). Объединенные органические слои промывали солевым раствором (500 мл×2), сушили над Na2SO4 и концентрировали. Остаток растирали с PE:EtOAc=15:1 (400 мл×3) и сушили с получением указанного в заголовке соединения. MS: m/z=223 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,25 (д, J=3,0 Гц, 1H), 7,95 (д, J=6,3 Гц, 1H), 7,25 (д, J=8,8 Гц, 1H), 6,73 (с, 1H), 1,51 (с, 9H).
Стадия B: трет-Бутил-(6-хлор-4-йодпиридин-3-ил)карбамат
трет-Бутиллитий (1,3M в гептанах, 111 мл, 144 ммоль) по каплям добавляли к раствору трет-бутил-(6-хлорпиридин-3-ил)карбамата (15,0 г, 65,6 ммоль) в безводном THF (300 мл) при -78°C в течение 30 мин в атмосфере N2. Полученную смесь перемешивали при -78°C в течение 1 ч, а затем при -10°C в течение 1 ч. Реакционную смесь охлаждали до -78°C, и добавляли раствор I2 (36,6 г, 144 ммоль) в безводном THF (100 мл). Полученную смесь нагревали до температуры окружающей среды и перемешивали в течение 18 ч. Избыток трет-бутиллития и I2 гасили добавлением насыщенного водного раствора NH4Cl (150 мл) и насыщенного водного раствора Na2S2O3 (500 мл), соответственно, и перемешивали полученную смесь в течение 30 мин. Органический слой разделяли, и экстрагировали водный слой EtOAc (250 мл×2). Объединенные органические слои промывали солевым раствором (250 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc:PE:Et3N=2:98:1, затем 2,5:97,5:1) с получением указанного в заголовке соединения. MS: m/z=355 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,93 (с, 1H), 7,72 (с, 1H), 6,64 (с, 1H), 1,53 (с, 9H).
Стадия C: трет-Бутил-(6-хлор-4-фенилпиридин-3-ил)карбамат
Дезоксигенированную смесь трет-бутил-(6-хлор-4-йодпиридин-3-ил)карбамата (5,30 г, 14,9 ммоль), фенилбороновой кислоты (2,00 г, 16,4 ммоль), Pd(OAc)2 (0,168 г, 0,747 ммоль), Ph3P (0,392 г, 1,49 ммоль) и водного раствора Na2CO3 (2M, 37,4 мл, 74,7 ммоль) в DME (150 мл) нагревали при 90°C в атмосфере N2 в течение 18 ч. Смесь охлаждали, добавляли силикагель (15 г), и концентрировали полученную смесь. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc:Et3N=95:5:1) с получением указанного в заголовке соединения. MS: m/z=305 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,08 (с, 1H), 7,48-7,55 (м, 3H), 7,34-7,36 (м, 2H), 7,16 (с, 1H), 6,35 (с, 1H), 1,45 (с, 9H).
Стадия D: трет-Бутил-(6-метил-4-фенилпиридин-3-ил)карбамат
Дезоксигенированную смесь трет-бутил-(6-хлор-4-фенилпиридин-3-ил)карбамата (2,30 г, 7,55 ммоль), Pd(Ph3P)4 (0,872 г, 0,755 ммоль), 2,4,6-триметил-1,3,5,2,4,6-триоксатриборинана (3,79 г, 30,2 ммоль) и K2CO3 (3,13 г, 22,6 ммоль) в диоксане (30 мл) нагревали при 110°C в атмосфере N2 в течение 18 ч. Смесь охлаждали и фильтровали через Celite®. Фильтрат концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE:EtOAc:Et3N=90:10:1, затем 80:20:1) с получением указанного в заголовке соединения. MS: m/z=285 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,05 (с, 1H), 7,43-7,51 (м, 3H), 7,34-7,36 (м, 2H), 6,99 (с, 1H), 6,26 (с, 1H), 2,53 (с, 3H), 1,44 (с, 9H).
Стадия E: 6-Метил-4-фенилпиридин-3-амин
Раствор HCl в EtOAc (4M, 11,3 мл, 45,0 ммоль) добавляли к раствору трет-бутил-(6-метил-4-фенилпиридин-3-ил)карбамата (1,28 г, 4,50 ммоль) в EtOAc (15 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 18 ч, а затем концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=185 (M+1).
Промежуточный продукт A2
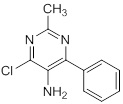
4-Хлор-2-метил-6-фенилпиримидин-5-амин
Дезоксигенированную смесь 4,6-дихлор-2-метилпиримидин-5-амина (536 мг, 3,01 ммоль), фенилбороновой кислоты (404 мг, 3,31 ммоль), Pd(PPh3)4 (174 мг, 0,151 ммоль) и водного раствора карбоната натрия (2M, 3,0 мл, 6,0 ммоль) в DMF (9 мл) нагревали при 110°C в условиях обработки микроволновым излучением в течение 1 ч. Смесь охлаждали и распределяли между водой (100 мл) и Et2O (3×70 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→50:50) с получением указанного в заголовке соединения. MS: m/z=220,2 (M+1).
Промежуточный продукт A3
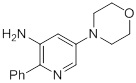
5-Морфолино-2-фенилпиридин-3-амин
Стадия A: Ди-трет-бутил-(5-хлор-2-фенилпиридин-3-ил)карбамат
Смесь 5-хлор-2-фенилпиридин-3-амина (3,0 г, 15 ммоль), Boc2O (3,5 г, 16 ммоль), TEA (4,0 мл, 29 ммоль) и DMAP (122 мг, 1,00 ммоль) в DCM (50 мл) нагревали при 30°C в течение 16 ч, а затем концентрировали. Остаток распределяли между DCM (100 мл) и водой (30 мл×2), органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=15:1) с получением указанного в заголовке соединения. MS: m/z=405 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,72 (д, J=2,4 Гц, 1H), 8,19 (д, J=2,4 Гц, 1H), 7,49 (м, 5H), 1,41 (с, 18H).
Стадия B: трет-Бутил-(5-морфолино-2-фенилпиридин-3-ил)карбамат
Дезоксигенированную смесь ди-трет-бутил-(5-хлор-2-фенилпиридин-3-ил)карбамата (2,0 г, 4,9 ммоль), морфолина (0,435 мл, 5,00 ммоль), t-BuONa (768 мг, 8,00 ммоль) и Pd2(dba)3 (100 мг, 0,11 ммоль) в диоксане (20 мл) нагревали при 80°C в течение 16 ч, а затем концентрировали. Остаток распределяли между DCM и H2O (50 мл×3), органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EA=3:1) с получением указанного в заголовке соединения. MS: m/z=356 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,24 (с, 1H), 7,98 (д, J=2,4 Гц, 1H), 7,52 (м, 5H), 7,44 (с, 1H), 3,87 (м, 4H), 3,29 (м, 4H) 1,47 (с, 9H).
Стадия C: 5-Морфолино-2-фенилпиридин-3-амин
Раствор HCl в EtOAc (4M, 5 мл, 20 ммоль) добавляли к раствору трет-бутил-(5-морфолино-2-фенилпиридин-3-ил)карбамата (160 мг, 0,40 ммоль) в EtOAc (2 мл), и перемешивали полученную смесь при 25°C в течение 2 ч. Смесь концентрировали, и распределяли остаток между DCM (50 мл) и водой (10 мл×3). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE:EtOAc=3:1) с получением указанного в заголовке соединения. MS: m/z=256 (M+1).
Схема реакций для промежуточного продукта A4

Промежуточный продукт A4
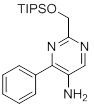
4-Фенил-2-(((триизопропилсилил)окси)метил)пиримидин-5-амин
Стадия A: 3-(Диметиламино)-2-нитро-1-фенилпроп-2-ен-1-он
Суспензию бензоилнитрометана (1,00 г, 6,06 ммоль) и N,N-диметилформамида диметилацеталя (0,877 мл, 6,60 ммоль) в DCM (12 мл) перемешивали при температуре окружающей среды в течение 5 суток. Смесь концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=221,2 (M+1).
Стадия B: (5-Нитро-4-фенилпиримидин-2-ил)метанол
Смесь 3-(диметиламино)-2-нитро-1-фенилпроп-2-ен-1-она (2,99 г, 13,6 ммоль), 2-гидроксиацетимидамида гидрохлорида (1,95 г, 17,6 ммоль) и этанолята натрия (2,77 г, 40,7 ммоль) в EtOH (27 мл) перемешивали при температуре окружающей среды в течение 3 ч. Реакционную смесь распределяли между водой (50 мл) и DCM (3×50 мл). Объединенные органические слои сушили над Na₂SO₄ и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=232,1 (M+1).
Стадия C: 5-Нитро-4-фенил-2-(((триизопропилсилил)окси)метил)пиримидин
TIPSOTf (1,29 мл, 4,76 ммоль) добавляли к раствору (5-нитро-4-фенилпиримидин-2-ил)метанола (1,00 г, 4,33 ммоль) и TEA (1,21 мл, 8,65 ммоль) в DCM (10 мл), и перемешивали полученную смесь при температуре окружающей среды в течение 0,5 ч. Реакционную смесь очищали непосредственно методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→30:70) с получением указанного в заголовке соединения. MS: m/z=388,3 (M+1).
Стадия D: 4-Фенил-2-(((триизопропилсилил)окси)метил)пиримидин-5-амин
К раствору 5-нитро-4-фенил-2-(((триизопропилсилил)окси)метил)пиримидина (117 мг, 0,302 ммоль) в MeOH (3 мл) добавляли взвесь 10% Pd/C (10 мг, 10 масс.%) в EtOAc (~0,2 мл), и перемешивали полученную смесь в атмосфере H2 при температуре окружающей среды в течение 3 ч. Суспензию фильтровали через Celite®, промывали MeOH (0,5 мл), и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=358,3 (M+1).
Схема реакций для промежуточного продукта A5
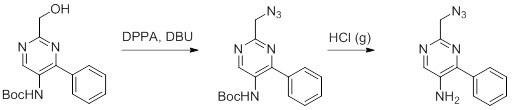
Промежуточный продукт A5
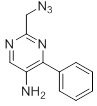
2-(Азидометил)-4-фенилпиримидин-5-амин
Стадия A: трет-Бутил-(2-(азидометил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамата (10,0 г, 33,2 ммоль) в 2-метилтетрагидрофуране (100 мл) при 0°C добавляли DPPA (8,58 мл, 39,8 ммоль), а затем DBU (6,00 мл, 39,8 ммоль). Полученную смесь перемешивали при 0°C в течение 30 мин, затем нагревали до 23°C и перемешивали в течение 4 ч. Содержащую продукт смесь распределяли между солевым раствором (200 мл) и EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток суспендировали в EtOAc (40 мл), и фильтровали полученное твердое вещество. Фильтрат концентрировали и повторяли последовательность суспендирования/фильтрования, после чего остаток очищали методом колоночной флэш-хроматографии (градиент гексаны→50% EtOAc в гексанах) с получением указанного в заголовке соединения. MS: m/z=327,2 (M+1).
Стадия B: 2-(Азидометил)-4-фенилпиримидин-5-амин
Раствор трет-бутил-(2-(азидометил)-4-фенилпиримидин-5-ил)карбамата (6,00 г, 18,4 ммоль) в EtOAc (200 мл) при 0°C насыщали газообразным HCl. Полученный раствор нагревали до 23°C и перемешивали в течение 1,5 ч, а затем концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=227,3 (M+1).
Схема реакций для промежуточного продукта A6
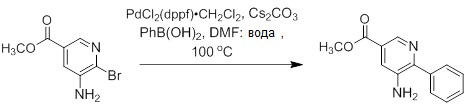
Промежуточный продукт A6
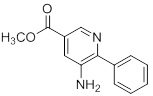
Метил-5-амино-6-фенилникотинат
Дезоксигенированную смесь метил-5-амино-6-бромникотината (950 мг, 4,11 ммоль), фенилбороновой кислоты (752 мг, 6,27 ммоль), PdCl2(dppf)⋅CH2Cl2 (336 мг, 0,411 ммоль) и карбоната цезия (4,02 г, 12,3 ммоль) в смеси DMF и воды (5:1, 20 мл) нагревали при 100°C в течение 6 ч. Смесь охлаждали и распределяли между водой (60 мл) и CH2Cl2 (3×80 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=229,4 (M+1).
Схема реакций для промежуточного продукта A7
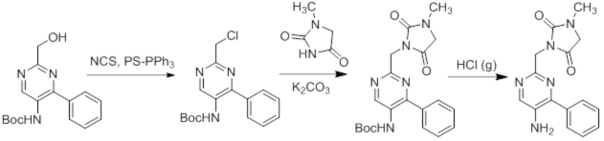
Промежуточный продукт A7

3-((5-Амино-4-фенилпиримидин-2-ил)метил)-1-метилимидазолидин-2,4-дион
Стадия A: трет-Бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат
NCS (576 мг, 4,31 ммоль) добавляли к суспензии трет-бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамата (1,00 г, 3,32 ммоль) и PS-трифенилфосфиновой смолы (3,11 г, 9,96 ммоль) в DCM (20 мл) при 23°C. Полученную смесь осторожно перемешивали в течение 20 мин, после чего дополнительно добавляли NCS (300 мг, 2,25 ммоль). Спустя 30 мин, смолу фильтровали и промывали DCM (2×50 мл). Объединенный фильтрат промывали насыщенным водным раствором бикарбоната натрия (2×100 мл), сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения. MS: m/z=320,2 (M+1).
Стадия B: трет-Бутил-(2-((3-метил-2,5-диоксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамат
Раствор трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (500 мг, 1,56 ммоль), 1-метилимидазолидин-2,4-диона (357 мг, 3,13 ммоль) и карбоната калия (432 мг, 3,13 ммоль) в смеси диоксана (5 мл) и воды (2 мл) перемешивали при 23°C в течение 60 ч. Содержащую продукт смесь распределяли между солевым раствором (75 мл) и EtOAc (2×75 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом колоночной флэш-хроматографии (градиент гексаны→100% EtOAc) с получением указанного в заголовке соединения. MS: m/z=398,3 (M+1)
Стадия C: 3-((5-Амино-4-фенилпиримидин-2-ил)метил)-1-метилимидазолидин-2,4-дион
Раствор трет-бутил-(2-((3-метил-2,5-диоксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамата (330 мг, 0,83 ммоль) в EtOAc (30 мл) при 0°C насыщали газообразным HCl. Полученный раствор нагревали до 23°C и перемешивали в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=298,2 (M+1).
Схема реакций для промежуточного продукта A8
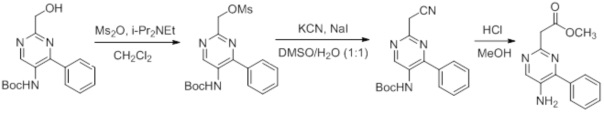
Промежуточный продукт A8
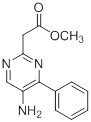
Метил-2-(5-амино-4-фенилпиримидин-2-ил)ацетат
Стадия A: (5-((трет-Бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метилметансульфонат
К раствору трет-бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамата (2,00 г, 6,64 ммоль) в CH2Cl2 (20 мл) при 23°C добавляли метансульфоновый ангидрид (2,54 г, 14,6 ммоль) и N,N-диизопропилэтиламин (1,73 мл, 9,96 ммоль), и перемешивали полученную смесь в течение 1,5 ч. Содержащую продукт смесь вливали в воду (20 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом колоночной флэш-хроматографии на SiO2 (картридж 120 г), элюируя 0-100% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=380,1 (M+1).
Стадия B: трет-Бутил-(2-(цианометил)-4-фенилпиримидин-5-ил)карбамат
(5-((трет-Бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метилметансульфонат (8,46 г, 22,3 ммоль) растворяли в CH2Cl2 (20 мл). Добавляли йодид натрия (334 мг, 2,23 ммоль) и цианид калия (1,9 г, 29 ммоль), а затем смесь DMSO и H2O (1:1, 500 мл). Полученную смесь нагревали при 50°C в течение 1 ч. Содержащую продукт смесь охлаждали, и разделяли органический слой. Водный слой экстрагировали CH2Cl2 (3×100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом колоночной флэш-хроматографии на SiO2 (картридж Isco Gold 80 г), элюируя 0-50% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=311,2 (M+1).
Стадия C: Метил-2-(5-амино-4-фенилпиримидин-2-ил)ацетат
К трет-бутил-(2-(цианометил)-4-фенилпиримидин-5-ил)карбамату (1,00 г, 3,22 ммоль) добавляли 3M раствор HCl в MeOH (80 мл, 240 ммоль), и нагревали полученную смесь при 60°C в течение 18 ч. Реакционную смесь концентрировали, и распределяли остаток между насыщенным водным раствором NaHCO3 (100 мл) и CH2Cl2 (100 мл×3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением указанного в заголовке соединения. MS: m/z=244,2 (M+1).
Схема реакций для промежуточного продукта A9
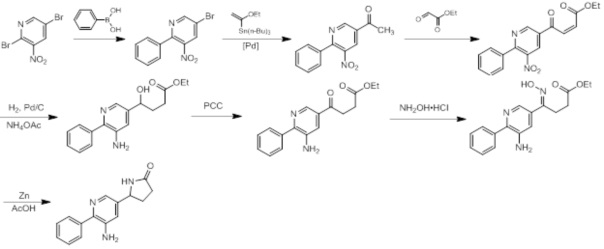
Промежуточный продукт A9
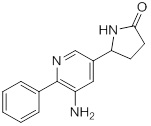
5-(5-Амино-6-фенилпиридин-3-ил)пирролидин-2-он
Стадия A: 5-Бром-3-нитро-2-фенилпиридин
К дезоксигенированному раствору 2,5-дибром-3-нитропиридина (25 г, 89 ммоль) в диоксане (200 мл) и воде (50 мл) добавляли фенилбороновую кислоту (11,9 г, 97,6 ммоль), карбонат калия (36,8 г, 266 ммоль) и PdCl2(dppf) (3,24 г, 4,43 ммоль). Полученную смесь нагревали при 100°C в течение 3 ч, а затем охлаждали и концентрировали. Остаток экстрагировали EtOAc (100 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии (PE/EtOAc=50/1→40/1→30/1) с получением указанного в заголовке соединения. MS: m/z=278,9 (M+1).
Стадия B: 1-(5-Нитро-6-фенилпиридин-3-ил)этанон
К дезоксигенированной смеси 5-бром-3-нитро-2-фенилпиридина (10,0 г, 35,8 ммоль) в диоксане (100 мл) добавляли трибутил(1-этоксивинил)станнан (19,1 г, 53,7 ммоль), йодид меди(I) (0,341 г, 1,79 ммоль) и дихлорбис(три-орто-толилфосфин)палладий(II) (2,82 г, 3,58 ммоль), и нагревали полученную смесь при 100°C в течение 3 ч. Добавляли 2н водный раствор HCl (30 мл), и перемешивали полученную смесь при 25°C в течение 1 ч. Добавляли насыщенный водный раствор фторида калия (15 мл), и фильтровали содержащую продукт смесь. Фильтрат подщелачивали до pH=9 добавлением насыщенного водного раствора K2CO3, а затем экстрагировали EtOAc (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EA=15/1, 10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=243,0 (M+1).
Стадия C: Этил-4-(5-нитро-6-фенилпиридин-3-ил)-4-оксобут-2-еноат
К раствору 1-(5-нитро-6-фенилпиридин-3-ил)этанона (0,500 г, 2,06 ммоль) в AcOH (5 мл) добавляли этил-2-оксоацетат (1,05 г, 10,3 ммоль), и перемешивали полученную смесь при 120°C в течение 12 ч. Содержащую продукт смесь охлаждали и распределяли между водой (10 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EA=3/1) с получением указанного в заголовке соединения. MS: m/z=327,0 (M+1).
Стадия D: Этил-4-(5-амино-6-фенилпиридин-3-ил)-4-гидроксибутаноат
К дезоксигенированному раствору этил-2-(5-нитро-6-фенилпиридин-3-ил)-4-оксобут-2-еноата (900 мг, 2,76 ммоль) в MeOH (20 мл) добавляли 10% Pd/C (29,4 мг, 0,276 ммоль) и NH4OAc (425 мг, 5,52 ммоль), и нагревали полученную смесь в атмосфере H2 (50 фунтов на кв.дюйм) при 30°C в течение 10 ч. Смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EA=8/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=301,1 (M+1).
Стадия E: Этил-4-(5-амино-6-фенилпиридин-3-ил)-4-оксобутаноат
К раствору этил-4-(5-амино-6-фенилпиридин-3-ил)-4-гидроксибутаноата (0,600 г, 2,00 ммоль) в CH2Cl2 (8 мл) добавляли PCC (1,72 г, 7,99 ммоль), и перемешивали полученную смесь при 25°C в течение 12 ч. Добавляли воду (5 мл), и фильтровали содержащую продукт смесь. Фильтрат экстрагировали EtOAc (5 мл×3), и концентрировали объединенные органические слои с получением указанного в заголовке соединения. MS: m/z=299,0 (M+1).
Стадия F: Этил-4-(5-амино-6-фенилпиридин-3-ил)-4-(гидроксиимино)бутаноат
К раствору этил-4-(5-амино-6-фенилпиридин-3-ил)-4-оксобутаноата (380 мг, 1,27 ммоль) в EtOH (4 мл) добавляли NH2OH⋅HCl (133 мг, 1,91 ммоль) и пиридин (0,308 мл, 3,82 ммоль), и нагревали полученную смесь при 80°C в течение 3 ч. Смесь распределяли между водой (5 мл) и EtOAc (5 мл×3). Объединенные органические фазы концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=314,0 (M+1).
Стадия G: 5-(5-Амино-6-фенилпиридин-3-ил)пирролидин-2-он
К перемешанному раствору этил-4-(5-амино-6-фенилпиридин-3-ил)-4-(гидроксиимино)бутаноата (150 мг, 0,479 ммоль) в AcOH (4 мл) добавляли цинковый порошок (156 мг, 2,39 ммоль), и нагревали полученную смесь при 80°C в течение 6 ч. Смесь охлаждали и фильтровали, и концентрировали фильтрат. Остаток распределяли между насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/2) с получением указанного в заголовке соединения. MS: m/z=254,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,95 (ушир. с, 1H), 7,56 (д, J=7,0 Гц, 2H), 7,40 (т, J=7,2 Гц, 2H), 7,34 (д, J=7,0 Гц, 1H), 6,93 (ушир. с, 1H), 6,65 (ушир. с, 1H), 4,67 (т, J=6,7 Гц, 1H), 3,95 (ушир. с, 2H), 2,57-2,49 (м, 1H), 2,46-2,32 (м, 2H), 1,99-1,91 (м, 1H).
Схема реакций для промежуточного продукта A10
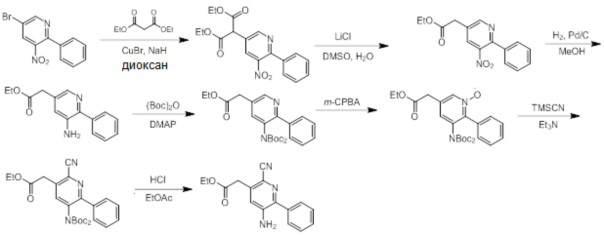
Промежуточный продукт A10
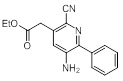
Этил-2-(5-амино-2-циано-6-фенилпиридин-3-ил)ацетат
Стадия A: Диэтил-2-(5-нитро-6-фенилпиридин-3-ил)малонат
К смеси 5-бром-3-нитро-2-фенилпиридина (5,00 г, 17,9 ммоль), диэтилмалоната (11,5 г, 71,7 ммоль) и бромида меди(I) (10,3 г, 71,7 ммоль) в диоксане (150 мл) порциями при 25°C добавляли NaH (60 масс.%, 3,15 г, 79,0 ммоль), и нагревали полученную смесь при 100°C в течение 16 ч. Реакционную смесь охлаждали и разбавляли насыщенным водным раствором NH4Cl (100 мл). Водную смесь экстрагировали EtOAc (100 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=50/1, 30/1, 10/1) с получением указанного в заголовке соединения. MS: m/z=359,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,84 (с, 1H), 8,41 (с, 1H), 7,54-7,45 (м, 5H), 6,11 (с, 1H), 4,27-4,18 (м, 4H), 1,29-1,23 (м, 6H).
Стадия B: Этил-2-(5-нитро-6-фенилпиридин-3-ил)ацетат
К перемешанному раствору диэтил-2-(5-нитро-6-фенилпиридин-3-ил)малоната (5,80 г, 16,2 ммоль) в DMSO (60 мл) и воде (3 мл) добавляли хлорид лития (2,10 г, 48,6 ммоль), и нагревали полученную смесь при 100°C в течение 5 ч. Смесь охлаждали и распределяли между водой (80 мл) и EtOAc (80 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=287,0 (M+1).
Стадия C: Этил-2-(5-амино-6-фенилпиридин-3-ил)ацетат
Дезоксигенированную смесь этил-2-(5-нитро-6-фенилпиридин-3-ил)ацетата (5,50 г, 19,2 ммоль) и 10% Pd/C (2,0 г, 1,9 ммоль) в MeOH (50 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 30°C в течение 5 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=257,0 (M+1).
Стадия D: Этил-2-(5-(ди-(трет-бутоксикарбонил)амино)-6-фенилпиридин-3-ил)ацетат
Смесь Boc2O (7,80 мл, 33,6 ммоль), триэтиламина (5,10 г, 50,3 ммоль), N,N-диметилпиридин-4-амина (2,10 г, 16,8 ммоль) и этил-2-(5-амино-6-фенилпиридин-3-ил)ацетата (4,30 г, 16,8 ммоль) в CH2Cl2 (50 мл) перемешивали при 30°C в течение 3 ч. Смесь концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=457,3 (M+1).
Стадия E: 3-(Ди-(трет-бутоксикарбонил)амино)-5-(2-этокси-2-оксоэтил)-2-фенилпиридин-1-оксид
К перемешанному раствору этил-2-(5-(ди-(трет-бутоксикарбонил)амино)-6-фенилпиридин-3-ил)ацетата (200 мг, 0,438 ммоль) в CHCl3 (4 мл) добавляли m-CPBA (267 мг, 1,31 ммоль), и перемешивали полученную смесь при 25°C в течение 2 ч. Содержащую продукт смесь распределяли между насыщенным водным раствором NaHCO3 (10 мл) и DCM (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=473,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1H), 8,03 (д, J=14,1 Гц, 1H), 7,94 (дд, J1=16,2 Гц, J2=7,6 Гц, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,43 (ушир. с, 2H), 7,21 (с, 1H), 4,21 (д, J=6,7 Гц, 2H), 3,62 (с, 2H), 1,34 (с, 18H), 1,29-1,27 (м, 3H).
Стадия F: Этил-2-(5-(ди-(трет-бутоксикарбонил)амино)-2-циано-6-фенилпиридин-3-ил)ацетат
К перемешанному раствору 3-(ди-(трет-бутоксикарбонил)амино)-5-(2-этокси-2-оксоэтил)-2-фенилпиридин-1-оксида (2,0 г, 4,2 ммоль) и Et3N (1,8 мл, 13 ммоль) в ацетонитриле (30 мл) добавляли TMSCN (2,3 мл, 17 ммоль), и нагревали полученную смесь при 80°C в течение 5 ч. Содержащую продукт смесь распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 1/1) с получением указанного в заголовке соединения. MS: m/z=482,3 (M+1).
Стадия G: Этил-2-(5-амино-2-циано-6-фенилпиридин-3-ил)ацетат
Раствор этил-2-(5-(ди-(трет-бутоксикарбонил)амино)-2-циано-6-фенилпиридин-3-ил)ацетата (500 мг, 1,00 ммоль) в 4н HCl в EtOAc (10 мл, 40,0 ммоль) перемешивали при 20°C в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=282,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,62 (д, J=7,0 Гц, 2H), 7,53-7,44 (м, 3H), 7,04 (с, 1H), 4,38 (ушир. с, 2H), 4,23 (кв, J=7,0 Гц, 2H), 3,81 (с, 2H), 1,31 (т, J=7,2 Гц, 3H).
Промежуточный продукт A11

Этил-2-(5-амино-6-фенилпиридин-3-ил)ацетат
К раствору этил-2-(5-нитро-6-фенилпиридин-3-ил)ацетата (400 мг, 0,699 ммоль) в THF (3 мл) и уксусной кислоте (1 мл) добавляли цинковый порошок (137 мг, 2,10 ммоль), и перемешивали полученную смесь при 25°C в течение 2 ч. Смесь распределяли между водой (20 мл) и этилацетатом (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=257,1 (M+1).
Схема реакций для промежуточного продукта A12

Промежуточный продукт A12
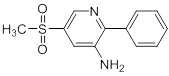
5-(Метилсульфонил)-2-фенилпиридин-3-амин
Стадия A: 5-Бром-2-фенилпиридин-3-амин
К раствору 5-бром-3-нитро-2-фенилпиридина (7,00 г, 22,6 ммоль) в этаноле (80 мл) добавляли хлорид олова (12,8 г, 67,7 ммоль), и нагревали полученную смесь при 90°C в течение 5 ч. Смесь распределяли между насыщенным водным раствором бикарбоната натрия (200 мл) и этилацетатом (200 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=249,1 (M+1).
Стадия B: 5-(Метилсульфонил)-2-фенилпиридин-3-амин
К раствору 5-бром-2-фенилпиридин-3-амина (800 мг, 3,21 ммоль) в диметилсульфоксиде (8 мл) добавляли натрия метансульфонат (393 мг, 3,85 ммоль), йодид меди(I) (61,2 мг, 0,321 ммоль), гидроксид натрия (25,7 мг, 0,642 ммоль) и L-пролин (73,9 мг, 0,642 ммоль). Полученную смесь нагревали при 70°C в течение 6 ч, затем охлаждали и распределяли между насыщенным водным раствором бикарбоната натрия (100 мл) и этилацетатом (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=249,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,57 (д, J=1,57 Гц, 1H), 7,67 (д, J=7,04 Гц, 2H), 7,54-7,42 (м, 4H), 4,18 (с, 2H), 3,11 (с, 3H).
Схема реакций для промежуточного продукта A13
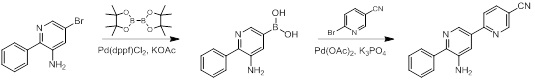
Промежуточный продукт A13
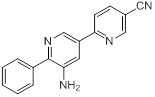
5'-Амино-6'-фенил-[2,3'-бипиридин]-5-карбонитрил
Стадия A: (5-Амино-6-фенилпиридин-3-ил)бороновая кислота
Дезоксигенированную смесь 5-бром-2-фенилпиридин-3-амина (5,00 г, 20,1 ммоль), PdCl2(dppf) (1,47 г, 2,01 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (5,10 г, 20,1 ммоль) и KOAc (5,91 г, 60,2 ммоль) в 1,4-диоксане (50 мл) нагревали при 100°C в течение 8 ч. Смесь охлаждали, а затем распределяли между водой (25 мл) и EtOAc (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=215,2 (M+1).
Стадия B: 5'-Амино-6'-фенил-[2,3'-бипиридин]-5-карбонитрил
Дезоксигенированную смесь (5-амино-6-фенилпиридин-3-ил)бороновой кислоты (500 мг, 2,34 ммоль), 6-бромникотинонитрила (428 мг, 2,34 ммоль), K3PO4 (1020 мг, 5,84 ммоль), Pd(OAc)2 (105 мг, 0,467 ммоль) и бутилди-1-адамантилфосфина (84 мг, 0,23 ммоль) в THF (5 мл) и воде (1 мл) нагревали при 100°C в течение 20 ч. Смесь распределяли между водой (5 мл) и EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=273,0 (M+1).
Схема реакций для промежуточного продукта A14
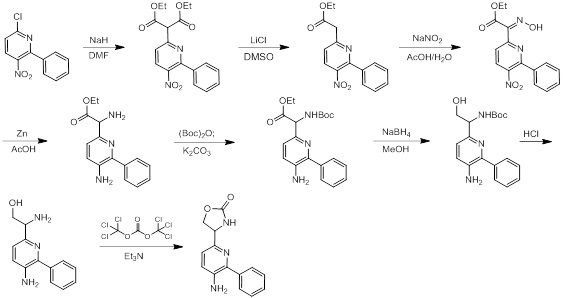
Промежуточный продукт A14

4-(5-Амино-6-фенилпиридин-2-ил)оксазолидин-2-он
Стадия A: Диэтил-2-(5-нитро-6-фенилпиридин-2-ил)малонат
К раствору диэтилмалоната (12,3 г, 76,8 ммоль) в DMF (120 мл) при 0°C добавляли NaH (60 масс.%, 3,84 г, 96 ммоль), и перемешивали смесь в течение 10 мин, после чего добавляли 6-хлор-3-нитро-2-фенилпиридин (15,0 г, 63,9 ммоль). Полученную смесь оставляли нагреваться до 20°C, после чего ее перемешивали в течение 5 ч. Содержащую продукт смесь вливали в воду со льдом (600 мл) и экстрагировали EtOAc (200 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=359,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,17 (д, J=8,0 Гц, 1H), 7,70 (д, J=8,5 Гц, 1H), 7,61-7,52 (м, 2H), 7,51-7,42 (м, 3H), 5,09 (с, 1H), 4,35-4,21 (м, 4H), 1,30 (т, J=7,0 Гц, 6H).
Стадия B: Этил-2-(5-нитро-6-фенилпиридин-2-ил)ацетат
К раствору диэтил-2-(5-нитро-6-фенилпиридин-2-ил)малоната (15,0 г, 41,9 ммоль) в DMSO (160 мл) добавляли хлорид лития (5,32 г, 126 ммоль) и воду (0,754 мл, 41,9 ммоль), и нагревали полученную смесь при 100°C в течение 18 ч. Реакционную смесь охлаждали и распределяли между водой (500 мл) и EtOAc (300 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=287,1 (M+1).
Стадия C: (E)-Этил-2-(гидроксиимино)-2-(5-нитро-6-фенилпиридин-2-ил)ацетат
К перемешанному раствору этил-2-(5-нитро-6-фенилпиридин-2-ил)ацетата (1,00 г, 3,49 ммоль) в AcOH (3 мл) по каплям добавляли раствор NaNO2 (0,265 г, 3,84 ммоль) в воде (1 мл), и перемешивали полученную смесь при 20°C в течение 2 ч. К реакционной смеси добавляли воду (3 мл), осадок фильтровали, промывали водой (20 мл), и сушили на воздухе с получением указанного в заголовке соединения. MS: m/z=316,2 (M+1).
Стадия D: Этил-2-амино-2-(5-амино-6-фенилпиридин-2-ил)ацетат
К перемешанному раствору (E)-этил-2-(гидроксиимино)-2-(5-нитро-6-фенилпиридин-2-ил)ацетата (720 мг, 2,28 ммоль) в AcOH (3 мл) добавляли цинковый порошок (747 мг, 11,4 ммоль), и нагревали полученную смесь при 80°C в течение 5 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=272,2 (M+1).
Стадия E: Этил-2-(5-амино-6-фенилпиридин-2-ил)-2-((трет-бутоксикарбонил)амино)ацетат
К перемешанному раствору этил-2-амино-2-(5-амино-6-фенилпиридин-2-ил)ацетата (460 мг, 1,70 ммоль) в THF (5 мл) и воде (3 мл) добавляли твердый Na2CO3 (539 мг, 5,09 ммоль), а затем Boc2O (0,787 мл, 3,39 ммоль). Полученную смесь перемешивали при 25°C в течение 1 ч, а затем распределяли между водой (20 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=372,2 (M+1).
Стадия F: трет-Бутил-(1-(5-амино-6-фенилпиридин-2-ил)-2-гидроксиэтил)карбамат
К перемешанному раствору этил-2-(5-амино-6-фенилпиридин-2-ил)-2-((трет-бутоксикарбонил)амино)ацетата (150 мг, 0,404 ммоль) в MeOH (3 мл) добавляли NaBH4 (76 мг, 2,0 ммоль), и перемешивали полученную смесь при 25°C в течение 4 ч. Смесь распределяли между водой (20 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 7,67 (д, J=7,4 Гц, 2H), 7,50 (т, J=7,4 Гц, 2H), 7,45-7,40 (м, 1H), 7,19-7,15 (м, 1H), 7,10-7,06 (м, 1H), 5,83 (д, J=6,7 Гц, 1H), 4,78 (ушир. с, 1H), 4,08 (д, J=8,2 Гц, 1H), 3,91 (д, J=3,9 Гц, 1H), 3,88 (д, J=3,9 Гц, 2H), 1,48-1,44 (м, 9H).
Стадия G: 2-Амино-2-(5-амино-6-фенилпиридин-2-ил)этанол
Раствор трет-бутил-(1-(5-амино-6-фенилпиридин-2-ил)-2-гидроксиэтил)карбамата (180 мг, 0,546 ммоль) в 4н HCl в EtOAc (5 мл, 20 ммоль) перемешивали при 25°C в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=230,0 (M+1).
Стадия H: 4-(5-Амино-6-фенилпиридин-2-ил)оксазолидин-2-он
К перемешанному раствору 2-амино-2-(5-амино-6-фенилпиридин-2-ил)этанола (78 мг, 0,26 ммоль) и Et3N (0,108 мл, 0,774 ммоль) в THF (3 мл) добавляли бис(трихлорметил)карбонат (230 мг, 0,774 ммоль). Полученную смесь перемешивали при 25°C в течение 2 ч, затем разбавляли водой (10 мл) и подщелачивали до pH=8 добавлением твердого Na2CO3. Содержащую продукт водную смесь экстрагировали EtOAc (10 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=256,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,58 (д, J=7,0 Гц, 2H), 7,48-7,40 (м, 3H), 7,36 (д, J=7,5 Гц, 1H), 7,13-7,09 (м, 1H), 7,07-7,02 (м, 1H), 6,00-5,92 (м, 1H), 4,75-4,68 (м, 1H), 4,40-4,32 (м, 1H).
Схема реакций для промежуточного продукта A15
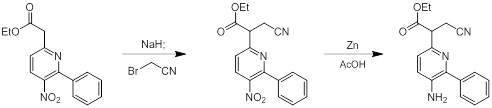
Промежуточный продукт A15
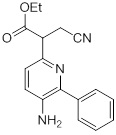
Этил-2-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат
Стадия A: Этил-3-циано-2-(5-нитро-6-фенилпиридин-2-ил)пропаноат
К раствору этил-2-(5-нитро-6-фенилпиридин-2-ил)ацетата (0,50 г, 1,7 ммоль) в тетрагидрофуране (10 мл) при 0°C добавляли гидрид натрия (60 масс.%, 70 мг, 1,7 ммоль). Смесь перемешивали при 0°C в течение 10 мин, после чего добавляли 2-бромацетонитрил (210 мг, 1,7 ммоль). Полученную смесь нагревали до 25°C и перемешивали в течение 1 ч. Содержащую продукт смесь распределяли между водой (20 мл) и этилацетатом (30 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=10/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8,31 (д, J=8,2 Гц, 1H), 7,65 (д, J=8,2 Гц, 1H), 7,54 (д, J=7,0 Гц, 2H), 7,50-7,41 (м, 3H), 4,19 (кв, J=6,8 Гц, 2H), 3,49 (ушир. с, 1H), 3,20 (с, 2H), 1,19 (т, J=7,0 Гц, 3H).
Стадия B: Этил-2-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат
К раствору этил-3-циано-2-(5-нитро-6-фенилпиридин-2-ил)пропаноата (0,40 г, 1,2 ммоль) в этаноле (20 мл) добавляли хлорид аммония (658 мг, 12,3 ммоль) и цинковый порошок (804 мг, 12,3 ммоль). Полученную смесь перемешивали при 0°C в течение 10 мин, а затем нагревали до 25°C и перемешивали в течение 1 ч. Содержащую продукт смесь фильтровали и концентрировали, и распределяли остаток между водой (20 мл) и этилацетатом (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=5/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 7,62 (д, J=7,0 Гц, 2H), 7,46 (т, J=7,4 Гц, 2H), 7,42-7,37 (м, 1H), 7,15-7,19 (м, 1H), 7,14-7,10 (м, 1H), 4,20-4,12 (м, 2H), 3,36 (д, J=3,1 Гц, 1H), 3,07 (дд, J1=7,4 Гц, J2=4,7 Гц,, 2H), 1,19 (т, J=7,2 Гц, 3H).
Схема реакций для промежуточного продукта A16
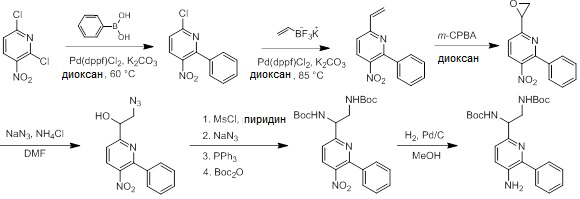
Промежуточный продукт A16
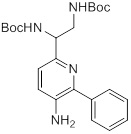
Ди-трет-бутил-(1-(5-амино-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамат
Стадия A: 6-Хлор-3-нитро-2-фенилпиридин
Дезоксигенированную смесь 2,6-дихлор-3-нитропиридина (45,0 г, 233 ммоль), фенилбороновой кислоты (31,3 г, 256 ммоль), Pd(dppf)Cl2 (3,41 г, 4,66 ммоль) и карбоната калия (97 г, 700 ммоль) в диоксане (500 мл) и воде (100 мл) нагревали при 60°C в течение 15 ч. Смесь фильтровали и концентрировали, и распределяли остаток между водой (500 мл) и EtOAc (400 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=20/1, 10/1) с получением указанного в заголовке соединения. 1H-ЯМР (CDCl3, 400 МГц) δ 8,10 (д, J=8,6 Гц, 1H), 7,53-7,60 (м, 2H), 7,45-7,50 (м, 3H), 7,43 (д, J=8,2 Гц, 1H).
Стадия B: 3-Нитро-2-фенил-6-винилпиридин
Дезоксигенированную смесь K2CO3 (4,42 г, 32,0 ммоль), PdCl2(dppf) (0,780 г, 1,06 ммоль), трифтор(винил)бората калия (1,43 г, 10,6 ммоль) и 6-хлор-3-нитро-2-фенилпиридина (2,50 г, 10,6 ммоль) в 1,4-диоксане (25 мл) и воде (5 мл) нагревали при 85°C в течение 5 ч. Смесь распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. 1H-ЯМР (CDCl3, 400 МГц) δ 8,13 (д, J=8,2 Гц, 1H), 7,59-7,55 (м, 2H), 7,49-7,45 (м, 3H), 7,41 (д, J=8,2 Гц, 1H), 6,91 (дд, J1=17,4 Гц, J2=10,8 Гц, 1H), 6,42 (д, J=17,2 Гц, 1H), 5,70 (д, J=11,0 Гц, 1H).
Стадия C: 3-Нитро-6-(оксиран-2-ил)-2-фенилпиридин
К перемешанному раствору 3-нитро-2-фенил-6-винилпиридина (10,0 г, 44,2 ммоль) в 1,4-диоксане (120 мл) добавляли 3-хлорпероксибензойную кислоту (18 г, 88 ммоль), и нагревали полученную смесь при 80°C в течение 5 ч. Смесь охлаждали, разбавляли водой (100 мл) и подщелачивали до pH=9 добавлением Na2CO3. Водный слой экстрагировали EtOAc (100 мл×3), объединенные органические слои сушили с Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=243,0 (M+1).
Стадия D: 2-Азидо-1-(5-нитро-6-фенилпиридин-2-ил)этанол
Смесь 3-нитро-6-(оксиран-2-ил)-2-фенилпиридина (6,00 г, 24,8 ммоль), NH4Cl (6,62 г, 124 ммоль) и NaN3 (4,83 г, 74,3 ммоль) в DMF (80 мл) нагревали при 80°C в течение 5 ч. Смесь разбавляли водой (100 мл) и подщелачивали до pH=11 добавлением K2CO3. Водный слой экстрагировали EtOAc (100 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=286,1 (M+1).
Стадия E: Ди-трет-бутил-(1-(5-нитро-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамат
К перемешанному раствору 2-азидо-1-(6-нитро-5-фенилпиридин-2-ил)этанола (2,00 г, 7,01 ммоль) и пиридина (4,09 мл, 50,6 ммоль) в CH2Cl2 (25 мл) добавляли метансульфонилхлорид (3,00 г, 26,2 ммоль), и перемешивали полученную смесь при 20°C в течение 5 ч. Смесь концентрировали, и растворяли остаток в DMF (25 мл). Полученный раствор охлаждали до 0°C, и добавляли NaN3 (4,00 г, 61,5 ммоль). Спустя 3 ч, добавляли PPh3 (7,36 г, 28,0 ммоль), и нагревали полученную смесь до 20°C и перемешивали в течение 2 ч. Смесь разбавляли водой (20 мл) и подщелачивали до pH=12 добавлением K2CO3. Водную смесь экстрагировали EtOAc (30 мл×3). К водному слою добавляли Boc2O (6,51 мл, 28,0 ммоль), и перемешивали полученную смесь при 20°C в течение 2 ч. Смесь экстрагировали EtOAc (30 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 8/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=459,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,11 (д, J=8,2 Гц, 1H), 7,53 (ушир. с, 3H), 7,49-7,42 (м, 5H), 3,58 (ушир. с, 3H), 1,43 (ушир. с, 18H).
Стадия F: Ди-трет-бутил-(1-(5-амино-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамат
К дезоксигенированному раствору ди-трет-бутил-(1-(6-нитро-5-фенилпиридин-2-ил)этан-1,2-диил)дикарбамата (350 мг, 0,763 ммоль) в MeOH (10 мл) добавляли 10% Pd/C (16 мг, 0,15 ммоль), и перемешивали полученную смесь в атмосфере H2 (50 фунт./кв.дюйм) при 20°C в течение 2 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=429,2 (M+1).
Схема реакций для промежуточного продукта A17
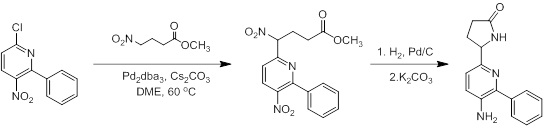
Промежуточный продукт A17
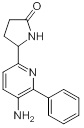
5-(5-Амино-6-фенилпиридин-2-ил)пирролидин-2-он
Стадия A: Метил-4-нитро-4-(5-нитро-6-фенилпиридин-2-ил)бутаноат
К дезоксигенированной смеси 6-хлор-3-нитро-2-фенилпиридина (0,10 г, 0,43 ммоль) в DME (1,5 мл) добавляли метил-4-нитробутаноат (0,13 г, 0,85 ммоль), Cs2CO3 (0,28 г, 0,85 ммоль), 2-(ди-трет-бутилфосфино)-2'-метилбифенил (53 мг, 0,17 ммоль) и Pd2(dba)3 (0,04 г, 0,04 ммоль). Полученную смесь нагревали при 60°C в течение 50 мин в условиях обработки микроволновым излучением. Реакционную смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом препаративной TLC (PE/EtOAc=3/1) с получением указанного в заголовке соединения. MS: m/z=346,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,36 (д, J=8,4 Гц, 1H), 7,74 (д, J=8,4 Гц, 1H), 7,55-7,46 (м, 5H), 6,02 (т, J=7,2 Гц, 1H), 3,63 (с, 3H), 2,82-2,79 (м, 1H), 2,65-2,60 (м, 1H), 2,48-2,45 (м, 2H).
Стадия B: 5-(5-Амино-6-фенилпиридин-2-ил)пирролидин-2-он
К дезоксигенированному раствору метил-4-нитро-4-(5-нитро-6-фенилпиридин-2-ил)бутаноата (80 мг, 0,23 ммоль) в MeOH (8 мл) добавляли 10% Pd/C (49 мг, 0,046 ммоль), и перемешивали полученную смесь в атмосфере H2 (30 фунт./кв.дюйм) при 30°C в течение 2 ч. Реакционную смесь фильтровали, и добавляли к фильтрату K2CO3 (64,0 мг, 0,46 ммоль). Полученную смесь нагревали при 60°C в течение 30 мин. После охлаждения, смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом препаративной TLC (100% EtOAc) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 7,62-7,58 (м, 2H), 7,51-7,42 (м, 3H), 7,23-7,21 (м, 1H), 7,15-7,13 (м, 1H), 4,79-4,75 (м, 1H), 2,56-2,38 (м, 3H), 2,10-2,08 (м, 1H).
Схема реакций для промежуточного продукта A18
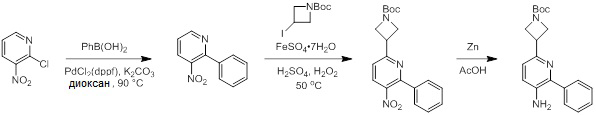
Промежуточный продукт A18

