ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По настоящей заявке испрашивается приоритет предварительной патентной заявки США № 61/643812, поданной 7 мая 2012 г., полное содержание которой таким образом приведено в качестве ссылки.
ЗАЯВЛЕНИЕ ПРАВ НА ИЗОБРЕТЕНИЯ, ВЫПОЛНЕННЫЕ В ФИНАНСИРУЕМЫХ ИЗ ФЕДЕРАЛЬНОГО БЮДЖЕТА ИССЛЕДОВАНИЯХ
[0002] Не предусмотрено.
ОБЛАСТЬ ТЕХНИКИ
[0003] Настоящее описание относится к области генной инженерии, в частности, направленной модификации генома клетки.
ПРЕДПОСЫЛКИ
[0004] Интеграция чужеродной ДНК в геном организмов и линий клеток представляет собой широко используемый способ для исследования биологических систем и манипуляций с ними. Традиционно, вставку трансгена направляют в специфический локус посредством предоставления плазмиды, несущей трансген, и имеющей существенную идентичность последовательности ДНК, фланкирующей желательный участок интеграции. Спонтанный разрыв хромосомы с последующей репарацией с использованием гомологичной области плазмидной ДНК в качестве матрицы приводит к переносу трансгена в геном. См., например, Roller et al. (1989) Proc. Nat’l. Acad. Sci. USA 86(22):8927-8931; Thomas et al. (1986) Cell 44(3): 419-428. Частоту этого типа управляемой гомологией направленной интеграции можно увеличивать вплоть до кратности 105 посредством преднамеренного создания двухцепочечного разрыва в окрестности намеченной области (Hockemeyer et al. (2009) Nature Biotech. 27(9):851-857; Lombardo et al. (2007) Nature Biotech. 25(11): 1298-1306; Moehle et al. (2007) Proc. Nat’l Acad. Sci. USA 104(9):3055-3060; Rouet et al. (1994) Proc. Nat’l Acad. Sci. USA 91(13):6064-6068.
[0005] Двухцепочечный разрыв (DSB) или пропуск можно получать посредством сайт-специфической нуклеазы, такой как нуклеаза с цинковыми пальцами (ZFN) или нуклеаза с эффекторным доменом TAL (TALEN), или с использованием системы CRISPR/Cas9 со сконструированной crРНК/tractРНК (одиночной руководящей РНК) для управления специфическим расщеплением. См., например, Burgess (2013) Nature Reviews Genetics 14:80-81, Urnov et al. (2010) Nature 435(7042):646-51; Публикации Патентов США 20030232410; 20050208489; 20050026157; 20050064474; 20060188987; 20090263900; 20090117617; 20100047805; 20110207221; 20110301073 и Международная публикация WO 2007/014275, полное содержание которых приведено в качестве ссылки для всех целей. Во многих организмах, вставку трансгена можно осуществлять посредством процесса направляемой гомологией репарации (HDR), требующего, чтобы вставленный трансген включал области гомологии с участком вставки (расщепления). Однако в некоторых организмах и линиях клеток отсутствует традиционный процесс HDR и направленная интеграция происходит в первую очередь посредством аппарата репарации ДНК независимым от гомологии соединением негомологичных концов (NHEJ). По существу, до настоящего времени в организмах и линиях клеток (например, клеток CHO), не подлежащих процессам HDR, только относительно короткие (<100 п.о.) олигонуклеотиды вставляли посредством независимых от гомологии путей после опосредованного нуклеазой расщепления локуса-мишени. См., например, Orlando et al. (2010) Nucleic Acids Res. 38(15):e152 и Публикацию Патента США № 20110207221.
[0006] Таким образом, остается необходимость композиций и способов независимой от гомологии направленной интеграции трансгенов, включая более крупные трансгены, непосредственно в участок расщепления, например, в организмы и линии клеток с отсутствием или дефицитом общепринятых зависимых от гомологии способов.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0007] В настоящем документе описаны способы и композиции для независимой от гомологии направленной интеграции трансгена.
[0008] В одном аспекте в настоящем документе описаны двухцепочечные донорные полинуклеотиды для интеграции в выбранный эндогенный локус после расщеплением in vivo донора с использованием по меньшей мере одной нуклеазы. Донорные полинуклеотиды включают экзогенную последовательность (трансген), подлежащие интеграции в эндогенный локус, и содержат по меньшей мере один участок-мишень для нуклеазы, например, два парных участка связывания нуклеазы, разделенные «спейсерной» последовательностью, разделяющей ближние концы участков связывания. Спейсер может иметь любой размер, например, между 4 и 20 парами оснований (или любое значение между ними). Доноры, имеющие множество участков-мишеней нуклеазы, могут иметь одинаковые или различные участки-мишени, например, два одинаковых парных участка, фланкирующих трансген, или два различных парных участка, фланкирующих трансген. Донорные нуклеотиды не обязательно требуют присутствия гомологичных плеч, фланкирующих последовательность трансгена. Единственной гомологией с хромосомой, которая может присутствовать в донорной последовательности является(являются) участок(участки) связывания нуклеазы. В вариантах осуществления, в которых участки-мишени нуклеазы обладают гомологией с геномом, гомология с геномом составляет в длину менее 50-100 (или любое количество пар оснований между 50 и 100) непрерывных пар оснований. В конкретных вариантах осуществления, где нуклеаза, используемая для расщепления донора не является такой же, как нуклеаза, используемая для расщепления хромосомы, может не присутствовать гомологии между хромосомным локусом, расщепляемым нуклеазой(нуклеазами), и донорной последовательностью. Кроме того, участок(участки)-мишень для нуклеазы не находится внутри трансгена и, как таковое, расщепление донорного полинуклеотида нуклеазой(нуклеазами), связывающейся(связывающимися) с участком(участками)-мишенью, не модифицирует трансген. В конкретных вариантах осуществления донорная нуклеиновая кислота содержит два участка-мишени, и спейсерная последовательность между двумя участками-мишенями не встречается в природе, например, когда спейсерная последовательность не встречается в геномной последовательности между двумя участками-мишенями, присутствующими в геноме. В конкретных вариантах осуществления, донорные молекулы интегрируют в эндогенный локус посредством независимых от гомологии механизмов (например, NHEJ). В других вариантах осуществления двухцепочечный донор содержит трансген длиной по меньшей мере 1 т.п.о. и участок(участки)-мишень для нуклеазы на 3’- и/или 5’-трансгена для расщепления in vivo. В конкретных вариантах осуществления, участок(участки)-мишень для нуклеазы, используемый для расщепления донора, не восстанавливается после интеграции трансгена, например, когда спейсер между парными участками-мишенями не присутствует в эндогенном локусе и/или не обладает гомологией с эндогенным локусом. Донорная молекула может представлять собой, например, плазмиду. В конкретных вариантах осуществления, донор интегрируют после опосредованного нуклеазой расщепления эндогенного локуса. В любой опосредованной нуклеазой интеграции донорной молекулы одна или более нуклеаз, использованных для расщепления донора, могут являться такими же, как одна или более нуклеаз, используемых для расщепления эндогенного локуса. Альтернативно, одна или более нуклеаз, использованных для расщепления донора, могут отличаться от одной или более нуклеаз, используемых для расщепления эндогенного локуса.
[0009] В некоторых вариантах осуществления донор содержится на плазмиде. Донор может быть интегрирован после опосредованного нуклеазой расщепления, где последовательность, подлежащая интеграции (донор или трансген), фланкирована в плазмиде по меньшей мере двумя участками расщепления нуклеазой. В других вариантах осуществления донор содержится на плазмиде, где донор может быть интегрирован после опосредованного нуклеазой расщепления, где последовательность, подлежащая интеграции (донор или трансген), представляет собой плазмиду, содержащую одиночный 5’-участок расщепления нуклеазой. В конкретных вариантах осуществления последовательность участков расщепления нуклеазой в донорной плазмиде является такой же, как последовательность участка расщепления нуклеазой в хромосомном локусе, предназначенном в качестве мишени. В вариантах осуществления, в которых участки расщепления являются одинаковыми в доноре и геноме, последовательности, разделяющие участки расщепления, могут являться одинаковыми или различными. В конкретных вариантах осуществления, последовательности, разделяющие участки расщепления (спейсеры), являются отличными в доноре по сравнению с геномом, так что после расщепления донора участки-мишени повторно не образуются, и донор невозможно расщепить снова той же самой нуклеазой(нуклеазами).
[0010] Представляющая интерес последовательность молекулы-донора может содержать одну или более последовательностей, кодирующих функциональный полипептид (например, кДНК), с промотором или без. В конкретных вариантах осуществления последовательность нуклеиновой кислоты содержит последовательность, кодирующую антитело, антиген, фермент, фактор роста, рецептор (поверхности клеток или ядерный), гормон, лимфокин, цитокин, репортер, ген устойчивости к насекомым, ген устойчивости к гербицидам, фактор транскрипции, белок секвестрации или функциональные фрагменты любого из вышеуказанных и комбинации вышеуказанных. Представляющая интерес последовательность вставки молекулы-донора может содержать одну или более последовательностей, кодирующих молекулу РНК, кодирующую функциональную или структурную РНК, например, РНКи, мРНКи, и/или мкРНКи. В вариантах осуществления, в которых последовательности, кодирующие функциональный полипептид, не имеют промоторов, экспрессию интегрированной последовательности затем обеспечивают посредством транскрипции, управляемой эндогенным промотором или другим контрольным элементом в представляющей интерес области. В других вариантах осуществления «тандемную» кассету интегрируют в выбранный участок таким образом, где первый компонент кассеты содержит не имеющую промотора последовательность, как описано выше, с последующей последовательностью терминации транскрипции, и вторую последовательность, кодирующую автономную экспрессирующую кассету. В донорную молекулу можно включать дополнительные последовательности (кодирующие или некодирующие последовательности), включая, но без ограничения, последовательности, кодирующие пептид 2A, участок SA, IRES и т.д. В конкретных вариантах осуществления донорная нуклеиновая кислота (трансген) содержит последовательности, кодирующие функциональные РНК, например, мкРНК или кшРНК.
[0011] Другой аспект, описанный в настоящем документе, относится к способам интеграции донорной нуклеиновой кислоты (например, донорной молекулы, как описано в настоящем документе) в геном клетки посредством независимых от гомологии механизмов. Способы включают создание двухцепочечного разрыва (DSB) в геноме клетки и расщепления молекулы-донора с использованием одной или более нуклеаз, так что донорную нуклеиновую кислоту интегрируют в участок DSB. В конкретных вариантах осуществления донорную нуклеиновую кислоту интегрируют посредством не зависимых от гомологии способов (например, NHEJ). Как отмечено выше, при расщеплении in vivo донорные последовательности можно интегрировать направленным способом в геном клетки в расположении DSB. Донорная последовательность может включать один или более одинаковых участков-мишеней для одной или более нуклеаз, использованных для получения DSB. Таким образом, донорную последовательность можно расщеплять одной или несколькими из одинаковых нуклеаз, использованных для расщепления эндогенного гена, в который желательна интеграция. В конкретных вариантах осуществления донорная последовательность включает участки-мишени для нуклеазы, отличной от нуклеаз, использованных для индукции DSB. DSB в геноме клетки-мишени можно получать посредством любого механизма. В конкретных вариантах осуществления DSB получают посредством одного или более (например, димеризующейся пары) нуклеаз с цинковыми пальцами (ZFN), слитых белков, содержащих связывающий домен с цинковыми пальцами, сконструированный для связывания последовательности внутри интересующей области, и расщепляющий домен или расщепляющий полудомен расщепления. В других вариантах осуществления DSB получают посредством одного или более ДНК-связывающих доменов TALE (природных или неприродных), слитых с доменом нуклеазы (TALEN). В следующих вариантах осуществления расщепление осуществляют с использованием системы нуклеаз, такой как CRISPR/Cas со сконструированной crРНК/tracrРНК.
[0012] Более того, в любом из способов, описанных в настоящем документе, первый и второй полудомены расщепления могут происходить из рестрикционной эндонуклеазы типа IIS, например, FokI или StsI. Более того, в любом из способов, описанных в настоящем документе, по меньшей мере один из слитых белков может содержать изменение в аминокислотной последовательности поверхности димеризации полудомена расщепления, например, такое, что формируются облигатные гетеродимеры полудоменов расщепления. Альтернативно, в любом из способов, описанных в настоящем документе, домен расщепления может представлять собой природную или неприродную (сконструированную) мегануклеазу.
[0013] В любом из способов, описанных в настоящем документе, клетка может представлять собой любую эукариотическую клетку, например, клетку растения или клетку млекопитающего, или линию клеток, включая клетки COS, CHO (например, CHO-S, CHO-K1, CHO-DG44, CHO-DUXB11, CHO-DUKX, CHOK1SV), VERO, MDCK, WI38, V79, B14AF28-G3, BHK, HaK, NSO, SP2/0-Agl4, HeLa, HEK293 (например, HEK293-F, HEK293-H, HEK293-T) и perC6, также как клетки насекомых, таких как Spodoptera fugiperda (Sf), или клетки грибов, таких как Saccharomyces, Pichia и Schizosaccharomyces. В конкретных вариантах осуществления линия клеток представляет собой линию клеток CHO, MDCK или HEK293. Пригодные клетки включают также стволовые клетки, например, такие как эмбриональные стволовые клетки, индуцированные плюрипотентные стволовые клетки, гематопоэтические стволовые клетки, нейрональные стволовые клетки и мезенхимальные стволовые клетки. Более того, клетки могут являться арестованными в фазе G2 клеточного цикла. В некоторых вариантах осуществления способов, описанных в настоящем документе, клетка может представлять собой клетку с отсутствием эффективной основанной на гомологии репарации ДНК, например, клетку CHO. В конкретных вариантах осуществления клетки могут являться первичными или не делящимися клетками, предпочтительно использующими путь репарации ДНК NHEJ. В некоторых вариантах осуществления клетка может представлять собой клетку растения или гриба. В других вариантах осуществления способы, описанные в настоящем документе, можно использовать в клетках с несеквенированными геномами. Эти клетки можно использовать для создания линий клеток и/или трансгенных организмов (например, животных или растений), несущих трансген(ы).
[0014] Другой аспект относится к трансгенным организмам (например, растениям или животным), содержащим трансген, интегрированный любым из способов, описанных в настоящем документе. В одном варианте осуществления конструируют клетку, линию клеток или трансгенный организм, несущий гетерозиготный генотип для выбранного гена, в то время как в другом варианте осуществления получают гомозиготную клетку, линию клеток или трансгенный организм, несущий две мутантные копии в обоих аллелях желаемого локуса.
[0015] Представлен также набор, включающий способы и композиции по изобретению. Набор может содержать нуклеазы, (например, гены, кодирующие молекулы РНК или ZFN, TALEN или систему CRISPR/Cas, содержащиеся в подходящем экспрессирующем векторе), или аликвоты белков нуклеаз, донорные молекулы, подходящие линии клеток-хозяев, инструкции для осуществления способов по изобретению и т.п. Набор может также содержать представляющие интерес донорные молекулы (например, репортерные гены, специфические трансгены и т.п.).
[0016] Эти и другие аспекты ясно очевидны специалисту в данной области в свете описания в целом.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
[0017] На фигуре 1, панели A-F, показан захват трансгена, расщепленного in vivo, в локусе AAVS1 в клетках K562. Фигура 1A представляет собой схематическое изображение иллюстративной донорной молекулы, имеющей два спаренных участка связывания (всего 4 участка связывания), где каждая пара отделена спейсерами), где сайты фланкируют трансген, подлежащий интеграции в геном. Участки-мишени могут являться одинаковыми или различными. Фигура 1B представляет собой схему, изображающую различные способы расщепления донора in vivo. В первом варианте осуществления, когда хромосома и донорная плазмида обе содержат участок расщепления ZFN (темно-серая область), расщепление донора и хромосомы является синхронизированным, позволяя эффективную интеграцию донора в хромосому. Интеграция может происходить как в прямой, так и в обратной ориентациях, названных «AB» и «BA», соответственно. Во втором варианте осуществления донор содержит более одного участка расщепления нуклеазой. Действие нуклеазы высвобождает линейный фрагмент ДНК, который интегрируют в хромосому. В третьем варианте осуществления хромосому расщепляют более, чем одной нуклеазой, и донор одной нуклеазой, что приводит к интеграции донора в делецию в хромосоме. В четвертом варианте осуществления как донорную ДНК, так и хромосомную ДНК расщепляют более чем одной нуклеазой, что приводит к интеграции линейного фрагмента в делецию в хромосоме. На фигуре 1C показано сравнение доноров, благоприятствующих прямой (AB) интеграции трансгена и воссозданию того же самого участка-мишени нуклеазы (верхние панели) с донорами, благоприятствующими обратной (BA) интеграции трансгена и не воссоздающими тот же самый участок-мишень нуклеазы (нижние панели). Последовательность между участками связывания (спейсерами) подчеркнута, и выступающие концы спейсеров дикого типа (верхние панели) и обратно комплементарных спейсеров (нижние панели), полученных после расщепления, показана справа верхних и нижних панелей. Последовательности, показанные вверху слева, представляют собой SEQ ID NO: 107 и 108. Последовательности, показанные вверху справа, представляют собой SEQ ID NO: 109 и 110. Последовательности, показанные внизу слева, представляют собой SEQ ID NO: 111 и 112, и последовательности, показанные внизу справа, представляют собой SEQ ID NO: 113 и 114. Фигура 1D представляет собой гель, показывающий детекцию направленной интеграции из доноров с последовательностями (или спейсерами) дикого типа («w.t.») между участками связывания ZFN (такими же последовательностями спейсеров, как в геноме), и обратно комплементарными («r.c.») последовательностями между участками связывания ZFN. Как показано, больший сигнал наблюдают в ориентации BA с выступающими обратно комплементарными нуклеотидами. Фигура 1E представляет собой гель, показывающий интеграцию донорной плазмиды в AAVS1 после расщепления in vivo специфическими для AAVS ZFN. Донорную плазмиду либо с содержанием, либо с отсутствием участка AAVS1 ZFN, совместно трансфицировали со специфическими для AAVS1 ZFN в клетки K562. Интеграцию как в AB, так и в BA ориентациях мониторировали посредством ПЦР участков стыковки хромосома-донор. При вставке в ориентации AB получают продукты ПЦР 391 и 423 п.о. для левого и правого участков стыковки, соответственно; при вставке в ориентации BA получают продукты ПЦР 369 и 471 п.о. для левого и правого участков стыковки, соответственно. «A1» относится к реакции ПЦР, разработанной для амплификации участка ZFN в AAVS1, и «NT» относится к реакциям с отсутствием ДНК-матрицы. На фигуре 1F изображены как схема триплоидного локуса AAVS1, так и результаты проверки направленной интеграции, как анализировали Саузерн-блоттингом. Три клона анализировали в двух повторах. Геномную ДНК из трех клонов либо расщепляли BglI и анализировали с помощью специфического для AAVS1 зонда (верхний гель), либо расщепляли AccI и анализировали с помощью специфического для трансгена зонда (bla).
[0018] На фигуре 2, панели A и B, показано, что расщепление донора in vivo способствует захвату трансгена в нескольких локусах двух различных типов клеток. На фигуре 2A показано, что направленная интеграция посредством NHEJ в участки в локусах IL2Rγ, CCR5 и глутаминсинтетазы (GS) является более эффективной, когда донорную плазмиду расщепляют совместно с хромосомой, вместо расщепления до трансфекции. Одну ПЦР участков стыковки для каждой ориентации проводили для всех трех локусов, левого участка стыковки для ориентации BA и правого участка стыковки для ориентации AB. Использованные условия эксперимента отмечены следующим образом «N» относится к трансфекциям, где у донора отсутствует участок ZFN; «ERV» относится к образцу, в котором донор предварительно расщепляли EcoRV до введения в клетку; «Y» относится к трансфекции, где донор и ген-мишень оба содержат участок ZFN; и «NT» относится к реакциям ПЦР без ДНК-матрицы. Размер ампликона, ожидаемый при амплификации ПЦР успешно интегрированных доноров, показан ниже каждой дорожки в парах оснований («Ожидаемый размер, п.о.»). Показанный рисунок представляет собой изображение с инвертированными цветами окрашенного бромидом этидия геля. На фигуре 2B изображено, что расщепление донора не обязательно должно быть выполнено с помощью таких же ZFN, как использованные для расщепления участка-мишени в хромосоме. Анализы специфической для участков стыковки ПЦР использовали для детекции интеграции трансгена в хромосомную мишень и для детекции ориентации интегрированных трансгенов. Эти анализы показали, что трансген можно интегрировать в любой ориентации после расщепления ZFN. Условия эксперимента отмечены следующим образом: GS, специфические для GS ZFN или донорная плазмида с участком расщепления ZFN GS; A1, специфические для AAVS1 ZFN или донор с участком расщепления ZFN AAVS1.
[0019] На фигуре 3, панели A-C, изображена направленная интеграция трансгена с высокой частотой в локус GS в клетках CHO-K1. Фигура 3А представляет собой схему локуса GS, показывающую трансген, интегрированный в ориентации BA. На фигуре 3B показан анализ Саузерн-блоттингом клонов клеток для направленной интеграции донора в локус GS. Экзонный зонд GS детектирует также два псевдогена GS. Ту же самую панель клонов анализировали по общей интеграции трансгена посредством анализа с зондом для гена bla E. coli (фигура 3C). Интеграцию трансгена в локус GS наблюдали вместе с интеграцией трансгена где-либо еще в геноме в трех из восьми анализированных клонов.
[0020] На фигуре 4, панели A и C, показано повреждение альфа-(1,6)-фукозилтрансферазы (FUT8) в клетках CHO-K1 посредством опосредованной ZFN и TALEN направленной вставки трансгена моноклонального антитела. На фигуре 4A изображены диаграммы Венна с пропорциональной площадью, показывающие соответствие между клонами, прошедшими скрининг по вставке трансгена посредством специфической для участков стыковки ПЦР и по экспрессии IgG. Фигура 4B представляет собой схему локуса FUT8, содержащего вставленный трансген (изображенный светло-серым), с отмеченной ориентацией трансгена в номенклатуре либо «AB» или «BA», и на фигуре 4C показано подтверждение Саузерн-блоттингом интеграции в FUT8. Отмечены интегранты, содержащие трансгены, вставленные в ориентациях BA и AB.
[0021] На фигуре 5, панели A-C, показана активность ZFN для экспериментов, описанных в примерах. На фигуре 5A показано расщепление ZFN в локусе AAVS1 (вверху) и в донорной плазмиде (внизу). Соответствующие дорожки на фигуре 5A из фигуры 1D указаны над гелем (например: «1,12»), как присутствие («Y») или отсутствие («N») фермента нуклеазы Surveyor™. Процент модифицированных молекул показан под дорожками с сигналом. На фигуре 5B показано расщепление ZFN в IL2Rγ, CCR5 и GS с использованием специфических для соответствующего гена ZFN. Как описано выше для 5A, соответствующие дорожки в 5B из фигуры 2A указаны над гелем, как присутствие («Y») или отсутствие («N») фермента нуклеазы Surveyor™. В то время как на гелях вверху фигуры 5B изображены результаты интеграции в локусы генов, на гелях внизу фигуры изображены результаты расщепления в донорах. На фигуре 5C показано расщепление ZFN в GS, где на геле слева изображены результаты для локуса гена в клетках CHO, в то время как на геле справа изображены результаты расщепления ZFN донора. Как выше, соответствующие дорожки на фигуре 5C из фигуры 2B указаны над гелем, как присутствие («Y») или отсутствие («N») фермента нуклеазы Surveyor™. Стрелками указаны ожидаемые продукты расщепления.
[0022] Фигура 6 представляет собой график, изображающий зависимую от гомологии направленную интеграцию кодирующего GFP трансгена в клетки HEK 293 и CHO-K1. Процент клеток, являющихся положительными по GFP, показан светло-серым (клетки HEK 293) или темно-серым (клетки CHO-K1). Количество использованного донора указано под каждой группой.
[0023] На фигуре 7 показана частичная последовательность ДНК продуктов ПЦР участков стыковки из клонов CHO K1, в которых расщепленные in vivo доноры были интегрированы в локус AAVS1. Хромосомная последовательность показана обычным текстом, и донорная последовательность показана курсивом. Участки связывания ZFN подчеркнуты и показаны жирным шрифтом. Микрогомология выделена серым. Ожидаемые последовательности аллелей в ориентации AB показаны сверху группы AB и определены как точное лигирование 5’-выступающих концов; ожидаемые последовательности аллелей в ориентации BA показаны сверху группы BA и определены как удаление 5’-выступающих концов с последующим лигированием. Идентификаторы последовательностей указаны на фигуре.
[0024] На фигуре 8, панели A-C, показаны последовательности ДНК из анализов продуктов ПЦР участков стыковки из пулов клеток CHO K1 с интеграцией трансгенов в AAVS1 (фигура 8A), CCR5 (фигура 8B), GS (фигура 8C), и IL2Rγ (фигура 8C). Хромосомная последовательность показана обычным текстом, донорная последовательность показана курсивом. Участки связывания ZFN подчеркнуты и выделены жирным шрифтом. Ожидаемые последовательности аллелей показаны, как выше, а также, как определено выше для фигуры 7. Идентичные последовательности, выделенные более одного раза, отмечены, и идентификаторы последовательности указаны на фигуре.
[0025] На фигуре 9 показаны последовательности ДНК после ПЦР участков стыковки из происходящих из отдельных клеток клонов GS. Хромосомная последовательность показана обычным текстом, донорная последовательность показана курсивом. Участки связывания ZFN подчеркнуты и выделены жирным шрифтом. Ожидаемые аллели в ориентации AB определены как точное лигирование 5’-выступающих концов; ожидаемые аллели в ориентации BA определены как удаление 5’-выступающих концов с последующим лигированием. Идентификаторы последовательности указаны на фигуре.
[0026] На фигуре 10, панели A и B, показаны последовательности ДНК после ПЦР участков стыковки происходящих из отдельных клеток клонов с интеграцией в FUT8. Хромосомная последовательность показана обычным текстом, донорная последовательность показана курсивом. На фигуре 10A показана интеграция после расщепления нацеленными на FUT8 ZFN (участки связывания ZFN подчеркнуты и выделены жирным шрифтом). На фигуре 10B показана интеграция после расщепления нацеленными на FUT8 TALEN (участки связывания TALEN подчеркнуты и выделены жирным шрифтом). Идентификаторы последовательности указаны на фигуре.
[0027] На фигуре 11 показана карта плазмиды pDAB109350.
[0028] На фигуре 12 показана карта плазмиды pDAB109360.
[0029] На фигуре 13 показана карта плазмиды pDAS000153.
[0030] На фигуре 14 показана карта плазмиды pDAS000150
[0031] На фигуре 15 показана карта плазмиды pDAS000143.
[0032] На фигуре 16 показана карта плазмиды pDAS000164.
[0033] На фигуре 17 показана карта плазмиды pDAS000433.
[0034] На фигуре 18 показана карта плазмиды pDAS000434.
[0035] На фигуре 19, панели A и B, изображено свободное от экзогенных маркеров, последовательное пакетирование трансгенов в эндогенном локусе AHAS в геноме пшеницы Triticum aestivum с использованием опосредованной ZFN, направленной NHEJ репарации ДНК. На фигуре 19A изображен первый пакет трансгенов; на фигуре 19B изображен второй пакет трансгенов.
[0036] На фигуре 20, панели A и B, изображено свободное от экзогенных маркеров, последовательное пакетирование трансгенов в эндогенном локусе AHAS в геноме пшеницы Triticum aestivum с использованием опосредованной ZFN, направляемой HDR репарации ДНК. На фигуре 20A изображен первый пакет трансгенов; на фигуре 20B изображен второй пакет трансгенов.
[0037] На фигуре 21 показана карта плазмиды pDAS000435.
[0038] На фигуре 22 показана карта плазмиды pDAB107827.
[0039] На фигуре 23 показана карта плазмиды pDAB107828.
[0040] На фигуре 24 показана карта плазмиды pDAS000340.
[0041] На фигуре 25 показана карта плазмиды pDAS000341.
[0042] На фигуре 26 показана карта плазмиды pDAS000342.
[0043] На фигуре 27 показана карта плазмиды pDAS000343.
[0044] На фигуре 28, панели A и B, показано расположение праймеров и их положение относительно стартового и стоп-кодона Fad3C. На фигуре 28A показано расположение участков праймеров для локуса Fad3C дикого типа. На фигуре 28B показано расположение участков праймеров для подтверждения интеграции донора и возможных ориентаций, посредством которых донор может интегрировать внутри локуса Fad3C.
[0045] На фигуре 29, панели A и B, показаны выравнивания последовательностей для различных направленных интеграций. На фигуре 29A показано выравнивание последовательностей, амплифицированных с участка стыковки кассеты tGFP из pDAS000341 с Fad3C в двухцепочечном разрыве после узнавания ZFN 28051-2A-28052. Показанные последовательности представляют собой SEQ ID NO:480-493 сверху вниз. «·» обозначает делеции, локализованные в участках разрезания. На фигуре 29B показано выравнивание последовательностей, амплифицированных с участка стыковки кассеты tGFP из pDAS000343 с Fad3C в двухцепочечном разрыве после узнавания ZFN 28051-2A-28052 и ZFN 28053-2A-28054. «·» обозначает делеции, локализованные в участках разрезания. Показанные последовательности представляют собой SEQ ID NO:494-507 сверху вниз.
[0046] На фигуре 30, панели A и B, показано выравнивание последовательностей, амплифицированных с участка стыковки кассеты hph из pDAS000340 с FAD3C в двухцепочечном разрыве после узнавания ZFN 28051-2A-28052. «·» обозначает делеции, локализованные в участках разрезания. Показанные последовательности представляют собой SEQ ID NO:508-523 сверху вниз. На фигуре 30A показаны последовательности для 5'-участка стыковки, и последовательности, показанные на фигуре 30B - для 3'-участка стыковки.
[0047] На фигуре 31, панели A и B, показано выравнивание последовательностей, амплифицированных с участка стыковки кассеты hph из pDAS00034 с FAD3C в двухцепочечном разрыве после узнавания ZFN 28051-2A-28052 и 28053-2A-28054. «·» обозначает делеции, локализованные в участках разрезания. Показанные последовательности представляют собой SEQ ID NO:524-532 сверху вниз. Последовательности, показанные на фигуре 31A - для 5'-участка стыковки, и последовательности, показанные на фигуре 31B - для 3'-участка стыковки.
[0048] На фигуре 32 показано отношение ZFN, сконструированных для связывания геномного локуса трансгенного события кукурузы DAS-59132. Шесть ZFN (E32ZFN1-6) идентифицированы из анализа в дрожжах, и четыре ZFN являлись преимущественными для оценки в растениях.
[0049] На фигуре 33 показана карта плазмиды pDAB105906.
[0050] На фигуре 34 показана карта плазмиды pDAB111809.
[0051] На фигуре 35 показана карта плазмиды pDAB100655.
[0052] На фигуре 36 изображен график, показывающий оценку временно экспрессированных ZFN в растениях. Четыре ZFN оценивали в каллюсе маиса по временно экспрессирующимся ZFN и внутреннему контролю - ZFN, нацеленной на ген IPPK2. После секвенирования нового поколения амплифицированных фрагментов ПЦР из области, окружающей участки расщепления ZFN, секвенированные амплифицированные фрагменты ПЦР оценивали в баллах по присутствию вариантов последовательности, возникающих в результате инделов. Показана относительная частота инделов для каждой из четырех пар E32 ZFN по сравнению с активностью IPPK2 ZFN. При трансгенном событии 32 ZFN6, содержащем связывающие домены с цинковыми пальцами 25716 (’716) и 25717 (’717), расщепление геномного локуса трансгенного события кукурузы DAS-59132 происходило в 380 раз эффективнее, чем для контрольной нуклеазы с цинковыми пальцами IPPK2.
[0053] На фигуре 37 изображен график повреждения ZFN локуса трансгенного события кукурузы DAS-59132.
[0054] Фигура 38 представляет собой схематическое изображение экспериментальной системы для интеграции донора в ELP генома маиса.
[0055] Фигура 39 представляет собой график, иллюстрирующий расщепление геномной ДНК-мишени посредством eZFN. ДНК выделена из каждой группы обработки (6 повторов для каждой), как указано. Анализы TAQMAN™ использовали для измерения расщепления ДНК-мишени. Активность расщепления eZFN представлена относительно обработок только донорной ДНК. Уровни eZFN (eZFN1 и eZFN3) составляли соотношения 1:1 или 1:10 относительно донорной ДНК. Статистические группы указаны строчными буквами.
[0056] Фигура 40 иллюстрирует участки связывания праймеров внутри локусов ELP генома кукурузы.
[0057] Фигура 41 иллюстрирует участки связывания праймеров фрагмента pDAB100651 для оценки количества копий.
[0058] На фигуре 42 показана активность расщепления eZFN относительно обработок только донорной ДНК. Уровни расщепления eZFN (eZFNl и eZFN3) составляли соотношения 1:1 или 1:10 относительно донорной ДНК. Статистические группы указаны строчными буквами.
[0059] На фигуре 43, панели A и B, показана последовательность участков стыковки из реакций внутренней-внешней ПЦР. Левая и правая последовательности представляют собой частичные последовательности AAD1 и ELP, соответственно. Последовательность, ожидаемая после инсерционного восстановления участка связывания eZFN, показана синим шрифтом. Участок связывания eZFN выделен зеленым, и делеции представлены черными штрихами. Показаны последовательности для прямой ориентации (фигура 43A) и обратной ориентации (фигура 43B). Последовательности находятся в блоках в соответствии с реакцией ПЦР, после которой они клонированы.
ПОДРОБНОЕ ОПИСАНИЕ
[0060] В настоящем документе описаны композиции и способы для опосредованной нуклеазой независимой от гомологии (например, захват NHEJ) направленной интеграции трансгена. В то время как вставку олигонуклеотидов можно проводить посредством простой совместной трансфекции ДНК с совместимыми 5’-выступающими концами, в настоящее время показано, что захват NHEJ фрагментов размера трансгена (например, >0,5 т.п.о.) сильно облегчается посредством опосредованного нуклеазой расщепления in vivo донорной плазмиды в дополнение к расщеплению хромосомы. Этим способом трансгены большего размера (например, длиной между 1 и 14 т.п.о. или длиннее) можно интегрировать направленным образом в организмы и линии клеток, такие как клетки яичника китайского хомяка (CHO), не поддающиеся основанной на HDR интеграции. Например, расщепление донора in vivo позволяло направленную интеграцию с высокой частотой (6%) в не подвергавшиеся селекции клетки CHO, тип клеток, в ином случае не поддающийся направленной вставке больших последовательностей ДНК.
[0061] Совместное расщепление хромосомы и содержащего трансген двухцепочечного донора, как описано в настоящем документе, приводит к успешной интеграции в любой эндогенный локус-мишень в выбранной клетке-хозяине. Способы и композиции, описанные в настоящем документе, позволяют эффективную независимую от гомологии направленную интеграцию, которая, как правило, недостижима простой совместной трансфекцией предварительно разрезанных доноров.
[0062] Таким образом, композиции и способы, описанные в настоящем документе, позволяют независимую от гомологии направленную интеграцию больших трансгенов в участки расщепления нуклеазой, включая делеции, образованные сконструированными нуклеазами, такими как ZFN и/или TALEN. Альтернативно, можно использовать донорную плазмиду с участками для нуклеазы, фланкирующими трансген, подлежащий интеграции, так что часть трансгена высвобождается после расщепления нуклеазой и эффективно интегрирует в намеченную локализацию. Кроме того, использование способов и композиций по изобретению позволяет опосредованное нуклеазой расщепление in vivo большой донорной молекулы, так что бактериальная или дрожжевая искусственная хромосома позволяет направленную интеграцию больших трансгенов в клетки млекопитающих и растений. Наконец, описанные композиции и способы для расщепления in vivo могут находить применение в направленной генетической модификации других организмов и клеток, особенно плохо осуществляющих направляемую гомологией репарацию ДНК.
Общее описание
[0063] В практическом осуществлении способов, также как в получении и применении композиций, описанных в настоящем документе, используют, если нет иных указаний, общепринятые способы молекулярной биологии, биохимии, структуры и анализа хроматина, компьютерной химии, культуры клеток, рекомбинантной ДНК и родственных областей, находящиеся в компетенции специалистов в данной области. Эти способы полностью объяснены в литературе. См., например, Sambrook et al. MOLECULAR CLONING: A LABORATORY MANUAL, Second edition, Cold Spring Harbor Laboratory Press, 1989 и Third edition, 2001; Ausubel et al., CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, New York, 1987 и периодические обновления; серии METHODS IN ENZYMOLOGY, Academic Press, San Diego; Wolffe, CHROMATIN STRUCTURE AND FUNCTION, Third edition, Academic Press, San Diego, 1998; METHODS IN ENZYMOLOGY, Vol. 304, «Chromatin» (P.M. Wassarman and A. P. Wolffe, eds.), Academic Press, San Diego, 1999; и METHODS IN MOLECULAR BIOLOGY, Vol. 119, «Chromatin Protocols» (P.B. Becker, ed.) Humana Press, Totowa, 1999.
Определения
[0064] Термины «нуклеиновая кислота», «полинуклеотид» и «олигонуклеотид» используют взаимозаменяемо, и они относятся к дезоксирибонуклеотидному или рибонуклеотидному полимеру, в линейной или кольцевой конформации, и либо в одноцепочечной, либо в двухцепочечной форме. Для целей по настоящему описанию, эти термины не предназначены в качестве ограничивающих по отношению к длине полимера. Термины могут включать известные аналоги природных нуклеотидов, так же как нуклеотиды, которые являются модифицированными в группах основания, сахара и/или фосфата (например, фосфоротиоатных остовов). Как правило, аналог конкретного нуклеотида обладает такой же специфичностью спаривания оснований; т.е. аналог основания A спаривается с основанием T.
[0065] Термины «полипептид», «пептид» и «белок» используют взаимозаменяемо для обозначения полимера из аминокислотных остатков. Термин относится также к аминокислотным полимерам, в которых одна или более аминокислот представляют собой химические аналоги или модифицированные производные соответствующих природных аминокислот.
[0066] «Связывание» относится к специфическому для последовательности нековалентному взаимодействию между макромолекулами (например, между белком и нуклеиновой кислотой). Не все компоненты взаимодействия связывания должны быть специфическими для последовательности (например, контактировать с фосфатными остатками в остове ДНК), при условии, что взаимодействие в целом является специфическим для последовательности. Такие взаимодействия, как правило, характеризуются константой диссоциации (Kd) 10-6 M-1 или ниже. «Аффинность» относится к силе связывания: увеличенная аффинность связывания коррелирует с более низкой Kd.
[0067] «Связывающий белок» представляет собой белок, способный нековалентно связываться с другой молекулой. Связывающий белок может связываться, например, с молекулой ДНК (ДНК-связывающий белок), с молекулой РНК (РНК-связывающий белок) и/или с молекулой белка (белок-связывающий белок). В случае белок-связывающего белка, он может связываться сам с собой (с формированием гомодимеров, гомотримеров и т.д.) и/или он может связываться с одной или несколькими молекулами отличного белка или белков. Связывающий белок может иметь более одного типа связывающей активности. Например, белки с цинковыми пальцами обладают активностью связывания ДНК, связывания РНК и связывания белка.
[0068] «Связывающий ДНК белок с цинковыми пальцами (или связывающий домен) представляет собой белок или домен внутри большего белка, который связывает ДНК специфическим для последовательности образом посредством одного или более цинковых пальцев, которые представляют собой области аминокислотной последовательности внутри связывающего домена, структура которых стабилизирована посредством координации с ионом цинка. Термин «связывающий ДНК белок с цинковыми пальцами» часто обозначают как белок с цинковыми пальцами или ZFP.
[0069] «Связывающий ДНК домен TALE» или «TALE» представляет собой полипептид, содержащий один или более доменов/единиц повтора TALE. Домены повторов вовлечены в связывание TALE с родственной ему последовательностью ДНК-мишени. Одна «единица повтора» (обозначенная также как «повтор») составляет, как правило, 33-35 аминокислоты в длину и обладает по меньшей мере некоторой гомологией последовательности с последовательностями другого повтора TALE внутри белка TALE. См., например, Публикацию патента США № 20110301073, полное содержание которой приведено в настоящем документе в качестве ссылки.
[0070] Домен цинковых пальцев и связывающий домен TALE можно «конструировать» для связывания с предопределенной нуклеотидной последовательностью, например, посредством конструирования (изменения одной или более аминокислот) спирального участка узнавания природного цинкового пальца. Подобным образом, TALE можно «конструировать» для связывания с предопределенной нуклеотидной последовательностью, например, посредством конструирования аминокислот, вовлеченных в связывание ДНК (область повтора двух вариабельных остатков или область RYD). Таким образом, сконструированные связывающие ДНК белки (цинковые пальцы или TALE) представляют собой белки, не встречающиеся в природе. Неограничивающими примерами способов конструирования связывающих ДНК белков являются дизайн и отбор. Сконструированный связывающий ДНК белок представляет собой белок, не встречающийся в природе, дизайн/композиция которого принципиально является результатом использования рациональных критериев. Рациональные критерии для дизайна включают применение правил замещения и компьютеризованных алгоритмов для переработки информации в хранящейся в базах данных информации о дизайне и данных связывания существующих ZFP и/или TALE. См., например, Патенты США 6140081; 6453242 и 6534261; см. также WO 98/53058; WO 98/53059; WO 98/53060; WO 02/016536 и WO 03/016496 и Патентные публикации США № 20110301073, 20110239315 и 20119145940.
[0071] «Отобранный» белок с цинковыми пальцами или TALE представляет собой белок, не обнаруженный в природе, получение которого в первую очередь происходит в результате эмпирического способа, такого как фаговый дисплей, ловушка взаимодействия или гибридный отбор. См., например, Патенты США № 5789538; US 5925523; US 6007988; US 6013453; US 6200759; WO 95/19431; WO 96/06166; WO 98/53057; WO 98/54311; WO 00/27878; WO 01/60970, WO 01/88197 и WO 02/099084, и Патентные публикации США № 20110301073, 20110239315 и 20119145940.
[0072] «Рекомбинация» относится к процессу обмена генетической информацией между двумя полинуклеотидами, включая, но без ограничения, захват донора посредством соединения негомологичных концов (NHEJ) и гомологичную рекомбинацию. Для целей по этому описанию, «гомологичная рекомбинация (HR)» относится к специализированной форме такого обмена, который имеет место, например, в ходе репарации двухцепочечных разрывов в клетках посредством механизмов направляемой гомологией репарации. Этот процесс требует гомологии нуклеотидной последовательности, использует «донорную» молекулу в качестве матрицы для репарации молекулы-«мишени» (т.е. молекулы, подвергнутой двухцепочечному разрыву), и является известным под разными названиями как «конверсия генов без кроссовера» или «конверсия коротких участков генов», поскольку он приводит к переносу генетической информации от донора к мишени. Без намерения быть связанными какой-либо конкретной теорией, такой перенос может вовлекать коррекцию неправильного спаривания гетеродуплекса ДНК, формирующегося между имеющими разрывы мишенью и донором, и/или «зависимой от синтеза гибридизации цепей», в которой донор используют для повторного синтеза генетической информации, которая становится частью мишени, и/или родственного процесса. Такая специализированная HR часто приводит к изменению последовательности молекулы-мишени, так что часть или вся из последовательности донорного полинуклеотида включена в полинуклеотид-мишень. Для направляемой HR интеграции, молекула-донор содержит по меньшей мере 2 области гомологии с геномом («плечи гомологии») длиной по меньшей мере 50-100 пар оснований. См., например, Публикацию патента США № 20110281361.
[0073] В способах из описания, одна или более направленных нуклеаз, как описано в настоящем документе, образует двухцепочечный разрыв в последовательности-мишени (например, клеточном хроматине) в предопределенном участке, и «донорный» полинуклеотид, обладающий гомологией с нуклеотидной последовательностью в области разрыва, можно вводить в клетку. Показано, что присутствие двухцепочечного разрыва облегчает интеграцию донорной последовательности. Донорная последовательность может быть физически интегрирована или, альтернативно, донорный полинуклеотид используют в качестве матрицы для репарации разрыва посредством гомологичной рекомбинации, приводящей к введению всей или части нуклеотидной последовательности в качестве донора в клеточный хроматин. Таким образом, первая последовательность в клеточном хроматине может быть изменена и, в конкретных вариантах осуществления, может быть конвертирована в последовательность, присутствующую в донорном полинуклеотиде. Таким образом, использование терминов «заменить» или «замена» можно понимать как представляющее замену одной нуклеотидной последовательности на другую (т.е. замену последовательности в информационном смысле), и оно не обязательно требует физической или химической замены одного полинуклеотида другим.
[0074] В любом из способов, описанных в настоящем документе, дополнительные пары белков с цинковыми пальцами или TALEN можно использовать для дополнительного двухцепочечного расщепления дополнительных участков-мишеней в клетке.
[0075] Любой из способов, описанных в настоящем документе, можно использовать для вставки донора любого размера и/или частичной или полной инактивации одной или более последовательностей-мишеней в клетке посредством направленной интеграции донорной последовательности, нарушающей экспрессию представляющего интерес гена(генов). Представлены также линии клеток с частично или полностью инактивированными генами.
[0076] Более того, способы направленной интеграции, как описано в настоящем документе, можно также использовать для интеграции одной или более экзогенных последовательностей. Экзогенная последовательность нуклеиновой кислоты может содержать, например, один или более генов или молекул кДНК, или любой тип кодирующей или некодирующей последовательности, так же, как один или более контрольных элементов (например, промоторов). Кроме того, экзогенная последовательность нуклеиновой кислоты (трансген) может образовывать одну или более молекул РНК (например, короткие шпилечные РНК (кшРНК), ингибирующие РНК (РНКи), микроРНК (мкРНК) и т.д.).
[0077] «Расщепление» относится к разрушению ковалентного остова молекулы ДНК. Расщепление можно инициировать множеством способов, включая, но без ограничения, ферментативный или химический гидролиз фосфодиэфирной связи. Возможны как одноцепочечное расщепление, так и двухцепочечное расщепление, и двухцепочечное расщепление может происходить в результате двух отдельных событий одноцепочечного расщепления. Расщепление ДНК может приводить к получению либо тупых концов, либо ступенчатых концов. В конкретных вариантах осуществления слитые полипептиды используют для направленного двухцепочечного расщепления ДНК.
[0078] «Полудомен расщепления» представляет собой полипептидную последовательность, которая, в сочетании с вторым полипептидом (либо идентичным, либо отличным) формирует комплекс, обладающий активностью расщепления (предпочтительно, активностью двухцепочечного расщепления). Термины «первый и второй полудомены расщепления;» «+ и - полудомены расщепления» и «правый и левый полудомены расщепления» используют взаимозаменяемо для обозначения пар полудоменов расщепления, которые димеризуются.
[0079] «Сконструированный полудомен расщепления» представляет собой полудомен расщепления, модифицированный так, чтобы формировать облигатные гетеродимеры с другим полудоменом расщепления (например, другим сконструированным полудоменом расщепления). См., также Публикации Патентов США № 2005/0064474, 20070218528 и 2008/0131962, полное содержание которых приведено в настоящем документе в качестве ссылки.
[0080] Термин «последовательность» относится к нуклеотидной последовательности любой длины, которая может представлять собой ДНК или РНК; может являться линейной, кольцевой или разветвленной и может являться либо одноцепочечной, либо двухцепочечной. Термин «донорная последовательность» или «трансген» относится к нуклеотидной последовательности, которую вставляют в геном. Донорная последовательность может иметь любую длину, например, длину между 2 и 100000000 нуклеотидами (или любое целое значение между ними или выше них), предпочтительно, длину между приблизительно 100 и 100000 нуклеотидами (или любое целое значение между ними), более предпочтительно, длину между приблизительно 2000 и 60000 нуклеотидов (или любое значение между ними) и даже более предпочтительно, между приблизительно 3 и 15 т.п.о. (или любое значение между ними).
[0081] «Хроматин» представляет собой нуклеопротеиновую структуру, содержащую геном клетки. Клеточный хроматин содержит нуклеиновую кислоту, преимущественно, ДНК, и белок, включая гистоны и не относящиеся к гистонам хромосомные белки. Большая часть хроматина эукариотической клетки существует в форме нуклеосом, где сердцевина нуклеосомы содержит приблизительно 150 пар оснований ДНК, связанные с октамером, содержащим по два каждого из гистонов H2A, H2B, H3 и H4; и линкерную ДНК (различной длины в зависимости от организма), протяженную между сердцевинами нуклеосом. Молекула гистона H1, как правило, связана с линкерной ДНК. Для целей по настоящему описанию, термин «хроматин» предназначен, чтобы включать все типы клеточного нуклеопротеина, как прокариотического, так и эукариотического. Клеточный хроматин включает как хромосомный, так и эписомальный хроматин.
[0082] «Хромосома» представляет собой комплекс хроматина, содержащий весь или часть генома клетки. Геном клетки часто характеризуют по его кариотипу, который представляет собой набор всех хромосом, содержащих геном клетки. Геном клетки может содержать одну или более хромосом.
[0083] «Эписома» представляет собой реплицирующуюся нуклеиновую кислоту, нуклеопротеиновый комплекс или другую структуру, содержащую нуклеиновую кислоту, которая не является частью хромосомного кариотипа клетки. Примеры эписом включают плазмиды и определенные вирусные геномы.
[0084] «Участок-мишень» или «последовательность-мишень» представляет собой последовательность нуклеиновой кислоты, определяющую часть нуклеиновой кислоты, с которой может связываться связывающая молекула, при условии существования подходящих условия для связывания.
[0085] «Экзогенная» молекула представляет собой молекулу, которая в норме не присутствует в клетке, но которую можно вводить в клетку посредством одного или более генетических, биохимических или других способов. «Нормальное присутствие в клетке» определяют по отношению к конкретной стадии развития и условий окружения клетки. Таким образом, например, молекула, присутствующая только в ходе эмбрионального развития мышцы, является экзогенной молекулой по отношению к клетке взрослой мышцы. Подобным образом, молекула, индуцированная тепловым шоком, является экзогенной молекулой по отношению к не подвергнутой тепловому шоку клетке. Экзогенная молекула может содержать, например, функциональный вариант неправильно функционирующей эндогенной молекулы или неправильно функционирующий вариант функционирующей в норме эндогенной молекулы.
[0086] Экзогенная молекула может представлять собой, среди других молекул, малую молекулу, такую как получают способом комбинаторной химии, или макромолекулу, такую как белок, нуклеиновая кислота, углевод, липид, гликопротеин, липопротеин, полисахарид, любое модифицированное производное вышеуказанных молекул, или любой комплекс, содержащий одну или более вышеуказанных молекул. Нуклеиновые кислоты включают ДНК и РНК, могут являться одно- или двухцепочечными; могут являться линейными, разветвленными или кольцевыми; и могут иметь любую длину. Нуклеиновые кислоты включают нуклеиновые кислоты, способные формировать дуплексы, также как формирующие триплекс нуклеиновые кислоты. См., например, Патенты США № 5176996 и 5422251. Белки включают, но без ограничения, ДНК-связывающие белки, факторы транскрипции, факторы ремоделирования хроматина, связывающие метилированную ДНК белки, полимеразы, метилазы, деметилазы, ацетилазы, деацетилазы, киназы, фосфатазы, интегразы, рекомбиназы, лигазы, топоизомеразы, гиразы и хеликазы.
[0087] Экзогенная молекула может относиться к такому же типу молекул, как эндогенная молекула, например, экзогенный белок или нуклеиновая кислота. Например, экзогенная нуклеиновая кислота может содержать инфекционный вирусный геном, плазмиду или эписому, введенные в клетку, или хромосому, которая в норме не присутствует в клетке. Способы введения экзогенных молекул в клетки известны специалистам в данной области и включают, но без ограничения, опосредованный липидами перенос (т.е., липосомы, содержащие нейтральные и катионные липиды), электропорацию, прямую инъекцию, слияние клеток, бомбардировку частицами, совместную преципитацию с фосфатом кальция, опосредованный DEAE-декстраном перенос и опосредованный вирусным вектором перенос. Экзогенная молекула может также относиться к такому же типу молекул, как эндогенная молекула, но являться выделенной из вида, отличного от того, из которого происходит клетка. Например, последовательность нуклеиновой кислоты человека можно вводить в линию клеток, исходно происходящую из клеток мыши или хомяка.
[0088] В отличие от этого, «эндогенная» молекула представляет собой молекулу, которая в норме присутствует в конкретной клетке на конкретной стадии развития в конкретных условиях внешней среды. Например, эндогенная нуклеиновая кислота может содержать хромосому, геном митохондрии, хлоропласта или другой органеллы, или природную эписомальную нуклеиновую кислоту. Дополнительные эндогенные молекулы могут включать белки, например, факторы транскрипции и ферменты.
[0089] «Слитая» молекула представляет собой молекулу, в которой две или более молекул субъединиц связаны, предпочтительно, ковалентно. Молекулы субъединиц могут представлять собой один и тот же химический тип молекул, или могут представлять собой различные химические типы молекул. Примеры первого типа слитых молекул включают, но без ограничения, слитые белки (например, слитый белок между ДНК-связывающим доменом ZFP или TALE и одним или несколькими активирующими доменами) и слитые нуклеиновые кислоты (например, нуклеиновая кислота, кодирующая слитый белок, описанный выше). Примеры второго типа слитой молекулы включают, но без ограничения, слитую молекулу между формирующей триплекс нуклеиновой кислотой и полипептидом, и слитую молекулу между связывающей малую бороздку молекулой и нуклеиновой кислотой.
[0090] Экспрессия слитого белка в клетке может происходить в результате доставки слитого белка в клетку или в результате доставки полинуклеотида, кодирующего слитый белок, в клетку, где полинуклеотид транскрибируется и транскрипт транслируется, для получения слитого белка. Транс-сплайсинг, расщепление полипептида и лигирование полипептида могут быть также вовлечены в экспрессию белка в клетке. Способы доставки полинуклеотида и полипептида в клетки представлены в другом месте в этом описании.
[0091] «Ген», для целей по настоящему описанию, включает область ДНК, кодирующую продукт гена (см. ниже), так же, как все области ДНК, регулирующие получение продукта гена, вне зависимости от того, являются или нет такие регуляторные последовательности соседними с кодирующими и/или транскрибируемыми последовательностями. Соответственно, ген включает, но без ограничения, промоторные последовательности, терминаторы, регуляторные последовательности для трансляции, такие как участки связывания рибосом и внутренние участки связывания рибосом, энхансеры, сайленсеры, инсуляторы, пограничные элементы, точки начала репликации, участки присоединения матрикса и контрольные области локусов.
[0092] «Экспрессия гена» относится к переводу информации, содержащейся в гене, в продукт гена. Продукт гена может представлять собой непосредственно продукт транскрипции гена (например, мРНК, тРНК, рРНК, антисмысловую РНК, рибозим, структурную РНК или любой другой тип РНК) или белок, полученный посредством трансляции мРНК. Продукты генов включают также РНК, модифицированные посредством таких процессов, как кэпирование, полиаденилирование, метилирование и редактирование, и белки, модифицированные, например, посредством метилирования, ацетилирования, фосфорилирования, убиквитинилирования, АДФ-рибозилирования, миристилирования и гликозилирования.
[0093] «Модуляция» экспрессии гена относится к изменению активности гена. Модуляция экспрессии может включать, но без ограничения, активацию гена и репрессию гена. Редактирование генома (например, расщепление, изменение, инактивацию, случайную мутацию) можно использовать для модуляции экспрессии. Инактивация гена относится к любому снижению экспрессии гена по сравнению с клеткой, не содержащей ZFP, как описано в настоящем документе. Таким образом, инактивация гена может являться частичной или полной.
[0094] «Представляющая интерес область» представляет собой любую область клеточного хроматина, например, такую как ген или некодирующая последовательность внутри гена или по соседству с геном, в которой желательно связывание экзогенной молекулы. Связывание может происходить с целью направленного расщепления ДНК и/или направленной рекомбинации. Представляющая интерес область может присутствовать, например, на хромосоме, эписоме, в геноме органеллы (например, митохондрии, хлоропласта) или в инфицирующем вирусном геноме. Представляющая интерес область может находиться внутри кодирующей области гена, внутри транскрибируемых некодирующих областей, например, таких как лидерные последовательности, трейлерные последовательности или интроны, или внутри нетранскрибируемых областей, либо выше, либо ниже кодирующей области. Представляющая интерес область может составлять настолько мало, как отдельная пара нуклеотидов, или вплоть до 2000 пар нуклеотидов в длину, или любое целое число пар нуклеотидов.
[0095] «Эукариотические» клетки включают, но без ограничения, клетки грибов (таких как дрожжи), клетки растений, клетки животных, клетки млекопитающих и клетки человека (например, T-клетки).
[0096] Термины «функциональная связь» и «функционально связанный» (или «оперативно связанный») используют взаимозаменяемо по отношению к смежному положению двух или более компонентов (таких как элементы последовательности), в котором компоненты организованы таким образом, что оба компонента функционируют нормально и обеспечивают возможность того, что по меньшей мере один из компонентов может опосредовать функцию, оказывающую воздействие по меньшей мере на один из других компонентов. В качестве иллюстрации, последовательность регуляции транскрипции, такая как промотор, является функционально связанной с кодирующей последовательностью, если последовательность регуляции транскрипции контролирует уровень транскрипции кодирующей последовательности в ответ на присутствие или отсутствие одного или более факторов регуляции транскрипции. Последовательность регуляции транскрипции, как правило, является функционально связанной в цис- с кодирующей последовательностью, но не обязательно должна непосредственно соседствовать с ней. Например, энхансер представляет собой последовательность регуляции транскрипции, функционально связанную с кодирующей последовательностью, даже если они не являются непрерывными.
[0097] По отношению к слитым полипептидам, термин «функционально связанный» может относиться к тому факту, что каждый из компонентов осуществляет такую же функцию в связи с другим компонентом, какую осуществлял бы в отсутствие такой связи. Например, по отношению к слитому полипептиду, в котором ДНК-связывающий домен ZFP, TALE или Cas слит с доменом активации, ДНК-связывающий домен ZFP, TALE или Cas и домен активации являются функционально связанными, если в слитом полипептиде часть ДНК-связывающего домена ZFP, TALE или Cas является способной связываться с его участком-мишенью и/или с его участком связывания, в то время как домен активации является способным осуществлять повышающую регуляцию экспрессии гена. Когда слитый полипептид, в котором ДНК-связывающий домен ZFP, TALE или Cas ДНК является слитым с доменом расщепления, ДНК-связывающий домен ZFP, TALE или Cas и домен расщепления являются функционально связанными, если в слитом полипептиде часть ДНК-связывающего домена ZFP, TALE или Cas является способной связываться с его участком-мишенью и/или с его участком связывания, в то время как домен расщепления является способным расщеплять ДНК вблизи участка-мишени.
[0098] «Функциональный фрагмент» белка, полипептида или нуклеиновой кислоты представляет собой белок, полипептид или нуклеиновую кислоту, последовательность которого не является идентичной полноразмерному белку, полипептиду или нуклеиновой кислоте, еще сохраняющий такую же функцию, как полноразмерный белок, полипептид или нуклеиновая кислота. Функциональный фрагмент может обладать большим, меньшим или одинаковым количеством остатков, как соответствующая нативная молекула, и/или может содержать одну или более замен аминокислот или нуклеотидов. Способы определения функции нуклеиновой кислоты, (например, кодирующей функции, способности гибридизоваться с другой нуклеиновой кислотой) хорошо известны в данной области. Подобным образом, способы определения функции белка хорошо известны. Например, функцию ДНК-связывания полипептида можно определять, например, посредством анализов связывания на фильтре, сдвига подвижности при электрофорезе или иммунопреципитации. Расщепление ДНК можно анализировать посредством электрофореза в геле. См. Ausubel et al., выше. Способность белка взаимодействовать с другим белком можно определять, например, посредством коиммунопреципитации, двухгибридных анализов или комплементации как генетической, так и биохимической. См., например, Fields et al. (1989) Nature 340:245-246; Патент США № 5585245 и PCT WO 98/44350.
[0099] «Вектор» является способным переносить последовательности генов к клеткам-мишеням. Как правило, «векторная конструкция», «экспрессирующий вектор» и «вектор для переноса генов» обозначает любую конструкцию нуклеиновой кислоты, которая способна управлять экспрессией представляющего интерес гена и которая может переносить последовательности генов к клеткам-мишеням. Таким образом, термин включает клонирующие и экспрессирующие векторы, так же как интегрирующие векторы.
[0100] «Репортерный ген» или «репортерная последовательность» относится к любой последовательности, образующей белковый продукт, который легко измерять, предпочтительно, хотя не обязательно, в общепринятом анализе. Пригодные репортерные гены включают, но без ограничения, последовательности, кодирующие белки, опосредующие устойчивость к антибиотику (например, устойчивость к ампициллину, устойчивость к неомицину, устойчивость к G418, устойчивость к пуромицину), последовательности, кодирующие окрашенные или флуоресцентные, или люминесцентные белки (например, зеленый флуоресцентный белок, улучшенный зеленый флуоресцентный белок, красный флуоресцентный белок, люциферазу), и белки, опосредующие усиленный рост клеток и/или амплификацию гена (например, дигидрофолатредуктазу). Эпитопы-метки включают, например, одну или более копий FLAG, His, myc, Tap, HA или любой поддающейся детекции аминокислотной последовательности. «Маркеры экспрессии» включают последовательности, кодирующие репортеры, которые могут являться функционально связанными с желательной последовательностью гена для мониторирования экспрессии представляющего интерес гена.
[0101] Локус «области безопасности» представляет собой локус внутри генома, куда можно вставлять ген без каких-либо неблагоприятных эффектов для клетки-хозяина. Наиболее преимущественным является локус области безопасности, в котором экспрессия последовательности представляющего интерес гена не нарушается посредством какой-либо сквозной экспрессии соседних генов. Неограничивающими примерами локусов области безопасности в клетках млекопитающих являются ген AAVS1 (см. Патент США № 8110379), ген CCR5 (см. Патентную публикацию США № 20080159996), локус Rosa (см. WO 2010/065123) и/или локус альбумина (Патентная заявка США № 13/624193). Неограничивающим примером локусов области безопасности в клетках растений является локус ZP15 (Патент США № 8329986).
Нуклеазы
[0102] В настоящем документе описаны композиции, в частности, нуклеазы, которые являются пригодными для расщепления in vivo донорной молекулы, несущей трансген, и нуклеазы для расщепления генома клетки, так что трансген интегрируют в геном направленным образом. В конкретных вариантах осуществления одна или более из нуклеаз являются природными. В других вариантах осуществления, одна или более из нуклеаз являются не природными, т.е. сконструированными в ДНК-связывающем домене и/или домене расщепления. Например, ДНК-связывающий домен природной нуклеазы можно изменять для связывания с выбранным участком-мишенью (например, мегануклеаза, сконструированная для связывания участка, отличного от родственного участка связывания). В других вариантах осуществления нуклеаза содержит гетерологичные ДНК-связывающий домен и домен расщепления (например, нуклеазы с цинковыми пальцами; ДНК-связывающие белки с эффекторным доменом TAL; ДНК-связывающие домены мегануклеазы с гетерологичными доменами расщепления).
A. ДНК-связывающие домены
[0103] В конкретных вариантах осуществления в композициях и способах, описанных в настоящем документе, используют ДНК-связывающий домен мегануклеазы (эндонуклеазы хоминга) для связывания с донорной молекулой и/или связывания с представляющей интерес областью в геноме клетки. Природные мегануклеазы узнают участки расщепления 15-40 пар оснований и в общем сгруппированы в четыре семейства: семейство LAGLIDADG, семейство GIY-YIG, семейство His-Cyst бокс и семейство HNH. Иллюстративные эндонуклеазы хоминга включают I-SceI, I-CeuI, PI-PspI, PI-Sce, I-SceIV, I-CsmI, I-PanI, I-SceII, I-PpoI, I-SceIII, I-CreI, I-TevI, I-TevII и I-TevIII. Их последовательности узнавания известны. См. также Патент США № 5420032; Патент США № 6833252; Belfort et al. (1997) Nucleic Acids Res. 25:3379-3388; Dujon et al. (1989) Gene 82:115-118; Perler et al. (1994) Nucleic Acids Res. 22, 1125-1127; Jasin (1996) Trends Genet. 12:224-228; Gimble et al. (1996) J. Mol. Biol. 263:163-180; Argast et al. (1998) J. Mol. Biol. 280:345-353 и каталог New England Biolabs.
[0104] В конкретных вариантах осуществления в способах и композициях, описанных в настоящем документе, используют нуклеазу, содержащую сконструированную (неприродную) эндонуклеазу хоминга (мегануклеазу). Последовательности узнавания эндонуклеаз хоминга и мегануклеаз, таких как I-SceI, I-CeuI, PI-PspI, PI-Sce, I-SceIV, I-CsmI, I-PanI, I-SceII, I-PpoI, I-SceIII, I-CreI, I-TevI, I-TevII и I-TevIII известны. См. также Патент США № 5420032; Патент США № 6833252; Belfort et al. (1997) Nucleic Acids Res. 25:3379-3388; Dujon et al. (1989) Gene 82:115-118; Perler et al. (1994) Nucleic Acids Res. 22, 1125-1127; Jasin (1996) Trends Genet. 12:224-228; Gimble et al. (1996) J. Mol. Biol. 263:163-180; Argast et al. (1998) J. Mol. Biol. 280:345-353 и каталог New England Biolabs. Кроме того, ДНК-связывающую специфичность эндонуклеаз и мегануклеаз хоминга можно конструировать для связывания неприродных участков-мишеней. См., например, Chevalier et al. (2002) Molec. Cell 10:895-905; Epinat et al. (2003) Nucleic Acids Res. 31:2952-2962; Ashworth et al. (2006) Nature 441:656-659; Paques et al. (2007) Current Gene Therapy 7:49-66; Публикацию патента США № 20070117128. ДНК-связывающие домены эндонуклеаз и мегануклеаз хоминга можно изменять в контексте нуклеазы в целом (т.е. так что нуклеаза содержит родственный домен расщепления) или можно сливать с гетерологичным доменом расщепления.