трет-Бутил-3-(5-амино-6-фенилпиридин-2-ил)азетидин-1-карбоксилат
Стадия A: 3-Нитро-2-фенилпиридин
К дезоксигенированной смеси 2-хлор-3-нитропиридина (10,0 г, 63,1 ммоль) в диоксане (150 мл) и воде (75 мл) добавляли PdCl2(dppf) (2,31 г, 3,15 ммоль), фенилбороновую кислоту (11,5 г, 95,0 ммоль), K2CO3 (17,4 г, 126 ммоль). Полученную смесь нагревали при 90°C в течение 16 ч, затем охлаждали, фильтровали и концентрировали. Остаток распределяли между водой (50 мл) и этилацетатом (50 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO, флэш-колонка SepaFlash® 120 г силикагеля, этилацетат в петролейном эфире: 0~20%, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,92-8,82 (м, 1H), 8,19-8,11 (м, 1H), 7,61-7,54 (м, 2H), 7,52-7,42 (м, 4H).
Стадия B: трет-Бутил-3-(5-нитро-6-фенилпиридин-2-ил)азетидин-1-карбоксилат
Водный 30% раствор H2O2 (1,31 мл, 15,0 ммоль) добавляли к перемешанному раствору 3-нитро-2-фенилпиридина (1,00 г, 5,00 ммоль), концентрированного водного раствора H2SO4 (0,53 мл, 10 ммоль), трет-бутил-3-йодазетидин-1-карбоксилата (2,83 г, 9,99 ммоль) и семиводного сульфата железа(II) (0,42 г, 1,5 ммоль) в DMSO (20 мл). Полученную смесь перемешивали при 27°C в течение 0,5 ч, после чего дополнительно добавляли семиводный сульфат железа(II) (0,42 г, 1,5 ммоль) и 30% H2O2 (1,31 мл, 15,0 ммоль). Перемешивание продолжали в течение 0,5 ч, после чего к смеси дополнительно добавляли 30% H2O2 (1,31 мл, 14,99 ммоль) и семиводный сульфат железа(II) (0,42 г, 1,50 ммоль). Полученную смесь затем нагревали при 50°C в течение 60 ч. Содержащую продукт смесь вливали в ледяной 15% водный раствор NaOH и корректировали до pH>10. Водную смесь фильтровали, и экстрагировали фильтрат этилацетатом (150 мл×3). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 40 г силикагеля, этилацетат в петролейном эфире=0-30%, 40 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. MS: m/z=356,1 (M+1).
Стадия C: трет-Бутил-3-(5-амино-6-фенилпиридин-2-ил)азетидин-1-карбоксилат
К раствору трет-бутил-3-(5-нитро-2-фенилпиридин-4-ил)азетидин-1-карбоксилата (48 мг, 0,14 ммоль) в AcOH (2 мл) добавляли цинковый порошок (53 мг, 0,81 ммоль), и перемешивали полученную смесь при 20°C в течение 16 ч. Смесь распределяли между насыщенным водным раствором NaHCO3 (50 мл) и этилацетатом (20 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=326,1 (M+1).
Схема реакций для промежуточного продукта A19
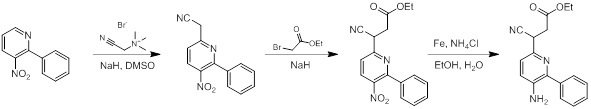
Промежуточный продукт A19
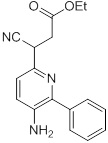
Этил-3-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат
Стадия A: 2-(5-Нитро-6-фенилпиридин-2-ил)ацетонитрил
К раствору 3-нитро-2-фенилпиридина (2,00 г, 9,99 ммоль) в DMSO (20 мл) при 0°C добавляли гидрид натрия (60 масс.%, 3,6 г, 90 ммоль) и 1-циано-N,N,N-триметилметанаминия бромид (5,37 г, 30,0 ммоль). Полученную смесь перемешивали в течение 35 мин, затем распределяли между водой (5 мл) и этилацетатом (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=240,2 (M+1).
Стадия B: Этил-3-циано-3-(5-нитро-6-фенилпиридин-2-ил)пропаноат
К дезоксигенированному раствору 2-(5-нитро-6-фенилпиридин-2-ил)ацетонитрила (400 мг, 1,67 ммоль) в THF (5 мл) при -78°C добавляли гидрид натрия (60 масс.%, 67 мг, 1,7 ммоль). Смесь перемешивали при -78°C в течение 5 мин, после чего добавляли этил-2-бромацетат (251 мг, 1,51 ммоль). Полученную смесь нагревали до 0°C и перемешивали в течение 1 ч. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=2/1) с получением указанного в заголовке соединения. MS: m/z=325,9 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,18 (д, J=8,2 Гц, 1H), 7,69 (д, J=8,6 Гц, 1H), 7,58-7,42 (м, 5H), 4,58 (т, J=6,8 Гц, 1H), 4,17 (кв, J=6,8 Гц, 2H), 3,33-3,25 (м, 1H), 3,19-3,10 (м, 1H), 1,23 (т, J=7,2 Гц, 3H).
Стадия C: Этил-3-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат
К раствору этил-3-циано-3-(5-нитро-6-фенилпиридин-2-ил)пропаноата (200 мг, 0,541 ммоль) в EtOH (3 мл) и воде (1 мл) добавляли хлорид аммония (87 мг, 1,6 ммоль) и железный порошок (91 мг, 1,6 ммоль). Полученную смесь нагревали при 80°C в течение 1 ч, затем охлаждали и фильтровали. Фильтрат распределяли между водой (10 мл) и EtOAc (10 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=296,1 (M+1).
Схема реакций для промежуточного продукта A20
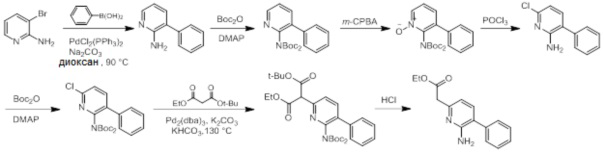
Промежуточный продукт A20
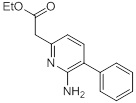
Этил-2-(6-амино-5-фенилпиридин-2-ил)ацетат
Стадия A: 3-Фенилпиридин-2-амин
К дезоксигенированному раствору 3-бромпиридин-2-амина (5,00 г, 28,9 ммоль), фенилбороновой кислоты (4,23 г, 34,7 ммоль) и карбоната натрия (9,2 г, 87 ммоль) в 1,4-диоксане (60 мл) и воде (30 мл) добавляли PdCl2(PPh3)2 (1,01 г, 1,44 ммоль). Полученную смесь нагревали с обратным холодильником в течение 4 ч, затем охлаждали, фильтровали и концентрировали. Остаток распределяли между солевым раствором (50 мл) и этилацетатом (30 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (этилацетат в петролейном эфире: 20-50%) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,09 (д, J=3,5 Гц, 1H), 7,51-7,43 (м, 4H), 7,41-7,35 (м, 2H), 6,76 (дд, J=7,0, 5,1 Гц, 1H), 4,61 (ушир. с, 2H).
Стадия B: 3-Фенилпиридин-2-(ди-трет-бутоксикарбонил)амин
К раствору 3-фенилпиридин-2-амина (10,0 г, 55,8 ммоль) и ди-трет-бутил-дикарбоната (48,7 г, 223 ммоль) в DCM (200 мл) порциями добавляли DMAP (13,6 г, 112 ммоль). Полученную смесь перемешивали при 26°C в течение 16 ч, затем промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 120 г силикагеля, этилацетат в петролейном эфире: 0-40%, 50 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,52 (д, J=3,1 Гц, 1H), 7,75 (дд, J=7,4, 1,6 Гц, 1H), 7,45-7,34 (м, 6H), 1,30 (с, 18H).
Стадия C: 2-(Ди-трет-бутоксикарбонил)амино-3-фенилпиридин 1-оксид
К раствору 3-фенилпиридин-2-(ди-трет-бутоксикарбонил)амина (17,5 г, 44,9 ммоль) в DCM (100 мл) добавляли m-CPBA (48,4 г, 224 ммоль), и перемешивали полученную смесь при 27°C в течение 16 ч. Смесь разбавляли насыщенным водным раствором Na2SO3 (200 мл), перемешивали в течение 10 мин, и распределяли между насыщенным водным раствором NaHCO3 (300 мл) и DCM (200 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 120 г силикагеля, этилацетат в петролейном эфире: 0-80%, 50 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,29 (дд, J=6,1, 1,4 Гц, 1H), 7,47-7,39 (м, 5H), 7,27-7,22 (м, 2H), 1,33 (с, 18H).
Стадия D: 6-Хлор-3-фенилпиридин-2-амин
К раствору 2-(ди-трет-бутоксикарбонил)амино-3-фенилпиридин-1-оксида (10,0 г, 25,9 ммоль) в DCM (80 мл) добавляли оксихлорид фосфора (20,8 мл, 223 ммоль), и нагревали полученную смесь при 35°C в течение 16 ч. Смесь концентрировали, и осторожно разбавляли остаток насыщенным водным раствором NaHCO3 (300 мл). Водный слой экстрагировали этилацетатом (100 мл×3). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (100 мл×3), сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 40 г силикагеля, этилацетат в петролейном эфире: 0-20%, 40 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 7,50-7,38 (м, 5H), 7,32 (д, J=7,4 Гц, 1H), 6,76 (д, J=7,8 Гц, 1H), 4,72 (ушир., 2H).
Стадия E: 6-Хлор-3-фенилпиридин-2-(ди-трет-бутоксикарбонил)амин
К раствору 6-хлор-3-фенилпиридин-2-амина (2,10 г, 9,75 ммоль) и ди-трет-бутил-дикарбоната (8,51 г, 39,0 ммоль) в DCM (30 мл) отдельными порциями добавляли DMAP (2,38 г, 19,50 ммоль), и перемешивали полученную смесь при 26°C в течение 16 ч. Полученную смесь промывали солевым раствором (10 мл×3), сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 40 г силикагеля, этилацетат в петролейном эфире: 0-15%, 40 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 7,71 (д, J=8,0 Гц, 1H), 7,45-7,37 (м, 6H), 1,30 (с, 18H).
Стадия F: 1-трет-Бутил-3-этил-2-(6-(ди-трет-бутоксикарбонил)амино-5-фенилпиридин-2-ил)малонат
Дезоксигенированную смесь три-трет-бутилфосфония тетрафторбората (0,34 г, 1,17 ммоль), гидрокарбоната калия (0,88 г, 8,8 ммоль), 6-хлор-3-фенилпиридин-2-(ди-трет-бутоксикарбонил)амина (2,50 г, 5,87 ммоль), трет-бутил-этил-малоната (3,33 мл, 17,6 ммоль), Pd(dba)2 (0,34 г, 0,59 ммоль) и K2CO3 (1,22 г, 8,80 ммоль) нагревали при 130°C в течение 1 ч. Смесь фильтровали и концентрировали с получением указанного в заголовке соединения, которое использовали на следующей стадии без очистки. MS: m/z=557,2 (M+1).
Стадия G: Этил-2-(6-амино-5-фенилпиридин-2-ил)ацетат
Смесь 1-трет-бутил-3-этил-2-(6-(ди-трет-бутоксикарбонил)амино-5-фенилпиридин-2-ил)малоната (чистота 39%, 5,20 г, 3,69 ммоль) в 4M растворе HCl в диоксане (12 мл) перемешивали при 20°C в течение 16 ч. Смесь распределяли между насыщенным водным раствором NaHCO3 (100 мл) и DCM (30 мл×3). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (30 мл×3), затем солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® 12 г силикагеля, этилацетат в петролейном эфире: 0-50%, 30 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 7,48-7,41 (м, 4H), 7,40-7,32 (м, 2H), 6,74 (д, J=7,4 Гц, 1H), 4,61 (ушир., 2H), 4,21 (кв, J=7,2 Гц, 2H), 3,69 (с, 2H), 1,31-1,26 (м, 3H).
Схема реакций для промежуточного продукта A21

Промежуточный продукт A21
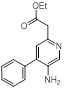
Этил-2-(5-амино-4-фенилпиридин-2-ил)ацетат
Стадия A: 2-Хлор-5-нитро-4-фенилпиридин
К дезоксигенированному раствору 2,4-дихлор-5-нитропиридина (3,00 г, 15,6 ммоль) в 1,4-диоксане (15 мл) и воде (3 мл) добавляли фенилбороновую кислоту (1,89 г, 15,6 ммоль), карбонат калия (4,30 г, 31,1 ммоль) и PdCl2(dppf) (0,57 г, 0,78 ммоль). Полученную смесь нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между водой (50 мл) и этилацетатом (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=235,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,96 (с, 1H), 7,58-7,54 (м, 3H), 7,51 (с, 1H), 7,43-7,37 (м, 2H).
Стадия B: Диэтил-2-(5-нитро-4-фенилпиридин-2-ил)малонат
К раствору диэтилмалоната (2,40 г, 14,9 ммоль) в DMF (20 мл) при 0°C порциями добавляли NaH (60 масс.%, 1,19 г, 29,8 ммоль). После перемешивания смеси в течение 10 мин, добавляли 2-хлор-5-нитро-4-фенилпиридин (3,50 г, 14,9 ммоль). Полученную смесь нагревали до 23°C и перемешивали в течение 2 ч. Смесь разбавляли водой со льдом (30 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=359,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,01 (с, 1H), 7,65 (с, 1H), 7,48 (дд, J1=1,5, J1=5,0 Гц, 3H), 7,40-7,33 (м, 2H), 5,05 (с, 1H), 4,33-4,22 (м, 4H), 1,30 (т, J=7,3 Гц, 6H).
Стадия C: Этил-2-(5-нитро-4-фенилпиридин-2-ил)ацетат
К раствору диэтил-2-(5-нитро-4-фенилпиридин-2-ил)малоната (2,5 г, 6,9 ммоль) в диметилсульфоксиде (5 мл) и воде (0,5 мл) добавляли хлорид лития (0,30 г, 6,9 ммоль). Полученную смесь нагревали при 100°C в течение 18 ч, затем охлаждали, и распределяли между водой (50 мл) и этилацетатом (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=287,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,02 (с, 1H), 7,50-7,45 (м, 3H), 7,42 (с, 1H), 7,35 (д, J=3,5 Гц, 2H), 4,22 (кв, J=7,0 Гц, 2H), 3,96 (с, 2H), 1,29 (т, J=7,0 Гц, 3H).
Стадия D: Этил-2-(5-амино-4-фенилпиридин-2-ил)ацетат
К дезоксигенированному раствору этил-2-(5-нитро-4-фенилпиридин-2-ил)ацетата (135 мг, 0,50 ммоль) в этаноле (10 мл) добавляли 10% Pd/C (50 мг, 0,05 ммоль), и перемешивали полученную смесь в атмосфере H2 (50 фунт./кв.дюйм) при 25-30°C в течение 18 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=257,2 (M+1).
Схема реакций для промежуточных продуктов A22 и A23
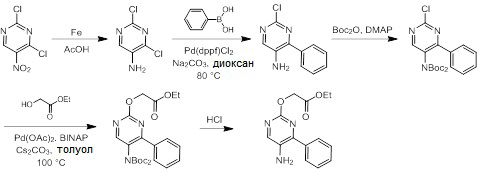
Промежуточный продукт A22
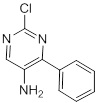
2-Хлор-4-фенилпиримидин-5-амин
Стадия A: 2,4-Дихлорпиримидин-5-амин
К перемешанному раствору 2,4-дихлор-5-нитропиримидина (55,0 г, 284 ммоль) в уксусной кислоте (800 мл) порциями добавляли железный порошок (47,5 г, 851 ммоль) при 23°C (экзотермичная реакция, температура реакционного раствора достигала 50°C). Смесь перемешивали еще в течение 1 ч без нагревания. Содержащую продукт смесь фильтровали через Celite®, и промывали осадок на фильтре этилацетатом (400 мл). Фильтрат концентрировали, и распределяли остаток между этилацетатом (400 мл) и водой (40 мл). Смесь перемешивали при 20°C в течение 5 мин и фильтровали. Органический слой разделяли, и экстрагировали водный слой этилацетатом (200 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перекристаллизовывали (петролейный эфир/этилацетат=10/1, 500 мл) с получением указанного в заголовке соединения. MS: m/z=163,9 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,02 (с, 1H), 4,08 (с, 2H).
Стадия B: 2-Хлор-4-фенилпиримидин-5-амин
К дезоксигенированной смеси 2,4-дихлорпиримидин-5-амина (37,5 г, 229 ммоль), фенилбороновой кислоты (22,9 г, 188 ммоль) и карбоната натрия (48,5 г, 457 ммоль) в диоксане (400 мл) и воде (200 мл) добавляли PdCl2(dppf) (1,67 г, 2,29 ммоль). Полученную смесь нагревали при 80°C в течение 10 ч в атмосфере азота. Содержащую продукт смесь охлаждали и концентрировали. Остаток распределяли между водой (200 мл) и этилацетатом (200 мл×3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=206,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,11 (с, 1H), 7,72 (д, J=6,3 Гц, 2H), 7,54-7,43 (м, 3H), 3,96 (с, 2H).
Промежуточный продукт A23
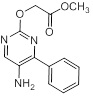
Метил-2-((5-амино-4-фенилпиримидин-2-ил)окси)ацетат
Стадия A: 2-Хлор-4-фенилпиримидин-5-(трет-бутоксикарбонил))амин
К смеси 2-хлор-4-фенилпиримидин-5-амина (19 г, 92 ммоль), триэтиламина (28 г, 280 ммоль) и N,N-диметилпиридин-4-амина (11 г, 92 ммоль) по каплям добавляли раствор ди-трет-бутилдикарбоната (60,5 г, 280 ммоль) в дихлорметане (20 мл). Полученную смесь перемешивали при 20°C в течение 1 ч, а затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=406,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1H), 7,72-7,64 (м, 2H), 7,53-7,43 (м, 3H), 1,26 (с, 18H).
Стадия B: Этил-2-((5-(бис(трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)окси)ацетат
К дезоксигенированному раствору 2-хлор-4-фенилпиримидин-5-(трет-бутоксикарбонил))амина (100 мг, 0,246 ммоль), этил-2-гидроксиацетата (77 мг, 0,74 ммоль) и карбоната цезия (161 мг, 0,493 ммоль) в толуоле (3 мл) добавляли Pd(OAc)2 (5,5 мг, 0,025 ммоль) и BINAP (31 мг, 0,049 ммоль). Смесь нагревали при 110°C в течение 4 ч. Содержащую продукт смесь распределяли между водой (5 мл) и этилацетатом (5 мл×3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения. MS: m/z=474,3 (M+1).
Стадия C: Метил-2-((5-амино-4-фенилпиримидин-2-ил)окси)ацетат
К перемешанному раствору этил-2-((5-(бис(трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)окси)ацетата (110 мг, 0,232 ммоль) в метаноле (1 мл) добавляли 4M раствор HCl в MeOH (4,0 мл, 16 ммоль). Смесь перемешивали при 15°C в течение 30 мин, а затем концентрировали. Остаток подщелачивали добавлением насыщенного раствора карбоната натрия (10 мл) и экстрагировали этилацетатом (10 мл×4). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=260,2 (M+1).
Схема реакций для промежуточного продукта A24
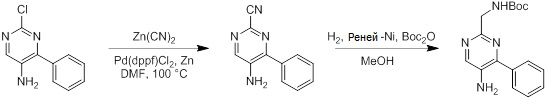
Промежуточный продукт A24
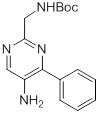
трет-Бутил-((5-амино-4-фенилпиримидин-2-ил)метил)карбамат
Стадия A: 5-Амино-4-фенилпиримидин-2-карбонитрил
К раствору 2-хлор-4-фенилпиримидин-5-амина (1,00 г, 4,86 ммоль) в диметилформамиде (20 мл) добавляли Zn(CN)2 (1,14 г, 9,73 ммоль), цинковый порошок (0,159 г, 2,43 ммоль) и PdCl2(dppf) (0,178 г, 0,243 ммоль). Полученную смесь нагревали при 145°C в течение 18 ч, а затем охлаждали и распределяли между водой (50 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,24 (с, 1H), 7,72-7,71 (д, J=4 МГц, 2H), 7,54-7,50 (т, J=7,2 МГц, 3H) 4,53 (с, 2H).
Стадия B: трет-Бутил-((5-амино-4-фенилпиримидин-2-ил)метил)карбамат
К дезоксигенированной смеси 5-амино-4-фенилпиримидин-2-карбонитрила (200 мг, 1,02 ммоль) в MeOH (10 мл) добавляли Boc2O (0,237 мл, 1,02 ммоль) и никель Ренея (598 мг, 1,02 ммоль), и перемешивали полученную смесь в атмосфере H2 (50 фунт./кв.дюйм) при 28-30°C в течение 2 ч. Содержащую продукт смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=301,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,2 (с, 1H), 7,74-7,72 (д, J=8 МГц, 2H), 7,52-7,45 (т, J=2,8 МГц, 3H), 4,5 (с, 2H), 1,45 (с, 9H).
Схема реакций для промежуточного продукта A25
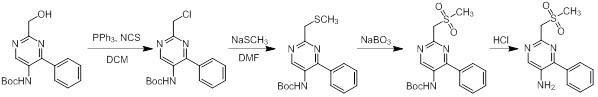
Промежуточный продукт A25
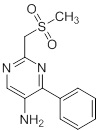
2-((Метилсульфонил)метил)-4-фенилпиримидин-5-амин
Стадия A: трет-Бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат
К смеси трет-бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамата (300 мг, 0,996 ммоль) в THF (5 мл) при 25°C добавляли 1-хлорпирролидин-2,5-дион (266 мг, 1,99 ммоль) и трифенилфосфин (610 мг, 1,99 ммоль). Смесь перемешивали при 25°C в течение 15 мин, затем распределяли между водой (5 мл) и EtOAc (10 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=320,0 (M+1).
Стадия B: трет-Бутил-(2-((метилтио)метил)-4-фенилпиримидин-5-ил)карбамат
К смеси трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат (250 мг, 0,782 ммоль) в DMF (5 мл) добавляли метантиолят натрия (82 мг, 1,2 ммоль), и нагревали полученную смесь при 70°C в течение 35 мин. Содержащую продукт смесь охлаждали и распределяли между водой (5 мл) и EtOAc (5 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z =322,1 (M+1).
Стадия C: трет-Бутил-(2-((метилсульфонил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-((метилтио)метил)-4-фенилпиримидин-5-ил)карбамата (100 мг, 0,302 ммоль) в AcOH (5 мл) добавляли NaBO3 (74,0 мг, 0,905 ммоль), и нагревали полученную смесь при 70°C в течение 25 мин. Содержащую продукт смесь охлаждали, затем распределяли между водой (5 мл) и DCM (5 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=364,1 (M+1).
Стадия D: 2-((Метилсульфонил)метил)-4-фенилпиримидин-5-амин
К раствору трет-бутил-(2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамата (115 мг, 0,257 ммоль) в EtOAc (3 мл) добавляли 4M раствор HCl в EtOAc (5,0 мл, 20 ммоль). Полученную смесь перемешивали при 25°C в течение 30 мин, затем концентрировали с получением указанного в заголовке соединения. MS: m/z=264,1 (M+1).
Схема реакций для промежуточного продукта A26
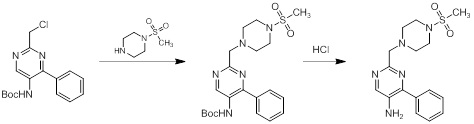
Промежуточный продукт A26

2-((Метилсульфонил)метил)-4-фенилпиримидин-5-амин
Стадия A: трет-Бутил-(2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (200 мг, 0,625 ммоль) в ацетонитриле (5 мл) добавляли карбонат калия (173 мг, 1,25 ммоль) и 1-(метилсульфонил)пиперазин (154 мг, 0,938 ммоль). Полученную смесь перемешивали при 25°C в течение 30 мин, затем фильтровали и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=448,3 (M+1).
Стадия B: 2-((4-(Метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-амин
К раствору трет-бутил-(2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамата (115 мг, 0,257 ммоль) в EtOAc (3 мл) добавляли 4M раствор HCl в EtOAc (5,0 мл, 20 ммоль). Полученную смесь перемешивали при 25°C в течение 30 мин, затем концентрировали с получением указанного в заголовке соединения. MS: m/z=348,1 (M+1).
Схема реакций для промежуточного продукта A27
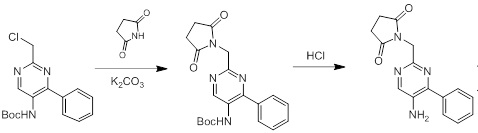
Промежуточный продукт A27
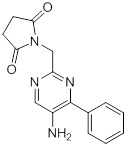
1-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион
Стадия A: трет-Бутил-(2-((2,5-диоксопирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (180 мг, 0,56 ммоль) в ацетонитриле (3 мл) добавляли пирролидин-2,5-дион (112 мг, 1,12 ммоль) и K2CO3 (233 мг, 1,68 ммоль). Полученную смесь перемешивали при 26°C в течение 5 ч, а затем распределяли между водой (5 мл) и EtOAc (10 мл×3). Объединенные органические слои концентрировали с получением указанного в заголовке соединения. MS: m/z=383,2 (M+1).
Стадия B: 1-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион
Смесь трет-бутил-(2-((2, 5-диоксопирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)карбамата (180 мг, 0,47 ммоль) в 4M растворе HCl в EtOAc (5,0 мл, 20 ммоль) перемешивали при 26°C в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=283,2 (M+1).
Схема реакций для промежуточного продукта A28
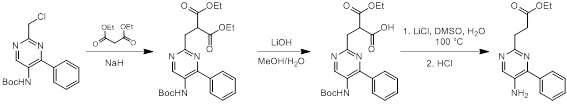
Промежуточный продукт A28
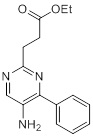
Этил-3-(5-амино-4-фенилпиримидин-2-ил)пропаноат
Стадия A: Диэтил-2-((5-((трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метил)малонат
К раствору диэтилмалоната (120 мг, 0,75 ммоль) в DMF (1 мл) при 0°C добавляли гидрид натрия (60 масс.%, 33 мг, 0,82 ммоль), и перемешивали полученную смесь в течение 20 мин. Раствор трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (60 мг, 0,19 ммоль) в DMF (1 мл) по каплям добавляли к описанной выше смеси, и перемешивали смесь при 0°C в течение 30 мин. Реакционный раствор разбавляли водой (2 мл), и экстрагировали водный слой EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=444,2 (M+1).
Стадия B: 2-((5-((трет-Бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метил)-3-этокси-3-оксопропановая кислота
К раствору диэтил-2-((5-((трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метил)малоната (92 мг, 0,21 ммоль) в этаноле (2 мл) и воде (0,1 мл) при 25°C добавляли LiOH (9,9 мг, 0,41 ммоль). Полученную смесь перемешивали в течение 30 мин, затем разбавляли водой (2 мл) и подкисляли до pH=5 добавлением 3M водного раствора HCl. Смесь экстрагировали EtOAc (5 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=416,4 (M+1).
Стадия C: Этил-3-(5-амино-4-фенилпиримидин-2-ил)пропаноат
К раствору 2-((5-((трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)метил)-3-этокси-3-оксопропановой кислоты (82 мг, 0,20 ммоль) в DMSO (4 мл) и воде (0,5 мл) добавляли LiCl (42 мг, 0,99 ммоль). Полученную смесь нагревали при 100°C в течение 6 ч, затем охлаждали, разбавляли водой (5 мл) и экстрагировали EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в 4M растворе HCl в диоксане (5 мл), полученный раствор нагревали при 50°C в течение 30 мин, затем охлаждали и концентрировали. Остаток распределяли между насыщенным водным раствором K2CO3 (3 мл) и EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=272,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,21 (с, 1H), 7,74 (д, J=7,2 Гц, 2H), 7,52-7,44 (м, 3H), 4,12 (кв, J=7,2 Гц, 2H), 3,55 (с, 2H), 3,25 (т, J=7,2 Гц, 2H), 2,85 (т, J=7,2 Гц, 2H), 1,22 (т, J=7,2 Гц, 3H).
Схема реакций для промежуточного продукта A29
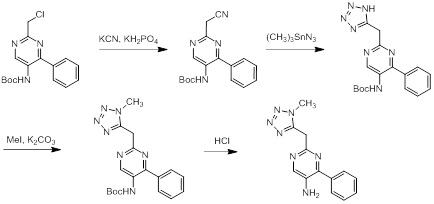
Промежуточный продукт A29
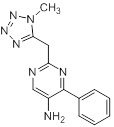
2-((1-Метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-амин
Стадия A: трет-Бутил-(2-(цианометил)-4-фенилпиримидин-5-ил)карбамат
К смеси цианида натрия (1,30 г, 26,6 ммоль) и KH2PO4 (4,34 г, 31,9 ммоль) в DMF (50 мл) добавляли трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат (1,70 г, 5,32 ммоль), и перемешивали полученную смесь при 18°C в течение 3 ч. Содержащую продукт смесь распределяли между водой и EtOAc (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=311,0 (M+1).
Стадия B: трет-Бутил-(2-((2H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(цианометил)-4-фенилпиримидин-5-ил)карбамата (200 мг, 0,644 ммоль) в толуоле (5 мл) добавляли азидотриметилстаннан (398 мг, 1,93 ммоль). Полученную смесь нагревали при 110°C в течение 8 ч в атмосфере азота. Содержащую продукт смесь охлаждали, и добавляли KF (200 мг). После перемешивания в течение 30 мин, смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом HPLC с обращенной фазой с получением указанного в заголовке соединения. MS: m/z=354,0 (M+1).
Стадия C: трет-Бутил-(2-((1-метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-((2H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)карбамата (100 мг, 0,283 ммоль) в ацетонитриле (3 мл) добавляли K2CO3 (78 мг, 0,57 ммоль) и CH3I (0,027 мл, 0,42 ммоль). Полученную смесь перемешивали при 17°C в течение 3 ч, а затем распределяли между водой и EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=368,1 (M+1). 1H-ЯМР (CDCl3, 400 МГц) δ 7,47 (м, 5H), 6,61 (с, 1H), 4,56 (м, 2H), 4,01 (с, 3H), 1,40 (с, 9H).
Стадия D: 2-((1-Метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-амин
К раствору трет-бутил-(2-((2-метил-2H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)карбамата (50 мг, 0,14 ммоль) в EtOAc (2 мл) добавляли 4M раствор HCl в EtOAc (2 мл). Полученную смесь перемешивали при 18°C в течение 1 ч и концентрировали с получением указанного в заголовке соединения. MS: m/z=268,1 (M+1).
Схема реакций для промежуточного продукта A30
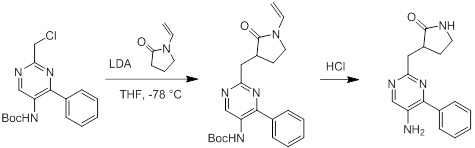
Промежуточный продукт A30
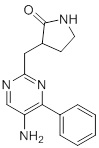
3-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2-он
Стадия A: трет-Бутил-(2-((2-оксо-1-винилпирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору 1-винилпирролидин-2-она (104 мг, 0,936 ммоль) в THF (5 мл) при -78°C добавляли LDA (134 мг, 1,25 ммоль). Смесь перемешивали при -78°C в течение 10 мин, после чего добавляли трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат (100 мг, 0,313 ммоль). Смесь нагревали до 26°C и перемешивали в течение 1 ч, а затем распределяли между водой (5 мл) и EtOAc (5 мл×3). Объединенные органические слои концентрировали с получением указанного в заголовке соединения. MS: m/z=395,2 (M+1).
Стадия B: 3-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2-он
Раствор трет-бутил-(2-((2-оксо-1-винилпирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)карбамата (100 мг, 0,254 ммоль) в 6н водном HCl (2 мл, 12 ммоль) перемешивали при 26°C в течение 5 ч. Смесь подщелачивали до pH=10 добавлением насыщенного водного раствора K2CO3 и экстрагировали EtOAc (5 мл×3). Объединенные органические слои концентрировали с получением указанного в заголовке соединения. MS: m/z=269,2 (M+1).
Схема реакций для промежуточного продукта A31

Промежуточный продукт A31
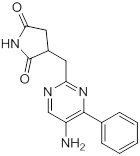
3-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион
Стадия A: 2-трет-Бутил-1,2-диметил-3-(5-((трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)пропан-1,2,2-трикарбоксилат
К раствору трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (350 мг, 1,09 ммоль) в ацетонитриле (10 мл) добавляли карбонат калия (454 мг, 3,28 ммоль) и 1-трет-бутил-1,2-диметилэтан-1,1,2-трикарбоксилат (809 мг, 3,28 ммоль). Полученную смесь перемешивали при 20°C в течение 5 ч, а затем распределяли между водой (10 мл) и EtOAc (10 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=7/1, 5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=530,3 (M+1).
Стадия B: 2-((5-Амино-4-фенилпиримидин-2-ил)метил)янтарная кислота
К раствору 2-трет-бутил-1,2-диметил-3-(5-((трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)пропан-1,2,2-трикарбоксилата (310 мг, 0,585 ммоль) в DCM (1 мл) добавляли 2,2,2-трифторуксусную кислоту (2,0 мл, 0,59 ммоль). Полученную смесь нагревали при 50°C в течение 30 мин, а затем охлаждали и концентрировали. Остаток растворяли в DMSO (2 мл), и добавляли хлорид лития (248 мг, 5,85 ммоль). Полученную смесь нагревали при 100°C в течение 5 ч. Содержащую продукт смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=302,0 (M+1).
Стадия C: N-(2-((2,5-Диоксотетрагидрофуран-3-ил)метил)-4-фенилпиримидин-5-ил)-2,2,2-трифторацетамид
К раствору 2-((5-амино-4-фенилпиримидин-2-ил)метил)янтарной кислоты (60 мг, 0,20 ммоль) в тетрагидрофуране (1 мл) добавляли 2,2,2-трифторуксусный ангидрид (418 мг, 1,99 ммоль). Полученную смесь перемешивали при 18°C в течение 30 мин, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=380,1 (M+1).
Стадия D: 3-((5-Амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион
К раствору N-(2-((2,5-диоксотетрагидрофуран-3-ил)метил)-4-фенилпиримидин-5-ил)-2,2,2-трифторацетамида (56 мг, 0,15 ммоль) в DMF (0,5 мл) и AcOH (0,5 мл) добавляли NH4OAc (57 мг, 0,74 ммоль). Полученную смесь нагревали при 140°C в течение 2 ч, затем охлаждали и очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=283,3 (M+1).
Схема реакций для промежуточного продукта A32
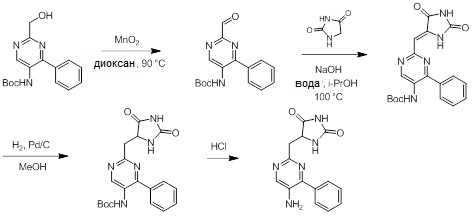
Промежуточный продукт A32
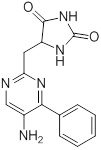
5-((5-Амино-4-фенилпиримидин-2-ил)метил)имидазолидин-2,4-дион
Стадия A: трет-Бутил-(2-формил-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамата (700 мг, 2,32 ммоль) в диоксане (8 мл) добавляли диоксид магния (2,02 г, 23,2 ммоль), и нагревали полученную смесь при 90°C в течение 2 ч. Содержащую продукт смесь охлаждали и фильтровали, и концентрировали фильтрат. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=3/1) с получением указанного в заголовке соединения. MS: m/z=299,2 (M+1).
Стадия B: трет-Бутил-(2-((2,5-диоксоимидазолидин-4-илиден)метил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-формил-4-фенилпиримидин-5-ил)карбамата (450 мг, 1,50 ммоль) в воде (5 мл) и изопропаноле (2,5 мл) при 23°C добавляли имидазолидин-2,4-дион (150 мг, 1,50 ммоль) и гидроксид натрия (60 мг, 1,5 ммоль). Полученную смесь нагревали при 100°C в течение 2 ч, затем охлаждали и подкисляли до pH=7 добавлением 3н водного раствора HCl. Осадок фильтровали и сушили с получением указанного в заголовке соединения. MS: m/z=382,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 10,16 (с, 1H), 9,08 (с, 1H), 8,73 (с, 1H), 7,75 (с, 2H), 7,46-7,53 (м, 4H), 6,29 (с, 1H), 1,24 (с, 9H).
Стадия C: трет-Бутил-(2-((2,5-диоксоимидазолидин-4-ил)метил)-4-фенилпиримидин-5-ил)карбамат
Смесь трет-бутил-(2-((2,5-диоксоимидазолидин-4-илиден)метил)-4-фенилпиримидин-5-ил)карбамата (100 мг, 0,262 ммоль) и 10% Pd/C (3 мг, 0,03 ммоль) в метаноле (20 мл) перемешивали в атмосфере водорода (50 фунт./кв.дюйм) при 23°C в течение 12 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=384,1 (M+1).
Стадия D: 5-((5-Амино-4-фенилпиримидин-2-ил)метил)имидазолидин-2,4-дион
К раствору трет-бутил-(2-((2,5-диоксоимидазолидин-4-ил)метил)-4-фенилпиримидин-5-ил)карбамата (90 мг, 0,24 ммоль) в диоксане (5 мл) добавляли 4M раствор HCl в диоксане (5 мл). Смесь перемешивали при 26°C в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=284,0 (M+1).
Схема реакций для промежуточного продукта A33