[0105] В других вариантах осуществления ДНК-связывающий домен одной или более нуклеаз, использованных в способах и композициях, описанных в настоящем документе, содержит природный или сконструированный (неприродный) ДНК-связывающий домен с эффектором TAL. См., например, Публикацию патента США № 20110301073, полное содержание которой приведено в настоящем документе в качестве ссылки. Известно, что патогенные бактерии растений рода Xanthomonas вызывают многие заболевания в важных сельскохозяйственных растениях. Патогенность Xanthomonas зависит от консервативной системы секреции типа III (T3S), инъецирующей более 25 различных эффекторных белков в клетку растения. Среди этих инъецируемых белков присутствуют подобные активатору транскрипции эффекторы (TAL), мимикрирующие активаторы транскрипции растений и манипулирующие транскриптомом растения (см. Kay et al. (2007) Science 318:648-651). Эти белки содержат ДНК-связывающий домен и домен активации транскрипции. Одним из наиболее хорошо охарактеризованных эффекторов TAL является AvrBs3 из Xanthomonas campestgris pv. Vesicatoria (см. Bonas et al Mol Gen Genet 218: 127-136 и WО2010079430). Эффекторы TAL содержат центральный домен из тандемных повторов, где каждый повтор содержит приблизительно 34 аминокислоты, являющиеся ключевыми для ДНК-связывающей специфичности этих белков. Кроме того, они содержат последовательность ядерной локализации и кислый домен активации транскрипции (обзор см. в Schomack S, et al. (2006) JPlant Physiol 163(3): 256-272). Кроме того, в фитопатогенных бактериях Ralstonia solanacearum обнаружены два гена, обозначенные brg11 и hpxl7, которые являются гомологичными семейству AvrBs3 Xanthomonas в R. solanacearum биоваре 1 штамме GMI1000 и в биоваре 4 штамме RS1000 (См. Heuer et al (2007) Appl and Envir Micro 73(13): 4379-4384). Эти гены являются на 98,9% идентичными друг другу по нуклеотидной последовательности, но отличаются делецией 1575 п.о. в домене повторов hpxl7. Однако продукты обоих генов обладают менее чем 40% идентичностью последовательности с семейством белков AvrBs3 Xanthomonas. См., например, Публикации патентов США № 20110239315, 20110145940 и 20110301073, полное содержание которых приведено в настоящем документе в качестве ссылки.
[0106] Специфичность этих эффекторов TAL зависит от последовательностей, обнаруженных в тандемных повторах. Последовательность повторов содержит приблизительно 102 п.о., и повторы являются, как правило, на 91-100% гомологичными друг другу (Bonas et al., там же). Полиморфизм повторов обычно локализован в положениях 12 и 13 и, по-видимому, существует взаимно однозначное соответствие между идентичностью двух гипервариабельных остатков в положениях 12 и 13 с идентичностью непрерывных нуклеотидов в последовательности-мишени эффекторов TAL (см. Moscou and Bogdanove, (2009) Science 326:1501 и Boch et al. (2009) Science 326:1509-1512). Экспериментально, природный код для узнавания ДНК этими эффекторами TAL определили так, что последовательность HD в положениях 12 и 13 приводит к связыванию с цитозином (C), NG связывается с T, NI с A, C, G или T, NN связывается с A или G, и ING связывается с T. Эти ДНК-связывающие повторы собирали в белки с новыми комбинациями и количеством повторов, для получения искусственных факторов транскрипции, способных взаимодействовать с новыми последовательностями и активировать экспрессию не эндогенного репортерного гена в клетках растений (Boch et al., там же). Сконструированные белки TAL связывали с полудоменом расщепления FokI для получения слитой с эффекторным доменом TAL нуклеазы (TALEN), обладающей активностью в анализе репортера в дрожжах (мишень на основе плазмиды). См., например, Публикацию патента США № 20110301073; Christian et al ((2010)< Genetics epub 10.1534/genetics. 110.120717).
[0107] В других вариантах осуществления нуклеаза представляет собой систему, содержащую систему нуклеазы CRISPR (короткие палиндромные повторы, расположенные группами, равномерно удаленными друг от друга)/Cas (ассоциированная с CRISPR). CRISPR/Cas представляет собой сконструированную систему нуклеазы, основанную на бактериальной системе, которую можно использовать для генной инженерии. Она основана на части адаптивного иммунного ответа многих бактерий и архей. Когда вирус или плазмида проникает в бактерию, фрагменты проникающей ДНК переводятся в CRISPR РНК (crРНК) посредством «иммунного» ответа. Затем эта crРНК связывается, через область частичной комплементарности, с другим типом РНК, называемой tracrРНК, для направления нуклеазы Cas9 на область, гомологичную crРНК в ДНК-мишени, называемую «протоспейсер». Cas9 расщепляет ДНК с получением тупых концов в DSB в участках, определяемых 20-нуклеотидной направляющей последовательностью, содержащейся в транскрипте crRNA. Cas9 требует как crРНК, так и tracrРНК для сайт-специфического узнавания и расщепления ДНК. Эта система в настоящее время сконструирована так, что crРНК и tracrРНК можно объединять в одну молекулу («одиночную руководящую РНК»), и эквивалентную crРНК часть одиночной руководящей РНК можно конструировать для направления нуклеазы Cas9 для нацеливания на любую желательную последовательность (см. Jinek et al. (2012) Science 337, p. 816-821, Jinek et al., (2013), eLife 2:e00471, и David Segal, (2013) eLife 2:e00563). Таким образом, систему CRISPR/Cas можно конструировать для создания DSB в желательной мишени в геноме, и на репарацию DSB можно влиять посредством использования ингибиторов репарации, чтобы вызвать увеличение допускающей ошибки репарации.
[0108] В конкретных вариантах осуществления ДНК-связывающий домен одной или более из нуклеаз, использованных для расщепления in vivo и/или направленного расщепления генома клетки, содержит белок с цинковыми пальцами. Предпочтительно, белок с цинковыми пальцами не является природным, в том, что он сконструирован для связывания с выбранным участком-мишенью. См., например, Beerli et al. (2002) Nature Biotechnol. 20:135-141; Pabo et al. (2001) Ann. Rev. Biochem. 70:313-340; Isalan et al. (2001) Nature Biotechnol. 19:656-660; Segal et al. (2001) Curr. Opin. Biotechnol. 12:632-637; Choo et al. (2000) Curr. Opin. Struct. Biol. 10:411-416; Патенты США № 6453242; 6534261; 6599692; 6503717; 6689558; 7030215; 6794136; 7067317; 7262054; 7070934; 7361635; 7253273; и Публикации патентов США № 2005/0064474; 2007/0218528; 2005/0267061, полное содержание всех из которых приведено в настоящем документе в качестве ссылки.
[0109] Сконструированный связывающий домен с цинковыми пальцами может обладать новой связывающей специфичностью, по сравнению с природным белком с цинковыми пальцами. Способы конструирования включают, но без ограничения, рациональный дизайн и различные типы отбора. Рациональный дизайн включает, например, использование баз данных, содержащих триплетные (или квадруплетные) нуклеотидные последовательности и аминокислотные последовательности отдельных цинковых пальцев, в которых каждая триплетная или квадруплетная нуклеотидная последовательность ассоциирована с одной или несколькими аминокислотными последовательностями цинковых пальцев, которые связывают конкретную триплетную или квадруплетную последовательность. См., например, принадлежащие тем же заявителям Патенты США 6453242 и 6534261, полное содержание которых приведено в настоящем документе в качестве ссылки.
[0110] Иллюстративные способы отбора, включая системы фагового дисплея и двугибридные системы, описаны в Патентах США 5789538; 5925523; 6007988; 6013453; 6410248; 6140466; 6200759; и 6242568; так же как в WO 98/37186; WO 98/53057; WO 00/27878; WO 01/88197 и GB 2338237. Кроме того, описано улучшение специфичности связывания связывающих доменов с цинковыми пальцами, например, в принадлежащей тем же заявителям WO 02/077227.
[0111] Кроме того, как описано в этих и других ссылках, домены с цинковыми пальцами и/или белки с множеством цинковых пальцев можно связывать вместе с использованием любых пригодных линкерных последовательностей, включая, например, линкеры длиной 5 или более аминокислот. См. также иллюстративные линкерные последовательности длиной 6 или более аминокислот в Патентах США № 6479626; 6903185; и 7153949. Белки, описанные в настоящем документе, могут включать любую комбинацию пригодных линкеров между отдельными цинковыми пальцами белка.
[0112] Выбор участков-мишеней; ZFP и способы дизайна и конструирования слитых белков (и кодирующих их полинуклеотидов) известны специалистам в данной области и подробно описаны в Патентах США № 6140081; 5789538; 6453242; 6534261; 5925523; 6007988; 6013453; 6200759; WO 95/19431; WO 96/06166; WO 98/53057; WO 98/54311; WO 00/27878; WO 01/60970 WO 01/88197; WO 02/099084; WO 98/53058; WO 98/53059; WO 98/53060; WO 02/016536 и WO 03/016496.
[0113] Кроме того, как описано в этих и других ссылках, домены с цинковыми пальцами и/или белки с множеством цинковых пальцев можно связывать вместе с использованием любых пригодных линкерных последовательностей, включая, например, линкеры длиной 5 или более аминокислот. См. также иллюстративные линкерные последовательности длиной 6 или более аминокислот в Патентах США № 6479626; 6903185; и 7153949. Белки, описанные в настоящем документе, могут включать любую комбинацию пригодных линкеров между отдельными цинковыми пальцами белка.
[0114] Локус CRISPR (короткие палиндромные повторы, расположенные группами, равномерно удаленными друг от друга), кодирующий РНК-компоненты системы, и локус cas (CRISPR-ассоциированный), кодирующий белки (Jansen et al., 2002. Mol. Microbiol. 43: 1565-1575; Makarova et al., 2002. Nucleic Acids Res. 30: 482-496; Makarova et al., 2006. Biol. Direct 1:7; Haft et al., 2005. PLoS Comput. Biol. 1: e60), составляют последовательности генов системы нуклеазы CRISPR/Cas. Локусы CRISPR в хозяевах-микроорганизмах содержат комбинацию CRISPR-ассоциированных (Cas) геов, так же, как некодирующих элементов РНК, способных программировать специфичность опосредованного CRISPR расщепления нуклеиновых кислот.
[0115] CRISPR типа II является одной из наиболее хорошо охарактеризованных систем и осуществляет направленный двухцепочечный разрыв ДНК за четыре последовательных стадии. Во-первых, две некодирующие РНК, массив пре-crРНК и tracrРНК, транскрибируются с локуса CRISPR. Во-вторых, tracrRNA гибридизуется с областями повторов пре-crРНК и опосредует процессинг пре-crРНК до зрелых crРНК, содержащих отдельные спейсерные последовательности. В третьих, зрелый комплекс crРНК:tracrРНК направляет Cas9 на ДНК-мишень посредством спаривания оснований по Уотсону-Крику между спейсером crРНК и протоспейсером на ДНК-мишени рядом с прилежащим к протоспейсеру мотивом (PAM), дополнительное требование для узнавания мишени. Наконец, Cas9 опосредует расщепление ДНК-мишени для создания двухцепочечного разрыва внутри протоспейсера. Активность системы CRISPR/Cas состоит из трех стадий: (i) вставка чужеродных последовательностей ДНК в массив CRISPR для предотвращения следующих атак, в процессе, называемом «адаптацией», (ii) экспрессия соответствующих белков, так же как экспрессия и процессинг массива, с последующей (iii) опосредованной РНК интерференцией чужеродной нуклеиновой кислоты. Таким образом, в бактериальной клетке, несколько так называемых белков «Cas» вовлечены в природную функцию системы CRISPR/Cas и играют роли в таких функциях, как вставка чужеродной ДНК и т.д.
[0116] В конкретных вариантах осуществления белок Cas может представлять собой «функциональное производное» природного белка Cas. «Функциональное производное» природной последовательности полипептида представляет собой соединение, обладающее качественным биологическим свойством, общим с природной последовательностью полипептида. «Функциональные производные» включают, но без ограничения, фрагменты природной последовательности и производные природной последовательности полипептида и их фрагменты, при условии, что они обладают биологической активностью, общей с соответствующей природной последовательностью полипептида. Биологическая активность, предусмотренная в настоящем документе, представляет собой способность функционального производного гидролизовать субстрат ДНК на фрагменты. Термин «производное» включает как варианты аминокислотной последовательности полипептида, ковалентные модификации, так и слитые с ней белки. Пригодные производные полипептида Cas или его фрагмента включают, но без ограничения, мутанты, слитые белки, ковалентные модификации белка Cas или его фрагмента. Белок Cas, который включает белок Cas или его фрагмент, так же как производные белка Cas или его фрагмента, можно получать из клетки или синтезировать химически или посредством комбинации этих двух способов. Клетка может представлять собой клетку, которая естественным образом продуцирует белок Cas, или клетку, которая естественным образом продуцирует белок Cas и подвергнута генетической инженерии для продукции эндогенного белка Cas с более высоким уровнем экспрессии или для продукции белка Cas с экзогенно введенной нуклеиновой кислотой, где нуклеиновая кислота кодирует Cas, который является таким же или отличным от эндогенного Cas. В некоторых случаях, клетка естественным образом не продуцирует белок Cas и подвергнута генетической инженерии для продукции белка Cas.
[0117] Таким образом, нуклеаза содержит ДНК-связывающий домен, так что специфически связывается с участком-мишенью в любом гене, в который является желательным вставить донор (трансген).
B. Домены расщепления
[0118] Любой пригодный домен расщепления можно оперативно связывать с ДНК-связывающим доменом для формирования нуклеазы. Например, ДНК-связывающие домены ZFP сливали с доменами нуклеазы для получения ZFN - функционального соединения, которое является способным узнавать предназначенную для него нуклеиновую кислоту-мишень посредством его сконструированного (ZFP) ДНК-связывающего домена и вызывать разрезание ДНК около участка связывания ZFP посредством активности нуклеазы. См., например, Kim et al. (1996) Proc Natl Acad Sci USA 93(3): 1156-1160. Более недавно, ZFN использовали для модификации генома во множестве организмов. См., например, Публикации патентов Соединенных Штатов 20030232410; 20050208489; 20050026157; 20050064474; 20060188987; 20060063231; и Международную публикацию WO 07/014275. Подобным образом, ДНК-связывающие домены TALE сливали с доменами нуклеазы для получения TALEN. См., например, Публикацию патента США № 20110301073.
[0119] Как указано выше, домен расщепления может являться гетерологичным для ДНК-связывающего домена, например, ДНК-связывающий домен с цинковыми пальцами и домен расщепления из нуклеазы или ДНК-связывающий домен и домен расщепления TALEN, или ДНК-связывающий домен мегануклеазы и домен расщепления из другой нуклеазы. Гетерологичные домены расщепления можно получать из любой эндонуклеазы или экзонуклеазы. Иллюстративные эндонуклеазы, из которых можно получать домен расщепления, включают, но без ограничения, эндонуклеазы рестрикции и эндонуклеазы хоминга. См., например, каталог 2002-2003, New England Biolabs, Beverly, MA; и Belfort et al. (1997) Nucleic Acids Res. 25:3379-3388. Известны дополнительные ферменты, расщепляющие ДНК (например, нуклеаза S1; нуклеаза золотистой фасоли; ДНКаза I поджелудочной железы; нуклеаза микрококков; эндонуклеаза дрожжей HO; см. также Linn et al. (eds.) Nucleases, Cold Spring Harbor Laboratory Press, 1993). Один или более из этих ферментов (или их функциональных фрагментов) можно использовать в качестве источника доменов расщепления и полудоменов расщепления.
[0120] Подобным образом, полудомен расщепления можно получать из любой нуклеазы или ее части, как указано выше, которая требует димеризации для активности расщепления. В общем, два слитых белка необходимо для расщепления, если слитые белки содержат полудомены расщепления. Альтернативно, можно использовать один белок, содержащий два полудомена расщепления. Два полудомена расщепления можно получать из одной и той же эндонуклеазы (или ее функциональных фрагментов), или каждый полудомен расщепления можно получать из различной эндонуклеазы (или ее функциональных фрагментов). Кроме того, участки-мишени для двух слитых белков предпочтительно расположены, по отношению друг к другу, так что связывание двух слитых белков с их соответствующими участками-мишенями помещает полудомены расщепления в пространственной ориентации друг к другу, позволяющей полудоменам расщепления формировать функциональный домен расщепления, например, посредством димеризации. Таким образом, в конкретных вариантах осуществления ближайшие границы участков-мишеней разделены 5-8 нуклеотидами или 15-18 нуклеотидами. Однако любое целое количество нуклеотидов или пар нуклеотидов может находиться между двумя участками-мишенями (например, от 2 до 50 пар нуклеотидов или более). Как правило, участок расщепления находится между участками-мишенями.
[0121] Рестрикционные эндонуклеазы (рестрикционные ферменты) присутствуют во многих видах и являются способными к специфическому для последовательности связыванию с ДНК (в участке узнавания) и расщеплению ДНК в участке связывания или около участка связывания. Конкретные рестрикционные ферменты (например, типа IIS) расщепляют ДНК в участках, удаленных от участка узнавания, и имеют отделяемые домены связывания и расщепления. Например, фермент типа IIS FokI катализирует двухцепочечное расщепление ДНК в 9 нуклеотидах от его участка узнавания на одной цепи и в 13 нуклеотидах от его участка узнавания на другой. См., например, Патенты США 5356802; 5436150 и 5487994; так же, как Li et al. (1992) Proc. Natl. Acad. Sci. USA 89:4275-4279; Li et al. (1993) Proc. Natl. Acad. Sci. USA 90:2764-2768; Kim et al. (1994a) Proc. Natl. Acad. Sci. USA 91:883-887; Kim et al. (1994b) J. Biol. Chem. 269:31,978-31,982. Таким образом, в одном варианте осуществления, слитые белки содержат домен расщепления (или полудомен расщепления) по меньшей мере из одного рестрикционного фермента типа IIS и один или более связывающих доменов с цинковыми пальцами, которые могут являться или могут не являться сконструированными.
[0122] Иллюстративным рестрикционным ферментом типа IIS, домен расщепления которого является отделяемым от его связывающего домена, является Fok I. Этот конкретный фермент является активным в форме димера. Bitinaite et al. (1998) Proc. Natl. Acad. Sci. USA 95: 10,570-10,575. Соответственно, для целей по настоящему описанию, часть фермента FokI, используемую в описанных слитых белках, рассматривают как полудомен расщепления. Таким образом, для направленного двухцепочечного расщепления и/или направленной замены клеточных последовательностей с использованием слитых белков с цинковыми пальцами-FokI, два слитых белка, каждый содержащий полудомен расщепления FokI, можно использовать для восстановления каталитически активного домена расщепления. Альтернативно можно использовать также одну полипептидную молекулу, содержащую связывающий домен с цинковыми пальцами и два полудомена расщепления FokI. Параметры направленного расщепления и направленного изменения последовательности с использованием слитых белков с цинковыми пальцами-FokI представлены в другом месте настоящего описания.
[0123] Домен расщепления или полудомен расщепления может представлять собой любую часть белка, которая сохраняет активность расщепления, или которая сохраняет способность к мультимеризации (например, димеризации) для формирования функционального домена расщепления.
[0124] Иллюстративные ферменты рестрикции типа IIS описаны в Международной патентной публикации WO 07/014275, полное содержание которой приведено в настоящем документе. Дополнительные ферменты рестрикции также содержат отделяемые домены связывания и расщепления, и они предусмотрены по настоящему описанию. См., например, Roberts et al. (2003) Nucleic Acids Res. 31:418-420.
[0125] В конкретных вариантах осуществления домен расщепления содержит один или более сконструированных полудоменов расщепления (обозначенных также как мутанты доменов димеризации), минимизирующих или предотвращающих гомодимеризацию, как описано, например, в Публикациях патентов США № 20050064474; 20060188987; 20070305346 и 20080131962, полное содержание всех из которых приведено в настоящем документе в качестве ссылки. Аминокислотные остатки в положениях 446, 447, 479, 483, 484, 486, 487, 490, 491, 496, 498, 499, 500, 531, 534, 537 и 538 FokI все являются мишенями для влияния на димеризацию полудоменов расщепления FokI.
[0126] Иллюстративные сконструированные полудомены расщепления Fok I, формирующие облигатные гетеродимеры, включают пару, в которой первый полудомен расщепления включает мутации в аминокислотных остатках в положениях 490 и 538 FokI, и второй полудомен расщепления включает мутации в аминокислотных остатках 486 и 499.
[0127] Таким образом, в одном варианте осуществления, мутация в 490 заменяет Glu (E) на Lys (K); мутация в 538 заменяет Iso (I) на Lys (K); мутация в 486 заменяет Gln (Q) на Glu (E); и мутация в положении 499 заменяет Iso (I) на Lys (K). Конкретно, сконструированные полудомены расщепления, описанные в настоящем документе, получали посредством введения мутаций в положениях 490 (E->K) и 538 (I->K) в один полудомен расщепления для получения сконструированного полудомена расщепления, обозначенного «E490K:I538K» и посредством введения мутаций в положениях 486 (Q->E) и 499 (I->L) в другой полудомен расщепления для получения сконструированного полудомена расщепления, обозначенного «Q486E:I499L». Сконструированные полудомены расщепления, описанные в настоящем документе, представляют собой мутанты облигатных гетеродимеров, в которых неправильное расщепление минимизировано или исключено. См., например, Публикацию патента США № 2008/0131962, полное содержание которой приведено в качестве ссылки для всех целей. В конкретных вариантах осуществления сконструированный полудомен расщепления содержит мутации в положениях 486, 499 и 496 (пронумерованы относительно FokI дикого типа), например, мутации, заменяющие остаток Gin (Q) дикого типа в положении 486 на остаток Glu (E), остаток Iso (I) дикого типа в положении 499 на остаток Leu (L) и остаток Asn (N) дикого типа в положении 496 на остаток Asp (D) или Glu (E) (обозначены также как «ELD» и «ELE» домены, соответственно). В других вариантах осуществления сконструированный полудомен расщепления содержит мутации в положениях 490, 538 и 537 (пронумерованы относительно FokI дикого типа), например, мутации, заменяющие остаток Glu (E) дикого типа в положении 490 на остаток Lys (K), остаток Iso (I) дикого типа в положении 538 на остаток Lys (K), и остаток His (H) дикого типа в положении 537 на остаток Lys (K) или остаток Arg (R) (обозначены также как домены «KKK» и «KKR», соответственно). В других вариантах осуществления сконструированный полудомен расщепления содержит мутации в положениях 490 и 537 (пронумерованы относительно FokI дикого типа), например, мутации, заменяющие остаток Glu (E) дикого типа в положении 490 на остаток Lys (K) и остаток His (H) дикого типа в положении 537 на остаток (K) или остаток Arg (R) (обозначены также как домены «KIK» и «KIR», соответственно). (См. Публикацию патента США № 20110201055). В других вариантах осуществления сконструированный полудомен расщепления содержит мутации «Sharkey» и/или «Sharkey’» (см. Guo et al., (2010) J. Mol. Biol. 400(1):96-107).
[0128] Сконструированные полудомены расщепления, описанные в настоящем документе, можно получать с использованием любого подходящего способа, например, посредством сайт-направленного мутагенеза полудоменов расщепления дикого типа (Fok I), как описано в Публикациях патентов США № 20050064474; 20080131962; и 20110201055.
[0129] Альтернативно, нуклеазы можно собирать in vivo в участке-мишени нуклеиновой кислоты с использованием так называемого способа «расщепленных ферментов» (см., например, Публикацию патента США № 20090068164). Компоненты таких расщепленных ферментов либо можно экспрессировать на отдельных экспрессирующих конструкциях, либо можно соединять в одной открытой рамке считывания, где индивидуальные компоненты разделены, например, посредством последовательности саморасщепляющегося пептида 2A или IRES. Компоненты могут представлять собой индивидуальные связывающие домены с цинковыми пальцами или домены связывающего нуклеиновую кислоту домена мегануклеазы.
[0130] Можно проводить скрининг нуклеаз по активности перед использованием, например, в системе на основе хромосомы дрожжей, как описано в WO 2009/042163 и 20090068164. Экспрессирующие нуклеазы конструкции можно легко разрабатывать с использованием способов, известных в данной области. См., например, Публикации Патентов Соединенных Штатов 20030232410; 20050208489; 20050026157; 20050064474; 20060188987; 20060063231; и Международную публикацию WO 07/014275. Экспрессия нуклеазы может происходить под контролем конститутивного промотора или индуцируемого промотора, например, промотора галактокиназы, который является активированным (дерепрессированным) в присутствии рафинозы и/или галактозы и репрессированным в присутствии глюкозы.
[0131] Связанная с Cas9 система CRISPR/Cas содержит два некодирующих РНК-компонента: tracrРНК и массив пре-crРНК, содержащих руководящие последовательности для нуклеазы (спейсеры) разделенные промежутками из идентичных прямых повторов (DR). Чтобы использовать систему CRISPR/Cas для осуществления геномной инженерии, обе функции этих РНК должны присутствовать (см. Cong et al., (2013) Sciencexpress 1/10,1126/science 1231143). В некоторых вариантах осуществления tracrРНК и пре-crРНК поставляют посредством отдельных экспрессирующих конструкций или в форме отдельных РНК. В других вариантах осуществления конструируют химерную РНК, где сконструированная зрелая crРНК (обеспечивающая специфичность к мишени) слита с tracrРНК (обеспечивающей взаимодействие с Cas9) для получения химерного гибрида cr-РНК-tracrРНК (называемого также одиночной руководящей РНК). (см. Jinek, там же, и Cong, там же).
Участки-мишени
[0132] Как подробно описано выше, домены ДНК можно разрабатывать для связывания с любой выбранной последовательностью. Сконструированный ДНК-связывающий домен может обладать новой специфичностью связывания, по сравнению с природным ДНК-связывающим доменом. Способы конструирования включают, но без ограничения, рациональный дизайн и различные типы отбора. Рациональный дизайн включает, например, использование баз данных, содержащих триплетные (или квадруплетные) нуклеотидные последовательности и аминокислотные последовательности отдельных цинковых пальцев, в которых каждая триплетная или квадруплетная нуклеотидная последовательность ассоциирована с одной или несколькими аминокислотными последовательностями цинковых пальцев, которые связывают конкретную триплетную или квадруплетную последовательность. См., например, принадлежащие тем же заявителям Патенты США 6453242 и 6534261, полное содержание которых приведено в настоящем документе в качестве ссылки. Можно осуществлять также рациональный дизайн эффекторных доменов TAL. См., например, Патентную публикацию США № 20110301073.
[0133] Иллюстративные способы отбора, применимые для ДНК-связывающих доменов, включая фаговый дисплей и двугибридные системы, описаны в Патентах США 5789538; 5925523; 6007988; 6013453; 6410248; 6140466; 6200759; и 6242568; так же как в WO 98/37186; WO 98/53057; WO 00/27878; WO 01/88197 и GB 2338237. Кроме того, описано улучшение специфичности связывания связывающих доменов с цинковыми пальцами, например, в принадлежащей тем же заявителям WO 02/077227.
[0134] Выбор участков-мишеней; нуклеаз и способов дизайна и конструирования слитых белков (и кодирующих их полинуклеотидов) известны специалистам в данной области и подробно описаны в Публикациях патентных заявок США № 20050064474 и 20060188987, полное содержание которых приведено в настоящем документе в качестве ссылки.
[0135] Кроме того, как описано в этих и других ссылках, ДНК-связывающие домены (например, белки с множеством цинковых пальцев) можно связывать вместе с использованием любых пригодных линкерных последовательностей, включая, например, линкеры из 5 или более аминокислот. См., например, иллюстративные линкерные последовательности длиной 6 или более аминокислот в Патентах США № 6479626; 6903185; и 7153949. Белки, описанные в настоящем документе, могут включать любую комбинацию пригодных линкеров между отдельными ДНК-связывающими доменами белка. См. также Публикацию патента США № 20110301073.
[0136] Как указано выше, ДНК-связывающие домены нуклеаз можно нацеливать на любой ген. В конкретных вариантах осуществления нуклеаза (компонент ДНК-связывающего домена) нацелен на локус «области безопасности», который включает, только в качестве примера, ген AAVS1 (см. Патент США № 8110379), ген CCR5 (см. Патентную публикацию США № 20080159996), локус Rosa (см. WO 2010/065123) и/или локус альбумина (см. Патентную заявку США № 13/624193).
Доноры
[0137] В настоящем документе описаны способы направленной вставки любых полинуклеотидов для вставки в выбранную локализацию. Полинуклеотиды для вставки можно также обозначать как «экзогенные» полинуклеотиды, «донорные» полинуклеотиды или молекулы, или «трансгены».
[0138] Неожиданно, показали, что двухцепочечные донорные нуклеотиды (например, плазмиды) без гомологичных плечей, фланкирующих экзогенную последовательность (трансген), можно эффективно интегрировать в выбранную область-мишень генома клетки после расщепления in vivo двухцепочечного донора. Таким образом, двухцепочечные доноры включают один или более участков связывания нуклеазы для расщепления донора in vivo (в клетке). В конкретных вариантах осуществления донор включает два участка связывания нуклеазы. В способах, в которых направленной интеграции достигают выполнением двухцепочечного разреза в области-мишени в геноме (см., например, Патенты США № 7888121; 7951925; 8110379 и Публикации патентов США № 20090263900; 20100129869 и 20110207221), одну или более из нуклеаз, использованных для расщепления области-мишени, можно использовать также для расщепления молекулы донора.
[0139] В конкретных вариантах осуществления двухцепочечный донор включает последовательности (например, кодирующие последовательности, обозначаемые также как трансгены) длиной более 1 т.п.о., например, между 2 и 200 т.п.о., между 2 и 10 т.п.о. (или любое значение между ними). Двухцепочечный донор включает также по меньшей мере один участок-мишень нуклеаз, например. В конкретных вариантах осуществления донор включает по меньшей мере 2 участка-мишени, например, для пары ZFN или TALEN. Как правило, участки-мишени нуклеаз находятся вне последовательности трансгена, например, на 5’- и/или 3’- от последовательности трансгена, для расщепления трансгена. Участок расщепления нуклеазой(нуклеазами) может быть предназначен для любой нуклеазы(нуклеаз). В конкретных вариантах осуществления участок(участки)-мишень нуклеаз, содержащийся в двухцепочечном доноре, предназначен для той же самой нуклеазы(нуклеаз), использованной для расщепления эндогенной мишени, в которую расщепленный донор интегрируют посредством независимых от гомологии способов.
[0140] Как указано выше, донор можно расщеплять in vivo и интегрировать в геном в прямой («AB») или в обратной («BA») ориентации. Направленная интеграция посредством расщепления донора in vivo, приводящая к точно лигированной вставке в ориентации AB, воссоздает спаренные участки связывания нуклеазы (например, ZFN или TALEN) с оригинальным расстоянием между участками. Такие воссозданные участки являются потенциальными субстратами для второго цикла расщепления нуклеазами. Расщепление нуклеазами в воссозданных участках может приводить к делеции ДНК в участках стыковки трансген-хромосома (в результате неточной репарации на основе NHEJ) или даже к вырезанию трансгена. В отличие от этого, вставки в обратной (BA) ориентации приводят к образованию двух различных пар участков связывания нуклеаз (например, гомодимеров левой и правой нуклеаз). Если используют облигатный гетеродимер (EL/KK, ELD/KKR и т.д.) доменов нуклеазы FokI, воссозданные участки BA не поддаются повторному расщеплению, поскольку воссозданные участки связывания оба являются гомодимерными участками. См. также фигуру 1C.
[0141] Более того, изменение нуклеотидов в спейсере нуклеазы донора трансгена, составляющего одноцепочечный 5'-выступающий конец по сравнению с последовательностью дикого типа (геномной), на обратно комплементарные последовательности дикого типа благоприятствует BA-ориентации вставки расщепленного донора (посредством спаривания оснований по Уотсону-Крику с выступающими концами на расщепленной хромосоме), которое создает не поддающуюся повторному вырезанию интеграцию трансгена (фигура 1C).
[0142] Трансгены, переносимые на донорных последовательностях, описанных в настоящем документе, можно выделять из плазмид, клеток или других источников с использованием общепринятых способов, известных в данной области, таких как ПЦР. Доноры для использования могут включать различные типы топологии, включая кольцевую суперскрученную, кольцевую релаксированную, линейную и т.п. Альтернативно, их можно химически синтезировать с использованием общепринятых способов синтеза олигонуклеотидов. Кроме того, доноры могут являться метилированными или лишенными метилирования. Доноры могут находиться в форме бактериальных или дрожжевых искусственных хромосом (BAC или YAC).
[0143] Двухцепочечные донорные полинуклеотиды, описанные в настоящем документе, могут включать один или более неприродных оснований и/или остовов. В частности, вставку донорной молекулы с метилированными остатками цитозина можно проводить с использованием способов, описанных в настоящем документе, для достижения состояния молчания транскрипции в представляющей интерес области.
[0144] Экзогенный (донорный) полинуклеотид может содержать любую представляющую интерес последовательность (экзогенную последовательность). Иллюстративные экзогенные последовательности включают, но без ограничения, любую кодирующую полипептид последовательность (например, кДНК), промоторные последовательности, энхансерные последовательности, эпитопы-метки, маркерные гены, участки узнавания ферментов расщепления и различные типы экспрессирующих конструкций. Маркерные гены включают, но без ограничения, последовательности, кодирующие белки, опосредующие устойчивость к антибиотику (например, устойчивость к ампициллину, устойчивость к неомицину, устойчивость к G418, устойчивость к пуромицину), последовательности, кодирующие окрашенные или флуоресцентные, или люминесцентные белки (например, зеленый флуоресцентный белок, улучшенный зеленый флуоресцентный белок, красный флуоресцентный белок, люциферазу), и белки, опосредующие усиленный рост клеток и/или амплификацию гена (например, дигидрофолатредуктазу). Эпитопы-метки включают, например, одну или более копий FLAG, His, myc, Tap, HA или любой поддающейся детекции аминокислотной последовательности.
[0145] В предпочтительном варианте осуществления экзогенная последовательность (трансген) содержит полинуклеотид, кодирующий любой полипептид, экспрессия которого в клетке является желательной, включая, но без ограничения, антитела, антигены, ферменты, рецепторы (поверхности клеток или ядерные), гормоны, лимфокины, цитокины, репортерные полипептиды, факторы роста, белки устойчивости к насекомым, факторы транскрипции и функциональные фрагменты любого из вышеуказанных. Кодирующие последовательности могут представлять собой, например, кДНК.
[0146] Например, экзогенная последовательность может содержать последовательность, кодирующую полипептид, отсутствующий или нефункциональный у субъекта, страдающего генетическим заболеванием, включая, но без ограничения, любое из следующих генетических заболеваний: ахондроплазия, ахроматопсия, дефицит кислой мальтазы, дефицит аденозиндезаминазы (OMIM № 102700), адренолейкодистрофия, синдром Экарди, дефицит антитрипсина альфа-1, альфа-талассемия, синдром нечувствительности к андрогенам, синдром Аперта, аритмогенная дисплазия правого желудочка, телеангиоэктатическая атаксия, синдром Барта, бета-талассемия, синдром голубых эластичных пузырчатых невусов, болезнь Канавана, хронические гранулематозные заболевания (CGD), синдром кошачьего крика, кистозный фиброз, болезнь Деркума, эктодермальная дисплазия, анемия Фанкони, прогрессирующая оссифицирующая фибродисплазия, синдром ломкой Х-хромосомы, галактоземия, болезнь Гоше, генерализованный ганглиозидоз (например, GM1), гемохроматоз, мутация гемоглобина С в 6-м кодоне бета-глобина (HbC), гемофилия, болезнь Гентингтона, синдром Гурлера, гипофосфатазия, синдром Клайнфельтера, болезнь Краббе, синдром Лангера-Гиедиона, нарушение адгезии лейкоцитов (LAD, OMIM № 116920), лейкодистрофия, синдром удлинения интервала QT, синдром Марфана, синдром Мебиуса, мукополисахаридоз (MPS), синдром ногтей-надколенника, несахарный почечный диабет, нейрофиброматоз, болезнь Ниманна-Пика, несовершенный остеогенез, порфирия, синдром Прадера-Вилли, прогерия, синдром Протея, ретинобластома, синдром Ретта, синдром Рубинштейна-Тэйби, синдром Санфилиппо, тяжелый комбинированный иммунодефицит (SCID), синдром Швачмана, заболевание серповидного изменения эритроцитов (серповидно-клеточная анемия), синдром Смита-Магениса, синдром Стиклера, болезнь Тея-Сакса, синдром тромбоцитопении с отсутствием лучевой кости (TAR), синдром Тричера-Коллинза, трисомия, туберозный склероз, синдром Тернера, нарушение цикла мочевины, болезнь фон Гиппеля-Линдау, синдром Ваарденбурга, синдром Вильямса, болезнь Вильсона, синдром Вискотта-Олдрича, X-сцепленный лимфопролиферативный синдром (XLP, OMIM № 308240).
[0147] Дополнительные иллюстративные заболевания, которые можно лечить посредством направленной интеграции, включают приобретенные иммунодефициты, заболевания лизосомального накопления (например, болезнь Гоше, GM1, болезнь Фабри и болезнь Тея-Сакса), мукополисахаридоз (например, болезнь Гунтера, болезнь Гурлера), гемоглобинопатии (например, заболевания серповидного изменения эритроцитов, HbC, α-талассемия, β-талассемия) и гемофилии.
[0148] В конкретных вариантах осуществления экзогенные последовательности могут содержать маркерный ген (описанный выше), позволяющий селекцию клеток, подвергнутых направленной интеграции, и связанную последовательность, кодирующую дополнительную функциональную молекулу. Неограничивающие примеры маркерных генов включают GFP, маркер(ы) для селекции с лекарственным средством и т.п.
[0149] Дополнительные последовательности генов, которые можно вставлять, могут включать, например, гены дикого типа для замены мутантных последовательностей. Например, последовательность гена фактора IX дикого типа можно вставлять в геном стволовой клетки, в которой эндогенная копия гена мутирована. Копию дикого типа можно вставлять в эндогенный локус, или можно, альтернативно, нацеливать на локус области безопасности.
[0150] В некоторых вариантах осуществления экзогенная последовательность нуклеиновой кислоты (трансген), содержащая представляющий интерес для агрономии ген или нуклеотидную последовательность, кодирующую представляющий интерес полипептид, может включать, в качестве неограничивающих примеров: ген, придающий устойчивость к вредителям или заболеваниям (См., например, Jones et al. (1994) Science 266:789 (клонирование гена Cf-9 томата для устойчивости к Cladosporium fulvum); Martin et al. (1993) Science 262:1432; Mindrinos et al. (1994) Cell 78:1089 (ген RSP2 для устойчивости к Pseudomonas syringae); Международная патентная публикация PCT № WO 1096/30517 (устойчивость к цистообразующей нематоде сои); PCT Международная патентная публикация № WO 93/19181); ген, кодирующий белок Bacillus thuringiensis, его производное или смоделированный на его основе синтетический полипептид (см., например, Geiser et al. (1986) Gene 48:109 (клонирование и нуклеотидная последовательность гена Bt δ-эндотоксина; кроме того, молекулы ДНК, кодирующие гены δ-эндотоксина можно закупать из American Type Culture Collection (Manassas, VA), например, под № доступа в ATCC 40098; 67136; 31995; и 31998)); ген, кодирующий лектин (см., например, Van Damme et al. (1994) Plant Molec. Biol. 24:25 (нуклеотидные последовательности нескольких генов связывающих маннозу лектинов Clivia miniata)); ген, кодирующий связывающий витамин белок, например, авидин (см. Международную патентную публикацию PCT № US93/06487 (использование авидина и гомологов авидина в качестве ларвицидов против насекомых-вредителей)); ген, кодирующий ингибитор фермента, например, ингибитор протеазы или протеиназы или ингибитор амилазы (См., например, Abe et al. (1987) J. Biol. Chem. 262:16793 (нуклеотидная последовательность ингибитора цистеиновой протеазы риса); Huub et al. (1993) Plant Molec. Biol. 21:985 (нуклеотидная последовательность кДНК, кодирующей ингибитор I протеиназы табака); Sumitani et al. (1993) Biosci. Biotech. Biochem. 57:1243 (нуклеотидная последовательность ингибитора альфа-амилазы Streptomyces nitrosporeus) и Патент США 5494813); ген, кодирующий специфический для насекомых гормон или феромон, например, экдистероидный или ювенильный гормон, его вариант, миметик на его основе или его антагонист или агонист (см., например, Hammock et al. (1990) Nature 344:458 (экспрессия в бакуловирусе клонированной эстеразы ювенильного гормона, инактиватора ювенильного гормона)); ген, кодирующий специфический для насекомых пептид или нейропептид, который после экспрессии нарушает физиологию пораженного вредителя (см., например, Regan (1994) J. Biol. Chem. 269:9 (экспрессионное клонирование образует ДНК, кодирующую рецептор диуретического гормона насекомого); Pratt et al. (1989) Biochem. Biophys. Res. Comm. 163:1243 (аллостатин в Diploptera puntata); и Патент США 5266317 (гены, кодирующие специфические для насекомых, паралитические нейротоксины)); ген, кодирующий специфический для насекомых яд, продуцируемый в природе змеей, осой или другим организмом (см., например, Pang et al. (1992) Gene 116:165 (гетерологичная экспрессия в растениях гена, кодирующего токсичный для скорпиона токсичный для насекомых пептид)); ген, кодирующий фермент, ответственный за повышенное накопление монотерпена, сесквитерпена, стероида, гидроксамовой кислоты, производного фенилпропаноида или другой небелковой молекулы с инсектицидной активностью; ген, кодирующий фермент, вовлеченный в модификацию, включая посттрансляционную модификацию, биологически активной молекулы, например, гликолитический фермент, протеолитический фермент, липолитический фермент, нуклеаза, циклаза, трансаминаза, эстераза, гидролаза, фосфатаза, киназа, фосфорилаза, полимераза, эластаза, хитиназа или глюканаза, независимо от того, являются ли они природными или синтетическими (см., например, Международную патентную публикацию PCT № WO 93/02197 (нуклеотидная последовательность гена каллазы); кроме того, молекулы ДНК, содержащие кодирующие хитиназу последовательности, можно получать, например, из ATCC, под № доступа 39637 и 67152; Kramer et al. (1993) Insect Biochem. Molec. Biol. 23:691 (нуклеотидная последовательность кДНК, кодирующей хитиназу бражника табачного); и Kawalleck et al. (1993) Plant Molec. Biol. 21:673 (нуклеотидная последовательность гена полиубиквитина ubi4-2 петрушки)); ген, кодирующий молекулу, стимулирующую передачу сигнала (см., например, Botella et al. (1994) Plant Molec. Biol. 24:757 (нуклеотид последовательности клонов кДНК кальмодулина золотистой фасоли); и Griess et al. (1994) Plant Physiol. 104:1467 (нуклеотидная последовательность клона кДНК кальмодулина маиса)); ген, кодирующий пептид с гидрофобным моментом (см., например, Международную патентную публикацию PCT № WO 95/16776 (пептидные производные тахиплезина, ингибирующие грибные патогены растений); и Международную патентную публикацию PCT № WO 95/18855 (синтетический противомикробные пептиды, придающие устойчивость к заболеваниям)); ген, кодирующий мембранную пермеазу, образователь каналов или блокатор каналов (см., например, Jaynes et al. (1993) Plant Sci 89:43 (гетерологичная экспрессии аналога цекропин-β-литического пептида для придания трансгенным растениям табака устойчивости к Pseudomonas solanacearum)); ген, кодирующий вирус-инвазивный белок или полученный из него комплексный токсин (см., например, Beachy et al. (1990) Ann, rev. Phytopathol. 28:451); ген, кодирующий специфическое для насекомых антитело или полученный из него иммунотоксин (см., например, Taylor et al., Abstract #497, Seventh Inf 1 Symposium on Molecular Plant-Microbe Interactions (Edinburgh, Scotland) (1994) (инактивация ферментов в трансгенном табаке посредством получения одноцепочечных фрагментов антител)); ген, кодирующий специфическое для вируса антитело (см., например, Tavladoraki et al. (1993) Nature 366:469 (трансгенные растения, экспрессирующие гены рекомбинантного антитела, защищены от вирусной атаки)); ген, кодирующий белок, вызывающий арест развития, продуцируемый в природе патогеном или паразитом (см., например, Lamb et al. (1992) Bio/Technology 10:1436 (эндо-1,4-D-полигалактуроназы грибов облегчают грибную колонизацию и высвобождение питательных веществ растений посредством солюбилизирующей клеточные стенки растения гомо-α-1,4-D-галактуроназы); Toubart et al. (1992) Plant J. 2:367 (клонирование и характеризация гена, кодирующего ингибирующий эндополигалактуроназу белок фасоли)); ген, кодирующий вызывающий арест развития белок, продуцируемый в природе растением (См., например, Logemann et al. (1992) Bio/Technology 10:305 (трансгенные растения, экспрессирующие ген, инактивирующий рибосомы, ячменя, имеют увеличенную устойчивость к грибковым заболеваниям)).
[0151] В некоторых вариантах осуществления нуклеиновые кислоты, содержащие представляющий интерес для агрономии ген или нуклеотидную последовательность, кодирующую представляющий интерес полипептид, могут также и/или альтернативно включать, в качестве неограничивающих примеров: гены, придающие устойчивость к гербициду, такому как гербицид, ингибирующий точку роста или меристему, например, имидазолинон или сульфонилмочевина (иллюстративные гены в этой категории кодируют мутантные ферменты ALS и AHAS, как описано, например, в Lee et al. (1988) EMBO J. 7:1241, и Miki et al. (1990) Theor. Appl. Genet. 80:449, соответственно); устойчивость к глифосату, какую придают, например, мутантные гены 5-енолпирувилшикимат-3-фосфатсинтазы (EPSP) (посредством введения рекомбинантных нуклеиновых кислот и/или различных форм мутагенеза in vivo нативных генов EPSP); гены aroA и глифосатацетилтрансферазы (GAT), соответственно); к другим фосфоносоединениям, таким как глуфосинат (гены фосфинотрицинацетилтрансферазы (PAT) из видов Streptomyces, включая Streptomyces hygroscopicus и Streptomyces viridichromogenes); и к пиридинокси- или феноксипропионовым кислотам и циклогексонам (кодирующие ингибитор ACC-азы гены). См., например, Патенты США 4940835 и 6248876 (нуклеотидные последовательности форм EPSP, которые могут придавать растению устойчивость к глифосату). Молекулу ДНК, кодирующую мутантный ген aroA, можно получать под номером доступа в ATCC 39256. См. также Патент США № 4769061 (нуклеотидная последовательность мутантного гена aroA). В Европейской патентной заявке № 0 333 033 и в Патенте США № 4975374 описаны нуклеотидные последовательности генов глутаминсинтетазы, которые могут придавать устойчивость к гербицидам, таким как L-фосфинотрицин. Нуклеотидные последовательности иллюстративных генов PAT представлены в Европейской патентной заявке № 0 242 246, и DeGreef et al. (1989) Bio/Technology 7:61 (получение трансгенных растений, экспрессирующих химерные гены bar, кодирующие активность PAT). Примеры генов, придающих устойчивость к феноксипропионовым кислотам и циклогексонам, таким как сетоксидим и галоксифоп, включают гены Acc1-S1, Acc1-S2 и Acc1-S3, описанные Marshall et al. (1992) Theor. Appl. Genet. 83:435. Гены GAT, способные придавать устойчивость к глифосату, описаны, например, в WO 2005012515. Гены, придающие устойчивость к 2,4-D, феноксипропионовой кислоте и пиридилоксиауксиновым гербицидам, описаны, например, в WO 2005107437.
[0152] Нуклеиновые кислоты, содержащие представляющий интерес для агрономии ген или нуклеотидную последовательность, кодирующую представляющий интерес полипептид, могут также включать, в качестве неограничивающих примеров: ген, придающий устойчивость к гербициду, ингибирующему фотосинтез, такому как триазин (гены psbA и gs+) или бензонитрил (ген нитрилазы). См., например, Przibila et al. (1991) Plant Cell 3:169 (трансформация Chlamydomonas плазмидами, кодирующим мутантные гены psbA). Нуклеотидные последовательности генов нитрилазы описаны в Патенте США 4810648, и молекулы ДНК, содержащие эти гены, доступны под № доступа в ATCC 53435; 67441; и 67442. См. также Hayes et al. (1992) Biochem. J. 285:173 (клонирование и экспрессия ДНК, кодирующей глутатион-S-трансферазу).
[0153] В некоторых вариантах осуществления нуклеиновые кислоты, содержащие представляющий интерес для агрономии ген или нуклеотидную последовательность, кодирующую представляющий интерес полипептид, могут также и/или альтернативно включать гены, придающие или вносящие вклад в признак добавленной ценности, в качестве неограничивающих примеров: модифицированный метаболизм жирных кислот, например, посредством трансформации растения антисмысловым геном стеарил-ACP-десатуразы для увеличения содержания стеариновой кислоты этого растения (см., например, Knultzon et al. (1992) Proc. Natl. Acad. Sci. U.S.A. 89:2624); уменьшенное содержание фитата, например, введение кодирующего фитазу гена может усиливать разрушение фитата, добавляя больше свободного фосфата трансформированному растению (см., например, Van Hartingsveldt et al. (1993) Gene 127:87 (нуклеотидная последовательность гена фитазы Aspergillus niger); ген можно вводить для уменьшения содержания фитата - в маисе, например, это можно осуществлять клонированием и затем повторным введением ДНК, ассоциированной с единственным аллелем, который может являться ответственным за мутанты маиса, характеризующиеся низкими уровнями фитиновой кислоты (см. Raboy et al. (1990) Maydica 35:383)); и модифицированный состав углеводов, на который влияют, например, трансформацией растений геном, кодирующим фермент, который изменяет характер ветвления крахмала (см., например, Shiroza et al. (1988) J. Bacteol. 170:810 (нуклеотидная последовательность мутантного гена фруктозилтрансферазы Streptococcus); Steinmetz et al. (1985) Mol. Gen. Genet. 20:220 (ген левансахараза); Pen et al. (1992) Bio/Technology 10:292 (α-амилаза); Elliot et al. (1993) Plant Molec. Biol. 21:515 (нуклеотидные последовательности генов инвертазы томата); Sogaard et al. (1993) J. Biol. Chem. 268:22480 (ген α-амилазы ячменя); и Fisher et al. (1993) Plant Physiol. 102:1045 (ветвящий фермент II крахмала эндосперма маиса)).
[0154] При конструировании таких экспрессирующих кассет, следуя способам из настоящего описания, используют способы, хорошо известные в данной области молекулярной биологии (см., например, Ausubel или Maniatis). Перед использованием экспрессирующей кассеты для получения трансгенного животного, способность экспрессирующей кассеты отвечать на индуктор стресса, ассоциированный с выбранными контрольными элементами, можно тестировать посредством введения экспрессирующей кассеты в пригодную линию клеток (например, линии первичных клеток, трансформированных клеток, или иммортализованных клеток).
[0155] Более того, хотя они не являются необходимыми для экспрессии, экзогенные последовательности (трансгены) могут также включать регуляторные последовательности транскрипции или трансляции, например, промоторы, энхансеры, инсуляторы, внутренние участки связывания рибосом, последовательности, кодирующие пептиды 2A и/или сигналы полиаденилирования. Кроме того, контрольные элементы представляющих интерес генов можно функционально связывать с репортерными генами для получения химерных генов (например, экспрессирующих репортер кассет).
[0156] Можно достигать также направленной вставки трансгена из некодирующей последовательности нуклеиновой кислоты. Трансгены, кодирующие антисмысловые РНК, РНКи, кшРНК и микроРНК (мкРНК), также можно использовать для направленных вставок.
[0157] В дополнительных вариантах осуществления донорная нуклеиновая кислота может содержать некодирующие последовательности, представляющие собой специфические участки-мишени для дизайна дополнительных нуклеаз. Затем дополнительные нуклеазы можно экспрессировать в клетках, так что оригинальную донорную молекулу расщепляют и модифицируют посредством вставки другой представляющей интерес донорной молекулы. Таким образом, можно получать повторяющиеся интеграции донорных молекул, позволяющих пакетирование признака в конкретном представляющем интерес локусе или в локусе области безопасности.
Способы направленной интеграции трансгена
[0158] Донорные молекулы, описанные в настоящем документе, интегрируют в геном клетки направленными, независимыми от гомологии способами. Для такой направленной интеграции геном расщепляют в желательной локализации (или локализациях) с использованием нуклеазы, например, слитого белка между ДНК-связывающим доменом (например, связывающий домен с цинковыми пальцами или эффекторный домен TAL конструируют для связывания участка в предопределенном участке расщепления или рядом с предопределенным участком расщепления) и доменом нуклеазы (например, домен расщепления или полудомен расщепления). В конкретных вариантах осуществления два слитых белка, каждый содержащий ДНК-связывающий домен и полудомен расщепления, экспрессируются в клетке и связываются с участками, расположенными по соседству таким образом, что реконструируется функциональный домен расщепления, и ДНК расщепляется поблизости от участка(участков)-мишени. В одном варианте осуществления расщепление происходит между участками связывания двух ДНК-связывающих доменов. Один или оба из ДНК-связывающих доменов могут являться сконструированными. См. также Патент США № 7888121; Патентную публикацию США 20050064474 и Международные патентные публикации WO05/084190, WO05/014791 и WO03/080809.
[0159] Нуклеазы, как описано в настоящем документе, можно вводить как полипептиды и/или полинуклеотиды. Например, два полинуклеотида, каждый содержащий последовательности, кодирующие один из вышеупомянутых полипептидов, можно вводить в клетку, и когда полипептиды экспрессируются, и каждый связывается с его последовательностью-мишенью, расщепление происходит в или около последовательности-мишени. Альтернативно, один полинуклеотид, содержащий последовательности, кодирующие оба слитых полипептида, вводят в клетку. Полинуклеотиды могут представлять собой ДНК, РНК или любые модифицированные формы или аналоги или ДНК и/или РНК.
[0160] После введения двухцепочечного разрыва в представляющую интерес область, трансген интегрируют в представляющую интерес область направленным образом посредством независимых от гомологии способов (например, соединение негомологичных концов (NHEJ)) после линеаризации двухцепочечной донорной молекулы, как описано в настоящем документе. Двухцепочечный донор предпочтительно линеаризуют in vivo с помощью нуклеазы, например, одной или более из таких же нуклеаз, какие использовали для введения двухцепочечного разрыва в геном, или отличных нуклеаз. Синхронизированное расщепление хромосомы и донора в клетке может ограничивать деградацию донорной ДНК (по сравнению с линеаризацией донорной молекулы перед введением в клетку). Участок(участки)-мишень нуклеаз, используемый для линеаризации донора, предпочтительно, не нарушает последовательность(последовательности) трансгена(трансгенов).
[0161] Трансген может интегрировать в геном в направлении, ожидаемом при простом лигировании выступающих концов после нуклеазы (обозначено «прямая» или «AB» ориентация) или в альтернативном направлении (обозначено «обратная» или «BA» ориентация). В конкретных вариантах осуществления трансген интегрирован после точного лигирования выступающих концов донора и хромосомы. В других вариантах осуществления интеграция трансгена либо в BA, либо в AB ориентации приводит к делеции нескольких нуклеотидов.
Доставка
[0162] Нуклеазы, полинуклеотиды, кодирующие эти нуклеазы, донорные полинуклеотиды и композиции, содержащие белки и/или полинуклеотиды, описанные в настоящем документе, можно доставлять in vivo или ex vivo посредством любых пригодных способов в любой тип клеток.
[0163] Пригодные клетки включают эукариотические (например, из животных или растений) и прокариотические клетки и/или линии клеток. Неограничивающие примеры таких клеток или линий клеток, полученных из таких клеток, включают клетки COS, CHO (например, CHO-S, CHO-K1, CHO-DG44, CHO-DUXB11, CHO-DUKX, CHOK1SV), VERO, MDCK, WI38, V79, B14AF28-G3, BHK, HaK, NS0, SP2/0-Ag14, HeLa, HEK293 (например, HEK293-F, HEK293-H, HEK293-T) и perC6, так же как клетки насекомых, таких как Spodoptera fugiperda (Sf), или клетки грибов, таких как Saccharomyces, Pichia и Schizosaccharomyces, так же, как клетки растений из однодольных или двудольных растений, включая, но без ограничения, маис, сою, хлопчатник, Arabidopsis, пшеницу, ячмень, овес, сахарный тростник, сорго, кормовые травы, люцерну, томат, табак, картофель, рис, подсолнечник и Brassica. В конкретных вариантах осуществления линия клеток представляет собой линию клеток CHO, MDCK или HEK293. Пригодные клетки включают также стволовые клетки, например, такие как эмбриональные стволовые клетки, индуцированные плюрипотентные стволовые клетки, гематопоэтические стволовые клетки, нейрональные стволовые клетки и мезенхимальные стволовые клетки. В конкретных вариантах осуществления клетки растений представляют собой, но без ограничения, суспензионные культуры, протопласты или организованные ткани, такие как зародыши, незрелые зародыши, листовые диски, семядоли, гипокотили и микроспоры. Способы доставки нуклеаз, как описано в настоящем документе, описаны, например, в Патентах США № 6453242; 6503717; 6534261; 6599692; 6607882; 6689558; 6824978; 6933113; 6979539; 7013219; и 7163824, полное содержание всех из которых приведено в настоящем документе в качестве ссылки.
[0164] Конструкции нуклеаз и/или донора, как описано в настоящем документе, можно также доставлять с использованием векторов, содержащих последовательности, кодирующие один или более белок(белков) с цинковыми пальцами. Можно использовать любые векторные системы, включая, но без ограничения, плазмидные векторы, ретровирусные векторы, лентивирусные векторы, аденовирусные векторы, поксвирусные векторы; векторы на основе вирусов герпеса и векторы на основе аденоассоциированных вирусов и т.д. См. также Патенты США № 6534261; 6607882; 6824978; 6933113; 6979539; 7013219; и 7163824, полное содержание которых приведено в настоящем документе в качестве ссылки. Более того, очевидно, что любые из этих векторов могут содержать одну или более последовательностей, необходимых для обработки. Таким образом, когда одну или более конструкций нуклеаз и донора вводят в клетку, полинуклеотиды нуклеаз и/или донора можно переносить на одном и том же векторе или на различных векторах. Когда используют несколько векторов, каждый вектор может содержать последовательность, кодирующую одну или множество конструкций нуклеаз и/или донора.
[0165] Общепринятые способы переноса генов на основе вирусов и не на основе вирусов можно использовать для введения нуклеиновых кислот, кодирующих конструкции нуклеаз и донора, в клетки (например, клетки млекопитающих) и ткани-мишени. Системы доставки невирусного вектора включают плазмидные ДНК, голую нуклеиновую кислоту и нуклеиновую кислоту в комплексе с носителем для доставки, таким как липосома или полоксамер. Системы доставки вирусного вектора включают ДНК- и РНК-вирусы, имеющие либо эписомальные, либо интегрированные геномы после доставки в клетку. Обзор доставки in vivo сконструированных ДНК-связывающих белков и слитых белков, содержащих эти связывающие белки, см., например, в Rebar (2004) Expert Opinion Invest. Drugs 13(7):829-839; Rossi et al. (2007) Nature Biotech. 25(12):1444-1454, также как в ссылках на доставку генов в общем, таких как Anderson, Science 256:808-813 (1992); Nabel & Feigner, TIBTECH 11:211-217 (1993); Mitani & Caskey, TIBTECH 11:162-166 (1993); Dillon, TIBTECH 11:167-175 (1993); Miller, Nature 357:455-460 (1992); Van Brunt, Biotechnology 6(10):1149-1154 (1988); Vigne, Restorative Neurology and Neuroscience 8:35-36 (1995); Kremer & Perricaudet, British Medical Bulletin 51(l):31-44 (1995); Fladdada et al., in Current Topics in Microbiology и Immunology Doerfler and Bohm (eds.) (1995); и Yu et al., Gene Therapy 1:13-26 (1994).
[0166] Способы невирусной доставки нуклеиновых кислот включают электропорацию, липофекцию, микроинъекцию, биолистику, виросомы, липосомы, иммунолипосомы, конъюгаты поликатион или липид:нуклеиновая кислота, голую ДНК, искусственные вирионы и усиленное агентами поглощение ДНК. Сонопорацию с использованием, например, системы Sonitron 2000 (Rich-Mar) также можно использовать для доставки нуклеиновых кислот.
[0167] Дополнительные иллюстративные системы доставки нуклеиновой кислоты включают системы, представленные Amaxa Biosystems (Cologne, Germany), Maxcytc, Inc. (Rockville, Maryland), BTX Molecular Delivery Systems (Holliston, MA) и Copernicus Therapeutics Inc, (см., например, US6008336). Липофекция описана, например, в Патентах США № 5049386; 4946787; и 4897355) и реагенты для липофекции поставляют коммерчески (например, Transfectam™ и Lipofectin™). Катионные и нейтральные липиды, пригодные для эффективной липофекции полинуклеотидов с узнаванием рецепторов, включают липиды из Feigner, WO 91/17424, WO 91/16024.
[0168] Получение комплексов липид: нуклеиновая кислота, включая направленные липосомы, такие как иммунолипидные комплексы, хорошо известно специалисту в данной области (см., например, Crystal, Science 270:404-410 (1995); Blaese et al., Cancer Gene Ther. 2:291-297 (1995); Behr et al., Bioconjugate Chem. 5:382-389 (1994); Remy et al., Bioconjugate Chem. 5:647-654 (1994); Gao et al., Gene Therapy 2:710-722 (1995); Ahmad et al., Cancer Res. 52:4817-4820 (1992); Патенты США № 4186183, 4217344, 4235871, 4261975, 4485054, 4501728, 4774085, 4837028 и 4946787).
[0169] Дополнительные способы доставки включают использование упаковки нуклеиновых кислот, подлежащих доставке, в носители для доставки EnGeneIC (EDV). Эти EDV специфически доставляют к тканям-мишеням с использованием биспецифических антител, где одно плечо антитела обладает специфичностью для ткани-мишени, и другое обладает специфичностью для EDV. Антитело приносит EDV к поверхности клетки-мишени, и затем EDV вводится в клетку посредством эндоцитоза. При попадании в клетку содержимое высвобождается (см. MacDiarmid et al. (2009) Nature Biotechnology 27(7):643).
[0170] При использовании систем на основе РНК- или ДНК-вирусов для доставки нуклеиновых кислот, кодирующих сконструированные ZFP, пользуются преимуществами прошедших длительную эволюцию процессов для нацеливания вируса на специфические клетки в организме и переноса нагрузки вируса в ядро. Вирусные векторы можно вводить непосредственно пациентам (in vivo), или их можно использовать для обработки клеток in vitro, и модифицированные клетки вводить пациентам (ex vivo). Общепринятые системы на основе вирусов для доставки ZFP включают, но без ограничения, ретровирусные, лентивирусные, аденовирусные векторы, векторы на основе аденоассоциированных вирусов, вирусов осповакцины и простого герпеса для переноса генов. Интеграция в геном хозяина возможна способами переноса генов с помощью ретровирусов, лентивирусов и аденоассоциированных вирусов, часто приводя к долгосрочной экспрессии вставленного трансгена. Кроме того, высокую эффективность трансдукции наблюдали во многих различных типах клеток и тканях-мишенях.
[0171] Тропизм ретровирусов можно изменять посредством введения чужеродных белков оболочки, расширяя потенциальную намеченную популяцию клеток-мишеней. Лентивирусные векторы представляют собой ретровирусные векторы, способные к трансдукции или инфекции неделящихся клеток и, как правило, продуцирующие высокие титры вирусов. Выбор ретровирусной системы для переноса генов зависит от ткани-мишени. Ретровирусные векторы содержат цис-действующие длинные концевые повторы с пакующей емкостью вплоть до 6-10 т.п.о. чужеродной последовательности. Минимум цис-действующих LTR достаточно для репликации и упаковки векторов, которые затем используют для интеграции терапевтического гена в клетку-мишень для обеспечения постоянной экспрессии трансген. Широко используемые ретровирусные векторы включают векторы на основе вируса лейкоза мышей Молони (MuLV), вируса лейкоза человекообразных гиббонов (GaLV), вируса иммунодефицита обезьян (SIV), вируса иммунодефицита человека (HIV) и их комбинаций (см., например, Buchscher et al., J. Virol. 66:2731-2739 (1992); Johann et al., J. Virol. 66:1635-1640(1992); Sommerfelt et al., Virol. 176:58-59; Wilson et al., J. Virol. 63:2374-2378 (1989); Miller et al., J. Virol. 65:2220-2224 (1991); PCT/US94/05700).
[0172] Для применений, в которых временная экспрессия является предпочтительной, можно использовать системы на основе аденовирусов. Векторы на основе аденовирусов являются способными к очень высокой эффективности трансдукции во многих типах клеток и не требуют деления клеток. С такими векторами получены высокие титры и высокие уровни экспрессии. Этот вектор можно получать в больших количествах в относительно простой системе. Векторы на основе аденоассоциированных вирусов («AAV») также используют для трансдукции клеток нуклеиновыми кислотами-мишенями, например, для продукции in vitro нуклеиновых кислот и пептидов, и для способов генотерапии in vivo и ex vivo (см., например, West et al., Virology 160:38-47 (1987); Патент США № 4797368; WO 93/24641; Kotin, Human Gene Therapy 5:793-801 (1994); Muzyczka, J. Clin. Invest. 94:1351 (1994). Конструирование рекомбинантных векторов AAV описаны в ряде публикаций, включая Патент США № 5173414; Tratschin et al., Mol. Cell. Biol. 5:3251-3260 (1985); Tratschin, et al., Mol. Cell. Biol. 4:2072-2081 (1984); Hermonat & Muzyczka, PNAS 81:6466-6470 (1984); и Samulski et al., J. Virol. 63:03822-3828 (1989).
[0173] По меньшей мере шесть способов для вирусных векторов доступны в настоящее время для переноса генов в клинических исследованиях, в которых используют комплементацию дефектных векторов генами, вставленными в линии клеток-помощников, для получения трансдуцирующего агента.
[0174] pLASN и MFG-S являются примерами ретровирусных векторов, использованных в клинических исследованиях (Dunbar et al., Blood 85:3048-305 (1995); Kohn et al., Nat. Med. 1:1017-102 (1995); Malech et al., PNAS 94:22 12133-12138 (1997)). PA317/pLASN являлся первым терапевтическим вектором, использованным в генотерапевтическом исследовании. (Blaese et al., Science 270:475-480 (1995)). Эффективность трансдукции 50% или более наблюдали для упакованных векторов MFG-S. (Ellem et al., Immunol Immunother. 44(1): 10-20 (1997); Dranoff et al., Hum. Gene Ther. 1:111-2 (1997).
[0175] Рекомбинантные векторы на основе аденоассоциированных вирусов (rAAV) представляют собой многообещающие альтернативные системы доставки генов на основе дефектного и непатогенного парвовируса - аденоассоциированного вируса типа 2. Все векторы получены из плазмиды, в которой сохранены только инвертированные концевые повторы AAV 145 п.о., фланкирующие экспрессирующую трансген кассету. Эффективный перенос генов и стабильная доставка трансгенов благодаря интеграции в геномы трансдуцированных клеток являются ключевыми признаками этой векторной системы. (Wagner et al., Lancet 351:9117 1702-3 (1998), Keams et al., Gene Ther. 9:748-55 (1996)). Другие серотипы AAV, включая AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9 и AAVrh.10, и любой новый серотип AAV, также можно использовать в соответствии с настоящим изобретением.
[0176] Дефектные по репликации рекомбинантные аденовирусные векторы (Ad) могут быть получены с высоким титром и легко инфицируют ряд различных типов клеток. Большинство аденовирусных векторов сконструировано так, что трансген заменяет гены Ad E1a, E1b, и/или E3; затем дефектный по репликации вектор размножают в клетках человека 293, обеспечивающих функцию делетированного гена в транс-. Векторами Ad можно трансдуцировать множество типов тканей in vivo, включая неделящиеся, дифференцированные клетки, такие как клетки, обнаруженные в печени, почке и мышце. Общепринятые векторы Ad обладают большой емкостью переноса. Пример использования вектора Ad в клинических исследованиях включает терапию полинуклеотидом для противоопухолевой иммунизации с помощью внутримышечной инъекции (Sterman et al., Hum. Gene Ther. 7:1083-9 (1998)). Дополнительные примеры использования аденовирусных векторов для переноса генов в клинических исследованиях включают Rosenecker et al., Infection 24:15-10 (1996); Sterman et al., Hum. Gene Ther. 9:7 1083-1089 (1998); Welsh et al., Hum. Gene Ther. 2:205-18 (1995); Alvarez et al., Hum. Gene Ther. 5:597-613 (1997); Topf et al., Gene Ther. 5:507-513 (1998); Sterman et al., Hum. Gene Ther. 7:1083-1089 (1998).
[0177] Упаковывающие клетки используют для формирования вирусных частиц, способных инфицировать клетку-хозяина. Такие клетки включают клетки 293, упаковывающие аденовирус, и клетки ψ2 или клетки PA17, упаковывающие ретровирус. Вирусные векторы, используемые в генотерапии, обычно получают в линии клеток-продуцентов, упаковывающих нуклеиновую кислоту вектора в вирусную частицу. Векторы, как правило, содержат минимальные вирусные последовательности, необходимые для упаковки и последующей интеграции в хозяина (если необходимо), где другие вирусные последовательности заменены экспрессирующей кассетой, кодирующей белок, подлежащий экспрессии. Отсутствующие функции вируса обеспечивает упаковывающая линия клеток в транс-. Например, векторы AAV, используемые в генотерапии, как правило, обладают только последовательностями инвертированных концевых повторов (ITR) из генома AAV, необходимыми для упаковки и интеграции в геном хозяина. Вирусную ДНК упаковывают в линии клеток, содержащей плазмиду-помощника, кодирующую другие гены AAV, а именно, rep и cap, но лишенную последовательностей ITR. Линию клеток инфицируют также аденовирусом в качестве помощника. Вирус-помощник стимулирует репликацию вектора AAV и экспрессию генов AAV с плазмиды-помощника. Плазмида-помощник не упаковывается в значительных количествах из-за отсутствия последовательностей ITR. Контаминацию аденовирусом можно снижать посредством, например, термической обработки, к которой аденовирус является более чувствительным, чем AAV.
[0178] Для многих генотерапевтических применений является желательным, чтобы вектор для генотерапии был доставлен с высокой степенью специфичности в конкретный тип ткани. Соответственно, можно модифицировать вирусный вектор для обладания специфичностью к данному типу клеток посредством экспрессии лиганда в форме белка, слитого с белком вирусной оболочки на внешней поверхности вируса. Выбирают лиганд, обладающий аффинностью для рецептора, как известно, присутствующего на клетках представляющего интерес типа. Например, Flan et al., Proc. Natl. Acad. Sci. USA 92:9747-9751 (1995), опубликовали, что вирус лейкоза мышей Молони можно модифицировать для экспрессии херегулина человека, слитого с gp70, и рекомбинантный вирус инфицирует конкретные клетки рака молочной железы, экспрессирующие рецептор эпидермального фактора роста человека. Этот принцип можно распространять на другие пары вирус - клетка-мишень, в которых клетка-мишень экспрессирует рецептор, и вирус экспрессирует слитый белок, содержащий лиганд для рецептора поверхности клеток. Например, можно сконструировать нитевидный фаг для экспонирования фрагментов антитела (например, FAB или Fv), обладающих специфической аффинностью связывания фактически для любого выбранного клеточного рецептора. Хотя приведенное выше описание относится к в первую очередь к вирусным векторам, те же принципы можно применять к невирусным векторам. Такие векторы можно конструировать с содержанием конкретных последовательностей для поглощения, благоприятствующих поглощению специфическими клетками-мишенями.
[0179] Векторы для генотерапии можно доставлять in vivo посредством введения индивидуальному пациенту, как правило, посредством системного введения (например: внутривенной, внутрибрюшинной, внутримышечной, подкожной или интракраниальной инфузии) или местного введения, как описано ниже. Альтернативно, векторы можно доставлять в клетки ex vivo, такие как клетки, эксплантированные от индивидуального пациента (например, лимфоциты, пунктат костного мозга, биоптат тканей), или гематопоэтические стволовые клетки универсального донора, с последующей повторной имплантацией клеток пациенту, как правило, после отбора клеток с включением вектора.
[0180] Векторы (например, ретровирусы, аденовирусы, липосомы и т.д.), содержащие конструкции нуклеаз и/или донора, можно также вводить непосредственно в организм для трансдукции клеток in vivo. Альтернативно, можно вводить голую ДНК. Введение происходит любым из способов, в норме используемых для приведения молекулы в первичный контакт с клетками крови или ткани, включая, но без ограничения, инъекцию, инфузию, местное введение и электропорацию. Пригодные способы введения таких нуклеиновых кислот доступны и хорошо известны специалистам в данной области, и хотя более одного способа можно использовать для введения конкретной композиции, конкретный способ часто может обеспечивать более незамедлительную и более эффективную реакцию, чем другой способ.
[0181] Векторы, пригодные для введения полинуклеотидов (например, кодирующих нуклеазу и/или двухцепочечных доноров), описанных в настоящем документе, включают неинтегрирующие лентивирусные векторы (IDLV). См., например, Ory et al. (1996) Proc. Natl. Acad. Sci. USA 93:11382-11388; Dull et al. (1998)/Virol. 72:8463-8471; Zuffery et al. (1998)/Virol. 72:9873-9880; Follenzi et al. (2000) Nature Genetics 25:217-222; Публикация патента США No 2009/054985.
[0182] Фармацевтически приемлемые носители частично определяются конкретной вводимой композицией, так же как конкретным способом, используемым для введения композиции. Соответственно, существует широкое множество пригодных составов фармацевтических композиций, доступных, как описано ниже (см., например, Remington’s Pharmaceutical Sciences, 17th ed., 1989).
[0183] Очевидно, что кодирующие нуклеазу последовательности и донорные конструкции можно доставлять с использованием одинаковых или различных систем. Например, нуклеазы и доноры может переносить один и тот же вектор. Альтернативно донорный полинуклеотид может переносить плазмида, в то время как одну или более нуклеаз может переносить вектор AAV. Более того, различные векторы можно вводить одинаковыми или различными способами (внутримышечной инъекцией, инъекцией в хвостовую вену, другой внутривенной инъекцией, внутрибрюшинным введением и/или внутримышечной инъекцией). Векторы можно доставлять одновременно или в любом последовательном порядке.
[0184] Таким образом, настоящее описание относится к лечению in vivo или ex vivo заболеваний и состояний, поддающихся вставке трансгенов, кодирующих терапевтический белок, например, к лечению гемофилий посредством опосредованной нуклеазой интеграции факторов свертывания крови, таких как фактор VIII (F8). Композиции вводят пациенту-человеку в количестве, эффективном для получения желаемой концентрации терапевтического полипептида в сыворотке или органе-мишени, или в клетках-мишенях. Введение можно осуществлять любыми способами, в которых полинуклеотиды доставляют к желательным клеткам-мишеням. Например, предусматривают способы как in vivo, так и ex vivo. Внутривенная инъекция в воротную вену является предпочтительным способом введения. Другие способы введения in vivo включают, например, прямую инъекцию в доли печени или желчный проток и внутривенную инъекцию в удалении от печени, включая введение посредством инфузии в печеночную артерию, прямую инъекцию в паренхиму печени, инъекцию через печеночную артерию, и/или ретроградную инъекцию через желчные протоки. Способы введения ex vivo включают трансдукцию in vitro вырезанных гепатоцитов или других клеток печени, с последующей инфузией трансдуцированных, вырезанных гепатоцитов обратно в портальную сосудистую сеть, паренхиму печени или желчные протоки пациента-человека, см., например, Grossman et al., (1994) Nature Genetics, 6:335-341.
[0185] Эффективное количество нуклеазы(нуклеаз) и донора, подлежащее введению, может меняться от пациента к пациенту и в соответствии с представляющим интерес терапевтическим полипептидом. Соответственно, эффективные количества лучше всего определять врачу, вводящему композиции, и соответствующие дозы может легко определить специалист в данной области. После предоставления достаточного времени для интеграции и экспрессии (как правило, например, 4-15 суток), анализом уровня в сыворотке или другой ткани терапевтического полипептида и сравнением с исходным уровнем до введения можно определять, является ли вводимое количество слишком низким, находится ли в правильном диапазоне или является ли слишком высоким. Пригодные режимы начального и последующих введений также можно менять, однако типичным примером является исходное введение с последующими введениями затем, если необходимо. Последующие введения можно осуществлять с различными интервалами, в диапазоне от ежесуточного до ежегодного, до раза в каждые несколько лет. Специалисту в данной области понятно, что пригодные способы иммуносупрессии могут быть рекомендованы для избегания ингибирования или блокады трансдукции посредством иммуносупрессии векторов для доставки, см., например, Vilquin et al., (1995)Human Gene Ther., 6:1391-1401.
[0186] Составы для введений как ex vivo, так и in vivo, включают суспензии в жидкости или эмульгированных жидкостях. Активные ингредиенты часто смешивают с наполнителями, которые являются фармацевтически приемлемыми и совместимыми с активным ингредиентом. Пригодные наполнители включают, например, воду, солевой раствор, декстрозу, глицерин, этанол или т.п., и их комбинации. Кроме того, композиция может содержать незначительные количества вспомогательных веществ, таких как увлажняющие или эмульгирующие средства, забуферивающие pH средства, стабилизаторы или другие реагенты, усиливающие эффективность фармацевтической композиции.
[0187] Доставку нуклеиновых кислот можно вводить в клетку растений в вариантах осуществления по изобретению любым способом, известным специалистам в данной области, включая, в качестве неограничивающих примеров: посредством трансформации протопластов (См., например, Патент США 5508184); посредством поглощения ДНК, опосредованного обезвоживанием/ингибированием (См., например, Potrykus et al. (1985) Mol. Gen. Genet. 199:183-8); посредством электропорации (См., например, Патент США 5384253); посредством встряхивания с волокнами карбида кремния (См., например, Патенты США 5302523 и 5464765); посредством опосредованной Agrobacterium трансформации (См., например, Патенты США 5563055, 5591616, 5693512, 5824877, 5981840 и 6384301); посредством ускорения покрытых ДНК частиц (См., например, Патенты США 5015580, 5550318, 5538880, 6160208, 6399861 и 6403865) и посредством наночастиц, наноносителей и проникающих в клетку пептидов (WO201126644A2; W02009046384A1; W02008148223A1) в способах доставки ДНК, РНК, пептидов и/или белков или комбинаций нуклеиновых кислот и пептидов в клетки растений.
[0188] Посредством применения таких способов, как эти, клетки фактически любых видов можно стабильно трансформировать. В некоторых вариантах осуществления трансформирующую ДНК интегрируют в геном клетки-хозяина. В случае многоклеточных видов, трансгенные клетки можно регенерировать в трансгенный организм. Любой из этих способов можно использовать для получения трансгенного растения, например, содержащего одну или более последовательностей нуклеиновых кислот по изобретению в геноме трансгенного растения.
[0189] Наиболее широко используемый способ введения экспрессирующего вектора в растения основан на природной системе трансформации Agrobacterium. A. tumefaciens и A. rhizogenes представляют собой патогенные почвенные бактерии, генетически трансформирующие клетки растений. Плазмиды Ti и Ri A. tumefaciens и A. rhizogenes, соответственно, несут гены, ответственные за генетическую трансформацию растений. Плазмиды Ti (индуцирующие опухоли) содержат большой фрагмент, известный как T-ДНК, переносимый к трансформированным растениям. Другой фрагмент плазмиды Ti, область vir, является ответственным за перенос T-ДНК. Область T-ДНК ограничена левой и правой границами, каждая из которых состоит из концевых повторяющихся нуклеотидных последовательностей. В некоторых модифицированных бинарных векторах индуцирующие опухоли гены делетируют, и функции области vir используют для переноса чужеродной ДНК, ограниченной последовательностями границ T-ДНК. T-область может также содержать, например, селективный маркер для эффективного восстановления трансгенных растений и клеток, и участок множественного клонирования для вставки последовательностей для переноса такой нуклеиновой кислоты, кодирующей слитый белок по изобретению.
[0190] Таким образом, в некоторых вариантах осуществления вектор для трансформации растений получен из плазмиды Ti A. tumefaciens (см., например, Патенты США № 4536475, 4693977, 4886937, и 5501967; и Европейский патент EP 0 122 791) или плазмиды Ri A. rhizogenes. Другие векторы для трансформации растений включают, в качестве неограничивающих примеров, векторы, описанные в Herrera-Estrella et al. (1983) Nature 303:209-13; Bevan et al. (1983), выше; Klee et al. (1985) Bio/Technol. 3:637-42; и в Европейском патенте EP 0 120 516, и векторы, полученные из любого из вышеописанных. Другие бактерии, такие как Sinorhizobium, Rhizobium и Mesorhizobium, которые естественным образом взаимодействуют с растениями, можно модифицировать, чтобы опосредовать перенос генов в ряд разнообразных растений. Эти ассоциированные с растениями симбиотические бактерии можно делать компетентными для переноса генов посредством поглощения как обезвреженной плазмиды Ti, так и пригодного бинарного вектора.
[0191] Следующие примеры относятся к иллюстративным вариантам осуществления настоящего описания, в которых нуклеаза содержит нуклеазу с цинковыми пальцами (ZFN) или TALEN. Следует понимать, что это только с целью примера, и что можно использовать другие нуклеазы, например, систему нуклеазы CRISPR/Cas или эндонуклеазы хоминга (мегануклеазы) со сконструированными ДНК-связывающими доменами и/или белки, слитые с ДНК-связывающими доменами природных или сконструированных эндонуклеаз хоминга (мегануклеаз) и гетерологичных доменов расщепления.
ПРИМЕРЫ
Пример 1: Материалы и методы
Рост клеток, трансфекция и анализ ZFN/TALEN.
[0192] Для трансфекции K562 (ATCC CCL-243) использовали Amaxa Solution V и программу Т-016; CHO-K1 (ATCC CC1-61), Amaxa Solution T и программу U-023. Все трансфекции содержали 106 клеток и следующие плазмиды: AAVS1, 3 мкг 2A-связанных ZFN и донора; IL2Rγ и CCR5, 2 мкг каждой несвязанной плазмиды с ZFN и 10 мкг донорной плазмиды; GS, FUT8, 3 мкг 2A-связанных ZFN и 10 мкг донорной плазмиды. Дополнительные подобности, включая дизайны ZFN, см. в Патенте США № 8110379 и Патентные публикации США № 20100129869 и 20090042250 и DeKelver et al. (2010) Genome Res. 20(8): 1133-1142; Liu et al. (2009) Biotechnol. Bioeng. 106(1):97-105; Malphettes et al. (2010) Biotechnology and Bioengineering 106(5):774-83; Perez et al. (2008) Nature Biotech. 26(7):808-816; Umov et al. (2005) Nature 435(7042):646-651.
[0193] Пара нуклеаз FUT8 TALE (SBS 101082 и SBS 101086) непосредственно перекрывает участок связывания ZFN в экзоне 10 FUT8 и сконструирована с использованием A152/+63 N- и С-концевых точек усечения (Miller et al. 2011). Участок связывания SBS 101082 FUT8 TALE представляет собой 5’-tgt atc tgg cca ctg at-3’ (SEQ ID NO:1); SBS 101086, 5’-ttt gtc ttt gcc tcc tt-3’ (SEQ ID NO:2).
Дизайн и конструирование донорной плазмиды
[0194] Олигонуклеотиды, содержащие участки-мишени ZFN для пар ZFN AAVS1 (5'-tgt ccc сtc cAC CCC АСА GTG Ggg cca cTA GGG АСА GGA Ttg gtg аса ga-3', SEQ ID NO:3), отделенного спейсерами инвертированного AAVS1 (5'-tgt ccc ctc cAC CCC АСА GTG Ggt ggc cTA GGG АСА GGA Ttg gtg аса ga-3', SEQ ID NO:541), GS (5'-gac cCC AAG CCC ATT CCT GGG Aac tgg aAT GGT GCA GGC Tgc cat acc aa-3', SEQ ID NO:4) и IL2Ry (5'-gtt tcg tgt tCG GAG CCG CTT Taa ccc ACT CTG TGG AAG tgc tca gca tt-3', SEQ ID NO:5) гибридизовали с обратно комплементарными им в 50 мМ NaCl, 10 мМ Трис pH 7,5, и 1 мМ ЭДТА. См. также Патентную публикацию США № 20100129869 и Патенты США № 7951925 и 8110379. Заглавные буквы обозначают участки связывания ZFN, в то время как строчные буквы обозначают фланкирующую и спейсерную последовательность.
[0195] Двухцепочечные продукты затем клонировали в участок EcoRV вектора pBluescript II KS- (Agilent). Донорную плазмиду CCR5 получали в результате вставки олигонуклеотидов участка-мишени CCR5 (5’-GTC АТС CTC АТС CTG АТА AAC TGC AAA AGa-3’, SEQ ID NO:6); 5-CTT TTG CAG TTT АТС AGG ATG AGG ATG ACa-3’ SEQ ID NO:7, см. также Патент США № 7951925) в pCR2.1 (Invitrogen). Вторую донорную плазмиду AAVS1 (см., фигуру 1E) получали посредством вставки вышеуказанных олигонуклеотидов участка-мишени AAVS1 в участок EcoRV плазмиды на основе pCR2.1, содержащей также открытую рамку считывания GFP под управлением промотора pGK.
[0196] Донорную плазмиду FUT8 получали посредством вставки участка связывания ZFN/TALEN (5’-ggc CGT GTA TCT GGC CAC TGA TGA CCC TTC TTt gtt aAA GGA GGC AAA GAC AAA Gta a-3’, SEQ ID NO:8) в донорную плазмиду, содержащую трансгены IgG и устойчивости к пуромицину (Moehle et al. (2007) Proc. Nat’l Acad. Sci. USA 104(9):3055-3060).
Анализ направленной интеграции
[0197] Все реакции ПЦР проводили с 100 нг геномной ДНК в качестве матрицы, с использованием полимеразы Accuprime HiFi™ (Invitrogen). Геномную ДНК очищали с помощью набора Masterpure™ (Epicentre).
[0198] Направленную интеграцию донорной плазмиды AAVS1 GFP в AAVS1 (известный также как PPP1R12C) анализировали во всех четырех возможных участках стыковки хромосома-донор посредством амплификации ПЦР. В реакциях ПЦР использовали температуру отжига 60°, время удлинения 30 секунд, 30 циклов амплификации и следующие праймеры: AB левый, AAVS1 CEL-I F (5’-ccc ctt acc tct cta gtc tgt gc-3’, SEQ ID NO:9) и участок стыковки AAVS1 R (5’-ggc gat taa gtt ggg taa cg-3’, SEQ ID NO:10); AB правый, участок стыковки AAVS1 F (5’-ggc сtc ttg gtc aag ttg tt-3’, SEQ ID NO:11) и AA VS1 CEL-I R (5’-сtc agg ttc tgg gag agg gta g-3’, SEQ ID NO:12); BA левый, AAVS1 CEL-I F и участок стыковки AAVS1 F; BA правый, участок стыковки AAVS1 R и AAVS1 CEL-I R. Для последовательностей на фигуре 8 A использовали следующие праймеры: AB левый, M13F (5’-gta aaa cga сgg cca gt-3’, SEQ ID N0:13) и AAVS1 CEL-F; BA правый, M13F и AAVS1 CEL-I R.
[0199] Направленную интеграцию в IL2Rγ, CCR5 и GS анализировали посредством амплификации ПЦР. В реакциях ПЦР использовали температуру отжига 58°, время удлинения 30 секунд, 5% DMSO, 26 циклов амплификации и праймер M13F в комбинации со следующими праймерами: IL2RγBA левый, IL2Rγ CEL-I F (5’-acc agt gag ttt tca tta gg-3’, SEQ ID NO:14); IL2RγAB правый, IL2Rγ CEL-I R (5’-tgg agс aaa aga cag tgg tg-3’, SEQ ID NO:15); CCR5 BA левый, R5F (5’-aag atg gat tat caa gtg tсa agt cc-3’, SEQ ID NO:16); CCR5 AB правый, R5R (5’-caa agt ccc act ggg cg-3’, SEQ ID NO:17); GS BA левый, GJC 172F (5’-atс cgc atg gga gat cat ct-3’, SEQ ID NO:18); GS AB правый, GJC 173R (5’-gtg tat gtt cgt tсa ccc ac-3’, SEQ ID NO:19).
[0200] Направленную интеграцию донора AAVS1 в GS (фигура 2B) анализировали посредством амплификации ПЦР. В реакциях ПЦР использовали температуру отжига 58°C, время удлинения 30 секунд, 5% DMSO, 26 циклов амплификации и следующие праймеры: AB левый, JcnlF (5’-caa ata gga ccc tgt gaa gga-3’, SEQ ID NO:20) и JcnlR (5’-gat taa gtt ggg taa cgc cag-3’, SEQ ID NO:21); BA левый, Jcn3F (5’-aat agg acс ctg tga agg a-3’, SEQ ID NO:22) и Jcn3R (5’-gtg tgg aat tgt gag сgg ata-3’, SEQ ID NO:23).
[0201] Направленную интеграцию донора IgG в FUT8 (фигура 4) анализировали посредством амплификации ПЦР. В реакциях ПЦР использовали температуру отжига 60°, время удлинения 30 секунд, 30 циклов амплификации (35 циклов для скрининга грубых лизатов), и следующие праймеры: AB левый участок стыковки, GJC 75F (5’-agt cca tgt cag acg cac tg-3’, SEQ ID NO:24) и SC seqpfzR (5’-aga gtg agg ctc tgt ctc aa-3’, SEQ ID NO:25); AB правый участок стыковки, донор FUT8 CELIF2 (5’-tac gta tag gсt gсg caa ct-3’, SEQ ID NO:26) и GJC 115R (5’-gca cat gta gtc ttt gat ttt g-3’, SEQ ID NO:27); BA левый участок стыковки, GJC75F и донор FUT8 CELIF2; BA правый участок стыковки, SC seqpfzR и GJC115R.
[0202] Саузерн-блоттинг интеграции донора AAVS1 GFP в AAVS1 анализировали, как описано ранее (DeKelver et al. 2010, ibid). Ожидаемые результаты этого Саузерн-блоттинга являются следующими: зонд AAVS1 будет гибридизоваться с полосой либо 2092, либо 6592 п.о. для ориентаций AB и BA, соответственно. Триплоидный локус AAVS1 дикого типа будут наблюдать как полосу 3287 п.о. Для Саузерн-блоттинга интеграции донора AAVS1 GFP где-либо еще в геноме в качестве зонда использовали полную открытую рамку считывания GFP. Для интеграции в AAVS1 будут получать полосы либо 3323, либо 4482 п.о. для ориентаций AB и BA, соответственно; для ненаправленных интеграций где-либо еще в геноме будут получать вторичные полосы не поддающегося детекции размера. Для Саузерн-блоттинга интеграции донора GS в GS в качестве зонда использовали фрагмент 424 п.о. гена GS, ограниченный 5’-ctg cag gtg aag аса gga tg-3’ и 5’-ccc act aga aag aac atg tt-3’. Интеграцию в GS можно выявлять как гибридизующуюся полосу 2933 п.о.; для локуса GS дикого типа будут получать полосу 1977 п.о. Для Саузерн-блоттинга интеграции донора GS где-либо еще в геноме в качестве зонда использовали фрагмент BsaI - ScaI гена bla E. coli. Для правильно интегрированных трансгенов будут получать полосу 2055 п.о.; для интеграции в псевдогены GS будут получать полосы 4878, 4214, 10080 и 9416 п.о. в зависимости от псевдогена и ориентации вставки; для других ненаправленных интеграций будут получать отдельную полосу непредсказуемого размера. Для Саузерн-блоттинга интеграции FIJT8 в качестве зонда использовали фрагмент HindIII-XmnI 407 п.о. локуса FUT8. Для обоих Саузерн-блоттингов для FUT8 геномную ДНК разрезали HindIII.
[0203] Контиги, содержащие псевдогены FUT8, GS и GS, выделяли из данных секвенирования полного генома CHO с использованием специально разработанного скрипта Python (Xu et al. (2011) Nat Biotechnol 29(8):735-41). FUT8 присутствует в контиге AFTD01065932.1; GS - в контиге AFTD01107178.1. Один псевдоген GS (содержащийся в AFTD01043599.1) обладает точным сохранением участков связывания ZFN, 120/128 (94%) п.о. гомологии с частью экзона 5 зонда, и ожидают его присутствия в фрагменте ScaI 6333 п.о. Второй псевдоген GS (содержащийся в AFTD01154859.1) обладает одним несовпадением в участках связывания ZFN, 116/128 (91%) п.о. гомологии с частью экзона 5 зонда, и ожидают его присутствия в фрагменте ScaI 13320 п.о.
[0204] Концентрации антител измеряли с использованием набора для анализа IgG Pierce Easy-Titer (23310) в соответствии с инструкциями производителя. Клоны с по меньшей мере двукратным превышением фона классифицировали как положительные.
Пример 2: Направленная интеграция после расщепления in vivo двухцепочечного донора
A. AAVS1
[0205] Для тестирования того, может ли расщепление трансгена in vivo с использованием такой же ZFN, которая расщепляет геномный участок-мишень, синхронизировать расщепление донора и хромосомы, минимизировать подверженность трансгена к деградации, клетки K562 трансфицировали нацеленными на AAVS1 ZFN и донорной плазмидой, содержащей участки-мишени AAVS1 ZFN для расщепления донорной плазмиды in vivo. Кратко, как описано в примере 1, авторы настоящего изобретения клонировали участок узнавания хорошо охарактеризованных и высокоактивных AAVS1 ZFN в донорную плазмиду, содержащую автономную экспрессирующую кассету GFP, но лишенную гомологии с локусом AAVS1. См. Патент США № 8110379 и DeKelver et al. (2010) Genome Res. 20(8):1133-1142. Донорную плазмиду (с участком-мишенью ZFN или без) совместно трансфицировали в клетки K562 вместе со второй плазмидой, кодирующей AAVS1 ZFN.
[0206] Вставку в хромосомный участок AAVS1 анализировали посредством амплификации ПЦР уникальных участков стыковки, сформированных посредством направленной интеграции донора, с геномной ДНК, выделенной через 3 суток после трансфекции, как описано выше в примере 1.
[0207] Как показано на фигуре 1, при совместной трансфекции родственными ZFN, может происходить одновременное расщепление содержащих участок ZFN как донорной плазмиды, так и хромосомы, позволяя вставку плазмиды в хромосому. Вставка донорной плазмиды в направлении, ожидаемом при простом лигировании выступающих концов после ZFN, обозначена как ориентация AB, альтернативное направление обозначено как ориентация BA. Как показано на фигуре 1D, ориентация BA (обратная) является преимущественной, когда нуклеотиды между участками-мишенями (спейсер) являются обратно комплементарными геномной последовательности (дикого типа).
[0208] Более того, как показано на фигурах 1D и 1E (см. дорожки 6, 8, 10, 17, 19 и 21 IE), в соответствии с успешным захватом расщепленной донорной ДНК, авторы настоящего изобретения детектировали ожидаемые 5’ и 3’ участки стыковки, сформированные посредством интеграции донора в обеих ориентациях AB и BA. Ориентация BA являлась преимущественной с обратно комплементарными спейсерами (фигура 1D). Интеграция донора требовала опосредованного ZFN расщепления, поскольку как (i) донор без участка AAVS1 ZFN не был интегрирован несмотря на эффективное расщепление локуса AAVS1 (см., фигуру 1E, дорожки 5, 7, 9, и дорожки 16, 18 и 20; фигура 5A), так и (ii) трансфекция донора без совместной трансфекции соответствующей ZFN также не смогла привести к направленной интеграции.
[0209] Клоны клеток получали посредством предельного разведения из пула, трансфицированного как ZFN, так и донором (дорожка 8/19). Три положительных по GFP и положительных по ПЦР участков стыковки клона анализировали в двух повторах Саузерн-блоттингом для подтверждения интеграции донорной плазмиды. Клоны попали в три класса: клон один содержит одну вставку AB; клон два содержит одну вставку BA; клон три содержит обе вставки AB и BA в дополнение к аллелю без вставки (фигура 1F).
[0210] Три клона анализировали также по интеграции вне мишеней посредством Саузерн-блоттинга со специфическим для GFP зондом. Клон один содержит только ожидаемую вставку в AAVS1, в то время как клоны 2 и 3 содержат вставку трансгена где-либо еще в геноме в дополнение к вставкам AAVS1 (Фигура 1F).
[0211] Ампликоны ПЦР участков стыковки интеграции хромосома-донор из этих трех линий клеток клонировали и секвенировали. Как показано на фигуре 7, клоны 1 и 3 содержали вставки AB с точным лигированием выступающих концов донора и хромосомы как в 5’-, так и в 3’-участках стыковки. Клоны 2 и 3 содержали вставки BA с аллелями, полученными посредством зависимой от микрогомологии репарации на левом, 5’-участке стыковки.
B. IL2Rv. GS и CCR
[0212] Чтобы продемонстрировать, что захват расщепленного донора не являлся ограниченным интеграцией в AAVS1 в клетках K562, авторы настоящего изобретения провели аналогичные эксперименты в трех других локусах (IL2Rγ, CCR и GS) в клетках K562 и CHO. Успешную направленную интеграцию мониторировали в одном участке стыковки хромосома-донор для каждой ориентации (AB и BA), как описано выше.
[0213] Сайт-специфическую интеграцию, направленную в участок расщепления ZFN, наблюдали для локусов IL2Rγ и CCR5 в клетках K562, и для локуса GS в клетках CHO-K1. (Фигура 2A, дорожки 3, 7, 11, 15, 19 и 23). Как и для интеграции в AAVS1, интеграция в IL2Rγ CCR5 и GS являлась зависимой от участка расщепления ZFN в донорной плазмиде (фигура 2A, дорожки 1, 5, 9, 13, 17 и 21) и совместной доставки самих ZFN. Активность ZFN, как на хромосомной мишени, так и на донорной плазмиде, являлась в основном одинаковой среди всех образцов (фигура 5B).
[0214] Секвенирование продуктов ПЦР участка стыковки хромосома-донор из этих локусов, так же как из аналогичного пула интегрантов AAVS1, выявило спектр событий вставки, согласующийся с правильной интеграцией в локус-мишень (фигура 8).
[0215] Таким образом, способность захватывать расщепленный in vivo трансгенный донор в DSB является общим свойством аппарата репарации ДНК млекопитающих и является независимой от конкретного участка-мишени или типа клеток.
Пример 3: Расщепление in vivo и in vitro
[0216] Чтобы подтвердить, что расщепление in vivo являлось необходимым для поддержания наблюдаемых уровней направленной вставки гена, авторы настоящего изобретения проводили прямое сравнение направленной интеграции с использованием расщепленных in vivo доноров и доноров, расщепленных in vitro с использованием EcoRV, как описано в примере 1.
[0217] Как показано на фигуре 2, в то время как интеграция предварительно расщепленной донорной плазмиды изредка поддавалась детекции, она являлась заметно менее эффективной по сравнению с расщепленными in vivo донорами (фигура 2A, сравни линии 2/3, 6/7, 10/11, 14/15, 18/19 и 22/23). Более того, для использования предварительно расщепленных донорных ДНК показан увеличенный диапазон размеров продуктов ПЦР участка стыковки, согласующийся с увеличенным уровнем деградации донорной ДНК перед хромосомным захватом (см., например, фигуру 2A, дорожку 22).
[0218] Для подтверждения того, что направленную интеграцию можно посредством использования двух различных нуклеаз (ZFN), авторы настоящего изобретения использовали пару GS ZFN (пример 1) для разрезания хромосомы клеток CHO-K1 и пару AAVS1 ZFN для разрезания донорной плазмиды в той же самой клетке. Интеграцию в GS детектировали со сходной частотой, как когда пара GS ZFN разрезает хромосому и донора (как на фигуре 2A), так и когда GS ZFN разрезает хромосому, в то время как AAVS1 ZFN разрезают донора (фигура 2B, дорожки 6 и 8, дорожки 15 и 17). Эффективность расщепления в GS снова являлась равномерной среди всех трансфицированных GS ZFN образцов (фигура 5C).
[0219] Таким образом, расщепление in vivo является более эффективным, чем предварительное расщепление донорной молекулы.
Пример 4: Независимая от гомологии направленная интеграция в клетки CHO
[0220] Направленная интеграция в клетки CHO имеет особенно важные применения в биотехнологии, при этом клетки CHO осуществляют основанную на HDR направленную интеграцию трансгенов из нескольких тысяч пар оснований очень плохо. Для прояснения этого пункта авторы настоящего изобретения сравнивали опосредованную HDR направленную интеграцию в клетках HEK-293 и клетках CHO-K1 с использованием системы, разработанной для доставки не имеющего промотора гена GFP в содержащий промотор акцепторный локус, в основном, как описано в Moehle et al. (2007) Proc. Nat’l Acad. Sci. USA 104(9):3055-3060). Направленная интеграция приводит к экспрессии GFP и позволяет количественную оценку посредством проточной цитометрии.
[0221] При трансфекции ZFN и содержащей гомологию донорной плазмидой (для интеграции посредством HDR), между 0,5 и 3% клеток HEK-293 становились положительными по GFP (фигура 6). В отличие от этого, никакое количество клеток CHO-K1 не становилось положительным по GFP при сходной трансфекции. Принимая во внимание, что клетки CHO плохо осуществляют основанную на HDR направленную интеграцию, и при этом доказана их полезность для продукции рекомбинантного белка, авторы настоящего изобретения затем поставили вопрос, можно ли использовать расщепление in vivo донорной ДНК для управления направленной интеграцией в клетках CHO.
[0222] Клетки CHO-K1 из пула, несущего направленную интеграцию в GS (фигура 2A, дорожки 19/23), клонировали посредством предельного разведения, и для происходящих из отдельных клеток клонов проводили скрининг посредством ПЦР по сайт-специфической интеграции. В отличие от отрицательных результатов, полученных с помощью способа на основе HDR, для независимого от гомологии захвата расщепленной in vivo донорной ДНК получено 10% (17/157) происходящих из отдельных клеток клонов, которые являлись положительными по ПЦР для левого участка стыковки хромосома-донор BA, 8% (13) положительных по правому участку стыковки BA, и 6% (10) положительных по обоим участкам стыковки BA. Восемь из этих десяти клонов случайным образом выбирали для анализа Саузерн-блоттингом. Все 8 содержали ожидаемую направленную вставку трансгена в участок-мишень GS, аллель GS дикого типа и два псевдогена GS (фигура 3). Только аллель GS дикого типа и псевдогены присутствуют в клетках CHO-K1 дикого типа (фигура 3, дорожка 9). Более того, при использовании в качестве зонда специфической для трансгена последовательности, показано, что пять из 8 клонов содержали только одну копию трансгена в GS, в то время как 3 содержали копию трансгена в GS вместе с одной или несколькими случайным образом интегрированными копиями в других участках генома CHO, один из которых соответствовал интеграции в псевдоген GS (фигура 3, дорожки 10, 12, 13, 16, 17 и дорожки 11, 14, 15, соответственно). Участки стыковки хромосома-трансген были секвенированы и показаны на фигуре 9.
Пример 5: Направленная интеграция в FUT8 и нарушение FUT8
[0223] Трансгены общепринятым образом вставляют в геном клеток CHO для получения биофармацевтических белков, в особенности, антител. В клетках CHO с делецией гена FUT8 получают фукозилированные антитела с в 100 раз более высокой антителозависимой клеточной цитотоксичностью (Malphettes et al. (2010) Biotechnology and Bioengineering 106(5):774-83; Yamane-Ohnuki et al. (2004) Biotech. Bioeng. 87(5):614-22). Более того, можно проводить отбор по нокауту экспрессии FUT8, таким образом, потенциально сочетая направленную интеграцию с этим поддающимся отбору признаком. Авторы настоящего изобретения, таким образом, использовали ранее описанные специфические для FUT8 ZFN для нарушения гена FUT8 посредством вставки расщепленной in vivo кассеты для продукции антител (Moehle et al. (2007) Proc. Nat 7 Acad. Sci. USA 104(9):3055-3060). Более того, авторы настоящего изобретения хотели определить, может ли происходить захват расщепленного in vivo донора в двухцепочечный разрыв, полученный посредством нуклеазы TALE (TALEN), специфической для FUT8.
[0224] Кратко, ZFN или TALEN, расщепляющие FUT8, совместно трансфицировали с экспрессирующей антитело плазмидой, содержащей участок FUT8 расщепления нуклеазой, как описано в примере 1. Проводили отбор трансфицированного пула по биаллельному нокауту FUT8 с использованием агглютинина Lens culinaris, и клетки клонировали посредством предельного разведения (см., Malphettes et al. (2010) Biotechnology and Bioengineering 106(5):774-83). Проводили скрининг клонов по секреции IgG и по вставке трансгена IgG посредством ПЦР как левого, так и правого участков стыковки трансген/хромосома.
[0225] Как показано на фигуре 4, для обработанных ZFN клонов, 25/96 (26%) клонов экспрессировали IgG, и 14/96 (15%) клонов являлись положительными по вставке полного трансгена IgG. Все кроме одного клоны, положительные по вставке по ПЦР, экспрессировали IgG. Сходные результаты получены для FUT8 TALEN: 35/171 (20%) клонов экспрессировали IgG, и все 16 (9%) клонов со вставкой полного трансгена экспрессировали IgG. Клоны с одним (но не с обоими) участками стыковки интеграции трансгена, поддающимися детекции посредством ПЦР, составляли значительную фракцию остальных экспрессирующих IgG клонов (Фигура 4A).
[0226] Эти эксперименты проводили также с использованием TALE-нуклеаз, нацеленных на FUT8. Интеграцию трансгена в FIJT8 подтверждали анализом Саузерн-блоттинга для десяти положительных по ПЦР и IgG клонов, полученных при опосредованной TALEN вставке трансгена (фигуры 4B и 4C). Кроме того, на фигуре 12 показаны последовательности, полученные секвенированием продуктов ПЦР участков стыковки интегрированных в FUT8 доноров с использованием ZFN (фигура 10A) или TALEN (фигура 10B).
[0227] Суммарно, способы и композиции, описанные в настоящем документе, относятся к простой и направленной интеграции больших трансгенов независимыми от гомологии способами, включая линии клеток (например, клетки CHO), являющиеся устойчивыми к управляемой гомологией интеграции.
Пример 6: Направленная интеграция в локусы AHAS пшеницы и повреждение локусов AHAS пшеницы. Характеризация и идентификация геномных последовательностей-мишеней AHAS
[0228] Транскрибируемые области для трех гомеологичных генов AHAS идентифицированы и определены, нуклеазы с цинковыми пальцами сконструированы для связывания и расщепления участков опосредованного NHEJ нацеливания донорной последовательности, как описано в Предварительном Патенте США № регистрации 61/809097, содержание которого приведено в настоящем документе в качестве ссылки. Эти новые последовательности перечислены как SEQ ID NO:116, SEQ ID NO:117 и SEQ ID NO:118. В предыдущих работах по секвенированию идентифицированы и генетически картированы гомеологичные копии генов AHAS из Triticum aestivum на длинных плечах хромосом 6A, 6B и 6D (Anderson et al., (2004) Weed Science 52:83-90; и, Li et al., (2008) Molecular Breeding 22:217-225). Анализ последовательностей маркеров экспрессируемой последовательности (EST) и геномных последовательностей, доступных в Genbank (номера доступа: AY210405.1, AY210407.1, AY210406.1, AY210408.1, FJ997628.1, FJ997629.1, FJ997631.1, FJ997630.1, FJ997627.1 и AY273827.1) использовали для определения транскрибируемой области для гомеологичных копий гена AHAS (SEQ ID NO: 116-118).
[0229] Новые, некодирующие последовательности гена AHAS, локализованные выше и ниже транскрибируемой области, охарактеризованы впервые. Дл полной характеризации этих некодирующих последовательностей, транскрибированные последовательности для каждой из трех гомеологичных копий гена AHAS использовали в качестве запросов BLASTN™ для скрининга несоставленных прочтений последовательностей на ROCHE 454™, полученных после секвенирования методом дробовика полного генома Triticum aestivum cv. Chinese Spring. Прочтения последовательностей на ROCHE 454™ Triticum aestivum cv. Chinese Spring получены для 5-кратного перекрывания последовательности. Составление последовательности завершали с использованием программного обеспечения SEQUENCHER™ (GeneCodes, Ann Arbor, MI) для ROCHE 454™. Прочтения последовательностей со значимым совпадением в BLASTN™ (значение E <0,0001) использовали для характеризации этой нетранскрибируемой области. Проводили повторяющиеся циклы анализа BLASTN™ и составления последовательностей. Каждый повтор включал составленную последовательность AHAS из предыдущего повтора, так что все последовательности скомпилировали в виде одной непрерывной последовательности. Всего охарактеризовано 4384, 7590 и 6205 геномных последовательностей для гомеологичных генов AHAS, локализованных на хромосомах 6A, 6B и 6D, соответственно (SEQ ID NO: 119-121).
Анализ последовательности генов AHAS, выделенных из Triticum aestivum cv. Bobwhite MPB26RH
[0230] Гомологичные копии гена AHAS клонировали и секвенировали из Triticum aestivum cv. Bobwhite MPB26RH для получения нуклеотидной последовательности, пригодной для конструирования специфических белков с цинковыми пальцами, которые могут связывать последовательности с высокой степенью специфичности. Анализ последовательности нуклеотидных последовательностей AHAS, полученных из Triticum aestivum cv. Bobwhite MPB26RH, являлся необходимым для подтверждения аннотации нуклеотидов, присутствующих в Genbank, и последовательностей гена AHAS из ROCHE 454™ и из-за аллельного разнообразия между cv. Bobwhite MPB26RH и другими сортами пшеницы, из которых получали последовательности из Genbank и ROCHE 454™.
[0231] Когорта праймеров для ПЦР разработана для амплификации генов AHAS (Таблица 1). Праймеры разработаны из консенсусной последовательности, полученной из выравниваний множества последовательностей, полученных с использованием CLUSTALW™ (Thompson et al., (1994) Nucleic Acids Research 22:4673-80). Выравнивания последовательности составлены из данных секвенирования cv. Chinese Spring, полученных при секвенировании на ROCHE 454™, завершенном с 5-кратным перекрыванием.
[0232] Как указано в таблице 1, праймеры для ПЦР сконструированы для амплификации всех трех гомеологичных последовательностей или для амплификации только отдельной гомеологичной последовательности. Например, праймеры для ПЦР, использованные для амплификации транскрибируемой области гена AHAS, разработаны для одновременной амплификации всех трех гомеологичных копий в одной мультиплексной реакции ПЦР. Праймеры для ПЦР, использованные для амплификации нетранскрибируемой области, сконструированы либо для амплификации всех трех гомеологичных копий, либо для амплификации только одной гомеологичной копии. Все праймеры для ПЦР сконструированы, чтобы составлять в длину между 18 и 27 нуклеотидами и иметь температуру отжига 60-65°C, оптимально, 63°C. Кроме того, несколько праймеров разработано для расположения предпоследнего основания (содержащего фосфоротиоатную связь и указанного в таблице 1 как звездочка [*]) над вариантом нуклеотидной последовательности, различающим копии гена из каждого субгенома пшеницы. В таблице 1 перечислены праймеры для ПЦР, которые были разработаны и синтезированы.
Последовательности праймеров, использованных для амплификации ПЦР последовательностей AHAS
transcribed
transcribed
transcribed
Кодирующая = праймеры, разработанные для транскрибируемых областей
Звездочкой (*) указано включение фосфоротиоатной последовательности
[0233] Специфической для субгенома амплификации достигали с использованием ПЦР вкл.-выкл. (Yang et al., (2005) Biochemical and Biophysical Research Communications 328:265-72) с праймерами, разработанными для расположения предпоследнего основания (содержащего фосфоротиоатную связь) над вариантом нуклеотидной последовательности, различающим копии гена из каждого субгенома пшеницы. Два различных набора условий ПЦР использовали для амплификации гомеологичных копий гена AHAS из cv. Bobwhite MPB26RH. Для транскрибируемых областей реакционная смесь для ПЦР содержала 0,2 мМ dNTP, буфер для ПЦР IXIMMOLASE™ (Bioline, Taunton, MA), 1,5 мМ MgCl2, 0,25 единиц ДНК-полимеразы IMMOLASE™ (Bioline, Taunton, MA), 0,2 мкМ каждый из прямого и обратного праймеров, и приблизительно 50 нг геномной ДНК. Реакционные смеси, содержащие праймеры AHAS_1F1 и AHAS_1R1, дополняли 8% (об./об.) DMSO. Для нетранскрибируемых областей реакционные смеси для ПЦР содержали 0,2 мМ dNTP, 1X буфер PHUSION GC™ (New England Biolabs Ipswich, MA), 0,5 единиц ДНК-полимеразы HOT-START PHUSION™ полимераза (New England Biolabs), 0,2 мкМ каждый из прямого и обратного праймеров, и приблизительно 50 нг геномной ДНК. ПЦР проводили в конечном объеме реакционной смеси с использованием термоциклера MJ PTC200® (BioRad, Hercules, CA). После циклов ПЦР продукты реакции очищали и клонировали с использованием вектора PGEM-T EASY™ (Promega, Madison, WI) в клетки E. coli JM109. Плазмидную ДНК выделяли с использованием набора для очистки плазмидной ДНК DNAEASY™ (Qiagen, Valencia, CA) и секвенировали по Сенгеру с использованием химических реактивов BIGDYE® v3.1 (Applied Biosystems, Carlsbad, CA) на автоматической платформе для капиллярного электрофореза ABI3730XL®. Анализ последовательности, проведенный с использованием программного обеспечения SEQUENCHER™ (GeneCodes, Ann Arbor, MI), использовали для получения консенсусной последовательности для каждой гомеологичной копии гена (SEQ ID NO:140, SEQ ID NO:141 и SEQ ID NO:142) из cv. Bobwhite MPB26RH. CLUSTALW™ использовали для получения выравнивания множества консенсусных последовательностей, по которым подтверждали разнообразие гомеологичных последовательностей, различающихся между копиями гена AHAS.
Дизайн связывающих доменов с цинковыми пальцами, специфических для последовательностей гена AHAS
[0234] Белки с цинковыми пальцами, направленные против идентифицированных последовательностей ДНК гомеологичных копий генов AHAS, конструировали, как описано ранее. См., например, Umov et al., (2005) Nature 435:646-551. Иллюстративная последовательность-мишень и спирали узнавания показаны в таблице 2 (дизайн областей спиралей узнавания) и в таблице 3 (участки-мишени). В таблице 3 нуклеотиды участка-мишени, контактирующие со спиралями узнавания ZFP, указаны заглавными буквами; не контактирующие нуклеотиды указаны строчными буквами. Участки-мишени нуклеазы с цинковыми пальцами (ZFN) разработаны для 4 областей в гене AHAS: области приблизительно 500 п.о. выше аминокислотного остатка серина 653, соседней области выше (в пределах 30 п.о.) аминокислотного остатка серина 653, соседней области ниже (в пределах 80 п.о.) аминокислотного остатка серина 653 и области приблизительно на 400 п.о. ниже аминокислотного остатка серина 653. Многочисленные виды дизайна ZFP разработаны и тестированы для идентификации пальцев, связывающихся с наивысшим уровнем эффективности с 22 различными участками-мишенями AHAS, идентифицированными в пшенице, как описано в Предварительном Патенте США № регистрации 61809097, содержание которого приведено в настоящем документе в качестве ссылки. Специфические спирали узнавания ZFP (таблица 2), связывающиеся с наивысшим уровнем эффективности с последовательностями узнавания цинковых пальцев, использовали для опосредованного NHEJ нацеливания и интеграции донорной последовательности (независимой от гомологии направленной интеграции) в локусе AHAS генома пшеницы.
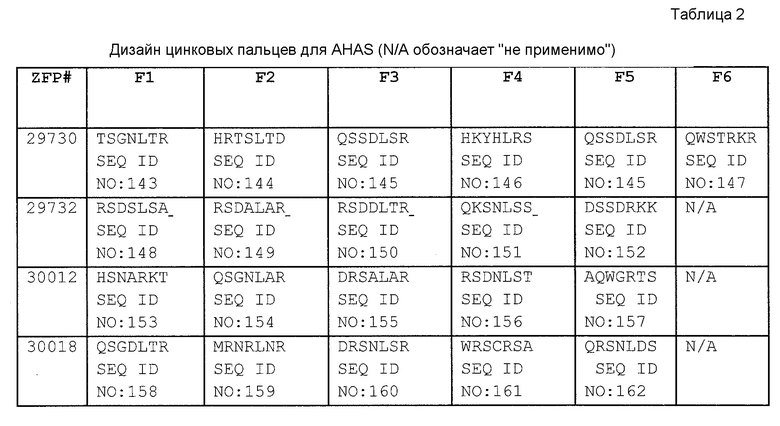
Участки-пишени цинковых пальцев в AHAS
[0235] Дизайны цинковых пальцев для AHAS включали в экспрессирующие векторы для цинковых пальцев, кодирующие белок, имеющий по меньшей мере один палец со структурой CCHC. См. Патентную публикацию США № 2008/0182332. В частности, последний палец в каждом белке имел остов CCHC для спирали узнавания. Последовательности, кодирующие неканонические цинковые пальцы, сливали с доменом нуклеазы рестрикционного фермента типа IIS FokI (аминокислоты 384-579 последовательности из Wah et al., (1998) Proc. Natl. Acad. Sci. USA 95:10564-10569) посредством линкера ZC из четырех аминокислот и сигналом ядерной локализации opaque-2, полученным из Zea mays, для формирования нуклеаз с цинковыми пальцами (ZFN) для AHAS. См. Патент США № 7888121.
[0236] Оптимальные цинковые пальцы верифицировали по активности расщепления с использованием системы на основе почкующихся дрожжей, как показано ранее, идентифицирующей активные нуклеазы. См., например, Патентную публикацию США № 2009/0111119; Doyon et al., (2008) Nat Biotechnology 26:702-708; Geurts et al., (2009) Science 325:433. Цинковые пальцы для различных функциональных доменов отбирали для использования in vivo. Из множества ZFN, которые были разработаны, получены и тестированы по связыванию с предположительным геномным полинуклеотидным участком-мишенью AHAS, ZFN, описанные в таблице 2 выше, идентифицированы как обладающие активностью in vivo на высоких уровнях и охарактеризованы как способные эффективно связывать и расщеплять уникальные геномные полинуклеотидные участки-мишени AHAS in planta.
Оценка расщепления генов AHAS нуклеазой с цинковыми пальцами с использованием анализов временной трансфекции
[0237] Сборка конструкции ZFN: Плазмидные векторы, содержащие экспрессирующие ген ZFN конструкции, идентифицированные с использованием анализа дрожжей, как описано ранее, разработаны и завершены с использованием квалификации и способов, общеизвестных в данной области (см., например, Ausubel или Maniatis). Каждую кодирующую ZFN последовательность сливали с последовательностью, кодирующей сигнал ядерной локализации opaque-2 (Maddaloni et al., (1989) Nuc. Acids Res. 17:7532), расположенный выше нуклеазы с цинковыми пальцами.
[0238] Экспрессией слитых белков управлял сильный конститутивный промотор из гена убиквитина Zea mays, (включающий 5’-нетранслируемую область (UTR) (Toki et al., (1992) Plant Physiology 100; 1503-07). Экспрессирующая кассета включала также 3'-UTR (содержащую терминатор транскрипции и участок полиаденилирования) из гена пероксидазы Zea mays (Per5) (Патентная публикация США № 2004/0158887). Кодирующую самогидролизующийся 2A нуклеотидную последовательность из вируса Thosea asigna (Szymczak et al., (2004) Nat Biotechnol. 22:760-760) добавляли между двумя слитыми белками нуклеаз с цинковыми пальцами, клонированными в конструкцию.
[0239] Плазмидные векторы собирали с использованием способа IN-FUSION™ Advantage (Clontech, Mountain View, CA). Рестрикционные эндонуклеазы получены из New England BioLabs (Ipswich, MA), и T4 ДНК-лигазу (Invitrogen, Carlsbad, CA) использовали для лигирования ДНК. Выделения плазмиды проводили с использованием набора для выделения плазмид NUCLEOSPIN® (Macherey-Nagel Inc., Bethlehem, PA) или набора для выделения плазмид Midi (Qiagen), следуя инструкциям производителей. Фрагменты ДНК выделяли с использованием набора для выделения из геля QIAQUICK™ (Qiagen) после электрофореза в агарозном геле с Трис-ацетатом. Скрининг колоний после всех реакций лигирования первоначально проводили посредством расщепления минипрепарата ДНК. Плазмидную ДНК избранных клонов секвенировали у коммерческого исполнителя секвенирования (Eurofins MWG Operon, Huntsville, AL). Данные о последовательности составляли и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
[0240] Репрезентативные плазмиды pDAB109350 и pDAB109360 показаны на фигуре 11 и фигуре 12 и подтверждены расщеплением рестрикционным ферментом и секвенированием ДНК.
Получение ДНК конструкций ZFN для трансфекции
[0241] Перед доставкой в протопласты Triticum aestivum плазмидную ДНК для каждой конструкции ZFN получали из культур E. coli с использованием системы PURE YIELD PLASMID MAXIPREP® (Promega Corporation, Madison, WI) или набора PLASMID MAXI® (Qiagen, Valencia, CA), следуя инструкциям производителей.
Выделение протопластов мезофилла пшеницы
[0242] Протопласты мезофилла из линии пшеницы cv. Bobwhite MPB26RH получали для трансфекции с использованием опосредованной полиэтиленгликолем (ПЭГ) доставки ДНК следующим образом.
[0243] Поверхность зрелых семян стерилизовали погружением в 80% (об./об.) этанол на 30 с, ополаскиванием дважды водопроводной водой с последующей промывкой 20% DOMESTOS® (0,8% об./об. активного хлора) в ротационном шейкере при 140 об./мин в течение 20 мин. DOMESTOS® удаляли декантацией, и семена ополаскивали четыре раза стерильной водой. Избыток воды удаляли помещением семян на фильтровальную бумагу WHATMAN™. Семена помещали в стерильную чашку ПЕТРИ™ на несколько листов увлажненной стерильной фильтровальной бумаги WHATMAN™ и инкубировали в течение 24 час при 24°C. После инкубации поверхность семян стерилизовали второй раз в 15% DOMESTOS® с встряхиванием 15 мин с последующим ополаскиванием стерильной водой, как описано ранее. Семена помещали на отвержденную среду Мурасиге и Скуга (MS) на 24 час при 24°C. Наконец, поверхность семян стерилизовали третий раз в 10% DOMESTOS® с встряхиванием 10 мин с последующим ополаскиванием стерильной водой, как описано ранее. Семена помещали, стороной бороздки вниз, на отвержденную среду MS с 10 семенами на чашку ПЕТРИ™ и проращивали в темноте при 24°C в течение 14-21 суток.
[0244] Приблизительно 2-3 грамм материала листьев из пророщенных семян нарезали длиной 2-3 см и помещали в предварительно взвешенную чашку ПЕТРИ™. Влагалища листьев и пожелтевший материал листьев выбрасывали. Приблизительно 10 мл ферментной смеси для расщепления листьев (0,6M маннит, 10 мМ MES, 1,5% масс./об. целлюлаза R10, 0,3% масс./об. мацерозим, 1 мМ CaCl2, 0,1% бычий сывороточный альбумин, 0,025% об./об. плюроновая кислота, 5 мМ β-меркаптоэтанол, pH 5,7) пипеткой вносили в чашку ПЕТРИ™, и материал листьев разрезали поперек на фрагменты 1-2 мм с использованием острого лезвия скальпеля. Материал листьев разрезали в присутствии смеси для расщепления листьев для предотвращения разрушения клеток в результате высыхания материала листьев. Дополнительную ферментную смесь для расщепления листьев добавляли в чашку ПЕТРИ™ до объема 10 мл на грамм массы свежего материала листьев и подвергали вакуумметрическому давлению (20 мм рт. ст.(2666 Па)) в течение 30 мин. Чашку ПЕТРИ™ герметично закрывали с помощью PARAFILM® и инкубировали при 28°C с осторожным ротационным встряхиванием в течение 4-5 часов.
[0245] Протопласты мезофилла, высвобожденные из фрагментов листьев в ферментную смесь для расщепления листьев, выделяли из растительного дебриса посредством пропускания расщепленной суспензии через сито 100 микрон и в 50 мл пробирку для сбора. Для максимизации выхода протопластов расщепленный материал листьев промывали три раза. Каждую промывку проводили добавлением 10 мл буфера для промывки (20 мМ KCl, 4 мМ MES, 0,6M маннит, pH 5,6) в чашку ПЕТРИ™, осторожным поворачиванием в течение 1 мин с последующим пропусканием буфера для промывки через сито 100 микрон в ту же самую 50 мл пробирку для сбора. Затем фильтрованную суспензию протопластов пропускали через сито 70 микрон, затем через сито 40 микрон. Затем аликвоты по 6 мл фильтрованной суспензии протопластов переносили в 12 мл круглодонные пробирки для центрифугирования с крышками и центирифугировали при 70 g и 12°C в течение 10 мин. После центрифугирования супернатант удаляли, и каждый из осадков протопластов ресуспендировали в 7 мл буфера для промывки. Протопласты осаждали второй раз центрифугированием, как описано выше. Каждый из осадков протопластов ресуспендировали в 1 мл буфера для промывки и объединяли до двух пробирок для центрифугирования. Объем буфера для промывки доводили до конечного объема 7 мл в каждой пробирке перед проведением центрифугирования, как описано выше. После удаления супернатанта осадки протопластов ресуспендировали в 1 мл буфера для промывки и объединяли до одной пробирки. Выход протопластов мезофилла оценивали с использованием гемоцитометра Нейбауэра. Краситель Эванса голубой использовали для определения соотношения выделенных живых клеток.
Опосредованная ПЭГ трансфекция протопластов мезофилла
[0246] Приблизительно 106 протопластов мезофилла добавляли в 12 мл круглодонную пробирку и осаждали центрифугированием при 70 g перед удалением супернатанта. Протопласты осторожно ресуспендировали в 600 мкл буфера для промывки, содержащего 70 мкг плазмидной ДНК. Плазмидная ДНК состояла из конструкций нуклеаз с цинковыми пальцами, описанных выше. Затем равный объем 40% раствора ПЭГ (40% масс./об. ПЭГ 4000, 0,8M маннит, 1M Ca(NO3)2, pH 5,6) медленно добавляли к суспензии протопластов с одновременным перемешиванием посредством осторожного вращения пробирки. Суспензии протопластов позволяли инкубироваться в течение 15 мин при комнатной температуре без какого-либо перемешивания.
[0247] Дополнительный объем 6 мл буфера для промывки медленно добавляли к суспензии протопластов в последовательных аликвотах 1 мл, 2 мл и 3 мл. Одновременное осторожное перемешивание использовали для поддержания гомогенной суспензии с каждой последующей аликвотой. Половину суспензии протопластов переносили во вторую 12 мл круглодонную пробирку, и дополнительный объем 3 мл буфера для промывки медленно добавляли в каждую пробирку с одновременным осторожным смешиванием. Протопласты осаждали центрифугированием при 70 g в течение 10 мин, и супернатант удаляли. Каждый из осадков протопластов ресуспендировали в 1 мл буфера для промывки перед объединением протопластов попарно из круглодонных пробирок в одну 12 мл пробирку. Дополнительные 7 мл буфера для промывки добавляли к объединенным протопластам перед центрифугированием, как описано выше. Супернатант полностью удаляли, и осадок протопластов ресуспендировали в 2 мл среды Цяо (0,44% масс./об. MS плюс витамины, 3 мМ MES, 0,0001% масс./об. 2,4-D, 0,6M глюкоза, pH 5,7). Суспензию протопластов переносили в стерильную 3 см чашку ПЕТРИ™ и инкубировали в темноте при 24°C в течение 72 час.
Выделение геномной ДНК из протопластов мезофилла
[0248] Трансфицированные протопласты переносили из 3 см чашки ПЕТРИ™ в 2 мл пробирку для микроцентрифугирования. Клетки осаждали центрифугированием при 70 g, и супернатант удаляли. Для максимизации выхода трансфицированных протопластов, чашку ПЕТРИ™ промывали три раза 1 мл буфера для промывки. Каждую промывку проводили поворачиванием буфера для промывки в чашке ПЕТРИ™ в течение 1 мин, с последующим переносом жидкости в ту же самую 2 мл пробирку для микроцентрифугирования. В конце каждой промывки клетки осаждали центрифугированием при 70 g, и супернатант удаляли. Осажденные протопласты быстро замораживали в жидком азоте перед лиофилизацией в течение 24 час в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированные клетки подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MINI (Qiagen), следуя инструкциям производителя, за исключением того, что разрушение тканей не требовалось, и клетки протопластов добавляли непосредственно в буфер для лизиса.
Анализ ПЦР геномной ДНК протопластов по расщеплению последовательности ZFN
[0249] Чтобы позволить исследование эффективности расщепления и специфичности для участков-мишеней ZFN, разработанных для локуса гена AHAS, разработаны праймеры для ПЦР для амплификации фрагмента вплоть до 300 п.о., внутри которого захвачены один или более участков-мишеней ZFN. Один из праймеров разработан, чтобы находиться в пределах окна 100 п.о. от захваченного участка-мишени(участков-мишеней) ZFN. Этот способ дизайна позволяет использование способа прочтения коротких фрагментов Illumina для оценки целостности участка-мишени в трансфицированных протопластах. Кроме того, праймеры для ПЦР разработаны для амплификации трех гомеологичных копий гена AHAS и для фиксирования варианта нуклеотидной последовательности, различающегося между гомеологами, так что прочтения последовательностей на Illumina можно однозначно приписывать субгеному, из которого они произошли.
[0250] Всего четыре набора праймеров для ПЦР разработано для амплификации локусов участков-мишеней ZFN (Таблица 4). Каждый набор праймеров синтезирован с последовательностями SP1 и SP2 Illumina на 5’-конце прямого и обратного праймера, соответственно, для обеспечения совместимости с химическими реактивами Illumina для секвенирования с коротким прочтением. Синтезированные праймеры содержали также фосфоротиоатную связь на предпоследних 5’- и 3’-нуклеотидах (указанных в таблице 4 как звездочка [*]). 5’-фосфоротиоатная связь позволяла защиту против деградации экзонуклеазами последовательностей SP1 и SP2 Illumina, в то время как 3’-фосфоротиоатная связь улучшала специфичность ПЦР для амплификации последовательностей-мишеней AHAS с использованием ПЦР вкл.-выкл. (Yang et al., (2005)). Все праймеры для ПЦР сконструированы, чтобы составлять в длину между 18 и 27 нуклеотидами и иметь температуру отжига 60-65°C, оптимально, 63°C.
[0251] В таблице 4 нуклеотиды, специфические для гена AHAS, указаны заглавным шрифтом; нуклеотиды, соответствующие последовательностям SP1 и SP2 Illumina, указаны строчным шрифтом. Каждый набор праймеров эмпирически тестировали по амплификации трех гомеологичных копий гена AHAS посредством секвенирования по Сенгеру продуктов амплификации ПЦР.
Последовательности праймеров, использованных для оценки эффективности и специфичности для участка-мишени расщепления AHAS ZFN
FN.F1
FN.R3
FN.F5
FN.R1
FN.F1
FN.R3
[0252] Амплификацию ПЦР локусов участков-мишеней ZFN с геномной ДНК, выделенной из трансфицированных протопластов мезофилла пшеницы, использовали для получения специфических для необходимых локусов молекул ДНК в правильном формате для способа секвенирования путем синтеза на основе Illumina. Каждый анализ ПЦР оптимизировали для работы с 200 нг исходной ДНК (приблизительно 12500 клеточных эквивалентов генома Triticum aestivum). Множество реакций проводили для трансфицированного образца для обеспечения анализа достаточного количества копий генома Triticum aestivum для надежной оценки эффективности и специфичности для участка-мишени ZFN. Приблизительно шестнадцать анализов ПЦР, эквивалентных 200000 копиям генома Triticum aestivum, взятым из отдельных протопластов, проводили для трансфицированного образца. Одну исходную смесь для ПЦР получали для каждого трансфицированного образца. Для обеспечения оптимальной амплификации ПЦР участка-мишени ZFN (т.е. для предотвращения того, чтобы реагенты для ПЦР становились лимитирующими, и для обеспечения того, чтобы ПЦР оставалась на стадии экспоненциальной амплификации) начальный анализ проводили с использованием способа количественной ПЦР для определения оптимального количества циклов для проведения для ткани-мишени. Начальную ПЦР проводили с необходимыми реакциями для отрицательного контроля на термоциклере MX3000P™ (Stratagene). По выходу данных, собранных в устройстве для количественной ПЦР, относительное увеличение флуоресценции наносили на график от цикла к циклу, и определяли количество циклов на анализ, которое может обеспечивать достаточную амплификацию, в то же время не позволяя реакции становиться лимитированной по реагентам, в попытке снизить излишнее количество циклов и сдвинутую амплификацию общераспространенных молекул. Неиспользованную исходную смесь оставляли на льду до завершения анализа количественной ПЦР и определения оптимального количества циклов. Затем оставшуюся исходную смесь аликвотировали на желаемое количество реакционных пробирок (приблизительно 16 на анализ ZFN), и амплификацию ПЦР проводили в течение оптимального количества циклов. После амплификации образцы одного и того же участка-мишени ZFN пулировали вместе, и по 200 мкл пулированного продукта на ZFN очищали с использованием набора для очистки продуктов ПЦР QIAQUICK MINIELUTE™ (Qiagen), следуя инструкциям производителя.