Промежуточный продукт A33
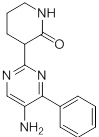
3-(5-Амино-4-фенилпиримидин-2-ил)пиперидин-2-он
Стадия A: 2-((5-(Ди-трет-бутоксикарбонил)амино-4-фенилпиримидин-2-ил))ацетат
К смеси метил-2-(5-амино-4-фенилпиримидин-2-ил)ацетата (5,00 г, 20,6 ммоль), Boc2O (14,4 мл, 61,9 ммоль), Et3N (8,26 мл, 59,3 ммоль) в DCM (30 мл) при 25°C добавляли DMAP (2,70 г, 22,1 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем распределяли между водой (20 мл) и DCM (50 мл×3). Объединенный органический слой сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=444,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1H), 7,67 (дд, J=6,3, 2,7 Гц, 2H), 7,45 (д, J=2,3 Гц, 3H), 4,10 (с, 2H), 3,73 (с, 3H), 1,30 (с, 18H).
Стадия B: Метил-2-(5-(бис(трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)-5-((трет-бутоксикарбонил)амино)пентаноат
К смеси 2-((5-(bis-трет-бутоксикарбонил)амино-4-фенилпиримидин-2-ил))ацетата (300 мг, 0,676 ммоль) и трет-бутил-(3-йодпропил)карбамата (231 мг, 0,812 ммоль) в THF (6 мл) при 25°C добавляли 2-метилпропан-2-олят калия (1,35 мл, 1,35 ммоль). Полученную смесь перемешивали при 25°C в течение 12 ч, а затем распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=601,4 (M+1).
Стадия C: 3-(5-Амино-4-фенилпиримидин-2-ил)пиперидин-2-он
Раствор метил-2-(5-(бис(трет-бутоксикарбонил)амино)-4-фенилпиримидин-2-ил)-5-((трет-бутоксикарбонил)амино)пентаноата (240 мг, 0,400 ммоль) в 4M растворе HCl в диоксане (12 мл, 48,0 ммоль) перемешивали при 25°C в течение 30 мин, а затем концентрировали. Остаток растворяли в MeOH (6 мл), и добавляли K2CO3 (442 мг, 3,20 ммоль). Полученную смесь перемешивали при 25°C в течение 12 ч, а затем фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc/MeOH=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=269,0 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,23 (с, 1H), 7,74 (д, J=6,7 Гц, 2H), 7,55-7,43 (м, 3H), 3,82 (дд, J=8,6, 7,0 Гц, 1H), 3,45-3,37 (м, 2H), 2,24-2,11 (м, 2H), 1,99-1,94 (м, 1H), 1,87-1,79 (м, 1H).
Схема реакций для промежуточного продукта A34
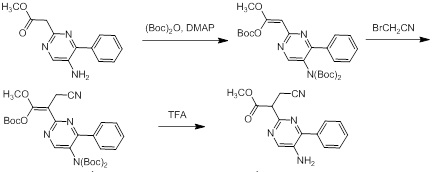
Промежуточный продукт A34
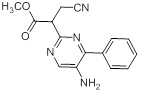
Метил-2-(5-Амино-4-фенилпиримидин-2-ил)-3-цианопропаноат
Стадия A: трет-Бутил-(2-(2-((бис-трет-бутоксикарбонил)окси)-2-метоксивинил)-4-фенилпиримидин-5-ил)карбамат
К смеси метил-2-(5-амино-4-фенилпиримидин-2-ил)ацетата (5,00 г, 20,6 ммоль), Boc2O (14,4 мл, 61,9 ммоль), Et3N (8,26 мл, 59,3 ммоль) в CH2Cl2 (30 мл) при 25°C добавляли DMAP (2,7 г, 22,10 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем распределяли между водой (20 мл) и CH2Cl2 (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=544,3 (M+1).
Стадия B: Ди-трет-бутил-(2-(1-((трет-бутоксикарбонил)окси)-3-циано-1-метоксипроп-1-ен-2-ил)-4-фенилпиримидин-5-ил)карбамат
К раствору трет-бутил-(2-(2-((ди-трет-бутоксикарбонил)окси)-2-метоксивинил)-4-фенилпиримидин-5-ил)карбамата (2,50 г, 4,60 ммоль) в диметилформамиде (30 мл) при 25°C добавляли K2CO3 (1,271 г, 9,20 ммоль) и бромацетонитрил (0,827 г, 6,90 ммоль). Полученную смесь перемешивали в течение 2 ч, а затем распределяли между водой (50 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=583,4 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,48 (с, 1H), 7,81 (дд, J1=6,53, J2=3,01 Гц, 2H), 7,57-7,52 (м, 3H), 5,08 (с, 1H), 3,77 (с, 3H), 1,45 (с, 9H), 1,23 (д, J=6,02 Гц, 18H).
Стадия C: Метил-2-(5-амино-4-фенилпиримидин-2-ил)-3-цианопропаноат
К раствору ди-трет-бутил-(2-(1-((трет-бутоксикарбонил)окси)-3-циано-1–метоксипроп-1-ен-2-ил)-4-фенилпиримидин-5-ил)карбамата (2,00 г, 3,09 ммоль) в ацетонитриле (4 мл) при 25°C добавляли TFA (5 мл). Смесь перемешивали в течение 2 ч, а затем распределяли между насыщенным водным раствором бикарбоната натрия (50 мл) и EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=282,9 (M+1).
Схема реакций для промежуточного продукта A35
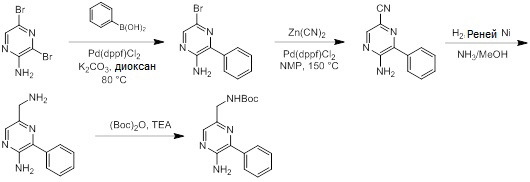
Промежуточный продукт A35
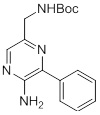
трет-Бутил-((5-амино-6-фенилпиразин-2-ил)метил)карбамат
Стадия A: 5-Бром-3-фенилпиразин-2-амин
К дезоксигенированному раствору 3,5-дибромпиразин-2-амина (5,00 г, 19,8 ммоль) в 1,4-диоксане (100 мл) добавляли карбонат калия (8,20 г, 59,3 ммоль), фенилбороновую кислоту (2,17 г, 17,8 ммоль) и Pd(dppf)Cl2 (1,45 г, 1,98 ммоль). Полученную смесь нагревали при 90°C в течение 4 ч, затем охлаждали и распределяли между водой (50 мл) и EtOAc (50 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=15/1, 10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=249,9 (M+1).
Стадия B: 5-Амино-6-фенилпиразин-2-карбонитрил
К дезоксигенированному раствору 5-бром-3-фенилпиразин-2-амина (1,00 г, 3,20 ммоль) в NMP (15 мл) добавляли дицианоцинк (1,13 г, 9,61 ммоль) и Pd(dppf)Cl2 (0,262 г, 0,320 ммоль). Полученную смесь нагревали при 150°C в течение 3 ч, а затем охлаждали и распределяли между водой (10 мл) и EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 2/1) с получением указанного в заголовке соединения. MS: m/z=196,9 (M+1).
Стадия C: 5-(Аминометил)-3-фенилпиразин-2-амин
К дезоксигенированному раствору 5-амино-6-фенилпиразин-2-карбонитрила (270 мг, 0,90 ммоль) в MeOH (15 мл) и 4M растворе NH3 в MeOH (10 мл) добавляли никель Ренея (5,3 мг, 0,090 ммоль). Полученную смесь перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 20°C в течение 30 мин. Содержащую продукт смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=201,0 (M+1).
Стадия D: трет-Бутил-((5-амино-6-фенилпиразин-2-ил)метил)карбамат
К раствору 5-(аминометил)-3-фенилпиразин-2-амина (260 мг, 1,30 ммоль) в CH2Cl2 (5 мл) добавляли триэтиламин (0,543 мл, 3,90 ммоль) и Boc2O (0,452 мл, 1,95 ммоль). Полученную смесь перемешивали в течение 30 мин при 20°C, а затем распределяли между водой (5 мл) и EtOAc (3 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 1/1) с получением указанного в заголовке соединения. MS: m/z=301,0 (M+1).
Схема реакций для промежуточного продукта A36
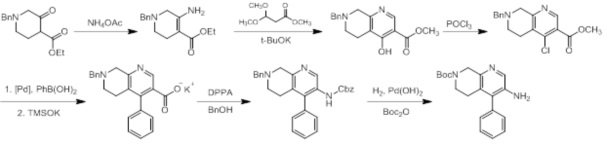
Промежуточный продукт A36
трет-Бутил-3-амино-4-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилат
Стадия A: Этил-5-амино-1-бензил-1,2,3,6-тетрагидропиридин-4-карбоксилат
К раствору этил-1-бензил-3-оксопиперидин-4-карбоксилата (30,0 г, 115 ммоль) в метаноле (200 мл) при 20°C добавляли ацетат аммония (44,2 г, 574 ммоль), и перемешивали полученную смесь в течение 18 ч. Содержащую продукт смесь концентрировали, и распределяли остаток между водой (200 мл) и этилацетатом (200 мл×2). Объединенный органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 7,47-7,21 (м, 5H), 4,09 (кв, J=7,3 Гц, 2H), 3,79 (с, 2H), 3,33 (с, 2H), 2,75 (т, J=6,1 Гц, 2H), 2,44 (т, J=5,9 Гц, 2H), 1,21 (т, J=7,0 Гц, 3H).
Стадия B: Метил-7-бензил-4-гидрокси-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилат
К раствору трет-бутоксида калия (34,9 г, 311 ммоль) в тетрагидрофуране (250 мл) при 20°C добавляли этил-5-амино-1-бензил-1,2,3,6-тетрагидропиридин-4-карбоксилат (27,0 г, 104 ммоль) и метил-3,3-диметоксипропаноат (46,1 г, 311 ммоль). Полученную смесь перемешивали в течение 18 ч, затем разбавляли водой со льдом (200 мл) и подкисляли до pH=5 добавлением 2M водного раствора HCl. Полученную смесь подщелачивали до pH=8 добавлением насыщенного водного раствора бикарбоната натрия и экстрагировали этилацетатом (200 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=299,1 (M+1).
Стадия C: Метил-7-бензил-4-хлор-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилат
Раствор метил-7-бензил-4-гидрокси-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилата (20,0 г, 67,0 ммоль) в трихлориде фосфора (61,7 г, 402 ммоль) нагревали при 80°C в течение 5 ч. Смесь охлаждали и осторожно вливали в воду (200 мл), и перемешивали водную смесь при 20°C в течение 1 ч, а затем подщелачивали до pH=8 добавлением насыщенного водного раствора карбоната калия. Смесь экстрагировали этилацетатом (200 мл×2), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=317,2 (M+1).
Стадия D: 7-Бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилат
К дезоксигенированному раствору метил-7-бензил-4-хлор-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилата (13,0 г, 41,0 ммоль) в диоксане (150 мл) и воде (15 мл) добавляли фенилбороновую кислоту (5,00 г, 41,0 ммоль), карбонат калия (5,67 г, 41,0 ммоль) и PdCl2(dppf) (30,0 г, 41,0 ммоль). Полученную смесь нагревали при 100°C в течение 18 ч, а затем охлаждали и распределяли между водой (100 мл) и этилацетатом (150 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением метил-7-бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилата. К раствору метил-7-бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилата (6,00 г, 16,74 ммоль) в тетрагидрофуране (150 мл) при 20°C добавляли триметилсиланолят калия (2,15 г, 16,7 ммоль), и перемешивали полученную смесь в течение 5 ч. Осадок фильтровали и сушили с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8,30 (с, 1H), 7,30-7,16 (м, 10H), 3,64-3,59 (м, 4H), 2,62-2,55 (м, 2H), 2,48 (д, J=5,5 Гц, 2H).
Стадия E: Бензил-(7-бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамат
К перемешанному раствору 7-бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-карбоксилата калия (4,50 г, 11,8 ммоль) в толуоле (50 мл) добавляли дифенилфосфорилазид (4,21 г, 15,3 ммоль) и бензиловый спирт (2,54 г, 23,5 ммоль). Полученную смесь нагревали при 100°C в течение 18 ч, затем охлаждали и распределяли между водой (50 мл) и этилацетатом (50 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=450,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,53 (с, 1H), 7,47-7,36 (м, 5H), 7,34-7,26 (м, 6H), 7,22 (д, J=7,4 Гц, 2H), 7,15 (д, J=6,7 Гц, 2H), 5,02 (с, 2H), 3,70 (д, J=8,6 Гц, 4H), 2,71-2,64 (м, 2H), 2,56-2,50 (м, 2H).
Стадия F: трет-Бутил-3-амино-4-фенил-5,6-дигидро-1,7-нафтиридин-7-(8H)-карбоксилат
К дезоксигенированному раствору бензил-(7-бензил-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамата (4,00 г, 8,90 ммоль) в метаноле (50 мл) добавляли 20% Pd(OH)2 на угле (0,63 г, 0,89 ммоль). Полученную смесь перемешивали в атмосфере водорода (55 фунт./кв.дюйм) при 20°C в течение 3 ч. Смесь фильтровали, и концентрировали фильтрат. Остаток растворяли в этилацетате (20 мл) и воде (20 мл) при 20°C, и добавляли к этому раствору ди-трет-бутилдикарбонат (2,09 г, 9,59 ммоль) и карбонат натрия (1,69 г, 16,0 ммоль). Полученную смесь перемешивали в течение 18 ч, а затем экстрагировали этилацетатом (30 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=326,3 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 7,94 (с, 1H), 7,48-7,57 (м, 2H), 7,40-7,45 (м, 1H), 7,21 (д, J=7,0 Гц, 2H), 4,52 (с, 2H), 3,50 (с, 2H), 2,40 (т, J=5,5 Гц, 2H), 1,46 (с, 9H).
Схема реакций для промежуточного продукта A37
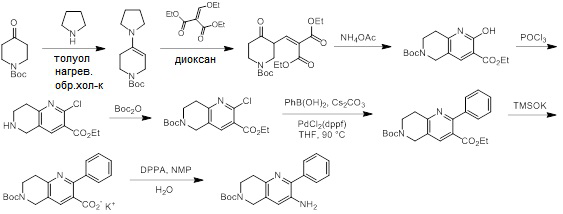
Промежуточный продукт A37
трет-Бутил-3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
Стадия A: 6-трет-Бутил-3-этил-2-гидрокси-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
Раствор трет-бутил-4-оксопиперидин-1-карбоксилата (35,0 г, 176 ммоль) и пирролидина (18,7 г, 263 ммоль) в толуоле (250 мл) нагревали с обратным холодильником в течение 18 ч с использованием аппарата Дина-Старка. Смесь концентрировали, и растворяли остаток в диоксане (250 мл). К этому раствору добавляли диэтил-2-(этоксиметилен)малонат (41,8 г, 193 ммоль). Полученную смесь нагревали с обратным холодильником в течение 6 ч, а затем охлаждали до 23°C. Добавляли ацетат аммония (23,0 г, 299 ммоль), и нагревали полученную смесь с обратным холодильником в течение 1 ч. Смесь охлаждали и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (этилацетат) с получением указанного в заголовке соединения. MS: m/z=323,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,10 (с, 1H), 4,39 (с, 2H), 4,32 (кв, J=7,2 Гц, 2H), 3,71 (т, J=5,0 Гц, 2H), 2,74 (т, J=5,5 Гц, 2H), 1,51 (с, 9H), 1,37 (т, J=7,0 Гц, 3H).
Стадия B: 6-трет-Бутил-3-этил-2-хлор-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
К суспензии 6-трет-бутил-3-этил-2-гидрокси-7,8-дигидро-1,6-нафтиридин-5,6(5H)-дикарбоксилата (40,0 г, 124 ммоль) в ацетонитриле (200 мл) по каплям добавляли оксихлорид фосфора (34,7 мл, 372 ммоль), и нагревали полученную смесь при 90°C в течение 12 ч. Смесь разбавляли водой (100 мл) и подщелачивали до pH=7 добавлением насыщенного водного раствора бикарбоната натрия. Смесь экстрагировали этилацетатом (100 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток растворяли в насыщенном водном растворе бикарбоната натрия (100 мл) и THF (100 мл), и добавляли к этому раствору ди-трет-бутил-дикарбонат (57,6 мл, 248 ммоль). Полученную смесь перемешивали при 18°C в течение 12 ч, а затем распределяли между водой (50 мл) и этилацетатом (100 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS m/z=341,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,91 (с, 1H), 4,60 (с, 2H), 4,40 (кв, J=7,0 Гц, 2H), 3,74 (т, J=5,5 Гц, 2H), 3,00 (с, 2H), 1,49 (с, 9H), 1,41 (т, J=7,24 Гц, 3H).
Стадия C: 6-трет-Бутил-3-этил-2-фенил-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
К дезоксигенированному раствору 6-трет-бутил-3-этил-2-хлор-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (6,40 г, 18,8 ммоль) и фенилбороновой кислоты (3,43 г, 28,2 ммоль) в THF (60 мл) и воде (6 мл) добавляли карбонат цезия (18,4 г, 56,3 ммоль) и PdCl2(dppf) (0,687 г, 0,939 ммоль). Полученную смесь нагревали при 90°C в течение 12 ч. Смесь охлаждали и фильтровали, и распределяли фильтрат между водой (20 мл) и этилацетатом (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=323,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,80 (с, 1H), 7,41 (д, J=3,13 Гц, 2H), 7,34 (д, J=5,09 Гц, 3H), 4,61 (с, 2H), 4,06 (кв, J=7,04 Гц, 2H), 3,72 (с, 2H), 3,02 (с, 2H), 1,44 (с, 9H) 0,96 (т, J=7,04 Гц, 3H).
Стадия D: 6-(трет-Бутоксикарбонил)-2-фенил-5,6,7,8-тетрагидро-1,6–нафтиридин-3-карбоксилат калия
К раствору 6-трет-бутил-3-этил-2-фенил-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (6,20 г, 16,2 ммоль) в THF (100 мл) добавляли триметилсиланолят калия (3,12 г, 24,3 ммоль), и перемешивали полученную смесь при 18°C в течение 12 ч. Содержащую продукт смесь разбавляли петролейным эфиром (100 мл), осадок фильтровали, промывали петролейным эфиром и сушили с получением указанного в заголовке соединения. MS: m/z=355,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 7,69 (д, J=6,6 Гц, 2H), 7,61 (с, 1H), 7,37 (м, 3H), 4,64 (с, 2H), 3,76 (с, 2H), 2,96 (т, J=5,7 Гц, 2H), 1,49 (с, 9H).
Стадия E: трет-Бутил-3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
К раствору 6-(трет-бутоксикарбонил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоксилата калия (5,00 г, 12,7 ммоль) в N-метил-2-пирролидоне (100 мл) при 20°C добавляли дифенилфосфорилазид (4,57 г, 16,6 ммоль). Полученную смесь нагревали при 90°C в течение 12 ч в атмосфере азота. Добавляли воду (10 мл), и нагревали смесь при 90°C еще в течение 12 ч. Смесь охлаждали и распределяли между насыщенным водным раствором бикарбоната натрия (30 мл) и этилацетатом (30 мл×3). Объединенные органические слои промывали водой (30 мл×2), а затем солевым раствором (30 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1) с получением указанного в заголовке соединения. MS: m/z=326,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 7,47 (м, 5H), 6,98 (с, 1H), 4,52 (с, 2H), 3,71 (м, 2H), 2,80 (с, 2H), 1,48 (с, 9H).
Схема реакций для промежуточного продукта A38
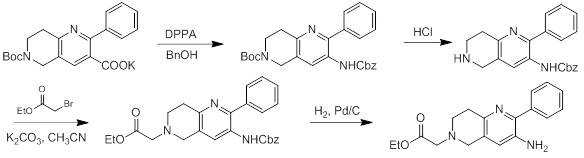
Промежуточный продукт A38
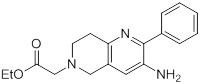
Этил-2-(3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат
Стадия A: трет-Бутил-3-(((бензилокси)карбонил)амино)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
К перемешанному раствору 6-(трет-бутоксикарбонил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоксилата калия (6,50 г, 16,6 ммоль) и бензилового спирта (7,16 г, 66,2 ммоль) в толуоле (100 мл) при 20°C добавляли DPPA (6,84 г, 24,8 ммоль). Полученную смесь нагревали при 110°C в течение 16 ч, затем охлаждали и концентрировали. Остаток распределяли между водой (100 мл) и EtOAc (100 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,26 (ушир., 1H), 7,48-7,47 (м, 5H), 7,43-7,42 (м, 4H), 7,17-7,15 (м, 1H), 6,77 (ушир., 1H), 5,15 (с, 2H), 4,63 (с, 2H), 3,75 (ушир., 2H), 2,97 (ушир., 2H), 1,49 (с, 9H).
Стадия B: Бензил-(2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)карбамат
Смесь трет-бутил-3-(((бензилокси)карбонил)амино)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (4,00 г, 8,70 ммоль) в 4M растворе HCl в EtOAc (50 мл, 200 ммоль) перемешивали при 20°C в течение 1 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=360,2 (M+1).
Стадия C: Этил-2-(3-(((бензилокси)карбонил)амино)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат
К раствору бензил-(2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)карбамата (2,80 г, 7,79 ммоль) в ацетонитриле (20 мл) добавляли K2CO3 (3,23 г, 23,4 ммоль) и этил-2-бромацетат (1,95 г, 11,7 ммоль). Полученную смесь перемешивали при 20°C в течение 1 ч, а затем распределяли между водой (10 мл) и EtOAc (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1, 3/1, 2/1, 1/1) с получением указанного в заголовке соединения. MS: m/z=446,3 (M+1).
Стадия D: Этил-2-(3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат
Смесь этил-2-(3-(((бензилокси)карбонил)амино)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат (1,40 г, 3,14 ммоль) и 10% Pd/C (0,33 г, 3,1 ммоль) в EtOH (20 мл) нагревали в атмосфере H2 (50 фунт./кв.дюйм) при 50°C в течение 6 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=312,1 (M+1).
Схема реакций для промежуточного продукта A39
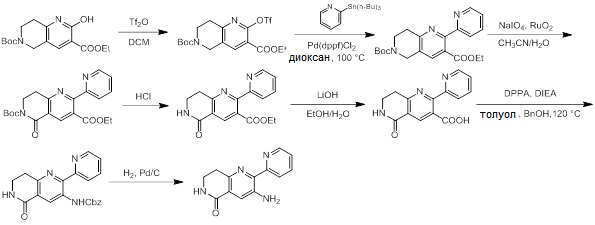
Промежуточный продукт A39
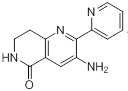
3-Амино-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
Стадия A: 6-трет-Бутил-3-этил-2-(((трифторметил)сульфонил)-окси)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
К раствору 6-трет-бутил-3-этил-2-гидрокси-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (10,0 г, 31,0 ммоль)) в CH2Cl2 (100 мл) при 25°C добавляли триэтиламин (9,42 г, 93,1 ммоль), а затем трифторметансульфоновый ангидрид (17,5 г, 62,0 ммоль). Полученную смесь перемешивали при 25°C в течение 18 ч, а затем распределяли между водой (100 мл) и CH2Cl2 (200 мл×3). Объединенные органические слои сушили над MgSO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ (с, 1H), 4,60 (с, 2H), 4,38 (кв, J=7,2 Гц, 2H), 3,70 (т, J=5,5 Гц, 2H), 2,94 (д, J=5,1 Гц, 2H), 1,43 (с, 9H), 1,36 (т, J=7,0 Гц, 3H).
Стадия B: 6-трет-Бутил-3-этил-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
К дезоксигенированному раствору 6-трет-бутил-3-этил-2-(((трифторметил)сульфонил)окси)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (1,00 г, 2,20 ммоль) в 1,4-диоксане (20 мл) добавляли 2-(трибутилстаннил)пиридин (0,900 г, 2,60 ммоль) и PdCl2(dppf) (0,10 г, 0,20 ммоль). Полученную смесь нагревали при 100°C в течение 15 ч, а затем охлаждали и распределяли между водой (30 мл) и этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=384,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,53 (д, J=4,3 Гц, 1H), 7,93 (д, J=7,0 Гц, 1H), 7,75 (т, J=7,0 Гц, 1H), 7,70 (с, 1H), 7,21-7,26 (м, 1H), 4,60 (с, 2H), 4,16 (кв, J=7,0 Гц, 2H), 3,72 (с, 2H), 3,02 (с, 2H), 1,42-1,46 (м, 9H), 1,06 (т, J=7,2 Гц, 3H).
Стадия C: 6-трет-Бутил-3-этил-5-оксо-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилат
К раствору 6-трет-бутил-3-этил-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (250 мг, 0,653 ммоль) в ацетонитриле (3 мл) и воде (3 мл) при 27°C добавляли перйодат натрия (279 мг, 1,30 ммоль) и оксид рутения(IV) (0,006 мл, 0,33 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем распределяли между водой (20 мл) и этилацетатом (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=4/1) с получением указанного в заголовке соединения. MS: m/z=398,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,68 (с, 1H), 8,61 (д, J=4,3 Гц, 1H), 8,09 (д, J=7,8 Гц, 1H), 7,84 (т, J=7,0 Гц, 1H), 7,37-7,32 (м, 1H), 4,28 (кв, J=7,0 Гц, 2H), 4,10 (т, J=6,3 Гц, 2H), 3,28 (т, J=6,3 Гц, 2H), 1,59 (с, 9H), 1,20 (т, J=7,2 Гц, 3H).
Стадия D: Этил-5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоксилат
Раствор 6-трет-бутил-3-этил-5-оксо-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-3,6(5H)-дикарбоксилата (260 мг, 0,655 ммоль) в 4M растворе HCl в диоксане (5 мл) перемешивали при 25°C в течение 30 мин. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=298,1 (M+1).
Стадия E: 5-Оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоновая кислота
К раствору этил-5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоксилата (180 мг, 0,606 ммоль) в этаноле (2 мл) и воде (0,50 мл) добавляли гидроксид лития (29 мг, 1,2 ммоль), и перемешивали полученную смесь 25°C в течение 2 ч. Смесь подкисляли до pH=4 добавлением 2M водного раствора HCl, а затем экстрагировали этилацетатом (20 мл). Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8,77 (с, 1H), 8,72 (д, J=4,7 Гц, 1H), 8,29-8,23 (м, 1H), 8,11 (д, J=7,8 Гц, 1H), 7,78-7,72 (м, 1H), 3,66 (т, J=6,8 Гц, 2H), 3,27-3,23 (м, 2H).
Стадия F: Бензил-(5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)карбамат
К раствору 5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-карбоновой кислоты (150 мг, 0,557 ммоль) в толуоле (5 мл) добавляли N,N-диизопропилэтиламин (0,29 мл, 1,7 ммоль) и дифенилфосфорилазид (307 мг, 1,11 ммоль), и нагревали полученную смесь при 80°C в течение 1 ч. Добавляли бензиловый спирт (0,12 мл, 1,1 ммоль), смесь нагревали при 100°C в течение 15 ч, а затем охлаждали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=375,2 (M+1).
Стадия G: 3-Амино-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-5(6H)-он
К дезоксигенированному раствору бензил-(5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)карбамата (70 мг, 0,19 ммоль) в метаноле (10 мл) добавляли 10% Pd/C (199 мг, 0,188 ммоль). Смесь перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 25°C в течение 10 ч, а затем фильтровали. Фильтрат концентрировали, и очищали остаток методом препаративной TLC (EtOAc/MeOH=10/1) с получением указанного в заголовке соединения. MS: m/z=241,0 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,62 (с, 1H), 8,48 (д, J=8,2 Гц, 1H), 7,93-7,87 (м, 1H), 7,67 (с, 1H), 7,35 (с, 1H), 3,57 (т, J=6,7 Гц, 2H), 3,06 (т, J=6,8 Гц, 2H).
Схема реакций для промежуточного продукта A40
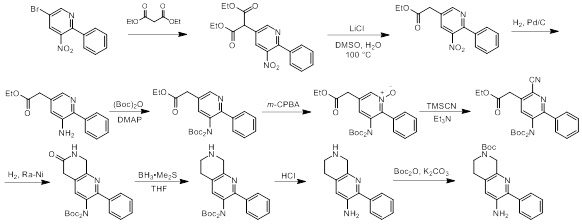
Промежуточный продукт A40