[0253] Для обеспечения возможности секвенирования образца с использованием способа прочтения коротких фрагментов Illumina, дополнительный цикл ПЦР проводили для введения последовательностей Illumina P5 и P7 в амплифицированные фрагменты ДНК, так же как последовательности индекса штрих-кода, который можно использовать для однозначного приписывания прочтений последовательностей образцу, из которого они произошли. Этого достигали с использованием праймеров, частично комплементарных последовательностям SP1 и SP2, добавленным на первом цикле амплификации, но также содержащих индекс образца и последовательности P5 и P7. Оптимальное количество циклов ПЦР, необходимых для добавления дополнительных последовательностей к матрице без избыточной амплификации общераспространенных фрагментов, определяли анализом циклов количественной ПЦР, как описано выше. После амплификации полученный продукт очищали с использованием магнитных бусин AMPURE® (Beckman-Coulter) с соотношением ДНК-к-бусинам 1:1,7. Очищенный фрагмент ДНК титровали для секвенирования способом прочтения коротких фрагментов Illumina с использованием набора на основе ПЦР для количественной оценки библиотек (КАРА) в соответствии с инструкциями производителя. Образцы подготавливали для секвенирования с использованием набора для формирования кластеров cBot (Illumina) и секвенировали на устройстве ILLUMINA GAIIx™ или HISEQ2000™ (Illumina) для получения прочтений парных концов 100 п.о. в соответствии с инструкциями производителя.
Анализ данных для детекции NHEJ в участках-мишенях ZFN
[0254] После получения данных прочтения коротких фрагментов Illumina для библиотек образцов, полученных для трансфицированных протопластов мезофилла, проводили биоинформатический анализ для идентификации делетированных нуклеотидов в участках-мишенях ZFN. Известно, что такие делеции являются показателями активности ZFN in planta, возникающей в результате репарации ДНК с соединением негомологичных концов (NHEJ).
[0255] Для идентификации прочтений последовательностей с делециями NHEJ поставленные производителем скрипты для переработки данных о последовательностях, полученных на устройстве HISEQ2000™ (Illumina), использовали для первого компьютерного приписывания прочтений коротких последовательностей образцу протопластов, из которого они произошли. Приписывание образцов было основано на последовательности индекса штрих-кода, которую вводили в ходе получения библиотеки, как описано ранее. Правильное приписывание образцов было обеспечено, поскольку индексы штрихкода 6 п.о., использованные для получения библиотек, отличались друг от друга по меньшей мере на две ступени различия последовательности.
[0256] После приписывания образцов, все последовательности пропускали через фильтр качества. Фильтр качества реализован в разработанном на заказ скрипте PERL. Прочтения последовательностей исключали, если присутствовало более трех сомнительных оснований, или если медиана показателя Phred составляла менее 20, или если присутствовало три или более последовательных оснований с показателем Phred менее 20, или если прочтение последовательности было короче, чем 40 нуклеотидов в длину.
[0257] Затем последовательности, прошедшие отсев по качеству, приписывали субгеному пшеницы, из которого они произошли. Этого достигали с использованием второго разработанного на заказ скрипта PERL, в котором приписывание субгеному пшеницы определяли по гаплотипу вариантов нуклеотидной последовательности, фиксированных посредством праймеров для ПЦР, использованных для амплификации трех гомеологичных копий гена AHAS, как описано выше.
[0258] Наконец, частоту делеций NHEJ в участке расщепления ZFN в приписанных субгеномам прочтениях последовательностей определяли для каждого образца с использованием третьего разработанного на заказ скрипта PERL и обработки данных вручную в Microsoft Excel 2010 (Microsoft Corporation). Этого достигали посредством подсчета частоты уникальных делеций NHEJ в каждом субгеноме в пределах каждого образца.
[0259] Два способа использовали для оценки эффективности расщепления и специфичности тестированных ZFN. Эффективность расщепления выражали (в частях на миллион прочтений) как долю приписанных субгеномам последовательностей, содержащих делецию NHEJ в участке-мишени ZFN. Порядок ранжирования ZFN по наблюдаемой для них эффективности расщепления использовали для идентификации ZFN с наилучшей активностью расщепления для каждой из четырех областей-мишеней генов AHAS специфическим для субгенома способом.
[0260] Для всех тестированных ZFN показано распределение размера делеций NHEJ, согласующееся с ожидаемой активностью ZFN in planta. Специфичность расщепления выражали как соотношение эффективностей расщепления, наблюдаемых среди трех субгеномов. Включение биологических повторов в анализ данных по существу не влияло на порядок ранжирования активности и специфичности расщепления тестированных ZFN.
[0261] По этим результатам ZFN, кодированные на плазмиде pDAB109350 (т.е. ZFN 29732 и 29730) и pDAB109360 (т.е. ZFN 30012 и 30018), выбраны для нацеливания in planta в последующих экспериментах, принимая во внимания их характеристики значительной активности расщепления геномной ДНК в каждом из трех субгеномов пшеницы.
Оценка дизайнов донора для опосредованного ZFN редактирования гена AHAS с использованием анализов временной трансфекции
[0262] Для исследования опосредованного ZFN геномного редактирования в эндогенном локусе гена AHAS в пшенице проводили серию экспериментов для оценки эффекта дизайна донора на эффективность направляемой соединением негомологичных концов (NHEJ) репарации ДНК. В этих экспериментах использовали анализы временной трансфекции для мониторирования эффективности опосредованного ZFN добавления описанной ранее мутации S653N, придающей устойчивость к гербицидам имидазолинонового класса (Li et al., (2008) Molecular Breeding 22:217-225), в эндогенном локусе гена AHAS в пшенице, или альтернативно, для опосредованного ZFN введения последовательности участка рестрикционной эндонуклеазы FcoRI в двухцепочечный разрыв ДНК, образованный в эндогенных генах AHAS посредством направленного расщепления ZFN.
Дизайны доноров для направляемой NHEJ репарации ДНК
[0263] Два типа дизайна донорной ДНК использовали для направляемой NHEJ репарации ДНК.
[0264] Первый тип дизайна донора представлял собой линейную, двухцепочечную молекулу ДНК, содержащую последовательность 41 п.о., разделяющую гомологию с эндогенными генами AHAS в пшенице. Разработаны две донорные молекулы ДНК, каждая для нацеливания на три гомеологичные копии гена AHAS. Обе донорные молекулы ДНК имели выступающие 5’- и 3’-концы для обеспечения выступающих концов для лигирования для облегчения опосредованной ZFN направляемой NHEJ репарации ДНК. Две донорные молекулы ДНК отличались последовательностью их выступающего 3’-конца. Первая донорная молекула ДНК, pDAS000152 (SEQ ID NO:175 и SEQ ID NO:176), разработана для обеспечения выступающих концов для лигирования, совместимых с выступающими концами, полученными расщеплением эндогенных генов AHAS ZFN 29732 и 29730 (кодированными на плазмиде pDAB109350), и для получения в результате вставки донорной молекулы 41 п.о. в эндогенный ген AHAS в участок двухцепочечного разрыва ДНК посредством направляемой NHEJ репарации ДНК. Вторая донорная молекула ДНК pDAS000149 (SEQ ID NO:177 и SEQ ID NO:178) разработана для обеспечения выступающих концов для лигирования, совместимых с выступающими концами, полученными двойным расщеплением эндогенных генов AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350) и ZFN 30012 и 30018 (кодированных на плазмиде pDAB 109360), и для получения в результате замены эндогенной последовательности AHAS, содержащейся между двумя двухцепочечными разрывами ДНК, образованными посредством ZFN, на донорную молекулу 41 п.о. посредством направляемой NHEJ репарации ДНК.
[0265] Второй тип донора представлял собой плазмидный ДНК-вектор, содержащий последовательность 41 п.о., не разделяющую гомологии с эндогенными генами AHAS в пшенице и фланкированную с обеих сторон последовательностью, узнаваемой ZFN, использованной(использованными) для получения двухцепочечных разрывов ДНК в эндогенных генах AHAS. Этот дизайн донора позволял высвобождение in planta уникальной последовательности 41 п.о. из молекулы плазмидной ДНК посредством той же (тех же) ZFN, использованных для расщепления участков-мишеней в эндогенных генах AHAS, и одновременное образование выступающих концов, пригодных для лигирования выступающих концов высвобожденной последовательности 41 п.о. в эндогенные гены AHAS посредством направляемой NHEJ репарации ДНК. Разработаны две плазмидные донорные молекулы ДНК, каждая для нацеливания на три гомеологичные копии гена AHAS. Первая плазмидная донорная молекула, pDAS000153 (SED ID NO:179 и SEQ ID NO:180) (фигура 13), разработана для обеспечения выступающих концов для лигирования на высвобожденном фрагменте ДНК 41 п.о., совместимых с выступающими концами, полученными расщеплением эндогенных генов AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350). Вторая плазмидная донорная молекула, pDAS000150 (SEQ ID NO:181 и SEQ ID NO:182) (фигура 14), разработана для обеспечения выступающих концов для лигирования на высвобожденном фрагменте ДНК 41 п.о., которые на одном конце являлись совместимыми с выступающими концами, полученными посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350), и на другом конце - совместимыми с выступающими концами, полученными посредством ZFN 30012 и 30018 (кодированных на плазмиде pDAB109360). Этот дизайн позволял замену эндогенной последовательности AHAS, содержащейся между двумя двухцепочечными разрывами ДНК, образованными посредством ZFN 29732 и 29730, и ZFN 30012 и 30018, на последовательность донорной молекулы 41 п.о.
Синтез донорной ДНК для направляемой NHEJ репарации ДНК
[0266] Стандартные способы клонирования, общеизвестные специалисту в данной области, использовали для получения плазмидных векторов. Перед доставкой в Triticum aestivum плазмидную ДНК для каждой донорной конструкции получали из культур E. coli с использованием системы PURE YIELD PLASMID MAXIPREP® (Promega Corporation, Madison, WI) или набора PLASMID MAXI KIT® (Qiagen, Valencia, CA), следуя инструкциям производителей.
[0267] Общепринятые фосфорамидитные химические реакции использовали для синтеза двухцепочечных донорных молекул ДНК (Integrated DNA Technologies, Coralville, IA). Для каждой донорной молекулы синтезировали пару комплементарных одноцепочечных олигомеров ДНК, каждый с двумя фосфоротиоатными связями на их 5’-концах для обеспечения защиты против деградации эндонуклеазами in planta. Одноцепочечные олигомеры ДНК очищали высокоэффективной жидкостной хроматографией для обогащения по полноразмерным молекулам и очищали от химических примесей после стадий синтеза с использованием Na+ обмена. Двухцепочечную донорную молекулу формировали посредством гибридизации эквимолярных количеств двух комплементарных одноцепочечных олигомеров ДНК с использованием стандартных способов, общеизвестных для специалиста в данной области. Перед доставкой в Triticum aestivum двухцепочечные молекулы ДНК разводили до необходимой концентрации в стерильной воде.
Выделение протопластов пшеницы, полученных из соматического эмбриогенного каллюса
[0268] Протопласты, полученные из соматического эмбриогенного каллюса (SEC) из донорной линии пшеницы cv. Bobwhite MPB26RH, подготавливали для трансфекции с использованием опосредованной полиэтиленгликолем (ПЭГ) доставки ДНК следующим образом:
[0269] Сеянцы донорной линии пшеницы выращивали в теплице в контролируемых условиях, поддерживаемых при 18/16°C (день/ночь) и длине светового дня 16/8 часов (день/ночь) с обеспечением освещения 800 ммоль на м2 в с. Колосья пшеницы собирали через 12-14 суток после цветения, и поверхность семян стерилизовали погружением на 1 мин в 70% (об./об.) этанол. Колосья обмолачивали, и незрелые семена стерилизовали в течение 15 мин в 17% (об./об.) отбеливателе с осторожным встряхиванием, с последующим ополаскиванием по меньшей мере три раза стерильной дистиллированной водой. Зародыши выделяли в асептических условиях из незрелых семян под препаровальной лупой. Ось зародыша удаляли с использованием острого скальпеля и выбрасывали. Щитки помещали в 9 см чашку ПЕТРИ™, содержащую среду 2-4 без TIMENTIN™, с неразрезанным щитком, ориентированным вверх. Всего 25 щитков высевали в каждую 9 см чашку ПЕТРИ™. Формирование соматического эмбриогенного каллюса (SEC) инициировали посредством инкубации в темноте при 24°C в течение 3 недель. Через 3 недели SEC отделяли от неэмбриогенного каллюса, помещали в свежую среду 2-4 без TIMENTIN™ и инкубировали в течение следующих 3 недель в темноте при 24°C. Субкультивирование SEC повторяли всего три раза перед использованием для получения протопластов.
[0270] Приблизительно один грамм SEC нарезали на куски 1-2 мм с использованием острого лезвия скальпеля в 10 см чашке ПЕТРИ™, содержащей приблизительно 10 мл смеси для расщепления каллюса пшеницы (2,5% масс./об. целлюлаза RS, 0,2% масс./об. пектолиаза Y23, 0,1% масс./об. DRISELASE®, 14 мМ CaCl2, 0,8 мМ MgSO4, 0,7 мМ KH2PО4, 0,6M маннит, pH 5,8) для предотвращения дегидратации каллюса. Дополнительную смесь для расщепления каллюса добавляли в чашку ПЕТРИ™ до объема 10 мл на грамм массы свежего каллюса и подвергали вакуумметрическому давлению (20 мм рт. ст.(2666 Па)) в течение 30 мин. Чашку ПЕТРИ™ герметично закрывали с помощью PARAFILM® и инкубировали при 28°C с осторожным ротационным встряхиванием при 30-40 об/мин в течение 4-5 часов.
[0271] Протопласты SEC, высвобожденные из каллюса, выделяли посредством пропускания расщепленной суспензии через сито 100 микрон и в 50 мл пробирку для сбора. Для максимизации выхода протопластов расщепленный материал каллюса промывали три раза. Каждую промывку проводили добавлением 10 мл буфера для промывки SEC (0,6M маннит, 0,44% масс./об. MS, pH 5,8) в чашку ПЕТРИ™, осторожным поворачиванием в течение 1 мин с последующим пропусканием буфера для промывки SEC через сито 100 микрон в ту же самую 50 мл пробирку для сбора. Затем фильтрованную суспензию протопластов пропускали через сито 70 микрон, затем через сито 40 микрон. Затем аликвоты по 6 мл фильтрованной суспензии протопластов переносили в 12 мл круглодонные пробирки для центрифугирования с крышками и центрифугировали при 70 g и 12°C в течение 10 мин. После центрифугирования супернатант удаляли, оставляя приблизительно 0,5 мл супернатанта, и каждый из осадков протопластов ресуспендировали в 7 мл 22% раствора сахарозы. На смесь сахарозы/протопластов осторожно наслаивали 2 мл буфера для промывки SEC, убеждаясь в отсутствии смешивания двух растворов. Протопласты центрифугировали второй раз, как описано выше. Полосу протопластов, видимую между буфером для промывки SEC и раствором сахарозы, собирали с использованием пипетки и помещали в чистую 12 мл круглодонную пробирку. Семь мл буфера для промывки SEC добавляли к протопластам, и пробирки центрифугировали, как описано выше. Супернатант удаляли, и протопласты SEC объединяли до одной пробирки и ресуспендировали в конечном объеме 1-2 мл буфера для промывки SEC. Выход протопластов SEC оценивали с использованием гемоцитометра Нейбауэра. Краситель Эванса голубой использовали для определения соотношения выделенных живых клеток.
Опосредованная ПЭГ трансфекция протопластов SEC
[0272] Приблизительно два миллиона протопластов SEC добавляли в 12 мл круглодонную пробирку и осаждали центрифугированием при 70 g перед удалением супернатанта. Протопласты осторожно ресуспендировали в 480 мкл буфера для промывки SEC, содержащего 70 мкг ДНК. ДНК состояла из конструкций ДНК нуклеаз с цинковыми пальцами и донорной ДНК, описанных выше, где каждая конструкция присутствует в молярном соотношении, необходимом для проведения эксперимента. Затем 720 мкл 50% раствора ПЭГ (50% масс./об. ПЭГ 4000, 0,8M маннит, 1M Ca(NO3)2, pH 5,6) медленно добавляли к суспензии протопластов с одновременным перемешиванием посредством осторожного вращения пробирки. Суспензии протопластов позволяли инкубироваться в течение 15 мин при комнатной температуре без какого-либо перемешивания.
[0273] Дополнительный объем 7 мл буфера для промывки SEC медленно добавляли к суспензии протопластов в последовательных аликвотах 1 мл, 2 мл и 3 мл. Одновременное осторожное перемешивание использовали для поддержания гомогенной суспензии с каждой последующей аликвотой. Половину суспензии протопластов переносили во вторую 12 мл круглодонную пробирку, и дополнительный объем 3 мл буфера для промывки SEC медленно добавляли в каждую пробирку с одновременным осторожным смешиванием. Протопласты осаждали центрифугированием при 70 g в течение 10 мин, и супернатант удаляли. Каждый из осадков протопластов ресуспендировали в 1 мл буфера для промывки SEC перед объединением протопластов попарно из круглодонных пробирок в одну 12 мл пробирку. Дополнительные 7 мл буфера для промывки добавляли к объединенным протопластам перед центрифугированием, как описано выше. Супернатант полностью удаляли, и осадок протопластов ресуспендировали в 2 мл среды Цяо. Суспензию протопластов переносили в стерильную 3 см чашку ПЕТРИ™ и инкубировали в темноте при 24°C в течение 72 час.
Выделение щитков из незрелых зиготных зародышей пшеницы
[0274] Щитки незрелых зиготных зародышей пшеницы из донорной линии пшеницы cv. Bobwhite MPB26RH подготавливали для трансфекции с использованием биолистически опосредованной доставки ДНК следующим образом.
[0275] Сеянцы донорной линии пшеницы выращивали в теплице в контролируемых условиях, поддерживаемых при 18/16°C (день/ночь) и длине светового дня 16/8 часов (день/ночь) с обеспечением освещения 800 ммоль на м2 в с. Колосья пшеницы собирали через 12-14 суток после цветения, и поверхность семян стерилизовали погружением на 1 мин в 70% (об./об.) этанол. Колосья обмолачивали, и незрелые семена стерилизовали в течение 15 мин в 17% (об./об.) отбеливателе с осторожным встряхиванием, с последующим ополаскиванием по меньшей мере три раза стерильной дистиллированной водой. Зародыши выделяли в асептических условиях из незрелых семян под препаровальной лупой. Ось зародыша удаляли с использованием острого скальпеля и выбрасывали. Щитки помещали в 9 см чашку ПЕТРИ™, содержащую осмотическую среду MS (E3 мальтоза), с неразрезанным щитком, ориентированным вверх. Всего 20 щитков высевали в каждую 9 см чашку ПЕТРИ™. Полученные зародыши предварительно культивировали в темноте при 26°C в течение минимум 4 час перед трансфекцией с использованием биолистически опосредованной доставки ДНК.
Трансфекция щитков из незрелых зиготных зародышей пшеницы посредством биолистически опосредованной доставки ДНК
[0276] Частицы золота для биолистически опосредованной доставки ДНК получали добавлением 40 мг частиц коллоидного золота 0,6 микрон (BioRad) к 1 мл стерильной воды в 1,5 мл микропробирке. Частицы золота ресуспендировали встряхиванием в течение 5 мин. Для получения достаточного количества материала для 10 бомбардировок, аликвоту 50 мкл суспензии частиц золота переносили в 1,5 мл микропробирку, содержащую 5 мкг ДНК, ресуспендированной в 5 мкл стерильной воды. После полного перемешивания встряхиванием, 50 мкл 2,5M CaCl2 и 20 мкл 0,1M спермидина добавляли в микропробирку, с полным перемешиванием после добавления каждого реагента. Покрытые ДНК частицы золота осаждали центрифугированием в течение 1 мин при максимальной скорости в настольной микроцентрифуге. Супернатант удаляли, и 1 мл 100% этанол добавляли для промывки и ресуспендирования частиц золота. Частицы золота осаждали центрифугированием, как описано выше, и супернатант выбрасывали. Покрытые ДНК частицы золота ресуспендировали в 110 мкл 100% этанола и сохраняли на льду. После краткого встряхивания 10 мкл раствора частиц золота посещали в центр мембраны макроносителя и позволяли высохнуть на воздухе.
[0277] Систему PDS-1000/HE PARTICLE GUN DELIVERY™ (BioRad) использовали для трансфекции щитков из незрелых зиготных зародышей пшеницы посредством биолистически опосредованной доставки ДНК. Доставку покрытых ДНК частиц золота осуществляли с использованием следующих установок: щель 2,5 см, апертура останавливающей пластины 0,8 см, расстояние до мишени 6,0 см, вакуум 91,4-94,8 кПа, скорость потока всасывания 5,0 и скорость потока выхода 4,5. Щитки из незрелых зиготных зародышей пшеницы подвергали бомбардировке с использованием разрывного диска 900 psi (6205 кПа). Каждую чашку ПЕТРИ™, содержащую 20 щитков, подвергали бомбардировке один раз. Щитки после бомбардировки инкубировали при 26°C в темноте в течение 16 час перед переносом на среду для индукции образования каллюса. Щитки культивировали на среде для индукции образования каллюса в темноте при 26°C в течение 7 суток.
Выделение геномной ДНК из протопластов SEC
[0278] Геномную ДНК выделяли из протопластов SEC с использованием способа, описанного ранее для протопластов мезофилла. Проводили дополнительную стадию очистки для уменьшения присутствия донорной ДНК, использованной для трансфекции. Этого достигали с использованием электрофореза в геле для отделения геномной ДНК из протопластов SEC от донорной ДНК, использованной для трансфекции. Выделенную ДНК подергали электрофорезу в течение 3 час в 0,5% агарозном геле с использованием 0,5X TBE. ДНК визуализировали посредством окрашивания SYBR® SAFE, и полосу, соответствующую геномной ДНК из протопластов SEC, вырезали. Геномную ДНК очищали из агарозного геля с использованием набора для очистки ДНК QIAQUICK™ (Qiagen), следуя инструкциям производителя, за исключением того, что колонку для очистки ДНК QIAQUICK™ ДНК заменяли колонкой для связывания ДНК из набора DNEASY PLANT DNA EXTRACTION MINI KIT™ (Qiagen).
Выделение геномной ДНК из щитков из незрелых зиготных зародышей
[0279] 20 трансфицированных щитков из незрелых зиготных зародышей пшеницы для каждой биолистически опосредованной доставки ДНК переносили в 15 мл пробирку и быстро замораживали в жидком азоте перед лиофилизацией в течение 24 час в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированные каллюсы подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MAXI™ KIT (Qiagen), следуя инструкциям производителя.
[0280] Проводили дополнительную стадию очистки для уменьшения присутствия донорной ДНК, использованной для трансфекции. Этого достигали с использованием электрофореза в геле для отделения геномной ДНК из каллюсов от донорной ДНК, использованной для трансфекции. Выделенную ДНК подергали электрофорезу в течение 3 час в 0,5% агарозном геле с использованием 0,5X TBE. ДНК визуализировали посредством окрашивания SYBR® SAFE, и полосу, соответствующую геномной ДНК из каллюсов, вырезали. Геномную ДНК очищали из агарозного геля с использованием набора для очистки ДНК QIAQUICK™ (Qiagen), следуя инструкциям производителя, за исключением того, что колонку для очистки ДНК QIAQUICK™ заменяли колонкой для связывания ДНК из набора DNEASY® PLANT DNA EXTRACTION MAXI™ (Qiagen).
Анализ ПЦР геномной ДНК для опосредованного ZFN редактирования AHAS
[0281] Для исследования для опосредованного ZFN геномного редактирования в эндогенных генах AHAS в пшенице с использованием направляемой NHEJ репарации ДНК и оценки эффекта дизайна донорной ДНК на эффективность каждого пути репарации ДНК, анализы ПЦР использовали для амплификации областей-мишеней AHAS из геномной ДНК трансфицированных клеток пшеницы. Анализы ПЦР проводили, как описано ранее, для получения специфических для необходимых локусов молекул ДНК в правильном формате для способа секвенирования путем синтеза на основе Illumina. Каждый анализ проводили с использованием ранее описанной пары праймеров (SEQ ID NO: 160 и SEQ ID NO: 170), разработанных для амплификации области-мишени для ZFN 29732 и 29730 (кодированных на плазмиде pDAB 109350), и ZFN 30012 и 30018 (кодированных на плазмиде pDAB109360) для каждой из трех гомеологичных копий генов AHAS. Множество реакций проводили для трансфицированного образца для обеспечения анализа достаточного количества копий генома Triticum aestivum для надежной оценки опосредованного ZFN редактирования генов. Для трансфицированных протопластов SEC вплоть до шестнадцати анализов ПЦР, эквивалентных 200000 копиям генома Triticum aestivum, полученным из отдельных протопластов, проводили для трансфицированного образца. Для трансфицированных щитков из незрелых зиготных зародышей, приблизительно сорок восемь (48) анализов ПЦР, эквивалентных 600000 копиям генома Triticum aestivum, полученным из отдельных протопластов, проводили для трансфицированного образца. Каждый трансфицированный образец подготавливали для секвенирования с использованием набора для формирования кластеров CBOT™ (Illumina) и секвенировали на устройстве ILLUMINA GAIIX™ или HISEQ2000™ (Illumina) для получения прочтений парных концов 100 п.о., как описано ранее.
Анализ данных для детекции опосредованного ZFN направляемого NHEJ редактирования генов AHAS
[0282] После получения данных прочтения коротких фрагментов Illumina для библиотек образцов, полученных для трансфицированных протопластов SEC и щитков из незрелых зиготных зародышей пшеницы, проводили анализы для идентификации доказательства на молекулярном уровне опосредованного ZFN направляемого NHEJ редактирования в участках-мишенях ZFN.
[0283] Для идентификации прочтений последовательностей с доказательством на молекулярном уровне направляемого NHEJ редактирования генов, сначала прочтения коротких последовательностей подвергали компьютерной обработке, как описано ранее, для приписывания каждого прочтения образцу и субгеному, из которого они произошли, и для проведения фильтрации по качеству, чтобы убедиться, что только последовательности высокого качества использованы для последующих анализов. Затем разработанные на заказ скрипты PERL и обработку данных вручную в Microsoft Excel 2010 (Microsoft Corporation) использовали для идентификации прочтений, содержащих последовательность как донорной молекулы ДНК, использованной для трансфекции, так и эндогенного локуса AHAS. Частоту редактирования (выраженную в частях на миллион прочтений) рассчитывали как долю приписанных субгеномам последовательностей, для которых показаны доказательства опосредованного ZFN направляемого NHEJ редактирования генов.
[0284] По результатам трех биологических повторов, выполненных для каждого дизайна линейной двухцепочечной ДНК-донора, получено доказательство на молекулярном уровне обогащения по прочтениям последовательностей, для которых показано опосредованное ZFN направляемое NHEJ редактирование в трех гомеологичных копиях эндогенных генов AHAS в пшенице (таблица 7 и таблица 8). Получены твердые доказательства на молекулярном уровне интеграции линейной, двухцепочечной донорной молекулы 41 п.о. в положении двухцепочечного разрыва ДНК, образованного посредством расщепления гомеологичных копий гена AHAS посредством ZFN 29732 и 29730 в образцах как протопластов SEC, так и щитков из незрелых зиготных зародышей, трансфицированных pDAB109350 и pDAS000152. Сходную эффективность редактирования наблюдали среди трех субгеномов пшеницы в этих образцах. В отличие от этого, в образцах протопластов SEC и щитков из незрелых зиготных зародышей, трансфицированных pDAB109350 и pDAS000153, показаны слабые доказательства опосредованного ZFN направляемого NHEJ редактирования генов, предположительно из-за необходимого условия высвобождения in planta донорной последовательности 41 п.о. из остова плазмиды. Доказательство на молекулярном уровне замены эндогенной последовательности AHAS на донорную молекулу 41 п.о. наблюдали как в протопластах SEC, так и в щитках из незрелых зиготных зародышей, трансфицированных pDAB109350, pDAB109360 и pDAS000149. Однако частота редактирования являлась значительно ниже той, которую наблюдали для трансфекций, проведенных с использованием pDAB109350 и pDAS000152, предположительно, из-за необходимости двойного расщепления ZFN эндогенной последовательности AHAS. Ограниченное доказательство получено для замены эндогенной последовательности AHAS на донорную молекулу 41-п.о., требующей высвобождения in planta из остова плазмиды в образцах протопластов SEC и щитков из незрелых зиготных зародышей, трансфицированных pDAB109350, pDAB109360 и pDAS000150.
Средняя частота редактирования NHEJ в частях на миллион (ppm) среди трех биологических повторов щитков, трансфицированных линейными двухцепочечными донорными ДНК различного дизайна. «na» обозначает «не применимо»
Средняя частота редактирования NHEJ в частях на миллион (ppm) среди трех биологических повторов протопластов SEC, трансфицированных линейными двухцепочечными донорными ДНК различного дизайна. «na» обозначает «не применимо»
[0285] Вместе результаты предоставляют твердое доказательство на молекулярном уровне точного опосредованного ZFN направляемого NHEJ редактирования в эндогенном локусе гена AHAS в пшенице. Эти результаты показывают, что на все три субгенома можно нацеливаться с помощью одной ZFN и донора. Результаты явно демонстрируют более высокую частоту редактирования для различного дизайна линейной донорной ДНК по сравнению с различным дизайном плазмидной донорной ДНК. Предположительно, эти результаты обусловлены необходимым условием линеаризации in planta плазмидных донорных молекул перед тем, как они смогут участвовать в направляемой NHEJ репарации ДНК. Результаты указывают также на то, что специфическое для субгенома опосредованное направляемое NHEJ редактирование гена облегчается двухцепочечным разрывом. ZFN, разработанные для индукции двухцепочечных разрывов ДНК, приводят к специфическому для субгенома опосредованному направляемому NHEJ редактированию гена при доставке с донорной ДНК в клетки растения Triticum aestivum.
Разработка системы трансформации для получения растений с редактированным AHAS
[0286] Эндогенный локус гена AHAS в пшенице выбран в качестве модельного локуса для разработки системы трансформации для получения растений с точными модификациями генома, индуцированными опосредованным ZFN редактированием гена. Эндогенный ген AHAS выбран в качестве модельного локуса из-за его способности образовывать поддающийся селекции фенотип (т.е. устойчивость к гербицидам группы В - ингибиторам ALS), известности необходимой информации о специфической для субгенома кодирующей последовательности гена и известности специфических мутаций, придающих устойчивость к гербицидам группы В, из характеризации пшеницы с химически индуцированными мутациями в генах AHAS. Мутация S653N, придающая устойчивость к гербицидам имидазолинонового класса, выбрана в качестве мишени для опосредованного ZFN редактирования гена из-за доступности коммерчески выпускаемых сортов пшеницы, несущих мутацию S653N, которые можно использовать в качестве положительного контроля для разработки системы химической селекции для обогащения по событиям точного редактирования.
Молекулярная характеризация Triticum aestivum cv. Clearfield Janz
[0287] Triticum aestivum cv. Clearfield Janz, коммерчески выпускаемый сорт пшеницы, несущий мутацию S653N в D-геноме, выбран для использования в качестве положительного контроля для разработки способа химической селекции для обогащения по растениям пшеницы с редактированным AHAS, полученными посредством опосредованного ZFN редактирования гена. Для получения генетически чистого исходного материала семян проводили скрининг 48 сеянцев с помощью 96 микросателлитных (SSR) маркеров с использованием способа ПЦР Multiplex-Ready (Hayden et al., (2008) BMC Genomics 9;80). Сеянцы с идентичными гаплотипами SSR использовали для получения семян, используемых в последующих экспериментах.
[0288] Чтобы убедиться, что растения пшеницы, используемые для получения семян, несут мутацию S653N, разработан анализ ПЦР для амплификации области гена AHAS, несущей мутацию, из D-генома пшеницы. Специфической для субгенома амплификации достигали с использованием ПЦР вкл.-выкл. (Yang et al., (2005) Biochemical and Biophysical Research Communications 328:265-72) с праймерами AHAS-PS-6DF2 и AHAS-PS-6DR2 (SEQ ID NO:183 и SEQ ID NO:184), разработанными для расположения предпоследнего основания (содержащего фосфоротиоатную связь) над вариантом нуклеотидной последовательности, различающимся между гомеологичными копиями генов AHAS. Праймеры для ПЦР сконструированы, чтобы составлять в длину между 18 и 27 нуклеотидами и иметь температуру отжига 60-65°C, оптимально, 63°C. Амплифицированные продукты ПЦР очищали с использованием набора для очистки продуктов ПЦР QIAQUICK MINIELUTE™ (Qiagen) и секвенировали с использованием способа прямого секвенирования по Сенгеру. Секвенируемые продукты очищали с помощью этанола, ацетата натрия и ЭДТА, следуя протоколу BIGDYE® v3.1 (Applied Biosystems), и электрофорез проводили на автоматической платформе для капиллярного электрофореза ABI3730XL®.
[0289] Анализ амплифицированных последовательностей гена AHAS с использованием SEQUENCHER v3.7™ (GeneCodes, Ann Arbor, MI) выявил сегрегацию по мутации S653N и позволил идентификацию растений, являвшихся гомозиготными (N653/N653) и гетерозиготными (N653/S653) по мутации S653N или гомозиготными (S653/S653) по аллелю чувствительности к гербициду. Сбор семян от отдельных растений обеспечивал источник семян, обладающих различным уровнем зиготности для мутации S653N на генетическом фоне cv. Clearfield Janz.
Оптимизация условий химической селекции на основе IMAZAMOX™
[0290] Проводили серию экспериментов для определения оптимальных условий селекции для регенерации растений пшеницы с редактированным AHAS. Эти эксперименты основаны на тестировании исходной переносимости IMAZAMOX™ донорной линии пшеницы cv. Bobwhite MPB26RH (генотип S653/S653) на стадиях индукции образования каллюса, регенерации растений и укоренения в разработанной системе трансформации пшеницы. Сходные эксперименты проводили для определения исходной переносимости и устойчивости генотипов cv. Clearfield Janz, несущих различные дозы мутации S653N; т.е., растений с генотипами N653/N653 и S653/S653.
[0291] Исходную переносимость донорной линии пшеницы cv. Bobwhite MPB26RH и исходную устойчивость генотипа (N653/N653) cv. Clearfield Janz для IMAZAMOX® на стадии индукции образования каллюса определяли следующим образом: щитки незрелых зиготных зародышей пшеницы из каждой линии пшеницы выделяли, как описано ранее, и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM, дополненную 0, 50, 100, 200, 300, 400 и 500 нМ IMAZAMOX®, соответственно. Двадцать щитков помещали в каждую чашку ПЕТРИ™. Всего 60 щитков для каждого из донорной линии пшеницы cv. Bobwhite MPB26RH и генотипа cv. Clearfield Janz тестировали по ответам исходной переносимости и исходной устойчивости, соответственно, при каждой концентрации IMAZAMOX®. После инкубации при 24°C в темноте в течение 4 недель регистрировали количество сформированного соматического эмбриогенного каллюса (SEC) при каждой концентрации IMAZAMOX®. Результаты показали, что формирование SEC для cv. Bobwhite MPB26RH являлось сниженным приблизительно на 70% при 100 нМ IMAZAMOX®, по сравнению с необработанными образцами. Формирование каллюса для генотипа cv. Clearfield Janz не было подвержено влиянию, по сравнению с контролем без обработки, при любых тестированных концентрациях IMAZAMOX®.
[0292] Исходную переносимость донорной линии пшеницы cv. Bobwhite MPB26RH для IMAZAMOX® на стадии регенерации растения определяли следующим образом: щитки незрелых зиготных зародышей пшеницы из каждой линии пшеницы выделяли, как описано ранее, и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM. Позволяли формирование соматического эмбриогенного каллюса посредством инкубации при 24°C в темноте в течение 4 недель. SEC переносили в 10 см чашки ПЕТРИ™, содержащие среду DRM, дополненную 0, 100, 200, 300, 400, 500 и 1000 нМ IMAZAMOX®, соответственно. Двадцать CIM помещали в каждую чашку ПЕТРИ™. Всего 60 CIM тестировали по ответам исходной переносимости при каждой концентрации IMAZAMOX®. После инкубации в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице, регистрировали ответ регенерации. Результаты показали, что регенерация растений являлась сниженной приблизительно на 80% при 200 нМ IMAZAMOX®, по сравнению с необработанными образцами.
[0293] Исходную переносимость генотипа cv. Clearfield Janz (S653/S653) и исходную устойчивость генотипа cv. Clearfield Janz (N653/N653) для IMAZAMOX® на стадии регенерации растения определяли с использованием модифицированного способа, поскольку для cv. Clearfield Janz наблюдали наличие слабого ответа регенерации растения (т.е., слабого эмбриогенеза) в культуре тканей. Семена для каждого генотипа cv. Clearfield Janz проращивали с использованием асептического способа, описанного выше для получения протопластов мезофилла пшеницы. Пророщенные сеянцы размножали in vitro посредством субкультивирования на среде для размножения. После размножения растения для каждого генотипа переносили в 10 см чашки ПЕТРИ™, содержащие среду для роста растений (MS +10 pM BA +0,8% агар), дополненную 0, 100, 300, 600, 900, 1200, 1500 и 3000 нМ IMAZAMOX®, соответственно. По десять растений помещали в каждую чашку ПЕТРИ™. Всего 30 растений на генотип тестировали по исходному ответу при каждой концентрации IMAZAMOX®. После инкубации в течение 3 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице регистрировали ответ роста. Результаты показали, что рост растений для генотипа (S653/S653) cv. Clearfield Janz являлся значительно сниженным на среде, содержащей по меньшей мере 200 нМ IMAZAMOX®, по сравнению с образцами без обработки. Этот ответ являлся сходным с ответом, наблюдаемым для генотипа (S653/S653) cv. Bobwhite MPB26RH. В отличие от этого, рост растений для генотипа (N653/N653) cv. Clearfield Janz не был сильно супрессирован, по сравнению с образцами без обработки, пока концентрация IMAZAMOX® не превышала 2000 нМ.
[0294] Исходную переносимость донорной линии пшеницы cv. Bobwhite MPB26RH для IMAZAMOX® на стадии укоренения растений определяли следующим образом: щитки незрелых зиготных зародышей пшеницы из донорной линии пшеницы выделяли, как описано ранее, и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM. Позволяли формирование соматического эмбриогенного каллюса посредством инкубации при 24°C в темноте в течение 4 недель. SEC переносили в 10 см чашки ПЕТРИ™, содержащие среду DRM, и инкубировали в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота), чтобы позволить произойти регенерации растений. Регенерированные растения переносили в 10 см чашки ПЕТРИ™, содержащие среду RM, дополненную 0, 100, 200, 300, 400, 500 нМ IMAZAMOX®, соответственно. По двадцать регенерированных растений помещали в каждую чашку ПЕТРИ™. Всего 60 регенерированных растений тестировали по исходному ответу переносимости при каждой концентрации IMAZAMOX®. После инкубации в течение 3 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице регистрировали ответ формирования корней. Результаты показали, что формирование корней являлось сильно ограниченными при всех тестированных концентрациях IMAZAMOX®, по сравнению с образцами без обработки.
[0295] Исходную переносимость генотипа (S653/S653) cv. Clearfield Janz и исходную устойчивость генотипа (N653/N653) cv. Clearfield Janz для IMAZAMOX® на стадии укоренения растений определяли с использованием модифицированного способа, поскольку для cv. Clearfield Janz наблюдали наличие слабого ответа регенерации растения (т.е. слабого эмбриогенеза) в культуре тканей. Семена для каждого генотипа cv. Clearfield Janz проращивали с использованием асептического способа, описанного выше для получения протопластов мезофилла пшеницы. Пророщенные сеянцы размножали in vitro посредством субкультивирования на среде для размножения. После размножения растения для каждого генотипа переносили в 10 см чашки ПЕТРИ™, содержащие среду для укоренения растений (1/2 MS, 0,5 мг/л NAA, 0,8% агар), дополненную 0, 50, 100, 200 и 250 нМ IMAZAMOX®, соответственно. По три растения помещали в каждую чашку ПЕТРИ™. Всего 6 растений на генотип тестировали по исходному ответу при каждой концентрации IMAZAMOX®. После инкубации в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице регистрировали ответ формирования корней.
[0296] Результаты показали, что формирование корней для генотипа (N653/N653) cv. Clearfield Janz являлось ограниченным, по сравнению с образцами без обработки, при 250 нМ IMAZAMOX®. формирование корней являлось серьезно ограниченным для генотипа (S653/S653) cv. Clearfield Janz при всех тестированных концентрациях IMAZAMOX®, по сравнению с образцами без обработки.
Дизайн и синтез донорной ДНК для опосредованного ZFN редактирования гена AHAS
[0297] Донорные молекулы ДНК разработаны для стимуляции точного опосредованного ZFN направляемого NHEJ редактирования гена в эндогенных генах AHAS в пшенице. Различный дизайн донора позволял введение мутации S653N, как известно, придающей переносимость гербицидов имидазолинонового класса (Li et al., (2008) Molecular Breeding 22:217-225).
[0298] Первый дизайн основан на интеграции двухцепочечной донорной молекулы 95 п.о. в положении двухцепочечного разрыва ДНК, образованного расщеплением гомеологичной копии эндогенного гена AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350). Донорная молекула ДНК, pDAS000267 (SEQ ID NO:423 и SEQ ID NO:424), содержала две части интегрирующего донорного полинуклеотида. 5’-конец содержал последовательность, почти идентичную эндогенному гену AHAS, кодированному в D-геноме, начиная от участка-мишени расщепления ZFN и заканчивая стоп-кодоном AHAS. Шесть целенаправленных мутаций введены в эту последовательность: две мутации, кодирующие мутацию S653N (AGC→AAT), и четыре мутации являлись синонимичными (в которых молчащая мутация введена в донорную последовательность). 3’-конец донорной молекулы содержал уникальную последовательность, которую можно использовать для диагностической ПЦР для детекции событий опосредованного ZFN направляемого NHEJ редактирования гена. Донорная молекула разработана с выступающими 5’- и 3’-концами для предоставления выступающих концов для лигирования для облегчения опосредованной ZFN направляемой NHEJ репарации ДНК.
[0299] Второй дизайн основан на замене эндогенной последовательности AHAS, локализованной между парой участков-мишеней ZFN, на двухцепочечную донорную молекулу 79 п.о. Конкретно, донор разработан для замены эндогенной последовательности AHAS, высвобожденной из хроматина после двойного расщепления гомеологичной копии гена AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB 109350) и ZFN 30012 и 30018 (кодированных на плазмиде pDAB109360). Донорная молекула, pDAS000268 (SEQ ID NO:425 и SEQ ID NO:426), содержала последовательность, почти идентичную эндогенному гену AHAS, кодированному в D-геноме, начиная от участка расщепления для ZFN 29732 и 29730, и заканчивая участком расщепления для ZFN 30012 и 30018. Десять целенаправленных мутаций введены в эту последовательность. Шесть мутаций являлись локализованными на 5’-конце донора: две мутации, кодирующие мутацию S653N (AGC→AAT), и четыре мутации являлись синонимичными. Четыре мутации являлись локализованными на 3’-конце донора и являлись локализованными в некодирующей последовательности. Донорная молекула разработана с выступающими 5’- и 3’-концами для предоставления выступающих концов для лигирования для облегчения опосредованной ZFN направляемой NHEJ репарации ДНК.
[0300] Общепринятые фосфорамидитные химические реакции использовали для синтеза двухцепочечных донорных молекул ДНК (Integrated DNA Technologies). Для каждой донорной молекулы синтезировали пару комплементарных одноцепочечных олигомеров ДНК, каждый с двумя фосфоротиоатными связями на их 5’-концах для обеспечения защиты против деградации эндонуклеазами in planta. Одноцепочечные олигомеры ДНК очищали высокоэффективной жидкостной хроматографией для обогащения по полноразмерным молекулам и очищали от химических примесей после стадий синтеза с использованием Na+ обмена. Двухцепочечную донорную молекулу формировали посредством гибридизации эквимолярных количеств двух комплементарных одноцепочечных олигомеров ДНК с использованием стандартных способов, общеизвестных для специалиста в данной области. Перед доставкой в Triticum aestivum двухцепочечные молекулы ДНК разводили до необходимой концентрации в стерильной воде.
Дизайн и получение бинарного вектора, кодирующего AHAS (S653N)
[0301] Стандартные способы клонирования использовали для конструирования бинарного вектора pDAS000143 (SEQ ID NO:185) (фигура 15). Кассета для экспрессии гена AHAS (S653N) состояла из промотора, 5'-нетранслируемой области и интрона из гена убиквитина (Ubi) из Zea mays (Toki et al., (1992) Plant Physiology 100; 1503-07) с последующей кодирующей последовательностью (1935 п.о.) гена AHAS из T. aestivum с мутацией пары оснований 1880 и 1181 от CG до AT для индукции замены аминокислоты с серина (S) на аспарагин (N) в аминокислотном остатке 653. Экспрессирующая кассета для AHAS содержала 3'-нетранслируемую область (UTR) гена нопалин-синтазы (nos) из A. tumefaciens pTi15955 (Fraley et al.,(1983) Proceedings of the National Academy of Sciences U.S.A. 80(15); 4803-4807). Кассета для селекции содержала промотор, 5'-нетранслируемую область и интрон из гена актина 1 (Act1) из Oryza sativa (McElroy et al., (1990) The Plant Cell 2(2); 163-171) с последующим синтетическим, оптимизированным для растений вариантом гена фосфинотрицинацетилтрансферазы (PAT), выделенного из Streptomyces viridochromogenes, кодирующим белок, придающий устойчивость к ингибиторам глутамин-синтетазы, включая фосфинотрицин, глуфосинат, и биалафос (Wohlleben et al., (1988) Gene 70(1); 25-37). Эта кассета заканчивалась 3'-UTR из гена 35S вируса мозаики цветной капусты (CaMV) (Chenault et al., (1993) Plant Physiology 101 (4); 1395-1396).
[0302] Кассету для селекции синтезировали у коммерческого исполнителя синтеза генов (GeneArt, Life Technologies) и клонировали в приспособленный для Gateway бинарный вектор с кассетой Gateway RfA, локализованной между геном убиквитина (Ubi) из Zea mays и 3'-нетранслируемой областью (UTR), содержащей терминатор транскрипции и участок полиаденилирования гена нопалин-синтазы (nos) из A. tumefaciens pTi15955. Кодирующую последовательность AHAS(S653N) амплифицировали с фланкирующих участков attB и субклонировали в pDONR221. Полученный клон ENTRY использовали в реакции LR CLONASEII™ (Invitrogen, Life Technologies) с приспособленным для Gateway бинарным вектором, кодирующим экспрессирующую кассету фосфинотрицинацетилтрансферазы (PAT). Первоначальный скрининг колоний клеток E. coli, трансформированных всеми реакционными смесями для лигирования, проводили по рестрикционному расщеплению минипрепарата ДНК. Рестрикционные эндонуклеазы получены из New England BioLabs и Promega. Выделение плазмид проводили с использованием набора QIAPREP SPIN MINIPREP™ или системы PURE YIELD PLASMID MAXIPREP™ (Promega Corporation, WI), следуя инструкциям производителя. Плазмидную ДНК выбранных клонов секвенировали с использованием ABI Sanger Sequencing и протокола циклического секвенирования BIG DYE TERMINATOR v3.1™ (Applied Biosystems, Life Technologies). Данные о последовательности составляли и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corporation, Ann Arbor, MI).
Система биолистически опосредованной трансформации для получения растений пшеницы с редактированным AHAS
[0303] Приблизительно 23000 щитков из незрелых зиготных зародышей донорной линии пшеницы cv. Bobwhite MPB26RH подготавливали для биолистически опосредованной доставки ДНК, как описано ранее. Покрытые ДНК частицы золота получали, как описано ранее, со следующими составами. Для трансфекций, проведенных с использованием pDAS000267, донорную ДНК смешивали в молярном соотношении 5:1 с плазмидной ДНК для pDAB109350 (кодирующей ZFN 29732 и 29730). Для трансфекций, проведенных с использованием pDAS000268, донорную ДНК смешивали в молярном соотношении 10:1:1 с плазмидной ДНК для pDAB109350 (кодирующей ZFN 29732 и 29730) и pDAB109360 (кодирующей ZFN 30012 и 30018). Трансфекции, проведенные с использованием pDAS000143, проводили с использованием частиц золота, покрытых только плазмидной ДНК для pDAS000143.
[0304] Биолистически опосредованные трансфекции проводили, как описано ранее. Всего 15620 щитков подвергали бомбардировке частицами золота, покрытыми ДНК, содержащей pDAS000267, всего 7310 щитков подвергали бомбардировке частицами золота, покрытыми ДНК, содержащей pDAS000268, и всего 2120 щитков подвергали бомбардировке частицами золота, покрытыми pDAS000143. После бомбардировки трансфицированные щитки инкубировали при 26°C в темноте в течение 16 час перед переносом на среду для индукции формирования каллюса.
[0305] Четыре различных способа химической селекции на основе IMAZAMOX® использовали для обогащения по регенерированным растениям пшеницы, имеющими мутацию S653N, точно интегрированную в одну или более гомеологичных копий эндогенного гена AHAS посредством опосредованного ZFN направляемого NHEJ редактирования гена. Четыре способа химической селекции описаны в таблице 9. Для каждого способа щитки культивировали в темноте на среде для индукции формирования каллюса при 24°C в течение 2 недель. Полученные каллюсы субкультивировали один раз в свежей среде для индукции формирования каллюса и сохраняли в тех е самых условиях в течение дополнительных двух недель. Соматический эмбриогенный каллюс (SEC) переносили в среду для регенерации растений и культивировали в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице. Регенерированные саженцы переносили в среду для укоренения и культивировали в тех же самых условиях в течение 2-3 недель. Для увеличения строгости селекции регенерированных растений, имеющих мутацию S653N, корни регенерированных растений удаляли, и растения снова субкультивировали на среде для укоренения в тех же самых условиях. Саженцы, укоренившиеся второй раз, переносили в почву и выращивали в условиях содержания в теплице. Семена T1 собирали от отдельных растений после помещения отдельных колосьев в пакеты для предотвращения ауткроссинга.
[0306] Эксплантаты щитков после бомбардировки частицами золота, покрытыми pDAS000143, использовали для мониторирования строгости селекции среди четырех способов химической селекции для регенерации растений пшеницы, несущих мутацию S653N AHAS. Растения, трансформированные pDAS000143, регенерировали с использованием способа, описанного выше.
Способы химической селекции, использованные для регенерации растений пшеницы, имеющих мутацию S653N, точно интегрированную в одну или более гомеологичных копий эндогенного гена AHAS посредством опосредованного ZFN направляемого NHEJ редактирования гена. (IMI = IMAZAMOX™)
[0307] Всего 14 растений пшеницы с предположительным опосредованным ZFN направляемым NHEJ редактированием AHAS выделили после трансфекции 22930 щитков из незрелых зиготных зародышей донорной линии пшеницы cv. Bobwhite MPB26RH. Растения с предположительным редактированием получены для всех четырех способов селекции для щитков после бомбардировки частицами золота, покрытыми ДНК, содержащей pDAS000267. Два растения с предположительным редактированием получено для второго способа селекции для щитков после бомбардировки частицами золота, покрытыми ДНК, содержащей pDAS000268. Всего 129 предположительно трансформированных растений пшеницы, несущих по меньшей мере одну случайным образом интегрированную копию донорного полинуклеотида AHAS (S653N), выделено для всех четырех способов химической селекции.
Молекулярная характеризация редактированных растений пшеницы
[0308] Растения пшеницы, полученные в результате бомбардировок донорным полинуклеотидом, кодирующим мутацию S653N, получали и характеризовали на молекулярном уровне для идентификации субгеномов пшеницы, содержащих интеграцию мутации S653N, происходящую в результате интеграции донора в геномный участок двухцепочечного расщепления. Проводили две серии бомбардировок. Первый набор экспериментов проводили с pDAS000143, и второй набор экспериментов проводили с pDAS000267 и pDAS000268. Отдельные растения пшеницы получали из обоих наборов экспериментов и анализировали посредством молекулярного способа для идентификации растений, содержащих интегрированную копию донорного полинуклеотида AHAS, кодирующего мутацию S653N.
[0309] Анализ гидролиза зонда (аналогичный анализу на основе TAQMAN®) для количественного анализа ПЦР использовали для подтверждения того, что восстановившиеся растения пшеницы, подвергнутые бомбардировке pDAS000143, несли по меньшей мере одну случайным образом интегрированную копию донорного полинуклеотида AHAS, кодирующего мутацию S653N. Подтверждение посредством анализа секвенирования по Сенгеру показало, что растения пшеницы, восстановившиеся после бомбардировок, проведенных с pDAS000267 и pDAS000268, содержали донорный полинуклеотид, содержащий S653N, по меньшей мере в одной из гомеологичных копий гена AHAS в положении, ожидаемом для опосредованного ZFN направляемого NHEJ редактирования гена.
Выделение геномной ДНК из регенерированных растений пшеницы
[0310] Геномную ДНК выделяли из лиофилизированной ткани листьев, собранной от каждого регенерированного растения пшеницы. Свежесобранную ткань листьев быстро замораживали в жидком азоте и лиофилизировали в течение 24 в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированный материал подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MINI KIT™ (Qiagen), следуя инструкциям производителя.
Анализ ПЦР для подтверждения случайной интеграции донорного полинуклеотида AHAS, кодирующего мутацию S653N
[0311] Для подтверждения того, что регенерированные растения пшеницы после бомбардировок, проведенных с pDAS000143, несли по меньшей мере одну случайным образом интегрированную копию донорного полинуклеотида AHAS, кодирующего мутацию S653N, дуплексный анализ гидролиза зонда qPCR (аналогичный TAQMAN®) использовали для амплификации эндогенного однокопийного гена пуроиндолина-b (Pinb) из D-генома гексаплоидной пшеницы (Gautier et al., (2000) Plant Science 153, 81-91; SEQ ID NO:186, SEQ ID NO:187 и SEQ ID NO:188 для последовательностей прямого и обратного праймеров и зонда, соответственно) и области промотора актина (Act1), присутствующей на pDAS000143 (SEQ ID NO:189, SEQ ID NO:190 и SEQ ID NO:191 для последовательностей прямого и обратного праймеров и зонда, соответственно). Анализы qPCR с гидролизом зонда проводили для 24 случайным образом выбранных растений пшеницы, выделенных для каждого из четырех способов химической селекции. Оценку присутствия и оценку количества копий pDAS00143 проводили способом, описанным в Livak and Schmittgen (2001) Methods 25(4):402-8.
[0312] Из результатов получено убедительное доказательство интеграции по меньшей мере одной копии донорного полинуклеотида AHAS, кодирующего мутацию S653N, в геном каждого из тестированных растений пшеницы. Эти результаты показывают, что четыре способа химической селекции обеспечивали строгую селекцию для восстановления растений, экспрессирующих мутацию S653N.
Анализ ПЦР геномной ДНК по опосредованному ZFN редактированию AHAS
[0313] Для характеризации субгеномной локализации и исходов опосредованного ZFN направляемого NHEJ редактирования гена в выделенных растениях пшеницы, ПЦР с праймерами AHAS_3F1 и AHAS_3R1 (SEQ ID NO:192 и SEQ ID NO:193) использовали для амплификации области-мишени с гомеологичных копий генов AHAS. Полученные продукты ПЦР клонировали в плазмидный вектор и секвенировали по Сенгеру с использованием химических реактивов BIGDYE® v3.1 (Applied Biosystems) на автоматической платформе для капиллярного электрофореза ABI3730XL®. Проводили секвенирование по Сенгеру вплоть до 120 независимых клонов плазмиды, чтобы убедиться, что каждый аллель в эндогенных гомеологах AHAS был секвенирован. Анализ последовательности, проведенный с использованием программного обеспечения SEQUENCHER™, использовали для получения консенсусной последовательности для каждого аллеля трех гомеологичных копий гена AHAS в каждом из выделенных растений пшеницы и для определения субгеномного происхождения и последовательности для каждого редактированного аллеля.
[0314] Из результатов убедительное доказательство точного опосредованного ZFN направляемого NHEJ редактирования гена в эндогенных локусах AHAS показано для 11 из 12 выделенных растений пшеницы, трансформированных с использованием pDAB109350 и pDAS000267 (таблица 10), и обоих выделенных растений пшеницы, трансформированных с использованием pDAB109350, pDAB109360 и pDAS000268 (таблица 11). Наблюдали растения с диапазоном исходов редактирования, включая: (1) независимые события с точным редактированием специфического для субгенома аллеля; (2) события с одиночным точным редактированием в A-геноме, B-геноме и D-геноме; (3) события с одновременным редактированием во множестве субгеномов; и (4) события, показывающие гемизиготное и гомозиготное редактирование специфического для субгенома аллеля. Впервые описан способ, который можно использовать для введения мутаций в локус гена во всех трех геномах растения пшеницы. Приведены примеры растений пшеницы, содержащих интегрированный донорный полинуклеотид AHAS, кодирующий мутацию S653N; интеграция последовательности полинуклеотида обеспечивает переносимость гербицидов имидазолинонового класса. Использование опосредованного ZFN геномного редактирования в эндогенном локусе гена в пшенице позволяет введение агрономических признаков (посредством мутации) без требующих больших затрат времени способов выведения пшеницы, требующих стадий обратного скрещивания и интрогрессии, в которых может быть увеличено количество времени, необходимое для интрогрессии признака во все три субгенома. Консенсусные последовательности по Сенгеру для аллелей, присутствующих в каждом субгеноме редактированных растений пшеницы, представлены как SEQ ID NO: 194-277 в таблицах 10 и 11.
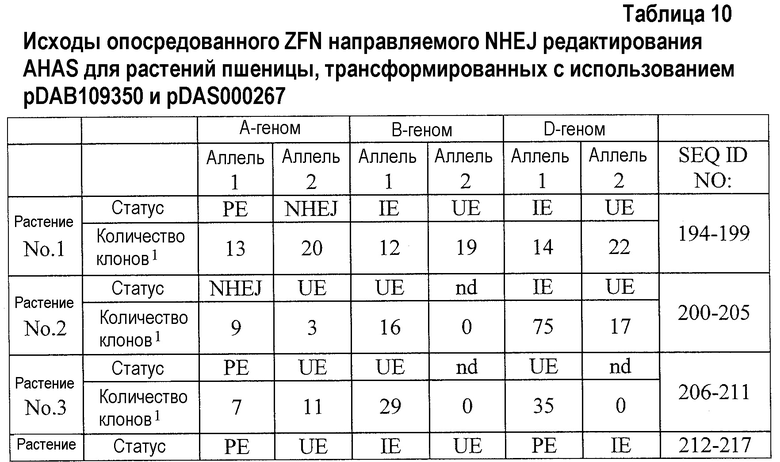
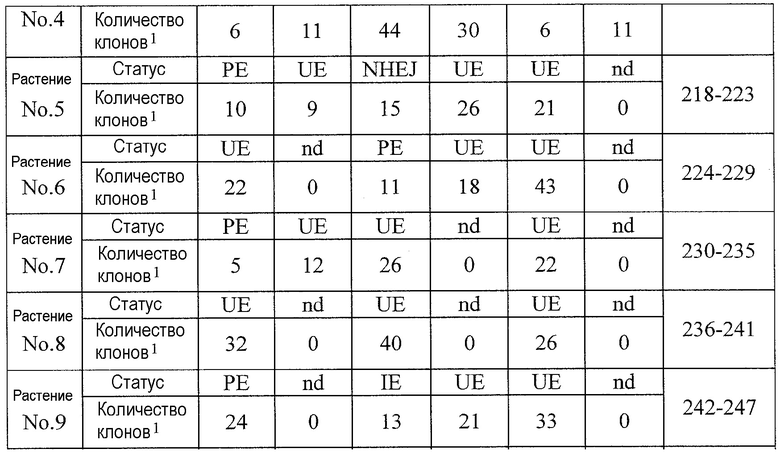

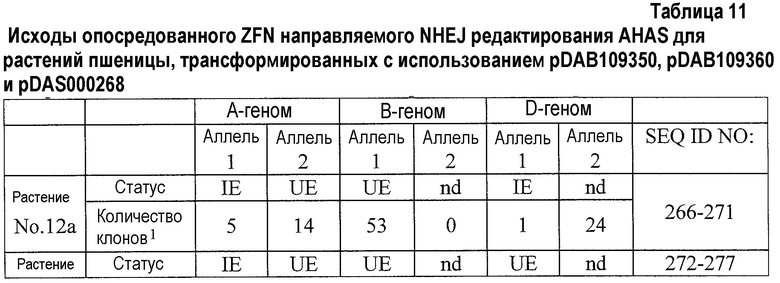

Дизайн связывающих доменов с цинковыми пальцами, специфических для области генов AHAS, кодирующей аминокислотный остаток P197
[0315] Белки с цинковыми пальцами, направленные против последовательности ДНК гомеологичных копий генов AHAS, разработаны, как описано ранее. Иллюстративная последовательность-мишень и спирали узнавания показаны в таблице 12 (дизайн областей спиралей узнавания) и в таблице 13 (участки-мишени). В таблице 13 нуклеотиды участка-мишени, контактирующие со спиралями узнавания ZFP, указаны заглавными буквами; не контактирующие нуклеотиды указаны строчными буквами. Участки-мишени нуклеазы с цинковыми пальцами (ZFN) разработаны выше (от 2 до 510 нуклеотидов выше) области гена AHAS, кодирующей аминокислотный остаток пролина 197 (P197).
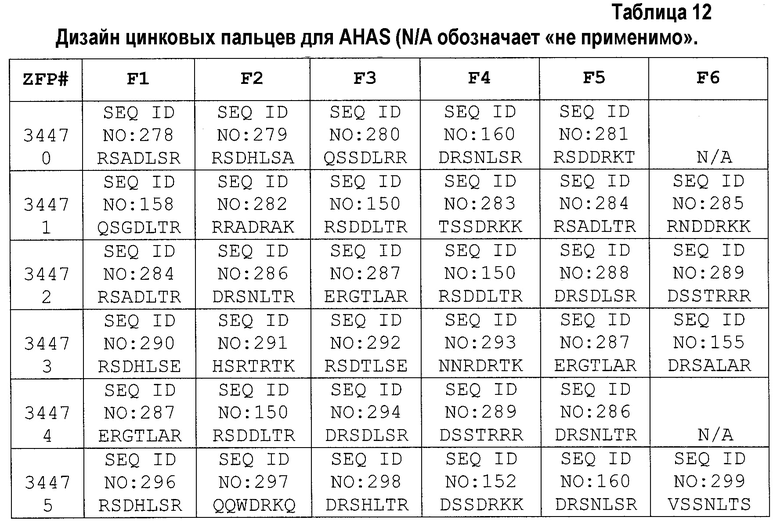
Участки-мишени цинковых пальцев в AHAS
cnGCGGCCATGGCGGCGGCGagggtttg
acCTCcCCCGCCGTCGCAttctcnggcg
ggCCGGACGCGCGGGCGtanccggacgc
cgTCGGCGTCTGCGTCGCCAcctccggc
acGCCGACGCGGCCgGACGCGcgggcgt
gcGTCGCCaCCTCCGGCCCGGgggccac
caGACGCCGACGCGGCCggacgcgcggg
gtCGCCACcTCCGGCCCGGGGgccacca
gcGACGCAGACGCCGACgcggccggacg
ccTCCGGCCCGGGGGCCaccaacctcgt
ggGATGGAGTCGAGGAGngcgtcngcga
tgGTCGCCATCACGGGCCAGgtcccccg
acCATGGGGATGGAGTCGAGgagngcgt
ccATСACGGGCCAGGTCccccgccgcat
caTCACGGGCCAGGTCCcccgccgcatg
[0316] Цинковые пальцы различного дизайна для AHAS вставляли в экспрессирующие цинковые пальцы векторы и верифицировали по активности расщепления с использованием системы почкующихся дрожжей, как описано ранее. Из множества ZFN, которые были разработаны, получены и тестированы по связыванию с предположительными геномными полинуклеотидными участками-мишенями AHAS, ZFN, описанные выше, идентифицированы как обладающие активностью in vivo на высоких уровнях и выбраны для дальнейших экспериментов. Эти ZFN разработаны для связывания трех гомеологичных AHAS и охарактеризованы как способные эффективно связывать и расщеплять уникальные геномные полинуклеотидные участки-мишени AHAS in planta.
Оценка расщепления генов AHAS нуклеазой с цинковыми пальцами с использованием анализов временной трансфекции
[0317] Сборка конструкции ZFN; Плазмидные векторы, содержащие экспрессирующие ZFN конструкции, верифицированные по активности расщепления с использованием системы дрожжей, разработаны и завершены, как описано ранее. Полученные плазмидные конструкции; pDAB111855 (ZFN 34470-2A-34471), pDAB111856 (ZFN 34472-2A-34473) и pDAB111857 (ZFN 34474-2A-34475) подтверждали расщеплением рестрикционными ферментами и секвенированием ДНК.
Получение ДНК конструкций ZFN для трансфекции
[0318] Перед доставкой в протопласты Triticum aestivum плазмидную ДНК для каждой конструкции ZFN получали из культур E. coli с использованием системы PURE YIELD PLASMID MAXIPREP® (Promega Corporation, Madison, WI) или набора PLASMID MAXI® (Qiagen, Valencia, CA), следуя инструкциям производителей.
Выделение и трансфекция протопластов мезофилла пшеницы
[0319] Протопласты мезофилла из донорной линии пшеницы cv. Bobwhite MPB26RH получали и трансфицировали с использованием опосредованной полиэтиленгликолем (ПЭГ) доставки ДНК, как описано ранее.
Анализ ПЦР геномной ДНК протопластов по расщеплению последовательности ZFN
[0320] Геномную ДНК выделяли из трансфицированных протопластов и использовали для анализов ПЦР для оценки эффективности расщепления и специфичности участков-мишеней ZFN, разработанных для области гена AHAS, кодирующей P197, как описано ранее. Пять наборов праймеров для ПЦР, содержащих фосфоротиоатную связь, как указано звездочкой [*], использовали для амплификации локусов участков-мишеней ZFN (таблица 14). Каждый набор праймеров конструировали согласно описанным ранее критериям.
Последовательности праймеров, использованных для оценки эффективности и специфичности для участка-мишени расщепления AHAS ZFN
Анализ данных для детекции NHEJ в участках-мишенях ZFN
[0321] После получения данных прочтения коротких фрагментов Illumina для библиотек образцов, полученных для трансфицированных протопластов мезофилла, проводили биоинформатический анализ (как описано выше) для идентификации делетированных нуклеотидов в участках-мишенях ZFN. Известно, что такие делеции являются показателями активности ZFN in planta, возникающей в результате репарации ДНК с соединением негомологичных концов (NHEJ).
[0322] Два способа использовали для оценки эффективности расщепления и специфичности тестированных ZFN. Эффективность расщепления выражали (в частях на миллион прочтений) как долю приписанных субгеномам последовательностей, содержащих делецию NHEJ в участке-мишени ZFN (таблица 15). Порядок ранжирования ZFN по наблюдаемой для них эффективности расщепления использовали для идентификации ZFN с наилучшей активностью расщепления для областей-мишеней генов AHAS специфическим для субгенома способом. Для всех тестированных ZFN показано распределение размера делеций NHEJ, согласующееся с ожидаемой активностью ZFN in planta. Специфичность расщепления выражали как соотношение эффективностей расщепления, наблюдаемых среди трех субгеномов.
[0323] По этим результатам ZFN, кодированные на плазмидах pDAB111855 (34470-2A-34471), pDAB111856 (34472-2A-34473) и pDAB111857 (34474-2A-34475), выбраны для нацеливания in planta в последующих экспериментах, принимая во внимания их характеристики значительной активности расщепления геномной ДНК в каждом из трех субгеномов пшеницы.
Получение доказательств на молекулярном уровне для опосредованного ZFN, свободного от экзогенных маркеров последовательного пакетирования трансгенов в эндогенном локусе AHAS с использованием анализов временной трансфекции
[0324] Получения доказательств на молекулярном уровне с использованием анализов временной трансфекции для опосредованного ZFN, последовательного, свободного от экзогенных маркеров пакетирования трансгенов в эндогенном локусе AHAS в геноме клеток Triticum aestivum посредством направляемой гомологией репарации ДНК достигали следующим образом.
[0325] Для растений пшеницы с редактированным AHAS (S653N), полученных посредством трансформации донором pDAS000267 и нуклеазой с цинковыми пальцами, кодированной на плазмиде pDAB109350, показана первая стадия последовательного, свободного от экзогенных маркеров пакетирования трансгенов в эндогенном локусе AHAS в геноме пшеницы. Эти редактированные растения используют для получения материала эксплантата (например, протопластов или щитков из незрелых зиготных зародышей) для трансфекции с использованием описанных ранее способов. Затем материал эксплантата подвергают совместной трансфекции с донорной молекулой ДНК и плазмидой, кодирующей ZFN (например, pDAB111855, pDAB111856 или pDAB111857), разработанную для нацеливания на участок связывания цинковых пальцев, локализованный в генах AHAS выше области, кодирующей аминокислотный остаток P197. ZFN расщепляет локус AHAS, и донорная молекула интегрирует в геном клеток Triticum aestivum посредством направляемой гомологией репарации. В результате опосредованной NHEJ интеграции донорной молекулы мутацию AHAS(P197S), придающую переносимость гербицидов класса сульфонилмочевины, вводят в эндогенную последовательность AHAS и одновременно, мутацию AHAS(S653N), введенную в первом цикле пакетирования трансгенов, удаляют. Следовательно, экспрессию эндогенного гена AHAS изменяют от придания переносимости имидазолинонов и чувствительности к сульфонилмочевинам (фенотип клеток пшеницы с правильным нацеливанием в первом цикле пакетирования трансгенов) до придания чувствительности к имидазолинонам и переносимости сульфонилмочевин, таким образом, позволяя регенерацию клеток с правильным нацеливанием с использованием средства для селекции сульфонилмочевины. Доказательство на молекулярном уровне интеграции донорной ДНК и получения клеток пшеницы с правильным нацеливанием подтверждают с использованием описанных ранее способов.
[0326] Специалистам в данной области понятно, что совместная трансформация клеток пшеницы донорной молекулой ДНК, содержащей один или более трансгенов, и плазмидой, кодирующей нуклеазу с цинковыми пальцами, позволяет параллельную (одновременную) или последовательную интеграцию трансгенов (пакетирование трансгенов) в геномы растений в точно такой же геномной локализации, включая одновременное редактирование множества аллелей в множестве геномов в полиплоидных видах растений.
Разработка системы трансформации для последовательного, свободного от экзогенных маркеров пакетирования трансгенов в эндогенных локусах AHAS в пшенице
[0327] Эндогенный ген AHAS в пшенице выбран в качестве модельного локуса для разработки опосредованной ZFN, свободной от экзогенных маркеров системы трансформации для получения растений с одним или несколькими трансгенами, точно расположенными в одной и той же геномной локализации. Система трансформации позволяет параллельное (одновременную интеграцию одного или более трансгенов) или последовательное пакетирование (последовательную интеграцию одного или более трансгенов) в точно такой же геномной локализации, включая одновременное параллельное или последовательное пакетирование множества аллелей в множестве субгеномов, посредством использования известных мутаций в гене AHAS, придающих переносимость гербицидов группы В. Опосредованную ZFN интеграцию донорной ДНК в локус AHAS дикого типа (чувствительного к гербицидам) используют для введения трансгена(трансгенов) и мутации в эндогенный ген AHAS, придающей переносимость имидазолинонов, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием средства для селекции имидазолинона. Пакетирования второго трансгена(трансгенов) в локусе AHAS достигают посредством интеграции донорной ДНК, которая вводит один или более дополнительных трансгенов и придает чувствительность к имидазолинонам, но переносимость сульфонилмочевин, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием средства для селекции сульфонилмочевины. Пакетирования третьего трансгена можно достигать посредством интеграции донорной молекулы, которая вводит следующий трансген(ы) и придает чувствительность к сульфонилмочевине и переносимость имидазолинонов, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием средства для селекции имидазолинона. В силу этого, продолжающиеся циклы последовательного пакетирования трансгенов возможны посредством использования донорной ДНК, которая вводит трансген(ы) и мутации в эндогенные гены AHAS для дифференциального чередования средств для селекции имидазолинона и сульфонилмочевины. Трансгены можно интегрировать в ген AHAS и пакетировать посредством пути NHEJ. Путь репарации и рекомбинации NHEJ можно определять дизайном донорного трансгена. В одном варианте осуществления трансгены, которые интегрируют и пакетируют в гене AHAS, можно разрабатывать, чтобы они содержали участки одиночного или двойного разрезания ZFN, фланкирующие переносимую область (например, мутацию AHAS и представляющий интерес ген). Соответственно, такой дизайн может использовать путь NHEJ для интеграции и пакетирования донорного полинуклеотида в хромосоме.
Получение низкокопийной, случайным образом интегрированной T-ДНК растений пшеницы с экспрессирующими AHAS(P197S) конструкциями
[0328] Бинарный вектор pDAS000164 (SEQ ID NO:326, фигура 16), содержащий кассеты для экспрессии AHAS(P197S) и селекции PAT, разрабатывали и собирали с использованием квалификации и способов, общеизвестных в данной области. Экспрессирующая кассета AHAS (P197S) состояла из промотора, 5'-нетранслируемой области и интрона из гена убиквитина (Ubi) из Zea mays (Toki et al., (1992) Plant Physiology, 100;1503-07) с последующей кодирующей последовательностью (1935 п.о.) гена AHAS из T. aestivum cv. Bobwhite MPB26RH с мутацией нуклеотида 511 от С до T для индукции замены аминокислоты с пролина (P) на серин (S). Экспрессирующая кассета для AHAS включала 3'-нетранслируемую область (UTR), содержащую ген нопалин-синтазы (nos) из A. tumefaciens pTi15955 (Fraley et al., (1983) Proceedings of the National Academy of Sciences U.S.A. 80(15): 4803-4807). Кассета для селекции содержала промотор, 5'-нетранслируемую область и интрон из гена актина 1 (Act1) из Oryza sativa (McElroy et al., (199) The Plant Cell 2(2): 163-171) с последующим синтетическим, оптимизированным для растений вариантом гена фосфинотрицинацетилтрансферазы (PAT), выделенного из Streptomyces viridochromogenes, кодирующим белок, придающий устойчивость к ингибиторам глутамин-синтетазы, включая фосфинотрицин, глуфосинат и биалафос (Wohlleben et al., (1988) Gene, 70(1): 25-37). Эта кассета заканчивалась 3'-UTR из гена 35S вируса мозаики цветной капусты (CaMV) (Chenault et al., (1993) Plant Physiology 101 (4): 1395-1396).
[0329] Кассету для селекции синтезировали у коммерческого исполнителя синтеза генов (например, GeneArt, Life Technologies и т.д.) и клонировали в приспособленный для GATEWAY® бинарный вектор с кассетой Gateway RfA, локализованной между геном убиквитина (Ubi) из Zea mays и 3’-нетранслируемой областью (UTR), содержащей терминатор транскрипции и участок полиаденилирования гена нопалин-синтазы (nos) из A. tumefaciens pTi15955. Кодирующую последовательность AHAS(P197S) амплифицировали с фланкирующих участков attB и субклонировали в pDONR221. Полученный клон ENTRY использовали в реакции LR CLONASEII® (Invitrogen, Life Technologies) с приспособленным для Gateway бинарным вектором, кодирующим экспрессирующую кассету фосфинотрицинацетилтрансферазы (PAT). Первоначальный скрининг колоний для всех собранных плазмид проводили по рестрикционному расщеплению минипрепарата ДНК. Рестрикционные эндонуклеазы получены из New England BioLabs (NEB; Ipswich, MA) и Promega (Promega Corporation, WI). Выделение плазмид проводили с использованием набора QIAPREP SPIN MINIPREP® (Qiagen, Hilden) или системы PURE YIELD PLASMID MAXIPREP® (Promega Corporation, WI), следуя инструкциям производителей. Плазмидную ДНК выбранных клонов секвенировали с использованием ABI Sanger Sequencing и протокола циклического секвенирования BIG DYE TERMINATOR V3.1® (Applied Biosystems, Life Technologies). Данные о последовательности составляли и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corporation, Ann Arbor, MI).
[0330] Полученным клоном pDAS000164 с бинарной экспрессией трансформировали Agrobacterium tumefaciens штамм EHA105. Трансгенные растения пшеницы со случайным образом интегрированной T-ДНК получали посредством опосредованной Agrobacterium трансформации с использованием донорной линии пшеницы cv. Bobwhite MPB26RH, следуя способу, сходному с способом Wu et al. (2008) Transgenic Research 17:425-436. Предположительные трансгенные объекты T0 для экспрессирующих AHAS (P197) экспрессирующих конструкций отбирали по переносимости фосфинотрицина (PPT), фенотипу, придаваемому селективным маркером PAT, и переносили в почву. Растения T0 выращивали в условиях содержания в теплице и получали семена T1.
[0331] Геномную ДНК из каждого растения T0 выделяли из ткани листьев, как описано ранее, и тестировали по присутствию Agrobacterium tumefaciens и по количеству интегрированных копий T-ДНК, кодирующих AHAS(P197S). Присутствие A. tumefaciens определяли с использованием дуплексного анализа гидролиза зонда qPCR (аналогичного TAQMAN™) для амплификации эндогенного гена убиквитина (SEQ ID NO:327, SEQ ID NO:328, и SEQ ID NO:329 для последовательностей прямого и обратного праймеров и зонда, соответственно) из генома пшеницы, и virC из pTiBo542 (SEQ ID NO:330, SEQ ID NO:331, и SEQ ID NO:332 для последовательностей прямого и обратного праймеров и зонда, соответственно). Количество интегрированных копий T-ДНК оценивали с использованием дуплексного анализа гидролиза зонда qPCR, как описано ранее, на основе пуроиндолина-b (Pinb) из D-генома гексаплоидной пшеницы и области промотора актина (Act1), присутствующей на pDAS000164. Всего получено 35 независимых объектов T0 с менее, чем тремя случайным образом интегрированными копиями T-ДНК.
Оптимизация условий химической селекции на основе сульфометуронметила
[0332] Проводили серии экспериментов для определения оптимальных условий селекции для регенерации растений пшеницы, экспрессирующих мутацию AHAS(P197S), придающую переносимость гербицидов класса сульфонилмочевины. Эти эксперименты основаны на тестировании исходной переносимости донорной линии пшеницы дикого типа cv. Bobwhite MPB26RH (генотип P197/P197, придающий чувствительность к сульфонилмочевине) на стадиях индукции образования каллюса, регенерации растений и укоренения в разработанной системе трансформации пшеницы. Сходные эксперименты проводили для определения исходной переносимости трансгенных объектов cv. Bobwhite MPB26RH, обладающих случайным образом интегрированной T-ДНК, экспрессирующей мутацию AHAS(P197), придающую переносимость средств для селекции сульфонилмочевин.
[0333] Исходную переносимость донорной линии пшеницы дикого типа для сульфометуронметила на стадии индукции образования каллюса определяли следующим образом: щитки незрелых зиготных зародышей пшеницы из каждой линии пшеницы выделяли, как описано ранее, и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM, дополненную 0, 100, 500, 1000, 1500 и 2000 нМ сульфометуронметила, соответственно. Двадцать щитков помещали в каждую чашку ПЕТРИ™. Всего 60 щитков тестировали при каждой концентрации сульфометуронметила. После инкубации при 24°C в темноте в течение 4 недель регистрировали количество сформированного соматического эмбриогенного каллюса (SEC) при каждой концентрации сульфометуронметила. Результаты показали, что трансформация SEC для cv. Bobwhite MPB26RH являлось сниженной приблизительно на 70% при 100 нМ сульфометуронметиле, по сравнению с необработанными образцами.
[0334] Исходную переносимость донорной линии пшеницы дикого типа для сульфометуронметила на стадии регенерации растения определяли следующим образом: щитки незрелых зиготных зародышей пшеницы из донорной линии пшеницы выделяли и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM. Позволяли формирование соматического эмбриогенного каллюса посредством инкубации при 24°C в темноте в течение 4 недель. SEC переносили в 10 см чашки ПЕТРИ™, содержащие среду DRM, дополненную 0, 100, 500, 1000, 1500, 2000, 2500 и 3000 нМ сульфометуронметила, соответственно. Двадцать CIM помещали в каждую чашку ПЕТРИ™. Всего 60 CIM тестировали по ответам исходной переносимости при каждой концентрации сульфометуронметила. После инкубации в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице, регистрировали ответ регенерации. Результаты показали, что регенерация растений являлась сниженной приблизительно на 80% при 2000 нМ сульфометуронметиле, по сравнению с необработанными образцами.
[0335] Исходную переносимость донорной линии пшеницы дикого типа для сульфометуронметила на стадии укоренения растений определяли следующим образом: щитки незрелых зиготных зародышей пшеницы выделяли и помещали в 10 см чашки ПЕТРИ™, содержащие среду CIM. Позволяли формирование соматического эмбриогенного каллюса посредством инкубации при 24°C в темноте в течение 4 недель. SEC переносили в 10 см чашки ПЕТРИ™, содержащие среду DRM, и инкубировали в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота), чтобы позволить произойти регенерации растений. Регенерированные растения переносили в 10 см чашки ПЕТРИ™, содержащие среду RM, дополненную 0, 100, 200, 250, 300, 400, 500, 1000 и 2000 нМ сульфометуронметила, соответственно. По десять регенерированных растений помещали в каждую чашку ПЕТРИ™. Всего 30 регенерированных растений тестировали по исходному ответу переносимости при каждой концентрации сульфометуронметила. После инкубации в течение 3 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице регистрировали ответ формирования корней. Результаты показали, что формирование корней являлось сильно ингибированным при концентрациях сульфометуронметила выше 400 нМ, по сравнению с образцами без обработки.
[0336] Исходную переносимость трансгенных объектов пшеницы, обладающей случайным образом интегрированной, низкокопийной (≤3) T-ДНК, экспрессирующей мутацию AHAS(P197S), для сульфометуронметила на стадии укоренения растений определяли следующим образом: четыре независимых трансгенных события выбирали случайным образом и размножали in vitro посредством субкультивирования на среде для размножения. После размножения, растения для каждого события переносили в 10 см чашки ПЕТРИ™, содержащие среду RM, дополненную 0, 400, 450, 500, 550 и 600 нМ сульфометуронметила, соответственно. Четыре растения (одно для каждого из четырех событий) помещали в каждую чашку ПЕТРИ™. Всего 3 растения на событие тестировали по исходной переносимости при каждой концентрации сульфометуронметила. После инкубации в течение 2 недель при 24°C при длине светового дня 16/8 часов (свет/темнота) в теплице регистрировали ответ формирования корней. Результаты показали, что формирование корней не являлось ограниченным, по сравнению с контролем без обработки, при любых тестированных концентрациях, что указывает на то, что мутация AHAS(P197S) придавала высокую переносимость сульфометуронметила.
Дизайн и синтез донорной ДНК для первого последовательного пакетирования трансгенов в эндогенном локусе AHAS с использованием направляемой NHEJ репарации ДНК
[0337] Донорная ДНК для первого цикла пакетирования трансгенов разработана для стимуляции точной интеграции донора в эндогенный локус AHAS посредством опосредованной ZFN направляемой NHEJ репарации. Дизайн основан на интеграции двухцепочечной донорной молекулы в положение двухцепочечного разрыва ДНК, образованного расщеплением гомеологичной копии эндогенного гена AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350). Донорная молекула (pDAS000433; SEQ ID NO:333, фигура 17) содержит несколько частей полинуклеотидных последовательностей. 5’-конец содержит последовательность, почти идентичную эндогенному гену AHAS, кодированному в D-геноме, начиная от участка-мишени расщепления ZFN и заканчивая стоп-кодоном AHAS. Семь целенаправленных мутаций введены в эту последовательность: две мутации, кодируют мутацию S653N, и пять оптимизированных по кодонам, синонимичных мутаций расположены на протяжении участка связывания ZFN 29732 для предотвращения повторного вырезания интегрированного донора. После стоп-кодона находится 316 п.о. некодирующей последовательности, соответствующей консервативной среди гомеологов AHAS 3’-нетранслируемой области (3’UTR). За последовательностью 3’UTR следуют участки связывания цинковых пальцев для ZFN 34480 и 34481 (кодированных на плазмиде pDAB111860) и ZFN 34482 и 34483 (кодированных на плазмиде pDAB111861). Эти участки связывания цинковых пальцев позволяют самовырезание происходящей из донора последовательности AHAS (кодирующей и 3’UTR), интегрированной в эндогенный локус, в ходе следующего цикла пакетирования трансгенов. За участками связывания цинковых пальцев для самовырезания следуют несколько дополнительных участков связывания цинковых пальцев (каждый из которых отделен 100 п.о. случайной последовательности), которые фланкируют два уникальных участка расщепления эндонуклеазой и которые позволяют вставку кассеты для экспрессии трансгена (например, экспрессирующей PAT кассеты, как описано ранее) в донорную молекулу. Дополнительные участки связывания цинковых пальцев позволяют будущее вырезание трансгенов, интегрированных в локус AHAS посредством последовательного свободного от маркеров пакетирования трансгенов, или продолжение последовательного пакетирования трансгенов в той же самой геномной локализации с использованием альтернативного способа пакетирования.
[0338] Донорную кассету синтезируют у коммерческого исполнителя синтеза генов (например, GeneArt, Life Sciences и т.д.) с коротким отрезком дополнительной фланкирующей последовательности на 5’- и 3’-концах для обеспечения возможности получения донорной молекулы с выступающими 5’- и 3’-концами, совместимыми с выступающими концами для лигирования, полученными посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350) при расщеплении эндогенного локуса AHAS. Донорную молекулу с выступающими 5’- и 3’-концами получают расщеплением плазмидной ДНК, содержащей донорную молекулу, рестрикционной эндонуклеазой BbsI с использованием общепринятых способов, известных специалисту в данной области.
Дизайн и синтез донорной ДНК для второго последовательного пакетирования трансгенов в эндогенном локусе AHAS с использованием направляемой NHEJ репарации ДНК
[0339] Донорная ДНК для второго цикла пакетирования трансгенов разработана для стимуляции точной интеграции донора в тот же самый локус AHAS, являвшийся мишенью первого пакетирования трансгенов, посредством опосредованной ZFN направляемой NHEJ репарации. Дизайн основан на интеграции двухцепочечной донорной молекулы в положение двухцепочечного разрыва ДНК, образованного расщеплением копии гена AHAS, содержащего первый пакетированный трансген, посредством ZFN 34480 и 34481 (кодированных на плазмиде pDAB111860) или ZFN 34482 и 34483 (кодированных на плазмиде pDAB111861). Донорная молекула (pDAS000434; SEQ ID NO:334, фигура 18) содержит несколько частей полинуклеотидных последовательностей. 5’-конец содержит последовательность, почти идентичную эндогенному гену AHAS, кодированному в D-геноме, начиная от участка-мишени расщепления ZFN и заканчивая стоп-кодоном AHAS. Несколько целенаправленных мутаций введены в эту последовательность: мутации, кодирующие мутацию P197S и оптимизированные по кодонам, синонимичные мутации, расположенные на протяжении участка связывания ZFN 34481 и 34483, для предотвращения повторного вырезания интегрированного донора. После стоп-кодона находится 316 п.о. некодирующей последовательности, соответствующей консервативной среди гомеологов AHAS 3’-нетранслируемой области (3’UTR). За последовательностью 3’UTR следуют участки связывания цинковых пальцев для ZFN 34474 и 34475 (кодированных на плазмиде pDAB111857) и ZFN 34476 и 34477 (кодированных на плазмиде pDAB111858). Эти участки связывания цинковых пальцев позволяют самовырезание происходящей из донора последовательности AHAS (кодирующей и 3’UTR), интегрированной в эндогенный локус, в ходе следующего цикла пакетирования трансгенов. За участками связывания цинковых пальцев для самовырезания следуют несколько дополнительных участков связывания цинковых пальцев (каждый из которых отделен 100 п.о. случайной последовательности), которые фланкируют уникальные участки расщепления эндонуклеазой и которые позволяют вставку кассеты для экспрессии трансгена (например, экспрессирующей DGT-28 кассеты, как описано в Патентной заявке номер 13757536). Дополнительные участки связывания цинковых пальцев позволяют будущее вырезание трансгенов, интегрированных в локус AHAS посредством последовательного свободного от маркеров пакетирования трансгенов, или продолжение последовательного пакетирования трансгенов в той же самой геномной локализации с использованием альтернативного способа пакетирования. Донорную кассету синтезируют у коммерческого исполнителя синтеза генов (например, Gene Art, Life Sciences) с коротким отрезком дополнительной фланкирующей последовательности на 5’- и 3’-концах для обеспечения возможности получения донорной молекулы с выступающими 5’- и 3’-концами, совместимыми с выступающими концами для лигирования, полученными посредством ZFN 34474 и 34475 (кодированных на плазмиде pDAB111857) или ZFN 34476 и 34477 (кодированных на плазмиде pDAB111858) при расщеплении эндогенного локуса AHAS. Донорную молекулу с выступающими 5’- и 3’-концами получают расщеплением плазмидной ДНК, содержащей донорную молекулу, рестрикционной эндонуклеазой BbsI с использованием общепринятых способов, известных специалисту в данной области.
Система трансформации для свободного от экзогенных маркеров, последовательного пакетирования трансгенов в эндогенном локусе AHAS в пшенице с использованием направляемой NHEJ репарации ДНК
[0340] Трансгенные объекты пшеницы с множеством трансгенов, пакетированных в одном и том же эндогенном локусе AHAS, получают посредством свободного от экзогенных маркеров, последовательного пакетирования трансгенов посредством трансформации донором pDAS000433 и ZFN 29732 и 29730 (кодированными на плазмиде pDAB109350). Точная опосредованная ZFN направляемая NHEJ интеграция донора вводит первый трансген и мутацию S653N, придающую переносимость имидазолинонов, в локус AHAS, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием IMAZAMOX® в качестве средства для селекции, как описано ранее. На фигуре 19a изображена интеграция. Последующая трансформация клеток пшеницы, полученных после первых событий пакетирования трансгена, донором pDAS000434 и ZFN 34480 и 34481 (кодированными на плазмиде pDAB111860) приводит к замене эндогенного хроматина, локализованного между участками связывания ZFN, расположенными выше P197 и в участке самовырезания, интегрированном в ходе первого пакетирования трансгена, на донорную молекулу. Это приводит к интеграции второго трансгена и мутации P197S, придающей переносимость сульфонилмочевины, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием сульфометуронметила в качестве средства для селекции. В то же самое время, интеграция второго донора удаляет мутацию S653N, таким образом, восстанавливая чувствительность к имидазолинонам (фигура 19B). Специалисту в данной области понятно, что пакетирования третьего трансгена можно достигать трансформацией подходящей нуклеазой с цинковыми пальцами и донора, который содержит дополнительный трансген и придает чувствительность к сульфонилмочевине и переносимость имидазолинонов, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием IMAZAMOX® в качестве средства для селекции. В силу этого, продолжающиеся циклы последовательного пакетирования трансгенов возможны посредством трансформации донорами, которые вводят трансгены и мутации в эндогенные гены AHAS для дифференциального чередования средств для селекции имидазолинона и сульфонилмочевины.
Дизайн и синтез донорной ДНК для первого последовательного пакетирования трансгенов в эндогенном локусе AHAS с использованием направляемой HDR репарации ДНК
[0341] Донорная ДНК для первого цикла пакетирования трансгенов разработана для стимуляции точной интеграции донора в эндогенный локус AHAS посредством опосредованной ZFN репарации. Дизайн основан на интеграции двухцепочечной донорной молекулы в положение двухцепочечного разрыва ДНК, образованного расщеплением гомеологичной копии эндогенного гена AHAS посредством ZFN 29732 и 29730 (кодированных на плазмиде pDAB109350). Донорная молекула (pDAS000435; SEQ ID NO: 335, Фигура 21) является идентичной по последовательности pDAS000433 (SEQ ID NO:333).
Система трансформации для свободного от экзогенных маркеров, последовательного пакетирования трансгенов в эндогенном локусе AHAS в пшенице с использованием направляемой HDR репарации ДНК
[0342] Трансгенные объекты пшеницы с множеством трансгенов, пакетированных в одном и том же эндогенном локусе AHAS, получают посредством свободного от экзогенных маркеров, последовательного пакетирования трансгенов посредством трансформации донором pDAS000435 и ZFN 29732 и 29730 (кодированными на плазмиде pDAB109350). Точная опосредованная ZFN направляемая HDR интеграция донора вводит первый трансген и мутацию S653N, придающую переносимость имидазолинонов, в локус AHAS, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием IMAZAMOX® в качестве средства для селекции, как описано ранее. На фигуре 20a изображена интеграция. Последующая трансформация клеток пшеницы, полученных после первых событий пакетирования трансгена, донором pDAS000436 и ZFN 34480 и 34481 (кодированными на плазмиде pDAB111860) приводит к замене эндогенного хроматина, локализованного между участками связывания ZFN, расположенными выше P197 и в участке самовырезания, интегрированном в ходе первого пакетирования трансгена, на донорную молекулу. Это приводит к интеграции второго трансгена и мутации P197S, придающей переносимость сульфонилмочевины, таким образом позволяя регенерацию растений с правильным нацеливанием с использованием сульфометуронметила в качестве средства для селекции. В то же самое время интеграция второго донора удаляет мутацию S653N, таким образом, восстанавливая чувствительность к имидазолинонам (Фигура 20b). Как очевидно специалисту в данной области, пакетирования третьего трансгена можно достигать трансформацией подходящей нуклеазой с цинковыми пальцами и донора, который содержит дополнительный трансген и придает чувствительность к сульфонилмочевине и переносимость имидазолинонов, таким образом, позволяя регенерацию растений с правильным нацеливанием с использованием IMAZAMOX® в качестве средства для селекции. В силу этого продолжающиеся циклы последовательного пакетирования трансгенов возможны посредством трансформации донорами, которые вводят трансгены и мутации в эндогенные гены AHAS для дифференциального чередования средств для селекции имидазолинона и сульфонилмочевины.
Искусственное скрещивание и молекулярный анализ для выделения трансгенных растений со специфическими комбинациями точных геномных модификаций
[0343] Объекты Triticum aestivum, полученные посредством трансформации конструкциями донорной ДНК и нуклеазы с цинковыми пальцами, имеют в результате интеграцию последовательности донорной молекулы в одну или более копий эндогенного локуса-мишени. Как показано ранее, опосредованная ZFN модификация генома может включать одновременное редактирование множества аллелей среди множества субгеномов. Искусственное скрещивание трансформированных объектов можно затем использовать для селекции специфических комбинаций точных геномных модификаций. Например, искусственное скрещивание полученных трансформированных объектов, имеющих гены AHAS с точной модификацией с мутацией S653N, можно использовать для получения растений пшеницы, имеющих мутацию S653N в конкретном субгеноме, в нескольких субгеномах или во всех трех субгеномах. Последующее искусственное скрещивание трансформированных объектов облегчает получение растений, имеющих специфические комбинации точных геномных модификаций. Специалист в данной области может использовать молекулярные анализы, такие как описано ранее, чтобы прослеживать наследование специфической геномной модификации в ходе искусственного скрещивания в последующих поколениях.
Пример 7: Направленная интеграция в ген десатуразы омега-3 жирных кислот (Fad3) Brassica napus и повреждение гена
Отбор связывающих доменов с цинковыми пальцами, специфических для Fad3C и Fad3A
[0344] Транскрибируемые области гомеологичных генов Fad3 идентифицированы и охарактеризованы, нуклеазы с цинковыми пальцами, разработанные для связывания и расщепления этих участков для опосредованного NHEJ нацеливания донорной последовательности, как описано в настоящем документе, разработаны и сконструированы. См. Предварительный Патент США с № подачи 61/697854, содержание которого приведено в настоящем документе в качестве ссылки. Белки с цинковыми пальцами (ZFP), направленные против последовательностей ДНК из гомеологов последовательностей Fad3, разработаны и тестированы, как описано ранее в Предварительном Патенте США с № подачи 61/697854. Для ZFN, обладающих целевой активностью, выбраны два белка с цинковыми пальцами, разрезающие мишень Fad3 с высокой эффективностью: ZFP 28051-2A-28052 узнает SEQ ID NO:336 5’-GCCCAAGGAACCCTTTTCTGGGCCATCTTCGTACTCGGCCACGACTGGTAATTTAAT-3’ и, как показано ранее, специфически связывает и расщепляет геномный локус Fad3C. Подобным образом, белок с цинковыми пальцами 28053-2A-28054 узнает SEQ ID NO:337 5’-AGCGAGAGAAAGCTTATTGCAACTTCAACTACTTGCTGGTCGATCGTGTTGGCCACTC-3’ и, как показано ранее, специфически связывает и расщепляет геномный локус Fad3A и Fad3C. Нуклеотиды в участках-мишенях, контактирующие со спиралями узнавания ZFP, показаны в таблице 16.


Дизайн и конструирование экспрессирующих векторов, кодирующих нуклеазы с цинковыми пальцами, специфические для Fad3C и Fad3A
[0345] Цинковые пальцы различного дизайна для Fad3 вставляли в экспрессирующие цинковые пальцы векторы, кодирующие белок, имеющий по меньшей мере один палец со структурой CCHC (Патентная публикация США № 2008/0182332). В частности, последний палец в каждом белке имел остов CCHC для спирали узнавания. Неканонические кодирующие цинковые пальцы последовательности сливали с доменом нуклеазы рестрикционного фермента типа IIS FokI (аминокислоты 384-579 последовательности из Wah et al., (1998) Proc. Natl. Acad. Sci. USA 95:10564-10569) посредством линкера ZC из четырех аминокислот и сигнала ядерной локализации sop2. Кодирующую самогидролизующийся 2A нуклеотидную последовательность из вируса Thosea asigna (Szymczak et al., 2004) добавляли между двумя слитыми белками нуклеаз с цинковыми пальцами, клонированными в конструкцию. Экспрессией ZFN управляли посредством сильного конститутивного промотора и 5'-нетранслируемой области (UTR) из вируса мозаики прожилок маниоки (Verdaguer et al, Plant Molecular Biology 1996, 31(6); 1129-1139), и она фланкирована 3'-UTR (включая терминатор транскрипции и участок полиаденилирования) из открытой рамки считывания 23 (ORF23) Agrobacterium tumefaciens pTi15955 (Barker et al., Plant Molecular Biology 1983, 2(6); 335-50).
[0346] Векторы собирали с использованием способа IN-FUSION™ Advantage (Clontech, Mountain View, CA). Рестрикционные эндонуклеазы получены из New England BioLabs (NEB; Ipswich, MA), и T4 ДНК-лигазу (Invitrogen) использовали для лигирования ДНК. Выделения плазмиды проводили с использованием набора для выделения плазмид NUCLEOSPIN® (Macherey-Nagel Inc., Bethlehem, PA) или набора для выделения плазмид MIDI™ (Qiagen), следуя инструкциям производителей. Фрагменты ДНК выделяли с использованием набора для выделения из геля QIAQUICK™ (Qiagen) после электрофореза в агарозном геле с Трис-ацетатом. Скрининг колоний собранных плазмид первоначально проводили посредством расщепления минипрепарата ДНК. Плазмидную ДНК избранных клонов секвенировали у коммерческого исполнителя секвенирования (Eurofins MWG Operon, Huntsville, AL). Данные о последовательности составляли и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes, Ann Arbor, MI). Полученные плазмидные конструкции: pDAB107827 (ZFN 28051-2A-28052, фигура 22, SEQ ID NO:350) и pDAB107828 (ZFN 28053-2A-28054, фигура 23, SEQ ID NO:351) подтверждали посредством расщепления рестрикционными ферментами и посредством секвенирования ДНК.
Дизайн и конструирование «донорных» векторов для направляемой NHEJ репарации ДНК
[0347] Выполняли два способа интеграции ДНК в Fad3; сплайсинг гена, где экспрессирующую кассету вставляли в один индуцированный ZFN двухцепочечный разрыв, и редактирование гена, где часть гена удаляли посредством использования двух индуцированных ZFN двухцепочечных разрывов и экспрессирующую кассету вставляли для репарации пропуска.
[0348] Для каждого способа интеграции, сплайсинга гена или редактирования гена, конструировали два вектора. Первый кодировал экспрессирующую кассету гена turboGFP (tGFP), и второй кодировал экспрессирующую кассету гена для придания устойчивости к антибиотику гигромицину. Экспрессирующая кассета tGFP состояла из промотора, 5'-нетранслируемой области и интрона из гена полиубиквитина 10 (UBQ10) из Arabidopsis thaliana (Norris et al, Plant Molecular Biology 1993, 21(5), 895-906) с последующей кодирующей последовательностью tGFP (Evrogen, Moscow, Russia). Кодирующая последовательность tGFP была оптимизирована по кодонам для экспрессии в двудольных растениях, и 3'-нетранслируемая область (UTR) содержала терминатор транскрипции и участок полиаденилирования из открытой рамки считывания 23 (ORF23) A. tumefaciens pTi15955 (Barker et al, Plant Molecular Biology 1983, 2(6), 335-50). Экспрессирующая кассета гена устойчивости к гигромицину состояла из промотора 19S, включая 5'-UTR, из вируса мозаики цветной капусты (CaMV) (Cook and Penon Plant Molecular Biology 1990 14(3), 391-405) с последующим геном гигромицинфосфотрансферазы (hph) (Raster et al Nucleic Acids Research 1983 11 (19), 6895-6911). Ген hph был оптимизирован по кодонам для экспрессии в двудольных растениях и фланкирован 3’UTR, содержащей терминатор транскрипции и участок полиаденилирования из открытой рамки считывания 1 (ORF1) A. tumefaciens pTi15955 (Barker et al, Plant Molecular Biology 1983, 2(6), 335-50). Обе кассеты синтезированы у коммерческого исполнителя синтеза генов (GeneArt, Life Technologies, Regensberg, Germany).
[0349] Векторы для сплайсинга гена конструировали посредством клонирования двух тандемных копий последовательности узнавания ZFN, являющейся мишенью для ZFN, кодированной в векторе pDAB10782. Векторы для редактирования гена конструировали посредством клонирования каждой из последовательностей узнавания ZFN, являющихся мишенью для ZFN, кодированных в векторах pDAB107827 и pDAB107828. В обоих случаях две последовательности узнавания ZFN разделены последовательностями узнавания рестрикционных эндонуклеаз ВаmHI и NotI. Кассеты tGFP и HPH клонировали в участки ВаmHI и NotI каждого вектора, получая в результате четыре «донорных» вектора: pDAS000340 (устойчивый к гигромицину донор для сплайсинга гена: SEQ ID NO:352, фигура 24), pDAS000341 (донор для сплайсинга с репортерным геном tGFP: SEQ ID NO:353, фигура 25), pDAS00342 (устойчивый к гигромицину донор для редактирования гена: SEQ ID NO:354, фигура 26) и pDAS000343 (донор для редактирования гена с репортерным геном tGFP: SEQ ID NO:355, фигура 27).
[0350] Первоначальный скрининг колоний собранных плазмид проводили по расщеплению рестрикционными эндонуклеазами ДНК, выделенной из ночных культур E. coli. Рестрикционные эндонуклеазы получены из New England BioLabs (NEB, Ipswich, MA) и Promega (Promega Corporation, WI). Выделение плазмид проводили с использованием набора QIAPREP SPIN MINIPREP™ (Qiagen, Hilden, Germany) или системы PURE YIELD PLASMID MAXIPREP SYSTEM™ (Promega Corporation, WI), следуя инструкциям производителей. После подтверждения рестрикционных фрагментов посредством электрофореза полученных фрагментов в агарозном геле, плазмидную ДНК выбранных клонов секвенировали с использованием ABI Sanger Sequencing и протокола циклического секвенирования BIG DYE TERMINATOR Y3.1™ (Applied Biosystems, Life Technologies). Данные о последовательности составляли и анализировали с использованием программного обеспечения Sequencher™ (Gene Codes, Ann Arbor, MI).
Поддержание растительного материала для выделения протопластов
[0351] Протопласты мезофилла выделяли из стерильных культур побегов Brassica napus (DFU0275) в возрасте трех недель. Соответствующие семена проращивали, следуя способам, описанным в настоящем документе. Поверхность семян стерилизовали с использованием 70% этанола в течение 1 минуты и осторожно встряхивали с последующими 3-4 ополаскиваниями в стерильной дважды дистиллированной воде. Затем семена стерилизовали с использованием 20% отбеливателя и 10 мкл Tween 20. Затем семена дополнительно обрабатывали отбеливателем в настольном встряхивателе приблизительно при 100 об/мин, в течение 15 минут с последующими 3-4 ополаскиваниями в стерильной дважды дистиллированной воде, семена осторожно переносили на стерильную фильтровальную бумагу для удаления избыточной влажности и высевали на среду для проращивания семян (1/2 концентрации MS/B5 витамины + 1% сахароза + 0,8% агар; pH 5,8.
[0352] Приблизительно 50-60 мл среды наливали в каждую чашку ПЕТРИ™ (15×100 мм) и помещали под небольшим углом с использованием подставки). Приблизительно по 50 семян помещали в чашку. Чашки инкубировали стоящими прямо при 22°C при свете 16 час/сутки (20 мкмоль м-2 с-1) в течение 6 суток. Фрагменты гипокотиля размером 0,5 см нарезали из сеянцев в возрасте шести суток и культивировали на среде для индукции образования побегов (MS/B5 витамины + 3% сахароза + 500 мг/л MES + BAP (13 мкМ) + зеатин (5 мкМ) + нитрат серебра (5 мг/л) + 0,8% агар (pH 5,8). Среду наливали в стерильную чашку ПЕТРИ™ 100×20 мм; приблизительно по 20 эксплантатов помещали в чашку. Меристему побегов, появившуюся через 3-4 недели, переносили в среду для удлинения побегов (MS/B5 витамины + 2% сахароза + 500 мг/л MES + BAP (2 мкМ) + GA-3 (0,1 мкМ) + 0,8% агар (pH 5,8) и разлита в 250 мл культуральные флаконы), и культуры поддерживали в этой среде в течение 4 недель с одним циклом субкультивирования между ними. Затем побеги высотой 2-3 см переносили в среду для инициации образования корней (1/2 концентрации MS/B5 витамины + 1% сахароза + 500 мг/л MES + IBA (2,5 мкМ) + 0,6% агар (pH 5,8) и разлита в 700 мл культуральные флаконы) для развития корней. Укоренившиеся побеги субкультивировали в свежей среде для инициации образования корней с интервалами 3-4 недели в качестве срезов побегов в течение двух-трех перед использованием. Культуры поддерживали на протяжении этого при 22°C при свете 16 час/сутки (30 мкмоль м-2 с-1).
Выделение и очистка протопластов мезофилла
[0353] Выращенные in vitro растения Brassica napus DH12075 использовали в качестве источника эксплантата для выделения протопластов мезофилла. Для выделения протопластов от 3-го до 4-го верхних полностью развившихся листьев из саженцев в возрасте 3-4 недель нарезали острым скальпелем на небольшие полоски (0,5-1 мм) для выделения протопластов. Ферментативное расщепление проводили посредством обработки 250-500 мг материала листьев 25 мл буфера для расщепления (1,2% (масс./об.) целлюлаза «ONOZUKA™» R10 и 0,2% (масс./об.) MACEROZYME® R10 (источник - Duchefa), растворенные в среде K4 (Spangenberg et al., 1998)). Чашку ПЕТРИ™, содержащую материал листьев и буфер для расщепления, герметично закрывали PARAFILM™ и инкубировали при комнатной температуре в течение 12-15 час в темноте. После инкубации в течение ночи расщепленный материал фильтровали через клеточное сито BD® (размер ячеек 70 мкм). На суспензии протопластов (5-6 мл), собранные в 14 мл круглодонные пробирки, наслаивали 1 мл буфера для промывки W5 (154 мМ NaCl, 125 мМ CaCl2, 5 мМ KCl и 5 мМ глюкоза; pH 5,8 Menzel et al. (1981)).
[0354] Суспензии далее центрифугировали при 400 об/мин в течение 10 мин. После центрифугирования протопласты, всплывшие в интерфазу, отбирали и промывали центрифугированием с использованием 10 мл буфера W5 при 400 об/мин в течение 10 мин. После последней промывки выделенные протопласты ресуспендировали с плотностью 1×106 протопластов на мл буфера W5 и инкубировали в течение 1 час перед трансфекциями.
Оценка выхода и жизнеспособности протопластов
[0355] Выход протопластов оценивали с использованием гемоцитометра, следуя способу Sambrook and Russell, (2006). Жизнеспособность клеток тестировали с использованием 400 мг/л красителя Эванса голубого, растворенного в 0,5M манните, как описано Huang et al. (1996) с незначительными модификациями способа.
Опосредованная ПЭГ 4000 доставка ДНК
[0356] Перед доставкой в протопласты B. napus, плазмидную ДНК каждой конструкции донора и ZFN получали из культур E. coli с использованием системы PURE YIELD PLASMID MAXIPREP® (Promega Corporation, Madison, WI), следуя инструкциям производителя. Аликвоты плазмидной ДНК донора и ZFN получали в трех молярных соотношениях: 1:1 (30 мкг каждой плазмиды), 5:1 (донорная плазмида к плазмиде ZFN, всего до 30 мкг плазмидной ДНК) и 10:1 (донорная плазмида к плазмиде ZFN, всего до 30 мкг плазмидной ДНК). Кроме того, аликвоты только донора и только ZFN (30 мкг) получали в качестве контроля. Количества ДНК, доставляемой в протопласты B. napus посредством опосредованной ПЭГ4000 трансформации, обобщены в таблице 17.
Количества ДНК ZFN и донорной ДНК, доставляемые в протопласты
[0357] Каждую аликвоту плазмидной ДНК вводили в один миллион протопластов (жизнеспособность ≥95), суспендированных в 100 мкл буфера для трансформации (15 мМ MgCl2, 0,1% (масс./об.) морфолиноэтансульфоновая кислота (MES) и 0,5M маннит; pH 5,8), затем в 150 мкл раствора ПЭГ (40% (масс./об.) ПЭГ 4000 в 0,4M манните и 0,1M Ca(NO3)2 (pH 6-7) Spangenberg and Potrykus (1995). После 10-15 мин инкубации при комнатной температуре, 5 мл буфера W5 добавляли по каплям и протопласты осторожно перемешивали. Другие 5 мл буфера W5 добавляли медленной струей к суспензии протопластов. Протопласты осторожно перемешивали и центрифугировали при 400 об./мин в течение 10 мин, и супернатант W5 осторожно удаляли, оставляя протопласты в форме осадка. Затем трансфицированные протопласты инкубировали в 1 мл буфера W5 при комнатной температуре, пока их не помещали в культуры типа бусин. Трансфицированные протопласты помещали в культуры, следуя способу с альгинатом натрия, как описано ниже.
Культивирование полученных из мезофилла протопластов для восстановления жизнеспособных микрокаллюсов
[0358] Перед помещением в культуру трансфицированные протопласты центрифугировали при 400 об/мин в течение 10 мин и буфер W5 осторожно удаляли. Затем протопласты ресуспендировали в 1,0 мл 0,5M маннита и инкубировали на льду. К этому добавляли равный объем 1,0% альгината натрия и осторожно перемешивали. Суспензию протопластов инкубировали на льду, пока их не помещали в культуры. Раствор для формирования бусин (0,4M маннит + 50 мМ СаСl2 (pH 5,8)) переносили в стерильный шестилуночный планшет (3-4 мл на лунку) с использованием серологической пипетки. Точно 1,0 мл суспензии протопластов добавляли по каплям с использованием 1 мл пипетки в раствор для формирования бусин, и каждый трансфицированный образец (приблизительно 5×105 протопластов) помещали в культуру в лунку. Суспензию протопластов инкубировали в течение 1-2 часов при комнатной температуре для формирования бусин альгината натрия. После периода инкубации раствор для формирования бусин осторожно удаляли и заменяли на 4-5 мл смеси 1:2 среды K3+H:A (Spangenberg et al., 1998), дополненной 1,5 мг/л гигромицина. Протопласты культивировали в течение 3-4 недель в темноте 22°C в встряхивателе (50 об/мин). После 3-4 недель устойчивые микрокаллюсы (0,5-1,0 мм) высвобождали обработкой буфером для деполимеризации (0,3M маннит + 20 мМ цитрат натрия (pH 5,8)). После удаления жидкой среды 3-4 мл буфера для деполимеризации добавляли в каждую лунку, содержащую культуры типа бусин, и инкубировали при комнатной температуре в течение 2 часов. С использованием стерильного пинцета бусины осторожно перемешивали для усиления эффективного высвобождения микрокаллюсов. Затем стерильную 1,0 мл пипетку использовали для осторожного перемешивания гелеобразующего средства, которое высвобождали в буфер для деполимеризации и затем удаляли. Микрокаллюсы промывали дважды с использованием 5 мл жидкой среды A, и микрокаллюсы ресуспендировали в достаточном количестве жидкой A (50 мл жидкой A использовали на один мл объема осажденных клеток (SCY: это измеряли после переноса всех высвобожденных микрокаллюсов в стерильную пробирку фалькон 50 или 15 мл и обеспечения возможности осаждения в течение 5 мин)). После равномерного перемешивания микрокаллюсов 0,5 мл микрокаллюсов, суспендированных в жидкой среде A, переносили в среду B1 (MS/MS витамины + 3,5% сахароза + 500 мг/л MES + BAP (5 мкМ) + NAA (5 мкМ) + 2,4-D (5 мкМ) + 1,5 мг/л гигромицина + 0,7% агарозы типа I (pH 6,0) и разливали в 100×20 мм стерильную чашку ПЕТРИ™), и с использованием 1-2 мл дополнительной жидкой среды A микрокаллюсы равномерно распределяли в среде B1, и избыток жидкой среды A осторожно удаляли из каждой чашки. Чашки запечатывали с использованием ленты из микропористого материала, что ускоряло созревание зародышей. Культуры поддерживали при 22°C при свете 16 час/сутки (30 мкмоль м-2 с-1).
Пролиферация и регенерация побегов из полученных из мезофилла протопластов
[0359] Устойчивые к гигромицину колонии скалывали со среды B1 (микрокаллюсы, полученные обоими способами SA и SP) после 2-3 недель инкубации и переносили в среду B2 (MS/MS витамины + 3,0% сахароза + 500 мг/л MES + 500 мг/л PVP + 5 мг/л нитрат серебра + 5 мг/л 2i P + NAA (0,5 мкМ) + GA-3 (0,3 мкМ) + 1,5 мг/л гигромицина + 0,7% агарозы типа I (pH 5,8) и разлита в 100×20 мм стерильную чашку ПЕТРИ™). Приблизительно по 25-30 каллюсов помещали в чашку, и чашки герметично запечатывали с использованием PARAFILM™ и инкубировали при 22°C при свете 16 час/сутки (30 мкмоль м-2 с-1). Затем устойчивые к гигромицину колонии выделяли после 5-6 циклов субкультивирования в среде B2 с интервалом две недели. Количество каллюсов на чашку уменьшали до 12-15 после третьего цикла субкультивирования. Примордий побегов, появившихся через 10-12 недель, осторожно выделяли вместе с остатками каллюсов и переносили в среду для удлинения побегов (MS/B5 витамины + 2% сахароза + 500 мг/л MES + BAP (2 мкМ) + GA-3 (0,1 мкМ) + 300 мг/л тиментин +1,5 мг/л гигромицин + 0,8% агар (pH 5,8) и разлита в 250 мл культуральные флаконы). Побеги, выжившие после 2-3 циклов селекции на гигромицине, переносили в среду для укоренения (1/2 концентрации MS/B5 витамины + 1% сахароза + 500 мг/л MES + IBA (2,5 мкМ) +1,5мг/л гигромицин + 0,6% агар (pH 5,8) и разлита в 700 мл культуральные флаконы).
Выделение геномной ДНК из протопластов мезофилла
[0360] Трансфицированные протопласты переносили из 3 см чашки ПЕТРИ™ в 2 мл пробирку для микроцентрифугирования. Клетки осаждали центрифугированием при 70 g, и супернатант удаляли. Для максимизации выхода трансфицированных протопластов, чашку ПЕТРИ™ промывали три раза 1 мл буфера для промывки. Каждую промывку проводили поворачиванием буфера для промывки в чашке ПЕТРИ™ в течение 1 минуты, с последующим переносом жидкости в ту же самую 2 мл пробирку для микроцентрифугирования. В конце каждой промывки клетки осаждали центрифугированием при 70 g, и супернатант удаляли. Осажденные протопласты быстро замораживали в жидком азоте перед лиофилизацией в течение 24 час в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированные клетки подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MINI (Qiagen), следуя инструкциям производителя, за исключением того, что разрушение тканей не требовалось, и клетки протопластов добавляли непосредственно в буфер для лизиса.
Выделение геномной ДНК из ткани каллюса
[0361] Отдельные каллюсы быстро замораживали в жидком азоте перед лиофилизацией в течение 24 час в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированные каллюсы подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MAXI (Qiagen, Hilden, Germany), следуя инструкциям производителя.
Выделение геномной ДНК из ткани листьев
[0362] Тридцать (30) мг ткани молодых листьев из регенерированных растений быстро замораживали в жидком азоте перед лиофилизацией в течение 24 час в LABCONCO FREEZONE 4.5® (Labconco, Kansas City, MO) при -40°C и давлении 133×10-3 мБар (13,3 Па). Лиофилизированные каллюсы подвергали выделению ДНК с использованием набора DNEASY® PLANT DNA EXTRACTION MAXI (Qiagen, Hilden, Germany), следуя инструкциям производителя.
Анализы ПЦР геномной ДНК по опосредованному NHEJ сплайсингу и редактированию Fad3C
[0363] Детекцию интеграции донорной ДНК в ген Fad3C B. napus выполняли посредством серий ПЦР, где по меньшей мере один праймер являлся специфическим для локуса Fad3C (таблица 18), и второй праймер являлся специфическим либо для промотора, либо для терминатора из кассеты gfp (таблица 18 и фигура 28A). Специфичность получали посредством разработки олигонуклеотидов, где последнюю пару оснований выравнивали с SNP, отличающим геномную последовательность Fad3C от других копий генов Fad3, и включающих фосфоротиоатную межнуклеотидную связь перед этой парой, как указано звездочкой [*]. Этот дизайн, используемый в сочетании с полимеразой, обладающей корректирующей активностью, направлял специфическую амплификацию каждого из аллелей Fad3C или Fad3A и исключал другие копии Fad3, как указано. Каждый набор праймеров эмпирически тестировали по амплификации правильных копий гена посредством секвенирования по Сенгеру продуктов амплификации ПЦР, полученных из B. napus дикого типа.
Последовательности олигонуклеотидов, использованных для детекции интеграции ДНК в индуцированные ZFN-двухцепочечные разрывы
Детекция добавления гена к Fad3C посредством соединения негомологичных концов в протопластах
[0364] Геномную ДНК выделяли из пулов протопластов (один миллион протопластов на пул), в которые донорную ДНК, кодирующую функциональную кассету репортера tGFP (pDAS000341 или pDAS000343), ДНК ZFN (pDAB107827 или pDAB107828) или смесь донорной ДНК и ДНК ZFN доставляли на двадцать четыре часа ранее. Количества ДНК, доставляемые на трансформацию, описаны выше. Продукты ПЦР клонировали в плазмидные векторы. Геномное редактирование происходит независимо в каждой клетке с образованием множества различных событий вставки, посредством клонирования в плазмидный вектор, каждое геномное редактирование можно секвенировать без неоднозначности. Несколько клонов секвенировали на автоматической платформе для капиллярного электрофореза ABI3730XL®. Анализ последовательностей генов выполняли с использованием программного обеспечения SEQUENCHER V5.0™ (GeneCodes, Ann Arbor, MI).
[0365] Доказательство добавления гена в локус Fad3C посредством редактирования или сплайсинга получали посредством амплификации как 5'-, так и 3'-участков стыковки Fad3C-кассета из геномной ДНК, выделенной из протопластов, с использованием праймеров, описанных в таблице 18. Продукты амплификации ПЦР с праймерами «FAD3CNHEJ-L4-F2» и «AtUbiNHEJ-R1» получали для амплификации 5'-участка стыковки кассеты tGFP и Fad3C. Амплификацию ПЦР с праймерами «FAD3CNHEJ-L4-R2» и «AtORF23tNHEJ-Fl» выполняли для амплификации 3'-участка стыковки кассеты tGFP и Fad3C. Амплификацию ПЦР с праймерами «FAD3CNHEJ-L4-F2» и «FAD3CNHEJ-L4-R2» выполняли для амплификации на протяжении двухцепочечных разрывов, индуцированных ZFN 28051-2A-28052. Не наблюдали амплификации для протопластов, в которые доставляли только плазмиду ZFN или донорную плазмиду. Все последовательности участков стыковки указывали на вставку кассеты tGFP в локус Fad3C посредством опосредованного NHEJ пути репарации. Наблюдали делеции различной длины из любого из генома и кассеты или из обоих, так же как добавление последовательностей, происходящих из остовов векторов (либо донора, либо ZFN), вставленных между геномом и кассетой.
Детекция добавления гена к Fad3C посредством соединения негомологичных концов в ткани каллюса, регенерированной из протопластов
[0366] Дополнительное доказательство сплайсинга и редактирования локуса Fad3C получали для ткани каллюса, регенерированной из протопластов при селекции (1,5 мг/л гигромицина, как описано выше), в которую доставляли донорную ДНК, кодирующую кассету hph (pDAS000340 или pDAS000342), только ДНК ZFN (pDABl07827 или pDABl07828) или донорную ДНК и ДНК ZFN (количества доставляемой ДНК приведены в таблице 17). ДНК выделяли из приблизительно 80 каллюсов для каждого соотношения, за исключением редактирования 1:1:1, для которого не выжило каллюсов, через четыре недели после трансфекции протопластов.
[0367] Интеграцию кассеты hph в геном B. napus (в Fad3C или случайным образом) подтверждали посредством TAQMAN™ qPCR с использованием праймеров (SEQ ID NO:402; F - 5-'CTTACATGCTTAGGATCGGACTTG-3', SEQ ID NO:403; R - 5'-AGTTCCAGCACCAGATCTAACG-3') и зонда (SEQ ID NO:404; 5'-CCCTGAGCCCAAGCAGCATСATCG-3'), специфических для гена hph. Эти пару праймеров и зонд использовали в дуплексной реакции с праймерами (SEQ ID NO:405; F - 5'-CGGAGAGGGCGTGGAAGG-3’, SEQ ID NO:406; R - 5'-TTCGATTTGCTACAGCGTСAAC-3') и зондом (SEQ ID NO:407; 5’-AGGCACCATCGCAGGCTTCGCT-3'), специфическими для белка I/I (HMG I/Y) группы белков с высокой подвижностью B. napus, присутствующего в форме отдельной копии в A-геноме (Weng et al., 2004, Plant Molecular Biology Reporter). Амплификацию проводили на термоциклере С1000 с системой REAL-TIME PCR DETECTION™ CFX96 или CF384 (BioRad, Hercules, CA). Результаты анализировали с использованием пакета программного обеспечения CFX MANAGER™ (BioRad). Относительное количество рассчитывали способом 2-ΔΔCt (Livak and Schmittgen, 2001), обеспечивающим определение количества копий кассеты hph, вставленной в геном. Доказательство опосредованного NHEJ сплайсинга и редактирования Fad3C получали проведением анализов ПЦР с одним праймером, специфическим для Fad3C, и вторым праймером, специфическим либо для промотора, либо для терминатора кассеты hph (таблица 17 и фигура 28B). Из-за ограниченных количеств ДНК, полученной из ткани каллюса, анализировали только интеграцию в смысловой ориентации. Продукты ПЦР очищали из геля с использованием набора для очистки продуктов ПЦР QIAQUICK MINIELUTE™ (Qiagen) и секвенировали с использованием способа прямого секвенирования по Сенгеру. Секвенируемые продукты очищали с помощью этанола, ацетата натрия и ЭДТА, следуя протоколу BIGDYE® v3.1 (Applied Biosystems) и секвенировали и анализировали, как выше.
[0368] Количество каллюсов, содержащих донорную кассету в каждом эксперименте, приведено в таблице 18. Доказательство добавления донорного гена в локус Fad3C посредством редактирования и/или сплайсинга получали посредством амплификации ПЦР (с праймерами, показанными в таблице 19) на протяжении участков разрезания ZFN и как 5'-, так и 3'-участков стыковки Fad3C-кассета hph. Амплификация ПЦР геномной ДНК, выделенной из ткани каллюса, восстановленной из контрольных протопластов, трансформированных только плазмидой hph (pDAS000340 и pDAS000342) или только плазмидой ZFN (pDAB107827 и pDAB107828), не привела к получению продуктов амплификации ПЦР.
[0369] Ампликоны ПЦР, полученные при амплификации 5'- и 3’-участков стыковки Fad3C-кассета hph, очищали из агарозного геля и секвенировали для подтверждения специфичности интеграции внутри геномного локуса Fad3C. Результаты анализа секвенирования продуктов ПЦР показали, что каждый выделенный каллюс, полученный из индивидуально трансформированного протопласта, образовывал отдельный продукт амплификации ПЦР и не содержал клеток смешанных генотипов.
[0370] В экспериментах по опосредованной NHEJ интеграции донорных последовательностей внутри геномного локуса Fad3C частота добавления в локус-мишень (как определено по любой части вектора донорной ДНК, амплифицированной с локуса-мишени), составляла 42%, 46% и 32% для концентраций ДНК 1:1, 5:1, и 10:1 (донорная ДНК: ДНК ZFN), соответственно. См. таблицу 20. Частоту целевого сплайсинга определяли посредством анализа того, поддавались ли амплификации оба участка стыковки с кассетой, и из секвенирования продуктов ПЦР. Эти результаты верифицировали, что кассета являлась вставленной в локус-мишень в правильной ориентации. Частоту интеграции рассчитывали как 4%, 3% и 3% для концентраций 1:1, 5:1 и 10:1 донорная плазмидная ДНК: плазмидная ДНК ZFN, соответственно. В экспериментах по редактированию гена частота добавления в локус-мишень, определенная по любой части вектора донорной ДНК, амплифицированной из локуса-мишени, составляла 66% и 65% для концентраций 5:1:1 и 10:1:1 донорная плазмидная ДНК: плазмидная ДНК ZFN, соответственно. См. таблицу 21. Частоту целевого редактирования определяли как по возможности амплификации обоих участков стыковки, так и по получению последовательности продуктов ПЦР. Эти результаты верифицировали, что кассета являлась вставленной в локус-мишень в правильной ориентации с частотой 3% и 6% для концентрации 5:1:1 и 10:1:1 донорная плазмидная ДНК: плазмидная ДНК ZFN, соответственно. Как наблюдали в анализах протопластов, либо пары оснований были делетированы, либо дополнительные основания вставлены между геномом и кассетой в результате расщепления геномного локуса посредством ZFN (фигуры 30-31).
[0371] В конкретных случаях получали продукты ПЦР в результате добавления нуклеотидных последовательностей внутри локуса-мишени, отсутствие продукта ПЦР или больший продукт ПЦР, чем наблюдали в образцах дикого типа. Эти результаты, полученные при амплификации ПЦР с использованием праймеров, фланкирующих участок разрезания, показали, что локус поврежден на обеих парах хромосом (фигуры 30-31). В некоторых случаях амплифицировали более одной полосы в участках стыковки после сплайсинга (фигуры 30-31), что указывает на то, что различные вставки происходили независимо в каждой копии генома.
Количество каллюсов, положительных по присутствию hph после четырех недель селекции
DAB107827
DAB107827
DAB107828
Количество каллюсов с hph, вставленным посредством сплайсинга в локус FadC в DSB, индуцированный посредством ZFN28051-2A-28052
+
DAB107827
Количество каллюсов с hph, вставленным посредством редактирования в локус FadC в учатски разрезания, индуцированного посредством ZFN28051-2A-28052 и ZFN28053-2A-28054
+
DAB107827
+
DAB107828
Детекция добавления гена к Fad3C посредством соединения негомологичных концов в растениях
[0372] ДНК выделяли из растений, которые регенерировали из протопластов и переносили в среду для посадки (как описано выше). Определили, что большинство восстановленных растений содержали только 1-2 копии кассеты hph, кодированной в донорной ДНК. Растения анализировали с помощью того же комплекса анализов, описанного для ткани каллюса, так же как с помощью анализов для определения того, вставлена ли кассета в антисмысловой ориентации, или интеграции донора в локус Fad3A.
Оцененное количество копий для растений, регенерированных из протопластов. Для каждого соотношения проводили три трансфекции одного миллиона протопластов
DAB107827
DAB107827
DAB107828
[0373] Частота целевого сплайсинга, где кассета hph являлась вставленной в Fad3C в любом направлении, составляла 51%, 32% и 56% для донорной ДНК: ДНК ZFN в концентрациях 1:1, 5:1 и 10:1, соответственно (таблица 23). Из этих результатов, 35% 32% и 50% (1:1, 5:1 и 10:1) были вставлены в прямой ориентации (таблица 23).
[0374] Частота целевого редактирования, где кассета hph являлась вставленной в Fad3C в любом направлении, заменяя область от локуса 4 до локуса 6, составляла 2% и 0% для донорной ДНК: ДНК ZFN: ДНК ZFN в концентрациях 5:1:1 и 10:1:1, соответственно (таблица 24). Кроме того, когда обе ZFN доставляли при 5:1:1, 2% вставлено после сплайсинга в локус 4, и 10% вставлено после сплайсинга в локус 6, и когда обе ZFN доставляли при 10:1:1 10% вставлено после сплайсинга в локус 4, и 15% вставлено после сплайсинга в локус 6.
[0375] Полученные полосы можно секвенировать для определения количества точных границ. Кроме того, можно проводить скрининг растений по не целевым вставкам для определения частоты интеграции hph в участках, отличных от Fad3, и частоты интеграции в Fad3A, а не в Fad3C.
Количество растений с hph, вставленным посредством сплайсинга в локус FadC в DSB, индуцированный посредством ZFN28051-2A-28052
обратная/
любая)
обратные/
любые)
+
DAB107827
Количество растений с hph, вставленным посредством редактирования в локус FadC в участки разрезания, индуцированного посредством ZFN28051-2A-28052 и ZFN28053-2A-28054
ZFN
обратная/
любая)
обратные/
любые)
+
DAB 107827
+
Пример 8: Направленная интеграция в трансгенное событие кукурузы DAS-59132 и его нарушение
Характеризация эндогенного геномного локуса для нацеливания гена
[0376] Геномный локус трансгенного события кукурузы DAS-59132 описан в Международной патентной заявке № WO 2009/100188 A2. Трансгенное событие кукурузы DAS-59132 содержит экспрессирующие кассеты трансгенов Cry34Abl, Cry35Abl и PAT. Эти экспрессирующие кассеты трансгенов интегрированы в область, происходящую из хромосомы 8 генома маиса B73 зародышевой плазмы маиса Hi-II (D. D. Songstad, W. L. Petersen, C. L. Armstrong, American Journal of Botany, Vol. 79, pp. 761-764, 1992), в форме полноразмерной вставки T-цепи. Кроме того, геномная ДНК, окружающая трансгенный локус, лишена каких-либо больших делеций относительно природной последовательности B73, и в основном лишена повторяющихся элементов, за исключением небольшого повторяющегося элемента.
[0377] Геномный локус, в который интегрировано трансгенное событие кукурузы DAS-59132, выбран в качестве эндогенного геномного локуса для нацеливания генов. Выбор этого эндогенного геномного локуса основан на характеризации трансгенного события кукурузы DAS-59132. Это событие произошло в результате интеграции T-цепи в эндогенный геномный локус и последующей экспрессии экспрессирующих кассет трех трансгенов. Кроме того, присутствовало минимальное изменение нормального роста и развития растений кукурузы, содержащих трансгенное событие кукурузы DAS-59132. Трансгенный объект сохранял агрономические и селекционные характеристики и являлся сравнимым по агрономической производительности с не трансформированными контрольными растениями.
[0378] Вариант осуществления описания включает полинуклеотидные последовательности, которые могут служить мишенями для интеграции трансгена. Полноразмерная молекула ДНК (PHI17662A), использованная для трансформации трансгенного события кукурузы DAS-59132, 3’-конец геномной фланкирующей последовательности и участок стыковки PHI17662A/3’-генома маиса описаны в описании Международной патентной заявки № WO 2009/100188 A2 и описаны при подаче этой заявки как SEQ ID NO:427, SEQ ID NO:428 и SEQ ID NO:429, соответственно. 5’-конец геномной фланкирующей последовательности и геномный локус, где трансгенное событие кукурузы DAS-59132 интегрировано в геном кукурузы, описано при подаче этой заявки как SEQ ID NO:430 и SEQ ID NO:431, соответственно. Геномный локус, описанный как SEQ ID NO:431, использовали для дизайна белков с цинковыми пальцами для нацеливания гена.
Получение белков с цинковыми пальцами, разработанных для связывания с геномным локусом для трансгенного события кукурузы DAS-59132
[0379] Белки с цинковыми пальцами, направленные против последовательностей ДНК, которые содержит геномный локус для трансгенного события кукурузы DAS-59132 (см. фигуру 32), разработаны, как описано ранее. См. например, Umov et al. (2005) Nature 435:646-651. Иллюстративная последовательность-мишень и спирали узнавания указаны в таблицах 25A (дизайн областей спиралей узнавания) и таблице 25B (участки-мишени). В таблице 25B, нуклеотиды в участке-мишени, контактирующие со спиралями узнавания ZFP, указаны заглавными буквами.