трет-Бутил-3-амино-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилат
Стадия A: Диэтил-2-(5-нитро-6-фенилпиридин-3-ил)малонат
К смеси 5-бром-3-нитро-2-фенилпиридина (10,0 г, 35,8 ммоль), диэтилмалоната (23,0 г, 143 ммоль), бромида меди(I) (20,6 г, 143 ммоль) в диоксане (60 мл) при 25°C порциями добавляли гидрид натрия (6,30 г, 158 ммоль). Полученную смесь нагревали при 100°C в течение 16 ч, а затем охлаждали и разбавляли насыщенным водным раствором NH4Cl (100 мл). Водную смесь экстрагировали EtOAc (100 мл×3), объединенные органические слои сушили Na2SO4 и концентрировали. Остаток очищали методом хроматографии на силикагеле (PE/EtOAc=20/1, 10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=359,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,81 (д, J=1,2 Гц, 1H), 8,33 (д, J=1,6 Гц, 1H), 7,53-7,59 (м, 2H), 7,44-7,49 (м, 3H), 4,24 (м, 4H), 1,29 (д, J=5,1 Гц, 6H).
Стадия B: Этил-2-(5-нитро-6-фенилпиридин-3-ил)ацетат
К перемешанному раствору диэтил-2-(5-нитро-6-фенилпиридин-3-ил)малоната (5,80 г, 16,2 ммоль) в DMSO (60 мл) и воде (3 мл) добавляли хлорид лития (2,10 г, 48,6 ммоль). Полученную смесь нагревали при 100°C в течение 5 ч, затем охлаждали и распределяли между водой (80 мл) и EtOAc (80 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 3/1) с получением указанного в заголовке соединения. MS: m/z=287,0 (M+1).
Стадия C: Этил-2-(5-((ди-трет-бутоксикарбонил)амино)-6-фенилпиридин-3-ил)ацетат
Дезоксигенированную смесь этил-2-(5-нитро-6-фенилпиридин-3-ил)ацетат (5,50 г, 19,2 ммоль) и 10% Pd/C (2,0 г, 1,9 ммоль) в MeOH (50 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 30°C в течение 5 ч. Смесь фильтровали, и концентрировали фильтрат. Остаток растворяли в CH2Cl2 (50 мл), и добавляли к этому раствору триэтиламин (5,10 г, 50,3 ммоль), DMAP (2,10 г, 16,8 ммоль) и Boc2O (7,80 мл, 33,6 ммоль). Полученную смесь перемешивали при 30°C в течение 3 ч, затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения.
Стадия D: 3-((Ди-трет-бутоксикарбонил)амино)-5-(2-этокси-2-оксоэтил)-2-фенилпиридин-1-оксид
К перемешанному раствору этил-2-(5-((ди-трет-бутоксикарбонил)амино)-6-фенилпиридин-3-ил)ацетата (200 мг, 0,438 ммоль) в CHCl3 (4 мл) добавляли m-CPBA (267 мг, 1,32 ммоль). Полученную смесь перемешивали при 25°C в течение 2 ч, затем распределяли между насыщенным водным раствором Na2SO3 (10 мл) и CH2Cl2 (15 мл×3). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (10 мл), сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=473,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1H), 8,03 (д, J=14,1 Гц, 1H), 7,94 (дд, J1=16,2 Гц, J2=7,6 Гц, 1H), 7,64 (д, J=7,8 Гц, 1H), 7,43 (ушир., 2H), 7,21 (с, 1H), 4,21 (д, J=6,7 Гц, 2H), 3,62 (с, 2H), 1,34 (с, 18H), 1,27-1,29 (м, 3H).
Стадия E: Этил-2-(5-((ди-трет-бутоксикарбонил)амино)-2-циано-6-фенилпиридин-3-ил)ацетат
К перемешанному раствору 3-((ди-трет-бутоксикарбонил)-амино)-5-(2-этокси-2-оксоэтил)-2-фенилпиридин-1-оксида (2,0 г, 4,2 ммоль) и Et3N (1,8 мл, 13 ммоль) в ацетонитриле (30 мл) добавляли TMSCN (2,3 мл, 17 ммоль). Полученную смесь нагревали при 80°C в течение 5 ч, затем охлаждали и распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1, 1/1) с получением указанного в заголовке соединения. MS: m/z=482,3 (M+1).
Стадия F: Ди-трет-бутил-(6-оксо-2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамат
Дезоксигенированную смесь этил-2-(5-((ди-трет-бутоксикарбонил)амино)-2-циано-6-фенилпиридин-3-ил)ацетата (1,0 г, 2,1 ммоль) и никеля Ренея (24 мг, 0,4 ммоль) в MeOH (20 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 25°C в течение 5 ч. Смесь фильтровали, и концентрировали фильтрат с получением неочищенного указанного в заголовке вещества. MS: m/z=440,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,49 (д, J=6,7 Гц, 2H), 7,40-7,32 (м, 4H), 7,29 (с, 1H), 4,65 (ушир., 2H), 3,66 (ушир., 2H), 1,23 (с, 18H).
Стадия G: Ди-трет-бутил-(2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамат
К перемешанному раствору ди-трет-бутил-(6-оксо-2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамата (200 мг, 0,46 ммоль) в THF (3 мл) при 25°C добавляли BH3⋅DMS (0,20 мл, 2,3 ммоль), и перемешивали полученную смесь в течение 5 ч. Добавляли MeOH (1 мл), и концентрировали смесь. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=467,3 (M+1+ MeCN).
Стадия I: 2-Фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-амин
Смесь ди-трет-бутил-(2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)карбамата (100 мг, 0,24 ммоль) в 4M растворе HCl в EtOAc (10 мл, 40,0 ммоль) перемешивали 25°C в течение 2 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=226,2 (M+1).
Стадия J: 2-Фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-амин
Смесь 2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-амина (50 мг, 0,22 ммоль), K2CO3 (92 мг, 0,67 ммоль) и Boc2O (0,10 мл, 0,44 ммоль) в THF (3 мл) и воде (1 мл) перемешивали при 30°C в течение 3 ч. Содержащую продукт смесь распределяли между водой (20 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=2/1) с получением указанного в заголовке соединения. MS: m/z=326,2 (M+1).
Схема реакций для промежуточного продукта A41
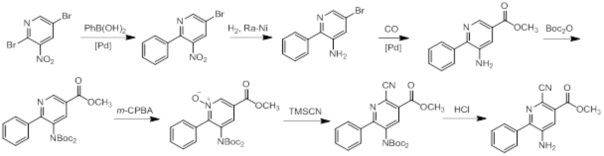
Промежуточный продукт A41
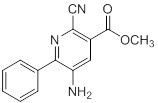
Метил-5-амино-2-циано-6-фенилникотинат
Стадия A: 5-Бром-3-нитро-2-фенилпиридин
К дезоксигенированному раствору 2,5-дибром-3-нитропиридина (50,0 г, 177 ммоль) в DME (800 мл) и воде (150 мл) добавляли фенилбороновую кислоту (23,8 г, 195 ммоль), K2CO3 (49,0 г, 355 ммоль) и PdCl2(dppf) (6,49 г, 8,87 ммоль). Полученную смесь нагревали при 100°C в течение 18 ч, а затем охлаждали и распределяли между водой (1000 мл) и EtOAc (500 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (100% PE, PE/EtOAc=50/1) с получением указанного в заголовке соединения. MS: m/z=279,1, 281,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,91 (д, J=2,0 Гц, 1H), 8,28 (д, J=2,0 Гц, 1H), 7,53-7,55 (м, 2H), 7,45-7,48 (м, 3H).
Стадия B: 5-Бром-2-фенилпиридин-3-амин
Смесь 5-бром-3-нитро-2-фенилпиридина (10,0 г, 35,8 ммоль) и никеля Ренея (3,07 г, 35,8 ммоль) в EtOAc (100 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 20°C в течение 4 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=249,1, 251,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,07 (д, J=2,0 Гц, 1H), 7,54-7,56 (м, 2H), 7,38-7,42 (м, 3H), 7,12 (с, 1H), 3,75 (ушир., 2H).
Стадия C: Метил-5-амино-6-фенилникотинат
К раствору 5-бром-2-фенилпиридин-3-амина (20 г, 80 ммоль) в MeOH (200 мл) добавляли PdCl2(dppf) (5,87 г, 8,03 ммоль) и Et3N (16,2 г, 161 ммоль). Смесь нагревали в атмосфере CO (3 мбар) при 120°C в течение 8 ч. Содержащую продукт смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом хроматографии на силикагеле (100% PE, PE/EA=20/1, 10/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8,42 (д, J=2,0 Гц, 1H), 7,74 (д, J=2,0 Гц, 1H), 7,60-7,62 (м, 2H), 7,46-7,50 (м, 3H), 3,91 (с, 3H).
Стадия D: Метил-5-((ди-трет-бутоксикарбонил)амино)-6-фенилникотинат
К раствору метил-5-амино-6-фенилникотината (12,0 г, 52,6 ммоль) в DCM (150 мл) добавляли DMAP (12,8 г, 105 ммоль), Et3N (10,6 г, 105 ммоль) и Boc2O (45,9 г, 210 ммоль). Полученную смесь перемешивали при 15°C в течение 1 ч, а затем распределяли между водой (100 мл) и DCM (100 мл×3). Объединенные органические слои промывали лимонной кислотой, сушили над Na2SO4 и концентрировали. Остаток очищали методом хроматографии на силикагеле (PE, PE/EtOAc=50/1, 20/1) с получением указанного в заголовке соединения. MS: m/z=429,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,21 (д, J=1,6 Гц, 1H), 8,10 (д, J=2,0 Гц, 1H), 7,64-7,62 (м, 2H), 7,44-7,40 (м, 3H), 3,98 (с, 3H), 1,27 (с, 18 H).
Стадия E: 3-((Ди-трет-бутоксикарбонил)амино)-5-(метоксикарбонил)-2-фенилпиридин-1-оксид
К раствору метил-5-((ди-трет-бутоксикарбонил)амино)-6-фенилникотината (21,0 г, 49,0 ммоль) в DCM (210 мл) добавляли m-CPBA (8,46 г, 49,0 ммоль). Полученную смесь перемешивали при 15°C в течение 6 ч, затем распределяли между водой (100 мл) и DCM (100 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом хроматографии на силикагеле (PE, PE/EtOAc=10/1, PE/EtOAc=5/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,05 (с, 1H), 7,95 (д, J=7,6 Гц, 1H), 7,64-7,45 (м, 5H), 3,99 (с, 3H), 1,41-1,25 (м, 18 H).
Стадия F: Метил-5-((ди-трет-бутоксикарбонил)амино)-2-циано-6-фенилникотинат
К раствору 3-((ди-трет-бутоксикарбонил)амино)-5-(метоксикарбонил)-2-фенилпиридин-1-оксида (14,0 г, 31,5 ммоль) в ацетонитриле (150 мл) добавляли Et3N (9,56 г, 945 ммоль) и триметилсиланкарбонитрил (6,25 г, 63,0 ммоль). Полученную смесь нагревали при 90°C в течение 4 ч. После охлаждения, смесь распределяли между водой (100 мл) и EtOAc (100×3 мл). Объединенные органические слои промывали лимонной кислотой, сушили над Na2SO4 и концентрировали. Остаток очищали методом хроматографии на силикагеле (PE, PE/EA=20/1, PE/EA=5/1) с получением указанного в заголовке соединения. MS: m/z=454,2 (M+1).
Стадия G: Метил-5-амино-2-циано-6-фенилникотинат
К раствору метил-5-((ди-трет-бутоксикарбонил)амино)-2-циано-6-фенилникотината (200 мг, 0,44 ммоль) в EtOAc (5 мл) добавляли 4M раствор HCl в EtOAc (0,11 мл, 0,44 ммоль), и перемешивали смесь при 20°C в течение 30 мин. Полученную взвесь концентрировали с получением указанного в заголовке соединения. MS: m/z=254,2 (M+1).
Схема реакций для промежуточного продукта A42
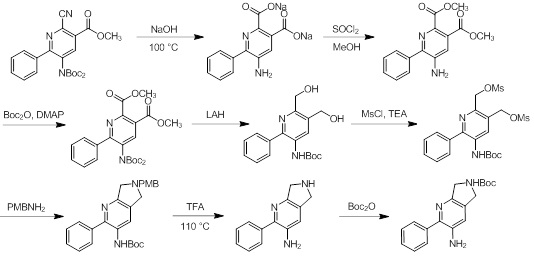
Промежуточный продукт A42
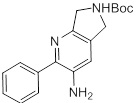
трет-Бутил-3-амино-2-фенил-5H-пирроло[3,4-b]пиридин-6(7H)-карбоксилат
Стадия A: 5-Амино-6-фенилпиридин-2,3-дикарбоксилат натрия
К раствору гидроксида натрия (10,6 г, 265 ммоль) в воде (100 мл) при 25°C добавляли метил-5-((ди-трет-бутоксикарбонил)-амино)-2-циано-6-фенилникотинат (10,0 г, 22,1 ммоль). Полученную смесь нагревали при 100°C в течение 5 ч, затем концентрировали с получением указанного в заголовке соединения в виде бис-натриевой соли. MS: m/z=259,2 (M+1).
Стадия B: Диметил-5-амино-6-фенилпиридин-2,3-дикарбоксилат
К смеси 5-амино-6-фенилпиридин-2,3-дикарбоксилата натрия (3,80 г, 12,6 ммоль) в метаноле (100 мл) при 25°C по каплям добавляли тионилхлорид (35,0 г, 294 ммоль). Полученную смесь нагревали при 80°C в течение 16 ч. Содержащую продукт смесь концентрировали, и разбавляли остаток водой (20 мл). Содержащую продукт водную смесь подщелачивали до pH=8-9 добавлением насыщенного раствора Na2CO3, а затем экстрагировали EtOAc (20 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=287,2 (M+1).
Стадия C: Диметил-5-((ди-трет-бутоксикарбонил)амино)-6-фенилпиридин-2,3-дикарбоксилат
К перемешанному раствору диметил-5-амино-6-фенилпиридин-2,3-дикарбоксилата (2,80 г, 9,78 ммоль) в дихлорметане (40 мл) добавляли триэтиламин (1,98 г, 19,6 ммоль), DMAP (2,39 г, 19,6 ммоль) и Boc2O (8,54 г, 39,1 ммоль). Полученную смесь перемешивали при 25°C в течение 1 ч, а затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1, 5/1) с получением указанного в заголовке соединения. MS: m/z=487,4 (M+1).
Стадия D: трет-Бутил-(5,6-бис(гидроксиметил)-2-фенилпиридин-3-ил)карбамат
К смеси диметил-5-((трет-бутоксикарбонил)амино)-6-фенилпиридин-2,3-дикарбоксилата (3,80 г, 7,81 ммоль) в THF (60 мл) при 0°C порциями добавляли LiAlH4 (1,19 г, 31,2 ммоль). Полученную смесь перемешивали при 0°C в течение 1 ч, и последовательно добавляли воду (1,2 мл) и 15% водный раствор гидроксида натрия (1,2 мл). Добавляли Mg2SO4 (5 г), смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=331,1 (M+1).
Стадия E: трет-Бутил-(6-(4-метоксибензил)-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)карбамат
К перемешанному раствору трет-бутил-(5,6-бис(гидроксиметил)-2-фенилпиридин-3-ил)карбамата (2,62 г, 7,93 ммоль) и Et3N (4,01 г, 39,7 ммоль) в DCM (50 мл) при 0°C по каплям добавляли MsCl (2,00 г, 17,5 ммоль). Полученную смесь перемешивали при 0°C в течение 30 мин, после чего добавляли раствор (4-метоксифенил)метанамина (1,31 г, 9,52 ммоль) в DCM (0,5 мл). Смесь нагревали до 25°C, после чего перемешивали ее в течение 16 ч, а затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=3/1) с получением указанного в заголовке соединения. MS: m/z=432,2 (M+1).
Стадия F: 2-Фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-амин
Смесь трет-бутил-(6-(4-метоксибензил)-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)карбамата (200 мг, 0,463 ммоль) в TFA (5 мл) нагревали при 110°C в течение 50 мин в условиях обработки микроволновым излучением. Реакционную смесь концентрировали с получением указанного в заголовке соединения. MS: m/z=212,2 (M+1).
Стадия G: трет-Бутил-3-амино-2-фенил-5H-пирроло[3,4-b]пиридин-6(7H)-карбоксилат
К раствору 2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-амина (100 мг, 0,473 ммоль) в DCM (2 мл) добавляли TEA (0,066 мл, 0,47 ммоль) и Boc2O (0,110 мл, 0,473 ммоль). Полученную смесь перемешивали при 25°C в течение 1 ч, а затем концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=312,2 (M+1).
Схема реакций для промежуточного продукта A43
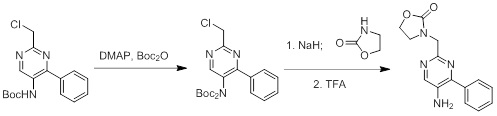
Промежуточный продукт A43
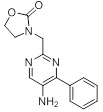
3-((5-Амино-4-фенилпиримидин-2-ил)метил)оксазолидин-2-он
Стадия A: Ди-трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамат
Раствор трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (573 мг, 1,79 ммоль), DMAP (438 мг, 3,58 ммоль) и Boc2O (0,832 мл, 3,58 ммоль) в CH2Cl2 (18 мл) перемешивали при 25°C в течение 1 ч. Содержащую продукт смесь распределяли между водой и 1,2-дихлорэтаном (2×). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали методом флэш-хроматографии (картридж 120 г SiO2), элюируя EtOAc в гексанах (0%→40%), с получением указанного в заголовке соединения. MS: m/z=420 (M+1).
Стадия B: 3-((5-Амино-4-фенилпиримидин-2-ил)метил)оксазолидин-2-он
К раствору ди-трет-бутил-(2-(хлорметил)-4-фенилпиримидин-5-ил)карбамата (147 мг, 0,350 ммоль) и 2-оксазолидона (305 мг, 3,50 ммоль) в DMF (4 мл) при 23°C добавляли NaH (60 масс.%, 140 мг, 3,50 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем распределяли между насыщенным водным раствором NH4Cl и этилацетатом (2×). Объединенные экстракты сушили над Na2SO4, фильтровали и концентрировали. Смесь остатка и TFA (0,027 мл, 0,35 ммоль) в DCM (4 мл) перемешивали при 23°C в течение 1 ч, а затем концентрировали. Остаток распределяли между насыщенным водным раствором бикарбоната натрия и DCM (2×). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Этот остаток очищали методом флэш-хроматографии (картридж 40 г SiO2), элюируя MeOH/CH2Cl2 (0%→15%), с получением указанного в заголовке соединения. MS: m/z=271 (M+1).
Схема реакций для промежуточного продукта B1
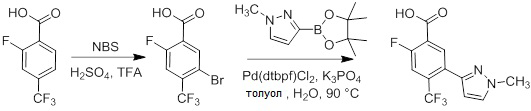
Промежуточный продукт B1
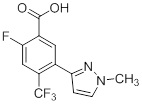
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
Стадия A: 5-Бром-2-фтор-4-(трифторметил)бензойная кислота
N-Бромсукцинимид (23,1 г, 130 ммоль) при 50°C добавляли порциями к смеси 2-фтор-4-(трифторметил)бензойной кислоты (15,0 г, 72,1 ммоль), серной кислоты (9,0 мл, 170 ммоль, 18 M) и TFA (50,0 мл, 650 ммоль), и перемешивали полученную смесь при 50°C в течение 18 ч. Дополнительно добавляли N-бромсукцинимид (3,0 г, 16 ммоль), и перемешивали смесь при 50°C в течение 4 ч. Реакционную смесь охлаждали, и добавляли воду (150 мл). Полученный осадок собирали и сушили с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 9,90 (с, 1H), 8,35 (д, J=6,3 Гц, 1H), 7,55 (д, J=10,3 Гц, 1H).
Стадия B: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
К дезоксигенированной смеси 5-бром-2-фтор-4-(трифторметил)бензойной кислоты (5,0 г, 17 ммоль), 1-(метил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,35 г, 20,9 ммоль) и K3PO4 (11,1 г, 52,3 ммоль) в толуоле (55 мл) и H2O (7 мл) добавляли 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия дихлорид (1,14 г, 1,74 ммоль). Полученную смесь нагревали при 90°C в течение 2 ч, а затем перемешивали при 50°C в течение 18 ч. Смесь охлаждали и фильтровали. Фильтрат концентрировали, и распределяли остаток между водой (200 мл) и EtOAc (300 мл). Водный слой подкисляли до pH=5 добавлением водного раствора HCl (1н), полученный осадок собирали и сушили с получением указанного в заголовке соединения. MS: m/z=289 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 13,85 (с, 1H), 8,11 (д, 1H), 7,82 (м, 2H), 6,45 (с, 1H), 3,92 (с, 3H).
Следующий промежуточный продукт получали способом, аналогичным описанной выше методике.
№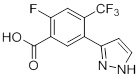
Промежуточный продукт B3
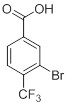
3-Бром-4-(трифторметил)бензойная кислота
Стадия A: Метил-3-бром-4-(трифторметил)бензоат
t-BuONO (79,0 г, 765 ммоль) при 0°C добавляли к раствору метил-3-амино-4-(трифторметил)бензоата (67,0 г, 306 ммоль) и CuBr (88,0 г, 612 ммоль) в ацетонитриле (1000 мл), полученную смесь нагревали до 25°C и перемешивали в течение 12 ч. Смесь затем вливали в EtOAc (600 мл) и фильтровали. Фильтрат промывали водным раствором HCl (1M, 200 мл×3), а затем солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EA=200:1) с получением указанного в заголовке соединения. MS: m/z=283, 285 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1H), 8,06 (д, J=8,0 Гц, 1H), 7,77 (д, J=8,0 Гц, 1H), 3,97 (с, 3H).
Стадия B: 3-Бром-4-(трифторметил)бензойная кислота
Смесь метил-3-бром-4-(трифторметил)бензоата (5,0 г, 17,7 ммоль) в водном растворе NaOH (1M, 100 мл) перемешивали при 25°C в течение 12 ч. Смесь подкисляли до pH=6 добавлением водного раствора HCl (1M), и экстрагировали полученную водную смесь EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=270 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1H), 8,14 (д, J=8,0 Гц, 1H), 7,83 (д, J=8,0 Гц, 1H).
Схема реакций для промежуточного продукта B4
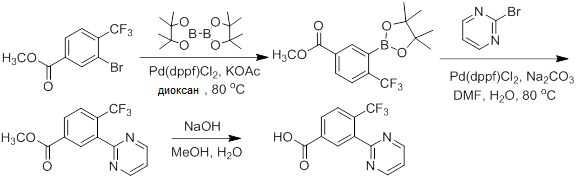
Промежуточный продукт B4
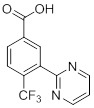
3-(Пиримидин-2-ил)-4-(трифторметил)бензоилхлорид
Стадия A: Метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)бензоат
К дезоксигенированной смеси метил-3-бром-4-(трифторметил)бензоата (20,0 г, 70,7 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (26,9 г, 106 ммоль) и ацетата калия (20,8 г, 212 ммоль) в диоксане (300 мл) добавляли PdCl2(dppf) (2,59 г, 3,50 ммоль), и нагревали полученную смесь при 80°C в течение 5 ч. Смесь охлаждали и фильтровали. Фильтрат концентрировали, и распределяли остаток между водой (100 мл) и EtOAc (200 мл). Органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=15:1) с получением указанного в заголовке соединения. MS: m/z=331 (M+1).
Стадия B: Метил-3-(пиримидин-2-ил)-4-(трифторметил)бензоат
К дезоксигенированной смеси метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)бензоата (12,0 г, 36,4 ммоль), 2-бромпиримидина (8,67 г, 54,5 ммоль) и карбоната натрия (11,6 г, 109 ммоль) в DMF (450 мл) и воде (60 мл) добавляли PdCl2(dppf) (1,3 г, 1,8 ммоль), и нагревали полученную смесь при 80°C в течение 5 ч. Смесь охлаждали и фильтровали. Фильтрат концентрировали, и распределяли остаток между водой (100 мл) и EtOAc (200 мл). Объединенный органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=5:1) с получением указанного в заголовке соединения. MS: m/z=283 (M+1).
Стадия C: 3-(Пиримидин-2-ил)-4-(трифторметил)бензойная кислота
Смесь метил-3-(пиримидин-2-ил)-4-(трифторметил)бензоата (7,0 г, 25 ммоль) и NaOH (3,0 г, 74 ммоль) в смеси MeOH и H2O (3:1, 120 мл) нагревали при 30°C в течение 16 ч. Реакционную смесь охлаждали, а затем распределяли между водой (30 мл) и MTBE (2×60 мл). Водный слой подкисляли до pH=4 добавлением водного раствора HCl (2н). Осадок фильтровали, промывали водой и сушили с получением указанного в заголовке соединения. MS: m/z=269 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,92 (д, J=5,0 Гц, 1H), 8,30 (м, 2H), 7,97 (д, J=8,0 Гц, 1H), 7,55 (т, J=4,9 Гц, 1H).
Следующие промежуточные продукты получали способом, аналогичным описанной выше методике.
№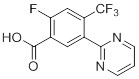
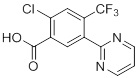
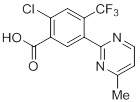
Схема реакций для промежуточного продукта B8
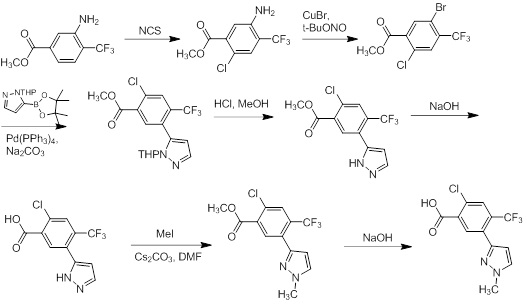
Промежуточный продукт B8
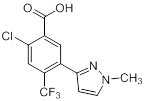
2-Хлор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
Стадия A: Метил-5-амино-2-хлор-4-(трифторметил)бензоат
N-Хлорсукцинимид (8,2 г, 61 ммоль) добавляли к раствору метил-3-амино-4-(трифторметил)бензоата (13,2 г, 60,0 ммоль) в ацетонитриле (200 мл), и нагревали полученную смесь при 80°C в течение 20 ч. После охлаждения, смесь распределяли между водой (500 мл) и EtOAc (2×300 мл). Объединенные органические слои промывали солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=6:1) с получением указанного в заголовке соединения. MS: m/z=254 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,49 (с, 1H), 7,17 (с, 1H), 3,92 (с, 3H).
Стадия B: Метил-5-бром-2-хлор-4-(трифторметил)бензоат
трет-Бутилнитрил (4,60 г, 44,5 ммоль) и метил-5-амино-2-хлор-4-(трифторметил)бензоат (4,50 г, 17,8 ммоль) добавляли порциями к суспензии бромида меди(I) (5,10 г, 35,6 ммоль) в DCM (100 мл). Полученную смесь нагревали при 60°C в течение 2 ч. После охлаждения, смесь разбавляли водой (50 мл) и водным раствором HCl (2M, 50 мл), а затем экстрагировали EtOAc (80 мл×2). Объединенные органические слои промывали водой (100 мл), затем солевым раствором (80 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (PE:EtOAc=50:1→30:1) с получением указанного в заголовке соединения. MS: m/z=319 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,15 (с, 1H), 7,77 (с, 1H), 3,97 (с, 3H).
Стадия C: Метил-2-хлор-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензоат
К дезоксигенированной смеси метил-5-бром-2-хлор-4-(трифторметил)бензоата (4,6 г, 14 ммоль), 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,86 г, 17,5 ммоль) и Na2CO3 (4,0 г, 44 ммоль) в DMF (150 мл) и H2O (24 мл) добавляли Pd(PPh3)4 (686 мг, 0,58 ммоль). Полученную смесь нагревали при 80°C в течение 5 ч, а затем охлаждали и фильтровали. Фильтрат концентрировали, и распределяли остаток между водой (200 мл) и EtOAc (300 мл). Органический слой промывали солевым раствором (100 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=389 (M+1).
Стадия D: Метил-2-хлор-5-(1H-пиразол-5-ил)-4-(трифторметил)бензоат
Раствор метил-2-хлор-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензоата (2,5 г, 6,4 ммоль) в растворе HCl в MeOH (4M, 50 мл) перемешивали при 15°C в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=305 (M+1).
Стадия E: 2-Хлор-5-(1H-пиразол-5-ил)-4-(трифторметил)бензойная кислота
Раствор NaOH (1,2 г, 0,030 моль) в H2O (15 мл) добавляли к раствору метил-2-хлор-5-(1H-пиразол-5-ил)-4-(трифторметил)бензоата (2,3 г, 7,6 ммоль) в MeOH (45 мл), и перемешивали полученную смесь при 15°C в течение 16 ч. Большую часть MeOH удаляли в условиях пониженного давления, и распределяли оставшуюся водную смесь между MTBE (50 мл) и водой (50 мл). Водный слой подкисляли до pH=5 добавлением водного раствора HCl (3н). Осадок фильтровали, промывали водой (50 мл×2) и сушили с получением указанного в заголовке соединения. MS: m/z=291 (M+1).
Стадия F: Метил-2-хлор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензоат и метил-2-хлор-5-(1-метил-1H-пиразол-5-ил)-4-(трифторметил)бензоат
Смесь 2-хлор-5-(1H-пиразол-5-ил)-4-(трифторметил)бензойной кислоты (500 мг, 1,72 ммоль), Cs2CO3 (1,7 г, 5,2 ммоль) и йодметана (0,54 мл, 8,6 ммоль) в DMF (15 мл) нагревали при 80°C в течение 2 ч. Реакционную смесь охлаждали и фильтровали, и концентрировали фильтрат. Остаток распределяли между водой (50 мл) и EtOAc (30 мл×3). Объединенные органические слои промывали H2O (50 мл×3), затем солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=319 (M+1).
Стадия G: 2-Хлор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
Раствор NaOH (414 мг, 10,4 ммоль) в H2O (5 мл) добавляли к смеси метил-2-хлор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензоата и метил-2-хлор-5-(1-метил-1H-пиразол-5-ил)-4-(трифторметил)бензоата (550 мг, 3,5 ммоль) в MeOH (15 мл). Полученную смесь перемешивали при 15°C в течение 16 ч. Большую часть MeOH удаляли в условиях пониженного давления, и распределяли полученный водный раствор между MTBE (30 мл) и водой (30 мл). Водный слой подкисляли до pH=4 добавлением водного раствора HCl (3н). Полученную суспензию затем экстрагировали EtOAc (50 мл×2). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали. Остаток перекристаллизовывали из MeOH (1 г/5 мл) с получением указанного в заголовке соединения. MS: m/z=305 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,36 (с, 1H), 7,86 (с, 1H), 7,48 (д, J=2,3 Гц, 1H), 6,59 (с, 1H), 4,15 (с, 3H).
Следующие промежуточные продукты получали аналогичным образом с использованием соответствующегго трибутилстаннанового реагента в катализируемой палладием реакции кросс-сочетания.
№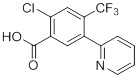
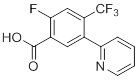
Промежуточный продукт B11
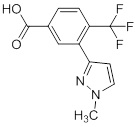
3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
Стадия A: 4-Бром-3-нитробензойная кислота
4-Бромбензойную кислоту (100 г, 0,5 моль) добавляли порциями к водному раствору HNO3 (16M, 200 мл), поддерживая температуру между 0 и 25°C, а затем при температуре окружающей среды по каплям добавляли водный раствор H2SO4 (18M, 240 мл). Полученную смесь перемешивали при температуре окружающей среды в течение 4 ч, а затем осторожно разбавляли 1,5 л воды. Осадок фильтровали, промывали водой и сушили с получением указанного в заголовке соединения. MS: m/z=246,0, 248,0 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 8,42 (с, 1H), 8,04 (с, 2H).
Стадия B: Метил-4-бром-3-нитробензоат
К раствору 4-бром-3-нитробензойной кислоты (115 г, 47,0 ммоль) в MeOH (600 мл) при температуре окружающей среды добавляли водный раствор H2SO4 (18M, 200 мл). Смесь нагревали с обратным холодильником в течение 2 ч, а затем охлаждали и фильтровали. Отфильтрованное твердое вещество промывали водой и сушили с получением указанного в заголовке соединения. MS: m/z=260, 262 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,48 (с, 3H), 8,09 (с, 2H), 3,91 (с, 3H).
Стадия C: Метил-3-нитро-4-(трифторметил)бензоат
К раствору метил-4-бром-3-нитробензоата (175 г, 0,670 моль) в безводном DMF (1,0 л) в атмосфере N2 добавляли CuI (140 г, 0,73 моль). После перемешивания при температуре окружающей среды в течение 10 мин, добавляли FSO2CF2CO2CH3 (185 мл, 0,730 моль), и нагревали вентилируемую смесь при 110°C в течение 3 ч до завершения процесса выделения газа. Смесь затем охлаждали и фильтровали через Celite®, промывая EtOAc. Фильтрат концентрировали, и распределяли остаток между водой (400 мл) и MTBE. Органический слой промывали водой, затем солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток перекристаллизовывали из DCM/MeOH (5/1) с получением указанного в заголовке соединения. Маточный раствор концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=20/1) с получением дополнительного количества указанного в заголовке соединения. MS: m/z=250,0 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,55 (ушир. с, 1H), 8,39 (д, J=7,5 Гц, 1H), 8,19 (д, J=8,0 Гц, 1H), 3,88-3,99 (м, 3H).
Стадия D: Метил-3-амино-4-(трифторметил)бензоат
Раствор метил-3-нитро-4-(трифторметил)бензоата (102 г, 0,410 моль) и 10% Pd/C (10 г, 10 масс.%) в MeOH (1,0 л) перемешивали в атмосфере H2 (35 фунт./кв.дюйм) при 30°C в течение 12 ч. Суспензию фильтровали через Celite®, промывая MeOH (30 мл×3). Фильтрат концентрировали с получением указанного в заголовке соединения. MS: m/z=220,0 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 7,40-7,50 (м, 2H), 7,09-7,15 (м, 1H), 5,92 (с, 2H), 3,82 (с, 3H).
Стадия E: Метил-3-бром-4-(трифторметил)бензоат
Метил-3-амино-4-(трифторметил)бензоат (40 г, 180 ммоль) при 0°C добавляли порциями к суспензии CuBr (53,0 г, 365 ммоль) и t-BuONO (47 г, 460 ммоль) в ацетонитриле (600 мл). Полученную смесь перемешивали при 0°C в течение 2 ч, а затем нагревали до 25°C и перемешивали в течение 16 ч. Смесь распределяли между EtOAc и водным раствором HCl (1M, 200 мл×4). Органический слой промывали солевым раствором (200 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=200/1) с получением указанного в заголовке соединения. MS: m/z=283, 285 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1H), 8,06 (д, J=8,0 Гц, 1H), 7,77 (д, J=8,0 Гц, 1H), 3,97 (с, 3H).
Стадия F: Метил-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензоат
Дезоксигенированную смесь метил-3-бром-4-(трифторметил)бензоата (5,0 г, 17 ммоль), 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (5,9 г, 21 ммоль), Pd(PPh3)4 (0,80 г, 0,69 ммоль) и водно8го раствора Na2CO3 (2M, 26 мл, 53 ммоль) в DMF (150 мл) нагревали при 70°C в атмосфере N2 в течение 2 ч. Смесь концентрировали, и распределяли остаток между EtOAc (200 мл) и водой (100 мл). Органический слой промывали солевым раствором (100 мл), затем сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=10/1) с получением указанного в заголовке соединения. MS: m/z=355,0 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 8,37 (с, 1H), 8,06 (д, J=8,0 Гц, 1H), 7,77 (д, J=8,0 Гц, 1H), 3,97 (с, 3H).
Стадия G: Метил-3-(1H-пиразол-5-ил)-4-(трифторметил)бензоат
К раствору метил-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензоата (5,0 г, 14 ммоль) в MeOH (100 мл) добавляли раствор HCl в MeOH (40 мл, 4 M). Смесь перемешивали при 10°C в течение 0,5 ч, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=271,0 (M+1).
Стадия H: Метил-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензоат и метил-3-(1-метил-1H-пиразол-5-ил)-4-(трифторметил)бензоат
К раствору метил-3-(1H-пиразол-5-ил)-4-(трифторметил)бензоата (7,0 г, 26 ммоль) в DMF (150 мл) добавляли Cs2CO3 (17 г, 52 ммоль) и CH3I (4,8 мл, 78 ммоль). Реакционную смесь нагревали при 80°C в течение 2 ч, а затем охлаждали и концентрировали. Остаток распределяли между водой (150 мл) и EtOAc (100 мл×3). Объединенные органические слои промывали солевым раствором (150 мл), сушили над Na2SO4 и концентрировали с получением смеси метил-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензоата и метил-3-(1-метил-1H-пиразол-5-ил)-4-(трифторметил)бензоата. MS: m/z=285,0 (M+1).
Стадия I: 3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
К раствору метил-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензоата и метил-3-(1-метил-1H-пиразол-5-ил)-4-(трифторметил)бензоата (6,5 г, 23 ммоль) в MeOH (100 мл) добавляли водный раствор NaOH (35 мл, 2 M). Смесь нагревали при 50°C в течение 50 мин, а затем охлаждали. Большую часть MeOH удаляли в условиях пониженного давления, и распределяли полученный водный раствор между EtOAc (100 мл) и водой (150 мл). Водный слой подкисляли до pH=5 добавлением водного раствора HCl (1н), а затем дополнительно экстрагировали EtOAc (150 мл×2). Объединенные органические слои промывали солевым раствором (150 мл), сушили над безводным Na2SO4 и концентрировали. Остаток очищали путем перекристаллизации из MeOH (1g/5 мл) с получением указанного в заголовке соединения. MS: m/z=271,0 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 13,43-13,68 (м, 1H) 8,18-8,24 (м, 1H), 8,05-8,12 (м, 1H), 7,92-7,99 (м, 1H), 7,77-7,84 (м, 1H), 6,43-6,52 (м, 1H), 3,93 (с, 3H).
Следующий промежуточный продукт получали способом, аналогичным описанной выше методике.
№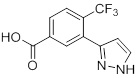
Промежуточный продукт B13
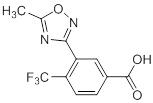
3-(5-Метил-1,2,4-оксадиазол-3-ил)-4-(трифторметил)бензойная кислота
Стадия A: Метил-3-циано-4-(трифторметил)бензоат
К смеси метил-3-амино-4-(трифторметил)бензоата (15 г, 0,073 моль) и водного раствора HCl (12M, 24 мл) в H2O (100 мл) при 0°C по каплям добавляли раствор NaNO2 (5,5 г, 0,080 моль) в H2O (30 мл). Реакционную смесь перемешивали при 0°C в течение 30 мин, а затем по каплям добавляли к взвеси CuCN (7,1 г, 0,080 моль) и KCN (8,4 г, 0,13 моль) в H2O (200 мл), поддерживая внутреннюю температуру 5-10°C. После завершения добавления, реакционную смесь нагревали при 80°C в течение 1 ч. Смесь охлаждали, и экстрагировали раствор EtOAc (200 мл×4). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (2% EtOAc в PE) с получением указанного в заголовке соединения. MS: m/z=230,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,46-8,53 (м, 1H), 8,33-8,42 (м, 1H), 7,87-7,95 (м, 1H), 4,01 (с, 3H).
Стадия B: Метил-3-(N'-гидроксикарбамимидоил)-4-(трифторметил)бензоат
К смеси метил-3-циано-4-(трифторметил)бензоата (1,6 г, 7,0 ммоль) и гидроксиламина гидрохлорида (0,98 г, 14 ммоль) в MeOH (20 мл) добавляли NaHCO3 (2,3 г, 28 ммоль). Полученную смесь нагревали при 85°C в течение 5 ч, а затем охлаждали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (40% EtOAc в PE) с получением указанного в заголовке соединения. MS: m/z=263,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,26 (с, 1H), 8,18-8,21 (д, J=8,4 Гц, 1H), 7,80-7,83 (д, J=8,0 Гц, 1H), 7,52 (с, 1H), 4,89 (с, 2H), 3,96 (с, 3H).
Стадия C: Метил-3-(N-ацетил-N'-гидроксикарбамимидоил)-4-(трифторметил)бензоат
К раствору метил-3-(N'-гидроксикарбамимидоил)-4-(трифторметил)бензоата (282 мг, 1,07 ммоль) и TEA (0,30 мл, 2,14 ммоль) в безводном DCM (20 мл) при 25°C добавляли AcCl (0,083 мл, 1,18 ммоль). Полученную смесь нагревали при 30°C в течение 20 мин, а затем охлаждали и концентрировали с получением указанного в заголовке соединения. MS: m/z=305,0 (M+1).
Стадия D: Метил-3-(5-метил-1,2,4-оксадиазол-3-ил)-4-(трифторметил)бензоат
Раствор метил-3-(N-ацетил-N'-гидроксикарбамимидоил)-4-(трифторметил)бензоата (0,28 г, 0,93 ммоль) в толуоле (10 мл) нагревали при 110°C в течение 2 ч, а затем охлаждали и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (30% EtOAc в PE) с получением указанного в заголовке соединения. MS: m/z=287,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,37-8,49 (м, 1H), 8,22-8,32 (м, 1H), 7,87-7,99 (м, 1H), 3,96 (с, 3H), 2,70 (с, 3H).
Стадия E: 3-(5-Метил-1,2,4-оксадиазол-3-ил)-4-(трифторметил)бензойная кислота
К раствору метил-3-(5-метил-1,2,4-оксадиазол-3-ил)-4-(трифторметил)бензоата (0,13 г, 0,45 ммоль) в MeOH (2,0 мл) добавляли водный раствор NaOH (2,0 мл, 1M). Полученную смесь нагревали при 50°C в течение 1 ч, а затем охлаждали и подкисляли до pH=5 добавлением водного раствора HCl (1M). Водную смесь экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=273,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,47 (с, 1H), 8,27 (д, J=8,0 Гц, 1H), 7,91 (д, J=8,8 Гц, 1H), 2,69 (с, 3H).
Промежуточный продукт B14

3-(1H-Пиразол-1-ил)-4-(трифторметил)бензойная кислота
Стадия A: Метил-3-(1H-пиразол-1-ил)-4-(трифторметил)бензоат
Смесь метил-3-бром-4-(трифторметил)бензоата (0,50 г, 1,8 ммоль), пиразола (0,18 г, 2,6 ммоль), Cs2CO3 (1,4 г, 4,4 ммоль), CuI (670 мг, 3,52 ммоль) и 1,10-фенантролина (0,13 г, 0,70 ммоль) в безводном толуоле (15 мл) нагревали при 140°C в течение 1 ч в условиях обработки микроволновым излучением. После охлаждения, реакционную смесь разбавляли EtOAc (50 мл) и фильтровали. Фильтрат концентрировали, и очищали остаток методом препаративной TLC (PE/EA=5/1) с получением указанного в заголовке соединения. MS: m/z=271,0 (M+1).
Стадия B: 3-(1H-Пиразол-1-ил)-4-(трифторметил)бензойная кислота
К раствору метил-3-(1H-пиразол-1-ил)-4-(трифторметил)бензоата (0,20 г, 0,74 ммоль) в MeOH (15 мл) добавляли водный раствор NaOH (3,0 мл, 2 M). Смесь нагревали при 50°C в течение 10 мин. Большую часть MeOH удаляли в условиях пониженного давления, и распределяли полученный водный раствор между EtOAc (30 мл) и водой (20 мл). Водный слой подкисляли до pH=5 добавлением водного раствора HCl (1M), а затем экстрагировали EtOAc (30 мл×2). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=257,0 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 8,19 (м, 1H), 8,13 (м, 1H), 8,07 (м, 1H), 7,97 (м, 1H), 7,78 (м, 1H), 6,55 (м, 1H).
Промежуточный продукт B15
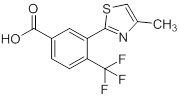
3-(4-Метилтиазол-2-ил)-4-(трифторметил)бензойная кислота
Стадия A: 3-Амино-4-(трифторметил)бензойная кислота
Смесь 3-нитро-4-(трифторметил)бензойной кислоты (1,0 г, 4,3 ммоль) и 10% Pd/C (0,20 г, 5 масс.%) в MeOH (20 мл) перемешивали в атмосфере H2 (15 фунт./кв.дюйм) при температуре окружающей среды в течение 12 ч. Катализатор фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=206,0 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 7,46 (с, 1H), 7,38-7,45 (м, 1H), 7,13 (д, J=8,3 Гц, 1H), 5,84 (с, 2H).
Стадия B: Метил-3-амино-4-(трифторметил)бензоат
Смесь 3-амино-4-(трифторметил)бензойной кислоты (3,4 г, 16 ммоль) и водного раствора H2SO4 (18M, 2,0 мл) в MeOH (20 мл) нагревали с обратным холодильником до расходования исходного вещества. Смесь охлаждали, а затем нейтрализовали до pH=7 добавлением водного раствора NaOH (1н). Водную смесь экстрагировали EtOAc (10 мл×3), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=220,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 7,46-7,52 (м, 1H), 7,42 (с, 2H), 4,30 (ушир. с, 2H), 3,92 (с, 3H).
Стадия C: Метил-3-циано-4-(трифторметил)бензоат
К смеси метил-3-амино-4-(трифторметил)бензоата (3,2 г, 15 ммоль) и водного раствора HCl (12M, 3,5 мл) в воде (20 мл) при 5°C по каплям добавляли раствор NaNO2 (1,2 г, 17 ммоль) в воде (7,0 мл). Полученную смесь перемешивали в течение 30 мин при 5°C, а затем по каплям добавляли взвесь CuCN (1,3 г, 15 ммоль) и KCN (1,6 г, 25 ммоль) в воде (4 мл), поддерживая внутреннюю температуру 5-10°C. Смесь перемешивали при 10°C в течение 30 мин, а затем нагревали при 80°C в течение 1 ч. После охлаждения, смесь экстрагировали DCM (30 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=230 (M +1). 1H-ЯМР (400 МГц, CDCl3) δ 8,45-8,53 (м, 1H), 8,33-8,40 (м, 1H), 7,91 (д, 1H, J=8,5 Гц), 4,01 (с, 3H).
Стадия D: Метил-3-карбамотиоил-4-(трифторметил)бензоат
Раствор метил-3-циано-4-(трифторметил)бензоата (0,10 г, 0,61 ммоль) и TEA (0,20 мл, 1,4 ммоль) в пиридине (10 мл) при температуре окружающей среды в течение 30 мин барботировали газообразным H2S. Смесь концентрировали, и распределяли остаток между водой и EtOAc (10 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=5:1) с получением указанного в заголовке соединения. MS: m/z=264,0 (M +1). 1H-ЯМР (400 МГц, CDCl3) δ 8,25-8,31 (м, 1H), 8,09-8,17 (м, 1H), 7,75 (д, J=8,0 Гц, 1H), 4,45-4,68 (м, 2H), 3,96 (с, 3H).
Стадия E: Метил-3-(4-гидрокси-4-метил-4,5-дигидротиазол-2-ил)-4-(трифторметил)бензоат
Смесь метил-3-карбамотиоил-4-(трифторметил)бензоата (100 мг, 0,38 ммоль), TEA (0,20 мл, 1,4 ммоль) и 1-хлорпропан-2-она (0,033 мл, 0,42 ммоль) в DMF (3,0 мл) нагревали при 120°C в течение 4 ч, а затем концентрировали. Остаток распределяли между водой и EtOAc (10 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EA=3:1) с получением указанного в заголовке соединения. MS: m/z=320,0 (M +1).
Стадия F: 3-(4-Метилтиазол-2-ил)-4-(трифторметил)бензойная кислота
Раствор метил-3-(4-гидрокси-4-метил-4,5-дигидротиазол-2-ил)-4-(трифторметил)бензоата в водном растворе NaOH (1M, 10 мл) перемешивали при температуре окружающей среды в течение 8 ч. Смесь подкисляли до pH=5 добавлением водного раствора HCl (1M), а затем экстрагировали EtOAc (10 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=288,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,23-8,34 (м, 1H), 8,06-8,17 (м, 1H), 7,68-7,83 (м, 1H), 6,97-7,10 (м, 1H), 2,50 (с, 3H).
Промежуточный продукт B16
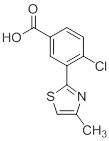
4-Хлор-3-(4-метилтиазол-2-ил)бензойная кислота
Стадия A: Метил-4-хлор-3-цианобензоат
К смеси метил-3-амино-4-хлорбензоата (10 г, 54 ммоль) и водного раствора HCl (12M, 15 мл) в воде (80 мл) при 0°C по каплям добавляли раствор NaNO2 (4,5 г, 60 ммоль) в воде (18 мл). Реакционную смесь перемешивали в течение 30 мин при 0°C, а затем по каплям добавляли к взвеси CuCN (4,9 г, 54 ммоль) и KCN (6,0 г, 92 ммоль) в воде (40 мл), поддерживая температуру 5-10°C. Реакционную смесь перемешивали при 10°C в течение 30 мин, а затем нагревали при 80°C в течение 1 ч. После охлаждения, смесь экстрагировали DCM. Органический слой промывали солевым раствором, сушили над Na2SO4, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=196,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,34 (д, J=2,0 Гц, 1H), 8,17-8,20 (м, 1H), 7,61 (д, J=8,4 Гц, 1H), 3,96 (с, 3H).
Стадия B: Метил-3-карбамотиоил-4-хлорбензоат
Раствор метил-4-хлор-3-цианобензоата (3,0 г, 15 ммоль) и TEA (2,13 мл, 15,3 ммоль) в пиридине (15 мл) при температуре окружающей среды в течение 1 ч барботировали газообразным H2S. Реакционную смесь концентрировали, и очищали остаток методом колоночной хроматографии (PE:EtOAc=10:1) с получением указанного в заголовке соединения. MS: m/z=230,0 (M +1). 1H-ЯМР (400 МГц, CDCl3) δ 8,29 (д, J=1,6 Гц, 1H), 7,95-7,97 (м, 2H), 7,45 (д, J=8,4 Гц, 1H), 7,26 (с, 1H), 3,92 (с, 3H).
Стадия C: Метил-4-хлор-3-(4-метилтиазол-2-ил)бензоат
Смесь метил-3-карбамотиоил-4-(трифторметил)бензоата (1,0 г, 4,3 ммоль), TEA (0,20 мл, 1,4 ммоль) и 1-хлорпропан-2-она (0,80 г, 8,6 ммоль) в DMF (10 мл) нагревали при 120°C в течение 4 ч, а затем концентрировали. Остаток распределяли между водой и EtOAc (10 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=3:1) с получением указанного в заголовке соединения. MS: m/z=268,0 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,29 (д, J=2,0 Гц, 1H), 7,97-8,00 (м, 1H), 7,76 (д, J=8,0 Гц, 1H), 7,09 (с, 1H), 3,92 (с, 3H), 2,56 (с, 3H).
Стадия D: 4-Хлор-3-(4-метилтиазол-2-ил)бензойная кислота
Смесь метил-4-хлор-3-(4-метилтиазол-2-ил)бензоата (0,40 г, 2,0 ммоль) в водном растворе NaOH (1M, 10 мл) перемешивали при температуре окружающей среды в течение 8 ч. Смесь подкисляли до pH=5 добавлением водного раствора HCl (2M), а затем экстрагировали EtOAc (10 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=254,0 (M+1).
Промежуточный продукт B17
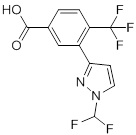
3-(1-(Дифторметил)-1H-пиразол-3-ил)-4-(трифторметил)бензойная кислота
Раствор метил-3-(1H-пиразол-5-ил)-4-(трифторметил)бензоата (50 мг, 0,18 ммоль), хлордифторацетата натрия (34 мг, 0,22 ммоль) и 18-краун-6 (9,8 мг, 0,037 ммоль) в ацетонитриле (1 мл) нагревали с обратным холодильником в течение 40 ч. Спустя 18 и 22 ч, дополнительно добавляли хлордифторацетат натрия (34 мг, 0,22 ммоль). Реакционную смесь охлаждали до температуры окружающей среды, и добавляли водный раствор NaOH (10M, 0,056 мл, 0,55 ммоль). Полученную смесь нагревали при 50°C в течение 2 ч. Смесь охлаждали, а затем фильтровали, промывая ацетонитрилом (1 мл) и DMF (1 мл). Фильтрат очищали методом HPLC с обращенной фазой (5-95% ацетонитрил + 0,1% трифторуксусная кислота в воде) с получением указанного в заголовке соединения. MS: m/z=307,0 (M+1).
Промежуточный продукт B18

3-(3-Метил-1H-пиразол-1-ил)-4-(трифторметил)бензойная кислота
Дезоксигенированный раствор 3-метил-1H-пиразола (0,120 мл, 1,49 ммоль), 3-бром-4-(трифторметил)бензойной кислоты (0,20 г, 0,74 ммоль), йодида меди(I) (28 мг, 0,15 ммоль), карбоната цезия (0,48 г, 1,5 ммоль) и транс-N,N'-диметилциклогексан-1,2-диамина (0,023 мл, 0,15 ммоль) в диоксане (1,0 мл) нагревали с обратным холодильником в течение 18 ч. Смесь охлаждали и фильтровали, промывая DMF (1,5 мл). Фильтрат очищали методом HPLC с обращенной фазой (5-95% ацетонитрил + 0,1% трифторуксусная кислота в воде) с получением указанного в заголовке соединения. MS: m/z=271,0 (M+1).
Промежуточный продукт B19