Геномный локус для дизайна связывающих цинковых пальцев для трансгенного события кукурузы DAS-59132
NO:533
NO:534
NO:535
NO:537
NO:143
NO:539
Последовательности-мишени для белков с цинковыми пальцами
[0380] Цинковые пальцы различного дизайна для трансгенного события кукурузы DAS-59132 вставляли в векторы, кодирующие белок, имеющий по меньшей мере один палец со структурой CCHC. См. Патентную публикацию США № 2008/0182332. В частности, последний палец в каждом белке имел остов CCHC для спирали узнавания. Неканонические кодирующие цинковые пальцы последовательности сливали с доменом нуклеазы рестрикционного фермента типа IIS FokI (аминокислоты 384-579 последовательности из Wah et al. (1998) Proc. Natl. Acad. Sci. USA 95:10564-10569) посредством линкера ZC из четырех аминокислот и сигнала ядерной локализации opaque-2, полученного из Zea mays, для формирования нуклеаз с цинковыми пальцами (ZFN) для трансгенного события кукурузы DAS-59132. Экспрессией слитых белков в бицистронной экспрессирующей конструкции с использованием сигнала остановки рибосомы 2A, как описано в Shukla et al. (2009) Nature 459:437-441, управляли посредством относительно сильного, конститутивного и эктопического промотора, такого как промотор CsVMV.
[0381] Оптимальные цинковые пальцы верифицировали по активности расщепления с использованием системы на основе почкующихся дрожжей, как показано ранее, идентифицирующей активные нуклеазы. См., например, Патентную публикацию США № 20090111119; Doyon et al. (2008) Nat Biotechnol. 26:702-708; Geurts et al. (2009) Science 325:433. Цинковые пальцы для различных функциональных доменов отбирали для использования in vivo. Из множества ZFN, которые были разработаны, получены и тестированы по связыванию с предположительными геномными полинуклеотидными участками-мишенями для трансгенного события кукурузы DAS-59132, четыре пары ZFN идентифицированы как обладающие активностью in vivo на высоких уровнях и выбраны для дальнейших экспериментов. См. таблицу 25A. Эти ZFN охарактеризованы как способные эффективно связывать и расщеплять четыре уникальных геномных полинуклеотидных участка-мишени для трансгенного события кукурузы DAS-59132 in planta.
[0382] На фигуре 1 показана геномная организация локуса трансгенного события кукурузы DAS-59132 по отношению к полинуклеотидным участкам связывания/мишеням ZFN четырех пар ZFN. Первые три пары ZFN (E32 ZFN1, E32 ZFN2 и E32 ZFN3) связываются выше вставки T-цепи трансгенного события кукурузы DAS-59132, вторые три пары ZFN (E32 ZFN4, E32 ZFN5 и E32 ZFN6) связываются ниже вставки T-цепи трансгенного события кукурузы DAS-59132. После тестирования пар ZFN в анализе почкующихся дрожжей, пары ZFN, которые оптимально связывают локус трансгенного события кукурузы DAS-59132, выдвинули для тестирования в анализе временной трансформации кукурузы.
Конструкции нуклеаз с цинковыми пальцами для экспрессии в маисе
[0383] Плазмидные векторы, содержащие экспрессирующие конструкции ZFN четырех иллюстративных нуклеаз с цинковыми пальцами, которые идентифицированы с использованием анализа дрожжей и описаны в примере 2, разрабатывали и выполняли с использованием квалификации и способов, общеизвестных в данной области. Каждую кодирующую цинковый палец последовательность сливали с последовательностью, кодирующей сигнал ядерной локализации opaque-2 (Maddaloni et al. (1989) Nuc. Acids Res. 17(18):7532), расположенный выше нуклеазы с цинковыми пальцами.
[0384] Затем слитую последовательность сигнал ядерной локализации opaque-2::нуклеаза с цинковыми пальцами спаривали с комплементарной слитой последовательностью сигнал ядерной локализации opaque-2::нуклеаза с цинковыми пальцами. В силу этого, каждая конструкция состояла из одной открытой рамки считывания, состоящей из двух слитых последовательностей сигнала ядерной локализации opaque-2::нуклеаза с цинковыми пальцами, разделенных последовательностью 2A из вируса Thosea asigna (Mattion et al. (1996) J. Virol. 70:8124-8127). Экспрессией кодирующей ZFN последовательности управляли посредством высоко экспрессирующего конститутивного промотора убиквитина 1 Zea mays (Christensen et al. (1992) Plant Mol. Biol. 18(4):675-89) и фланкировали ее 3’-поли A-нетранслируемой областью Per 5 Zea mays (Патент США № 6699984). Полученные четыре плазмидные конструкции подтверждали посредством расщепления рестрикционными ферментами и посредством секвенирования ДНК. На фигурах 33 и 34 приведено графическое представление завершенной плазмидной конструкции. ZFN, экспрессированная в плазмидной конструкции pDAB105906 (фигура 33), содержит «Fok-Mono», которая представляет собой эндонуклеазу FokI дикого типа. ZFN, экспрессированная в плазмидной конструкции pDAB111809 (фигура 34), содержит «FokI-ELD», которая представляет собой модифицированную эндонуклеазу FokI. Модифицированная эндонуклеаза FokI содержит изменения, как описано в Doyon Y., Vo Т., Mendel М., Greenberg S., Wang J., Xia D., Miller J., Umov F., Gregory P., и Holmes M. (2010) Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architecture. Nature Methods, 8(1); 74-79.
[0385] Донорная конструкция разработана для интеграции в расщепленную ZFN геномную ДНК геномного локуса трансгенного события кукурузы DAS-59132. На фигуре 35 проиллюстрирована донорная конструкция, pDAB100655, состоящая из экспрессирующей один ген кассеты. Экспрессирующей один ген кассетой управляют посредством промотора убиквитина 1 Zea mays (промотор ZmUbi1) :: кодирующая последовательность aad-1 (AAD1; Патент США № 7838733) :: и она терминирована 3’-нетранслируемой областью Per 5 Zea mays (ZmPer5 3’UTR). Конструкция содержит пару повторяющихся связывающих последовательностей E32 ZFN6, включенных ниже экспрессирующей кассеты гена aad-1. Различные генные элементы собраны в плазмиде с высоким количеством копий на основе pUC.
Временная трансформация маиса для определения эффективности ZFN
[0386] Эмбриогенные культуры маиса Hi-II получали, как описано в Патенте США № 7179902, и использовали для оценки и тестирования эффективности различных ZFN. Плазмидной ДНК, состоящей из pDAB105901, pDAB105902, pDAB105903, pDAB105904, pDAB105905 и pDAB105906, временно трансформировали клетки каллюса маиса для сравнения частоты разрезания различных ZFN против стандартной тестированной ZFN, pDAB7430, разработанной для локуса гена инозитол-полифосфат-киназы-2 в геноме маиса, как описано в Патентной заявке США № 2011/0119786.
[0387] По 12 мл объема осажденных клеток (PCV) из культур из ранее криоконсервированной линии клеток плюс 28 мл кондиционированной среды субкультивировали в 80 мл жидкой среды GN6 (среда N6 (Chu et al., (1975) Sci Sin. 18:659-668), 2,0 мг/л 2,4-D, 30 г/л сахароза, pH 6,0) в 500 мл колбе Эрленмейера, и помещали во встряхиватель при 125 об/мин при 28°C. Эту стадию повторяли два раза с использованием той же линии клеток, так что всего 36 мл PCV распределяли на три колбы. Через 24 часа содержимое выливали в стерильную чашку ПЕТРИ™, и жидкую среду GN6 удаляли. Слабо увлажненный каллюс переносили на круг диаметром 2,5 см на фильтровальную бумагу, содержащую твердую среду GN6 S/M (среда N6 (Chu et al., (1975) Sci Sin. 18:659-668), 2,0 мг/л 2,4-D, 30 г/л сахароза, 45,5 г/л сорбит, 45,5 г/л маннит, 100 мг/л мио-инозитол, 2,5 г/л Gelrite, pH 6,0). Чашки инкубировали в темноте в течение 4 часов при 28°C.
[0388] Микрочастицы золота (0,6 микрон, BioRad, Hercules, CA,) подготавливали для осаждения ДНК посредством взвешивания 21 мг в стерильную, силиконизированную 1,7 мл пробирку для микроцентрифугирования (Sigma-Aldrich, St. Louis, MO), и 350 мкл ледяного 100% этанола добавляли и встряхивали в течение 1 минуты. Золото осаждали центрифугированием при 10000 об/мин в течение 15 секунд с использованием центрифуги MINISPIN™ (Eppendorf, Hauppauge, NY). После удаления супернатанта добавляли 350 мкл ледяной, стерильной воды, перемешивали пипетированием вверх и вниз, и центрифугировали при 10000 об/мин в течение 15 секунд. Стадию промывки повторяли еще один раз перед суспендированием золота в 350 мкл ледяной, стерильной воды. Промытое золото затем хранили при -20°C до необходимости.
[0389] Для каждого осаждения ДНК аликвоту 3 мг золота в 50 мкл воды отбирали в силиконизированную 1,7 мл пробирку для микроцентрифугирования (Sigma-Aldrich, St. Louis, MO). Плазмидную ДНК (2,5 мкг E32 ZFN в плазмидах pDABl05901, pDAB105902, pDAB105903, pDAB105904, pDAB105905 или pDAB105906 и 2,5 мкг IPPK2 ZFN в плазмиде pDAB7430) предварительно смешивали в 0,6 мл пробирках для микроцентрифугирования (Fisher Scientific, Nazareth, PA) и добавляли к суспензии золота с осторожным пипетированием вверх и вниз 5-10 раз для полного смешивания. Затем добавляли двадцать микролитров (20 мкл) холодного 0,1M спермидина и осторожно перемешивали пипетированием вверх и вниз 5-10 раз. Пятьдесят микролитров (50 мкл) ледяного 2,5M хлорида кальция медленно добавляли и осторожно перемешивали пипетированием вверх и вниз 5-10 раз. Затем пробирку закрывали крышкой и позволяли инкубацию при комнатной температуре в течение 10 минут. После центрифугирования в течение 15 секунд при 10000 об/мин супернатант осторожно удаляли и добавляли 60 мкл ледяного, 100% этанола. Смесь золота и ДНК ресуспендировали осторожным пипетированием вверх и вниз 5-10 раз.
[0390] Для бомбардировки микрочастицами стерилизованные макроносители (BioRad, Hercules, CA) вставляли в держатели из нержавеющей стали (BioRad, Hercules, CA) и автоклавировали. Девять микролитров (9 мкл) суспензии золото/ДНК равномерно распределяли в центре макроносителя, обеспечивая пипетирование вверх и вниз, так чтобы поддерживать суспензию хорошо перемешанной между отбором аликвот. Затем макроносители помещали на кусок стерильной 125 мм фильтровальной бумаги Whatman #4 (GE Healthcare, Buckinghamshire, UK) на подложке из 8-меш DRIERITE™ (W.A Hammond Drierite Co., Xenia, OH) в стеклянной чашке ПЕТРИ™ 140×25 мм. Золоту/ДНК позволяли полностью высохнуть в течение приблизительно 5-10 минут. Разрывные диски (1100 psi (7584 кПа), BioRad, Hercules, CA) стерилизовали погружением на несколько секунд в изопропиловый спирт, затем загружали в колпачок распылителя устройства для бомбардировки микрочастицами (PDS-1000, BioRad, Hercules, CA). Автоклавированный останавливающий экран (BioRad, Hercules, CA) и загруженный макроноситель помещали в пусковой контейнер, крышку завинчивали и вставляли в камеру для бомбардировки непосредственно под распылитель. Чашку ПЕТРИ™, содержащую мишень, открывали и помещали в камеру для бомбардировки на 6 см ниже распылителя. Подключали вакуум (-0,9 бар (90 кПа)), и включали устройство. Стадии повторяли для каждой бомбардированной мишени. Мишени инкубировали в темноте при температуре 28°C в течение 24 часов в той же самой среде для бомбардировки. Клетки после бомбардировки переносили для восстановления на твердую среду для восстановления GN6 (среда N6 (Chu et al., (1975) Sci Sin. 18:659-668), 2,0 мг/л 2,4-D, 30 г/л сахароза, 2,5 г/л Gelrite, pH 6,0) и инкубировали в течение дополнительных 48 часов при 28°C в темноте. Через семьдесят два часа после бомбардировки клетки собирали в 2 мл пробирки EPPENDORF MICROFUGE SAFE LOCK™ и лиофилизировали в течение 48 часов в лиофилизаторе VIRTIS модель # 50L VIRTUAL XL-70™ (SP Scientific, Gardiner NY).
Анализ секвенированием нового поколения (NGS) временно трансформированного маиса
[0391] Ткань временно трансформированного каллюса маиса анализировали для определения эффективности расщепления белками - нуклеазами с цинковыми пальцами.
Подготовка образца:
[0392] Ткань каллюса маиса, временно трансформированного конструкциями ZFN и двумя контрольными векторами, pDAB100664 и pDAB100665, собирали в 2 мл пробирки EPPENDORF™ и лиофилизировали в течение 48 час. Геномную ДНК (гДНК) выделяли из лиофилизированной ткани с использованием набора QIAGEN PLANT DNA EXTRACTION™ (Valencia, CA), следуя инструкциям производителя. Выделенную гДНК ресуспендировали в 200 мкл воды, и концентрацию определяли с использованием спектрофотометра NANODROP® (Invitrogen, Carlsbad, CA). Целостность ДНК оценивали посредством анализа всех образцов в 0,8% агарозных E-гелях (Invitrogen). Все образцы гДНК нормализовали (25 нг/мкл) для амплификации ПЦР для получения ампликонов для анализа посредством секвенирования ILLUMINA™ (San Diego, CA).
[0393] Праймеры для ПЦР для амплификации геномных областей, проходящих через каждый тестируемый участок расщепления ZFN, и контрольные образцы закупали в Integrated DNA Technologies (Coralville, IA). Оптимальные условия амплификации для праймеров идентифицировали градиентной ПЦР с использованием 0,2 мкМ подходящих праймеров, ACCUPRIME PFX SUPERMIX™ (1,1 X, Invitrogen) и 100 нг геномной ДНК-матрицы в 23,5 мкл реакционной смеси. Параметры циклов представляли собой начальную денатурацию при 95°С (5 мин) с последующими 35 циклами денатурации (95°C, 15 с), отжига (55-72°C, 30 с), удлинения (68°C, 1 мин) и конечного удлинения (72°C, 7 мин). Продукты амплификации анализировали в 3,5% агарозных гелях с TAE. После идентификации оптимальной температуры отжига проводили препаративные реакции ПЦР для валидации каждого набора праймеров для ПЦР и для получения ампликона для секвенирования ILLUMINA™.
[0394] Для препаративной ПЦР, 8 отдельных реакций ПЦР в малом масштабе проводили для каждой матрицы с использованием условий, описанных выше, и полученные продукты ПЦР объединяли вместе и очищали из 3,5% агарозных гелей с использованием набора QIAGEN MINELUTE GEL EXTRACTION/PURIFICATION™, следуя инструкциям производителя. Концентрации очищенных из геля ампликонов определяли посредством NANODROP™, и образцы для секвенирования ILLUMINA™ получали объединением приблизительно 100 нг ампликонов ПЦР для мишеней ZFN и соответствующего контроля дикого типа. Праймеры, использованные для получения ампликонов ПЦР, показаны в таблице 26 ниже.
Олигонуклеотиды для амплификации участков связывания ZFN
SEQ ID NO:
Секвенирование и анализ ILLUMINA™:
[0395] ZFN разработаны для узнавания, связывания и модификации специфических последовательностей ДНК в геномном локусе трансгенного события кукурузы DAS-59132. Анализировали эффективность, с которой четыре ZFN расщепляли геномный локус, чтобы определить, какая ZFN расщепляет наиболее эффективно. Секвенирование ILLUMINA™ выполняли в Cofactor Genomics (St. Louis, MO), и последовательности анализировали с использованием скрипта для анализа последовательностей. Последовательности низкого качества отфильтровывали и оставшиеся последовательности разбирали в соответствии с уникальными идентификаторами последовательностей ДНК. Затем уникальные идентификаторы последовательностей ДНК выравнивали с контрольной последовательностью и оценивали в баллах по вставкам/делециям (инделам). Для определения уровня активности расщепления область, окружающую участок расщепления ZFN, оценивали в баллах по присутствию вариантов последовательности, возникающих в результате инделов. Активность расщепления для каждой ZFN в исследовании рассчитывали как количество последовательностей с инделами/1M последовательностей высокого качества или как процент последовательностей высокого качества с инделами. Затем уровни эффективности расщепления определяли посредством нормализации уровня активности расщепления ZFN по активности ZFN, направленной на ген IPP2-K, как описано в Патентной публикации США № 2011/0119786. На фигуре 36 и в таблице 27 представлена эффективность расщепления тестированных ZFN.
[0396] Событие 32 ZFN6, содержащее связывающие домены с цинковыми пальцами 25716 и 25717, расщепляло геномный локус трансгенного события кукурузы DAS-59132 с наиболее высокой эффективностью. Эта ZFN функционировала в 380 раз эффективнее контрольной нуклеазы с цинковыми пальцами IPPK2. Принимая во внимание неожиданно высокие уровни активности расщепления для события 32 ZFN6, эта ZFN выбрана для усовершенствования для тестирования интеграции донорного фрагмента ДНК в геномный локус посредством соединения негомологичных концов.
Эффективность расщепления тестированных ZFN
Трансформация протопластов ZFN
[0397] Разработана система нацеливания генов для нацеливания на эндогенный геномный локус трансгенного события кукурузы DAS-59132 и для оптимизации параметров нацеливания донора в маисе. Двухцепочечные разрывы получали в геноме трансгенного события кукурузы DAS-59132 и репарировали посредством либо соединения негомологичных концов (NHEJ), либо зависимой от гомологии репарации (HDR).
Выделение протопластов:
[0398] Эмбриогенные суспензионные культуры маиса Hi-II получали и поддерживали с расписанием поддержания 3,5 суток. В 50 мл стерильной конической пробирке растворы ферментов 10 мл раствора стерильной 6% (масс./об.) целлюлазы и 10 мл раствора стерильной 0,6% (масс./об.) пектолиазы пипетировали в конической пробирке с использованием наконечника пипетки 10 мл. Затем 4 объема осажденных клеток (PCV) суспензии клеток Hi-II добавляли в 50 мл пробирку, содержащую раствор для расщепления и обернутую парафильмом. Пробирки помещали на качающуюся платформу на ночь при комнатной температуре на ~16-18 час. На следующее утро пробирки удаляли из встряхивателя. В стерильную 50 мл коническую пробирку клетки и раствор фермента медленно фильтровали через клеточное сито 100 мкм. Затем клетки промывали с использованием клеточного сита 100 мкм пипетированием 10 мл среды W5 через сито. В стерильную 50 мл коническую пробирку клетки и раствор фермента медленно фильтровали через клеточное сито 70 мкм. За этой стадией фильтрования следовала вторая стадия фильтрования, где клетки и раствор фермента медленно фильтровали в 50 мл коническую пробирку через клеточное сито 40 мкм. С использованием наконечника пипетки 10 мл клеточное сито 40 мкм промывали 10 мл среды W5 для получения конечного объема 40 мл, и пробирку переворачивали. Очень медленно, 8 мл сахарозной подушки добавляли на дно раствора протопластов/фермента. С использованием центрифуги с качающимся бакет-ротором пробирки центрифугировали в течение 15 минут при 1500 об/мин. Клетки протопласты удаляли с использованием 5 мл наконечника пипетки узкого поперечного сечения. Эти клетки (7-8 мл), которые наблюдали в виде полосы протопластов, удаляли очень медленно и помещали в стерильную 50 мл коническую пробирку. Затем 25 мл среды W5 использовали для промывки пробирок. Добавляли среду W5, и пробирки медленно переворачивали и центрифугировали в течение 10 минут при 1500 об./мин. Супернатант удаляли, и 10 мл раствора MMG добавляли с медленным переворачиванием пробирки для ресуспендирования осадка протопластов. Плотность протопластов определяли с использованием гемоцитометра, для 4 PCV получали выход ~30 миллионов протопластов.
Трансформация протопластов:
[0399] Клетки протопласты разводили до 1,6 миллиона протопластов на мл с использованием раствора MMG. Протопласты осторожно ресуспендировали медленным переворачиванием пробирки. Затем 300 мкл протопластов (~500000 протопластов) добавляли в стерильную 2 мл пробирку, пробирки переворачивали для равномерного распределения клеток протопластов. Плазмидную ДНК в концентрации приблизительно 40-80 мкг, суспендированную в буфере ТЕ, добавляли к протопластам. Различные условия эксперимента описаны в таблице 28. Пробирки медленно поворачивали для суспендирования ДНК с протопластами, и пробирки инкубировали в течение 5-10 минут при комнатной температуре. Затем 300 мкл раствора ПЭГ добавляли к раствору протопластов/ДНК. После добавления всего раствора ПЭГ, раствор ПЭГ смешивали с раствором протопластов осторожным переворачиванием пробирки. Смесь инкубировали при комнатной температуре в течение 15-20 минут с периодическим переворачиванием пробирки(пробирок). После инкубации 1 мл раствора W5 медленно добавляли в пробирки, и пробирки осторожно переворачивали. Наконец, раствор центрифугировали при 1000 об/мин в течение 15 минут. Супернатант осторожно удаляли, так чтобы не потревожить осадок клеток. Наконец, добавляли 1 мл раствора для промывки/инкубации. Пробирки осторожно переворачивали, чтобы ресуспендировать осадок клеток. Пробирки накрывали алюминиевой фольгой для исключения любого воздействия света, и укладывали в штатив на бок для инкубации в течение ночи. Клетки собирали через 24 часа после трансформации для молекулярного анализа.
Различные группы обработки использовали для трансформации клеток протопластов. Различные концентрации ДНК, использованные для трансформации, описаны ниже.
(0 мкг)
(40 мкг)
(0 мкг)
(40 мкг)
(0 мкг)
(0 мкг)
(40 мкг)
(40 мкг)
(0 мкг)
(0 мкг)
(0 мкг)
(0 мкг)
(4 мкг)
(76 мкг)
(0 мкг)
(0 мкг)
(4 мкг)
(0 мкг)
(76 мкг)
(0 мкг)
(40 мкг)
(0 мкг)
(0 мкг)
(40 мкг)
(36 мкг)
(0 мкг)
(40 мкг)
(4 мкг)
(0 мкг)
(36 мкг)
Валидация нацеливания секвенированием
[0400] Результаты для активности расщепления ZFN в протопластах маиса подтверждали с использованием способа секвенирования нового поколения, описанного выше. Секвенированные амплифицированные ПЦР фрагменты оценивали в баллах по присутствию вариантов последовательности, возникающих в результате инделов. Трансгенное событие 32 ZFN6 расщепляло геномный локус трансгенного события кукурузы DAS-59132 с приблизительно 1,5% NHEJ/10 нг ампликона-мишени.
[0401] Нацеливание донорной кассеты AAD-1 на геномный локус трансгенного события кукурузы DAS-59132 в суспензиях трансгенных клеток маиса Hi-II посредством соединения негомологичных концов (NHEJ) подтверждали реакцией ПЦР вкл.-выкл. Выполняли реакцию ПЦР вкл.-выкл., где первая реакция ПЦР разработана для амплификации участка стыковки донора AAD-1 и геномного локуса трансгенного события кукурузы DAS-59132. Полученный ампликон подвергали второй реакции ПЦР, где праймеры были разработаны для связывания внутри первого ампликона. Комбинация двух независимых реакций ПЦР приводила к удалению неспецифических амплификаций, которые могли являться ложноположительными. Результаты ПЦР вкл.-выкл. в экспериментах трансформации протопластов показали, что на геномный локус трансгенного события кукурузы DAS-59132 можно воспроизводимо нацеливать 5,3 т.п.о. плазмидный донор AAD1 и нуклеазу с цинковыми пальцами E32 ZFN6 в соотношении 1:10 мкг ДНК (в присутствии и в отсутствие ДНК-наполнителя, состоящей либо из ДНК плазмиды pUC19, либо из ДНК спермы лосося). Нацеливание способом NHEJ доказывали посредством вставки донорной кассеты AAD-1 в обеих ориентациях. Данные о последовательности, полученные после реакций ПЦР, показали в результате три случая точной интеграции донорной ДНК. Таким образом, являлось возможным продемонстрировать нацеливание донора на эндогенный локус маиса с использованием ZFN посредством механизма репарации NHEJ-DSB.
Опосредованная WHISKERS™ стабильная трансформация ZFN и донором для направленной интеграции
[0402] Трансгенные события нацеливали на эндогенный геномный локус трансгенного события кукурузы DAS-59132. Конструкции, как описано в примере 2, включают донорную последовательность (pDAB100655) и трансгенное событие 32 ZFN 6 (pDAB105906).
[0403] Клетки каллюса маиса, состоящие из 12 мл объема осажденных клеток (PCV) из ранее криоконсервированной линии клеток, плюс 28 мл кондиционированной среды субкультивировали в 80 мл жидкой среды GN6 (среда N6 (Chu et al., (1975) Sci Sin. 18:659-668), 2,0 мг/л 2,4-D, 30 г/л сахароза, pH 5,8) в 500 мл колбе Эрленмейера, и помещали во встряхиватель при 125 об/мин при 28°C. Эту стадию повторяли два раза с использованием той же линии клеток, так что всего 36 мл PCV распределяли на три колбы. Через 24 часа жидкую среду GN6 удаляли и заменяли на 72 мл осмотической среды GN6 S/M (среда N6, 2,0 мг/л 2,4-D, 30 г/л сахароза, 45,5 г/л сорбит, 45,5 г/л маннит, 100 мг/л мио-инозитол, pH 6,0). Колбу инкубировали в темноте в течение 30-35 минут при 28°С с умеренным встряхиванием (125 об./мин). Во время периода инкубации получали суспензию 50 мг/мл карбида кремния WHISKERS™ (Advanced Composite Materials, LLC, Greer, SC) посредством добавления 8,1 мл жидкой среды GN6 S/M к 405 мг стерильного карбида кремния WHISKERS™.
[0404] После инкубации в осмотической среде GN6 S/M содержимое каждой колбы объединяли в 250 мл бутыли для центрифугирования. После осаждения всех клеток из колбы на дно, объем содержимого в избытке приблизительно 14 мл жидкой среды GN6 S/M отбирали и собирали в стерильной 1 л колбе для дальнейшего использования. Предварительно смоченную суспензию WHISKERS™ перемешивали на максимальной скорости во встряхивателе в течение 60 секунд и затем добавляли в бутыль для центрифугирования.
[0405] В этом примере 159 мкг pDAB100655 (донорной последовательности) и 11 мкг плазмидной ДНК pDAB10506 (ZFN) добавляли в каждую бутыль. После добавления плазмидной ДНК, бутыль немедленно помещали в модифицированный коммерческий смеситель для краски RED DEVIL 5400™ (Red Devil Equipment Co., Plymouth, MN) и встряхивали в течение 10 секунд. После встряхивания смесь клеток, среды, WHISKERS™ и плазмидной ДНК добавляли к содержимому 1 л колбы вместе с 125 мл свежей жидкой среды GN6 для снижения осмотичности. Клеткам позволяли восстановиться во встряхивателе, установленном на 125 об/мин, в течение 2 часов. 6 мл диспергированной суспензии фильтровали на фильтровальной бумаге Whatman #4 (5,5 см) с использованием стеклянного блока для сбора клеток, соединенного с встроенной вакуумной линией, так что получали 60 фильтров на бутыль. Фильтры помещали в 60×20 мм чашки с твердой средой GN6 (такой же, как жидкая среда GN6, за исключением добавления 2,5 г/л гелеобразующего средства Gelrite) и культивировали при 28°С в условиях темноты в течение 1 недели.
Идентификация и выделение предположительных целевых событий, интегрированных в геномный локус трансгенного события кукурузы DAS-59132
[0406] Через одну неделю после доставки ДНК, фильтровальную бумагу переносили в 60×20 мм чашки с селективной средой GN6 (1H) (среда N6, 2,0 мг/л 2,4-D, 30 г/л 5 сахароза, 100 мг/л мио-инозитол, 2,5 г/л Gelrite, pH 5,8), содержащей селективное средство. Эти селективные чашки инкубировали при 28°С в течение одной недели в темноте. После 1 недели селекции в темноте, ткань погружали в свежую среду посредством соскребания 1/2 клеток с каждой чашки в пробирку, содержащую 3,0 мл среды GN6 с агарозой, поддерживаемую при 37-38°С (среда N6, 2,0 мг/л 2,4-D, 30 г/л сахароза, 100 мг/л мио-инозитол, 7 г/л агароза SEAPLAQUE®, pH 5,8, автоклавирована в течение 10 минут при 121°C).
[0407] Смесь агароза/ткань разламывали шпателем и затем 3 мл смеси агароза/ткань равномерно наливали на поверхность 100×25 мм чашки ПЕТРИ™, содержащей среду GN6 (1H). Этот процесс повторяли для обеих половин каждой чашки. После покрытия всей ткани чашки инкубировали при 28°С в условиях темноты в течение вплоть до 10 недель. Предположительно трансформированные изоляты, которые росли в этих условиях селекции, удаляли из покрытых чашек и переносили в свежую селективную среду в 60×20 мм чашках. Если стабильный рост являлся очевидным после приблизительно 2 недель, событие считали устойчивым к используемому гербициду (селективному средству) и затем отбирали аликвоту клеток для анализа генотипа. В этом примере 24 события выделили из 6 обработанных бутылей. Эти события выдвинули для молекулярного анализа для подтверждения интеграции гена AAD-1 в геномный локус трансгенного события кукурузы DAS-59132.
Молекулярный анализ нацеливания NHEJ на геномный локус трансгенного события кукурузы DAS-59132
[0408] 24 трансгенных события, выделенные после опосредованной WHISKERS™ трансформации, как описано выше, анализировали с использованием нескольких различных инструментов молекулярного подтверждения. В результате анализа идентифицированы события, содержащие копию трансгена AAD-1, интегрированную в геномный локус трансгенного события кукурузы DAS-59132. Сначала подтвердили, что 24 различных события содержат копию трансгена AAD-1, затем события анализировали, чтобы определить, в геномном ли локусе трансгенного события кукурузы DAS-59132, что позволяет предполагать, что копия трансгена AAD-1 интегрирована посредством NHEJ в геном клеток маиса. События, идентифицированные как содержащие копию трансгена AAD-1 в геномном локусе трансгенного события кукурузы DAS-59132, дополнительно подтверждали посредством реакций внутренней-внешней ПЦР и Саузерн-блоттинга. Эти анализы подтвердили, что события содержали копию трансгена AAD-1, интегрированную в геномный локус трансгенного события кукурузы DAS-59132 посредством механизма NHEJ.
[0409] Выделение ДНК: ДНК выделяли из лиофилизированной ткани каллюса маиса с использованием набора для выделения ДНК QIAGEN BIOSPRINT 96™, следуя рекомендациям производителя. Предустановленную программу использовали для автоматического выделения, и ДНК элюировали в 200 мкл 1:1 буфера ТЕ/дистиллированной воды. По два микролитра (2 мкл) из каждого образца оценивали количественно на THERMOSCIENTIFIC NANODROP 8000™, и образцы нормализовали до 100 нг/мкл с использованием QIAGEN BIOROBOT 3000™. Нормализованную ДНК хранили при 4°C до дальнейшего анализа.
[0410] Оценка количества копий: Определение количества копий трансгена посредством анализа гидролиза зонда, аналогичного анализу TAQMAN®, проводили посредством ПЦР с детекцией в реальном времени с использованием системы LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN). Анализы разработаны для AAD-1 и внутреннего стандарта - гена инвертазы - с использованием программного обеспечения LIGHTCYCLER® Probe Design 2.0. Для амплификации исходную смесь зондов LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN) подготавливали в 1X конечной концентрации в мультиплексной реакционной смеси объемом 10 мкл, содержащей 0,4 мкМ каждого праймера и 0,2 мкМ каждого зонда (таблица 29). Проводили двухстадийную реакцию амплификации с удлинением при 60°C в течение 40 секунд с получением флюоресценции. Анализ данных ПЦР с детекцией в реальном времени по количеству копий проводили с использованием программного обеспечения LIGHTCYCLER® версии 1.5 с использованием модуля относительного количественного анализа и на основе способа ΔΔCt. Для этого образец гДНК калибратора с одной копией и известный контроль с двумя копиями включали в каждый прогон.
Последовательности праймеров/зондов для анализа гидролиза зонда AAD1 и внутреннего стандарта (Inv)
Анализ повреждения геномного локуса трансгенного события кукурузы DAS-59132:
[0411] Анализ повреждения геномного локуса трансгенного события кукурузы DAS-59132 проводили посредством ПЦР с детекцией в реальном времени с использованием системы LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN). Анализы разрабатывали для мониторирования специфичности, с которой при трансгенном событии 32 ZFN6 (25716/25717) связывались и расщеплялись геномные последовательности локуса трансгенного события кукурузы DAS-59132 и гена внутреннего стандарта IVF с использованием программного обеспечения LIGHTCYCLER® Probe Design 2.0. Для амплификации исходную смесь зондов LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN) подготавливали в 1X конечной концентрации в мультиплексной реакционной смеси объемом 10 мкл, содержащей 0,4 мкМ каждого праймера и 0,2 мкМ каждого зонда (таблица 30). Проводили двухстадийную реакцию амплификации с удлинением при 55°C в течение 30 секунд с получением флюоресценции. Анализ исследования повреждения проводили с использованием соотношения мишени к стандарту (фигура 37). Четыре из восьми событий идентифицировали как содержащие трансген AAD-1, интегрированный в геномный локус трансгенного события кукурузы DAS-59132. Следующие события, состоящие из: события 100655/105906[1]-001, события 100655/105906[5]-013, события 100655/105906[5]-015 и события 100655/105906[3]-018, выдвинуты для дальнейшего молекулярного анализа для подтверждения интеграции трансгена AAD-1 в геномный локус трансгенного события кукурузы DAS-59132.
Специфическая для локуса трансгенного события кукурузы DAS-59132 внутренняя-внешняя ПЦР:
[0412] Вставка донора AAD-1 в геномный локус трансгенного события кукурузы DAS-59132 посредством NHEJ может происходить в одной из двух ориентаций. Интеграцию трансгена AAD-1 и ориентацию этой интеграции подтверждали с помощью анализа внутренней-внешней ПЦР. В анализе внутренней-внешней ПЦР используют «внешний» праймер, разработанный для связывания последовательности-мишени геномного локуса трансгенного события кукурузы DAS-59132. Кроме того, «внутренний» праймер разработан для связывания с донорной последовательностью AAD-1. Реакции амплификации, которые проводили с использованием этих праймеров, амплифицируют только донорный ген, вставленный в участок-мишень. Результирующий ампликон ПЦР был получен с двух праймеров и состоял из последовательности, проходящей через участок стыковки вставки. Для каждого образца два набора праймеров для внутренней-внешней ПЦР включали в одну мультиплексную реакцию и использовали для детекции опосредованной NHEJ вставки донора, которая могла произойти в одной из двух различных ориентаций. Положительный и отрицательный контроль включали в анализ. Две плазмиды для положительного контроля, pDAB100664 и pDAB100665, конструировали для симуляции вставки донора в геномный локус трансгенного события кукурузы DAS-59132 посредством NHEJ в одной из двух различных ориентаций.
[0413] Интеркалирующий в ДНК краситель, SYTO-13, использовали в смеси для ПЦР для детекции амплификации в реальном времени в термоциклере с возможностью детекции флюоресценции. Кроме того, программу анализа температуры плавления (Tm) присоединяли к обычной программе ПЦР, так что амплифицированные продукты можно было анализировать по их профилям Tm. Сходство профиля Tm между неизвестным образцом и образцом положительного контроля дает веские основания предполагать, что неизвестный образец имеет такой же амплифицированный продукт, как положительный контроль. Реакции ПЦР проводили с использованием 10 нг геномной ДНК-матрицы, 0,2 мкМ dNTP, 0,2 мкМ прямого и обратного праймеров, 4 мкМ SYTO-13 и 0,15 мкл Ex Taq HS. Реакции проводили в двух стадиях: первая стадия состояла из одного цикла при 94°C (2 минуты) и 35 циклов при 98°C (12 секунд), 66°C (30 секунд) и 68°C (1,3 минуты); вторая стадия представляла собой программу Tm, покрывающую 60-95°C с последующими 65°C (30 секунд) и 72°C (10 минут) (таблица 30). Ампликоны посылали для секвенирования для подтверждения того, что ген AAD-1 интегрирован в геномный локус трансгенного события кукурузы DAS-59132.
[0414] Полученные в результате внутренней-внешней ПЦР с детекцией в реальном времени ампликоны визуализировали с использованием программного обеспечения ABI. Эти результаты дополнительно подтверждали с использованием анализа сдвига подвижности в геле, где ампликоны разделяли в 1,2% геле с TAE. Ожидаемые размеры ампликонов составляли ~1,8 т.п.о. для первой ориентации (как в pDAB100664) и ~2 т.п.о. для второй ориентации (как в pDAB100665). Результаты анализа сдвига подвижности в геле подтвердили данные реакции внутренней-внешней ПЦР с детекцией в реальном времени. Оба набора данных позволяли предполагать, что копия трансгена AAD-1 была интегрирована посредством NHEJ в геном клеток маиса в геномный локус трансгенного события кукурузы DAS-59132.
Праймеры для внутренней-внешней ПЦР для детекции опосредованного NHEJ нацеливания на трансгенное событие кукурузы DAS-59132 в клетках маиса
NJ-AAD1-Pri2
pDAB100664
NJ-AAD1-Pri2
pDAB100665
[0415] Анализ Саузерн-блоттингом: Трансгенные события для каллюса маиса, идентифицированные выше, подвергали дальнейшему скринингу с использованием анализа Саузерн-блоттингом. Этот анализ использовали для дальнейшего подтверждения того, что трансген AAD-1 интегрирован посредством NHEJ в геном клеток маиса в геномный локус трансгенного события кукурузы DAS-59132. В экспериментах анализа Саузерн-блоттингом получили данные, демонстрирующие интеграцию и целостность трансгена AAD-1 в геноме сои.
[0416] Выделение ДНК: Геномную ДНК выделяли из ткани каллюса, собранной для каждого индивидуального события. Сначала образцы ткани собирали в 2 мл пробирки и лиофилизировали в течение 2 суток. Мацерацию ткани проводили с помощью измельчителя тканей KLECO™ и гранул вольфрама (Kleco, Visalia, CA). После мацерации ткани геномную ДНК выделяли с использованием набора DNEASY PLANT MINI™ (Qiagen, Germantown, MD) в соответствии с протоколом, предложенным производителем.
[0417] Саузерн-блоттинг: Геномную ДНК (гДНК) оценивали количественно с использованием набора для анализа ДНК QUANT-IT PICO GREEN™ (Molecular Probes, Invitrogen, Carlsbad, CA). Оцененную количественно гДНК доводили до 4 мкг для анализа Саузерн-блоттингом. Затем образцы ДНК расщепляли с использованием рестрикционного фермента NcoI (New England BioLabs, Ipswich, MA) в течение ночи при 37°C с последующей очисткой с использованием QUICK-PRECIP™ (Edge BioSystem, Gaithersburg, MD) в соответствии с протоколом, предложенным производителем. ДНК ресуспендировали в 1X красителе и подвергали электрофорезу в течение 5 часов в 0,8% агарозном геле SEAKEM LE™ (Lonza, Rockland, ME) при 110 вольт в холодной комнате. Гель денатурировали, нейтрализовывали и затем переносили на нейлоновую заряженную мембрану (Millipore, Bedford, MA) в течение ночи, и ДНК сшивали с мембраной с использованием UV STRATA LINKER 1800™ (Stratagene, La Jolla, CA), и блоты подвергали предгибридизации с 20 мл PERFECTHYB PLUS™ (Sigma, St. Louis, MO). Зонд 226 п.о. (SEQ ID NO:464) метили с рассеянной затравкой с использованием PRIME-IT RMT™ (Stratagene, La Jolla, CA) в соответствии с протоколом, предложенным производителем, и очищали с использованием микроколонок PROBE QUANT G-50™ (GE Healthcare, Buckinghamshire, UK) в соответствии с протоколом, предложенным производителем. Приблизительно 20000000 cpm меченого зонда добавляли к блотам и инкубировали в течение ночи. Блоты промывали дважды в течение 15 минут на промывку и помещали на экран фосфоимиджера на 24 часа, и анализировали посредством сканера STORM 860™ (Molecular Dynamics).
[0418] Ожидаемые и наблюдаемые размеры фрагментов с расщеплением NcoI и зондом, на основании известных участков рестрикционного фермента AAD-1 и трансгенного события кукурузы DAS-59132, получены в результате Саузерн-блоттинга. Два фрагмента ДНК идентифицировали после этих расщеплений и гибридизаций. Саузерн-блоттинг, для которого получены результаты с полосами размеров приблизительно 2,9 и 5,5 т.п.о., указывал на то, что трансген AAD-1 интегрирован в геномный локус трансгенного события кукурузы DAS-59132 посредством механизма NHEJ.
Пример 9: Направленная интеграция и повреждение сконструированной «посадочной площадки» кукурузы
Характеризация геномной последовательности-мишени ELP
[0419] Геномный локус, в который интегрирована сконструированная «посадочная площадка» (ELP), выбран в качестве эндогенного геномного локуса для нацеливания гена. Конструирование последовательностей ELP, содержащих участки связывания цинковых пальцев (eZFN1 и eZFN3) и приблизительно 1,0 т.п.о. случайных искусственных последовательностей, фланкирующих участки связывания цинковых пальцев, в дополнение к белкам нуклеазам с цинковыми пальцами, описано в Международной патентной заявке WO2011091317, полное содержание которой приведено в настоящем документе в качестве ссылки. Для тестирования опосредованной NHEJ направленной интеграции в локусы ELP, сконструированы две донорные ДНК, обе из которых содержат одну из двух участков связывания eZFN в плазмиде 5,3 т.п.о., содержащей ген aad-1, придающий устойчивость к гербициду галоксифопу. Фигура 38 представляет репрезентативную схему интеграции.
Регенерация растений с трансгенными событиями, содержащими ELP
[0420] Четыре трансгенных события ELP, образованных при трансформации pDAB100640, pDAB100641 и pDAB106685 (два для каждого ELP), получены, как описано в Международной патентной заявке WO2011091317, полное содержание которой приведено в настоящем документе в качестве ссылки. События получали и подтверждали, что они являются однокопийными и содержат интактный PTU, содержащий ELP. Эти объекты регенерировали для получения донорного материала для нацеливания. Здоровую растущую ткань переносили сначала на 28(1H) (среда MS (Murashige and Skoog (1962) Physiol Plant 15:473-497), 0,025 мг/л 2,4-D, 5 мг/л BAP, 1,0 мг/л Herbiace, 30 г/л сахароза, 2,5 г/л gelrite, pH 5,7) и инкубировали при низкой освещенности (14 мкЭ/м2·с, длине светового дня 16 час) в течение 7 суток с последующей высокой освещенностью (89 мкЭ/м2·с, длине светового дня 16 час) в течение других 7 суток. Зеленеющие структуры переносили на 36(1H), которая является такой же, как 28(1H) минус BAP и 2,4-D, и инкубировали с высокой освещенностью (40 мкЭ/м2·с, длине светового дня 16 час), пока структуры побегов не развили достаточно корней для пересадки в теплицу. Растения выращивали до зрелости в теплице с использованием смеси 95% METRO-MIX 360® и 5% суглинистой почвы и опыляли в зависимости от состояния здоровья растения. Активно растущие растения подвергали самоопылению или опылению сибсами (растениями из того же самого события), и менее активные растения скрещивали с Hi-II или A188 для поддержания эмбриогенной емкости донорного материала. Семена T1 высевали в горшки 4” (10 см) и проводили скрининг проросших сеянцев по зиготности посредством qPCR. Сеянцы, определенные как гомозиготные по гену PAT, переносили в горшки по 5 галлонов (18,93 л), выращивали до стадии размножения, подвергали ауткроссингу с Hi-II, и зародыши T2 использовали для нацеливания посредством опосредованной NHEJ интеграции.
Нацеливание NHEJ в протопластах ELP
[0421] Протопласты Zea mays Hi-II получали и трансформировали с использованием описанного ранее способа трансформации протопластов. Донорной плазмидной ДНК pDAB100651 трансформировали с плазмидной ДНК ZFN pDAB105941. Подобным образом, донорной плазмидной ДНК pDAB100652 трансформировали с плазмидной ДНК ZFN pDAB105943. Донорными ДНК с нуклеазами с цинковыми пальцами трансформировали трансгенные растения ELP. После введения донорной ДНК и eZFN как донорную ДНК, так и локусы ELP, интегрированные внутри геномной ДНК-мишени, расщепляли. Затем донорную ДНК вставляли в геномную мишень. Вставка донорной ДНК в геномные локусы ELP может происходить в любой ориентаций. При вставке донорной ДНК в прямой ориентации в результате получают последовательность участка стыковки, соответствующую ожидаемой гибридизации и лигированию 4 п.о. одноцепочечных комплементарных концов, полученных при расщеплении ZFN. Вставки и делеции (инделы) в участках стыковки являются распространенными. При вставке донорной ДНК в обратной ориентации в результате получают фракции обоих участков стыковки, содержащие инделы.
[0422] Выделение протопластов: Эмбриогенные суспензионные культуры маиса Hi-II получали и поддерживали с расписанием поддержания 3,5 суток. В 50 мл стерильной конической пробирке растворы ферментов 10 мл раствора стерильной 6% (масс./об.) целлюлазы и 10 мл раствора стерильной 0,6% (масс./об.) пектолиазы пипетировали в конической пробирке с использованием наконечника пипетки 10 мл. Затем 4 объема осажденных клеток (PCV) суспензии клеток Hi-II добавляли в 50 мл пробирку, содержащую раствор для расщепления и обернутую парафильмом. Пробирки помещали на качающуюся платформу на ночь при комнатной температуре на ~16-18 час. На следующее утро пробирки удаляли из встряхивателя. В стерильную 50 мл коническую пробирку клетки и раствор фермента медленно фильтровали через клеточное сито 100 мкм. Затем клетки промывали с использованием клеточного сита 100 мкм пипетированием 10 мл среды W5 через сито. В стерильную 50 мл коническую пробирку клетки и раствор фермента медленно фильтровали через клеточное сито 70 мкм. За этой стадией фильтрования следовала вторая стадия фильтрования, где клетки и раствор фермента медленно фильтровали в 50 мл коническую пробирку через клеточное сито 40 мкм. С использованием наконечника пипетки 10 мл клеточное сито 40 мкм промывали 10 мл среды W5 для получения конечного объема 40 мл, и пробирку переворачивали. Очень медленно, 8 мл сахарозной подушки добавляли на дно раствора протопластов/фермента. С использованием центрифуги с качающимся бакет-ротором пробирки центрифугировали в течение 15 минут при 1500 об/мин. Клетки протопласты удаляли с использованием 5 мл наконечника пипетки узкого поперечного сечения. Эти клетки (7-8 мл), которые наблюдали в виде полосы протопластов, удаляли очень медленно и помещали в стерильную 50 мл коническую пробирку. Затем 25 мл среды W5 использовали для промывки пробирок. Добавляли среду W5, и пробирки медленно переворачивали и центрифугировали в течение 10 минут при 1500 об/мин. Супернатант удаляли, и 10 мл раствора MMG добавляли с медленным переворачиванием пробирки для ресуспендирования осадка протопластов. Плотность протопластов определяли с использованием гемоцитометра, для 4 PCV получали выход ~30 миллионов протопластов.
[0423] Трансформация протопластов: Клетки протопласты разводили до 1,6 миллиона протопластов на мл с использованием раствора MMG. Протопласты осторожно ресуспендировали медленным переворачиванием пробирки. Затем 300 мкл протопластов (~500000 протопластов) добавляли в стерильную 2 мл пробирку, пробирки переворачивали для равномерного распределения клеток протопластов. Плазмидную ДНК в концентрации приблизительно 80 мкг, суспендированную в буфере ТЕ, добавляли к протопластам. Трансформировали конструкциями как ZFN, так и донорной плазмиды. Экспрессирующие eZFN плазмиды (pDAB105941 и pDAB105943) добавляли по отдельности или в комбинации в соотношении 1:1 или 10:1 донорной ДНК (pDAB100651 и pDAB100652) к ДНК eZFN для каждой из обработок eZFN1 и eZFN3. Эффективность eZFN тестировали ранее, и они описаны более подробно в Международной патентной заявке WO2011091317, фигура 39 приведена в качестве представления eZFN относительно донорной ДНК. Пробирки медленно поворачивали для суспендирования ДНК с протопластами, и пробирки инкубировали в течение 5-10 минут при комнатной температуре. Затем 300 мкл раствора ПЭГ добавляли к раствору протопластов/ДНК. После добавления всего раствора ПЭГ, раствор ПЭГ смешивали с раствором протопластов осторожным переворачиванием пробирки. Смесь инкубировали при комнатной температуре в течение 15-20 минут с периодическим переворачиванием пробирки(пробирок). После инкубации 1 мл раствора W5 медленно добавляли в пробирки, и пробирки осторожно переворачивали. Наконец, раствор центрифугировали при 1000 об./мин в течение 15 минут. Супернатант осторожно удаляли, так чтобы не потревожить осадок клеток. Наконец, добавляли 1 мл раствора для промывки/инкубации. Пробирки осторожно переворачивали, чтобы ресуспендировать осадок клеток. Пробирки накрывали алюминиевой фольгой для исключения любого воздействия света, и укладывали в штатив на бок для инкубации в течение ночи. Клетки собирали через 24 часа после трансформации для молекулярного анализа для идентификации локусов ELP, содержащих донор, интегрированный посредством опосредованной NHEJ репарации ДНК.
Концентрации ДНК eZFN и донорной плазмидной ДНК, трансформированных в клетки маиса и интегрированных в локусы ELP
(мкг)
(pDAB)
(мкг)
(пг)
Молекулярное подтверждение нацеливания на ELP маиса посредством NHEJ в протопластах
[0424] Выделение ДНК: ДНК выделяли из ткани маиса автоматически с использованием робота Qiagen BIOSPRINT 96™, и ДНК элюировали в 200 мкл 1:1 буфера ТЕ/дистиллированной воды. ДНК из каждого образца оценивали количественно на THERMOSCIENTIFIC NANODROP 8000™, и образцы нормализовали до 100 нг/мкл с использованием QIAGEN BIOROBOT 3000™. Нормализованную ДНК хранили при 4°C до дальнейшего анализа.
[0425] Анализ повреждения локуса ELP: После сбора протопластов через 24 час, ДНК выделяли и анализировали с использованием анализ повреждения, анализа участков стыковки с использованием ПЦР и секвенирования ДНК, полученной посредством ПЦР участков стыковки. Анализ повреждения является косвенным показателем относительной активности расщепления eZFN. Анализом на основе TAQMAN™ определяют утрату интактных участков связывания eZFN в ДНК-мишени, как ожидают из-за неправильной репарации концов ДНК, которая может происходить при лигировании концов.
[0426] Данные анализа на основе TAQMAN™ позволяют предполагать, что eZFN1 обладает более высокой активностью, чем eZFN3, а также что присутствует значительное расщепление ДНК-мишени посредством eZFN, как показано по статистически значимому снижению сигнала в образцах eZFN по сравнению с образцами только донора.
[0427] Расщепление геномной ДНК-мишени посредством eZFN. ДНК выделяли для каждой группы обработки (6 повторов каждой), как указано. Анализы Taqman использовали для измерения расщепления ДНК-мишени. Анализ повреждения локуса ELP проводили посредством ПЦР с детекцией в реальном времени с использованием системы LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN). Анализы разрабатывали для мониторирования последовательностей связывания eZFN1 и eZFN3 внутри ELP1 и гена внутреннего стандарта IVF с использованием программного обеспечения LIGHTCYCLER® Probe Design 2.0. Для амплификации исходную смесь зондов LIGFITCYCLER®480 (Roche Applied Science, Indianapolis, IN) подготавливали в 1X конечной концентрации в мультиплексной реакционной смеси объемом 10 мкл, содержащей 0,4 мкМ каждого праймера и 0,2 мкМ каждого зонда (таблица 33). Проводили двухстадийную реакцию амплификации с удлинением при 55°C в течение 30 секунд с получением флюоресценции. Анализ исследования повреждения проводили с использованием соотношения мишени к стандарту. Локализация праймеров показана на фигуре 40.
Последовательности праймеров и зондов для анализа повреждения
[0428] Специфическая для локуса внутренняя-внешняя ПЦР: Вставка донора в ожидаемый участок ELP с использованием опосредованной NHEJ репарации может приводить к двум различным ориентациям. Две плазмиды для положительного контроля, pDAB100660 и pDAB100662, конструировали и трансформировали ими растения для симуляции вставки донора в участок ELP. Трансгенные растения, полученные с помощью контрольных конструкций pDAB100660 и pDAB100662, анализировали с использованием дизайна внутренней-внешней ПЦР, и условия внутренней-внешней ПЦР определяли на этом растительном материале.
[0429] В анализе внутренней-внешней ПЦР присутствует «внешний» праймер в последовательности ELP, в то время как «внутренний» праймер расположен в донорной последовательности, так что только когда донор вставлен в участок-мишень, два праймера могут амплифицировать последовательность, проходящую через участок стыковки вставки (таблица 34). Для каждого образца две реакции внутренней-внешней ПЦР использовали для детекции вставки донора в двух различных ориентациях. Положительный и отрицательный контроль включали в анализ.
[0430] Интеркалирующий в ДНК краситель, SYTO-13, использовали в смеси для ПЦР для детекции амплификации в реальном времени в термоциклере с возможностью детекции флюоресценции. Кроме того, программу анализа температуры плавления (Tm) присоединяли к обычной программе ПЦР, так что амплифицированные продукты можно было анализировать по их профилям Tm. Сходство профиля Tm между неизвестным образцом и образцом положительного контроля дает веские основания предполагать, что неизвестный образец имеет такой же амплифицированный продукт, как положительный контроль. Положительные по нацеливанию образцы, идентифицированные в анализе внутренней-внешней ПЦР с детекцией в реальном времени, дополнительно визуализировали с использованием анализа сдвига подвижности в геле. Ожидаемые размеры ампликонов составляли ~1,5 т.п.о. для ориентации 1 (как в pDAB100662) и ~1,4 т.п.о. для ориентации 2 (как в pDAB100660).
Праймеры для внутренней-внешней ПЦР для детекции опосредованного NHEJ нацеливания на ELP1 в маисе
контроль
NJ-AAD1-Pri2
SEQ ID NO:477 5’-GAC CAA GTC CTT GTC TGG GAC A-3’
pDAB100662
ELP2-PriR1
SEQ ID NO:479 5'-GAT GGT GGT TAT GAC AGG CTC CT-3’
pDAB100660
[0431] Реакции ПЦР проводили с использованием 10 нг геномной ДНК-матрицы, 0,2 мкМ dNTP, 0,2 мкМ прямого и обратного праймеров, 4 мкМ SYTO-13 и 0,15 мкл Ex Taq HS. Реакции проводили в двух стадиях: первая стадия состояла из одного цикла при 94°C (2 минуты) и 35 циклов при 98°C (12 секунд), 66°C (30 секунд) и 68°C (1,3 минуты); вторая стадия представляла собой программу Tm, покрывающую 60-95°C с последующими 65°C (30 секунд) и 72°C (10 минут). Продукты визуализировали с использованием программного обеспечения ABI, так же как посредством разделения в 1,2% геле с TAE.
[0432] Анализ внутренней-внешней ПЦР участков стыковки для eZFN1 и eZFN3 для опосредованного NHEJ нацеливания обработок донором, включающих только донорную ДНК, только eZFN или донорную ДНК с ДНК eZFN (в соотношении либо 1:1, либо 10:1) проводили в агарозных гелях. Результаты показали, что размер ампликона ПЦР для ДНК донора и eZFN являлся ожидаемым для направляемого NHEJ события.
[0433] Последовательность участков стыковки мишень/донор: Для нацеленных на ELP событий, подтвержденных посредством анализа внутренней-внешней ПЦР, продукты ампликонов ПЦР подтверждали секвенированием, и участки стыковки мишень-донор валидировали стандартным секвенированием по Сенгеру. Кратко, анализ ПЦР участков стыковки проводили для всех повторов в каждой группе обработки. Праймеры для ПЦР выбирали для амплификации одной стороны последовательностей участков стыковки вставки, которые находились либо в прямой, либо в обратной ориентации. Продукты ПЦР наблюдали в образцах, полученных из образцов ДНК eZFN и донора, но не из контрольных образцов, содержащих образцы только донорной ДНК или только eZFN. Продукты ПЦР выявлены в большинстве повторов образцов для обоих использованных соотношений ДНК eZFN и донора.
[0434] Репрезентативные образцы продуктов ПЦР клонировали и секвенировали. Как для прямой, так и для обратной ориентации, последовательности продуктов ПЦР из четырех различных реакций показаны на фигуре 41. Девять уникальных гаплотипов наблюдали для прямой ориентации вставки, как ожидали для неправильной репарации концов участков стыковки. Три из 16 последовательностей выровнены с последовательностью, ожидаемой при гибридизации и лигировании интактных концов вставленной донорной ДНК и последовательности-мишени. Все последовательности продуктов ПЦР в обратной ориентации имели инделы в участках стыковки, как ожидали, поскольку одноцепочечные концы донорной ДНК и ДНК-мишени не являются комплементарными.
Результаты опосредованного NHEJ нацеливания донора в протопластах маиса
[0435] Разработана система анализа временной трансформации маиса на основе протопластов, в которой показана высокая, воспроизводимая экспрессия репортерных генов. Протопласты получали из суспензионной культуры маиса, разработанной в DAS. Трансгенную линию маиса (линию маиса 106685[1]-007), несущую вставку ELP, использовали для опосредованной NHEJ интеграции донорной последовательности ДНК.
Нацеливание NHEJ на зародыши ELP посредством бомбардировки микрочастицами
[0436] За трое суток до бомбардировки микрочастицами зародыши 1,5-2,2 мм выделяли из колосьев со стерилизованной поверхностью и помещали (щитком вверх) на базовую среду N6 с витаминами (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л мио-инозитола и 4,25 мг/л нитрата серебра, отвержденную с помощью 2,5 г/л Gelzan (Phytotechnology Laboratories, Shawnee Mission, KS). За четыре часа до бомбардировки микрочастицами ~35-40 зародышей помещали (щитком вверх) в центр 100×15 мм чашки Петри, содержащей такую же среду с добавлением 36,4 г/л сорбита и 36,4 г/л маннита.
[0437] Микрочастицы золота (0,6 микрон, BioRad, Hercules, CA,) подготавливали для осаждения ДНК посредством взвешивания 15 мг в стерильную, силиконизированную 1,7 мл пробирку для микроцентрифугирования (Sigma-Aldrich, St. Louis, MO), и медленно добавляли 500 мкл ледяного 100% этанола. После 15 секунд обработки ультразвуком в ультразвуковой водяной бане FS-14 (Fisher Scientific, Nazareth, PA) золоту позволяли осесть в течение 30 минут при комнатной температуре перед центрифугированием при 3000 об./мин в течение 60 секунд с использованием MiniSpin (Eppendorf, Hauppauge, NY). После удаления супернатанта добавляли 1 мл ледяной, стерильной воды, перемешивали пипетированием вверх и вниз и позволяли осесть в течение 3-5 минут перед центрифугированием при 3000 об/мин в течение 60 секунд. Стадию промывки повторяли еще один раз перед суспендированием золота в 500 мкл ледяной, стерильной воды. Промытое золото затем разделяли на аликвоты в отдельные стерильные, силиконизированные 1,7 мл пробирки для микроцентрифугирования (50 мкл на пробирку), обеспечивая поддержание золота хорошо перемешанным посредством пипетирования вверх и вниз между пробирками. Промытое золото (~1,5 мг на 50 мкл) затем хранили при -20°C до необходимости.
[0438] Для осаждения ДНК одну пробирку, содержащую ~1,5 мг золота в 50 мкл воды, размораживали для каждых 10 мишеней, подлежащих бомбардировке, и обрабатывали ультразвуком в ультразвуковой водяной бане в течение 15 секунд, затем помещали в лед. Плазмидную ДНК (0,5 мкг ZFN + 4,5 мкг донора) предварительно смешивали в 0,6 мл пробирках для микроцентрифугирования (Fisher Scientific, Nazareth, PA) и добавляли к суспензии золота с осторожным пипетированием вверх и вниз несколько раз для полного смешивания. Добавляли пятьдесят мкл ледяного 2,5M хлорида кальция и осторожно перемешивали пипетированием вверх и вниз несколько раз. Затем добавляли двадцать микролитров холодного 0,1M спермидина и осторожно перемешивали пипетированием вверх и вниз несколько раз. Затем пробирку закрывали крышкой и помещали во встряхиватель Genie 2 (Scientific Instruments Inc., Bohemia, NY) и позволяли смешивание (установка на «встряхивание 2») в течение 10 минут, после чего смеси позволяли осесть в течение 3-5 минут. После центрифугирования в течение 15 секунд при 5000 об/мин супернатант осторожно удаляли и добавляли 250 мкл ледяного, 100% этанола, пробирку закрывали крышкой и энергично перемешивали вручную, чтобы разрыхлить осадок. После второго центрифугирования в течение 15 секунд при 5000 об/мин, добавляли 120 мкл ледяного, 100% этанола, пробирку закрывали крышкой и энергично перемешивали вручную, чтобы разрыхлить осадок.
[0439] Для бомбардировки микрочастицами стерилизованные макроносители (BioRad, Hercules, CA) вставляли в держатели из нержавеющей стали (BioRad, Hercules, CA) и автоклавировали. Десять мкл суспензии золото/ДНК равномерно распределяли в центре макроносителя, обеспечивая пипетирование вверх и вниз, так чтобы поддерживать хорошее перемешивание, затем помещали на кусок стерильной 125 мм фильтровальной бумаги Whatman #4 (GE Healthcare, Buckinghamshire, UK) на подложке из 8-меш Drierite (W.A Hammond Drierite Co., Xenia, OH) в стеклянной чашке Петри 140×25 мм. Золоту/ДНК позволяли полностью высохнуть в течение приблизительно 10 минут. Разрывные диски (650 psi (4482 кПа), BioRad, Hercules, CA) стерилизовали погружением на несколько секунд в изопропиловый спирт, затем загружали в колпачок распылителя устройства для бомбардировки микрочастицами (PDS-1000, BioRad, Hercules, CA). Автоклавированный останавливающий экран (BioRad, Hercules, CA) и загруженный макроноситель помещали в пусковой контейнер, крышку завинчивали и вставляли в камеру для бомбардировки непосредственно под распылитель. Чашку Петри, содержащую покрытый экраном лист мишени, открывали и помещали в камеру для бомбардировки на 6 см ниже распылителя. Подключали вакуум (-0,9 бар (90 кПа)), и включали устройство.
[0440] На следующие сутки (16-20 часов после бомбардировки), подвергнутые бомбардировке зародыши переносили (щитком вверх) на базовую среду N6 с витаминами (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л мио-инозитола и 4,25 мг/л нитрата серебра, отвержденную с помощью 2,5 г/л Gelzan (Phytotechnology Laboratories, Shawnee Mission, KS). Через 7 суток - 8 суток после бомбардировки, 11 суток от начала культивирования - зародыши переносили на селективную среду - базовую среду N6 с витаминами (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л мио-инозитола, 4,25 мг/л нитрата серебра и 0,0362 мг/л R-галоксифоп-кислоты, отвержденную с помощью 2,5 г/л Gelzan (Phytotechnology Laboratories, Shawnee Mission, KS). Через две недели зародыши переносили на свежую селективную среду - базовую среду N6 с витаминами (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л мио-инозитола, 4,25 мг/л нитрата серебра и 0,181 мг/л R-галоксифоп-кислоты, отвержденную с помощью 2,5 г/л Gelzan (Phytotechnology Laboratories, Shawnee Mission, KS), и через дополнительные две недели их переносили на свежую среду того же состава.
[0441] Образцы каллюса, растущего на 0,181 мг/л R-галоксифопа, отбирали для молекулярного анализа. Отбор образцов включал помещение либо ~50 мг в 1,2 мл кассетные пробирки для анализа ПЦР, либо ~200 мг в 2,0 мл пробирки Safe Lock (Eppendorf, Hauppauge, NY) для анализа Саузерн-блоттингом, окруженные сухим льдом для быстрого замораживания. Затем пробирки покрывали лентой из микропористого материала 3M (Fisher Scientific, Nazareth, PA) и лиофилизировали в течение 48 часов в Virtual XL-70 (VirTis, Gardiner, NY). Сразу после лиофилизации ткани пробирки закрывали крышкой и хранили при 8°C до анализа.
Молекулярное подтверждение нацеливания на ELP маиса посредством NHEJ в растениях
[0442] Выделение ДНК: ДНК выделяли из ткани маиса автоматически с использованием робота Qiagen BIOSPRINT 96™, и ДНК элюировали в 200 мкл 1:1 буфера ТЕ/дистиллированной воды. По два мкл из каждого образца оценивали количественно на THERMOSCIENTIFIC NANODROP 8000™, и образцы нормализовали до 100 нг/мкл с использованием QIAGEN BIOROBOT 3000™. Нормализованную ДНК хранили при 4°C до дальнейшего анализа.
[0443] Оценка количества копий: Определение количества копий трансгена посредством анализа гидролиза зонда, аналогичного анализу TAQMAN®, проводили посредством ПЦР с детекцией в реальном времени с использованием системы LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN). Анализы разработаны для трансгена aad-1 и внутреннего стандарта - гена инвертазы - с использованием программного обеспечения LIGHTCYCLER® PROBE DESIGN 2.0. Для амплификации исходную смесь зондов LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN) подготавливали в 1X конечной концентрации в мультиплексной реакционной смеси объемом 10 мкл, содержащей 0,4 мкМ каждого праймера и 0,2 мкМ каждого зонда (таблица 32). Проводили двухстадийную реакцию амплификации с удлинением при 60°C в течение 40 секунд с получением флюоресценции. Анализ данных ПЦР с детекцией в реальном времени по количеству копий проводили с использованием программного обеспечения LIGHTCYCLER® версии 1.5 с использованием модуля относительного количественного анализа и на основе способа ΔΔCt. Для этого образец гДНК калибратора с одной копией и известный контроль с 2 копиями включали в каждый прогон. Расположение праймеров показано на фигуре 41.