3-(4-Метилоксазол-2-ил)-4-(трифторметил)бензойная кислота
Дезоксигенированную смесь 3-бром-4-(трифторметил)бензойной кислоты (100 мг, 0,372 ммоль), 4-метилоксазола (0,061 мл, 0,74 ммоль), аддукта хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладия(II) и метил-трет-бутилового эфира (15,4 мг, 0,019 ммоль) и трет-бутоксид натрия (107 мг, 1,12 ммоль) в DMA (1,5 мл) нагревали при 110°C в условиях обработки микроволновым излучением в течение 18 ч. Смесь охлаждали и фильтровали, и очищали фильтрат методом HPLC с обращенной фазой (колонка C18, H2O:CH3CN:CF3CO2H=95:5:0,1→5:95:0,1) с получением указанного в заголовке соединения. MS: m/z=272,0 (M+1).
Промежуточный продукт B20
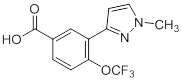
3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметокси)бензойная кислота
Стадия A: 3-Нитро-4-(трифторметокси)бензойная кислота
4-(Трифторметокси)бензойную кислоту (37,4 г, 0,181 моль) при 25°C порциями добавляли к водному раствору HNO3 (15M, 75 мл). Добавляли водный раствор H2SO4 (18M, 90 мл), и перемешивали полученную смесь в течение 18 ч. Реакционную смесь осторожно разбавляли водой (300 мл), осадок фильтровали, промывали водой и сушили с получением указанного в заголовке соединения. MS: m/z=252 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,54 (с, 1H), 8,32 (д, J=8,0 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H).
Стадия B: Метил-3-нитро-4-(трифторметокси)бензоат
Водный раствор H2SO4 (18M, 60 мл) при 0°C по каплям добавляли к раствору 3-нитро-4-(трифторметокси)бензойной кислоты (33,5 г, 0,135 моль) в MeOH (400 мл). Полученную смесь нагревали при 80°C в течение 2 ч, а затем охлаждали и концентрировали. Остаток разбавляли EtOAc и промывали водой (100 мл×3), водным раствором NaHCO3 (100 мл×3) и солевым раствором. Органический слой сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z: 266 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,54 (с, 1H), 8,32 (д, J=8,0 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 3,90 (с, 3H).
Стадия C: Метил-3-амино-4-(трифторметокси)бензоат
Смесь метил-3-нитро-4-(трифторметокси)бензоата (14 г, 0,053 моль) и 10% Pd/C (1,0 г, 10 масс.%) в MeOH (200 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 15°C в течение 24 ч. Суспензию фильтровали, и концентрировали фильтрат. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=5:1) с получением указанного в заголовке соединения. MS: m/z=236 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 7,47 (д, J=2,0 Гц, 1H), 7,19-7,25 (м, 1H), 7,11-7,17 (м, 1H), 5,71 (с, 2H), 3,82 (с, 3H).
Стадия D: Метил-3-бром-4-(трифторметокси)бензоат
Смесь CuBr (5,0 г, 34 ммоль) и t-BuONO (5,0 г, 43 ммоль) в ацетонитриле (60 мл) перемешивали при 0°C в течение 15 мин, а затем добавляли метил-3-амино-4-(трифторметокси)бензоат (4,0 г, 17 ммоль). Полученную смесь перемешивали при 0°C в течение 2 ч, а затем перемешивали при 15°C в течение 16 ч. Смесь фильтровали, и промывали осадок на фильтре EtOAc. Фильтрат промывали добавлением водного раствора HCl (1н), водой, а затем солевым раствором. Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc=20:1) с получением указанного в заголовке соединения. MS: m/z=298/300 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 8,14 (д, J=2,0 Гц, 1H), 7,96 (дд, J=8,7, 1,9 Гц, 1H), 7,55 (дд, J=8,7, 1,1 Гц, 1H), 3,84 (с, 3H).
Стадия E: Метил-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифтор-метокси)бензоат
Дезоксигенированную смесь метил-3-бром-4-(трифторметокси)бензоата (500 мг, 1,67 ммоль), 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (510 мг, 1,84 ммоль), Pd(PPh3)4 (50 мг, 0,05 ммоль) и Na2CO3 (530 мг, 5,0 ммоль) в DMF (5 мл) нагревали при 100°C в атмосфере N2 в течение 16 ч. Реакционную смесь охлаждали, а затем распределяли между водой (15 мл) и EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE:EtOAc=3:1) с получением указанного в заголовке соединения. MS: m/z=371 (M+1).
Стадия F: Метил-3-(1H-пиразол-5-ил)-4-(трифторметокси)бензоат
Раствор HCl в EtOAc (4M, 10 мл, 40 ммоль) добавляли к раствору метил-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметокси)бензоата (300 мг, 1,1 ммоль) в EtOAc (2 мл). Полученную смесь перемешивали при 15°C в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=287 (M+1).
Стадия G: Метил-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметокси)бензоат
Смесь метил-3-(1H-пиразол-5-ил)-4-(трифторметокси)бензоата (220 мг, 0,81 ммоль), CH3I (0,292 мл, 4,00 ммоль) и Cs2CO3 (780 мг, 2,4 ммоль) в DMF (5 мл) нагревали при 70°C в течение 1 ч. Смесь охлаждали, а затем распределяли между водой (10 мл) и EtOAc (10 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE:EtOAc=2:1) с получением указанного в заголовке соединения. MS: m/z=301 (M+1).
Стадия H: 3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметокси)бензойная кислота
Смесь метил-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметокси)бензоата (120 мг, 0,4 ммоль) и водного раствора NaOH (2M, 10 ммоль, 5 мл) нагревали при 50°C в течение 30 мин. Реакционную смесь охлаждали, подкисляли до pH=5 добавлением водного раствора HCl (1M), а затем экстрагировали EtOAc (10 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=287 (M+1).
Промежуточный продукт C1
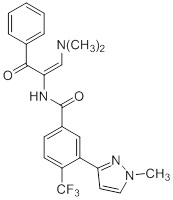
N-(1-(Диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: 3-(1-Метил-1H-пиразол-3-ил)-N-(2-оксо-2-фенилэтил)-4-(трифторметил)бензамид
Смесь 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (500 мг, 1,85 ммоль), 2-аминоацетофенона гидрохлорида (349 мг, 2,04 ммоль), HATU (844 мг, 2,22 ммоль) и DIEA (0,970 мл, 5,55 ммоль) в DMF (4 мл) перемешивали при температуре окружающей среды в течение 2 ч. Содержащую продукт смесь очищали непосредственно методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=388,2 (M+1).
Стадия B: N-(1-(Диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь 3-(1-метил-1H-пиразол-3-ил)-N-(2-оксо-2-фенилэтил)-4-(трифторметил)бензамида (88,0 мг, 0,227 ммоль) и N,N-диметилформамида диметилацеталя (0,036 мл, 0,27 ммоль) в диоксане (1 мл) перемешивали при температуре окружающей среды в течение 16 ч. Содержащую продукт смесь очищали непосредственно методом колоночной хроматографии на силикагеле (DCM:MeOH:NH4OH=100:0:0→90:10:1) с получением указанного в заголовке соединения. MS: m/z=443,2 (M+1).
Промежуточный продукт C2
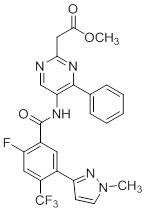
Метил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)ацетат
Смесь метил-2-(5-амино-4-фенилпиримидин-2-ил)ацетата (5,00 г, 20,6 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (5,92 г, 20,6 ммоль) растворяли в пиридине (68 мл), и охлаждали полученный раствор до 0°C. По каплям добавляли оксихлорид фосфора (2,10 мл, 22,6 ммоль). Полученную смесь перемешивали в течение 3 ч, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (60 мл). Водную смесь распределяли между солевым раствором (250 мл) и EtOAc (200×3 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Добавляли гептан (100 мл×2), и концентрировали смесь путем азеотропной перегонки для удаления остаточного пиридина. Остаток очищали методом колоночной флэш-хроматографии на SiO2 (картридж 330 г), элюируя 0-100% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=514,3 (M+1).
Промежуточный продукт C3
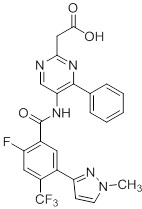
2-(5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)уксусная кислота
Метил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)ацетат (7,94 г, 15,5 ммоль) растворяли в диоксане (62 мл), и охлаждали полученный раствор до 0°C. Добавляли насыщенный водный раствор LiOH (46,3 мл, 247 ммоль), и перемешивали смесь при 23°C в течение 75 мин. Содержащую продукт смесь разбавляли водой (100 мл) и промывали CH2Cl2 (2×100 мл). Щелочной водный слой затем подкисляли путем добавления по каплям 12н водного раствора HCl (~20 мл). Полученную суспензию экстрагировали CH2Cl2 (3×100 мл), объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением указанного в заголовке соединения. MS: m/z=500,2 (M+1).
Промежуточный продукт C4
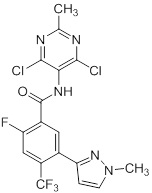
N-(4,6-Дихлор-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 5-амино-4,6-дихлор-2-метилпиримидина (3,00 г, 16,8 ммоль) в пиридине (140 мл) при −30°C добавляли 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойную кислоту (4,86 г, 16,8 ммоль) и оксихлорид фосфора (1,57 мл, 16,8 ммоль). Смесь перемешивали при 0°C в течение 0,5 ч, а затем при 23°C в течение 0,5 ч. Добавляли метанол (140 мл), а затем твердый K2CO3 (20 г), полученную смесь перемешивали в течение 10 мин, затем разбавляли CH2Cl2 (600 мл) и фильтровали через слой силикагеля, элюируя CH2Cl2. Фильтрат концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле, элюируя 10-30% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=447,8 (M+1).
Следующий промежуточный продукт получали способом, аналогичным описанным выше методикам.
№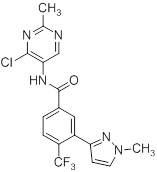
Схема реакций для промежуточного продукта C6
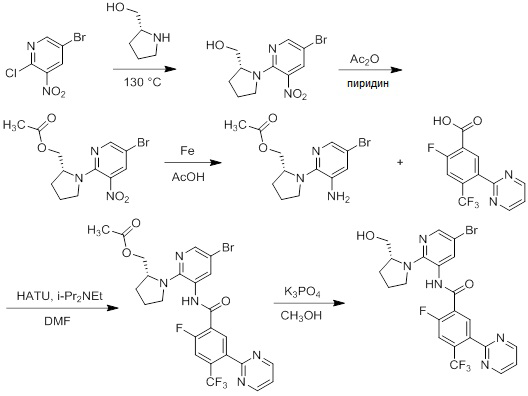
Промежуточный продукт C6
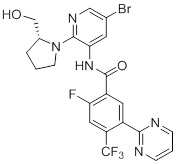
(R)-N-(5-Бром-2-(2-(гидроксиметил)пирролидин-1-ил)пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
Стадия A: (R)-(1-(5-Бром-3-нитропиридин-2-ил)пирролидин-2-ил)метанол
В колбу, содержащую 5-бром-2-хлор-3-нитропиридин (2 г, 8 ммоль), добавляли (R)-пирролидин-2-илметанол (1 г, 10 ммоль), а затем 2-метил-1-пропанол (34 мл). Полученный раствор переносили равными порциями в три флакона для обработки микроволнами емкостью 20 мл. Каждый флакон герметизировали, а затем нагревали при 130°C в микроволновом реакторе при перемешивании в течение 10 мин. После охлаждения, три отдельных реакционных смеси объединяли и разбавляли этилацетатом (200 мл), а затем промывали водой (200 мл). Водную фракцию экстрагировали этилацетатом (50 мл×2). Объединенные органические слои затем промывали солевым раствором (100 мл) и сушили над сульфатом натрия, а затем фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия B: (R)-(1-(5-Бром-3-нитропиридин-2-ил)пирролидин-2-ил)метилацетат
В колбу, содержащую раствор (R)-(1-(5-бром-3-нитропиридин-2-ил)пирролидин-2-ил)метанола (2,4 г, 8 ммоль) в пиридине (28 мл), добавляли уксусный ангидрид (1,6 мл, 17 ммоль), и перемешивали полученную смесь при 23°C в течение ночи. Содержащую продукт смесь затем распределяли между этилацетатом (200 мл) и водой (200 мл). Водный слой экстрагировали этилацетатом (100 мл×2). Объединенные органические фракции промывали солевым раствором (100 мл), затем сушили над сульфатом магния, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия C: (R)-(1-(3-Амино-5-бромпиридин-2-ил)пирролидин-2-ил)метилацетат
К (R)-(1-(5-бром-3-нитропиридин-2-ил)пирролидин-2-ил)метилацетату (2,9 г, 8 ммоль) добавляли уксусную кислоту (15 мл), а затем железо (2 г, 40 ммоль), которое добавляли двумя порциями. Полученную смесь перемешивали при 23°C в течение 20 ч. Содержащую продукт смесь разбавляли этилацетатом (100 мл) и водой (100 мл), и фильтровали полученную смесь через слой Celite® (толщина 1 см, 5 см в диаметре). После такого фильтрования, разделяли органический и водный слои. Водный слой экстрагировали этилацетатом (100 мл×2). Все органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом флэш-хроматографии (ISCO, колонка redisep 125 г, градиент 0-100% этилацетат в гексанах в течение 10 минут) с получением указанного в заголовке соединения. MS: m/z=314 (M+1). 1H-ЯМР (500 МГц, DMSO-d6) δ 7,51 (д, J=2,2 Гц, 1H), 7,04 (д, J=2,2 Гц, 1H), 5,02 (с, 2H), 4,42-4,37 (м, 1H), 3,91-3,84 (м, 2H), 3,67-3,62 (м, 1H), 2,91-2,87 (м, 1H), 2,06-2,01 (м, 1H), 1,9 (с, 3H), 1,76-1,61 (м, 2H).
Стадия D: (R)-(1-(5-Бром-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)пиридин-2-ил)пирролидин-2-ил)метилацетат
В колбу, содержащую (R)-(1-(3-амино-5-бромпиридин-2-ил)пирролидин-2-ил)метилацетат (980 мг, 3,1 ммоль), добавляли 2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензойную кислоту (930 мг, 3,3 ммоль) и HATU (2300 мг, 6,2 ммоль), а затем DMF (30 мл) и N,N-диизопропилэтиламин (1,6 мл, 9,4 ммоль). Полученный раствор перемешивали при 23°C в течение 20 ч. Содержащую продукт смесь распределяли между этилацетатом (200 мл) и водой (200 мл). Водный слой экстрагировали этилацетатом (50 мл×2). Объединенные органические слои промывали солевым раствором, затем сушили над сульфатом магния, а затем фильтровали и концентрировали. Остаток очищали методом флэш-хроматографии (ISCO, колонка redisep 120 г, градиент 0-100% (50/50, EtOH/EtOAc) в гексане в течение 15 минут) с получением указанного в заголовке соединения в виде основной компоненты смеси (4:1) с (R)-(1-(3-амино-5-бромпиридин-2-ил)пирролидин-2-ил)метилом.
MS: m/z=583 (M+1). 1H-ЯМР (500 МГц, DMSO-d6) δ 10,31 (с, 1H), 8,99 (д, J=4,9 Гц, 2H), 8,17-8,15 (м, 2H), 8,02 (д, J=10,2 Гц, 1H), 7,90 (д, J=2,4 Гц, 1H), 7,62 (т, J=4,9 Гц, 1H), 4,07-4,01 (м, 2H), 3,96-3,91 (м, 1H), 3,70-3,64 (м, 2H), 2,04-1,90 (м, 2H), 1,95 (с, 3H), 1,80-1,70 (м, 2H).
Стадия E: (R)-N-(5-Бром-2-(2-(гидроксиметил)пирролидин-1-ил)пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
К раствору (R)-(1-(5-бром-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)пиридин-2-ил)пирролидин-2-ил)метилацетата (775 мг, 1,06 ммоль) и метанола (8 мл) добавляли фосфат калия (3M, 0,710 мл, 2,13 ммоль). Смесь перемешивали при 23°C в течение 1 ч. Содержащую продукт смесь концентрировали, и распределяли остаток между водой и EtOAc. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом флэш-хроматографии (ISCO, колонка redisep 120 г, градиент 0-100% (50/50, EtOH/EtOAc) в гексане в течение 55 минут) с получением указанного в заголовке соединения. MS: m/z=540 (M+1). 1H-ЯМР (500 МГц, DMSO-d6) δ 10,35 (с, 1H), 8,99 (д, J=4,9 Гц, 2H), 8,16 (д, J=6,7 Гц, 1H), 8,12 (д, J=2,4 Гц, 1H), 8,02 (д, J=10,2 Гц, 1H), 7,91 (д, J=2,3 Гц, 1H), 7,61 (т, J=4,9 Гц, 1H), 5,15 (с, 1H), 4,55-4,49 (м, 2H), 4,34-4,29 (м, 1H), 3,60-3,55 (м, 1H), 3,48-3,44 (м, 1H), 1,93-1,84 (м, 2H), 1,79-1,69 (м, 2H).
Схема реакций для промежуточного продукта C7
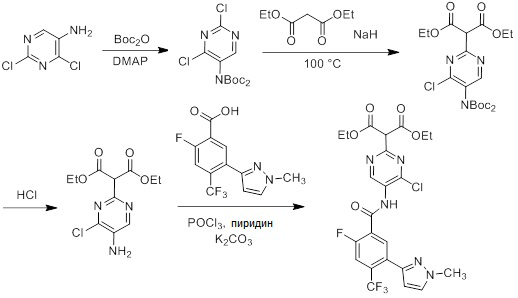
Промежуточный продукт C7
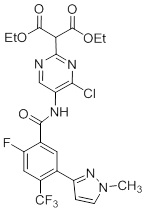
Диэтил-2-(4-хлор-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)пиримидин-2-ил)малонат
Стадия A: Ди-трет-бутил-(2,4-дихлорпиримидин-5-ил)карбамат
Смесь 5-амино-2,4-дихлорпиримидина (6,50 г, 39,6 ммоль), ди-трет-бутилдикарбоната (20,2 мл, 87,9 ммоль) и DMAP (0,968 г, 7,93 ммоль) в дихлорметане (250 мл) перемешивали при 23°C в течение 18 ч, а затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле, элюируя 10% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=386,0 (M+Na). 1H-ЯМР (400 МГц, CDCl3) δ 8,44 (с, 1H) 1,45 (с, 18H).
Стадия B: Диэтил-2-(5-((ди-трет-бутоксикарбонил)амино)-4-хлорпиримидин-2-ил)малонат
К смеси гидрида натрия (60 масс.%, 99 мг, 2,5 ммоль) в DMF (2 мл) при 23°C по каплям добавляли диэтилмалонат (0,38 мл, 2,5 ммоль). Полученную смесь перемешивали в течение 0,5 ч, после чего добавляли ди-трет-бутил-(2,4-дихлорпиримидин-5-ил)карбамат (300 мг, 0,824 ммоль) в DMF (1 мл). Смесь нагревали при 100°C в течение 3 ч, а затем охлаждали и подкисляли добавлением 1н водного раствора HCl. Содержащую продукт смесь распределяли между DCM и H2O. Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле, элюируя 0-35% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=488,1 (M+1).
Стадия C: Диэтил-2-(5-амино-4-хлорпиримидин-2-ил)малонат
К диэтил-2-(5-((ди-трет-бутоксикарбонил)амино)-4-хлорпиримидин-2-ил)малонату (280 мг, 0,574 ммоль) добавляли 4н раствор HCl в диоксане (1,43 мл, 5,74 ммоль). Полученную смесь перемешивали при 23°C в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=288,1 (M+1).
Стадия D: Диэтил-2-(4-хлор-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)пиримидин-2-ил)малонат
К раствору диэтил-2-(5-амино-4-хлорпиримидин-2-ил)малоната (180 мг, 0,626 ммоль) в пиридине (5,1 мл) при –30°C добавляли 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойную кислоту (180 мг, 0,626 ммоль), а затем оксихлорид фосфора (0,058 мл, 0,63 ммоль). Полученную смесь перемешивали при 0°C в течение 0,5 ч, а затем осторожно разбавляли насыщенным водным раствором бикарбоната натрия. Содержащую продукт водную смесь экстрагировали DCM (2×). Объединенные органические слои сушили над Na₂SO₄ и концентрировали. Остаток растворяли в метаноле, и добавляли твердый K2CO3 (избыток). Полученную смесь перемешивали при 23°C в течение 10 мин. Содержащую продукт смесь разбавляли DCM (600 мл), и фильтровали через слой силикагеля. Фильтрат концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле, элюируя 10-30% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=544,0 (M+1).
Схема реакций для промежуточного продукта D1
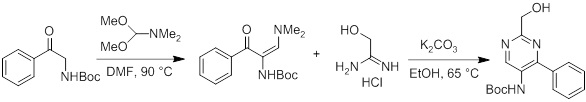
Промежуточный продукт D1
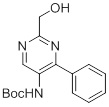
трет-Бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамат
Стадия A: (E)-трет-Бутил-(1-(диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)карбамат
Смесь трет-бутил-(2-оксо-2-фенилэтил)карбамата (1,00 г, 4,25 ммоль) и 1,1-диметокси-N,N-диметилметанамина (0,598 мл, 4,46 ммоль) в DMF (5 мл) нагревали при 90°C в течение 16 ч. Содержащую продукт смесь охлаждали и распределяли между EtOAc и водой. Органический слой промывали солевым раствором, сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения.
Стадия B: трет-Бутил-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)карбамат
Карбонат калия (6,57 г, 47,5 ммоль) при 25°C добавляли к смеси (E)-трет-бутил-(1-(диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)карбамата (4,60 г, 15,8 ммоль) и 2-гидроксиацетимидамида гидрохлорида (2,80 г, 25,3 ммоль) в этаноле (25 мл). Полученную смесь нагревали при 65°C в течение 24 ч, а затем охлаждали и концентрировали. Остаток распределяли между EtOAc (100 мл) и водой (2×100 мл). Органический слой промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток очищали методом колоночной флэш-хроматографии на силикагеле (градиент от 10% EtOAc в гексанах до 100% EtOAc) с получением указанного в заголовке соединения. MS: m/z=302,6 (M+1).
Схема реакций для примера 1
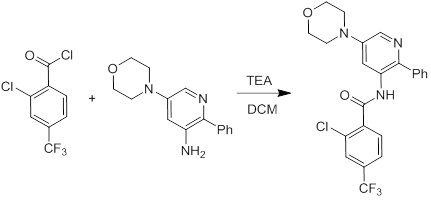
Пример 1
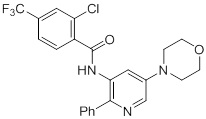
2-Хлор-N-(5-морфолино-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Смесь 5-морфолино-2-фенилпиридин-3-амина (30 мг, 0,12 ммоль), 2-хлор-4-(трифторметил)бензоилхлорида (49 мг, 0,20 моль) и TEA (0,050 мл, 0,36 ммоль) в DCM (2 мл) перемешивали при 15°C в течение 2 ч. Реакционную смесь концентрировали, и распределяли остаток между DCM (50 мл) и водой (10 мл×3). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой (10-40% ацетонитрил+0,75% трифторуксусная кислота в воде) с получением указанного в заголовке соединения. MS: m/z=462 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,30-8,25 (м, 2H), 7,82 (с, 1H), 7,74-7,70 (м, 1H), 7,69-7,63 (м, 3H), 7,59-7,54 (м, 3H), 3,92-3,88 (м, 4H), 3,47-3,42 (м, 4H).
Пример 2
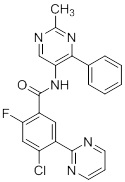
4-Хлор-2-фтор-N-(2-метил-4-фенилпиримидин-5-ил)-5-(пиримидин-2-ил)бензамид
Смесь 2-метил-4-фенилпиримидин-5-аминия хлорида (95 мг, 0,43 ммоль), пиридина (0,047 мл, 0,58 ммоль) и 4-хлор-2-фтор-5-(пиримидин-2-ил)бензоилхлорида (78 мг, 0,29 ммоль) в DMA (2,5 мл) перемешивали при 23°C в течение 16 ч. Реакционную смесь фильтровали, а затем очищали методом HPLC с обращенной фазой (5-95% ацетонитрил+0,1% трифторуксусная кислота в воде) с получением указанного в заголовке соединения. MS: m/z=420,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,69 (с, 1H), 8,90 (д, J=4,9 Гц, 2H), 8,59 (д, J=8,4 Гц, 1H), 7,66 (дд, J=6,6, 2,3 Гц, 2H), 7,56-7,60 (м, 4H), 7,36 (т, J=4,9 Гц, 1H), 2,80 (с, 3H).
Пример 3

3-(1-Метил-1H-пиразол-3-ил)-N-(6-метил-4-фенилпиридин-3-ил)-4-(трифторметокси)бензамид
Стадия A: 3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметокси)бензоилхлорид
Смесь 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметокси)-бензойной кислоты (85 мг, 0,30 ммоль) и оксалилхлорида (1,30 мл, 14,9 ммоль) в DCM (10 мл) нагревали при 50°C в течение 1 ч. Смесь охлаждали, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=301 (M+1, сложный метиловый эфир из реакции с MeOH).
Стадия B: 3-(1-Метил-1H-пиразол-3-ил)-N-(6-метил-4-фенилпиридин-3-ил)-4-(трифторметокси)бензамид
Смесь 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметокси)-бензоилхлорида (88 мг, 0,29 ммоль), 6-метил-4-фенилпиридин-3-амина (82 мг, 0,32 ммоль) и пиридина (0,047 мл, 0,58 ммоль) в DCM (10 мл) нагревали с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали, а затем распределяли между насыщенным раствором NaHCO3 (30 мл) и DCM (20 мл×3). Объединенные органические слои промывали водой (30 мл) и солевым раствором (30 мл), а затем сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой с получением указанного в заголовке соединения. MS: m/z=453 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,13 (с, 1H), 8,42 (д, J=2,2 Гц, 1H), 7,90 (с, 1H), 7,83 (дд, J=8,6, 2,4 Гц, 1H), 7,67-7,64 (м, 3H), 7,61-7,53 (м, 3H), 7,48 (дд, J=8,6, 1,3 Гц, 1H), 6,64 (д, J=2,2 Гц, 1H), 3,97 (с, 3H), 2,79 (с, 3H).
Пример 4
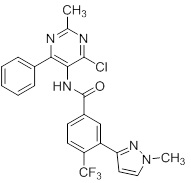
N-(4-Хлор-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
POCl3 (0,205 мл, 2,20 ммоль) при -15°C добавляли к смеси 4-хлор-2-метил-6-фенилпиримидин-5-амина (322 мг, 1,47 ммоль) и 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (396 мг, 1,47 ммоль) в пиридине (3 мл), и перемешивали полученную смесь в течение 2 ч. Содержащую продукт смесь очищали непосредственно методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=472,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,05 (с, 1H), 7,84-7,78 (м, 2H), 7,68-7,63 (м, 3H), 7,40-7,36 (м, 4H), 6,48 (с, 1H), 3,93 (с, 3H), 2,77 (с, 3H).
Схема реакций для примера 5
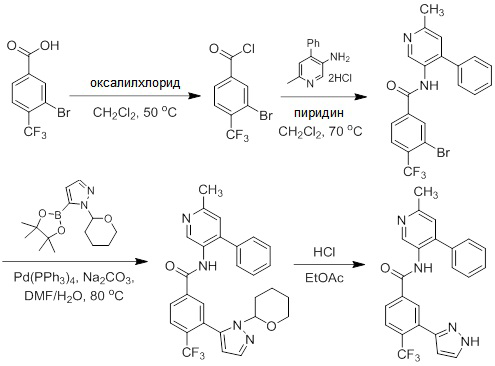
Пример 5
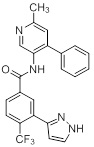
N-(6-Метил-4-фенилпиридин-3-ил)-3-(1H-пиразол-5-ил)-4-(трифторметил)бензамид
Стадия A: 3-Бром-4-(трифторметил)бензоилхлорид
Смесь 3-бром-4-(трифторметил)бензойной кислоты (400 мг, 1,49 ммоль) и оксалилхлорида (1,30 мл, 14,9 ммоль) в DCM (10 мл) нагревали при 50°C в течение 1 ч, а затем концентрировали с получением указанного в заголовке соединения. MS: m/z=283/285 (M+1, сложный метиловый эфир из реакции с MeOH).
Стадия B: 3-Бром-N-(6-метил-4-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Смесь 3-бром-4-(трифторметил)бензоилхлорида (427 мг, 1,49 ммоль), 6-метил-4-фенилпиридин-3-амина (421 мг, 1,64 ммоль) и пиридина (0,241 мл, 2,97 ммоль) в DCM (10 мл) нагревали с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали, а затем распределяли между насыщенным водным раствором NaHCO3 (30 мл) и DCM (20 мл×3). Объединенные органические слои промывали водой (30 мл) и солевым раствором (30 мл), а затем сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE:EtOAc:Et3N=80:20:1, 70:30:1, затем 50:50:1) с получением указанного в заголовке соединения. MS: m/z=435/437 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,54 (с, 1H), 8,14 (с, 1H), 7,89-7,84 (м, 2H), 7,48-7,38 (м, 6H), 2,60 (с, 3H).
Стадия C: N-(6-Метил-4-фенилпиридин-3-ил)-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензамид
Дезоксигенированную смесь 3-бром-N-(6-метил-4-фенилпиридин-3-ил)-4-(трифторметил)бензамида (200 мг, 0,460 ммоль), 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (192 мг, 0,689 ммоль), Pd(PPh3)4 (27 мг, 0,023 ммоль) и Na2CO3 (122 мг, 1,15 ммоль) в смеси DMF:вода (4:1, 7,5 мл) нагревали при 80°C в течение 18 ч. Реакционную смесь охлаждали, а затем распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои промывали водой (50 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=507 (M+1).
Стадия D: N-(6-Метил-4-фенилпиридин-3-ил)-3-(1H-пиразол-5-ил)-4-(трифторметил)бензамид
Раствор HCl в EtOAc (4M, 0,90 мл, 3,6 ммоль) добавляли к раствору N-(6-метил-4-фенилпиридин-3-ил)-3-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)-4-(трифторметил)бензамида (348 мг, 0,687 ммоль) в THF (10 мл), и перемешивали полученную смесь при температуре окружающей среды в течение 18 ч. Смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой с получением указанного в заголовке соединения. MS: m/z=423 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,09 (с, 1H), 8,02 (с, 1H), 7,91-7,98 (м, 2H), 7,88 (с, 1H), 7,73 (д, J=2,4 Гц, 1H), 7,61-7,64 (м, 2H), 7,50-7,57 (м, 3H), 6,50 (д, J=1,3 Гц, 1H), 2,79 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№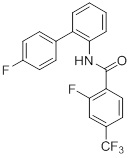





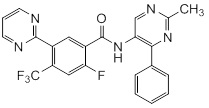
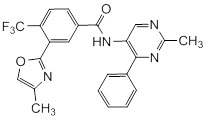
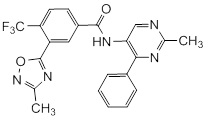
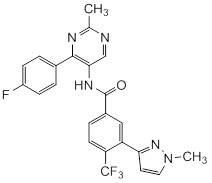
Схема реакций для примера 35
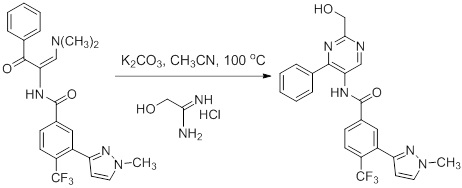
Пример 35
N-(2-(Гидроксиметил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь N-(1-(диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (2,04 г, 4,62 ммоль), 2-гидроксиацетимидамида гидрохлорида (0,818 г, 7,40 ммоль) и карбоната калия (1,92 г, 13,9 ммоль) в EtOH (8 мл) нагревали при 65°C в течение 2 ч, а затем при 80°C в течение 18 ч. Реакционную смесь охлаждали, а затем фильтровали, промывая EtOH и водой. Фильтрат разбавляли водой (50 мл) и экстрагировали DCM (3×50 мл). Объединенные органические слои сушили над Na₂SO₄ и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (гексаны/EtOAc=97:3→0:100) с получением указанного в заголовке соединения. MS: m/z=454,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,69 (с, 1H), 8,10 (с, 1H), 8,02 (с, 1H), 7,83-7,78 (м, 2H), 7,70-7,67 (м, 2H), 7,58-7,50 (м, 3H), 7,39 (д, J=2,3 Гц, 1H), 6,48 (с, 1H), 4,87 (д, J=4,8 Гц, 2H), 3,95 (с, 3H), 3,62 (т, J=4,8 Гц, 1H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№
Пример 39
N-(2-(Гидроксиметил)-4-фенилпиримидин-5-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамид
Стадия A: N-(4-Фенил-2-(((триизопропилсилил)окси)метил)-пиримидин-5-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамид
POCl3 (0,023 мл, 0,25 ммоль) при -15°C добавляли к раствору 4-фенил-2-(((триизопропилсилил)окси)метил)пиримидин-5-амина (59 мг, 0,16 ммоль), 3-(пиримидин-2-ил)-4-(трифторметил)бензойной кислоты (44 мг, 0,16 ммоль) и пиридина (0,080 мл, 0,99 ммоль) в DCM (1 мл), и перемешивали полученную смесь при температуре окружающей среды в течение 4,5 ч. Содержащую продукт смесь очищали непосредственно методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→50:50) с получением указанного в заголовке соединения. MS: m/z=608,4 (M+1).
Стадия B: N-(2-(Гидроксиметил)-4-фенилпиримидин-5-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамид
Смесь n-Bu4NF (0,118 мл, 0,118 ммоль, 1,0M в THF) и N-(4-фенил-2-(((триизопропилсилил)окси)метил)пиримидин-5-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамида (36 мг, 0,059 ммоль) в THF (1 мл) перемешивали при температуре окружающей среды в течение 2,5 ч. Содержащую продукт смесь очищали методом колоночной хроматографии на силикагеле (EtOAc:DCM=0:100→100:0) с получением указанного в заголовке соединения. MS: m/z=452,2 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 10,7 (с, 1H), 8,94 (д, J=4,9 Гц, 2H), 8,86 (с, 1H), 8,21 (с, 1H), 8,15 (д, J=8,9 Гц, 1H), 8,02 (д, J=8,4 Гц, 1H), 7,80-7,75 (м, 2H), 7,56 (т, J=4,9 Гц, 1H), 7,42-7,40 (м, 3H), 5,30 (т, J=6,6 Гц, 1H), 4,64 (д, J=6,3 Гц, 2H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№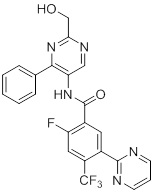
Пример 41
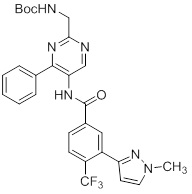
трет-Бутил-((5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метил)карбамат
Смесь N-(1-(диметиламино)-3-оксо-3-фенилпроп-1-ен-2-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (1,00 г, 2,26 ммоль), трет-бутил-(2-амино-2-иминоэтил)карбаматацетата (701 мг, 3,01 ммоль) и карбоната калия (0,937 г, 6,78 ммоль) в ацетонитриле (5 мл) нагревали при 100°C в течение 3,5 ч. Реакционную смесь охлаждали, а затем фильтровали, промывая ацетонитрилом (3×10 мл). Фильтрат концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (гексаны:EtOAc=97:3→0:100) с получением указанного в заголовке соединения. MS: m/z=553,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,66 (с, 1H), 8,06 (с, 1H), 8,01 (с, 1H), 7,81-7,78 (м, 2H), 7,68 (дд, J=7,3, 1,6 Гц, 2H), 7,56-7,49 (м, 3H), 7,39 (д, J=2,2 Гц, 1H), 6,47 (с, 1H), 5,62 (с, 1H), 4,62 (д, J=5,5 Гц, 2H), 3,95 (с, 3H), 1,46 (с, 9H).
Пример 42

N-(2-(Аминометил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор трет-бутил-((5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метил)карбамата (615 мг, 1,11 ммоль) в 4M HCl в диоксане (5,0 мл, 20 ммоль) перемешивали при температуре окружающей среды в течение 5 ч. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=453,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,50 (с, 1H), 8,61 (с, 2H), 8,04 (д, J=8,4 Гц, 1H), 7,97 (с, 1H), 7,82-7,78 (м, 2H), 7,68-7,60 (м, 2H), 7,49-7,42 (м, 3H), 7,42 (д, J=2,3 Гц, 1H), 6,49 (с, 1H), 4,47 (с, 2H), 3,95 (с, 3H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№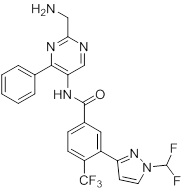
Пример 44
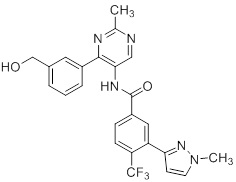
N-(4-(3-(Гидроксиметил)фенил)-2-метилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Дезоксигенированную смесь бис(три-трет-бутилфосфин)палладия (0,78 мг, 1,5 мкмоль), N-(4-хлор-2-метилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (12 мг, 0,030 ммоль), (3-(гидроксиметил)фенил)бороновой кислоты (14 мг, 0,091 ммоль) и водного раствора карбоната цезия (0,030 мл, 0,061 ммоль, 2M) в диоксане (0,4 мл) нагревали при 100°C в условиях обработки микроволновым излучением в течение 0,5 ч. Содержащую продукт смесь очищали методом колоночной хроматографии на силикагеле (DCM:MeOH:NH4OH=100:0:0→90:10:1) с получением указанного в заголовке соединения. MS: m/z=468,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,88 (с, 1H), 8,05 (с, 1H), 7,98 (д, J=8,2 Гц, 1H), 7,90 (д, J=8,2 Гц, 1H), 7,78 (с, 1H), 7,66-7,64 (м, 2H), 7,47-7,45 (м, 2H), 6,46 (с, 1H), 4,47 (д, J=1,2 Гц, 1H), 3,96 (с, 3H), 2,76 (с, 3H).
Пример 45
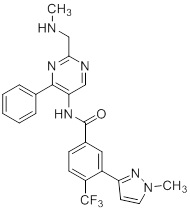
3-(1-Метил-1H-пиразол-3-ил)-N-(2-((метиламино)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
Стадия A: N-(2-Формил-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (452 мг, 0,997 ммоль) и перйодинана Десс-Мартина (507 мг, 1,20 ммоль) в DCM (5 мл) перемешивали при температуре окружающей среды в течение 18 ч. Дополнительно добавляли перйодинан Десс-Мартина (200 мг, 0,47 ммоль), и перемешивали смесь в течение 3 ч. Реакционную смесь фильтровали, промывая DCM. Фильтрат концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (DCM:EtOAc=100:0→0:100) с получением указанного в заголовке соединения. MS: m/z=452,2 (M+1).
Стадия B: 3-(1-Метил-1H-пиразол-3-ил)-N-(2-((метиламино)-метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
Смесь N-(2-формил-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (9 мг, 0,02 ммоль), метиламина (0,030 мл, 0,060 ммоль, 2M в THF), триацетоксиборгидрида натрия (5 мг, 0,03 ммоль) и уксусной кислоты (0,057 мл, 0,10 ммоль) в DCE (1 мл) перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой (колонка C18, H2O:CH3CN:CF3CO2H=95:5:0,1→5:95:0,1) с получением указанного в заголовке соединения в виде трифторацетата. MS: m/z=467,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,57 (с, 1H), 8,57 (с, 1H), 7,98 (с, 1H), 7,87-7,82 (м, 2H), 7,69-7,66 (м, 2H), 7,50-7,47 (м, 3H), 7,44 (д, J=2,4 Гц, 1H), 6,51 (с, 1H), 4,48 (с, 2H), 3,98 (с, 3H), 2,92 (с, 3H).
Пример 46
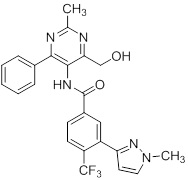
N-(4-(Гидроксиметил)-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(4-(((трет-Бутилдиметилсилил)окси)метил)-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Дезоксигенированную смесь N-(4-хлор-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (50 мг, 0,11 ммоль), трет-бутилдиметил((трибутилстаннил)метокси)силана (69 мг, 0,16 ммоль) и тетракис(трифенилфосфин)палладия (12 мг, 0,011 ммоль) в диоксане (0,6 мл) и нагревали при 150°C в условиях обработки микроволновым излучением в течение 2 ч. Реакционную смесь разбавляли DMF (1 мл) и очищали методом HPLC с обращенной фазой (колонка C18, H2O:CH3CN:CF3CO2H=95:5:0,1→5:95:0,1). Целевые фракции нейтрализовали путем распределения между насыщенным водным раствором NaHCO3 (10 мл) и DCM (3×10 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=582,3 (M+1).
Стадия B: N-(4-(Гидроксиметил)-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь n-Bu4NF (0,108 мл, 0,108 ммоль, 1M в THF) и N-(4-(((трет-бутилдиметилсилил)окси)метил)-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (21 мг, 0,036 ммоль) в THF (1 мл) перемешивали при температуре окружающей среды в течение 1 ч. Содержащую продукт смесь очищали методом колоночной хроматографии на силикагеле (DCM:EtOAc=97:3→0:100) с получением указанного в заголовке соединения. MS: m/z=468,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,03 (с, 1H), 7,84-7,78 (м, 2H), 7,76-7,63 (с, 1H), 7,60-7,55 (м, 2H), 7,42-7,36 (м, 4H), 6,47 (с, 1H), 4,69 (с, 2H), 3,92 (с, 3H), 2,81 (с, 3H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№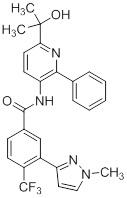
Схема реакций для примера 48

Пример 48
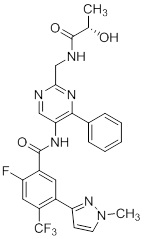
(S)-2-Фтор-N-(2-((2-гидроксипропанамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(2-(Азидометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Оксихлорид фосфора (1,38 мл, 14,8 ммоль) при 0°C по каплям добавляли к раствору 2-(азидометил)-4-фенилпиримидин-5-амина гидрохлорида (3,00 г, 11,4 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (4,28 г, 14,8 ммоль) в пиридине (30 мл). Полученную смесь перемешивали в течение 30 мин, а затем осторожно разбавляли насыщенным водным раствором бикарбоната натрия (75 мл). Большую часть пиридина удаляли в условиях пониженного давления, и распределяли оставшуюся водную смесь между водой (100 мл) и EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Карбонат калия (3,16 г, 22,8 ммоль) добавляли к раствору остатка в MeOH (50 мл), полученную суспензию перемешивали в течение 20 мин, а затем концентрировали. Остаток распределяли между водой (100 мл) и EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом колоночной флэш-хроматографии (градиент гексаны→100% EtOAc) с получением указанного в заголовке соединения. MS: m/z=497,3 (M+1).
Стадия B: N-(2-(Аминометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Суспензию N-(2-(азидометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (5,05 г, 10,2 ммоль) и PS-трифенилфосфиновой смолы (3,51 г, 30,5 ммоль) в 2-метилтетрагидрофуране (100 мл) осторожно перемешивали в течение 2 ч, после чего добавляли воду (1,83 мл, 102 ммоль). Смесь затем нагревали при 40°C в течение 60 ч, а затем фильтровали. Смолу промывали 2-метилтетрагидрофураном (2×50 мл), а затем DCM (2×50 мл). Объединенный фильтрат концентрировали с получением указанного в заголовке соединения. MS: m/z=471,3 (M+1).
Стадия C: (S)-2-Фтор-N-(2-((2-гидроксипропанамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь N-(2-(аминометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (1,00 г, 2,13 ммоль), (S)-2-гидроксипропановой кислоты (0,319 мл, 4,25 ммоль), EDC (0,815 г, 4,25 ммоль), HOBT (0,651 г, 4,25 ммоль) и Et3N (0,889 мл, 6,38 ммоль) в DMF (5 мл) перемешивали при 23°C в течение 1 ч. Содержащую продукт смесь распределяли между насыщенным водным раствором бикарбоната натрия (100 мл) и EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Карбонат калия (250 мг, 1,81 ммоль) добавляли к раствору остатка в MeOH (30 мл), полученную суспензию перемешивали в течение 30 мин, а затем концентрировали. Остаток распределяли между солевым раствором и EtOAc (2×50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом препаративной HPLC с обращенной фазой (градиент вода/CH3CN с 0,1% TFA в качестве модификатора) с получением указанного в заголовке соединения. MS: m/z=543,3 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 10,63 (с, 1H), 8,94 (с, 1H), 8,27 (м, 1H), 7,92 (д, J=6,8 Гц, 1H), 7,88 (д, J=10,3 Гц, 1H), 7,84-7,81 (м, 3H), 7,51 (ушир. м, 3H), 6,45 (с, 1H), 5,64 (д, J=5,1 Гц, 1H), 4,57 (д, J=5,6 Гц, 2H), 4,07 (м, 1H), 3,92 (с, 3H), 1,27 (д, J=6,6 Гц, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№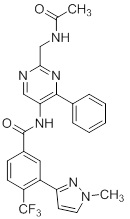
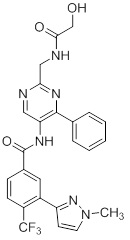
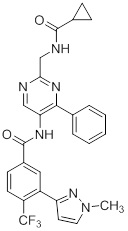
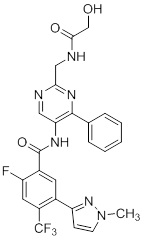
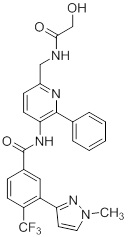
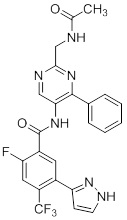
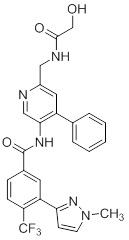

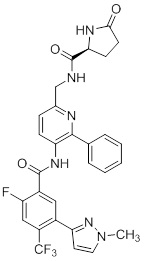
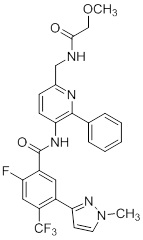
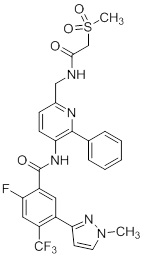
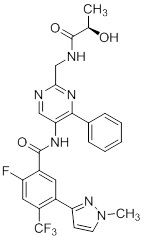
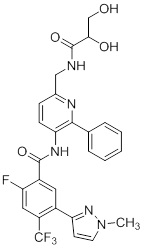
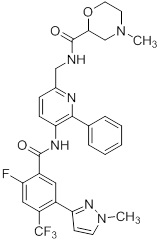
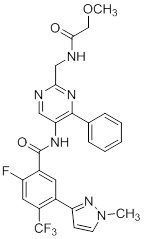
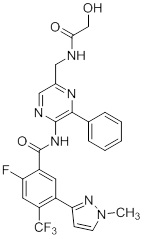
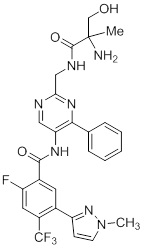
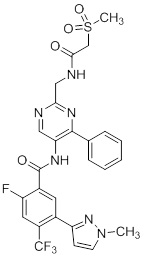


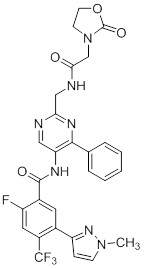
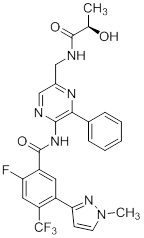
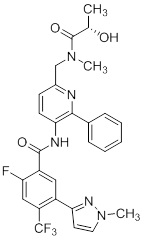
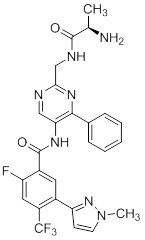
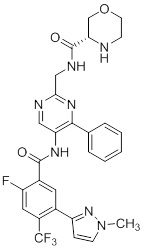
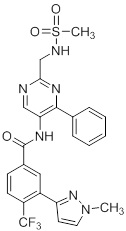
Пример 76
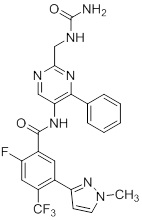
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(4-фенил-2-(уреидометил)пиримидин-5-ил)-4-(трифторметил)бензамид
Триметилсилилизоцианат (0,022 мл, 0,17 ммоль) добавляли к раствору N-(2-(аминометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (130 мг, 0,17 ммоль) в DMF (1,5 мл). Полученную смесь перемешивали в течение 30 мин, а затем очищали методом препаративной HPLC с обращенной фазой (градиент вода/CH3CN с 0,1% TFA в качестве модификатора) с получением указанного в заголовке соединения. MS: m/z=514,3 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 10,61 (с, 1H), 8,93 (с, 1H), 7,92 (д, J=6,6 Гц, 1H), 7,88 (д, J=10,5 Гц, 1H), 7,84-7,81 (м, 3H), 7,51 (ушир. м, 3H), 6,50 (ушир. м, 1H), 6,45 (с, 1H), 5,70 (ушир. с, 2H), 4,46 (д, J=5,9 Гц, 2H), 3,92 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№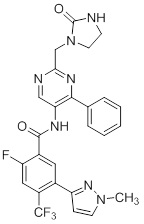
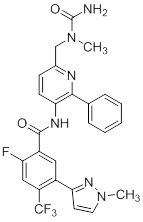

Пример 80
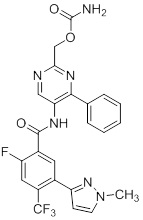
(5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метилкарбамат
Трихлорацетилизоцианат (0,177 мл, 1,48 ммоль) при 23°C добавляли к раствору 2-фтор-N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (500 мг, 1,06 ммоль) в хлороформе (10 мл), и перемешивали полученную смесь в течение 1 ч. Добавляли щелочной оксид алюминия (активность по Брокманну I, 5 г), суспензию перемешивали в течение 1 ч, а затем фильтровали и промывали 20% раствором MeOH в DCM (3×50 мл). Объединенный фильтрат промывали солевым раствором, сушили над сульфатом натрия и концентрировали. Остаток кристаллизовали из EtOAc (5 мл) с получением указанного в заголовке соединения. MS: m/z=515,3 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,11 (с, 1H), 7,97 (д, J=6,8 Гц, 1H), 7,82 (м, 2H), 7,69 (д, J=10,7 Гц, 1H), 7,66 (д, J=2,5 Гц, 1H), 7,51 (м, 3H), 6,44 (с, 1H), 5,28 (с, 2H), 3,96 (с, 3H).
Схема реакций для примера 81

Пример 81
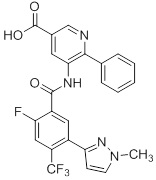
5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотиновая кислота
Стадия A: Метил-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотинат
Оксихлорид фосфора (0,19 мл, 1,3 ммоль) при -10°C по каплям добавляли к раствору метил-5-амино-6-фенилникотината (195 мг, 0,854 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (271 мг, 0,940 ммоль) в пиридине (2,9 мл). Полученную смесь перемешивали при 0°C в течение 2 ч, а затем осторожно разбавляли насыщенным водным раствором бикарбоната натрия (10 мл). Смесь разбавляли EtOAc (100 мл) и промывали водным раствором бикарбоната натрия (10 мл×3). Органический слой промывали солевым раствором (3 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (EtOAc:гексаны=0:100→80:20) с получением указанного в заголовке соединения. MS: m/z=499,1 (M+1).
Стадия B: 5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотиновая кислота
Смесь метил-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотината (390 мг, 0,78 ммоль) и водного раствора NaOH (1M, 0,78 ммоль, 0,78 мл) в 1,4-диоксане (2,5 мл) перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь подкисляли до pH=5 добавлением водного раствора HCl (1M), а затем экстрагировали EtOAc (50 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=485,2 (M+1).
Пример 82
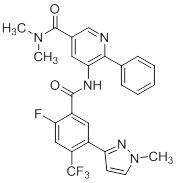
5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N,N-диметил-6-фенилникотинамид
HOBt (25 мг, 0,16 ммоль) добавляли к раствору 5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотиновой кислоты (65 мг, 0,13 ммоль), EDC (31 мг, 0,16 ммоль), диметиламина (1M в THF, 0,16 мл, 0,20 ммоль) и DIPEA (0,071 мл, 0,40 ммоль) в DMF (0,5 мл), и перемешивали полученную смесь при 25°C в течение 12 ч. Реакционную смесь разбавляли EtOAc (30 мл) и промывали водным раствором бикарбоната натрия (3 мл×2). Органический слой промывали солевым раствором (3 мл), сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой (5-80% ацетонитрил+0,75% трифторуксусная кислота в воде) с получением указанного в заголовке соединения. MS: m/z=512,2 (M +1). 1H-ЯМР (500 МГц, CDCl3) δ 9,22 (с, 1H), 8,81 (д, J=7,6 Гц, 1H), 8,68 (с, 1H), 8,38 (д, J=7,6 Гц, 1H), 7,61-7,51 (м, 5H), 7,48 (д, J=11,9 Гц, 1H), 7,43 (с, 1H), 6,46 (с, 1H), 4,00 (с, 3H), 3,20 (с, 3H), 3,17 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№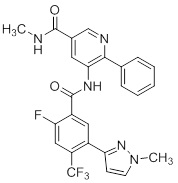
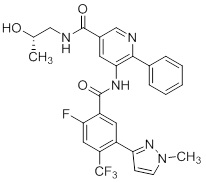
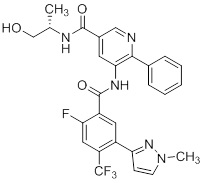
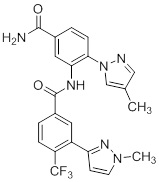
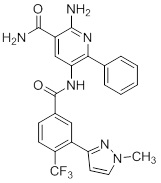
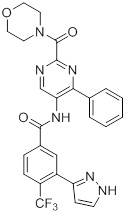
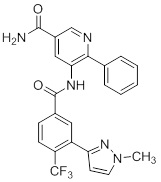

Пример 95
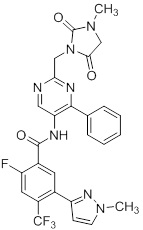
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((3-метил-2,5-диоксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
Оксихлорид фосфора (0,040 мл, 0,43 ммоль) при 0°C по каплям добавляли к раствору 3-((5-амино-4-фенилпиримидин-2-ил)метил)-1-метилимидазолидин-2,4-диона гидрохлорида (130 мг, 0,39 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (112 мг, 0,39 ммоль) в пиридине (5 мл). Полученную смесь перемешивали в течение 30 мин, а затем осторожно разбавляли насыщенным водным раствором бикарбоната натрия (50 мл). Большую часть пиридина удаляли в условиях пониженного давления, и экстрагировали оставшуюся водную смесь EtOAc (2×100 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом колоночной флэш-хроматографии (градиент гексаны→100% EtOAc), а затем препаративной HPLC с обращенной фазой (градиент вода/CH3CN с 0,1% TFA в качестве модификатора) с получением указанного в заголовке соединения. MS: m/z=568,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,76 (с, 1H), 8,62 (д, J=13,9 Гц, 1H), 8,44 (д, J=7,6 Гц, 1H), 7,64 (м, 2H), 7,56 (м, 3H), 7,47 (д, J=12,0 Гц, 1H), 7,40 (с, 1H), 7,51 (м, 3H), 6,44 (с, 1H), 5,01 (с, 2H), 4,01 (с, 2H), 3,96 (с, 3H), 3,06 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№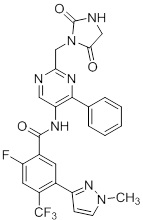
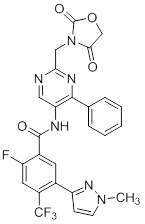
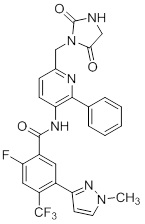
Пример 99
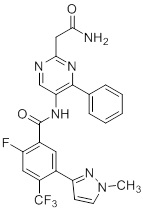
N-(2-(2-Амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор 2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)уксусной кислоты (439 мг, 0,879) в 1,4-диоксане (30 мл) насыщали газообразным аммиаком. Добавляли BOP (408 мг, 0,923 ммоль), и барботировали смесь аммиаком при быстром перемешивании. Спустя 20 мин, смесь распределяли между солевым раствором (75 мл) и EtOAc (75 мл×2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Остаток суспендировали в EtOAc в условиях обработки ультразвуком, затем фильтровали, и промывали отфильтрованное твердое вещество EtOAc с получением указанного в заголовке соединения. Объединенный фильтрат концентрировали, и очищали остаток методом колоночной флэш-хроматографии на SiO2 (градиент гексаны→100% EtOAc/EtOH=3/1) с получением дополнительного количества указанного в заголовке соединения. MS: m/z=499,3 (M+1). 1H-ЯМР (500 МГц, DMSO-d6): δ 10,60 (с, 1H), 8,92 (с, 1H), 7,92 (д, J=6,8 Гц, 1H), 7,89 (д, J=10,4 Гц, 1H), 7,81 (м, 3H), 7,60 (с, 1H), 7,52 (м, 3H), 7,07 (с, 1H), 6,45 (с, 1H), 3,93 (с, 3H), 3,83 (с, 2H).
Пример 100
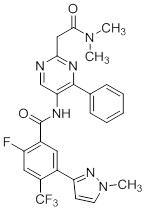
N-(2-(2-(Диметиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор диметиламина получали путем барботирования газообразным диметиламином 5 мл 1,2-дихлорэтана в течение 1 мин. При перемешивании во флакон помещали 2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)уксусную кислоту (25 мг, 0,050 ммоль) и HATU (20,9 мг, 0,055 ммоль), и добавляли полученный раствор диметиламина (0,5 мл). Спустя 10 мин, смесь разбавляли водой (10 мл) и экстрагировали CH2Cl2 (3×10 мл). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали. Остаток очищали методом колоночной флэш-хроматографии на SiO2, элюируя 0-40% (EtOAc/EtOH=3/1) в CH2Cl2, с получением указанного в заголовке соединения. MS: m/z=527,3 (M+1). 1H-ЯМР (500 МГц, DMSO-d6): δ 10,62 (с, 1H), 8,95 (с, 1H), 7,92 (д, J=6,6 Гц, 1H), 7,88 (д, J=10,2 Гц, 1H), 7,80 (м, 3H), 7,51 (м, 3H), 6,45 (с, 1H), 3,97 (с, 2H), 3,92 (с, 3H) 3,09 (с, 3H), 2,87 (с, 3H).
Пример 101
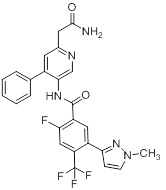
N-(6-(2-Амино-2-оксоэтил)-4-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-2-(5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-4–фенилпиридин-2-ил)ацетат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (169 мг, 0,585 ммоль) и этил-2-(5-амино-4-фенилпиридин-2-ил)ацетата (150 мг, 0,585 ммоль) в пиридине (3 мл) при 25°C по каплям добавляли POCl3 (0,109 мл, 1,17 ммоль). Полученную смесь перемешивали в течение 10 мин, затем разбавляли водой со льдом (20 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=527,7 (M+1).
Стадия B: N-(6-(2-Амино-2-оксоэтил)-4-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиридин-2-ил)ацетата (50 мг, 0,095 ммоль) в 4M растворе NH3 в EtOH (8 мл) перемешивали при 25°C в течение 36 ч. Содержащую продукт смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=498,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,66 (с, 1H), 8,51-8,39 (м, 2H), 7,56-7,49 (м, 3H), 7,46-7,31 (м, 5H), 7,23 (с, 1H), 6,42 (с, 1H), 5,42 (с, 1H), 3,95 (с, 3H), 3,76 (с, 2H).
Пример 102
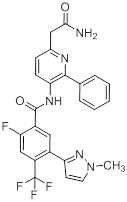
N-(6-(2-Амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)ацетат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (225 мг, 0,780 ммоль) и этил-2-(5-амино-6-фенилпиридин-2-ил)ацетата (200 мг, 0,780 ммоль) в пиридине (3 мл) при 32°C по каплям добавляли POCl3 (0,073 мл, 0,780 ммоль). Полученную смесь перемешивали в течение 10 мин, затем разбавляли водой (20 мл) и экстрагировали EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=2/1) с получением указанного в заголовке соединения. MS: m/z=507,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,85 (д, J=8,2 Гц, 1H), 8,63 (д, J=13,3 Гц, 1H), 8,41 (д, J=7,4 Гц, 1H), 7,61-7,36 (м, 8H), 6,44 (с, 1H), 4,20 (кв, J=7,0 Гц, 2H), 3,96 (с, 3H), 3,90 (с, 2H), 1,30-1,26 (м, 3H).
Стадия B: N-(6-(2-Амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)ацетата (50 мг, 0,095 ммоль) в MeOH (2 мл) при 25°C добавляли 3M раствор NH3 в MeOH (5 мл). Полученную смесь перемешивали в течение 24 ч. Содержащую продукт смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=498,1(M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,86 (д, J=8,2 Гц, 1H), 8,63 (д, J=13,7 Гц, 1H), 8,42 (д, J=7,8 Гц, 1H), 7,63-7,49 (м, 5H), 7,45 (д, J=11,7 Гц, 1H), 7,40 (д, J=2,0 Гц, 1H), 7,35 (д, J=8,6 Гц, 1H), 6,44 (с, 1H), 5,39 (с, 1H), 3,96 (с, 3H), 3,80 (с, 2H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№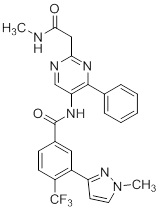
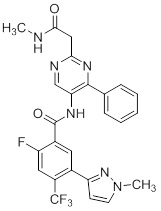
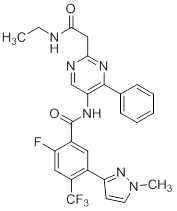
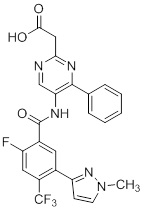
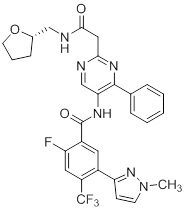
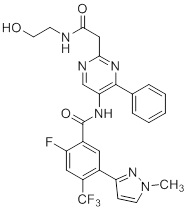
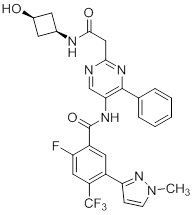
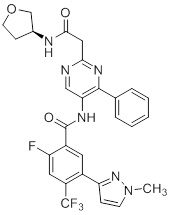
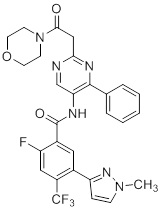
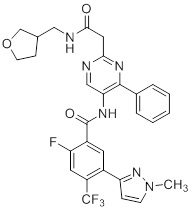
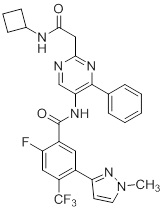
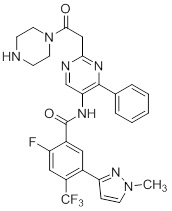
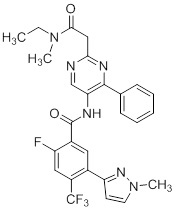


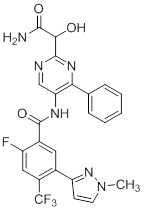


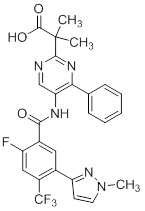

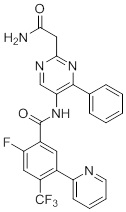

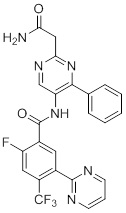
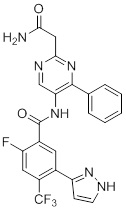
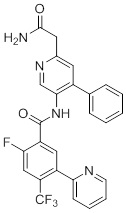
Пример 128
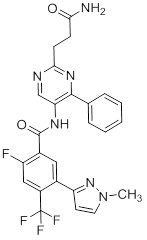
N-(2-(3-Амино-3-оксопропил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)пропаноат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (51 мг, 0,18 ммоль) в пиридине (1 мл) при 25°C добавляли POCl3 (0,020 мл, 0,21 ммоль). После перемешивания смеси в течение 15 мин, по каплям добавляли этил-3-(5-амино-4-фенилпиримидин-2-ил)пропаноат (40 мг, 0,15 ммоль) в пиридине (1 мл). Полученную смесь перемешивали в течение 10 мин, а затем распределяли между водой (5 мл) и EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=542,3 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,72 (с, 1H), 8,61-8,58 (м, 1H), 8,46 (д, J=8,0 Гц, 1H), 7,67-7,66 (м, 2H), 7,57-7,56 (м, 3H), 7,41 (с, 1H), 7,40 (с, 1H), 6,45 (с, 1H), 4,15 (кв, J=7,6 Гц, 2H), 3,97 (с, 3H), 3,37 (т, J=7,2 Гц, 2H ), 2,93 (т, J=7,2 Гц, 2H), 1,24 (т, J=7,2 Гц, 3H).
Стадия B: N-(2-(3-Амино-3-оксопропил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору этил-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)пропаноата (30 мг, 0,055 ммоль) в этаноле (0,5 мл) добавляли концентрированный водный раствор NH4OH (5 мл, 0,05 ммоль). Полученную смесь перемешивали при 25°C в течение 16 ч, а затем фильтровали и очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=513,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,02 (с, 1H), 7,96 (д, J=6,4 Гц, 1H), 7,82-7,81 (м, 2H), 7,71-7,66 (м, 2H), 7,51-7,50 (м, 3H), 6,45 (с, 1H), 3,97 (с, 3H), 3,34 (т, J=7,2 Гц, 2H), 2,83 (т, J=7,2 Гц, 2H).
Схема реакций для примера 129

Пример 129
N-(2-((4H-1,2,4-Триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: 2-Фтор-N-(2-(2-гидразинил-2-оксоэтил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К суспензии метил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)ацетата (800 мг, 1,56 ммоль) в MeOH (6 мл) добавляли гидразина моногидрат (0,38 мл, 7,8 ммоль), и нагревали полученную смесь до 60°C. Спустя 5 ч, смесь охлаждали и концентрировали. Остаток очищали методом хроматографии на силикагеле (RediSep-Rf 40 г, 100% EtOH) с получением указанного в заголовке соединения. 1H-ЯМР (500 МГц, CDCl3): δ 9,82 (с, 1H), 8,66 (д, J=14,2 Гц, 1H), 8,47 (д, J=7,8 Гц, 1H), 8,40 (с, 1H), 7,67-7,65 (м, 2H), 7,59-7,58 (м, 3H), 7,50 (с, 1H), 7,48 (с, 1H), 7,41 (д, J=2,3 Гц, 1H), 6,46 (с, 1H), 4,04 (с, 2H), 3,97-3,96 (м, 4H).
Стадия B: (E)-N'-(2,4-Диметоксибензил)-N,N-диметилформимидамид
(2,4-Диметоксифенил)метанамин (1,00 мл, 6,66 ммоль) и DMF-DMA (2,67 мл, 20,0 ммоль) объединяли в MeOH (20 мл), и нагревали полученную смесь с обратным холодильником. Спустя 3 ч, смесь охлаждали и концентрировали с получением указанного в заголовке соединения. 1H-ЯМР (500 МГц, CDCl3): δ 7,36 (с, 1H), 7,21 (д, J=8,1 Гц, 1H), 6,46-6,43 (м, 2H), 4,38 (с, 2H), 3,80 (с, 3H), 3,79 (с, 3H), 2,87 (с, 6H).
Стадия C: N-(2-((4-(2,4-Диметоксибензил)-4H-1,2,4-триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору (E)-N'-(2,4-диметоксибензил)-N,N-диметилформимидамида (162 мг, 0,730 ммоль) в CH3CN (5 мл) добавляли AcOH (0,100 мл, 1,75 ммоль), а затем 2-фтор-N-(2-(2-гидразинил-2-оксоэтил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид (300 мг, 0,584 ммоль). Полученную смесь перемешивали в течение 30 минут при 23°C, а затем нагревали до 80°C. Спустя 8 ч, смесь охлаждали, а затем разбавляли насыщенным водным раствором NH4Cl и экстрагировали EtOAc (3×). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали. Остаток очищали методом хроматографии на силикагеле (RediSep-Rf 24 г, 100% EtOAc/EtOH=3/1) с получением указанного в заголовке соединения. 1H-ЯМР (500 МГц, DMSO-d6): δ 10,61 (с, 1H), 8,91 (с, 1H), 8,38 (с, 1H), 7,87-7,91 (м, 1H), 7,81 (с, 1H), 7,73 (с, 2H), 7,50 (с, 3H), 7,00 (д, J=8,3 Гц, 1H), 6,52 (д, J=2,4 Гц, 1H), 6,45 (с, 1H), 6,42 (д, J=8,4 Гц, 1H), 5,14 (с, 2H), 4,52 (с, 2H), 3,92 (с, 3H), 3,73 (с, 3H), 3,72 (с, 3H).
Стадия D: N-(2-((4H-1,2,4-Триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
N-(2-((4-(2,4-Диметоксибензил)-4H-1,2,4-триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид (268 мг, 0,398 ммоль) растворяли в TFA (3 мл), и нагревали смесь до 60°C. Спустя 1 ч, смесь охлаждали, разбавляли MeOH и концентрировали. Остаток растворяли в DMSO (5 мл), фильтровали с использованием шприцевого фильтра (0,45 мкм PTFE), затем очищали методом препаративной HPLC с обращенной фазой (21×100 мм Phenomenex AXIA-Gemini-NX (0,1% TFA), градиент 10-50% CH3CN в воде в течение 18 мин при 20 мл/мин, 3 введения по ~1,7 мл каждое) с получением указанного в заголовке соединения. 1H-ЯМР (500 МГц, CDCl3): δ 9,90 (с, 1H), 8,73 (д, J=14,3 Гц, 1H), 8,45 (д, J=7,6 Гц, 1H), 8,20 (с, 1H), 7,67 (с, 3H), 7,62-7,61 (м, 3H), 7,50 (д, J=12,0 Гц, 1H), 7,42 (д, J=2,3 Гц, 1H), 6,46 (с, 1H), 4,79 (с, 2H), 3,99 (с, 3H). MS: m/z=523,3 (M+1).
Пример 130
N-(2-(1,2-Диамино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор NaNO2 (11 мг, 0,16 ммоль) в H2O (0,100 мл) при 23°C добавляли к смеси N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (50 мг, 0,100 ммоль) в AcOH (1 мл), и перемешивали полученную смесь в течение 20 ч. Дополнительно добавляли NaNO2 (6 мг, 0,09 ммоль) в H2O (0,050 мл), и продолжали перемешивание в течение 30 мин. Добавляли 10% Pd/C (11 мг, 0,010 ммоль), смесь помещали в атмосферу H2 (баллон) и быстро перемешивали при 23°C в течение 3 ч. Смесь фильтровали с использованием шприцевого фильтра (0,45 мкм PTFE), промывая AcOH, и концентрировали фильтрат. Остаток распределяли между насыщенным водным раствором NaHCO3 (20 мл) и DCM (3×30 мл). Объединенные органические слои фильтровали через слой Celite®, промывая DCM, и концентрировали. Остаток поглощали DMSO (0,5 мл), фильтровали с использованием шприцевого фильтра (0,45 мкм PTFE), а затем очищали методом препаративной HPLC с обращенной фазой (21×100 мм Phenomenex AXIA-Gemini-NX (0,1% TFA), градиент 5-50% CH3CN в воде в течение 18 мин при 20 мл/мин) с получением указанного в заголовке соединения в виде трифторацетата. 1H-ЯМР (500 МГц, DMSO-d6): δ 10,79 (с, 1H), 9,14 (с, 1H), 8,78 (с, 3H), 8,09 (с, 1H), 7,87-7,95 (м, 6H), 7,82 (д, J=2,2 Гц, 1H), 7,56 (д, J=5,7 Гц, 3H), 6,45 (с, 1H), 5,21 (с, 1H), 3,92 (с, 3H). MS: m/z=514,3 (M+1).
Пример 131
N-(2-(2,5-Диоксоимидазолидин-4-ил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору N-(2-(1,2-диамино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (50 мг, 0,097 ммоль) в 2-MeTHF (0,5 мл)) при 23°C добавляли CDI (20 мг, 0,12 ммоль). Полученную смесь нагревали до 80°C. Спустя 1 ч, смесь охлаждали и концентрировали. Остаток поглощали DMSO (1,0 мл), фильтровали с использованием шприцевого фильтра (0,45 мкм PTFE), а затем очищали методом препаративной HPLC с обращенной фазой (21×100 мм Phenomenex AXIA-Gemini-NX (0,1% TFA), градиент 20-65% CH3CN в воде в течение 18 мин при 20 мл/мин) с получением указанного в заголовке соединения. 1H-ЯМР (500 МГц, DMSO-d6): δ 11,86 (с, 1H), 10,30 (с, 1H), 8,52 (с, 1H), 7,88 (д, J=6,7 Гц, 1H), 7,79-7,84 (м, 5H), 7,52 (с, 1H), 7,47-7,51 (м, 3H), 6,43 (с, 1H), 3,92 (с, 3H). MS: m/z=540,2 (M+1).
Пример 132
N-(4-(3-(Ацетамидометил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(4-(3-(Ацетамидометил)фенил)-6-хлор-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор N-(4,6-дихлор-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (30 мг, 0,067 ммоль), фосфата калия (1M, 0,20 мл, 0,20 ммоль) и диоксана (0,5 мл) перемешивали при 23°C в течение 10 мин. Смесь добавляли во флакон для обработки микроволнами, содержащий (3-(ацетамидометил)фенил)бороновую кислоту (16 мг, 0,067 ммоль), комплекс Pd(dppf)Cl2-CH2Cl2 (10 мг, 0,012 ммоль) и мешальник. Флакон герметизировали и нагревали при 130°C в течение 5 мин в микроволновом реакторе. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения.
Стадия B: N-(4-(3-(Ацетамидометил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Во флакон для обработки микроволнами добавляли bis-pin (13 мг, 0,051 ммоль), ацетат калия (5,0 мг, 0,051 ммоль), комплекс Pd(dppf)Cl2-CH2Cl2 (20 мг, 0,024 ммоль) и раствор N-(4-(3-(ацетамидометил)фенил)-6-хлор-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида в 2-метил-1-пропаноле (1 мл). Флакон герметизировали и нагревали при 130°C в течение 5 мин в микроволновом реакторе. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали водой. Органический слой концентрировали, и очищали остаток методом HPLC с обращенной фазой (градиент CH3CN/вода 0,1% TFA в качестве модификатора) с получением указанного в заголовке соединения. MS: m/z=527,3 (M+1). 1H-ЯМР (500 МГц, DMSO-d6) δ 10,55 (с, 1H), 8,87 (с, 1H), 8,38 (с, 1H), 7,92-7,86 (м, 2H), 7,81 (д, J=2,3 Гц, 1H), 7,67 (д, J=9,9 Гц, 2H), 7,44 (т, J=7,6 Гц, 1H), 7,38 (д, J=7,7 Гц, 1H), 6,45 (с, 1H), 4,30 (д, J=5,9 Гц, 2H), 3,92 (с, 3H), 2,70 (с, 3H), 2,55 (с, 1H), 1,83 (с, 3H).
Следующие вещества получали способом, аналогичным описанным выше методикам. Для синтеза соединения 134, в начальной реакции катализируемого палладием кросс-сочетания использовали (2-(2-((трет-бутоксикарбонил)амино)этил)фенил)бороновую кислоту, а затем обрабатывали предпоследний продукт TFA в дихлорметане с удалением защитной Boc-группы.
№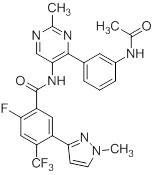
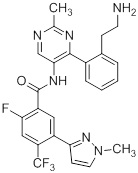
Пример 135
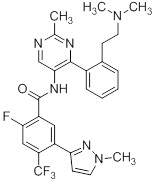
N-(4-(2-(2-(Диметиламино)этил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Формальдегид (0,003 мл, 0,04 ммоль) и ацетат натрия (2,1 мг, 0,026 ммоль) добавляли к раствору N-(4-(2-(2-аминоэтил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (13 мг, 0,026 ммоль) в MeOH (1 мл). Полученную смесь перемешивали при 23°C в течение 10 мин. Затем, добавляли цианборгидрид натрия (1,6 мг, 0,026 ммоль), и после перемешивания в течение 1 ч, смесь очищали методом HPLC с обращенной фазой (C-18), элюируя смесью ацетонитрил/вода+0,1% TFA, с получением указанного в заголовке соединения. MS: m/z=527,0 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,00 (с, 1H), 7,80 (м, 1H), 7,65 (м, 2H), 7,50 (м, 2H), 7,40 (м, 2H), 6,40 (с, 1H), 4,00 (с, 3H), 3,22 (т, 2H), 3,10 (т, 2H), 2,82 (с, 6H), 2,80 (с, 3H).
Схема реакций для примера 136
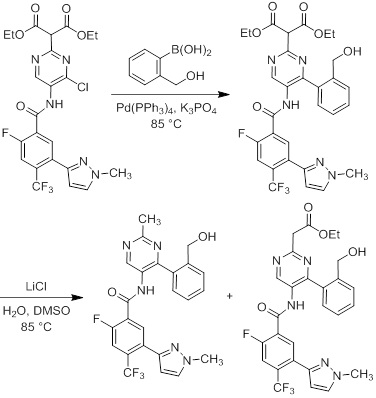
Пример 136

2-Фтор-N-(4-(2-(гидроксиметил)фенил)-2-метилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Диэтил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-(2-(гидроксиметил)фенил)пиримидин-2-ил)малонат
Дезоксигенированную смесь диэтил-2-(4-хлор-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)пиримидин-2-ил)малоната (0,38 г, 0,68 ммоль), Pd(dppf)Cl2-CH2Cl2 (0,028 г, 0,034 ммоль), 2-(гидроксиметил)фенилбороновой кислоты (0,16 г, 0,12 ммоль) и водного раствора фосфата калия (1н, 2,04 мл, 2,04 ммоль) в диоксане (5 мл) нагревали при 85°C в течение 1 ч. Смесь разбавляли DCM, сушили над Na₂SO₄ и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле, элюируя 10-90% EtOAc в гексанах, с получением указанного в заголовке соединения. MS: m/z=630,12 (M+1).
Стадия B: 2-Фтор-N-(4-(2-(гидроксиметил)фенил)-2-метилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид 2,2,2-трифторацетат
К раствору диэтил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-(2-(гидроксиметил)фенил)пиримидин-2-ил)малоната (390 мг, 0,619 ммоль) в DMSO (3,1 мл) добавляли H2O (0,067 мл, 3,7 ммоль) и хлорид лития (79 мг, 1,9 ммоль). Полученную смесь нагревали при 85°C в течение 1 ч, а затем охлаждали и разбавляли дихлорметаном. Полученную смесь промывали водой, сушили над Na₂SO₄ и концентрировали. Остаток очищали методом HPLC с обращенной фазой (C-18), элюируя смесью ацетонитрил/вода+0,1% TFA, с получением по отдельности этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-(2-(гидроксиметил)фенил)пиримидин-2-ил)ацетата и указанного в заголовке соединения. MS: m/z=486,0 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,20 (с, 1H), 7,90 (д, 1H), 7,65 (м, 3H), 7,52 (м, 1H), 7,40 (м, 1H), 7,38 (м, 1H), 6,40 (с, 1H), 4,55 (с, 2H), 3,98 (с, 3H), 2,75 (3H).
Пример 137
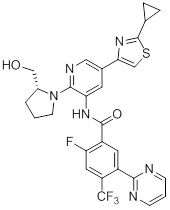
(R)-N-(5-(2-Циклопропилтиазол-4-ил)-2-(2-(гидроксиметил)пирролидин-1-ил)пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
(R)-N-(5-бром-2-(2-(гидроксиметил)пирролидин-1-ил)пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид (30 мг, 0,056 ммоль) и bis-pin (15 мг, 0,061 ммоль) добавляли во флакон емкостью 1 драм. Флакон переносили в перчаточный бокс. В перчаточном боксе, во флакон добавляли KOAc (16 мг, 0,17 ммоль), предкатализатор XPHOS 2-го поколения (4 мг, 0,006 ммоль) и безводный диоксан (0,3 мл). Полученную смесь нагревали при 100°C в течение 2 ч. В перчаточном боксе, в реакционный флакон добавляли 4-бром-2-циклопропилтиазол (14 мг, 0,067 ммоль), фосфат калия (2M, 0,083 мл, 0,17 ммоль) и PdCl2(dtbpf) (5 мг, 0,008 ммоль). Полученную смесь нагревали при 40°C в течение 2 часов. Смесь разбавляли этилацетатом и промывали водой. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой (CH3CN/вода с 0,1% TFA в качестве модификатора) с получением указанного в заголовке соединения. MS: m/z=585 (M+1). 1H-ЯМР (500 МГц, CD3OD) δ 8,94 (дд, J=5,0, 2,7 Гц, 2H), 8,50 (с, 2H), 8,29 (д, J=6,8 Гц, 1H), 7,88 (д, J=10,7 Гц, 1H), 7,65 (д, J=2,6 Гц, 1H), 7,57-7,55 (м, 1H), 4,60 (с, 1H), 3,76 (т, J=10,0 Гц, 2H), 3,71-3,63 (м, 2H), 2,42-2,39 (м, 1H), 2,17-2,09 (м, 3H), 2,00-1,97 (м, 2H), 1,21-1,18 (м, 2H), 1,10-1,08 (м, 2H).
Следующее соединение получали способом, аналогичным описанным выше методикам.
№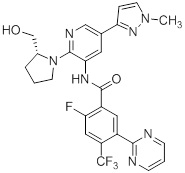
Пример 139
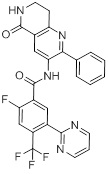
2-Фтор-N-(5-оксо-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
Стадия A: трет-Бутил-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилат
К раствору трет-бутил-3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (400 мг, 1,23 ммоль) и 2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензойной кислоты (352 мг, 1,23 ммоль) в пиридине (5 мл) при 19°C добавляли оксихлорид фосфора (0,229 мл, 2,46 ммоль). Полученную смесь перемешивали в течение 5 мин, а затем разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=594,3 (M+1).
Стадия B: 2-Фтор-N-(2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
К раствору трет-бутил-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (300 мг, 0,505 ммоль) в этилацетате (5 мл) при 18°C добавляли 4M раствор HCl в EtOAc (3 мл). Полученную смесь перемешивали в течение 1 ч, а затем концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=494,1 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 10,49 (с, 1H), 9,05 (с, 2H), 8,96 (д, J=4,7 Гц, 2H), 7,95 (м, 3H), 7,59 (м, 2H), 7,40 (д, J=7,0 Гц, 2H), 4,38 (с, 2H), 3,53 (с, 2H), 3,11 (с, 2H).
Стадия C: 2-Фтор-N-(5-оксо-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
К раствору трет-бутил-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксилата (200 мг, 0,337 ммоль) в ацетонитриле (3 мл) и воде (3 мл) при 20°C добавляли перйодат натрия (216 мг, 1,011 ммоль) и гидрат оксида рутения(IV) (10 мг, 0,067 ммоль). Полученную смесь перемешивали в течение 48 ч, а затем фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=508,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,34 (с, 1H), 8,87 (д, J=5,1 Гц, 2H), 8,64 (д, J=13,7 Гц, 1H), 8,57 (д, J=7,4 Гц, 1H), 7,63-7,59 (м, 2H), 7,57-7,49 (м, 4H), 7,35 (т, J=4,7 Гц, 1H), 6,02 (с, 1H), 3,69 (с, 2H), 3,24 (т, J=6,5 Гц, 2H).
Пример 140
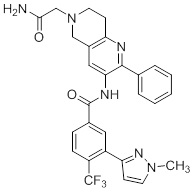
N-(6-(2-Амино-2-оксоэтил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-2-(3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат
К раствору 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (850 мг, 3,15 ммоль) в пиридине (5 мл) при 20°C добавляли этил-2-(3-амино-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетат (980 мг, 3,15 ммоль) и POCl3 (0,440 мл, 4,72 ммоль). Полученную смесь перемешивали в течение 0,5 ч, а затем разбавляли водой (5 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=564,3 (M+1).
Стадия B: 2-(3-(3-(1-Метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)уксусная кислота
К раствору этил-2-(3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)ацетата (200 мг, 0,355 ммоль) в THF (2 мл) и воде (2 мл) добавляли LiOH (17 мг, 0,70 ммоль). Полученную смесь перемешивали при 20°C в течение 30 мин, затем разбавляли воду (5 мл), подкисляли до pH=6 добавлением 6н водного раствора HCl, и экстрагировали EtOAc (5 мл×4). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного указанного в заголовке соединения. MS: m/z=536,2 (M+1).
Стадия C: N-(6-(2-Амино-2-оксоэтил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор 2-(3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)уксусной кислоты (174 мг, 0,325 ммоль) в DCM (2 мл) при 20°C подщелачивали до pH=8 добавлением DIEA. Добавляли HATU (148 мг, 0,384 ммоль) и NH4Cl (85 мг, 1,6 ммоль), и перемешивали полученную смесь в течение 1 ч. Содержащую продукт смесь распределяли между водой (2 мл) и EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=535,2 (M+1). 1H-ЯМР ( 400 МГц, CD3OD) δ 7,99 (с, 1H), 7,93-7,85 (м, 2H), 7,80 (с, 1H), 7,67 (д, J=2,0 Гц, 1H), 7,59 (д, J=7,0 Гц, 2H), 7,48-7,39 (м, 3H), 6,45 (с, 1H), 3,97 (с, 3H), 3,87 (с, 2H), 3,35 (с, 2H), 3,16-3,08 (м, 2H), 3,04-2,97 (м, 2H).
Пример 141
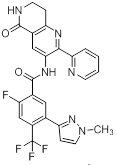
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамид
К раствору 3-амино-2-(пиридин-2-ил)-7,8-дигидро-1,6-нафтиридин-5(6H)-она (30 мг, 0,13 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (36,0 мг, 0,13 ммоль) в пиридине (2 мл) при 25°C добавляли оксихлорид фосфора (0,023 мл, 0,25 ммоль). Полученную смесь перемешивали в течение 10 мин, разбавляли водой со льдом (20 мл) и экстрагировали этилацетатом (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=511,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,77 (с, 1H), 8,77-8,71 (м, 2H), 8,47 (д, J=7,5 Гц, 1H), 7,99 (т, J=8,5 Гц, 1H), 7,67 (д, J=11,0 Гц, 1H), 7,48-7,43 (м, 2H), 6,54 (с, 1H), 6,06 (с, 1H), 4,04 (с, 3H), 3,79-3,72 (м, 2H), 3,30 (т, J=6,8 Гц, 2H).
Пример 142
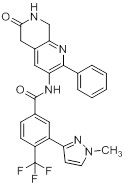
3-(1-Метил-1H-пиразол-3-ил)-N-(6-оксо-2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: Метил-2-(2-циано-5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)ацетат
К перемешанному раствору этил-2-(5-амино-2-циано-6-фенилпиридин-3-ил)ацетата (83 мг, 0,29 ммоль) и 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (80 мг, 0,28 ммоль) в пиридине (3 мл) при 25°C по каплям добавляли POCl3 (0,040 мл, 0,43 ммоль). Полученную смесь перемешивали при 25°C в течение 15 мин, затем осторожно разбавляли насыщенным водным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=534,2 (M+ 1).
Стадия B: 3-(1-Метил-1H-пиразол-3-ил)-N-(6-оксо-2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамид
Раствор этил-2-(2-циано-5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)ацетата (80 мг, 0,15 ммоль) и никеля Ренея (17,6 мг, 0,03 ммоль) в MeOH (10 мл) перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 25°C в течение 5 ч. Смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=492,2 (M+ H). 1H-ЯМР (400 МГц, CD3OD) δ 8,03 (с, 1H), 7,97 (с, 1H), 7,94-7,86 (м, 2H), 7,68 (д, J=2,5 Гц, 1H), 7,64 (д, J=6,5 Гц, 2H), 7,49-7,41 (м, 3H), 6,47 (с, 1H), 4,64 (с, 2H), 3,98 (с, 3H), 3,76 (с, 2H).
Пример 143
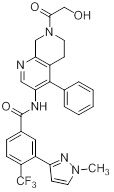
N-(7-(2-Гидроксиацетил)-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: трет-Бутил-3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилат
К раствору трет-бутил-3-амино-4-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилата (500 мг, 1,54 ммоль) и 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (415 мг, 1,54 ммоль) в пиридине (5 мл) при 20°C добавляли фосфорилтрихлорид (236 мг, 1,54 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем распределяли между водой (20 мл) и этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=578,3 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,59 (с, 1H), 7,84-7,77 (м, 2H), 7,73 (д, J=7,8 Гц, 1H), 7,64 (д, J=2,0 Гц, 1H), 7,48-7,41 (м, 2H), 7,38 (д, J=7,0 Гц, 1H), 7,26 (д, J=7,0 Гц, 2H), 6,40 (с, 1H), 4,71 (с, 2H), 3,94 (с, 3H), 3,59 (с, 2H), 2,59 (т, J=5,3 Гц, 2H), 1,48 (с, 9H).
Стадия B: 3-(1-Метил-1H-пиразол-3-ил)-N-(4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамид
К раствору трет-бутил-3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилата (200 мг, 0,346 ммоль) в диоксане (20 мл) при 20°C добавляли 4M раствор HCl в диоксане (20 мл). Полученную смесь перемешивали в течение 10 мин, а затем концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=478,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,54 (с, 1H), 7,82-7,76 (м, 2H), 7,71 (д, J=8,2 Гц, 1H), 7,63 (д, J=2,0 Гц, 1H), 7,47-7,40 (м, 2H), 7,39-7,32 (м, 1H), 7,24 (д, J=7,0 Гц, 2H), 6,39 (с, 1H), 4,08 (с, 2H), 3,94 (с, 3H), 3,01 (т, J=5,7 Гц, 2H), 2,56 (т, J=5,5 Гц, 2H).
Стадия C: N-(7-(2-Гидроксиацетил)-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь 2-гидроксиуксусной кислоты (29 мг, 0,38 ммоль), HATU (143 мг, 0,38 ммоль) и триэтиламина (0,053 мл, 0,38 ммоль) в DMF (5 мл) перемешивали при 20°C в течение 10 мин, после чего добавляли 3-(1-метил-1H-пиразол-3-ил)-N-(4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамид (90 мг, 0,19 ммоль). Полученную смесь перемешивали в течение 2 ч, а затем фильтровали и очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=536,2 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 10,08 (с, 1H), 8,46 (д, J=6,3 Гц, 1H), 7,85 (д, J=11,0 Гц, 2H), 7,80-7,69 (м, 2H), 7,43-7,28 (м, 3H), 7,23 (д, J=7,0 Гц, 2H), 6,38 (с, 1H), 4,69 (д, J=16,8 Гц, 3H), 4,20 (д, J=5,1 Гц, 2H), 3,88 (с, 3H), 3,64-3,51 (м, 2H), 2,56 (с, 2H).
Пример 144
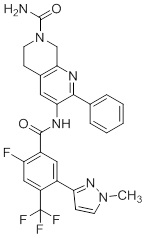
3-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксамид
Стадия A: трет-Бутил-3-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилат
К перемешанному раствору трет-бутил-3-амино-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилата (30 мг, 0,092 ммоль) и 2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензойной кислоты (26 мг, 0,090 ммоль) в пиридине (2 мл) при 25°C по каплям добавляли POCl3 (0,010 мл, 0,11 ммоль). Полученную смесь перемешивали в течение 15 мин, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (10 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=594,2 (M+1).
Стадия B: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамид
Раствор трет-бутил-3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксилата (70 мг, 0,12 ммоль) в 4M растворе HCl в диоксане (2 мл) перемешивали при 26°C в течение 2 ч. Содержащую продукт смесь концентрировали, и подщелачивали остаток добавлением насыщенного водного раствора K2CO3 (2 мл). Водную смесь экстрагировали DCM (4 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=496,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,24 (с, 1H), 7,92 (д, J=7,0 Гц, 1H), 7,68-7,62 (м, 4H), 7,46-7,51 (м, 3H), 6,44 (с, 1H), 4,44 (с, 2H), 3,97 (с, 3H), 3,61 (т, J=6,3 Гц, 2H), 3,24 (т, J=6,1 Гц, 2H).
Стадия C: 3-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамида (20 мг, 0,040 ммоль) в DCM (0,5 мл) при 28°C добавляли изоцианатотриметилсилан (9 мг, 0,08 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем распределяли между водой (1 мл) и DCM (2 мл×3). Объединенные органические слои объединяли, сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=539,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,09 (с, 1H), 7,89 (д, J=7,0 Гц, 1H), 7,67-7,60 (м, 4H), 7,50-7,43 (м, 3H), 6,43 (с, 1H), 4,67 (с, 2H), 3,96 (с, 3H), 3,75 (т, J=5,7 Гц, 2H), 2,98 (т, J=5,5 Гц, 2H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№
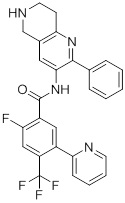
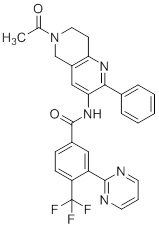
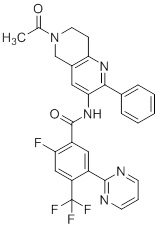
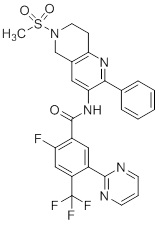
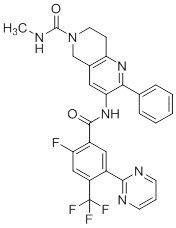
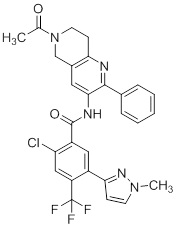
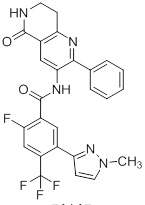
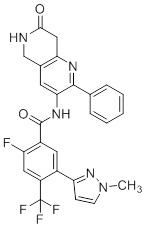
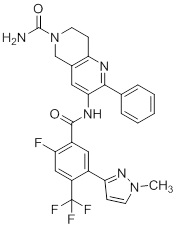
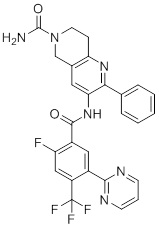
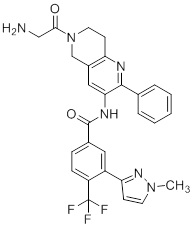