Последовательности праймеров/зондов для анализа гидролиза зонда AAD1 и внутреннего стандарта (Inv)
вание праймера
[0444] Анализ повреждения локуса ELP: Анализ повреждения локуса ELP проводили посредством ПЦР с детекцией в реальном времени с использованием системы LIGHTCYCLER®480 (Roche Applied Science, Indianapolis, IN), как описано ранее выше. Анализы разрабатывали для мониторирования последовательностей связывания eZFNl и eZFN3 внутри ELP1 и гена внутреннего стандарта IVF с использованием программного обеспечения LIGHTCYCLER® 2.0. Анализ для исследования повреждения проводили с использованием соотношения мишени к стандарту. Результаты показаны на фигуре 42.
[0445] Специфическая для локуса внутренняя-внешняя ПЦР: Анализ внутренней-внешней ПЦР проводили, как описано ранее выше. Анализ внутренней-внешней ПЦР участков стыковки для eZFN1 и eZFN3 при обработках для направляемого NHEJ нацеливания донора, включающих только донорную ДНК, только eZFN или донорную ДНК с ДНК eZFN (в соотношении либо 1:1, либо 10:1), проводили в агарозных гелях. Результаты показали, что размер ампликона ПЦР для ДНК донора и eZFN являлся ожидаемым для направляемого NHEJ события.
[0446] Последовательность участков стыковки мишень/донор: Для нацеленных на ELP событий, подтвержденных посредством анализа внутренней-внешней ПЦР, продукты ампликонов ПЦР подтверждали секвенированием, и участки стыковки мишень-донор валидировали стандартным секвенированием по Сенгеру. Кратко, анализ ПЦР участков стыковки проводили для всех повторов в каждой группе обработки. Праймеры для ПЦР выбирали для амплификации одной стороны последовательностей участков стыковки вставки, которые находились либо в прямой, либо в обратной ориентации. Продукты ПЦР наблюдали в образцах, полученных из образцов ДНК eZFN и донора, но не из контрольных образцов, содержащих образцы только донорной ДНК или только eZFN. Продукты ПЦР выявлены в большинстве повторов образцов для обоих использованных соотношений ДНК eZFN и донора.
[0447] Репрезентативные образцы продуктов ПЦР клонировали и секвенировали. Как для прямой, так и для обратной ориентации, последовательности продуктов ПЦР из четырех различных реакций показаны на фигуре 43. Девять уникальных гаплотипов наблюдали для прямой ориентации вставки, как ожидали для неправильной репарации концов участков стыковки. Три из 16 последовательностей выровнены с последовательностью, ожидаемой при гибридизации и лигировании интактных концов вставленной донорной ДНК и последовательности-мишени. Все последовательности продуктов ПЦР в обратной ориентации имели инделы в участках стыковки, как ожидали, поскольку одноцепочечные концы донорной ДНК и ДНК-мишени не являются комплементарными.
Анализ Саузерн-блоттингом: Каллюсы маиса, первоначально идентифицированные как содержащие донорную последовательность, интегрированную в локус ELP, посредством специфических для локуса анализа повреждения и внутренней-внешней ПЦР, выбраны для дальнейшего анализа посредством Саузерн-блоттинга для направленной интеграции (TI). Для Саузерн-блоттинга T1 ДНК расщепляли и анализировали зондами с помощью ферментов и зондов в локусе-мишени. Для выделения ДНК для Саузерн-блоттинга, образцы ткани собирали в 2 мл пробирки Эппендорф (Eppendorf) и лиофилизировали в течение 2 суток. Мацерацию ткани проводили с помощью измельчителя тканей Kleco и гранул вольфрама (Kleco, Visalia, CA). После мацерации ткани геномную ДНК выделяли с использованием набора DNEASY PLANT MINI™ (Qiagen, Germantown, MD) в соответствии с протоколом, предложенным производителем.
[0448] Геномную ДНК (гДНК) оценивали количественно посредством набора для анализа ДНК QUANT-IT PICO GREEN™ (Molecular Probes, Invitrogen, Carlsbad, CA). Оцененную количественно гДНК доводили до 4 мкг для анализа Саузерн-блоттингом. Затем образцы ДНК расщепляли с использованием рестрикционного фермента PmeI (New England BioLabs) в течение ночи при 37°C с последующей очисткой с использованием QUICK-PRECIP™ (Edge Bio System, Gaithersburg, MD) в соответствии с протоколом, предложенным производителем. Затем ДНК ресуспендировали в 1X красителе и подвергали электрофорезу в течение 5 часов в 0,8% агарозном геле SEAKEM LE (Lonza, Rockland, ME) при 110 вольт в холодной комнате. Гель денатурировали, нейтрализовывали и затем переносили на нейлоновую заряженную мембрану (Millipore, Bedford, MA) в течение ночи, и ДНК сшивали с мембраной с использованием UV STRATA ЛИНКЕР 1800™ (Stratagene, La Jolla, CA), и блоты подвергали предгибридизации с 20 мл PERFECTHYB PLUS™ (Sigma, St. Louis, MO). Зонд метили с рассеянной затравкой с использованием PRIME-IT RMT™ (Stratagene, La Jolla, CA) в соответствии с протоколом, предложенным производителем, и очищали с использованием микроколонок PROBE QUANT G-50™ 30 (GE Healthcare, Buckinghamshire, UK) в соответствии с протоколом, предложенным производителем. Приблизительно 20000000 cpm меченого зонда добавляли к блотам и инкубировали в течение ночи. Блоты промывали 2×15 минут на промывку, помещали на экран фосфоимиджера на 24 часа и анализировали посредством сканера STORM 860™ (Molecular Dynamics). Результаты показали ожидаемые полосы для направленной интеграции (~6,9 т.п.о.).
Результаты нацеливания на локусы ELP посредством опосредованной NHEJ интеграции
[0449] Результаты этого исследования показывают точную вставку в маис донорной плазмидной ДНК посредством NHEJ, вслед за вызванным ZFN расщеплением in vivo, ДНК-мишени и донорной ДНК. Нацеливание донорных ДНК происходило с использованием двух различных донорных ДНК (отличающихся тем, какие связывающие последовательности eZFN содержались в ELP) и двух различных eZFN в протопластах и зародышах маиса. Интеграция в локусы ELP посредством механизма репарации NHEJ происходило в обеих ориентациях. Вставку донорной ДНК детектировали в обеих из этих ориентаций в тестированных образцах.
[0450] Точное нацеливание генов с использованием механизма репарации NHEJ в тканях различных видов растений обеспечивает значительное преимущество над известными механизмами репарации, такими как опосредованная гомологичной рекомбинацией репарация. Механизм репарации NHEJ является преобладающим механизмом репарации, который действует в большинстве, если не во всех, тканях растений. Напротив, активность опосредованного гомологичной рекомбинацией пути репарации действует на протяжении фазы G2 клеточного цикла в растениях. В результате, многие ткани растений, способные к трансформации, не подвергаются активно делению клеток и, таким образом, не могут поддерживать нацеливание генов посредством опосредованного гомологичной рекомбинацией пути репарации. Другим преимуществом опосредованного NHEJ пути репарации для вставки донора в геном растений является то, что в отличие от опосредованного гомологичной рекомбинацией пути репарации, опосредованная NHEJ репарация не требует обширных областей гомологии, что уменьшает размер последовательностей донорной ДНК, необходимых для геномной вставки. Наконец, донорные последовательности ДНК более крупных размеров можно успешно вставлять в геномные локусы с использованием опосредованного NHEJ пути репарации по сравнению с опосредованным гомологичной рекомбинацией путем репарации. Опосредованная донорной ДНК интеграция в геномный локус посредством опосредованного гомологичной рекомбинацией пути репарации требует, чтобы ДНК-полимеразы копировали ДНК-матрицу, содержащуюся в донорной ДНК. В отличие от этого, опосредованный NHEJ путь репарации требует только взаимодействия донорной ДНК с ДНК-мишенью в двух точках (их концах) и не требует зависимого от матрицы синтеза ДНК. Соответственно, опосредованный NHEJ путь репарации можно использовать для интеграции донорных последовательностей ДНК более крупных размеров в геномный локус-мишень.
Пример 10: Иллюстративные последовательности






























































[0639] Полное содержание всех патентов, патентных заявок и публикаций, упомянутых в настоящем документе, таким образом, приведено в качестве ссылки.