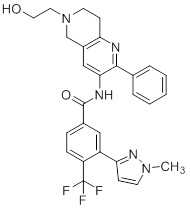
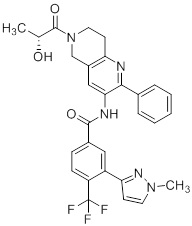
Пример 164
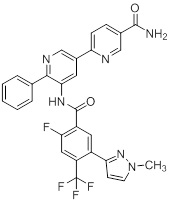
5'-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6'-фенил-[2,3'-бипиридин]-5-карбоксамид
Стадия A: N-(5-Cyano-6'-фенил-[2,3'-бипиридин]-5'-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (100 мг, 0,347 ммоль) в пиридине (2 мл) при 15°C добавляли 5'-амино-6'-фенил-[2,3'-бипиридин]-5-карбонитрил (94 мг, 0,35 ммоль), а затем POCl3 (53 мг, 0,35 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем разбавляли водой (10 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили с Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=543,2 (M+1).
Стадия B: 5'-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6'-фенил-[2,3'-бипиридин]-5-карбоксамид
К раствору N-(5-циано-6'-фенил-[2,3'-бипиридин]-5'-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (50 мг, 0,092 ммоль) в DMSO (2 мл) при 15°C добавляли LiOH (11 мг, 0,46 ммоль), а затем водный раствор H2O2 (37%, 0,008 мл, 0,09 ммоль). Полученную смесь перемешивали в течение 30 мин, а затем фильтровали и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=561,2 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 10,55 (ушир., 1H), 9,32 (ушир., 1H), 9,14 (ушир., 1H), 8,76 (ушир., 1H), 8,35 (д, J=8,2 Гц, 1H), 8,25 (д, J=7,4 Гц, 2H), 7,92-7,82 (м, 2H), 7,79 (д, J=1,6 Гц, 1H), 7,74 (д, J=6,7 Гц, 2H), 7,67 (ушир., 1H), 7,51-7,37 (м, 3H), 6,42 (ушир., 1H), 3,90 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№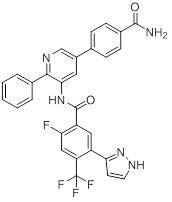
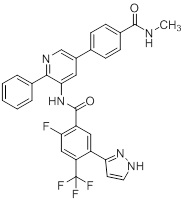

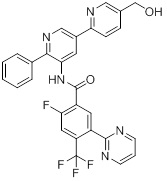
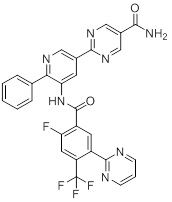
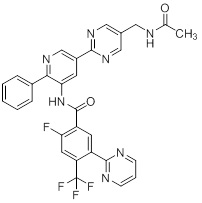
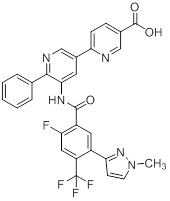



Пример 179
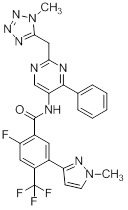
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((1-метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К раствору 2-((1-метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-амина (30 мг, 0,11 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (32 мг, 0,11 ммоль) в пиридине (2 мл) при 19°C добавляли POCl3 (0,021 мл, 0,22 ммоль). Полученную смесь перемешивали в течение 5 мин, а затем разбавляли водой (5 мл) и экстрагировали этилацетатом (5 мл×3). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=538,2 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 10,65 (с, 1H), 8,93 (с, 1H), 7,86 (м, 2 H), 7,78 (д, J=1,6 Гц, 1H), 7,72 (м, 2H), 7,47 (д, J=3,5 Гц, 3H), 6,41 (с, 1H), 4,77 (с, 2H), 4,06 (с, 3 H), 3,88 (с, 3H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№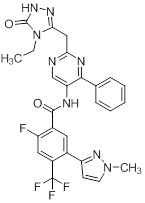
Пример 181
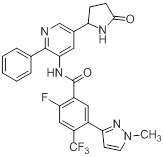
(R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-(5-оксопирролидин-2-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К перемешанному раствору 5-(5-амино-6-фенилпиридин-3-ил)пирролидин-2-она (50 мг, 0,20 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (57 мг, 0,20 ммоль) в пиридине (3 мл) при 20°C по каплям добавляли POCl3 (0,028 мл, 0,30 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем разбавляли водой (15 мл) и экстрагировали EtOAc (15 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (EtOAc/MeOH=20/1), а затем SFC (250 мм×30 мм, 5 мкм, OD колонка), элюируя 55% EtOH (0,1% NH3·H2O) со скоростью 80 мл/мин, с получением указанного в заголовке соединения (элюируемый вторым изомер согласно SFC). MS: m/z=524,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,48 (д, J=1,6 Гц, 1H), 8,31 (с, 1H), 7,91 (д, J=6,7 Гц, 1H), 7,66-7,60 (м, 4H), 7,50-7,44 (м, 3H), 6,42 (ушир. с, 1H), 4,99-4,96 (м, 1H), 3,95 (с, 3H), 2,70 (д, J=7,8 Гц, 1H), 2,52-2,48 (м, 2H), 2,05 (д, J=7,4 Гц, 1H).
Пример 182
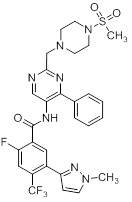
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К смеси 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (46 мг, 0,16 ммоль) в пиридине (5 мл) при 25°C добавляли 2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-амин (50 мг, 0,14 ммоль), а затем POCl3 (0,016 мл, 0,17 ммоль). Полученную смесь перемешивали в течение 15 мин, а затем разбавляли водой (5 мл) и экстрагировали DCM (10 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=618,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,13 (с, 1H), 7,95 (д, J=6,7 Гц, 1H), 7,79 (д, J=3,5 Гц, 2H), 7,71-7,64 (м, 2H), 7,54-7,48 (м, 3H), 6,43 (с, 1H), 3,98-3,90 (м, 5H), 3,26 (с, 4H), 2,83 (с, 3H), 2,76 (д, J=4,3 Гц, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№
Пример 185
N-(5,7-Диоксо-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
Стадия A: Метил-2-циано-5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилникотинат
К раствору 2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензойной кислоты (113 мг, 0,40 ммоль) в пиридине (3 мл) при 22°C добавляли POCl3 (0,18 мл, 2,0 ммоль), а затем метил-5-амино-2-циано-6-фенилникотинат (100 мг, 0,40 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем разбавляли водой (10 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили с Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=522,2 (M+1).
Стадия B: N-(5,7-Диоксо-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамид
К смеси метил-2-циано-5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилникотината (60 мг, 0,12 ммоль) в толуоле (3 мл) добавляли ацетальдегидоксим (7 мг, 0,1 ммоль) и тетрагидрат хлорида индия(III) (34 мг, 0,12 ммоль). Полученную смесь нагревали при 120°C в течение 24 ч, а затем распределяли между водой (10 мл) и EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=508,0 (M+1). 1H-ЯМР (400 МГц, DMSO-d6) δ 11,68 (с, 1H), 10,73 (с, 1H), 8,96 (д, J=4,7 Гц, 2H), 8,59 (ушир., 1H), 8,07 (д, J=5,9 Гц, 1H), 7,97 (д, J=10,2 Гц, 1H), 7,70 (ушир., 2H), 7,58 (т, J=4,9 Гц, 1H), 7,47 (ушир., 3H).
Пример 186

(R или S)-3-(1-Метил-1H-пиразол-3-ил)-N-(6-(2-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-3-циано-2-(5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)пропаноат
К раствору 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (201 мг, 0,698 ммоль) в пиридине (8 мл) при 25°C добавляли оксихлорид фосфора (0,10 мл, 1,1 ммоль) и этил-2-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат (220 мг, 0,75 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=5/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8,07 (д, J=8,2 Гц, 1H), 8,03 (ушир., 1H), 7,93 (д, J=7,8 Гц, 1H), 7,90-7,83 (м, 1H), 7,72-7,60 (м, 3H), 7,50 (д, J=8,2 Гц, 1H), 7,45-7,35 (м, 3H), 6,44 (с, 1H), 4,26-4,13 (м, 2H), 3,93 (с, 3H), 3,47 (с, 1H), 3,26-3,11 (м, 2H), 1,23-1,12 (м, 3H).
Стадия B: (R или S)-3-(1-Метил-1H-пиразол-3-ил)-N-(6-(2-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Смесь этил-3-циано-2-(5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)пропаноата (160 мг, 0,28 ммоль), триэтиламина (0,080 мл, 0,57 ммоль) и никеля Ренея (13 мг, 0,15 ммоль) в метаноле (30 мл) перемешивали в атмосфере водорода (50 фунт./кв.дюйм) при 20°C в течение 3 ч. Смесь фильтровали, и концентрировали фильтрат. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1), а затем SFC (колонка AD (250 мм×30 мм, 5 мкм); подвижная фаза A: сверхкритический CO2, B: EtOH (щелочн.), A/B=55/45 со скоростью 45 мл/мин; длина волны 220 нм) с получением указанного в заголовке соединения (элюируемый первым изомер согласно SFC). MS: m/z=506,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,10-8,00 (м, 2H), 7,93 (д, J=9,3 Гц, 2H), 7,68 (ушир., 3H), 7,44 (д, J=11,0 Гц, 4H), 6,48 (ушир., 1H), 4,06-3,89 (м, 4H), 3,60 (д, J=3,8 Гц, 1H), 3,49 (ушир., 1H), 2,62 (ушир., 2H).
Пример 187
N-(2-((2,5-диоксопирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (100 мг, 0,34 ммоль) в пиридине (5 мл) при 26°C добавляли POCl3 (0,040 мл, 0,52 ммоль) и 1-((5-амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион (98 мг, 0,34 ммоль). Полученную смесь перемешивали в течение 0,5 ч, а затем разбавляли водой (5 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=553,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,06 (с, 1H), 7,96 (д, J=7,0 Гц, 1H), 7,77 (д, J=3,5 Гц, 2H), 7,71-7,65 (м, 2H), 7,51 (ушир., 3H), 6,44 (с, 1H), 4,96 (с, 2H), 3,96 (с, 3H), 2,83 (с, 4H).
Пример 188
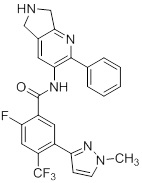
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: трет-Бутил-3-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5H-пирроло[3,4-b]пиридин-6(7H)-карбоксилат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (42 мг, 0,14 ммоль) в пиридине (2 мл) при 25°C добавляли POCl3 (22 мг, 0,14 ммоль) и трет-бутил-3-амино-2-фенил-5H-пирроло[3,4-b]пиридин-6(7H)-карбоксилат (45 мг, 0,14 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (2 мл) и экстрагировали EtOAc (2 мл×3). Объединенные органические слои сушили с Na2SO4 и концентрировали с получением указанного в заголовке соединения. MS: m/z=528,2 (M+1).
Стадия B: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-4-(трифторметил)бензамид
Раствор трет-бутил-3-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5H-пирроло[3,4-b]пиридин-6(7H)-карбоксилата (50 мг, 0,086 ммоль) в 4M растворе HCl в диоксане (10 мл, 40 ммоль) перемешивали при 26°C в течение 30 мин. Содержащую продукт смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=482,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,18-8,12 (м, 1H), 7,87 (д, J=6,7 Гц, 1H), 7,68-7,57 (м, 4H), 7,50-7,43 (м, 3H), 6,43 (ушир., 1H), 4,33 (с, 2H), 4,21 (с, 2H), 3,96 (с, 3H).
Пример 189
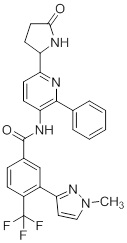
(R или S)-3-(1-Метил-1H-пиразол-3-ил)-N-(6-(5-оксопирролидин-2-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К раствору 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (25 мг, 0,093 ммоль) в пиридине (1 мл) при 30°C добавляли POCl3 (0,030 мл, 0,28 ммоль). Полученную смесь перемешивали в течение 15 мин, после чего по каплям добавляли раствор 5-(5-амино-6-фенилпиридин-2-ил)пирролидин-2-она (35 мг, 0,14 ммоль) в пиридине (2 мл). Полученную смесь перемешивали в течение 30 мин, а затем разбавляли водой (5 мл) и экстрагировали EtOAc (5 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток растворяли в смеси THF (2 мл) и насыщенного водного раствора K2CO3 (2 мл), и перемешивали полученную смесь при 30°C в течение 1 ч. Содержащую продукт смесь экстрагировали EtOAc (5 мл×3), объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (100% EtOAc), а затем SFC ((250 мм×30 мм, 5 мкм, колонка AD), элюируя 40% MeOH (0,1% NH4OH) + 60% CO2 со скоростью 50 мл/мин)), затем методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения (элюируемый вторым изомер согласно SFC). MS: m/z=506,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,07 (д, J=8,0 Гц, 1H), 8,02 (с, 1H), 7,91-7,89 (м, 2H), 7,66-7,65 (м, 3H), 7,48-7,42 (м, 4H), 6,45 (с, 1H), 4,95-4,92 (м, 1H), 3,96 (с, 3H), 2,67-2,50 (м, 1H), 2,49-2,42 (м, 2H), 2,19-2,17 (м, 1H).
Пример 190

(R или S)-N-(2-((2,5-Диоксоимидазолидин-4-ил)метил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (57 мг, 0,21 ммоль) в пиридине (3 мл) при 26°C добавляли POCl3 (39 мг, 0,25 ммоль). Полученную смесь перемешивали в течение 10 мин, после чего добавляли 5-((5-амино-4-фенилпиримидин-2-ил)метил)имидазолидин-2,4-дион (60 мг, 0,21 ммоль). После перемешивания в течение 20 мин, содержащую продукт смесь разбавляли водой (5 мл) и экстрагировали этилацетатом (3×5 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH), а затем SFC ((2,5 см×3 см, 10 мкм, AD колонка), элюируя 50% EtOH (0,1% NH3⋅H2O) + 50% CO2 со скоростью 80 мл/мин), с получением указанного в заголовке соединения (элюируемый вторым изомер согласно SFC). MS: m/z=536,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,90 (с, 1H), 8,08 (с, 1H), 8,01-7,95 (м, 1H), 7,92-7,87 (м, 1H), 7,77 (д, J=3,9 Гц, 2H), 7,65 (д, J=1,6 Гц, 1H), 7,46 (д, J=3,1 Гц, 3H), 6,46 (с, 1H), 4,76-4,66 (м, 1H), 3,95 (с, 3H), 3,60-3,52 (м, 1H), 3,46-3,36 (м, 1H).
Пример 191
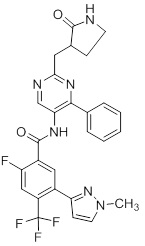
(R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксопирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (25 мг, 0,087 ммоль) в пиридине (2 мл) при 26°C добавляли POCl3 (20 мг, 0,13 ммоль) и 3-((5-амино-4-фенилпиримидин-2-ил)метил)пирролидин-2-он (23 мг, 0,086 ммоль). Полученную смесь перемешивали 0,5 ч, а затем разбавляли водой (3 мл) и экстрагировали CH2Cl2 (5 мл×3). Объединенные органические слои концентрировали, и очищали остаток методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA), а затем SFC ((250×30 мм, OJ колонка), элюируя 35% MeOH (0,1%NH3·H2O) + 65% CO2 со скоростью 60 мл/мин) с получением указанного в заголовке соединения (элюируемый первым изомер согласно SFC). MS: m/z=539,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,97 (с, 1H), 7,90 (д, J=7,0 Гц, 1H), 7,80-7,73 (м, 2H), 7,67-7,59 (м, 2H), 7,45 (д, J=3,1 Гц, 3H), 6,39 (с, 1H), 3,90 (с, 3H), 3,44 (дд, J1=14,5 Гц, J2=3,5 Гц, 1H), 3,29 (дд, J1=8,2 Гц, J2=5,5 Гц, 2H), 3,12-2,96 (м, 2H), 2,34-2,24 (м, 1H), 1,97-1,88 (м, 1H).
Пример 192
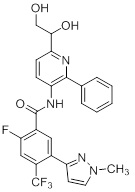
(R или S)-N-(6-(1,2-Дигидроксиэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(6-Хлор-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 6-хлор-2-фенилпиридин-3-амина (197 мг, 0,96 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (277 мг, 0,96 ммоль) в пиридине (5 мл) при 25°C добавляли POCl3 (211 мг, 1,44 ммоль). Полученную смесь перемешивали в течение 2 ч, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (20 мл). Органический слой сушили над Na2SO4 и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=3/1) с получением указанного в заголовке соединения. MS: m/z=475,2 (M+1).
Стадия B: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-6-винилпиридин-3-ил)-4-(трифторметил)бензамид
К дезоксигенированной смеси N-(6-хлор-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (70 мг, 0,15 ммоль), K2CO3 (41 мг, 0,30 ммоль) и трифтор(винил)бората калия (40 мг, 0,30 ммоль) в DME (7 мл) добавляли Pd(dppf)2Cl2 (30 мг). Полученную смесь нагревали при 100°C в течение 2 ч, а затем охлаждали и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=4/1) с получением указанного в заголовке соединения. MS: m/z=467,2 (M+1). 1H-ЯМР (400 МГц, DMSO) δ 8,19-8,09 (м, 1H), 8,05-7,87 (м, 3H), 7,84-7,75 (м, 1H), 7,69 (д, J=7,5 Гц, 1H), 7,59 (д, J=8,5 Гц, 1H), 7,47-7,32 (м, 3H), 6,89 (дд, J=10,8, 17,3 Гц, 1H), 6,46 (с, 1H), 6,27 (д, J=17,6 Гц, 1H), 5,52 (д, J=11,5 Гц, 1H), 3,92 (с, 3H).
Стадия C: (R или S)-N-(6-(1,2-Дигидроксиэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-6-винилпиридин-3-ил)-4-(трифторметил)бензамида (47 мг, 0,10 ммоль) и NMO (117 мг, 1,00 ммоль) в смеси THF (5 мл) и H2O (1 мл) при 25°C добавляли OsO4 (100 мг, 0,39 ммоль). Полученную смесь перемешивали в течение 2 ч, а затем распределяли между насыщенным водным раствором Na2SO3 (20 мл) и EtOAc (20 мл). Органический слой концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/4), а затем SFC ((250×30 мм, IC колонка), элюируя 35% EtOH (0,05% DEA) + 65% CO2 со скоростью 70 мл/мин), с получением указанного в заголовке соединения (элюируемый первым изомер согласно SFC). MS: m/z=501,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,14 (д, J=8,6 Гц, 1H), 7,80 (д, J=7,0 Гц, 1H), 7,71-7,56 (м, 5H), 7,44-7,29 (м, 3H), 6,34 (с, 1H), 4,74-4,71 (м, 1H), 3,87 (с, 3H), 3,81 (дд, J1=11,2 Гц, J2=4,1 Гц, 1H), 3,66 (дд, J1=11,3 Гц, J2=6,7 Гц, 1H).
Пример 193
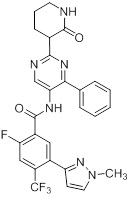
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксопиперидин-3-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (65 мг, 0,22 ммоль) и 3-(5-амино-4-фенилпиримидин-2-ил)пиперидин-2-она (60 мг, 0,22 ммоль) в пиридине (2 мл) при 25°C добавляли POCl3 (0,031 мл, 0,34 ммоль). Полученную смесь перемешивали в течение 0,5 ч, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (10 мл) и EtOAc (10 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток растворяли в смеси THF (5 мл) и воды (5 мл), и добавляли моногидрат гидроксида лития (28 мг, 0,67 ммоль). Полученную смесь перемешивали при 25°C в течение 10 мин, а затем экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=539,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,00 (с, 1H), 7,88 (д, J=6,7 Гц, 1H), 7,72 (д, J=3,5 Гц, 2H), 7,64-7,55 (м, 2H), 7,43 (д, J=3,1 Гц, 3H), 6,36 (с, 1H), 3,94 (т, J=7,8 Гц, 1H), 3,88 (с, 3H), 3,43-3,28 (м, 2H), 2,28-2,08 (м, 2H), 1,94 (д, J=4,7 Гц, 1H), 1,79 (td, J=4,6, 9,1 Гц, 1H).
Пример 194
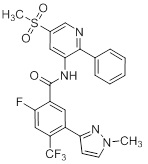
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-(метилсульфонил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (58 мг, 0,20 ммоль) и 5-(метилсульфонил)-2-фенилпиридин-3-амина (50 мг, 0,20 ммоль) в пиридине (3 мл) при 23°C добавляли оксихлорид фосфора (0,038 мл, 0,40 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×3). Объединенные органические слои сушили над безводным сульфатом натрия и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=519,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,51 (д, J=1,8 Гц, 1H), 9,03 (д, J=2,0 Гц, 1H), 8,91 (д, J=14,6 Гц, 1H), 8,48 (д, J=7,8 Гц, 1H), 7,68-7,59 (м, 5H), 7,50 (д, J=12,0 Гц, 1H), 7,44 (д, J=2,3 Гц, 1H), 6,48 (с, 1H), 4,00 (с, 3H), 3,24 (с, 3H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№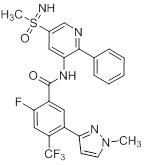
Пример 196
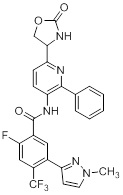
(R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксооксазолидин-4-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К перемешанному раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (43 мг, 0,15 ммоль) и 4-(5-амино-6-фенилпиридин-2-ил)оксазолидин-2-она (38 мг, 0,15 ммоль) в пиридине (2 мл) при 25°C по каплям добавляли POCl3 (0,021 мл, 0,22 ммоль). Полученную смесь перемешивали в течение 15 мин, а затем разбавляли 2M водным раствором LiOH (10 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=2/1), а затем SFC ((250×30 мм, AS колонка), элюируя 35% MeOH (0,05% DEA) + 65% CO2 со скоростью 80 мл/мин, → 100% MeOH) с получением указанного в заголовке соединения (элюируемый первым изомер согласно SFC). MS: m/z=526,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,22 (д, J=8,2 Гц, 1H), 7,82 (д, J=6,7 Гц, 1H), 7,60-7,55 (м, 4H), 7,44 (д, J=8,2 Гц, 1H), 7,41-7,34 (м, 3H), 6,34 (с, 1H), 5,03 (дд, J1=9,0 Гц, J2=5,5 Гц, 1H), 4,73-4,71 (м, 1H), 4,38 (дд, J1=8,4 Гц, J2=5,7 Гц, 1H), 3,87 (с, 3H).
Пример 197
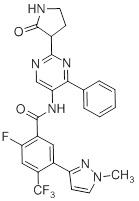
(R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксопирролидин-3-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
Стадия A: Метил-3-циано-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)пропаноат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (67 мг, 0,23 ммоль) и метил-2-(5-амино-4-фенилпиримидин-2-ил)-3-цианопропаноата (70 мг, 0,23 ммоль) в пиридине (3 мл) при 25°C добавляли POCl3 (0,043 мл, 0,47 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем разбавляли водой (20 мл) и экстрагировали EtOAc (10 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=574,9 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,86 (с, 1H), 8,71 (д, J=14,1 Гц, 1H), 8,46 (д, J=7,4 Гц, 1H), 7,68 (д, J=3,5 Гц, 2H), 7,58 (д, J=3,5 Гц, 3H), 7,48 (д, J=12,1 Гц, 1H), 7,39 (д, J=2,0 Гц, 1H), 6,44 (с, 1H), 4,42 (т, J=7,4 Гц, 1H), 3,96 (с, 3H), 3,77 (с, 3H), 3,29-3,14 (м, 2H).
Стадия B: Метил-4-амино-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)бутаноат
К дезоксигенированному раствору метил-3-циано-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)пропаноата (67 мг, 0,10 ммоль) в EtOAc (2 мл) при 25°C добавляли Et3N (0,044 мл, 0,31 ммоль) и никель Ренея (6 мг, 0,1 ммоль). Полученную смесь перемешивали в атмосфере H2 (50 фунт./кв.дюйм) при 25°C в течение 2 ч, а затем фильтровали и концентрировали с получением указанного в заголовке соединения. MS: m/z=557,2 (M+1).
Стадия C: (R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксопирролидин-3-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К раствору метил-4-амино-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)бутаноата (53 мг, 0,095 ммоль) в CH3CN (2 мл) при 28°C добавляли K2CO3 (40 мг, 0,29 ммоль). Полученную смесь нагревали при 80°C в течение 18 ч, а затем охлаждали, фильтровали и очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH), а затем SFC ((250×30 мм, AS колонка), элюируя 35% MeOH (0,05% DEA) + 65% CO2 со скоростью 80 мл/мин, → 100% MeOH) с получением указанного в заголовке соединения (элюируемый вторым изомер согласно SFC). MS: m/z=525,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 9,81 (с, 1H), 8,63 (д, J=13,7 Гц, 1H), 8,46 (д, J=4,3 Гц, 1H), 7,68 (д, J=3,5 Гц, 2H), 7,56 (с, 3H), 7,48 (д, J=11,7 Гц, 1H), 7,40 (с, 1H), 6,45 (с, 1H), 5,79 (с, 1H), 4,15-4,05 (м, 1H), 3,97 (с, 3H), 3,64 (с, 1H), 3,50 (д, J=0,8 Гц, 1H), 2,79-2,60 (м, 2H).
Пример 198
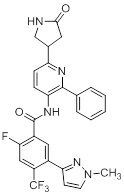
(R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(5-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-3-циано-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)пропаноат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (110 мг, 0,381 ммоль) в пиридине (5 мл) при 22°C добавляли POCl3 (0,10 мл, 1,1 ммоль). После перемешивания в течение 5 мин, добавляли этил-3-(5-амино-6-фенилпиридин-2-ил)-3-цианопропаноат (125 мг, 0,381 ммоль). Полученную смесь перемешивали в течение 20 мин, а затем разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=566,2 (M+1).
Стадия B: Этил-4-амино-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)бутаноат
Смесь этил-3-циано-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)пропаноата (30 мг, 0,048 ммоль) и никеля Ренея (0,4 мг, 0,005 ммоль) в EtOH (10 мл) перемешивали в атмосфере водорода (50 фунт./кв.дюйм) при 26°C в течение 2 ч. Смесь фильтровали, и концентрировали фильтрат с получением указанного в заголовке соединения. MS: m/z=570,3 (M+1).
Стадия C: (R или S)-2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(5-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К раствору этил-4-амино-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)бутаноата (10 мг, 0,012 ммоль) в EtOH (1 мл) при 22°C добавляли карбонат калия (5 мг, 0,04 ммоль). Полученную смесь перемешивали в течение 12 ч, а затем распределяли между водой (5 мл) и этилацетатом (5 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом препаративной TLC (CH2Cl2/MeOH=10/1), а затем SFC ((2,5 см×3 см, 5 мкм, AD колонка), элюируя 50% EtOH (0,1% NH4OH) + 50% CO2 со скоростью 50 мл/мин) с получением указанного в заголовке соединения (элюируемый первым изомер согласно SFC). MS: m/z=524,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,09 (д, J=8,2 Гц, 1H), 7,81 (д, J=6,7 Гц, 1H), 7,58 (д, J=6,3 Гц, 4H), 7,43-7,28 (м, 4H), 6,34 (с, 1H), 3,93-3,82 (м, 4H), 3,73 (т, J=9,0 Гц, 1H), 3,53 (дд, J=7,0, 9,8 Гц, 1H), 2,75-2,60 (м, 2H).
Пример 199
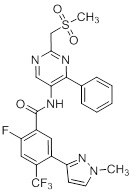
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((метилсульфонил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамид
К смеси 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (80 мг, 0,278 ммоль) в пиридине (3 мл) при 25°C добавляли POCl3 (0,039 мл, 0,41 ммоль), а затем 2-((метилсульфонил)метил)-4-фенилпиримидин-5-амин (88 мг, 0,33 ммоль). Полученную смесь перемешивали в течение 50 мин, а затем разбавляли водой (5 мл) и экстрагировали DCM (5 мл×2). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=534,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,23 (с, 1H), 7,99 (д, J=7,0 Гц, 1H), 7,81 (д, J=3,5 Гц, 2H), 7,68-7,64 (м, 2H), 7,51 (д, J=3,1 Гц, 3H), 6,44 (с, 1H), 4,82-4,72 (м, 2H), 3,94 (с, 3H), 3,20 (с, 3H).
Следующий пример получали способом, аналогичным описанным выше методикам.
№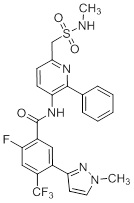
Пример 201
N-(2-((2,5-Диоксопирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (10 мг, 0,035 ммоль) в пиридине (0,2 мл) при 18°C добавляли фосфорилтрихлорид (8 мг, 0,05 ммоль). После перемешивания в течение 5 мин, добавляли 3-((5-амино-4-фенилпиримидин-2-ил)метил)пирролидин-2,5-дион (10 мг, 0,035 ммоль). Полученную смесь перемешивали в течение 20 мин, а затем концентрировали. Остаток очищали методом HPLC с обращенной фазой (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=553,2 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 9,01 (с, 1H), 7,95-7,93 (м, 1H), 7,80-7,65 (м, 4H), 7,58-7,45 (м, 3H), 6,43 (с, 1H), 3,94 (с, 3H), 3,48 (с, 3H), 2,93-2,89 (м, 1H), 2,69-2,49 (м, 1H).
Пример 202
N-(5-(2-Амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)ацетат
К раствору этил-2-(5-амино-6-фенилпиридин-3-ил)ацетата (103 мг, 0,321 ммоль) (80%) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (93 мг, 0,32 ммоль) в пиридине (3 мл) при 25°C добавляли оксихлорид фосфора (0,060 мл, 0,64 ммоль). Полученную смесь перемешивали в течение 10 мин, а затем распределяли между водой (20 мл) и этилацетатом (10 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=1/2) с получением указанного в заголовке соединения. MS: m/z=527,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,87 (с, 1H), 8,65 (д, J=13,7 Гц, 1H), 8,40 (с, 2H), 7,61-7,36 (м, 7H), 6,43 (с, 1H), 4,20 (кв, J=7,0 Гц, 2H), 3,95 (с, 3H), 3,72 (с, 2H), 1,29 (т, J=7,0 Гц, 3H).
Стадия B: 2-(5-(2-Фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6–фенил пиридин-3-ил)уксусная кислота
К раствору этил-2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)ацетата (60 мг, 0,11 ммоль) в этаноле (3 мл) и воде (1 мл) при 25°C добавляли гидроксид лития (8 мг, 0,3 ммоль). Полученную смесь перемешивали в течение 50 мин, а затем распределяли между водой (20 мл) и этилацетатом (10 мл×3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения. MS: m/z=499,1 (M+1).
Стадия C: N-(5-(2-Амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)уксусной кислоты (10 мг, 0,020 ммоль) в DMF (3 мл) при 25°C добавляли триэтиламин (8 мкл, 0,06 ммоль) и HATU (11 мг, 0,030 ммоль). Смесь перемешивали при 10 мин, после чего добавляли хлорид аммония (2 мг, 0,04 ммоль). Полученную смесь перемешивали в течение 1 ч, а затем фильтровали и очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=498,1 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,86 (с, 1H), 8,46-8,36 (м, 2H), 7,60-7,49 (м, 5H), 7,44 (д, J=11,9 Гц, 1H), 7,39 (с, 1H), 6,43 (с, 1H), 3,95 (с, 3H), 3,68 (с, 2H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№
Пример 205
N-(6-(2-Амино-2-оксоэтил)-3-фенилпиридин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Этил-2-(6-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-5-фенилпиридин-2-ил)ацетат
К раствору этил-2-(6-амино-5-фенилпиридин-2-ил)ацетата (400 мг, 1,56 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (495 мг, 1,72 ммоль) в пиридине (5 мл) при 20°C добавляли POCl3 (0,15 мл, 1,6 ммоль). Полученную смесь перемешивали в течение 30 мин, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (30 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 и солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом флэш-хроматографии на силикагеле (ISCO; флэш-колонка SepaFlash® с 12 г силикагеля, 0-50% EtOAc в петролейный эфире, 30 мл/мин, сухой ввод образца) с получением указанного в заголовке соединения. MS: m/z=527,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,42 (ушир., 1H), 8,16 (ушир., 1H), 7,75 (д, J=6,0 Гц, 1H), 7,69 (д, J=7,0 Гц, 1H), 7,52-7,42 (м, 4H), 7,41-7,36 (м, 2H), 6,43 (д, J=18,6 Гц, 2H), 3,98-3,91 (м, 4H), 1,25 (кв, J=7,0 Гц, 3H).
Стадия B: N-(6-(2-Амино-2-оксоэтил)-3-фенилпиридин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору этил-2-(6-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-5-фенилпиридин-2-ил)ацетата (200 мг, 0,380 ммоль) в MeOH (2 мл) при 20°C добавляли водный раствор гидроксида аммония (35%, 1 мл, 7 ммоль). Полученную смесь перемешивали в течение 16 ч, а затем распределяли между насыщенным водным раствором NaHCO3 (30 мл) и этилацетатом (20 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=498,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 7,84 (д, J=8,0 Гц, 1H), 7,79 (д, J=7,0 Гц, 1H), 7,67-7,61 (м, 2H), 7,52-7,47 (м, 2H), 7,46-7,40 (м, 3H), 7,38-7,33 (м, 1H), 6,42 (с, 1H), 3,96 (с, 3H), 3,78 (с, 2H).
Пример 206
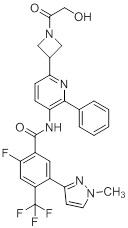
2-Фтор-N-(6-(1-(2-гидроксиацетил)азетидин-3-ил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: трет-Бутил-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)азетидин-1-карбоксилат
К раствору трет-бутил-3-(5-амино-6-фенилпиридин-2-ил)азетидин-1-карбоксилата (40 мг, 0,12 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (39 мг, 0,14 ммоль) в пиридине (2 мл) при 26°C добавляли фосфорилтрихлорид (19 мг, 0,12 ммоль). Полученную смесь перемешивали в течение 30 мин, а затем осторожно разбавляли насыщенным водным раствором NaHCO3 (30 мл) и этилацетатом (20 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом препаративной TLC (PE/EtOAc=3/1) с получением указанного в заголовке соединения. 1H-ЯМР (400 МГц, CDCl3) δ 8,70 (д, J=5,1 Гц, 1H), 8,36 (д, J=7,8 Гц, 1H), 8,03 (д, J=12,9 Гц, 1H), 7,57-7,47 (м, 4H), 7,42 (д, J=2,3 Гц, 4H), 6,47 (ушир., 1H), 4,32 (т, J=8,2 Гц, 2H), 4,07-3,93 (м, 6H), 1,46 (с, 9H).
Стадия B: N-(6-(Азетидин-3-ил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору трет-бутил-3-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)азетидин-1-карбоксилата (22 мг, 0,035 ммоль) в DCM (2 мл) при 26°C добавляли TFA (0,027 мл, 0,35 ммоль), и перемешивали полученную смесь в течение 1 ч. Содержащую продукт смесь распределяли между насыщенным водным раствором NaHCO3 (30 мл) и DCM (20 мл×3). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=496,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,89 (д, J=7,8 Гц, 1H), 8,74 (д, J=14,1 Гц, 1H), 8,42 (д, J=7,4 Гц, 1H), 7,66-7,55 (м, 5H), 7,48 (д, J=11,7 Гц, 1H), 7,42 (с, 1H), 6,46 (с, 1H), 4,47 (ушир., 4H), 4,29 (ушир., 1H), 3,98 (с, 3H).
Стадия C: 2-Фтор-N-(6-(1-(2-гидроксиацетил)азетидин-3-ил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 2-гидроксиуксусной кислоты (21 мг, 0,28 ммоль) в CH2Cl2 (4 мл) при 15°C добавляли HOBt (54 мг, 0,35 ммоль), DIEA (0,062 мл, 0,35 ммоль), EDC (60 мг, 0,31 ммоль) и N-(6-(азетидин-3-ил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид (70 мг, 0,14 ммоль). Полученную смесь перемешивали в течение 16 ч, а затем распределяли между водой (10 мл) и DCM (10 мл×4). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=554,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,97 (д, J=8,0 Гц, 1H), 8,72 (д, J=13,6 Гц, 1H), 8,39 (д, J=8,0 Гц, 1H), 7,66-7,53 (м, 5H), 7,47 (д, J=12,0 Гц, 1H), 7,43 (д, J=2,0 Гц, 1H), 7,38 (д, J=8,5 Гц, 1H), 6,46 (с, 1H), 4,58-4,34 (м, 4H), 4,20 (д, J=6,5 Гц, 1H), 4,08 (ушир. с, 2H), 4,00 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№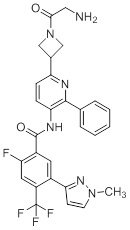
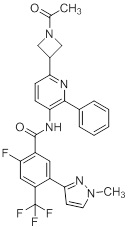
Пример 209
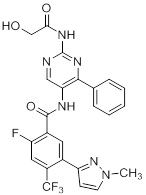
2-Фтор-N-(2-(2-гидроксиацетамидо)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(2-Хлор-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К перемешанному раствору 2-хлор-4-фенилпиримидин-5-амина (206 мг, 1,00 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (289 мг, 1,00 ммоль) в пиридине (4 мл) при 20°C добавляли фосфорилтрихлорид (184 мг, 1,20 ммоль). Полученную смесь перемешивали в течение 5 мин, а затем осторожно разбавляли насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали этилацетатом (5 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=476,2 (M+1).
Стадия B: 2-Фтор-N-(2-(2-гидроксиацетамидо)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
К дезоксигенированной смеси N-(2-хлор-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (110 мг, 0,231 ммоль), 2-гидроксиацетамида (52 мг, 0,69 ммоль) и гидрофосфата калия (121 мг, 0,694 ммоль) в диоксане (2 мл) добавляли Xantphos (13 мг, 0,023 ммоль) и Pd2(dba)3 (21 мг, 0,023 ммоль). Полученную смесь нагревали при 100°C в течение 15 мин в микроволновом реакторе, а затем охлаждали и распределяли между водой (5 мл) и этилацетатом (5 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали методом препаративной TLC (этилацетат), а затем методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=515,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,79 (с, 1H), 7,92 (д, J=6,8 Гц, 1H), 7,81 (д, J=3,5 Гц, 2H), 7,69 (д, J=10,6 Гц, 1H), 7,64 (д, J=2,0 Гц, 1H), 7,49 (д, J=2,3 Гц, 3H), 6,43 (с, 1H), 4,92 (с, 2H), 3,95 (с, 3H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№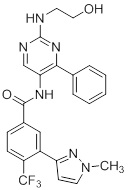
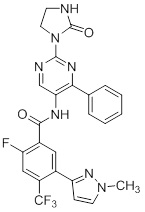
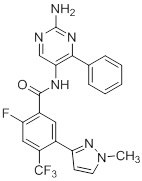
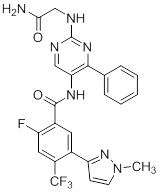
Пример 214
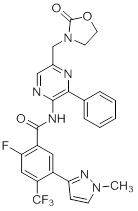
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-((2-оксооксазолидин-3-ил)метил)-3-фенилпиразин-2-ил)-4-(трифторметил)бензамид
Стадия A: трет-Бутил-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиразин-2-ил)метил)карбамат
К раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (202 мг, 0,700 ммоль) в пиридине (6 мл) при 20°C добавляли фосфорилтрихлорид (129 мг, 0,840 ммоль), а затем трет-бутил-((5-амино-6-фенилпиразин-2-ил)метил)карбамат (290 мг, 0,966 ммоль). Полученную смесь перемешивали в течение 30 мин, а затем разбавляли водой (3 мл) и экстрагировали EtOAc (5 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом колоночной хроматографии на силикагеле (PE/EtOAc=5/1, 2/1, 1/1) с получением указанного в заголовке соединения. MS: m/z=571,3 (M+1).
Стадия B: N-(5-(Аминометил)-3-фенилпиразин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь трет-бутил-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиразин-2-ил)метил)карбамата (220 мг, 0,355 ммоль) в 4M растворе HCl в диоксане (5 мл, 20 ммоль) нагревали при 50°C в течение 30 мин. Содержащую продукт смесь концентрировали. Остаток растворяли в воде (5 мл), подщелачивали эту смесь до pH=7 добавлением насыщенного водного раствора K2CO3 и экстрагировали EtOAc (4 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения. MS: m/z=471,2 (M+1).
Стадия C: 2-Хлорэтил-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиразин-2-ил)метил)карбамат
К раствору N-(5-(аминометил)-3-фенилпиразин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида (20 мг, 0,043 ммоль) в THF (1 мл) при 0°C добавляли триэтиламин (0,018 мл, 0,13 ммоль) и 2-хлорэтилкарбонохлоридат (12 мг, 0,085 ммоль). Полученную смесь перемешивали в течение 30 мин, а затем распределяли между водой (2 мл) и EtOAc (3 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения. MS: m/z/ 577,2 (M+1).
Стадия D: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-((2-оксооксазолидин-3-ил)метил)-3-фенилпиразин-2-ил)-4-(трифторметил)бензамид
К раствору 2-хлорэтил-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиразин-2-ил)метил)карбамата (45 мг, 0,058 ммоль) в THF (1 мл) при 0°C добавляли гидрид натрия (7 мг, 0,2 ммоль). Полученную смесь перемешивали в течение 2 ч, а затем разбавляли водой (1 мл) и экстрагировали EtOAc (2 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=541,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,40 (с, 1H), 7,72 (д, J=7,2 Гц, 3H), 7,61-7,55 (м, 2H), 7,39-7,32 (м, 3H), 6,34 (с, 1H), 4,60 (с, 2H), 4,32 (т, J=8,0 Гц, 2H), 3,86 (с, 3H), 3,67 (т, J=8,1 Гц, 2H).
Пример 215
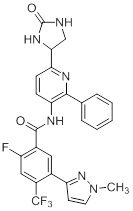
2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксоимидазолидин-4-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
Стадия A: Ди-трет-бутил-(1-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамат
К перемешанному раствору 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (200 мг, 0,693 ммоль) и ди-трет-бутил-(1-(5-амино-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамата (270 мг, 0,630 ммоль) в пиридине (3 мл) при 15°C по каплям добавляли POCl3 (0,088 мл, 0,95 ммоль). Полученную смесь перемешивали в течение 15 мин, а затем распределяли между насыщенным водным раствором LiOH (10 мл) и EtOAc (10 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=1/1) с получением указанного в заголовке соединения. MS: m/z=699,2 (M+1). 1H-ЯМР (400 МГц, CDCl3) δ 8,77 (д, J=8,0 Гц, 1H), 8,57 (д, J=13,3 Гц, 1H), 8,35 (д, J=7,4 Гц, 1H), 7,57-7,43 (м, 5H), 7,38 (д, J=11,7 Гц, 1H), 7,34-7,25 (м, 4H), 6,37 (с, 1H), 3,48 (д, J=5,3 Гц, 3H), 1,38 (с, 18H).
Стадия B: N-(6-(1,2-Диаминоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор ди-трет-бутил-(1-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)этан-1,2-диил)дикарбамата (280 мг, 0,401 ммоль) в 4M растворе HCl в EtOAc (10 мл) перемешивали при 15°C в течение 30 мин. Смесь концентрировали с получением указанного в заголовке соединения в виде гидрохлорида. MS: m/z=499,2 (M+1).
Стадия C: 2-Фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксоимидазолидин-4-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамид
К перемешанному раствору N-(6-(1,2-диаминоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида гидрохлорида (70 мг, 0,12 ммоль) в THF (5 мл) при 15°C добавляли CDI (39,7 мг, 0,245 ммоль). Смесь перемешивали в течение 15 мин, после чего добавляли K2CO3 (51 мг, 0,37 ммоль). Полученную смесь нагревали при 60°C в течение 2 ч, а затем охлаждали и распределяли между водой (30 мл) и EtOAc (30 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=525,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,30 (д, J=8,4 Гц, 1H), 7,91 (д, J=7,1 Гц, 1H), 7,69-7,64 (м, 4H), 7,59 (д, J=8,4 Гц, 1H), 7,51-7,43 (м, 3H), 6,43 (с, 1H), 5,01 (дд, J1=9,4 Гц, J2=6,3 Гц, 1H), 4,00-3,97 (м, 1H), 3,97 (с, 3H), 3,50 (дд, J1=9,2 Гц, J2=6,3 Гц, 1H).
Пример 216

N-(2-(2-Амино-2-оксоэтокси)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: Метил-2-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)окси)ацетат
К перемешанному раствору метил-2-((5-амино-4-фенилпиримидин-2-ил)окси)ацетата гидрохлорида (42 мг, 0,14 ммоль) и 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)-бензойной кислоты (41 мг, 0,14 ммоль) в пиридине (2 мл) при 15°C добавляли фосфорилтрихлорид (26 мг, 0,17 ммоль). Полученную смесь перемешивали в течение 15 мин, затем осторожно разбавляли насыщенным водным раствором бикарбоната натрия (5 мл) и экстрагировали этилацетатом (5 мл×3). Объединенные органические слои сушили над сульфатом натрия и концентрировали, и очищали остаток методом препаративной TLC (PE/EtOAc=2/3) с получением указанного в заголовке соединения. MS: m/z=530,2 (M+1).
Стадия B: N-(2-(2-Амино-2-оксоэтокси)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Раствор метил-2-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)окси)ацетата (53 мг, 0,10 ммоль) в 2M растворе NH3 в CH3OH (10 мл, 20 ммоль) перемешивали при 15°C в течение 10 ч. Содержащую продукт смесь концентрировали, и очищали остаток методом HPLC с обращенной фазой в щелочных условиях (градиент H2O/CH3CN с 0,05% NH4OH) с получением указанного в заголовке соединения. MS: m/z=514,1 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,81 (с, 1H), 7,93 (д, J=6,8 Гц, 1H), 7,86-7,79 (м, 2H), 7,71 (д, J=10,6 Гц, 1H), 7,66 (д, J=2,0 Гц, 1H), 7,53-7,47 (м, 3H), 6,45 (с, 1H), 4,94 (с, 2H), 3,96 (с, 3H).
Пример 217
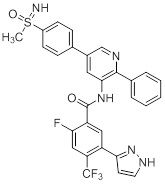
(R или S)-2-Фтор-N-(5-(4-(S-метилсульфонимидоил)фенил)-2-фенилпиридин-3-ил)-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамид
Стадия A: N-(5-бром-2-фенилпиридин-3-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамид
К раствору 5-бром-2-фенилпиридин-3-амина (1,5 г, 6,0 ммоль) и 2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (2,0 г, 6,0 ммоль) в пиридине (20 мл) при 25°C добавляли POCl3 (915 мг, 6,0 ммоль). Полученную смесь перемешивали 2 ч, а затем концентрировали. Остаток очищали методом колоночной хроматографии на силикагеле (PE/EtOAc=3/1) с получением указанного в заголовке соединения.
Стадия B: (R или S)-2-Фтор-N-(5-(4-(S-метилсульфонимидоил)фенил)-2-фенилпиридин-3-ил)-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамид
К дезоксигенированному раствору N-(5-бром-2-фенилпиридин-3-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида (150 мг, 0,30 ммоль) в диоксане (2 мл) добавляли энантиомерно чистый 4,4,5,5-тетраметил-2-(4-(S-метилсульфонимидоил)фенил)-1,3,2-диоксаборолан (S или R, второй пик согласно SFC) (100 мг, 0,36 ммоль), K2CO3 (102 мг, 0,75 ммоль) и Pd(dppf)Cl2 (22 мг, 0,03 ммоль). Полученную смесь нагревали при 80°C в течение 16 ч, а затем охлаждали и распределяли между водой (10 мл) и EtOAc (20 мл×3). Объединенные органические слои сушили над Na2SO4 и концентрировали. Остаток очищали методом HPLC с обращенной фазой в кислых условиях (градиент H2O/CH3CN с 0,1% TFA) с получением указанного в заголовке соединения. MS: m/z=580,0 (M+1). 1H-ЯМР (400 МГц, CD3OD) δ 8,90 (с, 1H), 8,78 (с, 1H), 8,28 (д, J=8,6 Гц, 2H), 8,18 (д, J=8,6 Гц, 2H), 7,96 (д, J=7,1 Гц, 1H), 7,79-7,66 (м, 4H), 7,57-7,43 (м, 3H), 6,50 (с, 1H), 3,73 (с, 3H).
Пример 218
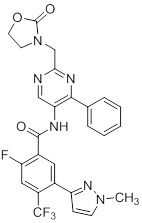
(R или S)-2-фтор-N-(2-(2-оксооксазолидин-3-ил)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамид
Смесь 3-((5-амино-4-фенилпиримидин-2-ил)метил)оксазолидин-2-она (35 мг, 0,13 ммоль), 2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензойной кислоты (41 мг, 0,14 ммоль), HATU (54 мг, 0,14 ммоль) и 2,6-лутидина (0,022 мл, 0,19 ммоль) в DMF (2 мл) перемешивали при 23°C в течение 3 суток. Содержащую продукт смесь очищали методом препаративной HPLC (Separation COE, обращенная фаза, SunFire C-18), элюируя смесью ацетонитрил/вода с 0,05% TFA (15%→65% в течение 15 мин, 50 мл/мин) с получением указанного в заголовке соединения в виде трифторацетата. MS: m/z=541,0 (M+1). 1H-ЯМР (500 МГц, DMSO-d6): δ 10,66 (с, 1H), 8,98 (с, 1H), 7,92-7,87 (м, 2H), 7,82-7,80 (м, 3H), 7,51-7,50 (м, 3H), 6,44 (с, 1H), 4,66 (с, 2H), 4,36 (т, J=8,0 Гц, 2H), 3,91 (с, 3H), 3,75 (т, J=8,0 Гц, 2H).
Следующие примеры получали способом, аналогичным описанным выше методикам.
№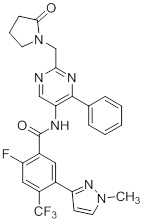
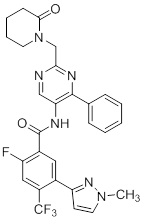
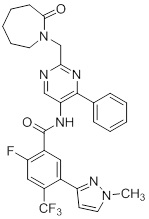

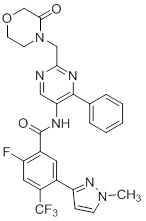
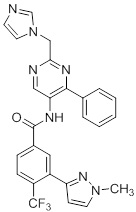
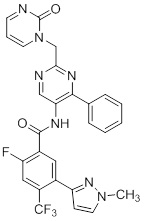
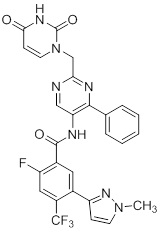
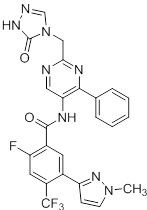
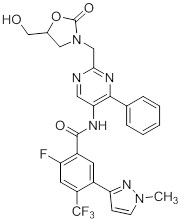
Биологическая полезность
Функциональную активность TrkA измеряли с использованием метода анализа DiscoverX PathHunter. В этом методе анализа клетки U2OS экспрессируют рецептор TrkA человека в виде слитого белка со слабо комплементарным фрагментом β-галактозидазы, который DiscoverX называет «Prolink (PK)»; кроме того, Shc1 слит с большим фрагментом, который называют «Enzyme Acceptor (EA)». Активация рецептора TrkA после присоединения NGF приводит к фосфорилированию киназного домена и к последующему активации белка Shc1-EA. Такая активация приводит к активному ферменту β-галактозидазе, который определяют путем добавления хемилюминесцентного субстрата. Белок p75NTR человека также экспрессируют в качестве корецептора для NGF.
Все реагенты приобретали в DiscoverX, за исключением агонистов рецептора (NGF, BDNF, NT3), которые приобретали в Peprotech. Клетки наращивали и замораживали в криофлаконах, хранили в газовой фазе жидкого азота, и размораживали сразу перед применением. Размороженные клетки вносили в 384-луночный планшет в количестве 7500 клеток на лунку и оставляли инкубироваться в течение ночи. На следующее утро добавляли соединение в различных концентрациях, и оставляли клетки инкубироваться в течение 1 часа. Затем, добавляли NGF в концентрации достаточной для получения ~80% максимального ответа, и оставляли инкубироваться в течение 3 часов при комнатной температуре. Затем добавляли проявляющий реагент DiscoverX PathHunter, и дополнительно инкубировали планшет в течение 1 часа в темноте. Далее проводили считывание люминесценции с планшета на Perkin Elmer Envision.
Для каждой концентрации соединения вычисляли процент ингибирования и определяли значение IC50 с использованием Уравнения 1, представленного ниже.
Уравнение 1:
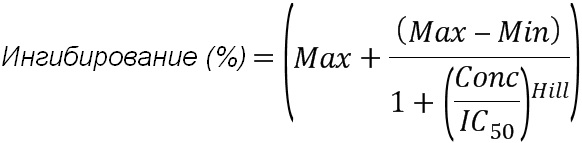
Значения IC50 из вышеупомянутого метода анализа для соединений согласно настоящему изобретения варьируют в пределах от 0,05 нМ до 10000 нМ. Значения IC50 для конкретных соединений согласно настоящему изобретению представлены ниже в Таблице 2.
Таблица 2
Хотя настоящее изобретение было описано и проиллюстрировано со ссылкой на его определенные конкретные варианты осуществления, специалистам в данной области техники следует понимать, что без выхода за рамки сущности и объема настоящего изобретения применительно к процедурам и протоколам могут быть сделаны различные адаптации, изменения, модификации, замены, исключения или добавления. Поэтому, подразумевается, что настоящее изобретение может быть определено объемом последующей формулы изобретения, и что приложенная формула изобретения может интерпретироваться так широко, насколько это целесообразно.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2013 |
|
RU2619465C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ГРИППА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2737190C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743360C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПРОИЗВОДНЫЕ [1,8]НАФТИРИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕПЛИКАЦИИ ВИРУСА HCV | 2006 |
|
RU2467007C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
Изобретение относится к соединению формулы I:
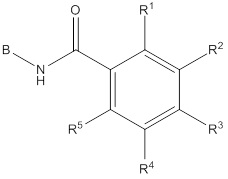 I
I
или его фармацевтически приемлемым солям. Значение радикалов приведено в формуле изобретения. Также предложены ряд конкретных соединений, фармацевтическая композиция и применение соединения формулы I. Соединения являются ингибиторами протеинкиназ семейства тропомиозин-зависимых рецепторных киназ (Trk), а потому применимы для лечения заболевания или нарушения, опосредованного Trk-рецепторами. 5 н. и 13 з.п. ф-лы, 2 табл., 218 пр.
1. Соединение формулы I:
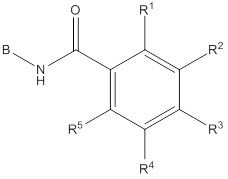 I
I
или его фармацевтически приемлемые соли, где:
B представляет собой шестичленный гетероцикл, выбранный из группы, состоящей из пиридила, пиримидинила, пирадизинила и пиразинила, причем указанный гетероцикл необязательно замещен 1-3 группами Ra;
R представляет собой водород, OH или C1-6алкил, причем упомянутый алкил необязательно замещен 1-3 группами Rf;
R1 и R5 независимо выбирают из группы, состоящей из водорода и галогена;
один из R2 и R4 представляет собой водород, а другой представляет собой необязательно замещенный (CHR)nC5-10гетероцикл, причем упомянутый гетероцикл необязательно замещен 1-3 группами Ra,
R3 представляет собой водород, C1-4галогеналкил, -OC1-4галогеналкил и галоген;
Ra выбирают из группы, состоящей из водорода, –CN, -(CH2)nC(O)ORf, -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CH2)nC3-6циклоалкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -(CH2)nC(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -O-, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, SO2Rd, (CH2)nNHSO2Rd, -(CH2)nSO2N(Rd)2, S(O)(NH)Rg, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNRC(O)NHRd, -(CH2)nNHC(O)ORd, (CHR)nC(O)N(Rd)2, -OC1-6алкила и –OH, причем упомянутые алкил, циклоалкил, арил или гетероцикл необязательно замещены 1-3 группами Rb, где если две группы Rd присоединены к атому азота, то они могут формировать вместе с этим атомом азота 4-8-членный гетероцикл, который необязательно замещен 1-3 группами Rf;
Rb выбирают из группы, состоящей из галогена, C1-6алкила, C1-6алкилOR, C1-4галогеналкила, -(CH2)nN(Rd)2, ORc, -O-, -CN, S(O)(NH)Rg, -SO2R, -SO2N(Rd)2, -(CH2)nC(O)N(Rd)2, -(CH2)nNHC(O)Rd, COR, C(O)OR, C3-6циклоалкила и C1-6алкилN(Rd)2, причем упомянутый алкил и гетероцикл необязательно замещены 1-3 группами Rf;
Rc выбирают из группы, состоящей из водорода, C1-6алкилORg и C1-6алкила;
Rd независимо выбирают из группы, состоящей из водорода, C1-6алкила и (CH2)nC3-6циклоалкила, причем упомянутые алкил, циклоалкил необязательно замещены 1-3 группами Rf; где если две группы Rd присоединены к атому азота, то они могут формировать вместе с этим атомом азота 4-8-членный гетероцикл, который необязательно замещен 1-3 группами Rf;
Rf выбирают из группы, состоящей из водорода, C1-6алкила, ORc, CN, -N(Rc)2, C(O)N(Rg)2, C(O)C1-6алкила, -SO2Rg, -O-, -C1-6алкилSO2Rg, -C1-6алкилORg, -C1-6алкилN(Rg)2,
Rg выбирают из группы, состоящей из водорода, C1-6алкила; и
n равно 0-6.
2. Соединение по п. 1, где B представляет собой незамещенный или замещенный пиридил.
3. Соединение по п. 1, где B представляет собой незамещенный или замещенный пиримидинил.
4. Соединение по п. 1, где гетероцикл в R2 или R4 выбирают из группы, состоящей из необязательно замещенного пиразолила, пиридила, тиазолила, триазолила, оксазолила, оксадиазолила и пиримидинила.
5. Соединение по п. 1, где R3 выбирают из группы, состоящей из водорода, CF3, OCF3, хлора и фтора.
6. Соединение по п. 5, где R3 представляет собой CF3.
7. Соединение по п. 1, где B представляет собой пиримидинил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутые алкил, арил или гетероцикл необязательно замещены 1-3 группами Rb, где n=0-2.
8. Соединение по п. 1, где B представляет собой пиридил, замещенный 1-3 группами Ra, выбранными из группы, состоящей из -C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутые алкил, арил или гетероцикл необязательно замещены 1-3 группами Rb, где n=0-2.
9. Соединение по п. 1, представленное структурной формулой Ia:
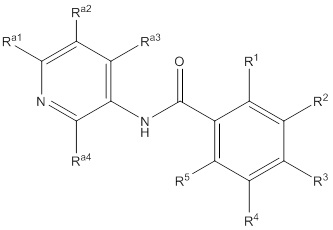 Ia
Ia
или его фармацевтическая соль, где значения R1, R2, R3, R4 и R5 описаны ранее, значения Ra1, Ra2, Ra3 и Ra4 все соответствуют Ra и значение Ra описано ранее.
10. Соединение по п. 9, где Ra1, Ra2, Ra3 и Ra4 независимо выбирают из водорода, C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутые алкил, арил или гетероцикл необязательно замещены 1-3 группами Rb, где по меньшей мере два из Ra1, Ra2, Ra3 и Ra4 на пиридиле представляют собой водород, а один из Ra1, Ra2, Ra3 и Ra4 представляет собой фенил, R1 и R5 независимо выбирают из водорода и галогена, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл.
11. Соединение по п. 1, представленное структурной формулой II:
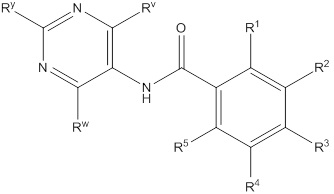 II
II
и его фармацевтически приемлемые соли, где значения R1, R2, R3, R4 и R5 описаны ранее и Rw, Rv и Ry = Ra.
12. Соединение по п. 11, где Rw, Rv и Ry независимо выбирают из водорода, C1-4галогеналкила, -OC1-4галогеналкила, C1-6алкила, -(CHR)nC6-10арила, -(CHR)nC4-10гетероцикла, -C(O)(CHR)nC4-10гетероцикла, -O-(CH2)nC6-10арила, -O-(CH2)nC4-10гетероцикла, -(CH2)nN(Rd)2, -(CH2)nC(O)NH(CH2)nC4-10гетероцикла, COR, -(CH2)nгалогена, -(CH2)nNHC(O)Rd, -(CH2)nNHC(O)NHRd, -(CH2)nNHC(O)ORd, -(CHR)nC(O)N(Rd)2, -(CH2)nNHSO2Rd и –OR, причем упомянутые алкил, арил или гетероцикл необязательно замещены 1-3 группами Rb, где по меньшей мере два из Rw, Rv и Ry на пиримидиниле представляют собой водород, а один из Rw, Rv и Ry представляет собой фенил, R1 и R5 независимо выбирают из водорода и галогена, R3 представляет собой CF3 или галоген, и один из R2 и R4 представляет собой водород, а другой представляет собой (CHR)nC5-10гетероцикл.
13. Соединение по п. 11, где Rw представляет собой фенил, R1 представляет собой водород, R3 представляет собой CF3, R5 представляет собой фтор, R2 представляет собой необязательно замещенный пиразолил и R4 представляет собой водород.
14. Соединение, которое выбирают из группы, состоящей из:
2-хлор-N-(5-морфолино-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
4-хлор-2-фтор-N-(2-метил-4-фенилпиримидин-5-ил)-5-(пиримидин-2-ил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(6-метил-4-фенилпиридин-3-ил)-4-(трифторметокси)бензамида,
N-(4-хлор-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-метил-4-фенилпиридин-3-ил)-3-(1H-пиразол-5-ил)-4-(трифторметил)бензамида,
2-фтор-N-(4'-фторбифенил-2-ил)-4-(трифторметил)бензамида,
N-[2-(5-метил-1H-пиразол-3-ил)фенил]-3-(1,3-тиазол-2-ил)бензамида,
2,3-дифтор-4-метил-N-(2-метил-4-фенилпиримидин-5-ил)бензамида,
2-хлор-5-(3,5-диметил-1H-пиразол-1-ил)-N-(2-метил-4-фенилпиримидин-5-ил)бензамида,
N-(6-фенил-3,3'-бипиридин-5-ил)-4-(трифторметил)бензамида,
2-хлор-5-(3,5-диметил-1H-пиразол-1-ил)-N-[2-(3-метилизоксазол-5-ил)фенил]бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(1,3-тиазол-4-ил)-4-(трифторметил)бензамида,
3-(3,5-диметил-1H-пиразол-1-ил)-4-фтор-N-(2-метил-4-фенилпиримидин-5-ил)бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-[5-(трифторметил)-1H-пиразол-1-ил]бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(2H-1,2,3-триазол-2-ил)-4-(трифторметил)бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(3-метил-1H-пиразол-1-ил)-4-(трифторметил)бензамида,
N-[2-метил-4-(3-метил-1H-пиразол-5-ил)пиримидин-5-ил]-3-(4-метил-1,3-тиазол-2-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-[4-(4-метил-1H-пиразол-1-ил)пиридин-3-ил]-4-(трифторметил)бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(1,3-оксазол-2-ил)-4-(трифторметил)бензамида,
N-(2-метил-4-фенилпиримидин-5-ил)-3-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(3-фенилпиридин-2-ил)-4-(трифторметил)бензамида,
2-хлор-N-(2-метил-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-хлор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(3-фенилпиразин-2-ил)-4-(трифторметил)бензамида,
N-(2,4-диметил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-метил-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-метил-4-фенилпиримидин-5-ил)-5-пиримидин-2-ил-4-(трифторметил)бензамида,
3-(4-метил-1,3-оксазол-2-ил)-N-(2-метил-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
3-(3-метил-1,2,4-оксадиазол-5-ил)-N-(2-метил-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-[4-(4-фторфенил)-2-метилпиримидин-5-ил]-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-[2-метил-4-(3-метилфенил)пиримидин-5-ил]-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-метил-4-фенилпиримидин-5-ил)-5-(5-метилтетрагидрофуран-2-ил)-4-(трифторметил)бензамида,
N-(2-(цианометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-3-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-гидроксиэтил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(2-(2-морфолиноэтил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамида,
N-(2-(гидроксиметил)-4-фенилпиримидин-5-ил)-3-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
трет-бутил-((5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метил)карбамат,
N-(2-(аминометил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(аминометил)-4-фенилпиримидин-5-ил)-3-(1-(дифторметил)-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(4-(3-(гидроксиметил)фенил)-2-метилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(2-((метиламино)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(4-(гидроксиметил)-2-метил-6-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-(2-гидроксипропан-2-ил)-2-фенилпиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-2-фтор-N-(2-((2-гидроксипропанамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(ацетамидометил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((2-гидроксиацетамидо)метил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(циклопропанкарбоксамидометил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-((2-гидроксиацетамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-((2-гидроксиацетамидо)метил)-2-фенилпиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(ацетамидометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-((2-гидроксиацетамидо)метил)-4-фенилпиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-2-фтор-N-(6-((2-гидроксипропанамидо)метил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-N-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)метил)-5-оксопирролидин-2-карбоксамида,
2-фтор-N-(6-((2-метоксиацетамидо)метил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-((2-(метилсульфонил)ацетамидо)метил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
(R)-2-фтор-N-(2-((2-гидроксипропанамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S или R)-N-(6-((2,3-дигидроксипропанамидо)метил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S или R)-N-(6-((2,3-дигидроксипропанамидо)метил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S или R)-N-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-2-ил)метил)-4-метилморфолин-2-карбоксамида,
2-фтор-N-(2-((2-метоксиацетамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-((2-гидроксиацетамидо)метил)-3-фенилпиразин-2-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S или R)-N-(2-((2-амино-3-гидрокси-2-метилпропанамидо)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-(метилсульфонил)ацетамидо)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-((2-гидроксиацетамидо)метил)-3-фенилпиридин-2-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-(2-оксооксазолидин-3-ил)ацетамидо)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
(R)-2-фтор-N-(5-((2-гидроксипропанамидо)метил)-3-фенилпиразин-2-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-2-фтор-N-(6-((2-гидрокси-N-метилпропанамидо)метил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(R)-N-(2-((2-аминопропанамидо)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-N-((5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метил)морфолин-3-карбоксамида,
3-(1-метил-1H-пиразол-3-ил)-N-(2-(метилсульфонамидометил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(4-фенил-2-(уреидометил)пиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-((1-метилуреидо)метил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(6-((3-(2-гидроксиэтил)-1-метилуреидо)метил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метилкарбамата,
5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотиновой кислоты,
5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N,N-диметил-6-фенилникотинамида,
5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N-метил-6-фенилникотинамида,
(S)-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N-(2-гидроксипропил)-6-фенилникотинамида,
(S)-5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N-(1-гидроксипропан-2-ил)-6-фенилникотинамида,
N-(5-карбамоил-2-(4-метил-1H-пиразол-1-ил)фенил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-амино-5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотинамида,
N-(2-(морфолин-4-карбонил)-4-фенилпиримидин-5-ил)-3-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
5-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотинамида,
5-(2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N-(1-метилазетидин-3-ил)-6-фенилникотинамида,
5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилникотинамида,
5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенил-N-(пиридин-3-илметил)никотинамида,
5-(2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилникотинамида,
5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6-фенилпиридазин-3-карбоксамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((3-метил-2,5-диоксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-((2,5-диоксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((2,4-диоксооксазолидин-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-((2,5-диоксоимидазолидин-1-ил)метил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-(диметиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-4-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(2-(2-(метиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-(метиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-(2-(этиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)уксусной кислоты,
(S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксо-2-(((тетрагидрофуран-2-ил)метил)амино)этил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-(2-((2-гидроксиэтил)амино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-(2-(((1s,3s)-3-гидроксициклобутил)амино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксо-2-((тетрагидрофуран-3-ил)амино)этил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-морфолино-2-оксоэтил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксо-2-(((тетрагидрофуран-3-ил)метил)амино)этил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-(2-(циклобутиламино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксо-2-(пиперазин-1-ил)этил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-(2-(этил(метил)амино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-(орто-толил)пиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-1-гидрокси-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(1-карбамоилциклопропил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(5-(2-амино-2-оксоэтил)-3-фенилпиразин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)-2-метилпропановая кислота,
N-(2-(1-амино-2-метил-1-оксопропан-2-ил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(пиридин-2-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(пиридин-2-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-4-фенилпиридин-3-ил)-2-фтор-5-(пиридин-2-ил)-4-(трифторметил)бензамида,
N-(2-(3-амино-3-оксопропил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((4H-1,2,4-триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(1,2-диамино-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(2,5-диоксоимидазолидин-4-ил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(4-(3-(ацетамидометил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(4-(3-ацетамидофенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(4-(2-(2-аминоэтил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(4-(2-(2-(диметиламино)этил)фенил)-2-метилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(4-(2-(гидроксиметил)фенил)-2-метилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(R)-N-(5-(2-циклопропилтиазол-4-ил)-2-(2-(гидроксиметил)пирролидин-1-ил)пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
(R)-2-фтор-N-(2-(2-(гидроксиметил)пирролидин-1-ил)-5-(1-метил-1H-пиразол-3-ил)пиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-оксо-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-оксо-2-(пиридин-2-ил)-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(6-оксо-2-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамида,
N-(7-(2-гидроксиацетил)-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-5,6-дигидро-1,7-нафтиридин-7(8H)-карбоксамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиридин-2-ил)-4-(трифторметил)бензамида,
N-(6-ацетил-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(пиримидин-2-ил)-4-(трифторметил)бензамида,
N-(6-ацетил-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
2-фтор-N-(6-(метилсульфонил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-N-метил-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксамида,
N-(6-ацетил-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-2-хлор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-оксо-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(7-оксо-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-4-(трифторметил)бензамида,
3-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксамида,
3-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-карбоксамида,
N-(6-глицил-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-(3-(3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-2-фенил-7,8-дигидро-1,6-нафтиридин-6(5H)-ил)уксусной кислоты,
N-(6-(2-гидроксиэтил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
3-(1-метил-1H-пиразол-3-ил)-N-(8-оксо-4-фенил-5,6,7,8-тетрагидро-1,7-нафтиридин-3-ил)-4-(трифторметил)бензамида,
N-(6-(2-гидроксиацетил)-4-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-оксо-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(6-(2-гидроксиацетил)-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
(R)-N-(6-(2-гидроксипропаноил)-2-фенил-5,6,7,8-тетрагидро-1,6-нафтиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
5'-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6'-фенил-[2,3'-бипиридин]-5-карбоксамида,
N-(5-(4-карбамоилфенил)-2-фенилпиридин-3-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-(4-(метилкарбамоил)фенил)-2-фенилпиридин-3-ил)-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-фенил-5-(пиридин-2-илокси)пиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-(гидроксиметил)-6'-фенил-[2,3'-бипиридин]-5'-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
2-(5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)пиримидин-5-карбоксамида,
N-(5-(5-(ацетамидометил)пиримидин-2-ил)-2-фенилпиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
5'-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-6'-фенил-[2,3'-бипиридин]-5-карбоновой кислоты,
2-фтор-N-(5-(2-гидроксипропан-2-ил)-6'-фенил-[2,3'-бипиридин]-5'-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-(5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)пиримидин-4-карбоксамида,
5'-(2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-N-метил-6'-фенил-[2,3'-бипиридин]-5-карбоксамида,
5-(5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)пиримидин-2-карбоксамида,
2-фтор-N-(2-фенил-5-(пиридин-4-илметокси)пиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
4-(5-(2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамидо)-6-фенилпиридин-3-ил)пиперазин-1-карбоксамида,
N-(5-(1-аминоэтил)-6'-фенил-[2,3'-бипиридин]-5'-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((1-метил-1H-тетразол-5-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-((4-этил-5-оксо-4,5-дигидро-1H-1,2,4-триазол-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-(5-оксопирролидин-2-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(6-((2-окса-6-азаспиро[3.3]гептан-6-ил)метил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(((3S,4R)-3,4-дигидроксипирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(5,7-диоксо-2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-2-фтор-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
(R или S)-3-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
N-(2-((2,5-диоксопирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-фенил-6,7-дигидро-5H-пирроло[3,4-b]пиридин-3-ил)-4-(трифторметил)бензамида,
(R или S)-3-(1-метил-1H-пиразол-3-ил)-N-(6-(5-оксопирролидин-2-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
(R или S)-N-(2-((2,5-диоксоимидазолидин-4-ил)метил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксопирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
(R или S)-N-(6-(1,2-дигидроксиэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксопиперидин-3-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-(метилсульфонил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-(S-метилсульфонимидоил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксооксазолидин-4-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксопирролидин-3-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(5-оксопирролидин-3-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((метилсульфонил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-((N-метилсульфамоил)метил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
N-(2-((2,5-диоксопирролидин-3-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(5-(2-амино-2-оксоэтил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(5-(2-амино-2-оксоэтил)-2-фенилпиридин-3-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(5-(оксазол-5-илметил)-2-фенилпиридин-3-ил)-5-(пиримидин-2-ил)-4-(трифторметил)бензамида,
N-(6-(2-амино-2-оксоэтил)-3-фенилпиридин-2-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(6-(1-(2-гидроксиацетил)азетидин-3-ил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(6-(1-глицилазетидин-3-ил)-2-фенилпиридин-3-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(6-(1-ацетилазетидин-3-ил)-2-фенилпиридин-3-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-N-(2-(2-гидроксиацетамидо)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((2-гидроксиэтил)амино)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-(2-оксоимидазолидин-1-ил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-амино-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((2-амино-2-оксоэтил)амино)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(5-((2-оксооксазолидин-3-ил)метил)-3-фенилпиразин-2-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(6-(2-оксоимидазолидин-4-ил)-2-фенилпиридин-3-ил)-4-(трифторметил)бензамида,
N-(2-(2-амино-2-оксоэтокси)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
(R или S)-2-фтор-N-(5-(4-(S-метилсульфонимидоил)фенил)-2-фенилпиридин-3-ил)-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксопирролидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксопиперидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксоазепан-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксо-1,3-оксазинан-3-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((3-оксоморфолино)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-((1H-имидазол-1-ил)метил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксопиримидин-1(2H)-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
N-(2-((2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)метил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((5-оксо-1,5-дигидро-4H-1,2,4-триазол-4-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида, и
(R или S)-2-фтор-N-(2-((5-(гидроксиметил)-2-оксооксазолидин-3-ил)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
или его фармацевтически приемлемая соль.
15. Соединение, которое выбирают из группы, состоящей из:
2-фтор-N-(2-((2-гидроксиацетамидо)метил)-4-фенилпиримидин-5-ил)-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-((2-гидроксиацетамидо)метил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(ацетамидометил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1H-пиразол-3-ил)-4-(трифторметил)бензамида,
N-(2-(ацетамидометил)-4-фенилпиримидин-5-ил)-3-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
2-фтор-5-(1-метил-1H-пиразол-3-ил)-N-(2-((2-оксоимидазолидин-1-ил)метил)-4-фенилпиримидин-5-ил)-4-(трифторметил)бензамида,
(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)метилкарбамата,
2-(5-(2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамидо)-4-фенилпиримидин-2-ил)уксусной кислоты, и
N-(2-(2-(этил(метил)амино)-2-оксоэтил)-4-фенилпиримидин-5-ил)-2-фтор-5-(1-метил-1H-пиразол-3-ил)-4-(трифторметил)бензамида,
или его фармацевтически приемлемая соль.
16. Фармацевтическая композиция для лечения заболевания или нарушения, опосредованного Trk-рецепторами, содержащая терапевтически эффективное количество соединения по п. 1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
17. Применение соединения по п. 1 или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя для производства лекарственного средства для лечения заболевания или нарушения, опосредованного Trk-рецепторами, где упомянутое заболевание или нарушение выбирают из группы, состоящей из боли, воспаления, злокачественной опухоли, рестеноза, атеросклероза, псориаза, тромбоза, заболевания, нарушения, повреждения или дисфункции, связанных с дисмиелинизацией или демиелинизацией, или заболевания или нарушения, ассоциированного с аномальной активностью рецепторной TrkA, связывающей фактор роста нервов (NGF).
18. Соединение по п. 1 для применения в лечении заболевания или нарушения, опосредованного Trk-рецепторами.
| NICHOLS D.A | |||
| et al, Structure-Based Design of Potent and Ligand-Efficient Inhibitors of CTX-M Class A β-Lactamase, J | |||
| Med | |||
| Chem., 2012, v | |||
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ изготовления окисных катодов | 1924 |
|
SU2163A1 |
| WO 2008156869 A2, 24.12.2008 | |||
| База данных REGISTRY [онлайн], RN 1571409-91-0, 21.03.2014, найдено в STN | |||
| База данных REGISTRY [онлайн], RN 1328343-21-0, 05.09.2011, найдено в STN | |||
| База данных REGISTRY [онлайн], RN 1328014-91-0, 04.09.2011, найдено в STN | |||
| База данных REGISTRY [онлайн], RN 1327875-68-2, 04.09.2011, найдено в STN | |||
| База данных REGISTRY [онлайн], RN 1328154-63-7, 04.09.2011, найдено в STN | |||
| US 20090163476 A1, 25.06.2009 | |||
| US 4638014 A, 20.01.1987 | |||
| US 6372767 B1, 16.04.2002 | |||
| WO 2014016434 A1, 30.01.2014 | |||
| RU 2011120345 A, 27.11.2012. | |||