[0640] Хотя описание представлено с некоторыми подробностями посредством иллюстраций и примеров с целью ясности для понимания, специалистам в данной области очевидно, что различные изменения и модификации можно осуществлять на практике без отклонения от сущности или объема описания. Соответственно, предшествующие описания и примеры не следует рассматривать как ограничивающие.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ВСТРАИВАНИЯ ЭКЗОГЕННОЙ ПОСЛЕДОВАТЕЛЬНОСТИ В ГЕНОМ РАСТЕНИЙ | 2014 |
|
RU2723130C2 |
| БЫСТРЫЙ НАПРАВЛЕННЫЙ АНАЛИЗ СЕЛЬСКОХОЗЯЙСТВЕННЫХ КУЛЬТУР ДЛЯ ОПРЕДЕЛЕНИЯ ДОНОРНОЙ ВСТАВКИ | 2014 |
|
RU2668819C2 |
| СКОНСТРУИРОВАННАЯ СПОСОБАМИ ИНЖЕНЕРИИ ПЛАТФОРМА ДЛЯ ВСТРАИВАНИЯ ТРАНСГЕНА (ETIP) ДЛЯ НАЦЕЛИВАНИЯ ГЕНОВ И СТЭКИНГА ПРИЗНАКОВ | 2013 |
|
RU2666916C2 |
| ЛОКУСЫ FAD3 ДЛЯ ВЫПОЛНЕНИЯ ОПЕРАЦИЙ И СООТВЕТСТВУЮЩИЕ СВЯЗЫВАЮЩИЕСЯ СО СПЕЦИФИЧЕСКИМИ САЙТАМИ-МИШЕНЯМИ БЕЛКИ, СПОСОБНЫЕ К ВЫЗОВУ НАПРАВЛЕННЫХ РАЗРЫВОВ | 2013 |
|
RU2665811C2 |
| ОБОГАЩЕНИЕ АКТИВИРУЕМОЙ ФЛУОРЕСЦЕНЦИЕЙ СОРТИРОВКИ КЛЕТОК (FACS) ДЛЯ СОЗДАНИЯ РАСТЕНИЙ | 2013 |
|
RU2679510C2 |
| ЛОКУСЫ ФУНКЦИОНАЛЬНОСТИ FAD2 И СООТВЕТСТВУЮЩИЕ СПЕЦИФИЧНЫЕ К УЧАСТКУ-МИШЕНИ СВЯЗЫВАЮЩИЕ БЕЛКИ, СПОСОБНЫЕ ИНДУЦИРОВАТЬ НАПРАВЛЕННЫЕ РАЗРЫВЫ | 2013 |
|
RU2656159C2 |
| УНИВЕРСАЛЬНАЯ ДОНОРНАЯ СИСТЕМА ДЛЯ НАПРАВЛЕННОГО ВОЗДЕЙСТВИЯ НА ГЕНЫ | 2014 |
|
RU2687369C2 |
| ОПТИМАЛЬНЫЕ ЛОКУСЫ СОИ | 2014 |
|
RU2719150C2 |
| ФУНКЦИОНАЛЬНЫЕ ЛОКУСЫ FAD2 И СООТВЕТСТВУЮЩИЕ СПЕЦИФИЧНЫЕ ДЛЯ САЙТА-МИШЕНИ СВЯЗЫВАЮЩИЕСЯ БЕЛКИ, СПОСОБНЫЕ ИНДУЦИРОВАТЬ НАПРАВЛЕННЫЕ РАЗРЫВЫ | 2013 |
|
RU2656158C2 |
| СПОСОБЫ ОБНАРУЖЕНИЯ ДНК ДЛЯ САЙТ-СПЕЦИФИЧЕСКОЙ НУКЛЕАЗНОЙ АКТИВНОСТИ | 2013 |
|
RU2678001C2 |





































































































































































































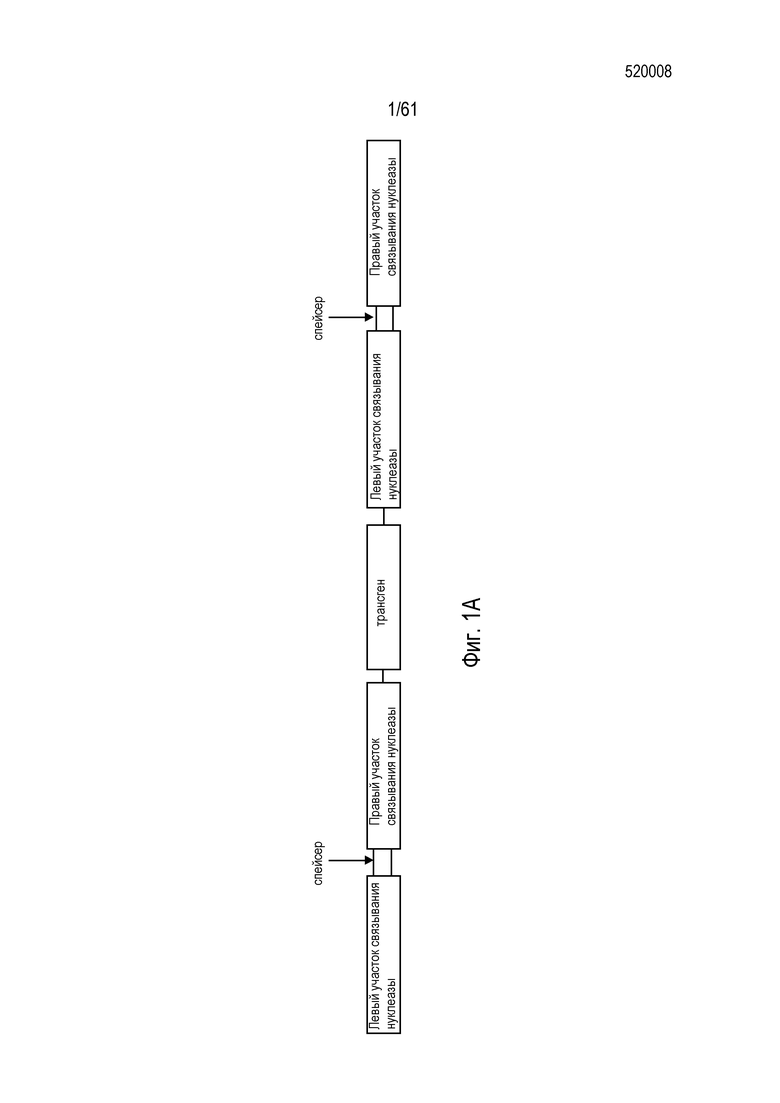
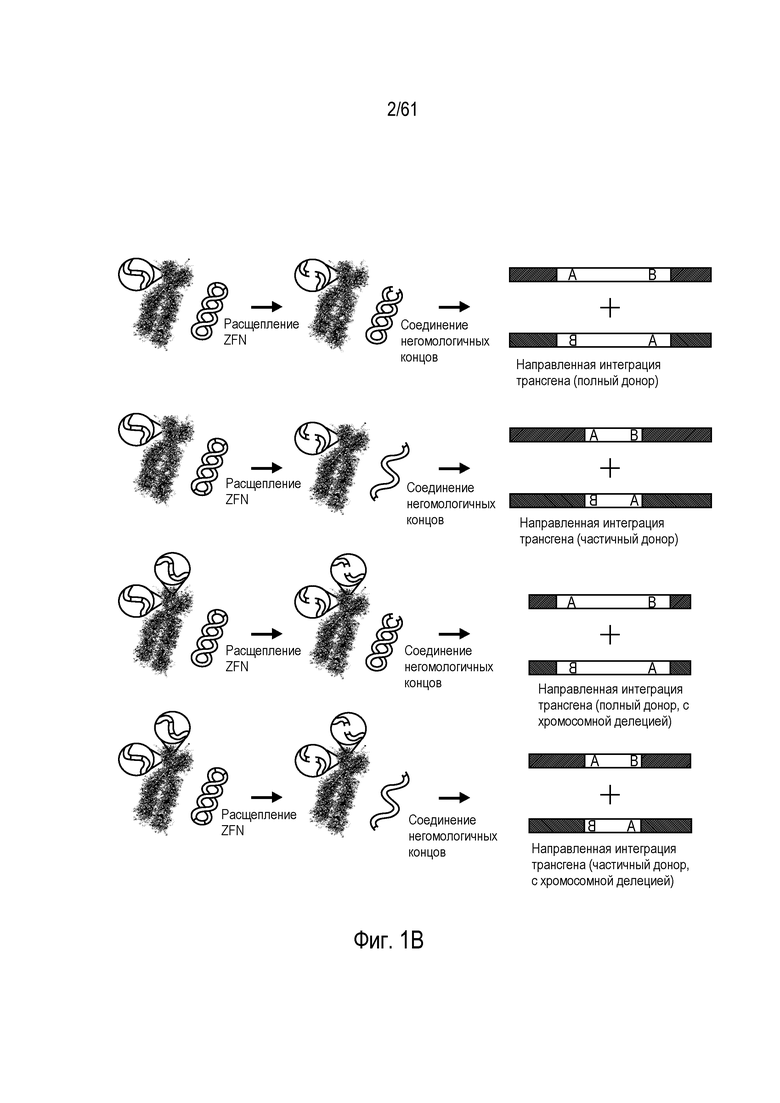
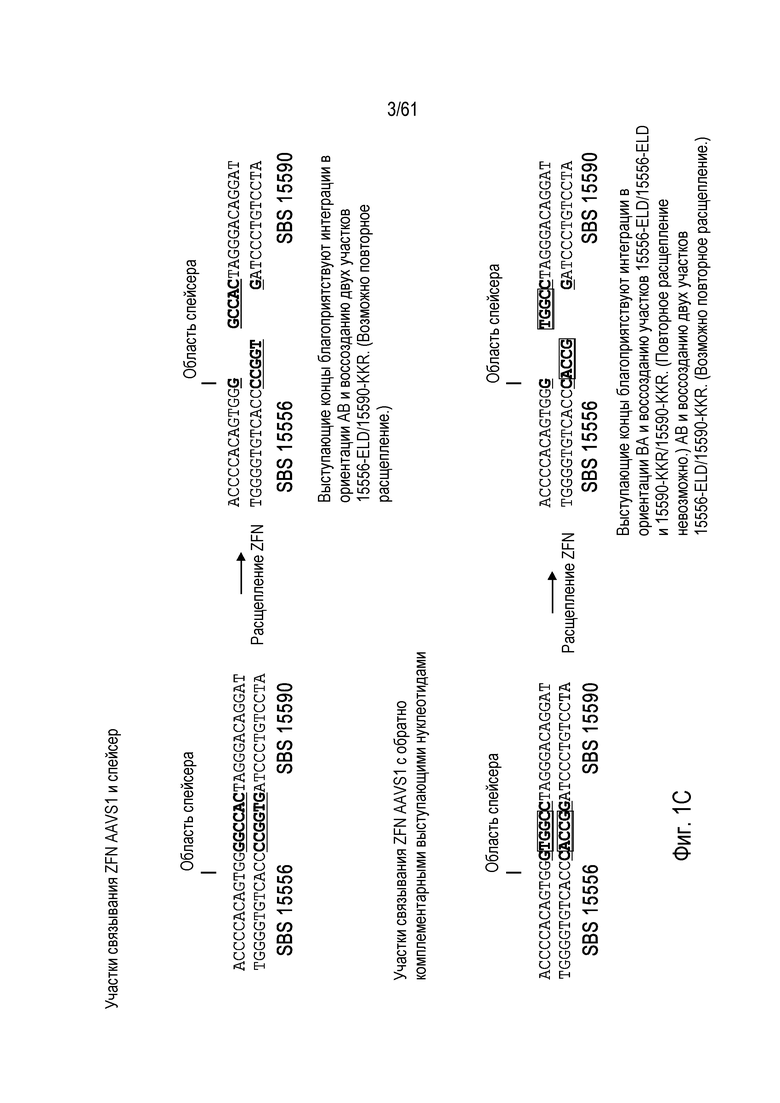
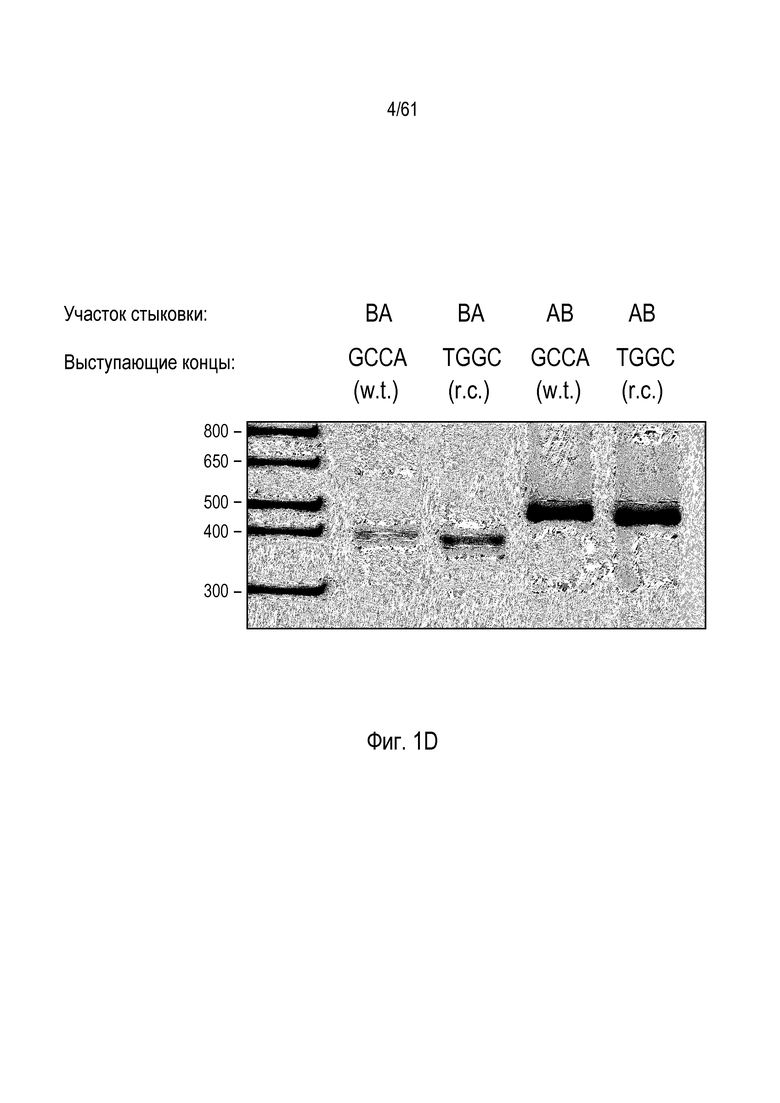
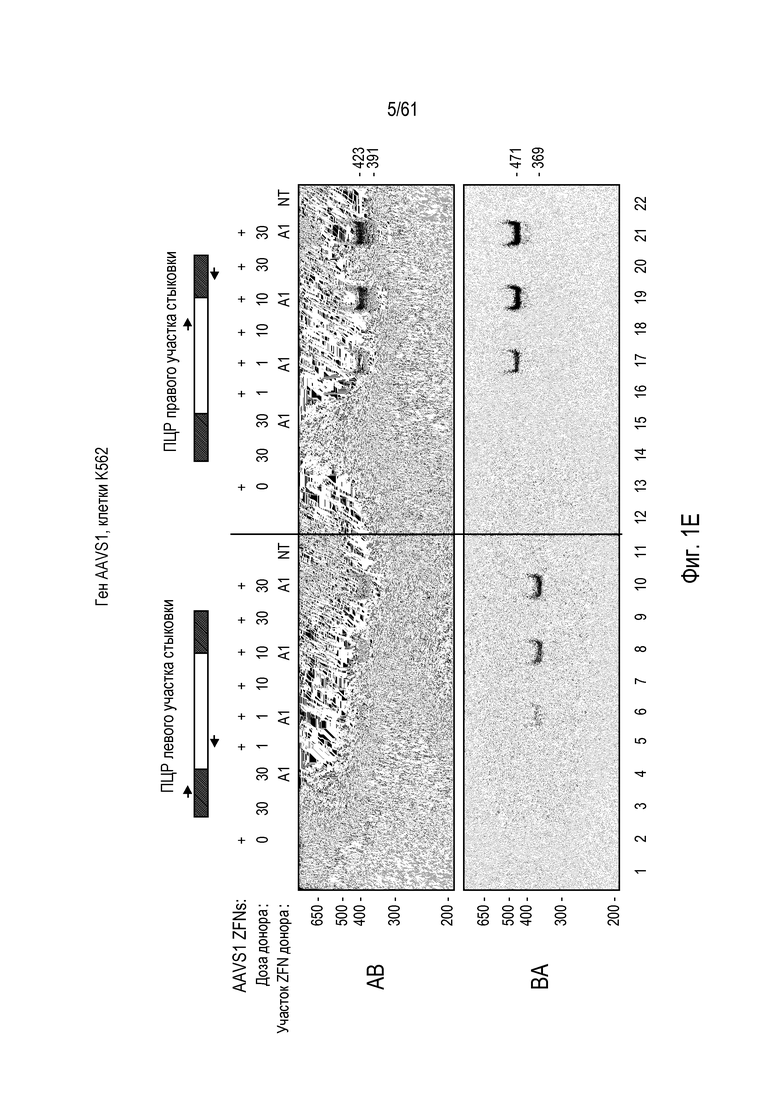
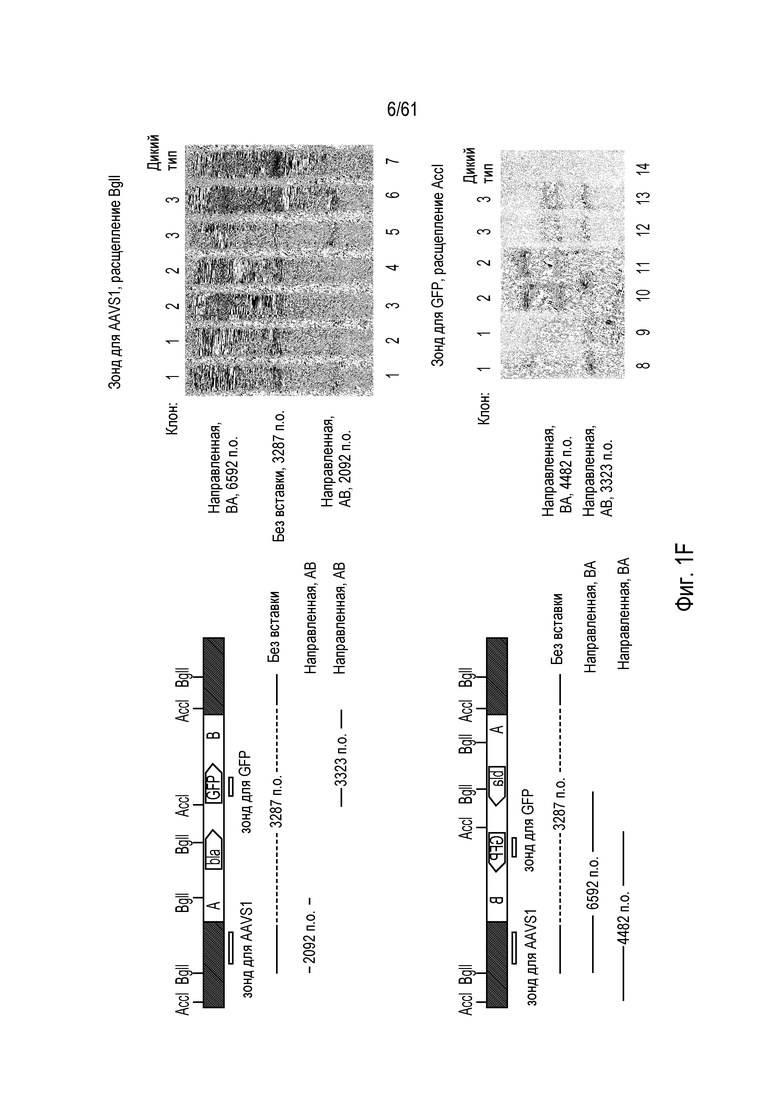
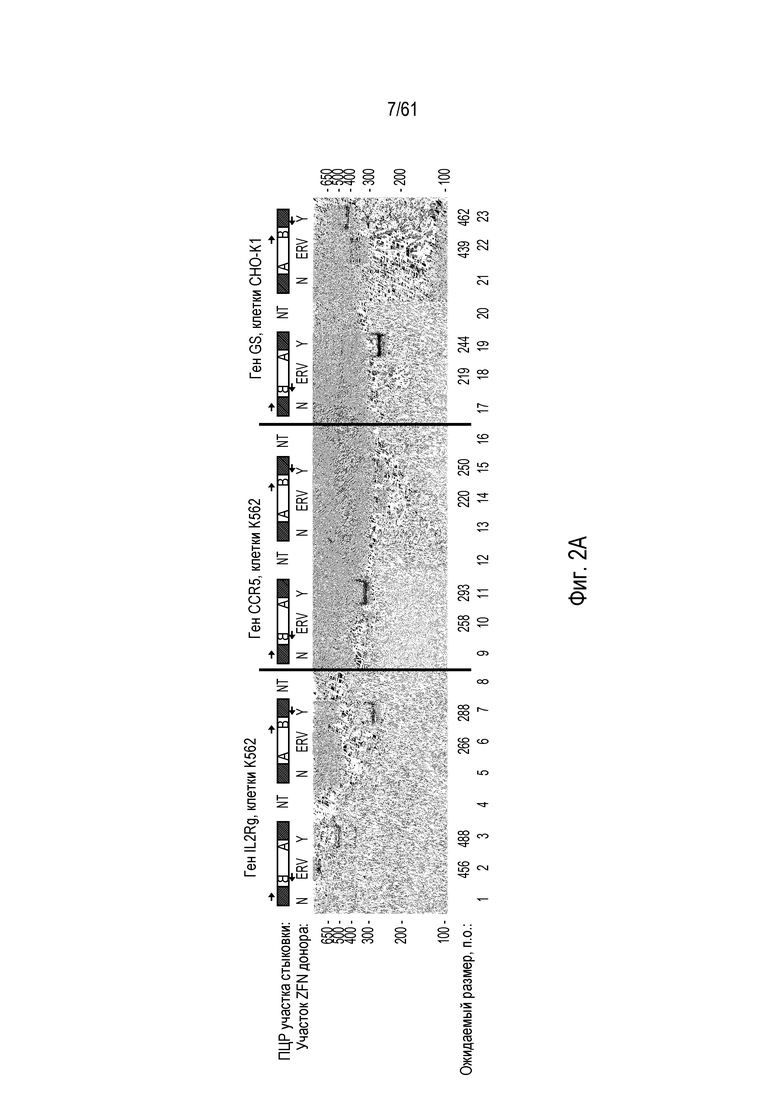
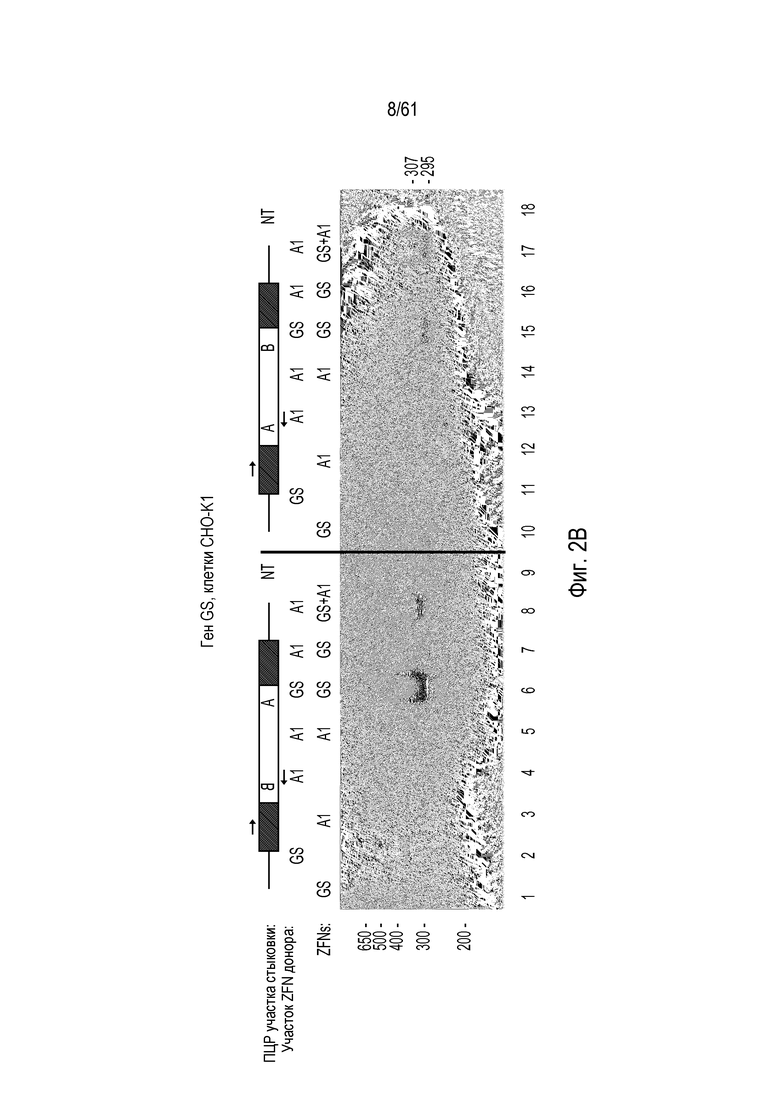
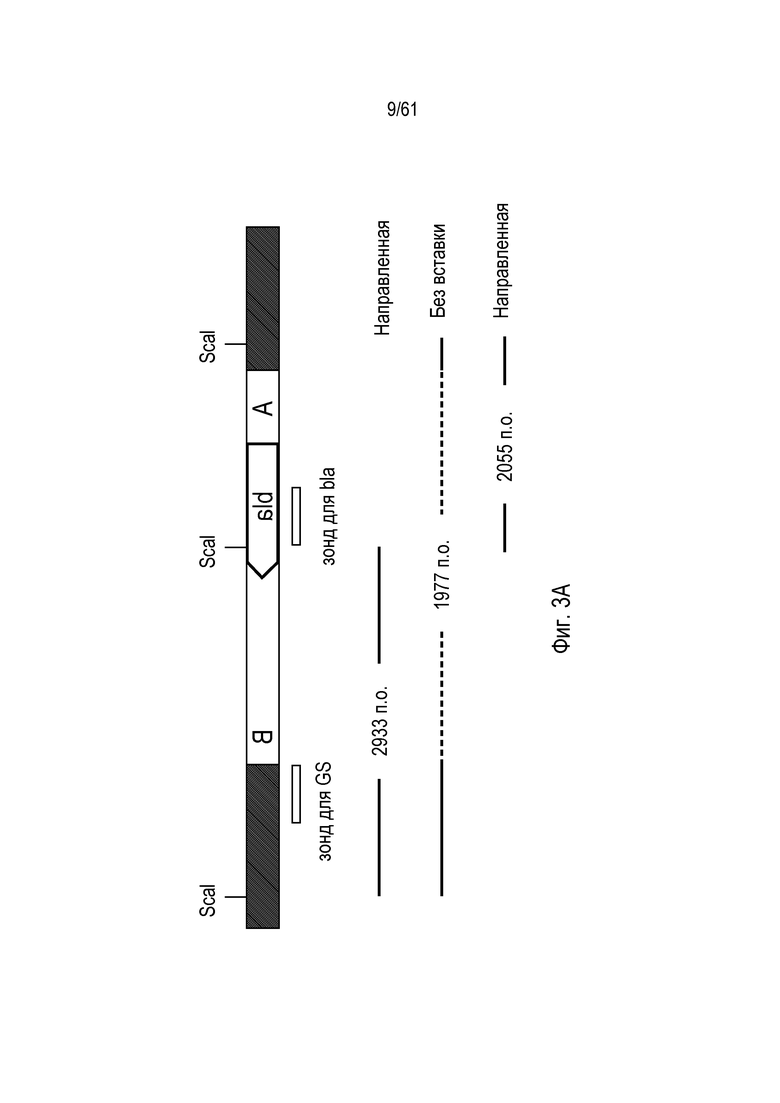
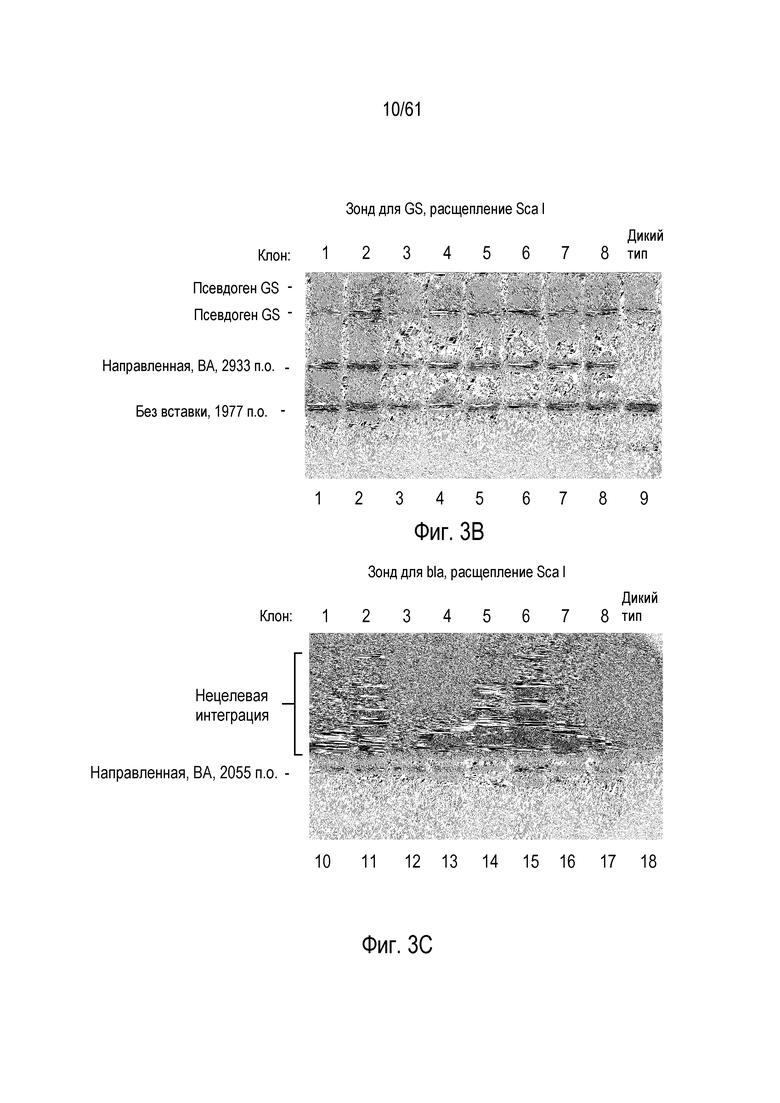
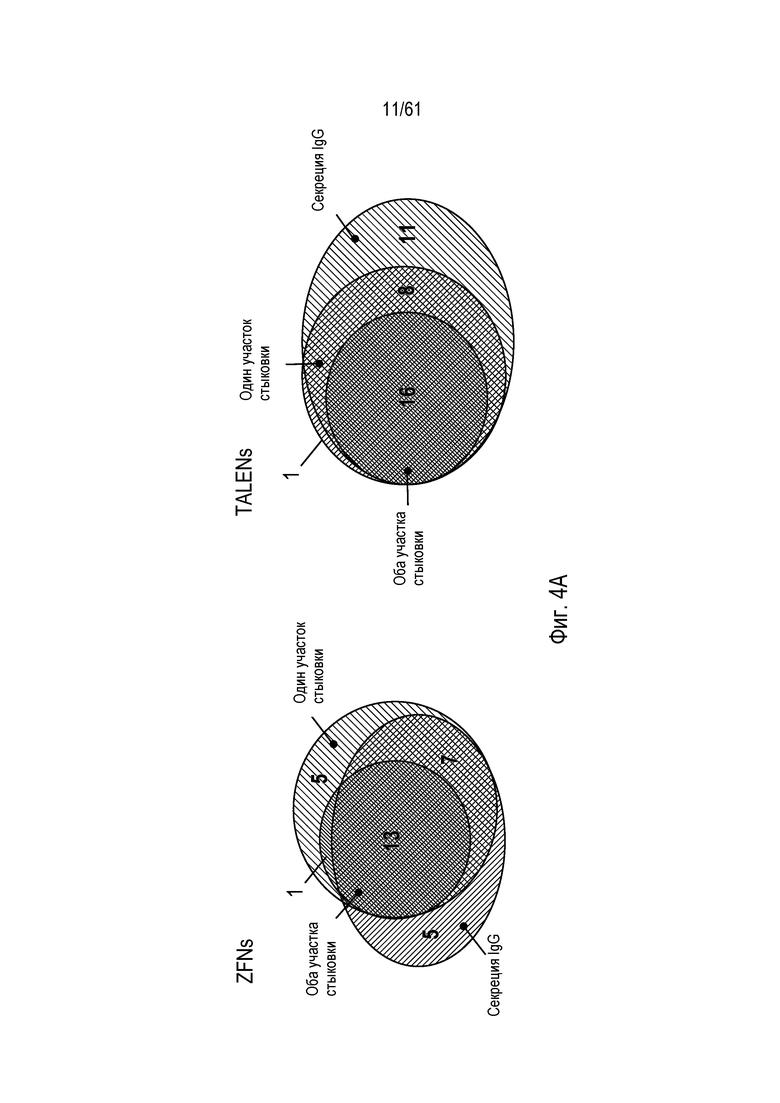
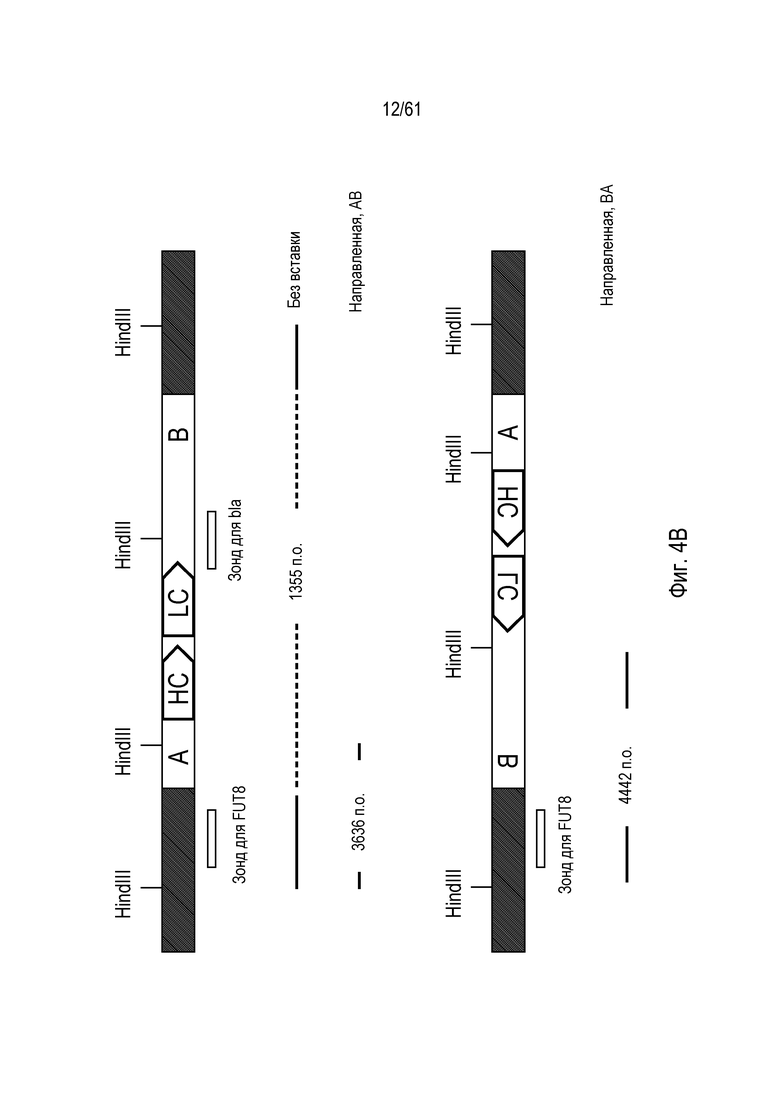
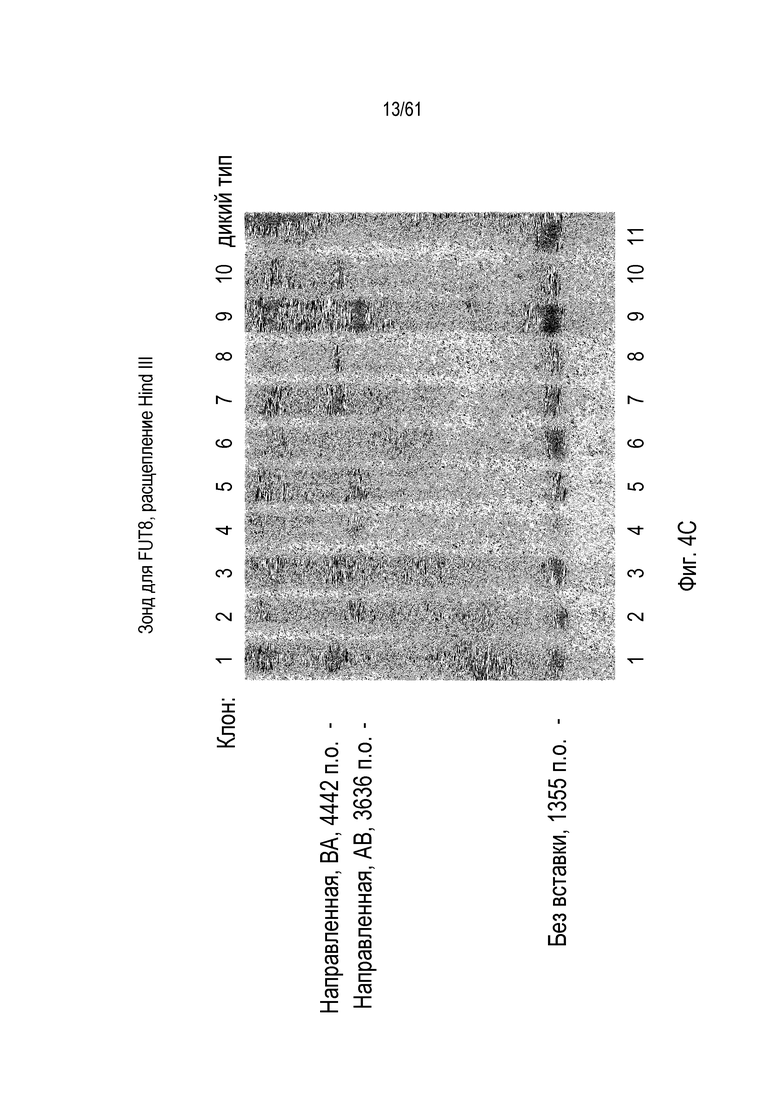
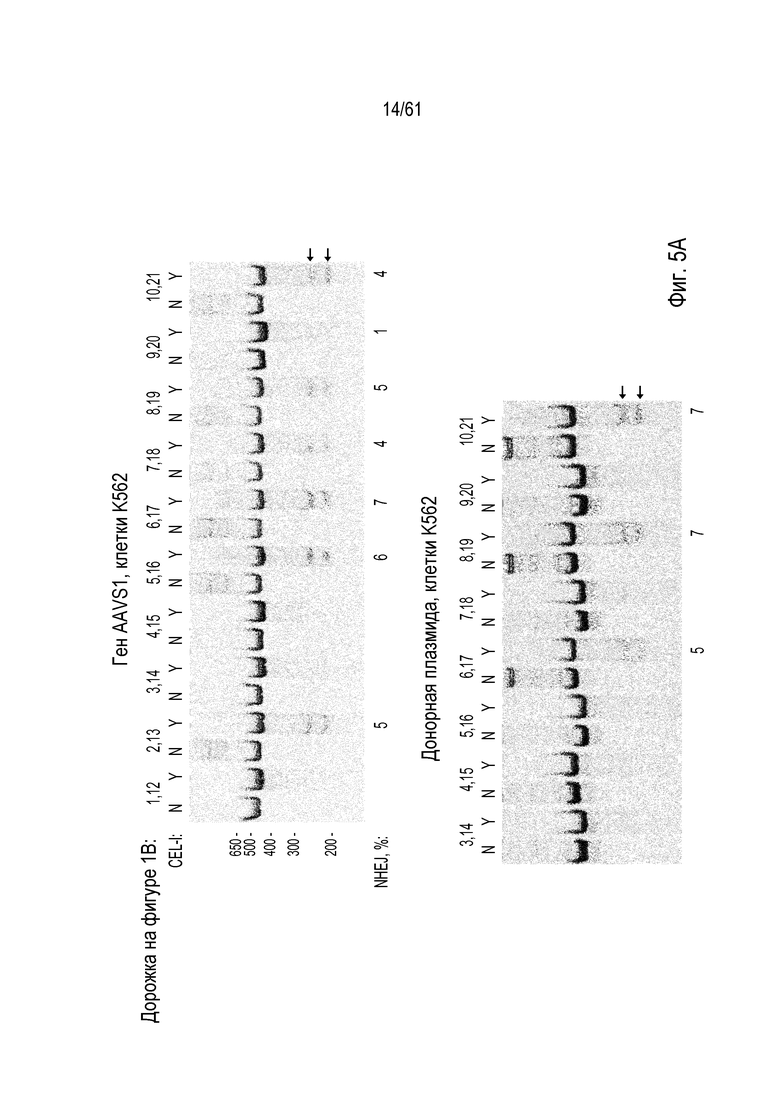


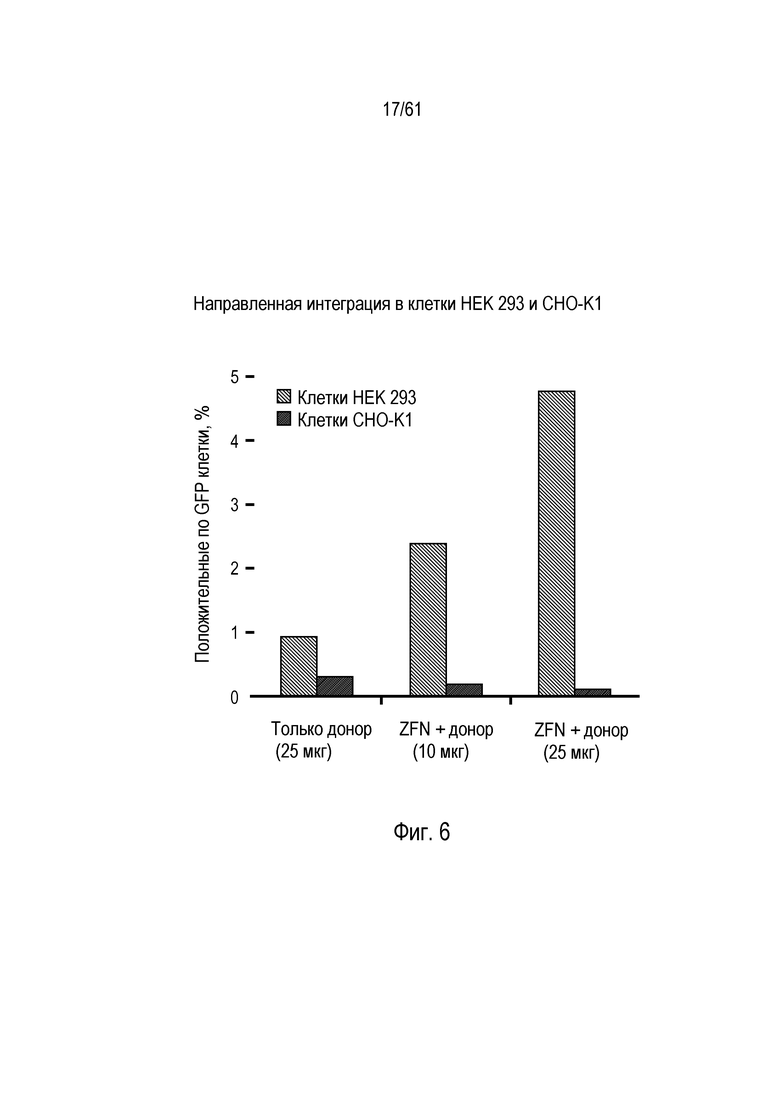

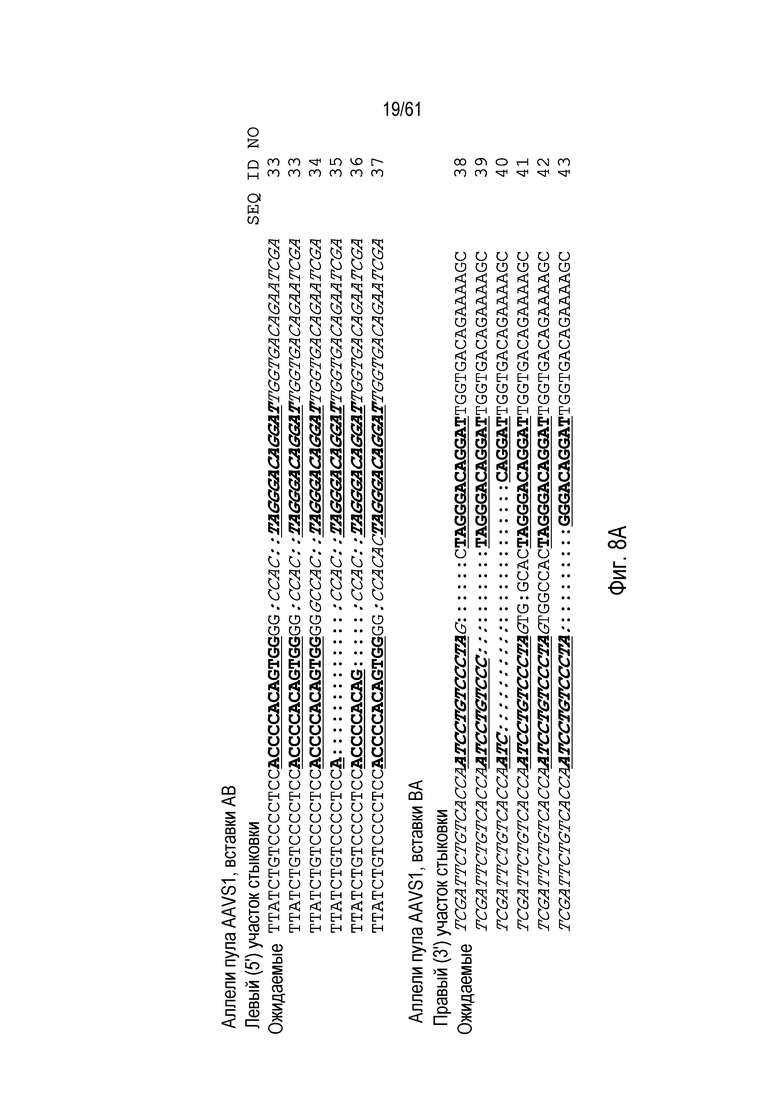
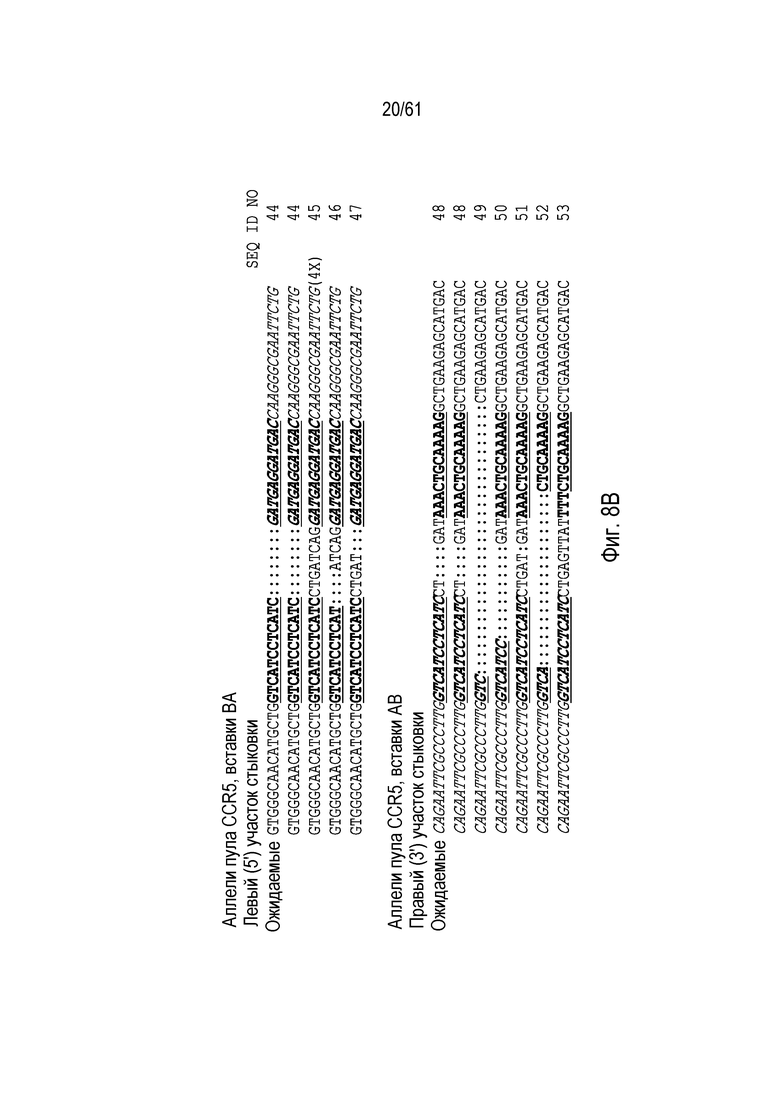
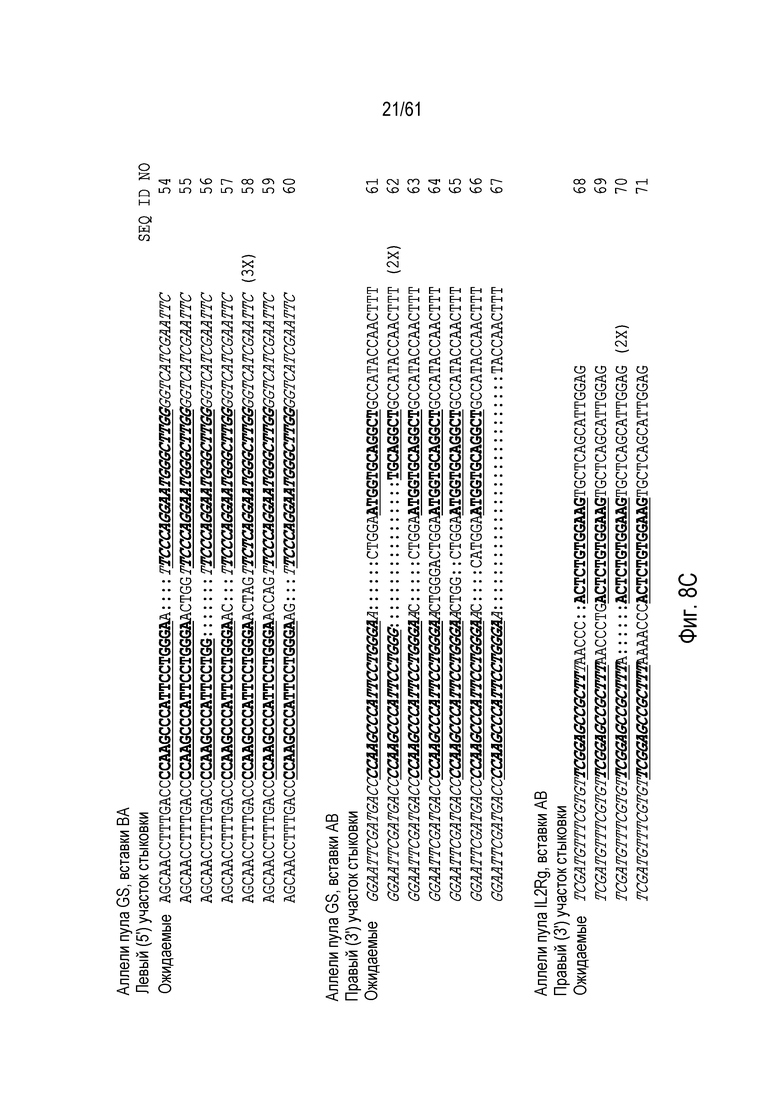

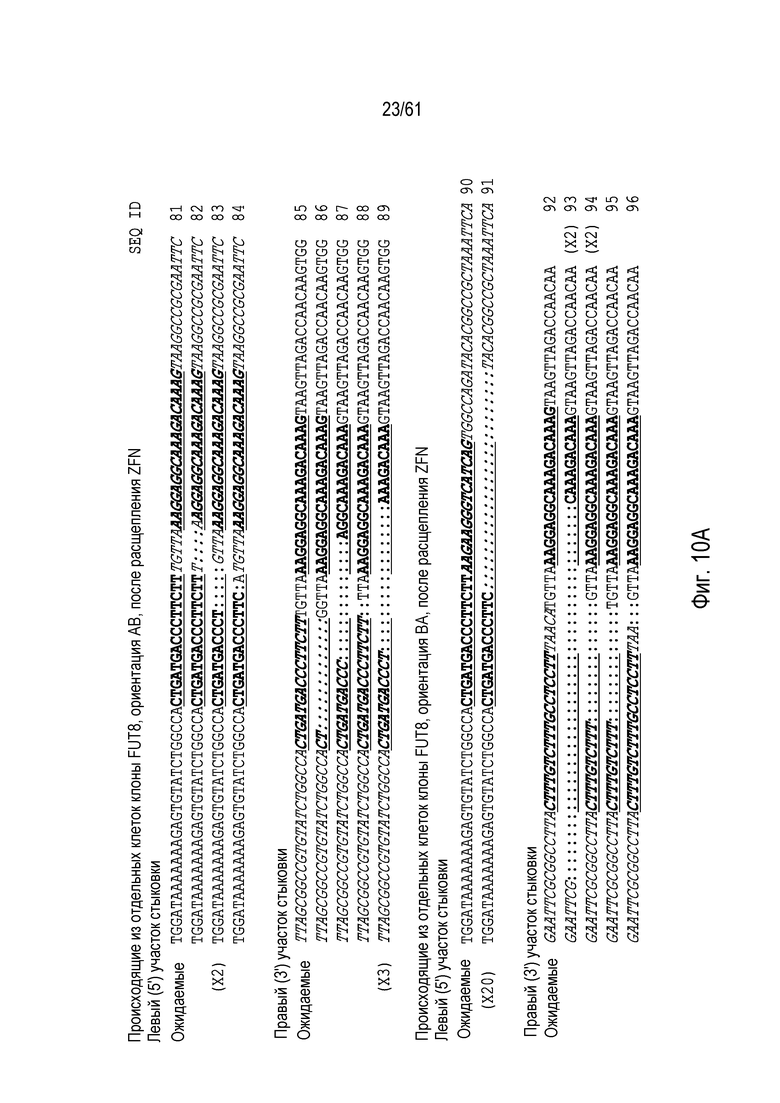

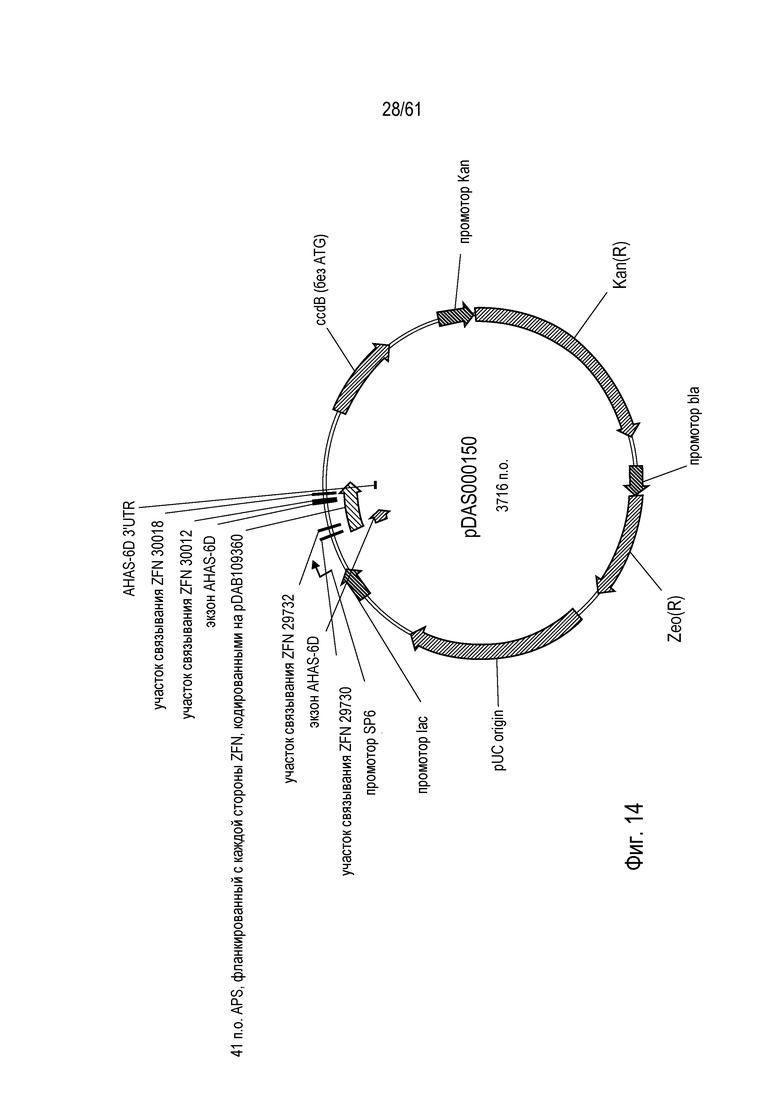
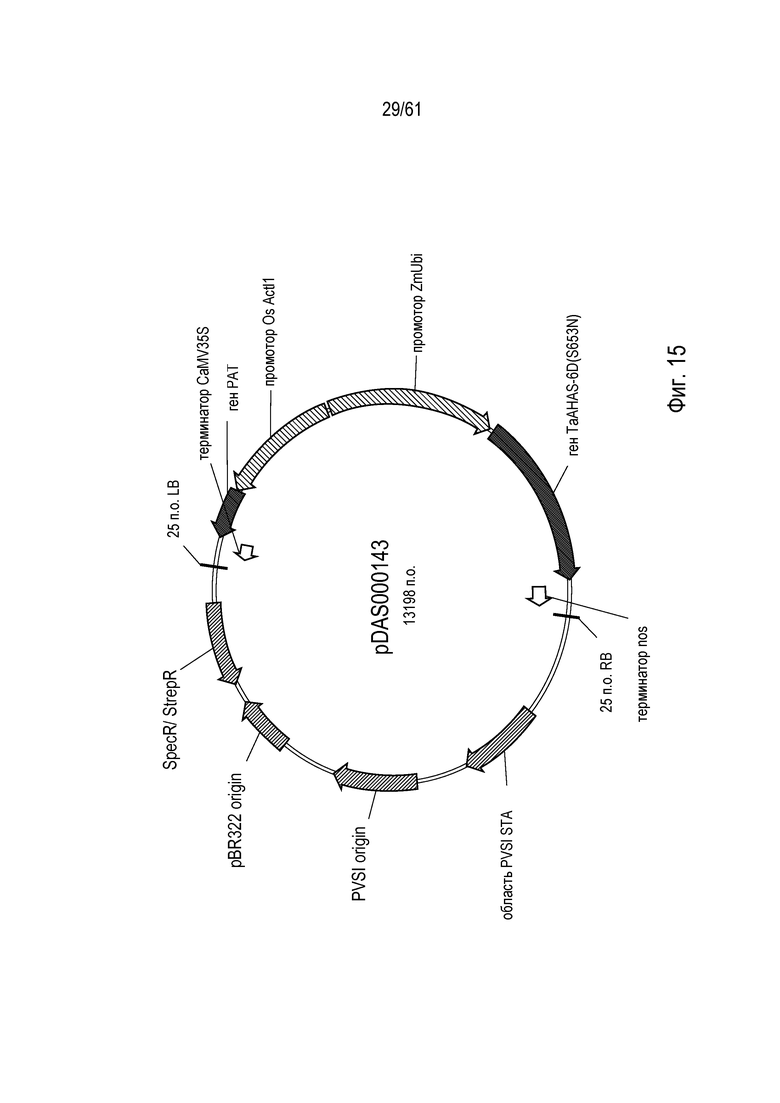
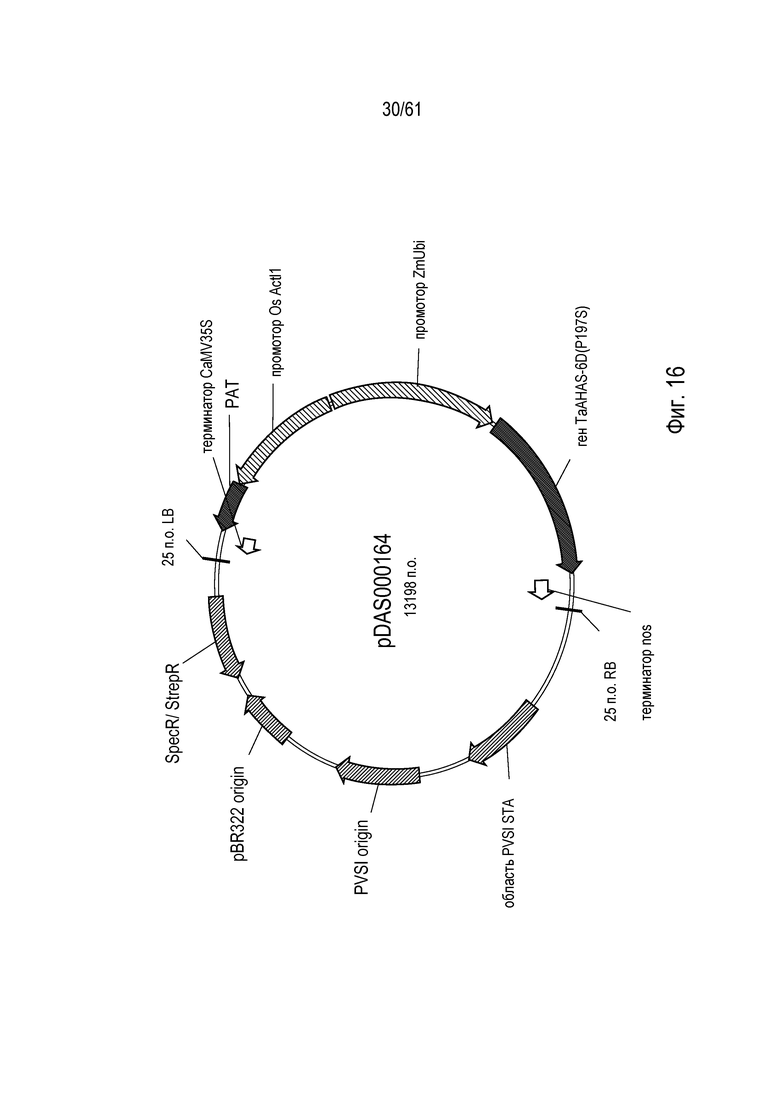
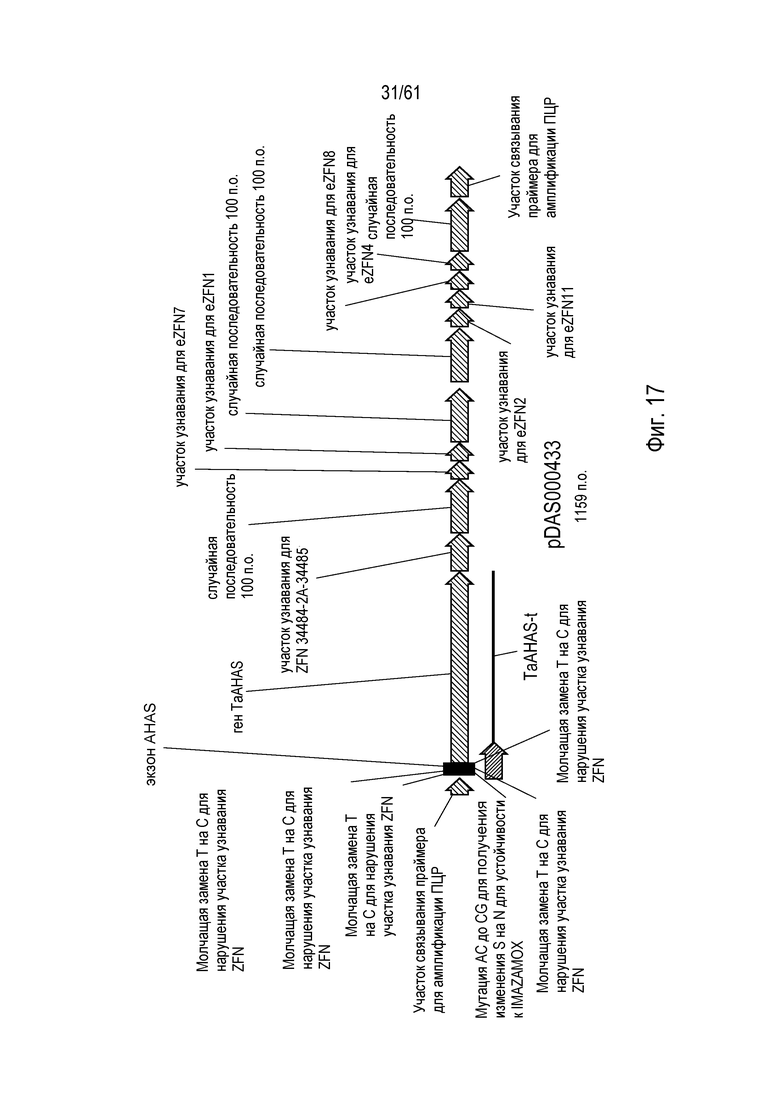
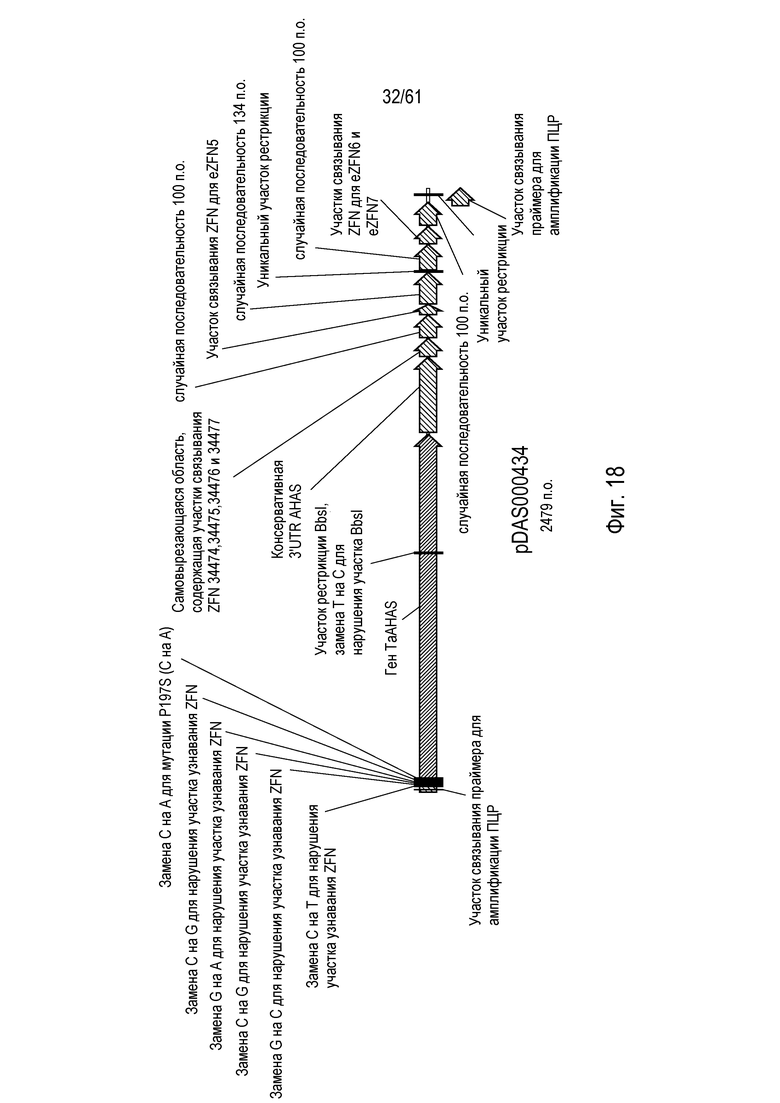
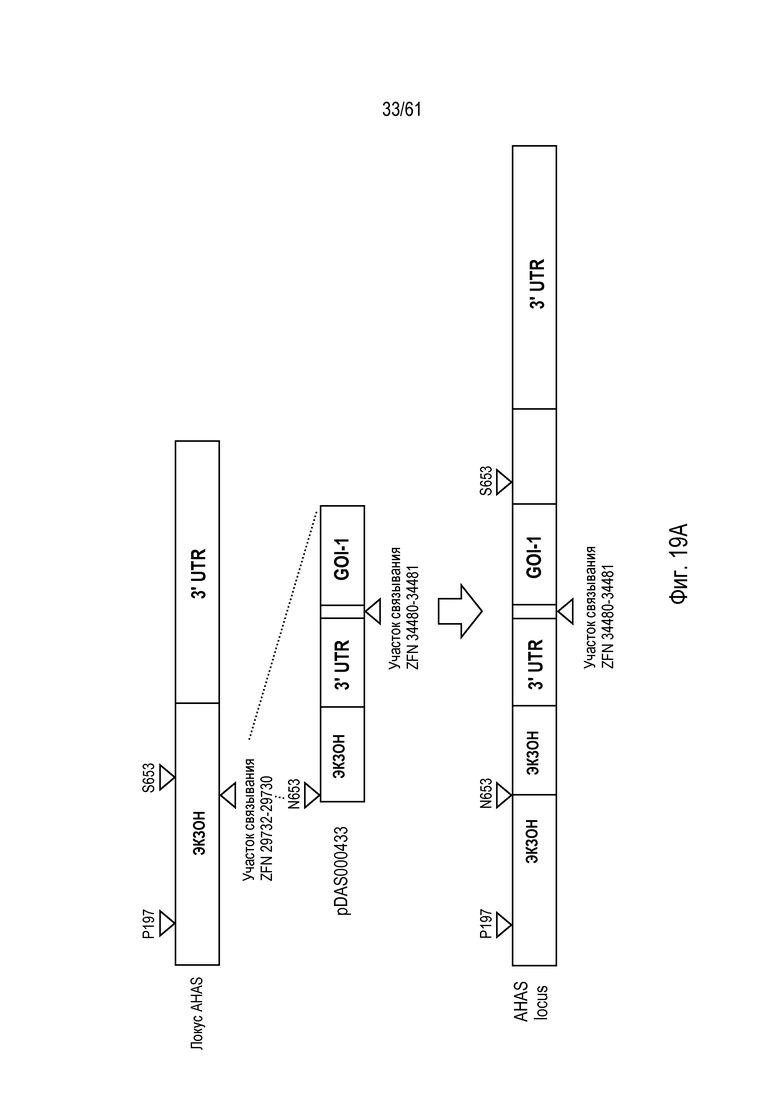

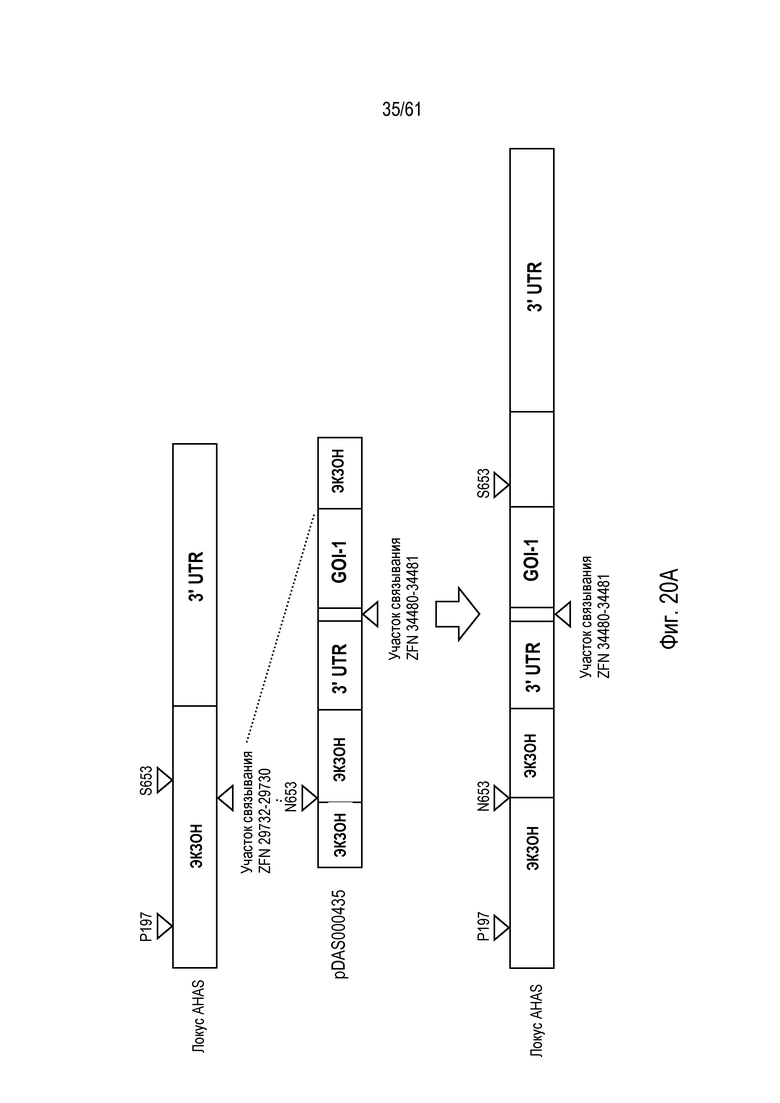
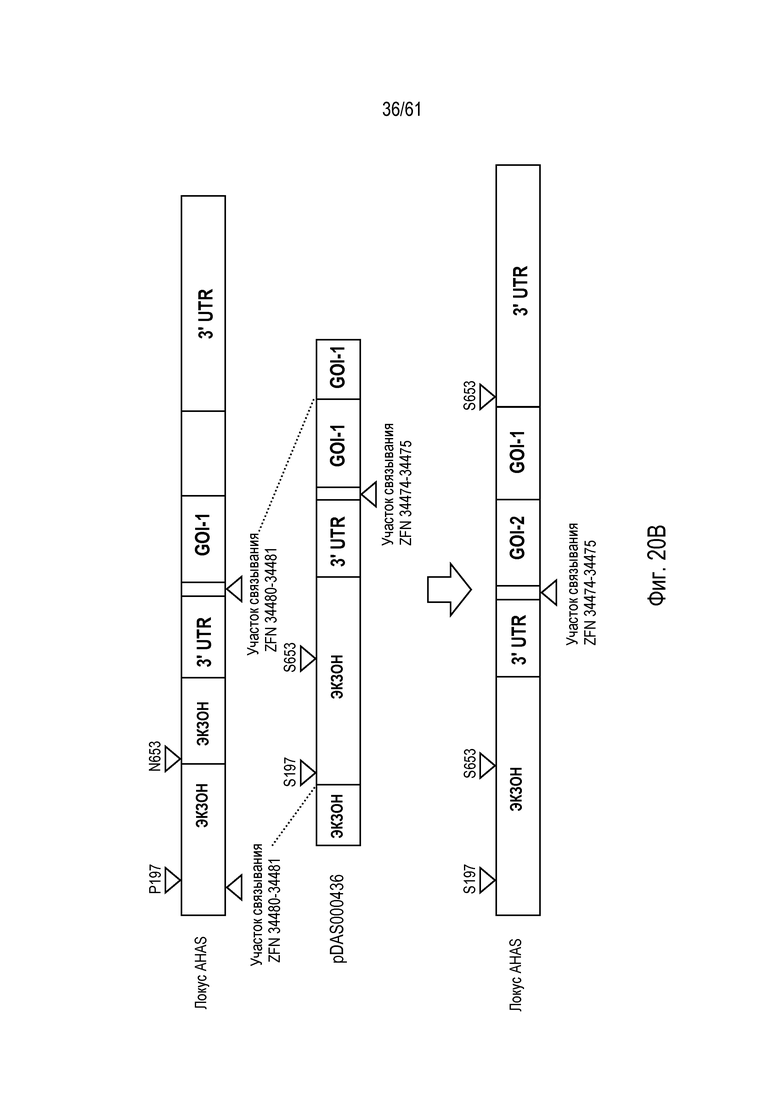
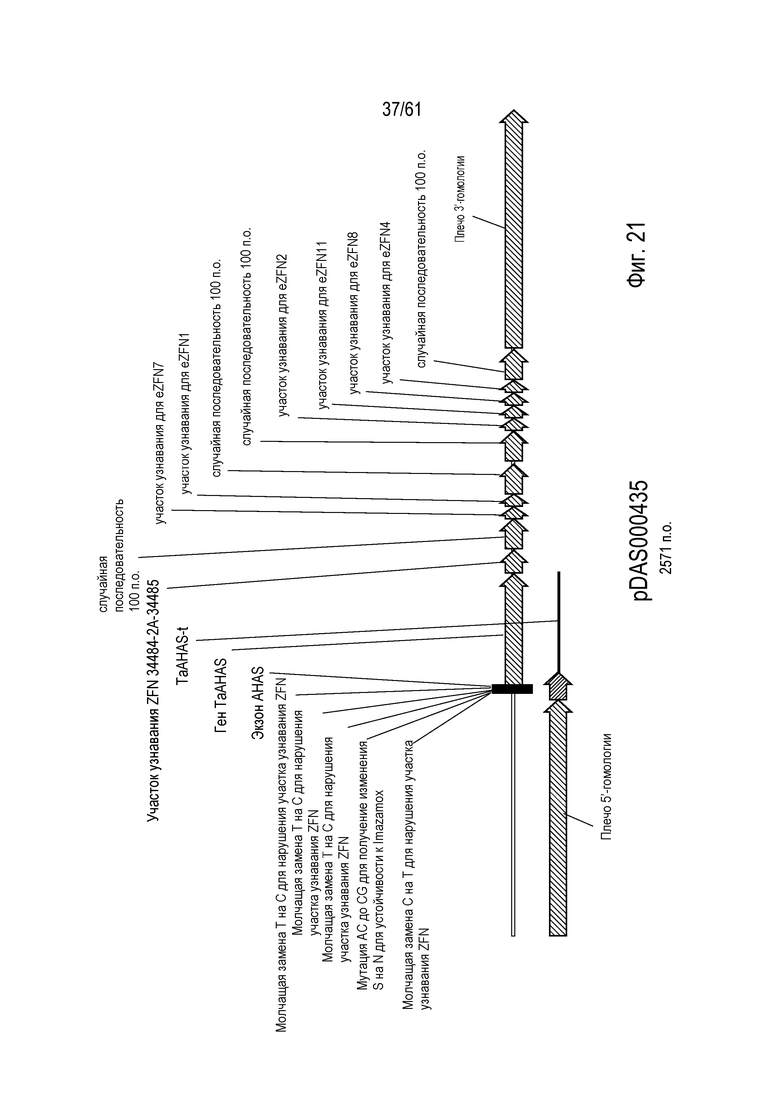

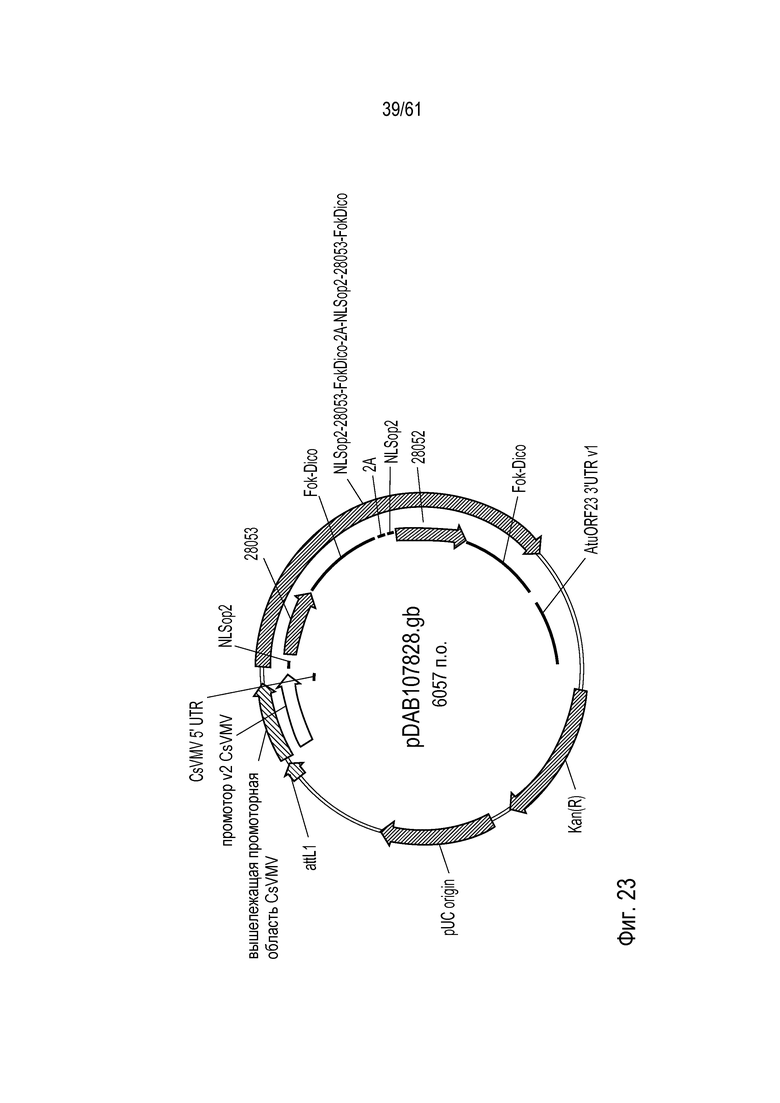
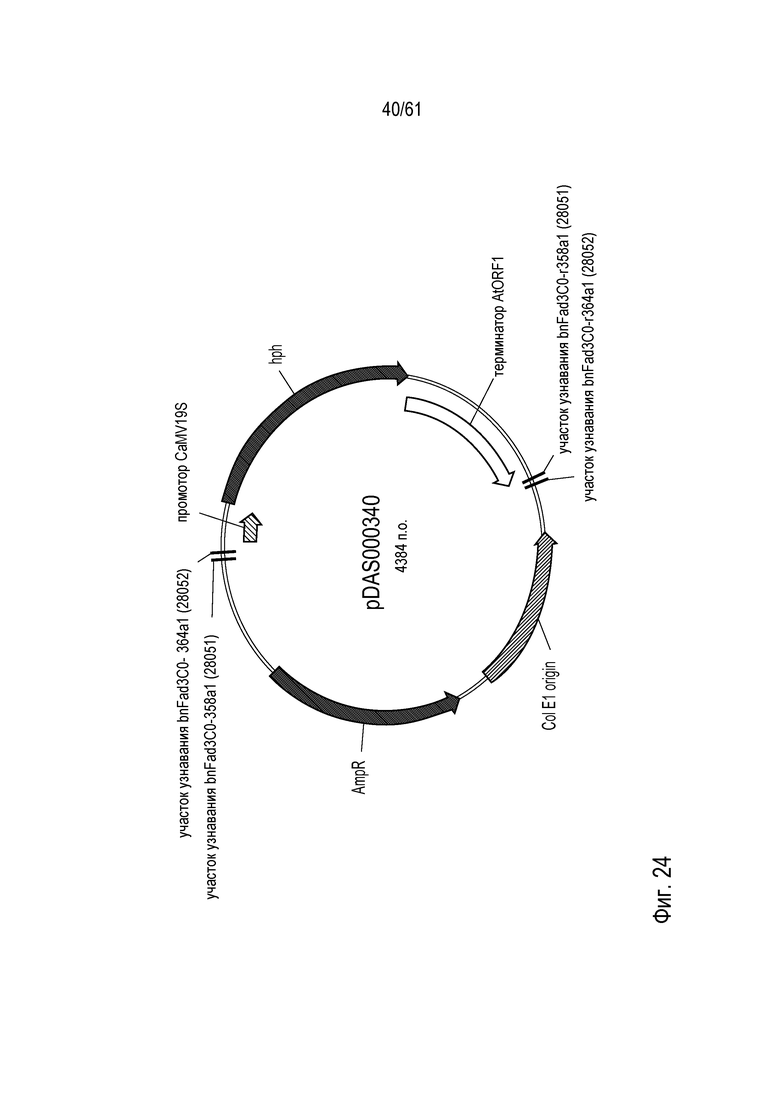
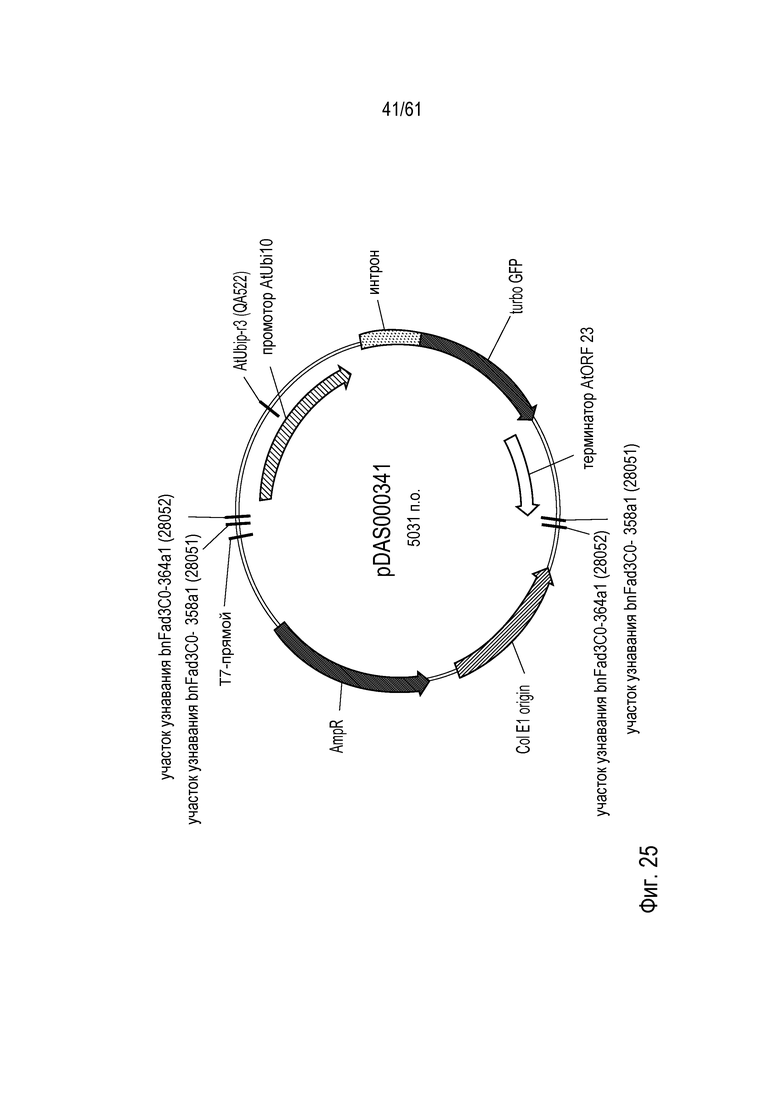
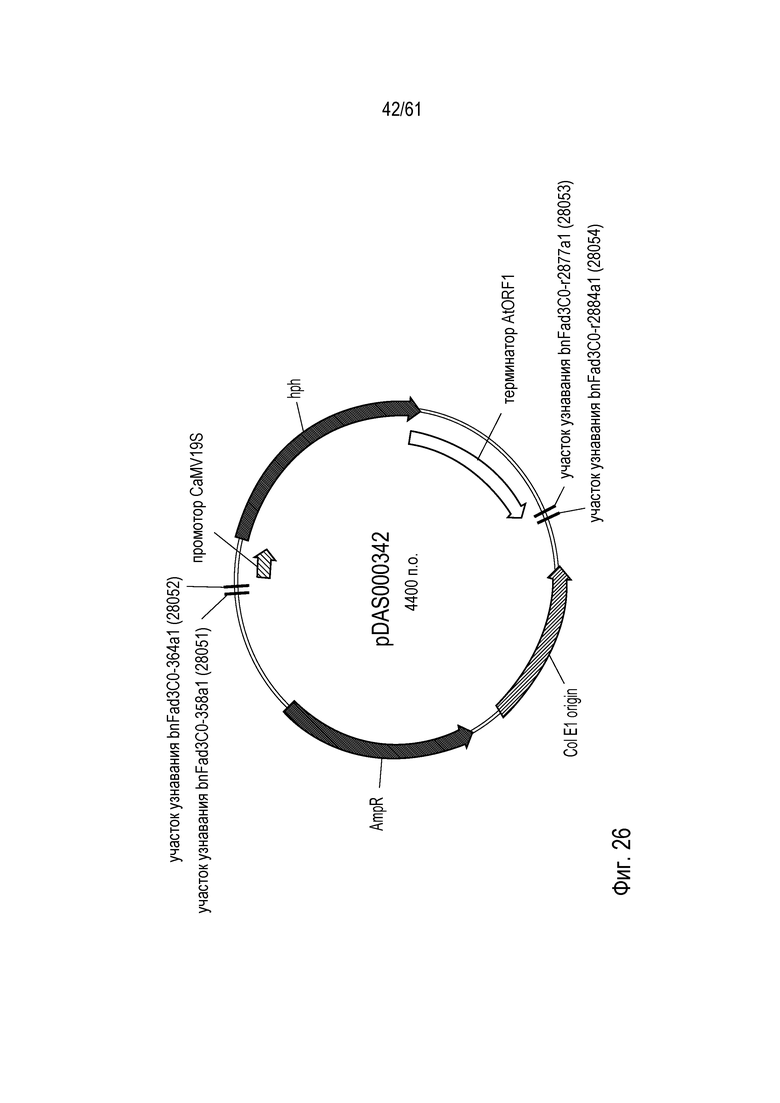
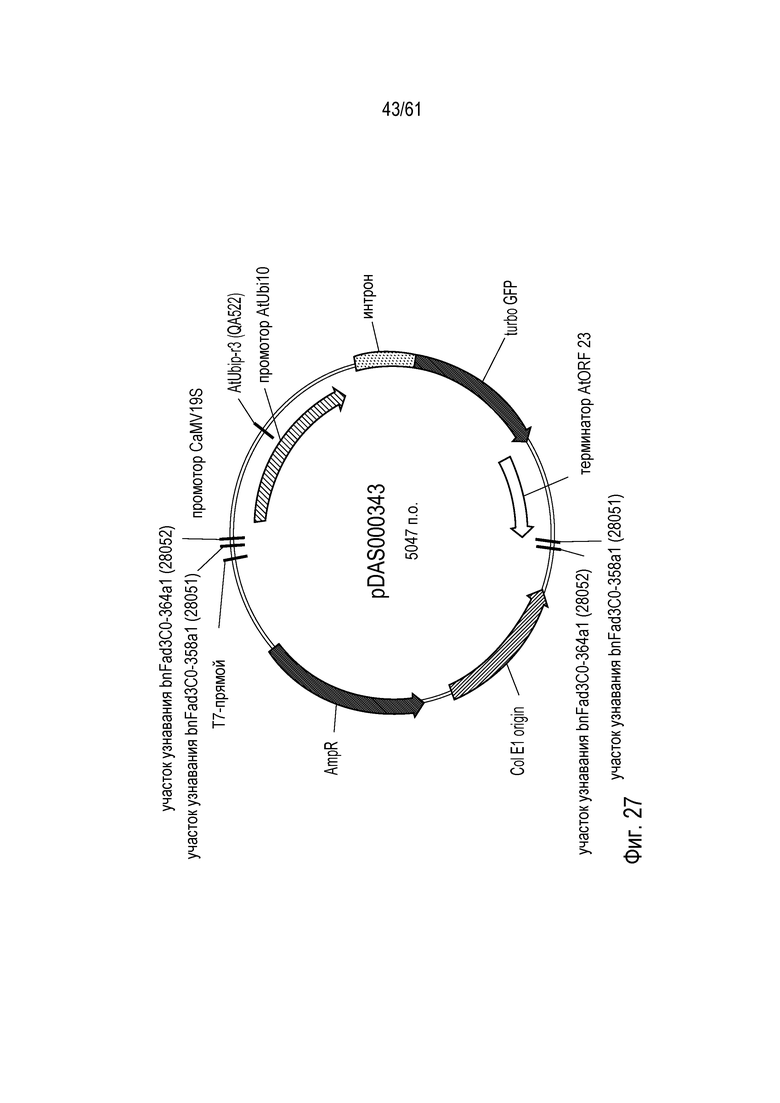
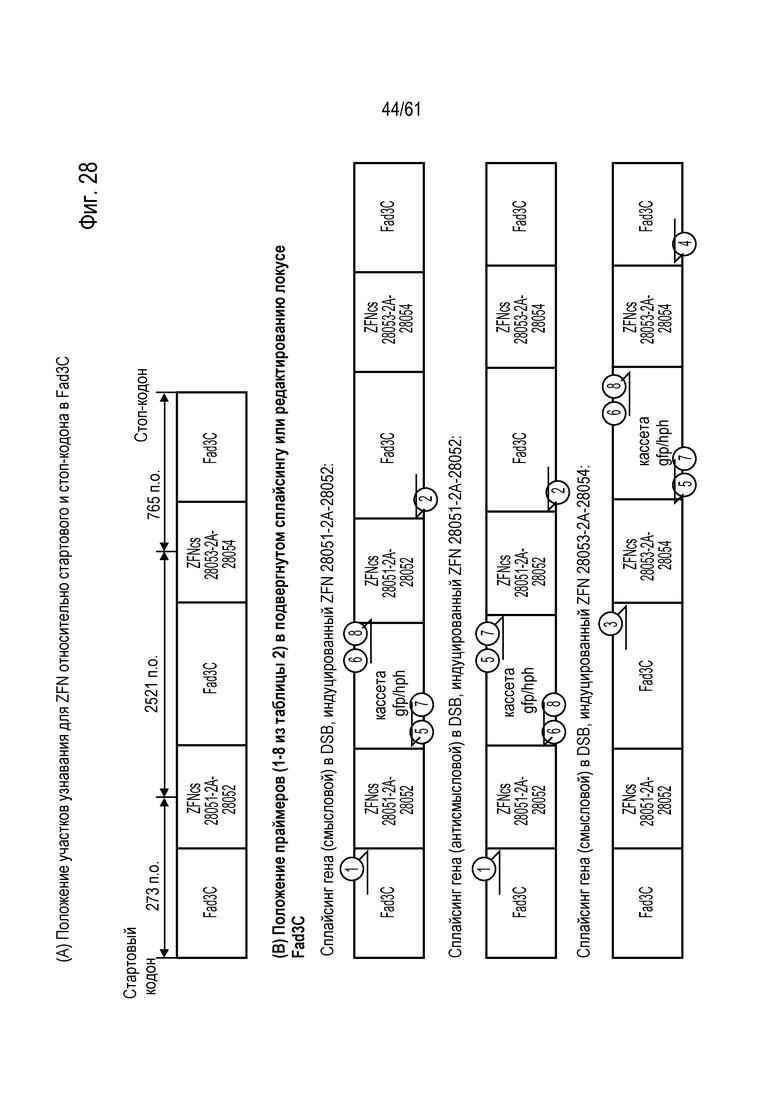
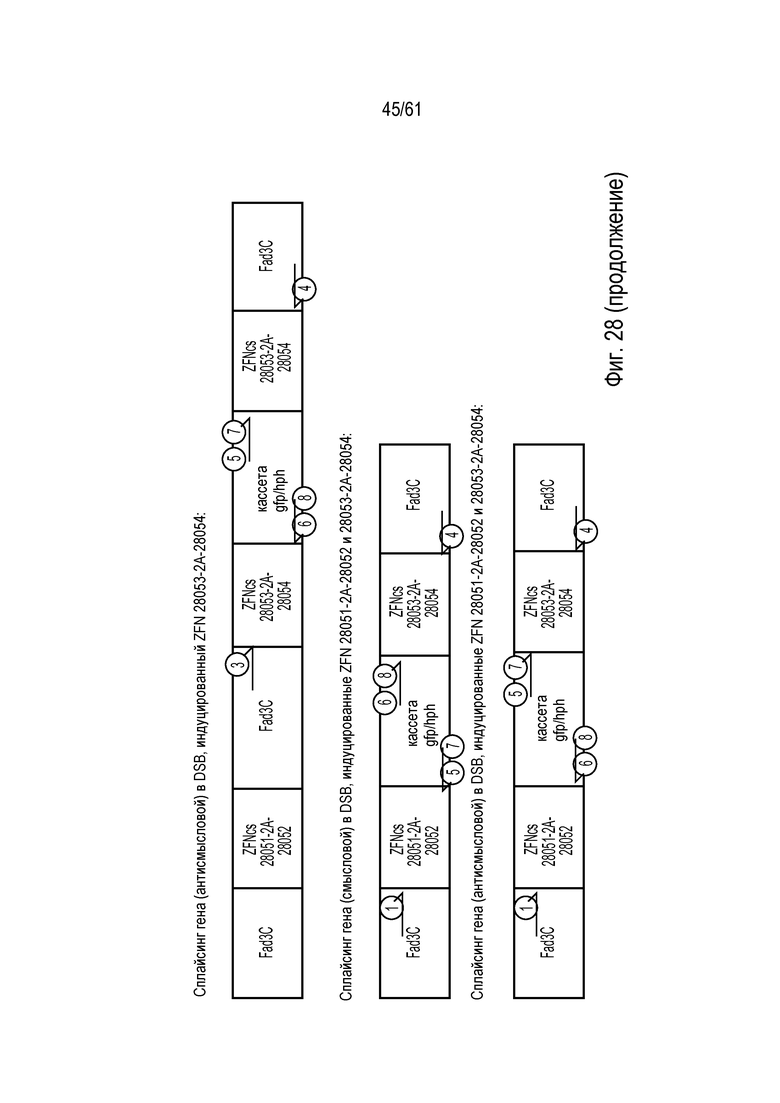


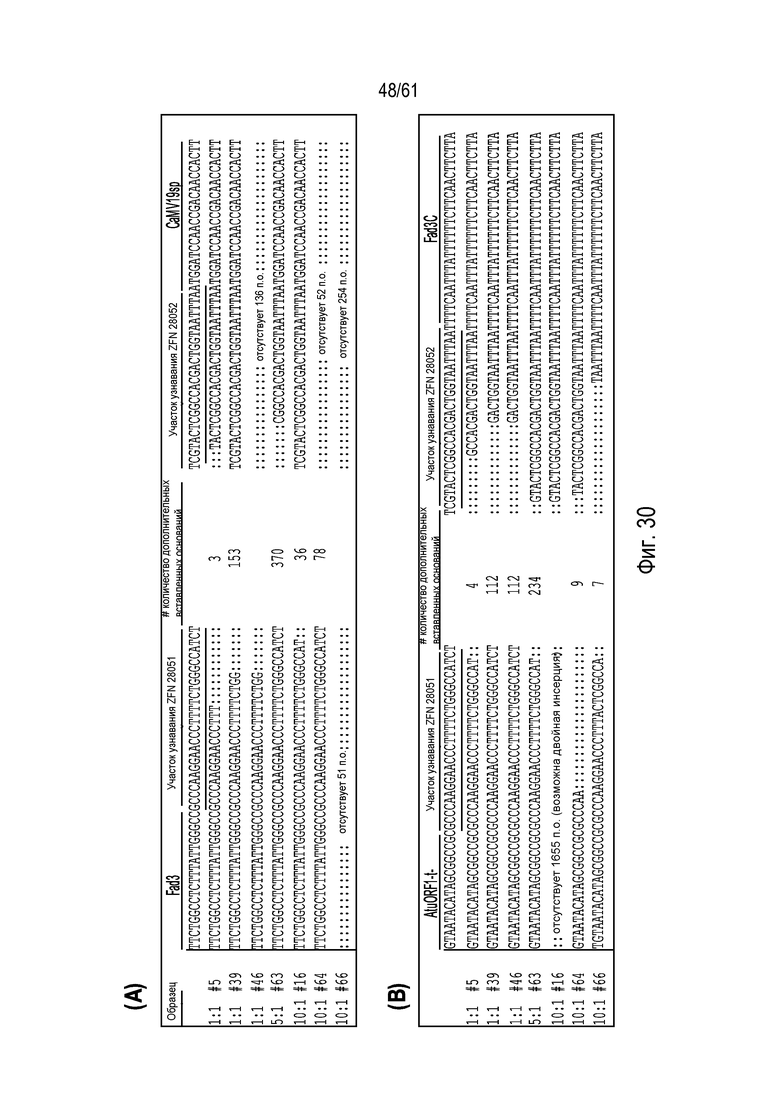

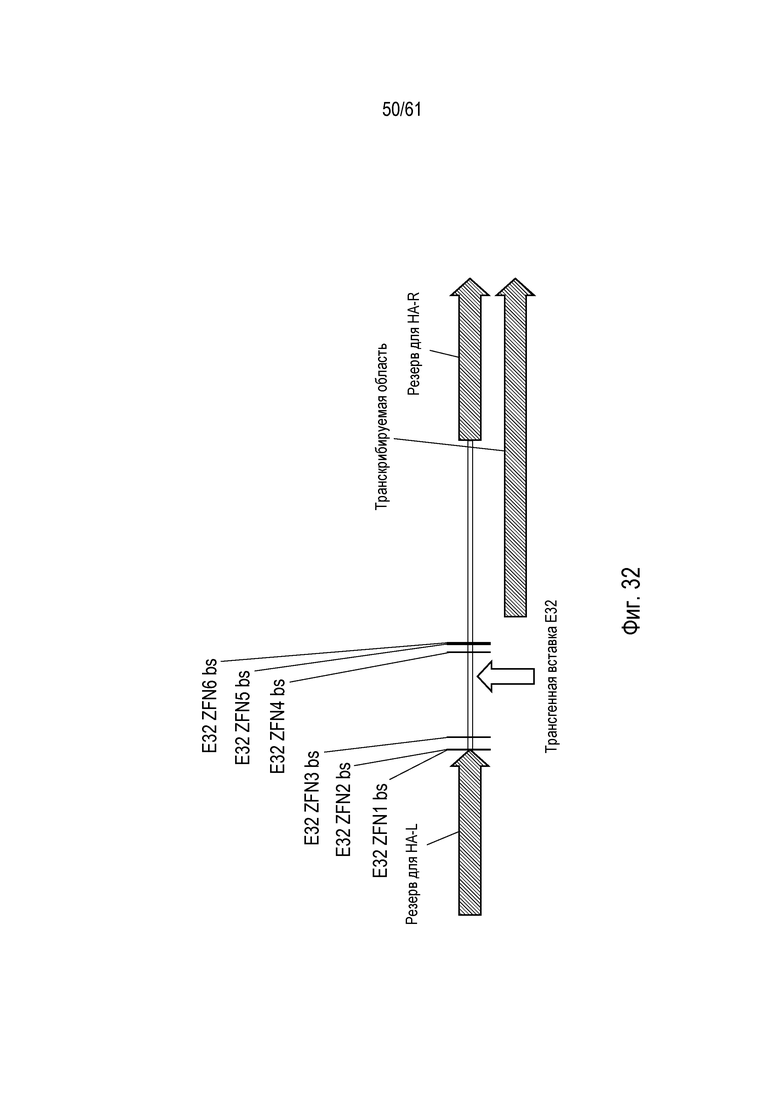
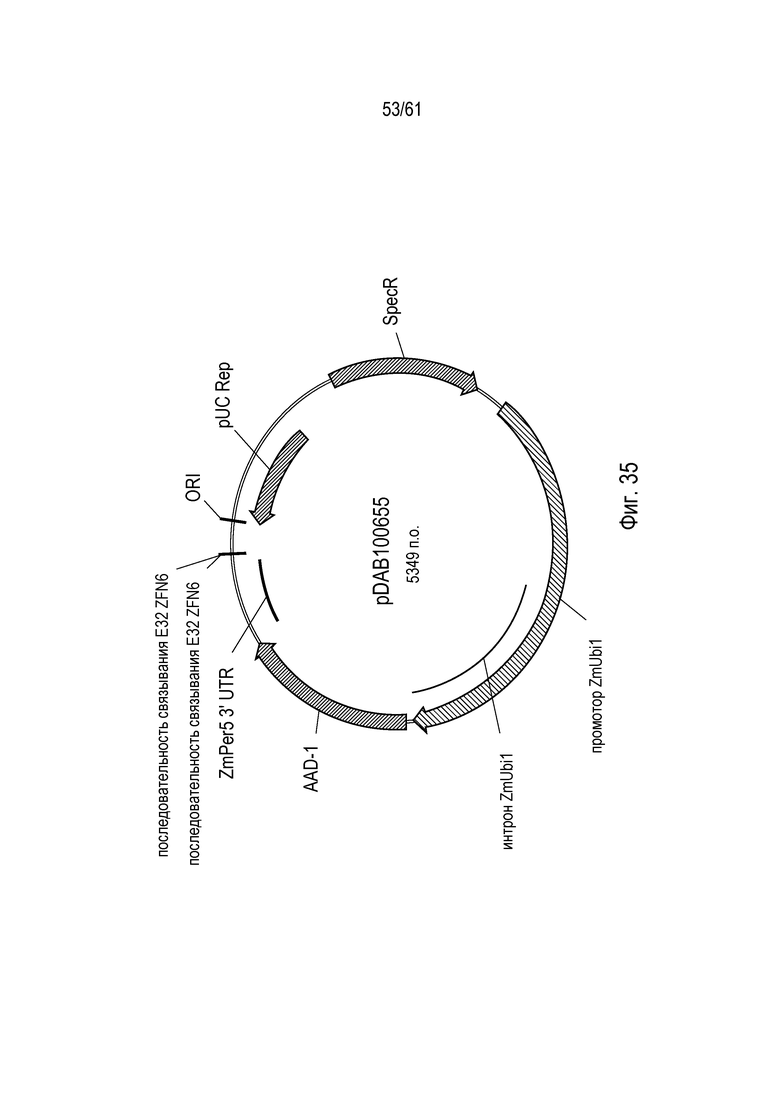
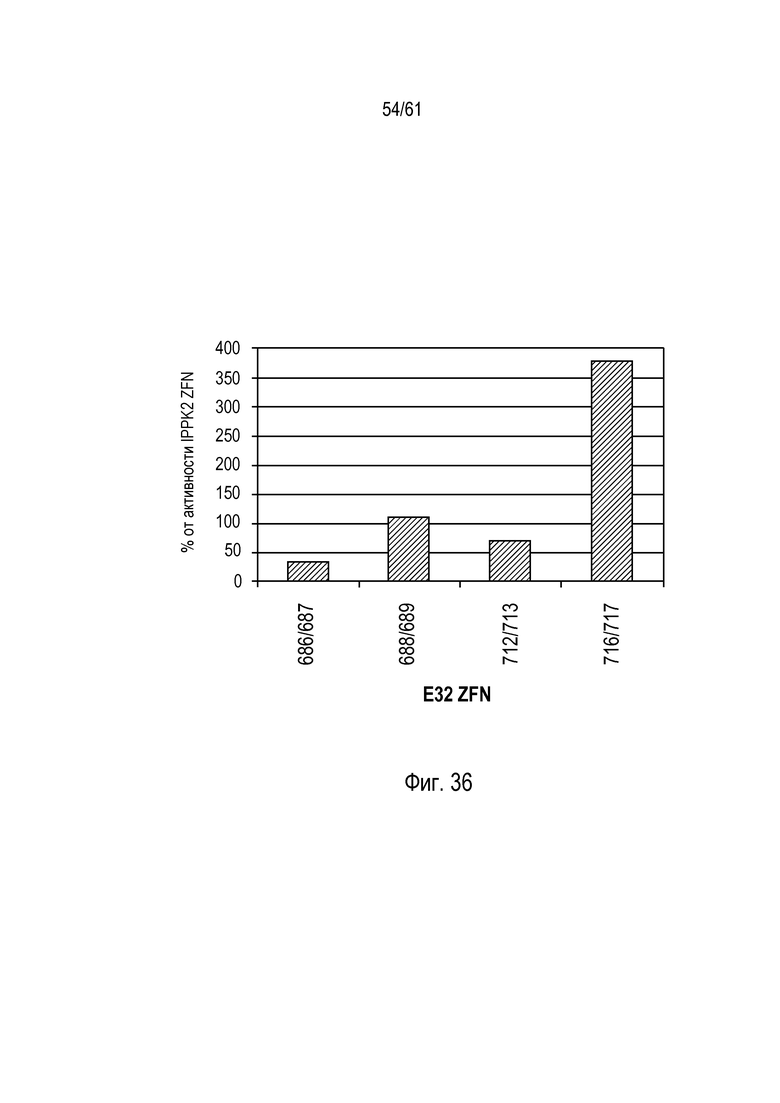
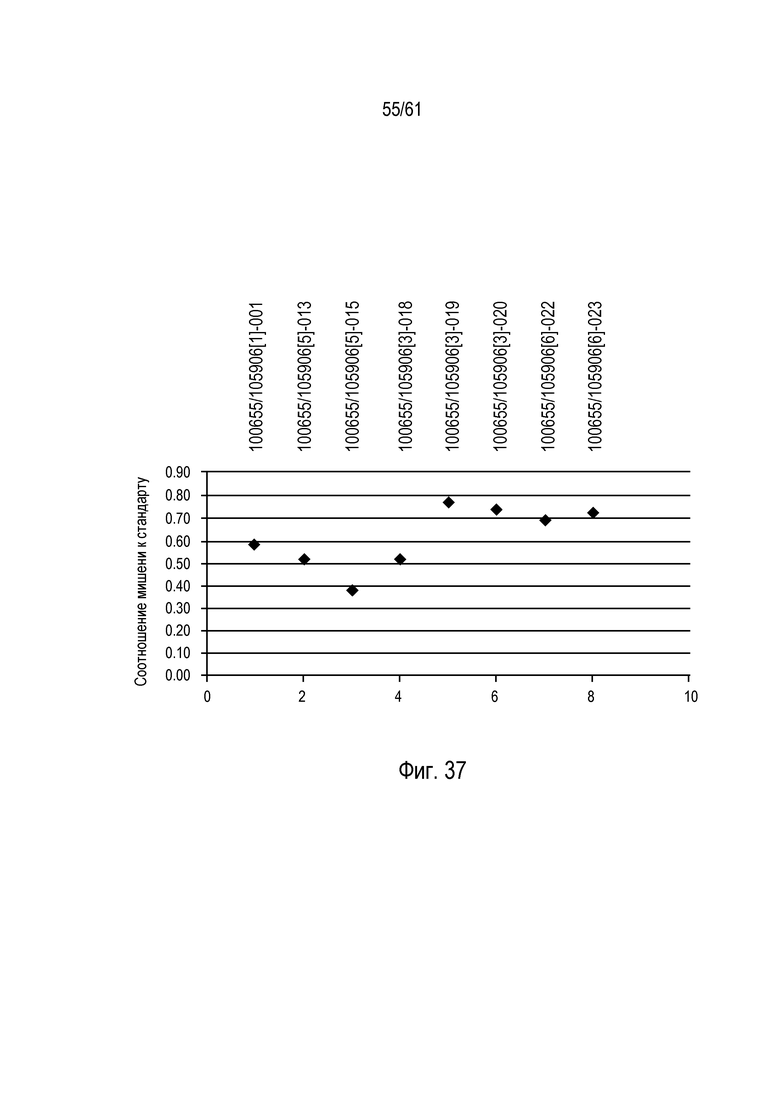
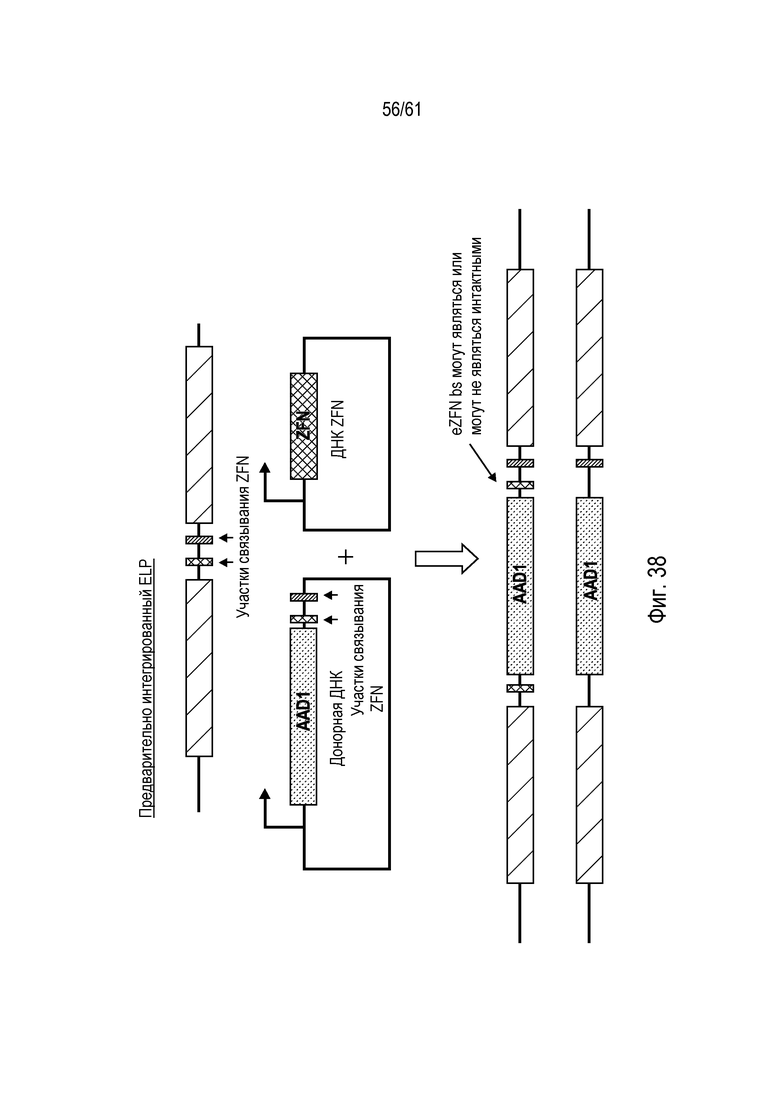
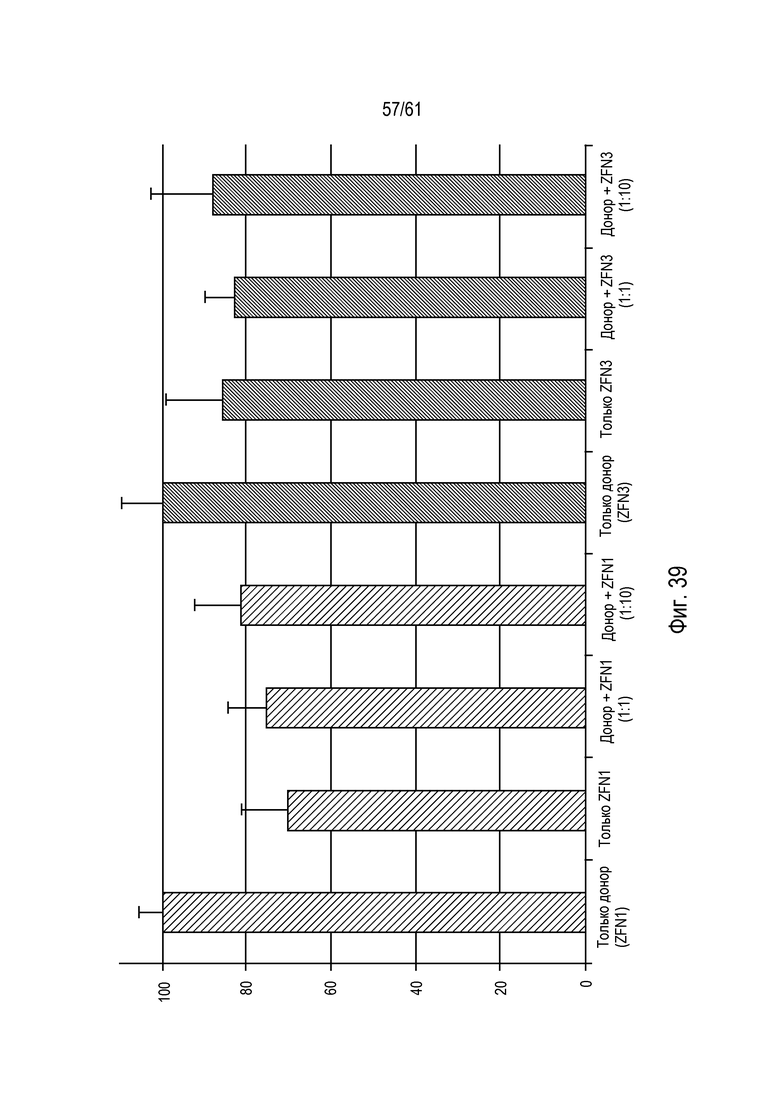
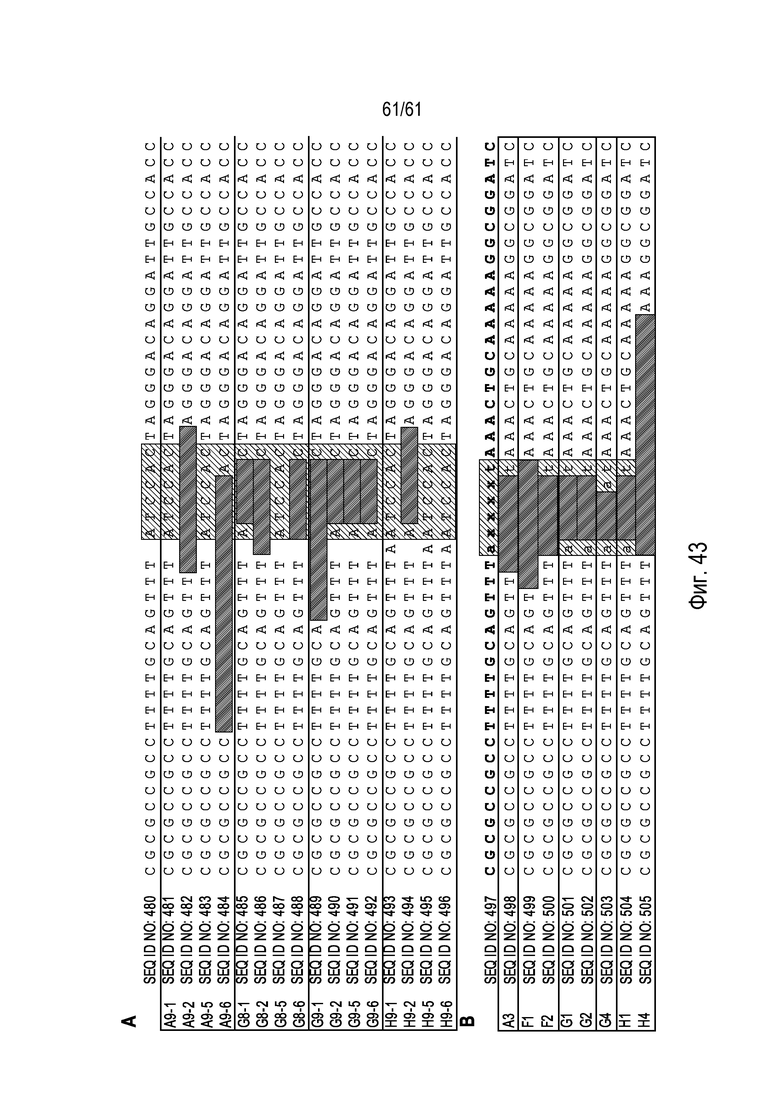
Изобретение относится к области биотехнологии. Предложены кольцевой двухцепочечный полинуклеотид для интеграции экзогенной нуклеиновой кислоты в геном клетки и способ интеграции экзогенной последовательности нуклеиновой кислоты в эндогенный локус клетки. Предложенный кольцевой двухцепочечный полинуклеотид содержит экзогенную последовательность нуклеиновой кислоты и две последовательности, фланкирующие указанную экзогенную последовательность. При этом фланкирующие последовательности содержат два или более участков-мишеней для двух или более не встречающихся в природе нуклеаз. Указанные сайты-мишени фланкируют спейсерную последовательность длиной 4-20 нуклеотидов, не встречающуюся в геноме указанной клетки. Предложенный двухцепочечный полинуклеотид не содержит плеч гомологии. Предложенные полинуклеотид и способ могут быть использованы в биотехнологии для независимой от гомологии направленной вставки донорных молекул в геном клетки. 2 н. и 14 з.п. ф-лы, 61 ил., 33 табл., 10 пр.
1. Кольцевой двухцепочечный полинуклеотид для интеграции экзогенной нуклеиновой кислоты в геном клетки, содержащий:
экзогенную последовательность нуклеиновой кислоты;
первую и вторую последовательности, фланкирующие указанную экзогенную последовательность нуклеиновой кислоты, где указанные первая и вторая последовательности содержат два или более участков-мишеней для двух или более не встречающихся в природе нуклеаз, которые расщепляют двухцепочечную ДНК, причем указанные сайты мишени фланкируют спейсерную последовательность длиной 4-20 нуклеотидов, где по меньшей мере одна спейсерная последовательность не встречается в геноме указанной клетки,
где один или более участков-мишеней находятся не внутри указанной последовательности экзогенной нуклеиновой кислоты и дополнительно где двухцепочечный полинуклеотид не содержит плеч гомологии.
2. Полинуклеотид по п.1, где экзогенная последовательность нуклеиновой кислоты составляет по меньшей мере 1 тысячу оснований в длину.
3. Полинуклеотид по п.1 или 2, где полинуклеотид с экзогенной последовательностью нуклеиновой кислоты представляет собой плазмиду.
4. Полинуклеотид по п.1 или 2, где экзогенная последовательность нуклеиновой кислоты кодирует белок.
5. Двухцепочечный полинуклеотид по п.1 или 2, где спейсерные последовательности имеют длину по меньшей мере 5 нуклеотидов.
6. Способ интеграции экзогенной последовательности нуклеиновой кислоты в эндогенный локус клетки, где способ включает:
введение полинуклеотида по любому из пп.1-5 в клетку;
введение одной или более нуклеаз в клетку, где нуклеазы расщепляют двухцепочечный полинуклеотид и расщепляют эндогенный локус, так что экзогенная последовательность нуклеиновой кислоты интегрируется в эндогенный локус.
7. Способ по п.6, где экзогенная последовательность нуклеиновой кислоты интегрируется в прямой ориентации.
8. Способ по п.6, где экзогенная последовательность нуклеиновой кислоты интегрируется в обратной ориентации.
9. Способ по любому из пп.6-8, где одинаковые нуклеазы расщепляют эндогенный локус и полинуклеотид.
10. Способ по любому из пп.6-8, где различные нуклеазы расщепляют эндогенный локус и полинуклеотид.
11. Способ по любому из пп.6-8, где экзогенная последовательность нуклеиновой кислоты интегрируется в эндогенный локус посредством независимых от гомологии механизмов.
12. Способ по любому из пп.6-8, где указанные участки-мишени, фланкирующие не встречающийся в природе спейсер, не воссоздаются после интеграции указанной экзогенной последовательности нуклеиновой кислоты.
13. Способ по любому из пп.6-8, где нуклеазы образуют делецию в эндогенном локусе, и экзогенная последовательность нуклеиновой кислоты интегрируется в делецию.
14. Способ по любому из пп.6-8, где клетка представляет собой эукариотическую клетку.
15. Способ по п.14, где клетка представляет собой клетку растения или млекопитающего.
16. Способ по п.15, где клетка растения представляет собой клетку двудольного или однодольного растения.
| WO 2011090804 A1, 28.07.2011 | |||
| WO 2011100058 A1, 18.08.2011 | |||
| WO 2011017315 A2, 10.02.2011 | |||
| ORLANDO S.J | |||
| ET AL | |||
| Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology | |||
| Nucleic Acids Research | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| WO 2005014791 A2, 17.02.2005 | |||
| WO 2011011767 A1, 27.01.2011 | |||
| СПОСОБ ВСТРАИВАНИЯ НУЖНОЙ ДНК В ГЕНОМ КЛЕТКИ МЛЕКОПИТАЮЩЕГО И ВЕКТОРНАЯ СИСТЕМА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1998 |
|
RU2233334C2 |

